UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | | | | |
| (Mark One) | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended: December 31, 2024
Or | | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-35060
PACIRA BIOSCIENCES, INC.
(Exact Name of Registrant as Specified in its Charter)
| | | | | |
| Delaware | 51-0619477 |
(State or Other Jurisdiction of
Incorporation or Organization) | (I.R.S. Employer
Identification No.) |
| |
| 5401 West Kennedy Boulevard, Suite 890 |
| Tampa, Florida 33609 |
| (Address and Zip Code of Principal Executive Offices) |
| |
| (813) 553-6680 |
| (Registrant’s Telephone Number, Including Area Code) |
Securities registered pursuant to Section 12(b) of the Act: | | | | | | | | | | | | | | |
| Title of each class | | Trading symbol | | Name of each exchange
on which registered |
| Common Stock, par value $0.001 per share | | PCRX | | Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | |
| Large accelerated filer | ☒ | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ |
| | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes x No o
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. o
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No x
The aggregate market value of the registrant’s voting stock held by non-affiliates of the registrant, based upon the closing sale price of the common stock as reported on the Nasdaq Global Select Market on June 28, 2024, the last trading day of the registrant’s most recently completed second fiscal quarter, of $28.61 per share was approximately $930.1 million. Shares of common stock held by each director and executive officer (and their respective affiliates) and by each person who owns 10 percent or more of the outstanding common stock or who is otherwise believed by the registrant to be in a control position have been excluded. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of February 25, 2025, 46,276,241 shares of the registrant’s common stock, $0.001 par value per share, were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this Annual Report on Form 10-K incorporates certain information by reference from the registrant’s proxy statement for the 2025 annual meeting of stockholders to be filed with the Securities and Exchange Commission no later than 120 days after the end of the registrant’s fiscal year ended December 31, 2024.
SUMMARY OF RISK FACTORS
This risk factor summary includes those risks most material to our business, financial condition, results of operations or prospects. A full discussion of the risks outlined in this summary, as well as those risks not outlined below, appear in Part I, Item 1A. Risk Factors in this Annual Report. • Our success depends primarily on our ability to successfully commercialize EXPAREL® (bupivacaine liposome injectable suspension) and ZILRETTA® (triamcinolone acetonide extended-release injectable suspension).
• Our efforts to successfully commercialize EXPAREL and ZILRETTA are subject to many internal and external challenges.
• That the commercial success of our products may be severely hindered if we are unable to achieve and maintain adequate levels of third-party payer coverage and reimbursement for the products we offer, on reasonable pricing terms.
• The significant competition we face from other pharmaceutical, medical device and biotechnology companies.
• The regulatory approval for any approved product being limited to those specific indications and conditions for which clinical safety and efficacy have been demonstrated, and risks related to allegations of our failure to comply with such approved indications.
• Our inability to establish and maintain effective marketing and sales capabilities or enter into agreements with third parties to market and sell our products.
• Our reliance on third parties to perform many essential services for EXPAREL, ZILRETTA and iovera®° and the fact that we will rely on third parties for any other products that we commercialize.
• That we may need to increase the size of our organization and effectively manage our sales force, and we may experience difficulties in managing such growth.
• Our inability to manage our business effectively if we are unable to attract and retain key personnel.
• The potential product liability exposure we may face.
• Our failure to manufacture our products in sufficient quantities and at acceptable quality and pricing levels, or to fully comply with CGMP (as defined below).
• That we may need to expand our manufacturing operations or outsource such operations to third parties.
• Our inability to continue manufacturing adequate quantities of our products.
• That our co-production and other agreements with Thermo Fisher (as defined below) may involve unanticipated expenses and delays.
• Our reliance on third parties for the timely supply of specified raw materials and equipment for the manufacture of EXPAREL, ZILRETTA and iovera°.
• Supply chain disruptions.
• Our dependence on the global supply chain.
• That our future growth depends—in part—on our ability to identify, develop, acquire or in-license products.
• That we make substantial investments in research and development and if those investments are unsuccessful, it could materially adversely affect our business, financial condition and results of operations.
• The use of hazardous materials in our business and that we must comply with environmental laws and regulations.
• That any collaboration arrangements that we may enter into in the future may not be successful.
• The expense, length and uncertain outcomes of our trials and if our trials fail to demonstrate the safety and efficacy of our drug products or medical devices, it could prevent or significantly delay obtaining regulatory approvals.
• Our dependence on contract research organizations.
• Our dependence on clinical investigators and clinical sites to enroll patients in our clinical trials and sometimes other third parties to manage the trials and to perform related data collection and analysis.
• Periodic litigation.
• Guidelines and recommendations published by various organizations could reduce the demand for or use of our products.
• If it is determined that we are promoting or have in the past promoted the “off-label” use of our products.
• Failure to receive regulatory approval for any of our product candidates, or any delay thereof.
• The regulatory clearance process, which may result in substantial delays, unexpected or additional costs and other unforeseen factors and limitations on the types and uses of products we would be able to commercialize.
• If it is determined that our products or any of our product candidates have undesirable side effects.
• The substantial penalties we could face if we do not comply with federal, state and foreign laws and regulations relating to the healthcare business.
• The highly regulated and technically complex design, development, manufacture, supply and distribution of our products.
• Our failure to comply with the extensive regulatory requirements to which we and our products are subject.
• If there is a failure to provide adequate coverage and payment rates for EXPAREL, ZILRETTA, iovera° or any future products, or if hospitals or ASCs (as defined below) choose to use alternative therapies that are less expensive.
• Public concern regarding the safety of drug and medical device products.
• That the patents and the patent applications that we have covering our pMVL (as defined below) products are limited and our market opportunity for our product candidates may be limited by the lack of patent protection for the active ingredient itself and other formulations and delivery technology and systems that may be developed by competitors.
• That the patents and the patent applications that we have covering ZILRETTA are limited.
• That the patents and the patent applications that we have covering iovera° are limited.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 1
• That the patents and the patent applications that we have covering PCRX-201 are limited.
• Our inability to protect our intellectual property rights and that all patents will eventually expire.
• If we are sued for infringing the intellectual property rights of third parties.
• That we may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
• Servicing our indebtedness, which requires a significant amount of cash, and that we may not have sufficient cash flow from our business to pay our substantial indebtedness.
• That our TLA Credit Agreement and the Indentures (each as defined below) each impose significant operating and financial restrictions on us and certain of our subsidiaries.
• Our inability to raise the funds necessary to settle conversions of the Notes (as defined below) in cash and our future indebtedness may contain limitations on our ability to pay cash upon conversion of the Notes or limitations on our ability to repurchase the Notes.
• Our indebtedness as well as the ability to meet payment obligations under our TLA Credit Agreement and the Notes.
• That despite our current level of indebtedness, we may be able to incur substantially more debt.
• The provisions of our charter documents and Delaware law that may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
• That the price of our common stock may be subject to significant fluctuations and volatility.
• Our intention to not pay dividends on our common stock for the foreseeable future.
• That future sales in the public market or issuances of our common stock could lower the market price for our common stock.
• That raising additional funds by issuing securities would cause dilution to existing stockholders and that raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.
• Changes in global economic conditions, including, but not limited to, those driven by inflation and tariffs.
• The significant losses we have incurred since our inception and that we may incur additional losses in the future.
• A material impairment in the carrying value of intangible assets or goodwill.
• That we may need additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
• The potential significant fluctuations in our quarterly operating results.
• Our inability to successfully integrate the businesses and personnel of acquired companies and businesses, and inability to realize the anticipated synergies and benefits of such acquisitions.
• Our inability to realize the benefits from the Flexion Acquisition (as defined below), being substantially dependent on the commercial success of ZILRETTA and the cost savings resulting from the timely and effective integration of the operations of Pacira and Flexion (as defined below).
• The use of our net operating loss carryforwards and research and development tax credits being limited.
• Risks related to cybersecurity threats and incidents.
• Our failure to maintain the privacy and security of personal and business information.
• Changes in data privacy and protection laws and regulations, particularly in Europe and the State of California.
• Risks and challenges related to the use of artificial intelligence.
• A pandemic, epidemic or outbreak of a contagious disease (such as the COVID-19 pandemic), or fear of such an event.
• That we may face risks related to corporate social responsibility issues.
• Significant changes in the global climate, extreme weather conditions and water availability.
• Our international operations, which expose us to numerous and sometimes conflicting legal and regulatory requirements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 2
PACIRA BIOSCIENCES, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2024
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 3
Cautionary Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K (the “Annual Report”) and certain other communications made by us contain forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the Private Securities Litigation Reform Act of 1995, including, without limitation, statements related to: ‘5x30’; our growth and business strategy; our future outlook; contributions of new directors and executives; our intellectual property and patent terms; the Flexion Acquisition (as defined below) and the costs and benefits thereof; the acquisition of GQ Bio Therapeutics GmbH; our growth and future operating results and trends; our strategy, plans, objectives, expectations (financial or otherwise) and intentions; future financial results and growth potential, including our plans with respect to the repayment of our indebtedness, anticipated product portfolio, development programs, development of products, strategic alliances and the Non-Opioids Prevent Addiction in the Nation (“NOPAIN”) Act. For this purpose, any statement that is not a statement of historical fact should be considered a forward-looking statement. We often use the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “project,” “should,” “will,” “would” and similar expressions to help identify forward-looking statements. We cannot assure you that our estimates, assumptions and expectations will prove to have been correct. Actual results may differ materially from these indicated by such forward-looking statements as a result of various important factors, including risks relating to, among others: the failure to realize the anticipated benefits any synergies from the acquisition of GQ Bio (as defined below); the ability to successfully integrate GQ Bio into our existing business; the commercial success of GQ Bio’s high-capacity adenovirus gene therapy vector platform; future opportunities and plans for GQ Bio and its product candidates, including uncertainty of the expected financial performance of GQ Bio and its product candidates; disruption from the acquisition of GQ Bio, making it more difficult to conduct business as usual or maintain relationships with customers, employees or suppliers; the possibility that if we do not achieve the perceived benefits of the transaction as rapidly or to the extent anticipated by financial analysts or investors, the market price of our common stock could decline; risks associated with acquisitions, such as the risk that the acquired businesses will not be integrated successfully, that such integration may be more difficult, time-consuming or costly than expected or that the expected benefits of the transaction will not occur; our manufacturing and supply chain, global and U.S. economic conditions (including inflation and rising interest rates), and our business, including our revenues, financial condition, cash flow and results of operations; the success of our sales and manufacturing efforts in support of the commercialization of EXPAREL, ZILRETTA and iovera°; the rate and degree of market acceptance of EXPAREL, ZILRETTA and iovera°; the size and growth of the potential markets for EXPAREL, ZILRETTA and iovera° and our ability to serve those markets; our plans to expand the use of EXPAREL, ZILRETTA and iovera° to additional indications and opportunities, and the timing and success of any related clinical trials for EXPAREL, ZILRETTA and iovera°; the commercial success of EXPAREL, ZILRETTA and iovera°; the related timing and success of U.S. Food and Drug Administration supplemental New Drug Applications and premarket notification 510(k)s; the related timing and success of European Medicines Agency Marketing Authorization Applications; our plans to evaluate, develop and pursue additional product candidates utilizing our proprietary multivesicular liposome (“pMVL”) drug delivery technology; the approval of the commercialization of our products in other jurisdictions; clinical trials in support of an existing or potential pMVL-based product; our commercialization and marketing capabilities; our ability to successfully complete capital projects; the outcome of any litigation; the recoverability of our deferred tax assets; assumptions associated with contingent consideration payments; assumptions used for estimated future cash flows associated with determining the fair value of the Company; the anticipated funding or benefits of our share repurchase program; and factors discussed in Part I-Item 1A. Risk Factors.
The forward-looking statements included in this Annual Report represent our views as of the filing date of this Annual Report. Important factors could cause our actual results to differ materially from those indicated or implied by forward-looking statements, and as such we anticipate that subsequent events and developments will cause our views to change. Except as required by applicable law, we undertake no intention or obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, and readers should not rely on the forward-looking statements as representing our views as of any date subsequent to the date of the filing of this Annual Report.
These forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to differ materially from those expressed or implied by these statements. These factors include the matters discussed and referenced in Part I-Item 1A. Risk Factors.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 4
PART I
Item 1. Business
References
Pacira BioSciences, Inc., a Delaware corporation, is the holding company for our California operating subsidiary named Pacira Pharmaceuticals, Inc. In March 2007, we acquired Pacira Pharmaceuticals, Inc. from SkyePharma Holdings, Inc. (now Vectura Group Limited, a subsidiary of Molex Asia Holdings, Ltd.), or Skyepharma (the “Skyepharma Acquisition”). In April 2019, we acquired MyoScience, Inc., a privately held medical technology company (the “MyoScience Acquisition”), in November 2021, we acquired Flexion Therapeutics, Inc., or Flexion, a publicly traded biopharmaceutical company (the “Flexion Acquisition”) and in February 2025, we acquired GQ Bio Therapeutics GmbH, a privately-held biopharmaceutical company (the “GQ Bio Acquisition”). Unless the context requires otherwise, references to “Pacira,” “we,” the “Company,” “us” and “our” in this Annual Report refers to Pacira BioSciences, Inc., a Delaware corporation, and its subsidiaries.
Corporate Information
We were incorporated in Delaware under the name Blue Acquisition Corp. in December 2006 and changed our name to Pacira, Inc. in June 2007. In October 2010, we changed our name to Pacira Pharmaceuticals, Inc. and in April 2019, we changed our name to Pacira BioSciences, Inc. Our principal executive offices and corporate headquarters are located in Tampa, Florida.
Trademarks and Service Marks
Pacira®, EXPAREL®, ZILRETTA®, iovera®°, the Pacira logo and other trademarks or service marks of Pacira appearing in this Annual Report are the property of Pacira, and when first used in each part of this Annual Report, include the ® symbol.
This Annual Report contains additional trade names, trademarks and service marks of other companies, which may or may not appear with the ® or ™ symbol. The absence of these symbols does not in any way imply that the respective owner(s) will not assert their rights to such marks to the fullest extent under applicable law. Our use of trademarks or trade names of other companies should not suggest any endorsement, sponsorship or other relationship with or by such companies.
Overview
Our mission is to deliver innovative, non-opioid pain therapies to transform the lives of patients. We have three commercialized non-opioid treatments: EXPAREL® (bupivacaine liposome injectable suspension), a long-acting, local analgesic currently approved for postsurgical pain management; ZILRETTA® (triamcinolone acetonide extended-release injectable suspension), an extended-release, intra-articular, or IA (meaning in the joint), corticosteroid injection indicated for the management of osteoarthritis, or OA, knee pain; and iovera®°, a novel, handheld device for delivering immediate, long-acting, drug-free pain control using precise, controlled doses of cold temperature to a targeted nerve. We are also advancing the development of PCRX-201 (enekinragene inzadenovec), a novel gene therapy vector platform enabling local administration of genetic medicines with the potential to treat large prevalent diseases like OA.
Strategy
In January 2025, we launched our new 5x30 strategic growth plan (“5x30”) to accelerate our transition into an innovative biopharmaceutical and therapeutic area leader in musculoskeletal pain and adjacencies. With 5x30, we intend to achieve the following five key objectives by 2030: (i) deliver our products to more than three million patients annually; (ii) grow product revenues by a double-digit compounded annual growth rate; (iii) achieve a five percentage-point improvement in gross margins over 2024; (iv) advance an innovative pipeline with at least five programs in clinical development and (v) establish five partnerships—including pipeline and commercial agreements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 5
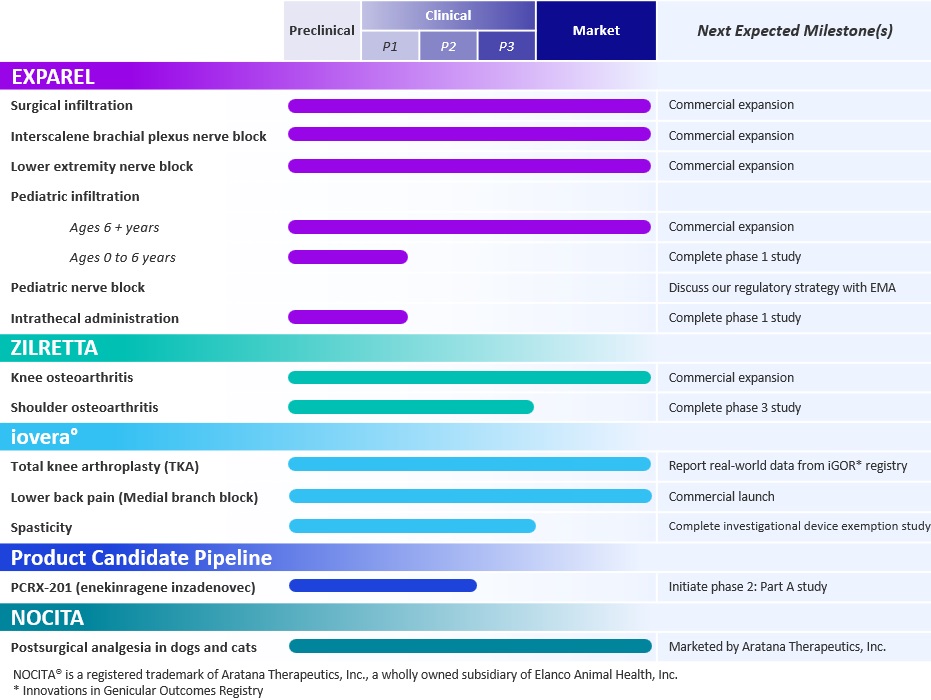
Product Portfolio and Product Candidate Pipeline
Our current product portfolio and product candidate pipeline, along with anticipated milestones over the next 12 to 18 months, are summarized in the table below:
Our Commercial Products
EXPAREL (bupivacaine liposome injectable suspension)
EXPAREL was approved by the FDA in October 2011 and was commercially launched in the U.S. in April 2012. EXPAREL is a long-acting, non-opioid proven to manage postsurgical pain. EXPAREL is the only product indicated for local analgesia via infiltration in patients aged six years and older and regional analgesia via interscalene brachial plexus nerve block, sciatic nerve block in the popliteal fossa, and adductor canal block in adults (safety and efficacy have not been established in other nerve blocks). In November 2020, the European Commission, or EC, granted marketing authorization for EXPAREL as a brachial plexus block or femoral nerve block for treatment of post-operative pain in adults, and as a field block for treatment of somatic post-operative pain from small- to medium-sized surgical wounds in adults and children aged six years and older.
EXPAREL consists of bupivacaine, an amide-type local anesthetic, encapsulated in our pMVL drug delivery technology, which delivers bupivacaine over time for extended analgesia. We believe EXPAREL addresses a significant medical need for a safe and effective long-acting non-opioid postsurgical analgesic and plays a significant role in opioid minimization strategies. EXPAREL is designed for recovery with minimal opioid use by: (i) delivering targeted local analgesia at the surgical site; (ii) reliably releasing bupivacaine over time for prolonged analgesia; (iii) eliminating the need for catheters and pumps that may hinder recovery and (iv) providing long-lasting pain control while reducing the need for opioids.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 6
Net product sales of EXPAREL were $549.0 million, $538.1 million and $536.9 million for the years ended December 31, 2024, 2023 and 2022, respectively. For the years ended December 31, 2024, 2023 and 2022, net product sales of EXPAREL accounted for 78%, 80% and 81% of our total revenues, respectively.
ZILRETTA (triamcinolone acetonide extended-release injectable suspension)
ZILRETTA is the first and only extended-release, single-shot corticosteroid injection for patients with OA knee pain. ZILRETTA uses a proprietary extended-release microsphere technology to slowly and continuously releases triamcinolone acetonide, or TA, a commonly administered, immediate-release corticosteroid into the knee for approximately three months to provide significant pain relief for 12 weeks, with some people experiencing pain relief through 16 weeks. ZILRETTA was approved by the FDA in October 2017 and launched in the U.S. shortly thereafter.
Net product sales of ZILRETTA were $118.1 million, $111.1 million and $105.5 million, for the years ended December 31, 2024, 2023 and 2022, respectively.
iovera°
The iovera° system is a non-opioid handheld cryoanalgesia device used to deliver precise, controlled doses of cold temperature to targeted nerves to produce an immediate, long-lasting neurolytic block that interrupts the pain-transmitting signals of a peripheral nerve. The effect on the nerve is temporary, providing months of pain relief until the nerve regenerates and function is restored over time. The structural components of the nerve are not affected by iovera° treatment.
It is FDA 510(k) cleared in the U.S., has a CE mark in the E.U. and is cleared for marketing in Canada for the blocking of pain. The iovera° system is highly complementary to EXPAREL and ZILRETTA as a locally administered, non-opioid therapy that alleviates pain using a non-pharmacological nerve block to disrupt pain signals being transmitted to the brain from the site of injury or surgery. It is also indicated for the relief of pain and symptoms associated with arthritis of the knee for up to 90 days. Net product sales of iovera° were $22.8 million, $19.7 million and $15.3 million for the years ended December 31, 2024, 2023 and 2022, respectively.
EXPAREL Clinical Benefits
We believe EXPAREL can replace the use of bupivacaine delivered via elastomeric pumps as the foundation of a multimodal regimen for long-acting postsurgical pain management. Based on our clinical data, EXPAREL:
•provides long-lasting local or regional analgesia;
•is a ready-to-use formulation;
•expands easily with saline or lactated Ringer’s solution to reach a desired volume;
•can be administered for local analgesia via infiltration and for regional analgesia via field block, as well as brachial plexus nerve block, sciatic nerve block in the popliteal fossa and adductor canal block; and
•facilitates treatment of a variety of surgical sites.
We believe EXPAREL is a key component of long-acting postsurgical pain management regimens that reduce the need for opioids. Based on our clinical studies as well as data from robust, retrospective real-world published studies, EXPAREL is associated with significantly reduced opioid use, fewer emergency department visits and shorter length of stay while improving postsurgical pain management.
In our Phase 3 hemorrhoidectomy trial, EXPAREL:
•delayed the median time to opioid rescue medication to 15 hours for patients treated with EXPAREL versus one hour for patients treated with placebo;
•significantly increased the percentage of patients requiring no opioid rescue medication through 72 hours post-surgery to 28%, compared to 10% for placebo;
•resulted in 45% less opioid usage through 72 hours post-surgery compared to placebo; and
•increased the percentage of patients who were pain free at 24 hours post-surgery compared to placebo.
In our Phase 3 trial as an interscalene brachial plexus nerve block for upper extremity surgeries, EXPAREL:
•decreased total opioid consumption by 78% (p<0.01) from zero to 48 hours after surgery;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 7
•reduced pain scores by 46% versus placebo (p<0.01); and
•allowed 13% of patients who received EXPAREL to remain opioid-free for 48 hours after surgery (p<0.01) compared to one opioid-free patient in the placebo arm.
In our Phase 3 trial as an adductor canal block in patients undergoing total knee arthroplasty, or TKA, EXPAREL:
•achieved the primary endpoint by significantly reducing cumulative pain scores from zero to 96 hours after surgery compared with bupivacaine HCl (p<0.01); and
•achieved its secondary endpoint with a statistically significant reduction in postsurgical opioid consumption through 96 hours (p<0.01) compared with bupivacaine HCl.
In our Phase 3 trial as a sciatic nerve block in the popliteal fossa in patients undergoing bunionectomy, EXPAREL:
•achieved the primary endpoint by significantly reducing cumulative pain scores from zero to 96 hours after surgery compared with bupivacaine HCl (p<0.01); and
•achieved its secondary endpoints with statistically significant reductions in postsurgical opioid consumption through 96 hours (p<0.01) and the percentage of opioid-free subjects (p<0.01) compared with bupivacaine HCl.
EXPAREL can improve patient satisfaction and outcomes. We believe EXPAREL:
•provides effective pain control without the need for expensive and difficult-to-use delivery technologies that extend the duration of action for bupivacaine, such as elastomeric pumps, or opioids administered through patient-controlled analgesia, or PCA, when used as part of a multimodal postsurgical pain regimen;
•reduces the need for patients to be constrained by elastomeric pumps and PCA systems, which are barriers to earlier ambulation and may introduce catheter-related issues, including infection; and
•promotes maintenance of early postsurgical pain management, which may reduce the time to discharge.
Key EXPAREL Markets
Orthopedics
EXPAREL is used across multiple orthopedic procedures, including joint reconstruction, shoulder, spine, extremity procedures, and hip fractures. In November 2023, the FDA approved our sNDA to expand the EXPAREL label to include administration in adults as an adductor canal block and a sciatic nerve block in the popliteal fossa. An adductor canal block is used for anesthesia and analgesia for surgeries of the knee, medial lower leg and ankle. A sciatic nerve block in the popliteal fossa is used for anesthesia and analgesia for foot, ankle, Achilles tendon and other lower leg surgeries. These new indications provide additional flexibility in the use of EXPAREL as a regional analgesic for more than three million lower extremity procedures annually, further increasing the utility of EXPAREL for major orthopedic procedures.
Total joint arthroplasties are expected to grow rapidly in the coming years with a significant migration of these procedures from the inpatient hospital setting to outpatient sites of care. EXPAREL-based regional analgesia as part of multimodal pain management protocols in enhanced recovery after surgery, or ERAS, pathways is supporting this surgical migration. The clinical and economic benefits of EXPAREL in total joint arthroplasty procedures have been demonstrated in clinical studies with EXPAREL use associated with significant reductions in opioid consumption, well-controlled pain management, shorter recovery time, same-day discharge to home and high patient satisfaction.
EXPAREL administered as a brachial plexus nerve block is a key and growing part of our business. An EXPAREL brachial plexus block provides pain coverage for the upper quadrant for use in rotator cuff, shoulder arthroplasty, elbow, wrist and hand procedures. Like other regional field blocks, anesthesiologists see the strong advantages of using interscalene brachial plexus blocks as a vehicle for shifting procedures to the outpatient setting by replacing antiquated pumps and catheters, which often become dislodged and prevent a procedure from taking place in a 23-hour site of care. Additionally, EXPAREL reimbursement is improving as payers and self-insured employers continue to drive the shift from inpatient to outpatient care for a variety of surgeries.
EXPAREL is being adopted in an increasing number of spine surgeries as a key component of a multimodal pain management solution enabling rapid recovery after surgery and a reduced reliance on opioids, which have been the mainstay in postsurgical pain control in the spine area for decades. Two important patient groups are driving the spine market: first, pediatric cases, like adolescent scoliosis patients, who are undergoing highly invasive surgeries and who until very recently
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 8
only had opioids available to treat their pain, and second, adult degenerative patients who are often coming into surgery opioid-tolerant and who may have already had multiple back surgeries. Managing postsurgical pain in these adult degenerative patients can be challenging due to their established opioid tolerance, but by utilizing EXPAREL, healthcare providers can control their pain with a non-opioid approach, and when feasible based on surgical intervention and patient characteristics, move many historical inpatient procedures to the 23-hour stay environment.
Abdominal and Colorectal
A variety of truncal blocks are used in abdominal and colorectal procedures. Transversus abdominis plane, or TAP, and erector spinae plane blocks represent a significant market where EXPAREL is providing long-acting pain control for abdominal and colorectal surgeries and supporting the migration of these procedures to the 23-hour setting. We expect EXPAREL field blocks will continue to be the foundation of enhanced recovery protocols across various abdominal and colorectal procedures.
Women’s Health
There is a significant and growing demand among women for managing pain with non-opioid options. Opioid addiction in women is growing at an alarming rate and studies have shown that women are 40 percent more likely than men to become newly persistent users of opioids following surgery. Women’s Health is a key target market as anesthesia-driven EXPAREL-based TAP and pectoralis blocks take hold as institutional protocol for Cesarean section, abdominoplasty, gynecologic oncology, mastectomy and breast reconstruction procedures.
Cardiothoracic
Cardiothoracic surgery is considered one of the most painful types of surgical procedures for both open and minimally invasive procedures. As a result, opioids are widely used but are often inadequate. Pain may persist for prolonged periods following surgery with 35 percent of patients reporting persistent thoracic pain one year post cardiac surgery. Opioids are used extensively after cardiothoracic surgery and nearly one in ten patients will continue to use opioids over 90 days after surgery. Regional anesthesia approaches have been evolving, with EXPAREL replacing thoracic epidurals as an alternative method of producing long-lasting postsurgical analgesia.
Pediatrics
In March 2021, the FDA approved our sNDA to expand the EXPAREL label to include use in patients six years of age and older for single-dose infiltration to produce postsurgical local analgesia. EXPAREL is the first and only FDA-approved long-acting local analgesic for the pediatric population as young as age six. In November 2022, both the EMA’s Committee for Medicinal Products for Human Use, or CHMP, and the Medicines and Healthcare Products Regulatory Agency, or MHRA, approved marketing authorization for an expanded indication of EXPAREL to include use in children aged six years and older as a field block for treatment of somatic post-operative pain from small- to medium-sized surgical wounds.
Opioids, short-acting local anesthetics and catheter-based devices have been the historical mainstay in pediatric postsurgical pain management despite safety implications and limited studies in children. The risks and complications of adult-based pain management approaches may be magnified in children with 50 percent of children reporting moderate to severe pain in the hospital after surgery and 20 percent of children reporting chronic pain 12 months after surgery.
EXPAREL is redefining the paradigm of care for postsurgical pain management in children as the market’s only clinically proven safe alternative for long-acting, non-opioid postsurgical pain control in children aged 6 and over. There are approximately one million pediatric procedures per year in the U.S. We are working with prominent thought leaders who are providing a rapid transfer of best-practice for establishing EXPAREL-based protocols as the new standard of care.
Third Molar (Wisdom Tooth) Procedures
Third molar (wisdom tooth) extractions are among the most common dental procedures in the U.S. and are performed in up to 5 million patients every year. Oral surgery, including third molar extraction, is associated with a defined period of pain and discomfort that traditionally leads to prescriptions for opioids. A large retrospective review of the Medicaid database found that of 2.8 million patients who underwent surgical tooth extraction, 1.2 million—or roughly 42 percent—filled a prescription for opioids within seven days after surgery, with a median of 120 morphine milligram equivalents dispensed per patient. A study of the effect of EXPAREL on postoperative opioid prescribing after third molar extraction showed that patients who received EXPAREL were prescribed significantly fewer opioids, including refills, compared to those who did not receive EXPAREL. The study, A Retrospective Cross-Sectional Study of the Effect of Liposomal Bupivacaine on Postoperative Opioid
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 9
Prescribing After Third Molar Extraction, was published in The Journal of Oral and Maxillofacial Surgery in July 2021. In this retrospective analysis, researchers reviewed data from 600 patients who underwent third molar extractions between 2012 and 2018. De-identified data from 300 patients who received EXPAREL were compared to data from 300 patients who did not receive an infiltration of EXPAREL. Data from two outpatient oral surgery centers were included in this analysis. Patients in the EXPAREL treatment group received:
•59 percent fewer opioids, including refills, compared to patients in the non-EXPAREL group (p<0.0001)
•Fewer additional opioid prescriptions compared to the non-EXPAREL group (3.3% of patients required a refill vs. 7.7% of patients, respectively)
In September 2022, we announced a joint initiative with Sevāredent Sourcing Solutions, or Sevāredent, a GPO that creates a competitive advantage for like-minded dental organizations through vendor partnerships that drive supply chain value and efficiencies, to provide expanded access to EXPAREL for patients undergoing oral and maxillofacial (OMFS) procedures ranging from third molar and full mouth extractions to dentures and implants. This collaboration, which advances Sevāredent’s goal of improving patient outcomes and reducing exposure to opioids and their associated risks, provides easy access to EXPAREL—including comprehensive product training and onboarding support from Pacira—for more than 1,800 dental offices across the U.S.
EXPAREL Economic Benefits
Retrospective, real-world studies highlight EXPAREL’s opioid-sparing and economic benefits:
•In October 2024, Surgery for Obesity and Related Diseases published a retrospective, cohort study of 4,298 patients undergoing laparoscopic sleeve gastrectomy. Patients who received EXPAREL were matched to patients who received non-EXPAREL analgesia. The EXPAREL cohort was associated with significantly lower opioid use and opioid related adverse events throughout their hospital stay, as well as a shorter length of hospital stay. Further, the odds of 30-day hospital readmission and incidence of opioid use disorder after discharge were significantly lower for the EXPAREL group.
•In June 2024, Plastic and Reconstructive Surgery Global Open published a retrospective, cohort study of 1,017 patients undergoing abdominal flap breast reconstruction. Patients who received EXPAREL were matched 2-to-1 with patients who received bupivacaine. The EXPAREL cohort was associated with significantly lower opioid use throughout their hospital stay. Further, three-month hospital readmission rates and outpatient clinic or office visits were also lower for the EXPAREL group.
•In May 2024, Surgery Open Science published results from a real-world study of 8,794 patients undergoing colorectal resection surgery. Patients who received EXPAREL were matched 1-to-1 with patients who did not. Average opioid utilization through six months after surgery, length of stay, outpatient or emergency department visits and hospital readmissions were all significantly lower in the EXPAREL cohort.
•In May 2024, The Spine Journal published results from a retrospective, comparative study of 1,524 patients undergoing outpatient spine surgery. Patients who received EXPAREL were matched 1-to-3 with patients who did not. The use of EXPAREL was associated with reduced opioid use at the hospital with significantly lower emergency department visits for 60 days after discharge.
ZILRETTA Clinical Benefits
ZILRETTA combines a commonly administered steroid, TA, with a proprietary, extended-release microsphere technology to administer extended therapeutic concentrations in the joint and persistent analgesic effect.
Based on the strength of its pivotal and other clinical trials, we believe that ZILRETTA represents an important treatment option for the millions of patients in the U.S. in need of safe and effective extended relief from OA knee pain. The pivotal Phase 3 trial showed that ZILRETTA significantly reduced OA knee pain for 12 weeks, with some people experiencing pain relief through 16 weeks. We believe that ZILRETTA holds the potential to become the corticosteroid of choice given its high patient satisfaction, with up to four months of reliable OA knee pain relief and fewer office visits. ZILRETTA also has a strong safety and pharmacokinetic profile. It remains localized in the knee, which allows for fewer systemic effects, including significantly lower hypoglycemia. This represents a meaningful opportunity as 14 percent of patients with OA also have diabetes. It is also the first and only extended-release corticosteroid on the market. In September 2021, the American Association of Orthopaedic Surgeons, or AAOS, updated its evidence-based clinical practice guidelines, finding ZILRETTA can improve patient outcomes over traditional immediate-release corticosteroids.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 10
We launched a Phase 3 registration study in 2024 that evaluates the safety and efficacy of ZILRETTA for the management of OA pain of the shoulder. If the study is successful, we plan to seek approval to expand the ZILRETTA label to include OA pain of the shoulder.
iovera° Clinical Benefits
There is a growing body of clinical data demonstrating success with iovera° treatment for a wide range of chronic pain conditions. Some of our strongest data relates directly to the improvement of OA pain of the knee. In a pivotal trial evaluating iovera° for knee OA pain, the majority of the patients suffering from OA pain of the knee experienced pain relief up to 150 days after being treated with iovera°.
Surgical intervention is typically a last resort for patients suffering from knee OA pain. Treatment with iovera° has also demonstrated effectiveness for managing pain associated with knee replacements. Specifically, findings demonstrated reductions in opioids, including:
•The daily morphine equivalent consumption in the per protocol group analysis was significantly lower at 72 hours (p<0.05), 6 weeks (p<0.05) and 12 weeks (p<0.05).
•Patients who were administered iovera° were far less likely to take opioids six weeks after surgery. The number of patients taking opioids six weeks after TKA in the control group was three times the number of patients taking opioids in the cryoanalgesia group (14 percent vs. 44 percent, p<0.01).
•Patients in the iovera° group demonstrated a statistically significant reduction in pain scores from their baseline pain scores at 72 hours (p<0.05) and at 12 weeks (p<0.05).
We believe these data validate iovera° as a clinically meaningful non-opioid alternative for patients with knee OA as well as those undergoing TKA, and that iovera° offers the opportunity to provide patients with non-opioid pain control well in advance of any necessary surgical intervention through a number of key product attributes:
•iovera° is safe and effective with immediate pain relief that can last for months as the nerve regenerates over time;
•iovera° is repeatable, with no diminishing effectiveness over time and repeat use;
•The iovera° technology does not risk damage to the surrounding tissue;
•iovera° is a convenient handheld device with a single-use procedure-specific Smart Tip; and
•iovera° can be delivered precisely using imaging guidance or an anatomical landmark.
A study published in 2021 that included 267 patients undergoing TKA (169 who underwent cryoneurolysis with iovera° compared to 98 patients who did not receive iovera° treatment) showed that patients who were treated with iovera° had 51% lower daily morphine milligram equivalents during their hospital stay and a 22% lower mean pain score versus those who were not. In addition, the iovera° group had greater function at discharge, a shorter length of hospital stay and received significantly fewer opioids, including discharge prescriptions at week 2 and week 6 after surgery.
In September 2021, the AAOS updated its evidence-based clinical practice guidelines, reporting that denervation therapy—including cryoneurolysis—may reduce knee pain and improve function in patients with symptomatic OA of the knee.
We are currently sponsoring a prospective, real-world registry called the Innovations in Genicular Outcomes Registry, or iGOR, which is a patient focused registry governed in collaboration with a steering committee of scientific experts that evaluates clinical, economic- and health-related patient-reported outcomes in patients who have received any treatment for knee OA pain, including TKA, for a minimum of 18 months. A unique feature of iGOR is that if patients receive additional treatments for OA, data capture resets so that outcomes of their treatment journey can be followed over multiple years. Unlike in clinical studies, treatment decisions in iGOR are decided by physicians and patients in a shared decision-making manner rather than being driven by treatment assignment, so that outcomes are truly those from real-world applications. The iGOR registry is tracking outcomes of iovera°, ZILRETTA and EXPAREL, as well as comparator treatments. Early outcomes from iGOR have shown that patients who receive iovera° prior to TKA have less pain, improved function and improved sleep for six months after surgery versus patients who do not receive iovera°. As of November 2024, more than 2,000 patients have enrolled in the iGOR registry across 12 participating sites in the U.S.
In addition, a pilot randomized control trial evaluating iovera° for the treatment of lower back pain showed that it had significantly greater improvements in pain and disability, and required fewer injections over a year, compared to patients who were treated with radiofrequency ablation.
Beyond treatment for pain, observational data has been presented at multiple congresses showing the effectiveness of iovera° for the treatment of upper limb spasticity over 90 days by targeting motor nerves. We currently have a pivotal trial underway to demonstrate the efficacy and safety of iovera° for treating spasticity.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 11
In December 2024, the FDA cleared our 510(k) submission to market a new Smart Tip designed to access the medial branch nerves to administer a medial branch block to manage chronic low back pain. Before the approval of this new Smart Tip, the iovera° portfolio consisted of either a three-pronged Smart Tip featuring 8.5mm long 27-gauge needles to treat superficial nerves or a single, 90mm long, 20-guage Smart Tip ideally suited to treat deeper nerves. It is most commonly used to treat knee pain, but frequently used to manage pain in the hip, shoulder, chest, foot and ankle, and more. Now, this new, 25-gauge 180mm Smart Tip allows for the treatment of deeper nerves, such as the medial branch nerve and is specifically designed so that it can relieve chronic low back pain associated with facet mediated pain. This longer-needle Smart Tip is uniquely designed for use through a cannula or introducer, providing the ability for ice ball formation at deeper peripheral nerves.
Chronic low back pain remains a pervasive health challenge in the U.S.:
•Back pain is the leading cause of disability nationwide.
•Back pain is also the most common reason for extended work absences.
•Chronic back pain is the number one indication for opioid prescriptions, often leading to dependency and abuse.
•Annually, 28 to 30 million Americans seek treatment for chronic back pain, yet only 2 to 3 million undergo interventional procedures.
With the introduction of this new iovera° Smart Tip, Pacira aims to address these gaps and elevate the standard of care. This FDA-cleared innovation offers a compelling alternative to conventional treatments such as radiofrequency ablation, or RFA, which has substantial limitations. With RFA, patients may not get the effects of pain relief until 1-2 weeks after treatment, further the intense heat can damage surrounding tissue and blood vessels, and tissue damage may lead to painful neuritis (inflammation in the nerves).
The Osteoarthritis Market
OA is the most common form of arthritis. It is also called degenerative joint disease and occurs most frequently in the hands, hips and knees. With OA, the cartilage within a joint begins to break down and the underlying bone begins to change. These changes usually develop slowly and worsen over time. OA can cause pain, stiffness and swelling. In some cases, it also causes reduced function and disability—some people are no longer able to do daily tasks or work. According to the CDC, OA affects over 32.5 million adults in the U.S.
The lifetime risk of developing symptomatic knee OA is 45 percent according to the Arthritis Foundation. The prevalence of symptomatic knee OA increases with each decade of life, with the annual incidence of knee OA being highest between age 55 and 64 years old. There are 14 million individuals in the U.S. who have symptomatic knee OA, and nearly two million are under the age of 45. Surgical intervention is typically a last resort for patients suffering from OA of the knee.
With ZILRETTA, we now offer clinicians the flexibility to individualize OA knee pain treatment with either ZILRETTA or a drug-free nerve block with iovera° based on patient factors and preference, physician training, site of care and reimbursement considerations.
Label and Global Activities
EXPAREL
•Pediatrics. We are launching a Phase 1 pharmacokinetic study after which we would launch a registration study to support expansion of the EXPAREL single-dose infiltration label to include patients under six years of age. If successful, we expect this study, followed by a Phase 3 study, will support expansion of the EXPAREL labels in the U.S. and E.U. We are also discussing with the FDA, EMA and Medicines and Healthcare Products Regulatory Agency (MHRA) our regulatory strategy for EXPAREL administered as a nerve block in the pediatric setting. We received notification from the FDA in October 2023 that our pediatric studies requirement had been waived for the indication of brachial plexus interscalene nerve block to produce postsurgical regional analgesia in pediatric patients as well as sciatic nerve block in the popliteal fossa and adductor canal block indications in October 2024.
•Global activities. Our products are currently only marketed in the U.S. We believe there is an opportunity in certain key markets outside of the U.S. where our products can be financially viable and deliver value. We are actively seeking potential commercial partners to realize that potential.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 12
ZILRETTA
We believe ZILRETTA’s extended-release profile may also provide effective treatment for OA pain of the shoulder, and in 2024 launched a Phase 3 trial investigating ZILRETTA as a treatment for OA pain of the shoulder. The shoulder study will compare ZILRETTA to immediate release TA.
iovera°
In 2022, we launched a next-generation iovera° handheld device, which we believe is more efficient, provides more consistent treatment, is easier for providers to use and is more durable. We have a plan to develop new iovera° Smart Tips for certain procedures and are near completion on developing a specific tip for a medial branch block for treating chronic low-back pain, as well as managing pain related to other spine surgical procedures. We are also preparing to launch an investigational device exemption (IDE) study to evaluate iovera° as an alternative treatment for the debilitating condition of spasticity. Additionally, we began selling iovera° in Europe through a contracted sales force in 2022.
We are also developing new iovera° Smart Tips to expand the use of iovera° to other chronic and acute pain applications, such as foot and ankle pain, elbow and wrist pain and pediatric care.
A pilot randomized controlled trial which compared 30 patients who underwent bilateral medial branch blocks with iovera° versus radiofrequency plus steroid injection showed a significant improvement in pain and disability index scores in patients who received an iovera° treatment versus radiofrequency. These data were presented at the 2024 North American Neuromodulation Society and the Association of Academic Physiatrists congresses.
Clinical Development Programs
PCRX-201
PCRX-201 (enekinragene inzadenovec) is our novel gene therapy vector platform in clinical development as a treatment for OA of the knee. Its innovative high-capacity adenovirus, or HCAd, design, manufacturing process, and local administration may solve many of the challenges that have made gene therapy inaccessible for common diseases like OA. Key features of the HCAd vector that support its use for treatments targeting large prevalent diseases like OA:
•Medicine where it matters. PCRX-201 is injected locally into the knee joint to boost cellular interleukin 1 receptor antagonist (IL-1Ra) production and block IL-1 pathway activation, significantly reducing chronic inflammation.
•Overcomes gene therapy pitfalls. PCRX-201’s innovative HCAd vector is more efficient at delivering genes into cells than other vectors, which means less medication needed to achieve the desired effect.
•Protein production only when needed. PCRX-201 uses an inflammation-responsive promoter to only produce IL-1Ra when needed, mimicking the body’s natural response to inflammation.
•Attractive cost of goods profile, Smaller doses, localized administration and scalable manufacturing processes translate to an attractive cost of goods profile because we could potentially make many thousands of doses in a single batch.
IL-1’s Role in Inflammation
The IL-1 pathway triggers inflammation in response to pathogens and cellular stress. IL-1Ra is a core regulator of this pathway and helps keep inflammation in balance by turning off the IL-1 pathway when it is not needed. As people age, their bodies have a more challenging time maintaining that balance. As a result, people develop chronic IL-1-driven inflammation that eventually causes joint damage and pain.
It has been well established that chronic IL-1 activation is an underlying driver of knee inflammation, joint degeneration and pain. By targeting it, we are able to target a root cause of disease rather than just alleviating only the symptoms of OA. FDA-approved drugs, such as anakinra and canakinumab, have successfully targeted the IL-1 pathway in other joint inflammatory conditions, proving that blocking its activation is a well-validated, de-risked strategy for reducing inflammation. Existing drugs that target IL-1 are not practical for OA because they would require very high doses or daily injections directly into the joint. The PCRX-201 mechanism of action could potentially solve the limitations experienced with existing drugs.
PCRX-201 Mechanism of Action
PCRX-201 is a gene therapy that boosts cellular IL-1Ra production, which blocks IL-1 pathway activation and dampens down chronic inflammation in the knee. It uses an inflammation-responsive promoter to only produce IL-1Ra when needed,
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 13
mimicking how the body naturally responds to inflammation. It is delivered directly into the knee joint capsule providing durable pain relief where it matters most and with no to minimal systemic exposure.
PCRX-201 Safety and Efficacy
In November 2024, we presented two-year safety and efficacy data at the American College of Rheumatology’s annual ACR Convergence meeting in Washington, DC. The new data is derived from an open-label, Phase 1 trial investigating the safety and efficacy of PCRX-201 administered via ultrasound-guided IA injection in 72 patients with moderate to severe osteoarthritis of the knee (OAK) graded at 2, 3, or 4 on the Kellgren-Lawrence scale, a semiquantitative method for evaluating the severity of OA on a scale of 0-4. Participants were broken into two cohorts. The first cohort received one of three doses of PCRX-201. The second cohort received concurrent pretreatment with an IA corticosteroid (methylprednisolone 40 mg), a technique common in gene therapy dosing to improve tolerability and gene transfer. Pain and function benefits were observed at all doses and across both cohorts over the full two years studied, with patients in the second cohort achieving greater pain reduction and fewer adverse events, or AEs. Additional results in the pretreated cohort, across all doses, included:
•48% to 65% improvement in pain from baseline, as measured by the Western Ontario and McMaster Universities Arthritis Index-A (WOMAC-A)
•53% to 72% improvement in stiffness from baseline, as measured by WOMAC-B:
◦Improvements in function from baseline, as measured by the KOOS Activities of Daily Living (ADL) scale, that were similar to improvements in WOMAC-A and WOMAC-B
•By 16 weeks more than 70% of participants achieved greater than 50% reductions from baseline pain.
No serious treatment-emergent AEs related to the treatment or procedure were reported regardless of steroid pretreatment or dose level administered. Treatment-related joint effusions (swelling) were the most common AE, occurring in 36% of patients who received steroid pretreatment versus 61% of patients who were not pretreated. The majority of effusions were mild to moderate in severity and resolved in a median of 33 days among patients in the pretreated group.
While other therapies typically provide relief for three to six months, PCRX-201 has already set a new standard with a year or more of sustained pain relief from a single injection. Given these highly encouraging Phase 1 data, we opened enrollment in a randomized, double-blind Phase 2 clinical study in knee OA in February 2025.
In February 2024, the FDA granted PCRX-201 a Regenerative Medicine Advanced Therapy, or RMAT, designation. Established under the 21st Century Cures Act, RMAT designation is a dedicated program designed to expedite the development and review processes for promising therapies, including genetic therapies, that are intended to treat, modify, reverse or cure a serious or life-threatening disease or condition, and for which preliminary clinical evidence indicates that the drug or therapy has the potential to address an unmet medical need. PCRX-201 is the first gene therapy product candidate to receive RMAT designation for OA.
HCAd Vector Platform
The HCAd vector platform solves many of the challenges in the field of genetic medicine that have prevented its utilization in treating common diseases like OA. Key features include:
•The HCAd vector is much more efficient at delivering genes into cells compared to many other gene therapies that rely on adenovirus associated virus, or AAV, vectors. As a result, the desired effect can be achieved with much smaller doses.
•The vector used in the HCAd platform can carry up to 30,000 base pairs of DNA, which enables gene therapy with multiple or larger genes compared to AAV vectors.
•Genetic medicines based on the HCAd platform can be administered locally and have the potential for redosing at therapeutically appropriate intervals.
•Lower dose levels and efficient delivery of genes into cells means that thousands of doses can potentially be produced in a single batch. As a result, therapies built on the HCAd platform are expected to have a commercially attractive and viable cost of goods profile.
In addition to PCRX-201 which is based on the HCAd vector platform, we have identified other well-validated cytokines that could be the basis for additional locally administered genetic therapies using the HCAd vector platform.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 14
External Innovation
In parallel to our internal clinical programs, we are pursuing innovative acquisition targets that are complementary to EXPAREL, ZILRETTA and iovera° and are of great interest to the surgical and anesthesia audiences we are already calling on today. We are using a combination of strategic investments, in-licensing and acquisition transactions to buildout a pipeline of innovation to improve patients’ journeys along the neural pain pathway. The strategic investments we have made to support promising early-stage platforms are summarized below:
| | | | | | | | | | | |
| Company | Development Stage | Description of Platform Technology | Potential Therapeutic Areas |
| CarthroniX, Inc. | Phase 1-Ready | CX-011, a small molecule modulator of gp130 formulated as an IA injection designed to slow joint degeneration by mediating IL-6 cytokines | Knee OA |
| Genascence Corporation | Phase 1b | Adeno-associated virus (AAV) based gene therapy engineered to deliver Interleukin-1 Receptor Antagonist (IL-1Ra) to target cells in joint(s) | Knee OA |
| Spine BioPharma, LLC | Phase 3 | SB-01, a 7-amino acid chain peptide that binds to and induces down regulation of transforming growth factor, beta 1 (TGFβ1) | Degenerative disc disease (DDD) |
Customer-Facing Organization
We have built our sales and marketing organization to commercialize our products. Our primary target audiences are healthcare practitioners who influence pain management decisions including anesthesiologists, surgeons, pharmacists and physician extenders (including physician assistants, nurse practitioners and registered nurses).
Our customer-facing team, consisting of sales representatives, account managers, scientific and medical affairs personnel and reimbursement and market access professionals, executes on a full range of activities to broaden the use of our non-opioid products for pain management, including:
•providing publications and abstracts showing clinical efficacy and safety, health outcomes and review articles;
•working in tandem with hospital staff, such as anesthesiologists, surgeons, heads of quality, pharmacists, executives and registered nurses, to provide access and resources for drug utilization or medication use evaluations and health outcomes studies, which provide retrospective and prospective analyses for our hospital customers using their own hospital data to demonstrate the true cost of opioid-based postsurgical pain control;
•working with KOLs and advisory boards to address topics of best practice techniques as well as guidelines and protocols for the use of our products, meeting the educational and training needs of our physician, surgeon, anesthesiologist, pharmacist and registered nurse customers;
•undertaking education initiatives such as center of excellence programs; preceptorship programs; opioid-sparing and ERAS pain protocols and predictive models for enhanced patient care; interactive discussion forums; patient education platforms leveraging public relations, advocacy partnerships and public affairs efforts where appropriate; web-based training and virtual launch programs;
•collaborating with healthcare providers towards improving the knowledge and management of pain in surgical and OA patients with a focus on opioid risk and non-opioid alternatives and engaging our field-based medical teams in system-wide partnerships to address the national opioid epidemic, with a goal of studying alternative postsurgical pain management options that focus on optimization and opioid alternative strategies; and
•facilitating reimbursement and the shift of procedures to hospital outpatient and ambulatory surgical center, or ASC, sites of care.
Other Agreements
Flexion Acquisition
On November 19, 2021, we completed the Flexion Acquisition pursuant to an Agreement and Plan of Merger (the “Flexion Merger Agreement”), dated as of October 11, 2021, by and among us, Oyster Acquisition Company Inc., a Delaware corporation and wholly owned subsidiary of Pacira (“Purchaser”), and Flexion. Following the completion of a successful tender offer for the shares of Flexion’s common stock, Purchaser merged with and into Flexion with Flexion surviving as a wholly
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 15
owned subsidiary of Pacira. We changed the name of Flexion to Pacira Therapeutics, Inc. after completing the merger. As part of the Flexion Acquisition, we acquired ZILRETTA, the first and only extended-release, IA (meaning in the joint) injection indicated for the management of OA knee pain. ZILRETTA is a non-opioid therapy that employs a proprietary microsphere technology to provide pain relief. The addition of ZILRETTA to our innovative non-opioid product portfolio directly aligned with our mission to deliver innovative, non-opioid pain therapies to transform the lives of patients.
Initially, the total consideration for the Flexion Acquisition was approximately $578.8 million consisting of: (i) $448.5 million of cash paid to Flexion shareholders and to settle restricted stock units and certain stock options; (ii) an $85.1 million cash payment of Flexion debt not assumed by Pacira and (iii) $45.2 million of estimated fair value of contingent consideration related to contingent value rights that were issued to Flexion shareholders and certain equity award holders in conjunction with the Flexion Acquisition. We funded the cash portion of the purchase price with cash on hand, and the consideration is subject to adjustments based on the estimated fair value of the potential milestone payments. As of December 31, 2024, these contingent value rights could aggregate up to a total of $372.3 million if certain regulatory and commercial milestones are met. For more information, see Note 11, Financial Instruments, to our consolidated financial statements included herein.
Research Development Foundation
Pursuant to an agreement with the Research Development Foundation, or RDF, we were required to pay RDF a low single-digit royalty on the collection of revenues from certain products for as long as certain patents assigned to us under the agreement remain valid. RDF has the right to terminate the agreement for an uncured material breach by us, in connection with our bankruptcy or insolvency or if we directly or indirectly oppose or dispute the validity of the assigned patent rights.
Our U.S. Patent No. 11,033,495 issued on June 15, 2021. Thereafter, RDF asserted that the issuance of that patent extends our royalty obligations under the agreement until 2041. We disagreed and explained that the royalty period under the agreement ended on December 24, 2021 with the expiration of our U.S. Patent No. 9,585,838. Because of the disagreement over the interpretation of this agreement, in December 2021, we filed a declaratory judgment lawsuit in the U.S. District Court for the District of Nevada (21-cv-02241). The lawsuit seeks a declaration from the court that we owe no royalties to RDF with respect to our EXPAREL product after December 24, 2021.
On August 8, 2023, the United States District Court, District of Nevada, granted our motion for partial summary judgment in respect to our claim for a declaration that we no longer owe royalties for EXPAREL made under the 45-liter manufacturing process as of December 24, 2021. As a result, we expect to receive $14.5 million from RDF, representing the royalties that we paid to RDF under protest after December 24, 2021 for EXPAREL made from the 45-liter manufacturing process. In November 2023, the United States District Court, District of Nevada conducted a mediation that did not result in a settlement.
During the pendency of the remaining lawsuit, we will continue to pay royalties associated with our enhanced larger-scale manufacturing process to RDF under protest. We are unable to predict the outcome of this action at this time.
For more information, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
Aratana Therapeutics, Inc.
In December 2012, we entered into an Exclusive License, Development and Commercialization Agreement and related Supply Agreement with Aratana Therapeutics, Inc., a wholly owned subsidiary of Elanco Animal Health, Inc., or Aratana. Under the agreements, we granted Aratana an exclusive royalty-bearing license, including the limited right to grant sublicenses, for the development and commercialization of our bupivacaine liposome injectable suspension product for use in animals. In August 2016, the FDA’s Center for Veterinary Medicine approved NOCITA® (bupivacaine liposome injectable suspension) as a local post-operative analgesia for cranial cruciate ligament surgery in dogs and in August 2018 expanded the NOCITA label to include its use as a peripheral nerve block to provide regional postoperative analgesia following onychectomy in cats. NOCITA is a registered trademark of Aratana.
Aratana pays us a tiered double-digit royalty on certain net sales made in the U.S., and we are eligible to receive up to $40.0 million upon the achievement of commercial milestones. If the product is approved by foreign regulatory agencies for sale outside of the U.S., Aratana will be required to pay us a tiered double-digit royalty on such net sales. Royalty rates will be reduced under certain circumstances. Either party has the right to terminate the license agreement in connection with certain events and unless terminated earlier pursuant to its terms, the license agreement is effective until July 2033, after which Aratana has the option to extend the agreement for an additional five-year term, subject to certain requirements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 16
GQ Bio Therapeutics GmbH
In April 2023, we entered into a process development agreement with GQ Bio Therapeutics GmbH, or GQ Bio, for the development of a commercially scalable manufacturing process for the production of PCRX-201. The agreement calls for us to pay GQ Bio upon the achievement of three milestones that can be achieved independently of each other. Milestone 1 includes a €0.5 million payment to GQ Bio for the execution and completion of a feasibility assessment proposal for scalable process to support milestone 2. Achieving milestone 2 is associated with the development of a qualified program process for PCRX-201 where GQ Bio would build a direct manufacturing cost of a PCRX-201 unit. Based on the direct manufacturing cost, we would pay GQ Bio a success fee within a scale of €0.5 million up to €7.5 million, plus royalties within a scale of 0.25% to 3.75% of net sales associated with PCRX-201. The achievement of milestone 3 requires us to pay GQ €0.5 million for delivering a validatable manufacturing process and validated analytical control package necessary for initiating process validation.
In February 2025, Pacira Therapeutics, Inc., a wholly-owned subsidiary of the Company, entered into a securities purchase agreement to acquire the remaining 81% of GQ Bio that was not already owned by us. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events, to our consolidated financial statements included herein. As of the date of this Annual Report, GQ Bio is a wholly-owned subsidiary of the Company.
Significant Customers
We had three wholesalers each comprising 10 percent or more of our total revenue for the year ended December 31, 2024: McKesson Drug Company, Cardinal Health, Inc. and AmerisourceBergen Health Corporation, which accounted for 34%, 23% and 20% of our total revenues, respectively. These wholesalers process orders for EXPAREL under a drop-ship program. EXPAREL is delivered directly to end-users without the wholesalers ever taking physical possession of the product. None of our customers of ZILRETTA or iovera° accounted for 10 percent or more of our total revenue for the year ended December 31, 2024.
Manufacturing and Research Facilities
Internal Facilities
We manufacture EXPAREL and iovera° handpieces at our facility in San Diego, California. We also have a mixed-use research and development, manufacturing and office facility which sits adjacent to our EXPAREL and iovera° handpiece manufacturing facility, and a warehouse located within five miles of these facilities. We refer to these three buildings as our Science Center Campus, and together they measure approximately 195,000 square feet. Our manufacturing facilities are inspected regularly and approved by the FDA, EMA, MHRA and the Environmental Protection Agency (EPA).
We purchase raw materials and components from third-party suppliers to manufacture EXPAREL, ZILRETTA and iovera°. In most instances, alternative sources of supply are available, although switching to an alternative source would, in some instances, take time and could lead to delays in manufacturing our product candidates. Suppliers may not sell these raw materials to us at the times that we need them or on commercially reasonable terms and we do not have direct control over the availability of these raw materials from our suppliers. In order to manage the risk related to raw material shortages, we strive to keep adequate supplies of key raw materials on hand and qualify additional sources of supply as appropriate.
All manufacturing of products, initial product release and stability testing are conducted by us and our manufacturing partners in accordance with Current Good Manufacturing Practices, or CGMP.
Our 84,000 square-foot EXPAREL manufacturing facility at the Science Center Campus is located on a five-acre site. It was custom built as a pharmaceutical research and development and manufacturing facility. Activities in this facility include the manufacture of EXPAREL bulk product on dedicated production lines and its fill/finish into vials, microbiological and quality control testing, product storage, development of analytical methods and manufacturing of development products. We recently expanded our EXPAREL manufacturing capacity at our Science Center Campus as we expect the future demand for EXPAREL will increase. This enhanced, large-scale manufacturing suite received FDA approval in February 2024.
Our 90,000 square-foot mixed-use research and development, manufacturing and office facility is located adjacent to our EXPAREL manufacturing facility and was completely renovated in 2020 to meet our specifications. We manufacture all of the iovera° handpieces at this facility. This building also houses our Science Center related research and development activities and general and administrative functions, as it includes both laboratories and the building infrastructure necessary to support the formulation, analytical testing, clinical and process development activities for manufacturing additional commercial product indications and new pipeline products. Our pilot plant suite for early-stage clinical product production is located in this building, and there is additional space for future expansion opportunities.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 17
We also occupy a 21,000 square-foot warehouse that serves as the main CGMP warehouse for our Science Center Campus operations, primarily being used for the storage of production materials. It contains ambient as well as cold temperature CGMP warehouse storage and also features a quality control clean room for sampling incoming materials.
Distribution of our pMVL products, including EXPAREL, requires cold-chain distribution, whereby a product must be maintained between specified temperatures. We have validated processes for continuous monitoring of temperature from manufacturing through delivery to the end-users.
Co-Production Facilities
Thermo Fisher Scientific Pharma Services
In April 2014, we entered into a Strategic Co-Production Agreement, Technical Transfer and Service Agreement and Manufacturing and Supply Agreement (the “EXPAREL Manufacturing and Supply Agreement”) with Thermo Fisher Scientific Pharma Services, or Thermo Fisher, to collaborate in the manufacture of EXPAREL. Thermo Fisher undertook certain technical transfer activities and construction services to prepare Thermo Fisher’s Swindon, United Kingdom, or U.K., facility for the manufacture of EXPAREL in a dedicated manufacturing suite. We provided Thermo Fisher with the equipment necessary to manufacture EXPAREL and paid fees to Thermo Fisher based on Thermo Fisher’s achievement of certain technical transfer and construction milestones. We also reimburse Thermo Fisher for certain nominal expenses and additional services. We are now using a second, larger-scale dedicated manufacturing suite that more than doubled our EXPAREL manufacturing capacity at the Thermo Fisher site. We began commercial production of EXPAREL out of that second dedicated manufacturing suite in August 2021.
The initial term of the EXPAREL Manufacturing and Supply Agreement is 10 years from the date of FDA approval of the first dedicated manufacturing suite, which was received in May 2018. We pay fees to Thermo Fisher for their operation of the manufacturing suite and the amount of EXPAREL produced by Thermo Fisher. We also reimburse Thermo Fisher for purchases made on our behalf, certain nominal expenses and additional services. We may terminate this agreement upon one month’s notice if a regulatory authority causes the withdrawal of EXPAREL from the U.S. or any other market that represents 80 percent of our overall sales, or at any time for convenience by providing 18 months’ notice. Either party may terminate the EXPAREL Manufacturing and Supply Agreement in the event of the breach or bankruptcy of the other party.
Prior to the Flexion Acquisition, in July 2015, Flexion and Thermo Fisher entered into a Manufacturing and Supply Agreement (the “ZILRETTA Manufacturing and Supply Agreement”) and a Technical Transfer and Service Agreement related to the manufacture of ZILRETTA at the same Thermo Fisher site in Swindon, U.K. where our EXPAREL suite is located. Thermo Fisher agreed to undertake certain transfer activities and construction services needed to prepare its facility for the commercial manufacture of ZILRETTA in dedicated manufacturing suites. Flexion provided Thermo Fisher with certain equipment and materials necessary to manufacture ZILRETTA. We make monthly payments to Thermo Fisher for such activities and reimburse Thermo Fisher for certain material, equipment and miscellaneous expenses and additional services.
The initial term of the ZILRETTA Manufacturing and Supply Agreement that we assumed as part of the Flexion Acquisition expires in October 2027. We pay a monthly base fee to Thermo Fisher for the operation of the manufacturing suites and a per product fee for each vial of ZILRETTA based upon a forecast of commercial demand. We also reimburse Thermo Fisher for purchases of materials and equipment made on our behalf, certain nominal expenses and additional services. The ZILRETTA Manufacturing and Supply Agreement will remain in full effect unless and until it expires or is terminated. We may terminate this agreement upon one month’s notice if a regulatory authority causes the withdrawal of ZILRETTA from the U.S. or any other market that represents 80 percent of our overall sales, or at any time for convenience by providing 24 months’ notice. Either party may terminate the ZILRETTA Manufacturing and Supply Agreement in the event of the breach or bankruptcy of the other party. Upon termination of the ZILRETTA Manufacturing Agreement (other than termination by us in the event that Thermo Fisher does not meet the manufacturing milestones or for a breach by Thermo Fisher), we will be obligated to pay for the costs incurred by Thermo Fisher associated with the removal of our manufacturing equipment and for Thermo Fisher’s termination costs up to a specified capped amount.
Carlisle Companies, Inc.
In January 2020, we and Carlisle Companies, Inc., or Carlisle, entered into a Manufacturing and Supply Agreement (the “Carlisle Agreement”) to collaborate in the manufacture of iovera° Smart Tips at Carlisle’s Tijuana, Mexico facility. The initial term of the Carlisle Agreement is five years with automatic one-year extensions unless either party provides prior notice in
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 18
writing. Under the Carlisle Agreement, we pay fees based on the amount of iovera° Smart Tips delivered by Carlisle. Since April 2022, all iovera° Smart Tips have been produced by Carlisle.
The Carlisle Agreement may be terminated by either party upon one years’ written notice without cause. We may terminate the Carlisle Agreement upon thirty days’ written notice in the event that iovera° is withdrawn from the market or no longer sold by us. Either party may terminate the Carlisle Agreement in the event of the breach or bankruptcy of the other party.
Intellectual Property and Exclusivity
We seek to protect our products, our product candidates and our technologies through a combination of patents, trade secrets, proprietary know-how, regulatory exclusivity and contractual restrictions on disclosure. We note that the patents and applications described below are only examples intended to highlight the variety of coverage provided by our existing and constantly developing portfolio.
Patents and Patent Applications
We seek to protect the proprietary position of our products and product candidates by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. As of December 31, 2024, there are over 13 families of patents and patent applications relating to various aspects of the pMVL drug delivery technology and 29 families of patents and patent applications relating to various aspects of the technology used by iovera°. There are two families of patents and patent applications relating to various aspects of the technology used by ZILRETTA. Patents have been issued in numerous countries, with an emphasis on the North American, European and Japanese markets. These utility patents generally have a term of 20 years from the date of the non-provisional filing unless claiming priority to an earlier filed non-provisional application. Our issued patents expire at various dates in the future, as discussed below, with the last currently issued patent for the pMVL drug delivery technology expiring in 2044, the last currently issued patent for ZILRETTA expiring in 2031 and the last currently issued utility patent for the iovera° technology expiring in 2040.
Patents and Patent Applications for our pMVL and pMVL Products
In June 2021, the United States Patent and Trademark Office, or USPTO, issued U.S. Patent No. 11,033,495 related to EXPAREL. The patent, “Manufacturing of Bupivacaine Multivesicular Liposomes,” claims composition of EXPAREL prepared by an improved manufacturing process developed in Swindon, U.K. In November 2021, the USPTO issued U.S. Patent Nos. 11,185,506 and 11,179,336, claiming the improved U.K. EXPAREL manufacturing process and EXPAREL composition, respectively. Eight U.S. patents relating to product and product-by-process in connection with the improved U.K. manufacturing process for EXPAREL were issued between March 2022 and November 2023, providing additional patent protection through 2041. In March 2022, the USPTO issued U.S. Patent No. 11,278,494, claiming EXPAREL composition. In April 2022, the USPTO issued U.S. Patent Nos. 11,304,904 and 11,311,486, claiming composition of EXPAREL prepared by an improved manufacturing process and EXPAREL composition, respectively. In June 2022, the USPTO issued U.S. Patent No. 11,357,727, claiming composition of EXPAREL prepared by the improved U.K. manufacturing process. In August 2022, the USPTO issued U.S. Patent No. 11,426,348, claiming EXPAREL batch compositions. In September 2022, the USPTO issued U.S. Patent No. 11,452,691, claiming EXPAREL batch compositions. In November 2023, the USPTO issued U.S. Patent Nos. 11,819,574 and 11,819,575, claiming batch compositions of EXPAREL prepared by the improved U.K. manufacturing process and compositions of EXPAREL, respectively. In March 2024, the USPTO issued U.S. Patent No. 11,925,706, claiming composition of EXPAREL. In November 2024, the USPTO issued U.S. Patent Nos. 12,144,890 and 12,151,024, claiming composition of EXPAREL prepared by the improved UK manufacturing process, and both composition of EXPAREL and composition of EXPAREL prepared by the improved UK manufacturing process, respectively. In December 2024, the USPTO issued U.S. Patent No. 12,178,909, claiming the improved EXPAREL UK manufacturing process.
All 15 patents issued between 2021 and 2024 have an expiration date of January 22, 2041. U.S. Patent Nos. 11,033,495, 11,179,336, 11,278,494, 11,304,904, 11,311,486, 11,357,727, 11,426,348, 11,452,691, 11,819,574, 11,819,575, 11,925,706, 12,144,890, and 12,151,024 are currently listed in the FDA’s “Approved Drug Products with Therapeutic Equivalence Evaluations” (the “Orange Book”).
In October 2021, we received a Notice Letter advising that eVenus Pharmaceutical Laboratories, Inc., or eVenus, of Princeton, New Jersey, submitted to the FDA an Abbreviated New Drug Application, or ANDA with a Paragraph IV certification seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (266 mg/20 mL) in the U.S. prior to the expiration of U.S. Patent No. 11,033,495. In August 2024, the U.S. District Court for the District of New Jersey issued its ruling in our patent infringement suit against eVenus and its parent company (Jiangsu Hengrui
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 19
Pharmaceuticals, Co. Ltd., or Jiangsu Hengrui) for infringement of EXPAREL U.S. Patent No. 11,033,495. The ruling found that this patent is not valid on the grounds of obviousness and anticipation. A notice of appeal was filed in September 2024 and remains pending. For information on this matter, as well as other subsequent patent litigation lawsuits against eVenus, Jiangsu Hengrui and Fresenius Kabi USA, LLC, or Fresenius, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
In December 2024, the USPTO also issued U.S. Patent No. 12,156,940 from a new family of patents related to EXPAREL produced by the Company’s enhanced, large-scale manufacturing process in San Diego, California, which received approval from the FDA in February 2024. The ’940 patent, entitled “Manufacturing of Bupivacaine Multivesicular Liposomes”, claims batch compositions of EXPAREL prepared the enhanced U.S. large-scale manufacturing process. The ’940 patent has an expiration date of July 2, 2044 and is currently listed in the FDA’s Orange Book.
Furthermore, the USPTO also issued U.S. Patent Nos. 11,918,565 and 11,931,459 in March 2024, relating to use of EXPAREL as a sciatic nerve block and the pediatric use of EXPAREL, respectively. U.S. Patent Nos. 11,918,565 and 11,931,459 have an expiration date of February 2, 2043 and March 17, 2042, respectively, both are currently listed in the FDA’s Orange Book.
We also own a family of U.S. and foreign patents on an alternative process to manufacture EXPAREL and other pMVL-based products. The process offers many advantages, including larger scale production and lower manufacturing costs. There are eight issued U.S. patents. Patents that claim the process and apparatus will expire at the latest in November 2033. One of the patents claims a product made by the process and expires in April 2031. As of December 31, 2024, we have four granted patents in China, one granted patent in Europe, one granted patent in Japan and one granted patent in Israel, protecting various aspects of the alternative process, including the methods of using the apparatus and the apparatus itself.
In October 2022, we filed a U.S. application and a Patent Cooperation Treaty, or PCT, application relating to compositions of matter, processes of making and methods of treatment in connection with dexamethasone sodium phosphate-pMVL product. In addition, a U.S. application and a PCT application were filed relating to compositions of matter, processes of making and methods of treatment in connection with a high-potency bupivacaine-pMVL product. In 2023, we filed several U.S. nonprovisional applications and PCT applications relating to the use of EXPAREL as a stellate ganglion block for managing cardiac arrhythmia and anxiety disorders, including electrical storm and post-traumatic stress disorder. In 2024, we filed a U.S. nonprovisional application relating to the use of EXPAREL as a stellate ganglion block for treating disorders associated with the sympathetic nervous system.
Patents and Patent Applications for ZILRETTA
A composition of matter patent has been issued by the USPTO for ZILRETTA, with a patent term into 2031. The USPTO has also issued two patents directed at the methods of manufacturing and using ZILRETTA with patent terms into 2031. Considerable expertise and effort were required to carry out the large body of original work underlying the formulation of ZILRETTA, including experimenting with, and observing the effects of over 50 steroid and poly lactic-co-glycolic acid, or PLGA, formulations. We believe our extensive know-how and trade secrets relating to the manufacturing process for ZILRETTA, including those that relate to precise pharmaceutical release profiles, represent a meaningful entry barrier.
We own three U.S. ZILRETTA patents as well as counterpart foreign patents and patent applications covering composition of matter, methods of manufacture, and methods of use. Our U.S. ZILRETTA patents have expiration dates in 2031. The ZILRETTA composition of matter invention is the result of several unique discoveries relating to a narrow drug load specification, a certain release profile of the copolymer, specific polymer component weights and ratios, and clinical efficacy observed within a dose-range. The U.S. patents directed to ZILRETTA’s composition of matter and methods of use are listed in the FDA Orange Book. We also have two U.S. patents directed at compositions of matter similar to ZILRETTA, as well as methods of making and using the same, with patent terms into 2031.
In 2022, we had one patent granted in Pakistan, further expanding our global intellectual property portfolio, which includes patents in the U.S., Australia, Canada, China, Europe, Hong Kong, Indonesia, India, Israel, Japan, Malaysia, Mexico, New Zealand, Pakistan, the Philippines, the Russian Federation, Saudi Arabia, Singapore, South Africa, South Korea, Taiwan and Ukraine. These foreign patents cover the composition of matter, methods of manufacturing, and methods of using ZILRETTA and are similar in scope to the protection in the U.S. described above.
In February 2024, we filed a PCT application relating to use ZILRETTA to treat OA pain in subpopulations of diabetic patients.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 20
We have also in-licensed intellectual property, owned by the Southwest Research Institute, or SwRI, which gives us exclusive rights to SwRI patents covering our proprietary microsphere manufacturing technology used in the production of ZILRETTA. These patents are scheduled to expire in September 2025.
Patents and Patent Applications for iovera°
Issued patents in the U.S. afford us a wide range of coverage of various aspects of the iovera° technology. For example, several of our earliest filed patents cover the structural aspects of a handheld cryogenic device with single needle and needle arrays, tissue-penetrating needle probes that may be detachable, fused silica tubing fluid delivery paths, methods of applying cryotherapy using the cryogenic device and methods for using replaceable needle probes. These patents are set to expire between 2025 and 2032. An important patent family specifically directed to systems and methods of treating pain offers both broad and variable coverage of cryogenic device features and methods of using the same for pain management, including single-use needle probes, particular needle sizes and shapes. Patents in this family are set to expire between 2025 and 2028. Another important patent family has broad disclosure and coverage of a variety of indications for treatment by cryogenic devices, including joint function and stiffness, OA, occipital neuralgia, spasticity, neuroma and other nerve entrapment indications and is set to expire between 2033 and 2037.
Additionally, there are several patents and pending patent applications directed to other important aspects of the iovera° technology. For example, patents covering the probe filtration system are set to expire in 2033 and patents on the Smart Tip technology are set to expire between 2034 and 2037. Other patents and applications cover methods of using needles with blunt tips and aspects of cryogenic devices coupled with a neurostimulator for locating nerves and are set to expire between 2035 and 2038. There are also eight utility and design patent families covering various features of commercial and developing next-generation technology, which are issued or pending in the North American, European, Japanese, Chinese and Brazilian markets, which could potentially prevent others from using commercial and/or next-generation cryogenic devices until at least 2040 for utility patents and 2046 for design patents.
In addition, we also filed a U.S. nonprovisional and a PCT application in 2023 covering the use of iovera° as a stellate ganglion block for managing cardiac arrhythmia, including electrical storm.
PCRX-201
In December 2017, Flexion acquired the global rights to PCRX-201 from GQ, including a direct exclusive license of certain foundational patents, patent applications, and other proprietary rights owned by the Baylor College of Medicine, or BCM, that are related to PCRX-201 for human applications. These patents generally cover the composition of matter and method of use of PCRX-201 in the treatment of OA. In 2019, the USPTO issued U.S. Patent No. 10,301,647, which covers the composition of matter and method of use of PCRX-201 in the treatment of OA with a term through January 2033. In addition, the BCM patents related to PCRX-201 are issued in Europe, with an expiry date in 2032, and in Australia, Canada, China, India, Japan and Eurasia with expiry dates in 2033. We are continuing to prosecute one BCM U.S. patent application related to PCRX-201. Further, we have entered the national phase in Brazil, China, Europe, Hong Kong, Japan and the U.S. based on a PCT application covering composition of matter and effective dosages of PCRX-201 in the treatment of OA in humans, which, if granted, are expected to provide protection until 2040.
We also have a family of patent applications pending in the U.S., Australia, Canada, China, Hong Kong, Europe, Japan and South Korea covering composition of matter and method of use of PCRX-201 for the treatment of degenerative disc disease, or DDD, which, if granted, are expected to provide patent protection until 2042. In February 2024, we also filed a PCT application covering compositions and method of use of PCRX-201 in combination with a corticosteroid.
In February 2025, as part of the GQ Bio Acquisition, we acquired an exclusive license of certain foundational patents and patent applications, and other proprietary rights owned by BCM related to an HCAd gene therapy vector encoding PRG4 gene for both human and veterinary applications. Issued European patent EP 2948553 B1 generally covers HCAd-PRG4 composition of matter and method of use in treating camptodactyly-arthropathy-coxa vara-pericarditis (CACP) syndrome, a musculoskeletal disorder, or a joint disorder. Issued U.S. Patent No.11,746,359 generally covers composition of matter comprising HCAd-PRG4 and HCAd-IL-1Ra. These patents expire in January 2034. In addition, we also acquired a PCT application owned by GQ Bio related to HCAd gene therapy for treating DDD using nucleic acid encoding SOX-9, GDF5, TIMP3, SIRT6, TBXT, GLP-1, ANGPTL3 in combination with nucleic acid encoding IL-1Ra or PRG4.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 21
Additional Intellectual Property
We have entered the national phase in Brazil, China, Europe and the U.S. based on a PCT application covering composition of matter, method of use, and method of manufacture for formulations of an anesthetic drug of amino amide group (lidocaine, bupivacaine and ropivacaine) formulated in a triblock copolymer component (one or more PLGA-polyethylene glycol-PLGA triblock copolymers), which if converted and granted, is expected to provide protection until 2042.
Trade Secrets and Proprietary Information
Trade secrets play an important role in protecting our pMVL-based products (including EXPAREL) and pipeline, ZILRETTA and iovera° and provide protection beyond patents and regulatory exclusivity. The scale-up and commercial manufacture of each of our products involve processes, custom equipment and in-process and release analytical techniques that we believe are unique to us. The expertise and knowledge required to understand the critical aspects of our pMVL manufacturing steps requires knowledge of both traditional and non-traditional emulsion processing and traditional pharmaceutical production, overlaid with all of the challenges presented by aseptic manufacturing. ZILRETTA is also manufactured using custom equipment and proprietary processes with respect to certain of the formulation and manufacturing techniques related to the TA-formulated PLGA microspheres in ZILRETTA, including those that relate to its precise pharmaceutical release profile. The iovera° system relies on custom manufacturing techniques that are able to provide the precision and tight tolerances required for a self-contained handheld cryogenic device. Additionally, the iovera° device includes proprietary software for device operations during cryotherapy treatments.
We seek to protect our proprietary information, including our trade secrets and proprietary know-how, by requiring our employees, consultants and other advisors to execute proprietary information and confidentiality agreements upon the commencement of their employment or engagement. These agreements generally provide that all confidential information developed or made known during the course of the relationship with us be kept confidential and not be disclosed to third parties except in specific circumstances. In the case of our employees, the agreements also typically provide that all inventions resulting from work performed for us, utilizing our property or relating to our business and conceived or completed during employment shall be our exclusive property to the extent permitted by law. Where appropriate, agreements we obtain with our consultants also typically contain similar assignment of invention obligations. Further, we require confidentiality agreements from third parties that receive our confidential data or materials.
Competition
The pharmaceutical industry is intensely competitive and subject to rapid and significant technological change. Our competitors include organizations such as major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies and generic drug companies. Many of our competitors have greater financial and other resources than we have, such as more commercial resources, larger research and development staffs and more extensive marketing and manufacturing organizations. As a result, these companies may obtain marketing approval more rapidly than we are able and may be more effective in developing, selling and marketing their products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies.
EXPAREL
Our competitors may succeed in developing, acquiring or licensing on an exclusive basis technologies and drug products that are more effective or less costly than EXPAREL or any other products that we are currently selling through partners or developing or that we may develop, which could render our products obsolete and noncompetitive. We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price and the availability of reimbursement from government and other third-party payers.
EXPAREL competes with well-established products with similar indications. Competing products available for postsurgical pain management include opioids such as morphine, fentanyl, meperidine and hydromorphone, each of which is available generically from several manufacturers, and several of which are available as proprietary products using novel delivery systems. Ketorolac, an NSAID, is also available generically in the U.S. from several manufacturers, and Caldolor (ibuprofen for injection), an NSAID, has been approved by the FDA for pain management and fever in adults. EXPAREL also competes with currently marketed non-opioid products such as bupivacaine, marcaine, ropivacaine and other anesthetics/analgesics, all of which are also used in the treatment of postsurgical pain and are available as either oral tablets, injectable dosage forms or administered using novel delivery systems. Additional products may be developed for the treatment of acute pain, including new injectable NSAIDs, oral NaV1.8 pain signal inhibitors, novel opioids, new formulations of currently
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 22
available opioids and NSAIDs, long-acting local anesthetics and new chemical entities as well as alternative delivery forms of various opioids and NSAIDs. Currently EXPAREL also competes with elastomeric pumps and catheter devices intended to provide bupivacaine over several days and with off-label combinations of other approved analgesics, called “cocktails,” that are combined by compound pharmacies in an attempt to extend the duration of pain control.
ZILRETTA
Immediate-release steroids and hyaluronic acid, or HA, injections are currently the two marketed classes of IA products that compete directly with ZILRETTA. Also available are stem cell and platelet rich plasma, or PRP, injections, but these require on-site preparation from tissue or blood taken from the patient and have generated questionable efficacy in controlled clinical trials. Because these are minimally manipulated autologous therapies, they do not require and have not received FDA review or approval. For that reason, they are generally not reimbursed by payers, and patients must pay out of pocket to receive these therapies. Furthermore, the American Association of Hip & Knee Surgeons (AAHKS) issued a position statement indicating that it cannot recommend biologic therapies, including stem cell and PRP injections, for the treatment of advanced hip or knee arthritis.
iovera°
The medical device industry is intensely competitive and subject to rapid and significant technological change. The cryotherapy pain management field in particular is a growing industry due to increased attention on opioid usage for pain, which has created a rapidly emerging market and has fueled an increased interest in opioid alternatives. Many of our competitors in our space have greater financial and other resources than we have, such as more commercial resources, larger research and development staffs and more extensive marketing and manufacturing organizations. As a result, these companies may obtain marketing approval more rapidly than we are able and may be more effective in developing, selling and marketing their products. The rise of various small and early-stage companies in the cryotherapy pain management field may also prove to be significant competitors, particularly if they enter into collaborative arrangements with large, established companies.
Our competitors are continuously engaged in trials and attempts to develop new products or approaches in hopes of capturing the pain management market. They may succeed in developing, acquiring or licensing on an exclusive basis, technologies that are more effective or less costly than the iovera° system, which could render the iovera° system obsolete and noncompetitive. As a result, it is critical that we continue to innovate and to increase marketing efforts in our primary markets. We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price and the availability of reimbursement from government and other third-party payers.
Besides pharmaceutical products for pain management, iovera° competes with medical devices that ablate or degenerate peripheral nerves to treat indications such as joint pain, neuralgia and OA pain. Competing products include cryotherapy devices as well as other devices such as cooled radio-frequency ablation devices that block or degenerate peripheral nerves involved in conducting pain signals. Avanos Medical, Inc. markets these medical devices in the U.S. Additional non-opioid products or entirely different approaches may also be developed for pain management by one or more of our competitors.
Government Regulation
In the U.S., prescription drug and medical device products are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the research, development, testing, manufacturing, distribution, safety, efficacy, approval, labeling, storage, record keeping, reporting, advertising and promotion of such products under the Federal Food, Drug and Cosmetic Act, or FDCA, and its implementing regulations. Outside the U.S., prescription drug and medical device products are regulated by comparable agencies (including the EMA and MHRA in the E.U. and U.K., respectively, as well as authorities in Canada and Latin America), laws and regulations. Failure to comply with applicable regulatory requirements may result in, among other things, refusal to approve pending applications, withdrawal of an approval, warning letters, clinical holds, civil or criminal penalties, recall or seizure of products, injunction, debarment, partial or total suspension of production or withdrawal of the product from the market. Any agency or judicial enforcement action could have a material adverse effect on us.
Regulatory Environment
Pharmaceuticals
In the U.S., generally the FDA must approve any new drug, including a new use of a previously approved drug, before marketing of the drug occurs in the U.S. This process generally involves:
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 23
•completion of preclinical laboratory and animal testing and formulation studies in compliance with the FDA’s Good Laboratory Practice regulations;
•submission to the FDA of an investigational new drug, or IND, application for human clinical testing, which must become effective before human clinical trials may begin for unapproved use in the U.S.;
•approval by an independent Institutional Review Board, or IRB, at each clinical trial site before each trial may be initiated;
•performance of adequate and well-controlled human clinical trials in accordance with the FDA’s Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed drug product for each intended use;
•completion of process validation, quality product release and stability;
•submission of a New Drug Application, or NDA, to the FDA;
•satisfactory completion of an FDA pre-approval inspection of the product’s manufacturing facility or facilities to assess compliance with CGMP requirements and to ensure that the facilities, methods and controls are adequate to preserve the drug’s identity, quality and purity;
•satisfactory completion of an FDA advisory committee review, if applicable; and
•review and approval by the FDA of the NDA.
The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that the FDA will grant approvals for any of our product candidates on a timely basis, if at all. Preclinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information, analytical data and a proposed clinical trial protocol and other information, are submitted as part of an IND application to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the trial on a clinical hold because of, among other things, concerns about the conduct of the clinical trial or about exposure of human research subjects to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Thus, submission of an IND does not by itself automatically result in FDA authorization to commence a clinical trial. In addition, the FDA requires us to amend an existing IND for each successive clinical trial conducted during product development. Further, an IRB covering each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial along with informed consent information for subjects before the clinical trial commences at that center. The IRB also must monitor the clinical trial until it is completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time, on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. We may also suspend or terminate a clinical trial based on evolving business objectives and/or the competitive climate.
Clinical trials involve the administration of the product candidate to healthy volunteers or patients having the disease being studied under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Sponsors of clinical trials generally must register at the National Institutes of Health-maintained website (www.clinicaltrials.gov) and report key findings and parameters. For purposes of an NDA submission and approval, typically, the conduct of human clinical trials occurs in the following three pre-market sequential phases, which may overlap or be combined:
•Phase 1: Sponsors initially conduct clinical trials in a limited population, either patients or healthy volunteers, to test the product candidate for safety, dose tolerance, absorption, metabolism, distribution, excretion and clinical pharmacology, and, if possible, to gain early evidence of effectiveness. In the cases of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing often is conducted only on patients having the specific disease.
•Phase 2: Sponsors conduct clinical trials generally in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted indications and to determine dose tolerance, optimal dosage and dosing schedule. Sponsors may conduct multiple Phase 2 clinical trials to obtain information prior to beginning larger and more extensive Phase 3 clinical trials.
•Phase 3: These include expanded controlled and uncontrolled trials, including pivotal clinical trials. When Phase 2 evaluations suggest the effectiveness of a dose range of the product and acceptability of such product’s safety profile, sponsors undertake Phase 3 clinical trials in larger patient populations to obtain additional information needed to evaluate the overall benefit and risk balance of the drug and to provide an adequate basis to develop labeling.
Some clinical trials may be overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 24
forward at designated check points based on access to certain data from the trial. The process of completing clinical testing and obtaining FDA approval for a new drug is likely to take a number of years and requires the expenditure of substantial resources. If an application is submitted, there can be no assurance that the FDA will review and approve the NDA. In addition, sponsors may elect to conduct, or be required by the FDA to, conduct post-approval clinical trials to further assess the drug’s safety or effectiveness after NDA approval, generate new data and best-practice administration techniques. Studies in an indication after approval are typically referred to as Phase 4 clinical trials.
The requirements for drug approval and the clinical trials that approvals are based on are similar in other countries, however each regulatory agency will have differing policies, procedures and processes that we must comply with in each market we wish to sell our products in. There also can be no assurance that approval or utilization of our products will be identical in different jurisdictions.
Medical Devices
In the U.S., the Medical Device Amendments of 1976 to the FDCA and its subsequent amendments regulate the design, manufacture and marketing of medical devices. Medical devices that require notification submitted as a 510(k) clearance request must be reviewed and cleared by the FDA before we can begin marketing them. To request 510(k) clearance, we must be able to demonstrate that the medical device is substantially equivalent to a previously cleared and legally marketed 510(k) medical device. Medical devices require extensive clinical testing which consists of safety and efficacy studies, followed by pre-market approval, or PMA, applications for specific surgical indications. The FDA’s Quality System Regulations, or QSRs, set forth standards for our product design and manufacturing processes, require the maintenance of certain records and provide for inspections of our facilities by the FDA. There are also certain requirements of state, local and foreign governments that must be complied with in the manufacture and marketing of our products. A new indication for 510(k) clearance may or may not require a clinical trial. Expanding the iovera° label to include the treatment of spasticity requires a clinical trial. We currently have a pivotal trial underway to demonstrate the efficacy and safety of iovera° for treating spasticity.
Review and Approval Process
Pharmaceuticals
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, sponsors submit the results of product development, preclinical studies and clinical trials to the FDA as part of an NDA requesting approval to market the product for one or more indications. NDAs must also contain extensive information relating to the product’s pharmacology, chemistry, manufacture, controls and proposed labeling, among other things. In addition, 505(b)(2) applications must contain a patent certification for each patent listed in the FDA’s Orange Book that covers the drug referenced in the application and upon which the third-party studies were conducted. For some drugs, regulatory agencies may require Risk Evaluation and Mitigation Strategies, or REMS, which could include medication guides, physician communication plans or restrictions on distribution and use, such as limitations on who may prescribe the drug or where it may be dispensed or administered. Currently, the FDA does not require a REMS for EXPAREL but the EMA and MHRA do.
If the FDA accepts a submission for substantive review, the FDA typically reviews the NDA in accordance with established timeframes. Under the Prescription Drug User Fee Act, or PDUFA, the FDA establishes goals for NDA review time through a two-tiered classification system: Priority Review and Standard Review. A Priority Review designation is given to drugs that address an unmet medical need by offering major advances in treatment or providing a treatment where no adequate therapy currently exists. Standard Review applies to all applications that are not eligible for Priority Review. The FDA aims to complete Standard Reviews of NDAs within 12 months of submission (ten months after the Day 60 filing date) and Priority Reviews within eight months of submission (six months after the Day 60 filing date). For an sNDA, the FDA aims to complete its Standard Review within 10 months of submission and Priority Reviews within six months of submission. Review processes may sometimes extend beyond these target completion dates due to FDA requests for additional information or clarification, difficulties scheduling an advisory committee meeting, negotiations regarding REMS or FDA workload issues, but in general under PDUFA the FDA is supposed to complete its reviews within the target timeframes despite these factors. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to the application’s approval. The recommendations of an advisory committee do not bind the FDA, but the FDA generally follows such recommendations.
Under PDUFA, NDA applicants must pay significant NDA user fees upon submission. In addition, manufacturers of approved prescription drug products must pay annual program fees, as we do for EXPAREL and ZILRETTA.
Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with CGMP requirements and are adequate to ensure consistent production of the product within required specifications. Additionally, the FDA will typically inspect one or more clinical sites to ensure compliance with GCP before approving an NDA.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 25
After the FDA evaluates the NDA and the manufacturing facilities, it may issue an approval letter or a Complete Response Letter, or CRL, to indicate that the review cycle for an application is complete and that the application is not ready for approval. CRLs generally outline the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we do. If the FDA requires a REMS plan, it could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may approve an NDA contingent on, among other things, changes to proposed labeling, a commitment to conduct one or more post-market studies or clinical trials and the correction of identified manufacturing deficiencies, including the development of adequate controls and specifications. If and when the deficiencies have been addressed to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Outside the U.S., although timelines vary as do specific regulatory procedures, the same general principals hold, including the potential for a REMS plan which could entail other requirements, including but not limited to patient registries and risk minimization tools.
Medical Devices
In the U.S., authorization to bring a medical device to market is generally obtained in one of two ways. The first pathway, a pre-market notification (the 510(k) process), requires demonstration that the new device is substantially equivalent to an already legally marketed medical device. The second pathway, a PMA, requires an independent demonstration that a medical device is safe and effective for its intended use. In general, PMAs require a much longer time horizon and can be much more expensive than obtaining clearance through the 510(k) process. A PMA must be submitted to the FDA if it is determined that the device is not eligible for the 510(k) clearance process. A PMA must be supported by extensive data including, but not limited to, technical, preclinical and clinical trials, manufacturing and labeling to demonstrate reasonable evidence of the device’s safety and efficacy to the FDA’s satisfaction.
To obtain 510(k) clearance, we must file with the FDA a pre-market notification demonstrating that our proposed device is substantially equivalent to a previously cleared and legally marketed 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a PMA. 510(k) clearance for a predecessor device to iovera° was first obtained in March 2009 when the focus of MyoScience was cosmetic applications (i.e., facial wrinkle reduction). MyoScience’s focus shifted to pain management in 2014, and since then there have been a number of advancements that led to three additional 510(k) submissions and clearances to support iovera° and the subsequent growth of the iovera° product line.
After a device receives 510(k) clearance or a PMA approval, it may be changed or modified. Any modification that could significantly affect its safety or effectiveness, or that would constitute a significant change in its intended use, will require a new clearance or approval. Regulations provide that the manufacturer initially determines when a specific modification requires notification to FDA. The FDA has issued draft guidance that, if finalized and implemented, will result in manufacturers needing to seek a significant number of new clearances for changes made to legally marketed devices. The FDA reviews the manufacturer’s decision to file a 510(k) or PMA for modifications during facility audits.
Section 505(b)(2) New Drug Applications
For pharmaceutical products, as an alternate path to FDA approval, particularly for modifications to drug products previously approved by the FDA, an applicant may submit an NDA under Section 505(b)(2) of the FDCA. Section 505(b)(2) was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984 (also known as the Hatch-Waxman Act), and permits the submission of an NDA where at least some of the information required for approval comes from preclinical and/or clinical trials not conducted by or for the applicant. The FDA interprets Section 505(b)(2) of the FDCA to permit the applicant to rely upon the FDA’s previous findings of safety and effectiveness for an approved product. The FDA may also require companies to perform additional clinical trials or measurements to support any change from the previously approved product. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
Applications under Section 505(b)(2) are subject to any non-patent exclusivity period applicable to the referenced product, which may delay approval of the 505(b)(2) application even if the FDA has completed its substantive review and determined the drug should be approved. In addition, 505(b)(2) applications must include patent certifications to any patents listed in the FDA’s Orange Book as covering the referenced product. If the 505(b)(2) applicant seeks to obtain approval before the expiration of an applicable listed patent, the 505(b)(2) applicant must provide notice to the patent owner and NDA holder of the referenced product. If the patent owner or NDA holder brings a patent infringement lawsuit within 45 days of such notice, the 505(b)(2) application cannot be approved for 30 months or until the 505(b)(2) applicant prevails, whichever is sooner. If the
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 26
505(b)(2) applicant loses the patent infringement suit, the FDA may not approve the 505(b)(2) application until the patent expires, plus any period of pediatric exclusivity.
In any future NDA submissions for our product candidates, we intend to follow the development and approval pathway permitted under the FDCA that we believe will maximize the commercial opportunities for these product candidates.
Post-Approval Requirements
Pharmaceuticals
After approval, the NDA sponsor must comply with comprehensive requirements governing, among other things, drug listing, recordkeeping, manufacturing, marketing activities, product sampling, distribution and annual reporting. Additionally, adverse events must be reported to the FDA in a timely fashion, and pharmacovigilance programs to proactively look for adverse events are mandated by the FDA. An adverse event is any undesirable experience associated with the use of a medical product in a patient. A serious adverse event is an adverse event that results in death, is life-threatening or results in hospitalization or disability, among other things. If the events suggest a new safety signal for the drug in question, that could lead to the need for additional safety statements in the labeling of the product or additional REMS. Additionally, adverse events found in other drugs could also mean that we have to abide by additional safety measures and include warnings in our labeling. Similar reporting and pharmacovigilance obligations exist with regulatory agencies outside the U.S.
If new safety issues are identified following approval, the FDA can require the NDA sponsor to revise the approved labeling to reflect the new safety information; conduct post-market studies or clinical trials to assess the new safety information and implement a REMS program to mitigate newly identified risks. The FDA may also require post-approval testing, including Phase 4 trials, and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the authority to prevent or limit further marketing of a product based on the results of these post-marketing programs. Drugs may be marketed only for approved indications and in accordance with the provisions of the FDA-approved label. Further, if we modify a drug, including any changes in indications, labeling or manufacturing processes or facilities, the FDA may require us to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with CGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from CGMP and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use.
If after approval the FDA determines that the product does not meet applicable regulatory requirements or poses unacceptable safety risks, the FDA may take other regulatory actions, including initiating suspension or withdrawal of the NDA approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products; or
•injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. These regulations include standards and restrictions for direct-to-consumer advertising, industry-sponsored scientific and educational activities, promotional activities involving the internet and off-label promotion. While physicians may prescribe for off-label uses, manufacturers may only promote for the approved indications and in accordance with the provisions of the approved label. The FDA has very broad enforcement authority under the FDCA, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing entities to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, which regulates the distribution of drugs and drug samples at the federal level and sets minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 27
pharmaceutical product samples and impose requirements to ensure accountability in distribution, including a drug pedigree which tracks the distribution of prescription drugs.
Medical Devices
The FDA has broad post‑market and regulatory obligations that we must adhere to. We are subject to unannounced inspections by the FDA to determine our compliance with QSRs and other rules and regulations.
After a medical device is placed on the market, numerous regulatory requirements apply. These include, but are not limited to:
•QSRs, which require manufacturers, including third‑party manufacturers, to follow stringent design, testing, documentation and other quality assurance procedures during product design and throughout the manufacturing process;
•Labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off‑label uses; and
•Medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur.
Failure to comply with regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
•restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical trials;
•the potential withdrawal of 510(k) clearance or other approvals that were previously granted;
•refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals;
•product seizure or detention, or refusal to permit the import or export of products;
•injunctions or the imposition of civil or criminal penalties; or
•requiring us to repair, replace and/or refund the cost of any medical device we have manufactured or distributed.
If any of these events were to occur, they could have a material adverse effect on our business.
International Regulation
In addition to regulations in the U.S., we are subject to a variety of foreign regulations governing clinical trials and the commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process and requirements vary from country to country, and the time may be longer or shorter than that necessary for FDA approval.
For example, in Europe, there are several tracks for marketing approval for pharmaceuticals, for product approval and post-approval regulatory processes, depending on the type of product for which approval is sought. Under the centralized procedure, a company submits a single application to the EMA. The marketing application is similar to the NDA in the U.S. and is evaluated by the CHMP, the expert scientific committee of the EMA. If the CHMP determines that the marketing application fulfills the requirements for quality, safety and efficacy, it will submit a favorable opinion to the EC. The CHMP opinion is not binding, but is typically adopted by the EC. A marketing application approved by the EC is valid in all E.U. member states and is recognized by the MHRA. The centralized procedure is required for all biological products, orphan medicinal products and new treatments for neurodegenerative disorders, and it is available for certain other products, including those which constitute a significant therapeutic, scientific or technical innovation.
As with FDA, EMA or MHRA approval, we may not be able to secure additional regulatory approvals in a timely manner, if at all. Additionally, as in the U.S., post-approval regulatory requirements, such as those regarding product manufacture, marketing or distribution would apply to any product that is approved in Europe, the U.K., Canada and Latin America, and failure to comply with such obligations could have a material adverse effect on our ability to successfully commercialize any product.
In addition to regulations in Europe and the U.S., we will be subject to regulations governing clinical trials, product approvals, and commercial distribution in the U.K, Canada, Latin America and any other jurisdictions in which EXPAREL, ZILRETTA, iovera° or any other future product is approved.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 28
Third-Party Payer Coverage and Reimbursement
The commercial success of our products and product candidates will depend, in part, upon the availability of coverage and reimbursement from third-party payers at the federal, state and private levels. Government payer programs, including Medicare and Medicaid, private health care insurance companies and managed care plans may deny coverage or reimbursement for a product or therapy in whole or in part if they determine that the product or therapy is not medically appropriate or necessary. Also, third-party payers have attempted to control costs by limiting coverage and the amount of reimbursement for particular procedures, medical devices or drug treatments. The U.S. Congress and state legislatures from time to time propose and adopt initiatives aimed at cost containment that could impact our ability to sell our products at a price level high enough to realize an appropriate return on our investment, which would materially impact our results of operations.
In March 2010, President Barack Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “Affordable Care Act”), a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. The Affordable Care Act revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates owed to states by pharmaceutical manufacturers for covered outpatient drugs. The Affordable Care Act also established a new Medicare Part D coverage gap discount program, in which drug manufacturers must agree to offer 50 percent point-of-sale discounts off negotiated prices of applicable brand name drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners. One such governmental program that was expanded as part of the Affordable Care Act is the 340B Drug Pricing Program, which requires pharmaceutical manufacturers that participate in Medicaid to enter into a pharmaceutical pricing agreement, or PPA, with the Secretary of Health and Human Services, and requires the manufacturer to extend discounts to entities covered under the 340B Drug Pricing Program. The 340B Drug Pricing Program aims to cover entities that have scarce financial resources to be able to reach the U.S.’s most financially vulnerable patient populations. There have been proposed in Congress a number of legislative initiatives regarding healthcare, including possible repeal of the Affordable Care Act. At this time, it remains unclear whether there will be any changes made to the Affordable Care Act. The full impact that the Affordable Care and other new laws will have on our business is uncertain. However, such laws appear likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Moreover, in the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our products.
In December 2022, the Non-Opioids Prevent Addiction In the Nation (“NOPAIN”) Act was signed into law as part of the Biden Administration’s Consolidated Appropriations Act of 2023. The NOPAIN Act is preventative legislation aimed at tackling the opioid crisis by incentivizing the use of non-opioids to manage surgical pain for Medicare patients treated in ASC and hospital outpatient department, or HOPD, settings. This federal mandate requires Medicare to reimburse for non-opioid products, such as EXPAREL and iovera°, used during all surgeries conducted in the ASC or HOPD setting. Specifically, the NOPAIN Act covers reimbursement for (i) all non-opioid medications indicated to reduce postoperative pain or produce postsurgical regional analgesia without acting upon the body’s opioid receptors; and (ii) all devices used to deliver a therapy, reduce postoperative pain or produce postsurgical or regional analgesia. Any drug or device that qualifies for reimbursement under the NOPAIN Act must have demonstrated the ability to reduce or avoid intraoperative opioid use or the quantity of opioids prescribed in a clinical trial or through data published in a peer-reviewed journal. The NOPAIN Act took effect on January 1, 2025. This policy eliminates the cost burden associated with providing Medicare patients best-in-class opioid-sparing strategies, allowing institutions the financial flexibility to treat more patients with no- and low-opioid pain management strategies.
In October 2024, the Centers for Medicare and Medicaid Services, or CMS, established a permanent product-specific Healthcare Common Procedure Coding System, or HCPCS, J-code for EXPAREL. The new J-code for EXPAREL (J0666) became effective January 1, 2025, and supersedes the prior C-code (C9290), which had been in place since 2019. In addition to the separate CMS reimbursement EXPAREL will receive in outpatient settings with the implementation of the NOPAIN Act in January 2025, this new J-code will also provide reimbursement when EXPAREL is used in the office setting and for office-based surgeries. J-codes are reimbursement codes used by commercial insurance plans, Medicare, Medicare Advantage, and other government payers for Medicare Part B drugs like EXPAREL. Claims submission and payment are standardized with a J-code, facilitating and streamlining billing and reimbursement. In addition, some commercial insurers require a J-code for payment.
Additionally, in November 2024, CMS confirmed that both EXPAREL and iovera° qualify as eligible non-opioid pain management products under the NOPAIN Act. HOPDs and ASCs, that use these products now receive additional Medicare reimbursement effective January 1, 2025. Additionally, separate reimbursement for iovera° will pay up to an additional $255.85 when providers administer iovera° in ASC and HOPD settings, using a new C-code created for iovera° (C9809). This new
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 29
Medicare payment is provided in addition to the current reimbursement available to HOPDs and ASCs when they perform a procedure with iovera°.
The marketability of our products may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the U.S. has increased, and we expect will continue to increase, the pressure on pharmaceutical and medical device pricing. Some third-party payers require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers that use such therapies, or place limits on the amount of reimbursement. Coverage policies and third-party payer reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for our products, less favorable coverage policies and reimbursement rates may be implemented in the future.
In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies. There can be no assurance that our products will be considered medically reasonable and necessary for a specific indication, that our products will be considered cost-effective by third-party payers or that an adequate level of reimbursement will be available so that the third-party payers’ reimbursement policies will not adversely affect our ability to sell our products profitably.
Marketing and Data Exclusivity
Market exclusivity provisions under the FDCA can delay the submission or approval of certain applications of other companies seeking to reference another company’s NDA. The FDA may grant three or five years of marketing exclusivity in the U.S. for the approval of new or supplemental NDAs, including Section 505(b)(2) NDAs, for, among other things, new indications, dosages or dosage forms of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an Abbreviated New Drug Application, or ANDA, or a Section 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, such an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. Additionally, six months of marketing exclusivity in the U.S. is available under Section 505A of the FDCA if, in response to a written request from the FDA, a sponsor submits and the agency accepts requested information relating to the use of the approved drug in the pediatric population. This six-month pediatric exclusivity period is not a standalone exclusivity period, but rather is added to any existing patent or non-patent exclusivity period for which the drug product is eligible. In the past, based on our clinical trial program for EXPAREL, the FDA granted three years of marketing exclusivity to EXPAREL, which expired in October 2014. In Europe, manufacturers qualify for 8 years of data exclusivity upon marketing authorization approval and an additional two years of market exclusivity, for a total of 10 years of regulatory exclusivity.
Manufacturing Requirements
We must comply with the FDA’s CGMP requirements and comparable regulations in other countries. The CGMP provisions include requirements relating to the organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. The manufacturing facilities for our products must meet CGMP requirements to the satisfaction of the FDA and other authorities pursuant to a pre-approval inspection before we can use them to manufacture our products. We and any third-party manufacturers we engage or with which we partner are also subject to periodic inspections of facilities by the FDA and other authorities, including procedures and operations used in the testing and manufacture of our products to assess our compliance with applicable regulations. Failure to comply with these and other statutory and regulatory requirements subjects a manufacturer to possible legal or regulatory action, including warning letters, the seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations and civil and criminal penalties. Adverse experiences with the product or product complaints must be reported and could result in the imposition of market restrictions through labeling changes or in product removal. Product approvals may be withdrawn if compliance with regulatory requirements is not maintained or if problems concerning safety or efficacy of the product occur following approval.
Regulations Pertaining to Sales and Marketing
We are subject to various federal and state laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. Anti-kickback laws generally prohibit a prescription drug or medical device manufacturer from soliciting, offering, receiving, or paying any remuneration to generate business, including the purchase or prescription of a
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 30
particular drug or device. Although the specific provisions of these laws vary, their scope is generally broad and there may be no regulations, guidance or court decisions that clarify how the laws apply to particular industry practices. There is therefore a possibility that our practices might be challenged under the anti-kickback or similar laws. False claims laws prohibit anyone from knowingly and willingly presenting, or causing to be presented for payment to third-party payers (including Medicare and Medicaid) claims for reimbursed drugs, procedures or services that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Violations of fraud and abuse laws may be punishable by criminal or civil sanctions, including fines and civil monetary penalties and exclusion from federal health care programs (including Medicare and Medicaid). In the U.S., federal and state authorities are paying increased attention to enforcement of these laws within the pharmaceutical and medical device industries and private individuals have been active in alleging violations of the laws and bringing suits on behalf of the government under the federal civil False Claims Act. If we were subject to allegations concerning, or were convicted of violating, these laws, our business could be harmed.
Laws and regulations have been enacted by the federal government and various states to regulate the sales and marketing practices of pharmaceutical and medical device manufacturers. The laws and regulations generally limit financial interactions between manufacturers and health care providers or require disclosure to the government and public of such interactions. The laws include the federal Physician Payment Sunshine Act, or “sunshine” provisions, enacted in 2010 as part of the Affordable Care Act. The sunshine provisions apply to pharmaceutical and medical device manufacturers with products reimbursed under certain government programs and require those manufacturers to disclose annually to the federal government (for re-disclosure to the public) certain payments made to physicians and certain other healthcare practitioners or to teaching hospitals. State laws may also require disclosure of pharmaceutical and medical device pricing information and marketing expenditures. Many of these laws and regulations contain ambiguous requirements. Given the lack of clarity in laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent federal and state laws and regulations. Outside the U.S., other countries have implemented requirements for disclosure of financial interactions with healthcare providers and additional countries may consider or implement such laws.
Regenerative Medicine Advanced Therapies
As part of the 21st Century Cures Act, Congress amended the FDCA to create the RMAT designation. The RMAT designation is intended to facilitate efficient development and expedite review of regenerative medicine advanced therapies, which are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition. RMAT covers cell therapies, gene therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products. A sponsor may request that the FDA designate a regenerative medicine advanced therapy concurrently with or at any time after submission of an IND. For example, in February 2024, the FDA granted an RMAT designation for PCRX-201. The FDA has 60 calendar days to determine whether the criteria are met, including whether there is preliminary clinical evidence indicating the potential to address unmet medical needs for a serious or life-threatening disease or condition. A Biologics License Application (BLA) for a regenerative medicine advanced therapy may be eligible for priority review or accelerated approval through surrogate or intermediate endpoints reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of clinical trial sites. Benefits of such designation also include early interactions with the FDA to discuss any potential surrogate or intermediate endpoint to be used to support accelerated approval. A regenerative medicine advanced therapy that is granted accelerated approval and is subject to post-approval requirements may fulfill such requirements through the submission of clinical evidence, clinical studies, patient registries, or other sources of real-world evidence, such as electronic health records; the collection of larger confirmatory data sets; or post-approval monitoring of all patients treated with such therapy prior to its approval.
Healthcare Privacy and Security Laws
We may be subject to, or our marketing activities may be limited by, the Health Insurance Portability and Accountability Act, or HIPAA and its implementing regulations, which established uniform standards for certain “covered entities” (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information. The American Recovery and Reinvestment Act of 2009, commonly referred to as the economic stimulus package, included sweeping expansion of HIPAA’s privacy and security standards called the Health Information Technology for Economic and Clinical Health Act, or HITECH, which became effective in February 2010. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 31
Environmental Matters
Our research and development processes and our manufacturing processes involve the controlled use of hazardous materials and chemicals and produce waste products, including pharmaceutical residues. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products, including those related to pharmaceutical residues. While we believe we are in compliance with applicable environmental regulations, the failure to fully comply with any such regulations could result in the imposition of penalties, fines and/or sanctions which could have a material adverse effect on our business. It is also possible that environmental issues may arise in the future which we cannot now predict.
We are working towards improving our sustainable footprint through key practices like waste reduction, water recycling, and using energy efficient equipment where possible. We have a focus on raising awareness and educating our employees on reducing our internal use of consumables and natural resources. In 2023, we achieved a certification whereby less than one percent of hazardous waste from our Science Center Campus in San Diego, California ended up in a landfill. In addition, we have a broad range of recycling and waste management initiatives at our manufacturing facilities and corporate offices. For example, at our internal manufacturing facilities we have addressed our use and recycling of paper products, aluminum cans, glass, electronics and plastic, as well as responsible disposal of non-recyclables and effective water management.
Cybersecurity
We face a number of cybersecurity risks in connection with our business. Although we have numerous controls to protect against common cybersecurity attacks, some attacks may still be effective. Our controls are designed to detect, triage and eradicate these attacks. Over the past three years, there have been no known material breaches, and no expenses related to the investigation of such breaches. For more information on our cybersecurity program, see Item 1C. Cybersecurity.
Corporate Citizenship
We are the industry leader in our commitment to deliver innovative, non-opioid pain therapies to transform the lives of patients. We are dedicated to the principles of social responsibility and good corporate governance. Our board of directors is comprised of industry leaders with extensive and diverse experience spanning business and scientific leadership. We hold ourselves to the highest standards and our Code of Business Conduct and Ethics reflects the business practices and principles of behavior that support this commitment. We are deeply invested in the welfare of our patients, employees, the environment and the communities where we live and work. We conduct our operations and manage our product and pipeline programs in a responsible manner and strive to comply with applicable laws, rules and regulations.
Over the past three years we provided support for charitable medical missions in Honduras, Ghana, Zambia, Guatemala, Ecuador, Mexico, India, Guyana, Palau, Nigeria and the Dominican Republic by donating EXPAREL to help support surgeries for patients in need; have supported the Louisiana State University Opioid Minimization Initiative as well as made a three-year commitment beginning in 2022 to donate EXPAREL to not-for-profit children’s hospitals each year.
Pacira Gives Back
As part of our ongoing commitment to support the communities where we live and work, we support a corporate giving campaign—Pacira Gives Back—which allows our employees to find local volunteer opportunities in their communities and help encourage use of a paid day off per year, known as our Community Day. Through Pacira Gives Back, employees can also make a donation to a not-for-profit organization of their choice with a dollar-for-dollar company match of up to $200 per employee annually.
Human Capital
Pacira Core Values
We are a team of dedicated and highly talented professionals focused on delivering innovative, non-opioid pain therapies to transform the lives of patients. We are an organization built on high ethical standards, an unwavering commitment to patients and transparent communications. We have a drive and a desire to improve the world around us and make a meaningful difference in the lives of patients, families, communities and society. Our core values guide our behaviors, choices and actions; foster our collaborative culture and further strengthen our organization.
The core values that underpin everything we do are:
•Every day, we are determined to achieve the extraordinary. We are bold enough to consistently do what others say cannot be done. Guided by our commitment to keep the patient at the center, we recognize that every day matters for a patient suffering in pain.
•Integrity is the foundation of who we are. We have the courage to tackle the biggest problems—and to do that, we know we have to follow the science. We believe in transparency, accountability and honesty, and we have the fortitude to make difficult decisions for the greater good.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 32
•We respect diverse talent and the collective power of a unified team. We recognize the role of teamwork toward tackling challenges and we believe in the value of different experiences and perspectives. We treat our people well—with fairness, equity and respect.
Corporate Sustainability Report
On an annual basis, we publish a Corporate Sustainability Report, or CSR, on our corporate website. The CSR report contains information about our people, our culture, patient and product safety, our commitment to our communities and opioid-sparing initiatives and our corporate governance and ethics. The foregoing reference to our CSR report is not intended to, nor shall it be deemed to, incorporate information in the CSR report or any other information contained on our corporate website into this Annual Report by reference.
Total Rewards
In order to attract and retain talent, we maintain broad-based benefits that are provided to all employees, including our 401(k) retirement plan with an employer matching contribution made each pay period, an employee stock purchase plan, flexible spending accounts, medical, dental and vision care plans, healthcare and dependent care savings accounts, life insurance, short- and long-term disability policies, paid vacation, paid sick time and paid company holidays. Additionally, we reward employees driving significant value creation with a variety of long-term and short-term incentives including a recognition platform, annual performance bonuses, stock options, restricted stock units and a long-term performance cash incentive. We also offer our executives the opportunity to participate in a deferred compensation plan with an employer match. We encourage our employees to give back in their communities and offer one paid day off per year to volunteer through our Community Day benefit. We regularly benchmark our rewards programs, adjusting as needed, to ensure our total rewards are competitive. We are committed to paying all our employees a fair and living wage.
Talent Management
We invest significantly in our future leaders by cultivating their growth and development. We regularly assess and identify our emerging talent and support their development with formal programs including classroom training, executive coaching, mentoring programs and “360-degree feedback” surveys geared towards our high-potential leaders. Many of our leaders participating in these programs advance to higher level positions within the organization. We are committed to soliciting employee feedback throughout their tenure with the organization, to shape organizational culture and to inform our people strategy. We conduct new hire surveys to solicit feedback on employees’ initial experiences with us to help ensure a successful onboarding and accelerate their assimilation into the organization and ability to contribute to our mission. We track turnover and employee engagement among other metrics, and conduct stay and exit interviews to ensure our talent strategy serves our goal of attracting, developing and retaining top talent to serve as our future leaders and stewards of our vision. In addition, we conduct mid-year and annual performance reviews for all employees to ensure regular discussions around performance, progress towards goals and professional development. We offer targeted selection training for interviewers to ensure a consistent methodology applied in identifying and hiring the best candidates for open positions and offer management skills trainings in live and virtual settings, along with online courses available to all employees through our learning platform.
Employee Wellbeing, Health and Safety
Pacira is committed to the total wellbeing of our employees and their families. We offer a range of benefits designed to meet individual needs and help employees and their families live healthy lives. This includes a variety of tools to promote total wellbeing in the areas of health, wealth, work and life to keep our employees and their families healthy, lower their healthcare costs and reduce stress. For example, we provide access to free biometric screenings, voluntary genetic testing (including enhanced support for cancer diagnoses), an employee assistance program, or EAP, and host in-person and webinar trainings on stress management and other EAP benefits, access to telemedicine including mental health visits and confidential counseling sessions for a number of needs, a health advocate service to help employees and their families navigate the healthcare system, a free consultation with an attorney for personal legal matters and discounted legal fees thereafter, activity challenges and more. We offer our eligible employees flexible work arrangements—including remote working opportunities, flexible schedules and reduced schedules to help achieve an appropriate work/life balance. Benefits that protect financial wellbeing are also provided, including but not limited to: a paid parental leave benefit, insurance to help protect assets during times of short- and long-term disability, life insurance and accidental death and dismemberment insurance, critical illness and accident insurance, financial education seminars on savings, debt and other financial topics, access to financial specialists, access to discounts on a variety of products and services and incentives to engage in a new or maintain a wellbeing activity. Additionally, our 401(k) retirement plan fiduciary is available to serve as a financial advisor to all employees with one-on-one personal financial planning. Furthermore, we maintain a recognition program based on our core values, known as Celebrate, through which we recognize each other’s commitment to making a meaningful difference for our patients and communities and create a shared culture where everyone is responsible for living and sustaining our core values. We also offer our employees and their covered family members in-network coverage of each of EXPAREL, ZILRETTA and iovera° through our employer-sponsored health benefit plans.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 33
We have a formal Environmental Health and Safety Program. It is our policy that everyone is entitled to a safe and healthful place to work. We recognize that accident prevention, employee wellness and efficiency of operations are directly related to quality, production and cost. Pacira operates its facilities in a manner that protects the health of its employees and minimizes the impact of its operations on the environment.
Diversity, Equity and Inclusion
We are committed to intentionally cultivating a culture of inclusion where all feel welcomed and valued for their backgrounds, perspectives and experiences. We hold one another accountable to promote trust and transparency in support of our communities and collective purpose. In support of our diversity, equity and inclusion vision, we have developed a strategy and multi-year roadmap, prioritizing education, training and diversity hiring, and developed a global labor and human rights policy. Our executive team and senior leaders have received Unconscious Bias and Inclusive Leadership training. We list our job postings on state job banks and distribute them to community engaged veteran, minority, women and diversity organizations as well as other targeted diversity sites. We are committed to evaluating our people processes to ensure we are attracting, developing, promoting and retaining diverse talent.
In 2018, we established P.O.W.E.R. (Preparing Our Women for Excellence and Results), an employee resource group open to all Pacira colleagues, focused on promoting leadership values, fostering a community of support and the advancement of women through professional development and networking opportunities. In 2020, we established a cross-functional diversity, equity and inclusion employee council to serve as an advisory board, comprised of employees who lead, advocate for, inform and communicate our corporate diversity, equity and inclusion strategic initiatives around four key areas: leadership development, diversity recruiting, culture and communications.
Employees
As of December 31, 2024, we had 790 employees, of which 788 are full-time and two are part-time. All of our employees are based in the U.S. except for seven employees based in the U.K. None of our employees are represented by a labor union, and we consider our current employee relations to be good.
Available Information
Our corporate website is located at www.pacira.com. We file reports and other information with the United States Securities and Exchange Commission, or SEC, as required by the Exchange Act, which are accessible on the SEC’s website at www.sec.gov. We also make available free of charge through our corporate website our Annual Report, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements and any amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act. We make these reports available through our corporate website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC. In addition, we regularly use our corporate website to post information regarding our business, product development programs and corporate governance, and we encourage investors to use our website, particularly the information in the sections entitled “Investors” and “News,” as a source of information about us. The foregoing references to our corporate website are not intended to, nor shall they be deemed to, incorporate information on our corporate website into this Annual Report by reference, and the inclusion of our corporate website address in this Annual Report is an inactive textual reference only and is not intended to be an active link to our corporate website.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 34
Item 1A. Risk Factors
In addition to the other information in this Annual Report, any of the factors set forth below could significantly and negatively affect our business, financial condition, results of operations or prospects. The trading price of our common stock may decline due to these risks. This section contains forward-looking statements. You should refer to the explanation of the qualifications and limitations on forward-looking statements beginning on page 1 of this Annual Report. These risk factors are not the only risks facing our Company. Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition or future results.
Risks Related to the Development and Commercialization of our Products and Product Candidates
Our success depends primarily on our ability to successfully commercialize EXPAREL and ZILRETTA.
We have invested a significant portion of our efforts and financial resources in the development and commercialization of our lead product, EXPAREL, which was first approved by the FDA on October 28, 2011 and commercially launched in April 2012. EXPAREL was approved by the EC (which included the U.K.) on November 16, 2020. During 2024, sales of EXPAREL accounted for 78% of our total revenue, and we expect EXPAREL sales will remain of primary importance for the foreseeable future. We added ZILRETTA to our product portfolio upon completing the Flexion Acquisition in November 2021 and it accounted for 17% of our total revenue in 2024. Our success primarily depends on our ability to continue to effectively commercialize EXPAREL and ZILRETTA. Our ability to effectively generate revenues from EXPAREL and ZILRETTA will depend on our ability to, among other things:
•create further market demand for EXPAREL and ZILRETTA through our marketing and sales activities and other arrangements established for their promotion;
•train, deploy and support a qualified sales force;
•secure formulary approvals for EXPAREL at a substantial number of targeted hospitals and ASCs;
•manufacture EXPAREL and ZILRETTA in sufficient quantities in compliance with requirements of regulatory agencies and at acceptable quality and pricing levels in order to meet commercial demand;
•implement and maintain agreements with wholesalers and distributors on commercially reasonable terms;
•appropriately help the market to take advantage of EXPAREL reimbursement at ASP plus 6 percent for Medicare patients receiving surgery in the outpatient setting;
•receive adequate levels of coverage and reimbursement for EXPAREL and ZILRETTA from commercial health plans and governmental health programs;
•maintain compliance with regulatory requirements;
•obtain regulatory approvals for additional indications and geographic expansion for the use of EXPAREL and ZILRETTA;
•ensure that our entire supply chain efficiently and consistently delivers EXPAREL and ZILRETTA to our customers; and
•maintain and defend our patent protection and regulatory exclusivity for EXPAREL and ZILRETTA, including our ongoing patent litigation lawsuits against eVenus, Jiangsu Hengrui and Fresenius. For more information on this matter, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
Any disruption in our ability to generate revenues from the sale of EXPAREL and ZILRETTA will have a material and adverse impact on our results of operations and financial condition.
Our efforts to successfully commercialize EXPAREL and ZILRETTA are subject to many internal and external challenges and if we cannot overcome these challenges in a timely manner, our future revenues and profits could be materially and adversely impacted.
EXPAREL has been a commercialized drug since April 2012. We continue to expend significant time and resources to train our sales force to be credible and persuasive in convincing physicians, hospitals and ASCs to use EXPAREL. In addition, we also must train our sales force to ensure that a consistent and appropriate message about EXPAREL is delivered to our potential customers. If we are unable to effectively train our sales force and equip them with effective materials, including medical and sales literature to help them inform and educate potential customers about the benefits and risks of EXPAREL and its proper administration, our efforts to successfully commercialize EXPAREL could be put in jeopardy, which could have a material adverse effect on our future revenues and profits.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 35
In addition to our extensive internal efforts, the successful commercialization of EXPAREL requires many third parties, over whom we have no control, to continue to utilize EXPAREL. These third parties include physicians and hospital pharmacy and therapeutics committees (“P&T committees”). Generally, before we can attempt to sell EXPAREL in a hospital, EXPAREL must be approved for addition to that hospital’s list of approved drugs, or formulary list, by the hospital’s P&T committee. A hospital’s P&T committee typically governs all matters pertaining to the use of medications within the institution, including the review of medication formulary data and recommendations for the appropriate use of drugs within the institution to the medical staff. The frequency of P&T committee meetings at hospitals varies considerably, and P&T committees often require additional information to aid in their decision-making process. Therefore, we may experience substantial delays in obtaining formulary approvals. Additionally, hospital pharmacists may be concerned that the cost of acquiring EXPAREL for use in their institutions will adversely impact their overall pharmacy budgets, which could cause pharmacists to resist efforts to add EXPAREL to the formulary, or to implement restrictions on the usage of EXPAREL or to encourage use of a lower cost dose than a surgeon or anesthesiologist would otherwise choose in order to control costs. Implementation of the NOPAIN Act in January 2025 now provides for separate reimbursement of qualifying non-opioids, like EXPAREL, administered during surgical procedures in the outpatient environment, is a significant policy advancement aimed at alleviating cost concerns for the Medicare population; however, we cannot guarantee that we will be successful in obtaining the approvals we need from enough P&T committees quickly enough to optimize hospital sales of EXPAREL. Even if we obtain hospital formulary approval for EXPAREL, physicians must still prescribe EXPAREL for its commercialization to be successful.
If EXPAREL does not achieve broader market acceptance, the revenues that we generate from its sales will be limited. The degree of market acceptance of EXPAREL also depends on a number of other factors, including:
•changes in the standard of care for the targeted indications for EXPAREL, which could reduce the marketing impact of any claims that we can make;
•the relative efficacy, convenience and ease of administration of EXPAREL;
•the prevalence and severity of adverse events associated with EXPAREL;
•the cost of treatment versus economic and clinical benefit, both in absolute terms and in relation to alternative treatments;
•the availability of adequate coverage or reimbursement by third parties, such as insurance companies and other healthcare payers, and by government healthcare programs, including Medicare and Medicaid, although implementation of the NOPAIN Act in January 2025 now provides Medicare coverage for separate reimbursement of qualifying non-opioids like EXPAREL in addition to its HCPCS J-code (J0666);
•the extent and strength of our marketing and distribution of EXPAREL;
•the safety, efficacy and other potential advantages over, and availability of, alternative treatments, including, in the case of EXPAREL, a number of products already used to treat pain in the hospital setting;
•potential future entrance into the market of a generic version of EXPAREL; and
•distribution and use restrictions imposed by regulatory agencies or to which we agree as part of a mandatory risk evaluation and mitigation strategy or voluntary risk management plan.
Our ability to effectively promote and sell EXPAREL and any product candidates that we may develop, license or acquire in the hospital or ASC marketplace will also depend on pricing and cost effectiveness, including our ability to produce a product at a competitive price and therefore achieve acceptance of the product onto hospital formularies, and our ability to obtain sufficient third-party coverage or reimbursement. We will also need to demonstrate acceptable evidence of safety and efficacy, as well as relative convenience and ease of administration. Market acceptance could be further limited depending on the prevalence and severity of any expected or unexpected adverse side effects associated with our product candidates, as well as products and therapies of competitors.
In addition, our approved labels for EXPAREL do not contain claims that EXPAREL is safer or more effective than competitive products and do not permit us to promote EXPAREL as being superior to competing products. Further, the availability of inexpensive generic forms of postsurgical pain management products may also limit acceptance of EXPAREL among physicians, patients and third-party payers. If EXPAREL does not achieve a broader level of acceptance among physicians, patients and third-party payers, we may not generate meaningful revenues from EXPAREL, and we may not remain profitable.
ZILRETTA is only approved for the management of OA pain of the knee for patients in the U.S. Successful commercialization of ZILRETTA is subject to many risks. Market acceptance of ZILRETTA will depend on a number of factors, including:
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 36
•the efficacy and safety as demonstrated in clinical trials;
•the ability to demonstrate the impact of real-world evidence;
•the timing and market introduction of competitive products;
•the product label and clinical indications for which the product is approved;
•acceptance by physicians, the medical community and patients of the product as a safe and effective treatment;
•the ability to distinguish safety and efficacy from existing, less expensive generic alternative therapies;
•the convenience of prescribing, administrating and initiating patients on the product;
•the potential and perceived advantages or value of the product over alternative treatments;
•the cost of treatment in relation to alternative treatments, including any similar generic treatments;
•the economics of a buy-and-bill product and discounts and rebates we offer;
•the availability of coverage and adequate reimbursement by third-party payers and government authorities to support pricing;
•the prevalence and severity of adverse side effects; and
•the effectiveness of sales and marketing efforts.
If ZILRETTA does not achieve a broader level of acceptance among physicians, patients and third-party payers, we may not generate meaningful revenues from ZILRETTA, and our business, financial condition and results of operations may suffer.
If we are unable to achieve and maintain adequate levels of third-party payer coverage and reimbursement for any product we may offer, on reasonable pricing terms, that product’s commercial success may be severely hindered.
ZILRETTA is a physician-administered product, and therefore physicians are required to purchase and manage the inventory of ZILRETTA, prior to administering the product to patients. Physicians obtain reimbursement for ZILRETTA from the applicable third-party payer, such as Medicare or a health insurance company, only after it has been administered to patients. This is called a “buy and bill” process. Because physicians are at financial risk for the cost of a “buy and bill” product until they have been reimbursed, concerns about reimbursement can impact a physician’s decision to use the product. The future growth of ZILRETTA depends on the availability of coverage and adequate reimbursement from third-party payers, including commercial payers, governmental healthcare programs, such as Medicare and Medicaid and managed care organizations, among others. EXPAREL reimbursement is subject to the same considerations in the ASC setting.
Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payers to reimburse all or part of the costs associated with their prescription drugs. Coverage and adequate reimbursement from third-party payers are critical to product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. The resulting reimbursement payment rates for EXPAREL and ZILRETTA might not be adequate or may require co-payments that patients find unacceptably high. If coverage and reimbursement for EXPAREL and ZILRETTA are not available or only available at limited levels, we may not be able to successfully commercialize EXPAREL and ZILRETTA, which could have a material adverse effect on our business, results of operations and financial condition.
We face significant competition from other pharmaceutical, medical device and biotechnology companies. Our operating results will suffer if we fail to compete effectively.
The pharmaceutical, medical device and biotechnology industries are intensely competitive and subject to rapid and significant technological change. Our major competitors include organizations such as major multinational pharmaceutical and medical device companies, established biotechnology companies and specialty pharmaceutical and generic drug companies. Many of our competitors have greater financial and other resources than we have, such as larger research and development staff, more extensive marketing, distribution, sales and manufacturing organizations and experience, more extensive clinical trial and regulatory experience, expertise in prosecution of intellectual property rights and access to development resources like personnel and technology. As a result, these companies may obtain regulatory approval more rapidly than we are able to and may be more effective in selling and marketing their products. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis technologies, drug products and medical devices that are more effective or less costly than EXPAREL, ZILRETTA, iovera° or any product candidate that we are currently developing or that we may develop, license or acquire, which could render our products obsolete and noncompetitive or significantly harm the commercial opportunity for EXPAREL, ZILRETTA, iovera° or any of our product candidates.
As a result of these factors, our competitors may obtain patent protection or other intellectual property rights that may limit our ability to develop other indications for, or commercialize, EXPAREL, ZILRETTA, iovera° or any of our product
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 37
candidates. Our competitors may also develop drugs or medical devices that are safer, more effective, useful or less costly than ours and may be more successful than us in manufacturing and marketing their products.
EXPAREL competes with well-established products with similar indications. Competing products available for postsurgical pain management include opioids such as morphine, fentanyl, meperidine and hydromorphone, each of which is available generically from several manufacturers, and several of which are available as proprietary products using novel delivery systems. Ketorolac, an NSAID, is also available generically in the U.S. from several manufacturers, and Caldolor (ibuprofen for injection), an NSAID, has been approved by the FDA for pain management and fever in adults. EXPAREL also faces competition from currently marketed non-opioid products such as bupivacaine, marcaine, ropivacaine and other anesthetics/analgesics, all of which are also used in the treatment of postsurgical pain and are available as either oral tablets, injectable dosage forms or administered using novel delivery systems. We are aware of at least one FDA-approved oral, non-opioid, highly selective NaV1.8 pain signal inhibitor that utilizes suzetrigine for the treatment of moderate-to-severe pain in adults. EXPAREL also competes with elastomeric pumps and catheter devices intended to provide bupivacaine over several days and with off-label combinations of other approved analgesics, called “cocktails”, that are combined by compound pharmacies in an attempt to extend the duration of pain control. Additional products may be developed for the treatment of acute pain, including new injectable NSAIDs, novel opioids, new formulations of currently available opioids and NSAIDs, long-acting local anesthetics and new chemical entities as well as alternative delivery forms of various opioids and NSAIDs. EXPAREL also competes with elastomeric bags and catheter devices intended to provide bupivacaine over several days.
ZILRETTA competes with immediate-release steroids and HA-containing products, as well as stem cell and PRP injections. Immediate-release TA and other injectable immediate-release steroids, which are the current IA standard of care for OA pain, are available in generic form and are therefore relatively inexpensive compared to the pricing for ZILRETTA. These generic steroids also have well-established market positions and familiarity with physicians, healthcare payers and patients. Although we believe the proven and extended pain relief evidenced in clinical trials demonstrate that ZILRETTA represents a clinically meaningful and highly efficacious option, it is possible that we will receive data from additional clinical trials or in a post-marketing setting from physician and patient experiences with the commercial product that does not continue to support such interpretations.
The iovera° system competes with cryotherapy devices as well as other devices such as cooled radio-frequency ablation devices that block or degenerate peripheral nerves involved in conducting pain signals.
Regulatory approval for any approved product is limited to those specific indications and conditions for which clinical safety and efficacy have been demonstrated, and allegations of our failure to comply with such approved indications could limit our sales efforts and have a material adverse effect on our business.
The marketing, labeling, advertising and promotion of prescription drugs and medical devices is strictly regulated. These regulations include standards and restrictions for direct-to-consumer advertising, industry-sponsored scientific and educational activities, promotional activities involving the internet and off-label promotion. Any regulatory approval granted is limited to those specific diseases and indications for which a product is deemed to be safe and effective by an appropriate regulatory agency. For example, the FDA-approved label for EXPAREL does not include an indication in obstetrical paracervical block anesthesia. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. If we are not able to obtain regulatory approval for any desired future indications for our products and product candidates, our ability to effectively market and sell our products may be reduced and our business may be adversely affected.
As an example, in the U.S. and Europe, while physicians may choose, and are generally permitted to prescribe drugs, medical devices or treatments for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical trials and approved by the regulatory authorities, our ability to promote the products is narrowly limited to those indications that are specifically approved by the FDA, EMA or MHRA. These “off-label” uses are common across medical specialties and may constitute an appropriate treatment for some patients in varied circumstances. Regulatory authorities generally do not regulate the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications by pharmaceutical and medical device companies on the subject of off-label use. In the U.S., although recent court decisions suggest that certain off-label promotional activities may be protected under the First Amendment of the U.S. Constitution, the scope of any such protection is unclear. If our promotional activities fail to comply with the FDA’s regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to issue warning letters or untitled letters, bring an enforcement action against us, suspend or withdraw an approved product from the market, require a recall or institute fines or civil fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our reputation and our business.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 38
If we are unable to establish and maintain effective marketing and sales capabilities or enter into agreements with third parties to market and sell our products, we may be unable to generate additional product revenues.
We are continuing to build our commercial infrastructure for the marketing, sale and distribution of pharmaceutical products. In order to continue commercializing our products effectively, we must continue to build our marketing, sales and distribution capabilities. The establishment, development and training of our sales force and related compliance plans to market our products is expensive and time consuming. In the event we are not successful in further developing our marketing and sales infrastructure, we may not be able to continue to successfully commercialize our products, including markets outside the U.S., which would limit our ability to generate additional product revenues.
In addition to our internal marketing and sales efforts, we have entered into agreements with third-party distributors to promote and sell EXPAREL in certain territories. For example, we previously had a co-promotion agreement with DePuy Synthes Sales, Inc. to market and promote the use of EXPAREL for orthopedic procedures in the U.S. market which we terminated effective January 2021. Additionally, in March 2020, Flexion entered into an exclusive license agreement with Hong Kong Tainuo Pharma Ltd., or HK Tainuo, and Jiangsu Tainuo Pharmaceutical Co. Ltd. for the development and commercialization (other than manufacturing) of ZILRETTA in Greater China. In July 2022, we submitted a letter to HK Tainuo associated with this license agreement seeking a mutual decision to end the licensing agreement and made a $13.0 million termination payment to HK Tainuo in January 2023. For more information, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
We may seek additional distribution arrangements in the future, including arrangements with third-party distributors to commercialize and sell our products in certain foreign countries, and there can be no assurance that such distributors and promoters will be successful in marketing and promoting our products. The use of distributors involves certain risks, including risks that such distributors will:
•not effectively distribute or support our products;
•not provide us with accurate or timely information regarding their inventories, the number of accounts using our products or complaints about our products;
•fail to comply with their obligations to us;
•fail to comply with laws and regulations to which they are subject, whether in the U.S. or in foreign jurisdictions;
•reduce or discontinue their efforts to sell or promote our products; or
•cease operations.
Any such failure may result in decreased sales and could lead us to incur other additional costs, which would have an adverse effect on our business.
We rely on third parties to perform many essential services for EXPAREL, ZILRETTA and iovera° and will rely on third parties for any other products that we commercialize. If these third parties fail to perform as expected or fail to comply with legal and regulatory requirements, our ability to commercialize EXPAREL, ZILRETTA and iovera° will be significantly impacted and we may be subject to regulatory sanctions.
We have entered into agreements with third-party service providers to perform a variety of functions related to the manufacture, sale and distribution of EXPAREL, ZILRETTA and iovera°, key aspects of which are out of our direct control. These service providers provide key services related to manufacturing our products, customer service support, warehousing and inventory program services, distribution services, contract administration and chargeback processing services, accounts receivable management and cash application services, financial management and information technology services. In addition, our finished goods inventory is stored at three warehouses maintained by two service providers. We substantially rely on these providers as well as other third-party providers that perform services for us, including entrusting our inventories of products to their care and handling. If these third-party service providers fail to comply with applicable laws and regulations, fail to meet expected deadlines or otherwise do not carry out their contractual duties to us, or encounter physical or natural damage at their facilities, our ability to deliver product to meet commercial demand would be significantly impaired. In addition, we may engage third parties to perform various other services for us relating to adverse event reporting, safety database management, fulfillment of requests for medical information regarding our product candidates and related services. If the quality or accuracy of the data maintained by these service providers is insufficient, we could be subject to regulatory sanctions.
Distribution of our pMVL-based products, including EXPAREL, requires cold-chain distribution provided by third parties, whereby the product must be maintained between specified temperatures. If a problem occurs in our cold-chain distribution processes, whether through our failure to maintain our products or product candidates between specified temperatures or because of a failure of one of our distributors or partners to maintain the temperature of the products or product candidates, the product or product candidate could be adulterated and rendered unusable. We have obtained limited inventory and cargo insurance coverage for our products. However, our insurance coverage may not reimburse us or may not be sufficient
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 39
to reimburse us for any expenses or losses we may suffer. This could have a material adverse effect on our business, financial condition, results of operations and reputation.
We may need to increase the size of our organization and effectively manage our sales force, and we may experience difficulties in managing growth.
As of December 31, 2024, we had 790 employees. We may need to expand our personnel resources in order to manage our operations and sales of EXPAREL, ZILRETTA, iovera°, any of our product candidates or products we acquire or in-license. Our management, personnel, systems and facilities currently in place may not be adequate to support this future growth. In addition, we may not be able to recruit and retain qualified personnel in the future, particularly in sales and marketing positions, due to competition for personnel among pharmaceutical and medical device businesses, and the failure to do so could have a significant negative impact on our future product revenues and results of operations. Our need to effectively manage our operations, growth and various projects requires that we:
•continue the hiring and training of an effective commercial organization for the commercialization of EXPAREL, ZILRETTA and iovera°, and establish appropriate systems, policies and infrastructure to support that organization;
•continue to establish and maintain effective relationships with distributors and commercial partners for the promotion and sale of our products;
•ensure that our distributors, partners, suppliers, consultants and other service providers successfully carry out their contractual obligations, provide high quality results and meet expected deadlines;
•manage our development efforts and clinical trials effectively;
•expand our manufacturing capabilities and effectively manage our co-production arrangements with Thermo Fisher and Carlisle;
•continue to carry out our own contractual obligations to our licensors and other third parties; and
•continue to improve our operational, financial and management controls, reporting systems and procedures.
We may be unable to successfully implement these tasks on a larger scale and, accordingly, may not achieve our development and commercialization goals. Additionally, these tasks may impose a strain on our administrative and operational infrastructure. If we are unable to effectively manage our growth, our product sales and resulting revenues will be negatively impacted.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel.
We may not be able to attract or retain qualified management and commercial, scientific and clinical personnel due to the intense competition for qualified personnel among biotechnology, pharmaceutical, medical device and other businesses, as well as universities, non-profit research organizations and government entities, particularly in and around the San Francisco Bay Area; San Diego, California; northern New Jersey/New York City metro and Tampa, Florida. If we are not able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our industry has experienced a high rate of turnover of management personnel in recent years. We are highly dependent on the development and manufacturing expertise for our products and pMVL drug delivery technology and the commercialization expertise of management. In particular, we are highly dependent on the skills and leadership of our senior management team. If we lose one or more of these key employees, our ability to successfully implement our business strategy could be seriously harmed. Replacing key employees may be difficult, costly and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire from this limited talent pool is intense, and we may be unable to hire, train, retain or motivate additional key personnel.
Competition for highly skilled personnel, including management and commercial, scientific and clinical personnel, is extremely competitive, particularly in and around the San Francisco Bay Area; San Diego, California; northern New Jersey/New York City metro and Tampa, Florida. While we offer remote work arrangements, which allows us to recruit employees residing outside of the geographic areas we operate in, we have experienced—and may continue to experience—some difficulty identifying and hiring qualified personnel, especially as we pursue our growth strategy. We may not be able to hire or retain such personnel at compensation or flexibility levels consistent with our existing policies. We periodically review our compensation levels and employee benefits to ensure they remain competitive and have increased them when we believe market conditions warrant it. We may need to further increase our existing compensation levels and employee benefits in response to competition or labor shortages, which would increase our operating costs and reduce our margins. Furthermore, a sustained labor shortage, lack of skilled labor, increased turnover or labor cost inflation (for example, such as that initially caused by the
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 40
recent COVID-19 pandemic), or as a result of general macroeconomic factors, could lead to increased costs, which could negatively affect our ability to efficiently operate our overall business and have other adverse effects on our results of operations and financial condition. Many of the companies with which we compete for experienced employees have greater resources than we have and may be able to offer more attractive terms of employment. In particular, candidates making employment decisions, specifically in our industry, often consider the value of any long-term incentive compensation, including stock-based compensation they may receive in connection with their employment. Any significant volatility in the price of our common stock may adversely affect our ability to attract or retain experienced, highly skilled and technical personnel.
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for EXPAREL, ZILRETTA, iovera° or any product candidates that we may develop and may have to limit their commercialization.
The use of EXPAREL, ZILRETTA, iovera° and any product candidates that we may develop, license or acquire in clinical trials and the sale of any products for which we obtain regulatory approval expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers or others using, administering or selling our products. We have been a party of these suits in the past and may be again in the future. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
•loss of revenue from decreased demand for our products and/or product candidates;
•impairment of our business reputation or financial stability;
•costs of any related litigation;
•substantial monetary awards to patients or other claimants;
•diversion of management attention;
•withdrawal of clinical trial participants and potential termination of clinical trial sites or entire clinical programs; and
•the inability to commercialize our products and/or product candidates.
We have limited product liability insurance coverage for our products and our clinical trials with a $10.0 million annual aggregate coverage limit. However, our insurance coverage may not reimburse us, or may not be sufficient to reimburse us, for any expenses or losses we may suffer, including our indemnification obligations to other parties. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage on acceptable terms, at a reasonable cost or in sufficient amounts to protect us against losses due to liability. We intend to expand our insurance coverage to include the sale of additional commercial products upon regulatory approval for our product candidates in development, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing, or at all. On occasion, large judgments have been awarded in class action lawsuits based on drugs or medical devices that had unanticipated side effects. A successful product liability claim or series of claims brought against us could cause the price of our common stock to fall and, if judgments exceed our insurance coverage, could decrease our cash balance and adversely affect our business.
If we fail to manufacture our products in sufficient quantities and at acceptable quality and pricing levels, or to fully comply with CGMP regulations, we may face delays in the commercialization of these products or be unable to meet market demand, and may lose potential revenues.
The manufacture of our products requires significant expertise and capital investment, including the development of advanced manufacturing techniques, process controls and the use of specialized processing equipment. We must comply with federal, state and foreign regulations, including the FDA’s regulations governing CGMP, enforced by the FDA through its facilities inspection program and by similar regulatory authorities in other jurisdictions where we do business. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. The FDA or similar foreign regulatory authorities at any time may implement new standards or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of our products. Any failure by us or our manufacturing partners to comply with applicable regulations may result in fines and civil penalties, suspension of production, product seizure or recall, operating restrictions, imposition of a consent decree, modification or withdrawal of product approval or criminal prosecution and would limit the availability of our product. Any manufacturing defect or error discovered after products have been produced and distributed also could result in significant consequences, including costly recall procedures, re-stocking costs, damage to our reputation and the potential for product liability claims.
The FDA requires manufacturers of medical devices to adhere to certain regulations, including the FDA’s QSRs, which requires periodic audits, design controls, quality control testing and documentation procedures, as well as complaint evaluations
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 41
and investigations. Regulations regarding the development, manufacture and sale of medical products are evolving and are subject to change in the future.
If we are unable to produce the required commercial quantities of our products to meet market demand those products on a timely basis or at all, or if we fail to comply with applicable laws for the manufacturing of our products, we will suffer damage to our reputation and commercial prospects, we will lose potential revenues and we may be required to expend significant time and resources to resolve any such issues.
We may need to expand our manufacturing operations or outsource such operations to third parties.
To successfully meet future customer demand for EXPAREL, ZILRETTA and iovera°, we may need to expand our existing commercial manufacturing facilities or establish larger-scale commercial manufacturing capabilities. For example, in February 2024 the FDA approved our new, larger-scale manufacturing suite at our Science Center Campus in San Diego, California. In addition, as our drug development pipeline increases and matures, we will have a greater need for clinical trial and commercial manufacturing capacity. As a result, we must continuously improve our manufacturing processes to allow us to reduce our production costs. We may not be able to manufacture our drugs and/or medical devices at a cost or in quantities necessary to be commercially successful.
The build-up or other expansion of our internal manufacturing capabilities for EXPAREL production at our Science Center Campus in San Diego, California and co-production capabilities for EXPAREL and ZILRETTA at Thermo Fisher’s Swindon, U.K. site, exposes us to significant up-front fixed costs. If market demand for our products does not align with our expanded manufacturing capacity, we may be unable to offset these costs or achieve economies of scale, and our operating results may be adversely affected as a result of high operating expenses, including overhead. Alternatively, if we experience demand for our products in excess of our estimates, our facilities may be insufficient to support higher production volumes, which could harm our customer relationships and overall reputation. Our ability to meet such excess demand could also depend on our ability to raise additional capital on terms acceptable to us and effectively scale our manufacturing operations.
In addition, the procurement time for the equipment that we use to manufacture EXPAREL and ZILRETTA requires long lead times. Therefore, we may experience delays, additional or unexpected costs and other adverse events in connection with our capacity expansion projects, including those associated with potential delays in the procurement of manufacturing equipment required to manufacture EXPAREL or ZILRETTA.
In addition to expanding our internal manufacturing facilities, we may enter into arrangements with third parties to supply, manufacture, package, test and/or store EXPAREL, ZILRETTA, iovera° or our product candidates, such as our manufacturing arrangements with Thermo Fisher and Carlisle. Entering into such arrangements requires testing and compliance inspections, regulatory agency approvals and development of the processes and facilities necessary for the production of our products. Such arrangements also involve additional risks, many of which would be outside of our control. Such risks include disruptions or delays in production, manufactured products that do not meet our required specifications, the failure of such third-party manufacturers to comply with CGMP regulations or other regulatory requirements, protection of our intellectual property and manufacturing processes, loss of control of our complex manufacturing processes, inabilities to fulfill our commercial needs and financial risks in connection with our investment in setting up a third-party manufacturing process, such as the substantial capital outlays that were required by us to assist in setting up our manufacturing process at Thermo Fisher’s facility in Swindon, U.K.
If we are unable to timely achieve and maintain satisfactory production yields and quality, whether through our internal manufacturing capabilities or arrangements with contract manufacturers, our relationships with customers and our reputation may be harmed and our revenues could decrease.
Our inability to continue manufacturing adequate quantities of our products could result in a disruption in the supply to our customers and partners, which could have a material adverse impact on our business and results of operations.
EXPAREL is currently manufactured at our facilities in San Diego, California; both EXPAREL and ZILRETTA are currently manufactured at the Thermo Fisher facility in Swindon, U.K., iovera° handpieces are currently manufactured at our facility in San Diego, California and iovera° Smart Tips are currently manufactured at Carlisle’s facility in Tijuana, Mexico. These facilities are the only currently approved sites in the world for manufacturing EXPAREL, ZILRETTA and iovera°. We may experience temporary or prolonged suspensions in production of our products due to issues in our manufacturing process that must be remediated or in response to inspections conducted by the FDA or similar foreign regulatory authorities, which could have a material adverse effect on our business, financial position and results of operations.
Our San Diego facilities in California, the Thermo Fisher facility in Swindon, U.K. and the Carlisle facility in Tijuana, Mexico are also subject to the risks of a natural or man-made disaster, including, but not limited to, storms, earthquakes, floods, fires or other business disruptions. In addition, we have obtained limited property and business interruption insurance coverage for our manufacturing sites in San Diego, U.K. and Mexico. However, our insurance coverage may not reimburse us, or may not be sufficient to reimburse us, for any expenses or losses we may suffer. There can be no assurance that we would be able to
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 42
meet our requirements for EXPAREL, ZILRETTA or iovera° if there were a catastrophic event or failure of our current manufacturing systems. If we are required to change or add a new manufacturer or supplier, the process would likely require prior FDA and/or equivalent foreign regulatory authority approval, would be very time consuming and could be expensive. An inability to continue manufacturing adequate supplies of EXPAREL, ZILRETTA or iovera° at our facilities could result in a disruption in the supply of these products to our customers and partners and a breach of our contractual obligations to such counterparties.
Our co-production and other agreements with Thermo Fisher may involve unanticipated expenses and delays.
We and Thermo Fisher have entered into a Co-Production Agreement, Technical Transfer and Service Agreement and Manufacturing and Supply Agreement. Under these agreements, Thermo Fisher undertook certain technical transfer activities and construction services to prepare their Swindon, U.K. facility for the manufacture of EXPAREL. We agreed with Thermo Fisher, among other things, to provide them with the process equipment necessary to manufacture EXPAREL at this facility.
Prior to the Flexion Acquisition, Flexion and Thermo Fisher entered into the ZILRETTA Manufacturing and Supply Agreement and the ZILRETTA Technical Transfer and Service Agreement related to the manufacture of ZILRETTA at the same Thermo Fisher site in Swindon, U.K. where our EXPAREL suites are located. Thermo Fisher agreed to undertake certain transfer activities and construction services needed to prepare its facility for the commercial manufacture of ZILRETTA in dedicated manufacturing suites. Flexion provided Thermo Fisher with certain equipment and materials necessary to manufacture ZILRETTA at this facility.
The Thermo Fisher facilities required regulatory approval prior to any production and manufacturing of EXPAREL and ZILRETTA. While we have anticipated and budgeted for additional capital expenditures associated with the Thermo Fisher suites for both EXPAREL and ZILRETTA, if the Thermo Fisher suites do not maintain their regulatory approvals (or fail to receive any additional regulatory approvals that may be needed in the future), this could have a material adverse effect on our business, financial position and results of operations.
Further, the production under these agreements involve additional risks, many of which would be outside of our control, such as disruptions or delays in production, manufactured products that do not meet our required specifications, the failure of Thermo Fisher to comply with CGMP regulations or other regulatory requirements, protection of our intellectual property and manufacturing processes, loss of control of our complex manufacturing processes and inabilities to fulfill our commercial needs.
We rely on third parties for the timely supply of specified raw materials and equipment for the manufacture of EXPAREL, ZILRETTA and iovera°. Although we actively manage these third-party relationships to provide continuity and quality, some events which are beyond our control could result in the complete or partial failure of these goods and services. Any such failure could have a material adverse effect on our financial condition and operations.
We purchase certain raw materials and equipment from various suppliers in order to manufacture our products. The acquisition of certain materials may require considerable lead times, and our ability to source such materials is also dependent on logistics providers. If we are unable to source the required raw materials and equipment on a timely basis or receive materials that do not meet our specifications, we may experience delays in manufacturing, which would have a material and adverse impact on our results of operations and financial condition as well as not being able to meet our customers’ or partners’ demands for our products. Additionally, we have some single sources of supply for certain materials and equipment used in our manufacturing processes. Should the need arise to qualify additional suppliers or change suppliers, we could bear substantial costs and could fail to maintain adequate production levels to meet demand for our products. In addition, we and our third-party suppliers must comply with federal, state and foreign regulations, including CGMP regulations, and any failure to comply with applicable regulations, or failure of government agencies to provide necessary authorizations, may harm our ability to manufacture and commercialize our products on a timely and competitive basis, which could result in decreased product sales and lower revenues.
We may also experience additional disruptions that could severely impact our supply chain, such as those caused by the recent COVID-19 pandemic, which would disrupt our clinical trials and commercialization efforts. To the extent that our vendors are unable to comply with their obligations under our agreements or cannot deliver goods or services timely, our ability to continue meeting commercial demand for our products or advancing development of our product candidates may become impaired. Furthermore, raw materials and supplies needed to manufacture COVID-19 vaccines were backed by government mandate orders, which previously impacted our suppliers’ ability to supply critical raw materials for our products. There can be no assurances that similar government mandates will not occur in the future or that critical raw materials will not be prioritized for other products.
Supply chain disruptions could interrupt product manufacturing and global logistics and increase product costs.
We rely on international shipping to receive certain raw materials and to transport our products to their various geographic markets. Delays in shipping may cause us to use more expensive expedited freight methods to ship our products or receive raw
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 43
materials. For example, the recent COVID-19 pandemic and related governmental actions caused delays in shipments. During the COVID-19 pandemic, we experienced increased lead-times for obtaining raw materials, including those caused by temporary closures and worker shortages. In addition, global inflation has contributed to already higher incremental freight costs and such inflation may continue to result in further increases in freight costs. Any tariffs imposed on imported goods may also raise the cost of certain raw materials and other property, plant and equipment costs. Failure to adequately produce and timely ship our products to customers could lead to lost potential revenue, failure to meet customer demand and strained relationships with customers—including wholesalers. Failure to adequately procure raw materials or equipment or produce and timely ship our products to customers could lead to lost potential revenue, failure to meet customer demand and strained relationships with our customers—including wholesalers. Despite our actions to mitigate these impacts and the pressures related to the COVID-19 pandemic having eased, we may still be impacted by global logistics challenges in the future.
Our operations are dependent on the global supply chain and impacts of supply chain constraints and inflationary pressure could adversely impact our operating results.
Our operations have been, and may continue to be, impacted by supply chain constraints and raw material shortages, resulting in increased material costs, longer lead times and increased freight costs first caused, in part, by the recent COVID-19 pandemic, the uncertain economic environment and macroeconomic trends. In addition, current or future governmental policies, including the imposition of tariffs, may increase the risk of inflation, which could further increase the costs of raw materials and components for our business. Tariffs can increase our manufacturing costs and if costs of goods continue to increase, our suppliers may seek price increases from us. If we are unable to mitigate the impact of supply chain constraints and inflationary pressure through price increases or other measures, our results of operations and financial condition could be negatively impacted. Even though we are working to alleviate supply chain constraints through various measures, we are unable to predict the impact of these constraints on the timing of revenue and operating costs of our business in the near future. Raw material supply shortages and supply chain constraints, including cost inflation, have impacted and could continue to negatively impact our ability to meet increased demand, which in turn could impact our net sales revenues and market share. We expect the situation to remain fluid as foreign exchange rates fluctuate and as inflationary pressure continues.
Our future growth depends—in part—on our ability to identify, develop, acquire or in-license products and if we do not successfully identify, develop, acquire or in-license related product candidates or integrate them into our operations, we may have limited growth opportunities.
An important part of our business strategy is to continue to develop a pipeline of product candidates by developing, acquiring or in-licensing products, businesses or technologies that we believe are a strategic fit with our focus on the hospital marketplace. However, these business activities may entail numerous operational and financial risks, including:
•significant capital expenditures;
•the difficulty or inability to secure financing to fund development activities for such development, acquisition or in-licensed products or technologies;
•the incurrence of substantial debt or dilutive issuances of securities to pay for the development, acquisition or in-licensing of new products and any related milestone or earn-out payments;
•the successful integration of acquired products, businesses or technologies into our operations, and achieving the expected benefits and synergies from such acquisitions;
•the disruption of our business and diversion of our management’s time and attention;
•higher than expected development, acquisition or in-license and integration costs;
•exposure to unknown liabilities;
•the difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel;
•the inability to retain key employees of any acquired businesses;
•the difficulty entering markets in which we have limited or no direct experience;
•the difficulty in managing multiple product development programs; and
•the inability to successfully develop new products or clinical failure.
We have limited resources to identify and execute the development, acquisition or in-licensing of products, businesses and technologies and integrate them into our current infrastructure. We may compete with larger pharmaceutical and medical device companies and other competitors, including public and private research organizations, academic institutions and government agencies, in our efforts to establish new collaborations and in-licensing opportunities. These competitors may have access to greater financial resources, research and development staffs and facilities than us and may have greater expertise in identifying and evaluating new opportunities. We may not be successful in locating and acquiring or in-licensing additional desirable
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 44
product candidates on acceptable terms or at all. We may also not be successful in developing or commercializing our current product candidates. Such efforts may require the dedication of significant financial and personnel resources, and any diversion of resources may also disrupt our management from expanding on EXPAREL, ZILRETTA or iovera° net product sales. Moreover, we may devote resources to potential development, acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts.
We make substantial investments in research and development and unsuccessful investments could materially adversely affect our business, financial condition and results of operations.
The industry in which we compete is characterized by intense competition, rapid technological change, changes in customer requirements, frequent new product introductions and enhancements, evolving industry standards and new therapeutic delivery methods. In order to remain competitive, we have made, and expect to continue to make, significant investments in research and development. If we fail to develop new and enhanced products and technologies, if we focus on products and technologies that do not become widely adopted, or if new competitive products and technologies that we do not support become widely accepted, demand for our products may be reduced. Increased investments in research and development or unsuccessful research and development efforts could cause our cost structure to fall out of alignment with the demand for our products, which would have a negative impact on our business, financial condition and results of operations.
Our business involves the use of hazardous materials and we must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our manufacturing activities involve the controlled storage, use and disposal of hazardous materials, including the components of our products, product candidates and other hazardous compounds. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling, release and disposal of, and exposure to, these hazardous materials. Violation of these laws and regulations could lead to substantial fines and penalties. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials or unintended failure to comply with these laws and regulations. In the event of an accident or failure to comply with these laws and regulations, federal, state or local authorities may curtail our use of these materials and interrupt our business operations. In addition, we could become subject to potentially material liabilities relating to the investigation and cleanup of any contamination, whether currently unknown or caused by future releases.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our product candidates.
Our business model is to commercialize our products in the U.S. while seeking collaboration arrangements with pharmaceutical or biotechnology companies for the development or commercialization of our products in other countries. Accordingly, we may enter into collaboration arrangements in the future on a selective basis. Any future collaboration arrangements that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaboration arrangements.
Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate(s) and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision-making authority.
Collaborations with pharmaceutical and/or medical device companies and other third parties often are terminated or allowed to expire by the other party. Any such termination or expiration would adversely affect us financially and could harm our business reputation.
Clinical trials are expensive, lengthy and have uncertain outcomes, and results of earlier studies and trials may not be predictive of future trial results. Clinical trials may fail to demonstrate the safety and efficacy of our drug products or medical devices, which could prevent or significantly delay obtaining regulatory approval.
Prior to receiving approval to commercialize any of our drug products or medical devices, we must demonstrate with scientifically appropriate and statistically sound evidence from well-controlled clinical trials, and to the satisfaction of the FDA and other regulatory authorities, that each of the products are both safe and effective. For each drug product, we will need to demonstrate its efficacy and monitor its safety throughout the process. Clinical trials are expensive and can take many years to complete, and their outcomes are inherently uncertain. If such development is unsuccessful, our business and reputation would be harmed and our stock price would be adversely affected.
All of our drug and medical device products are prone to the risks of failure inherent in development. Clinical trials of new drug and medical device products sufficient to obtain regulatory approval are expensive and take years to complete. We may not be able to successfully complete clinical testing within the time frame we have planned, or at all. We may experience
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 45
numerous unforeseen events during, or as a result of, the clinical trial process which could delay or prevent us from receiving regulatory approval or commercializing our products. In addition, the results of preclinical studies and early-stage clinical trials of our products do not necessarily predict the results of later-stage clinical trials. Later-stage clinical trials may fail to demonstrate that a product is safe and effective despite having progressed through initial clinical testing. Even if we believe the data collected from clinical trials of our products is promising, such data may not be sufficient to support approval by regulatory agencies. Preclinical and clinical data can be interpreted in different ways, and results generated in our completed clinical trials do not ensure that any future clinical trials will be successful or consistent with the results generated in previous trials.
Accordingly, regulatory authorities could interpret such data in different ways than we or our partners do, which could delay, limit or prevent regulatory approval. Regulatory authorities, our institutional review boards, our contract research organizations, or CROs, or we ourselves may suspend or terminate our clinical trials for our drug products and medical devices. Any failure or significant delay in completing clinical trials for our drug products or medical devices, or in receiving regulatory approval for the sale of any of our drugs or medical devices, may severely harm our business and reputation. Even if we receive regulatory approvals, our drug and medical device products may later exhibit adverse effects that may limit or prevent their widespread use, may cause a regulatory authority to revoke, suspend or limit their approval, or may force us to withdraw products derived from those drug or medical device products from the market.
Our dependence on contract research organizations could result in delays in and additional costs for our drug or medical device development efforts.
We may rely on CROs to perform preclinical testing and clinical trials for drug or medical device candidates that we choose to develop without a collaborator. If the CROs that we hire to perform our preclinical testing and clinical trials or our collaborators or licensees do not meet deadlines, do not follow proper procedures or a conflict arises between us and our CROs, our preclinical testing and clinical trials may take longer than expected, may be delayed or may be terminated. If we were forced to find a replacement CRO to perform any of our preclinical testing or clinical trials, we may not be able to find a suitable replacement on favorable terms, if at all. Even if we were able to find another CRO to perform a preclinical test or clinical trial, any material delay in a test or clinical trial may result in significant additional expenditures that could adversely affect our operating results. Events such as these may also delay regulatory approval for our drug or medical device candidates or our ability to commercialize our products.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials and sometimes other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays outside of our control.
We rely on clinical investigators and clinical sites to enroll patients and sometimes third parties to manage our trials and to perform related data collection and analysis. However, we may be unable to control the amount and timing of resources that the clinical sites which conduct the clinical testing may devote to our clinical trials.
Our clinical trials may be delayed or terminated due to the inability of our clinical investigators to enroll enough qualified patients. Patient enrollment depends on many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites and the eligibility criteria for the trial. If our clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to enroll them on our planned schedule, we may face increased costs, delays or termination of the trials, which could delay or prevent us from obtaining regulatory approvals for our product candidates.
Our agreements with clinical investigators and clinical sites for clinical testing and for trial management services place substantial responsibilities on these parties, which could result in delays in, or termination of, our clinical trials if these parties fail to perform as expected. For example, if any of our clinical trial sites fail to comply with FDA-approved GCPs, we may be unable to use the data gathered at those sites. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our product candidates.
We are subject to periodic litigation, which could result in losses or unexpected expense of time and resources.
From time to time, we are called upon to defend ourselves against lawsuits relating to our business and intellectual property. Due to the inherent uncertainties of litigation, we cannot accurately predict the ultimate outcome of any such proceedings. An unfavorable outcome in these or other proceedings could have an adverse impact on our business, financial condition and results of operations. In addition, any significant litigation in the future, regardless of its merits, could divert management’s attention and resources from our operations that are needed to successfully run our business and also result in substantial legal fees. In addition, if our stock price is volatile, we may become involved in securities class action lawsuits in the future.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 46
For information about our legal proceedings, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
Guidelines and recommendations published by various organizations could reduce the demand for or use of our products.
Government agencies promulgate regulations and guidelines directly applicable to us and to our products and product candidates. In addition, professional societies, practice management groups, private health and science foundations and other organizations from time to time may publish papers, guidelines or recommendations to the healthcare and patient communities with respect to specific products or classes of products. Recommendations of government agencies or these other groups or organizations may relate to such matters as usage, dosage, route of administration and use of concomitant therapies. Recommendations or guidelines that do not recognize a product, suggest limitations or inadequacies of a product or suggest the use of competitive or alternative products as the standard of care to be followed by patients and healthcare providers could result in decreased use or adoption of any of our products which could have an adverse impact on our business, financial condition and results of operations.
Regulatory Risks
Our business could be materially adversely affected if a regulatory or enforcement agency determines that we are promoting or have in the past promoted the “off-label” use of our products.
The marketing, labeling, advertising and promotion of prescription drugs and medical devices is strictly regulated. These regulations include standards and restrictions for direct-to-consumer advertising, industry-sponsored scientific and educational activities, promotional activities involving the internet and off-label promotion. According to these regulations, companies may not promote drugs or medical devices for “off-label” uses—that is—uses that are not consistent with the product’s labeling and that differ from those that were approved by the FDA, EMA, MHRA or other regulatory agency. For example, the FDA-approved label for EXPAREL does not include an indication in obstetrical paracervical block anesthesia. In addition to the FDA approval required for new formulations or device enhancements, any new indication for an approved product also requires FDA approval. If we are not able to obtain regulatory approval for any desired future indications for our products and product candidates, our ability to effectively market and sell our products may be reduced and our business may be adversely affected.
As an example, while physicians may choose, and are generally permitted to prescribe drugs and/or medical devices for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical trials and approved by a regulatory authority, our ability to promote the products is narrowly limited to those indications that are approved by the FDA or other regulatory agency. “Off-label” uses are common across medical specialties and may constitute an appropriate treatment for some patients in varied circumstances. Regulatory authorities generally do not regulate the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications by pharmaceutical and medical device companies on the subject of off-label use. Although recent court decisions suggest that certain off-label promotional activities may be protected under the First Amendment of the U.S. Constitution, the scope of such protection is unclear. Moreover, while we promote our products consistent with what we believe to be the approved indication for our drugs and medical devices, regulators may disagree. If a regulatory agency determines that our promotional activities fail to comply with their regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow rules and guidelines relating to promotion and advertising may cause a regulatory body to issue warning letters or untitled letters, bring an enforcement action against us, suspend or withdraw an approved product from the market, require a recall or institute fines or civil fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our reputation and our business.
For example, in September 2014, we received a warning letter from the FDA’s Office of Prescription Drug Promotion (OPDP) pertaining to certain promotional aspects of EXPAREL. We took actions to immediately address the FDA’s concerns and minimize further disruption to our business. Ultimately, however, in September 2015, we, along with two independent physicians, filed a lawsuit in federal court against the FDA and other governmental defendants seeking to exercise our lawful rights to communicate truthful and non-misleading information about EXPAREL. The complaint outlined our belief that the FDA’s warning letter received in September 2014 and regulations restricting our truthful and non-misleading speech about EXPAREL violated the Administrative Procedure Act and the First and Fifth Amendments of the U.S. Constitution. The lawsuit sought a declaration and injunctive relief to permit us to promote EXPAREL consistent with its approved indication and pivotal trials that supported FDA approval. In December 2015, we announced that the FDA had formally withdrawn the September 2014 Warning Letter via a “Rescission Letter,” and that the FDA and Pacira had reached an amicable resolution of the lawsuit. As part of the resolution of this matter, the FDA confirmed that EXPAREL was broadly approved for “administration into the surgical site to produce postsurgical analgesia” in a variety of surgeries not limited to those studied in its pivotal trials. The FDA also approved a labeling supplement for EXPAREL that further clarified that EXPAREL was not limited to any specific surgery type or site, that the proper dosage and administration of EXPAREL is based on various patient and procedure-specific factors, that there was a significant treatment effect for EXPAREL compared to placebo over the first 72 hours in the pivotal hemorrhoidectomy trial and that EXPAREL may be admixed with bupivacaine, provided certain
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 47
medication ratios are observed. The Warning Letter and labeling supplement only applied to the infiltration indication that was approved at that time, and does not apply to the interscalene brachial plexus nerve block indication subsequently approved by the FDA in April 2018, the use of EXPAREL in patients six years of age and older for single-dose infiltration to produce postsurgical local analgesia that was approved by the FDA in March 2021 and the indications for adductor canal block and sciatic nerve block in the popliteal fossa that were FDA approved in November 2023. We and the FDA agreed that, in future interactions, the parties will deal with each other in an open, forthright and fair manner.
In April 2015, we received a subpoena from the U.S. Department of Justice, U.S. Attorney’s Office for the District of New Jersey, requiring the production of a broad range of documents pertaining to marketing and promotional practices related to EXPAREL. In July 2020, we formally entered into settlement agreements that resolved all outstanding investigations and claims by the U.S. Department of Justice, the U.S. of Health and Human Services, various States Attorneys’ General and a private plaintiff (the “Plaintiffs”). This agreement concluded a five-year investigation related to the sale and marketing of EXPAREL. Under the various settlement agreements, we paid a global settlement of $3.5 million. As part of the settlement, we admitted to no wrongdoing and explicitly denied the Plaintiffs’ allegations. We have been given assurances that this settlement concluded the investigation that originated from the U.S. Department of Justice subpoena in April 2015.
We are unable to predict whether any future regulatory actions will have an effect on our product sales, and even if such actions are ultimately resolved favorably, our sales may suffer due to reputational or other concerns. We can make no assurances that we will not receive warning letters in the future from the FDA or other regulatory authority or be subject to other regulatory action. As noted above, any regulatory violation or allegations of a violation may have a material adverse effect on our reputation and business.
We may not receive regulatory approval for any of our product candidates, or the approval may be delayed for various reasons, including successful challenges to the FDA’s interpretation of Section 505(b)(2), which would have a material adverse effect on our business and financial condition.
We may experience delays in our efforts to obtain regulatory approval from the FDA for any of our product candidates, and there can be no assurance that such approval will not be delayed, or that the FDA will ultimately approve these product candidates. Although the FDA’s longstanding position has been that the agency may rely upon prior findings of safety or effectiveness to support approval of a 505(b)(2) application, this policy has been controversial and subject to challenge in the past. If the FDA’s policy is successfully challenged administratively or in court, we may be required to seek approval of our products via full NDAs that contain a complete data package demonstrating the safety and effectiveness of our product candidates, which would be time-consuming, expensive and would have a material adverse effect on our business and financial condition.
The FDA, as a condition of the EXPAREL NDA approval on October 28, 2011, has required us to study EXPAREL in pediatric patients as a post-marketing requirement. We have agreed to a trial timeline where we will study successive pediatric patient subpopulations. In December 2019, we announced positive results for our extended pharmacokinetic and safety study for local analgesia in children aged 6 to 17 undergoing cardiovascular or spine surgeries. Those positive results provided the foundation for an sNDA submission which was approved by the FDA in March 2021. Additionally, we are in negotiations with the FDA and EMA for clarity on other pediatric study obligations for children aged zero to less than six years old. In October 2023, we received notification from the FDA that our pediatric studies requirement had been waived for the indication of brachial plexus interscalene nerve block to produce postsurgical regional analgesia in pediatric patients and in October 2024, we received notification from the FDA that our pediatric studies requirement had been waived for the indications of sciatic nerve block in the popliteal fossa and adductor canal block indications. These trials will be expensive and time consuming and we are required to meet the timelines for submission of protocols and data and for completion as agreed with the FDA and EMA, and we may be delayed in meeting such timelines. We are required to conduct these trials even if we believe that the costs and potential benefits of conducting the trials are not warranted from a scientific or financial perspective. The failure to conduct these pediatric trials or to meet applicable deadlines could result in the imposition of sanctions, including, among other things, issuance of warnings letters or imposition of seizures or injunctions. For more information regarding our pediatric study obligations, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
For iovera° and any other potential medical device, we must obtain clearance or approval from the FDA or other regulatory authorities prior to introducing a new product or a modification to an existing product. The regulatory clearance process may result in substantial delays, unexpected or additional costs and other unforeseen factors and limitations on the types and uses of products we would be able to commercialize, any of which could have a material adverse effect on our business and financial condition.
In the U.S., before we are able to market a new medical device, or a new use, claim for, or significant modification to an existing medical device, we generally must first receive clearance or approval from the FDA and certain other regulatory authorities. Many foreign jurisdictions outside the U.S. also require clearance, approval or compliance with certain standards before a medical device or other product can be marketed. The process of obtaining regulatory clearances and approvals to market a medical device can be costly, time consuming, involve rigorous preclinical and clinical testing, require changes in
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 48
products or result in limitations on the indicated uses of products. There can be no assurance that these clearances and approvals will be granted on a timely basis, if at all. In addition, once a medical device has been cleared or approved, a new clearance or approval may be required before the medical device may be modified, its labeling changed or marketed for a different use. Medical devices are cleared or approved for one or more specific intended uses and promoting a device for an off-label use could result in government enforcement action. Furthermore, a product approval or clearance can be withdrawn or limited due to unforeseen problems with the medical device or issues relating to its application. The regulatory clearance and approval process may result in, among other things, delayed, if at all, realization of product net sales, substantial additional costs and limitations on the types of products we may bring to market or their indicated uses, any one of which could have a material adverse effect on our business, results of operations, financial condition and cash flows.
A regulatory authority may determine that our products or any of our product candidates have undesirable side effects.
If concerns are raised regarding the safety of a new product candidate as a result of undesirable side effects identified during clinical testing, a regulatory authority may decline to approve the drug or medical device or issue a letter requesting additional data or information prior to making a final decision regarding whether or not to approve the product. Undesirable side effects caused by our products or any product candidate could also result in the inclusion of unfavorable information in our product labeling, imposition of distribution or use restrictions, a requirement to conduct post-market studies or to implement a risk evaluation and mitigation strategy, denial, suspension or withdrawal of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us from commercializing and generating revenues from the sale of EXPAREL, ZILRETTA, iovera° or any product candidate.
For example, the side effects observed in the EXPAREL clinical trials completed to date include nausea and vomiting. In addition, the class of drugs that EXPAREL belongs to has been associated with nervous system and cardiovascular toxicities at high doses. We cannot be certain that these side effects and others will not be observed in the future, or that regulatory authorities will not require additional trials or impose more severe labeling restrictions due to these side effects or other concerns. The active component of EXPAREL is bupivacaine, and bupivacaine infusions have been associated with the destruction of articular cartilage, or chondrolysis. Chondrolysis has not been observed in clinical trials of EXPAREL, but we cannot be certain that this side effect will not be observed in the future.
Following approval of EXPAREL, ZILRETTA, iovera° or any of our product candidates, if we or others later identify previously unknown undesirable side effects caused by such products, if known side effects are more frequent or severe than in the past, or if we or others detect unexpected safety signals for such products or any products perceived to be similar to such products:
•regulatory authorities may require the addition of unfavorable labeling statements, specific warnings or contraindications (including boxed warnings);
•regulatory authorities may suspend or withdraw their approval of the product, or require it to be removed from the market;
•regulatory authorities may impose restrictions on the distribution or use of the product;
•we may be required to change the way the product is administered, conduct additional clinical trials, reformulate the product, change the labeling of the product or change or obtain re-approvals of manufacturing facilities;
•sales of the product may be significantly decreased versus projected sales;
•we may be subject to government investigations, product liability claims and litigation; and
•our reputation may suffer.
Any of these events could prevent us from achieving or maintaining market acceptance of our products or any of our product candidates and could substantially increase our commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from its sale.
If we do not comply with federal, state and foreign laws and regulations relating to the health care business, we could face substantial penalties.
We and our customers are subject to extensive regulation by the federal government, and the governments of the states and foreign countries in which we may conduct our business. In the U.S., the laws that directly or indirectly affect our ability to operate our business include the following:
•the Federal Anti-Kickback Law, which prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration—directly or indirectly—in cash or in kind, to induce either the referral of an individual or furnishing or arranging for a good or service for which payment may be made under federal health care programs such as Medicare and Medicaid;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 49
•other Medicare laws and regulations that prescribe the requirements for coverage and payment for services performed by our customers, including the amount of such payment;
•the Federal False Claims Act, which imposes civil and criminal liability on individuals and entities who submit, or cause to be submitted, false or fraudulent claims for payment to the government;
•the Federal False Statements Act, which prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with delivery of or payment for health care benefits, items or services; and
•various state laws that impose similar requirements and liability with respect to state healthcare reimbursement and other programs.
If our operations are found to be in violation of any of the laws and regulations described above or any other law or governmental regulation to which we or our customers are or will be subject, we may be subject to civil and criminal penalties, damages, fines, exclusion from the Medicare and Medicaid programs and the curtailment or restructuring of our operations. Similarly, if our customers are found to be non-compliant with applicable laws, they may be subject to sanctions, which could also have a negative impact on us. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business and our financial results. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
The design, development, manufacture, supply and distribution of our products are highly regulated and technically complex.
The design, development, manufacture, supply and distribution of our products are all highly regulated. We, along with our third-party providers, must comply with all applicable regulatory requirements of the FDA and foreign regulatory authorities. In addition, the facilities used to manufacture, store and distribute our products are subject to inspection by regulatory authorities at any time to determine compliance with applicable regulations.
The manufacturing techniques and facilities used for the manufacture and supply of our products must be operated in conformity with CGMP and other FDA, EMA and MHRA regulations, including potentially prior regulatory approval. In addition, any expansion of our existing manufacturing facilities or the introduction of any new manufacturing facilities, including the manufacturing suites at the Thermo Fisher and Carlisle facilities, also require conformity with CGMP and other FDA, EMA and MHRA regulations. In complying with these requirements, we, along with our co-production partners and suppliers, must continually expend time, money and effort in production, record keeping and quality assurance and control to ensure that our products meet applicable specifications and other requirements for safety, efficacy and quality. In addition, we, along with our co-production partners and suppliers, are subject to unannounced inspections by the FDA, EMA, MHRA and other regulatory authorities.
Any failure to comply with regulatory and other legal requirements applicable to the manufacture, supply and distribution of our products could lead to remedial action (such as recalls), civil and criminal penalties and delays in manufacture, supply and distribution of our products.
The design, development, manufacture, supply and distribution of our products are all highly complex. If we are unable to manufacture our products in compliance with our highly complex specifications in the future, we may be subject to product exchanges, significant costs and charges, supply constraints or other corrective measures.
If we fail to comply with the extensive regulatory requirements to which we and our products are subject, such products could be subject to restrictions or withdrawal from the market and we could be subject to penalties.
The testing, manufacturing, quality control, labeling, safety, effectiveness, advertising, promotion, storage, sales, distribution, import, export and marketing, among other things, of EXPAREL, ZILRETTA, iovera° and our product candidates are subject to extensive regulation by governmental authorities in the U.S. and elsewhere throughout the world. Quality control and manufacturing procedures regarding our products and product candidates must conform to CGMP. Regulatory authorities, including but not limited to the FDA, EMA and MHRA, periodically inspect manufacturing facilities to assess compliance with CGMP. Our failure, or the failure of any contract manufacturers with whom we may work in the future, to comply with the laws administered by the FDA, EMA, the MHRA or other governmental authorities could result in, among other things, any of the following:
•product recall or seizure;
•suspension or withdrawal of an approved product from the market;
•interruption of production;
•reputational concerns of our customers or the medical community;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 50
•operating restrictions;
•warning letters;
•injunctions;
•refusal to permit import or export of an approved product;
•refusal to approve pending applications or supplements to approved applications that we submit;
•denial of permission to file an application or supplement in a jurisdiction;
•consent decrees;
•suspension or termination of ongoing clinical trials;
•fines and other monetary penalties;
•criminal prosecutions; and
•unanticipated expenditures.
If the government or third-party payers fail to provide adequate coverage and payment rates for EXPAREL, ZILRETTA, iovera° or any future products, or if hospitals or ASCs choose to use alternative therapies that are less expensive, our revenue and prospects for profitability will be limited.
In both domestic and foreign markets, sales of our existing products and any future products will depend in part upon the availability of coverage and reimbursement from third-party payers. Such third-party payers include government health programs such as Medicare and Medicaid, managed care providers, private health insurers and other organizations. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Assuming coverage is approved, the resulting reimbursement payment rates might not be adequate. In particular, many U.S. hospitals and ASCs receive a fixed reimbursement amount per procedure for certain surgeries and other treatment therapies they perform. Because this amount may not be based on the actual expenses the hospital or ASC incurs, these sites may choose to use therapies which are less expensive when compared to our product candidates. Although hospitals and ASCs may receive separate reimbursement for EXPAREL, ZILRETTA, iovera° or any product candidates that we may develop, in-license or acquire, if approved, will face competition from other therapies and drugs for these limited hospital and ASC financial resources. We may need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to the satisfaction of hospitals, ASCs, other target customers and their third-party payers. Such studies might require us to commit a significant amount of management time, financial and other resources. Our future products might not ultimately be considered cost-effective. Adequate third-party coverage and reimbursement might not be available to enable us to maintain price levels sufficient to realize an appropriate return on investment in product development.
Third-party payers, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. For example, the 340B Drug Pricing Program requires pharmaceutical manufacturers that participate in Medicaid to enter into a PPA with the Secretary of Health and Human Services. Under the PPA, the manufacturer agrees to provide front-end discounts on covered outpatient drugs purchased by specified providers, called “covered entities,” that serve the nation’s most vulnerable patient populations. Any expansion of such covered entities or changes to the Medicaid rebate formula may cause the required 340B discount to increase, resulting in increased revenue leakage.
Additionally, third-party payers may limit the indications or circumstances for which our products will be reimbursed to a smaller set of indications or circumstances than we believe is appropriate. In addition, in the U.S., no uniform policy of coverage and reimbursement for drug or medical device products exists among third-party payers. Therefore, coverage and reimbursement for drug products can differ significantly from payer to payer.
Further, barring separate reimbursement for qualifying non-opioids administered to Medicare surgical patients in the outpatient setting as mandated by NOPAIN, we believe that future coverage and reimbursement will likely be subject to increased restrictions both in the U.S. and in international markets, as federal, state and foreign governments continue to propose and pass new legislation designed to reduce or contain the cost of healthcare. Third-party coverage and reimbursement for our products or product candidates for which we receive regulatory approval may not be available or adequate in either the U.S. or international markets, which could have a negative effect on our business, results of operations, financial condition and prospects.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 51
Public concern regarding the safety of drug products such as EXPAREL and ZILRETTA and medical device products such as iovera° could result in the inclusion of unfavorable information in our labeling, or require us to undertake other activities that may entail additional costs.
In light of widely publicized events concerning the safety risk of certain drug products, the FDA, members of Congress, the Government Accountability Office, medical professionals and the general public have raised concerns about potential drug and medical device safety issues. These events have resulted in the withdrawal of drug and medical device products, revisions to labeling that further limits use of the drug and medical device products and the establishment of risk management programs that may, for example, restrict distribution of drug or medical device products after approval. The Food and Drug Administration Amendments Act of 2007, or FDAAA, grants significant expanded authority to the FDA, much of which is aimed at improving the safety of drug and medical device products before and after approval. In particular, the FDAAA authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to product labeling to reflect new safety information and require risk evaluation and mitigation strategies for certain drugs and medical devices, including certain currently approved drugs and medical devices. The FDAAA also significantly expands the federal government’s clinical trial registry and results databank, which we expect will result in significantly increased government oversight of clinical trials. Under the FDAAA, companies that violate these and other provisions of the new law are subject to substantial civil monetary penalties, among other regulatory, civil and criminal penalties. The increased attention to drug safety issues may result in a more cautious approach by the FDA in its review of data from our clinical trials. Data from clinical trials may receive greater scrutiny, particularly with respect to safety, which may make the FDA or other regulatory authorities more likely to require additional preclinical studies or clinical trials. If the FDA or any other regulatory agency requires us to provide additional clinical or preclinical data for EXPAREL, ZILRETTA or iovera°, the indications for which these products were approved may be limited or there may be specific warnings or limitations on dosing, and our efforts to commercialize EXPAREL, ZILRETTA or iovera° may be otherwise adversely impacted.
Risks Related to Intellectual Property
The patents and the patent applications that we have covering our pMVL products are limited to specific injectable formulations, processes and uses of drugs encapsulated in our pMVL drug delivery technology and our market opportunity for our product candidates may be limited by the lack of patent protection for the active ingredient itself and other formulations and delivery technologies and systems that may be developed by competitors.
The active ingredient in EXPAREL is bupivacaine. Patent protection for the bupivacaine molecules themselves has expired and generic immediate-release products are available. As a result, competitors who obtain the requisite regulatory approval can offer products with the same active ingredient as EXPAREL so long as the competitors do not infringe any process, use or formulation patents that we have developed for drugs encapsulated in our pMVL drug delivery technology.
For example, we are aware of at least one FDA-approved long-acting instillable bupivacaine product on the market which utilizes an alternative delivery system to EXPAREL. Such a product is similar to EXPAREL in that it also extends the duration of effect of bupivacaine, but achieves this clinical outcome using a completely different drug delivery system as compared to our pMVL drug delivery technology.
The number of patents and patent applications covering products in the same field as EXPAREL indicates that competitors have sought to develop and may seek to market competing formulations that may not be covered by our patents and patent applications. The commercial opportunity for EXPAREL could be significantly harmed if competitors are able to develop and commercialize alternative formulations of bupivacaine that are long-acting but outside the scope of our patents.
For instance, because EXPAREL has been approved by the FDA, one or more third parties may challenge the patents covering this product, as described below, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. For example, if a third-party files an ANDA for a generic drug product containing bupivacaine and relies in whole or in part on studies conducted by or for us, the third-party will be required to certify to the FDA that either: (i) there is no patent information listed in the FDA’s Orange Book with respect to our NDA for EXPAREL; (ii) the patents listed in the Orange Book have expired; (iii) the listed patents have not expired, but will expire on a particular date and approval is sought after patent expiration or (iv) the listed patents are invalid or will not be infringed by the manufacture, use or sale of the third-party’s generic drug product. A certification that the new product will not infringe the Orange Book-listed patents for EXPAREL, or that such patents are invalid, is called a Paragraph IV certification. If the third-party submits a Paragraph IV certification to the FDA, a notice of the Paragraph IV certification must also be sent to us once the third-party’s ANDA is accepted for filing by the FDA. We may then initiate a lawsuit to defend the patents identified in the notice. The filing of a patent infringement lawsuit within 45 days of receipt of the notice automatically prevents the FDA from approving the third-party’s ANDA until the earliest of 30 months or the date on which the patent expires, the lawsuit is settled or the court reaches a decision in the infringement lawsuit in favor of the third-party. If we do not file a patent infringement lawsuit within the required 45-day period, the third-party’s ANDA will not be subject to the 30-month stay. Litigation or other proceedings to enforce or defend intellectual property rights are often very complex in nature, may be very expensive and time-consuming,
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 52
may divert our management’s attention from our core business and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with our products.
For example, in October 2021, we received a Notice Letter advising that eVenus Pharmaceutical Laboratories, Inc., or eVenus, of Princeton, New Jersey, submitted to the FDA an ANDA with a Paragraph IV certification seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (266 mg/20 mL) in the U.S. prior to the expiration of U.S. Patent No. 11,033,495 (the ’495 patent).
In November 2021, we filed a patent infringement suit against eVenus and its parent company (Jiangsu Hengrui Pharmaceuticals Co. Ltd., or Jiangsu Hengrui) in the U.S. District Court for the District of New Jersey (21-cv-19829) asserting infringement of the ’495 patent. This triggered an automatic 30-month stay of final approval of the eVenus ANDA. In January 2022, eVenus filed an Answer with counterclaims to the Complaint, alleging the ’495 patent is invalid and/or not infringed through the manufacture, sale, or offer for sale of the product described in product described in eVenus’s ANDA submission.
In December 2021, we received a second Notice Letter advising that eVenus submitted to the FDA an amendment to its ANDA with a Paragraph IV Certification seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (133 mg/10 mL) in the U.S. prior to the expiration of the ’495 patent. In the Notice Letter, eVenus also advised that it submitted a Paragraph IV Certification to the FDA seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (266 mg/20 mL and 133 mg/10 mL) in the U.S. prior to the expiration of U.S. Patent No. 11,179,336 (the ’336 patent). eVenus further alleges in the Notice Letter that both the ’495 patent and the ’336 patent are invalid and/or not infringed.
In February 2022, we filed a second patent infringement suit against eVenus and Jiangsu Hengrui in the U.S. District Court for the District of New Jersey (22-cv-00718) asserting that the 133 mg/10 mL ANDA product will infringe the ’495 and ’336 patents and that the 266 mg/20 mL ANDA product will infringe the ’336 patent. This filing triggered a second automatic 30-month stay of final approval for the 133 mg/10 mL ANDA product.
In February 2023, eVenus filed its first amended answer to the first amended complaint, alleging patent invalidity, non-infringement and inequitable conduct. We have denied the allegations in eVenus’s first amended answer. We have subsequently voluntarily dismissed our claims with respect to the ’336 Patent. The trial on the remaining patent was conducted in February 2024, and in August 2024, the U.S. District Court for the District of New Jersey issued its ruling in the patent infringement suit for the ‘495 patent. The ruling found that claim 7 of the ‘495 patent is not valid on the grounds of obviousness and anticipation. A notice of appeal was filed in September 2024 and is ongoing.
In April 2023, we filed a third patent infringement suit against eVenus, Jiangsu Hengrui, and Fresenius Kabi USA, LLC, in the U.S. District Court for the District of New Jersey (23-cv-2367) asserting that the 133 mg/10 mL and 266 mg/20 mL ANDA products will infringe U.S. Patent No. 11,426,348 (the ’348 patent). In July 2023, eVenus filed its answer with claims for declaratory judgment, alleging patent invalidity, non-infringement and inequitable conduct with respect to the ’348 patent as well as our other patents, U.S. Patent Nos. 11,278,494; 11,304,904; 11,311,486; 11,357,727 and 11,452,691.The parties have subsequently dismissed all patents other than the ’348 patent from this litigation.
For more information on these litigations and other legal proceedings we are involved in, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
We are unable to predict the outcome of these litigations at this time.
The patents and the patent applications that we have covering ZILRETTA are limited to specific injectable formulations, uses and processes of manufacturing and our market opportunity for our product candidate may be limited by the lack of patent protection for the active ingredient itself and other formulations and delivery technologies and systems that may be developed by competitors.
The active ingredient in ZILRETTA is triamcinolone acetonide, or TCA. Patent protection for TCA has expired and generic immediate-release products are available. As a result, competitors who obtain the requisite regulatory approval can offer products with the same active ingredient as ZILRETTA so long as the competitors do not infringe any process, use or formulation patents covering ZILRETTA. The commercial opportunity for ZILRETTA could be significantly harmed if competitors are able to develop and commercialize alternative extended release formulations of TCA that are outside the scope of our patents.
Furthermore, because ZILRETTA has been approved by the FDA, one or more third parties may challenge the patents covering this product, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. For example, if a third-party files an ANDA for a generic version of ZILRETTA and relies in whole or in part on studies conducted by or for us, the third-party will be required to certify to the FDA that either: (i) there is no patent information listed in the FDA’s Orange Book with respect to our NDA for ZILRETTA; (ii) the patents listed in the Orange Book have expired; (iii) the listed patents have not expired, but will expire on a particular date and approval is sought after patent expiration or (iv) the listed patents are invalid or will not be infringed by the manufacture, use or sale of the third-party’s generic drug product. A
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 53
certification that the new product will not infringe the Orange Book-listed patents for ZILRETTA, or that such patents are invalid, is called a Paragraph IV certification. If the third-party submits a Paragraph IV certification to the FDA, a notice of the Paragraph IV certification must also be sent to us once the third-party’s ANDA is accepted for filing by the FDA. We may then initiate a lawsuit to defend the patents identified in the notice. The filing of a patent infringement lawsuit within 45 days of receipt of the notice automatically prevents the FDA from approving the third-party’s ANDA until the earliest of 30 months or the date on which the patent expires, the lawsuit is settled or the court reaches a decision in the infringement lawsuit in favor of the third-party. If we do not file a patent infringement lawsuit within the required 45-day period, the third-party’s ANDA will not be subject to the 30-month stay. Litigation or other proceedings to enforce or defend intellectual property rights are often very complex in nature, may be very expensive and time-consuming, may divert our management’s attention from our core business and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with ZILRETTA.
The patents and the patent applications that we have covering our iovera° products are primarily limited to specific handheld cryogenic needle devices that are cooled by a cryogen and methods for applying cryotherapy to nerve tissue using the cryogenic devices. Our market opportunity for our product candidates may be limited by gaps in patent coverage for the cryogenic devices, methods of use and other cryotherapy technology and systems that may be developed by competitors.
The iovera° cryogenic device is a compact, self-contained handheld device with a replaceable cryogen cartridge that delivers a cryogen through internal supply tubes to needle lumens of a replaceable needle probe, so as to cool the needle probe and thereby cool a surrounding target nerve tissue. We also have secured patents covering particular cryotherapy methods and pain treatments that provide what we deem to be optimal treatment using the iovera° cryogenic device.
Although we have patents that are broad enough to cover various alternative designs and methods, much of our patent coverage is tailored to cover the iovera° device and methods of use. It is thus possible that competitors may attempt to design around many of our patents. For example, we are aware of competitors developing cryogenic systems that are not self-contained handheld devices, or cryogenic systems that deliver cryotherapy through different mechanisms. It is also possible that competitors may attempt to develop and market cryotherapy devices and methods not covered by our patents, for example, basic cryotherapy treatment systems that are off-patent or cryoanalgesia for other nerve entrapment treatments.
The commercial opportunity for iovera° could be significantly harmed if competitors are able to develop and commercialize alternative designs and methods outside the scope of our patents.
Furthermore, the earliest patent family for iovera° is scheduled to expire in December 2025, thereby opening the door for competitors to copy some of our early iovera° technology. This early patent family is primarily focused on treating cosmetic defects that are no longer the focus of iovera°, but the underlying technology is nonetheless relevant enough for there to be appreciable overlap.
Finally, one or more third parties may challenge the patents covering the iovera° product, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. Litigation or other proceedings to defend or enforce intellectual property rights are often very complex in nature, may be very expensive and time-consuming, may divert our management’s attention from our core business and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with our products.
The patents and the patent applications that we have covering PCRX-201 are limited to composition of matter and uses of an adenoviral vector containing a nucleic acid encoding specific gene and our market opportunity for our product candidate may be limited by the patent protection and other gene therapies for treating OA that may be developed by competitors.
We reply upon a combination of intellectual property rights, including patent rights, trade secret protection, know-how and confidentiality agreements to protect intellectual property related to PCRX-201. We have in-licensed certain issued patents covering PCRX-201 from BCM that expire between 2032 and 2033. If we are unable to obtain additional patent protection with later expiration dates, we may not be able to compete effectively in our markets.
The intellectual property landscape around adenoviral vector delivery and expression systems is highly dynamic, and third parties may initiate and prevail in legal proceedings alleging that the patents that we in-license or owned are invalid or that we are infringing, misappropriating, or otherwise violating their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Furthermore, certain inventions which we have in-licensed from BCM may have been discovered through government funded programs and thus may be subject to federal regulations under the Bayh-Dole Act of 1980. The U.S. government has certain rights also referred to as “march-in rights.” In addition, the U.S. government also has the right to take title to these inventions if certain disclosure and reporting requirements were not met. In addition, the U.S. government requires that any products embodying the subject invention or produced through the use of the subject invention be manufactured substantially in the U.S. The manufacturing preference requirement can be waived if the owner of the intellectual property can show that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 54
to manufacture substantially in the U.S. or that under the circumstances domestic manufacture is not commercially feasible. This preference for U.S. manufacturers may limit our ability to contract with non-U.S. product manufacturers for products covered by such intellectual property.
Because it is difficult and costly to protect our proprietary rights, we may not be able to ensure their protection and all patents will eventually expire.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection for EXPAREL, ZILRETTA, iovera°, our pMVL drug delivery technology and for any product candidates that we may develop, license or acquire and the methods we use to manufacture them, as well as successfully defending these patents and trade secrets against third-party challenges. We will only be able to protect our technologies from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them.
The patent positions of pharmaceutical, medical device and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceutical, medical device or biotechnology patents has emerged to date in the U.S. Patent positions and policies outside the U.S. are even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
•we may not have been the first to make the inventions covered by each of our pending patent applications and issued patents;
•we may not have been the first to file patent applications for these inventions;
•others may independently develop similar or alternative technologies or duplicate any of our product candidates or technologies;
•it is possible that none of the pending patent applications will result in issued patents;
•the issued patents covering our product candidates may not provide a basis for commercially viable active products, may not provide us with any competitive advantages, may not have sufficient scope or strength to protect the technologies they were intended to protect or may be challenged by third parties;
•others may design around our patent claims to produce competitive products that fall outside the scope of our patents;
•we may not develop or in-license additional proprietary technologies that are patentable;
•patents of others may have an adverse effect on our business; or
•competitors may infringe our patents and we may not have adequate resources to enforce our patents.
Patent applications in the U.S. are maintained in confidence for at least 18 months after their earliest effective filing date. Consequently, we cannot be certain we were the first to invent or the first to file patent applications on EXPAREL, ZILRETTA, iovera°, our pMVL drug delivery technology or any product candidates that we may develop, license or acquire. In the event that a third-party has also filed a U.S. patent application relating to our product candidates or a similar invention, we may have to participate in interference proceedings declared by the USPTO to determine priority of invention in the U.S. The costs of these proceedings could be substantial and it is possible that our efforts would be unsuccessful, resulting in a material adverse effect on our U.S. patent position. Furthermore, we may not have identified all U.S. and foreign patents or published applications that affect our business either by blocking our ability to commercialize our drugs or medical devices or by covering similar technologies that affect our drug or medical device markets.
In addition, some countries, including many in Europe, do not grant patent claims directed to methods of treating humans, and in these countries patent protection may not be available at all to protect our product candidates. Even if patents are issued, we cannot guarantee that the claims of those patents will be valid and enforceable or provide us with any significant protection against competitive products, or otherwise be commercially valuable to us. Furthermore, while we generally apply for patents in those countries where we intend to make, have made, use or sell patented products, we may not accurately predict all of the countries where patent protection will ultimately be desirable. If we fail to timely file a patent application in any such country, we may be precluded from doing so at a later date. We also cannot assure you that the patents issuing as a result of our foreign patent applications will have the same scope of coverage as our U.S. patents.
Some of our older patents have already expired. In the case of EXPAREL, the European and U.S. patents protecting the formulation of EXPAREL expired in 2018. An existing formulation patent for EXPAREL expired in November 2013. An existing formulation patent for EXPAREL expired in the U.S. in 2013 and its equivalents in Canada, Germany, France, Spain,
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 55
Italy and the U.K. expired in 2014. In Europe, manufacturers qualify for 8 years of data exclusivity upon marketing authorization approval and an additional two years of market exclusivity, for a total of 10 years of regulatory exclusivity. Our earliest patent family for iovera° is scheduled to expire in December 2025, though that patent family is primarily focused on treating cosmetic defects that are no longer the focus of iovera°. Once our patents covering EXPAREL, ZILRETTA and iovera° have expired, we will be more reliant on trade secrets to protect against generic competition.
We also rely on trade secrets to protect our technology, particularly where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. While we use reasonable efforts to protect our trade secrets through confidentiality and non-disclosure agreements, our licensors, employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Policing unauthorized use of our trade secrets or enforcing a claim that a third-party illegally obtained and is using our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, trade secret laws in other countries may not be as protective as they are in the U.S. Thus, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
In order to protect the goodwill associated with our company and product names, we rely on trademark protection for our marks. We have registered the “Pacira,” “EXPAREL,” “ZILRETTA,” “iovera°” and other marks with the USPTO. A third-party may assert a claim that one of our marks is confusingly similar to its mark, and such claims or the failure to timely register a mark or objections by the FDA or other regulatory agency could force us to select a new name for one of our product candidates, which could cause us to incur additional expense or delay the commercialization of such product.
If we fail to obtain or maintain patent, trade secret and/or trademark protection for EXPAREL, ZILRETTA, iovera°, our pMVL drug delivery technology or any product candidate that we may develop, license or acquire, third parties could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate revenues and remain profitable.
If we are sued for infringing the intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in any litigation would harm our business.
Our ability to develop, manufacture, market and sell EXPAREL, ZILRETTA, iovera°, our pMVL drug delivery technology or any product candidates that we may develop, license or acquire depends upon our ability to avoid infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the general fields of pain management and cancer treatment and cover the use of numerous compounds, formulations and medical devices in our targeted markets. Because of the uncertainty inherent in any patent or other litigation involving proprietary rights, we and our licensors may not be successful in defending intellectual property claims by third parties, which could have a material adverse effect on our results of operations. Regardless of the outcome of any litigation, defending the litigation may be expensive, time-consuming and distracting to management. In addition, because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that EXPAREL, ZILRETTA or iovera° may infringe. There could also be existing patents of which we are not aware that EXPAREL, ZILRETTA or iovera° may inadvertently infringe.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology, biopharmaceutical and medical device industries in general. If a third-party claims that we infringe on their products or technology, we could face a number of issues, including:
•infringement and other intellectual property claims which, with or without merit, can be expensive and time consuming to litigate and can divert management’s attention from our core business;
•substantial damages for past infringement which we may have to pay if a court decides that our product infringes on a competitor’s patent;
•a court prohibiting us from selling or licensing our product unless the patent holder licenses the patent to us, which it would not be required to do;
•if a license is available from a patent holder, we may have to pay substantial royalties or grant cross licenses to our patents; and
•redesigning our processes so they do not infringe, which may not be possible or could require substantial expenditures and time.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology, pharmaceutical and medical device industries, we employ individuals who were previously employed at other biotechnology, pharmaceutical and medical device companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 56
we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to our Financial Condition, Indebtedness and our Common Stock
Servicing our indebtedness requires a significant amount of cash, and we may not have sufficient cash flow from our business to pay our substantial indebtedness.
Our ability to make payments of the principal of, to pay interest on or to refinance our indebtedness, including the TLA Term Loan (as defined below), the 0.750% convertible senior notes due 2025, or 2025 Notes, issued in our private offering completed on July 10, 2020, and the 2.125% convertible senior notes due 2029, or 2029 Notes, issued in our private offering completed on May 14, 2024, and, together with the 2025 Notes, the Notes, each as described below, or to make cash payments in connection with any conversion of the 2025 Notes or 2029 Notes (if applicable) depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not generate sufficient cash flow from operations in the future to service our indebtedness and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring indebtedness or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
In December 2021, we entered into a term loan credit agreement (the “TLB Credit Agreement”) with JPMorgan Chase Bank, N.A., as administrative agent and the initial lender. The term loan issued under the TLB Credit Agreement (the “TLB Term Loan”) was issued at a 3.00% discount and allowed for a single-advance term loan B facility in the principal amount of $375.0 million, which was secured by substantially all of the Company’s and each subsidiary guarantor’s assets. On March 31, 2023, we entered into a credit agreement (as amended, the “TLA Credit Agreement”) with JPMorgan Chase Bank, N.A., as administrative agent, and certain lenders, to refinance the indebtedness outstanding under our then-existing TLB Credit Agreement. The term loan issued under the TLA Credit Agreement (the “TLA Term Loan”) was issued at a 0.30% discount and provides for a single-advance term loan A facility in the principal amount of $150.0 million, which is secured by substantially all of our and any subsidiary guarantor’s assets and is scheduled to mature on March 31, 2028, subject to certain exceptions set forth in the TLA Credit Agreement.
On July 10, 2020, we completed a private placement of $402.5 million in aggregate principal amount of 2025 Notes, and entered into an indenture, or 2025 Indenture, with respect to the 2025 Notes. In May 2024, we used $191.4 million of the net proceeds of the 2029 Notes (as described below) to repurchase $200.0 million aggregate principal amount of the 2025 Notes. The 2025 Notes accrue interest at a fixed rate of 0.750% per year, payable semiannually in arrears on February 1 and August 1 of each year and mature on August 1, 2025.
On May 14, 2024, we completed a private placement of $287.5 million in aggregate principal amount of 2029 Notes, and entered into an indenture, or 2029 Indenture, and, together with the 2025 Indenture, the Indentures, with respect to the 2029 Notes. The 2029 Notes accrue interest at a fixed rate of 2.125% per year, payable semiannually in arrears on May 15 and November 15 of each year and mature on May 15, 2029.
As of December 31, 2024, our total consolidated gross indebtedness was $595.3 million, which consisted of $202.5 million of principal outstanding on the 2025 Notes, $287.5 million of principal outstanding on the 2029 Notes and $105.3 million of principal outstanding on the TLA Term Loan. See Note 10, Debt, to our consolidated financial statements included herein for more information. Additionally, our subsidiaries had no indebtedness (excluding trade payables, intercompany liabilities and income tax-related liabilities).
Our TLA Credit Agreement and the Indentures each impose significant operating and financial restrictions on us and certain of our subsidiaries, which may prevent us from capitalizing on business opportunities. A breach of any of those restrictive covenants may cause us to be in default under the TLA Credit Agreement and/or the Indentures, and our lenders could foreclose on our assets.
Our TLA Credit Agreement requires us to maintain certain financial covenants. A decline in our operating performance could negatively impact our ability to meet these financial covenants. If we breach any of these restrictive covenants, the lenders could either refuse to lend funds to us or accelerate the repayment of any outstanding borrowings under the TLA Credit Agreement. We may not have sufficient funds to repay such indebtedness upon a default or be unable to receive a waiver of the default from the lenders. If we are unable to repay the indebtedness, the lenders could initiate a bankruptcy proceeding or collection proceedings with respect to our assets, all of which secure our indebtedness under the TLA Credit Agreement.
The TLA Credit Agreement and the Indentures also contain certain restrictive covenants that limit, and in some circumstances prohibit, our ability to, among other things: incur additional debt or issue preferred stock; sell, lease or transfer
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 57
our assets; pay dividends on, and make other distributions on, or redeem or repurchase, our common stock; make certain capital expenditures and investments; guarantee debt or obligations; create certain liens; enter into transactions with our affiliates; and enter into certain merger, consolidation or other reorganization transactions. These restrictions could limit our ability to obtain future financing, incur or guarantee additional debt, incur certain liens, enter into transactions with affiliates, transfer or sell certain assets, make acquisitions or needed capital expenditures, withstand potential downturns in our business, or the economy in general, conduct operations or otherwise take advantage of business opportunities that may arise, any of which could place us at a competitive disadvantage relative to our competitors. The terms of any future indebtedness we may incur could include more restrictive covenants. We cannot assure you that we will be able to maintain compliance with these covenants in the future and, if we fail to do so, that we will be able to obtain waivers from the lenders and/or amend the covenants. Our failure to comply with the restrictive covenants described above as well as other terms of our indebtedness could result in an event of default, which, if not cured or waived, could result in our being required to repay these borrowings before their due date. If we are forced to refinance these borrowings on less favorable terms or cannot refinance these borrowings, our results of operations and financial condition could be adversely affected.
We may not have the ability to raise the funds necessary to settle conversions of the Notes in cash to the extent elected or to repurchase the Notes upon a fundamental change, and our future indebtedness may contain limitations on our ability to pay cash upon conversion of the Notes or limitations on our ability to repurchase the Notes.
Holders of the Notes will have the right to require us to repurchase their Notes upon the occurrence of a fundamental change at a repurchase price equal to 100% of their principal amount, plus accrued and unpaid interest, if any. We have the option to pay the principal in cash, shares of our common stock, or any combination thereof. While it is our intention to pay the principal in cash, upon conversion of the Notes we will be required to make cash payments for each $1,000 in principal amount of Notes converted of at least the lesser of $1,000 and the sum of the daily conversion values. However, we may not have enough available cash or be able to obtain financing at the time we are required to make repurchases of Notes surrendered therefor or Notes being converted. The TLA Credit Agreement limits—and any credit facility or other agreement that we may enter into may limit—our ability to make cash payments at the time of a fundamental change or upon conversion of the Notes. Further, our ability to repurchase the Notes or to pay cash upon conversions of the Notes may be limited by law, by regulatory authority or by agreements governing our future indebtedness. Our failure to repurchase Notes at a time when the repurchase is required by the applicable indenture or to pay any cash payable on future conversions of the Notes as required by the Indenture would constitute a default under the applicable indenture. A default under the applicable indenture or the fundamental change itself could also lead to a default under agreements governing our TLA Credit Agreement or future indebtedness. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and repurchase the Notes or make cash payments upon conversions thereof.
Our indebtedness could adversely affect our business, financial condition, and results of operations, as well as the ability to meet payment obligations under our TLA Credit Agreement and the Notes.
As of December 31, 2024, our total consolidated gross indebtedness was $595.3 million, which consisted of $202.5 million of principal outstanding on the 2025 Notes, $287.5 million of principal outstanding on the 2029 Notes and $105.3 million of principal outstanding on the TLA Term Loan. See Note 10, Debt, to our consolidated financial statements included herein for more information. Subject to the limits contained in the TLA Credit Agreement and the Indentures, we may be able to incur substantial additional debt from time to time. If we do so, the risks related to our level of debt could increase. Specifically, our level of debt could have important consequences, including the following:
• making it more difficult for us to meet our obligations with respect to our debt;
• limiting our ability to obtain additional financing to fund future working capital, capital expenditures, acquisitions or other general corporate purposes;
• requiring a substantial portion of our cash flows to be dedicated to debt service payments instead of other purposes, thereby reducing the amount of cash flows available for future working capital, capital expenditures, acquisitions or other general corporate purposes;
• increasing our vulnerability to general adverse economic and industry conditions;
• exposing us to the risk of increased interest rates as certain of our borrowings are at variable rates of interest;
• placing us at a disadvantage compared to other, less leveraged competitors;
• increasing our cost of borrowing; and
• limiting our flexibility in planning for changes in our business and reacting to changes in the industry in which we compete.
Furthermore, if we are unable to meet our debt service obligations or should we fail to comply with our financial and other negative covenants contained in the agreements governing our indebtedness, we may be required to refinance all or part of
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 58
our debt, sell important strategic assets at unfavorable prices, incur additional indebtedness or issue common stock or other equity securities. We may not be able to, at any given time, refinance our debt, sell assets, incur additional indebtedness or issue equity securities on terms acceptable to us, in amounts sufficient to meet our needs. If we are able to raise additional funds through the issuance of equity securities, such issuance would also result in dilution to our stockholders. Our inability to service our obligations or refinance our debt could have a material and adverse effect on our business, financial condition or operating results. In addition, our debt obligations may limit our ability to make required investments in capacity, technology, or other areas of our business, which could have a material adverse effect on our business, financial condition, or operating results.
Any of these factors could have an adverse effect on our business, financial condition and results of operations and our ability to meet our debt payment obligations.
Despite our current level of indebtedness, we may be able to incur substantially more debt, which could increase the risks to our financial condition described above.
We may be able to incur substantial additional indebtedness in the future. Although certain of the agreements governing our existing indebtedness contain restrictions on the incurrence of additional indebtedness and entering into certain types of other transactions, these restrictions are subject to a number of qualifications and exceptions, including compliance with various financial conditions. Additional indebtedness incurred in compliance with our existing debt instruments could be substantial. To the extent new debt is added to our current debt levels, the substantial leverage risks described in the immediately preceding risk factor would increase.
As of December 31, 2024, our total consolidated gross indebtedness was $595.3 million, which consisted of $202.5 million of principal outstanding on the 2025 Notes, $287.5 million of principal outstanding on the 2029 Notes and $105.3 million of principal outstanding on the TLA Term Loan. See Note 10, Debt, to our consolidated financial statements included herein for more information on our indebtedness.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our restated certificate of incorporation and our bylaws, as well as provisions of the Delaware General Corporation Law, or DGCL, could make it more difficult for a third-party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions include:
•authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval;
•prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders;
•eliminating the ability of stockholders to call a special meeting of stockholders; and
•establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings.
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management. In addition, we are subject to Section 203 of the DGCL, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by our board of directors. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders.
Our common stock price may be subject to significant fluctuations and volatility.
Our stock price is volatile, and from February 3, 2011, the first day of trading of our common stock, to February 26, 2025, the trading prices of our stock have ranged from $6.16 to $121.95 per share.
Our stock could be subject to wide fluctuations in price in response to various factors, including the following:
•the commercial success of EXPAREL, ZILRETTA and iovera°;
•our ability to execute on our business strategy;
•results of clinical trials of our products, product candidates or those of our competitors;
•changes or developments in laws or regulations applicable to our products or product candidates;
•introduction of competitive products or technologies;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 59
•failure to meet or exceed financial projections we provide to the public;
•actual or anticipated variations in quarterly operating results;
•failure to meet or exceed the estimates and projections of the investment community;
•the perception of the pharmaceutical, biotechnological and medical device industry by the public, legislatures, regulators and the investment community;
•regulatory concerns or government actions;
•general economic and market conditions and overall fluctuations in U.S. equity markets and the impact of macroeconomic developments, such as general political, health and economic conditions, economic slowdowns, recessions, inflation, rising interest rates and the tightening of credit markets;
•increased interest rates and their generally negative effect on U.S. equity markets;
•developments concerning our sources of manufacturing supply;
•disputes or other developments relating to patents, intellectual property or other proprietary rights;
•additions or departures of key scientific or management personnel;
•the extent to which we acquire or invest in products, businesses and technologies;
•issuances of debt, equity or convertible securities;
•changes in the market valuations of similar companies;
•evolving investor expectations and concerns regarding environmental, social and corporate governance issues; and
•the other factors described in this “Risk Factors” section.
In addition, the stock market in general, and the market for pharmaceutical, biotechnology and medical device companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. Fluctuations in our stock price could, among other things, adversely impact the trading price of our shares.
We do not intend to pay dividends on our common stock for the foreseeable future.
We have never declared or paid any dividends on our common stock. We currently intend to retain any future earnings to finance the future development and expansion of our business, and as such we do not expect to pay any cash dividends on our common stock in the foreseeable future. The payment of future dividends, if any, will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in future financing instruments, provisions of applicable law and any other factors our board of directors deems relevant.
Future sales in the public market or issuances of our common stock could lower the market price for our common stock.
In the future, we may sell additional shares of our common stock to raise capital. Except under limited circumstances, we are not restricted from issuing additional common stock, including securities that are convertible into or exchangeable for, or that represent the right to receive, common stock. The issuance of additional shares of our common stock or convertible securities, including upon exercise of our outstanding options, vesting of our restricted stock units or otherwise, will dilute the ownership interest of our common stockholders. In addition, our stockholders, particularly but not limited to those that own 5% or more of the Company may sell a substantial number of their shares in the public market, which could also affect the market price for our common stock.
In addition, certain of our executive officers and directors have established or may establish trading plans under Rule 10b5-1 of the Exchange Act (a “10b5-1 trading plan”), which provide for sales of shares of our common stock from time to time. Under a 10b5-1 trading plan, a broker executes trades pursuant to parameters established by the executive officer or director when entering into the plan, without further direction from the executive officer or director. A 10b5-1 trading plan may be amended or terminated in some circumstances. Our executive officers and directors also may buy or sell additional shares outside of a 10b5-1 trading plan when they are not in possession of material, nonpublic information. Refer to Item 9B. Other Information, for more information.
We cannot predict the size of future sales or issuances of our common stock or the effect, if any, that they may have on the market price for our common stock. The issuance and/or sale of substantial amounts of common stock, or the perception that such issuances and/or sales may occur, could adversely affect the market price of our common stock and impair our ability to raise capital through the sale or issuance of additional equity or debt securities.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 60
Raising additional funds by issuing securities would cause dilution to existing stockholders and raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital by issuing equity securities, our existing stockholders’ ownership would be diluted. If we raise additional funds through licensing arrangements, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us. Any debt financing we enter into may involve covenants that restrict our operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets as well as prohibitions on our ability to create liens, pay dividends, redeem our stock or make investments.
Changes in global economic conditions, including, but not limited to, those driven by inflation and tariffs, may adversely affect spending and the financial health of our customers and others with whom we do business, which may adversely affect our financial condition, results of operations and cash flows.
Uncertainty about current and future global economic conditions, inflation and tariffs may cause patients to defer or cancel medical procedures. Our financial success is sensitive to changes in general economic conditions, both globally and in specific markets, that may adversely affect the demand for our products including recessionary economic cycles, higher interest rates, higher fuel and other energy costs, increased labor costs, declines in asset values, inflation, the imposition of tariffs, increases in commodity prices, higher levels of unemployment, higher consumer debt levels, higher tax rates and other changes in tax laws, public health issues (such as the recent COVID-19 pandemic), or other economic factors, certain of which effects, including cost inflation and higher interest rates, we experienced in 2023 and 2024 and expect to continue to experience in 2025.
If global economic and financial market conditions deteriorate or remain weak for an extended period of time, the following factors, among others, could have a material adverse effect on our financial condition, results of operations and cash flows:
•Changes in foreign currency exchange rates relative to the U.S. dollar.
•The imposition of tariffs and other import restrictions by the U.S. or foreign governments.
•Slower consumer spending that may result in our inability to maintain or increase our sales to new and existing customers, reduce patient volumes, cause reduced product orders or product order delays or cancellations from wholesale accounts that are directly impacted by fluctuations in the broader economy, difficulties managing inventories, higher discounts and lower product margins.
•A decrease in liquidity or credit available to our customers, product suppliers and other service providers.
•If our customers experience diminished liquidity, we may experience a reduction in product orders, an increase in customer order cancellations, and/or the need to extend customer payment terms, which could lead to larger balances and delayed collection of our accounts receivable, reduced cash flows, greater expenses for collection efforts and increased risk of nonpayment of our accounts receivable.
•If we are unable to mitigate the impact of supply chain constraints and inflationary pressure through price increases or other measures, our results of operations and financial condition could be negatively impacted. Furthermore, even if we are able to raise the prices of our products, consumers might react negatively to such price increases, which could have a material adverse effect on, among other things, our brands, reputation, and sales.
Certain of the foregoing could also result in lower levels of healthcare insurance coverage and/or depress consume confidence, any of which could limit the ability of some customers to purchase our products and reduce consumer spend on certain elective medical procedures in both the short- and medium-term.
The U.S. Federal Reserve previously raised interest rates multiple times in response to concerns about inflation, though it recently lowered interest rates multiple times and further interest rate changes remain uncertain. If higher interest rates return, this, coupled with reduced government spending and volatility in financial markets may also increase economic uncertainty and negatively affect consumer spending. Similarly, the ongoing war in Ukraine and the Israel-Hamas war have created extreme volatility in the global capital markets and is expected to continue to have further global economic consequences, including disruptions of the global supply chain and energy markets. Any such volatility and disruptions may adversely affect our business or the third parties on whom we rely. If the equity and credit markets deteriorate, including as a result of political unrest or war, it may make any necessary debt or equity financing (or refinancing) more difficult to obtain in a timely manner, or on favorable terms, more costly or more dilutive. Increased inflation rates have already, and may continue to, adversely affect us by increasing our costs, including labor costs, service costs and employee benefit costs. In addition, higher inflation and macro turmoil and uncertainty could also adversely affect our customers, which could reduce demand for our products.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 61
Rising international tariffs, including any tariffs applied to goods traded between the U.S. and China, the U.S. and Mexico and the U.S. and Canada, could materially and adversely affect our business and results of operations. For example, in February 2025, the current U.S. presidential administration imposed tariffs on foreign imports into the United States, including an additional 10% tariff on all imports from China and an additional 25% tariff on all imports from Mexico and Canada (except a 10% Canadian tariff on energy imports which do not apply to us), which tariffs imports from Mexico and Canada were subsequently suspended for 30 days in order to facilitate negotiations. As of the date of this Annual Report, the proposed tariffs on all imports from Mexico remain suspended. Our iovera° tips are made in Mexico, we have suppliers of raw materials for each of our commercialized products from China and some of our manufacturing equipment is fabricated in Canada. These tariffs will increase the cost of these products and equipment and negatively impact our results of operations. At this time, it remains unclear what additional actions, if any, will be taken by the U.S. or other governments with respect to international trade agreements, the imposition of additional tariffs on goods imported into the U.S., tax policy related to international commerce, increased export control, sanctions and investment restrictions, or other trade matters. Although the ultimate scope and timing of any such actions is currently indeterminable, if implemented, they could have a material impact on our financial condition and results of operations.
Other effects of these changes, including impacts on the price of raw materials and equipment, responsive or retaliatory actions from governments, such as retaliatory tariffs on imports into China, Mexico or Canada from the U.S. and the opportunity for competitors not subject to such changes to establish a presence in markets where we participate, could also have significant impacts on our results of operations, though whether any of the foregoing actions will be taken remains unclear. Furthermore, we may not be able to increase prices for our products enough to offset tariffs, which could impact our margins. If we raise prices in response to tariffs, the demand for our products may go down, which could have a negative impact on our sales. We cannot predict what further action may be taken with respect to export restrictions, tariffs or trade relations between the U.S. and other governments, and any further changes in U.S. or international trade policy could have an adverse impact on our business, financial condition and results of operations.
Cumulatively, we have incurred significant losses since our inception and may incur additional losses in the future.
To date, we have focused primarily on developing and commercializing EXPAREL, and have since acquired ZILRETTA and iovera°. We recorded a net loss of $99.6 million for the year ended December 31, 2024, which includes a goodwill impairment of $163.2 million based upon an assessment that the fair value of goodwill is less than its carrying value (for more information, see Note 8, Goodwill and Intangible Assets, to our consolidated financial statements included herein), and recorded net income of $42.0 million and $15.9 million for the years ended December 31, 2023 and 2022, respectively. As of December 31, 2024, we had an accumulated deficit of $206.4 million. Prior losses, among other things, have had an adverse effect on stockholders’ equity and working capital. We incurred significant pre-commercialization expenses as we prepared for the commercial launch of EXPAREL, and we continue to incur significant sales, marketing and manufacturing expenses, as well as ongoing development expenses related to the commercialization of EXPAREL, ZILRETTA and iovera°. As a result, we had not been profitable prior to 2015 and incurred net losses from 2016 through 2019 and again in 2024. Because of the numerous risks and uncertainties associated with developing and commercializing pharmaceutical products and medical devices, we are unable to predict the extent of future losses, if any.
A material impairment in the carrying value of our intangible assets or goodwill could negatively affect our results of operation and financial condition.
A significant portion of our total assets is comprised of intangible assets. Pursuant to U.S. generally accepted accounting principles, we are required to assess our indefinite-lived intangible assets for impairment. Intangible assets with definite lives are amortized on a straight-line basis over their estimated useful lives and are recorded at cost, net of accumulated amortization. Indefinite-lived intangible assets are not amortized and are tested for impairment at least annually or when a triggering event occurs that could indicate a potential impairment exists. Impairment charges are recognized to the extent the carrying value exceeds its fair value. At December 31, 2024, the carrying value of our intangible assets, net of accumulated amortization, was $426.0 million. If the carrying value of these assets exceeds their current estimated fair value, the assets would be considered impaired, and this would result in a noncash charge to our statement of operations, which could be material. Events and conditions that could result in an impairment include but are not limited to: changes in assumptions regarding future revenue or cash flow forecasts, increased competition or loss of market share, obsolescence, product claims that result in a significant loss of sales or profitability over the product life, deterioration in macroeconomic conditions or declining financial performance in comparison to projected results.
For example, in 2022, we recognized a $26.1 million impairment charge related to an intangible asset for acquired in-process research and development related to ZILRETTA for the treatment of OA pain of the shoulder, driven by facts and circumstances revealed in the fourth quarter of 2022 that suggested the fair value reduction in this intangible asset was driven by later timelines for the completion of clinical trials impacting revenue forecasts, among other factors. For additional information, see Note 8, Goodwill and Intangible Assets, to our consolidated financial statements included herein. Further
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 62
changes to the assumptions regarding the future fair values of our intangible assets could result in additional impairment charges in the future, which could be significant.
Previously, a significant portion of our total assets was comprised of goodwill. Pursuant to U.S. generally accepted accounting principles, we are required to assess our goodwill and indefinite-lived intangible assets for impairment. Goodwill is not amortized but is subject to impairment testing at least annually or when a triggering event occurs that could indicate a potential impairment exists. In July 2024, the FDA approved a generic competitor to EXPAREL and in August 2024, a U.S. District Court ruled that one of our EXPAREL patents was not valid. We determined that these events, combined with a subsequent decrease in our common stock price, indicated that it was more likely than not that the fair value of goodwill may be less than its carrying value, which required us to perform a quantitative impairment test. This quantitative impairment test resulted in our carrying value exceeding the fair value of the Company by more than the goodwill balance. As a result, our goodwill balance of $163.2 million was fully impaired during the three months ended September 30, 2024. For more information, see Note 8, Goodwill and Intangible Assets, to our consolidated financial statements included herein. In February 2025, we completed the GQ Bio Acquisition and will assess the fair value of goodwill related to this transaction in the first quarter of 2025. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events, to our consolidated financial statements included herein.
It is possible that we may have goodwill again in the future, and, if so, events and conditions that could result in an impairment include but are not limited to: changes in assumptions regarding future revenue or cash flow forecasts, a sustained drop in the market price of our common stock, increased competition or loss of market share, obsolescence, product claims that result in a significant loss of sales or profitability over the product life, deterioration in macroeconomic conditions or declining financial performance in comparison to projected results.
We may need additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
Developing and commercializing products for use in the hospital or ASC settings, conducting clinical trials, establishing outsourced manufacturing relationships and successfully manufacturing and marketing drugs and medical devices that we may develop is expensive. We may need to raise additional capital to:
•continue to fund our operations;
•continue our efforts to hire additional personnel and build a commercial infrastructure to commercialize EXPAREL, ZILRETTA and iovera°;
•qualify, outsource or build additional commercial-scale manufacturing of our products in accordance with CGMP;
•acquire, in-license and develop additional product candidates; and
•refinance our Notes and our TLA Term Loan.
We may not have sufficient financial resources to continue our operations or meet all of our objectives, which could require us to postpone, scale back or eliminate some, or all, of these objectives. Our future funding requirements will depend on many factors, including, but not limited to:
•the costs of maintaining a commercial organization to sell, market and distribute EXPAREL, ZILRETTA and iovera°;
•the success of the commercialization of EXPAREL, ZILRETTA and iovera°;
•the cost and timing of manufacturing sufficient quantities of EXPAREL, ZILRETTA and iovera° to meet customer demand, including the cost of expanding our manufacturing facilities to produce EXPAREL, ZILRETTA and iovera°;
•the rate of progress and costs of our efforts to prepare for the submission of an IND, NDA, sNDA or 510(k) pre-market notification for any product candidates that we may develop, in-license or acquire in the future, and the potential that we may need to conduct additional clinical trials to support applications for regulatory approval;
•the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights associated with our products and product candidates, including any such costs we may be required to expend if our licensors are unwilling or unable to do so;
•the effect of competing technological and market developments;
•the terms and timing of any collaborative, licensing, co-promotion or other arrangements that we may establish;
•the potential that we may be required to file a lawsuit to defend our patent rights or regulatory exclusivities from challenges by companies seeking to market generic versions of extended-release liposome injections of
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 63
bupivacaine, long-acting injections of triamcinolone or a cryoanalgesic device that infringes on the various patents covering iovera°; and
•restrictions contained in the agreements governing our indebtedness.
Future capital requirements will also depend on the extent to which we acquire or invest in additional complementary businesses, products and technologies.
Unless and until we can generate sufficiently more revenue from our products, we expect to primarily finance or supplement future cash needs through public or private equity offerings, debt financings, stock option exercises, royalties, collaboration and licensing arrangements, as well as through interest income earned on our cash and investment balances. If needed, we cannot be certain that additional funding will be available on acceptable terms, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs or our commercialization efforts.
Our quarterly operating results may fluctuate significantly.
We expect our operating results to be subject to quarterly fluctuations. Our operating results will be affected by numerous factors, including:
•the level of underlying hospital and ASC demand for EXPAREL, ZILRETTA and iovera° and end-user buying patterns;
•maintaining our existing manufacturing facilities for EXPAREL, ZILRETTA and iovera° and expanding their manufacturing capacities;
•our execution of other collaborative, licensing, distribution, manufacturing or similar arrangements and the timing of payments we may make or receive under these arrangements;
•variations in the level of expenses related to our future development programs;
•any product liability or intellectual property infringement lawsuit in which we may become involved;
•regulatory developments, lawsuits and investigations affecting EXPAREL, ZILRETTA, iovera°, our product candidates or the products and product candidates of our competitors; and
•the impact of macroeconomic developments, such as general political, health and economic conditions, including those resulting from the war in Ukraine and the Israel-Hamas war, economic slowdowns, recessions, inflation, rising interest rates and tightening of credit markets on our business.
If our quarterly or annual operating results fall below the expectations of our investors or securities analysts, the price of our common stock could substantially decline. Furthermore, any quarterly or annual fluctuations in our operating results may in turn cause the price of our common stock to fluctuate substantially. We believe that quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
We may be unable to successfully integrate the businesses and personnel of acquired companies and businesses, and may not realize the anticipated synergies and benefits of such acquisitions.
From time to time, we may complete acquisitions of companies and certain businesses of companies, and we may not realize the expected benefits from such acquisitions because of integration difficulties or other challenges. For example, in April 2019, we completed the MyoScience Acquisition and in November 2021, we completed the Flexion Acquisition.
The success of any acquisitions will depend, in part, on our ability to realize all or some of the anticipated synergies and other benefits from integrating the acquired businesses with our existing businesses. The integration process may be complex, costly and time-consuming. The potential difficulties we may face in integrating the operations of our acquisitions include, among others:
•failure to implement our business plans for the combined businesses and consolidation or expansion of production capacity as planned and where applicable;
•unexpected losses of key employees, customers or suppliers of our acquired companies and businesses;
•unanticipated issues in conforming our acquired companies’ and businesses’ standards, processes, procedures and internal controls with our operations;
•coordinating new product and process development;
•increasing the scope, geographic diversity and complexity of our operations;
•diversion of management’s attention from other business concerns;
•adverse effects on our or our acquired companies’ and businesses’ existing business relationships;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 64
•unanticipated changes in applicable laws and regulations;
•risks inherent in our acquired companies’ and businesses’ industry and operations;
•unanticipated expenses and liabilities;
•potential unfamiliarity with our acquired companies and businesses technology, products and markets, which may place us at a competitive disadvantage; and
•other difficulties in the assimilation of our acquired companies and businesses operations, technologies, products and systems.
If MyoScience, Flexion, or any other acquired companies and businesses have unanticipated or larger than anticipated liabilities for patent and trademark infringement claims, violations of laws, commercial disputes, taxes and other known and unknown types of liabilities, there may be liabilities that we underestimated or did not discover in the course of performing our due diligence investigation of our acquired companies and businesses. We may have no recourse or limited recourse under the applicable acquisition-related agreement to recover damages relating to the liabilities of our acquired companies and businesses.
We may not be able to maintain or increase the levels of revenue, earnings or operating efficiency that each of the acquired companies and businesses and Pacira had historically achieved or might achieve separately. In addition, we may not accomplish the integration of any acquired companies and businesses smoothly, successfully or within the anticipated costs or timeframe. If we experience difficulties with the integration process or if the business of any acquired companies or businesses deteriorates, the anticipated cost savings, growth opportunities and other synergies of any acquired companies and businesses may not be realized fully or at all, or may take longer to realize than expected. If any of the above risks occur, our business, financial condition, results of operations and cash flows may be materially and adversely impacted; we may fail to meet the expectations of investors or analysts; and our stock price may decline as a result.
Our ability to realize the benefits from the Flexion Acquisition is substantially dependent on the commercial success of ZILRETTA and the cost savings resulting from the timely and effective integration of the operations of Pacira and Flexion.
Our ability to realize the benefits from the Flexion Acquisition is substantially dependent on our ability to successfully commercialize ZILRETTA. Combining with Pacira may not accelerate the growth and success of ZILRETTA. If we are unsuccessful at convincing health care providers to increase their rate of adoption of ZILRETTA, our sales could be adversely affected, and our business and financial condition could suffer.
Further, our ability to realize the benefits from the Flexion Acquisition is substantially dependent on the cost savings resulting from the timely and effective integration of the operations Pacira and Flexion. The process of integrating the operations of Pacira and Flexion could encounter unexpected costs and delays, which include but are not limited to: the loss of key personnel; the loss of key customers; the loss of key suppliers; integrating the products, services and related assets, as well as internal controls into our business operations; and unanticipated issues in integrating the sales, marketing and administrative functions. If we are unable to timely and effectively integrate the operations of Pacira and Flexion, our results of operations could be adversely affected, and our business could suffer. Further, even if the integration is timely and effective, we may never realize the cost savings expected from the integration and synergies of the operations of the two companies.
The use of our net operating loss carryforwards and research and development tax credits will be limited.
We have significant Federal and state net operating loss, or NOL, carryforwards and federal and state research and development tax credit carryforwards. Our NOL carryforwards and research and development tax credits may expire and not be used. Our state NOL carryforwards will begin expiring in 2028 if we have not used them prior to that time. We have non-U.S. NOLs that do not expire. Additionally, our ability to use certain NOLs to offset taxable income in the future will be limited under Internal Revenue Code Section 382 because we experienced cumulative changes in ownership of more than 50% within a three-year period. Such ownership changes were triggered by the cumulative ownership changes arising as a result of the initial acquisition of the Company’s stock in 2007 and the completion of our initial public offering in February 2011 and our other financing transactions. Additionally, in November 2021, we completed the Flexion Acquisition which also triggered an ownership change. Because of these ownership changes, we will be limited regarding the amount of NOL carryforwards that we can utilize annually in the future to offset taxable income. Such an annual limitation may significantly reduce the utilization of the NOLs before they expire.
Risks Related to Information Technology, Cybersecurity and Data Privacy
We face risks related to cybersecurity threats and incidents.
We regularly face attempts by others to gain unauthorized access through the internet, or to introduce malicious software, to our Information Technology, or IT, systems. Individuals or organizations, including malicious hackers and insider threats including employees and third-party service providers, or intruders into our physical facilities, at times attempt to gain unauthorized access to our software, network and services. We could also be a target of malicious attackers who attempt to gain
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 65
access to our network or data centers; steal proprietary information related to our business, products, employees, suppliers and customers; interrupt our systems and services or those of our suppliers, customers, or others; or demand a ransom to return control of such systems and services. Such attempts—including but not limited to—social engineering or “phishing” attempts, denial of service attacks and malware (including viruses, trojans and keyloggers) are increasing in number, intensity and in technical sophistication, and are increasingly difficult to detect for periods of time, especially as they relate to attacks on third-party vendors, and, if successful, expose us and any affected parties to risk of loss or misuse of proprietary or confidential information or disruptions of our business operations, including our manufacturing operations. These attacks are often carried out by motivated and highly skilled actors, who are increasingly well-resourced. Our IT infrastructure also includes services provided by third parties, and these service providers can experience breaches of their systems and products that impact the security of our systems and our proprietary or confidential information. In addition, certain factors, such as growth through acquisitions, rapid technology evolution, including increased adoption of artificial intelligence, and geopolitical events, have increased cybersecurity risks. A substantial breach of our or one of our service providers’ systems could result in the loss of revenues, the misuse of confidential data, manufacturing challenges or disruption, diversion of management attention, litigation, regulatory action and damage to our relationships with vendors, business partners and customers, and we may incur significant expenses to resolve such issues.
Finally, the SEC has adopted rules that require us to provide greater disclosures around cybersecurity risk management, strategy and governance, as well as disclose the occurrence of material cybersecurity incidents. We cannot yet predict or estimate the amount of additional costs we will incur in order to comply with these rules or the timing of such costs. These rules and regulations may also require us to report a cybersecurity incident before we have been able to fully assess its impact or remediate the underlying issue. Efforts to comply with such reporting requirements could divert management’s attention from our incident response and could potentially reveal system vulnerabilities to threat actors. Failure to timely report incidents under these or other similar rules could also result in monetary fines, sanctions or subject us to other forms of liability. This regulatory environment is increasingly challenging and may present material obligations and risks to our business, including significantly expanded compliance burdens, costs and enforcement risks.
If we do not maintain the privacy and security of personal and business information, we could damage our reputation with customers and employees, incur substantial additional costs and become subject to litigation.
We receive, retain and transmit personal information about our customers and employees as well as confidential or proprietary information of ours and our customers, vendors and other business partners, and entrust that information to third-party suppliers, including cloud service-providers, software-as-a-service (“SaaS”) solutions, platform-as-a-service (“PaaS”) solutions, data hosting and processing facilities, artificial intelligence, tools and other hardware, software (including open-source software) technical applications and platforms and managed services, including some that are managed, hosted, provided and/or used by third-party vendors, to operate our business that perform activities for us. Our business depends upon the secure transmission of encrypted confidential information over public networks, including information permitting payments. A compromise of our security systems or defects within our hardware or software, or those of our suppliers, that results in our customers’ or our employees’ information being obtained by unauthorized persons, could adversely affect our reputation with our customers and others, as well as our operations, results of operations, financial condition and liquidity, and could result in litigation, government actions, or the imposition of penalties. In addition, a breach could disrupt our operations and require that we expend significant additional resources related to the security of our information systems.
The use of data by our business is regulated at the national and state or local level in all of our operating countries. Privacy and information-security laws and regulations change, and compliance with them may result in cost increases due to, among other things, systems changes and the development of new processes. If we or those with whom we share information fail to comply with these laws and regulations, our reputation could be damaged, possibly resulting in lost future business, and we could be subjected to additional legal risk as a result of non-compliance.
We have security measures and controls to protect personal and business information and continue to make investments to secure access to our information technology network. These measures may be undermined, however, due to the actions of outside parties, employee error, internal or external malfeasance, or otherwise, and, as a result, an unauthorized party may obtain access to our data systems and misappropriate business and personal information. Because the techniques used to obtain unauthorized access, disable or degrade service, or sabotage systems change frequently and may not immediately produce signs of intrusion, we may be unable to anticipate these techniques, timely discover or counter them, or implement adequate preventative measures. Any such breach or unauthorized access could result in significant legal and financial exposure, damage to our reputation, and potentially have an adverse effect on our business and results of operations.
Changes in data privacy and protection laws and regulations, particularly in Europe and the State of California, or any failure to comply with such laws and regulations, could adversely affect our business and financial results.
We are subject to a variety of continuously evolving and developing laws and regulations globally regarding privacy, data protection and data security, including those related to the collection, storage, handling, use, disclosure, transfer and security of personal data. Significant uncertainty exists as privacy and data protection laws may be interpreted and applied differently from
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 66
country to country and may create inconsistent or conflicting requirements. These laws apply to transfers of information among our affiliates, as well as to transactions we enter into with third-party vendors.
For example, the E.U. adopted a comprehensive General Data Privacy Regulation, or GDPR, in May 2016 that replaced the then-current E.U. Data Protection Directive and related country-specific legislation in May 2018. GDPR requires companies to satisfy new requirements regarding the handling of personal and sensitive data, including its use, protection and the ability of persons whose data is stored to correct or delete such data about themselves. Failure to comply with GDPR requirements could result in penalties of up to 4% of total worldwide revenue.
Additionally, the California Consumer Privacy Act, or CCPA, became effective in January 2020 and imposed new responsibilities on us for the handling, disclosure and deletion of personal information for our employees and consumers who reside in California. The CCPA permits California to assess potentially significant fines for violating CCPA and creates a right for individuals to bring class action suits seeking damages for violations. We have also implemented more stringent privacy regulations related to the California Privacy Rights Act, which was an amendment to the CCPA.
Furthermore, legislators and regulators in the U.S. are proposing new and more robust cybersecurity rules in light of the recent broad-based cyberattacks at a number of companies. Our efforts to comply with GDPR, the CCPA and other privacy and data protection laws may impose significant costs and challenges that are likely to increase over time and may require us to revise certain of our business practices. These and similar initiatives around the world could increase the cost of developing, implementing or securing our servers and require us to allocate more resources to improved technologies, adding to our information technology and compliance costs. In addition, enforcement actions and investigations by regulatory authorities related to data security incidents and privacy violations continue to increase. The enactment of more restrictive laws, rules, regulations, or future enforcement actions or investigations could impact us through increased costs or restrictions on our business, and noncompliance could result in substantial regulatory penalties and significant legal liability or litigation related to violation of existing or future data privacy laws and regulations.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, human error, unauthorized access, natural or man-made disasters, intentional acts of vandalism, terrorism, war and network, telecommunication and electrical failures. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our manufacturing operations or product development programs. For example, the loss of clinical trial data from completed clinical trials for our products could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability, reputation damage and harm to our business operations.
Artificial intelligence presents risks and challenges that can impact our business including by posing security risks to our confidential information, proprietary information, and personal data.
Issues in the development and use of artificial intelligence, combined with an uncertain regulatory environment, may result in reputational harm, liability or other adverse consequences to our business operations. As with many technological innovations, artificial intelligence presents risks and challenges that could impact our business. We may adopt and integrate generative artificial intelligence tools into our systems for specific use cases reviewed by legal and information security. Our vendors may incorporate generative artificial intelligence tools into their offerings without disclosing this use to us, and the providers of these generative artificial intelligence tools may not meet existing or rapidly evolving regulatory or industry standards with respect to privacy and data protection and may inhibit our or our vendors’ ability to maintain an adequate level of service and experience. If we, our vendors or our third-party partners experience an actual or perceived breach or privacy or security incident because of the use of generative artificial intelligence, we may lose valuable intellectual property and confidential information and our reputation and the public perception of the effectiveness of our security measures could be harmed. Further, bad actors around the world use increasingly sophisticated and rapidly evolving methods, including the use of artificial intelligence, to engage in illegal activities involving the theft and misuse of personal information, confidential information and intellectual property. Any of these outcomes could damage our reputation, result in the loss of valuable property and information, and adversely impact our business.
General Risk Factors
A pandemic, epidemic or outbreak of a contagious disease (such as the recent novel coronavirus (COVID-19) pandemic), or fear of such an event, could have a material adverse effect on our business, operating results and financial condition.
A pandemic, epidemic or outbreak of an infectious disease, including the lingering impact of the COVID-19 pandemic (despite the end of the federal COVID-19 public health emergency declaration in May of 2023), or other public health crisis, could have a material adverse effect on our business, financial condition and operations, including but not limited to our
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 67
revenue and cash flows, including potential decreases in sales, manufacturing issues, supply chain issues, including, but not limited to, staffing shortages, cost inflation and shipping delays, and delays in payments by our customers. For example, during 2020, our net product sales were negatively impacted by the COVID-19 pandemic due to the significant postponement or suspension in the scheduling of elective surgical procedures resulting from public health guidance and government directives. New or prolonged suspensions of elective surgeries by governmental restrictions or action would cause net sales of our products to decrease. In addition, health concerns from a pandemic, epidemic or outbreak of an infectious disease or negative economic conditions, could cause patients and clinicians to cancel or defer elective procedures or otherwise avoid medical treatment, which would result in reduced patient volumes and revenues and could potentially continue over an extended period of time.
Business disruptions could include disruptions or restrictions to our workforce, including the ability of our sales teams to interact with our customers and healthcare professionals to educate them on the benefits of our products and perform typical sales activities. For example, the COVID-19 pandemic previously had significantly impacted the ability of our sales representatives to access customers and healthcare professionals through personal interactions within the healthcare setting, including hospitals and ASCs. In addition, any temporary closures of our manufacturing facilities, the facilities of our suppliers and contract manufacturers (and the resulting impact on production or our products) or the workforce at such facilities, or those of our distributors, could cause delays in the shipment or production of our products. If our customers experience disruptions to their businesses and cash flows, we could experience delays or difficulties with the collection of our accounts receivable. Any sustained impacts and business disruptions to our facilities or workforce, our customers, our suppliers, or our contract manufacturers would likely adversely impact our cash flows, sales and operating results.
The significant increase in the number of our employees who may return to working remotely as a result of a future pandemic, and an extended period of remote work arrangements and subsequent reintroduction into the workplace could introduce operational risk, strain our business continuity plans, negatively impact productivity and/or collaboration, and give rise to claims by employees or otherwise adversely affect our business. Additionally, a pandemic or other public health emergencies could require new or modified processes, procedures and controls to respond to changes in our business environment. We may take further actions as may be required by government authorities or that we determine are in the best interests of our employees, customers and business partners. There is no certainty that such measures will be sufficient to mitigate the risks posed by a pandemic or other public health emergencies.
In addition, a pandemic or other public health emergency could, among other things, cause global macroeconomic uncertainty, disrupt consumer spending and supply chains, contribute to various global shipping delays and port congestions and create significant volatility and disruption of financial markets.
Ultimately, the extent to which future public health crises could impact our business is difficult to predict and will depend on many factors beyond our control, including the speed of contagion, the development and implementation of effective preventative measures and possible treatments, the scope of governmental and other restrictions on elective surgeries, travel and other activity through quarantines/social distancing and other measures, the timing of effective vaccines becoming widely available and accepted by the public, public reactions to these factors and more.
Corporate social responsibility, or CSR, issues may have an adverse effect on our business, financial condition and results of operations and damage our reputation.
There is an increasing focus from certain investors, customers, consumers, employees, lawmakers, regulators (such as the SEC) and other stakeholders concerning CSR matters, including particular focus on climate-related risks. Additionally, public interest and legislative pressure related to public companies’ CSR practices continue to grow. The landscape related to such regulation, compliance and reporting is constantly evolving, including expanding in scope and complexity. We may experience significant future cost increases associated with regulatory compliance, including fees, licenses, reporting, auditing, and the cost of capital improvements for our operating facilities to meet sustainability and/or environmental regulatory requirements. If our CSR practices fail to meet regulatory requirements or investor, customer, consumer, employee or other stakeholders’ evolving expectations and standards for responsible corporate citizenship in areas including environmental stewardship, support for local communities, board of director and employee diversity, human capital management, employee health and safety practices, product quality, supply chain management, corporate governance and transparency, our reputation, brand and employee retention may be negatively impacted, and our customers and suppliers may be unwilling to continue to do business with us.
From time to time, we communicate certain CSR initiatives and goals to market participants and our customers and business partners. Any corporate responsibility disclosure we make may include our policies, practices, initiatives and goals on a variety of social and ethical matters, corporate governance, environmental compliance, sustainability, employee health and safety practices, human capital management, product quality, supply chain management and workforce inclusion and diversity. Although we have undertaken significant efforts to improve and implement our CSR initiatives, it is possible that the aforementioned parties may not be satisfied with such disclosures, our CSR practices or the speed with which we adopt, implement and/or disclose our plans. Furthermore, some stakeholders may disagree with our goals and there is also a risk that stakeholders may change their views on these topics over time. Our various stakeholders or regulators may also have divergent opinions on these types of matters as well as conflicting expectations regarding our culture, values, goals and business, which
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 68
makes it difficult to achieve a consistently positive perception amongst all of our various stakeholders. Moreover, we may determine that it is in the best interest of our Company and our stockholders to prioritize other business, social, governance or sustainable investments over the achievement of our current goals based on economic, technological developments, regulatory and social factors, business strategy or pressure from investors, activists or other stakeholders.
If our CSR practices do not meet investor or other stakeholder expectations and standards, which continue to evolve, or if we are perceived or deemed to have not appropriately responded to the growing concern for CSR issues, regardless of whether there is a legal requirement to do so, we may suffer from reputational damage from stakeholders and consumers and our business and financial condition could be materially and adversely affected. We may also incur additional costs or require additional resources to monitor such stakeholder expectations and standards and to meet our targets and commitments.
Significant changes in the global climate, extreme weather conditions, water availability and other climate related risks could adversely affect our business or operations.
We could experience adverse impacts to our business if climate change, storms, or other extreme weather conditions and/or water availability challenges adversely affect our operations or the operations of our suppliers, distributors and customers. There is mounting scientific evidence, as well as concern from the general public, that emissions of greenhouse gases and contributing human activities have caused and will continue to cause significant changes in global temperatures and weather patterns and increase the frequency or severity of storms and other weather events, extreme heat, hurricanes, wildfires and flooding. While such conditions cannot be predicted, if such conditions were to impact our manufacturing sites or otherwise alter production schedules, including those of our third-party suppliers of raw materials, our manufacturing equipment, or our distributors, we could experience a disruption in the supply of EXPAREL, ZILRETTA or iovera° to our customers and partners, or we could see an unfavorable impact on the cost or availability of our raw or packaging materials. Disruptions to the operations of our customers could also adversely impact the demand for our products. Regulations in response to climate change could result in increased manufacturing costs associated with increased compliance and water and energy costs.
The effects of climate change, natural disasters such as earthquakes, wildfires, hurricanes, tornadoes, droughts, tsunamis or other adverse weather events and climate conditions, whether occurring in the U.S. or abroad, and the consequences and effects thereof, including damage to our supply chain, such as availability of raw materials, increased manufacturing costs and disruptions to productivity of our manufacturing operations, changes in consumer preferences or spending priorities, and energy shortages, have in the past and could in the future harm or disrupt our operations or the operations of our vendors, other suppliers, or customers, or result in economic instability that may negatively impact our operating results and financial condition. Additionally, certain catastrophes may not be covered by our general insurance policies, which could result in significant unrecoverable losses. Many governmental and other regulatory bodies worldwide are enacting regulations to mitigate the impacts of climate change. If we, our suppliers, or others in our supply chain are required to comply with these laws and regulations, or if we choose to take additional voluntary steps to reduce or mitigate our impact on the climate, we may experience increased costs for energy, production, transportation and raw materials, increased capital expenditures, or increased insurance premiums and deductibles, each of which could adversely impact our operations. In addition, inconsistent regulations among jurisdictions may also affect our cost to comply with such laws and regulations. Any assessment of the potential impact of future climate change legislation, regulations or industry standards, as well as any international treaties and accords, is uncertain given the wide scope of potential regulatory change in the countries in which we operate.
Our international operations expose us to numerous and sometimes conflicting legal and regulatory requirements, the compliance of which could be costly and time consuming and violation of these regulations could adversely affect our business or operations.
We are subject to numerous, and sometimes conflicting, legal requirements on matters as diverse as pharmaceutical and medical device marketing, product liability, anti-corruption, data protection and privacy, compliance, taxation, accounting and financial reporting, employment laws, wage-and-hour standards, labor relations and human rights, among others. The global nature of our operations may increase the difficulty and cost of compliance with various regulations and laws, as compliance with diverse legal requirements is costly, time-consuming and requires significant resources. Violations of one or more of these regulations in the conduct of our business could result in significant fines, enforcement actions or criminal sanctions against us and/or our employees, prohibitions on doing business and damage to our reputation.
In addition to these legal and regulatory requirements, there are risks inherent in doing business internationally, including but not limited to:
•different or more restrictive privacy, data protection, data localization, and other laws that could require us to make changes to our products, services and operations, such as mandating that certain types of data collected in a particular country be stored and/or processed within that country;
•difficulties in developing, staffing, and simultaneously managing our foreign operations as a result of geographic distance, language, and cultural differences;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 69
•stringent local labor laws and regulations;
•profit repatriation and foreign currency exchange restrictions;
•geopolitical events, including natural disasters, acts of war and terrorism, and public health emergencies, including divergent governmental responses thereto across the jurisdictions in which we operate;
•import or export regulations;
•trade barriers and changes in trade regulations; including impositions of tariffs on imported raw materials and property, plant and equipment;
•compliance with the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act, and laws and regulations of other jurisdictions prohibiting corrupt payments to government officials and other third parties;
•antitrust and competition regulations;
•delays associated with the manufacture, transportation and delivery of products, including delays related to global port backlog or congestion;
•increased transportation costs due to distance, energy prices, inflation or other factors;
•potentially adverse tax developments;
•political or social unrest, including but not limited to the war in Ukraine and the Israel-Hamas war, economic instability, repression, or human rights issues; and
•risks related to other government regulation or required compliance with local laws.
In addition, we are subject to customs laws and regulations with respect to our export and import activity, which are complex and vary within legal jurisdictions in which we operate. We cannot ensure that there will not be a control failure around customs enforcement despite the precautions we take. We are currently subject to audits by customs authorities. Any failure to comply with customs laws and regulations could be discovered during a U.S. or foreign government customs audit, or customs authorities may disagree with our tariff treatments, and such actions could result in substantial fines and penalties, which could have an adverse effect on our business and financial results. In addition, changes to U.S. trade laws or the imposition of tariffs may adversely impact our operations. These changes and any changes to the trade laws of other countries may add additional compliance costs and obligations and subject us to significant fines and penalties for non-compliance. Compliance with these and other foreign legal regimes may have a material adverse impact on our business and results of operations. Furthermore, as a global company, we are subject to foreign and U.S. laws and regulations designed to combat governmental corruption, including the U.S. Foreign Corrupt Practices Act and the U.K. Bribery Act. Violations of these laws and regulations could result in fines and penalties; criminal sanctions against us, our directors, our officers, or our employees; prohibitions on the conduct of our business and on our ability to offer our products and services in one or more countries; and a materially negative effect on our brands and our operating results. Although we have implemented policies and procedures designed to ensure compliance with these foreign and U.S. laws and regulations, including the U.S. Foreign Corrupt Practices Act and the U.K. Bribery Act, there can be no assurance that our employees, business partners, or agents will not violate our policies.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 70
Item 1B. Unresolved Staff Comments
None.
Item 1C. Cybersecurity
We are subject to cybersecurity threats that could have a material adverse impact on our results of operations, financial condition and cash flows, as well as our operations—including our manufacturing and marketing capabilities. We operate a risk-based cybersecurity program which is designed to: (i) ensure the security, confidentiality, integrity and availability of our information and systems; (ii) protect against anticipated or actual cyber threats to our information and systems; and (iii) protect against unauthorized access and/or use of our information and systems. Overall cybersecurity risk reporting is integrated with our enterprise risk management program, is included in discussions with the Audit Committee of our board of directors and disclosed where appropriate. Our information technology and cybersecurity function is headed by our Chief Administrative Officer, or CAO, and Vice President of Information Technology, who are responsible for managerial oversight of our cybersecurity program. Our CAO reports directly to our Chief Executive Officer and our Vice President of Information Technology reports directly to our CAO and has over 15 years of experience in cybersecurity.
We utilize a layered approach in assessing, identifying, evaluating and managing material risks from cybersecurity threats, and leverage outside partners to gain intelligence on threats. Highlighted risks are integrated into our Enterprise Risk Management process. We take input from industry activities, third party assessments and internal simulations and continuously adjust our protection mechanisms to be effective. We also assess operational and data security risks associated with our use of third-party service providers, understanding where failure points may exist within our supply chain operations and data protections. If we learn of a cybersecurity incident at a third-party service provider, our information technology department will maintain communication with that third-party service provider and communicate any cybersecurity incidents to the Vice President of Information Technology and CAO. All Pacira employees receive information security training (including data protection and fraud awareness) on an annual basis, and we use state-of-the-art technology to monitor systems for anomalous behavior. We also require employees in certain roles to complete additional role-based, specialized cybersecurity trainings. In the event an incident were to occur, a Security Incident Response Team would be convened that consists of members from many functions, including legal counsel, the Vice President of Information Technology and the CAO.
Our board of directors has the ultimate oversight of the Company’s risks—including cybersecurity risks—with our Audit Committee assisting the board in their oversight of cyber and information security risks. Members of management that possess information security certifications and many years of experience work with our legal, finance and corporate governance functions to identify, define and report cybersecurity risks, policies and procedures and incident response plans. The Audit Committee receives updates on our cybersecurity program from management on a quarterly basis and more frequently as determined to be necessary or advisable, and the full board receives a cybersecurity program update at least annually. Updates to the Audit Committee include policies, processes, procedures and any significant developments related to the identification, mitigation and remediation of cybersecurity risks, as well as effectiveness and changes in our ability to monitor, protect, detect and respond to incidents, risk reviews and industry news briefings. The Audit Committee also ensures that management provides a cyber and information security update to the board at least annually. Finally, in the event a material cybersecurity incident were to occur, the CAO and Vice President of Information Technology would brief the Audit Committee which would then be responsible for assessing the materiality of the incident and making the determination of materiality and any related disclosure.
We face a number of cybersecurity risks and threats in connection with our business. Although we have numerous controls to protect against common attacks, some attacks may still be effective. Our controls are designed to detect, triage and eradicate these attacks. While we carry a cyber insurance policy to help cover investigation and mitigation expenses, it may be subject to limitations and be insufficient to cover all expenses that may result from a cybersecurity incident. Although the risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, have not materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations or financial condition, such incidents could have a material adverse effect in the future as cyberattacks continue to increase in frequency and sophistication.
For more information about the cybersecurity risks and other information technology and data privacy risks we face, see Item 1A. Risk Factors and the subsection titled Risks Related to Information Technology, Cybersecurity and Data Privacy.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 71
Item 2. Properties
We occupy three facilities totaling approximately 195,000 square feet at our Science Center Campus in San Diego, California. We use these facilities for research and development, manufacturing, general and administrative purposes and the storage of inventory and raw materials. Our manufacturing facility on this site where we produce EXPAREL and the handpieces for iovera° and our mixed-use research and development property leases both expire in June 2030 and our warehouse lease expires in August 2030.
Our Pacira Innovation and Training Center in Tampa, Florida, is an approximately 13,000 square-foot facility that supports a full range of educational events to advance clinician understanding of the latest local, regional and field block approaches for managing pain and reducing or eliminating exposure to opioids. Our principal executive offices and corporate headquarters are also located here, and our lease expires in December 2026.
In addition, we maintain two administrative, commercial and business development offices—one in Parsippany, New Jersey, where we occupy approximately 53,000 square feet under a lease expiring in March 2028 and one in Brisbane, California, where we occupy approximately 20,000 square feet under a lease expiring in May 2026.
In February 2025, Pacira Therapeutics, Inc., a wholly-owned subsidiary of the Company, entered into a securities purchase agreement to acquire the remaining 81% of GQ Bio that was not already owned by us. GQ Bio maintains its main administrative office in Hamburg, Germany, has a research and development lab in Luckenwalde, Germany and maintains smaller administrative offices in each of Eupen and Liège, Belgium. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events, to our consolidated financial statements included herein.
We believe that our research and development and manufacturing facilities at our Science Center Campus, Thermo Fisher and Carlisle sites (as discussed in Item 1—Business above) will be sufficient for our current commercial and pipeline development needs. We also may add new facilities or expand existing facilities as we add employees, expand our geographic markets and if demand for EXPAREL, ZILRETTA and iovera° increases and we believe that suitable additional or substitute space will be available as needed to accommodate any such expansion of our operations.
Item 3. Legal Proceedings
We are subject to legal proceedings and claims that have not been fully resolved and that have arisen in the ordinary course of business. For information related to Item 3. Legal Proceedings, refer to Note 19, Commitments and Contingencies, to our consolidated financial statements included herein.
Item 4. Mine Safety Disclosures
Not applicable.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 72
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Our common stock is listed and traded under the ticker symbol “PCRX” on the Nasdaq Global Select Market. As of February 26, 2025, we had nine holders of record of our common stock. The number of record holders is based on the actual number of holders registered on the books of our transfer agent and does not reflect the substantially greater amount of holders of shares in “street name,” whose shares are held of record by banks, brokers and other financial institutions.
Performance Graph
The following graph and table shows the value of an investment of $100.00 made on December 31, 2019—the last trading day of 2019—in each of Pacira BioSciences, Inc. (PCRX), the Nasdaq Composite Index (^IXIC), the Nasdaq Biotechnology Index (^NBI) and the S&P Pharmaceuticals Select Index (^SPSIPH). The three indices included are for comparative purposes only and do not necessarily reflect management’s opinion that such indices are an appropriate measure of the relative performance of our common stock. All results assume the reinvestment of dividends, if any, and are calculated as of December 31st of each year. The historical stock price performance of our common stock and the indices shown in this performance graph is not necessarily indicative of future stock price performance.
Comparison of Five-Year Cumulative Total Returns Among
Pacira BioSciences, Inc., the Nasdaq Composite Index,
the Nasdaq Biotechnology Index and the S&P Pharmaceuticals Select Index
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Cumulative Total Return as of December 31, |
| 2019 | | 2020 | | 2021 | | 2022 | | 2023 | | 2024 |
| Pacira BioSciences, Inc. (PCRX) | $ | 100.00 | | | $ | 132.10 | | | $ | 132.83 | | | $ | 85.23 | | | $ | 74.48 | | | $ | 41.59 | |
| Nasdaq Composite Index (^IXIC) | $ | 100.00 | | | $ | 143.64 | | | $ | 174.36 | | | $ | 116.65 | | | $ | 167.30 | | | $ | 215.22 | |
| Nasdaq Biotechnology Index (^NBI) | $ | 100.00 | | | $ | 125.69 | | | $ | 124.89 | | | $ | 111.27 | | | $ | 115.42 | | | $ | 113.84 | |
| S&P Pharmaceuticals Select Index (^SPSIPH) | $ | 100.00 | | | $ | 113.59 | | | $ | 100.83 | | | $ | 89.53 | | | $ | 90.76 | | | $ | 93.69 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 73
Dividend Policy
We have never declared or paid any dividends on our common stock. We currently intend to retain any future earnings to finance the future development and expansion of our business, and as such we do not expect to pay any dividends on our common stock in the foreseeable future. The payment of any future dividends would be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in the agreements governing our indebtedness, provisions of applicable law and any other factors our board of directors deems relevant.
Purchases of Equity Securities by the Registrant
None.
Item 6. Reserved
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Management’s Discussion and Analysis of Financial Condition and Results of Operations is based upon our consolidated financial statements, which have been prepared in accordance with generally accepted accounting principles in the United States of America, or GAAP, and in accordance with the rules and regulations of the United States Securities and Exchange Commission, or SEC. We operate and report our financial information in one segment. The following discussion of our financial condition and results of operations should be read in conjunction with the other sections of this Annual Report, including our consolidated financial statements and the notes to those consolidated financial statements appearing in Part IV, Item 15, of this Annual Report. This discussion contains forward-looking statements that involve significant risks and uncertainties. As a result of many factors, such as those set forth under “Risk Factors” in Part I, Item 1A. of this Annual Report, our actual results may differ materially from those anticipated in these forward-looking statements. Certain defined terms have been brought forward from Part I of this Annual Report.
This section of this Annual Report discusses year-to-year comparisons between 2024 and 2023, as well as other discussions of 2024 and 2023 items. We have omitted discussion of the year ended December 31, 2022 (the earliest of the three years covered by our consolidated financial statements presented in this Annual Report) as permitted by SEC regulations. The complete Management’s Discussion and Analysis of Financial Condition and Results of Operations for year-to-year comparisons between 2023 and 2022 and other discussions of 2022 items can be found within Part II, Item 7, to our Annual Report for the year ended December 31, 2023, filed with the SEC on February 29, 2024, which is available on the SEC’s website at www.sec.gov and our corporate website at www.pacira.com. The foregoing reference to our corporate website is not intended to, nor shall it be deemed to, incorporate information on our corporate website into this Annual Report by reference, and the inclusion of our corporate website address in this Annual Report is an inactive textual reference only and is not intended to be an active link to our corporate website. Overview
Our stated corporate mission is to deliver innovative, non-opioid pain therapies to transform the lives of patients. We are also developing innovative interventions to address debilitating conditions involving the sympathetic nervous system, such as cardiac electrical storm, chronic pain and spasticity. Our long-acting, local analgesic EXPAREL® (bupivacaine liposome injectable suspension) utilizes our unique pMVL drug delivery technology that encapsulates drugs without altering their molecular structure and releases them over a desired period of time. In the U.S., EXPAREL is a long-acting, non-opioid option proven to manage postsurgical pain. EXPAREL is the only product indicated for local analgesia via infiltration in patients aged six years and older and regional analgesia via interscalene brachial plexus nerve block, sciatic nerve block in the popliteal fossa and adductor canal block in adults. In Europe, EXPAREL is approved as a brachial plexus block or femoral nerve block for treatment of post-operative pain in adults, and as a field block for treatment of somatic post-operative pain from small- to medium-sized surgical wounds in adults and children aged six years and older. We drop-ship EXPAREL directly to end-users based on orders placed to wholesalers or directly to us, and there is no product held by wholesalers. With the acquisition of Flexion Therapeutics, Inc. in November 2021 (the “Flexion Acquisition”), we acquired ZILRETTA® (triamcinolone acetonide extended-release injectable suspension), the first and only extended-release, intra-articular injectable therapy that can provide major relief for OA knee pain for three months and has the potential to become an alternative to hyaluronic acid, or HA, and platelet rich plasma, or PRP, injections or other early intervention treatments. With the acquisition of MyoScience, Inc. in April 2019 (the “MyoScience Acquisition”), we acquired iovera®°, a handheld cryoanalgesia device used to deliver a precise, controlled application of cold temperature to targeted nerves, which we sell directly to end users. EXPAREL, ZILRETTA and the iovera° system are highly complementary products as long-acting, non-opioid therapies that alleviate pain. We are also advancing the development of PCRX-201 (enekinragene inzadenovec), a novel gene therapy vector platform enabling local administration of genetic medicines with the potential to treat large prevalent diseases like OA. In February 2025, we acquired
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 74
GQ Bio Therapeutics GmbH, a privately-held biopharmaceutical company (the “GQ Bio Acquisition”). PCRX-201 is the lead program from this platform.
We expect to continue to pursue the expanded use of EXPAREL, ZILRETTA and iovera° in additional procedures; progress our product candidate pipeline, including the development of PCRX-201; advance regulatory activities for EXPAREL, ZILRETTA, iovera° and our other product candidates; invest in sales and marketing resources for EXPAREL, ZILRETTA and iovera°; expand and enhance our manufacturing capacity for EXPAREL, ZILRETTA and iovera°; invest in products, businesses and technologies; and support legal matters.
Recent Highlights
•In November 2024, we presented two-year safety and efficacy data at the American College of Rheumatology’s annual ACR Convergence meeting in Washington, DC. The new data is derived from an open-label, Phase 1 trial investigating the safety and efficacy of PCRX-201 administered via ultrasound-guided intraarticular injection in 72 patients with moderate to severe osteoarthritis of the knee (OAK) graded at 2, 3, or 4 on the Kellgren-Lawrence scale, a semiquantitative method for evaluating the severity of OA on a scale of 0-4. Participants were broken into two cohorts. The first cohort received one of three doses of PCRX-201. The second cohort received concurrent pretreatment with an IA corticosteroid (methylprednisolone 40 mg), a technique common in gene therapy dosing to improve tolerability and gene transfer. Pain and function benefits were observed at all doses and across both cohorts over the full two years studied, with patients in the second cohort achieving greater pain reduction and fewer AEs. Given these highly encouraging Phase 1 data, we opened enrollment in a randomized, double-blind Phase 2 clinical study in knee OA in February 2025.
•In December 2024, we announced the receipt of U.S. Patent No. 12,156,940 (the ‘940 patent) from the USPTO. The ‘940 patent—entitled “Manufacturing of Bupivacaine Multivesicular Liposomes”—protects the chemical composition of EXPAREL. This patent is the first patent from a new family of patents related to EXPAREL produced by our enhanced larger-scale manufacturing process in San Diego, California, which received approval from the FDA in February 2024. We expect the ‘940 patent to provide protection into July 2044. Additionally, the ‘940 patent is listed in the FDA’s “Approved Drug Products with Therapeutic Equivalence Evaluations” (the “Orange Book”). Patents that are eligible for Orange Book listing are those that have claims covering the active ingredient, the drug product (formulation and composition) or the approved method of use. Additionally, the USPTO recently issued U.S. Patent Nos. 12,151,024 (the ‘024 patent) and 12,144,890 (the ‘890 patent). The ‘024 and ‘890 patents belong to the ‘574 and ‘495 family of patents, respectively, and are listed in the Orange Book with an expiration date of January 21, 2041. We continue to prosecute patent applications and anticipate that additional patents are forthcoming.
•In January 2025, we announced the receipt of clearance from the FDA to market a new Smart Tip designed to access the medial branch nerves to manage chronic low back pain. This new, 25-gauge 180 mm Smart Tip allows for the treatment of deeper nerves, such as the medial branch nerve, and is specifically designed so that it can relieve chronic low back pain associated with facet mediated pain. This longer-needle Smart Tip is uniquely designed for use through a cannula or introducer, providing the ability for ice ball formation at deeper peripheral nerves. This FDA-cleared innovation offers a compelling alternative to conventional treatments such as RFA, which has substantial limitations. With RFA, patients may not get the effects of pain relief until one-to-two weeks after treatment. Further the intense heat can damage surrounding tissue and blood vessels, and tissue damage may lead to painful neuritis (inflammation in the nerves).
•In January 2025, Laura Brege was appointed Chair of the Board, following the retirement of former Chair, Paul J. Hastings, and Andreas Wicki, PhD. These changes align with our ongoing commitment to board refreshment. With these changes, the board is composed of nine directors, eight of whom are independent and five of whom have joined since October 2023. Each director brings expertise in areas important to our business to support our 5x30 growth-oriented plan including executive leadership, mergers and acquisitions, research and development, operations, commercialization, manufacturing and supply chain.
•In February 2025, we announced a definitive purchase agreement to acquire all outstanding equity interests of GQ Bio not already owned by us for approximately $32 million, net of working capital and other transaction adjustments to equity holders other than us. The net purchase price includes $18 million of cash paid at closing, $8 million to be paid over three years pursuant to a key employee holdback agreement and a post-closing indemnity holdback of $6 million. This acquisition adds a novel locally administered gene therapy vector platform and pipeline of preclinical assets with disease-modifying potential for prevalent musculoskeletal diseases and will advance our 5x30 growth strategy to transition into an innovative biopharmaceutical organization. PCRX-201 is the lead program from this platform. It
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 75
provides financial benefits with the elimination of future development and sales-based milestones including a $4.5 million milestone payment due upon initiation of the Phase 2 clinical trial of PCRX-201 that opened enrollment in February 2025. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events, to our consolidated financial statements included herein.
Global Economic Conditions, Inflation and Tariffs
Direct and indirect effects of global economic conditions have in the past, and may continue to, negatively impact our business, financial condition and results of operations. Such impacts may include the effect of prolonged periods of inflation or the imposition of tariffs, which could, among other things, result in higher costs for labor, raw materials, equipment and other goods and services; cause our patients to defer or cancel medical procedures, thereby adversely impacting our revenues; and negatively impact our suppliers which could cause longer lead-times or the inability to secure a sufficient supply of materials. The current macroeconomic environment remains dynamic and subject to rapid and possibly material changes. Additional negative impacts may also arise that we are unable to foresee. The nature and extent of such impacts will depend on future developments, which are highly uncertain and cannot be predicted.
Results of Operations
Comparison of the Years Ended December 31, 2024 and 2023
Revenues
Net product sales consist of sales of (i) EXPAREL in the U.S., E.U., and U.K.; (ii) ZILRETTA in the U.S.; (iii) iovera° in the U.S., Canada and the E.U. and (iv) sales of our bupivacaine liposome injectable suspension product for veterinary use. Royalty revenues are related to a collaborative licensing agreement from the sale of our bupivacaine liposome injectable suspension for veterinary use.
The following table provides information regarding our revenues during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| Net product sales: | | | | | |
| EXPAREL | $ | 548,962 | | | $ | 538,120 | | | 2% |
| ZILRETTA | 118,089 | | | 111,098 | | | 6% |
| iovera° | 22,813 | | | 19,685 | | | 16% |
| Bupivacaine liposome injectable suspension | 7,322 | | | 3,342 | | | 100%+ |
| Total net product sales | 697,186 | | | 672,245 | | | 4% |
| Royalty revenue | 3,780 | | | 2,733 | | | 38% |
| | | | | |
| Total revenues | $ | 700,966 | | | $ | 674,978 | | | 4% |
EXPAREL revenue increased 2% in 2024 versus 2023. A 4% increase in gross vial volume was partially offset by a shift in vial mix. EXPAREL revenue was also impacted by a 1% increase in selling price per unit related to a price increase, net of increases in sales related allowances as a result of group purchasing organization contracting.
ZILRETTA revenue increased 6% in 2024 versus 2023, primarily due to a 4% increase in net selling price per unit and a 2% increase in kit volume. The increase in net selling price per unit is related to two price increases and favorable sales related allowances.
Net product sales of iovera° increased 16% in 2024 versus 2023 primarily due to a 20% increase in Smart Tip volume, partially offset by increased sales related allowances and accruals.
Bupivacaine liposome injectable suspension revenue increased more than 100% in 2024 versus 2023, and the related royalties increased 38%, primarily due to the sales mix of vial sizes and the timing of orders placed for veterinary use.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 76
Cost of Goods Sold
Cost of goods sold primarily relates to the costs to produce, package and deliver our products to customers. These expenses include labor, raw materials, manufacturing overhead and occupancy costs, depreciation of facilities, royalty payments, quality control and engineering.
The following table provides information regarding cost of goods sold and gross margin during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| Cost of goods sold | $ | 170,428 | | | $ | 184,669 | | | (8)% |
| Gross margin | 76% | | 73% | | |
Gross margin increased three percentage points in 2024 versus 2023 primarily due to lower EXPAREL product costs as a result of higher production volumes and the absence of a step-up of ZILRETTA fixed assets and inventory to fair value in accordance with purchase accounting that existed in the prior period, partially offset by higher EXPAREL and ZILRETTA inventory reserves.
Research and Development Expense
Research and development expenses primarily consist of costs related to clinical trials and related outside services, product development and other research and development costs, including trials that we are conducting to generate new data for EXPAREL, ZILRETTA and iovera°, clinical trials for PCRX-201 and stock-based compensation expense. Clinical and preclinical development expenses include costs for clinical personnel, clinical trials performed by third-parties, toxicology studies, materials and supplies, database management and other third-party fees. Product development and manufacturing capacity expansion expenses include development costs for our products, which include personnel, research equipment, materials and contractor costs for process development and product candidates, development costs related to significant scale-ups of our manufacturing capacity and facility costs for our research space. Regulatory and other expenses include regulatory activities related to unapproved products and indications, medical information and scientific communication expenses, expenses related to our iGOR registry study and related personnel. Stock-based compensation expense relates to the costs of stock option grants, awards of restricted stock units, or RSUs, and our employee stock purchase plan, or ESPP.
The following table provides a breakout of our research and development expenses during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| Clinical and preclinical development | $ | 33,696 | | | $ | 24,471 | | | 38% |
| Product development and manufacturing capacity expansion | 30,803 | | | 33,365 | | | (8)% |
| Regulatory and other | 9,697 | | | 9,727 | | | (0)% |
| Stock-based compensation | 7,381 | | | 8,694 | | | (15)% |
| Total research and development expense | $ | 81,577 | | | $ | 76,257 | | | 7% |
| % of total revenue | 12% | | 11% | | |
Total research and development expense increased 7% in 2024 versus 2023.
Clinical and preclinical development expense increased 38% in 2024 versus 2023 due to site start-up and ongoing enrollment in a ZILRETTA shoulder trial, an iovera° spasticity trial and an EXPAREL pediatric trial, as well as start-up expenses related to the PCRX-201 Phase 2 trial for knee OA. These increases were partially offset by the winding down of a PCRX-201 Phase 1 trial for knee OA as two-year follow-up visits of subjects were completed in November 2023. This Phase 1 trial remains on track for completion by November 2026, the last year of the follow-up period for the last patient dosed. In addition, toxicology studies for product candidates were completed in 2023.
Product development and manufacturing capacity expansion expense decreased 8% in 2024 versus 2023, primarily attributable to the completion of pre-commercial scale-up activities of our larger-scale EXPAREL manufacturing capacity at
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 77
our Science Center Campus in San Diego, California, which the FDA approved in February 2024 and was subsequently placed into service in July 2024. The decrease was partially offset by ongoing product development costs related to PCRX-201 and development costs for the iovera° medial branch Smart Tip which received FDA clearance in January 2025.
Regulatory and other research and development expenses remained flat in 2024 versus 2023. Increased enrollment and additional sites related to our iGOR registry study were offset by the reduction of international regulatory activities and headcount vacancies.
Stock-based compensation decreased 15% in 2024 versus 2023 primarily due to fewer equity awards granted to research and development personnel and headcount vacancies as well as the acceleration of stock-based compensation awards related to a terminated executive in 2023.
Selling, General and Administrative Expense
Sales and marketing expenses primarily consist of compensation and benefits for our sales force and personnel that support our sales, marketing, medical and scientific affairs operations, expenses related to communicating the health outcome benefits of our products, investments in provider-level market access and patient reimbursement support and educational programs for our customers. General and administrative expenses consist of compensation and benefits for legal, finance, regulatory activities related to approved products and indications, compliance, information technology, human resources, business development, executive management and other supporting personnel. It also includes professional fees for legal, audit, tax and consulting services. Stock-based compensation expense relates to the costs of stock option grants, RSU awards and our ESPP.
The following table provides information regarding selling, general and administrative expense during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| Sales and marketing | $ | 172,015 | | | $ | 153,040 | | | 12% |
| General and administrative | 87,227 | | | 82,737 | | | 5% |
| Stock-based compensation | 34,857 | | | 33,664 | | | 4% |
| Total selling, general and administrative expense | $ | 294,099 | | | $ | 269,441 | | | 9% |
| % of total revenue | 42% | | 40% | | |
Total selling, general and administrative expense increased 9% in 2024 versus 2023.
Sales and marketing expense increased 12% in 2024 versus 2023, driven by investments in programs to drive awareness and education for our customers and enhance our marketing, market access and reimbursement teams and value creation for the implementation of separate Medicare reimbursement for EXPAREL at average sales price plus 6 percent in HOPD settings and iovera° at up to an additional $255.85 when providers administer iovera° in ASC and HOPD settings beginning in January 2025 as part of the NOPAIN Act. Investments in these programs continued through the end of 2024 as we launched our national campaign—Make the NOPAIN Pact—which targets hospital pharmacists, administrators, clinicians and revenue management teams and is focused on ensuring these audiences are ready for the commencement of the NOPAIN Act which took effect on January 1, 2025. We also expanded the size of our sales force in the second half of 2024 in order to better extend our reach on each of our commercial products. These increases were partially offset by the impact of a February 2024 restructuring plan designed to ensure we are well positioned for long-term growth (for more information, see Note 17, Contingent Consideration Gains, Restructuring Charges and Other, to our consolidated financial statements included herein).
General and administrative expense increased 5% in 2024 versus 2023 primarily driven by third-party management consulting to assess strategic opportunities and market assessments for our products and compensatory costs associated with the transition to our new Chief Executive Officer effective January 2, 2024, which included compensation related to the current Chief Executive Officer and to the former Chief Executive Officer who remains an advisor to the Company in a consulting capacity.
Stock-based compensation increased 4% in 2024 versus 2023 primarily due to greater equity awards granted to personnel, including the initial stock option and restricted stock unit grants to our new Chief Executive Officer, Frank D. Lee, in January 2024 and our new Chief Financial Officer, Shawn M. Cross, in November 2024.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 78
Amortization of Acquired Intangible Assets
The following table provides a summary of the amortization of acquired intangible assets during the years indicated, including percent changes (dollar amounts in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, | | % Increase / (Decrease) |
| 2024 | | 2023 | |
| Amortization of acquired intangible assets | $ | 57,288 | | | $ | 57,288 | | | —% |
As part of the Flexion Acquisition and the MyoScience Acquisition, we acquired intangible assets consisting of developed technology intangible assets and customer relationships, with estimated useful lives between 9 and 14 years. For more information, see Note 8, Goodwill and Intangible Assets, to our consolidated financial statements included herein.
Goodwill Impairment
The following table provides a summary of goodwill impairments during the periods indicated, including percent changes (dollar amounts in thousands):
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, | | % Increase / (Decrease) |
| 2024 | | 2023 | |
| Goodwill impairment | $ | 163,243 | | | $ | — | | | N/A |
During the three months ended September 30, 2024, the FDA approved a generic competitor to EXPAREL and a U.S. District Court ruled that one of our patents was not valid (for more information, see Note 19, Commitments and Contingencies, to our consolidated financial statements included herein). Due to these events and a subsequent decrease in our common stock price, it was determined these qualitative factors indicated it was more likely than not that the fair value of goodwill may be less than its carrying value. Accordingly, we performed a quantitative assessment through a discounted cash flow model (or income approach), which resulted in the carrying value of the Company exceeding its fair value by more than the goodwill balance. As a result, the goodwill balance of $163.2 million was recorded as fully impaired during the three months ended September 30, 2024.
Contingent Consideration Gains, Restructuring Charges and Other
The following table provides a summary of the costs (gains) related to contingent consideration, restructuring charges, acquisition-related charges and other activities during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, | | % Increase / (Decrease) |
| 2024 | | 2023 | |
| Contingent consideration | $ | (4,457) | | | $ | (3,424) | | | 30% |
| Acquisition-related charges | 1,462 | | | 1,963 | | | (26)% |
| Restructuring charges | 8,532 | | | 1,109 | | | 100%+ |
| Loss on lease termination | 2,165 | | | — | | | N/A |
| Total contingent consideration gains, restructuring charges and other | $ | 7,702 | | | $ | (352) | | | N/A |
In 2024, we recognized contingent consideration gains of $4.5 million due to adjustments reflecting the probability of achieving the remaining Flexion regulatory milestone by December 31, 2030—the milestone expiration date, partially offset by revisions to our weighted average cost of capital and the latest discount rates. In 2023, we recognized contingent consideration gains of $3.4 million due to a decrease in the fair value of the Flexion contingent consideration. The decrease was primarily due to adjustments in the assumption for the long-term forecasts which reduced the probability of meeting the sales-based contingent consideration milestones by December 31, 2030—the expiration date for achieving the milestones. The impact of this assumption on the fair value was partially offset by a decrease to the assumed discount rate based on a significant improvement in our incremental borrowing rate resulting from the TLA Credit Agreement entered into in March 2023. For more information, see Note 11, Financial Instruments, to our consolidated financial statements included herein.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 79
In 2024 and 2023, we recognized acquisition-related charges of $1.5 million and $2.0 million, respectively, primarily related to vacant and underutilized Flexion leases that were assumed from the Flexion Acquisition.
In 2024, we initiated a restructuring plan designed to ensure that we are well positioned for long-term growth. The restructuring plan included, among other things: (i) reshaping the Company’s executive team; (ii) reallocating efforts and resources from our ex-U.S. and certain early-stage development programs to our commercial portfolio in the U.S. market and (iii) reprioritizing investments to focus on other commercial initiatives. As a result, in 2024, we recognized restructuring charges of $8.5 million related to employee termination benefits, such as the acceleration of share-based compensation, severance, and, to a lesser extent, other employment-related termination costs, as well as contract termination costs. In 2023, we recognized restructuring charges of $1.1 million that included a restructuring plan in an effort to improve our operational efficiencies and recognized one-time employee termination benefits through a reduction of headcount.
For more information, see Note 17, Contingent Consideration Gains, Restructuring Charges and Other, to our consolidated financial statements included herein.
In 2024, we recognized a loss of $2.2 million associated with exiting a lease to a training center located in Houston, Texas. See Note 7, Leases, to our consolidated financial statements included herein.
Other Income (Expense), Net
The following table provides information regarding other income (expense), net during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| Interest income | $ | 19,689 | | | $ | 11,444 | | | 72% |
| Interest expense | (16,569) | | | (20,306) | | | (18)% |
| Gain (loss) on early extinguishment of debt | 7,518 | | | (16,926) | | | N/A |
| Other, net | (373) | | | (186) | | | 100%+ |
| Total other income (expense), net | $ | 10,265 | | | $ | (25,974) | | | N/A |
Total other income, net was $10.3 million in 2024 versus total other expense, net of $26.0 million in 2023.
Interest income increased 72% in 2024 versus 2023 due to higher overall investment balances.
The 18% decrease in interest expense was primarily driven by the lower outstanding principal associated with the TLA Term Loan (as defined below) that was entered into in March 2023 which replaced our then-outstanding TLB Term Loan that had a higher principal balance and interest rate, partially offset by issuing the 2029 Notes (as defined below) in May 2024.
In 2024, we recognized a $7.5 million gain on early extinguishment of debt in conjunction with the repurchase of $200.0 million aggregate principal of our 2025 Notes (as defined below). The partial repurchase of the 2025 Notes was completed with our net proceeds from the issuance of the 2029 Notes (as defined below). In 2023, in conjunction with the entry into the TLA Credit Agreement (as defined below), we incurred a $16.9 million loss on early extinguishment of debt recognized as a result of the retirement of $287.5 million aggregate principal of our TLB Term Loan.
For more information, See Note 10, Debt, to our consolidated financial statements herein.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 80
Income Tax Expense
The following table provides information regarding our income tax expense during the years indicated, including percent changes (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | | % Increase / (Decrease) |
| | 2024 | | 2023 | |
| (Loss) income before income taxes | $ | (63,106) | | | $ | 61,701 | | | N/A |
| Income tax expense | $ | 36,454 | | | $ | 19,746 | | | 85% |
| Effective tax rate | (58)% | | 32% | | |
We recorded income tax expense of $36.5 million and $19.7 million for the years ended December 31, 2024 and 2023, respectively. The effective tax rate of (58)% for the year ended December 31, 2024 differed from the U.S. statutory tax rate of 21% primarily due to non-deductible goodwill impairment charges during the three months ended September 30, 2024 and costs related to non-deductible executive compensation and stock-based compensation, mainly related to expired stock options. The effective tax rate of 32% for the year ended December 31, 2023 differed from the U.S. statutory tax rate of 21% primarily due to non-deductible stock-based and executive compensation and non-U.S. valuation allowances, partially offset by tax credits.
Liquidity and Capital Resources
Since our inception in 2006, we have devoted most of our cash resources to manufacturing, research and development and selling, general and administrative activities related to the development and commercialization of EXPAREL. In addition, we acquired ZILRETTA as part of the Flexion Acquisition in November 2021 and iovera° as part of the MyoScience Acquisition in April 2019. We are primarily dependent on the commercial success of EXPAREL and ZILRETTA. We have financed our operations primarily with the proceeds from the sale of convertible senior notes and other debt, common stock, product sales and collaborative licensing and milestone revenue. As of December 31, 2024, we had an accumulated deficit of $206.4 million, cash and cash equivalents and available-for-sale investments of $484.6 million and working capital of $435.2 million.
We expect that our cash and cash equivalents and available-for-sale investments on hand will be adequate to cover our short-term liquidity needs, and that we would be able to access other sources of financing should the need arise.
Summary of Cash Flows
The following table summarizes our cash flows from operating, investing and financing activities for the years ended December 31, 2024 and 2023 (in thousands): | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| Consolidated Statements of Cash Flows Data: | | 2024 | | 2023 |
| Net cash provided by (used in): | | | | |
| Operating activities | | $ | 189,389 | | | $ | 154,649 | |
| Investing activities | | (83,276) | | | 77,541 | |
| Financing activities | | 17,363 | | | (183,031) | |
| Net increase in cash and cash equivalents | | $ | 123,476 | | | $ | 49,159 | |
Operating Activities
In 2024, net cash provided by operating activities was $189.4 million compared to $154.6 million in 2023. The increase of $34.7 million was primarily attributable to increased revenue with favorable gross margins, lower interest paid and the impact of a $13.0 million payment made in the prior year for a termination fee relating to a licensing agreement, partially offset by an increased investment in inventory levels.
Investing Activities
In 2024, net cash used in investing activities was $83.3 million, which reflected $72.6 million of available-for-sale investment purchases (net of sales) and $10.6 million of capital expenditures for manufacturing product fill lines and our EXPAREL capacity expansion project at our Science Center Campus in San Diego, California, partially offset by an increased investment in inventory levels.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 81
In 2023, net cash provided by investing activities was $77.5 million, which reflected $99.5 million of available-for-sale investment maturities (net of purchases), purchases of fixed assets of $15.2 million for manufacturing product fill lines for our products and equipment for our EXPAREL capacity expansion project at our Science Center Campus in San Diego, California and purchases of debt investments of $6.8 million in external complementary development stage product candidates.
Financing Activities
In 2024, net cash provided by financing activities was $17.4 million, which primarily consisted of $287.5 million in proceeds from the issuance of the 2029 Notes. We used the majority of the proceeds from the 2029 Notes to make a partial repurchase of the 2025 Notes in the amount of $191.0 million, enter into a capped call transaction for $26.7 million, repurchase $25.0 million of treasury stock, and pay debt issuance and financing costs of $9.4 million. Additionally, we made $11.3 million of voluntary prepayments associated with the TLA Term Loan and paid the remaining $8.6 million of 3.375% convertible senior notes due 2024 assumed from the Flexion Acquisition (the “Flexion 2024 Notes”) upon their maturity. See Note 10, Debt, to our condensed consolidated financial statements included herein for further discussion on the Flexion 2024 Notes, 2025 Notes, 2029 Notes, the capped call transaction and the TLA Term Loan. There was also $2.3 million of proceeds from the issuance of common stock through our ESPP.
In 2023, net cash used in financing activities was $183.0 million, which consisted of a $296.9 million repayment of TLB Term Loan principal as well as a $5.8 million prepayment penalty in connection with the retirement of the TLB Term Loan facility and $33.4 million repayments of TLA Term Loan principal, partially offset by the net proceeds from the TLA Term Loan of $149.6 million, $2.8 million from the issuance of common stock through our ESPP and proceeds from the exercise of stock options of $1.9 million.
Equity Financings
From our inception in December 2006 through December 31, 2024, we have raised $344.5 million of net proceeds from the sale of common stock and other equity securities via public offerings.
Stock Repurchase Program
In May 2024, we announced that our board of directors approved a share repurchase program which was effective immediately and authorizes us to repurchase up to an aggregate of $150.0 million of our outstanding common stock. Repurchases under this program may be made at management’s discretion on the open market or through privately negotiated transactions. The share repurchase program may be suspended or discontinued at any time by us and has an expiration date of December 31, 2026.
During the year ended December 31, 2024, concurrently with the pricing of the offering of the 2029 Notes, we entered into separate privately negotiated agreements with certain of the initial purchasers of the 2029 Notes or their respective affiliates and/or certain other financial institutions to repurchase 837,240 shares of our common stock for a total cost of $25.1 million, inclusive of $0.1 million of accrued excise tax. During the year ended December 31, 2023, we did not repurchase any shares of our common stock.
As of December 31, 2024, we had remaining authorization to repurchase approximately $125.0 million of our common stock, subject to restrictions under the TLA Credit Agreement and the Indentures.
Debt
2028 Term Loan A Facility
On March 31, 2023, we entered into a credit agreement (as amended, the “TLA Credit Agreement”) to refinance the indebtedness outstanding under our TLB Credit Agreement. The term loan issued under the TLA Credit Agreement (the “TLA Term Loan”) was issued at a 0.30% discount and provides for a single-advance term loan A facility in the principal amount of $150.0 million, which is secured by substantially all of our and any subsidiary guarantor’s assets and matures on March 31, 2028. We may elect to borrow either (i) alternate base rate borrowings or (ii) term benchmark borrowings or daily simple SOFR (as defined in the TLA Credit Agreement) borrowings. Each term loan borrowing which is an alternate base rate borrowing bears interest at a rate per annum equal to (i) the Alternate Base Rate (as defined in the TLA Credit Agreement), plus (ii) a spread based on our Senior Secured Net Leverage Ratio ranging from 2.00% to 2.75%. Each term loan borrowing which is a term benchmark borrowing or daily simple SOFR borrowing bears interest at a rate per annum equal to (i) the Adjusted Term SOFR Rate or Adjusted Daily Simple SOFR (as each is defined in the TLA Credit Agreement), plus (ii) a spread based on our Senior Secured Net Leverage Ratio ranging from 3.00% to 3.75%. During the year ended December 31, 2024, we made $11.3 million of voluntary principal prepayments. During the year ended December 31, 2023, we made a scheduled principal payment of $2.8 million as well as $30.6 million of voluntary principal prepayments. As of December 31, 2024, borrowings
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 82
under the TLA Term Loan consisted entirely of term benchmark borrowings at a rate of 7.43%. At December 31, 2024, the outstanding principal on the TLA Term Loan was $105.3 million.
The TLA Credit Agreement requires us to, among other things, maintain (i) a Senior Secured Net Leverage Ratio (as defined in the TLA Credit Agreement), determined as of the last day of each fiscal quarter, of no greater than 3.00 to 1.00 and (ii) a Fixed Charge Coverage Ratio (as defined in the TLA Credit Agreement), determined as of the last day of each fiscal quarter, of no less than 1.50 to 1.00. The TLA Credit Agreement requires us to maintain an unrestricted cash and cash equivalents balance of at least $500.0 million less any prepayments of the 2025 Notes at any time from 91 days prior to the maturity date through the earlier of (i) the latest maturity date of the 2025 Notes and (ii) the date on which there is no outstanding principal amount of the 2025 Notes. The TLA Credit Agreement also contains customary affirmative and negative covenants, financial covenants, representations and warranties, events of default and other provisions. As of December 31, 2024, we were in compliance with all financial covenants under the TLA Credit Agreement. See Note 10, Debt, to our consolidated financial statements included herein for further discussion.
2029 Convertible Senior Notes
In May 2024, we completed a private placement of $287.5 million in aggregate principal amount of our 2.125% convertible senior notes due 2029, or 2029 Notes, and entered into an indenture with respect to the 2029 Notes. The 2029 Notes accrue interest at a fixed rate of 2.125% per annum, payable semiannually in arrears on May 15th and November 15th of each year, and mature on May 15, 2029.
In May 2024, we used part of the net proceeds from the issuance of the 2029 Notes to repurchase $200.0 million aggregate principal amount of the 2025 Notes in privately negotiated transactions at a discount for $191.4 million in cash (including accrued interest). The partial repurchase of the 2025 Notes resulted in a $7.5 million gain on early extinguishment of debt.
At December 31, 2024, all $287.5 million of principal was outstanding on the 2029 Notes. See Note 10, Debt, to our consolidated financial statements included herein for further discussion.
2025 Convertible Senior Notes
In July 2020, we completed a private placement of $402.5 million in aggregate principal amount of our 2025 Notes, and entered into an indenture with respect to the 2025 Notes. The 2025 Notes accrue interest at a fixed rate of 0.750% per annum, payable semiannually in arrears on February 1st and August 1st of each year, and mature on August 1, 2025. At December 31, 2024, the outstanding principal on the 2025 Notes was $202.5 million. See Note 10, Debt, to our consolidated financial statements included herein for further discussion.
Future Capital Requirements
We believe that our existing cash and cash equivalents, available-for-sale investments and cash received from product sales will be sufficient to enable us to fund our operating expenses, capital expenditure requirements and payment of the interest and principal on our TLA Term Loan, 2025 Notes and 2029 Notes through the next 12 months. Our future use of operating cash and capital requirements will depend on many forward-looking factors, including, but not limited to:
•the cost and timing of the potential milestone payments to former Flexion stockholders, which could be up to an aggregate of $372.3 million if certain regulatory and commercial milestones are met. See Note 11, Financial Instruments, to our consolidated financial statements included herein for more information;
•the impact of global economic conditions—including the impact of inflation and tariffs—on our product, material and labor costs, supply chain, longer lead-times, an inability to secure a sufficient supply of materials, our operating expenses and our business strategy;
•the timing of and extent to which the holders of our 2025 Notes and 2029 Notes elect to convert their 2025 Notes and 2029 Notes, the timing of principal and interest payments on our TLA Term Loan and the timing and impact of increases to the variable interest rate on our TLA Term Loan borrowings in accordance with the terms of the TLA Credit Agreement;
•the costs and our ability to successfully continue to expand the commercialization of EXPAREL, ZILRETTA and iovera°;
•the cost and timing of expanding and maintaining our manufacturing facilities;
•the cost and timing of additional strategic investments, including additional investments under existing agreements;
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 83
•the costs related to legal and regulatory matters, including those to develop and defend our intellectual property;
•the costs of performing additional clinical trials for our products, including the additional pediatric trials required by the FDA and EMA as a condition of the approval of EXPAREL;
•the costs for the development and commercialization of other product candidates;
•the costs and timing of future payments under our employee benefit plans, including but not limited to our cash long-term incentive plan and non-qualified deferred compensation plan;
•the timing and the number of shares of our common stock repurchased through our share repurchase program; and
•the extent to which we acquire or invest in products, businesses and technologies.
We may require additional debt or equity financing to meet our future operating and capital requirements. We have no committed external sources of funds, and additional equity or debt financing may not be available on acceptable terms, if at all. In particular, capital market disruptions or negative economic conditions may hinder our access to capital.
Contractual Obligations
We had two convertible senior notes outstanding as of December 31, 2024, for which $202.5 million in aggregate principal amount is due on our 2025 Notes in August 2025 and $287.5 million in aggregate principal amount is due on our 2029 Notes in May 2029. The remaining interest payments on our 2025 Notes is $1.5 million, all of which is due in 2025. The remaining interest payments on our 2029 Notes is $27.3 million, of which $6.1 million is due in 2025. We also have the TLA Term Loan with $105.3 million in outstanding principal. Due to voluntary principal prepayments of $30.6 million made during the year ended December 31, 2023 and $11.3 million made during December 31, 2024, we are not contractually obligated to make principal payments in 2025, although we retain the option to do so. As of December 31, 2024, there are contractually obligated principal payments of $4.1 million in 2026, $15.0 million in 2027 and $86.3 million in 2028. The remaining interest payments of the TLA Term Loan are approximately $25.0 million based on the current interest rate, of which $7.9 million is due in 2025.
In the normal course of business, we enter into various lease agreements for manufacturing, research and development and corporate activities, which are typically classified as operating leases under the provisions of Financial Accounting Standards Board Accounting Standards Codification Topic 842, Leases. As of December 31, 2024, we had net minimum commitments of $64.0 million, of which $12.3 million are due in 2025. For more information, refer to Note 7, Leases, to our consolidated financial statements included herein.
In addition, we have approximately $59.9 million of minimum, non-cancelable contractual commitments for contract manufacturing services as of December 31, 2024, of which $20.3 million is due in each of 2025 and 2026, $17.9 million is due in 2027 and the remaining $1.4 million is due in 2028. We have approximately $6.5 million of minimum, non-cancelable contractual commitments for the purchase of certain raw materials as of December 31, 2024, all of which are due in 2025. We had $1.8 million of other minimum, non-cancelable contractual commitments as of December 31, 2024, of which $1.4 million is due in 2025, and the remaining $0.4 million is due thereafter.
As part of the Flexion Acquisition, there are up to $372.3 million in potential payments if all regulatory and commercial milestones are met. For more information, see Note 11, Financial Instruments, to our consolidated financial statements included herein.
In February 2025, we completed the GQ Bio Acquisition. The net purchase price includes approximately $8 million to be paid over three years pursuant to a key employee holdback agreement as well as a post-closing indemnity holdback of approximately $6 million. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events, to our consolidated financial statements included herein.
Critical Accounting Estimates
We have based our Management’s Discussion and Analysis of our Financial Condition and Results of Operations on our financial statements that have been prepared in accordance with GAAP in the U.S. The preparation of these financial statements requires us to make estimates that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, contingent consideration, impairment of intangible assets and goodwill, inventory costs, liabilities and accruals, clinical trial expenses, stock-based compensation and the valuation of deferred tax assets. We base our estimates on historical experience, contract
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 84
terms and on other factors we believe to be appropriate under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are more fully discussed in Note 2, Summary of Significant Accounting Policies, to our consolidated financial statements included herein. The following accounting policies, which may include significant judgments and estimates, were used in the preparation of our consolidated financial statements.
Revenue Recognition
Revenues from sales of products are recorded net of returns allowances, prompt payment discounts, service fees, government rebates, volume rebates and chargebacks. These reserves are based on estimates of the amounts earned or to be claimed on the related sales. These amounts are treated as variable consideration, estimated and recognized as a reduction of the transaction price at the time of the sale, using the most likely amount method, except for returns, which is based on the expected value method. We include these estimated amounts in the transaction price to the extent it is probable that a significant reversal of cumulative revenue recognized for such transaction will not occur, or when the uncertainty associated with the variable consideration is resolved. The calculation of some of these items requires management to make estimates based on sales data, historical return data, contracts, statutory requirements and other related information that may become known in the future. The adequacy of these provisions is reviewed on a quarterly basis. If our assessments, experiences or judgments are not accurate estimates of future results, our results of operations could be affected. The sensitivity of our estimates varies by program. Estimates associated with chargebacks and government programs have the greatest risk of being subject to adjustment because of the time delay between recording the accrual and the final settlement. Historically, adjustments to these estimates to reflect actual results or updated expectations have not been material.
The summary of activity with respect to our sales related allowances and accruals for the years ended December 31, 2024, 2023 and 2022 appears in Note 4, Revenue, to our consolidated financial statements included herein.
Contingent Consideration
Subsequent to an acquisition, we measure contingent consideration arrangements at fair value for each period with changes in fair value recognized in the consolidated statements of operations as contingent consideration gains, restructuring charges and other. Changes in contingent consideration can result from changes in the assumed achievement and timing of estimated sales and regulatory approvals. In the absence of new information, changes in fair value reflect the impact of the passage of time towards the potential achievement of the milestones.
The following table includes the key assumptions used in the valuation of our contingent consideration milestones:
| | | | | | | | |
| Assumption | | Ranges
Utilized as of
December 31, 2024 |
| Discount rates | | 7.8% to 8.1% |
| Probability of payment for remaining regulatory milestones | | 0% |
| | |
The maximum remaining potential payments related to contingent consideration from the Flexion Acquisition is $372.3 million as of December 31, 2024. Changes to assumptions may result in a material impact to the calculated amounts. Additionally, the forecasted revenue annual growth rates are key assumptions in the contingent consideration valuations associated with our commercial milestones. The impact of a hypothetical 10 percent increase in the forecasted annual growth rates would have increased the value of our contingent consideration liability associated with the Flexion Acquisition as of December 31, 2024 by $6.3 million. The impact of a hypothetical 100 basis point increase in the discount rate would have reduced the value of our contingent consideration liability associated with the Flexion Acquisition as of December 31, 2024 by $0.9 million.
Goodwill
Goodwill represents the excess of the purchase price over the estimated fair value of the net assets acquired in a business combination and is subject to impairment testing at least annually or upon the occurrence of a triggering event that could indicate a potential impairment. We have historically tested goodwill for impairment by performing a qualitative assessment in order to determine whether facts and circumstances support a determination that reporting unit fair values are greater than their carrying values. This has historically been performed using readily available market data and company-specific factors.
If we determine that it is more likely than not that the fair value of the Company is less than its carrying value, a quantitative test is required. This is performed by comparing the fair value of the Company with its carrying value. If the
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 85
estimated fair value of the reporting unit is less than the carrying amount of the reporting unit, impairment is indicated, requiring recognition of a goodwill impairment charge up to the carrying value of goodwill. The fair value of the Company would be calculated through an income approach. Under the income approach, we calculate the fair value based on the present value of estimated future cash flows. Considerable management judgment is necessary to evaluate the impact of operating and macroeconomic changes and to estimate the future cash flows used to assume fair value. Our estimates of future cash flows consider past performance, current and anticipated market conditions and internal projections and operating plans which incorporate estimates for sales growth and future margins. Additional assumptions would include estimated discount rates and the probability of success for our product pipeline candidate products. We believe such assumptions would reflect current and anticipated market conditions and are consistent with those that would be used by other marketplace participants for similar valuation purposes. Such assumptions are subject to change due to changing economic and competitive conditions.
In July 2024, the FDA approved a generic competitor to EXPAREL and in August 2024, a U.S. District Court ruled that one of our EXPAREL patents was not valid. We determined that these events, combined with a subsequent decrease in our common stock price, indicated that it was more likely than not that the fair value of goodwill may be less than its carrying value, which required us to perform a quantitative impairment test. This quantitative impairment test resulted in our carrying value exceeding the fair value of the Company by more than the goodwill balance. As a result, the goodwill balance of $163.2 million was fully impaired during the three months ended September 30, 2024. For more information, see Note 8, Goodwill and Intangible Assets, to our consolidated financial statements included herein.
Recent Accounting Pronouncements
See Note 3, Recent Accounting Pronouncements, to our consolidated financial statements included herein for further discussion of recent accounting pronouncements.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
The primary objective of our cash equivalents and investment activities is to preserve principal while at the same time maximizing the income that we receive from our investments without significantly increasing risk. We invest in corporate bonds, commercial paper, asset-backed securities and U.S. Treasury and other government agency notes for purposes other than trading which are reported at fair value. These securities are subject to interest rate risk and credit risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with a fixed interest rate at the then-prevailing rate and the interest rate later rises, we expect that the fair value of our investment will decline. A hypothetical 100 basis point increase in interest rates would have reduced the fair value of our available-for-sale securities at December 31, 2024 by approximately $0.8 million.
The fair value of our 2025 Notes is impacted by both the fair value of our common stock and interest rate fluctuations. As of December 31, 2024, the estimated fair value of the 2025 Notes was $971 per $1,000 principal amount. See Note 10, Debt, to our consolidated financial statements included herein for further discussion of our 2025 Notes, which bears interest at a fixed rate. At December 31, 2024, $202.5 million of principal remains outstanding on the 2025 Notes.
The fair value of our 2029 Notes is impacted by both the fair value of our common stock and interest rate fluctuations. As of December 31, 2024, the estimated fair value of the 2029 Notes was $861 per $1,000 principal amount. See Note 10, Debt, to our consolidated financial statements included herein for further discussion of our 2029 Notes, which bears interest at a fixed rate. At December 31, 2024, all $287.5 million of principal remains outstanding on the 2029 Notes.
The TLA Term Loan provides for a single-advance term loan in the principal amount of $150.0 million and is scheduled to mature on March 31, 2028. Each term loan borrowing that is a term benchmark borrowing or daily simple SOFR borrowing bears interest at a rate per annum equal to (i) the Adjusted Term SOFR Rate or Adjusted Daily Simple SOFR (as each is defined in the TLA Credit Agreement), plus (ii) a spread based on our Senior Secured Net Leverage Ratio ranging from 3.00% to 3.75%. At December 31, 2024, the outstanding principal on the TLA Term Loan was $105.3 million. As of December 31, 2024, borrowings under the TLA Term Loan consisted entirely of term benchmark borrowings at a rate of 7.43%. A hypothetical 100 basis point increase in interest rates would increase interest expense over the next 12 months by approximately $1.1 million, based on the balances outstanding for these borrowings as of December 31, 2024.
We have agreements with certain vendors and partners that operate in foreign jurisdictions. The more significant transactions are primarily denominated in the U.S. Dollar, subject to an annual adjustment based on changes in currency exchange rates.
Additionally, our accounts receivable are primarily concentrated with three large wholesalers of pharmaceutical products. In the event of non-performance or non-payment, there may be a material adverse impact on our financial condition, results of operations or net cash flow.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 86
Item 8. Financial Statements and Supplementary Data
Our consolidated financial statements required by this item, together with the report of our independent registered public accounting firm, begin on page F-1 of this Annual Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures, as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, which are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Based on their evaluation as of December 31, 2024, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2024.
Management’s Report on Internal Control over Financial Reporting
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with GAAP. Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2024, based on the criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based upon the results of the evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2024.
The effectiveness of our internal control over financial reporting as of December 31, 2024 was audited by KPMG LLP, our independent registered public accounting firm, which expressed an unqualified opinion on the effectiveness of our internal control over financial reporting as of December 31, 2024.
Changes in Internal Control over Financial Reporting
During the quarter ended December 31, 2024, there have been no changes in our internal control over financial reporting that occurred that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
During the quarter ended December 31, 2024, no director or executive officer of the Company adopted or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in Item 408(a) of Regulation S-K.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
None.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 87
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Information required by this item will be included in the proxy statement for our 2025 annual stockholders’ meeting and is incorporated by reference into this report.
Item 11. Executive Compensation
Information required by this item will be included in the proxy statement for our 2025 annual stockholders’ meeting and is incorporated by reference into this report.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholders Matters
Information required by this item will be included in the proxy statement for our 2025 annual stockholders’ meeting and is incorporated by reference into this report.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information required by this item will be included in the proxy statement for our 2025 annual stockholders’ meeting and is incorporated by reference into this report.
Item 14. Principal Accountant Fees and Services
Information required by this item will be included in the proxy statement for our 2025 annual stockholders’ meeting and is incorporated by reference into this report.
PART IV
Item 15. Exhibits, Financial Statement Schedules
(a)Documents filed as part of this Annual Report on Form 10-K:
(1) Financial Statements
| | | | | | | | |
| Index to the Consolidated Financial Statements | | Page # |
| | |
| | |
| | |
| | |
| | |
| | |
The report of our independent registered accounting firm, KPMG LLP, with respect to the above-referenced financial statements and on internal control over financial reporting, is included in this Annual Report on Form 10-K. Their consent appears as Exhibit 23.1 of this Annual Report on Form 10-K.
(2) Schedules
All financial statement schedules have been omitted because they are not required, are not applicable or the information is included in the consolidated financial statements or related notes thereto.
(3) Exhibits
The following exhibits are filed with, or incorporated by reference in this Annual Report on Form 10-K.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 88
EXHIBIT INDEX | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Incorporation By Reference From |
Exhibit
Number | | Description | | Form | | Exhibit | | Date
Filed |
| | Agreement and Plan of Merger, dated March 4, 2019, by and among Pacira Pharmaceuticals, Inc., PS Merger, Inc., MyoScience, Inc., and Fortis Advisors LLC, as the securityholders’ representative. # † | | 8-K | | 2.1 | | 3/5/2019 |
| | Agreement and Plan of Merger, dated as of October 11, 2021, by and among Flexion Therapeutics, Inc., Pacira BioSciences, Inc. and Oyster Acquisition Company Inc. | | 8-K | | 2.1 | | 10/12/2021 |
| | Amended and Restated Certificate of Incorporation. | | 8-K | | 3.1 | | 2/11/2011 |
| | Certificate of Amendment to the Amended and Restated Certificate of Incorporation, dated April 9, 2019. | | 8-K | | 3.1 | | 4/9/2019 |
| | Second Amended and Restated Bylaws. | | 8-K | | 3.2 | | 4/9/2019 |
| | Specimen Certificate Evidencing Shares of Common Stock.* | | | | | | |
| | Indenture (including form of 0.750% Convertible Senior Notes due 2025), dated July 10, 2020, between the Registrant and Computershare Corporate Trust, National Association, as trustee (formerly Wells Fargo Bank, National Association). | | 8-K | | 4.1 | | 7/10/2020 |
| | Indenture (including form of 2.125% Convertible Senior Notes due 2029), dated May 14, 2024, between the Registrant and Computershare Corporate Trust, National Association, as trustee. | | 8-K | | 4.1 | | 5/14/2024 |
| | Description of Securities. | | 10-K | | 4.3 | | 2/21/2020 |
| | Amended and Restated 2011 Stock Incentive Plan.*** | | 8-K | | 10.1 | | 6/20/2023 |
| | Form of Nonstatutory Stock Option Agreement under the Amended and Restated 2011 Stock Incentive Plan for grants made prior to February 1, 2022.*** | | 8-K | | 10.3 | | 6/4/2014 |
| | Form of Nonstatutory Stock Option Agreement (Employees) under the Amended and Restated 2011 Stock Incentive Plan for grants made on or after February 1, 2022.*** | | 10-K | | 10.3 | | 2/28/2022 |
| | Form of Nonstatutory Stock Option Agreement (Non-Employee Directors) under the Amended and Restated 2011 Stock Incentive Plan for grants made on or after February 1, 2022.*** | | 10-K | | 10.4 | | 2/28/2022 |
| | Form of Restricted Stock Unit Award Agreement (Employees) under the Amended and Restated 2011 Stock Incentive Plan for grants made prior to February 1, 2022.*** | | 10-K | | 10.5 | | 2/29/2024 |
| | Form of Restricted Stock Unit Award Agreement (Non-Employee Directors) under the Amended and Restated 2011 Stock Incentive Plan for grants made prior to February 1, 2022.*** | | 10-K | | 10.6 | | 2/29/2024 |
| | Amended and Restated 2014 Inducement Plan.*** | | 8-K | | 10.1 | | 1/21/2025 |
| | Form of Nonstatutory Stock Option Agreement under the Amended and Restated 2014 Inducement Plan.*** | | 8-K | | 10.2 | | 1/21/2025 |
| | Form of Restricted Stock Unit Award Agreement under the Amended and Restated 2014 Inducement Plan.*** | | 8-K | | 10.3 | | 1/21/2025 |
| | Amended and Restated 2014 Employee Stock Purchase Plan.*** | | 8-K | | 10.1 | | 6/10/2022 |
| | Assignment Agreement, dated February 9, 1994, amended April 15, 2004, between the Registrant and Research Development Foundation. | | S-1/A | | 10.4 | | 12/3/2010 |
| | Stock Purchase Agreement, dated January 8, 2007, between SkyePharma, Inc. and the Registrant. | | S-1/A | | 10.5 | | 12/3/2010 |
| | Employment Agreement by and between Pacira Pharmaceuticals, Inc. and David Stack.*** | | S-1/A | | 10.21 | | 12/3/2010 |
| | Amendment No. 1 to Executive Employment Agreement, dated March 13, 2013, by and between Pacira Pharmaceuticals, Inc. and David Stack.*** | | 8-K | | 99.3 | | 3/18/2013 |
| | Amendment No. 2 to Executive Employment Agreement, dated June 30, 2015, by and between Pacira Pharmaceuticals, Inc. and David Stack.*** | | 10-Q | | 10.2 | | 7/30/2015 |
| | Transition and Retirement Agreement, dated September 20, 2023, by and between Pacira Pharmaceuticals, Inc. and David Stack.*** | | 8-K | | 10.1 | | 9/26/2023 |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 89
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Incorporation By Reference From |
Exhibit
Number | | Description | | Form | | Exhibit | | Date
Filed |
| | Employment Agreement, dated December 20, 2023, by and between Pacira Pharmaceuticals, Inc. and Frank D. Lee.*** | | 8-K | | 10.1 | | 12/21/2023 |
| | Employment Agreement, dated October 21, 2024, by and between Pacira Pharmaceuticals, Inc. and Shawn M. Cross.* *** | | | | | | |
| | Employment Agreement, dated November 29, 2012, by and between Pacira Pharmaceuticals, Inc. and Kristen Williams.*** | | 10-Q | | 10.2 | | 4/30/2015 |
| | Amendment No. 1 to Employment Agreement, dated March 13, 2013, by and between Pacira Pharmaceuticals, Inc. and Kristen Williams.*** | | 10-Q | | 10.3 | | 4/30/2015 |
| | Amendment No. 2 to Employment Agreement, dated June 30, 2015, by and between Pacira Pharmaceuticals, Inc. and Kristen Williams.*** | | 10-Q | | 10.5 | | 7/30/2015 |
| | Amendment No. 3 to Employment Agreement, dated as of October 31, 2024, by and between Pacira Pharmaceuticals, Inc. and Kristen Williams.*** | | 8-K | | 10.3 | | 11/6/2024 |
| | Executive Employment Agreement, dated May 2, 2016, by and between Pacira Pharmaceuticals, Inc. and Charles A. Reinhart, III.*** | | 10-Q | | 10.1 | | 8/4/2016 |
| | Non-Healthcare Professional Consulting Agreement, effective October 1, 2024, by and between Pacira Pharmaceuticals, Inc. and Charles. A. Reinhart, III* *** | | 10-Q | | 10.5 | | 11/6/2024 |
| | Amended and Restated Executive Employment Agreement, dated as of November 4, 2024, by and between Pacira Pharmaceuticals, Inc. and Lauren Riker.*** | | 8-K | | 10.4 | | 11/6/2024 |
| | Form of Indemnification Agreement between the Registrant and its directors and officers.*** | | 10-K | | 10.22 | | 2/29/2024 |
| | Commercial Outsourcing Services Agreement entered into as of February 26, 2024 by the Registrant and Integrated Commercialization Solutions, Inc.†† | | 10-K | | 10.23 | | 2/29/2024 |
| | Pacira BioSciences, Inc. Deferred Compensation Plan.*** | | 8-K | | 10.1 | | 6/11/2020 |
| | Amendment No. 1 to Pacira BioSciences, Inc. Deferred Compensation Plan.*** | | 10-K | | 10.25 | | 2/28/2023 |
| | Amendment No. 2 to Pacira BioSciences, Inc. Deferred Compensation Plan.* *** | | | | | | |
| | Pacira BioSciences, Inc. Long-Term Incentive Plan.* *** | | | | | | |
| | Strategic Co-Production Agreement dated April 4, 2014,by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.† | | 10-Q | | 10.1 | | 7/31/2014 |
| | Manufacturing and Supply Agreement dated April 4, 2014,by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.† | | 10-Q | | 10.2 | | 7/31/2014 |
| | First Amendment to Manufacturing and Supply Agreement dated April 2, 2019 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Second Amendment to Manufacturing and Supply Agreement dated October 31, 2024 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Technical Transfer and Service Agreement dated April 4, 2014, by and between by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.† | | 10-Q | | 10.3 | | 7/31/2014 |
| | First Amendment to Technical Transfer and Service Agreement dated November 15, 2016 by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.* ## | | | | | | |
| | Second Amendment to Technical Transfer and Service Agreement dated May 1, 2018 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Third Amendment to Technical Transfer and Service Agreement dated April 2, 2019 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Fourth Amendment to Technical Transfer and Service Agreement dated August 30, 2019 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Fifth Amendment to Technical Transfer and Service Agreement dated December 11, 2019 by and between Pacira Limited and Patheon UK Limited.* †† ## | | | | | | |
| | Side Letter dated June 5, 2023, to the Manufacturing and Supply Agreement by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.†† | | 10-Q | | 10.2 | | 8/2/2023 |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 90
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Incorporation By Reference From |
Exhibit
Number | | Description | | Form | | Exhibit | | Date
Filed |
| | Manufacturing and Supply Agreement dated July 31, 2015, between Flexion Therapeutics, Inc. and Patheon UK Limited, as amended to date.†† | | 10-K | | 10.36 | | 2/28/2022 |
| | First Amendment to Manufacturing and Supply Agreement dated May 8, 2019, between Flexion Therapeutics, Inc. and Patheon UK Limited.†† | | 10-K | | 10.32 | | 2/29/2024 |
| | Second Amendment to Manufacturing and Supply Agreement dated June 17, 2019, between Flexion Therapeutics, Inc. and Patheon UK Limited.†† | | 10-K | | 10.33 | | 2/29/2024 |
| | Third Amendment to Manufacturing and Supply Agreement dated December 1, 2023, by and between by and between Pacira Pharmaceuticals, Inc. and Patheon UK Limited.†† | | 10-K | | 10.34 | | 2/29/2024 |
| | Technical Transfer and Service Agreement dated July 31, 2015, between Flexion Therapeutics, Inc. and Patheon UK Limited, as amended to date.†† | | 10-K | | 10.37 | | 2/28/2022 |
| | Side Letter to the Manufacturing and Supply Agreement between Flexion Therapeutics, Inc. and Patheon UK Limited, dated as of April 8, 2020.†† | | 10-K | | 10.38 | | 2/28/2022 |
| | Amended and Restated Consulting Agreement, dated April 3, 2012, by and between Pacira Pharmaceuticals, Inc. and Gary Pace.*** | | 10-Q | | 10.1 | | 5/9/2012 |
| | Second Amended and Restated Consulting Agreement, dated August 17, 2012, by and between Pacira Pharmaceuticals, Inc. and Gary Pace.*** | | 10-Q | | 10.1 | | 11/1/2012 |
| | Third Amendment to Consulting Agreement, dated September 11, 2013, by and between Pacira Pharmaceuticals, Inc. and Gary Pace.*** | | 10-Q | | 10.3 | | 10/31/2013 |
| | Fourth Amendment to Consulting Agreement, dated November 25, 2015, by and between Pacira Pharmaceuticals, Inc. and Gary Pace.*** | | 10-K | | 10.57 | | 2/25/2016 |
| | Fifth Amendment to Consulting Agreement, dated June 12, 2024, by and between Pacira Pharmaceuticals, Inc. and Gary Pace.*** †† | | 10-Q | | 10.3 | | 7/30/2024 |
| | Contingent Value Right Agreement, dated as of November 19, 2021, by and between the Registrant and American Stock Transfer & Trust Company, LLC. | | 8-K | | 10.1 | | 11/19/2021 |
| | Credit Agreement, dated as of March 31, 2023, by and among the Registrant, the lenders from time to time party thereto and JPMorgan Chase Bank, N.A., as administrative agent.## | | 8-K | | 10.1 | | 4/3/2023 |
| | First Amendment, dated as of May 8, 2024, to Credit Agreement, dated as of March 31, 2023, by and among the Registrant, the lenders from time to time party thereto and JPMorgan Chase Bank, N.A., as administrative agent.## | | 8-K | | 10.1 | | 5/8/2024 |
| | Executive Employment Agreement, dated May 4, 2020, by and between Pacira Pharmaceuticals, Inc. and Jonathan Slonin.*** | | 10-Q | | 10.1 | | 5/4/2022 |
| | Amendment No. 1 to Executive Employment, dated as of October 31, 2024, by and between Pacira Pharmaceuticals, Inc. and Jonathan Slonin.*** | | 8-K | | 10.2 | | 11/6/2024 |
| | Executive Employment Agreement, dated June 17, 2019, by and between Pacira Pharmaceuticals, Inc. and Daryl Gaugler.*** | | 10-Q | | 10.1 | | 5/3/2023 |
| | Amendment No. 1 to Executive Employment Agreement, dated as of October 31, 2024, by and between Pacira Pharmaceuticals, Inc. and Daryl Gaugler.*** | | 8-K | | 10.1 | | 11/6/2024 |
| | Form of Capped Call Transaction Confirmation. | | 8-K | | 10.1 | | 5/14/2024 |
| | Pacira BioSciences, Inc. Insider Trading Policy.* | | | | | | |
| | Subsidiaries of the Registrant.* | | | | | | |
| | Consent of KPMG LLP.* | | | | | | |
| | Certification of Chief Executive Officer pursuant to Exchange Act Rule 13a-14(a).* | | | | | | |
| | Certification of Chief Financial Officer pursuant to Exchange Act Rule 13a-14(a).* | | | | | | |
| | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.** | | | | | | |
| | Incentive Compensation Recovery Policy.*** | | 10-K | | 97 | | 2/29/2024 |
| | | | | | | | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 91
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | Incorporation By Reference From |
Exhibit
Number | | Description | | Form | | Exhibit | | Date
Filed |
| 101.INS* | | Inline XBRL Instance Document.* | | | | | | |
| 101.SCH* | | Inline XBRL Taxonomy Schema Document.* | | | | | | |
| 101.CAL* | | Inline XBRL Taxonomy Calculation Linkbase Document.* | | | | | | |
| 101.LAB* | | Inline XBRL Taxonomy Label Linkbase Document.* | | | | | | |
| 101.PRE* | | Inline XBRL Taxonomy Presentation Linkbase Document.* | | | | | | |
| 101.DEF* | | Inline XBRL Taxonomy Extension Definition Linkbase Document.* | | | | | | |
| 104* | | Cover Page Interactive Data File (Formatted as Inline XBRL and contained in Exhibit 101). | | | | | | |
| | | | | | | | |
| * | | Filed herewith. |
| ** | | Furnished herewith. |
| *** | | Denotes management contract or compensatory plan or arrangement. |
| † | | Confidential treatment has been requested or granted as to certain portions, which portions were omitted and filed separately with the Securities and Exchange Commission pursuant to a Confidential Treatment Request. |
| †† | | Certain portions of the exhibit have been omitted pursuant to Rule 601(b)(10) of Regulation S-K. The omitted information (i) is not material and (ii) is the type that the Registrant treats as private or confidential. |
| # | | Certain schedules have been omitted pursuant to Item 601(b)(2) of Regulation S-K under the Securities Exchange Act of 1934, as amended. The Company hereby undertakes to supplementally furnish copies of any omitted schedules to the Securities and Exchange Commission upon request. |
| ## | | Schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Company hereby undertakes to supplementally furnish copies of any omitted schedules and exhibits to the Securities and Exchange Commission upon request. |
Item 16. Form 10-K Summary
None.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 92
| | |
| SIGNATURES |
|
| Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized. |
| | | | | | | | | | | | | | |
| | | | PACIRA BIOSCIENCES, INC.
/s/ FRANK D. LEE |
| Date: | February 27, 2025 | By: | | Frank D. Lee Chief Executive Officer and Director |
| | |
| Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated and on February 27, 2025. |
| | | | | | | | | | | | | | | | | |
| Principal Executive Officer | | | Directors |
| | | | | |
| /s/ FRANK D. LEE | | | /s/ MARCELO BIGAL | | /s/ ABRAHAM CEESAY |
| Frank D. Lee | | | Marcelo Bigal | | Abraham Ceesay |
| Chief Executive Officer and Director | | | | | |
| | | /s/ CHRISTOPHER J. CHRISTIE | | /s/ MARK FROIMSON |
| Principal Financial Officer | | | Christopher J. Christie | | Mark Froimson |
| | | | | |
| /s/ SHAWN M. CROSS | | | /s/ MARK KRONENFELD | | /s/ MICHAEL YANG |
| Shawn M. Cross | | | Mark Kronenfeld | | Michael Yang |
| Chief Financial Officer | | | | | |
| | | /s/ ALETHIA YOUNG | | /s/ LAURA BREGE |
| Principal Accounting Officer | | | Alethia Young | | Laura Brege |
| | | | | Chair of the Board of Directors |
| /s/ LAUREN RIKER | | | | | |
| Lauren Riker | | | | | |
| Senior Vice President, Finance | | | | | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page 93
PACIRA BIOSCIENCES, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2024
INDEX TO THE CONSOLIDATED FINANCIAL STATEMENTS | | | | | | | | | | | | | | |
| | | | Page # |
| |
| Auditor Name: | KPMG LLP | | |
| Auditor Location: | Short Hills, NJ | | |
| Auditor Firm ID: | 185 | | |
| |
| |
| |
| |
| |
| |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors
Pacira BioSciences, Inc.:
Opinions on the Consolidated Financial Statements and Internal Control Over Financial Reporting
We have audited the accompanying consolidated balance sheets of Pacira BioSciences, Inc. and subsidiaries (the Company) as of December 31, 2024 and 2023, the related consolidated statements of operations, comprehensive (loss) income, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2024, and the related notes (collectively, the consolidated financial statements). We also have audited the Company’s internal control over financial reporting as of December 31, 2024, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2024, in conformity with U.S. generally accepted accounting principles. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2024 based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
Basis for Opinions
The Company’s management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s consolidated financial statements and an opinion on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-2
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Fair value measurement of the contingent consideration liability associated with the acquisition of Flexion
As discussed in Notes 2 and 11 to the consolidated financial statements, the Company recognized a contingent consideration liability at its estimated fair value on the acquisition date, in connection with the acquisition of Flexion Therapeutics, Inc. (Flexion). Subsequent changes to the fair value of the contingent consideration liability are recorded in the consolidated statement of operations in the period of change. The Company estimates the fair value using a Monte Carlo simulation. The fair value of the Flexion contingent consideration as of December 31, 2024 was $20.2 million.
We identified the evaluation of the fair value measurement of the contingent consideration liability related to achieving commercial milestones associated with the acquisition of Flexion as a critical audit matter. Evaluating the fair value measurement of the contingent consideration liability required significant auditor judgment, due to the high degree of subjectivity inherent in certain assumptions with unobservable inputs that were used in the model. In particular, the fair value measurement was sensitive to management’s forecasts of revenues, volatility, and discount rates. In addition, the audit effort associated with the evaluation of the Company’s volatility and discount rates involved the use of valuation professionals with specialized skills and knowledge.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of certain internal controls related to the Company’s fair value measurement process for the contingent consideration liability related to achieving commercial milestones. This included controls related to the development of the assumptions for forecasted revenues, volatility, and discount rates. We evaluated the forecasted revenues and certain commercial milestone assumptions used in the Company’s models by comparing them to historical data, industry benchmarks and other third-party market data that were assessed to be relevant and reliable. We involved valuation professionals with specialized skills and knowledge, who assisted in developing an independent estimate of the discount rates and volatility assumptions using inputs from publicly available market data and comparing the results to the Company’s discount rates and volatility assumptions.
Impairment of Goodwill
As discussed in Notes 2 and 8 to the consolidated financial statements, the Company performs goodwill impairment testing on an annual basis or upon the occurrence of a triggering event that could indicate a potential impairment. During the three months ended September 30, 2024, the Company determined that certain triggering events, combined with a subsequent decrease in common stock price, indicated that it was more likely than not that the fair value of goodwill may be less than its carrying value, which required the Company to perform a quantitative impairment test. The fair value of the Company’s reporting unit was calculated through a discounted cash flow model (or income approach), based on the present value of estimated future cash flows including assumptions around forecasted revenue growth rates and estimated discount rate. As a result of the quantitative impairment test, the goodwill balance of $163.2 million was fully impaired during the three months ended September 30, 2024.
We identified the evaluation of the fair value of the Company’s reporting unit as a critical audit matter. Evaluating the fair value of the reporting unit was complex and required significant auditor judgment due to the high degree of subjectivity in evaluating certain assumptions used to estimate the fair value of the reporting unit. In particular, the fair value measurement was sensitive to management’s forecast of revenue growth rates and the discount rate assumptions. Changes in these assumptions could have a significant impact on the fair value of the reporting unit. In addition, the audit effort associated with the evaluation of the Company’s revenue growth rates and discount rate required specialized skills and knowledge.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of certain internal controls related to the Company’s calculation of the fair value of its reporting unit. This included controls related to the development of assumptions for forecasted revenue growth rates and discount rate. We evaluated the Company’s forecasted revenue growth rates by comparing them to historical actual growth rates and industry
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-3
data that was assessed to be relevant and reliable. We involved valuation professionals with specialized skills and knowledge, who assisted in evaluating the projected revenue growth rates by comparing them to guideline public companies and developing an independent estimate of the discount rate using inputs from publicly available market data and comparing the results to the Company's discount rate assumption.
| | | | | | | | |
| /s/ KPMG LLP | | |
| | |
| We have served as the Company’s auditor since 2015. | |
| | |
| Short Hills, New Jersey | |
| February 27, 2025 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-4
PACIRA BIOSCIENCES, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts) | | | | | | | | | | | |
| | December 31, |
| 2024 | | 2023 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 276,774 | | | $ | 153,298 | |
| Short-term available-for-sale investments | 207,841 | | | 125,283 | |
| Accounts receivable, net | 113,304 | | | 105,556 | |
| Inventories, net | 125,282 | | | 104,353 | |
| Prepaid expenses and other current assets | 21,929 | | | 21,504 | |
| Total current assets | 745,130 | | | 509,994 | |
| Noncurrent available-for-sale investments | — | | | 2,410 | |
| Fixed assets, net | 167,169 | | | 173,927 | |
| Right-of-use assets, net | 49,222 | | | 61,020 | |
| Goodwill | — | | | 163,243 | |
| Intangible assets, net | 425,970 | | | 483,258 | |
| Deferred tax assets | 130,376 | | | 144,485 | |
| Investments and other assets | 35,649 | | | 36,049 | |
| | | |
| Total assets | $ | 1,553,516 | | | $ | 1,574,386 | |
| | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 19,133 | | | $ | 15,698 | |
| Accrued expenses | 80,124 | | | 64,243 | |
| Lease liabilities | 8,887 | | | 8,801 | |
| Current portion of convertible senior notes, net | 201,776 | | | 8,641 | |
| | | |
| | | |
| | | |
| | | |
| | | |
| Total current liabilities | 309,920 | | | 97,383 | |
| Convertible senior notes, net | 279,334 | | | 398,594 | |
| Long-term debt, net | 104,211 | | | 115,202 | |
| Lease liabilities | 44,645 | | | 54,806 | |
| | | |
| Contingent consideration | 20,241 | | | 24,698 | |
| Other liabilities | 16,817 | | | 13,573 | |
| Total liabilities | 775,168 | | | 704,256 | |
| Commitments and contingencies (Note 19) | | | |
| Stockholders’ equity: | | | |
| Preferred stock, par value $0.001; 5,000,000 shares authorized; none issued and outstanding at December 31, 2024 and 2023 | — | | | — | |
| Common stock, par value $0.001; 250,000,000 shares authorized; 47,077,844 shares issued and 46,240,604 shares outstanding at December 31, 2024 and 46,481,174 shares issued and outstanding at December 31, 2023 | 47 | | | 46 | |
| Treasury stock, at cost, 837,240 and zero shares at December 31, 2024 and 2023, respectively, inclusive of excise tax | (25,121) | | | — | |
| Additional paid-in capital | 1,009,435 | | | 976,633 | |
| Accumulated deficit | (206,356) | | | (106,796) | |
| Accumulated other comprehensive income | 343 | | | 247 | |
| Total stockholders’ equity | 778,348 | | | 870,130 | |
| Total liabilities and stockholders’ equity | $ | 1,553,516 | | | $ | 1,574,386 | |
See accompanying notes to consolidated financial statements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-5
PACIRA BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts) | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Revenues: | | | | | |
| Net product sales | $ | 697,186 | | | $ | 672,245 | | | $ | 664,150 | |
| Royalty revenue | 3,780 | | | 2,733 | | | 2,673 | |
| | | | | |
| Total revenues | 700,966 | | | 674,978 | | | 666,823 | |
| Operating expenses: | | | | | |
| Cost of goods sold | 170,428 | | | 184,669 | | | 199,295 | |
| Research and development | 81,577 | | | 76,257 | | | 84,797 | |
| Selling, general and administrative | 294,099 | | | 269,441 | | | 254,516 | |
| Amortization of acquired intangible assets | 57,288 | | | 57,288 | | | 57,288 | |
| Goodwill impairment | 163,243 | | | — | | | — | |
| Contingent consideration gains, restructuring charges and other | 7,702 | | | (352) | | | 10,903 | |
| Total operating expenses | 774,337 | | | 587,303 | | | 606,799 | |
| (Loss) income from operations | (73,371) | | | 87,675 | | | 60,024 | |
| Other income (expense): | | | | | |
| Interest income | 19,689 | | | 11,444 | | | 4,542 | |
| Interest expense | (16,569) | | | (20,306) | | | (39,976) | |
| Gain (loss) on early extinguishment of debt | 7,518 | | | (16,926) | | | — | |
| Other, net | (373) | | | (186) | | | (11,288) | |
| Total other income (expense), net | 10,265 | | | (25,974) | | | (46,722) | |
| (Loss) income before income taxes | (63,106) | | | 61,701 | | | 13,302 | |
| Income tax (expense) benefit | (36,454) | | | (19,746) | | | 2,607 | |
| Net (loss) income | $ | (99,560) | | | $ | 41,955 | | | $ | 15,909 | |
| | | | | |
| Net (loss) income per share: | | | | | |
| Basic net (loss) income per common share | $ | (2.15) | | | $ | 0.91 | | | $ | 0.35 | |
| Diluted net (loss) income per common share | $ | (2.15) | | | $ | 0.89 | | | $ | 0.34 | |
| Weighted average common shares outstanding: | | | | | |
| Basic | 46,245 | | | 46,222 | | | 45,521 | |
| Diluted | 46,245 | | | 51,979 | | | 46,538 | |
See accompanying notes to consolidated financial statements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-6
PACIRA BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE (LOSS) INCOME
(In thousands) | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Net (loss) income | $ | (99,560) | | | $ | 41,955 | | | $ | 15,909 | |
| Other comprehensive income (loss): | | | | | |
| Net unrealized gain (loss) on investments, net of tax | 66 | | | 647 | | | (662) | |
| Foreign currency translation adjustments | 30 | | | (20) | | | 115 | |
| Total other comprehensive income (loss) | 96 | | | 627 | | | (547) | |
| Comprehensive (loss) income | $ | (99,464) | | | $ | 42,582 | | | $ | 15,362 | |
See accompanying notes to consolidated financial statements
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-7
PACIRA BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR THE YEARS ENDED DECEMBER 31, 2024, 2023 AND 2022
(In thousands) | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Number of shares outstanding | | | | | | Additional
Paid-In
Capital | | Accumulated
Deficit | | Accumulated
Other
Comprehensive
Income (Loss) | | Total |
| | Common Shares | | Treasury Shares | | Common Stock | | Treasury Stock | | | | |
| Balance at December 31, 2021 | 44,734 | | | — | | | $ | 45 | | | $ | — | | | $ | 942,091 | | | $ | (211,895) | | | $ | 167 | | | $ | 730,408 | |
| Reclassification of the equity component of convertible senior notes to liabilities upon adoption of Accounting Standards Update 2020-06 | — | | | — | | | — | | | — | | | (96,468) | | | 47,235 | | | — | | | (49,233) | |
| Exercise of stock options | 690 | | | — | | | 1 | | | — | | | 24,386 | | | — | | | — | | | 24,387 | |
| Vested restricted stock units | 331 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Common stock issued under employee stock purchase plan | 71 | | | — | | | — | | | — | | | 2,954 | | | — | | | — | | | 2,954 | |
| Stock-based compensation | — | | | — | | | — | | | — | | | 48,092 | | | — | | | — | | | 48,092 | |
| Issuance of common stock upon conversion of 2022 convertible senior notes | 102 | | | — | | | — | | | — | | | 3,040 | | | — | | | — | | | 3,040 | |
| Other comprehensive loss (Note 12) | — | | | — | | | — | | | — | | | — | | | — | | | (547) | | | (547) | |
| Net income | — | | | — | | | — | | | — | | | — | | | 15,909 | | | — | | | 15,909 | |
| Balance at December 31, 2022 | 45,928 | | | — | | | 46 | | | — | | | 924,095 | | | (148,751) | | | (380) | | | 775,010 | |
| Exercise of stock options | 63 | | | — | | | — | | | — | | | 1,939 | | | — | | | — | | | 1,939 | |
| Vested restricted stock units | 404 | | | — | | | — | | | — | | | (1) | | | — | | | — | | | (1) | |
| Common stock withheld for employee withholding tax liabilities on vested restricted stock units | (4) | | | — | | | — | | | — | | | (106) | | | — | | | — | | | (106) | |
| Common stock issued under employee stock purchase plan | 90 | | | — | | | — | | | — | | | 2,811 | | | — | | | — | | | 2,811 | |
| Stock-based compensation | — | | | — | | | — | | | — | | | 47,895 | | | — | | | — | | | 47,895 | |
| Other comprehensive income (Note 12) | — | | | — | | | — | | | — | | | — | | | — | | | 627 | | | 627 | |
| Net income | — | | | — | | | — | | | — | | | — | | | 41,955 | | | — | | | 41,955 | |
| Balance at December 31, 2023 | 46,481 | | | — | | | 46 | | | — | | | 976,633 | | | (106,796) | | | 247 | | | 870,130 | |
| | | | | | | | | | | | | | | |
| Vested restricted stock units | 501 | | | — | | | 1 | | | — | | | — | | | — | | | — | | | 1 | |
| Common stock withheld for employee withholding tax liabilities on vested restricted stock units | (19) | | | — | | | — | | | — | | | (491) | | | — | | | — | | | (491) | |
| Common stock issued under employee stock purchase plan | 115 | | | — | | | — | | | — | | | 2,298 | | | — | | | — | | | 2,298 | |
| Stock-based compensation | — | | | — | | | — | | | — | | | 51,171 | | | — | | | — | | | 51,171 | |
| Purchase of treasury stock, inclusive of excise tax | — | | | (837) | | | — | | | (25,121) | | | — | | | — | | | — | | | (25,121) | |
| Purchase of capped call transaction, net of tax | — | | | — | | | — | | | — | | | (20,176) | | | — | | | — | | | (20,176) | |
| Other comprehensive income (Note 12) | — | | | — | | | — | | | — | | | — | | | — | | | 96 | | | 96 | |
| Net loss | — | | | — | | | — | | | — | | | — | | | (99,560) | | | — | | | (99,560) | |
| Balance at December 31, 2024 | 47,078 | | | (837) | | | $ | 47 | | | $ | (25,121) | | | $ | 1,009,435 | | | $ | (206,356) | | | $ | 343 | | | $ | 778,348 | |
See accompanying notes to consolidated financial statements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-8
PACIRA BIOSCIENCES, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands) | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Operating activities: | | | | | |
| Net (loss) income | $ | (99,560) | | | $ | 41,955 | | | $ | 15,909 | |
| Adjustments to reconcile net (loss) income to net cash provided by operating activities: | | | | | |
| Goodwill impairment | 163,243 | | | — | | | — | |
| Loss on lease terminations | 2,165 | | | — | | | — | |
| Deferred taxes | 20,621 | | | 15,615 | | | (7,945) | |
| Depreciation of fixed assets and amortization of intangible assets | 78,785 | | | 75,574 | | | 91,501 | |
| Amortization of debt issuance costs | 3,130 | | | 2,996 | | | 4,400 | |
| Amortization of debt discount | 92 | | | 752 | | | 2,807 | |
| | | | | |
| (Gain) loss on early extinguishment of debt | (7,518) | | | 16,926 | | | — | |
| Stock-based compensation | 51,171 | | | 47,895 | | | 48,092 | |
| Changes in contingent consideration | (4,457) | | | (3,424) | | | (29,476) | |
| Impairment of indefinite-lived intangible asset | — | | | — | | | 26,134 | |
| Impairment of investment | — | | | — | | | 10,000 | |
| | | | | |
| Other net losses | 236 | | | 2,137 | | | 285 | |
| Changes in operating assets and liabilities: | | | | | |
| Accounts receivable, net | (7,748) | | | (7,159) | | | (2,079) | |
| Inventories, net | (20,929) | | | (8,290) | | | 2,486 | |
| Prepaid expenses and other assets | (7,360) | | | (9,639) | | | (2,699) | |
| Accounts payable | 3,084 | | | 916 | | | 6,272 | |
| Accrued expenses and income taxes payable | 11,720 | | | (22,039) | | | (19,857) | |
| Other liabilities | 2,714 | | | 434 | | | (556) | |
| | | | | |
| | | | | |
| Net cash provided by operating activities | 189,389 | | | 154,649 | | | 145,274 | |
| Investing activities: | | | | | |
| | | | | |
| Purchases of fixed assets | (10,636) | | | (15,161) | | | (30,076) | |
| | | | | |
| Purchases of available-for-sale investments | (252,212) | | | (137,608) | | | (387,685) | |
| Sales of available-for-sale investments | 179,572 | | | 237,068 | | | 237,576 | |
| Payment of contingent consideration | — | | | — | | | (32,000) | |
| | | | | |
| Purchases of debt and equity investments | — | | | (6,758) | | | (13,000) | |
| Net cash (used in) provided by investing activities | (83,276) | | | 77,541 | | | (225,185) | |
| Financing activities: | | | | | |
| Proceeds from exercises of stock options | — | | | 1,939 | | | 24,387 | |
| Proceeds from shares issued under employee stock purchase plan | 2,298 | | | 2,811 | | | 2,954 | |
| Payment of employee withholding taxes on restricted stock unit vests | (491) | | | (106) | | | — | |
| Purchase of treasury stock | (25,000) | | | — | | | — | |
| Proceeds from 2029 convertible senior notes | 287,500 | | | — | | | — | |
| Proceeds from Term loan A facility | — | | | 149,550 | | | — | |
| | | | | |
| | | | | |
| | | | | |
| Repayment of 2022 convertible senior notes | — | | | — | | | (156,960) | |
| | | | | |
| Repayment of 2024 convertible senior notes | (8,641) | | | — | | | (192,609) | |
| Repayment of 2025 convertible senior notes | (190,994) | | | — | | | — | |
| Repayment of Term loan B facility | — | | | (296,875) | | | (78,125) | |
| Repayment of Term loan A facility | (11,250) | | | (33,437) | | | — | |
| Purchase of capped call transactions | (26,709) | | | — | | | — | |
| Debt extinguishment costs | — | | | (5,750) | | | — | |
| Payment of debt issuance and financing costs | (9,350) | | | (1,163) | | | (1,175) | |
| | | | | |
| | | | | |
| Net cash provided by (used in) financing activities | 17,363 | | | (183,031) | | | (401,528) | |
| Net increase (decrease) in cash and cash equivalents | 123,476 | | | 49,159 | | | (481,439) | |
| Cash and cash equivalents, beginning of year | 153,298 | | | 104,139 | | | 585,578 | |
| Cash and cash equivalents, end of year | $ | 276,774 | | | $ | 153,298 | | | $ | 104,139 | |
| See accompanying notes to consolidated financial statements. |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-9
| | | | | | | | | | | | | | | | | |
PACIRA BIOSCIENCES, INC. CONSOLIDATED STATEMENTS OF CASH FLOWS (CONTINUED) (In thousands) |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Supplemental cash flow information: | | | | | |
| Cash paid for interest | $ | 15,418 | | | $ | 27,635 | | | $ | 33,295 | |
| Cash paid for income taxes, net of refunds | $ | 11,021 | | | $ | 4,366 | | | $ | 7,398 | |
| Non-cash investing and financing activities: | | | | | |
| Issuance of common stock from conversion of 2022 convertible senior notes | $ | — | | | $ | — | | | $ | 3,040 | |
| Fixed assets included in accounts payable and accrued liabilities | $ | 5,565 | | | $ | 1,982 | | | $ | 5,888 | |
| | | | | |
| Excise tax on share repurchases included in accrued liabilities | $ | 121 | | | $ | — | | | $ | — | |
See accompanying notes to consolidated financial statements.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-10
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1—DESCRIPTION OF BUSINESS
Pacira BioSciences, Inc. and its subsidiaries (collectively, the “Company” or “Pacira”) delivers innovative, non-opioid pain therapies to transform the lives of patients. The Company’s long-acting, local analgesic, EXPAREL® (bupivacaine liposome injectable suspension), was commercially launched in the United States, or U.S., in April 2012 and approved in select European countries and the United Kingdom, or U.K., in November 2021. EXPAREL utilizes the Company’s proprietary multivesicular liposome, or pMVL, drug delivery technology that encapsulates drugs without altering their molecular structure and releases them over a desired period of time. EXPAREL is currently indicated to produce postsurgical local analgesia via infiltration in patients aged 6 years and older, and postsurgical regional analgesia via an interscalene brachial plexus block in adults, a sciatic nerve block in the popliteal fossa in adults, and an adductor canal block in adults for postsurgical pain management (the safety and effectiveness of EXPAREL have not been established to produce postsurgical regional analgesia via other nerve blocks besides an interscalene brachial plexus nerve block, a sciatic nerve block in the popliteal fossa, or an adductor canal block). In November 2021, the Company acquired Flexion Therapeutics, Inc., or Flexion (the “Flexion Acquisition”), and added ZILRETTA® (triamcinolone acetonide extended-release injectable suspension) to its product portfolio. ZILRETTA is the first and only extended-release, intra-articular (meaning in the joint) injection indicated for the management of osteoarthritis, or OA, knee pain. In April 2019, the Company added iovera®° to its commercial offering with the acquisition of MyoScience, Inc., or MyoScience (the “MyoScience Acquisition”). The iovera° system is a handheld cryoanalgesia device that delivers immediate, long-acting, drug-free pain control using precise, controlled doses of cold temperature to a targeted nerve. The Company is also advancing the development of PCRX-201 (enekinragene inzadenovec), a novel gene therapy vector platform enabling local administration of genetic medicines with the potential to treat large prevalent diseases like OA. In February 2025, the Company acquired the remaining equity interest in GQ Bio Therapeutics GmbH, or GQ Bio (the “GQ Bio Acquisition”) a privately-held biopharmaceutical company with a novel, high-capacity, local-delivery platform that makes genetic medicines more efficient and enables the use of large and multiple gene constructs. PCRX-201 is the lead program from this platform. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events.
Pacira is subject to risks common to companies in similar industries and stages, including, but not limited to, competition from larger companies and potential generic entrants, reliance on revenue from three products, reliance on a limited number of wholesalers, reliance on a limited number of manufacturing sites, new technological innovations, dependence on key personnel, reliance on third-party service providers and sole source suppliers, protection of proprietary technology, compliance with government regulations and risks related to cybersecurity.
NOTE 2—SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Principles of Consolidation
These consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America, or GAAP, and in accordance with the rules and regulations of the U.S. Securities and Exchange Commission, or SEC. The accounts of the Company’s wholly owned subsidiaries are included in these consolidated financial statements. All intercompany balances and transactions have been eliminated in consolidation. Certain reclassifications from previously issued financial statements have been made to conform to the current presentation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires the use of estimates and assumptions that affect the reported amounts of assets and liabilities, including disclosure of contingent assets and contingent liabilities, at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Estimates are used for, among other things, revenue recognition, valuation of acquired assets and liabilities, stock-based compensation, inventory costs, impairments of equity investments, long-lived assets, goodwill and other intangible assets, liabilities and accruals, including contingent consideration, and the valuation of deferred tax assets. Because of the uncertainty of factors surrounding the estimates or judgments used in the preparation of the consolidated financial statements, actual results could differ from these estimates.
Revenue From Contracts With Customers
The Company’s net product sales consist of (i) EXPAREL in the U.S., European Union, or E.U., and the U.K.; (ii) ZILRETTA in the U.S.; (iii) iovera° in the U.S., Canada and Europe and (iv) sales of its bupivacaine liposome injectable suspension for veterinary use. Royalty revenues are related to a collaborative licensing agreement from the sale of the Company’s bupivacaine liposome injectable suspension product for veterinary use. See Note 4, Revenue, for further information on the Company’s accounting policies related to revenue from contracts with customers.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-11
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Royalty Revenue
Royalties are estimated and recognized as revenue when sales to the Company’s commercial partners occur, unless some constraint exists, as the royalties predominately relate to a supply agreement. Royalties are based on sales of the Company’s bupivacaine liposome injectable suspension product for veterinary use.
Concentration of Major Customers
The Company sells EXPAREL through a drop-ship program under which orders are processed through wholesalers (including AmerisourceBergen Health Corporation, Cardinal Health, Inc. and McKesson Drug Company), but shipments of the product are sent directly to individual accounts, such as hospitals, ambulatory surgery centers and individual physicians. The Company also sells EXPAREL directly to ambulatory surgery centers and individual physicians. The Company sells ZILRETTA primarily to specialty distributors and specialty pharmacies, who then subsequently resell ZILRETTA to physicians, clinics and certain medical centers or hospitals. The Company also contracts directly with healthcare providers and intermediaries such as Group Purchasing Organizations, or GPOs. The Company sells iovera° directly to end users and its bupivacaine liposome injectable suspension product for veterinary use to a third-party licensee in the U.S.
The table below includes the percentage of revenues comprised by the Company’s three largest wholesalers in each period presented: | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Largest wholesaler | 34 | % | | 33 | % | | 31 | % |
| Second largest wholesaler | 23 | % | | 24 | % | | 23 | % |
| Third largest wholesaler | 20 | % | | 20 | % | | 22 | % |
| Total | 77 | % | | 77 | % | | 76 | % |
Revenue from outside the U.S. accounted for less than 1% of the Company’s total revenue for each of the years ended December 31, 2024, 2023 and 2022.
Research and Development Expenses
Research and development expenditures are expensed as incurred. These include both internal and external costs, of which a significant portion of development activities are outsourced to third parties, including contract research organizations, or CROs. Clinical trial costs are accrued over the service periods specified in contracts and adjusted as necessary based on an ongoing review of the level of effort and actual costs incurred by the CROs. Research and development costs are presented net of any reimbursements from commercial partners.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to basis differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
The Company accrues interest and penalties on underpayments of income taxes, including those related to unrecognized tax benefits, as a component of income tax expense in its consolidated statements of operations.
Stock-Based Compensation
The Company’s stock-based compensation consists of grants of stock options and restricted stock units, or RSUs, to employees, consultants and non-employee directors, in addition to the opportunity for employees to participate in an employee stock purchase plan. The expense associated with these programs is recognized in the Company’s consolidated statements of operations based on their fair values as they are earned under their applicable vesting terms or the length of an offering period.
In calculating the estimated fair value of stock options and employee stock purchase plan share options granted, the Company uses the Black-Scholes option valuation model, or Black-Scholes model, which requires the consideration of the
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-12
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
following variables for purposes of estimating fair value in addition to the closing price of the Company’s common stock on the date of grant:
•Expected term of the option
•Expected volatility
•Expected dividends
•Risk-free interest rate
The Company utilizes its historical volatility data to determine expected volatility over the expected term of the option. The Company uses an expected term based on its historical stock option activity data for stock option grants and the length of an offering period for employee stock purchase plan share option grants. The risk-free interest rate is based on the implied yield on U.S. Department of the Treasury zero-coupon bonds for periods commensurate with the expected term of the options. The dividend yield on the Company’s common stock is estimated to be zero as the Company has not declared or paid any dividends since inception, nor does it have any intention to do so in the foreseeable future. Additionally, the Company’s ability to declare and pay a dividend in the future could be limited per the agreements governing its indebtedness. The Company records forfeitures of grants as they occur rather than estimating forfeitures during each reporting period.
Cash and Cash Equivalents
All highly liquid investments with maturities of 90 days or less when purchased are considered cash equivalents. Cash equivalents include money market funds. As of December 31, 2024, the carrying value of the Company’s money market funds was $269.4 million. As of December 31, 2023, the carrying value of money market funds was $26.1 million. The carrying values approximate fair value as of December 31, 2024 and 2023.
Short-Term and Noncurrent Available-For-Sale Investments
Available-for-sale investments may consist of asset-backed securities collateralized by credit card receivables, investment grade commercial paper, corporate bonds, federal agency bonds, government and Yankee bonds, and other bonds issued in the U.S. (and denominated in the U.S. dollar) by foreign entities. Current available-for-sale investments are those with maturities of greater than three months, but less than one year. Noncurrent available-for-sale investments hold maturities greater than one year. The Company evaluates the classification of its investments at the time of purchase and re-evaluates such determination at each balance sheet date, which includes an assessment of the intent to hold the available-for-sale securities. The Company’s investment policy sets minimum credit quality criteria and maximum maturity limits on its investments to provide for preservation of capital, liquidity and a reasonable rate of return. The Company classifies its investments as available-for-sale. Available-for-sale securities are recorded at fair value, based on current market valuations. Unrealized holding gains and losses on available-for-sale securities (except for credit losses) are excluded from net (loss) income and are reported as a separate component of accumulated other comprehensive income until realized. Realized gains and losses are included in interest income in the consolidated statements of operations and are derived using the specific identification method for determining the cost of the securities sold. The Company evaluates whether a credit loss exists, and in the event a credit loss does exist, the credit loss is recognized in the consolidated statements of operations based on the amount that the fair value is less than the amortized cost.
Inventories
Inventories consist of finished goods held for sale and distribution, raw materials and work in process. Inventories are stated at the lower of cost, which includes amounts related to material, labor and overhead, or net realizable value, and is determined using the first-in, first-out method. The Company periodically reviews its inventory to identify obsolete, slow-moving, or otherwise unsalable inventories, and establishes allowances for situations in which the cost of the inventory is not expected to be recovered.
Fixed Assets
Fixed assets are recorded at cost, net of accumulated depreciation and amortization. The Company reviews its property, plant and equipment assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
Depreciation of fixed assets is provided over their estimated useful lives on a straight-line basis. The Company periodically reviews these useful lives relative to physical factors, economic factors and industry trends. If there are changes in the planned use of property or equipment, the useful lives assigned to these assets may need to be shortened, resulting in the
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-13
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
recognition of accelerated depreciation expense in future periods. Leasehold improvements are amortized on a straight-line basis over the shorter of their estimated useful lives or the related remaining lease terms. Useful lives by asset category are as follows:
| | | | | | | | |
| Asset Category | | Useful Life |
| Computer equipment and software | | 1 to 3 years |
| Office furniture and equipment | | 5 years |
| Manufacturing and laboratory equipment | | 5 to 15 years |
Asset Retirement Obligations
The Company has contractual obligations stemming from certain of its lease agreements to return leased space to its original condition upon termination of such lease agreements. The Company records its asset retirement obligations, or ARO, along with a corresponding capital asset in an amount equal to the estimated fair value of the ARO, based on the present value of expected future cash flows. In subsequent periods, the Company records expense to accrete the ARO to its full value. Each ARO capital asset is depreciated over the depreciable term of the associated fixed asset.
Leases
The Company recognizes right-of-use, or ROU, assets and lease liabilities at the commencement of its lease agreements. The leases are evaluated at commencement to determine whether they should be classified as operating or financing leases. Lease costs associated with operating leases are recognized on a straight-line basis, while lease costs for financing leases are recognized over the lease term using the effective interest method. The Company does not currently have any financing leases. The amount of ROU assets and lease liabilities to be recognized is impacted by the type of lease payments, the lease term and the incremental borrowing rate. Variable lease payments are not included at commencement and are recognized in the period in which they are incurred.
The Company has elected to net the amortization of its ROU assets and the reduction of the lease liability principal in other liabilities in the consolidated statement of cash flows.
The lease term is based on the contractual term and is adjusted for any renewal options or termination rights that are reasonably certain to be exercised. The incremental borrowing rate is based on the rate the Company estimates it would pay on a collateralized basis over a similar term in a similar economic environment.
Acquisitions
In a business combination, the acquisition method of accounting requires that the assets acquired and liabilities assumed be recorded as of the date of the acquisition at their respective fair values, with some exceptions. Assets acquired and liabilities assumed in a business combination that arise from contingencies are generally recognized at fair value. If fair value can be determined, the asset or liability is recognized; if fair value is not determinable, then no asset or liability is recognized. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an “exit price”) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date.
Acquired in-process research and development, or IPR&D, is recognized at fair value and initially characterized as an indefinite-lived intangible asset, irrespective of whether the acquired IPR&D has an alternative future use. If the acquired net assets do not constitute a business under the acquisition method of accounting, the transaction is accounted for as an asset acquisition and no goodwill is recognized. In an asset acquisition, the amount allocated to acquired IPR&D with no alternative future use is recorded as an expense at the acquisition date.
Any excess of the purchase price (consideration transferred) over the estimated fair values of net assets acquired is recorded as goodwill. Transaction costs and costs to restructure the acquired company are expensed as incurred. The operating results of the acquired business are reflected in the Company’s consolidated financial statements after the closing date of the acquisition.
Contingent Consideration
Subsequent to an acquisition, the Company measures contingent consideration arrangements at fair value at each reporting period, with changes in fair value recognized in the consolidated statements of operations. Changes in contingent consideration
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-14
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
can result from changes in the assumed achievement and timing of estimated sales and regulatory approvals. In the absence of new information, changes in fair value reflect the passage of time towards achievement or expiration of the milestones, and are accreted to the period in which payments are expected to be made.
Goodwill
Goodwill represents the excess of the purchase price over the estimated fair value of the net assets acquired in a business combination and is subject to impairment testing at least annually or upon the occurrence of a triggering event that could indicate a potential impairment. The Company has historically tested goodwill for impairment by performing a qualitative assessment in order to determine whether facts and circumstances support a determination that reporting unit fair values are greater than their carrying values. This has historically been performed using readily available market data and company-specific factors.
If the Company determines that it is more likely than not that the fair value of the Company is less than its carrying value, a quantitative test is required. This is performed by comparing the fair value of the Company with its carrying value. If the estimated fair value of the reporting unit is less than the carrying amount of the reporting unit, impairment is indicated, requiring recognition of a goodwill impairment charge up to the carrying value of goodwill. The fair value of the Company would be calculated through an income approach. Under the income approach, the Company calculates the fair value based on the present value of estimated future cash flows. Considerable management judgment is necessary to evaluate the impact of operating and macroeconomic changes and to estimate the future cash flows used to assume fair value. The Company’s estimates of future cash flows consider past performance, current and anticipated market conditions and internal projections and operating plans which incorporate estimates for sales growth and future margins. Additional assumptions would include forecasted growth rates, estimated discount rates and the probability of success for the Company’s product pipeline candidate products. The Company believes such assumptions would reflect current and anticipated market conditions and are consistent with those that would be used by other marketplace participants for similar valuation purposes. Such assumptions are subject to change due to changing economic and competitive conditions.
Intangible Assets
Intangible assets with definite useful lives are amortized on a straight-line basis over their estimated useful lives and are recorded at cost, net of accumulated amortization. Indefinite-lived intangible assets are tested for impairment at least annually or when a triggering event occurs that could indicate a potential impairment exists. Impairment charges are recognized to the extent the carrying value exceeds its fair value.
Equity Investments
The Company holds investments in equity securities without a readily determinable fair value which are recognized at cost less any impairments, plus or minus any changes resulting from observable price changes in orderly transactions for a similar investment.
Impairments of Long-Lived Assets
Management reviews long-lived assets, including fixed assets and finite-lived intangible assets, for impairment whenever events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured as the amount by which the carrying amount of the assets exceeds the fair value of the assets.
Per Share Data
Basic net (loss) income per common share is computed by dividing net (loss) income available (attributable) to common stockholders by the weighted average number of shares of common stock outstanding during the period.
Diluted net (loss) income per common share is calculated by dividing net (loss) income available (attributable) to common stockholders as adjusted for the effect of dilutive securities, if any, by the weighted average number of shares of common stock and dilutive common stock outstanding during the period.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-15
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Potential common shares include the shares of common stock issuable upon the exercise of outstanding stock options, the vesting of RSUs and the purchase of shares from the Company’s employee stock purchase plan (using the treasury stock method), if applicable. Potential common shares associated with convertible senior notes are treated under the if-converted method and adjustments are made to the diluted net (loss) income per common share calculation as if the Company had converted the convertible senior notes on the first day of each period presented. Adjustments to the numerator are made to add back the interest expense associated with the convertible senior notes on a post-tax basis. Adjustments to the denominator reflect the number of shares assumed to be convertible at the beginning of the period.
Treasury Stock
Repurchases of the Company’s common stock are accounted for at cost and recorded as treasury stock. The excise tax on repurchases of the Company’s common stock is recorded as a cost of acquiring treasury stock. Any reissued treasury stock is accounted for at average cost. Gains or losses on reissued treasury stock arising from the difference between the average cost and the fair value of the award is recorded in additional paid-in capital in the consolidated balance sheets.
Foreign Currencies
The balance sheet accounts of the Company’s foreign subsidiaries with functional currencies other than the U.S. Dollar are translated using the exchange rate at each respective balance sheet date. Revenues and expenses are translated using the average exchange rates for each calendar month during the year. Translation adjustments are recorded as a component of accumulated other comprehensive income in the consolidated financial statements. Gains or losses from foreign currency exchanges are recorded in other, net in the consolidated statements of operations.
NOTE 3—RECENT ACCOUNTING PRONOUNCEMENTS
Recently Adopted Accounting Pronouncements
In November 2023, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, 2023-07, Segment Reporting (Topic 280), Improvements to Reportable Segment Disclosures. The ASU amendment improves reportable segment disclosure requirements, primarily through enhanced disclosures about significant segment expenses on an interim and annual basis. The new segment disclosure requirements apply for entities with a single reportable segment. The Company adopted the standard for its annual reporting which was applied retrospectively for all prior years presented. The ASU's amendment is effective for interim periods in fiscal years beginning after December 15, 2024. Refer to Note 20, Segment Information, for more information.
Recently Issued Accounting Pronouncements Not Adopted as of December 31, 2024
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740), Improvements to Income Tax Disclosures. The ASU amendment addresses investor requests for more transparency about income tax information through improvements to income tax disclosures primarily related to the rate reconciliation and income taxes paid information. The ASU’s amendments are effective for fiscal years beginning after December 15, 2024 and may be adopted on a prospective or retrospective basis. The Company is currently evaluating the impact of adopting ASU 2023-09 on its consolidated financial statements.
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40), Disaggregation of Income Statement Expenses. The ASU amendment improves financial reporting by requiring public business entities disclose additional information about specific expense categories in the notes to financial statements at interim and annual reporting periods. The ASU’s amendments are effective for annual reporting periods beginning after December 31, 2026 and interim periods beginning after December 15, 2027, with early adoption permitted. This ASU amendment can be applied on a prospective basis or retrospectively. The Company is currently evaluating the impact of adopting ASU 2024-03 on its footnote disclosures.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-16
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 4—REVENUE
The Company’s sources of revenue are detailed in Note 2, Summary of Significant Accounting Policies. The Company does not consider revenue from sources other than sales of EXPAREL and ZILRETTA to be material sources of its consolidated revenue. As such, the following disclosure is limited to revenue associated with net product sales of EXPAREL and ZILRETTA.
Net Product Sales
The Company sells EXPAREL through a drop-ship program under which orders are processed through wholesalers based on orders of the product placed by end-users, namely hospitals, ambulatory surgery centers and healthcare provider offices. EXPAREL is delivered directly to the end-user without the wholesaler ever taking physical possession of the product. The Company primarily sells ZILRETTA to specialty distributors and specialty pharmacies, who then subsequently resell ZILRETTA to physicians, clinics and certain medical centers or hospitals. The Company also contracts directly with healthcare providers and intermediaries such as GPOs. Product revenue is recognized when control of the promised goods are transferred to the customer, in an amount that reflects the consideration the Company expects to be entitled to in exchange for transferring those goods. EXPAREL and ZILRETTA revenue is recorded at the time the products are transferred to the customer.
Revenues from sales of products are recorded net of returns allowances, prompt payment discounts, service fees, government rebates, volume rebates and chargebacks. These reserves are based on estimates of the amounts earned or to be claimed on the related sales. These amounts are treated as variable consideration, estimated and recognized as a reduction of the transaction price at the time of the sale, using the most likely amount method, except for returns, which is based on the expected value method. The Company includes these estimated amounts in the transaction price to the extent it is probable that a significant reversal of cumulative revenue recognized for such transaction will not occur, or when the uncertainty associated with the variable consideration is resolved.
Chargebacks for fees and discounts represent the estimated obligations resulting from contractual commitments to sell products to Department of Veteran Affairs hospitals, participating GPO members, 340B qualified entities and other contracted customers at prices lower than the list price. The 340B Drug Discount Program is a U.S. federal government program that requires participating drug manufacturers to provide outpatient drugs to eligible health care organizations and covered entities at reduced prices. Customers claim the difference between the amount invoiced and the discounted selling price through a chargeback issued by a wholesaler. Reserves are established in the same period that the related revenue is recognized, resulting in a reduction of product revenue and trade receivables, net. Chargeback amounts are determined at the time of sale and the Company generally issues credits for such amounts within weeks of receiving notification from a wholesaler. Reserves for chargebacks consist of anticipated credits the Company expects to issue based on expected units sold and chargebacks that customers have claimed for which credits have not yet been issued.
The calculation for some of these items requires management to make estimates based on sales data, historical return data, contracts, statutory requirements and other related information that may become known in the future. The adequacy of these provisions is reviewed on a quarterly basis.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-17
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The following table provides a summary of activity with respect to the Company’s sales related allowances and accruals related to EXPAREL and ZILRETTA for the years ended December 31, 2024, 2023 and 2022 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Returns Allowances | | Prompt Payment Discounts | | Service
Fees | | Volume Rebates and Chargebacks | | Government Rebates | | Total |
| Balance at December 31, 2021 | $ | 3,361 | | | $ | 1,178 | | | $ | 3,636 | | | $ | 3,494 | | | $ | 761 | | | $ | 12,430 | |
| Provision | 1,390 | | | 11,145 | | | 16,866 | | | 48,890 | | | 1,641 | | | 79,932 | |
| Payments | (3,060) | | | (11,136) | | | (17,309) | | | (46,932) | | | (1,616) | | | (80,053) | |
| Balance at December 31, 2022 | 1,691 | | | 1,187 | | | 3,193 | | | 5,452 | | | 786 | | | 12,309 | |
| Provision | 1,335 | | | 11,970 | | | 18,129 | | | 92,009 | | | 2,176 | | | 125,619 | |
| Payments | (1,158) | | | (11,849) | | | (17,625) | | | (91,591) | | | (1,787) | | | (124,010) | |
| Balance at December 31, 2023 | 1,868 | | | 1,308 | | | 3,697 | | | 5,870 | | | 1,175 | | | 13,918 | |
| Provision | 2,260 | | | 12,697 | | | 21,022 | | | 115,087 | | | 2,155 | | | 153,221 | |
| Payments | (2,528) | | | (12,697) | | | (19,844) | | | (116,094) | | | (1,623) | | | (152,786) | |
| Balance at December 31, 2024 | $ | 1,600 | | | $ | 1,308 | | | $ | 4,875 | | | $ | 4,863 | | | $ | 1,707 | | | $ | 14,353 | |
Accounts Receivable
The majority of accounts receivable arise from product sales and represent amounts due from wholesalers, hospitals, ambulatory surgery centers, specialty distributors, specialty pharmacies and individual physicians. Payment terms generally range from zero to four months from the date of the transaction, and accordingly, there is no significant financing component.
Performance Obligations
A performance obligation is a promise in a contract to transfer a distinct good or service to the customer and is the unit of account in Accounting Standards Codification, or ASC, 606. A contract’s transaction price is allocated to each distinct performance obligation and recognized as revenue when, or as, the performance obligation is satisfied.
At contract inception, the Company assesses the goods promised in its contracts with customers and identifies a performance obligation for each promise to transfer to the customer a good that is distinct. When identifying individual performance obligations, the Company considers all goods promised in the contract regardless of whether explicitly stated in the customer contract or implied by customary business practices. The Company’s contracts with customers require it to transfer an individual distinct product, which represents a single performance obligation. The Company’s performance obligation with respect to its product sales is satisfied at a point in time, which transfers control upon delivery of EXPAREL and ZILRETTA to its customers. The Company considers control to have transferred upon delivery because the customer has legal title to the asset, physical possession of the asset has been transferred, the customer has significant risks and rewards of ownership of the asset and the Company has a present right to payment at that time.
Disaggregated Revenue
The following table represents disaggregated net product sales in the periods presented as follows (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Net product sales: | | | | | |
| EXPAREL | $ | 548,962 | | | $ | 538,120 | | | $ | 536,899 | |
| ZILRETTA | 118,089 | | | 111,098 | | | 105,517 | |
| iovera° | 22,813 | | | 19,685 | | | 15,258 | |
| Bupivacaine liposome injectable suspension | 7,322 | | | 3,342 | | | 6,476 | |
| Total net product sales | $ | 697,186 | | | $ | 672,245 | | | $ | 664,150 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-18
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 5—INVENTORIES
The components of inventories, net are as follows (in thousands): | | | | | | | | | | | |
| | December 31, |
| | 2024 | | 2023 |
| Raw materials | $ | 50,800 | | | $ | 54,099 | |
| Work-in-process | 27,384 | | | 31,215 | |
| Finished goods | 47,098 | | | 19,039 | |
| Total | $ | 125,282 | | | $ | 104,353 | |
NOTE 6—FIXED ASSETS
Fixed assets, net, summarized by major category, consist of the following (in thousands): | | | | | | | | | | | |
| | December 31, |
| | 2024 | | 2023 |
Machinery and equipment (1) (2) | $ | 160,643 | | | $ | 121,773 | |
Leasehold improvements (2) | 86,034 | | | 61,826 | |
Computer equipment and software (2) | 23,473 | | | 17,186 | |
| Office furniture and equipment | 1,952 | | | 2,543 | |
Construction in progress (2) | 27,996 | | | 105,905 | |
| Total | 300,098 | | | 309,233 | |
Less: accumulated depreciation (1) | (132,929) | | | (135,306) | |
| Fixed assets, net | $ | 167,169 | | | $ | 173,927 | |
|
For information on useful lives by asset category, refer to Note 2, Summary of Significant Accounting Policies. |
(1) During the year ended December 31, 2024, the Company disposed of $19.0 million of fully depreciated machinery and equipment associated with its 45-liter EXPAREL manufacturing process at its contract manufacturing facility located in Swindon, U.K. The Company continues to operate its enhanced, larger-scale EXPAREL manufacturing process at the same facility.
(2) During the year ended December 31, 2024, an enhanced, large-scale EXPAREL manufacturing suite at the Company’s Science Center Campus in San Diego, California was placed into service, for which approximately $76.1 million was reclassified from construction in progress to machinery and equipment, leasehold improvements and computer equipment and software.
Depreciation expense for the years ended December 31, 2024, 2023 and 2022 was $21.5 million, $18.3 million and $34.2 million, respectively. In 2022, the Company accelerated $10.5 million of depreciation expense for certain machinery and equipment for which no future economic benefit was identified. During the years ended December 31, 2024, 2023 and 2022, the Company capitalized interest on the construction of manufacturing sites of $2.1 million, $3.5 million and $4.1 million, respectively.
As of December 31, 2024 and 2023, total fixed assets, net, includes manufacturing process equipment and leasehold improvements located outside of the U.S. in the amount of $51.1 million and $36.8 million, respectively.
As of December 31, 2024 and 2023, the Company had AROs of $4.2 million and $4.3 million, respectively, included in accrued expenses and other liabilities on its consolidated balance sheets, for costs associated with returning leased spaces to their original condition upon the termination of their lease agreements.
During the three months ended September 30, 2024, the U.S. Food and Drug Administration, or FDA, approved a generic competitor to EXPAREL and a U.S. District Court ruled that one of the Company’s patents was not valid (for more information, see Note 19, Commitments and Contingencies). The Company determined that these events and a subsequent decrease in the Company’s common stock price constituted impairment indicators under ASC 360 Property, Plant and Equipment. As of September 30, 2024, the Company performed a quantitative recoverability test of the carrying values of its asset group. The Company estimated the undiscounted future cash flows expected to result from the use of its asset group and determined the carrying amount of the asset group was recoverable.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-19
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 7—LEASES
The Company leases all of its facilities, including its EXPAREL and iovera° handpiece manufacturing facility at its Science Center Campus in San Diego, California. The Company also has two embedded leases with Thermo Fisher Scientific Pharma Services, or Thermo Fisher, for the use of their manufacturing facility in Swindon, U.K. for the production of EXPAREL and ZILRETTA. A portion of the associated monthly base fees has been allocated to the lease components based on a relative fair value basis.
Since July 2022 and February 2023, the Company has recognized sublease income for laboratory space leased in Woburn, Massachusetts and a portion of office space leased in Burlington, Massachusetts, respectively, from leases that were assumed as part of the Flexion Acquisition. In February 2024, the lease and sublease term concluded for the laboratory space in Woburn, Massachusetts.
During 2023, the Company partially exited its Burlington, Massachusetts office space lease that had been assumed as part of the Flexion Acquisition through a one-time termination fee of $0.8 million, which released its obligation of $1.6 million in future cash payments for the respective proportion of square footage exited. The partial lease termination resulted in a nominal gain which was recorded within contingent consideration gains, restructuring charges and other in the consolidated statements of operations.
In December 2024, the Company exited a lease for a training facility in Houston, Texas. The Company recognized a loss of $2.2 million during the year ended December 31, 2024 associated with exiting the lease, which was recorded within contingent consideration gains, restructuring charges and other in the consolidated statements of operations. The loss resulted from the derecognition of the right-of-use asset, its related lease liability and a termination payment of $1.3 million.
The operating lease costs for these facilities include lease and non-lease components, such as common area maintenance and other common operating expenses, along with executory costs such as insurance and real estate taxes. Total operating lease costs are as follows (in thousands): | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| Operating Lease Costs | | 2024 | | 2023 | | 2022 |
| Fixed lease costs | | $ | 13,876 | | | $ | 14,344 | | | $ | 13,949 | |
| Variable lease costs | | 1,889 | | | 1,952 | | | 1,988 | |
| Sublease income | | (304) | | | (657) | | | (253) | |
| Total | | $ | 15,461 | | | $ | 15,639 | | | $ | 15,684 | |
Supplemental cash flow information related to operating leases is as follows (in thousands): | | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Cash paid for operating lease liabilities, net of lease incentives | | $ | 12,991 | | | $ | 14,259 | | | $ | 14,357 | |
| Right-of-use assets recorded in exchange for lease obligations | | $ | — | | | $ | — | | | $ | 3,324 | |
The weighted average remaining lease terms and the weighted average discount rates are summarized as follows: | | | | | | | | | | | | | | |
| | December 31, |
| | 2024 | | 2023 |
| Weighted average remaining lease term | | 5.19 years | | 6.04 years |
| Weighted average discount rate | | 6.89% | | 7.02% |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-20
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
As of December 31, 2024, maturities of the Company’s operating lease liabilities are as follows (in thousands): | | | | | | | | |
| Year | | Aggregate Minimum
Payments Due |
| 2025 | | $ | 12,285 | |
| 2026 | | 12,319 | |
| 2027 | | 12,072 | |
| 2028 | | 10,924 | |
| 2029 | | 10,812 | |
| Thereafter | | 5,614 | |
| Total future lease payments | | 64,026 | |
| Less: imputed interest | | (10,494) | |
| Total operating lease liabilities | | $ | 53,532 | |
NOTE 8—GOODWILL AND INTANGIBLE ASSETS
Goodwill
The Company’s goodwill resulted from the acquisition of Pacira Pharmaceuticals, Inc. (the Company’s California operating subsidiary) from SkyePharma Holding, Inc. (now Vectura Group Limited, a subsidiary of Molex Asia Holdings Ltd.) in 2007, the MyoScience Acquisition in 2019 and the Flexion Acquisition in 2021. The goodwill balance at December 31, 2023 was $163.2 million.
Goodwill represents the excess of the purchase price over the estimated fair value of the net assets acquired in a business combination and is subject to impairment testing at least annually or upon the occurrence of a triggering event that could indicate a potential impairment. During the three months ended September 30, 2024, the FDA approved a generic competitor to EXPAREL and a U.S. District Court ruled that one of the Company’s patents was not valid (for more information, see Note 19, Commitments and Contingencies). The Company determined that these events, combined with a subsequent decrease in the Company’s common stock price, indicated that it was more likely than not that the fair value of goodwill may be less than its carrying value, which required the Company to perform a quantitative impairment test. This was performed by comparing the fair value of the Company with its carrying value. If the estimated fair value of the reporting unit is less than the carrying amount of the reporting unit, impairment is indicated, requiring recognition of a goodwill impairment charge up to the carrying value of goodwill. The fair value of the Company was calculated through an income approach, in which the Company calculated the fair value based on the present value of estimated future cash flows. Considerable management judgment is necessary to evaluate the impact of operating and macroeconomic changes and to estimate the future cash flows used to assume fair value. The Company’s estimates of future cash flows consider past performance, current and anticipated market conditions, internal projections and operating plans which incorporate estimates for sales growth and future margins. Additional assumptions include forecasted growth rates, estimated discount rates and the probability of success for the Company’s product pipeline candidate products. The assumptions also reflect current and anticipated market conditions and are consistent with those that would be used by other marketplace participants for similar valuation purposes. Such assumptions are subject to change due to changing economic and competitive conditions. The conclusion of the income approach as of September 30, 2024 resulted in the carrying value of the Company exceeding its fair value by more than the goodwill balance. As a result, the goodwill balance of $163.2 million was fully impaired during the three months ended September 30, 2024 and the Company had no remaining goodwill balance at December 31, 2024.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-21
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Intangible Assets
Intangible assets, net, consists of the developed technology and IPR&D from the Flexion Acquisition and developed technology and customer relationships from the MyoScience Acquisition and are summarized as follows (dollar amounts in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2024 | | Gross
Carrying Value | | Accumulated
Amortization | | Intangible
Assets, Net | | Weighted-Average
Useful Lives |
| Developed technologies | | $ | 590,000 | | | $ | (198,934) | | | $ | 391,066 | | | 10 years, 5 months |
| Customer relationships | | 90 | | | (52) | | | 38 | | | 10 years |
| Total finite-lived intangible assets, net | | 590,090 | | | (198,986) | | | 391,104 | | | |
| Acquired IPR&D | | 34,866 | | | — | | | 34,866 | | | |
| Total intangible assets, net | | $ | 624,956 | | | $ | (198,986) | | | $ | 425,970 | | | |
| | | | | | | | |
| December 31, 2023 | | Gross
Carrying Value | | Accumulated
Amortization | | Intangible
Assets, Net | | Weighted-Average
Useful Lives |
| Developed technology | | $ | 590,000 | | | $ | (141,655) | | | $ | 448,345 | | | 10 years, 5 months |
| Customer relationships | | 90 | | | (43) | | | 47 | | | 10 years |
| Total finite-lived intangible assets, net | | 590,090 | | | (141,698) | | | 448,392 | | | |
| Acquired IPR&D | | 34,866 | | | — | | | 34,866 | | | |
| Total intangible assets, net | | $ | 624,956 | | | $ | (141,698) | | | $ | 483,258 | | | |
Amortization expense on intangible assets was $57.3 million for each of the years ended December 31, 2024 and 2023.
Assuming no changes in the gross carrying amount of these intangible assets, the future estimated amortization expense on the finite-lived intangible assets will be $57.3 million each year from 2025 to 2030, $37.4 million in 2031, $7.9 million in 2032 and $2.2 million in 2033.
The Company reviews its indefinite-lived intangible assets for impairment annually and whenever an event or change in circumstances arises that indicates the carrying amount of an indefinite-lived intangible asset is at risk of not being recoverable. During the year ended December 31, 2024 and 2023, the Company conducted the annual impairment assessment for its acquired IPR&D and concluded there was no impairment as of that date. During the year ended December 31, 2022, the annual impairment assessment of ZILRETTA acquired IPR&D for an indication for the treatment of OA pain of the shoulder was conducted through a recoverability test at December 31, 2022 by comparing the $60.0 million carrying value of the asset against the fair value through a discounted cash flow model of $33.9 million based on new facts and circumstances. The change in fair value was primarily driven by later timelines for the completion of clinical trials impacting revenue forecasts, among other factors. An impairment of $26.1 million was recognized within contingent consideration gains, restructuring charges and other in the consolidated statements of operations for the year ended December 31, 2022 based on the amount its previous carrying value exceeded its updated fair value.
NOTE 9—ACCRUED EXPENSES
Accrued expenses consist of the following (in thousands): | | | | | | | | | | | |
| | December 31, |
| | 2024 | | 2023 |
| Accrued selling, general and administrative expenses | $ | 15,622 | | | $ | 12,811 | |
| Accrued research and development expenses | 4,501 | | | 5,141 | |
| Accrued cost of goods sold | 8,411 | | | 10,180 | |
| Other accrued operating expenses | 9,301 | | | 3,953 | |
| Compensation and benefits | 28,811 | | | 21,682 | |
| | | |
| | | |
| Accrued royalties | 1,451 | | | 561 | |
| Accrued interest | 1,418 | | | 1,389 | |
| Product returns and wholesaler service fees | 10,609 | | | 8,526 | |
| Total | $ | 80,124 | | | $ | 64,243 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-22
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 10—DEBT
The carrying value of the Company’s outstanding debt is summarized as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2024 | | 2023 |
| Term loan A facility maturing March 2028 | $ | 104,211 | | | $ | 115,202 | |
| 2.125% Convertible senior notes due May 2029 | 279,334 | | | — | |
| 0.750% Convertible senior notes due August 2025 | 201,776 | | | 398,594 | |
3.375% Convertible senior notes due May 2024 (1) | — | | | 8,641 | |
| Total | $ | 585,321 | | | $ | 522,437 | |
(1) The 3.375% convertible senior notes due May 2024 matured and were repaid on May 1, 2024.
2028 Term Loan A Facility
On March 31, 2023, the Company entered into a credit agreement (as amended and/or restated to date, the “TLA Credit Agreement”) with JPMorgan Chase Bank, N.A., as administrative agent, and certain lenders, to refinance the indebtedness outstanding under the Company’s then-existing TLB Credit Agreement (as defined and discussed below). The term loan issued under the TLA Credit Agreement (the “TLA Term Loan”) was issued at a 0.30% discount and provides for a single-advance term loan A facility in the principal amount of $150.0 million, which is secured by substantially all of the Company’s and any subsidiary guarantor’s assets. Subject to certain conditions, the Company may, at any time, on one or more occasion, add one or more new classes of term facilities and/or increase the principal amount of the loans of any existing class by requesting one or more incremental term facilities. The net proceeds of the TLA Term Loan were approximately $149.6 million after deducting an original issue discount of $0.4 million.
On May 8, 2024, the Company, JPMorgan Chase Bank, N.A., as administrative agent, and certain lenders entered into a first amendment (the “First TLA Amendment”) to the TLA Credit Agreement. The First TLA Amendment, among other things, permits the Company’s $150.0 million share repurchase program and the Capped Call Transactions (as defined and described below).
The total debt composition of the TLA Term Loan is as follows (in thousands):
| | | | | | | | | | | |
| December 31, |
| 2024 | | 2023 |
| Term loan A facility maturing March 2028 | $ | 105,313 | | | $ | 116,563 | |
| Deferred financing costs | (821) | | | (988) | |
| Discount on debt | (281) | | | (373) | |
| Total debt, net of debt discount and deferred financing costs | $ | 104,211 | | | $ | 115,202 | |
The TLA Term Loan matures on March 31, 2028 and the TLA Credit Agreement requires quarterly repayments of principal in the amount of $2.8 million which commenced on June 30, 2023, increasing to $3.8 million commencing March 31, 2025, with a remaining balloon payment of approximately $85.3 million due at maturity. Due to voluntary principal prepayments, the Company is not required to make further principal payments until September 2026, although the Company retains the option to do so.
The TLA Credit Agreement requires the Company to, among other things, maintain (i) a Senior Secured Net Leverage Ratio (as defined in the TLA Credit Agreement), determined as of the last day of each fiscal quarter, of no greater than 3.00 to 1.00 and (ii) a Fixed Charge Coverage Ratio (as defined in the TLA Credit Agreement), determined as of the last day of each fiscal quarter, of no less than 1.50 to 1.00. The TLA Credit Agreement requires the Company to maintain an unrestricted cash and cash equivalents balance of at least $300.0 million ($500.0 million less a $200.0 million prepayment of the 2025 Notes in the year ended December 31, 2024) less any additional prepayments of the 2025 Notes (as defined below) at any time from 91 days prior to the maturity date through the earlier of (i) the latest maturity date of the 2025 Notes and (ii) the date on which there is no outstanding principal amount of the 2025 Notes. The TLA Credit Agreement also contains customary affirmative and negative covenants, financial covenants, representations and warranties, events of default and other provisions. As of December 31, 2024, the Company was in compliance with all financial covenants under the TLA Credit Agreement.
Pacira BioSciences, Inc. | 2024 Form 10-K | Page F-23
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The Company may elect to borrow either (i) alternate base rate borrowings or (ii) term benchmark borrowings or daily simple SOFR (as defined in the TLA Credit Agreement) borrowings. Each term loan borrowing that is an alternate base rate borrowing bears interest at a rate per annum equal to (i) the Alternate Base Rate (as defined in the TLA Credit Agreement), plus (ii) a spread based on the Company’s Senior Secured Net Leverage Ratio ranging from 2.00% to 2.75%. Each term loan borrowing that is a term benchmark borrowing or daily simple SOFR borrowing bears interest at a rate per annum equal to (i) the Adjusted Term SOFR Rate or Adjusted Daily Simple SOFR (as each is defined in the Credit Agreement), plus (ii) a spread based on the Company’s Senior Secured Net Leverage Ratio ranging from 3.00% to 3.75%. During the year ended December 31, 2024, the Company made $11.3 million of voluntary principal prepayments. During the year ended December 31, 2023, the Company made a scheduled principal payment of $2.8 million as well as $30.6 million of voluntary principal prepayments. As of December 31, 2024, borrowings under the TLA Term Loan consisted entirely of term benchmark borrowings at a rate of 7.43%.
2026 Term Loan B Facility
In December 2021, the Company entered into a term loan credit agreement (the “TLB Credit Agreement”) with JPMorgan Chase Bank, N.A., as administrative agent and the initial lender. The term loan issued under the TLB Credit Agreement (the “TLB Term Loan”) was issued at a 3.00% discount and allowed for a single-advance term loan B facility in the principal amount of $375.0 million, which was secured by substantially all of the Company’s and each subsidiary guarantor’s assets. The net proceeds of the TLB Term Loan were approximately $363.8 million after deducting an original issue discount of $11.2 million.
On March 31, 2023, the Company used the $149.6 million of net borrowings under the TLA Credit Agreement and cash on hand to repay the $296.9 million then-outstanding principal under the TLB Credit Agreement and concurrently terminated the TLB Credit Agreement, which resulted in a $16.9 million loss on early extinguishment of debt. The Company incurred a prepayment fee of 2.00% of the outstanding principal balance of the TLB Term Loan in connection with the termination.
Convertible Senior Notes Due 2029
In May 2024, the Company completed a private placement of $287.5 million in aggregate principal amount of its 2.125% convertible senior notes due 2029, or 2029 Notes, and entered into an indenture with Computershare Corporate Trust, N.A., or 2029 Indenture, with respect to the 2029 Notes. The 2029 Notes accrue interest at a fixed rate of 2.125% per year, payable semiannually in arrears on May 15th and November 15th of each year. The 2029 Notes mature on May 15, 2029.
The total debt composition of the 2029 Notes is as follows (in thousands):
| | | | | | | |
| December 31, |
| 2024 | | |
| 2.125% convertible senior notes due May 2029 | $ | 287,500 | | | |
| Deferred financing costs | (8,166) | | | |
| Total debt, net of deferred financing costs | $ | 279,334 | | | |
Holders may convert the 2029 Notes prior to the close of business on the business day immediately preceding November 15, 2028, only if certain circumstances are met, including, but not limited to, if during the previous calendar quarter, the last reported sales price of the Company’s common stock was greater than 130% of the conversion price then applicable for at least 20 out of the last 30 consecutive trading days of the quarter. During the quarter ended December 31, 2024, the conditions for conversion were not met. On or after November 15, 2028, until the close of business on the second scheduled trading day immediately preceding May 15, 2029, holders may convert their 2029 Notes at any time.
Upon conversion, holders will receive the principal amount of their 2029 Notes and any excess conversion value, calculated based on the per share volume-weighted average price for each of the 50 consecutive trading days during the observation period (as more fully described in the 2029 Indenture). For the principal, the Company will settle in cash per the terms of the 2029 Notes. For any excess conversion value, holders may receive cash, shares of the Company’s common stock or a combination of cash and shares of the Company’s common stock, at the Company’s option. The initial conversion rate for the 2029 Notes is 25.2752 shares of common stock per $1,000 principal amount, which is equivalent to an initial conversion price of $39.56 per share of the Company’s common stock. The conversion rate will be subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. The initial conversion price of the 2029 Notes represents a premium of approximately 32.5% to the closing sale price of $29.86 per share of the Company’s common stock on the Nasdaq Global Select Market on May 9, 2024, the date that the Company priced the private offering of the 2029 Notes.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-24
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
As of December 31, 2024, the 2029 Notes had a market price of $861 per $1,000 principal amount. In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2029 Notes will be paid pursuant to the terms of the 2029 Indenture. In the event that all of the 2029 Notes are converted, the Company would be required to repay the $287.5 million in principal value in cash, whereas any conversion premium would be required to be repaid in any combination of cash and shares of its common stock (at the Company’s option).
Prior to the close of business on the business day immediately preceding November 15, 2028, the 2029 Notes are convertible only under the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on June 30, 2024 (and only during such calendar quarter), if the last reported sale price of the Common Stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is equal to or greater than 130% of the conversion price on each applicable trading day; (2) during the five business-day period after any five consecutive trading-day period (the “measurement period”) in which the trading price per $1,000 principal amount of the 2029 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate on each such trading day; (3) upon the occurrence of specified corporate events; or (4) upon a Company redemption. On or after November 15, 2028, until the close of business on the second scheduled trading day immediately preceding May 15, 2029, holders of the 2029 Notes may convert all or a portion of their 2029 Notes, at any time. No sinking fund is provided for the 2029 Notes.
On or after May 17, 2027 and on or before the 50th scheduled trading day immediately before the maturity date, the Company may redeem for cash all or part of the 2029 Notes if (i) the 2029 Notes are “freely tradable” (as defined in the 2029 Indenture) and any accrued and unpaid additional interest has been paid as of the date the Company sends the related notice of the redemption and (ii) the last reported sales price of the Company’s common stock exceeds 130% of the conversion price then in effect for (1) each of at least 20 trading days (whether or not consecutive) during any 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related notice of the redemption; and (2) the trading day immediately before the date the Company sends such notice. The redemption price of each 2029 Note to be redeemed will be the principal amount of such 2029 Note, plus accrued and unpaid interest, if any. In addition, calling any 2029 Notes for redemption will constitute a make-whole fundamental change, in which case the conversion rate applicable to those 2029 Notes, if converted in connection with the redemption, will be increased in certain circumstances. Upon the occurrence of a “make-whole fundamental change” (as defined in the 2029 Indenture), subject to a limited exception for certain cash mergers, holders may require the Company to repurchase all or a portion of their 2029 Notes for cash at a price equal to 100% of the principal amount of the 2029 Notes to be repurchased plus any accrued and unpaid interest.
While the 2029 Notes are currently classified on the Company’s condensed consolidated balance sheet at December 31, 2024 as long-term debt, the future convertibility and resulting balance sheet classification of this liability is monitored at each quarterly reporting date and is analyzed dependent upon market prices of the Company’s common stock during the prescribed measurement periods. In the event that the holders of the 2029 Notes have the election to convert the 2029 Notes at any time during the prescribed measurement period, the 2029 Notes would then be considered a current obligation and classified as such.
On May 9, 2024, in connection with the pricing of the 2029 Notes, and on May 10, 2024, in connection with the exercise in full by the initial purchasers of the 2029 Notes (the “Initial Purchasers”) of their option to purchase additional 2029 Notes, the Company entered into privately negotiated capped call transactions (the “Capped Call Transactions”) with certain of the Initial Purchasers of the 2029 Notes and/or their respective affiliates and/or other financial institutions (the “Option Counterparties”). The Capped Call Transactions are expected to cover, subject to anti-dilution adjustments substantially similar to those applicable to the 2029 Notes, the number of shares of the Company’s common stock underlying the 2029 Notes.
The Capped Call Transactions are expected to reduce the potential dilution to the Company’s common stock upon any conversion of the 2029 Notes and/or offset any potential cash payments the Company is required to make in excess of the principal amount of converted 2029 Notes, as the case may be, upon any conversion of the 2029 Notes, with such reduction and/or offset subject to a cap. The cap price of the Capped Call Transactions will initially be approximately $53.75 per share, representing a premium of approximately 80% over the closing price of $29.86 per share of the Company’s common stock on May 9, 2024, and is subject to certain adjustments under the terms of the Capped Call Transactions. The capped call was recorded as a reduction to additional paid-in capital at its cost of $26.7 million, partially offset by a $6.5 million deferred tax asset.
The Capped Call Transactions are separate transactions entered into by the Company with the Option Counterparties, are not part of the terms of the 2029 Notes and will not affect any holder’s rights under the 2029 Notes. Holders of the 2029 Notes will not have any rights with respect to the Capped Call Transactions.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-25
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Convertible Senior Notes Due 2025
In July 2020, the Company completed a private placement of $402.5 million in aggregate principal amount of 0.750% convertible senior notes due 2025, or 2025 Notes, and entered into an indenture with Computershare Corporate Trust, N.A. (formerly Wells Fargo Bank, N.A.), or 2025 Indenture, with respect to the 2025 Notes. The 2025 Notes accrue interest at a fixed rate of 0.750% per year, payable semiannually in arrears on February 1st and August 1st of each year. The 2025 Notes mature on August 1, 2025.
In May 2024, the Company used part of the net proceeds from the issuance of the 2029 Notes to repurchase $200.0 million aggregate principal amount of the 2025 Notes in privately negotiated transactions at a discount for $191.4 million in cash (including accrued interest). The partial repurchase of the 2025 Notes resulted in a $7.5 million gain on early extinguishment of debt.
The total debt composition of the 2025 Notes is as follows (in thousands): | | | | | | | | | | | |
| December 31, |
| 2024 | | 2023 |
| 0.750% convertible senior notes due August 2025 | $ | 202,500 | | | $ | 402,500 | |
| Deferred financing costs | (724) | | | (3,906) | |
| | | |
| Total debt, net of deferred financing costs | $ | 201,776 | | | $ | 398,594 | |
| | | |
| | | |
| | | |
| | | |
The net proceeds from the issuance of the 2025 Notes were approximately $390.0 million, after deducting commissions and the offering expenses paid by the Company. A portion of the net proceeds from the 2025 Notes was used by the Company to repurchase $185.0 million in aggregate principal amount of its then-outstanding 2.375% convertible senior notes due 2022 in privately-negotiated transactions for a total of $211.1 million of cash (including accrued interest). The Company’s transaction costs of approximately $12.5 million related to the issuance of the 2025 Notes are amortized to interest expense over the five-year term of the 2025 Notes.
Holders were able to convert the 2025 Notes at any time prior to the close of business on the business day immediately preceding February 3, 2025, only under the following circumstances: (i) during any calendar quarter (and only during such calendar quarter), if the last reported sales price of the Company’s common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; (ii) during the five-business day period immediately after any five consecutive trading day period (the “measurement period”) in which the trading price (as defined in the 2025 Indenture) per $1,000 principal amount of notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of the Company’s common stock and the conversion rate on each such trading day; (iii) upon the occurrence of specified corporate events, including a merger or a sale of all or substantially all of the Company’s assets; or (iv) if the Company calls the 2025 Notes for redemption, until the close of business on the business day immediately preceding the redemption date. The conditions for conversion were not met during the calendar quarter ended December 31, 2024.
As of February 3, 2025, until the close of business on the second scheduled trading day immediately preceding August 1, 2025, holders may convert their 2025 Notes at any time.
Upon conversion, holders will receive the principal amount of their 2025 Notes and any excess conversion value, calculated based on the per share volume-weighted average price for each of the 40 consecutive trading days during the observation period (as more fully described in the 2025 Indenture). For both the principal and excess conversion value, holders may receive cash, shares of the Company’s common stock or a combination of cash and shares of the Company’s common stock, at the Company’s option. The initial conversion rate for the 2025 Notes is 13.9324 shares of common stock per $1,000 principal amount, which is equivalent to an initial conversion price of $71.78 per share of the Company’s common stock. The conversion rate will be subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. The initial conversion price of the 2025 Notes represents a premium of approximately 32.5% to the closing sale price of $54.17 per share of the Company’s common stock on the Nasdaq Global Select Market on July 7, 2020, the date that the Company priced the private offering of the 2025 Notes.
As of December 31, 2024, the 2025 Notes had a market price of $971 per $1,000 principal amount. In the event of conversion, holders would forgo all future interest payments, any unpaid accrued interest and the possibility of further stock price appreciation. Upon the receipt of conversion requests, the settlement of the 2025 Notes will be paid pursuant to the terms
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-26
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
of the 2025 Indenture. In the event that all of the 2025 Notes are converted, the Company would be required to repay the remaining $202.5 million in principal value and any conversion premium in any combination of cash and shares of its common stock (at the Company’s option).
Since August 1, 2023 (but, in the case of a redemption of less than all of the outstanding 2025 Notes, no later than the 40th scheduled trading day immediately before the maturity date), the Company may redeem for cash all or part of the 2025 Notes if the last reported sale price (as defined in the 2025 Indenture) of the Company’s common stock has been at least 130% of the conversion price then in effect for (i) each of at least 20 trading days (whether or not consecutive) during any 30 consecutive trading days ending on, and including, the trading day immediately before the date the Company sends the related notice of redemption and (ii) the trading day immediately before the date the Company sends such notice. The redemption price will equal the sum of (i) 100% of the principal amount of the 2025 Notes being redeemed, plus (ii) accrued and unpaid interest, including additional interest, if any, to, but excluding, the redemption date. In addition, calling the 2025 Notes for redemption will constitute a “make-whole fundamental change” (as defined in the 2025 Indenture) and will, in certain circumstances, increase the conversion rate applicable to the conversion of such notes if it is converted in connection with the redemption. No sinking fund is provided for the 2025 Notes.
If the Company undergoes a fundamental change, as defined in the 2025 Indenture, subject to certain conditions, holders of the 2025 Notes may require the Company to repurchase for cash all or part of their 2025 Notes at a repurchase price equal to 100% of the principal amount of the 2025 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. In addition, if a make-whole fundamental change occurs prior to August 1, 2025, the Company will, in certain circumstances, increase the conversion rate for a holder who elects to convert its notes in connection with the make-whole fundamental change.
The 2025 Notes are the Company’s general unsecured obligations that rank senior in right of payment to all of its indebtedness that is expressly subordinated in right of payment to the 2025 Notes, and equal in right of payment to the Company’s unsecured indebtedness. The 2025 Notes are also effectively junior in right of payment to any of the Company’s secured indebtedness to the extent of the value of the assets securing such indebtedness, and are structurally subordinated to any debt or other liabilities (including trade payables) of the Company’s subsidiaries.
Prior to January 1, 2022, under the previous ASC 470-20, Debt with Conversion and Other Options, an entity used to separately account for the liability and equity components of convertible debt instruments that may be settled entirely or partially in cash upon conversion in a manner that reflects the issuer’s economic interest cost. The liability component of the instrument used to be valued in a manner that reflected the market interest rate for a similar nonconvertible instrument at the date of issuance. The initial carrying value of the liability component of $314.7 million was calculated using a 5.78% assumed borrowing rate. The equity component of $87.8 million, which represented the conversion option, was determined by deducting the fair value of the liability component from the par value of the 2025 Notes and was recorded in additional paid-in capital on the consolidated balance sheet at the issuance date. The equity component used to be treated as a discount on the liability component of the 2025 Notes, which was amortized over the five-year term of the 2025 Notes using the effective interest rate method.
Resulting from ASU 2020-06, ASC 470-20 was revised effective January 1, 2022 which eliminated the requirement to separately account for the embedded conversion features that are not clearly and closely related to the debt, that meet the definition of a derivative and that do not qualify for the scope exception from derivative accounting and convertible debt instruments issued with substantial premiums for which the premiums were recorded as paid in capital. Effective January 1, 2022, the 2025 Notes debt discount carrying value of $64.7 million was eliminated and there was a $1.7 million increase in deferred financing costs offset by additional paid-in capital, accumulated deficit and deferred tax assets.
The 2025 Notes do not contain any financial or operating covenants or any restrictions on the payment of dividends, the issuance of other indebtedness or the issuance or repurchase of securities by the Company. The 2025 Indenture contains customary events of default with respect to the 2025 Notes, including that upon certain events of default, 100% of the principal and accrued and unpaid interest on the 2025 Notes will automatically become due and payable.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-27
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Convertible Senior Notes Due 2024 Assumed from the Flexion Acquisition
Prior to the Flexion Acquisition, in May 2017, Flexion issued an aggregate of $201.3 million principal amount of 3.375% convertible senior notes due May 1, 2024 (the “Flexion 2024 Notes”), pursuant to an indenture between Flexion and Computershare Corporate Trust, N.A. (formerly Wells Fargo Bank, N.A.), as trustee (the “Flexion Trustee”), as supplemented by the First Supplemental Indenture, dated as of November 19, 2021, between Flexion and the Flexion Trustee. Interest was payable semi-annually on May 1st and November 1st of each year. Upon the Flexion Acquisition, the principal was assumed and recorded at fair value by the Company.
In December 2021, as a result of the Flexion Acquisition and in accordance with the indenture governing the Flexion 2024 Notes, Flexion provided a Fundamental Change Company Notice (as defined in this indenture) and offered to repurchase all of the outstanding Flexion 2024 Notes at a repurchase price in cash equal to 100% of the principal amount plus accrued and unpaid interest.
On January 7, 2022, following the expiration of the offer to purchase, the Company accepted the $192.6 million aggregate principal amount of Flexion 2024 Notes that were validly tendered (and not validly withdrawn). No Flexion 2024 Notes were converted in connection with the Notice. The remaining principal of $8.6 million was repaid at maturity on May 1, 2024.
Convertible Senior Notes Due 2022
In March 2017, the Company completed a private placement of $345.0 million in aggregate principal amount of 2.375% convertible senior notes due 2022, or 2022 Notes, and entered into an indenture with respect to the 2022 Notes. On April 1, 2022, the 2022 Notes matured and the Company settled the remaining outstanding principal balance of $160.0 million and a conversion premium of $4.8 million through a cash payment of $156.9 million and the issuance of 101,521 shares of the Company’s common stock, which increased additional paid-in capital by $3.0 million.
Interest Expense
The following table sets forth the total interest expense recognized in the periods presented (dollar amounts in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Contractual and other interest expense | $ | 15,447 | | | $ | 20,082 | | | $ | 36,880 | |
| Amortization of debt issuance costs | 3,130 | | | 2,996 | | | 4,400 | |
| Amortization of debt discount | 92 | | | 752 | | | 2,807 | |
| Capitalized interest and other (Note 6) | (2,100) | | | (3,524) | | | (4,111) | |
| Total | $ | 16,569 | | | $ | 20,306 | | | $ | 39,976 | |
| | | | | |
| Effective interest rate on total debt | 2.97 | % | | 3.74 | % | | 5.47 | % |
NOTE 11—FINANCIAL INSTRUMENTS
Fair Value Measurements
Fair value is defined as the price that would be received to sell an asset or be paid to transfer a liability in the principal or most advantageous market in an orderly transaction. To increase consistency and comparability in fair value measurements, the FASB established a three-level hierarchy which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The three levels of fair value measurements are:
•Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
•Level 2: Observable prices that are based on inputs not quoted on active markets, but corroborated by market data.
•Level 3: Unobservable inputs that are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-28
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The carrying value of financial instruments including cash and cash equivalents, accounts receivable and accounts payable approximate their respective fair values due to the short-term nature of these items. The fair value of the Company’s convertible senior notes and its TLA Term Loan are calculated utilizing market quotations from an over-the-counter trading market for these notes (Level 2). The fair value of the Company’s acquisition-related contingent consideration is reported at fair value on a recurring basis (Level 3). The carrying amounts of equity investments and convertible notes receivable without readily determinable fair values have not been adjusted for either an impairment or upward or downward adjustments based on observable transactions, whereas an equity investment was fully impaired during the year ended December 31, 2022.
At December 31, 2024, the carrying values and fair values of the Company’s financial assets and liabilities were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Carrying
Value | | Fair Value Measurements Using |
| | | Level 1 | | Level 2 | | Level 3 |
| Financial Assets and Financial Liabilities Measured at Fair Value on a Recurring Basis: | | | | | | | | |
| Financial Assets: | | | | | | | | |
| Equity investments | | $ | 15,877 | | | $ | — | | | $ | — | | | $ | 15,877 | |
| Convertible notes receivable | | $ | 11,898 | | | $ | — | | | $ | — | | | $ | 11,898 | |
| Financial Liabilities: | | | | | | | | |
| Acquisition-related contingent consideration | | $ | 20,241 | | | $ | — | | | $ | — | | | $ | 20,241 | |
| | | | | | | | |
| Financial Liabilities Measured at Amortized Cost: | | | | | | | | |
| Term loan A facility due March 2028 | | $ | 104,211 | | | $ | — | | | $ | 104,786 | | | $ | — | |
2.125% convertible senior notes due 2029 (1) | | $ | 279,334 | | | $ | — | | | $ | 247,609 | | | $ | — | |
0.750% convertible senior notes due 2025 (2) | | $ | 201,776 | | | $ | — | | | $ | 196,678 | | | $ | — | |
(1) The closing price of the Company’s common stock as reported on the Nasdaq Global Select Market was $18.84 per share on December 31, 2024, compared to a conversion price of $39.56 per share. At December 31, 2024, as the conversion price was above the stock price, the requirements for conversion have not been met.
(2) The closing price of the Company’s common stock as reported on the Nasdaq Global Select Market was $18.84 per share on December 31, 2024, compared to a conversion price of $71.78 per share. At December 31, 2024, as the conversion price was above the stock price, the requirements for conversion have not been met. The maximum conversion on the principal that could have been due on the 2025 Notes is 2.8 million shares of the Company’s common stock, which assumes no increase in the conversion rate for certain corporate events.
Certain assets and liabilities are measured at fair value on a non-recurring basis, including assets and liabilities acquired in a business combination and long-lived assets, which would be recognized at fair value if deemed impaired or if reclassified as assets held for sale. The fair value in these instances would be determined using Level 3 inputs.
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
Equity and Convertible Note Investments
The Company holds strategic investments in clinical and preclinical stage privately-held biotechnology companies in the form of equity and convertible note investments. The following investments have no readily determinable fair value and are recorded at cost minus impairment, if any, plus or minus observable price changes of identical or similar investments (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Equity Investments | | Convertible Notes Receivable | | Total |
| Balance at December 31, 2022 | | $ | 15,877 | | | $ | 5,315 | | | $ | 21,192 | |
| Purchases | | — | | | 6,758 | | | 6,758 | |
| Foreign currency adjustments | | — | | | 61 | | | 61 | |
| Balance at December 31, 2023 | | 15,877 | | | 12,134 | | | 28,011 | |
| Foreign currency adjustments | | — | | | (236) | | | (236) | |
| Balance at December 31, 2024 | | $ | 15,877 | | | $ | 11,898 | | | $ | 27,775 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-29
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
During the year ended December 31, 2022, an impairment of an equity investment of $10.0 million was recorded in other, net in the consolidated statements of operations.
Acquisition-Related Contingent Consideration
The Company has recognized contingent consideration related to the Flexion Acquisition in the amount of $20.2 million and $24.7 million as of December 31, 2024 and 2023, respectively. Refer to Note 17, Contingent Consideration Gains, Restructuring Charges and Other, for more information.
The Company’s contingent consideration obligations are recorded at their estimated fair values and are revalued each reporting period if and until the related contingencies are resolved. The Company has measured the fair value of its contingent consideration using a Monte Carlo simulation. These inputs include, as applicable, estimated forecasts of revenue and costs and the discount rates used to calculate the present value of estimated future payments. Significant changes may increase or decrease the probabilities of achieving the related commercial and regulatory events, shorten or lengthen the time required to achieve such events, or increase or decrease estimated forecasts.
In November 2021, the Company completed the Flexion Acquisition, which provided for contingent consideration related to contingent value rights that were issued to Flexion shareholders and certain equity award holders which could aggregate up to a total of $372.3 million if certain regulatory and commercial milestones are met. The aggregate amount was initially $425.5 million prior to the Company’s September 2022 decision to formally discontinue further development of Flexion’s investigational product candidate, PCRX-301. The Company’s obligation to make milestone payments is limited to those milestones achieved through December 31, 2030, and are to be paid within 60 days of the end of the fiscal quarter of achievement. For the year ended December 31, 2024, the Company recorded gains of $4.5 million due to adjustments reflecting the probability of achieving the remaining Flexion regulatory milestone by December 31, 2030—the milestone expiration date, partially offset by revisions to the Company’s weighted average cost of capital and the latest discount rates. For the year ended December 31, 2023, the Company recorded gains of $3.4 million due to a decrease in the fair value of the Flexion contingent consideration. The decrease was primarily due to adjustments in the assumption for the long-term forecasts which reduced the probability of meeting the sales-based contingent consideration milestones by December 31, 2030—the expiration date for achieving the milestones. The impact of this assumption on the fair value was partially offset by a decrease to the assumed discount rate based on a significant improvement in the Company’s incremental borrowing rate resulting from the TLA Credit Agreement entered into in March 2023. These gains were recorded as contingent consideration gains, restructuring charges and other in the consolidated statements of operations. At December 31, 2024, the weighted average discount rate was 7.9%.
The following table includes the key assumptions used in the valuation of the Company’s contingent consideration: | | | | | | | | | | |
| Assumption | | Ranges Utilized as of December 31, 2024 | | |
| Discount rates | | 7.8% to 8.1% | | |
| Probability of payment for remaining regulatory milestones | | 0% | | |
The change in the Company’s contingent consideration recorded at fair value using Level 3 measurements is as follows (in thousands): | | | | | | | | |
| | Contingent
Consideration
Fair Value |
| Balance at December 31, 2022 | | $ | 28,122 | |
| | |
| Fair value adjustments and accretion | | (3,424) | |
| | |
| Balance at December 31, 2023 | | 24,698 | |
| Fair value adjustments and accretion | | (4,457) | |
| Balance at December 31, 2024 | | $ | 20,241 | |
Available-for-Sale Investments
Short-term investments consist of asset-backed securities collateralized by credit card receivables, investment grade commercial paper and corporate, federal agency, government and Yankee bonds with maturities greater than three months, but less than one year. Noncurrent investments consist of asset-backed securities collateralized by credit card receivables and contain maturities greater than one year but less than three years. Net unrealized gains and losses (excluding credit losses, if
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-30
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
any) from the Company’s short-term investments are reported in other comprehensive (loss) income. At December 31, 2024 and 2023, all of the Company’s short-term and noncurrent investments are classified as available-for-sale investments and are determined to be Level 2 instruments, with the exception of U.S. government bonds, which are measured at fair value using standard industry models with observable inputs. The fair value of the commercial paper is measured based on a standard industry model that uses the three-month U.S. Treasury bill rate as an observable input. The fair value of the asset-backed securities and corporate bonds is principally measured or corroborated by trade data for identical issues in which related trading activity is not sufficiently frequent to be considered a Level 1 input or that of comparable securities. The fair value of U.S. government bonds is based on level 1 trading activity. At the time of purchase, all available-for-sale investments had an “A” or better rating by Standard & Poor’s.
The following summarizes the Company’s available-for-sale investments at December 31, 2024 and 2023 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2024 Investments: | | Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | | | Fair Value
(Level 2) |
| Current: | | | | | | | | | | |
| Asset-backed securities | | $ | 21,626 | | | $ | 43 | | | $ | — | | | | | $ | 21,669 | |
| Commercial paper | | 142,556 | | | 120 | | | (55) | | | | | 142,621 | |
| Corporate bonds | | 32,502 | | | 25 | | | (5) | | | | | 32,522 | |
| U.S. federal agency bonds | | 5,996 | | | 8 | | | — | | | | | 6,004 | |
| | | | | | | | | | |
| Yankee bond | | 5,012 | | | 13 | | | — | | | | | 5,025 | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| Total | | $ | 207,692 | | | $ | 209 | | | $ | (60) | | | | | $ | 207,841 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| December 31, 2023 Investments: | | Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value
(Level 1) | | Fair Value
(Level 2) |
| Current: | | | | | | | | | | |
| Asset-backed securities | | $ | 9,539 | | | $ | 1 | | | $ | — | | | $ | — | | | $ | 9,540 | |
| Commercial paper | | 77,941 | | | 103 | | | — | | | — | | | 78,044 | |
| | | | | | | | | | |
| U.S. federal agency bonds | | 22,849 | | | — | | | (29) | | | — | | | 22,820 | |
| U.S. government bonds | | 14,899 | | | — | | | (20) | | | 14,879 | | | — | |
| Subtotal | | 125,228 | | | 104 | | | (49) | | | 14,879 | | | 110,404 | |
| Noncurrent: | | | | | | | | | | |
| Asset-backed securities | | 2,403 | | | 7 | | | — | | | — | | | 2,410 | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
| Subtotal | | 2,403 | | | 7 | | | — | | | — | | | 2,410 | |
| Total | | $ | 127,631 | | | $ | 111 | | | $ | (49) | | | $ | 14,879 | | | $ | 112,814 | |
At December 31, 2024, there were no investments available for sale that were materially less than their amortized cost.
The Company elects to recognize its interest receivable separate from its available-for-sale investments. At December 31, 2024 and 2023, the interest receivable from its available-for-sale investments recognized in prepaid expenses and other current assets was $0.5 million and $0.4 million, respectively.
Credit Risk
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents, short-term and long-term available-for-sale investments and accounts receivable. The Company maintains its cash and cash equivalents with high-credit quality financial institutions. Such amounts may exceed federally-insured limits.
As of December 31, 2024, three wholesalers each accounted for over 10% of the Company’s accounts receivable, at 34%, 18% and 16%. As of December 31, 2023, three wholesalers each accounted for over 10% of the Company’s accounts receivable, at 37%, 19% and 16%. For additional information regarding the Company’s wholesalers, see Note 2, Summary of Significant Accounting Policies. EXPAREL and ZILRETTA revenues are primarily derived from major wholesalers and specialty distributors that generally have significant cash resources. The Company performs ongoing credit evaluations of its customers as warranted and generally does not require collateral. Allowances for credit losses on the Company’s accounts receivable are maintained based on historical payment patterns, current and estimated future economic conditions, aging of
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-31
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
accounts receivable and its write-off history. As of December 31, 2024, there were $0.4 million of allowances for credit losses on its accounts receivable. As of December 31, 2023, the Company did not deem any allowances for credit losses on its accounts receivable necessary.
NOTE 12—STOCKHOLDERS’ EQUITY
Common Stock
The Company is authorized to issue up to 250,000,000 shares of common stock, of which 47,077,844 shares were issued and 46,240,604 shares were outstanding at December 31, 2024 and 46,481,174 shares were issued and outstanding at December 31, 2023.
Preferred Stock
The Company is authorized to issue up to 5,000,000 shares of preferred stock. No preferred stock was issued or outstanding at either December 31, 2024 or 2023.
Accumulated Other Comprehensive Income (Loss)
The following table illustrates the changes in the balances of the Company’s accumulated other comprehensive income (loss) for the periods presented (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | Net Unrealized Gains
(Losses) From Available-For-Sale Investments | | Unrealized Foreign Currency Translation | | Accumulated Other Comprehensive Income (Loss) |
| | | | | | |
| Balance at December 31, 2021 | | $ | 139 | | | $ | 28 | | | $ | 167 | |
Net unrealized loss on investments, net of tax (1) | | (662) | | | — | | | (662) | |
| Foreign currency translation adjustments | | — | | | 115 | | | 115 | |
| | | | | | |
| Balance at December 31, 2022 | | (523) | | | 143 | | | (380) | |
Net unrealized gain on investments, net of tax (1) | | 647 | | | — | | | 647 | |
| Foreign currency translation adjustments | | — | | | (20) | | | (20) | |
| | | | | | |
| Balance at December 31, 2023 | | 124 | | | 123 | | | 247 | |
Net unrealized gain on investments, net of tax (1) | | 66 | | | — | | | 66 | |
| Foreign currency translation adjustments | | — | | | 30 | | | 30 | |
| | | | | | |
| Balance at December 31, 2024 | | $ | 190 | | | $ | 153 | | | $ | 343 | |
(1) Net of a nominal tax expense for the year ended December 31, 2024, and a $(0.2) million and $0.2 million tax (expense) benefit for the years ended December 31, 2023 and 2022, respectively.
Stock Repurchase Program
On May 7, 2024, the Company announced that its board of directors approved a share repurchase program, effective immediately, which authorizes the Company to repurchase up to an aggregate of $150.0 million of its outstanding common stock. Repurchases under this program may be made at management’s discretion on the open market or through privately negotiated transactions. The share repurchase program may be suspended or discontinued at any time by the Company and has an expiration date of December 31, 2026.
On May 9, 2024, concurrently with the pricing of the offering of the 2029 Notes, the Company entered into separate, privately negotiated agreements with certain of the initial purchasers of the 2029 Notes or their respective affiliates and/or certain other financial institutions to repurchase 837,240 shares of the Company’s common stock for a total cost of $25.1 million, inclusive of $0.1 million of accrued excise tax. The repurchase occurred on May 10, 2024.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-32
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 13—STOCK PLANS
Stock Incentive Plans
The Pacira BioSciences, Inc. Amended and Restated 2011 Stock Incentive Plan, or 2011 Plan, was originally adopted by its board of directors and approved by its stockholders in June 2011 and was amended and restated in June 2014, June 2016, June 2019, June 2021 and June 2023. The June 2023 amendment and restatement and approval by the Company’s stockholders increased the number of shares of common stock authorized for issuance as equity awards under the 2011 Plan by 3,300,000 shares, which allows for the granting of incentive stock options, non-statutory stock options, restricted stock units and other stock-based awards.
In April 2014, the Company’s board of directors approved and adopted the Company’s 2014 Inducement Plan (the “2014 Inducement Plan”), pursuant to which awards could be made to new employees under the 2014 Inducement Plan for up to 175,000 shares of the Company’s common stock as a material inducement to such persons entering into employment with the Company.
In December 2023, the board of directors, upon recommendation of the compensation committee of the board of directors, adopted the Pacira BioSciences, Inc. Amended and Restated 2014 Inducement Plan (as amended and restated, the “First A&R Inducement Plan”) such that, among other things, an additional 642,093 shares of the Company’s common stock would be available for grant. In September 2024, the board of directors, upon the recommendation of the compensation committee of the board of directors, adopted the Pacira BioSciences, Inc. Amended and Restated 2014 Inducement Plan (as amended and restated, the “Second A&R Inducement Plan”) to add an additional 707,907 shares of the Company’s common stock to bring the total amount of shares reserved for issuance under the Inducement Plan to 1,525,000, of which, as of December 31, 2024, 125,107 shares remained available for issuance, and the term of the Inducement Plan was extended such that it will now expire on September 3, 2034.
In January 2025, the board of directors, upon the recommendation of the compensation committee of the board of directors, adopted the Pacira BioSciences, Inc. Amended and Restated 2014 Inducement Plan (as amended and restated to date, the “Inducement Plan”) to add an additional 785,000 shares of the Company’s common stock to bring the total amount of shares reserved for issuance under the Inducement Plan to 2,310,000 and extend the term of the Inducement Plan such that it will now expire on January 18, 2035. The Inducement Plan allows for the granting of nonstatutory stock options, restricted stock awards and other stock-based awards.
The Inducement Plan was adopted by the board of directors without stockholder approval pursuant to Rule 5635(c)(4) of the Nasdaq Listing Rules. In accordance with Rule 5635(c)(4) of the Nasdaq Listing Rules, awards under the Inducement Plan may only be made to an employee who has not previously been an employee or member of the board of directors or the board of directors or any parent or subsidiary, or following a bona fide period of non-employment by the Company or a parent or subsidiary, if he or she is granted such award in connection with his or her commencement of employment with the Company or a subsidiary and such grant is an inducement material to his or her entering into employment with the Company or such subsidiary.
Inducement Awards
From time to time, the board of directors, upon recommendation of the compensation committee, has approved individually negotiated grants of options and restricted stock units for certain of the Company’s officers in connection with their respective appointments, in each case, pursuant to the inducement plan in effect at such time.
Equity Grants
The Company’s stock option grants have an exercise price equal to the closing price of the Company’s common stock on the date of grant, generally have a 10-year contractual term and vest in increments (typically over four years from the date of grant, although the Company may occasionally grant stock options with different vesting terms, including grants made to its non-employee directors). The Company also grants RSUs to employees generally vesting in equal, annual increments over four years from the date of grant, except for such grants made to non-employees and non-employee directors. The Company uses authorized but unissued shares of its common stock to satisfy its obligations under these plans.
Employee Stock Purchase Plan
The Company’s Amended and Restated 2014 Employee Stock Purchase Plan, or ESPP, was originally adopted by its board of directors in April 2014, approved by the Company’s stockholders in June 2014 and amended and restated in June
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-33
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
2022. The June 2022 amendment and restatement increased the number of shares of common stock that may be sold under the plan by an additional 500,000 shares from the originally provided 500,000 shares. The purpose of the ESPP is to provide a vehicle for eligible employees to purchase shares of the Company’s common stock at a discounted price and to help retain and motivate current employees as well as attract new talent. Under the ESPP, up to 1,000,000 shares of common stock may be sold. The ESPP expires in June 2032. The ESPP is intended to qualify as an “employee stock purchase plan” within the meaning of Section 423 of the Internal Revenue Code, or IRC. The maximum fair market value of stock which can be purchased by a participant in a calendar year is $25,000. Six-month offering periods begin on January 1st and July 1st of each year. During an offering period, eligible employees have the opportunity to elect to purchase shares of the Company’s common stock on the purchase dates of June 30 and December 31 (or the last trading day of an offering period). The per share purchase price is equal to 85% of the fair market value of the Company’s common stock on either the offering date or the purchase date, whichever is lesser. During the year ended December 31, 2024, 114,405 shares were purchased and issued through the ESPP.
The following tables contain information about the Company’s stock incentive plans at December 31, 2024:
| | | | | | | | | | | | | | | | | | | | |
| Stock Incentive Plan | | Awards Reserved For Issuance | | Awards
Issued | | Awards Available For Grant |
| 2011 Plan | | 17,731,701 | | | 15,659,387 | | | 2,072,314 | |
2014 Inducement Plan (1) | | 1,525,000 | | | 1,399,893 | | | 125,107 | |
| Total | | 19,256,701 | | | 17,059,280 | | | 2,197,421 | |
| | | | | | |
| Employee Stock Purchase Plan | | Shares Reserved
For Purchase | | Shares
Purchased | | Shares Available
For Purchase |
| ESPP | | 1,000,000 | | | 685,071 | | | 314,929 | |
(1) In January 2025, the board of directors approved an amendment to the 2014 Inducement Plan to authorize an additional 785,000 shares reserved for issuance.
Stock-Based Compensation
Compensation expense for stock options and RSUs is based on the estimated grant date fair value of an award recognized over the requisite service period on a straight-line expense attribution method. Compensation expense for ESPP share options is based on the estimated grant date fair value of the ESPP shares and the number of shares that can be purchased as of the grant date, which is recognized as expense on a straight-line expense attribution method over the length of an offering period.
The Company recognized stock-based compensation expense in its consolidated statements of operations for the years ended December 31, 2024, 2023 and 2022 as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| | | 2024 | | 2023 | | 2022 |
| Cost of goods sold | | $ | 5,331 | | | $ | 5,537 | | | $ | 5,967 | |
| Research and development | | 7,381 | | | 8,694 | | | 6,594 | |
| Selling, general and administrative | | 34,857 | | | 33,664 | | | 35,531 | |
| Contingent consideration gains, restructuring charges and other | | 3,602 | | | — | | | — | |
| Total | | $ | 51,171 | | | $ | 47,895 | | | $ | 48,092 | |
| | | | | | |
| Stock-based compensation from: | | | | | | |
| Stock options | | $ | 21,730 | | | $ | 24,005 | | | $ | 26,800 | |
| RSUs | | 28,656 | | | 22,974 | | | 20,310 | |
| ESPP share options | | 785 | | | 916 | | | 982 | |
| Total | | $ | 51,171 | | | $ | 47,895 | | | $ | 48,092 | |
| | | | | | |
| Related income tax benefit | | $ | 10,506 | | | $ | 10,186 | | | $ | 10,219 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-34
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
The following table summarizes the Company’s stock option activity and related information for the year ended December 31, 2024:
| | | | | | | | | | | | | | | | | | | | | | | | |
| Number of
Stock Options | | Weighted
Average
Exercise Price (Per Share) | | Weighted Average
Remaining
Contractual
Term (Years) | | Aggregate
Intrinsic Value
(in Thousands) | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
| Outstanding at December 31, 2023 | 7,079,748 | | | $ | 49.40 | | | 6.03 | | $ | 863 | | |
| Granted | 1,472,223 | | | 27.81 | | | | | | |
| | | | | | | | |
| Forfeited | (292,061) | | | 44.61 | | | | | | |
| Expired | (1,414,292) | | | 59.15 | | | | | | |
| Outstanding at December 31, 2024 | 6,845,618 | | | $ | 42.95 | | | 5.72 | | $ | 804 | | |
| Exercisable at December 31, 2024 | 4,420,578 | | | $ | 47.50 | | | 4.32 | | $ | — | | |
| Vested and expected to vest as of December 31, 2024 | 6,845,618 | | | $ | 42.95 | | | 5.72 | | $ | 804 | | |
As of December 31, 2024, $30.8 million of total unrecognized compensation cost related to unvested stock options is expected to be recognized over a remaining weighted average period of 2.4 years. The Company’s stock options have a maximum expiration date of ten years from the date of grant.
The weighted average fair value of stock options granted for the years ended December 31, 2024, 2023 and 2022 was $11.77, $15.92 and $25.60 per share, respectively. The fair values of stock options granted were estimated using the Black-Scholes model with the following weighted average assumptions:
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| Black-Scholes Weighted Average Assumption | | 2024 | | 2023 | | 2022 |
| Expected dividend yield | | None | | None | | None |
| Risk-free interest rate | | 3.55% - 4.58% | | 3.05% - 4.81% | | 1.37% - 4.17% |
| Expected volatility | | 40.6% | | 41.3% | | 45.1% |
| Expected term of options | | 5.15 years | | 4.90 years | | 4.92 years |
The following table summarizes the Company’s RSU activity and related information for the year ended December 31, 2024:
| | | | | | | | | | | | | | | | | |
| | Number of
Restricted Stock Units | | Weighted
Average Grant
Date Fair Value (Per Share) | | Aggregate
Intrinsic Value
(in Thousands) |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| | | | | |
| Unvested at December 31, 2023 | 1,364,618 | | | $ | 47.66 | | | $ | 46,042 | |
| Granted | 2,198,365 | | | 27.29 | | | |
| Vested | (500,785) | | | 48.78 | | | |
| Forfeited | (292,470) | | | 40.04 | | | |
| Unvested and expected to vest as of December 31, 2024 | 2,769,728 | | | $ | 32.07 | | | $ | 52,182 | |
As of December 31, 2024, $70.6 million of total unrecognized compensation cost related to unvested RSUs is expected to be recognized over a weighted average period of 1.7 years. The Company’s RSUs have a maximum vest date of four years from the date of grant. The fair values of RSUs awarded are equal to the closing price of the Company’s common stock on the date of grant.
The fair values of the ESPP share options granted were estimated using the Black-Scholes model with the following weighted average assumptions:
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-35
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
| | | | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| Black-Scholes Weighted Average Assumption | | 2024 | | 2023 | | 2022 |
| ESPP share option fair value | | $8.20 - $8.25 | | $10.00 - $10.34 | | $15.26 - $15.86 |
| Expected dividend yield | | None | | None | | None |
| Risk-free interest rate | | 5.33% - 5.26% | | 4.77% - 5.53% | | 0.22% - 2.52% |
| Expected volatility | | 40.5% | | 35.4% | | 39.5% |
| Expected term of ESPP share options | | 6 months | | 6 months | | 6 months |
NOTE 14—NET (LOSS) INCOME PER COMMON SHARE
Potential common shares are excluded from the diluted net (loss) income per common share computation to the extent that they would be antidilutive. Since the Company reported a net loss for the year ended December 31, 2024, no potentially dilutive securities were included in the computation of diluted net loss per share for that period. As discussed in Note 10, Debt, the Company has the option to pay cash for the aggregate principal amount due upon the conversion of its 2025 Notes. For additional information on the Company’s computation of its net (loss) income per common share, see Note 2, Summary of Significant Accounting Policies.
The following table sets forth the computation of basic and diluted net (loss) income per common share for the years ended December 31, 2024, 2023 and 2022 (in thousands, except per common share amounts):
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Numerator: | | | | | |
| Net (loss) income—basic | $ | (99,560) | | | $ | 41,955 | | | $ | 15,909 | |
| 2025 Notes if-converted method adjustment | — | | | 4,114 | | | — | |
| | | | | |
| | | | | |
| | | | | |
| Adjusted net (loss) income—diluted | $ | (99,560) | | | $ | 46,069 | | | $ | 15,909 | |
| Denominator: | | | | | |
| Weighted average common shares outstanding—basic | 46,245 | | | 46,222 | | | 45,521 | |
| Computation of diluted securities: | | | | | |
| 2025 Notes if-converted method adjustment | — | | | 5,608 | | | — | |
| Dilutive effect of stock options | — | | | 51 | | | 787 | |
| Dilutive effect of RSUs | — | | | 96 | | | 226 | |
| | | | | |
| Dilutive effect of ESPP share options | — | | | 2 | | | 4 | |
| Weighted average common shares outstanding—diluted | 46,245 | | | 51,979 | | | 46,538 | |
| Net (loss) income per common share: | | | | | |
| Basic net (loss) income per common share | $ | (2.15) | | | $ | 0.91 | | | $ | 0.35 | |
| Diluted net (loss) income per common share | $ | (2.15) | | | $ | 0.89 | | | $ | 0.34 | |
The following table summarizes the outstanding stock options, RSUs, ESPP share options and convertible senior notes that were excluded from the diluted net (loss) income per common share calculation because the effects of including these potential shares were antidilutive in the periods presented (in thousands): | | | | | | | | | | | | | | | | | |
| | Year Ended December 31, |
| | 2024 | | 2023 | | 2022 |
| Weighted average number of stock options | 7,217 | | | 6,251 | | | 2,821 | |
| 2025 Notes | 3,843 | | | — | | | 6,206 | |
| Weighted average number of RSUs | 2,139 | | | 1,033 | | | 417 | |
| Weighted average ESPP share options | 54 | | | 20 | | | 7 | |
| Total | 13,253 | | | 7,304 | | | 9,451 | |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-36
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 15—INCOME TAXES
(Loss) income before income taxes and the related tax expense (benefit) is as follows (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| (Loss) income before income taxes: | | | | | |
| Domestic | $ | (65,325) | | | $ | 66,257 | | | $ | 21,068 | |
| Foreign | 2,219 | | | (4,556) | | | (7,766) | |
| Total (loss) income before income taxes | $ | (63,106) | | | $ | 61,701 | | | $ | 13,302 | |
| | | | | |
| Current taxes: | | | | | |
| Federal | $ | 9,295 | | | $ | 1,686 | | | $ | — | |
| State | 6,527 | | | 2,444 | | | 5,309 | |
| Foreign | 11 | | | 1 | | | 29 | |
| Total current taxes | $ | 15,833 | | | $ | 4,131 | | | $ | 5,338 | |
| Deferred taxes: | | | | | |
| Federal | $ | 18,784 | | | $ | 16,790 | | | $ | (2,781) | |
| State | 1,837 | | | (1,175) | | | (5,164) | |
| | | | | |
| Total deferred taxes | $ | 20,621 | | | $ | 15,615 | | | $ | (7,945) | |
| | | | | |
| Total income tax expense (benefit) | $ | 36,454 | | | $ | 19,746 | | | $ | (2,607) | |
A reconciliation of income tax expense (benefit) at the U.S. federal statutory rate to the provision for income taxes is as follows (dollars in thousands): | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, |
| | | 2024 | | 2023 | | 2022 |
| | Amount | | Tax Rate | | Amount | | Tax Rate | | Amount | | Tax Rate |
| U.S. statutory rate applied to (loss) income before taxes | | $ | (13,252) | | | 21.00 | % | | $ | 12,957 | | | 21.00 | % | | $ | 2,793 | | | 21.00 | % |
| State taxes | | 7,028 | | | (11.14) | % | | 1,770 | | | 2.87 | % | | 508 | | | 3.82 | % |
| Foreign taxes | | (104) | | | 0.16 | % | | (1,798) | | | (2.91) | % | | 248 | | | 1.86 | % |
| Change in valuation allowance | | (589) | | | 0.93 | % | | 2,192 | | | 3.55 | % | | 2,871 | | | 21.58 | % |
| Executive compensation | | 2,426 | | | (3.84) | % | | 3,171 | | | 5.14 | % | | 2,188 | | | 16.45 | % |
| Stock-based compensation | | 9,918 | | | (15.72) | % | | 4,070 | | | 6.60 | % | | (2,715) | | | (20.41) | % |
| Tax credits | | (5,674) | | | 8.99 | % | | (3,327) | | | (5.39) | % | | (3,245) | | | (24.39) | % |
| | | | | | | | | | | | |
| Reserves | | 2,661 | | | (4.22) | % | | 389 | | | 0.63 | % | | 984 | | | 7.40 | % |
| Goodwill impairment | | 34,281 | | | (54.32) | % | | — | | | — | % | | — | | | — | % |
| Non-Taxable or Nondeductible Items: | | | | | | | | | | | | |
| Interest expense | | — | | | — | % | | — | | | — | % | | (3,477) | | | (26.14) | % |
| Contingent consideration | | (936) | | | 1.48 | % | | (719) | | | (1.17) | % | | (3,841) | | | (28.88) | % |
| Nondeductible expenses | | 564 | | | (0.89) | % | | 975 | | | 1.58 | % | | 1,164 | | | 8.75 | % |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| Other | | 131 | | | (0.20) | % | | 66 | | | 0.10 | % | | (85) | | | (0.64) | % |
| Income tax expense (benefit) and effective tax rate | | $ | 36,454 | | | (57.77) | % | | $ | 19,746 | | | 32.00 | % | | $ | (2,607) | | | (19.60) | % |
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-37
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Deferred taxes reflect the tax effects of the differences between the amounts recorded as assets and liabilities for financial reporting purposes and the comparable amounts recorded for income tax purposes. At each reporting date, the Company considers new evidence, both positive and negative, that could affect its view of the future realization of deferred tax assets. The Company records a valuation allowance on U.S. capital losses and on foreign net deferred tax balances as it is more-likely-than-not the tax benefits are not realizable.
Significant components of the Company’s deferred tax assets and liabilities at December 31, 2024 and 2023 are as follows (in thousands):
| | | | | | | | | | | |
| | December 31, |
| | 2024 | | 2023 |
| Deferred tax assets: | | | |
| Net operating loss carryforwards | $ | 124,536 | | | $ | 132,265 | |
| Federal and state credits | 12,351 | | | 24,018 | |
| Accruals and reserves | 16,114 | | | 18,936 | |
| Stock based compensation | 23,078 | | | 27,520 | |
| | | |
| Inventory reserves | 2,639 | | | 2,149 | |
| Interest | 5,716 | | | — | |
| Other | 5,126 | | | 7,143 | |
| Total deferred tax assets | 189,560 | | | 212,031 | |
| Deferred tax liabilities: | | | |
| Depreciation and amortization | (34,368) | | | (42,026) | |
| | | |
| Total deferred tax liabilities | (34,368) | | | (42,026) | |
| Deferred tax assets, net of deferred tax liabilities | 155,192 | | | 170,005 | |
| Less: valuation allowance | (24,816) | | | (25,520) | |
| Net deferred tax assets | $ | 130,376 | | | $ | 144,485 | |
On May 9, 2024, and on May 10, 2024, the Company entered into privately negotiated Capped Call Transactions related to the 2029 Notes. See Note 10, Debt, for further discussion of the Capped Call Transactions. The capped call was recorded as a reduction to additional paid-in-capital at its cost of $26.7 million. A related deferred tax asset of $6.5 million was recorded with an offset to additional paid-in-capital and will be amortized as a current tax deduction over a 60-month period. As of December 31, 2024, $5.7 million of the related capped call deferred tax asset remains in the Company’s deferred tax balances.
As of December 31, 2024, the Company’s federal net operating losses, or NOLs, and federal tax credit carryforwards totaled $451.2 million and $4.9 million, respectively. The Company also had state NOLs and state tax credit carryforwards of $499.4 million and $7.5 million, respectively, which are subject to change on an annual basis due to variations in the Company’s annual state apportionment factors. The state NOLs will begin to expire in 2028. The Company had non-U.S. NOLs of $3.5 million at December 31, 2024, which do not expire.
Since the Company had cumulative changes in ownership of more than 50% within a three-year period, under IRC sections 382 and 383, the Company’s ability to use certain NOLs, tax attributes and credit carryforwards to offset taxable income or tax will be limited. Such ownership changes were triggered by the initial acquisition of the Company’s stock in 2007 as well as cumulative ownership changes arising as a result of the completion of the Company’s initial public offering, other financing transactions and the Flexion Acquisition in 2021. As a result of these ownership changes, the Company has $451.2 million of federal NOLs subject to annual limitations and are available as follows: $32.5 million will become available in 2025, $28.3 million in 2026, $6.9 million in 2027 and thereafter.
In accordance with ASC Topic 740, the Company establishes a valuation allowance for deferred tax assets that, in its judgment, are not more-likely-than-not realizable. These judgments are based on projections of future income—including tax-planning strategies—by individual tax jurisdictions. In each reporting period, the Company assesses the likelihood that its deferred tax assets will be realized and determines if adjustments to its valuation allowance are appropriate. The Company had a net decrease in its valuation allowance of $0.7 million in the year ended December 31, 2024 and a net increase of $2.6 million for the year ended December 31, 2023. The $0.7 million net decrease in the current year valuation allowance is primarily
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-38
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
related to foreign net deferred tax assets. The Company continues to maintain a full valuation allowance against foreign net deferred tax assets since it is more-likely-than-not the tax benefit related to the foreign losses are not realizable.
In 2024, the Company recorded a $2.7 million net increase to unrecognized tax benefits, or UTBs of which a $1.5 million increase is related to tax credit and filing positions taken during the year, and a $1.2 million increase related to prior year tax credit and filing positions. The Company’s UTB liability at December 31, 2024 was $9.4 million. The change in the Company’s UTBs for the year ended December 31, 2024 is summarized as follows (in thousands):
| | | | | | | | |
| | Unrecognized
Tax Benefit |
| | |
| Balance at December 31, 2022 | | $ | 6,322 | |
| | |
| | |
| | |
| Additions for current year positions | | 553 | |
| Reduction for prior year positions | | (164) | |
| | |
| Balance at December 31, 2023 | | 6,711 | |
| Additions for prior year positions | | 1,189 | |
| | |
| | |
| Additions for current year positions | | 1,472 | |
| | |
| | |
| Balance at December 31, 2024 | | $ | 9,372 | |
The UTBs as of December 31, 2024, 2023 and 2022 would, if subsequently recognized, favorably impact the effective income tax rate.
The Company regularly assesses the likelihood of additional tax assessments by jurisdiction and, if necessary, adjusts its reserve for UTBs based on new information or developments. Of the UTB balance at December 31, 2024, $4.3 million was recorded as a reduction to the Company’s deferred tax assets. The remaining $5.1 million of the UTB balance at December 31, 2024 was recorded to long-term income taxes payable within other liabilities on the consolidated balance sheet and relates to both the utilization of tax credits and filing positions. Any potential deficiency would not result in a tax liability, therefore, no interest or penalties were recognized in income tax expense for the years ended December 31, 2024, 2023 and 2022 for positions recorded to the Company’s deferred tax assets and long-term income taxes payable.
The Company is currently subject to audit by the U.S. Internal Revenue Service, or IRS, for the years 2019 through 2024, and state tax jurisdictions for the years 2018 through 2024. However, the IRS or states may still examine and adjust an NOL arising from a closed year to the extent it is utilized in a year that remains subject to audit. The Company’s previously filed income tax returns are not presently under audit by the IRS. The Company is under a state audit for the years 2020 through 2021.
As of December 31, 2024, $0.7 million of current federal income taxes payable were recorded to other accrued operating expenses within the accrued expenses line item of the consolidated balance sheets.
NOTE 16—EMPLOYEE BENEFIT PLANS
401(k) Salary Savings Plan
The Company’s 401(k) Salary Savings Plan, or 401(k) Plan, is a deferred salary arrangement under section 401(k) of the IRC. Under the 401(k) Plan, participating U.S. employees may defer a portion of their pre-tax earnings which are eligible for a discretionary percentage match as defined in the 401(k) Plan and determined by the Company’s board of directors (up to the maximum amount permitted by the IRC). The Company recognized $5.8 million, $4.8 million and $3.4 million of related compensation expense for its 401(k) Plan discretionary match for the years ended December 31, 2024, 2023 and 2022, respectively.
Deferred Compensation Plan
The Company intends that its Deferred Compensation Plan, or DCP, constitute, and be construed and administered as, an unfunded plan of deferred compensation within the meaning of the Employee Retirement Income Security Act of 1974, as amended, and the IRC of 1986, as amended, under which eligible participants may elect to defer the receipt of current compensation. Eligible participants include select management and highly compensated employees of the Company, including the Company’s named executive officers. Pursuant to the DCP, subject to any minimum and maximum deferral requirements that the administrator of the DCP may establish, participants may elect to defer their base salary and annual incentive awards. In
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-39
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
addition to elective deferrals, the DCP permits the Company to make matching and certain other discretionary contributions to the participants. The company contributes assets to a rabbi trust to accumulate funds to pay benefits under the DCP. Funds held in the rabbi trust must be used to pay benefits to DCP participants, except in the case of the Company’s bankruptcy or insolvency, in which case, they become subject to claims by the Company’s creditors. The Company recognized a $0.1 million credit to the related compensation expense for the Company’s discretionary match due to forfeitures related to the departure of executives exceeding its discretionary match for the year ended December 31, 2024. The Company recognized $0.3 million of related compensation expense for its DCP discretionary match for each of the years ended December 31, 2023 and 2022. The carrying value of assets held in the rabbi trust equaled the DCP liability as of both December 31, 2024 and 2023. As of December 31, 2024, the carrying value of the assets held in the rabbi trust was $7.2 million, of which $1.0 million is classified as current. As of December 31, 2023, the carrying value of the assets held in the rabbi trust was $6.9 million, of which $0.7 million was classified as current. The rabbi trust’s current and noncurrent assets are classified within prepaid expenses and other assets and investments and other assets, respectively, within the consolidated balance sheets. The DCP current and noncurrent liabilities are classified within accrued expenses and other liabilities, respectively, within the consolidated balance sheets.
Cash Long-Term Incentive Plan
The Company’s cash long-term incentive plan, or LTIP, is focused on pre-determined and objective performance goals during each applicable performance period from January 1 through December 31 of each calendar year. Award amounts ranging from 0% to 225% of the target cash award can be earned based on achievement of two equally weighted financial metrics: net revenue and adjusted earnings before interest, taxes, depreciation and amortization (EBITDA), with a relative total shareholder return modifier based on the Company’s stock price performance relative to the companies comprising the S&P Pharmaceuticals Select Industry Index. The performance period for these metrics is one year, with an additional three years of time-vesting following the performance period. For the years ended December 31, 2024 and 2023, the Company recognized $1.8 million and $0.5 million of related compensation expense under the LTIP, respectively. Amounts recognized in 2024 related to the 2024 and 2021 performance periods, and amounts recognized in 2023 only related to the 2021 performance period. Amounts earned for the 2024 performance year are payable to eligible participants in January 2028 after a three-year vesting period concludes. As of December 31, 2024 and 2023, there was $1.5 million and $2.7 million included in other liabilities in the condensed balance sheets, respectively. At December 31, 2024, $3.0 million was included in accrued expenses in the consolidated balance sheets, which was paid to eligible participants in January 2025 for the 2021 performance period after their three-year vesting period concluded.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-40
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 17—CONTINGENT CONSIDERATION GAINS, RESTRUCTURING CHARGES AND OTHER
Contingent consideration gains, restructuring charges and other for the years ended December 31, 2024, 2023 and 2022 are summarized below (in thousands): | | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Contingent consideration gains: | | | | | |
| Flexion contingent consideration | $ | (4,457) | | | $ | (3,424) | | | $ | (18,292) | |
| MyoScience contingent consideration | — | | | — | | | (11,184) | |
| Total contingent consideration gains | (4,457) | | | (3,424) | | | (29,476) | |
| | | | | |
| Acquisition-related charges: | | | | | |
| Acquisition-related expenses | 1,462 | | | 1,963 | | | 1,032 | |
| Severance-related expenses | — | | | — | | | 4,494 | |
| Other acquisition expenses | — | | | — | | | 5,719 | |
| Total acquisition-related charges | 1,462 | | | 1,963 | | | 11,245 | |
| | | | | |
| Restructuring charges | 8,532 | | | 1,109 | | | — | |
| Loss on lease termination | 2,165 | | | — | | | — | |
| Impairment of acquired IPR&D | — | | | — | | | 26,134 | |
| Termination of license agreement | — | | | — | | | 3,000 | |
| Total contingent consideration gains, restructuring charges and other | $ | 7,702 | | | $ | (352) | | | $ | 10,903 | |
Flexion Acquisition Contingent Consideration
For the years ended December 31, 2024, 2023 and 2022 the Company recognized contingent consideration gains of $4.5 million, $3.4 million and $18.3 million, respectively, due to a decrease in the fair value of its contingent consideration related to the Flexion Acquisition. See Note 11, Financial Instruments, for information regarding the method and key assumptions used in the fair value measurements of the Company’s contingent consideration and more information regarding the changes in fair value.
MyoScience Acquisition Contingent Consideration
The Company recognized contingent consideration gains related to the MyoScience Acquisition of $11.2 million for the year ended December 31, 2022. See Note 11, Financial Instruments, for information regarding the method and the changes in fair value.
Acquisition-Related Charges
The Company recognized acquisition-related charges of $1.5 million and $2.0 million during the years ended December 31, 2024 and 2023 primarily related to vacant and underutilized Flexion leases that were assumed from the Flexion Acquisition. The Company recognized acquisition-related charges and other costs of $11.2 million during the year ended December 31, 2022 primarily for severance, legal fees, third-party services and other one-time charges related to the Flexion Acquisition.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-41
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Restructuring Charges
In February 2024, the Company initiated a restructuring plan to ensure it is well positioned for long-term growth. The restructuring plan includes: (i) reshaping the Company’s executive team, (ii) reallocating efforts and resources from the Company’s ex-U.S. and certain early-stage development programs to its commercial portfolio in the U.S. market and (iii) reprioritizing investments to focus on commercial readiness for the implementation of separate Medicare reimbursement for EXPAREL at average sales price plus 6 percent in outpatient settings and iovera° up to an additional $255.85 when providers administer iovera° in ambulatory surgical centers and outpatient settings beginning in January 2025 as part of the Non-Opioids Prevent Addiction In the Nation (“NOPAIN”) Act and broader commercial initiatives in key areas, such as strategic national accounts, marketing and market access and reimbursement. The Company recognized $8.5 million of restructuring charges for the year ended December 31, 2024, related to employee termination benefits, such as the acceleration of share-based compensation, severance, and, to a lesser extent, other employment-related termination costs, as well as contract termination costs.
The Company’s restructuring charges as of December 31, 2024, including the beginning and ending liability balances, are summarized below (in thousands):
| | | | | | | | | | | | | | | | | |
| Employee Termination Benefits (1) | | Contract Termination Costs | | Total |
| Balance at December 31, 2023 | $ | — | | | $ | — | | | $ | — | |
Charges incurred (1) | 3,220 | | | 1,709 | | | 4,929 | |
| Cash payments made / settled | (1,985) | | | (20) | | | (2,005) | |
| Balance at December 31, 2024 | $ | 1,235 | | | $ | 1,689 | | | $ | 2,924 | |
(1) During the year ended December 31, 2024, there were $3.6 million of employee termination benefits related to share-based compensation excluded from the table above as they are non-cash and recorded against additional paid-in capital. |
In June 2023, the Company implemented a restructuring plan in an effort to improve its operational efficiencies. The restructuring charges were predominantly related to one-time employee termination benefits through a reduction of headcount, such as severance and related costs. During the year ended December 31, 2023, the Company recognized $1.1 million of restructuring charges.
Loss on Lease Termination
The Company recognized a loss of $2.2 million during the year ended December 31, 2024 associated with exiting a lease for a training facility in Houston, Texas. The loss resulted from the derecognition of the right-of-use asset, its lease liability and a termination payment of $1.3 million.
Impairment of Acquired IPR&D
For the year ended December 31, 2022, an impairment of $26.1 million for an acquired IPR&D intangible asset related to ZILRETTA for the treatment of OA pain of the shoulder was recognized based on the amount its previous carrying value of $60.0 million exceeded its fair value of $33.9 million. See Note 8, Goodwill and Intangible Assets, for more information.
Termination of License Agreement
The Company recognized expense of $3.0 million during the year ended December 31, 2022 related to the termination of a license agreement. See Note 19, Commitments and Contingencies, for more information.
NOTE 18—COMMERCIAL PARTNERS AND OTHER AGREEMENTS
Thermo Fisher Scientific Pharma Services
In April 2014, the Company and Thermo Fisher entered into a Strategic Co-Production Agreement, a Technical Transfer and Service Agreement (the “EXPAREL Technical Transfer and Service Agreement”) and a Manufacturing and Supply Agreement to collaborate in the manufacture of EXPAREL. Under the terms of the EXPAREL Technical Transfer and Service Agreement, Thermo Fisher undertook certain technical transfer activities and construction services needed to prepare its Swindon, U.K. facility for the manufacture of EXPAREL in dedicated manufacturing suites. The Company is now utilizing a second, larger-scale dedicated manufacturing suite. Under these agreements, the Company makes monthly base fee payments to
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-42
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Thermo Fisher. Unless earlier terminated by providing 18 months’ notice (other than termination by the Company in the event of a material breach by Thermo Fisher), this agreement will expire in May 2028.
Prior to the Flexion Acquisition, in July 2015, Flexion and Thermo Fisher entered into a Manufacturing and Supply Agreement (the “ZILRETTA Manufacturing and Supply Agreement”) and a Technical Transfer and Service Agreement related to the manufacture of ZILRETTA at the same Thermo Fisher site in Swindon, U.K where the Company’s EXPAREL manufacturing suite is located. Thermo Fisher agreed to undertake certain transfer activities and construction services needed to prepare its facility for the commercial manufacture of ZILRETTA in dedicated manufacturing suites. Flexion provided Thermo Fisher with certain equipment and materials necessary to manufacture ZILRETTA. The Company pays a monthly base fee to Thermo Fisher for the operation of the manufacturing suites and a per product fee for each vial of ZILRETTA based upon a forecast of commercial demand. The Company also reimburses Thermo Fisher for purchases of materials and equipment made on its behalf, certain nominal expenses and additional services. Unless earlier terminated (other than termination by the Company in the event that Thermo Fisher does not meet specified milestones or for a breach by Thermo Fisher), the Company will be obligated to pay for the costs incurred by Thermo Fisher associated with the removal of its manufacturing equipment and for Thermo Fisher’s termination costs up to a specified capped amount.
The initial term of the ZILRETTA Manufacturing and Supply Agreement that the Company assumed as part of the Flexion Acquisition expires in October 2027. The ZILRETTA Manufacturing and Supply Agreement will remain in full effect unless and until it expires or is terminated. The Company may terminate this agreement upon one month’s notice if a regulatory authority causes the withdrawal of ZILRETTA from the U.S. or any other market that represents 80 percent of its overall sales, or at any time for convenience by providing 24 months’ notice. Either party may terminate the ZILRETTA Manufacturing and Supply Agreement in the event of the breach or bankruptcy of the other party.
Aratana Therapeutics, Inc.
In December 2012, the Company entered into a worldwide license, development and commercialization agreement with Aratana Therapeutics, Inc., a wholly owned subsidiary of Elanco Animal Health, Inc., or Aratana. Under the agreement, the Company granted Aratana an exclusive royalty-bearing license, including the limited right to grant sublicenses, for the development and commercialization of the Company’s bupivacaine liposome injectable suspension product for veterinary use. Under the agreement, Aratana developed and obtained FDA approval for the use of the product in veterinary surgery to manage postsurgical pain. The Company is eligible to receive from Aratana up to an aggregate of $40.0 million upon the achievement of commercial milestones. Aratana is required to pay the Company a tiered double-digit royalty on certain net sales made in the U.S. If the product is approved by foreign regulatory agencies for sale outside of the U.S., Aratana will be required to pay the Company a tiered double-digit royalty on such net sales. Royalty rates will be reduced by a certain percentage upon the entry of a generic competitor for animal health indications into certain jurisdictions or if Aratana must pay royalties to third parties under certain circumstances. Unless terminated earlier pursuant to its terms, the license agreement is effective until July 2033, after which Aratana has the option to extend the agreement for an additional five-year term, subject to certain requirements.
Aratana began purchasing bupivacaine liposome injectable suspension product in 2016, which they market under the trade name NOCITA® (a registered trademark of Aratana) for veterinary use.
Carlisle Companies, Inc.
In January 2020, the Company and Carlisle Companies, Inc., or Carlisle, entered into a Manufacturing and Supply Agreement (the “Carlisle Agreement”) to collaborate in the manufacture of iovera° Smart Tips at Carlisle’s Tijuana, Mexico facility. The initial term of the Carlisle Agreement is five years with automatic one-year extensions unless either party provides prior notice in writing. Under the Carlisle Agreement, the Company pays fees based on the amount of iovera° Smart Tips delivered by Carlisle. Since April 2022, all iovera° Smart Tips have been produced by Carlisle.
The Carlisle Agreement may be terminated by either party upon one years’ written notice without cause. The Company may terminate the Carlisle Agreement upon thirty days’ written notice in the event that iovera° is withdrawn from the market or no longer sold by us. Either party may terminate the Carlisle Agreement in the event of the breach or bankruptcy of the other party.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-43
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
NOTE 19—COMMITMENTS AND CONTINGENCIES
Legal Proceedings
From time to time, the Company has been and may again become involved in legal proceedings arising in the ordinary course of its business, including those related to its patents and intellectual property, product liability and government investigations. Except as described below, the Company is not presently a party to any legal proceedings that it believes to be material, and is not aware of any pending or threatened litigation against the Company which it believes could have a material adverse effect on its business, operating results, financial condition or cash flows. The Company is not in a position to assess the likelihood of any potential losses or adverse effect on its financial condition or to estimate the amount or range of potential losses, if any, from the following actions at this time.
MyoScience Milestone Litigation
In August 2020, the Company and its subsidiary, Pacira CryoTech, Inc. (“Pacira CryoTech”), filed a lawsuit in the Court of Chancery of the State of Delaware against Fortis Advisors LLC (“Fortis”), solely in its capacity as representative for the former securityholders of MyoScience, and certain other defendants, seeking declaratory judgment with respect to certain terms of the merger agreement for the MyoScience Acquisition (the “MyoScience Merger Agreement”), specifically related to the achievement of certain milestone payments under the MyoScience Merger Agreement. In addition, the Company and Pacira CryoTech sought general, special and compensatory damages against the other defendants related to breach of fiduciary duties in connection with the purported achievement of milestone payments under the MyoScience Merger Agreement, and breach of the MyoScience Merger Agreement and certain other agreements with the defendants. In October 2020, Fortis filed an answer and counterclaim against the Company and Pacira CryoTech seeking to recover certain milestone payments under the MyoScience Merger Agreement.
A trial was conducted in September 2023. In January 2025, the Court issued its decision, finding that the disputed milestones were not met and therefore granted judgment to the Company in full. A final order will be entered in the coming months.
eVenus Pharmaceutical Laboratories Litigations
In October 2021, the Company received a Notice Letter advising that eVenus Pharmaceutical Laboratories, Inc., or eVenus, of Princeton, New Jersey, submitted to the FDA an Abbreviated New Drug Application, or ANDA with a Paragraph IV certification seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (266 mg/20 mL) in the U.S. prior to the expiration of U.S. Patent No. 11,033,495 (the ’495 patent).
In November 2021, the Company filed a patent infringement suit against eVenus and its parent company (Jiangsu Hengrui Pharmaceuticals, Co. Ltd., or Jiangsu Hengrui) in the U.S. District Court for the District of New Jersey (21-cv-19829) asserting infringement of the ’495 patent. This triggered an automatic 30-month stay of final approval of the eVenus ANDA which expired on July 1, 2024. On January 6, 2022, eVenus filed an Answer with counterclaims to the Complaint, alleging the ’495 patent is invalid and/or not infringed through the manufacture, sale, or offer for sale of the product described in product described in eVenus’s ANDA submission.
In December 2021, the Company received a second Notice Letter advising that eVenus submitted to the FDA an amendment to its ANDA with a Paragraph IV Certification seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (133 mg/10 mL) in the U.S. prior to the expiration of the ’495 patent. In the second Notice Letter, eVenus also advised that it submitted a Paragraph IV Certification to the FDA seeking authorization for the manufacturing and marketing of a generic version of EXPAREL (266 mg/20 mL and 133 mg/10 mL) in the U.S. prior to the expiration of U.S. Patent No. 11,179,336 (the ’336 patent). eVenus further alleges in the Notice Letter that both the ’495 patent and the ’336 patent are invalid and/or not infringed.
In February 2022, the Company filed a second patent infringement suit against eVenus and its parent company in the U.S. District Court for the District of New Jersey (22-cv-00718) asserting that the 133 mg/10 mL ANDA product will infringe the ’495 and ’336 patents and that the 266 mg/20 mL ANDA product will infringe the ’336 patent. This filing triggered a second automatic 30-month stay of final approval for the 133 mg/10 mL ANDA product which expired on July 1, 2024. The first and second patent infringement suits were consolidated.
In February 2023, eVenus filed its first amended answer to the first amended complaint, alleging patent invalidity, non-infringement and inequitable conduct. The Company has denied the allegations in eVenus’s first amended answer. The Company has subsequently voluntarily dismissed its claims with respect to the ’336 Patent. The trial on the remaining patent
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-44
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
was conducted in February 2024, and in August 2024,the U.S. District Court for the District of New Jersey issued its ruling in the patent infringement suit for the ‘495 patent. The ruling found that claim 7 of the ‘495 patent is not valid on the grounds of obviousness and anticipation. A notice of appeal was filed in September 2024 and is ongoing.
In April 2023, the Company filed a third patent infringement suit against eVenus, its parent company, and Fresenius Kabi USA, LLC, or Fresenius, in the U.S. District Court for the District of New Jersey (23-cv-2367) asserting that the 133 mg/10 mL and 266 mg/20 mL ANDA products will infringe U.S. Patent No. 11,426,348 (the ’348 patent). In July 2023, eVenus filed its answer with claims for declaratory judgment, alleging patent invalidity, non-infringement and inequitable conduct with respect to the ’348 patent as well as the Company’s other patents, U.S. Patent Nos. 11,278,494; 11,304,904; 11,311,486; 11,357,727 and 11,452,691. The parties have subsequently dismissed all patents other than the ’348 patent from this litigation. This action has been stayed pending resolution of the appeal of the U.S. District Court’s ruling on the ‘495 patent.
In May 2024, the Company filed a fourth patent infringement suit against eVenus, its parent company and Fresenius in the U.S. District Court for the District of New Jersey (24-cv-6294) asserting that the 133 mg/10 mL and 266 mg/20 mL ANDA products will infringe U.S. Patent Nos. 11,819,574 (the ‘574 patent) and 11,819,575 (the ‘575 patent). The Company subsequently filed a First Amended Complaint alleging infringement only with respect to the ‘574 patent. The Defendants filed an answer and counterclaim on September 25, 2024, with counterclaims alleging non-infringement and invalidity of the ‘574 patent, invalidity of the ‘575 patent and U.S. Patent No. 11,925,706 (the ‘706 patent) and inequitable conduct with respect to the ‘574, ‘575 and ‘706 patents. This action is in the discovery stage.
In July 2024, the Company filed a fifth patent infringement suit against eVenus, its parent company and Fresenius in the U.S. District Court for the District of New Jersey (24-cv-7680) asserting that the 133 mg/10 mL and 266 mg/20 mL ANDA products will infringe the ‘706 patent. The Company voluntarily filed a stipulation of dismissal without prejudice on September 9, 2024.
In July 2024, eVenus received FDA approval of a generic version of EXPAREL—the Company’s bupivacaine liposome injectable suspension product.
In December 2024, the Company filed a sixth patent infringement suit against Fresenius and eVenus’s parent company in the Northern District of Illinois (24-cv-12416) asserting that the 133 mg/10 mL and 266 mg/20 mL ANDA products will infringe U.S. Patent No. 12,156,940 (the ‘940 patent). This patent is the first patent from a new family of patents related to EXPAREL produced by the Company’s enhanced larger-scale manufacturing process in San Diego, California. This action is in the pleadings stage.
Also in December 2024, eVenus, its parent company, and Fresenius filed an action for declaratory judgment of non-infringement and invalidity with respect to the ‘940 patent in the District Court of New Jersey (24-cv-11014). This action is in the pleadings stage.
The Company is unable to predict the outcome of these litigations at this time.
Argentum Request for Ex Parte Reexamination of ‘495 Patent
On October 3, 2024, Argentum Pharmaceuticals LLC, or Argentum, filed a Request for Ex Parte Reexamination of the ‘495 patent. Specifically, Argentum alleged that claims 1, 7 and 8 of the ‘495 patent are obvious and cite to U.S. Patent No. 9,585,838 and the Physician’s Desk Reference in support of its allegation. The Company is unable to predict the outcome of this proceeding at this time.
Securities Class Action
On January 13, 2025, Leandro Alvarez filed a putative class action on behalf of Company shareholders between August 2, 2023 and August 8, 2024 against the Company and certain of its officers, in the District Court of New Jersey (25-cv-322). The complaint alleges that the Company made materially false and misleading statements and/or concealed material adverse facts concerning EXPAREL patents. The case is in the pleadings stage and the Company is unable to predict the outcome of this litigation at this time.
Research Development Foundation
Pursuant to an agreement with the Research Development Foundation, or RDF, the Company was required to pay RDF a low single-digit royalty on the collection of revenues from certain products, for as long as certain patents assigned to the Company under the agreement remain valid. RDF has the right to terminate the agreement for an uncured material breach by
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-45
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
the Company, in connection with its bankruptcy or insolvency or if it directly or indirectly opposes or disputes the validity of the assigned patent rights. The Company’s ’495 patent was issued on June 15, 2021. Thereafter, RDF asserted that the issuance of that patent extends the Company’s royalty obligations under the agreement until 2041. The Company believes that the royalty period under the agreement ended on December 24, 2021 with the expiration of its U.S. Patent No. 9,585,838. Because of the disagreement over the interpretation of the agreement, in December 2021, the Company filed a declaratory judgment lawsuit in the U.S. District Court for the District of Nevada (21-cv-02241). The lawsuit seeks a declaration from the court that the Company owes no royalties to RDF with respect to its EXPAREL product after December 24, 2021.
On August 8, 2023, the U.S. District Court, District of Nevada, granted the Company’s motion for partial summary judgment in respect to the Company’s claim for a declaration that it no longer owes royalties for EXPAREL made under the 45-liter manufacturing process as of December 24, 2021. As a result, the Company expects to receive $14.5 million from RDF, representing the royalties that the Company paid to RDF under protest after December 24, 2021 for EXPAREL made from the 45-liter manufacturing process. Once it becomes probable that the settlement amount will be received, the Company will record a settlement gain within other operating expenses in the condensed consolidated statement of operations. In November 2023, the United States District Court, District of Nevada conducted a mediation that did not result in a settlement. During the pendency of the remaining lawsuit, the Company will continue to pay royalties associated with an enhanced, large-scale manufacturing process to RDF under protest. The Company is unable to predict the outcome of this action at this time.
Purchase Obligations
The Company has approximately $59.9 million of minimum, non-cancelable contractual commitments for contract manufacturing services as of December 31, 2024, of which $20.3 million is due in each of 2025 and 2026, $17.9 million is due in 2027 and the remaining $1.4 million is due in 2028. The Company has approximately $6.5 million of minimum, non-cancelable contractual commitments for the purchase of certain raw materials as of December 31, 2024, all of which are due in 2025. The Company has $1.8 million of other minimum, non-cancelable contractual commitments as of December 31, 2024, of which $1.4 million is due in 2025 and the remaining $0.4 million is due thereafter.
Other Commitments and Contingencies
Termination of License Agreement
Prior to the Flexion Acquisition, in March 2020, Flexion entered into an exclusive license agreement with Hong Kong Tainuo Pharma Ltd., or HK Tainuo, and Jiangsu Tainuo Pharmaceutical Co. Ltd., a subsidiary of China Shijiazhuang Pharmaceutical Co, Ltd., for the development and commercialization of ZILRETTA in Greater China (consisting of mainland China, Hong Kong, Macau and Taiwan). Under the terms of the agreement, HK Tainuo paid Flexion an upfront payment of $10.0 million during the year ended December 31, 2020 which was recorded as deferred revenue as of December 31, 2021. The Company was also eligible to receive up to $32.5 million in aggregate development, regulatory and commercial sales milestone payments under the exclusive license agreement. HK Tainuo was responsible for the clinical development, product registration and commercialization of ZILRETTA in Greater China. The Company was solely responsible for the manufacture and supply of ZILRETTA to HK Tainuo for all clinical and commercial activities. The terms related to product manufacturing and supply, including pricing and minimum purchase requirements agreed to in the license agreement, were to be covered by a separate supply agreement which was never finalized.
In July 2022, the Company submitted notice to HK Tainuo of its intent to pursue termination of the license agreement. The $13.0 million related to the termination of the license agreement was paid in January 2023.
Pediatric Trial Commitments
The FDA, as a condition of EXPAREL approval, has required the Company to study EXPAREL for infiltration and as a brachial plexus block in pediatric patients. The Company was granted deferrals for the required pediatric trials until after the indications were approved in adults. Similarly, in Europe, the Company agreed with the European Medicines Agency, or EMA, on a Pediatric Investigation Plan as a prerequisite for submitting a Marketing Authorization Application (MAA) in the E.U. Despite the U.K.’s withdrawal from the E.U., the agreed pediatric plan is applicable in the U.K.
The Company received notification from the FDA that its pediatric studies requirement had been waived for the indications of brachial plexus interscalene nerve block, lower extremity nerve block, sciatic nerve block in the popliteal fossa and adductor canal block indications to produce postsurgical regional analgesia in pediatric patients. The Company is still working with the FDA, EMA and Medicines and Healthcare Regulatory Agency (MHRA) to finalize the regulatory pathways for its remaining pediatric commitments.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-46
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
Contingent Milestone Payments
Refer to Note 11, Financial Instruments, for information on potential contingent milestone payments related to the Flexion Acquisition.
PCRX-201
PCRX-201 (enekinragene inzadenovec) is a novel, locally administered gene therapy vector platform product candidate that boosts cellular production of the anti-inflammatory protein interleukin-1 receptor antagonist (IL-1Ra) for treating OA pain in the knee, was added to the Company’s portfolio as part of the Flexion Acquisition in November 2021.
Prior to the Flexion Acquisition, in 2017, Flexion entered into an agreement with GQ Bio to acquire the global rights to PCRX-201, a gene therapy product candidate. As part of the agreement, up to an aggregate of $56.0 million of payments could have become due upon the achievement of certain development and regulatory milestones, including up to $4.5 million through initiation of a Phase 2 clinical trial and up to an additional $51.5 million in development and global regulatory approval milestone payments. In February 2025, Pacira Therapeutics, Inc., a wholly-owned subsidiary of the Company, completed the GQ Bio Acquisition and acquired the remaining 81% of GQ Bio that was not already owned by the Company. For more information on the GQ Bio Acquisition, see Note 21, Subsequent Events. As of the date hereof, GQ Bio is a wholly-owned subsidiary of the Company.
Also in 2017, in an agreement between The Baylor College of Medicine, or BCM, and GQ Bio, the Company (through the Flexion Acquisition) became the direct licensee of certain underlying BCM patents and other proprietary rights related to PCRX-201. The license agreement grants the Company an exclusive, royalty-bearing, world-wide right and license under its patent and other proprietary rights directly related to PCRX-201. The license agreement with BCM includes a low single-digit royalty on net product sales of PCRX-201. Milestone payments range from $0.1 million up to $0.6 million based on the completion of a Phase 1 FDA trial up to a Phase 3 clinical trial.
In February 2024, the FDA granted a Regenerative Medicine Advanced Therapy (RMAT) designation to PCRX-201 for the treatment of OA pain of the knee.
NOTE 20—SEGMENT INFORMATION
The Company is managed and operated as a single business focused on the development, manufacture, marketing, distribution and sale of non-opioid pain management and regenerative health solutions. The Company is managed by a single management team, and, consistent with its organizational structure, the Chief Executive Officer—who is the Company’s chief operating decision maker, or CODM—manages and allocates resources at a consolidated level. Accordingly, the Company views its business as one operating segment and one reportable segment to evaluate its performance, allocate resources, set operational targets and forecast its future financial results.
The key measure of the Company is GAAP net income. The CODM uses this measure to evaluate its performance, allocate resources, set operational targets and forecast its future financial results.
There are significant expense categories and amounts that are regularly provided to the CODM. These expense categories differ from what is disclosed in the Company’s financial results. The table below reconciles the significant expense categories provided to the CODM to the Company’s expenses as disclosed under U.S. GAAP (in thousands):
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-47
PACIRA BIOSCIENCES, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (CONTINUED)
| | | | | | | | | | | | | | | | | |
| Year Ended December 31, |
| 2024 | | 2023 | | 2022 |
| | | | | |
| Revenues | $ | 700,966 | | | $ | 674,978 | | | $ | 666,823 | |
| Less: | | | | | |
| Adjusted cost of goods sold | 165,097 | | | 173,980 | | | 174,856 | |
| Adjusted research and development | 74,196 | | | 67,563 | | | 78,203 | |
| Adjusted selling and marketing | 172,015 | | | 153,040 | | | 144,996 | |
| Adjusted general and administrative | 86,385 | | | 82,737 | | | 73,989 | |
| | | | | |
| Goodwill impairment | 163,243 | | | — | | | — | |
| Stock-based compensation | 51,171 | | | 47,895 | | | 48,092 | |
| Amortization of acquired intangible assets | 57,288 | | | 57,288 | | | 57,288 | |
| Changes in the fair value of contingent consideration | (4,457) | | | (3,424) | | | (29,476) | |
| Impairment of acquired IPR&D | — | | | — | | | 26,134 | |
| Accelerated depreciation | — | | | — | | | 10,545 | |
Other (1) | 9,399 | | | 8,224 | | | 22,172 | |
| Total operating expenses | 774,337 | | | 587,303 | | | 606,799 | |
| Adjusted other income (expense) | 2,747 | | | (9,048) | | | (46,722) | |
| Gain (loss) on early extinguishment of debt | 7,518 | | | (16,926) | | | — | |
| Total other income (expense), net | 10,265 | | | (25,974) | | | (46,722) | |
| (Loss) income before income taxes | (63,106) | | | 61,701 | | | 13,302 | |
| Income tax (expense) benefit | (36,454) | | | (19,746) | | | 2,607 | |
| Net (loss) income | $ | (99,560) | | | $ | 41,955 | | | $ | 15,909 | |
(1) During the year ended December 31, 2024, the remaining other operating expenses relates to restructuring charges, a loss on lease termination and acquisition-related charges. During the years ended December 31, 2023 and 2022, the remaining other operating expenses relates to step-up of acquired Flexion fixed assets and inventory to fair value and acquisition-related charges.
The Company’s long-lived assets are primarily located in the U.S. For information on the Company’s fixed assets located outside of the U.S., refer to Note 6, Fixed Assets.
NOTE 21—SUBSEQUENT EVENTS (UNAUDITED)
Acquisition of GQ Bio Therapeutics GmbH
As of December 31, 2024, the Company owned approximately 19% of GQ Bio. On February 25, 2025, Pacira Therapeutics, Inc., a wholly-owned subsidiary of the Company, entered into a securities purchase agreement to acquire the remaining 81% of GQ Bio for approximately $32 million, net of working capital and other transaction adjustments. The net purchase price includes $8 million to be paid over three years pursuant to a key employee holdback agreement and a post-closing indemnity holdback of $6 million, whereas the remaining $18 million was paid at closing using cash on hand.
Brisbane, California Office Lease
In February 2025, the Company signed a lease for an office in Brisbane, California to be used for selling, general and administrative purposes. As a result, the Company recorded $0.5 million of right-of-use assets in exchange for lease obligations in the first quarter of 2025.
Pacira BioSciences, Inc. | 2024 Annual Report on Form 10-K | Page F-48