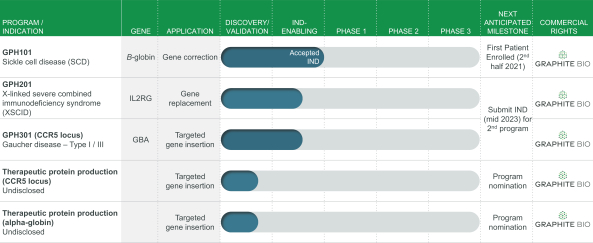
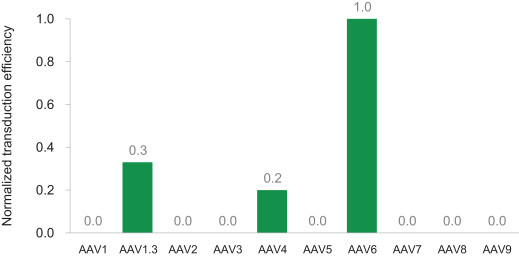
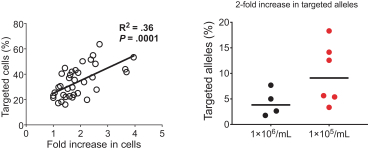
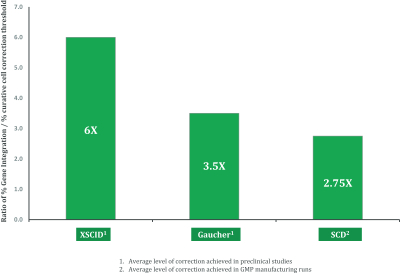
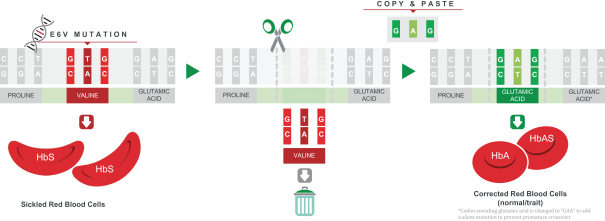
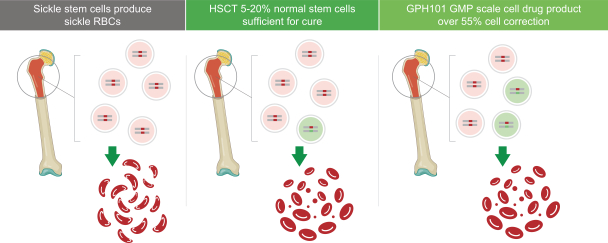
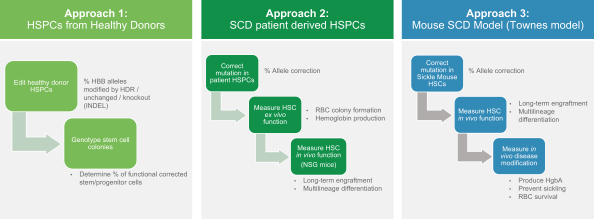
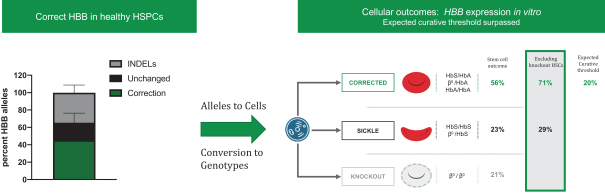
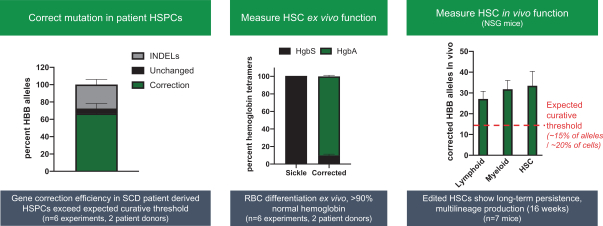
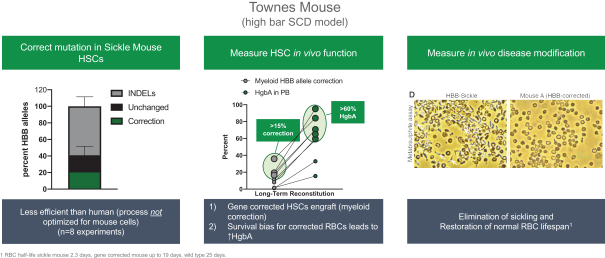
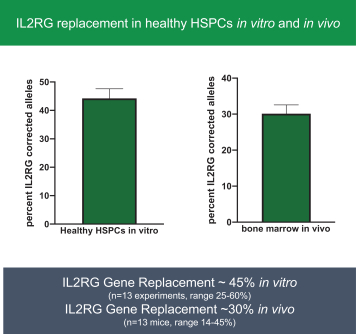
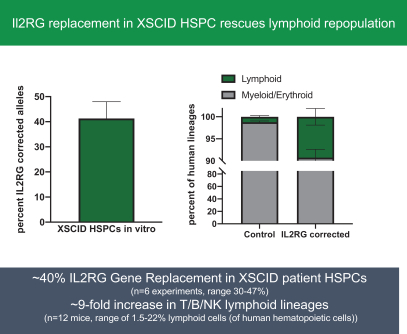
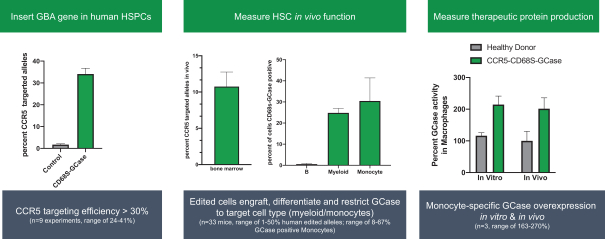
regulatory milestone payments. To date, we have not paid any amounts to IDT under the IDT License Agreement, but the $3.0 million upfront payment is due within 90 days from June 7, 2021 and receipt of an invoice from IDT. Each regulatory milestone payment is payable once on an indication-by-indication basis. In addition, we have agreed to pay IDT a low single-digit royalty on the net sales of products, subject to reductions in specified circumstances, and a low double digit percentage payment for certain sublicense revenue, which is also subject to reduction in the event a sublicense includes other patent rights that are not patents licensed from IDT.
The IDT License Agreement remains in effect on a country-by-country and product-by-product basis until the expiration of the royalty term for such product in such jurisdiction. We and IDT each have the right to terminate the IDT License Agreement for the other party’s material breach of its obligations under the IDT License Agreement, subject to specified rights to cure. Additionally, we may terminate the IDT License Agreement for any reason.
Our current in-licensed patent and patent applications from IDT, if the appropriate maintenance fees are paid, are expected to expire in 2037, excluding any additional term for patent term adjustments or patent term extensions.
The IDT License Agreement includes customary representations and warranties by each party as are customarily found in transactions of this nature, including as to the licensed intellectual property. The IDT License Agreement also provides for certain mutual indemnities for breaches of representations, warranties and covenants.
Government Regulation
The FDA and other regulatory authorities at federal, state and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring and post-approval reporting of biologics such as those we are developing.
U.S. Biologics Regulation
In the United States, the FDA regulates biological products under the Federal Food, Drug, and Cosmetic Act (FDCA), and the Public Health Service Act (PHSA), and their implementing regulations. Biological products are also subject to other federal, state, local and foreign statutes and regulations. We, along with third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may result in delays to the conduct of a study, regulatory review and approval or subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, license suspension or revocation, refusal to allow an applicant to proceed with clinical trials, imposition of a clinical hold, issuance of untitled or warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations or penalties.
Our product candidates must be approved by the FDA through the Biologics License Application (BLA), process before they may be legally marketed in the United States. The process required by the FDA before biological product candidates may be marketed in the United States generally involves the following:
| | • | | completion of extensive preclinical laboratory tests and animal studies performed in accordance with applicable regulations, including the FDA’s Good Laboratory Practices (GLPs), regulations and standards; |
| | • | | submission to the FDA of an IND application, which must become effective before clinical trials may begin; |
| | • | | approval by an independent institutional review board (IRB) or ethics committee representing each clinical site before the trial is commenced; |
146