| PROSPECTUS | Filed Pursuant to Rule 424(b)(3) Registration No. 333-232510 |

APTORUM GROUP LIMITED
27,765,821 Class A Ordinary Shares
This prospectus relates to the resale from time to time of up to 27,765,821 Class A Ordinary Shares of Aptorum Group Limited by the selling shareholders identified in this prospectus under the caption “Selling Shareholders,” including their transferees, pledgees or donees or their respective successors. We are registering these shares on behalf of the selling shareholders, to be offered and sold by them from time to time. We will not receive any proceeds from the sale of the shares offered by this prospectus.
We have agreed to bear all of the expenses incurred in connection with the registration of these shares. The selling shareholders will pay or assume discounts, commissions, fees of underwriters, selling brokers or dealer managers and similar expenses, if any, incurred for the sale of Class A Ordinary Shares.
The selling shareholders identified in this prospectus, or their respective transferees, pledgees, donees or other successors-in-interest, may sell these shares from time to time in the principal market on which our Class A Ordinary Shares are traded at the prevailing market price, in negotiated transactions, or through any other means described in the section titled “Plan of Distribution.” For additional information on the methods of sale that may be used by the selling shareholders, see the section entitled “Plan of Distribution” on page 136. For a list of the selling shareholders, see the section entitled “Selling Shareholders” on page 134.
We may amend or supplement this prospectus from time to time by filing amendments or supplements as required. You should read the entire prospectus and any amendments or supplements carefully before you make your investment decision.
Our Class A Ordinary Shares are traded on the Nasdaq Global Market under the symbol “APM.” On October 29, 2019, the closing sale price of our Class A Ordinary Shares on the Nasdaq Global Market was $15.73 per share. You are urged to obtain current market quotations for our Class A Ordinary Shares.
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012 and will therefore be subject to reduced public company reporting requirements.
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 13 for a discussion of information that should be considered in connection with an investment in our securities.
Neither the SEC nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
This prospectus does not constitute, and there will not be, an offering of securities to the public in the Cayman Islands.
Prospectus dated October 31, 2019
TABLE OF CONTENTS
i
We have not authorized any person to provide you with information different from that contained in this prospectus or any related free-writing prospectus that we authorize to be distributed to you. This prospectus is not an offer to sell, nor is it seeking an offer to buy, these securities in any jurisdiction where the offer or sale is not permitted. The information in this prospectus speaks only as of the date of this prospectus unless the information specifically indicates that another date applies, regardless of the time of delivery of this prospectus or of any sale of the securities offered hereby.
For investors outside of the United States: We have not done anything that would permit this Offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than the United States. Persons outside of the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the Offering and the distribution of this prospectus outside of the United States.
This prospectus includes statistical and other industry and market data that we obtained from industry publications and research, surveys and studies conducted by third parties. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe these industry publications and third-party research, surveys and studies are reliable, you are cautioned not to give undue weight to this information.
All references in this prospectus to “$,” “U.S.$,” “U.S. dollars,” “dollars,” “US$,” and “USD” mean United States dollars unless otherwise noted. All references to the “PRC” or “China” in this prospectus refer to the People’s Republic of China. All references to “Hong Kong” or “H.K.” in this prospectus refer to Hong Kong Special Administrative Region of the People’s Republic of China. All references to the “United States,” “U.S.” or “US” refer to the United States of America.
| ● | “Acticule” refers to Acticule Life Sciences Limited, an 80% owned subsidiary of Aptorum Group. |
| ● | “Aeneas” refers to AENEAS CAPITAL LIMITED, a wholly-owned subsidiary of Aeneas Group Limited, which is an indirect wholly-owned subsidiary of Jurchen Investment Corporation through Aeneas Limited. Because Mr. Huen, our CEO, holds 100% equity interest in Jurchen Investment Corporation, we refer to Aeneas as a fellow subsidiary of Aptorum Group. |
| ● | “AGL” refers to Aeneas Group Limited, a wholly-owned subsidiary of Aeneas Limited and we refer to AGL as a fellow subsidiary of Aptorum Group. |
| ● | “AL” refers to Aeneas Limited, an entity 80% owned by Jurchen Investment Corporation and we refer to AL as a fellow subsidiary of Aptorum Group. |
| ● | “AML” refers to Aptorum Medical Limited, a 94% owned-subsidiary of Aptorum Group. |
| ● | “AML Clinic” refers to an outpatient medical clinic operated by AML under the name of Talem Medical. |
| ● | “APD” refers to Aptorum Pharmaceutical Development Limited, a wholly-owned subsidiary of Aptorum Group. |
| ● | “Aptorum Group,” “Company,” “we,” “Group” and “us” refer to Aptorum Group Limited, a Cayman Islands exempted company with limited liability whose principal place of business is in Hong Kong. |
ii
| ● | “Aptorum Non-Therapeutics Group” refers to the Company’s non-therapeutics segment that encompasses: (i) the development of surgical robotics and medical devices, which is operated through Signate Life Sciences Limited and (ii) AML Clinic. |
| ● | “Aptorum Therapeutics Group” refers to the Company’s therapeutics segment that is operated through its wholly-owned subsidiary, Aptorum Therapeutics Limited, a Cayman Islands exempted company with limited liability, whose principal place of business is in Hong Kong and its indirect subsidiary companies, whose principal places of business are in Hong Kong. |
| ● | “Bond” refers to the $15,000,000 convertible bond the Company originally issued to Peace Range (as hereinafter defined) in the Bond Offering, but which has since been repurchased by one of the Company’s wholly owned subsidiary, Aptorum Investment Holding Limited, pursuant to that certain Bond Repurchase Agreement dated April 24, 2019 between the Company, Peace Range Limited and Aptorum Investment Holding Limited. |
| ● | “Bond Offering” refers to the Company’s private offering of the Bond that closed on April 25, 2018. |
| ● | “Boustead” refers to Boustead Securities, LLC. |
| ● | “cGCP” refers to Current Good Clinical Practice as adopted by the applicable regulatory authority. |
| ● | “cGLP” refers to Current Good Laboratory Practice as adopted by the applicable regulatory authority. |
| ● | “cGMP” refers to Current Good Manufacturing Practice as adopted by the applicable regulatory authority. |
| ● | “Class A Ordinary Shares,” “Ordinary Shares,” or “Shares” refers to the Company’s Class A Ordinary Shares, par value $1.00 per share. |
| ● | “CMC” refers to chemical, manufacturing and control. |
| ● | “Covar” refers to Covar Pharmaceuticals Incorporated, a contract research organization engaged by the Company. |
| ● | “CROs” refers to contract research organizations. |
| ● | “EMA” refers to the European Medicines Agency. |
| ● | “EMEA” refers to Europe, the Middle East and Africa. |
| ● | “EPO” refers to the European Patent Organization or the European Patent Office operated by it. |
| ● | “European Patent” refers to patents issuable by the EPO. |
| ● | “Exchange Act” refers to the U.S. Securities Exchange Act of 1934, as amended. |
| ● | “FDA” refers to U.S. Food and Drug Administration. |
| ● | “FDCA” refers to the U.S. Federal Food, Drug and Cosmetic Act. |
| ● | “HKD” refers to Hong Kong Dollars. |
| ● | “Hong Kong” or “H.K.” refers to Hong Kong Special Administrative Region of the People’s Republic of China. |
| ● | “Hong Kong Doctors” refers to the doctors in Hong Kong under the employment of AML Clinic. |
iii
| ● | “IND” refers to Investigational New Drugs. |
| ● | “IP” refers to intellectual property. |
| ● | “IPO” means the initial public offering by the Company of 761,419 Class A Ordinary Shares consummated on December 17, 2018. |
| ● | “Jurchen” refers to Jurchen Investment Corporation, a company wholly-owned by our CEO, Ian Huen, and a holding company of Aptorum Group. |
| ● | “Lead Projects” refers to three of the Company’s therapeutic projects ALS-1, ALS-4 and NLS-1. |
| ● | “Major Patent Jurisdictions” refers to the United States, member states of the European Patent Organization and the People’s Republic of China. |
| ● | “Nativus” refers to Nativus Life Sciences Limited, a wholly-owned subsidiary of Aptorum Group. |
| ● | “NMPA” refers to China’s National Medical Products Administration and its predecessor, the China Food and Drug Administration. |
| ● | “NDA” refers to a New Drug Application issued by the FDA. |
| ● | “Offering” refers to the resale of the Ordinary Shares offered by the Selling Shareholders included herein. |
| ● | “PRC” and “China” refer to the People’s Republic of China. |
| ● | “Restructure” refers to the Company’s change from an investment fund with management shares and non-voting participating redeemable preference shares to a holding company with operating subsidiaries, effective as of March 1, 2017. |
| ● | “R&D” refers to research and development. |
| ● | “R&D Center” refers to an in-house pharmaceutical development center operated by APD. |
| ● | “Securities Exchange Commission,” “SEC,” “Commission” or similar terms refer to the Securities Exchange Commission. |
| ● | “Sarbanes-Oxley Act” refers to the Sarbanes-Oxley Act of 2002. |
| ● | “Securities Act” refers to the U.S. Securities Act of 1933, as amended. |
| ● | “Selling Shareholders” refers to our pre-existing shareholders who are selling their Class A Ordinary Shares pursuant to the F-1. |
| ● | “Series A Notes” refers to Series A convertible notes, at a purchase price of $10,000 per note, sold in the Series A Note Offering. |
| ● | “Series A Note Investors” refers to the investors who purchased Series A Notes. |
| ● | “Series A Note Offering” refers to the private offering of Series A Notes, pursuant to Regulation S or Regulation D, as promulgated under the Securities Act that closed on May 15, 2018. |
| ● | “Signate” refers to Signate Life Sciences Limited, a wholly-owned subsidiary of Aptorum Group. |
| ● | “UK” refers to the United Kingdom. |
| ● | “Underwriter Warrants” refers to warrants issued to the underwriters of the IPO which have now been fully exercised on a cashless basis. |
| ● | “United States,” “U.S.” and “US” refer to the United States of America. |
| ● | “Videns” refers to Videns Incorporation Limited, a wholly-owned subsidiary of Aptorum Group. |
| ● | “$,” “U.S. $,” “U.S. dollars,” “dollars,” “US$” and “USD” refer to the United States dollars. |
iv
This summary highlights information contained elsewhere in this prospectus and does not contain all of the information that you should consider in making your investment decision. Before investing in our Class A Ordinary Shares, you should carefully read the entire prospectus, including our financial statements and the related notes included elsewhere in this prospectus. You should also consider, among other things, the matters described under “Risk Factors” and “Management’s Discussion and Analysis” in each case appearing elsewhere in this prospectus. Unless otherwise stated, all references to “us,” “our,” “Aptorum,” “we,” the “Company,” the “group” and similar designations refer to Aptorum Group Limited, a Cayman Islands exempted company with limited liability,
Overview
We are a pharmaceutical company currently in the preclinical stage, dedicated to developing and commercializing a broad range of therapeutic and diagnostic technologies to tackle unmet medical needs. We have obtained exclusive licenses for our technologies. In addition, we are also developing certain proprietary technologies as product candidates. We are pursuing therapeutic and diagnostic projects (including projects seeking to use extracts or derivatives from natural substances to treat diseases) in neurology, infectious diseases, gastroenterology, oncology and other disease areas. We also have projects focused on surgical robotics. (See “Our Business – Lead Projects and Other Projects under Development – Lead Projects”) Also, we opened a medical clinic, AML Clinic, in June 2018.
Although none of our drug or device candidates has yet been approved for testing in humans, our goal is to develop a broad range of early stage novel therapeutics and diagnostics across a wide range of disease/therapeutic areas. Key components of our strategy for achieving this goal include: (for details of our strategy, See “Our Business – Our Strategy”)
| ● | Developing therapeutic and diagnostic innovations across a wide range of disease/therapeutic areas; | |
| ● | Selectively expanding our portfolio with potential products that may be able to attain orphan drug designation and/or satisfy current unmet medical needs; | |
| ● | Collaborating with leading academic institutions and CROs; | |
| ● | Expanding our in-house pharmaceutical development center; | |
| ● | Leveraging our management’s expertise, experience and commercial networks; | |
| ● | Strategically developing opportunities in Hong Kong to promote access to the PRC market; and | |
| ● | Obtaining and leveraging government grants to fund project development. |
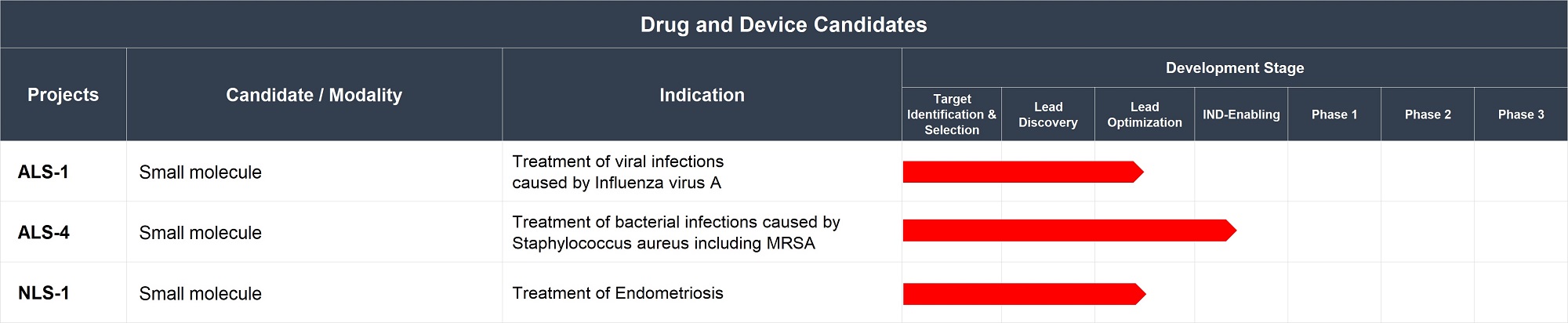
We have devoted a portion of the proceeds from our IPO, to three therapeutic projects (“Lead Projects”). The drug candidates being advanced as the Lead Projects are ALS-1, ALS-4 and NLS-1, described in further detail below. If the results of the remaining preclinical studies of these drug candidates are positive, we expect to be able to submit by 2020 or 2021 an Investigational New Drug Application (“IND”) for at least one of these candidates to the U.S. Food and Drug Administration (“FDA”) or an equivalent application to the regulatory authorities in one or more other jurisdictions such as the China’s National Medical Products Administration (“NMPA”) and/or the European Medicines Agency (“EMA”). Acceptance of these applications by the relevant regulatory authority would enable the Company to begin testing that drug candidate in humans in that jurisdiction. Our ability to obtain any approval of such applications is entirely dependent upon the results of our preclinical studies, none of which have yet been completed.
1
Our current business consists of “therapeutics” and “non-therapeutics” segments. However, our focus is on the therapeutics segments. Because of the risks, costs and extended development time required for successful drug development, we have determined to pursue projects within our non-therapeutics segments, such as AML Clinic, to provide some interim revenue and medical robots that may be brought to market and generate revenue more quickly.
Therapeutics Segment. In our therapeutics segment (“Aptorum Therapeutics Group”), we are currently seeking to develop various drug molecules (including projects seeking to use extracts or derivatives from natural substances to treat diseases) and certain technologies for the treatment (“therapeutics”) and diagnosis (“diagnostics”) of human disease conditions in neurology, infectious diseases, gastroenterology, oncology and other disease areas. In addition, we are seeking to identify additional prospects which may qualify for potential orphan drug designation (e.g., rare types of cancer) or which address other current unmet medical needs. Aptorum Therapeutics Group is operated through Aptorum’s wholly-owned subsidiary, Aptorum Therapeutics Limited, a Cayman Islands exempted company with limited liability, whose principal place of business is in Hong Kong and its indirect subsidiary companies (who we sometimes refer to herein as project companies), whose principal places of business are also in Hong Kong.
Non-Therapeutics Segment. The non-therapeutics segment (“Aptorum Non-Therapeutics Group”) encompasses two businesses: (i) the development of surgical robotics and medical devices and (ii) AML Clinic. The development of surgical robotics and medical devices business is operated through Signate Life Sciences Limited, a subsidiary of Aptorum Therapeutics Limited. The outpatient clinic is operated through our subsidiary, Aptorum Medical Limited. Effective as of March 2018, we leased office space in Central, Hong Kong as the home to AML Clinic. AML Clinic commenced operations under the name of Talem Medical in June 2018. The estimated general administrative expenses and other operating expenses of the AML Clinic is expected to be no more than USD120,000 per month. The clinic is expected to reach operating profit in 18 months from the clinic reaching its full operating capacity upon (i) the successful recruitment of a minimum of six full time physicians (AML Clinic currently has one full time physician and six part time physicians) and (ii) establishing steady patients flow via brand development. (See “Our Business – Lead Projects and Other Projects under Development – Other Projects under Development – Aptorum Medical Limited - AML Clinic”)
The Company has already obtained opportunities resulting in our existing licensing agreements from various contractual relationships that we have entered into, including service/consulting agreements with some of the world’s leading specialists and clinicians in our areas of interest, with academic institutions and organizations, and with CROs. We anticipate that these relationships will generate additional licensing opportunities in the future. In addition, we have established and are continuing to expand our in-house research facilities (collectively, the “R&D Center”) to develop some of our drug and device candidates internally and to collaborate with third-party researchers.
Prior to March 2017, the Company had pursued passive healthcare related investments in early stage companies primarily in the United States. However, we have since ceased pursuing further passive investment operations and intend to exit all such portfolio investments over an appropriate timeframe to focus resources on our current business.
Aptorum’s Lead Projects
Based on our evaluation of preliminary data and our consideration of a number of factors including substantial unmet needs, benefits over existing therapies, potential market size, competition in market, the Company have decided to prioritize our resources in developing our three Lead Projects, namely, ALS-1, ALS-4 and NLS-1, among all our projects under development. Overall, our rationale for selecting Lead Projects was not based on any mechanical formula or rigid selection criteria, but instead focused on a combination of the factors and individual attributes of the Lead Projects themselves.
2
For the definition of different stages of development, such as Target Identification & Selection, Lead Discovery, Lead Optimization, etc., please refer to page 72.
ALS-1: Small molecule intended for the treatment of viral infections caused by Influenza virus A
Professor Richard Kao (Inventor of ALS-1, Founder and Principal Investigator of Acticule) was the first to identify nucleoprotein (“NP”) as an effective drug target (Nature Biotechnology. 28:600-605) for the treatment of viral infections caused by Influenza virus A. It is hypothesized that Influenza A NP is an essential protein for the proliferation of the influenza virus. ALS-1 is a novel small drug molecule which targets viral NP and triggers the aggregation of NP, which prevents the aggregated NP from entering the nucleus. ALS-1 is designed to target a broad range of NP variants. We are exploring ALS-1 as a potential treatment for viral infections caused by Influenza virus A. It is currently at the Lead Optimization Stage to optimize its drug-like properties.
Market size of target indication
Influenza can cause severe illness or death especially in people with high risk. Globally, the annual epidemics are estimated to result in about 3 to 5 million cases of severe influenza infections, causing about 290,000 to 650,000 deaths each year1. The market for influenza drugs is huge, the total influenza therapeutics market is expected to swell to US$1.2 billion globally by 2025, from US$600 million in 20162. Specifically, ALS-1 is a drug that targets Influenza virus A, a highly contagious respiratory illness caused by infection with virus. Around 50%-80% of influenza infections are type A3.
Significant unmet medical needs and benefits over existing treatments
The emergence of antiviral drug resistance in influenza virus is a major concern for treatment of same. ALS-1 targets a broad range of NP variants. As NP is essential to the replication of influenza virus, by disrupting NP, ALS-1 aims to suppress viral replication. Compared with the currently marketed antiviral drugs for which the viruses have acquired extensive resistance, ALS-1 acts on a completely different therapeutic target.
| 1 | World Health Organization, “Influenza (Seasonal)”, World Health Organization, 31 January 2018, http://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) |
| 2 | Bloomberg News, “New Drugs Are Coming to Fight Nasty Flu”, February 9, 2018, https://www.bloomberg.com/news/articles/2018-02-08/flu-relief-is-coming-as-successors-to-aging-tamiflu-near-market |
| 3 | World Health Organization, “Global circulation of influenza viruses”, Influenza Laboratory Surveillance Information generated on 28/08/2018 21:28:47 UTC by the Global Influenza Surveillance and Response System (GISRS), http://apps.who.int/flumart/Default?ReportNo=6 |
ALS-4: Small molecule for the treatment of bacterial infections caused by Staphylococcus aureus including Methicillin-resistant Staphylococcus aureus (“MRSA”)
ALS-4 is a small drug molecule which appears to target the products produced by bacterial genes that facilitate the successful colonization and survival of the bacterium in the body or that cause damage to the body’s systems. These products of bacterial genes are referred to as “virulence expression”. Targeting bacterial virulence is an alternative approach to antimicrobial therapy that offers promising opportunities to overcome the emergence and increasing prevalence of antibiotic-resistant bacteria.
3
ALS-4 is directed to a novel drug target, an enzyme essential for Staphylococcus aureus (including MRSA) survival in vivo. We believe that the product of this enzyme promotes Staphylococcus aureus (including MRSA) survival by shielding the bacteria from the attack by the immune system. ALS-4 may have particular value if it can be shown to be an effective therapy in situations where a Staphylococcus aureus infection is resistant to available antibiotics (i.e., where the pathogen is MRSA).
ALS-4 is at present under active development for the treatment of bacterial infections caused by Staphylococcus aureus including MRSA. It is currently at the IND enabling stage to optimize its drug-like properties.The development of ALS-4 candidate has been progressing well and the first series of GLP toxicology studies have been completed through an appointed North American based contract research organization (CRO). In particular, ALS-4 candidate did not show any mutagenicity in the in vitro Ames tests. ALS-4 development is on our proposed track and we target submitting the related IND in the first half year of 2020 and a hybrid Phase 1 clinical study is currently planned in North America with both healthy volunteers and patients to obtain preliminary efficacy readout.
Market size of target indication
Staphylococcus aureus is a commensal bacterium, meaning that it infects about 30% of the human population without causing symptoms or harm. A study shows that as many as 53 million people worldwide carry MRSA, which is one of the most commonly identified antibiotic-resistant pathogens.4 In its symptomatic form, it is one of the five most common causes of hospital-acquired infections and is often the cause of wound infections following surgery. For example, in the U.S. alone, approximately 126,000 hospitalizations are due to MRSA yearly, where severe MRSA infections occur in approximately 94,000 people each year and are associated with approximately 19,000 deaths5. Global MRSA drugs market generated US$2.97 billion of revenue in globally 2016, and the global market for MRSA drugs is estimated to reach US$3.91 billion by the end of 20256.
Significant unmet medical needs and benefits over existing treatments
Staphylococcus aureus commonly causes skin infections including abscesses, respiratory infections such as sinusitis, and food poisoning.Existing treatments forStaphylococcus aureus consist of antibiotics administered to kill the bacteria. However, MRSA is a type of Staphylococcus aureus that causes particularly difficult-to-treat infections in humans. Nicknamed to be a “Super Bug,” MRSA is a major hospital acquired pathogen that causes severe morbidity and mortality worldwide. MRSA has developed resistance to many common antibiotics that once destroyed it. It is now resistant to methicillin, amoxicillin, penicillin, oxacillin, and many other common antibiotics and may overtime develops resistance to other new antibiotics.
ALS-4 does not work as a typical antibiotic but instead targets virulence expression without direct bactericidal properties to kill the bacteria. It presents an alternative treatment to antimicrobial therapy and offers promising opportunities to overcome the emergence and increasing prevalence of antibiotic-resistant bacteria. Specifically, ALS-4 appears to inhibit the production of staphyloxanthin (i.e., a virulence factor that would escape from the host immune system) without killing the bacteria, and thereby enabling the immune system to clear MRSA, which may provide a novel treatment forStaphylococcus aureus infections and MRSA.
| 4 | Roche, “Roche’s Annual Report 2017”, https://www.roche.com/dam/jcr:78519d71-10af-4e02-b490-7b4648a5edb8/en/ar17e.pdf |
| 5 | Charles Patrick Davis, Melissa Conrad Stöppler, “MRSA”, November 9, 2017, eMedicineHealth, https://www.emedicinehealth.com/mrsa_infection/article_em.htm#how_common_is_mrsa |
| 6 | Market Research Reports Search Engine (MRRSE), “Global Methicillin-resistant Staphylococcus Aureus (MRSA) Drugs Market Analysis and Forecast Predictions”, HEALTHCAREDIVE, https://www.healthcaredive.com/press-release/20180405-global-methicillin-resistant-staphylococcus-aureus-mrsa-drugs-market-anal/ |
4
NLS-1: A Derivative of Epigallocatechin-3-Gallate (“Pro-EGCG”) for the treatment of Endometriosis
NLS-1, a drug molecule derived from natural products (green tea), is currently under development for the treatment of endometriosis, a disease in which the tissue that normally lines the uterus (endometrium) grows outside the uterus. It can grow on the ovaries, fallopian tubes, bowels, or bladder. Rarely, it grows in other parts of the body. Many studies have assessed the applications of EGCG, a naturally occurring molecule extracted from green tea, for the treatment of endometriosis in vitro and in animal models. (Hum Reprod. 2014 29(8):1677; Hum Reprod. 2013 28(1):178; Fertil Steril. 2011 96(4):1021). For example, in a mouse model, Ricci et al (Hum Reprod. 2013 28(1):178) demonstrated that EGCG brought a statistically significant reduction in the mean number and the volume of established lesions compared with the control group without treatment. The treatment diminished cell proliferation in a statistically significant manner, reduced vascular density and increased apoptosis within the lesions. EGCG induced reduction in human EEC proliferation and increased apoptosis in primary cultures. Matsuzaki and Darcha (Hum Reprod. 2014 29(8):1677) also showed that EGCG prevented the progression of fibrosis in endometriosis in an animal model.
However, the attractiveness of EGCG as a drug candidate has been diminished by its chemical and metabolic instability (Hum Reprod. 2014 29(8):1677; Angiogenesis. 2013 16(1):59). The Company’s drug candidate, NLS-1 is supposed to overcome these challenges. NLS-1 is an EGCG derivative synthesized by acetylation of the reactive hydroxyl groups, which appears to prevent generation of reactive phenoxide anions and radicals for dimerization and metabolism, thereby overcoming the chemical and metabolic instability of EGCG.
NLS-1 is under active development for the treatment of endometriosis. It is currently under lead optimization to optimize its drug-like properties.
Market size of target indication
Endometriosis affects an estimated approximately 176 million women in the world (approximately 1 in 10 women during their reproductive years)7. It is estimated that 30-40% of women with endometriosis are subject to risk of infertility and may develop complications during pregnancy8,9. The market for the treatment of endometriosis across the seven major countries (U.S., France, Germany, Italy, Spain, the U.K., and Japan), is approximately $1.72 billion in 2015. It is expected to grow to just over $2 billion across the seven major countries by 202510.
Significant unmet medical needs and benefits over existing treatments
Endometriosis is a condition where tissue that normally lines inside of uterus grows outside of it, and would often cause pelvic pain and infertility to the patient. At present, endometriosis is usually treated with hormonal therapy (including Gonadotropin-releasing hormone), which is a non-invasive therapy toslow growth of endometrial tissue growth and prevent new implants of endometrial tissue.However, hormone-based therapy often causes adverse side effect, such as menopausal symptoms, infertility, bone density loss, higher risk of osteoporosis, mood swings, hair loss, etc., and is not a permanent cure as the symptoms may return after stopping of treatment.Surgery can be effective to remove endometriosis lesions and scar tissue, but success rates are dependent on the extent of disease and are invasive.
In view of the above, a non-invasive drug without the current side effects of hormone-based therapy are highly desirable to the market, and NLS-1 could address this unmet need. NLS-1 is a drug molecule derived from natural extracts and offers a potential non-hormonal treatment of endometriosis. As demonstrated in animal models, NLS-1 exhibits the following attributes in these studies: (i) statistically significant inhibition against development, growth and angiogenesis of uterine tissue, and (ii) statistically significant reduction of lesions compared with EGCG and other conventional hormone-based therapy.
| 7 | ENDOMETRIOSIS.org, “Facts about endometriosis”, http://endometriosis.org/resources/articles/facts-about-endometriosis/ |
| 8 | Washington University Physicians, “Endometriosis”, https://fertility.wustl.edu/getting-started-infertility/infertility-factors/endometriosis/ |
| 9 | J. Fisher M. Kirkman, “Endometriosis and fertility: women’s accounts of healthcare”, Human Reproduction, Volume 31, Issue 3, March 1, 2016, Pages 554–562, January 11, 2016, https://doi.org/10.1093/humrep/dev337 |
| 10 | GlobalData, “Endometriosis Market Expected to Surpass $2 Billion by 2025”, November 11, 2016, R&D, https://www.rdmag.com/news/2016/11/endometriosis-market-expected-surpass-2-billion-2025 |
5
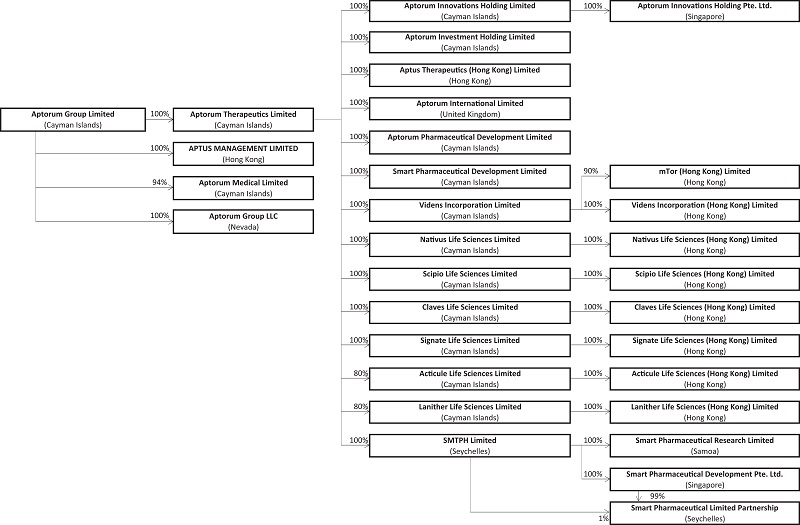
Our Structure
The following diagram illustrates our corporate structure as of the date of this prospectus. For more details regarding our corporate history and current structure, please refer to “Corporate History and Background” appearing on page 88 of this prospectus.
Controlled Company
As long as our officers and directors, either individually or in theaggregate, own at least 50% of the voting power of our Company, we will be a “controlled company” as defined under NASDAQ Marketplace Rules. However, even if we qualify as a “controlled company,” we do not intend to rely on the controlled company exemptions provided under NASDAQ Marketplace Rules. To that extent, we have set up the Audit Committee, the Compensation Committee, and the Nominating and Corporate Governance Committee, all of which consist solely of independent directors and adopted a charter for each committee
6
For so long as we are a controlled company under that definition, we are permitted however to elect to rely, and may rely, on certain exemptions from corporate governance rules, including:
| ● | an exemption from the rule that a majority of our board of directors must be independent directors; | |
| ● | an exemption from the rule that the compensation of our chief executive officer must be determined or recommended solely by independent directors; and | |
| ● | an exemption from the rule that our director nominees must be selected or recommended solely by independent directors. |
As a result, you will not have the same protection afforded to shareholders of companies that are subject to these corporate governance requirements.
Although we do not intend to rely on the “controlled company” exemption under the Nasdaq listing rules, we could elect to rely on this exemption in the future. If we elect to rely on the “controlled company” exemption, a majority of the members of our board of directors might not be independent directors and our nominating and corporate governance and compensation committees might not consist entirely of independent directors. (See “Risk Factors – Risks Related to Our Corporate Structure –As a “controlled company” under the rules of the NASDAQ Global Market, we may choose to exempt our company from certain corporate governance requirements that could have an adverse effect on our public shareholders.”)
Risks Associated with Our Business
Investing in our Class A Ordinary Shares involves risks. You should carefully consider the risks described in “Risk Factors” beginning on page 13 of this prospectus before making a decision to purchase Class A Ordinary Shares. If any of these risks actually occurs, our business, financial condition or results of operations would likely be materially adversely affected. In such case, the trading price of our Class A Ordinary Shares would likely decline, and you may lose all or part of your investment.
Recent Events
Master Collaboration Agreement
On April 24, 2019, we signed an agreement with Aeneas, and A*ccelerate Technologies Pte. Ltd, the enterprise office of the Agency for Science, Technology and Research (“A*STAR”), to co-create local deep tech startups. This agreement, which is part of A*ccelerate’s venture co-creation (“VCC”) initiative, commits all parties to the co-creation of local startups in the healthcare and life science sector (the “Master Collaboration Agreement”).
The goal is to create a total of up to 20 deep tech ventures in Singapore will be created by this partnership over the next 5 years. These enterprises will leverage technologies co-developed by both A*STAR’s research institutes and Aptorum Group, as well as technologies identified and collaborated on worldwide by both institutions. As part of this agreement, all parties will also actively seek expertise, and nurture entrepreneurs to accelerate the growth of its ventures through Singapore and its worldwide partnerships.
Under the Master Collaboration Agreement, the parties will set up a panel consisting of 1 nominee from A*STAR and 1 nominee from the Company, Aeneas, or Aptorum Innovations Holding Pte Limited (“Aptorum Innovations”) (which will act as the holding company for all subsequent venture creation or joint commercialization center activities including the development of the startups), to review and approve the business plan of all suitable startups. Each startup shall have a valuation mutually agreed upon between all parties, and shall have A*STAR and Aptorum Innovations as founding shareholders.
A*STAR shall contribute a total of up to $30,000,000 to any suitable startups, at their discretion. The Company will set up a healthcare and life science strategic investment fund (“Fund”) to be managed by Aeneas Capital Limited. Through the Fund, it will contribute a total of up to $30,000,000 to any suitable startups at their discretion with a focus on (i) securing pilot customers; (ii) incorporation of the startups as companies and financial commitments of such customers; (iii) capital raising and capital market plans; (iv) recruiting and building of the startup teams; (v) equipment and infrastructure; and (vi) licensing of IP to the startups under the separate technology license agreements.
7
The Master Collaboration Agreement shall continue for a period of 5 years, unless otherwise terminated or extended by the parties.
Bond Repurchase
On April 24, 2019, the Company’s wholly-owned subsidiary, Aptorum Investment Holding Limited, repurchased convertible bonds (the “Bonds”) from Peace Range Limited (“Peace Range”), a wholly owned subsidiary of Adamas Ping An Opportunities Fund LP. The Bonds were originally issued on April 25, 2018, in the principal amount of $15,000,000 (minus a structuring fee equal to 2% of the principal amount of the Bonds). As part of the original subscription for the Bonds, the bondholder was granted certain rights to subscribe for additional ordinary shares of the Company, in an amount up to the principal amount of the Bonds at a price of US$12.17 (subject to adjustment) on or before December 17, 2019 (“Subscription Right”). The total consideration of the repurchase of Bonds and the Subscription Rights was US$13.6 million in cash, excluding accrued interest. In connection with the repurchase, various other agreements initially entered into when the Bond was originally issued to Peace Range were terminated.
Establishment of Smart-ACTTM Platform
On April 24, 2019, the Company announced the establishment of Smart Pharma (“Smart Pharma”), which operates novel computational repurposed drug discovery, modeling and validation platform, referred to as the Smart-ACTTM platform. Smart Pharma is controlled by SMTPH Limited, an International Business Company incorporated in Seychelles and a wholly-owned subsidiary of Aptorum Therapeutics Limited (“SMTPH”).
Smart-ACTTM stands for “Accelerated Commercialization of Therapeutics” and encompasses state-of-the-art technology in systematic screening of existing approved drug molecules against selected therapeutic targets. Specifically, the Smart-ACTTM platform comprises of a network of modules and processes that simulate the effectiveness of drug molecules against diseases for outcome prediction and selection. The Smart-ACTTM platform initially focuses on screening drug molecules for orphan diseases or for fulfillment of unmet medical needs.
To date, the Smart-ACTTMplatform has completed computational screening based on structural affinity and scoring analysis of around 1,600 approved drugs against 3 therapeutic target proteins related to poor prognosis of neuroblastoma (i.e., a type of cancer that forms in certain types of nerve tissue and most frequently in the adrenal glands as well as spine, chest, abdomen or neck). Among the 1,600 drugs that have been screened, around 40 have been identified for further evaluation under wet lab validation.
We are currently evaluating the shortlisted compounds via wet lab validation to confirm their efficacy in cell-based and animal models for treating neuroblastoma. The validation is being conducted in vitro and in vivo validation in collaboration with Aptorum Group Limited to assess and validate the compounds’ usage for the new indication.
Smart Pharma’s current funding needs include funding for validation and assessment of candidates, operation and improvement of the platform, legal/professional services and exchanges-listing. In an effort to raise funds for the development and operation of the Smart-ACTTMplatform, Smart Pharma is conducting a Smart Pharma Token (“SMPT token”) offering (See “Prospectus Summary – Recent Events – Smart Pharma Token”).
Until Smart Pharma becomes self-sustaining, the proceeds from the token offering, if any, will most likely be insufficient to fully fund Smart Pharma’s current and future operations. In such case, it could have a material adverse effect on SMTPH’s ability to fund its objectives and carry out its related business plans, and its ability to develop the Smart-ACTTMplatform may be limited.
Therefore, Smart Pharma will likely require funding from Aptorum Group to subsidize and support its operations. Nonetheless, ifSmart Pharma is unable to obtain adequate funding from other sources, or if the internal funding from Aptorum Group is insufficient, Smart Pharma might be required to decrease or eliminate expenditure on the Smart-ACTTMplatform altogether.
Smart Pharma Token
On April 24, 2019, the SMPT token was announced to be launched. The SMPT tokens are issued by Smart Pharmaceutical Limited Partnership (“SPLP”), a limited partnership registered in Seychelles, which is managed by SMTPH as its sole general partner. SMTPH is a wholly-owned subsidiary of Aptorum Therapeutics Limited. Aptorum Group Limited is not involved with the offer and sale of the SMPT token in any way, other than the potential indirect benefit it will receive as a result of its subsidiary, Smart Pharma, from drug candidates developed by the Smart-ACTTM platform.
The SMPT token is not for sale in the U.S. and is not offered, available for sale and/or otherwise transferrable to any U.S. persons; Aptorum Group Limited is not involved with the sale of the SMPT token, other than the indirect benefit it will receive as a result of its subsidiary, Smart Pharma receiving profit from the sale of such tokens.
8
The SMPT token is an ERC-1404 security compliant token with ERC-20 and ERC233 compliance on the Ethereum blockchain, which tokenizes rights to a portion of sales-based royalties (as described below), non-royalty sublicensing income (as described below), as well as additional cash flow (as described below) (if applicable) (collectively referred to as “Commercialization Income”), derived from the subsequent commercialization of intellectual property rights of drug candidates discovered under our Smart-ACTTM platform. SMPT token is backed by SPLP’s assets, including intellectual property rights of drug candidates created through the Smart-ACTTM platform and Commercialization Income. SPLP acts as the intellectual property holding company of Smart Pharma, and holds all title, rights and ownership interest of the intellectual property rights developed by Smart-ACTTM(“Project IP”).
Specifically, Smart Pharma has the right to commercialize the drug candidates discovered under the Smart-ACTTM platform (e.g. through direct commercialization by affiliates, sublicensing to third parties, collaboration with third parties, and assignment to others) and distribute a portion of the Commercialization Income to the token holders, as set forth below.
First, for drug candidates that are directly commercialized by Smart Pharma or its affiliates (i.e., the licensee), Smart Pharma will set aside a 3% (minimum) to 5% (maximum) of the net sales of the products (on a product-by-product basis and as determined by Smart Pharma) to SPLP for distribution to the SMPT token holders. This type of consideration is hereby known as “sales-based royalties”.
Second, for drug candidates commercialized by third parties, instead of paying the sales-based royalties discussed above, Smart Pharma (i.e., the licensee) will receive from its sublicensee on account of the grant of the sublicense (on a product-by-product basis, such consideration may include royalties, upfront fees and milestone payments). Smart Pharma will set aside 10% of the consideration it receives to SPLP for distribution to the SMPT token holders. This type of consideration is hereby known as “sublicensing income”.
Therefore, depending on how a product is commercialized, Smart Pharma will set aside either sales-based royalties or sublicensing income to SPLP for distribution to the SMPT token holders. The above percentages for sales-based royalties or sublicensing income are subject to adjustment upon further notice to SMPT token holders.
Third, at its discretion, in addition to sales-based royalties and sublicensing income, Smart Pharma may occasionally set aside other additional amounts to SPLP for distribution to SMPT token holders (such as rebates, price protection, performance bonus, other discount and incentives), as applicable on a product-by-product basis. This type of consideration is hereby known as “additional cash flow”.
Accordingly, the amount of distributions payable to token holders is not tied to any funding we may provide to Smart Pharma and any such funding will not impact the amount of distributions so payable. The amount of distributions payable is also not correlated with the number of tokens sold or the amount of proceeds, if any raised through the token offering.
Once any of the above aforementioned distribution is accrued and set aside to SPLP by Smart Pharma, SPLP is not required to immediately distribute any such distribution to the SMPT token holders, but may choose to make a distribution at any time it deems it best to do so.
Currently, as the drug efficacy validation process has just begun, SPLP does not expect to distribute any sales-based royalties, sublicensing income or additional cash flow generated by drug candidates developed by the Smart-ACTTM platform at any time for the near future, although it will be accruing same.
In the event of liquidation, dissolution or winding up of SPLP, the SMPT token holders will be entitled to certain liquidation rights. Specifically, SPLP will, after payment of its debts and obligations, distribute any “relevant assets”1 of SPLP to the SMPT token holders, with equal priority pro rata among the SMPT token holders, ratably and in proportion to the full amount of the relevant assets. Given that SMPT token holders are not shareholders of SPLP, they are only entitled to collect the relevant assets but not other assets held by SPLP. Therefore, in the event of liquidation, dissolution or winding up of SPLP, the total distribution issuable to the token holders will not exceed the “relevant assets”.
There is no assurance that any or all of the SMPT tokens will be sold and SPLP can elect in their discretion not to issue any SMPT tokens for any reason.
The SMPT tokens are not offered for sale to citizens, nationals, residents (tax or otherwise), green card holders and/or companies domiciled in the following jurisdictions: (a) the United States of America; (b) Singapore; (c) Hong Kong (except for Professional Investors); (d) the People’s Republic of China; (e) Samoa, (f) Seychelles, (g) sanctioned countries under the OFAC and (h) any other jurisdiction which prohibits the possession, dissemination or communication of tokens. The SMPT token is not registered under the U.S. Securities Act and we have no intention of doing so. The SMPT tokens may not be offered or resold in the U.S. or to U.S. persons unless registered under the Securities Act or pursuant to an exemption therefrom. The SMPT tokens may not be transferred to a U.S person, as such term is defined in Regulation S of the Securities Act, except and unless in accordance with the provisions of U.S. securities laws, particularly they must either be registered or comply with an exemption from registration. Further, hedging transactions with regard to the SMPT tokens may not be conducted unless in compliance with the Securities Act.
1 The relevant assets of SPLP shall be limited to all accrued sales-based royalties, sublicensing income and/or additional cash flow generated by the drug candidates developed by the Smart-ACTTM platform, set aside by Smart Pharma and not yet distributed to the SMPT token holders. The relevant assets are secured by way of a floating charge against the Project IP (See “Risk Factors – Risks Related to the SMPT tokens - SMPT Tokenholders’ security interest in the intellectual property rights may affect our shareholder’s interest in the Company”).
9
Currently, Smart Pharma has no immediate plan to register the SMPT tokens under the Securities Act or to offer the SMPT tokens in the U.S.
Since July 2019, the SMPT tokens have been listed for trading on two cryptocurrency exchange platforms, IDAX and LATOKEN.
This document shall not constitute an offer to sell or the solicitation of an offer to buy SMPT tokens.
Establishment novel therapeutic platform
On May 6, 2019, Claves Life Sciences Limited, a wholly owned subsidiary of Aptorum Group Limited announced the establishment of a novel therapeutic platform intended for the treatment of various diseases via modulation of the chemical signaling relating to gut microbiota.
Hong Kong Micro Cap Exchange
Effective Tuesday, May 28, 2019, the Company shall be added to the Morgan Stanley Capital International MSCI (“MSCI”) Hong Kong Micro Cap Index. MSCI Hong Kong Micro Cap Index is an international equity benchmark recognized by institutional investors. Inclusion of constituent companies is based on excellence of performance and potential of development. We believe that our inclusion in the MSCI Hong Kong Micro Cap Index will help expand Aptorum’s investor base and enhance its corporate image and market presence.
Our Securities
Our authorized share capital is divided into Class A Ordinary Shares and Class B Ordinary Shares. Holders of Class A Ordinary Shares and Class B Ordinary Shares have the same rights except for voting and conversion rights. In respect of matters requiring a shareholder vote, each Class A Ordinary Share will be entitled to one vote and each Class B Ordinary Share will be entitled to ten votes. Due to the Class B Ordinary Share’s voting power, the holders of Class B Ordinary shares currently and may continue to have a concentration of voting power, which limits the holders of Class A Ordinary Shares’ ability to influence corporate matters.(See “Risk Factors–Risks Related to our securities –Our Class B Ordinary Shares have stronger voting power than our Class A Ordinary Shares and certain existing shareholders have substantial influence over our Company and their interests may not be aligned with the interests of our other shareholders.”)Each Class B Ordinary Share is convertible into one Class A Ordinary Share at any time by the holder thereof. Class A Ordinary Shares are not convertible into Class B Ordinary Shares under any circumstances. (See “Description of Share Capital”)
Corporate Information
Our principal executive office is located on the 17th Floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong. Our telephone number is +852 2117 6611.
Our website is www.aptorumgroup.com.The information on our website is not part of this prospectus.
Implications of Being an Emerging Growth Company
We qualify as an “emerging growth company” as defined in the JOBS Act. As an emerging growth company, we may take advantage of specified reduced disclosure and other requirements that are otherwise applicable generally to public companies. These provisions include:
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley Act”); |
10
| ● | the ability to include only two years of audited financial statements in addition to any required interim financial statements and correspondingly reduced disclosure in management’s discussion and analysis of financial condition and results of operations in the registration statement for this Offering of which this prospectus forms a part; and |
| ● | to the extent that we no longer qualify as a foreign private issuer, (1) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements; and (2) exemptions from the requirements of holding a non-binding advisory vote on executive compensation, including golden parachute compensation. |
We may take advantage of these exemptions for up to five years or such earlier time that we are no longer an emerging growth company. We would cease to be an emerging growth company upon the earliest to occur of (1) the last day of the fiscal year in which we have more than $1.07 billion in annual revenue; (2) the date we qualify as a “large accelerated filer” with at least $700 million of equity securities held by non-affiliates; (3) the issuance, in any three-year period, by our Company of more than $1.0 billion in non-convertible debt securities; and (4) the last day of the fiscal year ending after the fifth anniversary of the IPO. We may choose to take advantage of some but not all of these exemptions. For example, Section 107 of the JOBS Act provides that an emerging growth company can use the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. We are choosing to elect to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(2) of the JOBS Act, which allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold equity securities.
Implications of Being a Foreign Private Issuer
We are also considered a “foreign private issuer.” In our capacity as a foreign private issuer, we are exempted from certain rules under the U.S. Securities Exchange Act of 1934, as amended (“Exchange Act”), that impose certain disclosure obligations and procedural requirements for proxy solicitations under Section 14 of the Exchange Act. In addition, our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules under the Exchange Act with respect to their purchases and sales of our Class A Ordinary Shares. Moreover, we are not required to file periodic reports and financial statements with the U.S. Securities and Exchange Commission (“SEC”), as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. In addition, we are not required to comply with Regulation FD, which restricts the selective disclosure of material information.
We may take advantage of these exemptions until such time as we are no longer a foreign private issuer. We would cease to be a foreign private issuer at such time when more than 50% of our outstanding voting securities are held by U.S. residents and any of the following three circumstances applies: (1) the majority of our executive officers or directors are U.S. citizens or residents; (2) more than 50% of our assets are located in the United States; or (3) our business is administered principally in the United States.
We have taken advantage of certain reduced reporting and other requirements in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold equity securities.
Notes on Prospectus Presentation
Numerical figures included in this prospectus have been subject to rounding adjustments. Accordingly, numerical figures shown as totals in various tables may not be arithmetic aggregations of the figures that precede them. Certain market data and other statistical information contained in this prospectus is based on information from independent industry organizations, publications, surveys and forecasts. Some market data and statistical information contained in this prospectus are also based on management’s estimates and calculations, which are derived from our review and interpretation of the independent sources listed above, our internal research and our knowledge of pharmaceutical industry. While we believe such information is reliable, we have not independently verified any third-party information and our internal data has not been verified by any independent source.
11
Accordingly, actual events or circumstances may differ materially from events and circumstances that are assumed in this information and you are cautioned not to give undue weight to such data.
Offering Summary
| Issuer: | Aptorum Group Limited | |
| Securities being Offered by Selling Shareholders | Up to 27,765,821 Class A Ordinary Shares.The Selling Shareholders may sell their Class A Ordinary Shares at prevailing market prices or privately negotiated prices. We will not receive any proceeds from the sales by the Selling Shareholders. | |
| Symbol | Our Class A Ordinary Shares trade on the NASDAQ Global Market under the symbol APM. | |
| Transfer Agent | Continental Stock Transfer & Trust Company |
| Risk Factors | Investing in our Class A Ordinary Shares involves a high degree of risk and purchasers of our Class A Ordinary Shares may lose part or all of their investment. See “Risk Factors” for a discussion of factors you should carefully consider before deciding to invest in our Class A Ordinary Shares beginning on Page 13. | |
| Use of Proceeds | We will not receive any proceeds from the sale of the Class A Ordinary Shares offered hereby. |
12
Investing in our Class A Ordinary Shares involves a high degree of risk. You should carefully consider the following risks and all other information contained in this prospectus, including our financial statements, consolidated financial statements and the related notes, before making an investment decision regarding our securities. The risks and uncertainties described below are those significant risk factors, currently known and specific to us that we believe are relevant to an investment in our securities. If any of these risks materialize, our business, financial condition or results of operations could suffer, the price of our Class A Ordinary Shares could decline and you could lose part or all of your investment.
Risks Related to the Preclinical and Clinical Development of Our Drug Candidates
We currently do not generate revenue from product sales and may never become profitable; unless we can raise more capital through additional financings, of which there can be no guarantee, our principal source of revenue will be from AML Clinic, which may not be substantial.
Our ability to generate revenue and become profitable depends upon our ability to successfully complete the development of, and obtain the necessary regulatory approvals for, the drug candidates in our Lead Projects and any future drug candidates we may develop, as we do not currently have any drugs that are available for commercial sale. We expect to continue to incur losses before commercialization of our drug candidates and any future drug candidates. None of our drug candidates has been approved for marketing in the U.S., Europe, the PRC or any other jurisdictions and may never receive such approval. Our ability to generate revenue and achieve profitability is dependent on our ability to complete the development of our drug candidates and any future drug candidates we develop in our portfolio, obtain necessary regulatory approvals, and have our drugs products under development manufactured and successfully marketed, of which there can be no guarantee. Although AML Clinic commenced operations in June 2018 and we expect to receive some revenue from such operations, even at full capacity, AML Clinic may not bring enough revenue to support our operation and R&D. Thus, we may not be able to generate a profit until our drug candidates become profitable.
13
Even if we receive regulatory approval and marketing authorization for one or more of our drug candidates or one or more of any future drug candidates for commercial sale, a potential product may not generate revenue at all unless we are successful in:
| ● | developing a sustainable and scalable manufacturing process for our drug candidates and any approved products, including establishing and maintaining commercially viable supply relationships with third parties; | |
| ● | launching and commercializing drug candidates following regulatory approvals and marketing authorizations, either directly or with a collaborator or distributor; | |
| ● | obtaining market acceptance of our drug candidates as viable treatment options; | |
| ● | addressing any competing technological and market developments; | |
| ● | negotiating and maintaining favorable terms in any collaboration, licensing or other arrangement into which we may enter to commercialize drug candidates for which we have obtained required approvals and marketing authorizations; and | |
| ● | maintaining, protecting and expanding our portfolio of IP rights, including patents, trade secrets and know-how. |
In addition, our ability to achieve and maintain profitability depends on timing and the amount of expenses we will incur. Our expenses could increase materially if we are required by the FDA, NMPA, EMA or other comparable regulatory authorities to perform studies in addition to those that we currently have anticipated. Even if our drug candidates are approved for commercial sale, we anticipate incurring significant costs associated with the commercial launch of these products.
Our ability to become and remain profitable depends on our ability to generate revenue. Even if we are able to generate revenues from AML Clinic or the sale or sublicense of any products we may develop or license, we may not become profitable on a sustainable basis or at all. Our failure to become and remain profitable would decrease the value of our Company and adversely affect the market price of our Class A Ordinary Shares, which could impair our ability to raise capital, expand our business or continue our operations.
AML Clinic’s operations may be our principal source of revenue for the foreseeable future and most likely, without additional financing, such revenue will not be sufficient for us to carry out all of our plans.
As stated above, we have not generated any revenue and do not foresee generating any revenue from our drug candidates in the near future. Effective as of March 2018, we leased the property in Central, Hong Kong that is the home to AML Clinic, which commenced operations in June 2018.
Until our therapeutic candidates produce revenue, our principal source of revenue shall be from AML Clinic, but we cannot guarantee that it will provide the expected revenue, and even if expected revenue is realized, it will not be sufficient by itself to fund our other operations. We believe that available cash, together with the efforts from management plans and actions described elsewhere in this prospectus, should enable the Company to meet presently anticipated cash needs for at least the next 12 months after the date that the financial statements are issued and the Company has prepared the consolidated financial statements on a going concern basis. However, the Company continues to have ongoing obligations and it expects that it will require additional capital in order to execute its longer-term development plan. If the Company encounters unforeseen circumstances that place constraints on its capital resources, management will be required to take various measures to conserve liquidity, which could include, but not necessarily be limited to, deferring some of its research and seeking to dispose of marketable securities. Management cannot provide any assurance that the Company will raise additional capital if needed.
14
We depend substantially on the success of the drug candidates being researched as our current Lead Projects, which are in the preclinical stage of development. The preclinical development, IND-enabling, and clinical trials of our drug candidates may not be successful. If we are unable to license or sublicense, sell or otherwise commercialize our drug candidates, or experience significant delays in doing so, our business will be materially harmed.
Our business and the ability to generate revenue related to product sales, if ever achieved, will depend on the successful development, regulatory approval and licensing or sublicensing or other commercialization of our drug candidates or any other drug candidates we may develop. We have invested a significant amount of financial resources in the development of our drug candidates and we expect to invest in other drug candidates. The success of our drug candidates and any other potential drug candidates will depend on many factors, including but not limited to:
| ● | successful enrollment in, and completion of, studies in animals and clinical trials; | |
| ● | other parties’ ability in conducting our clinical trials safely, efficiently and according to the agreed protocol; | |
| ● | receipt of regulatory approvals from the FDA, NMPA, EMA and other comparable regulatory authorities for our drug candidates; | |
| ● | our ability to establish commercial manufacturing capabilities by making arrangements with third-party manufacturers; | |
| ● | reliance on other parties to conduct our clinical trials swiftly and effectively; | |
| ● | launch of commercial sales of our drug candidates, if and when approved; | |
| ● | obtaining and maintaining patents, trade secrets and other IP protection and regulatory exclusivity, as well as protecting our rights in our own IP; | |
| ● | ensuring that we do not infringe, misappropriate or otherwise violate patents, trade secrets or other IP rights of other parties; | |
| ● | obtaining acceptance of our drug candidates by doctors and patients; | |
| ● | obtaining reimbursement from third-party payors for our drug candidates, if and when approved; | |
| ● | our ability to compete with other drug candidates and drugs; and | |
| ● | maintenance of an acceptable safety profile for our drug candidates following regulatory approval, if and when received. |
We may not achieve regulatory approval and commercialization in a timely manner or at all. Significant delays in obtaining approval for and/or to successfully commercialize our drug candidates would materially harm our business and we may not be able to generate sufficient revenues and cash flows to continue our operations.
We may not be successful in our efforts to identify or discover additional drug candidates. Due to our limited resources and access to capital, we must continue to prioritize development of certain drug candidates; such decisions may prove to be wrong and may adversely affect our business.
Although we intend to explore other therapeutic opportunities in addition to the drug candidates that we are currently developing, we may fail to identify other drug candidates for a number of reasons. For example, our research methodology may be unsuccessful in identifying potential drug candidates or those we identify may be shown to have harmful side effects or other undesirable characteristics that make them unmarketable or unlikely to receive regulatory approval.
15
Research programs to pursue the development of our drug candidates for additional indications and to identify new drug candidates and disease targets require substantial technical, financial and human resources whether or not we ultimately are successful. Our research programs may initially show promise in identifying potential indications and/or drug candidates, yet fail to yield results for clinical development for a number of reasons, including but not limited to:
| ● | the research methodology used may not be successful in identifying potential indications and/or drug candidates; | |
| ● | potential drug candidates may, after further study, be shown to have harmful adverse effects or other characteristics that indicate they are unlikely to be effective drugs; or | |
| ● | it may take greater human and financial resources to identify additional therapeutic opportunities for our drug candidates or to develop suitable potential drug candidates through internal research programs than we will possess, thereby limiting our ability to diversify and expand our drug portfolio. |
Because we have limited financial and managerial resources, we have chosen to focus at present on our three Lead Projects, which may ultimately prove to be unsuccessful. As a result of this focus, we may forego or delay pursuit of opportunities with other drug candidates, or for other indications that later prove to have greater commercial potential or a greater likelihood of success. Even if we determine to pursue alternative therapeutic or diagnostic drug candidates, these other drug candidates or other potential programs may ultimately prove to be unsuccessful. In short, our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities.
Accordingly, there can be no assurance that we will ever be able to develop suitable potential drug candidates through internal research programs. This could materially adversely affect our future growth and prospects.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
While we have not commenced any clinical trials and do not expect to start our first clinical trials until at least 2020 or 2021, assuming we obtain approval to do so from at least one regulatory authority, of which there can be no assurance, timely completion of clinical trials in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who meet the trial criteria and remain in the trial until its conclusion. We may experience difficulties enrolling and retaining appropriate patients in our clinical trials for a variety of reasons, including but not limited to:
| ● | the size and nature of the patient population; | |
| ● | patient eligibility criteria defined in the clinical protocol; | |
| ● | the size of study population required for statistical analysis of the trial’s primary endpoints; | |
| ● | the proximity of patients to trial sites; | |
| ● | the design of the trial and changes to the design of the trial; | |
| ● | our ability to recruit clinical trial investigators with the appropriate competencies and experience; | |
| ● | competing clinical trials for similar therapies or other new therapeutics exist and will reduce the number and types of patients available to us; |
16
| ● | clinicians’ and patients’ perceptions as to the potential advantages and side effects of the drug candidate being studied in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are investigating; | |
| ● | our ability to obtain and maintain patient consents; | |
| ● | patients enrolled in clinical trials may not complete a clinical trial; and | |
| ● | the availability of approved therapies that are similar to our drug candidates. |
Even if we are able to enroll a sufficient number of patients in our clinical trials, delays in patient enrollment may result in increased costs or may affect the timing or outcome of the planned clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our drug candidates.
Clinical drug development involves a lengthy and expensive process and could fail at any stage of the process. We have limited experience in conducting clinical trials and results of earlier studies and trials may not be reproduced in future clinical trials.
For our drug candidates, clinical testing is expensive and can take many years to complete, while failure can occur at any time during the clinical trial process. The results of studies in animals and early clinical trials of our drug candidates may not predict the results of later-stage clinical trials. Drug candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through studies in animals and initial clinical trials. In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same drug candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations (including genetic differences), patient adherence to the dosing regimen and the patient dropout rate. Results in later trials may also differ from earlier trials due to a larger number of clinical trial sites and additional countries and languages involved in such trials. In addition, the design of a clinical trial can determine whether its results will support approval of a drug candidate, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced and significant expense has been incurred.
A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of demonstrated efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts. Furthermore, if the trials we conduct fail to meet their primary statistical and clinical endpoints, they will not support the approval from the FDA, NMPA, EMA or other comparable regulatory authorities for our drug candidates. If this occurs, we would need to replace the failed study with new trials, which would require significant additional expense, cause substantial delays in commercialization and materially adversely affect our business, financial condition, cash flows and results of operations. (See “We are subject to risks related to the carrying out and outcome of clinical trials of medical devices”)
If clinical trials of our drug candidates fail to demonstrate safety and efficacy to the satisfaction of the FDA, NMPA, EMA or other comparable regulatory authorities, or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our drug candidates.
Before applying for and obtaining regulatory approval for the sale of any of our drug candidates, we must conduct extensive clinical trials to demonstrate the safety and efficacy of our drug candidates in humans. Clinical testing is expensive, difficult to design and implement, can take many years to complete and may fail. A failure of one or more of our clinical trials can occur at any stage of testing and successful interim results of a clinical trial do not necessarily predict successful final results.
We and our CROs are required to comply with current Good Clinical Practices (“cGCP”) requirements, which are regulations and guidelines enforced by the FDA, NMPA, EMA and other comparable regulatory authorities for all drugs in clinical development. Regulatory authorities enforce these cGCP through periodic inspections of trial sponsors, principal investigators and trial sites. Compliance with cGCP can be costly and if we or any of our CROs fail to comply with applicable cGCP, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, NMPA, EMA or comparable regulatory authorities may require us to perform additional clinical trials before approving our marketing applications.
17
We may experience numerous unexpected events during, or as a result of, clinical trials that could delay or prevent our ability to receive regulatory approval or commercialize our drug candidates, including but not limited to:
| ● | regulators, institutional review boards (“IRBs”) or ethics committees may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; | |
| ● | clinical trials of our drug candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon drug development programs; | |
| ● | the number of patients required for clinical trials of our drug candidates may be larger than we anticipate, enrollment may be insufficient or slower than we anticipate or patients may drop out at a higher rate than we anticipate; | |
| ● | our contractors and investigators may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; | |
| ● | we might have to suspend or terminate clinical trials of our drug candidates for various reasons, including a lack of clinical response or a determination that participants are being exposed to unacceptable health risks; | |
| ● | regulators, IRBs or ethics committees may require that we or our investigators suspend or terminate clinical research for various reasons, including non-compliance with regulatory requirements; | |
| ● | the cost of clinical trials of our drug candidates may be greater than we anticipate; | |
| ● | the supply or quality of our drug candidates or other materials necessary to conduct clinical trials of our drug candidates may be insufficient or inadequate; and | |
| ● | our drug candidates may cause adverse events, have undesirable side effects or other unexpected characteristics, causing us, our investigators, or regulators to suspend or terminate the trials. |
If we are required to conduct additional clinical trials or other testing of our drug candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our drug candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if they raise safety concerns, we may:
| ● | be delayed in obtaining regulatory approval for our drug candidates; | |
| ● | not obtain regulatory approval at all; | |
| ● | obtain approval for indications that are not as broad as intended; | |
| ● | have a drug removed from the market after obtaining regulatory approval; | |
| ● | be subject to additional post-marketing testing requirements; | |
| ● | be subject to restrictions on how a drug is distributed or used; or | |
| ● | be unable to obtain reimbursement for use of a drug. |
Delays in testing or approvals may result in increases in our drug development costs. We do not know whether any clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, or at all. Clinical trials may produce negative or inconclusive results. Moreover, these trials may be delayed or proceed less quickly than intended. Delays in completing our clinical trials will increase our costs, slow down our drug candidate development and approval process, and jeopardize our ability to commence product sales and generate revenues and we may not have sufficient funding to complete the testing and approval process. Any of these events may significantly harm our business, financial condition and prospects, lead to the denial of regulatory approval of our drug candidates or allow our competitors to bring drugs to market before we do, impairing our ability to commercialize our drugs if and when approved.
18
Significant clinical trial delays also could shorten any periods during which we have the exclusive right to commercialize our drug candidates or allow our competitors to bring products to market before we do, impair our ability to commercialize our drug candidates and may harm our business and results of operations.
We may in the future conduct clinical trials for our drug candidates in sites outside the U.S. and the FDA may not accept data from trials conducted in such locations.
We may in the future conduct certain of our clinical trials outside the U.S. Although the FDA may accept data from clinical trials conducted outside the U.S. for our New Drug Application (“NDA”), acceptance of this data is subject to certain conditions imposed by the FDA. There can be no assurance the FDA will accept data from any of the clinical trials we conduct outside the U.S. If the FDA does not accept the data from any of our clinical trials conducted outside the U.S., it would likely result in the need for additional clinical trials in the U.S., which would be costly and time-consuming and could delay or prevent the commercialization of any of our drug candidates.
Risks Related to Obtaining Regulatory Approval for Our Drug Candidates
The regulatory approval processes of the FDA, NMPA, EMA and other comparable regulatory authorities are lengthy, time-consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our current drug candidates or any future drug candidates we may develop, our business will be substantially harmed.
We cannot commercialize drug candidates without first obtaining regulatory approval to market each drug from the FDA, NMPA, EMA or comparable regulatory authorities. Before obtaining regulatory approvals for the commercial sale of any drug candidate for a target indication, we must demonstrate in studies in animals and well-controlled clinical trials, and, with respect to approval in the United States and other regulatory agencies, to the satisfaction of the FDA, NMPA, EMA or comparable regulatory authorities, that the drug candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls are adequate.
The time required to obtain approval from the FDA, NMPA, EMA and other comparable regulatory authorities is unpredictable but typically takes many years following the commencement of studies in animals and clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities.
In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval can differ among regulatory authorities and may change during the course of the development of a drug candidate. We have not obtained regulatory approval for any drug candidate. It is possible that neither our existing drug candidates nor any drug candidates we may discover or acquire for development in the future will ever obtain regulatory approval. Even if we obtain regulatory approval in one jurisdiction, we may not obtain it in other jurisdictions.
Our drug candidates could fail to receive regulatory approval from any of the FDA, NMPA, EMA or other comparable regulatory authorities for many reasons, including but not limited to:
| ● | disagreement with regulators regarding the design or implementation of our clinical trials; | |
| ● | failure to demonstrate that a drug candidate is safe and effective or safe, pure and potent for its proposed indication; | |
| ● | failure of clinical trial results to meet the level of statistical significance required for approval; |
19
| ● | failure to demonstrate that a drug candidate’s clinical and other benefits outweigh its safety risks; | |
| ● | disagreement with regulators regarding our interpretation of data from studies in animals or clinical trials; | |
| ● | insufficiency of data collected from clinical trials of our drug candidates to support the submission and filing of a New Drug Application (“NDA”), or other submission or to obtain marketing approval; | |
| ● | the FDA, NMPA, EMA or a comparable regulatory authority’s finding of deficiencies related to the manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies; and | |
| ● | changes in approval policies or regulations that render our preclinical studies and clinical data insufficient for approval. |
Any of the FDA, NMPA, EMA or other comparable regulatory authorities may require more information, including additional preclinical studies or clinical data, to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program. If we were to obtain approval, regulatory authorities may approve any of our drug candidates for fewer or more limited indications than we request. Regulatory authorities also may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a drug candidate with a label that is not desirable for the successful commercialization of that drug candidate. In addition, if our drug candidate produces undesirable side effects or involves other safety issues, the FDA may require the establishment of a Risk Evaluation Mitigation Strategy (“REMS”), or NMPA, EMA or other comparable regulatory authorities may require the establishment of a similar strategy. Such a strategy may, for instance, restrict distribution of our drug candidates, require patient or physician education, or impose other burdensome implementation requirements on us.
Regulatory approval may be substantially delayed or may not be obtained for one or all of our drug candidates if regulatory authorities require additional time or studies to assess the safety or efficacy of our drug candidates.
We currently do not have any drug candidates that have gained approval for sale by the FDA, NMPA or EMA or other regulatory authorities in any other country, and we cannot guarantee that we will ever have marketable drugs. Our business is substantially dependent on our ability to complete the development of, obtain marketing approval for and successfully commercialize drug candidates in a timely manner. We cannot commercialize drug candidates without first obtaining marketing approval from the FDA, NMPA, EMA and comparable regulatory authorities. In the U.S., we hope to file INDs for the drug candidates from our Lead Projects and, subject to the approval of IND, Phase 1 clinical trials in humans. Even if we are permitted to commence such clinical trials, they may not be successful and regulators may not agree with our conclusions regarding the data generated by our clinical trials.
We may be unable to complete development of our drug candidates or initiate or complete development of any future drug candidates we may develop on our projected schedule. While we believe that our existing cash will likely enable us to complete the preclinical development of at least one of our current Lead Projects, even assuming we can complete such preclinical studies for any drug candidate by 2021, the full clinical development, manufacturing and launch of that drug candidate, will take significant additional time and likely require funding beyond the existing cash. In addition, if regulatory authorities require additional time or studies to assess the safety or efficacy of our drug candidates, we may not have or be able to obtain adequate funding to complete the necessary steps for approval for our drug candidates or any future drug candidates.
20
Preclinical studies in animals and clinical trials in humans to demonstrate the safety and efficacy of our drug candidates are time-consuming, expensive and take several years or more to complete. Delays in preclinical or clinical trials, regulatory approvals or rejections of applications for regulatory approval in the U.S., Europe, the PRC or other markets may result from many factors, including but not limited to:
| ● | our inability to obtain sufficient funds required to conduct or continue a trial, including lack of funding due to unforeseen costs or other business decisions; | |
| ● | regulatory reports for additional analysts, reports, data, preclinical studies and clinical trials; | |
| ● | failure to reach agreement with, or inability to comply with conditions imposed by the FDA, NMPA, EMA or other regulators regarding the scope or design of our clinical trials; | |
| ● | regulatory questions regarding interpretations of data and results and the emergence of new information regarding our drug candidates or other products; | |
| ● | delay or failure in obtaining authorization to commence a clinical trial or inability to comply with conditions imposed by a regulatory authority regarding the scope or design of a clinical trial; | |
| ● | withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials; | |
| ● | unfavorable or inconclusive results of clinical trials and supportive non-clinical studies, including unfavorable results regarding effectiveness of drug candidates during clinical trials; | |
| ● | difficulty in maintaining contact with patients during or after treatment, resulting in incomplete data; | |
| ● | our inability to obtain approval from IRBs or ethics committees to conduct clinical trials at their respective sites; | |
| ● | our inability to enroll and retain a sufficient number of patients who meet the inclusion and exclusion criteria in a clinical trial; | |
| ● | our inability to conduct a clinical trial in accordance with regulatory requirements or our clinical protocols; | |
| ● | clinical sites and investigators deviating from trial protocol, failing to conduct the trial in accordance with regulatory requirements, withdrawing from or dropping out of a trial, or becoming ineligible to participate in a trial; | |
| ● | failure of our clinical trial managers to satisfy their contractual duties or meet expected deadlines; | |
| ● | manufacturing issues, including problems with manufacturing or timely obtaining from third parties sufficient quantities of a drug candidate for use in a clinical trial; | |
| ● | ambiguous or negative interim results, or results that are inconsistent with earlier results; | |
| ● | feedback from the FDA, NMPA, EMA, an IRB, data safety monitoring boards, or comparable entities, or results from earlier stage or concurrent studies in animals and clinical trials, regarding our drug candidates, including which might require modification of a trial protocol; | |
| ● | unacceptable risk-benefit profile or unforeseen safety issues or adverse side effects; and | |
| ● | a decision by the FDA, NMPA, EMA, an IRB, comparable entities, or the Company, or recommendation by a data safety monitoring board or comparable regulatory entity, to suspend or terminate clinical trials at any time for safety issues or for any other reason. |
Changes in regulatory requirements and guidance may also occur, and we may need to amend clinical trial protocols submitted to applicable regulatory authorities to reflect these changes. Amendments may require us to resubmit clinical trial protocols to IRBs or ethics committees for re-examination, which may increase the costs or time required to complete a clinical trial.
21
If we experience delays in the completion of, or the termination of, a clinical trial, of any of our drug candidates, the commercial prospects of our drug candidates will be harmed, and our ability to generate product sales revenues from any of those drug candidates will be delayed. In addition, any delay in completing our clinical trials will increase our costs, slow down our drug candidate development and approval process, and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our drug candidates.
If we are required to conduct additional clinical trials or other studies with respect to any of our drug candidates beyond those that we initially contemplated, if we are unable to successfully complete our clinical trials or other studies or if the results of these studies are not positive or are only modestly positive, we may be delayed in obtaining regulatory approval for that drug candidate, we may not be able to obtain regulatory approval at all or we may obtain approval for indications that are not as broad as intended. Our product development costs will also increase if we experience delays in testing or approvals, and we may not have sufficient funding to complete the testing and approval process. Significant clinical trial delays could allow our competitors to bring their products to market before we do and impair our ability to commercialize our drugs, if and when approved. If any of this occurs, our business will be materially harmed.
Our drug candidates may cause undesirable adverse events or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any regulatory approval.
Undesirable adverse events caused by our drug candidates or any future drug candidates we may develop could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, NMPA, EMA or other comparable regulatory authorities. Results of our potential clinical trials could reveal a high and unacceptable severity or prevalence of adverse effects. In such event, our trials could be suspended or terminated and the FDA, NMPA, EMA or other comparable regulatory authorities could order us to cease further development of, or deny approval of, our drug candidates for any or all target indications. Drug-related adverse events could also affect patient recruitment or the ability of enrolled subjects to complete the trial, could result in potential product liability claims and may harm our reputation, business, financial condition and business prospects significantly.
Additionally, if any of our current or future drug candidates receives regulatory approval, and we or others later identify undesirable side effects caused by such drugs, a number of potentially significant negative consequences could result, including but not limited to:
| ● | suspending the marketing of the drug; | |
| ● | having regulatory authorities withdraw approvals of the drug; | |
| ● | adding warnings on the label; | |
| ● | developing a REMS for the drug or, if a REMS is already in place, incorporating additional requirements under the REMS, or to develop a similar strategy as required by a comparable regulatory authority; | |
| ● | conducting post-market studies; | |
| ● | being sued and held liable for harm caused to subjects or patients; and | |
| ● | damage to our reputation. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular drug candidate, if approved, and could significantly harm our business, results of operations and prospects.
22
Even if we receive regulatory approval for our drug candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our drug candidates.
If our drug candidates or any future drug candidates we develop are approved, they will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, efficacy, and other post-market information, including both federal and state requirements in the United States and requirements of comparable regulatory authorities outside of the United States.
Manufacturers and manufacturers’ facilities are required to comply with extensive requirements from the FDA, NMPA, EMA and comparable regulatory authorities, including, in the United States, ensuring that quality control and manufacturing procedures conform to cGMP regulations. As such, our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any NDA, other marketing application, and previous responses to inspection observations. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control.
Any regulatory approvals that we receive for our drug candidates may be subject to limitations on the approved indicated uses for which the drug may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials and surveillance to monitor the safety and efficacy of the drug candidate. The regulatory authorities may also require risk management plans or programs as a condition of approval of our drug candidates (such as REMS of the FDA and risk-management plan of the EMA), which could entail requirements for long-term patient follow-up, a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA, NMPA, EMA or a comparable regulatory authority approves our drug candidates, we will have to comply with requirements including, for example, submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGCP and cGMP, for any clinical trials that we conduct post-approval.
The FDA may impose consent decrees or withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the drug reaches the market. Later discovery of previously unknown problems with our drug candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of our drug candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; | |
| ● | fines, untitled or warning letters, or holds on clinical trials; | |
| ● | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals; | |
| ● | product seizure or detention, or refusal to permit the import or export of our drug candidates; and | |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Companies may promote drugs only for the approved indications and in accordance with the provisions of the approved label and may not promote drugs for any off-label use, such as uses that are not described in the product’s labeling and that differ from those approved by the regulatory authorities. However, physicians may prescribe drug products for off-label uses and such off-label uses are common across some medical specialties. Thus, they may, unbeknownst to us, use our product for an “off label” indication for a specific treatment recipient. The FDA, NMPA, EMA and other regulatory authorities actively enforce the laws and regulations prohibiting the promotion of off-label uses, and if we are found to be out of compliance with the requirements and restrictions imposed on us under those laws and restrictions, we may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions, and the off-label use of our products may increase the risk of product liability claims. In addition, management’s attention could be diverted from our business operations and our reputation could be damaged.
23
The policies of the FDA, NMPA, EMA and other regulatory authorities may change and we cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any regulatory approval that we may have obtained and we may not achieve or sustain profitability.
We may be subject to government regulations for dietary supplements
The Company may develop some of the molecules under development in formulations intended as dietary supplements. The FDA regulates dietary supplements and drugs under different regulatory schemes, and the Company’s dietary supplement formulations will also be subject to other government regulation, including regulation by the Centers for Medicare & Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services, state and local governments and the foreign equivalents of the FDA and these other agencies.
For example, the FDA regulates the research, development, preclinical and clinical testing, safety, effectiveness, record keeping, reporting, labeling, storage processing, formulation, safety, manufacturing, packaging, labeling, advertising and distribution import and export of pharmaceutical products under various regulatory provisions. If any dietary supplements we develop are tested or marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not we have obtained FDA approval for a given product and its uses. Such foreign regulation may be equally or more demanding than corresponding U.S. regulation.
In addition, the regulatory policies of the agencies in the U.S. or other countries may change and additional government regulations may be issued that could prevent, limit, or delay regulatory approval of our dietary supplement candidates, or impose more stringent product labeling and post-marketing testing and other requirements.
Risks Related to Commercialization of Our Drug Candidates
Even if any of our drug candidates receive regulatory approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
After we complete clinical trials and receive regulatory approval for any of our drug candidates, which may not happen for some time, we recognize that such candidate(s) may ultimately fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. We may not be able to achieve or maintain market acceptance of our products over time if new products or technology are introduced that are more favorably received than our products, are more cost effective or render our drug obsolete. We will face competition with respect to our drug candidates from other pharmaceutical companies developing products in the same disease/therapeutic area and specialty pharmaceutical and biotechnology companies worldwide. Many of the companies against which we may be competing have significantly greater financial resources and expertise in research and development, manufacturing, animal testing, conducting clinical trials, obtaining regulatory approvals and marketing approval for drugs than we do. Physicians, patients and third-party payors may prefer other novel products to ours, which means that we may not generate significant sales revenues for that product and that product may not become profitable. The degree of market acceptance of our drug candidates, if approved for commercial sale, will depend on a number of factors, including but not limited to:
| ● | clinical indications for which our drug candidates are approved; | |
| ● | physicians, hospitals, and patients considering our drug candidates as a safe and effective treatment; |
24
| ● | the potential and perceived advantages of our drug candidates over alternative treatments; | |
| ● | the prevalence and severity of any side effects; | |
| ● | product labeling or product insert requirements of the FDA, NMPA, EMA or other comparable regulatory authorities; | |
| ● | limitations or warnings contained in the labeling approved by the FDA, NMPA, EMA or other comparable regulatory authorities; | |
| ● | the timing of market introduction of our drug candidates as well as competitive drugs; | |
| ● | the cost of treatment in relation to alternative treatments and their relative benefits; | |
| ● | the availability of adequate coverage, reimbursement and pricing by third-party payors and government authorities; | |
| ● | lack of experience and financial and other limitations on our ability to create and sustain effective sales and marketing efforts or ineffectiveness of our sales and marketing partners; and | |
| ● | changes in legislative and regulatory requirements that could prevent or delay regulatory approval of our drug candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any drug candidates for which we obtain regulatory approval. |
Risks Related to Our IP
A significant portion of our IP portfolio currently includes pending patent applications that have not yet been issued as granted patents and if the pending patent applications covering our product candidates fail to be issued, our business will be adversely affected. If we or our licensors are unable to obtain and maintain patent protection for our technology and drugs, our competitors could develop and commercialize technology and drugs similar or identical to ours, and our ability to successfully commercialize our technology and drugs may be adversely affected.
Our success depends largely on our ability to obtain and maintain patent protection and other forms of IP rights for the composition of matter, method of use and/or method of manufacture for each of our drug candidates. Failure to obtain, maintain protection, enforce or extend adequate patent and other IP rights could materially adversely affect our ability to develop and market one or more of our drug candidates. We also rely on trade secrets and know-how to develop and maintain our proprietary and IP position for each of our drug candidates. Any failure to protect our trade secrets and know-how with respect to any specific drug and device candidate could adversely affect the market potential of that potential product.
As of the date hereof, the Company has, through its licenses, obtained rights to patents and patent applications covering some or all its drug and device candidates that have been filed in major jurisdictions such as the United States, member states of the European Patent Organization (the “EPO”) and the PRC (collectively, “Major Patent Jurisdictions”), as well as in other countries.
As of the date hereof, we are the exclusive licensee of 13 U.S. patents and 6 pending U.S. non-provisional applications, as well as corresponding patents and patent applications internationally. In addition, we are the exclusive licensee of 3 international patent applications under the Patent Cooperation Treaty (the “PCT”) which we have filed and/or plan to file nationally in member states of the EPO, PRC and other jurisdictions before the expiration of the time limits for entry of national stage application. We have also filed a number of provisional applications to establish earlier filing dates for certain of our other ongoing researches, the specifics of which are currently proprietary and confidential. To the extent we do not seek or obtain patent protection in a particular jurisdiction, we may not have commercial incentive to seek marketing authorization in such jurisdiction. Nonetheless, other parties might enter those markets with generic versions or copies of our products and received regulatory approval without having significantly invested in their own research and development costs compared to the Company’s investment. For more information about our IP portfolio, please refer to the Intellectual Property section below.
25
With respect to issued patents in certain jurisdictions, for example in the U.S. and under the EPO, we may be entitled to obtain a patent term extension to extend the patent expiration date provided we meet the applicable requirements for obtaining such patent term extensions. We have sought to support our proprietary position by working with our licensors in filing patent applications in the names of the licensors in the United States and through the PCT, related to the Lead Projects and certain other drug candidates. In the future, we intend to file patent applications on supplemental or improvement IP derived from the licensed technologies, where those IP would be solely or jointly owned by the Company pursuant to the terms of respective license agreements. Filing patents covering multiple technologies in multiple countries is time-consuming and expensive, and we may not have the resources file and prosecute all necessary or desirable patent applications in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
We cannot be certain that patents will be issued or granted with respect to patent applications that are currently pending, or that issued or granted patents will not later be found to be invalid or unenforceable.
The patent position of biotechnology and pharmaceutical companies is generally uncertain because it involves complex legal and factual considerations. The standards applied by the EPO, the U.S. Patent and Trademark Office, or USPTO, and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology and pharmaceutical patents. Consequently, patents may not issue from our pending patent applications and even if they do issue, such patents may not issue in a form that effectively prevents others from commercializing competing products. As such, we do not know the degree of future protection that we will have on our proprietary products and technology.
Additionally, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Even if patents do successfully issue and even if such patents cover our drug candidates, other parties may initiate, for patents filed before March 16, 2013 (i.e., the enactment of the America Invents Act), interference or re-examination proceedings, for patents filed on or after March 16, 2013, post-grant review,inter partes review, nullification or derivation proceedings, in court or before patent offices, or similar proceedings challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated. Successful defense of its patents can constitute a material factor in a company’s expenses. According to an August 2017 article published by Bloomberg News (https://www.bna.com/cost-patent-infringement-n73014463011/), depending on the value at stake, the American Intellectual Property Law Association’s “2017 Report of the Economic Survey” reported the average cost of a patent litigation in 2017 to be $1.7 million.
In addition, the fact that the Company has exclusive rights to prevent others from using a patented invention does not necessarily mean that the Company itself will have the unrestricted right to use that invention. Other parties may obtain ownership or licenses to patents or other IP rights that cover the manufacture, use or sale of our current or future products (or elements thereof). This may enable such other parties to enforce their patents or IP rights against us, and may, as a result, affect the commercialization of our products or exploitation of our own technology. We endeavor to identify early patents and patent applications which may block development of a product or technology and minimize this risk by conducting prior art searches before and during the projects. However, relevant documents may be overlooked, yet-to-be published or missed, which may in turn impact on the freedom to commercialize the relevant asset. In such cases, we may not be in a position to develop or commercialize products or drug candidates unless we successfully pursue litigation to nullify or invalidate the other IP rights concerned, or enter into a license agreement with the IP right holder, if available on commercially reasonable terms.
If we are unable to obtain and maintain the appropriate scope for our patents, our competitors could develop and commercialize technology and drugs similar or identical to ours, and our ability to successfully commercialize our technology and drugs may be adversely affected.
We may not obtain sufficient claim scope in those patents to prevent another party from competing successfully with our drug and device candidates. Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technology or drug and device candidates in a non-infringing manner. The issuance of a patent is not conclusive as to its scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in patent claims being narrowed, invalidated or held unenforceable, which could limit our ability to stop or prevent us from stopping others from using or commercializing similar or identical technology and drug and device candidates, or limit the duration of the patent protection of our technology and drug and device candidates. Given the amount of time required for the development, testing and regulatory review of new drug and device candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing drug and device candidates similar or identical to ours.
26
Further, the issuance, scope, validity, enforceability and commercial value of our and our current or future licensors’ or collaboration partners’ patent rights are highly uncertain. Our and our licensors’ pending and future patent applications may not result in patents being issued which protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products.
We may not be able to protect and enforce our IP rights throughout the world.
Our commercial success will depend, in part, on our ability to maintain IP protection for our drug candidates in which we seek to develop and commercialize. While we rely primarily upon a combination of patents, trademarks, trade secrets and other contractual obligations to protect the IP related to our brands, products and other proprietary technologies, these legal means may afford only limited protection.
Filing and prosecuting patents on drug candidates and defending the validity of the same (if challenged) in all countries throughout the world could be prohibitively expensive for us, and our IP rights in countries outside the Major Patent Jurisdictions can be less extensive than those in the Major Patent Jurisdictions. In addition, the laws of some countries in the rest of the world such as India do not protect IP rights to the same extent as laws in the Major Patent Jurisdictions. Consequently, we may not be able to prevent other parties from practicing our inventions in the rest of the world. Competitors may use our technology in jurisdictions where we have not or not yet obtained patent protection to develop their own drugs and further, may export otherwise infringing drugs to non-U.S. jurisdictions where we have patent protection.
Our, our licensors’ or collaboration partners’ patent applications cannot be enforced against other parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then only to the extent the issued claims cover the technology. In addition, patents and other IP rights also will not protect our technology, drug candidates if another party, including our competitors, design around our protected technology, drug candidates without infringing, misappropriating or otherwise violating our patents or other IP rights.
Moreover, currently and as our R&D continues to progress, some of our patents and patent applications are or may be co-owned with another party. Some of our licenses already provide that future-developed technologies (and any resulting patents) will be co-owned with the licensors and other patents for technologies we may acquire or develop with other parties may also be jointly owned. If we are unable to obtain an exclusive license to any such co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other persons, including our competitors, and our competitors could market competing products and technology, and we will be unable to transfer or grant exclusive rights to potential purchasers or development partners of such co-owned technologies. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against other parties, and such cooperation may not be provided to us. Any of the foregoing could limit the revenue we might generate from our patents or patent applications and thus have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Because patent applications are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we or our licensors or collaborators were or will be the first to file any patent application related to a drug or device candidate. Furthermore, in the United States, if patent applications of other parties have an effective filing date before March 16, 2013, an interference proceeding can be initiated by such other party to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. If patent applications of other parties have an effective filing date on or after March 16, 2013, in the United States a derivation proceeding can be initiated by such other parties to determine whether our invention was derived from theirs.
27
Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing our invention where the other party can show that they used the invention in commerce before our filing date or the other party benefits from a compulsory license. In addition, we may be subject to other challenges regarding our exclusive ownership of our IP. If another party were successful in challenging our exclusive ownership of any of our IP, we may lose our right to use such IP, such other party may be able to license such IP to other parties, including our competitors, and our competitors could market competing products and technology. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Many companies have encountered significant problems in protecting and defending IP rights in jurisdictions outside Major Patent Jurisdictions. The legal systems of some countries do not favor the enforcement of patents, trade secrets and other IP, which could make it difficult in those jurisdictions for us to stop the infringement or misappropriation of our patents or other IP rights, or the marketing of competing drugs in violation of our proprietary rights generally.
To date, we have not sought to enforce any issued patents in any jurisdictions. Proceedings to enforce our patent and other IP rights in any jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business.
Furthermore, such proceedings could put our patents at risk of being invalidated, held unenforceable, or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke other parties to assert claims of infringement or misappropriation against us. We may not prevail in any lawsuits that we initiate in jurisdictions where opposition proceedings are available and the damages or other remedies awarded, if any, may not be commercially meaningful. The requirements for patentability may differ in certain countries, particularly developing countries. Certain countries in Europe, the PRC, and developing countries including India, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to other parties. In those countries, we and our licensors may have limited remedies if patents are infringed or if we or our licensors are compelled to grant a license to another party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our IP rights around the world may be inadequate to obtain a significant commercial advantage from the IP that we develop.
We may become involved in lawsuits to protect or enforce our IP, which could be expensive, time-consuming and unsuccessful. Our patent rights relating to our drug and device candidates could be found invalid or unenforceable if challenged in court or before the USPTO or comparable non-U.S. authority.
Competitors may infringe our patent rights or misappropriate or otherwise violate our IP rights. To counter infringement or unauthorized use, litigation may be necessary in the future to enforce or defend our IP rights, to protect our trade secrets or determine the validity and scope of our own IP rights or the proprietary rights of others. This can be expensive and time-consuming. Any claim that we assert against perceived infringers could also provoke these parties to assert counterclaims against us alleging that we infringe their IP rights. Many of our current and potential competitors have the ability to dedicate substantially greater resources to enforce and/or defend their IP rights than we can. Accordingly, despite our efforts, we may not be able to prevent other parties from infringing upon or misappropriating our IP. Litigation could result in substantial costs and diversion of management resources, which could harm our business and financial results. In addition, in an infringement proceeding, a court may decide that patent rights or other IP rights owned by us are invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patent rights or other IP rights do not cover the technology in question. An adverse result in any litigation proceeding could put our patent, as well as any patents that may issue in the future from our pending patent applications, at risk of being invalidated, held unenforceable or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with IP litigation, there is risk that some of our confidential information could be compromised by disclosure during this type of litigation.
28
If we initiate legal proceedings against another party to enforce our patent, or any patents that may be issued in the future from our patent applications, that relates to one of our drug and device candidates, the defendant could counterclaim that such patent rights are invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace, and there are numerous grounds upon which another party can assert invalidity or unenforceability of a patent. Parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include ex parte re-examination,inter partes review, post-grant review, derivation and equivalent proceedings in non-U.S. jurisdictions, such as opposition proceedings. Such proceedings could result in revocation or amendment to our patents in such a way that they no longer cover and protect our drug and device candidates. With respect to the validity of our patents, for example, there may be invalidating prior art of which we, our patent counsel, and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our drug and device candidates. Such a loss of patent protection could have a material adverse impact on our business.
We may not be able to prevent misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States. Furthermore, because of the substantial amount of discovery required in connection with IP litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
We may be subject to claims challenging the inventorship of our patents and other IP.
Although we are not currently experiencing any claims challenging the inventorship of our patents or ownership of our IP, we may in the future be subject to claims that former employees, collaborators or other parties have an interest in our patents or other IP as inventors or co-inventors. For example, we may have inventorship disputes arise from conflicting obligations of consultants or others who are involved in developing our drug and device candidates and who have not clearly contracted to transfer or assign any rights they may have to the Company. In addition, for our licensed patents, although a majority of our licensors have procured assignment forms and records from inventors to affirm their ownership in the licensed IP, another party or former employee or collaborator of our licensors not named in the patents may challenge the inventorship of claim an ownership interest in one or more of our or our licensors’ patents. Litigation may be necessary to defend against these and other claims challenging inventorship. If we fail in defending any such claims, in addition to paying monetary damages, we may lose rights such as exclusive ownership of, or right to use, our patent rights or other IP. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
If we are sued for infringing IP rights of other parties, such litigation could be costly and time-consuming and could prevent or delay us from developing or commercializing our drug candidates, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends in part on our avoiding infringement of the patents and other IP rights of other parties. There is a substantial amount of litigation involving patent and other IP rights in the biotechnology and pharmaceutical industries. Numerous issued patents, provisional patents and pending patent applications, which are owned by other parties, exist in the fields in which we are developing drug candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our drug candidates may give rise to claims of infringement of the patent rights of others.
Other parties may assert that we are employing their proprietary technology without authorization. There may be other patents of which we are currently unaware with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our drug candidates. Because patent applications can take many years to issue, there may be currently pending patent applications or provisional patents which may later result in issued patents that our drug candidates may infringe. In addition, other parties may obtain patents in the future and claim that use of our technology infringes upon these patents. If any other patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our drug candidates, any molecules formed during the manufacturing process or any final drug itself, the holders of any such patents may be able to prevent us from commercializing such drug candidate unless we obtain a license under the applicable patents, or until such patents expire or they are finally determined to be held invalid or unenforceable. Similarly, if any other patent were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy or patient selection methods, the holders of any such patent may be able to block our ability to develop and commercialize the applicable drug candidate unless we obtain a license, limit our uses, or until such patent expires, or is finally determined to be held invalid or unenforceable. In either case, such a license may not be available on commercially reasonable terms or at all.
29
Other parties who bring successful claims against us for infringement of their IP rights may obtain injunctive or other equitable relief, which could prevent us from developing and commercializing one or more of our drug candidates. Defense of these claims, regardless of their merits, would involve substantial litigation expense and be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement or misappropriation against us, we may have to pay substantial damages, including treble damages and attorneys’ fees in the case of willful infringement, obtain one or more licenses from other parties, pay royalties or redesign our infringing drug candidates, which may be impossible or require substantial time and monetary expenditure. In the event of an adverse result in any such litigation, or even in the absence of litigation, we may need to obtain licenses from other parties to advance our research or allow commercialization of our drug candidates. Any required license may not be available at all, or may not be available on commercially reasonable terms. In the event that we are unable to obtain such a license, we would be unable to further develop and commercialize one or more of our drug candidates, which could harm our business significantly. We may also elect to enter into license agreements in order to settle patent infringement claims or resolve disputes prior to litigation, and any such license agreements may require us to pay royalties and other fees that could significantly reduce our profitability for any product related to that patent and thus harm our business.
Even if resolved in our favor, litigation or other legal proceedings relating to IP claims may cause us to incur significant expenses, and could distract our technical personnel, management personnel, or both from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our Class A Ordinary Shares. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing or distribution activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
There may be patent applications pending of which we are not aware, but which cover similar products to the ones we are attempting to license or develop, which may result in lost time and money, as well as litigation.
It is possible that we have failed to identify relevant outstanding patents or applications. For example, U.S. applications filed before November 29, 2000 and certain U.S. applications filed after that date that will not be filed outside the United States remain confidential until patents are issued. Patent applications filed in the United States after November 29, 2000 and generally filed elsewhere are published approximately 18 months after the earliest filing for which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Therefore, patent applications covering our products could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our products or the use of our products. Holders of any such unanticipated patents or patent applications may actively bring infringement claims against us, with the same potential litigation consequences as alluded to elsewhere in this prospectus. Any of these events could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
30
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and other patent agencies in several stages over the lifetime of the patent. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions during the patent application process. Although an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees, and failure to properly submit documents requesting an extension of time. In any such event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
The terms of our patents may not be sufficient to effectively protect our drug and device candidates and business.
In most countries in which we file, including the United States, the term of an issued patent is generally 20 years from the earliest claimed filing date of a non-provisional patent application in the applicable country. Although various extensions may be available, the life of a patent and the protection it affords is limited. For example, depending upon the timing, duration and specifics of the FDA regulatory approval for our drug candidates, one or more of our U.S. patents, if issued, might be eligible for limited patent term restoration under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent term extension of up to five years as compensation for patent term lost during drug development and the FDA regulatory review process. Patent term extensions, however, cannot extend the remaining term of a patent beyond a total of 14 years from the date of drug approval by the FDA, and only one patent can be extended for a particular drug. The application for patent term extension is subject to approval by the USPTO, in conjunction with the FDA. We may not be granted an extension because of, for example, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain a patent term extension for a given patent or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our drug will be that of the originally issued patents themselves.
Even if patents covering one of our drug candidates are obtained, thereby giving us a period of exclusivity for manufacturing and marketing that drug, we will not be able to assert such patent rights upon the expiration of the issued patents against potential competitors who may begin marketing generic copies of our medications, and our business and results of operations may be adversely affected.
Changes in patent law in the United States could diminish the value of patents in general, thereby impairing our ability to protect our drug and device candidates.
The United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained, if any. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents in the United States could change in unpredictable ways that would weaken our ability to obtain new patents, or to enforce our existing patents and patents that we might obtain in the future. For example, in a recent case,Assoc. for Molecular Pathology v. Myriad Genetics, Inc., the U.S. Supreme Court held that certain claims to naturally-occurring substances are not patentable. Although we do not believe that any of the patents owned or licensed by us will be found invalid based on this decision, future decisions by the courts, the U.S. Congress or the USPTO may impact the value of our patent rights. There could be similar changes in the laws of foreign jurisdictions that may impact the value of our patent rights or our other IP rights.
31
In addition, recent patent reform legislation in the U.S., including the Leahy-Smith America Invents Act, or the America Invents Act, could increase those uncertainties and costs. The America Invents Act was signed into law on September 16, 2011, and many of the substantive changes became effective on March 16, 2013. The America Invents Act reforms U.S. patent law in part by changing the U.S. patent system from a “first to invent” system to a “first inventor to file” system, expanding the definition of prior art, and developing a post-grant review system, thus changing the U.S. patent law in a way that may weaken our ability to obtain patent protection in the U.S. for those applications filed after March 16, 2013. Further, the America Invents Act created new procedures to challenge the validity of issued patents in the U.S., including post-grant review andinterpartes review proceedings, which some other parties have been using to cause the cancellation of selected or all claims of issued patents of competitors. For a patent with an effective filing date of March 16, 2013 or later, a petition for post-grant review can be filed by another party in a nine-month window from issuance of the patent. A petition forinter partes review can be filed immediately following the issuance of a patent if the patent has an effective filing date prior to March 16, 2013. A petition forinter partes review can be filed after the nine-month-period for filing a post-grant review petition has expired for a patent with an effective filing date of March 16, 2013 or later. Post-grant review proceedings can be brought on any ground of invalidity, whereasinter partes review proceedings can only raise an invalidity challenge based on published prior art and patents. These adversarial actions at the USPTO review patent claims without the presumption of validity afforded to U.S. patents in lawsuits in U.S. federal courts, and use a lower burden of proof than used in litigation in U.S. federal courts. Therefore, it is generally considered easier for a competitor or other party to have a U.S. patent invalidated in a USPTO post-grant review orinter partes review proceeding than invalidated in a litigation in a U.S. federal court. If any of our patents are challenged by another party in such a USPTO proceeding, there is no guarantee that we or our licensors or collaborators will be successful in defending the patent, which would result in our loss of the challenged patent right.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to our issued patents, provisional patent, and pending patent applications, we expect to rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position and protect our drug and device candidates. We seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties that have access to them, such as our employees, corporate collaborators, outside scientific collaborators, sponsored researchers, contract manufacturers, consultants, advisors and other parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. However, any of these parties may breach such agreements and disclose our proprietary information, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. If trade secrets which are material to our business were to be obtained by a competitor, our competitive position would be harmed.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed IP, including trade secrets or other proprietary information, of any such employee’s former employer. In addition, while we typically require our employees, consultants and contractors who may be involved in the development of IP to execute agreements assigning such IP to us, we may be unsuccessful in executing such an agreement with each party who in fact develops IP that we regard as our own, which may result in claims by or against us related to the ownership of such IP. We are not aware of any threatened or pending claims that any of our projects involve misappropriated IP or other proprietary information, but in the future litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable IP rights. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
32
We may be unable to execute on the optimal development plan for one or more of our existing product candidates if we are unable to obtain or maintain necessary rights for some aspect of the developing technology through acquisitions or licenses.
Our existing programs currently use or may in the future use additional technologies subject to proprietary rights held by others, such as particular compositions or methods of manufacture, treatment or use. The licensing and acquisition of IP rights is a competitive area, and more established companies may pursue strategies to license or acquire such IP rights that we may consider necessary or useful. These established companies may have a competitive advantage over us due to their size, cash resources and greater capabilities in clinical development and commercialization.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire IP rights on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain or maintain licenses or other rights from other parties to use IP of those parties, our business, financial condition and prospects for growth could suffer.
If we fail to comply with our obligations in the agreements under which we license IP rights from other parties or otherwise experience disruptions to our business relationships with our licensors, we could be required to pay monetary damages or could lose license rights that are important to our business.
Many of our projects (including our Lead Projects) are based on IP which we have licensed from other parties. (See “Our Business – Intellectual Property”) Certain of these license agreements impose diligence, development or commercialization obligations on us, such as obligations to pay royalties on net product sales of our drug and device candidates once commercialized by us, to pay a percentage of sublicensing revenues if the licensed product is sublicensed, to make other specified milestone and/or annual payments relating to our drug candidates or to pay license maintenance and other fees, as well as obligations to pursue commercialization with due diligence. Specifically, a number of our license agreements also require us to meet development timelines in order to maintain the related license(s). In spite of our efforts, our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore seek to terminate the license agreements. If one of our licensors, despite our efforts, were to be successful in terminating its agreement with us, we would not be able to continue to develop, manufacture or market any drug candidate under that license agreements, and we could face claims for monetary damages or other penalties under that agreement. Such an occurrence would diminish or eliminate the value of that project to our Company, even if we are able to negotiate new or reinstated agreements, which may have less favorable terms. Depending on the importance of the IP and the related project, any such development could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| ● | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; | |
| ● | the sublicensing of patent and other rights under our collaborative development relationships; | |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; | |
| ● | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and | |
| ● | the priority of invention of patented technology. |
In addition, the agreements under which we currently license intellectual property or technology from other parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which (depending on the importance of the IP and the related project) could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangement for a project on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected drug or device candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects.
33
We may not have complete control of the preparation, filing and prosecution of patent applications, or to maintain patents, licensed by us from other parties.
The Company has in-licensed, and expects in the future to in-license patents owned or controlled by others for our use as part of our development plans. We also may out-license or sublicense patents which we own or control in collaborations with others for development and commercialization of our products. In either case, the continuing right to control the preparation, filing and prosecution of patent applications, or to maintain the patents, covering technology under development is a matter for negotiation and we may not always be the party that obtains such control, in which case we will be reliant on our licensors, collaboration partners or sublicensees for determining strategies with respect to those patents. For our existing licenses, while we have an understanding with most of the licensors who maintain control over patent prosecution and we have jointly appointed and engaged patent agents nominated by us under one or more of our licenses, we cannot guarantee that such licensors or collaborators will always accept prosecution strategies proposed by us and/or our patent agents. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. If our current or future licensors or collaboration partners fail to establish, maintain or protect such patents and other IP rights, such rights may be reduced or eliminated. If our licensors or joint development partners are not fully cooperative or disagree with us as to the prosecution, maintenance or enforcement of any patent rights, such patent rights could be compromised.
Risks Related to Our Reliance on Unrelated Parties
We rely on unrelated parties to conduct discovery and further improvement of our innovations and licensed technologies, as well as our preclinical studies and clinical trials. If these unrelated parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our drug candidates, and our business could be substantially harmed.
We have relied upon and plan to continue to rely upon CROs and collaborating institutions to monitor and manage data for our ongoing preclinical studies and programs. We rely on these parties for execution of preclinical studies and clinical trials, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, and regulatory requirements and scientific standards, and our reliance on the CROs and collaborating institutions does not relieve us of our regulatory responsibilities. If CROs, collaborating institutions or clinical investigators do not successfully carry out their contractual duties or obligations or meet expected deadlines, development of our product candidates could be delayed and our business could be adversely affected.
In addition, our CROs and collaborating institutions, are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and waste. In the event of contamination or injury resulting from our use of hazardous materials, we might be held liable for any resulting damages, and any liability could exceed our resources. We could also be subject to civil or criminal fines and penalties, and significant associated costs.
If the Company obtains approval of an IND for one of our drug candidates and moves into human clinical trials requiring significantly larger quantities of the candidate to be tested, we expect to rely on unrelated parties to manufacture supplies of that candidate. If those unrelated parties fail to provide us with sufficient quantities of clinical supply on that candidate or fail to do so at acceptable quality levels or prices, or fail to maintain required cGMP licenses, we may not be able to manufacture that candidate in sufficient quantities to conduct the necessary human trials. Should the failure by the CRO occur in anticipation of or after marketing approval of that candidate, we may be unable to generate as much revenue as rapidly (and such revenue may not be as profitable) as we had anticipated.
The manufacture of many drug products, particularly in commercial quantities, can be complex and may require significant expertise and capital investment, particularly if the development of advanced manufacturing techniques and process controls are required. If we obtain approval of an IND for any of our drug candidates, of which there can be no assurance, we intend to contract with outside contractors to manufacture clinical supplies and process our drug candidates. We have not yet had our drug candidates to be manufactured or processed on a commercial scale and may not be able to do so for any of our drug candidates.
34
As we expect to engage contract manufacturers, the Company will be exposed to the following risks:
| ● | we might be unable to identify manufacturers on acceptable terms or at all because the FDA, NMPA, EMA or other comparable regulatory authorities must approve any manufacturers we determine to use and any potential manufacturer may be unable to satisfy federal, state or international regulatory standards; | |
| ● | although we would be choosing manufacturers with the type of experience most suitable for our drug candidates, it is possible that our contract manufacturers may not be able to execute unique manufacturing procedures and other logistical support requirements we have developed and they might require a significant amount of support from us to implement and maintain the infrastructure and processes required to manufacture our particular drug candidates; | |
| ● | our contract manufacturers might be unable to reproduce the quantity and quality of the drugs we need to meet our clinical and commercial needs within the time frames when we require those drugs; | |
| ● | our contract manufacturers may breach their contracts with us, including by not performing as agreed or not devoting sufficient resources to our drug candidates, or they may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products; | |
| ● | even if initially accepted by regulatory authorities, a manufacturer remains subject to ongoing periodic unannounced inspection by regulatory authorities to ensure strict compliance with cGMP and other government regulations, and our contract manufacturers may fail to comply with these regulations and requirements, resulting in rescission of cGMP licenses and our inability to continue using their services, requiring us to find a replacement manufacturer; | |
| ● | depending on the terms of our agreement with a manufacturer, we may not own, or may have to share, the IP rights to any improvements made by the manufacturer in the manufacturing process for our drug candidates; and | |
| ● | our contract manufacturers may have unacceptable or inconsistent product quality success rates and yields. |
Each of these risks could delay or prevent the completion of our clinical trials or the approval of any of our drug candidates by the FDA, NMPA, EMA or other comparable regulatory authorities, result in higher costs or adversely impact commercialization of our drug candidates.
We are also responsible for quality control by our manufacturers. We intend to rely on those unrelated-party manufactures to perform certain quality assurance tests on our drug candidates prior to delivery to patients. If these tests are not appropriately done and test data are not reliable, patients could be put at risk of serious harm and the FDA, NMPA, EMA or other comparable regulatory authorities could place significant restrictions on our Company until deficiencies are remedied.
35
Manufacturers of drug products often encounter difficulties in production, particularly in scaling up or out, validating the production process, and assuring high reliability of the manufacturing process (including the absence of contamination). These problems include logistics and shipping, difficulties with production costs and yields, quality control, including stability of the product, product testing, operator error, availability of qualified personnel, as well as compliance with strictly enforced federal, state and non-U.S. regulations. Furthermore, if contaminants are discovered in our supply of our drug candidates or in the manufacturing facilities, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. It is possible that stability failures or other issues relating to the manufacture of our drug candidates may occur in the future. Additionally, our manufacturers may experience manufacturing difficulties due to resource constraints, or as a result of labor disputes or unstable political environments. If our manufacturers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, our ability to provide our drug candidate to patients in clinical trials would be jeopardized. Any delay or interruption in the manufacturing of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to begin new clinical trials with additional costs or terminate clinical trials completely.
Review of changes in the manufacturing process of our drug candidates could cause delays resulting from the need for additional regulatory approvals.
Changes in a process or procedure for manufacturing one of our drug candidates, including a change in the location where the drug candidate is manufactured or a change of a contract manufacturer, could require prior review by the FDA, NMPA, EMA or other comparable regulatory authorities and approval of the manufacturing process and procedures in accordance with the FDA, NMPA or EMA’s regulations, or comparable requirements. This review may be costly and time-consuming and could delay or prevent the launch of a product. The new facility will also be subject to pre-approval inspection. In addition, we would have to demonstrate that the product made at the new facility is equivalent to the product made at the former facility by physical and chemical methods, which are costly and time-consuming. It is also possible that the FDA, NMPA, EMA or other comparable regulatory authorities may require clinical testing as a way to prove equivalency, which would result in additional costs and delay.
Risks Related to AML Clinic
Failure to comply with all laws and regulations applicable to the business of AML Clinic could have a material, adverse impact on the Company’s business.
Operation of AML Clinic subjects the Company to a variety of Hong Kong laws and regulations specific to companies and professionals in the business of delivering medical care. We and our employees will be subject to licensing and professional qualifications that do not apply to our other businesses. Breach of any of these laws, regulations or licensing requirements could subject the Company to significant fines and other penalties and possibly damage the Company’s reputation, which could have a material adverse effect on the Company’s business.
Risks Related to Our Device Candidates
We are subject to risks related to obtaining regulatory approval for device candidates.
The Company’s device candidates (including those being developed under SLS-1), are likely to be regulated as medical devices. Medical devices are subject to extensive regulations, supervised by regulatory authorities around the world, including the FDA, NMPA and applicable national authorities in relevant European countries. The regulatory framework related to medical devices covers research, development, design, manufacturing, safety, reporting, testing, labeling, packaging, storage, installation, servicing, marketing, sales and distribution. The Company is and may also be, in addition to these industry-specific regulations, subject to numerous other ongoing regulatory obligations, such as data protection, environmental, health and safety laws and restrictions. The costs of compliance with applicable regulations, requirements or guidelines could be substantial. Furthermore, the regulatory environment has generally become more stringent and extensive over time. Failure to comply with these regulations could result in sanctions including fines, injunctions, civil penalties, denial of applications for marketing approval of the Company’s products, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions, partial suspension or total shutdown of production and criminal prosecutions, any of which could significantly increase the Company’s costs, delay the development and commercialization of its device candidates.
36
We are subject to risks related to the carrying out and outcome of clinical trials of medical devices.
The Company may sponsor studies on human participants in clinical studies of its device candidates. Such clinical studies are performed to support regulatory approvals for market access or to generate evidence relating to clinical benefits and cost benefits of using such device candidates. Clinical studies are costly and time consuming and associated with risks such as finding trial sites, recruitment of suitable patients, the actual cost per patient exceeding budget and inadequacies in the execution of the trials. There is also a risk of delays in the performance of clinical studies, which can occur for a variety of reasons. For example, delays in obtaining regulatory approval to commence a trial, reaching agreements on acceptable terms with prospective contract research organizations (“CROs”) and clinical investigational sites, obtaining institutional review board approval at each site, difficulties in patient enrolment, patients failing to complete a trial or return for follow-up, adding new sites or obtaining sufficient supplies of products or clinical sites dropping out of a trial. If delays persist, there is a risk that studies eventually are suspended or terminated if the delays occur due to circumstances that a sponsor of a clinical trial has difficulties controlling, or is unable to control, or if the measures required for conducting the studies further are deemed too costly or extensive in relation to the scopes and goals of the studies.
There are many factors which may affect patient enrollment. Amongst these are the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical study and competing clinical studies. Furthermore, clinicians’ and patients’ perceptions as to the potential advantages of the product being studied in relation to other available therapies, including any new products that may be approved for the indications the company is investigating. Clinical studies may also be suspended or terminated if participating subjects are exposed to unacceptable health risks or undesired side-effects.
Furthermore, there is a risk that clinical studies may not demonstrate the required clinical benefit for the prospective indication the trial is aimed at. Failure in premarketing clinical studies could lead to market clearance or approvals not being obtained which could delay or jeopardize the Company’s ability to develop, market and sell the device candidates being studied. At any stage of the development, the Company may discontinue device candidate based on review of available preclinical and clinical data, the estimated costs of continued development, market considerations and other factors. Furthermore, with respect to the clinical studies of device candidates conducted by CROs and others, the Company may have less control over their timing or outcome.
Risks Related to Our Industry, Business and Operation
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and clinic operations involve the use of hazardous materials, chemicals and various radioactive compounds/radiation and AML Clinic may create medical waste and radiation. Our R&D Center may maintain quantities of various flammable and toxic chemicals in our facilities that are required for our research, development and manufacturing activities. We are subject to local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials and of medical waste at the jurisdictions where we operate our clinic and research facilities, which are currently limited to Hong Kong. We believe our procedures for storing, handling and disposing of these materials comply with the relevant guidelines and laws of the jurisdictions in which our facilities are located. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials and medical waste.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties, if we violate any of these laws or regulations.
37
Our future success depends on our ability to retain our Chief Executive Officer, our scientific and clinical advisors, and other key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on Ian Huen, our Chief Executive Officer, as well as, other principal members of our management teams, scientific teams as well as scientific and clinical advisors. Although we have formal employment agreements, which we refer to as appointment letters, with all of our executive officers, these agreements do not prevent our executives from terminating their employment with us at any time, subject to applicable notice periods. Nevertheless, the loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives.
To induce valuable employees to remain at our Company, in addition to salary and cash incentives, we plan to provide share incentive grants that vest over time. The value to employees of these equity grants that vest over time may be significantly affected by movements in the price of our Class A Ordinary Shares that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies. Although we have appointment letters with our key employees, any of our employees could resign at any time, with 1-month to 3-months prior written notice or with payment in lieu of notice.
Recruiting and retaining qualified officers, scientific, clinical, sales and marketing personnel or consultants will also be critical to our success. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our discovery and preclinical studies development and commercialization strategy. The loss of the services of our executive officers or other key employees and consultants could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy.
Furthermore, replacing executive officers and key employees or consultants may be difficult and may take an extended period of time, because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize drug and device candidates. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel or consultants on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel.
We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain high quality personnel, our ability to pursue our growth strategy will be limited.
38
We will need to increase the size and capabilities of our organization, and we may experience difficulties in managing our growth.
As of the date hereof, we have 41 employees, including 39 full-time employees and 2 part-time employees. Of these, 15 are engaged in full-time research and development and laboratory operations, 19 are engaged in general and administrative functions, 5 are full-time employees engaged in the clinic operation and 2 part-time employees are engaged in sponsored research and development, laboratory operations and legal clerical support. As of the date of hereof, 41 of our employees are located in Hong Kong. In addition, we have engaged and may continue to engage 33 independent contracted consultants and advisors to assist us with our operations. As our development and commercialization plans and strategies develop, and as we have transitioned into operating as a public company, we will need to establish and maintain effective disclosure and financial controls and make changes in our corporate governance practices. We will need to add a significant number of additional managerial, operational, sales, marketing, financial and other personnel with the appropriate public company experience and technical knowledge and we may not successfully recruit and maintain such personnel. Future growth will impose significant added responsibilities on members of management, including:
| ● | identifying, recruiting, integrating, maintaining and motivating additional employees; | |
| ● | managing our internal development efforts effectively, including clinical, the FDA or other comparable regulatory authority review process for our drug and device candidates, while complying with our contractual obligations to contractors and others; and | |
| ● | improving our operational, financial and management controls, reporting systems and procedures. |
Our future financial performance and our ability to commercialize our drug candidates will depend, in part, on our ability to effectively manage our future growth, and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors and consultants for significant input in selecting and evaluating new products to pursue. These independent organizations, advisors and consultants may not continue to be available to us on a timely basis when needed, and in such case, we may not have the ability to find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities, or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval of our drug candidates or otherwise advance our business. Furthermore, we may not be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, if at all.
If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize our drug and device candidates and, accordingly, may not achieve our research, development and commercialization goals.
We intend to seek additional collaborations, strategic alliances or acquisitions or enter into royalty-seeking or sublicensing arrangements in the future, but we may not realize the benefits of these arrangements.
We intend to form or seek strategic alliances, create joint ventures or collaborations, acquire complimentary products, IP rights, technology or businesses or enter into additional licensing arrangements with unrelated parties that we determine may complement or augment our development and commercialization efforts with respect to our drug and device candidates. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing shareholders, or disrupt our management and business.
We will face significant competition in seeking appropriate strategic partners and the negotiation process is likely to be time-consuming, costly and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or another alternative arrangement for any of our drug and device candidates because their state of development may be deemed to be too early for collaborative effort and others may not view our drug and device candidates as having the requisite potential to demonstrate safety and efficacy. If and when we enter into an agreement with a collaboration partner or sublicensee for development and commercialization of a drug or device candidate, we can expect to relinquish some or all of the control over the future success of that drug and device candidate to the unrelated-party.
Further, even if we enter into a collaboration involving any of our drug and device candidates, the arrangement will be subject to numerous risks, which may include the following:
| ● | the collaborators will likely have significant discretion in determining the efforts and resources that they will apply to a collaboration; | |
| ● | the collaborator may ultimately choose not pursue development and commercialization of our drug candidates or may elect not to continue or renew development or commercialization programs, based on clinical trial results, changes in their strategic focus due to the acquisition of competitive drugs, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; |
39
| ● | the collaborator may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a drug or device candidate, repeat or conduct new clinical trials, or require a new formulation of a drug or device candidate for clinical testing; | |
| ● | the collaborator could independently develop, or develop with unrelated parties, drugs that compete directly or indirectly with our drugs or drug and device candidates; | |
| ● | the collaborator with marketing and distribution rights to one or more drugs may not commit sufficient resources to their marketing and distribution; | |
| ● | the collaborator may not properly maintain or defend our IP rights or may use our IP or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our IP or proprietary information or expose us to potential liability; | |
| ● | disputes may arise between us and the collaborator that cause the delay or termination of the research, development or commercialization of our drug and device candidates, or that result in costly litigation or arbitration that diverts management attention and resources; | |
| ● | the collaboration may be terminated and, if terminated, may result the Company needing additional capital to pursue further development or commercialization of the applicable drug and device candidates; | |
| ● | the collaborator may own or co-own IP covering our drugs that results from our collaborating with them, and in such cases, we would not have the exclusive right to commercialize such IP; | |
| ● | the collaboration may result in increased operating expenses or the assumption of indebtedness or contingent liabilities; and | |
| ● | the collaboration arrangement may result in the loss of key personnel and uncertainties in our ability to maintain key business relationships. |
As a result, if we enter into collaboration agreements and strategic partnerships or license our drugs, we may not be able to realize the benefit of such transactions, which could delay our timelines or otherwise adversely affect our business. Following a strategic transaction or license, we may not achieve the revenue or specific net income that justifies such transaction. If we are unable to reach agreements with a suitable collaborator on a timely basis, on acceptable terms, or at all, we may have to curtail the development of a drug or device candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense.
If we fail to enter into collaborations, we may seek to fund and undertake development or commercialization activities on our own, but we may not have sufficient funds or expertise to undertake the necessary development and commercialization activities. In such a case, we may not be able to further develop our drug and device candidates or bring them to market and generate product sales revenue, which would harm our business prospects, financial condition and results of operations.
40
Our employees, independent contractors, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements.
We are exposed to the risk of fraud, misconduct or other illegal activity by our employees, independent contractors, consultants, commercial partners and vendors. Misconduct by these parties could include intentional, reckless and negligent conduct that fails to: comply with the laws of the FDA and other similar non-U.S. regulatory authorities; provide true, complete and accurate information to the FDA and other similar non-U.S. regulatory authorities; comply with manufacturing standards we have established; comply with healthcare fraud and abuse laws in the United States and similar non-U.S. fraudulent misconduct laws; or report financial information or data accurately or to disclose unauthorized activities to us. If we obtain the FDA approval for any of our drug and device candidates and begin commercializing those drugs in the United States, our potential exposure under U.S. laws will increase significantly and our costs associated with compliance with such laws are also likely to increase. These laws may impact, among other things, our current activities with principal investigators of our sponsored researches and research patients and our use of information obtained in the course of patient recruitment for clinical trials, as well as proposed and future sales, marketing and education programs. In particular, the promotion, sales and marketing of healthcare items and services, as well as certain business arrangements in the healthcare industry, are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally.
It is not always possible to identify and deter misconduct by employees and other parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses, or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
Our disclosure controls and procedures are designed to reasonably assure that information required to be disclosed by us in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC.
We believe that any disclosure controls and procedures, or internal controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected, which would likely cause investors to lose confidence in our reported financial information. This could in turn limit our access to capital markets, harm our results of operations, and lead to a decline in the trading price of our Class A Ordinary Shares. Additionally, ineffective internal control over financial reporting could expose us to increased risk of fraud or misuse of corporate assets and subject us to potential delisting from the stock exchange on which we list, regulatory investigations and civil or criminal sanctions. We may also be required to restate our financial statements from prior periods.
If we fail to establish and maintain proper internal financial reporting controls, our ability to produce accurate financial statements or comply with applicable regulations could be impaired.
Pursuant to Section 404 of the Sarbanes-Oxley Act, we are required to file a report by our management on our internal control over financial reporting, including an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm and due to a transition period established by rules of the SEC for newly public companies, we are not required to include a report of management’s assessment regarding internal control over financial reporting in this prospectus. The presence of material weaknesses in internal control over financial reporting could result in financial statement errors which, in turn, could lead to errors in our financial reports and/or delays in our financial reporting, which could require us to restate our operating results. In connection with the audit of our financial statements for the period January 1, 2017 through February 28, 2017, the period March 1, 2017 through December 31, 2017 and the year ended December 31, 2018, we and our independent registered public accounting firm identified one material weakness in our internal control over financial reporting, as defined in the standards established by the Public Company Accounting Oversight Board of the United States, as of December 31, 2018. The material weakness identified was the lack of dedicated resources to take responsibility for the finance and accounting functions and the preparation of financial statements in compliance with generally accepted accounting principles in the United States, or U.S. GAAP.
41
We have already taken some steps and have continued to implement measures to remediate the material weakness identified, including but not limited to providing trainings to staff, changing to a new and well-established accounting system, and continuing to monitor the internal control over financial reporting. However, we cannot assure you that we will not identify additional material weaknesses or significant deficiencies in the future.
Due to the material weakness in our internal controls over financial reporting, we conclude that our internal controls over financial reporting are ineffective and therefore investors may lose confidence in our operating results, the price of the Class A Ordinary Shares could decline and we may be subject to litigation or regulatory enforcement actions. In addition, if we are unable to meet the requirements of Section 404 of the Sarbanes-Oxley Act, the Class A Ordinary Shares may not be able to remain listed on the NASDAQ Global Market.
We may market our products, if approved, globally; if we do, we will be subject to the risk of doing business internationally.
We operate and expect to operate in various countries, and we may not be able to market our products in, or develop new products successfully for, these markets. We may also encounter other risks of doing business internationally including but not limited to:
| ● | unexpected changes in, or impositions of, legislative or regulatory requirements; | |
| ● | efforts to develop an international sales, marketing and distribution organization may increase our expenses, divert our management’s attention from the acquisition or development of drug candidates or cause us to forgo profitable licensing opportunities in these geographies; | |
| ● | the occurrence of economic weakness, including inflation or political instability; | |
| ● | the effects of applicable non-U.S. tax structures and potentially adverse tax consequences; | |
| ● | differences in protection of our IP rights including patent rights of other parties; | |
| ● | the burden of complying with a variety of foreign laws including difficulties in effective enforcement of contractual provisions; | |
| ● | delays resulting from difficulty in obtaining export licenses, tariffs and other barriers and restrictions, potentially longer payment cycles, greater difficulty in accounts receivable collection and potentially adverse tax treatment; and | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad. |
In addition, we are subject to general geopolitical risks in foreign countries where we operate, such as political and economic instability and changes in diplomatic and trade relationships, which could affect, among other things, customers’ inventory levels and consumer purchasing, which could cause our results to fluctuate and our net sales to decline. The occurrence of any one or more of these risks of doing business internationally, individually or in the aggregate, could materially and adversely affect our business and results of operations.
42
If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our shareholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks.
We may evaluate various acquisitions and strategic partnerships, including licensing or acquiring complementary products, IP rights, technology or businesses. Any potential acquisition or strategic partnership may entail numerous risks, including, but not limited to:
| ● | increase in operating expenses and cash requirements; | |
| ● | the assumption of additional indebtedness or contingent liabilities; | |
| ● | the issuance of our equity securities; | |
| ● | assimilation of operations, IP and products of an acquired company, including difficulties associated with integrating new personnel; | |
| ● | the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition; | |
| ● | retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships; | |
| ● | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing drugs or drug and device candidates and regulatory approvals; and | |
| ● | our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
In addition, if we undertake acquisitions, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
If we fail to comply with the U.S. Foreign Corrupt Practices Act (“FCPA”), or other anti-bribery laws, our reputation may be harmed and we could be subject to penalties and significant expenses that have a material adverse effect on our business, financial condition and results of operations.
We are subject to the FCPA. The FCPA generally prohibits us from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. We are also subject to the anti-bribery laws of other jurisdictions, particularly the PRC. As our business expands, the applicability of the FCPA and other anti-bribery laws to our operations will increase. Our procedures and controls to monitor anti-bribery compliance may fail to protect us from reckless or criminal acts committed by our employees or agents. If we, due to either our own deliberate or inadvertent acts or those of others, fail to comply with applicable anti-bribery laws, our reputation could be harmed and we could incur criminal or civil penalties, other sanctions and/or significant expenses, which could have a material adverse effect on our business, including our financial condition, results of operations, cash flows and prospects.
If we commence clinical trials of one of our drug or device candidates, and product liability lawsuits are brought against us, we may incur substantial liabilities and the commercialization of such drug or device candidates may be affected.
If any of our drug or device candidates enter clinical trials, we will face an inherent risk of product liability suits and will face an even greater risk if we obtain approval to commercialize any drugs. For example, we may be sued if our drug candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the drug, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our drug candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our drugs; | |
| ● | injury to our reputation; |
43
| ● | withdrawal of clinical trial participants and inability to continue clinical trials; | |
| ● | initiation of investigations by regulators; | |
| ● | costs to defend the related litigation; | |
| ● | a diversion of management’s time and our resources; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | loss of revenue; | |
| ● | exhaustion of any available insurance and our capital resources; | |
| ● | the inability to commercialize any drug candidate; and | |
| ● | a decline in the price of our Class A Ordinary Shares. |
We shall seek to obtain the appropriate insurance once our candidates are ready for clinical trial. However, our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of drugs we develop, alone or with collaborators. We currently do not have in place product liability insurance and although we plan to have in place such insurance as and when the products are ready for commercialization, as well as insurance covering clinical trials, the amount of such insurance coverage may not be adequate, we may be unable to maintain such insurance, or we may not be able to obtain additional or replacement insurance at a reasonable cost, if at all. Our insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
Additionally, we may be sued if the products that we commercialize, market or sell cause or are perceived to cause injury or are found to be otherwise unsuitable, and may result in:
| ● | decreased demand for those products; | |
| ● | damage to our reputation; | |
| ● | costs incurred related to product recalls; | |
| ● | limiting our opportunities to enter into future commercial partnership; and | |
| ● | a decline in the price of our Class A Ordinary Shares. |
Our insurance coverage may be inadequate to protect us against losses.
We currently maintain property insurance for our office premises (including one unit of server and accessories). We hold employer’s liability insurance generally covering death or work-related injury of employees; we maintain “Office Care Plan Insurance” for those persons working in our offices and “Medical Plan” for our employee. We hold public liability insurance covering certain incidents involving unrelated parties that occur on or in the premises of the Company. We do have directors and officers liability insurance. We do not have key-man life insurance on any of our senior management or key personnel, or business interruption insurance. Our insurance coverage may be insufficient to cover any claim for product liability, damage to our fixed assets or employee injuries. If any claims for damage are brought against us, or if we experience any business disruption, litigation or natural disaster, we might incur substantial costs and diversion of resources.
44
Fluctuations in exchange rates could result in foreign currency exchange losses
Our operations and equity are funded in U.S. dollars and we currently incur the majority of our expenses in U.S. dollars or in H.K. dollars. H.K. dollar is currently pegged to the U.S. dollar; however, we cannot guarantee that such peg will continue to be in place in the future. Our exposure to foreign exchange risk primarily relates to the limited cash denominated in currencies other than the functional currencies of each entity and limited revenue contracts dominated in H.K. dollars in certain Hong Kong operating entities. We do not believe that we currently have any significant direct foreign exchange risk and have not hedged exposures denominated in foreign currencies or any other derivative financial instruments.
If we are exposed to foreign currency exchange risk as our results of operations, cash flows maybe subject to fluctuations in foreign currency exchange rates. For example, if a significant portion of our clinical trial activities may be conducted outside of the United States, and associated costs may be incurred in the local currency of the country in which the trial is being conducted, which costs could be subject to fluctuations in currency exchange rates. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the U.S. dollar. A decline in the value of the U.S. dollar against currencies in countries in which we conduct clinical trials could have a negative impact on our research and development costs. Foreign currency fluctuations are unpredictable and may adversely affect our financial condition, results of operations and cash flows.
Our investments are subject to risks that could result in losses.
We had unrestricted cash of $4.47 million, $12.01 million and $16.25 million as of June 30, 2019, December 31, 2018 and December 31, 2017, respectively. We may invest our cash in a variety of financial instruments. All of these investments are subject to credit, liquidity, market and interest rate risk. Such risks, including the failure or severe financial distress of the financial institutions that hold our cash, cash equivalents and investments, may result in a loss of liquidity, impairment to our investments, realization of substantial future losses, or a complete loss of the investments in the long-term, which may have a material adverse effect on our business, results of operations, liquidity and financial condition. While we believe our cash position does not expose us to excessive risk, future investments may be subject to adverse changes in market value.
We are exposed to risks associated with our computer hardware, network security and data storage.
Similar to all other computer network users, our computer network system is vulnerable to attack of computer virus, worms, trojan horses, hackers or other similar computer network disruptive problems. Any failure in safeguarding our computer network system from these disruptive problems may cause breakdown of our computer network system and leakage of confidential information of the Company. Any failure in the protection of our computer network system from external threat may disrupt our operation and may damage our reputation for any breach of confidentiality to our customers, which in turn may adversely affect our business operation and performance. In the event that our confidential information is stolen and misused, we may become exposed to potential risks of losses from litigation and possible liability.
In addition, we are highly dependent on our IT infrastructure to store research data and information and manage our business operations. We do not backup all data on a real-time basis and the effectiveness of our business operations may be materially affected by any failure in our IT infrastructure. If our communications and IT systems do not function properly, or if there is any partial or complete failure of our systems, we could suffer financial losses, business disruption or damage to our reputation.
45
Business disruptions could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our research institution collaborators, CROs, suppliers and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, damage from computer viruses, material computer system failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions. In addition, we partially rely on our research institution collaborators for conducting research and development of our drug candidates, and they may be affected by government shutdowns or withdrawn funding. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on contract manufacturers to produce and process our drug candidates. Our ability to obtain clinical supplies of our drug candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption. A large portion of our contract manufacturer’s operations is located in a single facility. Damage or extended periods of interruption to our corporate or our contract manufacturer’s development or research facilities due to fire, natural disaster, power loss, communications failure, unauthorized entry or other events could cause us to cease or delay development of some or all of our drug candidates.
Although we do not currently conduct any business in the PRC, we may in the future; in doing so we would be exposed to various risks related to doing business in the PRC.
Although we currently do not conduct any business in the PRC, we are the exclusive licensee to certain PRC patents directed to our drug candidates such as ALS-1, NLS-1, NLS-2 and SPLS-1, and we intend to file application for certain products in the PRC. The pharmaceutical industry in the PRC is subject to comprehensive government regulation and supervision, encompassing the approval, registration, manufacturing, packaging, licensing and marketing of new drugs. (See “Our Business – Government Regulation – PRC Regulations”). In recent years, the regulatory framework in the PRC regarding the pharmaceutical industry has undergone significant changes, and we expect that it will continue to undergo significant changes. Any such changes or amendments may result in increased compliance costs on our business or cause delays in or prevent the successful development or commercialization of our drug candidates in the PRC and reduce the current benefits that we believe are available to us from developing and manufacturing drugs in the PRC. Chinese authorities have become increasingly vigilant in enforcing laws in the pharmaceutical industry and any failure by us or our partners to maintain compliance with applicable laws and regulations or obtain and maintain required licenses and permits may result in the suspension or termination of our business activities in the PRC. We believe our strategy and approach is aligned with the PRC government’s policies, but we cannot ensure that our strategy and approach will continue to be aligned.
If in the future, we commence business or operation in the PRC, changes in the political and economic policies of the PRC government may materially and adversely affect our business, financial condition and results of operations and may result in our inability to sustain our growth and expansion strategies. Once we start doing business in the PRC, our financial condition and results of operation in the PRC could be materially and adversely affected by government control over capital investments or changes in tax regulations that are applicable to us, and consequently have a material adverse effect on our businesses, financial condition and results of operations.
The SEC could take the position that we are an “investment company” subject to the extensive requirements of the Investment Company Act of 1940. Such a characterization and the associated compliance requirements could have a material adverse effect on our business, financial condition, and results of operations.
Our business had historically included passive healthcare related investments in early stage companies primarily in the United States. Although we are in the process of liquidating those securities that remain in our portfolio, we still hold some such investments and these are included as assets of our Company on a consolidated basis. As part of the Restructure, we resolved to exit such portfolio investments over an appropriate timeframe and focus our resources on our current business. Since the date of the Restructure, we have not held ourselves out as an investment company and we do not believe we are an “investment company” under the Investment Company Act of 1940. If the SEC or a court, however, were to disagree with us, we could be required to register as an investment company. This would subject us to disclosure and accounting rules geared toward investment companies, rather than operating companies, which may limit our ability to borrow money, issue options, issue multiple classes of stock and debt, and engage in transactions with affiliates, and may require us to undertake significant costs and expenses to meet the disclosure and regulatory requirements to which we would be subject as a registered investment company.
46
If we are classified as a passive foreign investment company for U.S. federal income tax purposes, United States holders of our Class A Ordinary Shares may be subject to adverse United States federal income tax consequences.
A non-U.S. corporation will be a passive foreign investment company (“PFIC”) for U.S. federal income tax purposes, for such year, if either
| ● | At least 75% of its gross income for such year is passive income; or | |
| ● | The average percentage of our assets (determined at the end of each quarter) during such year which produce passive income or which are held for the production of passive income is at least 50%. |
Passive income generally includes dividends, interests, rents and royalties (other than rents or royalties derived from the active conduct of a trade or business) and gains from the disposition of passive assets.
A separate determination must be made after the close of each taxable year as to whether a non-U.S. corporation is a PFIC for that year. For purposes of the PFIC analysis, in general, a non-U.S. corporation is deemed to own its pro rata share of the gross income and assets of any entity in which it is considered to own at least 25% of the equity by value. Based on the current and anticipated value of our assets, we believe we were a PFIC for U.S. federal income tax purposes for our taxable year ending December 31, 2017, and we may be a PFIC for U.S. federal income tax purposes for our current taxable year ending December 31, 2018.
In determining whether we are a PFIC, cash is considered by the U.S. Internal Revenue Service (“IRS”) to be a passive asset. During our taxable year ending December 31, 2018, we believe that the amount of restricted and unrestricted cash we had on hand was greater than 50% of our total assets. The composition of our assets during the current taxable year may cause us to continue to be classified as a PFIC. The determination of whether we will be a PFIC for our current taxable year or a future year may depend in part upon how quickly we spend our liquid assets, and on the value of our goodwill and other unbooked intangibles not reflected on our balance sheet, which may depend upon the market value of our Class A Ordinary Shares from time to time. Further, while we will endeavor to use a classification methodology and valuation approach that is reasonable, the IRS may challenge our classification or valuation of our goodwill and other unbooked intangibles for purposes of determining whether we are a PFIC in the current or one or more future taxable years.
If we are a PFIC for any taxable year during which a U.S. Holder owns our Class A Ordinary Shares, certain adverse U.S. federal income tax consequences could apply to such U.S. Holder. As discussed under “Taxation – Material U.S. Federal Income Tax Considerations for U.S. Holders – Passive Foreign Investment Company Rules”, a U.S. Holder may be able to make certain tax elections that would lessen the adverse impact of PFIC status; however, in order to make such elections the U.S. holder will usually have to have been provided information about the company by us, and there is no assurance that the company will provide such information.
For a more detailed discussion of the application of the PFIC rules to us and the consequences to U.S. holders if we were determined to be a PFIC. (See “Taxation – Material U.S. Federal Income Tax Considerations for U.S. Holders – Passive Foreign Investment Company Rules”)
Risks Related to Our Corporate Structure
Our CEO has control over key decision making as a result of his control of a majority of our voting shares.
Our Founder, CEO, and our Executive Director, Mr. Ian Huen, and his affiliates, over which he is deemed to have control and/or have substantial influence, has voting rights with respect to an aggregate of 17,836,077 ordinary shares, on an as converted basis (1,774,608 Class A Ordinary Shares and 16,061,469 Class B Ordinary Shares), representing approximately 70% of the voting power of our outstanding ordinary shares as of the date hereof. As a result, Mr. Huen has the ability to control the outcome of matters submitted to our shareholders for approval, including the election of directors and any merger, consolidation, or sale of all or substantially all of our assets. In addition, Mr. Huen has the ability to control the management and affairs of our company as a result of his position as our CEO and his ability to control the election of our directors. Additionally, in the event that Mr. Huen controls our company at the time of his death, control may be transferred to a person or entity that he designates as his successor. As a board member and officer, Mr. Huen owes a fiduciary duty to our shareholders and must act in good faith in a manner he reasonably believes to be in the best interests of our shareholders. As a shareholder, even a controlling shareholder, Mr. Huen is entitled to vote his shares, and shares over which he has voting control as a result of voting agreements, in his own interests, which may not always be in the interests of our shareholders generally.
47
The dual class structure of our ordinary shares has the effect of concentrating voting control with our CEO, directors and their affiliates.
Each Class B Ordinary Share has ten votes per share and each Class A Ordinary Share has one vote per share. Shareholders who hold shares of Class B Ordinary Shares, including our executive officers and their affiliates who hold such shares, hold approximately 97 % of the voting power of our outstanding ordinary shares as of the date hereof. Because of the ten-to-one voting ratio between our Class B and Class A Ordinary Shares, the holders of our Class B Ordinary Shares collectively will continue to control a majority of the combined voting power of our ordinary share and therefore be able to control all matters submitted to our shareholders for approval so long as the shares of Class B Ordinary Shares represent at least 9.1% of all outstanding shares of our Class A Ordinary Shares and Class B Ordinary Shares. This concentrated control will limit your ability to influence corporate matters for the foreseeable future.
Future transfers by holders of Class B Ordinary Shares will generally result in those shares converting to Class A Ordinary Shares, subject to limited exceptions, such as certain transfers effected for estate planning purposes. The conversion of Class B Ordinary Shares to Class A Ordinary Shares will have the effect, over time, of increasing the relative voting power of those holders of Class B Ordinary Shares who retain their shares in the long term. If, for example, Mr. Huen retains a significant portion of his holdings of Class B Ordinary Share for an extended period of time, he could, in the future, continue to control a majority of the combined voting power of our Class A Ordinary Shares and Class B Ordinary Shares.
As a “controlled company” under the rules of the NASDAQ Global Market, we may choose to exempt our company from certain corporate governance requirements that could have an adverse effect on our public shareholders.
Our directors and officers beneficially own a majority of the voting power of our outstanding Class A Ordinary Shares. Under the Rule 4350(c) of the NASDAQ Global Market, a company of which more than 50% of the voting power is held by an individual, group or another company is a “controlled company” and may electnot to comply with certain corporate governance requirements, including the requirement that a majority of our directors be independent, as defined in the NASDAQ Global Market Rules, and the requirement that our compensation and nominating and corporate governance committees consist entirely of independent directors. Although we do not intend to rely on the “controlled company” exemption under the Nasdaq listing rules, we could elect to rely on this exemption in the future. If we elect to rely on the “controlled company” exemption, a majority of the members of our board of directors might not be independent directors and our nominating and corporate governance and compensation committees might not consist entirely of independent directors. Accordingly, during any time while we remain a controlled company relying on the exemption and during any transition period following a time when we are no longer a controlled company, you would not have the same protections afforded to shareholders of companies that are subject to all of the NASDAQ Global Market corporate governance requirements. Our status as a controlled company could cause our Class A Ordinary Share to look less attractive to certain investors or otherwise harm our trading price.
Risks Related to our Securities
Shares eligible for future sale may adversely affect the market price of our Class A Ordinary Shares if the shares are successfully listed on NASDAQ or other stock markets, as the future sale of a substantial amount of outstanding Class A Ordinary Shares in the public marketplace could reduce the price of our Class A Ordinary Shares.
The market price of our Class A Ordinary Shares could decline as a result of sales of substantial amounts of our Class A Ordinary Shares in the public market, or the perception that these sales could occur. In addition, these factors could make it more difficult for us to raise funds through future offerings of our Class A Ordinary Shares. An aggregate of 6,597,362 Class A Ordinary Shares are outstanding as of the date of this prospectus. We are including 27,765,821 Resale Shares in this prospectus, all of which, once sold by the Selling Shareholders pursuant to this prospectus upon and after its effectiveness, will be freely tradable. All of the Class A Ordinary Shares sold in our initial public offering are freely transferable without restriction or further registration under the Securities Act. The remaining Class A Ordinary Shares will be “restricted securities” as defined in Rule 144. These Class A Ordinary Shares may be sold without registration under the Securities Act to the extent permitted by Rule 144 or other exemptions under the Securities Act.
48
A sale or perceived sale of a substantial number of our Ordinary Shares may cause the price of our Class A Ordinary Shares to decline.
If our shareholders sell substantial amounts of our Class A Ordinary Shares in the public market, the market price of our Class A Ordinary Shares could fall. Moreover, the perceived risk of this potential dilution could cause shareholders to attempt to sell their shares and investors to short our Class A Ordinary Shares. These sales also may make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate.
Issuances by us of additional securities, whether in traditional or token format, could affect ownership and voting rights over us. In addition, the issuance of preferred shares, or options or warrants to purchase those preferred shares, could negatively impact the value of the Ordinary Shares as the result of preferential dividend rights, conversion rights, redemption rights and liquidation provisions granted to the stockholders of such preferred shares.
From time to time, we may issue in public or private sales additional securities to third party investors. Such securities may provide holders with ownership and voting rights that could provide the holders thereof with substantial influence over our business. Any preferred shares that may be issued shall have such rights, preferences, privileges and restrictions as may be designated from time-to-time by our board, including preferential dividend rights, voting rights, conversion rights, redemption rights and liquidation provisions. There cannot be any assurance that we will not issue preferred securities with rights and preferences that are more beneficial than those provided to our Ordinary Shares.
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our shares.
We have never paid any cash dividends on our Class A Ordinary Shares and do not anticipate paying any cash dividends on our Class A Ordinary Shares in the foreseeable future, and any return on investment may be limited to the value of our Class A Ordinary Shares. We plan to retain any future earnings to finance growth.
Our dividend policy is subject to the discretion of our Board of Directors and will depend on, among other things, our earnings, financial condition, capital requirements and other factors. There is no assurance that our Board of Directors will declare dividends even if we are profitable. Under Cayman Islands law, dividends may be declared and paid only out of funds legally available therefor, namely out of either profit or our share premium account, and provided further that a dividend may not be paid if this would result in our Company being unable to pay its debts as they fall due in the ordinary course of business and the realizable value of assets of our Company will not be less than the sum of our total liabilities, other than deferred taxes as shown on our books of account, and our capital.
Our Class B Ordinary Shares have stronger voting power than our Class A Ordinary Shares and certain existing shareholders have substantial influence over our Company and their interests may not be aligned with the interests of our other shareholders.
We have a dual-class voting structure consisting of Class A Ordinary Shares and Class B Ordinary Shares. Under this structure, holders of Class A Ordinary Shares are entitled to one vote per share, and holders of Class B Ordinary Shares are entitled to ten votes per share, which can cause the holders of Class B Ordinary Shares to have an unbalanced, higher concentration of voting power. Our management team as a group beneficially owns over 18 million Class B Ordinary Shares representing 80% voting power. As a result, until such time as their collective voting power is below 50%, our management team as a group of controlling shareholders have substantial influence over our business, including decisions regarding mergers, consolidations and the sale of all or substantially all of our assets, election of directors and other significant corporate actions. They may take actions that are not in the best interests of us or our other shareholders. These corporate actions may be taken even if they are opposed by our other shareholders.Further, concentration of ownership of our Class B Ordinary Shares may discourage, prevent or delay the consummation of change of control transactions that shareholders may consider favorable, including transactions in which shareholders might otherwise receive a premium for their shares. Future issuances of Class B Ordinary Shares may also be dilutive to the holders of Class A Ordinary Shares. As a result, the market price of our Class A Ordinary Shares could be adversely affected.
49
Shareholders who hold shares of Class B Ordinary Shares, including our executive officers and their affiliates, hold approximately 97% of the voting power of our outstanding ordinary shares. Because of the ten-to-one voting ratio between our Class B and Class A Ordinary Shares, the holders of our Class B Ordinary Shares will collectively continue to control a majority of the combined voting power of our Ordinary Shares and therefore be able to control all matters submitted to our shareholders for approval, so long as the Class B Ordinary Shares represent at least 9.1% of all outstanding shares of our Ordinary Shares.
Raising additional capital may cause dilution to our shareholders, restrict our operations or require us to relinquish rights to our technology or drug and device candidates.
We may seek additional funding through a combination of equity offerings, debt financings, collaborations, licensing arrangements, strategic alliances and marketing or distribution arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a holder of our Class A Ordinary Shares. The incurrence of additional indebtedness or the issuance of certain equity securities could result in increased fixed payment obligations, and could also result in certain additional restrictive covenants, such as limitations on our ability to incur additional debt or issue additional equity, limitations on our ability to acquire or license IP rights and other operating restrictions that could adversely impact our ability to conduct our business. In addition, issuance of additional equity securities, or the possibility of such issuance, may cause the market price of our Class A Ordinary Shares to decline. In the event that we enter into collaborations or licensing arrangements to raise capital, we may be required to accept unfavorable terms, including relinquishing or licensing to another party on unfavorable terms our rights to technology or drug and device candidates that we otherwise would seek to develop or commercialize ourselves or potentially reserve for future potential arrangements when we might be able to achieve more favorable terms.
Resales of our Class A Ordinary Shares in the public market by the Selling Shareholders may cause the market price of our Class A Ordinary Shares to decline.
Sales of Resale Shares could result in resales of our Class A Ordinary Shares by our current shareholders concerned about the potential dilution of their holdings. In turn, these resales could have the effect of depressing the market price for our Class A Ordinary Shares.
Since we are a Cayman Islands exempted company, the rights of our shareholders may be more limited than those of shareholders of a company organized in the United States.
Our corporate affairs are governed by our Second Amended and Restated Memorandum and Articles of Association (as may be amended from time to time) (“Memorandum and Articles”), the Companies Law (2018 Revision) of the Cayman Islands (the “Companies Law”) and the common law of the Cayman Islands.The rights of shareholders to take action against the directors, actions by minority shareholders and the fiduciary responsibilities of our directors are to a large extent governed by the common law of the Cayman Islands. This common law is derived in part from comparatively limited judicial precedent in the Cayman Islands as well as from English common law, which has persuasive, but not binding, authority on a court in the Cayman Islands.Under the laws of some jurisdictions in the United States, majority and controlling shareholders generally have certain fiduciary responsibilities to the minority shareholders. Shareholder action must be taken in good faith, and actions by controlling shareholders which are obviously unreasonable may be declared null and void. Cayman Islands law protecting the interests of minority shareholders may not be as protective in all circumstances as the law protecting minority shareholders in some U.S. jurisdictions. In addition, the circumstances in which a shareholder of a Cayman Islands company may sue the company derivatively, and the procedures and defenses that may be available to the company, may result in the rights of shareholders of a Cayman Islands company being more limited than those of shareholders of a company organized in the United States. Accordingly, shareholders may have fewer alternatives available to them if they believe that corporate wrongdoing has occurred. The Cayman Islands courts are also unlikely to recognize or enforce judgments from U.S. courts based on certain liability provisions of U.S. securities laws that are penal in nature. There is no statutory recognition in the Cayman Islands of judgments obtained in the United States, although the courts of the Cayman Islands will generally recognize and enforce non-penal judgment of a foreign court of competent jurisdiction for a liquidated sum without retrial on its merits which is not obtained in a manner contrary to public policy in the Cayman Islands and in respect of which there are no concurrent proceedings in the Cayman Islands. This means, even if shareholders were to sue us successfully, they may not be able to recover anything to make up for the losses suffered.
50
Furthermore, our directors have the power to take certain actions without shareholder approval which would require shareholder approval under the laws of most U.S. jurisdictions. For example, the directors of a Cayman Islands company, without shareholder approval, may implement a sale of any assets, property, part of the business, or securities of the Company.
While Cayman Islands law allows a dissenting shareholder to express the shareholder’s view that a court sanctioned reorganization of a Cayman Islands company would not provide fair value for the shareholder’s shares, Cayman Islands statutory law does not specifically provide for shareholder appraisal rights on a merger or consolidation of a company. This may make it more difficult for you to assess the value of any consideration you may receive in a merger or consolidation or to require that the acquirer gives you additional consideration if you believe the consideration offered is insufficient. However, Cayman Islands’ statutory law does provide a mechanism for a dissenting shareholder in a merger or consolidation to apply to the Grand Court for a determination of the fair value of the dissenter’s shares, if it is not possible for the Company and the dissenter to agree a fair price within the time limits prescribed.
Shareholders of Cayman Islands exempted companies, such as our Company, have no general rights under Cayman Islands’ law to inspect corporate records and accounts or to obtain copies of lists of shareholders. Our directors have discretion under our Memorandum and Articles to determine whether or not, and under what conditions, our corporate records may be inspected by our shareholders, but are not obliged to make them available to our shareholders. This may make it more difficult for you to obtain the information needed to establish any facts necessary for a shareholder motion or to solicit proxies from other shareholders in connection with a proxy contest.
Lastly, under the law of the Cayman Islands, there is little statutory law for the protection of minority shareholders. The principal protection under statutory law is that shareholders may bring an action to enforce the constituent documents of the corporation, our Memorandum and Articles. Shareholders are entitled to have the affairs of the company conducted in accordance with the general law and the memorandum and articles of association.
There are common law rights for the protection of shareholders that may be invoked, largely dependent on English company law, since the common law of the Cayman Islands for business companies is limited. Under the general rule pursuant to English company law known as the rule in Foss v. Harbottle, a court will generally refuse to interfere with the management of a company at the insistence of a minority of its shareholders who express dissatisfaction with the conduct of the company’s affairs by the majority or the board of directors. However, every shareholder is entitled to have the affairs of the company conducted properly according to law and the constituent documents of the company. As such, if those who control the company have persistently disregarded the requirements of company law or the provisions of the company’s memorandum and articles of association, then the courts will grant relief. Generally, the areas in which the courts will intervene are the following: (1) an act complained of which is outside the scope of the authorized business or is illegal or not capable of ratification by the majority; (2) acts that constitute fraud on the minority where the wrongdoers control the company; (3) acts that infringe on the personal rights of the shareholders, such as the right to vote; and (4) where the company has not complied with provisions requiring approval of a special or extraordinary majority of shareholders, which are more limited than the rights afforded minority shareholders under the laws of many states in the United States subject to limited exceptions, under Cayman Islands Law a minority shareholder may not bring a derivative action against directors. Our Cayman Islands’ counsel has advised us that they are aware of one recent as yet unreported derivative action having been brought in a Cayman Islands’ court. Class actions are not recognized in the Cayman Islands, but groups of shareholders with identical interests may bring representative proceedings, which are similar.
As a result, you may be limited in your ability to protect your interests if you are harmed in a manner that would otherwise enable you to sue in a United States federal court. In addition, shareholders of Cayman Islands companies may not have standing to initiate a shareholder derivative action in U.S. federal courts.
As a result of all of the above, shareholders of our Company may have more difficulty in protecting their interests in the face of actions taken by management, members of the board of directors or controlling shareholders than they would have as shareholders of a public U.S. company.
You may face difficulties in protecting your interests, and your ability to protect your rights through the U.S. federal courts may be limited because we are incorporated under Cayman Islands law, we currently conduct substantially all of our operations outside the United States and some of our directors and executive officers reside outside the United States.
We are incorporated in the Cayman Islands and currently conduct substantially all of our operations outside the United States through our subsidiaries. Some of our directors and executive officers reside outside the United States and a substantial portion of their assets are located outside of the United States. As a result, it may be difficult or impossible for you to bring an action against us or against these individuals in the Cayman Islands or in Hong Kong, in the event that you believe that your rights have been infringed under the securities laws of the United States or otherwise. Even if you are successful in bringing an action of this kind, the laws of the Cayman Islands and Hong Kong may render you unable to enforce a judgment against our assets or the assets of our directors and officers. There is no statutory recognition in the Cayman Islands of judgments obtained in the United States or Hong Kong, although the courts of the Cayman Islands will generally recognize and enforce a non-penal judgment of a foreign court of competent jurisdiction without retrial on the merits if such judgment is final, for a liquidated sum, not in the nature of taxes, a fine or penalty, is not inconsistent with a Cayman Islands’ judgment in respect of the same matters, and was not obtained in a manner which is contrary to public policy. In addition, a Cayman Islands court may stay proceedings if concurrent proceedings are being brought elsewhere.
51
We are an emerging growth company within the meaning of the Securities Act and will take advantage of certain reduced reporting requirements.
We are an “emerging growth company,” as defined in the JOBS Act and take advantage of certain exemptions from various requirements applicable to other public companies that are not emerging growth companies including, most significantly, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act for so long as we are an emerging growth company. As a result, if we elect not to comply with such auditor attestation requirements, our investors may not have access to certain information they may deem important.
The JOBS Act also provides that an emerging growth company does not need to comply with any new or revised financial accounting standards until such date that a private company is otherwise required to comply with such new or revised accounting standards. The Company has elected to use the extended transition period for complying with new or revised accounting standard under Section 102(b)(2) of the Jobs Act, that allows the Company to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies.
Risks Related to the SMPT tokens
There is no assurance that purchasers of the SMPT tokens will receive a return on their investment.
Each Token will entitle its holder (each, a “Tokenholder”) to receive certain sales-based royalties, sublicensing income or additional cash flow generated by drug candidates developed by the Smart-ACTTM platform (the “Token Distribution”) (See “Prospectus Summary – Recent Events – Smart Pharma Token”).
As identified in the aforementioned risk factors, a pharmaceutical company’s ability to generate revenue and achieve profitability is dependent on its ability to complete the development of drug candidates and any future drug candidates one develops in its portfolio, obtain necessary regulatory approvals, and have our drugs products under development manufactured and successfully marketed, of which there can be no guarantee. Furthermore, the research methodology used may be unsuccessful in identifying potential drug candidates, or those drug candidates identified may have harmful side effects or other undesirable characteristics that make them unmarketable or unlikely to receive regulatory approval (See “Risk Factors – Risks Related to the Preclinical and Clinical Development of Our Drug Candidates - We currently do not generate revenue from product sales and may never become profitable; unless we can raise more capital through additional financings, of which there can be no guarantee, our principal source of revenue will be from AML Clinic, which may not be substantial” and “We may not be successful in our efforts to identify or discover additional drug candidates. Due to our limited resources and access to capital, we must continue to the prioritize development of certain drug candidates; such decisions may prove to be wrong and may adversely affect our business”).
Therefore, we cannot guarantee that any drug candidates currently and subsequently developed by SMTPH using the Smart-ACTTMplatform will generate any revenue that would derive any sales-based royalties, sublicensing income or additional cash flow for distribution to Tokenholders.
Accordingly, there is no assurance that purchasers of SMPT tokens will realize any return on their investments or that their entire investments will not be lost.
SMPT Tokenholders’ security interest in the intellectual property rights may affect our shareholder’s interest in the Company.
SPLP acts as the intellectual property holding company of Smart Pharma, and holds all title, rights and ownership interest of the intellectual property rights developed by Smart-ACTTM (“Project IP”). The SMPT tokens are secured by way of a floating charge against the Project IP to guarantee the distribution of accrued sales-based royalties, sublicensing income or additional cash flow generated by drug candidates developed by the Smart-ACTTM platform (See “Prospectus Summary – Recent Events – Smart Pharma Token”).
Therefore, regardless of the number of the SMPT tokens sold and the amount of proceeds raised from the token sales, Tokenholders will only be eligible to receive a Token Distribution if any sales-based royalties, sublicensing income or additional cash flow is generated by drug candidates developed by the Smart-ACTTM platform, as and when SPLP declares the distribution.
Because the Token Distribution is secured by a security interest in such intellectual property rights, if and when SPLP defaults in its distribution obligations to the Tokenholders, or in the event of liquidation, dissolution or winding up of SPLP, the floating charge may crystallize into a fixed charge over the charged assets (i.e., the Project IP owned by SPLP), while a receiver may be appointed by the Tokenholders to sell off the Project IP. If this were to occur, the disposal of the Project IP by an appointed receiver may trigger a breach of any commercialization agreements between Smart Pharma and third parties with respect to the repurposed drug project, which may in turn affect our business, revenue and reputation.
The distributions to SMPT Tokenholders are not correlated with the number of SMPT tokens sold or net proceeds raised through the SMPT token sales.
SMTPH intends to use all of the proceeds raised from the SMPT token sales towards the development and operation of the Smart-ACTTMplatform. If the issuance of the SMPT tokens does not result in substantial proceeds, it could have a material adverse effect on SMTPH’s ability to fund these objectives and carry out its related business plans, its ability to develop the Smart-ACTTMplatform may be limited.
Aptorum Group anticipates that the net proceeds from the sales of the SMPT tokens will not be sufficient to fully fund Smart Pharma's current and future operations until it becomes self-sustaining. Smart Pharma’s current funding needs include funding for validation and assessment of candidates, operation and improvement of the platform, legal/professional services and exchanges-listing.
Therefore, Smart Pharma will likely require funding from Aptorum Group or other sources to subsidize and support its operations. The presence and level of funding support from Aptorum Group or other sources will not affect the aggregate distribution entitled by the Tokenholders, as the aggregate distribution is dependent on the ability for Smart-ACTTMto develop drug candidates that can generate sales-based royalties, sublicensing income or additional cash flow and the extent of commercial success of such candidate.
Therefore, the distributions to SMPT Tokenholders would not necessarily be correlated with the number of SMPT tokens sold or the net proceeds raised through the SMPT token sales. The dollar value of the aggregate distributions will not be affected by proceeds from the SMPT token sales, regardless of whether the proceeds greatly exceed or are significantly lower than the actual costs for funding Smart Pharma’s current and future operations.
52
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus, including the sections titled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Our Business” contains forward-looking statements that are based on our management’s belief and assumptions and on information currently available to our management. Although we believe that the expectations reflected in these forward-looking statements are reasonable, these statements relate to future events or our future financial performance, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. Forward-looking statements in this prospectus include, but are not limited to, statements about:
| ● | the initiation, timing, progress and results of our preclinical and clinical trials, and our research and development programs; |
| ● | our ability to advance our drug candidates into, and successfully complete, clinical trials; |
| ● | our ability to identify and develop new drug and device candidates; |
| ● | our reliance on the success of our drug candidates currently undergoing preclinical development; in particular, our Lead Project candidates; |
| ● | the timing or likelihood of regulatory filings and approvals; |
| ● | the commercialization of our drug and device candidates, if approved; |
| ● | our ability to develop sales and marketing capabilities; |
| ● | the pricing and reimbursement of our drug candidates, if approved; |
| ● | the implementation of our business model, strategic plans for our business and technology; |
| ● | the scope of protection we are able to establish and maintain for IP rights covering our drug and device candidates and technology; |
| ● | our ability to operate our business without infringing the IP rights and proprietary technology of other parties; |
| ● | costs associated with defending IP infringement, product liability and other claims; |
| ● | regulatory development in the U.S., Europe and PRC and other jurisdictions; |
| ● | estimates of our expenses, future revenues, capital requirements and our needs for additional financing; |
| ● | the potential benefits of strategic collaboration agreements and our ability to enter into strategic arrangements; |
| ● | our ability to maintain and establish collaborations or obtain additional grant funding; the rate and degree of market acceptance of our drug and device candidates; |
| ● | developments relating to our competitors and industry, including competing therapies; |
| ● | our ability to effectively manage our anticipated growth; |
| ● | our ability to attract and retain qualified employees and key personnel; |
| ● | our expectations regarding the period during which we qualify as an emerging growth company under the JOBS Act; |
| ● | statements regarding future revenue, hiring plans, expenses, capital expenditures, capital requirements and share performance; |
| ● | the future trading price of our Class A Ordinary Shares and impact of securities analysts’ reports on these prices; and |
| ● | other risks and uncertainties, including those listed under the caption “Risk Factors.” |
In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue” or the negative of these terms or other comparable terminologies. These statements are only predictions. You should not place undue reliance on forward-looking statements because they involve known and unknown risks, uncertainties and other factors, which are, in some cases, beyond our control and which could materially affect results. Factors that may cause actual results to differ materially from current expectations include, among other things, those listed under “Risk Factors” and elsewhere in this prospectus. If one or more of these risks or uncertainties occur, or if our underlying assumptions prove to be incorrect, actual events or results may vary significantly from those implied or projected by the forward-looking statements. No forward-looking statement is a guarantee of future performance. You should read this prospectus and the documents that we reference in this prospectus and have filed with the SEC as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from any future results expressed or implied by these forward-looking statements.
The forward-looking statements in this prospectus represent our views as of the date of this prospectus. We anticipate that subsequent events and developments will cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we have no current intention of doing so except to the extent required by applicable law. You should therefore not rely on these forward-looking statements as representing our views as of any date subsequent to the date of this prospectus.
This prospectus contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. While we believe the market position, market opportunity and market size information included in this prospectus is generally reliable, such information is inherently imprecise.
53
TRADEMARKS, SERVICE MARKS AND TRADENAMES
This prospectus contains trademarks, service marks and trade names of others, which are the property of their respective owners. Solely for convenience, the trademarks, service marks, logos and trade names referred to in this prospectus are included without the ® and ™ symbols. All trademarks, service marks and trade names appearing in this prospectus are, to our knowledge, the property of their respective owners. We do not intend our use or display of other companies’ trademarks, service marks, copyrights or trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies or unrelated parties.
We will not receive any proceeds from the sale of the Class A Ordinary Shares by the selling shareholders pursuant to this prospectus. All proceeds from the sale of the shares will be for the account of each of the selling shareholders. The selling shareholders may sell these shares in the open market or otherwise, at market prices prevailing at the time of sale, at prices related to the prevailing market price, or at negotiated prices.
The selling shareholders will pay any underwriting discounts and commissions and expenses incurred by the selling shareholder for brokerage or legal services or any other expenses incurred by the selling shareholder in disposing of the shares included in this prospectus. We will bear all other costs, fees and expenses incurred in effecting the registration of the shares covered by this prospectus, including all registration and filing fees and fees and expenses of our counsel and accountants.
We have never declared or paid cash dividends to our shareholders, and we do not intend to pay cash dividends in the foreseeable future. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our Board of Directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, our strategic goals and plans to expand our business, applicable law and other factors that our Board of Directors may deem relevant.
Under Cayman Islands law, dividends may be declared and paid only out of funds legally available therefor, namely out of either profit or our share premium account, and provided further that a dividend may not be paid if this would result in our Company being unable to pay its debts as they fall due in the ordinary course of business.
(See “Risk Factors – We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our shares” and “Description of Share Capital – Dividends”)
The table below sets forth our capitalization and indebtedness as of June 30, 2019:
| ● | on an actual basis (column 1); and | |
| ● | on a pro forma basis as adjusted basis, to give effect to the issuance of 22,437,754 Class A Ordinary Shares issuable upon conversion of the Class B Ordinary Shares; (See “Description of Share Capital”) (column 2). | |
| ● | The table does not include any shares underlying the outstanding options. |
This table should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements, consolidated financial statements and related notes included elsewhere in this prospectus.
| June 30, 2019 | ||||||||
| 1 | 2 | |||||||
| Actual | Pro Forma | |||||||
| US$ | US$ | |||||||
| Equity | ||||||||
| Class A Ordinary Shares | 6,597,362 | 29,035,116 | ||||||
| Class B Ordinary Shares | 22,437,754 | - | ||||||
| Additional paid-in capital | 23,857,814 | 23,857,814 | ||||||
| Accumulated other comprehensive income | 7,345 | 7,345 | ||||||
| Accumulated deficit | (27,957,689 | ) | (27,957,689 | ) | ||||
| Non-controlling interests | (920,298 | ) | (920,298 | ) | ||||
| Total equity | 24,022,288 | 24,022,288 | ||||||
| Total capitalization | 24,022,288 | 24,022,288 | ||||||
The information above is illustrative only.
54
The following summary consolidated balance sheets (successor basis) as of December 31, 2018 and 2017, consolidated statements of operations and comprehensive loss (successor basis) for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, as well as the statement of operations (predecessor basis) for the period January 1, 2017 through February 28, 2017, have been derived from our audited financial statements included elsewhere in this prospectus. The related consolidated balance sheet (successor basis) as of June 30, 2019, consolidated statements of operations and comprehensive loss for the six months ended June 30, 2019 and 2018 have been derived from our unaudited financial statements included elsewhere in this prospectus. You should read this data together with our audited consolidated financial statements and related notes appearing elsewhere in this prospectus and the information under the captions “Capitalization” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Our historical results are not necessarily indicative of our future results. Our consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States, or U.S. GAAP.
You should not view our historical results as an indicator of our future performance.
The following table presents our summary consolidated statements of operations and comprehensive loss (successor basis) for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017.
Selected Consolidated Statements of Operations and Comprehensive Loss (Successor Basis)
(In U.S. Dollars, except number of shares)
Six months | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Revenue: | ||||||||||||||||
| Healthcare services income | $ | 239,792 | $ | 26,662 | $ | 383,450 | $ | - | ||||||||
| Expenses: | ||||||||||||||||
| Cost of healthcare services | (371,218 | ) | (22,749 | ) | (318,011 | ) | - | |||||||||
| Research and development expenses | (2,714,217 | ) | (1,342,179 | ) | (3,101,432 | ) | (2,560,323 | ) | ||||||||
| General and administrative fees | (3,232,916 | ) | (2,238,025 | ) | (4,919,626 | ) | (1,480,093 | ) | ||||||||
| Legal and professional fees | (2,008,774 | ) | (1,063,032 | ) | (1,811,770 | ) | (1,395,490 | ) | ||||||||
| Other operating expenses | (120,788 | ) | (235,413 | ) | (560,709 | ) | (257,177 | ) | ||||||||
| Total expenses | (8,447,913 | ) | (4,901,398 | ) | (10,711,548 | ) | (5,693,083 | ) | ||||||||
| Other (loss) income: | ||||||||||||||||
| Gain on investments in marketable securities, net | 315,977 | - | 501,522 | 3,912,500 | ||||||||||||
| Gain on non-marketable investments | 1,147,199 | - | - | - | ||||||||||||
| Gain (loss) on investments in derivatives, net | 310,195 | (359,844 | ) | (974,444 | ) | (827,501 | ) | |||||||||
| Realized gain on use of digital currencies | 12,334 | - | - | - | ||||||||||||
| Changes in fair value of warrant liabilities | (866,300 | ) | - | 124,726 | - | |||||||||||
| Gain on extinguishment of convertible debts | 1,198,490 | - | - | - | ||||||||||||
| Interest (expense) income, net | (3,678,566 | ) | (301,362 | ) | (4,458,191 | ) | 44,269 | |||||||||
| Dividend income | - | - | - | 2,308 | ||||||||||||
| Sundry income | 128,444 | - | - | - | ||||||||||||
| Total other (loss) income, net | (1,432,227 | ) | (661,206 | ) | (4,806,387 | ) | 3,131,576 | |||||||||
| Net loss | (9,640,348 | ) | (5,535,942 | ) | (15,134,485 | ) | (2,561,507 | ) | ||||||||
| Less: net loss attributable to non-controlling interests | (551,877 | ) | (47,570 | ) | (302,762 | ) | (14,045 | ) | ||||||||
| Net loss attributable to Aptorum Group Limited | $ | (9,088,471 | ) | $ | (5,488,372 | ) | $ | (14,831,723 | ) | $ | (2,547,462 | ) | ||||
| Net loss per share – basic and diluted* | $ | (0.31 | ) | $ | (0.20 | ) | $ | (0.53 | ) | $ | (0.09 | ) | ||||
| Weighted-average shares outstanding – basic and diluted | 28,978,151 | 27,864,135 | 27,909,788 | 26,963,435 | ||||||||||||
| Net loss | $ | (9,640,348 | ) | $ | (5,535,942 | ) | $ | (15,134,485 | ) | $ | (2,561,507 | ) | ||||
| Other comprehensive income (loss) | ||||||||||||||||
| Unrealized loss on investments in available-for-sale securities | - | (178,027 | ) | (1,122,251 | ) | (367,782 | ) | |||||||||
| Exchange differences on translation of foreign operations | 2,000 | 167 | 5,345 | - | ||||||||||||
| Other comprehensive income (loss) | 2,000 | (177,860 | ) | (1,116,906 | ) | (367,782 | ) | |||||||||
| Comprehensive loss | (9,638,348 | ) | (5,713,802 | ) | (16,251,391 | ) | (2,929,289 | ) | ||||||||
| Less: comprehensive loss attributable to non-controlling interests | (551,877 | ) | (47,570 | ) | (302,762 | ) | (14,045 | ) | ||||||||
| Comprehensive loss attributable to the shareholders of Aptorum Group Limited | $ | (9,086,471 | ) | $ | (5,666,232 | ) | $ | (15,948,629 | ) | $ | (2,915,244 | ) | ||||
| * | The shares and per share data are presented at a weighted average basis to reflect the nominal share issuance. |
55
The following table presents our summary statements of operations (predecessor basis) for the period January 1, 2017 through February 28, 2017.
Selected Statement of Operations (Predecessor Basis)
(In U.S. Dollars)
| January 1, 2017 through February 28, 2017 | ||||
| Investment income: | ||||
| Interest income | $ | 3,011 | ||
| Total investment income | 3,011 | |||
| Expenses | ||||
| General and administrative fees | 17,516 | |||
| Management fees | 108,958 | |||
| Legal and professional fees | 98,646 | |||
| Other operating expenses | 1,907 | |||
| Total expenses | 227,027 | |||
| Net investment loss | $ | (224,016 | ) | |
| Realized and unrealized losses | ||||
| Net realized losses on investments in unaffiliated issuers | $ | (15,327 | ) | |
| Net change in unrealized depreciation on investments | (386,741 | ) | ||
| Net realized and unrealized losses | (402,068 | ) | ||
| Net decrease in net assets resulting from operations | $ | (626,084 | ) | |
The following table presents our summary consolidated balance sheets (successor basis) as of June 30, 2019, December 31, 2018 and December 31, 2017.
| As of June 30, 2019 | As of December 31, 2018 | As of December 31, 2017 | ||||||||||
| (Unaudited) | ||||||||||||
| Cash, restricted cash and marketable securities | $ | 6,135,837 | $ | 27,121,576 | $ | 18,698,455 | ||||||
| Total current assets | 7,742,644 | 28,722,941 | 20,283,399 | |||||||||
| Total assets | 24,740,370 | 45,074,640 | 31,559,982 | |||||||||
| Total current liabilities | 597,141 | 12,184,865 | 1,330,734 | |||||||||
| Total liabilities | 718,082 | 12,328,738 | 1,330,734 | |||||||||
| Total equity attributable to the shareholders of Aptorum Group Limited | 24,942,586 | 33,114,435 | 30,243,293 | |||||||||
| Non-controlling interests | (920,298 | ) | (368,533 | ) | (14,045 | ) | ||||||
| Total equity | 24,022,288 | 32,745,902 | 30,229,248 | |||||||||
| Total liabilities and equity | $ | 24,740,370 | $ | 45,074,640 | $ | 31,559,982 | ||||||
56
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with the section titled “Selected Financial Data” and the financial statements, consolidated financial statements and related notes included elsewhere in this prospectus. This discussion contains forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those discussed in the section titled “Risk Factors” and in other parts of this prospectus. Our financial statements and consolidated financial statements have been prepared in accordance with U.S. GAAP.
Overview
We are a pharmaceutical company currently in the preclinical stage, dedicated to developing and commercializing a broad range of therapeutic and diagnostic technologies to tackle unmet medical needs. We have obtained exclusive licenses for our technologies. In addition, we are also developing certain proprietary technologies as product candidates. We are pursuing therapeutic and diagnostic projects (including projects seeking to use extracts or derivatives from natural substances to treat diseases) in neurology, infectious diseases, gastroenterology, oncology and other disease areas. We also have projects focused on surgical robotics. (See “Our Business – Lead Projects and Other Projects under Development”) Also, we opened a medical clinic, AML Clinic, in June 2018.
Although none of our drug or device candidates has yet been approved for testing in humans, our goal is to develop a broad range of early stage novel therapeutics and diagnostics across a wide range of disease/therapeutic areas. Key components of our strategy for achieving this goal include: (for details of our strategy, See “Our Business – Our Strategy”)
| ● | Developing therapeutic and diagnostic innovations across a wide range of disease/therapeutic areas; | |
| ● | Selectively expanding our portfolio with potential products that may be able to attain orphan drug designation and/or satisfy current unmet medical needs; | |
| ● | Collaborating with leading academic institutions and CROs; | |
| ● | Expanding our in-house pharmaceutical development center; | |
| ● | Leveraging our management’s expertise, experience and commercial networks; | |
| ● | Strategically developing opportunities in Hong Kong to promote access to the PRC market; and | |
| ● | Obtaining and leveraging government grants to fund project development. |
We have begun to devote a significant percentage of our resources, including a substantial portion of the proceeds to three therapeutic projects (“Lead Projects”). The drug candidates being advanced as the Lead Projects are ALS-1, ALS-4 and NLS-1, described in further detail below. If the results of the remaining preclinical studies of these drug candidates are positive, we expect to be able to submit by 2020 or 2021 an Investigational New Drug Application (“IND”) for at least one of these candidates to the U.S. Food and Drug Administration (“FDA”) or an equivalent application to the regulatory authorities in one or more other jurisdictions such as the China’s National Medical Products Administration (“NMPA”) and/or the European Medicines Agency (“EMA”). Acceptance of these applications by the relevant regulatory authority would enable the Company to begin testing that drug candidate in humans in that jurisdiction. Our ability to obtain any approval of such applications is entirely dependent upon the results of our preclinical studies, none of which have yet been completed.
57
Our current business consists of “therapeutics” and “non-therapeutics” segments. However, our focus is on the therapeutics segments. Because of the risks, costs and extended development time required for successful drug development, we have determined to pursue projects within our non-therapeutics segments, such as AML Clinic, to provide some interim revenue and medical robots that may be brought to market and generate revenue more quickly.
Therapeutics Segment. In our therapeutics segment (“Aptorum Therapeutics Group”), we are currently seeking to develop various drug molecules (including projects seeking to use extracts or derivatives from natural substances to treat diseases) and certain technologies for the treatment (“therapeutics”) and diagnosis (“diagnostics”) of human disease conditions in neurology, infectious diseases, gastroenterology, oncology and other disease areas. In addition, we are seeking to identify additional prospects which may qualify for potential orphan drug designation (e.g., rare types of cancer) or which address other current unmet medical needs. Aptorum Therapeutics Group is operated through Aptorum’s wholly-owned subsidiary, Aptorum Therapeutics Limited, a Cayman Islands exempted company with limited liability, whose principal place of business is in Hong Kong and its indirect subsidiary companies (who we sometimes refer to herein as project companies), whose principal places of business are also in Hong Kong.
Non-Therapeutics Segment. The non-therapeutics segment (“Aptorum Non-Therapeutics Group”) encompasses two businesses: (i) the development of surgical robotics and medical devices and (ii) AML Clinic. The development of surgical robotics and medical devices business is operated through Signate Life Sciences Limited, a subsidiary of Aptorum Therapeutics Limited. The outpatient clinic is operated through our subsidiary, Aptorum Medical Limited. Effective as of March 2018, we leased office space in Central, Hong Kong as the home to our medical clinic (“AML Clinic”). AML Clinic commenced operations under the name of Talem Medical in June 2018. The estimated general administrative expenses and other operating expenses of AML Clinic is expected to be no more than USD120,000 per month. The clinic is expected to reach operating profit in 18 months from the clinic reaching its full operating capacity upon (i) the successful recruitment of a minimum of six full time physicians (AML Clinic currently has one full time physician and six part time physicians) and (ii) establishing steady patients flow via brand development. (See “Our Business – Lead Projects and Other Projects under Development – Other Projects under Development – Aptorum Medical Limited - AML Clinic”)
The Company has already obtained opportunities resulting in our existing licensing agreements from various contractual relationships that we have entered into, including service/consulting agreements with some of the world’s leading specialists and clinicians in our areas of interest, with academic institutions and organizations, and with CROs. We anticipate that these relationships will generate additional licensing opportunities in the future. In addition, we have established and are continuing to expand our in-house research facilities (collectively, the “R&D Center”) to develop some of our drug and device candidates internally and to collaborate with third-party researchers.
Prior to March 2017, the Company had pursued passive healthcare related investments in early stage companies primarily in the United States. However, we have since ceased pursuing further passive investment operations and intend to exit all such portfolio investments over an appropriate timeframe to focus resources on our current business.
Critical Accounting Policies, Estimates and Assumptions
Basis of presentation
The consolidated financial statements are prepared in accordance with U.S. GAAP. Before March 1, 2017, the Company was an investment company under U.S. GAAP for the purposes of financial reporting. U.S. GAAP for an investment company requires investments to be recorded at estimated fair value and the unrealized gains and/or losses in an investment’s fair value are recognized on a current basis in the statements of operations. In addition, the Company did not consolidate its subsidiaries, since they were operating companies and not investment companies. Such entities were fair valued in accordance with ASC Topic 946 (“ASC 946”) and ASC Topic 820 (“ASC 820”).
As of March 1, 2017, after the change of business purpose, legal form and substantive activities, the Company’s status changed to an operating company from an investment company since it no longer met the criteria to qualify as an investment company under the ASC 946. The Company discontinued applying the guidance in ASC 946 and began to account for the change in status prospectively by accounting for its investments in accordance with other U.S. GAAP topics. The consolidated financial statements include the financial statements of the Company and its subsidiaries. All significant intercompany transactions and balances have been eliminated in consolidation.
58
Principles of consolidation
The consolidated financial statements of the Group are presented on the accrual basis of accounting in accordance with U.S. GAAP and include the accounts of the Company, its direct and indirect wholly and majority owned subsidiaries and a variable interest entity. All material intercompany balances and transactions have been eliminated in preparation of the consolidated financial statements. Non-controlling interests represent the equity interest that is not owned by the Group.
Use of estimates
The preparation of the consolidated financial statements on successor basis in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of increases and decreases in net assets from operations as well as income and expenses during the reporting period. Significant accounting estimates reflected in the Group’s consolidated financial statements include valuing equity securities, fair value of investments in securities, convertible debts and finance lease, the useful lives of intangible assets and property, plant and equipment, impairment of long-lived assets, valuation allowance for deferred tax assets, and collectability of receivables. Actual results could differ from those estimates.
Foreign currency translation and transaction
USD is the reporting currency. The functional currency of subsidiaries in the Cayman Islands is USD, the functional currency of subsidiaries in Hong Kong is Hong Kong Dollars (“HKD”), the functional currency of a subsidiary in Macao is Macanese Pataca (“MOP”) and the functional currency of a subsidiary in the United Kingdom is Great British Pound (“GBP”). An entity’s functional currency is the currency of the primary economic environment in which it operates, normally that is the currency of the environment in which it primarily generates and expends cash. The management considered various indicators, such as cash flows, market expenses, financing and inter-company transactions and arrangements in determining the Group’s functional currency.
In the consolidated financial statements, the financial information of the Company and its subsidiaries, which use HKD, MOP and GBP as their functional currency, has been translated into USD. Assets and liabilities are translated from each subsidiary’s functional currency at the exchange rates on the balance sheet date, equity amounts are translated at historical exchange rates, and revenues, expenses, gains, and losses are translated using the average rate for the year. Translation adjustments are reported as cumulative translation adjustments and are shown as a separate component of other comprehensive income or loss in the statement of operations and comprehensive loss.
Fair value measurement
Fair value is defined as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required or permitted to be recorded at fair value, the Group considers the principal or most advantageous market in which it would transact its business, and it considers assumptions that market participants would use when pricing the asset or liability.
As a basis for considering such assumptions, a three-tier fair value hierarchy prioritizes the inputs utilized in measuring fair value as follows:
| ● | Level 1 applies to assets or liabilities for which there are quoted prices in active markets for identical assets or liabilities. | |
| ● | Level 2 applies to assets or liabilities for which there are inputs other than quoted prices included within Level 1 that are observable for the asset or liability such as quoted prices for similar assets or liabilities in active markets; quoted prices for identical assets or liabilities in markets with insufficient volume or infrequent transactions (less active markets); or model-derived valuations in which significant inputs are observable or can be derived principally from, or corroborated by, observable market data. | |
| ● | Level 3 applies to assets or liabilities for which there are unobservable inputs to the valuation methodology that are significant to the measurement of the fair value of the assets or liabilities. |
59
Impairment of long-lived assets
The Group reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may no longer be recoverable. When these events occur, the Group measures impairment by comparing the carrying value of the long-lived assets to the estimated undiscounted future cash flows expected to result from the use of the assets and their eventual disposition. If the sum of the expected undiscounted cash flow is less than the carrying amount of the assets, the Group would recognize an impairment loss, which is the excess of carrying amount over the fair value of the assets, using the expected future discounted cash flows.
Convertible debts
The Group determines the appropriate accounting treatment of its convertible debts in accordance with the terms in relation to the conversion feature, call and put option, beneficial conversion feature (“BCF”) and settlement feature. On December 17, 2018, the Group concluded that the contingency of BCF was effectively resolved upon the completion of the IPO and recognized BCF according to the agreement.
Convertible debts are subsequently measured at amortized cost, using the effective interest rate method. Amortized cost is calculated by taking into account any discount or premium on acquisition and fees or costs that are an integral part of the effective interest rate. The effective interest rate amortization is included in interest expense in the condensed consolidated statements of operations.
Amortized cost is calculated by taking into account any discount or premium on acquisition and fees or costs that are an integral part of the effective interest rate. The effective interest rate amortization is included in finance costs in the condensed consolidated statements of operations.
The repurchasing of convertible debts is considered an extinguishment and the difference between the repurchasing price of debt, the net carrying amount of the extinguished debt and the intrinsic value of BCF is recognized in the condensed consolidated statement of operations. The intrinsic value of BCF at the extinguishment date decrease to additional paid-in capital. On April 24, 2019, the Group repurchased its convertible debts at $13.6 million with carrying amount of $13.5 million and the intrinsic value of BCF is $1.3 million with a gain on extinguishment of convertible debts of $1.2 million.
Revenue recognition
Revenue is recognized when (or as) the Company satisfies performance obligations by transferring a promised goods or services to a customer. Revenue is measured at the transaction price which is based on the amount of consideration that the Company expects to receive in exchange for transferring the promised goods or services to the customer. Contracts with customers are comprised of invoices and written contracts. Revenue from healthcare services is measured upon the provision of the relevant services.
Income taxes
The Group accounts for income taxes under the asset and liability method. Under this method, deferred income taxes are determined based on differences between the financial carrying amounts of existing assets and liabilities and their tax bases. Income taxes are provided for in accordance with the laws of the relevant taxing authorities.
A valuation allowance is provided for deferred tax assets if it is more likely than not that these items will either expire before the Group is able to realize their benefits, or that future deductibility is uncertain. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount expected to be realized.
RESULTS OF OPERATION (SUCCESSOR BASIS)
Explanatory Note
After the Restructure, the results of operations and cash flows of the Group for period beginning March 1, 2017 and subsequent balance sheet dates are referred to herein as “Successor” consolidated financial information.
Financial statements and information are presented for the year ended December 31, 2018 (Successor) and the ten months ended December 31, 2017 (Successor), which may not be comparable with amounts shown in each year/period.
60
The following table summarizes our results of operations for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017.
Six months ended | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Revenue: | ||||||||||||||||
| Healthcare services income | $ | 239,792 | $ | 26,662 | $ | 383,450 | $ | - | ||||||||
| Expenses: | ||||||||||||||||
| Cost of healthcare services | (371,218 | ) | (22,749 | ) | (318,011 | ) | - | |||||||||
| Research and development expenses | (2,714,217 | ) | (1,342,179 | ) | (3,101,432 | ) | (2,560,323 | ) | ||||||||
| General and administrative fees | (3,232,916 | ) | (2,238,025 | ) | (4,919,626 | ) | (1,480,093 | ) | ||||||||
| Legal and professional fees | (2,008,774 | ) | (1,063,032 | ) | (1,811,770 | ) | (1,395,490 | ) | ||||||||
| Other operating expenses | (120,788 | ) | (235,413 | ) | (560,709 | ) | (257,177 | ) | ||||||||
| Total expenses | (8,447,913 | ) | (4,901,398 | ) | (10,711,548 | ) | (5,693,083 | ) | ||||||||
| Other (loss) income: | ||||||||||||||||
| Gain on investments in marketable securities, net | 315,977 | - | 501,522 | 3,912,500 | ||||||||||||
| Gain on non-marketable investments | 1,147,199 | - | - | - | ||||||||||||
| Gain (loss) on investments in derivatives, net | 310,195 | (359,844 | ) | (974,444 | ) | (827,501 | ) | |||||||||
| Realized gain on use of digital currencies | 12,334 | - | - | - | ||||||||||||
| Changes in fair value of warrant liabilities | (866,300 | ) | - | 124,726 | - | |||||||||||
| Gain on extinguishment of convertible debts | 1,198,490 | - | - | - | ||||||||||||
| Interest (expense) income, net | (3,678,566 | ) | (301,362 | ) | (4,458,191 | ) | 44,269 | |||||||||
| Dividend income | - | - | - | 2,308 | ||||||||||||
| Sundry income | 128,444 | - | - | - | ||||||||||||
| Total other (loss) income, net | (1,432,227 | ) | (661,206 | ) | (4,806,387 | ) | 3,131,576 | |||||||||
| Net loss | $ | (9,640,348 | ) | $ | (5,535,942 | ) | $ | (15,134,485 | ) | $ | (2,561,507 | ) | ||||
Revenue
Healthcare services income was $239,792, $26,662 and $383,450 for the six months ended June 30, 2019 and 2018, and the year ended December 31, 2018, respectively, which related to the service income derived from the AML Clinic. We had no healthcare services income for the period March 1, 2017 to December 31, 2017.
Research and development expenses
Research and development expenses comprised of costs incurred related to research and development activities, including payroll expenses to our research and development staff, sponsored research programs with various universities and research institutions and costs in acquiring IP rights which did not meet the criteria of capitalization under the U.S. GAAP. We currently maintain a system to keep track of costs spent by each project. The following table sets forth a summary of our research and development expenses for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017. The increase in research and development expenses was primarily due to the incurred expenses for new sponsored research entered into with Universities in the current period, and the full operation of research and development was started in the second half of 2018 which led to depreciation and increase in payroll expenses since second half of 2018.
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Research and Development Expenses: | ||||||||||||||||
| Payroll expenses | $ | 810,166 | $ | 575,968 | $ | 1,363,740 | $ | 95,078 | ||||||||
| Sponsored research | 844,575 | 306,822 | 796,943 | 1,327,247 | ||||||||||||
| Amortization and depreciation | 384,782 | 160,502 | 437,453 | 58,903 | ||||||||||||
| Consultation | 544,722 | 192,107 | 298,315 | 92,129 | ||||||||||||
| General R&D expense | 122,472 | 106,780 | 174,981 | 186,910 | ||||||||||||
| Research grant | 7,500 | - | 30,000 | 800,056 | ||||||||||||
| Total Research and Development Expenses | $ | 2,714,217 | $ | 1,342,179 | $ | 3,101,432 | $ | 2,560,323 | ||||||||
61
General and administrative fees
The following table sets forth a summary of our general and administrative expenses for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017. The increase in general and administration fees was mainly due to the increased headcount in the Group to support the business development, and the higher amounts of insurance expense incurred after the Company listed its securities on NASDAQ.
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| General and Administrative Fees: | ||||||||||||||||
| Administrative fees | $ | - | $ | 384,615 | $ | 448,718 | $ | 640,932 | ||||||||
| Payroll expenses | 1,604,933 | 1,060,950 | 2,510,331 | 306,967 | ||||||||||||
| Rent and rates | 285,037 | 296,074 | 681,502 | 49,518 | ||||||||||||
| Travelling expenses | 431,287 | 126,286 | 414,696 | 175,671 | ||||||||||||
| Amortization and depreciation | 200,919 | 48,765 | 244,839 | 344 | ||||||||||||
| Insurance | 341,614 | 88,649 | 199,698 | 23,412 | ||||||||||||
| Recruitment expenses | 1,316 | 29,665 | 50,476 | 125,535 | ||||||||||||
| Other expenses | 367,810 | 203,021 | 369,366 | 157,714 | ||||||||||||
| Total General and Administrative Fees | $ | 3,232,916 | $ | 2,238,025 | $ | 4,919,626 | $ | 1,480,093 | ||||||||
Management fees/Administrative fees
AENEAS CAPITAL LIMITED, formerly known as APTUS CAPITAL LIMITED, a related company of the Group/Company, provided management and administrative services to the Group and incurred pre-determined management fees. For the six months ended June 30, 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, the administrative fees of $384,615, $448,718 and $640,932, respectively, has been reclassified to general and administrative fees due to the Restructure and since the Company has become a pharmaceutical company, so the management fees are no longer determined by net asset value since then. The services were terminated in July 2018.
Legal and professional fees
For the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, the legal and professional fees were $2,008,774, $1,063,032, $1,811,770 and $1,395,490, respectively. The increase in legal and professional fees was mainly due to the increasing need for consultancy services on various projects.
62
Other operating expenses
The following table sets forth a summary of our other operating expenses for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017. The increase in other operating expenses in 2018 was mainly due to more corporate events held to promote the Company and business expansion.
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Other Operating Expenses: | ||||||||||||||||
| Event and meeting expenses | $ | 52,507 | $ | 131,926 | $ | 385,483 | $ | 83,288 | ||||||||
| Commission expenses | 1,759 | - | 1,517 | 55,726 | ||||||||||||
| Other expenses | 66,522 | 103,487 | 173,709 | 118,163 | ||||||||||||
| Total Other Operating Expenses | 120,788 | 235,413 | 560,709 | 257,177 | ||||||||||||
Other (loss) income
The following table sets forth a summary of our other (loss) income for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017.
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Other (loss) income: | ||||||||||||||||
| Gain on investments in marketable securities, net | $ | 315,977 | $ | - | $ | 501,522 | $ | 3,912,500 | ||||||||
| Gain on non-marketable investment | 1,147,199 | - | - | - | ||||||||||||
| Gain (loss) on investments in derivatives, net | 310,195 | (359,844 | ) | (974,444 | ) | (827,501 | ) | |||||||||
| Realized gain on use of digital currencies | 12,334 | - | - | - | ||||||||||||
| Changes in fair value of warrant liabilities | (866,300 | ) | - | 124,726 | - | |||||||||||
| Gain on extinguishment of convertible debts | 1,198,490 | - | - | - | ||||||||||||
| Interest (expense) income, net | (3,678,566 | ) | (301,362 | ) | (4,458,191 | ) | 44,269 | |||||||||
| Dividend income | - | - | - | 2,308 | ||||||||||||
| Sundry income | 128,444 | - | - | - | ||||||||||||
| Total other (loss) income | (1,432,227 | ) | (661,206 | ) | (4,806,387 | ) | 3,131,576 | |||||||||
Net loss attributable to Aptorum Group Limited
For the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, net loss attributable to Aptorum Group Limited (excluding net loss attributable to non-controlling interests) was $9,088,471, $5,488,372, $14,831,723 and $2,547,462, respectively.
RESULTS OF OPERATION (PREDECESSOR BASIS)
Explanatory Note
Before March 1, 2017, Aptorum Group Limited was incorporated as an exempted company with limited liability in the Cayman Islands and operated as an open ended investment company, which would own and oversee the management, operations and investments of its subsidiaries. On February 21, 2017, a special resolution was passed at a directors’ meeting, and on March 1, 2017, a resolution also was passed at a shareholders’ meeting. According to which, the Company changed from an investment fund with management shares and non-voting participating redeemable preference shares to a holding company with operating subsidiaries (the “Restructure”). After the Restructure, the Company has become a Hong Kong based pharmaceutical company currently in the preclinical stage. The results of operations and cash flows of the Company for the periods ended on or prior to February 28, 2017, and its financial position as of balance sheet date on or prior to February 28, 2017 are referred to as “Predecessor” financial information.
Financial statements and information are presented for the two months ended February 28, 2017 (Predecessor).
General and administrative fees
For the period January 1, 2017 to February 28, 2017, the general and administrative fees were $17,516 which are miscellaneous expenses.
Management fees
AENEAS CAPTIAL LIMITED, formerly known as APTUS CAPITAL LIMITED, a related company of the Group/Company, provided management and administrative services to the Group and incurred pre-determined management fees. For the period January 1, 2017 to February 28, 2017, AENEAS CAPITAL LIMITED was entitled to receive a management fee which was equal to 2.5% per annum of the net asset value of the Company.
63
Legal and professional fees
For the period January 1, 2017 to February 28, 2017, the legal and professional fees were $98,646.
Other operating expenses
For the period January 1, 2017 to February 28, 2017, other operating expenses were $1,907.
Other income
The Company met the assessment of an investment company under the Financial Accounting Standards Board (the “FASB”) Accounting Standards Codification Topic 946 (“ASC 946”) and was an investment company under U.S. GAAP for the purposes of financial reporting for the period January 1, 2017 to February 28, 2017, which the interest income was $3,011.
Realized and unrealized losses on investments
Realized and unrealized losses on investments mainly consist of net realized loss on investments in unaffiliated issuers and net unrealized depreciation on investments in unaffiliated issuers. For the period January 1, 2017 to February 28, 2017, the realized and unrealized losses were $402,068.
Net decrease in net assets resulting from operations
For the period January 1, 2017 to February 28, 2017, the net decrease in net assets resulting from operations was $626,084.
LIQUIDITY AND CAPITAL RESOURCES
The Company reported a net loss of $9,640,348 and net operation cash outflow of $7,335,009 for the six months ended June 30, 2019, respectively. In addition, the Company had an accumulated deficit of $27,957,689 as of June 30, 2019. The Company’s operating results for future periods are subject to numerous uncertainties and it is uncertain if the Company will be able to reduce or eliminate its net losses for the foreseeable future. If management is not able to generate significant revenues from its product candidates currently in development, the Company may not be able to achieve profitability.
The Company’s principal sources of liquidity have been cash and marketable securities. As of the date of issuance of the consolidated financial statements, the Company has approximately $3 million of cash and marketable securities. In addition, based upon the current market price of the Company’s marketable securities, it anticipates it can liquidate such marketable securities for greater than its carrying amount, if necessary. On August 13, 2019, the Company entered into financing arrangements with Aeneas Group Limited, a related party, and Jurchen Investment Corporation, the ultimate parent of the Group, allowing the Group to access up to a total $15.0 million in line of credit debt financing.
The Company believes that available cash and marketable securities, together with signed loan facilities, should enable the Company to meet presently anticipated cash needs for at least the next 12 months after the date that the financial statements are issued and the Company has prepared the consolidated financial statements on a going concern basis. If the Company encounters unforeseen circumstances that place constraints on its capital resources, management will be required to take various measures to conserve liquidity, which could include, but not necessarily be limited to, deferring some of its research and seeking to dispose of marketable securities.
CONTRACTUAL OBLIGATIONS
The following table sets forth our contractual obligations as of June 30, 2019.
| Payment Due by Period | ||||||||||||
| Total | One to three years | Three to five years | ||||||||||
| US$ | US$ | US$ | ||||||||||
| Operating lease commitments | 1,399,332 | 1,399,332 | - | |||||||||
64
Operating lease commitments
We have several operating leases, primarily for offices. Our principal executive offices are located in Hong Kong; we also have offices in London and Jersey City. Payments under operating leases are expensed on a straight-line basis over the periods of the respective leases, and the terms of the leases do not contain rent escalation, contingent rent, and renewal or purchase options. The aggregate future minimum payment under these non-cancelable operating leases are summarizes in the table above.
CONTINGENT PAYMENT OBLIGATIONS
The Group has additional contingency payment obligations under each of the license agreements, such as milestone payments, royalties, research and development funding, if certain condition or milestone is met.
Milestone payments are to be made upon achievements of certain conditions, such as Investigational New Drugs (“IND”) filing or U.S. Food and Drug Administration (“FDA”) approval, first commercial sale of the licensed products, or other achievements. The aggregate amount of the milestone payments that the Group are required to pay up to different achievements of conditions and milestones for all the license agreements signed as of June 30, 2019 are below:
| Amount | ||||
| Drug molecules: up to the conditions and milestones of | ||||
| Preclinical to IND filing | $ | 372,564 | ||
| From entering phase 1 to before first commercial sale | 24,216,410 | |||
| First commercial sale | 15,656,410 | |||
| Net sales amount more than certain threshold in a year | 75,769,231 | |||
| Subtotal | 116,014,615 | |||
| Surgical robotics and medical devices: up to the conditions and milestones of | ||||
| Before FDA approval | 270,000 | |||
| FDA approval obtained | 200,000 | |||
| Subtotal | 470,000 | |||
| Total | $ | 116,484,615 | ||
For the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, the Group incurred $nil, $nil, $30,000 and $nil milestone payments, respectively. For the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, the Group did not incur any royalties or research and development funding, respectively. As of June 30, 2019, no other milestone payments had been triggered under any of the existing license agreements.
CONDENSED SUMMARY OF OUR CASH FLOWS (SUCCESSOR BASIS)
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Net cash used in operating activities | $ | (7,335,009 | ) | $ | (6,315,706 | ) | $ | (10,035,531 | ) | $ | (5,782,695 | ) | ||||
| Net cash provided by (used in) investing activities | (678,566 | ) | (2,779,328 | ) | (6,061,987 | ) | 12,802,718 | |||||||||
| Net cash (used in) provided by financing activities | (13,626,922 | ) | 15,296,425 | 25,478,949 | 9,082,001 | |||||||||||
| Net (decrease) increase in cash and restricted cash | (21,640,497 | ) | 6,201,391 | 9,381,431 | 16,102,024 | |||||||||||
65
Operating activities
Net cash used in operating activities amounted to $7.3 million for the six months ended June 30, 2019. During the period, the Group had net loss of $9.6 million. Meanwhile, the Group had gain on extinguishment of convertible debts of $1.2 million, gain on non-marketable investments of $1.1 million, and interest expense and accretion of convertible debts of $3.7 million.
Net cash used in operating activities amounted to $6.3 million for the six months ended June 30, 2018. During the period, the Group had net loss of $5.5 million. Meanwhile, the Group had an increase of long-term prepayments of $1.6 million, an increase of accounts payable and accrued expenses of $0.2 million, loss on investments in derivatives of $0.4 million and amortization and depreciation expenses of $0.2 million.
Net cash used in operating activities amounted to $10.0 million for the year ended December 31, 2018. During the year, the Group had net loss of $15.1 million. Meanwhile, the Group had interest expense and accretion of convertible debts of $4.6 million.
Net cash used in operating activities amounted to $5.8 million for the period March 1, 2017 to December 31, 2017. During the period, the Group had net loss of $2.6 million. Meanwhile, the Group had gain on investments in marketable securities of $3.9 million, loss on investments in derivatives of $0.8 million and an increase of other receivables and prepayments of $0.3 million.
Investing activities
Net cash provided by investing activities amounted to $0.7 million for the six months ended June 30, 2019. During the period, the Group had purchases of property, plant and equipment of $0.7 million, proceeds from sale of marketable securities of $0.8 million, net loan to a third party of $0.6 million.
Net cash used in investing activities amounted to $2.8 million for the six months ended June 30, 2018. During the period, the Group had purchases of property, plant and equipment of $2.5 million and purchases of intangible assets of $0.2 million.
Net cash used in investing activities amounted to $6.1 million for the year ended December 31, 2018. During the year, the Group had purchases of property, plant and equipment of $5.6 million and purchases of intangible assets of $0.4 million.
Net cash provided by investing activities amounted to $12.8 million for the period March 1, 2017 to December 31, 2017. During the period, the Group had proceeds from sales of investment securities of $16.0 million, purchases of intangible assets of $1.0 million and purchases of equipment of $2.1 million.
Financing activities
Net cash used in financing activities amounted to $13.6 million for the six months ended June 30, 2019. During the period, the Group had repurchased of convertible debts of $13.6 million.
Net cash provided by financing activities amounted to $15.3 million for the six months ended June 30, 2018. During the period, the Group had proceeds from issuance of convertible debts of $16.1 million and payments for debt issuance costs of $0.9 million.
Net cash provided by financing activities amounted to $25.5 million for the year ended December 31, 2018. During the year, the Group had proceeds from issuance of convertible debts of $16.1 million, proceeds from issuance of shares of $11.1 million, payments of debt issuance costs of $1.1 million and payments of initial public offering costs of $0.5 million.
Net cash provided by financing activities amounted to $9.1 million for the period March 1, 2017 to December 31, 2017. During the period, the Group had proceeds from issuance of shares of $8.6 million and proceeds from issuance of convertible debts of $0.5 million.
CONDENSED SUMMARY OF OUR CASH FLOWS (PREDECESSOR BASIS)
Operating activities
Net cash used in operating activities amounted to $0.3 million for the period January 1, 2017 to February 28, 2017. During the period, the Company had net decrease in net assets resulting from operations of $0.6 million and unrealized depreciation on investments of $0.4 million.
Investing activities
No cash flow from investing activities for the period January 1, 2017 to February 28, 2017.
Financing activities
Net cash flow from financing activities was nil for the period January 1, 2017 to February 28, 2017.
CAPITAL EXPENDITURES
Our capital expenditures were $0.7 million, $2.8 million, $6.0 million and 3.1 million for the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, respectively. We did not incur capital expenditures for the period January 1, 2017 to February 28, 2017. These capital expenditures were incurred primarily for investments in facilities, leasehold improvements, equipment and technology.
66
RECENT ACCOUNTING PRONOUNCEMENTS
Recently adopted accounting pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”), which was subsequently modified in August 2015 by ASU 2015-14, Revenue from Contracts with Customers: Deferral of the Effective Date. We adopted this standard effective January 1, 2019 using the modified retrospective approach, in which case the cumulative effect of applying the standard would be recognized at the date of initial application. The adoption does not have a material impact to the condensed consolidated financial statements.
In January 2016, the FASB issued Accounting Standards Update No. 2016-01 (ASU 2016-01) "Financial Instruments-Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities," which amends various aspects of the recognition, measurement, presentation, and disclosure of financial instruments. We adopted ASU 2016-01 as of January 1, 2019 using the modified retrospective method for our marketable equity securities and the prospective method for our non-marketable equity securities. The following table summarizes the changes to our condensed consolidated balance sheet for the adoption of ASU 2016-01:
| December 31, 2018 | Adjustment due to ASU 2016-01 | January 1, 2019 | ||||||||||
| Accumulated deficit | $ | (17,379,185 | ) | $ | (1,490,033 | ) | $ | (18,869,218 | ) | |||
| Accumulated other comprehensive loss | $ | (1,484,688 | ) | $ | 1,490,033 | $ | 5,345 | |||||
We have elected to use the measurement alternative for our non-marketable equity securities, defined as cost adjusted for changes from observable transactions for identical or similar investments of the same issuer, less impairment. The adoption of ASU 2016-01 increases the volatility of our other income (expense), net, as a result of the unrealized gain or loss from the remeasurement of our equity securities.
Recently issued accounting standards which have not yet been adopted
In February 2016, the FASB issued ASU 2016-02, Leases (“ASU 2016-02”), which requires a lessee to recognize a right-of-use asset and a lease liability for operating leases, initially measured at the present value of the future lease payments, in the balance sheet. ASU 2016-02 also requires a lessee to recognize a single lease cost, calculated so that the cost of the lease is allocated over the lease term, generally on a straight-line basis. This new guidance is effective for fiscal years beginning after December 15, 2019. Early adoption is permitted. The Company is currently evaluating the potential effects of adopting the provisions of ASU 2016-02 on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, Disclosure Framework – Changes to the Disclosure Requirements for Fair Value Measurement, which amends ASC 820, Fair Value Measurement. This ASU modifies the disclosure requirements for fair value measurements by removing, modifying, or adding certain disclosures. The effective date is the first quarter of fiscal year 2021, with early adoption permitted for the removed disclosures and delayed adoption until fiscal year 2021 permitted for the new disclosures. The removed and modified disclosures will be adopted on a retrospective basis and the new disclosures will be adopted on a prospective basis. The adoption will not have a material effect on the Company’s financial statements.
In June 2016, the FASB issued ASU 2016-13, Financial Instruments—Credit Losses ("ASU 2016-13"). Subsequently, the FASB issued ASU 2019-05, Financial Instruments- Credit Losses (Topic 326): Targeted Transition Relief. The amendments in ASU 2016-13 update guidance on reporting credit losses for financial assets. These amendments affect loans, debt securities, accounts receivables, net investments in leases, off balance sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. The amendments are effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. The Company is currently evaluating the impact on its financial statements of adopting this guidance.
67
The Group does not believe other recently issued but not yet effective accounting standards, if currently adopted, would have a material effect on the consolidated financial position, statements of operations and cash flows.
RESEARCH AND DEVELOPMENT
As of June 30, 2019, the Company has obtained 11 exclusively licensed technologies in neurology, infectious diseases, gastroenterology, oncology, surgical robotics and natural health and is in the process of developing two “in-house” projects in the neurology area. For the six months ended June 30, 2019 and 2018, the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, the Group incurred $2,714,217, $1,342,179, $3,101,432 and $2,560,323, respectively, on research and development expenses. We did not incur research and development expenses for the period January 1, 2017 to February 28, 2017.
OFF-BALANCE SHEET ARRANGEMENTS
As at June 30, 2019, the Company did not have any off-balance sheet debt, nor do we have any transactions, arrangements or relationships with any special purpose entities.
Overview
We are a pharmaceutical company currently in the preclinical stage, dedicated to developing and commercializing a broad range of therapeutic and diagnostic technologies to tackle unmet medical needs. We have obtained exclusive licenses for our technologies. In addition, we are also developing certain proprietary technologies as product candidates. We are pursuing therapeutic and diagnostic projects (including projects seeking to use extracts or derivatives from natural substances to treat diseases) in neurology, infectious diseases, gastroenterology, oncology and other disease areas. We also have projects focused on surgical robotics. (See “Lead Projects and Other Projects under Development – Lead Projects”) Also, we opened a medical clinic, AML Clinic, in June 2018.
Although none of our drug or device candidates has yet been approved for testing in humans, our goal is to develop a broad range of early stage novel therapeutics and diagnostics across a wide range of disease/therapeutic areas. Key components of our strategy for achieving this goal include: (for details of our strategy, See “Our Strategy”)
| ● | Developing therapeutic and diagnostic innovations across a wide range of disease/therapeutic areas; | |
| ● | Selectively expanding our portfolio with potential products that may be able to attain orphan drug designation and/or satisfy current unmet medical needs; | |
| ● | Collaborating with leading academic institutions and CROs; | |
| ● | Expanding our in-house pharmaceutical development center; | |
| ● | Leveraging our management’s expertise, experience and commercial networks; | |
| ● | Strategically developing opportunities in Hong Kong to promote access to the PRC market; and | |
| ● | Obtaining and leveraging government grants to fund project development. |
We have devoted a portion of the proceeds from our IPO, to three therapeutic projects (“Lead Projects”). The drug candidates being advanced as the Lead Projects are ALS-1, ALS-4 and NLS-1, described in further detail below. If the results of the remaining preclinical studies of these drug candidates are positive, we expect to be able to submit by 2020 or 2021 an Investigational New Drug Application (“IND”) for at least one of these candidates to the U.S. Food and Drug Administration (“FDA”) or an equivalent application to the regulatory authorities in one or more other jurisdictions such as the China’s National Medical Products Administration (“NMPA”) and/or the European Medicines Agency (“EMA”). Acceptance of these applications by the relevant regulatory authority would enable the Company to begin testing that drug candidate in humans in that jurisdiction. Our ability to obtain any approval of such applications is entirely dependent upon the results of our preclinical studies, none of which have yet been completed.
68
Our current business consists of “therapeutics” and “non-therapeutics” segments. However, our focus is on the therapeutics segments. Because of the risks, costs and extended development time required for successful drug development, we have determined to pursue projects within our non-therapeutics segments, such as AML Clinic, to provide some interim revenue and medical robots that may be brought to market and generate revenue more quickly.
Therapeutics Segment. In our therapeutics segment (“Aptorum Therapeutics Group”), we are currently seeking to develop various drug molecules (including projects seeking to use extracts or derivatives from natural substances to treat diseases) and certain technologies for the treatment (“therapeutics”) and diagnosis (“diagnostics”) of human disease conditions in neurology, infectious diseases, gastroenterology, oncology and other disease areas. In addition, we are seeking to identify additional prospects which may qualify for potential orphan drug designation (e.g., rare types of cancer) or which address other current unmet medical needs. Aptorum Therapeutics Group is operated through Aptorum’s wholly-owned subsidiary, Aptorum Therapeutics Limited, a Cayman Islands exempted company with limited liability, whose principal place of business is in Hong Kong and its indirect subsidiary companies (who we sometimes refer to herein as project companies), whose principal places of business are also in Hong Kong.
Non-Therapeutics Segment. The non-therapeutics segment (“Aptorum Non-Therapeutics Group”) encompasses two businesses: (i) the development of surgical robotics and medical devices and (ii) AML Clinic. The development of surgical robotics and medical devices business is operated through Signate Life Sciences Limited, a subsidiary of Aptorum Therapeutics Limited. The outpatient clinic is operated through our subsidiary, Aptorum Medical Limited. Effective as of March 2018, we leased office space in Central, Hong Kong as the home to AML Clinic. AML Clinic commenced operations under the name of Talem Medical in June 2018. The estimated general administrative expenses and other operating expenses of the AML Clinic is expected to be no more than USD120,000 per month. The clinic is expected to reach operating profit in 18 months from the clinic reaching its full operating capacity upon (i) the successful recruitment of a minimum of six full time physicians (AML Clinic currently has one full time physician and six part time physicians) and (ii) establishing steady patients flow via brand development. (See “Lead Projects and Other Projects under Development – Other Projects under Development – Aptorum Medical Limited - AML Clinic”)
The Company has already obtained opportunities resulting in our existing licensing agreements from various contractual relationships that we have entered into, including service/consulting agreements with some of the world’s leading specialists and clinicians in our areas of interest, with academic institutions and organizations, and with CROs. We anticipate that these relationships will generate additional licensing opportunities in the future. In addition, we have established and are continuing to expand our in-house research facilities (collectively, the “R&D Center”) to develop some of our drug and device candidates internally and to collaborate with third-party researchers.
Prior to March 2017, the Company had pursued passive healthcare related investments in early stage companies primarily in the United States. However, we have since ceased pursuing further passive investment operations and intend to exit all such portfolio investments over an appropriate timeframe to focus resources on our current business.
Our Strategy
Although we plan to continue the development and improvement of a broad range of novel therapeutics and diagnostics across a wide range of disease/therapeutic areas, over the next 24-36 months we plan to concentrate on development of our Lead Projects, while also allocating some resources to develop SLS-1 and maintaining our AML Clinic.
We believe that execution of this strategy will position the Company to catalyze the development and improvement of a broad range of early-staged novel therapeutics and diagnostics across a wide range of disease/therapeutic areas. Failure to achieve positive results in at least one of the programs for a Lead Project could have a material adverse effect on the Company’s prospects and business.
69
To achieve this goal, we are implementing the following strategies:
| ● | Developing therapeutic and diagnostic innovations across a wide range of disease/therapeutic areas.We are currently developing drug and device candidates in several disease/therapeutic areas. We believe that by diversifying our research efforts, it would increase the likelihood that at least one of our projects will achieve clinical success and therefore add value to the Company. As of date hereof, we have obtained 11 exclusively licensed technologies across the areas of neurology, infectious diseases, gastroenterology, oncology, surgical robotics and natural health. Our initial focus will be on developing our Lead Projects, but intend to continue developing our other current projects and seeking new licensing opportunities where we determine that the market potential justifies the additional commitment of our limited resources. | |
| ● | Selectively expanding our portfolio with potential products that may be able to attain orphan drug designation and/or satisfy current unmet medical needs.We have selected innovations for development which we believe are of superior scientific quality, whilst taking into account the potential market size and demand for same, for example, taking into consideration whether the relevant product can satisfy significant unmet medical needs. In particular, Aptorum Therapeutics Limited has established a Scientific Assessment Committee, which helped us to select our current projects and which we expect will provide input from a scientific perspective towards any future opportunities for acquiring or licensing life science innovations. We intend to continue expanding our line of projects under development, and subject to our financial and other resource limitations, exploring acquisitions or licenses of additional products which may be able to attain orphan drug designations (e.g., rare types of cancer) or satisfy significant unmet medical needs and that show strong preclinical and/or early clinical data to provide promising opportunities for clinical and commercial success. | |
| ● | Collaborating with leading academic institutions and CROs.In building and developing our product portfolio, we believe that accessing external innovation, expertise and technology through collaboration with leading academic institutions and CROs is a vital and cost-efficient strategy. We have established strong relationships with leading academic institutions around the world and expect to continue to strengthen our collaborations by, for example, seeking to provide their affiliated Principal Investigators resources through sponsorship to conduct further research in specialty fields of interest and association with personnel connected to our current project companies, in exchange for obtaining for the Company the first right to negotiate for an exclusive license to any resulting innovations. In addition, we have entered and will continue to actively source arrangements with pharmaceutical companies, in most cases in roles as CROs, to streamline the development of our projects. This may include outsourcing part of the preclinical, clinical studies and clinical supplies manufacturing to externally accredited cGLP, cGMP and cGCP standard CROs or laboratories in order to attain the required studies for submission to the regulatory authorities as part of the clinical development plan. (See “Arrangements with Other Parties”) | |
| ● | Expanding our in-house pharmaceutical development center.We believe collaborations between the R&D Center operated by APD and the scientists engaged in work for our project companies will enhance clinical and commercial potential of the projects. In addition, APD will assist the project companies by engaging external pharmaceutical companies and/or CROs to outsource any part of the preclinical or clinical development work that cannot be performed by the R&D Center in order to obtain the resources necessary for our development process. | |
| ● | Leveraging our management’s expertise, experience and commercial networks.We believe the combination of our management’s expertise and experience, with their academic and commercial networks make us an effective platform for advancing healthcare innovations towards clinical studies and commercialization in key global markets. We have assembled a management team with global experience and an extensive record of accomplishments in medical research, consulting and financing, and identification and acquisition of pharmaceutical and biopharmaceutical drug and device candidates. Our Head of Research and Development also has extensive experiences in drug development. We also employ key management personnel with banking and financial experience, which enhances our capability to establish the most efficient financial structure for the development of our programs. |
70
| ● | Strategically developing opportunities in Hong Kong to provide access to the PRC market.The PRC is the world’s second largest healthcare market (https://seekingalpha.com/article/4038677-opportunities-chinas-healthcare-market) and we plan to market our products there in the future as part of our overall growth strategy. In October 2017, the PRC government announced that the country is planning to accept trial data gathered overseas to speed up drug approvals (https://www.reuters.com/article/us-china-pharmaceuticals/china-to-accept-overseas-trial-data-in-bid-to-speed-up-drug-approvals-idUSKBN1CE080 and http://www.lawinfochina.com/display.aspx?id=26778&lib=law), which is a potential boon for foreign pharmaceutical companies. We believe strategically locating our principal businesses in Hong Kong, as a Special Administrative Region of the PRC, may provide us distinctive advantages in accessing the PRC healthcare market. Two of our key collaborators, The University of Hong Kong (the “HKU”) and the Chinese University of Hong Kong (the “CUHK”) have received clinical drug trial accreditation by the NMPA for their clinical trial units/centers (http://www.crmo.med.cuhk.edu.hk/en-us/nmpaaccreditation.aspx and https://www.ctc.hku.hk/assurance_cfda.php). | |
| ● | Obtaining and leveraging government grants to fund project development.The Hong Kong government pays close attention to the development of the biotechnology sector in Hong Kong and provides support and funding. We intend to aggressively seek government support from Hong Kong for our product development and to facilitate the development of some of our projects. |
Arrangements with Other Parties
As mentioned above, part of our business model includes collaborating with research entities such as academic institutions and CROs, as well as highly regarded experts in their respective fields. We engage these entities and researchers either for purposes of exploring new innovations or advancing preclinical studies of our existing licensed drug candidates. Although the financial cost of these arrangements does not represent a material expense to the Company, the relationships we can access through, specifically, sponsored research arrangements (“SRAs”) with academic institutions and organizations can provide significant value for our business; for example, we may decide whether to continue development of certain early-staged projects and/or out-license a project based on the data and results from research governed by SRAs. However, as of the date hereof, we do not consider the particulars of any of our SRAs to be material to the success of our current business plans.
Our drug discovery programs are based upon licenses from universities and are mainly conducted in universities via SRAs. As for the development of our drug candidates, our R&D Center conducts part of the CMC work. However, since our current facilities are not cGMP, cGLP or cGCP qualified, we will have to rely on CROs to conduct that type of work, if and when our drug candidates reach the level of development that requires such qualification.
Lead Projects and Other Projects under Development
We are actively operating and managing the development of our drug and device candidates through various subsidiaries. Each candidate is being researched in a subsidiary with a medical/scientific area of focus related to the drug and device candidate in development. We refer to these as our “Project Companies” and their products or areas of focus as either our Lead Projects (i.e., ALS-1, ALS-4 and NLS-1) or Other Projects under Development (as defined below). The selection of a drug and device candidate is based on our estimate of the market potential for that candidate, the scientific expertise required to develop it, and our overall corporate strategy, including our ability to commit personnel and future investment to that candidate.
71
To pursue a number of our current projects, our Project Companies have entered into standard license agreements with various university and licensing entities customized to the nature of each project. These license agreements largely contain the same terms, as is typically seen in license agreements for an early-stage life science invention; such terms include a worldwide license with licensed field comprising indications in the intended treatment areas, having upfront payments, certain royalty rates, sublicensing royalties, as well as provisions for payments upon occurrence of development and/or regulatory milestones. Under the license agreements, the Project Company must also adhere to certain diligence obligations and may or may not be required to obtain prior consent from the licensor to sublicense the invention. The license terms of our Lead Projects are discussed in detail below.
Generally speaking, pharmaceutical development consists of preclinical and clinical phases. Our immediate efforts would be on the preclinical phase which can further sub-divided into the following stages:
Target Identification & Selection: The target is the naturally existing cellular or modular structure that appears to have an important role in a particular disease pathway and will be targeted by the drug that will subsequently be developed. Target validation techniques for different disease areas can be very different but typically include from in vitro and in silico methods through to the use of whole animal models.
Lead Discovery: Following “Target Identification & Selection,” compound screening assays are developed as part of the Lead Discovery. ‘Lead’ molecules can mean slightly different things to different researches or companies, but in this prospectus, we refer to Lead Discovery as the process of identifying one or more small molecules with the desired activity against the identified targets. Leads can be identified through one or more approaches, which can depend on the target and what, if any, previous knowledge exists.
Lead Optimization: In this stage of the drug discovery process, the aim is to produce a preclinical drug candidate by maintaining the desired and favorable properties in the lead compounds, while repairing or reducing deficiencies in their structures. For example, to optimize the chemical structures to improve, among others, efficacy, reduce toxicity, improve metabolism, absorption and pharmacokinetic properties.
IND-Enabling Studies: Includes all the essential studies such as GLP toxicology studies, pharmacology and efficacy, pharmacokinetics, in vitro metabolism, CMC studies, and the data of which are used for IND submission.
72
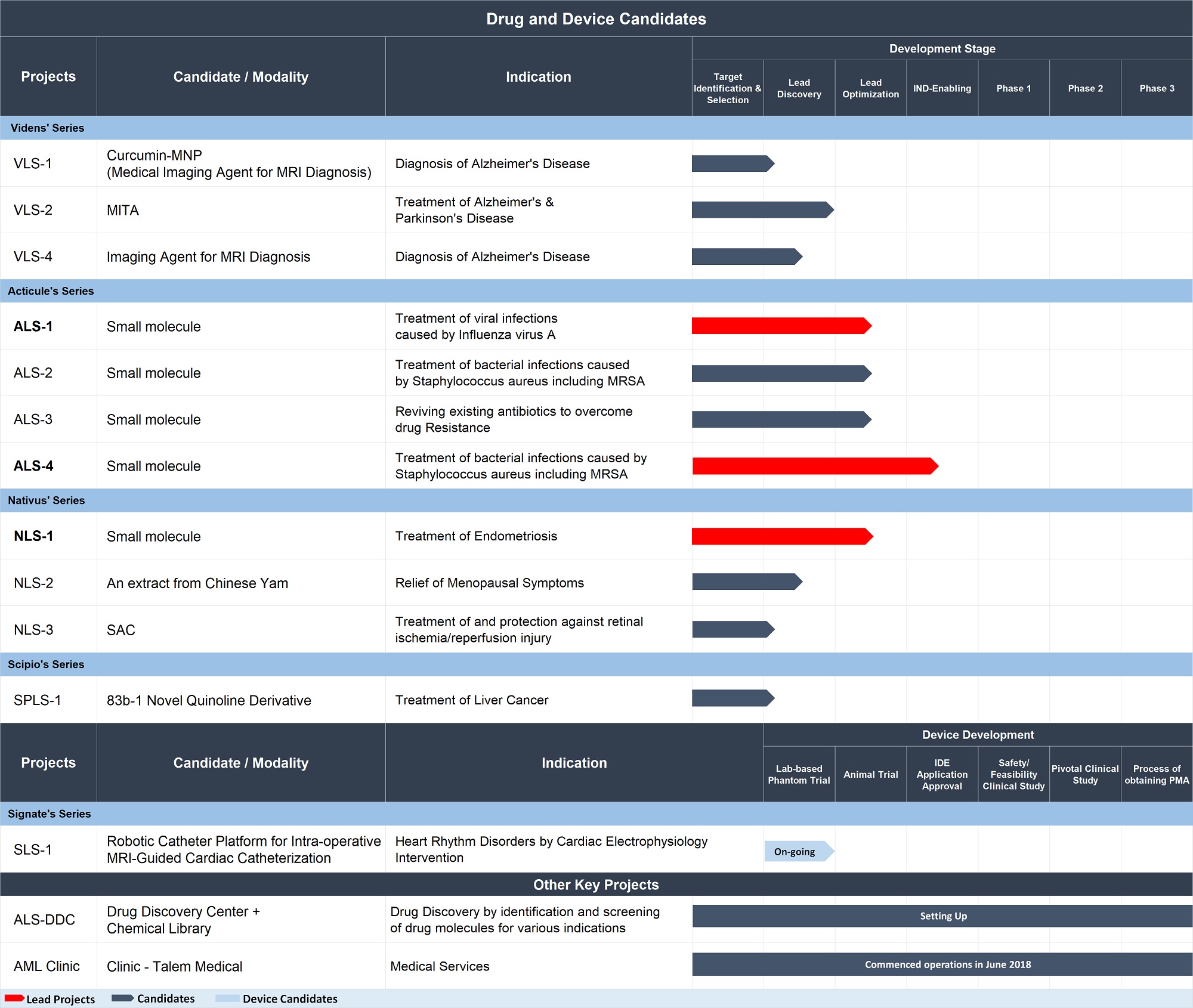
*See “Explanatory Note” and “Incorporation Of Certain Information By Reference”
Another subsidiary, Aptorum Medical Limited (“AML”)1, is our vehicle for developing our business of delivering medical services in the form of AML Clinic.
We anticipate allocating approximately 20% of our resources to develop projects other than our Lead Projects (such other projects being referred to herein as “Other Projects under Development”), with a strong focus on SLS-1 and AML Clinic. As a device candidate, SLS-1 may not need to undergo the same regulatory approval process as a drug candidate and therefore we may be able to bring it to market sooner. AML Clinic is expected to provide us with a modest amount of revenue. Even if SLS-1 achieves commercial sales, of which there can be no assurance, revenue from these products alone will not be sufficient for us to carry out all of our plans, but it will assist with name recognition and supplement our income while we develop our Lead Projects.
| 1 | Clark Cheng, our Chief Medical Officer and an Executive Director, owns 6% of Aptorum Medical Limited as of the date hereof; Mr. Cheng is also an Executive Director of Smart Pharmaceutical Development PTE. Ltd, which is also a limited partner of Smart Pharmaceutical Limited Partner. Dr. Cheng also serves as a director of several other of our subsidiaries. |
73
Lead Projects

ALS-1: Small molecule intended for the treatment of viral infections caused by Influenza virus A
Professor Richard Kao (Inventor of ALS-1, Founder and Principal Investigator of Acticule) was the first to identify NP as an effective drug target (Nature Biotechnology. 28:600-605). We are exploring ALS-1 as a potential treatment for viral infections caused by Influenza virus A (“IVA”).
Two widely prescribed antiviral drug classes for the treatment of influenza are neuraminidase inhibitors (“NI”) and M2 protein inhibitors. Zanamivir is a second-generation neuraminidase inhibitor for the treatment of both Influenza A and B in adults and children (5 years old and above). Oseltamivir is a third-generation neuraminidase inhibitor for the treatment of Influenza A and B in individuals older than 1 year of age. Amantadine and rimantadine are M2 membrane protein inhibitors that block the M2 ion channel activity of Influenza A but have no effect on Influenza B. Given the widespread resistance to M2 inhibitors, amantadine and rimantadine are no longer recommended for the treatment of Influenza A.
It is our hypothesis that Influenza A NP is an essential protein for the proliferation of the influenza virus. ALS-1 targets NP and triggers the aggregation of NP and this prevents the aggregated NP from entering the nucleus. In a paper published by the inventor, Prof. Richard Kao, in Nature Biotechnology (28 (6): 600, 2010), ALS-1 inhibited infection of MDCK cells by the Influenza A/WSN/33, H3N2 (clinical isolate) and Vietnam/1194/04 (H5N1) viruses with an IC50 (IC50 is defined as the concentration of a drug which inhibits half of the maximal response of a biochemical process. In this case, inhibition of the growth of PFU = plaque-forming units is the response) of 0.069 ± 0.003 μM, 0.16 ± 0.01 μM and 0.33 ± 0.04 μM in plaque reduction assay (PRA), respectively (Figure 1A). In this study, oseltamivir (sold under the brand name Tamiflu®) was also included as a control. In this cell culture, ALS-1 outperformed oseltamivir with a lower IC50 (Figure 1A). ALS-1 inhibited viral growth even when added within 6 hours after infection of the MDCK cells with the virus (Figure 1B), indicating that the antiviral activities of ALS-1 arise from post-entry and post-nuclear events, suggesting that multiple processes involving NP may be affected, although only the nuclear import process of NP can be readily observed.
In the treatment-free control group, all mice died 7 days after inoculation. After treating with ALS-1, 50% of the mice receiving two doses of ALS-1 (100 μl of 2.3 mg/ml ALS-1) per day for 7 days survived for more than 21 days. Three mice were sacrificed from each treated and untreated group on the 6th day after infection and their lungs tested for live virus by a plaque reduction assay. About a 10x reduction of viral load in the lungs of the ALS-1-treated mice was observed compared to the untreated control group. The animal study results suggest that ALS-1 has the potential to be developed into a useful anti-influenza therapeutic.
ALS-1 is designed to target a broad range of NP variants, a novel therapeutic target. Compared with the currently marketed antiviral drugs for which the viruses have acquired extensive resistance, ALS-1 acts on a completely different therapeutic target. ALS-1 is currently undergoing Lead Optimization to optimize its drug-like properties.
74
| Figure 1A | Figure 1B | |
 |  |
Figure 1A: ALS-1 is shown to cause a greater reduction in the number of infectious virus particles of human H1N1, H3N2 and H5N1 Influenza viruses. MDCK cells were infected with different strains of virus and antiviral activities of different treatments were determined by plaque reduction assay (PRA). Oseltamivir (curve in red) was included for comparisons of in vitro efficacies. The PRA assay was conducted in triplicate and repeated twice for confirmation. PFU = plaque-forming units, a measure of number of infectious virus particles Nucleozin = ALS-1 (Adapted from Nature Biotechnology (28 (6): 600, 2010).
Figure 1B: Efficacies of ALS-1 added at various time points. The experiments were carried out in triplicate and repeated twice for confirmation. The mean value is shown with s.d.; PFU = plaque-forming units, a measure of number of infectious virus particulates (Adapted from Nature Biotechnology (28 (6): 600,2010)).
Patent License
On October 18, 2017, the Company’s subsidiary, Acticule, entered into an exclusive license agreement with Versitech Limited, the licensing entity of HKU, for the rights to ALS-1. Subsequently on June 7, 2018, the parties entered into a first amendment to the license agreement, and on July 10, 2019, the parties entered into a second amendment to the license agreement.
Under the exclusive license agreement, we were granted an exclusive, royalty-bearing, sublicensable license to develop, make, have made, use, sell, offer for sale and import products that are covered by the licensed patents (as described below). The territory of the license is worldwide and the field of the license is for treatment or prevention of viral infections including influenza.
We paid an upfront fee upon entering into the license agreement. We are required to pay less than 10% of the net sales of the licensed products sold by us or our affiliates as royalties, as well as a low teens percentage of sublicense royalties that we receive from our sublicensees, if any. In addition, we agreed to pay to the licensor aggregate regulatory milestones of up to US$1 million subject to the following achievements: submission of investigational new drug application; completion of phase 1, 2 and 3 clinical trials; and submission of new drug application; grant of regulatory approval. We also agreed to pay to the licensor aggregate sales milestones of up to US$7.8 million subject to the following achievement: first commercial sale; and annual net sales exceeding US$100 million in one jurisdiction.
75
Pursuant to the license agreement, Acticule became the exclusive licensee of 1 U.S. patent, 1 European Patent, 1 PRC patent and 1 German patent. The claimed invention is described as: “Antiviral Compounds and Methods of Making and Using Thereof.”
Acticule has the right to grant sublicenses under the license agreement without prior approval from Versitech Limited and to assign the agreement to any successor to the business related to the license. In the event that Acticule makes an improvement to the licensed technologies, so long as the improvement does not incorporate any licensed patents, Acticule will be the owner of such improvement, subject to a non-exclusive royalty-free license being granted back to Versitech Limited for academic and research purposes only.
The exclusive license agreement shall be in effect until the expiration of all licensed patents. Acticule may terminate the license at any time with 6-month written notice in advance. Either party may terminate the agreement upon a material breach by other party.
ALS-4: Small molecule for the treatment of bacterial infections caused by Staphylococcus aureus including Methicillin-resistant Staphylococcus aureus (“MRSA”)
Just as certain strains of viruses, such as human immunodeficiency virus (“HIV”) and influenza have developed resistance to drugs developed to treat them, certain bacteria such asStaphylococcus aureus,Mycobacterium tuberculosis andPseudomonas aeruginosahave become “superbugs”, having developed resistance to many, if not all, of the existing drugs available to treat them, rendering those treatments ineffective in many instances. MRSA is one such bacterium, a gram-positive bacterium that is genetically different from other strains of Staphylococcus aureus. Staphylococcus aureus and MRSA can cause a variety of problems ranging from skin infections and sepsis to pneumonia and bloodstream infections. It is estimated that about one out of every three people (33%) carry Staphylococcus aureus in their nose, usually without any illness; about two in a hundred (2%) carry MRSA (source: https://www.cdc.gov/mrsa/tracking/index.html). Both adults and children may carry MRSA.
Most MRSA infections occur in people who have been in hospital or other health care settings, such as nursing homes and dialysis centers (source: https://www.mayoclinic.org/diseases-conditions/mrsa/symptoms-causes/syc-20375336), which is known as Healthcare-Associated MRSA (“HA-MRSA”). HA-MRSA infections are typically associated with invasive procedures or devices, such as surgeries, intravenous tubing or artificial joints. Another type of MRSA infection, known as Community-Associated MRSA (“CA-MRSA”), has occurred in wider community among healthy people. It often begins as a painful skin boil and spreads by skin-to-skin contact. About 85% of serious, invasive MRSA infections are healthcare associated infections (https://www.cdc.gov/media/pressrel/2007/r071016.htm). The incidence of CA-MRSA varies according to population and geographic location. In the U.S., more than 94,000 people develop serious MRSA infection and about 19,000 patients die as a result each year (https://www.cdc.gov/media/pressrel/2007/r071016.htm). According to the US Centers for Disease Control and Prevention (“CDC”), Staphylococcus aureus, including MRSA, caused about 11% of healthcare-associated infections in 2011 (source: http://www.healthcommunities.com/mrsa-infection/incidence.shtml). Each year in the U.S., around one out of every twenty-five hospitalized patients contracts at least one infection in the hospital (N Engl J Med. 2014, 27;370(13):1198-208). In the U.S., there were over 80,000 invasive MRSA infections and 11,285 related deaths in 2011 (source: https://edition.cnn.com/2013/06/28/us/mrsa-fast-facts/index.html). Indeed, severe MRSA infections most commonly occur during or soon after inpatient medical care. More than 290,000 hospitalized patients are infected with Staphylococcus aureus and of these staphylococcal infections, approximately 126,000 are related to MRSA (source: http://www.healthcommunities.com/mrsa-infection/incidence.shtml).
ALS-4 is a small drug molecule which appears to target the products produced by bacterial genes that facilitate the successful colonization and survival of the bacterium in the body or that cause damage to the body’s systems. These products of bacterial genes are referred to as “virulence expression.” Targeting bacterial virulence is an alternative approach to antimicrobial therapy that offers promising opportunities to overcome the emergence and increasing prevalence of antibiotic-resistant bacteria.
76
Professor Richard Kao from The University of Hong Kong (who is also the Founder and Principal Investigator of Acticule and Inventor of ALS-2, ALS-3 and ALS-4) initiated a high throughput approach for screening compounds which are active against virulence expression, which resulted in the discovery of ALS-2, ALS-3 and ALS-4.
ALS-4 targets an enzyme essential for Staphylococcus aureus (including MRSA) survival in vivo. This enzyme is involved in the production of Staphyloxanthin, a carotenoid pigment produced by Staphylococcus aureus including MRSA, and is responsible for the characteristic golden color. This pigment has proven to be an important factor in promoting bacterial invasion as well as rendering the bacteria resistant to attack from reactive oxygen species (ROS) and neutrophils. In other words, pigmented bacteria have increased resistance to the host’s immune defenses. ALS-4 may have particular value if it can be shown to be an effective therapy in situations where a Staphylococcus aureus infection is resistant to available antibiotics (i.e., where the pathogen is MRSA).
In a recent study by the inventor, Prof. Richard Kao, ALS-4 demonstrates potent activity against Staphylococcus aureus pigment formation in vitro, as indicated in Figure 2, with an IC50 (IC50 is defined as the concentration of a drug which inhibits half of the maximal response of a biochemical process. In this case, inhibition of the formation of the golden pigment is the response) equal to 20nM.
Figure 2

Figure 2: In vitro pigment inhibition by compound ALS-4.
(A) Inhibition of wild-type (WT) Staphylococcus aureus pigmentation in the presence of increasing concentrations of ALS-4.
(B) Pigment inhibition by ALS-4; the IC50for pigment formation is roughly 300 nM.
All data represent mean values ±SD.
77
NP16 = ALS-4
This assay was conducted in triplicate and repeated twice for confirmation
(Adapted from mBio (8(5): e01224, 2017))
By employing a systemic Staphylococcus aureus mouse infection model, the treatment (1mM of ALS-4 twice daily) and control groups (vehicle) were compared. In both acute treatment and delayed treatment groups, the bacterial counts in the kidneys of mice treated with compound ALS-4 were significantly lower than those of the no treatment group.
Figure 3
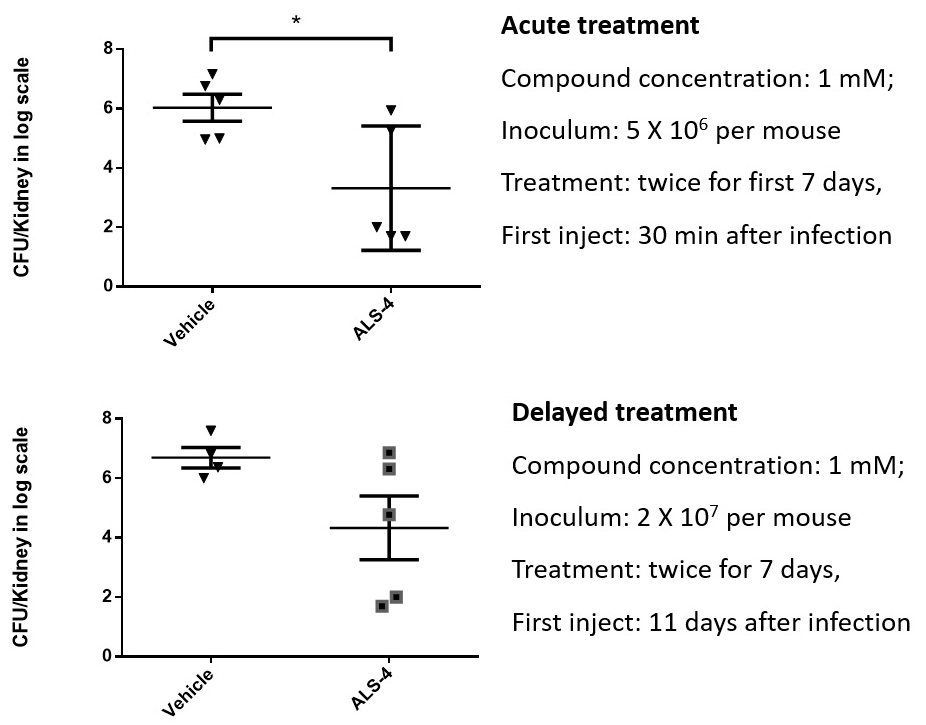
Figure 3: ALS-4 is observed to reduce bacterial load in mice
CFU = Colony Forming Unit, a unit used to estimate the number of viable bacteria in a sample
78
ALS-4 is currently undergoing IND enabling stage to optimize its drug-like properties.
Patent License
On October 18, 2017, the Company’s subsidiary, Acticule, entered into an exclusive license agreement with Versitech Limited, the licensing entity of HKU, for ALS-4. Subsequently on June 7, 2018, the parties entered into a first amendment to the exclusive license agreement.
On January 11, 2019, Acticule and Versitech Limited entered into a second license agreement for ALS-4, where Acticule exclusively licensed the intellectual property rights on certain HKU-owned improvements to the original licensed invention.
Under the exclusive license agreements, we were granted an exclusive, royalty-bearing, sublicensable licenses to develop, make, have made, use, sell, offer for sale and import products that are covered by the licensed patents (as described below). The territory of the licenses is worldwide and the field of the licenses is for treatment or prevention of bacterial infections caused by Staphylococcus aureus including MRSA and bacterial virulence.
We paid an upfront fee upon entering into the license agreements. We are required to pay less than 10% of the net sales of the licensed products sold by us or our affiliates as royalties, as well as a low teens percentage of sublicense royalties that we receive from our sublicensees, if any. In addition, we agreed to pay to the licensor aggregate regulatory milestones of up to US$1 million subject to the following achievements: submission of investigational new drug application; completion of phase 1, 2 and 3 clinical trials; and submission of new drug application; grant of regulatory approval. We also agreed to pay to the licensor aggregate sales milestones of up to US$7.8 million subject to the following achievement: first commercial sale; and annual net sales exceeding US$100 million in one jurisdiction.
Pursuant to the license agreements, Acticule became the exclusive licensee of 2 pending U.S. non-provisional patent applications and 2 PCT applications. With respect to the PCT applications, we plan to enter national phase in member states of the EPO, in PRC and other jurisdictions before the deadline on January 23, 2021. The claimed inventions are described as: “Compounds Affecting Pigment Production and Methods for Treatment of Bacterial Diseases.”
Acticule has the right to grant sublicenses to third parties under the license agreements without prior approval from Versitech Limited and to assign the agreements to any successor to the business related to the licenses. In the event that Acticule makes an improvement to the licensed technologies, so long as the improvement does not incorporate any licensed patents, Acticule will be the owner to such improvement, subject to a non-exclusive royalty-free license being granted back to Versitech Limited for academic and research purposes only.
The exclusive license agreements shall be in effect until the expiration of all licensed patents. Acticule may terminate the licenses at any time with 6-month written notice in advance. Either party may terminate the agreements upon a material breach by other party.
NLS-1: A Derivative of Epigallocatechin-3-Gallate (“Pro-EGCG”) for the treatment of Endometriosis
NLS-1, a drug molecule derived from natural products (green tea), is currently under development for the treatment of endometriosis, a disease in which the tissue that normally lines the uterus (endometrium) grows outside the uterus. It can grow on the ovaries, fallopian tubes, bowels, or bladder. Rarely, it grows in other parts of the body. Many studies have assessed the applications of EGCG, a naturally occurring molecule extracted from green tea, for the treatment of endometriosisin vitro and in animal models (Hum Reprod. 2014 29(8):1677; Hum Reprod. 2013 28(1):178; Fertil Steril. 2011 96(4):1021). For example, in a mouse model, Ricci et al (Hum Reprod. 2013 28(1):178) demonstrated that EGCG brought a statistically significant reduction in the mean number and the volume of established lesions compared with the control group without treatment. The treatment diminished cell proliferation in a statistically significant manner, reduced vascular density and increased apoptosis within the lesions. EGCG induced reduction in human EEC proliferation and increased apoptosis in primary cultures. Matsuzaki and Darcha (Hum Reprod. 2014 29(8):1677) also showed that EGCG prevented the progression of fibrosis in endometriosis in an animal model.
79
However, the attractiveness of epigallocatechin-3-gallate as a drug candidate has been diminished by its chemical and metabolic instability (Hum Reprod. 2014 29(8):1677; Angiogenesis. 2013 16(1):59). The Company’s drug candidate, NLS-1 or EGCG octaacetate, is supposed to overcome these challenges. NLS-1 is an EGCG derivative synthesized by acetylation of the reactive hydroxyl groups, which appears to prevent generation of reactive phenoxide anions and radicals for dimerization and metabolism, thereby overcoming the chemical and metabolic instability of EGCG.
Despite different hypotheses proposed for the pathogenesis of endometriosis, it is widely accepted that endometriosis is an angiogenesis-dependent disorder, and that angiogenesis plays an essential role in the growth and survival of endometriotic lesions. Endometriotic lesions require new vessel formation to deliver oxygen and nutrients that are essential to the development and progression of endometriosis. Dense vascularization is a typical pathological feature of endometriosis. Numerous peritoneal blood vessels can be observed around the endometriotic lesions during laparoscopy, and ectopic endometrium is highly vascularized under histological examination. Researchers have confirmed in animal models that angiogenesis occurs in endometriosis, by demonstrating the development of adjacent blood vessels from the surrounding vasculature into the endometriotic implants. Anti-angiogenesis therapy offers a potential novel treatment of endometriosis.
In a paper published by the inventors in Angiogenesis (16:59, 2013), NLS-1 brought a statistically significantly reduction in the lesion size and weight compared with EGCG and the control without any treatment in an experimental endometriosis mouse model (Student t-test, p <0.05) (Figure 4A & B). In addition, the inhibition by NLS-1 in all of the angiogenesis parameters was statistically significantly greater than that by EGCG (Student t-test, p < 0.05) (Figure 5A & B). In addition, NLS-1 significantly (Student t-test, p < 0.05) reduces the lesion size in both prevention and treatment group compared with both saline and EGCG groups (Figure 6). Moreover, NLS-1 also had better bioavailability and greater antioxidation and anti-angiogenesis capacities compared with EGCG.
In addition, regarding a safety study in mice, no signs of stress to NLS-1 administration were observed during the treatment period. No significant weight change was observed over the course of the experiment. Histological examination revealed no obvious reproductive effects on ovarian follicles and endometrial glands under NLS-1 treatments (Figure 7). Also, vascularization of the ovaries and the uterus was not affected in the NLS-1 treatment group.
Figure 4

Figure 4A & B
NLS-1 (Pro-EGCG) limits the development of experimental endometriosis in mice. Upper panels show the endometrial implants developed in the right ventral abdominal wall under laparotomy. Arrows indicate the greatest length and perpendicular width of the lesions for lesion size calculation. Lower panels show the sandwich structures of outer skin and subcutaneous layers (s), middle endometriotic lesions with endometrial glands (g) and endometrial cyst-like structures (c), and inner abdominal muscle and peritoneum (m). Scale bars: 0.5 mm. b Bar charts of the lesion size and weight in different groups and representative lesion pictures are shown. Mean ± SEM, student’s t test, *P < 0.05 compared with saline group; P < 0.05 compared with EGCG group.
The sample size was 4 (N=4) for each group.
(Adapted from Angiogenesis (16:59, 2013))
80
Figure 5
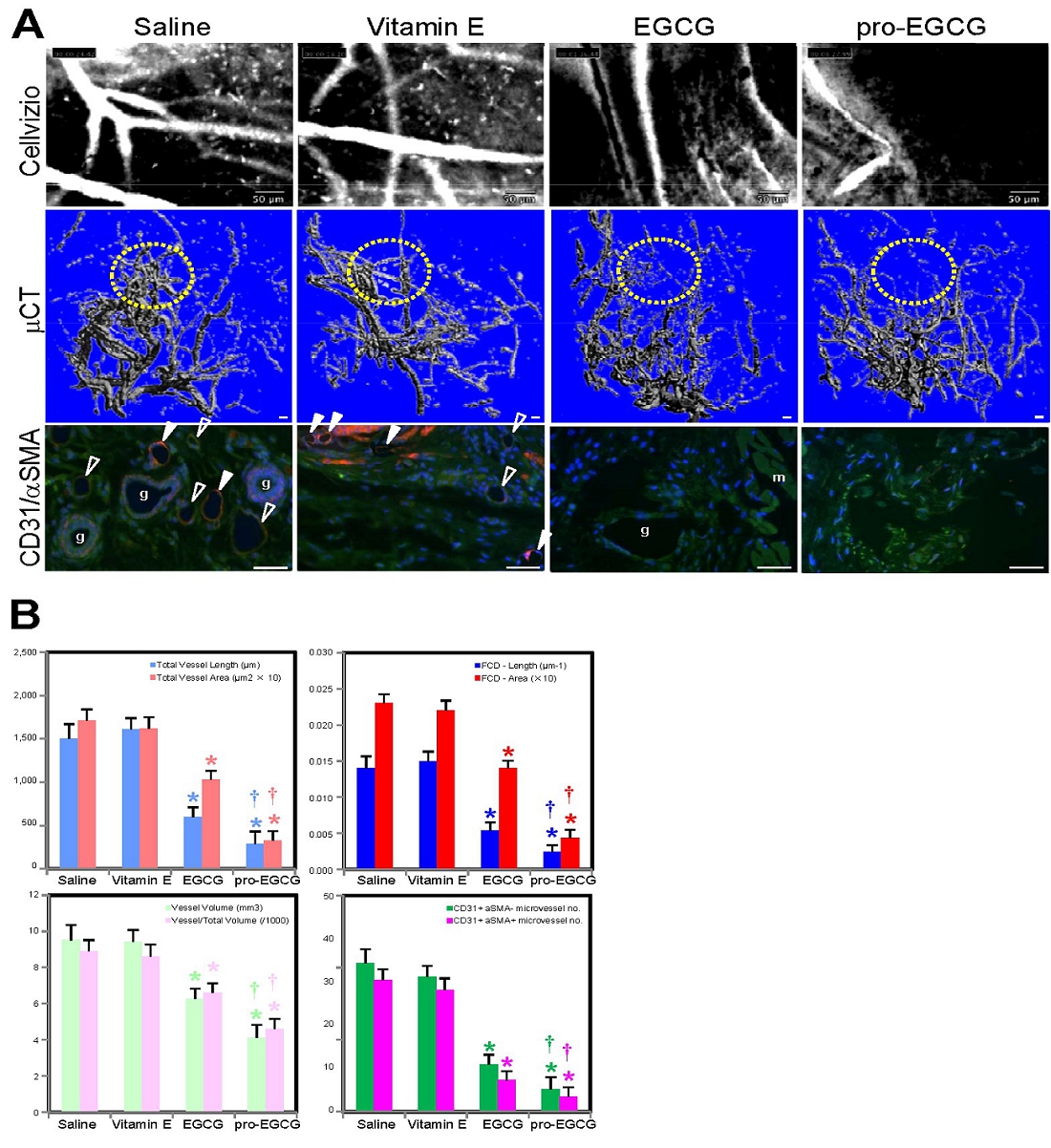
81
Figure 5A & B
NLS-1 inhibits the angiogenesis of experimental endometriosis in mice. Upper panels: Microvessels in the endometriotic implants were perfused with FITC-Dextran and captured by Cellvizio (white colour) (N=8). Middle panels: Microvessel architectures surrounding the lesions and within the lesions were perfused with microfil contrast medium and captured by lCT (yellow dots) (N=4). Lower panels: Microvessels in the endometriotic lesions were determined by specific antimouse antibodies CD31 for endothelial cells in red, aSMA for smooth muscles in green, and DAPI for nuclei in blue (N=4). New microvessels are CD31-positively and aSMA-negatively stained (closed arrows), old microvessels are CD31-positively and aSMA-positively stained (opened arrows). g: endometrial glands; c: endometrial cyst-like structures; m: abdominal muscle. Representative images in different groups are shown. Scale bars: 10 lm. b Bar charts of the lesion microvessel parameters in different groups are presented. Mean ± - SEM, student’s t test, *P < 0.05 compared with saline group; P < 0.05 compared with EGCG group. (Adapted from Angiogenesis (16:59, 2013)). In addition, NLS-1 significantly (p < 0.05) reduces the lesion size in both prevention and treatment group compared with both saline and EGCG groups.
Figure 6
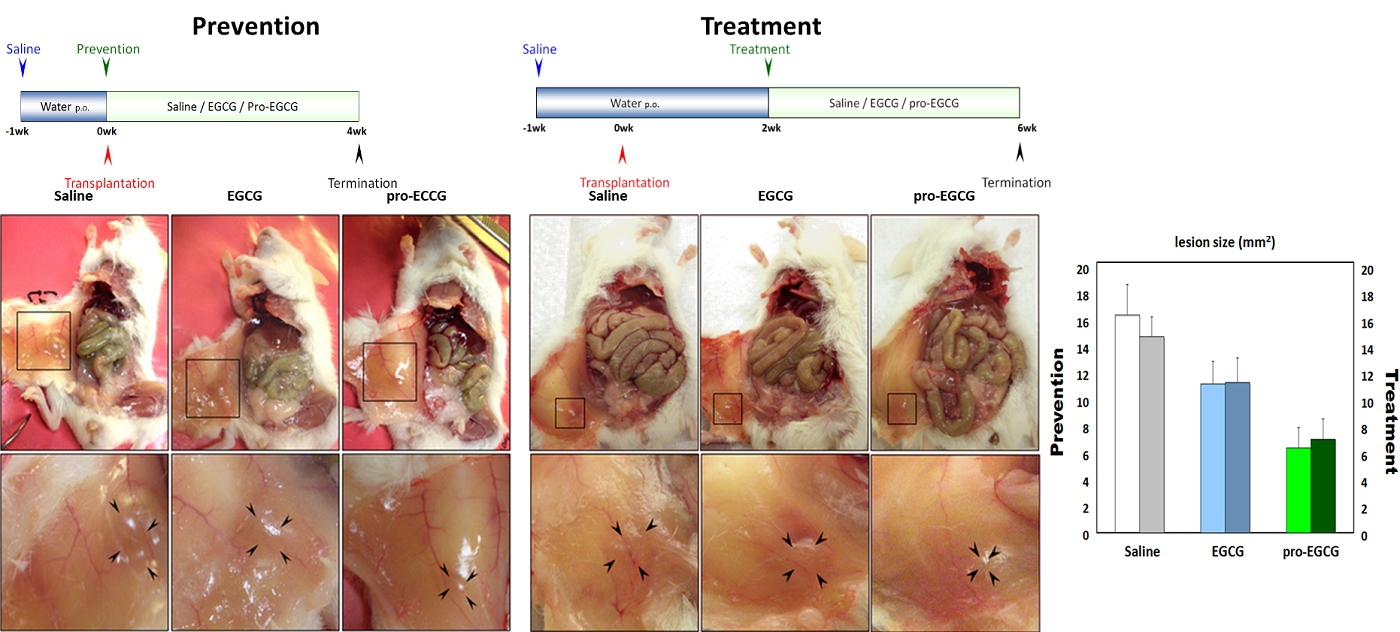
Figure 6: NLS-1 reduces the lesion size in both prevention and treatment groups
82
Figure 7
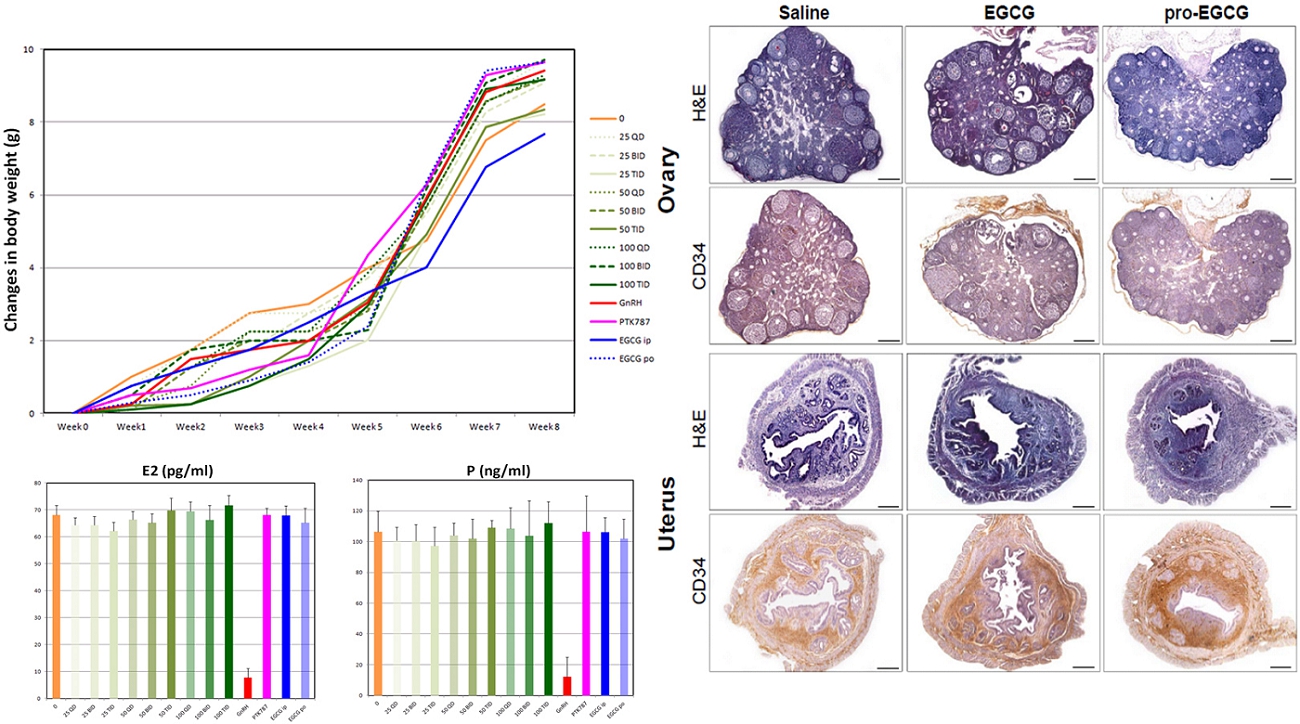
Figure 7
NLS-1 does not cause any weight loss in mice (Upper figure in the left)
NLS-1 does not reduce any estrogen and progesterone level in mice (Lower figures in the left) NLS-1 preserves normal ovarian follicles and endometrial glands. Ovarian follicles and endometrial glands were determined by H&E staining and microvessels in ovarian and endometrial stroma were determined by anti-mouse CD34 immunostaining in ovaries (upper panels in the right) and uterus (lower panels in the right). Representative images in different groups are shown. Scale bars: 0.5 mm.
N=8 was conducted for each group.
(Adapted from Angiogenesis (16:59, 2013)).
As a follow-up study in an animal model of endometriosis, orally administered NLS-1 reduced the lesion size significantly better than oral EGCG (p<0.05-0.001 at week 3- 8, ANOVA) and other hormone-based therapy such as intramuscular GnRH analog (p<0.05 at week 4-8, ANOVA) and other synthetic anti-angiogenesis agents such as intraperitoneal PTK787 (p<0.05-0.01 at week 4-8, ANOVA), as reflected in Figure 8.
83
Figure 8

Figure 8
Comparison of the efficacy of different treatment in an experimental endometriosis model
The current approved treatment for endometriosis is hormonal therapy, which can cause severe undesirable side effects. At present, there are only a few non-hormonal therapeutics with different mechanisms than NLS-1 that are under preclinical or clinical development, such as:
| 1) | BAY 1128688, which is a non-hormonal approach developed by Bayer HealthCare for endometriosis and which entered Phase 2 study in Spain in 2017 (https://adisinsight.springer.com/drugs/800041929); and, |
| 2) | Small molecules co-developed by Bayer and Evotec that have entered Phase 1 studies (Source:https://www.businesswire.com/news/home/20180417006820/en/Evotec-Bayer-Advance-Endometriosis-Programme-Phase-Clinical). |
NLS-1 is under active development for the treatment of endometriosis. It is currently at the Lead Optimization stage to optimize its drug-like properties.
Patent License
On July 3, 2017, the Company’s subsidiary, Aptorum Therapeutics Limited, entered into an exclusive license agreement with PolyU Technology and Consultancy Limited, The Royal Institution for the Advancement of Learning/McGill University, Wayne State University, H. Lee Moffitt Cancer Center and Research Institute Inc. and CUHK (all representing the licensors) for NLS-1.
We paid an upfront fee upon entering into the license agreement. We are required to pay less than 10% of the net sales of the licensed products sold by us or our affiliates as royalties, as well as a percentage of sublicense royalties that do not exceed 30% from what we receive from our sublicensees, if any. In addition, we agreed to pay the licensor aggregate regulatory and development milestones of up to HK$41.9 million (approximately US$5.37 million) for the first drug product subject to the following achievements: submission of investigational new drug application; commencement of phase 1, 2 and 3 clinical trials; submission of new drug application; and grant of first, second and third regulatory approval among the FDA, EMA and NMPA. We also agreed to pay the licensor aggregate sales milestones of up to HK$80 million (approximately US$10.26 million) subject to the following achievements: first commercial sale; and annual net sales exceeding US$100 million in one jurisdiction.
84
Further, for each of the second and third drug products, we agreed to pay aggregate regulatory development milestones of up to HK$9 million (approximately US$1.15 million) and aggregate sales milestone of up to HK$40 million (approximately US$5.13 million) subject to achievement of similar milestones for the first drug product. We have also agreed to pay certain one-time payments for non-drug product upon the commercialization and market launch of such non-drug product. In addition, following the filing of the IND, the Company has to pay an immaterial annual fee to the licensors.
Pursuant to the license agreement, Aptorum Therapeutics Limited became the exclusive licensee of 6 U.S. patents, 1 European Patent, 1 PRC patent, 1 Indian patent and 1 Japanese patent, as well as 1 pending US patent application, 1 pending PRC patent application and 1 pending Hong Kong patent application. Two technologies are claimed in the patents: “Epigallocatechin Gallate Derivatives for Inhibiting Proteasome,” which is jointly owned by PolyU Technology and Consultancy Limited, The Royal Institution for the Advancement of Learning/McGill University, Wayne State University and H. Lee Moffitt Cancer Center and Research Institute Inc. and “Pro-EGCG for Use in the Treatment of Endometriosis,” which is jointly owned by PolyU Technology and Consultancy Limited and CUHK. The licensors have nominated PolyU Technology and Consultancy Limited to represent them and take the lead in negotiating and managing the license.
Aptorum Therapeutics Limited has the right to grant sublicenses under the license agreement with prior consent from the licensors, and such approval shall not be unreasonably withheld. In the event that Aptorum Therapeutics Limited develops any improvements or new development, such licensee inventions are to be jointly owned by the licensors and Aptorum Therapeutics Limited, so that both owners will have the right to use any such inventions for any purpose. In such a case, the Company expects to negotiate a separate agreement with the licensors governing the terms on which the licensors may use such inventions.
In addition, Aptorum Therapeutics Limited also committed to providing HK$3 million (US$384,615) of research funding before July 3, 2020 to sponsor research carried out by the three principal individual inventors upon their request with respect to further R&D on the licensed technologies. The research funding shall be in the form of matching funds provided by the Innovation Technology Fund (“ITF”). The ITF is administered by the Innovation and Technology Commission of the Government of Hong Kong and encompasses a scheme where the Hong Kong government offers matching grant for joint researches to foster collaboration between private companies and public research institutions. If an ITF application is approved, the Hong Kong government will provide a grant that matches the contribution by the private company in the research projects. Since the ITF funding is merit-based and there is no guarantee that an ITF application will be granted, Aptorum Therapeutics’ obligation to contribute to the research fund under the agreement will be contingent on the successful application of ITF scheme granting HK$3 million fund that matches our proposed contribution. In the event that an ITF application related to NLS-1 is not successful, the parties have agreed to negotiate for and agree to enter into new funding terms to support the ongoing research. As of today, the inventors have not filed such ITF application.
During the term of the license agreement and for two years thereafter, Aptorum Therapeutics Limited undertakes not to develop or commercialize any product that directly competes with any marketed product that is covered by the licensed technology.
The exclusive license agreement shall be in effect until the later of (1) the expiry of the term of the last to expire licensed patent set forth in the agreement, (2) final disposition of the last of the pending patent application set forth in the agreement, and (3) ten years following the first commercial sale of the product. Either party may terminate the agreement upon a material breach by or insolvency of the other party. Further, the Licensors may terminate the agreement if the licensee commits any act or omission that could tarnish the reputation of any licensors.
85
Statistical Significance
The term statistical significance is to define the probability that a measured difference between two groups (e.g. two treatment groups, treatment versus control groups) is the result of a real difference in the tested variations and not the result of chance. It means that the result of a test does not appear randomly or by chance, but because of a specific change that is tested, so it can be attributed to a specific cause.
The confidence level indicates to what percentage the test results will not commit a type 1 error, the false positive. A false positive occurs when a change in the result is due to randomness (or other noise) and not the change in variations. At a 95% confidence level (p = 0.05), there is a 5% chance that the test results are due to a type 1 error. 95% has become the standard and usually be the minimum confidence level for the tests. To make the test more stringent, a 99% confidence level (p = 0.01) is also commonly employed, which means that there is a 1% chance that the test results are due to a type 1 error.
In other words, a p value represents the confidence level. For example, if the p-value for a test is < 0.05, it means that there is less than 5% chance the difference between two groups is due to random error or by chance. If the p-value is < 0.01, it means that there is less than 1% chance the difference between two groups is due to random error or by chance.
We employed statistical testing to compare different treatment groups in animal studies simply for proof of concept and to aid internal decision making for further development. We do not intend to use this standard for any regulatory submission. The US FDA or other regulatory agencies may not necessarily employ the same statistical standard to assess the efficacy in clinical trials, the results of which would be submitted for regulatory approval. Although a p-value of 0.05 has become the standard, the US FDA or other regulatory agencies may also individualize their efficacy standard for different clinical programs based on the indications, the purpose of a clinical trial, among others.
FDA Application Status
As of the date hereof, we have not submitted any applications for investigational new drugs (“IND”) to the US Food and Drug Administration (“FDA”). By 2020 or 2021, we expect to be in a position to submit at least one application for one of our drug candidates to commence trials in humans (INDs to the FDA or an equivalent application to the regulatory authorities in another jurisdiction such as the China’s National Medical Products Administration (the “NMPA”) or the European Medicines Agency (“EMA”)). However, there can be no assurance we will be able to make any such application by such time. Should we be delayed in making such filing or should such filing not be approved, our business will be adversely affected.
Other Projects under Development
The following provides additional detail regarding Other Projects under Development:
VLS-1: Curcumin-conjugated superparamagnetic iron oxide nanoparticles (“Curcumin-MNP”) for MRI (“magnetic resonance imaging”) imaging of amyloid beta plaques in Alzheimer’s disease (“AD”)
VLS-1 is an MRI contrast agent, which the Company believes may enable superior imaging for identifying amyloid beta plaques in Alzheimer’s disease. VLS-1 differs from other existing contrast agents for amyloid imaging, such as Amyvid (Eli Lilly), Vizamyl (GE Healthcare) and Neuraceq (Piramal Healthcare), in the following respects: 1) utilization of a natural compound, curcumin, with a known high amyloid beta binding affinity and proven safety; 2) a nanoparticle-based system to enhance delivery efficiency to the brain; and 3) the combination of curcumin with iron oxide, known to be an effective MRI contrast agent. VLS-1 is currently at the Lead Discovery stage.
86
VLS-2: mTOR-independent transcription factor EB activator (“MITA”) as autophagy activator for treatment of neurodegenerative diseases
Autophagy is an endogenous cellular mechanism for clearing multiple pathological protein aggregates including tau, the presence of which is believed to account for neurodegeneration in AD and other neurodegenerative diseases. mTOR is part of a biological pathway that is a central regulator of mammalian metabolism and physiology. Inhibition of mTOR activity is associated with various side effects, such as immunosuppression. Many other molecules that activate autophagy also inhibit mTOR activity. VLS-2 is a small drug molecule that appears to activate autophagy without inhibiting mTOR function. VLS-2 is currently at the Lead Discovery stage.
VLS-4: Other contrast agents for MRI diagnostics
In addition to VLS-1, the Company is actively developing a new class of MRI contrast agents for diagnosis of neurodegenerative diseases. The design of these agents takes into consideration the physicochemical properties that need to be optimized for best imaging performance, and the novel agents are currently undergoing rigorous evaluation. VLS-4 is currently at the Lead Discovery stage.
ALS-2: Small molecule for the treatment of bacterial infections caused by Staphylococcus aureus including MRSA
ALS-2 is a next generation small molecule targeting bacterial virulence for the treatment of bacterial infections caused by Staphylococcus aureus including MRSA. In a recent paper published by the inventor, Professor Richard Kao from The University of Hong Kong (also the Founder and Principal Investigator of Acticule), in PNAS (115(310: 8003, 2018), ALS-2 suppresses the expression of multiple virulence factors in Staphylococcus aureus simultaneously. In a lethal infection mouse model, compared with the vehicle group, ALS-2 protected against Staphylococcus aureus for all the mice in the group, with significant differences between the treatment and control groups [P = 0.0057, by log-rank (Mantel-Cox) test].
ALS-2 is currently at the Lead Optimization stage to optimize its drug-like properties.
ALS-3: Small molecule acting synergistically with certain existing antibiotics
ALS-3 is a novel small molecule that is at present under investigation to combine with certain classes of existing antibiotics to overcome drug resistance. We are exploring ALS-3 for the treatment of bacterial infections including MRSA. ALS-3 is currently at the Lead Optimization stage to optimize its drug-like properties.
NLS-2: An extract from Chinese Yam for relief of menopausal symptoms
NLS-2 is an extract isolated from Chinese Yam, Dioscorea opposita Thunb. In development for the treatment of menopausal syndrome, we expect NLS-2 is to be formulated into an oral dosage form or nasal spray for administration. Each therapy cycle is expected to last for 3 months. Menopausal syndrome refers to the symptoms experienced by women during menopause, such as hot flashes, mood disorders, night sweats, depression, nervous tension and insomnia that are related to estrogen deficiency. Our research suggests that NLS-2 stimulates estradiol biosynthesis in rat ovarian granulosa cells; induces estradiol and progesterone secretion in aged rats by upregulating expressions of follicle-stimulating hormone receptor and ovarian aromatase; counteracts the progression of osteoporosis and augments bone mineral density; and improves cognitive functioning by upregulating protein expressions of brain-derived neurotrophic factor and TrkB receptors in the prefrontal cortex. Furthermore, NLS-2 does not appear to stimulate the proliferation of breast cancer and ovarian cancer cells, which suggests that it could be a more efficacious and safer alternative to hormone replacement therapy (Sci Rep. 2015 5:10179). NLS-2 is currently at the Lead Discovery stage. We are also evaluating whether the yam extract is suited for production as dietary supplement.
NLS-3: Extract from garlic for the treatment of and protection against retinal ischemia/reperfusion injury
NLS-3 is based on S-Allyl L-Cysteine (“SAC”), an active organosulfur compound in aged garlic extract which has been reported to possess antioxidative activity. In macrophages and endothelium, it has been shown that SAC possesses potent antioxidative effects involving the scavenging of superoxide radicals, hydroxyl radicals and hydrogen peroxide. Central/branch retinal artery/vein occlusion, glaucoma and, possibly, age related macular degeneration (“AMD”) are conditions associated with retinal ischemia. All these diseases may lead to severe complications or after-effects. Furthermore, after retinal ischemia/reperfusion (“I/R”), large amounts of reactive oxygen species (“ROS”) are produced, which attack nearby cells and cause tissue damage. Therefore, management of retinal ischemia is vital and NLS-3 is being developed for the treatment of and protection against ischemia/reperfusion injury. NLS-3 is currently at the Lead Discovery stage.
87
SPLS-1: A quinoline derivate for liver cancer treatment
SPLS-1, a novel quinoline derivative from Ephedra pachyclada, is at present under active investigation for the treatment of liver cancer. It is currently at the Lead Discovery stage.
SLS-1: Robotic Catheter Platform for Intra-operative MRI-guided Cardiac Catheterization
SLS-1 is our robotic catheter platform for MRI-guided cardiovascular intervention for the treatment of arrhythmia. The platform consists of a magnetic resonance imaging-guided (“MRI-guided”) robotic electrophysiology (“EP”) catheter system, an MR-based positional tracking unit, and a navigation interface. This platform has the potential to offer a major step toward achievement of several clinical goals: (i) enhancing catheter manipulation and lesion ablation, which we believe will decrease the chance of arrhythmia recurrence; (ii) improving the safety of catheter navigation, thereby decreasing the rates of undesired or inadvertent tissue damage; and (iii) enhancing catheter control, thus facilitating shorter learning curves for surgeons and better treatment in more complex patient cases. Should such goals be demonstrated, patient outcomes should be improved, compensating for the cost of using MRI and reducing the overall expenditure.
To date, a product prototype has been developed. Lab-based experiments have been conducted to verify the performance of the robot towards an image-guided pulmonary vein isolation (“PVI”) task. The MR-based tracking unit has also been developed and validated in MRI scanners. The next step is to test the robotic catheterization using a dynamic heart phantom simulated with the pulsatile liquid flow. Preclinical trials can then be conducted with all the components ready. Radiofrequency ablation will be conducted in a live porcine model, prepared with arrhythmia. If all the results are positive, we will approach the US FDA or other regulatory agencies to apply for conducting clinical trials on the equipment.
SLS-1 is currently in Lab-based Phantom Trial and it will follow the regulatory pathway for approval as indicated in the table in Page 73.
Aptorum Medical Limited - AML Clinic
Incorporated in August 2017, Aptorum Medical Limited is a Hong Kong-based company incorporated in Cayman Islands focused on delivering premium healthcare and clinic services. AML can draw on the expertise of many of the region’s most experienced medical practitioners, and is committed to providing a comprehensive cross-functional facility for healthcare professionals to practice evidence-based medicine and offer high-quality medical services to their patients. We also intend that AML will offer to conduct clinical trials of both the Company’s and third parties’ new drug and device products.
Effective as of March 2018, we leased office space in Central, Hong Kong, the commercial and financial heart of Hong Kong, as the home to AML Clinic. We operate the AML Clinic under the name of Talem Medical. AML Clinic commenced operation in June 2018.
The recently renovated medical center is staffed by our group of medical professionals and offers state-of-the-art facilities. Initially we expect to focus our expertise on treatment of chronic diseases resulting from modern sedentary lifestyles and an aging population.
Corporate History and Background
Aptorum was incorporated under the laws of the Cayman Islands on September 13, 2010. Our share capital is $100,000,000.00 divided into 60,000,000 Class A Ordinary Shares with a nominal or par value of $1.00 each and 40,000,000 Class B Ordinary Shares with a nominal or par value of $1.00 each.
88
APTUS CAPITAL LIMITED, which has since been renamed to AENEAS CAPITAL LIMITED and which we refer to herein as Aeneas, was always under the direct ownership of Jurchen and not under the ownership chain of Aptorum Group. However, Aptus Asia Financial Holdings Limited (“AAFH”), which has since been renamed to Aeneas Group Limited, was transferred out of the Aptorum Group on November 10, 2017 to be held directly by Jurchen Investment Corporation and that subsequently, APTUS CAPITAL LIMITED was then transferred to be under AAFH.
On May 4, 2017, Mr. Huen transferred all of the ordinary shares in the Company he owned (in the amount of 22,307,596) to Jurchen, a company incorporated in the British Virgin Islands and wholly-owned by Mr. Huen. On October 13, 2017, as part of the Conversions (as defined below) the ordinary shares held by Jurchen were redesignated as 2,230,760 Class A Ordinary Shares and 20,076,836 Class B Ordinary Shares.
On February 21, 2017 the sole director of the Company and on March 1, 2017, the Company’s board of directors and shareholders respectively, resolved to restructure the Company from an investment fund with management shares and non-voting participating redeemable preference shares to a holding company with operating subsidiaries, respectively (the “Restructuring Plan”).
According to the Restructuring Plan, the 256,571.12 issued participating shares with par value of $0.01 (“Participating Shares”) were redeemed and 4,743,418.88 unissued Participating Shares were cancelled; following such redemption and cancellation, we no longer have any Participating Shares authorized or issued. Additionally, the Company authorized a class of shares consisting of 100,000,000 ordinary shares, par value $1.00 per share (“Ordinary Shares”) and issued 25,657,110 Ordinary Shares to our original investors.
During the period March 1, 2017 through October 13, 2017, an aggregate of 2,207,025 Ordinary Shares were issued at a price of approximately $3.90 per share in a private placement we described as a “Series A” offering. Each investor of the Series A offering, in addition to a subscription agreement, signed a shareholder agreement, which set forth the basic governance terms of the Company, as well as our capital structure. The shareholders agreement was terminated in October 2017.
On October 13, 2017, ordinary resolutions were passed at an extraordinary general meeting of the Company approving (the “Conversions”): (i) converting 72,135,865 of authorized but unissued Ordinary Shares into 54,573,620 authorized but unissued Class A Ordinary Shares, par value of $1.00 per share and 17,562,245 authorized but unissued Class B Ordinary Shares, par value of $1.00 per share (“Class B Ordinary Shares”), respectively; (ii) converting 24,930,839 Ordinary Shares held by three shareholders into an aggregate of 2,493,085 Class A Ordinary Shares and 22,437,754 Class B Ordinary Shares; and (iii) converting 2,933,296 Ordinary Shares held by 24 shareholders into an aggregate 2,933,296 Class A Ordinary Shares. Following these issuances, we had 27 shareholders of record.
89
On October 19, 2017, we changed our name from APTUS Holdings Limited to our current name, Aptorum Group Limited.
On March 23, 2018, Jurchen transferred 446,152 Class A Ordinary Shares and 4,015,367 Class B Ordinary Shares to CGY Investments Limited, a company incorporated in Hong Kong and which we deem Mr. Darren Lui controls and/or of which he has substantial influence on the disposition rights and voting rights of such shares. Following this transfer, Jurchen owns approximately 33% and 72% of our Class A Ordinary Shares and Class B Ordinary Shares, respectively.
On December 17, 2018, the Company consummated its IPO of 761,419 Class A Ordinary Shares; the related registration statement was declared effective by the U.S. Securities and Exchange Commission on December 3, 2018 (the “Effective Date”). The shares were sold at a price of $15.80 per share, generating gross proceeds to the Company of approximately $12,030,420. Immediately following the consummation of the IPO and automatic conversion of the Notes and Bonds, there were an aggregate of 6,537,269 Class A Ordinary Shares issued and outstanding.
Please see the chart illustrating our current corporate structure, under the heading of “Our Structure” in the Prospectus Summary, included earlier in this prospectus.
Intellectual Property
The technologies underlying our various research and development projects are the subject of various patents and patent applications claiming, in certain instances, composition of matter and, in other instances, methods of use. Prosecution, maintenance and enforcement of these patents, as well as those on any future protectable technologies we may acquire, are and will continue to be an important part of our strategy to develop and commercialize novel medicines and medical devices, as described in more detail below. Through entering into license agreements with their owners, we have obtained exclusive rights to these patents, applications and related know-how in the U.S. and certain other countries to develop, manufacture and commercialize the products using or incorporating the protected inventions that are described in this prospectus, and that are expected to contribute significant value to our business. The technologies protected by these patents may also for the basis for the development of other products.
In addition to licensed intellectual property, our in-house science team has been actively developing our own proprietary intellectual property. No patent applications have yet been filed in the Company’s own name for the Lead Projects. We have, however, filed a number of provisional applications to establish earlier filing dates for certain of our other ongoing researches, the specifics of which are currently proprietary and confidential.
The U.S. patent system permits the filing of provisional and non-provisional patent applications (i.e., a regular patent application). A non-provisional patent application is examined by the USPTO, and can mature into a patent once the USPTO determines that the claimed invention meets the standards for patentability. On the other hand, a provisional patent application is not examined for patentability, and automatically expires 12 months after its filing date. As a result, a provisional patent application cannot mature into a patent.
Provisional applications are often used, among other things, to establish an earlier filing date for a subsequent non-provisional patent application. The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained.
The effective filing date of a non-provisional patent application is used by the USPTO to determine what information is prior art when it considers the patentability of a claimed invention. If certain requirements are satisfied, a non-provisional patent application can claim the benefit of the filing date of an earlier filed provisional patent application. As a result, the filing date accorded by the provisional patent application may supersede information that otherwise could preclude the patentability of an invention.
A provisional patent application is not eligible to become an issued patent unless, among other things, we file a non-provisional patent application within 12 months of the filing date of the provisional patent application. If we do not timely file a non-provisional patent application claiming priority to said provisional application, we may lose our priority date with respect to our provisional patent applications. Further, if any (self or by others) publication of the invention is made after such priority date, and if we do not file a non-provisional application claiming priority to said provisional application, our invention may become unpatentable.
Moreover, we cannot predict whether such future patent applications will result in the issuance of patents that effectively protect any of our product candidates or will effectively prevent others from commercializing competitive products.
We do not expect to incur material expenses in the prosecution of the provisional applications or other licensed patent applications. We expect to fund the patent costs from our cash and restricted cash.
90
The value of our drug and device products will depend significantly on our ability to obtain and maintain patent and other proprietary protection for those products, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of other parties.
As of the date hereof, in addition to a number of provisional patent applications discussed above, we are the exclusive licensee of 13 U.S. patents and 6 pending U.S. non-provisional applications, as well as corresponding patents and patent applications internationally. In addition, we are the exclusive licensee of 3 international patent applications under the Patent Cooperation Treaty (the “PCT”) which we have filed and/or plan to file nationally in member states of the EPO, PRC and other jurisdictions before the expiration of the time limits for entry of national stage application. The following table sets forth a list related to our Lead Projects of our patent rights under the exclusive licenses as of the date hereof:
| Project Company / Project name | License Agreement | Licensor (s) | Licensee | Licensed / IP Rights | Patent Expiration Dates | |||||
| Acticule / ALS-1 | Exclusive Patent License Agreement, dated October 18, 2017
First Amendment to Exclusive License Agreement, dated June 7, 2018
| Versitech Limited | Acticule Life Sciences Limited | Exclusive licensee: 1 U.S. patent (US9212177), 1 European Patent (EP2462138B1), 1 PRC patent (CN102596946B), 1 German patent (DE60 2010 019 171.0) | The licensed IP rights include granted patents in the U.S., Switzerland, Germany, Great Britain and PRC.
The U.S. patent will expire in 2031; the European Patent in 2030; the PRC patent in 2030 and the German patent in 2030.
| |||||
| Acticule / ALS-4 | Exclusive Patent License Agreement, dated October 18, 2017
First Amendment to Exclusive License Agreement, dated June 7, 2018
Exclusive Patent License Agreement dated January 11, 2019
| Versitech Limited | Acticule Life Sciences Limited | Exclusive licensee: 2 pending U.S. applications (16/041,836 and US 16/041,838), and 2 pending PCT applications (PCT/IB2018/055458, PCT/IB2018/055459)2 | The licensed IP rights include pending patent applications in the U.S. and under the PCT.
Any patent based on the application, if granted, will have a 20-year patent term from 2018.
|
91
| Nativus / NLS-1 | 1) Exclusive License Agreement, dated July 3, 2017
2) Addendum to License Agreement, dated February 9, 2018
| 1) PolyU Technology and Consultancy Company Limited
2) McGill University
3) Wayne State University
4) H. Lee Moffitt Cancer Center and Research Institute Inc.
5) The Chinese University of Hong Kong
| Aptorum Therapeutics Limited | Exclusive licensee: 6 U.S. patents (US9713603, US7544816, US8193377, US8710248, US9169230, US10188629), 1 European Patent (EP1778663), 1 PRC patent (CN101072764B), 1 Indian patent (IN263365) and 1 Japanese patent (JP5265915), as well as 1 pending U.S. application (US16/259,620), 1 pending PRC application (CN104703596A), and 1 pending Hong Kong application (HK15111955.3) | The licensed IP rights include granted patents in the U.S., Germany, Great Britain, France, Italy, Spain, PRC, India and Japan, as well as pending patent applications in the U.S., PRC and Hong Kong. We cannot predict whether such future patent applications will result in the issuance of patents that effectively protect the candidate.
The U.S., European and PRC patents covering the compound will expire in 2025; the indication U.S. patent will not expire until 2033.
|
2 We intend to file national stage applications in at least PRC and before the EPO prior to the 30-month entry deadline of the PCT application falling on January 2021.
Because of the extensive time required for clinical development and regulatory review of a drug we may develop, it is possible that, before any of our drug and device candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of any such patent. If appropriate, the Company may seek to extend the period during which it has exclusive rights to a product by pursuing patent term extensions and marketing exclusivity periods that are available from the regulatory authorities of certain countries (including the United States) and the EPO.
Even though the Company has certain patent rights, the ability to obtain and maintain protection of biotechnology and pharmaceutical products and processes such as those we intend to develop and commercialize involves complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in such patents has emerged to date in the U.S. The scope of patent protection outside the United States is even more uncertain. Changes in the patent laws or in interpretations of patent laws in the United States and other countries have diminished (and may further diminish) our ability to protect our inventions and enforce our IP rights and, more generally, could affect the value of IP.
While we have already secured rights to a number of issued patents directed to our drug candidates, we cannot predict the breadth of claims that may issue from the pending patent applications and provisional patents that we have licensed or that we have filed. Substantial scientific and commercial research has been conducted for many years in the areas in which we have focused our development efforts, which has resulted in other parties having a number of issued patents, provisional patents and pending patent applications relating to such areas. The patent examiner in any particular jurisdiction may take the view that prior issued patents and prior publications render our patent claims “obvious” and therefore unpatentable or require us to reduce the scope of the claims for which we are seeking patent protection.
In addition, patent applications in the United States and elsewhere generally are not available to the public until at least 18 months from the priority date, and the publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made. Therefore, patent applications relating to drugs and devices similar to our drug and device candidates may have already been filed, which (if they result in issued patents) could restrict or prohibit our ability to commercialize our drug and device candidates.
92
The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other IP rights. Our ability to prevent competition for our drug and device candidates and technologies will depend on our success in obtaining patents containing substantial and enforceable claims for those candidates and enforcing those claims once granted. With respect to any applications which have not yet resulted in issued patents, there can be no assurance that meaningful claims will be obtained. Even issued patents may be challenged or invalidated. If others have prepared and filed patent applications in the United States that also claim technology to which we have filed patent applications or otherwise wish to challenge our patents, we may have to participate in interferences, post-grant reviews, inter parties reviews, derivation or other proceedings in the USPTO and other patent offices to determine issues such as priority of claimed invention or validity of such patent applications as well as our own patent applications and issued patents. Patents may also be circumvented, and our competitors may be able to independently develop and commercialize similar drugs or mimic our technology, business model or strategy without infringing our patents. The rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology.
We may rely, in some limited circumstances, on unpatented trade secrets and know-how to protect aspects of our technology. However, it is challenging to monitor and prevent the disclosure of trade secrets. We seek to protect our proprietary trade secrets and know-how, in part, by entering into confidentiality agreements with consultants, scientific advisors and contractors and invention assignment agreements with our employees. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, giving our competitors knowledge of our trade secrets and know-how, and we may not have adequate remedies for any such breach, in which case our business could be adversely affected. Our trade secrets will not prevent our competitors from independently discovering or developing the same know-how. Although our agreements with our consultants, contractors or collaborators require them to provide us only original work product and prohibit them from incorporating or using IP owned by others in their work for us, if they breach these obligations, disputes may arise as to the rights in any know-how or inventions that arise from their work.
Our commercial success will also depend in part on not infringing the proprietary rights of other parties. Although we seek to review the patent landscape relevant to our technologies on an ongoing basis, we may become aware of a new patent which has been issued to others with claims covering or related to aspects of one of our drug or device candidate. The issuance of such a patent could require us to alter our development plans for that candidate, redesign the candidate, obtain a license from the patent holder or cease development. Our inability to obtain a license to proprietary rights that we may require to develop or commercialize any of our drug and device candidates would have a material adverse impact on us.
(See “Risk Factors – Risks Related to Our IP”)
Trademarks
As of the date hereof, we have a total of 26 trademark registrations covering the trade names and logos of Aptorum and our subsidiaries, including but not limited to “APTORUM THERAPEUTICS,” “VIDENS LIFE SCIENCES,” “ACTICULE LIFE SCIENCES,” “TALEM,” “Talem in Chinese characters,” in jurisdictions such as U.S., Hong Kong, EU, the United Kingdom and China. Furthermore, we are in the process of applying for registration of trademarks in jurisdictions including the U.S., PRC and Hong Kong.
We also own certain unregistered trademark rights or have submitted applications for trademarks for our and our subsidiaries’ trade names and logos.
All other trade names, trademarks and service marks of other companies appearing in this prospectus are the property of their respective holders. Solely for convenience, the trademarks and trade names in this prospectus are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend our use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
93
Important Advisors and Consultants to the Company
In addition to Company management, the following individuals provide the Company with significant advice and insight in their respective fields:
Scientific Advisory Board
We are in the process of reorganizing the Scientific Assessment Committee into a newly formed Scientific Advisory Board. The Scientific Advisory Board shall help the Company sharpen its focus on innovation and technological advancements and address critical scientific challenges in our research and development; it will provide overall advice on the scientific development of the company. As of the date hereof, we only have one member of the Board – Dr. Keith Chan, but anticipate adding more members in the near future.
DR. KEITH CHAN
The appointment of Dr. Chan is through a Consultancy Agreement by and between the Company and GloboAsia LLC, a firm based in Rockville, Maryland (“GloboAsia”), where Dr. Chan serves as Director of International Affairs.
Dr. Chan is currently a Senior Advisor of Cornerstone Intellectual Property Foundation in Taiwan. He is also serving as an adjunct professor at the Graduate Institute of Intellectual Property, College of Commerce, National Chengchi University and adjunct professor and advisor at the Research Center for Drug Discovery, National Yang Ming University in Taipei, Taiwan.
Dr. Chan co-founded GloboMax LLC, a drug development organization, in Hanover, Maryland, in July 1997, and served as a consultant for numerous multi-national pharmaceutical and biotech firms in the U.S, Europe and Asia. GloboMax LLC was acquired by ICON, plc. in August 2003, and Dr. Chan exited the operation. Prior to that, he joined the FDA in 1995 as a Director of Division of Bioequivalence, Office of Generic Drugs, responsible for managing and approval of generic drugs in the States. Dr. Chan had worked for Ciba-Geigy Corporation in Ardsley, New York, for 15 years, and held various senior and management positions. Dr. Chan also has extensive experience in new and generic drug development in executing preclinical animal studies, bioassay development, Phases I to VI Pharmacokinetics, pharmacodynamics, bioavailability, bioequivalence studies, outside contract, regulatory submission, advanced drug delivery systems, and all phases of new drug development. In addition, he has served as Professor/adjunct Professor at the School of Pharmacy, University of Maryland at Baltimore during 1996-2009 and also as Adjunct Professor and National Board of Advisor, College of Pharmacy, University of Minnesota during 1984 - 2006. He has published more than 150 abstracts and research articles in peer-reviewed journals and delivered over 200 professional presentations. He was elected as Fellow of the American Association of Pharmaceutical Scientists (“AAPS”) in 1995 for his scientific accomplishments on drug absorption in humans.
Although much of his career was based in the United States, Dr. Chan has been assisting Asian pharmaceutical and biotech companies for over 14 years. He has organized numerous workshops and conferences in the PRC, Taiwan, Hong Kong, Singapore and Korea. He lectures frequently in Asia and serves as a scientific advisor for many regulatory agencies in Asia. Over the last several years, he has successfully assisted many Asian companies in their technology transfers and licensing deals to and from the U.S., as well as with numerous regulatory submissions to the FDA.
Dr. Chan obtained his Ph.D. degree in Pharmaceutics from the University of Minnesota in January 1980.
94
Senior Clinical Advisor of Aptorum Therapeutics Limited
DR. NISHANT AGRAWAL
Dr. Agrawal, MD, has been serving as the Director of Head and Neck Surgical Oncology, and Professor of Surgery at The University of Chicago School of Medicine since October 2015. He is specialized in management of patients with benign and malignant tumors of the head and neck, and has been practicing Otolaryngology - Head and Neck Surgery, at The University of Chicago Medicine, and Center for Advanced Medicine, both in Chicago since 2009.
Dr. Agrawal’s work has achieved international recognition in the field of head and neck surgical oncology, as well as head and neck cancer genetics. Under his leadership, a team of researchers completed a landmark study that examined the genome of head and neck squamous cell carcinoma. His team has published extensively in the genomic landscapes of major head and neck cancers, including esophageal squamous cell carcinoma, esophageal adenocarcinoma, medullary thyroid cancer, adenoid cystic carcinoma, and mucoepidermoid carcinoma. Dr. Agrawal then applied these findings to identify tumor DNA as a biomarker that improves cancer diagnostics in the saliva and plasma of patients with head and neck squamous cell carcinoma. His researches focus on the application of cancer genetics to design diagnostic approaches to reduce morbidity and mortality from head and neck cancer.
In addition to his clinical and research contributions, Dr. Agrawal is an accomplished educator-teaching medical students, residents, and fellows about the management of patients with head and neck cancer. Prior to joining the University of Chicago, Dr. Agrawal was an associate professor at Johns Hopkins University, where he completed his medical training in 2001, followed by internship and residency.
In addition, Dr. Agrawal was granted fellowships from the Memorial Sloan Kettering Cancer Center, New York (Head and Neck Surgical Oncology), and from Johns Hopkins University School of Medicine, Baltimore (Molecular Genetics). He holds numerous Memberships from accredited American medical associations and institutions.
Specifically, as a Senior Clinical Advisor, Dr. Agrawal supports our efforts to identify, develop and commercialize novel therapies for patients and the healthcare industry. He provides a diverse collection of academic, industrial and regulatory expertise.
Senior Advisors of Aptorum Therapeutics Limited
DR. HENRY CHAN LIK YUEN
Dr. Chan has been serving as the Associate Dean (Global Engagement) at the Faculty of Medicine at CUHK since 2018, and served as the Assistant Dean (External Affairs) at the Faculty of Medicine at CUHK and the Head of Division of Gastroenterology and Hepatology, Department of Medicine and Therapeutics from 2013 to 2018.
Dr. Chan specializes in Gastroenterology and Hepatology. Key areas of his research interest include viral hepatitis, liver fibrosis, liver cancer, anti-viral therapy, and fatty liver disease. Currently, Dr. Chan is also the Director at the Institute of Digestive Disease, the Director at the Centre for Liver Health, and the Director at the Office of Global Engagement. His other honorary appointments include the Chairman for the Strategic and Technical Advisory Committee for Viral Hepatitis at the Western Pacific Regional Office of World Health Organization (“WHO”).
Dr. Chan is a key investigator in over 30 Phase 1 to Phase 4 international trials on antiviral treatment of chronic hepatitis B and C, and is the global lead author in publications on peginterferon-alfa, peginterferon-lambda, telbivudine, tenofovir disoproxil fumarate, and tenofovir alafenamide for the treatment of viral hepatitis B. He has received numerous local, national, and international research awards including the Excellent Research Award by the Food and Health Bureau, Hong Kong in 2010 and 2014, and the National Award for Science and Technology Progress in 2012. He has published over 400 papers in peer-reviewed journals.
Dr. Chan graduated in medicine and completed his doctoral degree at CUHK in August 2001, where he was also appointed to a full professorship in the Department of Medicine and Therapeutics in August 2008. He is currently a Fellow of the Hong Kong College of Physicians, a Fellow of the Royal College of Physicians of Edinburgh and London, a fellow of the American Association for the Study of Liver Diseases, and an Honorary Consultant at the Prince of Wales Hospital. Prior to this, he joined the staff of the Prince of Wales Hospital in July 1993.
95
As an expert in Gastroenterology and Hepatology, Dr. Chan is responsible for providing advice for Claves, our project company specializing in modulating microbiota in the gastrointestinal tract for curing various diseases; and Scipio, our project company specializing in the development of oncology products. At preclinical stage, Dr. Chan is providing advice as to different indications that our product candidates are targeting for. With extensive experience in conducting clinical trials for pharmaceutical companies, Dr. Chan will provide advice on the design of clinical trials for our products candidates at clinical stages.
DR. PHILIP WY CHIU
Dr. Philip Chiu has been a Professor of Department of Surgery, Institute of Digestive Disease since August 2010; the Director of CUHK Jockey Club Minimal Invasive Surgical Skills Center since November 2011; the Director of CUHK Chow Yuk Ho Technology Center for Innovative Medicine and the Assistant Dean (External Affairs), Faculty of Medicine, CUHK, since 2013.
Dr. Chiu graduated from the Faculty of Medicine, CUHK in 1994 with two scholarships. He became a fellow of the Royal College of Surgeons of Edinburgh, Hong Kong Academy of Medicine in 2001 and received his Doctor of Medicine at CUHK in 2009. Dr. Chiu was the first to perform endoscopic submucosal dissection (“ESD”) for the treatment of early GI cancers in Hong Kong. In 2010, he performed the first Per-oral Endoscopic Myotomy (“P.O.E.M.”) in Hong Kong. His research interests include upper gastrointestinal bleeding, esophageal cancer and minimally invasive and robotic esophagectomy, novel endoscopic technology for diagnosis of early GI cancers, ESD and novel endoscopic procedures as well as Natural Orifices Transluminal Endoscopic Surgery (“NOTES”).
Currently Dr. Chiu is an honorary treasurer of the College of Surgeons of Hong Kong. He published over 100 peer-reviewed journals and four book chapters. He has received numerous prestigious awards including State Scientific Technology and Progress Award from the PRC in 2007 and 2nd class award in Technological Advancement, Ministry of Education of the PRC in 2011. Recently his research on P.O.E.M. was awarded the best of DDW 2011 and the first prize of ASGE world cup of endoscopy 2012. He is currently an associate editor of Digestive Endoscopy and co-editor of Endoscopy.
As a surgeon by training, especially in minimally invasive and robotic surgery, Dr. Chiu is providing advice for Signate, our project company specializing in the development of surgical robots. Specifically, Dr. Chiu will provide advice on the design of surgical robots for minimally invasive and robotic surgeries and on the design of clinical trials of Signate at a later stage.
DR. VINCENT MOK CHUNG TONG
Dr. Vincent Mok has been serving as the Assistant Dean (Admissions) at the Faculty of Medicine, CUHK since January 2014, the Head of Division of Neurology of the Department of Medicine and Therapeutics, since December 2016 and has been appointed as the Mok Hing Yiu Professor of Medicine since November 2017. He has also been serving in Master Programme in Stroke and Clinical Neurosciences since July 2007.
Dr. Mok specializes in Neurology, Dementia, and Movement disorders. Key areas of research interest include Vascular Cognitive Impairment, Cerebral Small Vessel Disease, Neuroimaging in Cognitive Impairment, and Parkinson’s Disease. Dr. Mok has also been serving as a Convener of Lui Che Woo Institute of Innovative Medicine - Brain Theme since January 2017, the Director of Therese Pei Fong Chow Research Centre for Prevention of Dementia since May 2016, and Executive Committee Member of Chow Yuk Ho Technology Center for Innovative Medicine since January 2015.
Dr. Mok’s qualifications include: Doctor of Medicine at CUHK (December 2005), Fellow of the Royal College of Physicians (Edinburgh) (July 2007), Fellow of the Hong Kong Academy of Medicine (December 2000), Fellow of the Hong Kong College of Physicians (July 2000), Member of the Royal College of Physicians (November 1996), and Bachelor of Medicine and Bachelor of Surgery (University of Sydney) (April 1993).
96
As an expert in neurology, Dr. Mok is responsible for providing advice for Videns, our project company specializing in the development of neurology products. At preclinical stage, Dr. Mok is providing directions in product development targeting different indications related to neurodegenerative diseases. At a later stage, he will be involved in the clinical design once our candidates enter clinical trials.
External Experts serve as the members of Aptorum Therapeutics Limited’s Scientific Assessment Committee
The members of our Scientific Assessment Committee work together to provide valuable input from a scientific perspective towards target acquisition or in-licensing opportunities of life science innovations. This committee, together with the Senior Clinical Advisors and internal experts, performs an 8-Dimensions assessment process (Pharmacology/Animal efficacy, Medicinal Chemistry, Chemistry, Manufacturing & Control (CMC), Drug Metabolism and Pharmacokinetics (DMPK), Clinical Trials, Technology, Legal/IPs and Business/Financing) that helps to evaluate the market potential and scientific quality of our product portfolio. After collecting all the advice from different advisors and internal experts, the chairs of the Committee will jointly make decisions on licensing.
DR. WILLIAM WU KA KEI
Dr. William Wu has been an Assistant Professor in the Department of Anaesthesia and Intensive Care at CUHK from December 2014 to August 2018, and he was promoted to Associate Professor in August 2018. Prior to this, Dr. Wu was appointed as a Research Assistant Professor in the Institute of Digestive Diseases at CUHK from December 2011 to November 2014. He is an expert in molecular pharmacology and toxicology. He has published extensively in cancer biomarkers and novel therapeutics, with over 220 peer-reviewed journals published on international journals, includingNature Communications,Molecular Biology and Evolution,Autophagy,Cell Research, andCancer Research, and six book chapters with citations over 6,000 and an h-index of 40 (Scopus). His research has been recognized both nationally and internationally. He has earned his Fellowship of the Royal College of Pathologists (FRCPath) from his original works in toxicology, and has been conferred the Young Research Award by CUHK, the First-Class Higher Education Outstanding Scientific Research Output Award (Natural Science) by the Ministry of Education of the PRC, and the Second-Class State Natural Science Award.
Dr. Wu obtained his Ph.D. in Medical Sciences in December 2009 and received post-doctoral training from 2009 to 2011 in the Institute of Digestive Diseases both from CUHK.
As a basic scientist in cancer biology, Dr. Wu is responsible for assessing pharmacology/animal efficacy of our potential drug candidates, especially candidates for Scipio, our project company specializing in the development of oncology products.
DR. JASON Y. K. CHAN
Dr. Jason Chan has been an Assistant Professor in the Department of Otorhinolaryngology, Head & Neck Surgery at CUHK since September 2014. Between June 2013 and September 2014, Dr. Jason Chan satisfied the Hong Kong Medical Council’s requirements to practice medicine in Hong Kong and obtained his American Board certification while continuing his research interests. Dr. Chan graduated from Guy’s, King’s and St Thomas’ School of Medicine in London in July 2005, followed by completion of specialist training in Otolaryngology, Head and Neck surgery at the Johns Hopkins School of Medicine with advanced training in head and neck surgery on microvascular reconstruction and robotics in June 2013.
His research interests include genomics, microbiome, diagnosis, treatment and surveillance of head and neck cancers and the development of novel robotic applications for head and neck surgery.
Dr. Chan’s qualifications include: Licentiate of Medical Council of Hong Kong, Bachelor of Medicine and Bachelor of Surgery (London), Diplomate American Board of Otolaryngology, Head and Neck Surgery, Fellow of the Hong Kong College of Otorhinolaryngology, Fellow of the Hong Kong Academy of Medicine (Otorhinolaryngology), and Fellow of the Royal College of Surgeons Edinburgh (Otolaryngology).
97
As an experienced user of surgical robots in conducting head and neck surgery, Dr. Chan is responsible for assessing technology related to surgical robotics, especially candidates for Signate, our project company specializing in the development of surgical robots.
DR. KA-WAI KWOK
Dr. Ka-Wai Kwok has been serving as Assistant Professor in Department of Mechanical Engineering, HKU since August 2014. He has also been serving as an Adjunct Assistant Professor in the School of Science and Engineering at The Chinese University of Hong Kong, Shenzhen (“CUHK SZ”), since October 2016.
His research interests focus on surgical robotics, intra-operative medical image processing, and their uses of high-performance computing techniques. To date, he has been involving in various designs of surgical robotic devices and interfaces for endoscopy, laparoscopy, stereotactic and intra-cardiac catheter interventions. His works have been highly recognized and winning several awards from IEEE international conferences in robotics and computing, including ICRA’18, RCAR’17, ICRA’17, ICRA’14, IROS’13 and FCCM’11, Hamlyn Symposium’12 and ’08, and Surgical Robot Challenge’16. He also became the recipient of Early Career Awards 2015/16 offered by Research Grants Council of Hong Kong. He currently serves as associate editor for an academic journal, “Frontier in Robotics and AI.”
He obtained his Ph.D. in The Hamlyn Centre for Robotic Surgery, Department of Computing, Imperial College London in March 2012, where he continued research on surgical robotics as a postdoctoral fellow between March 2012 and May 2013 and obtained the Croucher Foundation Fellowship between August 2013 and August 2014. This subsequently supported his research jointly hosted by The University of Georgia and Brigham and Women’s Hospital - Harvard Medical School.
As a mechanical engineer specializing in surgical robotics, Dr. Kwok is responsible for assessing technology related to surgical robotics, especially candidates for Signate, our project company specializing in the development of surgical robots.
DR. KENNY YU KWOK HEI
Dr. Kenny Yu was appointed as the NIHR Academic Clinical Lecturer at the University of Manchester in the United Kingdom in 2017.
Dr. Kenny Yu commenced specialist training in Neurosurgery at Salford Royal Hospital, United Kingdom in 2008 and attained his FRCS (Surgical Neurology) specialist qualification in 2018. His research interests are in myeloid cell infiltration in malignant gliomas, intra-tumoral delivery of therapeutics and in the application of advanced data analytical technology for biological and clinical datasets. He completed his Ph.D. in 2016 at the Stem Cell and Neurotherapies Laboratory at the University of Manchester under Prof Brian Bigger and subsequently completed a post-doctoral research fellowship at Dr. Peter Dirks laboratory in Toronto, Canada.
Dr. Yu’s key areas of research interests include Neurosurgery, Neuro-oncology, Cancer Inflammation and Cancer Immunology.
As both a basic and clinical scientist, Dr. Yu is responsible for assessing pharmacology/animal efficacy and clinical trials of our potential drug candidates, especially candidates for Scipio, our project company specializing in the development of oncology products; and Videns, our project company specializing in the development of neurology products.
DR. OWEN KO HO
Dr. Ko has been serving as a principal investigator and executive committee member at the Gerald Choa Neuroscience Center since 2017, a principal investigator at the Li Ka Shing Institute of Health Sciences and a clinical lecturer (from 2016 to 2018) and Assistant Professor (since 2018) in the Department of Medicine and Therapeutics, with all appointments at CUHK. Leading a team with diverse expertise in biology, chemistry and engineering, his research work focuses on the principles by which neural circuits mediate sensory perception and learning, as well as development of novel neuroimaging techniques.
Dr. Ko was admitted to the Bachelor of Medicine and Bachelor of Surgery Programme (MBChB) at CUHK in 2005. After completing his second year of studies, he pursued a one-year Intercalated Bachelor of Medical Sciences (BMedSci), followed by a three-year Ph.D. program in neuroscience at University College London (“UCL”) in the UK under the guidance of Professor Thomas Mrsic-Flogel. In 2012, Dr. Ko returned to Hong Kong and completed clinical training in 2015. He has published two first-authored Nature papers and one first-authored Nature Neuroscience paper from his Ph.D. studies. His breakthrough research has led to his runner-up award of the 2014 Eppendorf & Science Prize for Neurobiology, as the first awardee in Hong Kong.
98
As a neuroscientist and medical doctor, Dr. Ko is responsible for assessing pharmacology/animal efficacy and clinical trials of our potential drug candidates, especially candidates for Videns, our project company specializing in the development of neurology products.
DR. SUNNY WONG HEI
Dr. Sunny Wong has been an Assistant Professor in the Department of Medicine and Therapeutics, and an investigator at the laboratory of the Li Ka Shing Institute of Health Science at CUHK since December 2013. He is also the leader of the Clinical Metagenomics Research Group, with a focus to study the mechanistic role and translational potential of host-microbial interactions in diseases.
Dr. Wong has been a specialist in Gastroenterology and Hepatology since September 2016, and is a Physician-Scientist with expertise in genomics and molecular microbiology. He has published over 60 peer-reviewed journals in international journals, including the New England Journal of Medicine, Nature Genetics, Nature Communications, Gastroenterology and Gut as of December 2017. He has been an investigator in epidemiological studies and clinical trials, and a member of the local clinical research ethics committee since 2016.
Dr. Wong’s qualifications include: MBChB(Hons)(CUHK) (June 2006); DPhil (Oxon) (June 2010); MRCP (UK) (February 2012); FHKCP (September 2016); FHKAM (Medicine) (December 2016); FRCP (Edin) (September 2017); FRCPath (February 2018).
As both a basic scientist and Gastroenterologist, Dr. Wong is responsible for assessing pharmacology/animal efficacy and clinical trials of our potential drug candidates, especially candidates for Claves, our project company specializing in modulating microbiota in the gastrointestinal tract for curing various diseases.
99
Competition
Our industry is highly competitive and subject to rapid and significant change. While we believe that our development and commercialization experience, scientific knowledge and industry relationships provide us with competitive advantages, we face competition from pharmaceutical and biotechnology companies, including specialty pharmaceutical companies, and generic drug companies, academic institutions, government agencies and research institutions.
There are a number of large pharmaceutical and biotechnology companies that currently market and sell drugs or are pursuing the development of drugs and devices for the diagnosis and treatment of diseases for which we are developing products or technology. Moreover, a number of additional drugs are currently in clinical trials and may become competitors if and when they receive regulatory approval.
Many of our competitors have longer operating histories, better name recognition, stronger management capabilities, better supplier relationships, a larger technical staff and sales force and greater financial, technical or marketing resources than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer or less costly than our current drug candidates, or any future drug candidates we may develop, or obtain regulatory approval for their products more rapidly than we may obtain approval for our current drug candidates or any such future drug candidates. Our success will be based in part on our ability to identify, develop and manage a portfolio of drug and device candidates that are safer and more effective than competing products.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries extensively regulate, among other things, the research and clinical development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing, pricing, export and import of drug and device products (“Regulated Products”), such as those we are developing. Generally, before a new Regulated Product can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized to address the requirements of and in the format specific to each regulatory authority, submitted for review and approved by the regulatory authority. This process is very lengthy and expensive, and success is uncertain.
Regulated Products are also subject to other federal, state and local statutes and regulations in the United States and other countries, as applicable. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable regulatory requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the regulatory authority’s refusal to approve pending applications, withdrawal of an approval, clinical holds, untitled or warning letters, voluntary product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution, injunctions, disbarment, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any such administrative or judicial enforcement action could have a material adverse effect on us.
As the Company’s principal place of business is in Hong Kong, and because AML Clinic is located there, the Company is subject to various Hong Kong laws and regulation covering its business activities there, described in further detail below. Also, the Company anticipates that, if it obtains marketing approval for any of its drug and device candidates, it intends to focus its marketing and sales efforts primarily in three regions: the United States, Europe and PRC. The regulatory framework for each of these regions is described below.
U.S. Drug Development Process
The process of obtaining regulatory approvals and maintaining compliance with appropriate federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions or lead to voluntary product recalls. Administrative or judicial sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of non-clinical laboratory tests, preclinical studies according to cGLP and manufacturing of clinical supplies according to cGMP; |
100
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin; |
| ● | approval by an independent IRB, at each clinical site before each trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials according to cGCP, to establish the safety and efficacy of the proposed product for its intended use; |
| ● | preparation and submission to the FDA of an NDA, for a drug; |
| ● | satisfactory completion of an FDA advisory committee review, if applicable; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product, or components thereof, are produced to assess compliance with cGMP; and |
| ● | payment of user fees and the FDA review and approval of the NDA. |
Devices are subject to different forms of testing and approval, but (except for certain laboratory-developed diagnostic tests) still require satisfaction of various FDA requirements in order to be brought to market. As of the date hereof, the device candidate currently under development is SLS-1. We do not currently have a commercialization timeline for SLS-1 and cannot assure you that SLS-1 will ever be ready for commercialization.
The testing and approval process requires substantial time, effort and financial resources and we cannot be certain that any approvals for our drug candidates, or any future drug candidates we may develop, will be granted on a timely basis, if at all.
Once a drug candidate is identified for development, it enters the non-clinical testing stage. Non-clinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as preclinical studies. An IND sponsor must submit the results of the non-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND prior to commencing any testing in humans. An IND sponsor must also include a protocol detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some non-clinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to a proposed clinical trial and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns or non-compliance, and may be imposed on all products within a certain class of products. The FDA also can impose partial clinical holds, for example, prohibiting the initiation of clinical trials for certain duration or for certain doses.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with cGCP regulations. These regulations include the requirement that all research subjects provide informed consent in writing before their participation in any clinical trial. Further, an IRB representing each institution participating in a clinical trial must review and approve the plan for any clinical trial before it commences at that institution, and the IRB must conduct continuing review and reapprove the study at least annually. An IRB is responsible for protecting the rights of clinical trial subjects and considers, among other things, whether the risks to individuals participating in the clinical trial are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Each new clinical protocol and any amendments to the protocol must be submitted to the FDA for review, and to the IRBs for approval. Protocol detail, among other things, includes the objectives of the clinical trial, testing procedures, sublease selection and exclusion criteria, and the parameters to be used to monitor subject safety.
101
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. Phase 1 includes the initial introduction of an investigational new drug into humans. These studies are closely monitored and may be conducted in patients, but are usually conducted in healthy volunteer subjects. These studies are designed to determine the metabolic and pharmacologic actions of the drug in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. During Phase 1, sufficient information about the drug’s pharmacokinetics and pharmacological effects should be obtained to permit the design of well-controlled, scientifically valid, Phase 2 studies. Phase 1 studies also evaluate drug metabolism, structure-activity relationships, and the mechanism of action in humans. These studies also determine which investigational drugs are used as research tools to explore biological phenomena or disease processes. The total number of subjects included in Phase 1 studies varies with the drug, but is generally in the range of twenty to eighty. |
| ● | Phase 2. Phase 2 includes the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness of the drug for a particular indication or indications in patients with the disease or condition. This phase of testing also helps determine the common short-term side effects and risks associated with the drug. Phase 2 studies are typically well-controlled, closely monitored, and conducted in a relatively small number of patients, usually involving several hundred people. |
| ● | Phase 3.Phase 3 studies are expanded controlled and uncontrolled trials. They are performed after preliminary evidence suggesting effectiveness of the drug has been obtained in Phase 2, and are intended to gather the additional information about effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug. Phase 3 studies are designed to provide an adequate basis for extrapolating the results to the general population and transmitting that information in the physician labeling. Phase 3 studies usually include several hundred to several thousand people. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and clinical investigators within 15 calendar days for serious and unexpected suspected adverse events, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator’s brochure, or any findings from other studies or animal or in vitro testing that suggest a significant risk in humans exposed to the drug candidate. Additionally, a sponsor must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction no later than 7 calendar days after the sponsor’s receipt of the information. There is no assurance that Phase 1, Phase 2 and Phase 3 testing can be completed successfully within any specified period, or at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product has been associated with unexpected serious harm to subjects.
Concurrent with clinical trials, companies usually complete additional preclinical studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product drug and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product drug does not undergo unacceptable deterioration over its shelf life.
102
The results of product development, non-clinical studies and clinical trials, together with other detailed information regarding the manufacturing process, analytical tests conducted on the product, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA requesting approval to market the new drug. The FDA reviews all NDAs submitted within 60 days of submission to ensure that they are sufficiently complete for substantive review before it accepts them for filing. If the submission is accepted for filing, the FDA begins an in-depth substantive review.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive, and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA that must be satisfactorily addressed before it can be approved. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application or request an opportunity for a hearing.
If after such review a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. Any products for which we receive the FDA approval would be subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with the FDA promotion and advertising requirements. In addition, the FDA may require post-approval studies, including Phase 4 clinical trials, to further assess a product’s safety and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA also may conclude that an NDA may only be approved with a Risk Evaluation and Mitigation Strategy designed to mitigate risks through, for example, a medication guide, physician communication plan, or other elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
Post-Approval Requirements
Any products for which we receive the FDA approval are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with the FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally require prior the FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further the FDA review and approval.
The FDA may withdraw a product approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product’s marketing or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, untitled or warning letters, holds on clinical trials, product seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or consent decrees, or civil or criminal penalties, or may lead to voluntary product recalls.
103
Patent Term Restoration and Marketing Exclusivity
Because drug approval can take an extended period of time, there may be limited remaining life for the patents covering the approved drug, meaning that the company has limited time to use the patents to protect the sponsor’s exclusive rights to make, use and sell that drug. In such a case, U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date.
In addition, the FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (“ANDA”) or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval.
In the future, if appropriate, we intend to apply for restorations of patent term and/or marketing exclusivity for some of our products; however, there can be no assurance that any such extension or exclusivity will be granted to us.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of the FDA-regulated products, including drugs are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Pharmaceutical Coverage, Pricing and Reimbursement
Much of the revenue generated by new Regulated Products depends on the willingness of third-party payors to reimburse the price of the product. Significant uncertainty exists as to the coverage and reimbursement status of any products for which we may obtain regulatory approval. In the United States, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payors. Third-party payors include government authorities, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the reimbursement rate that the payor will pay for the product. Third-party payors may limit coverage to specific products on an approved list, or formulary, which is not required to include all of the FDA-approved products for a particular indication. Moreover, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
104
Third-party payors are increasingly challenging the price and examining the medical necessity and cost- effectiveness of medical products and services, in addition to their safety and efficacy. To obtain coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of any products, in addition to the costs required to obtain regulatory approvals. Our product candidates may not be considered medically necessary or cost-effective. If third-party payors do not consider a product to be cost-effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
The U.S. government and state legislatures have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future. Unfavorable coverage or reimbursement policies regarding any of the Company’s products would have a material adverse impact on the value of that product.
Other Healthcare Laws and Compliance Requirements
If we obtain regulatory approval of our products, we may be subject to various federal and state laws targeting fraud and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business.
Patient Protection and the Affordable Care Act
The Affordable Care Act, enacted in March 2010, includes measures that have or will significantly change the way health care is financed in the United States by both governmental and private insurers. Among the provisions of the Affordable Care Act of greatest importance to the pharmaceutical industry are the following:
| ● | The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. The Affordable Care Act increased pharmaceutical manufacturers’ rebate liability on most branded prescription drugs from 15.1% of the average manufacturer price to 23.1% of the average manufacturer price, added a new rebate calculation for line extensions of solid oral dosage forms of branded products, and modified the statutory definition of average manufacturer price. The Affordable Care Act also expanded the universe of Medicaid utilization subject to drug rebates by requiring pharmaceutical manufacturers to pay rebates on Medicaid managed care utilization and expanding the population potentially eligible for Medicaid drug benefits. |
| ● | In order for a pharmaceutical product to receive federal reimbursement under the Medicare Part B and Medicaid programs or to be sold directly to U.S. government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The Affordable Care Act expanded the types of entities eligible to receive discounted 340B pricing. |
| ● | The Affordable Care Act imposed a requirement on manufacturers of branded drugs to provide a 50% discount off the negotiated price of branded drugs dispensed to Medicare Part D patients in the coverage gap (i.e., the “donut hole”). |
| ● | The Affordable Care Act imposed an annual, non-deductible fee on any entity that manufactures or imports certain branded prescription drugs, apportioned among these entities according to their market share in certain government healthcare programs, although this fee does not apply to sales of certain products approved exclusively for orphan indications. |
105
In addition to these provisions, the Affordable Care Act established a number of bodies whose work may have a future impact on the market for certain pharmaceutical products. These include the Patient-Centered Outcomes Research Institute, established to oversee, identify priorities in, and conduct comparative clinical effectiveness research, the Independent Payment Advisory Board, which has authority to recommend certain changes to the Medicare program to reduce expenditures by the program, and the Center for Medicare and Medicaid Innovation within the Centers for Medicare and Medicaid Services, to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
These and other laws may result in additional reductions in healthcare funding, which could have a material adverse effect on customers for our product candidates, if we gain approval for any of them. Although we cannot predict the full effect on our business of the implementation of existing legislation or the enactment of additional legislation pursuant to healthcare and other legislative reform, we believe that legislation or regulations that would reduce reimbursement for, or restrict coverage of, our products could adversely affect how much or under what circumstances healthcare providers will use our product candidates if we gain approval for any of them.
U.S. Medical Device Regulatory Approval Process
Medical Devices are subject to different forms of testing and approval, and require satisfaction of various FDA requirements including the Food, Drug and Cosmetic Act (FDCA) in order to be brought to market.
The two primary types of FDA marketing authorization applicable to a medical device are premarket notification, also called 510(k) clearance, and premarket approval. The type of marketing authorization is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes — Class I, Class II or Class III — based on the degree of risk the FDA determines to be associated with a device and the level of regulatory control deemed necessary to ensure the device’s safety and effectiveness. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are deemed to pose the least risk and are subject only to general controls applicable to all devices, such as requirements for device labeling, premarket notification, and adherence to the FDA’s Good Manufacturing Practices. Class II devices are intermediate risk devices that are subject to general controls and may also be subject to special controls such as performance standards, product-specific guidance documents, special labeling requirements, patient registries, or post-market surveillance. Class III devices are those for which insufficient information exists to assure safety and effectiveness solely through general controls or if the device is a life-sustaining, life-supporting or a device of substantial importance in preventing impairment of human health, or which presents a potential, unreasonable risk of illness or injury and special controls are not adequate to assure safety and effectiveness.
Most Class I devices and some Class II devices are exempted by regulation from the 510(k) clearance requirement and can be marketed without prior authorization from the FDA. Most Class II devices (and certain Class I devices that are not exempt) are eligible for marketing through the 510(k) clearance pathway. By contrast, devices placed in Class III generally require premarket approval or 510(k) de novo clearance prior to commercial marketing. The premarket approval process is more stringent, time-consuming, and expensive than the 510(k) clearance process. However, the 510(k) clearance process has also become increasingly stringent and expensive.
510(k) Clearance Pathway. When a 510(k) clearance is required, a premarket notification must be submitted to the FDA demonstrating that a proposed device is “substantially equivalent” to a previously cleared and legally marketed 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a premarket approval application, which is commonly known as the “predicate device.” A device is substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (i) the same technological characteristics or (ii) different technological characteristics and the information submitted demonstrates that the device is as safe and effective as a legally marked device and does not raise different questions of safety or effectiveness. By law, the FDA is required to clear or deny a 510(k) premarket notification within 90 days of submission of the application. As a practical matter, clearance often takes significantly longer. The FDA may require further information, including clinical data, to make a determination regarding substantial equivalence. If the FDA determines that the device, or its intended use, is not substantially equivalent to a previously-cleared device or use, the FDA will issue a not substantially equivalent decision. This means the device cannot be cleared through the 510k process and will require marketing authorization through the premarket approval pathway.
106
Premarket Approval Pathway. A premarket approval application must be submitted to the FDA if the device cannot be cleared through the 510(k) process. The premarket approval application process is much more demanding than the 510(k) premarket notification process and requires the payment of significant user fees. A premarket approval application must be supported by valid scientific evidence, which typically requires extensive data, including but not limited to technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction reasonable evidence of safety and effectiveness of the device. The FDA has 45 days from its receipt of a premarket approval application to determine whether the application will be accepted for filing based on the FDA’s threshold determination that it is sufficiently complete to permit substantive review. After the FDA determines that the application is sufficiently complete to permit a substantive review, the FDA will accept the application and begin its in-depth review. The FDA has 180 days to review an “accepted” premarket approval application, although this process typically takes significantly longer and may require several years to complete. During this review period, the FDA may request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with quality system regulations. The FDA may delay, limit or deny approval of a premarket approval application for many reasons, including:
| ● | failure of the applicant to demonstrate that there is reasonable assurance that the medical device is safe or effective under the conditions of use prescribed, recommended or suggested in the proposed labeling; |
| ● | insufficient data from the preclinical studies and clinical trials; |
| ● | the manufacturing processes, methods, controls or facilities used for the manufacture, processing, packing or installation of the device do not meet applicable requirements. If the FDA evaluations of both the premarket approval application and the manufacturing facilities are favorable, the FDA will either issue an approval order or an approvable letter, which usually contains a number of conditions that must be met in order to secure final approval of the premarket approval application. If the FDA’s evaluation of the premarket approval application or manufacturing facilities is not favorable, the FDA will deny approval of the premarket approval application or issue a not approvable letter. A not approvable letter will outline the deficiencies in the application and, where practical, will identify what is necessary to make the premarket approval application. The FDA may also determine that additional clinical trials are necessary, in which case the premarket approval application may be delayed for several months or years while the trials are conducted and then the data submitted in an amendment to the premarket approval application. Once granted, a premarket approval application may be withdrawn by the FDA if compliance with post approval requirements, conditions of approval or other regulatory standards is not maintained or problems are identified following initial marketing. |
Clinical Trials. Clinical trials are almost always required to support premarket approval and are sometimes required for 510(k) clearance. In the United States, these trials generally require submission of an application for an Investigational Device Exemption, or IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing it is safe to test the device in humans and that the testing protocol is scientifically sound. The FDA must approve the IDE in advance of trials for a specific number of patients unless the product is deemed a non-significant risk device eligible for more abbreviated IDE requirements or the clinical investigation is exempt from the IDE regulations. Clinical trials for significant risk devices may not begin until the IDE application is approved by the FDA and the appropriate institutional review boards, or IRBs, at the clinical trial sites. The applicant, the FDA or the IRB at each site at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the benefits. Even if a trial is completed, the results of clinical testing may not demonstrate the safety and efficacy of the device, may be equivocal or may otherwise not be sufficient to obtain approval or clearance of the product.
107
Both the 510(k) and premarket approval processes can be expensive and lengthy and require the payment of significant fees, unless an exemption applies. The FDA’s 510(k) clearance process usually takes from approximately three to 12 months, but may take longer. The process of obtaining a premarket approval is much more costly and uncertain than the 510(k) clearance process and generally takes from approximately one to five years, or longer, from the time the application is submitted to the FDA until an approval is obtained. The process of obtaining regulatory clearances or approvals to market a medical device can be costly and time consuming, and the applicant may not be able to obtain these clearances or approvals on a timely basis, if at all.
As of the date hereof, our sole device candidate currently under development is SLS-1, which is a platform for the dexterous manipulation of cardiovascular robotic surgical catheter, conventionally classified as a cardiovascular steerable catheter, in the MRI environment. We do not currently have a commercialization timeline for SLS-1 and cannot assure you that SLS-1 will ever be ready for commercialization. If we are ready to seek regulatory approval for the SLS-1 device in the U.S., we expect that the FDA will classify it as a Class II non-exempted device requiring premarket clearance under Section 510(k) of the FDCA. If our device cannot clear through the 510(k) process, we will need to obtain marketing authorization through the premarket approval pathway, which will be more costly, lengthy and uncertain.
European Union Regulation
Regulation in the European Union
The process governing approval of medicinal products in the EU generally follows the same lines as in the United States. It entails satisfactory completion of pharmaceutical development, non-clinical studies and adequate and well-controlled clinical trials to establish the safety and efficacy of the medicinal product for each proposed indication. It also requires the submission to relevant competent authorities for clinical trials authorization and to the European Medicines Authority, or EMA, for a marketing authorization application, or MAA, and granting of a marketing authorization by these authorities before the product can be marketed and sold in the EU.
Clinical Trial Approval
Pursuant to the currently applicable Clinical Trials Directive 2001/20/EC and the Directive 2005/28/EC on cGCP, a system for the approval of clinical trials in the EU (the equivalent of the IND process in the United States) has been implemented through national legislation of the EU member states. Under this system, an applicant must obtain approval from the competent national authority of an EU member state in which the clinical trial is to be conducted or in multiple EU member states if the clinical trial is to be conducted in a number of EU member states. Furthermore, the applicant may only start a clinical trial at a specific study site after the independent ethics committee has issued a favorable opinion. The clinical trial application, or CTA, must be accompanied by an investigational medicinal product dossier with supporting information prescribed by Directive 2001/20/EC and Directive 2005/28/EC and corresponding national laws of the EU member states and further detailed in applicable guidance documents.
In April 2014, the EU adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current Clinical Trials Directive 2001/20/EC. It is expected that the new Clinical Trials Regulation will apply in 2019. It will overhaul the current system of approvals for clinical trials in the EU. Specifically, the new regulation, which will be directly applicable in all EU member states, aims at simplifying and streamlining the approval of clinical trials in the EU. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure using a single entry point and strictly defined deadlines for the assessment of clinical trial applications.
Marketing Authorization
To obtain a marketing authorization for a product under the EU regulatory system (the equivalent of the NDA process in the United States), an applicant must submit an MAA, either under a centralized procedure administered by the EMA or one of the procedures administered by competent authorities in EU member states (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization may be granted only to an applicant established in the EU. Regulation (EC) No. 1901/2006 provides that prior to obtaining a marketing authorization in the EU, an applicant must demonstrate compliance with all measures included in an EMA-approved Pediatric Investigation Plan, or PIP, covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, class waiver, or a deferral for one or more of the measures included in the PIP.
108
The centralized procedure provides for the grant of a single marketing authorization by the European Commission that is valid for all EU member states. Pursuant to Regulation (EC) No. 726/2004, the centralized procedure is compulsory for specific products, including for medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy products and products with a new active substance indicated for the treatment of certain diseases, including products for the treatment of cancer. For products with a new active substance indicated for the treatment of other diseases and products that are highly innovative or for which a centralized process is in the interest of patients, the centralized procedure may be optional.
Under the centralized procedure, the Committee for Medicinal Products for Human Use, or the CHMP, established by the EMA is responsible for conducting the assessment of a product to define its risk/benefit profile. Under the centralized procedure, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions of the CHMP. Accelerated evaluation may be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation.
If the CHMP accepts such a request, the time limit of 210 days will be reduced to 150 days, but it is possible that the CHMP may revert to the standard time limit for the centralized procedure if it determines that it is no longer appropriate to conduct an accelerated assessment.
Periods of Authorization and Renewals
A marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a reevaluation of the risk benefit balance by the EMA or by the competent authority of the authorizing Member State. To that end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal period. Any authorization that is not followed by the placement of the drug on the EU market (in the case of the centralized procedure) or on the market of the authorizing Member State within three years after authorization ceases to be valid.
Regulatory Requirements after Marketing Authorization
Following approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of the medicinal product. These include compliance with the EU’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. In addition, the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other regulatory bodies in the EU, which mandate the methods, facilities and controls used in manufacturing, processing and packing of drugs to assure their safety and identity. Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising directed toward the prescribers of drugs and/or the general public, are strictly regulated in the EU under Directive 2001/83EC, as amended.
Orphan Drug Designation and Exclusivity
Regulation (EC) No. 141/2000 and Regulation (EC) No. 847/2000 provide that a product can be designated as an orphan drug by the European Commission if its sponsor can establish: that the product is intended for the diagnosis, prevention or treatment of (1) a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons in the EU when the application is made, or (2) a life-threatening, seriously debilitating or serious and chronic condition in the EU and that without incentives it is unlikely that the marketing of the drug in the EU would generate sufficient return to justify the necessary investment. For either of these conditions, the applicant must demonstrate that there exists no satisfactory method of diagnosis, prevention, or treatment of the condition in question that has been authorized in the EU or, if such method exists, the drug has to be of significant benefit compared to products available for the condition.
109
An orphan drug designation provides a number of benefits, including fee reductions, regulatory assistance and the possibility to apply for a centralized EU marketing authorization. Marketing authorization for an orphan drug leads to a ten-year period of market exclusivity. During this market exclusivity period, neither the EMA nor the European Commission or the EU member states can accept an application or grant a marketing authorization for a “similar medicinal product.” A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan drug designation because, for example, the product is sufficiently profitable not to justify market exclusivity.
European Medical Device Regulatory Approval Process
As in the United States, there is a separate regulatory framework for approval of medical devices. If the Company determines to commercialize SLS-1 or another medical device, it will become subject to all of the requirements for approval required by those regulations.
PRC Regulation
In order to protect our potential market in the PRC, we have obtained an exclusive license of certain PRC patents directed to certain of the drug candidates that we are developing and are currently seeking approval of additional patent and other IP filings in the PRC. We do not otherwise conduct business in the PRC. Seeking IP approval in the PRC subjects us to some of the rules and practices of the PRC government. Since the Company intends eventually to market its products in the PRC, at least some of our drug candidates may become subject to regulatory approval and marketing authorization in the PRC.
Hong Kong Regulation
The operations of AML Clinic in Hong Kong are subject to certain general laws and regulations in relation to clinic medical professionals, trade description and safety of consumer goods, medical advertisement and importation, exportation, dealing in and sale of pharmaceutical products and drugs.
Medical Clinics Ordinance
The Medical Clinics Ordinance provides for the registration, control and inspection of medical clinics. It requires a medical clinic to be registered, with name and address and other prescribed particulars. “Medical clinic” means any premises used or intended to be used for the medical diagnosis or treatment of persons suffering from, or believed to be suffering from, any disease, injury or disability of mind or body, with specific exceptions, including private consulting rooms used exclusively by registered medical practitioners in the course of their practice on their own account and not bearing any title or description which includes the word “clinic” or “polyclinic” in the English language.
The application of registration may be refused if:
| (i) | the income derived or to be derived from the establishment or operation of the clinic is not, or will not be, applied solely towards the promotion of the objects of the clinic; or | |
| (ii) | any portion of such income, except payment of remuneration to employed registered medical practitioners, nurses and menial servants, will be paid by way of dividend, bonus or otherwise howsoever by way of profit to the applicant himself, or to any persons properly so employed, or to any other persons howsoever. |
110
We do not believe that the Medical Clinic Ordinance is applicable to the business of our Company and its subsidiaries, having considered, among others, the following:
| (iii) | the legislative intent behind the Medical Clinics Ordinance was to provide for registration of non-profit making clinics; |
| (iv) | the Food and Health Bureau of Hong Kong published a consultation document, “Regulation of Private Healthcare Facilities” in 2014 which specifically states that the Medical Clinics Ordinance and the Code of Practice For Clinics Registered Under The Medical Clinics Ordinance (Chapter 343 of the Laws of Hong Kong) set out the regulatory framework for non-profit-making medical clinics and that other private healthcare facilities, such as ambulatory medical centers and clinics operated by medical groups or individual medical practitioners, are not subject to direct statutory control beyond the regulation of an individual’s professional practice; and |
| (v) | our business is one which makes and intends to continue making profit as a listed entity. The payment of bonuses to some of our Hong Kong Doctors is clearly a reflection of the profit-making nature of our business. |
Hence, we do not believe that AML Clinic is required to be registered under the Medical Clinics Ordinance.
Waste Disposal Ordinance
The Waste Disposal Ordinance (Chapter 354 of the Laws of Hong Kong) (“WDO”) and the Waste Disposal (Clinical Waste) (General) Regulation (Chapter 354O of the Laws of Hong Kong) (the “WDR”) provide for, among others, the control and regulation of the production, storage, collection and disposal of clinical waste.
Under the WDO, clinical waste means waste consisting of any substance, matter or thing generated in connection with:
| ● | a dental, medical, nursing or veterinary practice; | |
| ● | any other practice, or establishment (howsoever described), that provides medical care and services for the sick, injured, infirm or those who require medical treatment; | |
| ● | dental, medical, nursing, veterinary, pathological or pharmaceutical research; or | |
| ● | a dental, medical, veterinary or pathological laboratory practice, |
and which consists wholly or partly of any of the materials specified in one or more of the groups listed below:
| ● | used or contaminated sharps;
| |
| ● | laboratory waste;
| |
| ● | human and animal tissues;
| |
| ● | infectious materials;
| |
| ● | dressings; and
| |
| ● | such other wastes as specified by the Director of the Environmental Protection Department (“EPD”) of Hong Kong. |
Given the medical services provided by AML Clinic and the research works in our R&D Center may produce used or contaminated sharps such as syringes and needles as well as dressings, we are subject to WDO, WDR and the Code of Practice.
111
Rest of the World Regulation
For other countries in the world, the requirements governing the conduct of clinical trials, medical product licensing, pricing and reimbursement vary from country to country. In all cases if clinical trials are required, they must be conducted in accordance with cGCP requirements and the applicable regulatory requirements and the ethical principles having their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Employees
As of the date hereof, we have 41 employees, including 39 full-time employees and 2 part-time employees. Of these, 15 are engaged in full-time research and development and laboratory operations, 19 are engaged in general and administrative functions, 5 are full-time employees engaged in the clinic operation and 2 part-time employees are engaged in sponsored research and development, laboratory operations and legal clerical support. As of the date hereof, 41 of our employees are located in Hong Kong. In addition, we have engaged and may continue to engage 33 independent contracted consultants and advisors to assist us with our operations. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have never experienced any employment related work stoppages, and we consider our relations with our employees to be good.
Facilities
We have several operating leases, primarily for offices. Our principal executive offices are located in Hong Kong; we also have offices in London and Jersey City.
Our facilities in Hong Kong consists of: (i) 638 square foot lab space under a lease that commenced in December 2017 and expires in December 2020, that carries a monthly rent of $2,127 and which is used for the center run by APD (the “R&D Center”); (ii) 851 square foot office space under a lease that commenced in December 2017 and expires in December 2020 that carries a monthly rent of $2,509, which is also used for the center run by APD (the “HKSTP Office Space”); (iii) 3,250 square foot office space under a lease that commenced in February 2018 and expires in January 2021 and that carries a monthly rent of $16,667 (the “Guangdong Investment Tower Lease”) (See “Transactions with Related Persons – Leased Facilities”); and (iv) 3,173 square foot space under a lease that commenced in March 2018 and expires in March 2022 (the “AML Lease”, which is home to AML Clinic).
Pursuant to the lease agreement for the R&D Center, we are also obligated to pay $1,090 per month as service charges (this is an increase, as of April 2018, from $1,058 per month with one month notice from landlord in accordance with the lease).
Pursuant to the lease agreement for the HKSTP Office Space, we are also obligated to pay $666 per month as service charges (this is an increase, as of April 2018, from $633 per month with one month notice from landlord in accordance to the lease).
Pursuant to the lease agreement for AML Lease, it carries a monthly rent of approximately $29,500 for the first year; the monthly rent increases each year thereafter by about $810 per month. We must also pay a monthly management fee and air conditioning fee of about $3,540 per month (this is an increase, as of March 2019, from $3,370 per month subject to revision from time to time in accordance with the AML lease). Pursuant to the terms of the AML Lease, if we fail to pay any sums that are due thereunder, we shall incur interest at the rate of 2% per calendar month until full payment is made of all owed sums. The landlord maintains the right to terminate the AML Lease with no less than six months’ notice if the landlord enters into a contract to sell the building of which the property we rented forms a part or determines to redevelop or renovate the building. The AML Lease is guaranteed by Clark Cheng, our Chief Medical Officer and one of our Executive Directors, who is also an executive director of AML and Smart Pharmaceutical Development PTE. Ltd. Dr. Cheng also serves as a director of several other of our subsidiaries.
112
Our office space in London consists of approximately 172 square feet under a lease that commenced in August 2019, expires in March 2020 and has a rent of $2,611 per month. Our office space in Cambridge, Massachusetts consists of approximately 120 square feet under a lease that commenced in September 2019 and expires in March 2020 and has a rent of $2,211 per month.
Payments under operating leases are expensed on a straight-line basis over the periods of the respective leases, and the terms of the leases do not contain rent escalation, contingent rent, and renewal or purchase options.
We believe our current facilities are sufficient to meet our needs.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Directors and Executive Officers
Below is a list of our directors and executive officers, as of the date of this prospectus, and a brief account of the business experience of each of them. The business address for the directors and officers of Aptorum Group Limited is 17th floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong.
On October 10, 2019, Mr. Lui resigned from his position as Chief Business Officer.
| Name | Age | Position | ||
| Executive Officers | ||||
| Ian Huen | 39 | Founder, Chief Executive Officer and Executive Director | ||
| Darren Lui | 38 | President and Executive Director | ||
| Clark Cheng | 39 | Chief Medical Officer and Executive Director | ||
| Sabrina Khan | 38 | Chief Financial Officer | ||
| Thomas Lee Wai Yip | 46 | Head of Research and Development | ||
| Angel Siu Yan Ng | 38 | Chief Operating Officer | ||
| Non-Management Directors | ||||
| Charles Bathurst | 64 | Independent Non-Executive Director and Chair of Audit Committee | ||
| Mirko Scherer | 51 | Independent Non-Executive Director | ||
| Justin Wu | 49 | Independent Non-Executive Director and Chair of Compensation Committee | ||
| Douglas Arner | 49 | Independent Non-Executive Director and Chair of Nominating and Corporate Governance Committee |
Executive Officers
MR. IAN HUEN, Founder, Chief Executive Officer and Executive Director
Mr. Ian Huen is the Founder, Chief Executive Officer and Executive Director of Aptorum Group Limited. Mr. Huen is also Co-Founder of a Hong Kong company, AENEAS CAPITAL LIMITED, a licensed corporation regulated by the Hong Kong Securities & Futures Commission as a Type 9 Asset Manager, since 2005. He has over 15 years of global asset management experience and previously covered the U.S. healthcare sector as an equity research analyst at Janus Henderson Group plc (formerly known as Janus Capital). Mr. Huen was the financial advisor in the sale of Seng Heng Bank Limited (Macau) to Industrial and Commercial Bank of China in 2007 and was appointed as the vice president of the Board of General Meeting in Industrial and Commercial Bank of China (Macau) Capital Limited in March 2007 for a term of 12 years until March 2019.
113
As a trustee board member of the Dr. Stanley Ho Medical Development Foundation, Mr. Huen facilitates advisory, development funding, access to research resources across Asia and continues to establish relationships with leading academic institutions to propel innovations in healthcare.
Mr. Huen graduated from Princeton University with an A.B. degree in Economics in June 2001, earned a MA in Comparative and Public History from CUHK in June 2016. Mr. Huen is also a Chartered Financial Analyst (“CFA”).
MR. DARREN LUI,President and Executive Director
Mr. Darren Lui is the President and an Executive Director of Aptorum Group Limited. Mr. Lui is also an Executive Director and Co-Founder of AENEAS CAPITAL LIMITED, a licensed corporation regulated by the Hong Kong Securities & Futures Commission as a Type 9 Asset Manager.
Mr. Lui was previously the founder, director and responsible officer of Varengold Capital Securities Limited and Varengold Capital Asset Management Limited in Hong Kong, with subsidiaries operating brokerage, asset management, and investment businesses in Asia established since January 2015.
Prior to this, he was a Director within the Fixed Income Group of Barclays Capital, where he spent over nine years from September 2005 to February 2014 developing and establishing their London, Singapore and New York structuring teams. From September 2002 to August 2005 he was qualified as a Chartered Accountant with Ernst & Young LLP (London), specializing in capital markets advisory.
Mr. Lui graduated with First-Class Honors from Imperial College, London with a BSc degree in Biochemistry in June 2002. He is a Chartered Accountant (ICAS), a CFA, and an Associate of Chartered Institute of Securities & Investments (UK).
DR. CLARK CHENG, Chief Medical Officer and Executive Director, Aptorum Group Limited
Executive Director, Aptorum Medical Limited
Dr. Clark Cheng is the Chief Medical Officer and Executive Director of Aptorum Group Limited; he is also an executive director of AML and Smart Pharmaceutical Development PTE. Ltd, which is also a limited partner of Smart Pharmaceutical Limited Partner; Dr. Cheng also serves as a director of several other of our subsidiaries. Prior to this appointment, Dr. Cheng served as the Operations Director since 2009 of Raffles Medical Group, and the company’s Deputy General Manager since 2011, representing an expanded role in the region. During his employment with Raffles Medical Group, he practiced as a full-time medical administrator to overlook Raffles Medical Hong Kong operations and supported its development in the PRC.
Dr. Cheng received his medical training at the University College London, UK, in 2005 and completed his foundation year training at The Royal Free Hospital in 2007. Pursuing his career in surgery, he obtained his membership of the Royal College of Surgeons of Edinburgh in 2009 and commenced his training in Orthopaedics where he practiced as Specialist Registrar at the National University Hospital, Singapore, with special interest in Traumatology of the lower limbs. In 2011, he also obtained his Master in Business & Administration with distinction from Tippie College of Business, University of Iowa, US.
Dr. Cheng is an active member of the Singapore Chamber of Commerce, and appears regularly as a guest speaker for The Open University of Hong Kong, The Airport Authority Hong Kong and other corporate events.
114
MISS SABRINA KHAN, Chief Financial Officer
Miss Sabrina Khan is the Chief Financial Officer of Aptorum Group Limited. She leads the Company’s financial strategy and operations, as well as Investor Relations. She has extensive experience working at KPMG (Hong Kong) and Ernst & Young LLP (Hong Kong). She was recently the regional financial controller in Asia for St. James’s Place Wealth Management (Hong Kong), which St. James’s Place Wealth Management Group (LON: STJ) is a FTSE100 company with £103.5 billion of client funds under management. Prior to that, she served as the senior finance manager of Neo Derm Group, a leading medical aesthetic group in Asia, in charge of its finance-related matters and expansion in the PRC. From August 2009 to May 2013, she served as the senior finance manager of Global Cord Blood Corporation (formerly known as China Cord Blood Corporation (NYSE: CO)), which was a subsidiary of Golden Meditech Holdings Limited (HK: 801), where she played an important role with the NYSE listing filings, investor relations and post IPO reporting. During her employment with Global Cord Blood Corporation, she was actively involved in the issuance of convertible bonds to Kohlberg Kravis Roberts and various merger and acquisition projects, facilitated and liaised with investment banks on due diligence, deal structuring, and also involved in commercial negotiation with respect to major contract terms.
Miss Khan qualified as certified public accountant and graduated with a BBA (Hons) in Accounting & Finance at The University of Hong Kong in 2003. She was qualified as an Advanced China Certified Taxation Consultant in 2015.
DR. THOMAS LEE WAI YIP, Head of Research and Development
Dr. Thomas Lee is the Head of Research & Development of Aptorum Group Limited. He served as Chief Executive Officer and Chief Scientific Officer of Aptorum Therapeutics Limited, a wholly-owned therapeutics subsidiary of Aptorum Group Limited from January 2018 to March 2019. Prior to that, Dr. Lee served as an Assistant Professor in the School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong from August 2013 to January 2018. Dr. Lee’s key area of research involves drug delivery with specialties including: formulation development of poorly soluble compounds, oral delivery, Nanotechnology, and similar fields.
Prior to academia, Dr. Lee accumulated big-pharma experience from the decade he spent at two multinational pharmaceutical companies in the U.S. From November 2008 to July 2013, Dr. Lee worked at Celgene Corporation as a Senior Scientist of the Formulations Research & Development. From June 2003 to November 2008, Dr. Lee worked at Novartis Pharmaceuticals Corporation, as a Principal Scientist.
Dr. Lee graduated with B.Pharm. (Hons) Degree from The Chinese University of Hong Kong in December 1995, and received his Ph.D. in Pharmaceutical Sciences (Drug Delivery) from the University of Wisconsin-Madison in the U.S in May 2003.
DR. ANGEL SIU YAN NG, Chief Operating Officer
Dr. Angel Ng is the Chief Operating Officer of Aptorum Group Limited. She served as the Chief Operating Officer (“COO”) of Aptorum Therapeutics Limited, a wholly-owned therapeutics subsidiary of Aptorum Group Limited from September 2017 to March 2019. During this time, Dr. Ng led Aptorum Therapeutics Limited and its subsidiaries’ operations and business strategies. Dr. Ng has extensive experience in project management with Innovation and Technology government funds and academic institutions.
Since September 2016, Dr. Ng works as a Research Officer cum Project Manager at The University of Hong Kong (“HKU”) in project management for various research projects including government funded project of novel medical device. During this time, Dr. Ng led the research team towards cadaveric trial for a novel soft robotics medical device and coordinated all research related agreements. During December 2014 to September 2015, Dr. Ng served as Project Manager at Hong Kong Science & Technology Parks Corporation (“HKSTP”), where she worked on technology transfer and commercialization for research and development projects through partnerships between local universities and the worldwide network and expertise of the Oxford University commercial arm. Dr. Ng also worked for The Chinese University of Hong Kong (“CUHK”) as Project Manager from September 2007 to January 2009. She managed a HK$60M government funded R & D project with a team of specialists in CUHK where she kept close liaison with industry and government authorities. Dr. Ng was in the precision chemical machining industry from 2003 to 2007, where she managed the manufacturing team and business operations in PRC.
115
Dr. Ng serves as a Director of Tecford Trading & Technology Company Limited since December 2017. Dr. Ng graduated with a B.Sc (Hons) from Department of Chemistry at HKU in December 2002, received her M.Sc in Composite Materials from Imperial College London in November 2003 and obtained her Ph.D. in Mechanical Engineering from HKU in December 2015.
Independent Non-Executive Directors
MR. CHARLES BATHURST
Mr. Bathurst is an Independent Non-Executive Director of Aptorum Group Limited. He has over 40 years’ experience of management and senior executive roles primarily in financial services. In 2011, he set up his own independent consultancy service, Summerhill Advisors Limited, advising on management structure, business development, financial reporting, internal audit controls and compliance to both emerging and multinational companies. Today he holds Non-Executive and Advisory board positions on fast-growing companies in healthcare, technology and financial services.
Prior to establishing Summerhill, he served as a Director for J.O. Hambro Investment Management from September 2008 to August 2011, where he oversaw the restructuring and commercialization a range of in-house investment funds. He was appointed to the management board and supervised reporting teams including Business development, accounting teams, regulatory reporting teams and internal controls.
From April 2004 to March 2008, Mr. Bathurst served in multiple roles at Old Mutual Asset Managers (UK), including being a member of the senior management team and head of international sales. Duties included business development, launching new investment funds, recruitment, establishing and supervision of regulatory and financial reporting teams, as well as ensuring compliance with funds’ regulatory requirements and corporate governance standards.
Prior to this, Mr. Bathurst was an advisor to Lion Capital Advisors Limited from April 2003 to March 2004, and from June 2002 to March 2003 business development reporting to the board of management of LCF Rothschild Asset Management Limited.
From April 1995 to March 2002, Mr. Bathurst joined a newly formed alternative investment management team at Credit Agricole Asset Management, establishing the London Branch as the Managing Director in 1998. He was responsible for the recruitment and development strategy for marketing, sales, investment, financial reporting, compliance and regulatory controls and investor relations.
Between the period of September 1989 and December 1994, Mr. Bathurst worked for GNI, the largest futures and options execution and clearing broker on the London International Financial Futures Exchange, where he focused on marketing to European and Middle East financial institutions. In 1991, he joined a new management team to launch a series of specialist investment funds while serving as the Head of Sales and Product Development.
Mr. Bathurst graduated from the Royal Military Academy Sandhurst in November 1974 and commissioned into the British Army serving in the UK and Germany.
DR. MIRKO SCHERER
Dr. Mirko Scherer is an Independent Non-Executive Director of Aptorum Group Limited. Dr. Scherer has been serving as the Chief Executive Officer at CoFeS China (formerly known as “TVM Capital China”) in Hong Kong since March 2015. CoFeS China focuses on cross-border activities in the life science industry between China and the West. CoFeS China acts as a bridge between China and the West, assisting Chinese investors and pharmaceutical companies accessing western innovations, while collaborating with innovative life science companies from the West to enter the fast-growing China market.
Dr. Mirko Scherer has served on the Board of the Frankfurt Stock Exchange from 2005 to 2007 and has been a board member of the Stichting Preferente Aandelen QIAGEN since 2004. From August 2016 through July 2018, Dr. Scherer served as a Non-Executive board member of Quantapore Inc. and from April 2015 through September 2017, he was a director of China BioPharma Capital I, (GP).
116
Dr. Scherer is an experienced biotechnology executive and has led numerous financing M&A and licensing transactions, in both public and private markets, in Europe and the U.S. for over 20 years. He consulted MPM Capital for the period between July 2012 and December 2014. Dr. Scherer was also a co-founder and partner of KI Kapital from November 2008 to February 2014, a company which was specialized in providing consultation in life science industry.
Prior to working in the venture capital industry, Dr. Scherer co-founded GPC Biotech (Munich and Princeton, NJ) and served as the Chief Financial Officer from October 1997 to December 2007. GPC Biotech engaged in numerous pharmaceutical alliances with companies such as Sanofi Aventis, Boehringer Ingelheim, Altana (now part of Takeda), Yakult, and Pharmion (now part of Celgene). Over the past 20 years, Dr. Scherer has established an extensive network in the U.S., European, and China’s biotechnology and venture capital industry. Prior to his time at GPC Biotech, Dr. Scherer worked as a consultant from May 1993 to June 1994 at the Boston Consulting Group.
Dr. Scherer earned a Doctorate in Finance from the European Business School in Oestrich-Winkel/Germany in 1998, a MBA from Harvard Business School in June 1996, and a degree in Business Administration from the University of Mannheim/Germany in February 1993.
DR. JUSTIN WU
Dr. Justin Wu is an Independent Non-Executive Director of Aptorum Group Limited. He also has been serving as the Chief Operating Officer of CUHK Medical Centre since August 2018. He served as the Associate Dean (Development) of the Faculty of Medicine at CUHK from July 2014 to June 2018 and the Associate Dean (Clinical) of the Faculty of Medicine at CUHK from December 2012 to July 2014, and has been serving a Professor in the Department of Medicine and Therapeutics since 2009, also the Director of the S. H. Ho Center for Digestive Health, a research center specializing in functional gastrointestinal diseases, reflux and motility disorders, and digestive endoscopy. Active in research publications and assessments, Dr. Wu served as the International Associate Editor of American Journal of Gastroenterology (“AJG”), and Managing Editor of Journal of Gastroenterology and Hepatology (“JGH”). He is also the Secretary General of the Asian Neurogastroenterology and Motility Association (“ANMA”), and Secretary General of the Asia Pacific Association of Gastroenterology (“APAGE”).
Dr. Wu has won a number of awards including the Emerging Leader in Gastroenterology Award by the JGH Foundation, and the Vice Chancellor’s Exemplary Teaching Award at CUHK. Aside from his expertise in gastroenterology, Dr. Wu has an extensive interest in the development of Integrative Medicine in Hong Kong. He is the Founding Director of the Hong Kong Institute of Integrative Medicine, working closely with the School of Chinese Medicine to develop an integrative model at an international level. The institute aims at maximizing the strength of Western and Chinese medicine to provide a safe and effective integrative treatment to patients.
Dr. Wu served as a consultant and an advisory board member for Takeda Pharmaceutical, AstraZeneca, Menarini, Reckitt Benckiser and Abbott Laboratory. He earned his Bachelor of Medicine and Bachelor of Surgery Degree (1993), and his Doctor of Medicine Degree (2000) from CUHK. Additionally, he attained Fellowships of the Royal College of Physicians of Edinburgh and London in 2007 and 2012 respectively, Fellowship of the Hong Kong College of Physicians in 2002, Fellowship of the Hong Kong Academy of Medicine in 2002, and has been an American Gastroenterological Association Fellow since 2012.
117
PROFESSOR DOUGLAS ARNER
Professor Douglas W. Arner is an Independent Non-Executive Director of Aptorum Group Limited. He is the Kerry Holdings Professor in Law at the University of Hong Kong and one of the world’s leading experts on financial regulation, particularly the intersection between law, finance and technology. At HKU, he is Faculty Director of the Faculty of Law’s LLM in Compliance and Regulation, LLM in Corporate and Financial Law and Law, Innovation, Technology and Entrepreneurship (LITE) Programmes. He is a Senior Visiting Fellow of Melbourne Law School, University of Melbourne, and an Executive Committee Member of the Asia Pacific Structured Finance Association. He led the development of the world’s largest massive open online course (MOOC): Introduction to FinTech, launched on edX in May 2018, now with over 35,000 learners spanning every country in the world. From 2006 to 2011, he was the Director of HKU’s Asian Institute of International Financial Law, which he co-founded in 1999, and from 2012 to 2018, he led a major research project on Hong Kong’s future as a leading international financial center. He was an inaugural member of the Hong Kong Financial Services Development Council, of which he was a member from 2013-2019. Douglas served as Head of the HKU Department of Law from 2011 to 2014 and as Co-Director of the Duke University-HKU Asia-America Institute in Transnational Law from 2005 to 2016. He has published fifteen books and more than 150 articles, chapters and reports on international financial law and regulation, including most recently Reconceptualising Global Finance and its Regulation (Cambridge 2016) (with Ross Buckley and Emilios Avgouleas). The RegTech Book (forthcoming 2019, with Janos Barberis and Ross Buckley). His recent papers are available on SSRN at https://papers.ssrn.com/sol3/cf_dev/AbsByAuth.cfm?per_id=524849, where he is among the top 150 authors in the world by total downloads.
Douglas has served as a consultant with, among others, the World Bank, Asian Development Bank, APEC, Alliance for Financial Inclusion, and European Bank for Reconstruction and Development, and has lectured, co-organized conferences and seminars and been involved with financial sector reform projects around the world. He has been a visiting professor or fellow at Duke, Harvard, the Hong Kong Institute for Monetary Research, IDC Herzliya, McGill, Melbourne, National University of Singapore, University of New South Wales, Shanghai University of Finance and Economics, and Zurich, among others. Since March 1, 2018, Professor Arner is the Senior Regulatory & Strategic Advisor of AENEAS CAPITAL LIMITED, a licensed corporation regulated by the Hong Kong Securities & Futures Commission as a Type 9 Asset Manager.
He holds a BA from Drury College (where he studied literature, economics and political science) in 1992, a JD (cum laude) from Southern Methodist University in 1995, an LLM (with distinction) in banking and finance law from the University of London (Queen Mary College) in 1996, and a PhD from the University of London in 2005.
Corporate Governance
As long as our officers and directors, either individually or in theaggregate, own at least 50% of the voting power of our Company, we will be a “controlled company” as defined under NASDAQ Marketplace Rules (specifically, as defined in Rule 5615(c)). We have no current intention to rely on the controlled company exemptions afforded to a controlled company under the NASDAQ Marketplace Rules.
Composition of Our Board of Directors
Our Board of Directors currently consists of seven members, all of whom were elected pursuant to our current Memorandum and Articles. Our nominating and governance committee and board of directors will consider a broad range of factors relating to the qualifications and background of nominees, which may include diversity and is not limited to race, gender or national origin. We have no formal policy regarding board diversity. Our nominating and governance committee’s and board of directors’ priority in selecting board members is identification of persons who will further the interests of our shareholders through his or her established record of professional accomplishment, the ability to contribute positively to the collaborative culture among board members, knowledge of our business, understanding of the competitive landscape and professional and personal experiences and expertise relevant to our growth strategy.
There is no Cayman Islands law requirement that a director must hold office for a certain term and stand for re-election unless the resolutions appointing the director impose a term on the appointment. The Memorandum and Articles provide that our directors will be elected annually to serve a term of one year, or until his or her earlier resignation or removal. We do not have any age limit requirements relating to our director’s term of office.
Our Memorandum and Articles also provide that our directors may be removed by the directors or ordinary resolution of the shareholders, and that any vacancy on our Board of Directors, including a vacancy resulting from an enlargement of our Board of Directors (which shall not exceed any maximum number stated therein), may be filled by ordinary resolution or by vote of a majority of our directors then in office.
118
Director Independence
Our Board of Directors has determined that Justin Wu, Mirko Scherer, Douglas Arner and Charles Bathurst are independent, as determined in accordance with the rules of the NASDAQ Global Market. In making such independence determination, our Board of Directors considered the relationships that each such non-employee director has with us and all other facts and circumstances that the board of directors deemed relevant in determining their independence, including the beneficial ownership of our share capital by each non-employee director and the transactions involving them described in the section titled “Transactions with Related Persons.” We believe that the composition and functioning of our Board of Directors and each of our committees comply with all applicable requirements of the NASDAQ Global Market and the rules and regulations of the SEC. There are no family relationships among any of our directors or executive officers.
Board’s Role in Risk Oversight
Our Board of Directors oversees the management of risks inherent in the operation of our business and the implementation of our business strategies. Our Board of Directors performs this oversight role by using several different levels of review. In connection with its reviews of our operations and corporate functions, our Board of Directors addresses the primary risks associated with those operations and corporate functions. In addition, our Board of Directors reviews the risks associated with our business strategies periodically throughout the year as part of its consideration of undertaking any such business strategies.
Each of our board committees also oversees the management of our risk that falls within the committee’s areas of responsibility. In performing this function, each committee has full access to management, as well as the ability to engage advisors. Our Chief Financial Officer reports to the audit committee and is responsible for identifying, evaluating and implementing risk management controls and methodologies to address any identified risks. In connection with its risk management role, our audit committee meets privately with representatives from our independent registered public accounting firm and our Chief Financial Officer. The audit committee oversees the operation of our risk management program, including the identification of the primary risks associated with our business and periodic updates to such risks, and reports to our Board of Directors regarding these activities.
Board Committees
Our Board of Directors has established an audit committee, a compensation committee and a nominating and corporate governance committee, each of which operates pursuant to a separate charter adopted by our Board of Directors. The composition and functioning of all of our committees will comply with all applicable requirements of the Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the NASDAQ Global Market and SEC rules and regulations. Our Board of Directors may establish other committees from time to time.
Audit Committee
Charles Bathurst, Douglas Arner and Justin Wu currently serve on the audit committee, which is chaired by Charles Bathurst. Our Board of Directors has determined that each member of the audit committee is “independent” for audit committee purposes as that term is defined in the rules of the SEC and the applicable rules of the NASDAQ Global Market. The audit committee’s responsibilities include:
| ● | selecting and appointing our independent registered public accounting firm, and approving the audit and permitted non-audit services to be provided by our independent registered public accounting firm; | |
| ● | evaluating the performance and independence of our independent registered public accounting firm; | |
| ● | monitoring the integrity of our financial statements and our compliance with legal and regulatory requirements as they relate to our financial statements or accounting matters; | |
| ● | reviewing the adequacy and effectiveness of our accounting and internal control policies and procedures; |
119
| ● | establishing procedures for the receipt, retention and treatment of accounting-related complaints and concerns; | |
| ● | reviewing and discussing with the independent registered public accounting firm the results of our year-end audit, and recommending to our Board of Directors, based upon such review and discussions, whether our financial statements shall be included in our Annual Report on Form 20-F; | |
| ● | reviewing all related party transactions for potential conflict of interest situations and approving all such transactions; and | |
| ● | reviewing the type and presentation of information to be included in our earnings press releases, as well as financial information and earnings guidance provided by us to analysts and rating agencies. |
Audit Committee Financial Expert
We have one financial expert as of the date hereof. Our Board of Directors has determined that Mr. Charles Bathurst, Chair of our audit committee, qualifies as an “audit committee financial expert” as defined in the SEC rules and satisfies the financial sophistication requirements of The NASDAQ Global Market.
Compensation Committee
Charles Bathurst, Douglas Arner and Justin Wu currently serve on the compensation committee, which is chaired by Justin Wu. Our Board of Directors has determined that each member of the compensation committee is “independent” as that term is defined in the applicable rules of the NASDAQ Global Market. The compensation committee’s responsibilities include:
| ● | reviewing the goals and objectives of our executive compensation plans, as well as our executive compensation plans in light of such goals and objectives; | |
| ● | evaluating the performance of our executive officers in light of the goals and objectives of our executive compensation plans and recommending to our Board of Directors with respect to the compensation of our executive officers; | |
| ● | reviewing the goals and objectives of our general compensation plans and other employee benefit plans as well as our general compensation plans and other employee benefit plans in light of such goals and objectives; | |
| ● | retaining and approving the compensation of any compensation advisors; | |
| ● | reviewing all equity-compensation plans to be submitted for shareholder approval under the NASDAQ listing rules, and reviewing and approving all equity-compensation plans that are exempt from such shareholder approval requirement; | |
| ● | evaluating the appropriate level of compensation for board and board committee service by non-employee directors; and | |
| ● | reviewing and approving description of executive compensation included in our Annual Report on Form 20-F. |
120
Nominating and Corporate Governance Committee
Charles Bathurst, Douglas Arner and Justin Wu currently serve on the nominating and corporate governance committee, which is chaired by Professor Arner. Our Board of Directors has determined that each member of the nominating and corporate governance committee is “independent” as that term is defined in the applicable rules of the NASDAQ Global Market. The nominating and corporate governance committee’s responsibilities include:
| ● | assisting our Board of Directors in identifying prospective director nominees and recommending nominees for election by the shareholders or appointment by our Board of Directors; | |
| ● | advising the board of directors periodically with respect to significant developments in the law and practice of corporate governance as well as our compliance with applicable laws and regulations, and making recommendations to our Board of Directors on all matters of corporate governance and on any corrective action to be taken; | |
| ● | overseeing the evaluation of our Board of Directors; and | |
| ● | recommending members for each board committee of our Board of Directors. |
Scientific Advisory Board
We are in the process of reorganizing the Scientific Assessment Committee into a newly formed Scientific Advisory Board. The Scientific Advisory Board shall help the Company sharpen its focus on innovation and technological advancements and address critical scientific challenges in our research and development; it will provide overall advise on the scientific development of the company. As of the date hereof, we only have one member of the Board – Dr. Keith Chan, but anticipate adding more members in the near future.
Code of Business Conduct and Ethics
Our board has adopted a code of business conduct and ethics that applies to our directors, officers and employees. A copy of this code is available on our website:www.aptorumgroup.com. We intend to disclose on our website or in a current report on Form 6-K, any amendments to the Code of Business Conduct and Ethics and any waivers of the Code of Business Conduct and Ethics that apply to our principal executive officer, principal financial officer, principal accounting officer, controller, or persons performing similar functions.
Duties of Directors
Under Cayman Islands law, our directors have a duty to act honestly, in good faith and with a view to our best interests. Our directors also have a duty to exercise the care, diligence and skills that a reasonably prudent person would exercise in comparable circumstances. (See “Description of Share Capital – Differences in Corporate Law”) In fulfilling their duty of care to us, our directors must ensure compliance with our Memorandum and Articles. We have the right to seek damages if a duty owed by our directors is breached.
The functions and powers of our Board of Directors include, among others:
| ● | appointing officers and determining the term of office of the officers; |
| ● | authorizing the payment of donations to religious, charitable, public or other bodies, clubs, funds or associations as deemed advisable; |
| ● | exercising the borrowing powers of the company and mortgaging the property of the company; |
| ● | executing checks, promissory notes and other negotiable instruments on behalf of the company; and |
| ● | maintaining or registering a register of mortgages, charges or other encumbrances of the company. |
121
Interested Transactions
So long as it does not adversely affect such person’s performance of duties or responsibilities to the Company and so long as it is not in direct competition with the Company and the Company’s business, no director or officer shall be disqualified by his office from contracting and/or dealing with the Company as vendor, purchaser or otherwise; nor shall any such contract or any contract or arrangement entered into by or on behalf of the Company in which any director or officer shall be in any way interested be or be liable to be avoided; nor shall any director or officer so contracting or being so interested be liable to account to the Company for any profit realized by any such contract or arrangement by reason of such director or officer holding that office or the fiduciary relationship thereby established. However, any such transaction that would reasonably be likely to affect a director status as an “Independent Director,” or that would constitute a “related party transaction” pursuant to the laws or rules promulgated by the SEC or the stock exchange on which our shares are then listed, shall require the review and approval of the Audit Committee. The nature of the director’s interest must be disclosed by him at the meeting of the directors at which the contract or arrangement is considered if his interest then exists, or in any other case, at the first meeting of the directors after the acquisition of his interest. A director, having disclosed his interest as aforesaid, shall not be counted in the quorum and shall refrain from voting as a director in respect of any contract or arrangement in which he is as interested as aforesaid.
A director must promptly disclose the interest to all other directors after becoming aware of the fact that he or she is interested in a transaction we have entered into or are to enter into. A general notice or disclosure to the board or otherwise contained in the minutes of a meeting or a written resolution of the board or any committee of the board that a director is a shareholder, director, officer or trustee of any specified firm or company and is to be regarded as interested in any transaction with such firm or company will be sufficient disclosure, and, after such general notice, it will not be necessary to give special notice relating to any particular transaction.
Qualification
The shareholding qualification for directors may be fixed by the Company in general meeting, and unless and until so fixed no qualification shall be required.
122
Compensation of Executive Officers and Directors
The following table sets forth all cash compensation paid by us, as well as certain other compensation paid or accrued, in fiscal 2018 to each of the following named executive officers. The total amount was $1.2 million in 2018. This amount does not include business travel, relocation, professional and business association dues and expenses reimbursed to such persons, and other benefits commonly reimbursed or paid by companies in our industry. No options were awarded in 2018.
| Name and Principal Position | Fiscal Year | Salary ($)(1) | Bonus ($) | Option Awards ($) | Non-Equity Incentive Plan Compensation ($) | Change in Pension Value and Nonqualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | ||||||||||||||||||||||
| Ian Huen(2) | 2018 | 276,923 | 23,077 | - | - | 2,308 | - | 302,308 | ||||||||||||||||||||||
| (CEO) | ||||||||||||||||||||||||||||||
| Darren Lui(3) | 2018 | 230,769 | 19,231 | - | - | 2,308 | - | 252,308 | ||||||||||||||||||||||
| (CBO, President) | ||||||||||||||||||||||||||||||
| Clark Cheng(4) | 2018 | 261,636 | - | - | - | 2,308 | 526 | (7) | 264,470 | |||||||||||||||||||||
| (CMO) | ||||||||||||||||||||||||||||||
| Keith Chan(5) | 2018 | 120,000 | (8) | - | - | - | - | - | 120,000 | |||||||||||||||||||||
| (CSO) | ||||||||||||||||||||||||||||||
| Sabrina Khan(6) | 2018 | 180,769 | 105,502 | - | - | 2,308 | - | 288,579 | ||||||||||||||||||||||
| (CFO) | ||||||||||||||||||||||||||||||
| (1) | The Appointment Letters provide salaries in HKD; for purposes of this table, we used a conversion ratio of HKD7.80 to USD1.00 to determine the salary in USD. |
| (2) | Mr. Huen is the founder and was appointed as the Chief Executive Officer of Aptorum Group on October 1, 2017. Before that, he was a director of the Company. |
123
| (3) | Mr. Lui was appointed as the Chief Business Officer and President of Aptorum Group on October 1, 2017 and resigned from such position on October 10, 2019. |
| (4) | Dr. Cheng was appointed as the Chief Medical Officer of Aptorum Group on January 2, 2018. |
| (5) | Dr. Chan served as the Company’s Chief Scientific Officer from August 18, 2017 until March 31, 2019. As of April 1, 2019, Dr. Chan serves us as a member of our Scientific Advisory Board. |
| (6) | Miss Khan was appointed as the Chief Financial Officer of Aptorum Group on October 16, 2017; as per an addendum to her appointment letter that we entered into with Miss Khan on April 24, 2018, her monthly salary increased to HKD122,500 as of April 1, 2018. |
| (7) | Pursuant to his appointment letter, Dr. Cheng also received a share bonus of 526 ordinary shares of AML, representing 5% of AML’s issued and outstanding ordinary shares (the “Share Bonus”). Based on the Company’s financial position and Dr. Cheng’s performance, on each anniversary of Dr. Cheng’s employment commencement date, the Share Bonus is eligible to increase by 1% of AML’s then issued and outstanding ordinary share count per year up to a maximum additional amount of 5% of AML’s then issued and outstanding ordinary share count by the 5th anniversary from his employment commencement date. As of the date hereof, Dr. Cheng received a total of 638 ordinary shares of AML, representing 6% of AML’s issued and outstanding ordinary shares; during fiscal 2018, Dr. Cheng received 526 ordinary shares of AML, the cash value of which is USD526. |
| (8) | As described elsewhere in this prospectus, we were party to a consulting agreement dated August 18, 2017 with GloboAsia, LLC, for which Dr. Chan serves as the Director of International Affairs. All fees payable to Dr. Chan for services provided to us as Chief Scientific Officer were paid to GloboAsia, LLC, pursuant to the consulting agreement and appointment letter with Dr. Chan. Following Dr. Chan’s resignation in March 2019, the consulting agreement was terminated effective as of March 31, 2019. (See “Transactions with Related Persons – Consulting Arrangements”) |
Effective as of January 1, 2019, the base salary of Mr. Huen, Mr. Lui and Dr. Cheng shall increase by 4%. The Board believes this increase is in line with the market average salary payable to persons in similar positions at similarly situated companies and general inflation. The Board also determined to issue Dr. Cheng a discretionary cash bonus equal to one-month of his base salary.
Dr. Lee has entered into an employment agreement, effective April 1, 2019 which amended his original agreement with the Company to reflect his new position as of Head of R&D. Other than his compensation, which shall now be USD242,667 per annum, all other terms of the original agreement remain the same.
Dr. Ng entered into an employment agreement effective April 1, 2019 which amended her original agreement with the Company to reflect her new position as of COO; other than her compensation, which shall now be USD104,000 per annum, all other terms of her original agreement remain the same.
124
Compensation of Directors
The following table sets forth information for the fiscal year ended December 31, 2018 regarding the compensation of our directors who at December 31, 2018, were not also named executive officers. No options were awarded in 2018.
| Name | Fees Earned or Paid in Cash ($) | Stock Awards ($) | Option Awards ($) | Non-Equity ($) | Non-qualified Deferred Compensation Earnings ($) | All Other Compensation ($) | Total ($) | |||||||||||||||||||||
| Charles Bathurst(1) | 47,880 | (2) | - | - | - | - | - | 47,880 | ||||||||||||||||||||
| Mirko Scherer(3) | 30,000 | - | - | - | - | - | 30,000 | |||||||||||||||||||||
| Justin Wu(4) | 30,000 | - | - | - | - | - | 30,000 | |||||||||||||||||||||
| Douglas Arner(5) | 25,000 | - | - | - | - | - | 25,000 | |||||||||||||||||||||
| (1) | Mr. Bathurst was appointed as one of our directors as of October 2017 and pursuant to his appointment letter, is entitled to receive $47,880 annually for his combined services as a director and a committee member. |
| (2) | Mr. Bathurst’s appointment Letter provides his salary in GBP. For purposes of this table, we used a conversion ratio of GBP0.75 to USD1.00 to determine his salary in USD; however, the ultimate amount paid is based on the actual rate as of the relevant pay day at the end of each month. |
| (3) | Dr. Scherer was appointed as one of our directors as of October 2017 and pursuant to his appointment letter, is entitled to receive $30,000 annually for his services as a director. |
| (4) | Dr. Wu was appointed as one of our directors as of October 2017 and pursuant to his appointment letter, is entitled to receive $30,000 annually for his combined services as a director and a committee member. |
| (5) | Professor Arner’s appointment as one of our directors became effective as of April 1, 2018. Pursuant to his appointment letter, Professor Arner is entitled to receive $30,000 annually for his combined services as a director and a committee member; he also received a signing bonus of $2,500. |
According to our Memorandum and Articles, the remuneration to be paid to the directors shall be such remuneration as the directors shall determine and as is in accordance with the Charter of the Compensation Committee, as applicable and the Company’s other corporate governance documents. Such remuneration shall be deemed to accrue from day to day. The directors also may be paid travelling, hotel and other expenses properly incurred by them in attending and returning from meetings of the directors or any committee of the directors or general meetings of the Company or in connection with the business of the Company or the discharge of their duties as a director, or receive a fixed allowance in respect thereof as may be determined by the directors from time to time or a combination of partly of one such method and partly the other. The directors may provide benefits, whether by the payment of gratuities or pensions or by insurance or otherwise, for any existing director or any director who has held but no longer holds any executive office or employment with the Company or with any entity which is or has been a subsidiary of the Company or a predecessor in business of the Company or of any such subsidiary, and for any member of his family (including a spouse and a former spouse) or any person who is or was dependent on him, and may (as well before as after he ceases to hold such office or employment) contribute to any fund and pay premiums for the purchase or provision of any such benefit.
Omnibus Incentive Plan
On October 13, 2017, we adopted the Option Plan. Under the Option Plan, up to an aggregate of 5,500,000 Class A Ordinary Shares (subject to subsequent adjustments described more fully below) may be issued pursuant to awards under the Option Plan. Subsequent adjustments include that on each January 1, starting with January 1, 2020, an additional number of shares equal to the lesser of (A) 2% of the outstanding number of Class A Ordinary Shares (on a fully diluted basis) on the immediate preceding December 31, and (B) such lower number of Class A Ordinary Shares as may be determined by the board of directors, subject in all cases to adjustments as provided in Section 10 of the Aptorum Group Limited 2017 Share Option Plan. Awards will be made pursuant to agreements and may be subject to vesting and other restrictions as determined by the board of directors.
We adopted the Option Plan to provide additional incentives to selected directors, officers, employees and consultants, and enable our Company to obtain and retain the services of these individuals. The Option Plan will enable us to grant options, restricted shares or other awards to our directors, employees and consultants. Awards will be made pursuant to agreements and may be subject to vesting and other restrictions as determined by the board of directors.
125
As of the date hereof, we have granted options that can be exercised for an aggregate of 218,610 Class A Ordinary Shares. These options were all granted on March 15, 2019. One-half of each option grant vests on January 1, 2020 and the other half vests on January 1, 2021. The exercise price is $12.91 per share, which was based on the closing price of the shares traded on the NASDAQ stock exchange on the trading day preceding the grant date.
Limitation on Liability and Other Indemnification Matters
The Companies Law does not limit the extent to which a company’s memorandum and articles of association may provide for indemnification of officers and directors, except to the extent any such provision may be held by the Cayman Islands courts to be contrary to public policy, such as to provide indemnification against civil fraud or the consequences of committing a crime. Our Memorandum and Articles permit indemnification of officers and directors for actions, proceedings, claims, losses, damages, costs, liabilities and expenses (“Indemnified Losses”) incurred in their capacities as such unless such Indemnified Losses arise from dishonesty of such directors or officers. This standard of conduct is generally the same as permitted under the Delaware General Corporation Law for a Delaware corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us under the foregoing provisions, we have been informed that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
TRANSACTIONS WITH RELATED PERSONS
The following discussion is a brief summary of certain material arrangements, agreements and transactions we have with related parties since January 1, 2016, other than the compensation and shareholding arrangements we describe in “Management” and “Principal Shareholders.” We also engage in other transactions with related parties that we do not perceive as material.
Line of Credit
On August 13, 2019 (the “Effective Date”), Aptorum Therapeutics Limited (“ATL”), entered into two separate Promissory Notes and Line of Credit Agreements (the “Agreements”) with AGL and Jurchen. The AGL Agreement and Jurchen Agreement provide ATL with a line of credit up to twelve million dollars ($12,000,000) and three million dollars ($3,000,000), respectively (collectively, the “Line of Credit”), representing the maximum aggregate amount of the advances of funds from the Line of Credit that may be outstanding at any time under the Line of Credit (the “Principal Indebtedness”). ATL may draw down from the Line of Credit at any time through the day immediately preceding the third anniversary of the Effective Date (the “Maturity Date”). Interest will be payable on the outstanding Principal Indebtedness at the rate of eight percent (8%) per annum, payable semi-annually in arrears on February 12 and August 12 in each year. ATL may pre-pay in whole or in part, the Principal Indebtedness of the Line of Credit, and all interest accrued at any time prior to the Maturity Date, without penalty. Under the Agreements, in addition to certain standard covenants, we are also not permitted, without the prior written consent of AGL and Jurchen to (i) liquidate, dissolve or wind-up our business and affairs; (ii) effect any merger or consolidation transaction; (iii) sell, lease, transfer, license or otherwise dispose, in a single transaction or series of related transactions, all or substantially all of our assets; or (iv) consent to any of the foregoing. The Agreements are subject to standard events of default, which if not cured within the agreed upon cure period, permits AGL or Jurchen, as applicable, to declare the outstanding Principal Indebtedness immediately due and payable, to exercise any other remedy provided for in the Agreements or any other right available to AGL or Jurchen as provided at law or in equity. Jurchen and AGL also maintain the right to set-off during the term of the Agreements. As of the date hereof, the Company has drawn down $0.9 million from the Line of Credit.
Sales and Purchases of Securities
Share Issuances
During the period of March 2017 through December 2017, we issued an aggregate of 2,207,025 Ordinary Share at a purchase price of approximately $3.90 per share, in a private placement we described as a “Series A” offering. Each investor of the Series A offering, in addition to a subscription agreement, signed a shareholder agreement, which set forth the basic governance terms of the Company, as well as our capital structure. The shareholders agreement was terminated in October 2017.
On October 13, 2017, ordinary resolutions were passed at an extraordinary general meeting of the Company approving: (i) converting 72,135,865 of authorized but unissued Ordinary Shares into 54,573,620 authorized but unissued Class A ordinary shares, par value of $1.00 per share (“Class A Ordinary Shares”) and 17,562,245 authorized but unissued Class B ordinary shares, par value of $1.00 per share (“Class B Ordinary Shares”), respectively; (ii) converting 24,930,839 Ordinary Shares held by three shareholders into an aggregate of 2,493,085 Class A Ordinary Shares and 22,437,754 Class B Ordinary Shares; and (iii) converting 2,933,296 Ordinary Shares held by 24 shareholders into an aggregate 2,933,296 Class A Ordinary Shares. Following these issuances, we had 27 shareholders of record.
KHE Holdings Limited, which is owned by Dr. Kenny Yu’s family, purchased $200,000 Series A Notes in our private Note offering, which closed on May 15, 2018; such notes automatically converted into 28,776 Class A Ordinary Shares upon the closing of the IPO.
A total of 5,504 shares were purchased in the IPO by related persons.
126
Share Transfer: Change in direct substantial shareholders of the Company
On May 4, 2017, Mr. Huen transferred all of the ordinary shares in the Company he owned (in the amount of 22,307,596) to Jurchen, a company incorporated in the British Virgin Islands and wholly-owned by Mr. Huen. On October 13, 2017, the ordinary shares held by Jurchen were redesignated as 2,230,760 Class A Ordinary Shares and 20,076,836 Class B Ordinary Shares.
On March 23, 2018, Jurchen transferred 446,152 Class A Ordinary Shares and 4,015,367 Class B Ordinary Shares to CGY Investments Limited, a company incorporated in Hong Kong and which we deem Mr. Darren Lui controls and/or of which he has substantial influence on the disposition rights and voting rights of such shares. Following this transfer, Jurchen owns approximately 33% and 72% of our Class A Ordinary Shares and Class B Shares, respectively.
Consulting Arrangements
With respect to the consulting agreements entered into between the Company and GloboAsia, LLC, Aeneas, and Covar Pharmaceuticals Incorporated (the “Consultants”), none of the Consultants are associated or affiliated with any FINRA members.
| 1. | GloboAsia, LLC |
We entered into a consulting agreement with GloboAsia effective as of August 18, 2017 (the “2017 GA Agreement”); GloboAsia is not associated or affiliated with any FINRA members. However, the 2017 GA Agreement was terminated when Dr. Chan resigned from his position as our Chief Scientific Officer in March 2019. Dr. Chan serves as the Director of International Affairs of GloboAsia.
Effective as of April 1, 2019, GloboAsia, through Dr. Chan, shall serve as a member on our Scientific Advisory Board. To formalize such service we entered into that certain consulting agreement with GloboAsia dated March 13, 2019 (the “2019 GA Agreement”). Pursuant to the 2019 GA Agreement, GloboAsia provides advisory and management services to us and as a member of the Scientific Advisory Board, they provide advice to us regarding research and development, the scientific merit of licenses or products and other related scientific issues. We agreed to pay GloboAsia an hourly rate of USD300 for work actually performed. The initial term of 2019 GA Agreement is until December 31, 2020 and shall thereafter be automatically renewed for successive one year terms, unless earlier terminated by either party upon three months’ notice prior to the end of the then applicable term; either party may also terminate the agreement upon 2 months written notice and the Company may terminate the agreement if Dr. Chan is no longer with GloboAsia or if GloboAsia commits any act of fraud or dishonesty.
| 2. | Aeneas |
a. We entered into a Management Agreement with Aeneas in October 2010, as amended (the “Management Agreement”). Our CEO, Mr. Huen is the sole beneficial owner and one of the executive directors of Aeneas, which is a Hong Kong incorporated licensed corporation regulated by the Securities & Futures Commission for asset management activities. Prior to the Restructure, Aeneas was the Company’s asset manager. Pursuant to the Management Agreement, we were to pay Aeneas certain management fees and performance fees. In February 2017, as part of the Restructure, the Management Agreement was terminated and no further fees were payable pursuant thereto. Prior to termination, we paid Aeneas an aggregate of $4.8 million pursuant to the terms of the Management Agreement.
b. In March 2017, we entered into a new Management Agreement with Aeneas (the “2017 Agreement”), pursuant to which Aeneas will provide certain management and administrative functions, as well as investment functions related to the Company, IP acquisitions and other investor relations services (the “Services”). In consideration for the Services, we agreed to pay Aeneas HK$500,000 per month (approximately US$64,103 per month), payable on the last day of each month. The 2017 Agreement was terminated in July 2018. Prior to the termination, we paid Aeneas an aggregate of $1.1 million pursuant to the terms of the 2017 Agreement.
c. On April 24, 2019, we signed aMaster Collaboration Agreementwith Aeneas and A*ccelerate Technologies Pte. Ltd. (See “Prospectus Summary – Master Collaboration Agreement”)
127
d. On January 1, 2019, Aptus Management Limited (one of our wholly-owned subsidiaries) (“Aptus Management”) entered into an Administrative consultant Services Agreement with Aeneas Management Limited (a subsidiary of Aeneas Limited). Pursuant to this agreement, Aeneas shall provide certain business and financial services to Aptus Management Limited; Aeneas shall be paid a monthly service fee of HK$452,000 per month (approximately US$57,949 per month), payable by the 25th day of each month during the term of the agreement, which is until December 31, 2019. The agreement may be terminated by either party providing 3-months written notice to the other party.
e. On January 1, 2019, Aenco Limited (“Aenco”) (a subsidiary of AGL) and Aptus Management entered into a Secondment Agreement. Pursuant to this agreement, Aenco shall assign certain of its employees to Aptus Management from time to time to assist Aptus Management with information technology development and maintenance activities for Aptus Management’s affiliates; such employees shall be integrated into Aptus Management’s organization only to the extent necessary to carry out such employees specific duties for Aptus Management. Aptus Management shall pay all salary and benefits up to HK$540,000 per month (approximately US$69,231 per month); Aenco shall be responsible for the costs associated with any employee relocation required as a result of this agreement. The agreement shall terminate on December 31, 2019, although either party may terminate the agreement upon giving the other party 3-months written notice.
f. In July 2019, Smart Pharmaceutical Limited Partnership, (“SPLP”), a wholly owned subsidiary of the Group, transferred 100,000,000 Smart Pharma Tokens (“SMPT token”) to Aenco Solutions Limited, a related party, in exchange of the service to deal with the token offering.
Aeneas is wholly-owned by Aeneas Group Limited (“AGL”), which in turn is wholly-owned by Aeneas Limited (“AL”). AL is 80% owned by Jurchen, which is wholly-owned by Mr. Huen, our CEO. Mr. Huen and Mr. Lui both serve as the executive directors of Aeneas and Professor Arner, one of our directors, is a Senior Regulatory and Strategic Advisor for Aeneas. Under his agreement with AGL dated March 12, 2018, Professor Arner shall, among other services, advise the board of AGL with its management, execution of business, and regulatory initiatives of AGL and AL, assist AGL with access to expert networks as appropriate and required. Professor Arner’s compensation thereunder is HK$234,000 per year (approximately US$30,000 per year) and Professor Arner is entitled to participate in AGL’s share option plans.
In addition, AGL was one of the selected dealers for our IPO.
3. Covar Pharmaceuticals Incorporated - Covar Pharmaceuticals Incorporated (“Covar”), an advisor, through a wholly-owned subsidiary, Videns, which is one of our project companies and is developing three of our projects.
Aptorum Therapeutics Limited and Covar entered into a Master Service Agreement on May 15, 2019 (the “Covar Agreement”), pursuant to which Covar shall provide certain program management services, consultancy and other related pharmaceutical research and development services, as more fully set forth in the Covar Agreement to Aptorum Therapeutics Limited. Covar received a $30,000 retainer for the services to be provided under the Covar Agreement. Additionally, Covar shall receive fees for their services in amounts to be determined by the parties at the time of each service; however, the Covar Agreement does mandate specific hourly rates for certain personnel and subcontractors ranging from $70-$250 per hour, depending on the service and expertise to be provided. Unless terminated earlier, the Covar Agreement shall have a three year term. Aptorum Therapeutics Limited may terminate the Covar Agreement at any time with 60 days’ prior written notice. Either party can terminate the Covar Agreement for a material breach of the Covar Agreement if such breach is not cured within 30 days’ notice of same, or 14 days’ notice in the event of a failure to pay for Covar’s services. The parties are entitled to terminate specific work orders too.
According to the Cover Agreement, all applicable records, notes, materials and other intellectual property or physical property created by Covar or its subcontractors (as defined in the Covar Agreement) used by Covar exclusively for Aptorum Therapeutics Limited (“Deliverables”) shall be deemed “works for hire” and the exclusive property of Aptorum Therapeutics Limited to the fullest extent possible. If any of Covar’s employees would be considered a co-inventor to any patents for Deliverables or inventions, such employees will be named as (co)-inventors in the patent application, where required by law, but Aptorum Therapeutics Limited will be the sole owner of such patents.
128
Previously, Covar had agreements with one of our subsidiaries, Videns, pursuant to which Covar would act as a product and clinical development advisor to Videns with respect to the development of contrast agents for MRI imaging applicable to the human brain (VLS-1). Pursuant to such agreements, two employees of Covar, Mr. Austin Freedman and Dr. Kwok Chow were to provide certain technical services and later, because the services provided by Covar, Mr. Freedman and Dr. Chow expanded beyond the original scope of services, we appointed Mr. Freedman and Dr. Chow as two of our Senior Clinical Development Managers, effective as of August 1, 2017 pursuant to an Appointment Letter and Addendum to Service Agreement with each of Dr. Chow and Mr. Freedman. All of these prior agreements between Covar and Videns, including the appointment letters with Mr. Freedman and Dr. Chow, have now terminated by their own terms. Pursuant to such agreements, Covar, Mr. Freedman and Dr. Chow received an aggregate of $524,000. Dr. Chow and Mr. Freedman may provide services under the current Covar Agreement and will be compensated for same.
Appointment Letters
We have entered into Appointment Letters with each of our executive officers. The terms of the Appointment Letters for each of our executive officers are consistent with each other, except with regard to the individual’s compensation, term of employment and duties and responsibilities, the latter of which coincides with the standard functions normally associated with the given position. Below, we set forth the specific compensation and term of employment terms of each of our executive officer’s appointment letter, as in effect as of the date hereof:
| ● | Ian Huen - Chief Executive Officer and Executive Director- US$24,000 (HKD187,200) per month payable in an equivalent amount of thirteen (13) months per calendar year with no set term of employment. | |
| ● | Darren Lui - President and Executive Director- US$20,000 (HKD156,000) per month payable in an equivalent amount of thirteen (13) months per calendar year with no set term of employment | |
| ● | Dr. Clark Cheng - Chief Medical Officer and Executive Director- US$23,275 (HKD181,542) per month payable in twelve (12) instalments per calendar year with no set term of employment. Dr. Cheng is also entitled to receive a share bonus of 5% of Aptorum Medical Limited’s ordinary shares upon commencement of employment, which shall be increased by 1% annually up to a maximum additional amount of 5% of issued ordinary share capital of Aptorum Medial Limited. The Board also determined to issue Dr. Cheng a discretionary cash bonus equal to one-month of his base salary. | |
| ● | Dr. Keith Chan – Scientific Advisory Board Member – Dr. Chan’s Appointment Letter is in connection with the Consultancy Agreement we entered into with Globo Asia LLC, for which Dr. Chan serves as Director of International Affairs. Pursuant to this agreement, we shall pay Globo Asia LLC $300 an hour for services Dr. Chan renders to us. The agreement terminates on December 31, 2020, subject to satisfactory performance on an annual basis as determined by the Company and shall thereafter automatically renew for successive one year terms until otherwise terminated. | |
| ● | Sabrina Khan - Chief Financial Officer- US$16,333 (HKD127,400) per month payable in an equivalent amount of twelve (12) months per calendar year with no set term of employment. | |
| ● | Dr. Thomas Lee Wai Yip – Head of Research & Development - US$18,667 (HKD145,600) per month payable in an equivalent amount of thirteen (13) months per calendar year with no set term of employment. | |
| ● | Dr. Angel Siu Yan Ng – Chief Operating Officer – US$8,000 (HKD 62,400) per month payable in an equivalent amount of thirteen (13) months per calendar year with no set term of employment. |
Remaining material terms of the appointment agreements are described below.
We may terminate employment for cause, at any time, without advance notice or remuneration, for certain acts of the executive officer, such as conviction or plea of guilty to a felony or any crime involving moral turpitude, negligent or dishonest acts to our detriment, or misconduct or a failure to perform agreed duties. We may also terminate an executive officer’s employment without cause upon three-month advance written notice. In such case of termination by us, we will provide severance payments to the executive officer as expressly required by applicable law of the jurisdiction where the executive officer is based. The executive officer may resign at any time with three-month advance written notice.
129
Each executive officer has agreed to hold, both during and after the termination or expiration of his or her Appointment Letter, in strict confidence and not to use, except as required in the performance of his or her duties in connection with the employment or pursuant to applicable law, any of our confidential information or trade secrets, any confidential information or trade secrets of our clients or prospective clients, or the confidential or proprietary information of any third-party received by us and for which we have confidential obligations.
In addition, each executive officer has agreed to be bound by non-solicitation and non-compete restrictions during the term of his or her employment and typically for one year following the last date of employment. Specifically, each executive officer has agreed not to (i) solicit or entice away from the Company, any person, firm, company or organization that is or shall have been at any time within 12 months prior to termination of employee a customer, client, identified prospective customer or client of the Company or in the habit of dealing with the Company; (ii) employ, solicit or entice away from the Company any person who is or shall have been on the date of or within 12 months prior to termination of employment an employee of the Company; or (iii) assume employment with or provide services to, or otherwise engage in income generating activities with any of our competitors, or engage, whether as principal, partner, licensor or otherwise, any of our competitors, without our express consent.
Some of our Appointment Letters also provide for the executive officer to participate in our mandatory provident fund, which is similar to a pension fund.
Leased Facilities
Our lease for our office at Guangdong Investment Tower is a Sub-Tenancy Agreement between Jurchen Investment Corporation and Aptus Management Limited, which is one of our wholly-owned subsidiaries.
Other Relationships
As stated elsewhere in this prospectus, Dr. Cheng serves as our Chief Medical Officer and one of our Executive Directors, who is also an Executive Director of Aptorum Medical. Dr. Cheng is also the guarantor on the AML Lease.
The Series A Note Offering
On May 15, 2018, we closed a private financing with certain investors (the “Series A Note Investors”) who purchased an aggregate of $1,600,400 Series A convertible notes, at a purchase price of $10,000 per note (the “Series A Notes”), pursuant to a note purchase agreement. Some of the Series A Note Investors are either affiliates of the Company or “related persons,” as such term is defined in Item 404 of Regulation S-K (See “Transactions with Related Persons”). We refer to this private placement transaction as the “Series A Note Offering.” The Series A Note Investors entered into a lock-up agreement, pursuant to which they agreed not to sell or otherwise transfer or dispose the Series A Notes or the Class A Ordinary Shares underlying the Series A Notes during the six-month period commencing on the date our Class A Ordinary Shares commence trading on NASDAQ Global Market, which has now expired. The Series A Notes automatically converted into 230,252 Class A Ordinary Shares at the closing of the IPO and at the commencement of trading our Class A Ordinary Shares on NASDAQ Global Market at a conversion price equal to a 56% discount to the actual price per Class A Ordinary Share (“Conversion Price”). Accordingly, the Series A Notes converted into, and we issued an aggregate of 230,252 shares of Class A Ordinary Shares after the IPO closed.
One of the underwriters in the IPO also served as a placement agent for the Series A Note Offering and received: (i) a cash success fee of $68,516 and (ii) warrants to purchase 12,663 Class A Ordinary Shares, at an exercise price of $6.95 per share, subject to adjustment (the “Series A Note PA Warrants”). All of the Series A Note PA Warrants have now been exercised on a cashless basis.
130
The issuance and sale of Series A Notes, and the underlying Class A Ordinary Shares to the Series A Note Investors in the Series A Note Offering were made in reliance on an exemption from registration contained in either Regulation D or Regulation S of the Securities Act of 1933, as amended (the “Securities Act”). The securities sold in the Series A Note Offering are not registered by the registration statement and have not been registered under the Securities Act, and may be offered or sold only pursuant to an effective registration statement or pursuant to an available exemption from the registration requirements of the Securities Act. However, the Series A Note Investors have piggyback registration rights with respect to the Class A Ordinary Shares underlying the Series A Notes that entitle the Series A Note Investors to request their securities be included in a future Securities Act registration statement, after our IPO, subject to certain exceptions and conditions. However, we decided to include the Class A Ordinary Shares underlying the Series A Notes in the registration statement for our IPO.
Employment Agreements
See “Appointment Letters” above.
PRINCIPAL AND SELLING SHAREHOLDERS
The following table sets forth information with respect to the beneficial ownership of our Class A Ordinary Shares (including Class A Ordinary Shares issuable upon the conversion of outstanding Class B Ordinary Shares), subject to certain assumptions set forth in the footnotes and as adjusted to reflect the sale of our Class A Ordinary Shares offered in this Offering for:
| ● | each person or group of affiliated persons known by us to own beneficially 5% or more of our outstanding Class A Ordinary Shares or Class B Ordinary Shares; |
| ● | each of our directors and named executive officers individually; and |
| ● | all of our executive officers and directors as a group. |
The beneficial ownership of our Class A Ordinary Shares is determined in accordance with the rules of the SEC and generally includes any shares over which a person exercises sole or shared voting or investment power, and includes the Class A Ordinary Shares issuable upon the conversion of the outstanding Class B Ordinary Shares and the Class A Ordinary Shares issuable pursuant to share options that are exercisable within 60 days of the date of this prospectus. Class A Ordinary Shares issuable pursuant to share options are deemed outstanding for computing the percentage of the person holding such options but are not outstanding for computing the percentage of any other person. As of the date of this prospectus, there were no Class A Ordinary Shares issuable pursuant to share options exercisable within 60 days thereof.
Beneficial ownership includes voting or investment power with respect to the securities. Except as indicated below, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all Ordinary Shares shown as beneficially owned by them. Percentage of beneficial ownership of each listed person is based on 6,597,362 Class A Ordinary Shares and 22,437,754 Class B Ordinary Shares outstanding as of October 31, 2019.
131
Except where otherwise indicated, we believe, based on information furnished to us by such owners, that the beneficial owners of the Class A Ordinary Shares and Class B Ordinary Shares listed below have sole investment and voting power with respect to such shares.
| Name and Address of Beneficial Owner | Class A Ordinary Shares Beneficially Owned | Class B Ordinary Shares Beneficially Owned | Percentage of Total Class A and Class B Ordinary Shares(1) | Percentage of Total Voting Power(2) | ||||||||||||
| Ian Huen(3) | 1,774,608 | 16,061,469 | 61.43 | % | 70.31 | % | ||||||||||
| Darren Lui(4) | 250,755 | 2,141,333 | 8.24 | % | 9.38 | % | ||||||||||
| Clark Cheng(5) | - | - | - | - | ||||||||||||
| Sabrina Khan(6) | * | - | * | * | ||||||||||||
| Thomas Lee Wai Yip(7) | * | - | * | * | ||||||||||||
| Angel Ng Siu Yan(8) | * | - | * | * | ||||||||||||
| Charles Bathurst(9) | - | - | - | - | ||||||||||||
| Mirko Scherer(10) | - | - | - | - | ||||||||||||
| Justin Wu(11) | 206,560 | - | 0.71 | % | 0.09 | % | ||||||||||
| Douglas Arner(12) | - | - | - | - | ||||||||||||
| All directors and executive officers as a group (10 persons) | 2,231,923 | 18,202,802 | 70.38 | % | 79.77 | % | ||||||||||
| 5% Beneficial Owner | ||||||||||||||||
| Jurchen Investment Corporation(3) | 1,774,608 | 16,061,469 | 61.43 | % | 70.31 | % | ||||||||||
| Sui Fong Isabel Huen Ng(3) | 211,986 | 1,907,870 | 7.30 | % | 8.35 | % | ||||||||||
| CGY Investments Limited(13) | 471,809 | 4,015,367 | 15.45 | % | 17.59 | % | ||||||||||
| * | Less than 1%. |
| (1) | For each person and group included in this column, percentage ownership is calculated by dividing the number of Class A Ordinary Shares and Class B Ordinary Shares beneficially owned by such person or group, including shares that such person or group has the right to acquire within 60 days after the date of this prospectus, by the sum of Class A Ordinary Shares and Class B Ordinary Shares, and the number of Class A Ordinary Shares that such person or group has the right to acquire beneficial ownership within 60 days after the date of this prospectus, 2019. Following the IPO, each Class B Ordinary Share can be converted at any time on a one-for-one basis into Class A Ordinary Shares at the discretion of the holder. |
| (2) | For each person and group included in this column, percentage of total voting power represents voting power based on both Class A Ordinary Shares and Class B Ordinary Shares beneficially owned by such person or group with respect to all of our outstanding Class A Ordinary Shares and Class B Ordinary Shares as one single class. Holders of Class A Ordinary Shares are entitled to one vote per share and holders of Class B Ordinary Shares are entitled to ten votes per share on all matters subject to a shareholders’ vote. |
| (3) | Includes 1,774,608 Class A Ordinary Shares and 16,061,469 Class B Ordinary Shares held by Jurchen Investment Corporation, a company wholly-owned by Mr. Huen. Mr. Huen maintains sole voting control over the shares held by Jurchen, the principal office address of which is at 17th Floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong. Does not include 20,107 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Mr. Huen pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (4) | Includes (i) 14,850 Class A Ordinary Shares and 133,649 Class B Ordinary Shares held by DSF Investment Holdings Limited, which is 29.5% held by Mr. Lui and 70.5% held by Eternal Clarity Holdings Limited, which is wholly-owned by Mr. Lui’s mother, Ms. Emily Woo, and is located at Flat A2, 11th Floor, Wing Hang Insurance Building, 11 Wing Kut Street, Hong Kong and (ii) 235,905 Class A Ordinary Shares and 2,007,684 Class B Ordinary Shares held by CGY Investments Limited, which is 50% held by Seng Fun Yee (Mr. Lui’s spouse), 25% held by Mandy Lui (Mr. Lui’s sister) and 25% held by Adrian Lui (Mr. Lui’s brother). Mr. Lui only controls and/or has substantial influence on the disposition and voting rights of 29.5% of the Aptorum shares DSF owns; Mr. Lui controls and/or has substantial influence on the disposition and voting rights of the shares held by his spouse, but no such control over the shares held by his sister or brother regarding the CGY shares. Does not include 20,107 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Mr. Lui pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
132
| (5) | Dr. Cheng does not directly own any Company shares; however, pursuant to his appointment letter, Dr. Cheng received a stock bonus of 6% of Aptorum Medical Limited’s ordinary shares as of the date hereof. Does not include 20,107 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Dr. Cheng pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (6) | Does not include 9,498 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Miss Khan pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (7) | Does not include 20,107 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Dr. Lee pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (8) | Does not include 1,551 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Dr. Ng pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (9) | Does not include 2,011 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Mr. Bathurst pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (10) | Does not include 2,011 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Dr. Scherer pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (11) | Includes (i) 129,589 Class A Ordinary Shares held by Chi Ling Lily Heung, the wife of Dr. Wu and (ii) 76,971 Class A Ordinary Shares held by Dr. Wu. Does not include 2,011 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Dr. Wu pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (12) | Does not include 2,011 Class A Ordinary Shares issuable upon exercise of outstanding options issued on March 15, 2019 to Professor Arner pursuant to the Option Plan, since such options have not vested and will not be exercisable within 60 days of the date of this prospectus. |
| (13) | CGY Investments Limited is 50% held by Seng Fun Yee (Mr. Lui’s spouse), 25% held by Mandy Lui (Mr. Lui’s sister) and 25% held by Adrian Lui (Mr. Lui’s brother). Mr. Lui controls and/or has substantial influence on the disposition and voting rights of the shares held by his spouse, but no such control over the shares held by his sister or brother. |
133
Selling Shareholders
We are registering for resale our certain Class A Ordinary Shares issued in private transactions (the “Resale Shares”). The securities listed herein were issued in accordance with the exemption from the registration provisions of the Securities Act of 1933, as amended, provided by Section 4(a)(2) of such Act for issuances not involving any public offering and Rule 506 of Regulation D promulgated thereunder and/or Regulation S promulgated thereunder. We are registering the shares to permit the Selling Shareholders and their pledgees, donees, transferees and other successors-in-interest that receive their shares from a Selling Shareholder as a gift, partnership distribution or other non-sale related transfer after the date of this prospectus to resell the shares when and as they deem appropriate in the manner described in the “Plan of Distribution.” As of the date hereof, there are 6,597,362 Class A Ordinary Shares and 22,437,754 Class B Ordinary Shares issued and outstanding.
The following table sets forth:
| ● | the name of the Selling Shareholders; | |
| ● | the number of our Class A Ordinary Shares that the Selling Shareholders beneficially owned prior to the Offering for resale of the shares under this prospectus; | |
| ● | the maximum number of our Class A Ordinary Shares that may be offered for resale for the account of the Selling Shareholders under this prospectus, including the Class A Ordinary Shares underlying such Selling Shareholder’s Class B Ordinary Shares, if any, since each Class B Ordinary Share can be converted at any time on a one-for-one basis into Class A Ordinary Share at the discretion of the holder (See “Description of Share Capital”); and | |
| ● | the number and percentage of our Class A Ordinary Shares beneficially owned by the Selling Shareholders after the Offering of the shares (assuming all of the offered shares are sold by the Selling Shareholders), is based on 29,035,116 Class A Ordinary Shares outstanding as of the date hereof. |
Except as noted in the footnotes below, we have not had a material relationship with any of the Selling Shareholders within the last three years and all of the Selling Shareholders named below received their securities in connection with private transactions.
None of the Selling Shareholders is a broker dealer or an affiliate of a broker dealer. None of the Selling Shareholders has any agreement or understanding to distribute any of the shares being registered.
Each Selling Shareholder may offer for sale all or part of the shares from time to time. The table below assumes that the Selling Shareholders will sell all of the shares offered for resale. A Selling Shareholder is under no obligation, however, to sell any shares pursuant to this prospectus.
Unless otherwise noted, the address for all Selling Shareholders is 17th Floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong.
| Name of Selling Shareholder | Class A Ordinary Shares Beneficially Owned Prior to Offering | Maximum Number of Class A Ordinary Shares to be Sold | Number of Class A Ordinary Shares Owned After Offering(1) | Percentage Ownership After Offering (2) | ||||||||||
| Jurchen Investment Corporation(3) | 17,836,077 | 17,836,077 | 0 | * | ||||||||||
| Diocesan Boy’s School Foundation Limited(4) | 10,000 | 10,000 | 0 | * | ||||||||||
| Sui Fong Isabel Huen Ng(5) | 2,119,856 | 2,119,856 | 0 | * | ||||||||||
| Kevin Haoyu Sun | 760,484 | 760,484 | 0 | * | ||||||||||
| DSF Investment Holdings Limited(6) | 503,387 | 503,387 | 0 | * | ||||||||||
| KHE Holdings Limited(7) | 335,979 | 256,571 | 79,408 | * | ||||||||||
| Long Boom Ventures Limited(8) | 192,014 | 192,014 | 0 | * | ||||||||||
| Dennis Saihong Lam | 166,771 | 166,771 | 0 | * | ||||||||||
| Heung, Chi Ling Lily(9) | 129,589 | 128,285 | 1,304 | * | ||||||||||
| Wong Shui Yan Matthew | 98,314 | 98,314 | 0 | * | ||||||||||
| Wu, Che Yuen Justin(10) | 76,971 | 76,971 | 0 | * | ||||||||||
| Eton Finance Services Limited(11) | 51,314 | 51,314 | 0 | * | ||||||||||
| Yu Chengen | 51,314 | 51,314 | 0 | * | ||||||||||
| Chak Tung Wong(12) | 38,485 | 38,485 | 0 | * | ||||||||||
| Widevision Electronics Limited(13) | 32,841 | 32,841 | 0 | * | ||||||||||
| Shih Yiu Man Veronica | 25,657 | 25,657 | 0 | * | ||||||||||
| CGY Investments Limited(14) | 4,487,176 | 4,487,176 | 0 | * | ||||||||||
| Chan Kin Wai(15) | 43,597 | 25,657 | 17,940 | * | ||||||||||
| Chan She Sam(16) | 33,597 | 25,657 | 7,940 | * | ||||||||||
| Adrian Hassan | 25,657 | 25,657 | 0 | * | ||||||||||
| David Lorne Levy | 25,657 | 25,657 | 0 | * | ||||||||||
| Jeffrey Hartman Zielinski | 25,657 | 25,657 | 0 | * | ||||||||||
| Yau Chung Wai | 20,525 | 20,525 | 0 | * | ||||||||||
| Chit Ming Chng | 12,828 | 12,828 | 0 | * | ||||||||||
| Wu Ka Kei(17) | 10,262 | 10,262 | 0 | * | ||||||||||
| Ho Ko(18) | 10,262 | 10,262 | 0 | * | ||||||||||
| Marvel Link Limited(19) | 748,142 | 748,142 | 0 | * | ||||||||||
| * | Represents beneficial ownership of less than one percent of our outstanding shares (assuming all of the offered shares are sold by the Selling Shareholders). |
134
| (1) | Since we do not have the ability to control how many, if any, of their shares each of the Selling Shareholders will sell, we have assumed that the Selling Shareholders will sell all of the shares offered herein for purposes of determining how many shares they will own after the Offering and their percentage of ownership following the Offering. |
| (2) | All percentages have been rounded up to the nearest one hundredth of one percent. |
| (3) | Includes (i) 1,784,608 Class A Ordinary Shares and 16,061,469 Class B Ordinary Shares held by Jurchen Investment Corporation, a company wholly-owned by Mr. Huen. Mr. Huen maintains sole voting control over the shares held by Jurchen, the principal office address of which is at 17th Floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong. |
| (4) | The person having voting, dispositive or investment powers over Diocesan Boy’s School Foundation Limited is its board of directors. |
| (5) | Includes (i) 211,986 Class A Ordinary Shares and 1,907,870 Class B Ordinary Shares held by Sui Fong Isabel Huen Ng, the mother of Mr. Huen. |
| (6) | Includes (i) 50,339 Class A Ordinary Shares and 453,048 Class B Ordinary Shares held by DSF Investment Holdings Limited, which is owned by Mr. Lui (29.5%) and Eternal Clarity Holdings Limited (70.5%), which is wholly-owned by Mr. Lui’s mother, Ms. Emily Woo. The address for DSF Investment Holdings Limited is Flat A2, 11th Floor, Wing Hang Insurance Building, 11 Wing Kut Street, Hong Kong. |
| (7) | Includes (i) 28,776 Class A Ordinary Share KHE received upon the automatic conversion of its Series A Note and 50,632 Class A Ordinary Shares the shareholder purchased in the IPO, all of which are registered on our registration statement on Form F-1 (File No. 333-227198), which was declared effective by the SEC on December 3, 2018 and (ii) 256,571 Class A Ordinary Shares received in private transaction and which we are seeking to register pursuant hereto. The person having voting, dispositive or investment powers over KHE Holdings Limited is Yu Kuen Ying Denny and Choi Myung Sung Teresa, family members to Dr. Yu. Dr. Yu is a member of our Scientific Assessment Committee. The address for KHE Holdings Limited is Unit 1310, 13/F, Capital Centre, 151 Gloucester Road, Hong Kong. |
| (8) | The person having voting, dispositive or investment powers over Long Boom Ventures Limited is Kenrick Fok, a director of Aptus Management Limited and SMTPH Limited, which is the general partner of Smart Pharmaceutical Limited Partnership and one of our wholly-owned subsidiaries. The address for Long Boom Ventures Limited is PO Box 957, Offshore Incorporations Centre, Road Town, Tortola, British Virgin Islands. |
| (9) | Ms. Heung is the wife of one of our directors, Dr. Wu. |
| (10) | This is one of our directors. |
| (11) | The person having voting, dispositive or investment powers over Eton Finance Services Limited is Eliza R. Valtos. The address for Eton Finance Services Limited is c/o ICCP, 17th Floor, Robinsons Summit Center, 6783 Ayala Avenue, Makati City 1226, Philippines. |
| (12) | Mr. Wong is the father of Dr. Sunny Wong Hei, a member of the Company’s Scientific Assessment Committee. |
| (13) | The person having voting, dispositive or investment powers over Widevision Electronics Limited is Mr. Hui Ka Ming. The address for Widevision Electronics Limited is Flat/Rm E, Block 3, Camelpaint Building, 60 Hoi Yuen Road, Kwun Tong, Hong Kong. |
| (14) | Includes (i) 471,809 Class A Ordinary Shares and 4,015,367 Class B Ordinary. CGY Investments is 50% held by Seng Fun, Mr. Lui’s spouse, 25% held by Mandy, Mr. Lui’s sister and 25% held by Adrian, Mr. Lui’s brother. Due to the spousal relationship, Mr. Lui is deemed to control and/or have substantial influence on the disposition rights and voting rights over the shares held by Seng Fun based on Section 13(d) of the Exchange Act, but Mr. Lui does not have such control and/or have substantial influence on the disposition rights and voting rights over the shares held by his sister or brother. The principal business address of the CGY Investments is Unit A 3/F Cheong Sun Tower, 116-118 Wing Lok St, Sheung Wan, Hong Kong. |
| (15) | Includes (i) 2,877 Class A Ordinary Shares the shareholder received upon the automatic conversion of its Series A Note and 5,063 Class A Ordinary Shares the shareholder purchased in the IPO, all of which are registered on our registration statement on Form F-1 (File No. 333-227198), which was declared effective by the SEC on December 3, 2018 and (ii) 25,657 Class A Ordinary Shares received in private transaction and which we are seeking to register pursuant hereto. |
| (16) | Includes (i) 2,877 Class A Ordinary Shares the shareholder received upon the automatic conversion of its Series A Note and 5,063 Class A Ordinary Shares the shareholder purchased in the IPO, all of which are registered on our registration statement on Form F-1 (File No. 333-227198), which was declared effective by the SEC on December 3, 2018 and (ii) 25,657 Class A Ordinary Shares received in private transaction and which we are seeking to register pursuant hereto. Chan She Sam is the father of Jason Chan, a member of our Scientific Assessment Committee. |
| (17) | This is a member of our Scientific Assessment Committee. |
| (18) | This is a member of our Scientific Assessment Committee. |
| (19) | The person having voting, dispositive or investment powers over Marvel Link Limited is Mr. Michael Hamilton TIMSO. The address for Marvel Link Limited is 21/F, 1 Lan Kwai Fong, Central, Hong Kong. |
135
The Selling Shareholders and any of their pledgees, donees, transferees, assignees and successors-in-interest may, from time to time, sell any or all of their Resale Shares at prevailing market prices or privately negotiated prices. The Selling Shareholders may use any one or more of the following methods when selling Resale Shares:
| ● | ordinary brokerage transactions and transactions in which the broker-dealer solicits investors; | |
| ● | block trades in which the broker-dealer will attempt to sell the Class A Ordinary Shares as agent but may position and resell a portion of the block as principal to facilitate the transaction; | |
| ● | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; | |
| ● | an exchange distribution in accordance with the rules of the applicable exchange; | |
| ● | privately negotiated transactions; | |
| ● | to cover short sales made after the date that the registration statement, to which this prospectus is a part, is declared effective by the SEC; | |
| ● | broker-dealers may agree with the Selling Shareholders to sell a specified number of such Resale Shares at a stipulated price per share; | |
| ● | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; | |
| ● | a combination of any such methods of sale; and | |
| ● | any other method permitted pursuant to applicable law. |
The Selling Shareholders may also sell Resale Shares under Rule 144 under the Securities Act, if all of the conditions in Rule 144(i)(2) are satisfied at the time of the proposed sale, rather than under this prospectus.
In connection with the sale of the Resale Shares or interests therein, the Selling Shareholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the Resale Shares in the course of hedging the positions they assume. The Selling Shareholders may also sell the Resale Shares short and deliver these securities to close out their short positions, or loan or pledge the Resale Shares to broker-dealers that in turn may sell these securities. The Selling Shareholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of Resale Shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
Broker-dealers engaged by the Selling Shareholders may arrange for other brokers-dealers to participate in sales. Broker-dealers may receive commissions or discounts from the Selling Shareholders (or, if any broker-dealer acts as agent for the purchaser of shares, from the purchaser) in amounts to be negotiated. The Selling Shareholders do not expect these commissions and discounts to exceed what is customary in the types of transactions involved.
The Selling Shareholders may from time to time pledge or grant a security interest in some or all of the Resale Shares owned by them and, if they default in the performance of their secured obligations, the amendment or supplement to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act of 1933 will be filed amending the list of Selling Shareholders to include the pledgee, transferee or other successors in interest as Selling Shareholders under this prospectus and the pledgees or secured parties may offer and sell Resale Shares from time to time under the supplement or amendment to this prospectus.
136
The Selling Shareholders also may transfer the Resale Shares in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
The Selling Shareholders and any broker-dealers or agents that are involved in selling the Resale Shares may be deemed to be “underwriters” within the meaning of the Securities Act in connection with such sales. In such event, any commissions received by such broker-dealers or agents and any profit on the resale of the Resale Shares purchased by them may be deemed to be underwriting commissions or discounts under the Securities Act. Discounts, concessions, commissions and similar selling expenses, if any, that can be attributed to the sale of Resale Shares will be paid by the Selling Shareholder and/or the purchasers. Each Selling Shareholder has represented and warranted to the Company that it acquired the securities subject to the registration statement, to which this prospectus forms a part, in the ordinary course of such Selling Shareholder’s business and, at the time of its purchase of such securities such Selling Shareholder had no agreements or understandings, directly or indirectly, with any person to distribute any such securities.
FINRA Rule 5110 requires FINRA member firms (unless an exemption applies) to satisfy the filing requirements of Rule 5110 in connection with the resale, on behalf of Selling Shareholders, of the securities on a principal or agency basis. NASD Notice to Members 88-101 states that in the event a Selling Shareholder intends to sell any of the shares registered for resale in this prospectus through a member of FINRA participating in a distribution of our securities, such member is responsible for insuring that a timely filing, if required, is first made with the Corporate Finance Department of FINRA and disclosing to FINRA the following:
| ● | it intends to take possession of the registered securities or to facilitate the transfer of such certificates; | |
| ● | the complete details of how the Selling Shareholders’ shares are and will be held, including location of the particular accounts; | |
| ● | whether the member firm or any direct or indirect affiliates thereof have entered into, will facilitate or otherwise participate in any type of payment transaction with the Selling Shareholders, including details regarding any such transactions; and | |
| ● | in the event any of the securities offered by the Selling Shareholders are sold, transferred, assigned or hypothecated by any Selling Shareholder in a transaction that directly or indirectly involves a member firm of FINRA or any affiliates thereof, that prior to or at the time of said transaction the member firm will timely file all relevant documents with respect to such transaction(s) with the Corporate Finance Department of FINRA for review. |
We have advised each Selling Shareholder that it may not use shares registered on the registration statement, to which this prospectus forms a part, to cover short sales of Resale Shares made prior to the date on which the registration statement, to which this prospectus forms a part, shall have been declared effective by the SEC. If a Selling Shareholder uses this prospectus for any sale of the Resale Shares, it will be subject to the prospectus delivery requirements of the Securities Act. The Selling Shareholders will be responsible to comply with the applicable provisions of the Securities Act and Exchange Act, and the rules and regulations thereunder promulgated, including, without limitation, Regulation M, as applicable to such Selling Shareholders in connection with resales of their respective Resale Shares under the registration statement, to which this prospectus forms a part.
We are required to pay all fees and expenses incident to the registration of the Resale Shares, but the Company will not receive any proceeds from the sale of the Resale Shares. The Company has agreed to indemnify the Selling Shareholders against certain losses, claims, damages and liabilities, including liabilities under the Securities Act.
137
We are a Cayman Islands exempted company with limited liability and our affairs are governed by our Memorandum and Articles, the Companies Law, the common law of the Cayman Islands, our corporate governance documents and rules and regulations of the stock exchange on which are shares are traded.
As of the date hereof, the authorized share capital of the Company is $100,000,000, consisting of 60,000,000 Class A Ordinary Shares, par value $1.00 each and 40,000,000 Class B Ordinary Shares, par value $1.00 each. As of the date hereof, 6,597,362 Class A Ordinary Shares and 22,437,754 Class B Ordinary Shares are issued and outstanding. All of our issued and outstanding Class A Ordinary Shares and Class B Ordinary Shares are fully paid.
Ordinary Shares
The following are summaries of material provisions of our Memorandum and Articles, corporate governance policies and the Companies Law insofar as they relate to the material terms of our Class A Ordinary Shares and Class B Ordinary Shares.
Objects of Our Company
Under our Memorandum and Articles, the objects of our Company are unrestricted and we have the full power and authority to carry out any object not prohibited by the law of the Cayman Islands.
Share Capital
Our authorized share capital is divided into Class A Ordinary Shares and Class B Ordinary Shares. Holders of our Class A Ordinary Shares and Class B Ordinary Shares will have the same rights except for voting rights and conversion rights.
The holders of Class A Ordinary Shares are entitled to one vote for each such share held and shall be entitled to notice of any shareholders’ meeting, and, subject to the terms of Memorandum and Articles, to vote thereat. The Class A Ordinary Shares are not redeemable at the option of the holder and are not convertible into shares of any other class.
The holders of Class B Ordinary Shares shall have the right to ten votes for each such share held, and shall be entitled to notice of any shareholders’ meeting and, subject to the terms of the Memorandum and Articles, to vote thereat. The Class B Ordinary Shares are not redeemable at the option of the holder but are convertible into Class A Ordinary Shares at any time after issue at the option of the holder on a one to one basis.
Dividends
The holders of our Class A Ordinary Shares and Class B Ordinary Shares are entitled to such dividends as may be declared by our Board of Directors subject to the Companies Law and to our Memorandum and Articles.
Voting Rights
In respect of all matters subject to a shareholders’ vote, each Class B Ordinary Share is entitled to ten votes, and each Class A Ordinary Share is entitled to one vote, voting together as one class. Voting at any shareholders’ meeting is by show of hands unless a poll is demanded by the chairman or persons holding certain amounts of shares as set forth in the Memorandum and Articles. Actions that may be taken at a general meeting also may be taken by a unanimous resolution of the shareholders in writing.
No business shall be transacted at any general meeting unless a quorum of members is present at the time when the meeting proceeds to business; two members present in person or by proxy, one of whom shall be the holder of the majority of the shares in the Company, shall be a quorum provided always that if the Company has one member of record the quorum shall be that one member present in person or by proxy. An ordinary resolution to be passed at a general meeting requires the affirmative vote of a simple majority of the votes cast, while a special resolution requires the affirmative vote of at least two-thirds of votes cast at a general meeting. A special resolution will be required for important matters.
A special resolution of members is required to change the name of the Company, approve a merger, wind up the Company, amend the Memorandum and Articles and reduce the share capital.
138
Conversion
Class A Ordinary Shares are not convertible. Each Class B Ordinary Share shall be convertible, at the option of the holder thereof, into such number of fully paid and non-assessable Class A Ordinary Shares on the basis that one Class B Ordinary Share shall be converted into one Class A Ordinary Share (being a 1:1 ratio and hereafter referred to as the “Conversion Rate”), subject to adjustment.
Transfer of Ordinary Shares
Subject to the restrictions set out below, any of our shareholders may transfer all or any of his, its or her Class A Ordinary Shares or Class B Ordinary Shares by an instrument of transfer in the usual or common form or any other form approved by our Board of Directors or in a form prescribed by the stock exchange on which our shares are then listed.
Our Board of Directors may, in its sole discretion, decline to register any transfer of any Class A Ordinary Shares or Class B Ordinary Shares whether or not it is fully paid up to the total consideration paid for such shares. Our directors may also decline to register any transfer of any Class A Ordinary Shares or Class B Ordinary Shares if (a) the instrument of transfer is not accompanied by the certificate covering the shares to which it relates or any other evidence as our Board of Directors may reasonably require to prove the title of the transferor to, or his/her right to transfer the shares; or (b) the instrument of transfer is in respect of more than one class of shares.
If our directors refuse to register a transfer, they shall, within two months after the date on which the instrument of transfer was lodged, send to the transferee notice of such refusal.
The registration of transfers may be suspended and the register closed at such times and for such periods as our Board of Directors may from time to time determine, provided, however, that the registration of transfers shall not be suspended nor the register closed for more than 30 days in any year.
Winding-Up/Liquidation
On a return of capital on winding up or otherwise (other than on conversion, redemption or purchase of shares), a liquidator may be appointed to determine how to distribute the assets among the holders of the Class A Ordinary Shares and Class B Ordinary Shares. If our assets available for distribution are insufficient to repay all of the paid-up capital, the assets will be distributed so that the losses are borne by our shareholders proportionately; a similar basis will be employed if the assets are more than sufficient to repay the whole of the capital at the commencement of the winding up.
Calls on Ordinary Shares and Forfeiture of Ordinary Shares
Our Board of Directors may from time to time make calls upon shareholders for any amounts unpaid on their Class A Ordinary Shares or Class B Ordinary Shares in a notice served to such shareholders at least 14 days prior to the specified time and place of payment. The shares that have been called upon and remain unpaid on the specified time are subject to forfeiture.
Redemption of Shares
We may issue shares on terms that are subject to redemption, at our option or at the option of the holders, on such terms and in such manner as may be determined by our Board of Directors.
Variations of Rights of Shares
All or any of the special rights attached to any class of shares may, be varied with the resolution of at least two thirds of the issued shares of that class or a resolution passed at a general meeting of the holders of the shares of that class present in person or by proxy or with the consent in writing of the holders of at least two-thirds of the issued shares of that class.
139
Inspection of Books and Records
Directors shall from time to time determine whether and to what extent and at what times and places and under what conditions or regulations the accounts and books of the Company or any of them shall be open to the inspection of members not being Directors and no member (not being a Director) shall have any right of inspecting any account or book or document of the Company except as conferred by Companies Law or authorized by the Directors or by the Company in a general meeting. However, the Directors shall from time to time cause to be prepared and to be laid before the Company in a general meeting, profit and loss accounts, balance sheets, group accounts (if any) and such other reports and accounts as may be required by Companies Law. (See “Where You Can Find More Information”)
Issuance of Additional Shares
Our Memorandum and Articles authorize our Board of Directors to issue additional Class A Ordinary Shares or Class B Ordinary Shares from time to time as our Board of Directors shall determine, to the extent there are available authorized but unissued shares.
Our Memorandum and Articles also authorizes our Board of Directors to establish from time to time one or more series of preferred shares and to determine, subject to compliance with the variation of rights of shares provision in the Memorandum and Articles, with respect to any series of preferred shares, the terms and rights of that series, including:
| ● | the designation of the series; | |
| ● | the number of shares of the series; | |
| ● | the dividend rights, dividend rates, conversion rights, voting rights; and | |
| ● | the rights and terms of redemption and liquidation preferences. |
Our Board of Directors may, issue preferred shares without action by our shareholders to the extent there are authorized but unissued shares available. Issuance of additional shares may dilute the voting power of holders of Class A Ordinary Shares and Class B Ordinary Shares. However, our Memorandum of Association provides for authorized share capital comprising Class A Ordinary Shares and Class B Ordinary Shares and to the extent the rights attached to any class may be varied, the Company must comply with the provisions in the Memorandum and Articles relating to variations to rights of shares.
Anti-Takeover Provisions
Some provisions of our Memorandum and Articles may discourage, delay or prevent a change of control of our Company or management that shareholders may consider favorable, including provisions that:
| ● | authorize our Board of Directors to issue preferred shares in one or more series and to designate the price, rights, preferences, privileges and restrictions of such preferred shares without any further vote or action by our shareholders (subject to variation of rights of shares provisions in our Memorandum and Articles); and | |
| ● | limit the ability of shareholders to requisition and convene general meetings of shareholders. Our Memorandum and Articles allow our shareholders holding shares representing in aggregate not less than ten percent of our paid up share capital (as to the total consideration paid for such shares) in issue to requisition an extraordinary general meeting of our shareholders, in which case our directors are obliged to call such meeting and to put the resolutions so requisitioned to a vote at such meeting. |
However, under Cayman Islands law, our directors may only exercise the rights and powers granted to them under our Memorandum and Articles for a proper purpose and for what they believe in good faith to be in the best interests of our Company.
140
General Meetings of Shareholders and Shareholder Proposals
Our shareholders’ general meetings may be held in such place within or outside the Cayman Islands as our Board of Directors considers appropriate.
As a Cayman Islands exempted company, we are not obliged by the Companies Law to call shareholders’ annual general meetings. However, our Memorandum and Articles provide that we shall hold a general meeting in each year as our annual general meeting other than the year in which the Memorandum and Articles were adopted at such time and place as determined by the directors. The directors may, whenever they think fit, convene an extraordinary general meeting.
Shareholders’ annual general meetings and any other general meetings of our shareholders may be convened by a majority of our Board of Directors. Our Board of Directors shall give not less than seven days’ written notice of a shareholders’ meeting to those persons whose names appear as members in our register of members on the date the notice is given (or on any other date determined by our directors to be the record date for such meeting) and who are entitled to vote at the meeting.
Cayman Islands law provides shareholders with only limited rights to requisition a general meeting, and does not provide shareholders with any right to put any proposal before a general meeting. However, these rights may be provided in a company’s articles of association. Our Memorandum and Articles allow our shareholders holding shares representing in aggregate not less than ten percent of our paid up share capital (as to the total consideration paid for such shares) in issue to requisition an extraordinary general meeting of our shareholders, in which case our directors are obliged to call such meeting and to put the resolutions so requisitioned to a vote at such meeting; otherwise, our Memorandum and Articles do not provide our shareholders with any right to put any proposals before annual general meetings or extraordinary general meetings not called by such shareholders.
Exempted Company
We are an exempted company with limited liability under the Companies Law. The Companies Law distinguishes between ordinary resident companies and exempted companies. A Cayman Islands exempted company:
| ● | is a company that conducts its business mainly outside of the Cayman Islands; | |
| ● | is exempted from certain requirements of the Companies Law, including the filing an annual return of its shareholders with the Registrar of Companies or the Immigration Board; | |
| ● | does not have to make its register of members open for inspection; | |
| ● | does not have to hold an annual general meeting; | |
| ● | may issue negotiable or bearer shares or shares with no par value (subject to the provisions of the Companies Law); | |
| ● | may obtain an undertaking against the imposition of any future taxation (such undertakings are usually given for 20 years in the first instance); and | |
| ● | may register by way of continuation in another jurisdiction and be deregistered in the Cayman Islands. |
“Limited liability” means that the liability of each shareholder is limited to the amount unpaid by the shareholder on the shares of the company (except in exceptional circumstances, such as involving fraud, the establishment of an agency relationship or an illegal or improper purpose or other circumstances in which a court may be prepared to pierce or lift the corporate veil).
141
Register of Members
Under Cayman Islands law, we must keep a register of members and there should be entered therein:
| ● | the names and addresses of the members, a statement of the shares held by each member, and of the amount paid or agreed to be considered as paid, on the shares of each member; | |
| ● | the date on which the name of any person was entered on the register as a member; and | |
| ● | the date on which any person ceased to be a member. |
Under Cayman Islands law, the register of members of our Company is prima facie evidence of the matters set out therein (i.e. the register of members will raise a presumption of fact on the matters referred to above unless rebutted) and a member registered in the register of members is deemed as a matter of Cayman Islands law to have legal title to the shares as set against its name in the register of members. Once our register of members has been updated, the shareholders recorded in the register of members are deemed to have legal title to the shares set against their name.
If the name of any person is incorrectly entered in, or omitted from, our register of members, or if there is any default or unnecessary delay in entering on the register the fact of any person having ceased to be a member of our Company, the person or member aggrieved (or any member of our Company or our Company itself) may apply to the Cayman Islands Grand Court for an order that the register be rectified, and the Court may either refuse such application or it may, if satisfied of the justice of the case, make an order for the rectification of the register.
Indemnification of Directors and Executive Officers and Limitation of Liability
Cayman Islands law does not limit the extent to which a company’s memorandum and articles of association may provide for indemnification of officers and directors, except to the extent any such provision may be held by the Cayman Islands courts to be contrary to public policy, such as to provide indemnification against civil fraud or the consequences of committing a crime. Our Memorandum and Articles require us to indemnify our officers and directors for actions, proceedings, claims, losses, damages, costs, liabilities and expenses (“Indemnified Losses”) incurred in their capacities as such unless such Indemnified Losses arise from dishonesty of such directors or officers. This standard of conduct is generally the same as permitted under the Delaware General Corporation Law for a Delaware corporation.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us under the foregoing provisions, we have been informed that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Convertible Bond
As of the date hereof, we have a $13,500,000 Bond outstanding. Pursuant to the conversion terms of the Bond, we issued an aggregate of 119,217 shares of Class A Ordinary Shares to the original Bond holder after the IPO closed. The following are summaries of material terms of the Subscription Agreement for the Bond and the Bond itself; such summaries do not purport to be a complete description of the terms, the Subscription Agreement or any transactions contemplated by the Subscription Agreement and you are urged to read the agreements in their entirety.
Conversion
Upon the closing of the IPO, 10% of the then-outstanding Principal Amount automatically converted into 119,217 Class A Ordinary Shares. The holders of the Bond also maintain the right to convert the balance of the Bond until the earlier of: (i) the date falling 12 calendar months after the Maturity Date or the Extended Maturity Date (as such terms are defined in the Bond), as the case may be (both days inclusive); and (ii) the date falling 12 calendar months after the closing of the IPO.
On April 25, 2019, we have repurchased $13,500,000 Bond outstanding. The holders of the Bond have the right to convert the balance of the Bond at any time up to seven days prior to the Maturity Date or the Extended Maturity Date (as such terms are defined in the Bond), as the case may be (both days inclusive).
142
Interest
The Bond accrue interest at the rate of 8% per annum, payable semi-annually in arrears in equal instalments of $10,000 per Calculation Amount (as defined below) on the date falling on the 6th calendar month following the Issue Date and the date falling on the 12th calendar month following the Issue Date, and if the Maturity Date is extended to the Extended Maturity Date, on the date falling on the 18th calendar month following the Issue Date.
Interest in respect of any Bond shall be calculated per $250,000 in principal amount of the Bond (the “Calculation Amount”). The amount of interest payable per Calculation Amount for any period shall, save as provided above in relation to equal instalments, be equal to the product of 8% per annum, the Calculation Amount and the day-count fraction (as determined in accordance with the formula set forth in Schedule 1 to the Subscription Agreement) for the relevant period, rounding the resulting figure to the nearest cent ($0.005 being rounded upward).
The interest rate shall increase upon the occurrence of an event of default, as set forth in Schedule 1 to the Subscription Agreement (the “Default Interest Rate”).
Event of Default
Upon the occurrence of an event of default, the holder of the Bond may give us and Jurchen notice that the Bond are, and they shall immediately become, due and repayable at their principal amount together with interest, at the Default Interest Rate.
Events of default include non-payment of any sums due under the Bond, failure to deliver any shares due under the Bond, breach of our covenants, representations and warranties, breach of our obligations under other liabilities or indebtedness, enforcement proceedings, our insolvency, if the holders of the Bond cease to have adequate control over the Debt Service Reserve Account and if the guarantee or security rights are not enforceable. The Subscription Agreements allows for some cure periods and/or notice prior to declaring the Bond due.
Negative Covenants
The Subscription Agreement contains certain negative covenants, including our inability to incur certain liabilities before and after the closing of this Offering without the Bond holders’ prior written consent, limits on our ability to issue equity securities and limits on certain corporate actions.
Differences in Corporate Law
The Companies Law is modeled after that of English law but does not follow many recent English law statutory enactments. In addition, the Companies Law differs from laws applicable to United States corporations and their shareholders. Set forth below is a summary of some of the significant differences between the provisions of the Companies Law applicable to us and the laws applicable to companies incorporated in the State of Delaware.
Mergers and Similar Arrangements.The Companies Law permits mergers and consolidations between Cayman Islands companies and between Cayman Islands companies and non-Cayman Islands companies. For these purposes, a “merger” means the merging of two or more constituent companies and the vesting of their undertaking, property and liabilities in one of such companies as the surviving company, and a “consolidation” means the combination of two or more constituent companies into a consolidated company and the vesting of the undertaking, property and liabilities of such companies to the consolidated company.
In order to effect a merger or consolidation, the directors of each constituent company must approve a written plan of merger or consolidation, which must then be authorized by a special resolution of the shareholders of each constituent company, and such other authorization, if any, as may be specified in such constituent company’s articles of association.
143
The plan of merger or consolidation must be filed with the Registrar of Companies of the Cayman Islands together with a declaration as to: the solvency of the consolidated or surviving company, the merger or consolidation being bona fide and not intended to defraud creditors, no petition or other proceeding, order or resolution to wind up the Company, no receiver, administrator or similar having been appointed over assets or property and no scheme or other arrangement having been entered into with creditors; a list of the assets and liabilities of each constituent company and an undertaking that a copy of the certificate of merger or consolidation will be given to the members and creditors of each constituent company; and that notification of the merger and consolidation will be published in the Cayman Islands Gazette. The non-surviving constituent company must have resigned from any fiduciary office held or will do so and each constituent company having complied with any applicable regulatory laws. Dissenting shareholders have the right to be paid the fair value of their shares if they follow the required procedures under the Companies Law subject to certain exceptions. The fair value of the shares will be determined by the Cayman Islands court if it cannot be agreed among the parties. Court approval is not required for a merger or consolidation effected in compliance with these statutory procedures.
In addition, there are statutory provisions that facilitate the reconstruction and amalgamation of companies, provided that the arrangement is approved by a majority in number of each class of shareholders and creditors with whom the arrangement is to be made, and who must in addition represent three-fourths in value of each such class of shareholders or creditors, as the case may be, that are present and voting either in person or by proxy at a meeting, or meetings, convened for that purpose. The convening of the meetings and subsequently the arrangement must be sanctioned by the Grand Court of the Cayman Islands.
While a dissenting shareholder has the right to express to the court the view that the transaction ought not to be approved, the court can be expected to approve the arrangement if it determines that:
| ● | the statutory provisions as to the required majority vote have been met; | |
| ● | the shareholders have been fairly represented at the meeting in question; | |
| ● | the arrangement is such that an intelligent and honest man of that class acting in respect of his interest would reasonably approve; and | |
| ● | the arrangement is not one that would more properly be sanctioned under some other provision of the Companies Law or that would amount to a “fraud on the minority.” |
When a take-over offer is made and accepted by holders of not less than 90% of the shares within four months, the offer, or may, within a two-month period commencing on the expiration of such four months period, require the holders of the remaining shares to transfer such shares on the terms of the offer. An objection can be made to the Grand Court of the Cayman Islands but this is unlikely to succeed unless there is evidence of fraud, bad faith or collusion.
If the arrangement and reconstruction is thus approved, the dissenting shareholder would have no rights comparable to appraisal rights, which would otherwise ordinarily be available to dissenting shareholders of United States corporations, providing rights to receive payment in cash for the judicially determined value of the shares.
Shareholders’ Suits. In principle, we will normally be the proper plaintiff to sue for a wrong done to us as a company and as a general rule a derivative action may not be brought by a minority shareholder. However, based on English authorities, which would in all likelihood be of persuasive authority in the Cayman Islands, there are exceptions to the foregoing principle, including when:
| ● | a company acts or proposes to act illegally or ultra vires and is therefore incapable of ratification by the shareholders; | |
| ● | the act complained of, although not ultra vires, could only be duly effected if authorized by more than a simple majority vote that has not been obtained; and | |
| ● | those who control the company are perpetrating a “fraud on the minority.” |
144
Indemnification of Directors and Executive Officers and Limitation of Liability. The Companies Law does not limit the extent to which a company’s memorandum and articles of association may provide for indemnification of officers and directors, except to the extent any such provision may be held by the Cayman Islands courts to be contrary to public policy, such as to provide indemnification against civil fraud or the consequences of committing a crime. As stated above, our Memorandum and Articles permit indemnification of officers and directors for actions, proceedings, claims, losses, damages, costs, liabilities and expenses (“Indemnified Losses”) incurred in their capacities as such unless such losses or damages arise from dishonesty of such directors or officers. This standard of conduct is generally the same as permitted under the Delaware General Corporation Law for a Delaware corporation. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us under the foregoing provisions, we have been informed that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Directors’ Fiduciary Duties. Under Delaware corporate law, a director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components: the duty of care and the duty of loyalty. The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director acts in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Should such evidence be presented concerning a transaction by a director, the director must prove the procedural fairness of the transaction, and that the transaction was of fair value to the corporation. As a matter of Cayman Islands law, a director of a Cayman Islands company is in the position of a fiduciary with respect to the company and therefore it is considered that he or she owes the following duties to the company: a duty to act bona fide in the best interests of the company, a duty not to make a profit based on his or her position as director (unless the company permits him or her to do so) and a duty not to put himself or herself in a position where the interests of the company conflict with his or her personal interest or his or her duty to a third-party. Our Memorandum and Articles do not disqualify a director from acting or from contacting with the Company as a vendor, purchaser or otherwise provided that it does not adversely affect his or her performance of duties or responsibilities and the nature of the interest is disclosed at the meeting at which the contract or arrangement is considered (if not previously disclosed), and having disclosed such interest the director is not counted in the quorum and must refrain from voting on the contract or arrangement. A director of a Cayman Islands company also owes to the company a duty to exercise the powers for the purpose for which they were given and the duty to act with skill and care. It was previously considered that a director need not exhibit in the performance of his or her duties a greater degree of skill than may reasonably be expected from a person of his or her knowledge and experience. However, courts are moving towards an objective standard with regard to the required skill and care and these authorities are likely to be followed in the Cayman Islands.
Shareholder Action by Written Consent. Under the Delaware General Corporation Law, a corporation may eliminate the right of shareholders to act by written consent by amendment to its certificate of incorporation. Cayman Islands law and our Memorandum and Articles provide that shareholders may approve corporate matters by way of a unanimous written resolution signed by or on behalf of each shareholder who would have been entitled to vote on such matter at a general meeting without a meeting being held.
Shareholder Proposals. Under the Delaware General Corporation Law, a shareholder has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings. The Companies Law provides shareholders with only limited rights to requisition a general meeting and does not provide shareholders with any right to put any proposal before a general meeting. However, these rights may be provided in articles of association. Our Memorandum and Articles allow our shareholders holding not less than 1/10 of all voting power of our (paid up) share capital in issue to requisition a shareholder’s meeting. Other than this right to requisition a shareholders’ meeting, our Memorandum and Articles do not provide our shareholders other rights to put proposal before a meeting. As an exempted Cayman Islands company, we are not obliged by law to call shareholders’ annual general meetings although our Memorandum and Articles provide for same.
145
Cumulative Voting.Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation specifically provides for it. Cumulative voting potentially facilitates the representation of minority shareholders on a board of directors since it permits the minority shareholder to cast all the votes to which the shareholder is entitled on a single director, which increases the shareholder’s voting power with respect to electing such director. There are no prohibitions in relation to cumulative voting under the Companies Law but our Memorandum and Articles do not provide for cumulative voting.
Removal of Directors.Under the Delaware General Corporation Law, a director of a corporation with a may be removed with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. Under our Memorandum and Articles, directors may be removed with or without cause, by the directors or by an ordinary resolution of our shareholders.
Transactions with Interested Shareholders. The Delaware General Corporation Law contains a business combination statute applicable to Delaware corporations whereby, unless the corporation has specifically elected not to be governed by such statute by amendment to its certificate of incorporation, it is prohibited from engaging in certain business combinations with an ���interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or a group who or which owns or owned 15% or more of the target’s outstanding voting share within the past three years. This has the effect of limiting the ability of a potential acquirer to make a two-tiered bid for the target in which all shareholders would not be treated equally. The statute does not apply if, among other things, prior to the date on which such shareholder becomes an interested shareholder, the board of directors approves either the business combination or the transaction which resulted in the person becoming an interested shareholder. This encourages any potential acquirer of a Delaware corporation to negotiate the terms of any acquisition transaction with the target’s board of directors. The Cayman Islands has no comparable statute. As a result, we cannot avail ourselves of the types of protections afforded by the Delaware business combination statute. However, although Cayman Islands law does not regulate transactions between a company and its significant shareholders, it does provide that such transactions must be entered into bona fide in the best interests of the company and for a proper corporate purpose and not with the effect of constituting a fraud on the minority shareholders. Our Memorandum and Articles, as well as our Code of Business Conduct and Ethics that applies to our officers, directors and employees outlines how to handle these types of transactions and other potential conflicts of interest.
Dissolution; Winding up. Under the Delaware General Corporation Law, unless the board of directors approves the proposal to dissolve, dissolution must be approved by shareholders holding 100% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board. Under the Companies Law, a company may be wound up by either an order of the courts of the Cayman Islands or by a special resolution of its members or, if the company is unable to pay its debts as they fall due, by an ordinary resolution of its members. The court has authority to order winding up in a number of specified circumstances including where it is, in the opinion of the court, just and equitable to do so. Under the Companies Law a company may be dissolved, liquidated or wound up by a special resolution of our shareholders; however, under our Memorandum and Articles, only our Directors have power to present a winding up petition in the name of the Company and/or to apply for the appointment of provisional liquidators in respect of the Company.
Variation of Rights of Shares. Under the Delaware General Corporation Law, a corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. Under the Companies Law and our Memorandum and Articles, if our share capital is divided into more than one class of shares, we may vary the rights attached to any class with the written consent of the holders of two-thirds of the issued shares of that class or with the sanction of a special resolution passed at a separate general meeting of the holders of the shares of that class.
146
Amendment of Governing Documents. Under the Delaware General Corporation Law, a corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. As permitted by the Companies Law, each of our Memorandum of Association and Articles of Association may only be amended with a special resolution of our shareholders.
Rights of Non-resident or Foreign Shareholders. There are no limitations imposed by our Memorandum and Articles on the rights of non-resident or foreign shareholders to hold or exercise voting rights on our shares. In addition, there are no provisions in our Memorandum and Articles governing the ownership threshold above which shareholder ownership must be disclosed.
Rule 144
Shares Held for Six Months
In general, under Rule 144 as currently in effect, and subject to the terms of any lock-up agreement, commencing 90 days after the closing of the IPO, a person (or persons whose shares are aggregated), including an affiliate, who has beneficially owned our Class A Ordinary Shares for six months or more, including the holding period of any prior owner other than one of our affiliates (i.e., commencing when the shares were acquired from our Company or from an affiliate of our Company as restricted securities), is entitled to sell our shares, subject to the availability of current public information about us. In the case of an affiliate shareholder, the right to sell is also subject to the fulfillment of certain additional conditions, including manner of sale provisions and notice requirements, and to a volume limitation that limits the number of shares to be sold thereby, within any three-month period, to the greater of:
| ● | 1% of the number of Class A Ordinary Shares then outstanding; or | |
| ● | the average weekly trading volume of our Class A Ordinary Shares on the NASDAQ Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale. |
The six-month holding period of Rule 144 does not apply to sales of unrestricted securities. Accordingly, persons who hold unrestricted securities may sell them under the requirements of Rule 144 described above without regard to the six-month holding period, even if they were considered our affiliates at the time of the sale or at any time during the 90 days preceding such date.
Shares Held by Non-Affiliates for One Year
Under Rule 144 as currently in effect, a person (or persons whose shares are aggregated) who is not considered to have been one of our affiliates at any time during the 90 days preceding a sale and who has beneficially owned the shares proposed to be sold for at least one year, including the holding period of any prior owner other than one of our affiliates, is entitled to sell his, her or its shares under Rule 144 without complying with the provisions relating to the availability of current public information or with any other conditions under Rule 144. Therefore, unless subject to a lock-up agreement or otherwise restricted, such shares may be sold immediately upon the closing of the IPO.
147
The following summary contains a description of certain Cayman Islands and U.S. federal income tax consequences of the acquisition, ownership and disposition of Class A Ordinary Shares. Please note that this summary should not be considered a comprehensive description of all the tax considerations that may be relevant to the decision to purchase Class A Ordinary Shares. The summary is based upon the tax laws of the Cayman Islands and regulations thereunder and on the tax laws of the United States and regulations thereunder as of the date hereof, which are subject to change.
Cayman Islands Tax Considerations
The Cayman Islands currently levies no taxes on individuals or corporations based upon profits, income, gains or appreciation and there is no taxation in the nature of inheritance tax or estate duty. There are no other taxes likely to be material to us levied by the government of the Cayman Islands except for stamp duties which may be applicable on instruments executed in, or brought within, the jurisdiction of the Cayman Islands. The Cayman Islands is not party to any double tax treaties which are applicable to any payments made by or to our Company. There are no exchange control regulations or currency restrictions in the Cayman Islands.
Payments of dividends and capital in respect of our Class A Ordinary Shares will not be subject to taxation in the Cayman Islands and no withholding will be required on the payment of a dividend or capital to any holder of our Class A Ordinary Shares, nor will gains derived from the disposal of our Class A Ordinary Shares be subject to Cayman Islands income or corporation tax.
No stamp duty is payable in respect of the issue of our Class A Ordinary Shares or on an instrument of transfer in respect of our Class A Ordinary Shares except on instruments executed in, or brought within, the jurisdiction of the Cayman Islands.
Material U.S. Federal Income Tax Considerations for U.S. Holders
The following is a description of the material U.S. federal income tax consequences to U.S. Holders (as defined below) of purchasing, owning and disposing of Class A Ordinary Shares. It is not a comprehensive description of all U.S. federal income tax considerations that may be relevant to a particular person’s decision to acquire Class A Ordinary Shares. This discussion applies only to a U.S. Holder that holds a Class A Ordinary Share as a capital asset for U.S. federal income tax purposes (generally, property held for investment). In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder’s particular circumstances, including state and local tax consequences, non-U.S. tax consequences, federal estate or gift tax consequences, alternative minimum tax consequences, the potential application of the provisions of the Code known as the Medicare Contribution Tax, and tax consequences applicable to U.S. Holders subject to special rules, such as:
| ● | banks and other financial institutions; |
| ● | insurance companies; | |
| ● | dealers or traders in securities who use a mark-to-market method of tax accounting; |
| ● | persons holding Class A Ordinary Shares as part of a hedging transaction, “straddle,” wash sale, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to the Class A Ordinary Shares; |
| ● | persons whose “functional currency” for U.S. federal income tax purposes is not the U.S. dollar; |
| ● | tax exempt entities, including “individual retirement accounts” and “Roth IRAs”; |
| ● | former citizens or long-term residents of the United States; |
| ● | entities or arrangements classified as partnerships for U.S. federal income tax purposes; |
| ● | regulated investment companies or real estate investment trusts; |
| ● | persons who acquired our Class A Ordinary Shares pursuant to the exercise of an employee share option or otherwise as compensation; |
| ● | persons that own or are deemed to own ten percent or more of our shares; and |
| ● | persons holding Class A Ordinary Shares in connection with a trade or business conducted outside the United States. |
148
If an entity or arrangement that is classified as a partnership for U.S. federal income tax purposes holds Class A Ordinary Shares, the U.S. federal income tax treatment of such partnership and each partner thereof will generally depend on the status of the partner and the activities of the partnership. Partnerships holding Class A Ordinary Shares and partners in such partnerships are encouraged to consult their tax advisors as to the particular U.S. federal income tax consequences of purchasing, holding and disposing of Class A Ordinary Shares.
The discussion is based on the Code, the Treasury Regulations issued thereunder, and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect, or to different interpretation. Such change could materially and adversely affect the tax consequences described below.
For purposes of this discussion, a “U.S. Holder” is a holder who, for U.S. federal income tax purposes, is a beneficial owner of Class A Ordinary Shares and that is:
| (1) | an individual citizen or resident of the United States; | |
| (2) | a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia; | |
| (3) | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or | |
| (4) | a trust, (i) if a court within the United States is able to exercise primary supervision over its administration and one or more “U.S. persons” (within the meaning of the Code) have the authority to control all of its substantial decisions, or (ii) if a valid election is in effect for the trust to be treated as a U.S. person. |
U.S. Holders are encouraged to consult their tax advisors concerning the U.S. federal, state, local and foreign tax consequences of purchasing, owning and disposing of Class A Ordinary Shares in their particular circumstances.
Taxation of Distributions
Subject to the discussion below under “Passive Foreign Investment Company Rules,” a U.S. Holder will be required to include in gross income as dividend income the gross amount of any distributions paid on Class A Ordinary Shares (including any amount of taxes withheld), other than certain pro rata distributions of Class A Ordinary Shares, to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Distributions in excess of our current and accumulated earnings and profits would be treated as a non-taxable return of capital to the extent of the U.S. Holder’s adjusted tax basis in the Class A Ordinary Shares and thereafter as a gain from the sale of the Class A Ordinary Shares. However, because we do not calculate our earnings and profits under U.S. federal income tax principles, we expect that distributions generally will be reported to U.S. Holders as dividends.
In case of a U.S. Holder that is a corporation, dividends paid on the Class A Ordinary Shares will be subject to regular corporate rates and will not be eligible for the “dividends received” deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations.
Dividends received by an individual, trust or estate will be subject to taxation at standard tax rates. A reduced income tax rate applies to dividends paid by a “qualified foreign corporations” (if certain holding period requirements and other conditions are met). A non-U.S. corporation generally will be considered to be a qualified foreign corporation (i) if it is eligible for the benefits of a comprehensive tax treaty with the United States which includes an exchange of information program or (ii) with respect to any dividend it pays on stock which is readily tradable on an established securities market in the United States. US. Treasury Department guidance indicates that our Class A Ordinary Shares, which will be listed on the NASDAQ Global Market will be readily tradable on an established securities market in the United States. There can be no assurance, however, that our Class A Ordinary Shares will be considered readily tradable on an established securities market in later years.
149
Non-corporate U.S. Holders will not be eligible for reduced rates of taxation on any dividends received from us if we are a PFIC in the taxable year in which such dividends are paid or in the preceding taxable year (see “Passive Foreign Investment Company Rules” below).
A U.S. Holder may be eligible, subject to a number of complex limitations, to claim a foreign tax credit in respect of any foreign withholding taxes imposed on dividends received on the Class A Ordinary Shares. A U.S. Holder who does not elect to claim a foreign tax credit for foreign income tax withheld may instead claim a deduction for U.S. federal income tax purposes in respect of such withholding, but only for a year in which such investor elects to do so for all creditable foreign income taxes. For purposes of calculating the foreign tax credit limitation, dividends paid by us will, depending on the circumstances of the U.S. Holder, be either general or passive income.
While we do not expect to pay dividends in the near future, in the event any dividends are paid and if a dividend is paid in non-U.S. currency, it must be included in a U.S. Holder’s income as a U.S. dollar amount based on the exchange rate in effect on the date such dividend is actually or constructively received, regardless of whether the dividend is in fact converted into U.S. dollars. If the dividend is converted to U.S. dollars on the date of receipt, a U.S. Holder generally will not recognize a foreign currency gain or loss. If the non-U.S. currency is converted into U.S. dollars on a later date, however, the U.S. Holder must include in income any gain or loss resulting from any exchange rate fluctuations. Such gain or loss will generally be ordinary income or loss and will be from sources within the United States for foreign tax credit limitation purposes. U.S. Holders should consult their own tax advisors regarding the tax consequences to them if we pay dividends in non-U.S. currency.
Sale or Other Taxable Disposition of Shares
Subject to the discussion below under “Passive Foreign Investment Company Rules,” gain or loss realized on the sale or other taxable disposition of Class A Ordinary Shares will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the Class A Ordinary Shares for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the Class A Ordinary Shares disposed of and the amount realized on the disposition. Long-term capital gain of a non-corporate U.S. Holder is generally taxed at preferential rates. This gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes. The deductibility of capital losses is subject to limitations. U.S. Holders are urged to consult their tax advisors regarding the tax consequences if a foreign tax is imposed on the disposition of Class A Ordinary Shares, including the availability of the foreign tax credit under an investor’s own particular circumstances.
A U.S. Holder that receives non-U.S. currency on the disposition of the Class A Ordinary Shares will realize an amount equal to the U.S. dollar value of the foreign currency received on the date of disposition (or in the case of cash basis and electing accrual basis taxpayers, the settlement date) whether or not converted into U.S. dollars at that time. Very generally, the U.S. Holder will recognize currency gain or loss if the U.S. dollar value of the currency received on the settlement date differs from the amount realized with respect to the Class A Ordinary Shares. Any currency gain or loss on the settlement date or on any subsequent disposition of the foreign currency generally will be U.S.-source ordinary income or loss.
Passive Foreign Investment Company Rules
Special U.S. federal income tax rules apply to a U.S. Holder that holds stock in a foreign corporation classified as a PFIC for U.S. federal income tax purposes. In general, a non-U.S. corporation will be classified as a PFIC for any taxable year in which, after applying certain look-through rules, either:
| ● | at least 75% of its gross income for such taxable year is passive income (e.g., dividends, interest, capital gains and rents derived other than in the active conduct of a rental business); or |
| ● | at least 50% of its gross assets (determined on the basis of a quarterly average) is attributable to assets that produce passive income or are held for the production of passive income. |
We will be treated as owning our proportionate share of the assets and earning our proportionate share of the income of any other corporation in which we own, directly or indirectly, 25% or more (by value) of the equity.
150
A separate determination must be made after the close of each taxable year as to whether we are a PFIC for that year. As a result, our PFIC status may change. In particular, the total value of our assets generally will be calculated using the market price of our Class A Ordinary Shares, which may fluctuate considerably. Fluctuations in the market price of our Class A Ordinary Shares may result in our being a PFIC for any taxable year.
Due to the amount of restricted and unrestricted cash that we had on hand during our year ending December 31, 2018 and 2017, we believe that we were classified as a PFIC for that tax year. Depending on the future composition and value of our assets, we may be classified as a PFIC for future years.
If we were to be classified as a PFIC, a U.S. Holder would be subject to different taxation rules depending on whether the U.S. Holder (i) takes no action, (ii) makes an election to treat us as a “Qualified Electing Fund” (a “QEF election”) or (iii) if permitted, makes a “mark-to-market” election with respect to our Class A Ordinary Shares. A U.S. Holder of our Class A Ordinary Shares will also be required under applicable Treasury Regulations to file an annual information return (Form 8621) containing information regarding our company. Additional explanations of the PFIC rules are set forth below: this material is complex and may affect different U.S. Holders differently. Accordingly, U.S. Holders should consult their own tax advisors about the consequences of our company being classified as a PFIC and about what steps, if any, they might take to lessen the tax impact of our PFIC status on them.
A U.S. Holder who does not make a timely QEF or mark-to-market election (a “Non-Electing Holder,”), as discussed below, will be subject to special tax rules with respect to any “excess distribution” that you receive and any gain you realize from a sale or other disposition (including a pledge) of Class A Ordinary Shares. Distributions you receive in a taxable year that are greater than 125% of the average annual distributions you received during the shorter of the three preceding taxable years or your holding period for the Class A Ordinary Shares will be treated as an excess distribution. Under these special tax rules:
| ● | the excess distribution or gain will be allocated ratably over your holding period for the Class A Ordinary Shares; |
| ● | the amount allocated to the current taxable year, and any taxable year prior to the first taxable year in which we became a PFIC, will be treated as ordinary income; and |
| ● | the amount allocated to each other year will be subject to the highest tax rate in effect for that year and the interest charge generally applicable to underpayments of tax will be imposed on the resulting tax attributable to each such year. |
It should be noted that, until such time as we make a distribution, there are no tax consequences to Non-Electing Holders. However, if we ever did make a distribution it would in all likelihood be an excess distribution (because we would not have previously made any distributions to holders of Class A Ordinary Shares). At that point, and for all subsequent distributions, the rules described above would apply to Non-Electing Holders. The tax liability for amounts allocated to years prior to the year of disposition or “excess distribution” cannot be offset by any net operating losses for such years, and gains (but not losses) realized on the sale of the Class A Ordinary Shares cannot be treated as capital, even if you hold the Ordinary Shares as capital assets.
Certain elections may be available that would result in alternative treatments. The adverse consequences of owning stock in a PFIC could be mitigated if a U.S. Holder makes a valid QEF election (a U.S. Holder which we refer to as an “Electing Holder”) which, among other things, would require the Electing Holder to include currently in income its pro rata share of the PFIC’s net capital gain and ordinary earnings, if any, for our taxable year that ends with or within the taxable year of the Electing Holder, regardless of whether or not the Electing Holder actually received distributions from us. When an Electing Holder makes a QEF election, its adjusted tax basis in our Class A Ordinary Shares is increased to reflect taxed but undistributed earnings and profits. Distributions of earnings and profits that had been previously taxed will result in a corresponding reduction in the adjusted tax basis in our Class A Ordinary Shares and will not be taxed again once distributed. An Electing Holder would generally recognize capital gain or loss on the sale, exchange or other disposition of our Class A Ordinary Shares.
A U.S. Holder can make a QEF election with respect to any year that we are a PFIC by filing IRS Form 8621 with its U.S. federal income tax return. This election must be made by the deadline (including extensions) for filing the U.S. Holder’s federal tax return for the year in question. U.S. Holders should discuss their election alternatives with their own tax advisors. Once an election is made, the Electing Holder is subject to the QEF rules for as long as we are a PFIC.
151
It should be noted that in order to make a QEF election a U.S. Holder needs information from us concerning our PFIC status and our financial results for the year. We cannot assure our U.S. Holders that we will provide such information.
As an alternative to making a QEF election, a U.S. Holder may make a “mark-to-market” election with respect to our Class A Ordinary Shares provided our Class A Ordinary Shares are treated as “marketable stock.” The Class A Ordinary Shares generally will be treated as marketable stock if they are regularly traded on a “qualified exchange or other market” (within the meaning of applicable Treasury Regulations) on at least 15 days during each calendar quarter (other than in de minimis amounts).
If a U.S. Holder makes an effective mark-to-market election, for each taxable year that we are a PFIC, the U.S. Holder will include as ordinary income the excess of the fair market value of its Class A Ordinary Shares at the end of the year over its adjusted tax basis in the Class A Ordinary Shares. You will be entitled to deduct as an ordinary loss in each such year the excess of your adjusted tax basis in the Class A Ordinary Shares over their fair market value at the end of the year, but only to the extent of the net amount previously included in income as a result of the mark-to-market election. A U.S. Holder’s adjusted tax basis in the Class A Ordinary Shares will be increased by the amount of any income inclusion and decreased by the amount of any deductions under the mark-to-market rules. In addition, upon the sale or other disposition of your Class A Ordinary Shares in a year that we are PFIC, any gain will be treated as ordinary income and any loss will be treated as ordinary loss, but only to the extent of the net amount of previously included income as a result of the mark-to-market election.
If a U.S. Holder makes a mark-to-market election, it will be effective for the taxable year for which the election is made and all subsequent taxable years unless the Class A Ordinary Shares are no longer regularly traded on a qualified exchange or other market, or the IRS consents to the revocation of the election. You are urged to consult your tax advisor about the availability of the mark-to-market election, and whether making the election would be advisable in your particular circumstances.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries generally are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding.
Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the holder’s U.S. federal income tax liability and may entitle it to a refund, provided that the required information is timely furnished to the IRS.
Information with Respect to Foreign Financial Assets
Certain U.S. Holders may be required to report information relating to the Class A Ordinary Shares, subject to certain exceptions (including an exception for Class A Ordinary Shares held in accounts maintained by certain U.S. financial institutions). U.S. Holders should consult their tax advisors regarding their reporting obligations with respect to their purchase, ownership and disposition of the Class A Ordinary Shares.
152
Set forth below is an itemization of our total expenses, which are expected to be incurred in connection with the offer and sale of the Class A Ordinary Shares by us. With the exception of the SEC registration fee, all amounts are estimates.
| Securities and Exchange Commission registration fee | $ | 87,496 | ||
| Legal fees and expenses | $ | 40,000 | ||
| Other professional fees | $ | 8,000 | ||
| Total | $ | 135,496 |
The validity of the Class A Ordinary Shares being offered by this prospectus and other legal matters concerning the Resale Shares relating to Cayman Islands law will be passed upon for us by Campbells. Certain legal matters in connection with the Resale Shares with respect to the United States federal securities law and New York law will be passed upon for us by Hunter Taubman Fischer & Li LLC, New York, New York.
The financial statements appearing in this prospectus for the year ended December 31, 2018 have been audited by Marcum Bernstein & Pinchuk LLP, an independent registered public accounting firm, as set forth in their report thereon appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
ENFORCEMENT OF CIVIL LIABILITIES
We are incorporated under the laws of the Cayman Islands as an exempted company with limited liability. We incorporated in the Cayman Islands because of certain benefits associated with being a Cayman Islands corporation, such as political and economic stability, an effective judicial system, a favorable tax system, the absence of foreign exchange control or currency restrictions and the availability of professional and support services. However, the Cayman Islands have a less developed body of securities laws that provide significantly less protection to investors as compared to the securities laws of the United States. In addition, Cayman Islands companies may not have standing to sue before the federal courts of the United States.
All of our assets are located in Hong Kong. In addition, some of our directors and officers are residents of jurisdictions other than the United States and all or a substantial portion of their assets are located outside the United States. As a result, it may be difficult for investors to effect service of process within the United States upon us or our directors and officers, or to enforce against us or them judgments obtained in United States courts, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state in the United States.
According to our local Cayman Islands’ counsel, there is uncertainty with regard to Cayman Islands law relating to whether a judgment obtained from the United States or Hong Kong courts under civil liability provisions of the securities laws will be determined by the courts of the Cayman Islands as penal or punitive in nature. If such a determination is made, the courts of the Cayman Islands will not recognize or enforce the judgment against a Cayman Islands’ company. The courts of the Cayman Islands in the past determined that disgorgement proceedings brought at the instance of the Securities and Exchange Commission are penal or punitive in nature and such judgments would not be enforceable in the Cayman Islands. Other civil liability provisions of the securities laws may be characterized as remedial, and therefore enforceable but the Cayman Islands’ Courts have not yet ruled in this regard. Our Cayman Islands’ counsel has further advised us that a final and conclusive judgment in the federal or state courts of the United States under which a sum of money is payable other than a sum payable in respect of taxes, fines, penalties or similar charges, may be subject to enforcement proceedings as a debt in the courts of the Cayman Islands.
153
As of the date of this prospectus, no treaty or other form of reciprocity exists between the Cayman Islands and Hong Kong governing the recognition and enforcement of judgments.
Cayman Islands’ counsel further advised that although there is no statutory enforcement in the Cayman Islands of judgments obtained in the United States or Hong Kong, a judgment obtained in such jurisdictions will be recognized and enforced in the courts of the Cayman Islands at common law, without any re-examination of the merits of the underlying dispute, by an action commenced on the foreign judgment debt in the Grand Court of the Cayman Islands, provided such judgment (1) is given by a foreign court of competent jurisdiction, (2) imposes on the judgment debtor a liability to pay a liquidated sum for which the judgment has been given, (3) is final, (4) is not in respect of taxes, a fine or a penalty, and (5) was not obtained in a manner and is of a kind the enforcement of which is contrary to natural justice or the public policy of the Cayman Islands.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form F-1 under the Securities Act relating to this Offering of our Class A Ordinary Shares. This prospectus does not contain all of the information contained in the registration statement. The rules and regulations of the SEC allow us to omit certain information from this prospectus that is included in the registration statement. Statements made in this prospectus concerning the contents of any contract, agreement or other document are summaries of all material information about the documents summarized, but are not complete descriptions of all terms of these documents. If we filed any of these documents as an exhibit to the registration statement, you may read the document itself for a complete description of its terms.
You may read and copy the registration statement, including the related exhibits and schedules, and any document we file with the SEC without charge at the SEC’s public reference room at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You may also obtain copies of the documents at prescribed rates by writing to the Public Reference Section of the SEC at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. The SEC also maintains an Internet website that contains reports and other information regarding issuers that file electronically with the SEC. Our filings with the SEC are also available to the public through the SEC’s website at http://www.sec.gov.
We are subject to the information reporting requirements of the Exchange Act that are applicable to foreign private issuers, and under those requirements file reports with the SEC. Those other reports or other information may be inspected without charge at the locations described above. As a foreign private issuer, we will be exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders will be exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we will not be required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. However, we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm, and will submit to the SEC, on Form 6-K, unaudited quarterly financial information for the first three quarters of each fiscal year.
We maintain a corporate website at www.aptorumgroup.com. Information contained on, or that can be accessed through, our website does not constitute a part of this prospectus.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to “incorporate by reference” information into this document. This means that we can disclose important information to you by referring you to another document filed separately with the SEC. The information incorporated by reference is considered to be a part of this document, except for any information superseded by information that is included directly in this document.
This prospectus incorporates by reference the documents listed below:
| (1) | our Annual Report onForm 20-F for the fiscal year ended December 31, 2018, filed with the SEC on April 15, 2019; | |
| (2) | the amendment to our Annual Report onForm 20-F/A for the fiscal year ended December 31, 2018, filed with the SEC on April 22, 2019; | |
| (3) | the financial information included in the Report onForm 6-K filed with the SEC dated on April 15, 2019; | |
| (4) | Exhibit 99.1 and 99.3 to the Report onForm 6-K filed with the SEC on September 9, 2019; | |
| (5) | Exhibit 99.1 to the Report onForm 6-K filed with the SEC on October 25, 2019; and, | |
| (5) | the description of our Ordinary Shares contained in our Registration Statement onForm 8-A filed with the SEC on December 14, 2018, including any amendments and reports filed for the purpose of updating such description. |
We will provide a copy of the documents we incorporate by reference, at no cost, to any person who receives this prospectus. To request a copy of any or all of these documents, you should write or telephone us at 17th Floor, Guangdong Investment Tower, 148 Connaught Road Central, Hong Kong, Attention: Sabrina Khan, Chief Financial Officer, +852 2117 6611. Additionally, copies of the documents incorporated herein by reference may be accessed at our website at www.aptorumgroup.com. The reference to our website address does not constitute incorporation by reference of the information contained on or accessible through our website, and you should not consider the contents of our website in making an investment decision with respect to our Class A Ordinary Shares.
154
Financial Statements
Table of Contents
F-1

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Aptorum Group Limited
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets (successor basis) of Aptorum Group Limited (the “Company”) as of December 31, 2018 and 2017, the related consolidated statements (successor basis) of operations and comprehensive loss, equity and cash flows for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, and the statements (predecessor basis) of operations, changes in net assets, and cash flows for the period January 1, 2017 through February 28, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for the year ended December 31, 2018, the period March 1, 2017 through December 31, 2017 and the period January 1, 2017 through February 28, 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.

We have served as the Company’s auditor since 2017.
New York, New York
April 15, 2019

NEW YORK OFFICE ● 7 Penn Plaza ● Suite 830 ● New York, New York ● 10001
Phone 646.442.4845 ● Fax 646.349.5200 ● www.marcumbp.com
F-2
CONSOLIDATED BALANCE SHEETS (SUCCESSOR BASIS)
December 31, 2018 and 2017
(Stated in U.S. Dollars)
December 31, 2018 | December 31, 2017 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 12,006,624 | $ | 16,245,807 | ||||
| Restricted cash | 14,100,614 | 480,000 | ||||||
| Accounts receivable | 2,827 | - | ||||||
| Inventories | 30,642 | - | ||||||
| Marketable securities, at fair value | 1,014,338 | 1,972,648 | ||||||
| Investments in derivatives | 115,721 | 1,095,122 | ||||||
| Amounts due from related parties | 169,051 | - | ||||||
| Due from brokers | 818,968 | 179,492 | ||||||
| Other receivables and prepayments | 464,156 | 310,330 | ||||||
| Total current assets | 28,722,941 | 20,283,399 | ||||||
| Property, plant and equipment, net | 4,260,602 | 346,587 | ||||||
| Non-marketable investments | 7,094,712 | 7,394,713 | ||||||
| Intangible assets, net | 1,409,540 | 1,472,707 | ||||||
| Amounts due from related parties | 50,000 | 304,820 | ||||||
| Long-term deposits | 3,417,178 | 1,757,756 | ||||||
| Other non-current asset | 119,667 | - | ||||||
| Total Assets | $ | 45,074,640 | $ | 31,559,982 | ||||
| LIABILITIES AND EQUITY | ||||||||
| LIABILITIES | ||||||||
| Current liabilities: | ||||||||
| Amounts due to related parties | $ | 33,417 | $ | 197,386 | ||||
| Accounts payable and accrued expenses | 1,247,147 | 653,348 | ||||||
| Finance lease payable, current portion | 43,877 | - | ||||||
| Warrant liabilities | 753,118 | - | ||||||
| Convertible debts | 10,107,306 | 480,000 | ||||||
| Total current liabilities | 12,184,865 | 1,330,734 | ||||||
| Finance lease payable, non-current portion | 143,873 | - | ||||||
| Total Liabilities | $ | 12,328,738 | $ | 1,330,734 | ||||
| Commitments and contingencies | - | - | ||||||
| EQUITY | ||||||||
| Class A Ordinary Shares ($1.00 par value; 60,000,000 shares authorized, 6,537,269 shares issued and outstanding at December 31, 2018 and 5,426,381 shares issued and outstanding at December 31, 2017, respectively) | $ | 6,537,269 | $ | 5,426,381 | ||||
| Class B Ordinary Shares ($1.00 par value; 40,000,000 shares authorized, 22,437,754 shares issued and outstanding as at December 31, 2018 and 2017) | 22,437,754 | 22,437,754 | ||||||
| Additional paid-in capital | 23,003,285 | 5,294,402 | ||||||
| Accumulated other comprehensive loss | (1,484,688 | ) | (367,782 | ) | ||||
| Accumulated deficit | (17,379,185 | ) | (2,547,462 | ) | ||||
| Total equity attributable to the shareholders of Aptorum Group Limited | 33,114,435 | 30,243,293 | ||||||
| Non-controlling interests | (368,533 | ) | (14,045 | ) | ||||
| Total equity | 32,745,902 | 30,229,248 | ||||||
| Total Liabilities and Equity | $ | 45,074,640 | $ | 31,559,982 | ||||
See accompanying notes to the consolidated financial statements.
F-3
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(SUCCESSOR BASIS)
For Year Ended December 31, 2018 and the Period March 1, 2017 through December 31, 2017
(Stated in U.S. Dollars)
| Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||
| Revenue | ||||||||
| Healthcare service income | $ | 383,450 | $ | - | ||||
| Operating expenses | ||||||||
| Cost of healthcare service | (318,011 | ) | - | |||||
| Research and development expenses | (3,101,432 | ) | (2,560,323 | ) | ||||
| General and administrative fees | (4,919,626 | ) | (1,480,093 | ) | ||||
| Legal and professional fees | (1,811,770 | ) | (1,395,490 | ) | ||||
| Other operating expenses | (560,709 | ) | (257,177 | ) | ||||
| Total expenses | (10,711,548 | ) | (5,693,083 | ) | ||||
| Other (loss) income | ||||||||
| Gain on investments in marketable securities, net | 501,522 | 3,912,500 | ||||||
| Loss on investments in derivatives, net | (974,444 | ) | (827,501 | ) | ||||
| Changes in fair value of warrant liabilities | 124,726 | - | ||||||
| Interest (expense) income, net | (4,458,191 | ) | 44,269 | |||||
| Dividend income | - | 2,308 | ||||||
| Total other (loss) income, net | (4,806,387 | ) | 3,131,576 | |||||
| Net loss | (15,134,485 | ) | (2,561,507 | ) | ||||
| Less: net loss attributable to non-controlling interests | (302,762 | ) | (14,045 | ) | ||||
| Net loss attributable to Aptorum Group Limited | $ | (14,831,723 | ) | $ | (2,547,462 | ) | ||
| Net loss per share – basic and diluted | $ | (0.53 | ) | $ | (0.09 | ) | ||
| Weighted-average shares outstanding – basic and diluted | 27,909,788 | 26,963,435 | ||||||
| Net loss | $ | (15,134,485 | ) | $ | (2,561,507 | ) | ||
| Other Comprehensive loss | ||||||||
| Unrealized loss on investments in available-for-sale securities | (1,122,251 | ) | (367,782 | ) | ||||
| Exchange differences on translation of foreign operations | 5,345 | - | ||||||
| Other Comprehensive loss | (1,116,906 | ) | (367,782 | ) | ||||
| Comprehensive loss | (16,251,391 | ) | (2,929,289 | ) | ||||
| Less: comprehensive loss attributable to non-controlling interests | (302,762 | ) | (14,045 | ) | ||||
| Comprehensive loss attributable to the shareholders of Aptorum Group Limited | (15,948,629 | ) | (2,915,244 | ) | ||||
See accompanying notes to the consolidated financial statements.
F-4
STATEMENT OF OPERATIONS (PREDECESSOR BASIS)
For the Period January 1, 2017 through February 28, 2017
(Stated in U.S. Dollars)
January 1, February 28, | ||||
| Investment income | ||||
| Dividend income from unaffiliated issuers | $ | - | ||
| Interest income | 3,011 | |||
| Total investment income | 3,011 | |||
| Expenses | ||||
| General and administrative fees | 17,516 | |||
| Management fees | 108,958 | |||
| Legal and professional fees | 98,646 | |||
| Other operating expenses | 1,907 | |||
| Total expenses | 227,027 | |||
| Net investment loss | $ | (224,016 | ) | |
| Realized and unrealized losses | ||||
| Net realized losses on investments in unaffiliated issuers | $ | (15,327 | ) | |
| Net change in unrealized depreciation on investments | ||||
| Aptorum Therapeutics - related party | (98,434 | ) | ||
| Unaffiliated issuers | (288,307 | ) | ||
| Net realized and unrealized losses | (402,068 | ) | ||
| Net decrease in net assets resulting from operations | $ | (626,084 | ) | |
See accompanying notes to the consolidated financial statements.
F-5
CONSOLIDATED STATEMENTS OF EQUITY (SUCCESSOR BASIS)
For Year Ended December 31, 2018 and the Period March 1, 2017 through December 31, 2017
(Stated in U.S. Dollars)
| Net assets to allocate to equity at February 28, 2017 | $ | 24,488,662 |
| Ordinary shares | Class A Ordinary Shares | Class B Ordinary Shares | Additional Paid-in Capital | Accumulated deficit | Accumulated other comprehensive loss | Non- controlling interests | Total | |||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Amount | Amount | Amount | Amount | Amount | ||||||||||||||||||||||||||||||||||
| Balance, March 1, 2017 | 25,657,110 | $ | 25,657,110 | - | $ | - | - | $ | - | $ | (1,168,448 | ) | $ | - | $ | - | $ | - | $ | 24,488,662 | ||||||||||||||||||||||||
| Proceeds from issuance of shares | 2,207,025 | 2,207,025 | - | - | - | - | 6,394,976 | - | - | - | 8,602,001 | |||||||||||||||||||||||||||||||||
| Converted from ordinary shares | (27,864,135 | ) | (27,864,135 | ) | 5,426,381 | 5,426,381 | 22,437,754 | 22,437,754 | - | - | - | - | - | |||||||||||||||||||||||||||||||
| Unrealized loss on investments in available-for-sale securities | - | - | - | - | - | - | - | - | (367,782 | ) | - | (367,782 | ) | |||||||||||||||||||||||||||||||
| Gain on disposal of entity under common control | - | - | - | - | - | - | 67,874 | - | - | - | 67,874 | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | (2,547,462 | ) | - | (14,045 | ) | (2,561,507 | ) | ||||||||||||||||||||||||||||||
| Balance, December 31, 2017 | - | $ | - | 5,426,381 | $ | 5,426,381 | 22,437,754 | $ | 22,437,754 | $ | 5,294,402 | $ | (2,547,462 | ) | $ | (367,782 | ) | $ | (14,045 | ) | $ | 30,229,248 | ||||||||||||||||||||||
| Issuance of initial public offering of ordinary shares on December 17, 2018 at $15.8 per share, net of underwriting discount and offering expenses | - | - | 761,419 | 761,419 | - | - | 9,536,631 | - | - | - | 10,298,050 | |||||||||||||||||||||||||||||||||
| Proceeds from non-controlling interest | - | - | - | - | - | - | 51,727 | - | - | (51,726 | ) | 1 | ||||||||||||||||||||||||||||||||
| Converted from convertible debts | - | - | 349,469 | 349,469 | - | - | 2,683,839 | - | - | - | 3,033,308 | |||||||||||||||||||||||||||||||||
| Unrealized loss on investments in available-for-sale securities | - | - | - | - | - | - | - | - | (1,122,251 | ) | - | (1,122,251 | ) | |||||||||||||||||||||||||||||||
| Exchange difference on translation of foreign operation | - | - | - | - | - | - | - | - | 5,345 | - | 5,345 | |||||||||||||||||||||||||||||||||
| Beneficial conversion feature | - | - | - | - | - | - | 5,436,686 | - | - | - | 5,436,686 | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | - | (14,831,723 | ) | - | (302,762 | ) | (15,134,485 | ) | ||||||||||||||||||||||||||||||
| Balance, December 31, 2018 | - | $ | - | 6,537,269 | $ | 6,537,269 | 22,437,754 | $ | 22,437,754 | $ | 23,003,285 | $ | (17,379,185 | ) | $ | (1,484,688 | ) | $ | (368,533 | ) | $ | 32,745,902 | ||||||||||||||||||||||
See accompanying notes to the consolidated financial statements.
F-6
STATEMENT OF CHANGES IN NET ASSETS (PREDECESSOR BASIS)
For the Period January 1, 2017 through February 28, 2017
(Stated in U.S. Dollars)
| January 1, 2017 through February 28, 2017 | ||||
| Operations | ||||
| Net investment losses | $ | (224,016 | ) | |
| Net realized losses | (15,327 | ) | ||
| Net change in unrealized depreciation | (386,741 | ) | ||
| Net decrease in net assets resulting from operations | (626,084 | ) | ||
| Distributions to shareholders | ||||
| Equalization payable | 9,663 | |||
| Return of capital | (9,663 | ) | ||
| Total distributions | - | |||
| Total decrease in net assets | (626,084 | ) | ||
| Net assets | ||||
| Beginning of period | 25,114,746 | |||
| End of period | $ | 24,488,662 | ||
See accompanying notes to the consolidated financial statements.
F-7
CONSOLIDATED STATEMENTS OF CASH FLOWS (SUCCESSOR BASIS)
For Year Ended December 31, 2018 and the Period March 1, 2017 through December 31, 2017
(Stated in U.S. Dollars)
| Year Ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (15,134,485 | ) | $ | (2,561,507 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities | ||||||||
| Amortization and depreciation | 682,292 | 58,903 | ||||||
| Gain on investments in marketable securities, net | (501,522 | ) | (3,912,500 | ) | ||||
| Loss on investments in derivatives, net | 974,444 | 827,501 | ||||||
| Changes in fair value of warrant liabilities | (124,726 | ) | - | |||||
| Interest income | (108,512 | ) | - | |||||
| Interest expense and accretion of convertible debts | 4,559,714 | - | ||||||
| Accretion of capital lease obligation | 6,989 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | (2,827 | ) | - | |||||
| Inventories | (30,642 | ) | - | |||||
| Other receivables and prepayments | (45,911 | ) | (303,925 | ) | ||||
| Other non-current asset | (179,500 | ) | - | |||||
| Long-term deposits | (111,951 | ) | (20,092 | ) | ||||
| Due from brokers | 751 | (54,158 | ) | |||||
| Due from related parties | (79,204 | ) | - | |||||
| Due to related parties | 1,004 | - | ||||||
| Accounts payable and accrued expenses | 58,555 | 183,083 | ||||||
| Net cash used in operating activities | (10,035,531 | ) | (5,782,695 | ) | ||||
| Cash flows from investing activities | ||||||||
| Advances to/payments received from related parties | - | (186,898 | ) | |||||
| Purchases of intangible assets | (417,794 | ) | (968,730 | ) | ||||
| Purchases of property, plant and equipment | (5,646,505 | ) | (2,090,721 | ) | ||||
| Proceeds from sales of investment securities | 2,312 | 16,049,067 | ||||||
| Disbursement of a loan to a third party | (3,000,000 | ) | - | |||||
| Repayment of a loan from a third party | 3,000,000 | - | ||||||
| Net cash (used in) provided by investing activities | (6,061,987 | ) | 12,802,718 | |||||
| Cash flows from financing activities | ||||||||
| Proceeds from issuance of convertible debts | 16,120,400 | 480,000 | ||||||
| Proceeds from issuance of shares | 11,054,319 | 8,602,001 | ||||||
| Payments of initial public offering costs | (538,122 | ) | - | |||||
| Payments for debt issuance costs | (1,099,316 | ) | - | |||||
| Payment of finance lease obligations | (58,332 | ) | - | |||||
| Net cash provided by financing activities | 25,478,949 | 9,082,001 | ||||||
| Net increase in cash and restricted cash | 9,381,431 | 16,102,024 | ||||||
| Cash and restricted cash – Beginning of period | 16,725,807 | 623,783 | ||||||
| Cash and restricted cash – End of period | 26,107,238 | $ | 16,725,807 | |||||
| Supplemental disclosures of cash flow information | ||||||||
| Interest paid | $ | 606,989 | $ | - | ||||
| Income taxes paid | $ | - | $ | - | ||||
| Proceeds in broker accounts | $ | 640,227 | $ | - | ||||
| Non-cash investing and financing activities: | ||||||||
| Net settlement of related party balances | $ | 164,973 | $ | - | ||||
| Equipment acquired through finance lease | $ | 239,093 | $ | - | ||||
| Conversion of convertible debts | $ | 3,033,308 | $ | - | ||||
| Reconciliation of cash and restricted cash | ||||||||
| Cash | $ | 12,006,624 | $ | 16,245,807 | ||||
| Restricted cash | 14,100,614 | 480,000 | ||||||
| Total cash and restricted cash shown in the consolidated statements of cash flows | $ | 26,107,238 | $ | 16,725,807 | ||||
See accompanying notes to the consolidated financial statements.
F-8
STATEMENT OF CASH FLOWS (PREDECESSOR BASIS)
For the Period January 1, 2017 through February 28, 2017
(Stated in U.S. Dollars)
January 1, 2017 February 28, | ||||
| Cash flows from operating activities | ||||
| Net decrease in net assets resulting from operations | $ | (626,084 | ) | |
| Adjustments to reconcile net decrease in net assets resulting from operations to net cash used in operating activities: | ||||
| Net change in unrealized depreciation on investments | 386,741 | |||
| Net realized loss on sales of investments in unaffiliated issuers | 15,327 | |||
| Proceeds from sales of investment securities | 28,425 | |||
| Increase in interest receivable | (5,099 | ) | ||
| Increase in due from brokers | (28,438 | ) | ||
| Decrease in other receivable and prepayments | 2,520 | |||
| Increase in accounts payable and accrued expenses | 13,778 | |||
| Decrease in management fees payable - related party | (58,830 | ) | ||
| Net cash used in operating activities | (271,660 | ) | ||
| Net decrease in cash | (271,660 | ) | ||
| Cash - Beginning of period | 301,643 | |||
| Cash - End of period | $ | 29,983 | ||
| Supplemental disclosures of cash flow information | ||||
| Interest paid | $ | - | ||
| Income taxes paid | $ | - | ||
See accompanying notes to the consolidated financial statements.
F-9
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
1. ORGANIZATION
The consolidated financial statements include the financial statements the Aptorum Group Limited (the “Company”) and its subsidiaries. The Company and its subsidiaries are hereinafter collectively referred to as the “Group”.
The Company, formerly known as APTUS Holdings Limited and STRIKER ASIA OPPORTUNITIES FUND CORPORATION, is a company incorporated on September 13, 2010 under the laws of the Cayman Islands with limited liability.
Before March 1, 2017, the Company was incorporated as an exempted open-ended investment company with limited liability in the Cayman Islands, which would own and oversee the management, operations and investments of its subsidiaries. The Company was managed by AENEAS CAPITAL LIMITED, formerly known as APTUS CAPITAL LIMITED or Guardian Capital Management Limited (the “Manager”), with its objective to generate long-term capital appreciation by acquiring, holding and/or investing in, by itself or through one or more of its subsidiaries or other investment vehicles, a wide range of investments, assets and/or rights, with a focus on the healthcare industry. Since March 1, 2017, the Manager enters into a new Management Agreement with the Company to manage certain investment and reinvestment.
On February 21, 2017, a special resolution was passed at the directors’ meeting and on March 1, 2017, a resolution was passed at the shareholders’ meeting. According to which, the Company changed from an investment fund with management shares and non-voting participating redeemable preference shares to a holding company with operating subsidiaries (the “Restructure”).
On March 3, 2017, an ordinary resolution passed at the extraordinary general meeting of the Company and approved by the Cayman Islands Government General Registry changed the name of the Company from STRIKER ASIA OPPORTUNITIES FUND CORPORATION to APTUS Holdings Limited.
On October 13, 2017, a special resolution passed at the extraordinary general meeting of the Company, and on October 19, 2017 it was approved by the Cayman Islands Government General Registry changing the name of the Company from APTUS Holdings Limited to Aptorum Group Limited.
After the Restructure as on March 1, 2017, the Company has become a Hong Kong based pharmaceutical company currently in the preclinical stage. The Company researches and develops life science and biopharmaceutical products within its wholly-owned subsidiary, Aptorum Therapeutics Limited, formerly known as APTUS Therapeutics Limited (“Aptorum Therapeutics”) and its indirect subsidiary companies (collectively, “Aptorum Therapeutics Group”).
F-10
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Below summarizes the list of the subsidiaries consolidated as of December 31, 2018:
| Name | Incorporation date | Ownership | Place of incorporation | Principle activities | ||||||
| Aptorum Therapeutics Limited | June 30, 2016 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| APTUS MANAGEMENT LIMITED | May 16, 2017 | 100% | Hong Kong | Provision of management services to its holding company and fellow subsidiaries | ||||||
| Aptus Therapeutics (Hong Kong) Limited | June 30, 2016 | 100% | Hong Kong | Research and development of life science and biopharmaceutical products | ||||||
| APTUS BIOTECHNOLOGY (MACAO) LIMITED | June 6, 2016 | 99% | Macao | Inactive | ||||||
| Videns Incorporation Limited (Formerly named Videns Biosciences Limited and VIDENS CORPORATION) | March 2, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| mTOR (Hong Kong) Limited | November 4, 2016 | 90% | Hong Kong | Research and development of life science and biopharmaceutical products | ||||||
| Videns Incorporation (Hong Kong) Limited | July 3, 2017 | 100% | Hong Kong | Inactive | ||||||
| Nativus Life Sciences Limited | July 7, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Scipio Life Sciences Limited | July 19, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Claves Life Sciences Limited | August 2, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Nativus Life Sciences (Hong Kong) Limited | August 8, 2017 | 100% | Hong Kong | Inactive | ||||||
| Scipio Life Sciences (Hong Kong) Limited | August 10, 2017 | 100% | Hong Kong | Inactive | ||||||
| Signate Life Sciences (Hong Kong) Limited | August 10, 2017 | 100% | Hong Kong | Inactive | ||||||
| Claves Life Sciences (Hong Kong) Limited | August 22, 2017 | 100% | Hong Kong | Inactive | ||||||
| Aptorum Pharmaceutical Development Limited | August 28, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Aptorum Medical Limited | August 28, 2017 | 95% | Cayman Islands | Provision of medical clinic services | ||||||
| Signate Life Sciences Limited | August 28, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Acticule Life Sciences Limited | June 30, 2017 | 80% | Cayman Islands | Research and development of life science and biopharmaceutical products | ||||||
| Acticule Life Sciences (Hong Kong) Limited | July 27, 2017 | 100% | Hong Kong | Inactive | ||||||
| Forum Property Holding Limited | March 6, 2018 | 100% | Cayman Islands | Inactive | ||||||
| APTORUM INTERNATIONAL LIMITED | March 26, 2018 | 100% | United Kingdom | Inactive | ||||||
| Lanither Life Sciences Limited | April 4, 2018 | 80% | Cayman Islands | Inactive | ||||||
| Lanither Life Sciences (Hong Kong) Limited | May 25, 2018 | 100% | Hong Kong | Inactive | ||||||
Initial public offering
On December 17, 2018, the Group completed an initial public offering (the “IPO” or “Offering”) with new issuance of 761,419 ordinary shares at $15.80 for total offering size of approximately $12.0 million before deducting commissions and expenses. The net proceeds from the IPO was approximately $10.3 million, net off underwriting discount approximately $1.2 million, including warrant issued $0.2 million, and offering costs approximately $0.5 million. The ordinary shares began trading on the NASDAQ Global Market on December 17, 2018 under the ticker symbol “APM”.
F-11
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Deferred offering costs
Deferred offering costs consist principally of legal, printing and registration costs in connection with the Group’s IPO. Such costs are deferred until the closing of the Offering, at which time the deferred costs are offset against the offering proceeds. Deferred offering costs as of December 31, 2018 and 2017 amounted to $nil on the consolidated balance sheets. At the completion of the IPO, US$1,732,229 offering costs was charged to additional paid-in capital.
2. LIQUIDITY
The Company reported a net loss of $15,134,485 and net operation cash outflow of $10,035,531 for the year ended December 31, 2018, respectively. In addition, the Company had an accumulated deficit of $17,379,185 as of December 31, 2018. The Company’s operating results for future periods are subject to numerous uncertainties and it is uncertain if the Company will be able to reduce or eliminate its net losses for the foreseeable future. If management is not able to generate significant revenues from its product candidates currently in development, the Company may not be able to achieve profitability.
The Company’s principal sources of liquidity have been cash and marketable securities. As of the date of issuance of the consolidated financial statements, the Company has approximately $7 million of unrestricted cash. In addition, based upon the current market price of the Company’s marketable securities, it anticipates it can liquidate such marketable securities for greater than its carrying amount, if necessary. In addition, the Company will need to maintain its operating costs at a level which will not exceed such aforementioned sources of funds in order to continue as a going concern for a period within one year after the issuance of its consolidated financial statements.
The Company believes that available cash, together with the efforts from aforementioned management plan and actions, should enable the Company to meet presently anticipated cash needs for at least the next 12 months after the date that the financial statements are issued and the Company has prepared the consolidated financial statements on a going concern basis. However, the Company continues to have ongoing obligations and it expects that it will require additional capital in order to execute its longer-term development plan. If the Company encounters unforeseen circumstances that place constraints on its capital resources, management will be required to take various measures to conserve liquidity, which could include, but not necessarily be limited to, deferring some of its research and seeking to dispose of marketable securities. Management cannot provide any assurance that the Company will raise additional capital if needed.
F-12
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of presentation
The consolidated financial statements are prepared in accordance with U.S. GAAP. Before March 1, 2017, the Company was an investment company under U.S. GAAP for the purposes of financial reporting. U.S. GAAP for an investment company requires investments to be recorded at estimated fair value and the unrealized gains and/or losses in an investment’s fair value are recognized on a current basis in the statements of operations. In addition, the Company did not consolidate its subsidiaries, since they were operating companies and not investment companies. Such entities were fair valued in accordance with ASC Topic 946 (“ASC 946”) and ASC Topic 820 (“ASC 820”).
As of March 1, 2017, after the change of business purpose, legal form and substantive activities, the Company’s status changed to an operating company from an investment company since it no longer met the criteria to qualify as an investment company under the ASC 946. The Company discontinued applying the guidance in ASC 946 and began to account for the change in status prospectively by accounting for its investments in accordance with other U.S. GAAP topics.
This change in status and the accounting policies affect the comparability of the financial statements. As such, for the period January 1, 2017 through February 28, 2017, statements of operations, changes in net assets, and cash flows have been presented on the predecessor basis of accounting as an investment company, and on the basis of accounting as an operating company since March 1, 2017. The consolidated balance sheets as of December 31, 2018 and 2017 have been presented on the successor basis.
Principles of consolidation
The consolidated financial statements of the Group are presented on the accrual basis of accounting in accordance with U.S. GAAP and include the accounts of the Company, its direct and indirect wholly and majority owned subsidiaries and a variable interest entity. All material intercompany balances and transactions have been eliminated in preparation of the consolidated financial statements. Non-controlling interests represent the equity interest that is not owned by the Group.
Use of estimates
The preparation of the consolidated financial statements on successor basis in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of increases and decreases in net assets from operations as well as income and expenses during the reporting period. Significant accounting estimates reflected in the Group’s consolidated financial statements include valuation of warrants, fair value of investments in securities, convertible debts and finance lease, the useful lives of intangible assets and property, plant and equipment, impairment of long-lived assets, valuation allowance for deferred tax assets, and collectability of receivables. Actual results could differ from those estimates.
F-13
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Foreign currency translation and transaction
USD is the reporting currency. The functional currency of subsidiaries in the Cayman Islands is USD, the functional currency of subsidiaries in Hong Kong is Hong Kong Dollars (“HKD”), the functional currency of a subsidiary in Macao is Macanese Pataca (“MOP”) and the functional currency of a subsidiary in the United Kingdom is Great British Pound (“GBP”). An entity’s functional currency is the currency of the primary economic environment in which it operates, normally that is the currency of the environment in which it primarily generates and expends cash. The management considered various indicators, such as cash flows, market expenses, financing and inter-company transactions and arrangements in determining the Group’s functional currency.
In the consolidated financial statements, the financial information of the Company and its subsidiaries, which use HKD, MOP and GBP as their functional currency, has been translated into USD. Assets and liabilities are translated from each subsidiary’s functional currency at the exchange rates on the balance sheet dates, equity amounts are translated at historical exchange rates, and revenues, expenses, gains, and losses are translated using the average exchange rates for the year. Translation adjustments are reported as cumulative translation adjustments and are shown as a separate component of other comprehensive income or loss in the statements of operations and comprehensive loss.
Cash
Cash consists of cash on hand and bank deposits and cash denominated in foreign currencies, which is unrestricted as to withdrawal and use.
Restricted Cash
Restricted cash relates to cash deposited into the escrow account from investors for the purpose of the subscription of convertible debts.
Inventories
Inventories are stated at lower of cost or net realizable value. Cost is determined using the weighted average method.
Where there is evidence that the utility of inventories, in their disposal in the ordinary course of business, will be less than cost, whether due to physical deterioration, obsolescence, changes in price levels, or other causes, the inventories are written down to net realizable value.
Accounts receivable
Accounts receivable are stated at the original amount less an allowance for doubtful receivables, if any, based on a review of all outstanding amounts at period end. An allowance is also made when there is objective evidence that the Group will not be able to collect all amounts due according to the original terms of the receivables. The Group analyzes the aging of the customer accounts, historical and current economic trends and the age of the receivables when evaluating the adequacy of the allowance for doubtful accounts.
Marketable Securities
Marketable Securities are accounted for as trading securities or available-for-sale securities based on the trading purpose, which are measured at fair value. Gains or losses from changes in fair value of trading securities are recorded through earnings. Gains or losses from changes in the fair value of available-for-sale securities are recorded in accumulated other comprehensive income, until the investment is sold or otherwise disposed of, or until the investment is determined to be other-than-temporarily impaired, at which time the cumulative gain or loss previously reported in equity is included in income. The specific identification method is used to determine the realized gain or loss on investments sold or otherwise disposed.
F-14
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
The Group measures the investments in marketable securities at fair value based on quoted market prices. Gains from the marketable securities amounting to $501,522 and $3,912,500, respectively, were recognized in the consolidated statements of operations and comprehensive loss for the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017. The Group recognized the unrealized loss on investments in available-for-sale securities amounting to $1,122,251 and $367,782, respectively, for the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017.
For the year ended December 31, 2018, the Group disposed the available-for-sale securities with sales proceeds of $637,582 received and recognized a gain of $501,522 in the consolidated statements of operations.
During the period March 1, 2017 to December 31, 2017, the Group disposed the trading securities and available-for-sale securities, with sales proceeds of $15,738,517 and $310,550 received, and recognized a gain of $3,917,046 and a loss of $4,546 on the consolidated statements of operations.
Investments in derivatives
Investments in derivatives consisted of warrants, which are measured at fair value, with gains or losses from changes in fair value recorded through earnings. The fair value of these warrants have been determined using the Black-Scholes pricing mode. The Black-Scholes pricing model provides for assumptions regarding volatility, call and put features and risk-free interest rates within the total period to maturity.
For the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017, the Group disposed of warrants with proceeds of $4,957 and $nil received, respectively. Loss on the warrants amounted to $974,444 and $827,501, respectively, was recognized in the consolidated statements of operations for the year ended December 31, 2018 and the period March 1, 2017 to December 31, 2017.
Non-marketable investments
Non-marketable investments are comprising of investments in non-redeemable preferred shares of privately-held companies accounted for under the cost method and are not required to be consolidated under the variable interest or voting models. Non-marketable investments are classified as non-current assets on the consolidated balance sheets as those investments do not have stated contractual maturity dates. Non-marketable equity investments are measured at purchase cost with appropriate consideration given to impairment.
The Group periodically review the equity investments for impairment. The Group considers impairment indicators such as negative changes in industry and market conditions, financial performance, business prospects, and other relevant events and factors. If any impairment is considered other-than-temporary, the fair value of the securities is below the carrying amount, the Group will write down the securities to fair value. As of December 31, 2018 and 2017, the Group believes no impairment charge is necessary.
As of December 31, 2018 and 2017, investments accounted for under the cost method had a carrying value of $7,094,712 and $7,394,713 respectively.
Fair value measurement
Fair value is defined as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required or permitted to be recorded at fair value, the Group considers the principal or most advantageous market in which it would transact its business, and it considers assumptions that market participants would use when pricing the asset or liability.
F-15
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
As a basis for considering such assumptions, a three-tier fair value hierarchy prioritizes the inputs utilized in measuring fair value as follows:
| ● | Level 1 applies to assets or liabilities for which there are quoted prices in active markets for identical assets or liabilities. | |
| ● | Level 2 applies to assets or liabilities for which there are inputs other than quoted prices included within Level 1 that are observable for the asset or liability such as quoted prices for similar assets or liabilities in active markets; quoted prices for identical assets or liabilities in markets with insufficient volume or infrequent transactions (less active markets); or model-derived valuations in which significant inputs are observable or can be derived principally from, or corroborated by, observable market data. | |
| ● | Level 3 applies to assets or liabilities for which there are unobservable inputs to the valuation methodology that are significant to the measurement of the fair value of the assets or liabilities. |
The hierarchy requires the Group to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. A financial instrument’s categorization within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. The Group has estimated the fair value amounts of its financial instruments using the available market information and valuation methodologies considered to be appropriate and has determined that the carrying value of the Group’s cash, restricted cash, due from brokers, other receivables and prepayments, amounts due from/to related parties, and accounts payable and accrued expenses as of December 31, 2018 and 2017 approximate fair value due to the short-term nature of these assets and liabilities.
Property, plant and equipment
Property, plant and equipment is stated at cost less accumulated depreciation. Cost represents the purchase price of the asset and other costs incurred to bring the asset into its existing use. Maintenance, repairs and betterments, including replacement of minor items, are charged to expense; major additions to physical properties are capitalized.
Depreciation of property, plant and equipment is provided using the straight-line method over their estimated useful lives:
| Building | 29 years | |
| Computer equipment | 3 years | |
| Furniture, fixture, and office and medical equipment | 5 years | |
| Leasehold improvements | Shorter of the remaining lease terms or 5 years | |
| Laboratory equipment | 5 years | |
| Motor vehicle | 5 years |
Upon sale or disposal, the applicable amounts of asset cost and accumulated depreciation are removed from the accounts and the net amount less proceeds from disposal is charged or credited to income.
Other non-current asset
Other non-current asset represents laboratory supplies that can be used for more than one year. Cost represents the purchase price of the supplies.
Amortization of other non-current asset is provided using the straight-line method over their estimated useful lives. The amortization expenses for the year ended December 31, 2018 is $59,833.
F-16
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Intangible assets
Indefinite-lived intangible assets are tested for impairment at least annually and are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Indefinite-lived intangible assets are impaired if their estimated fair values are less than their carrying values.
Finite-lived intangible assets are initially recorded at fair value when acquired, in which the finite intangible assets are amortized over their estimated useful life, which is the period over which the assets are expected to contribute directly or indirectly to the future cash flows of the Group. These intangible assets are tested for impairment at the time of a triggering event, if one were to occur. Finite-lived intangible assets may be impaired when the estimated undiscounted future cash flows generated from the assets are less than their carrying amounts.
The Group may rely on a qualitative assessment when performing its intangible asset impairment test. Otherwise, the impairment evaluation is performed at the lowest level of identifiable cash flows independent of other assets.
The Group’s intangible assets mainly consist of computer software, exclusive rights in prepaid patented and unpatented licenses. The prepaid patented licenses are for clinical purpose or further development into other products. Prepaid unpatented license is for further development, once the associated research and development efforts are completed, the prepaid unpatented license will be reclassified as a finite-lived asset and is amortized over its useful life. The estimated useful life of the exclusive rights in using patents is generally the remaining patent life from the acquisition date to expiration date under the law, which is 17 to 20 years, the Group will reassess the remaining patent life on annual basis, and the Group will assess the intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may no longer be recoverable.
Impairment of long-lived assets
The Group reviews its long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may no longer be recoverable. When these events occur, the Group measures impairment by comparing the carrying value of the long-lived assets to the estimated undiscounted future cash flows expected to result from the use of the assets and their eventual disposition. If the sum of the expected undiscounted cash flow is less than the carrying amount of the assets, the Group would recognize an impairment loss, which is the excess of carrying amount over the fair value of the assets, using the expected future discounted cash flows.
Convertible debts
The Group determines the appropriate accounting treatment of its convertible debts in accordance with the terms in relation to the conversion feature, call and put option, beneficial conversion feature (“BCF”) and settlement feature. After considering the impact of such features, the Group concludes that, as of December 31, 2017, the convertible debts contained a contingent beneficial conversion, which shall not be recognized in earnings until the contingency is resolved, and therefore accounts for such instrument as a liability in its entirety.
Convertible debts were subsequently measured at amortized cost, using the effective interest rate method. Amortized cost is calculated by taking into account any discount or premium on acquisition and fees or costs that are an integral part of the effective interest rate. The effective interest rate amortization is included in interest expense in the consolidated statements of operations.
Amortized cost is calculated by taking into account any discount or premium on acquisition and fees or costs that are an integral part of the effective interest rate. The effective interest rate amortization is included in finance costs in the condensed consolidated statements of operations.
F-17
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Management concluded that the contingency was effectively resolved upon the completion of the IPO on December 17, 2018 so that part of the convertible debts were converted automatically accordingly. The BCF derecognized upon automatic conversion was recorded as interest expense with a corresponding increase to additional paid-in capital. The remaining BCF was recorded as debt discount, which was amortized through the maturity of the convertible debts, with a corresponding increase to additional paid-in capital.
Finance lease
Leases that transfer substantially all the rewards and risks of ownership of assets to the Group, other than legal title, are accounted for as finance leases. At the inception of a finance lease, the cost of the leased asset is capitalized at the present value of the minimum lease payments and recorded together with the obligation, excluding the interest element, to reflect the purchase and financing. Assets held under capitalized finance leases are included in property, plant and equipment, and depreciated over the shorter of the lease terms and the estimated useful lives of the assets. The interest expenses of such leases are charged to the consolidated statements of operations so as to provide a constant periodic rate of charge over the lease terms.
Warrant liabilities
For warrants that are not indexed to the Group’s shares, the Group records the fair value of the issued warrants as liabilities at each balance sheet date and records changes in the estimated fair value as a non-cash gain or loss in the consolidated statements of operations and comprehensive loss. The warrant liabilities are recognized in the consolidated balance sheets at the fair value (level 3). The fair value of these warrants have been determined using the Black-Scholes pricing mode. The Black-Scholes pricing model provides for assumptions regarding volatility, call and put features and risk-free interest rates within the total period to maturity.
Revenue recognition
Dividend income is recorded on the ex-dividend date, and interest income is recorded on an accrual basis.
Healthcare service income is recognized when persuasive evidence of the services is rendered, the services price is fixed or determinable and collectability of the receivable is reasonably assured.
Cost of healthcare service
Cost of healthcare service rendered represents cost in relation to the medical services provided including the cost of pharmaceutical supplies and medicine.
Research and development expenses
Research and development costs are expensed as incurred. Research and development expenses are comprised of costs incurred in performing research and development activities, including amortization of the patent license, depreciation of laboratory equipment, external costs of outside vendors engaged to conduct preclinical development activities and trials.
F-18
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Income taxes
The Group accounts for income taxes under the asset and liability method. Under this method, deferred income taxes are determined based on differences between the financial carrying amounts of existing assets and liabilities and their tax bases. Income taxes are provided for in accordance with the laws of the relevant taxing authorities.
A valuation allowance is provided for deferred tax assets if it is more likely than not that these items will either expire before the Group is able to realize their benefits, or that future deductibility is uncertain. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount expected to be realized.
Uncertain tax positions
The Group accounts for uncertainty in income taxes using a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon settlement. Interest and penalties related to uncertain tax positions are recognized and recorded as necessary in the provision for income taxes. The Group recognizes interest on non-payment of income taxes and penalties associated with tax positions when a tax position does not meet more likely than not thresholds be sustained under examination. The tax returns of the Group’s Hong Kong subsidiaries and variable interest entity (“VIE”) are subject to examination by the relevant tax authorities. According to the Hong Kong Inland Revenue Department, the statute of limitation is six years if any company chargeable with tax has not been assessed or has been assessed at less than the proper amount, the statute of limitation is extended to ten years if the underpayment of taxes is due to fraud or willful evasion. According to United Kingdom’s policy, trading losses are available to be carried forward indefinitely. The Group did not have any material interest or penalties associated with tax positions for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, and did not have any significant unrecognized uncertain tax positions as of December 31, 2018 and 2017. The Group does not believe that its assessment regarding unrecognized tax benefits will materially change over the next twelve months.
Comprehensive income or loss
U.S. GAAP generally requires that recognized revenue, expenses, gains and losses be included in net income or loss. Although certain changes in assets and liabilities are reported as separate components of the equity section of the consolidated balance sheets, such items, along with net income, are components of comprehensive income or loss. The components of other comprehensive income or loss consist of unrealized gain or loss on available-for-sale short-term investments and exchange differences on translation of foreign operations.
Loss per share
Basic loss per share is computed by dividing net loss attributable to ordinary shareholders by the weighted average number of ordinary shares outstanding during the period. Diluted loss per share reflects the potential dilution that could occur if securities or other contracts to issue ordinary shares were exercised or converted into ordinary shares. Potential dilutive securities are excluded from the calculation of diluted loss per share in loss periods as their effect would be anti-dilutive.
F-19
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Recently issued accounting standards
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”), which was subsequently modified in August 2015 by ASU 2015-14, Revenue from Contracts with Customers: Deferral of the Effective Date. This guidance will be effective for fiscal years (and interim reporting periods within those years) beginning after December 15, 2017. The core principle of ASU 2014-09 is that companies should recognize revenue when the transfer of promised goods or services to customers occurs in an amount that reflects what the company expects to receive. It requires additional disclosures to describe the nature, amount, timing and uncertainty of revenue and cash flows from contracts with customers. In 2016, the FASB issued additional ASUs that clarify the implementation guidance on principal versus agent considerations (ASU 2016-08), on identifying performance obligations and licensing (ASU 2016-10), and on narrow-scope improvements and practical expedients (ASU 2016-12) as well as on the revenue recognition criteria and other technical corrections (ASU 2016-20). In 2017, the FASB issued Accounting Standards Update (ASU) 2017-05, Other Income—Gains and Losses from the Derecognition of Nonfinancial Assets (Subtopic 610-20), which was originally issued in ASU 2014-09. The amendments in this Update require that an entity to initially measure a retained non-controlling interest in a nonfinancial asset at fair value consistent with a how a retained non-controlling interest in a business is measured.
Under Topic 606, an entity recognizes revenue when the consultation service was completed, the control of the medicine was delivered to the patients and the appointment of the laboratory test was made, in an amount that reflects the consideration which the entity expects to receive in exchange for those goods or services. It also impacts certain other areas, such as the accounting for costs to obtain or fulfill a contract. The standard also requires disclosure of the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers.
The Group is an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2010 (the “JOBS Act”). Under the JOBS Act, emerging growth companies (“EGCs”) can delay adopting new or revised accounting standards issued subsequent to the enactment of the JOBS Act until such time as those standards apply to private companies. Management has adopted this standard effective January 1, 2019 using the modified-retrospective approach, in which case the cumulative effect of applying the standard would be recognized at the date of initial application. The Group also estimates there will not be a material impact to the beginning balance of retained earnings.
In January 2016, the FASB issued ASU No. 2016-01, Financial Instruments—Overall: Recognition and Measurement of Financial Assets and Financial Liabilities (ASU 2016-01), which requires equity investments to be measured at fair value through net income. Equity investments that are accounted for under the equity method are not impacted. ASU 2016-01 provides that equity investments without readily determinable fair values can be valued at cost minus impairment using a simplified impairment assessment that utilizes qualitative assessments. ASU 2016-01 requires separate presentation of the financial assets and liabilities by category and form. ASU 2016-01 should be applied prospectively and will be effective for fiscal years beginning after December 15, 2017, and for interim periods within those fiscal years. As an EGC, the Group chose to delay the adoption of the update for one year. The Group adopted the new standard on January 1, 2019. The most significant impact to the consolidated financial statements relates to the recognition and measurement of equity investments at fair value in the consolidated statements of operations. The management has elected to use the measurement alternative, defined as cost, less impairments, adjusted by observable price changes. The management anticipates that the adoption of ASU 2016-01 will increase the volatility of the other (loss) income, net, as a result of the remeasurement of the equity securities upon the occurrence of observable price changes and impairments.
In February 2016, the FASB issued ASU 2016-02, Leases (“ASU 2016-02”), which requires a lessee to recognize a right-of-use asset and a lease liability for operating leases, initially measured at the present value of the future lease payments, in the balance sheet. ASU 2016-02 also requires a lessee to recognize a single lease cost, calculated so that the cost of the lease is allocated over the lease term, generally on a straight-line basis. This new guidance is effective for fiscal years beginning after December 15, 2019. Early adoption is permitted. The Group is currently evaluating the potential effects of adopting the provisions of ASU 2016-02 on its consolidated financial statements.
F-20
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
In August 2018, the FASB issued ASU No. 2018-13, Disclosure Framework – Changes to the Disclosure Requirements for Fair Value Measurement, which amends ASC 820, Fair Value Measurement. This ASU modifies the disclosure requirements for fair value measurements by removing, modifying, or adding certain disclosures. The effective date is the first quarter of fiscal year 2021, with early adoption permitted for the removed disclosures and delayed adoption until fiscal year 2021 permitted for the new disclosures. The removed and modified disclosures will be adopted on a retrospective basis and the new disclosures will be adopted on a prospective basis. The adoption will not have a material effect on the Group’s financial statements.
The Group does not believe other recently issued but not yet effective accounting standards, if currently adopted, would have a material effect on the consolidated financial position, statements of operations and cash flows.
4. CHANGE IN STATUS
Prior to the March 1, 2017 change in status as an investment company, the Company recorded its investments at fair value and recorded the changes in the fair value as unrealized gain or loss. In addition, the Company recorded its direct and indirect wholly and majority owned subsidiaries at fair value since they were operating companies not providing services to the Company and not investment companies.
Upon the effective date of the change in status, the fair value accounting as an investment company was no longer applicable to the Company, rather the Company began presenting such subsidiaries on a consolidated basis. The investments in unaffiliated issuers are measured at fair value or cost, less impairment. The Company’s initial carrying value of the net assets of the investments in subsidiaries was the fair value on the effective date of the change in status determined as follows:
| Fair value of subsidiaries as of the effective date of the change in status on March 1, 2017 | $ | 757,647 | ||||||
| Total net assets of the combined properties | ||||||||
| Intangible assets, net | $ | 194,146 | ||||||
| Cash | 593,800 | |||||||
| Prepayments | 256 | |||||||
| An amount due to a related party | (28,717 | ) | ||||||
| Accounts payable and accrued expenses | (207,692 | ) | 551,793 | |||||
| Increase to the initial carrying value of the net assets on the effective date of the change in status on March 1, 2017 | $ | 205,854 |
5. VARIABLE INTEREST ENTITY
On July 28, 2017, the Group, through one of its subsidiaries, Aptorum Therapeutics Limited, entered into a convertible loan agreement (the “Agreement”) with Acticule Life Sciences Limited (“Acticule”), at interest rate of 0% but no amount or maturity limits.
Acticule was incorporated by an individual on June 30, 2017, with paid-in capital of $1. Acticule mainly engaged in research and development of life science and biopharmaceutical products. From July 28, 2017 to December 22, 2017, Acticule has drawn down the loan in aggregate amount of $1,000,000. Other than that, Acticule has not obtained any financial support for its business operation.
After evaluation of the design of Acticule as the basis for determining its variability in applying the variable interest entity model, the Group believes that Acticule was a VIE, and the Group is the primary beneficiary, due to the Group has the power to ultimately direct the activities and significantly affect its economic performance, as well as the obligation to absorb losses or the right to receive benefit from Acticule that could potentially be significant to Acticule. Therefore, the financial statement of Acticule was consolidated by the Group since the first loan drawn down to Acticule on July 28, 2017.
F-21
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
On December 22, 2017, Acticule accepted the election made by the Group to convert the entire loan of $1,000,000 into shares in Acticule. After the conversion, the Group held approximately 100% equity interest of Acticule, which ceased to be a VIE but consolidated by the Group under the voting interest entity model thereafter.
From July 28, 2017 to December 22, 2017, Acticule was consolidated under the VIE model, and its operating expense and net loss are listed below:
| July 28, 2017 through December 22, 2017 | ||||
| Total expense | $ | 559,850 | ||
| Net loss | $ | 559,850 | ||
6. FAIR VALUE MEASUREMENT
The following table provides the assets and liabilities carried at fair value measured on a recurring basis as of December 31, 2018 and 2017:
| December 31, 2018 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Current Assets | ||||||||||||||||
| Marketable securities – Available-for-sale securities | ||||||||||||||||
| Common stocks | $ | 813,728 | $ | 200,610 | $ | - | $ | 1,014,338 | ||||||||
| Investments in derivatives | ||||||||||||||||
| Warrants | - | - | 115,721 | 115,721 | ||||||||||||
| Total assets at fair value | $ | 813,728 | $ | 200,610 | $ | 115,721 | $ | 1,130,059 | ||||||||
December 31, 2017 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Current Assets | ||||||||||||||||
| Marketable securities – Available-for-sale securities | ||||||||||||||||
| Common stocks | $ | - | $ | 1,972,648 | $ | - | $ | 1,972,648 | ||||||||
| Investments in derivatives | ||||||||||||||||
| Warrants | 24,182 | - | 1,070,940 | 1,095,122 | ||||||||||||
| Total assets at fair value | $ | 24,182 | $ | 1,972,648 | $ | 1,070,940 | $ | 3,067,770 | ||||||||
The following is a reconciliation of Level 3 assets during the year ended December 31, 2018:
| Warrants | ||||
| Balance at January 1, 2018 | $ | 1,070,940 | ||
| Change in unrealized depreciation | (955,219 | ) | ||
| Balance at December 31, 2018 | $ | 115,721 | ||
| Net change in unrealized depreciation relating to investments still held at December 31, 2018 | (955,219 | ) | ||
F-22
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
The following is a reconciliation of Level 3 assets for the period February 28, 2017 through December 31, 2017:
Aptorum Therapeutics– related party | Common Stocks | Preferred Stocks | Warrants | Convertible Notes | Total | |||||||||||||||||||
| Balance at February 28, 2017 | $ | 757,647 | $ | 7,920,000 | $ | 4,314,998 | $ | 1,907,470 | $ | 3,082,020 | $ | 17,982,135 | ||||||||||||
| Transfer out of Level 3 due to change in status – consolidated subsidiary (a) | (757,647 | ) | - | - | - | - | (757,647 | ) | ||||||||||||||||
| Transfer out of fair value leveling since recorded as cost method (b) | - | (7,920,000 | ) | (4,314,998 | ) | - | - | (12,234,998 | ) | |||||||||||||||
| Balance at March 1, 2017 | $ | - | $ | - | $ | - | $ | 1,907,470 | $ | 3,082,020 | $ | 4,989,490 | ||||||||||||
| Reclassification between different investment type (c) | - | - | 3,079,715 | - | (3,079,715 | ) | - | |||||||||||||||||
| Transfer out of fair value leveling since recorded as cost method (c) | - | - | (3,079,715 | ) | - | - | (3,079,715 | ) | ||||||||||||||||
| Change in unrealized depreciation | - | - | - | (836,530 | ) | (2,305 | ) | (838,835 | ) | |||||||||||||||
| Balance at December 31, 2017 | $ | - | $ | - | $ | - | $ | 1,070,940 | $ | - | $ | 1,070,940 | ||||||||||||
| Net change in unrealized depreciation relating to investments still held at December 31, 2017 | - | - | - | (836,530 | ) | - | (836,530 | ) | ||||||||||||||||
| a. | Upon the effective date of the change in status, March 1, 2017, the subsidiaries were no longer recognized at fair value and were instead consolidated when preparing the financial statements. |
| b. | The equity investments of common stock and preferred stock were non-marketable investments under cost method upon change in status. Subsequently, Athenex Inc. was listed on the NASDAQ stock exchange on June 14, 2017 and common stock with an amount of $7,920,000 has been transferred to common stock in Level 1 with amount of $7,920,000, which was subsequently sold in December 2017 with a gain from the marketable securities of $3,722,234 recognized. |
| c. | On March 9, 2017, the convertible promissory notes (including its accrued interest, totally $520,822) of Centrexion Therapeutics Corporation was converted into preferred stock (Series C) of the same company. On May 25, 2017, the convertible promissory notes (including its accrued interest, totaling $2,558,893) of Alzheon Inc., was converted into preferred stock (Series B) of the same company. The preferred stocks are considered non-marketable investments and were therefore reclassified out of the fair value hierarchy to be reported under cost method. |
F-23
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
The following table presents the quantitative information about the Group’s Level 3 fair value measurements of investment as of December 31, 2018 and 2017, which utilized significant unobservable internally-developed inputs:
| December 31, 2018 | Valuation technique | Unobservable input | Range (weighted average) | Sensitivity of fair value to input | ||||
| Warrants | Black-Scholes Model | Estimated time to exit Historical Volatility | 12-30 months 73% - 188% | 10% increase (decrease) in volatility would result in increase (decrease) in fair value by $19,691 |
December 31, | Valuation technique | Unobservable input | Range (weighted average) | Sensitivity of fair value to input | ||||
| Warrants | Black-Scholes Model | Estimated time to exit Historical Volatility | 24-42 months 97% - 136% | 10% increase (decrease) in volatility would result in increase (decrease) in fair value by $122,664 |
Warrants
As of December 31, 2018 and 2017, the volume of the Group’s derivative activities based on their notional amount and number of contracts, categorized by primary underlying risk, are as follows:
| Long Exposure | ||||||||||||
| December 31, 2018 | December 31, 2017 | |||||||||||
| Primary underlying risk | Notional Amounts | Number of Contracts | Notional Amounts | Number of Contracts | ||||||||
| Equity Price | ||||||||||||
| Warrants | $ | 218,270 | 2,257,682 | $ | 2,261,530 | 2,338,290 | ||||||
The following table identifies the fair value amounts of derivative instruments included in the consolidated balance sheets as derivative contracts, categorized by primary underlying risk, at December 31, 2018 and 2017. The following table also identifies the net gain and loss amounts included in the consolidated statement of operations as net unrealized gain from derivative contracts, categorized by primary underlying risk, for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017:
| Year ended December 31, 2018 | ||||||||||||||||
| Primary underlying risk | Derivative assets | Derivative liabilities | Realized loss | Unrealized loss | ||||||||||||
| Equity Price | ||||||||||||||||
| Warrants | $ | 115,721 | $ | - | $ | (19,225 | ) | $ | (955,219 | ) | ||||||
| March 1, 2017 through December 31, 2017 | ||||||||||||||||
| Primary underlying risk | Derivative assets | Derivative liabilities | Realized loss | Unrealized loss | ||||||||||||
| Equity Price | ||||||||||||||||
| Warrants | $ | 1,095,122 | $ | - | $ | (7,094 | ) | $ | (820,407 | ) | ||||||
F-24
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
7. OTHER RECEIVABLES AND PREPAYMENTS
Other receivables and prepayments as of December 31, 2018 and 2017 consisted of:
December 31, 2018 | December 31, 2017 | |||||||
| Prepaid insurance | $ | 147,864 | $ | 107,842 | ||||
| Prepaid service fee | 75,224 | 91,002 | ||||||
| Rental deposits | 8,576 | 61,333 | ||||||
| Prepaid rental expenses | 46,948 | 11,910 | ||||||
| Prepaid research and development expenses | 41,614 | - | ||||||
| Other receivables | 109,435 | 16,186 | ||||||
| Others | 34,495 | 22,057 | ||||||
| $ | 464,156 | $ | 310,330 | |||||
8. PROPERTY, PLANT AND EQUIPMENT, NET
Property, plant and equipment as of December 31, 2018 and 2017 consisted of:
December 31, 2018 | December 31, 2017 | |||||||
| Building | $ | 1,488,396 | $ | - | ||||
| Computer equipment | 64,911 | 14,057 | ||||||
| Furniture, fixture, and office and medical equipment | 262,819 | - | ||||||
| Leasehold improvements | 664,713 | - | ||||||
| Laboratory equipment | 2,045,034 | 339,000 | ||||||
| Motor vehicle | 239,093 | - | ||||||
| 4,764,966 | 353,057 | |||||||
| Less: accumulated depreciation | 504,364 | 6,470 | ||||||
| Property, plant and equipment, net | $ | 4,260,602 | $ | 346,587 | ||||
Depreciation expenses for property, plant and equipment amounted to $497,908 and $6,470 for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, respectively.
F-25
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
9. INTANGIBLE ASSETS, NET
December 31, 2018 | December 31, 2017 | |||||||
| Gross carrying amount | ||||||||
| Prepaid unpatented license | $ | 200,000 | $ | 200,000 | ||||
| Prepaid patented licenses | 1,322,820 | 1,322,820 | ||||||
| Computer software | 61,384 | 2,320 | ||||||
| 1,584,204 | 1,525,140 | |||||||
| Less: accumulated amortization | ||||||||
| Prepaid patented licenses | 155,026 | 52,433 | ||||||
| Computer software | 19,638 | - | ||||||
| 174,664 | 52,433 | |||||||
| Intangible assets, net | ||||||||
| Prepaid unpatented license | 200,000 | 200,000 | ||||||
| Prepaid patented licenses | 1,167,794 | 1,270,387 | ||||||
| Computer software | 41,746 | 2,320 | ||||||
| Intangible assets, net | $ | 1,409,540 | $ | 1,472,707 | ||||
As of December 31, 2018 and 2017, the Group has capitalized seven of the exclusive licenses which includes seven patented and one unpatented technologies in the areas of neurology, infectious diseases, gastroenterology, oncology, surgical robotics and natural health. Pursuant to the license agreements, the Group paid upfront payments and became the exclusive licensee to prosecute certain patents developed or licensed under the applicable agreements.
The Group recognized the prepaid unpatented license to reflect the fair value of the subsidiaries as of the date of the change in status from an investment company. The Group capitalizes the prepaid patented license for the exclusive rights with completed filing of patents in certain jurisdictions (e.g., the United States of America and Europe) and alternative future uses.
Prepaid unpatented license is indefinite-lived intangible assets which are tested for impairment annually. Prepaid patented licenses and computer software are finite-lived intangible assets which are amortized over their estimated useful life. Amortization expenses for finite-lived intangible assets amounted to $124,551 and $52,433 for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, respectively. The Group wrote off the cost and the related amortization of $2,320 after the expiration of the computer software for the year ended December 31, 2018.
The Group expects amortization expense related to its finite-lived intangible assets for the next five years and thereafter to be as follows as of December 31, 2018:
| For the years ending December 31, | Amount | |||
| 2019 | $ | 133,095 | ||
| 2020 | 111,588 | |||
| 2021 | 104,842 | |||
| 2022 | 102,593 | |||
| 2023 | 102,593 | |||
| Thereafter | 654,829 | |||
| Total | $ | 1,209,540 | ||
F-26
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
10. LONG-TERM DEPOSITS
Long-term deposits as of December 31, 2018 and 2017 consisted of:
December 31, 2018 | December 31, 2017 | |||||||
| Rental deposits | $ | 132,043 | $ | 20,092 | ||||
| Prepayments for equipment | 3,285,135 | 1,737,664 | ||||||
| $ | 3,417,178 | $ | 1,757,756 | |||||
11. ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses as of December 31, 2018 and 2017 consisted of:
December 31, 2018 | December 31, 2017 | |||||||
| License agreements payable | $ | - | $ | 356,410 | ||||
| Healthcare consultation service payable | 40,139 | - | ||||||
| Professional fees payable | 178,117 | 154,429 | ||||||
| Research and development expenses payable | 398,899 | 104,013 | ||||||
| Interest payable | 223,802 | - | ||||||
| Payables for leasehold improvement and equipment | 73,864 | - | ||||||
| Salary payable | 183,065 | - | ||||||
| Deferred rent | 58,810 | - | ||||||
| Others | 90,451 | 38,496 | ||||||
| $ | 1,247,147 | $ | 653,348 | |||||
12. INCOME TAXES
The Company and its subsidiaries file tax returns separately.
Income taxes
Cayman Islands: under the current laws of the Cayman Islands, the Company and its subsidiaries in the Cayman Islands are not subject to taxes on their income and capital gains.
Hong Kong: in accordance with the relevant tax laws and regulations of Hong Kong, a company registered in Hong Kong is subject to income taxes within Hong Kong at the applicable tax rate on taxable income. In March 2018, the Hong Kong Government introduced a two-tiered profit tax rate regime by enacting the Inland Revenue (Amendment) (No.3) Ordinance 2018 (the “Ordinance”). Under the two-tiered profits tax rate regime, the first $2 million of assessable profits of qualifying corporations is taxed at 8.25% and the remaining assessable profits at 16.5%. The Ordinance is effective from the year of assessment 2018-2019. According to the policy, if no election has been made, the whole of the taxpaying entity’s assessable profits will be chargeable to Profits Tax at the rate of 16.5% or 15%, as applicable. Because the preferential tax treatment is not elected by the Group, all the subsidiaries registered in Hong Kong are subject to income tax at a rate of 16.5%. The subsidiaries registered in Hong Kong did not have assessable profits that were derived Hong Kong during the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017. Therefore, no Hong Kong profit tax has been provided for in the periods presented.
Macao: Taxpayers in Macao are divided into Group A and Group B, Group A taxpayers are companies that have maintained proper accounting books and records, with capital of MOP1,000,000 and above or average assessed annual taxable profits in the past three years of more than MOP500,000, those who do not meet the criteria of Group A taxpayers are assigned to Group B. Group B taxpayers are assessed by the Macao Finance Bureau on a deemed profit basis, and Group B taxpayers are unable to carry forward tax losses. The capital of the subsidiary in Macao is MOP100,000 and it is assigned to Group B taxpayer. The tax loss of subsidiary in Macao cannot be utilized.
F-27
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
United Kingdom: in accordance with the relevant tax laws and regulations of United Kingdom, a company registered in the United Kingdom is subject to income taxes within United Kingdom at the applicable tax rate on taxable income. All the United Kingdom subsidiaries that are not entitled to any tax holiday were subject to income tax at a rate of 19%. The subsidiary in United Kingdom did not have assessable profits that were derived United Kingdom during the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017. Therefore, no United Kingdom profit tax has been provided for in the periods presented.
The components of the provision for income taxes expenses are:
| Year ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||
| Current | $ | - | $ | - | ||||
| Deferred | - | - | ||||||
| Total income taxes expense | $ | - | $ | - |
The reconciliation of income taxes expenses computed at the Hong Kong statutory tax rate applicable to income tax expense is as follows:
| Year ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||
| Net loss before tax | $ | (15,134,485 | ) | $ | (2,561,507 | ) | ||
| Provision for income taxes at Hong Kong statutory income tax rate (16.5%) | (2,497,190 | ) | (422,649 | ) | ||||
| Impact of different tax rates in other jurisdictions | (3,066 | ) | - | |||||
| Non-taxable income | (95,018 | ) | - | |||||
| Non-deductible expenses | 540,893 | - | ||||||
| Prior year tax effect | - | (576,970 | ) | |||||
| Change in valuation allowance | 2,054,381 | 999,619 | ||||||
| Effective income tax expense | $ | - | $ | - | ||||
F-28
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Deferred tax asset, net
Deferred tax assets and deferred tax liabilities reflect the tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purpose and the tax bases used for income tax purpose. The following represents the tax effect of each major type of temporary difference.
| December 31, 2018 | December 31, 2017 | |||||||
| Deferred tax asset: | ||||||||
| Tax loss carry forward | $ | 3,499,428 | $ | 1,249,900 | ||||
| Deferred tax liability: | ||||||||
| Depreciation and amortization | (445,428 | ) | (250,281 | ) | ||||
| Net deferred tax assets before valuation allowance | 3,054,000 | 999,619 | ||||||
| Valuation allowance | (3,054,000 | ) | (999,619 | ) | ||||
| Deferred tax asset, net | $ | - | $ | - | ||||
As of December 31, 2018 and 2017, the Group had net operating loss carry-forwards of $21,191,279 and $7,575,154, respectively, including its Hong Kong and United Kingdom operations, which are available to reduce future taxable income; and all of these losses can be carried forward indefinitely.
Valuation allowance was provided against deferred tax assets in entities where it was determined, it was more likely than not that the benefits of the deferred tax assets will not be realized. The Group had deferred tax assets which consisted of tax loss carry forward, which can be carried forward to offset future taxable income. The Group maintains a full valuation allowance on its net deferred tax assets. The management determines it is more likely than not that all of its deferred tax assets will not be utilized. The valuation allowance increased by $2,054,381 and $999,619, respectively, for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017.
During the preparation of the Group’s financial statements for the year ended December 31, 2018, the Group made adjustments to the previously issued consolidated financial statements included in Form F-1 filed with the SEC related to: 1) deferred tax assets and valuation allowance; and 2) deferred tax liabilities, due to that the Cayman Islands subsidiaries which are registered as non-Hong Kong companies in Hong Kong may subject to income taxes within Hong Kong at the applicable tax rate on taxable income. The Company evaluated the materiality of this adjustment and concluded that its impact was not material on its financial statements taken as a whole and did not affect the March 1, 2017 and December 31, 2017 balance sheets, statements of operations, stockholder’s equity and cash flows for the periods ended March 1, 2017 and December 31, 2017. The Company elected to adjust the balances in the following tables.
The reconciliation of income taxes expenses computed at the Hong Kong statutory tax rate applicable to income tax expense is as follows:
| March 1, 2017 through December 31, 2017 | ||||||||||||
| As previously reported | Adjustments | As adjusted | ||||||||||
| (audited) | ||||||||||||
| Net loss before tax | (2,561,507) | - | (2,561,507) | |||||||||
| Provision for income taxes at Hong Kong statutory income tax rate (16.5%) | (422,649 | ) | - | (422,649 | ) | |||||||
| Previous year tax effect | - | (576,970 | ) | (576,970 | ) | |||||||
| Impact of different tax rates in other jurisdictions | 393,217 | (393,217 | ) | - | ||||||||
| Changes in valuation allowance | 29,432 | 970,187 | 999,619 | |||||||||
| Effective income tax expense | - | - | - | |||||||||
F-29
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Deferred tax asset, net
| December 31, 2017 | ||||||||||||
| As previously reported | Adjustments | As adjusted | ||||||||||
| (audited) | ||||||||||||
| Deferred tax asset: | ||||||||||||
| Tax loss carry forward | 29,432 | 1,220,468 | 1,249,900 | |||||||||
| Deferred tax liability | ||||||||||||
| Depreciation and amortization | - | (250,281 | ) | (250,281 | ) | |||||||
| Net deferred tax assets before valuation allowance | 29,432 | 970,187 | 999,619 | |||||||||
| Valuation allowance | (29,432 | ) | (970,187 | ) | (999,619 | ) | ||||||
| Deferred tax asset, net | - | - | - | |||||||||
13. RELATED PARTY BALANCES AND TRANSACTIONS
The following is a list of a director and related parties to which the Group has transactions with:
| (a) | Ian Huen, the Chief Executive Officer and Executive Director of the Group; |
| (b) | AENEAS CAPITAL LIMITED, an entity controlled by Darren Lui, the Executive Director of the Group; |
| (c) | Aeneas Limited, formerly known as Aptus Financial Holdings Limited, an entity controlled by Ian Huen; |
| (d) | Aeneas Group Limited, formerly known as Aptus Asia Financial Holdings Limited, an entity controlled by Ian Huen. |
| (e) | Aeneas Management Limited, an entity controlled by Ian Huen. |
| (f) | Jurchen Investment Corporation, the holding company and an entity controlled by Ian Huen. |
| (g) | Clark Cheng, the Executive Director of the Group |
| (h) | Sabrina Khan, the Chief Financial Officer of the Group |
Amounts due from related parties
Amounts due from related parties consisted of the following as of December 31, 2018 and 2017:
| December 31, 2018 | December 31, 2017 | |||||||
| Current | ||||||||
| AENEAS CAPITAL LIMITED | $ | 169,051 | $ | - | ||||
| Non-current | ||||||||
| AENEAS CAPITAL LIMITED | - | 106,942 | ||||||
| Aeneas Limited | - | 190,427 | ||||||
| Aeneas Group Limited | - | 7,451 | ||||||
| Jurchen Investment Corporation | 50,000 | - | ||||||
| Total | $ | 50,000 | $ | 304,820 | ||||
F-30
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Amounts due to related parties
Amounts due to related parties consisted of the following as of December 31, 2018 and 2017:
| December 31, 2018 | December 31, 2017 | |||||||
| AENEAS CAPITAL LIMITED | $ | - | $ | 197,386 | ||||
| Ian Huen | 2,545 | - | ||||||
| Clark Cheng | 8,893 | - | ||||||
| Sabrina Khan | 21,979 | - | ||||||
| Total | $ | 33,417 | $ | 197,386 | ||||
Related party transactions
Related party transactions consisted of the following for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017:
| Year ended December 31, 2018 | March 1, 2017 | |||||||
| A borrowing from a related party (Note 1) | ||||||||
| - Ian Huen | $ | - | $ | 6,410 | ||||
| Payments on behalf of the Group (Note 2) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | - | $ | 64,038 | ||||
| - Aeneas Management Limited | $ | 156,961 | $ | - | ||||
| Expense reimbursement (Note 2) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | 7,331 | $ | 66,881 | ||||
| - Aeneas Management Limited | $ | 156,961 | $ | - | ||||
| Payments on behalf of related parties (Note 3) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | 22,934 | $ | 109,025 | ||||
| - Aeneas Limited | $ | - | $ | 132,074 | ||||
| - Aeneas Group Limited | $ | - | $ | 1,853 | ||||
| Repayments from related parties (Note 3) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | 132,128 | $ | - | ||||
| - Aeneas Limited | $ | 190,427 | $ | - | ||||
| - Aeneas Group Limited | $ | 7,451 | - | |||||
| Management and administrative fees (Note 4) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | 448,718 | $ | 640,932 | ||||
| Settlement of Management fees (Note 4) | ||||||||
| - AENEAS CAPITAL LIMITED | $ | 705,128 | $ | - | ||||
| Rental expense (Note 5) | ||||||||
| - Jurchen Investment Corporation | $ | 207,841 | $ | - | ||||
| Settlement of rental expense (Note 5) | ||||||||
| - Jurchen Investment Corporation | $ | 207,841 | $ | - | ||||
| Payment for rental deposit (Note 5) | ||||||||
| - Jurchen Investment Corporation | $ | 50,000 | $ | - | ||||
Note 1: The non-interest-bearing loan was borrowed from management for operation purpose and the loan was due on demand.
F-31
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Note 2: AENEAS CAPITAL LIMITED has paid the audit fee and legal fee on behalf of the Group and received the expense reimbursement. Some of the amounts were repaid during the periods. The balances were non-interest bearing.
Aeneas Management Limited has paid the operation fee on behalf of the Group and received the expense reimbursement. The balances were non-interest bearing.
Note 3: The Group has paid the expenses on behalf of AENEAS CAPITAL LIMITED, Aeneas Limited and Aeneas Group Limited, and the balances were non-interest bearing. There was no further payment on behalf transactions since April 2018.
Note 4: AENEAS CAPITAL LIMITED provides certain management and administrative services to the Group. For the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, AENEAS CAPITAL LIMITED was entitled to receive a fixed amount of administrative fees of HKD500,000 (approximately $64,103) per calendar month. On July 31, 2018, the agreement was mutually agreed to be terminated.
Note 5: Jurchen Investment Corporation entered into a sub-tenancy agreement with a subsidiary of the Group for the rental arrangement of an office in Hong Kong. For the period February 1, 2018 through January 31, 2021, Jurchen Investment Corporation was entitled to receive a fixed amount of rental fee of HK$130,000 (approximately USD16,667) per calendar month.
On November 11, 2017, the Group sold 100% of the ownership of Aeneas Limited and its subsidiary, Aeneas Group Limited, to Jurchen Investment Corporation for cash proceeds of $1. The Group recognized a gain on disposal of entity under common control of $67,874, net of net liabilities of Aeneas Limited and its subsidiary of $67,874 in consolidated statement of equity.
On April 3, 2018, Aptorum Medical Limited issued 526 shares to Clark Cheng, decreasing the equity interest of the Company from 100% to 95%.
In April 2018, the Group, AENEAS CAPITAL LIMITED, Aeneas Management Limited and Aeneas Group Limited entered into a net settlement agreement to offset the amounts due from related parties against the amounts due to related parties. Thereby, the Group is released from obligation for a total amount of $164,973, netting off receivables of total amount of $197,878 and collected remaining balance of $32,905.
F-32
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
14.CONVERTIBLE Debts
Convertible promissory notes
As of December 31, 2017, the Group issued an aggregated amounted of $480,000 of convertible promissory notes (the “Notes”). The Notes will be redeemed by the Group on the earlier of (i) the twelve months anniversary of the issuance date; and (ii) the date that the Group redeems the Notes if it has not consummated the IPO within twelve months of the issuance date. Interest on the Notes is accrued at a rate of 1% per annum and shall be compounded annually. The Notes are convertible into the Class A Ordinary Shares of the Company at a price of 56% discount to the actual price per Class A Ordinary Share to be issued in the IPO at the time that the Group consummates an initial closing of the IPO. The Group concludes that the convertible debts contained a contingent beneficial conversion, which shall not be recognized in earnings until the contingency is resolved.
One of the underwriters in this Offering, Boustead, also served as a placement agent for the Notes and received (i) a cash success fee of $68,516 and (ii) warrants to purchase 12,663 Class A Ordinary Shares, at an exercise price of $6.95 per share, subject to adjustment (the “Series A Note PA Warrants”). The Bond PA Warrants are exercisable on a cashless basis. For the year ended December 31, 2018, $130,935 was recorded as expense for the warrants after the completion of the performance obligation, which is the success of IPO.
In 2018, the Group has additionally issued $1,120,400 of convertible promissory notes under the same terms above and $1,600,400 of convertible promissory notes were issued accumulatively, and an unamortized debt issuance costs and discounts of $22,935 was remaining before the IPO. For the year ended December 31, 2018, the interest accretion and the contractual interest coupon of the Notes was $26,380 and $8,802, respectively.
In accordance with Accounting Standards Codification (“ASC”) 470-20-30-8, the Group should record a charge equal to the lower amount of either i) the Intrinsic Value of the BCF or ii) the proceeds realized upon the issuance of the Notes. The Group completed its IPO on December 17, 2018. Pursuant to the terms of the Notes, all of the outstanding principal amount of the Notes was automatically converted into 230,252 Class A Ordinary Shares. The intrinsic value of the BCF was determined to be $1,600,400. The Group concluded that the contingency was effectively resolved upon the automatic conversion, and recorded a one-time charge to interest expense of $1,600,400 with a corresponding increase to additional paid-in capital.
Convertible bonds
On April 6, 2018, the Group has entered into a subscription agreement (the “Bond Subscription Agreement”) with Peace Range Limited (“Peace Range”). Pursuant to the Bond Subscription Agreement, the Group issued Peace Range a $15,000,000 convertible bond (the “Bond” and the “Bond Offering”), minus a structuring fee equal to 2% of the principal amount of the Bond, on April 25, 2018. The Group also agreed to pay certain expenses, up to an aggregate limit of $250,000, incurred by Peace Range in connection with the Bond Offering. The Bond earns interest at the rate of 8% per annum, payable semi-annually. The payment of the Bond is guaranteed by the holding company, Jurchen Investment Corporation. In addition, the repayment of the principal of the Bond and interest payables is secured by a fund the Group set aside in a debt service reserve account, with the funds in the debt service reserve account to be released in an amount pro rata to the principal amount of the Bond being converted. The Bond shall mature on the twelfth calendar month following the issuance date, or with prior written consent of the holders of the Bond, the business day falling six calendar months thereafter. 10% of the principal amount of the Bond shall be automatically converted into our Class A Ordinary Shares upon the closing of the IPO and the rest of the Bond is convertible at the option of the holder commencing on the closing of the IPO until the earlier of the date falling 12 calendar months after the maturity of the Bond and the date falling 12 calendar months after the closing of the IPO, at a price offered at the IPO with a discount ranging from 19% to 22% depending on the date of the IPO occurred. The Group closed the Bond Offering on April 25, 2018 and issued a Bond to Peace Range pursuant to the Bond Subscription Agreement. The contingent beneficial conversion is contained in convertible bonds, which shall not be recognized in earnings until the contingency event, initial closing of the IPO, is resolved. The Group has determined that the conversion feature embedded in the convertible loan should not be bifurcated, and therefore, accounted as a liability in its entirety before the IPO.
F-33
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
One of the underwriters in this Offering, Boustead, also served as a placement agent for the Bond Offering and received (i) a cash success fee of $600,000 and (ii) warrants to purchase 67,790 Class A Ordinary Shares, at an exercise price of $12.17 per share, subject to adjustment (the “Bond PA Warrants”). The Bond PA Warrants are exercisable on a cashless basis. For the year ended December 31, 2018, $528,762 was recorded as expense for the warrants after the completion of the performance obligation, which is the success of IPO. China Renaissance also served as a placement agent for the Bond Offering; for such services, China Renaissance received a cash success fee of $150,000.
The Group completed its IPO on December 17, 2018. Pursuant to the terms of the Bond, 10% of the outstanding principal amount of the Bond was automatically converted into 119,217 Class A Ordinary Shares. Upon the automatic conversion, the contingency was effectively resolved, and the value of the 10% of the BCF of $383,629 was recorded as additional interest expense with a corresponding increase to additional paid-in capital. The remaining BCF of $3,452,657 was recorded as debt discount, which was amortized through the maturity of the convertible debts, with a corresponding increase to additional paid-in capital. For the year ended December 31, 2018, the interest accretion of the BCF was $374,707.
As of December 31, 2018, the remaining principal amount of the Bond was $13,500,000 and the remaining unamortized debt issuance costs and discounts was $314,744. The aggregate effective interest rate on the Bond is approximately 16.65% per annum. For the year ended December 31, 2018, the interest accretion and the contractual interest coupon of the Bond was $691,099 and $815,000, respectively.
The following lists the components of the ending balance of convertible debts as of December 31, 2018 and 2017, respectively:
December 31, 2018 | December 31, 2017 | |||||||
| Gross convertible debts | $ | 13,500,000 | $ | 480,000 | ||||
| Less: Discount on issuance cost | 314,744 | - | ||||||
| Discount on BCF | 3,077,950 | - | ||||||
| Convertible debts, net | $ | 10,107,306 | $ | 480,000 | ||||
15. FINANCE LEASE
On May 14, 2018, the Group leased a vehicle for its operation with a lease term of 54 months, and the lease was classified as a finance lease. The following lists the components of the net present value of capital leases obligation:
| December 31, 2018 | ||||
| Gross capital lease obligation | $ | 210,891 | ||
| Less: Discount on capital lease obligation | 23,141 | |||
| 187,750 | ||||
| Less: Current portion of capital lease obligation | 43,877 | |||
| Net present value of capital lease obligation, net of current portion | $ | 143,873 | ||
The present value of the net minimum payments on capital lease as of December 31, 2018 is as follows:
| 2019 | 2020 | 2021 | 2022 | Total | ||||||||||||||||
| Minimum lease payments | $ | 53,844 | $ | 53,845 | $ | 53,845 | $ | 49,357 | $ | 210,891 | ||||||||||
| Less: Amortization of discount | 9,967 | 7,290 | 4,449 | 1,435 | 23,141 | |||||||||||||||
| Capital lease obligation | $ | 43,877 | $ | 46,555 | $ | 49,396 | $ | 47,922 | $ | 187,750 | ||||||||||
F-34
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
16. ORDINARY SHARES
According to the Restructuring Plan, the ten management shares of par value of $0.01 have been cancelled, and the 256,571 issued participating shares of par value of $0.01 have been compulsorily redeemed and 4,743,419 unissued participating shares of par value of $0.01 each have been cancelled. Meanwhile, the Company has an authorized share capital consisting of 100,000,000 ordinary shares (the “Ordinary Shares”), par value $1.00 per share, and 25,657,110 shares was issued to the original investors.
During the period March 1, 2017 through October 13, 2017, 2,207,025 of the Company’s Ordinary Shares were issued at a price of $3.90 per share.
On October 13, 2017, a resolution was passed at a general meeting of the Company that: (i) 72,135,865 of authorized but unissued Ordinary Shares of the Company were replaced with 54,573,620 Class A ordinary shares (the “Class A Ordinary Shares”) of par value of $1.00 per share and 17,562,245 Class B ordinary shares (the “Class B Ordinary Shares”) of par value of $1.00 per share, respectively; (ii) 24,930,839 issued Ordinary Shares, which were issued to three shareholders, were converted into 2,493,085 Class A Ordinary Shares of par value of $1.00 per share and 22,437,754 Class B Ordinary Shares of par value of $1.00 per share; and (iii) 2,933,296 issued Ordinary Shares, which were issued to 24 shareholders, were converted into 2,933,296 Class A Ordinary Shares of par value of $1.00 per share.
On December 17, 2018, the Group consummated its IPO of 761,419 Class A Ordinary Shares. The shares were sold at a price of $15.80 per share, generating gross proceeds to the Group of approximately $12,030,420. At the completion of the IPO, $1,732,229 offering costs was charged to additional paid-in capital. Following the consummation of the IPO and automatic conversion of the Notes and the Bonds (see Note 14), there were an aggregate of 6,537,269 Class A Ordinary Shares issued and outstanding as of December 31, 2018.
Holders of Class A Ordinary Shares and Class B Ordinary Shares have the same rights except for the following: (i) each Class A Ordinary Share is entitled to one vote while each Class B Ordinary Share is entitled to ten votes; and (ii) each Class B Ordinary Share is convertible into one Class A Ordinary Share at any time while Class A Ordinary Shares are not convertible under any circumstances.
Share option plan
A total of 5,500,000 Class A Ordinary Shares (subject to subsequent adjustments described more fully below) may be issued pursuant to awards under the 2017 Omnibus Incentive Plan (the “2017 Share Option Plan”). Subsequent adjustments include that on each January 1, starting with January 1, 2020, an additional number of shares equal to the lesser of (i) 2% of the outstanding number of Class A Ordinary Shares (on a fully diluted basis) on the immediate preceding December 31, and (ii) such lower number of Class A Ordinary Shares as may be determined by the board of directors, subject in all cases to adjustments as provided in Section 10 of the 2017 Share Option Plan. Awards will be made pursuant to agreements and may be subject to vesting and other restrictions as determined by the board of directors. As of December 31, 2018, 5,500,000 shares were available for future grant under the 2017 Share Option Plan.
17. NON-CONTROLLING INTEREST
As of December 31, 2018, non-controlling interest related to the 1% equity interest in APTUS BIOTECHNOLOGY (MACAO) LIMITED, 10% equity interest in mTOR (Hong Kong) Limited, 5% equity interest in Aptorum Medical Limited, 20% equity interest in Acticule, and 20% equity interest in the Lanither Life Sciences Limited in the consolidated balance sheets was deficit of $368,533 in total.
As of December 31, 2017, non-controlling interest related to the 1% equity interest in APTUS BIOTECHNOLOGY (MACAO) LIMITED and 10% equity interest in mTOR (Hong Kong) Limited in the consolidated balance sheets was deficit of $14,045 in total.
18.Warrants
On November 30, 2018 and December 17, 2018, the Company entered into several agreements with underwriter. In return for the underwriter’s services, the Company issued an aggregate of 80,453 and 38,071 warrants to purchase the same number of the Company’s ordinary shares, for the convertible debts and the IPO, respectively. The shares were fully vested upon the IPO completion date and the fair value of the warrants was $659,697 and $218,147, respectively, which was calculated using the Black-Scholes pricing model, with the following weighted-average assumptions.
| December 31, 2018 | As of the date of issuance | |||||||
| Expected volatility | 58.18 | % | 65.70 | % | ||||
| Risk-free interest rate | 2.820%-2.822% | 2.820%-2.822% | ||||||
| Expected term from grant date (in years) | 2.43 | 2.50 | ||||||
| Dividend rate | - | - | ||||||
| Fair value | $ | 4.60-9.48 | $ | 5.73-10.34 | ||||
Expected Volatility
The expected volatility used for the year ended December 31, 2018 is based upon the Company’s peer group trading history.
F-35
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Risk-Free Interest Rate
The risk-free interest rate assumption is based on U.S. Treasury instruments with a term consistent with the contractual term of the warrants issued for the year ended December 31, 2018.
Expected Term
The expected term of the warrants issued during the year ended December 31, 2018, represents the remaining contractual term of the warrants.
Dividend Yield
The Company has never declared or paid any cash dividends and does not plan to pay cash dividends in the foreseeable future, and therefore, used an expected dividend yield of zero in the valuation model.
The movement of the warrants for the year ended December 31, 2018 is as following:
| Warrants | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term in Years | ||||||||||
| Outstanding, January 1, 2018 | - | $ | - | - | ||||||||
| Granted | 118,524 | $ | 13.79 | 2.50 | ||||||||
| Outstanding, December 31, 2018 | 118,524 | $ | 13.79 | 2.43 | ||||||||
The Group analyzed the warrants issued in the IPO and the convertible debts in accordance with ASC Topic 815 “Derivatives and Hedging”. In accordance with ASC Topic 815, the Group determined that the warrants should not be considered index to its own stock, as the strike price of the warrants is dominated in a currency (USD) other than the primary economy environment currency of the Group (HKD). As a result, the warrants does not meet the scope exception of ASC Topic 815, therefore, should be accounted for as derivative liabilities and measure at fair value with changes in fair value be recorded in earnings in each reporting period.
F-36
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
19. NET LOSS PER SHARE
The following table sets forth the computation of basic and diluted loss per share:
| Year ended December 31, 2018 | March 1, 2017 through December 31, 2017 | |||||||
| Numerator: | ||||||||
| Net loss attributable to Aptorum Group Limited | $ | (14,831,723 | ) | $ | (2,547,462 | ) | ||
| Denominator: | ||||||||
| Basic and diluted weighted average common shares outstanding | 27,909,788 | 26,963,435 | ||||||
| Basic and diluted loss per share | $ | (0.53 | ) | $ | (0.09 | ) | ||
Basic loss per share is computed by dividing net loss attributable to ordinary shareholders by the weighted average number of ordinary shares outstanding during the period. Diluted loss per share reflects the potential dilution that could occur if securities or other contracts to issue ordinary shares were exercised or converted into ordinary shares. Potential dilutive securities are excluded from the calculation of diluted loss per share in loss periods as their effect would be anti-dilutive.
20. PRINCIPAL RISK
MARKET RISK
Market risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market variables such as interest rate, foreign exchange rates and equity prices.
The maximum risk resulting from financial instruments equals their fair value.
(a) Interest rate risk
Interest rate risk arises from the possibility that changes in interest rates will affect future cash flows or the fair values of financial instruments.
Interest rate risk sensitivity analysis
The Group’s cash held with the Cash Custodian and the Custodian are exposed to interest rate risk. However, Management considers the risk to be minimal as they are short-term with terms less than one month.
(b) Currency risk
Currency risk is the risk that the value of financial assets or liabilities will fluctuate due to changes in foreign exchange rates.
Currency risk sensitivity analysis
At December 31, 2018 and 2017, the Group has no significant foreign currency risk because its business is principally conducted in Hong Kong and most of the transactions are denominated in Hong Kong dollar. Since the Hong Kong dollar is pegged to the United States dollar, the Group’s exposure to foreign currency risk in respect of the balances denominated in Hong Kong dollars is considered to be minimal.
F-37
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
(c) Equity price risk
Equity price risk is the risk of unfavorable changes in the fair values of equities or equity-linked derivatives as the result of changes in the levels of equity indices and the value of individual shares. The Group has been exposed to price risk on all of its equities investments and equities-linked derivatives.
Management’s best estimate of the effect on net assets and profit due to a reasonably possible change of relevant benchmarks, with all other variables held constant is as follows. In practice, the actual trading results may differ from the sensitivity analysis below and the difference could be material.
LIQUIDITY RISK
Liquidity risk is the risk that the Group will encounter difficulty in raising funds to meet commitments associated with financial assets and liabilities. Liquidity risk may result from an inability to sell a financial asset quickly at an amount close to its fair value.
The Group invests in private equities which are generally unquoted and not readily marketable. The Group manages its liquidity risk by setting investment limits on unlisted securities that cannot be readily disposed of. Investment of the Group’s assets in unquoted securities may restrict the ability of the Group to dispose of its investment at a price and time it wishes to do so.
CREDIT RISK
Financial assets which potentially subject the Group to concentrations of credit risk consist principally of bank deposits and balances, and assets held with the Custodian/Prime Broker.
The Custodian/Prime Broker provides the clearing and depository operations for the Group’s security transactions. The Custodian/Prime Broker also provides loans and financing to the Group and assets held by the Custodian/Prime Brokers will be charged as a continuing security for the payment and discharge of all liabilities of the Group.
The Group is exposed to credit risk on the cash held with the Custodian/Prime Broker amounting to $112,746 and $122,127, respectively, as of December 31, 2018 and 2017. The credit rating ascribed by Standard and Poor’s to Credit Suisse as of December 31, 2018 and 2017 was A.
Furthermore, the Group takes on exposure to credit risk on cash and restricted cash balances held with HSBC, DBS Bank Ltd, Hong Kong Branch, Industrial and Commercial Bank of China (Macao) Limited, Bank of China (Hong Kong) Limited and Mitsubishi UFJ Financial Group for the purposes of payments of Group expenses.
All transactions in listed securities are settled or paid for upon delivery using approved and reputable brokers. The risk of default is considered minimal, as delivery of securities sold is only made when the broker has received payment. Payment is made on a purchase when the securities have been received by the broker. The trade will fail if either party fails to meet its obligation. The Group limits its exposure to credit risk by transacting all of its securities and contractual commitment activities with broker-dealers, banks and regulated exchanges with high credit ratings and that the Group considers to be well established.
F-38
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
CONCENTRATION RISK
The table below analyses the Group’s concentration of equity price risk by country and region, and industry:
December 31, 2018 | December 31, 2017 | |||||||
| Country and Region | ||||||||
| United States of America | $ | 8,224,771 | $ | 10,462,483 | ||||
| Total | $ | 8,224,771 | $ | 10,462,483 | ||||
| Industry | ||||||||
| Pharmaceutical and biotechnology | $ | 8,224,285 | $ | 10,443,175 | ||||
| Healthcare | 486 | 19,308 | ||||||
| Total | $ | 8,224,771 | $ | 10,462,483 | ||||
INVESTMENTS IN DERIVATIVES RISK
Warrants
Since warrants have a limited life, as the expiration date of a warrant approaches, the time value of a warrant will decline. In addition, if the stock underlying the warrant declines in price, the intrinsic value of an “in the money” warrant will decline. Further, if the price of the stock underlying the warrant does not exceed the strike price of the warrant on the expiration date, the warrant will expire worthless. As a result, there is the potential for the Group to lose its entire investment in a warrant. The Group is exposed to counterparty risk from the potential failure of an issuer to settle its exercised warrants. The maximum risk of loss from counterparty risk to the Group is the fair value of the contracts and the purchase price of the warrants.
21. COMMITMENTS AND CONTINGENCIES
Lease Commitments
The total future minimum lease payments under the non-cancellable operating leases with respect to the offices and thelaboratoryas of December 31, 2018 are as follows:
| For the years ending December 31, | Amount | |||
| 2019 | $ | 600,978 | ||
| 2020 | 626,277 | |||
| 2021 | 397,842 | |||
| 2022 | 75,174 | |||
| 2023 and thereafter | - | |||
| Total | $ | 1,700,271 | ||
Rental expenses for the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017 were $591,546, and $49,518, respectively.
Contingent Payment Obligations
The Group has entered into agreements with independent third parties for purchasing office and laboratory equipment. As of December 31, 2018, the Group had non-cancellable purchase commitments of $487,930.
The Group has additional contingency payment obligations under each of the license agreements, such as milestone payments, royalties, research and development funding, if certain condition or milestone is met.
F-39
APTORUM GROUP LIMITED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(Stated in U.S. Dollars)
Milestone payments are to be made upon achievements of certain conditions, such as Investigational New Drugs (“IND”) filing or U.S. Food and Drug Administration (“FDA”) approval, first commercial sale of the licensed products, or other achievements. The aggregate amount of the milestone payments that the Group are required to pay up to different achievements of conditions and milestones for all the license agreements signed as of December 31, 2018 are below:
| Amount | ||||
| Drug molecules: up to the conditions and milestones of | ||||
| Preclinical to IND filing | $ | 372,564 | ||
| From entering phase 1 to before first commercial sale | 24,216,410 | |||
| First commercial sale | 15,656,410 | |||
| Net sales amount more than certain threshold in a year | 75,769,231 | |||
| Subtotal | 116,014,615 | |||
| Surgical robotics and medical devices: up to the conditions and milestones of | ||||
| Before FDA approval | 270,000 | |||
| FDA approval obtained | 200,000 | |||
| Subtotal | 470,000 | |||
| Total | $ | 116,484,615 | ||
For the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, the Group incurred $30,000 and $nil milestone payments, respectively. For the year ended December 31, 2018 and the period March 1, 2017 through December 31, 2017, the Group did not incur any royalties or research and development funding, respectively. As of December 31, 2018, no other milestone payments had been triggered under any of the existing license agreements.
22. SEGMENT REPORTING
The Group’s chief operating decision maker, the Chief Executive Officer, reviews the consolidated results when making decisions about allocating resources and accessing performance of the Group as a whole and hence, the Group has only one reportable segment. The Group does not distinguish between markets or segments for the purpose of internal reporting. The Group’s long-lived assets are substantially all located in Hong Kong and substantially all of the Group’s expense is derived from within Hong Kong. Therefore, no geographical segments are presented.
23. SUBSEQUENT EVENTS
The Group has evaluated subsequent events through the date of issuance of the consolidated financial statements, and except for the following events with material financial impact on the Group’s consolidated financial statements, no other subsequent event is identified that would have required adjustment or disclosure in the consolidated financial statements.
On March 15, 2019, the Company granted 215,795 share options to employees, external consultants and advisors of the Group in accordance to the 2017 Share Option Plan with an exercise price of $12.91.
On March 29, 2019, Aptorum Medical Limited issued 112 shares to a director of the Company, decreasing the equity interest of the Company from 95% to 94%.
F-40
APTORUM GROUP LIMITED
Financial Statements
Table of Contents
F-41
CONDENSED CONSOLIDATED BALANCE SHEETS
June 30, 2019 and December 31, 2018
(Stated in U.S. Dollars)
| As of June 30, 2019 | As of December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 4,466,741 | $ | 12,006,624 | ||||
| Restricted cash | - | 14,100,614 | ||||||
| Digital currencies | 117,482 | - | ||||||
| Accounts receivable | 8,367 | 2,827 | ||||||
| Inventories | 33,911 | 30,642 | ||||||
| Marketable securities, at fair value | 1,669,096 | 1,014,338 | ||||||
| Investments in derivatives | 425,916 | 115,721 | ||||||
| Amounts due from related parties | - | 169,051 | ||||||
| Due from brokers | 109,134 | 818,968 | ||||||
| Other receivables and prepayments | 911,997 | 464,156 | ||||||
| Total current assets | 7,742,644 | 28,722,941 | ||||||
| Property, plant and equipment, net | 5,777,657 | 4,260,602 | ||||||
| Non-marketable investments | 7,112,180 | 7,094,712 | ||||||
| Intangible assets, net | 1,347,594 | 1,409,540 | ||||||
| Amounts due from related parties | 50,000 | 50,000 | ||||||
| Long-term prepayments | 2,048,570 | 3,417,178 | ||||||
| Loan receivable | 571,975 | - | ||||||
| Other non-current asset | 89,750 | 119,667 | ||||||
| Total Assets | $ | 24,740,370 | $ | 45,074,640 | ||||
| LIABILITIES AND EQUITY | ||||||||
| LIABILITIES | ||||||||
| Current liabilities: | ||||||||
| Amounts due to related parties | $ | 3,512 | $ | 33,417 | ||||
| Accounts payable and accrued expenses | 548,433 | 1,247,147 | ||||||
| Finance lease payable, current portion | 45,196 | 43,877 | ||||||
| Warrant liabilities | - | 753,118 | ||||||
| Convertible debts | - | 10,107,306 | ||||||
| Total current liabilities | 597,141 | 12,184,865 | ||||||
| Finance lease payable, non-current portion | 120,941 | 143,873 | ||||||
| Total Liabilities | $ | 718,082 | $ | 12,328,738 | ||||
| Commitments and contingencies | - | - | ||||||
EQUITY | ||||||||
| Class A Ordinary Shares ($1.00 par value; 60,000,000 shares authorized, 6,597,362 shares issued and outstanding as at June 30, 2019 and 6,537,269 shares issued and outstanding as at December 31, 2018, respectively) | $ | 6,597,362 | $ | 6,537,269 | ||||
| Class B Ordinary Shares ($1.00 par value; 40,000,000 shares authorized, 22,437,754 shares issued and outstanding as at June 30, 2019 and December 31, 2018) | 22,437,754 | 22,437,754 | ||||||
| Additional paid-in capital | 23,857,814 | 23,003,285 | ||||||
| Accumulated other comprehensive income (loss) | 7,345 | (1,484,688 | ) | |||||
| Accumulated deficit | (27,957,689 | ) | (17,379,185 | ) | ||||
| Total equity attributable to the shareholders of Aptorum Group Limited | 24,942,586 | 33,114,435 | ||||||
| Non-controlling interests | (920,298 | ) | (368,533 | ) | ||||
| Total equity | 24,022,288 | 32,745,902 | ||||||
| Total Liabilities and Equity | $ | 24,740,370 | $ | 45,074,640 | ||||
See accompanying notes to the condensed consolidated financial statements.
F-42
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
For the six months ended June 30, 2019 and 2018
(Stated in U.S. Dollars)
| For the six months ended June 30, | ||||||||
| 2019 | 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Revenue | ||||||||
| Healthcare services income | $ | 239,792 | $ | 26,662 | ||||
Operating expenses | ||||||||
| Costs of healthcare services | (371,218 | ) | (22,749 | ) | ||||
| Research and development expenses | (2,714,217 | ) | (1,342,179 | ) | ||||
| General and administrative fees | (3,232,916 | ) | (2,238,025 | ) | ||||
| Legal and professional fees | (2,008,774 | ) | (1,063,032 | ) | ||||
| Other operating expenses | (120,788 | ) | (235,413 | ) | ||||
| Total operating expenses | (8,447,913 | ) | (4,901,398 | ) | ||||
| Other loss | ||||||||
| Gain on investments in marketable securities, net | 315,977 | - | ||||||
| Gain on non-marketable investment | 1,147,199 | - | ||||||
| Gain (loss) on investments in derivatives, net | 310,195 | (359,844 | ) | |||||
| Realized gain on use of digital currencies | 12,334 | - | ||||||
| Changes in fair value of warrant liabilities | (866,300 | ) | - | |||||
| Gain on extinguishment of convertible debts | 1,198,490 | - | ||||||
| Interest expense, net | (3,678,566 | ) | (301,362 | ) | ||||
| Sundry income | 128,444 | - | ||||||
| Total other loss, net | (1,432,227 | ) | (661,206 | ) | ||||
| Net loss | $ | (9,640,348 | ) | $ | (5,535,942 | ) | ||
| Less: net loss attributable to non-controlling interests | (551,877 | ) | (47,570 | ) | ||||
| Net loss attributable to Aptorum Group Limited | $ | (9,088,471 | ) | $ | (5,488,372 | ) | ||
| Net loss per share – basic and diluted | $ | (0.31 | ) | $ | (0.20 | ) | ||
| Weighted-average shares outstanding – basic and diluted | 28,978,151 | 27,864,135 | ||||||
| Net loss | $ | (9,640,348 | ) | $ | (5,535,942 | ) | ||
Other Comprehensive income (loss) | ||||||||
| Unrealized loss on investments in available-for-sale securities | - | (178,027 | ) | |||||
| Exchange differences on translation of foreign operations | 2,000 | 167 | ||||||
| Other Comprehensive income (loss) | 2,000 | (177,860 | ) | |||||
Comprehensive loss | (9,638,348 | ) | (5,713,802 | ) | ||||
| Less: comprehensive loss attributable to non-controlling interests | (551,877 | ) | (47,570 | ) | ||||
| Comprehensive loss attributable to the shareholders of Aptorum Group Limited | (9,086,471 | ) | (5,666,232 | ) | ||||
See accompanying notes to the condensed consolidated financial statements.
F-43
CONDENSED CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
For the six months ended June 30, 2019 and 2018
(Stated in U.S. Dollars)
| Class A Ordinary Shares | Class B Ordinary Shares | Additional Paid-in Capital | Accumulated deficit | Accumulated other comprehensive income (loss) | Non-controlling interests | Total | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Amount | Amount | Amount | Amount | Amount | ||||||||||||||||||||||||||||
| Balance, December 31, 2018 | 6,537,269 | $ | 6,537,269 | 22,437,754 | $ | 22,437,754 | $ | 23,003,285 | $ | (17,379,185 | ) | $ | (1,484,688 | ) | $ | (368,533 | ) | $ | 32,745,902 | |||||||||||||||||
| Adjustment to opening balance of equity | - | - | - | - | - | (1,490,033 | ) | 1,490,033 | - | - | ||||||||||||||||||||||||||
| Balance, January 1, 2019 | 6,537,269 | 6,537,269 | 22,437,754 | 22,437,754 | 23,003,285 | (18,869,218 | ) | 5,345 | (368,533 | ) | 32,745,902 | |||||||||||||||||||||||||
| Issuance of share to non-controlling interest | - | - | - | - | (112 | ) | - | - | 112 | - | ||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (9,088,471 | ) | - | (551,877 | ) | (9,640,348 | ) | ||||||||||||||||||||||||
| Reacquisition of convertible bonds | - | - | - | - | (1,298,490 | ) | - | - | - | (1,298,490 | ) | |||||||||||||||||||||||||
| Share-based compensation | - | - | - | - | 593,806 | - | - | - | 593,806 | |||||||||||||||||||||||||||
| Exercise of warrants | 60,093 | 60,093 | - | - | 1,559,325 | - | - | - | 1,619,418 | |||||||||||||||||||||||||||
| Exchange difference on translation of foreign operations | - | - | - | - | - | - | 2,000 | - | 2,000 | |||||||||||||||||||||||||||
| Balance, June 30, 2019 (Unaudited) | 6,597,362 | $ | 6,597,362 | $ | 22,437,754 | $ | 22,437,754 | $ | 23,857,814 | $ | (27,957,689 | ) | $ | 7,345 | $ | (920,298 | ) | $ | 24,022,288 | |||||||||||||||||
| Balance, January 1, 2018 | 5,426,381 | $ | 5,426,381 | 22,437,754 | $ | 22,437,754 | $ | 5,294,402 | $ | (2,547,462 | ) | $ | (367,782 | ) | $ | (14,045 | ) | $ | 30,229,248 | |||||||||||||||||
| Proceeds from non-controlling interest | - | - | - | - | 51,727 | - | - | (51,726 | ) | 1 | ||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (5,488,372 | ) | - | (47,570 | ) | (5,535,942 | ) | ||||||||||||||||||||||||
| Unrealized loss on investments in available-for-sale securities | - | - | - | - | - | - | (178,027 | ) | - | (178,027 | ) | |||||||||||||||||||||||||
| Exchange difference on translation of foreign operations | - | - | - | - | - | - | 167 | - | 167 | |||||||||||||||||||||||||||
| Balance, June 30, 2018 (Unaudited) | 5,426,381 | $ | 5,426,381 | $ | 22,437,754 | $ | 22,437,754 | $ | 5,346,129 | $ | (8,035,834 | ) | $ | (545,642 | ) | $ | (113,341 | ) | $ | 24,515,447 | ||||||||||||||||
See accompanying notes to the condensed consolidated financial statements.
F-44
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
For the six months ended June 30, 2019 and 2018
(Stated in U.S. Dollars)
| Six months ended June 30, 2019 | Six months ended June 30, 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (9,640,348 | ) | $ | (5,535,942 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Amortization and depreciation | 585,701 | 209,267 | ||||||
| Share-based compensation | 593,806 | - | ||||||
| Gain on investments in marketable securities, net | (315,977 | ) | - | |||||
| Gain on non-marketable investment | (1,147,199 | ) | - | |||||
| (Gain) loss on investments in derivatives, net | (310,195 | ) | 359,844 | |||||
| Changes in fair value of warrant liabilities | 866,300 | - | ||||||
| Realized gain on use of digital currencies | (12,334 | ) | - | |||||
| Utilization of digital currencies | 94,852 | - | ||||||
| Gain on extinguishment of convertible debts | (1,198,490 | ) | - | |||||
| Interest income | (61,791 | ) | (105,118 | ) | ||||
| Interest expense and accretion of convertible debts | 3,735,027 | 405,430 | ||||||
| Accretion of capital lease obligation | 5,309 | 1,050 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | (5,540 | ) | (9,835 | ) | ||||
| Inventories | (3,269 | ) | (3,741 | ) | ||||
| Other receivables and prepayments | (386,069 | ) | (8,492 | ) | ||||
| Other non-current asset | - | (179,500 | ) | |||||
| Long-term prepayments | 55,429 | (1,631,105 | ) | |||||
| Due from brokers | 709,834 | (258 | ) | |||||
| Due from related parties | 169,051 | - | ||||||
| Due to related parties | (29,905 | ) | 17,612 | |||||
| Accounts payable and accrued expenses | (1,039,201 | ) | 165,082 | |||||
| Net cash used in operating activities | (7,335,009 | ) | (6,315,706 | ) | ||||
| Cash flows from investing activities | ||||||||
| Disbursement of a loan to a third party | (1,400,000 | ) | (3,000,000 | ) | ||||
| Repayment of a loan from a third party | 828,025 | 3,000,000 | ||||||
| Purchases of intangible assets | (10,743 | ) | (237,289 | ) | ||||
| Purchases of property, plant and equipment | (686,798 | ) | (2,542,039 | ) | ||||
| Proceeds from sale of marketable securities | 790,950 | - | ||||||
| Purchase of digital currencies | (200,000 | ) | - | |||||
| Net cash provided by (used in) investing activities | (678,566 | ) | (2,779,328 | ) | ||||
Cash flows from financing activities | ||||||||
| Payment for settlement of convertible debts | (13,600,000 | ) | - | |||||
| Payment of finance lease obligations | (26,922 | ) | (31,409 | ) | ||||
| Advances to/payments received from related parties | - | 107,434 | ||||||
| Proceeds from issuance of convertible debts | - | 16,120,400 | ||||||
| Payments for debt issuance costs | - | (900,000 | ) | |||||
| Net cash (used in) provided by financing activities | (13,626,922 | ) | 15,296,425 | |||||
| Net (decrease) increase in cash and restricted cash | (21,640,497 | ) | 6,201,391 | |||||
| Cash and restricted cash- Beginning of period | 26,107,238 | 16,725,807 | ||||||
| Cash and restricted cash - End of period | $ | 4,466,741 | $ | 22,927,198 | ||||
Supplemental disclosures of cash flow information | ||||||||
| Interest paid | $ | 557,333 | $ | 1,050 | ||||
| Income taxes paid | $ | - | $ | - | ||||
| Non-cash investing and financing activities: | ||||||||
| Net settlement of related party balances | $ | - | $ | 164,976 | ||||
| Reconciliation of cash and restricted cash | ||||||||
| Cash | $ | 4,466,741 | $ | 6,727,200 | ||||
| Restricted cash | - | 16,199,998 | ||||||
| Total cash and restricted cash shown on the consolidated statements of cash flows | $ | 4,466,741 | $ | 22,927,198 | ||||
See accompanying notes to the condensed consolidated financial statements.
F-45
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
1. ORGANIZATION
The Company, formally known as APTUS Holdings Limited and STRIKER ASIA OPPORTUNITIES FUND CORPORATION, is a company incorporated on September 13, 2010 under the laws of the Cayman Islands with limited liability.
The condensed consolidated financial statements include the financial statements of Aptorum Group Limited (the Company”) and its subsidiaries. The Company and its subsidiaries are hereinafter collectively referred to as the “Group”.
On March 1, 2017, the Company changed from an investment fund with management shares and non-voting participating redeemable preference shares to a holding company with operating subsidiaries. After that, the Company has become a biopharmaceutical company currently in the preclinical stage. The Company researches and develops life science and biopharmaceutical products within its wholly-owned subsidiary, Aptorum Therapeutics Limited, formerly known as APTUS Therapeutics Limited (“Aptorum Therapeutics”) and its indirect subsidiary companies (collectively, “Aptorum Therapeutics Group”).
Below summarizes the list of the subsidiaries consolidated as of June 30, 2019:
| Name | Incorporation date | Ownership | Place of incorporation | Principle activities | |||||
| Aptorum Therapeutics Limited | June 30, 2016 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| APTUS MANAGEMENT LIMITED | May 16, 2017 | 100% | Hong Kong | Provision of management services to its holding company and fellow subsidiaries | |||||
| Aptorum Medical Limited | August 28, 2017 | 94% | Cayman Islands | Provision of medical clinic services | |||||
| Aptorum Innovations Holding Limited | April 15, 2019 | 100% | Cayman Islands | Investment holding company | |||||
| Aptorum Innovations Holding Pte. Ltd. | June 5, 2019 | 100% | Singapore | Research and development of life science and biopharmaceutical products | |||||
| Aptorum Investment Holding Limited | March 29, 2019 | 100% | Cayman Islands | Investment holding company | |||||
| Aptorum Group LLC | August 14, 2019 | 100% | Nevada | Provision of public relation services to its holding company | |||||
| Aptus Therapeutics (Hong Kong) Limited | June 30, 2016 | 100% | Hong Kong | Research and development of life science and biopharmaceutical products | |||||
| APTUS BIOTECHNOLOGY (MACAO) LIMITED* | June 6, 2016 | 99% | Macao | Inactive | |||||
| APTORUM INTERNATIONAL LIMITED | March 26, 2018 | 100% | United Kingdom | Inactive | |||||
| Aptorum Pharmaceutical Development Limited | August 28, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Smart Pharmaceutical Development Limited (Formerly known as “Forum Property Holding Limited”) | March 6, 2018 | 100% | Cayman Islands | Inactive | |||||
F-46
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
| Name | Incorporation date | Ownership | Place of incorporation | Principle activities | |||||
| Videns Incorporation Limited (Formerly named Videns Biosciences Limited and VIDENS CORPORATION) | March 2, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| mTOR (Hong Kong) Limited | November 4, 2016 | 90% | Hong Kong | Research and development of life science and biopharmaceutical products | |||||
| Videns Incorporation (Hong Kong) Limited | July 3, 2017 | 100% | Hong Kong | Inactive | |||||
| Nativus Life Sciences Limited | July 7, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Nativus Life Sciences (Hong Kong) Limited | August 8, 2017 | 100% | Hong Kong | Inactive | |||||
| Scipio Life Sciences Limited | July 19, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Scipio Life Sciences (Hong Kong) Limited | August 10, 2017 | 100% | Hong Kong | Inactive | |||||
| Claves Life Sciences Limited | August 2, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Claves Life Sciences (Hong Kong) Limited | August 22, 2017 | 100% | Hong Kong | Inactive | |||||
| Signate Life Sciences Limited | August 28, 2017 | 100% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Signate Life Sciences (Hong Kong) Limited | August 10, 2017 | 100% | Hong Kong | Inactive | |||||
| Acticule Life Sciences Limited | June 30, 2017 | 80% | Cayman Islands | Research and development of life science and biopharmaceutical products | |||||
| Acticule Life Sciences (Hong Kong) Limited | July 27, 2017 | 80% | Hong Kong | Inactive | |||||
| Lanither Life Sciences Limited | April 4, 2018 | 80% | Cayman Islands | Inactive | |||||
| Lanither Life Sciences (Hong Kong) Limited | May 25, 2018 | 80% | Hong Kong | Inactive | |||||
| SMTPH Limited | April 18, 2019 | 100% | Seychelles | Investment holding company | |||||
| Smart Pharmaceutical Research Limited | April 24, 2019 | 100% | Samoa | Pharmaceutical research and analysis | |||||
| Smart Pharmaceutical Development Pte. Ltd. | May 10, 2019 | 100% | Singapore | Research and development of life science and biopharmaceutical products | |||||
| Smart Pharmaceutical Limited Partnership | June 7, 2019 | 100% | Seychelles | Issuance of asset backed securities | |||||
| * | The subsidiary was deregistered on August 20, 2019. |
Initial public offering
On December 17, 2018, the Group completed an initial public offering (the “IPO” or “Offering”) with new issuance of 761,419 ordinary shares at $15.80 per share for total offering size of approximately $12.0 million before deducting commissions and expenses. The net proceeds from the IPO was approximately $10.3 million, net of an underwriting discount of approximately $1.2 million, including $0.2 million warrant issued, and offering costs of approximately $0.5 million. The Class A Ordinary Shares began trading on the NASDAQ Global Market on December 17, 2018 under the ticker symbol “APM”.
Deferred offering costs
Deferred offering costs consist principally of legal, printing and registration costs in connection with the Group’s IPO. Such costs are deferred until the closing of the Offering, at which time the deferred costs are offset against the offering proceeds. Deferred offering costs as of December 31, 2018 and 2017 amounted to $nil on the consolidated balance sheets. At the completion of the IPO, US$1,732,229 offering costs was charged to additional paid-in capital.
F-47
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
2. LIQUIDITY
The Company reported a net loss of $9,640,348 and net operation cash outflow of $7,335,009 for the six months ended June 30, 2019, respectively. In addition, the Company had an accumulated deficit of $27,957,689 as of June 30, 2019. The Company’s operating results for future periods are subject to numerous uncertainties and it is uncertain if the Company will be able to reduce or eliminate its net losses for the foreseeable future. If management is not able to generate significant revenues from its product candidates currently in development, the Company may not be able to achieve profitability.
The Company’s principal sources of liquidity have been cash and marketable securities. As of the date of issuance of the consolidated financial statements, the Company has approximately $3 million of cash and marketable securities. In addition, based upon the current market price of the Company’s marketable securities, it anticipates it can liquidate such marketable securities, if necessary. On August 13, 2019, the Company entered into financing arrangements with Aeneas Group Limited, a related party, and Jurchen Investment Corporation, the ultimate parent of the Group, allowing the Group to access up to a total $15.0 million in line of credit debt financing. (See Note 17)
The Company believes that available cash and marketable securities, together with signed loan facilities, should enable the Company to meet presently anticipated cash needs for at least the next 12 months after the date that the financial statements are issued and the Company has prepared the consolidated financial statements on a going concern basis. If the Company encounters unforeseen circumstances that place constraints on its capital resources, management will be required to take various measures to conserve liquidity, which could include, but not necessarily be limited to, deferring some of its research and seeking to dispose of marketable securities.
3. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of consolidation
The condensed consolidated financial statements of the Group are presented on the accrual basis of accounting in accordance with U.S. GAAP and include the accounts of the Company, its direct and indirect wholly and majority owned subsidiaries. All material intercompany balances and transactions have been eliminated in preparation of the condensed consolidated financial statements. Non-controlling interests represent the equity interest that is not owned by the Group.
Use of estimates
The preparation of the condensed consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of increases and decreases in net assets from operations as well as income and expenses during the reporting period. Significant accounting estimates reflected in the Group’s condensed consolidated financial statements include valuing equity securities, fair value of investments in securities, convertible debts and finance lease, the useful lives of intangible assets and property, plant and equipment, impairment of long-lived assets, valuation allowance for deferred tax assets, and collectability of receivables. Actual results could differ from those estimates.
F-48
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
Digital currencies
Digital currencies are included in current assets in the accompanying condensed consolidated balance sheets. Digital currencies purchased are recorded at cost.
Digital currencies held are accounted for as intangible assets with indefinite useful lives. An intangible asset with an indefinite useful life is not amortized but assessed for impairment annually, or more frequently, when events or changes in circumstances occur indicating that it is more likely than not that the indefinite-lived asset is impaired. Impairment exists when the carrying amount exceeds its fair value, which is measured using the quoted price of the digital currency at the time its fair value is being measured. In testing for impairment, the Company has the option to first perform a qualitative assessment to determine whether it is more likely than not that an impairment exists. If it is determined that it is not more likely than not that an impairment exists, a quantitative impairment test is not necessary. If the Company concludes otherwise, it is required to perform a quantitative impairment test. To the extent an impairment loss is recognized, the loss establishes the new cost basis of the asset. Subsequent reversal of impairment losses is not permitted.
Purchases of digital currencies by the Group are included within investing activities in the accompanying condensed consolidated statements of cash flows. The utilization of digital currencies in exchange of services are included within operating activities in the accompanying condensed consolidated statements of cash flows and any realized gains or losses from such use are included in other income (expense) in the condensed consolidated statements of operations. The Company accounts for its gains or losses in accordance with the first in first out (FIFO) method.
Marketable Securities
Marketable Securities are publicly traded stocks measured at fair value and classified within Level 1 and 2 in the fair value hierarchy because the Group uses quoted prices for identical assets in active markets or inputs that are based upon quoted prices for similar instruments in active markets.
Gain on investments in marketable securities, net, amounting to $315,977 and $nil, respectively, were recognized in the condensed consolidated statements of operations for the six months ended June 30, 2019 and 2018.
During the six months ended June 30, 2019, the Group disposed of marketable securities, with sales proceeds of $790,950 received and recorded in due from brokers, and recognized a realized gain of $627,014 in the condensed consolidated statements of operations, respectively. No disposal was recorded during the period from January 1, 2018 to June 30, 2018.
Investments in Derivatives
Investments in derivatives consisted of warrants, which are measured at fair value, with gains or losses from changes in fair value recorded through earnings. The fair value of these warrants have been determined using the Black-Scholes pricing mode. The Black-Scholes pricing model provides for assumptions regarding volatility, call and put features and risk-free interest rates within the total period to maturity.
No disposal was recorded during the six months ended June 30, 2019 and 2018. Unrealized gain on the investments in derivatives amounted to $310,195 was recognized in the condensed consolidated statements of operations for the six months ended June 30, 2019. Unrealized loss on the investments in derivatives amounted to $359,844 was recognized in the condensed consolidated statements of operations for the six months ended June 30, 2018.
Non-marketable investments
Non-marketable investments are comprising of investments in non-redeemable preferred shares of privately-held companies are not required to be consolidated under the variable interest or voting models. Non-marketable investments are classified as non-current assets on the condensed consolidated balance sheets as those investments do not have stated contractual maturity dates.
Effective January 1, 2019, the non-marketable equity securities not accounted for under the equity method are either carried at fair value or under the measurement alternative upon the adoption of ASU 2016-01. Under the measurement alternative, the carrying value is measured at cost, less any impairment, plus or minus changes resulting from observable price changes in orderly transactions for identical or similar investments of the same issuer. Adjustments are determined primarily based on a market approach as of the transaction date.
F-49
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
Convertible debts
The Group determines the appropriate accounting treatment of its convertible debts in accordance with the terms in relation to the conversion feature, call and put option, beneficial conversion feature (“BCF”) and settlement feature. On December 17, 2018, the Group concluded that the contingency of BCF was effectively resolved upon the completion of the IPO and recognized BCF according to the agreement.
The repurchasing of convertible debts is considered an extinguishment and the difference between the repurchasing price of debt, the net carrying amount of the extinguished debt and the intrinsic value of BCF is recognized in the condensed consolidated statements of operations. The intrinsic value of BCF of $1.3 million at the extinguishment date was recorded as a reduction of additional paid-in capital. On April 24, 2019, the Group repurchased its convertible debts at $13.6 million with carrying amount of $13.5 million and the intrinsic value of BCF is $1.3 million with a gain on extinguishment on convertible debts of $1.2 million.
Revenue recognition
Revenue is recognized when (or as) the Company satisfies performance obligations by transferring a promised goods or services to a customer. Revenue is measured at the transaction price which is based on the amount of consideration that the Company expects to receive in exchange for transferring the promised goods or services to the customer. Contracts with customers are comprised of invoices and written contracts. Revenue from healthcare services is measured upon the provision of the relevant services.
Recently adopted accounting pronouncements
In May 2014, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”), which was subsequently modified in August 2015 by ASU 2015-14, Revenue from Contracts with Customers: Deferral of the Effective Date. We adopted this standard effective January 1, 2019 using the modified retrospective approach, in which case the cumulative effect of applying the standard would be recognized at the date of initial application. The adoption does not have a material impact to the condensed consolidated financial statements.
In January 2016, the FASB issued Accounting Standards Update No. 2016-01 (ASU 2016-01) "Financial Instruments-Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities," which amends various aspects of the recognition, measurement, presentation, and disclosure of financial instruments. We adopted ASU 2016-01 as of January 1, 2019 using the modified retrospective method for our marketable equity securities and the prospective method for our non-marketable equity securities. The following table summarizes the changes to our condensed consolidated balance sheet for the adoption of ASU 2016-01:
| December 31, 2018 | Adjustment due to ASU 2016-01 | January 1, 2019 | ||||||||||
| Accumulated deficit | $ | (17,379,185 | ) | $ | (1,490,033 | ) | $ | (18,869,218 | ) | |||
| Accumulated other comprehensive loss | $ | (1,484,688 | ) | $ | 1,490,033 | $ | 5,345 | |||||
We have elected to use the measurement alternative for our non-marketable equity securities, defined as cost adjusted for changes from observable transactions for identical or similar investments of the same issuer, less impairment. The adoption of ASU 2016-01 increases the volatility of our other income (expense), net, as a result of the unrealized gain or loss from the remeasurement of our equity securities.
Recently issued accounting standards which have not yet been adopted
In June 2016, the FASB issued ASU 2016-13, Financial Instruments—Credit Losses ("ASU 2016-13"). Subsequently, the FASB issued ASU 2019-05, Financial Instruments- Credit Losses (Topic 326): Targeted Transition Relief. The amendments in ASU 2016-13 update guidance on reporting credit losses for financial assets. These amendments affect loans, debt securities, accounts receivables, net investments in leases, off balance sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. The amendments are effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. The Company is currently evaluating the impact on its financial statements of adopting this guidance.
F-50
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
4. REVENUE
The Company adopted ASC 606 using the modified retrospective method as applied to customer contracts that were not completed as of January 1, 2019. As a result, financial information for reporting periods beginning after January 1, 2019 are presented under ASC 606, while comparative financial information has not been adjusted and continues to be reported in accordance with the Company’s historical accounting policy for revenue recognition prior to the adoption of ASC 606.
For the six months ended June 30, 2019, all revenue is come from provision of healthcare services in Hong Kong.
5. FAIR VALUE MEASUREMENT
The following table provides the assets and liabilities carried at fair value measured on a recurring basis as of June 30, 2019 and December 31, 2018:
| June 30, 2019 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Current Assets | ||||||||||||||||
| Marketable securities | ||||||||||||||||
| Common stocks | $ | 379,110 | $ | 1,289,986 | $ | - | $ | 1,669,096 | ||||||||
| Investments in derivatives | ||||||||||||||||
| Warrants | - | - | 425,916 | 425,916 | ||||||||||||
| Total assets at fair value | $ | 379,110 | $ | 1,289,986 | $ | 425,916 | $ | 2,095,012 | ||||||||
| December 31, 2018 | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Current Assets | ||||||||||||||||
| Marketable securities – Available-for-sale securities | ||||||||||||||||
| Common stocks | $ | 813,728 | $ | 200,610 | $ | - | $ | 1,014,338 | ||||||||
| Investments in derivatives | ||||||||||||||||
| Warrants | - | - | 115,721 | 115,721 | ||||||||||||
| Total assets at fair value | $ | 813,728 | $ | 200,610 | $ | 115,721 | $ | 1,130,059 | ||||||||
The following is a reconciliation of Level 3 assets for the six months ended June 30, 2019:
| Warrants | ||||
| Balance at January 1, 2019 | $ | 115,721 | ||
| Change in unrealized appreciation | 310,195 | |||
| Balance at June 30, 2019 | $ | 425,916 | ||
| Net change in unrealized appreciation relating to investments still held at June 30, 2019 | 310,195 | |||
The following is a reconciliation of Level 3 assets for the six months ended June 30, 2018
| Warrants | ||||
Balance at January 1, 2018 | $ | 1,070,940 | ||
| Change in unrealized depreciation | (359,836 | ) | ||
| Balance at June 30, 2018 | $ | 711,104 | ||
| Net change in unrealized depreciation relating to investments still held at June 30, 2018 | (359,836 | ) | ||
F-51
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
The following table presents the quantitative information about the Group’s Level 3 fair value measurements of investment as of June 30, 2019 and December 31, 2018, which utilized significant unobservable internally-developed inputs:
| June 30, 2019 | Valuation technique | Unobservable input | Range (weighted average) | Sensitivity of fair value to input | ||||
| Warrants | Black-Scholes Model | Estimated time to exit Historical Volatility | 6-24 months 75% - 350% | 10% increase (decrease) in volatility would result in increase (decrease) in fair value by $8,157 |
| December 31, 2018 | Valuation technique | Unobservable input | Range (weighted average) | Sensitivity of fair value to input | ||||
| Warrants | Black-Scholes Model | Estimated time to exit Historical Volatility | 12-30 months 73% - 188% | 10% increase (decrease) in volatility would result in increase (decrease) in fair value by $19,691 |
Warrants
As of June 30, 2019 and December 31, 2018, the volume of the Group’s derivative activities based on their notional amount and number of contracts, categorized by primary underlying risk, are as follows:
| Long Exposure | ||||||||||||||||
| June 30, 2019 | December 31, 2018 | |||||||||||||||
| Primary underlying risk | Notional Amounts | Number of Warrants | Notional Amounts | Number of Warrants | ||||||||||||
| Equity Price | ||||||||||||||||
| Warrants | $ | 481,794 | 2,257,682 | $ | 218,270 | 2,257,682 | ||||||||||
The following table identifies the fair value amounts of derivative instruments included in the statement of financial condition as derivative contracts, categorized by primary underlying risk, at June 30, 2019 and December 31, 2018. The following table also identifies the net gain and loss amounts included in the statements of operations as net unrealized gain from derivative contracts, categorized by primary underlying risk, for the six months ended June 30, 2019 and 2018:
| June 30, 2019 | December 31, 2018 | |||||||||||||||
| Primary underlying risk | Derivative assets | Derivative liabilities | Derivative assets | Derivative liabilities | ||||||||||||
| Equity Price | ||||||||||||||||
| Warrants | $ | 425,916 | $ | - | $ | 115,721 | $ | - | ||||||||
| For the six months ended June 30, | ||||||||||||||||
| 2019 | 2018 | |||||||||||||||
| Primary underlying risk | Realized gain (loss) | Unrealized gain (loss) | Realized gain (loss) | Unrealized gain (loss) | ||||||||||||
| Equity Price | ||||||||||||||||
| Warrants | $ | - | $ | 310,195 | $ | - | $ | (359,844 | ) | |||||||
Non-marketable equity securities remeasured during the six months ended June 30, 2019 are classified within Level 3 in the fair value hierarchy because we estimate the value based on valuation methods using the observable transaction price at the transaction date and other unobservable inputs including volatility, rights, and obligations of the securities we hold.
F-52
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
The following is a summary of unrealized gains and losses recorded in other income (expense), net, and included as adjustments to the carrying value of non-marketable investments held as of June 30, 2019:
| For the six months ended June 30, 2019 | ||||
| Upward adjustments | $ | 1,017,468 | ||
| Total unrealized gain for non-marketable investments | $ | 1,017,468 | ||
The following table summarizes the total carrying value of our non-marketable investments held as of June 30, 2019 including cumulative unrealized upward and downward adjustments made to the initial cost basis of the investments:
| June 30, 2019 | ||||
| Initial cost basis | $ | 6,094,712 | ||
| Upward adjustments | 1,017,468 | |||
| Total carrying value at the end of the period | $ | 7,112,180 | ||
6. OTHER RECEIVABLES AND PREPAYMENTS
Other receivables and prepayments as of June 30, 2019 and December 31, 2018 consisted of:
| June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Prepaid insurance | $ | 192,011 | $ | 147,864 | ||||
| Prepaid service fee | 100,871 | 75,224 | ||||||
| Rental deposits | 8,575 | 8,576 | ||||||
| Prepaid rental expenses | 48,345 | 46,948 | ||||||
| Prepaid research and development expenses | 417,413 | 41,614 | ||||||
| Other receivables | 119,735 | 109,435 | ||||||
| Others | 25,047 | 34,495 | ||||||
| $ | 911,997 | $ | 464,156 | |||||
7.Digital Currencies
The following table presents additional information about digital currencies:
June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Beginning balance | $ | - | $ | - | ||||
| Purchase of digital currencies | 200,000 | - | ||||||
| Utilization of digital currencies | (94,852 | ) | - | |||||
| Realized gain on use of digital currencies | 12,334 | - | ||||||
| Ending balance | $ | 117,482 | $ | - | ||||
8. PROPERTY, PLANT AND EQUIPMENT, NET
Property, plant and equipment as of June 30, 2019 and December 31, 2018 consisted of:
June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Building | $ | 1,488,396 | $ | 1,488,396 | ||||
| Computer equipment | 73,611 | 64,911 | ||||||
| Furniture, fixture, and office and medical equipment | 268,653 | 262,819 | ||||||
| Leasehold improvements | 665,546 | 664,713 | ||||||
| Laboratory equipment | 4,029,640 | 2,045,034 | ||||||
| Motor vehicle | 239,093 | 239,093 | ||||||
| 6,764,939 | 4,764,966 | |||||||
| Less: accumulated depreciation | 987,282 | 504,364 | ||||||
| Property, plant and equipment, net | $ | 5,777,657 | $ | 4,260,602 | ||||
Depreciation expenses for property, plant and equipment amounted to $482,925 and $124,245 for the six months ended June 30, 2019 and 2018, respectively.
F-53
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
9. LONG-TERM PREPAYMENTS
Long-term prepayments as of June 30, 2019 and December 31, 2018 consisted of:
June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Rental deposits | $ | 132,043 | $ | 132,043 | ||||
| Prepayments for equipment | 1,916,527 | 3,285,135 | ||||||
| $ | 2,048,570 | $ | 3,417,178 | |||||
10. ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses as of June 30, 2019 and December 31, 2018 consisted of:
June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Healthcare consultation service payable | $ | 29,870 | $ | 40,139 | ||||
| Professional fees payable | 58,630 | 178,117 | ||||||
| Research and development expenses payable | 162,319 | 398,899 | ||||||
| Interest payable | 8,802 | 223,802 | ||||||
| Payables for leasehold improvement and equipment | 26,779 | 73,864 | ||||||
| Salaries payable | 154,589 | 183,065 | ||||||
| Deferred rent | 55,856 | 58,810 | ||||||
| Others | 51,588 | 90,451 | ||||||
| $ | 548,433 | $ | 1,247,147 | |||||
11. INCOME TAXES
The Company and its subsidiaries file tax returns separately.
Income taxes
Cayman Islands: under the current laws of the Cayman Islands, the Company and its subsidiaries in the Cayman Islands are not subject to taxes on their income and capital gains.
Hong Kong: in accordance with the relevant tax laws and regulations of Hong Kong, a company registered in Hong Kong is subject to income taxes within Hong Kong at the applicable tax rate on taxable income. All the Hong Kong subsidiaries that are not entitled to any tax holiday were subject to income tax at a rate of 16.5%. The subsidiaries of the Group in Hong Kong did not have assessable profits that were derived Hong Kong during the six months ended June 30, 2019 and 2018. Therefore, no Hong Kong profit tax has been provided for in the periods presented.
United Kingdom: in accordance with the relevant tax laws and regulations of United Kingdom, a company registered in the United Kingdom is subject to income taxes within the United Kingdom at the applicable tax rate on taxable income. All the United Kingdom subsidiaries that are not entitled to any tax holiday were subject to income tax at a rate of 19%. The subsidiary of the Group in the United Kingdom did not have assessable profits that were derived from the United Kingdom during the six months ended June 30, 2019 and 2018. Therefore, no United Kingdom profit tax has been provided for in the periods presented.
On a semi-annually basis, the Group evaluates the realizability of deferred tax assets by jurisdiction and assesses the need for a valuation allowance. In assessing the realizability of deferred tax assets, the Company considers historical profitability, evaluation of scheduled reversals of deferred tax liabilities, projected future taxable income and tax-planning strategies. Valuation allowances have been provided on deferred tax assets where, based on all available evidence, it was considered more likely than not that some portion or all of the recorded deferred tax assets will not be realized in future periods. After consideration of all positive and negative evidence, the Company believes that as of June 30, 2019, it is more likely than not the deferred tax assets will not be realized.
F-54
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
12. RELATED PARTY BALANCES AND TRANSACTIONS
The following is a list of a director and related parties to which the Group has transactions with:
| (a) | Ian Huen, the Chief Executive Officer and Executive Director of the Group; |
| (b) | Darren Lui, the Executive Director of the Group; |
| (c) | Aenco Limited, an entity controlled by Ian Huen; |
| (d) | AENEAS CAPITAL LIMITED, an entity controlled by Ian Huen; |
| (e) | Aeneas Management Limited, an entity controlled by Ian Huen. |
| (f) | Jurchen Investment Corporation, the holding company and an entity controlled by Ian Huen. |
| (g) | Clark Cheng, the Executive Director of the Group |
| (h) | Sabrina Khan, the Chief Financial Officer of the Group |
Amounts due from related parties
Amounts due from related parties consisted of the following as of June 30, 2019 and December 31, 2018:
| June 30, 2019 | December 31, 2018 | |||||||
| Current | (Unaudited) | |||||||
| AENEAS CAPITAL LIMITED | $ | - | $ | 169,051 | ||||
| Non-current | ||||||||
| Jurchen Investment Corporation | 50,000 | 50,000 | ||||||
| Total | $ | 50,000 | $ | 50,000 | ||||
Amounts due to related parties
Amounts due to related parties consisted of the following as of June 30, 2019 and December 31, 2018:
| June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Ian Huen | $ | - | $ | 2,545 | ||||
| Darren Lui | 2,732 | - | ||||||
| Clark Cheng | - | 8,893 | ||||||
| Sabrina Khan | 780 | 21,979 | ||||||
| Total | $ | 3,512 | $ | 33,417 | ||||
F-55
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
Related party transactions
Related party transactions consisted of the following for the six months ended June 30, 2019 and 2018:
| For the six months ended June 30, | ||||||||
| 2019 | 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Payments on behalf of the Group (Note I) | ||||||||
| - AENEAS CAPITAL LIMITED (d) | $ | 5,057 | $ | - | ||||
| - Aeneas Management Limited (e) | 5,372 | 8,064 | ||||||
| Expense reimbursement (Note I) | ||||||||
| - AENEAS CAPITAL LIMITED (d) | $ | 5,057 | $ | 7,331 | ||||
| - Aeneas Management Limited (e) | 5,372 | 8,064 | ||||||
| Payments on behalf of related parties (Note II) | ||||||||
| - AENEAS CAPITAL LIMITED (d) | $ | - | $ | 22,933 | ||||
| Repayments from related parties (Note II) | ||||||||
| - AENEAS CAPITAL LIMITED (d) | $ | 169,051 | $ | 330,005 | ||||
| Consultant, management and administrative fees (Note III) | ||||||||
| - AENEAS CAPITAL LIMITED (d) | $ | - | $ | 384,615 | ||||
| - Aenco Limited (c) | 415,385 | - | ||||||
| - Aeneas Management Limited (e) | 347,692 | - | ||||||
| Settlement of consultant, management and administrative fees (Note III) | ||||||||
| - Aenco Limited (c) | $ | 415,385 | $ | - | ||||
| - Aeneas Management Limited (e) | 347,692 | - | ||||||
| Rental expense (Note IV) | ||||||||
| - Jurchen Investment Corporation (f) | $ | 113,572 | $ | 94,304 | ||||
| Settlement of rental expense (Note IV) | ||||||||
| - Jurchen Investment Corporation (f) | $ | 113,572 | $ | 94,304 | ||||
F-56
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
Note I: AENEAS CAPITAL LIMITED has paid the audit fee and legal fee on behalf of the Group and received the expense reimbursement. The balances were non-interest bearing.
Aeneas Management Limited has paid the operation fee on behalf of the Group and received the expense reimbursement. The balances were non-interest bearing.
Note II: The Group has paid the expenses on behalf of AENEAS CAPITAL LIMITED, of which the whole amounts were non-interest bearing. There was no further payment on behalf transactions since April 2018.
Note III: AENEAS CAPITAL LIMITED provided certain management and administrative services to the Group. For the six months ended June 30, 2018, AENEAS CAPITAL LIMITED was entitled to receive a fixed amount of administrative fees of HKD500,000 (approximately $64,103) per calendar month. On July 31, 2018, the agreement was mutually agreed to be terminated.
Aenco Limited provided certain information technology services to the Group. For the six months ended June 30, 2019, Aenco Limited was entitled to receive a fixed amount of services fees of HKD540,000 (approximately $69,231) per calendar month. The agreement will expire on December 31, 2019.
Aeneas Management Limited provided certain documentation and administrative services to the Group. For the six months ended June 30, 2019, Aenco Limited was entitled to receive a fixed amount of services fees of HKD452,000 (approximately $57,949) per calendar month. The agreement will expire on December 31, 2019.
Note IV: Jurchen Investment Corporation entered into a sub-tenancy agreement with a subsidiary of the Group for the rental arrangement of an office in Hong Kong. For the period February 1, 2018 through January 31, 2021, Jurchen Investment Corporation was entitled to receive a fixed amount of rental fee of HKD130,000 (approximately $16,667) per calendar month.
On April 3, 2018, Aptorum Medical Limited issued 526 shares to Clark Cheng, decreasing the equity interest of the Company from 100% to 95%. On April 1, 2019, Aptorum Medical Limited further issued 112 shares to Clark Cheng in according to the appointment agreement, decreasing the equity interest of the Company from 95% to 94%.
In April 2018, the Group, AENEAS CAPITAL LIMITED, Aeneas Management Limited and Aeneas Group Limited entered into a net settlement agreement to offset the amount due from related parties against the amount due to related parties. Thereby, the Group is released from obligation for a total amount of $164,973, netting off receivables of total amount of $197,878 and collected remaining balance of $32,905.
13.CONVERTIBLE debts
Convertible bonds
On April 6, 2018, the Group entered into a subscription agreement (the “Bond Subscription Agreement”) with Peace Range Limited (“Peace Range”). Pursuant to the Bond Subscription Agreement, the Group issued Peace Range a $15,000,000 convertible bond (the “Bond” and the “Bond Offering”) on April 25, 2018.
The Group completed its IPO on December 17, 2018. Pursuant to the terms of the Bond, 10% of the outstanding principal amount of the Bond was automatically converted into 119,217 Class A Ordinary Shares. Upon the automatic conversion, the contingency was effectively resolved, and the value of the 10% of the BCF of $383,629 was recorded as additional interest expense with a corresponding increase to additional paid-in capital. The remaining BCF of $3,452,657 was recorded as debt discount, which was amortized through the maturity of the convertible debts, with a corresponding increase to additional paid-in capital. For the year ended December 31, 2018, the interest amortization of the BCF was $374,707.
F-57
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
The following lists the components of the ending balance of convertible debts as of June 30, 2019 and December 31, 2018, respectively:
June 30, 2019 | December 31, 2018 | |||||||
| (Unaudited) | ||||||||
| Gross convertible debts | $ | - | $ | 13,500,000 | ||||
| Less: Discount on issuance cost | - | 314,744 | ||||||
| Discount on BCF | - | 3,077,950 | ||||||
| Convertible debts, net | $ | - | $ | 10,107,306 | ||||
For the six months ended June 30, 2019 and 2018, the amortization of BCF and interest accretion of convertible debts were $3,392,694 and $186,763 respectively. The contractual interest for the six months ended June 30, 2019 and 2018 were $342,333 and $218,667 respectively
The repurchasing of convertible debts is considered an extinguishment and the difference between the repurchasing price of debt, and the net carrying amount of the extinguished debt and the intrinsic value of BCF is recognized on the condensed consolidated statements of operations. The intrinsic value of BCF at the extinguishment date decrease to additional paid-in capital. On April 24, 2019, the Group repurchased its convertible debts at $13.6 million with carrying amount of $13.5 million and the intrinsic value of BCF is $1.3 million with a gain on extinguishment on convertible debts of $1.2 million.
14. SHARE BASED COMPENSATION EXPENSES
Share option plan
A total of 5,500,000 Class A Ordinary Shares (subject to subsequent adjustments described more fully below) may be issued pursuant to awards under the 2017 Omnibus Incentive Plan (the “2017 Share Option Plan”). Subsequent adjustments include that on each January 1, starting with January 1, 2020, an additional number of shares equal to the lesser of (i) 2% of the outstanding number of Class A Ordinary Shares (on a fully diluted basis) on the immediate preceding December 31, and (ii) such lower number of Class A Ordinary Shares as may be determined by the board of directors, subject in all cases to adjustments as provided in Section 10 of the 2017 Share Option Plan. Awards will be made pursuant to agreements and may be subject to vesting and other restrictions as determined by the board of directors.
On March 15, 2019, the Company granted 218,610 share options to directors, employees, external consultants and advisors of the Group in accordance to the 2017 Share Option Plan with an exercise price of $12.91.
A summary of the option activity as of June 30, 2019 and changes during the period is presented below:
Options granted to employees
| Number of share options | Weighted average exercise price $ | Remaining contractual term in years | ||||||||||
| Outstanding, January 1, 2019 | - | - | - | |||||||||
| Granted | 218,610 | 12.91 | 12.31 | |||||||||
| Outstanding, June 30, 2019 | 218,610 | 12.91 | 12.01 | |||||||||
| Exercisable, June 30, 2019 | - | - | - | |||||||||
F-58
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
Intrinsic value is calculated as the amount by which the current market value of a share of common stock exceeds the exercise price multiplied by the number of option. The aggregate intrinsic value of options outstanding as of June 30, 2019 was approximately $2,669,000.
The fair value of each stock option award is estimated on the date of grant using the Black-Scholes option pricing model under the following assumptions.
| Date of grant | |||
| Expected volatility | 95.02%-95.15% | ||
| Risk-free interest rate | 2.46%-2.49% | ||
| Expected term from grant date (in years) | 6.29-7.29 | ||
| Dividend rate | - | ||
| Dilution factor | 0.9962 | ||
| Fair value | $10.10-10.52 |
In connection with the grant of share options to employees and non-employees, the Group recorded share-based compensation charges of $435,967 and $157,839, respectively.
15.Warrants
On November 30, 2018 and December 17, 2018, the Company entered into several agreements with underwriter. In return for the underwriter’s services, the Company issued an aggregate of 80,453 and 38,071 warrants to purchase the same number of the Company’s ordinary shares, for the convertible debts and the IPO, respectively. The shares were fully vested upon the IPO completion date and the fair value of the warrants was $659,697 and $218,147, respectively, which was calculated using the Black-Scholes pricing model, with the following weighted-average assumptions.
The Group analyzed the warrants issued in the IPO and the convertible debts in accordance with ASC Topic 815 “Derivatives and Hedging”. In accordance with ASC Topic 815, the Group determined that the warrants should not be considered index to its own stock, as the strike price of the warrants is dominated in a currency (USD) other than the primary economy environment currency of the Group (HKD). As a result, the warrants do not meet the scope exception of ASC Topic 815, therefore, should be accounted for as derivative liabilities and measure at fair value with changes in fair value be recorded in earnings in each reporting period.
All warrants were exercised on June 19, 2019 on a cashless basis. $866,300 loss in changes in fair value of warrant liabilities was recorded in condensed consolidated statements of operations.
| June 30, 2019 | December 31, 2018 | ||||||
| Expected volatility | - | % | 58.18% | ||||
| Risk-free interest rate | - | 2.820%-2.822% | |||||
| Expected term from grant date (in years) | - | 2.43 | |||||
| Dividend rate | - | - | |||||
| Fair value | $ | - | $4.60-9.48 | ||||
Expected Volatility
The expected volatility used for the year ended December 31, 2018 is based upon the Company’s peer group trading history.
Risk-Free Interest Rate
The risk-free interest rate assumption is based on U.S. Treasury instruments with a term consistent with the contractual term of the warrants issued for the year ended December 31, 2018.
Expected Term
The expected term of the warrants issued during the year ended December 31, 2018, represents the remaining contractual term of the warrants.
Dividend Yield
The Company has never declared or paid any cash dividends and does not plan to pay cash dividends in the foreseeable future, and therefore, used an expected dividend yield of zero in the valuation model.
F-59
APTORUM GROUP LIMITED
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
(Stated in U.S. Dollars)
The movement of the warrants for the six months ended June 30, 2019 is as follows:
| Warrants | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term in Years | ||||||||||
| Outstanding, January 1, 2019 | 118,524 | $ | 13.79 | 2.43 | ||||||||
| Exercised | 118,524 | $ | 13.79 | 1.96 | ||||||||
| Outstanding, June 30, 2019 | - | $ | - | - | ||||||||
16. NET LOSS PER SHARE
The following table sets forth the computation of basic and diluted loss per share:
| For the six months ended June 30, | ||||||||
| 2019 | 2018 | |||||||
| (Unaudited) | (Unaudited) | |||||||
| Numerator: | ||||||||
| Net loss attributable to Aptorum Group Limited | $ | (9,088,471 | ) | $ | (5,488,372 | ) | ||
| Denominator: | ||||||||
| Basic and diluted weighted average common shares outstanding | 28,978,151 | 27,864,135 | ||||||
| Basic and diluted loss per share | $ | (0.31 | ) | $ | (0.20 | ) | ||
Basic loss per share is computed by dividing net loss attributable to ordinary shareholders by the weighted average number of ordinary shares outstanding during the period. Diluted earnings per share reflects the potential dilution that could occur if securities or other contracts to issue ordinary shares were exercised or converted into ordinary shares. Potential dilutive securities are excluded from the calculation of diluted EPS in loss periods as their effect would be anti-dilutive.
17. SUBSEQUENT EVENTS
The Group has evaluated subsequent events through the date of issuance of the condensed consolidated financial statements, and except for the following events with material financial impact on the Group’s condensed consolidated financial statements, no other subsequent event is identified that would have required adjustment or disclosure in the condensed consolidated financial statements.
On August 13, 2019, the Group entered into financing arrangements with Aeneas Group Limited, a related party, and Jurchen Investment Corporation, the ultimate parent of the Group, allowing the Group to access up to a total $15.0 million in line of credit debt financing. The line of credit will mature on August 12, 2022 and the interest on the outstanding principal indebtedness will be at the rate of 8% per annum. As of the date of issuance of the condensed consolidated financial statements, the Company has drawn down $0.4 million from this line of credit.
In July 2019, Smart Pharmaceutical Limited Partnership, (“SPLP”), a wholly owned subsidiary of the Group, transferred 100,000,000 Smart Pharma Token (“SMPT token”) to Aenco Solutions Limited, a related party, in exchange of the service to deal with the token offering.
The SMPT token tokenizes rights to a portion of sales-based royalties, non-royalty sublicensing income and additional cash flow, derived from the subsequent commercialization of intellectual property rights of drug candidates discovered under our Smart-ACTTMplatform. SMPT token is backed by SPLP’s assets, including intellectual property rights of drug candidates created through the Smart-ACTTM platform and commercialization income. SPLP acts as the intellectual property holding company of Smart Pharma, and holds all title, rights and ownership interest of the intellectual property rights developed by Smart-ACTTM. As of June 30, 2019 and through the date of issuance of the condensed consolidated financial statements, SPLP has no substantial assets and liabilities.
F-60
Aptorum Group Limited
APTORUM GROUP LIMITED
27,765,821 Class A Ordinary Shares
PROSPECTUS
October 31, 2019
Through and including November 25, 2019 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this Offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to their unsold allotments or subscriptions.
No dealer, salesperson or any other person is authorized to give any information or make any representations in connection with this offering other than those contained in this prospectus and, if given or made, the information or representations must not be relied upon as having been authorized by us. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any security other than the securities offered by this prospectus, or an offer to sell or a solicitation of an offer to buy any securities by anyone in any jurisdiction in which the offer or solicitation is not authorized or is unlawful.