| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| Delaware | 82-3827296 | |
| (State or Other Jurisdiction of Incorporation or Organization) | | (I.R.S. Employer Identification Number) |
| 30 Technology Drive, Warren, NJ | 07059 | |
| (Address of Principal Executive Offices) | (Zip Code) |
| Title of each class | Name of each exchange on which registered | |
| Common Stock, par value $0.001 per share | NASDAQ Global Market |
Large accelerated filer ☐ | Accelerated filer ☐ |
Non-accelerated filer ☒ | Smaller reporting company ☒ |
Emerging growth company ☒ |
| Page No. | ||
| 3 | ||
| Item 1. | 3 | |
| Item 1A. | 29 | |
| Item 1B. | 66 | |
| Item 2. | 66 | |
| Item 3. | 66 | |
| Item 4. | 69 | |
| 69 | ||
| Item 5. | 69 | |
| Item 6. | 71 | |
| Item 7. | 72 | |
| Item 7A. | 84 | |
| Item 8. | 84 | |
| Item 9. | 85 | |
| Item 9A. | 85 | |
| Item 9B. | 85 | |
| 86 | ||
| Item 10. | 86 | |
| Item 11. | 86 | |
| Item 12. | 86 | |
| Item 13. | 86 | |
| Item 14. | 86 | |
| 87 | ||
| Item 15. | 87 |
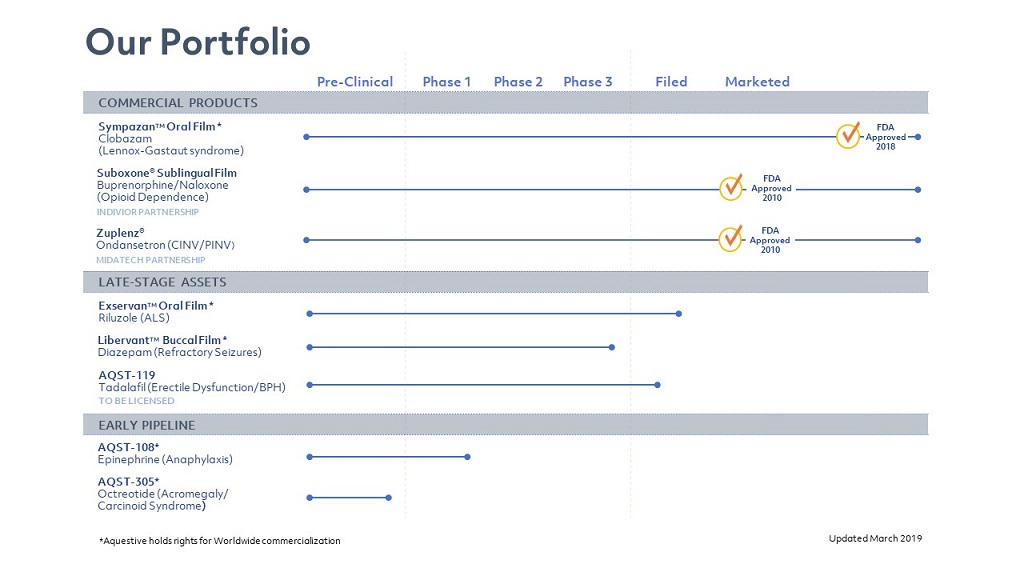
| · | Sympazan – an oral soluble film formulation of clobazam, a benzodiazepine used as an adjunctive therapy for seizures associated with LGS. We developed Sympazan as an alternative to Onfi brand and clobazam generic, which were previously only available in either tablet form or liquid suspensions. LGS patients often have difficulty swallowing pills and large volume suspensions leading to uncertain and inconsistent dosing. These challenges increase the burden of care, particularly for patients that have difficulty swallowing or who may be combative or resistant during treatment administration. We believe that Sympazan addresses these treatment obstacles because it is mucoadhesive, dissolves rapidly in existing saliva and is swallowed along with a patient’s natural saliva production, and therefore cannot be easily spit out. In clinical trials, Sympazan has demonstrated bioequivalence to Onfi. Following approval by the FDA, we launched Sympazan in December 2018. |
| · | Libervant – a buccally, or inside of the cheek, administered soluble film formulation of diazepam, a benzodiazepine intended for rapid treatment of acute uncontrolled seizures in selected, refractory patients with epilepsy on stable regimens of AEDs who require intermittant use of diazapam to control bouts of increased seizure activity. Libervant has received orphan drug designation from the FDA. We are developing Libervant as an alternative to Diastat (diazepam rectal gel), the current standard of care rescue therapy for patients with refractory epilepsy, which as a rectal gel, is invasive, inconvenient, and difficult to administer. As a result, a large portion of the patient population does not receive adequate treatment or foregoes treatment altogether. We believe that Libervant will enable a larger share of patients to receive more appropriate treatment by providing consistent therapeutic dosing in a non-invasive and innovative treatment form for epileptic seizures. Certain epilepsy monitoring unit studies were completed for Libervant in 2018, we expect to complete additional clinical studies during the year and commence a rolling NDA submission. |
| · | Exservan, formerly AQST-117 – an oral soluble film formulation of riluzole, a small molecule glutamate antagonist used as an adjunctive therapy in the treatment of ALS, which has been shown to slow disease progression, increase lifespan and improve quality of life. However, because ALS patients typically have difficulty swallowing, tablet and liquid administration is challenging. Exservan has received orphan drug designation from the FDA. We are developing Exservan as an alternative to Rilutek and Tiglutik (riluzole), which is currently available only in tablet or liquid form in order to achieve an easier, more reliable and accurate dosing. This may allow patients to continue therapy even after their ability to swallow has become compromised. Exservan addresses these treatment obstacles because it is mucoadhesive and dissolves easily on the tongue without the need for water and without a substantial increase in salivary flow. In clinical trials, Exservan has demonstrated bioequivalence to Rilutek. We began the NDA submission process for Exservan in January 2019. |
| · | AQST-108 – a sublingual film formulation of epinephrine that we are developing for the treatment of anaphylaxis, a severe and potentially life-threatening allergic reaction, and potentially for treatment of allergic reactions in patients at risk of anaphylaxis. Epinephrine is the standard of care in the treatment of anaphylaxis and is currently administered via intramuscular injection. The current market leader is EpiPen, a single-dose, pre-filled automatic injection device. As a result of its administration via intra-muscular injection, many patients and their caregivers are reluctant to use currently available products, resulting in increased hospital visits and overall cost of care to treat anaphylactic events. We are designing AQST-108 to be the first oral delivery form of epinephrine used to treat anaphylaxis as well as other acute allergic reactions. We believe that, as a result of its sublingual administration, AQST-108 will improve patient compliance and lower the total cost of care. AQST-108 has shown promising results in early human proof of concept trials. We are currently expecting to begin our next Phase 1 proof of concept study with our enhanced prototype in the first half of 2019, with completion planned within this year. |
| · | AQST-305 – a sublingual film formulation of octreotide, a small peptide that has a similar pharmacological profile to natural somatostatin, for the treatment of acromegaly, as well as severe diarrhea and flushing associated with carcinoid syndrome. Acromegaly is a hormone disorder that results from the overproduction of growth hormone in middle-aged adults. Octreotide is the standard of care for the treatment of acromegaly. The current market leader, Sandostatin, is administered via deep subcutaneous or intramuscular injections once a month. This monthly treatment regimen can result in loss of efficacy towards the end of the monthly treatment cycle. We are developing AQST-305 as a non-invasive, pain-free alternative to Sandostatin to reduce treatment burden, healthcare costs and the potential loss of efficacy over the treatment cycle. AQST-305 has shown promising preclinical results. We began a human proof of concept study in Canada during the third quarter of 2018, and, based on anticipated enhancements that may be indicated by that study, we are planning to start additional human proof of concept trials late in 2019. |
| · | Suboxone – a sublingual film formulation of buprenorphine and naloxone that is marketed in the United States and internationally for the treatment of opioid dependence. Suboxone Sublingual Film was launched by licensee Indivior Inc., or Indivior, in 2010. Suboxone Sublingual Film is the most prescribed branded product in its category and is the first sublingual film product for the treatment of opioid dependence with a greater than 50% market share despite multiple competitors, including alternative dosing formulations. We are the sole and exclusive supplier and manufacturer of Suboxone Sublingual Film to Indivior and Sandoz authorized generic. Since launch of this product in 2010, we have produced approximately 2 billion doses of Suboxone. Recently, on February 20, 2019, Dr. Reddy’s Labs and Alvogen, Inc. launched competing generic formulations of this product at risk, and on February 22, 2019, Mylan Pharmaceuticals, Inc. announced its launch of a similar generic formulation, also at risk. |
| · | Zuplenz – an oral soluble film formulation of ondansetron, a 5-HT3 antagonist approved for the treatment of nausea and vomiting associated with chemotherapy and post-operative recovery. Ondansetron is available as intravenous injections, intramuscular injections, orally dissolving tablets, oral solution, tablets, and film. Generic and branded products are available, with the branded product marketed as Zofran by GlaxoSmithKline. We licensed commercial rights for Zuplenz to Midatech Pharma in the United States, Canada, and China. Midatech launched Zuplenz in the United States in 2015. We are the sole and exclusive manufacturer of Zuplenz for Midatech. |
| · | APL-130277 – a sublingual film formulation of apomorphine, which is a dopamine agonist in development to treat episodic off-periods in Parkinson’s disease. APL-130277 is being developed by our licensee as a sublingual alternative to injectable form of apomorphine. We licensed intellectual property for APL-130277 to Cynapsus Therapeutics, Inc., a company that was acquired by Sunovion Pharmaceuticals Inc., or Sunovion. Sunovion received a complete response letter on January 29, 2019 and announced that no additional clinical trials were necessary to submit a revised NDA. Assuming FDA approval for this product candidate, we intend to explore royalty monetization opportunities for the expected royalty and milestone revenue streams from this product which, if successful, could lead to additional non-dilutive capital for the Company. |

| · | preferred alternative to more invasive drug administration methods such as injection, rectal or nasal applications; |
| · | faster, or at least equivalent, onset of action; |
| · | ease of administration and availability (no device required, no gel to transport); |
| · | direct absorption into the bloodstream reducing or avoiding “first pass” effects in the liver; |
| · | reduced gastrointestinal, or GI, side effects; |
| · | positive dosing outcomes, especially for patients with physical (e.g., dysphagia) or psychological barriers to other methods of drug administration; |
| · | stable, durable, portable and quick-dissolving (with or without water); |
| · | customizable delivery routes for tailored pharmacokinetic, or PK, profiles (buccal, sublingual or lingual); and |
| · | customizable taste profiles. |
| · | Advance our late stage proprietary portfolio of CNS product candidates to solve critical healthcare problems and make a meaningful improvement in the lives of patients and caregivers. We have focused development efforts on three proprietary CNS product candidates. These product candidates address treatment challenges associated with epilepsy and ALS. We have received FDA approval and subsequently began, in December 2018, distribution and sales of Sympazan. We expect to submit an NDA for Libervant during 2019. We began the NDA submission process for Exservan in January 2019. |
| · | Scale our commercial platform to maximize the value of our proprietary product candidates. In order to maximize the value of our proprietary product candidates, we are continuing our plan to self-commercialize our late stage CNS and other proprietary product candidates through a dedicated and focused commercial organization. We have recently built expertise in marketing, sales, payor and market access management and medical affairs in anticipation of multiple product launches, the first of which began in December 2018. Based on overlapping prescriber call points for our initial CNS product candidates, we believe an efficient and dedicated sales force can effectively cover the vast majority of targeted prescribers. |
| · | Exploit our technology and know-how to develop oral versions of more complex injectable drugs to address unmet patient needs. Based on promising preclinical and early clinical results, we intend to continue to develop oral transmucosal versions of epinephrine and octreotide, products that are currently available only in injectable form. |
| · | Continue to identify product opportunities within CNS and other markets to expand our proprietary product pipeline. We intend to identify additional product candidates that provide clinical differentiation and solve unmet needs. In the CNS space, we will leverage our relationships with key stakeholders including patients, caregivers, key opinion leaders and patient advocacy groups to identify new product opportunities. Additionally, we will continue to evaluate other therapeutic areas, indications and products where we believe that our expertise and know-how can create differentiation and value. |
| · | Acquire and market products, or establish licensing relationships to develop and manufacture products, utilizing new chemical entities. We intend to continue to strategically expand our product portfolio by developing products that incorporate new chemical entities to treat disorders with high unmet need. |
| · | Continue to expand and solidify our intellectual property portfolio for our products, product candidates and manufacturing processes. Our global intellectual property portfolio is a significant source of competitive advantage, the strength of which has been demonstrated through multiple successful patent defenses. We have built a two-tier patent estate consisting of composition-of-matter and method of manufacture patents and patent applications. We intend to expand our intellectual property estate as we advance our PharmFilm® and other technologies and as we develop new and existing product candidates. |
| · | completion of preclinical laboratory and animal testing and formulation studies in compliance with the FDA’s current good laboratory practice, or GLP, regulations; |
| · | submission to the FDA of an Investigational New Drug, or IND, application for human clinical testing which must become effective before human clinical trials may begin in the United States; |
| · | approval by an independent institutional review board, or IRB, at each clinical trial site before each trial may be initiated; |
| · | performance of adequate and well-controlled human clinical trials in accordance with current good clinical practices, or GCP, to establish the safety and efficacy of the proposed drug product for each intended use; |
| · | submission to the FDA of an NDA; |
| · | satisfactory completion of an FDA pre-approval inspection of the facility or facilities at which the product is manufactured to assess compliance with the FDA’s cGMP regulations to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| · | satisfactory completion of a potential review by an FDA advisory committee, if applicable; and |
| · | FDA review and approval of the NDA. |
| Phase 1: | In Phase 1, through the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses, and, if possible, early evidence on effectiveness. |
| Phase 2: | Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, dosage tolerance and optimum dosage, and to identify common adverse effects and safety risks. |
| Phase 3: | Phase 3 trials are undertaken to obtain the additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. In most cases, the FDA requires two adequate and well controlled Phase 3 clinical trials to demonstrate the efficacy of the drug. A single Phase 3 trial with other confirmatory evidence may be sufficient in rare instances where the study is a large multicenter trial demonstrating internal consistency and a statistically persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible. |
| · | conducting clinical trials of our product candidates; |
| · | seeking regulatory approval for any of our product candidates that successfully complete clinical development; |
| · | commercialization activities, including product sales, marketing, manufacturing and distribution, for our products, if approved; |
| · | maintaining, expanding and protecting our intellectual property portfolio; |
| · | acquiring or in-licensing new technologies or development-stage or approved products; |
| · | adding clinical, scientific, operational, financial and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts and to support our transition to operations as a public company; and |
| · | experiencing incremental costs due to delays or encountering any issues with any of the above, including, but not limited to, failed trials, complex results, safety issues or other regulatory challenges. |
| · | the timing of FDA or any other regulatory authority approvals; |
| · | the timing of process validation for particular product candidates; |
| · | the timing of product launches and market acceptance of such products launched; |
| · | changes in the amount we spend to research, develop, acquire, license or promote new product candidates; |
| · | the outcome of our research, development and clinical trial programs; |
| · | serious or unexpected health or safety concerns related to our product candidates; |
| · | the introduction of new products by others that render our product candidates obsolete or noncompetitive; |
| · | our ability to maintain selling prices and gross margins on our commercial products and product candidates; |
| · | our ability to comply with complex governmental regulations applicable to many aspects of our business; |
| · | changes in coverage and reimbursement policies of health plans and other health insurers, including changes to Medicare, Medicaid and similar government healthcare programs; |
| · | increases in the cost of raw materials used to manufacture our commercial products and product candidates; |
| · | manufacturing and supply interruptions, including product rejections or recalls due to failure to comply with manufacturing specifications; |
| · | timing of revenue recognition related to our collaboration agreements; |
| · | our ability to protect our intellectual property and avoid infringing the intellectual property of others and any adverse developments in any related legal proceeding; and |
| · | the outcome and cost of possible litigation with third parties. |
| · | we may have difficulty satisfying our obligations with respect to our existing indebtedness including the repayment of such indebtedness; |
| · | we may have difficulty obtaining financing in the future for working capital, capital expenditures, acquisitions or other purposes; |
| · | we will need to use a substantial portion of our available cash flow to pay interest and principal on our debt, which will reduce the amount of money available to finance our operations and other business activities; |
| · | we may be more vulnerable to general economic downturns and adverse industry conditions; |
| · | if cash flows from product sales are insufficient to satisfy our obligations with respect to our existing indebtedness, we may be forced to sell assets or seek additional capital, which we may not be able to accomplish on favorable terms, if at all; |
| · | we could be limited in our flexibility in planning for, or reacting to, changes in our business and in our industry in general; |
| · | we could be placed at a competitive disadvantage compared to our competitors that have less debt; |
| · | our failure to comply with the financial and other restrictive covenants in our debt instruments which, among other things, require us to maintain specified financial covenants and limit our ability to incur debt and sell assets, could result in an event of default that, if not cured or waived, could have a material adverse effect on our business or prospects; and |
| · | our tangible and intangible assets, including our intellectual property, are subject to first priority liens and may be used to satisfy our outstanding debt. |
| · | the timing of market introduction of the product candidate as well as competitive products; |
| · | the clinical indications for which the product candidate is approved; |
| · | the potential and perceived advantages of such product candidate over alternative treatments; |
| · | favorable pricing and the availability of coverage and adequate reimbursement by third-party payors and government authorities; |
| · | relative convenience and ease of administration; |
| · | any negative publicity related to our or our competitors’ products that include the same active ingredient; |
| · | the prevalence and severity of adverse side effects, including limitations or warnings contained in a product’s FDA-approved labeling; and |
| · | the effectiveness of sales and marketing efforts. |
| · | the efficacy and safety of our products and product candidates, including marketed products and product candidates in development by third parties; |
| · | the time it takes for our product candidates to complete clinical development and receive marketing approval; |
| · | our ability to maintain a good relationship with regulatory authorities; |
| · | our ability to commercialize and market any of our product candidates that receive regulatory approval; |
| · | the price of our products relative to pricing of branded or generic competitors; |
| · | whether coverage and adequate levels of reimbursement are available under private and governmental health insurance plans, including Medicare and Medicaid; |
| · | our ability to protect intellectual property rights related to our products and product candidates; |
| · | our ability to manufacture on a cost-effective basis and sell commercial quantities of our existing products and product candidates that may receive regulatory approval in the future; and |
| · | acceptance by physicians and other healthcare providers of any of our products and product candidates that receive regulatory approval. |
| · | a covered benefit under its health plan; |
| · | appropriate and medically necessary for the specific condition or disease; |
| · | cost effective; and |
| · | neither experimental nor investigational. |
| · | the federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or indirectly, overtly or covertly, in cash or in kind, in return for the purchase, recommendation, leasing or furnishing of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand, and prescribers, purchasers and formulary managers on the other. The Patient Protection and Affordable Care Act, as amended, or the PPACA, amended the intent requirement of the federal Anti-Kickback Statute. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it; |
| · | federal civil and criminal false claims laws, including, without limitation, the False Claims Act, and civil monetary penalty laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid or other government payors that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. The PPACA provides, and recent government cases against pharmaceutical and medical device manufacturers support, the view that federal Anti-Kickback Statute violations and certain marketing practices, including off-label promotion, may implicate the False Claims Act; |
| · | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit a person from knowingly and willfully executing a scheme or making false or fraudulent statements to defraud any healthcare benefit program, regardless of the payor (e.g., public or private); |
| · | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information without appropriate authorization on entities subject to the rule, such as health plans, health care clearinghouses and certain health care providers, and their respective business associates who provide services involving the creation, use or disclosure of HIPAA protected health information; |
| · | federal transparency laws, including the federal Physician Payments Sunshine Act, which is part of the PPACA, that require certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to: (i) payments or other “transfers of value” made to physicians and teaching hospitals; and (ii) ownership and investment interests held by physicians and their immediate family members, with such information being made publicly available through a searchable website; |
| · | state and foreign law equivalents of each of the above federal laws; state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers, marketing expenditures, or pricing information; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or to adopt compliance programs as prescribed by state laws and regulations, or that otherwise restrict payments that may be made to healthcare providers; and state and local laws that require the registration of pharmaceutical sales representatives; and |
| · | state and foreign laws that govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
| · | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs, although this fee does not apply to sales of certain products approved exclusively for orphan indications; |
| · | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| · | expansion of manufacturers’ rebate liability under the Medicaid Drug Rebate Program by increasing the minimum rebate for both branded and generic drugs and revising the definition of “average manufacturer price,” or AMP, for calculating and reporting Medicaid drug rebates on outpatient prescription drug prices and extending rebate liability to prescriptions for individuals enrolled in Medicare Advantage plans; |
| · | addition of more entity types eligible for participation in the Public Health Service the 340B drug pricing program, or the 340B program; |
| · | established the Medicare Part D coverage gap discount program by requiring manufacturers to provide a 50% point-of-sale-discount off the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D; |
| · | the Bipartisan Budget Act of 2018, or BBA, that among other things, increased the manufacturer’s subsidy under this program from 50% to 70% of the negotiated price, beginning in 2019; |
| · | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and |
| · | established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. |
| · | we may be required to undertake the expenditure of substantial operational, financial and management resources; |
| · | we may be required to issue equity securities that would dilute our stockholders’ percentage of ownership; |
| · | we may be required to assume substantial actual or contingent liabilities; |
| · | strategic collaborators could terminate the arrangement or allow it to expire, which would delay the development and may increase the cost of developing our product candidates; |
| · | business combinations of a strategic collaborator or significant changes in a strategic collaborator’s business strategy may affect a strategic collaborator’s willingness or ability to complete its obligations under any arrangement; and |
| · | strategic collaborators could decide to move forward with a competing product candidate developed either independently or in collaboration with others, including our competitors. |
| · | reductions in the payment of royalties or other payments we believe are due pursuant to the applicable collaborative arrangement; |
| · | actions taken by a partner inside or outside our collaboration which could negatively impact our rights or benefits under our collaboration; and |
| · | unwillingness on the part of a partner to keep us informed regarding the progress of its development and commercialization activities or to permit public disclosure of the results of those activities. |
| · | impairment of our business reputation; |
| · | withdrawal of clinical study participants; |
| · | costs due to related litigation; |
| · | distraction of management’s attention from our primary business; |
| · | substantial monetary awards to patients or other claimants; |
| · | the inability to commercialize our product candidates; and |
| · | decreased demand for our product candidates, if approved for commercial sale. |
| · | comply with FDA regulations or the regulations applicable in other jurisdictions; |
| · | provide accurate information to the FDA and other regulatory authorities; |
| · | comply with healthcare fraud and abuse laws and regulations in the United States and abroad; |
| · | report financial information or data accurately; or |
| · | disclose unauthorized activities to us. |
| · | inability to raise or delays in raising funding necessary to initiate or continue a trial; |
| · | delays in obtaining regulatory approval to commence a trial; |
| · | delays in reaching agreement with the FDA on final trial design; |
| · | imposition of a clinical hold for safety reasons or following an inspection of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| · | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites, or failure by such CROs to carry out the clinical trial at each site in accordance with the terms of our agreements with them; |
| · | delays in obtaining required institutional review board, or IRB, approval at each site; |
| · | difficulties or delays in having patients complete participation in a trial or return for post-treatment follow-up; |
| · | clinical sites electing to terminate their participation in one of our clinical trials, which would likely have a detrimental effect on subject enrollment; or |
| · | time required to add new clinical sites. |
| · | severity of the disease under investigation; |
| · | design of the trial protocol; |
| · | size of the patient population; |
| · | eligibility criteria for the trial in question; |
| · | perceived risks and benefits of the product candidate under trial; |
| · | proximity and availability of clinical trial sites for prospective patients; |
| · | availability of competing therapies and clinical trials; |
| · | efforts to facilitate timely enrollment in clinical trials; |
| · | patient referral practices of physicians; and |
| · | ability to monitor patients adequately during and after treatment. |
| · | regulatory authorities may withdraw their approval of the product or impose restrictions on its distribution; |
| · | the FDA may require implementation of a Risk Evaluation and Mitigation Strategy, or REMS; |
| · | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| · | we may be required to change the way the product is administered or conduct additional clinical studies; |
| · | we could be sued and held liable for harm caused to patients; or |
| · | our reputation may suffer. |
| · | the FDA or comparable foreign regulatory authorities may disagree that our changes to branded reference drugs meet the criteria for the 505(b)(2) regulatory pathway or foreign regulatory pathways; |
| · | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective or comparable to its branded reference product for its proposed indication; |
| · | the results of any clinical trials we conduct may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| · | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| · | we or third-party API or component manufacturers with which we may contract may be unable to maintain a compliance status acceptable to the FDA or comparable foreign regulatory authorities or the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes identified in our marketing application; and |
| · | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may change significantly in a manner rendering our clinical data insufficient for approval. |
| · | issue warning letters or untitled letters asserting that we are in violation of the law; |
| · | impose restrictions on the marketing or manufacturing of the product; |
| · | seek an injunction or impose civil, criminal and/or administrative penalties, damages, assess monetary fines, require disgorgement, consider exclusion from participation in Medicare, Medicaid and other federal healthcare programs and require curtailment or restructuring of our operations; |
| · | suspend or withdraw regulatory approval; |
| · | suspend any ongoing clinical trials; |
| · | refuse to approve a pending NDA or supplements to an NDA submitted by us; |
| · | seize product; or |
| · | refuse to allow us to enter into government contracts. |
In an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees from our core business. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
| · | others may be able to make products that are similar to our products or product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed; |
| · | we or any potential future licensors might not have been the first to file patent applications covering certain of our inventions; |
| · | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| · | it is possible that our pending patent applications will not lead to issued patents; |
| · | issued patents that we own or have exclusively licensed may be held invalid or unenforceable as a result of legal challenges by our competitors; |
| · | issued patents that we own or have exclusively licensed may not provide coverage for all aspects of our products or product candidates in all countries; |
| · | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| · | we may not develop additional proprietary technologies that are patentable; and |
| · | the patents of others may have an adverse effect on our business. |
| · | sales of our approved products; |
| · | results of clinical trials of our current and any future product candidates or those of our competitors; |
| · | the success of competitive drugs or therapies; |
| · | regulatory or legal developments in the United States and other countries; |
| · | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| · | the recruitment or departure of key personnel; |
| · | the level of expenses related to our current and any future product candidates or clinical development programs; |
| · | the results of our efforts to discover, develop, acquire or in-license additional product candidates; |
| · | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| · | our inability to obtain or delays in obtaining adequate drug supply for any approved drug or inability to do so at acceptable prices; |
| · | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| · | significant lawsuits, including patent or stockholder litigation; |
| · | variations in our financial results or those of companies that are perceived to be similar to us; |
| · | changes in the structure of healthcare payment systems; |
| · | market conditions in the pharmaceutical and biotechnology sectors; |
| · | general economic, industry and market conditions; and |
| · | the other factors described in this “Risk Factors” section. |
| · | whether the FDA requires us to complete additional, unanticipated studies, trials or other activities prior to approving any of our current and future product candidates, which would likely delay any such approval; |
| · | our execution of other collaborative, licensing or similar arrangements and the timing of payments we may make or receive under these arrangements; |
| · | variations in the level of expenses related to our future development programs; |
| · | any product liability or intellectual property infringement lawsuit in which we may become involved; |
| · | regulatory developments affecting any of our other current and future product candidates, or the product candidates of our competitors; and |
| · | if any of our current or future product candidates receive regulatory approval, the level of underlying demand for such product candidate and wholesaler buying patterns. |
| · | authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
| · | limiting the removal of directors by the stockholders; |
| · | creating a classified board of directors; |
| · | establishing a supermajority stockholder vote requirement for amending certain provisions of our amended and restated certificate of incorporation or our amended and restated bylaws; |
| · | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
| · | eliminating the ability of stockholders to call a special meeting of stockholders; and |
| · | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
| · | Sandoz. By court order in August 2016, our ANDA patent litigation case against Sandoz has been dismissed without prejudice for lack of subject matter jurisdiction because Sandoz is no longer pursuing a Paragraph IV certification for its proposed generic version of Suboxone Sublingual Film, and therefore is no longer challenging the validity or infringement of our Orange Book-listed patents. |
| · | Mylan. The case against Mylan was settled and the Court signed a Consent Judgment in September 2017 disposing of the entire case. |
| · | Par. All cases against Par were resolved pursuant to a May 2018 settlement agreement between us, Indivior, and Par and certain of its affiliates. |
| · | The first, a declaratory judgment action brought by BDSI against Indivior and Aquestive, seeks declarations of invalidity and non-infringement of U.S. Patents Nos. 7,897,080, or the ’080 patent, 8,652,378, or the ’378 patent, and 8,475,832, or the ’832 patent. This case stayed pending inter partes review of the ’832 patent and reexamination of the ’080 patent. |
| · | The second was filed by us and Indivior related to BDSI’s infringing Bunavail product, and alleges infringement of our patent, U.S. Patent No. 8,765,167, or the ’167 patent. This case was initially filed in September 2014 in the U.S. District Court for the District of New Jersey but was transferred to North Carolina. Shortly after the case was filed, BDSI filed an IPR challenging the asserted ’167 patent. On March 24, 2016, the Patent Trial and Appeal Board, or the PTAB, issued a final written decision finding the ’167 patent was not unpatentable. The case was stayed in May 2016 pending the final determination of the ’167 patent IPR proceedings. Following the PTAB’s February 7, 2019 decisions on remand denying institution (discussed further below), we and Indivior submitted a notice to the Court on February 15, 2019 notifying the Court that the stay should be lifted as result of the PTAB’s decisions. BDSI also sent a letter to the Court on February 13, 2019, indicating its intent to appeal the PTAB’s decisions. The parties are awaiting further action from the Court. |
Year Ended December 31, | ||||||||||||
| (In thousands, except per share data amounts) | 2018 | 2017 | 2016 | |||||||||
| Revenues | $ | 67,430 | $ | 66,918 | $ | 51,785 | ||||||
| Costs and expenses: | ||||||||||||
| Manufacture and supply | 20,988 | 19,820 | 16,378 | |||||||||
| Research and development | 23,112 | 22,133 | 15,450 | |||||||||
| Selling, general and administrative | 72,264 | 25,078 | 20,804 | |||||||||
| Total costs and expenses | 116,364 | 67,031 | 52,632 | |||||||||
| Loss from operations | (48,934 | ) | (113 | ) | (847 | ) | ||||||
| Other expenses: | ||||||||||||
| Interest expense | (7,711 | ) | (7,707 | ) | (6,143 | ) | ||||||
| Interest income | 552 | — | — | |||||||||
| Loss on extinguishment of debt | — | — | (757 | ) | ||||||||
| Loss on impairment of investment | — | — | (1,006 | ) | ||||||||
| Change in fair value of warrant | (5,278 | ) | (1,123 | ) | (750 | ) | ||||||
| Other expense | (5 | ) | — | (99 | ) | |||||||
| Net loss before income taxes | (61,376 | ) | (8,943 | ) | (9,602 | ) | ||||||
| Income taxes | — | — | — | |||||||||
| Net loss | (61,376 | ) | (8,943 | ) | (9,602 | ) | ||||||
| Dividends on redeemable preferred interests | — | (2,480 | ) | (2,342 | ) | |||||||
| Net loss attributable to common shares/ members’ interests | (61,376 | ) | (11,423 | ) | (11,944 | ) | ||||||
| Comprehensive net loss | $ | (61,376 | ) | $ | (11,423 | ) | $ | (11,944 | ) | |||
| Net loss per share – basic and diluted | $ | (2.96 | ) | |||||||||
| Weighted-average number of common shares outstanding - basic and diluted | 20,725,526 | |||||||||||
| December 31, | ||||||||||||
| Selected Balance Sheet Data: | 2018 | 2017 | 2016 | |||||||||
| Cash and cash equivalents | $ | 60,599 | $ | 17,379 | $ | 9,209 | ||||||
| Working capital | 41,249 | 12,813 | 12,526 | |||||||||
| Total assets | 86,851 | 43,116 | 39,389 | |||||||||
| Loans Payable, net | 47,203 | 45,507 | 38,650 | |||||||||
| Total liabilities | 76,771 | 69,611 | 56,965 | |||||||||
| Accumulated deficit | (61,376 | ) | (120,093 | ) | (108,670 | ) | ||||||
| Total shareholders’ equity/members’ deficit | 10,080 | (68,596 | ) | (57,197 | ) | |||||||
| · | Libervant, a buccal soluble film formulation of diazepam used as a rescue therapy for breakthrough epileptic seizures and an adjunctive therapy for use in recurrent convulsive seizures, for which a pre-NDA meeting was held in December 2018 with the FDA. The results of this meeting resulted in a plan to complete a small single-dose crossover study comparing Libervant to the reference listed drug, Diastat. The Company is developing a plan for a rolling NDA submission to begin in 2019; and |
| · | Exservan, an oral soluble film formulation of riluzole for the treatment of Amyotrophic Lateral Sclerosis, or ALS, for which we have submitted an NDA in the first quarter of 2019. |
| · | AQST-108, a sublingual soluble film formulation for the treatment of anaphylaxis intended to provide an alternative to injection treatments such as EpiPen. After the Company’s first human proof of concept trials, a re-formulated and more advanced prototype has been developed, for which we began additional phase 1 proof of concept trials in early 2019; and |
| · | AQST-305, a sublingual soluble film formulation of octreotide for the treatment of acromegaly and neuroendocrine tumors, for which we are undertaking human proof of concept trials at this time; we expect to do additional formulation work, and to re-enter proof of concept trials in 2019. |
| · | fund commercialization investments for Sympazan (launched in December 2018) and Libervant, our epilepsy products, and ALS product, Exservan; |
| · | continue clinical development of our complex molecules, AQST-108 and AQST-305; |
| · | identify and evaluate new pipeline candidates in CNS diseases and other indications; and |
| · | fund working capital requirements and expected capital expenditures as a result of the launch of proprietary products and related growth. |
| · | the costs necessary to successfully complete our development efforts of our proprietary product candidates; |
| · | continued revenue from our licensed products; |
| · | the levels and timing of revenues and costs of commercialization of our late stage CNS product candidates; and |
| · | the infrastructure costs to support a public company. |
| · | employee-related expenses, including compensation, benefits, share-based compensation and travel expense; |
| · | external research and development expenses incurred under arrangements with third parties, such as contract research organizations, investigational sites and consultants; |
| · | the cost of acquiring, developing and manufacturing clinical study materials; and |
| · | costs associated with preclinical and clinical activities and regulatory operations. |
| Change | ||||||||||||||||
| 2018 | 2017 | $ | % | |||||||||||||
| (In thousands, except %) | ||||||||||||||||
| Manufacture and supply revenue | $ | 37,319 | $ | 40,092 | $ | (2,773 | ) | (7 | %) | |||||||
| License and royalty revenue | 24,699 | 23,133 | 1,566 | 7 | % | |||||||||||
| Co-development and research fees | 5,184 | 3,693 | 1,491 | 40 | % | |||||||||||
Proprietary product sales, net | 228 | - | 228 | NM | ||||||||||||
| Revenues | 67,430 | 66,918 | 512 | 1 | % | |||||||||||
| Change | ||||||||||||||||
| 2018 | 2017 | $ | % | |||||||||||||
| (In thousands, except %) | ||||||||||||||||
| Manufacturing and supply | $ | 20,988 | $ | 19,820 | $ | 1,168 | 6 | % | ||||||||
| Research and development | 23,112 | 22,133 | 979 | 4 | % | |||||||||||
| Selling, general and administrative | 72,264 | 25,078 | 47,186 | 188 | % | |||||||||||
| Interest expense | 7,711 | 7,707 | 4 | 0 | % | |||||||||||
| Interest income | (552 | ) | - | 552 | NM | |||||||||||
| Other | 5,283 | 1,123 | 4,160 | 370 | % | |||||||||||
| Year Ended December 31, | ||||||||
| (In thousands) | 2018 | 2017 | ||||||
| Clinical Trials | $ | 8,526 | $ | 10,486 | ||||
| Labor - R&D staff | 7,620 | 5,114 | ||||||
| Regulatory Submission Costs & Support | - | 2,330 | ||||||
| All Other R&D | 6,966 | 4,203 | ||||||
| Total | $ | 23,112 | $ | 22,133 | ||||
| · | $50,000 from debt facilities further described below; |
| · | $77,457 from pre-IPO equity financings, with most of these proceeds received in 2008 and prior years and |
| · | $63,482 from our IPO including the over-allotment option. |
| (In thousands) | 2018 | 2017 | ||||||
| Net cash (used for) provided by operating activities | $ | (12,991 | ) | $ | 5,824 | |||
| Net cash used for investing activities | (1,824 | ) | (2,068 | ) | ||||
| Net cash provided by financing activities | 58,035 | 4,414 | ||||||
| Net increase in cash and cash equivalents | $ | 43,220 | $ | 8,170 | ||||
| · | continued funding of our commercialization costs for Sympazan, our first proprietary product launched in December 2018, |
| · | continued funding of our development and pre-launch commercialization of CNS products Libervant, Exservan and our other proprietary product candidates; |
| · | continued revenue from our proprietary and licensed products, and; |
| · | the infrastructure and administrative costs to support being a public company. |
| Contractual Obligations | Total | Less than one year | One to three years | Four to five years | After five years | |||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Perceptive debt principal and interest | $ | 60,550 | $ | 10,403 | $ | 50,147 | $ | — | $ | — | ||||||||||
| Operating lease obligations | 3,675 | 1,233 | 2,377 | 65 | — | |||||||||||||||
| Total contractual obligations | $ | 64,225 | $ | 11,636 | $ | 52,524 | $ | 65 | $ | — | ||||||||||
Number | Description | |
Amended and Restated Certificate of Incorporation of Aquestive Therapeutics, Inc., dated as of July 27, 2018 (filed as Exhibit 3.1 to the Current Report on Form 8-K of the Company, as filed on July 27, 2018, and incorporated by reference herein). | ||
| Amended and Restated Bylaws of Aquestive Therapeutics, Inc. (filed as Exhibit 3.6 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Form of Common Stock Certificate of Aquestive Therapeutics, Inc. (filed as Exhibit 4.1 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Warrant to Purchase 11,625,437 senior common equity interests to Perceptive Credit Holdings, LP, dated as of January 1, 2018 (filed as Exhibit 4.2 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Registration Rights Agreement, dated as of June 24, 2018, by and between Aquestive Partners, LLC and certain of the holders of its membership interests (filed as Exhibit 4.3 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Form of Indemnification Agreement, by and between Aquestive Therapeutics, Inc and its directors and officers (filed as Exhibit 10.1 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Credit Agreement and Guaranty, dated as of August 16, 2016, by and among MonoSol Rx, LLC, the Subsidiary Guarantors from time to time parties thereto, the Lenders from time to time parties thereto and Perceptive Credit Holdings, LP, as administrative agent and collateral agent for the Lenders (filed as Exhibit 10.2 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Omnibus Amendment No. 1 to Credit Agreement and Guaranty, dated as of January 1, 2018, by and among MonoSol Rx, LLC, the Lenders party thereto and Perceptive Credit Holdings, LP, as administrative agent and collateral agent for the Lenders (filed as Exhibit 10.3 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Amendment No. 2 to Credit Agreement and Guaranty and Consent, dated as of May 21, 2018, by and among Aquestive Therapeutics, Inc., the Lenders party thereto and Perceptive Credit Holdings, LP, as administrative agent and collateral agent for the Lenders (filed as Exhibit 10.4 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
Executive Employment Agreement, dated as of June 30, 2018, by and between Aquestive Therapeutics, Inc. and Keith J. Kendall (filed as Exhibit 10.5 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924), and incorporated by reference herein). | ||
| Executive Employment Agreement, dated as of June 26, 2018, by and between Aquestive Therapeutics, Inc. and Daniel Barber (filed as Exhibit 10.6 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). |
| Executive Employment Agreement, dated as of June 26, 2018, by and between Aquestive Therapeutics, Inc. and John T. Maxwell (filed as Exhibit 10.7 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
Executive Employment Agreement, dated as of July 9, 2018, by and between Aquestive Therapeutics, Inc. and A. Mark Schobel (filed as Exhibit 10.8 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924), and incorporated by reference herein). | ||
| Commercial Exploitation Agreement, by and between MonoSol Rx, LLC and Reckitt Benckiser Pharmaceuticals Inc., dated as of August 15, 2008 (as amended on August 19, 2009, November 13, 2009, March 30, 2010, October 13, 2010, December 15, 2010, December 9, 2011, December 1, 2012, October 14, 2013 (by Addendum A), July 30, 2014 (by Addendum B), and January 12, 2017) (filed as Exhibit 10.9 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Agreement, by and between MonoSol Rx, LLC and Indivior UK Limited, dated as of September 24, 2017 (filed as Exhibit 10.10 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Agreement to Terminate CLA, by and between MonoSol Rx, LLC and KemPharm, Inc., dated as of March 20, 2012 (filed as Exhibit 10.11 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| License Agreement, by and between MonoSol Rx, LLC and Cynapsus Therapeutics Inc., dated as of April 1, 2016 (filed as Exhibit 10.12 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
| Industrial Lease Agreement, by and between Ashland Northwest Partners, L.P. and MonoSol Rx, LLC, dated as of October 24, 2006 (as amended on October 24, 2011 and February 8, 2018) (filed as Exhibit 10.13 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
Aquestive Therapeutics, Inc. 2018 Equity Incentive Plan (filed as Exhibit 10.14 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924) and incorporated by reference herein). | ||
Aquestive Therapeutics, Inc. Employee Stock Purchase Plan (filed as Exhibit 10.15 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924) and incorporated by reference herein). | ||
| Form of Stock Option Agreement (filed as Exhibit 10.16 to the Registration Statement on Form S-1 of the Company (File No. 333-225924), as filed on June 27, 2018, and incorporated by reference herein). | ||
Form of Stock Option Agreement under the Aquestive Therapeutics, Inc. 2018 Equity Incentive Plan (filed as Exhibit 10.17 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924) and incorporated by reference herein). | ||
Form of Restricted Stock Unit Agreement under the Aquestive Therapeutics, Inc. 2018 Equity Incentive Plan (filed as Exhibit 10.18 to the Pre-Effective Amendment No. 1, as filed on July 16, 2018, to the Registration Statement on Form S-1 of the Company (File No. 333-225924) and incorporated by reference herein). | ||
| Executive Employment Agreement, dated as of September 10, 2018, by and between Aquestive Therapeutics, Inc. and Lori J. Braender (filed as Exhibit 10.4 to the Quarterly Report on Form 10-Q of the Company, as filed on November 6, 2018, and incorporated by reference herein). | ||
| Consent of KPMG LLP, Independent Registered Public Accounting Firm (filed herewith). | ||
| Certification of Principal Executive Officer pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (filed herewith). | ||
| Certification of Principal Financial Officer pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (filed herewith). | ||
| Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (furnished herewith). | ||
| 32.2* | Certification of Principal Accounting Officer Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (furnished herewith). | |
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document |
| † | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment that has been granted by the Securities and Exchange Commission. |
| * | Furnished herewith and not deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and shall not be deemed to be incorporated by reference to any filing under the Securities Act of 1933, as amended, or the Exchange Act (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing. |
| + | Indicates a management contract or compensatory plan. |
| AQUESTIVE THERAPEUTICS, INC. | ||
| Date: March 14, 2019 | By: | /s/ Keith J. Kendall |
| Keith J. Kendall | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) | ||
| Signature | Title | Date | ||
| /s/ Keith J. Kendall | President, Chief Executive Officer and Member of the Board of Directors | March 14, 2019 | ||
| Keith J. Kendall | (Principal Executive Officer) | |||
/s/John T. Maxwell | Chief Financial Officer | March 14. 2019 | ||
| John T. Maxwell | (Principal Financial Officer and Principal Accounting Officer) | | ||
| /s/ Douglas Bratton | Member of the Board of Directors | March 14, 2019 | ||
| Douglas Bratton | ||||
| /s/ Gregory Brown | Member of the Board of Directors | March 14, 2019 | ||
| Gregory Brown, M.D. | ||||
| /s/ John Cochran | Member of the Board of Directors | March 14, 2019 | ||
| John Cochran | ||||
| /s/ Santo Costa | Chairman of the Board of Directors | March 14, 2019 | ||
| Santo Costa | ||||
| /s/ Nancy Lurker | Member of the Board of Directors | March 14, 2019 | ||
| Nancy Lurker | ||||
| /s/ James S. Scibetta | Member of the Board of Directors | March 14, 2019 | ||
| James S. Scibetta |
Page Number | ||
| Report of Independent Registered Public Accounting Firm | ||
| Consolidated Balance Sheets as of December 31, 2018 and 2017 | ||
| Consolidated Statements of Operations and Comprehensive Loss for the Years Ended December 31, 2018 and 2017 | ||
Consolidated Statements of Changes in Shareholders’ Equity/Members’ Deficit for the Years Ended December 31, 2018 and 2017 | ||
| Consolidated Statements of Cash Flows for the Years Ended December 31, 2018 and 2017 | ||
| Notes to the Consolidated Financial Statements |
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 60,599 | $ | 17,379 | ||||
| Trade and other receivables, net | 6,481 | 6,179 | ||||||
| Inventories | 5,441 | 4,014 | ||||||
| Prepaid expenses and other current assets | 1,680 | 591 | ||||||
| Total current assets | 74,201 | 28,163 | ||||||
| Property and equipment, net | 12,207 | 13,460 | ||||||
| Intangible assets, net | 204 | 254 | ||||||
| Other assets | 239 | 1,239 | ||||||
| Total assets | $ | 86,851 | $ | 43,116 | ||||
| Liabilities and shareholders’ equity/members’ deficit | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 20,436 | $ | 9,601 | ||||
| Accrued expenses | 7,195 | 4,402 | ||||||
| Deferred revenue | 721 | 1,347 | ||||||
| Loans payable, current | 4,600 | — | ||||||
| Total current liabilities | 32,952 | 15,350 | ||||||
| Loans payable, net | 42,603 | 45,507 | ||||||
| Warrant liability | — | 7,673 | ||||||
| Asset retirement obligations | 1,216 | 1,081 | ||||||
| Total liabilities | 76,771 | 69,611 | ||||||
| Commitments and contingencies (note 18) | ||||||||
| Redeemable Preferred A-3 interests and accrued dividends | — | 5,896 | ||||||
| Redeemable Preferred A-2 interests and accrued dividends | — | 36,205 | ||||||
| Shareholders/members’ equity/(deficit): | ||||||||
| Preferred A interests, no par value. Authorized 100,000,000 units; 16,886,750 units issued and outstanding at December 31, 2017 | — | 16,887 | ||||||
| Preferred A-1 interests, no par value. Authorized 100,000,000 units; 21,526,850 units issued and outstanding at December 31, 2017 | — | 21,883 | ||||||
| Common interests, no par value. Authorized 500,000,000 units; 121,228,353 units issued and outstanding at December 31, 2017 | — | 12,727 | ||||||
| Common stock, $.001 par value. Authorized 250,000,000 shares; 24,957,309 shares issued and outstanding at December 31, 2018 | 25 | — | ||||||
| Additional paid-in capital | 71,431 | — | ||||||
| Accumulated deficit | (61,376 | ) | (120,093 | ) | ||||
| Total shareholders’ equity/members’ (deficit) | 10,080 | (68,596 | ) | |||||
| Total liabilities and shareholders’ equity/ members’ deficit | $ | 86,851 | $ | 43,116 | ||||
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Revenues | $ | 67,430 | $ | 66,918 | ||||
| Costs and expenses: | ||||||||
| Manufacture and supply | 20,988 | 19,820 | ||||||
| Research and development | 23,112 | 22,133 | ||||||
| Selling, general and administrative | 72,264 | 25,078 | ||||||
| Total costs and expenses | 116,364 | 67,031 | ||||||
| Loss from operations | (48,934 | ) | (113 | ) | ||||
| Other income (expenses): | ||||||||
| Interest expense | (7,711 | ) | (7,707 | ) | ||||
| Interest income | 552 | — | ||||||
| Change in fair value of warrant | (5,278 | ) | (1,123 | ) | ||||
| Other expense | (5 | ) | — | |||||
| Net loss before income taxes | (61,376 | ) | (8,943 | ) | ||||
| Income taxes | — | — | ||||||
| Net loss | (61,376 | ) | (8,943 | ) | ||||
| Dividends on redeemable preferred interests | — | (2,480 | ) | |||||
| Net loss attributable to common shares/ members’ interests | (61,376 | ) | (11,423 | ) | ||||
| Comprehensive loss | $ | (61,376 | ) | $ | (11,423 | ) | ||
| Net loss per share – basic and diluted | $ | (2.96 | ) | |||||
| Weighted-average number of common shares outstanding - basic and diluted | 20,725,526 | |||||||
Preferred A Interests | Preferred A-1 Interests | Common Interests | Common Stock | Additional Paid-in Capital | Accumulated Deficit | Total Shareholders’ Equity/ Members’ Deficit | ||||||||||||||||||||||||||||||||||||||
Units | Amount | Units | Amount | Units | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2016 | 16,886,750 | $ | 16,887 | 21,526,850 | $ | 21,883 | 118,785,104 | $ | 11,243 | — | $ | — | 1,460 | $ | (108,670 | ) | $ | (57,197 | ) | |||||||||||||||||||||||||
| Dividends on preferred interests | — | — | — | — | — | — | — | — | — | (2,480 | ) | (2,480 | ) | |||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | (8,943 | ) | (8,943 | ) | |||||||||||||||||||||||||||||||
| Issuance of common interests upon exercise of warrants | — | — | — | — | 2,443,249 | 1,484 | — | — | (1,460 | ) | — | 24 | ||||||||||||||||||||||||||||||||
| Balance at December 31, 2017 | 16,886,750 | $ | 16,887 | 21,526,850 | $ | 21,883 | 121,228,353 | $ | 12,727 | — | $ | — | $ | — | $ | (120,093 | ) | $ | (68,596 | ) | ||||||||||||||||||||||||
| Reorganization to C-Corporation | (16,886,750 | ) | (16,887 | ) | (21,526,850 | ) | (21,883 | ) | (121,228,353 | ) | (12,727 | ) | 5,000 | — | (26,495 | ) | 120,093 | 42,101 | ||||||||||||||||||||||||||
| Effect of Stock Split | — | — | — | — | — | — | 15,072,647 | 15 | (15 | ) | — | — | ||||||||||||||||||||||||||||||||
| Common Stock issued to performance unit plan participants | — | — | — | — | — | — | 4,922,353 | 5 | 19,118 | — | 19,123 | |||||||||||||||||||||||||||||||||
| Reclassification of warrant liability to equity | — | — | — | — | — | — | — | — | 12,952 | — | 12,952 | |||||||||||||||||||||||||||||||||
Cash received for warrant exercise | — | — | — | — | — | — | — | — | 116 | — | 116 | |||||||||||||||||||||||||||||||||
| Common Stock issued related to initial public offering | — | — | — | — | — | — | 4,925,727 | 5 | 68,709 | — | 68,714 | |||||||||||||||||||||||||||||||||
| Issuance costs related to initial public offering | — | — | — | — | — | — | — | — | (5,232 | ) | — | (5,232 | ) | |||||||||||||||||||||||||||||||
| Share-based compensation | — | — | — | — | — | — | 31,582 | — | 2,278 | — | 2,278 | |||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | (61,376 | ) | (61,376 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2018 | — | $ | — | — | $ | — | — | $ | — | 24,957,309 | $ | 25 | $ | 71,431 | $ | (61,376 | ) | $ | 10,080 | |||||||||||||||||||||||||
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (61,376 | ) | $ | (8,943 | ) | ||
| Adjustments to reconcile net loss to net cash (used for) provided by operating activities: | ||||||||
| Depreciation and amortization | 3,186 | 3,750 | ||||||
| Change in fair value of warrant | 5,278 | 1,123 | ||||||
| Share-based compensation | 29,940 | — | ||||||
| Asset retirement obligation accretion | 130 | 122 | ||||||
| Amortization of intangible | 50 | 51 | ||||||
| Amortization of debt issuance costs and discounts | 1,696 | 1,860 | ||||||
| Non-cash interest expense | — | 33 | ||||||
| Bad debt provision (recovery) | 3 | (53 | ) | |||||
Other, net | 104 | — | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Trade receivables and other receivables | (409 | ) | 4,691 | |||||
| Inventories, net | (1,427 | ) | (1,128 | ) | ||||
| Prepaid expenses and other current assets | (1,140 | ) | (171 | ) | ||||
| Accounts payable | 11,319 | 2,943 | ||||||
| Accrued expenses | 281 | | 1,001 | |||||
| Deferred revenue | (626 | ) | 545 | |||||
| Net cash (used for) provided by operating activities | (12,991 | ) | 5,824 | |||||
| Cash flows from investing activities: | ||||||||
| Capital expenditures | (1,824 | ) | (2,068 | ) | ||||
| Net cash (used for) investing activities | (1,824 | ) | (2,068 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from initial offering of common stock | 68,714 | — | ||||||
| Proceeds from warrant exercise | 116 | 24 | ||||||
| Proceeds from issuance of debt | — | 5,000 | ||||||
| Payments for deferred financing costs | (4,768 | ) | (610 | ) | ||||
| Payments for taxes on share-based compensation | (6,027 | ) | — | |||||
| Net cash provided by financing activities | 58,035 | 4,414 | ||||||
| Net increase in cash and cash equivalents | 43,220 | 8,170 | ||||||
| Cash and cash equivalents: | ||||||||
| Beginning of period | 17,379 | 9,209 | ||||||
| End of period | $ | 60,599 | $ | 17,379 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash payments for interest | $ | 6,049 | $ | 5,814 | ||||
| Net increase in capital expenditures included in accounts payable and accrued expenses: | 104 | 20 | ||||||
| Net (decrease) increase in offering costs included in accounts payable and accrued expenses | (588 | ) | 588 | |||||
| Accrued Series A-2 and A-3 preferred dividends | — | 2,480 | ||||||
| Accrued withholding tax for share based compensation | 2,515 | — | ||||||
| Deferred financing costs charged to additional paid in capital | 5,232 | — | ||||||
| Noncash component of warrants exercised | 12,591 | — | ||||||
December 31, 2017 | ||||
| Redeemable Preferred A-3 Interests | 5,055,000 | |||
| Redeemable Preferred A-2 Interests | 82,071,200 | |||
| Nonredeemable A-1 interests | 21,526,850 | |||
| Nonredeemable A interests | 16,886,750 | |||
| Common Interests | 121,228,353 | |||
| 246,768,153 | ||||
| (i) | increase the authorized number of capital stock from 25,000 to 350,000,000 shares, |
| (ii) | authorize the Non-Voting Common Stock, and |
| (iii) | affect a stock split of the Company’s common stock, par value $0.001 per share, such that each share be subdivided and reclassified into 37,212 shares of Voting Common Stock, par value $0.001 per share. |
| · | Level 1 — Quoted prices in active markets for identical assets or liabilities. Cash and cash equivalents consisted of cash in bank checking and savings accounts and money market funds which are all Level 1 assets. |
| · | Level 2 — Observable inputs (other than Level 1 quoted prices), such as quoted prices in active markets for similar assets or liabilities, quoted prices in markets that are not active for identical or similar assets or liabilities, or other inputs that are observable or can be corroborated by observable market data. The Company currently has no Level 2 assets or liabilities. |
| · | Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to determining the fair value of the assets or liabilities, including pricing models, discounted cash flow methodologies and similar techniques. As of December 31, 2018, the Company has no level 3 assets or liabilities. |
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Manufacture and supply revenue | $ | 37,319 | $ | 40,092 | ||||
| License and royalty revenue | 24,699 | 23,133 | ||||||
| Co-development and research fees | 5,184 | 3,693 | ||||||
| Proprietary product sales, net | 228 | — | ||||||
| Revenues | $ | 67,430 | $ | 66,918 | ||||
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| United States | $ | 64,565 | $ | 63,710 | ||||
| Ex-United States | 2,865 | 3,208 | ||||||
| Revenues | $ | 67,430 | $ | 66,918 | ||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Accounts receivable | $ | 6,610 | $ | 6,156 | ||||
| Other receivables | 33 | 78 | ||||||
| Less: allowance for bad debt | (58 | ) | (55 | ) | ||||
| Less: sales-related allowances | (104 | ) | — | |||||
| Trade and other receivables, net | $ | 6,481 | $ | 6,179 | ||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Allowance for doubtful accounts at beginning of year | $ | 55 | $ | 108 | ||||
| Additions charged to bad debt expense | 53 | — | ||||||
| Write-downs charged against the allowance | (50 | ) | — | |||||
| Recoveries of amounts previously reserved | — | (53 | ) | |||||
| Allowance for doubtful accounts at end of year | $ | 58 | $ | 55 | ||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Balance at December 31, 2017 | $ | — | $ | — | ||||
| Provision related to sales in 2018 | 104 | — | ||||||
| Credits and payments | — | — | ||||||
| Balance at December 31, 2018 | $ | 104 | $ | — | ||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Raw material | $ | 1,283 | $ | 725 | ||||
| Packaging material | 2,975 | 2,225 | ||||||
| Finished goods | 1,183 | 1,064 | ||||||
| Total inventory | $ | 5,441 | $ | 4,014 | ||||
| December 31, | |||||||||
| Useful Lives | 2018 | 2017 | |||||||
| Machinery | 3-15 yrs | $ | 20,681 | $ | 20,056 | ||||
| Furniture and fixtures | 3-15 yrs | 1,150 | 1,109 | ||||||
| Leasehold improvements | (a) | 21,333 | 21,271 | ||||||
| Computer, network equipment and software | 3-7 yrs | 2,579 | 2,108 | ||||||
| Construction in progress | 1,655 | 921 | |||||||
| 47,398 | 45,465 | ||||||||
| Less: accumulated depreciation and amortization | (35,191 | ) | (32,005 | ) | |||||
| Total property and equipment, net | $ | 12,207 | $ | 13,460 | |||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Purchase technology-based intangible | $ | 2,358 | $ | 2,358 | ||||
| Purchased patent | 509 | 509 | ||||||
| 2,867 | 2,867 | |||||||
| Less: accumulated amortization | (2,663 | ) | (2,613 | ) | ||||
| Intangible assets, net | $ | 204 | $ | 254 | ||||
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Accrued Compensation | $ | 3,604 | $ | 3,805 | ||||
| Accrued withholding tax for share-based compensation | 2,515 | — | ||||||
| Real estate and personal property taxes | 388 | 372 | ||||||
| Accrued distribution expenses | 481 | — | ||||||
| Other | 207 | 225 | ||||||
| Total accrued expenses | $ | 7,195 | $ | 4,402 | ||||
| Balance as of December 31, 2016 | $ | 6,550 | ||
| Changes in fair value recognized | 1,123 | |||
| Balance as of December 31, 2017 | 7,673 | |||
| Changes in fair value recognized | 5,278 | |||
| Exercise of warrants | (12,951 | ) | ||
| Balance as of December 31, 2018 | $ | — |
| Balance at December 31, 2016 | $ | 959 | ||
| Additions | — | |||
| Accretion | 122 | |||
| Balance at December 31, 2017 | 1,081 | |||
| Additions | 5 | |||
| Accretion | 130 | |||
| Balance at December 31, 2018 | $ | 1,216 |
Year Ended December 31, 2018 | ||||
| Numerator: | ||||
| Net loss | $ | (61,376 | ) | |
| Denominator: | ||||
| Weighted-average number of common shares – basic and diluted | 20,725,526 | |||
| Loss per common share – basic and diluted | $ | (2.96 | ) | |
| Expense classification: | Year Ended December 31, 2018 | |||
| Manufacturing and supply | $ | 414 | ||
| Research and development | 2,583 | |||
| Selling, general and administrative | 26,943 | |||
| Total share-based compensation expenses | $ | 29,940 | ||
| Share-based compensation from: | ||||
| Non-voting common shares (A) | $ | 27,298 | ||
| Restricted Stock Units (B) | 1,085 | |||
| Stock Options (B) | 1,557 | |||
| Total share-based compensation expenses | $ | 29,940 | ||
| Valuation assumptions: | |
| Discount for lack of marketability | 34% |
| Volatility | 90% |
| Weighted average cost of capital | 27.5% |
Number of Units | Weighted Average Grant Date Fair Value Per Share | |||||||
| (In thousands) | ||||||||
| Unvested at Plan adoption | — | $ | — | |||||
| Granted | 265 | 14.83 | ||||||
| Vested | (60 | ) | 15.03 | |||||
| Unvested, December 31, 2018 | 205 | $ | 14.77 | |||||
| (in 000s, except share price data) | Number of Options | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term in Years | Aggregate Intrinsic Value | ||||||||||||
| Outstanding at January 1, 2018 | — | |||||||||||||||
| Granted | 1,033 | $ | 14.72 | 9.55 | $ | — | ||||||||||
| Exercised, Forfeited, Expired | — | |||||||||||||||
| Outstanding at December 31, 2018 | 1,033 | $ | 14.72 | 9.55 | $ | — | ||||||||||
| Vested or expected to vest at December 31, 2018 | 962 | $ | 14.72 | 9.55 | $ | — | ||||||||||
| Exercisable at December 31, 2018 | 87 | $ | 15.02 | 9.56 | $ | — | ||||||||||
| Expected dividend yield | 0% |
| Expected volatility | 90% |
| Expected term (years) | 5.8 - 6.1 |
| Risk-free interest rate | 2.8 - 2.9% |
| December 31, | ||||||||
| 2018 | 2017 | |||||||
| Deferred tax assets: | ||||||||
| Accounts receivable | $ | 16 | $ | 14 | ||||
| Inventory | 120 | 49 | ||||||
| Accrued expenses | 15 | 12 | ||||||
| NOL carryforwards | 10,899 | 1,330 | ||||||
| Interest limitation imposed by the TJCA | 2,124 | — | ||||||
| Stock Compensation | 1,224 | — | ||||||
| Other | 260 | 319 | ||||||
| Property and equipment | 1,380 | 1,145 | ||||||
| Orphan Drug and R&D Tax Credits | 3,917 | 113 | ||||||
| 19,955 | 2,982 | |||||||
| Deferred tax liabilities: | ||||||||
| Intangible assets | (39 | ) | (45 | ) | ||||
| Prepaid expenses | (407 | ) | (148 | ) | ||||
| (446 | ) | (193 | ||||||
| Valuation Allowance | (19,509 | ) | (2,789 | ) | ||||
| Net deferred tax asset/(liability) | $ | — | $ | — | ||||
Year Ended December 31, | ||||||||
| 2018 | 2017 | |||||||
| Income taxes at statutory rate | 21.00 | % | 34.00 | % | ||||
| Increase (decrease) resulting from: | ||||||||
| State income tax | 7.04 | 4.06 | ||||||
| Permanent differences | (7.09 | ) | (8.90 | ) | ||||
| Research & development credit | 4.40 | 1.72 | ||||||
| Return to provision | 1.48 | — | ||||||
| Effect of state rate change | 0.41 | — | ||||||
| Valuation allowance | (27.24 | ) | (13.54 | ) | ||||
| Effect of the deferred rate change | — | (17.34 | ) | |||||
| Effective tax rate | 0.00 | % | 0.00 | % | ||||
| Amount | ||||
| 2019 | $ | 1,233 | ||
| 2020 | 881 | |||
| 2021 | 815 | |||
| 2022 | 681 | |||
| 2023 | 65 | |||
| Thereafter | 0 | |||
| Total | $ | 3,675 | ||
| · | Sandoz. By court order in August 2016, our ANDA patent litigation case against Sandoz has been dismissed without prejudice for lack of subject matter jurisdiction because Sandoz is no longer pursuing a Paragraph IV certification for its proposed generic version of Suboxone Sublingual Film, and therefore is no longer challenging the validity or infringement of our Orange Book-listed patents. |
| · | Mylan. The case against Mylan was settled and the Court signed a Consent Judgment in September 2017 disposing of the entire case. |
| · | Par. All cases against Par were resolved pursuant to a May 2018 settlement agreement between Aquestive, Indivior, and Par and certain of its affiliates. |
| · | The first, a declaratory judgment action brought by BDSI against Indivior and Aquestive, seeks declarations of invalidity and non-infringement of U.S. Patents Nos. 7,897,080, or the ’080 patent, 8,652,378, or the ’378 patent, and 8,475,832, or the ’832 patent. This case stayed pending inter partes review of the ’832 patent and reexamination of the ’080 patent. |
| · | The second was filed by the Company and Indivior related to BDSI’s infringing Bunavail product, and alleges infringement of our patent, U.S. Patent No. 8,765,167, or the ’167 patent. This case was initially filed in September 2014 in the U.S. District Court for the District of New Jersey but was transferred to North Carolina. Shortly after the case was filed, BDSI filed an IPR challenging the asserted ’167 patent. On March 24, 2016, the Patent Trial and Appeal Board, or the PTAB, issued a final written decision finding the ’167 patent was not unpatentable. The case was stayed in May 2016 pending the final determination of the ’167 patent IPR proceedings. Following the PTAB’s February 7, 2019, decisions on remand denying institution (discussed further below), Aquestive and Indivior submitted a notice to the Court on February 15, 2019, notifying the Court that the stay should be lifted as result of the PTAB’s decisions. BDSI also sent a letter to the Court on February 13, 2019, indicating its intent to appeal the PTAB’s decisions. The parties are awaiting further action from the Court. |
| Three Months Ended | |||||||||||||||
March 31, 2018 | June 30, 2018 | September 30, 2018 | December 31, 2018 | ||||||||||||
| Revenues | $ | 23,411 | $ | 13,928 | $ | 13,267 | $ | 16,824 | |||||||
| Manufacture and supply | 5,636 | 4,973 | 5,592 | 4,787 | |||||||||||
| Total costs and expenses | 18,106 | 46,614 | 22,471 | 29,173 | |||||||||||
| Net income (loss) | 4,099 | (36,493) | (15,038) | (13,944) | |||||||||||
| Basic and diluted net income (loss) per common share | $ | 0.27 | $ | (1.90) | $ | (0.64) | $ | (0.56) | |||||||
| Three Months Ended | |||||||||||||||
March 31, 2017 | June 30, 2017 | September 30, 2017 | December 31, 2017 | ||||||||||||
| Revenues | $ | 16,436 | $ | 11,142 | $ | 27,146 | $ | 12,194 | |||||||
| Manufacture and supply | 4,184 | 5,141 | 4,880 | 5,615 | |||||||||||
| Total costs and expenses | 15,655 | 15,201 | 16,725 | 19,450 | |||||||||||
| Net income (loss) | (1,457) | (5,897) | 8,451 | (10,040) | |||||||||||