As filed with the Securities and Exchange Commission on September 9, 2024
Registration No. 333-281204
Delaware | | | 2834 | | | 26-1622110 |
(State or other jurisdiction of incorporation or organization) | | | (Primary Standard Industrial Classification Code Number) | | | (I.R.S. Employer Identification No.) |
Large accelerated filer | | | ☐ | | | Accelerated filer | | | ☒ |
Non-accelerated filer | | | ☐ | | | Smaller reporting company | | | ☒ |
| | | | | Emerging growth company | | | ☐ |
| • | 5,544,719 shares of Common Stock issuable upon the conversion of 166,341.592 outstanding shares of Series A Preferred Stock; |
| • | 2,937,903 shares of Common Stock issuable upon the conversion of 2,937,903 outstanding shares of Series B Preferred Stock; |
| • | 1,981,189 shares of Common Stock issuable upon the exercise of outstanding stock options at a weighted-average exercise price of $9.99; |
| • | 448,211 shares of Common Stock issuable upon the vesting of outstanding restricted stock units; |
| • | 974,954 shares of Common Stock issuable upon the exercise of outstanding warrants; |
| • | 27,270 shares of Common Stock reserved for issuance under the Cartesian Therapeutics, Inc. Amended and Restated 2016 Incentive Award Plan (the “Old Cartesian Plan”); |
| • | 3,511,101 shares of Common Stock reserved for issuance under our Amended and Restated 2016 Incentive Award Plan (the “2016 Plan”); |
| • | 253,377 shares of Common Stock reserved for issuance under our Amended and Restated 2018 Employment Inducement Incentive Award Plan (the “2018 Plan”); and |
| • | 45,795 shares of Common Stock reserved for issuance pursuant to our 2016 Employee Stock Purchase Plan (the “2016 ESPP”). |
| • | We may fail to obtain stockholder approval of the conversion of our Series B Preferred Stock. |
| • | We are a development-stage company and we expect to incur losses for the foreseeable future and may never achieve or maintain profitability. |
| • | We will need substantial additional funding in order to complete development of our product candidates and commercialize our products, if approved. If we are unable to raise capital when needed and on terms favorable to us, we could be forced to delay, reduce or eliminate our product development programs or commercialization efforts. |
| • | We develop our mRNA-based product candidates by leveraging our proprietary technology and our manufacturing platform, RNA Armory®, which is an unproven approach to the treatment of autoimmune disease. We are early in most of our clinical development efforts and may not be successful in our efforts to build a pipeline of product candidates and develop marketable drugs. |
| • | Clinical drug development is inherently risky and involves a lengthy and expensive process, which is subject to a number of factors, many of which are outside of our control. We may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates. |
| • | We expect to continue to grow our manufacturing capabilities and resources and we must incur significant costs to develop this expertise and/or rely on third parties to manufacture our products. |
| • | We rely, and expect to continue to rely, on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including by failing to meet deadlines for the completion of such trials. |
| • | If we or our licensors are unable to adequately protect our proprietary technology, or obtain and maintain issued patents that are sufficient to protect our product candidates, others could compete against us more directly, which would negatively impact our business. |
| • | We have been in the past and may in the future be subject to stockholder litigation. |
| • | The failure to successfully integrate the businesses of Selecta and Old Cartesian in the expected timeframe would adversely affect the Company’s future results. |
| • | We have identified a material weakness in our internal control over financial reporting and may identify additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls, which may result in material misstatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations. |
| • | design, initiation and completion of preclinical studies and clinical trials with positive results; |
| • | reliance on third parties, including but not limited to collaborators, licensees, clinical research organizations and contract manufacturing organizations; |
| • | receipt of marketing approvals from applicable regulatory authorities; |
| • | obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates and not infringing or violating patents or other intellectual property of third parties; |
| • | manufacturability, manufacturing, logistics, and stability of our cell therapies, including autologous cell therapies; |
| • | growing our internal cGMP manufacturing capabilities to support commercial manufacturing or making arrangements with third-party manufacturers; |
| • | launching commercial sales of our products, if and when approved, whether alone or in collaboration with others; |
| • | acceptance of our products, if and when approved, by patients and the medical community; |
| • | effectively competing with other therapies; |
| • | obtaining and maintaining coverage and adequate reimbursement by third-party payors, including government payors, for our products, if approved; |
| • | maintaining an acceptable safety profile of our products following approval; and |
| • | maintaining and growing an organization of scientists and businesspeople who can develop and commercialize our product candidates and technology. |
| • | clinical trials of our product candidates may produce unfavorable, incomplete or inconclusive results; |
| • | we may be unable to manufacture our product candidates, which in some cases such as mRNA CAR-T, are manufactured on a patient-by-patient basis; |
| • | regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site or may place a clinical hold on existing clinical trials; |
| • | we may experience delays in reaching, or fail to reach, agreement on acceptable terms with contract research organizations (“CROs”), or clinical trial sites; |
| • | we may be unable to recruit suitable patients to participate in a clinical trial, the number of patients required for clinical trials of our product candidates may be larger than we expect, enrollment in these clinical trials may be slower than we expect or participants may drop out of these clinical trials at a higher rate than we expect, or enrollment could be affected by the ongoing conflicts in Ukraine and the Middle East; |
| • | the number of clinical trial sites required for clinical trials of our product candidates may be larger than we expect; |
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; |
| • | we may have to suspend or terminate clinical trials of our product candidates for various reasons, including a finding that the participants are being exposed to unacceptable health risks; |
| • | investigators, regulators, data safety monitoring boards or institutional review boards may require that we or our investigators suspend or terminate clinical research, or we may decide to do so ourselves; |
| • | investigators may deviate from the trial protocol, fail to conduct the trial in accordance with regulatory requirements or misreport study data; |
| • | the cost of clinical trials of our product candidates may be greater than we expect or we may have insufficient resources to pursue or complete certain aspects of our clinical trial programs or to do so within the timeframe we planned; |
| • | the supply or quality of raw materials or manufactured product candidates (whether provided by us or third parties) or other materials necessary to conduct clinical trials of our product candidates may be insufficient, inadequate or not available at an acceptable cost, or in a timely manner, or we may experience interruptions in supply; |
| • | laboratories that we rely upon to perform certain quality control tests may become unavailable, or their services could be delayed; |
| • | regulators may revise the requirements for approving our product candidates, or such requirements may not be as we expect; |
| • | the FDA or comparable foreign regulatory authorities may disagree with our clinical trial design or our interpretation of data from preclinical studies and clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design of our clinical trials; |
| • | regarding trials managed by our existing or any future collaborators, our collaborators may face any of the above issues, and may conduct clinical trials in ways they view as advantageous to them but potentially suboptimal for us; and |
| • | geopolitical events may affect international and overseas trial sites in ways beyond our control. |
| • | be delayed in obtaining marketing approval for our product candidates, if at all; |
| • | obtain marketing approval in some countries and not in others; |
| • | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| • | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| • | be subject to additional post-marketing testing requirements; or |
| • | have a product removed from the market after obtaining marketing approval. |
| • | foreign regulatory requirements that could burden or limit our ability to conduct our clinical trials; |
| • | increased costs and heightened supply constraints associated with the acquisition of standard of care drugs and/or combination or comparator agents for which we may bear responsibility in certain jurisdictions; |
| • | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| • | foreign exchange fluctuations; |
| • | more burdensome manufacturing, customs, shipment and storage requirements; |
| • | cultural differences in medical practice and clinical research; |
| • | lack of consistency in standard of care from country to country; |
| • | diminished protection of intellectual property in some countries; |
| • | changes in country or regional regulatory requirements; and |
| • | geopolitical instability or wars in regions outside of the United States where we conduct clinical trials may impact ongoing clinical trials. |
| • | regulatory authorities may withdraw approvals of such product; |
| • | regulatory authorities may require the addition of labeling statements, such as a boxed warning or a contraindication; |
| • | regulatory authorities may impose additional restrictions on the marketing of, or the manufacturing processes for, the particular product; |
| • | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
| • | we could be sued and held liable for harm caused to patients, or become subject to fines, injunctions or the imposition of civil or criminal penalties; |
| • | our reputation may suffer; and |
| • | we could be required to develop a risk evaluation and mitigation strategies (“REMS”), plan to prevent, monitor and/or manage a specific serious risk by informing, educating and/or reinforcing actions to reduce the frequency and/or severity of the event. |
| • | we or our current or future collaborators may not be able to initiate or continue clinical trials of product candidates that are under development; |
| • | we or our current or future collaborators may be delayed in submitting regulatory applications, or receiving regulatory approvals, for our product candidates; |
| • | we may lose the cooperation of our collaborators; |
| • | our facilities and those of our CMOs, and our products could be the subject of inspections by regulatory authorities that could have a negative outcome and result in delays in supply; |
| • | we may be required to cease distribution or recall some or all batches of our products or take action to recover clinical trial material from clinical trial sites; and |
| • | ultimately, we may not be able to meet the clinical and commercial demands for our products. |
| • | the efficacy, safety and potential advantages compared to alternative treatments; |
| • | our ability to manufacture and distribute cell therapies in a timely and secure manner; |
| • | our ability to offer our products for sale at competitive prices; |
| • | the convenience and ease of administration compared to alternative treatments; |
| • | product labeling or product insert requirements of the FDA or foreign regulatory authorities, including any limitations or warnings contained in a product’s approved labeling, including any black box warning or REMS; |
| • | the willingness of the target patient population to try new treatments and of physicians to prescribe these treatments; |
| • | our ability to hire and retain a sales force; |
| • | the strength of marketing and distribution support; |
| • | the availability of third-party coverage and adequate reimbursement for our product candidates, once approved; |
| • | the prevalence and severity of any side effects; and |
| • | any restrictions on the use of our products together with other medications. |
| • | regulatory investigations, product recalls or withdrawals, or labeling, marketing or promotional restrictions; |
| • | decreased demand for any product candidates or products that we may develop; |
| • | injury to our reputation and significant negative media attention; |
| • | loss of clinical trial participants or increased difficulty in enrolling future participants; |
| • | significant costs to defend the related litigation or to reach a settlement; |
| • | substantial payments to trial participants or patients; |
| • | loss of revenue; |
| • | reduced resources of our management to pursue our business strategy; |
| • | the inability to commercialize any products that we may develop; |
| • | distraction of management’s attention from our primary business; and |
| • | substantial monetary awards to patients or other claimants. |
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| • | the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government claims for payment that are false or fraudulent. Private individuals (e.g., whistleblowers) can bring these actions on behalf of the government; in addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| • | the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which imposes criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH”), and their respective implementing regulations, which also impose obligations, including mandatory contractual terms, on certain types of people and entities with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| • | the federal Physician Payments Sunshine Act (the “Sunshine Act”), which requires applicable manufacturers of certain products for which payment is available under a federal healthcare program to report annually to the government information related to certain payments or other “transfers of value” made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other health care professionals beginning in 2022, and teaching hospitals, as well as ownership and investment interests held by the physicians and their immediate family members; |
| • | analogous state laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by third-party payors, including private insurers; and requirements to comply with federal and pharmaceutical industry compliance guidelines; |
| • | state data privacy and price transparency laws, many of which differ from each other in significant ways and often are broader than and not preempted by HIPAA or the Sunshine Act, thus complicating compliance efforts; by way of example, the California Consumer Privacy Act (“CCPA”), which went into effect January 1, 2020, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context; and |
| • | similar healthcare laws and regulations in the EU and other jurisdictions, including reporting requirements detailing interactions with and payments to healthcare providers and laws governing the privacy and security of certain protected information, such as the General Data Protection Regulation (“GDPR”), which imposes obligations and restrictions on the collection and use of personal data relating to individuals located in the EU (including health data); in addition, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the EU will be regulated. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom’s departure from the EU. |
| • | continue the research and development of our product candidates; |
| • | increase and develop our manufacturing and distribution capacities; |
| • | discover and develop additional product candidates; |
| • | seek to maintain and enter into collaboration, licensing and other agreements, including, but not limited to research and development, and/or commercialization agreements; |
| • | seek regulatory approvals for any product candidates that successfully complete clinical trials; |
| • | potentially establish a sales, marketing and distribution infrastructure and scale up internal manufacturing capabilities to commercialize any products for which we may obtain regulatory approval; |
| • | maintain, expand and protect our intellectual property portfolio, including through licensing arrangements; |
| • | add clinical, scientific, operational, financial and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts; |
| • | experience any delays or encounter any issues with any of the above, including, but not limited to, failed studies, complex results, safety issues or other regulatory, manufacturing or scale-up challenges; and |
| • | are exposed to broad macroeconomic conditions including inflation and supply chain tightness which could result in us paying more, or being unable, to access goods and services. |
| • | the scope, progress, results and costs of our clinical trials, preclinical development, manufacturing, laboratory testing and logistics; |
| • | the number of product candidates that we pursue and the speed with which we pursue development; |
| • | our headcount growth and associated costs; |
| • | the costs, timing and outcome of regulatory review of our product candidates; |
| • | the costs and timing of future commercialization activities, including manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval; |
| • | the revenue, if any, from commercial sales of our product candidates for which we receive marketing approval; |
| • | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; |
| • | the effect of competing technological and market developments; and |
| • | the extent to which we acquire or invest in businesses, products and technologies, including entering into licensing or collaboration arrangements for product candidates. |
| • | multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| • | failure by us to obtain and maintain regulatory approvals for the use of our product candidates in various countries; |
| • | additional potentially relevant third-party patent rights; |
| • | complexities and difficulties in obtaining protection of and enforcing our intellectual property rights; |
| • | difficulties in staffing and managing foreign operations; |
| • | complexities associated with managing multiple-payor reimbursement regimes, government payors or patient self-pay systems; |
| • | limits on our ability to penetrate international markets; |
| • | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our product candidates and exposure to foreign currency exchange rate fluctuations, which could result in increased operating expenses and reduced revenues; |
| • | natural disasters, political and economic instability, including wars, events of terrorism and political unrest, outbreak of disease, including the COVID-19 pandemic, boycotts, curtailment of trade and other business restrictions, economic sanctions, and economic weakness, including inflation; |
| • | changes in diplomatic and trade relationships; |
| • | challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| • | restriction on cross-border investment, including enhanced oversight by the Committee on Foreign Investment in the United States and substantial restrictions on investment from China; |
| • | certain expenses including, among others, expenses for travel, translation and insurance; |
| • | legal risks, including use of the legal system by the government to benefit itself or affiliated entities at our expense, including expropriation of property; |
| • | regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the FCPA its books and records provisions, or its anti-bribery provisions; and |
| • | risks that we may suffer reputational harm as a result of our operations in Russia. |
| • | disruption in our relationships with future customers or with current or future distributors or suppliers as a result of such a transaction; |
| • | unexpected liabilities related to acquired companies; |
| • | difficulties integrating acquired personnel, technologies and operations into our existing business; |
| • | diversion of management time and focus from operating our business to acquisition integration challenges; |
| • | increases in our expenses and reductions in our cash available for operations and other uses; |
| • | possible write-offs or impairment charges relating to acquired businesses; and |
| • | inability to develop a sales force for any additional product candidates. |
| • | the success of competitive products or technologies; |
| • | results or progress, or changes in approach or timelines, of clinical trials of our product candidates or those of our competitors; |
| • | failure or discontinuation of any of our development programs; |
| • | commencement of, termination of, or any development related to any collaboration or licensing arrangement; |
| • | regulatory or legal developments in the United States and other countries; |
| • | development of new product candidates that may address our markets and make our product candidates less attractive; |
| • | changes in physician, hospital or healthcare provider practices that may make our product candidates less useful; |
| • | announcements by us, our collaborators or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
| • | announcement or market expectation of additional financing efforts; |
| • | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| • | the recruitment or departure of key personnel; |
| • | the level of expenses related to any of our product candidates or clinical development programs; |
| • | failure to meet or exceed financial estimates, projections or development timelines of the investment community or that we provide to the public; |
| • | the results of our efforts to discover, develop, acquire or in-license additional product candidates or products; |
| • | actual or expected changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| • | variations in our financial results or those of companies that are perceived to be similar to us; |
| • | changes in the structure of healthcare payment systems; |
| • | sale of Common Stock by us or our stockholders in the future as well as the overall trading volume of our Common Stock; |
| • | changes in the composition of our stockholder base; |
| • | activity in the options market for shares of our Common Stock; |
| • | market conditions in the pharmaceutical and biotechnology sectors; |
| • | general economic, industry and market conditions; and |
| • | the other factors described in this “Risk Factors” section. |
| • | stockholder approval of the conversion rights of the Series B Preferred Stock; |
| • | our expectations regarding the conversion of the Series B Preferred Stock and our Series A Non-Voting Convertible Preferred Stock, par value $0.0001 per share (“Series A Preferred Stock”), into Common Stock; |
| • | any future payouts under the contingent value right (“CVR”), issued to our holders of record as of the close of business on December 4, 2023; |
| • | our future results of operations and financial position, business strategy, and the length of time that we believe our existing cash resources will fund our operations; |
| • | our market size and our potential growth opportunities; |
| • | our preclinical and future clinical development activities; |
| • | the efficacy and safety profile of our product candidates; |
| • | the potential therapeutic benefits and economic value of our product candidates; |
| • | the timing and results of preclinical studies and clinical trials; |
| • | the expected impact of macroeconomic conditions, including inflation, increasing interest rates and volatile market conditions, current or potential bank failures; |
| • | global events, including the ongoing conflicts between Russia and Ukraine and between Hamas and Israel and geopolitical tensions in China on our operations; |
| • | the receipt and timing of potential regulatory designations, approvals and commercialization of product candidates; |
| • | our ability to prevent or minimize the effects of litigation and other contingencies; |
| • | our status as a preclinical and development-stage company and our expectation to incur losses in the future, and the possibility that we never achieve or maintain profitability; |
| • | uncertainties with respect to our ability to access future capital; |
| • | our ability to maximize the value of our pipeline of product candidates; |
| • | our unproven approach to therapeutic intervention; |
| • | our ability to enroll patients in clinical trials, timely and successfully complete those trials and receive necessary regulatory approvals; |
| • | our ability to continue to grow our manufacturing capabilities and resources; |
| • | our ability to manufacture our product candidates, which in some cases are manufactured on a patient-by-patient basis; |
| • | our ability to access manufacturing facilities and to receive or manufacture sufficient quantities of our product candidates; |
| • | our ability to maintain our existing or future collaborations or licenses and to seek new collaborations, licenses or partnerships; |
| • | the impact of resurgence of the COVID-19 pandemic on our operations, the continuity of our business, including our preclinical studies and clinical trials, and general economic conditions; |
| • | our ability to protect and enforce our intellectual property rights; |
| • | federal, state, and foreign regulatory requirements, including U.S. Food and Drug Administration (“FDA”) regulation of our product candidates; |
| • | our ability to obtain and retain key executives and retain qualified personnel; and |
| • | developments relating to our competitors and our industry, including the impact of government regulation. |
| • | advance Descartes-08 for MG into Phase 3 development; |
| • | continue to develop our preclinical and clinical-stage product candidates; |
| • | seek regulatory approvals for any product candidates that successfully complete clinical trials; and |
| • | maintain, expand and protect our intellectual property portfolio, including through licensing arrangements. |
| • | the scope, progress, results and costs of our clinical trials, preclinical development, manufacturing, laboratory testing and logistics; |
| • | the number of product candidates that we pursue and the speed with which we pursue development; |
| • | our headcount growth and associated costs; |
| • | the costs, timing and outcome of regulatory review of our product candidates; |
| • | the costs and timing of future commercialization activities, including manufacturing, marketing, sales and distribution, for any of our product candidates for which we receive marketing approval; |
| • | the revenue, if any, from commercial sales of our product candidates for which we receive marketing approval; |
| • | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; |
| • | the effect of competing technological and market developments; and |
| • | the extent to which we acquire or invest in businesses, products and technologies, including entering into licensing or collaboration arrangements for product candidates. |
| | | Six Months Ended June 30, | ||||
(In thousands) | | | 2024 | | | 2023 |
Cash (used in) and provided by: | | | | | ||
Operating activities | | | $(30,363) | | | $(18,660) |
Investing activities | | | (2,189) | | | 28,112 |
Financing activities | | | 43,151 | | | (2,437) |
Effect of exchange rate changes on cash | | | 9 | | | (49) |
Net change in cash, cash equivalents, and restricted cash | | | $10,608 | | | $6,966 |
| | | Year Ended December 31, | |||||||
(In thousands) | | | 2023 | | | 2022 | | | 2021 |
Cash (used in) and provided by: | | | | | | | |||
Operating activities | | | $(51,161) | | | $(31,631) | | | $(60,382) |
Investing activities | | | 34,609 | | | (15,002) | | | (17,140) |
Financing activities | | | (13,145) | | | 39,215 | | | 52,897 |
Effect of exchange rate changes on cash | | | (53) | | | 20 | | | (3) |
Net change in cash, cash equivalents, and restricted cash | | | $(29,750) | | | $(7,398) | | | $(24,628) |
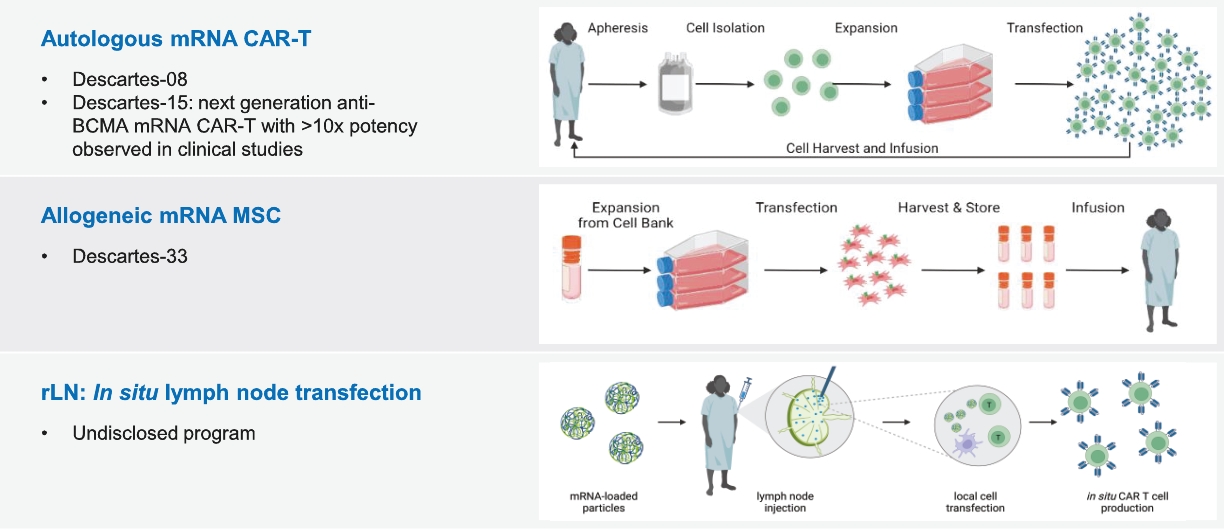

| Phase 1. | The biological product candidate is evaluated in a limited population of patients or healthy volunteers to identify the maximum tolerated dose, recommended Phase 2 dose, possible adverse effects and safety risks. For the types of products and therapeutic areas we focus on, Phase 1 studies will generally be done in patients and not healthy volunteers. |
| Phase 2. | The biological product candidate is evaluated in a broader population to evaluate safety further and preliminarily evaluate the efficacy of the product for specific targeted diseases, and to determine the optimal dosing schedule. |
| Phase 3. | Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical study sites. These clinical trials are intended to establish the overall risk/benefit ratio of the product candidate and provide an adequate basis for product labeling. |
| • | The IRA requires manufacturers to pay rebates for Medicare Part B and Part D drugs whose price increases exceed inflation. |
| • | The IRA eliminates the so-called “donut hole” under Medicare Part D beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and requiring manufacturers to subsidize, through a newly established manufacturer discount program, 10% of Part D enrollees’ prescription costs for brand drugs below the out-of-pocket maximum and 20% once the out-of-pocket maximum has been reached. |
| • | The IRA delays the rebate rule that would require pass through of pharmacy benefit manager rebates to beneficiaries. |
| • | The IRA directs the Centers for Medicare and Medicaid Services (“CMS”), to engage in price-capped negotiation for certain Medicare Part B and Part D products. Specifically, the IRA’s Price Negotiation Program applies to high-expenditure single-source drugs and biologics that have been approved for at least seven or 11 years, respectively, among other negotiation selection criteria, beginning with 10 high-cost drugs paid for by Medicare Part D starting in 2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and beyond. The negotiated prices will be capped at a statutorily determined ceiling price. There are certain statutory exemptions from the IRA’s Price Negotiation Program, such as for a drug that has only a single orphan drug designation and is approved only for an indication or indications within the scope of such designation. The IRA’s Price Negotiation Program is currently the subject of legal challenges. |
Plan category | | | Number of securities to be issued upon exercise of outstanding stock options, warrants and rights | | | Weighted-average exercise price of outstanding options, warrants and rights(1) | | | Number of securities remaining available for future issuance under equity compensation plans(2) |
| | | (a) | | | (b) | | | (c) | |
Equity compensation plans approved by security holders(3) | | | —(4) | | | $—(4) | | | 795,941(5) |
Equity compensation plans not approved by security holders(6) | | | 790,977.299(7) | | | $4.34 | | | 278,360(8) |
Total | | | 790,977.299 | | | $4.34 | | | 1,074,301 |
| (1) | Represents the weighted-average exercise price of outstanding options and is calculated without taking into account outstanding RSUs. |
| (2) | Pursuant to the terms of the 2016 Plan, the number of shares of Common Stock available for issuance under the 2016 Plan automatically increases on each January 1, until and including January 1, 2034, by an amount equal to the lesser of: (a) 4% of the number of shares of the Company’s Common Stock outstanding on the last day of the applicable preceding calendar year and (b) such smaller number of shares as is determined by our Board of Directors. Pursuant to the terms of the 2016 ESPP, the number of shares of Common Stock available for issuance under the 2016 ESPP automatically increases on each January 1, until and including January 1, 2026, by an amount equal to the lesser of: (a) 1% of the number of shares of the Company’s Common Stock outstanding on the last day of the applicable preceding calendar year and (b) such smaller number of shares as is determined by our Board of Directors. |
| (3) | Includes the 2016 Plan and the 2016 ESPP. |
| (4) | There were no outstanding stock options, warrants or rights under the 2016 Plan and the 2016 ESPP as of December 31, 2023. |
| (5) | Represents 750,146 shares of Common Stock available for issuance under the 2016 Plan and 45,795 shares of Common Stock available for issuance under the 2016 ESPP. |
| (6) | Includes the 2018 Plan and the Old Cartesian Plan. 1,247,268 shares of Common Stock are issuable upon exercise of outstanding stock options under the Old Cartesian Plan at a weighted-average exercise price of $2.76. See Note 13 to our consolidated audited financial statements as of and for the year ended December 31, 2023 included elsewhere in the registration statement of which this prospectus forms a part for a description of the material features of the 2018 Plan and the Old Cartesian Plan. |
| (7) | Includes outstanding options to purchase 776,865 shares of Common Stock and to purchase 14,112.299 shares of Series A Preferred Stock, convertible to 470,403 shares of Common Stock, under the Old Cartesian Plan and no outstanding stock options, warrants or rights under the 2018 Plan as of December 31, 2023. Following the automatic conversion of the majority of our Series A Preferred Stock into Common Stock on April 8, 2024 (the “Series A Preferred Stock Automatic Conversion”), the options exercisable for 14,112.299 shares of Series A Preferred Stock became exercisable for Common Stock. |
| (8) | Represents 150,043 shares of Common Stock available for issuance under the 2018 Plan and 128,317 shares of Common Stock available for issuance under the Old Cartesian Plan. |
Name of Director | | | Age | | | Served as a Director Since | | | Position(s) with Cartesian |
Class I Directors: | | | | | | | |||
Michael Singer, M.D., Ph.D. | | | 50 | | | 2023 | | | Director |
Timothy A. Springer, Ph.D. | | | 76 | | | 2016 | | | Director |
Patrick Zenner | | | 77 | | | 2017 | | | Director |
Class II Directors: | | | | | | | |||
Carrie S. Cox | | | 66 | | | 2019 | | | Chairman of the Board |
Murat Kalayoglu, M.D., Ph.D. | | | 51 | | | 2023 | | | Director |
Kemal Malik, MBBS | | | 61 | | | 2024 | | | Director |
Class III Directors: | | | | | | | |||
Timothy C. Barabe | | | 71 | | | 2016 | | | Director |
Carsten Brunn, Ph.D. | | | 54 | | | 2018 | | | President and Chief Executive Officer, Director |
Nishan de Silva, M.D., M.B.A. | | | 51 | | | 2021 | | | Director |
| | | | Female | | | Male | | | Non-Binary | | | Did Not Disclose Gender | | |
| | Directors | | | 1 | | | 7 | | | 0 | | | 1 | |
| | Number of Directors Who Identify in Any of the Categories Below | | | | ||||||||||
| | African American or Black | | | 0 | | | 0 | | | 0 | | | 0 | |
| | Alaskan Native or Native American | | | 0 | | | 0 | | | 0 | | | 0 | |
| | Asian | | | 0 | | | 2 | | | 0 | | | 0 | |
| | Hispanic or Latinx | | | 0 | | | 0 | | | 0 | | | 0 | |
| | Native Hawaiian or Pacific Islander | | | 0 | | | 0 | | | 0 | | | 0 | |
| | White | | | 1 | | | 4 | | | 0 | | | 0 | |
| | Two or More Races or Ethnicities | | | 0 | | | 0 | | | 0 | | | 0 | |
| | LGBTQ+ | | | 0 | | | 0 | | | 0 | | | 0 | |
| | Did Not Disclose Demographic Background | | | 0 | | | 1 | | | 0 | | | 1 | |
Name | | | Audit | | | Compensation | | | Nominating and Corporate Governance | | | Science, IP and Quality |
Timothy C. Barabe | | | Chair | | | — | | | X | | | — |
Carsten Brunn, Ph.D. | | | — | | | — | | | — | | | — |
Carrie S. Cox | | | X | | | Chair | | | — | | | — |
Nishan de Silva, M.D., M.B.A. | | | X | | | — | | | — | | | X |
Murat Kalayoglu, M.D., Ph.D. | | | — | | | — | | | — | | | Chair |
Kemal Malik, MBBS | | | — | | | X | | | — | | | X |
Michael Singer, M.D., Ph.D. | | | — | | | — | | | X | | | X |
Timothy A. Springer, Ph.D. | | | — | | | — | | | — | | | X |
Patrick Zenner | | | X | | | X | | | Chair | | | — |
| • | appointing, approving the compensation of, and assessing the independence of our independent registered public accounting firm; |
| • | overseeing the work of our independent registered public accounting firm, including through the receipt and consideration of reports from such firm; |
| • | reviewing and discussing with management and the independent registered public accounting firm our annual and quarterly financial statements and related disclosures; |
| • | monitoring our internal control over financial reporting, disclosure controls and procedures and code of business conduct and ethics; |
| • | discussing our risk management policies and conducting regular risk assessments related to all matters affecting the enterprise, including cybersecurity, and receives periodic reports on our cybersecurity risks and activities; |
| • | establishing policies regarding hiring employees from the independent registered public accounting firm and procedures for the receipt and retention of accounting related complaints and concerns; |
| • | meeting independently with our internal auditing staff, if any, independent registered public accounting firm and management; |
| • | reviewing and approving or ratifying any related person transactions; and |
| • | preparing the Audit Committee report required by the SEC rules. |
| • | annually reviewing and approving corporate goals and objectives relevant to Chief Executive Officer compensation; |
| • | reviewing and approving, or making recommendations to our Board of Directors with respect to, the compensation of our Chief Executive Officer and other executive officers; |
| • | overseeing an evaluation of our senior executives; |
| • | administering our cash and equity incentive plans; |
| • | reviewing and making recommendations to our Board of Directors with respect to director compensation; |
| • | reviewing and discussing annually with management our “Compensation Discussion and Analysis”; and |
| • | preparing the annual compensation committee report, if required by SEC rules. |
| • | identifying individuals qualified to become board members; |
| • | recommending to our Board of Directors the persons to be nominated for election as directors and to each board committee; |
| • | reviewing and making recommendations to our Board of Directors with respect to management succession planning; |
| • | developing and recommending to our Board of Directors corporate governance principles; and |
| • | overseeing a periodic assessment of our Board of Directors. |
| • | reviewing the Company’s research and development strategy as well as the Company’s long-term strategic goals and objectives, and monitoring the Company’s progress in achieving such goals and objectives; |
| • | advising the Board of Directors on scientific, technological, and research and development matters, and on strategic issues associated with the Company’s product pipeline and technology; |
| • | reviewing and discussing the effectiveness and competitiveness of the Company’s position and strategies in relation to emerging scientific and technology trends and activities relevant to the success of the Company’s product pipeline and technology; |
| • | reviewing the quality, direction, and competitiveness of the Company’s research and development programs, and product pipeline; |
| • | reviewing the organization, resources and capabilities of the Company’s research, analytical, chemistry, manufacturing, and controls, and clinical departments; |
| • | reviewing strategies and approaches to acquiring, in licensing, out licensing, and maintaining innovation and technology positions; |
| • | advising the Board of Directors on the scientific, medical, and technical aspects of significant acquisitions, in licenses, out licenses, and other strategic business development transactions; |
| • | assisting the Company in reviewing, as requested, the capabilities of the Company’s current and prospective key scientific, clinical, medical, or technical personnel and engagement with key opinion leaders, and the depth and breadth of the Company’s scientific resources; |
| • | advising the Board of Directors, and the committees of the Board of Directors, as requested, with regard to performance and succession planning of the Company’s officers and other leadership of the research and development, manufacturing, medical, and other technical or scientific functions within the Company; |
| • | reviewing and opining on the strategy for the Company’s intellectual property portfolio; |
| • | providing counsel and know-how to the Company’s management in the area of research and development and the Company’s product pipeline and technology; and |
| • | carrying out other tasks or providing other advice related to the Company’s product pipeline and technology as may be requested by the Board of Directors. |
Name | | | Age | | | Position |
Carsten Brunn, Ph.D.(1) | | | 54 | | | President and Chief Executive Officer, Director |
Blaine Davis | | | 50 | | | Chief Financial Officer |
Metin Kurtoglu, M.D., Ph.D. | | | 46 | | | Chief Technology Officer |
Chris Jewell, Ph.D. | | | 43 | | | Chief Scientific Officer |
Milos Miljkovic, M.D. | | | 40 | | | Chief Medical Officer |
Emily English, Ph.D. | | | 44 | | | Senior Vice President, Head of Manufacturing |
| (1) | For Dr. Brunn’s biographical information, see “Directors” above. |
| • | Carsten Brunn, Ph.D., our President and Chief Executive Officer (our “CEO”); |
| • | Blaine Davis, our Chief Financial Officer (our “CFO”); |
| • | Metin Kurtoglu, M.D., Ph.D., our Chief Technology Officer; |
| • | Chris Jewell, Ph.D., our Chief Scientific Officer; |
| • | Peter G. Traber, M.D., our former Chief Medical Officer; and |
| • | Lloyd Johnston, Ph.D., our former Chief Operations Officer. |
| • | On November 13, 2023, we announced our Merger with Old Cartesian, a clinical-stage biotechnology company pioneering mRNA cell therapies for autoimmune diseases. In connection with the Merger, we also announced a $60.25 million private placement financing led by Timothy A. Springer, Ph.D., a member of our Board of Directors. Since consummating the Merger, we have been focused on continuing to advance our pipeline of innovative cell therapies, with several clinical milestones expected in 2024. |
| • | In August 2023, we announced a strategic initiative designed to maximize stockholder value associated with our legacy product candidate, SEL-212. As part of the initiative, we announced plans to halt further investments in our pipeline programs outside of SEL-212 and Xork and to stop or discontinue non-essential activities. SEL-212 is a combination of our ImmTOR immune tolerance platform and a therapeutic uricase enzyme (pegadricase). In March 2023, we and our SEL-212 development partner, Sobi, reported positive Phase 3 data from the DISSOLVE I and II Phase 3, placebo-controlled, randomized clinical trials of SEL-212 for the treatment of patients with chronic refractory gout. Both trials met their primary endpoint, and SEL-212 was observed to be well-tolerated. In October 2023, we announced that we entered into an agreement to transition the manufacturing and development rights and remaining clinical operations of ImmTOR for SEL-212 to Sobi. Under the terms of the agreement, 15 of our employees transferred their employment to full-time positions at Sobi. |
Named Executive Officer | | | 2023 Base Salary Increase from 2022 | | | 2023 Annual Bonus Target as a Percentage of Base Salary | | | 2023 Annual Time- Based Stock Options (# of shares) | | | 2023 Annual Time- Based RSU Awards (# of shares) |
Carsten Brunn, Ph.D. | | | 5.4% | | | 55% | | | 42,499 | | | 9,426 |
Blaine Davis(1) | | | — | | | 40% | | | — | | | — |
Metin Kurtoglu, M.D., Ph.D.(2) | | | — | | | 40% | | | — | | | — |
Chris Jewell, Ph.D.(3) | | | — | | | 40% | | | — | | | — |
Peter G. Traber, M.D.(4) | | | 9.0% | | | 40% | | | 29,999 | | | 6,666 |
Lloyd Johnston, Ph.D.(5) | | | 4.5% | | | 40% | | | 15,833 | | | 3,333 |
| (1) | Mr. Davis was appointed Chief Financial Officer on November 28, 2022, and per the terms of his employment agreement was not eligible for a base salary increase, annual time-based stock options, or annual time-based RSU awards in 2022. Mr. Davis received a one-time grant of 41,666 options upon his hiring on November 28, 2022. |
| (2) | Dr. Kurtoglu was appointed Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (3) | Dr. Jewell was appointed Chief Scientific Officer on November 13, 2023. |
| (4) | Dr. Traber ceased to serve as our Chief Medical Officer on November 13, 2023. |
| (5) | Dr. Johnston ceased to serve as our Chief Operations Officer on November 13, 2023. |
Named Executive Officer | | | Title | | | Total Pay (2023) | | | Percentage of Pay (Fixed) | | | Percentage of Pay (Variable) |
Carsten Brunn, Ph.D. | | | President and Chief Executive Officer | | | $6,111,883 | | | 10.1% | | | 89.9% |
Blaine Davis | | | Chief Financial Officer | | | $2,494,759 | | | 17.6% | | | 82.4% |
Metin Kurtoglu, M.D., Ph.D.(1) | | | Chief Technology Officer | | | $71,243 | | | 70.6% | | | 29.4% |
Chris Jewell, Ph.D.(2) | | | Chief Scientific Officer | | | $53,100 | | | 70.6% | | | 29.4% |
Peter G. Traber, M.D.(3) | | | Former Chief Medical Officer | | | $3,780,920 | | | 12.1% | | | 87.9% |
Lloyd Johnston, Ph.D.(4) | | | Former Chief Operations Officer | | | $2,697,590 | | | 16.0% | | | 84.0% |
| (1) | Dr. Kurtoglu was appointed Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (2) | Dr. Jewell was appointed Chief Scientific Officer on November 13, 2023. |
| (3) | This row represents Dr. Traber’s total pay at target. Upon his departure as a full-time employee in December 2023, Dr. Traber forfeited his 2023 annual target bonus opportunity of $200,000. However, as discussed under “—Post-Employment Compensation” below, Dr. Traber was paid an amount of $200,000 as a term of his separation agreement, which was approved separately from the cash bonus plan for 2023 (the “2023 Bonus Plan”) by the Compensation Committee. |
| (4) | This row represents Dr. Johnston’s total pay at target. Upon his departure as a full-time employee in December 2023, Dr. Johnston forfeited his 2023 annual target bonus opportunity of $173,888. However, as discussed under “—Post-Employment Compensation” below, Dr. Johnston was paid an amount of $173,888 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee. |
| • | each option to acquire shares of Common Stock (a “Company Stock Option”) and each RSU award with respect to shares of Common Stock, in each case that was outstanding and unvested immediately prior to the Effective Time (as defined in the Merger Agreement), vested in full at the Effective Time; |
| • | each Company Option was canceled at the Effective Time, and in exchange therefor, former holders of such canceled Company Stock Options became entitled to receive (without interest), in consideration of the cancellation of such Company Stock Option, an amount in cash (less applicable tax withholdings) equal to the product of (A) the total number of shares of Common Stock subject to the unexercised portion of such Company Stock Option immediately prior to the Effective Time (determined after giving effect to the accelerated vesting) multiplied by (B) the excess, if any, of $2.06 (the “Cash-out Amount”) over the applicable exercise price per share of Common Stock under such Company Stock Option; provided, however, that, if the exercise price per share of Common Stock of any Company Stock Option was equal to or greater than the Cash-out Amount, such Company Stock Option was canceled and terminated without any consideration in respect thereof; and |
| • | each RSU award with respect to shares of Common Stock was cancelled at the Effective Time, and the former holder of such canceled RSU became entitled, in exchange therefor, to receive (without interest) an amount in cash (less applicable tax withholdings) equal to the product of (A) the total number of shares of Common Stock deliverable under such RSU immediately prior to the Effective Time (determined after giving effect to the accelerated vesting) multiplied by (B) the Cash-out Amount. |
Named Executive Officer | | | Company Stock Options Subject to Accelerated Vesting | | | RSUs Subject to Accelerated Vesting | | | Company Stock Options Canceled | | | RSUs Canceled | | | Cash Received for Canceled Company Stock Options | | | Cash Received for Canceled RSUs |
Carsten Brunn, Ph.D. | | | 2,154,651 | | | 521,125 | | | 5,569,100 | | | 521,125 | | | $1,309,750 | | | $1,073,518 |
Blaine Davis | | | 1,250,000 | | | — | | | 1,250,000 | | | — | | | $987,500 | | | — |
Peter G. Traber, M.D. | | | 1,339,155 | | | 291,200 | | | 2,250,300 | | | 291,200 | | | $837,000 | | | $599,872 |
Lloyd Johnston, Ph.D. | | | 738,752 | | | 173,000 | | | 1,667,357 | | | 173,000 | | | $497,550 | | | $356,380 |
Metin Kurtoglu, M.D., Ph.D. | | | — | | | — | | | — | | | — | | | — | | | — |
Chris Jewell, Ph.D. | | | — | | | — | | | — | | | — | | | — | | | — |
| • | the number of shares of Common Stock subject to each Old Cartesian Option assumed by the Company was determined by multiplying (A) the number of shares of Old Cartesian common stock that were subject to such Old Cartesian Option, as in effect immediately prior to the First Effective Time, by (B) the Exchange Ratio (as defined in the Merger Agreement), and rounding the resulting number down to the nearest whole number of shares of Common Stock; |
| • | the per-share exercise price for Common Stock issuable upon exercise of each Old Cartesian Option assumed by the Company was determined by dividing (A) the per share exercise price of Old Cartesian common stock subject to such Old Cartesian Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio and rounding the resulting exercise price up to the nearest whole cent; |
| • | the number of shares of Series A Preferred Stock subject to each Continuing Officer Option assumed by the Company was determined by multiplying (A) the number of shares of Old Cartesian common stock that were subject to such Continuing Officer Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio (as defined in the Merger Agreement), and (C) dividing such resulting number by 1,000 and rounding the resulting number down to the nearest 1/1000th of a share of Series A Preferred Stock; |
| • | the per share exercise price for Series A Preferred Stock issuable upon exercise of each Continuing Officer Option assumed by the Company was determined by dividing (A) the per share exercise price of Old Cartesian common stock subject to such Continuing Officer Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio, and (C) multiplying the resulting number by 1,000 and rounding the resulting exercise price up to the nearest whole cent; and |
| • | any restriction on the exercise of any Old Cartesian Option assumed by the Company, including the Continuing Officer Options, will continue in full force and effect and, except as expressly provided in the Merger Agreement, the term, exercisability, vesting schedule and other provisions of such Old Cartesian Option will otherwise remain unchanged. |
Named Executive Officer | | | Number of Securities Underlying Unexercised Options Exercisable(1) | | | Number of Securities Underlying Unexercised Options Not Yet Exercisable(1) | | | Option Exercise Price | | | Option Expiration Date |
Metin Kurtoglu, M.D., Ph.D. | | | 213,820 | | | — | | | $1.41 | | | 11/6/2026 |
| | 14,254 | | | — | | | $3.23 | | | 4/25/2031 | ||
| | 3,563 | | | 10,691 | | | $3.23 | | | 2/29/2032 | ||
Chris Jewell, Ph.D. | | | 84,638 | | | 57,909 | | | $3.23 | | | 1/15/2033 |
| (1) | The Continuing Officer Options were exercisable initially only for shares of Series A Preferred Stock. Following the conversion of the majority of the Company’s outstanding shares of Series A Preferred Stock, the Continuing Officer Options were converted into options to purchase shares of Common Stock. The numbers of shares presented in this column are shares of Common Stock. |
| ✔ | Compensation Committee Independence - Our Board of Directors maintains a Compensation Committee comprised solely of independent directors who have established effective means for communicating with our stockholders regarding their executive compensation ideas and concerns. |
| ✔ | Compensation Committee Advisor Independence - The Compensation Committee engages and retains its own advisors. In 2023, the Compensation Committee engaged Compensia as compensation consultant to assist with its responsibilities. Compensia performs no consulting or other services for the Company. |
| ✔ | Annual Compensation Review - The Compensation Committee conducts an annual review of our executive compensation philosophy and strategy, including a review of the compensation peer group used for comparative purposes. |
| ✔ | Compensation-Related Risk Assessment - We conduct an annual evaluation of our compensation programs, policies, and practices to ensure that they reflect an appropriate level of risk-taking but do not encourage our employees to take excessive or unnecessary risks that could have a material adverse impact on our Company. |
| ✔ | Emphasize Long-Term Equity Compensation - The Compensation Committee uses equity awards to |
| ✔ | Limited Executive Perquisites - We provide only modest amounts of perquisites or other personal benefits that serve a sound business purpose to the Named Executive Officers. In addition, the Named Executive Officers participate in our health and welfare benefit programs on the same terms as all of our employees. |
| ✔ | “Double-Trigger” Change in Control Arrangements - The post-employment compensation arrangements for our executive officers, including our Named Executive Officers, are based on a “double-trigger” arrangement that provides for the receipt of payments and benefits only in the event of (i) a change in control of the Company and (ii) a qualifying termination of employment. |
| ✔ | Executive Clawback Policy - Effective as of October 2, 2023, we instituted a new executive clawback policy, compliant with new SEC rules, which allows the Board of Directors to recover any incentive awards from any executive officer in the event the Company is required to file a restatement of its financial reporting as a result of that executive officer’s fraud or misconduct. |
| ✔ | Reasonable Change-in-Control Arrangements - We believe the post-employment compensation arrangements for our executive officers, including our Named Executive Officers, provide for amounts and multiples that are within reasonable market norms. |
| ✔ | Prohibition on Hedging and Pledging - Under our Insider Trading Policy, we prohibit our employees from hedging any Company securities and from pledging any Company securities as collateral for a loan. |
| ✔ | Succession Planning - Our Board of Directors reviews the risks associated with our key executive positions on an annual basis so that we continue to evaluate an adequate succession strategy. |
| ✘ | Retirement Programs - Other than our Section 401(k) plan generally available to all employees, we do not offer defined benefit or contribution retirement plans or arrangements or nonqualified deferred compensation plans or arrangements for our executive officers, including our Named Executive Officers. |
| ✘ | No Dividends - We do not pay dividends or dividend equivalents on unvested or unearned RSUs and performance-based RSU awards. |
| ✘ | No Stock Option Repricing - We do not reprice options to purchase shares of our Common Stock without stockholder approval. |
| • | the recommendations of our CEO (except with respect to his own compensation) as described below; |
| • | our corporate growth and other elements of financial and operational performance; |
| • | our corporate and individual achievements against one or more short-term and long-term performance objectives; |
| • | the individual performance of each executive officer against his management objectives; |
| • | a review of the relevant competitive market analysis prepared by its compensation consultant (as described below); |
| • | the expected future contribution of the individual executive officer; |
| • | historical compensation awards we have made to our executive officers; and |
| • | internal pay equity based on the impact on our business and performance. |
| • | developed and subsequently updated the compensation peer group; |
| • | provided advice with respect to compensation best practices and market trends for executive officers and members of our Board of Directors; |
| • | conducted an analysis of the levels of overall compensation and each element of compensation for of our executive officers and non-executive employees; |
| • | conducted an analysis of the levels of overall compensation and each element of compensation for the members of our Board of Directors; and |
| • | provided ad hoc advice and support throughout the year. |
| • | the company’s stage of clinical development; |
| • | the company’s area(s) of therapeutic focus; |
| • | the company’s market capitalization; |
| • | the comparability of the company’s organizational complexities and growth attributes; |
| • | the comparability of the company’s business focus and corporate strategy; and |
| • | the comparability of the company’s operational performance (for consistency with our strategy and future performance expectations). |
4D Molecular Therapeutics | | | Dyne Therapeutics | | | Lineage Cell Therapeutics |
Aldeyra Therapeutics | | | Editas Medicine | | | MeiraGTx |
Allogene Therapeutics | | | Genelux | | | REGENXBIO |
Alpine Immune Sciences | | | Gritstone Bio | | | Replimune Group |
AnaptysBio | | | Inovio Pharmaceuticals | | | Sana Biotechnology |
Cabaletta Bio | | | iTeos Therapeutics | | | Viridian Therapeutics |
Cogent Biosciences | | | Kymera Therapeutics | | |
Compensation Element | | | What This Element Rewards | | | Purpose and Key Features of Element |
Base salary | | | Individual performance, level of experience, expected future performance and contributions. | | | Provides competitive level of fixed compensation determined by the market value of the position, with actual base salaries established based on the facts and circumstances of each executive officer and each individual position. |
Annual cash bonuses | | | Achievement of pre-established corporate and individual performance objectives (for 2023, focused on the advancement of our pipeline, corporate strategy and business development, culture and engagement, and the company’s financials, as well as management objectives). | | | Motivate executive officers to achieve certain corporate objectives and drive the company’s value. Generally, performance levels are established to incent our executive officers to achieve or exceed performance objectives. For 2023, payouts for corporate performance objectives were not weighted individually, and the Compensation Committee had discretion to determine payouts based on overall achievement of the corporate goals as a group, taking into account overachievement on certain objectives, if applicable. Payouts for individual performance objectives could range from 0% to an undetermined percentage. |
Retention bonuses | | | Maintain a stable management team and retain key Company talent through short-term market volatility and changes in corporate strategy. | | | Designed to stabilize the executive leadership team and reduce the possibility of turnover. These non-recurring retention bonuses awarded during the year ended December 31, 2023 were subject to repayment in the event the award recipient resigned other than for “Good Reason” or was earlier terminated by the Board of Directors prior to |
Compensation Element | | | What This Element Rewards | | | Purpose and Key Features of Element |
| | | | | March 31, 2024 (in the cases of Drs. Traber and Johnston) or June 30, 2024 (in the cases of Dr. Brunn and Mr. Davis). | ||
Long-term incentives/equity awards | | | Achievement of corporate and individual performance objectives designed to enhance long-term stockholder value and attract, retain, motivate, and reward executive officers over extended periods for achieving important corporate objectives. Time-based vesting requirements promote retention of highly-valued executive officers. | | | Annual equity awards that vest over four years and provide a variable “at risk” pay opportunity. Because the ultimate value of these equity awards is directly related to the market price of our Common Stock, and the awards are only earned over an extended period of time subject to vesting, they serve to focus management on the creation and maintenance of long-term stockholder value and also provide retentive value to key employees. |
Named Executive Officer | | | 2022 Base Salary | | | 2023 Base Salary | | | Percentage Adjustment |
Carsten Brunn, Ph.D. | | | $592,072 | | | $624,000 | | | 5.4% |
Blaine Davis | | | $440,000 | | | $440,000 | | | — |
Metin Kurtoglu, M.D., Ph.D.(1) | | | — | | | $402,500 | | | — |
Christopher Jewell, Ph.D.(2) | | | — | | | $300,000 | | | — |
Peter Traber, M.D.(3) | | | $458,920 | | | $500,000 | | | 9.0% |
Lloyd Johnston, Ph.D.(4) | | | $416,000 | | | $434,720 | | | 4.5% |
| (1) | Dr. Kurtoglu was appointed to serve as the Company’s Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (2) | Dr. Jewell was appointed to serve as the Company’s Chief Scientific Officer on November 13, 2023. |
| (3) | Dr. Traber ceased to serve as our Chief Medical Officer on November 13, 2023. |
| (4) | Dr. Johnston ceased to serve as our Chief Operations Officer on November 13, 2023. |
Named Executive Officer | | | Annual Base Salary | | | Target Bonus Opportunity (as a percentage of base salary) | | | Target Bonus Opportunity | | | Actual Bonus Payout |
Carsten Brunn, Ph.D. | | | $624,000 | | | 55% | | | $343,200 | | | $343,200 |
Blaine Davis | | | $440,000 | | | 40% | | | $176,000 | | | $176,000 |
Metin Kurtoglu, M.D., Ph.D.(1) | | | $402,500 | | | 40% | | | $161,000 | | | $20,930 |
Christopher Jewell, Ph.D.(2) | | | $300,000 | | | 40% | | | $120,000 | | | $15,600 |
Peter Traber, M.D. | | | $500,000 | | | 40% | | | $200,000 | | | —(3) |
Lloyd Johnston, Ph.D. | | | $434,720 | | | 40% | | | $173,888 | | | —(4) |
| (1) | Dr. Kurtoglu was appointed to serve as the Company’s Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. Dr. Kurtoglu received a pro-rated bonus based on employment with the Company since November 13, 2023. |
| (2) | Dr. Jewell was appointed to serve as the Company’s Chief Scientific Officer on November 13, 2023. Dr. Jewell received a pro-rated bonus based on employment with the Company since November 13, 2023. |
| (3) | Upon his departure as a full-time employee in December 2023, Dr. Traber forfeited his 2023 annual target bonus opportunity of $200,000. However, as discussed under “—Post-employment Compensation” below, Dr. Traber was paid an amount of $200,000 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee. |
| (4) | Upon his departure as a full-time employee in December 2023, Dr. Johnston forfeited his 2023 annual target bonus opportunity of $173,888. However, as discussed under “—Post-employment Compensation” below, Dr. Johnston was paid an amount of $173,888 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee. |
Corporate Performance Measure | | | 2023 Target Achievement % |
Pipeline Development | | | 100% |
Corporate Strategy and Business Development | | | 100% |
Finance | | | 100% |
Named Executive Officer | | | Target Annual Cash Bonus Opportunity | | | Amount Related to Corporate Performance Objectives | | | Actual Annual Cash Bonus Payment | | | Percentage of Target Annual Cash Bonus Opportunity |
Carsten Brunn, Ph.D. | | | $343,200 | | | $343,200 | | | $343,200 | | | 100% |
Blaine Davis | | | $176,000 | | | $176,000 | | | $176,000 | | | 100% |
Metin Kurtoglu, M.D., Ph.D.(1) | | | $161,000 | | | $161,000 | | | $20,930 | | | 13% |
Christopher Jewell, Ph.D.(2) | | | $120,000 | | | $120,000 | | | $15,600 | | | 13% |
Peter G. Traber, M.D.(3) | | | $200,000 | | | $200,000 | | | — | | | — |
Lloyd Johnston, Ph.D.(4) | | | $173,888 | | | $173,888 | | | — | | | — |
| (1) | Dr. Kurtoglu was appointed to serve as the Company’s Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. Dr. Kurtoglu received a pro-rated bonus based on employment with the Company since November 13, 2023. |
| (2) | Dr. Jewell was appointed to serve as the Company’s Chief Scientific Officer on November 13, 2023. Dr. Jewell received a pro-rated bonus based on employment with the Company since November 13, 2023. |
| (3) | Upon his departure as a full-time employee in December 2023, Dr. Traber forfeited his 2023 annual target bonus opportunity of $200,000. However, as discussed under “—Post-employment Compensation” below, Dr. Traber was paid an amount of $200,000 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee. |
| (4) | Upon his departure as a full-time employee in December 2023, Dr. Johnston forfeited his 2023 annual target bonus opportunity of $173,888. However, as discussed under “—Post-employment Compensation” below, Dr. Johnston was paid an amount of $173,888 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee. |
Named Executive Officer | | | Stock Options (Number of Shares) | | | RSU Awards (Number of Shares) |
Carsten Brunn, Ph.D. | | | 42,499 | | | 9,426 |
Blaine Davis(1) | | | — | | | — |
Peter G. Traber, M.D. | | | 29,999 | | | 6,666 |
Lloyd Johnston, Ph.D. | | | 15,833 | | | 3,333 |
Metin Kurtoglu, M.D., Ph.D.(2) | | | — | | | — |
Chris Jewell, Ph.D.(3) | | | — | | | — |
| (1) | Mr. Davis was appointed Chief Financial Officer on November 28, 2022, and did not receive options to purchase shares of our Common Stock or RSUs in January 2023. Mr. Davis received a one-time grant of an option to purchase 41,666 shares of our Common Stock upon his hiring on November 28, 2022. |
| (2) | Dr. Kurtoglu was appointed Chief Operations Officer on November 13, 2023, and did not receive options to purchase shares of our Common Stock or RSUs in January 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (3) | Dr. Jewell was appointed Chief Scientific Officer on November 13, 2023, and did not receive options to purchase shares of our Common Stock or RSUs in January 2023. |
| • | Carsten Brunn, Ph.D., our President and Chief Executive Officer; |
| • | Blaine Davis, our Chief Financial Officer; |
| • | Christopher Jewell, Ph.D., our Chief Scientific Officer who served in that position beginning November 13, 2023; |
| • | Metin Kurtoglu, M.D., Ph.D., who served as our Chief Operations Officer from November 13, 2023 until March 28, 2024, and then continued to serve as our Chief Technology Officer; |
| • | Peter G. Traber, M.D., our former Chief Medical Officer; and |
| • | Lloyd Johnston, Ph.D., our former Chief Operations Officer. |
Name and principal position | | | Year | | | Salary ($)(1) | | | Bonus ($)(2) | | | Stock awards ($)(3) | | | Option awards ($)(3) | | | Non-equity incentive plan compensation ($)(4) | | | All other compensation ($)(5) | | | Total ($) |
Carsten Brunn, Ph.D. President and Chief Executive Officer | | | 2023 | | | 618,228 | | | 1,323,000 | | | 319,564 | | | 1,123,658 | | | 343,200 | | | 2,384,233 | | | 6,111,883 |
| | 2022 | | | 588,043 | | | — | | | 749,053 | | | 2,628,525 | | | 325,655 | | | 966 | | | 4,292,242 | ||
Blaine Davis(6) Chief Financial Officer | | | 2023 | | | 440,000 | | | 880,000 | | | — | | | — | | | 176,000 | | | 998,759 | | | 2,494,759 |
| | 2022 | | | 33,846 | | | — | | | — | | | 1,228,750 | | | — | | | 1,110 | | | 1,263,706 | ||
Peter G. Traber, M.D.(7) Former Chief Medical Officer | | | 2023 | | | 458,920 | | | 850,000 | | | 226,000 | | | 793,170 | | | — | | | 1,452,830 | | | 3,780,920 |
| | 2022 | | | 455,573 | | | — | | | 298,562 | | | 1,035,133 | | | 183,568 | | | 31,147 | | | 2,003,983 | ||
Lloyd Johnston, Ph.D.(8) Former Chief Operations Officer | | | 2023 | | | 431,336 | | | 869,000 | | | 113,000 | | | 418,618 | | | — | | | 865,636 | | | 2,697,590 |
| | 2022 | | | 413,169 | | | — | | | 238,320 | | | 846,225 | | | 166,400 | | | 10,245 | | | 1,674,359 | ||
Metin Kurtoglu, M.D., Ph.D.(9) Chief Technology Officer | | | 2023 | | | 50,313 | | | — | | | — | | | — | | | 20,930 | | | — | | | 71,243 |
| | 2022 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | ||
Chris Jewell, Ph.D.(10) Chief Scientific Officer | | | 2023 | | | 37,500 | | | — | | | — | | | — | | | 15,600 | | | — | | | 53,100 |
| | 2022 | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
| (1) | These amounts represent actual earnings for the calendar year, which may be impacted by, among other things, hire date and the timing of any salary increases made during the year. |
| (2) | These amounts include one-time bonuses of $75,000 and $100,000 for Dr. Brunn and Dr. Traber, respectively, for efforts to deliver the positive topline data for the Company’s DISSOLVE I and DISSOLVE II Phase 3 clinical trials and retention bonuses. |
| (3) | Represents the aggregate grant date fair value of stock and option awards computed in accordance with ASC Topic 718, excluding the effect of estimated forfeitures. For a description of the assumptions used in valuing these awards, see Note 13 to our consolidated audited financial statements included elsewhere in the registration statement of which this prospectus forms a part. |
| (4) | Represents amounts earned under our annual performance-based bonus program. For additional information, see “—Performance Bonuses” below. |
| (5) | For Dr. Brunn, the amount for 2022 includes $966 representing payments on Dr. Brunn’s term life insurance policy. The amount for 2023 includes $2,383,268 for settlement of outstanding awards as of November 13, 2023 in connection with the Merger and $965 representing payments on Dr. Brunn’s term life insurance policy. For Mr. Davis, the amount for 2022 includes $35 representing payments on Mr. Davis’ term life insurance policy, $75 representing reimbursement for cell phone expenses, and $1,000 representing our 401(k) matching contributions. The amount for 2023 includes $987,500 for settlement of outstanding awards as of November 13, 2023 in connection with the Merger, $459 representing payments on Mr. Davis’ term life insurance policy, $900 representing reimbursement for cell phone expenses, and $9,900 representing our 401(k) matching contributions. For Dr. Traber, the amount for 2022 includes $21,097 representing payments on Dr. Traber’s term life insurance policy, $900 representing reimbursement for cell phone expenses, and $9,150 representing our 401(k) matching contributions. The amount for 2023 includes $1,436,872 for settlement of outstanding awards as of November 13, 2023 in connection with the Merger, $5,158 representing payments on Dr. Traber’s term life insurance policy, $900 representing reimbursement for cell phone expenses, and $9,900 representing our 401(k) matching contributions. |
| (6) | Mr. Davis was appointed Chief Financial Officer on November 28, 2022. |
| (7) | Dr. Traber ceased service as our Chief Medical Officer effective as of November 13, 2023. Upon his departure in December 2023, Dr. Traber forfeited his 2023 annual target bonus opportunity of $200,000. However, Dr. Traber was paid an amount of $200,000 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee, in 2024. |
| (8) | Dr. Johnston ceased service as our Chief Operations Officer effective as of November 13, 2023. Upon his departure as a full-time employee in December 2023, Dr. Johnston forfeited his 2023 annual target bonus opportunity of $173,888. However, Dr. Johnston was paid an amount of $173,888 as a term of his separation agreement, which was approved separately from the 2023 Bonus Plan by the Compensation Committee, in 2024. |
| (9) | Dr. Kurtoglu was appointed Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (10) | Dr. Jewell was appointed Chief Scientific Officer on November 13, 2023. |
| | | Option Awards | ||||||||||||||||
Name | | | Grant date(1) | | | Vesting Commencement Date | | | Number of securities underlying unexercised options (#) exercisable(2) | | | Number of securities underlying unexercised options (#) unexercisable(2) | | | Option exercise price ($) | | | Option expiration date |
Metin Kurtoglu, M.D., Ph.D.(3) | | | 11/7/2016 | | | 11/7/2016 | | | 6,414.682(4) | | | —(4) | | | 46.77 (4) | | | 11/6/2026 |
| | 4/26/2021 | | | 4/26/2021 | | | 427.645(5) | | | —(5) | | | 107.59 (5) | | | 4/25/2031 | ||
| | 3/1/2022 | | | 3/1/2022 | | | 106.911(6) | | | 320.735(6) | | | 107.59 (6) | | | 2/29/2032 | ||
Chris Jewell, Ph.D.(7) | | | 1/16/2023 | | | 1/16/2023 | | | 2539.177(8) | | | 1737.277(8) | | | 107.59(8) | | | 1/15/2033 |
| (1) | On November 13, 2023, we acquired Old Cartesian in the Merger. Options to purchase Old Cartesian common stock held by the reporting person were converted into options to purchase shares of Series A Preferred Stock in connection with the Merger. |
| (2) | As of December 31, 2023, these options were exercisable for Series A Preferred Stock. On March 27, 2024, we held a special meeting of stockholders (the “March 2024 Special Meeting”). At the March 2024 Special Meeting, our stockholders approved, and our Board of Directors subsequently implemented, a 1-for-30 reverse stock split of all then-outstanding shares of our Common Stock and the conversion of our Series A Preferred Stock into shares of Common Stock. Following the Series A Preferred Stock Automatic Conversion on April 8, 2024, options previously exercisable for shares of Series A Preferred Stock may be exercised solely for shares of Common Stock. |
| (3) | Dr. Kurtoglu was appointed Chief Operations Officer on November 13, 2023. On March 28, 2024, Dr. Kurtoglu’s title was changed to Chief Technology Officer. |
| (4) | Following the Series A Preferred Stock Automatic Conversion, these options are fully exercisable for 213,820 shares of Common Stock at an exercise price of $1.41. |
| (5) | Following the Series A Preferred Stock Automatic Conversion, these options are fully exercisable for 14,254 shares of Common Stock at an exercise price of $3.23. |
| (6) | Following the Series A Preferred Stock Automatic Conversion, these options are currently exercisable for 7,127 shares of Common Stock and become exercisable for the remaining 7,127 shares of Common Stock in two equal tranches on March 1, 2025 and March 1, 2026 at an exercise price of $3.23. |
| (7) | Dr. Jewell was appointed Chief Scientific Officer on November 13, 2023. |
| (8) | Following the Series A Preferred Stock Automatic Conversion, these options are currently exercisable for 114,328 shares of Common Stock and become exercisable for an additional 2,969 shares of Common Stock each month until they are fully exercisable on June 16, 2025 at an exercise price of $3.23. |
| • | annual director fee of $40,000; |
| • | chairman of the Board of Directors, $30,000 and lead independent director, $20,000; |
| • | chairman of the Audit Committee, $15,000; |
| • | Audit Committee member other than the chairman, $7,500; |
| • | chairman of the Compensation Committee, $12,000; |
| • | Compensation Committee member other than the chairman, $6,000; |
| • | chairman of the Nominating and Corporate Governance Committee, $9,000; |
| • | Nominating and Corporate Governance Committee member other than the chairman, $4,500; |
| • | chairman of the now-disbanded Research and Development Committee, $12,000; and |
| • | Research and Development Committee member other than the chairman, $6,000. |
| • | Increasing the annual retainer fee for the lead independent director to $25,000. |
| • | Increasing the annual retainer fee for the chairman of the Nominating and Corporate Governance Committee to $10,000 and to $5,000 for a Nominating and Corporate Governance committee member other than the chairman. |
| • | Increasing the annual equity awards from options to purchase 3,166 shares of Common Stock to options to purchase 4,000 shares of Common Stock for the Chairman of the Board of Directors and from options to purchase 2,500 shares of Common Stock to options to purchase 3,800 shares of Common Stock for each other non-employee director, and increasing the initial grant from options to purchase 3,166 shares of Common Stock to options to purchase 8,266 shares of Common Stock for the Chairman of the Board of Directors and from options to purchase 2,500 shares of Common Stock to options to purchase 7,600 shares of Common Stock for each other non-employee director. |
| • | Establishing a framework for granting RSUs to members of the Board of Directors, such that each Board of Directors member received a one-time award of 5,933 RSUs, to vest in three equal annual installments, and such that subsequent RSU awards shall also be provided to each new member of the Board of Directors upon the commencement of their service, and further such that each Board of Directors member, on the first business day of January of each year, shall receive a one-time award of 2,966 RSUs, also to vest in three equal installments. |
| • | Following the disbanding of the Research & Development Committee and the formation of the Science, IP and Quality Committee, an annual retainer in the amount of $12,000 was established for the chairman of the Science, IP and Quality Committee, and in the amount of $6,000 for each member of the Committee other than the chairman. |
Name | | | Fees earned or paid in cash ($)(1) | | | Option awards ($)(2) | | | All other compensation ($)(3) | | | Total ($) |
Göran Ando, M.D.(4) | | | 477 | | | 65,325 | | | — | | | 65,802 |
Timothy C. Barabe | | | 55,340 | | | 76,213 | | | 81,375 | | | 212,928 |
Carrie S. Cox | | | 83,908 | | | 93,633 | | | 114,775 | | | 292,316 |
Nishan de Silva, M.D., M.B.A. | | | 53,500 | | | 65,325 | | | 69,750 | | | 188,575 |
Murat Kalayoglu, M.D., Ph.D.(5) | | | 5,370 | | | — | | | — | | | 5,370 |
Scott D. Myers(6) | | | 50,136 | | | 76,213 | | | 81,375 | | | 207,724 |
Aymeric Sallin(7) | | | 46,000 | | | 65,325 | | | 69,750 | | | 181,075 |
Michael Singer, M.D., Ph.D.(8) | | | 5,712 | | | — | | | — | | | 5,712 |
Timothy A. Springer, Ph.D. | | | 57,651 | | | 65,325 | | | 69,750 | | | 192,726 |
Patrick Zenner | | | 56,976 | | | 65,325 | | | 69,750 | | | 192,051 |
| (1) | Represents cash retainers earned for services rendered as members of the Board of Directors and related committees. |
| (2) | The value of option awards represents the aggregate grant date fair value of stock options computed in accordance with ASC Topic 718, excluding the effect of estimated forfeitures. For a description of the assumptions used in valuing these awards, see Note 11 to our consolidated financial statements included elsewhere in the registration statement of which this prospectus forms a part. |
| (3) | Options outstanding as of the Merger date were canceled in the Merger in exchange for a cash payment representing the difference between the exercise price of the option and $2.06, the Cash-out Amount as applied in the Merger. |
| (4) | Dr. Ando resigned from the Board of Directors on January 4, 2023. |
| (5) | Dr. Kalayoglu was appointed to the Board of Directors on November 13, 2023. |
| (6) | Mr. Myers resigned from the Board of Directors on November 21, 2023. |
| (7) | Mr. Sallin resigned from the Board of Directors on February 28, 2024. |
| (8) | Dr. Singer was appointed to the Board of Directors on November 13, 2023. |
Name | | | Options outstanding at fiscal year end(1) |
Göran Ando(2) | | | — |
Timothy C. Barabe | | | — |
Carrie S. Cox | | | — |
Nishan de Silva, M.D., M.B.A. | | | — |
Murat Kalayoglu, M.D., Ph.D.(3) | | | — |
Scott D. Myers(4) | | | — |
Aymeric Sallin(5) | | | — |
Michael Singer, M.D., M.B.A.(6) | | | — |
Timothy A. Springer, Ph.D. | | | — |
Patrick Zenner | | | — |
| (1) | Options outstanding as of the Merger date, except with respect to Dr. Ando, whose options were cancelled on April 4, 2023 in connection with his retirement from the Board of Directors, were canceled in the Merger in exchange for a cash payment, representing the difference between the exercise price of the option and $2.06, the Cash-out Amount as applied in the Merger. |
| (2) | Dr. Ando resigned from the Board of Directors on January 4, 2023. |
| (3) | Dr. Kalayoglu was appointed to the Board of Directors on November 13, 2023. |
| (4) | Mr. Myers resigned from the Board of Directors on November 21, 2023. |
| (5) | Mr. Sallin resigned from the Board of Directors on February 28, 2024. |
| (6) | Dr. Singer was appointed to the Board of Directors on November 13, 2023. |
| | | No Conversion of Preferred Stock | | | Full Conversion of Preferred Stock (subject to beneficial ownership limitations) | |||||||
Name and address of beneficial owner | | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned | | | Number of Shares Beneficially Owned | | | Percentage of Shares Beneficially Owned |
5% Stockholders: | | | | | | | | | ||||
Entities affiliated with Timothy A. Springer, Ph.D.(1) | | | 6,482,256 | | | 30.1% | | | 8,841,756 | | | 34.4% |
Entities affiliated with Murat Kalayoglu, M.D., Ph.D.(2) | | | 3,539,442 | | | 16.6% | | | 5,077,513 | | | 19.9% |
FMR, LLC(3) | | | 2,722,656 | | | 12.7% | | | 2,722,656 | | | 10.7% |
Named Executive Officers and Directors: | | | | | | | | | ||||
Carsten Brunn, Ph.D.(4) | | | 8,731 | | | * | | | 8,731 | | | * |
Blaine Davis | | | — | | | * | | | — | | | * |
Chris Jewell, Ph.D.(5) | | | 120,266 | | | * | | | 120,266 | | | * |
Metin Kurtoglu, M.D., Ph.D.(6) | | | 235,201 | | | 1.1% | | | 235,201 | | | * |
Peter G. Traber, M.D.(7) | | | 5,301 | | | * | | | 5,301 | | | * |
Lloyd Johnston, Ph.D.(8) | | | 2,983 | | | * | | | 2,983 | | | * |
Carrie S. Cox(9) | | | 10,486 | | | * | | | 10,486 | | | * |
Göran Ando, M.D.(10) | | | — | | | * | | | — | | | * |
Timothy C. Barabe(11) | | | 17,944 | | | * | | | 17,944 | | | * |
Nishan de Silva, M.D., M.B.A.(12) | | | 2,344 | | | * | | | 2,344 | | | * |
Murat Kalayoglu, M.D., Ph.D.(2) | | | 3,539,442 | | | 16.6% | | | 5,077,513 | | | 19.9% |
Kemal Malik, MBBS(13) | | | 844 | | | * | | | 844 | | | * |
Scott D. Myers(14) | | | 3,350 | | | * | | | 3,350 | | | * |
Aymeric Sallin(15) | | | — | | | * | | | — | | | * |
Michael Singer, M.D., Ph.D.(16) | | | 770,800 | | | 3.6% | | | 770,800 | | | 3.0% |
Timothy A. Springer, Ph.D.(1) | | | 6,482,256 | | | 30.1% | | | 8,841,756 | | | 34.4% |
Patrick Zenner(17) | | | 3,952 | | | * | | | 3,952 | | | * |
All executive officers and directors as a group (14 persons)(18) | | | 11,200,550 | | | 51.1% | | | 15,098,121 | | | 58.0% |
| * | Represents beneficial ownership of less than one percent. |
| (1) | Based on a Schedule 13D/A filed with the SEC on July 5, 2024 and other information known to us, consists of (i) 4,373,966 shares of Common Stock held directly by Timothy A. Springer, Ph.D., a member of our Board of Directors, (ii) 1,636,832 shares of Common Stock issuable upon conversion of shares of Series B Preferred Stock held directly by Timothy A. Springer, Ph.D., (iii) 2,111 shares of Common Stock issuable upon exercise of outstanding options within 60 days of September 3, 2024 and held directly by Timothy A. Springer, Ph.D., (iv) 1,927,630 shares of Common Stock held by TAS Partners LLC (“TAS”) directly, (v) 721,361 shares of Common Stock issuable upon conversion of shares of Series B Preferred Stock held by TAS directly, (vi) 167,040 shares of Common Stock issuable upon exercise of underlying warrants exercisable within 60 days of September 3, 2024 held by TAS directly, (vii) 11,509 shares of Common Stock held by Dr. Chafen Lu, Dr. Springer’s wife, and (viii) 1,307 shares of Common Stock issuable upon conversion of shares of Series B Preferred Stock held by Dr. Chafen Lu. Dr. Springer is the sole managing member of TAS. Dr. Springer exercises sole voting and dispositive power over the shares held by him directly and the shares held by TAS. Dr. Springer disclaims beneficial ownership of the shares held by TAS. Dr. Lu exercises sole voting and dispositive power over the shares held by her directly. The principal business address of each of Dr. Springer, TAS, and Dr. Lu is 36 Woodman Road, Newton, MA, 02467. |
| (2) | Based on a Schedule 13D/A filed with the SEC on April 10, 2024 and other information known to us, consists of (i) 500,444 shares of Common Stock held directly by Murat Kalayoglu, M.D., Ph.D., a member of our Board of Directors, (ii) 2,111 shares of Common Stock issuable upon exercise of outstanding options within 60 days of September 3, 2024 and held directly by Murat Kalayoglu, M.D., Ph.D., (iii) 3,036,887 shares of Common Stock held by Seven One Eight Three Four Irrevocable Trust directly, and (iv) up to 3,398,448 shares of Common Stock issuable upon conversion of shares of Series A Preferred Stock held by Seven One Eight Three Four Irrevocable Trust. The trustees of Seven One Eight Three Four Irrevocable Trust are Elizabeth Hoge and Sinan Kalayoglu, each of whom has shared voting and dispositive control over the shares of Common Stock and Series A Preferred Stock held by Seven One Eight Three Four Irrevocable Trust. Dr. Kalayoglu has the power to remove and appoint new trustees of Seven One Eight Three Four Irrevocable Trust and, pursuant to a right of substitution, to acquire from Seven One Eight Three Four Irrevocable Trust the shares of Common Stock and Series A Preferred Stock held by Seven One Eight Three Four Irrevocable Trust in exchange for assets with an equal value to such shares. Accordingly, Dr. Kalayoglu may be deemed to have sole voting and dispositive power of the shares of Common Stock and Series A Preferred Stock held by Seven One Eight Three Four Irrevocable Trust. The ability of the shares of Series A Preferred Stock held by Dr. Kalayoglu and Seven One Eight Three Four Irrevocable Trust to convert into shares of Common Stock is subject to a beneficial ownership limitation, such that neither Dr. Kalayoglu nor Seven One Eight Three Four Irrevocable Trust may convert shares of Series A Preferred Stock into Common Stock to the extent that doing so would result in such holder beneficially owning greater than 19.9% of the Company’s outstanding Common Stock after giving effect to such conversion. Accordingly, the numbers of shares of Common Stock presented in this row include only a total of 1,538,071 shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock held by Seven One Eight Three Four Irrevocable Trust, and assume Seven One Eight Three Four Irrevocable Trust does not convert any shares of Series A Preferred Stock beyond such limitation. |
| (3) | Based on a Schedule 13G/A filed with the SEC on August 12, 2024 and other information known to us, consists of 2,722,656 shares of Common Stock owned by funds or accounts managed by direct or indirect subsidiaries of FMR LLC, all of which shares are beneficially owned, or may be deemed to be beneficially owned, by FMR LLC, certain of its subsidiaries and affiliates, and other companies. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of these funds and accounts is 245 Summer Street, Boston, MA 02210. |
| (4) | Consists of 8,731 shares of Common Stock held by Dr. Brunn directly. |
| (5) | Consists of 120,266 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024 held by Dr. Jewell directly. |
| (6) | Consists of 235,201 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024 held by Dr. Kurtoglu directly. |
| (7) | Consists of 5,301 shares of Common Stock held by Dr. Traber directly. Dr. Traber ceased service as our Chief Medical Officer effective November 13, 2023 and ceased full-time employment with the Company effective December 31, 2023. |
| (8) | Consists of 2,983 shares of Common Stock held by Dr. Johnston directly. Dr. Johnston ceased service as our Chief Operations Officer effective November 13, 2023 and ceased full-time employment with the Company effective December 31, 2023. |
| (9) | Consists of (i) 7,096 shares of Common Stock held by Ms. Cox directly, (ii) 1,094 shares of Common Stock issuable upon exercise of underlying warrants exercisable within 60 days of September 3, 2024, and (iii) 2,296 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024. |
| (10) | We are not aware of any beneficial ownership of Common Stock by Dr. Ando, who retired from the Board of Directors on January 4, 2023. |
| (11) | Consists of (i) 15,833 shares of Common Stock held by Mr. Barabe directly and (ii) 2,111 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024. |
| (12) | Consists of (i) 233 shares of Common Stock held by Dr. de Silva directly and (ii) 2,111 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024. |
| (13) | Consists of 844 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024 held by Dr. Malik directly. |
| (14) | Consists of (i) 2,694 shares of Common Stock held by Mr. Myers directly and (ii) 656 shares of Common Stock issuable upon exercise of underlying warrants exercisable within 60 days of September 3, 2024. Mr. Myers resigned from the Board of Directors on November 21, 2023. |
| (15) | We are not aware of any beneficial ownership of Common Stock by Mr. Sallin directly. Mr. Sallin resigned from the Board of Directors on February 28, 2024. |
| (16) | Based on a Schedule 13D/A filed with the SEC on April 10, 2024 and other information known to us, consists of (i) 113,821 shares of Common Stock held by Dr. Singer directly, (ii) 383,796 shares of Common Stock held by Thirsty Brook 2010 Irrevocable Trust, a trust for which Dr. Singer is a trustee, that Dr. Singer has the right to acquire pursuant to a right of substitution in exchange for assets with |
| (17) | Consists of 1,841 shares of Common Stock held by Mr. Zenner directly and (ii) 2,111 shares of Common Stock underlying outstanding stock options exercisable within 60 days of September 3, 2024. |
| (18) | Includes (i) 10,661,143 shares of Common Stock owned directly or beneficially by our executive officers or members of our Board of Directors, (ii) 539,407 shares of Common Stock underlying outstanding stock options and warrants exercisable within 60 days of September 3, 2024, (iii) 1,538,071 shares of Common Stock issuable upon conversion of the shares of Series A Preferred Stock, and (iv) 2,359,500 shares of Common Stock issuable upon conversion of shares of Series B Preferred Stock. |
| • | Each Company Stock Option and each RSU award with respect to shares of Common Stock, in each case that was outstanding and unvested immediately prior to the Effective Time (as defined in the Merger Agreement), vested in full at the Effective Time; |
| • | each Company Option was canceled at the Effective Time, and in exchange therefor, former holders of such canceled Company Stock Options became entitled to receive (without interest), in consideration of the cancellation of such Company Stock Option, an amount in cash (less applicable tax withholdings) equal to the product of (A) the total number of shares of Common Stock subject to the unexercised portion of such Company Stock Option immediately prior to the Effective Time (determined after giving effect to the accelerated vesting) multiplied by (B) the excess, if any, of the Cash-out Amount over the applicable exercise price per share of Common Stock under such Company Stock Option; provided, however, that, if the exercise price per share of Common Stock of any Company Stock Option was equal to or greater than the Cash-out Amount, such Company Stock Option was canceled and terminated without any consideration in respect thereof; and |
| • | each RSU award with respect to shares of Common Stock was cancelled at the Effective Time, and the former holder of such canceled RSU became entitled, in exchange therefor, to receive (without interest) an amount in cash (less applicable tax withholdings) equal to the product of (A) the total number of shares of Common Stock deliverable under such RSU immediately prior to the Effective Time (determined after giving effect to the accelerated vesting) multiplied by (B) the Cash-out Amount. |
| • | the number of shares of Common Stock subject to each Old Cartesian Option assumed by the Company will be determined by multiplying (A) the number of shares of Old Cartesian common stock that were subject to such Old Cartesian Option, as in effect immediately prior to the First Effective Time, by (B) the Exchange Ratio (as defined in the Merger Agreement), and rounding the resulting number down to the nearest whole number of shares of Common Stock; |
| • | the per-share exercise price for Common Stock issuable upon exercise of each Old Cartesian Option assumed by the Company will be determined by dividing (A) the per share exercise price of Old Cartesian common stock subject to such Old Cartesian Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio and rounding the resulting exercise price up to the nearest whole cent; |
| • | the number of shares of Series A Preferred Stock subject to each Continuing Officer Option assumed by the Company will be determined by multiplying (A) the number of shares of Old Cartesian common stock that were subject to such Continuing Officer Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio (as defined in the Merger Agreement), and (C) dividing such resulting number by 1,000 and rounding the resulting number down to the nearest 1/1000th of a share of Series A Preferred Stock; |
| • | the per share exercise price for Series A Preferred Stock issuable upon exercise of each Continuing Officer Option assumed by the Company will be determined by dividing (A) the per share exercise price of Old Cartesian common stock subject to such Continuing Officer Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio, and (C) multiplying the resulting number by 1,000 and rounding the resulting exercise price up to the nearest whole cent; and |
| • | any restriction on the exercise of any Old Cartesian Option assumed by the Company, including the Continuing Officer Options, will continue in full force and effect and, except as expressly provided in the Merger Agreement, the term, exercisability, vesting schedule and other provisions of such Old Cartesian Option will otherwise remain unchanged. |
Name | | | Shares of Series A Preferred Stock Purchased | | | Total Aggregate Purchase Price |
Timothy A. Springer, Ph.D. | | | 123,925.407 | | | $50,000,000 |
TAS Partners, LLC (affiliate of Timothy A. Springer, Ph.D.) | | | 24,785.081 | | | $10,000,000 |
Seven One Eight Three Four Irrevocable Trust (affiliate of Murat Kalayoglu, M.D., Ph.D.) | | | 619.627 | | | $250,000 |
Name | | | Shares of Series B Preferred Stock Purchased | | | Total Aggregate Purchase Price |
Timothy A. Springer, Ph.D. | | | 1,636,832 | | | $32,736,640 |
TAS Partners, LLC (affiliate of Timothy A. Springer, Ph.D.) | | | 721,361 | | | $14,427,220 |
Chafen Lu, Ph.D. | | | 1,307 | | | $26,140 |
Name of Selling Stockholders(1) | | | Common Stock Beneficially Owned Before Offering(2) | | | Common Stock that May Be Offered Pursuant to Prospectus | | | Common Stock Beneficially Owned After Offering(2) | |||
| | | | | | | Number | | | Percentage (%) | |||
Entities affiliated with Timothy A. Springer, Ph.D.(3) | | | 8,841,756 | | | 2,359,500 | | | 6,482,256 | | | 27.1% |
Schooner Century Fund LLC(4) | | | 1,608,709 | | | 737,500 | | | 871,209 | | | 4.0% |
Fidelity Growth Company Commingled Pool(5) | | | 461,147 | | | 461,147 | | | — | | | * |
Fidelity Select Portfolios: Biotechnology Portfolio(6) | | | 884,159 | | | 375,000 | | | 509,159 | | | 2.4% |
Citadel CEMF Investments Ltd.(7) | | | 375,000 | | | 375,000 | | | — | | | * |
Fidelity Select Portfolios: Select Health Care Portfolio(8) | | | 368,992 | | | 368,992 | | | — | | | * |
Fidelity Mt. Vernon Street Trust: Fidelity Growth Company Fund(9) | | | 341,346 | | | 341,346 | | | — | | | * |
Invus Public Equities, L.P.(10) | | | 573,962 | | | 250,000 | | | 323,962 | | | 1.5% |
Fidelity Advisor Series VII: Fidelity Advisor Health Care Fund(11) | | | 230,254 | | | 230,254 | | | — | | | * |
HBM Healthcare Investments (Cayman) Ltd.(12) | | | 200,000 | | | 200,000 | | | — | | | * |
Entities affiliated with Great Point Partners LLC(13) | | | 200,000 | | | 200,000 | | | — | | | * |
Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund(14) | | | 167,950 | | | 167,950 | | | — | | | * |
Fidelity Mt. Vernon Street Trust: Fidelity Growth Company K6 Fund(15) | | | 108,576 | | | 108,576 | | | — | | | * |
Armistice Capital, LLC(16) | | | 100,000 | | | 100,000 | | | — | | | * |
Fidelity Mt. Vernon Street Trust: Fidelity Series Growth Company Fund(17) | | | 88,931 | | | 88,931 | | | — | | | * |
Variable Insurance Products Fund IV: VIP Health Care Portfolio(18) | | | 56,954 | | | 56,954 | | | — | | | * |
Schonfeld Global Master Fund L.P.(19) | | | 50,000 | | | 50,000 | | | — | | | * |
683 Capital Partners, LP(20) | | | 30,000 | | | 30,000 | | | — | | | * |
| * | Less than 1% |
| (1) | To our knowledge, unless otherwise indicated, all persons named in the table above have sole voting and investment power with respect to their shares of Common Stock. Unless otherwise indicated, the address of each beneficial owner listed below is 704 Quince Orchard Road, Gaithersburg, Maryland 20878. |
| (2) | “Beneficial ownership” is a term broadly defined by the SEC in Rule 13d-3 under the Exchange Act, and includes more than the typical form of stock ownership, that is, stock held in the person’s name. The term also includes what is referred to as “indirect ownership,” meaning ownership of shares as to which a person has or shares investment power. Notwithstanding the foregoing, the beneficial ownership amounts assume the sale of all Common Stock that may be offered pursuant to this prospectus without taking into account certain limitations, including that a holder of Series B Preferred Stock is prohibited from converting shares of Series B Preferred Stock into shares of Common Stock (i) prior to stockholder approval of the Conversion Proposal is obtained, or (ii) if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than a specified percentage (established by the holder between 0.0% and 19.9%) (the “Beneficial Ownership Limitation”) of the total number of shares of Common Stock issued and outstanding immediately after giving effect to such conversion. |
| (3) | Based on information known to us, and consists of (i) 4,373,966 shares of Common Stock held directly by Timothy A. Springer, Ph.D., a member of our Board of Directors, (ii) 1,636,832 shares of Common Stock issuable upon conversion of shares of Series B Preferred Stock held directly by Timothy A. Springer, Ph.D., (iii) 2,111 shares of Common Stock issuable upon exercise of outstanding options within 60 days of September 3, 2024 and held directly by Timothy A. Springer, Ph.D., (iv) 1,927,630 shares of Common Stock held by |
| (4) | Based on information known to us, the Resale Shares that are registered for resale hereby consists of 176,385 shares of Common Stock and 561,115 shares of Common Stock underlying Series B Preferred Stock held directly by Schooner Century Fund LLC. Schooner Century Fund LLC also holds 871,209 shares of Common Stock and 2,146,271 shares of Common Stock underlying outstanding shares of Series A Preferred Stock that are not registered for resale hereby. Schooner Capital LLC is the Sole Manager of Schooner Century Fund LLC. Schooner Capital LLC is managed by Stephen D. Maiocco, Edward D. Henderson, and Peter K. Binas, serving as its Managing Partners. These Managing Partners, along with Vincent J. Ryan as majority member of the Sole Manager share the sole voting discretion and dispositive power with respect to all shares of Cartesian Therapeutics, Inc. held by Schooner Century Fund LLC. The ability of the shares of Series A Preferred Stock or Series B Preferred Stock held by Schooner Century Fund LLC to convert into shares of Common Stock are subject to beneficial ownership limitations, such that Schooner Century Fund LLC may not convert shares of Series A Preferred Stock or Series B Preferred Stock into Common Stock to the extent that doing so would result in such holder beneficially owning greater than 4.9% of the Company's outstanding Common Stock after giving effect to any such conversion. |
| (5) | Based on information known to us, and consists of 461,147 shares of Common Stock held by Fidelity Growth Company Commingled Pool. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (6) | Based on information known to us, and consists of 375,000 shares of Common Stock held by Fidelity Select Portfolios: Biotechnology Portfolio. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (7) | Based on information known to us, and consists of 369,621 shares of Common Stock and 5,379 shares of Common Stock underlying Series B Preferred Stock held directly by Citadel CEMF Investments Ltd. Citadel Advisors LLC is the portfolio manager of Citadel CEMF Investments LTD. Citadel Advisors Holdings LP (“CAH”) is the sole member of Citadel Advisors LLC. Citadel GP LLC (“CGP”) is the general partner of CAH. Kenneth Griffin owns a controlling interest in CGP. Mr. Griffin, as the owner of a controlling interest in CGP, may be deemed to have shared power to vote or direct the vote of, and/or shared power to dispose or direct the disposition over, these securities. The address of Citadel CEMF Investments Ltd. is c/o Citadel Enterprise Americas LLC, Southeast Financial Center, 200 S. Biscayne Blvd., Suite 3300, Miami, FL 33131. |
| (8) | Based on information known to us, and consists of 368,992 shares of Common Stock held by Fidelity Select Portfolios: Select Health Care Portfolio. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (9) | Based on information known to us, and consists of 341,346 shares of Common Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Growth Company Fund. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (10) | Based on information known to us, and consists of 246,413 shares of Common Stock and 3,587 shares of Common Stock underlying Series B Preferred Stock held directly by Invus Public Equities, L.P. (“IPE”). Additionally, IPE holds 279,989 shares of Common Stock and 43,973 shares of Common Stock underlying warrants that are exercisable within 60 days of the date hereof that are not registered for resale hereby. Invus Public Equities Advisors, LLC (“IPEA”) controls IPE, as its general partner and accordingly, may be deemed to beneficially own the shares held by IPE. Invus Global Management, LLC (“IGM”) controls IPEA, as its managing member and accordingly, may be deemed to beneficially own the shares that IPEA may be deemed to beneficially own. Siren, L.L.C. (“Siren”) controls IGM, as its managing member and accordingly, may be deemed to beneficially own the shares that IGM may be deemed to beneficially own. Mr. Raymond Debbane, as the managing member of Siren, controls Siren and accordingly, may be deemed to beneficially own the shares that Siren may be deemed to beneficially own. The address of Invus Public Equities, L.P. is 750 Lexington |
| (11) | Based on information known to us, and consists of 230,254 shares of Common Stock held by Fidelity Advisor Series VII: Fidelity Advisor Health Care Fund. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (12) | Based on information known to us, and consists of 197,130 shares of Common Stock and 2,870 shares of Common Stock underlying Series B Preferred Stock held directly by HBM Healthcare Investments (Cayman) Ltd. Voting and investment power over the shares held by HBM Healthcare Investments (Cayman) Ltd. is exercised by the board of directors of HBM Healthcare Investments (Cayman) Ltd. (the “HBM Board”). The HBM Board consists of Jean-Marc Lesieur, Richard H. Coles, Sophia Harris, Dr. Andreas Wicki, Mark Kronenfeld, M.D. and Richard Paul Woodhouse, none of whom has individual voting or investment power with respect to the shares. The address of HBM Healthcare Investments (Cayman) Ltd. is Governor’s Square, 23 Lime Tree Bay Ave., PO Box 30852, Grand Cayman, KY1-1204, Cayman Islands. |
| (13) | Based on information known to us, and consists of (i) 108,816 shares of Common Stock and 1,584 shares of Common Stock underlying Series B Preferred Stock held by Biomedical Value Fund, L.P. (“BMVF”), (ii) 74,909 shares of Common Stock and 1,091 shares of Common Stock underlying Series B Preferred Stock held by Biomedical Offshore Value Fund, Ltd. (“BOVF”), and (iii) 13,405 shares of Common Stock and 195 shares of Common Stock underlying Series B Preferred Stock held by Cheyne Select Master Fund ICAV – Cheyne Global Equity Fund ((“CGEF”) and together with BMVF and BOVF, the “GPP Entities”)). Great Point Partners LLC (“GPP LLC”) is the investment manager of BMVF and BOVF and the sub-advisor to CGEF, and by virtue of such status may be deemed to be the beneficial owner of the securities held by the GPP Entities. Each of Dr. Jeffrey R. Jay, M.D., as Senior Managing Member of GPP LLC, and Mr. Ortav Yehudai, as Managing Director of GPP LLC, has voting and investment power with respect to securities held by the GPP Entities, and therefore may be deemed to be the beneficial owner of the securities held by the GPP Entities. Notwithstanding the above, GPP LLC, Dr. Jay and Mr. Yehudai disclaim beneficial ownership of the securities held by the GPP Entities except to the extent of their respective pecuniary interests. The address of the GPP Entities is 165 Mason Street, 3rd Floor, Greenwich, CT 06830. |
| (14) | Based on information known to us, and consists of 167,950 shares of Common Stock held by Fidelity Advisor Series VII: Fidelity Advisor Biotechnology Fund. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (15) | Based on information known to us, and consists of 108,576 shares of Common Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Growth Company K6 Fund. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (16) | Based on information known to us, and consists of 98,565 shares of Common Stock and 1,435 shares of Common Stock underlying shares of Series B Preferred Stock. The securities are directly held by Armistice Capital Master Fund Ltd., a Cayman Islands exempted company (the “Master Fund”), and may be deemed to be beneficially owned by: (i) Armistice Capital, LLC (“Armistice Capital”), as the investment manager of the Master Fund; and (ii) Steven Boyd, as the Managing Member of Armistice Capital. The address of Armistice Capital Master Fund Ltd. is c/o Armistice Capital, LLC, 510 Madison Avenue, 7th Floor, New York, NY 10022. |
| (17) | Based on information known to us, and consists of 88,931 shares of Common Stock held by Fidelity Mt. Vernon Street Trust: Fidelity Series Growth Company Fund. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (18) | Based on information known to us, and consists of 56,954 shares of Common Stock held by Variable Insurance Products Fund IV: VIP Health Care Portfolio. This fund is managed by direct or indirect subsidiaries of FMR LLC. Abigail P. Johnson is a Director, the Chairman and the Chief Executive Officer of FMR LLC. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. The address of this fund is 245 Summer Street, Boston, MA 02210. |
| (19) | Based on information known to us, and consists of 49,283 shares of Common Stock and 717 shares of Common Stock underlying Series B Preferred Stock held directly by Schonfeld Global Master Fund L.P. Ryan Tolkin, the CEO and CIO of Schonfeld Strategic |
| (20) | Based on information known to us, and consists of 29,570 shares of Common Stock and 430 shares of Common Stock underlying shares of Series B Preferred Stock. Ari Zweiman has voting and investment power with respect to these securities and therefore may be deemed to be the beneficial owner thereof. The address of 683 Capital Partners, LP is 1700 Broadway, Suite 4200, New York, NY 10019. |
| • | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| • | block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
| • | to or through underwriters or purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| • | an exchange distribution in accordance with the rules of the applicable exchange; |
| • | privately negotiated transactions; |
| • | settlement of short sales entered into after the effective date of the registration statement of which this prospectus is a part; |
| • | broker-dealers may agree with the Selling Stockholders to sell a specified number of such Resale Shares at a stipulated price per share; |
| • | through the writing or settlement of options or other hedging transactions, whether such options are listed on an options exchange or otherwise; |
| • | a combination of any such methods of sale; and |
| • | any other method permitted pursuant to applicable law. |
| • | the title and stated value; |
| • | the number of shares offered; |
| • | the liquidation preference per share; |
| • | the purchase price per share; |
| • | the dividend rate(s), period(s) and/or payment date(s) or method(s) of calculation for dividends; |
| • | whether dividends are cumulative or non-cumulative and, if cumulative, the date from which dividends will accumulate; |
| • | our right, if any, to defer payment of dividends and the maximum length of such deferral period; |
| • | the procedures for auction and remarketing, if any; |
| • | the provisions for a sinking fund, if any; |
| • | the provision for redemption or repurchase, if applicable, and any restrictions on our ability to exercise those redemption and repurchase rights; |
| • | any listing of the preferred stock on any securities exchange or market; |
| • | the terms and conditions, if applicable, upon which the preferred stock will be convertible into Common Stock, including the conversion price (or manner of calculation) and conversion period; |
| • | whether the preferred stock will be exchangeable into debt securities, and, if applicable, the exchange price, or how it will be calculated, and the exchange period; |
| • | voting rights, if any, of the preferred stock; |
| • | preemptive rights, if any; |
| • | restrictions on transfer, sale or other assignment, if any; |
| • | whether interests in the preferred stock will be represented by depositary shares; |
| • | a discussion of any material and/or special U.S. federal income tax considerations applicable to the preferred stock; |
| • | the relative ranking and preferences of the preferred stock as to dividend rights and rights upon the liquidation, dissolution or winding up of our affairs; |
| • | any limitations on issuance of any class or series of preferred stock ranking senior to or on a parity with the class or series of preferred stock as to dividend rights and rights upon liquidation, dissolution or winding up of our affairs; and |
| • | any other specific terms, preferences, rights, limitations or restrictions of the preferred stock. |
| | | Page | |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| | | Page | |
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | | ||
| | |
| | | June 30, 2024 | | | December 31, 2023 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $87,227 | | | $76,911 |
Accounts receivable | | | 32,039 | | | 5,870 |
Unbilled receivables | | | 3,472 | | | 2,981 |
Prepaid expenses and other current assets | | | 2,044 | | | 4,967 |
Total current assets | | | 124,782 | | | 90,729 |
Non-current assets: | | | | | ||
Property and equipment, net | | | 6,672 | | | 2,113 |
Right-of-use asset, net | | | 13,852 | | | 10,068 |
In-process research and development assets | | | 150,600 | | | 150,600 |
Goodwill | | | 48,163 | | | 48,163 |
Long-term restricted cash | | | 1,669 | | | 1,377 |
Investments | | | 2,000 | | | 2,000 |
Total assets | | | $347,738 | | | $305,050 |
Liabilities, convertible preferred stock, and stockholders’ deficit | | | | | ||
Current liabilities: | | | | | ||
Accounts payable | | | $2,862 | | | $3,150 |
Accrued expenses and other current liabilities | | | 10,954 | | | 15,572 |
Lease liability | | | 2,523 | | | 2,166 |
Deferred revenue | | | — | | | 2,311 |
Warrant liabilities | | | 1,205 | | | 720 |
Contingent value right liability | | | 8,571 | | | 15,983 |
Forward contract liabilities | | | — | | | 28,307 |
Total current liabilities | | | 26,115 | | | 68,209 |
Non-current liabilities: | | | | | ||
Lease liability, net of current portion | | | 12,344 | | | 8,789 |
Deferred revenue, net of current portion | | | — | | | 3,538 |
Warrant liabilities, net of current portion | | | 8,055 | | | 5,674 |
Contingent value right liability, net of current portion | | | 386,829 | | | 342,617 |
Deferred tax liabilities, net | | | 15,853 | | | 15,853 |
Total liabilities | | | 449,196 | | | 444,680 |
Commitments and contingencies (Note 18) | | | | | ||
Series A Preferred Stock, $0.0001 par value; no and 548,375 shares authorized as of June 30, 2024 and December 31, 2023, respectively; no and 435,120.513 shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | | | — | | | 296,851 |
Options for Series A Preferred Stock | | | — | | | 3,703 |
Stockholders’ deficit: | | | | | ||
Series A Preferred Stock, $0.0001 par value; 180,455.753 and no shares authorized as of June 30, 2024 and December 31, 2023, respectively; 166,341.592 and no shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | | | — | | | — |
Preferred stock, $0.0001 par value; 9,819,544.247 and 9,451,625 shares authorized as of June 30, 2024 and December 31, 2023, respectively; no shares issued and outstanding as of June 30, 2024 and December 31, 2023 | | | — | | | — |
| | | June 30, 2024 | | | December 31, 2023 | |
Common stock, $0.0001 par value; 350,000,000 shares authorized as of June 30, 2024 and December 31, 2023; 17,816,238 and 5,397,597 shares issued and outstanding as of June 30, 2024 and December 31, 2023, respectively | | | 2 | | | 1 |
Additional paid-in capital | | | 560,766 | | | 179,062 |
Accumulated deficit | | | (657,635) | | | (614,647) |
Accumulated other comprehensive loss | | | (4,591) | | | (4,600) |
Total stockholders’ deficit | | | (101,458) | | | (440,184) |
Total liabilities, convertible preferred stock, and stockholders’ deficit | | | $347,738 | | | $305,050 |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | |||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | |
Revenue: | | | | | | | | | ||||
Collaboration and license revenue | | | $33,271 | | | $5,249 | | | $39,111 | | | $11,187 |
Grant revenue | | | 174 | | | — | | | 174 | | | — |
Total revenue | | | 33,445 | | | 5,249 | | | 39,285 | | | 11,187 |
Operating expenses: | | | | | | | | | ||||
Research and development | | | 12,661 | | | 17,782 | | | 22,399 | | | 36,406 |
General and administrative | | | 7,027 | | | 6,105 | | | 16,477 | | | 11,800 |
Total operating expenses | | | 19,688 | | | 23,887 | | | 38,876 | | | 48,206 |
Operating income (loss) | | | 13,757 | | | (18,638) | | | 409 | | | (37,019) |
Investment income | | | 1,195 | | | 1,394 | | | 2,359 | | | 2,725 |
Foreign currency transaction, net | | | — | | | 23 | | | — | | | 42 |
Interest expense | | | — | | | (752) | | | — | | | (1,560) |
Change in fair value of warrant liabilities | | | (3,908) | | | 6,341 | | | (2,866) | | | 2,262 |
Change in fair value of contingent value right liability | | | 2,500 | | | — | | | (36,800) | | | — |
Change in fair value of forward contract liabilities | | | — | | | — | | | (6,890) | | | — |
Other income, net | | | 292 | | | 245 | | | 800 | | | 500 |
Net income (loss) | | | $13,836 | | | $(11,387) | | | $(42,988) | | | $(33,050) |
Other comprehensive income (loss): | | | | | | | | | ||||
Foreign currency translation adjustment | | | 14 | | | (27) | | | 9 | | | (49) |
Unrealized gain on marketable securities | | | — | | | — | | | — | | | 11 |
Total comprehensive income (loss) | | | $13,850 | | | $(11,414) | | | $(42,979) | | | $(33,088) |
Net income (loss) | | | 13,836 | | | (11,387) | | | (42,988) | | | (33,050) |
Less: Undistributed earnings allocable to participating securities | | | (4,208) | | | — | | | — | | | — |
Net income (loss) allocable to shares of common stock - basic and diluted | | | 9,628 | | | (11,387) | | | (42,988) | | | (33,050) |
Net income (loss) per share allocable to common stockholders: | | | | | | | | | ||||
Basic | | | $0.58 | | | $(2.23) | | | $(3.88) | | | $(6.46) |
Diluted | | | $0.54 | | | $(2.23) | | | $(3.88) | | | $(6.46) |
Weighted-average common shares outstanding: | | | | | | | | | ||||
Basic | | | 16,723,479 | | | 5,114,747 | | | 11,068,749 | | | 5,113,213 |
Diluted | | | 17,791,143 | | | 5,114,747 | | | 11,068,749 | | | 5,113,213 |
| | | Series A Preferred Stock | | | Options for Series A Preferred Stock | | | Series A Preferred Stock | | | Common stock | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Stockholders’ equity (deficit) | ||||||||||
| | | Shares | | | Amount | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2023 | | | 435,120.513 | | | $296,851 | | | $3,703 | | | — | | | $— | | | 5,397,597 | | | $1 | | | $179,062 | | | $(614,647) | | | $(4,600) | | | $(440,184) |
Issuance of Series A Preferred Stock in connection with private placement and settlement of related forward contract | | | 99,140.326 | | | 75,197 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Transfer of Series A Preferred Stock and options for Series A Preferred Stock to permanent equity | | | (534,260.839) | | | (372,048) | | | (3,703) | | | 534,260.839 | | | — | | | — | | | — | | | 375,751 | | | — | | | — | | | 375,751 |
Issuance of common stock upon exercise of options | | | — | | | — | | | — | | | — | | | — | | | 52,558 | | | — | | | 154 | | | — | | | — | | | 154 |
Issuance of common stock upon exercise of warrants | | | — | | | — | | | — | | | — | | | — | | | 65,681 | | | — | | | 2,877 | | | — | | | — | | | 2,877 |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,431 | | | — | | | — | | | 1,431 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (5) | | | (5) |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (56,824) | | | — | | | (56,824) |
Balance at March 31, 2024 | | | — | | | $— | | | $— | | | 534,260.839 | | | $— | | | 5,515,836 | | | $1 | | | $559,275 | | | $(671,471) | | | $(4,605) | | | $(116,800) |
Conversion of Series A Preferred Stock to common stock | | | — | | | — | | | — | | | (367,919.247) | | | — | | | 12,263,951 | | | 1 | | | (1) | | | — | | | — | | | — |
Issuance of common stock upon exercise of options | | | — | | | — | | | — | | | — | | | — | | | 36,451 | | | — | | | 120 | | | — | | | — | | | 120 |
Equity offering costs | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (219) | | | — | | | — | | | (219) |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,591 | | | — | | | — | | | 1,591 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 14 | | | 14 |
Net income | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 13,836 | | | — | | | 13,836 |
Balance at June 30, 2024 | | | — | | | $— | | | $— | | | 166,341.592 | | | $— | | | 17,816,238 | | | $2 | | | $560,766 | | | $(657,635) | | | $(4,591) | | | $(101,458) |
| | | Common stock | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Stockholders’ equity (deficit) | ||||
| | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2022 | | | 5,101,459 | | | $1 | | | $493,322 | | | $(394,937) | | | $(4,558) | | | $93,828 |
Issuance of common stock under Employee Stock Purchase Plan | | | 3,584 | | | — | | | 149 | | | — | | | — | | | 149 |
Issuance of vested restricted stock units | | | 9,226 | | | — | | | — | | | — | | | — | | | — |
Stock-based compensation expense | | | — | | | — | | | 2,276 | | | — | | | — | | | 2,276 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | (22) | | | (22) |
Unrealized gain on marketable securities | | | — | | | — | | | — | | | — | | | 11 | | | 11 |
Net loss | | | — | | | — | | | — | | | (21,663) | | | — | | | (21,663) |
Balance at March 31, 2023 | | | 5,114,269 | | | $1 | | | $495,747 | | | $(416,600) | | | $(4,569) | | | $74,579 |
Issuance of vested restricted stock units | | | 20 | | | — | | | — | | | — | | | — | | | — |
Stock-based compensation expense | | | — | | | — | | | 2,283 | | | — | | | — | | | 2,283 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | (27) | | | (27) |
Net loss | | | — | | | — | | | — | | | (11,387) | | | — | | | (11,387) |
Balance at June 30, 2023 | | | 5,114,289 | | | $1 | | | $498,030 | | | $(427,987) | | | $(4,596) | | | $65,448 |
| | | Six Months Ended June 30, | ||||
| | | 2024 | | | 2023 | |
Cash flows from operating activities | | | | | ||
Net loss | | | $(42,988) | | | $(33,050) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | ||
Depreciation and amortization | | | 379 | | | 382 |
Amortization of premiums and discounts on marketable securities | | | — | | | (79) |
Non-cash lease expense | | | 1,113 | | | 842 |
Loss on disposal of property and equipment | | | 2 | | | — |
Stock-based compensation expense | | | 3,022 | | | 6,059 |
Non-cash interest expense | | | — | | | 533 |
Warrant liabilities revaluation | | | 2,866 | | | (2,262) |
Contingent value right liability revaluation | | | 36,800 | | | — |
Forward contract liabilities revaluation | | | 6,890 | | | — |
Changes in operating assets and liabilities: | | | | | ||
Accounts receivable | | | (26,169) | | | 1,211 |
Unbilled receivable | | | (491) | | | 2,107 |
Prepaid expenses, deposits and other assets | | | 2,848 | | | 815 |
Accounts payable | | | (290) | | | (49) |
Deferred revenue | | | (5,849) | | | 8,504 |
Accrued expenses and other liabilities | | | (8,496) | | | (3,673) |
Net cash used in operating activities | | | (30,363) | | | (18,660) |
Cash flows from investing activities | | | | | ||
Proceeds from maturities of marketable securities | | | — | | | 28,254 |
Purchases of property and equipment | | | (2,189) | | | (142) |
Net cash (used in) provided by investing activities | | | (2,189) | | | 28,112 |
Cash flows from financing activities | | | | | ||
Repayments of principal, final payment fee, and prepayment penalty on debt | | | — | | | (2,586) |
Proceeds from exercise of common warrants | | | 2,877 | | | — |
Proceeds from issuance of Series A Preferred Stock, gross in private placement | | | 40,000 | | | — |
Proceeds from exercise of stock options | | | 274 | | | — |
Proceeds from issuance of common stock under Employee Stock Purchase Plan | | | — | | | 149 |
Net cash provided by (used in) financing activities | | | 43,151 | | | (2,437) |
Effect of exchange rate changes on cash | | | 9 | | | (49) |
Net change in cash, cash equivalents, and restricted cash | | | 10,608 | | | 6,966 |
Cash, cash equivalents, and restricted cash at beginning of period | | | 78,288 | | | 108,038 |
Cash, cash equivalents, and restricted cash at end of period | | | $88,896 | | | $115,004 |
Supplemental cash flow information | | | | | ||
Cash paid for interest | | | $— | | | $1,242 |
Noncash investing and financing activities | | | | | ||
Stock-based compensation expense in accrued liabilities | | | $— | | | $1,500 |
Purchase of property and equipment not yet paid | | | $2,879 | | | $48 |
Equity offering costs in accrued liabilities | | | $219 | | | $— |
Forward contract to issue common stock | | | $2,713 |
Forward contract to issue Series A Preferred Stock | | | 155,308 |
Stock options allocated to consideration paid | | | 10,444 |
Total consideration | | | $168,465 |
| | | As of November 13, 2023 | |
Assets acquired: | | | |
Cash and cash equivalents | | | $6,561 |
Prepaid expenses and other current assets | | | 309 |
Property and equipment, net | | | 215 |
Right-of-use asset, net | | | 915 |
In-process research and development assets | | | 150,600 |
Goodwill | | | 48,163 |
| | | $206,763 | |
Liabilities assumed | | | |
Accrued expenses and other current liabilities | | | $2,530 |
Lease liability | | | 292 |
Lease liability, net of current portion | | | 623 |
Deferred tax liability | | | 34,853 |
| | | $38,298 | |
Net assets acquired | | | $168,465 |
| | | Acquisition Date Fair Value | |
Descartes-08 for MG | | | $93,900 |
Descartes-08 for SLE | | | 56,700 |
Total in-process research and development assets | | | $150,600 |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | |||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | |
Numerator: | | | | | | | | | ||||
Net income (loss) | | | $13,836 | | | $(11,387) | | | $(42,988) | | | $(33,050) |
Less: Undistributed earnings allocable to participating securities | | | (4,208) | | | — | | | — | | | — |
Net income (loss) allocable to shares of common stock - basic and diluted | | | $9,628 | | | $(11,387) | | | $(42,988) | | | $(33,050) |
Denominator: | | | | | | | | | ||||
Weighted-average common shares outstanding - basic | | | 16,723,479 | | | 5,114,747 | | | 11,068,749 | | | 5,113,213 |
Dilutive effect of employee equity incentive plans | | | 1,067,664 | | | — | | | — | | | — |
Weighted-average common shares outstanding - diluted | | | 17,791,143 | | | 5,114,747 | | | 11,068,749 | | | 5,113,213 |
Net income (loss) per share allocable to common stockholders: | | | | | | | | | ||||
Basic | | | $0.58 | | | $(2.23) | | | $(3.88) | | | $(6.46) |
Diluted | | | $0.54 | | | $(2.23) | | | $(3.88) | | | $(6.46) |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | |||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | |
Common stock options, restricted stock units and ESPP shares | | | 740,211 | | | 753,590 | | | 2,344,017 | | | 753,590 |
Warrants to purchase common stock | | | 975,132 | | | 1,040,943 | | | 975,132 | | | 1,040,943 |
Series A Preferred Stock | | | 5,544,719 | | | — | | | 5,544,719 | | | — |
Total | | | 7,260,062 | | | 1,794,533 | | | 8,863,868 | | | 1,794,533 |
| | | June 30, 2024 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $39,181 | | | $39,181 | | | $— | | | $— |
Total assets | | | $39,181 | | | $39,181 | | | $— | | | $— |
Liabilities: | | | | | | | | | ||||
Warrant liabilities | | | $9,260 | | | $— | | | $— | | | $9,260 |
Contingent value right liability | | | $395,400 | | | $— | | | $— | | | $395,400 |
Total liabilities | | | $404,660 | | | $— | | | $— | | | $404,660 |
| | | December 31, 2023 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $41,161 | | | $41,161 | | | $— | | | $— |
Total assets | | | $41,161 | | | $41,161 | | | $— | | | $— |
Liabilities: | | | | | | | | | ||||
Warrant liabilities | | | $6,394 | | | $— | | | $— | | | $6,394 |
Contingent value right liability | | | $358,600 | | | $— | | | $— | | | $358,600 |
Forward contract liabilities | | | $28,307 | | | $— | | | $28,307 | | | $— |
Total liabilities | | | $393,301 | | | $— | | | $28,307 | | | $364,994 |
| | | June 30, | ||||
| | | 2024 | | | 2023 | |
Cash and cash equivalents | | | $87,227 | | | $112,027 |
Short-term restricted cash | | | — | | | 1,600 |
Long-term restricted cash | | | 1,669 | | | 1,377 |
Total cash, cash equivalents, and restricted cash | | | $88,896 | | | $115,004 |
| | | June 30, 2024 | | | December 31, 2023 | |
Risk-free interest rate | | | 5.33% | | | 4.79% |
Dividend yield | | | — | | | — |
Expected life (in years) | | | 0.49 | | | 0.98 |
Expected volatility | | | 110.52% | | | 83.67% |
| | | June 30, 2024 | | | December 31, 2023 | |
Risk-free interest rate | | | 4.52% | | | 4.01% |
Dividend yield | | | — | | | — |
Expected life (in years) | | | 2.78 | | | 3.28 |
Expected volatility | | | 87.23% | | | 84.09% |
| | | Warrant liabilities | |
Fair value as of December 31, 2023 | | | $6,394 |
Change in fair value | | | 2,866 |
Fair value as of June 30, 2024 | | | $9,260 |
| • | 100% of all milestone payments, royalties and other amounts paid to the Company or its controlled affiliates, or the Company Entities, under the Sobi License or, following certain terminations of the Sobi License, any agreement a Company Entity enters into that provides for the development and commercialization of SEL-212; and |
| • | 100% of all cash consideration and the actual liquidation value of any and all non-cash consideration of any kind that is paid to or is actually received by any Company Entity prior to the Termination Date pursuant to an agreement relating to a sale, license, transfer or other disposition of any transferable asset of the Company existing as of immediately prior to the Merger, other than those exclusively licensed under the Sobi License or which the Company Entities are required to continue to own in order to comply with the Sobi License. |
| | | June 30, 2024 | |
Estimated cash flow dates | | | 2024-2038 |
Estimated probability of success | | | 95.0% - 100.0% |
Expected volatility of future revenues | | | 22.0% |
| | | December 31, 2023 | |
Estimated cash flow dates | | | 2024 - 2038 |
Estimated probability of success | | | 95.0% |
Risk-adjusted discount rate | | | 13.7% |
| | | CVR liability | |
Fair value as of December 31, 2023 | | | $358,600 |
Change in fair value | | | 36,800 |
Fair value as of June 30, 2024 | | | $395,400 |
| | | Forward contract liabilities | |
Fair value as of December 31, 2023 | | | $28,307 |
Settlements | | | (35,197) |
Change in fair value | | | 6,890 |
Fair value as of June 30, 2024 | | | $— |
| | | June 30, 2024 | | | December 31, 2023 | |
Laboratory equipment | | | $6,944 | | | $6,280 |
Computer equipment and software | | | 621 | | | 702 |
Leasehold improvements | | | 61 | | | 61 |
Furniture and fixtures | | | 462 | | | 452 |
Office equipment | | | 196 | | | 196 |
Construction in process | | | 4,417 | | | 150 |
Total property and equipment | | | 12,701 | | | 7,841 |
Less: Accumulated depreciation | | | (6,029) | | | (5,728) |
Property and equipment, net | | | $6,672 | | | $2,113 |
| | | June 30, 2024 | | | December 31, 2023 | |
Payroll and employee related expenses | | | $1,402 | | | $4,390 |
Accrued patent fees | | | 689 | | | 472 |
Accrued external research and development costs | | | 3,467 | | | 4,896 |
Accrued professional and consulting services | | | 1,871 | | | 4,331 |
Property and equipment | | | 2,877 | | | 128 |
Other | | | 309 | | | 516 |
Accrued expenses | | | $10,615 | | | $14,733 |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | | | ||||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | | |||
Operating lease cost | | | $956 | | | $696 | | | $1,731 | | | $1,392 | | ||
Variable lease cost | | | 352 | | | 270 | | | 749 | | | 412 | | ||
Short-term lease cost | | | 1 | | | 2 | | | 4 | | | 5 | | ||
Less: Sublease income | | | (250) | | | (251) | | | (760) | | | (506) | | ||
Total lease cost | | | $1,059 | | | $717 | | | $1,724 | | | $1,303 | | ||
| | | June 30, 2024 | |
2024 (remainder) | | | $1,239 |
2025 | | | 4,350 |
2026 | | | 4,471 |
2027 | | | 4,276 |
2028 | | | 2,243 |
Thereafter | | | 3,409 |
Total future minimum lease payments | | | 19,988 |
Less: Imputed interest | | | 5,121 |
Total operating lease liabilities | | | $14,867 |
| | | Six Months Ended June 30, | ||||
| | | 2024 | | | 2023 | |
Cash paid for amounts included in the measurement of lease liabilities: | | | $1,604 | | | $1,319 |
| | | June 30, | ||||
| | | 2024 | | | 2023 | |
Weighted-average remaining lease term | | | 4.9 years | | | 4.9 years |
Weighted-average discount rate | | | 11.5% | | | 9.7% |
| | | Number of Warrants | | | Weighted- average exercise price | |||||||
| | | Equity classified | | | Liability classified | | | Total | | |||
Outstanding at December 31, 2023 | | | 74,420 | | | 966,393 | | | 1,040,813 | | | $45.98 |
Exercises | | | (65,681) | | | — | | | (65,681) | | | 43.80 |
Outstanding at June 30, 2024 | | | 8,739 | | | 966,393 | | | 975,132 | | | $46.12 |
Exercise of warrants | | | 975,132 |
Shares available for future stock incentive awards | | | 3,930,990 |
Unvested restricted stock units | | | 454,456 |
Outstanding common stock options | | | 1,889,561 |
Series A Preferred Stock | | | 5,544,719 |
Total | | | 12,794,858 |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | |||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | |
Research and development | | | $776 | | | $2,677 | | | $1,488 | | | $3,869 |
General and administrative | | | 815 | | | 1,106 | | | 1,534 | | | 2,190 |
Total stock-based compensation expense | | | $1,591 | | | $3,783 | | | $3,022 | | | $6,059 |
| | | Three Months Ended June 30, | | | Six Months Ended June 30, | |||||||
| | | 2024 | | | 2023 | | | 2024 | | | 2023 | |
Risk-free interest rate | | | 4.53% | | | 3.38% | | | 4.04% | | | 3.95% |
Dividend yield | | | — | | | — | | | — | | | — |
Expected term (in years) | | | 6.18 | | | 6.00 | | | 6.20 | | | 5.94 |
Expected volatility | | | 93.57% | | | 94.40% | | | 95.09% | | | 94.64% |
Weighted-average fair value of common stock | | | $22.08 | | | $39.30 | | | $20.14 | | | $34.50 |
| | | Number of common stock options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Outstanding at December 31, 2023 | | | 776,865 | | | $2.97 | | | 6.50 | | | $13,760 |
Granted | | | 761,556 | | | $20.14 | | | | | ||
Converted from options for Series A Preferred Stock | | | 470,403 | | | $2.40 | | | | | ||
Exercised | | | (89,009) | | | $3.08 | | | | | ||
Forfeited | | | (30,254) | | | $14.84 | | | | | ||
Outstanding at June 30, 2024 | | | 1,889,561 | | | $9.56 | | | 7.24 | | | $33,081 |
Vested at June 30, 2024 | | | 944,490 | | | $2.74 | | | 5.43 | | | $22,925 |
Vested and expected to vest at June 30, 2024 | | | 1,743,749 | | | $8.66 | | | 7.12 | | | $32,070 |
| | | Number of Series A Preferred Stock options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Outstanding at December 31, 2023 | | | 14,112.299 | | | $79.94 | | | 5.91 | | | $8,601 |
Converted to options for common stock | | | (14,112.299) | | | $79.94 | | | | | ||
Outstanding at June 30, 2024 | | | — | | | $— | | | |
| | | Number of shares | | | Weighted-average grant date fair value ($) | |
Unvested at December 31, 2023 | | | — | | | $— |
Granted | | | 471,104 | | | 19.80 |
Forfeited | | | (16,648) | | | 19.80 |
Unvested at June 30, 2024 | | | 454,456 | | | $19.80 |
| | | Balance at beginning of period | | | Additions | | | Deductions | | | Balance at end of period | |
Six Months Ended June 30, 2024 | | | | | | | | | ||||
Contract liabilities: | | | | | | | | | ||||
Deferred revenue | | | $5,849 | | | $— | | | $(5,849) | | | $— |
Total contract liabilities | | | $5,849 | | | $— | | | $(5,849) | | | $— |
Name | | | Shares of Series A Preferred Stock purchased | | | Total aggregate purchase price |
Timothy A. Springer, Ph.D. | | | 99,140.326 | | | $40,000,000 |
| | | Beginning Balance December 31, 2023 | | | Charges | | | Payments | | | Ending Balance June 30, 2024 | |
Severance liability | | | $3,896 | | | $805 | | | $(4,527) | | | $174 |
| | | Valuation of in-process research and development acquired in a business combination | |
Description of the Matter | | | As described in Note 3, on November 13, 2023, the Company acquired Cartesian Therapeutics, Inc. in a stock for stock transfer, which was accounted for as a business combination using the acquisition method of accounting. The acquired intangible assets consisted of in-process research and development which had estimated acquisition-date fair values of $150.6 million. Auditing the acquisition date fair value of the in-process research and development was complex due to the significant judgment required in estimating the fair value. In particular, the fair value estimate required the use of valuation methodologies that were sensitive to significant assumptions (e.g., projected revenue growth rates, including forecasted selling prices and unit volumes, and discount rates applied to the in-process research and development), which are affected by expected future market or economic conditions. |
| | | ||
How We Addressed the Matter in Our Audit | | | To test the estimated fair value of the acquired in-process research and development intangible assets, our audit procedures included, among others, assessing the appropriateness of the valuation methodology and testing the significant assumptions discussed above and the completeness and accuracy of the underlying data used by the Company. For example, we evaluated the reasonableness of assumptions used to determine the projected revenue growth rates by comparing the forecasted assumptions to projected industry growth rates, and other factors considered by management in developing the model. We involved our valuation specialist to assist in evaluating the valuation methodologies and discount rates used to value in-process research and development intangible assets. We also performed sensitivity analyses to evaluate the changes in the fair value of the acquired in-process research and development intangible assets that would result from changes in the significant assumptions. |
| | | December 31, 2023 | | | December 31, 2022 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $76,911 | | | $106,438 |
Marketable securities | | | — | | | 28,164 |
Accounts receivable | | | 5,870 | | | 6,596 |
Unbilled receivables | | | 2,981 | | | 3,162 |
Prepaid expenses and other current assets | | | 4,967 | | | 3,778 |
Total current assets | | | 90,729 | | | 148,138 |
Non-current assets: | | | | | ||
Property and equipment, net | | | 2,113 | | | 2,794 |
Right-of-use asset, net | | | 10,068 | | | 11,617 |
In-process research and development assets | | | 150,600 | | | — |
Goodwill | | | 48,163 | | | — |
Long-term restricted cash | | | 1,377 | | | 1,311 |
Investments | | | 2,000 | | | 2,000 |
Other assets | | | — | | | 26 |
Total assets | | | $305,050 | | | $165,886 |
Liabilities, convertible preferred stock, and stockholders’ (deficit) equity | | | | | ||
Current liabilities: | | | | | ||
Accounts payable | | | $3,150 | | | $316 |
Accrued expenses and other current liabilities | | | 15,572 | | | 14,084 |
Loan payable | | | — | | | 8,476 |
Lease liability | | | 2,166 | | | 1,608 |
Deferred revenue | | | 2,311 | | | 593 |
Warrant liabilities | | | 720 | | | — |
Contingent value right liability | | | 15,983 | | | — |
Forward contract liabilities | | | 28,307 | | | — |
Total current liabilities | | | 68,209 | | | 25,077 |
Non-current liabilities: | | | | | ||
Loan payable, net of current portion | | | — | | | 17,786 |
Lease liability, net of current portion | | | 8,789 | | | 10,055 |
Deferred revenue, net of current portion | | | 3,538 | | | — |
Warrant liabilities, net of current portion | | | 5,674 | | | 19,140 |
Contingent value right liability, net of current portion | | | 342,617 | | | — |
Deferred tax liabilities, net | | | 15,853 | | | — |
Total liabilities | | | 444,680 | | | 72,058 |
Commitments and contingencies (Note 19) | | | | | ||
Series A Preferred Stock, $0.0001 par value; 548,375 and no shares authorized as of December 31, 2023 and December 31, 2022, respectively; 435,120.513 and no shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively | | | 296,851 | | | — |
Options for Series A Preferred Stock | | | 3,703 | | | — |
Stockholders’ (deficit) equity: | | | | | ||
Preferred stock, $0.0001 par value; 9,451,625 and 10,000,000 shares authorized as of December 31, 2023 and December 31, 2022, respectively; no shares issued and outstanding as of December 31, 2023 and December 31, 2022 | | | — | | | — |
| | | December 31, 2023 | | | December 31, 2022 | |
Common stock, $0.0001 par value; 350,000,000 shares authorized as of December 31, 2023 and December 31, 2022; 161,927,821 and 153,042,435 shares issued and outstanding as of December 31, 2023 and December 31, 2022, respectively | | | 16 | | | 15 |
Additional paid-in capital | | | 179,047 | | | 493,308 |
Accumulated deficit | | | (614,647) | | | (394,937) |
Accumulated other comprehensive loss | | | (4,600) | | | (4,558) |
Total stockholders’ (deficit) equity | | | (440,184) | | | 93,828 |
Total liabilities, convertible preferred stock, and stockholders’ (deficit) equity | | | $305,050 | | | $165,886 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Collaboration and license revenue | | | $26,004 | | | $110,777 | | | $85,077 |
Operating expenses: | | | | | | | |||
Research and development | | | 71,839 | | | 72,377 | | | 68,736 |
General and administrative | | | 40,581 | | | 23,862 | | | 20,938 |
Total operating expenses | | | 112,420 | | | 96,239 | | | 89,674 |
Operating (loss) income | | | (86,416) | | | 14,538 | | | (4,597) |
Investment income | | | 4,964 | | | 2,073 | | | 44 |
Foreign currency transaction gain (loss), net | | | 38 | | | (22) | | | — |
Interest expense | | | (2,833) | | | (3,031) | | | (2,844) |
Change in fair value of warrant liabilities | | | 12,746 | | | 20,882 | | | (2,339) |
Change in fair value of contingent value right liability | | | (18,300) | | | — | | | — |
Change in fair value of forward contract liabilities | | | (149,600) | | | — | | | — |
Other income, net | | | 691 | | | 330 | | | 15 |
(Loss) income before income taxes | | | (238,710) | | | 34,770 | | | (9,721) |
Income tax benefit (expense) | | | 19,000 | | | 609 | | | (15,966) |
Net (loss) income | | | $(219,710) | | | $35,379 | | | $(25,687) |
| | | | | | | ||||
Other comprehensive (loss) income: | | | | | | | |||
Foreign currency translation adjustment | | | (53) | | | 18 | | | (2) |
Unrealized gain (loss) on marketable securities | | | 11 | | | (10) | | | (1) |
Total comprehensive (loss) income | | | $(219,752) | | | $35,387 | | | $(25,690) |
| | | | | | | ||||
Net (loss) income per share: | | | | | | | |||
Basic | | | $(1.66) | | | $0.24 | | | $(0.22) |
Diluted | | | $(1.66) | | | $0.10 | | | $(0.22) |
Weighted-average common shares outstanding: | | | | | | | |||
Basic | | | 155,109,561 | | | 144,758,555 | | | 114,328,798 |
Diluted | | | 155,109,561 | | | 145,874,889 | | | 114,328,798 |
| | | Series A Preferred stock | | | Options for Series A Preferred Stock | | | Common stock | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Stockholders’ (Deficit) Equity | |||||||
| | | Shares | | | Amount | | | Amount | | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2020 | | | — | | | $— | | | $— | | | 108,071,249 | | | $11 | | | $391,175 | | | $(404,629) | | | $(4,563) | | | $(18,006) |
Issuance of common stock under Employee Stock Purchase Plan | | | — | | | — | | | — | | | 58,794 | | | — | | | 161 | | | — | | | — | | | 161 |
Issuance of common stock upon exercise of options | | | — | | | — | | | — | | | 447,492 | | | — | | | 778 | | | — | | | — | | | 778 |
Issuance of vested restricted stock units | | | — | | | — | | | — | | | 201,250 | | | — | | | — | | | — | | | — | | | — |
Issuance of common stock through at-the-market offering, net | | | — | | | — | | | — | | | 13,767,511 | | | 1 | | | 51,933 | | | — | | | — | | | 51,934 |
Issuance of common stock upon exercise of warrants | | | — | | | — | | | — | | | 1,076,669 | | | — | | | 5,624 | | | — | | | — | | | 5,624 |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | 7,720 | | | — | | | — | | | 7,720 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (2) | | | (2) |
Unrealized loss on marketable securities | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (1) | | | (1) |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | (25,687) | | | — | | | (25,687) |
Balance at December 31, 2021 | | | — | | | $— | | | $— | | | 123,622,965 | | | $12 | | | $457,391 | | | $(430,316) | | | $(4,566) | | | $22,521 |
Issuance of common stock under Employee Stock Purchase Plan | | | — | | | — | | | — | | | 120,877 | | | — | | | 189 | | | — | | | — | | | 189 |
Issuance of common stock upon exercise of options | | | — | | | — | | | — | | | 71,190 | | | — | | | 156 | | | — | | | — | | | 156 |
Issuance of vested restricted stock units | | | — | | | — | | | — | | | 131,430 | | | — | | | — | | | — | | | — | | | — |
Issuance of common stock through at-the-market offering, net | | | — | | | — | | | — | | | 774,544 | | | — | | | 2,121 | | | — | | | — | | | 2,121 |
Issuance of common stock and common warrants | | | — | | | — | | | — | | | 27,428,572 | | | 3 | | | 21,477 | | | — | | | — | | | 21,480 |
Issuance of common stock, license agreement | | | — | | | — | | | — | | | 892,857 | | | — | | | 1,000 | | | — | | | — | | | 1,000 |
Reclassification of warrant liabilities | | | — | | | — | | | — | | | — | | | — | | | 780 | | | — | | | — | | | 780 |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | 10,194 | | | — | | | — | | | 10,194 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 18 | | | 18 |
Unrealized loss on marketable securities | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (10) | | | (10) |
Net income | | | — | | | — | | | — | | | — | | | — | | | — | | | 35,379 | | | — | | | 35,379 |
Balance at December 31, 2022 | | | — | | | $— | | | $— | | | 153,042,435 | | | $15 | | | $493,308 | | | $(394,937) | | | $(4,558) | | | $93,828 |
| | | Series A Preferred stock | | | Options for Series A Preferred Stock | | | Common stock | | | Additional paid-in capital | | | Accumulated deficit | | | Accumulated other comprehensive loss | | | Stockholders’ (Deficit) Equity | |||||||
| | | Shares | | | Amount | | | Amount | | | Shares | | | Amount | | ||||||||||||
Issuance of Series A Preferred Stock in private placement | | | 619.627 | | | 250 | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Issuance of Series A Preferred Stock in connection with the Merger and settlement of related forward contract | | | 384,930.724 | | | 261,753 | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Issuance of Series A Preferred Stock in connection with private placement and settlement of related forward contract | | | 49,570.162 | | | 34,848 | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Issuance of common stock under Employee Stock Purchase Plan | | | — | | | — | | | — | | | 186,044 | | | — | | | 231 | | | — | | | — | | | 231 |
Issuance of vested restricted stock units | | | — | | | — | | | — | | | 636,418 | | | — | | | — | | | — | | | — | | | — |
Issuance of common stock forward in connection with the Merger | | | — | | | — | | | — | | | — | | | — | | | 2,713 | | | — | | | — | | | 2,713 |
Issuance of common stock in connection with the Merger and settlement of related forward contract | | | — | | | — | | | — | | | 6,723,639 | | | 1 | | | (1) | | | — | | | — | | | — |
Issuance of replacement options in Merger | | | — | | | — | | | 3,643 | | | — | | | — | | | 6,801 | | | — | | | — | | | 6,801 |
Issuance of common stock, license agreement | | | — | | | — | | | — | | | 1,339,285 | | | — | | | 1,500 | | | — | | | — | | | 1,500 |
Settlement of outstanding equity awards at Merger | | | — | | | — | | | — | | | — | | | — | | | (6,169) | | | — | | | — | | | (6,169) |
Distribution of contingent value rights | | | — | | | — | | | — | | | — | | | — | | | (340,300) | | | — | | | — | | | (340,300) |
Stock-based compensation expense | | | — | | | — | | | 60 | | | — | | | — | | | 20,964 | | | — | | | — | | | 20,964 |
Currency translation adjustment | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (53) | | | (53) |
Unrealized gain on marketable securities | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 11 | | | 11 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | (219,710) | | | — | | | (219,710) |
Balance at December 31, 2023 | | | 435,120.513 | | | $296,851 | | | $3,703 | | | 161,927,821 | | | $16 | | | $179,047 | | | $(614,647) | | | $(4,600) | | | $(440,184) |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
| | | (Amounts in thousands) | |||||||
Cash flows from operating activities | | | | | | | |||
Net (loss) income | | | $(219,710) | | | $35,379 | | | $(25,687) |
Adjustments to reconcile net (loss) income to net cash used in operating activities: | | | | | | | |||
Depreciation and amortization | | | 843 | | | 1,287 | | | 1,252 |
Amortization of premiums and discounts on marketable securities | | | (79) | | | (375) | | | 57 |
Non-cash lease expense | | | 1,754 | | | 1,337 | | | 1,119 |
Impairment of Right of use asset | | | 710 | | | — | | | — |
Loss (gain) on disposal of property and equipment | | | 477 | | | (147) | | | — |
Stock-based compensation expense | | | 22,524 | | | 11,194 | | | 7,720 |
Non-cash interest expense | | | 455 | | | 953 | | | 1,012 |
Warrant liabilities revaluation | | | (12,746) | | | (20,882) | | | 2,339 |
Contingent value right liability revaluation | | | 18,300 | | | — | | | — |
Forward contract liabilities revaluation | | | 149,600 | | | — | | | — |
Loss on extinguishment of debt | | | 740 | | | — | | | — |
Provision (benefit) for deferred taxes | | | (19,000) | | | — | | | — |
Changes in operating assets and liabilities: | | | | | | | |||
Accounts receivable | | | 726 | | | 3,318 | | | (2,690) |
Unbilled receivable | | | 181 | | | (3,162) | | | — |
Prepaid expenses, deposits and other assets | | | (1,265) | | | 2,471 | | | (1,451) |
Accounts payable | | | 2,834 | | | 92 | | | (219) |
Income taxes payable | | | — | | | (601) | | | 601 |
Deferred revenue | | | 5,256 | | | (64,707) | | | (45,496) |
Accrued expenses and other liabilities | | | (2,761) | | | 2,212 | | | 1,061 |
Net cash used in operating activities | | | (51,161) | | | (31,631) | | | (60,382) |
Cash flows from investing activities | | | | | | | |||
Cash assumed in acquisition of Old Cartesian | | | 6,561 | | | — | | | — |
Proceeds from maturities of marketable securities | | | 28,254 | | | 19,700 | | | 16,400 |
Payment made for investments | | | — | | | — | | | (2,000) |
Purchases of marketable securities | | | — | | | (33,501) | | | (30,455) |
Purchases of property and equipment | | | (206) | | | (1,201) | | | (1,085) |
Net cash provided by (used in) investing activities | | | 34,609 | | | (15,002) | | | (17,140) |
Cash flows from financing activities | | | | | | | |||
Proceeds from issuance of Series A Preferred Stock, gross in private placement | | | 20,250 | | | — | | | — |
Repayments of principal, final payment fee, and prepayment penalty on debt | | | (27,457) | | | — | | | — |
Debt amendment fee included in debt discount | | | — | | | (110) | | | — |
Net proceeds from issuance of common stock- at-the-market offering | | | — | | | 2,121 | | | 51,958 |
Net proceeds from issuance of common stock and common warrants | | | — | | | 36,859 | | | — |
Settlement of outstanding equity awards at Merger | | | (6,169) | | | — | | | — |
Proceeds from exercise of stock options | | | — | | | 156 | | | 778 |
Proceeds from issuance of common stock under Employee Stock Purchase Plan | | | 231 | | | 189 | | | 161 |
Net cash (used in) provided by financing activities | | | (13,145) | | | 39,215 | | | 52,897 |
Effect of exchange rate changes on cash | | | (53) | | | 20 | | | (3) |
Net change in cash, cash equivalents, and restricted cash | | | (29,750) | | | (7,398) | | | (24,628) |
Cash, cash equivalents, and restricted cash at beginning of period | | | 108,038 | | | 115,436 | | | 140,064 |
Cash, cash equivalents, and restricted cash at end of period | | | $78,288 | | | $108,038 | | | $115,436 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
| | | (Amounts in thousands) | |||||||
Supplement cash flow information | | | | | | | |||
Cash paid for interest | | | $1,853 | | | $2,248 | | | $2,002 |
Non-cash investing and financing activities | | | | | | | |||
Issuance of common stock, license agreement in stock-based compensation expense | | | $1,500 | | | $1,000 | | | $— |
Cashless warrant exercise | | | $— | | | $— | | | $5,624 |
Reclassification of warrant liability to equity | | | $— | | | $780 | | | $— |
Purchase of property and equipment not yet paid | | | $128 | | | $17 | | | $224 |
Forward contract to issue common stock | | | $2,713 |
Forward contract to issue Series A Preferred Stock | | | 155,308 |
Stock options allocated to consideration paid | | | 10,444 |
Total consideration | | | $168,465 |
| | | As of November 13, 2023 | |
Assets acquired: | | | |
Cash and cash equivalents | | | $6,561 |
Prepaid expenses and other current assets | | | 309 |
Property and equipment, net | | | 215 |
Right-of-use asset, net | | | 915 |
In-process research and development assets | | | 150,600 |
Goodwill | | | 48,163 |
| | | $206,763 | |
Liabilities assumed | | | |
Accrued expenses and other current liabilities | | | $2,530 |
Lease liability | | | $292 |
Lease liability, net of current portion | | | $623 |
Deferred tax liability | | | $34,853 |
| | | $38,298 | |
Net assets acquired | | | $168,465 |
| | | Acquisition Date Fair Value | | | Impairment | | | Carrying Value at December 31, 2023 | |
Descartes-08 for MG | | | $93,900 | | | $— | | | $93,900 |
Descartes-08 for SLE | | | 56,700 | | | — | | | 56,700 |
Total in-process research and development assets | | | $150,600 | | | $— | | | $150,600 |
| | | Year Ended December 31, | ||||
| | | 2023 | | | 2022 | |
Revenue | | | $26,004 | | | $112,226 |
Net (loss) income | | | $(232,259) | | | $29,607 |
| | | Amortized cost | | | Unrealized gains | | | Unrealized losses | | | Fair value | |
December 31, 2022 | | | | | | | | | ||||
U.S. government agency securities and treasuries | | | $13,566 | | | $— | | | $(9) | | | $13,557 |
Corporate bonds | | | $1,953 | | | $— | | | $(2) | | | $1,951 |
Commercial paper | | | 12,656 | | | — | | | — | | | 12,656 |
Total | | | $28,175 | | | $— | | | $(11) | | | $28,164 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Numerator: | | | | | | | |||
Net (loss) income | | | $(219,710) | | | $35,379 | | | $(25,687) |
Less: CVR distribution to participating securities | | | (37,550) | | | — | | | — |
Net (loss) income allocable to shares of common stock - basic | | | (257,260) | | | 35,379 | | | (25,687) |
Less: Change in fair value of warrants | | | — | | | (20,882) | | | — |
Net (loss) income allocable to shares of common stock - diluted | | | $(257,260) | | | $14,497 | | | $(25,687) |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Denominator: | | | | | | | |||
Weighted-average common shares outstanding - basic | | | 155,109,561 | | | 144,758,555 | | | 114,328,798 |
Dilutive effect of employee equity incentive plans and outstanding warrants | | | — | | | 1,116,334 | | | — |
Weighted-average common shares used in per share calculations - diluted | | | 155,109,561 | | | 145,874,889 | | | 114,328,798 |
Net (loss) income per share: | | | | | | | |||
Basic | | | $(1.66) | | | $0.24 | | | $(0.22) |
Diluted | | | $(1.66) | | | $0.10 | | | $(0.22) |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Warrants to purchase common stock | | | 31,224,703 | | | 213,339 | | | 10,735,980 |
Series A Preferred Stock | | | 435,120,513 | | | — | | | — |
Forward contract to issue Series A Preferred Stock | | | 99,140,326 | | | — | | | — |
Common stock options, RSUs and ESPP shares | | | 23,306,661 | | | 17,800,034 | | | 11,492,002 |
Series A Preferred Stock options | | | 14,112,299 | | | — | | | — |
Total | | | 602,904,502 | | | 18,013,373 | | | 22,227,982 |
| | | December 31, 2023 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $41,161 | | | $41,161 | | | $— | | | $— |
Total assets | | | $41,161 | | | $41,161 | | | $— | | | $— |
| | | | | | | | | |||||
Liabilities: | | | | | | | | | ||||
Warrant liabilities | | | $6,394 | | | $— | | | $— | | | $6,394 |
Contingent value right liability | | | 358,600 | | | — | | | — | | | 358,600 |
Forward contract liabilities | | | 28,307 | | | — | | | 28,307 | | | — |
Total liabilities | | | $393,301 | | | $— | | | $28,307 | | | $364,994 |
| | | December 31, 2022 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $53,552 | | | $53,552 | | | $— | | | $— |
Marketable securities: | | | | | | | | | ||||
U.S. government agency securities and treasuries | | | 13,557 | | | — | | | 13,557 | | | — |
Corporate bonds | | | 1,951 | | | — | | | 1,951 | | | — |
Commercial paper | | | 12,656 | | | — | | | 12,656 | | | — |
Total assets | | | $81,716 | | | $53,552 | | | $28,164 | | | $— |
| | | | | | | | | |||||
| | | December 31, 2022 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Liabilities: | | | | | | | | | ||||
Warrant liabilities | | | $19,140 | | | $— | | | $— | | | $19,140 |
Total liabilities | | | $19,140 | | | $— | | | $— | | | $19,140 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Cash and cash equivalents | | | $76,911 | | | $106,438 | | | $114,057 |
Short-term restricted cash | | | — | | | 289 | | | — |
Long-term restricted cash | | | 1,377 | | | 1,311 | | | 1,379 |
Total cash, cash equivalents, and restricted cash | | | $78,288 | | | $108,038 | | | $115,436 |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Risk-free interest rate | | | 4.79% | | | 4.74% |
Dividend yield | | | — | | | — |
Expected life (in years) | | | 0.98 | | | 1.98 |
Expected volatility | | | 83.67% | | | 79.92% |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Risk-free interest rate | | | 4.01% | | | 4.22% |
Dividend yield | | | — | | | — |
Expected life (in years) | | | 3.28 | | | 4.28 |
Expected volatility | | | 84.09% | | | 98.05% |
| | | Warrant liabilities | |
Fair value as of December 31, 2022 | | | $19,140 |
Change in fair value | | | (12,746) |
Fair value as of December 31, 2023 | | | $6,394 |
| • | 100% of all milestone payments, royalties and other amounts paid to the Company or its controlled affiliates, or the Company Entities, under the Sobi License or, following certain terminations of the Sobi License, any agreement a Company Entity enters into that provides for the development and commercialization of SEL-212; and |
| • | 100% of all cash consideration and the actual liquidation value of any and all non-cash consideration of any kind that is paid to or is actually received by any Company Entity prior to the Termination Date pursuant to an agreement relating to a sale, license, transfer or other disposition of any transferable asset of the Company existing as of immediately prior to the Merger, other than those exclusively licensed under the Sobi License or which the Company Entities are required to continue to own in order to comply with the Sobi License. |
| | | December 31, 2023 | | | At Issuance November 13, 2023 | |
Estimated cash flow dates | | | 2024 - 2038 | | | 2024 - 2038 |
Estimated probability of success | | | 95.0% | | | 95.0% |
Risk-adjusted discount rate | | | 13.7% | | | 14.4% |
| | | CVR liability | |
Fair value as of December 31, 2022 | | | $— |
Issuances | | | 340,300 |
Change in fair value | | | 18,300 |
Fair value as of December 31, 2023 | | | $358,600 |
| | | Forward contract liabilities | |
Fair value as of December 31, 2022 | | | $— |
Issuances | | | 155,308 |
Settlements | | | (276,601) |
Change in fair value | | | 149,600 |
Fair value as of December 31, 2023 | | | $28,307 |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Laboratory equipment | | | $6,280 | | | $6,001 |
Computer equipment and software | | | 702 | | | 697 |
Leasehold improvements | | | 61 | | | 57 |
Furniture and fixtures | | | 452 | | | 453 |
Office equipment | | | 196 | | | 192 |
Construction in process | | | 150 | | | 599 |
Total property and equipment | | | 7,841 | | | 7,999 |
Less accumulated depreciation | | | (5,728) | | | (5,205) |
Property and equipment, net | | | $2,113 | | | $2,794 |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Payroll and employee related expenses | | | $4,390 | | | $4,242 |
Accrued patent fees | | | 472 | | | 696 |
Accrued external research and development costs | | | 4,896 | | | 7,274 |
Accrued professional and consulting services | | | 4,331 | | | 985 |
Accrued interest | | | — | | | 222 |
Other | | | 644 | | | 665 |
Accrued expenses | | | $14,733 | | | $14,084 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Operating lease cost | | | $2,828 | | | $2,276 | | | $2,023 |
Variable lease cost | | | 965 | | | 910 | | | 834 |
Short-term lease cost | | | 8 | | | 11 | | | 10 |
Less sublease income | | | (1,172) | | | (176) | | | — |
Total lease cost | | | $2,629 | | | $3,021 | | | $2,867 |
| | | December 31, 2023 | |
2024 | | | $3,077 |
2025 | | | 3,164 |
2026 | | | 3,248 |
2027 | | | 3,017 |
2028 | | | 946 |
Thereafter | | | — |
Total future minimum lease payments | | | 13,452 |
Less: Imputed interest | | | 2,497 |
Total operating lease liabilities | | | $10,955 |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Cash paid for amounts included in the measurement of lease liabilities: | | | $2,696 | | | $2,048 |
| | | December 31, | ||||
| | | 2023 | | | 2022 | |
Weighted-average remaining lease term | | | 4.3 years | | | 5.4 years |
Weighted-average discount rate | | | 9.9 % | | | 9.7 % |
| | | As of December 31, 2023 | |
Shares reserved for issuance in November 2023 Private Placement | | | 99,140.326 |
Outstanding Series A Preferred Stock options | | | 14,112.299 |
Total | | | 113,252.625 |
| | | Number of Warrants | | | ||||||||
| | | Equity classified | | | Liability classified | | | Total | | | Weighted average exercise price | |
Outstanding at December 31, 2021 | | | 292,469 | | | 10,443,511 | | | 10,735,980 | | | $1.62 |
Issuance | | | — | | | 20,571,429 | | | 20,571,429 | | | 1.55 |
Canceled | | | (79,130) | | | — | | | (79,130) | | | $17.71 |
Reclassification of warrant liability to equity on modification | | | 2,022,987 | | | (2,022,987) | | | — | | | $1.46 |
Outstanding at December 31, 2022 | | | 2,236,326 | | | 28,991,953 | | | 31,228,279 | | | $1.53 |
Canceled | | | (3,576) | | | — | | | (3,576) | | | 16.77 |
Outstanding at December 31, 2023 | | | 2,232,750 | | | 28,991,953 | | | 31,224,703 | | | $1.53 |
| | | As of December 31, 2023 | |
Exercise of warrants | | | 31,224,703 |
Shares available for future stock incentive awards | | | 35,836,268 |
Outstanding common stock options | | | 23,306,661 |
Total | | | 90,367,632 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Research and development | | | $12,985 | | | $5,061 | | | $3,204 |
General and administrative | | | 12,793 | | | 6,133 | | | 4,516 |
Total stock-based compensation expense | | | $25,778 | | | $11,194 | | | $7,720 |
| | | Common Stock | | | Series A Preferred Stock | |
Risk-free interest rate | | | 4.83 % | | | 4.92 % |
Dividend yield | | | — | | | — |
Expected term | | | 3.59 | | | 3.29 |
Expected volatility | | | 83.77 % | | | 83.87 % |
Weighted-average fair value of common stock or Series A Preferred Stock, as applicable | | | $0.40 | | | $403.47 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Risk-free interest rate | | | 3.95 % | | | 2.24 % | | | 0.79 % |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Dividend yield | | | — | | | — | | | — |
Expected term | | | 5.94 | | | 6.02 | | | 6.03 |
Expected volatility | | | 94.64 % | | | 92.21 % | | | 95.04 % |
Weighted-average fair value of common stock | | | $1.15 | | | $2.63 | | | $3.58 |
| | | Number of Common Stock options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Employees | | | | | | | | | ||||
Outstanding at December 31, 2022 | | | 15,578,412 | | | $3.44 | | | 7.57 | | | $4 |
Granted | | | 5,477,200 | | | $1.15 | | | | | ||
Assumed in connection with Merger | | | 23,306,661 | | | $0.10 | | | | | ||
Exercised | | | — | | | $— | | | | | ||
Forfeited | | | (2,215,020) | | | $2.68 | | | | | ||
Cancelled/settled in connection with the Merger | | | (18,840,592) | | | $2.86 | | | | | ||
Outstanding at December 31, 2023 | | | 23,306,661 | | | $0.10 | | | 6.50 | | | $13,760 |
Vested at December 31, 2023 | | | 18,067,999 | | | $0.10 | | | 6.13 | | | $10,725 |
Vested and expected to vest at December 31, 2023 | | | 23,306,661 | | | $0.10 | | | 6.50 | | | $13,760 |
| | | | | | | | | |||||
Non-employee consultants | | | | | | | | | ||||
Outstanding at December 31, 2022 | | | 266,239 | | | $8.05 | | | 5.08 | | | $— |
Forfeited | | | — | | | $— | | | | | ||
Cancelled/settled in connection with the Merger | | | (266,239) | | | $8.05 | | | | | ||
Outstanding at December 31, 2023 | | | — | | | $— | | | — | | | $— |
| | | Number of Series A Preferred Stock options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Employees | | | | | | | | | ||||
Outstanding at December 31, 2022 | | | — | | | $— | | | — | | | $— |
Assumed in connection with Merger | | | 14,112.299 | | | $79.94 | | | | | ||
Outstanding at December 31, 2023 | | | 14,112.299 | | | $79.94 | | | 5.91 | | | $8,601 |
| | | Number of Series A Preferred Stock options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Vested at December 31, 2023 | | | 10,860.441 | | | $71.67 | | | 5.15 | | | $6,709 |
Vested and expected to vest at December 31, 2023 | | | 14,112.299 | | | $79.94 | | | 5.91 | | | $8,601 |
| | | Number of shares | | | Weighted average grant date fair value ($) | |
Unvested at December 31, 2022 | | | 1,705,558 | | | $2.62 |
Granted | | | 1,054,600 | | | 1.13 |
Vested | | | (636,418) | | | 2.40 |
Forfeited | | | (446,108) | | | 1.91 |
Cancelled/settled in connection with the Merger | | | (1,677,632) | | | 1.96 |
Unvested at December 31, 2023 | | | — | | | $— |
| | | Balance at beginning of period | | | Additions | | | Deductions | | | Balance at end of period | |
Contract liabilities: | | | | | | | | | ||||
Deferred revenue | | | $593 | | | $10,500 | | | $(5,244) | | | $5,849 |
Total contract liabilities | | | $593 | | | $10,500 | | | $(5,244) | | | $5,849 |
Name | | | Shares of Series A Preferred Stock purchased | | | Total aggregate purchase price |
Timothy A. Springer, Ph.D. | | | 24,785.081 | | | $10,000,000 |
TAS Partners LLC (affiliate of Timothy A. Springer, Ph.D.) | | | 24,785.081 | | | $10,000,000 |
Seven One Eight Three Four Irrevocable Trust (affiliate of Murat Kalayoglu, MD, Ph.D.) | | | 619.627 | | | $250,000 |
Name | | | Shares of Common Stock purchased | | | 2022 Warrants purchased | | | Total aggregate purchase price |
TAS Partners LLC (affiliate of Timothy A. Springer, Ph.D.) | | | 6,681,600 | | | 5,011,200 | | | $9,421,056 |
| | | Year Ended December 31, | |||||||
| | | 2023 | | | 2022 | | | 2021 | |
Statutory U.S. federal rate | | | 21.0 % | | | 21.0 % | | | 21.0 % |
State income taxes - net of federal benefit | | | 2.3 % | | | 1.6 % | | | (166.0)% |
Permanent items | | | (1.6)% | | | (18.6)% | | | 8.3 % |
Research tax credits | | | 0.6 % | | | (3.2)% | | | 55.0 % |
Deferred revenue | | | — % | | | — % | | | 156.5 % |
Other | | | — % | | | — % | | | (3.7)% |
Change in fair value of forward contract liabilities | | | (13.2)% | | | — % | | | — % |
Valuation allowance, net | | | 2.8 % | | | (4.4)% | | v | (230.1)% |
Stock-based compensation | | | (3.9)% | | | 1.8 % | | | (5.2)% |
Effective income tax rate | | | 8.0% | | | (1.8)% | | | (164.2)% |
| | | Year Ended December 31, | ||||
| | | 2023 | | | 2022 | |
Deferred Tax Assets | | | | | ||
Net operating loss carryforwards | | | $29,841 | | | $17,015 |
Research and development credits | | | 5,649 | | | 2,806 |
Stock-based compensation expense | | | 8 | | | 5,892 |
Other expenses | | | 705 | | | 1,697 |
Deferred revenue | | | 84,626 | | | 83,417 |
Operating lease liabilities | | | 2,718 | | | 3,186 |
R&E Capitalization | | | 19,778 | | | 9,588 |
Patent and license costs | | | 9,140 | | | 7,472 |
Gross deferred tax assets | | | 152,465 | | | 131,073 |
Deferred Tax Liabilities | | | | | ||
Intangible assets | | | $(41,144) | | | $— |
Depreciation | | | (128) | | | (81) |
Operating lease right-of-use assets | | | (2,751) | | | (3,174) |
Gross deferred tax liabilities | | | (44,023) | | | (3,255) |
Net deferred tax assets before valuation allowance | | | 108,442 | | | 127,818 |
Valuation allowance | | | (124,295) | | | (127,818) |
Net deferred tax assets/(liabilities) | | | $(15,853) | | | $— |
| | | Beginning Balance December 31, 2022 | | | Charges | | | Payments | | | Ending Balance December 31, 2023 | |
Severance liability | | | $— | | | $6,431 | | | $2,535 | | | $3,896 |
| | | September 30, 2023 | | | December 31, 2022 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $6,875 | | | $12,001 |
Accounts receivable | | | 994 | | | 994 |
Payroll tax credit receivable | | | 248 | | | 351 |
Prepaid expenses and other current assets | | | 51 | | | 59 |
Total current assets | | | $8,168 | | | $13,405 |
Non-current assets: | | | | | ||
Property and equipment, net | | | 228 | | | 197 |
Right-of-use asset, net | | | 891 | | | 983 |
Security deposit | | | 25 | | | 25 |
Total assets | | | $9,312 | | | $14,610 |
Liabilities, preferred stock and stockholders' deficit | | | | | ||
Current liabilities: | | | | | ||
Lease liability | | | $273 | | | $228 |
NIH liability | | | 569 | | | 461 |
Accrued expenses and other current liabilities | | | 1,513 | | | 949 |
Total current liabilities | | | $2,355 | | | $1,638 |
Non-current liabilities: | | | | | ||
Lease liability, net of current | | | 743 | | | 880 |
Total liabilities | | | $3,098 | | | $2,518 |
Commitments and contingencies (Note 10) | | | | | ||
Series A Preferred Stock; $0.01 par value, 220 authorized, 219.125 issued and outstanding as of September 30, 2023 and December 31, 2022 | | | 9,623 | | | 9,623 |
Series B Preferred Stock; $0.01 par value, 110 authorized, 109.267 issued and outstanding as of September 30, 2023 and December 31, 2022 | | | 7,128 | | | 7,128 |
Series B-1 Preferred Stock; $0.01 par value, 77 authorized, 65.017 issued and outstanding as of September 30, 2023 and December 31, 2022 | | | 3,162 | | | 3,162 |
Series B-2 Preferred Stock; $0.01 par value, 195 authorized, 193.644 issued and outstanding as of September 30, 2023 and December 31, 2022 | | | 12,144 | | | 12,144 |
Series B-2 Preferred Stock Subscription Receivable | | | — | | | (1,333) |
Stockholders' deficit: | | | | | ||
Common stock, $0.01 par value, 3,200 authorized, 1,244.625 issued and outstanding as of September 30, 2023 and 1,240.625 issued and outstanding as of December 31, 2022 | | | — | | | — |
Additional paid-in capital | | | 7,985 | | | 7,432 |
Accumulated deficit | | | (33,828) | | | (26,064) |
Total stockholders’ deficit | | | $(25,843) | | | $(18,632) |
Total liabilities, preferred stock and stockholders' deficit | | | $9,312 | | | $14,610 |
| | | Nine Months Ended September 30, | ||||
| | | 2023 | | | 2022 | |
Grant revenue: | | | $— | | | $1,035 |
Operating expenses: | | | | | ||
Research and development | | | 6,965 | | | 5,273 |
General and administrative | | | 1,286 | | | 1,069 |
Total operating expenses | | | 8,251 | | | 6,342 |
Loss from operations | | | (8,251) | | | (5,307) |
Other income, net: | | | | | ||
Interest income | | | 311 | | | 20 |
Other income, net | | | 176 | | | 101 |
Total other income | | | 487 | | | 121 |
Net loss | | | $(7,764) | | | $(5,186) |
| | | Series A Preferred Stock | | | Series B Preferred Stock | | | Series B-1 Preferred Stock | | | Series B-2 Preferred Stock | | | Series B-2 Preferred Stock Subscription Receivable | | | Series A Preferred Stock | | | Series B Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders' Deficit | ||||||||||||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2022 | | | 219.125 | | | $9,623 | | | 109.267 | | | $7,128 | | | 65.017 | | | $3,162 | | | 193.644 | | | $12,144 | | | $(1,333) | | | — | | | $— | | | — | | | $— | | | 1,240.625 | | | $— | | | $7,432 | | | $(26,064) | | | $(18,632) |
Subscription Receivable from preferred stockholders | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 1,333 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 461 | | | — | | | 461 |
Exercise of options to purchase common stock | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 4.000 | | | — | | | 92 | | | — | | | 92 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (7,764) | | | (7,764) |
Balance at September 30, 2023 | | | 219.125 | | | $9,623 | | | 109.267 | | | $7,128 | | | 65.017 | | | $3,162 | | | 193.644 | | | $12,144 | | | $— | | | — | | | $— | | | — | | | $— | | | 1,244.625 | | | $— | | | $7,985 | | | $(33,828) | | | $(25,843) |
| | | Series A Preferred Stock | | | Series B Preferred Stock | | | Series B-1 Preferred Stock | | | Series B-2 Preferred Stock | | | Series B-2 Preferred Stock Subscription Receivable | | | Series A Preferred Stock | | | Series B Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders' Deficit | ||||||||||||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2021 | | | 219.125 | | | $9,623 | | | 109.267 | | | $7,128 | | | 65.017 | | | $3,162 | | | — | | | $— | | | $— | | | — | | | $— | | | — | | | $— | | | 1,237.625 | | | $— | | | $6,644 | | | $(19,609) | | | $(12,965) |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 579 | | | — | | | 579 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (5,186) | | | (5,186) |
Balance at September 30, 2022 | | | 219.125 | | | $9,623 | | | 109.267 | | | $7,128 | | | 65.017 | | | $3,162 | | | — | | | $— | | | $— | | | — | | | $— | | | — | | | $— | | | 1,237.625 | | | $— | | | $7,223 | | | $(24,795) | | | $(17,572) |
| | | Nine Months Ended September 30, | ||||
| | | 2023 | | | 2022 | |
Cash flows from operating activities | | | | | ||
Net loss | | | $(7,764) | | | $ (5,186) |
Adjustments to reconcile net loss to net cash used in operating activities | | | | | ||
Depreciation expense | | | 69 | | | 88 |
Non-cash lease expense | | | 92 | | | 157 |
Stock-based compensation expense | | | 461 | | | 579 |
Changes in operating assets and liabilities: | | | | | ||
Accounts receivable | | | — | | | 2,377 |
Payroll tax credit receivable | | | 103 | | | (99) |
Prepaid expenses and other current assets | | | 8 | | | 15 |
Operating lease liability | | | (92) | | | (120) |
Deferred revenue | | | — | | | 54 |
NIH liability | | | 108 | | | 39 |
Accrued expenses and other current liabilities | | | 514 | | | 240 |
Net cash used in operating activites | | | (6,501) | | | (1,856) |
Cash flows from investing activities | | | | | ||
Purchases of property and equipment | | | (50) | | | (151) |
Net cash used in investing activities | | | (50) | | | (151) |
Cash flows from financing activities | | | | | ||
Net proceeds from issuance of Series B-2 Preferred Stock | | | 1,333 | | | — |
Proceeds from exercise of stock options | | | 92 | | | — |
Net cash provided by financing activities | | | 1,425 | | | — |
Net change in cash and cash equivalents | | | (5,126) | | | (2,007) |
Cash and cash equivalents at beginning of period | | | 12,001 | | | 4,735 |
Cash and cash equivalents at end of period | | | $6,875 | | | $2,728 |
Noncash investing and financing activities | | | | | ||
Purchase of equipment not yet paid | | | $50 | | | $— |
| | | September 30, 2023 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $6,531 | | | $6,531 | | | $— | | | $— |
Total assets | | | $6,531 | | | $6,531 | | | $— | | | $— |
| | | | | | | | | |||||
Liabilities: | | | | | | | | | ||||
Contingent payment to NIH | | | $569 | | | $— | | | $— | | | $569 |
Total liabilities | | | $569 | | | $— | | | $— | | | $569 |
| | | December 31, 2022 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $1,004 | | | $1,004 | | | $— | | | $— |
Certificates of deposits (included in cash equivalents) | | | 25 | | | 25 | | | — | | | — |
Total assets | | | $1,029 | | | $1,029 | | | $— | | | $— |
| | | | | | | | | |||||
Liabilities: | | | | | | | | | ||||
Contingent payment to NIH | | | $461 | | | $— | | | $— | | | $461 |
Total liabilities | | | $461 | | | $— | | | $— | | | $461 |
| | | Total | |
Balance at December 31, 2022 | | | $ 461 |
Change in fair value of contingent payment to NIH | | | 108 |
Balance at September 30, 2023 | | | $ 569 |
| | | September 30, 2023 | | | December 31, 2022 | |
Laboratory equipment | | | $879 | | | $779 |
Less accumulated depreciation | | | (651) | | | (582) |
Property and equipment, net | | | $228 | | | $197 |
| | | September 30, 2023 | | | December 31, 2022 | |
Accrued external research and development costs | | | $1,317 | | | $758 |
Accrued professional and consulting services | | | 48 | | | 60 |
Accrued payroll | | | 42 | | | 98 |
Accrued equipment | | | 50 | | | — |
Other current liabilities | | | 56 | | | 33 |
Accrued expenses and other current liabilities | | | $1,513 | | | $949 |
| | | Nine Months Ended September 30, | ||||
| | | 2023 | | | 2022 | |
Operating lease cost | | | $227 | | | $224 |
Variable lease cost | | | 143 | | | 113 |
Total lease cost | | | $370 | | | $337 |
| | | September 30, 2023 | |
2023 | | | $82 |
2024 | | | 336 |
2025 | | | 346 |
2026 | | | 346 |
2027 | | | 28 |
Thereafter | | | — |
Total future minimum lease payments | | | 1,138 |
Less imputed interest | | | (122) |
Total operating lease liabilities | | | $1,016 |
| | | September 30, | ||||
| | | 2023 | | | 2022 | |
Cash paid for amounts included in the measurement of lease liabilities: | | | $227 | | | $187 |
| | | September 30, | ||||
| | | 2023 | | | 2022 | |
Weighted-average remaining lease term | | | 3.03 years | | | 4.33 years |
Weighted-average discount rate | | | 7.09 % | | | 7.34 % |
| | | Nine Months Ended September 30, | ||||
| | | 2023 | | | 2022 | |
Research and development | | | $461 | | | $579 |
General and administrative | | | — | | | — |
Total stock-based compensation expense | | | $461 | | | $579 |
| | | Nine Months Ended September 30, | ||||
| | | 2023 | | | 2022 | |
Risk-free interest rate | | | 3.6 – 4.0% | | | 1.3% – 2.0% |
Dividend yield | | | — | | | — |
Expected term | | | 6.20 - 6.25 | | | 5.0 - 6.25 |
Expected volatility | | | 95% | | | 95% |
Fair value of common stock | | | $18,505 | | | $23,005 |
| | | Number of options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Outstanding at December 31, 2022 | | | 152 | | | $18,727 | | | 6.90 | | | $425 |
Granted | | | 29 | | | $23,005 | | | | | ||
Exercised | | | (4) | | | $23,005 | | | | | ||
Forfeited | | | (4) | | | $23,005 | | | | | ||
Outstanding at September 30, 2023 | | | 173 | | | $19,246 | | | 6.60 | | | $425 |
| | | | | | | | | |||||
Vested at September 30, 2023 | | | 119 | | | $17,541 | | | 5.72 | | | $425 |
Vested and expected to vest at September 30, 2023 | | | 173 | | | $19,246 | | | 6.60 | | | $425 |
| • | 100% of all milestone payments, royalties and other amounts paid to the Company or its controlled affiliates (the “Company Entities”) under the Sobi License or, following certain terminations of the Sobi License, any agreement a Company Entity enters into that provides for the development and commercialization of SEL-212; and |
| • | 100% of all cash consideration and the actual liquidation value of any and all non-cash consideration of any kind that is paid to or is actually received by any Company Entity prior to the Termination Date pursuant to an agreement relating to a sale, license, transfer or other disposition of any transferable asset of the Company existing as of immediately prior to the Merger, other than those exclusively licensed under the Sobi License or which the Company Entities are required to continue to own in order to comply with the Sobi License. |
| • | Each option to acquire shares of Common Stock and each restricted stock unit award with respect to shares of Common Stock, in each case that was outstanding and unvested immediately prior to the Merger, was accelerated and vested in full at the effective time of the First Merger; |
| • | each option to acquire shares of Common Stock was canceled and in exchange therefor, former holders became entitled to receive an amount in cash equal to the product of (A) the total number of shares of Common Stock subject to the unexercised portion the stock option (determined after giving effect to the accelerated vesting) multiplied by (B) the excess, if any, of $2.06 (the “Cash-out Amount”) over the applicable exercise price per share of Common Stock under such stock option; and |
| • | each restricted stock unit award with respect to shares of Common Stock was cancelled and the former holder of such canceled restricted stock unit became entitled, in exchange therefor, to receive an amount in cash equal to the product of (A) the total number of shares of Common Stock deliverable under such restricted stock unit (determined after giving effect to the accelerated vesting) multiplied by (B) the Cash-out Amount. |
| | | Selecta Biosciences, Inc. | | | Cartesian Therapeutics, Inc. (Old Cartesian) | | | Transaction Adjustments | | | Notes | | | Pro Forma Combined | |
ASSETS | | | | | | | | | | | |||||
Current assets: | | | | | | | | | | | |||||
Cash and cash equivalents | | | $79,603 | | | $6,875 | | | $(9,423) | | | B | | | $137,305 |
| | | | | | | 60,250 | | | G | | | ||||
Accounts receivable | | | 4,898 | | | 994 | | | — | | | | | 5,892 | |
Unbilled receivables | | | 1,875 | | | — | | | — | | | | | 1,875 | |
Prepaid expenses and other current assets | | | 3,493 | | | 299 | | | — | | | | | 3,792 | |
Total current assets | | | 89,869 | | | 8,168 | | | 50,827 | | | | | 148,864 | |
Non-current assets: | | | | | | | | | | | |||||
Property and equipment, net | | | 2,421 | | | 228 | | | — | | | | | 2,649 | |
Right-of-use asset, net | | | 10,339 | | | 891 | | | — | | | | | 11,230 | |
Intangible assets | | | — | | | — | | | 150,700 | | | F | | | 150,700 |
Goodwill | | | — | | | — | | | 48,062 | | | F | | | 48,062 |
Other assets | | | 3,405 | | | 25 | | | — | | | | | 3,430 | |
TOTAL ASSETS | | | $106,034 | | | $9,312 | | | $249,589 | | | | | $364,935 | |
LIABILITIES, PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | | | | | | | | | |||||
Current liabilities: | | | | | | | | | | | |||||
Accounts payable and accrued expenses | | | $14,012 | | | $2,082 | | | $4,895 | | | A | | | $20,989 |
Lease liability | | | 1,787 | | | 273 | | | — | | | | | 2,060 | |
Deferred revenue | | | 4,140 | | | — | | | — | | | | | 4,140 | |
Total current liabilities | | | 19,939 | | | 2,355 | | | 4,895 | | | | | 27,189 | |
Non-current liabilities: | | | | | | | | | | | |||||
Lease liability | | | 8,694 | | | 743 | | | — | | | | | 9,437 | |
Deferred revenue | | | 3,981 | | | — | | | — | | | | | 3,981 | |
Warrant liabilities | | | 13,091 | | | — | | | — | | | | | 13,091 | |
Deferred tax liability | | | — | | | — | | | 34,853 | | | F | | | 15,854 |
| | | | | | | (18,999) | | | J | | | ||||
Contingent value right obligation | | | — | | | — | | | 340,300 | | | H | | | 340,300 |
Total liabilities | | | 45,705 | | | 3.098 | | | 361,049 | | | | | 409,852 | |
Commitments and contingencies | | | | | | | | | | | |||||
Convertible Preferred Stock | | | — | | | 32,057 | | | 155,308 | | | F | | | 215,558 |
| | | | | | | 60,250 | | | G | | | ||||
| | | | | | | (32,057) | | | I | | | ||||
Stockholders’ equity (deficit): | | | | | | | | | | | |||||
Common stock | | | 15 | | | — | | | — | | | F I | | | 15 |
Additional paid-in capital | | | 501,919 | | | 7,985 | | | 6,977 | | | B | | | 182,372 |
| | | | | | | 619 | | | D | | | ||||
| | | | | | | 13,157 | | | F | | | ||||
| | | | | | | (340,300) | | | H | | | ||||
| | | | | | | (7,985) | | | I | | | ||||
Accumulated deficit | | | (436,989) | | | (33,828) | | | (4,895) | | | A | | | (438,246) |
| | | | | | | (16,400) | | | B | | | ||||
| | | | | | | (619) | | | D | | | ||||
| | | | | | | 35,486 | | | I | | | ||||
| | | | | | | 18,999 | | | J | | | ||||
Accumulated other comprehensive loss | | | (4,616) | | | — | | | — | | | | | (4,616) | |
Total stockholders’ equity (deficit) | | | 60,329 | | | (25,843) | | | (294,961) | | | | | (260,475) | |
TOTAL LIABILITIES, PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | $106,034 | | | $9,312 | | | $249,589 | | | | | $364,935 |
| | | Selecta Biosciences, Inc. | | | Cartesian Therapeutics, Inc. (Old Cartesian) | | | Transaction Adjustments | | | Notes | | | Pro Forma Combined | |
Revenue: | | | | | | | | | | | |||||
Collaboration and license revenue | | | $110,777 | | | $— | | | $— | | | | | $110,777 | |
Grant revenue | | | — | | | 1,449 | | | — | | | | | 1,449 | |
Total revenue | | | 110,777 | | | 1,449 | | | — | | | | | 112,226 | |
Operating expenses: | | | | | | | | | | | |||||
Research and development | | | 72,377 | | | 6,841 | | | 7,462 | | | B | | | 88,488 |
| | | | | | | 619 | | | D | | | ||||
| | | | | | | 1,189 | | | E | | | ||||
General and administrative | | | 23,862 | | | 1,244 | | | 4,895 | | | A | | | 38,939 |
| | | | | | | 8,938 | | | B | | | ||||
Total operating expenses | | | 96,239 | | | 8,085 | | | 23,103 | | | | | 127,427 | |
Operating income (loss) | | | 14,538 | | | (6,636) | | | (23,103) | | | | | (15,201) | |
Investment income | | | 2,073 | | | 35 | | | — | | | | | 2,108 | |
Foreign currency transaction, net | | | (22) | | | — | | | — | | | | | (22) | |
Interest (expense) income, net | | | (3,031) | | | — | | | — | | | | | (3,031) | |
Change in fair value of warrant liabilities | | | 20,882 | | | — | | | — | | | | | 20,882 | |
Other income, net | | | 330 | | | 146 | | | (108) | | | C | | | 368 |
Income (loss) before income taxes | | | 34,770 | | | (6,455) | | | (23,211) | | | | | 5,104 | |
Income tax benefit | | | 609 | | | — | | | 18,999 | | | J | | | 19,608 |
Net income (loss) | | | 35,379 | | | (6,455) | | | (4,212) | | | | | 24,712 | |
Other comprehensive income (loss) | | | | | | | | | | | |||||
Foreign currency translation adjustment | | | 18 | | | — | | | — | | | | | 18 | |
Unrealized gain on marketable securities | | | (10) | | | — | | | — | | | | | (10) | |
Total comprehensive income (loss) | | | $35,387 | | | $(6,455) | | | $(4,212) | | | | | $24,720 | |
Net (loss) income per share | | | | | | | | | | | |||||
Basic | | | $0.24 | | | | | | | K | | | $(0.08) | ||
Diluted | | | $0.10 | | | | | | | K | | | $(0.22) | ||
Weighted-average common shares outstanding | | | | | | | | | | | |||||
Basic | | | 144,758,555 | | | | | | | K | | | 151,482,194 | ||
Diluted | | | 145,874,889 | | | | | | | K | | | 152,282,286 |
| | | Selecta Biosciences, Inc. | | | Cartesian Therapeutics, Inc. (Old Cartesian) | | | Transaction Adjustments | | | Notes | | | Pro Forma Combined | |
Collaboration and license revenue | | | $17,738 | | | $— | | | $— | | | | | $17,738 | |
Operating expenses: | | | | | | | | | | | |||||
Research and development | | | 49,408 | | | 6,965 | | | 684 | | | E | | | 57,057 |
General and administrative | | | 18,414 | | | 1,286 | | | — | | | | | 19,700 | |
Total operating expenses | | | 67,822 | | | 8,251 | | | 684 | | | | | 76,757 | |
Operating loss | | | (50,084) | | | (8,251) | | | (684) | | | | | (59,019) | |
Investment income | | | 4,024 | | | 311 | | | — | | | | | 4,335 | |
Foreign currency transaction, net | | | 39 | | | — | | | — | | | | | 39 | |
Interest expense | | | (2,833) | | | — | | | — | | | | | (2,833) | |
Change in fair value of warrant liabilities | | | 6,049 | | | — | | | — | | | | | 6,049 | |
Other income, net | | | 753 | | | 176 | | | 108 | | | C | | | 1,037 |
Loss before income taxes | | | (42,052) | | | (7,764) | | | (576) | | | | | (50,392) | |
Income tax (expense) benefit | | | — | | | — | | | — | | | | | — | |
Net loss | | | (42,052) | | | (7,764) | | | (576) | | | | | (50,392) | |
Other comprehensive income (loss): | | | | | | | | | | | |||||
Foreign currency translation adjustment | | | (69) | | | — | | | — | | | | | (69) | |
Unrealized gain on marketable securities | | | 11 | | | — | | | — | | | | | 11 | |
Total comprehensive loss | | | $(42,110) | | | $(7,764) | | | $(576) | | | | | $(50,450) | |
Net loss per share | | | | | | | | | | | |||||
Basic | | | $(0.27) | | | | | | | | | $(0.31) | |||
Diluted | | | $(0.27) | | | | | | | | | $(0.31) | |||
Weighted-average common shares outstanding | | | | | | | | | | | |||||
Basic | | | 153,870,912 | | | | | | | F K | | | 160,594,551 | ||
Diluted | | | 153,870,912 | | | | | | | F K | | | 160,594,551 |
| | | Amounts | |
Total purchase consideration | | | |
Common Stock | | | $2,713 |
Series A Preferred Stock | | | 155,308 |
Assumption of Cartesian stock options | | | 10,444 |
Total purchase price | | | $168,465 |
Allocation of the purchase consideration | | | |
Tangible assets | | | $8,000 |
Liabilities assumed | | | (3,444) |
Intangible assets | | | 150,700 |
Deferred tax liabilities | | | (34,853) |
Goodwill | | | 48,062 |
Total purchase price allocation | | | $168,465 |
| A | To accrue additional $4.9 million of transaction costs incurred by Selecta subsequent to September 30, 2023. |
| B | Recognize total research and development expense of $7.5 million and general and administrative expense of $8.9 million associated with the modification of Selecta stock options and restricted stock units to accelerate the vesting of all awards upon the Merger and the cash settlement of certain awards. |
| C | An in-license agreement held by Old Cartesian included a payment to the licensor that is contingent upon certain corporate transactions. In connection with the Merger, a payment in the amount of $0.6 million was due to the licensor and fully accrued as of September 30, 2023. The Company accounted for the obligation as a derivative which was remeasured at fair value at the end of each reporting period. The expense related to the remeasurement of the contingent liability which is recorded in other income, net for the nine months ended September 30, 2023 ($0.1 million) was removed. The expense has been reflected in the year ended December 31, 2022, as the Merger is assumed to have occurred on January 1, 2022, for pro forma purposes. |
| D | In connection with the Merger, one Old Cartesian employee had a pre-existing provision in the employee’s stock option agreement, which provided for an acceleration of vesting upon a change in control, which was triggered as a result of the Merger. The additional expense of $0.6 million will be included in Old Cartesian’s pre-acquisition net loss, upon the Merger. This amount is included as a pro forma adjustment as the expense is not included in the historical financial statements presented. |
| E | To record stock compensation expense for the assumed unvested stock option awards (valued at approximately $2.6 million) that is to be recorded prospectively over the remaining service period of the awards. Total expense of $1.2 million and $0.7 million was classified as research and development expense during the year ended December 31, 2022 and the nine months ended September 30, 2023, respectively. There are no awards related to general and administrative activities. |
| F | To record purchase consideration and acquired intangible assets, goodwill and deferred tax liabilities. |
| G | To reflect the $60.25 million Financing associated with the issuance of Series A Preferred Stock under the Securities Purchase Agreement. |
| H | In connection with the Merger, the Company entered into the CVR Agreement to distribute the rights to future cash flows associated with certain licensed products and other assets to its stockholders. One CVR was distributed with respect to each share of Common Stock outstanding as of December 4, 2023 and each share of Common Stock underlying the Selecta Warrants issued on April 11, 2022. Further, one CVR will be distributed in respect of each share of Common Stock underlying the other Selecta Warrants, in each case if and to the extent each such Selecta Warrant is exercised in the future in accordance with its own terms. Each CVR was valued at $1.83 per Common Stock equivalent. The aggregate fair value of the CVR obligation on November 13, 2023 (the date that the CVR dividend was declared) was $340.3 million, which is recognized as a liability with the dividend recognized to additional paid in capital. |
| I | To eliminate the historical equity of Cartesian Therapeutics, Inc. (Old Cartesian). |
| J | To recognize the tax benefit associated with the deferred tax liability recorded as part of the purchase price allocation. |
| K | The Series A Preferred Stock and the Selecta Warrants issued on April 11, 2022 are considered participating securities and therefore the Company follows the two-class method when computing pro forma net loss (income) per share. During periods of net loss, there is no allocation of undistributed earnings required under the two-class method since the participating securities do not have a contractual obligation to fund the losses of the Company. The following represents the pro forma calculation of basic EPS for the year ended December 31, 2022: |
Net income | | | $24,712 |
Less: CVR distribution to participating securities | | | (37,550) |
Net loss allocable to shares of common stock, basic | | | (12,838) |
Net loss per share, basic | | | $(0.08) |
Weighted-average shares of common stock outstanding, basic | | | 151,482,194 |
Net loss allocable to shares of common stock, basic | | | $(12,838) |
Less: change in fair value of dilutive warrants | | | (21,029) |
Net loss allocable to shares of common stock, diluted | | | (33,867) |
Net loss per share, diluted | | | $(0.22) |
Weighted-average shares of common stock outstanding, diluted | | | 152,282,286 |
| | | September 30, 2023 | | | December 31, 2022 | |
Warrants to purchase Common Stock | | | 31,224,703 | | | 22,807,755 |
Series A preferred stock issued to Cartesian stockholders | | | 384,930,724 | | | 384,930,724 |
Series A preferred stock issued in Financing | | | 149,330,115 | | | 149,330,115 |
Common Stock options | | | 23,306,661 | | | 23,306,661 |
Series A Preferred Stock options | | | 14,112,299 | | | 14,112,299 |
Total | | | 602,904,502 | | | 594,487,554 |
| • | Exercise professional judgment and maintain professional skepticism throughout the audit. |
| • | Identify and assess the risks of material misstatement of the financial statements, whether due to fraud or error, and design and perform audit procedures responsive to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. |
| • | Obtain an understanding of internal control relevant to the audit in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control. Accordingly, no such opinion is expressed. |
| • | Evaluate the appropriateness of accounting policies used and the reasonableness of significant accounting estimates made by management, as well as evaluate the overall presentation of the financial statements. |
| • | Conclude whether, in our judgment, there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for a reasonable period of time |
| | | December 31, 2022 | | | December 31, 2021 | |
Assets | | | | | ||
Current assets: | | | | | ||
Cash and cash equivalents | | | $12,001 | | | $4,735 |
Accounts receivable | | | 994 | | | 3,129 |
Payroll tax credit receivable | | | 351 | | | 225 |
Prepaid expenses and other current assets | | | 59 | | | 50 |
Total current assets | | | $13,405 | | | $8,139 |
Non-current assets: | | | | | ||
Property and equipment, net | | | 197 | | | 309 |
Right-of-use asset, net | | | 983 | | | 1,195 |
Security deposit | | | 25 | | | 25 |
Total assets | | | $14,610 | | | $9,668 |
Liabilities, preferred stock and stockholders' deficit | | | | | ||
Current liabilities: | | | | | ||
Lease liability | | | $228 | | | $172 |
Deferred revenue | | | — | | | 117 |
NIH liability | | | 461 | | | — |
Accrued expenses and other current liabilities | | | 949 | | | 978 |
Total current liabilities | | | $1,638 | | | $1,267 |
Non-current liabilities: | | | | | ||
NIH liability | | | — | | | 345 |
Lease liability, net of current | | | 880 | | | 1,108 |
Total liabilities | | | $2,518 | | | $2,720 |
Commitments and contingencies (Note 11) | | | | | ||
Series A Preferred Stock; $0.01 par value, 220 authorized, 219.125 issued and outstanding as of December 31, 2022 and December 31, 2021 | | | 9,623 | | | 9,623 |
Series B Preferred Stock; $0.01 par value, 110 authorized, 109.267 issued and outstanding as of December 31, 2022 and December 31, 2021 | | | 7,128 | | | 7,128 |
Series B-1 Preferred Stock; $0.01 par value, 77 authorized, 65.017 issued and outstanding as of December 31, 2022 and December 31, 2021 | | | 3,162 | | | 3,162 |
Series B-2 Preferred Stock; $0.01 par value, 195 authorized, 193.644 issued and outstanding as of December 31, 2022 and none authorized, issued and outstanding as of December 31, 2021 | | | 12,144 | | | — |
Series B-2 Preferred Stock Subscription Receivable | | | (1,333) | | | — |
Stockholders' deficit: | | | | | ||
Common stock, $0.01 par value, 3,200 authorized, 1,240.625 issued and outstanding as of December 31, 2022 and 1,237.625 issued and outstanding as of December 31, 2021 | | | — | | | — |
Additional paid-in capital | | | 7,432 | | | 6,644 |
Accumulated deficit | | | (26,064) | | | (19,609) |
Total stockholders’ deficit | | | $(18,632) | | | $(12,965) |
Total liabilities, preferred stock and stockholders' deficit | | | $14,610 | | | $9,668 |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Grant revenue: | | | $1,449 | | | $3,337 |
Operating expenses: | | | | | ||
Research and development | | | 6,841 | | | 6,090 |
General and administrative | | | 1,244 | | | 1,006 |
Total operating expenses | | | 8,085 | | | 7,096 |
Loss from operations | | | (6,636) | | | (3,759) |
Other income, net: | | | | | ||
Interest income | | | 35 | | | 3 |
Other income, net | | | 146 | | | 116 |
Total other income | | | 181 | | | 119 |
Net loss | | | $ (6,455) | | | $ (3,640) |
| | | Series A Preferred Stock | | | Series B Preferred Stock | | | Series B-1 Preferred Stock | | | Series B-2 Preferred Stock | | | Series B-2 Preferred Stock Subscription Receivable | | | Series A Preferred Stock | | | Series B Preferred Stock | | | Common Stock | | | Additional Paid-In Capital | | | Accumulated Deficit | | | Total Stockholders' Deficit | ||||||||||||||||||||||
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | ||||||||||||
Balance at December 31, 2020 | | | — | | | $— | | | — | | | $— | | | — | | | $— | | | — | | | $— | | | $— | | | 169.125 | | | $ — | | | 109.267 | | | $ — | | | 1,287.625 | | | $ — | | | $20,909 | | | $ (15,319) | | | $5,590 |
Issuance of Series B-1 Preferred Stock, net of $16 of issuance costs | | | — | | | — | | | — | | | — | | | 65.017 | | | 4,207 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Exchange of Common Stock to Series A Preferred Stock | | | 50.000 | | | 2,196 | | | — | | | — | | | — | | | (1,045) | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (50.000) | | | — | | | (500) | | | (650) | | | (1,150) |
Reclassification of Series A and Series B Preferred Stock | | | 169.125 | | | 7,427 | | | 109.267 | | | 7,128 | | | — | | | — | | | — | | | — | | | — | | | (169.125) | | | — | | | (109.267) | | | — | | | — | | | — | | | (14,555) | | | — | | | (14,555) |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 790 | | | — | | | 790 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (3,640) | | | (3,640) |
Balance at December 31, 2021 | | | 219.125 | | | $ 9,623 | | | 109.267 | | | $ 7,128 | | | 65.017 | | | $3,162 | | | — | | | $— | | | $— | | | — | | | $ — | | | — | | | $ — | | | 1,237.625 | | | $ — | | | $6,644 | | | $ (19,609) | | | $ (12,965) |
Issuance of Series B-2 Preferred Stock, net of $24 of issuance costs | | | — | | | — | | | — | | | — | | | — | | | — | | | 193.644 | | | 12,144 | | | (1,333) | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — |
Stock-based compensation expense | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 719 | | | — | | | 719 |
Exercise of options to purchase common stock | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | 3.000 | | | — | | | 69 | | | — | | | 69 |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (6,455) | | | (6,455) |
Balance at December 31, 2022 | | | 219.125 | | | $ 9,623 | | | 109.267 | | | $ 7,128 | | | 65.017 | | | $3,162 | | | 193.644 | | | $ 12,144 | | | $ (1,333) | | | — | | | $ — | | | — | | | $ — | | | 1,240.625 | | | $ — | | | $7,432 | | | $ (26,064) | | | $ (18,632) |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Cash flows from operating activities | | | | | ||
Net loss | | | $(6,455) | | | $(3,640) |
Adjustments to reconcile net loss to net cash used in operating activities | | | | | ||
Depreciation expense | | | 112 | | | 123 |
Non-cash lease expense | | | 212 | | | 128 |
Stock-based compensation expense | | | 719 | | | 790 |
Changes in operating assets and liabilities: | | | | | ||
Accounts receivable | | | 2,135 | | | (2,135) |
Payroll tax credit receivable | | | (126) | | | (72) |
Prepaid expenses and other current assets | | | (9) | | | (51) |
Operating lease liability | | | (172) | | | (108) |
Deferred revenue | | | (117) | | | 117 |
NIH liability | | | 116 | | | 79 |
Accrued expenses and other current liabilities | | | 122 | | | (32) |
Net cash used in operating activites | | | (3,463) | | | (4,801) |
Cash flows from investing activities | | | | | ||
Purchases of property and equipment | | | (151) | | | — |
Net cash used in investing activities | | | (151) | | | — |
Cash flows from financing activities | | | | | ||
Net proceeds from issuance of Series B-1 Preferred Stock | | | — | | | 4,207 |
Net proceeds from issuance of Series B-2 Preferred Stock | | | 10,811 | | | — |
Proceeds from exercise of stock options | | | 69 | | | — |
Net cash provided by financing activities | | | 10,880 | | | 4,207 |
Net change in cash and cash equivalents | | | 7,266 | | | (594) |
Cash and cash equivalents at beginning of period | | | 4,735 | | | 5,329 |
Cash and cash equivalents at end of period | | | $12,001 | | | $4,735 |
Noncash investing and financing activities | | | | | ||
Issuance of Series B-2 Preferred Stock subscription | | | $1,333 | | | $— |
Purchase of equipment not yet paid | | | $— | | | $151 |
Increase in right-of-use asset due to lease modification | | | $— | | | $893 |
Increase in lease liability due to lease modification | | | $— | | | $893 |
| | | December 31, 2022 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $1,004 | | | $1,004 | | | $— | | | $— |
Certificates of deposit (included in cash equivalents) | | | 25 | | | 25 | | | — | | | — |
Total assets | | | $1,029 | | | $1,029 | | | $— | | | $— |
Liabilities: | | | | | | | | | ||||
Contingent payment to NIH | | | $461 | | | $— | | | $— | | | $461 |
Total liabilities | | | $461 | | | $— | | | $— | | | $461 |
| | | December 31, 2021 | ||||||||||
| | | Total | | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | ||||
Money market funds (included in cash equivalents) | | | $4,502 | | | $4,502 | | | $— | | | $— |
Certificates of deposits (included in cash equivalents) | | | 25 | | | 25 | | | — | | | — |
Total assets | | | $4,527 | | | $4,527 | | | $— | | | $— |
Liabilities: | | | | | | | | | ||||
Contingent payment to NIH | | | $345 | | | $— | | | $— | | | $345 |
Total liabilities | | | $345 | | | $— | | | $— | | | $345 |
| | | Total | |
Balance at December 31, 2020 | | | $266 |
Change in fair value of contingent payment to NIH | | | 79 |
Balance at December 31, 2021 | | | $ 345 |
Change in fair value of contingent payment to NIH | | | 116 |
Balance at December 31, 2022 | | | $461 |
| | | December 31, | ||||
| | | 2022 | | | 2021 | |
Laboratory equipment | | | $779 | | | $779 |
Less accumulated depreciation | | | (582) | | | (470) |
Property and equipment, net | | | $197 | | | $309 |
| | | December 31, | ||||
| | | 2022 | | | 2021 | |
Accrued external research and development costs | | | $758 | | | $600 |
Accrued professional and consulting services | | | 60 | | | 72 |
Accrued payroll | | | 98 | | | 115 |
Accrued equipment | | | — | | | 151 |
Other current liabilities | | | 33 | | | 40 |
Accrued expenses and other current liabilities | | | $949 | | | $978 |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Operating lease cost | | | $299 | | | $191 |
Variable lease cost | | | 147 | | | 57 |
Total lease cost | | | $446 | | | $248 |
| | | December 31, 2022 | |
2023 | | | $300 |
2024 | | | 309 |
2025 | | | 318 |
2026 | | | 328 |
2027 | | | 28 |
Thereafter | | | — |
Total future minimum lease payments | | | 1,283 |
Less imputed interest | | | (175) |
Total operating lease liabilities | | | $1,108 |
| | | December 31, | ||||
| | | 2022 | | | 2021 | |
Cash paid for amounts included in the measurement of lease liabilities: | | | $260 | | | $172 |
| | | December 31, | ||||
| | | 2022 | | | 2021 | |
Weighted-average remaining lease term | | | 4.1 years | | | 5.08 years |
Weighted-average discount rate | | | 7.3 % | | | 7.3 % |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Research and development | | | $719 | | | $790 |
General and administrative | | | — | | | — |
Total stock-based compensation expense | | | $719 | | | $790 |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Risk-free interest rate | | | 1.13% - 1.96% | | | 0.85% - 1.45% |
Dividend yield | | | — | | | — |
Expected term | | | 1.0 - 7.0 | | | 5.0 - 7.0 |
Expected volatility | | | 95 % | | | 95 % |
Fair value of common stock | | | $23,005 | | | $23,005 - 64,962 |
| | | Number of options | | | Weighted-average exercise price ($) | | | Weighted-average remaining contractual term (in years) | | | Aggregate intrinsic value (in thousands) | |
Outstanding at December 31, 2021 | | | 153 | | | $18,755 | | | 7.88 | | | $650 |
Granted | | | 9 | | | $23,005 | | | | | ||
Exercised | | | (3) | | | $23,005 | | | | | ||
Forfeited | | | (7) | | | $23,005 | | | | | ||
Outstanding at December 31, 2022 | | | 152 | | | $18,727 | | | 6.90 | | | $425 |
Vested at December 31, 2022 | | | 110 | | | $17,094 | | | 6.25 | | | $425 |
Vested and expected to vest at December 31, 2022 | | | 152 | | | $18,727 | | | 6.90 | | | $425 |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Current: Federal | | | $ — | | | $— |
State | | | — | | | — |
Deferred: Federal | | | — | | | — |
State | | | — | | | — |
Total | | | $— | | | $— |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Loss before Income Tax | | | $ (6,455) | | | $ (3,640) |
Tax provision (benefit) at federal statutory rate | | | (1,356) | | | (764) |
State tax (net of federal benefit) | | | (421) | | | (237) |
Stock Based Compensation | | | 197 | | | 216 |
Non-deductible items and other permanent differences | | | — | | | (60) |
Deferred Adjustments | | | — | | | — |
Valuation Allowance | | | 2,096 | | | 845 |
Research and development credit | | | (516) | | | — |
Total Income Tax Provision | | | $— | | | $— |
| | | Year Ended December 31, | ||||
| | | 2022 | | | 2021 | |
Deferred Tax Assets | | | | | ||
Net operating loss carryforwards | | | $4,711 | | | $5,012 |
Intangibles | | | 7 | | | 7 |
Operating lease right-of-use liabilities | | | 305 | | | 352 |
Stock based compensation | | | 45 | | | 44 |
Research and development expenses | | | 1,293 | | | — |
Charitable contribution carryforward | | | 10 | | | 41 |
Accrual to cash | | | 63 | | | —- |
Research and development credit carryforward | | | 784 | | | 268 |
Gross deferred tax assets | | | $7,218 | | | $5,724 |
Deferred Tax Liabilities | | | | | ||
Fixed Assets | | | $(54) | | | $(85) |
Accrual to cash | | | — | | | (513) |
Operating lease right-of-use assets | | | (271) | | | (329) |
Gross deferred tax liabilities | | | (325) | | | (927) |
Net deferred tax assets before valuation allowance | | | 6,894 | | | 4,798 |
Valuation allowance | | | (6,894) | | | (4,798) |
Net deferred tax assets | | | $— | | | $— |
| Item 13. | Other Expenses of Issuance and Distribution |
SEC registration fee | | | $15,958 |
Printing and engraving | | | 50,000 |
Legal fees and expenses | | | 370,000 |
Accounting fees and expenses | | | 60,000 |
Miscellaneous expenses | | | 54,042 |
Total | | | $550,000 |
| Item 14. | Indemnification of Officers and Directors |
| Item 15. | Recent Sales of Unregistered Securities |
| Item 16. | Exhibits and Financial Statement Schedules |
EXHIBIT NUMBER | | | DESCRIPTION OF EXHIBIT |
2.1*^ | | | Agreement and Plan of Merger, dated November 13, 2023, by and among Selecta Biosciences, Inc. Sakura Merger Sub I, Inc., Sakura Merger Sub II, LLC and Cartesian Therapeutics, Inc. |
3.1^ | | | Restated Certificate of Incorporation of Selecta Biosciences, Inc. |
3.2^ | | | Certificate of Amendment to the Restated Certificate of Incorporation of Selecta Biosciences, Inc., dated June 21, 2022 |
3.3^ | | | Certificate of Amendment to the Restated Certificate of Incorporation of Selecta Biosciences, Inc., dated November 13, 2023 |
3.4^ | | | Certificate of Amendment to the Restated Certificate of Incorporation, as amended, of Cartesian Therapeutics, Inc., dated March 28, 2024 |
3.5^ | | | Amended and Restated By-laws of Cartesian Therapeutics, Inc. |
3.6^ | | | Certificate of Designation of Preferences, Rights and Limitations of Series A Non-Voting Convertible Preferred Stock |
3.7^ | | | Certificate of Amendment to the Certificate of Designation of Series A Non-Voting Convertible Preferred Stock, dated March 26, 2024 |
3.8^ | | | Certificate of Designation of Preferences, Rights and Limitations of Series B Non-Voting Convertible Preferred Stock |
4.1^ | | | Form of Registration Rights Agreement |
4.2^ | | | Form of Specimen Certificate Representing Common Stock |
4.3^ | | | Form of Warrant to Purchase Shares of Series D Preferred Stock, dated August 9, 2013 or July 25, 2014, issued by the Registrant to Oxford Finance LLC and Square One Bank, together with a schedule of warrant holders |
4.4^ | | | Form of Warrant to Purchase Shares of Series E Preferred Stock, dated December 31, 2015, issued by the Registrant to Oxford Finance LLC and Square One Bank, together with a schedule of warrant holders |
4.5^ | | | Common Stock Purchase Warrant, dated June 27, 2017, by and between the Registrant and Timothy Springer, Ph.D. |
4.6^ | | | Registration Rights Agreement, dated December 23, 2019, by and among the Registrant and the Investors named therein |
EXHIBIT NUMBER | | | DESCRIPTION OF EXHIBIT |
4.7^ | | | Registration Rights Agreement, dated as of June 11, 2020, by and between the Registrant and Swedish Orphan Biovitrum AB (Publ) |
4.8^ | | | Registration Rights Agreement, dated as of June 11, 2020, by and between the Registrant and Swedish Orphan Biovitrum AB (Publ), as amended on November 4, 2020 |
4.9^ | | | Form of Common Stock Purchase Warrant, dated December 23, 2019 |
4.10^ | | | Form of Amendment No. 1 to Common Stock Purchase Warrant by and between Selecta Biosciences, Inc. and certain Directors, dated December 20, 2022 |
4.11^ | | | Form of Warrant to Purchase Stock, dated August 31, 2020, issued by Selecta Biosciences, Inc. to Oxford Finance LLC and Silicon Valley Bank, together with a schedule of warrants. |
4.12^ | | | Form of Common Stock Purchase Warrant, dated April 11, 2022 |
4.13^ | | | Form of Contingent Value Rights Agreement |
4.14^ | | | Registration Rights Agreement, by and among Selecta Biosciences, Inc. and certain purchasers party thereto, dated as of November 13, 2023 |
5.1^ | | | Opinion of Covington & Burling LLP |
10.1†^ | | | Amended and Restated 2016 Incentive Award Plan and form of award agreements thereunder |
10.2†^ | | | 2016 Employee Stock Purchase Plan |
10.3†^ | | | Amended and Restated Cartesian Therapeutics, Inc. 2018 Employment Inducement Incentive Award Plan, and forms of award agreements thereunder |
10.4†^ | | | 2008 Stock Incentive Plan and form of award agreements thereunder |
10.5†^ | | | Cartesian Therapeutics, Inc. 2016 Stock Incentive Plan, and forms of award agreements thereunder |
10.6†^ | | | Non-Employee Director Compensation Program |
10.7†^ | | | Form of Indemnification Agreement for Directors and Officers |
10.8#^ | | | Amended and Restated License Agreement, dated as of May 31, 2017, by and between the Registrant and Shenyang Sunshine Pharmaceutical Co., Ltd. |
10.9#^ | | | Manufacturing Services Agreement, dated as of August 1, 2014, by and between the Registrant and Shenyang Sunshine Pharmaceutical Co., Ltd. |
| | | Lease Agreement by and between BRE-BMR Grove LLC and Selecta Biosciences, Inc. dated July 23, 2019 | |
| | | First Amendment to Lease by and between BRE-BMR Grove LLC and Selecta Biosciences, Inc. dated September 1, 2022 | |
10.12#^ | | | Lease Agreement by and between 704 Quince Orchard Owner, LLC and Cartesian Therapeutics, Inc. dated May 11, 2018 |
10.13#^ | | | First Amendment to Lease Agreement by and between 704 Quince Orchard Owner, LLC and Cartesian Therapeutics, Inc. dated March 22, 2021 |
10.14#^ | | | Second Amendment to Lease Agreement by and between 704 Quince Orchard Owner, LLC and Cartesian Therapeutics, Inc. dated May 3, 2021 |
10.15†^ | | | Employment Agreement, dated as of September 25, 2018, by and between the Registrant and Carsten Brunn, Ph.D. |
10.16†^ | | | Employment Agreement, dated as of November 9, 2022, by and between the Registrant and Blaine Davis |
10.17#^ | | | License and Development Agreement, dated as of June 11, 2020, by and between the Registrant and Swedish Orphan Biovitrum AB (Publ) |
10.18#^ | | | Amendment No. 1 to License and Development Agreement, dated as of October 31, 2023, by and between the Registrant and Swedish Orphan Biovitrum AB (Publ) |
10.19#^ | | | Patent License Agreement, between Cartesian Therapeutics, Inc. and the U.S. Department of Health and Human Services, as represented by the National Cancer Institute of the National Institutes of Health, dated September 16, 2019 |
10.20#^ | | | Patent License Agreement by and between Biogen MA, Inc. and Cartesian Therapeutics, Inc., dated September 8, 2023 |
| | | Securities Purchase Agreement, dated June 26, 2017, by and between the Registrant and Timothy Springer, Ph.D. |
EXHIBIT NUMBER | | | DESCRIPTION OF EXHIBIT |
| | | Stock Purchase Agreement, dated August 19, 2019, by and among the Registrant and the Investors named therein | |
| | | Loan and Security Agreement, dated August 31, 2020, between Selecta Biosciences, Inc., Oxford Finance LLC, as Collateral Agent and as a lender, and Silicon Valley Bank, as a lender. | |
| | | First Amendment to Loan and Security Agreement, dated September 7, 2021, by and among Selecta Biosciences, Inc., Oxford Finance LLC, and Silicon Valley Bank | |
| | | Second Amendment to Loan and Security Agreement, dated March 21, 2022, between Selecta Biosciences, Inc., Oxford Finance LLC, as Collateral Agent and as a lender, and Silicon Valley Bank, as a lender | |
| | | Third Amendment to Loan and Security Agreement, dated September 20, 2022, between Selecta Biosciences, Inc., Oxford Finance LLC, as Collateral Agent and as a Lender, and Silicon Valley Bank, as a Lender | |
| | | Fourth Amendment to Loan and Security Agreement, dated March 31, 2023, between Selecta Biosciences, Inc., Oxford Finance LLC, as Collateral Agent and as a lender, and Silicon Valley Bank, a division of First-Citizens Bank & Trust Company (successor by purchase to the Federal Deposit Insurance Corporation as Receiver for Silicon Valley Bridge Bank, N.A. (as successor to Silicon Valley Bank)), as a lender | |
| | | License and Development Agreement, dated January 8, 2023, by and between Selecta Biosciences, Inc. and Audentes Therapeutics, Inc. | |
| | | Form of Retention Bonus Letter | |
| | | Securities Purchase Agreement, dated as of November 13, 2023, by and among Selecta Biosciences, Inc. and each purchaser identified on Annex A thereto | |
| | | Employment Agreement, dated as of March 26, 2024, by and between the Registrant and Christopher Jewell, Ph.D. | |
| | | Employment Agreement, dated as of March 28, 2024, by and between the Registrant and Metin Kurtoglu, M.D., Ph.D. | |
| | | Lease Agreement by and between 7495 RP, LLC and Cartesian Therapeutics, Inc. dated February 28, 2024 | |
| | | First Amendment to Lease Agreement by and between 7495 RP, LLC and Cartesian Therapeutics, Inc. dated May 7, 2024 | |
| | | Securities Purchase Agreement, dated as of July 2, 2024, by and between Cartesian Therapeutics, Inc. and each purchaser identified on Annex A thereto | |
21.1^ | | | Subsidiaries of the Registrant |
| | | Consent of Ernst & Young LLP | |
| | | Consent of BDO USA, P.C. | |
23.3^ | | | Consent of Covington & Burling LLP (filed as part of Exhibit 5.1) |
24.1^ | | | Power of Attorney (included on the signature page to the registration statement) |
101.INS | | | Inline XBRL Instance Document. |
101.SCH | | | Inline XBRL Taxonomy Extension Schema Document. |
101.CAL | | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
101.DEF | | | Inline XBRL Taxonomy Extension Definition Linkbase Document. |
101.LAB | | | Inline XBRL Taxonomy Extension Labels Linkbase Document. |
101.PRE | | | Inline XBRL Taxonomy Extension Presentation Linkbase Document. |
104 | | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
107^ | | | Filing Fee Table |
| † | Indicates a management contract or compensatory plan. |
| * | Certain annexes, schedules and exhibits have been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Registrant agrees to furnish supplementally a copy of any omitted attachment to the SEC on a confidential basis upon request. |
| # | Certain confidential information contained in this exhibit, marked by brackets and asterisks, has been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K because the information (i) is not material and (ii) is the type of information that the Registrant both customarily and actually treats as private and confidential. |
| ^ | Previously filed. |
| Item 17. | Undertakings |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | to include any prospectus required by Section 10(a)(3) of the Securities Act; |
| (ii) | to reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Filing Fee Tables” or “Calculation of Registration Fee” table, as applicable, in the effective registration statement; and |
| (iii) | to include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
| (2) | That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability under the Securities Act to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use. |
| (5) | Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers, and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer, or controlling person of the Registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. |
| | | CARTESIAN THERAPEUTICS, INC. | ||||
| | | By: | | | /s/ Carsten Brunn, Ph.D. | |
| | | | | Name: Carsten Brunn, Ph.D. President and Chief Executive Officer | ||
Signature | | | Title | | | Date |
| | | | | |||
/s/ Carsten Brunn, Ph.D. | | | President and Chief Executive Officer, Director (Principal Executive Officer) | | | September 9, 2024 |
Carsten Brunn, Ph.D. | | |||||
| | | | | |||
/s/ Blaine Davis | | | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) | | | September 9, 2024 |
Blaine Davis | | |||||
| | | | | |||
* | | | Chairman of the Board | | | September 9, 2024 |
Carrie S. Cox | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Timothy C. Barabe | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Nishan de Silva, M.D. | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Murat Kalayoglu, M.D., Ph.D. | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Kemal Malik, MBBS | | |||||
| | | | | |||
* | | | Director | | | September 9,2024 |
Michael Singer, M.D., Ph.D. | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Timothy Springer, Ph.D. | | |||||
| | | | | |||
* | | | Director | | | September 9, 2024 |
Patrick Zenner | |
* By: | | | /s/ Carsten Brunn, Ph.D. | | | |
| | | Carsten Brunn, Ph.D. | | | ||
| | | Attorney-in-Fact | | |