SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | |
| | For the fiscal year ended December 31, 2012 |
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | |
| | For the transition period from to |
Commission file number 001-34641 FURIEX PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | | 27-1197863 |
(State or other jurisdiction of incorporation or organization) | | (IRS Employer Identification No.) |
3900 Paramount Parkway, Suite 150
Morrisville, North Carolina 27560
(Address of principal executive offices, including zip code)
(919) 456-7800
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | |
| | (Name of each exchange on which registered) |
| Common Stock, par value $0.001 per share | | Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the
Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its Corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | | ¨ | | Accelerated filer | | x |
| | | | |
| Non-accelerated filer | | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the common stock held by non-affiliates of the registrant was approximately $149.8 million as of June 29, 2012, the last business day of the most recently completed second fiscal quarter, based on the closing price of the Common Stock on that date on the NASDAQ Global Market. Shares of common stock held by each executive officer and director and by each person who owns 10% or more of the outstanding common stock have been excluded in that such person might be deemed to be an affiliate. This determination of affiliate status might not be conclusive for other purposes.
As of February 28, 2013, there were 10,015,297 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The Company’s definitive Proxy Statement for its 2013 Annual Meeting of Stockholders (certain parts, as indicated in Part III).
| Part I. | |
| | | | |
| | Item 1. | Business | 3 |
| | | | |
| | Item 1A. | Risk Factors | 13 |
| | | | |
| | Item 1B. | Unresolved Staff Comments | 23 |
| | | | |
| | Item 2. | Properties | 23 |
| | | | |
| | Item 3. | Legal Proceedings | 23 |
| | | | |
| | Item 4. | Mine Safety Disclosures | 23 |
| | | | |
| | | Executive Officers of the Registrant | 24 |
| | | | |
| Part II. | | | |
| | | | |
| | Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 25 |
| | | | |
| | Item 6. | Selected Financial Data | 27 |
| | | | |
| | Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 28 |
| | | | |
| | Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 38 |
| | | | |
| | Item 8. | Financial Statements and Supplementary Data | 38 |
| | | | |
| | Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | 38 |
| | | | |
| | Item 9A. | Controls and Procedures | 39 |
| | | | |
| | Item 9B. | Other Information | 41 |
| | | | |
| Part III. | | | |
| | | | |
| | Item 10. | Directors, Executive Officers and Corporate Governance | 42 |
| | | | |
| | Item 11. | Executive Compensation | 42 |
| | | | |
| | Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 42 |
| | | | |
| | Item 13. | Certain Relationships and Related Transactions, and Director Independence | 43 |
| | | | |
| | Item 14. | Principal Accounting Fees and Services | 43 |
| | | | |
| Part IV. | | | |
| | | | |
| | Item 15. | Exhibits, Financial Statement Schedules | 44 |
| | | | |
| | | Signatures | 47 |
This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. These forward-looking statements are subject to risks and uncertainties, including those set forth under “Item 1A. Risk Factors” and “Cautionary Statement” included in “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this report, that could cause actual results to differ materially from historical results or anticipated results. Unless otherwise indicated or required by the context, the terms “we,” “our,” “us” and the “Company” refer to Furiex Pharmaceuticals, Inc. and all of its subsidiaries.
Item 1. Business
Our Business
About Furiex Pharmaceuticals
We are a drug development collaboration company that uses innovative clinical development strategies to increase the value of partnered pharmaceutical assets and accelerate their development timelines. We collaborate with pharmaceutical and biotechnology companies to increase the value of their drug candidates by applying our novel approach to drug development, which we believe expedites research and development decision-making and can shorten drug development timelines. We share the risk with our collaborators by conducting and financing drug development programs, and in exchange, we share the potential rewards, receiving milestone and royalty payments for successful drug candidates. This business model is designed to help feed compound pipelines and deliver therapies to improve lives.
Our Company continues the compound partnering business started by Pharmaceutical Product Development, Inc., or PPD, in 1998. We became an independent publicly traded company on June 14, 2010, when PPD spun-off its compound partnering business through a tax-free, pro-rata dividend distribution of all of the shares of the Company to PPD shareholders. PPD does not have any ownership or other form of equity interest in the Company following the spin-off. The Company's operations are headquartered in Morrisville, North Carolina. Our website address is www.furiex.com. Information on our website is not incorporated herein by reference. We make available free of charge through our website press releases, Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after we have electronically filed with, or furnished to, the Securities and Exchange Commission.
Business Description
Our goal is to fully in-license from or form strategic alliances with pharmaceutical and biotechnology businesses to develop and commercialize therapeutics in which the risks and rewards are shared. We seek to collaborate with pharmaceutical and biotechnology companies to increase the value of early stage drug candidates by applying our novel approach to drug development, which we believe expedites research and development decision-making and can shorten drug development timelines. Furiex’s team is staffed with key PPD team members who demonstrated proven success in the drug development collaboration business while at PPD, as well as highly-qualified additional members. Our strategy is to invest in drug candidates that have a relatively straightforward path to regulatory approval and a large addressable market. Every drug candidate we review is subjected to our rigorous due diligence process by our team of experts who possess experience in all aspects of the drug development process.
Once we fully in-license or form an alliance, we use our drug development experience and financial resources to advance the drug candidate through clinical development. We apply a novel approach that we believe shortens drug development timelines and transforms research and development into revenues more rapidly than the typical development cycle for such collaborations. Specifically, we set the development strategy based on a product candidate’s best market position, design and manage non-clinical and clinical studies, manage the drug manufacturing programs and evaluate the efficacy and safety data necessary to obtain regulatory approvals for the drug candidate. We use service providers to execute the tasks needed to develop and commercialize our product candidates.
Most of our collaborations involve late-stage development and commercialization agreements with large pharmaceutical companies. Typically, if our collaborators are unable or unwilling to execute on late stage development and commercialization, then we have the option to seek new collaborators. In exchange for our drug development efforts and sharing the risk with our collaborator, we are entitled to receive milestone payments and royalties based on the continued development and commercialization success of the drug candidate, as described in more detail under “Our Portfolio” below.
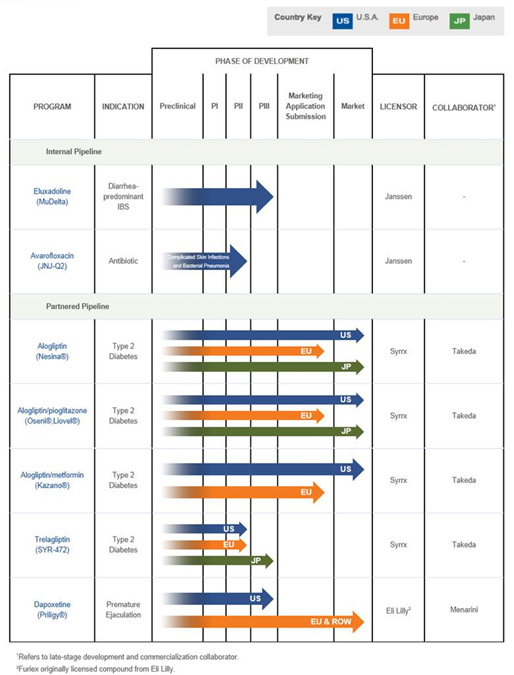
Currently, we have rights to numerous compounds in various stages of development and commercialization, including:
| · | Rights to royalties and regulatory and sales-based milestone payments from Takeda Pharmaceuticals Company Limited, or Takeda, for alogliptin, alogliptin combination products and SYR-472 (trelagliptin) for the treatment of Type-2 diabetes. In Japan, Takeda currently markets alogliptin under the name Nesina® and two fixed dose combination tablets of Nesina and Actos® (pioglitazone) under the name Liovel®. In January 2013, Takeda confirmed the approval from the U.S. Food and Drug Administration, or FDA, of Nesina (alogliptin) and the fixed-dose combination therapies, Oseni® (alogliptin and pioglitazone) and Kazano® (alogliptin and metformin) for treatment of Type-2 diabetes. In September 2011, Takeda commenced Phase III clinical trials for trelagliptin in Japan for treatment of Type-2 diabetes. Takeda estimates the study will complete in the second half of 2013. |
| · | Rights to royalties, launch-based and sales-based milestones from Berlin-Chemie AG (Menarini Group), or Menarini, for Priligy® (dapoxetine), the first approved treatment in the world for premature ejaculation. In May 2012, we entered into an agreement whereby Alza Corporation, or Alza, and Janssen Pharmaceutica, NV, or Janssen (an affiliate of Johnson & Johnson), transferred worldwide Priligy rights to us. In 2012, we also entered into a license agreement with Menarini to commercialize Priligy in Europe, most of Asia, Africa, Latin America and the Middle East. Priligy will continue to be made available to patients, and we will still receive royalties, under a sales service agreement with Janssen, until the marketing authorizations are transferred to Menarini. Priligy is currently marketed in 15 countries in Europe, Asia-Pacific and Latin America. |
| · | Eluxadoline1, a mu opioid receptor agonist and delta opioid receptor antagonist, which we call MuDelta, currently in Phase III development for the treatment of diarrhea-predominant irritable bowel syndrome, or IBS-d. We licensed MuDelta from Janssen in November 2009 and acquired full exclusive license rights to develop and commercialize MuDelta under our existing development and license agreement in November 2011. |
| · | Avarofloxacin1, a Phase III-ready fluoroquinolone antibiotic, which we call JNJ-Q2, for the treatment of acute bacterial skin and skin structure infections, or ABSSSI, and community-acquired bacterial pneumonia, or CABP. We licensed JNJ-Q2 from Janssen in November 2009 and acquired full exclusive license rights to develop and commercialize JNJ-Q2 under our existing development and license agreement in April 2011. |
The following chart summarizes the status of our pipeline of compounds:
1 United States Adopted Names Council (USAN) adopted, International Nonproprietary Names (INN) approval pending.
Our Solution
The drug development industry is under increasing economic pressure to develop new products more quickly and efficiently. To address this industry issue, we have developed what we believe is a novel approach to drug development. Our approach to drug development involves applying proven solutions from our extensive global drug development experience to reduce development timelines and expedite the decision-making cycle, planning for success and bridging steps in development by conducting earlier elements of a program while simultaneously planning for later phases of development.
In order to obtain regulatory approval from the FDA and other regulatory agencies around the world to market a drug, certain data about the safety and efficacy of the drug is required. To obtain such data, drug developers frequently choose to run studies sequentially. For example, they might run one study, wait to see the results, and then they run the next study. Developers prefer this approach primarily to limit upfront expenditures since the success of any given study is not known and the decision might be made not to move forward due to negative data. However, this sequential approach slows down the development process.
We approach drug development by minimizing the time it takes to bring products to the market. Our novel approach manages drug development with parallel processing and efficient decision-making. We use our drug development experience to predict possible outcomes of a study and take risks based on those predictions. By assuming success at each critical decision point in advance, as opposed to waiting for results, development time is reduced. In addition, we seek to mitigate risks by contingency planning for potential problems. As a result, we can accelerate the development process by bridging steps across the developmental program as well as between studies. Additionally, we focus our efforts on only those essential studies necessary for regulatory approval. This helps to shorten developmental timelines.
Two key elements to our approach are our due diligence process and our planning for the success of each compound. Before we enter into a collaboration for a compound, we subject it to an intense due diligence review covering every step in the development process, from preclinical and clinical studies through marketing approval. We generally look for, and enter into collaborations with respect to, compounds that have the following characteristics:
| | • | | address medical conditions with a significant unmet need; |
| | • | | a reasonable development time; |
| | • | | reasonable predictability of non-clinical models; |
| | • | | clinical evidence no later than Phase Ib; |
| | • | | acceptable estimated cost of goods; and |
| | • | | attractive economic terms with the compound’s innovator and ultimate commercial collaborator. |
If a compound passes our rigorous diligence review hurdles, we then plan the entire development timeline upfront, using a set of assumptions. Part of the upfront planning involves initiating long-term studies, such as carcinogenicity studies, earlier than usual. We also use real-time data analysis tools to monitor the clinical study data of a drug candidate. By initiating long-term studies earlier and reviewing data in real time, we can significantly reduce the time needed after the conclusion of clinical studies to complete the necessary documentation for regulatory filing.
We believe this approach works well because the core development team is empowered to make decisions, real-time technology tools facilitate rapid data review, development programs are designed to optimize market position and timelines are driven by science and “must have” studies. The resulting ability to reduce development timelines in turn allows us to capitalize more quickly on our investment. We believe our success evolves from our development efficiency.
According to the Tufts University Center for the Study of Drug Development Outlook 2009, since 2002 the average time from the filing of an Investigational New Drug application, or IND, to the filing of a New Drug Application, or NDA, is over eight years. By contrast, we advanced alogliptin as a treatment for Type-2 diabetes (for the monotherapy program) from IND to NDA in only 39 months.
Our Business Strategy
Our strategy is to fully in-license and develop novel early stage drug candidates that address medical conditions with significant unmet need. We invest in innovative early stage drug candidates whose targets have scientific or clinical validation, and in disease areas that have a relatively straightforward path to regulatory approval. We leverage our extensive drug development expertise to implement efficient and high quality development programs that accelerate time to market. We progress drug candidates to key value inflection points and form strategic collaborations with commercial pharmaceutical companies in exchange for milestones and royalties. We subject each potential drug candidate we consider to a rigorous review process by our due diligence team, which has expertise in all aspects of drug development, as well as in intellectual property and commercial assessment. This approach has enabled us to build a diversified portfolio of drug candidates and commercialized products that offer value to patients, our investors and collaborators.
Our Portfolio
We have exclusive license rights to one compound that is in Phase III development (MuDelta) and another (JNJ-Q2) that is Phase III-ready. We also have one compound, trelagliptin (SYR-472), in Phase III development with a collaborator, and three products that are commercialized by collaborators, for which we are eligible to receive regulatory or launch-based milestone payments plus worldwide sales-based royalty and sales-based milestone payments. The commercialized products, Nesina, Liovel and Priligy, are currently marketed outside of the United States, and we have no further development obligations for any of these products or SYR-472.
Compounds in Clinical Development
MuDelta (eluxadoline) for diarrhea-predominant irritable bowel syndrome
Diarrhea-predominant irritable bowel syndrome affects approximately 28 million patients in the United States and the five major European Union countries, and is characterized by chronic abdominal pain and frequent diarrhea. Studies have demonstrated that IBS-d is associated with work absenteeism, high medical costs and low quality of life. We believe the market for prescription treatments for IBS-d is underserved due to the limited number of available treatments and the adverse side effects associated with those treatments.
MuDelta is a novel, orally active, investigational agent in Phase III development, with combined mu opioid receptor agonist and delta opioid receptor antagonist activity. The compound's dual opioid activity is designed to treat diarrhea and pain symptoms of IBS-d, without causing the constipating side effects that occur with mu opioid agonists. MuDelta acts locally in the gut and has very low oral bioavailability, thus limiting the potential for systemic side effects, such as sedation. In January 2011, the FDA granted Fast Track designation to the MuDelta IBS-d program. Fast Track is a process for facilitating the development and expediting the review of drugs to treat serious diseases and fill unmet medical needs, with the goal of bringing important new drugs to patients earlier.
In 2011, we completed a large multicenter randomized-double-blind Phase II Proof-of-Concept trial in patients with IBS-d, which demonstrated that MuDelta has a favorable efficacy and safety profile. Key findings from the Phase II Proof-of-Concept study results are summarized below:
The study reached statistical significance for the primary endpoint of improvement in stool consistency and abdominal pain at week four of treatment, which was developed prior to the release of the FDA's IBS guidance in 2010, as well as secondary endpoints of adequate relief of IBS-d symptoms at weeks 4, 8 and 12. Importantly, the favorable efficacy results were supported by a post hoc responder analysis, using the composite endpoint of improvement in daily pain and diarrheal symptoms over 12 weeks, consistent with the FDA 2012 final guidance, MuDelta showed statistically and clinically meaningful differences compared with placebo at both the 100 mg BID and 200 mg BID doses.
At our End of Phase II Meeting and also through written communications, the FDA agreed that the aforementioned endpoint is an acceptable primary endpoint for Phase III pivotal studies; we believe this provides a clear regulatory path for the program. We also believe that our favorable Phase II study results with this endpoint bodes well for the Phase III program.
In June 2012, we began two Phase III pivotal studies in parallel. Both trials have the same overall design and efficacy endpoints, but differ in overall duration due to the need to treat and monitor safety in a sufficient number of patients to satisfy regulatory requirements. Each study captures both the 12-week FDA endpoint as well as longer term efficacy data for up to 30 weeks, the latter of which might be used to support a European regulatory submission. Each study will have three treatment arms, placebo, 75 mg twice a day and 100 mg twice a day, with approximately 375 patients per arm. Our total enrollment target is 2,250 patients.
In November 2011, we acquired full exclusive license rights to develop and commercialize MuDelta under our existing development and license agreement with Janssen. We acquired these rights as a result of Janssen's decision not to exercise its option under the agreement to continue development of MuDelta. Based on our existing agreement, we will continue developing and commercializing the compound and Janssen may receive up to $45.0 million in regulatory milestone payments and, if approved for marketing, up to $75.0 million in sales-based milestone payments and sales-based royalties increasing from the mid- to upper-single digit percentages as sales volume increases. Royalties are to be paid for a period of ten years after the first commercial sale or, if later, the expiration of the last valid patent claim or the expiration of patent exclusivity.
Recent events: As of February 2013, we had recruited over 55% of patients needed to complete the Phase III trials. We have more than 650 study sites in North America and more than 50 study sites in the United Kingdom. We are actively exploring both partnering and funding opportunities to support the Phase III development of MuDelta.
JNJ-Q2 (avarofloxacin) for skin and lung infections
Community-acquired bacterial pneumonia and acute bacterial skin and skin structure infections are important public-health concerns due to increasing drug resistance of established antibiotics to causative pathogens. Due to the emerging resistance to established antibiotics, there is a large unmet need for antibiotics such as JNJ-Q2 that cover a broad range of pathogens, including resistant Staphylococcus (“Staph”) and Streptococcus (“Strep”), and that have the potential for both intravenous and oral use. Bacterial infections are a major cause of morbidity and mortality. Global microbiological surveillance suggests that approximately 40% of Staph infections in the U.S., Latin America and Asia Pacific are methicillin-resistant Staphylococcus “Staph” aureus, or MRSA. According to Global Data, the global MRSA market was valued at $900 million in 2010 and is projected to exceed $1.0 billion by 2017. The pneumonia therapeutics market was valued at $2.0 billion in 2010 with $1.8 billion value forecast for 2018 due to expected patent expirations.
JNJ-Q2 is a novel broad-spectrum fluoroquinolone antibiotic that is Phase III-ready for two indications: ABSSSI and CABP. JNJ-Q2 has a low propensity to cause drug-resistance in vitro and has potent activity against two important drug-resistant pathogens: MRSA and drug-resistant Streptococcus pneumonia. In addition, JNJ-Q2 is highly active against other common and difficult to treat bacteria, including those that are gram-positive, atypical and anaerobic, as well as some gram negative bacteria. This broad bactericidal spectrum gives JNJ-Q2 an advantage over many other antibiotics, which do not reliably treat polymicrobial skin and wound infections or such a wide variety of respiratory pathogens. JNJ-Q2 is active against resistant pathogens that might be used in bioterrorism and also against drug-resistant gonorrhea. It can be dosed both intravenously and orally, differentiating it from many other MRSA treatments that are only dosed intravenously.
In April 2011, we acquired full exclusive license rights to develop and commercialize JNJ-Q2 under our existing development and license agreement with Janssen. We acquired rights to JNJ-Q2 as a result of Janssen's decision not to exercise its option under the agreement, which gave Janssen the opportunity to continue development of JNJ-Q2. This decision was related to Janssen's 2011 announcement that it would not be investing in new antibacterial therapies. Based on our existing agreement, Janssen may receive up to $50.0 million in regulatory milestone payments, and if approved for marketing, up to $75.0 million in sales-based milestone payments and sales-based royalties increasing from the mid- to upper-single digit percentages as sales volume increases. Royalties would be paid for a period of ten years after the first commercial sale or, if later, the expiration of the last valid patent claim or the expiration of patent exclusivity.
We are continuing to seek to partner or out-license JNJ-Q2; however, the partnering environment for antibacterial agents remains challenging. We have forecasted minimal expenditures for this compound in 2013.
Recent events: In February 2013, JNJ-Q2 was granted a Qualified Infectious Disease Product and Fast Track designation from the FDA. These designations should enable us and/or any future collaborator with respect to the compound to benefit from certain incentives for the development of new antibiotics, including priority review and additional five-year market exclusivity, as provided under the Generating Antibiotic Incentives Now (GAIN) Act, which is incorporated within the FDA Safety and Innovation Act of 2012.
Marketed Products
Nesina ® (alogliptin) for Type-2 diabetes
Type-2 diabetes is the most common form of diabetes and has reached worldwide epidemic proportions. The global health care expenditures to treat and prevent diabetes and its complications were estimated at $471.0 billion in 2012. By 2030, this number is projected to exceed $595.0 billion. In addition to diet and exercise, diabetic patients often require multiple medicines to help manage their condition.
Nesina, which is marketed by Takeda, is the trade name for alogliptin. Nesina is a highly selective orally-active dipeptidyl peptidase-4, or DPP-4, inhibitor that slows the inactivation of hormones known as incretins (glucagon-like peptide-1 and glucose-dependent insulinotropic peptide), which play a major role in regulating blood sugar levels and might improve pancreatic function. Approximately 8,500 patients with Type-2 diabetes have been treated with Nesina in 14 randomized, double-blind, controlled clinical trials. Pivotal trials demonstrated that Nesina was well-tolerated when given as a single daily dose and it significantly improved glycemic control in Type-2 diabetes patients without raising the incidence of hypoglycemia. Additionally, Nesina has been shown to enhance glycemic control when used in combination with other commonly prescribed diabetes drugs. Nesina is approved for combination use in Japan with a variety of other diabetes drugs including glucosidase inhibitors, sulfonylureas, biguanides, and Actos, and is also marketed in Japan as a fixed-dose combination with Actos, known as Liovel. Takeda is conducting a prospective study of alogliptin known as the “EXAMINE” trial, which is a cardiovascular outcomes study of subjects with Type-2 diabetes and a history of acute coronary syndrome.
We have no further financial obligations under our agreement with Takeda. Under this agreement, we will be entitled to receive up to $10.0 million in future regulatory milestone payments for E.U. marketing authorization, and up to $33.0 million in sales-based milestone payments. In addition, we are entitled to receive payments on worldwide sales of Nesina based on royalty rates of 7% to 12% in the U.S., 4% to 8% in Europe and Japan and 3% to 7% in regions other than the U.S., Europe or Japan. Royalty payments are subject to a reduction of up to 0.5% for a portion of payments by Takeda to a licensor for intellectual property related to Nesina (except in Japan, where there is currently no intellectual property that would cause such a reduction).
Royalties are to be paid for the later of ten years following the first commercial sale or two years following the expiration of the last to expire patent. For combination product sales outside the U.S., Takeda must pay us royalties based on the proportion of Nesina's average sales price compared to that of the combination product. For sales in the U.S, the royalties for combination product sales are based on an agreed-upon percent of product sales.
Recent events: On January 25, 2013, Takeda confirmed the approval from the FDA of three new Type-2 diabetes therapies, Nesina (alogliptin) and the fixed-dose combination therapies, Oseni (alogliptin and pioglitazone) and Kazano (alogliptin and metformin), for the treatment of Type-2 diabetes. Oseni is the first DPP4 inhibitor and thiazolidinedione combination product approved in the U.S. These approvals triggered a $25.0 million milestone payment to us, which we received on February 27, 2013, as well as potential royalties on sales in the United States and potential sales-based milestones. The E.U. marketing application, which Takeda submitted in the second quarter of 2012, continues to progress, and Takeda expects a regulatory decision about marketing approval in the latter half of 2013. Additional marketing applications are under review in the emerging markets.
Priligy ® (dapoxetine) for premature ejaculation
The reported percentage of men affected with premature ejaculation, or PE, at some point during their lives ranges from 4% to 30%, depending on the methodology and criteria used. Priligy is the trade name for dapoxetine, a drug in tablet form specifically indicated for the “on-demand” treatment of PE.
Priligy is a unique, short-acting, selective serotonin reuptake inhibitor, or SSRI, designed to be taken only when needed, one to three hours before sexual intercourse, rather than every day. Priligy has been studied in over 12,000 patients in clinical trials, including five randomized, placebo-controlled Phase III clinical trials involving more than 6,000 men with PE. It is the first oral medication to be approved for this condition. Priligy is approved in over 55 countries and currently marketed in 15 countries in Europe, Asia-Pacific and Latin America.
In May 2012, we entered into an agreement whereby Alza and Janssen transferred worldwide Priligy rights to us. In May 2012, we also entered into a license agreement with Menarini to commercialize Priligy in Europe, most of Asia, Africa, Latin America and the Middle East. We will retain full development and commercialization rights in the United States, Japan and Canada. Janssen will continue to make Priligy available to patients, until marketing authorizations are transferred to Menarini. Janssen will also manufacture and manage certain clinical and regulatory activities for Priligy for pre-defined periods. Under our license agreement with Menarini, we received a $15.0 million upfront payment and $10.0 million of regulatory milestone payments, and are eligible to receive up to $10.0 million in launch milestones and up to $40.0 million in sales-based milestones, plus tiered royalties ranging from the mid-teens to mid-twenties in percentage terms. We are obligated to pay $15.0 million to Janssen for transition services. As of December 31, 2012, we had paid $11.25 million related to these transition services and will owe the remaining $3.75 million in the first quarter of 2013. We must also pay Janssen fees for the product sales and distribution activities that they will perform as part of a sales service agreement. In addition, we may incur up to $1.0 million for reasonable out-of-pocket expenses over the transition period. Menarini is obligated to pay for the current on-going clinical studies. We now hold the U.S. IND for Priligy.
At this time, we do not have plans to re-submit the NDA that Janssen filed in 2004, as our primary development focus is our MuDelta program. Furthermore, the FDA has not provided guidance on patient-reported outcomes that it considers acceptable for studies of premature ejaculation. We will continue to monitor the situation and might reassess our strategy should circumstances change.
We originally acquired patents for Priligy from Eli Lilly and Company, or Lilly, and are obligated to pay Lilly a royalty of 5% on annual sales in excess of $800.0 million. In addition, under the terms of the license agreement with Menarini, we remain responsible for payment of royalties to Lilly, except Menarini will pay the portion of the royalties owed to Lilly in each country where Menarini is licensed to sell Priligy, where Lilly is eligible for payments, and where we are no longer eligible for payments from Menarini.
Recent Events: The transition of Priligy is progressing as planned; country-specific marketing authorization licenses are continuing to transfer from Janssen to Menarini, and Menarini is now marketing Priligy in some countries. We anticipate that in 2013, Menarini should be able to launch or re-launch Priligy in key commercial markets.
Compound in Clinical Development by Collaborator
Trelagliptin for Type-2 diabetes
Trelagliptin (SYR-472) is part of the DPP-4 inhibitor portfolio that Takeda purchased from PPD and Syrrx in 2005. Trelagliptin has the same mechanism of action as alogliptin. However, in contrast to alogliptin, which is a once-daily oral therapy, trelagliptin is a once-weekly oral agent. Takeda completed two Phase II studies in early 2008, the primary endpoint of which was HbA (1C) at 12 weeks. In September 2011, Takeda commenced Phase III clinical trials for trelagliptin in Japan for treatment of Type-2 diabetes. Takeda is currently conducting: (1) a 52-week open-label study in Japan in approximately 600 subjects who are not well-controlled on diet and exercise or another oral anti-diabetic drug; and (2) a 24-week double-blind study of trelagliptin compared to placebo or alogliptin. Currently, all available DPP-4 inhibitors are dosed once-daily. A once-weekly treatment, such as trelagliptin, should provide patients with a convenient therapeutic alternative, and has the potential to improve treatment compliance. If trelagliptin is approved, then we would be eligible to receive sales-based royalty payments at the same rates as for Nesina, as described above. Under our agreement with Takeda, we would be entitled to receive regulatory milestone payments for trelagliptin or Nesina, whichever compound achieves the milestone(s) first. We would also be entitled to receive sales-based milestone payments based on global product sales.
Our Drug Development Capabilities
The drug development capabilities of our executive officer team embodies over 50 years of research and development experience. This experience includes a deep understanding of the biological causes of human diseases and the factors that impact all aspects of successful drug development such as manufacturing, formulation, the cause of drug side effects, drug interactions and drug pharmacokinetics. We believe that our drug development capability and proven success rate will continue to provide a pipeline of unique compounds. Depending upon the availability of our development resources, other drug candidates might be added to our own internal clinical pipeline, or out-licensed to other companies for clinical development and commercialization.
Our Patents and Other Proprietary Rights
Patents and other proprietary rights are important to our business. It is our policy to seek patent protection for our assets, and also to rely upon trade secrets, know-how and licensing opportunities to develop and maintain our competitive position.
We own or have exclusively licensed 11 issued U.S. patents and have approximately 221 U.S. and non-U.S. pending patent applications. We have a policy to seek worldwide patent protection for our products and have foreign patent rights corresponding to most of our U.S. patents.
We license the rights to the following material patents related to our product candidates:
| | • | | MuDelta. Licensed from Janssen. The license continues as long as we meet our obligations to Janssen and we have marketing rights to the compound. As of December 31, 2012, 64 U.S. and foreign patents have been issued to Janssen in this patent family. Additional U.S. and foreign patent applications are still pending. |
| | • | | JNJ-Q2. Licensed from Janssen. The license continues as long as we meet our obligations to Janssen and we have marketing rights to the compound. As of December 31, 2012, 60 U.S. and foreign patents have been issued to Janssen in this patent family. Additional U.S. and foreign patent applications are still pending. |
Pursuant to the terms of the Uruguay Round Agreements Act, patents issued from applications filed on or after June 8, 1995, have a term of 20 years from the date of filing, no matter how long it takes for the patent to issue. Because patent applications in the pharmaceutical industry often take a long time to issue, this method of patent term calculation can result in a shorter period of patent protection afforded to us compared to the prior method of term calculation, which was 17 years from the date of issue. Our issued U.S. patents expire between 2023 and 2029, excluding any potential patent term extension available under U.S. federal law. We actively seek full patent term adjustment following allowance of a patent. We also actively seek patent term extensions following marketing approval. Under the Drug Price Competition and Patent Term Restoration Act of 1984 and the Generic Animal Drug and Patent Term Restoration Act of 1988, a patent that claims a product, use or method of manufacture covering drugs may be extended for up to five years to compensate the patent holder for a portion of the time required for FDA review.
While we file and prosecute patent applications to protect our inventions, our pending patent applications might not result in the issuance of patents or our issued patents might not provide competitive advantages. Also, our patent protection might not prevent others from developing competitive products using related or other technology.
In addition to seeking the protection of patents and licenses, we also rely upon trade secrets, know-how and continuing technological innovation, which we seek to protect, in part, by confidentiality agreements with employees, consultants, suppliers and licensees.
The scope, enforceability and effective term of issued patents can be highly uncertain and often involve complex legal and factual questions. No consistent policy has emerged regarding the breadth of claims in pharmaceutical patents, so that even issued patents might later be modified or revoked by the relevant patent authorities or courts. Moreover, the issuance of a patent in one country does not assure the issuance of a patent with similar claim scope in another country, and claim interpretation and infringement laws vary among countries, so we are unable to predict the extent of patent protection in any country. The patents we obtain and the unpatented proprietary technology we hold might not afford us significant commercial protection. Additional information regarding risks associated with our patents and other proprietary rights that affect our business is contained under the headings “We must protect our patent and other intellectual property rights to succeed” and “We might need to obtain patent licenses from others in order to manufacture or sell our potential products and we might not be able to obtain these licenses on terms acceptable to us or at all” under the heading “Risk Factors”.
Manufacturing and Supply
We currently rely on our collaborators and contract manufacturers to produce drug substances and drug products required for our clinical trials under current good manufacturing practices, with oversight by our internal managers.
We plan to continue to rely upon contract manufacturers and collaboration partners to manufacture commercial quantities of our drug candidates if and when approved for marketing by the applicable regulatory agency. We generally rely on one manufacturer for the active pharmaceutical ingredient and another manufacturer for the formulated drug product for each of our drug candidate programs. At the early stage of clinical studies, we do not believe that we are substantially dependent on any supplier, or that additional manufacturers would be beneficial due the possibility of changes in the method of manufacturing of the drug candidate. As a drug candidate moves to later stages of development and the drug formulation method is established, we, or our collaboration partners, might then seek additional manufacturers for the drug.
Sales and Marketing
We currently have no marketing, sales or distribution capabilities. We plan to rely on third party collaborators to market our products, like Alza and Menarini for Priligy and Takeda for Nesina and related products, and therefore we are subject to the strategic marketing decisions of such third parties. We generally plan to out-license our commercial rights in a territory to a third party with marketing, sales and distribution capabilities in exchange for one or more of the following: up-front payments; research funding; development funding; milestone payments; and royalties on drug sales. In some instances, and at some point in the future, we might choose to develop our own staff for marketing, sales or distribution.
Government Regulation
The manufacturing, testing, labeling, approval and storage of our products are subject to rigorous regulation by numerous governmental authorities in the United States and other countries at the federal, state and local level, including the FDA. The process of obtaining approval for a new pharmaceutical product or for additional therapeutic indications within this regulatory framework requires expenditure of substantial resources and usually takes several years. Companies in the pharmaceutical and biotechnology industries, including us, have suffered significant setbacks in various stages of clinical trials, even in advanced clinical trials after promising results had been obtained in earlier trials.
The process for obtaining FDA approval of drug candidates customarily begins with the filing of an IND with the FDA for the use of a drug candidate to treat a particular indication. If the FDA accepts the IND, we would then start human clinical trials to determine, among other things, the proper dose, safety and efficacy of the drug candidate in the stated indication. The clinical trial process is customarily divided into three phases—Phase I, Phase II and Phase III. Each successive phase is generally larger and more time-consuming and expensive than the preceding phase. Throughout each phase we are subject to extensive regulation and oversight by the FDA. Even after a drug is approved and being marketed for commercial use, the FDA may require that we conduct additional trials, including Phase IV trials, to further study safety or efficacy.
As part of the regulatory approval process, we, or our collaboration partners, must demonstrate to the FDA the ability to manufacture a pharmaceutical product before we receive marketing approval. Our products must conform to rigorous standards regarding manufacturing and quality control procedures in order to receive FDA approval. The validation of these procedures is a costly endeavor. Pharmaceutical manufacturers are subject to inspections by the FDA and local authorities as well as inspections by authorities of other countries. To supply pharmaceutical products for use in the United States, foreign manufacturers must comply with these FDA-approved guidelines. These foreign manufacturers are also subject to periodic inspection by the FDA or by corresponding regulatory agencies in these countries under reciprocal agreements with the FDA. Moreover, state, local and other authorities may also regulate pharmaceutical product manufacturing facilities. Before we are able to manufacture commercial products, we, or our collaboration partners, as the case may be, must meet FDA guidelines.
Both before and after marketing approval is obtained, a pharmaceutical product, its manufacturer and the holder of the Biologics License Application, or BLA, or NDA for the pharmaceutical product are subject to comprehensive regulatory oversight. The FDA may deny approval to a BLA or NDA if applicable regulatory criteria are not satisfied. Moreover, even if regulatory approval is granted, such approval may be subject to limitations on the indicated uses for which a product may be marketed. Further, marketing approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems with the pharmaceutical product occur following approval. In addition, under a BLA or NDA, the manufacturer of the product continues to be subject to facility inspections and the applicant must assume responsibility for compliance with applicable pharmaceutical product and establishment standards. Violations of regulatory requirements at any stage may result in various adverse consequences, which may include, among other adverse actions, withdrawal of the previously approved pharmaceutical product or marketing approvals or the imposition of criminal penalties against the manufacturer or BLA or NDA holder.
For the development of pharmaceutical products outside the United States, we and our collaborator partners are subject to foreign regulatory requirements, and the ability to market a drug is contingent upon receiving marketing authorizations from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. In countries other than European Union countries, foreign marketing authorizations are applied for at a national level. Within the European Union, procedures are available to companies wishing to market a product in more than one European Union member state. Clinical trial applications must be filed with the relevant regulatory authority in each country in which we would want to conduct a clinical trial. Assuming approval and the success of any clinical trial, we would then need to seek marketing approval for the drug. The process for obtaining marketing approval of drug candidates in the European Union begins with the filing with the European Medicines Agency, or EMA, of a Marketing Authorization Application for the use of a drug candidate to treat a particular indication. Similar processes and outcomes of such human clinical trials that are required by the FDA are also required by the EMA including testing for dose, safety and efficacy in three phases.
Similar to the FDA, the European regulators subject us to extensive regulation and oversight throughout each phase. Even after a drug is approved and being marketed for commercial use, the EMA may require that additional trials be conducted, including Phase IV trials, to further study safety or efficacy. As a result, the EMA regulatory approval process includes all of the risks associated with FDA approval set forth above.
If and when necessary, we, or our collaboration partners, will choose the appropriate route of European or other international regulatory filing to accomplish the most rapid regulatory approvals. Requirements relating to manufacturing, conduct of clinical trials and product licensing vary widely in different countries, and the chosen regulatory strategy might not secure regulatory approvals or approvals of our chosen product indications. In addition, if a particular product to be used outside of the United States is manufactured in the United States, FDA requirements and U.S. export provisions will apply.
Outside of the United States, many countries require that a pricing approval be obtained in addition to regulatory approval prior to launching the product in the approving country. We, or our collaboration partners, might encounter difficulties or unanticipated costs or price controls in our respective efforts to secure necessary governmental approvals. Failure to obtain pricing approval in a timely manner or approval of pricing which would support an adequate return on investment or generate a sufficient margin to justify the economic risk might delay or prohibit the commercial launch of the product in those countries.
The marketing and sale of approved pharmaceutical products is subject to strict regulation. Promotional materials and activities must comply with the approving agency’s regulations and other guidelines. Physicians may prescribe pharmaceutical or biologic products for uses that are not described in a product’s labeling or differ from those approved by the approving agency. While such “off-label” uses are common and regulatory agencies do not regulate physicians’ choice of treatments, many approving agencies restrict a company’s communications on the subject of “off-label” use. Companies cannot promote approved pharmaceutical or biologic products for off-label uses. If any advertising or promotional activities we, or our collaboration partners, undertake fail to comply with applicable regulations or guidelines regarding “off-label” use, we, or our collaboration partners, may be subject to warnings or enforcement action.
Competition
The pharmaceutical industry is highly competitive. Many of our competitors are global companies with substantially greater resources than we have to develop and commercialize their drugs and drug candidates. Potential competitors have developed and are developing compounds for treating the same indications as our product candidates. In addition, a number of academic and commercial organizations are actively pursuing similar technologies and several companies have developed or might develop technologies that might compete with our compounds.
Priligy, indicated for premature ejaculation, competes with Cromadyn, a generic paroxetine sold by More Pharmaceuticals in Mexico. Additional competition includes “off-label” treatment with chronically dosed SSRIs (e.g. paroxetine, fluoxetine). We are aware of two other compounds in development for premature ejaculation: (1) PD502 (Phase III) a novel formulation of lidocaine and prilocaine being developed by Shionogi that is administered topically; and (2) Zertane (Phase III) a re-purposed formulation of tramadol (a centrally acting oral opioid mu receptor agonist with serotonin and norepinephrine reuptake inhibitory activities) being developed by Ampio Pharma.
Nesina competes in the Type-2 diabetes space with three DPP-4 inhibitors currently on the market, Bristol-Myers Squibb/AstraZeneca’s Onglyza® (saxagliptin), Boehringer Ingelheim/Lilly’s Tradjenta™ (linagliptin) and Merck’s Januvia® (sitagliptin). Merck also markets Janumet® and Janumet XR®, fixed-dose combinations of sitagliptin and metformin; Boehringer Ingelheim/Lilly also market Jentadueto™, a combination of linagliptin and metformin and Bristol-Myers Squibb/AstraZeneca also market Kombiglyze®, a combination of saxagliptin and extended release metformin.
In Japan, there are two additional DPP-4 inhibitors on the market, Mitsubishi Tanabe/Daiichi-Sankyo’s Tenelia® (tenagliptin) and Sanwa/Kowa’s Suiny® (anagliptin). Novartis markets the DPP-4 inhibitor Galvus® (vildagliptin) and Eucreas® (vildagliptin and metformin) in Europe. Other marketed oral anti-diabetic competitors include generic metformin, generic sulfonylureas and generic thiazolidinediones.
The diabetes pipeline is crowded, with, to our knowledge, over 100 compounds in Phase I development, approximately 90 in Phase II development and approximately 60 in Phase III development or preregistration. In addition to DPP-4 inhibitors, competitors are also developing GLP-1 agonists, SGLT-2 antagonists, PPAR agonists and compounds with other mechanisms for treatment of diabetes. Other companies with DPP-4 inhibitors in clinical development of which we are aware include Amgen/Servier, Arisaph Pharmaceuticals, Dong-A Pharmaceuticals (South Korea), Dainippon Sumitomo Pharma, Phenomix, Glenmark Pharmaceuticals, Kyorin Pharmaceuticals and LG Life Sciences (South Korea).
If approved, JNJ-Q2 will compete with other fluoroquinolones currently on the market, including Johnson and Johnson’s Levaquin® (levofloxacin), Bayer/Merck’s Avelox® (moxifloxacin), Bayer/Merck’s Cipro® (ciprofloxacin) and Cornerstone Therapeutics’s Factive® (gemifloxacin). Generic versions of ciprofloxacin and levofloxacin are currently available, and generic versions of moxifloxacin will likely become available when the patents covering these products expire in 2014. If JNJ-Q2 is found to be effective against MRSA infections, it would compete with Pfizer’s Zyvox® (linezolid), Cubist’s Cubicin (daptomycin), Wyeth’s Tygacil (tigecycline), Theravance’s Vibativ® (telavancin), Forest’s Teflaro™ (ceftaroline) and the generic drug vancomycin.
Companies developing compounds to treat MRSA infections in clinical trials include Baselia, Nabriva, Trius, Paratek, Cempra, Durata, Affinium, e-Therapeutics, FAB Pharma, Medicines Company, Novexel (now AstraZeneca), Phico Therapeutics, PolyMedix, Rib-X Pharmaceuticals, TaiGen, Theravance and Wockhardt (India). The Rib-X and Wockhardt compounds are both fluoroquinolones. In addition, MerLion Pharmaceuticals is developing a fluoroquinolone in Phase II. Merck and Biota Pharmaceuticals are both developing vaccines against staphylococcus aureus.
If approved, MuDelta will compete with Lotronex® (alosetron), marketed by Prometheus Laboratories. The following generic opiates and/or antispasmodic agents are also used for diarrhea predominant IBS: loperamide (Imodium®, an over the counter anti-diarrheal), diphenoxylate/atropine (an opiate/anticholinergic agent) and dicyclomine. In the Asia Pacific region, ramosetron (a 5HT3 antagonist) is marketed under various brand names, as a treatment for diarrhea-predominant IBS. The pipeline for diarrhea-predominant IBS includes: asimadoline, which is being developed by Tioga Pharmaceuticals and is in Phase III; rifaximin, an antibiotic approved product for hepatic encephalopathy that is the subject of a supplemental NDA by Salix Pharmaceuticals; AST-120, currently in Phase II development by Ocera; ibodutant (NK 2 antagonist), currently in Phase II development by Menarini Group; dextofisopam, currently in Phase II development by Pharmos Corporation; and LX1031 (a serotonin synthesis inhibitor), currently in Phase II development by Lexicon Pharmaceuticals.
Competitors might succeed in more rapidly developing and marketing technologies and products that are more effective than our products or that would render our products or technology obsolete or noncompetitive. Our collaborators might also independently develop products that are competitive with products that we have licensed to them. Any product that we or our collaborators succeed in developing and for which regulatory approval is obtained must then compete for market acceptance and market share. The relative speed with which we and our collaborators can develop products, complete clinical testing and approval processes, and supply commercial quantities of the products to the market compared to competitive companies will affect market success. In addition, the amount of marketing and sales resources, and the effectiveness of the marketing used with respect to a product will affect its success. In addition, some clinical research organizations, or CRO, service providers and private equity funds are developing risk sharing models to finance the pharmaceutical industry’s pipeline. NovaQuest is active in this business. As these types of business models evolve, there will be increasing competition for compounds and funds that will affect our ability to add to our portfolio.
Other competitive factors affecting our business generally include:
| | • | | product efficacy and safety; |
| | • | | timing and scope of regulatory approval; |
| | • | | product availability, marketing and sales capabilities; |
| | • | | reimbursement coverage; |
| | • | | the amount of clinical benefit of our product candidates relative to their cost; |
| | • | | method of and frequency of administration of any of our product candidates which may be commercialized; |
| | • | | patent protection of our product candidates; |
| | • | | the capabilities of our collaborators; and |
| | • | | the ability to hire qualified personnel. |
Employees
We have 24 full-time employees, a majority of whom are engaged in research and development activities. Our success depends in large part on our ability to attract and retain skilled and experienced employees. None of our employees are covered by a collective bargaining agreement. We consider our relations with our employees to be good.
Item 1A. Risk Factors
Our business operations face a number of risks. These risks should be read and considered with other information provided in this report.
Risks Relating to Furiex’s Business
We anticipate that we will incur additional losses. We might never achieve or sustain profitability. If additional capital is not available, we might have to curtail or cease operations.
Our business has experienced significant net losses. We had net losses of $54.7 million, $49.0 million and $42.9 million in 2010, 2011 and 2012, respectively. We will continue to incur additional net losses until revenues from all sources reach a level sufficient to support our on-going operations. Because we or our collaborators or licensees might not successfully develop additional products, obtain required regulatory approvals, manufacture products at an acceptable cost or with appropriate quality, or successfully market products with desired margins, our expenses might continue to exceed any revenues we receive. Our commitment of resources to the continued development of our products might require significant additional funds for development. Our operating expenses also might increase if we:
| | • | | move our earlier stage potential products into later stage clinical development, as we have done with MuDelta; |
| | • | | encounter problems during clinical development that require a change in scope and/or timelines; |
| | • | | pursue clinical development of our potential products in new indications; |
| | • | | increase the number of patents we are prosecuting or otherwise expend additional resources on patent prosecution or defense; or |
| | • | | invest in or acquire additional technologies, product candidates or businesses. |
In the absence of substantial licensing, milestone and other revenues from third-party collaborators, royalties on sales of products under our agreements with collaborators, future revenues from our products in development or other sources of revenues, we will continue to incur operating losses and might require additional capital to fully execute our business strategy. The likelihood of reaching, and time required to reach, sustained profitability are highly uncertain.
Although we expect that we will have sufficient cash to fund our operations and working capital requirements for at least the next 12 months based on current operating plans, we might need to raise additional capital in the future to:
| | • | | fund our research and development programs; |
| | • | | acquire complementary businesses or technologies; |
| | • | | respond to competitive pressures; or |
| | • | | commercialize our product candidates. |
Our future capital needs depend on many factors, including:
| | • | | the scope, duration and expenditures associated with our research and development programs; |
| | • | | continued progress in these programs; |
| | • | | the outcome of potential licensing transactions, if any; |
| | • | | competing technological developments; |
| | • | | the regulatory approval process for our product candidates; and |
| | • | | the cost of attracting and retaining employees. |
We might seek to raise additional funds through public or private equity offerings, debt financings or additional collaborations and licensing arrangements. We might not be able to obtain additional financing on terms favorable to us, if at all. General market conditions might make it difficult for us to seek financing from the capital markets. We might have to relinquish rights to our technologies or product candidates, or grant licenses on terms that are not optimal to us, in order to raise additional funds. If adequate funds are not available, we might have to delay, reduce or eliminate one or more of our research or development programs and reduce overhead expenses, or restructure or cease operations. These actions might reduce the market price of our common stock.
Our near-term revenue is largely dependent on the success of Nesina and Priligy and we cannot be certain of the extent that our collaborators will be able to obtain regulatory approval for or obtain commercial success of drugs.
We currently are relying on Nesina and related products, and Priligy to generate revenue. While Priligy is approved for marketing outside of the U.S., it has not been approved in the U.S. and the FDA issued a “not approvable” letter with respect to the compound in October 2005. We currently do not have plans to re-submit the NDA. While Nesina is approved for marketing in Japan and was approved for marketing in January 2013 for the U.S., Takeda, our collaborative partner, continues to seek approval in Europe. We have also invested a significant amount of time and financial resources in the development of MuDelta. Our future success might depend in part on our, or our collaborator’s, ability to successfully complete Phase III clinical trials for MuDelta, which began in June 2012. We have also invested a significant amount of time and financial resources in the development of JNJ-Q2. FDA guidance for developing drugs to treat community-acquired bacterial pneumonia includes challenging requirements for the drug developer. Our future success might depend in part on our collaborator’s ability to successfully complete the Phase III trial for this pneumonia indication using JNJ-Q2 in view of the FDA guidelines. We anticipate that our success will depend largely on the receipt of regulatory approval and successful commercialization of these drug candidates. The future success of these drug candidates will depend on several factors, including the following:
| | • | | our ability to provide acceptable evidence of their safety and efficacy; |
| | • | | receipt of marketing approval from the FDA and any similar foreign regulatory authorities; |
| | • | | our or our partners ability to obtain and maintain commercial manufacturing arrangements with third-party manufacturers or establishing commercial-scale manufacturing capabilities; |
| | • | | collaborating with pharmaceutical companies or contract sales organizations to further develop, market and sell any approved drug; and |
| | • | | acceptance of any approved drug in the medical community and by patients and third-party payors. |
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will be able to continue generating revenues through the sale of Priligy, Nesina and related products or generate any revenue from the sale of other product candidates.
Our ability to continue to develop and commercialize our late stage product candidates depends on our ability to find new collaborators.
Our ability to succeed in our drug development business by advancing our late stage product candidates through Phase III clinical trials will depend on our ability to successfully find collaborators able to fund and execute late-stage development and commercialization of our product candidates. We generally conduct our drug development business in two stages. During the first stage, we in-license a product candidate from a collaborator and develop that candidate through Phase II clinical trials. If the product candidate successfully completes Phase II testing, we would likely enter a second stage during which we seek a collaborator, which might or might not be the same collaborator as in the first stage, for the continued late stage development and ultimate commercialization of the product candidate. Janssen, our original collaborator for MuDelta and JNJ-Q2, has elected not to continue development of these two product candidates. If we cannot find a collaborator for final development and commercialization, we might not be able to complete the development and commercialization on our own due to the significant costs associated with these activities. As a result, we may not be able to recoup all or any part of our investment in the product candidate.
Our milestone and royalty payments from collaborators and the successful development and marketing of our product candidates depends on our collaborators continuing to develop and commercialize the product candidates. If our collaborators are not successful or choose not to develop these compounds, we might not receive future payment.
The drug development industry is under increasing economic pressure. The third parties with which we collaborate might not perform their obligations as expected or they might breach or terminate their agreements with us or otherwise fail to conduct their collaborative activities successfully or in a timely manner. Further, parties collaborating with us might not devote sufficient resources to the development, manufacture, regulatory strategy and approvals, marketing or sale of these product candidates. If the parties to our collaborative agreements do not fulfill their obligations, elect not to develop a candidate or fail to devote sufficient resources to it, our business could be materially and adversely affected. In these circumstances, our ability to further develop potential products could be severely limited. An example of the way we rely on our collaborators is continued cooperation from Alza and Janssen under our May 2012 agreement to transfer the existing regulatory licenses and license applications as set forth in the agreement.
We have agreements under which we rely on collaborators to manufacture our product candidates and essential components for those product candidates, design and conduct clinical trials, compile and analyze the data received from these trials, obtain regulatory approvals and, if approved, market these products. As a result, we might have limited or no control over the manufacturing, development and marketing of these potential products. In addition, the performance of our collaborators might not be sufficient or appropriate for regulatory review and approval for our product candidates. Further, we often rely on one manufacturer or other collaborator for such services, the loss of which could significantly delay the development of any of our product candidates. Our milestone and royalty payments rely on the performance of our collaborators and would be impacted by any delay or termination by our collaborators.
Our collaborators can terminate our collaborative agreements under certain conditions. A collaborator may terminate its agreement with us or separately pursue alternative products, therapeutic approaches or technologies as a means of developing treatments for the diseases targeted by us, or our collaborative effort. Even if a collaborator continues to contribute to the arrangement, it might nevertheless decide not to actively pursue the development or commercialization of any resulting products. In these circumstances, our ability to further develop potential products could be severely limited. While we generally seek non-compete terms in our agreements with our collaborators for the products we are developing, the enforcement of a non-compete can be expensive and difficult to monitor and enforce and might be subject to being invalidated by a court or judge.
Continued funding and participation by collaborators will depend on the continued timely achievement of our research and development objectives, the retention of key personnel performing work under those agreements and on each collaborator’s own financial, competitive, marketing and strategic capabilities and priorities. These considerations include:
| | • | | the commitment of each collaborator’s management to the continued development of the licensed products or technology; |
| | • | | the relationships among the individuals responsible for the implementation and maintenance of the development efforts; and |
| | • | | the relative advantages of alternative products or technology being marketed or developed by each collaborator or by others, including their relative patent and proprietary technology positions, and their ability to manufacture potential products successfully. |
The willingness of our existing collaborators to continue development of our potential products and our ability to enter into new relationships depends upon, among other things, our patent position with respect to such products. If we are unable to successfully obtain and maintain patents, we might be unable to collect royalties on existing licensed products or enter into additional agreements.
If we are unable to enter into agreements with third parties to market and sell our drug candidates or are unable to establish our own sales and marketing capabilities, we might be unable to generate product revenue.
We do not currently have the resources to sell, market or distribute any pharmaceutical products. In order to market any of our products that receive regulatory approval, we must make arrangements with third parties to perform these services, or build our sales, marketing, managerial and other non-technical capabilities, which would be difficult, expensive and time-consuming. If we are unable to do so, we might not be able to generate product revenue and might not become profitable.
We might obtain future financing through the issuance of debt or equity or other forms of financing, which might have an adverse effect on our shareholders or otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity or other forms of financing, any debt securities or preferred stock issued will have rights, preferences and privileges senior to those of holders of our common stock in the event of liquidation. In such event, there is a possibility that once all senior claims are settled, there might be no assets remaining to pay out to the holders of our common stock. In addition, if we raise funds through the issuance of additional equity, whether through private placements or public offerings, such an issuance would dilute the ownership of our then current shareholders.
The terms of debt securities might also impose restrictions on our operations, which might include limiting our ability to incur additional indebtedness, to pay dividends on or repurchase our capital stock, or to make certain acquisitions or investments. For example, under our loan and security agreement with MidCap Funding III, LLC, MidCap Funding RE Holdings, LLC and Silicon Valley Bank, we are subject to various affirmative and negative covenants, including that we may not enter into a merger or consolidation or certain change of control events or incur additional indebtedness, in each case subject to certain customary exceptions. In addition, we might be subject to covenants requiring us to satisfy certain financial tests and ratios, and our ability to satisfy such covenants may be affected by events outside of our control.
Our operating expenses and results and any revenue likely will fluctuate in future periods.
Our revenues and expenses are unpredictable and likely will fluctuate from quarter to quarter due to, among other things, the timing and the unpredictable nature of clinical trials and related expenses, including payments owed by us and to us under collaborative agreements for reimbursement of expenses, future milestone revenues under collaborative agreements, royalties owed to us related to sales of Priligy and Nesina and related products and any royalty payments which may be owed by us under collaborative or other agreements. For example, under our agreement with Takeda for Nesina, we may be responsible for royalty payments in Japan up to 0.5%, subject to a quarterly cap, for payments made by Takeda to a licensor for patent(s), if granted, in Japan covering intellectual property related to Nesina. In addition, the recognition of clinical trial and other expenses that we otherwise would recognize over a period of time under applicable accounting principles might be accelerated or expanded in certain circumstances. In such a case, it might cause our expenses during that period to be higher than they otherwise would have been had the circumstances not occurred. For example, if we terminate a clinical trial for which we paid non-refundable upfront fees to a clinical research organization and in which we did not accrue all of the patient costs, the recognition of the expense associated with those fees that we were recognizing as we accrued patient costs would be accelerated and recognized in the period in which the termination occurred.
We are dependent on the performance of service providers.
We rely on service providers, such as contract manufacturers, clinical research organizations, medical institutions and clinical investigators, including physician sponsors, to conduct nearly all of our clinical trials, including recruiting and enrolling patients in the trials. In connection with the spin-off, we entered into a Master Development Services Agreement with PPD pursuant to which PPD provides us clinical development services at discounted rates on a preferred provider basis. If PPD or any of these other parties do not successfully carry out their contractual duties or meet expected deadlines, we might be delayed or may not obtain regulatory approval for or be able to commercialize our product candidates. If any of the third parties upon whom we rely to conduct our clinical trials do not comply with applicable laws, successfully carry out their obligations or meet expected deadlines, our clinical trials might be extended, delayed or terminated.
If the quality or accuracy of the clinical data obtained by third party contractors is compromised due to their failure to adhere to applicable laws or our clinical protocols, or for other reasons, we might not obtain regulatory approval for or successfully commercialize any of our product candidates. If our relationship with any of these organizations or individuals terminates, replacing any of these third parties could delay our clinical trials and could jeopardize our ability to obtain regulatory approvals and commercialize our product candidates on a timely basis, if at all. For example, we rely on our clinical research organizations’ computer programming and data management capabilities to run and support our clinical programs in such a manner to meet all necessary regulatory and compliance standards.
Risks Relating to Our Operations
We might not successfully operate the compound partnering business as an independent entity.
It takes many years for a drug development business like ours to generate revenue and income. Although we have experience operating our compound partnering business within PPD’s Discovery Sciences segment since 1998, we might not be successful in operating this business as a stand-alone company. Generating revenue and income, consistently or at all, from our drug development business and compound partnering activities depends on our ability to:
| | • | | develop products internally or obtain rights to them from others on favorable terms; |
| | • | | successfully complete non-clinical and clinical studies; |
| | • | | obtain clinical trial materials of sufficient quality or quantity; |
| | • | | obtain and maintain intellectual property rights to these products; |
| | • | | obtain and maintain regulatory approvals; |
| | • | | enter into agreements with third parties to continue the development and commercialization of drug candidates; and |
| | • | | enter into arrangements with third parties to manufacture products on our behalf and to provide sales and marketing functions. |
We must attract and retain key employees in order to succeed.
To be successful, our unique business model requires that our personnel have extensive experience in designing and implementing drug development programs that will run faster than typical studies in the industry. We also require qualified personnel, experienced at building and maintaining relationships with our collaborators. We rely on the services of our senior management, particularly our President and Chief Medical Officer, June Almenoff, our Senior Vice President—Research, Gail McIntyre, and our Senior Vice President—Clinical Development and Operations, Paul Covington, as well as our Chief Financial Officer, Marshall Woodworth, our Vice President—Legal Affairs, Nadine Chien, and our Vice President—Strategic Development, Sailash Patel, the loss of any of whom could adversely impact our operations. We do not carry key man insurance on any of these individuals or any of our other officers or employees. Any inability to hire additional qualified personnel might also require an increase in the workload for both existing and new personnel. We might not be successful in attracting new scientists or management, or in retaining or motivating our existing personnel. The shortage of experienced scientists and managers capable of working within our unique business model might lead to increased recruiting, relocation and compensation costs for these professionals, which might exceed our forecasts. If we are unable to attract and retain any of these personnel, our ability to execute our business plan will be adversely affected.
If our product identification efforts are not successful, we might not be able to effectively develop new products.
Our product candidates are in various stages of development. Some or all of our product candidates might never be developed for any number of reasons, including failure to meet clinical trial tests and failure to receive regulatory approval. For example, we suspended our PPD-10558 program due to unfavorable efficacy data from the Phase II clinical trial. To maintain our business, we need to have a sufficient pipeline of product candidates. Our success in identifying new product candidates depends upon our ability to identify and validate new targets through in-licensing or collaborative arrangements. In order to increase the possibilities of identifying compounds with a reasonable chance for success in clinical studies, part of our business strategy is to identify a higher number of potential targets than we expect to be able to progress through clinical development. If we are unsuccessful in our efforts to identify or obtain rights to new product candidates that lead to the required regulatory approvals and the successful commercialization of products, our business could be harmed.
We or our collaborators might not be able to attract a sufficient number of sites or enroll a sufficient number of patients in a timely manner in order to complete our clinical trials.
The rate of completion of clinical trials is significantly dependent upon the rate of patient enrollment. Patient enrollment is a function of many factors, including:
| | • | | changing regulatory requirements; |
| | • | | the size of the patient population; |
| | • | | perceived risks and benefits of the drug under study; |
| | • | | availability of competing therapies, including those in clinical development; |
| | • | | availability of clinical drug supply; |
| | • | | participation of qualified clinical trial sites; |
| | • | | availability and willingness of potential participants to enroll in clinical trials; |
| | • | | design of the protocol; |
| | • | | proximity of and access by patients to clinical sites; |
| | • | | patient referral practices of physicians; |
| | • | | eligibility criteria for the study in question; and |
| | • | | efforts of the sponsor of and clinical sites involved in the trial to facilitate timely enrollment. |
For example, patient enrollment for our Phase II Proof-of-Concept trial of JNJ-Q2 in hospitalized pneumonia patients was slower than expected. We might have difficulties obtaining sufficient patient enrollment or clinician support to conduct our clinical trials as planned, and we might need to expend additional funds to obtain access to resources or delay or modify our plans significantly. These considerations might result in our being unable to successfully achieve our projected development timelines, or potentially even lead us to consider the termination of on-going clinical trials or development of a product for a particular indication.
Many of our drug candidates are in development and we or our collaborators might not be able to obtain regulatory approval for our product candidates.
The development and commercialization of pharmaceutical products are subject to extensive governmental regulation in the United States and foreign countries. Government approvals are required to develop, market and sell the potential drug candidates we develop alone or with others under our risk-sharing arrangements. Especially for the early-stage compounds we target for in-licensing, obtaining government approval to develop, market and sell drug candidates is time-consuming and expensive. Further, clinical trial results for a particular drug candidate might not satisfy requirements to obtain government approvals. For example, in late 2005, Janssen, our collaborator at the time on dapoxetine, received a “not approvable” letter from the FDA. In addition, governmental approvals might not be received in a timely manner, if at all, and we and our collaborative partners might not be able to meet other regulatory requirements for our products. In addition, governmental approvals might not be received in a timely manner, if at all, and we and our collaborative partners might not be able to meet other regulatory requirements for our products. Finally, even if we are successful in obtaining all required approvals to market and sell a drug candidate, post-approval requirements and the failure to comply with other regulations could result in suspension or limitation of government approvals.
In connection with drug development activities outside the United States, we and our collaborators will be subject to foreign regulatory requirements governing the testing, approval, manufacture, labeling, marketing and sale of pharmaceutical products. These requirements vary from country to country. Even if approval has been obtained for a product in the United States, approvals in foreign countries must be obtained prior to marketing the product in those countries. The approval process in foreign countries could be more or less rigorous and the time required for approval might be longer or shorter than that required in the United States. Clinical studies conducted outside of any particular country might not be accepted by that country, and the approval of a pharmaceutical product in one country does not assure that the product will be approved in another country.
The failure to gain market acceptance of our product candidates among the medical community would adversely affect our future revenue.
Even if approved, our product candidates might not gain market acceptance among physicians, patients, third-party payors and the medical community. We might not achieve market acceptance even if clinical trials demonstrate safety and efficacy and we obtain the necessary regulatory and reimbursement approvals. The degree of market acceptance of any product candidates that we develop will depend on a number of factors, including:
| | • | | establishment and demonstration of clinical efficacy and safety; |
| | • | | cost-effectiveness of our product candidates versus competing products; |
| | • | | their potential advantage over alternative treatment methods; |
| | • | | pricing requirements in various markets; |
| | • | | reimbursement policies of government and third-party payors; and |
| | • | | marketing and distribution support for our product candidates, including the efforts of our collaborators where they have marketing and distribution responsibilities. |
Physicians will not recommend our products until clinical data or other factors demonstrate the safety and efficacy of our product as compared to conventional drug and other treatments. Even if we establish the clinical safety and efficacy of our product candidates, physicians might elect not to use our product for any number of other reasons, including whether the mode of administration of our products is effective for certain indications. The failure of our product candidates to achieve significant market acceptance would materially harm our business, financial condition and results of operations.
We might face additional regulation in the U.S. if our drug candidate MuDelta is classified as a controlled substance by the Drug Enforcement Agency.
The Drug Enforcement Agency, or DEA, regulates drugs that are controlled substances. Controlled substances are those drugs that appear on one of the five schedules promulgated and administered by the DEA under the Controlled Substances Act, or CSA. Any drug that acts on the central nervous system has the potential to become a controlled substance, and scheduling by the DEA is an independent process that might delay the commercial launch of a drug even after FDA approval of the NDA. The CSA governs, among other things, the inventory distribution, recordkeeping, handling, security and disposal of controlled substances.
MuDelta is a novel, orally active, investigational agent in Phase III development, with combined mu opioid receptor agonist and delta opioid receptor antagonist activity. Because it likely acts on the central nervous system, MuDelta has the potential to be scheduled as a controlled substance by the DEA. However, our animal and clinical studies indicate MuDelta is not absorbed into the blood in an appreciable amount via an oral route of administration, thus limiting delivery to the central nervous system. If the DEA schedules MuDelta as a controlled substance, we will be subject to periodic and on-going inspections by the DEA and similar state drug enforcement authorities to assess our on-going compliance with the DEA’s regulations. Any failure to comply with these regulations could lead to a variety of sanctions, including the revocation, or a denial of renewal, of any DEA registrations, injunctions, or civil or criminal penalties. Additionally, if the DEA schedules a drug because it is addictive, doctors might be reluctant to prescribe that drug. It is possible that the DEA will schedule MuDelta as a controlled substance, and, based on the type of scheduling, doctors might not prescribe MuDelta as frequently as they would otherwise, which could negatively impact our revenues.
We face significant competition.
We face significant competition, including from entities that have substantially greater resources and more experience in the commercialization and marketing of pharmaceuticals than we have. Potential competitors in the United States and other countries include major pharmaceutical and biotechnology companies and specialized pharmaceutical companies. These entities have developed and are developing compounds that might compete with our products in development. These competitors might succeed in more rapidly developing and marketing technologies and products that are more effective than our product candidates or technologies or that would render any future commercialized products or technology obsolete or noncompetitive. Our product candidates and any future commercialized products might also face significant competition from both brand-name and generic manufacturers that could adversely affect any future sales of our products.
Any product that we or our collaborators succeed in developing and for which regulatory approval is obtained must then compete for market acceptance and market share. The relative speed with which we and our collaborators can develop products, complete the clinical testing and approval processes, and supply commercial quantities of the products to the market compared to competitive companies will affect market success. In addition, the amount of marketing and sales resources and the effectiveness of the marketing used with respect to a product will affect its marketing success. Other factors affecting the ability of our products to compete include their efficacy and safety, the manner and frequency of their administration, and the extent of any reimbursement coverage.
In addition, some CRO services providers and private equity funds are developing risk sharing models to finance the pharmaceutical industry’s pipeline. As these types of business models evolve, there will be increasing competition for compounds and funds to develop those compounds.
We must protect our patent and other intellectual property rights to succeed.
Our success is dependent in part on our ability to develop and protect patent and other intellectual property rights and operate without infringing the intellectual property rights of others.
Our pending patent applications might not result in the issuance of valid patents or the claim scope of our issued patents might not provide competitive advantages. Also, our patent protection might not prevent others from developing competitive products using related or other technology that does not infringe our patent rights. In addition, our patent for Priligy is for method of use and not composition of matter. Further, patent applications are confidential for a period of time after filing. We therefore might not know that a competitor has filed a patent application covering subject matter similar to subject matter in one of our patent applications or that we were the first to invent the innovation we seek to patent. This might lead to disputes including interference proceeding or litigation to determine rights to patentable subject matter. These disputes are often expensive and might result in our being unable to patent an innovation.
The scope, enforceability and effective term of patents can be highly uncertain and often involve complex legal and factual questions and proceedings. No consistent policy has emerged regarding the breadth of claims in pharmaceutical or biotechnology patents, so that even issued patents might later be modified or revoked by the relevant patent authorities or courts. These proceedings could be expensive, last several years and either prevent issuance of additional patents to us or result in a significant reduction in the scope or invalidation of our patents. Any limitation in claim scope could reduce our ability to negotiate future collaborative research and development agreements based on these patents. Moreover, the issuance of a patent in one country does not assure the issuance of a patent with similar claim scope in another country, and claim interpretation and infringement laws vary among countries, so we are unable to predict the extent of patent protection in any country. Although we might obtain patents for our intellectual property, third parties could challenge the patents before they expire. Further, the grant of a patent does not mean that the issued patent will necessarily be held valid and enforceable by a court. If a governmental agency or court determines that a patent we hold is invalid, noninfringed or unenforceable, the patent will not provide us with market exclusivity, and third parties could therefore enter the market with competing products prior to the expiration of the patent.
In certain cases, we rely upon our collaborator to file, negotiate and maintain patents covering a licensed product. Our collaborators might fail to adequately obtain and maintain such patents.
In addition to seeking the protection of patents and licenses, we also rely upon trade secrets, know-how and continuing technological innovation that we seek to protect, in part, by confidentiality agreements with employees, consultants, suppliers and licensees. If these agreements are not honored, we might not have adequate remedies for any breach. Additionally, our trade secrets might otherwise become known or patented by our competitors.
We might need to obtain patent licenses from others in order to manufacture or sell our potential products and we might not be able to obtain these licenses on terms acceptable to us or at all.
Other companies, universities and research institutions might obtain patents that could limit our ability to use, import, manufacture, market or sell our products or impair our competitive position. As a result, we might need to obtain licenses from others before we could continue using, importing, manufacturing, marketing, or selling our products. We might not be able to obtain required licenses on terms acceptable to us, if at all. If we do not obtain required licenses, we might encounter significant delays in product development while we redesign potentially infringing products or methods or we might not be able to market our products at all.
Changes in the U.S. and international healthcare industry, including reimbursement rates, could adversely affect the commercial value of our development product candidates.
The U.S. and international healthcare industry is subject to changing political, economic and regulatory influences that may significantly affect the purchasing practices and pricing of pharmaceuticals. The laws and regulations governing and issued by applicable regulatory agencies may change and additional government regulations might be enacted, which could prevent or delay regulatory approval of our product candidates. The U.S. Congress adopted healthcare reform and might adopt other legislation that could have the effect of putting downward pressure on the prices that pharmaceutical and biotechnology companies can charge for prescription drugs. Cost-containment measures, whether instituted by healthcare providers or imposed by government health administration regulators or new regulations, could result in greater selectivity in the purchase of drugs. As a result, third-party payors might challenge the price and cost effectiveness of our products. In addition, in many major markets outside the United States, pricing approval is required before sales may commence. As a result, significant uncertainty exists as to the reimbursement status of approved healthcare products.
We might not be able to obtain or maintain our desired price for the products we develop. Any product we introduce might not be considered cost-effective relative to alternative therapies. As a result, adequate third-party reimbursement might not be available to enable us to obtain or maintain prices sufficient to realize an appropriate return on our investment in product development. Also, the trend towards managed healthcare in the United States and the concurrent growth of organizations such as health maintenance organizations, as well as legislative proposals to reform healthcare or reduce government insurance programs, might all result in lower prices, reduced reimbursement levels and diminished markets for our products. These factors will also affect the products that are marketed by our collaborators and licensees. We cannot predict the likelihood, nature or extent of adverse government regulation that might arise from legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our future products and our business could suffer.
Manufacturing changes might result in delays in obtaining regulatory approval or marketing for our products.
If we or our partners make changes in the manufacturing process for any of our products, we or our partners might be required to demonstrate to the applicable regulatory agencies that the changes have not caused the resulting drug material to differ significantly from the drug material previously produced. Further, any significant manufacturing changes for the production of our product candidates could result in delays in development or regulatory approval or in the reduction or interruption of commercial sales of our product candidates. We or our partners contract manufacturers’ inability to maintain manufacturing operations in compliance with applicable regulations within our planned time and cost parameters could materially harm our business, financial condition and results of operations.
We have made manufacturing changes and could make additional manufacturing changes for the production of our products currently in clinical development. These manufacturing changes or an inability to immediately show comparability between the older material and the newer material after making manufacturing changes could result in delays in development or regulatory approvals or in reduction or interruption of commercial sales and could impair our competitive position.
Our business might be harmed if we cannot obtain sufficient quantities of raw materials.
We depend on outside vendors for the supply of raw materials used to produce our product candidates for use in clinical trials. Once a supplier’s materials have been selected for use in the manufacturing process, the supplier in effect becomes a sole or limited source of that raw material due to regulatory compliance procedures. If the third-party suppliers were to cease production or otherwise fail to supply us with quality raw materials and we were unable to contract on acceptable terms for these services with alternative suppliers, our ability to produce our products and to conduct preclinical testing and clinical trials of product candidates would be adversely affected. This could impair our competitive position.
We must comply with extensive government regulations and laws.
We and our collaboration partners are subject to extensive regulation by federal government, state governments, and the foreign countries in which we conduct our business.
In particular, we are subject to extensive and rigorous government regulation as a developer of drug candidates. For example, the FDA regulates, among other things, the development, testing, research, manufacture, record-keeping, labeling, storage, approval, quality control, adverse event reporting, advertising, promotion, sale and distribution of pharmaceutical products. Our product candidates are subject to extensive regulation by foreign governments. The regulatory review and approval process, which includes preclinical studies and clinical trials of each product candidate, is lengthy, expensive and uncertain.
We must rely on our contract manufacturers and third-party suppliers for regulatory compliance and adhering to the FDA’s current Good Manufacturing Practices, or cGMP, requirements. If these manufacturers or suppliers fail to comply with applicable regulations, including FDA pre-or post-approval inspections and cGMP requirements, then the FDA could sanction us. These sanctions could include fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delay, suspension or withdrawal of approvals, license revocation, product seizures or recalls, operational restrictions or criminal prosecutions, any of which could significantly and adversely affect our operating results.
If our operations are found to violate any applicable law or other governmental regulations, we might be subject to civil and criminal penalties, damages and fines. Similarly, if the hospitals, physicians or other providers or entities with which we do business are found non-compliant with applicable laws, they might be subject to sanctions, which could also have a negative impact on us. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations, and additional legal or regulatory change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
We expend a significant amount of resources on compliance efforts and such expenses are unpredictable and might adversely affect our operating results. Changing laws, regulations and standards might also create uncertainty and increase insurance costs. We are committed to compliance and maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment might result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities.
We might incur significant costs in order to comply with environmental regulations or to defend claims arising from accidents involving the use of hazardous materials.
We are subject to federal, state and local laws and regulations governing the use, discharge, handling and disposal of materials and wastes used in our operations. As a result, we might be required to incur significant costs to comply with these laws and regulations. We cannot eliminate the risk of accidental contamination or injury from these materials. In the event of such an accident, we could be held liable for any resulting damages and incur liabilities, which exceed our resources. In addition, we cannot predict the extent of the adverse effect on our business or the financial and other costs that might result from any new government requirements arising out of future legislative, administrative or judicial actions.
We might be subject to product liability claims, and our insurance coverage and indemnification rights might not be adequate to cover these claims.
We face an inherent business risk of exposure to product liability and other types of claims in the event that the use of products during research and development efforts or after commercialization results in death, personal injury or other adverse effects.
Pre-clinical and clinical trials are conducted during the development of our drug candidates to determine the safety and efficacy of potential products for use by humans following approval by regulatory authorities. Despite our efforts to determine the safety of our drug candidates in pre-clinical studies and use of clinical study protocols approved by regulators, unanticipated negative side effects might become evident only when the drug candidates have been delivered to humans during clinical trials or used by patients in the marketplace.
This risk exists even with respect to any products that receive regulatory approval for commercial sale. While we will procure and maintain liability insurance with coverage up to $15.0 million per occurrence and in the aggregate, and generally have indemnification rights under our collaboration agreements, our insurance might not be sufficient to satisfy any or all liabilities that may arise and our indemnification rights might not apply or be sufficient to cover such claims. Also, adequate insurance coverage might not be available in the future at acceptable cost, if at all.
Our operations might be affected by the occurrence of a natural disaster or other catastrophic event.
We depend on our collaboration partners, service providers and other facilities for the continued operation of our business. Natural disasters or other catastrophic events, including terrorist attacks, pandemic flu, hurricanes and ice storms, could disrupt our operations or those of our collaboration partners, which could also affect us. Even though we carry business interruption insurance policies and typically have provisions in our contracts that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our insurance policies or for which we do not have coverage. Any natural disaster or catastrophic event affecting us, or our collaboration partners, could have a significant negative impact on our operations and financial performance.
We, and the clinical research organizations who conduct our clinical trials, must protect electronic information and assets to succeed.
We rely on critical and sensitive data, such as personally identifiable patient information, trade secrets, intellectual property and corporate strategic plans. Security of this type of data is exposed to increasing external threats. We, and our CROs, are also subject to various standards for the protection of personally identifiable information. Failure to implement appropriate safeguards to adequately protect against any unauthorized or unintentional access, acquisition, use, modification, loss or disclosure of this critical or sensitive data may adversely affect our operations.
Risks Resulting from Our Spin-Off From PPD
Our historical financial information is not necessarily indicative of our future financial position, future results of operations or future cash flows and does not reflect what our financial position, results of operations or cash flows would have been as a stand-alone company during the periods presented.
Our historical financial information included in this Form 10-K does not necessarily reflect what our financial position, results of operations or cash flows would have been as a stand-alone publicly traded company during the periods presented prior to June 2010. In addition, it is not necessarily indicative of our future financial position, future results of operations or future cash flows. This is primarily a result of the following factors:
| | • | | Prior to our separation, our business was operated by PPD as part of its broader corporate organization and we did not operate as a stand-alone company; |
| | • | | Most general administrative functions were performed by PPD for the combined entity, so although our historical combined financial statements reflect allocations of costs for services shared with PPD, these allocations may differ from the costs we will incur for these services as an independent company; |
| | • | | After the completion of our separation, the cost of capital for our business might be higher than PPD’s cost of capital prior to our separation; and |
| | • | | Prior to the separation, our financial statements include revenues and expenses of services that we did not continue subsequent to the separation. |
We have a limited history operating as an independent company upon which you can evaluate us.
We have a limited operating history as a stand-alone entity. While our compound partnering business has constituted a part of the historic operations of PPD since 1998, we have only operated as a stand-alone company without the CRO Business since June 2010. Following the spin-off, as an independent company, our ability to satisfy our obligations and achieve profitability will be solely dependent upon the future performance of our compound partnering business, and we will not be able to rely upon the capital resources and cash flows of the CRO Business remaining with PPD.
We might have received better terms from unaffiliated third parties than the terms we receive in our agreements with PPD.
The agreements we entered into with PPD in connection with the spin-off, including the Master Development Services Agreement, the sublease, the Employee Matters Agreement and the Transition Services Agreement, were negotiated while we were still part of PPD. The terms of these agreements relate to, among other things, drug development services to be provided to us by PPD, the subleasing of our offices, employee benefit matters and the provision of transition services to us by PPD. The Master Development Services Agreement requires us to use PPD for specified drug development services for three years contingent on PPD’s expertise and capabilities to provide the needed services. While we believe the terms and conditions of these agreements with PPD are reasonable and acceptable to us, they might not reflect the same terms and conditions that we could have obtained had we sought competitive bids from and negotiated with unaffiliated parties.
Risks Relating to Our Common Stock
Various factors could negatively affect the market price or market of our common stock, which has traded publicly since June 2010.
Our stock has a limited trading history because we only became a separate public company in June 2010, and our trading volume is lower than many companies, which could make investing in our stock riskier than more established companies. In addition, market prices for securities of pharmaceutical companies have been highly volatile, and we expect such volatility to continue for the foreseeable future, so that investment in our securities involves substantial risk. Additionally, the stock market from time to time has experienced significant price and volume fluctuations unrelated to the operating performance of particular companies. The following are some of the factors that might have a significant effect on the market price of our common stock:
| | • | | developments or disputes as to patent or other proprietary rights; |
| | • | | approval or introduction of competing products and technologies; |
| | • | | results of clinical trials; |
| | • | | failures or unexpected delays in timelines for our potential products in development, including the obtaining of regulatory approvals; |
| | • | | delays in manufacturing or clinical trial plans; |
| | • | | fluctuations in our operating results; |
| | • | | market reaction to announcements by other biotechnology or pharmaceutical companies; |
| | • | | initiation, termination or modification of agreements with our collaborators or disputes or disagreements with collaborators; |
| | • | | litigation or the threat of litigation; |
| | • | | public concern as to the safety of drugs developed by us; |
| | • | | sales of our common stock held by our directors and executive officers; and |
| | • | | comments and expectations of results made by securities analysts or investors. |
If any of these factors causes us to fail to meet the expectations of securities analysts or investors, or if adverse conditions prevail or are perceived to prevail with respect to our business, the price of our common stock would likely drop significantly. A significant drop in the price of a company’s common stock often leads to the filing of securities class action litigation against such a company. This type of litigation against us could result in substantial costs and a diversion of management’s attention and resources.
Your percentage ownership in Furiex might be diluted in the future.
Your percentage ownership in Furiex might be diluted in the future because of equity awards that we expect will be granted to our directors, officers and employees, as well as any future equity financing.
Provisions in our amended and restated certificate of incorporation and bylaws and of Delaware law might prevent or delay an acquisition of our Company, which could decrease the trading price of our common stock.
Our amended and restated certificate of incorporation, bylaws and Delaware law contain provisions that are intended to deter coercive takeover practices and inadequate takeover bids by making such practices or bids unacceptably expensive to the raider and to encourage prospective acquirors to negotiate with our Board rather than to attempt a hostile takeover. These provisions include, among others:
| | • | | no right of our shareholders to act by written consent; |
| | • | | procedures requiring advance notice of shareholder proposals or nominations for directors for election at shareholder meetings; |
| | • | | the right of our Board to issue preferred stock without shareholder approval; and |
| | • | | no shareholder rights to call a special shareholders meeting. |
Delaware law also imposes some restrictions on mergers and other business combinations between us and any holder of 15% or more of our outstanding common stock.
We believe these provisions protect our shareholders from coercive or otherwise unfair takeover tactics by requiring potential acquirors to negotiate with our Board and by providing our Board with more time to assess any acquisition proposal. These provisions are not intended to make our company immune from takeovers. However, these provisions apply even if the offer might be considered beneficial by some shareholders and could delay or prevent an acquisition that our Board determines is not in the best interests of our Company and our shareholders.
Item 1B. Unresolved Staff Comments
None.
Our headquarters is located in Morrisville, North Carolina, where we occupy approximately 4,650 square feet of office space under a lease expiring in June 2013. We are negotiating to renew this lease, and believe adequate alternatives are readily available. We own substantially all of the equipment used in our facilities.
Item 3. Legal Proceedings
In the normal course of business, we might be a party to various claims and legal proceedings. As of the date of this Annual Report, there are no outstanding claims that management believes will have a material effect upon our financial condition, results of operations or cash flows.
Item 4. Mine Safety Disclosures
Not applicable.
Executive Officers of the Registrant
The following table sets forth information regarding individuals who serve as our executive officers, including their positions.
| | Age | | | |
| June S. Almenoff | | 56 | | | President and Chief Medical Officer |
| Gail F. McIntyre | | 50 | | | Senior Vice President-Research |
| Paul S. Covington | | 57 | | | Senior Vice President-Clinical Development and Operations |
| Marshall H. Woodworth | | 55 | | | Chief Financial Officer, Treasurer and Assistant Secretary |
June S. Almenoff has served as the president and chief medical officer of Furiex since its inception in 2010 and is the principal executive officer of the Company. She has served on the Board of Directors since 2012. Dr. Almenoff joined Furiex after a successful 12-year career at GlaxoSmithKline, or GSK. She was vice president in the clinical safety and pharmacovigilance organization at GSK, where she served on the company’s senior governing medical boards and managed a diverse therapeutic portfolio supporting numerous regulatory approvals. She led the GSK teams that developed three pioneering systems for minimizing risk in early- and late-stage drug development; these have been widely implemented by pharmaceutical companies and regulatory agencies and their impact on the industry has been recognized by the Wall Street Journal Technology Innovation Award and several other prestigious awards. During her tenure at GSK, Dr. Almenoff chaired the Pharma-FDA working group on safety signal detection and was lead author on its influential position paper. She also led the scientific diligence effort for the acquisition of Stiefel Laboratories and established a licensing program for a drug development unit. Prior to joining GSK, Dr. Almenoff was on the faculty of Duke University Medical Center, where she is currently a Consulting Professor of Medicine. She is an author on more than 50 publications. Dr. Almenoff earned a bachelor's degree, cum laude, from Smith College. She graduated from the M.D.-Ph.D. program at Mt. Sinai School of Medicine and completed a residency in internal medicine and a fellowship in infectious diseases at Stanford University Medical Center. She is a board-certified Fellow of the American College of Physicians with 10 years of clinical practice experience.
Gail F. McIntyre has served as senior vice president of research of Furiex since its inception in 2010. She is responsible for all nonclinical activities as well as for working with the clinical team to move programs from the preclinical arena into the clinical phase. Dr. McIntyre has more than 19 years of experience in the drug discovery and development industry. Her experience covers multiple therapeutic areas including oncology, infectious diseases, central nervous system and metabolic/endocrine as well as various therapies including small drugs, treatment vaccines, immunomodulators, antibodies, immunoconjugates and peptide mimetics. Dr. McIntyre has prepared more than 30 regulatory submissions and ushered compounds through the lead optimization phase to early drug development and from early drug development through the investigational new drug and new drug application phases. Dr. McIntyre earned a bachelor's degree in biology from Merrimack College. Both her master's degree and doctorate are in biochemistry and biophysics from the University of North Carolina at Chapel Hill. Dr. McIntyre is board certified in clinical pathology (hematology and clinical chemistry) and toxicology. She is a member of the American College of Toxicology, the American Society of Clinical Pathologists, the Drug Information Association and the American Association for the Advancement of Science.
Paul S. Covington has served as senior vice president of clinical development and operations of Furiex since its inception in 2010. He is responsible for overseeing all regulatory and clinical operations of Furiex, including design and implementation of clinical trials for compounds in Furiex's pipeline. Dr. Covington joined PPD in 1991 and has more than 17 years of drug development experience. As PPD's executive vice president and chief medical officer, he designed and implemented the development programs for all PPD's compound partnering alliances. Dr. Covington was responsible for the successful Phase I and Phase II development of Priligy and alogliptin, both of which were partnered to large pharmaceutical companies following completion of the Proof-of-Concept studies. As part of his contribution to PPD's compound partnering programs, Dr. Covington also participated in joint development committees with each alliance partner. At PPD, Dr. Covington also oversaw all aspects of medical and regulatory affairs services for quality drug development including pharmacovigilance, medical writing and program management. He was at the forefront of establishing monitoring processes for patient safety and data integrity for complex studies involving extremely ill patients. Dr. Covington is well respected in the pharmaceutical industry and was often sought by clients of PPD for his knowledge in clinical trial design and development. He has managed more than 100 clinical trials across a variety of indications, and is an expert in protocol design and clinical operations. During his career at PPD, Dr. Covington advised numerous pharmaceutical companies on drug development and regulatory strategies and successfully supported companies at key FDA milestone meetings. Prior to joining PPD, Dr. Covington served in various medical roles in both hospital and private practice settings, where he was lead investigator in multiple protocols. He was medical director at Future HealthCare Research Centers in Birmingham, Ala., and chief of staff, director of cardio respiratory and director of critical care at Central Alabama Community Hospital. He completed his residency at Carraway Methodist Medical Center in Birmingham. Dr. Covington received his bachelor's and medical degrees from the University of Alabama in Birmingham.
Marshall H. Woodworth has served as Chief Financial Officer, Treasurer and Assistant Secretary of Furiex since its inception in 2010 and is the principal financial officer of the Company. His responsibilities include the areas of finance, accounting, treasury, human resources and purchasing. Mr. Woodworth has more than 24 years of financial experience of which more than 14 years were in pharmaceutical and life science-related companies, including various finance positions at Eli Lilly, Dow AgroSciences and Monsanto. Prior to joining Furiex, he served as vice president of finance at Xerium Technologies Inc. Mr. Woodworth earned a bachelor's degree in science from the University of Maryland and an M.B.A. from the Indiana University at Bloomington.
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is traded under the symbol “FURX” and is quoted on the NASDAQ Global Market. The following table sets forth the high and low sales prices for shares of our common stock, as reported by NASDAQ for the periods indicated.
| | | 2012 | |
| | | High | | | Low | |
| First Quarter | | $ | 25.00 | | | $ | 15.75 | |
| Second Quarter | | $ | 23.96 | | | $ | 12.16 | |
| Third Quarter | | $ | 22.47 | | | $ | 17.90 | |
| Fourth Quarter | | $ | 19.90 | | | $ | 17.50 | |
| | | 2011 | |
| | | High | | | Low | |
| First Quarter | | $ | 17.41 | | | $ | 14.00 | |
| Second Quarter | | $ | 19.55 | | | $ | 13.76 | |
| Third Quarter | | $ | 19.26 | | | $ | 13.58 | |
| Fourth Quarter | | $ | 18.87 | | | $ | 12.25 | |
The information required by Item 5 of Form 10-K regarding shares subject to outstanding options or warrants to purchase common stock is incorporated herein by reference to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.”
Holders
As of February 28, 2013 there were 3,402 stockholders of record, which excludes stockholders whose shares were held in nominee or street name by brokers.
On February 28, 2013 the closing price for the common stock as reported on the NASDAQ Global Market was $37.06.
Equity Compensation Plans
The information required by Item 5 of Form 10-K regarding equity compensation plans is incorporated herein by reference to “Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.”
Performance Graph
The following graph compares our cumulative total stockholder return from May 28, 2010, when our common stock began trading on a “when issued” basis, with those of the NASDAQ Global Market Composite Index (NQGM) and the NASDAQ Biotechnology Index (NBI). The graph assumes that U.S. $100 was invested on May 28, 2010 in (1) our common stock, (2) the NASDAQ Global Market Composite Index and (3) the NASDAQ Biotechnology Index. The measurement points utilized in the graph consist of the last trading day in each calendar year, which closely approximates the last day of the respective fiscal year of the Company. The historical stock performance presented below is not intended to and may not be indicative of future stock performance.
| | | 5/28/2010 | | | 12/31/2010 | | | 12/30/2011 | | | 12/31/2012 | | | 2/28/2013 | |
| FURX | | $ | 100 | | | $ | 83 | | | $ | 95 | | | $ | 110 | | | $ | 212 | |
| Nasdaq Global Market Composite Index | | $ | 100 | | | $ | 118 | | | $ | 101 | | | $ | 117 | | | $ | 126 | |
| Nasdaq Biotech Index | | $ | 100 | | | $ | 116 | | | $ | 129 | | | $ | 171 | | | $ | 184 | |
Dividends
We have never declared or paid cash dividends on our common stock. We currently expect to retain future earnings, if any, for use in the operation and expansion of the business and do not anticipate paying any cash dividends in the foreseeable future.
Item 6. Selected Financial Data
The tables below set forth selected historical financial information of the Company that has been derived from the audited financial statements as of December 31, 2008, 2009, 2010, 2011 and 2012, and for the five years in the period ended December 31, 2012. For all periods presented prior to December 31, 2010, the weighted-average shares outstanding are calculated based on the 9,881,340 shares issued in connection with the spin-off on June 14, 2010.
The selected historical financial data should be read in conjunction with the combined and consolidated financial statements and related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, included elsewhere in this Form 10-K.
Combined and Consolidated Statements of Operations Data:
| | | Year Ended December 31, | |
| (in thousands, except per share data) | | 2008 | | | 2009 | | | 2010 | | | 2011 | | | 2012 | |
| Total revenue | | $ | 18,419 | | | $ | 6,312 | | | $ | 8,983 | | | $ | 4,490 | | | $ | 40,508 | |
| | | | | | | | | | | | | | | | | | | | | |
| Operating expenses | | | 11,645 | | | | 14,621 | | | | 58,504 | | | | 53,046 | | | | 80,852 | |
| | | | | | | | | | | | | | | | | | | | | |
Income (loss) from operations (1) | | | 6,774 | | | | (8,309 | ) | | | (49,521 | ) | | | (48,556 | ) | | | (40,344 | ) |
| Interest expense | | | — | | | | — | | | | — | | | | 413 | | | | 2,508 | |
| Other income, net | | | 14 | | | | 10 | | | | 9 | | | | 2 | | | | 1 | |
| Provision for income taxes | | | — | | | | — | | | | 14 | | | | 14 | | | | 14 | |
| | | | | | | | | | | | | | | | | | | | | |
| Income (loss) from continuing operations, net | | | 6,788 | | | | (8,299 | ) | | | (49,526 | ) | | | (48,981 | ) | | | (42,865 | ) |
Discontinued operations, net (2) | | | (976 | ) | | | (632 | ) | | | (5,133 | ) | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Net income (loss) | | $ | 5,812 | | | $ | (8,931 | ) | | $ | (54,659 | ) | | $ | (48,981 | ) | | $ | | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Income (loss) from continuing operations, net of income taxes per basic and diluted share | | $ | 0.69 | | | $ | (0.84 | ) | | $ | (5.01 | ) | | $ | (4.96 | ) | | $ | (4.29 | ) |
| Loss from discontinued operations, net of income taxes per basic and diluted share | | $ | (0.10 | ) | | $ | (0.06 | ) | | $ | (0.52 | ) | | $ | — | | | $ | — | |
| Net income (loss) per basic and diluted share | | $ | 0.59 | | | $ | (0.90 | ) | | $ | (5.53 | ) | | $ | (4.96 | ) | | $ | (4.29 | ) |
| Weighted-average shares used to compute net income (loss) per basic and diluted share: | | | 9,881 | | | | 9,881 | | | | 9,881 | | | | 9,884 | | | | 9,984 | |
Combined and Consolidated Balance Sheet Data:
| | | As of December 31, | |
| (in thousands) | | 2008 | | | 2009 | | | 2010 | | | 2011 | | | 2012 | |
| Total assets | | $ | 61,138 | | | $ | 55,877 | | | $ | 132,559 | | | $ | 95,124 | | | $ | 94,755 | |
| Total debt | | | — | | | | — | | | | — | | | | 10,000 | | | | 40,000 | |
| Total shareholders’ equity | | | — | | | | — | | | | 118,504 | | | | 74,323 | | | | 37,597 | |
PPD net investment (3) | | | 55,524 | | | | 49,270 | | | | — | | | | — | | | | — | |
| (1) | Impairments of intangible assets are included in income (loss) from operations. For 2009, the impairment of intangible asset was related to in-process research and development for the MAG-131 compound obtained through the acquisition of Magen Biosciences, Inc. For 2008, the impairment of intangible asset related to the remaining unamortized value of our royalty interest in SinuNase and other Accentia antifungal products. |
| (2) | In 2009, PPD completed dispositions of Piedmont Research Center, LLC and PPD Biomarker Discovery Sciences, LLC. Results of operations for these dispositions are included in discontinued operations. In May 2010, PPD closed the dermatology therapeutic discovery unit, PPD Dermatology, Inc., formerly Magen Biosciences, Inc. |
| (3) | Prior to June 14, 2010, the financial statements of the Company represent a combination of various components of PPD comprising the Discovery Sciences segment. Because a direct ownership relationship did not exist among all the components comprising the Company prior to the spin-off, PPD’s net investment in the Company is shown within the statements of shareholders’ equity in the combined and consolidated financial statements prior to December 31, 2010. The net investment account represents the cumulative investments in, distributions from and earnings (loss) of the Company. |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This Form10-K includes forward-looking statements. All statements other than statements of historical facts are forward-looking statements, including any projections of milestones, royalties or other financial items, any statements of the plans and objectives of management for future operations, any statements concerning research and development, clinical development timelines, proposed new products or licensing or collaborative arrangements, any statements regarding future economic conditions or performance, and any statement of assumptions underlying any of the foregoing. In some cases, forward-looking statements can be identified by the use of terminology such as “believes”, “might”, “will”, “expects”, “plans”, “anticipates”, “estimates”, “potential” or “continue”, or the negative thereof or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements contained in this Form 10-K are reasonable, there can be no assurance that such expectations or any of the forward-looking statements will prove to be correct, and actual results could differ materially from those projected or assumed in the forward-looking statements. Our future financial condition and results of operations, as well as any forward-looking statements, are subject to inherent risks and uncertainties, including the risk factors set forth in Item 1A, and for the reasons described elsewhere in this Form 10-K, any of which could significantly adversely impact our business. All forward-looking statements and reasons why results might differ included in this Form 10-K are made as of the date hereof, and we assume no obligation to update these forward-looking statements or reasons why actual results might differ.
Results of Operations
Our business consists solely of compound development and collaborative activities. Accordingly, we operate in one reportable business segment. Historically, our revenues consisted primarily of milestone and royalty payments from collaborators. For the year ended December 31, 2011, our total revenue of $4.5 million was comprised of royalty revenue from the sale of Nesina, Liovel and Priligy by our collaborators. For the year ended December 31, 2012, our total revenue of $40.5 million consisted of $10.0 million related to a regulatory milestone payment from Takeda related to Nesina, $10.0 million of regulatory milestone payments from Menarini related to Priligy and $20.5 million of royalty revenue from the sale of Nesina, Liovel and Priligy by our collaborators.
We incurred research and development expenses of $44.2 million and $69.5 million for the years ended December 31, 2011 and 2012, respectively. Our research and development expenses include Phase III costs associated with the continued development of MuDelta, a $1.0 million development milestone payment to Ranbaxy Laboratories, Ltd., or Ranbaxy, in connection with the completion of the Phase II final study report for PPD-10558 and a $5.0 million development milestone payment to Janssen related to the dosing of the fifth patient in the on-going Phase III trial for MuDelta.
We expense all research and development costs for our drug candidates and external collaborations as incurred. Our forecasted total research and development expenses for the next 12 months are expected to run between $90.0 million and $100.0 million, comprised almost entirely of Phase III study costs, manufacturing costs, non-clinical costs and Phase I study costs associated with MuDelta. We are actively exploring various partnering options with respect to MuDelta. Any partnering arrangement we enter into could alter our forecasted research and development expenses for MuDelta.
For the years ended December 31, 2011 and 2012, we reported net loss of $49.0 million and $42.9 million, respectively. We expect to continue to incur net losses until revenues from all sources reach a level sufficient to support our on-going operations.
Our business is subject to various risks and uncertainties. See “Risk Factors” described in Item 1A for information on these risks and uncertainties.
Basis of Accounting
We have prepared the combined and consolidated financial statements in accordance with accounting principles generally accepted in the United States of America, or GAAP, and they include the accounts of Furiex Pharmaceuticals, Inc. and its subsidiaries. All intercompany balances and transactions have been eliminated in consolidation. The preparation of financial statements in conformity with GAAP requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
The accompanying combined financial statements for periods prior to June 14, 2010, have been derived from the combined financial statements and accounting records of PPD, and from the historical cost basis of the assets and liabilities of the various activities that reflect the combined results of operations, financial condition and cash flows of the Discovery Sciences segment of PPD. All the business components of the Discovery Sciences segment have been included in the historical statements because they were managed by common PPD segment management, and because they reflected historical performance of segment management.
In May 2010, PPD discontinued operations of its wholly owned subsidiary PPD Dermatology, Inc., formerly Magen Biosciences, Inc., due to unfavorable efficacy data associated with the MAG-131 program. This business unit is recorded as discontinued operations in the statements of operations. Additionally, the Discovery Sciences segment included pre-clinical consulting services not offered by us. All rights and obligations related to pre-clinical consulting services and the definitive purchase agreements related to PPD Dermatology, Inc. have been retained by PPD.
For periods prior to the June 14, 2010 spin-off, we were allocated expenses from PPD such as executive oversight, risk management, accounting, tax, legal, investor relations, human resources, information technology, facilities and depreciation, but were not allocated the underlying productive assets, such as information systems equipment, and furniture and facilities that were not assigned to us, but from which we benefited. We reflected such expenses in the combined and consolidated financial statements as expense allocations from PPD. The basis of these allocations included full-time equivalent employees for the respective periods presented and square footage of occupied space. See Note 14 to our combined and consolidated financial statements for further discussion of the allocations.
Management believes that the assumptions and allocations underlying the combined and consolidated financial statements are reasonable. For periods prior to the June 14, 2010 spin-off, the financial information in these combined and consolidated financial statements does not include all expenses that would have been incurred had we been a separate, stand-alone publicly traded entity. For periods prior to the June 14, 2010 spin-off, the combined and consolidated financial statements include assets, liabilities and operations for PPD Dermatology, Inc. and pre-clinical consulting services that are not included in our operations after the spin-off. As a result, the financial information herein does not reflect our financial position, results of operations or cash flows had we been a separate, stand-alone entity prior to June 14, 2010.
Year Ended December 31, 2011 versus Year Ended December 31, 2012
The following table sets forth amounts from our combined and consolidated financial statements for the year ended December 31, 2011 compared to the year ended December 31, 2012.
| | | Year Ended December 31, | |
| (in thousands) | | 2011 | | | 2012 | |
| Revenue: | | | | | | | | |
| Milestones | | $ | — | | | $ | 20,000 | |
| Royalties | | | 4,490 | | | | 20,508 | |
| | | | | | | | | |
| Total revenue | | | 4,490 | | | | 40,508 | |
| | | | | | | | | |
| Research and development expenses | | | 44,202 | | | | 69,505 | |
| Selling, general and administrative expenses | | | 8,761 | | | | 11,261 | |
| Depreciation and amortization | | | 83 | | | | 86 | |
| | | | | | | | | |
| Total operating expenses | | | 53,046 | | | | 80,852 | |
| | | | | | | | | |
| Operating loss | | | (48,556 | ) | | | (40,344 | ) |
| Interest expense | | | 413 | | | | 2,508 | |
| Other income, net | | | 2 | | | | 1 | |
| | | | | | | | | |
| Loss before provision for income taxes | | | (48,967 | ) | | | (42,851 | ) |
| Provision for income taxes | | | 14 | | | | 14 | |
| | | | | | | | | |
| Net loss | | $ | (48,981 | ) | | $ | (42,865 | ) |
Revenue
Total revenue increased $36.0 million to $40.5 million for the year ended December 31, 2012 from 2011. This increase was due to a $10.0 million regulatory milestone payment from Takeda with respect to the EMA acceptance of the submission of a Marketing Authorization Application for alogliptin in May 2012, $10.0 million of regulatory milestone payments from Menarini upon the closing of the new license agreement related to Priligy in July 2012, an increase in royalty revenue related to the sale of Nesina and Liovel in Japan and an increase in royalty revenue related to sales of Priligy in various countries outside the United States.
Expenses
Research and development, or R&D, expenses increased $25.3 million to $69.5 million for the year ended December 31, 2012 from 2011. The increase in R&D expense was due predominantly to Phase III costs associated with the continued development of MuDelta, a $1.0 million development milestone payment to Ranbaxy in connection with the completion of the Phase II final study report for PPD-10558 and a $5.0 million development milestone payment to Janssen related to the dosing of the fifth patient in the on-going Phase III trial for MuDelta, partially offset by decreases in spending for the discontinued PPD-10558 program and completion of the Phase II work related to MuDelta and JNJ-Q2.
The following table sets forth amounts from our combined and consolidated statements of operations for R&D expenses along with the dollar amount of the changes for the year ended December 31, 2011 compared to the year ended December 31, 2012.
| | | Year Ended December 31, | | | $ Inc (Dec) | |
| (in thousands) | | 2011 | | | 2012 | | | | |
| R&D expense by project: | | | | | | | | | | | | |
| MuDelta | | $ | 15,954 | | | $ | 64,652 | | | $ | 48,698 | |
| JNJ-Q2 | | | 12,787 | | | | 814 | | | | (11,973 | ) |
| PPD-10558 | | | 12,873 | | | | 1,201 | | | | (11,672 | ) |
| Other R&D expense | | | 2,588 | | | | 2,838 | | | | 250 | |
| | | | | | | | | | | | | |
| Total R&D expense | | $ | 44,202 | | | $ | 69,505 | | | $ | 25,303 | |
R&D expenses will likely fluctuate significantly from period to period for a variety of reasons, including the number of compounds under development, the stages of development and changes in development plans. In 2011, we acquired the rights to the JNJ-Q2 and MuDelta compounds as a result of Janssen's decision not to exercise its option under the collaborative agreements for those compounds. We are continuing to seek to partner or out-license JNJ-Q2, with minimal expenditures forecasted in 2013. We are actively exploring partnering options for MuDelta. Any partnering arrangement we enter into could alter our forecasted research and development expenses for MuDelta. Our forecasted total research and development expenses for the next 12 months are expected to run between $90.0 million and $100.0 million, comprised almost entirely of Phase III study costs, manufacturing costs, non-clinical costs and Phase I study costs associated with MuDelta.
Selling, general and administrative, or SG&A, expenses increased $2.5 million to $11.3 million for the year ended December 31, 2012 from 2011. The increase in SG&A expenses was due primarily to increases in non-cash stock compensation expense of $1.1 million, including additional grants issued to employees during 2012 and the mark-to-market adjustment for non-vested consultant options, and increases in consulting and legal expenses of $0.9 million.
Interest expense of $2.5 million for the year ended December 31, 2012 related entirely to our loan agreement with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank.
Income Taxes
During 2011 and 2012, we did not record a tax benefit related to our operating losses because we have provided full valuation allowances against our assets based on our history of operating losses. We anticipate that we will require a full valuation allowance against any deferred tax assets until such time as we are able to demonstrate a consistent pattern of profitability. For the years ended December 31, 2011 and 2012, we recorded an insignificant amount of income tax expense. This amount relates to the adjustment of a deferred tax liability associated with historical goodwill, which we amortize and deduct for tax purposes, but treat as an indefinite-lived intangible asset for financial reporting purposes.
Results of Operations
Net loss of $42.9 million for the year ended December 31, 2012 represents a $6.1 million decrease from net loss of $49.0 million during 2011. This decrease in our net loss resulted primarily from the $36.0 million increase in revenue, partially offset by the $25.3 million increase in R&D expenses, a $2.5 million increase in SG&A expenses and the $2.1 million increase in interest expense, as described above.
Year Ended December 31, 2010 versus Year Ended December 31, 2011
The following table sets forth amounts from our combined and consolidated financial statements for the year ended December 31, 2010 compared to the year ended December 31, 2011.
| | | Year Ended December 31, | |
| (in thousands) | | 2010 | | | 2011 | |
| Revenue: | | | | | | | | |
| Milestones | | $ | 7,500 | | | $ | — | |
| Royalties | | | 1,330 | | | | 4,490 | |
| Service | | | 75 | | | | — | |
| Other | | | 78 | | | | — | |
| | | | | | | | | |
| Total revenue | | | 8,983 | | | | 4,490 | |
| | | | | | | | | |
| Direct expenses | | | 21 | | | | — | |
| Research and development expenses | | | 50,112 | | | | 44,202 | |
| Selling, general and administrative expenses | | | 8,262 | | | | 8,761 | |
| Depreciation and amortization | | | 109 | | | | 83 | |
| | | | | | | | | |
| Total operating expenses | | | 58,504 | | | | 53,046 | |
| | | | | | | | | |
| Operating loss | | | (49,521 | ) | | | (48,556 | ) |
| Interest expense | | | — | | | | 413 | |
| Other income, net | | | 9 | | | | 2 | |
| | | | | | | | | |
| Loss from continuing operations before provision for income taxes | | | (49,512 | ) | | | (48,967 | ) |
| Provision for income taxes | | | 14 | | | | 14 | |
| | | | | | | | | |
| Loss from continuing operations, net of income taxes | | | (49,526 | ) | | | (48,981 | ) |
| Loss from discontinued operations, net of income taxes | | | (5,133 | ) | | | — | |
| | | | | | | | | |
| Net loss | | $ | (54,659 | ) | | $ | (48,981 | ) |
Revenue
Total revenue decreased $4.5 million to $4.5 million for the year ended December 31, 2011 from 2010. The decrease in total revenue was primarily attributable to a $7.5 million decrease in milestone revenue earned in 2010 as a result of regulatory and pricing approvals of Nesina in Japan, partially offset by an increase of $3.2 million in royalty revenue from 2010 based on the sale of approved products by our collaborators. For the year ended December 31, 2011, we received royalties of $4.5 million from sales of Priligy in various countries outside the United States, and from the sale of Nesina and Liovel in Japan.
Expenses
R&D expenses decreased $5.9 million to $44.2 million for the year ended December 31, 2011 from 2010. The decrease in R&D expense was due predominantly to reduced development costs for the MuDelta and JNJ-Q2 compounds, offset by increased spending related to the PPD-10558 compound. As of December 31, 2011, we had substantially completed the Phase II clinical trials for the MuDelta and JNJ-Q2 compounds.
The following table sets forth amounts from our combined and consolidated statements of operations for R&D expenses along with the dollar amount of the changes for the year ended December 31, 2010 compared to the year ended December 31, 2011.
| | | Year Ended December 31, | | | $ Inc (Dec) | |
| (in thousands) | | 2010 | | | 2011 | | | | |
| R&D expense by project: | | | | | | | | | | | | |
| MuDelta | | $ | 24,670 | | | $ | 15,954 | | | $ | (8,716 | ) |
| JNJ-Q2 | | | 22,668 | | | | 12,787 | | | | (9,881 | ) |
| PPD-10558 | | | 1,197 | | | | 12,873 | | | | 11,676 | |
| Other R&D expense | | | 1,577 | | | | 2,588 | | | | 1,011 | |
| | | | | | | | | | | | | |
| Total R&D expense | | $ | 50,112 | | | $ | 44,202 | | | $ | (5,910 | ) |
On April 18, 2011, Janssen announced that in connection with a broad strategic review of its portfolio of infectious disease programs, it will be redirecting its research and development efforts toward antivirals and vaccines, and will not be investing in the development of new antibacterial therapies. As a result, Janssen elected not to exercise its option to continue the development of the JNJ-Q2 compound. On April 19, 2011, we announced that we had acquired full exclusive license rights to develop and commercialize the JNJ-Q2 compound under our existing development and license agreement with Janssen. On November 1, 2011, we announced we had acquired full exclusive license rights to develop and commercialize the MuDelta compound under our existing development and license agreement with Janssen. We acquired these rights as a result of Janssen’s decision not to exercise its option under the agreement to continue development of MuDelta.
In December 2011, we announced top-line results from the Phase II trial of PPD-10558. Based on these results, we have discontinued further spending on the PPD-10558 program and have terminated the license agreement with Ranbaxy in accordance with the terms of the agreement. R&D expenses may fluctuate significantly from period to period for a variety of reasons, including the number of compounds under development, the stages of development and changes in development plans.
Selling, general and administrative, or SG&A expenses, increased $0.5 million to $8.8 million for the year ended December 31, 2011 from 2010. The increase in SG&A expenses was due primarily to increases in non-cash stock compensation expense.
Income Taxes
During 2010 and 2011, we did not record a tax benefit related to our operating losses because we have provided full valuation allowances against our assets based on our history of operating losses. Additionally, with the exception of the pre-acquisition federal and state tax filings for Magen BioSciences, Inc. and certain separate state filings, through the June 14, 2010 spin-off, our operations were included in the consolidated federal and combined state tax returns of PPD, and the resulting tax attributes have been fully utilized by PPD and are no longer available to us for future use. Subsequent to June 14, 2010, we have filed federal and state returns separately from PPD and can use our tax attributes accordingly. However, we anticipate that we will require a full valuation allowance against any deferred tax assets until such time as we are able to demonstrate a consistent pattern of profitability. For the years ended December 31, 2010 and 2011, we recorded an insignificant amount of income tax expense. This amount relates to the adjustment of a deferred tax liability associated with historical goodwill, which is amortized and deductible for tax purposes, but is an indefinite-lived intangible asset for financial reporting purposes.
Results of Operations
Net loss of $49.0 million in 2011 represents a $5.7 million decrease from net loss of $54.7 million in 2010. This decrease in net loss resulted primarily from discontinued operations, in addition to changes in revenue and R&D expenses, as described above. In May 2010, PPD discontinued operations of its wholly owned subsidiary PPD Dermatology, Inc. due to unfavorable efficacy data associated with the MAG-131 program. As a result, this business unit is shown as discontinued operations for 2010. Loss from discontinued operations was $5.1 million for the year ended December 31, 2010.
Liquidity and Capital Resources
As of December 31, 2012, we had $25.7 million of cash and cash equivalents. In addition, we had $11.7 million of accounts receivable and $16.8 million of accounts payable and accrued expenses. Included in the year end accrued expense balance is $3.75 million owed to Janssen associated with Priligy transition services. The primary source of our current cash, cash equivalents and investments is related to upfront, milestone and royalty payments received since the spin-off from PPD and cash from the issuance of debt.
On August 18, 2011, we entered into a loan agreement with MidCap Funding III, LLC and Silicon Valley Bank. This initial borrowing in the amount of $10.0 million had a fixed interest rate of 10.25% per annum and was initially due August 1, 2015. On August 2, 2012, we entered into an amended loan agreement with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank for an additional $30.0 million. This new agreement amended the prior agreement by resetting the maturity date to August 2, 2016 for a total amount of $40.0 million, bearing interest at a fixed rate of 10.00%, subject to adjustment under specified conditions. Interest accrues daily and is payable on the first day of the following month, in arrears. Principal payments are due on a ratable monthly basis from August 1, 2013 until maturity. As part of this amended agreement, we are required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $7.5 million through August 2, 2014, $5.0 million through August 2, 2015 and $2.5 million through August 2, 2016. As of December 31, 2012, this amount is presented in investments within the consolidated balance sheets. In addition, we must maintain our primary deposit and investment accounts with Silicon Valley Bank, consisting of at least 50% of our total cash, cash equivalents and investments balance.
We have incurred losses and negative cash flows from operations since the spin-off and will continue to incur operating losses until revenues from all sources reach a level sufficient to support our on-going operations. Our long-term liquidity needs will largely be determined by the success of our products already being commercialized by collaborators, key development and regulatory events that might impact our ability to out-license our development compounds and expenses associated with research and development efforts. Based on current forecasts, we believe we have sufficient liquidity to continue our planned operations for at least the next 12 months. However, if one or more of our forecasted milestone events and / or the expected growth in royalty payments fails to meet our expectations, or if research and development expenses increase because of slower than expected enrollment rates or other factors, we will likely need to find additional sources of financing to support our Phase III development efforts for MuDelta. Depending upon the success and timing of receipt of various milestone payments and royalties, it might be necessary to do one or more of the following in the next 12 months: (a) raise additional capital; (b) reduce spending on research and development; or (c) restructure our operations. On an on-going basis we evaluate our financial position with respect to current and future financing activities. The financing activities could take the form of additional debt or equity as dictated by our needs and our view toward our overall capital structure. We currently expect that additional funding, if necessary or desired, would come from potential sources including, but not limited to, partnering income, royalty financing, and debt and / or equity issuances.
While we believe we have adequate sources of liquidity to fund our operations for at least the next 12 months based on our current operating projections, our liquidity over that time period could be affected by, among other things: costs related to our development efforts, regulatory approval and commercialization of our compound candidates which could affect milestone and royalty receipts; changes in regulatory compliance requirements; reliance on existing collaborators and the potential need to enter into additional collaborative arrangements; our ability to raise additional funds through equity, debt or other financing alternatives; or other factors described under Item 1A. “Risk Factors".
The timing and amount of any future expenses, trial completion dates and revenues related to our compounds are subject to significant uncertainty. We do not know if we will be successful in developing any of our drug candidates. The timing and amount of our research and development expenses will depend upon the costs associated with the present and potential future clinical trials and non-clinical studies of our drug candidates, any related expansion of our research and development organization, changes in regulatory requirements and manufacturing costs. There are numerous risks and uncertainties associated with the duration and cost of clinical trials, which vary significantly over the life of a project as a result of events arising during clinical development. For example, if the FDA, or another regulatory authority, were to require us or one of our collaborators to conduct clinical trials beyond those we currently anticipate to complete clinical development of a drug candidate, or if we experience significant delays in enrollment in any of our clinical trials, we would be required to expend significant additional financial resources and time on the completion of clinical development. The timing and amount of revenues, if any, are dependent upon the success of the clinical trials as well as the commercial success of these products in the marketplace, all of which are subject to a variety of risks and uncertainties.
Our future capital requirements will depend on numerous factors, including, among others: the cost and expense of continuing the research and development activities of our existing candidates; new collaborative agreements that we might enter into in the future; progress of compound candidates in clinical trials as it relates to the cost of development and the receipt of future milestone payments, if any; the ability of our licensees and collaborators to obtain regulatory approval and successfully manufacture and market collaboration products; the continued or additional support by our collaborators or other third parties of research and development efforts and clinical trials; time required to gain regulatory approvals; the demand for our potential products, if and when approved; potential acquisitions of technology, product candidates or businesses by us; and the costs of defending or prosecuting any patent opposition or litigation necessary to protect our proprietary technologies. In order to develop and obtain regulatory approval for our potential product candidates we might raise additional funds through equity or debt financings or from other sources, collaborative arrangements, the use of sponsored research efforts or other means. However, additional financing might not be available on acceptable terms, if at all, and such financing might only be available on terms dilutive or otherwise detrimental to our stockholders or our business.
For the year ended December 31, 2012, our operating activities used $40.6 million in cash as compared to $49.0 million used for the same period in 2011. The decrease in net cash used in operating activities of $8.4 million was due primarily to changes in operating assets and liabilities, an increase in revenue of $36.0 million and an increase in R&D expense of $25.3 million from 2011.
For the year ended December 31, 2012, our investing activities provided $2.4 million in cash as compared to $10.0 million of cash used for the same period in 2011. The change in our investing activities from the prior year was due primarily to the purchase and sale of investments.
For the year ended December 31, 2012, our financing activities provided $30.3 million in cash due primarily to the loan agreement with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank for $30.0 million and $0.6 million from the issuance of common stock related to option exercises by consultants, partially offset by additions to deferred financing costs of $0.3 million. For the year ended December 31, 2011, our financing activities provided $10.0 million in cash under the previous loan agreement with MidCap Funding III, LLC and Silicon Valley Bank and $0.6 million from the issuance of common stock related to option exercises by employees and consultants.
Contractual Obligations
On August 18, 2011, we entered into a Loan and Security Agreement (the “Agreement”) with MidCap Funding III, LLC and Silicon Valley Bank. This initial borrowing in the amount of $10.0 million had a fixed interest rate of 10.25% per annum and was initially due August 1, 2015. Interest accrued monthly and was payable on the first day of the following month, in arrears. Principal payments of the initial borrowing were to be paid on a ratable monthly basis from August 1, 2012 until maturity. However, on August 2, 2012, we entered into an Amended and Restated Loan and Security Agreement (the “Amended Agreement”) with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank, or collectively, the Lenders. The Amended Agreement modified the prior Agreement by providing an additional $30.0 million in borrowings and reset the maturity date of the initial Agreement to August 2, 2016.
Under the terms of the Amended Agreement, the total amount due to the Lenders of $40.0 million has a maturity date of August 2, 2016. The Amended Agreement bears interest at a fixed rate of 10.00%, subject to adjustment under specified conditions. Interest accrues daily and is payable on the first day of the following month, in arrears. Principal payments are due on a ratable monthly basis from August 1, 2013 until maturity.
As part of this new agreement, we are required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $7.5 million through August 2, 2014, $5.0 million through August 2, 2015 and $2.5 million through August 2, 2016. In addition, we must maintain our primary deposit and investment accounts with Silicon Valley Bank, consisting of at least 50% of our total cash, cash equivalents and investments balance. Under the initial Agreement we were required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $10.0 million until mid-January 2012. We intend to use the proceeds from the loan to support research and development for our MuDelta compound.
A final payment fee is due to the Lenders in an amount equal to 2.5% of the total loan commitment, payable at the maturity date or earlier prepayment of the loan. We may prepay the loan subject to a prepayment fee of between one and four percent of the amount borrowed, depending on the time of the prepayment. The amount of interest expense related to the initial Agreement and Amended Agreement included in the statements of operations for the years ended December 31, 2011 and 2012 was $0.4 million and $2.5 million, respectively. Included in these amounts are the ratable accrual of the final payment fee and the amortization of deferred financing costs over the term of the loan, which are included in other long-term liabilities and other assets within the consolidated balance sheets, respectively.
Under the Amended Agreement, we are subject to affirmative covenants, including the obligations to maintain good standing, provide certain notices to the Lenders, deliver financial statements to the Lenders, maintain insurance, discharge all taxes, protect intellectual property and protect collateral. We are also subject to negative covenants, including that we may not enter into a merger or consolidation or certain change of control events, incur liens on the collateral, incur additional indebtedness, dispose of any property, change our jurisdictions of organization or organizational structures or types, declare or pay dividends (other than dividends payable solely in common stock), make certain investments or acquisitions, and enter into certain transactions with affiliates, in each case subject to certain customary exceptions, including exceptions that allow us to acquire additional compounds and to enter into licenses and similar agreements providing for the use and collaboration of our intellectual property provided certain conditions are met. Our assets serve as collateral for the loan.
The Amended Agreement provides that events of default include failure to make payment of principal or interest on the loan when required, failure to perform certain obligations under the Amended Agreement and related documents, defaults in certain other indebtedness and certain other events including certain adverse actions taken by the FDA or other governmental authorities. Upon events of default, our obligations under the Amended Agreement may, or in the event of insolvency or bankruptcy, will automatically be accelerated. Upon the occurrence of any event of default, our obligations under the Amended Agreement will bear interest at a rate equal to the lesser of (a) 4% above the rate of interest applicable to such obligations immediately prior to the occurrence of the event of default or (b) the maximum rate allowable under law.
As of December 31, 2012, future minimum payments on all of our contractual obligations for fiscal years ended subsequent to December 31, 2012 were as follows related to our Amended Loan and Security Agreement, Priligy transition service payments owed Janssen and operating leases in Morrisville, North Carolina and Wilmington, North Carolina:
Years Ended December 31 (in thousands) | | | | | | | | | | | | | | | |
Long-term Debt: Principal | | | | | | | | | | | | | | | | | | | | |
Long-term Debt: Interest and Fees | | | | | | | | | | | | | | | | | | | 9,638 | |
Janssen Transition Service Payments | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | 94 | |
Total Contractual Obligations | | | | | | | | | | | | | | | | | | | | |
As of December 31, 2012, we were contingently obligated under collaboration agreements that have not been included in the table above due to the inherent uncertainty in the amounts and timing of payments.
As of December 31, 2012, we had two collaborations that involve potential future expenditures. The first is our collaboration with Alza and Janssen for Priligy.
On May 14, 2012, we entered into a license and asset transfer agreement with Alza and Janssen, whereby Alza and Janssen transferred to us worldwide rights for Priligy. To facilitate a uniform transition, Janssen will continue to manufacture and manage certain clinical and regulatory activities with respect to Priligy for a pre-defined period after the closing date of the agreement. This transaction became effective on July 30, 2012. Under the terms of this transaction, we are obligated to pay Janssen for transition services provided to us in the amount of $15.0 million, with $7.5 million paid within 45 days of closing, and $3.75 million due within 10 business days of the beginning of each of the following two calendar quarters. In addition, we are obligated to pay Janssen up to $19.0 million in potential on-going clinical study costs and up to $1.0 million for reasonable out-of-pocket expenses over the transition period. We must also pay Janssen fees related to Priligy sales and distribution activities that Janssen will perform for us during the transition period pursuant to a sales services agreement. We believe the transition period will be completed within 12 months from the contract date. Priligy will continue to be made available to patients under the sales service agreement until the marketing authorizations are transferred, at which time commercialization of the product will transition to us or our licensee. The term of the license and asset transfer agreement will expire on the latest of the completion of the transition of the product rights to us, the expiration of our last payment obligation to Janssen, or the expiration of the last-to-expire ancillary agreement.
Our collaboration with Alza and Janssen for Priligy, as described above, is associated with an out-license agreement under which we did receive, and are eligible to receive, additional payments. On May 14, 2012, we entered into a license agreement with Menarini by which we will license to Menarini exclusive rights to commercialize Priligy in Europe, most of Asia, Africa, Latin America and the Middle East. This transaction became effective on July 30, 2012. We will retain full development and commercialization rights in the United States, Japan and Canada. Menarini will assume responsibility for commercialization activities in the licensed territories and will fund the on-going clinical studies being performed by Janssen. Under the license agreement with Menarini, we received a $15.0 million upfront payment and $10.0 million of regulatory milestone payments during the third quarter of 2012, and are eligible to receive up to $19.0 million to fund potential costs of on-going clinical studies being performed by Janssen, up to $10.0 million in launch-based milestones and up to $40.0 million in sales-based milestones, plus tiered royalties ranging from the mid-teens to mid-twenties in percentage terms. During the third quarter of 2012, we recognized milestone revenue of $10.0 million related to this agreement related to regulatory submissions. The $15.0 million upfront payment received during the third quarter of 2012 was not recorded as milestone revenue, nor will the potential up to $19.0 million for on-going clinical study costs, based on the terms of the agreement described below. The term of the license agreement (and the period during which Menarini must pay us royalties in a particular country for a particular product) will end, on a country-by-country basis, upon the latest of (i) the expiration of a valid relevant patent claim in that country, (ii) the expiration of marketing and data exclusivity in that country, or (iii) the market entry of an approved product in that country containing dapoxetine for use, on an as needed basis, for premature ejaculation.
In connection with the license and asset transfer agreement with Alza and Janssen, and the license agreement with Menarini, we have assessed the indicators associated with agent and principal considerations for each transaction. Based on the terms of the underlying agreements, the nature of the related cash payments and receipts (regarding the $15.0 million up-front and transition service payments, the up to $19.0 million to fund potential on-going clinical study costs and the fees related to Priligy sales and distribution activities performed by Janssen), and the determination that we are not the primary obligor for the underlying activities that are associated with these payments, we have recorded these amounts on a net basis within the combined and consolidated statements of operations.
For the year ended December 31, 2012, we have paid amounts to Janssen, and correspondingly received equal payments from Menarini, totaling $2.0 million related to on-going clinical study costs for Priligy. In addition, we have paid to Janssen approximately $0.2 million in fees associated with sales and distribution activities for Priligy during the transition period. We have also considered the contractual right-of-offset related to these payments, for which we are effectively an intermediary, and have recorded these amounts on a gross basis within the consolidated balance sheets.
As of December 31, 2012, the remaining amounts due to Janssen associated with Priligy transition services of $3.75 million are reflected in accrued expenses. As of December 31, 2012, approximately $1.7 million is reflected in both accounts receivable and accrued expenses related to the on-going Priligy clinical study costs being performed by Janssen.
We originally acquired patents for Priligy from Eli Lilly and Company, or Lilly, and are obligated to pay Lilly a royalty of 5% on annual sales in excess of $800.0 million. In addition, under the terms of the license agreement with Menarini, we remain responsible for payment of royalties to Lilly, except Menarini will pay the portion of the royalties owed to Lilly in each country where Menarini is licensed to sell Priligy, where Lilly is eligible for payments, and where we are no longer eligible for payments from Menarini. The term of the license agreement with Lilly (and the period during which we or Menarini must pay Lilly royalties in a particular country) will end, on a country-by-country basis, upon the later of (i) the last to expire valid patent claim licensed to Lilly in that country, (ii) the expiration of data exclusivity in that country, or (iii) the tenth anniversary of the first date of sale of Priligy in that country.
The second collaboration involving future expenditures is associated with the two compounds in-licensed from Janssen: JNJ-Q2 and MuDelta. On April 18, 2011, Janssen announced that in connection with a broad strategic review of its portfolio of infectious disease programs, it will be redirecting its research and development efforts toward antivirals and vaccines, and will not be investing in the development of new antibacterial therapies. As a result, Janssen elected not to exercise its option to continue the development of the JNJ-Q2 compound. On April 19, 2011, we announced that we had acquired full exclusive license rights to develop and commercialize the JNJ-Q2 compound under our existing development and license agreement with Janssen. On November 1, 2011, we announced that we had acquired full exclusive license rights to develop and commercialize the MuDelta compound under our existing development and license agreement with Janssen. We acquired these rights as a result of Janssen's decision not to exercise its option under the agreement to continue development of MuDelta.
We plan to continue evaluating other partnering and funding opportunities for both the JNJ-Q2 and MuDelta compounds. We may be obligated to pay Janssen: up to $50.0 million in regulatory milestone payments for the JNJ-Q2 compound; up to $45.0 million in regulatory milestone payments for the MuDelta compound; and, if approved for marketing, for both the JNJ-Q2 and MuDelta compounds, individually, up to $75.0 million in sales-based milestone payments and sales-based royalties increasing from the mid- to upper-single digit percentages as sales volume increases. Royalties would be paid for a period of ten years after the first commercial sale or, if later, the expiration of the last valid patent claim or the expiration of patent exclusivity.
Off-balance Sheet Arrangements
We have no off-balance sheet arrangements except for operating leases entered into in the normal course of business.
Critical Accounting Policies and the Use of Estimates
The preparation of our combined and consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in our combined and consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates. The items in our combined and consolidated financial statements requiring significant estimates and judgments are as follows:
Revenue Recognition
We generate revenue in the form of upfront payments, development and regulatory milestone payments, royalties, and launch-based and sales-based milestone payments in connection with the out-license or sale of compounds. The receipt of future milestone payments and royalties depends on the success of our compound development and our collaborators' success in developing and commercializing compounds. Upfront payments are generally paid within a short period of time following the execution of an out-license or collaboration agreement. Development and regulatory milestone payments are typically one-time payments to us triggered by the collaborator's achievement of specified development and regulatory events such as the commencement of Phase III trials or regulatory submission approval. Royalties are payments received by us based on net product sales of a collaborator. Launch-based milestone payments are one-time payments to us triggered when a collaborator first introduces for sale an out-licensed product in a new geographical region. Sales-based milestone payments are typically one-time payments to us triggered when aggregate net sales of product by a collaborator for a specified period (for example, an annual period) reach an agreed upon threshold amount. We recognize upfront payments, development and regulatory milestone payments, royalty payments, and launch-based and sales-based milestone payments from our collaborators when the event that triggers the obligation of payment has occurred, there is no further obligation on our part in connection with the payment and collection is reasonably assured.
We assess each collaboration agreement we enter into for potential indicators associated with agent and principal considerations for each related cash payment and receipt. Based on the terms of the underlying agreement and the determination of which party to the transaction is the primary obligor, we record the underlying activities that are associated with contractual payments within the combined and consolidated statements of operations and consolidated balance sheets on a net or gross basis, accordingly.
Goodwill
We review goodwill for impairment annually on October 1 and whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. In performing the annual impairment test, the fair value of the Company was determined using a combination of the income and market approaches. We have a single reporting unit. For purposes of the income approach, fair value was determined based on the present value of estimated future cash flows, discounted at an appropriate risk-adjusted rate. The market approach considers recent comparable transactional valuation multiples for pharmaceutical and biotechnology companies.
For the income approach, we made assumptions about the amount and timing of future expected cash flows, probability of future compound development and appropriate discount rates. The compound development estimates are highly subjective due to the uncertainty associated with the amounts and timing of expected milestone and royalty payments. We base the amount and timing of future cash flows within our analysis on our most recent operational budgets, long-range strategic plans and other estimates. Actual results may differ from those we assume in our forecasts, which could have a material impact on our combined and consolidated financial statements. We use estimates of market participant weighted-average cost of capital as a basis for determining the discount rates to apply to our future expected cash flows, adjusted for the risks and uncertainty inherent in our industry generally and in our internally developed forecasts. Based on our review as of October 1, 2012, our calculated fair value of equity was in excess of carrying value by a substantial margin.
The fair value of goodwill could be materially impacted by future adverse changes such as future declines in operating results, a decline in the valuation of pharmaceutical and biotechnology company stocks, including the valuation of the Company's own common stock, a slowdown in the worldwide economy or the pharmaceutical and biotechnology industry, failure to meet the performance projections included in forecasted operating results, or the delay or abandonment of any research and development programs.
Share-Based Compensation
We recognize compensation expense using a fair-value based method related to stock options and other share-based compensation. We measure the expense based on the grant date fair value of the awards that are expected to vest and record the expense over the applicable requisite service period. In the absence of an observable market price for a share-based award, we base the fair value upon a valuation methodology that takes into consideration various factors, including the exercise price of the award, the expected term of the award, the current price of the underlying shares, the expected volatility of the underlying share price based on peer companies, the expected dividends on the underlying shares and the risk-free interest rate.
We calculated our income tax provision for the periods prior to June 14, 2010 using the separate return basis as if we had filed separate income tax returns under our existing structure. We determined the provision for income taxes subsequent to the spin-off using the asset and liability approach of accounting for income taxes. Under this approach, deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. The provision for income taxes represents income taxes paid or payable for the year, plus the change in deferred taxes during the year. Deferred taxes result from differences between the financial reporting and tax basis of our assets and liabilities. We measure deferred tax assets and liabilities using the currently enacted tax rates that apply to taxable income in effect for the years in which we expect those tax attributes to be recovered or paid, and adjust these measurements for changes in tax rates and tax laws when enacted. We record valuation allowances to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized. The assessment of whether or not a valuation allowance is required often requires significant judgment, including the long-range forecast of future taxable income and the evaluation of tax planning initiatives. Adjustments to the deferred tax valuation allowances are made to earnings in the period when such assessments are made. Due to the historical losses from our operations, we have recorded a full valuation allowance on our deferred tax assets.
Recent Accounting Pronouncements
In September 2011, the Financial Accounting Standards Board issued an update to the accounting standard that permits an entity to make a qualitative assessment of whether it is more likely than not that a reporting unit's fair value is less than its carrying value before applying the two-step goodwill impairment model. If it is determined through the qualitative assessment that a reporting unit's fair value is, more likely than not, greater than its carrying value, the remaining impairment steps would be unnecessary. The qualitative assessment is optional, allowing companies to proceed directly to the quantitative assessment. This guidance was effective for interim and annual reporting periods beginning January 1, 2012. Our adoption of this standard on January 1, 2012 did not have a material impact on our combined and consolidated financial statements as we used the quantitative two-step goodwill impairment model as of the October 1, 2012 annual impairment test.
Income Taxes
Except for the pre-acquisition federal and state tax filings for Magen BioSciences, Inc. and certain separate state filings through the June 14, 2010 spin-off, our operations prior to June 14, 2010 have been included in the consolidated federal and combined state tax returns of PPD. As such, except for the pre-acquisition tax attributes of Magen BioSciences, Inc., and some losses from certain separate filing states, the tax attributes of our operations prior to June 14, 2010 have been utilized or paid by PPD. Thus, the tax attributes which have been included in PPD’s combined returns have not been accounted for in the results of our operations. Subsequent to June 14, 2010, we have filed federal and state returns separately from PPD and can use our tax attributes accordingly.
Potential Volatility of Annual Operating Results
Our annual operating results have fluctuated in the past, and we expect that they will continue to fluctuate in the future. Factors that could cause these fluctuations to occur include:
| | • | | the success of achieving milestones and the timing of our milestone payments or other revenue, if any; |
| | • | | our dependence on a small number of compounds and collaborations; |
| | • | | the success or failure of clinical trials and other aspects of developing and commercializing our product candidates; |
| | • | | our ability to properly manage our growth; |
| | • | | the timing and amount of costs associated with research and development and compound partnering collaborations; |
| | • | | our ability to recruit and retain experienced personnel; |
| | • | | the timing and extent of new government regulations; and |
| | • | | intellectual property risks. |
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Under our current investment policies, we invest our cash, cash equivalents and investments in money market funds that invest in short-term U.S. Treasury securities with insignificant rates of return. Due to the short-term nature of our investments, we do not believe that a decrease in market rates would have a significant negative impact on the value of our cash, cash equivalents and investments.
Under our Amended and Restated Loan and Security Agreement with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank, we have $40.0 million of outstanding debt due August 2, 2016. This debt bears interest at a fixed rate of 10.00%, subject to adjustment under specified conditions unrelated to current market interest rates. Due to the fixed rate associated with this outstanding debt, an increase in market rates would have no impact on our future obligations as of December 31, 2012.
The net sales associated with Nesina and related combination products and Priligy that generate our current stream of royalty payments are denominated in the respective foreign currency of the underlying location of the sale of the respective product. The fluctuations of foreign currencies with respect to the U.S. dollar have historically been insignificant and have not exposed us to material risk. However, this exposure could become material in future periods based upon fluctuations in currency rates.
Our purchases of raw materials and finished goods are denominated primarily in U.S. dollars; purchases denominated in currencies other than the U.S. dollar are insignificant. Additionally, our net assets denominated in currencies other than the U.S. dollar are insignificant and have not historically exposed us to material risk associated with fluctuations in currency rates.
Given these facts, we have not yet considered it necessary to use foreign currency contracts or other derivative instruments to manage changes in currency rates. However, we continually evaluate the potential need to do so and may use derivative financial instruments for speculative or trading purposes in the future.
Item 8. Financial Statements and Supplementary Data
The information required by this Item is set forth in the Combined and Consolidated Financial Statements and Notes thereto beginning at page F-1 of this Report.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
Disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e)) are designed only to provide reasonable assurance that information to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our management, including our President and Chief Medical Officer (our principal executive officer) and our Chief Financial Officer, Treasurer and Assistant Secretary (our principal financial and accounting officer), of the effectiveness of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b). Based upon that evaluation, our President and Chief Medical Officer and our Chief Financial Officer, Treasurer and Assistant Secretary have concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report to provide the reasonable assurance discussed above.
Internal Control Over Financial Reporting
No change to our internal control over financial reporting occurred during the last fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining effective internal control over financial reporting as defined in Rules 13a-15(f) under the Securities Exchange Act of 1934. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of management and our Board of Directors and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
A control system, no matter how well designed and operated, can only provide reasonable, not absolute, assurance that the objectives of the control system are met and must reflect the fact that there are resource constraints that require management to consider the benefits of internal controls relative to their costs. Because of these inherent limitations, management does not expect that our internal controls over financial reporting will prevent all errors and all fraud. Also, internal controls might become inadequate because of changes in business conditions or a decline in the degree of compliance with our policies or procedures.
Management, with the participation of our President and Chief Medical Officer and our Chief Financial Officer, Treasurer and Assistant Secretary, assessed the effectiveness of our internal control over financial reporting as of December 31, 2012. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework. Based on our assessment, we believe that, as of December 31, 2012, our internal control over financial reporting was effective based on those criteria.
Deloitte & Touche, LLP, the registered public accounting firm that audited the financial statements included in this annual report, has also issued an opinion on our internal control over financial reporting.
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
Furiex Pharmaceuticals, Inc.
Morrisville, North Carolina
We have audited the internal control over financial reporting of Furiex Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2012, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed by, or under the supervision of, the company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the company’s Board of Directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2012, based on the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements as of and for the year ended December 31, 2012, of the Company and our report dated March 18, 2013, expressed an unqualified opinion on those financial statements.
/s/ DELOITTE & TOUCHE LLP
Raleigh, North Carolina
March 18, 2013
Item 9B. Other Information
None.
Item 10. Directors, Executive Officers and Corporate Governance
Information required by this Item concerning our directors is incorporated by reference from the section captioned “Proposal No. 1—Election of Directors” contained in our proxy statement related to the 2013 Annual Meeting of Stockholders scheduled to be held on May 24, 2013 which we intend to file with the SEC within 120 days of the end of our fiscal year pursuant to General Instruction G(3) of Form 10-K.
The Board of Directors has determined that the members of the Audit Committee are independent as defined in Rule 5605(a)(2) of the NASDAQ listing standards. The Board of Directors has also determined that Committee Chair Robert P. Ruscher is an “audit committee financial expert” as defined in Item 401(h) of Regulation S-K.
Our Board of Directors adopted a code of conduct that applies to all of our directors and employees. Our Board also adopted a separate code of ethics for our President (principal executive officer), Chief Financial Officer (principal financial and accounting officer), and Corporate Controller, or persons performing similar functions. We will provide copies of our code of conduct and code of ethics without charge upon request. To obtain a copy of our code of conduct and code of ethics, please send your written request to Furiex Pharmaceuticals, Inc., 3900 Paramount Parkway, Suite 150, Morrisville, North Carolina 27560, Attn: Investor Relations. In addition, you can find those codes on our website at http://www.furiex.com/investors/corporate-governance/.
The information required by this Item concerning executive officers of the Registrant is set forth at the end of Part I of this report.
The information required by this Item concerning compliance with Section 16(a) of the United States Securities Exchange Act of 1934, as amended, is incorporated by reference from the section of the proxy statement captioned “—Section 16(a) Beneficial Ownership Reporting Compliance.”
Item 11. Executive Compensation
The information required by this Item is incorporated by reference to the information under the sections captioned “—Compensation for Non-Employee Directors,” “—Compensation Discussion and Analysis,” “—Summary Compensation Table,” “—Grants of Plan Based Awards in Fiscal 2012,” “—Outstanding Equity Awards at Fiscal Year End 2012,” “—Compensation Committee Report,” and “—Compensation Committee Interlocks and Insider Participation” contained in the proxy statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Equity Compensation Plans
The following table sets forth the indicated information as of December 31, 2012 with respect to our equity compensation plans:
| | Number of securities to be issued upon exercise of outstanding options, warrants and rights | | | Weighted-average exercise price of outstanding options, warrants and rights | | | Number of securities remaining available for future issuance under equity compensation plans | |
| Equity compensation plans approved by our shareholders | | | 1,643,349 | | | $ | 12.33 | | | | 1,335 | |
| Equity compensation plans not approved by our shareholders | | | — | | | | — | | | | — | |
| Total | | | 1,643,349 | | | $ | 12.33 | | | | 1,335 | |
Our equity compensation plan consists of the 2010 Stock Plan, which was approved by our stockholders. We do not have any equity compensation plans or arrangements that have not been approved by our stockholders.
The other information required by this Item is incorporated by reference to the information under the section captioned “—Security Ownership of Management and Certain Beneficial Owners” contained in the proxy statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference to the information under the section captioned “—Related Party Transactions” and “Proposal No. 1—Election of Directors—Information About the Board of Directors and its Committees” contained in the proxy statement.
Item 14. Principal Accounting Fees and Services
The information required by this Item is incorporated by reference to the information under the section captioned “—Report of the Audit Committee” and “—Fees Paid to the Independent Registered Public Accounting Firm” contained in the proxy statement.
Item 15. Exhibits, Financial Statement Schedules
(a) Financial Statements
Our combined and consolidated financial statements filed as part of this report are listed in the attached Index to Combined and Consolidated Financial Statements. There are no schedules to our combined and consolidated financial statements.
| Exhibit No. | | Exhibit Description | | Registrant’s Form | | Dated | | Exhibit Number | | Filed Herewith |
| 2.1 | | Separation and Distribution Agreement by and between Furiex Pharmaceuticals, Inc. and Pharmaceutical Product Development, Inc. | | 8-K | | 6/18/10 | | 2.1 | | |
| | | | | | | | | | | |
| 3.1 | | Amended and Restated Certificate of Incorporation. | | 10-12B | | 2/24/10 | | 3.1 | | |
| | | | | | | | | | | |
| 3.2 | | Amended and Restated Bylaws. | | 10-12B | | 2/24/10 | | 3.2 | | |
| | | | | | | | | | | |
| 10.1 | | Sublease Agreement dated as of June 14, 2010 by and between Furiex Pharmaceuticals, Inc. and PPD Development, LP. | | 8-K | | 6/18/10 | | 10.2 | | |
| | | | | | | | | | | |
| 10.2 | | Tax Sharing Agreement dated as of June 14, 2010 by and between Furiex Pharmaceuticals, Inc. and Pharmaceutical Product Development, Inc. | | 8-K | | 6/18/10 | | 10.3 | | |
| | | | | | | | | | | |
| 10.3 | | Employee Matters Agreement dated as of June 14, 2010 by and between Furiex Pharmaceuticals, Inc. and Pharmaceutical Product Development, Inc. | | 8-K | | 6/18/10 | | 10.4 | | |
| | | | | | | | | | | |
| 10.4 | | Transition Services Agreement dated as of June 14, 2010 by and between Furiex Pharmaceuticals, Inc. and Pharmaceutical Product Development, Inc. | | 8-K | | 6/18/10 | | 10.5 | | |
| | | | | | | | | | | |
| 10.5† | | Master Development Services Agreement dated as of June 14, 2010 by and between Furiex Pharmaceuticals, Inc. and PPD Development, LP. | | 8-K | | 6/18/10 | | 10.6 | | |
| | | | | | | | | | | |
| 10.6† | | MuDelta Development and License Agreement dated as of November 16, 2009 by and between Janssen Pharmaceutica, N.V. and PPD Therapeutics, Inc., as amended February 9, 2010. | | 10-12B/A | | 5/14/10 | | 10.6 | | |
| | | | | | | | | | | |
| 10.7† | | MuDelta Master Services Agreement dated as of November 16, 2009 by and between Janssen Pharmaceutica, N.V. and PPD Therapeutics, Inc. | | 10-12B | | 2/24/10 | | 10.7 | | |
| | | | | | | | | | | |
| 10.8† | | Topo Development and License Agreement, dated as of November 16, 2009 by and between Janssen Pharmaceutica, N.V. and PPD Therapeutics, Inc., as amended February 15, 2010. | | 10-12B/A | | 5/14/10 | | 10.8 | | |
| | | | | | | | | | | |
| 10.9† | | Topo Master Services Agreement dated as of November 16, 2009 by and between Janssen Pharmaceutica, N.V. and PPD Therapeutics, Inc. | | 10-12B | | 2/24/10 | | 10.9 | | |
| | | | Registrant’s Form | | Dated | | Exhibit Number | | Filed Herewith |
| 10.10† | | License Agreement dated as of January 2, 2001 by and among Pharmaceutical Product Development, Inc., GenuPro, Inc. and Alza Corporation, as amended December 26, 2003 and October 16, 2009. | | 10-12B/A | | 5/25/10 | | 10.10 | | |
| | | | | | | | | | | |
| 10.11† | | Agreement between Takeda San Diego, Inc., Takeda Pharmaceutical Company Limited, Development Partners LLC, and Pharmaceutical Product Development, Inc., dated as of July 13, 2005, as amended October 10, 2005. | | 10-12B/A | | 5/27/10 | | 10.11 | | |
| | | | | | | | | | | |
| 10.12† | | Termination and License Agreement dated as of December 18, 2003 by and among Eli Lilly and Company, Pharmaceutical Product Development, Inc., GenuPro, Inc. and APBI Holdings, LLC. | | 10-12B | | 2/24/10 | | 10.12 | | |
| | | | | | | | | | | |
| 10.13† | | Option and License Agreement effective as of December 15, 2006 among Pharmaco Investments, Inc. and Ranbaxy Laboratories Ltd. | | 10-12B/A | | 5/25/10 | | 10.13 | | |
| | | | | | | | | | | |
| 10.16 | | Employment Agreement effective as of March 16, 2010 between Furiex Pharmaceuticals, Inc. and June S. Almenoff, M.D. Ph.D. | | 10-12B/A | | 5/14/10 | | 10.16 | | |
| | | | | | | | | | | |
| 10.17 | | Employment Agreement effective as of April 1, 2010 between Furiex Pharmaceuticals, Inc. and Gail McIntyre. | | 10-12B/A | | 5/14/10 | | 10.17 | | |
| | | | | | | | | | | |
| 10.18 | | Employment Agreement effective as of January 15, 2010 between Furiex Pharmaceuticals, Inc. and Paul S. Covington, M.D. | | 10-12B/A | | 5/14/10 | | 10.18 | | |
| | | | | | | | | | | |
| 10.19 | | Employment Agreement effective as of January 29, 2010 between Furiex Pharmaceuticals, Inc. and Marshall Woodworth. | | 10-12B/A | | 5/14/10 | | 10.19 | | �� |
| | | | | | | | | | | |
| 10.20 | | Form of Severance Agreement between Furiex Pharmaceuticals, Inc. and various individuals. | | 10-12B | | 2/24/10 | | 10.20 | | |
| | | | | | | | | | | |
| 10.21 | | 2010 Stock Plan. | | 10-12B | | 2/24/10 | | 10.21 | | |
| | | | | | | | | | | |
| 10.22 | | Consulting Agreement by and between Furiex Pharmaceuticals, Inc., Elk Mountain Consulting, LLC, and Fredric N. Eshelman. | | 8-K | | 6/18/10 | | 10.7 | | |
| | | | | | | | | | | |
| 10.23† | | Loan and Security Agreement dated August 18, 2011 with Midcap Funding III, LLC and Silicon Valley Bank (the Lenders). | | 10-Q | | 11/10/11 | | 10.23 | | |
| | | | | | | | | | | |
| 10.24 | | Pledge Agreement dated August 18, 2011 with the Lenders. | | 10-Q | | 11/10/11 | | 10.24 | | |
| | | | | | | | | | | |
| 10.25 | | Secured Promissory Note dated August 18, 2011 to the Lenders. | | 10-Q | | 11/10/11 | | 10.25 | | |
| | | | | | | | | | | |
| 10.26† | | License and Asset Transfer Agreement, dated as of May 14, 2012, by and between Furiex Pharmaceuticals, Inc. and Genupro, Inc. and Alza Corporation and Janssen Pharmaceutica, NV. | | 10-Q | | 8/7/12 | | 10.26 | | |
| | | | | | | | | | | |
| 10.27† | | Priligy License Agreement, dated as of May 14, 2012, by and between Genupro, Inc. and Berlin Chemie AG (Menarini Group). | | 10-Q | | 8/7/12 | | 10.27 | | |
| | | | Registrant’s Form | | Dated | | Exhibit Number | | Filed Herewith |
| 10.28† | | Amended and Restated Loan and Security Agreement dated August 2, 2012 by and among MidCap Funding III, LLC, Furiex Pharmaceuticals, Inc., APBI Holdings, LLC, Development Partners, LLC and Genupro, Inc. | | 10-Q/A | | 2/12/13 | | 10.28 | | |
| | | | | | | | | | | |
| 10.29 | | Omnibus Amendment and Reaffirmation Agreement dated August 2, 2012 by and among MidCap Funding III, LLC, Furiex Pharmaceuticals, Inc., APBI Holdings, LLC, Development Partners, LLC and Genupro, Inc. | | 10-Q | | 11/6/12 | | 10.29 | | |
| | | | | | | | | | | |
| 21.1 | | Subsidiaries of Furiex Pharmaceuticals, Inc. | | | | | | | | X |
| | | | | | | | | | | |
| 23.1 | | Consent of Independent Registered Public Accounting Firm. | | | | | | | | X |
| | | | | | | | | | | |
| 31.1 | | Certification by the principal executive officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| | | | | | | | | | | |
| 31.2 | | Certification by the principal financial officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| | | | | | | | | | | |
| 32.1 | | Certification by the principal executive officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| | | | | | | | | | | |
| 32.2 | | Certification by the principal financial officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| | | | | | | | | | | |
| 101 | | Financial information from the Company’s Annual Report on Form 10-K for the period ended December 31, 2012 formatted in eXtensible Business Reporting Language (XBRL). | | | | | | | | X |
��
| † | The registrant has requested confidential treatment with respect to portions of this exhibit. These portions have been omitted from the exhibit and filed separately with the U.S. Securities and Exchange Commission. |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | |
| | | | | FURIEX PHARMACEUTICALS, INC. |
| | | | |
| Date: March 18, 2013 | | | | By: | | / S / JUNE S. ALMENOFF |
| | | | | Name: | | June S. Almenoff |
| | | | | Title: | | President |
| | | | | | | (Principal Executive Officer) |
| | | | |
| | | | | By: | | / S / MARSHALL H. WOODWORTH |
| | | | | Name: | | Marshall H. Woodworth |
| | | | | Title: | | Chief Financial Officer |
| | | | | | | (Principal Financial Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | President and Director | | March 18, 2013 |
| June S. Almenoff | | (Principal Executive Officer) | | |
| | | |
/ S / MARSHALL H. WOODWORTH | | Chief Financial Officer | | March 18, 2013 |
| Marshall H. Woodworth | | (Principal Financial and Accounting Officer) | | |
| | | |
/ S / FREDRIC N. ESHELMAN | | Chairman | | March 18, 2013 |
| Fredric N. Eshelman | | | | |
| | | |
| | Director | | March 18, 2013 |
| Peter B. Corr | | | | |
| | | |
| | Director | | March 18, 2013 |
| Wendy L. Dixon | | | | |
| | | |
| | Director | | March 18, 2013 |
| Stephen W. Kaldor | | | | |
| | | |
| | Director | | March 18, 2013 |
| Robert P. Ruscher | | | | |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
INDEX TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
| | | Page | |
| Report of Independent Registered Public Accounting Firm | | | F-2 | |
| Combined and Consolidated Statements of Operations for the Years Ended December 31, 2010, 2011 and 2012 | | | F-3 | |
| Consolidated Balance Sheets as of December 31, 2011 and 2012 | | | F-4 | |
| Combined and Consolidated Statements of Shareholders’ Equity for the Years Ended December 31, 2010, 2011 and 2012 | | | F-5 | |
| Combined and Consolidated Statements of Cash Flows for the Years Ended December 31, 2010, 2011 and 2012 | | | F-6 | |
| Notes to Combined and Consolidated Financial Statements | | | F-7 | |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
Furiex Pharmaceuticals, Inc.
Morrisville, North Carolina
We have audited the accompanying consolidated balance sheets of Furiex Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2011 and 2012, and the related combined and consolidated statements of operations, shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2012. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such combined and consolidated financial statements present fairly, in all material respects, the financial position of Furiex Pharmaceuticals, Inc. and subsidiaries as of December 31, 2011 and 2012, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2012, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2012, based on the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 18, 2013 expressed an unqualified opinion on the Company’s internal control over financial reporting.
| |
| /s/ DELOITTE & TOUCHE LLP |
| Raleigh, North Carolina |
| |
| March 18, 2013 |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
COMBINED AND CONSOLIDATED STATEMENTS OF OPERATIONS FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(in thousands, except per share data)
| | | 2010 | | | 2011 | | | 2012 | |
| Revenue: | | | | | | | | | | | | |
| Milestones | | $ | 7,500 | | | $ | — | | | $ | 20,000 | |
| Royalties | | | 1,330 | | | | 4,490 | | | | 20,508 | |
| Service | | | 75 | | | | — | | | | — | |
| Other | | | 78 | | | | — | | | | — | |
| | | | | | | | | | | | | |
| Total revenue | | | 8,983 | | | | 4,490 | | | | 40,508 | |
| | | | | | | | | | | | | |
| Direct expenses | | | 21 | | | | — | | | | — | |
| Research and development expenses | | | 50,112 | | | | 44,202 | | | | 69,505 | |
| Selling, general and administrative expenses | | | 8,262 | | | | 8,761 | | | | 11,261 | |
| Depreciation and amortization | | | 109 | | | | 83 | | | | 86 | |
| | | | | | | | | | | | | |
| Total operating expenses | | | 58,504 | | | | 53,046 | | | | 80,852 | |
| | | | | | | | | | | | | |
| Operating loss | | | (49,521 | ) | | | (48,556 | ) | | | (40,344 | ) |
| Interest expense | | | — | | | | 413 | | | | 2,508 | |
| Other income, net | | | 9 | | | | 2 | | | | 1 | |
| | | | | | | | | | | | | |
| Loss from continuing operations before provision for income taxes | | | (49,512 | ) | | | (48,967 | ) | | | (42,851 | ) |
| Provision for income taxes | | | 14 | | | | 14 | | | | 14 | |
| | | | | | | | | | | | | |
| Loss from continuing operations, net of income taxes | | | (49,526 | ) | | | (48,981 | ) | | | (42,865 | ) |
| Loss from discontinued operations, net of income taxes | | | (5,133 | ) | | | — | | | | — | |
| | | | | | | | | | | | | |
| Net loss | | $ | (54,659 | ) | | $ | (48,981 | ) | | $ | | ) |
| | | | | | | | | | | | | |
| Loss from continuing operations, net of income taxes per basic and diluted share | | $ | (5.01 | ) | | $ | (4.96 | ) | | $ | (4.29) | |
| | | | | | | | | | | | | |
| Loss from discontinued operations, net of income taxes per basic and diluted share | | $ | (0.52 | ) | | $ | — | | | $ | — | |
| | | | | | | | | | | | | |
| Net loss per basic and diluted share | | $ | (5.53 | ) | | $ | (4.96 | ) | | $ | (4.29) | |
| | | | | | | | | | | | | |
| Weighted-average shares used to compute net loss per basic and diluted share: | | | 9,881 | | | | 9,884 | | | | 9,984 | |
The accompanying notes are an integral part of these combined and consolidated financial statements.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS AS OF DECEMBER 31, 2011 AND 2012
(in thousands, except share data)
| | | 2011 | | | 2012 | |
| Assets | | | | | | | | |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 33,628 | | | $ | 25,718 | |
| Short-term investments | | | 10,000 | | | | — | |
| Accounts receivable, net | | | 1,985 | | | | 11,745 | |
| Prepaid expenses | | | 214 | | | | 320 | |
| | | | | | | | | |
| Total current assets | | | 45,827 | | | | 37,783 | |
| Property and equipment, net | | | 181 | | | | 118 | |
| Investments | | | — | | | | 7,500 | |
| Goodwill | | | 49,116 | | | | 49,116 | |
| Other assets | | | — | | | | 238 | |
| | | | | | | | | |
| Total assets | | $ | 95,124 | | | $ | 94,755 | |
| | | | | | | | | |
| Liabilities and Shareholders’ Equity | | | | | | | | |
| Current liabilities: | | | | | | | | |
| Accounts payable | | $ | 147 | | | $ | 6,604 | |
| Accrued expenses | | | 10,422 | | | | 10,230 | |
| Current portion of long-term debt | | | 1,351 | | | | 5,405 | |
| | | | | | | | | |
| Total current liabilities | | | 11,920 | | | | 22,239 | |
| Long-term debt, net | | | 8,649 | | | | 34,595 | |
| Other long-term liabilities | | | 232 | | | | 324 | |
| | | | | | | | | |
| Total liabilities | | | 20,801 | | | | 57,158 | |
| | | | | | | | | |
| Commitments and contingencies (Note 13) | | | | | | | | |
| Common stock, $0.001 par value, 40,000,000 shares authorized; 9,949,422 and 10,015,297 shares issued and outstanding, respectively | | | 10 | | | | 10 | |
| Preferred stock, $0.001 par value, 10,000,000 shares authorized; no shares issued or outstanding | | | — | | | | — | |
| Paid-in capital | | | 158,438 | | | | 164,577 | |
| Accumulated deficit | | | (84,125 | ) | | | (126,990 | ) |
| | | | | | | | | |
| Total shareholders’ equity | | | 74,323 | | | | 37,597 | |
| | | | | | | | | |
| Total liabilities and shareholders’ equity | | $ | 95,124 | | | $ | 94,755 | |
The accompanying notes are an integral part of these combined and consolidated financial statements.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
COMBINED AND CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(in thousands)
| | | Common Stock | | | Paid-in capital | | | Accumulated deficit | | | Parent Company Investment | | | Total | |
| | | Shares | | | Par value | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance January 1, 2010 | | | — | | | $ | — | | | $ | — | | | $ | — | | | $ | 49,270 | | | $ | 49,270 | |
| Net transfers from parent | | | — | | | | — | | | | — | | | | — | | | | 16,046 | | | | 16,046 | |
| Net liability retained by parent | | | — | | | | — | | | | — | | | | — | | | | 6,637 | | | | 6,637 | |
| Stock compensation expense | | | — | | | | — | | | | 1,210 | | | | — | | | | — | | | | 1,210 | |
| Contribution of cash and cash equivalents from parent | | | — | | | | — | | | | — | | | | — | | | | 100,000 | | | | 100,000 | |
| Contribution of net operating assets and liabilities to Furiex Pharmaceuticals, Inc. and issuance of common shares to Pharmaceutical Product Development, Inc. shareholders | | | 9,881 | | | | 10 | | | | 152,428 | | | | — | | | | (152,438 | ) | | | — | |
| Net loss | | | — | | | | — | | | | — | | | | (35,144 | ) | | | (19,515 | ) | | | (54,659 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance December 31, 2010 | | | 9,881 | | | $ | 10 | | | $ | 153,638 | | | $ | (35,144 | ) | | $ | — | | | $ | 118,504 | |
| Exercise of common stock options | | | 68 | | | | — | | | | 620 | | | | — | | | | — | | | | 620 | |
| Stock compensation expense | | | — | | | | — | | | | 4,180 | | | | — | | | | — | | | | 4,180 | |
| Net loss | | | — | | | | — | | | | — | | | | (48,981 | ) | | | — | | | | (48,981 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance December 31, 2011 | | | 9,949 | | | $ | 10 | | | $ | 158,438 | | | $ | (84,125 | ) | | $ | — | | | $ | 74,323 | |
| Exercise of common stock options | | | 66 | | | | — | | | | 600 | | | | — | | | | — | | | | 600 | |
| Stock compensation expense | | | — | | | | — | | | | 5,539 | | | | — | | | | — | | | | 5,539 | |
| Net loss | | | — | | | | — | | | | — | | | | (42,865 | ) | | | — | | | | | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance December 31, 2012 | | | 10,015 | | | $ | 10 | | | $ | 164,577 | | | $ | (126,990 | ) | | $ | — | | | $ | 37,597 | |
The accompanying notes are an integral part of these combined and consolidated financial statements.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
COMBINED AND CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(in thousands)
| | | 2010 | | | 2011 | | | 2012 | |
| Cash flows from operating activities: | | | | | | | | | | | | |
| Net loss | | $ | (54,659 | ) | | $ | (48,981 | ) | | $ | (42,865 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
| Depreciation and amortization | | | 1,169 | | | | 83 | | | | 86 | |
| Stock compensation expense | | | 1,210 | | | | 4,180 | | | | 5,539 | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | |
| Accounts receivable, net | | | 302 | | | | (1,726 | ) | | | (9,760 | ) |
| Prepaid expenses and other current assets | | | 1,038 | | | | 752 | | | | (106 | ) |
| Other assets | | | — | | | | — | | | | 62 | |
| Accounts payable | | | 21 | | | | (3 | ) | | | 6,511 | |
| Accrued expenses | | | 7,446 | | | | (3,345 | ) | | | (192 | ) |
| Deferred rent | | | (43 | ) | | | — | | | | — | |
| Other long-term liabilities | | | 192 | | | | 40 | | | | 92 | |
| | | | | | | | | | | | | |
| Net cash used in operating activities | | | (43,324 | ) | | | (49,000 | ) | | | (40,633 | ) |
| | | | | | | | | | | | | |
| Cash flows from investing activities: | | | | | | | | | | | | |
| Purchases of property and equipment | | | (683 | ) | | | (22 | ) | | | (77 | ) |
| Purchases of investments | | | — | | | | (10,000 | ) | | | (7,500 | ) |
| Proceeds from sale of investments | | | — | | | | — | | | | 10,000 | |
| Net proceeds from sale of businesses | | | 3,464 | | | | — | | | | — | |
| | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | 2,781 | | | | (10,022 | ) | | | 2,423 | |
| | | | | | | | | | | | | |
| Cash flows from financing activities: | | | | | | | | | | | | |
| Proceeds from borrowings on long-term debt | | | — | | | | 10,000 | | | | 30,000 | |
| Deferred financing costs | | | — | | | | — | | | | (300 | ) |
| Proceeds from issuance of common stock | | | — | | | | 620 | | | | 600 | |
| Net change in investment from parent | | | 22,567 | | | | — | | | | — | |
| Cash contributed by parent | | | 100,000 | | | | — | | | | — | |
| | | | | | | | | | | | | |
| Net cash provided by financing activities | | | 122,567 | | | | 10,620 | | | | 30,300 | |
| | | | | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | | 82,024 | | | | (48,402 | ) | | | (7,910 | ) |
| Cash and cash equivalents, beginning of the year | | | 6 | | | | 82,030 | | | | 33,628 | |
| | | | | | | | | | | | | |
| Cash and cash equivalents, end of the year | | $ | 82,030 | | | $ | 33,628 | | | $ | 25,718 | |
| | | | | | | | | | | | | |
| Supplemental Disclosure of Cash Flow Information: | | | | | | | | | | | | |
| Interest paid | | $ | — | | | $ | 299 | | | $ | 2,020 | |
The accompanying notes are an integral part of these combined and consolidated financial statements.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
1. Summary of Operations and Significant Accounting Policies
Organization and Business Description
Furiex Pharmaceuticals, Inc., a Delaware corporation (“Furiex” or the “Company”), is a drug development company that continues the compound partnering business started by Pharmaceutical Product Development, Inc. (“PPD”) in 1998. On June 14, 2010, PPD effected the spin-off of Furiex through a tax-free, pro-rata dividend distribution of all of the shares of the Company to PPD shareholders. PPD transferred the compound partnering business (previously part of the Discovery Science segment of PPD), including assets, employees, intellectual property rights and liabilities comprising that business, and $100.0 million in cash as of the spin-off date. PPD does not have any ownership or other form of equity interest in the Company following the spin-off.
The goal of the Company is to in-license compounds from, or form strategic alliances with, pharmaceutical and biotechnology companies to share the risks and rewards of developing therapeutics. The Company's operations are headquartered in Morrisville, North Carolina.
The Company has incurred losses and negative cash flows from operations since the spin-off. Based on current forecasts, the Company believes it has sufficient liquidity to continue its planned operations for at least the next 12 months. The Company's long-term liquidity needs will largely be determined by the success of its products already being commercialized by collaborators, key development and regulatory events that might impact the Company's ability to out-license its development compounds, and expenses associated with research and development efforts. Depending upon the success and timing of receipt of various milestone payments and royalties, it might be necessary to do one or more of the following in the next 12 months: (a) raise additional capital through equity or debt financings or from other sources; (b) reduce spending on research and development; or (c) restructure the Company's operations. The Company currently receives on-going revenue from royalties on sales of Nesina®, Liovel® and Priligy®. The Company will continue to incur operating losses unless revenues from all sources reach a level sufficient to support its on-going operations.
Basis of Accounting
The accompanying combined and consolidated financial statements, through the date of the spin-off from PPD, have been derived from the combined financial statements and accounting records of PPD from the historical cost basis of the assets and liabilities of the various activities that reflect the combined results of operations, financial condition and cash flows of the Discovery Sciences segment of PPD. All the business components of the Discovery Sciences segment have been included in the historical statements because they were managed by common segment management, and because they reflect the historical performance of PPD segment management.
PPD’s net investment in the Company is shown in lieu of shareholders’ equity in the combined financial statements prior to the spin-off as a direct ownership relationship did not exist among all the components comprising the Company. The net investment account represents the cumulative investments in, distributions from and earnings (losses) of the Company. Prior to the spin-off, all cash was held and managed by PPD. Accordingly, cash used to pay the Company’s expenses or cash collected from collaboration agreements, royalties or customer contracts by PPD on behalf of the Company was recorded as an increase or decrease in PPD’s net investment.
In May 2010, PPD discontinued the operations of its wholly owned subsidiary, PPD Dermatology, Inc., due to unfavorable efficacy data associated with its MAG-131 program. This business unit is recorded as discontinued operations in the accompanying combined and consolidated financial statements. Additionally, the Discovery Sciences segment of PPD included pre-clinical consulting services not offered by Furiex after the spin-off. As such, the accompanying financial information does not reflect the results of operations or cash flows of the Company had it been a separate, stand-alone entity during the periods presented prior to the spin-off. All rights and obligations related to pre-clinical consulting services and the definitive purchase agreements related to PPD Dermatology, Inc. were retained by PPD.
The Company was allocated expenses from PPD, such as executive oversight, risk management, accounting, tax, legal, investor relations, human resources, information technology, stock compensation, and facilities services and depreciation, but was not allocated the underlying productive assets, such as information systems equipment, furniture and facilities that were not assigned to the Company, but from which the Company benefited. Such expenses have been included in the combined and consolidated financial statements as expense allocations from PPD for periods prior to the spin-off. The basis of these allocations included full-time equivalent employees for the respective periods presented and square footage of occupied space. See Note 15 for further discussion of the allocations.
Management believes that the assumptions and allocations underlying the combined and consolidated financial statements are reasonable. However, the financial information in these combined and consolidated financial statements does not include all of the expenses that would have been incurred had the Company been a separate, stand-alone publicly traded entity prior to the spin-off.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
Principles of Combination and Consolidation
The Company prepared the accompanying combined and consolidated financial statements in accordance with accounting principles generally accepted in the United States of America, or GAAP, and they include the accounts of Furiex Pharmaceuticals, Inc. and its subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates in Preparation of the Financial Statements
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
The Company calculates net loss per basic and diluted share by dividing net loss by the weighted-average number of shares outstanding during the reporting period. The calculation of net loss per diluted share is the same as net loss per basic share since the inclusion of any potentially dilutive securities would be anti-dilutive for the years ended December 31, 2010, 2011 and 2012. All potentially dilutive securities relate to stock options issued as part of the Company's share-based compensation plan. Potentially dilutive securities totaling approximately 839,000, 1,511,000 and 1,644,000 options for the years ended December 31, 2010, 2011 and 2012, respectively, were excluded from the calculation of diluted loss per share because of their anti-dilutive effect.
Separation Costs
In 2010, the Company incurred legal, tax and other costs specifically associated with the spin-off, which are recorded as a component of selling, general and administrative expenses. These amounts for the year ended December 31, 2010 were $2.6 million.
Comprehensive Income (Loss)
There are no items of comprehensive loss other than net loss for any periods presented.
Revenue Recognition
The Company generates revenue in the form of upfront payments, development and regulatory milestone payments, royalties, and launch-based and sales-based milestone payments in connection with the out-license or sale of compounds. The receipt of future milestone payments and royalties depends on the success of the Company's compound development and the Company's collaborators' success in developing and commercializing compounds. Upfront payments are generally paid within a short period of time following the execution of an out-license or collaboration agreement. Development and regulatory milestone payments are typically one-time payments to the Company triggered by the collaborator's achievement of specified development and regulatory events such as the commencement of Phase III trials or regulatory submission approval. Royalties are payments received by the Company based on net product sales of a collaborator. Launch-based milestone payments are one-time payments to the Company triggered when a collaborator first introduces for sale an out-licensed product in a new geographical region. Sales-based milestone payments are typically one-time payments to the Company triggered when aggregate net sales of product by a collaborator for a specified period (for example, an annual period) reach an agreed upon threshold amount. The Company recognizes upfront payments, development and regulatory milestone payments, royalty payments, and launch-based and sales-based milestone payments from its collaborators when the event that triggers the obligation of payment has occurred, there is no further obligation on the Company's part in connection with the payment and collection is reasonably assured.
The Company assesses each collaboration agreement it enters into for potential indicators associated with agent and principal considerations for each related cash payment and receipt. Based on the terms of the underlying agreement and the determination of which party to the transaction is the primary obligor, the Company records the underlying activities that are associated with contractual payments within the combined and consolidated statements of operations and consolidated balance sheets on a net or gross basis, accordingly.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
Concentration of Credit Risk
The Company's collaborators, which are its current sources of revenue, are primarily pharmaceutical companies. A concentration of credit risk with respect to revenue exists due to the small number of collaborators. Three collaborators accounted for all of the Company's revenue for the years ended December 31, 2010, 2011 and 2012. The first collaborator accounted for $7.9 million, $3.6 million and $28.1 million of total revenue for the years ended December 31, 2010, 2011 and 2012, respectively. The second collaborator accounted for $1.0 million, $0.8 million and $2.4 million of total revenue for the years ended December 31, 2010, 2011 and 2012, respectively. The third collaborator accounted for $10.0 million of total revenue for the year ended December 31, 2012. The Company had no revenue from the third collaborator for the years ended December 31, 2010 and 2011.
The December 31, 2011 and 2012 balance of accounts receivable relates to royalty receivables related to Nesina related products and Priligy based on net product sales by the Company's collaborators and, as described in Note 13, $1.7 million due from a collaborator to fund on-going clinical study costs associated with Priligy. One collaborator accounted for the majority of the accounts receivable balance as of December 31, 2011 and 2012, respectively.
Research and Development Expenses
Research and development costs consist primarily of costs associated with pre-clinical studies, non-clinical studies and the clinical trials of the Company's product candidates, development materials, labor and related benefit charges associated with personnel performing research and development work, supplies associated with this work and consulting services. Research and development costs include clinical research services, pre-clinical testing, non-clinical testing and clinical drug manufacturing provided by third parties, the direct cost of the Company's personnel managing the programs and upfront and milestone payments to the Company's collaborators. The Company charges research and development costs to operations as incurred and discloses them in the combined and consolidated statements of operations.
The Company calculated its income tax provision for the periods prior to June 14, 2010 using the separate return basis as if the Company had filed separate income tax returns under its existing structure. The provision for income taxes subsequent to the spin-off has been determined using the asset and liability approach of accounting for income taxes. Under this approach, deferred taxes represent the future tax consequences expected to occur when the reported amounts of assets and liabilities are recovered or paid. The provision for income taxes represents income taxes paid or payable for the year, plus the change in deferred taxes during the year. Deferred taxes result from differences between the financial reporting and tax basis of the Company's assets and liabilities. Deferred tax assets and liabilities are measured using the currently enacted tax rates that apply to taxable income in effect for the years in which those tax attributes are expected to be recovered or paid, and are adjusted for changes in tax rates and tax laws when enacted.
Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized. The assessment of whether or not a valuation allowance is required often requires significant judgment, including the long-range forecast of future taxable income and the evaluation of tax planning initiatives. Adjustments to the deferred tax valuation allowances are made to earnings in the period when such assessments are made. Due to the historical losses from the Company's operations, a full valuation allowance on deferred tax assets has been recorded.
The Company recognizes compensation expense using a fair-value based method related to stock options and other share-based compensation. The expense is measured based on the grant date fair value of the awards that are expected to vest and is recorded over the applicable requisite service period. In the absence of an observable market price for a share-based award, the fair value is based upon a valuation methodology that takes into consideration various factors, including the exercise price of the award, the expected term of the award, the current price of the underlying shares, the expected volatility of the underlying share price based on peer companies, the expected dividends on the underlying shares and the risk-free interest rate.
The Company records as goodwill the excess of the purchase price of a business acquired over the fair value of net tangible assets and identifiable intangible assets at the date of the acquisition. The Company evaluates goodwill for impairment on an annual basis each October 1 or more frequently if events or changes in circumstances indicate that goodwill might be impaired. Any impairment could have a material adverse effect on the Company's financial condition and results of operations.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
Cash, Cash Equivalents, Short-Term Investments and Investments
Cash and cash equivalents consist of unrestricted cash accounts that are not subject to withdrawal restrictions or penalties, and all highly liquid investments that have a maturity of three months or less at the date of purchase.
Short-term investments and investments consist of restricted cash accounts and money market funds that hold short-term U.S. Treasury securities that are subject to contractual withdrawal restrictions and penalties. As described in Note 8, under the Loan and Security Agreement with MidCap Funding III, LLC and Silicon Valley Bank, the Company was required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $10.0 million until mid-January 2012. As of December 31, 2011, this amount was reflected in current assets within the consolidated balance sheets as this requirement expired within 12 months. As also described in Note 8, under the Amended and Restated Loan and Security Agreement with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank, the Company is required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $7.5 million through August 2, 2014, $5.0 million through August 2, 2015 and $2.5 million through August 2, 2016. As of December 31, 2012, this amount is reflected in total assets within the consolidated balance sheets as this requirement will expire August 2, 2016. In addition, the Company must maintain its primary deposit and investment accounts with Silicon Valley Bank, consisting of at least 50% of the Company's total cash and cash equivalents balance.
Realizability of Carrying Value of Long-Lived Assets
The Company reviews the recoverability of long-lived and finite-lived intangible assets when circumstances indicate that the carrying amount of assets might not be recoverable. The Company bases this evaluation on various analyses, including undiscounted cash flow projections. In the event undiscounted cash flow projections indicate impairment, the Company would record an impairment based on the fair value of the assets at the date of the impairment. Property and Equipment
The Company records property and equipment at cost less accumulated depreciation. The Company records depreciation using the straight-line method, based on the following estimated useful lives:
| | | Years |
| Furniture and equipment | | | 5 | – | 10 | |
| Computer equipment and software | | | 2 | – | 5 | |
Operating Leases
The Company records rent expense for operating leases on a straight-line basis over the term of the lease. The Company begins amortization on the date of initial possession, which is generally when the Company enters the space and begins to make improvements in preparation for its intended use. The Company accounts for the difference between rent expense and rent paid as deferred rent. The Company records a deferred rent liability at the inception of the lease term and amortizes the deferred rent over the term of the lease as a reduction to rent expense.
Recent Accounting Pronouncements
In September 2011, the Financial Accounting Standards Board issued an update to the accounting standard that permits an entity to make a qualitative assessment of whether it is more likely than not that a reporting unit's fair value is less than its carrying value before applying the two-step goodwill impairment model. If it is determined through the qualitative assessment that a reporting unit's fair value is, more likely than not, greater than its carrying value, the remaining impairment steps would be unnecessary. The qualitative assessment is optional, allowing companies to proceed directly to the quantitative assessment. This guidance was effective for interim and annual reporting periods beginning January 1, 2012. The Company's adoption of this standard on January 1, 2012 had no material impact on the Company's combined and consolidated financial statements as the Company used the quantitative two-step goodwill impairment model as of the October 1, 2012 annual impairment test.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
2. Spin-off from Pharmaceutical Product Development Inc.
On June 14, 2010, PPD spun off its compound partnering business through the spin-off of Furiex. PPD contributed substantially all of the compound partnering business components of the Discovery Sciences segment and $100.0 million of cash to Furiex. All outstanding shares of Furiex were then distributed to PPD shareholders of record on June 1, 2010 as a pro-rata, tax-free dividend of one share of Furiex common stock for every twelve shares of PPD’s common stock.
In connection with the spin-off, the Company and PPD entered into a series of agreements, including a separation and distribution agreement, transition services agreement, sublease and license agreements, employee matters agreement, tax sharing agreement and a master development services agreement.
The total amount of the Furiex contribution of $152.4 million was based on the book value of the net assets that were transferred to Furiex in connection with the spin-off, as follows:
| | | 2010 | |
| Net book value of assets transferred: | | | | |
| Cash | | $ | 100,000 | |
| Accounts receivable | | | 7,705 | |
| Prepaid expenses | | | 100 | |
| Property and equipment, net | | | 18 | |
| Goodwill | | | 49,116 | |
| Accounts payable | | | (758 | ) |
| Accrued expenses and other current liabilities | | | (3,542 | ) |
| Long-term liabilities | | | (201 | ) |
| | | | | |
| Net assets transferred | | $ | 152,438 | |
3. Discontinued Operations
In April 2009, PPD acquired 100 percent of the outstanding equity interests of Magen BioSciences, Inc., a biotechnology company focused on the development of dermatologic therapies. This business unit, which became known as PPD Dermatology, Inc., was included in the Discovery Sciences segment of PPD.
In May 2010, PPD discontinued the operations of its wholly owned subsidiary, PPD Dermatology, Inc., due to unfavorable efficacy data associated with its MAG-131 program. This business unit, and the respective loss from discontinued operations of $5.1 million for the year ended December 31, 2010, is recorded as discontinued operations in the accompanying combined and consolidated financial statements. All rights and obligations related to PPD Dermatology, Inc. were retained by PPD after the spin-off on June 14, 2010.
Accounts receivable consisted of the following amounts on the dates set forth below:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Royalties | | $ | 1,985 | | | $ | 10,045 | |
| Priligy on-going clinical study costs (Note 13) | | | — | | | | 1,700 | |
| | | | | | | | | |
| | | $ | 1,985 | | | $ | 11,745 | |
The Company did not record a provision for doubtful accounts as of December 31, 2011 or 2012 based on its assessment of collection risks. The December 31, 2012 balance of accounts receivable relate to royalty receivables related to Nesina, Liovel and Priligy based on net product sales by the Company's collaborators and, as described in Note 13, amounts due from a collaborator to fund on-going clinical study costs associated with Priligy.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
5. Property and Equipment
Property and equipment, stated at cost, consisted of the following amounts on the dates set forth below:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Furniture and equipment | | $ | 97 | | | $ | 98 | |
| Computer equipment and software | | | 223 | | | | 245 | |
| | | | | | | | | |
| Total property and equipment | | | 320 | | | | 343 | |
| Less accumulated depreciation | | | (139 | ) | | | (225 | ) |
| | | | | | | | | |
| Total property and equipment, net | | $ | 181 | | | $ | 118 | |
Noncash investing activity related to liabilities that remain unpaid for the acquisition of property and equipment as of December 31, 2011 and 2012 were approximately $0.05 million and zero, respectively.
6. Goodwill
The Company reviews goodwill for impairment annually on October 1 and whenever events or changes in circumstances indicate that the carrying amount of goodwill might not be recoverable. This analysis utilizes both the income and market approaches. For the income approach, the Company uses a discounted cash flow method using the expected future inflows and outflows of the business and an appropriate discount rate. The market approach considers recent comparable transactional valuation multiples for pharmaceutical and biotechnology companies. Based on the review as of October 1, 2012, the Company's calculated fair value of its sole goodwill reporting unit was in excess of carrying value.
The fair value of goodwill could be materially impacted by future adverse changes such as future declines in operating results, a decline in the valuation of pharmaceutical and biotechnology company stocks, including the valuation of the Company's own common stock, a slowdown in the worldwide economy or the pharmaceutical and biotechnology industry, failure to meet the performance projections included in forecasted operating results, or the delay or abandonment of any research and development programs.
7. Accrued Expenses
Accrued expenses consisted of the following amounts on the dates set forth below:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Salaries, wages, benefits and related costs | | $ | 1,346 | | | $ | 1,181 | |
| Research and development costs | | | 8,681 | | | | 3,143 | |
| Professional fees | | | 200 | | | | 101 | |
| Interest | | | 88 | | | | 344 | |
| Janssen transition service payments (Note 13) | | | — | | | | 3,750 | |
| Priligy on-going clinical study costs (Note 13) | | | — | | | | 1,700 | |
| Other | | | 107 | | | | 11 | |
| | | | | | | | | |
| | | $ | 10,422 | | | $ | 10,230 | |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
8. Long-Term Debt
On August 18, 2011, the Company and its subsidiaries entered into a Loan and Security Agreement (the “Agreement”) with MidCap Funding III, LLC and Silicon Valley Bank. This initial borrowing in the amount of $10.0 million had a fixed interest rate of 10.25% per annum and was initially due August 1, 2015. Interest accrued monthly and was payable on the first day of the following month, in arrears. Principal payments of the initial borrowing were to be paid on a ratable monthly basis from August 1, 2012 until maturity. However, on August 2, 2012, the Company and its subsidiaries entered into an Amended and Restated Loan and Security Agreement (the “Amended Agreement”) with MidCap Funding III, LLC, Midcap Funding RE Holdings, LLC and Silicon Valley Bank, or collectively, the Lenders. The Amended Agreement modified the prior Agreement by providing an additional $30.0 million in borrowings and reset the maturity date of the initial Agreement to August 2, 2016.
Under the terms of the Amended Agreement, the total amount due to the Lenders of $40.0 million has a maturity date of August 2, 2016. The Amended Agreement bears interest at a fixed rate of 10.00%, subject to adjustment under specified conditions. Interest accrues daily and is payable on the first day of the following month, in arrears. Principal payments are due on a ratable monthly basis from August 1, 2013 until maturity.
As part of this new agreement, the Company is required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $7.5 million through August 2, 2014, $5.0 million through August 2, 2015 and $2.5 million through August 2, 2016. In addition, the Company must maintain its primary deposit and investment accounts with Silicon Valley Bank, consisting of at least 50% of the Company's total cash and cash equivalents balance. Under the initial Agreement the Company was required to maintain a cash balance with Silicon Valley Bank in an amount of not less than $10.0 million until mid-January 2012. The Company intends to use the proceeds from the loan to support research and development for its MuDelta compound.
A final payment fee is due to the Lenders in an amount equal to 2.5% of the total loan commitment, payable at the maturity date or earlier prepayment of the loan. The Company may prepay the loan subject to a prepayment fee of between one and four percent of the amount borrowed, depending on the time of the prepayment. The amount of interest expense related to the initial Agreement and Amended Agreement included in the statements of operations for the years ended December 31, 2011 and 2012 was $0.4 million and $2.5 million, respectively. Included in these amounts are the ratable accrual of the final payment fee and the amortization of deferred financing costs over the term of the loan, which are included in other long-term liabilities and other assets within the consolidated balance sheets, respectively.
Under the Amended Agreement, the Company and its subsidiaries are subject to affirmative covenants, including the obligations to maintain good standing, provide certain notices to the Lenders, deliver financial statements to the Lenders, maintain insurance, discharge all taxes, protect intellectual property and protect collateral. The Company and its subsidiaries are also subject to negative covenants, including that each may not enter into a merger or consolidation or certain change of control events, incur liens on the collateral, incur additional indebtedness, dispose of any property, change its jurisdictions of organization or organizational structures or types, declare or pay dividends (other than dividends payable solely in common stock), make certain investments or acquisitions, and enter into certain transactions with affiliates, in each case subject to certain customary exceptions, including exceptions that allow the Company and its subsidiaries to acquire additional compounds and to enter into licenses and similar agreements providing for the use and collaboration of the Company's and its subsidiaries' intellectual property provided certain conditions are met. The Company's assets serve as collateral for the loan.
The Amended Agreement provides that events of default include failure to make payment of principal or interest on the loan when required, failure to perform certain obligations under the Amended Agreement and related documents, defaults in certain other indebtedness and certain other events including certain adverse actions taken by the U.S. Food and Drug Administration or other governmental authorities. Upon events of default, the Company's obligations under the Amended Agreement may, or in the event of insolvency or bankruptcy, will automatically be accelerated. Upon the occurrence of any event of default, the Company's obligations under the Amended Agreement will bear interest at a rate equal to the lesser of (a) 4% above the rate of interest applicable to such obligations immediately prior to the occurrence of the event of default or (b) the maximum rate allowable under law. The Company is currently in compliance with its obligations under the Amended Agreement.
As of December 31, 2011 and 2012, long-term debt outstanding was as follows:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Total debt | | $ | 10,000 | | | $ | 40,000 | |
| Less current portion of long-term debt | | | (1,351 | ) | | | (5,405 | ) |
| | | | | | | | | |
| Long-term debt, net | | $ | 8,649 | | | $ | 34,595 | |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
As of December 31, 2012, maturities of debt per the Amended Agreement for each of the next four years were as follows:
| 2013 | | $ | 5,405 | |
| 2014 | | | 12,973 | |
| 2015 | | | 12,973 | |
| 2016 | | | 8,649 | |
| | | | | |
| | | $ | 40,000 | |
9. Lease Obligations
The Company is currently obligated under an operating lease and a sublease for two locations relating to office space and associated building expenses. These leases expire at different dates in 2013, with renewal terms for one location for one year. As of December 31, 2012, future minimum payments for all lease obligations for the subsequent year were $0.09 million.
Prior to the spin-off, the Company recognized operating lease expense for leases which were acquired as part of the Magen BioSciences, Inc., acquisition. However, these operating lease obligations remained with PPD as of the spin-off date. Rental expense related to operating leases has been recorded in the combined and consolidated statements of operations in the amounts of $0.09 million, $0.2 million and $0.2 million for the years ended December 31, 2010, 2011 and 2012, respectively.
10. Share-Based Compensation
Equity Compensation Plan—Furiex Plan
The Company has adopted an equity incentive plan, the Furiex Pharmaceuticals, Inc. 2010 Stock Plan (the “Plan”). The Company is authorized to issue a total of 1,778,641 shares under the Plan. The Plan is intended to provide incentives to employees, directors and consultants through the issuance of common stock-based awards, including restricted stock, stock options, stock appreciation rights and other equity-based awards. The plan is administered by a committee designated by its Board of Directors.
During the years ended December 31, 2010, 2011 and 2012, the Company granted 839,642, 742,234 and 198,000 stock options to employees, directors, and consultants, with a weighted-average exercise price of $9.11, $13.87 and $17.95, respectively. All options were granted with an exercise price equal to the fair value of the Company's common stock on the grant date. The fair value of the Company's common stock on the grant date is equal to the most recent NASDAQ closing price of the Company's stock on the date of grant.
The Company recognizes compensation expense using a fair-value based method related to stock options and other share-based compensation. The expense is measured based on the grant date fair value of the awards that are expected to vest and is recorded over the applicable requisite service period on a straight-line basis. The options granted vest per one of the following schedules: (1) after a period of approximately one year (or less in the case of certain 2011 Director grants); (2) ratably over three years on the anniversary date of grant; or (3) one-third vest on grant date and the remaining ratably over two years on the anniversary date of grant. The options expire on the earlier of ten years from the date of grant, or within specified time limits following termination of employment, retirement or death. Shares are issued from authorized, but unissued stock. The Company does not pay dividends on unexercised options.
The weighted-average grant date fair value per share was determined using the Black-Scholes option-pricing method. The weighted-average grant date fair value per share of options granted during the years ended December 31, 2010, 2011 and 2012 was $6.28, $8.67 and $10.84, respectively.
The amount of stock compensation expense related to consultant option grants, classified in selling, general and administrative expenses within the combined and consolidated statements of operations, is marked to market at the end of each financial reporting period until such options vest using the Black-Scholes option-pricing method and the period end closing stock price. For the years ended December 31, 2010, 2011 and 2012, amounts reflected in the combined and consolidated statements of operations related to consultant stock compensation expense, including the mark to market adjustment, was $0.4 million, $1.1 million and $1.6 million, respectively. These non-employee grants relate to a consulting agreement executed with the Company’s founding Chairman, Dr. Fred Eshelman. The terms of this consulting agreement provided for a grant of stock options to purchase shares of the Company’s common stock equal to 2.0% of the Company’s common stock outstanding immediately after the completion of the spin-off, and additional stock options for an additional 1.0% on or about the second anniversary of the spin-off date. All options related to this consulting agreement had been granted as of December 31, 2011.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
For the years ended December 31, 2010, 2011 and 2012, stock-based compensation cost for the Company’s employees, directors and consultants under the Plan totaled $1.2 million, $4.2 million and $5.5 million, respectively, and is included in the accompanying combined and consolidated financial statements. For the year ended December 31, 2010, no cash was received by the Company from the exercise of stock options granted by the Company as no options vested during the year. For each of the years ended December 31, 2011 and 2012, the Company received $0.6 million of cash from the exercise of stock options.
A summary of option activity for the Plan as of December 31, 2010, 2011 and 2012, and changes during the years, is presented below:
| | | Shares | | | Weighted-Average Exercise Price | | | Weighted-Average Remaining Contractual Life (Years) | | | Aggregate Intrinsic Value | |
| Outstanding at January 1, 2010 | | | — | | | $ | — | | | | | | | | | |
| Granted | | | 840 | | | | 9.11 | | | | | | | | | |
| Forfeited | | | (1 | ) | | | 9.11 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at December 31, 2010 | | | 839 | | | $ | 9.11 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Exercisable at December 31, 2010 | | | — | | | $ | — | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at January 1, 2011 | | | 839 | | | $ | 9.11 | | | | | | | | | |
| Granted | | | 742 | | | | 13.87 | | | | | | | | | |
| Exercised | | | (68 | ) | | | 9.11 | | | | | | | | | |
| Forfeited | | | (1 | ) | | | 9.11 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at December 31, 2011 | | | 1,512 | | | $ | 11.45 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Exercisable at December 31, 2011 | | | 343 | | | $ | 10.58 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at January 1, 2012 | | | 1,512 | | | $ | 11.45 | | | | | | | | | |
| Granted | | | 198 | | | | 17.95 | | | | | | | | | |
| Exercised | | | (66 | ) | | | 9.11 | | | | | | | | | |
| Forfeited | | | — | | | | — | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at December 31, 2012 | | | 1,644 | | | $ | 12.33 | | | | 8.2 | | | $ | 11,396 | |
| | | | | | | | | | | | | | | | | |
| Exercisable at December 31, 2012 | | | 842 | | | $ | 11.44 | | | | 8.0 | | | $ | 6,581 | |
| | | | | | | | | | | | | | | | | |
| Vested or expected to vest at December 31, 2012 | | | 1,596 | | | $ | 12.29 | | | | 8.2 | | | $ | 11,134 | |
The following table summarizes information about stock options outstanding for the Company as of December 31, 2012:
| | | | Options Outstanding | | | Options Exercisable | |
Range of | | | Number Outstanding | | | Weighted-Average Remaining Contractual Life (Years) | | | Weighted- Average Exercise Price | | | Number Exercisable | | | Weighted- Average Exercise Price | |
| $9.11 | — | 13.00 | | | | 703 | | | | 7.5 | | | $ | 9.11 | | | | 448 | | | $ | 9.11 | |
| $13.01 | — | 15.00 | | | | 741 | | | | 8.6 | | | | 13.87 | | | | 394 | | | | 14.09 | |
| $15.01 | — | 17.00 | | | | 2 | | | | 9.0 | | | | 16.71 | | | | — | | | | — | |
| $17.01 | — | 18.22 | | | | 198 | | | | 9.2 | | | | 17.95 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | 1,644 | | | | 8.2 | | | $ | 12.33 | | | | 842 | | | $ | 11.44 | |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
The aggregate fair value of options granted during the years ended December 31, 2010, 2011 and 2012 was $5.3 million, $6.4 million and $2.1 million, respectively. The total intrinsic value (the amount by which the market value of the Company’s common stock exceeded the exercise price of the options on the date of exercise) of options exercised during the year ended December 31, 2010 was zero as no options vested, and none were exercised during the year. The total intrinsic value of options exercised during the years ended December 31, 2011 and 2012 was $0.5 million and $0.8 million, respectively.
A summary of the status of unvested options held by the Company’s employees, directors and consultants as of December 31, 2012, and changes during the year then ended, is presented below:
| | Shares | | | Weighted- Average Grant Date Fair Value | |
| Unvested at January 1, 2012 | | | 1,168 | | | $ | 7.62 | |
| Granted | | | 198 | | | | 10.84 | |
| Vested | | | (564 | ) | | | 7.54 | |
| Forfeited | | | — | | | | — | |
| | | | | | | | | |
| Unvested at December 31, 2012 | | | 802 | | | $ | 8.47 | |
The total fair value of shares vested during the years ended December 31, 2011 and 2012 was $3.3 million and $5.0 million, respectively. As of December 31, 2012, unrecognized compensation expense related to the unvested portion of the Company’s stock options granted to employees, directors and consultants was approximately $4.6 million, and will be recognized over a weighted-average period of 1.4 years. There was no associated income tax benefit recognized for the years ended December 31, 2010, 2011 or 2012 based on the Company’s valuation allowance that is recorded against its net deferred tax assets.
The following table indicates the assumptions used in estimating fair value of each Plan option granted to employees and directors for the years ended December 31, 2010, 2011 and 2012.
| | | 2010 | | | 2011 | | | 2012 | |
| Expected term (years) | | | 5.50 | — | 6.00 | | | | 5.50 | — | 6.00 | | | | 5.50 | — | 6.00 | |
| Dividend yield (%) | | | | — | | | | | | — | | | | | | — | | |
| Risk-free interest rate (%) | | | 2.19 | — | 2.40 | | | | 1.10 | — | 2.18 | | | | 0.84 | — | 1.15 | |
| Expected volatility (%) | | | 70.99 | — | 71.94 | | | | 65.09 | — | 69.97 | | | | 68.28 | — | 68.96 | |
The following table indicates the assumptions used in estimating fair value of each Plan option granted to consultants for the years ended December 31, 2010 and 2011. There were no consultant option grants during the year ended December 31, 2012.
| | | 2010 | | | 2011 | |
| Expected term (years) | | | 10.00 | | | | 10.00 | |
| Dividend yield (%) | | | — | | | | — | |
| Risk-free interest rate (%) | | | 3.27 | | | | 1.92 | |
| Expected volatility (%) | | | 73.81 | | | | 67.05 | |
Expected option lives were based on the simplified method and volatilities used in fair valuation calculations are based on a benchmark of peer companies with similar expected lives. The Company does not currently intend to pay dividends on common stock; as a result, no dividend yield has been utilized in the fair valuation calculation. The risk-free interest rate is based on the rate at the date of grant for actively traded non-inflation-indexed issues adjusted to constant maturities with a term that approximates the expected term of the option.
As of December 31, 2010, 2011 and 2012, the Company had options outstanding to purchase an aggregate of approximately 839,000, 1,511,000 and 1,644,000 shares of its common stock, respectively.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
Equity Compensation Plan—PPD Plan
For the periods prior to June 14, 2010, some Company employees participated in PPD’s equity compensation plan (the “PPD Plan”). The PPD Plan provided for the grant of incentive stock options, non-qualified stock options, restricted stock and other types of equity awards to its directors, officers, employees and consultants. The plan was administered by a committee designated by PPD’s Board of Directors. Some employees of the Company historically received awards from PPD. Accordingly, the following information regarding share-based compensation has been derived from the equity awards granted to Company employees by PPD prior to June 14, 2010. All unvested options granted under the PPD Plan to Company employees were forfeited as of the spin-off date.
The exercise price of each option granted under the PPD Plan was equal to the market price of PPD’s common stock on the date of grant, and the maximum exercise term of each option granted did not exceed ten years. Options were granted upon approval of the compensation committee of the Board of Directors of PPD. The majority of the options vested ratably over a period of three years. The options expire on the earlier of ten years from the date of grant, or within specified time limits following termination of employment, retirement or death. Shares were issued from authorized, but unissued stock. PPD did not pay dividends on unexercised options.
The fair value of each PPD Plan option grant was estimated on the grant date using the Black-Scholes option-pricing model. No PPD Plan options were granted during the year ended December 31, 2010. For the year ended December 31, 2010, stock-based compensation cost for the Company’s employees under the PPD Plan totaled $0.1 million and is included in the accompanying combined and consolidated financial statements. For the year ended December 31, 2010, PPD received no cash from the exercise of PPD stock options granted to the Company’s employees as no options were exercised.
A summary of option activity under PPD’s plan for the Company’s employees as of December 31, 2010, and changes during the year, is presented below:
| | | Shares | | | Weighted-Average Exercise Price | | | Weighted-Average Remaining Contractual Life (Years) | | | Aggregate Intrinsic Value | |
| Outstanding at January 1, 2010 | | | 237 | | | $ | 29.02 | | | | | | | | | |
| Forfeited | | | (100 | ) | | | 28.88 | | | | | | | | | |
| Expired | | | (1 | ) | | | 37.42 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding at December 31, 2010 | | | 136 | | | $ | 27.38 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Exercisable at December 31, 2010 | | | 136 | | | $ | 27.38 | | | | 4.8 | | | $ | (191 | ) |
| | | | | | | | | | | | | | | | | |
| Vested at December 31, 2010 | | | 136 | | | $ | 27.38 | | | | 4.8 | | | $ | (191 | ) |
The following table summarizes information about PPD’s stock options outstanding for the Company’s employees as of December 31, 2010:
| | | | Options Outstanding | | | Options Exercisable | |
Range of | | | Number Outstanding | | | Weighted-Average Remaining Contractual Life (Years) | | | Weighted- Average Exercise Price | | | Number Exercisable | | | Weighted- Average Exercise Price | |
| $19.94 | — | 21.00 | | | | 71 | | | | 5.9 | | | $ | 20.45 | | | | 71 | | | $ | 20.45 | |
| $21.01 | — | 34.00 | | | | 46 | | | | 3.8 | | | | 31.29 | | | | 46 | | | | 31.29 | |
| $34.01 | — | 43.26 | | | | 19 | | | | 6.7 | | | | 42.26 | | | | 19 | | | | 42.26 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | 136 | | | | 4.8 | | | $ | 27.38 | | | | 136 | | | $ | 27.38 | |
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
All PPD Plan options were granted with an exercise price equal to the fair value of PPD’s common stock on the grant date. The fair value of PPD’s common stock on the grant date was equal to the NASDAQ closing price of the stock on the date of grant. A summary of the status of unvested PPD options held by the Company’s employees as of December 31, 2010, and changes during the year then ended, is presented below:
| | Shares | | | Weighted-Average Grant Date Fair Value | |
| Unvested at January 1, 2010 | | | 83 | | | $ | 7.80 | |
| Vested | | | (25 | ) | | | 8.88 | |
| Forfeited | | | (58 | ) | | | 7.34 | |
| | | | | | | | | |
| Unvested at December 31, 2010 | | | — | | | $ | — | |
As of December 31, 2010, there was no unrecognized compensation cost related to unvested PPD stock options held by the Company’s employees as all unvested PPD Plan options which were not vested as of the spin-off date were forfeited. The total fair value of shares vested during the year ended December 31, 2010 was $0.2 million.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
11. Income Taxes
Taxes computed at the statutory U.S. federal income tax rate of 35% are reconciled to the provision for income taxes as follows:
| | | Year Ended December 31, | |
| | | 2010 | | | 2011 | | | 2012 | |
| Effective tax rate | | | 0 | % | | | 0 | % | | | 0 | % |
| | | | | | | | | | | | | |
| Statutory rate of 35% | | $ | (17,329 | ) | | $ | (17,138 | ) | | $ | (14,998 | ) |
| State taxes, net of federal benefit | | | (2,031 | ) | | | (284 | ) | | | (121 | ) |
| Permanent differences | | | 111 | | | | 371 | | | | 377 | |
| Change in valuation allowance | | | 10,119 | | | | 17,065 | | | | 13,934 | |
| Adjustment to deferred state tax rate | | | — | | | | — | | | | 822 | |
| Net operating loss and related items offset by former Parent Company consolidated group | | | 9,144 | | | | — | | | | — | |
| | | | | | | | | | | | | |
| Provision for income taxes | | $ | 14 | | | $ | 14 | | | $ | 14 | |
Components of the current deferred tax assets (liabilities) were as follows:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Accrued expenses | | $ | 435 | | | $ | 360 | |
| Valuation allowance | | | (435 | ) | | | (360 | ) |
| | | | | | | | | |
| Total current deferred tax asset (liability) | | $ | — | | | $ | — | |
Components of the long-term deferred tax assets (liabilities) were as follows:
| | | December 31, | |
| | | 2011 | | | 2012 | |
| Depreciation and amortization | | $ | 2,399 | | | $ | 2,283 | |
| Stock options | | | 1,290 | | | | 2,567 | |
| Future benefit of carry forward losses | | | 29,853 | | | | 42,688 | |
| Valuation allowance | | | (33,748 | ) | | | (47,758 | ) |
| | | | | | | | | |
| Total long-term deferred tax asset (liability) | | $ | (206 | ) | | $ | (220 | ) |
For the years ended December 31, 2010, 2011 and 2012, the Company recorded an insignificant amount of income tax expense. These amounts relate to the adjustment of a deferred tax liability associated with historical goodwill, which is amortized and deductible for tax purposes, but is an indefinite-lived intangible asset for financial reporting purposes. The amount reflected in the statements of operations for the years ended December 31, 2010, 2011 and 2012 is the tax effect of the tax amortization of this item. Because the associated deferred tax liability relates to an indefinite-lived intangible, the Company does not consider this item in computing the valuation allowance related to the Company's net deferred tax assets. As of December 31, 2011 and 2012, the deferred tax liability associated with this intangible asset, reflected in other long-term liabilities within the consolidated balance sheets, was approximately $0.2 million, respectively.
The Company has determined that any uncertain tax positions for the tax years open for examination would have no material impact on the combined and consolidated financial statements of the Company.
The Company has federal operating loss carry forwards of approximately $117.6 million that will begin to expire in 2030. The Company also has state operating loss carry forwards of approximately $33.5 million that will begin to expire in 2013.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
12. Employee Savings Plan
For the periods prior to June 14, 2010, Company employees were eligible to participate in PPD’s 401(k) Retirement Savings Plan. PPD’s plan matched 50% of an employee’s savings up to 6% of pay and those contributions vested ratably over a four-year period. PPD’s contributions to the plan, net of forfeitures, were $0.09 million for the year ended December 31, 2010.
For the periods after June 14, 2010, Company employees are eligible to participate in the Furiex 401(k) Retirement Savings Plan. The Company’s plan matches 100% of an employee’s savings up to 4% of the employee’s deferral, and those contributions vest immediately. The Company’s contributions to the plan, net of forfeitures, were $0.06 million, $0.14 million and $0.15 million for the years ended December 31, 2010, 2011 and 2012, respectively.
13. Commitments and Contingencies
The Company is involved in compound development and commercialization collaborations. The Company developed a risk-sharing research and development model with pharmaceutical and biotechnology companies to advance compounds to commercialization. Through collaborative arrangements based on this model, the Company shares with its collaborators the risks and potential rewards associated with the development and commercialization of drugs. As of December 31, 2012, the Company's four main collaborations were with Janssen Pharmaceutica, NV, or Janssen (an affiliate of Johnson & Johnson), related to the JNJ-Q2 and MuDelta compounds and the product Priligy; Alza Corporation, or Alza, related to the product Priligy; Berlin-Chemie AG (Menarini Group), or Menarini, related to the product Priligy; and Takeda Pharmaceuticals Company Limited, or Takeda, related to the Trelagliptin compound and the products Nesina and Liovel.
As of December 31, 2012, the Company had two collaborations that involve potential future expenditures. The first is its collaboration with Alza and Janssen for Priligy.
On May 14, 2012, the Company and its wholly-owned subsidiary Genupro, Inc. entered into a license and asset transfer agreement with Alza and Janssen, whereby Alza and Janssen transferred to the Company worldwide rights for Priligy. To facilitate a uniform transition, Janssen will continue to manufacture and manage certain clinical and regulatory activities with respect to Priligy for a pre-defined period after the closing date of the agreement. This transaction became effective on July 30, 2012. Under the terms of this transaction, the Company is obligated to pay Janssen for transition services provided to the Company in the amount of $15.0 million, with $7.5 million paid within 45 days of closing, and $3.75 million due within 10 business days of the beginning of each of the following two calendar quarters. In addition, the Company is obligated to pay Janssen up to $19.0 million in potential on-going clinical study costs and up to $1.0 million for reasonable out-of-pocket expenses over the transition period. The Company must also pay Janssen fees related to Priligy sales and distribution activities that Janssen will perform for the Company during the transition period pursuant to a sales services agreement. The Company believes the transition period will be completed within 12 months from the contract date. Priligy will continue to be made available to patients under the sales service agreement until the marketing authorizations are transferred, at which time commercialization of the product will transition to the Company or its licensee. The term of the license and asset transfer agreement will expire on the latest of the completion of the transition of the product rights to the Company, the expiration of its last payment obligation to Janssen, or the expiration of the last-to-expire ancillary agreement.
The Company's collaboration with Alza and Janssen for Priligy, as described above, is associated with an out-license agreement under which the Company did receive, and is eligible to receive, additional payments. On May 14, 2012, Genupro entered into a license agreement with Menarini by which the Company will license to Menarini exclusive rights to commercialize Priligy in Europe, most of Asia, Africa, Latin America and the Middle East. This transaction became effective on July 30, 2012. The Company will retain full development and commercialization rights in the United States, Japan and Canada. Menarini will assume responsibility for commercialization activities in the licensed territories and will fund the on-going clinical studies being performed by Janssen. Under the license agreement with Menarini, the Company received a $15.0 million upfront payment and $10.0 million of regulatory milestone payments during the third quarter of 2012, and is eligible to receive up to $19.0 million to fund potential on-going clinical study costs being performed by Janssen, up to $10.0 million in launch-based milestones and up to $40.0 million in sales-based milestones, plus tiered royalties ranging from the mid-teens to mid-twenties in percentage terms. During the third quarter of 2012, the Company recognized milestone revenue of $10.0 million related to this agreement related to regulatory submissions. The $15.0 million upfront payment received during the third quarter of 2012 was not recorded as milestone revenue, nor will the potential up to $19.0 million for on-going clinical study costs, based on the terms of the agreement described below. The term of the license agreement (and the period during which Menarini must pay the Company royalties in a particular country for a particular product) will end, on a country-by-country basis, upon the latest of (i) the expiration of a valid relevant patent claim in that country, (ii) the expiration of marketing and data exclusivity in that country, or (iii) the market entry of an approved product in that country containing dapoxetine for use, on an as needed basis, for premature ejaculation.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
In connection with the license and asset transfer agreement with Alza and Janssen, and the license agreement with Menarini, the Company has assessed the indicators associated with agent and principal considerations for each transaction. Based on the terms of the underlying agreements, the nature of the related cash payments and receipts (regarding the $15.0 million up-front and transition service payments, the up to $19.0 million to fund potential on-going clinical study costs and the fees related to Priligy sales and distribution activities performed by Janssen), and the determination that the Company is not the primary obligor for the underlying activities that are associated with these payments, the Company has recorded these amounts on a net basis within the combined and consolidated statements of operations. For the year ended December 31, 2012, the Company has paid amounts to Janssen, and correspondingly received equal payments from Menarini, totaling $2.0 million related to on-going clinical studies for Priligy. In addition, the Company has paid to Janssen approximately $0.2 million in fees associated with sales and distribution activities for Priligy during the transition period. The Company has also considered the contractual right-of-offset related to these payments, for which the Company is effectively an intermediary, and has recorded these amounts on a gross basis within the consolidated balance sheets. As of December 31, 2012, the remaining amounts due to Janssen associated with Priligy transition services of $3.75 million are reflected in accrued expenses. As of December 31, 2012, approximately $1.7 million is reflected in both accounts receivable and accrued expenses related to the on-going Priligy clinical study costs being performed by Janssen.
The Company originally acquired patents for Priligy from Eli Lilly and Company, or Lilly, and is obligated to pay Lilly a royalty of 5% on annual sales in excess of $800.0 million. In addition, under the terms of the license agreement with Menarini, the Company remains responsible for payment of royalties to Lilly, except Menarini will pay the portion of the royalties owed to Lilly in each country where Menarini is licensed to sell Priligy, where Lilly is eligible for payments, and where the Company is no longer eligible for payments from Menarini. The term of the license agreement with Lilly (and the period during which the Company or Menarini must pay Lilly royalties in a particular country) will end, on a country-by-country basis, upon the later of (i) the last to expire valid patent claim licensed to Lilly in that country, (ii) the expiration of data exclusivity in that country, or (iii) the tenth anniversary of the first date of sale of Priligy in that country.
The second collaboration involving future expenditures is associated with the two compounds in-licensed from Janssen: JNJ-Q2 and MuDelta. On April 18, 2011, Janssen announced that in connection with a broad strategic review of its portfolio of infectious disease programs, it will be redirecting its research and development efforts toward antivirals and vaccines, and will not be investing in the development of new antibacterial therapies. As a result, Janssen elected not to exercise its option to continue the development of the JNJ-Q2 compound. On April 19, 2011, the Company announced it had acquired full exclusive license rights to develop and commercialize the JNJ-Q2 compound under its existing development and license agreement with Janssen. On November 1, 2011, the Company announced it had acquired full exclusive license rights to develop and commercialize the MuDelta compound under its existing development and license agreement with Janssen. The Company acquired these rights as a result of Janssen's decision not to exercise its option under the agreement to continue development of MuDelta.
The Company plans to continue evaluating other partnering and funding opportunities for both the JNJ-Q2 and MuDelta compounds. The Company may be obligated to pay Janssen up to $50.0 million in regulatory milestone payments for the JNJ-Q2 compound, up to $45.0 million in regulatory milestone payments for the MuDelta compound, and, if approved for marketing, for both the JNJ-Q2 and MuDelta compounds, individually, up to $75.0 million in sales-based milestone payments and sales-based royalties increasing from the mid- to upper-single digit percentages as sales volume increases. Royalties would be paid for a period of ten years after the first commercial sale or, if later, the expiration of the last valid patent claim or the expiration of patent exclusivity.
The Company currently maintains insurance for risks associated with the operation of its business. These policies provide coverage for a variety of potential losses, including loss or damage to property, bodily injury, general commercial liability and product liability. The Company might be a party to various claims and legal proceedings in the normal course of business. As of December 31, 2012, there are no outstanding claims or legal proceedings that management believes will have a material effect upon the Company’s financial condition, results of operations or cash flows.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
14. Fair Value of Financial Instruments
Cash and Cash Equivalents, Short-Term Investments, Accounts Receivable, Investments, Accounts Payable and Accrued Expenses
The carrying amounts of cash and cash equivalents, short-term investments, accounts receivable, investments, accounts payable and accrued expenses approximates fair value because of the short maturity of these items. The Company considers all cash on deposit and money market accounts with original maturities of three months or less at time of purchase to be cash and cash equivalents. The Company's cash and cash equivalents, short-term investments and investments represent cash accounts and money market funds that invest in short-term U.S. Treasury securities with insignificant rates of return and are considered Level 1 investments. Level 1 investments are investments where there are quoted prices in active markets available for identical assets or liabilities.
Long-Term Debt
The fair value of long-term debt approximates its carrying value because there have been no significant changes in interest rates or the creditworthiness of the Company since the debt was incurred in August 2012.
15. Related Party Transactions
Pharmaceutical Product Development, Inc. Net Investment
The following table reflects a summary of the transfers to parent included in the combined and consolidated statements of shareholders’ equity related to changes in PPD’s net investment for the periods prior to the spin-off date:
| | | 2010 | |
| Corporate overhead allocations | | $ | 1,007 | |
| Research and development services | | | 8,376 | |
| Transfer of proceeds from sale of businesses | | | (3,464 | ) |
| Transfers to parent, net | | | 10,127 | |
| Total | | $ | 16,046 | |
Corporate Overhead Allocations
For the periods prior to the June 14, 2010 spin-off, the Company’s operations were fully integrated with PPD, including executive services, finance, treasury, corporate income tax, human resources, information technology, facilities, legal services and investor relations services. The accompanying combined and consolidated financial statements reflect the application of estimates and allocations of operating expenses. Management believes the methods used to allocate these operating expenses were reasonable. The allocation methods included relative time devoted by executive management to the Company’s business, and the related benefit received by the Company for other services.
Expense allocations for these services of $0.6 million associated with continuing operations and $0.5 million associated with discontinued operations for the year ended December 31, 2010 are reflected in the accompanying combined and consolidated statements of operations.
Research and Development Services
PPD performed drug development work for the Company as a related party prior to June 14, 2010 and the expenses related to these services are included in research and development expenses in the accompanying combined and consolidated financial statements. These amounts were $8.4 million for the year ended December 31, 2010.
The Company was provided services by PPD after the spin-off on June 14, 2010. One member of the Company’s Board of Directors previously held a Board position with PPD. Expenses paid by the Company to PPD for the years ended December 31, 2010 and 2011 were approximately $24.5 million and $30.6 million, respectively.
Transfer of Proceeds from Sale of Business
In 2009, PPD disposed of Piedmont Research Center, LLC and PPD Biomarker Discovery Sciences, LLC. The cash proceeds received from these transactions in 2009 were transferred to PPD. Cash proceeds of $3.5 million received from these transactions in 2010, for the payment of an outstanding escrow account, were also transferred to PPD.
FURIEX PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO COMBINED AND CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2010, 2011 AND 2012
(dollars and shares in tables in thousands)
16. Segment Information
The Company’s business consists solely of compound development and collaboration activities. Accordingly, the Company operates in one reportable business segment.
17. Quarterly Financial Data (unaudited)
| | | First | | | Second | | | Third | | | Fourth | | | Total | |
| | | | | | | | | | | | | | | |
| Total revenue | | $ | 361 | | | $ | 872 | | | $ | 1,272 | | | $ | 1,985 | | | $ | 4,490 | |
| Operating loss | | | (14,359 | ) | | | (14,876 | ) | | | (12,871 | ) | | | (6,450 | ) | | | (48,556 | ) |
| Net loss | | | (14,365 | ) | | | (14,882 | ) | | | (13,013 | ) | | | (6,721 | ) | | | (48,981 | ) |
| Net loss per basic and diluted share | | $ | (1.45 | ) | | $ | (1.51 | ) | | $ | (1.32 | ) | | $ | (0.68 | ) | | $ | (4.96 | ) |
| | | | | | | | | | | | | | | |
| Total revenue (a) | | $ | 2,645 | | | $ | 13,075 | | | $ | 15,577 | | | $ | 9,211 | | | $ | 40,508 | |
| Operating loss | | | (9,477 | ) | | | (20,489 | ) | | | (2,452 | ) | | | (7,926 | ) | | | (40,344 | ) |
| Net loss | | | (9,758 | ) | | | (20,769 | ) | | | (3,286 | ) | | | (9,052 | ) | | | (42,865 | ) |
| Net loss per basic and diluted share | | $ | (0.98 | ) | | $ | (2.09 | ) | | $ | (0.33 | ) | | $ | (0.90 | ) | | $ | (4.29 | ) |
| (a) | The second quarter of 2012 includes a $10.0 million regulatory milestone related to the acceptance of the submission of a Marketing Authorization Application by the European Medicines Agency for alogliptin in May 2012. The third quarter of 2012 includes a $10.0 million launch-based milestone related to the Priligy license agreement with Menarini. Total net loss per basic and diluted share for 2012 differs from the sum of quarterly amounts due to rounding. |
18. Subsequent Events
On January 25, 2013, Takeda confirmed the approval from the U.S. Food and Drug Administration of Nesina (alogliptin) and the fixed-dose combination therapies, Oseni® (alogliptin and pioglitazone) and Kazano® (alogliptin and metformin), for the treatment of Type-2 diabetes. Under the agreement with Takeda, the Company received a $25.0 million milestone payment on February 27, 2013 as a result of this approval.
F-23