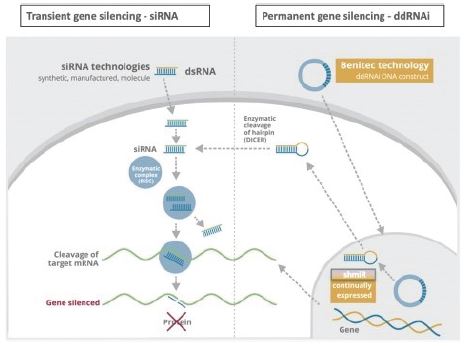
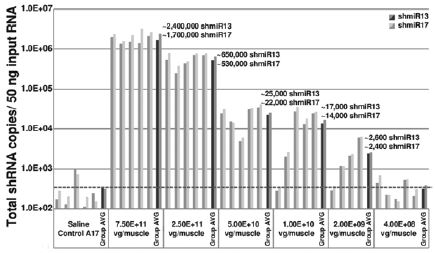
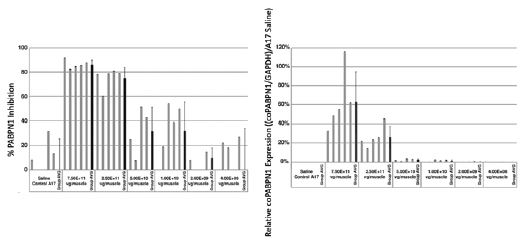
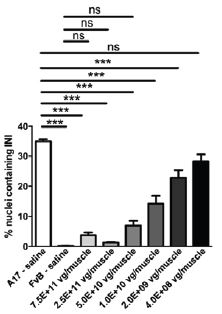
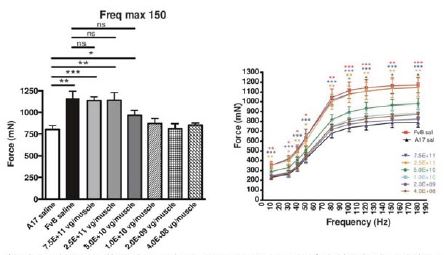
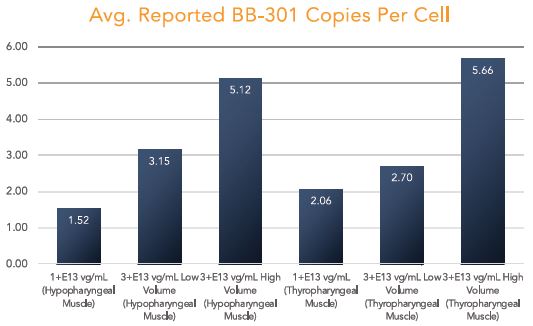
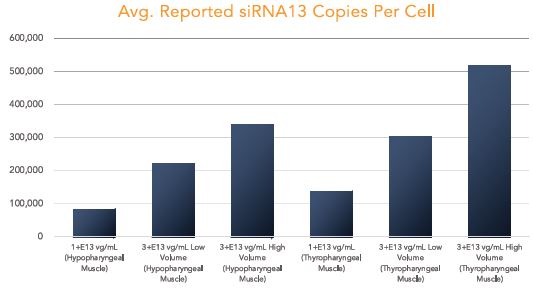
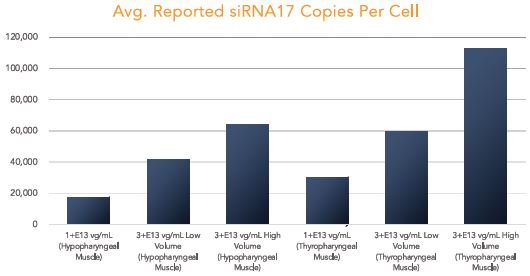
The biopharmaceutical industry is characterized by intense and dynamic competition to develop new technologies and proprietary therapies.
Any product candidates that we successfully develop and commercialize will have to compete with existing therapies and new therapies that may become available in the future. While we believe that our proprietary technology and scientific expertise in gene silencing using ddRNAi provide us with competitive advantages, we face potential competition from many different sources, including larger and better-funded pharmaceutical, specialty pharmaceutical and biotechnology companies, as well as from academic institutions and governmental agencies and public and private research institutions that may develop potentially competitive products or technologies. We are aware of several companies focused on developing gene therapy or gene silencing product candidates, including Alnylam, Arbutus and Arrowhead.
We are not aware of any companies developing a gene therapy or gene silencing approach for OPMD. Our product candidates, if approved, would also compete with treatments that have already been approved and accepted by the medical community, patients and third-party payers.
Many of our competitors and potential competitors, alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of treatments and the commercialization of those treatments. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and subject registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new products enter the market and advanced technologies become available. We expect any treatments that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, convenience of administration and delivery, price, the level of competition and the availability of reimbursement from government and other third party-payers.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, we expect that our therapeutic products, if approved, will be priced at a significant premium over competitive products and our ability to compete may be affected in many cases by insurers or other third-party payers seeking to encourage the use of competitive products including biosimilar or generic products.
This increasingly competitive landscape may compromise the development of our product candidates.
As a pharmaceutical and biological product company that wishes to conduct clinical trials and ultimately obtain marketing approval in the United States, we are subject to extensive regulation by the FDA, and other federal, state, and local regulatory agencies. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, the Public Health Service Act, or PHS Act, and their implementing regulations set forth, among other things, requirements for the research, testing, development, manufacture, quality control, safety, effectiveness, approval, labeling, storage, record keeping, reporting, distribution, import, export, advertising and promotion of our products. A failure to comply explicitly with any requirements during the product development, approval, or post-approval
26