FORBION EUROPEAN ACQUISITION CORP.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS (UNAUDITED)
Note 1 — Organization, Business Operation, Going Concern and Liquidity
Forbion European Acquisition Corp. (the “Company”) was incorporated as a Cayman Islands exempted company on August 9, 2021. The Company was incorporated for the purpose of effecting a merger, share exchange, asset acquisition, share purchase, reorganization or similar Business Combination with one or more businesses or entities (the “Business Combination”).
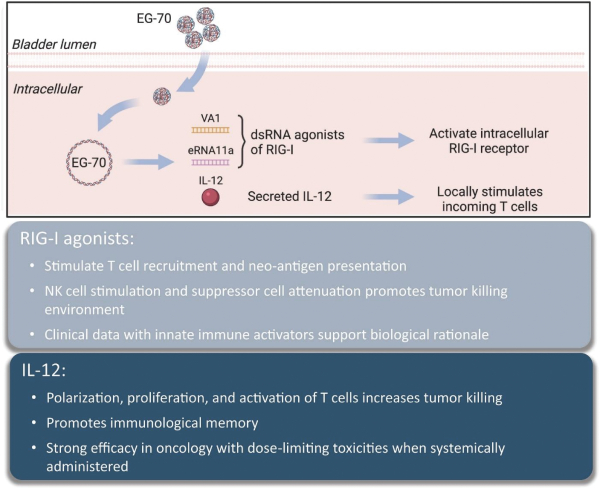
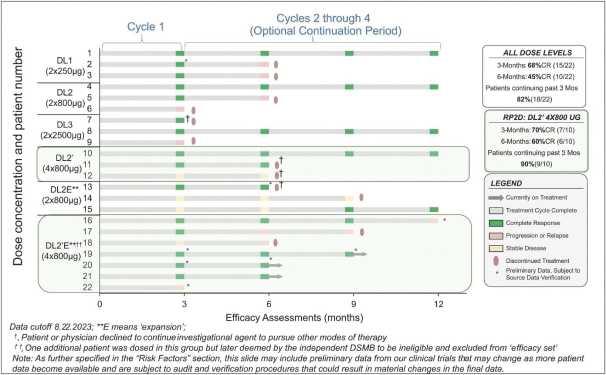
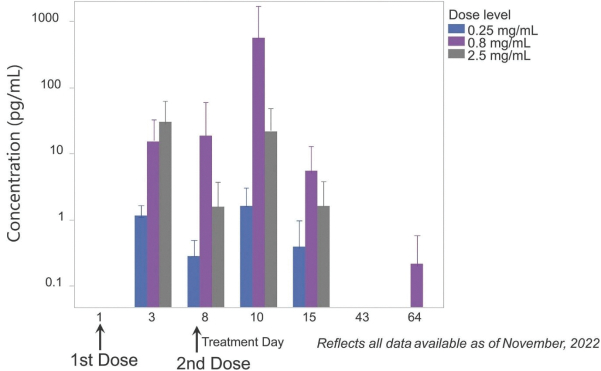
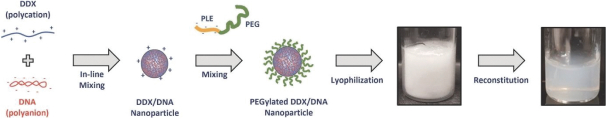
The Company has one wholly owned subsidiary, 15333881 Canada Inc, a Canadian private limited liability company incorporated on September 5, 2023 in anticipation of the consummation of the contemplated business combination between the Company, enGene, Inc. and enGene Holdings Inc. announced on May 16, 2023.
As of September 30, 2023, the Company had not commenced any operations. All activity through September 30, 2023 relates to the Company’s formation and the Initial Public Offering (the “IPO” or “Public Offering”) which is described below, and the Company’s completion of a Business Combination. The Company will not generate any operating revenues until after the completion of its initial Business Combination, at the earliest. The Company will generate
non-operating
income the form of interest income on cash and cash equivalents from the proceeds derived from the Public Offering.
The Company’s Sponsor is Forbion Growth Sponsor FEAC I B.V., a Dutch private limited liability company (the “Sponsor”).
The registration statement for the Company’s IPO was declared effective on December 9, 2021 (the “Effective Date”). On December 14, 2021, the Company’s commenced the IPO of 11,000,000 units (or 12,650,000 units if the underwriters’ over-allotment option is exercised in full) at $10.00 per unit (the “Units”), which is discussed in Note 3. Each Unit consists of one Class A ordinary share and
one-third
of one redeemable warrant (the “Public Warrants”). Each whole warrant entitles the holder to purchase one Class A ordinary share at a price of $11.50 per share. On December 15, 2021, the underwriters exercised their full over-allotment option and purchased the additional Units available to them. The aggregate Units sold in the IPO and subsequent over-allotment were 12,650,000 and generated gross proceeds of $126,500,000.
Simultaneously with the consummation of the IPO, the Company consummated the private placement of 4,700,000 warrants (or 5,195,000 warrants when the underwriters’ over-allotment option was fully exercised on December 15, 2021) (the “Private Placement Warrants”) to the Sponsor, at a price of $1.50 per Private Placement Warrant. The sale of the Private Placement Warrants in connection with the IPO and subsequent over-allotment option exercise generated gross proceeds of $7,792,500.
Transaction costs related to the IPO amounted to $5,793,160 consisting of $1,800,000 of underwriting commissions, $3,150,000 of deferred underwriting commissions, and $843,160 of other offering costs. The underwriters’ exercise of their full over-allotment option generated an additional $907,500 in transaction costs for aggregate transaction costs of $6,700,660 consisting of $2,130,000 of underwriting commissions, $3,727,500 of deferred underwriting commissions and $843,160 of other offering costs. In addition, $1,641,236 of cash was held outside of the Trust Account (as defined below) and is available for working capital purposes.
On December 15, 2021, the underwriter fully exercised the over-allotment option and purchased an additional 1,650,000 Units for additional gross proceeds of $16,500,000. Simultaneously with the exercise of the over-allotment option, the Sponsor purchased an additional 495,000 Private Placement Warrants for additional gross proceeds of $742,500.
The underwriters’ exercise of their full over-allotment option generated an additional $907,500 in transaction costs for aggregate transaction costs of $6,700,660 consisting of $2,130,000 of underwriting commissions, $3,727,500 of deferred underwriting commissions and $843,160 of other offering costs.
F-88