As filed with the Securities and Exchange Commission on April 7, 2022
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-3
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
INMED PHARMACEUTICALS INC.
(Exact name of registrant as specified in its charter)
| British Columbia | 98-1428279 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S Employer Identification No.) |
Suite 310 – 815 West Hastings Street
Vancouver, British Columbia V6C 1B4
Canada
Telephone: (604) 669-7207
(Address, including zip code and telephone number, including area code, of registrant’s principal executive offices)
Registered Agent Solutions, Inc.
1100 H Street NW
Suite 840
Washington, DC 20005
Telephone: (888) 705-7274
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Brenda Edwards Interim Chief Financial Officer InMed Pharmaceuticals Inc. Suite 310 – 815 West Hastings Street Vancouver, British Columbia V6C 1B4 Canada Telephone: (604) 669-7207 | Brian P. Fenske Norton Rose Fulbright US LLP 1301 McKinney, Suite 5100 Houston, Texas 77010 Telephone: (713) 651-5557 |
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this registration statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box. ☐
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, other than securities offered only in connection with the dividend or interest reinvestment plans, check the following box. ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a registration statement pursuant to General Instruction I.D. or a post-effective amendment thereto that shall become effective upon filing with the Commission pursuant to Rule 462(e) under the Securities Act, check the following box. ☐
If this Form is a post-effective amendment to a registration statement filed pursuant to General Instruction I.D. filed to register additional securities or additional classes of securities pursuant to Rule 413(b) under the Securities Act, check the following box. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |||
| Emerging growth company | ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. The selling shareholders may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED [______], 2022
PRELIMINARY PROSPECTUS
6,139,727 Common Shares

InMed Pharmaceuticals Inc.
This prospectus relates to the offering and resale by the selling shareholders identified herein of up to 6,139,727 of our common shares. The common shares being offered by the selling shareholders consist of 6,139,727 common shares of the company (“common shares”), issuable upon the exercise of the following previously issued share purchase warrants that remain outstanding: 1,780,000 public warrants issued in November 2020 in connection with our initial public offering (“IPO”), 323,400 common share purchase warrants issued in February 2021 in connection with a private placement offering (the “February Private Placement”), and 4,036,327 Series A warrants issued in July 2021 in connection with a private placement offering (the “July Private Placement”).
We will not receive any proceeds from the sale of common shares by the selling shareholders.
The selling shareholders may sell all or a portion of the common shares beneficially owned by it and the common shares are offered hereby from time to time directly or through one or more underwriters, broker-dealers or agents. Please see “Plan Of Distribution” on page 104 of this prospectus for more information. For more information regarding the selling shareholders, see “Selling Shareholders” on page 101 of this prospectus.
Our common shares are quoted on the Nasdaq Capital Market under the symbol “INM”. We are an “emerging growth company” as defined by the Jumpstart Our Business Startups Act of 2012 and, as such, we have elected to comply with certain reduced public company reporting requirements for this prospectus and future filings. On April 6, 2022, the closing price of our common share was $0.89 per share.
Investing in our common shares involves a high degree of risk. Please read “Risk Factors” beginning on page 56 of this prospectus, contained in any applicable prospectus supplement, and in the documents incorporated by reference herein and therein for a discussion of the factors you should carefully consider before deciding to purchase our securities.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Prospectus dated , 2022
Table of Contents
i
This prospectus is part of a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, or the “SEC,” using a “shelf” registration process for the delayed offering and sale of securities pursuant to Rule 415 under the Securities Act. Under this shelf process, this prospectus may be used from time to time by the selling shareholders to sell up to 6,139,727 shares of our common shares as described herein and under the heading “Selling Shareholders.” The common shares being offered by the selling shareholders pursuant to this prospectus are the underlying shares issuable upon the exercise of the following previously issued share purchase warrants that remain outstanding: 1,780,000 public warrants issued in November 2020 in connection with our IPO, 323,400 common share purchase warrants issued in the February 2021 Private Placement, and 4,036,327 Series A warrants issued in the July 2021 Private Placement. Any prospectus supplement may include a discussion of any risk factors or other special considerations that apply to our common shares. The prospectus supplement may also add, supplement, change, update, supersede, or amend information included in this prospectus. You should carefully read both this prospectus, any prospectus supplement, any free writing prospectus that we authorize to be distributed to you, and any information incorporated by reference into the foregoing, together with additional information described under the headings “Incorporation of Certain Documents by Reference” and “Where You Can Find More Information” before investing in any of the securities offered under this prospectus.
We have not authorized anyone to give you any additional information different from that contained in this prospectus, any accompanying prospectus supplement or any free writing prospectus provided in connection with an offering. We take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. We are not making an offer to sell these securities in any jurisdiction where the offer is not permitted.
This prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by reference to the actual documents.
The information contained in this prospectus, in any prospectus supplement, or in any document incorporated by reference in the foregoing is accurate only as of the date of such document containing such information, regardless of when this prospectus or any prospectus supplement is delivered or when any sale of our securities occurs. Our business, operating results, cash flows, financial condition, or prospects may have changed since that date. If there is any inconsistency between the information in this prospectus, in any prospectus supplement, or in any document incorporated by reference in the foregoing, you should rely on the information in the document with the most recent date.
This prospectus is not an offer to sell or solicitation of an offer to buy our securities in any circumstances under which or jurisdiction in which the offer or solicitation is unlawful. Unless the context indicates otherwise, as used in this prospectus, the terms “InMed,” “InMed Pharmaceuticals,” “BayMedica Inc.”, “BayMedica”, “BayMedica LLC”, “we,” “us,” “our,” “our company” and “our business” refer to InMed Pharmaceuticals Inc. The phrase “this prospectus” refers to this prospectus and any applicable prospectus supplement, unless the context otherwise requires. In this prospectus, we sometimes refer to the common share as the “securities.”
ii
Overview
We are a clinical stage pharmaceutical company developing a pipeline of prescription-based products, including rare cannabinoids and novel cannabinoid analogs, targeting the treatment of diseases with high unmet medical needs (“Product Candidates”) as well as developing proprietary manufacturing technologies to produce rare cannabinoids for sale in the health and wellness industry (“Products”).
We are developing multiple manufacturing approaches for synthesizing rare cannabinoids for potential use in pharmaceutical Product Candidates as well as serving as a business to business (B2B) supplier to wholesalers and end-product manufacturers / marketers in the health and wellness sector. This includes traditional approaches such as chemical synthesis and biosynthesis, as well as a proprietary, integrated manufacturing approach called IntegraSynTM. We are dedicated to delivering new therapeutic alternatives to patients and consumers who may benefit from cannabinoid-based products. Our approach leverages on the several thousand years’ history of health benefits attributed to the Cannabis plant and brings this anecdotal information into the 21st century by applying tried, tested and true scientific approaches to establish non-plant-derived (synthetically manufactured), individual cannabinoid compounds as Product Candidates in important market segments including clinically proven, FDA-approved medicines and non-prescription, over-the-counter consumer products via B2B supply relationships with wholesalers and end-product manufacturers. While our activities do not involve direct use of Cannabis nor extracts from the plant, we note that the U.S. Food and Drug Administration (“FDA”) has, to date, not approved any marketing application for Cannabis for the treatment of any disease or condition and has approved only one Cannabis-derived and three Cannabis-related drug products. Our ingredients are synthetically made and, therefore, we have no interaction with the Cannabis plant. We do not grow nor utilize Cannabis nor its extracts in any of our Products or Product Candidates; our current pharmaceutical drug Product Candidates are applied topically (not inhaled nor ingested); and, we do not utilize THC or CBD, the most common cannabinoid compounds that are typically extracted from the Cannabis plant, in any of our Products or Product Candidates. The active pharmaceutical ingredient (“API”) under development for our initial two drug candidates, INM-755 for Epidermolysis bullosa (“EB”) and INM-088 for glaucoma, is cannabinol (“CBN”). Additional uses of both INM-755 and INM-088 are being explored, as well as the application of additional rare cannabinoids to treat diseases including but not limited to neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s.
We believe we are positioned to develop multiple pharmaceutical Product Candidates in diseases which may benefit from medicines based on rare cannabinoid compounds. Most currently approved cannabinoid therapies are based specifically on CBD and/or THC and are often delivered orally, which has limitations and drawbacks, such as side effects (including the intoxicating effects of THC). Currently, we intend to deliver our rare cannabinoid pharmaceutical drug candidates through various topical formulations (cream for dermatology, eye drops for ocular diseases) as a way of enabling treatment of the specific disease at the site of disease while seeking to minimize systemic exposure and any related unwanted systemic side effects, including any drug-drug interactions and any metabolism of the active pharmaceutical ingredient by the liver. The cannabinoids sold through our B2B raw material supply business are integrated into various product formats by the companies who then further commercializes such products. We plan to access rare cannabinoids via all non-extraction approaches, including chemical synthesis, biosynthesis and our proprietary integrated IntegraSynTM approach, thus negating any interaction with or exposure to the Cannabis plant.
1

On October 13, 2021, we acquired BayMedica Inc., now named BayMedica LLC (“BayMedica”). Upon closing of the transaction, BayMedica became a wholly-owned subsidiary of InMed.
Corporate Information
We were originally incorporated in the Province of British Columbia, under the BCBCA, on May 19, 1981 and we have undergone a number of corporate name and business sector changes since this incorporation, ultimately changing our name to “InMed Pharmaceuticals Inc.” on October 6, 2014 to signify our intent to specialize in cannabinoid pharmaceutical product development. Our principal executive offices are located at Suite 310 – 815 W Hastings Street, Vancouver, BC, Canada, V6C 1B4 and our telephone number is +1-604-669-5699. Our internet address is https://www.inmedpharma.com/.
Employees and Human Capital
Our management team is comprised of highly experienced pharmaceutical and biotechnology executives with successful track records in researching, developing, gaining approval for and commercializing novel medicines to treat serious diseases. Each member of our management team has over 20 to 30 years of industry experience, including our CEO, CFO, General Manager, and (Sr.) Vice Presidents of Clinical and Regulatory Affairs, of Preclinical Research and Development, of Chemistry, Manufacturing and Controls, of Discovery Research, of Chemistry, of Synthetic Biology, of Sales & Marketing and of Commercial Operations. Together, this team has covered the spectrum of pharmaceutical drug discovery, preclinical research, formulation development, manufacturing, human clinical trials, regulatory submissions and approval, and global commercialization. Additionally, the team has significant experience in company formation, capital raises, mergers/acquisitions, business development, and sales and marketing in the pharmaceutical industry. Our Board is constituted by individuals with significant experience in the pharmaceutical and biotechnology industries. As of April 5, 2022, including our management team, we had 19 full time employees and we also utilize the services of several consultants. None of our employees are represented by a collective bargaining agreement, nor have we experienced any work stoppage. We believe that our relations with our employees are good.
2
We are committed to growing our business over the long-term. As a result of the competitive nature of the industry in which we operate, employees have significant career mobility and as a result, the competition for experienced employees is great. The existence of this competition, and the need for talented and experienced employees to realize our business objectives, underlies the design and implementation of our compensation programs. At the same time, we seek to keep our approach to compensation simple and streamlined to reflect the still relatively moderate size of our company. We have compensation, leave and benefits programs necessary to attract and retain the talented and experienced employees necessary to develop our business including competitive salaries, stock options awards to permanent employees, both upon initial hiring and annually thereafter, and pay annual bonuses to permanent employees based on the achievement of corporate and/or personal objectives. We have developed an Employee Handbook that contains all corporate policies and guidelines for professional behavior. Our policies and practices apply to all employees, regardless of title. These guidelines include our Code of Business Conduct as well as our policies for corporate disclosure, insider trading and whistle blower, all of which are
In response to the COVID-19 pandemic, commencing in March 2020, we implemented a work-from-home mandate and ceased all non-essential business travel. In the recent months, some employees have recommenced limited business travel and some have transitioned back to working on-site in conjunction with the implementation of additional safety and infection prevention measures including enhanced cleaning, additional personal protective equipment, and contact tracing protocols. We continue to provide our employees with the option to work from home.
Rationale for Use of CBN in Pharmaceutical Drug Development
CBN is one of several non-intoxicating rare cannabinoids naturally produced in the Cannabis plant, albeit at significantly lower levels relative to the more commonly known THC and CBD. Despite their common origin, different cannabinoids have been observed to have distinct physiological properties, we are specifically exploring these unique effects of CBN, as well as other rare cannabinoids, and their therapeutic potential to treat disease.
Rare vs. Major Cannabinoids: Types, Prevalence & Application

Extensive preclinical testing undertaken by us has identified several unique properties of CBN that outperformed both THC and CBD in various disease-related assays and models. CBN can act with higher potency when interacting with some receptor systems in the body, while acting with lower potency for others.
3
Physical and Chemical Properties of Active Pharmaceutical Ingredient CBN
CBN is a stable, highly lipophilic cannabinoid compound. It is insoluble in water, but soluble in organic solvents.
| International Non-proprietary Name: | Cannabinol (abbreviated CBN) |
| International Union of Pure and Applied Chemistry Name: | 6,6,9-trimethyl-3-pentyl-benzo[c]chromen-1-ol |
| Chemical Abstracts Service Registration Number: | 521-35-7 |
| United States Adopted Name: | Cannabinol |
The molecular formula is C21H26O2 and the molecular weight is 310.43 g/mol. CBN has no chiral centers.
Figure 1 Structural Formula of CBN
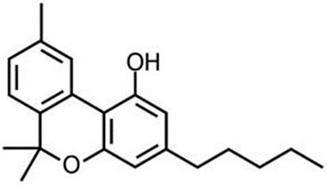
CBN occurs naturally as a trace component of Cannabis, or as a degradation product of D9-THC. However, our Product Candidates utilizing CBN contain highly purified synthetic CBN, rather than a biological extract.
CBN as our Lead API
As the API in our lead therapeutic programs in dermatology (INM-755) and ocular disease (INM-088), CBN has several compelling features, including:
| ● | A rare cannabinoid with unique physiological properties; |
| ● | A natural compound, but designated as a new chemical entity, or “NCE” for pharmaceutical development; |
| ● | Found in trace amounts in the plant and impractical to extract; and |
| ● | Our preclinical studies show therapeutic potential for dermatology and ocular diseases. |
We believe that we offer a differentiated approach to selecting and delivering rare cannabinoids vis-à-vis other current competitors, many of whom are exclusively focused on THC and/or CBD as their therapeutic agents. We believe that rare cannabinoids in general, and CBN in particular, represent significant opportunities to treat a wide spectrum of diseases with high unmet medical need. In our preclinical testing, CBN has demonstrated therapeutic potential beyond CBD for several symptoms and disease-modifying effects for dermatological conditions and has demonstrated benefits beyond CBD and THC for ocular diseases. We believe that a topical application of CBN may maximize the clinical benefit at the disease site (skin, eye) while minimizing the systemic exposure and any corresponding adverse effects.
INM-755, our lead product candidate, is being developed as a topical skin cream formulation containing CBN for the treatment of symptoms related to EB, a rare genetic skin disease characterized by fragile skin that blisters easily from minimal friction that causes shearing of the skin layers. The blisters become open wounds that do not heal well.
In addition to relief of symptoms, inflammation, pain, and others, we believe INM-755 may impact the underlying disease by enhancing skin integrity in a subset of EB patients. We have completed more than 30 preclinical pharmacology and toxicology studies to investigate the effects of CBN. Several of these nonclinical studies explored the effect on important symptoms such as pain and inflammation. In in vitro pharmacology studies, CBN demonstrated activity in reducing markers of prolonged inflammation. CBN upregulated expression of a type of keratin called keratin 15, or “K15”, which might lead to skin strengthening and reduced blister formation in EB simplex, or “EBS”, patients with mutations in another keratin called keratin 14, or “K14”. The anti-inflammatory activity of CBN may be beneficial in healing chronic wounds caused by prolonged inflammation.
4
Following a review of our toxicology studies, a regulatory application to support our first Phase I clinical study in healthy volunteers with INM-755 (755-101-HV) was submitted November 4, 2019 and approved December 6, 2019 in the Netherlands. The initial Phase I clinical study evaluated the safety, tolerability, and pharmacokinetics of INM-755 cream in healthy volunteers with normal, intact skin; the volunteers had cream applied once daily for a period of 14 days. All subjects in this first clinical trial completed treatment and evaluations by March 27, 2020. A regulatory application was approved April 17, 2020, for a second Phase I clinical study of healthy volunteers to test the local safety and tolerability of applying sterile INM-755 cream to small wounds once daily for 14 days. As with the initial Phase I trial, the second trial (755-102-HV) was conducted with two different drug concentrations and a vehicle control. Enrollment began in early July 2020 and the clinical trial completed treatment and evaluations at the end of September 2020. The safety of INM-755 will continue to be assessed throughout its clinical development.
INM-755 cream was well tolerated in the two Phase I clinical studies in healthy volunteers and, based upon this outcome, we advanced the product candidate into a Phase II clinical trial in patients with EB (Study 755-201-EB). The 755-201-EB study is designed to enroll up to 20 patients using a within-patient design in which matched index areas are randomized to INM-755 cream or vehicle (no drug) cream in a blinded manner. InMed will evaluate the safety of INM-755 (cannabinol) cream and its preliminary efficacy in treating symptoms and wound healing over a 28-day treatment period, the longest period supported by nonclinical toxicology. All four subtypes of inherited EB; EB Simplex, Dystrophic EB, Junctional EB, and Kindler Syndrome are eligible for this study.
Regulatory applications to support this global trial were filed for review by the National Competent Authorities and Ethics Committees in 8 countries for 13 clinical sites. Approvals were obtained in all countries (Austria, France, Germany, Israel, Italy, Serbia, and Spain) as of March 2022. Enrollment and patient treatment began in December 2021 and are expected to complete during the calendar year 2022.
CBN is also the active pharmaceutical ingredient in our second pharmaceutical drug candidate, INM-088, which is in preclinical studies as a potential treatment for glaucoma. Current treatments for glaucoma primarily focus on decreasing fluid build-up in the eye. We are conducting preclinical studies to test INM-088’s ability to provide both neuroprotection and reduce intraocular pressure in the eye. We compared several cannabinoids, including CBD and THC, to determine which cannabinoid was the best drug candidate for the treatment of glaucoma. Of all the cannabinoids examined in preclinical studies, CBN demonstrated the most optimal neuroprotective effect. Notably, exposure of retinal neurons, called retinal ganglion cells (“RGCs”) to increasing concentrations of several cannabinoids, including THC and CBD, resulted in dose dependent cytotoxicity, or cell death, over time. Importantly, CBN-exposed RGCs demonstrated the lowest level of toxicity among the cannabinoids used in these experiments. We also verified that CBN has an anti-apoptotic effect on differentiated RGCs when subjected to elevated hydrostatic pressure.
Furthermore, CBN also exhibited intraocular pressure reduction capability. INM-088 is in advanced formulation development. We selected a final delivery technology (MiDrops®, EyeCRO LLC) based on the extensive data collected from these assessments that included solubility, drug delivery localization and sustained effect.
For all current and future pharmaceutical Product Candidates we intend to submit new drug applications (NDAs) (or their international equivalents) in most major jurisdictions, including the U.S. either alone or with development/commercial partners.
We are actively establishing a broad patent portfolio to protect our commercial interests in utilizing CBN and other rare cannabinoids across these and other diseases. We have also filed multiple patent applications for our integrated, biosynthesis-based manufacturing approach. If granted, these patents may confer meaningful protection to the commercial potential for these technologies.
5
Rare Cannabinoid Products in the Health and Wellness Sector
We are a world leader in the manufacturing and commercialization of the rare cannabinoid named cannabichromene (CBC) as a B2B supplier to wholesalers and end-product manufacturers / marketers in the health and wellness sector. Since sales began at the end of 2019, manufacturing has scaled to the 200kg level and the predecessor company, BayMedica Inc., had cumulative revenues of $2.4 million for the 21-month period ending September 30, 2021. Since October 13, 2021, the date of acquisition, to December 31, 2021, BayMedica had revenues of approximately $265,000. We plan to leverage our existing synthetic chemistry manufacturing capabilities to produce other non-intoxicating rare cannabinoids of high interest in the health and wellness segment, such as CBDV and THCV. Over time, we will continue to improve margins on these and other products by improving on manufacturing techniques, approaches and scale.
Our Business Strategy
Our goal is to develop a pipeline of prescription-based Product Candidates targeting treatments for diseases with high unmet medical needs as well as to develop proprietary manufacturing technologies to produce rare cannabinoid Products for sale in the health and wellness industry and to produce their novel analogs for our use in the pharmaceutical industry, by pursuing the following:
| ● | Advance INM-755 and INM-088 through preclinical and clinical development, thereby establishing important human proof-of-concept in multiple therapeutic applications |
| ● | Expand portfolio and revenues of rare cannabinoids into existing distribution network and to end-product manufacturers of specialty health and wellness products |
| ● | Develop and produce novel cannabinoid analogs for use in our drug development program and/or licensing, partnering or sale to external companies. |
These activities are well underway, at various stages, for both INM-755 for diseases of the skin and INM-088 for diseases of the eye. Building upon preclinical data sets, we have the internal capabilities to design and execute, together with multiple external vendors, the preclinical data sets and clinical studies required to advance pharmaceutical drug candidates towards commercialization. We will continue to build out and sell a catalog of rare cannabinoids to end-product manufacturers in the health and wellness sector as well as continue our internal development of novel cannabinoids and their analogs.
| ● | Establishing partnerships for our various technologies, at different stages of development, to expedite their path towards commercialization in a resource-efficient manner. |
We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products. With respect to the commercialization of each Product Candidate, we may rely on either i) a “go-it-alone” commercialization effort; ii) out-licensing to third parties; or, iii) co-promotion agreements with strategic collaborators for our Product Candidates. Any decision on a “go-it-alone” commercialization effort versus out-licensing to third parties will depend on various factors including, but not limited to, the complexity, the expertise required and related cost of building any such infrastructure for our Product Candidates. For INM-755 in EB, we could oversee the clinical trials, given the relatively small patient sizes expected for such trials, and build the requisite internal commercialization infrastructure to self-market the product to EB clinics, which are limited in number and provide direct access to the vast majority of EB patients. For INM-088 in glaucoma, because of the potentially large number of clinical trial participants (possibly several thousand) and the extensive sales effort required to reach a large number of prescribing physicians, we may consider exploring partnership opportunities early in the development process.
| ● | Develop multiple cost-efficient manufacturing processes for high quality rare cannabinoids as APIs for our core internal drug candidate pipeline, for licensing opportunities of non-core drug candidates, as well as a source for rare cannabinoids in the health and wellness sector. |
We are developing an integrative cannabinoid synthesis approach designed to produce bio-identical, economical, pharmaceutical-grade cannabinoids in a cost-efficient manner, called IntegraSynTM. IntegraSynTM is designed to offer superior yield, control, consistency and quality of rare cannabinoids when compared to alternative methods. Additionally, we continue to develop cost-effective manufacturing techniques to supply rare cannabinoids to end-product manufacturers and wholesalers in the health and wellness sector via our wholly-owned subsidiary BayMedica.
| ● | Continue to invent, manufacture and research the potential of a wide array of rare cannabinoid analogs to treat diseases based on our significant history in cannabinoid research and lead drug candidate identification. |
6
Individual cannabinoids affect a range of different receptors in the human body, including, but not limited to, known endocannabinoid receptors. As such, they are responsible for a wide variety of pharmacological effects. However, due to the limited research into these varying effects, a full understanding of the role of each cannabinoid compound remains elusive. As a company, we have been formally investigating the utility of cannabinoids in treating disease for over 6 years.
At the core of our activities, we are a pharmaceutical drug development company and a developer and supplier of rare, naturally occurring cannabinoids and their analogs that is focused on commercializing important cannabinoid-based medicines to treat diseases with high unmet medical needs and, as a B2B supplier, selling rare cannabinoids to the health and wellness segments.
Our Strengths
We are the only clinical-stage company with both multiple cannabinoid drug candidates, in multiple therapeutic categories, that also is currently supplying rare cannabinoids to manufacturers in the health and wellness sector and that has internal expertise in multiple manufacturing approaches including chemical synthesis, biosynthesis and a proprietary, integrated biosynthesis-based manufacturing approach, called IntegraSynTM, to meet the needs of the rapidly evolving markets for rare cannabinoids. Key strengths include:
Experienced executive team and board of directors with proven track records.
One key critical success factor in the field of pharmaceutical drug development is the experience and skill set of the individuals leading the company. We have been successful in attracting and retaining executive and directors with extensive (20+ years) experience in all facets of the pharmaceutical industry, including fundamental research and development, multiple manufacturing techniques, drug formulation, clinical trial execution, regulatory approvals, pharmaceutical commercialization, company and capital formation, business development, legal, and corporate governance. Our leadership team is well-poised to lead us through all facets of drug development and product commercialization, either internally or externally via partnerships. It is this group of individuals that will help optimize our chances for success.
Multiple manufacturing approaches.
The combined manufacturing technologies from InMed and BayMedica provide us with a competitive advantage to utilize the most cost-efficient methodology (i.e. chemical synthesis, biosynthesis, IntegraSynTM) for the development and commercialization of new Products and Product Candidates and provision of rare bio-identical cannabinoids or their analogs to a wide spectrum of markets.
Early mover status as a B2B supplier of rare cannabinoids to the consumer health and wellness sector.
As demonstrated by the launch of CBC into the health and wellness sector, the team at BayMedica has substantial expertise in the commercial manufacturing scale-up to produce rare cannabinoids at large scale. This know-how is important to establishing an early-mover status and to maintain cost leadership with regards to specific rare cannabinoids.
Leading experts in the therapeutic potential of the rare cannabinoid CBN.
We have invested significant time and effort in understanding the characteristics and therapeutic potential of our first rare cannabinoid drug candidate, CBN. As such, we are positioning ourselves to be a world leader in the pharmaceutical development of this rare cannabinoid. We anticipate that CBN will be the first of several such drug candidates.
7
Targeting medical applications of rare cannabinoids to treat diseases with high unmet medical needs.
Significant investment in understanding the therapeutic potential of CBN has provided us with important insight as to how best to develop this class of compounds for treating various diseases. We intend to apply this know-how across several diseases that may benefit from cannabinoid-based medicines.
Diverse portfolio of patent applications covering a spectrum of commercial opportunities.
Success in pharmaceutical markets often rests with the strength of intellectual property, including patents, to protect our commercialization interests. We have filed several patents on our novel findings and expect to continue to do so. The acquisition of BayMedica brought several additional new patent families to enrichen our manufacturing as well as drug development opportunities.
Our Business Strategy
Our goal is to become a global leader in the manufacturing, supply and clinical development of rare cannabinoids and their analogs while continuing to avoid any direct interaction with the Cannabis plant. Our strategies to accomplish this include:
Advance INM-755 and INM-088 through preclinical and clinical development, thereby establishing important human proof-of-concept in multiple therapeutic applications.
These activities are well underway, at various stages, for both INM-755 for diseases of the skin and INM-088 for diseases of the eye. We have the internal capabilities to design and execute, together with multiple external vendors, the preclinical data sets and clinical studies required to advance pharmaceutical drugs towards regulatory submission.
Establishing partnerships for our various technologies, at different stages of development, to expedite their path towards commercialization in a resource-efficient manner.
We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products. With respect to the commercialization of each Product Candidate, we may rely on i) a “go-it-alone” commercialization effort; ii) out-licensing to third parties; or iii) co-promotion agreements with strategic collaborators for of our Product Candidates. To develop the appropriate commercial infrastructure internally, we would have to invest financial and management resources, some of which would have to be deployed prior to any confirmation our Product Candidates will be approved by regulatory authorities. Any decision on a “go-it-alone” commercialization effort versus out-licensing to / partnering with third parties will depend on various factors including, but not limited to, the complexity, the expertise required and related cost of building any such infrastructure for our Product Candidates. For INM-755 in EB, it is conceivable that we could oversee the clinical trials, given the relatively small patient sizes expected for such trials, and build the requisite internal commercialization infrastructure to self-market the product to EB clinics, which are limited in number and provide direct access to the vast majority of EB patients. For INM-088 in glaucoma, because of the potentially large clinical trial patient enrollees (possibly several thousand) and the extensive sales effort required to reach the many thousand prescribing physicians, we may consider exploring partnership opportunities early in the development process.
Develop multiple cost-efficient manufacturing sources for high quality rare cannabinoids as API for our core internal drug candidate pipeline, for licensing opportunities of non-core drug candidates, as well as a supply source, on a B2B basis, for rare cannabinoids in the health and wellness segment.
Extraction of rare cannabinoids from the plant is economically impractical for commercial applications. Modern approaches to product manufacturing, including chemical synthesis and biosynthesis, can be deployed in a targeted fashion once three criteria have been established: which cannabinoid is desired, what quality is required (pharmaceutical grade vs. food grade), and how large is the target production quantity. For early-stage supply to the health and wellness sector, chemical synthesis may be the most efficient pathway to early revenues. Over time, as quantities increase, biosynthesis may be most beneficial from a cost standpoint. For pharmaceutical drug applications, our integrative approach, called IntegraSynTM, may be most beneficial from a cost perspective. The cannabinoids that will be produced from our multiple approaches are targeted to be bio-identical to the naturally occurring cannabinoids. Our manufacturing approaches are designed to offer superior yield, control, consistency and quality of rare cannabinoids.
8
Continue to explore the potential of a wide array of rare cannabinoids and their analogs/variants to treat diseases based on our significant history in cannabinoid research and lead drug candidate identification.
Individual cannabinoids affect a range of different receptors in the human body, including, but not limited to, known endocannabinoid receptors. As such, they are responsible for a wide variety of pharmacological effects. However, due to the limited research into these varying effects, a full understanding of the role of each cannabinoid compound remains elusive. As a company, we have been formally investigating the utility of cannabinoids in treating disease for over seven years.
We have numerous options for commercializing our various technologies. At the core of our activities, we are a drug development company focused on commercializing important cannabinoid-based medicines to treat diseases with high unmet medical needs.
Cannabinoid Science Overview
Cannabinoids are a class of compounds that exist throughout nature and can be found in significant numbers and varying quantities in the Cannabis plant. The two predominant, or major, cannabinoids in the Cannabis plant are THC and CBD. These two exist in relatively large quantities in the plant and can be easily extracted, which has led to significant research into these two compounds over the previous several decades. Nevertheless, there are over 140 additional cannabinoid compounds found in the plant, referred to as minor or rare cannabinoids. Each cannabinoid has one or more specific chemical differences that may confer unique physiological properties in humans.
Cannabinoid receptors are found throughout the body and are involved in many different functions, such as pain perception, memory, immune function and sleep. Cannabinoids act as messengers that bind to cannabinoid receptors, as well as other receptors, signaling the endocannabinoid system into action. The relevance of the endocannabinoid system on many important physiological processes has made cannabinoids an important target to potentially treat a number of diseases and symptoms.
Two cannabinoid receptors in the human body are the endocannabinoid receptor 1 (CB1), which is more significant to the central nervous system, and endocannabinoid receptor 2 (CB2), which is more common with the immune system. Scientific literature suggests that CBN has a greater effect on the immune system than on the central nervous system; however, information on the effects of CBN on the endocannabinoid system is limited. We continue to research the effects of CBN and how it interacts and modulates receptors in the body.
9
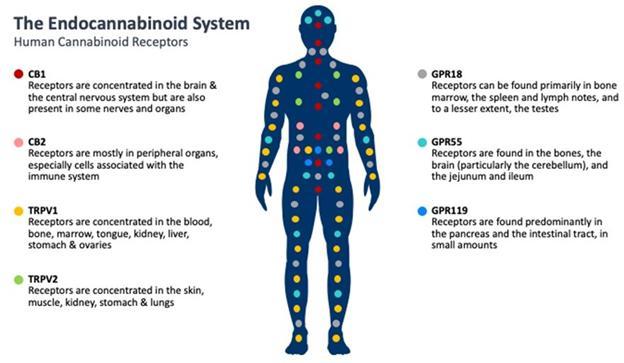
Significant investigation is currently underway to determine the role of cannabinoids in affecting other receptor systems in the human body.
Our Products, Product Candidates and Technologies
Development of a Flexible Suite of Processes for the Manufacturing of Cannabinoids
Introduction:
While there are over 140 different individual cannabinoids in the Cannabis plant, the two most well-known and studied compounds are also the two that occur in the largest quantities: THC and CBD. Due to their relative abundance in the Cannabis plant, it is also only THC and CBD that can currently be extracted economically; this also now includes genetically modified plants designed to significantly increase quantities of CBG in harvested crops. Among other challenges, the expense of extraction – or that of synthetic manufacturing – of the remaining minor or rare cannabinoids, may be orders of magnitude greater than that of THC, CBD and CBG.
Nevertheless, like the major cannabinoids THC, CBD and CBG, these rare cannabinoids may hold very important physiological benefits in humans. The challenge, and opportunity, that we have identified, and seek to solve, is selecting and engineering the most appropriate manufacturing approach to making a specific rare cannabinoid, at the desired quantity and requisite quality, which is cost-efficient and consistently yields bio-identical cannabinoids as compared to the compounds found in nature, among several other benefits. We believe that providing this solution will be a critical success factor not only for our own drug development strategy, but also for other pharmaceutical and health and wellness companies.
Development of InMed’s biosynthesis and IntegraSynTM technologies:
In 2015, we commenced the development of a biosynthesis process for the manufacturing of cannabinoids through a research collaboration with Dr. Vikramaditya Yadav from the Department of Biological and Chemical Engineering at the University of British Columbia. Utilizing the basis of a specific vector created for us, Dr. Yadav initiated a Research and Development Project titled “The Metabolic Engineering of yeast and bacteria for synthesis of cannabinoids and Cannabis-derived terpenoids” under a collaborative research agreement. Subsequently, we signed a Technology Assignment Agreement with the University of British Columbia whereby we retain sole worldwide rights to all patents emergent from the technology under development in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology and a single digit royalty on any sub-licensing revenues. Other than the 1% royalty, we do not have any ongoing financial commitments under these arrangements with the University of British Columbia.
10
Microorganisms do not naturally produce cannabinoids nor the enzymes required for their assembly. However, utilizing genome engineering to modify their metabolism, we have systematically introduced different aspects of the Cannabis plant’s metabolic pathways into a bacteria (E. coli), referred to as a host, and have reported what we believe to be the first-of-its-kind production of fully differentiated cannabinoids in this bacteria. This research served as the basis for the subsequent development of a new, integrated approach to cannabinoid manufacturing that we refer to as IntegraSynTM. IntegraSynTM is a flexible, integrative cannabinoid synthesis approach utilizing novel enzyme(s) to efficiently produce bio-identical, economical, pharmaceutical-grade cannabinoids without the risk and high-resource requirements of an agriculture growing operation.
In early research, we utilized the specific gene sequences from the Cannabis plant that encode the instructions to make specific enzymes that enable cannabinoid synthesis and subsequently transferred these genes into E. coli. This intervention converts the bacterium into a manufacturing system that produces substantial quantities of the target cannabinoids. This technology may provide an opportunity for industrial-scale manufacturing of cannabinoids, which we believe would be a significant improvement over existing manufacturing platforms. Specifically, direct extraction is quite cumbersome, time-consuming and relatively low yielding for all but a few of the cannabinoid compounds. In contrast, the use of microorganisms for manufacturing cannabinoids eliminates the need for an agricultural-centric process, including planting, growing, harvesting and extraction. There are also economic and environmental advantages including substantially reduced resource requirements (e.g., water, electricity, manpower, etc.). Furthermore, the agricultural approach has several hard-to-remove impurities (e.g., pesticides, etc.), potentially presenting safety issues. As with all crops, yield fluctuations influenced by the environment present an additional risk. Only a few of the 100+ cannabinoids can currently be extracted from the plant in sufficient quantities to make the process economically viable. For certain cannabinoids, chemical synthesis, by comparison, can be challenging and expensive due to the complexity of these molecules and the ability to economically scale the process. For these reasons, we believe that a modified biosynthetic approach may be superior to both of these alternatives for pharmaceutical grade production of certain cannabinoids.
Cannabinoids are prenylated polyketides that are derived from fatty acid and terpenoid precursors. The biosynthesis of these molecules involves four metabolic pathways, two of which originate from central carbon metabolism. The first pathway (the Terpenoid pathway referenced in Figure 1 below) culminates with the synthesis of geranyl pyrophosphate, or “GPP”, and neryl pyrophosphate, or “NPP”. These molecules are terpenoid building blocks, or precursors. The second cannabinoid biosynthetic pathway, or the Polyketide pathway, is a version of a polyketide biosynthetic pathway and results in the second requisite precursor, either: olivetolic acid, or “OA”, and/or divarinic acid, or “DVA”. The polyketide precursors subsequently combine with the terpenoid precursors in the third pathway, which comprises a single, specialized enzyme in the plant, to yield the ‘gateway’ cannabinoids, the cannabinoids that act as precursor molecules for further differentiation into all others. For instance, OA combines with GPP to yield the gateway cannabinoid cannabigerolic acid, or “CBGA”. The gateway cannabinoids are subsequently modified in the fourth pathway to yield cannabinoids such as tetrahydrocannabinolic acid and cannabidiolic acid. We refer to the fourth pathway as the down-stream pathway involving the transformation of the acid form of the cannabinoids into the non-acid form via enzymes called synthases. Synthesis of CBGA is the most dominant pathway in the plant, resulting in high quantities of the down-stream cannabinoids THC and CBD. Other combinations of the various precursors result in different gateway cannabinoids which, in turn, leads to diversification into more than 140 different individual cannabinoids.
11
Figure 1:
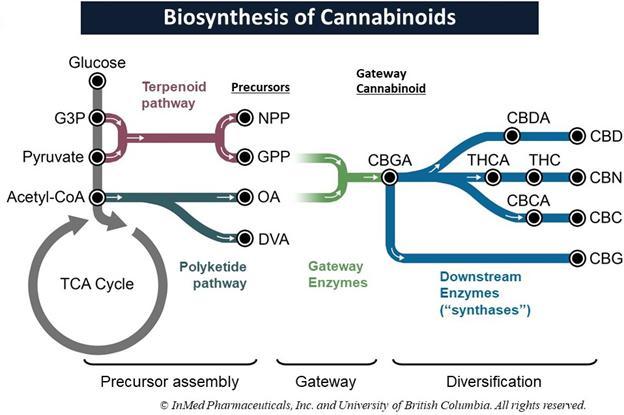
Figure 1: Synthesis of the gateway cannabinoid CBGA is the most prevalent pathway in the Cannabis plant, leading to high levels of both THC and CBD. Our technology, IntegraSynTM, is designed to mimic the natural biosynthesis of cannabinoids starting with an E. coli biofermentation process combined with additional common pharmaceutical manufacturing technologies.
Our earlier research led to the successful construction of the terpenoid biosynthetic pathway and the gateway pathway for synthesis of CBGA and the down-stream diversification pathways for synthesis of other cannabinoids.
Our goal for the biosynthesis program has always been to achieve the simplest, most efficient, scalable, flexible and economical solution with the least steps and fastest production cycle, to make bio-identical cannabinoids to those found in nature. While developing our bacterial biosynthesis system, we further optimized the fermentation conditions and the purification processes. Working with various contract development and manufacturing organizations (“CDMOs”), we have continued development and optimization of our manufacturing processes that led to the development of IntegraSynTM.
IntegraSynTM integrates various pharmaceutical manufacturing processes to maximize yield and minimize the cost of cannabinoid synthesis, in particular for pharmaceutical-grade products. We utilize proprietary, high efficiency enzymes produced via the E. coli biofermentation portion of the IntegraSynTM approach for the production of a cannabinoid. Our enzymes are used in combination with cost-effective yet sophisticated substrates (or, starting materials) to produce a cannabinoid in bulk via a biotransformation process, which is then further processed with downstream purification steps including separation, purification and drying. This cannabinoid can be inventoried in bulk and used either as a finished API cannabinoid product or as a starting material for other cannabinoids. This further differentiation can utilize any one of several well-established manufacturing approaches – including enzymatic biotransformation and traditional chemical synthesis – to optimize yield, time and cost.
12
IntegraSynTM makes cost-efficient use of sophisticated starting materials, requires fewer costly steps from precursor substrates all the way through to end-product, and is designed as a high-yield manufacturing process for pharmaceutical use. Furthermore, this manufacturing method is flexible in shifting production from one cannabinoid to another under GMP conditions. Our initial data demonstrated a substantial increase in cannabinoid production yield per fermentation batch compared to our traditional biosynthesis method. The final cost of goods for individual cannabinoids is driven by several factors including, among others: efficiency of the enzyme(s) used; number of manufacturing steps; type of manufacturing equipment / processes used; and, final yield of the entire manufacturing process.
Targeted Benefits of IntegraSynTM:
| A. | Improved yields beyond other standard manufacturing methods for various cannabinoids at pharmaceutical-grade |
| B. | Cost-efficient due to minimization of expensive manufacturing steps and cost-effective use of sophisticated raw materials |
| C. | Flexible, modular approach, able to shift from production of one cannabinoid to another |
| D. | Scalable to meet market demand of cannabinoids for pharmaceutical products or other purposes |
| E. | Sustainable approach with less environmental impact than plant-grow-harvest-extract-purify methods |
Next steps in the further development of IntegraSynTM, all of which are currently ongoing, include:
| ● | Continue to optimize and scale-up the IntegraSynTM process to larger vessels, whereby protocols will be developed to optimize the manufacturing parameters associated with the entire process with the Almac Group (UK); |
| ● | Conduct analytical assays to support batch production |
| ● | Scale-up process to be GMP ready; |
| ● | Continue efforts to optimize pathways to further diversify the number of cannabinoids produced using our technology; and |
| ● | Identify potential commercial partnership opportunities |
We currently view our options for achieving GMP production capabilities as three-fold: (a) building our own dedicated biosynthesis facility; (b) accessing existing manufacturing capacity via leases with third parties; or (c) licensing our process/know-how to a CDMO with existing infrastructure to produce the requisite preclinical, clinical and commercial-scale supply of our Product Candidates.
BayMedica’s Chemical Synthesis and Biosynthesis Technologies for the Development and Production of Cannabinoids, Their Variants and Analogs
BayMedica, Inc. was founded by Shane Johnson, MD, Philip Barr, PhD and Charles Marlowe, PhD in September of 2016 with the objective of manufacturing cannabinoids and novel cannabinoid compounds for use in health and wellness and pharmaceutical markets. BayMedica set out to develop cannabinoid manufacturing techniques that are ‘method agnostic’, utilizing the most practicable, expeditious and cost-effective means to produce any particular cannabinoid or novel cannabinoid compound.
Chemical Synthesis for the Development and Production of Cannabinoids
Chemical synthesis is a well-established, long-standing, robust and reliable method for the production of myriad compounds for use in both consumer and pharmaceutical products, including such commonly used medications as vitamin-D and acetaminophen. The production of cannabinoids, in particular CBD and THC, by synthetic methods was first described in 1965 by Mechoulam, et. al. Although yields were typically less than 10% and the scales were small, this work paved the way for methods to synthesize these common cannabinoids as well as many rare and novel cannabinoid compounds. Today several companies such as Noramco, Purisys, Biovectra, Kinetochem and Benuvia reliably manufacture a wide variety of common and rare cannabinoids to pharmaceutical API standards. However, because price points for these compounds have remained high, their utility in non-pharmaceutical applications has historically been limited.
13
Pharmaceutical chemistry methods have also been of paramount utility in the creation of combinatorial libraries of new chemical entities for drug discovery. We believe these pharmaceutical chemistry methods will also be of great value in the preparation of novel cannabinoid compounds with enhanced pharmacological activities and have leveraged our expertise to this end. Using combinations of pharmaceutical chemistry and biosynthesis, the BayMedica team has generated a wide variety of these compounds with the primary focus being on modifying the pentyl side chain found on many naturally occurring cannabinoids. We believe that unlike a traditional drug discovery approach using combinatorial libraries, our approach, because it does not change the core structure of each cannabinoid type, will result in a larger proportion of novel cannabinoids showing pharmacological activity, thus increasing the probability of a successful drug candidate. Prior to the acquisition by InMed, BayMedica delivered multiple novel CBN-based compounds to InMed for evaluation. Since that time, we have developed additional New Chemical Entities (“NCEs”) with the ability to expand through existing and novel methods currently under development.
Chemical Synthesis-Derived Cannabinoids Under Development or Commercialized BayMedica
Cannabichromene (CBC)
The historically high cost of goods for cannabinoids manufactured by chemical synthesis has largely precluded their widespread adoption for non-pharmaceutical applications. BayMedica is a world-leader and has successfully manufactured and commercialized a rare cannabinoid, CBC, for sale to distributors into the health and wellness industry. The development of a scalable process for the manufacturing of CBC began in 2018 using well established chemical synthesis protocols.
In calendar 1Q2019, a Material Services Agreement was completed with a multinational contract research, development and manufacturing organization (“Chemistry CDMO”) to facilitate the optimization and scale-up of BayMedica’s proprietary CBC manufacturing process using commercially available starting materials sourced from various manufacturers. We scaled to a batch size of greater than 1kg by calendar 3Q2019 at which time we contracted a leading U.S. manufacturer to provide the final purification of CBC to greater than 95% purity, a product we call ProDiol™ CBC. This manufacturer also operates a North American (NA) based toll-processing facility with the capability to process from 10kg to metric ton quantities of our crude CBC material under food-grade GMP conditions. By late 4Q2019 our Chemistry CDMO had scaled the process to greater than 10kg, and by year end 2019 to almost 30kg with final purification at the NA contractor. We commenced commercial sales of ProDiol™ CBC in November 2019.
Large scale manufacturing of crude CBC began at our Chemistry CDMO in 1Q2020 at >40kg. The emergence of the Covid-19 pandemic significantly impacted sales in calendar 1H2020. However, purchase interest returned in 3Q2020. Large scale production continued with several batches of over 100kg and 200kg in 2021.
Cannabicitran (CBT)
We have developed a process for the efficient chemical synthesis of CBT through both in-house R&D efforts and via our CDMO. We began scaling this process and conducted downstream processing and purification trials in late calendar 2H2021. We received initial purchase orders for, and commenced commercial sales of, CBT in calendar 1Q2022.
Cannabidivarin (CBDV)
In calendar 1Q2021, BayMedica acquired a license to utilize novel chemical synthesis technologies to produce and distribute the rare, non-intoxicating “varin” cannabinoid, CBDV, exclusively in the USA and non-exclusively worldwide. BayMedica began R&D and scale up with our Chemistry CDMO in calendar 2Q2021. Through that partnership, we have developed a novel and efficient method allowing for the scalable preparation of CBDV. In calendar 4Q2021, via the Chemistry CDMO, we successfully scaled CBDV synthesis to commercial quantities and initiated procurement of starting materials sufficient to meet expected customer demand. Originally scheduled for calendar 1Q2022,due to global supply chain and shipping delays, the commercial launch for CBDV is now scheduled for calendar 2Q2022.
14
Tetrahydrocannabivarin (THCV)
As part of the calendar 1Q2021 license for techniques to synthesize and produce CBDV, we also acquired the rights to synthesize, sell, and distribute the non-intoxicating rare cannabinoid THCV exclusively in the USA and non-exclusively worldwide. In calendar 3Q2021, we began researching and developing processes to convert CBDV to THCV. In conjunction with our Chemistry CDMO and our in-house team, we have developed a robust pilot-scale process that produces THCV. We are currently developing a purification process to produce the finished THCV material. We have engaged a North American-based manufacturer with the equipment and capabilities to produce commercial scale quantities of THCV. We began scale-up of our novel process with this CDMO in calendar 1Q2022 with anticipated commercial launch in calendar 2Q2022.
Analogs of Cannabinoids / New Chemical Entities
In the field of pharmaceutical drug development, the term analog is used to describe structural and functional similarity between an original (or parent) molecule and one that has been somewhat modified. While any company researching a naturally occurring compound, like cannabinoids, cannot own a patent on the molecule itself for commercial exclusivity, a modified molecule, which has certain structural and pharmacological similarities with the original compound, can be patented. As well, modifications of the original molecule (ie, the analog) can be designed to confer certain improvement in activity of the parent, such as an elevation of the desired physiological effects, a decrease in unwanted side effects, improvement in aspects related to drug delivery to targeted tissues, etc., or a combination of these targeted outcomes. We have filed patents covering numerous structural additions and modification of the naturally occurring cannabinoids. Each individual modification to each individual cannabinoid represents a New Chemical Entity (“NCE”) which can be patented. If issued, this patent family will confer market exclusivity to us for the analogs that we intend to develop into pharmaceutical Product Candidates, license, partner or sell to interested external parties.
BayMedica Yeast Biosynthesis for Cannabinoid Development and Production
BayMedica utilizes the yeast S. cerevisiae as a “biofactory” to produce cannabinoids and cannabinoid analogs. Compared to other heterologous systems, we believe that S. cerevisiae is an attractive host for development of a cannabinoid biosynthesis platform. S. cerevisiae is a generally recognized as a safe organism that has a long history of successful use in the biotechnology industry for the large-scale production of a variety of different fermentation products. In particular, S. cerevisiae has previously been engineered to produce high levels of terpenes and polyketides – the building blocks of all cannabinoids. Genetic manipulation of S. cerevisiae is straightforward and advanced genetic tools enable rapid strain engineering for optimized cannabinoid production. Moreover, our team has deep knowledge and experience engineering S. cerevisiae for the heterologous production of natural products by fermentation.
Unlike in the Cannabis plant, our flexible yeast platform allows for the specific production of a single precursor or cannabinoid. This process negates the need for additional downstream steps to isolate and purify the desired precursor or cannabinoid from other naturally produced compounds found in a plant-based extraction process. To that end we have engineered strains that produce the precursors and cannabinoids OA, DVA, CBGA, CBGVA and THCVA at concentrations supporting further scale-up and development for future commercialization. At benchtop scale, we have developed simple downstream processes for the purification of these compounds. We have evaluated multiple CDMOs capable of both the biosynthesis scale-up and downstream processing.
In addition to the natural cannabinoids above, we have leveraged our expertise in pharmaceutical chemistry and yeast biosynthesis to produce a number of novel cannabinoid analogs and variants of pharmaceutical interest.
15
Hybrid Methods as a Platform for Cannabinoid Development and Production
Unlike other groups, our expertise in the areas of yeast engineering, biosynthesis and pharmaceutical chemistry allows us to develop in-house technologies for the production of novel cannabinoid compounds as well as the conversion of “common” precursors or cannabinoids into rare natural cannabinoids. For instance, we have employed methods where we take a biosynthetic starting material and convert it into CBC via chemical synthesis.
We have also developed technologies whereby chemical synthesis is used to make novel precursor compounds that are then converted biosynthetically into novel cannabinoids. As part of an announced collaborative research agreement prior to the acquisition of BayMedica by InMed Pharmaceuticals, BayMedica delivered several novel CBN analogs to InMed Pharmaceuticals for preclinical evaluation.
Competitive Conditions:
Other companies deploy a diversified number of cannabinoid synthesis manufacturing techniques, including:
| ● | Biosynthesis (generation of the final compound inside a single system) using yeast, non-E. coli bacteria, or other approaches (algae, etc.) as a host organism; |
| ● | Synthetic chemistry; and |
| ● | Combinations of these above-listed technologies |
Several companies (see chart below) are active in the cannabinoid manufacturing space including BioVectra, CB Therapeutics, Cellibre, Cronos, Ginko Bioworks, Hyasynth, Intrexon, KinetoChem, Librede, and Purisys, among several others.

Key Milestones:
On May 21, 2015, we commenced the development of our biosynthesis process for the manufacturing of cannabinoids through a research collaboration with Dr. Vikramaditya Yadav from the Department of Biological and Chemical Engineering at the University of British Columbia under a project titled “The Metabolic Engineering of yeast and bacteria for synthesis of cannabinoids and Cannabis derived terpenoids”. On May 31, 2017, we signed a Technology Assignment Agreement with the University of British Columbia whereby we retain sole worldwide rights to all patents emergent from the technology under development in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology and a single digit royalty on sub-licensing revenues. Royalties are payable, on a country-by-country basis, until such time as there is no longer a patent pending, unexpired patent or issued patent derived from the transfer technology, in any country. On May 15, 2018, we extended our Collaborative Research Agreement, which may be terminated by either party upon 30 calendar days written notice, with the University of British Columbia for an additional three years. Other than the 1% royalty, we do not have any ongoing financial commitments under these arrangements with the University of British Columbia.
16
We, in conjunction with our collaboration partners at the University of British Columbia, continue to advance the production platform for the biofermentation of cannabinoids. Optimization of the vector continued in parallel with the identification of optimal fermentation conditions and down-stream purification processes with third party contract manufacturing organizations. Optimization of the fermentation conditions was a project conducted with the National Research Council Canada at their dedicated fermentation facility in Montreal, Quebec. While we do not anticipate any new intellectual property arising from this venture, under the terms of this research agreement, the National Research Council of Canada owns all new IP and we have a sole, fully-paid-up license to all commercialization rights of such IP. This project was initiated in October 2018 and concluded in the second half of 2019.
In February 2019, we entered into a separate process development collaboration by way of a Master Service Agreement with the Almac Group (UK), or “Almac”, a seasoned GMP pharmaceutical CDMO. Almac was initially tasked to develop a down-stream purification process to support the fermentation optimization activities at the National Research Council of Canada. In addition, we also engaged Almac to assist in the development of an “alternative” manufacturing process for cannabinoids which integrates the best available technologies across the spectrum of pharmaceutical drug production. This process is now referred to as IntegraSynTM. In May 2020, we announced our working relationship with Almac on an integrated approach to augment current biosynthesis-based methods for cannabinoid production. The companies have been engaged in developing a streamlined cannabinoid manufacturing process, specifically optimizing the upstream cannabinoid assembly processes as well as downstream purification processes, to achieve cost-efficient, GMP-grade active pharmaceutical ingredients for prescription-based cannabinoid medications. Almac is an international, privately-owned organization which has grown organically over the past five decades now employing over 5,600 highly skilled personnel across 18 facilities including Europe, the US and Asia. We retain all rights to this new process while Almac retains certain rights-of-first refusal on the production and supply of certain precursors, or starting materials, for this alternative process.
Other Milestones Include:
| ● | September 12, 2017 – We announced the filing of a provisional patent application entitled, “Metabolic Engineering of E. coli for the Biosynthesis of Cannabinoid Products” (#62/554,494) pertaining to our biosynthesis program for the manufacture of cannabinoids that are identical to those found in nature. We expect that this patent application, since converted into an application pursuant to the Patent Cooperation Treaty, or a “PCT Application”, and pursued in key jurisdictions throughout the world, will provide significant commercial protection for our E. coli-based expression system to manufacture any of the 100+ cannabinoid compounds that may have a medical impact on important human diseases. This is the first in a series of patent applications directed to various aspects of our biosynthesis program. See “Intellectual Property”. |
| ● | September 25, 2017 – We announced an update on the significant advancements in our technology for the microbial biosynthesis of cannabinoids. We have successfully demonstrated an ability to selectively produce various gateway cannabinoids using genetically engineered microorganisms. These molecules can be functionalized further to produce any of the 100+ down-stream cannabinoids, or those formed from an enzymatic reaction with the gateway cannabinoid CBGA, found naturally in the Cannabis plant. We are actively employing this production chassis to synthesize compounds for certain pharmaceutical research programs. Our biosynthesis program has resulted in what we believe to be two significant firsts |
| o | new metabolic pathway for manufacturing the terpenoid family of cannabinoid precursors that is much more robust than other microbial expression systems tested by us; and |
| o | first-ever production of any fully assembled down-stream cannabinoids in E. coli, beginning with genetic material to produce precursors, enzymes, and synthases. |
17
| ● | September 10, 2018 – We announced the filing of a PCT Application for biosynthesis which claims a priority date from September 5, 2017 (PCT/CA2018/051074). The PCT Application filing is a conversion from the provisional patent filed in September 2017. |
| ● | March 18, 2019 – We announced the publication of the first in a series of pending patent applications directed to our biosynthesis platform technology for the manufacturing of pharmaceutical-grade cannabinoids. International Patent Application International Patent Application No. PCT/CA2018/051074, which published as WO2019046941, entitled “METABOLIC ENGINEERING OF E. COLI FOR THE BIOSYNTHESIS OF CANNABINOID PRODUCTS”, addresses the enablement and maximization of cannabinoid production through optimization of the precursor substrates needed to support specific cannabinoid synthesis. This application, as well as two more recently filed U.S. provisional patent applications, covers various elements required to enable functional cannabinoid synthase production in an E. coli system. We will actively seek to convert these two follow-on provisional applications, and subsequent provisional patents from new patent families, into additional PCT Applications in all major commercial jurisdictions, in due course. See “Intellectual Property”. |
| ● | May 19, 2020 – We announced the filing of a key Patent Cooperation Treaty (“PCT”) patent application directed to our biosynthesis platform technology for the manufacturing of pharmaceutical-grade cannabinoids. The PCT patent application entitled “Compositions and Methods for Biosynthesis of Terpenoids or Cannabinoids in a Heterologous System”. This application” was initially filed as two separate United States Provisional Patent applications and further addresses the enablement and maximization of cannabinoid production through optimization of the precursor substrates needed to support specific cannabinoid synthesis |
| ● | September 22, 2020 – We announced the filing of a PCT patent application as part of a growing portfolio of intellectual property related to the IntegraSyn™ manufacturing approach for producing low-cost, pharmaceutical-grade cannabinoids. |
| ● | April 26, 2021 – We announced that the IntegraSyn™ cannabinoid manufacturing approach has achieved a level of 2g/L cannabinoid yield, a milestone that signals commercial viability and supports advancement to large-scale production in the coming months. Having achieved a 2g/L yield level, we will now focus on manufacturing scale-up to larger batch sizes while continuing process and enzyme optimization, targeting increased cannabinoid yield and further reducing the overall cost of goods. In parallel, we continue to prepare the manufacturing process to be Good Manufacturing Practice (GMP)-ready for pharmaceutical quality production. The next stage of large-scale production is to produce a batch with a target output of one kilogram of the selected cannabinoid in the second half of calendar 2021 via a GMP-ready process |
| ● | June 17, 2021 – We announced that we increased cannabinoid yield to 5 g/L with IntegraSyn™ in advance of commercial-scale production, a milestone that significantly reduces the overall cost of rare cannabinoid manufacturing. |
Research and Development Pipeline of Therapeutic Drug Candidates
INM-755 for the Treatment of Epidermolysis bullosa (“EB”)
Introduction
INM-755 (CBN) cream is being developed as a proprietary, topical, single-cannabinoid product candidate intended as a therapy in dermatological diseases. The first clinical indication under development is EB. EB is a collective name for a group of genetic disorders of connective tissues characterized by skin fragility leading to extensive blistering and wounding. It affects skin and mucous membranes, particularly of the gastrointestinal tract, genitourinary and respiratory systems. EB is a debilitating disease affecting a small proportion of people in the United States, thus earning it an orphan-disease status. The disease has no definitive cure and all current treatments are directed towards symptom relief. There are, however, a number of products, mainly gene therapies, currently in clinical trials, in which a cure is being explored, according to several recent scientific publications. Our preclinical research has identified a specific cannabinoid, CBN, that may prove beneficial to patients: first, by addressing certain key disease hallmarks (which may include wound healing, infection, pain, inflammation); and second, by regulating the expression of various proteins (keratins) that may compensate for reduced expression of others.
18
The active ingredient in INM-755, CBN, is an agonist for both cannabinoid (CB) 1 and CB2 receptors, with a higher affinity for CB2, which means it should have a greater effect on the immune system than on the central nervous system. The distribution of CB1 and CB2 receptors in sensory nerves and inflammatory cells in the skin make it an attractive pharmaceutical agent for dermal treatments in medical conditions characterized by inflammation and pain.
In preclinical pharmacology studies, CBN demonstrated activity as an anti-inflammatory and antinociceptive agent. CBN upregulated expression of keratin 15 (K15), which might lead to skin strengthening and reduced blister formation in EBS patients with keratin 14 (K14) mutations. At the cream concentrations chosen for clinical development, it does not appear to impede wound healing of partial-thickness wounds. Its anti-inflammatory activity may be beneficial in healing chronic wounds caused by prolonged inflammation.
We have completed 20 safety pharmacology and toxicology studies to investigate the effects of CBN. We have also completed three Phase 1 safety and tolerability studies in healthy volunteers, two studies of which were conducted with varying concentrations of INM-755 cream and one study of which examined the non-CBN components of the cream base for INM-755. In December 2021, we began patient treatment in a Phase 2 clinical trial for INM-755 in EB.
The Science Behind EB
At the most basic level, the hallmark of EB is poor anchorage of the epidermis to the dermis such that the skin and mucous membranes of the affected individuals tend to shear and blister on minimal friction. This is due to the genetically inherited defect in certain genes (multiple genes have been shown to be associated with the different subtypes of EB) that code for some specific proteins that are concerned with maintaining the integrity of skin and mucous membranes.
There are four main subtypes of the condition. Each of these subtypes can display a spectrum of phenotypic severity reflecting the types of mutations in different genes, together with modifying environmental factors. The types of mutations also determine the mode of inheritance, either autosomal dominant or autosomal recessive. The following table shows the pattern of inheritance and the defective genes and proteins in each:
Classification of EB Types

19
(a) EBS
This is the most common form of EB and is characterized by a lack of adhesion of the skin directly above the basement membrane (the basal layer). An estimated 55% of people with EB have EBS resulting from a genetic defect of the keratins K5 and K14, with the incidence between the two defects estimated to be essentially equal. The most common form of EBS manifests itself as blistering confined to the hands and feet while in others blistering can occur all over the body. Blistering generally appears during the neonatal period but it can also manifest itself in later childhood (or even in adult life). Painful skin blisters are accentuated by friction, especially on the feet where footwear causes increased irritation. Friction injuries tend to occur more commonly in warm weather and secondary infections are common.
(b) Junctional EB
Junctional EB is characterized by a lack of adhesion of the skin through the basement membrane and affects some 5% of those with EB. The generalized type of junctional disease (about half of cases of junctional EB) is usually fatal in infancy. This is often as a result of anemia and malnutrition due to poor feeding caused by the serious blistering in the pharynx and esophagus. The milder form of the disease can cause life-long pain and disability.
(c) Dystrophic EB, or “DEB”
DEB is characterized by a lack of adhesion of the skin under the basement membrane. Approximately 30% of people with EB have DEB. Patients with DEB tend to develop blisters that heal with fibrosis, leading to joint contracture, fusion of the fingers, contractures of the mouth membranes and narrowing of the esophagus. Often the dominant inherited type of DEB is the least severe type and the patient can lead an almost normal life. However, the severity of the condition does increase with age due to scarring, syndactyly and generalized skin atrophy. Those with recessive DEB have a high chance of developing a squamous cell carcinoma, often before the age of 35.
(d) Kindler Syndrome
This type of EB is rare and usually becomes apparent at birth or soon after. This condition is called mixed type because blisters appear across the skin layers. The condition usually improves with time and can disappear. It is the only type that causes patchy discoloring (mottling) of skin exposed to the sun. Kindler syndrome is recessive.
(e) Epidermolysis bullosa acquisita
Epidermolysis bullosa acquisita is a rare type that is not inherited. The blisters result from the immune system attacking healthy tissue by mistake. It’s similar to another immune system disorder called bullous pemphigoid. It tends to cause blisters on the hands, feet and mucous membranes.
Epidemiology, Morbidity and Mortality
The most reliable figures on prevalence and incidence of EB are derived from the National EB Registry, or “NEBR”, which collected cross-sectional and longitudinal data on about 3,300 EB patients in the United States from 1986 through 2002. The prevalence of EB was estimated to be approximately 11 per million and the incidence approximately 20 per million live births. In the United States, assuming that mild cases of EBS are reported only 10% of the time, the affected population in the United States is approximately 12,500. Other sources cite populations of up to 25,000 in the United States.
Generalized blistering caused by any subtype may be complicated by infection, sepsis, and death especially in infancy. Severe forms of EB increase the mortality risk during infancy. In patients with EB that survive childhood, the most common cause of death is metastatic squamous cell carcinoma. This skin cancer occurs most frequently in patients with recessively inherited DEB who are aged 15-35 years. In contrast, dominantly inherited EBS and DEB and milder forms of junctional EB may not affect a patient’s life expectancy adversely. Onset of EB is at birth or shortly after. The exception occurs in mild cases of EBS, which may remain undetected until adulthood or remain undiagnosed. The disease appears to have equal incidences in both sexes.
20
Current Treatments
As a genetic disease, EB has no cure and, as a designated orphan-disease, there are no approved products specifically to treat this indication. Effective management of EB patients involves a collaborative approach between several specialists, including surgeons, dermatologists, ophthalmologists, dentists, psychologists, podiatrists, physiotherapists and geneticists. The aim is to provide support to the patient by alleviating symptoms and managing complications; in particular, the patient caregivers must assess and act daily to treat the wound and enable wound healing, address the current level of pain and itch, provide adequate antimicrobial protection, reduce inflammation (as a source of depressed wound healing abilities) and address the emotional state of the patient.
Current medications are employed in control of pain (various types of analgesics including nonsteroidal anti-inflammatory drugs, or “NSAIDS”, tricyclic antidepressants, gabapentin, and narcotics) and pruritus (antihistamines, etc.) and to address complications such as local infection and septicemia (local and systemic antibiotics). Steroids and phenytoin are also used in managing dysphagia-associated pain. Tetracycline is considered to be beneficial in improving the blistering and epithelial disadhesion. The complications of these classes of medications are well known and the drugs are most likely to further complicate the patients’ conditions since they will be used on long-term basis.
The newer products currently in research also have challenges. For example, the use of bone marrow was being researched by the University of Minnesota with some promising results. However, the severe immunosuppression that bone marrow transplantation requires causes a significant risk of serious infections in patients with large scale blisters and skin erosions.
Competitive Landscape
We are developing INM-755, our proprietary, topical, single cannabinoid product candidate, as a first-line therapy in all EB patients for symptom relief and may explore its effects in EBS with a K14 genetic mutation as a therapy to potentially strengthen skin integrity via up-regulation of the keratin K15.
There are no therapies approved specifically for the treatment of EB. This lack of treatment options creates a significant unmet medical need in this devastating condition. For those products currently envisioned or in clinical trials as topical treatments, wound healing and symptom relief are the primary endpoints.
According to public information, several topical investigational drug formulations are currently at various stages of clinical development for the treatment of EB, including:
| ● | Amryt Pharma’s investigational drug, Oleogel-S10, which is a topical product incorporating a betulin-based active ingredient formulated with sunflower oil. AP101 causes the keratinocytes to migrate faster and to differentiate into mature epithelial skin cells. This product is currently approved in some jurisdictions for the treatment of partial-thickness wounds in adults. Oleogel-S10 was investigated in a Phase III registration clinical trial. The Phase III double blind study is complete and met its primary endpoint on time to first target wound closure (p=0.013). The 24-month open label extension study is ongoing (NCT03068780). The product does not currently have regulatory approval to treat EB but has been submitted to the FDA for approval. In June 2021, Amryt received confirmation from the FDA that its NDA for Oleogel-S10 had been accepted and granted priority review and in November 2021 the FDA extended the review period. The submission of additional information by Amryt was determined by the FDA to constitute a Major Amendment to the NDA, resulting in an extension of the PDUFA goal date by three months to February 28, 2022; and |
| ● | Castle Creek Biosciences has a Phase III Clinical trial ongoing. The purpose of this study is to determine whether administration of FCX-007 in addition to standard of care improves wound healing as compared to standard of care alone (control) in children, adolescents, and adults with Recessive Dystrophic Epidermolysis Bullosa. DEFI-RDEB is a multi-center, intra-patient randomized, controlled, open-label, Phase III study of FCX-007 for the treatment of persistent non-healing and recurrent RDEB wounds in approximately 24 subjects. |
21
Other approaches have shown promise and are under investigation for the treatment of EB:
| ● | Gene therapies; |
| ● | Skin grafts with gene-modified epidermal sheets; |
| ● | Stem cell transplants |
| ● | Intravenous replacement of recombinant collagen VII (for RDEB); |
| ● | Topical/intradermal gentamicin to restore laminin beta3 (JEB/DEB with nonsense mutations); and |
| ● | Granulocyte colony-stimulating factor (DEB). |
Additionally, several companies are pursuing the symptomatic relief for EB patients, including the patient advocacy organization DEBRA, which is sponsoring a trial using oral cannabinoids (THC, CBD) to mitigate pain and itch.
Regulatory Perspectives
According to the National Epidermolysis Bullosa Registry, the overall incidence is about 20 per million live births and prevalence is 11 per million in the United States. EB is designated as an “orphan disease”, and we plan to seek regulatory designation of INM-755 as such in the U.S. and similar designations in various jurisdictions. The FDA defines orphan products as “those intended for the safe and effective treatment, diagnosis or prevention of rare diseases/disorders that affect fewer than 200,000 people in the United States, or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug”. The EMA has its own definition of orphan disease and, under the European definition, EB is also an orphan disease.
The mission of the FDA Office of Orphan Products Development, or “OOPD”, is to advance the evaluation and development of products (drugs, biologics, devices, or medical foods) that demonstrate promise for the diagnosis and/or treatment of rare diseases or conditions. This arm of the agency evaluates scientific and clinical data to identify and designate products as promising for rare diseases and to further advance scientific development of such promising medical products. The OOPD also works on rare disease issues with the medical and research communities, professional organizations, academia, governmental agencies, industry, and rare disease patient groups. The OOPD provides incentives for sponsors to develop products for rare diseases. The Orphan-Drug Designation program, which is administered by the OOPD, provides orphan status to drugs and biologics which are defined using the FDA definition above. The Orphan Products Grants Program, which is administered by the OOPD, provides funding for clinical research that tests the safety and efficacy of drugs, biologics, medical devices and medical foods in rare diseases or conditions.
It is worth noting that there is a common pathway for application of orphan status for a product to both the FDA and EMA, and applicants to the FDA are advised to use the common application platform. With regards to the data to be used in the application, it is expected that applicants demonstrate that there is “promise” that the drug will be effective in treating said disease. “Promise” is interpreted to include either data from clinical trials, data from case studies/reports, data from appropriate animal models or, on rare occasions where there is no appropriate animal, data from in vitro experiments in addition to supporting information.
22
Regulatory Incentives for Orphan Product Development

Data Summary of Preclinical Studies for INM-755
INM-755 is a topical, single cannabinoid cream formulation that is being developed to: (i) strengthen skin integrity in some patients with EBS (the most common form of EB), and (ii) to treat symptoms of the disease in all patients with EB.
We have conducted several preclinical studies to identify potential drug development pathways for a product in EB. The following data has been generated in support of these cannabinoids as a potential therapy in EB:
(a) Enhancing skin integrity and skin regeneration:
A desirable treatment outcome for all subtypes of EB would be enhanced skin integrity to prevent new wounds from forming. For patients with EBS, an estimated half of them will have a mutation in K14. The goal of modifying keratin production is to target the upregulation of a potentially compensatory K15. Under normal conditions, K5 and K14 combine (dimerize) to form adhesion at the basal layer within the epidermis. In EBS, one or both of these keratins are damaged. Our investigational hypothesis is that K15 may be able to compensate by replacing K14 in this equation and combining with K5 to form the adhesive properties needed for normal skin structure.
CBN was studied in a panel of cannabinoids to determine its ability to regulate keratin expression. CBN induced upregulation of K15 in 2 of the 3 experiments. Concentrations of 0.1 µM and 1 µM produced similar effects (approximately 6 to 17-fold increase in K15 expression). The highest concentration of 10 µM did not increase the size of the effect (approximately 3 to 13-fold increase). Lack of a dose-response may mean a threshold was exceeded, above which no further effect can occur.
23
Relative K15 Expression in Human Keratinocytes (HaCaT), Post-Confluence (48 hours)
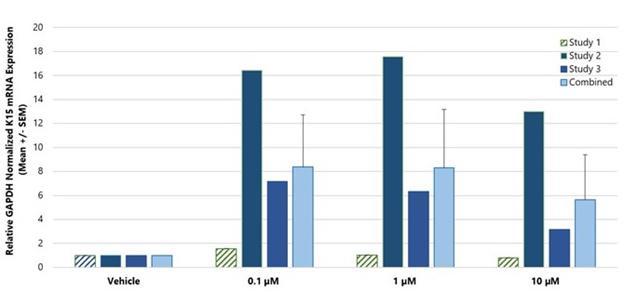
Study 1 did not exhibit an important effect. The reason for this is uncertain, with one hypothesis being that the cells tested had been through too many passages.
Despite the variation observed across these three studies, these results are encouraging as INM-755 cream may help create stronger skin by upregulating K15.
Hemidesmosome formation also occurs during normal differentiation of keratinocytes as they mature from the basal layer, not only in a wound-healing situation. Through the upregulation of K15, INM-755 cream applied to intact skin might gradually strengthen the skin and reduce the number of blisters and eventual wounds. For this effect, it could also be applied to wounds that have completed the initial re-epithelialization stage.
(b) Effects on Wound Healing
Cutaneous wound healing is a complex process with four main phases: inflammation, re-epithelialization, tissue formation, and tissue remodeling. In EB wounds, all four phases of cutaneous wound healing can be impacted, leading to chronic non-healing wounds. The wounds of EB patients are found primarily at or close to the junction of the epidermal and dermal layers. In these partial-thickness wounds, wound closure is achieved primarily by re-epithelialization rather than through granulation.
One major disease symptom in EB is the extensive wounds that can be generated throughout any day by simple friction on the skin, even as simple as clothes rubbing the skin. In addition to increasing the skin integrity via K15 up-regulation, another key goal would be facilitating accelerated wound healing via rapid skin regeneration and wound closure. E-Cadherin is major component of epithelium integrity. During wound healing, transforming growth factor beta, or TGF-ß, causes a reduction in E-Cadherin, allowing keratinocyte migration across the open wound. This is then followed by a return to normal levels of E-Cadherin to rebuild the integrity of the skin. CBN may play a role in the second phase of wound healing by accelerating the normalization of E-Cadherin expression. Additional studies are warranted to further explore this effect.
Inflammation is an important early step in wound healing and several of our studies demonstrated CBN has anti-inflammatory activity. Therefore, we conducted studies to evaluate the effect of CBN on the normal wound healing process. While an early in vitro assay indicated that high concentration of CBN could cause delays or prevent one of the first steps in wound healing, a subsequent study conducted with the INM-755 cream formulation did not hinder cell viability, cell migration, or wound closure. This was demonstrated in a wound-healing experiment conducted in 3-dimensional reconstructed human epidermis, or “RHE”, models with fully differentiated skin layers. Punch biopsy wounds were treated with INM-755 creams at three strengths, which included the intended cream concentrations for the first studies in healthy volunteers. No delay or inhibition of re-epithelialization was shown in CBN-treated models; the untreated control healed slightly slower in the first 5 days.
24
A composite of pictures showing 2D photographic images of the punch biopsy wounds as they heal over time. The re-epithelialization of the wound is shown by migration and growth of keratinocytes from the outside edge of the wound over time, migrating/growing to the center of the wound until the wound is closed:
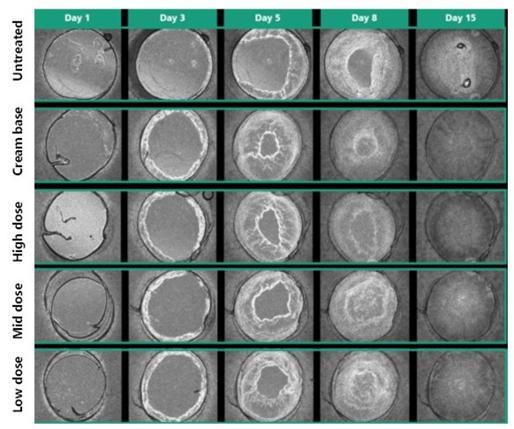
One more nonclinical study was conducted to explore the potential of CBN to interfere with early stage wound healing. In this study, superficial partial thickness wounds were introduced by a dermatome in an in vivo animal model and treated for 7 days with INM-755 creams at the same three strengths as used in the RHE models. Wound healing assessments included clinical observations, quantitative wound area measurements on photographic images and histopathologic examination. Treatment with INM-755 creams at the strengths intended for clinical development did not cause any delays in wound healing.
25

Lastly, to complete the investigation of effects on wound healing before applying INM-755 cream to open wounds in patients with EB, a clinical study (755-102-HV) was conducted in healthy human volunteers to test the safety of INM-755 cream. In this study in 8 subjects, a within-subject comparison was made for two concentrations of INM-755 cream, vehicle cream, and no treatment. INM-755 cream was well tolerated and did not impair normal wound healing in small epidermal (blister) wounds in this study.
(c) Reducing inflammation:
CBN was tested on two important markers of inflammation: IL-8 and MMP-9, because of their suspected links with blister formation in EBS and chronic cutaneous inflammation.
Interleukin-8, or IL-8, is the most potent chemoattractant for blood neutrophils and important mediator of angiogenesis, or the formation of new blood vessels. Chronic IL-8 production and neutrophil activation in a skin wound is an unfavorable element of skin pathology as it leads to extensive inflammation.
Matrix metalloproteinases, or “MMPs”, are part of the zinc-dependent endo-proteases family which modulate homeostasis of the extracellular matrix in skin. In response to skin damage and inflammation, metalloproteinases, including MMP-9, are often up-regulated. Specifically, exposure of keratinocytes, such as HaCaT cells, to TNF-α induces expression of the inflammatory-related factors such as IL-8 and MMP-9.
IL-8 and MMP-9 are upregulated in blisters of EBS patients and both are suspected to be contributing to blister formation. Both IL-8 and MMP-9 have been identified as targets for treatment of cutaneous inflammation in EBS. Therefore, reducing one or both might be helpful for controlling/reducing chronic skin inflammation in EBS.
While inflammation is an important first step in healing a new cutaneous wound, prolonged inflammation will interfere with the later stages of wound healing.
Persistent inflammatory activity, which may occur with infection or re-injury, often interferes with healing EB wounds.
26
Dose-Related Reduction in Relative IL-8 Expression in Human Keratinocytes (HaCaT)
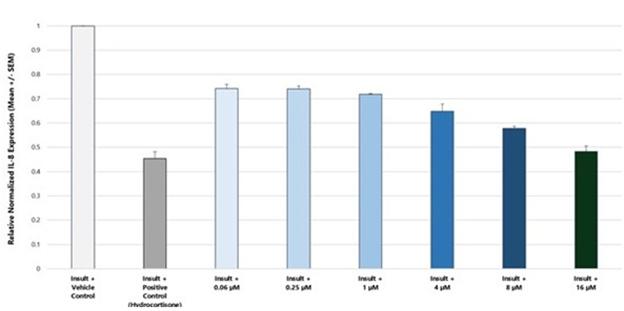
Insult = Tumor Necrosis Factor α (TNFα) and Interferon g (IFNg)
For IL-8: CBN produced a clear dose response with 35% reduction of IL-8 expression at 4 µM, 42% at 8 µM and 52% at 16 µM. Therefore, the IC50 was 16 µM. By comparison, hydrocortisone at 10 µM caused a 54% reduction in IL-8 expression. CBN was similar to hydrocortisone with respect to anti-inflammatory activity in this model.
Dose-Related Reduction in Relative MMP-9 Expression in Human Keratinocytes (HaCaT)
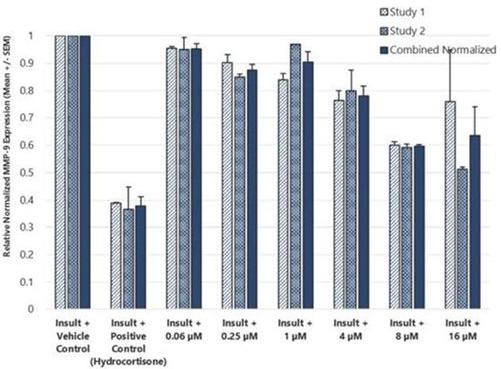
27
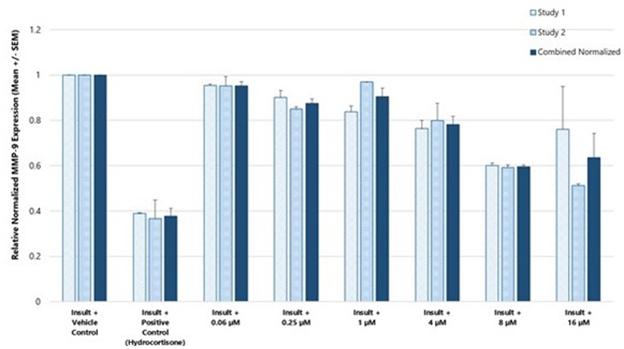
Insult = Tumor Necrosis Factor α (TNFα) and Interferon g (IFNg)
For MMP-9: Consistent results in both studies with a dose-related reduction of MMP-9 expression. The consistency in direction and magnitude of effect provides convincing evidence for down regulation of MMP-9 by CBN under insult conditions. The reduction was 22% at 4 µM and about 40% at both 8 and 16 µM. CBN showed a little less anti-inflammatory activity than hydrocortisone in this model, but still an important reduction.
(d) Pain reduction:
One pharmacodynamic endpoint that was studied was pain. Pain is one of the key symptoms in EB and requires significant effort to monitor and treat. CBN has demonstrated positive pain-relieving effects in NGF-induced in an in vivo pain model. To further demonstrate this, we utilized in vivo electrophysiology where CBN blocked the pain signals in the neurons.
In an in vivo of myofascial pain, nerve growth factor, or “NGF”, was injected into the masseter muscle, resulting in local mechanical sensitization lasting about 5 days. On Day 3, CBN was injected into the masseter muscle and the mechanical withdrawal threshold was assessed with a rigid von Frey hair. The mechanical force was gradually increased until the animal moved its head away from the stimulus.
28
Behavioral Effects of CBN in In vivo Model of Myofascial Pain

Adapted from Wong H, Cairns BE. Arch. Oral Biol. 2019;104:33-9.
CBN injected into the masseter muscle significantly reversed NGF induced mechanical sensitization at 10 minutes post-injection. (Behavioral study)
In parallel, electrophysiology recordings of single ganglion neurons that innervate the craniofacial muscles were performed (33 masticatory muscle mechanoreceptors). The electrophysiology effects parallel the behavioral effects. CBN significantly increased the relative mechanical threshold at 30- and 60-minutes post-injection. The results of this study have been published.
29
Electrophysiological Effects of CBN in In vivo Models of Myofascial Pain
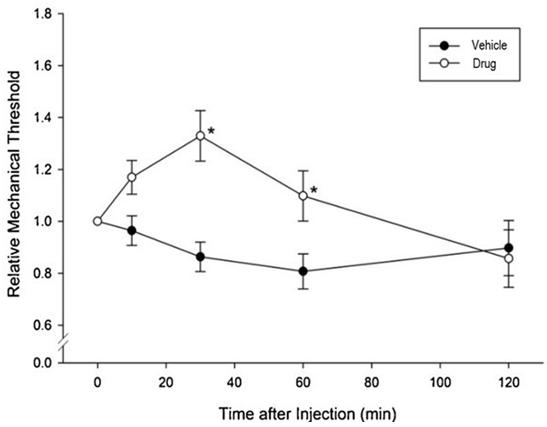
Adapted from Wong H, Cairns BE. Arch. Oral Biol. 2019;104:33-9.
(e) Antimicrobial activity:
In the literature, certain cannabinoid compounds have been shown to have potent antibacterial properties including against various strains of multidrug-resistant bacteria, including methicillin-resistant S. aureus, or “MRSA”. We have screened a number of cannabinoid compounds by standard methods against a broad range of Gram-positive and Gram-negative aerobic and anaerobic bacteria. Results of this third-party research demonstrated potent antimicrobial activity for all tested cannabinoid compounds, particularly against Gram-positive isolates. While these cannabinoids may provide some localized antibacterial benefit, it is unlikely that such effects would encourage cessation of broad-spectrum, systemic antibiotic usage.
(f) EBS formulation prototype development:
Careful attention must be paid to any topical product to be administered for the treatment of EB for several reasons. Our target product is designed to be applied over major portions of the body (if not the entire body), once each day. As such, the patients, who are often children, will be exposed to the active drug as well as the excipients of the skin cream, possibly for the duration of their lives. Accordingly, great care must be given that these components will be safe over the long-term and that they will not add to the already painful condition that the patients are suffering.
The excipients are safe for extensive body surface area exposure for a long duration of time
The API (cannabinoid) is dosed at the appropriate level – high enough to provide optimal clinical effect at the treatment site but low enough to minimize any systemic exposure; and
The final formulation can be administered daily with minimal friction to the skin.
30
We have utilized the Franz Cell diffusion method to assess skin penetration rates and depth for a proposed topical formulation for INM-755. The formulation is applied to skin samples and measurements are taken of how much drug penetrates to which depths in the skin. Using this method, a preliminary formulation of INM-755 achieved drug delivery to the epidermis and dermis layers as needed. Working with well-characterized excipients, we have tested several slight variations in formulation to achieve the desired concentration of drug in the skin while simultaneously avoiding high drug concentrations in systemic circulation (in the blood).
Starting in mid-2017 to present, we worked with several leading, international preclinical contract research organization to: (i) develop a final formulation used in INM-755; and (ii) initiate work of an Investigational New Drug Application, or “IND” enabling pharmacology and toxicology studies that are required before INM-755 could be used in future clinical studies.
Toxicology and Safety Pharmacology Studies of CBN
The investigational medicinal product, INM-755 (CBN) cream is for topical application on the skin. The cream base has a simple formulation with known pharmaceutical-grade excipients. It is a pluronic lecithin organogel. Pluronic lecithin organogels have been widely used by compounding pharmacists for topical preparations since the early 1990s. Therefore, the focus of the toxicology program has been to characterize effects of the active agent.
CBN is a new molecular entity, or “NME”, not yet approved for medical use in any country. Therefore, we are required to perform thorough safety testing prior to human administration. The intended route of administration for INM-755 is topical and is anticipated to result in low systemic exposure via the bloodstream. Despite only nominal risk of meaningful systemic exposure, regulatory authorities still require that we examine the consequences of systemic exposure on key biological functions and organ systems. For this purpose, the drug was administered by subcutaneous (SC) injection to achieve high in blood circulation. Topical safety studies using the intended route of administration and the clinical cream formulation were also conducted. These nonclinical toxicology studies included:
| ● | Tropical 28-day safety, in vivo; |
| ● | Systemic 28-day safety study, in vivo, with SC administration; |
| ● | Genotoxicity – standard battery of required tests for NMEs, including |
| o | In vitro bacterial mutagenicity study (classically the Ames assay) [Organisation for Economic Cooperation and Development test guideline 471 (OECD 471)], |
| o | In vitro micronucleus study in Chinese Hamster Ovary cells [OECD 487], and |
| ● | In vivo mammalian erythrocyte micronucleus study [OECD 474]; |
| ● | Phototoxicity – required because CBN has some absorbance in the UVB range; in vitro neutral red dye uptake study in cells from BALB/c 3T3 mice [OECD 432]; |
| ● | EpiOcular, in vitro eye irritation study [OECD 492]; |
| ● | Non-adjuvant Buehler method skin sensitization study, in vivo [OECD 406]; and |
| ● | In vivo drug distribution study with SC injection of radiolabeled drug. |
In the 28-day in vivo dermal toxicity study, INM-755 cream was given as topical daily doses applied to 10% body surface area. The quantity of cream applied resulted in a thick layer of cream, much more than a typical clinical application. After each daily cream application, the application sites were covered with a hypoallergenic, waterproof, breathable dressing for 24 hours and then scored for local tolerance. In this GLP study, systemic toxicity was also fully investigated by standard parameters. Based on clinical and histopathologic review, no CBN-related dermal toxicity was demonstrated in this study. Systemic exposure was minimal due to the topical route of administration and no systemic toxicities occurred either. The No Adverse Effect Level, or “NOAEL”, was determined to be the highest concentration of cream tested.
31
In the 28-day in vivo systemic toxicity study, CBN was given as daily SC injections up to the solubility-driven maximum feasible dose. No adverse drug-related effects were noted on clinical signs, clinical pathology parameters, ophthalmic evaluations, gross necropsy, organ weights, or histopathology. CBN was well tolerated at all doses, despite considerable systemic exposure. The NOAEL was determined to be the highest dose tested.
The standard battery of genotoxicity studies was conducted with CBN (2 in vitro and 1 in vivo) and all were negative. CBN did not cause phototoxicity in vitro. INM-755 cream at low and mid dose levels did not cause eye irritation in vitro. INM-755 cream at the highest tested dose did not cause a sensitization reaction in the in vivo sensitization model.
In summary, we have completed 20 safety pharmacology and toxicology studies to investigate the effects of CBN. We have also completed three Phase 1 safety and tolerability studies in healthy volunteers, two studies of which were conducted with varying concentrations of INM-755 cream and one study of which examined the non-CBN components of the cream base for INM-755.
Toxicity to Central Nervous System
Due to the well-documented intoxicating effects of THC, all cannabinoid compounds need to be tested for their psychoactive/intoxicating potential. In a standardized safety pharmacology study, we tested exceptionally high dose levels of CBN (more than 10,000 times the expected systemic exposure after topical dosing). No central nervous system adverse effects were observed even at the highest dose. 108 different central nervous system criteria were measured.

The toxicology and safety pharmacology data package covered a broad range of drug concentrations and was designed to support other clinical programs to treat topical skin conditions.
32
Summary of Completed and Contemplated Clinical Development Plans
A regulatory application to support our first Phase I clinical trial in healthy volunteers with INM-755 (755-101-HV) was submitted November 4, 2019 and approved December 6, 2019 in the Netherlands. The initial Phase I clinical trial evaluated the safety, tolerability, and pharmacokinetics of INM-755 cream in 22 healthy volunteers with normal, intact skin; the volunteers had cream applied once daily for a period of 14 days. All subjects in this first clinical trial completed treatment and evaluations by March 27, 2020. Database completion and data analyses were delayed by pandemic restrictions. Study results were reported November 25, 2020. A blinded interim safety review from the first 16 subjects in the Phase I study were included in a regulatory application that was approved April 17, 2020, for a second Phase I clinical trial of 8 healthy volunteers to test the local safety and tolerability of applying sterile INM-755 cream to small wounds once daily for 14 days. As with the initial Phase I trial, the second clinical trial (755-102-HV) was conducted with two different drug concentrations and a vehicle control. Enrollment began in early July 2020 and the clinical trial completed treatment and evaluations at the end of September 2020. Study results were reported January 8, 2021. The safety of INM-755 will continue to be assessed throughout its clinical development.
INM-755 cream was well tolerated in the two Phase I clinical trials in healthy volunteers and the next clinical trial will study INM-755 cream in patients with EB (Study 755-201-EB). Regulatory applications to support this global trial were filed for review by the National Competent Authorities and Ethics Committees in 8 countries for 13 clinical sites. Approvals were obtained in all countries (Austria, France, Germany, Israel, Italy, Serbia, and Spain) as of March 2022. Enrollment and patient treatment began in December 2021 and are expected to complete during the calendar year 2022. The goal of this Phase II study is to obtain safety and preliminary efficacy of INM-755 cream in treating symptoms and wound healing in patients with EB, using a within-patient design in which matched index areas are randomized to INM-755 cream or vehicle (no drug) cream in a blinded manner. Up to 20 patients will be enrolled and treatment will be for 28 days, the longest period supported by nonclinical toxicology studies.
We can make certain scope-estimates in terms of potential clinical trial sizes, timing and endpoints based on the recent clinical pathway followed by another phytochemical-based topical product for EB, ZorblisaTM (Amicus Therapeutics). The key finding from our review of publicly available information for the ZorblisaTM development program is that a clinical program is very focused for an orphan indication and the clinical trials do not include large numbers of patients. It would not be feasible to conduct large trials for such a rare disease. Therefore, the clinical studies need to be carefully designed and controlled to allow suitable assessment of the safety and efficacy of a new therapy in a small number of patients. Broad multicenter trials would be needed to recruit patients as quickly as possible. We will work closely with regulatory authorities and clinical experts in developing the clinical program for INM-755.
On average, it takes at least ten years to complete the development of an investigational drug from its initial discovery to the marketplace, with clinical trials alone taking six to seven years on average. It is not possible with any degree of certainty to estimate how long it will take to complete clinical trials and potentially obtain marketing approval for INM-755. To the extent that INM-755 may potentially be designated as either a Fast Track drug, a Breakthrough Therapy, or eligible for Priority/Accelerated Review, our timeline to any potential marketing approval may be shorter than might otherwise be the case.
Subject to COVID-related delays and other external factors, we plan to complete enrollment in Study 755-201-EB during calendar year 2022:
Key Milestones for the EB Program:
| ● | August 6, 2015 – We reported positive response from preclinical research on several cannabinoids (one of which was CBN), tested in various in vitro assays. By modulating the expression of various keratin genes that are responsible for cytoskeleton intermediate filaments and/or wound healing using different cannabinoids, we sought to alleviate the EBS symptoms. We believe that these preliminary results validated our approach as the cannabinoids displayed modulation of expression of various keratin genes. |
| ● | May 18, 2016 – We reported additional preclinical results demonstrating positive pain-relieving effects of cannabinoids in animal models. This animal data demonstrated a reduction in both acute and chronic pain (CBN was one of the cannabinoids tested in this study). |
| ● | May 4, 2017 – We filed an application with the Canadian Intellectual Property Office a PCT Application, Serial No. CA2017050546 titled, “A Cannabinoid-Based Topical Therapy for Diseases and Conditions Associated with Intermediate Filament Dysfunction”. |
33
| ● | March 13, 2019 – We announced that we will conduct all future development with a single cannabinoid skin cream, now designated INM-755. We determined that the clinical development path forward with its investigational drug candidate for the treatment of EB, previously referred to as INM-750, will be optimized by transitioning to an alternative formulation. INM-755 is formulated based on one of the two cannabinoids that comprised INM-750. We believe that pursuing a single-agent formulation, rather than a combination product, will ultimately improve the probability of development and regulatory success in this complex and rare disease. |
| ● | December 9, 2019 – We received clinical trial application approval for study 755-101-HV, a randomized, double-blind, vehicle-controlled Phase I study designed to evaluate the local and systemic safety, tolerability, and pharmacokinetics of INM-755 applied daily on intact skin in healthy volunteers. Two strengths of INM-755 cream, plus vehicle-only, will be evaluated in 22 adult subjects over a 14-day treatment period. |
| ● | January 20, 2020 – We revealed that the active ingredient in INM-755 and INM-088 is the rare cannabinoid, CBN. We are the first company to conduct human clinical trials with CBN. Extensive preclinical program to support the INM-755 program was exhibited at the EB2020 World Congress in London UK. |
| ● | April 30, 2020 – We announced clinical trial application approval in the Netherlands for Study 755-102-HV, a randomized, double-blind, vehicle-controlled Phase I study designed to evaluate the safety and tolerability of INM-755 (two strengths) applied daily for 14 days on epidermal wounds in 8 healthy volunteers. |
| ● | November 25, 2020 – We announced the top-line results of Study 755-101-HV (“Study 101”). Study 101 was a randomized, vehicle-controlled, double-blind, Phase 1 trial, that examined the safety and tolerability of two strengths of INM-755 cream on intact skin in 22 healthy adult volunteers over a 14-day treatment period. The Study 101 results indicate that INM-755 was safe and well-tolerated on intact skin, caused no systemic or serious adverse effects. In addition, there were no subject withdrawals due to adverse events. Drug concentrations in the blood were very low, as expected. |
| ● | January 8, 2021 – We announced the top-line results of Study 755-102-HV (“Study 102”). Study 102 was a randomized, double-blind, vehicle controlled, single-center study, in 8 healthy adult volunteers to test the tolerability of 14 days of application of the INM-755 cream on epidermal wounds under treatment procedures designed to simulate wound care for Epidermolysis Bullosa (“EB”) patients with open wounds. Results of Study 102 indicate that INM-755 cream was safe and well-tolerated on induced open epidermal wounds, caused no systemic or serious adverse effects. In addition, there were no subject withdrawals due to adverse events. These data from Study 101 and Study 102 support moving forward into clinical trials in patients with EB. |
| ● | April 28, 2021 – We announced that we filed Clinical Trial Applications (“CTAs”) in Austria, Israel and Serbia as part of a Phase 2 clinical trial of INM-755 (cannabinol) cream in Epidermolysis Bullosa (“EB”). Additional CTAs for 755-201-EB (the ‘201 study) will be submitted to National Competent Authorities (“NCAs”) and Ethics Committees (“ECs”) in France, Germany, Greece, and Italy in the coming weeks. Responses from the NCAs and ECs are expected throughout July and August 2021; timing will vary slightly by country due to differences in local procedures. |
| ● | On September 30, 2021 - We announced commencement of a Phase II clinical trial, the 755-201-EB study, of INM-755 (cannabinol) cream in the treatment of EB, marking the first time cannabinol has advanced to a Phase II clinical trial to be studied as a therapeutic option to treat a disease. The 755-201-EB study is designed to enroll up to 20 patients. InMed will evaluate the safety of INM-755 (cannabinol) cream and its preliminary efficacy in treating symptoms and wound healing over a 28-day treatment period. All four subtypes of inherited EB; EB Simplex, Dystrophic EB, Junctional EB, and Kindler Syndrome are eligible for this study. |
34
Additional Indications for INM-755
Once a company has gone to the significant investments of bringing a new chemical entity into human clinical trials, the traditional approach is to investigate as many therapeutic uses of that product in different indications, or specific diseases. We intend to pursue this strategy as a way to leverage our knowledge of CBN and investment in the development of INM-755 as a topical skin cream. Under the assumption that we would use the same formulation for other dermatological indications, there should be no need for further Phase I safety studies allowing us to proceed directly to Phase II safety and preliminary efficacy studies in humans, since the toxicology and initial human safety studies have been completed; however, the adequacy of the nonclinical and human safety data to support new dermatologic indications will be determined by the appropriate health authority. We intend to engage with dermatologists to discuss which diseases might best benefit from INM-755, outside of EB.
INM-088 for the Treatment of Glaucoma
Introduction
Glaucoma is a chronic optic neuropathy that is typically characterized by high intraocular pressure. The cause of glaucoma is understood to be inadequate or obstructed drainage of the fluid in the eye, or “aqueous humor”, through a drainage membrane called the trabecular meshwork, or “TM”, increasing the fluid pressure within the front part of the eye, or “anterior chamber”, and subsequently leading to pressure at the back part of the eye, or “posterior chamber”. The increased intraocular pressure exacts a toll on the nerve cells, called neurons, located at the back of the eye in the retina, thinning the mesh-like tissue in this region and resulting in damage to the neurons and specifically to the optic nerve, which provides the impulses of sight to the brain. This damage leads to blindness. Glaucoma is currently the second leading cause of blindness world-wide and is estimated to affect a population of about 76 million worldwide.

Current glaucoma therapies generally act to lower intraocular pressure either by reducing the aqueous humor production by the cells around the eye, or the “ciliary epithelial cells”, or by increasing fluid drainage through the TM. Nevertheless, we believe that there is considerable room for improvement of existing drugs, most of which are formulated as eye drops, in terms of increasing the amount of drug that can be safely delivered to increase its effect, improving the delivery of the drug into the eye, and reducing the common effect in currently used therapies that, over time, their efficacy diminishes as the body becomes tolerant to these classes of drugs. Studies have shown that when drugs are delivered as eye drops, less than 5% of the dose penetrates into the eye, indicating that 95% of the administered drug never reaches its desired target as it is wiped away upon blinking. Thus, there is much room for improvement on the drug delivery as a means of increasing clinical efficacy.
CBN is the key API in our second drug candidate, INM-088, which is in preclinical studies as a potential treatment for glaucoma. We conducted studies to test the ability of CBN to provide protection to the neurons at the back of the eye, referred to as “neuroprotection”, and reduce the intraocular pressure in the eye. We compared several cannabinoids, including CBD and THC, to determine which cannabinoid was the best drug candidate for the treatment of glaucoma. Of all of the cannabinoids examined, CBN demonstrated the most optimal effect of neuroprotection. Furthermore, CBN also exhibited intraocular pressure reduction capability.
35
Science behind Glaucoma
Glaucoma is a group of eye diseases which results in degeneration of neurons, damage to the optic nerve and vision loss. The most common type is open-angle glaucoma, or “OAG”, with less common types including closed-angle glaucoma, or “CAG”, and normal-tension (i.e., no increase in intraocular pressure) glaucoma. OAG develops slowly over time and the patients normally don’t experience pain. If left untreated, side vision may begin to decrease followed by central vision, resulting in blindness. CAG can present gradually or suddenly. The sudden presentation may involve severe eye pain, blurred vision, mid-dilated pupil, redness of the eye and nausea. Vision loss from glaucoma, once it has occurred, is permanent.
Risk factors for glaucoma include increased pressure in the eye, the thinness of the cornea, a family history of the condition, age over 40 years in African Americans, and age over 60 years for other ethnic groups (especially Mexican Americans). High intraocular pressure (those with a value of greater than 21 mmHg or 2.8 kPa) is often associated with a greater risk of glaucoma. However, some people may have high eye pressure for years and never develop damage. Conversely, neurodegeneration and optic nerve damage may occur with normal pressure, known as normal-tension glaucoma. The mechanism of OAG is believed to be slow exit of aqueous humor through the trabecular meshwork while in CAG the iris blocks the TM. Diagnosis is typically made by a dilated eye examination.
If treated early, it is possible to slow or stop the progression of the disease with medication, laser treatment, or surgery. Currently, the goal of these treatments is to decrease eye pressure. A number of different classes of glaucoma medication are available. Laser treatments may be effective in both OAG and CAG. Several of types of glaucoma surgeries may be used in people who do not respond sufficiently to other measures. Treatment of CAG is a medical emergency.
Epidemiology
The global prevalence of glaucoma for population aged 40–80 years is 3.54%, of which 75% is OAG. As of 2010, there were 44.7 million people in the world with OAG of which 2.8 million were in the United States. By 2020, the prevalence is projected to increase to 80 million worldwide and 3.4 million the United States. It occurs more commonly among older people. CAG is more common in women. Both internationally and in the United States, glaucoma is the second-leading cause of blindness.
Current Treatments in Glaucoma
Current treatments for glaucoma include medication, laser treatment and surgery. The goals of glaucoma management are to avoid glaucomatous damage, nerve damage and preserve visual field and total quality of life for patients, with minimal side effects. This requires appropriate diagnostic techniques and follow-up examinations, and judicious selection of treatments for the individual patient. Although intraocular pressure is only one of the major risk factors for glaucoma, lowering it via various pharmaceuticals and/or surgical techniques is currently the mainstay of glaucoma treatment.
Treatment Considerations based on Glaucoma Severity
Treatment considerations for glaucoma span the therapeutic spectrum from drug intervention to surgery.
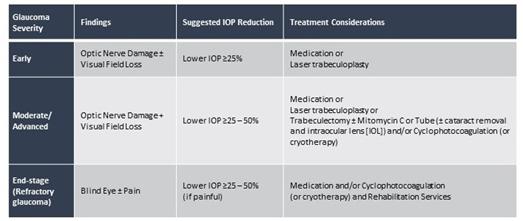
36
Pharmaceutical drug intervention has the greatest potential to generate direct competition for INM-088.
Medicines for Glaucoma Treatment (Intraocular Pressure-Lowering Drugs)
Current prescription eyedrop medications targeting intraocular pressure reduction include:
| ● | Prostaglandins and prostaglandin analogs (“PGA”) such as latanoprost, bimatoprost and travoprost to increase the outflow of fluid from the eye and, thereby, reduce ocular pressure. The adverse effects of PGAs include conjunctival hyperemia, or eye redness, irreversible change in iris color, discoloration of the skin around the eyes, and droopiness of eyelids caused by the loss of orbital fat; |
| ● | Beta blockers, most commonly prescribed as drugs to treat hypertension, are also prescribed for glaucoma. With their MOA designed to inhibit aqueous production, they are one of the oldest approved drug classes for the reduction of IOP. The most commonly used drug in this class is timolol. Beta blockers are less effective than PGAs in terms of IOP reduction and are typically used twice daily. Beta blockers are the most commonly used non-PGA drug. They are used as an initially prescribed monotherapy and as an adjunctive therapy to PGAs when the efficacy of PGAs is insufficient. Beta blocker eye drops have contraindications in their label as a result of potential systemic exposures from the topical application of the eye drops, potentially leading to cardio-pulmonary events such as bronchospasm, arrhythmia and heart failure. Other possible side effects include wheezing or difficulty breathing, slowed heart rate, lower blood pressure, impotence and fatigue; |
| ● | Alpha agonists, with their MOA designed to inhibit aqueous production plus their effect on uveoscleral outflow, are less effective than PGAs and need to be dosed three times daily in order to obtain the desired IOP reduction. In clinical studies, the most frequently reported adverse reactions that occurred in individuals receiving brimonidine ophthalmic solution, a commonly prescribed alpha agonist, included allergic conjunctivitis, conjunctival hyperemia, eye pruritus, burning sensation, conjunctival folliculosis, hypertension, ocular allergic reaction, oral dryness and visual disturbance. |
| ● | Alpha-adrenergic agonists such as apraclonidine and brimonidine, both reduce the production of aqueous humor and increase the outflow of fluid from the eye. Side effects may include dry mouth, red eyes or eyelids, fatigue, low or high blood pressure, blurred vision and light sensitivity; and |
| ● | Carbonic anhydrase inhibitors such as dorzolamide and brinzolamide reduce the production of fluid in the eye, but they are associated with blurred vision, bitter metallic taste in the mouth, dry eyes, red/irritated eyes, headache, and upset stomach. |
Despite their modest efficacy, safety and tolerability profiles, the requirement for two to three doses per day, and the fact that they do not target the diseased tissue in glaucoma, beta blocker, carbonic anhydrase inhibitor and alpha agonist products account for a significant portion of the total prescription volume for the treatment of glaucoma based on historical prescription patterns, with beta blocker timolol being the most widely prescribed non-PGA drug. This is driven by the PGA products not being sufficiently effective as monotherapy for up to half of all glaucoma patients. Often patients need to take a combination of different drugs and multiple eye drops throughout the day. Fixed-dose combination glaucoma products are also currently marketed in the United States, including Cosopt®, the combination of a beta blocker with a carbonic anhydrase inhibitor, and Combigan®, the combination of a beta blocker with an alpha agonist. There are no fixed-dose combinations of PGAs with other glaucoma drugs currently available in the United States.
Given side effect profiles, many patients do not take their medications properly. Surgery and laser therapies are intended to physically improve the drainage of fluid from the eyes and lowering of the intraocular pressure. Patients with OAG can have clogged channels in the TM opened with laser therapy, filtering surgery (trabeculectomy) or electrocautery. In other cases, small drainage tubes may be implanted in the eye. Possible complications include pain, redness, infection, inflammation, bleeding, abnormally high or low eye pressure and loss of vision. Some types of eye surgery may accelerate the development of cataracts. Additional procedures may be needed if eye pressure continues to increase.
37
Emerging Competition for INM-088 in Glaucoma
Due to the large medical need and potentially significant commercial opportunity, the competitive landscape of glaucoma is intense. As such, there are currently over 10 medications approved by the FDA for the treatment of glaucoma, which are summarized in the table below, according to drug class. In addition to the currently approved medications, there are a multitude of other therapies being evaluated in clinical trials, and many others at the preclinical stage. Finally, it should be noted that there are several laser surgeries, and other forms of surgical procedures that are currently being performed to treat glaucoma, which also serve as a source of competition to the therapeutic alternatives.
In December 2017, the FDA approved RHOPRESSA® as the first in a new class of glaucoma treatments known as Rho Kinase inhibitors. RHOPRESSA® is indicated for the reduction of elevated intraocular pressure in patients with open-angle glaucoma or ocular hypertension.
New eye drops for the treatment of glaucoma continue to be developed by our competitors. The following table, which may not be exhaustive, outlines publicly disclosed development programs for the treatment of glaucoma:
| New Mechanisms Of Action (MOA) | ||||
| Brand | MOA / Dosing | Status | ||
| Rhopressa® | ROCK inhibitor (qd) | US: Marketed; launched in Apr 2018 Europe: Centralized MA granted in Nov 2019 Japan: Phase 3 | ||
| Rocklatan® | ROCK inhibitor + PGA (qd) | US: Marketed; launched in May 2019 Europe: Centralized MA granted in Jan 2021 | ||
| New PGAs | ||||
| Brand | MOA / Dosing | Status | ||
| Vyzulta® (Bausch) | NO donating latanoprost (qd) | US: Marketed | ||
| XelprosTM (Sun) | Latanoprost, without BAK (qd) | US: Marketed | ||
| DE-117 (Santen) | EP2 agonist (qd) | US: Phase 3 Japan: launched in Nov 2018 | ||
| DE-126 (Santen) | FP/EP3 agonist (qd) | US and Japan: Phase 2b | ||
| NCX-470 (Nicox) | NO donating bimatoprost (qd) | US: Phase 3 | ||
qd – once-per-day dosing (latin: quaque die)
| ||||
| INM-088 is envisioned as a once- or twice-a-day eye drop medication to compete with treatment modalities in the medicines category if approved for commercialization. | ||||
In addition to INM-088, we are aware of only one other pharmaceutical-grade cannabinoid-based therapy being evaluated for the treatment of glaucoma. Specifically, Skye Biosciences Inc. (“Skye”, formerly Emerald Biosciences) is developing NB1111 (THC-Val-HS) for the treatment of glaucoma. NB1111 is a THC prodrug, which has demonstrated intraocular pressure-lowering efficacy in preclinical models. On August 10, 2021 Skye successfully completed genotoxicity studies of THCVHS required prior to beginning its planned Phase I clinical study in Australia.
Investigational Glaucoma Treatments
Despite the treatments available for lowering the intraocular pressure, there are some individuals for whom these treatments are either not tolerated due to side effects or in whom the intraocular pressure is not sufficiently lowered. In these situations, both glaucoma patient and physician look for alternative therapies.
While some experimental glaucoma medications explore new ways of controlling intraocular pressure, other treatments are directed at protecting the optic nerve (neuroprotection) to prevent eye damage, potential vision loss or even blindness. Many ongoing clinical studies are trying to find neuroprotective agents that might benefit the optic nerve and certain retinal cells in glaucoma.
38
Some investigational treatments are undergoing FDA clinical trials to prove safety and effectiveness. Other potential glaucoma treatments are strictly in experimental stages and may be years away from the possibility of being available on the marketplace.
Cannabis (THC) to treat Glaucoma
Decades of anecdotal evidence suggests that the use of Cannabis may play a role in lowering intraocular pressure in glaucoma. However, no such products have been formally investigated in clinical trials and none is currently approved for the treatment of this disease. The neuroprotective role of cannabinoids has not heretofore been utilized as a therapeutic strategy in glaucoma, primarily due to great difficulties associated with the targeted delivery of cannabinoids to intraocular tissues. This class of compound is also relatively poorly bioavailable due to its low aqueous solubility.
Previously reported attempts for topical delivery of cannabinoids, in particular, the intoxicating drug THC, to the ocular tissues used formulations based on mineral oil. Until very recently, studies on novel topical ophthalmic formulations of cannabinoids have been largely non-existent. Nevertheless, the use of marijuana to treat glaucoma has extensive anecdotal evidence and some supporting clinical data. It has been definitively demonstrated and widely appreciated, that smoking marijuana lowers intraocular pressure in both normal individuals and in those with glaucoma. Certain drawbacks are associated with the use of (smoked) marijuana to treat glaucoma:
| ● | Marijuana has a short duration of action (only 3-4 hours), meaning that to lower the intraocular pressure around the clock it would have to be smoked every three hours; |
| ● | Marijuana’s mood-altering and intoxicating effects, almost exclusively via the chemical THC, would prevent the patient who is using it from driving, operating heavy machinery, and functioning at maximum mental capacity; and |
| ● | Marijuana cigarettes also contain hundreds of compounds that damage the lungs, and the deleterious effect of chronic, frequent use of marijuana upon the brain is well established. |
Other means of administering THC include oral, sublingual, and eye drop instillation. The first two modalities avoid the deleterious effect of marijuana smoke on the lungs but are limited by the other systemic side effects. Other side effects associated with systemic use of THC for glaucoma include: impaired lung function, psychosis, anxiety dependence, tolerance, acute cardiac events and central nervous system-related adverse effects. In one study in which doctors offered some of their patients with worsening glaucoma the option of pills containing THC and/or smoking marijuana, all of them experienced side effects and 4 of 9 patients had discontinued use by either or both methods within 9 months due to side effects. Given that glaucoma is a lifelong disease, commonly requiring treatment for decades, these results strongly suggest that systemic use of THC is not a reasonable treatment option for such patients. The use of eye drops containing THC, or related compounds, has been investigated, but it has not yet been possible to formulate an eye drop that is able to introduce the drug into the eye in sufficient concentrations due to the low poor water solubility of the active ingredients.
Although marijuana may lower the intraocular pressure temporarily, that intraocular pressure-lowering effect is only one consideration in slowing the optic nerve damage of glaucoma. For instance, there is a growing body of evidence that inadequate blood supply to the optic nerve may contribute to glaucoma-related damage. Since marijuana given systemically is known to lower blood pressure, it is possible that such an effect could be damaging to the optic nerve in glaucoma, possibly reducing or eliminating whatever beneficial effect that would be conferred by lowering intraocular pressure. For this reason, marijuana, or its components administered systemically, cannot be recommended without a long-term trial which evaluates the health of the optic nerve.
39
An exciting finding is the discovery of receptors for cannabinoids in the tissues of the eye itself, suggesting that local administration has the possibility of being effective. Furthermore, there is evidence from research in the brain that there may be properties of the cannabinoids that protect nerve cells like those in the optic nerve. This raises the hope that cannabinoids could protect the optic nerve not only through intraocular pressure-lowering but also through a neuroprotective mechanism. However, unless a well-tolerated formulation of a marijuana-related compound with a much longer duration of action is demonstrated in rigorous clinical testing to reduce optic nerve damage and preserve vision, there is no scientific basis for use of these agents in the treatment of glaucoma.
The wide variety of topically effective anti-glaucoma drugs that are available today, and a few others in the developmental stage, represent significant advancement in ocular therapeutics. While these topical ophthalmic preparations have reduced the risk of systemic toxicity to some extent, their long-term use causes systemic as well as ocular toxicity. Many ophthalmologists generally select the drugs individually and replace them regularly in order to prevent the habituation phenomenon (reduction in effect of the drug over time due to tolerance) and negative side effects.
Drug Discovery Process
To date, we have utilized several preclinical investigations to:
| ● | Compile a list of genes that are associated with development of glaucoma disease from our own in-house curated disease analysis. We grouped these selected genes based on the glaucoma disease hallmarks such as trabecular meshwork remodeling, retinal ganglion cell survival and genes involved in extracellular matrix; and |
| ● | Better understand the relationship among selected glaucoma disease genes, we constructed a protein-protein interaction network and the graphic view of the interaction network was built for further discovery. |
Glaucoma is a neurodegenerative disease in which various triggers (such as elevated intraocular pressure) induce cascades of events, which ultimately lead to apoptotic retinal ganglion cell death and result in irreversible loss of vision. However, as mentioned above, the goal of all current glaucoma therapies is to reduce intraocular pressure without including any strategies of neuroprotective treatment. In fact, some patients often fail to show much improvement even after intraocular pressure reduction, whereas others develop glaucoma in the absence of elevated intraocular pressure.
Key Preclinical Results for CBN as a Drug Candidate to Treat Glaucoma
INM-088 is an eye-drop CBN formulation being developed for the treatment of glaucoma. The preclinical development program for INM-088 has included a number of studies comparing a number of cannabinoids, including CBN, THC and CBD, among others, to determine which cannabinoid holds the greatest potential to treat glaucoma. This preclinical research to date is comprised of both in vitro and in vivo studies and led to the selection of CBN as the lead drug candidate for further development.
The scope of the in vitro studies to date include the following:
| 1) | Evaluation of the neuroprotective effects of selected cannabinoids on the differentiated retinal ganglion cells, or “RGCs”, a thin layer of neurons responsible for relaying visual signals in the eye, under normal atmosphere pressure and elevated pressure conditions. |
Notably, exposure of RGCs to increasing concentrations of several cannabinoids, including THC and CBD resulted in dose dependent cytotoxicity, or cell death, over time. Importantly, CBN-exposed RGCs demonstrated the lowest level of toxicity among the cannabinoids used in these experiments.
40
Cytotoxicity and neuroprotective effects of selected cannabinoids
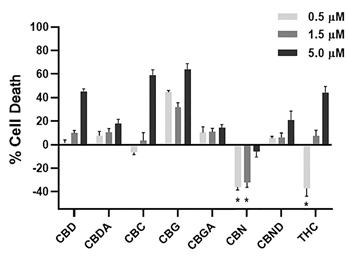
661W cells were treated with selected cannabinoids (0.5, 1.5 and 5 µM) at normal atmospheric pressure for 72 hrs. Data normalized to vehicle under normal pressure (NP-VC) was considered as 0% cell death. Note, CBN (0.5 - 5 µM) treatment under normal pressure did not result in cytotoxicity. Other cannabinoids, including CBD, CBDA, CBC, CBG, CBGA, and CBND, displayed significant toxicity. Enhanced cell proliferation in the presence of a low concentration of ∆9-THC (0.5 µM) was observed in contrast to increased cell death at higher concentrations (1.5 and 5 µM). Experiments were performed in triplicates, two independent times. The data is presented as mean ± SEM. *p < 0.05 against NP-VC (one-way ANOVA).
In addition, exposure of the RGCs to elevated pressure in a cell-based model for glaucoma (without exposure to cannabinoids) for 72 hours resulted in high level of cytotoxicity, whereas exposure of these cells to both an elevated pressure (20-40 mmHg) plus CBN, within the same time-period, resulted in cell survival in a dose dependent fashion. A neuroprotective effect of CBN was also observed under elevated pressure conditions in the pressurized chamber that is designed to mimic the clinical situation of increased intraocular pressure in glaucoma. CBN performed better than both CBD and THC in this preclinical model under identical testing conditions.
Comparison of CBN versus THC-mediated inhibition of elevated pressure-induced cell death
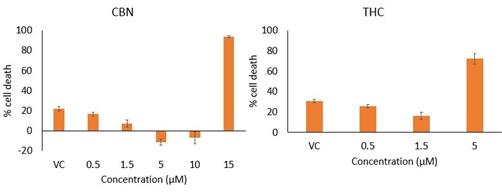
Significant neuroprotective effect of CBN on differentiated 661W cells when cultured for 72 hours under a hydrostatic pressure of approximately 20 to 25 mmHg as compared to ∆9-THC. Cell were treated with CBN at 0.5, 1.5, 5, 10 and 15 µM and ∆9-THC at 0.5, 1.5 and 5 µM respectively. Vehicle Control (VC) contained 0.15% ethanol. Data presented as Cell Death (%) vs normal pressure Vehicle Control (taken as 0% cell death). The data is presented as mean ± SEM.
41
CBN mediated inhibition of elevated pressure-induced cell death
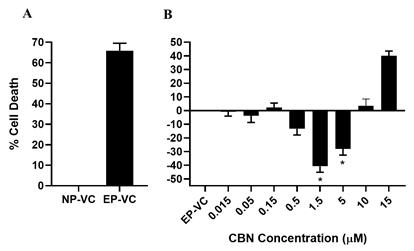
CBN increases cell survival under elevated pressure. 661W cells were treated with CBN in a dose-dependent manner at increased atmospheric pressure (40 mmHg) for 72 hours. A) Significant cytotoxicity was observed in untreated cells under elevated pressure in the pressure chamber (EP-VC) compared to normal pressure vehicle control (NP-VC). Data normalized to normal pressure (NP-VC) was considered as 0% cell death. B) CBN treatment under elevated pressure protected 661W cells from cell death at 0.5, 1.5 and 5 µM. Data normalized to elevated pressure vehicle control (EP-VC) that was considered as 0% cell death. Protective effect was significantly different from pressure chamber vehicle control (EP-VC) at 1.5 and 5 µM (p<0.0001 and p<0.001). CBN treatment of 661W cells at 15 µM under elevated pressure results in cell death. Data presented as mean ± SEM of duplicate independent runs (n=6). Data analyzed statistically by one-way ANOVA.
| 2) | Evaluation of anti-apoptotic effects of CBN on the differentiated RGCs when exposed to elevated pressure conditions. |
Using the same in vitro model described above, we also looked at a specific, natural self-destruction process called programed cell death, or apoptosis. We verified that CBN has an anti-apoptotic effect on differentiated RGCs when subjected to elevated hydrostatic pressure. Exposure of these cells to high-pressure levels in the pressure chamber apparatus, without exposure to cannabinoids, for 6 hours resulted in an induction of apoptosis ranging from 30-60% (n=3). Exposure of these cells under the same conditions concurrently with CBN prevented apoptosis and resulted in a higher level of cell survival.
42
CBN mediated attenuation of elevated pressure-induced apoptosis

CBN mediated attenuation of elevated pressure-induced apoptosis. 661W cells were treated in the presence of the indicated concentrations of CBN at increased atmospheric pressure (~20–25 mm Hg) for 6 h. Significant apoptosis was observed under elevated pressure in the pressure chamber (EP-VC) when compared to NP-VC. CBN at higher concentrations (0.5, 1.5 and 5 μM) but not at lower concentrations (0.015–0.15 μM) significantly inhibited pressure-induced apoptosis of 661W cells. Experiments were performed in triplicates, two independent times. The data is presented as mean ± SEM. #p < 0.05 against NP-VC analyzed by using an unpaired t-test; *p < 0.05 against EP-VC analyzed statistically by one-way ANOVA.
| 3) | Evaluation of CBN impact on the expression of specific extracellular matrix (ECM) markers on primary human trabecular meshwork (TM) cells under basal condition and following stress-induction with Transforming Growth Factor Beta 2 (TGF-ß2), a cytokine used to alter extracellular matrix metabolism. |
A key risk factor for the development and progression of glaucoma is elevated IOP, the result of increased resistance to aqueous humor outflow through the TM. Increased outflow resistance has been strongly correlated with aberrantly elevated levels of TGF-ß2, a cytokine used to alter extracellular matrix metabolism of the TM of glaucoma patients compared to healthy individual. Therefore, evaluation of CBN effects on the TM observed under elevated TGF-ß2conditions mimics the clinical presentation of IOP in glaucoma and is relevant in the clinical context of the disease. Using human primary TM cells derived from various donors and propagated in vitro at different cell passages, we were able to demonstrate that several extra-cellular matrix proteins, or “ECM” markers, were upregulated by TGF-ß2 induced condition. Furthermore, CBN treated TM cells under basal condition or TGF-ß2 induced conditions for a duration of 72 hours resulted in reduction in the expression of several of these ECM protein markers.
43
CBN attenuated TGF-β2 induced changes on TM markers Fibronectin (FN)
and Collagen 1A (COL1A) in hTM cells
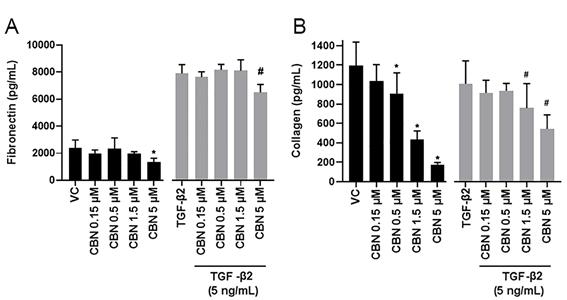
CBN attenuated TGF-β2 induced changes in TM markers Fibronectin (FN) and Collagen 1A (COL1A) in hTM cells. hTM cells were treated with CBN (0.15, 0.5, 1.5 and 5 μM) for 72 h with or without TGF-β2 (5 ng/mL). Post-treatment, cell lysates were collected, total protein was quantified by Bradford assay, and ELISA was performed according to the manufacturer’s instructions. (A) TGF-β2 induced significant upregulation of FN expression. Note, a significant reduction in basal or TGF-β2 induced FN levels was observed in the presence of CBN (5 μM). (B) TGF-β2 did not induce upregulation of COL1A level in hTM cells, whereas CBN in a dose dependent manner (0.5–5 μM) attenuated COL1A levels in the presence or absence of TGF-β2. Experiments were performed in triplicates, two independent times. The data is presented as mean ± SEM. Changes in the treatment groups were compared to the vehicle control (VC) group (*) or TGF-β2 induced group (#), and data were analyzed by using one-way ANOVA (p < 0.05).
Role of CBN in the inhibition of TGF-β2 induced changes in expression of α-SMA and pERK1/2

44
Figure 6: Role of CBN in the inhibition of TGF-β2 induced changes in expression of α-SMA and pERK1/2. Representative immunoblots showing CBN mediated changes in expression of α-SMA and phospho-ERK1/2 in the presence or absence of TGF-β2. TGF-β2 stimulated α-SMA expression and increased the level of ERK1/2 phosphorylation when compared to vehicle control. Note the inhibition of α-SMA expression and ERK1/2 phosphorylation in the presence of CBN in comparison to vehicle control and/or TGF-β2 treatment. GAPDH was used as the loading control. Histograms in the lower panel represent densitometry analysis of the Western blots (n=3–4). Changes in the treatment groups were compared to the vehicle control (VC) group (*) or TGF-β2 group (#), and data were analyzed statistically by unpaired t-test (p < 0.05).
| 4) | Evaluation of CBN pharmacokinetic profile in the eye and plasma of a preclinical model (rat) by direct intravitreal (IVT) injection into the eye. |
Our first in vivo study was designed to determine the pharmacokinetic profile of CBN in preclinical models, specifically measuring CBN levels in the eye and plasma following direct bilateral IVT injection. This means that individual injections were made directly into the vitreous humor (fluid of the central cavity of the eye). Following IVT delivery, CBN levels from the plasma (n=3 per time point) and the whole eye (n=6 per time point) were measured at several timepoints using a qualified LC-MS/MS method. CBN levels in the plasma samples were below the detection limit of the assay (0.05 ng/mL). Furthermore, CBN levels in the rat whole eye were shown to be slowly cleared from the eye with a projected half-life (t1⁄2) of approximately 33 hrs.
Percentage of CBN remaining in the whole eye following single IVT injection at 10 µM initial dose

Percentage of CBN remaining in the whole eye following single IVT injection at 10 μM initial dose. Each point represents the mean of left and right eye measurements. The concentration-over-time elimination profile of CBN from IVT injection in the whole eye indicates a slow clearance rate of this cannabinoid, with approximately 11% of the total CBN remaining at 72 h termination time-point (n = 6 eyes per time-point; values presented as mean±SEM).
45
Time dependent elimination of CBN from the whole eye following
single IVT injection at 10 μM initial dose
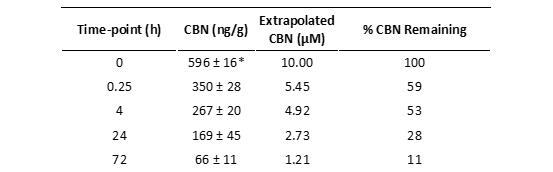
Concentration-over-time elimination profile of CBN from IVT injection in the whole eye indicates a slow clearance rate of this cannabinoid, with approximately 11% of the total CBN remaining at 72 hrs termination time-point. Rats received a single bilateral IVT injection (5 μL) of CBN (50 μM) to target a final concentration of 10 μM inside the eye. Values measured as ng/g ocular tissue or μM and presented as mean ± SEM. n = 6 eyes per time-point. *596 ng/g represents an extrapolated initial dose intended for delivery inside the eye calculated based on the actual whole eye tissue weight.
Pharmacokinetic parameters of CBN in the whole eye following
single IVT injection at 10 μM initial dose

CBN was quantified by a qualified LC-MS/MS method in the whole eye homogenates. The analysis of pharmacokinetic parameters was performed using Pharmacokinetic Solver 2.0 Software. To produce an output, a Non-Compartmental Analysis using the linear trapezoid rule after extravascular input was performed. CBN was cleared at a slow rate from the ocular tissue with CLivt = ~0.04 mL/h, favorable AUC0-inf ocular exposure at 250 μmol/mL * h, and relatively long ocular t1/2 =~33 h.
| 5) | Evaluation of CBN neuroprotective and IOP-lowering effects in a rat preclinical glaucoma model (Rats) by IVT injection. |
We conducted a preclinical efficacy study to evaluate neuroprotective and IOP lowering effects of CBN following IVT injection in a rat episcleral vein laser photocoagulation model of glaucoma. The laser photocoagulation of episcleral veins was done unilaterally on the oculus dexter (OD) eye of the animals on day 0 and day 7. The contralateral oculus sinister (OS) eye served as control and was not subjected to lasering. The rats were randomized into 4 treatment groups based on their baseline pERG amplitudes measured at day - 4: Group 1: vehicle control (0.5% DMSO-PBS); n=11. Group 2: CBN low dose at 5 µM final concentration inside the eye; n=13. Group 3: CBN high dose at 50 µM final concentration inside the eye; n=11. Group 4: brimonidine tartrate (Alphagan®, 0.5% ophthalmic eye drops, a registered trademark of Allergan PLC) a selective α2-adrenoceptor agonist clinical reference drug; n=14. Animals in Groups 1, 2 and 3 were administered either vehicle or CBN by IVT injection at 5 µl volume on day 0, 7 and 15. Animals in Group 4 were topically instilled with brimonidine twice daily at 5 µl volume per eye 4 days before laser photocoagulation and throughout the study duration. The study was terminated on day 21. High IOP was induced unilaterally by laser photocoagulation of episcleral veins (to approximately 19 mmHg). CBN was delivered by IVT injection after episcleral laser photocoagulation on three occasions on day 0, immediately after the lasering, and on days 7 and 15 post-lasering. IOP and pERG were monitored at specific time points throughout the study. Significant reduction in IOP (to approximately 13 mmHg) was observed for the CBN high dose treated group on Days 7 and 17 and significant improvement of pERG amplitudes for CBN low dose treated group was observed on Day 21 (-39.8% from baseline for vehicle control group, -23.1% from baseline for the active control (brimonidine tartrate) group, -16.6% from baseline for the CBN low dose group and -41.2% from baseline for the CBN high dose group). These were the measured outcomes that are useful in evaluating candidates for a potential glaucoma treatment. In summary, data from this study demonstrated a reduction of IOP and improvement of pERG function following IVT injection of CBN in the Rat episcleral vein laser photocoagulation model of glaucoma.
46
CBN mediated effect on IOP in the rat episcleral vein laser photocoagulation model of glaucoma

CBN mediated effect on IOP in the rat episcleral vein laser photocoagulation model of glaucoma. The IOP lowering effect of CBN was analyzed in the lasered eyes of different treatment groups versus the vehicle control group. At a low dose (5 μM), CBN treatment did not reduce IOP. At a high dose (50 μM), CBN significantly reduced IOP on days 7 and 17. On day 21, IOP values in all groups normalized to the baseline levels. Data analyses are presented for the time points with a statistically significant difference. Brimonidine (Alphagan®) was used as a reference control. Data were analyzed statistically by unpaired t-test and presented as mean ± SEM. *p < 0.05 against vehicle control (n =11–14 rats per treatment group).
47
CBN mediated changes in the pattern electroretinogram (pERG) in the rat episcleral vein laser
photocoagulation model of glaucoma
A.

B.
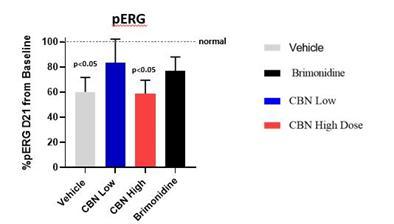
The pERG amplitudes (µV) decreased in all treatment groups after the IOP elevation induced by episcleral vein laser photocoagulation. (A) pERG baseline-corrected Δ-Amplitude values (μV) (mean ± SEM) of animals in each treatment group were recorded at baseline before lasering and on days 7, 14 and 21 post-lasering. (B) %pERG reduction from 100% normal baseline values at Day 21. Significant RGC functional impairment was observed in the vehicle group on day 21 and in the CBN high dose (50 µM) group on days 14 and 21 when compared to baseline. The pERG amplitudes in the low dose CBN (5 μM) and brimonidine (Alphagan®) groups did not differ significantly from the baseline on both follow-up days 14 and 21, indicating that CBN confers neuroprotective effect on RGCs. Data is presented as mean ± SEM of baseline-corrected values and analyzed statistically by two-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05 against the baseline values; n=11-14 rats per treatment group).
48
Ocular Formulation Development for INM-088
There are a wide variety of topically effective anti-glaucoma drugs that are available today and others in the developmental stage that represent significant advancements for ocular therapeutics. Ophthalmologists typically prescribe drugs individually and then switch to different classes of drugs on a regular basis in order to prevent the habituation phenomenon (reduction in effect of the drug over time) and negative side effects. There is an opportunity for new therapies with low systemic toxicity and those which may not exhibit habituation.
Until very recently, studies on novel topical ophthalmic formulations of cannabinoids have been largely non-existent. Designing an ideal delivery system for any ocular disease depends on the molecular properties of the drug substance and incorporating it into the formulation while taking into consideration parameters such as size, charge, and affinity towards various ocular tissues and pigments.
For all delivery technologies under examination as candidates for INM-088, key design criteria include, among others:
| ● | Biocompatibility and biodegradability of the formulation; |
| ● | Viscous fluid behavior while inside the container (to facilitate ease of manufacturing, handling and dosing); |
| ● | Characterized and defined drug release, absorption and subsequent carrier degradation; |
| ● | Optimized particle size and surface charge to avoid irritation upon application to the eye and to facilitate ocular penetration; and |
| ● | Stable final drug product to ensure drug product quality storage over time. |
One of the delivery technologies under development as a potential delivery vehicle for CBN in ocular disease is our proprietary, stimulus-responsive, nanoparticle-laden hydrogel vehicle for spatiotemporal and dosage-controlled release of cannabinoids into the aqueous humor of the eye. This hydrogel is envisioned to be packaged as a liquid and is intended for application as an eye drop. We investigated the compatibility and effectiveness of our hydrogel formulation with CBN as compared to other third-party ocular drug delivery technologies such as EyeCRO’s MiDROPs® microemulsion. We conducted an in vivo study that compared both the hydrogel and MiDROPs® formulated with CBN and showed that a similar level of CBN was measured in the retina and retinal pigmented epithelium tissues following topical administration of each formulation. In early December 2020, we selected a final delivery technology based on the extensive data collected from these assessments that included solubility, drug delivery localization and sustained effect. This selection resulted in a licensing agreement with EyeCRO LLC for its proprietary MiDROPs® technology. Through this agreement, InMed has secured an exclusive, global commercial rights for the utilization of MiDROPs® for all cannabinoids, cannabinoid analogs and their variants. One key benefit for our INM-088 program by working with EyeCRO is that their product development and testing with MiDROPs® is already well advanced, having been previously reviewed by the US FDA during a pre-IND meeting.
Distribution of INM-088 formulation in Ocular Tissue
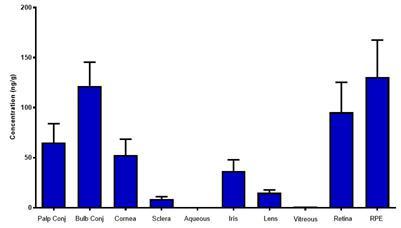
49
Ocular pharmacokinetic study of INM-088 following 5 days of bilateral ocular topical administration in New Zealand White Rabbits. INM-088 was formulated as 0.1% CBN-MiDROPS™ and dosed BID at 50 µl/dose for 5 days via bilateral topical ocular instillation in 3 naïve NZW rabbits. CBN level was measured 1 hour following the final dose administration on Day 5, Administration of INM-088 formulation resulted in measurable CBN concentrations in each of the ocular tissues analyzed in this study. The study was performed independently by EyeCRO LLC.
Next Steps for the INM-088 in Glaucoma Program:
Subject to COVID-related delays and other external factors, we plan to accomplish the following tasks for the INM-088 in Glaucoma program in calendar year 2022:
| ● | Process and analytical development and scale-up of INM-088 formulation, MiDROPs® with CBN, to enable pre-clinical and clinical supply; |
| ● | Conduct additional preclinical studies; and |
| ● | Initiate and complete IND/CTA-enabling toxicology studies. |
Key Milestones:
| ● | October 24, 2017 – We announced results from a study co-sponsored by us and University of British Columbia. We believe that this InMed-University of British Columbia study was the first ever to report hydrogel-mediated cannabinoid nanoparticle delivery into the eye, resulting in enhanced drug uptake via the cornea and lens. This study further evidences our capacity to conduct a wide spectrum of drug development activities, including: packaging the cannabinoid as a nanoparticle; formulation of a cannabinoid drug candidate into a novel, tissue specific delivery vehicle; and confirmation of drug delivery and diffusion into a target tissue. |
| ● | May 14, 2018 – We announced the filing of a PCT Application (PCT/CA2018/050548) for a cannabinoid-based topical therapy for glaucoma, which includes the protection of our technology in several countries, including the United States, and claims a priority date from May 8, 2017 (PCT/CA2018/050548). The PCT Application filing is a conversion from the provisional patent filed in May 2017. |
| ● | January 20, 2020 – We revealed that the active ingredient in INM-755 and INM-088 is the rare cannabinoid, CBN and that we are the first company to conduct human clinical trials with CBN. |
| ● | May 12, 2020 – We announced filing of a PCT application entitled “Compositions and Methods for Use of Cannabinoids for Neuroprotection”. This application was initially filed as a provisional patent application pertaining to the potential of cannabinoids in the prevention of neuron damage associated with glaucoma. |
| ● | December 3, 2020 – We announced the selection of the final formulation for INM-088, and we secured an exclusive, worldwide license from EyeCRO LLC for its Microemulsion Drug Ocular Penetration System (“MiDROPS®”) eyedrop delivery technology targeting effective, topical administration of cannabinoids to the eye. |
| ● | December 20, 2021 – We announced that a peer-reviewed scientific article entitled “Cannabinol Modulates Neuroprotection and Intraocular Pressure: A Potential Multi-Target Therapeutic Intervention for Glaucoma”, has been published in Biochimica et Biophysical Acta (BBA - Molecular Basis of Disease), a leading international journal focused on biochemistry and molecular genetics of disease processes and models of human disease in the area of aging, cancer, metabolic-, neurological-, and immunological-based diseases. The peer-reviewed article highlights research evaluating the use of cannabinol, or CBN, as a potential treatment option for glaucoma. Several studies were conducted to evaluate the survival of retinal ganglion cells, modulation of intraocular pressure and its effects on extracellular matrix proteins using in vitro and in vivo glaucoma models. |
50
Additional indications in ocular disease
Similar to the strategy being pursued with INM-755, we intend to fully investigate the potential for CBN in INM-088 to treat a wide array of ocular diseases, in particular, the potential for CBN to provide neuroprotection across several diseases where blindness is the ultimate outcome. We are currently pursuing preclinical models to more closely study this effect and will leverage the toxicology and Phase I safety studies across these new indications, if deemed applicable. In addition, we are actively working with our collaborators to test selected potential CBN-analogs, produced by BayMedica, to demonstrate neuroprotection in other ocular indications such as Age-Related Macular Degeneration (AMD).
Other Research and Development Programs
There is a need to find alternatives to treat chronic and severe pain that are non-addictive and have limited side effects. We have conducted limited preclinical investigations of the potential of non-THC cannabinoids to treat pain using a topical approach. In September 2018, we filed a PCT Application in the United States for INM-405 as cannabinoid-based topical therapies for the treatment of pain, which is an important step in protecting our intellectual and commercial property. The patent cites a range of cannabinoids, alone or in combination, applied topically to treat various types of pain—muscle, nerve, arthritis-induced joint pain, etc.
Key In Vivo Results for our Pain Program
Important data from our research program for pain medications were published in the European Journal of Pain (2017) and the Archives of Oral Biology (2019). Both publications specifically cited data on the use of THC and certain other cannabinoids, alone and in combination, at varying ratios, in a preclinical pain model. Findings from the published studies include:
| ● | Expression of cannabinoid receptors on masseter ganglion neurons. Both CB1 and CB2 receptor expression was observed in the trigeminal ganglion neurons that innervate the masseter muscle, as well as in the neuronal fibers in the muscle itself. This confirms that these peripheral nerves may be appropriate targets for a cannabinoid therapy; |
| ● | Effect of intramuscular injections of THC and certain other cannabinoids, alone and in combination, on nerve growth factor, or “NGF”, induced sensitization. NGF, if injected into a target tissue (muscle), makes the tissue more sensitive to pain, as can be measured by a mechanical threshold, or “MT”, scale. On this scale, a lower number represent a lower pain threshold, or a lower ability to tolerate a painful stimulus. NGF injection resulted in a lowering of the MT score. Applications of THC and certain other cannabinoids, either alone or in combination, were associated with an increase of MT, meaning a higher ability to tolerate pain. It should be noted that the NGF-induced reduction in MT model mimics the type of pain reported by sufferers of temporomandibular disorders, or “TMD”. Importantly, these cannabinoids only affected the muscle into which it was injected; there was no effect on surrounding tissue; and |
| ● | In a behavioral analysis in these studies, test subjects treated with peripheral application of THC, the leading intoxicating component in marijuana, and certain other cannabinoids did not exhibit any effect on motor function. This indicates that the dose of THC used did not achieve sufficient circulatory distribution to reach the brain where it may exhibit intoxicating effects. However, repeat applications of THC may still have potential to induce significant undesirable central effect. |
Our INM-405 research program is at an early-stage and its continued development is subject to available resources and/or our ability to find funding or strategic partners. Continued investment in our INM-405 research program is under review and we will make a determination as to its future development based on several strategic factors, including other research priorities, in due course.
Other areas of our early research focus have included Chronic Obstructive Pulmonary Disease, or “COPD”, neurodegenerative diseases such as Huntington’s Disease, and breast cancer. Recently, we have conducted a broad range of research and development activities to explore other uses of cannabinoids in treating human diseases with unmet medical needs. Our studies have demonstrated the potential for the use of a rare cannabinoid to improve neuronal function and provide neuroprotection for treating neurodegenerative disorders.
51
In October 2021, we filed a PCT Application patent application demonstrating neuroprotection and enhanced neuronal function using a rare cannabinoid for the potential treatment of neurodegenerative diseases such as Alzheimer’s Disease, Parkinson’s Disease, Huntington’s Disease and others. This PCT patent application advances our strategy directed towards researching and developing rare cannabinoids as potential pharmaceutical therapeutics for diseases with high unmet medical needs. Expanding our patent portfolio to include, in addition to CBN, an incremental rare cannabinoid for the potential treatment of major neurodegeneration indications.
These programs are at various early stages of development and, as non-core assets, their continued development is subject to available resources and/or our ability to find funding or strategic partners. Continued investment in each program is under review and we will make determinations as to which programs to continue based on several strategic factors. In addition, we may choose to partner some or all of these programs with external parties.
Acquisition of BayMedica Inc.
On October 13, 2021, we announced the closing of the transaction acquiring BayMedica Inc., renamed as BayMedica LLC, (“BayMedica”) a private company based in the USA specializing in the manufacture and commercialization of rare cannabinoids. We acquired 100% of BayMedica in exchange for 2.05 million our common shares issued to BayMedica’s equity and convertible debt holders, subject to a six-month contractual hold period, and a cash component of US$1 million. The entirety of this cash consideration is held in escrow, subject to cancellation, to satisfy certain potential post-closing indemnification and other claims that we may have under the definitive agreement in the six- and twelve-month periods following the closing.
With this transaction, we are positioned to be a global leader in the manufacturing of rare cannabinoids, with expertise in three distinct and complementary cannabinoid manufacturing approaches. Our proprietary cannabinoid manufacturing process, IntegraSyn™, combined with BayMedica’s synthetic biology and chemical synthesis capabilities, provides us with complete manufacturing flexibility to select the most appropriate, cost-effective method based on the target cannabinoid and appropriate quality specifications for the desired market segment. In parallel to cannabinoid manufacturing, the combined company will continue to explore the therapeutic potential of cannabinoids and novel cannabinoid analogs for pharmaceutical drug development, as well as expand commercial sales of rare cannabinoids to the consumer health and wellness sector.
BayMedica is a revenue-stage biotechnology company leveraging its significant expertise in synthetic biology and pharmaceutical chemistry to develop efficient, scalable, and proprietary manufacturing approaches to produce high quality, regulatory-compliant rare cannabinoids for consumer applications. BayMedica is currently commercializing the rare cannabinoid CBC (cannabichromene) as a B2B supplier to distributors and manufacturers marketing products in the health and wellness sector. Revenues of BayMedica’s initial rare cannabinoid product, Prodiol® CBC (cannabichromene), have grown steadily since sales commenced in December 2019; the predecessor company, BayMedica Inc., had cumulative revenues of $2.4 million for the 21-month period ending September 30, 2021. Since October 13, 2021, the date of acquisition, to December 31, 2021, BayMedica had revenues of approximately $265,000. Based on publicly known data, we believe BayMedica leads the industry in large batch production of CBC with current batch sizes of more than 200kg and an ability to increase to metric ton quantities as market demand increases. BayMedica is focused on the wholesale to consumer health and wellness markets, including nutraceuticals, cosmetic, functional food and beverage, as well as animal health markets. In addition to CBC, BayMedica has received initial purchase orders for, and has commenced commercial sales of, CBT and has several high value non-intoxicating rare cannabinoids in various stages of commercial manufacturing scale-up including CBDV, THCV, CBGV and CBN for the health and wellness markets.
52
Manufacturing of Our Active Pharmaceutical Ingredients (API)
The CBN used in INM-755 and INM-088 is currently sourced from either contract manufacturers or, for smaller quantities, from research material suppliers, that typically utilize synthetic chemistry. Changes in contract manufacturers or suppliers may require additional verification of the vendor’s quality systems, compliance, manufacturing process, testing and equivalency to the currently supplied CBN prior to use. This is intended to be an interim step to enable us to proceed with developing its formulations, execute preclinical toxicology studies and progress through Phase I and II clinical trials. Thereafter, we may be able to utilize our IntegraSynTM system for GMP APIs. Bridging studies consisting of chemical analysis and, possibly, animal bioavailability studies may be required in order to switch our API from the current external manufacturing sources to our internal IntegraSynTM based APIs.
We expect that the final formulations (API + excipients + packaging) of INM-755 topical cream and the INM-088 eye drop formulation will be manufactured by contract manufacturers and sub-component fabricators. The contract manufacturers and sub-component fabricators will be selected based on their specific competencies in manufacturing, quality standards, and materials. FDA regulations require that products be produced under current cGMP.
Intellectual Property
A patent is a monopoly granted by a government for a period of up to 20 years. A patent provides an enforceable legal right to prevent others from exploiting an invention being a product, device, system, substance, process or method in the country of grant. For an invention to be patentable, it must be novel, involve an inventive step and useful at the time of filing the initial patent application for that invention. At 18 months from the initial patent application, the detailed description of the invention is published. In order to secure patent protection, a patent application is filed with the patent office in each country of interest, the application is considered under the patent laws of that country, and a patent will issue if the application meets the patentability criteria of that country. After a patent expires or lapses, anyone can then use the invention.
The grant of a patent does not guarantee validity and a patent may be challenged by third parties at a patent office by re-examination in some countries or through the courts by revocation proceedings. The grant of a valid patent does not mean that the invention may be exploited in a given country without infringing third party intellectual property rights in that country.
The owner of a patent has the exclusive right to prevent others from making, selling, importing or otherwise using the patented invention for the life of the patent. Patent infringement occurs when someone makes, hires, uses, imports or sells the patented invention, or a product made by a patented method, or offers to do these things, within the country covered by the patent without the permission of the owner of the patent.
Patent applications and patents are subject to payment of renewal fees over the life of the patent in order to maintain patent rights. If the renewal fees are not paid then the application or patent may lapse.
Adequate protection of intellectual property is a means to ensure that we can commercialize our intellectual property and reduce the likelihood of imitation by competitors. We intend to utilize patents available to protect its IP wherever possible. In addition, we also rely on trade-secrets and process know-how to protect our intellectual property. While we cannot patent the naturally occurring individual cannabinoids used in our Products and Product Candidates, there are a number of other approaches to protect our inventions. These include:
| ● | patents on individual or combinations of cannabinoids that provide novel methods for treating diseases; |
| ● | cannabinoid delivery technology, formulations designed specifically to increase the safety and efficacy of drug treatments; and |
| ● | manufacturing processes for cannabinoids. |
The patent methodologies listed above will be designed with the intention to maximize the protection of our multi-faceted approach to developing novel cannabinoid medicines. We typically file patent applications in US, Canada, EU and other selected commercially significant foreign jurisdictions.
53
InMed Patent Portfolio, January 2022 | ||||||
| Subject Matter | Scope | Owner- ship/ Origin | Filing Status/ Filing Date | Patent Reference Number | Earliest Potential/Patent Expiry3 | Jurisdictions |
| Metabolic engineering of E. coli for the biosynthesis of cannabinoid products | Manufacturing Process | InMed, UBC1 | PCT Application filed 09/05/2018 | WO/2019/046941 | 2038 | AU, CA, CN, EP, IL, IN, JP, KR, SG, US |
| Compositions and methods for biosynthesis of terpenoids or cannabinoids in a heterologous system | Manufacturing Process | InMed, UBC1 | PCT Application filed 3/6/2020 | WO/2020/176998 | 2040 | AU, CA, CN, EP, IL, IN, JP, KR, SG, US |
Ocular drug delivery formulation (Hydrogel) | Formulation, Use | InMed | PCT Application filed 05/08/2018 | WO/2018/205022
| 2038 | AU, CA, CN, EP, IL, IN, JP, KR, MX, SG, US, ZA |
| Compositions and methods for use of cannabinoids for neuroprotection | Use | InMed | PCT Application filed 04/24/2020 | WO/2020/215164 | 2040 | AU, CA, CN, EP, IL, IN, JP, KR, MX, SG, US, ZA |
| Topical formulations of cannabinoids and use thereof in the treatment of pain | Formulation, Use | InMed | PCT Application filed 09/21/2018 | WO/2019/056123 | 2038 | EP, US |
| Use of topical formulations of cannabinoids in the treatment of epidermolysis bullosa and related connective tissue disorders | Use | InMed | PCT Application filed 05/04/2017 | WO/2017/190249 | 2037 | AU, CA, CN, EP, IL, JP, SG, US |
| Compositions and methods for treating neuronal disorders with cannabinoids | Use | InMed | PCT Application filed 10/21/2021 | PCT/CA2021/051487 | 2041 | Currently pending at international Stage |
| Recombinant production systems for prenylated polyketides of the cannabinoid family | Manufacturing Process | BM2 | PCT Application filed 05/10/2018
| WO/2018/209143 | 2038 | AU, CA, CN, EP, IN, MX, US |
| Cannabinoid analogs and methods for their preparation | New Chemical Entity; Manufacturing Process | BM2 | PCT Application filed 10/31/2019 | WO/2020/092823 | 2039 | AU, BR, CA, CN, EP, IN, IL, JP, MX, US |
| Use of Type I and Type II polyketide synthases for the production of cannabinoids and cannabinoid analogs | Manufacturing Process | BM2 | PCT Application filed 11/13/2019 | PCT/US2019/061289 | 2039 | US |
| Preparation of cannabichromene and related cannabinoids | Manufacturing Process | BM2 | PCT Application filed 12/23/2020 | PCT/US2020/066965 | 2040 | Currently pending at international Stage |
| Genetically modified yeast for the production of cannabigerolic acid, cannabichromenic acid and related cannabinoids | Manufacturing Process | BM2 | PCT Application filed 01/20/2021 | PCT/US2021/014226 | 2041 | Currently pending at international Stage |
| Acyl activating enzymes for preparation of cannabinoids | Manufacturing Process | BM2 | PCT Application filed 01/20/2022 | PCT/US2022/013140 | 2042 | Currently pending at international Stage |
1 UBC is a co-inventor and has assigned all commercial rights to InMed in exchange for a royalty of less than 1% on sales revenues from products utilizing cannabinoids manufactured using the technology and a single digit royalty on any sub-licensing revenues. 2 BM = BayMedica 3 Patents typically expire 20 years from their filing dates, if granted, the patent expiry may be extended by patent agencies and/or health regulatory authorities.
PCT = Patent Cooperation Treaty. Members in this treaty includes over 150 countries including USA, Canada, Europe and others. | ||||||
54
As of January 1, 2022, we have thirteen patent families covering manufacturing, Product Candidates, novel methods for treating diseases including two for our INM-755 program (WO/2017/190249 and WO/2019/056123), one for our INM-088 program (PCT/CA2020/050547)) and one for treating neurodegenerative diseases (PCT/CA2021/051487). If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2037-2042. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions.
As of January 1, 2022, we have one patent family covering cannabinoid delivery technology for the ocular program (WO/2018/205022). If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2038. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions.
As of January 1, 2022, we have eight patent families covering manufacturing process for cannabinoids of interest (WO/2019/046941, WO/2020/176998, WO/2018/209143, WO/2020/092823, PCT/US2019/061289, PCT/US2020/066965, PCT/US2021/014226 and PCT/US2022/013140). If these patents applications are granted and all maintenance fees or annuities are paid, these patents are expected to expire in 2038-2042. In some situations, the patent may be eligible for adjustment or extension of the patent terms due to delay in the patent office during the prosecution phase. The expiration date above does not include the adjustments or extensions.
The Patent Cooperation Treaty, or “PCT”, is an international patent law treaty, which provides a unified procedure for filing patent applications to protect inventions in each of its member states. There are 151 member countries within the PCT, enabling near-global patent coverage through successful patent prosecution in the U.S., Japan, Europe, Canada, Australia, New Zealand, China, Brazil, Russia, India and many other countries. We have several filed patent applications currently either in the provisional stage or PCT stage of review as shown above. None have been granted to date. We retain the full commercial rights to all of these patents with any exceptions noted in the above table.
55
Investing in our common shares involves a high degree of risk. You should carefully consider each of the following risks, together with all other information set forth in this prospectus, including the consolidated financial statements and the related notes, before making a decision to buy our common shares. If any of the following risks actually occurs, our business could be harmed. In that case, the trading price of our common shares could decline, and you may lose all or part of your investment. You should also carefully consider the risk factors set forth under “Risk Factors” in our Annual Report on Form 10-K for the year ended June 30, 2021, filed with the SEC on September 24, 2021 (which document is incorporated by reference herein), as well as other risk factors described under the caption “Risk Factors” in any accompanying prospectus supplement and any documents we incorporate by reference into this prospectus, including all future filings we make with the SEC pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) that are so incorporated, before deciding to invest in our common stock. See “Incorporation By Reference.”
Summary of Risk Factors
The following is a summary of material risks that could affect our business. This summary may not contain all of our material risks, and it is qualified in its entirety by the more detailed risk factors set forth below.
| ● | Our IntegraSynTM or BayMedica yeast biosynthesis manufacturing approaches may prove unsuccessful in achieving yields and/or cost levels required to be economically competitive with alternative methods of manufacturing. |
| ● | Our prospects depend on the success of our Product Candidates which are at early-stages of development with a statistically high probability of failure and are subject to lengthy, time-consuming and inherently unpredictable regulatory processes. |
| ● | Our Products and Product Candidates contain compounds that may be classified as “controlled substances”, the use of which may generate public controversy and restrict their development or commercialization. |
| ● | The FDA or particular states may ultimately prohibit the sale of some or all dietary supplements or conventional foods containing cannabinoid ingredients and our downstream B2B customers may be required to submit a New Dietary Ingredient notification to the FDA, which may not be accepted without objection. |
| ● | U.S. Regulatory Framework for (non-THC) Cannabinoid Related products is rapidly evolving and changes could delay or prevent commercialization and result in materially adverse effects on our business. |
| ● | The COVID-19 coronavirus could adversely impact our business, including several key activities that are critical to our success. |
| ● | The market prices for our common shares are volatile and will fluctuate. |
| ● | Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or Product Candidates. |
| ● | If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common shares. |
| ● | In connection with the audit of our financial statements as of and for the years ended June 30, 2021 and 2020, material weaknesses in our internal control over financial reporting were identified and we may identify additional material weaknesses in the future. |
56
| ● | We have incurred, and will continue to incur, increased costs as a result of operating as a public company, and our management has been required, and will continue to be required, to devote substantial time to new compliance initiatives. |
| ● | We have incurred significant losses since our inception, we anticipate that we will continue to incur losses in the future, we have had limited commercial revenue and we may never become profitable. |
| ● | We may become subject to claims or become involved in lawsuits related to intellectual property. |
| ● | We rely heavily on contract manufacturers over whom we have limited control and our existing collaboration agreements and any that we may enter into in the future may not be successful. |
| ● | We are dependent upon our key personnel to achieve our business objectives. |
| ● | Our insurance may be insufficient to cover losses that may occur as a result of our operations. |
Risk Factors
Risks Related to our Business and Industry
Risks related to pharmaceutical drug development activities
Our prospects depend on the success of our Product Candidates which are at early stages of development with a statistically high probability of failure.
Given the early-stage of development, we can make no assurance that our research and development programs will result in regulatory approval or commercially viable products. To achieve profitable operations, we, alone or with others, must successfully develop, gain regulatory approval, and market our future products. We currently have no products that have been approved by the FDA, HC, or any similar regulatory authority. To obtain regulatory approvals for our Product Candidates being developed and to achieve commercial success, clinical trials must demonstrate that the Product Candidates are safe for human use and that they demonstrate efficacy. We have no products or technologies which are currently in human clinical trials. Additionally, we have no pharmaceutical products for commercial sale or licensed for commercial sale, nor do we expect to have any such products for the next several years.
Many potential pharmaceuticals products never reach the stage of clinical testing and even those that do have only a small chance of successfully completing clinical development and gaining regulatory approval. Our Product Candidates may fail for a number of reasons, including, but not limited to, being unsafe for human use or due to the failure to provide therapeutic benefits equal to or better than the standard of treatment at the time of testing. Positive results of early preclinical research may not be indicative of the results that will be obtained in later stages of preclinical or clinical research. Similarly, positive results from early-stage clinical trials may not be indicative of favorable outcomes in later-stage clinical trials. We can make no assurance that any future studies, if undertaken, will yield favorable results.
The early stage of our product development makes it particularly uncertain whether any of our product development efforts will prove to be successful and meet applicable regulatory requirements, and whether any of our Product Candidates will receive the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be successfully marketed. If we are successful in developing our current and future Product Candidates into approved products, we will still experience many potential obstacles, such as the need to develop or obtain manufacturing, marketing and distribution capabilities. If we are unable to successfully commercialize any of our products, our financial condition and results of operations may be materially and adversely affected.
57
Our IntegraSynTM or BayMedica yeast biosynthesis manufacturing approaches may prove unsuccessful in achieving yields and/or cost levels required to be economically competitive with alternative methods of manufacturing.
Given the early stage of development of the IntegraSynTM and BayMedica biosynthesis programs and the risks inherent in research and development, it is too early to project the commercial viability of cannabinoids produced via one or both of these processes. Potential negative outcomes from these programs include but are not limited to:
| ● | the technology fails to produce sufficient quantities of cannabinoids or ones for which we or others have a need; or |
| ● | the cost structure of the technology is such that it is not commercially competitive with alternate methods of cannabinoid manufacturing leading to the technology having no value proposition nor incremental value to the Company. |
Even if our Product Candidates advance through preclinical studies and clinical trials, we may experience difficulties in managing our growth and expanding our operations.
We have limited resources to carry out objectives for our current and future preclinical studies and clinical trials. Since our inception as a pharmaceutical company in October 2014, we have conducted numerous preclinical experiments and are currently conducting early-stage clinical trials, which is a time-consuming, expensive and uncertain process. In addition, while we have experienced management and expect to contract out many of the activities related to conducting these programs, we are a small company with less than 20 employees and, therefore, have limited internal resources both to conduct preclinical studies and clinical trials and to monitor third-party providers. As our Product Candidates advance through preclinical studies and clinical trials, we will need to expand our development, regulatory and manufacturing operations, either by expanding our internal capabilities or contracting with other organizations to provide these capabilities for us. In the future, we expect to have to manage additional relationships with collaborators or partners, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls, reporting systems and procedures.
If we have difficulty enrolling patients in clinical trials, the completion of the trials may be delayed or cancelled.
As our Product Candidates advance from preclinical testing to clinical testing, and then through progressively larger and more complex clinical trials, we will need to enroll an increasing number of patients that meet the eligibility criteria for those trials. The factors that affect our ability to enroll patients are largely uncontrollable and include, but are not limited to, the following:
| ● | size and nature of the patient population; |
| ● | inclusion and exclusion criteria for the trial; |
| ● | design of the study protocol; |
| ● | competition with other companies for clinical sites or patients; |
| ● | the perceived risks and benefits of the product candidate under study; |
| ● | the patient referral practices of physicians; and |
| ● | the number, availability, location and accessibility of clinical trial sites. |
As a result of the foregoing factors, we may have difficulty enrolling or maintaining the enrollment of patients in any clinical trials conducted for our products, which may result in the delay or cancellation of such trials. The delay or cancellation of any clinical trials could shorten any periods during which we may have the exclusive right to commercialize our Product Candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our Product Candidates and may harm our financial condition, results of operations and prospects.
58
If clinical trials of our Product Candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we would incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our Product Candidates.
Before obtaining marketing approval from regulatory authorities for the sale of our Product Candidates, we must conduct preclinical studies in animals and extensive clinical trials in humans to demonstrate the safety and efficacy of the Product Candidates. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of preclinical studies and early clinical trials may not predict the success of later clinical trials and interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. We do not know whether the clinical trials we may conduct will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our Product Candidates in any jurisdiction. A product candidate may fail for safety or efficacy reasons at any stage of the testing process. A major risk we face is the possibility that none of our Product Candidates under development will successfully gain market approval from the FDA or other regulatory authorities, resulting in us being unable to derive any commercial revenue from them after investing significant amounts of capital in multiple stages of preclinical and clinical testing.
If we experience delays in clinical testing, we will be delayed in commercializing our Product Candidates, and our business may be substantially harmed.
We cannot predict whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could shorten any periods during which we may have the exclusive right to commercialize our Product Candidates or allow our competitors to bring products to market before us, which would impair our ability to successfully commercialize our Product Candidates and may harm our financial condition, results of operations and prospects. The commencement and completion of clinical trials for our products may be delayed for a number of reasons, including delays related, but not limited, to:
| ● | failure by regulatory authorities to grant permission to proceed or placing the clinical trial on hold; |
| ● | import/export and research restrictions for cannabinoid-based pharmaceuticals may delay or prevent clinical trials in various geographical jurisdictions; |
| ● | patients failing to enroll or remain in our trials at the rate we expect; |
| ● | suspension or termination of clinical trials by regulators for many reasons, including concerns about patient safety or failure of our contract manufacturers to comply with current good manufacturing practice, or “cGMP”, requirements; |
| ● | any changes to our manufacturing process that may be necessary or desired; |
| ● | delays or failure to obtain clinical supply from contract manufacturers of our products necessary to conduct clinical trials; |
| ● | Product Candidates demonstrating a lack of safety or efficacy during clinical trials; |
| ● | patients choosing an alternative treatment for the indications for which we are developing any of our Product Candidates or participating in competing clinical trials and/or scheduling conflicts with participating clinicians; |
| ● | patients failing to complete clinical trials due to dissatisfaction with the treatment, side effects or other reasons; |
59
| ● | reports of clinical testing on similar technologies and products raising safety and/or efficacy concerns; |
| ● | clinical investigators not performing our clinical trials on their anticipated schedule, dropping out of a trial, or employing methods not consistent with the clinical trial protocol, regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
| ● | failure of our CROs to satisfy their contractual duties or meet expected deadlines; |
| ● | inspections of clinical trial sites by regulatory authorities or Institutional Review Boards, or “IRBs”, or ethics committees finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study; |
| ● | one or more IRBs or ethics committees rejecting, suspending or terminating the study at an investigational site, precluding enrollment of additional subjects, or withdrawing its approval of the trial; or |
| ● | failure to reach agreement on acceptable terms with prospective clinical trial sites. |
Our product development costs will increase if we experience delays in testing or approval or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to regulatory authorities or IRBs or ethics committees for re-examination, which may impact the cost, timing or successful completion of that trial. Delays or increased product development costs may have a material adverse effect on our business, financial condition and prospects.
Negative results from clinical trials or studies of others and adverse safety events involving the targets of our products may have an adverse impact on our future commercialization efforts.
From time to time, studies or clinical trials on various aspects of pharmaceutical products are conducted by academic researchers, competitors or others. The results of these studies or trials, when published, may have a significant effect on the market for the pharmaceutical product that is the subject of the study. The publication of negative results of studies or clinical trials or adverse safety events related to our Product Candidates, or the therapeutic areas in which our Product Candidates compete, could adversely affect the price of our common shares and our ability to finance future development of our Product Candidates, and our business and financial results could be materially and adversely affected.
We intend to expend our limited resources to pursue our Product Candidates for certain indications and may fail to capitalize on other Product Candidates or other indications for our Product Candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we are focusing on research programs relating to our Product Candidates for certain indications, primarily for the treatment of EB, which concentrates the risk of product failure in the event our Product Candidates prove to be unsafe or ineffective or inadequate for clinical development or commercialization. As a result, we may forego or delay pursuit of opportunities with other Product Candidates or for other indications that could later prove to have greater commercial potential. We may also deem it advisable to refocus our clinical development programs based on clinical trial results.
The regulatory approval processes of the FDA, HC, the EMA and other comparable foreign regulatory authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our Product Candidates, our business will be substantially harmed.
We are not permitted to market our Product Candidates in any jurisdiction until we receive formal approval from the appropriate regulatory authorities. For example, prior to submitting an NDA to the FDA or an MAA to the EMA for approval of our Product Candidates, we will need to complete our preclinical studies and clinical trials. Successfully completing our clinical program and obtaining approval of an application seeking commercialization approval is a complex, lengthy, expensive and uncertain process, and the regulatory authorities may delay, limit or deny approval of our Product Candidates for many reasons, including, among others, because:
| ● | we may not be able to demonstrate that our Product Candidates are safe and effective in treating patients to the satisfaction of the regulatory authorities such as the FDA, HC or EMA; |
60
| ● | the results of our clinical trials may not meet the level of statistical or clinical significance required by the regulatory authorities for marketing approval; |
| ● | the regulatory authorities may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| ● | the regulatory authorities may require that we conduct additional clinical trials; |
| ● | the regulatory authorities or other applicable foreign regulatory authorities may not approve the formulation, labeling or specifications of our Product Candidates; |
| ● | the contract manufacturing organizations and other contractors that we may retain to conduct our clinical trials may take actions outside of our control that materially adversely impact our clinical trials; |
| ● | the regulatory authorities may find the data from clinical studies and clinical trials insufficient to demonstrate that our Product Candidates are safe and effective for their proposed indications; |
| ● | the regulatory authorities may disagree with our interpretation of data from our preclinical studies and clinical trials; |
| ● | the regulatory authorities may not accept data generated at our clinical trial sites or may disagree with us over whether to accept efficacy results from clinical trial sites outside the United States, Canada or outside the European Union, as applicable, where the standard of care is potentially different from that in the United States, Canada or in the European Union, as applicable; |
| ● | if our applications are submitted to the regulatory authorities, the regulatory authorities may have difficulties scheduling the necessary review meetings in a timely manner, may recommend against approval of our application or may recommend or require, as a condition of approval, additional preclinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| ● | the FDA may require development of a Risk Evaluation and Mitigation Strategy which would use risk minimization strategies to ensure that the benefits of certain prescription drugs outweigh their risks, as a condition of approval or post-approval, and the EMA may grant only conditional marketing authorization or impose specific obligations as a condition for marketing authorization, or may require us to conduct post-authorization safety studies; |
| ● | the FDA, DEA, HC, EMA or other applicable foreign regulatory agencies may not approve the manufacturing processes or facilities of third-party manufacturers with which we contract or DEA or other applicable foreign regulatory agency quotas may limit the quantities of controlled substances available to our manufacturers; or |
| ● | the FDA, HC, EMA or other applicable foreign regulatory agencies may change their approval policies or adopt new regulations. |
In the United States, our activities are potentially subject to additional regulation by various federal, state and local authorities in addition to the FDA, including, among others, the Centers for Medicare and Medicaid Services, other divisions of the United States Department of Health and Human Services, or “HHS”, (for example, the Office of Inspector General), the Department of Justice, or “DOJ”, and individual United States Attorney offices within the DOJ, and state and local governments. Because of the breadth of these laws and the narrowness of available statutory and regulatory exemptions, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including criminal and significant civil monetary penalties, damages, fines, imprisonment, exclusion from participation in government programs, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre marketing product approvals, private “qui tam” actions brought by individual whistleblowers in the name of the government or refusal to allow us to enter into supply contracts, including government contracts, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws, and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
61
Any of these factors, many of which are beyond our control, could increase development costs, jeopardize our ability to obtain regulatory approval for and successfully market our Product Candidates and generate product revenue.
We intend to conduct clinical trials for our Product Candidates in several international jurisdictions, and acceptance by all regulatory authorities for such “international” data is not certain.
We intend to conduct clinical trials for our Product Candidates both inside and outside the United States. To date, all of our clinical development has been conducted outside of the United States. Ultimately, we plan to submit NDAs for our Product Candidates to the FDA and other regulatory authorities upon completion of all requisite clinical trials. As an example, although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the clinical trial must be conducted in accordance with FDA regulations relating governing human subject protection and the conduct of clinical trials, which are referred to as “Good Clinical Practice”, or “GCP” requirements and the FDA must be able to validate the data from the clinical trial through an onsite inspection if it deems such inspection necessary. Where data from foreign clinical trials are intended to serve as the sole basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless those data are considered applicable to the U.S. patient population and U.S. medical practice, the clinical trials were performed by clinical investigators of recognized competence, and the data is considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. In addition, such clinical trials would be subject to the applicable local laws of the foreign jurisdictions where the clinical trials are conducted. There can be no assurance the FDA or any other regulatory authorities will accept data from clinical trials conducted outside of the United States or other international jurisdictions. If the FDA or any other regulatory authorities does not accept any such data, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay aspects of our development plan.
In addition, the conduct of clinical trials outside the United States could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| ● | foreign regulatory requirements that could burden or limit our ability to conduct our clinical trials; |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; |
| ● | foreign currency fluctuations which could negatively impact our financial condition since certain payments are paid in local currencies; |
| ● | manufacturing, customs, shipment and storage requirements; |
| ● | cultural differences in medical practice and clinical research; and |
| ● | diminished protection of intellectual property in some countries. |
62
Our Product Candidates contain compounds that may be classified as “controlled substances”, the use of which may generate public controversy and restrict their development or commercialization.
If a drug has a potential for abuse, the NDA or other regulatory submission must include a description and analysis of studies or information related to abuse of the drug, including a proposal for scheduling (for example, in the U.S. under the federal Controlled Substances Act, or “CSA”). A description of any studies related to overdosage is also required, including information on dialysis, antidotes, or other treatments, if known. While we believe there would be relatively minimal abuse potential with our Product Candidates given the low drug concentration and topical route of administration, we could be incorrect or they may be perceived as having the potential for substance abuse. In either case, there may be a negative effect on our ability to successfully develop or commercialize our Product Candidates. Since our Product Candidates contain purified substances that are chemically identical to those occurring in nature, they may, therefore, be classified as “controlled substances”, and their regulatory approval may generate public controversy. Political and social pressures and adverse publicity could lead to delays in approval of, and increased expenses for, our Product Candidates. These pressures could also limit or restrict the introduction and marketing of our Product Candidates. Despite that fact that our APIs, which are the ingredients that give medicines their effects, are synthetically made and, therefore, we have no interaction with the Cannabis plant, adverse publicity from Cannabis misuse or adverse side effects from Cannabis or other cannabinoid products may adversely affect the commercial success or market penetration achievable for our Product Candidates. The nature of our business attracts a high level of public and media interest, and in the event of any resultant adverse publicity, our reputation may be harmed. Furthermore, if our Product Candidates are classified as “controlled substances”, they may be subject to import/export and research restrictions that could delay or prevent the development of our products in various geographical jurisdictions. The successful commercialization of our Product Candidates may require permits or approvals from regulatory bodies, such as the DEA, that regulate controlled substances.
Research restrictions, product shipment delays or prohibitions could have a material adverse effect on our business, results of operations and financial condition.
Research on and the shipment, import and export of our Product Candidates and the API used in our Product Candidates will require research permits, import and export licenses by many different authorities. For instance, in the United States, the FDA, U.S. Customs and Border Protection, and the DEA; in Canada, the Canada Border Services Agency, and HC; in Europe, the EMA and the European Commission; in Australia and New Zealand, the Australian Customs and Border Protection Service, the Therapeutic Goods Administration, the New Zealand Medicines and Medical Device Safety Authority and the New Zealand Customs Service; and in other countries, similar regulatory authorities, regulate the research on and import and export of pharmaceutical products that contain controlled substances. Specifically, the import and export process requires the issuance of import and export licenses by the relevant controlled substance authority in both the importing and exporting country. We may not be granted, or if granted, maintain, such licenses from the authorities in certain countries. Even if we obtain the relevant licenses, shipments of API and our Product Candidates may be held up in transit, which could cause significant delays and may lead to product batches being stored outside required temperature ranges. Inappropriate storage may damage the product shipment resulting in delays in clinical trials or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our Product Candidates. Once shipment is complete, we or the research contractors we are working with may also suffer further delays or restrictions as a result of regulations governing research on cannabinoids. A delay in a clinical trial or, upon commercialization, a partial or total loss of revenue from one or more shipments of API or our Product Candidates could have a material adverse effect on our business, results of operations and financial condition. The aforementioned examples and lists of various authorities that may currently, or in the future, affect our ability to conduct research on or import or export our Product Candidates and/or API, should not be construed as exhaustive or comprehensive in any way.
Healthcare legislation, including potentially unfavorable pricing regulations or other healthcare reform initiatives, may increase the difficulty and cost for us to obtain marketing approval of and commercialize our Product Candidates.
Particularly in the United States but also in other jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our Product Candidates, restrict or regulate post-approval activities or affect our ability to profitably sell any Product Candidates for which we obtain marketing approval. One such regulation is the U.S. federal Patient Protection and Affordable Care Act (P.L. 111-148), or “PPACA”, also referred to as the “Affordable Care Act” or “ACA”, was signed March 23, 2010, as amended by the Health Care and Education Reconciliation Act, signed March 31, 2010. The act contains many provisions, with various effective dates. Provisions included in the ACA are intended to expand access to insurance, increase consumer protections, emphasize prevention and wellness, improve quality and system performance, expand the health workforce, and curb rising health care costs. The ACA aims to extend health insurance coverage to about 32 million uninsured Americans by expanding both private and public insurance.
63
We expect that the Affordable Care Act, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria, new payment methodologies and in additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenue. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may compromise our ability to generate revenue, attain profitability or commercialize our products.
Increased scrutiny on drug pricing or changes in pricing regulations could restrict the amount that we are able to charge for our Product Candidates, which could adversely affect our revenue and results of operations.
Drug pricing by pharmaceutical companies is currently under increased scrutiny and is expected to continue to be the subject of intense political and public debate in the United States and other jurisdictions. Specifically, there have been several recent U.S. Congressional inquiries and hearings with respect to pharmaceutical drug pricing practices, including in connection with the investigation of specific price increases by several pharmaceutical companies. Additionally, several states have recently passed laws designed to, among other things, bring more transparency to drug pricing, and other states may pursue similar initiatives in the future. We cannot predict the extent to which our business may be affected by these or other potential future legislative or regulatory developments. However, increased scrutiny on drug pricing, negative publicity related to the pricing of pharmaceutical drugs generally, or changes in pricing regulations could restrict the amount that we are able to charge for our Product Candidates, which could have a material adverse effect on our revenue and results of operations.
Even if we are able to commercialize our Product Candidates, they may not receive coverage and adequate reimbursement from third-party payors, which could harm our business.
The availability of reimbursement by governmental and private payors is essential for most patients to be able to afford their treatments. Sales of our Product Candidates, if approved, will depend substantially on the extent to which the costs of these Product Candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our Product Candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
In the United States, the Medicare Modernization Act, established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could decrease the coverage available for any of our approved products. Furthermore, private payors often follow Medicare in setting their own coverage policies. Therefore, any reduction in coverage that results from the Medicare Modernization Act may result in a similar reduction from private payors.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services, or “CMS”, an agency within the HHS, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree.
The intended use of a drug product by a physician can also affect pricing. For example, CMS could initiate a National Coverage Determination administrative procedure, by which the agency determines which uses of a therapeutic product would and would not be reimbursable under Medicare. This determination process can be lengthy, thereby creating a long period during which the future reimbursement for a particular product may be uncertain.
64
Outside the United States, particularly in EU Member States, the pricing of prescription drugs is subject to governmental control. In these countries, pricing negotiations or the successful completion of Health Technology Assessment, or “HTA”, procedures with governmental authorities can take considerable time after receipt of marketing authorization for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Certain countries allow companies to fix their own prices for medicines but monitor and control company profits. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU Member States and parallel distribution, or arbitrage between low-priced and high-priced EU member states, can further reduce net realized prices. In some countries, we or our collaborators may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our Product Candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of any product candidate approved for marketing is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations or prospects could be adversely affected.
Our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse, federal exclusion or debarment, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation and prescription of any Product Candidates for which we obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. As a pharmaceutical company, even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. Restrictions under applicable federal and state healthcare laws and regulations that may affect our ability to operate include the following:
| ● | the U.S. federal healthcare Anti-Kickback Statute impacts our marketing practices, educational programs, pricing policies and relationships with healthcare providers or other entities, by prohibiting, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment of government funds (including through reimbursement by Medicare or Medicaid or other federal health care programs), which has been applied to impermissible promotion of pharmaceutical products for off-label uses, or making a false statement or record to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | the U.S. Health Insurance Portability and Accountability Act, or “HIPPA”, as amended by the Health Information Technology for Economic and Clinical Health Act, or “HITECH Act”, among other things, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services; |
| ● | the U.S. federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, requires applicable manufacturers of covered drugs, devices, biologics and medical supplies to report annually to HHS information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; |
| ● | analogous state laws and regulations, such as state anti-kickback laws, false claims laws and privacy and security of health information laws, may apply to sales or marketing arrangements, claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers, or health information; and |
65
| ● | certain state laws require pharmaceutical companies to adopt codes of conduct consistent with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; restrict certain marketing-related activities including the provision of gifts, meals, or other items to certain health care providers; and/or require drug manufacturers to report information related to payments and other transfers of value to physicians and certain other healthcare providers or marketing expenditures. |
Comparable laws and regulations exist in the countries within the European Economic Area, or “EEA”. Although such laws are partially based upon European Union, or “EU”, law, they may vary from country to country. Healthcare specific, as well as general EU and national laws, regulations and industry codes constrain, for example, our interactions with government officials and healthcare professionals, and the collection and processing of personal health data. Non-compliance with any of these laws or regulations could lead to criminal or civil liability.
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any physicians or other healthcare providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Failure to comply with the U.S. Foreign Corrupt Practices Act, or “FCPA”, the Canadian Corruption of Foreign Public Officials Act, or “CFPOA”, and other global anti-corruption and anti-bribery laws could subject us to penalties and other adverse consequences.
The FCPA and the CFPOA, as well as any other applicable domestic or foreign anti-corruption or anti-bribery laws to which we are or may become subject generally prohibit corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity and requires companies to maintain accurate books and records and internal controls, including at foreign-controlled subsidiaries. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity.
Compliance with these anti-corruption laws and anti-bribery laws may be expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, these laws present particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and physicians and other hospital employees are considered to be foreign officials. Certain payments by other companies to hospitals in connection with clinical trials and other work have been deemed to be improper payments to governmental officials and have led to FCPA enforcement actions.
Our internal control policies and procedures may not protect us from reckless or negligent acts committed by our employees, future distributors, licensees or agents. We are currently working to get policies and processes in place to monitor compliance with the FCPA and CFPOA. We can make no assurance that they will not engage in prohibited conduct, and we may be held liable for their acts under applicable anti-corruption and anti-bribery laws. Noncompliance with these laws could subject us to investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension or debarment from contracting with certain persons, the loss of export privileges, whistleblower complaints, reputational harm, adverse media coverage, and other collateral consequences. Any investigations, actions or sanctions or other previously mentioned harm could have a material negative effect on our business, operating results and financial condition.
66
Recent federal legislation and actions by state and local governments may permit reimportation of drugs from/to foreign countries where the drugs are sold at lower prices than in the country of origination, which could materially adversely affect our business and financial condition.
We may face competition for our Product Candidates, if approved, from cheaper generics and/or cannabinoid therapies sourced from foreign countries that have placed price controls on pharmaceutical products. This is referred to as parallel importation. For instance, the Medicare Modernization Act contains provisions that may change U.S. importation laws and expand pharmacists’ and wholesalers’ ability to import cheaper versions of an approved drug and competing products from Canada, where there are government price controls. These changes to U.S. importation laws will not take effect unless and until the Secretary of HHS certifies that the changes will pose no additional risk to the public’s health and safety and will result in a significant reduction in the cost of products to consumers. The Secretary of HHS has so far declined to approve a reimportation plan. Proponents of drug reimportation, including certain state legislatures, may attempt to pass legislation that would directly allow reimportation under certain circumstances. Legislation or regulations allowing the reimportation of drugs, if enacted, could decrease the price we receive for any products that we may develop, including our Product Candidates, and adversely affect our future revenues and prospects for profitability.
Risks related to general corporate administration
We are dependent upon our key personnel to achieve our business objectives.
We depend on key personnel, the loss of any of whom could harm our business. Our future performance and development will depend to a significant extent on the efforts and abilities of its executive officers, key employees, and consultants. The loss of the services of one or more of these individuals could harm our business. Our success will depend largely on our continuing ability to attract, develop and retain skilled employees and consultants in our business. Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in our field is intense. Due to this intense competition, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel. Any delay in replacing such persons, or an inability to replace them with persons of similar expertise, would have a material adverse effect on our business, financial condition and results of operations.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could subject us to significant liability and harm our reputation.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with regulations of domestic or foreign regulatory authorities. In addition, misconduct by employees could include intentional failures to comply with certain development standards, to report financial information or data accurately, or to disclose unauthorized activities to us. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. While prohibited, it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
Our directors’ and officers’ liability insurance may be insufficient to cover losses that may occur as a result of our operations.
We currently maintain directors’ and officers’ liability insurance, clinical trial insurance and property and general liability insurance and intend in the future to obtain shipping and storage insurance for Product Candidates. This insurance may not remain available to us or be obtainable by us at commercially reasonable rates, and the amount of our coverage may not be adequate to cover any liability we incur. Future increases in insurance costs, coupled with the increase in deductibles, will result in higher operating costs and increased risk. If we were to incur substantial liability and such damages were not covered by insurance or were in excess of policy limits, or if we were to incur such liability at a time when we were not able to obtain liability insurance, our business, results of operations and financial condition could be materially adversely affected.
67
There may be changes in laws, regulations and guidelines which are detrimental to our business.
Our operations are subject to a variety of laws, regulations and guidelines relating to pharmacology, cannabinoids and drug delivery, as well as laws and regulations relating to health and safety, the conduct of operations, and the protection of the environment. While, to the knowledge of our management, we are currently in compliance with all such laws, changes to such laws, regulations and guidelines due to matters beyond our control may cause adverse effects to our operations and financial condition. These changes may require us to incur substantial costs associated with legal and compliance fees and ultimately require us to alter our business plan. In addition, if the governments of Canada or the United States were to enact or amend laws relating to our industry, it may decrease the size of, or eliminate entirely, the market for our Product Candidates, may introduce significant new competition into the market and may otherwise potentially materially and adversely affect our business, results of operations and financial condition.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
The research and development that we carry out either directly or through third-parties involves, and may in the future involve, the use of potentially hazardous materials and chemicals. Our operations may produce hazardous waste products. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by local, state and federal laws and regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations and fire and building codes. Although we maintain workers’ compensation insurance as prescribed by the Province of British Columbia to cover us for costs and expenses we may incur due to injuries to our employees, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Our proprietary information, or that of our customers, suppliers and business partners, may be lost or we may suffer security breaches.
In the ordinary course of our business, we may collect and store sensitive data, including intellectual property, data from preclinical studies, clinical trial data, our proprietary business information and that of our customers, suppliers and business partners, and personally identifiable information of our customers, clinical trial subjects and employees, in our data centers and on our networks. The secure processing, maintenance and transmission of this information is critical to our operations. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Although to our knowledge we have not experienced any such material security breach to date, any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. Any such access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, regulatory penalties, disrupt our operations, damage to our ability to obtain patent protection for our Product Candidates, damage to our reputation, and cause a loss of confidence in our products and our ability to conduct clinical trials, which could adversely affect our business and reputation and lead to delays in gaining regulatory approvals.
We expect to face intense competition, often from companies with greater resources and experience than we have.
The pharmaceutical industry is highly competitive and subject to rapid change. The industry continues to expand and evolve as an increasing number of competitors and potential competitors enter the market. Many of these competitors and potential competitors have substantially greater financial, technological, managerial and research and development resources and experience than we have. Some of these competitors and potential competitors have more experience than we have in the development of pharmaceutical products, including validation procedures and regulatory matters. Other companies researching in the same disease areas may develop products that are competitive or superior to our Product Candidates. Other companies working in cannabinoid research may develop products targeting the same diseases that we are focused on that are competitive or superior to our Product Candidates. In addition, there are non-FDA approved Cannabis / cannabinoid preparations being made available from companies in the so-called “medical marijuana” industry, which may be competitive to our products. If we are unable to compete successfully, our commercial opportunities will be reduced and our business, results of operations and financial conditions may be materially harmed.
68
The development, production and supply of rare cannabinoids to the health and wellness industries is also highly competitive and subject to rapid change. The industry continues to expand and grow as an increasing number of competitors and potential competitors enter the market. Many of these competitors and potential competitors have substantially greater financial, technological, managerial and research and development resources and experience than we have in the development and scale up of rare cannabinoids. These companies are developing and scaling natural cannabinoids that are competitive to ours and will present increased pricing pressure and competition. If we are unable to compete successfully, our commercial opportunities will be reduced and our business, results of operations and financial conditions may be materially harmed
If we receive regulatory approvals, we intend to market our Product Candidates in multiple jurisdictions where we have limited or no operating experience and may be subject to increased business and economic risks that could affect our financial results.
If we receive regulatory approvals, we may plan to market our Product Candidates in jurisdictions where we have limited or no experience in marketing, developing and distributing our products. Certain markets have substantial legal and regulatory complexities that we may not have experience navigating. We are subject to a variety of risks inherent in doing business internationally, including risks related to the legal and regulatory environment in non-U.S. jurisdictions, including with respect to privacy and data security, trade control laws and unexpected changes in laws, regulatory requirements and enforcement, as well as risks related to fluctuations in currency exchange rates and political, social and economic instability in foreign countries. If we are unable to manage our international operations successfully, our financial results could be adversely affected.
Risks related to governmental legislation and regulation for Cannabis and Cannabinoids
U.S. Regulatory Framework for (non-THC) Cannabinoid Related products is rapidly evolving and changes could delay or prevent commercialization and result in materially adverse effects on our business.
The Agricultural Improvement Act of 2018, known as the “2018 Farm Bill” was enacted in the U.S. on December 20, 2018. The 2018 Farm Bill, among other things, redefined U.S. hemp as “the plant Cannabis sativa L. and any part of that plant, including the seeds thereof and all derivatives, extracts, cannabinoids, isomers, acids, salts, and salts of isomers, whether growing or not, with a delta-9 tetrahydrocannabinol concentration of not more than 0.3 percent on a dry weight basis” and removed as controlled substances its derivatives, extracts and cannabinoids, including CBD, from the definition of “marijuana” as defined under the Controlled Substances Act (CSA). The 2018 Farm Bill also amended the Agricultural Marketing Act of 1946 to allow for production and sale of U.S. hemp and its derivatives in the U.S.
The 2018 Farm Bill tasks the US Department of Agriculture (“USDA”) with promulgating regulations in relation to the cultivation and production of U.S. hemp. The 2018 Farm Bill also directs the USDA to promulgate federal regulations that would apply to the production of U.S. hemp in every state which does not put forth a state U.S. hemp plan for approval by the USDA. There remains uncertainty concerning the timing and manner of implementation of the 2018 Farm Bill at the federal level.
Under the 2018 Farm Bill, the FDA has retained authority over the Federal Food, Drug, and Cosmetic Act-regulated products (e.g., drugs, food, dietary supplements and cosmetics) containing U.S. hemp and U.S. hemp-derived ingredients, including CBD. Moreover, states have retained regulatory authority through their own legislation analogous to the Federal Food, Drug and Cosmetic Act, and the states may diverge from the federal treatment of the use of U.S. hemp as, or in, food, dietary supplements or cosmetic products.
The FDA has consistently taken the position that CBD, whether derived from U.S. hemp or U.S. Schedule I cannabis, is prohibited from use as an ingredient in food and dietary supplements. This stems from its interpretation of the exclusionary clauses in the Federal Food Drug & Cosmetic Act because CBD has been approved as a prescription drug and is the subject of substantial clinical investigations as a drug, which have been made public. The exclusionary clauses under the Federal Food Drug & Cosmetic Act provide that a substance that has been approved and/or has been subject to substantial clinical investigations as a drug may not be used in a food or dietary supplement, unless the substance was first marketed in a food or dietary supplement prior to the initiation of substantial clinical investigations of the substance as a drug.
69
The FDA has not issued regulations that elaborate on the exclusionary clauses and the FDA has not taken any enforcement action in the courts asserting a violation of the exclusionary clauses. To date, the FDA has issued a number of warning letters to companies unlawfully marketing CBD products. In many of these cases, the manufacturer made unsubstantiated claims about the product being able to treat medical conditions (e.g., cancer, Alzheimer’s disease, opioid withdrawal and anxiety) and had not obtained drug approvals. Others were issued to companies marketing CBD products as dietary supplements despite those products which contain CBD not meeting the definition of a dietary supplement, or adding CBD to human and animal foods and marketing CBD products for infants and children and other vulnerable populations. Some of these letters were co-signed with the FTC and cited the companies for making claims about the efficacy of CBD which were not substantiated by competent and reliable scientific evidence. Recently, the FDA has issued warning letters against dietary supplement manufacturers for manufacturing CBD supplements in licensed facilities in addition to various other violations. Importantly, several of these recent warning letters did not object to the CBD dietary supplements on the basis of any claims made - instead, the FDA cited the manufacturer on the basis that CBD was not a permissible dietary supplement ingredient. In 2021 the FDA formally rejected two New Dietary Ingredient (NDI) applications seeking to establish full spectrum hemp extract with CBD as a lawful dietary supplement. It remains to be seen whether or how these precedents may apply to other cannabinoids that have not been the subject of substantial clinical investigations drugs and/or have been marketed in foods and/or dietary supplements.
In November 2019, the FDA published a revised “Consumer Update” on CBD. The update noted that, as at the time of the Consumer Update, the FDA has approved only one CBD product, a prescription drug product to treat two rare, severe forms of epilepsy. The update also stated that it is illegal to market CBD by adding it to a food or labeling it as a dietary supplement, that the FDA has seen only limited data about CBD safety and these data point to real risks that need to be considered before taking CBD for any reason and that some CBD products are being marketed with unproven medical claims and are of unknown quality. Lastly, the FDA stated that it continues to evaluate the regulatory frameworks that apply to certain cannabis-derived products that are intended for non-drug uses, including whether and/or how they might consider updating their regulations, as well as whether potential legislation might be appropriate.
The FDA has stated that it recognizes the potential opportunities and significant interest in drug and other consumer products containing CBD, is committed to evaluating the agency’s regulatory policies related to CBD and has established a dedicated internal working group to explore potential pathways for various types of CBD products to be lawfully marketed. The rules and regulations and enforcement in this area continue to evolve and develop which could have a material adverse impact on our business, financial condition and results of operations.
States have the independent authority to regulate hemp, hemp derived cannabinoids and associated products. As such, many states have adopted their own hemp laws and regulations, in many cases creating a state-legal framework for the cultivation of hemp and the manufacture of products containing hemp derived extracts and cannabinoids. Changes by states regarding the transport, research, shipment, sales or resale of our Products could delay or prevent commercialization in such states and result in materially adverse effects on our business.
We operate in highly regulated sectors where the regulatory environment is rapidly developing and we may not always succeed in complying fully with applicable regulatory requirements in all jurisdictions where we carry on business.
Our business and activities are heavily regulated in all jurisdictions where we carry on business. Our operations are subject to various laws, regulations and guidelines by governmental authorities, including Health Canada, The European Medicines Authority, the US Food and Drug Administration (“FDA”), the Drug Enforcement Administration (DEA), US Customs and Border Protection (CBP) and analogous federal, provincial, state and local regulatory agencies around the globe relating to the manufacture, import, export, marketing, management, transportation, storage, sale, pricing, etc. of materials used in manufacturing, precursor compounds, intermediates, pharmaceutical, and non-pharmaceutical products. This also extends to laws, regulations and guidelines relating to health and safety, insurance coverage, the conduct of operations and the protection of the environment (including relating to emissions and discharges to water, air and land, the handling and disposal of hazardous and non- hazardous materials and wastes). Our operations may also be affected in varying degrees by government regulations with respect to, but not limited to, commercialization, manufacturing, import/export controls etc. Laws, regulations and guidelines, applied generally, grant government agencies and self-regulatory bodies broad administrative discretion over our activities, including the power to limit or restrict business activities as well as impose additional disclosure requirements on our products and services.
70
Achievement of our business objectives is contingent, in part, upon compliance with regulatory requirements enacted by these governmental authorities and obtaining all necessary regulatory approvals for the production, storage, transportation, sale, import and export, as applicable, of our products. The effect of relevant governmental authorities’ administration, application and enforcement of their respective regulatory regimes and delays in obtaining, or failure to obtain, applicable regulatory approvals which may be required may significantly delay or impact the development of markets, products and sales initiatives and could have a material adverse effect on our business, financial condition and results of operations.
The regulatory environment for our non-pharmaceutical rare cannabinoid Products business is rapidly developing, and the need to build and maintain robust systems to comply with different and changing regulations in multiple and, in some cases, overlapping jurisdictions increases the possibility that we may inadvertently violate one or more applicable requirements. Since rare cannabinoids are substances related to the cannabis plant they may now or in the future be classified as “Cannabis” or “controlled substances” by one or more regulatory agencies or jurisdictions. Political and social pressures, adverse publicity, misuse, for example, may lead to increasingly complex regulatory considerations. These complexities may also limit or restrict the introduction, marketing and sale of new cannabinoid Products, or force removal of certain Products from the market. The nature of our business attracts a high level of public and media interest, and in the event of any resultant adverse publicity, our reputation may be harmed. Furthermore, if any of our target cannabinoids are classified as “controlled substances” by any regulatory authority, they may be subject to import, export, transport, research or resale restrictions that could delay or prevent commercialization in select jurisdictions or even worldwide.
While we endeavor to comply with all relevant laws, regulations and guidelines, any failure to comply with the regulatory requirements applicable to our operations could subject us to negative consequences, including, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, asset seizures, revocation or imposition of additional conditions on licenses to operate our business, the denial of regulatory applications, including, in the U.S., by other regulatory regimes that rely on the positions of the DEA and FDA in the application of their respective regimes, or the imposition of additional or more stringent inspection, import, export, testing or reporting requirements, any of which could materially adversely affect our business and financial results. In the U.S., failure to comply with any FDA requirements, or analogous state agencies, for both our pharmaceutical and non-pharmaceutical products may result in, among other things, injunctions, product withdrawals, recalls, product seizures, fines and criminal prosecutions. The outcome of any regulatory or agency proceedings, investigations, audits, and other contingencies could harm our reputation, require us to take, or refrain from taking, actions that could harm our operations or require us to pay substantial amounts of money, harming our financial condition. There can be no assurance that any pending or future regulatory or agency proceedings, investigations and audits will not result in substantial costs or a diversion of management’s attention and resources, negatively impact our future growth plans and opportunities or have a material adverse impact on our business, financial condition and results of operations.
If our U.S. rare cannabinoid raw materials supply business activities are found to be in violation of any of U.S. federal, state or local laws or any other governmental regulations, in addition to the items described above:
| ● | we may be subject to “Warning Letters,” fines, penalties, administrative sanctions, settlements, injunctions, product recalls and/or other enforcement actions arising from civil, administrative or other proceedings initiated that could adversely affect our business, financial condition, operating results, liquidity, cash flow and operational performance; and |
| ● | our suppliers, service providers and distributors may elect, at any time, to breach or otherwise cease to participate in supply, service or distribution agreements, or other relationships, on which our operations rely. |
71
Changes in the laws, regulations and guidelines governing Cannabis and (U.S.) hemp may adversely impact our business.
While our current operations are not specifically subject to various laws, regulations and guidelines by governmental authorities (including, in Canada, Health Canada and, in the U.S., the FDA, DEA, CBP, FTC and PTO) relating to the marketing, acquisition, manufacture, packaging, labeling, management, transportation, import, export, storage, sale or disposal of Cannabis or U.S. hemp, supplying non-intoxicating rare cannabinoids to companies that further market products to the health and wellness sector may be considered to be ’Cannabis adjacent’ and may be negatively impacted by emerging changes to policies and determinations of these governmental authorities. No assurance can be given that new laws, regulations and guidelines will not be enacted or that existing laws, regulations and guidelines will not be amended, repealed, interpreted or applied in a manner which could require extensive changes to our operations, increase compliance costs, give rise to material liabilities or a revocation of our licenses and other permits, restrict the growth opportunities that we currently anticipate or otherwise limit or curtail our operations. Amendments to current laws, regulations and guidelines governing the production, sale and use of Cannabis, hemp, Cannabis-, hemp- and cannabinoid-based products, more stringent implementation or enforcement thereof or other unanticipated events, including changes in political regimes or political instability, supply chain disruptions, currency controls, fluctuations in currency exchange rates and rates of inflation, labor unrest, changes in taxation laws, regulations and policies, restrictions on foreign exchange and repatriation, changing political conditions and governmental regulations relating to foreign investment and the Cannabis business more generally, and changes in attitudes toward Cannabis, are beyond our control and could require extensive changes to our operations, which in turn may result in a material adverse effect on our business, financial condition and results of operations.
Controlled substance legislation may differ in other jurisdictions and could restrict our ability to market our products internationally, which would result in increased business and economic risks that could affect our financial results.
Controlled substance legislation may differ in other jurisdictions and could restrict our ability to market our products internationally. Most countries are parties to the Single Convention on Narcotic Drugs 1961, which governs international trade and domestic control of narcotic substances, including Cannabis extracts. Countries may interpret and implement their treaty obligations in a way that creates a legal obstacle to our obtaining marketing approval for Product Candidates in those countries. These countries may not be willing or able to amend or otherwise modify their laws and regulations to permit our Product Candidates to be marketed or achieving such amendments to the laws and regulations may take a prolonged period of time. We would be unable to market our Product Candidates in countries with such obstacles in the near future or perhaps at all without modification to laws and regulations.
Risks associated with commercial operations in the consumer health and wellness sector
We operate in a developing industry and will be subject to all associated regulatory risks.
Our business must be evaluated considering the problems, delays, uncertainties and complications encountered in connection with establishing a cannabinoid-based synthesis business that incorporates both chemical synthesis and synthetic biology.
There is a possibility that none of our discoveries under development in the future will be found to be safe and effective, that we will be unable to meet regulatory requirements or receive necessary regulatory approvals in order to commercialize them, or that evolving regulatory guidelines or regulatory approvals will be too narrow in scope to yield a commercially viable opportunity.
Any failure to successfully develop products that meet regulatory requirements, or to obtain any necessary regulatory approval for products would have a material adverse effect on our business, financial condition and results of operations.
Scale up, marketing and commercialization processes will be expensive and time consuming, and their outcomes uncertain.
Before we can obtain can launch any new non-pharmaceutical raw material products, we will be required to complete extensive scale-up processes, and ultimately provide adequate sales and marketing educational support. Commercialization is expensive and can be difficult to achieve. Commercialization is also time-consuming and can often be subject to unexpected delays.
72
The rare cannabinoid raw materials sector is highly competitive and the success of any of these competitors could materially adversely impact our business, financial condition and results of operations.
The rare cannabinoid health and wellness and pharmaceutical industries are competitive and subject to rapid change; the rare cannabinoid raw materials business specifically is highly competitive and involves a high degree of risk. While the rare cannabinoid raw materials industry continues to expand and evolve, an increasing number of competitors and potential competitors are entering the market. Many of these competitors and potential competitors have substantially greater financial, technological, managerial and research and development resources and experience than we have, with the potential to manufacture and sell similar products on a worldwide basis. Additionally, some of these competitors and potential competitors have more experience than we do in the development of pharmaceutical and Cannabis products, including validation procedures and regulatory matters. We may not be able to enter into supply agreements necessary to establish longer terms distribution channels or execute material downstream B2B partnerships. Our competitors may develop advantages over us in terms of price, technology, intellectual property, quality or purity of product, product efficacy, supply chain, scale, regulatory compliance, business relationships, or other factors, any of which could materially adversely impact our business, financial condition and results of operations.
In our non-pharmaceutical raw materials business, we compete with cannabinoid producers using traditional plant-based extraction. We also compete with traditional chemical manufacturers, developers of new methods for cannabinoid production and companies that have initiated biosynthetic cannabinoid programs. These competitors include companies who have invested in cultured cannabinoid production. There are also biotechnology companies that have announced partnerships that are targeting biosynthetic production of cannabinoids. The success of any of these competitors could materially adversely impact our business, financial condition and results of operations.
There is limited long-term data with respect to the efficacy and side effects of our products and future clinical research studies on the effects of Cannabis, hemp and cannabinoids may lead to conclusions that dispute or conflict with our understanding and belief regarding their benefits, viability, safety, efficacy, dosing and social acceptance.
Research in Canada, the U.S. and internationally regarding the benefits, viability, safety, efficacy, dosing and social acceptance of Cannabis, U.S. hemp or isolated cannabinoids in dietary supplements, food, or cosmetic products remains in early stages. There have been relatively few clinical trials on the benefits of Cannabis, hemp or isolated cannabinoids to date and there is limited long-term data with respect to efficacy, side effects and/or interaction of these substances with human or animal biochemistry. As a result, our products targeting the health and wellness sector could have unexpected side effects and safety concerns, the discovery of which could lead to civil litigation, regulatory actions and even possibly criminal enforcement actions. In addition, if the products we sell do not or are not perceived to have the effects intended by the end user, this could have a material adverse effect on our business, financial condition and results of operations. See also “- We may be subject to, or prosecute, litigation in the ordinary course of business.”, “- We may be subject to product liability claims.” and “- Our products may in the future be subject to recalls.”
The statements made by us, including in this document, concerning the potential benefits of isolated rare cannabinoids in the health and wellness segment are based on published articles and reports and therefore are subject to the experimental parameters, qualifications and limitations in such studies that have been completed. Although we believe that the existing public scientific literature generally supports our beliefs regarding the benefits, viability, safety, efficacy, dosing and social acceptance of rare cannabinoids, future research and clinical trials may cast doubt or disprove such beliefs, or could raise or heighten concerns regarding, and perceptions relating to rare cannabinoids, which could have a material adverse effect on the demand for our products with the potential to lead to a material adverse effect on our business, financial condition and results of operations. Given these risks, uncertainties and assumptions, undue reliance should not be placed on such literature. As an example, the FDA has raised several questions regarding the safety of CBD and gaps in the public scientific literature supporting the use of CBD by the general population.
73
The FDA or particular states may ultimately prohibit the sale of some or all dietary supplements or conventional foods containing cannabinoid ingredients and our downstream B2B customers may be required to submit a New Dietary Ingredient notification to the FDA, which may not be accepted without objection.
Under the 2018 Farm Bill, the FDA has retained authority over the Federal Food, Drug, and Cosmetic Act-regulated products (e.g., drugs (human and animal), food (human and animal), dietary supplements and cosmetics) containing U.S. hemp and U.S. hemp-derived ingredients, including CBD. The FDA has consistently taken the position that CBD, whether derived from U.S. hemp or U.S. Schedule 1 Cannabis, is prohibited from use as an ingredient in food and dietary supplements. This stems from its interpretation of the exclusionary clauses in the Federal Food Drug & Cosmetic Act because CBD is the active ingredient in a drug that has been approved as a prescription drug and is the subject of substantial clinical investigations as a drug, which have been made public. The exclusionary clauses under the Federal Food Drug & Cosmetic Act provide that a substance that has been approved and/or has been subject to substantial clinical investigations as a drug may not be used in a food or dietary supplement, unless the substance was first marketed in a food or dietary supplement prior to the initiation of substantial clinical investigations of the substance as a drug.
The FDA has not issued regulations that elaborate on the exclusionary clauses, and the FDA has not taken any enforcement action in the courts asserting a violation of the exclusionary clauses due to the marketing of U.S. hemp, U.S. hemp extracts, or CBD. To date, the FDA has issued several “Warning Letters” to companies unlawfully marketing CBD products. Some of these letters were co-signed with the FTC and cited the companies for making claims about the efficacy of CBD which were not substantiated by competent and reliable scientific evidence.
Until the FDA formally adopts regulations with respect to cannabinoid products, or announces an official position with respect to cannabinoid products, there is a risk that the FDA could take enforcement action (e.g., “Warning Letter,” seizure, injunction) against the Company’s U.S. hemp-derived cannabinoid products sold in the U.S.
Moreover, states have retained regulatory authority through their own analogues to the Federal Food, Drug and Cosmetic Act, and the states may diverge from the federal treatment of the use of U.S. hemp as, or in, food, dietary supplements or cosmetic products. The FDA or applicable states (under their CSA and Federal Food, Drug, and Cosmetic Act analogues) may ultimately not permit the sale of non-pharmaceutical products containing hemp-derived cannabinoids, or bio-equivalent cannabinoids not derived directly from hemp, which would have a material adverse impact on our business, financial condition and results of operations.
Even if the exclusionary clause issue discussed above is resolved in a manner favorable to us, our downstream customers could be required to submit a New Dietary Ingredient Notification (“NDIN”) to the FDA with respect to U.S. hemp-derived or other cannabinoid ingredients used in dietary supplement products. This could depend on whether we can establish that a particular ingredient was marketed as a dietary ingredient in a dietary supplement prior to any meaningful clinical research being conducted or is otherwise currently in the food supply in the same chemical form as used in such dietary supplement products. If the FDA objects to any such NDIN notification, this could reduce or eliminate any market for our cannabinoid products, which would have a material adverse impact on our business, financial condition and results of operations.
We may be subject to, or prosecute, litigation in the ordinary course of our marketing, distribution and sale of our products.
We are subject to litigation, claims and other legal and regulatory proceedings from time to time in the ordinary course of our marketing, distribution and sale of our products, some of which may adversely affect our business, financial condition and results of operations. As an example, several companies in the U.S. hemp-derived CBD industry have recently become party to an increasing number of purported class actions lawsuits relating to their food and dietary supplement products containing U.S. hemp-derived CBD. Should we or our B2B customers face similar class actions, plaintiffs in such class action lawsuits, as well as in other lawsuits against us, may seek very large or indeterminate amounts, including punitive damages, which may remain unknown for substantial periods of time. Should any litigation in which we become involved be determined against us, such a decision could adversely affect our ability to continue operating, adversely affect the market price for the common shares and require the use of significant resources. Even if we are involved in litigation and win, litigation can redirect significant resources. Litigation may also create a negative perception of our brands, which could have an adverse effect on our business, financial condition and results of operations.
74
Product liability lawsuits against us could cause us to incur substantial liabilities.
As a manufacturer and distributor of products designed to be ingested or otherwise used by humans, we face an inherent risk of exposure to product liability claims, regulatory action and litigation if our products are alleged to have caused significant loss or injury. Previously unknown adverse reactions resulting from human consumption of any cannabinoid-containing products alone or in combination with other medications or substances could occur. We may be subject to various product liability claims, including, among others, that our products caused injury or illness, include inadequate instructions for use or include inadequate warnings concerning possible side effects or interactions with other substances. A product liability claim or regulatory action against us could result in increased costs, could adversely affect our reputation with our clients and consumers generally, and could have a material adverse effect on our business, financial condition and results of operations.
There can be no assurances that we will be able to obtain or maintain product liability insurance on acceptable terms or with adequate coverage against potential liabilities. Such insurance is expensive and may not be available in the future on acceptable terms, or at all. The inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of products.
Our use of our Product Candidates in clinical trials and the sale of our Product Candidates, if approved, exposes us to the risk of product liability claims. Product liability claims might be brought against us by patients, healthcare providers or others selling or otherwise coming into contact with our Product Candidates. For example, we may be sued if any product we develop allegedly causes injury or is alleged to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, including as a result of interactions with alcohol or other drugs, negligence, strict liability, and a breach of warranties. Claims could also be asserted under local jurisdiction consumer protection acts. If we become subject to product liability claims and cannot successfully defend ourselves against them, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
| ● | withdrawal of patients from our clinical trials; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | decreased demand for our Product Candidates following marketing approval, if obtained; |
| ● | damage to our reputation and exposure to adverse publicity; |
| ● | increased FDA warnings on product labels or increased warnings imposed by the EMA or other regulatory authorities; |
| ● | litigation costs; |
| ● | distraction of management’s attention from our primary business; |
| ● | loss of revenue; and |
| ● | the inability to successfully commercialize our Product Candidates, if approved. |
Our current clinical trial liability insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If we obtain marketing approval for our Product Candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial, particularly in light of the size of our business and financial resources. A product liability claim or series of claims brought against us could cause our share price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, results of operations, business and prospects could be materially adversely affected.
75
Our products may, in the future, be subject to recalls.
Manufacturers and distributors of products are sometimes subject to the recall or return of their products for a variety of reasons, including product defects, such as contamination, unintended harmful side effects or interactions with other substances, packaging safety and inadequate or inaccurate labelling disclosure. If one or more of our products are recalled due to an alleged product defect or for any other reason, we could be required to incur the unexpected expense of the recall and any legal proceedings that might arise in connection with the recall. We may lose significant sales and may not be able to replace those sales at an acceptable margin, or at all. In addition, a product recall may require significant management attention. Although we have detailed procedures in place for testing finished products, there can be no assurance that any quality, potency or contamination problems will be detected in time to avoid unforeseen product recalls, regulatory action or lawsuits. Additionally, if one or more of our products were subject to recall, the public perception of that product and us could be harmed. A recall for any of the foregoing reasons could lead to decreased demand for products produced by us and could have a material adverse effect on our business, financial condition and results of operations. Additionally, product recalls may lead to increased scrutiny of our operations by regulatory authorities (Health Canada, the FDA, the DEA or other regulatory agencies), requiring further management attention and potential legal fees and other expenses. Furthermore, any product recall affecting the cannabinoid industry more broadly could lead consumers to lose confidence in the safety and security of the products sold by participants in this industry generally, which could have a material adverse effect on our business, financial condition and results of operations.
Our insurance coverage may not adequately coverall of the risks we face; the insurance premiums for such insurance may not continue to be commercially justifiable and there may be coverage limitations and other exclusions which may not be sufficient to cover our potential liabilities.
Our insurance coverage may not address all material risks to which we are exposed in our current state of operations, such insurance may be subject to coverage limits and exclusions and may not be available for the risks and hazards to which we are exposed. For example, certain wholesalers, distributors, retailers and other service providers may require suppliers of U.S. cannabinoid products to provide an indemnification from liability in connection with such products, which may not be covered by insurance. In addition, no assurance can be given that such insurance will be adequate to cover our liabilities or will be generally available in the future or, if available, that premiums will be commercially justifiable. If we were to incur substantial liability and such damages were not covered by insurance or were in excess of policy limits, or if we were to incur such liability at a time when we are not able to obtain liability insurance, there could be a material adverse effect on our business, financial condition and results of operations.
Furthermore, we manufacture and distribute raw materials that are incorporated into products designed to be ingested by humans. Such businesses face an inherent risk of exposure to product liability claims, regulatory action and litigation if products are alleged to have caused significant loss or injury. In addition, previously unknown adverse reactions resulting from human consumption of cannabinoids alone or in combination with other medications or substances could occur. While we maintain product liability coverage, no assurance can be given that such insurance would cover a specific product liability claim, or that if covered, such a claim would not be in excess of policy limits. As such, a product liability claim (or related regulatory action against us) could result in increased costs, adversely affect our reputation and have a material adverse effect on the results of our operations and financial condition.
76
Risks related to Information Technology
Failure to protect our information technology infrastructure against cyber-based attacks, network security breaches, service interruptions, or data corruption could significantly disrupt our operations and adversely affect our business and operating results.
We rely on information technology, telephone networks and systems, including the internet, to process and transmit sensitive electronic information and to manage or support a variety of business processes and activities. We use enterprise information technology systems to record, process and summarize financial information and results of operations for internal reporting purposes and to comply with regulatory, financial reporting, legal and tax requirements. Despite the implementation of security measures, our information technology systems, and those of our third-party contractors and consultants, are vulnerable to a cyber-attack, malicious intrusion, breakdown, destruction, loss of data privacy or other significant disruption. Any such successful attacks could result in the theft of intellectual property or other misappropriation of assets, or otherwise compromise our confidential or proprietary information and disrupt our operations. Cyber-attacks are becoming more sophisticated and frequent, and our systems could be the target of malware and other cyber-attacks. We have invested in our systems and the protection of our data to reduce the risk of an intrusion or interruption, and we monitor our systems on an ongoing basis for any current or potential threats. Nonetheless, our computer systems are subject to penetration and our data protection measures may not prevent unauthorized access. We can give no assurances that these measures and efforts will prevent interruptions or breakdowns. If we are unable to detect or prevent a security breach or cyber-attack or other disruption from occurring, then we could incur losses or damage to our data, or inappropriate disclosure of our confidential information or that of others; and we could sustain damage to our reputation, suffer disruptions to our research and development and incur increased operating costs including increased cybersecurity and other insurance premiums, costs to mitigate any damage caused and protect against future damage, and be exposed to additional regulatory scrutiny or penalties and to civil litigation and possible financial liability. For instance, the loss of preclinical or clinical data could result in delays in our development and regulatory filing efforts and significantly increase our costs.
Our failure to comply with data protection laws and regulations could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results.
We are subject to various domestic and international data protection laws and regulations (i.e., laws and regulations that address privacy and data security). The legislative and regulatory landscape for data protection continues to evolve, and in recent years there has been an increasing focus on privacy and data security issues. Numerous laws, including data breach notification laws, health information privacy laws and consumer protection laws, govern the collection, use and disclosure of health-related and other personal information. In addition, we may obtain health information from third parties (e.g., healthcare providers who prescribe our products) that are subject to privacy and security requirements under HIPAA regulations.
EU Member States, Australia and other countries have also adopted data protection laws and regulations, which impose significant compliance obligations. For example, the collection and use of personal data in the EU is governed by the provisions of the General Data Protection Regulation, or “GDPR”. The GDPR and the national implementing legislation of the EU Member States impose strict obligations and restrictions on the ability to collect, analyze and transfer personal data, including health data from clinical trials and adverse event reporting. In particular, these obligations and restrictions concern the consent of the individuals to whom the personal data relates, the information provided to the individuals, the rights of individuals to control personal data and the security and confidentiality of the personal data. In addition, the Australian Privacy Act 1988 (Cth), and other laws in the states and territories in Australia where we conduct certain of our clinical trials, apply similar restrictions on our ability to collect, analyze and transfer medical records and other patient data.
A claim or series of claims brought against us alleging a failure to comply with these laws, or changes in the way in which these laws are implemented, could lead to government enforcement actions and significant penalties against us, and adversely impact our operating results and could cause our share price to decline and, if we are unsuccessful in defending such a claim or claims and the resulting judgments exceed our insurance coverage, our financial condition, results of operations, business and prospects could be materially adversely affected.
The COVID-19 coronavirus pandemic, or other pandemics, could adversely impact our business, including several key activities that are critical to our success.
The global outbreak of COVID-19 continues to rapidly evolve. As a result, businesses have closed and limits have been placed on travel. The extent to which COVID-19 may impact our business will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate impact of the disease on specific geographies, the duration of the outbreak, travel restrictions and social distancing in the United States, Canada and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States, Canada and other countries to contain and treat the disease.
77
The spread of COVID-19 throughout the world has also created global economic uncertainty, which may cause partners, suppliers and potential customers to closely monitor their costs and reduce their spending budget. Any of the foregoing could materially adversely affect our research and development activities, clinical trials, supply chain, financial condition and cash flows.
If the COVID-19 outbreak continues to spread, we may need to limit operations or implement other limitations on our activities. There is a risk that countries or regions outside the United States and Canada may be less effective at vaccinations and containing COVID-19, in which case the risks described herein could be elevated significantly.
Risks Related to our Securities
The market prices for our common shares are volatile and will fluctuate.
The market price for our common shares may be volatile and subject to wide fluctuations in response to numerous factors, many of which are beyond our control, including the following: (i) actual or anticipated fluctuations in our quarterly financial results; (ii) recommendations by securities research analysts; (iii) changes in the economic performance or market valuations of other issuers that investors deem comparable to ours; (iv) addition or departure of our executive officers or members of our Board and other key personnel; (v) release or expiration of lock-up or other transfer restrictions on outstanding common shares; (vi) sales or perceived sales of additional common shares; (vii) liquidity of the common shares; (viii) significant acquisitions or business combinations, strategic partnerships, joint ventures or capital commitments by or involving us or our competitors; and (ix) news reports relating to trends, concerns, technological or competitive developments, regulatory changes and other related issues in our industry or target markets. Financial markets often experience significant price and volume fluctuations that affect the market prices of equity securities of public entities and that are, in many cases, unrelated to the operating performance, underlying asset values or prospects of such entities. Accordingly, the market price of our common shares may decline even if our operating results, underlying asset values or prospects have not changed. Additionally, these factors, as well as other related factors, may cause decreases in asset values that are deemed to be other than temporary, which may result in impairment losses. As well, certain institutional investors may base their investment decisions on consideration of our environmental, governance and social practices and performance against such institutions’ respective investment guidelines and criteria, and failure to meet such criteria may result in limited or no investment in our common shares by those institutions, which could materially adversely affect the trading price of our common shares. There can be no assurance that continuing fluctuations in price and volume will not occur. If such increased levels of volatility and market turmoil continue for a protracted period of time, our operations could be materially adversely impacted and the trading price of our common shares may be materially adversely affected.
In connection with the audit of our financial statements as of and for the years ended June 30, 2021 and 2020, material weaknesses in our internal control over financial reporting were identified and we may identify additional material weaknesses in the future.
In connection with the preparation and audits of our financial statements as of and for the years ended June 30, 2021 and 2020, material weaknesses (as defined under the Exchange Act and by the auditing standards of the U.S. Public Company Accounting Oversight Board, or “PCAOB”), were identified in our internal control over financial reporting. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual financial statements will not be prevented or detected on a timely basis. The identified material weaknesses arose from a lack of resources in our finance function that resulted in an overstatement of the valuation of warrants issued as part of a financing.
In light of the identified material weaknesses, it is possible that, had we performed a formal assessment of our internal control over financial reporting or had our independent registered public accounting firm performed an audit of our internal control over financial reporting in accordance with PCAOB standards, additional control deficiencies may have been identified.
We have begun taking measures, and plan to continue to take measures, to remediate these material weaknesses. However, the implementation of these measures may not fully address these material weaknesses in our internal control over financial reporting, and, if so, we would not be able to conclude that they have been fully remedied. Our failure to correct these material weaknesses or our failure to discover and address any other control deficiencies could result in inaccuracies in our financial statements and could also impair our ability to comply with applicable financial reporting requirements and make related regulatory filings on a timely basis. As a result, our business, financial condition, results of operations and prospects, as well as the trading price of our common shares, may be materially and adversely affected.
78
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our technologies or Product Candidates.
We may seek additional capital through a combination of private and public equity offerings, debt financings, strategic partnerships and alliances and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, existing ownership interests will be diluted and the terms of such financings may include liquidation or other preferences that adversely affect the rights of existing shareholders. Debt financings may be coupled with an equity component, such as warrants to purchase shares, which could also result in dilution of our existing shareholders’ ownership. The incurrence of indebtedness would result in increased fixed payment obligations and could also result in certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business and may result in liens being placed on our assets and intellectual property. If we were to default on such indebtedness, we could lose such assets and intellectual property. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our Product Candidates or grant licenses on terms that are not favorable to us.
Future offerings of debt or equity securities may rank senior to common shares.
If we decide to issue debt or equity securities in the future ranking senior to our common shares or otherwise incur additional indebtedness, it is possible that these securities or indebtedness will be governed by an indenture or other instrument containing covenants restricting our operating flexibility and limiting our ability to pay dividends to shareholders. Additionally, any convertible or exchangeable securities that we issue in the future may have rights, preferences and privileges, including with respect to dividends, more favorable than those of common shares and may result in dilution to shareholders. Because our decision to issue debt or equity securities in any future offering or otherwise incur indebtedness will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing or nature of our future offerings or financings, any of which could reduce the market price of our common shares and dilute their value.
Investors in our securities may face adverse tax consequences. In particular, we may be considered a “passive foreign investment company” which may have adverse United States federal income tax consequences for United States holders.
Prospective investors should be aware that the purchase of any of our securities may have tax consequences in the United States, Canada and other jurisdictions. Prospective investors should consult with their own independent tax advisor before purchasing any of our securities.
In particular, investors in our securities who are subject to United States federal taxation should be aware that we believe we may be classified as a passive foreign investment company, or “PFIC”, during the tax year ended June 30, 2021, and based on the nature of our business, the projected composition of our gross income and the projected composition and estimated fair market value of our assets, we may be classified as a PFIC for the current tax year ending June 30, 2022 and may be a PFIC in subsequent tax years. If we are a PFIC for any year during a United States holder’s holding period, then such United States holder generally will be required to treat any gain realized upon a disposition of securities, or any so-called “excess distribution” received on securities, as ordinary income, and to pay an interest charge on a portion of such gain or distributions, unless the holder makes a timely and effective “qualified electing fund” election, or a QEF election, or a “mark-to-market” election. Subject to certain limitations, a QEF election may be made with respect to the common shares, pre-funded warrants and warrant shares. Subject to certain limitations, such mark-to-market election may be made with respect to the common shares and warrant shares. A United States holder who makes a QEF election generally must report on a current basis its share of our net capital gain and ordinary earnings for any year in which we are a PFIC, whether or not we distribute any amounts to securityholders. A United States holder who makes the mark-to-market election generally must include as ordinary income each year the excess of the fair market value of the common shares or warrant shares over the taxpayer’s basis therein. Each United States holder should consult its own tax advisor regarding the United States federal, United States local, and foreign tax consequences of the PFIC rules and the acquisition, ownership, and disposition of our securities.
79
Future sales of common shares by officers and directors may negatively impact the market price for our common shares.
Subject to compliance with applicable securities laws, our directors and officers and their affiliates may sell some or all of their common shares in the future. No prediction can be made as to the effect, if any, such future sales of common shares may have on the market price of the common shares prevailing from time to time. However, the future sale of a substantial number of common shares by our directors and officers and their affiliates, or the perception that such sales could occur, could adversely affect prevailing market prices for our common shares.
We do not currently pay dividends on our common shares and have no intention to pay dividends on our common shares for the foreseeable future.
No dividends on our common shares have been paid by us to date. We do not intend to declare or pay any cash dividends in the foreseeable future. Payment of any future dividends will be at the discretion of our Board, after taking into account a multitude of factors appropriate in the circumstances, including our operating results, financial condition and current and anticipated cash needs. In addition, the terms of any future debt or credit facility may preclude us from paying any dividends unless certain consents are obtained and certain conditions are met.
We are exposed to risks related to currency exchange rates.
We currently hold the majority of our cash, cash equivalents and short-term investments in U.S. dollars which is our functional currency. A portion of our current operations is conducted in Canadian dollars. Exchange rate fluctuations between other currencies and the U.S. dollar create risk in several ways, including the following:
| ● | weakening of the Canadian dollar may decrease the value of our Canadian dollar cash, cash equivalents and short-term investments; |
| ● | weakening of the U.S. dollar may increase the cost of operations and products/services sourced in Canada; |
| ● | the exchange rates on non-U.S. dollar transactions and cash deposits can distort our financial results; and |
| ● | commercial product pricing and profit margins are affected by currency fluctuations. |
For as long as we are an “emerging growth company” we intend to take advantage of reduced disclosure and governance requirements applicable to emerging growth companies, which could result in our common shares being less attractive to investors and could make it more difficult for us to raise capital as and when we need it.
We are an “emerging growth company,” as defined in the JOBS Act, and we have taken advantage, and intend to continue to take advantage, of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
Investors may find our common shares less attractive because we rely on these exemptions, which could contribute to a less active trading market for our common shares or volatility in our share price. In addition, we may be less attractive to investors and it may be difficult for us to raise additional capital as and when we need it. Investors may be unable to compare our business with other companies in our industry if they believe that our financial accounting is not as transparent as other companies in our industry. If we are unable to raise additional capital as and when we need it, our financial condition and results of operations may be materially and adversely affected.
We may take advantage of these reporting exemptions until we are no longer an emerging growth company.
80
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence in us and, as a result, the value of our common shares.
We will be required, under Section 404 of the Sarbanes-Oxley Act, to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment includes disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting that results in more than a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. Section 404 of the Sarbanes-Oxley Act also generally requires an attestation from our independent registered public accounting firm on the effectiveness of our internal control over financial reporting. However, for as long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of the exemption permitting us not to comply with the independent registered public accounting firm attestation requirement.
Our compliance with Section 404 will require that we incur substantial accounting expense and expend significant management efforts. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. This may expose us, including individual executives, to potential liability which could significantly affect our business. If we are unable to conclude that our internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in our internal control over financial reporting once that firm begins its audits of internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common shares could decline, and we could be subject to sanctions or investigations by Nasdaq, the SEC, or other regulatory authorities. Failure to remedy any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Securities Exchange Act of 1934 is accumulated and communicated to management, recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements or insufficient disclosures due to error or fraud may occur and not be detected.
Deficiencies in disclosure controls and procedures and internal control over financial reporting could result in a material misstatement in our financial statements.
We could be adversely affected if there are deficiencies in our disclosure controls and procedures or in our internal controls over financial reporting. The design and effectiveness of our disclosure controls and procedures and our internal controls over financial reporting may not prevent all errors, misstatements or misrepresentations. Consistent with other entities in similar stages of development, we have a limited number of employees currently in the accounting group, limiting our ability to provide for segregation of duties and secondary review. A lack of resources in the accounting group could lead to material misstatements resulting from undetected errors occurring from an individual performing primarily all areas of accounting with limited secondary review. Deficiencies in internal controls over financial reporting which may occur could result in material misstatements of our results of operations, restatements of financial statements, other required remediations, a decline in the price of our common shares, or otherwise materially adversely affect our business, reputation, results of operations, financial condition or liquidity.
81
We have incurred, and will continue to incur, increased costs as a result of operating as a public company, and our management has been required, and will continue to be required, to devote substantial time to new compliance initiatives.
As a public company, we have incurred and are continuing to incur significant legal, accounting and other expenses and these expenses may increase even more after we are no longer an “emerging growth company.” We are subject to the reporting requirements of the Exchange Act and the rules adopted, and to be adopted, by the SEC. Our management and other personnel devote a substantial amount of time to these compliance initiatives.
Moreover, these rules and regulations have substantially increased our legal and financial compliance costs and made some activities more time-consuming and costly. The increased costs have increased our net loss. These rules and regulations may make it more difficult and more expensive for us to maintain sufficient director’s and officer’s liability insurance coverage. We cannot predict or estimate the amount or timing of additional costs we may continue to incur to respond to these requirements. The ongoing impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our Board, our Board committees or as executive officers.
Future sales and issuances of our common shares or rights to purchase common shares pursuant to our equity incentive plan could result in additional dilution of the percentage ownership of our shareholders and may cause our share price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To raise capital, we may sell substantial amounts of common shares or securities convertible into or exchangeable for common shares. These future issuances of common shares or common share-related securities, together with the exercise of outstanding options and any additional shares issued in connection with acquisitions, if any, may result in material dilution to our investors. Such sales may also result in material dilution to our existing shareholders, and new investors could gain rights, preferences and privileges senior to those of holders of our common shares.
Pursuant to our 2017 Amended and Restated Stock Option Plan, and as amended at our Annual General Meeting in November 2020, our compensation committee is authorized to grant equity-based incentive awards in the form of options to purchase common shares to our directors, executive officers and other employees and service providers. As of June 30, 2021, there were 493,387 options to purchase common shares available for future grant under our stock option plan. Future equity incentive grants under our stock option plan may result in material dilution to our shareholders and may have an adverse effect on the market price of our common shares.
Provisions in our corporate charter documents and certain Canadian laws could delay or deter a change of control.
Provisions in our articles and our by-laws, as well as certain provisions under the BCBCA and applicable Canadian securities laws, may discourage, delay or prevent a merger, acquisition, tender offer or other change in control of us that some shareholders may consider favorable. In addition, because our Board is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it more difficult for shareholders to replace members of our Board. As well, our preferred shares are available for issuance from time to time at the discretion of our Board, without shareholder approval. Our articles allow our Board, without shareholder approval, to determine the special rights to be attached to our preferred shares, and such rights may be superior to those of our common shares.
In addition, limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act in Canada. This legislation permits the Commissioner of Competition of Canada, or “Commissioner”, to review any acquisition of a significant interest in us. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada. The Investment Canada Act subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets, as calculated pursuant to the legislation, exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to result in a net benefit to Canada. Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
82
If securities or industry analysts publish inaccurate or unfavorable research about our business, our share price and trading volume may decline.
The trading market for our common shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who cover us downgrade our shares or publish inaccurate or unfavorable research about our business, our shares price may decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our shares may decrease, which may cause our shares price and trading volume to decline.
We are incorporated in Canada, with our assets and officers primarily located in Canada, with the result that it may be difficult for U.S. investors to enforce judgments obtained against us or some of our officers.
We are a company organized and existing under the laws of British Columbia, Canada. Many of our directors and officers and the experts named in this Annual Form on 10-K are residents of Canada or otherwise reside outside the United States, and all or a substantial portion of their assets, and a substantial portion of our assets, are located outside the United States. It may be difficult for holders of common shares who reside in the United States to effect service within the United States upon those directors, officers and experts who are not residents of the United States. It may also be difficult for holders of securities who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, officers and experts under the U.S. federal securities laws. Our Canadian counsel has advised us that there is doubt as to the enforceability in Canada against us or against our directors, officers and experts who are not residents of the United States, in original actions or in actions for enforcement of judgments of courts of the United States, of liabilities predicated solely upon U.S. federal or state securities laws.
Conversely, some of our directors and officers reside outside Canada and some of our assets are also located outside Canada. Therefore, it may not be possible for you to enforce in Canada against our assets or those directors and officers residing outside Canada, judgments obtained in Canadian courts based upon the civil liability provisions of the Canadian securities laws or other laws of Canada.
Risks Related to our Financial Position and Capital Needs
We have incurred significant losses since our inception and anticipate that we will continue to incur losses in the future.
Since our inception as a pharmaceutical company in October 2014, we have devoted substantially all of our resources to the development of our proprietary Product Candidates. We have generated significant operating losses since our inception with an accumulated deficit to December 31, 2021 of approximately $82.1 million. Our accumulated deficit increased between 2014, when we began focusing on the development of cannabinoid-derived pharmaceuticals following the acquisition of Biogen Science Inc., and December 31, 2021 is approximately $53.3 million. Our comprehensive losses for the fiscal years ended June 30, 2021 and 2020 were approximately $9.8 million and $9.4 million, respectively, and approximately $7.3 million and $3.4 million for the six month periods ended December 31, 2021 and 2020, respectively. Substantially all of our losses have resulted from expenses incurred in connection with our research and development programs and from general and administrative costs associated with our operations.
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate these losses will increase as we continue the research and development of, and clinical trials for, our Product Candidates. In addition to budgeted expenses, we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business. If our Product Candidates fail in preclinical or clinical trials, or do not gain regulatory approval, or even if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Due to our limited operating history and history of losses, any predictions about our future success, performance or viability may not be accurate.
83
We will require additional capital to fund our operations and if we fail to obtain necessary financing, we will not be able to complete the development and commercialization of our Product Candidates.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial and increasing amounts to conduct further research and development, preclinical testing and clinical trials of our Product Candidates, to seek regulatory approvals and reimbursement for our Product Candidates and to launch and commercialize any Product Candidates for which we receive regulatory approval.
As at December 31, 2021, we had approximately $11.3 million in cash, cash equivalents and short-term investments, which, we currently estimate funds our operations until approximately into the first quarter of fiscal 2023. Our ability to develop our research and development programs beyond these specific activities, which are expected to be substantially completed by the end of our current fiscal year, is subject to accessing additional capital, including through the sale of equity, partnership revenues, and out-licensing activities. There is no assurance that we will be successful in these efforts.
The progress of our Product Candidates for both current and prospective target indication(s) is uncertain because it is difficult to predict our spending for our Product Candidates up to the time that we seek FDA approval due to numerous factors, including, without limitation, the rate of progress of clinical trials, the results of preclinical studies and clinical trials for such indication, the costs and timing of seeking and obtaining FDA and other regulatory approvals for clinical trials and FDA guidance regarding clinical trials for such indication. Moreover, changing circumstances may cause us to expend cash significantly faster than we currently anticipate, and we may need to spend more cash than currently expected because of circumstances beyond our control. For these reasons, we are unable to state unequivocally the actual funds we will require for development and any approved marketing and commercialization activities. Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timing, costs and results of preclinical studies and clinical trials for our Product Candidates; |
| ● | any change in the clinical development plans or target indications for these Product Candidates; |
| ● | the number and characteristics of Product Candidates that we develop or may in-license; |
| ● | the terms of any collaboration agreements we may choose to execute; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the Drug Enforcement Administration, or “DEA”, the FDA, the European Medicines Agency, or “EMA”, Health Canada, or “HC”, or other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending intellectual property disputes, including patent infringement actions brought by third parties against us; |
| ● | the effect of competing product and market developments; |
| ● | the costs and timing of the implementation of commercial scale manufacturing activities; and |
| ● | the cost of establishing, or outsourcing, sales, marketing and distribution capabilities for any Product Candidates for which we may receive regulatory approval in regions where we choose to commercialize our products on our own. |
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our Product Candidates or one or more of our other research and development initiatives.
84
Any doubt about our ability to continue as a going concern may materially and adversely affect the price of our common shares, and it may be more difficult for us to obtain financing. Any doubt about our ability to continue as a going concern may also adversely affect our relationships with current and future collaborators, contract manufacturers and investors, who may become concerned about our ability to meet our ongoing financial obligations. If potential collaborators decline to do business with us or potential investors decline to participate in any future financings due to such concerns, our ability to increase our financial resources may be limited. We have prepared our financial statements on a going concern basis, which assumes that we will be able to meet our commitments, realize our assets and discharge our liabilities in the normal course of business. Our consolidated financial statements do not include any adjustment to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
We currently have limited commercial revenue and may never become profitable.
In addition to the on-going revenues from our bulk supply business, our ability to generate revenue and become profitable depends upon our ability to obtain regulatory approval for, and successfully commercialize, our Product Candidates that we may develop, in-license or acquire in the future.
Even if we are able to successfully achieve regulatory approval for these Product Candidates, we do not know what the reimbursement status of our Product Candidates will be or when any of these products will generate revenue for us, if at all. We have not generated, and do not expect to generate, any product revenue for the foreseeable future, and we expect to continue to incur significant operating losses for the foreseeable future due to the cost of research and development, preclinical studies and clinical trials and the regulatory approval process for our Product Candidates. The amount of future losses is uncertain and will depend, in part, on the rate of growth of our expenses.
Our ability to generate revenue and become profitable depends upon a number of additional factors, including our ability to:
| ● | successfully complete development activities, including the remaining preclinical studies and ongoing and planned clinical trials for our Product Candidates; |
| ● | in-license or acquire in the future, Product Candidates and other potential lines of business that we may develop; |
| ● | complete and submit NDAs to the FDA and Marketing Authorization Applications, or “MAAs”, to the EMA, and obtain regulatory approval for indications for which there is a commercial market; |
| ● | complete and submit applications to, and obtain regulatory approval from, other foreign regulatory authorities; |
| ● | manufacture any approved products in commercial quantities and on commercially reasonable terms; |
| ● | develop a commercial organization, or find suitable partners, to market, sell and distribute approved products in the markets in which we have retained commercialization rights; |
| ● | achieve acceptance among patients, clinicians and advocacy groups for any products we develop; |
| ● | obtain coverage and adequate reimbursement from third parties, including government payors; and |
| ● | set a commercially viable price for any products for which we may receive approval. |
| ● | develop, scale and sell existing and new rare cannabinoids and their analogs to the health/wellness sectors |
85
We are unable to predict the timing or amount of increased expenses, or when or if we will be able to achieve or maintain profitability. Even if we are able to complete the processes described above, we anticipate incurring significant costs associated with commercializing our Product Candidates.
Changes in tax laws and unanticipated tax liabilities could adversely affect our effective income tax rate and ability to achieve profitability.
We are subject to income taxes in the United States and Canada. As our operations expand, we may become subject to income tax in jurisdictions outside of the United States and Canada. Our effective income tax rate in the future could be adversely affected by a number of factors including changes in the mix of earnings (losses) in countries with differing statutory tax rates, changes in the valuation of deferred tax assets and liabilities and changes in tax laws. We regularly assess all of these matters to determine the adequacy of our tax provision which is subject to discretion. If our assessments are incorrect, it could have an adverse effect on our business and financial condition. There can be no assurance that income tax laws and administrative policies with respect to the income tax consequences generally applicable to us or to our subsidiaries will not be changed in a manner which adversely affects our shareholders.
Our ability to use our net operating loss carryforwards and other tax attributes may be limited.
As of our last fiscal year end, we had non-capital loss, or “NOL”, carry-forwards of approximately $50.9 million available to offset future taxable income in Canada. These NOL carry-forwards begin to expire in 2026.
Our NOL carryforwards could expire unused and be unavailable to offset future income tax liabilities. Under provisions in the Canadian Income Tax Act, and corresponding provisions of Canadian provincial law, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change, by value, the corporation’s ability to use its pre-change Canadian NOLs and other pre-change tax attributes, such as research and development tax credits, to offset its post-change income may be limited. Specifically, NOLs from a business before the change of control may be carried forward to taxation years after the change of control, but only if the same business is carried forward on after the change in control with a reasonable expectation of profit, and only to offset income from that business or a similar business. We have not performed any analyses under the applicable provisions in the Canadian Income Tax Act and cannot forecast or otherwise determine our ability to derive benefit from our various federal or provincial tax attribute carryforwards. As a result, if we earn net taxable income, our ability to use our pre-change NOL carryforwards to offset Canadian federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the provincial level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase provincial taxes owed.
In addition, we may experience ownership changes in the future as a result of subsequent shifts in our share ownership, including in any future offerings, some of which may be outside of our control. If we determine that an ownership change has occurred and our ability to use our NOL carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
Changes to accounting standards may adversely impact the manner in which we report our financial position and operating results.
There are ongoing projects conducted by the Financial Accounting Standards Board in the United States that are expected to result in new pronouncements that continue to evolve, which could adversely impact the manner in which we report our financial position and operating results.
86
Risks Related to our Intellectual Property
Our success is highly dependent upon our patents, proprietary technology, and other intellectual property.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. Patents and other proprietary rights are essential to our business. We rely on trade secret, patent, copyright and trademark laws, and confidentiality and other agreements with employees and third parties, all of which offer only limited protection. Our general policy has been to file patent applications to protect our inventions and improvements to our inventions that are considered important to the development of our business. In certain cases, we have chosen to protect our intellectual property by treating it as confidential internal know-how. Our success will depend in part on our ability to obtain patents, defend patents, maintain internal know-how/trade secret protection and operate without infringing on the proprietary rights of others. Interpretation and evaluation of pharmaceutical patent claims present complex legal and factual questions. Further, patent protection may not be available for some of the products or technology we are developing. If we are placed in a position where we must spend significant time and money defending or enforcing our patents, designing around patents held by others or licensing patents or other proprietary rights held by others, our business, results of operations and financial condition may be harmed. In seeking to protect our inventions using patents it is important to note that we have no assurance that:
| ● | patent applications will result in the issuance of patents; |
| ● | additional proprietary products developed will be patentable; |
| ● | patents issued will provide adequate protection or any competitive advantages; |
| ● | patents issued will not be successfully challenged by third parties; |
| ● | commercial exploitation of our inventions does not infringe the patents or intellectual property of others; or |
| ● | we will be able to obtain any extensions of the patent term. |
A number of pharmaceutical, biotechnology and medical device companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to our business. Some of these technologies, applications or patents could limit the scope of the patents, if any, that we may be able to obtain. It is also possible that these technologies, applications or patents may preclude us from obtaining patent protection for our inventions. Further, there may be uncertainty as to whether we may be able to successfully defend any challenge to our patent portfolio. Moreover, we may have to participate in derivation proceedings, inter partes review proceedings, post-grant review proceedings, or opposition proceedings in the various jurisdictions around the world. An unfavorable outcome in a derivation proceeding, an inter partes review proceeding, a post-grant review proceeding, or an opposition proceeding could preclude us or our collaborators or licensees from making, using or selling products using the technology, or require us to obtain license rights from third parties. It is not known whether any prevailing party would offer a license on commercially acceptable terms, if at all. Further, any such license could require the expenditure of substantial time and resources and could harm our business. If such licenses are not available, we could encounter delays or prohibition of the development or introduction of our product. In the case of intellectual property where we have chosen to protect it by treating it as internal knowhow, there can be no assurance that others with greater expertise or access to greater resources do not develop similar or superior technology that impairs the competitive value of our internal know-how.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The U.S. Patent and Trademark Office, or “PTO”, and various foreign national or international patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. Periodic maintenance fees on any issued patent are due to be paid to the PTO and various foreign national or international patent agencies in several stages over the lifetime of the patent. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of patent rights include, but are not limited to, failure to timely file national and regional stage patent applications based on our international patent application, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our Product Candidates, our competitors might be able to enter the market, which would have a material adverse effect on our business.
87
We may become subject to claims by third parties asserting that we or our employees have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Our commercial success depends upon our ability to develop, manufacture, market and sell our Product Candidates, and to use our related proprietary technologies without violating the intellectual property rights of others. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our Product Candidates, including interference or derivation proceedings before the PTO or other international patent offices. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue commercializing our Product Candidates. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Under certain circumstances, we could be forced, including by court order, to cease commercializing the applicable product candidate. In addition, in any such proceeding or litigation, we could be found liable for monetary damages. A finding of infringement could prevent us from commercializing our Product Candidates or force us to cease some of our business operations, which could materially harm our business. Any claims by third parties that we have misappropriated their confidential information or trade secrets could have a similar negative impact on our business.
While our preclinical studies are ongoing, we believe that the use of our Product Candidates in these preclinical studies fall within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the United States, which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our Product Candidates progress toward clinical trials and, ultimately, commercialization, the possibility of a patent infringement claim against us increases. We attempt to ensure that our Product Candidates and the methods we employ to manufacture them, as well as the methods for their uses we intend to promote, do not infringe other parties’ patents and other proprietary rights. There can be no assurance they do not, however, and competitors or other parties may assert that we infringe their proprietary rights in any event.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful and have a material adverse effect on the success of our business.
Competitors may infringe our patents or misappropriate or otherwise violate our intellectual property rights. To counter infringement or unauthorized use, litigation may be necessary in the future to enforce or defend our intellectual property rights, to protect our trade secrets or to determine the validity and scope of our own intellectual property rights or the proprietary rights of others. Also, third parties may initiate legal proceedings against us to challenge the validity or scope of intellectual property rights we own. These proceedings can be expensive and time consuming. Many of our current and potential competitors have the ability to dedicate substantially greater resources to defend their intellectual property rights than we can. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that a patent owned by us is invalid or unenforceable or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common shares.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be significantly diminished.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our current and former employees, consultants, outside scientific collaborators, sponsored researchers, contract manufacturers, vendors and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, we cannot guarantee that we have executed these agreements with each party that may have or have had access to our trade secrets. Any party with whom we or they have executed such an agreement may breach that agreement and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches.
88
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they disclose such trade secrets, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third-party, our competitive position would be harmed.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our Products and Product Candidates throughout the world would be prohibitively expensive. Therefore, we have filed applications and/or obtained patents only in key markets such as the United States, Canada, Japan and Europe. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may be able to export otherwise infringing products to territories where we have patent protection but where enforcement is not as strong as that in the United States. These products may compete with our Products and Product Candidates in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, an April 2016 report from the Office of the United States Trade Representative identified a number of countries, including India and China, where challenges to the procurement and enforcement of patent rights have been reported. Several countries, including India and China, have been listed in the report every year since 1989. As a result, proceedings to enforce our patent rights in certain foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business and could be unsuccessful.
Patent terms may be inadequate to protect our competitive position on our Product Candidates for an adequate amount of time.
Given the amount of time required for the development, testing and regulatory review of new Product Candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. We expect to seek extensions of patent terms in the United States and, if available, in other countries where we are prosecuting patents. In the United States, the Drug Price Competition and Patent Term Restoration Act of 1984 permits a patent term extension of up to five years beyond the normal expiration of the patent, which is limited to the approved indication (or any additional indications approved during the period of extension). However, the applicable authorities, including the FDA and the PTO, and any equivalent regulatory authorities in other countries, may not agree with our assessment of whether such extensions are available, and may refuse to grant extensions to our patents, or may grant more limited extensions than we request. If this occurs, our competitors may be able to take advantage of our investment in development and clinical trials by referencing our clinical and preclinical data and launch their product earlier than might otherwise be the case.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. For example:
| ● | others may be able to make compounds that are the same as or similar to our Product Candidates but that are not covered by the claims of the patents that we own; |
89
| ● | we might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own; |
| ● | we might not have been the first to file patent applications covering certain of our inventions; |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| ● | it is possible that our pending patent applications will not lead to issued patents; |
| ● | issued patents that we own may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
| ● | our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; or |
| ● | the patents of others may have an adverse effect on our business. |
Risks Related to our Third Parties
We rely heavily on contract manufacturers over whom we have limited control. If we are subject to quality, cost or delivery issues with the preclinical and clinical grade materials supplied by contract manufacturers, our business operations could suffer significant harm.
We currently have no manufacturing capabilities and rely on contract development and manufacturing organizations, or “CDMOs”, to manufacture our Product Candidates for preclinical studies and clinical trials, and our non-pharmaceutical Products for the health and wellness sector. We rely on CDMOs for manufacturing, filling, packaging, testing, storing and shipping of drug products in compliance with cGMP, regulations applicable to our products. We also rely on CDMOs for the scale up, manufacturing, downstream processing, filling, packaging, and testing of our non-pharmaceutical raw materials and material Products. The FDA and other regulatory agencies ensure the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities and controls used in manufacturing, processing and packaging of a drug product. If our CDMOs increase their prices or fail to meet our quality standards, or those of regulatory agencies such as the FDA, and cannot be replaced by other acceptable CDMOs, our ability to obtain regulatory approval for and commercialize our Product Candidates may be materially adversely affected. Furthermore, if our CDMOs increase their prices or fail to meet our quality standards and cannot be replaced in a timely manner by other acceptable CDMOs, our business may be materially adversely affected.
Additionally, our CDMOs, in turn, rely on certain precursors, substrates, reagents, and other materials or ingredients to be available to manufacture our products. Any disruption to the supply chain, change in regulatory status, change in state, provincial, federal, or national position on the import, export, or transport, or availability of these materials, or price volatility, could materially adversely impact our business.
The raw materials and APIs used in all of our Products and Product Candidates are currently sourced from either contract manufacturers or, for smaller quantities, from research material suppliers, that typically utilize synthetic chemistry as their manufacturing method. For our Products, synthetic chemistry is the most expeditious approach for introducing new rare cannabinoids into the market. For our Product Candidates, this is intended to be an interim step to enable us to proceed with developing our formulation, execute preclinical toxicology studies and progress through Phase I and II clinical trials, after which time we anticipate that we will have been able to successfully scale-up our IntegraSynTM manufacturing approach or one of our other technologies so that it will be cGMP ready at pharmaceutical grade. For Product Candidates, bridging studies consisting of chemical analysis and, possibly, animal studies may be required if we decide to switch our APIs from the current external manufacturing sources. There is no guarantee that we will be successful in scaling up either of our biosynthesis-based approaches (yeast biosynthesis and IntegraSynTM) for cannabinoids, or successfully complete any required bridging studies, or be able to successfully transfer these manufacturing processes to a CDMO. The key risks and challenges associated with the development of the biosynthesis-based processes include: failure to continue optimization and development of the process manufacturing steps from the current scale while maintaining the same or greater output of the selected cannabinoid; equipment and techniques may not be able to be scaled up using existing commercial processing equipment; supply of the key starting materials for the process may not be secured to ensure stability and security of commercial supply; and, failure of the large scale process to consistently produce the selected cannabinoid within set specifications and meeting the process parameters and in process controls to enable the manufacturing process to be validated for GMP commercial production of an API, among others. Failing to accomplish these or other criteria for the biosynthesis-based manufacturing process with a CDMO may mean that we are not able to produce certain cannabinoids in a cost-effective manner. This could result in us not being able to successfully commercialize or utilize our APIs in our Product Candidates, if any, that may obtain regulatory approval.
90
Our existing collaboration agreements and any that we may enter into in the future may not be successful.
We also have relationships with scientific collaborators at academic and other institutions, some of whom conduct research at our request or assist us in formulating our research and development strategies. These scientific collaborators are not our employees and may have commitments to, or consulting or advisory contracts with, companies that conflict in interests with and pose a competitive threat to us. Moreover, to the extent that we decide to enter into collaboration agreements, we will face significant competition in seeking appropriate collaborators. Collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish, implement and maintain collaborations or other alternative arrangements if we choose to enter into such arrangements and our selected partners may be given, and may exercise, a right to terminate their agreement with us without cause. Our Collaborative Research Agreement with the University of British Columbia may be terminated by either party upon 30 calendar days written notice. The terms of any collaboration or other arrangements that we may establish may not be favorable to us.
We are vulnerable to third-party, supply, manufacturing and transportation risks.
We depend on a global supply chain of chemical compounds, international manufacturers and fast and efficient international courier services to obtain Products and distribute our Products to our customers. Any prolonged disruption of any of these services may have materially adverse effect on our business, financial condition and results of operations. Rising costs or changes in policies regarding cannabinoids associated with any of these services used by us to manufacture, supply or ship our Products may also have a material adverse effect on our business, financial condition and results of operations.
We rely on third-party testing and analytical methods, some of which are not yet validated and some are validated but not yet standardized.
We test our non-pharmaceutical raw material products in various jurisdictions such as the U.S. with independent third-party testing laboratories for, among other things, cannabinoid levels. However, testing methods and analytical assays for cannabinoid levels of detection vary among different testing laboratories. The detected and reported cannabinoid content in our raw material products therefore can differ depending on the laboratory and testing methods (analytical assays) used. Variations in reported cannabinoid content will likely continue until the relevant regulatory agencies and independent certification bodies (e.g., ISO, USP) collaborate to develop, publish and implement standardized testing approaches for Cannabis (including U.S. hemp), cannabinoids and their derivative products. Such differences could cause confusion with our consumers which could lead to a negative perception of us and our products, increase the risk of litigation regarding cannabinoid content and regulatory enforcement action and could make it more difficult for us to comply with regulatory requirements regarding contents of ingredients and packaging and labeling.
For all of the aforesaid reasons and others set forth in our Annual Report on Form 10-K, an investment in our common shares and any other securities that we may offer from time to time involves a certain degree of risk. Any person considering an investment in our common shares or any other of our securities should be aware of these and other factors set forth in our 10-K and should consult with his or her legal, tax and financial advisors prior to making an investment in our common shares or any other of our securities that may be offered from time to time. Our common shares and any other securities that we may offer from time to time should only be purchased by persons who can afford to lose all of their investment.
91
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Exchange Act. We may, in some cases, use words such as “anticipate”, “believe”, “could”, “estimate”, “expect”, “future”, “intend”, “may”, “plan”, “predict”, “project”, “will”, “would”, and similar expressions, and variations or negatives of these words, that convey uncertainty of future events or outcomes to identify these forward-looking statements. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements. Forward-looking statements in this prospectus include, but are not limited to, statements about:
| ● | Our researching, developing, manufacturing and commercializing cannabinoid-based biopharmaceutical products will treat diseases with high unmet medical needs; |
| ● | The continued optimization of cannabinoid manufacturing approaches; |
| ● | Our success in initiating discussions with potential partners for licensing various aspects of our Product Candidates; |
| ● | Our ability to commercialize and, where required, register Product Candidates and Products in the United States and other jurisdictions; |
| ● | Our ability to successfully access existing manufacturing capacity via leases with third-parties or to transfer our manufacturing processes to a contract manufacturing organizations; |
| ● | Our belief that our manufacturing approaches that we are developing are robust and effective and will result in high yields of cannabinoids and will be a significant improvement upon existing manufacturing platforms; |
| ● | Our belief that that INM-755 offers specific advantages and will prove to provide the extensive relief symptomology with the added potential of addressing the underlying disease in EB; |
| ● | The structure and timing of future INM-755 studies including that we will complete patient enrollment into our Phase II study in EB in 2022; |
| ● | The ability of the IntegraSynTM approach to introduce a revenue stream to us before the expected commercial approval of our therapeutic programs; |
| ● | Our ability to successfully scale up our IntegraSynTM or other cost effective approaches so that it will be commercial-scale ready after Phase II clinical trials are completed, after which time we may no longer need to source APIs from API manufacturers; |
| ● | The success of the key next steps in our manufacturing approaches, including continuing efforts to diversify the number of cannabinoids produced, scaling-up the processes to larger vessels and identifying external vendors to assist in the commercial scale-up of the process; |
| ● | Our ability to potentially grow existing BayMedica sales revenues from existing and new cannabinoid Products; |
| ● | Our ability to successfully make determinations as to which research and development programs to continue based on several strategic factors; |
| ● | Our ability to monetize our IntegraSynTM manufacturing approach to the broader pharmaceutical industry; |
| ● | Our ability to take an opportunistic approach in the rapidly emerging sector of cannabinoid pharmaceutical development and the sale of cannabinoids in the health and wellness sectors to maximize the return to investors/shareholders; |
92
| ● | Successfully developing CBDV to meet expected customer demand in early 2022; |
| ● | Our ability to continue to outsource the majority of our research and development activities through scientific collaboration agreements and arrangements with various scientific collaborators, academic institutions and their personnel; |
| ● | The success of work to be conducted under the research and development collaboration between us and various contract development and manufacturing organizations (“CDMOs”); |
| ● | Our ability to develop our therapies through early human testing; |
| ● | Our ability to evaluate the financial returns on various commercialization approaches for our Product Candidates, such as a ‘go it-alone’ commercialization effort, out-licensing to third parties, or co-promotion agreements with strategic collaborators; |
| ● | Our ability to oversee clinical trials for INM-755 in EB and building the requisite internal commercialization infrastructure to self-market the product to EB clinics; |
| ● | Our ability to find a partnership early in the development process for INM-088 in glaucoma; |
| ● | Our IntegraSynTM or BayMedica derived products being bio-identical to the naturally occurring cannabinoids, and offering superior ease, control and quality of manufacturing when compared to alternative methods; |
| ● | Our ability to scale-up our IntegraSynTM manufacturing approaches to Good Manufacturing Practice (“GMP”) batch size for pharmaceutical use; |
| ● | Our ability to explore our manufacturing technologies as processes which may confer certain benefits, either cost, yield, speed, or all of the above, when pursuing specific types of cannabinoids, and filing a provisional patent application for same; |
| ● | Plans regarding our next steps, options, and targeted benefits of our manufacturing technologies; |
| ● | Our ability to potentially earn revenue from our IntegraSynTM approach by (i) becoming a supplier of APIs to the pharmaceutical industry and/or (ii) providing pharmaceutical-grade ingredients to the non-pharmaceutical market; |
| ● | Our plans to work closely with regulatory authorities and clinical experts in developing the clinical program for INM-755; |
| ● | Our ability to successfully prosecute patent applications; |
| ● | Our ability to complete formulation development and scale-up, conduct additional preclinical studies, and initiate and complete IND/CTA-enabling toxicology studies in calendar 2022 for INM-088; |
| ● | INM-088 being a once-a-day or twice-a-day eye drop medication that will compete with treatment modalities in the medicines category, and with the potential of INM-088 assisting in reducing the high rate of non-adherence with current glaucoma therapies; |
| ● | Developing a purification process to produce finished THCV and commencing scale-up of our novel process with a CDMO in calendar 1Q2022; |
| ● | Our belief that with a novel delivery system, the reduction of IOP and/or providing neuroprotection in glaucoma patients by topical (eye drop) application of cannabinoids will hold significant promise as a new therapy; |
| ● | The potential for any of our patent applications to provide intellectual property protection for us; |
93
| ● | Our ability to secure insurance coverage for shipping and storage of Product Candidates, and clinical trial insurance; |
| ● | Our ability to expand our insurance coverage to include the commercial sale of Products and Product Candidates; |
| ● | Developing patentable New Chemical Entities (“NCE”) which, if issued, will confer market exclusivity to us for the potential development into pharmaceutical Product Candidates, license, partner or sell to interested external parties; |
| ● | Our ability to initiate discussions and conclude strategic partnerships to assist with development of certain programs; |
| ● | Our ability to position ourselves to achieve value-driving, near term milestones for our Product Candidates with limited investment; |
| ● | Our ability to execute our business strategy; |
| ● | Critical accounting estimates; |
| ● | Management’s assessment of future plans and operations; |
| ● | The outlook of our business and the global economic and geopolitical conditions; and |
| ● | The competitive environment in which we and our business units operate. |
Undue reliance should not be placed on our forward-looking statements. Although forward-looking statements reflect our good faith beliefs, reliance should not be placed on forward-looking statements because they involve known and unknown risks, uncertainties and other factors, which may cause our actual results, performance or achievements to differ materially from anticipated future results, performance or achievements expressed or implied by such forward-looking statements. We undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events, changed circumstances or otherwise, except to the extent law requires.
94
The common shares to be offered and sold pursuant to this prospectus will be offered and sold by the selling shareholders. We will not receive any proceeds in connection with the sale of any common shares offered by the selling shareholders. The selling shareholders will pay expenses of the selling shareholders incurred in connection with the sale of common shares from time to time.
95
General
Our authorized share capital consists of an unlimited number of common shares without par value and an unlimited number of preferred shares without par value. As of the date of this registration statement, we had 14,362,160 common shares issued and outstanding and no preferred shares issued and outstanding.
The description of our securities contained herein is a summary only and may be exclusive of certain information that may be important to you. For more complete information, you should read our Amended and Restated Articles (the “Articles”), which have been filed with the SEC as an exhibit to our annual report on Form 10-K.
Common Shares
Each common share entitles the holder thereof to one vote at all meetings of shareholders.
There are no limitations on the rights of non-Canadian owners to hold or vote common shares.
In the event of our liquidation, dissolution or winding-up, whether voluntary or involuntary, or other distribution of our assets among shareholders for the purpose of winding up our affairs, subject to the rights, privileges and restrictions attaching to any preferred shares that may then be outstanding, the shareholders shall be entitled to receive our remaining property.
The shareholders are entitled to receive dividends, as and when declared by our Board, subject to the rights, privileges and restrictions attaching to our securities, which may be paid in money, property or by the issue of fully paid shares in our capital.
However, we do not anticipate paying any cash dividends for the foreseeable future, and instead intend to retain future earnings, if any, for use in the operation and expansion of our business.
Certain Takeover Bid Requirements
Unless such offer constitutes an exempt transaction, an offer made by a person to acquire outstanding shares of a Canadian entity that, when aggregated with the offeror’s holdings (and those of persons or companies acting jointly with the offeror), would constitute 20% or more of the outstanding shares, would be subject to the take-over provisions of Canadian securities laws. The foregoing is a limited and general summary of certain aspects of applicable securities law in the provinces and territories of Canada, all in effect as of the date hereof.
In addition to the take-over bid requirements noted above, the acquisition of shares may trigger the application of additional statutory regimes including amongst others, the Investment Canada Act and the Competition Act.
This summary is not a comprehensive description of relevant or applicable considerations regarding such requirements and, accordingly, is not intended to be, and should not be interpreted as, legal advice to any prospective purchaser and no representation with respect to such requirements to any prospective purchaser is made. Prospective investors should consult their own Canadian legal advisors with respect to any questions regarding securities law in the provinces and territories of Canada.
Actions Requiring a Special Majority
Under the BCBCA, unless otherwise stated in the Articles, certain corporate actions require the approval of a special majority of shareholders, meaning holders of shares representing 66 2/3% of those votes cast in respect of a shareholder vote addressing such matter. Those items requiring the approval of a special majority generally relate to fundamental changes with respect to our business, and include amongst others, resolutions: (i) removing a director prior to the expiry of his or her term; (ii) altering the Articles, (iii) approving an amalgamation; (iv) approving a plan of arrangement; and (v) providing for a sale of all or substantially all of our assets.
96
Transfer Agent and Registrar
The transfer agent and registrar for our common shares is Computershare Investor Services Inc., 100 University Avenue, 9th Floor, Toronto, Ontario, Canada M5J 2Y1.
Reports to Shareholders
We are subject to the periodic reporting requirements of the Exchange Act and in accordance therewith file periodic reports, including, but not limited to, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports, proxy statements and other information filed or furnished with the Securities and Exchange Commission pursuant to Section 13(a) or 15(d) of the Exchange Act. We plan to furnish our shareholders with an annual report for each fiscal year beginning for the fiscal year ending June 30, 2021 containing financial statements audited by our independent registered public accounting firm. You may read and copy (at prescribed rates) any such reports, proxy statements and other information at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference room. Our SEC filings will also be available to you on the SEC’s website at http://www.sec.gov. Our website address is https://www.inmedpharma.com. The information contained in or accessible from our website is not incorporated into this prospectus, and you should not consider it part of this prospectus. We have included our website address in this prospectus solely as an inactive textual reference.
Market Price of and Dividends on the Our Common Shares
Our common shares are currently quoted under the symbol “INM” on the Nasdaq Capital Market.
While there are no restrictions on the payment of dividends, we have never declared nor paid any cash dividends on our common shares, and we presently have no intention of paying any cash dividend in the foreseeable future. Our current policy is to retain earnings, if any, to finance the expansion of our business. The future payment of dividends will depend on our results of operations, financial condition, capital expenditure plans and other factors that we deem relevant and will be at the sole discretion of our Board.
Holders
As of December 31, 2021, there were 3,878 holders of record of our issued and outstanding common shares.
Description of the Warrants
Public Warrants Issued in the IPO
General. On November 16, 2020, we issued public warrants to purchase an aggregate of 1,780,000 common shares to the public in connection with our IPO. Each public warrant has an exercise price of $5.11 per share, is upon issuance exercisable and will expire on the sixth anniversary of the original issuance date. The following is a brief summary of the public warrant and is subject in all respects to the provisions contained in the form of warrant. The public warrants are not publicly traded and we do not plan to list the public warrants on the Nasdaq Capital Market, any other national securities exchange or any other nationally recognized trading system. As of April 6, 2022, there were 1,780,000 public warrants outstanding and 1,780,000 underlying common shares that are reserved for these warrants.
Fractional Shares; Rights as a Stockholder. No fractional common shares will be issued in connection with the exercise of a public warrant. Warrant amounts will be rounded up to the next whole share. A public warrant may be transferred by a holder, upon surrender of the warrant, properly endorsed (by the holder executing an assignment in the form attached to the warrant). Prior to the exercise of any warrants to purchase common shares, holders of the warrants will not have any of the rights of holders of the common shares purchasable upon exercise, including the right to vote, except as set forth therein.
97
Duration and Exercise Price. Each whole public warrant represents the right to purchase one common share at an exercise price equal to $5.11, subject to adjustment as described below. Each public warrant may be exercised on or after the closing date of the IPO through and including the close of business on the sixth anniversary of the date of issuance. Each public warrant will have a cashless exercise right in the event that the common shares underlying such warrants are not covered by an effective registration statement at the time of such exercise.
Adjustments; Fundamental Transaction. The exercise price and the number of shares underlying the public warrants are subject to appropriate adjustment in the event of stock splits, stock dividends on our common shares, stock combinations or similar events affecting our common shares. In addition, in the event we consummate a merger or consolidation with or into another person or other reorganization event in which our common shares are converted or exchange for securities, cash or other property, or we sell, lease, license, assign, transfer, convey or otherwise dispose of all or substantially all of our assets or we or another person acquire 50% or more of our outstanding common shares, then following such event, the holders of the warrants will be entitled to receive upon exercise of the warrants the same kind and amount of securities, cash or property which the holders would have received had they exercised the warrants immediately prior to such fundamental transaction. Any successor to us or surviving entity will assume the obligations under the warrants. Additionally, as more fully described in the form of public warrant, in the event of certain fundamental transactions, the holders of the public warrant will be entitled to receive consideration in an amount equal to the Black Scholes value of the public warrants on the date of consummation of such transaction.
Amendment and Waiver. Amendments and waivers of the terms of the public warrants require the written consent of the holder of such warrant and us.
Common Share Purchase Warrants issued in February 2021 Private Placement
General. On February 11, 2021, we issued common share purchase warrants to purchase an aggregate of 693,000 common shares to a group of institutional investors in connection with a private placement offering. Pursuant to an Amendment of Purchase Agreement and Common Stock Purchase Warrant, by and among holders of common shares purchase warrants and the Company, dated March 21, 2022, the exercise price for these warrants issued in the February 2021 Private Placement is $0.45 per share, subject to adjustment as discussed below. The warrants issued in the February 2021 Private Placement will expire on March 31, 2023. As of April 6, 2022, there were 323,400 common share purchase warrants outstanding and 323,400 underlying common shares that are reserved for these warrants.
Exercise. Following stockholder approval, the warrants issued in the February 2021 Private Placement may be exercised by providing an executed notice of exercise form followed by full payment of the exercise price or on a cashless basis. The selling shareholders do not have the rights or privileges of holders of common shares or any voting rights with respect to the common shares represented by the warrants issued in the February 2021 Private Placement until such selling shareholder exercises such warrant and receives its common shares. After the issuance of common shares upon exercise of the warrants issued in the February 2021 Private Placement, the selling shareholders will be entitled to one vote for each share held of record on all matters to be voted on by stockholders generally.
Beneficial Ownership Limitation. Each selling shareholder will be subject to a requirement that they will not have the right to exercise the warrants issued in the February 2021 Private Placement to the extent that, after giving effect to such exercise, such selling shareholder (together with its affiliates) would beneficially own in excess of 4.99%/9.99%, as applicable, of the common shares of the Company outstanding immediately after giving effect to such exercise.
Anti-Dilution Protection. If the number of outstanding common shares: (i) is increased by a stock dividend payable in common shares; (ii) is increased by a split-up of common shares; (iii) is decreased by a combination of outstanding common shares; or (iv) is reclassified by the issuance of any common shares of the Company then, on the effective date of such event, the exercise price of the warrant will be multiplied by a fraction of which the numerator is (x) the number of common shares outstanding immediately prior to such event and the denominator is (y) the number of common shares outstanding immediately after such event, and the number of common shares issuable upon exercise of the warrant will be proportionately adjusted such that the aggregate exercise price will remain unchanged. Such adjustment will be effective immediately after the record date for the determination of stockholders entitled to receive such dividend or distribution and will be effective immediately after the effective date in the case of a subdivision, combination or re-classification.
98
In addition, if the Company, at any time while the warrants issued in the February 2021 Private Placement are outstanding and unexpired, grants, issues or sells any: (i) securities of the Company or its subsidiaries which would entitle the holder thereof to acquire at any time common shares, including, without limitation, any debt, preferred stock, right, option, warrant or other instrument that is at any time convertible into or exercisable or exchangeable for, or otherwise entitles the holder thereof to receive, common shares; or (ii) rights to purchase stock, warrants, securities or other property pro rata to the record holders of any class of common shares (the “Purchase Rights”), then the selling shareholders will be entitled to acquire, upon the terms applicable to such Purchase Rights, the aggregate Purchase Rights which it could have acquired if it had held the number of common shares acquirable upon complete exercise of the warrants issued in the Private Placement immediately before the date on which a record is taken or the record holders are determined for the grant, issuance or sale of such Purchase Rights.
If the Company, at any time while the warrants issued in the February 2021 Private Placement are outstanding and unexpired, declares or makes any dividend or other distribution of assets to holders of common shares, by way of return of capital or otherwise, at any time after the issuance of the warrants issued in the Private Placement, then the selling shareholders shall be entitled to participate in such distribution to the same extent that it would have participated therein had it held the number of common shares acquirable upon complete exercise of the warrants immediately before the date of which a record is taken or the record holders are determined for such distribution.
Fundamental Transaction. In the event of a “fundamental transaction” then, upon a subsequent exercise of the warrants issued in the February 2021 Private Placement, the selling shareholders will have the right to purchase and receive the same kind and amount of consideration receivable by the stockholders of the Company in such fundamental transaction. The Company will cause the surviving company in a fundamental transaction to assume the obligations of the Company under the warrants issued in the February 2021 Private Placement. For purposes of the warrants issued in the February 2021 Private Placement, a “fundamental transaction” includes, subject to certain exceptions: (i) any reclassification, reorganization or recapitalization of the common shares of the Company; (ii) any merger or consolidation of the Company with or into another corporation; (iii) any sale, lease, license, assignment, transfer, conveyance or other disposition of all or substantially all of the Company’s assets in one or more transactions; (iv) any, direct or indirect, purchase offer, tender offer or exchange offer is completed pursuant to which stockholders are permitted to sell, tender or exchange their shares for other securities, cash or property and has been accepted by the holders of 50% or more of the outstanding common shares of the Company; or (v) the Company, directly or indirectly, in one or more related transactions consummates a stock or share purchase agreement or other business combination with another person whereby such other person acquires more than 50% of the outstanding common shares of the Company.
Amendments. The warrants issued in the February 2021 Private Placement provide that the terms of the warrants issued in the February 2021 Private Placement may be amended only in writing signed by the Company and such selling shareholders.
Series A Common Shares Purchase Warrants issued in July 2021 Private Placement
General. On July 2, 2021, we issued Series A warrants to purchase up to 4,036,327 common shares to an institutional investor in connection with a private placement offering. The Series A warrants issued in the July 2021 Private Placement entitle the selling shareholder the option to purchase at an exercise price of $2.848 per share, subject to adjustment as discussed below. The Series A warrants will expire on July 2, 2026. As of April 6, 2022, there were 4,036,327 Series A warrants outstanding and 4,036,327 underlying common shares that are reserved for the Series A warrants.
Exercise. The Series A warrants are exercisable immediately and may be exercised by providing an executed notice of exercise form followed by full payment of the exercise price or on a cashless basis, if applicable. The selling shareholder does not have the rights or privileges of holders of common shares or any voting rights with respect to the common shares represented by the warrants until such selling shareholder exercises such warrant and receives its common shares. After the issuance of common shares upon exercise of the warrants, the selling shareholder will be entitled to one vote for each share held of record on all matters to be voted on by stockholders generally.
Beneficial Ownership Limitation. The selling shareholder is subject to a requirement that it cannot exercise its Series A warrants to the extent that, after giving effect to such exercise, the selling shareholder (together with its affiliates) would beneficially own in excess of 4.99% of the common shares of the Company outstanding immediately after giving effect to such exercise.
99
Anti-Dilution Protection. If the number of outstanding common shares: (i) is increased by a stock dividend payable in common shares; (ii) is increased by a split-up of common shares; (iii) is decreased by a combination of outstanding common shares; or (iv) is reclassified by the issuance of any common shares of the Company then, on the effective date of such event, the exercise price of the warrant will be multiplied by a fraction of which the numerator is (x) the number of common shares outstanding immediately prior to such event and the denominator is (y) the number of common shares outstanding immediately after such event, and the number of common shares issuable upon exercise of the warrant will be proportionately adjusted such that the aggregate exercise price will remain unchanged. Such adjustment will be effective immediately after the record date for the determination of stockholders entitled to receive such dividend or distribution and will be effective immediately after the effective date in the case of a subdivision, combination or re-classification.
In addition, if the Company, at any time while the Series A warrants are outstanding and unexpired, grants, issues or sells any: (i) securities of the Company or its subsidiaries which would entitle the holder thereof to acquire at any time common shares, including, without limitation, any debt, preferred stock, right, option, warrant or other instrument that is at any time convertible into or exercisable or exchangeable for, or otherwise entitles the holder thereof to receive, common shares; or (ii) rights to purchase stock, warrants, securities or other property pro rata to the record holders of any class of common shares (the “Purchase Rights”), then the selling shareholder will be entitled to acquire, upon the terms applicable to such Purchase Rights, the aggregate Purchase Rights which it could have acquired if it had held the number of common shares acquirable upon complete exercise of the Series A warrants immediately before the date on which a record is taken or the record holders are determined for the grant, issuance or sale of such Purchase Rights.
If the Company, at any time while the Series A warrants are outstanding and unexpired, declares or makes any dividend or other distribution of assets to holders of common shares, by way of return of capital or otherwise, at any time after the issuance of the Series A warrants, then the selling shareholder shall be entitled to participate in such distribution to the same extent that it would have participated therein had it held the number of common shares acquirable upon complete exercise of the Series A warrants immediately before the date of which a record is taken or the record holders are determined for such distribution.
Fundamental Transaction. In the event of a “fundamental transaction” then, upon a subsequent exercise of the Series A warrants, the selling shareholder will have the right to purchase and receive the same kind and amount of consideration receivable by the stockholders of the Company in such fundamental transaction. The Company will cause the surviving company in a fundamental transaction to assume the obligations of the Company under the Series A warrants. For purposes of the Series A warrants, a “fundamental transaction” includes, subject to certain exceptions: (i) any reclassification, reorganization or recapitalization of the common shares of the Company; (ii) any merger or consolidation of the Company with or into another corporation; (iii) any sale, lease, license, assignment, transfer, conveyance or other disposition of all or substantially all of the Company’s assets in one or more transactions; (iv) any, direct or indirect, purchase offer, tender offer or exchange offer is completed pursuant to which stockholders are permitted to sell, tender or exchange their shares for other securities, cash or property and has been accepted by the holders of 50% or more of the outstanding common shares of the Company; or (v) the Company, directly or indirectly, in one or more related transactions consummates a stock or share purchase agreement or other business combination with another person whereby such other person acquires more than 50% of the outstanding common shares of the Company.
Amendments. The Series A warrants provide that they may be amended only in writing signed by the Company and such selling shareholder.
100
The following table sets forth information as of April 1, 2022 about the beneficial ownership of our common shares by the selling shareholders both before and immediately after the offering.
We believe, based on the information furnished to us, that the selling shareholders have sole voting and investment power with respect to all of the common shares reported, subject to community property laws where applicable, unless otherwise indicated.
The percent of beneficial ownership for the selling shareholders is based on 14,362,160 common shares issued and outstanding as of April 1, 2022. Unless otherwise stated in the footnotes to the table below and the information incorporated herein by reference, to our knowledge, and based upon information provided by the selling shareholders, none of the selling shareholders has had a material relationship with us other than as a shareholder at any time within the past three years or has ever been one of our officers or directors.
The percentages of common shares owned after completion of the offering are based on the number of common shares that were outstanding as of April 1, 2022, unless otherwise stated in the footnotes to the table below. In addition, the selling shareholders may have sold, transferred or otherwise disposed of some or all of their common shares since the date on which the information reflected herein was provided to us and may in the future sell, transfer or otherwise dispose of some or all of their common shares in private placement transactions exempt from or not subject to the registration requirements of the Securities Act.
Pursuant to Rules 13d-3 and 13d-5 of the Exchange Act, beneficial ownership includes any shares of our common share as to which a shareholder has sole or shared voting power or investment power, and also any shares of our common share which the shareholder has the right to acquire within 60 days, including upon exercise of stock options or other rights to purchase shares of our common share.
The common shares being offered pursuant to this prospectus may be offered for sale from time to time during the period the registration statement of which this prospectus is a part remains effective, by or for the account of the selling shareholders. After the date of effectiveness, the selling shareholders may have sold or transferred, in transactions covered by this prospectus or in transactions exempt from the registration requirements of the Securities Act, some or all of their common shares.
Except as described below, the selling shareholders are neither broker-dealers nor affiliates, as defined in Rule 405, of broker-dealers. The common shares being offered by the selling shareholders pursuant to this prospectus were acquired upon the consummation of the Merger. The common shares issued in the Merger were privately placed under Section 4(a)(2) of the Securities Act and remain subject to certain lock-up restrictions until April 11, 2022. In connection with the acquisition, we entered into a registration rights agreement pursuant to which we agreed to file the registration statement of which this prospectus is a part.
Other information about the selling shareholders may also change over time. Any changed information will be set forth in an amendment to the registration statement or supplement to this prospectus, to the extent required by law.
101
| Share Beneficially Owned Prior to this Offering | Shares Offered by this Prospectus | Share Beneficially Owned After this Offering | ||||||||||||||||||
| Name of Selling Shareholder | Number | Percentage | Number | Percentage | ||||||||||||||||
| Armistice Capital Master Fund, Ltd. | 4,635,871 | 32.3 | % | 4,635,871 | - | * | ||||||||||||||
| Empery Asset Master Ltd | 118,045 | * | 118,045 | - | * | |||||||||||||||
| Empery Tax Efficient, LP | 86,915 | * | 86,915 | - | * | |||||||||||||||
| Empery Tax Efficient III, LP | 239,484 | 1.7 | % | 239,484 | - | * | ||||||||||||||
| Hudson Bay Master Fund Ltd. | 444,444 | 3.1 | % | 444,444 | - | * | ||||||||||||||
| Roth Capital Partners, LLC | 37,330 | * | 37,330 | - | * | |||||||||||||||
| Arcadia Securities LLC | 6,588 | * | 6,588 | - | * | |||||||||||||||
| Dillon Capital, LLC | 10,000 | * | 10,000 | - | * | |||||||||||||||
| TEC Opportunities Fund I LP | 5,000 | * | 5,000 | - | * | |||||||||||||||
| Mark Mays | 33,112 | * | 33,112 | - | * | |||||||||||||||
| Robert Gehl | 22,222 | * | 22,222 | - | * | |||||||||||||||
| Stephen Cook | 22,222 | * | 22,222 | - | * | |||||||||||||||
| Davide Bianchi | 22,222 | * | 22,222 | - | * | |||||||||||||||
| Jeffrey P Roberts | 22,222 | * | 22,222 | - | * | |||||||||||||||
| Espen Lund | 16,000 | * | 16,000 | - | * | |||||||||||||||
| Ulf Harring | 15,778 | * | 15,778 | - | * | |||||||||||||||
| Steven McConnell | 11,111 | * | 11,111 | - | * | |||||||||||||||
| Andrew Reid | 11,111 | * | 11,111 | - | * | |||||||||||||||
| Even Lund | 7,500 | * | 7,500 | - | * | |||||||||||||||
| Warberg WFVIII LP | 5,000 | * | 5,000 | - | * | |||||||||||||||
| Steve Rubinstien | 1,000 | * | 1,000 | - | * | |||||||||||||||
| Fred Goldman | 25,000 | * | 25,000 | - | * | |||||||||||||||
| Richard Molinksy | 3,750 | * | 3,750 | - | * | |||||||||||||||
| Iroquois Master Fund, Ltd. | 35,000 | * | 35,000 | - | * | |||||||||||||||
| Iroquois Capital Investmnet Group, LLC | 15,000 | * | 15,000 | - | * | |||||||||||||||
| Mikhail Guervich | 2,500 | * | 2,500 | - | * | |||||||||||||||
| FirstFire Global Opportunities Fund LLC | 10,000 | * | 10,000 | - | * | |||||||||||||||
| Revere Securities | 10,000 | * | 10,000 | - | * | |||||||||||||||
| RFMF PARTNERS LLC | 6,000 | * | 6,000 | - | * | |||||||||||||||
| RMB WEALTH MANAGEMENT LLC | 2,000 | * | 2,000 | - | * | |||||||||||||||
| HRM WEALTH MANAGEMENT | 2,000 | * | 2,000 | - | * | |||||||||||||||
102
| Share Beneficially Owned Prior to this Offering | Shares Offered by this Prospectus | Share Beneficially Owned After this Offering | ||||||||||||||||||
| Name of Selling Shareholder | Number | Percentage | Number | Percentage | ||||||||||||||||
| PATRICIA WINTER | 61,300 | * | 61,300 | - | * | |||||||||||||||
| ORLANDO SANTA CRUZ R/O IRA AXOS CLEARING CUST | 3,100 | * | 3,100 | - | * | |||||||||||||||
| TIMOTHY C DAVIS | 2,500 | * | 2,500 | - | * | |||||||||||||||
| RBC CAPITAL MARKETS LLC CUSTODIAN THOMAS J GUERIN IRA | 6,700 | * | 6,700 | - | * | |||||||||||||||
| RBC CAPITAL MARKETS LLC CUSTODIAN MATT FOWLES ROTH IRA | 675 | * | 675 | - | * | |||||||||||||||
| RBC CAPITAL MARKETS LLC CUSTODIAN JULIE ZOELLIN IRA | 925 | * | 925 | - | * | |||||||||||||||
| RBC CAPITAL MARKETS LLC CUSTODIAN WILLIAM ZOELLIN IRA | 800 | * | 800 | - | * | |||||||||||||||
| IBH CAPITAL | 2,000 | * | 2,000 | - | * | |||||||||||||||
| NORTH WOODS CAPITAL INC | 900 | * | 900 | - | * | |||||||||||||||
| NORTH WOODS CAPITAL INC | 100 | * | 100 | - | * | |||||||||||||||
| TMB PACIFIC GLOBAL LLC | 2,000 | * | 2,000 | - | * | |||||||||||||||
| TMB PACIFIC GLOBAL LLC | 100 | * | 100 | - | * | |||||||||||||||
| TIMOTHY BECKETT | 100 | * | 100 | - | * | |||||||||||||||
| MARIA BECKETT | 100 | * | 100 | - | * | |||||||||||||||
| MARIA BECKETT | 100 | * | 100 | - | * | |||||||||||||||
| PEREGRINE GLOBAL MGMT LLC | 500 | * | 500 | - | * | |||||||||||||||
| JJL CAPITAL LLC | 100 | * | 100 | - | * | |||||||||||||||
| JOHN C LOWE | 100 | * | 100 | - | * | |||||||||||||||
| RIO NORTE | 1,000 | * | 1,000 | - | * | |||||||||||||||
| RIO NORTE CAPITAL | 100 | * | 100 | - | * | |||||||||||||||
| KENNETH LANDE | 100 | * | 100 | - | * | |||||||||||||||
| MISTHOS INVESTMENT GROUP LLC | 1,000 | * | 1,000 | - | * | |||||||||||||||
| ORCHARD GROUP INC | 1,000 | * | 1,000 | - | * | |||||||||||||||
| BIRD RESOURCES INC | 100 | * | 100 | - | * | |||||||||||||||
| PRO SPORTS MARKETING LLC | 200 | * | 200 | - | * | |||||||||||||||
| JESSE ROGGEN | 200 | * | 200 | - | * | |||||||||||||||
| BELLAGIRL MEDIA LLC | 200 | * | 200 | - | * | |||||||||||||||
| COLPETTO CAPITAL LLC | 500 | * | 500 | - | * | |||||||||||||||
| BJI FINANCIAL GROUP INC | 500 | * | 500 | - | * | |||||||||||||||
| Intracoastal Capital, LLC | 115,000 | * | 115,000 | - | * | |||||||||||||||
| Lind Global Macro Fund, LP | 52,800 | * | 52,800 | - | * | |||||||||||||||
| TOTAL | 6,139,227 | 6,139,227 | 0 | |||||||||||||||||
| * | Less than 1%. | |
| (1) | Includes Nil common shares which the stockholder has the right to acquire within 60 days upon exercise of stock options. |
103
The selling shareholders (the “Selling Shareholders”) of the securities and any of its pledgees, assignees and successors-in-interest may, from time to time, sell any or all of their securities covered hereby on the principal trading market or any other stock exchange, market or trading facility on which the securities are traded or in private transactions. These sales may be at fixed or negotiated prices. The Selling Shareholders may use any one or more of the following methods when selling securities:
| ● | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| ● | block trades in which the broker-dealer will attempt to sell the securities as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
| ● | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| ● | an exchange distribution in accordance with the rules of the applicable exchange; |
| ● | privately negotiated transactions; |
| ● | settlement of short sales; |
| ● | in transactions through broker-dealers that agree with the Selling Shareholders to sell a specified number of such securities at a stipulated price per security; |
| ● | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| ● | a combination of any such methods of sale; or |
| ● | any other method permitted pursuant to applicable law. |
The Selling Shareholders may also sell securities under Rule 144 or any other exemption from registration under the Securities Act of 1933, as amended (the “Securities Act”), if available, rather than under this prospectus.
Broker-dealers engaged by the Selling Shareholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the Selling Shareholders (or, if any broker-dealer acts as agent for the purchaser of securities, from the purchaser) in amounts to be negotiated, but, except as set forth in a supplement to this prospectus, in the case of an agency transaction not in excess of a customary brokerage commission in compliance with FINRA Rule 2121; and in the case of a principal transaction a markup or markdown in compliance with FINRA Rule 2121.
In connection with the sale of the securities or interests therein, the Selling Shareholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the securities in the course of hedging the positions they assume. The Selling Shareholders may also sell securities short and deliver these securities to close out its short positions, or loan or pledge the securities to broker-dealers that in turn may sell these securities. The Selling Shareholders may also enter into option or other transactions with broker-dealers or other financial institutions or create one or more derivative securities which require the delivery to such broker-dealer or other financial institution of securities offered by this prospectus, which securities such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The Selling Shareholders, and any broker-dealers or agents that are involved in selling the securities, may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the securities purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. The Selling Shareholders have informed the Company that it does not have any written or oral agreement or understanding, directly or indirectly, with any person to distribute the securities.
104
The Company is required to pay certain fees and expenses incurred by the Company incident to the registration of the securities.
We agreed to keep this prospectus effective until the earlier of (i) the date on which the securities may be resold by the Selling Shareholders without registration and without regard to any volume or manner-of-sale limitations by reason of Rule 144, without the requirement for the Company to be in compliance with the current public information under Rule 144 under the Securities Act or any other rule of similar effect or (ii) all of the securities have been sold pursuant to this prospectus or Rule 144 under the Securities Act or any other rule of similar effect. The resale securities will be sold only through registered or licensed brokers or dealers if required under applicable state securities laws. In addition, in certain states, the resale securities covered hereby may not be sold unless they have been registered or qualified for sale in the applicable state or an exemption from the registration or qualification requirement is available and is complied with.
Under applicable rules and regulations under the Exchange Act, any person engaged in the distribution of the resale securities may not simultaneously engage in market making activities with respect to the common shares for the applicable restricted period, as defined in Regulation M, prior to the commencement of the distribution. In addition, the Selling Shareholders will be subject to applicable provisions of the Exchange Act and the rules and regulations thereunder, including Regulation M, which may limit the timing of purchases and sales of the common shares by the Selling Shareholders or any other person. We will make copies of this prospectus available to the Selling Shareholders and have informed it of the need to deliver a copy of this prospectus to each purchaser at or prior to the time of the sale (including by compliance with Rule 172 under the Securities Act).
105
WHERE YOU CAN FIND MORE INFORMATION
We are currently subject to the information requirements of the Exchange Act and in accordance therewith file periodic reports, current reports, proxy statements, and other information with the SEC. Our SEC filings are available on the SEC’s website at www.sec.gov. Our website address is www.inmedpharma.com. Information on our website is not incorporated into this prospectus or our other securities filings and is not a part of this prospectus or any prospectus supplement.
We have filed with the SEC a registration statement on Form S-3 under the Securities Act with respect to the securities that may be offered under this prospectus. This prospectus, which forms part of the registration statement, does not contain all of the information in the registration statement. We have omitted certain parts of the registration statement, as permitted by the rules and regulations of the SEC. For further information about us and our securities, please see the registration statement and our other filings with the SEC, including our annual, quarterly, and current reports and proxy statements.
We will provide to each person, including any beneficial owner, to whom a prospectus is delivered, upon a request for such information in writing or by telephone, without charge, a copy of any or all of the information incorporated by reference into this prospectus. Any such request should be directed to:
Corporate Secretary
InMed Pharmaceuticals, Inc.
310-815 W. Hastings Street
Vancouver, BC
V6C 1B4
Canada
+1-604-669-7207
106
INCORPORATION OF CERTAIN DOCUMENTS BY REFERENCE
The SEC allows us to “incorporate by reference” certain information into this prospectus, which means that we can disclose important information about us by referring you to another document filed separately with the SEC. The information incorporated by reference is considered to be a part of this prospectus. Because we are incorporating by reference future filings with the SEC, this prospectus is continually updated and those future filings may modify or supersede some of the information included or incorporated in this prospectus. This means that you must carefully review all of the SEC filings that we incorporate by reference to determine if any of the statements in this prospectus or in any document previously incorporated by reference have been modified or superseded. However, we undertake no obligation to update or revise any statements we make, except as required by law.
This prospectus incorporates by reference the documents listed below and any filings we make with the SEC under Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act of 1934, as amended (the “Exchange Act”) (in each case, other than those documents or the portions of those documents furnished and not filed with the SEC, including information furnished under Item 2.02 or Item 7.01 of Form 8-K and any corresponding information furnished with respect to such Items under Item 9.01 or as an exhibit) prior to the termination of the offering covered by this prospectus and any prospectus supplement:
| ● | the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2021, filed with the SEC on September 24, 2021; |
| ● | the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2021 and December 31, 2021, filed with the SEC on November 10, 2021 and February 14, 2022; |
| ● | the Company’s Current Reports on Form 8-K, filed with the SEC on September 13, 2021, October 1, 2021, October 13, 2021, October 28, 2021, November 3, 2021, November 10, 2021, November 23, 2021, December 20, 2021, December 22, 2021, February 23, 2022, March 18, 2022, and March 22, 2022 (except, in each case, any information, including exhibits, furnished to the SEC pursuant Items 2.02 and 7.01); |
| ● | the description of our common shares in our Registration Statement on Form 8-A filed on November 5, 2020 and any subsequent amendment thereto filed for the purpose of updating such description. |
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus will be deemed to be modified or superseded to the extent that a statement contained in any subsequently filed document which is or is deemed to be incorporated by reference in this prospectus modifies or supersedes that statement. Any statement so modified or superseded will not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
107
Norton Rose Fulbright US LLP, which has acted as our United States counsel in connection with this offering, will pass on certain legal matters with respect to United States federal law in connection with this offering. Norton Rose Fulbright Canada LLP, which has acted as our Canadian counsel in connection with this offering, will pass on certain legal matters with respect to Canadian law in connection with this offering. Any underwriters, dealers, or agents will be advised about other issues relating to any offering by their own legal counsel named in the applicable prospectus supplement.
108
The consolidated financial statements of InMed Pharmaceuticals Inc. as of June 30, 2021 and 2020, and for each of the years in the two-year period ended June 30, 2021 have been incorporated by reference herein in reliance upon the report of KPMG LLP, independent registered public accounting firm, incorporated by reference herein, and upon the authority of said firm as experts in accounting and auditing. The audit report covering the June 30, 2021 consolidated financial statements contains an explanatory paragraph that states that the Company has incurred recurring losses and negative cash flows and has an accumulated deficit that raise substantial doubt about its ability to continue as a going concern. The consolidated financial statements do not include any adjustments that might result from the outcome of that uncertainty.
109
6,139,727 Common Shares
PROSPECTUS
[●], 2022
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 14. Other Expenses of Issuance and Distribution.
Set forth below is an estimate (except in the case of the registration fee) of the amount of fees and expenses to be incurred in connection with the issuance and distribution of the offered securities, other than underwriting discounts and commissions. The selling shareholders will not bear any portion of the below expenses.
| SEC Registration fee | $ | 843 | ||
| Printing expenses | * | |||
| Legal fees and expenses | * | |||
| Accounting fees and expenses | * | |||
| Transfer agent’s fees and expenses | * | |||
| Miscellaneous fees and expenses | * | |||
| Total | * |
| * | These fees are calculated based on the number of issuances and amount of securities offered and accordingly cannot be estimated at this time. |
II-1
Item 15. Indemnification of Directors and Officers.
We are subject to the provisions of Part 5, Division 5 of the Business Corporations Act (British Columbia), or “BCBCA”. Under Section 160 of the BCBCA, we may, subject to
Section 163 of the BCBCA:
| 1. | indemnify an individual who: |
| ● | is or was a director or officer of our company; |
| ● | is or was a director or officer of another corporation (i) at a time when such corporation is or was an affiliate of our company; or (ii) at our request, or |
| ● | at our request, is or was, or holds or held a position equivalent to that of, a director or officer of a partnership, trust, joint venture or other unincorporated entity, and including, subject to certain limited exceptions, the heirs and personal or other legal representatives of that individual (collectively, an “eligible party”), against all eligible penalties to which the eligible party is or may be liable; and |
| 2. | after final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by an eligible party in respect of that proceeding, where: |
| ● | “eligible penalty” means a judgment, penalty or fine awarded or imposed in, or an amount paid in settlement of, and eligible proceeding. |
| ● | “eligible proceeding” means a proceeding in which an eligible party or any of the heirs and personal or other legal representatives of the eligible party, by reason of the eligible party being or having been a director or officer of, or holding or having held a position equivalent to that of a director or officer of, our company or an associated corporation (a) is or may be joined as a party, or (b) is or may be liable for or in respect of a judgment, penalty or fine in, or expenses related to, the proceeding. |
| ● | “proceeding” includes any legal proceeding or investigative action, whether current, threatened, pending or completed. Under Section 161 of the BCBCA, and subject to Section 163 of the BCBCA, we must, after the final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by an eligible party in respect of that proceeding if the eligible party (a) has not been reimbursed for those expenses, and (b) is wholly successful, on the merits or otherwise, in the outcome of the proceeding or is substantially successful on the merits in the outcome of the proceeding. |
Under Section 162 of the BCBCA, and subject to Section 163 of the BCBCA, we may pay, as they are incurred in advance of the final disposition of an eligible proceeding, the expenses actually and reasonably incurred by an eligible party in respect of the proceeding, provided that we must not make such payments unless we first receive from the eligible party a written undertaking that, if it is ultimately determined that the payment of expenses is prohibited under Section 163 of the BCBCA, the eligible party will repay the amounts advanced.
Under Section 163 of the BCBCA, we must not indemnify an eligible party against eligible penalties to which the eligible party is or may be liable or pay the expenses of an eligible party in respect of that proceeding under Sections 160, 161 or 162 of the BCBCA, as the case may be, if any of the following circumstances apply:
| ● | if the indemnity or payment is made under an earlier agreement to indemnify or pay expenses and, at the time that the agreement to indemnify or pay expenses was made, we were prohibited from giving the indemnity or paying the expenses by our memorandum or articles; |
| ● | if the indemnity or payment is made otherwise than under an earlier agreement to indemnify or pay expenses and, at the time that the indemnity or payment is made, we are prohibited from giving the indemnity or paying the expenses by our memorandum or articles; |
II-2
| ● | if, in relation to the subject matter of the eligible proceeding, the eligible party did not act honestly and in good faith with a view to the best interests of our company or the associated corporation, as the case may be; or |
| ● | in the case of an eligible proceeding other than a civil proceeding, if the eligible party did not have reasonable grounds for believing that the eligible party’s conduct in respect of which the proceeding was brought was lawful. |
If an eligible proceeding is brought against an eligible party by or on behalf of our company or by or on behalf of an associated corporation, we must not either indemnify the eligible party against eligible penalties to which the eligible party is or may be liable, or pay the expenses of the eligible party under Sections 160, 161 or 162 of the BCBCA, as the case may be, in respect of the proceeding.
Under Section 164 of the BCBCA, and despite any other provision of Part 5, Division 5 of the BCBCA and whether or not payment of expenses or indemnification has been sought, authorized or declined under Part 5, Division 5 of the BCBCA, on application of our company or an eligible party, the Supreme Court of British Columbia may do one or more of the following:
| ● | order us to indemnify an eligible party against any liability incurred by the eligible party in respect of an eligible proceeding; |
| ● | order us to pay some or all of the expenses incurred by an eligible party in respect of an eligible proceeding; |
| ● | order the enforcement of, or payment under, an agreement of indemnification entered into by us; |
| ● | order us to pay some or all of the expenses actually and reasonably incurred by any person in obtaining an order under Section 164 of the BCBCA; or |
| ● | make any other order the court considers appropriate. |
Section 165 of the BCBCA provides that we may purchase and maintain insurance for the benefit of an eligible party or the heirs and personal or other legal representatives of the eligible party against any liability that may be incurred by reason of the eligible party being or having been a director or officer of, or holding or having held a position equivalent to that of a director or officer of, our company or an associated corporation.
Under our articles, and subject to the BCBCA, we must indemnify our directors, former directors or alternate directors and his or her heirs and legal personal representatives against all eligible penalties to which such person is or may be liable, and we must, after the final disposition of an eligible proceeding, pay the expenses actually and reasonably incurred by such person in respect of that proceeding. Each director and alternate director is deemed to have contracted with our company on the terms of the indemnity contained in our articles.
Under our articles, and subject to the BCBCA, we may agree to indemnify and may indemnify any person (including an eligible party) against eligible penalties and pay expenses incurred in connection with the performance of services by that person for us. We have entered into indemnity agreements with our directors and certain of our officers.
Pursuant to our articles, the failure of an eligible party to comply with the BCBCA or our articles does not, of itself, invalidate any indemnity to which he or she is entitled under our articles.
Under our articles, we may purchase and maintain insurance for the benefit of any person (or his or her heirs or legal personal representatives) who:
| ● | is or was our director, alternate director, officer, employee or agent; |
| ● | is or was a director, alternate director, officer, employee or agent of a corporation at a time when the corporation is or was our affiliate; |
| ● | at our request, is or was a director, alternate director, officer, employee or agent of a corporation or of a partnership, trust, joint venture or other unincorporated entity; or |
| ● | at our request, holds or held a position equivalent to that of a director, alternate director or officer of a partnership, trust, joint venture or other unincorporated entity; |
| ● | against any liability incurred by him or her as such director, alternate director, officer, employee or agent or person who holds or held such equivalent position. |
In addition, we have entered into an indemnification agreement with each of our directors and our Chief Financial Officer, which requires us to indemnify them.
II-3
ITEM 16. Exhibits.
(a) Exhibits
The following is a list of exhibits filed as part of this registration statement.
| * | To be filed as an Exhibit to a Current Report on Form 8-K or in a post-effective amendment to this registration statement. |
| ** | Filed herewith. |
| ^ | Portions of this exhibit have been omitted pursuant to Rule 601(b)(10) of Regulation S-K. |
II-4
(b) Financial Statement
Schedules None
ITEM 17. Undertakings.
(a) The undersigned registrant hereby undertakes:
| (i) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (1) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933; |
| (2) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and |
| (3) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement. |
provided, however, that the undertakings set forth in paragraphs (a)(i)(1), (a)(i)(2) and (a)(i)(3) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in the registration statement or is contained in a form of prospectus filed pursuant to Rule 424(b) that is a part of the registration statement.
| (ii) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (iii) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (iv) | That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser: |
| (a) | Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and |
II-5
| (b) | Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date. |
| (b) | The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the registrant’s annual report pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual or transition report pursuant to Section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (c) | The undersigned registrant hereby undertakes that: (i) for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of the registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of the registration statement as of the time it was declared effective; and (ii) for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (d) | Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is therefore unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933, and will be governed by the final adjudication of such issue. |
II-6
Pursuant to the requirements of the Securities Act, InMed Pharmaceuticals Inc., certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-3 and has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Vancouver, British Columbia, Canada, on April 7, 2022.
| INMED PHARMACEUTICALS INC. | ||
| By: | /s/ Eric A. Adams | |
| Eric A. Adams | ||
| President and Chief Executive Officer | ||
Signature Page to S-3
II-7
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below, the undersigned officers and directors of InMed Pharmaceuticals Inc., hereby severally constitute and appoint Eric A. Adams and Brenda Edwards, and each of them singly (with full power to each of them to act alone), his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution in each of them for him and in his name, place and stead, and in any and all capacities, to sign any and all amendments (including post-effective amendments) to this Registration Statement on Form S-3 and any registration statement for the same offering filed pursuant to Rule 462 under the Securities Act, as amended (the “Securities Act”), and to file the same, with all exhibits thereto and other documents in connection therewith, with the Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act, this Registration Statement has been signed by the following persons in the capacities and on April 7, 2022.
| Name | Title | |
/s/ Eric A. Adams | President, Chief Executive Officer and Director | |
| Eric A. Adams | (principal executive officer) | |
/s/ Brenda Edwards | Interim Chief Financial Officer | |
| Brenda Edwards | (principal financial officer and principal accounting officer) | |
| /s/ William J. Garner | Director | |
| William J. Garner | ||
/s/ Janet Grove | Director | |
| Janet Grove | ||
| /s/ Adam Cutler | Director | |
| Adam Cutler | ||
/s/ Andrew Hull | Director | |
| Andrew Hull | ||
Signature Page to S-3
II-8
AUTHORIZED U.S. REPRESENTATIVE
Pursuant to the requirements to Section 6(a) of the Securities Act of 1933, the undersigned has signed this registration statement solely in the capacity of the duly authorized representative of InMed Pharmaceuticals Inc. in the United States on April 7, 2022.
| BayMedica, LLC | ||
| By: | /s/ Eric A. Adams | |
| Eric A. Adams | ||
| Director | ||
Authorized U.S. Representative Signature Page to S-3
II-9