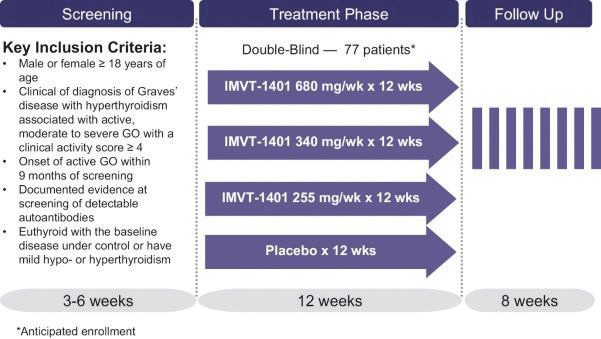
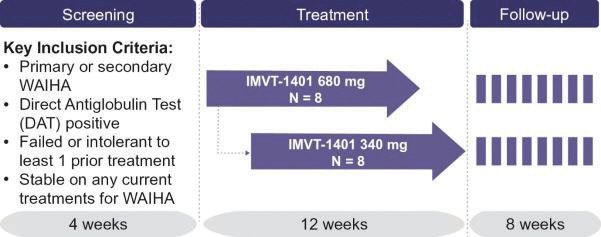
In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S.non-provisional filing date, as the term of a patent granted on a utility patent application filed after June 8, 1995 expires 20 years after thenon-provisional U.S. filing date (or any earlier filing date relied upon under 35 U.S.C. 120, 121, or 365(c)), with the timely payment of maintenance fees. In certain instances, the patent term may be adjusted to add additional days to compensate for certain delays incurred by the U.S. Patent and Trademark Office (“USPTO”) in the examination process, issuing the patent and/or the patent term may be extended for a period of time to compensate for at least a portion of the time a product candidate was undergoing FDA regulatory review. However, the patent extension granted for FDA regulatory review is only applied to a single patent that covers either the product candidate or a method of using or manufacturing the same which has not expired at the time of FDA approval. Additionally, the period of time the patent is extended may not exceed five years, and the total patent term, including the period of time the patent is extended, must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effectivenon-provisional filing date. The protection afforded by a patent with respect to a particular product varies on aproduct-by-product basis, from country to country, and depends upon many factors, including the type of patent, its coverage, the availability of regulatory-related extensions, the availability of legal remedies in the particular country and the validity and enforceability of the patent under the local laws. ISG owns a trademark for IMMUNOVANT, the corporate logo, and a composite trademark for its corporate logo with the IMMUNOVANT mark. As of May 14, 2020, this trademark portfolio included registration of the Immunovant trademark in the United States, Argentina, Brazil, Chile, China, Colombia, EU, Hong Kong Israel, the International Register, Japan, Mexico, Monaco, New Zealand, Norway, Philippines, Russian Federation, Saudi Arabia, Serbia, Singapore, South Korea and Switzerland, plus registration of the Immunovant logo trademark in Switzerland, and registration of the composite trademark in Chile, EU, the International Register, Monaco, Saudi Arabia, Switzerland and the United Kingdom. Additionally, trademark applications are pending for the Immunovant trademark in 22 foreign jurisdictions and for the composite trademark in the United States and 19 foreign jurisdictions. Under the HanAll Agreement, we have the right to market IMVT-1401 in the Licensed Territory under the trademark(s) of our choice, subject to regulatory approval. However, upon termination of the HanAll Agreement, we must assign to HanAll all right, title and interest in and to any and all trademarks we use in the development, manufacture or commercialization of the licensed products.
Furthermore, we rely upon trade secrets andknow-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, by using confidentiality and invention assignment agreements with its commercial partners, collaborators, employees and consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant it ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resultingknow-how and inventions.
Our commercial success will also depend in part on not infringing upon the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter its development or commercial strategies for our product candidates or processes, or to obtain licenses or cease certain activities. Our breach of any license agreements or failure to obtain a license to proprietary rights that it may require to develop or commercialize its future products may have an adverse impact on us. If third parties prepare and file patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference or derivation proceedings in the USPTO to determine priority of invention.
Government Regulation
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of biologics such as those we are developing. We, along with third-party contractors, will be required to navigate the various nonclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of IMVT-1401 or any future product candidate.
FDA Drug Approval Process
In the United States, the FDA regulates biologics under both the Federal Food, Drug and Cosmetic Act and the Public Health Services Act and their implementing regulations. The process required by the FDA before biologic product candidates may be marketed in the United States generally involves the following:
| | • | | completion of preclinical laboratory tests and animal studies performed in accordance with applicable regulations, including the FDA’s current Good Laboratory Practices (“GLP”) regulations and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| | • | | submission to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| | • | | approval by an institutional review board (“IRB”) or ethics committee at each clinical site before the trial is commenced; |
28