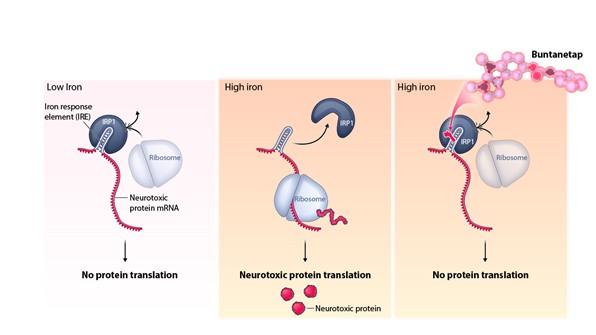
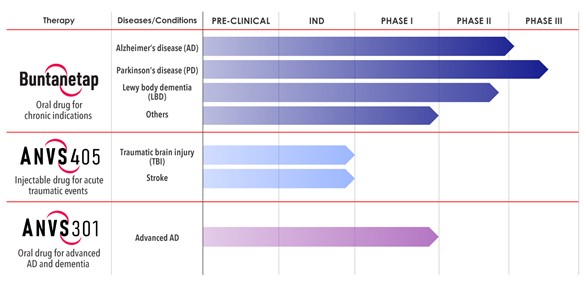
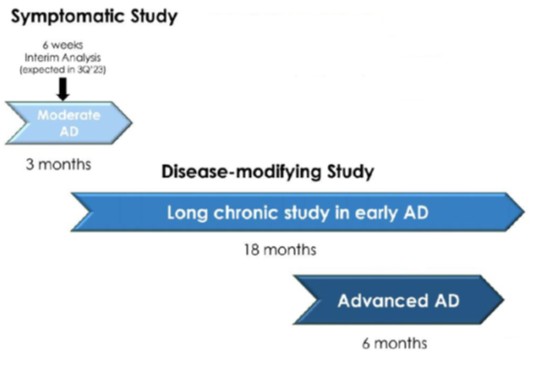
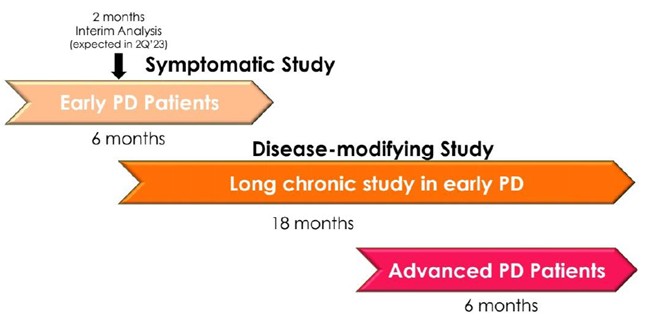
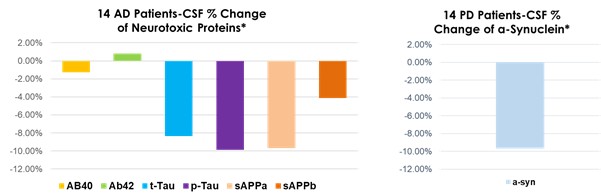
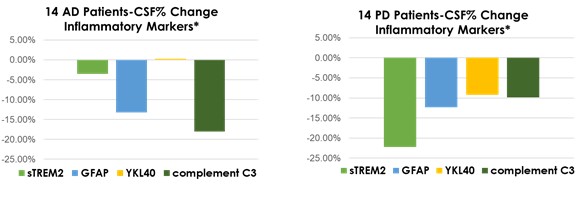
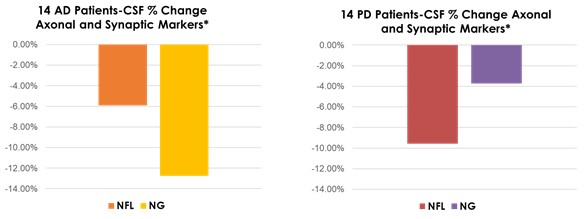
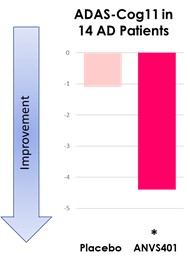
Interim “top-line” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are further subject to audit and verification procedures that could result in material changes in the final data. Furthermore, undue reliance on interim analyses may be detrimental to our long-term clinical development plans, which could harm our business, operating results, prospects or financial condition.
From time to time, we may publish interim “top-line” or preliminary data from our clinical studies, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to evaluate all data fully and carefully. Preliminary or “top-line” data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. Further, disclosure of interim data by us or by our competitors could result in volatility in the price of our common stock.
In addition, others, including regulatory authorities, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. Moreover, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, product candidate or our business. If the interim “topline” or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, Buntanetap and any future product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
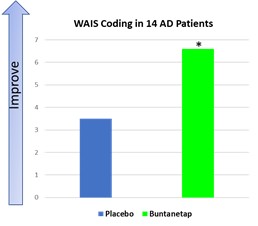
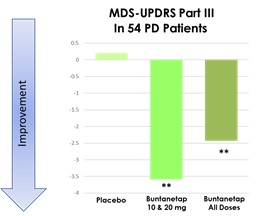
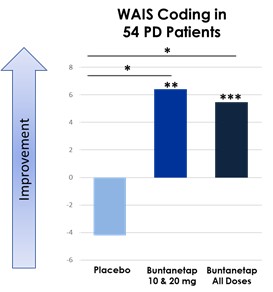
Furthermore, our Phase 3 PD Study and Phase 2/3 AD Study each included interim analyses. Our Phase 3 PD Study incorporated an interim analysis at two months, the results of which were disclosed on March 31, 2023. Based on the results of the interim analysis, we proceeded with the Phase 3 PD Study as planned in accordance with the previously established protocol. We disclosed the results of the interim analysis for our Phase 2/3 AD Study on October 23, 2023, and as for the PD study based on the outcome of the interim analysis we proceeded with the study as planned. The Phase 2/3 AD study was completed on February 13, 2024. We plan to consult with the FDA following completion of the full analyses of the two studies, to obtain feedback on our planned AD and PD studies, including conducting disease-modifying studies and open label extensions. Accordingly, if the interim data upon which we have relied on is shown to be materially different from the final, complete study results, the basis upon which we set forth our development plans may be called into question and our ability to obtain approval for, and commercialize, Buntanetap may be harmed, which could harm our business, operating results, prospects or financial condition.
Our product candidates may cause serious adverse events or undesirable side effects, which may delay or prevent marketing approval, or, if approved, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
As is the case with biopharmaceuticals generally, it is likely that there may be adverse side effects associated with Buntanetap or any future product candidates’ use. Serious adverse events or undesirable side effects caused by Buntanetap or any other product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Results of any clinical trial we conduct could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. Patients treated with Buntanetap to date, at high doses have experienced adverse events that include nausea, vomiting and dizziness.
If unacceptable side effects arise in the development of our product candidates, we, the FDA or the IRBs at the institutions in which our studies are conducted, or the DSMB, if constituted for our clinical trials, could recommend a suspension or termination of our clinical trials, or the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of a product candidate for any or all targeted indications. In addition, drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete a trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel