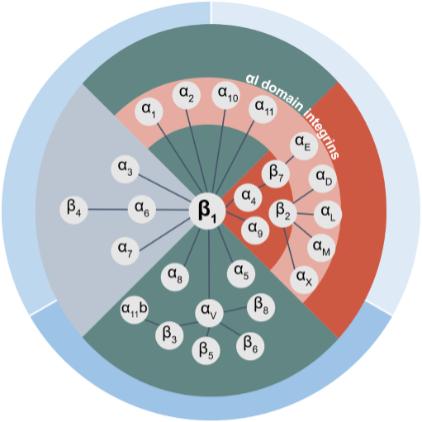
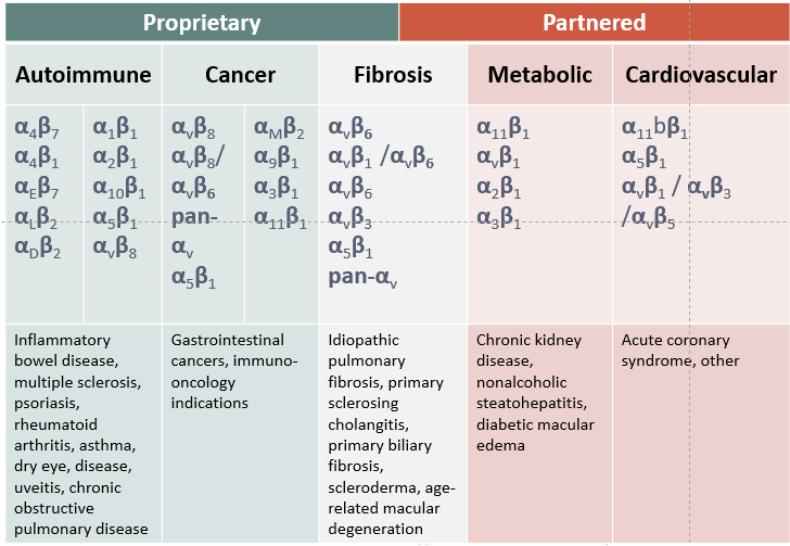
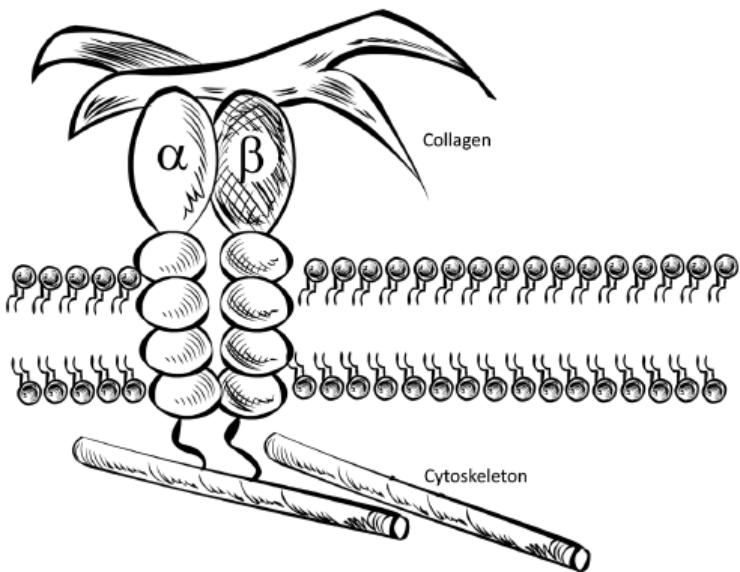
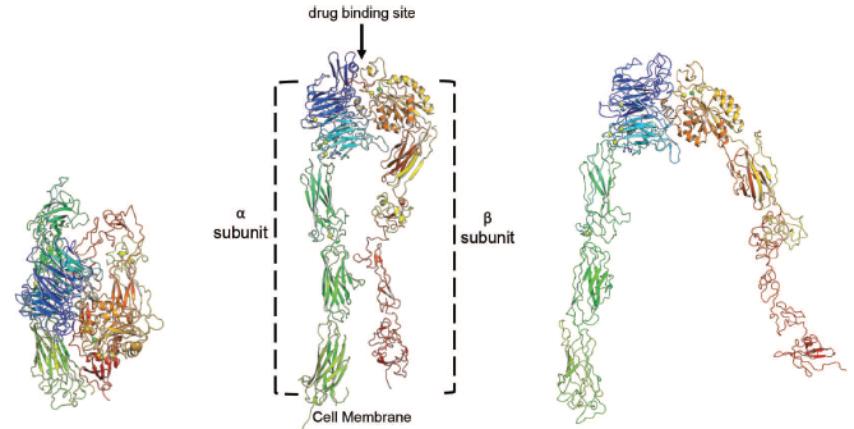
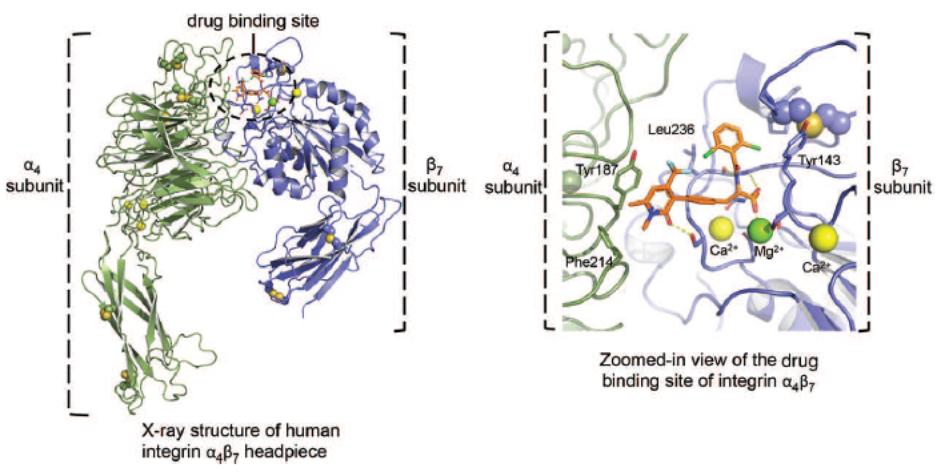
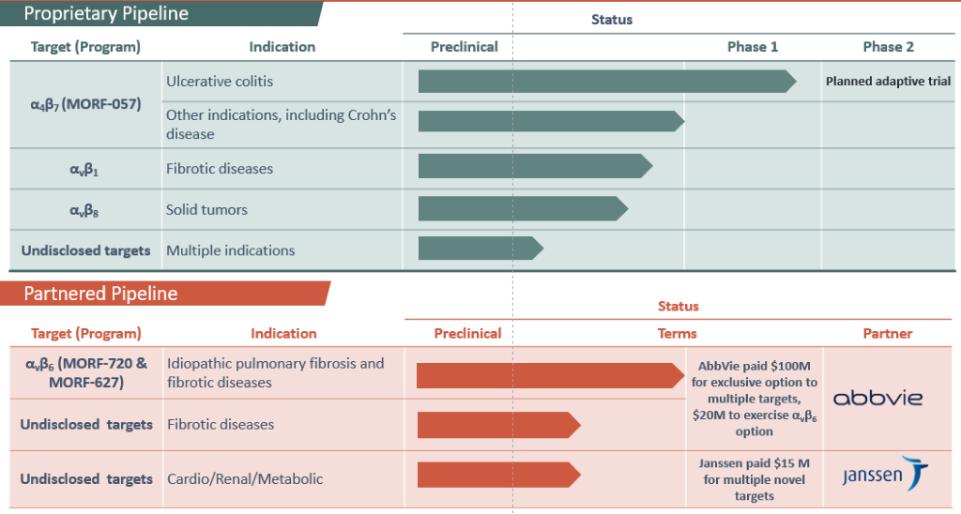
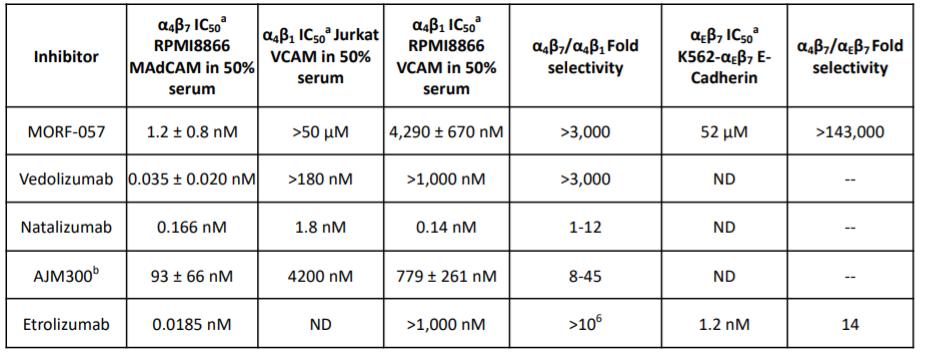
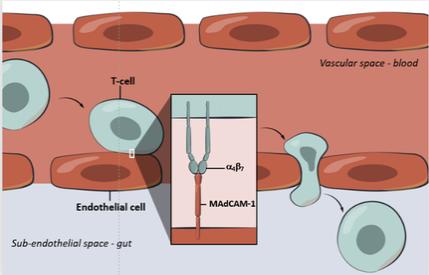
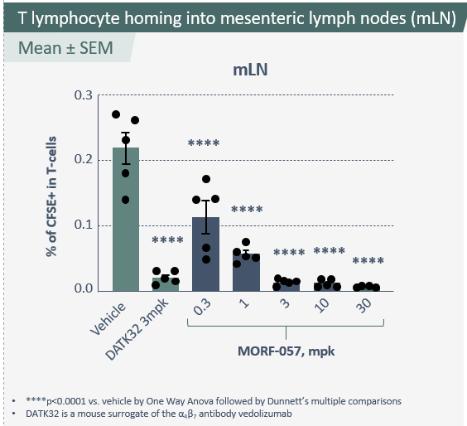
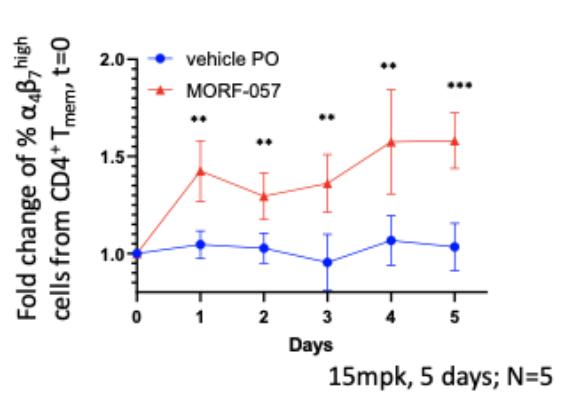
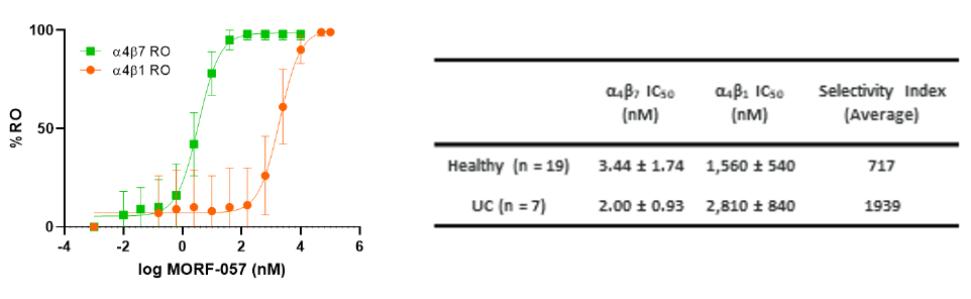
Biogen and Pfizer, in addition to other major pharmaceutical companies, against which our product candidate may compete, if approved. Further, Takeda Pharmaceutical Company Ltd. currently markets Entyvio, which is an α4β7 monoclonal antibody to treat ulcerative colitis and Crohn’s disease. In addition, we are aware of IBD treatments in clinical development by AbbVie, Johnson & Johnson, Pfizer, Gilead Sciences, Inc., Eli Lilly and Company, Bristol-Myers Squibb Company, Boehringer Ingelheim, Theravance, and Arena Pharmaceuticals, in addition to other pharmaceutical companies. Further, Protagonist Therapeutics, Inc. has a Phase 2 gut-restricted α4β7 program in Phase 2 development for ulcerative colitis.
Our αvβ6-specific integrin inhibitor program is under development for the treatment of IPF by our collaboration partner AbbVie, and if any of our compounds licensed to AbbVie is approved for IPF, would face competition from approved IPF treatments marketed by Roche Holding AG and Boehringer Ingelheim GmbH. In addition, we are aware of IPF treatments in development by Galapagos NV, FibroGen, Inc., Galecto Biotech, Roche Holding AG, Bristol-Myers Squibb Company, Kadmon Holdings, Inc. and Liminal BioSciences, Inc., in addition to other pharmaceutical companies. Further, we are aware of programs targeting αvβ6 that are currently being investigated in clinical trials by companies including Pliant Therapeutics, Inc. In September 2019, Biogen Inc. announced the termination of a Phase 2 study of its monoclonal antibody targeting αvβ6, citing safety concerns. Many of our competitors have significantly greater financial resources and expertise than we do in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Additionally, our competitors may also include companies that are or will be developing therapies for the same therapeutic areas that we are targeting, including autoimmune, cardiovascular and metabolic diseases, fibrosis and cancer.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries and jurisdictions extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products. The processes for obtaining regulatory approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
FDA Approval Process
In the United States, pharmaceutical products are subject to extensive regulation by FDA. The Federal Food, Drug, and Cosmetic Act, or FD&C Act, and other federal and state statutes and regulations govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Pharmaceutical product development for a new product or certain changes to an approved product in the U.S. typically involves preclinical laboratory and animal tests, the submission to FDA of an investigational new drug application, or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including good laboratory practices. The results of preclinical testing are submitted to FDA as part of an IND along with other information, including information about product chemistry,