compared to the rate of inflation (based on the CPI-U published by the United States Department of Labor). This calculation is based on the baseline pricing data for the first full quarter of sales associated with a branded drug’s NDA, and baseline data cannot generally be reset, even on transfer of the NDA to another manufacturer. This “additional rebate” calculation can, in some cases where price increases have been relatively high versus the first quarter of sales of the NDA, result in Medicaid rebates up to 100% of a drug’s “average manufacturer price” and 340B prices of one penny. Separately, subject to the control of Directive 89/105/EEC, pricing and reimbursement in the EU/EEA (“European Economic Area”) is governed by national rules and policies and may vary from Member State to Member State.
Also, the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) outlines several federal crimes, including health care fraud and false statements relating to health care matters. Most healthcare providers who are expected to prescribe our products and from whom we obtain patient health information are subject to privacy and security requirements under HIPAA. Although we are not directly subject to HIPAA, we could be subject to criminal penalties if we knowingly obtain individually identifiable health information from a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
The privacy and protection of consumer information remains a developing area and we continue to monitor legislative and regulatory developments both in the United States as well as Europe. For example, the California Consumer Privacy Act (“CCPA”) became effective on January 1, 2020 and, as enacted, requires us to make new disclosures to consumers about our data collection, use, and sharing practices. It also provides a cause of action for data breaches. Beyond California, many other states are developing their own data privacy protections, which, along with the CCPA, could create liability for us or increase our cost of doing business. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information. For example, the General Data Protection Regulation (Regulation (EU) 2016/679), the U.K.’s Data Protection Act 2018 and the Swiss Federal Data Protection Act and Data Protection Ordinance, regulate the processing of personal data within the U.K., the EU and between countries in the EU, U.K. and countries outside of the EU and U.K., including the U.S. Failure to provide adequate privacy protections and maintain compliance with the EU, U.K. and Swiss Privacy Laws, could jeopardize business transactions across borders and result in significant penalties. Similar to the impact of the CCPA or other U.S. state frameworks, these European laws could create liability for us or increase our cost of doing business.
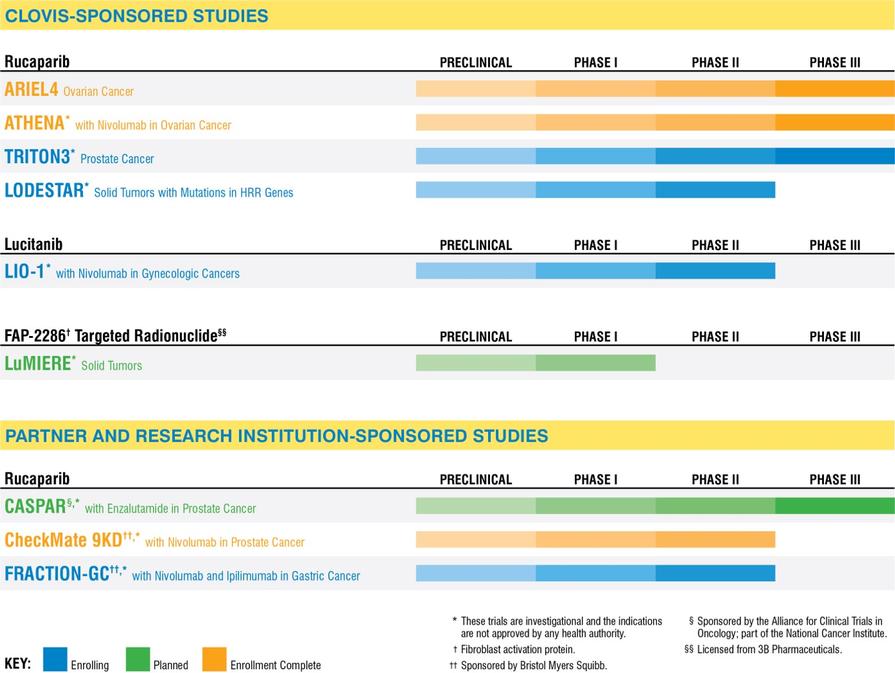
Regulation of Diagnostic Tests
In the United States, the FDCA and its implementing regulations, and other federal and state statutes and regulations govern, among other things, medical device design and development, non-clinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. Diagnostic tests are classified as medical devices under the FDCA. Unless an exemption applies, diagnostic tests require marketing clearance or approval from the FDA prior to commercial distribution, depending on their classification by FDA.
In the United States, devices are classified into one of three classes (Class I, II, or III) based on the controls deemed necessary by the FDA to reasonably ensure their safety and effectiveness. Class I and II devices are subject to general controls including, but not limited to, performance standards, premarket notification, also called 510(k) clearance, and post market surveillance. Class III devices are those that either support or sustains human life, are of substantial importance in preventing impairment of human health, or present a potential, unreasonable risk of illness or injury. Class III devices are subject to more rigorous review and approval requirements than Class I or II, known as a premarket approval, or PMA approval. Because the diagnostic tests being developed by our third-party collaborators are of substantial importance in preventing impairment of human health, they are considered Class III devices, subject to the PMA approval process.
PMA applications must be supported by valid scientific evidence, which typically requires extensive data, including technical, non-clinical, clinical and manufacturing data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. For diagnostic tests, a PMA application typically includes data regarding analytical and clinical validation studies. As part of its review of the PMA, the FDA will conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with the Quality System Regulation, or QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures. FDA review of an initial PMA application is required by statute to take between six to ten months, although the process typically takes longer, and may require several years to complete. If the FDA evaluations of both the PMA application and the