| ITEM 2. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited interim condensed consolidated financial statements and related notes thereto appearing elsewhere in this Quarterly Report on Form 10-Q (this “Quarterly Report”) and with the audited financial statements and notes thereto of the Company as of and for the year ended December 31, 2022 on Form 10-K, filed with the Securities and Exchange Commission, or SEC, on March 28, 2023.
Cautionary Note Regarding Forward-Looking Statements
This Quarterly Report contains forward-looking statements (including within the meaning of Section 21E of the United States Securities Exchange Act of 1934, as amended, and Section 27A of the United States Securities Act of 1933, as amended) concerning the Company and other matters. These statements may discuss goals, intentions and expectations as to future plans, trends, events, results of operations or financial condition, or otherwise, based on current beliefs of the Company’s management, as well as assumptions made by, and information currently available to, management. Forward-looking statements generally include statements that are predictive in nature and depend upon or refer to future events or conditions, and include words such as “may,” “will,” “should,” “would,” “expect,” “anticipate,” “plan,” “likely,” “believe,” “estimate,” “project,” “intend,” “forecast,” “guidance”, “outlook” and other similar expressions among others. Forward-looking statements are based on current beliefs and assumptions that are subject to risks and uncertainties and are not guarantees of future performance. Actual results could differ materially from those contained in any forward-looking statement as a result of various factors, including, without limitation:
| ● | the Company’s ability to protect its intellectual property rights; |
| ● | the Company’s anticipated capital requirements, including the Company’s anticipated cash runway and the Company’s current expectations regarding its plans for future equity financings; |
| ● | the Company’s dependence on additional financing to fund its operations and complete the development and commercialization of its clinical candidates, and the risks that raising such additional capital may restrict the Company’s operations or require the Company to relinquish rights to the Company’s technologies or clinical candidates; |
| ● | the Company’s limited operating history in the Company’s current line of business, which makes it difficult to evaluate the Company’s prospects, the Company’s business plan or the likelihood of the Company’s successful implementation of such business plan; |
| ● | the timing for the Company or its partners to initiate the planned clinical trials for PDS0101, PDS0103, PDS0203 and other Versamune and Infectimune based clinical candidates and the future success of such trials; |
| ● | the successful implementation of the Company’s research and development programs and collaborations, including any collaboration trials concerning the Company’s Versamune and Infectimune based clinical candidates and the Company’s interpretation of the results and findings of such programs and collaborations and whether such results are sufficient to support the future success of the Company’s clinical candidates; |
| ● | the success, timing and cost of the Company’s ongoing clinical trials and anticipated clinical trials for the Company’s current clinical candidates, including statements regarding the timing of initiation, pace of enrollment and completion of the trials (including our ability to fully fund our disclosed clinical trials, which assumes no material changes to our currently projected expenses), futility analyses, presentations at conferences and data reported in an abstract, and receipt of interim results (including, without limitation, any preclinical results or data), which are not necessarily indicative of the final results of the Company’s ongoing clinical trials; |
| ● | expectations for the clinical and preclinical development, manufacturing, regulatory approval, and commercialization of our clinical candidates; |
| ● | any Company statements about its understanding of clinical candidates’ mechanisms of action and interpretation of preclinical and early clinical results from its clinical development programs and any collaboration trials; the acceptance by the market of the Company’s clinical candidates, if approved; |
| ● | the timing of and the Company’s ability to obtain and maintain U.S. Food and Drug Administration or other regulatory authority approval of, or other action with respect to, the Company’s clinical candidates; and |
| ● | other factors, including legislative, regulatory, political and economic developments not within the Company’s control, including unforeseen circumstances or other disruptions to normal business operations arising from or related to those listed under Part II, Item 1A. Risk Factors. |
Any forward-looking statements in this Quarterly Report reflect our current views with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, whether as a result of new information, future events or otherwise.
In this Quarterly Report, unless otherwise stated or the context otherwise indicates, references to “PDS Biotech,” “the Company,” “we,” “us,” “our” and similar references refer to PDS Biotechnology Corporation, a Delaware corporation.
Company Overview
We are a clinical-stage immunotherapy company developing a growing pipeline of targeted cancer and infectious disease immunotherapies based on our Versamune®, Versamune® plus IL12 fused anti-body drug conjugate (ADC) PDS01ADC (formerly PDS0301/M9241) and Infectimune® T cell-activating platforms and PDS01ADC tumor targeting immunocytokine. We believe our targeted immunotherapies have the potential to overcome the limitations of current immunotherapy approaches through the activation of the right type, quantity and potency of T cells. Versamune, and Versamune plus PDS01ADC are utilized for treatments in oncology and Infectimune, for treatments in infectious disease. When paired with an antigen, which is a disease-related protein that is recognizable by the immune system, Versamune and Infectimune have both been shown to induce, in vivo, large quantities of high-quality, highly potent polyfunctional CD4 helper and CD8 killer T cells, a specific sub-type of T cell that is more effective at killing infected or target cells. Infectimune is also designed to promote the induction of disease-specific neutralizing antibodies. PDS01ADC is an investigational tumor targeting IL-12 that enhances the proliferation, potency and longevity of T cells in the tumor microenvironment. Versamune plus PDS01ADC enhances the proliferation, potency and longevity of antigen specific multifunctional CD8 T cells in the tumor microenvironment and works synergistically to overcome tumor immune suppression.
In December 2022, we executed an exclusive global license agreement with Merck KGaA, Darmstadt, Germany for the tumor targeting IL12 fused antibody drug conjugate, M9241, which joined our pipeline as PDS01ADC. PDS01ADC is a novel investigational tumor-targeting fusion protein of Interleukin 12 that enhances the proliferation, potency, infiltration and longevity of T cells in the tumor microenvironment and is therefore designed to overcome the limitations of cytokine therapy which today have resulted in high toxicity and limited therapeutic potential. The proprietary combination of Versamune plus PDS01ADC is designed to overcome tumor immune suppression utilizing a different mechanism from immune checkpoint inhibitors (ICI). The ownership of both assets we believe will streamline the registrational process and its use. The combination of Versamune® and IL-12 to overcome immune suppression is patented by PDS Biotech. In a Phase 2 National Cancer Institute (NCI)-led clinical trial in ICI resistant patients, the combination of PDS0101 and PDS01ADC administered with an investigational bi-functional ICI resulted in a median overall survival of approximately 20 months, which compares favorably to the historical median survival of 3-4 months reported in ICI resistant HPV-positive cancers when treated with ICIs and best reported median survival to date with systemic therapy of 8.2 months in ICI resistant head and neck cancer.
In February 2023, we announced a successful completion of a Type B meeting with the FDA for the triple combination of PDS0101 and PDS01ADC with an FDA-approved immune checkpoint inhibitor for the treatment of recurrent/metastatic, ICI resistant head and neck cancer that is positive for the human papilloma virus (HPV) type 16. In recent interactions with the FDA, we confirmed the required contents of a clinical protocol for the potential registrational trial.
In June 2023, an abstract was presented at the 2023 American Society of Clinical Oncology: Abstract number 6012, Safety and Efficacy of Immune Checkpoint Inhibitor (ICI) Naïve Cohort from Study of PDS0101 and Pembrolizumab in HPV16-Positive Head and Neck Squamous Cell Carcinoma (HNSCC). The abstract was also selected as one of the featured posters reviewed by an expert panel in the Head and Neck Cancer discussion session.
In September 2023, data on our investigational universal flu vaccine, PDS0202, were presented at the 9th European Scientific Working Group on Influenza (ESWI) conference. These data demonstrated broad neutralization across multiple influenza strains and provided protection against infection after challenging animals not previously exposed to flu with lethal doses of the pandemic H1N1 flu virus.
In October 2023, data demonstrating PDS0101 in combination with standard-of-care (SOC) chemoradiotherapy was associated with a rapid decline in human papillomavirus circulating cell-free DNA (ctHPV-DNA), a potential predictive biomarker of treatment response. The data from the IMMUNOCERV Phase 2 clinical trial were featured in an oral presentation at the American Society for Radiation Oncology Annual Meeting.
In October 2023, updated interim data based on an August 2nd cut off from our VERSATILE-002 Phase 2 clinical trial evaluating the combination of PDS0101 in combination with Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab) which is the FDA-approved standard of care for first-line treatment of recurrent/ metastatic head and neck cancer was presented. The data was presented at a Company-sponsored key opinion leader roundtable.
In October 2023, interim safety and immune response data was presented for the first-in-human Phase1/2 clinical trial evaluating PDS01ADC in combination with current SOC chemotherapy, docetaxel, to treat metastatic castration sensitive and castration resistant prostate cancer. The data was featured in an oral presentation at the 11th Annual Meeting of the International Cytokine & Interferon Society.
In October 2023, immune response data from a preliminary analysis of a subset if patients in our VERSATILE-002 Phase 2 clinical trial was presented at the European Society for Medical Oncology Congress 2023.
In November 2023, we announced updated interim survival data from our NCI-led Phase 2 triple combination study.
In November 2023, preclinical data from our NCI-led trial including PDS0101, PDS01ADC and an HDAC inhibitor in ICI-resistant HPV-16 positive cancer was presented during a poster presentation at the Society for Immunotherapy of Cancer 38th Annual Meeting.
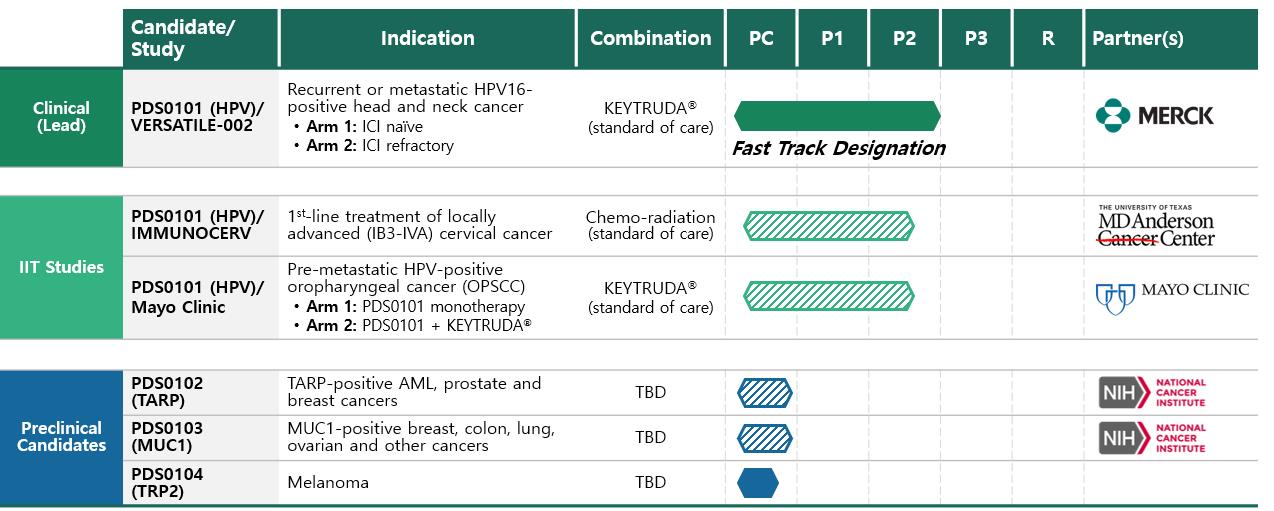
Clinical Candidate Pipeline
VERSATILE-002: PDS0101 + KEYTRUDA®
In November 2020, our VERSATILE-002 Phase 2 clinical trial evaluating the combination of PDS0101 in combination with Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab) which is the FDA-approved standard of care for first-line treatment of recurrent/ metastatic head and neck cancer commenced. Enrollment in stage 2 of 2 for the ICI naïve arm and the ICI resistant arms are complete. The clinical trial will evaluate the efficacy and safety of this therapeutic combination as a first and second line treatment in patients with recurrent or metastatic head and neck cancer and high-risk human papillomavirus-16 (HPV16) infection.
In this PDS Biotech-sponsored trial patients whose cancer has returned following initial treatment or spread will be treated with the combination of PDS0101 and KEYTRUDA® to evaluate if the addition of PDS0101 might improve the efficacy reported in published studies of KEYTRUDA® alone. Patients in the trial will receive a total of 5 cycles of combination therapy in the context of standard of care KEYTRUDA® therapy administered every three weeks until disease progression. The primary endpoint of VERSATILE-002 is the objective response rate– or ORR – at six months following initiation of treatment. There are two cohorts in the trial. Cohort 1 is for patients who have yet to be treated with an immune checkpoint inhibitor (ICI naïve) and cohort 2 which consists of patients who have failed immune checkpoint inhibitor therapy (ICI resistant).
In February 2022, we achieved the preliminary efficacy milestone of at least four or more objective responses of the first 17 patients in the ICI naïve arm that allowed that arm to proceed to full enrollment. We also announced detailed preliminary safety data which showed that the combination is well tolerated without evidence of enhanced or significant toxicity in the first 18 patients in the ICI naïve arm. We have completed enrollment in Stage 1 of the ICI resistant arm and we are waiting for sufficient follow up to conduct the futility analysis.
In June 2022, we presented additional preliminary efficacy and safety data from this trial at the ASCO Annual Meeting (Weiss J et al. J Clin Oncol 40, 2022 (suppl 16; abstr 6041)). The abstract provided preliminary data on 19 patients (safety) with available imaging data for 17 of the 19 patients (efficacy). Data on 17 patients was presented. Highlights from the abstract were as follows:
| ● | Confirmed and unconfirmed response rates thus far (tumor shrinkage greater than 30%) seen in 7/17 (41.2%) patients in comparison to the published results of approximately 19% for approved ICIs, used as monotherapy for recurrent or metastatic head and neck cancer, with 2 of the 7 having complete responses (CR) |
| ● | Stable disease (SD) was reported in 6/17 (35.3%) patients, with 4 of the 6 (67%) experiencing tumor shrinkage of less than 30% |
| ● | Clinical efficacy (ORR + SD) was seen in 13/17 (76.5%) patients |
| ● | Progressive/ongoing disease was reported in 4/17 (23.5%) patients
|
| ● | Patients had received a median of 4/5 doses of PDS0101 (range 1-5) and 9/35 doses of KEYTRUDA® (range 1-18) |
| ● | There were no treatment-related adverse events greater than or equal to Grade 3 (N=19) |
| ● | No patients required dose interruption or reduction on the combination treatment |
| ● | No patients discontinued the combination treatment |
| ● | At 9 months of follow up (median not yet achieved): |
| ● | Progression free survival (PFS) rate was 55.2% |
| ● | Overall survival (OS) rate was 87.2% |
| ● | No control or comparative studies have been conducted between ICIs and PDS0101 |
In May 2022, we expanded this trial into Europe and in June 2022, as described above, we received Fast Track designation from the US FDA for PDS0101 in combination with pembrolizumab.
In August 2022, our independent Data Monitoring Committee (DMC) met and evaluated data from 43 patients and noted there were no Grade 3 or greater treatment-related adverse events attributed to the combination. The DMC recommended continuing the trial with no modifications.
In October 2022, we announced the results of an end-of-phase 2 meeting with the FDA for PDS0101 in combination with KEYTRUDA®. We have completed the plan for the Phase 3 clinical program that will support the submission of a BLA for PDS0101 and have submitted to the FDA.
In December 2022, we completed the first stage of enrollment in the ICI resistant arm. Cohort follow-up is in progress that will permit a futility analysis to determine progression to stage 2 enrollment is in progress.
In May 2023, we completed enrollment in the ICI naïve arm. We filed our amended IND with the FDA in the third quarter of 2023. In October 2023, we received feedback from the FDA on the amended IND. Later in October, as part of business development discussions, we received insights from a potential business partners on the Phase 3 clinical protocol. The clinical and medical teams are currently evaluating this feedback and therefore plan to initiate a Phase 3 trial, VERSATILE-003 in the first quarter of 2024.
In June 2023, an abstract was presented at the 2023 American Society of Clinical Oncology: Abstract number 6012, Safety and Efficacy of Immune Checkpoint Inhibitor (ICI) Naïve Cohort from Study of PDS0101 and Pembrolizumab in HPV16-Positive Head and Neck Squamous Cell Carcinoma (HNSCC). The abstract was also selected as one of the featured posters to be reviewed by an expert panel in the Head and Neck Cancer discussion session. Data on 34 patients was presented. The data from the abstract is as follows:
| ● | Estimated 12-month overall survival rate was 87.1%. Published results are 36-50% with approved ICIs used alone. |
| ● | Median progression-free survival was 10.4 months (95% CI 4.2, 15.3). Published results are median PFS of 2-3 months for approved ICIs when used as monotherapy in patients with similar PD-L1 levels. |
| ● | A disease control rate (disease stabilization or tumor shrinkage) of 70.6% (24/34) |
| ● | Confirmed and unconfirmed objective response rate is 41.2% (14/34 patients), which is identical to the preliminary response rate data PDS Biotech previously reported at ASCO 2022 (7/17 patients). To date these responses have been confirmed in nine of the 34 patients (26.5%), including one complete response. |
| ● | 15/34 patients (44.1%) had stable disease. |
| ● | 9/34 patients (26.5%) had progressive disease. |
| ● | 4/48 (8.3%) of patients had a Grade 3 treatment-related adverse event (TRAE). No Grade 4 or higher TRAEs were observed. |
In October 2023, at a key opinion roundtable updated interim data was presented based on an August 2nd cut off from our VERSATILE-002 Phase 2 clinical trial evaluating the combination of PDS0101 in combination with Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab) which is the FDA-approved standard of care for first-line treatment of recurrent/ metastatic head and neck cancer. Data on 52 patients was presented. The data from the roundtable based on investigator assessment were as follows:
Highlights from the ICI naïve cohort include:
| ● | 24-month overall survival (OS) rate is 74%; published 24-month survival rate of less than 30% for approved ICI. |
| ● | 12-month OS rate is 80%; published results of 30-50% with approved ICIs1. |
| ● | Tumor shrinkage seen in 60% (31/52) of patients. |
| ● | Confirmed overall response rate (ORR) is 27% (14/52) to date. |
| ● | Median progression-free survival (PFS) is 8.1 months to date; published results of 2-3 months PFS with approved ICIs. |
| ● | 13% (8/62) of patients experienced Grade 3 treatment-related adverse events (TRAE) and 0% (0/62) experienced Grade 4 or 5 TRAE; published results report 13-17% Grade 3-5 TRAE with approved ICI monotherapy. |
| ● | 60% (33/55) of patients have CPS score of 1-19 (who generally have a weaker response to KEYTRUDA®), and 40% (22/55) have CPS score >20 (who generally have a higher response to KEYTRUDA®). |
Highlights form the ICI refractory cohort include:
| ● | The 12-month OS rate is 56%. The published median 12-month OS rate is 17% with no salvage chemotherapy following tumor progression on ICI (ICI Resistant). |
| ● | 0% (0/21) confirmed ORR suggests that PDS0101’s impact on survival does not appear to be dependent on tumor shrinkage. |
| ● | 4% (1/25) of patients experienced Grade 3 TRAE and 0% (0/21) patients experienced Grade 4 and 5 TRAE. |
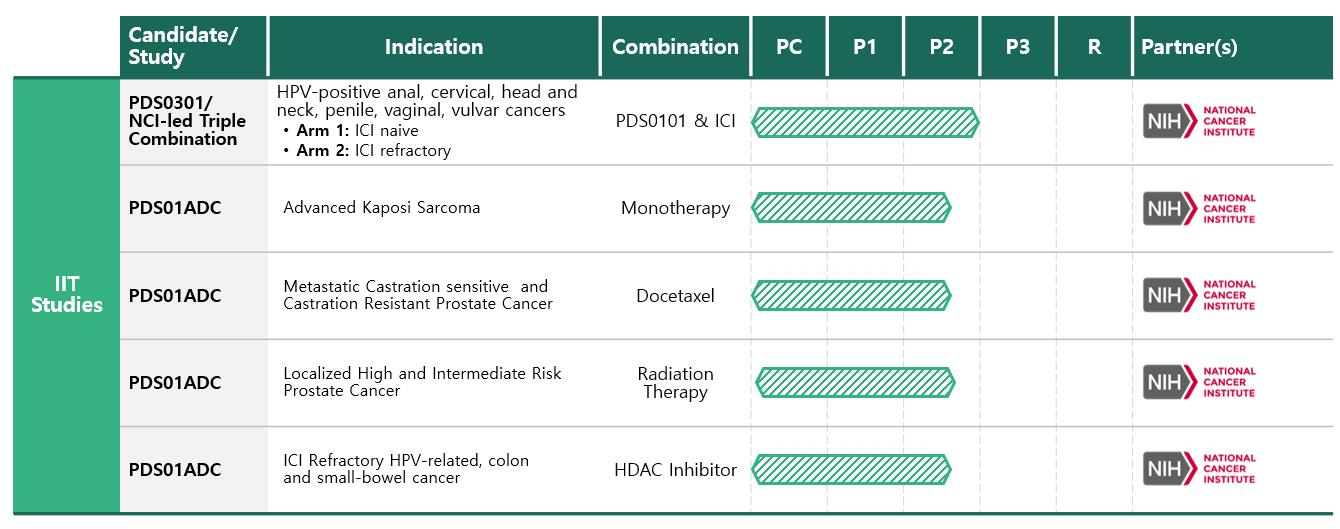
National Cancer Institute: PDS0101+ M9241 (now PDS01ADC) +Bintrafusp Alfa
In June 2020, the first patient was dosed under a PDS0101 Cooperative Research and Development Agreement (CRADA), in the NCI led Phase 2 investigator-initiated trial evaluating PDS0101 with an IL-12 ADC now PDS01ADC, and M7824 (Bintrafusp alfa), which is owned by EMD Serono (Merck KGaA) in patients with advanced HPV-positive cancers who have failed prior treatment. In February 2021, the NCI’s Phase 2 clinical trial of PDS0101 for the treatment of advanced HPV-positive cancers had achieved its preliminary objective response target in patients naïve to check point inhibitors which allowed for full enrollment of approximately 20 patients in this group. In addition, based on promising results in the ICI naïve arm, the trial was amended to allow enrollment of a separate cohort of IC -resistant patients for assessment of safety and activity of the triple combination. The trial has been closed for enrollment. Preliminary efficacy assessment of the triple combination in this added group of 29 ICI resistant patients has been completed and evaluation of long-term patient survival ongoing.
Preclinical study results arising from this CRADA were published in the Journal for ImmunoTherapy of Cancer, Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine (Journal for ImmunoTherapy of Cancer2020;8:e000612. Doi:10.1136/ jitc-2020-000612), and indicate that PDS0101 generated both HPV-specific T cells and an associated antitumor response when used as a monotherapy. When PDS0101 was combined with the two other novel clinical-stage anti-cancer agents, Bintrafusp Alfa and M9241 (which is now owned by us and referred to as PDS01ADC), the preclinical data suggested that all three therapeutic agents worked synergistically to provide superior tumor T cell responses and subsequent tumor regression when compared to any of the agents alone or the 2-component combinations. The published preclinical data demonstrating powerful activity of the triple combination appears to be corroborated in the Phase 2 trial, and this triple combination could form the basis of a unique platform providing improved cancer treatments across multiple cancers.
In June 2022, at the 2022 ASCO Annual Meeting, the NCI provided an update to the preliminary data presented at the 2021 meeting (Strauss J et al. J Clin Oncol 40, 2022 [suppl 16; abstr 2518]). This included data from 30 HPV16-positive patients and highlights were as follows:
| ● | Objective response (OR = >30% tumor reduction) was seen in 88% (7/8) of patients with ICI naive disease; 4/7 (57%) patients’ responses are ongoing (median 17 months). |
| ● | With ICI resistant patients: PDS0301 dosing appears to affect response rates, with 5/8 (63%) patients receiving PDS0301 at 16.8 mcg/kg achieving an OR compared to 1/14 (7%) patients who received PDS0301 at 8 mcg/kg achieving an OR; 4/6 (67%) patients’ responses are ongoing (median 12 months). |
| ● | Tumor reduction was seen in 45% (10/22) of patients with ICI resistant disease, including patients receiving high or low dose PDS0301. |
| ● | In ICI resistant patients treated with high or low dose PDS0301, survival outcomes were similar (p=0.96 by Kaplan Meier analysis). At a median of 12 months of follow up 17/22 (77%) of patients were alive. |
| ● | In ICI naïve patients 6/8 (75%) were alive at median 17 months of follow up. |
| ● | Similar OR and survival were seen across all types of HPV16-positive cancers. |
| ● | Preliminary safety data: 13/30 (43%) of patients experienced Grade 3 treatment-related adverse events (AEs), and 2/30 patients (7%) experienced Grade 4 AEs. There were no grade 5 treatment-related AEs. |
We believe the study results to date strongly suggest, in agreement with the published preclinical studies, that all 3 drugs contribute to the clinical outcomes.
In September 2022, we determined, in agreement with the NCI, to select the ICI resistant patients as the preferred treatment group in the on-going PDS0101-based triple combination therapy in advanced HPV-positive cancers and the trial was closed to further enrollment given the ICI resistant arm had been fully recruited.
In October 2022, we presented additional interim data as follows:
| ● | Survival data: 66% (19/29) of HPV16-positive ICI resistant patients in the cohort were alive at a median follow up of 16 months. |
| ● | Safety profile: 48% (24/50) patients experienced Grade 3 treatment-related adverse events (AEs), and 4% (2/50) patients experienced Grade 4 AEs. There were no Grade 3 treatment-related AEs. |
| ● | HPV16-positive ICI naïve patients: 75% (6/8) were alive at a median follow up of 25 months and 38% (3/8) of responders had a complete response. |
In December 2022, we presented interim data as follows:
| ● | Median OS was 21 months in 29 checkpoint inhibitor resistant patients who received the triple combination. The reported historical median OS in patients with ICI resistant disease is 3-4 months seen with checkpoint inhibitors and best reported median survival to date with systemic therapy of 8.2 months in ICI resistant head and neck cancer. |
| ● | In ICI naïve subjects, 75% remain alive at a median follow-up of 27 months. As a result, median OS had not yet been reached. Historically median OS for similar patients with platinum experienced ICI naïve disease is 7-11 months. |
| ● | Objective response rate (ORR) in ICI resistant patients who received the optimal dose of the triple combination is 63% (5/8). In current approaches ORR is reported to be less than 10%. |
| ● | ORR in ICI naïve patients with the triple combination is 88%. In current approaches ORR is reported to be less than 25% with FDA-approved ICIs in HPV-positive cancers. |
| ● | Safety data had not changed since October’s update. 48% (24/50) of patients experienced Grade 3 (moderate) treatment-related adverse events (AEs), and 4% (2/50) patients experienced Grade 4 (severe) AEs, compared with approximately 70% of patients receiving the combination of ICIs and chemotherapy reporting Grade 3 and higher treatment-related AEs. |
In February 2023, we announced the successful completion of a Type B meeting with the FDA for the combination therapy of PDS0101, PDS01ADC, and an FDA-approved immune checkpoint inhibitor for the treatment of recurrent/metastatic HPV-positive ICI-resistant head and neck cancer. We confirmed the required contents of the trial design for a potential registrational trial of the combination.
In November 2023, we released updated interim survival data as follows:
| ● | 75% of immune checkpoint inhibitor (ICI) naïve patients remain alive at 36 months; published median overall survival (OS) in similar patients is 7-11 months |
| ● | 12-month survival rate in (ICI) resistant patients is 72% |
| ● | Median OS in ICI resistant HPV-positive patients is approximately 20 months; published median OS is 3.4 months |
MD Anderson Cancer Center (IMMUNOCERV): PDS0101+ Chemoradiotherapy
In October 2020, another PDS0101 Phase 2 IIT was initiated with The University of Texas MD Anderson Cancer Center and is actively recruiting patients. This clinical trial is investigating the safety and anti-tumor efficacy of PDS0101 in combination with standard-of-care chemo-radiotherapy, or CRT, and their correlation with critical immunological biomarkers in patients with locally advanced cervical cancer. We believe that Versamune has strong T cell induction with the potential to enhance efficacy of the current standard of care CRT treatment in this indication with the FDA at this meeting.
In November 2022, data from this trial was included in a poster presentation at the 2022 SITC Annual Meeting which included the following:
| ● | 9 of the 17 patients had completed a Day 170 post-treatment Positron Emission Tomography, Computed Tomography (PET CT) scan to assess the status of the cancer. This included 78% (7/9) of treated patients with advanced cervical cancer (FIGO stage III or IV). |
| ● | 100% (9/9) of patients treated with the combination of PDS0101 and CRT had an objective response. |
| ● | 89% (8/9) of patients treated with the combination of PDS0101 and CRT demonstrated a complete response (CR) on Day 170 by PET CT. One patient who received 3 of the 5 scheduled doses of PDS0101 showed signs of residual disease. One patient who had a CR died from an event unrelated to either their underlying disease or treatment. |
| ● | 1-year disease-free survival and 1-year overall survival of 89% (8/9) in patients treated with the combination of PDS0101 and CRT. |
| ● | As previously reported, data confirm PDS0101 treatment activates HPV16-specific CD8 T cells. This increase was not seen in patients who did not receive PDS0101. The increase in HPV16-specific T cells generated by the treatment is positively correlated with tumor cell death, suggesting cytotoxic CD8 T cells are important mediators of antigen-specific immunity. |
| ● | The data affirm that PDS0101 activates Type 1 interferon pathway in humans, mimicking the mechanism previously demonstrated in preclinical studies in animal models. |
| ● | Toxicity of PDS0101 remains limited to low-grade local injection site reactions. |
In October 2023, data demonstrating PDS0101 in combination with standard-of-care (SOC) chemoradiotherapy was associated with a rapid decline in human papillomavirus circulating cell-free DNA (ctHPV-DNA), a potential predictive biomarker of treatment response. The data from the IMMUNOCERV Phase 2 clinical trial were featured in an oral presentation at the American Society for Radiation Oncology Annual Meeting which included the following:
| ● | Earlier and greater proportion of ctDNA clearance with PDS0101 plus chemoradiation (CRT) vs. SOC CRT alone (81.3% clearance after 3 weeks vs. 30.3% with SOC (p=0.0018), and 91.7% of clearance at 5 weeks vs. 53.1% with SOC (p=0.0179). |
| ● | Baseline ctDNA levels correlated with the International Federation of Gynecology and Obstetrics (FIGO) stage and lymph node involvement; 100% of patients treated with PDS0101 had cancer that had spread to the lymph nodes. |
Mayo Clinic: PDS0101 Monotherapy and in combination with KEYTRUDA®
In February 2022, we initiated an Investigator-Initiated Trial (ITT), MC200710, for PDS0101 alone or in combination with the immune checkpoint inhibitor, KEYTRUDA®, in patients with HPV-positive oropharyngeal cancer (HPV(+)OPSCC) at high risk of recurrence. The trial is being led by Drs. David Routman, Katharine Price, Kathryn Van Abel, and Ashish Chintakuntlawar at Mayo Clinic, a nationally and internationally recognized center of excellence for the treatment of head and neck cancers. We believe that this trial not only broadens our addressable patient population of those affected by the increasing incidence of HPV(+)OPSCC, but also allows us to better understand the activity of PDS0101 alone or in combination with KEYTRUDA® in earlier stages of disease. This trial is currently open for enrollment.
In this trial, treatment will be administered before patients proceed to transoral robotic surgery (TORS) with curative intent. Treatment in this setting is referred to as neoadjuvant treatment. PDS0101 has been shown to induce killer T cells that target and kill HPV-positive cancers, either alone or in combination with ICIs in preclinical studies, and in combination in clinical studies of patients with advanced recurrent/metastatic HPV-positive cancers. This trial will explore whether PDS0101 with or without checkpoint inhibition may increase HPV-specific anti-tumor responses, potentially resulting in tumor shrinkage, pathologic regression, and decreases in circulating tumor DNA (ctDNA).
PDS0102
PDS0102 is an investigational immunotherapy utilizing tumor-associated and immunologically active T cell receptor gamma alternate reading framed protein (TARP) from the NCI. PDS0102 is designed to treat TARP-associated cancers including, acute myeloid leukemia (AML), prostate and breast cancer. In our preclinical work, in the administration of PDS0102, the Versamuine+TARP antigen combination led to the induction of large numbers of tumor targeted killer T cells. In addition, the TARP tumor antigen alone has already been studied at the NCI in men with prostate cancer and been shown to be safe, immunogenic with slowing tumor growth rates (NCT00972309). We are evaluating the next steps in the clinical development of PDS0102 and are seeking nondilutive financings to move the program into clinical trials.
In April 2020, the above mentioned PDS Biotech-NCI CRADA was expanded beyond PDS0101 to include clinical and preclinical development of PDS0103. PDS0103 is an investigational immune therapy owned by PDS Biotech and designed to treat cancers associated with the mucin-1, or MUC1, oncogenic protein. These include cancers such as ovarian, breast, colorectal and lung cancers. PDS0103 combines Versamune with novel highly immunogenic agonist epitopes of MUC1 developed by the NCI and licensed by PDS. PDS0103 is currently in the tech transfer and clinical scale up and manufacturing stage.
MUC1 is highly expressed in several types of cancer and has been shown to be associated with drug resistance and poor disease prognosis in breast, colorectal, lung and ovarian cancers, for which PDS0103 is being developed. Expression of MUC1 is often associated with poor disease prognosis, due in part to drug resistance. In preclinical studies, and similarly to PDS0101, PDS0103 demonstrated the ability to generate powerful MUC1-specific CD8 killer T cells. In the first quarter of 2022, we held a pre-IND meeting with the FDA on PDS0103 and we are prepared to submit our IND package by the first half of 2024. However, the actual submission date may potentially be impacted by the allocation of resources to initiate the pivotal trial for PDS0101. Our primary goal is commercialization of PDS0101, and allocation of resources to implement an earlier than planned start of a registrational trial may delay PDS0103 initiation.
Our current pipeline of Versamune based therapies is as follows:
IL-12 Oncology Immunocytokine Pipeline
PDS01ADC (formerly known as PDS0301/M9241 and NHS-IL-12 ) is a novel investigational IL12 fused antibody drug conjugate (IgG1), tumor-targeting interleukin 12 (IL-12) immune-cytokine that enhances the proliferation, potency and longevity of T cells in the tumor microenvironment. Together with Versamune based immunotherapies PDS01ADC works synergistically to overcome tumor immune suppression and to promote a targeted T cell attack against cancers. As with Versamune, PDS01ADC is given by a simple subcutaneous injection. Clinical data suggests the addition of PDS01ADC to Versamune based immunotherapies may demonstrate significant disease control in advanced cancer patients by shrinking tumors and/or prolonging life.
With the exclusive global license agreement with Merck KGaA, Darmstadt, Germany for PDS01ADC, we believe we have simplified our registrational pathway for the NCI-led triple combination by owning both PDS0101 and PDS01ADC and combining these agents with an FDA approved ICI. PDS01ADC has been designed to overcome the limitations of cytokine therapy as explained above, and based on extensive preclinical studies performed at the NCI evaluating PDS01ADC as a monotherapy and also in combinations with established standard of care treatments for cancer, we believe that PDS01ADC has significant potential as a cytokine therapy independent of Versamune. Based on the informative preclinical studies, a number of ITT Phase 2 trials are currently in progress at the NCI, some of which are outlined below:
| ● | Phase II Study Evaluating ICI Naïve and Resistant Patients with HPV-positive malignancies treated with PDS01ADC, PDS0101 and bintrafusp alfa. |
| ● | A Phase II Study Evaluating T-Cell Clonality After Stereotactic Body Radiation Therapy Alone and in Combination with the Immunocytokine PDS01ADCin Localized High and Intermediate Risk Prostate Cancer Treated with Androgen Deprivation Therapy |
| ● | A Phase I/II Study of PDS01ADC in Combination with Docetaxel in Adults with Metastatic Castration Sensitive and Castration Resistant Prostate Cancer |
| ● | Phase I/II of PDS01ADC going forward as a Monotherapy in Advanced Kaposi Sarcoma |
| ● | Phase I/II of PDS01ADC in Combination of with a Histone Deacetylase (HDAC) Inhibitor in ICI resistant MUC1-positive colon and bladder cancers among others |
In October 2023, interim safety and immune response data was presented for the first-in-human Phase1/2 clinical trial evaluating PDS01ADC in combination with current SOC chemotherapy, docetaxel, to treat metastatic castration sensitive and castration resistant prostate cancer. The data was featured in an oral presentation at the 11th Annual Meeting of the International Cytokine & Interferon Society. Data presented as follows:
| ● | Decrease in PSA levels was seen in all patients at all three tested doses of PDS0301 and 61% of patients had at least a 60% decrease in PSA levels. |
| ● | All doses of the combination were well-tolerated with one patient experiencing Grade 4 neutropenia. |
| ● | Administration of the combination was associated with decreases in T reg cells and increases in activated natural killer (NK) cells, memory CD8 T cells, proliferating CD4 and CD8 T cells and cytokines INF-γ and Interleukin 10 (IL-10). |
| ● | The changes in immune responses with the combination were independent of the PDS0301 dose. |
We are working closely with the NCI to determine the best pathway forward for the prioritized PDS01ADC studies, as well as evaluating the use of PDS01ADC in combination with other Versamune based clinical candidates.
Our current pipeline of IL12 fused antibody conjugated PDS01ADC based therapies is as follows:
Infectimune Development Strategy
We believe that the key differentiating attributes of the Infectimune platform technology are strong induction of CD8 and CD4 T cells as well as antibodies which can be leveraged to improve treatment and preventive options in several infectious disease indications. In January 2022, we presented preclinical data on our universal flu program sponsored by the National Institute of Allergy and Infectious Disease (NIAID) demonstrating the potential of the Infectimune technology with computationally designed influenza proteins developed by the laboratory of Dr. Ted Ross at the University of Georgia to generate broadly protective anti-influenza immune responses across multiple strains of influenza. This data has provided a unique opportunity to highlight Infectimune’s potentially transformative utility in the development of more broadly effective and longer lasting protective vaccines. Current preventive and prophylactic vaccine approaches and technologies predominantly focus on creating strong induction of antibody responses. However, the induction of T cell responses, in addition to antibody responses, provides more durable and broad protection against infectious diseases.
Based on the promising data with the universal seasonal flu vaccine and the current focus of the NIAID in developing more effective flu vaccines, we have decided to opportunistically focus our near-term infectious disease activities to align with the interests of the NIAID Collaborative Influenza Vaccine Innovation Centers (CIVICs) program. This will involve development of a universal seasonal flu vaccine and the potential development of a universal pandemic influenza vaccine based on similar computationally designed antigens as have shown promise with Infectimune.
In July 2022, universal flu vaccine preclinical data for PDS0202 at the 41st American Society of Virology meeting: Abstract number 3733830, Infectimune enhances antibodies elicited by COBRA hemagglutinin influenza vaccine. We are evaluating the next steps in the clinical development and funding for PDS0202.
The results for Infectimune based vaccines were published in two separate articles in the peer reviewed journal Viruses in February 2023: 1. preclinical studies demonstrating complete protection against sickness after lethal challenge with live SARS-CoV-2 or influenza viruses (Gandhapudi SK et al. Viruses 2023, 15, 432) and 2. Dramatically enhanced CD4 T cell responses to recombinant influenza proteins compared to leading commercial vaccine adjuvants (Henson TR et al. Viruses 2023, 15, 538).
In September 2023, data on our investigational universal flu vaccine, PDS0202, were presented at the 9th European Scientific Working Group on Influenza (ESWI) conference. These data demonstrated active neutralization across multiple influenza viruses and provided protection against infection and weight loss after challenging with high doses of H1N1 viruses not previously exposed to flu.
Liquidity
We have never been profitable and have incurred net losses in each year since inception. Our net losses were $32.0 million, and $21.7 million for the nine months ended September 30, 2023 and 2022, respectively. As of September 30, 2023, we had an accumulated deficit of $133.6 million. Substantially all of our net losses have resulted from costs incurred in connection with our research and development programs and from general and administrative costs associated with these operations.
As of September 30, 2023, we had $54.3 million in cash and cash equivalents.
Our future funding requirements will depend on many factors, including the following:
| ● | the timing and costs of our planned clinical trials; |
| ● | the timing and costs of our planned preclinical studies of our Versamune® platform; |
| ● | the outcome, timing and costs of seeking regulatory approvals; |
| ● | the terms and timing of any future collaborations, licensing, consulting or other arrangements that we may enter into; |
| ● | the amount and timing of any payments we may be required to make in connection with the licensing, filing, prosecution, maintenance, defense and enforcement of any patents or patent applications or other intellectual property rights; and |
| ● | the extent to which we license or acquire other products and technologies. |
SELECTED FINANCIAL OPERATIONS OVERVIEW
Revenue
We have not generated any revenues from commercial product sales and do not expect to generate any such revenue in the near future. We may generate revenue in the future from a combination of research and development payments, license fees and other upfront payments or milestone payments.
Research and Development Expenses
Research and development expenses include employee-related expenses, licensing fees to use certain technology in our research and development projects, costs of acquiring, developing and manufacturing clinical trial materials, as well as fees paid to consultants and various entities that perform certain research and testing on our behalf. Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued expenses. Costs incurred in connection with research and development activities are expensed as incurred.
We expect that our research and development expenses will increase significantly over the next several years as we advance our platforms including Versamune based immuno-oncology, IL12 Fused antibody drug candidates in oncology, and Infectimune based infectious disease candidates into and through clinical trials, pursue regulatory approval of our investigational candidates and prepare for a possible commercial launch, all of which will also require a significant investment in contract and internal manufacturing and inventory related costs.
The process of conducting clinical trials necessary to obtain regulatory approval is costly and time consuming. We may never succeed in achieving marketing approval. The probability of successful commercialization of our drug candidates may be affected by numerous factors, including clinical data obtained in future trials, competition, manufacturing capability and commercial viability. As a result, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of any of our product candidates.
Results of Operations
The following table summarizes the results of our operations for the three months ended September 30, 2023 and 2022:
| | | Three Months Ended September 30, | | | Increase (Decrease)
| |
| | | 2023 | | | 2022 | | | $ Amount | | | % | |
| | | (in thousands) | | | | | | | |
| Operating expenses: | | | | | | | | | | | | |
| Research and development expenses | | $ | 6,449 | | | $ | 4,353 | | | $ | 2,096 | | | | 48 | % |
| General and administrative expenses | | | 4,071 | | | | 2,926 | | | | 1,145 | | | | 39 | % |
| Total operating expenses | | | 10,520 | | | | 7,279 | | | | 3,241 | | | | 45 | % |
| Loss from operations | | | (10,520 | ) | | | (7,279 | ) | | | (3,241 | ) | | | 45 | % |
| Interest income (expense), net | | | (329 | ) | | | (145 | ) | | | (184 | ) | | | 127 | % |
| Net loss and comprehensive loss | | $ | (10,849 | ) | | $ | (7,424 | ) | | $ | (3,425 | ) | | | 46 | % |
Research and Development Expenses
Research and development (R&D) expenses increased to $6.4 million for the three months ended September 30, 2023 from $4.4 million for the three months ended September 30, 2022. The increase of $2.0 million is primarily attributable to an increase of $1.3 million in clinical trials, and $0.7 million in personnel costs, including $0.3 million in non-cash stock-based compensation.
General and Administrative Expenses
General and administrative expenses increased to $4.1 million for the three months ended September 30, 2023 from $2.9 million for the three months ended September 30, 2022. The increase of $1.2 million is primarily attributable to an increase of $0.7 million in personnel costs, including $0.5 million in non-cash stock-based compensation, and $0.5 million in investor relations costs.
Comparison of the Nine months September 30, 2023 and 2022
The following table summarizes the results of our operations for the nine months September 30, 2023 and 2022:
| | | Nine Months Ended September 30, | | | Increase (Decrease) | |
| | | 2023 | | | 2022 | | | $ Amount | | | % | |
| | | (in thousands) | | | | | | | |
| Operating expenses: | | | | | | | | | | | | |
| Research and development expenses | | $ | 20,297 | | | $ | 13,276 | | | $ | 7,021 | | | | 53 | % |
| General and administrative expenses | | | 12,341 | | | | 9,575 | | | | 2,766 | | | | 29 | % |
| Total operating expenses | | | 32,638 | | | | 22,851 | | | | 9,787 | | | | 43 | % |
| Loss from operations | | | (32,638 | ) | | | (22,851 | ) | | | (9,787 | ) | | | 43 | % |
| Interest income (expense), net | | | (812 | ) | | | (65 | ) | | | (747 | ) | | | 1,149 | % |
| Benefit from income taxes | | | 1,406 | | | | 1,199 | | | | 207 | | | | 17 | % |
| Net loss and comprehensive loss | | $ | (32,044 | ) | | $ | (21,717 | ) | | $ | (10,327 | ) | | | 48 | % |
Research and Development Expenses
Research and development (R&D) expenses increased to $20.3 million for the nine months September 30, 2023 from $13.3 million for the nine months ended September 30, 2022. The increase of $7.0 million was primarily attributable to an increase in personnel costs of $2.0 million, including $1.0 million in non-cash stock-based compensation, clinical trials of $3.4 million, manufacturing expenses of $1.4 million and professional fees and facilities costs of $0.2 million.
General and Administrative Expenses
General and administrative expenses increased to $12.3 million for the nine months September 30, 2023 from $9.6 million for the nine months ended September 30, 2022. The increase of $2.7 million was primarily attributable to an increase in personnel costs of $2.0 million, including $1.4 million in non-cash stock-based compensation, and $0.7 million in investor relations costs.
Benefit from Income Taxes
Income tax benefit was $1.4 million for the nine months ended September 30, 2023 and $1.2 million for the nine months ended September 30, 2022. The increase of $0.2 million was due to an increase in the amount of New Jersey NOL carryforwards sold when compared to the comparable period.
Liquidity and Capital Resources
In April 2022, we received approximately $1.2 million from the net sale of tax benefits to an unrelated, profitable New Jersey corporation pursuant our participation in the New Jersey Technology Business Tax Certificate Transfer NOL program for tax year 2020.
In August 2022, we filed a shelf registration statement, or the 2022 Shelf Registration Statement, with the SEC for the issuance of common stock, preferred stock, warrants, rights, debt securities, and units, up to an aggregate amount of $150 million, $50 million of which covers the offer, issuance and sale by us of our common stock under the Sales Agreement (as discussed below). The 2022 Shelf Registration Statement was declared effective on September 2, 2022.
In August 2022, we entered into an At Market Issuance Sales Agreement, or the Sales Agreement, with B. Riley Securities, Inc. and BTIG, LLC, each an Agent and collectively the Agents, with respect to an at-the-market offering program under which we may offer and sell, from time to time at our sole discretion, shares of our common stock, having an aggregate offering price of up to $50 million, or the Placement Shares, through or to the Agents, as sales agents or principals. Upon delivery of a placement notice and subject to the terms and conditions of the Sales Agreement, the Agents may sell the Placement Shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act of 1933, as amended, including, without limitation, sales made through The Nasdaq Capital Market or on any other existing trading market for our common stock. The Agents will use commercially reasonable efforts to sell the Placement Shares from time to time, based upon our instructions (including any price, time or size limits or other customary parameters or conditions we may impose). We will pay the Agents a commission equal to three percent (3%) of the gross sales proceeds of any Placement Shares sold through the Agents under the Sales Agreement, and also have provided the Agents with customary indemnification and contribution rights. We are not obligated to make any sales of our common stock under the Sales Agreement. The offering of Placement Shares pursuant to the Sales Agreement will terminate upon the earlier of (i) the sale of all Placement Shares subject to the Sales Agreement or (ii) termination of the Sales Agreement in accordance with its terms. For the year ended December 31, 2022, we sold 1,238,491 shares of our common stock with a net value of $9.9 million pursuant to the Sales Agreement. During the three and nine months ended September 30, 2023, we sold 139,575 and 736,037 shares of our common stock with a net value of $0.8 million and $5.7 million, respectively, pursuant to the Sales Agreement.
In August 2022, we entered into a venture loan and security agreement, or the Loan and Security Agreement, with Horizon Technology Finance Corporation, as lender and collateral agent for itself and the other lenders. The Loan and Security Agreement provides for the following 6 separate and independent term loans: (a) a term loan in the amount of $7,500,000, or Loan A, (b) a term loan in the amount of $10,000,000, or Loan B, (c) a term loan in the amount of $3,750,000, or Loan C, (d) a term loan in the amount of $3,750,000, or Loan D, (e) a term loan in the amount of $5,000,000, or Loan E, and (f) a term loan in the amount of $5,000,000, or Loan F, (with each of Loan A, Loan B, Loan C, Loan D, Loan E, and Loan F, individually a Loan and, collectively, the Loans). Loan A, Loan B, Loan C, and Loan D were delivered to us on August 24, 2022. Loan E and Loan F were uncommitted Loans that could have been advanced by the Lenders upon the parties agreement prior to July 31, 2023 upon the satisfaction by the Company of certain agreed upon conditions. At this time the option has expired and Loan E and Loan F are no longer available to the Company under the Loan and Security Agreement . We may only use the proceeds of the Loans for working capital or general corporate purposes.
Each Loan matures on the 48-month anniversary following the applicable funding date unless accelerated pursuant to agreed upon events of default. Payments on the principal balance begin on October 1, 2024 and are paid monthly in the succeeding 24 months. The principal balance of each Loan bears a floating interest. The interest rate is calculated initially and, thereafter, each calendar month as the sum of (a) the per annum rate of interest from time to time published in The Wall Street Journal as contemplated by the Loan and Security Agreement, or any successor publication thereto, as the “prime rate” then in effect, plus (b) 5.75%; provided that, in the event such rate of interest is less than 4.00%, such rate shall be deemed to be 4.00% for purposes of calculating the interest rate.
Interest is payable on a monthly basis based on each Loan principal amount outstanding the preceding month. We, at our option upon at least ten (10) business days’ written notice to the lenders, may prepay all (and not less than all) of the outstanding Loan by simultaneously paying to each lender an amount equal to (i) any accrued and unpaid interest on the outstanding principal balance of the Loans; plus (ii) an amount equal to (A) if such Loan is prepaid on or before the Loan Amortization Date (as defined in the Loan and Security Agreement) applicable to such Loan, 3% of the then outstanding principal balance of such Loan, (B) if such Loan is prepaid after the Loan Amortization Date applicable to such Loan, but on or before the date that is 12 months after such Loan Amortization Date, 2% of the then outstanding principal balance of such Loan, or (C) if such Loan is prepaid more than 12 months after the Loan Amortization Date but prior to the stated maturity date applicable to such Loan, 1% of the then outstanding principal balance of such Loan; plus (iii) the outstanding principal balance of such Loan; plus (iv) all other sums, if any, that shall have become due and payable thereunder. No prepayment premium will be applied to any outstanding balance of any Loan paid on the stated maturity date.
In connection with the Loan and Security Agreement, we issued Horizon Technology Finance Corporation and Powerscourt Investments XXV, LP warrants to purchase an aggregate total of 381,625 shares of our common stock at an initial exercise price of $3.6685 per share. Each warrant is classified as equity and is exercisable at any time for a period beginning on the date of grant and ending on the earlier of (A) 10 years from the date of grant, and (B) the closing of (A) (i) the sale, lease, exchange, conveyance or other disposition of all or substantially all of the our property or business, or (ii) its merger into or consolidation with any other corporation (other than a wholly-owned subsidiary of the Company), or any transaction (including a merger or other reorganization) or series of related transactions, in which more than 50% of the voting power of the Company is disposed of, in each case, for cash or for marketable securities meeting certain requirements as described in the applicable warrants. The key assumptions used in Black-Scholes option pricing model were (i) expected term of 10 years, (ii) a risk-free rate of 3.11%, (iii) expected volatility of 93.8%, and (iv) no estimated dividend yield.
In April 2023, we received approximately $1.4 million from the net sale of tax benefits to an unrelated, profitable New Jersey corporation pursuant our participation in the New Jersey Technology Business Tax Certificate Transfer NOL program for tax year 2021.
As of September 30, 2023, we had $54.3 million in cash and cash equivalents. Our primary uses of cash are to fund operating expenses, primarily research and development expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.
We plan to continue to fund our operations and capital funding needs through equity and/or debt financings. However, we cannot be certain that additional financing will be available when needed or that, if available, financing will be obtained on terms favorable to us or our existing stockholders. We may also enter into government funding programs and consider selectively partnering for clinical development and commercialization. The sale of additional equity would result in additional dilution to our stockholders. Incurring debt financing would result in debt service obligations, and the instruments governing such debt could provide for operating and financing covenants that would restrict our operations. If we are unable to raise additional capital in sufficient amounts or on acceptable terms, we may be required to delay, limit, reduce, or terminate our clinical development or future commercialization efforts or grant rights to develop and market immunotherapies that we would otherwise prefer to develop and market ourselves. Any of these actions could harm our business, results of operations and prospects.
We evaluated whether there are any conditions and events, considered in the aggregate, that raise substantial doubt about our ability to continue as a going concern within one year after the filing of this Quarterly Report on Form 10-Q. While we intend to finance our cash needs principally through collaborations, strategic alliances, or license agreements with third parties and/or debt or equity financings, there is no assurance that new financing will be available to us on commercially acceptable terms or in the amounts required, if at all. As such, we have concluded that substantial doubt exists about our ability to continue as a going concern for a period of at least 12 months from the date of the issuance of these unaudited condensed consolidated financial statements. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
Cash Flows
The following table shows a summary of our cash flows for each of the periods indicated (in thousands):
| | | Nine Months Ended September 30, | |
| | | 2023 | | | 2022 | |
Net cash used in operating activities | | $ | (25,179 | ) | | $ | (18,181 | ) |
| Net cash provided by financing activities | | | 5,610 | | | | 24,581 | |
| Net increase in cash and cash equivalents | | $ | (19,569 | ) | | $ | 6,400 | |
Net Cash Used in Operating Activities
Net cash used in operating activities was $25.2 million and $18.2 million for the nine months ended September 30, 2023 and 2022, respectively. The increase in net cash used in operating activities of $7.0 million was primarily due to an increase in net loss of $10.3 million, reduced by the increase in the non-cash stock-based compensation expense of $2.4 million, offset by changes in the timing of working capital requirements, including changes in prepaid expenses and other assets, accrued expenses and accounts payable. The increase in accounts payable, specifically, is primarily the result of a dispute with a certain vendor and we are working towards resolving this dispute.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the nine months ended September 30, 2023 was due to the receipt of net proceeds of $5.6 million due to the sale of common stock under the Sales Agreement. Net cash provided by financing activities for the nine months ended September 30, 2022 was primarily due to the receipt of net proceeds of $24.6 million due to the Venture Loan.
Operating Capital Requirements
To date, we have not generated any product revenue. We do not know when, or if, we will generate any product revenue and we do not expect to generate significant product revenue unless and until we obtain regulatory approval and commercialize one of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our vaccine candidates, and begin to commercialize any approved vaccine candidates. We are subject to all of the risks incident to the development of new products, and may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may harm our business. We expect to incur additional costs associated with operating as a public company and anticipate that we will need substantial additional funding in connection with our continuing operations.
We evaluated whether there are any conditions and events, considered in the aggregate, that raise substantial doubt about our ability to continue as a going concern within one year after the filing of this Quarterly Report. Our budgeted cash requirements in 2023 and beyond include expenses related to continuing development and clinical studies. Until we can generate significant cash from our operations, we expect to continue to fund our operations with available financial resources. These financial resources may not be adequate to sustain our operations. While we intend to finance our cash needs principally through collaborations, strategic alliances, or license agreements with third parties and/or debt or equity financings, there is no assurance that new financing will be available to us on commercially acceptable terms or in the amounts required, if at all. As such, we have concluded that substantial doubt exists about our ability to continue as a going concern for a period of at least 12 months from the date of the issuance of these unaudited condensed consolidated financial statements.
We have based our projections of operating capital requirements on assumptions that may prove to be incorrect and we may use all of our available capital resources sooner than we expect. Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timing, costs and results of our planned clinical trials; |
| ● | the effects of health epidemics, pandemics, or outbreaks of infectious diseases, on our business operations, financial condition, results of operations and cash flows; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the U.S. Food and Drug Administration, or FDA, the European Medicines Agency, or EMA, and other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending potential intellectual property disputes, including patent infringement actions brought by third parties against us now or in the future; |
| ● | the effect of competing technological and market developments; |
| ● | the cost of establishing sales, marketing and distribution capabilities in regions where we choose to commercialize our products on our own; and |
| ● | the initiation, progress, timing and results of our commercialization of our clinical candidates, if approved, for commercial sale. |
Please see the section titled “Risk Factors” elsewhere in the Quarterly Report and Annual Report for additional risks associated with our operations.
Purchase Commitments
We have no material non-cancelable purchase commitments with service providers as we have generally contracted on a cancelable, purchase order basis.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. GAAP. Our accounting policies are more fully described in Note 2 to the consolidated financial statements included in this Quarterly Report on Form 10-Q. As described in Note 2, the preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue generated and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Estimates are assessed each period and updated to reflect current information. Actual results may differ from these estimates under different assumptions or conditions. We believe that the discussion in our management’s discussion and analysis addresses our most critical accounting policies, which are those that are most important to the portrayal of our financial condition and results of operations and require management’s most difficult, subjective and complex judgments.
There have been no material changes to our critical accounting policies and estimates during the nine months ended September 30, 2023 from those disclosed in our Annual Report on Form 10-K for the year ended December 31, 2022.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Smaller Reporting Company
As of January 1, 2021, we were no longer an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. However, we remain a “smaller reporting company,” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended. We will cease to be a smaller reporting company if we have a non-affiliate public float in excess of $250 million and annual revenues in excess of $100 million, or a non-affiliate public float in excess of $700 million, determined on an annual basis. As a smaller reporting company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not smaller reporting companies. We will continue to take advantage of some or all of the available exemptions.
| ITEM 3: | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
We are exposed to market risks in the ordinary course of our business and from changes in the interest rate on our debt borrowings. These market risks are principally limited to interest rate fluctuations. As of September 30, 2023, our cash equivalents consisted of bank deposits and money market accounts and our debt is a variable interest rate instrument. Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio and debt agreement, we do not believe an immediate 100 basis point increase in interest rates would have a material effect on the fair market value of our portfolio, and, accordingly, we do not expect our operating results or cash flows to be materially affected by a sudden change in market interest rates.
Inflation generally affects us by increasing our cost of labor and pricing of contracts. We do not believe that inflation has had a material effect on our business, financial condition, or results of operations during the three months ended September 30, 2023.
| ITEM 4. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
An evaluation was carried out, under the supervision of and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15 (e)) under the Securities Exchange Act of 1934, or the Exchange Act, as of the end of the period covered by this report. Based on the evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that the information required to be disclosed by us in the reports we file or submit under the Exchange Act was recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as such term is defined in Rule 13a-15(f) under the Exchange Act) identified in connection with the evaluation identified above that occurred during the quarter ended September 30, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| PART II. | OTHER INFORMATION |
The information in Note 9 to the Condensed Consolidated Financial Statements contained in Part I, Item 1 of this Quarterly Report on Form 10-Q is incorporated herein by reference. There are no matters which constitute material pending legal proceedings to which we are a party other than those incorporated into this item by reference from Note 9 to our Condensed Consolidated Financial Statements for the quarter ended September 30, 2023 contained in this Quarterly Report on Form 10-Q.
With the exception of the risk factor noted below, there have been no material changes from our risk factors as previously reported in our Annual Report on Form 10-K for the year ended December 31, 2022. However, any investment in our business involves a high degree of risk. Before making an investment decision, you should carefully consider the information we include in this Quarterly Report on Form 10-Q, including our unaudited interim condensed consolidated financial statements and accompanying notes, our Annual Report on Form 10-K for the year ended December 31, 2022 filed on March 28, 2023, including our financial statements and related notes contained therein, and the additional information in the other reports we file with the Securities and Exchange Commission. These risks may result in material harm to our business and our financial condition and results of operations. In this event, the market price of our common stock may decline and you could lose part or all of your investment. Additional risks that we currently believe are immaterial may also impair our business operations. Our business, financial conditions and future prospects and the trading price of our common stock could be harmed as a result of any of these risks.
We have identified conditions and events that raise substantial doubt regarding our ability to continue as a going concern.
We have incurred net losses and utilized cash in operations since inception. In addition, as of September 30, 2023, we had approximately $54.3 million in cash and cash equivalents, and during the nine months ended September 30, 2023, we used $18.2 million of cash in operations and expect to continue to incur significant cash outflows and incur future additional losses to execute our operating plan. While we intend to finance our cash needs principally through collaborations, strategic alliances, or license agreements with third parties and/or debt or equity financings, there is no assurance that new financing will be available to us on commercially acceptable terms or in the amounts required, if at all. Due to the uncertainty in securing additional funding, and the insufficient amount of cash and cash equivalents as of September 30, 2023, we have concluded that substantial doubt exists about our ability to continue as a going concern within one year after the date of the filing of this Quarterly Report. If we are unsuccessful in securing sufficient financing, we may need to delay, reduce, or eliminate our research and development programs, which could adversely affect our business prospects, or cease operations.
Our unaudited condensed consolidated financial statements included in this Quarterly Report have been prepared on a going concern basis under which an entity is able to realize its assets and satisfy its liabilities in the ordinary course of business. The unaudited condensed consolidated financial statements do not give effect to any adjustments relating to the carrying values and classification of assets and liabilities that would be necessary should we be unable to continue as a going concern within one year after the date that the financial statements are issued.
Our future operations are dependent upon the successful entry into collaborations, strategic alliances, or license agreements with third parties and/or on the identification and successful completion of equity or debt financing and the achievement of profitable operations at an indeterminate time in the future. There can be no assurances that we will be successful in completing these collaborations or alliances, equity or debt financing or in achieving profitability. As such, there can be no assurance that we will be able to continue as a going concern.
Substantial doubt about our ability to continue as a going concern may materially and adversely affect the price per share of our common stock, and it may be more difficult for us to obtain financing. If potential collaborators decline to do business with us or potential investors decline to participate in any future financings due to such concerns, our ability to increase our cash position may be limited. The perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations. If we are unable to continue as a going concern, you could lose all or part of your investment in our Company.
| ITEM 2. | UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS |
None
| ITEM 3. | DEFAULTS UPON SENIOR SECURITIES |
None.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.