Use these links to rapidly review the document
TABLE OF CONTENTS
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | |
| (Mark One) | | |
ý |
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2014 |
or |
o |
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
|
Commission file number 001-10865
AMAG Pharmaceuticals, Inc.
(Exact Name of Registrant as Specified in Its Charter)
| | |
Delaware
(State or Other Jurisdiction of
Incorporation or Organization) | | 04-2742593
(I.R.S. Employer
Identification No.) |
1100 Winter Street
Waltham, Massachusetts
(Address of Principal Executive Offices) |
|
02451
(Zip Code) |
(617) 498-3300
(Registrant's Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| | |
Title of each class | | Name of each exchange on which registered |
|---|
| Common Stock, par value $0.01 per share | | NASDAQ Global Select Market |
| Preferred Share Purchase Rights | | |
Securities registered pursuant to Section 12(g) of the Act:None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No o
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of "accelerated filer," "large accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer o | | Accelerated filer ý | | Non-accelerated filer o
(Do not check if a
smaller reporting company) | | Smaller Reporting Company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No ý
The aggregate market value of the registrant's voting stock held by non-affiliates as of June 30, 2014 was approximately $453,000,000 based on the closing price of $20.72 of the Common Stock of the registrant as reported on the NASDAQ Global Select Market on such date. As of February 4, 2015, there were 25,615,978 shares of the registrant's Common Stock, par value $0.01 per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Definitive Proxy Statement that the registrant intends to file in connection with the solicitation of proxies for the Annual Meeting of Stockholders within 120 days of the end of the fiscal year ended December 31, 2014 are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
AMAG PHARMACEUTICALS, INC.
FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2014
TABLE OF CONTENTS
| | | | |
| | PART I | | |
Item 1. | | Business | | 2 |
Item 1A. | | Risk Factors | | 39 |
Item 1B. | | Unresolved Staff Comments | | 72 |
Item 2. | | Properties | | 72 |
Item 3. | | Legal Proceedings | | 73 |
Item 4. | | Mine Safety Disclosures | | 75 |
| | PART II | | |
Item 5. | | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | | 76 |
Item 6. | | Selected Financial Data | | 79 |
Item 7. | | Management's Discussion and Analysis of Financial Condition and Results of Operations | | 81 |
Item 7A. | | Quantitative and Qualitative Disclosures About Market Risk | | 115 |
Item 8. | | Financial Statements and Supplementary Data | | 117 |
Item 9. | | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | | 179 |
Item 9A. | | Controls and Procedures | | 179 |
Item 9B. | | Other Information | | 179 |
| | PART III | | |
Item 10. | | Directors, Executive Officers and Corporate Governance | | 180 |
Item 11. | | Executive Compensation | | 180 |
Item 12. | | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | | 180 |
Item 13. | | Certain Relationships and Related Transactions, and Director Independence | | 180 |
Item 14. | | Principal Accountant Fees and Services | | 180 |
| | PART IV | | |
Item 15. | | Exhibits and Financial Statement Schedules | | 181 |
Table of Contents
PART I
Except for the historical information contained herein, the matters discussed in this Annual Report on Form 10-K may be deemed to be forward-looking statements that involve risks and uncertainties. We make such forward-looking statements pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 and other federal securities laws. In this Annual Report on Form 10-K terminology such as "may," "will," "could," "should," "would," "expect," "anticipate," "continue," "believe," "plan," "estimate," "intend" or other similar words and expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements.
Unless the context suggests otherwise, references to "Feraheme" refer to both Feraheme (the trade name for ferumoxytol in the U.S. and Canada) and Rienso (the trade name for ferumoxytol in the EU and Switzerland).
Examples of forward-looking statements contained in this report include, without limitation, statements regarding the following: plans to pursue opportunities to make new advancements in patients' health and to enhance treatment accessibility; plans to diversify and grow our product portfolio; expectations and plans as to regulatory and commercial developments and activities, including with regard to label changes for Feraheme and our plan to work with the FDA to finalize the Feraheme label, the pursuit, if any, of a broader indication for Feraheme, commercialization efforts, if any, for Feraheme outside of the U.S., requirements and initiatives for clinical trials and studies, post-approval commitments for our products and the lifecycle management program for Makena; expectations as to what impact recent regulatory developments will have on our business and competition, including recent changes to our product information and label, and other risk minimization measures in the EU; the market opportunities for each of our products; the amount of resources that we intend to dedicate to the commercialization of Feraheme; expected transitioning activities with Takeda Pharmaceutical Company Limited ("Takeda") and the impact of Takeda's withdrawal of the application for Type II Variation to vary the marketing authorization for Rienso in the EU or our mutual decision with Takeda to initiate withdrawal of Rienso's current marketing authorizations in the EU and Switzerland; our expectations regarding the results of discussions with Health Canada, including our belief that approval of the broader indication for Feraheme in such territory is unlikely without additional clinical data and the possibility that Health Canada will impose additional restrictions on the current Feraheme CKD indication; beliefs about compounding pharmacies and the impact of recent legislation focused on compounding pharmacies; beliefs regarding possible entry of generic competitors, including timing, for both Makena and Feraheme; plans regarding our sales and marketing initiatives, including our contracting strategy and efforts to increase patient compliance and access; the impact of government regulations on our business and the pricing and reimbursement for our products, including the Branded Drug Fee under the Healthcare Reform Act and the Medicare reimbursement rate and estimates for Medicaid rebates; our expectations regarding the timing for enrollment in and commencement of our clinical trials and studies; our expectation of costs to be incurred in connection with and revenue sources to fund our future operations; our expectation for the patient populations for Makena and Feraheme; our expectations regarding the contribution of Makena and Feraheme sales to the funding of our on-going operations; the magnitude of costs and timing of integrating Lumara Health into our current business; expectations regarding the manufacture of all drug substance and drug products at our third-party manufacturers; plans to increase headcount; our expectations regarding customer returns and other revenue-related reserves and accruals; estimates regarding our net operating loss carryforwards and other tax attributes; initiatives to improve the reputation of Makena and educate industry participants on the benefits of Makena; the impact of accounting pronouncements; the effect of product price increases; expected increases in research and development expenses; expectations regarding our financial results, including revenues, cost of product sales, selling, general and administrative expenses, restructuring costs and net income (expense); the impact on revenues from the termination of our license arrangement with Takeda; our investing activities; expectations regarding our cash, cash equivalents and investments balances and capital needs; the impact and outcomes of our legal proceedings; our beliefs regarding the validity of our
1
Table of Contents
ferumoxytol patent portfolio; estimates and beliefs related to our debt, including our Convertible Notes and the Term Loan Facility; expected customer mix and utilization rates for our products; the impact of volume rebates and other incentives; provider purchase patterns and use of competitive products; the valuation of certain intangible assets, goodwill, contingent consideration, debt and other assets and liabilities, including our methodology and assumptions regarding fair value measurements; our gross to net sales adjustments; our expectations regarding competitive pressures and the impact on growth on our product sales; our plans regarding manufacturing; the timing of our planned research and development projects; the manner in which we intend or are required to settle the conversion of our Convertible Notes; plans to submit the NOL Amendment to our Rights Plan to our shareholders for approval; and our expectations for our cash, revenue, cash equivalents and investments balances and information with respect to any other plans and strategies for our business. Our actual results and the timing of certain events may differ materially from the results discussed, projected, anticipated or indicated in any forward-looking statements.
Any forward-looking statement should be considered in light of the factors discussed in Part I, Item 1A below under "Risk Factors" and elsewhere in this Annual Report on Form 10-K. We caution readers not to place undue reliance on any such forward-looking statements, which speak only as of the date they are made. We disclaim any obligation, except as specifically required by law and the rules of the U.S. Securities and Exchange Commission, to publicly update or revise any such statements to reflect any change in company expectations or in events, conditions or circumstances on which any such statements may be based, or that may affect the likelihood that actual results will differ from those set forth in the forward-looking statements.
ITEM 1. BUSINESS:
Overview
AMAG Pharmaceuticals, Inc., a Delaware corporation, was founded in 1981. We are a specialty pharmaceutical company with a focus on maternal health, anemia and cancer supportive care. We currently market Makena® (hydroxyprogesterone caproate injection), Feraheme® (ferumoxytol) Injection for Intravenous ("IV") use and MuGard® Mucoadhesive Oral Wound Rinse. The primary goal of our company is to bring to market therapies that provide clear benefits and improve patients' lives.
Currently, our two primary sources of revenue are from the sale ofMakena andFeraheme. On November 12, 2014, we acquired Lumara Health Inc. ("Lumara Health"), a privately held pharmaceutical company specializing in women's health, for approximately $600.0 million in upfront cash consideration (subject to finalization of certain adjustments related to Lumara Health's financial position at the time of closing, including adjustments related to net working capital, net debt and transaction expenses as set forth in the definitive agreement with Lumara Health (the "Lumara Agreement")) and approximately 3.2 million shares of our common stock having a fair value of approximately $112.0 million at the time of closing. The Lumara Agreement includes future contingent payments of up to $350.0 million in cash (or upon mutual agreement between us and the former Lumara Health security holders, future contingent payments may also be made in common stock or some combination thereof) payable by us to the former Lumara Health security holders based upon the achievement of certain sales milestones through calendar year 2019. In connection with the acquisition of Lumara Health, we acquiredMakena, a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. We sellMakena to specialty pharmacies and distributors, who, in turn sellMakena to healthcare providers, hospitals, government agencies and integrated delivery systems. Additional details regarding the Lumara Agreement can be found in Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K.
2
Table of Contents
Feraheme was approved for marketing in the U.S. in June 2009 by the U.S. Food and Drug Administration (the "FDA") for use as an IV iron replacement therapy for the treatment of iron deficiency anemia ("IDA") in adult patients with chronic kidney disease ("CKD"). We began sellingFeraheme in the U.S. in July 2009 through our commercial organization, including a specialty sales force. We sellFeraheme to authorized wholesalers and specialty distributors, who in turn, sellFeraheme to healthcare providers who administerFeraheme primarily within hospitals, hematology and oncology centers, and nephrology clinics.
In addition to continuing to pursue opportunities to make new advancements in patients' health and to enhance treatment accessibility, we intend to continue to expand and diversify our portfolio through the in-license or purchase of additional pharmaceutical products or companies. We are seeking complementary products that will leverage our corporate infrastructure, sales force call points and commercial expertise, with a particular focus on maternal health specialists, hematology and oncology centers, nephrology clinics and hospitals. We are evaluating and plan to pursue commercial products as well as late-stage development assets. In addition, we are contemplating transactions that allow us to realize cost synergies to increase cash flows, as well as transactions that potentially optimize after-tax cash flows.
In June 2014, we proposed changes to the FDA related to our current U.S. label ofFeraheme based on a review of global post-marketing data to strengthen the warnings and precautions section of the label and mitigate the risk of serious hypersensitivity reactions, including anaphylaxis, in order to enhance patient safety. After considering our June 2014 submission and other information, in January 2015, the FDA notified us that it believes new safety information should be included in the labeling forFeraheme, including, among other things, a boxed warning to highlight the risks of serious hypersensitivity/anaphylaxis reactions and revisions thatFeraheme should only be administered through an IV infusion (i.e., not by IV injection) and should be contraindicated for patients with any known history of drug allergy. The FDA's recommended label changes go beyond what we proposed in June 2014. We plan to work with the FDA to finalize an updated U.S.Feraheme label.
In December 2012, we submitted a supplemental new drug application ("sNDA") to the FDA seeking approval forFeraheme for the treatment of IDA in adult patients who had failed or could not use oral iron. In January 2014, we received a complete response letter from the FDA for the sNDA informing us that our sNDA could not be approved in its present form and stating that we have not provided sufficient information to permit labeling ofFeraheme for safe and effective use for the proposed broader indication. The FDA indicated that its decision was based on the cumulative ferumoxytol data, including the global Phase III IDA program and global post-marketing safety reports for the currently indicated CKD patient population. The FDA suggested, among other things, that we submit additional clinical trial data in the proposed broad IDA patient population with a primary composite safety endpoint of serious hypersensitivity/anaphylaxis, cardiovascular events and death, events that are included in the labels ofFeraheme and other IV irons and that have been reported in the post-marketing environment forFeraheme. Additionally, the FDA proposed potentially evaluating alternative dosing and/or administration ofFeraheme as well as potential changes to labeling that would be intended to reduce the risk of serious hypersensitivity reactions associated withFeraheme. In June 2014, we met with the FDA to discuss our proposed approach to resolving the points that were raised in the complete response letter. Based on the FDA's feedback, we submitted a revised proposal that includes the design of a potential clinical trial, a safety endpoint for such trial and alternative methods of administration ofFeraheme. We expect to receive feedback from the FDA during 2015 and expect thereafter to be able to assess and determine the path forward, if any, forFeraheme in the broad IDA patient population in the U.S., including the related timing and cost of any clinical trials.
3
Table of Contents
Further, in October 2014, we filed with the FDA a prior approval supplement to the originalMakena New Drug Application ("NDA") seeking approval of a 1 mL preservative-free vial ofMakena and we are seeking to expandMakena's formulations and drug delivery technologies as part of the product's lifecycle management program.
Outside of the U.S., ferumoxytol has been granted marketing approval in the European Union ("EU"), Canada and Switzerland for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD. In March 2010, we entered into a License, Development and Commercialization Agreement (the "Takeda Agreement"), which was amended in June 2012 (the "Amended Takeda Agreement") with Takeda. On December 29, 2014, we entered into an agreement with Takeda to terminate the Amended Takeda Agreement and we will regain all worldwide development and commercialization rights forFeraheme following the transfer of marketing authorizations from Takeda to us (the "Takeda Termination Agreement"). Under the Amended Takeda Agreement, Takeda had an exclusive license to market and sell ferumoxytol in the EU, Canada, and Switzerland, as well as certain other geographic territories. The EU marketing authorization forRienso is valid in the 28 EU Member States as well as in Iceland, Liechtenstein and Norway. The trade name for ferumoxytol in Canada isFeraheme and outside of the U.S. and Canada the trade name isRienso. Additional details regarding the Takeda Termination Agreement can be found in Note R, "Collaborative Agreements," to our consolidated financial statements included in this Annual Report on Form 10-K.
Sales ofFeraheme/Rienso outside of the U.S. do not and are not expected to materially contribute to our revenues. As such, and in light of the Takeda Termination Agreement, we have been assessing various commercialization strategies forRienso in the EU and Switzerland andFeraheme in Canada. A number of considerations influence our analysis of our commercialization opportunities outside of the U.S., including (i) regulatory developments and the potential cost of post-approval clinical trial commitments and post-marketing obligations required by regulatory authorities outside of the U.S., (ii) the product's commercial viability (sales potential relative to the cost of maintaining the product on the market) in light of the current CKD label, the possible impact of future label changes, including any impact in the U.S., and the competitive landscape, and (iii) possible approaches in different geographies, which may include seeking a licensing or distribution partner or commercializing the product ourselves. Based on these considerations, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
In the future, we may decide to seek to obtain a new marketing authorization for ferumoxytol in the EU, particularly if we generate additional clinical data to support potential approval in the broader IDA indication. There can be no assurance that we will be able to develop an approach that would be economically viable for us or a commercialization partner.
In February 2014, we issued $200.0 million of 2.5% convertible senior notes due February 15, 2019 (the "Convertible Notes"). Interest is payable semi-annually in arrears on February 15 and August 15 of each year, beginning on August 15, 2014. The initial conversion rate is 36.9079 shares of our common stock per $1,000 principal amount of the Convertible Notes, which corresponds to an initial conversion price of approximately $27.09 per share of our common stock and represents a conversion premium of approximately 35% based on the last reported sale price of our common stock of $20.07 on February 11, 2014, the date the Convertible Notes offering was priced. In addition, in connection with the pricing of the Convertible Notes and in order to reduce the potential dilution to our common stock and/or offset cash payments due upon conversion of the Convertible Notes, we also entered into convertible bond hedge and warrant transactions in February 2014. The initial exercise price of the warrants is $34.12 per share, subject to adjustment upon certain events, which is 70% above the last reported sale price of our common stock of $20.07 on February 11, 2014.
4
Table of Contents
On November 12, 2014, in connection with the acquisition of Lumara Health, we entered into the Term Loan Facility, which provides for term loans in the aggregate principal amount of $340.0 million (the "Term Loan Facility"). We used $327.5 million of the Term Loan Facility proceeds to partially finance the $600.0 million cash portion of the Lumara Health acquisition. The Term Loan Facility bears interest, at our option, at either the Eurodollar rate plus a margin of 6.25% or the prime rate plus a margin of 5.25%. The Eurodollar rate is subject to a 1.00% floor and the prime rate is subject to a 2.00% floor. As of December 31, 2014, the stated interest rate was 7.25%. We must repay the Term Loan Facility in installments of (a) $8.5 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2015 through the quarter ending December 31, 2015, and (b) $12.8 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2016 through the quarter ending September 30, 2020, with the balance due in a final installment on November 12, 2020. The Term Loan Facility matures on November 12, 2020, except that the Term Loan Facility will mature on September 30, 2018 if:
- (a)
- more than $25.0 million in aggregate principal amount of our Convertible Notes remain outstanding and not converted to common stock or refinanced and replaced with debt that matures following, and has no amortization prior to, the date that is six and one half years following the closing date; and
- (b)
- the aggregate principal amount of the Term Loan Facility (including all undrawn incremental commitments) is greater than $50.0 million on and as of such date.
See Note S, "Debt," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information regarding the Convertible Notes, the bond hedge and warrant transactions, as well as the Term Loan Facility.
Our common stock trades on the NASDAQ Global Select Market ("NASDAQ") under the trading symbol "AMAG."
5
Table of Contents
Products
The following table summarizes the current uses and, subject to regulatory approval, potential uses of our products, the current U.S. and foreign regulatory status, and the primary markets for our products.
| | | | | | |
Product | | Uses/Potential Uses | | U.S. Regulatory Status | | Foreign
Regulatory Status |
|---|
| Makena® (hydroxyprogesterone caproate injection) (5 mL multi-use vial) | | A progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. | | Approved and marketed. | | Not approved outside of the U.S. |
Makena® (hydroxyprogesterone caproate injection) (1 mL vial, preservative-free, single dose) |
|
A progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. |
|
Prior approval supplement submitted to the FDA in October 2014.
Decision from the FDA expected in the second quarter 2015. |
|
Not approved outside of the U.S. |
Feraheme® (ferumoxytol) |
|
IV iron replacement therapeutic agent for the treatment of IDA in adult patients with CKD. |
|
Approved and marketed. |
|
Approved and marketed asFeraheme in Canada.
Approved and marketed asRienso in the EU.*
Approved in Switzerland and not currently marketed.* |
Feraheme® (ferumoxytol) |
|
IV iron replacement therapeutic agent for the treatment of patients with a history of unsatisfactory oral iron therapy or in whom oral iron cannot be used. |
|
sNDA filed December 2012. Complete Response Letter received January 2014.
Submitted proposal to FDA in 2014 that included the design of a potential clinical trial and we are awaiting feedback. |
|
Application for Type II Variation filed with the European Medicines Agency ("EMA") in 2013 and withdrawn in January 2015.
Decision from Health Canada on sNDS expected in the second half of 2015. |
MuGard® Mucoadhesive Oral Wound Rinse |
|
Management of oral mucocitis/stomatiits and all types of oral wounds. |
|
Cleared and marketed. |
|
We license only the U.S. commercial rights from PlasmaTech. |
- *
- As discussed above, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. Our licensing arrangement with Takeda, and its termination, is discussed below under the heading "Collaboration, License and Other Material Agreements—Takeda."
For a discussion of the substantive regulatory requirements applicable to the development and regulatory approval process in the U.S. and other countries, see "Government Regulation" below.
Makena
On November 12, 2014, we acquired Lumara Health, a privately held pharmaceutical company specializing in women's health, including its marketed drug productMakena, the only FDA-approved drug indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth.Makena is administrated intramuscularly by a healthcare professional at a dose of 250 mg (1 mL) weekly with treatment beginning between 16 weeks and
6
Table of Contents
20 weeks and six days and continuing until 37 weeks (through 36 weeks and six days) of pregnancy or delivery, whichever happens first.
Makena was approved by the FDA in February 2011 and was granted orphan drug exclusivity through February 3, 2018. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, defined, in part, as a patient population of fewer than 200,000 in the U.S. In the U.S., the company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug for a period of seven years. This orphan drug exclusivity prevents the FDA from approving another application for the "same drug" for the same orphan indication during the exclusivity period, except in very limited circumstances. A designated orphan drug may not receive orphan drug exclusivity for an approved indication if that indication is for the treatment of a condition broader than that for which it received orphan designation. In addition, orphan drug exclusivity marketing rights in the U.S. may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
Preterm birth is defined as a birth prior to 37 weeks of pregnancy. According to the Centers for Disease Control and Prevention ("CDC"), in 2012, preterm births affected more than 450,000 babies, or one of every nine infants born in the U.S. Although, the causes of preterm births are not fully understood, certain women are at a greater risk for preterm birth, including those who have had a previous preterm birth, are pregnant with multiples or have certain uterine or cervical problems.Makena is indicated only for women with a history of spontaneous singleton preterm birth who are pregnant with a singleton, which accounts for approximately 140,000 pregnancies annually in the U.S. High blood pressure, pregnancy complications (such as placental problems) and certain other health or lifestyle factors may also be contributing factors. The last few weeks of a woman's pregnancy are important to the full development of many major organ systems, including the brain, lungs, and liver. Preterm births can increase the risk of infant death and can also result in serious long-term health issues for the child, including respiratory problems, gastrointestinal conditions, cerebral palsy, developmental delays, and vision and hearing impairments. According to a 2007 report by the Institute of Medicine (US) Committee on Understanding Premature Birth and Assuring Healthy Outcome, the annual societal economic cost associated with preterm birth is at least $26.2 billion and includes medical and healthcare costs for the baby, labor and delivery costs for the mother, early intervention and special education services, and costs associated with lost work and pay.
Makena was approved under the provisions of the FDA's "Subpart H" Accelerated Approval regulations. The Subpart H regulations allow certain drugs, for serious or life-threatening conditions, to be approved on the basis of surrogate endpoints or clinical endpoints other than survival or irreversible morbidity. As a condition of approval under Subpart H, the FDA required thatMakena's sponsor perform certain adequate and well-controlled post-marketing clinical studies to verify and describe clinical benefit ofMakena as well as fulfill certain other post-approval commitments. We are currently conducting the following clinical studies; (a) an ongoing efficacy and safety clinical study ofMakena; (b) an ongoing follow-up study of the babies born to mothers from the efficacy and safety clinical study; and (c) a completed pharmacokinetic study of women takingMakena. Given the patient population (i.e., women pregnant who are at an increased high risk for recurrent preterm delivery) and the informed risk of receiving a placebo instead of the active approved drug in the U.S., the pool of prospective subjects for such clinical trials in the U.S. is small and we are therefore seeking enrollment on a global scale.
7
Table of Contents
We are pursuing a lifecycle management program forMakena, some elements of which may provide new intellectual property or data exclusivity beyond February 2018 by exploring new routes of administration and the use of new delivery technologies, as well as reformulation technologies. As part of this program, in October 2014, a prior approval supplement for a preservative-free, single-dose (1 mL) vial forMakena was filed with and is under review by the FDA. We expect a decision in the second quarter of 2015.Makena is currently available in a 5-dose (5 mL) vial.
Feraheme for the treatment of IDA in patients with CKD
In June 2009,Feraheme was approved for marketing in the U.S. by the FDA for use as an IV iron replacement therapy for the treatment of IDA in adult patients with all stages of CKD, Stage 1 through Stage 5 (end-stage renal disease). In July 2009, we began to market and sellFeraheme in the U.S. WhileFeraheme is approved for IDA in all stages of CKD, beginning in 2010, due to changes in the way the federal government reimburses providers for the care of dialysis patients, the utilization ofFeraheme shifted to non-dialysis patients. The non-dialysis CKD IDA market is made up of a range of healthcare providers who administer IV iron, including nephrologists, hematologists, oncologists, hospitals and other end-users who treat patients with CKD. We anticipate the majority of allFeraheme utilization in the U.S. will continue to be in the non-dialysis CKD patient population if and untilFeraheme receives a broader label to include non-CKD patients.
In June 2014, we proposed changes to the FDA related to our current U.S. label ofFeraheme based on a review of global post-marketing data to strengthen the warnings and precautions section of the label and mitigate the risk of serious hypersensitivity reactions, including anaphylaxis, in order to enhance patient safety. After considering our June 2014 submission and other information, on January 7, 2015, the FDA notified us that it believes new safety information should be included in the labeling forFeraheme, including, among other things, a boxed warning to highlight the risks of serious hypersensitivity/anaphylaxis reactions and revisions thatFeraheme should only be administered through an IV infusion (i.e., not by IV injection) and should be contraindicated for patients with any known history of drug allergy. The FDA's recommended label changes go beyond what we proposed in June 2014. We plan to work with the FDA to finalize an updated U.S.Feraheme label.
In Europe, Takeda has been commercializing ferumoxytol since its approval in June 2012 under the trade nameRienso, currently in nine EU countries.Rienso is subject to periodic review by the EMA's Pharmacovigilance Risk Assessment Committee ("PRAC") and in February 2014 Takeda, as the marketing authorization holder (the "MAH") forRienso, submitted to PRAC a Periodic Safety Update Report ("PSUR") concerningRienso as part of such review. A PSUR is a pharmacovigilance document submitted by the MAH at defined intervals and is intended to provide a safety update permitting an evaluation of the risk-benefit balance of a medicinal product while it is commercialized.
As part of its assessment of the PSUR, PRAC reviewed various data, including the rate of hypersensitivity reactions with fatal outcomes withRienso. Following that assessment, and in agreement with the EMA, Takeda issued a Direct Healthcare Professional Communication ("DHPC") letter in May 2014 to remind physicians in the EU of the existing risk minimization measures for all IV iron products to manage and minimize the risk of serious hypersensitivity reactions that were included in the special warnings and precautions sections of theRienso label.
In July 2014 and again in January 2015, also in connection with the PSUR evaluation, PRAC confirmed that the benefit/risk balance ofRienso in the currently approved CKD indication remains favorable. These confirmations were subject to a number of proposed changes to the product information and label and other risk minimization measures, including, among others, thatRienso
8
Table of Contents
should be administered to patients by infusion over at least 15 minutes (replacing injection) and that it should be contraindicated in patients with any known history of drug allergy (the "July Recommendations"), that the label should caution that elderly patients or patients with multiple co-morbidities who experience a serious hypersensitivity reaction due toRienso may have more severe outcomes (the "January Recommendations"), and related variations to the Summary of Product Characteristics ("SmPC"). The PRAC's recommendations were subsequently endorsed by the EMA's Committee for Medicinal Products for Human Use ("CHMP"). Takeda updated the product's label to reflect the July Recommendations and in August 2014 issued a DHPC letter informing physicians of these changes.
In December 2011, ferumoxytol was granted marketing approval in Canada, under the trade nameFeraheme, for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD. In August 2012, ferumoxytol was granted marketing approval in Switzerland under the trade nameRienso for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD, but has subsequently been withdrawn from the market. As discussed above, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
Chronic kidney disease, anemia, and iron deficiency
CKD is the gradual and permanent loss of kidney function. It is a progressive illness that contributes to the development of many complications, including anemia, hypertension, fluid and electrolyte imbalances, acid/base abnormalities, bone disease and cardiovascular disease. According to the National Kidney Foundation, 26 million Americans are living with CKD and millions of others are at risk. Anemia, a common condition among CKD patients, is associated with cardiovascular complications, decreased quality of life, hospitalizations, and increased mortality. Anemia develops early during the course of CKD and worsens with advancing kidney disease. Patients with anemia can look pale, feel fatigued, experience shortness of breath, low energy, headaches, palpitations or chest pains, and have a loss of appetite, trouble sleeping and trouble concentrating. Anemia in CKD patients is most often considered to be caused by an insufficient production of erythropoietin, a hormone made by the kidneys which tells the body to produce red blood cells, and iron deficiency, due to inadequate iron intake, blood loss or because the body cannot use iron stores. Regardless of the cause of the iron deficiency, iron replacement therapy is essential to increase iron stores and raise hemoglobin levels. Iron is also essential for effective treatment with erythropoiesis stimulating agents ("ESAs"), which are commonly used in anemic patients to stimulate red blood cell production. Based on data contained in a 2009 publication in theJournal of the American Society of Nephrology, we estimate that there are approximately 1.6 million adults in the U.S. diagnosed with IDA and stages 3 through 5 CKD, who are patients in the mid to later stages of CKD but not yet on dialysis and could therefore benefit from receiving iron.
Currently there are two methods used to treat IDA in CKD patients: oral iron supplements and IV iron. The National Kidney Foundation's Kidney Disease Outcomes Quality Initiative guidelines recommend either oral or IV iron for peritoneal dialysis patients and non-dialysis patients with stages 1 through 5 CKD. Oral iron is currently the first-line iron replacement therapy of choice of most physicians in both the U.S. and abroad. However, oral iron supplements are poorly absorbed by many patients, which may adversely impact their effectiveness, and are associated with certain side effects, such as constipation, diarrhea, and cramping, that may adversely affect patient compliance in using such products. In addition, it can take an extended time for hemoglobin levels to improve following the initiation of oral iron treatment, and even then the targeted hemoglobin levels may not be reached. Conversely, iron given intravenously allows larger amounts of iron to be provided to patients while avoiding many of the side effects and treatment compliance issues associated with oral iron, and can result in faster rises in hemoglobin levels. The administration of IV iron has been shown to be effective
9
Table of Contents
in treating anemia either when used alone or in combination with an ESA. Current U.S. treatment guidelines indicate that treating first with iron alone may delay or reduce the need for ESA therapy. Iron supplementation is widely used in CKD patients to treat iron deficiency, prevent its development in ESA-treated patients, raise hemoglobin levels in the presence or absence of ESA treatment, and reduce ESA doses in patients receiving ESA treatment. We believe that a small fraction of non-dialysis CKD patients in the U.S. who are diagnosed with IDA are currently being treated with IV iron, and thus a significant opportunity remains to grow the market for IV iron in this patient population.
We have initiated a randomized, active-controlled pediatric study ofFeraheme for the treatment of IDA in pediatric CKD patients to meet our FDA post-approval Pediatric Research Equity Act requirement to support pediatric labeling ofFeraheme in the U.S. The study covers both dialysis-dependent and non-dialysis dependent CKD pediatric patients and will assess the safety and efficacy ofFeraheme treatment as compared to oral iron in approximately 288 pediatric patients.
Our pediatric investigation plan, which was a requirement for submission of the marketing authorization application for ferumoxytol, was approved by the EMA in December 2009 and amended in 2012 and 2014. It includes the pediatric study, as described above, and two additional pediatric studies requested by the EMA. These additional studies include a rollover extension study in pediatric CKD patients and a study in pediatric patients with IDA regardless of the underlying cause. The rollover study is open for enrollment. The pediatric IDA study will commence once the appropriate dose ofFeraheme is determined from the study data resulting from the pediatric study ofFeraheme, described above.
As part of our post-approval commitments to the EMA, we are conducting a global multi-center clinical trial to determine the safety and efficacy of repeat doses of ferumoxytol for the treatment of IDA in patients with hemodialysis-dependent CKD. As part of the commitment we made to the EMA as a condition of the approval of the marketing authorization for ferumoxytol in the EU, this study includes a treatment arm with iron sucrose using a magnetic resonance imaging sub-analysis to evaluate the potential for iron to accumulate in the body following treatment with IV iron, specifically in the heart and liver, and, where possible, other major organs following repeated IV iron administration over a two year period (the "hd-CKD Study"). Enrollment has been completed.
We have assumed any post-marketing obligations of Takeda as part of the Takeda Termination Agreement, including costs that otherwise would have been Takeda's obligation under the Amended Takeda Agreement for the ongoing pediatric studies and the ongoing multi-center clinical trial discussed above. In connection with our decision to withdraw the marketing authorization forRienso in the EU and Switzerland, we may modify or terminate clinical trials being conducted as part of our post-approval commitments to the EMA.
Feraheme for the treatment of IDA in a broad range of patients
IDA not associated with CKD is widely prevalent in many different patient populations. For many of these patients, treatment with oral iron is unsatisfactory. In the U.S., approximately 900,000 grams of IV iron were administered for the treatment of non-dialysis patients with IDA in 2014. We believe that approximately half, or 450,000 grams, of the IV iron administered in the U.S. was for the treatment of non-dialysis patients with CKD and the other half was for non-CKD patients with IDA due to other causes, including patients with gastrointestinal diseases or disorders, abnormal uterine bleeding, inflammatory diseases, and chemotherapy-induced anemia. It is estimated that more than 4.5 million patients in the U.S. have IDA (CKD and non-CKD). We estimate that approximately 5% to 10% of these patients are currently treated with IV iron.
10
Table of Contents
As discussed above, in December 2012, we submitted an sNDA to the FDA seeking approval forFeraheme for the treatment of IDA in adult patients who had failed or could not use oral iron. The sNDA included data from two controlled, multi-center Phase III clinical trials ("IDA-301 and IDA-302"), including more than 1,400 patients, which evaluated the safety and efficacy of ferumoxytol for the treatment of IDA in this broader patient population. Both studies met the primary efficacy endpoints related to improvements in hemoglobin. In these studies no new safety signals were observed withFeraheme treatment and the types of reported adverse events were consistent with those seen in previous studies and those contained in the approved U.S. package insert forFeraheme. In addition, patients from IDA-301 were eligible to enroll in an open-label extension study ("IDA-303") and receive treatment withFeraheme, as defined in the protocol.
In January 2014, we received a complete response letter from the FDA for the sNDA informing us that our sNDA could not be approved in its present form and stating that we have not provided sufficient information to permit labeling ofFeraheme for safe and effective use for the proposed broader indication. The FDA indicated that its decision was based on the cumulative ferumoxytol data, including the global Phase III IDA program and global post-marketing safety reports for the currently indicated CKD patient population. The FDA suggested, among other things, that we submit additional clinical trial data in the proposed broad IDA patient population with a primary composite safety endpoint of serious hypersensitivity/anaphylaxis, cardiovascular events and death, events that are included in the labels ofFeraheme and other IV irons and that have been reported in the post-marketing environment forFeraheme. Additionally, the FDA proposed potentially evaluating alternative dosing and/or administration ofFeraheme as well as potential changes to labeling that would be intended to reduce the risk of serious hypersensitivity reactions associated withFeraheme. In June 2014, we met with the FDA to discuss our proposed approach to resolving the points that were raised in the complete response letter. Based on the FDA's feedback, we submitted a revised proposal that includes the design of a potential clinical trial, a safety endpoint for such trial and alternative methods of administration ofFeraheme. We expect to receive feedback from the FDA during 2015 and expect thereafter to be able to assess and determine the path forward, if any, forFeraheme in the broad IDA patient population in the U.S., including the related timing and cost of any clinical trials.
In June 2013, Takeda filed an application for Type II Variation to vary the marketing authorization forRienso in the EU with the EMA to extend the therapeutic indication from adult patients with IDA associated with CKD to adult patients with iron deficiency from any underlying cause. During the course of CHMP's review of the Type II Variation, Takeda received inquiries and reports from regulators indicating that approval of the Type II Variation would be unlikely without additional confirmative clinical data. As a result, in January 2015, we and Takeda mutually agreed that Takeda withdraw the Type II Variation.
In addition, in October 2013, Takeda filed an sNDS with Health Canada seeking marketing approval forFeraheme for the treatment of IDA in a broad range of patients. In October 2014, Takeda received inquiries from Health Canada and in January 2015, we submitted a response to these inquiries. Based on these inquiries and interactions, we believe that approval in the broader indication is unlikely in Canada without additional clinical data. We believe that we will receive Health Canada's final decision on the sNDS in the second half of 2015, however we cannot guarantee that Health Canada will issue a final decision on the expected timeline. In addition, until we have further conversations with Health Canada, we cannot predict whether their concerns with regard to approval of the broader IDA indication, including with regard to the need for additional clinical data, will cause Health Canada to impose additional restrictions on the current CKD indication.
As discussed above, we are in the process of regaining all worldwide development and commercialization rights forFeraheme from Takeda and have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
11
Table of Contents
MuGard
In June 2013, we entered into the License Agreement with PlasmaTech Biopharmaceuticals, Inc. ("PlasmaTech") (formerly known as Access Pharmaceuticals, Inc.), under which we acquired the U.S. commercial rights toMuGard for the management of oral mucositis (the "MuGard License Agreement").MuGard was launched in the U.S. by PlasmaTech in 2010 after receiving 510(k) clearance from the FDA and is indicated for the management of oral mucositis/stomatitis (that may be caused by radiotherapy and/or chemotherapy) and all types of oral wounds (mouth sores and injuries), including aphthous ulcers/canker sores and traumatic ulcers, such as those caused by oral surgery or ill-fitting dentures or braces. Mucositis is the painful inflammation and ulceration of the mucous membranes of the mouth and gastrointestinal tract that can be caused by high-dose chemotherapy and/or radiotherapy. Oral mucositis is a common and often debilitating complication of cancer treatment that may impair oral nutritional intake or result in delays, unplanned breaks or decreases in dose for chemotherapy and/or radiation treatments, leading to sub-optimal cancer treatment results. In the U.S., there are approximately 400,000 people per year who experience oral mucositis and approximately 80% of patients with mucositis experience severe oral pain. The incidence rate and severity of symptoms depends on the type of anti-cancer treatment and patient-related risk factors. For example, based on data reported in a 2001 article inCA: A Cancer Journal for Clinicians, the incidence of oral mucositis for patients undergoing radiation for the treatment of head and neck cancer could approximate 80%. The incidence of oral mucositis for bone marrow transplant patients undergoing high dose chemotherapy and/or radiation pre-conditioning and patients undergoing conventional chemotherapy is approximately 70% and 40%, respectively.
There are few effective treatments for oral mucositis and the products in this market remain mostly undifferentiated. There are a number of approaches used to treat or manage oral mucositis, including the use of ice chips during chemotherapy treatments, various medicinal mouthwashes, topical anesthetics and analgesics, and oral gel treatments. We sellMuGard through a distribution network of specialty pharmacies and wholesalers, who in turn supply it to hospitals or hematology/oncology clinics. Currently,MuGard is used by a small percentage of the oral mucositis patients in the U.S., which represents a significant opportunity for us to address an unmet medical need and grow the sales ofMuGard in the oral mucositis market.
Our Core Proprietary Technology
Our core proprietary technology for ferumoxytol is based on coated superparamagnetic iron oxide particles and their characteristic properties. Our core competencies for ferumoxytol include the ability to design such particles for particular applications and to manufacture the particles in controlled sizes. Our technology and expertise enable us to synthesize, sterilize and stabilize these iron oxide particles in a manner necessary for use in pharmaceutical products such as IV iron replacement therapeutics.
Our iron oxide particles are composed of bioavailable iron that is easily utilized by the body and incorporated into the body's iron stores. As a result, our core technology for ferumoxytol is well-suited for use as an IV iron replacement therapy product.
Our rights to the technology are derived from and/or protected by license agreements, patents, patent applications and trade secret protections. See "Patents and Trade Secrets" below. There are no patents coveringMakena. Our rights toMuGard are governed by the MuGard License Agreement. See Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K.
12
Table of Contents
Collaboration, License and Other Material Agreements
Takeda
In March 2010, we entered into the Takeda Agreement, as amended in June 2012, under which we granted exclusive rights to Takeda to develop and commercializeFeraheme as a therapeutic agent in certain agreed-upon territories. In February 2014, we entered into the Supply Agreement with Takeda, which provides the terms under which we sellFeraheme to Takeda in order for Takeda to meet its requirements for commercial use ofFeraheme in its licensed territories. On December 29, 2014, we entered into the Takeda Termination Agreement, under which the Amended Takeda Agreement will be terminated and we will regain all worldwide development and commercialization rights forFeraheme following the transfer of the outstanding marketing authorizations. Pursuant to the Takeda Termination Agreement, we and Takeda have agreed to effectuate the termination of the Amended Takeda Agreement on a rolling basis, whereby the termination will be effective for a particular geographic territory (e.g., countries under the regulatory jurisdictions of Health Canada, the EMA and SwissMedic) upon the earlier of effectiveness of the transfer to us or a Withdrawal (as defined below) of the marketing authorization for such territory, with the final effective termination date to be on the third such effective date ("Termination Date").
In connection with each Termination Date and in accordance with the terms of the Takeda Termination Agreement, Takeda is obligated, with respect to the applicable terminated territory, to transfer and assign to us all applicable regulatory materials and approvals and certain product data, unlabeled inventory, third party contracts intellectual property rights and know-how to us, and to grant us an exclusive license for certain Takeda technology used and applied to commercializeFeraheme in the applicable territory. The Takeda Termination Agreement also details the regulatory activities each party is required to perform in connection with transferring the marketing authorization from Takeda to us in each of the territories and the allocation of the costs of such activities. We and Takeda have agreed to use commercially reasonable efforts to transfer all required activities to us on a territory-by-territory basis within 60 days after the applicable Termination Date (subject to a 30-day extension upon our request and Takeda's consent). In addition, Takeda is obligated pursuant to the Takeda Termination Agreement to provide transition assistance to us, at no cost to us, for up to 180 days after each Termination Date for the applicable termination territory. With Takeda's consent (which shall not be unreasonably withheld or delayed), we may extend the transition services period for a terminated territory for a period of time reasonably necessary to complete any services that cannot be reasonably transitioned to us during the initial 180-day period, which extension will not exceed an additional 180 days. If we request, and Takeda agrees to conduct, additional transition services after the end of the applicable transition services period, as may be extended, we will reimburse Takeda's fully burdened costs for such additional services plus 5%.
The Takeda Termination Agreement also provides that if the marketing authorization for the product is suspended in a particular territory and the parties are prevented from completing the transfer of such marketing authorization to us within 120 days after such suspension due to applicable laws or any regulatory requirements or restrictions, or if we do not fulfill our obligations to initiate marketing authorization transfer by the agreed-upon, territory-specific deadline, Takeda will have the right, in Takeda's sole discretion, to withdraw such marketing authorization (a "Withdrawal").
In consideration for the early termination of the Amended Takeda Agreement and the activities to be performed by us earlier than contemplated under the Amended Takeda Agreement, and in lieu of any future cost-sharing and milestone payments contemplated by the Amended Takeda Agreement, Takeda agreed to make certain payments to us, subject to certain terms and conditions, including up to approximately $6.7 million in connection with clinical study obligations, pharmacovigilance activities, regulatory filings and support, commercialization and back-office support and distribution expenditures and a $3.0 million milestone payment payable subject to certain regulatory conditions.
13
Table of Contents
Additionally, the Supply Agreement, which continues in effect until the expiration or termination of the Amended Takeda Agreement, will also terminate as of the respective Termination Date in the applicable geographic territory.
We have assumed any post-marketing obligations of Takeda as part of the Takeda Termination Agreement, including costs that otherwise would have been Takeda's obligation under the Amended Takeda Agreement for the ongoing pediatric studies, and the hd-CKD Study. As discussed above, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada. In connection with our decision to withdraw the marketing authorization forRienso in the EU and Switzerland, we may modify or terminate clinical trials being conducted as part of our post-approval commitments to the EMA.
Additional details regarding the Takeda Termination Agreement and related revenue can be found in Note R, "Collaborative Agreements," to our consolidated financial statements included in this Annual Report on Form 10-K.
PlasmaTech
In June 2013, we entered into the MuGard License Agreement under which PlasmaTech granted us an exclusive, royalty-bearing license, with the right to grant sublicenses, to certain intellectual property rights, including know-how, patents and trademarks, to use, import, offer for sale, sell, manufacture and commercializeMuGard in the U.S. and its territories (the "U.S. Territory") for the management of all diseases or conditions of the oropharyngeal cavity, including mucositis.
In consideration for the license, we paid PlasmaTech an upfront license fee of $3.3 million in June 2013. We are required to pay royalties to PlasmaTech on net sales ofMuGard until the later of (a) the expiration of the licensed patents or (b) the tenth anniversary of the first commercial sale ofMuGard in the U.S. Territory (the "Royalty Term"). These tiered, double-digit royalty rates decrease after the expiration of the licensed patents and are subject to off-set against certain of our expenses. After the expiration of the Royalty Term, the license shall become a fully paid-up, royalty-free and perpetual license in the U.S. Territory.
PlasmaTech remains responsible for the manufacture ofMuGard and we have entered into a quality agreement and a supply agreement with PlasmaTech under which we purchaseMuGard inventory from PlasmaTech. Our inventory purchases are at the price actually paid by PlasmaTech to purchase it from a third-party plus a mark-up to cover administration, handling and overhead.
PlasmaTech is responsible for maintenance of the licensed patents at its own expense, and we retain the first right to enforce any licensed patent against third party infringement. The MuGard License Agreement terminates at the end of the Royalty Term, but is subject to early termination by us for convenience and by either party upon an uncured breach by or bankruptcy of the other party.
3SBio
In 2008, we entered into the 3SBio License Agreement and the 3SBio Supply Agreement with 3SBio Inc. ("3SBio") for the development and commercialization ofFeraheme as an IV iron replacement therapeutic agent in China. In consideration of the grant of the license, we received an upfront payment of $1.0 million. In late January 2014, we mutually terminated the agreement with 3SBio, effective immediately, due to the fact that, despite the best efforts of the parties, regulatory approval in China could not be obtained within the agreed upon time period.
14
Table of Contents
Manufacturing
We currently rely solely on third parties for the manufacture ofFeraheme andMakena for our commercial and clinical use. Our third-party contract manufacturing facilities forFeraheme andMakena are subject to current good manufacturing practices ("cGMP"), regulations enforced by the FDA and equivalent foreign regulatory agencies through periodic inspections to confirm such compliance. Although we are currently working to establish and qualify alternative manufacturing facilities for both drug substance and drug product ofFeraheme and drug product forMakena, we do not currently have alternative manufacturers for ourFeraheme andMakena drug substance and drug product, as applicable. In addition, we currently do not have a supply agreement forMakena drug substance and, until we do, we plan to obtainMakena drug substance on a purchase order basis. We target to maintain sufficient inventory levels throughout our supply chain to meet our projected U.S. near-term demand ofFeraheme andMakena drug product in order to minimize risks of supply disruption at points in our single source supply chain. We intend to continue to outsource the manufacture and distribution ofFeraheme andMakena for the foreseeable future, and we believe this manufacturing strategy will enable us to direct more of our financial resources to the commercialization of our products. Under the terms of the MuGard License Agreement, PlasmaTech is responsible for all aspects of manufacturingMuGard. We have entered into a quality agreement and a supply agreement with PlasmaTech under which we purchaseMuGard inventory from PlasmaTech.
To support the commercialization of our products, we have developed a fully integrated manufacturing support system, including quality assurance, quality control, regulatory affairs and inventory control policies and procedures. These support systems are intended to enable us to maintain high standards of quality for our products.
We have also established certain testing and release specifications with the FDA and other foreign regulatory agencies. This release testing must be performed in order to allow the finished product to be used for commercial sale.
Makena
TheMakena drug product for our commercial and clinical use is currently manufactured by Hospira Worldwide, Inc. ("Hospira") under a Development and Supply Agreement, originally dated September 17, 2009, by and between Hologic, Inc. (from whom Lumara Health, then-named K-V Pharmaceutical Company ("K-V Pharmaceutical") originally purchased the worldwide rights toMakena) and Hospira, which was fully assigned to K-V Pharmaceutical in December 2012, and was amended on March 28, 2014 (as amended, the "Hospira Agreement"). Under the terms of the Hospira Agreement, Hospira was manufacturingMakena at certain agreed-upon pricing through December 31, 2014 and currently Hospira can increase the price (subject to certain limitations) ofMakena for both commercial and clinical uses, upon advance written notice to us. In addition, under the terms of the Hospira Agreement we are obligated to make certain minimum purchase requirements. The term of the Hospira Agreement applies to the manufacture of certain dosage forms and provides for an option to extend the term based on the occurrence, timing and amount of certain forecasts and purchase orders related to other dosage forms. We cannot make any guarantees that we will be able to extend the term of the Hospira Agreement on favorable terms, if at all.
Lumara Health, as our wholly owned subsidiary following consummation of the acquisition, is subject to certain continuing obligations under a Consent Decree of Permanent Injunction (the "Consent Decree") among the FDA, Lumara Health's predecessor company, K-V Pharmaceutical and certain former officers and affiliates of K-V Pharmaceutical. In particular, Lumara Health is bound by a number of provisions and requirements in the Consent Decree including, but not limited to, inspection of Lumara Health's places of business by the FDA without prior notice, and notification of FDA of particular actions and events. If Lumara Health fails to comply with applicable provisions in the
15
Table of Contents
Consent Decree, the Federal Food, Drug, and Cosmetic Act (the "FDC Act") or the FDC Act's implementing regulations, the FDA may impose specific sanctions including, but not limited to, the requirement to cease any of Lumara Health's manufacturing operations, the imposition of substantial financial penalties, and the requirement to implement additional corrective actions.
Feraheme
We currently have the following contracts in place related to the manufacture ofFeraheme:
In August 2012, we entered into a Commercial Supply Agreement, as amended in October 2013 and December 2014, with Sigma-Aldrich, Inc. ("SAFC") pursuant to which SAFC agreed to manufacture and we agreed to purchase from SAFC, the active pharmaceutical ingredient ("API") or the drug product intermediate ("DPI") for use in the finished product of ferumoxytol for U.S. commercial sale, for sale outside of the U.S., as well as for use in clinical trials (as amended, the "SAFC Agreement"). Subject to certain conditions, the SAFC Agreement provides that we purchase from SAFC certain minimum quantities of API or DPI each year, but we are not obligated to use SAFC as our sole supplier of API or DPI. In addition, the prices for each batch will decline as batches are produced in greater quantities throughout each year of the agreement. The SAFC Agreement has an initial term that ends December 31, 2020, which may be automatically extended thereafter for additional two year periods, unless cancelled by us or SAFC within an agreed-upon notice period.
The amendments to the SAFC Agreement provide updated pricing terms beginning on a certain date in the future, which are based on the amount of product produced by SAFC in a given calendar year. If SAFC is unable to offer these agreed-upon prices, we may terminate our minimum purchase commitments. In addition, if SAFC is unable to meet our actual demand requirements other than due to our acts, omissions or default, our minimum purchase commitment will be suspended for such period. Further, if after a certain date in the future, SAFC is unable to match abona fide offer from a third party to manufacture and supply product to us on better terms than provided by SAFC pursuant to the SAFC Agreement then a reduced minimum purchase commitment will apply. We have the right to terminate the SAFC Agreement and any purchase orders under certain conditions and subject to certain notice requirements. The SAFC Agreement also specifies cost-sharing arrangements relating to future process changes or capital improvements to the manufacturing process forFeraheme under the SAFC Agreement.
Patheon, Inc. (formerly DSM Pharmaceuticals, Inc.)
In January 2010, we entered into a Pharmaceutical Manufacturing and Supply Agreement, as amended in July 2014, with Patheon, Inc. (formerly DSM Pharmaceuticals, Inc.) ("Patheon") pursuant to which Patheon agreed to manufacture ferumoxytol finished drug product for U.S. commercial sale, for sale outside of the U.S., as well as for use in clinical trials at a fixed price per vial (as amended, the "Patheon Agreement"). The Patheon Agreement will continue in force until December 31, 2015. The Patheon Agreement may be terminated at any time upon mutual written agreement by us and Patheon or at any time by us subject to certain notice requirements and early termination fees. In addition, the Patheon Agreement may be terminated by either us or Patheon in the event of a material breach of the agreement by the other party provided that the breaching party fails to cure such breach within an agreed-upon notice period.
Raw Materials
We and our third-party manufacturers currently purchase certain raw and other materials used to manufactureFeraheme andMakena from third-party suppliers and, at present, do not have long-term
16
Table of Contents
supply contracts with most of these third parties. Although certain of our raw or other materials are readily available, others may be obtained only from qualified suppliers. Certain materials used inFeraheme andMakena may from time to time be procured from a single source without a qualified alternative supplier of the high-quality standards imposed on our raw and other materials used to manufactureFeraheme, we may not be able to obtain such materials of the quality required to manufactureFeraheme orMakena. The qualification of an alternative source may require repeated testing of the new materials and generate greater expenses to us if materials that we test do not perform in an acceptable manner. In addition, we or our third-party manufacturers sometimes obtain raw or other materials from one vendor only, even where multiple sources are available, to maintain quality control and enhance working relationships with suppliers, which could make us susceptible to price inflation by the sole supplier, thereby increasing our production costs. As a result of the high-quality standards imposed on our raw or other materials, we or our third-party manufacturers may not be able to obtain such materials of the quality required to manufactureFeraheme orMakena from an alternative source on commercially reasonable terms, or in a timely manner, if at all.
Patents and Trade Secrets
We consider the protection of our technology to be material to our business. Because of the substantial length of time and expense associated with bringing new products through development and regulatory approval to the marketplace, we place considerable importance on obtaining patent and trade secret protection for our products. Our success depends, in large part, on our ability to maintain the proprietary nature of our technology and other trade secrets. To do so, we must prosecute and maintain existing patents, obtain new patents and ensure trade secret protection. We must also operate without infringing the proprietary rights of third parties or allowing third parties to infringe our rights.
Our policy is to aggressively protect our competitive technology position by a variety of means, including applying for patents in the U.S. and in appropriate foreign countries. We currently hold a number of U.S. and foreign patents, which expire at various times through 2023. One of our U.S.Feraheme patents is subject to a patent term extension under U.S. patent law and FDA regulations and will expire in June 2023. There are no patents coveringMakena. We have a license to two U.S. patents relating toMuGard, that each expire in 2022. Our foreign patents may also be eligible for extension in accordance with applicable law in certain countries.
We also have patent applications pending in the U.S. and have filed counterpart patent applications in certain foreign countries directed toFeraheme. Although further patents may be issued on pending applications, we cannot be sure that any such patents will be issued on a timely basis, if at all. In addition, any issued patents may not provide us with competitive advantages or may be challenged by others, and the existing or future patents of third parties may limit our ability to commercializeFeraheme. For example, in July 2010, Sandoz GmbH ("Sandoz") filed with the European Patent Office (the "EPO") an opposition to a previously issued patent which covers ferumoxytol in EU jurisdictions. In October 2012, at an oral hearing, the Opposition Division of the EPO revoked this patent. In December 2012, our notice of appeal of that decision was recorded with the EPO, which suspended the revocation of our patent. On May 13, 2013, we filed a statement of grounds of appeal and on September 27, 2013, Sandoz filed a response to that statement. We filed a reply to that response on March 17, 2014 and oral proceedings for the appeal are scheduled for June 16, 2015. We continue to defend the validity of this patent throughout the appeals process, which we expect to take two to three years. However, in the event that we do not experience a successful outcome from the appeals process, under EU regulations ferumoxytol would still be entitled to eight years of data protection and ten years of market exclusivity from the date of approval, which we believe would create barriers to entry for any generic version of ferumoxytol into the EU market until sometime between 2020 and 2022.
17
Table of Contents
Our licensed patent rights toMuGard may not prevent competitors from independently developing and marketing a competing product that does not infringe our licensed patents or other intellectual property. Further, there are no patents coveringMakena and thus the successful commercialization ofMakena is significantly reliant on our ability to take advantage of its orphan drug exclusivity.
Frequently, the unpredictable nature and significant costs of patent litigation leads the parties to settle to remove any uncertainty related to the status of their patents. Settlement agreements between branded companies and generic applicants may allow, among other things, a generic product to enter the market prior to the expiration of any or all of the applicable patents covering the branded product, either through the introduction of an authorized generic or by providing a license to the applicant for the patents in suit.
Competition
The pharmaceutical and biopharmaceutical industries are intensely competitive and subject to rapid technological change. Many of our competitors forFeraheme are large, well-known pharmaceutical companies and may benefit from significantly greater financial, sales and marketing capabilities, greater technological or competitive advantages, and other resources. ForMakena, most of our competition comes from pharmacies that compound non-FDA approved formulations of HPC (defined below), which are sold at a lower cost thanMakena. In addition, genericFeraheme andMakena competitors could enter the market through approval of abbreviated new drug applications ("ANDAs") that useFeraheme orMakena as a reference listed drug, which would allow generic competitors to rely onFeraheme's orMakena's safety and efficacy trials instead of conducting their own studies. Because entry into the market can occur upon the expiration of the reference listed drug's exclusivity, we could face such competition in the near-term asFeraheme's U.S. market exclusivity expired in June 2014 andMakena's orphan drug exclusivity expires in February 2018. Our existing or potential new competitors forFeraheme andMakena may develop products that are more widely accepted than ours and may receive patent protection that dominates, blocks or adversely affects our product development or business.
Makena
AlthoughMakena is the only FDA-approved drug indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth, it competes for market share with compounding pharmacies. Hydroxyprogesterone caproate ("HPC") is the active ingredient inMakena. Compounding pharmacies have been manufacturing formulations of HPC (which compounded formulations we refer to as "c17P") for many years and c17P formulations will likely remain available even thoughMakena has been granted orphan drug exclusivity until February 2018. We estimate that between approximately 40% and 50% of the at-risk patient population is treated with c17P.Makena currently has between approximately 20% and 30% of the market share of the at-risk patient population with at least 30% of the at-risk patient population being treated either with other therapies that are not approved for women pregnant with a singleton with a prior history of spontaneous preterm birth of a singleton, or not treated at all.
In March 2011, the FDA issued a press release announcing that, in order to ensure continued access for patients, the FDA intended to refrain from taking enforcement action with respect to compounding pharmacies producing c17P in response to individual prescriptions for individual patients, resulting in a reduction in commercial value ofMakena's orphan exclusivity protection and in the loss of substantial market share to compounding pharmacies. In June 2012, the FDA recommended using FDA-approvedMakena instead of a compounded drug except when there is a specific medical need (e.g., an allergy) that cannot be met by the approved drug. In July 2014, the FDA issued another public statement affirming the position it took in its June 2012 press release recommending use of FDA-approvedMakena, except when there is a specific need for a compounded drug. The FDA also
18
Table of Contents
stated that when it identifies a pharmacist that compounds regularly or in inordinate amounts of any drug products that are essentially copies ofMakena, the FDA intends to take enforcement action as it deems appropriate. Despite recent negative publicity regarding compounding pharmacies, including the 2012 meningitis outbreak involving compounded drugs, the November 2013 enactment of the federal Drug Quality and Security Act ("DQSA") and recent enforcement actions against compounders violating the FDC Act,Makena will likely continue to face competition from c17P, especially in light of the long-standing availability of such compounded products, their lower cost and the criticism Lumara Health received in the past in connection with the pricing ofMakena, as discussed below.
Lumara Health was criticized for the initial list pricing ofMakena in numerous news articles and internet postings following the FDA's February 2011 approval ofMakena. Although the list price ofMakena was subsequently reduced in March 2011,Makena is still priced at a premium to c17P, which has negatively impacted coverage ofMakena by some state Medicaid programs and by certain commercial payers. Although we are undertaking efforts to educate physicians and patients about progress made toward expanding coverage ofMakena and about the benefits of FDA-approvedMakena, certain doctors continue to choose to prescribe non-FDA approved purported substitute products made by pharmaceutical compounders in lieu of prescribingMakena. In addition, efforts to appropriately respond to future concerns raised by media, professional societies, advocacy groups, policymakers or regulatory agencies regarding patient access toMakena are costly and may not be successful.
Additionally, in 1956, the FDA-approved the drug Delalutin, which contained the same active ingredient asMakena. Delalutin was approved for conditions other than reducing the risk of preterm birth and was marketed by Bristol-Myers Squibb ("BMS"). BMS stopped marketing and manufacturing the FDA-approved product and it was withdrawn from the market in 1999. In 2010, in response to a citizen petition, the FDA determined that Delalutin was not withdrawn from sale for reasons of safety or effectiveness. As such, generic drug applications may reference the withdrawn Delalutin NDA.
Thus, before the expiration ofMakena's orphan exclusivity, the FDA could determine that it has the authority to approve ANDAs that reference Delalutin so long as the ANDAs meet all relevant legal and regulatory requirements for approval and are labeled for the same indications as Delalutin (i.e., not for the risk of preterm birth). If such an approval is granted, doctors may elect to prescribe such approved drug off-label (i.e., outside of FDA-approved indications) forMakena's orphan-protected indication, which could have an adverse impact on our business and results of operations.
Moreover, if one or more generic applicants were to receive approval to sell a generic or follow-on version ofMakena for the orphan-protected indication, those generic products could potentially be approved as early as February 3, 2018 (the date on whichMakena's orphan exclusivity ends) and we would become subject to increased competition at that time.
For a detailed discussions regarding the risks and uncertainties related to competition forMakena, please refer to our Risk Factor, "Our ability to successfully commercialize Makena is dependent upon a number of factors, including maintaining the benefits of Makena's orphan drug exclusivity and the length of time before competitors begin selling generic versions of Makena."
Feraheme
AlthoughFeraheme is approved in the U.S. for the treatment of IDA in adult patients with CKD, including both dialysis and non-dialysis CKD patients, our U.S. commercial strategy is entirely focused on growing the utilization ofFeraheme in non-dialysis dependent adult CKD patients who are diagnosed with IDA. We believe there is a significant opportunity in the U.S. forFeraheme for the treatment of IDA in CKD patients not yet on dialysis. The U.S. non-dialysis IV iron market is comprised primarily of three sites of care where a substantial number of CKD patients are treated: hospitals, hematology and oncology centers, and nephrology clinics.
19
Table of Contents
Feraheme currently competes with the following IV iron replacement therapies in the U.S. for the treatment of IDA in CKD patients:
- •
- Venofer®, an iron sucrose complex, which is approved for use in hemodialysis, peritoneal dialysis, non-dialysis dependent CKD patients and pediatric CKD patients and is marketed in the U.S. by Fresenius Medical Care North America and American Regent Laboratories, Inc. ("American Regent") a subsidiary of Luitpold Pharmaceuticals, Inc. Venofer® is typically administered as a slow intravenous injection over two to five minutes in doses of 100 to 200 milligrams, thus requiring five to ten physician visits to reach a standard one gram therapeutic course;
- •
- Injectafer®, a ferric carboxymaltose injection, which is known as Ferinject® in Europe, was approved in the U.S. in July 2013 to treat IDA in adult patients who have intolerance to oral iron or have had an unsatisfactory response to oral iron. Injectafer® is also indicated for IDA in adult patients with non-dialysis dependent CKD. Injectafer® is marketed in the U.S. by American Regent, the same distributor of Venofer®. The labeled administration of Injectafer® is two slow injections or infusion of 750 milligrams each separated by at least seven days for a total cumulative dose of 1,500 milligrams, or one and a half grams per therapeutic course;
- •
- Ferrlecit®, a sodium ferric gluconate, which is marketed by Sanofi-Aventis U.S. LLC, is approved for use only in hemodialysis patients. The recommended dose of Ferrlecit® and the generic version of Ferrlecit® is 125 milligrams administered by intravenous infusion over one hour per dialysis session or undiluted as a slow intravenous injection per dialysis session, thus requiring eight physician visits to reach a standard one gram therapeutic course;
- •
- A generic version of Ferrlecit® marketed by Watson Pharmaceuticals, Inc. ("Watson");
- •
- INFeD®, an iron dextran product marketed by Watson, which is approved in the U.S. for the treatment of patients with documented iron deficiency in whom oral iron administration is unsatisfactory or impossible. The recommended dose of INFeD® is a slow push in 100 milligram doses, which would require up to ten physician visits to receive a standard one gram therapeutic course; and
- •
- Dexferrum®, an iron dextran product marketed by American Regent, which is approved in the U.S. for the treatment of patients with documented iron deficiency in whom oral iron administration is unsatisfactory or impossible. The recommended dose of INFeD® and Dexferrum® is a slow push in 100 milligram doses, which would require up to ten physician visits to receive a standard one gram therapeutic course.
As compared to the dosing regimens described above forFeraheme's U.S. competitors,Feraheme is currently administered as a 510 milligram injection or infusion followed by a second 510 milligram injection or infusion three to eight days later. In June 2014, we proposed changes to the FDA related to our current U.S. label ofFeraheme based on a review of global post-marketing data to strengthen the warnings and precautions section of the label and mitigate the risk of serious hypersensitivity reactions, including anaphylaxis, in order to enhance patient safety. After considering our June 2014 submission and other information, in January 2015, the FDA notified us that it believes new safety information should be included in the labeling forFeraheme, including, among other things, a boxed warning to highlight the risks of serious hypersensitivity/anaphylaxis reactions and revisions thatFeraheme should only be administered through an IV infusion (i.e., not by IV injection) and should be contraindicated for patients with any known history of drug allergy. These or any future changes to the label/package could adversely impact our ability to successfully compete in the U.S. IV iron market.
Pharmacosmos A/S ("Pharmacosmos") the producer of another IV iron, Monofer® (iron isomaltoside 1000), which is approved and marketed in Europe, is also conducting clinical trials in the U.S. and may try to gain regulatory approval in the U.S. for Monofer®. In January 2015, the Helsinn
20
Table of Contents
Group and Pharmacosmos announced that they entered into an agreement for the exclusive U.S. commercialization rights to Monofer®.
Outside of the U.S.,Feraheme also competes with a number of branded IV iron replacement products, including Venofer®, Ferrlecit®, Monofer®, Ferinject® (ferric carboxymaltose injection) (the brand name for Injectafer® outside the U.S.) and certain other iron dextran and iron sucrose products. Venofer® and Ferrlecit®, described above, have been marketed in many countries throughout the world, including most of Europe and Canada, for many years. Monofer® is an injectable iron preparation developed by Pharmacosmos, which is currently approved for marketing in approximately 30 countries, primarily in Europe, for the treatment of IDA. Ferinject® is an IV iron replacement therapy developed by Vifor Pharma, the pharmaceuticals business unit of the Galenica Group, and is currently approved for marketing in approximately 62 countries worldwide, for the treatment of iron deficiency where oral iron is ineffective or cannot be used.
Currently, all other IV iron products approved and marketed in the EU are approved for marketing to a broader group of patients with IDA.Rienso was approved only for use in CKD patients. In January 2015, we and Takeda mutually agreed to withdraw the application of Type II Variation forRienso in the EU to extend the therapeutic indication from adult patients with IDA associated with CKD to adult patients with iron deficiency from any underlying cause. The limitation ofRienso's approved indication to CKD patients may putFeraheme at a competitive disadvantage if we were to pursue commercialization efforts in the EU with the product's currently labelled indicated patient population. In addition, based on PRAC's July and January Recommendations, Takeda issued a DHPC letter providing that, among other measures,Riesno be administered to patients by infusion over at least 15 minutes (replacing injection) and that it be contraindicated in patients with any known history of drug allergy, and we and Takeda are in the process of updating the label to caution that elderly patients or patients with multiple co-morbidities who experience a serious hypersensitivity reaction due toRienso may have more severe outcomes, and related variations to the Summary of Product Characteristics ("SmPC"). These or any future changes toFeraheme's current indication could further put it at a disadvantage to its competitors. As discussed above, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
Feraheme may also face competition from generic IV iron replacement therapy products that achieve commercial success. For example, in 2011, Watson launched a generic version of Ferrlecit® in the U.S. which is approved for marketing in the U.S. for the treatment of IDA in adult patients and in pediatric patients age six years and older undergoing chronic hemodialysis who are receiving supplemental epoetin therapy. Sagent Pharmaceuticals, Inc. has also indicated its intention to introduce a generic iron sucrose in the U.S. in the future. Outside the U.S., there is currently a generic version of Venofer®.
The Drug Price Competition and Patent Term Restoration Act of 1984, as amended (the "Hatch-Waxman Act") requires an applicant whose subject drug is a drug listed in the FDA publication, "Approved Drug Products with Therapeutic Equivalence Evaluations," also known as the "Orange Book," to notify the patent-holder of their application and potential infringement of their patent rights. If an applicant for ferumoxytol notifies us of such application, we would have 45 days upon receipt of that notice to bring a patent infringement suit in federal district court against the applicant seeking approval of a product. If such a suit is commenced, the FDA is generally prohibited from granting approval of an application until the earliest of 30 months from the date the FDA accepted the application for filing, the conclusion of litigation in the generic's favor, or expiration of the patent(s). If the litigation is resolved in favor of the applicant or the challenged patent expires during the 30-month stay period, the stay is lifted and the FDA may thereafter approve the application based on the applicable standards for approval.
21
Table of Contents
A generic version ofFeraheme can be marketed only with the approval of the FDA of the respective application for such generic version. In December 2012, the FDA issued draft guidance making recommendations regarding establishing bioequivalence withFeraheme, pursuant to which a party could seek approval of a generic version ofFeraheme through an ANDA. The FDA generally publishes product-specific bioequivalence guidance after it has received an inquiry from a generic drug manufacturer about submitting an ANDA for the product in question; thus, it is possible that a generic drug manufacturer has approached the FDA requesting guidance about submitting an ANDA for ferumoxytol, the active ingredient inFeraheme, and that such an ANDA may be filed in the near future. The ANDA process is discussed in more detail below under the heading "U.S. Approval Process—Abbreviated New Drug Application."
Companies that manufacture generic products typically invest far fewer resources in research and development than the manufacturers of branded products and can therefore price their products significantly lower than those branded products already on the market. Therefore, competition from generic IV iron products could limit our sales.
We believe that our ability to successfully compete with other IV iron products depends on a number of factors, including the actual or perceived safety and efficacy profile ofFeraheme as compared to alternative iron replacement therapeutics, current and future limitations onFeraheme's approved indications and patient populations, our ability to obtain and maintain favorable pricing, insurance coverage and reimbursement rates and terms forFeraheme, our ability to implement effective marketing programs, the effectiveness of our sales force, our ability to maintain favorable patent protection forFeraheme, market acceptance ofFeraheme, and our ability to manufacture sufficient quantities ofFeraheme at commercially acceptable costs. For additional details on the risks and uncertainties regardingFeraheme's competition, see our Risk Factor, "Market acceptance of Feraheme may suffer as a result of the widespread use of competing iron replacement therapy products, including Injectafer®, and as a result of the approval of generic drug products in the near-term, which would have a material adverse effect on our operations and our profitability."
Based on sales data provided to us in January 2015 by IMS Health Incorporated ("IMS"), we estimate that the size of the total 2014 U.S. non-dialysis IV iron replacement therapy market was approximately 900,000 grams, which represents an increase of approximately 6% over 2013. Based on this IMS data, the following represents the 2014 and 2013 U.S. market share allocation of the total non-dialysis IV iron market based on the volume of IV iron administered:
| | | | | | | |
| | 2014 U.S. Non-dialysis
IV Iron Market
(900,000 grams) | | 2013 U.S. Non-dialysis
IV Iron Market
(851,000 grams) | |
|---|
Venofer® | | | 43 | % | | 46 | % |
INFeD® | | | 20 | % | | 22 | % |
Feraheme | | | 16 | % | | 15 | % |
Generic sodium ferric gluconate | | | 10 | % | | 10 | % |
Injectafer® | | | 6 | % | | <1 | % |
Ferrlecit® | | | 5 | % | | 6 | % |
Dexferrum® | | | <1 | % | | <1 | % |
The market share data listed in the table above is not necessarily indicative of the market shares in dollars due to the variations in selling prices among the IV iron products.
MuGard
Up to 50% of certain new cancer patients develop oral mucositis each year for which there are currently few effective treatments. The market for treating oral mucositis is driven primarily by convenience, price and reimbursement and the products in this market remain mostly undifferentiated.
22
Table of Contents
There are a number of approaches used to treat or manage oral mucositis, including the use of ice chips during chemotherapy treatments, various medicinal mouthwashes, topical anesthetics and analgesics, and oral gel treatments. For example, many physicians use what is commonly known as "magic mouthwash", which may currently be the most commonly prescribed medication to manage oral mucositis or treat the pain associated with mucositis caused by radiation therapy or chemotherapy. Magic mouthwash is a combination of generic ingredients which are typically compounded in a pharmacy and is preferred by many physicians because of the availability of less expensive generic ingredients used to formulate the mouthwash. However, there is no clinical trial data to support the efficacy or safety of magic mouthwash. The efficacy ofMuGard has been supported by a randomized, Phase IV multicenter, double-blind, sham-controlled trial.
There are a number of companies in the U.S. commercializing products for the management or treatment of oral mucositis that may compete withMuGard, including the following marketed products:
- •
- NeutraSal® (supersaturated calcium phosphate rinse), a prescription mouth rinse marketed by Invado Pharmaceuticals, LLC and indicated to treat the painful symptoms associated with oral mucositis;
- •
- Caphosol®, a supersaturated calcium phosphate artificial saliva marketed by Jazz Pharmaceuticals, PLC, which is indicated as an adjunct to standard oral care in treating oral mucositis caused by radiation or high dose chemotherapy; and
- •
- Kepivance® (palifermin), an IV human growth factor manufactured by Amgen and marketed by Swedish Orphan Biovitrum AB, which is used to reduce the chances of developing severe mucositis and to shorten the time with severe mucositis in patients with cancer who receive high doses of chemotherapy and radiation therapy.
Further, there are several marketed products available which are indicated for the management of pain associated with oral mucositis including the following products:
- •
- Episil®, marketed by Cangene BioPharma, Inc., is indicated for the management of pain and relief from pain, by adhering to the mucosal surface of the mouth, soothing oral lesions of various etiologies, including oral mucositis/stomatitis that may be caused by chemotherapy or radio therapy;
- •
- Gelclair®, marketed by DARA BioSciences, Inc., is a viscous, concentrated, bio adherent oral gel, indicated for the management of painful symptoms of mucositis of the oropharyngeal cavity caused by chemo-radiotherapy; and
- •
- GelX® Oral Gel, marketed by Praelia Pharmaceuticals, Inc., is an oral gel indicated for the relief and management of pain by adhering to the mucosal surface of the mouth and soothing oral lesions of various etiologies, including oral mucositis/stomatitis (may be caused by chemotherapy or radiotherapy), irritation due to oral surgery, aging, and traumatic ulcers caused by braces or ill-fitting dentures, medication, or disease.
Based on data provided to us in January 2015 by IMS we estimate that the total number of prescriptions ("TRx's") filled in the U.S. in 2014 for the treatment or management of oral mucositis was approximately 16,500. The following represents the 2014 market share allocation based on TRx data to treat or manage oral mucositis, which accounts for approximately 75% of the total oral
23
Table of Contents
mucositis business. These figures do not include products purchased by hospitals or outpatient clinics, such as Kepivance®:
| | | | | | | |
| | 2014 Oral Mucositis Market
(16,500 TRx) | | 2013 Oral Mucositis Market
(14,900 TRx) | |
|---|
Neutrasal® | | | 49 | % | | 46 | % |
Caphosol® | | | 16 | % | | 23 | % |
Gelclair® | | | 14 | % | | 3 | % |
MuGard | | | 12 | % | | 13 | % |
Episil® | | | 7 | % | | 11 | % |
GelX® | | | 2 | % | | 4 | % |
The market share data listed in the table above is not necessarily indicative of the market shares in dollars due to the variations in selling prices among the oral mucositis products.
Sales, Marketing and Distribution
Makena
In November 2014, we completed our acquisition of Lumara Health, including its commercialized drugMakena, a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. In connection with the acquisition, we retained theMakena commercial team, including 88 members of the Lumara Health sales team dedicated exclusively to the OB/GYN subspecialty. We sellMakena to specialty pharmacies and distributors, who, in turn sellMakena to healthcare providers, hospitals, government agencies and integrated delivery systems.
We estimate thatMakena is currently used to treat between 20% and 30% of the at-risk patient population, allowing for significant potential to increase its market share. Our sales and marketing teams use a variety of common pharmaceutical marketing strategies and methods to promoteMakena, including dedicating a separate reimbursement team to focus on health plans, both commercial and managed Medicaid as well as fee-for-service Medicaid programs.
In addition, we offer customer support through the Makena Care Connection, a support program for patients and healthcare providers that provides administrative, financial assistance and treatment support forMakena. Administrative and treatment support includes insurance benefit investigation, reimbursement and patient assistance programs. Because specialty injectable products likeMakena are not typically carried by retail pharmacies, the process for facilitating prescriptions forMakena is managed by this dedicated customer support center. In December 2013, the Makena Care Connection initiated a pilot program in California designed to improve overall customer satisfaction by reducing the time from the prescription to the initiation of therapy, increasing the average number of injections per patient and increasing the number of paying patients. Favorable results from the pilot project led to a national roll out of the customer service initiative in 2014.
We also operate a patient assistance program forMakena that provides co-pay assistance (for insured patients), and financial assistance (for uninsured patients). Under the program, patients with a household income of $120,000 or less pay $20 or less per injection ofMakena. This encompasses 85% of the U.S. based on 2009 U.S. census data. Clinically eligible patients who are uninsured and whose financial need is greatest will receiveMakena at no cost. There are no upper-level income caps to qualify for the patient assistance program.
In early 2015, we plan to launch a telephonic 24/7 nursing services program to increase patient compliance (i.e., following a weekly injection regime) via education and awareness of preterm birth andMakena's benefits. The program will provide a registered nurse to each expectant mother who will be
24
Table of Contents
available to answer patient questions and guide the patient to her provider for necessary care ensuring all the patient's questions and concerns are addressed.
Feraheme
In July 2009, we began U.S. commercial sale ofFeraheme, which is being marketed and sold in the U.S. through our commercial organization, including a specialty sales force. We sellFeraheme to authorized wholesalers and specialty distributors who, in turn, sellFeraheme to healthcare providers who administerFeraheme primarily within hospitals, hematology and oncology centers and nephrology clinics. Since many hospitals and hematology, oncology and nephrology practices are members of group purchasing organizations ("GPOs"), which leverage the purchasing power of a group of entities to obtain discounts based on the collective bargaining power of the group, we also routinely enter into pricing agreements with GPOs in these markets so the members of the GPOs have access toFeraheme and to the related discounts or rebates.
Our sales and marketing organization uses a variety of common pharmaceutical marketing strategies and methods to promoteFeraheme including sales calls to purchasing entities, such as hospitals, hematology and oncology centers and nephrology practices in addition to individual physicians or other healthcare professionals, medical education symposia, personal and non-personal promotional materials, local and national educational programs, scientific meetings and conferences and informational and disease state awareness websites. In addition, we provide customer service and other related programs forFeraheme including physician reimbursement support services, a patient assistance program for uninsured or under-insured patients and a customer service call center.
Our commercial strategy currently focuses on the non-dialysis dependent CKD market in the U.S. We believe there is a significant opportunity in this market to provide IV iron to non-dialysis CKD patients, and our sales team has been working to educate physicians who treat CKD patients on the benefits of IV iron and the dosing profile ofFeraheme in order to change existing treatment paradigms and expand the IV iron use in physicians' offices, clinics, and hospitals where CKD patients are treated for IDA.
Feraheme has been granted marketing approval in the EU, Canada, Iceland, Liechtenstein, Norway and Switzerland for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD and was commercially launched in the EU, Canada, and Switzerland in 2012. In December 2014, we entered into the Takeda Termination Agreement with Takeda, under which we are in the process of regaining all worldwide development and commercialization rights forFeraheme. Prior to the Takeda Termination Agreement, Takeda was solely responsible forFeraheme commercialization efforts in these areas, including the deployment of a specialized sales force, pricing and reimbursement negotiations with national, provincial or local health authorities and customers, and development of market access strategies. Sales ofFeraheme outside of the U.S. do not and are not expected to materially contribute to our revenues. As such, and in light of the Takeda Termination Agreement, we and Takeda have come to the mutual decision to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
MuGard
In June 2013, we acquired the U.S. rights toMuGard from PlasmaTech. We began comprehensive promotional activities related toMuGard in the third quarter of 2013, including training our sales force and developing new marketing materials, such as healthcare provider brochures, patient materials, reimbursement information and starter kits. To optimize the sales potential of both of our commercial products, our initial call targets forMuGard included currentFeraheme prescribers as well as other high prescribing clinicians, including radiation oncologists who manage head and neck cancer patients undergoing radiation therapy where the incidence of oral mucositis could approximate 80%. Our current commercial strategy forMuGard includes differentiatingMuGard from other currently used approaches for treating and managing oral mucositis, targeting oral mucositis prescribers and expanding reimbursement coverage forMuGard.
25
Table of Contents
Our sales and marketing teams use a variety of common pharmaceutical marketing strategies and methods to promoteMuGard, including sales calls to providing entities, such as hospitals and hematology and oncology centers. In addition, other tactical programs may include personal and non-personal promotional materials to individual physicians or other healthcare professionals, sponsoring local and national educational programs, participation in scientific meetings and conferences and implementing informational product specific websites.
We market and sellMuGard to wholesalers and specialty pharmacies. Patients primarily receiveMuGard through specialty pharmacies, which receive prescriptions from either ourMuGard patient reimbursement and support center (the "HUB") or from physicians directly. We utilize the HUB as a centralized patient intake and referral management center to process insurance coverage issues and administer our patient assistance and copayment programs. In order to provideMuGard to patients as soon as possible, we have implemented a robust program that delivers a starter kit to clinicians, including a sample bottle and all pertinent information that the patient or caregiver needs to immediately beginMuGard therapy.
Product Supply Chain
We outsource a number of our product supply chain services for our products to third-party logistics providers, including services related to warehousing and inventory management, distribution, chargeback processing, accounts receivable management, sample distribution to our sales force, and customer service call center management.
Major Customers
The following table sets forth customers who represented 10% or more of our total revenues for 2014, 2013, and 2012. Revenues from Takeda includeFeraheme collaboration revenue, milestone payments, revenues from product sales to Takeda and royalty payments, in each case in connection with the Amended Takeda Agreement.
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
AmerisourceBergen Drug Corporation | | | 34% | | | 41% | | | 34% | |
McKesson Corporation | | | 21% | | | 24% | | | 17% | |
Cardinal Health, Inc. | | | 15% | | | 16% | | | 12% | |
Takeda Pharmaceuticals Company Limited | | | 11% | | | 11% | | | 31% | |
In addition, approximately 26%, 30% and 32% of ourFeraheme end-user demand in 2014, 2013 and 2012, respectively, was generated by members of a single GPO with which we have contracted.
The loss of any of these customers would have a material adverse effect on our business.
Government Regulation
Overview
Our activities are subject to extensive regulation by numerous governmental authorities in the U.S. and abroad. In the U.S., the FDC Act and other federal and state statutes and regulations govern, among other things, the research and development, manufacturing, quality control, labeling, recordkeeping, approval, storage, distribution, and advertising and promotion of pharmaceutical products and medical devices. Our activities outside of the U.S. are also subject to regulatory requirements governing the testing, approval, safety, effectiveness, manufacturing, labeling, and marketing ofFeraheme.
Failure to comply with any of the applicable U.S. or foreign regulatory requirements may result in a variety of administrative or judicially imposed sanctions including, among other things, the regulatory
26
Table of Contents
agency's refusal to approve pending applications, suspension, variations or withdrawals of approval, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of operations, injunctions, fines, civil penalties, or criminal prosecution.
U.S. Approval Process
Before we may market a new human drug product in the U.S., we must obtain FDA approval of a NDA for that product. The FDA may approve an NDA if the safety and effectiveness of the drug candidate can be established based on the results of clinical trials.
Clinical testing proceeds in three phases. Phase I trials seek to establish initial data about safety, tolerability, and optimal dosing of the drug candidate in humans. The goal of Phase II trials is to provide evidence about the desired therapeutic efficacy of the product candidate in limited studies with small numbers of carefully selected subjects. Phase III trials generally consist of expanded, large-scale, randomized, double-blind, multi-center studies of the safety and effectiveness of the product in the target patient population.
Although we currently have no new unapproved drugs in development and our intention is to expand our portfolio with additional commercial-stage specialty products, we would be required to comply with the requirements for drug approval if we develop new or acquire earlier-stage products.
Following the successful completion of clinical trials, the sponsor submits the results to the FDA as part of an NDA. The NDA must also include the results of pre-clinical tests and studies, information related to the preparation and manufacturing of the drug candidate, analytical methods, and proposed packaging and labeling. Pursuant to the Prescription Drug User Fee Act ("PDUFA"), the FDA has a goal of acting on most original NDAs within six months or ten months of the application filing date, depending on the nature of the drug. For drugs candidates intended to treat serious and life-threatening conditions, the FDA has a number of programs intended to help expedite testing, review, and approval. For example, under the provisions of the FDA's Subpart H accelerated approval is permitted for a new drug that is intended to treat a serious or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint.
If the FDA's evaluations of the NDA and of the sponsor's manufacturing facilities are favorable, the FDA will issue an approval letter, and the sponsor may begin marketing the drug in the U.S. for the approved indications, subject to any post-approval requirements described further below. If the FDA determines it cannot approve the NDA in its current form, it will issue a complete response letter indicating that the application will not be approved in its current form. The complete response letter usually describes the specific deficiencies that the FDA identified in the application and may require additional clinical or other data or impose other conditions that must be met in order to obtain approval of the NDA. Addressing the deficiencies noted by the FDA could be impractical and it is possible that approval may not be obtained, or may be costly and may result in significant delays prior to approval.
Where a sponsor wishes to expand the originally approved prescribing information, such as adding a new indication, it must submit and obtain approval of an sNDA. Changes to an indication generally require additional clinical studies, which can be time-consuming and require the expenditure of substantial additional resources. Under PDUFA, the target timeframe for the review of an sNDA to add a new clinical indication is ten months from the date of filing. As with an NDA, if the FDA determines that it cannot approve an sNDA in its current form, it will issue a complete response letter as discussed above. See the discussion above under"Feraheme for the treatment of IDA in a broad range of patients" for our ongoing post-marketing activities forFeraheme.
27
Table of Contents
An ANDA is filed when approval is sought to market a generic equivalent of a drug product previously approved under an NDA and listed in the Orange Book. Rather than directly demonstrating the product's safety and effectiveness, as is required of an NDA, an ANDA must show that the proposed generic product is the same as the previously approved product in terms of active ingredient(s), strength, dosage form, route of administration and bioavailability. In addition, with certain exceptions, the generic product must have the same labeling as the product to which it refers.
NDA applicants and NDA holders must provide certain information about patents related to the branded drug for listing in the Orange Book. When an ANDA application is submitted, it must contain one of several possible certifications regarding each of the patents listed in the Orange Book for the branded product that is the reference listed drug. A certification that a listed patent is invalid or will not be infringed by the sale of the proposed product is called a Paragraph IV Certification.
The FDA requires a sponsor to submit reports of certain information on side effects and adverse events ("AEs") associated with its products that occur either during clinical trials or after marketing approval. These requirements include specific and timely notification of certain serious, unexpected and/or frequent AEs, as well as regular periodic reports summarizing adverse drug experiences. Failure to comply with these FDA safety reporting requirements may result in FDA regulatory action that may include civil action or criminal penalties. In addition, as a result of these reports, the FDA could create a Tracked Safety Issue for a product in the FDA's Document Archiving, Reporting and Regulatory Tracking System, place additional limitations on an approved product's use, such as through labeling changes, or, potentially, could require withdrawal or suspension of the product from the market.
Even if initial approval of an NDA or sNDA is granted, such approval may be subject to post-market regulatory requirements, any or all of which may adversely impact a sponsor's ability to effectively market and sell the approved product. The FDA may require the sponsor to conduct Phase IV clinical trials, also known as post-marketing requirements or post-marketing commitments, to provide additional information on safety and efficacy. The results of such post-market studies may be negative and could lead to limitations on the further marketing of a product. Also, under the Pediatric Research Equity Act, the FDA may require pediatric assessment of certain drugs unless waived or deferred due to the fact that necessary studies are impossible or highly impractical to conduct or where the drug is not likely to be used in a substantial number of pediatric patients, for example. In addition, the FDA may require a sponsor to implement a Risk Evaluation Mitigation Strategy ("REMS"), a strategy to manage a known or potential serious risk associated with the product. Failure to comply with REMS requirements may result in civil penalties. Further, if an approved product encounters any safety or efficacy issues, including drug interaction problems, the FDA has broad authority to force the sponsor to take any number of actions, including but not limited to, undertaking post-approval clinical studies, implementing labeling changes, adopting a REMS, issuing DHPC letters, or removing the product from the market.
The FDA also regulates all advertising and promotional activities for prescription drugs, both prior to and after approval. Approved drug products must be promoted in a manner consistent with their terms and conditions of approval, including the scope of their approved use. The FDA may take enforcement action against a company for promoting unapproved uses of a product, or off-label promotion, or for other violations of its advertising and labeling laws and regulations. Failure to comply with these requirements could lead to, among other things, adverse publicity, product seizures, civil or
28
Table of Contents
criminal penalties, or regulatory letters, which may include warnings and require corrective advertising or other corrective communications to healthcare professionals.
Under the Subpart H regulations, until theMakena confirmatory post-marketing clinical trial is completed, we are subject to a special 30-day promotional material review by the FDA's Office of Promotional Drug Products ("OPDP"). This extra requirement means that there is a longer lead time before we are able to introduce new promotional material to the market forMakena and we are subject to increased scrutiny prior to using promotional pieces to ensure fair balance.
Manufacturing procedures and quality control for approved drugs must conform to cGMP. Domestic manufacturing establishments must follow cGMP at all times, and are subject to periodic inspections by the FDA in order to assess, among other things, cGMP compliance. In addition, prior to approval of an NDA or sNDA, the FDA will perform a pre-approval inspection of the sponsor's manufacturing facility, including its equipment, facilities, laboratories and processes, to determine the facility's compliance with cGMP and other rules and regulations. Vendors that supply finished products or components to the sponsor that are used to manufacture, package, and label products are subject to similar regulation and periodic inspections. If the FDA identifies deficiencies during an inspection, it may issue a formal notice, which may be followed by a warning letter if observations are not addressed satisfactorily. FDA guidelines specify that a warning letter be issued only for violations of "regulatory significance" for which the failure to adequately and promptly achieve correction may be expected to result in an enforcement action. For example, as discussed above, Lumara Health is subject to certain continuing obligations under the Consent Decree, including, but not limited to, inspection of Lumara Health's places of business by the FDA without prior notice, and notification of FDA of particular actions and events. If Lumara Health fails to comply with applicable provisions in the Consent Decree, the FDC Act, or the FDC Act's implementing regulations, the FDA may impose specific sanctions including, but not limited to, the requirement to cease any of Lumara Health's manufacturing operations, the imposition of substantial financial penalties, and the requirement to implement additional corrective actions.
Product approval may be delayed or denied due to cGMP non-compliance or other issues at the sponsor's manufacturing facilities or contractor sites or suppliers included in the NDA or sNDA, and the complete resolution of these inspectional findings may be beyond the sponsor's control. If the FDA determines that the sponsor's equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against the sponsor, including suspension of its manufacturing operations.
To supply products for use outside of the U.S., our third-party manufacturers must comply with cGMP and are subject to periodic inspection by the FDA or by regulatory authorities in certain other countries. In complying with these requirements, manufacturers, including a drug sponsor's third-party contract manufacturers, must continue to expend time, money, and effort in the area of production and quality to ensure compliance. Failure to maintain compliance with cGMP regulations and other applicable manufacturing requirements of various regulatory agencies could result in fines, unanticipated compliance expenditures, recall, total or partial suspension of production, suspension, variation or withdrawal of the marketing authorization for the product, suspension of the FDA's review of future sNDAs, enforcement actions, injunctions, or criminal prosecution.
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, defined, in part, as a patient population of fewer than 200,000 in the U.S. In the U.S., the company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug
29
Table of Contents
for a period of seven years. This orphan drug exclusivity prevents the FDA from approving another application for the "same drug" for the same orphan indication during the exclusivity period, except in very limited circumstances. In addition, orphan drug exclusivity marketing rights in the U.S. may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition.
In November 2013, the DQSA legislation was implemented to amend the FDC Act with respect to the regulation and monitoring of the manufacturing of compounding drugs. Among other provisions of the DQSA, compounding pharmacies may now elect to register as an "outsourcing facility" under FDC Act 503B. Registration as an outsourcing facility requires that drugs be compounded according to cGMP standards; that facilities report adverse events to the FDA; and that facilities be subject to a risk-based inspection schedule, among other requirements. Additionally, FDC Act 503A describes the conditions that must be satisfied for drug products compounded by a licensed pharmacist or licensed physician to be exempt from approval, labeling, and cGMP requirements. To qualify for these exemptions, a compounded drug product must, among other things, be compounded for an identified patient based on a valid prescription or in limited quantities before the receipt of a prescription for such individual patient in certain circumstances. Under both 503A and 503B of the FDC Act, compounding pharmacies may not compound regularly or in inordinate amounts any drug products that are "essentially copies of commercially available drug products." Depending on how aggressively the FDA enforces this provision of the statute, pharmacy compounders may be significantly restricted in their future ability to make drug products that are copies or near-copies of FDA approved drugs.
Our general operations, and the research, development, manufacture, sale, and marketing of our products, are subject to extensive federal and state regulation, including but not limited to FDA regulations, the Federal Anti-Kickback Statute ("AKS"), the Federal False Claims Act ("FCA"), and the Foreign Corrupt Practices Act, and their state analogues, and similar laws in countries outside of the U.S., laws governing sampling and distribution of products and government price reporting laws.
- •
- The AKS makes it illegal to knowingly and willfully solicit, offer, receive, or pay any remuneration, directly or indirectly, in cash or in kind, in exchange for, or to induce, purchasing, ordering, arranging for, or recommending the purchase or order of any item or service, including the purchase or prescription of a particular drug, that is reimbursed by a federal healthcare program. Liability may be established without proving actual knowledge of the statute or specific intent to violate it. In addition, federal law now provides that the government may assert that a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the FCA, described below. Violations of the AKS carry potentially significant civil and criminal penalties, including imprisonment, fines, and exclusion from participation in federal healthcare programs. Many states have enacted similar anti-kickback laws, including in some cases laws that prohibit paying or receiving remuneration to induce a referral or recommendation of an item or service reimbursed by any payer, including private payers.
- •
- The FCA prohibits, among other things, anyone from knowingly presenting, or causing to be presented, claims for reimbursement of drugs or services to third-party payers such as Medicare or Medicaid, or other claims for payment of government funds, where those claims are false or fraudulent. The FCA also prohibits knowingly making, using, or causing to be made or used a false record or statement material to an obligation to pay money to the government or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. The FCA permits a private individual acting
30
Table of Contents
as a "whistleblower" to bring an action on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. Government enforcement agencies and private whistleblowers have asserted liability under the FCA for, among other things, claims for items or services not provided as claimed or for medically unnecessary items or services, kickbacks, promotion of off-label uses, and misreporting of drug prices to federal agencies. Many states have enacted similar false claims laws, including in some cases laws that apply where a claim is submitted to any third-party payer, not just government programs.
- •
- The Foreign Corrupt Practices Act prohibits companies and their intermediaries from making, or offering or promising to make improper payments to non-U.S. officials for the purpose of obtaining or retaining business or otherwise seeking favorable treatment. Similar anti-bribery laws exist in other countries where we intend to commercializeFeraheme. For example, the U.K. Bribery Act imposes significant potential fines and other penalties for, among other things, giving, offering, or promising bribes in the public and private sectors, and bribing a foreign public official or private person.
Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. Federal and state authorities continue to devote significant attention and resources to enforcement of these laws within the pharmaceutical industry, and private individuals have been active in bringing lawsuits on behalf of the government under the FCA. We have developed and implemented a corporate compliance program based on what we believe are current best practices in the pharmaceutical industry. However, these laws are broad in scope and there may not be regulations, guidance, or court decisions that definitively interpret these laws in the context of particular industry practices. We cannot guarantee that we, our employees, our consultants, or our contractors are or will be in compliance will all federal, state, and foreign regulations. If we or our representatives fail to comply with any of these laws or regulations, a range of fines, penalties, and/or other sanctions could be imposed on us, including, but not limited to, restrictions on how we market and sell our products, significant fines, exclusions from government healthcare programs, including Medicare and Medicaid, litigation, or other sanctions. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could also have an adverse effect on our business, financial condition and results of operations. Such investigations or suits may also result in related shareholder lawsuits, which can also have an adverse effect on our business.
In the EU, the advertising and promotion of our products are subject to EU level and EU Member States' national laws governing promotion of medicinal products, interactions with physicians, misleading and comparative advertising and unfair commercial practices. These laws require that promotional materials and advertising in relation to medicinal products comply with the product's SmPC. The SmPC is the document that provides information to physicians concerning the safe and effective use of the medicinal product. It forms an intrinsic and integral part of the marketing authorization granted for the medicinal product. Promotion of a medicinal product that does not comply with the SmPC is considered to constitute off-label promotion. The off-label promotion of medicinal products is prohibited in the EU. The applicable laws at EU level and in the individual EU Member States also prohibit the direct-to-consumer advertising of prescription-only medicinal products. Violations of the rules governing the promotion of medicinal products in the EU could be penalized by administrative measures, fines and imprisonment. These laws may further limit or restrict the advertising and promotion of our products to the general public and may also impose limitations on our promotional activities with healthcare professionals.
In recent years, several states have enacted legislation requiring pharmaceutical companies operating within the state to establish marketing and promotional compliance programs or codes of conduct and/or file periodic reports with the state or make periodic public disclosures on sales,
31
Table of Contents
marketing, pricing, clinical trials and other activities. In addition, as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (the "Health Care Reform Act") manufacturers of drugs are required to publicly report gifts and other payments or transfers of value made to U.S. physicians and teaching hospitals. Several states have also adopted laws that prohibit certain marketing-related activities, including the provision of gifts, meals or other items to certain healthcare providers. Many of these requirements are new and uncertain, and the likely extent of future enforcement for failure to comply with these requirements is unclear. However, compliance with these laws is difficult, time-consuming, and costly, and if we are found not to be in full compliance with these laws, we may face enforcement actions, fines, and other penalties, and we could receive adverse publicity which could have an adverse effect on our business, financial condition, and results of operations.
We are also subject to laws and regulations covering data privacy and the protection of health-related and other personal information. We obtain patient health information from most healthcare providers who prescribe our products and research institutions we collaborate with, and they are subject to privacy and security requirements that may affect us. Claims that we have violated individuals' privacy rights or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Foreign Regulatory Process
In our efforts to market and sellFeraheme outside of the U.S., we are subject to foreign regulatory requirements. Approval of a drug by applicable regulatory agencies of foreign countries must be secured prior to the marketing of such drug in those countries. The regulatory approval process in countries outside of the U.S. vary widely from country to country and may in some cases be more rigorous than requirements in the U.S. Certain foreign regulatory authorities may require additional studies or studies designed with different clinical endpoints and/or comparators than those which we are conducting or have already completed. In addition, any adverse regulatory action taken by the FDA with respect to an approved product, or a product under review, in the U.S. may affect the regulatory requirements or decisions made by certain foreign regulatory bodies with regard to the regulatory approval of products outside of the U.S.
To obtain regulatory approval of a drug in the EU, marketing authorizations may be submitted through a centralized, mutual recognition or decentralized procedure or national procedure (single country). The legislators, policymakers and healthcare insurance funds in the EU Member States continue to propose and implement cost-containing measures to keep healthcare costs down, due in part to the attention being paid to healthcare cost containment and other austerity measures in the EU. Certain of these changes could impose limitations on the prices pharmaceutical companies are able to charge for their products. The amounts of reimbursement available from governmental agencies or third-party payers for these products may increase the tax obligations on pharmaceutical companies such as ours, or may facilitate the introduction of generic competition with respect to our products. Furthermore, an increasing number of EU Member States and other foreign countries use prices for medicinal products established in other countries as "reference prices" to help determine the price of the product in their own territory. Consequently, a downward trend in prices of medicinal products in some countries could contribute to similar downward trends elsewhere. In addition, the ongoing budgetary difficulties faced by a number of EU Member States have led and may continue to lead to substantial delays in payment and payment partially with government bonds rather than cash for medicinal products. Moreover, in order to obtain reimbursement of our medicinal products in some countries, including some EU Member States, sponsors may be required to conduct Health Technology Assessments ("HTAs") that compare the cost-effectiveness of the sponsors' products to other available therapies.
32
Table of Contents
The Canadian pharmaceutical industry is subject to federal regulation by Health Canada, the public health department of the Canadian government charged with overseeing healthcare-related regulatory matters, pursuant to the Canadian federal Food and Drugs Act. Health Canada's criteria for obtaining and maintaining marketing approval is generally similar to that of the FDA. Health Canada is also empowered to compel information, recall unsafe therapeutic products, disclose confidential business information and direct label change/package modification to address safety issues. In December 2011,Feraheme was granted marketing approval by Health Canada for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD and commercially launched in late 2012.
The pharmaceutical industry in Switzerland is subject to federal regulation by Swissmedic. In August 2012,Rienso was granted marketing approval by Swissmedic and commercially launched in late 2012.Rienso is not currently being marketed in Switzerland. We are currently unable to predict when or ifRienso will be reintroduced into the Swiss market.
Medical Device Regulation
Medical devices, such asMuGard, are similarly subject to FDA approval and extensive post-approval regulation under the FDC Act. Authorization to commercially distribute a new medical device in the U.S. is generally received in one of two ways. The first, known as premarket notification, or the 510(k) process, requires a sponsor to demonstrate that the new medical device is substantially equivalent to a legally marketed medical device that is not subject to premarket approval. The second, more rigorous process, known as premarket approval, requires a sponsor to independently demonstrate that the new medical device is safe and effective.
Both before and after a device is commercially released, there are ongoing responsibilities under FDA regulations. The FDA reviews design and manufacturing practices, labeling and record keeping, and manufacturers' required reports of adverse experiences and other information to identify potential problems with marketed medical devices, similar to the reviews conducted in connection with drug product discussed above. If the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our medical devices are ineffective or pose an unreasonable health risk, the FDA could require us to notify health professionals and others that the devices present unreasonable risks of substantial harm to the public health, order a recall, repair, replacement, or refund of such devices, detain or seize adulterated or misbranded medical devices, or ban such medical devices. The FDA may also impose operating restrictions, enjoin and/or restrain certain conduct resulting in violations of applicable law pertaining to medical devices and assess civil or criminal penalties against our officers, employees, or us.
MuGard was launched in the U.S. by PlasmaTech in 2010 after receiving 510(k) clearance from the FDA. Under the terms of the MuGard License Agreement, PlasmaTech continues to hold the 510(k).MuGard is categorized as a pre-amendments device. This type of device has not been classified per se, but continues to be subject to regulatory review under the 510(k) premarket clearance process.
Pharmaceutical Pricing and Reimbursement
In both the U.S. and foreign markets, our ability to successfully commercialize our products is dependent, in significant part, on the availability and extent of reimbursement to end-users from third-party payers for the use of our products, including governmental payers, health maintenance organizations ("HMOs"), managed care organizations, and private health insurers. In the U.S., the federal government provides health insurance for people who are 65 or older, certain younger people with disabilities, and people with End-Stage Renal Disease through the Medicare program, and certain prescription drugs, includingFeraheme andMakena, are covered under Medicare Part B. Medicaid, another program in the U.S., is a health insurance program for low-income children, families, pregnant women, and people with disabilities that is jointly funded by the federal and state governments, but administered by the states. In general, state Medicaid programs are required to cover drugs and biologicals of manufacturers that have entered into a Medicaid Drug Rebate Agreement, although such products and biologicals may be subject to prior authorization or other utilization controls. Both Medicare and Medicaid are administered by the Centers for Medicare and Medicaid Services ("CMS").
33
Table of Contents
We participate in and have certain price reporting obligations to the Medicaid Drug Rebate program, and we have obligations to report Average Sales Price ("ASP") for the Medicare program. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to CMS. These data include the average manufacturer price and, in the case of innovator products such asFeraheme andMakena, the best price for each drug.
Federal law also requires that a company that participates in the Medicaid program report ASP information to CMS for certain categories of drugs that are paid under Part B of the Medicare program, such asFeraheme andMakena. This ASP information forms the basis for reimbursement for the majority of our currentFeraheme business, and to a lesser extent, for ourMakena business. Manufacturers calculate ASP based on a statutorily defined formula and interpretations of the statute by CMS as to what should or should not be considered in computing ASP. An ASP for each National Drug Code for a product that is subject to the ASP reporting requirement must be submitted to CMS no later than 30 days after the end of each calendar quarter. CMS uses these submissions to determine payment rates for drugs under Medicare Part B. Changes affecting the calculation of ASP could affect the ASP calculations for our products and the resulting Medicare payment rate, and could negatively impact our results of operations.
Federal law requires that any company that participates in the Medicaid rebate program also participate in the Public Health Service's 340B drug pricing discount program in order for federal funds to be available for the manufacturer's drugs under Medicaid and Medicare Part B. The 340B pricing program requires participating manufacturers to agree to charge statutorily defined covered entities no more than the 340B "ceiling price" for the manufacturer's covered outpatient drugs. These 340B covered entities include a variety of community health clinics and other entities that receive health services grants from the Public Health Service, as well as hospitals that serve a disproportionate share of low-income patients. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid rebate program. Changes to the definition of average manufacturer price and the Medicaid rebate amount under the Healthcare Reform Act, as discussed below, and CMS's issuance of final regulations implementing those changes also could affect our 340B ceiling price calculations and negatively impact our results of operations.
The Healthcare Reform Act also expanded the Public Health Service's 340B drug pricing program to include additional types of covered entities: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by the Healthcare Reform Act. For example, the percentage ofFeraheme sold to 340B institutions has grown from 11% in 2011 to 17% in 2014. Since these institutions are granted lower prices than those offered to our other customers, any further growth in our 340B business may have a negative impact on our sales price per gram and operating margins.
The Healthcare Reform Act exempts "orphan drugs," such asMakena, from the ceiling price requirements for the covered entity types newly added to the program by the Healthcare Reform Act. On July 21, 2014, the Health Resources and Services Administration ("HRSA"), which administers the 340B program, issued an interpretive rule to implement the orphan drug exception which interprets the orphan drug exception narrowly. It exempts orphan drugs from the ceiling price requirements for the newly eligible entities only when the orphan drug is used for its orphan indication. The newly eligible entities are entitled to purchase orphan drugs at the ceiling price when the orphan drug is not used for its orphan indication. A manufacturer trade group has filed a lawsuit challenging the interpretive rule as inconsistent with the statutory language. That challenge remains ongoing. The uncertainty regarding how the statutory orphan drug exception will be applied will increase the complexity of compliance, will
34
Table of Contents
make compliance more time-consuming, and could negatively impact our results of operations. If HRSA's narrow interpretation of the scope of the orphan drug exemption prevails, it could potentially negatively impact the price we are paid forMakena by certain entities and increase the complexity of compliance with the 340B program.
In order to be eligible to have our products paid for with federal funds under the Medicaid program and purchased by certain federal agencies and grantees, we participate in the Department of Veterans Affairs ("VA") Federal Supply Schedule ("FSS") pricing program. To participate, we are required to enter into an FSS contract with the VA, under which we must make our innovator "covered drugs" available to the "Big Four" federal agencies, including the VA, the Department of Defense ("DoD"), the Public Health Service, and the Coast Guard, at pricing that is capped pursuant to a statutory federal ceiling price ("FCP") formula set forth in Section 603 of the Veterans Health Care Act of 1992 ("VHCA"). The FCP is based on a weighted average non-federal average manufacturer price ("Non-FAMP"), which manufacturers are required to report on a quarterly and annual basis to the VA. If a company misstates Non-FAMPs or FCPs it must restate these figures. Pursuant to the VHCA, knowing provision of false information in connection with a Non-FAMP filing can subject a manufacturer to penalties of $100,000 for each item of false information.
FSS contracts are federal procurement contracts that include standard government terms and conditions, separate pricing for each product, and extensive disclosure and certification requirements. All items on FSS contracts are subject to a standard FSS contract clause that requires FSS contract price reductions under certain circumstances where pricing is reduced to an agreed "tracking customer." Further, in addition to the "Big Four" agencies, all other federal agencies and some non-federal entities are authorized to access FSS contracts. FSS contractors are permitted to charge FSS purchasers other than the Big Four agencies "negotiated pricing" for covered drugs that is not capped by the FCP; instead, such pricing is negotiated based on a mandatory disclosure of the contractor's commercial "most favored customer" pricing. We offer single pricing on our FSS contract.
In addition, pursuant to regulations issued by the DoD TRICARE Management Activity, now the Defense Health Agency, to implement Section 703 of the National Defense Authorization Act for Fiscal Year 2008, each of our covered drugs is listed on a Section 703 Agreement under which we have agreed to pay rebates on covered drug prescriptions dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. Companies are required to list their innovator products on Section 703 Agreements in order for those products to be eligible for DoD formulary inclusion. The formula for determining the rebate is established in the regulations and our Section 703 Agreement and is based on the difference between the annual Non-FAMP and the FCP (as described above, these price points are required to be calculated by us under the VHCA).
Reimbursement by third-party payers depends on a number of factors, including the third-party's determination that the product is competitively priced, safe and effective, appropriate for the specific patient, and cost-effective. Third-party payers are increasingly challenging the prices charged for pharmaceutical products and have instituted and continue to institute cost containment measures to control or significantly influence the purchase of pharmaceutical products. For example, to reduce expenditures associated with pharmaceutical products, many third-party payers use cost containment methods, including: (a) formularies, which limit coverage for drugs not included on a predetermined list; (b) variable co-payments, which may make a certain drug more expensive for patients as compared with a competing drug; (c) utilization management controls, such as requirements for prior authorization before the payer will cover the drug; and (d) other coverage policies that limit access to certain drugs for certain uses based on the payer-specific coverage policy.
For example, prior to the implementation of the DQSA, as discussed above, the reimbursement ofMakena was often difficult to obtain in light of the less expensive compounding products. As a result of the provisions under the DQSA and efforts by Lumara Health to work with individual states, including
35
Table of Contents
entering into supplement rebate agreements, access toMakena has since expanded. However, Lumara Health has had to use the legal system to defend reimbursement practices related to Lumara Health. For example, in 2012, Lumara Health sued the Georgia Department of Community Health ("DCH") because they were requiring patients to provide documentation of medical necessity to approveMakena in favor of compounded versions of the active ingredient ofMakena. During 2014, a permanent order was issued stating that DCH and their managed Medicaid plans must reimburse forMakena when prescribed by physicians for an on-label patient. Although, this case remains in the appeals process, this ruling would aid as precedent for other states to comply with current Medicaid laws.
In addition, U.S. and many foreign governments continue to attempt to curb healthcare costs through legislation, including legislation aimed at reducing the pricing and reimbursement of pharmaceutical products. The Healthcare Reform Act was enacted in the U.S. in March 2010 and includes certain cost containment measures including an increase to the minimum rebates for products covered by Medicaid programs from 15.1% to 23.1% of the average manufacturer price for most innovator products, and the expansion of the 340B Drug Discount Program under the Public Health Service Act. Effective March 2010, the Healthcare Reform Act expanded manufacturer rebate liability under the Medicaid program from fee-for-service Medicaid utilization to include the utilization of Medicaid managed care organizations as well. In addition, the Healthcare Reform Act and subsequent legislation changed the definition of average manufacturer price. Further, federal budgetary concerns could result in the implementation of significant federal spending cuts, including cuts in Medicare and other health related spending in the near-term. For example, recent legislative enactments have resulted in Medicare payments being subject to a two percent reduction, referred to as sequestration, until 2024. Finally, the Healthcare Reform Act required pharmaceutical manufacturers of branded prescription drugs to pay a branded prescription drug fee to the federal government. Each individual pharmaceutical manufacturer pays a prorated share of the branded prescription drug fee of $3.0 billion in 2015, based on the dollar value of its branded prescription drug sales to certain federal programs identified in the law. Sales of orphan drugs, such asMakena are excluded from the determination.
Some of the Healthcare Reform Act's significant reforms do not take effect until 2015. In 2012, CMS, issued proposed regulations to implement the changes to the drug rebate components of the Medicaid program under the Healthcare Reform Act but has not yet issued final regulations. CMS is currently expected to release the final regulations in 2015.
In addition, the heightened focus on the healthcare industry by the federal government could result in the implementation of significant federal spending cuts including cuts in Medicare and other health related spending in the near-term. In recent years, some states have also passed legislation to control the prices of drugs as well as begun a move toward managed care to relieve some of their Medicaid cost burden. These and any future changes in government regulation or private third-party payers' reimbursement policies may reduce the extent of reimbursement for our products and adversely affect our future operating results. For example, since almost half ofMakena patients are Medicaid beneficiaries, the impact of future legislative changes may have a significant impact onMakena sales. Payers also are increasingly considering new metrics as the basis for reimbursement rates, such as average sales price, average manufacturer price and actual acquisition cost. The existing data for reimbursement based on these metrics is relatively limited, although certain states have begun to survey acquisition cost data for the purpose of setting Medicaid reimbursement rates. CMS has begun posting drafts of this retail survey price information on at least a monthly basis in the form of draft National Average Drug Acquisition Cost ("NADAC") files, which reflect retail community pharmacy invoice costs, and National Average Retail Price ("NARP") files, which reflect retail community pharmacy prices to consumers. In July 2013, CMS suspended the publication of draft NARP data, pending funding decisions. In November 2013, CMS moved to publishing final rather than draft NADAC data and has since made updated NADAC data publicly available on a weekly basis. Therefore, it may be difficult to project the impact of these evolving reimbursement mechanics on the willingness of payers
36
Table of Contents
to cover our products. Any failure to cover our products appropriately, in addition to legislative and regulatory changes and others that may occur in the future, could impact our ability to maximize revenues in the federal marketplace.
Currently, in U.S. physician clinic and hospital settings, Medicare Part B generally reimburses for physician-administered drugs at a rate of 106% of the drug's ASP. ASP is defined by statute based on sales and price concession data, including rebates and chargebacks, for a defined period of time. As noted above, we submit the required information to CMS on a quarterly basis. In advance of the quarter in which the payment limit for drugs reimbursed under Medicare Part B program will go into effect, CMS calculates and publishes the payment limit. Under this methodology, payment rates change on a quarterly basis, and significant downward fluctuations in ASP, and therefore reimbursement rates, could negatively impact sales of a product. Because the ASP-based payment rate is defined by statute, and changes to Medicare payment methodologies require legislative change, it is unclear if and when ASP reimbursement methodology will change for the physician office setting. While the statute requires Medicare Part B payments for most drugs furnished in the physician office setting to be at 106% of ASP, the statute does not have a similar requirement for hospital outpatient departments. For that setting, the Medicare payment for many covered Part B drugs also is at 106% of ASP, but CMS could change that through regulations, without any intervening legislation. While Medicare is the predominant payer forMakena andFeraheme for treatment of patients with CKD, Medicare payment policy, in time, can also influence pricing and reimbursement in the non-Medicare markets, as private third-party payers and state Medicaid plans frequently adopt Medicare principles in setting reimbursement methodologies. We cannot predict the impact that any changes in reimbursement policies may have on our ability to compete effectively.
For example, in the U.S. hospital inpatient setting, most drugs are not reimbursed separately within the Medicare prospective payment system, based largely on the drug costs, but are bundled as a per discharge reimbursement based on the diagnosis and/or procedure rather than actual costs incurred in patient treatments, thereby increasing the incentive for a hospital to limit or control expenditures. As a result, we do not expect premium priced products, such asFeraheme, to be broadly used in the hospital inpatient setting.
In countries outside of the U.S., market acceptance may also depend, in part, upon the availability of reimbursement within existing healthcare payment systems. Generally, in the EU and other countries outside of the U.S., the government sponsored healthcare system is the primary payer of healthcare costs of patients and therefore enjoys significant market power. Some foreign countries also set prices for pharmaceutical products as part of the regulatory process, and we cannot guarantee that the prices set by such governments will be sufficient to generate substantial revenues or allow sales ofFeraheme to be profitable in those countries. Moreover, in order to obtain reimbursement of our medicinal products in some countries, including some EU Member States, we may be required to conduct HTAs that compare the cost-effectiveness of our products to other available therapies. In addition, we may be unable to obtain favorable pricing and reimbursement approvals in certain EU Member States.
The legislators, policymakers and healthcare insurance funds in the EU Member States continue to propose and implement cost-containing measures to keep healthcare costs down, due in part to the attention being paid to healthcare cost containment and other austerity measures in the EU. Certain of these changes could impose limitations on the prices pharmaceutical companies are able to charge for their products. The amounts of reimbursement available for these products from governmental agencies or third-party payers, may increase the tax obligations on pharmaceutical companies such as ours, or may facilitate the introduction of generic competition with respect to our products. Furthermore, an increasing number of EU Member States and other foreign countries use prices for medicinal products established in other countries as "reference prices" to help determine the price of the product in their own territory. Consequently, a downward trend in prices of medicinal products in some countries could contribute to similar downward trends elsewhere. In addition, the ongoing budgetary difficulties faced
37
Table of Contents
by a number of EU Member States have led and may continue to lead to substantial delays in payment and payment partially with government bonds rather than cash for medicinal drug products, which could negatively impact our revenues and profitability. Moreover, in order to obtain reimbursement of our medicinal products in some countries, including some EU Member States, we may be required to conduct HTAs that compare the cost-effectiveness of our products to other available therapies. There can be no assurance that our medicinal products will obtain favorable reimbursement status in any country.
If adequate reimbursement levels are not maintained by government and other third-party payers for our products, our ability to sell our products may be limited and/or our ability to establish acceptable pricing levels for our products may be impaired, thereby reducing anticipated revenues and our profitability.
Backlog
We had a $4.3 million and $0.9 million product sales backlog as of December 31, 2014 and 2013, respectively. We expect to recognize the $4.3 million in 2015. These backlogs were largely due to timing of orders received from our third-party logistics providers. Generally, product orders from our customers are fulfilled within a relatively short time of receipt of a customer order.
Employees
As of February 4, 2015, we had 257 employees, including 108 employees of Lumara Health who accepted employment with us following our November 2014 acquisition of Lumara Health. We also utilize consultants and independent contractors on a regular basis to assist in the development and commercialization of our products. Our success depends to a significant extent on our ability to continue to attract, retain and motivate qualified sales, technical operations, managerial, scientific and medical personnel of all levels. Although we believe we have been relatively successful to date in obtaining and retaining such personnel, we may not be successful in the future. During 2014 and 2013, we expanded our leadership team and strengthened our commercial organization and medical affairs teams. We expect to continue these efforts in 2015 in support of the growth in our business.
None of our employees is represented by a labor union, and we consider our relationship with our employees to be good.
Foreign Operations
We have no foreign operations. Revenues from customers outside of the U.S. amounted to approximately 12%, 11% and 32% of our total revenues for 2014, 2013 and 2012, respectively, and were principally related to collaboration revenues recognized in connection with our agreement with Takeda, which is headquartered in Japan. During 2012, our revenues from customers outside of the U.S included approximately $20.0 million related to the recognition of upfront payments and milestones achieved under the Amended Takeda Agreement, which we entered into the Takeda Termination Agreement to terminate. Sales ofFeraheme outside of the U.S. do not and are not expected to materially contribute to our revenues. As such, and in light of the Takeda Termination Agreement, we and Takeda have come to the mutual decision to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada. We have no plans to commercializeMakena outside of the U.S.
Research and Development
We have dedicated a significant portion of our resources to our efforts to develop our products and product candidates, particularlyFeraheme. We incurred research and development expenses of $24.2 million, $20.6 million, and $33.3 million during 2014, 2013 and 2012, respectively. We expect our
38
Table of Contents
research and development expenses to increase in 2015 due to the timing of expenses related to our pediatric clinical studies and our hd-CKD Study as well as current clinical trials related toMakena's post approval commitments and its lifecycle management program. In addition, research and development expenses could increase further and significantly depending on the outcome of discussions with the FDA on the regulatory path forward forFeraheme in the broad indication and any resulting clinical trials or development efforts that we may undertake.
Segment Reporting
We conduct our operations in one business segment as further described in Note P, "Business Segments," to our consolidated financial statements included in this Annual Report on Form 10-K.
Code of Ethics
Our Board of Directors has adopted a code of ethics that applies to our officers, directors and employees. We have posted the text of our code of ethics on our website athttp://www.amagpharma.com in the "Investors" section. We will provide to any person without charge a copy of such code of ethics, upon request in writing to Investor Relations, AMAG Pharmaceuticals, Inc., 1100 Winter Street, Waltham, MA 02451. In addition, should any changes be made to our code of ethics, we intend to disclose within four business days, on our website (or in any other medium required by law or the NASDAQ): (a) the date and nature of any amendment to our code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (b) the nature of any waiver, including an implicit waiver, from a provision of our code of ethics that is granted to one of these specified officers, the name of such person who is granted the waiver, and the date of the waiver.
Available Information
Our internet website address ishttp://www.amagpharma.com. Through our website, we make available, free of charge, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy and registration statements, and all of our insider Section 16 reports (and any amendments to such filings), as soon as reasonably practicable after such material is electronically filed with, or furnished to, the U.S. Securities and Exchange Commission (the "SEC"). These SEC reports can be accessed through the "Investors" section of our website. The information found on our website is not part of this or any other report we file with, or furnish to, the SEC. Paper copies of our SEC reports are available free of charge upon request in writing to Investor Relations, AMAG Pharmaceuticals, Inc., 1100 Winter Street, Waltham, MA 02451. The content on any website referred to in this Form 10-K is not incorporated by reference into this Form 10-K unless expressly noted.
ITEM 1A. RISK FACTORS:
The following information sets forth material risks and uncertainties that may affect our business, including our future financial and operational results and could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Annual Report on Form 10-K and elsewhere as discussed in the introduction to Part I above. You should carefully consider the risks described below, in addition to the other information in this Annual Report on Form 10-K, before making an investment decision. The risks and uncertainties described below are not the only ones we face. Additional risks not presently known to us or other factors not perceived by us to present material risks to our business at this time also may impair our business operations.
Unless the context suggests otherwise, references to "Feraheme" refer to both Feraheme (the trade name for ferumoxytol in the U.S. and Canada) and Rienso (the trade name for ferumoxytol in the EU and Switzerland).
39
Table of Contents
Risks Related to Our Products
We are primarily dependent on revenues from our two principal products.
We currently derive substantially all of our revenue from sales ofMakena andFeraheme. Although we may introduce additional products for commercialization to our product portfolio, we may be substantially dependent on sales ofMakena andFeraheme for many years. Our financial condition will be materially adversely affected, we may have to restructure our current operations, and our business prospects will be limited if we experience any negative developments relating toMakena orFeraheme, including the following:
- •
- Actual or perceived safety or efficacy issues;
- •
- Restrictions on current or future labels;
- •
- The introduction or greater acceptance of competing products, including generic products, products that may be prescribed off-label and products made by compounding pharmacies;
- •
- Constraints on product pricing or price increases; and
- •
- Changes in reimbursement policies or adverse regulatory or legislative developments.
In the U.S., ifMakena orFeraheme face any safety or efficacy issues, including drug interaction problems, under the Federal Food, Drug and Cosmetic Act (the "FDC Act"), the U.S. Food and Drug Administration ("FDA") has broad authority to force us to take any number of actions, including, but not limited to the following:
- •
- Requiring us to conduct post-approval clinical studies to assess known risks or signals of serious risks, or to identify unexpected serious risks;
- •
- Mandating labeling changes to a product;
- •
- Requiring us to implement a risk evaluation and mitigation strategy ("REMS") where necessary to assure safe use of the drug; or
- •
- Removing an already approved product from the market.
Similar laws and regulations exist in countries outside of the U.S. In addition, actual or perceived safety or drug interaction problems could result in product recalls, restrictions on the product's permissible uses, changes to the product label, a negative impact on regulatory applications (including supplemental new drug applications ("sNDAs") and applications for variations to the marketing authorization), suspension, variation or withdrawal of the marketing authorization for the product, or withdrawal of the product from the U.S. and/or foreign markets. Any such actions would adversely affect our results of operations.
The commercial success of our products depends upon the level of market adoption and continued use by physicians, hospitals, patients, and healthcare payers, including government payers, health maintenance organizations ("HMOs"), managed care organizations, group purchasing organizations ("GPOs") and specialty pharmacies. Our products might not be adopted if perceived to be no safer, less safe, no more effective, less effective, no more convenient, or less convenient than currently available products. In addition, the pricing and/or reimbursement rates and terms of our products may not be viewed as advantageous to potential prescribers and payers as the pricing and/or reimbursement rates and terms of other available products, including, in the case ofMakena, compounded products. If our products do not achieve or maintain an adequate level of market adoption for any reason, our profitability and our future business prospects will be adversely impacted.
40
Table of Contents
Competition in the pharmaceutical and biopharmaceutical industries, including from companies marketing generic products, is intense. If we fail to compete effectively, our business and market position will suffer.
The pharmaceutical and biopharmaceutical industries are intensely competitive and subject to rapid technological change. Many of our competitors forFeraheme are large, well-known pharmaceutical companies and may benefit from significantly greater financial, sales and marketing capabilities, greater technological or competitive advantages, and other resources. ForMakena, most of our competition comes from pharmacies that compound a non-FDA approved version ofMakena, which is sold at a much lower cost thanMakena. Our existing or potential new competitors forFeraheme andMakena may develop products that are more widely accepted than ours and may receive patent protection that dominates, blocks or adversely affects our product development or business.
In addition, generic versions ofFeraheme andMakena could enter the market through approval of abbreviated new drug applications ("ANDAs") that useFeraheme orMakena as a reference listed drug, which would allow generic competitors to rely onFeraheme's orMakena's safety and efficacy trials instead of conducting their own studies. Further, there are no patents coveringMakena.
For example, in December 2012, the FDA published a draft guidance containing product-specific bioequivalence recommendations for drug products containing ferumoxytol. The FDA generally publishes product-specific bioequivalence guidance after it has received an inquiry from a generic drug manufacturer about submitting an ANDA for the product in question; thus, it is possible that a generic drug manufacturer has approached the FDA requesting guidance about submitting an ANDA for ferumoxytol, the active ingredient inFeraheme, and that such an ANDA may be filed in the near future. The published bioequivalence guidance could encourage a generic entrant seeking a path to approval of a generic ferumoxytol to file an ANDA. As a result, we could face generic competition in the near-term or have to engage in extensive litigation with a generic competitor to protect our patent rights, either of which could adversely affect our business and results of operations. Companies that manufacture generic products typically invest far fewer resources in research and development and marketing efforts than the manufacturers or marketers of branded products and can therefore price their products significantly lower than those branded products already on the market. As a result, competition from generic IV iron products could limit our sales, which would have an adverse impact on our business and results of operations.
The introduction by our competitors of alternatives toFeraheme orMakena that would be, or are perceived to be, more efficacious, safer, less expensive, easier to administer, available for a broader patient population, or provide more favorable insurance coverage or reimbursement, could reduce our revenues and the value of our product development efforts. For more information onFeraheme andMakena specific competition risks, please see Risk Factors "Market acceptance of Feraheme may suffer as a result of the widespread use of competing iron replacement therapy products, including Injectafer®, and as a result of the approval of generic drug products in the near-term, which would have a material adverse effect on our operations and our profitability" and "Our ability to successfully commercialize Makena is dependent upon a number of factors, including maintaining the benefits of Makena's orphan drug exclusivity and the length of time before competitors begin selling generic versions of Makena."
The success of our products depends on our ability to maintain the proprietary nature of our technology.
We rely on a combination of patents, trademarks and copyrights in the conduct of our business. The patent positions of pharmaceutical and biopharmaceutical firms are generally uncertain and involve complex legal and factual questions. We may not be successful or timely in obtaining any patents for which we submit applications. The breadth of the claims obtained in our patents may not provide sufficient protection for our technology. The degree of protection afforded by patents for proprietary or licensed technologies or for future discoveries may not be adequate to preserve our ability to protect or commercially exploit those technologies or discoveries. The patents issued to us may provide us with
41
Table of Contents
little or no competitive advantage. In addition, there is a risk that others will independently develop or duplicate similar technology or products or circumvent the patents issued to us.
One of our U.S.Feraheme patents is subject to a patent term extension under U.S. patent law and FDA regulations and will expire in June 2023. Our other U.S. patents relating toFeraheme expire in 2020. These and any other patents issued to or acquired by us may be contested or invalidated. There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. We may become a party to patent litigation and other proceedings, including interference and reexamination proceedings declared by the United States Patent and Trademark Office. There are no patents coveringMakena and thus the successful commercialization ofMakena is significantly reliant on our ability to take advantage of its orphan drug exclusivity, which risks are described in the Risk Factor "Our ability to successfully commercialize Makena is dependent upon a number of factors, including maintaining the benefits of Makena's orphan drug exclusivity and the length of time before competitors begin selling generic versions of Makena."
In addition, claims of infringement or violation of the proprietary rights of others may be asserted against us. If we are required to defend against such claims or to protect our own proprietary rights against others, it could result in substantial financial and business costs, including the distraction of our management. An adverse ruling in any litigation or administrative proceeding could result in monetary damages, injunctive relief or otherwise harm our competitive position, including by limiting our marketing and selling activities, increasing the risk for generic competition, limiting our development and commercialization activities or requiring us to obtain licenses to use the relevant technology (which licenses may not be available on commercially reasonable terms, if at all).
We also rely upon unpatented trade secrets and improvements, unpatented know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with our corporate licensees, collaborators, contract manufacturers, employees and consultants. These agreements, however, may be breached. We may not have adequate remedies for any such breaches, and our trade secrets might otherwise become known or might be independently discovered by our competitors. In addition, we cannot be certain that others will not independently develop substantially equivalent or superseding proprietary technology, or that an equivalent product will not be marketed in competition with our products, thereby substantially reducing the value of our proprietary rights.
Further, the laws of foreign countries may not protect our intellectual property rights to the same extent as do the laws of the U.S. and therefore our intellectual property rights may be subject to increased risk abroad, including opposition proceedings before the patent offices for other countries, such as the European Patent Office, or similar adversarial proceedings, regarding intellectual property rights with respect toRienso.
We may not be able to further expand our product portfolio by entering into additional business development transactions, such as in-licensing arrangements, acquisitions, or collaborations or, if such arrangements are entered into, we may not realize the anticipated benefits and they could disrupt our business, decrease our profitability, result in dilution to our stockholders or cause us to incur significant additional debt or expense.
As part of our business strategy to expand our product portfolio, we are seeking to in-license or acquire additional pharmaceutical products or companies that leverage our corporate infrastructure and commercial expertise, such as our recent acquisition of Lumara Health Inc. ("Lumara Health"). We have limited experience with respect to these business development activities and there can be no assurance that we will be able to identify or complete any additional transactions in a timely manner, on a cost-effective basis, or at all.
Further, the valuation methods that we use for any acquired product or business requires significant judgment and assumptions. Actual results and performance of the products or businesses
42
Table of Contents
that we may acquire, including anticipated synergies and other financial benefits, could differ significantly from our original assumptions, especially during the periods immediately following the closing of the transaction. In addition, acquisitions may cause significant changes to our current organization and operations, may subject us to more rigid or constraining regulations or government oversight and may have negative tax and accounting consequences. These results could have a negative impact on our financial position or results of operations and result in significant charges in future periods.
In addition, proposing, negotiating and implementing collaborations, in-licensing arrangements or acquisition agreements is a lengthy and complex process. Other companies, including those with substantially greater financial, marketing and sales resources, may compete with us for these arrangements, and we may not be able to enter into such arrangements on acceptable terms or at all. Further, any such strategic transactions by us could result in large and immediate write-offs or the incurrence of additional debt and contingent liabilities, each of which may contain restrictive covenants that could adversely impact or limit our ability to grow our business, enter into new agreements, and adversely affect our operating results. Management of a license arrangement, collaboration, or other strategic arrangement and/or integration of an acquired asset or company may also disrupt our ongoing business, require management resources that otherwise would be available for ongoing development of our existing business, including our commercialization ofFeraheme andMakena.
In addition, our cash, cash equivalents and investments may not be sufficient to finance any additional strategic transactions, and we may choose to issue shares of our common or preferred stock as consideration, which would result in dilution to our stockholders. Alternatively, it may be necessary for us to raise additional funds through public or private financings, and such additional funds may not be available on terms that are favorable to us, if at all, and our stockholders may experience significant dilution. Our Term Loan Facility, which provided us with $340.0 million to finance our acquisition of Lumara Health (the "Term Loan Facility") contains restrictions on our ability to acquire additional pharmaceutical products and companies, to enter into exclusive licensing arrangements, to incur additional indebtedness and will require us to use a portion of our free cash flow to repay indebtedness on an annual basis. These provisions may limit our ability to pursue attractive business development opportunities. If we are unable to successfully obtain rights to suitable products or if any acquisition or in-license arrangement we make is not successful, our business, financial condition and prospects for growth could suffer.
Further, even if we do acquire additional products or businesses, the integration of the operations of such acquired products or businesses requires significant efforts, including the coordination of information technologies, sales and marketing, operations, manufacturing, safety and pharmacovigilance, medical and finance. These efforts result in additional expenses and involve significant amounts of management's time. In addition, we may have to rely on the other parties with whom we may enter into a future agreement to perform certain regulatory filings, oversee certain functions, such as pharmacovigilance or the manufacture of the product we license from them, and any failure of such party to perform these functions for any reason, including ceasing doing business, could have a material effect on our ability to commercialize the licensed product. Similarly, we are relying on theMakena commercial team and other key Lumara Health personnel to assist with the integration and operations of Lumara Health and the commercialization ofMakena. We may not realize the anticipated benefits of Lumara Health or any future acquisition, license or collaboration, any of which involves numerous risks including those discussed above and the following:
- •
- Entry into markets in which we have no or limited direct prior experience, such as markets where we compete with non-traditional drug manufacturers, such as compounding pharmacies, and where competitors in such markets have stronger market positions;
43
Table of Contents
- •
- Our ability to train our sales force, and the ability of our sales force, to successfully incorporate new products into their call points, or to successfully integrate and leverage sales forces that we retain, such as theMakena commercial team;
- •
- Additional legal, compliance and/or accounting risks associated with such acquisitions, including liabilities assumed as part of the acquisition, which may be unknown or contingent; and
- •
- The introduction or wider acceptance of competitive products.
If we cannot successfully integrate the Lumara Health business, or other businesses or products we may acquire or in-license, into our company, we may experience material negative consequences to our business, financial condition or results of operations. We cannot be certain that, following any such acquisitions or in-licenses, including Lumara Health, we will achieve the expected synergies and other benefits that justify the purchase price of such transaction.
We are completely dependent on third parties to manufacture our commercial products and any difficulties, disruptions or delays, or the need to find alternative sources, could adversely affect our profitability and future business prospects.
We do not currently own or operate, and currently do not plan to own or operate, facilities for the manufacture of our products, and we do not plan to own or operate facilities for the manufacture of any commercial products we may acquire or in-license. We currently rely solely on third-party contract manufacturers to manufactureFeraheme andMakena for our commercial and clinical use. We do not currently have an alternative manufacturer for ourFeraheme drug substance and finished drug product nor do we have an alternative manufacturer forMakena drug substance or drug product, and we may not be able to enter into agreements with second source manufacturers whose facilities and procedures comply with current good manufacturing practices ("cGMP") regulations and other regulatory requirements on a timely basis and with terms that are favorable to us, if at all.
Our ability to have our products manufactured in sufficient quantities and at acceptable costs to meet our commercial demand and clinical development needs is dependent on the uninterrupted and efficient operation of our third-party contract manufacturing facilities. Any difficulties, disruptions or delays in the manufacturing process could result in product defects or shipment delays, suspension of manufacturing or sale of the product, recall or withdrawal of product previously shipped for commercial or clinical purposes, inventory write-offs or the inability to meet commercial demand in a timely and cost-effective manner. Furthermore, our current third-party manufacturers do not manufacture for us exclusively and may exhaust some or all of their resources meeting the demand of other customers. In addition, securing additional third-party contract manufacturers forFeraheme orMakena will require significant time for transitioning the necessary manufacturing processes, gaining regulatory approval, and for having the appropriate oversight and may increase the risk of certain problems, including cost overruns, process reproducibility, stability issues, the inability to deliver required quantities of product that conform to specifications in a timely manner, or the inability to manufactureFeraheme orMakena in accordance with cGMP.
Further, we and our third-party manufacturers currently purchase certain raw and other materials used to manufactureFeraheme andMakena from third-party suppliers and, at present, do not have long-term supply contracts with most of these third parties. These third-party suppliers may cease to produce the raw or other materials used inFeraheme andMakena or otherwise fail to supply these materials to us or our third-party manufacturers or fail to supply sufficient quantities of these materials to us or our third-party manufacturers in a timely manner for a number of reasons, including but not limited to the following:
- •
- Unexpected demand for or shortage of raw or other materials;
- •
- Adverse financial developments at or affecting the supplier;
44
Table of Contents
- •
- Regulatory requirements or action;
- •
- An inability to provide timely scheduling and/or sufficient capacity;
- •
- Manufacturing difficulties;
- •
- Changes to the specifications of the raw materials such that they no longer meet our standards;
- •
- Lack of sufficient quantities or profit on the production of raw materials to interest suppliers;
- •
- Labor disputes or shortages; or
- •
- Import or export problems.
Any other interruption in our third-party supply chain could adversely affect our ability to satisfy commercial demand and our clinical development needs forFeraheme. In addition, there is only one FDA-approved supplier of the drug substance forMakena and, currently, we do not have a long-term supply agreement with that supplier. The supplier of drug substance may determine that it is not financially attractive for them to continue to supply drug substance forMakena at current prices, or at all, based on our expected purchasing volumes. The qualification of an alternative source may require repeated testing of the new materials and generate greater expenses to us if materials that we test do not perform in an acceptable manner. In addition, we or our third-party manufacturers sometimes obtain raw or other materials from one vendor only, even where multiple sources are available, to maintain quality control and enhance working relationships with suppliers, which could make us susceptible to price inflation by the sole supplier, thereby increasing our production costs. As a result of the high-quality standards imposed on our raw or other materials, we or our third-party manufacturers may not be able to obtain such materials of the quality required to manufactureFeraheme orMakena from an alternative source on commercially reasonable terms, or in a timely manner, if at all.
If we are unable to haveFeraheme orMakena manufactured on a timely or sufficient basis because of the factors discussed above, we may not be able to meet commercial demand or our clinical development needs forFeraheme orMakena, or may not be able to manufactureMakena orFeraheme in a cost-effective manner. As a result, we may lose sales, fail to generate increased revenues or suffer regulatory setbacks, any of which could have an adverse impact on our profitability and future business prospects.
We rely on third parties in the conduct of our business, including our clinical trials and product distribution, and if they fail to fulfill their obligations, our commercialization and development plans may be adversely affected.
We rely on and intend to continue to rely on third parties, including clinical research organizations ("CROs"), third-party logistics providers, packaging, storage and labeling providers, wholesale distributors and certain other important vendors and consultants in the conduct of our business. In addition, we have contracted and plan to continue to contract with certain third parties to provide certain services, including site selection, enrollment, monitoring, data management and other services, in connection with the conduct of our clinical trials and the preparation and filing of our regulatory applications. We have limited experience conducting clinical trials outside the U.S., and, therefore, we are also largely relying on third parties such as CROs to manage, monitor and carry out these clinical trials. Although we depend heavily on these parties, we do not control them and, therefore, we cannot be assured that these third parties will adequately and timely perform all of their contractual obligations to us. If our third-party service providers cannot adequately fulfill their obligations to us in a timely and satisfactory manner, if the quality and accuracy of our clinical trial data or our regulatory submissions are compromised due to poor quality or failure to adhere to our protocols or regulatory requirements or if such third parties otherwise fail to adequately discharge their responsibilities or meet
45
Table of Contents
deadlines, our current and future development plans and regulatory submissions, or our commercialization efforts in current indications, may be delayed, terminated, limited or subject to additional expense, which would adversely impact our ability to generate revenues.
Further, in most cases, we do not currently have back-up suppliers or service providers to perform these tasks. If any of these third parties experience significant difficulties in their respective processes, fail to maintain compliance with applicable legal or regulatory requirements, fail to meet expected deadlines or otherwise do not carry out their contractual duties to us, or encounter physical or natural damages at their facilities, our ability to deliver our products to meet commercial demand could be significantly impaired. The loss of any of our third-party providers, together with a delay or inability to secure an alternate distribution source for end-users in a timely manner, could cause the distribution of our products to be delayed or interrupted, which would have an adverse effect on our business, financial condition and results of operations.
Additionally, we have limited experience independently commercializing multiple pharmaceutical products, including managing and maintaining a supply chain and distribution network for multiple products, and we are placing substantial reliance on third parties to perform this expanded network of product supply chain and distribution services for us. Any failure on our part to effectively execute on our multi-product commercial plans or to effectively manage our supply chain and distribution network would have an adverse impact on our business.
We depend, to a significant degree, on the availability and extent of reimbursement from third-party payers for the use of our products, and a reduction in the availability or extent of reimbursement could adversely affect our sales revenues and results of operations.
Our ability to successfully commercialize our products is dependent, in significant part, on the availability and extent of reimbursement to end-users from third-party payers for the use of our products, including governmental payers, HMOs, managed care organizations and private health insurers. Reimbursement by third-party payers depends on a number of factors, including the third-party's determination that the product is competitively priced, safe and effective, appropriate for the specific patient, and cost-effective. Third-party payers are increasingly challenging the prices charged for pharmaceutical products and have instituted and continue to institute cost containment measures to control or significantly influence the purchase of pharmaceutical products, such as through the use of prior authorizations and step therapy. If these entities do not provide coverage and reimbursement for our products or provide an insufficient level of coverage and reimbursement, physicians and other healthcare providers may choose to use alternative products, which would have an adverse effect on our ability to generate revenues.
In addition, U.S. and many foreign governments continue to propose and pass legislation designed to reduce the cost of healthcare for patients. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (the "Healthcare Reform Act") includes certain cost containment measures including an increase to the minimum rebates for products covered by Medicaid programs, the extension of such rebates to drugs dispensed to Medicaid beneficiaries enrolled in Medicaid managed care organizations and the expansion of the 340B Drug Discount Program under the Public Health Service Act. In addition, federal budgetary concerns could result in the implementation of significant federal spending cuts, including cuts in Medicare and other health related spending in the near-term. Please see our discussion above under the heading, "Pharmaceutical Pricing and Reimbursement" in Item 1. Business for a more detailed discussion of such changes. The magnitude of the impact of these laws on our business is uncertain. Further, in recent years, some states have also passed legislation to control the prices of drugs as well as begun a move toward managed care to relieve some of their Medicaid cost burden. Given that almost half ofMakena patients are Medicaid beneficiaries, the impact of future legislative changes may have a significant and adverse impact onMakena sales. Further, while Medicare is the predominant payer forFeraheme,
46
Table of Contents
Medicare payment policy, in time, can also influence pricing and reimbursement in the non-Medicare markets, as private third-party payers and state Medicaid plans frequently adopt Medicare principles in setting reimbursement methodologies. These and any future changes in government regulation or private third-party payers' reimbursement policies may reduce the extent of reimbursement for our products and adversely affect our future operating results.
Risks Related toMakena
Our ability to successfully commercialize Makena is dependent upon a number of factors, including maintaining the benefits of Makena's orphan drug exclusivity and the length of time before competitors begin selling generic versions of Makena.
Makena has been granted orphan drug exclusivity in the U.S. until February 3, 2018 for prevention of recurrent preterm birth in singleton pregnancies. Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug intended to treat a rare disease or condition, defined, in part, as a patient population of fewer than 200,000 in the U.S. In the U.S., the company that first obtains FDA approval for a designated orphan drug for the specified rare disease or condition receives orphan drug marketing exclusivity for that drug for a period of seven years. This orphan drug exclusivity prevents the FDA from approving another application for the "same drug" for the same orphan indication during the exclusivity period, except in very limited circumstances. In addition, orphan drug exclusivity marketing rights in the U.S. may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantity of the drug to meet the needs of patients with the rare disease or condition. Finally, FDA may approve a subsequent drug that is the same as a currently approved orphan drug for the same orphan indication during the exclusivity period if the sponsor of the subsequent drug can demonstrate that the drug is clinically superior to the already approved drug. According to the FDA, clinical superiority may be demonstrated by showing that a drug is significantly more effective in a clinical trial, safer in a substantial portion of the target population, or provides a major contribution to patient care relative to the currently approved drug.
Additionally, in 1956, the FDA-approved the drug Delalutin, which contained the same active ingredient asMakena. Delalutin was approved for conditions other than reducing the risk of preterm birth and was marketed by Bristol-Myers Squibb ("BMS"). BMS stopped marketing and manufacturing the FDA-approved product and it was withdrawn from the market in 1999. In 2010, in response to a citizen petition, the FDA determined that Delalutin was not withdrawn from sale for reasons of safety or effectiveness. As such, generic drug applications may reference the withdrawn Delalutin New Drug Application ("NDA"). Thus, before the expiration ofMakena's orphan exclusivity, the FDA could determine that it has the authority to approve ANDAs that reference Delalutin so long as the ANDAs meet all relevant legal and regulatory requirements for approval and are labeled for the same indications as Delalutin. If such an approval is granted, doctors may elect to prescribe such approved drug off-label (i.e., outside of FDA-approved indications) forMakena's orphan-protected indication, which could have an adverse impact on our business and results of operations.
Moreover, if one or more ANDA filers or a generic manufacturer were to receive approval to sell a generic or follow-on version ofMakena for the orphan indication, those generic products could potentially be approved as early as February 3, 2018 (the date on whichMakena's orphan exclusivity ends) and we would become subject to increased competition at that time.
47
Table of Contents
Further, our ability to successfully commercializeMakena depends on a number of additional factors, including but not limited to the following:
- •
- The possibility that the benefit of the remaining exclusivity period resulting from the designation ofMakena as an orphan drug may not be realized as a result of off-label use by physicians of current or future FDA-approved drugs in the market whereMakena competes;
- •
- The level of enforcement by the FDA to ensure compounded copies of commercially available FDA-approved products manufactured by compounding pharmacies, including compounded copies of hydroxyprogesterone caproate ("HPC") that are in violation of the federal Drug Quality and Security Act ("DQSA"), as well as other relevant provisions of the FDC Act, are not distributed to patients;
- •
- The size of the pool of patients who may be eligible to receiveMakena;
- •
- Actual or perceived safety and efficacy ofMakena;
- •
- Our ability to increase patient compliance in line with the current label;
- •
- The successful integration and retention of theMakena commercial sales team and any other key employees into our business structure; and
- •
- Our ability to successfully leverage Lumara Health's commercial organizations and distribution networks in marketing, selling and supplyingMakena.
Failure to achieve any or all of these commercial objectives could have an adverse material effect on the growth ofMakena and our ability to achieve our revenue forecasts which could impact our financial condition or results of operations.
We have no experience facing competition from compounded products and if we are unsuccessful in differentiating Makena from compounded HPC products, sales of Makena, and thus our profitability, could be materially adversely affected.
We are aware that formulations of HPC have been available from compounding pharmacies for many years (which compounded formulations of HPC we refer to as "c17P") and will likely remain available even thoughMakena has been granted orphan drug exclusivity until February 3, 2018, and we have no prior experience with facing such competition. In March 2011, the FDA communicated to Lumara Health and also separately issued a press release that, in order to ensure continued access for patients, the FDA intended to refrain from taking enforcement action with respect to compounding pharmacies producing c17P in response to individual prescriptions for individual patients. The FDA's statement had an adverse effect on Lumara Health's ability to realize the benefit of orphan drug exclusivity and its ability to grow sales ofMakena following the launch of the product in March 2011. The failure by the FDA to take enforcement action against compounding pharmacies resulted in substantial sales of compounded copies ofMakena and the effective loss of the value of marketing exclusivity for the affected period of time. In June 2012, the FDA recommended using an FDA-approved drug product, such asMakena, instead of a compounded drug except when there is a specific medical need (e.g., an allergy) that cannot be met by the approved drug. In July 2014, the FDA issued another public statement affirming the position it took in its June 2012 press release recommending use of FDA-approvedMakena except when there is a specific need for a compounded drug. The FDA also stated that when it identifies a pharmacist that compounds regularly or in inordinate amounts of any drug products that are essentially copies ofMakena, the FDA intends to take enforcement action as it deems appropriate. Despite recent negative publicity regarding compounding pharmacies, including the 2012 meningitis outbreak involving compounded drugs, the November 2013 enactment of the DQSA and recent enforcement actions against compounders violating the FDC Act,Makena may continue to face competition from c17P, especially in light of the
48
Table of Contents
long-standing availability of such compounded products, their lower cost and the criticism Lumara Health received in the past in connection with the pricing ofMakena, as discussed below. Further, if any safety or efficacy concerns arise with respect to the c17P products, it may negatively impact sales ofMakena if healthcare providers and patients do not distinguish between the compounded product and the FDA-approvedMakena.
We may not be successful in implementing Makena's lifecycle management program, which could have a negative impact on our business.
In October 2014, we filed with the FDA a prior approval supplement to the originalMakena NDA seeking approval of a 1 mL preservative-free vial ofMakena (the "Single Dose Vial") and we are seeking to expandMakena's formulations and drug delivery technologies as part of the product's lifecycle management program. The lifecycle management program forMakena is an important strategy for our maternal health business, especially in light of the expiration ofMakena's orphan drug exclusivity in February 2018. We have limited experience in the development of alternative formulations forMakena and in developing and implementing lifecycle management programs. We can make no assurance that our prior approval supplement for the Single Dose Vial will be approved on the expected timeline, or at all, or that our other lifecycle management activities will be successful in supporting our maternal health business. Further, the Single Dose Vial will not, and future activities may not, extend or grant exclusivity or provide patent protection, which will likely increase competition. If we are not successful in implementingMakena's lifecycle management program, or if such activities cannot be completed on anticipated timelines, our business will suffer.
The commercial success and growth prospects for Makena will be dependent upon perceptions related to pricing and access.
Lumara Health was criticized for the initial list pricing ofMakena in numerous news articles and internet postings following the FDA's February 2011 approval ofMakena for the prevention of recurrent preterm birth in certain at-risk women. Although the list price ofMakena was subsequently reduced in March 2011,Makena is still priced at a premium to c17P, which has negatively impacted coverage ofMakena by some state Medicaid programs and by certain commercial payers. Although we are undertaking efforts to educate physicians and patients about progress made toward expanding coverage ofMakena and about the benefits of FDA-approvedMakena, certain doctors continue to prescribe non-FDA approved purported substitute products made by pharmaceutical compounders in lieu of prescribingMakena. In addition, efforts to appropriately respond to future concerns about pricing and access raised by media, professional societies, advocacy groups, policymakers or regulatory agencies regarding patient access toMakena, are costly and may not be successful. If we are unable to increase usage ofMakena by physicians and strengthen relationships with professional societies, advocacy groups, policymakers and regulatory agencies, some of whom have been previously critical of Lumara Health, our sales ofMakena may suffer, which would have a materially adverse impact on revenues and our results of operations.
The FDA has required post-marketing studies to verify and describe the clinical benefit of Makena, and the FDA may limit further marketing of the product based on the results of these post-marketing studies, failure to complete these trials in a timely manner or evidence of safety risks or lack of effectiveness.
Makena was approved by the FDA in February 2011 under the provisions of the FDA's "Subpart H" Accelerated Approval regulations. The Subpart H regulations allow certain drugs, for serious or life-threatening conditions, to be approved on the basis of surrogate endpoints or clinical endpoints other than survival or irreversible morbidity. As a condition of approval under Subpart H, the FDA required thatMakena's sponsor perform certain adequate and well-controlled post-marketing clinical studies to verify and describe the clinical benefit ofMakena as well as fulfill certain other
49
Table of Contents
post-marketing commitments. Given the patient population (i.e., women pregnant and at an increased high risk for recurrent preterm delivery) and the informed risk of receiving a placebo instead of the active approved drug in the U.S., the pool of prospective subjects for such clinical trials in the U.S. is small, and we have therefore sought enrollment on a global scale. These factors make the enrollment process slow, difficult, time-consuming and costly. If the required post-marketing studies fail to verify the clinical benefit of the drug, if a sufficient number of participants cannot be enrolled, or if the applicant fails to perform the required post-marketing studies with due diligence, the FDA has the authority to withdraw approval of the drug following a hearing conducted under the FDA's regulations, which would have a materially adverse impact on our business. We cannot be certain of the results of the confirmatory clinical studies or what action the FDA may take if the results of those studies are not as expected based on clinical data that FDA has already reviewed or if such studies are not completed in a timely manner.
Risks Related toFeraheme
The market for Feraheme is limited because Feraheme is only indicated for the treatment of IDA in adult patients with CKD. Significant safety or drug interaction problems, or the evaluation or reevaluation of existing or future data by the FDA or other regulators, could have an adverse impact on Feraheme in this indication, which would adversely impact our future business prospects.
The market forFeraheme is limited becauseFeraheme is only indicated for the treatment of iron deficiency anemia ("IDA") in adult patients with chronic kidney disease ("CKD"). Although we intend to continue to dedicate significant resources to the commercialization ofFeraheme, it may never receive approval for a broader indication and we may not be successful in our efforts to continue to successfully commercializeFeraheme in its current market, which would have a materially adverse effect on our results of operations and future business prospects.
Sales in the current indication may be limited or may decrease if label changes require us to provide additional warnings and/or restrictions related toFeraheme's current or future indications or impose limitations or changes to the method of administering the drug, thereby giving rise to increased competitive pressures ifFeraheme is viewed as less safe than other IV iron products. Significant safety or drug interaction problems with respect toFeraheme, including an increase in the severity or frequency of known adverse events or the discovery of previously unknown adverse events, or the evaluation or reevaluation of data, including pharmacovigilance data, by the FDA, the European Medicines Agency ("EMA"), the competent authorities of the European Union ("EU") Member States or other regulators, could result in lawsuits and increased regulatory scrutiny or a variety of adverse regulatory actions, including changes to the product label, the implementation of a REMS or any other enforcement actions. For example, the Committee for Medicinal Products for Human Use ("CHMP") recently issued opinions that, among other measures,Rienso be administered to patients by infusion over at least 15 minutes (replacing injection), that it be contraindicated in patients with any known history of drug allergy, that the label caution that elderly patients or patients with multiple co-morbidities who experience a serious hypersensitivity reaction due toRienso may have more severe outcomes, and that related variations to the Summary of Product Characteristics ("SmPC") be implemented. Similarly, in June 2014, we proposed changes to FDA related to our current U.S. label ofFeraheme based on a review of global post-marketing data to strengthen the warnings and precautions section of the label and mitigate the risk of serious hypersensitivity reactions, including anaphylaxis, in order to enhance patient safety. After considering our June 2014 submission and other information, in January 2015, the FDA notified us that it believes new safety information should be included in the labeling forFeraheme, including, among other things, a boxed warning to highlight the risks of serious hypersensitivity/anaphylaxis reactions and revisions thatFeraheme should only be administered through an IV infusion (i.e., not by IV injection) and should be contraindicated for patients with any known history of drug allergy. We plan to work with the FDA to finalize an updated U.S.Feraheme label.
50
Table of Contents
These or any future changes to the label/package could adversely impact our ability to successfully compete in the IV iron market and could have an adverse impact on potential sales ofFeraheme and our future business prospects.
In addition, regulators may require us to conduct additional post-approval clinical trials or undertake other activities in order to maintainFeraheme's current indication. For example, CHMP recommended that amendments be made to the Risk Management Plan, including a Post Authorization Safety Study to be conducted to further characterize the risk of hypersensitivity withRienso in patients with CKD and a non-clinical mechanistic study of hypersensitivity reactions. Pursuit of these or other studies are time-consuming and costly and the resulting data might not be as desired or expected, which could further limit the market forFeraheme.
Non-compliance with any recommendations or requirements from regulators could result in product recalls, restrictions on the product's permissible uses, changes to the product label, a negative impact on regulatory applications, suspension, variation or withdrawal of the marketing authorization for the product, or withdrawal of the product from the U.S. and/or foreign markets. Our business could be adversely affected if any such results occur.
Moreover, new safety or drug interaction issues may arise asFeraheme is used over longer periods of time by a wider group of patients, some of whom may be taking other medicines or have additional underlying health problems, which may require us to, among other things, provide additional warnings and/or restrictions on the label/package insert, including a boxed warning in the U.S. or similar warnings outside of the U.S., notify healthcare providers of new safety information, narrow our approved indications, change the rate or method of administration, alter or terminate current or future trials for additional uses ofFeraheme, or even removeFeraheme from the market, any of which could have a significant adverse impact on potential sales ofFeraheme or require us to expend significant additional funds. In the EU,Rienso is subject to additional monitoring by the EMA and the competent authorities of the EU Member States. In addition, if and as we conduct and complete other clinical trials forFeraheme, new safety issues may be identified, which could negatively impact our ability to successfully complete these studies and which could also negatively impact the use and/or regulatory status ofFeraheme for the treatment of IDA in patients with CKD.
For additional details regarding these and other regulatory developments forFeraheme's current indication, please see the discussion under the heading "Feraheme for the treatment of IDA in patients with CKD—Overview" in Item 1. Business.
We may never receive regulatory approval to market and sell Feraheme to the broader IDA patient population.
As discussed above in Item 1. Business under the heading "Feraheme for the treatment of IDA in a broad range of patients—Overview", in January 2014, we received a complete response letter from the FDA informing us that our sNDA for the broad IDA indication could not be approved in its present form. In the letter, the FDA stated that we have not provided sufficient information to permit labeling ofFeraheme for safe and effective use for the proposed indication. This decision by the FDA represents a significant set-back in our efforts to obtain U.S. approval forFeraheme for a broader indication as the issues raised and information requested by the FDA may be costly and time-consuming to address and generate. Further, there is no guarantee that any efforts that we decide to undertake will meet the FDA's requirements, and we may not receive approval at all forFeraheme in a broader indication despite such efforts.
Although we are continuing to work with the FDA, we may decide not to pursue regulatory approval for the broader indication. If we continue to pursue approval in the U.S. for the commercial marketing and sale ofFeraheme for the broad IDA indication, we will have to demonstrate, through the submission of clinical study reports and data sets from one or more controlled clinical trials, that the benefit ofFeraheme use in the proposed population would warrant the risks associated withFeraheme,
51
Table of Contents
including the potential for adverse events, including anaphylaxis, cardiovascular events, and death. The FDA has substantial discretion in the approval process and may decide that the results of any such additional trials and the information we submit seeking approval in the broader patient population or other information reviewed, such as post-marketing safety data, including reports of serious anaphylaxis, cardiovascular events, and death, or any information we provide in response to FDA requests, are insufficient for approval or thatFeraheme is not effective or safe for the proposed broader indication. We have submitted proposed protocols for a clinical study to the FDA for potential pursuit of the broader indication and are awaiting the FDA's feedback. There is no guarantee that the FDA will support any protocols we propose or determine that the results of any clinical trials we undertake ofFeraheme for the treatment of IDA in adult patients who have failed or could not tolerate oral iron will adequately support approval ofFeraheme in this broader patient population, or any of the individual subpopulations of IDA patients.
If we do not obtain U.S. approval to market and sellFeraheme for the treatment of IDA in a broad range of patients, or if we experience additional significant delays or setbacks in obtaining approval, or if we receive approval with significant restrictions, or are required to incur significant costs as post-marketing commitments, our cash position, our ability to increase revenues, our ability to leverage our product portfolio, our profitability, and the future prospects of our business could be materially adversely affected.
Efforts to pursue a broader indication could also have a negative impact on the commercialization ofFeraheme in its current indication if information submitted for purposes of the broader indication and any reevaluation of existing data, such as reports of serious anaphylaxis, cardiovascular events, and death, results in requirements to provide additional warnings and/or restrictions on ourFeraheme label/package insert, change the rate or method of administration ofFeraheme, notify healthcare providers of changes to the label/package insert, narrow the current indication, alter or terminate current or future trials forFeraheme or incur significant costs related to post-marketing requirements/commitments. Such adverse developments could put us at a disadvantage to our competitors and cause healthcare providers to choose to treat all of their IDA patients with competing IV irons based on the actual or perceived safety and efficacy ofFeraheme in light of such activities.
Market acceptance of Feraheme may suffer as a result of the widespread use of competing iron replacement therapy products, including Injectafer®, and as a result of the approval of generic drug products in the near-term, which would have a material adverse effect on our operations and our profitability.
Market acceptance ofFeraheme may suffer as a result of competing iron replacement therapy products, in part because most of these products have been on the market longer and are currently widely used by physicians in the U.S. and abroad, and because certain of these products are approved for the treatment of IDA in a broader group of patients. For example, in July 2013, Injectafer® was approved by the FDA for the treatment of IDA in adult patients who have an unsatisfactory response to oral iron or who have intolerance to oral iron, which is a broader indication than our currentFeraheme indication. Injectafer® is approved in the U.S. with a recommended dose of two slow injections or infusions of 750 milligrams each separated by at least seven days apart for a total of 1,500 milligrams. Given potential label changes in the U.S., which could provide, among other changes, thatFeraheme be administered to patients by infusion over at least 15 minutes (replacing injection),Feraheme could lose a competitive advantage to Injectafer® and other IV irons. Further, we may not be able to offer discounts, incentives or rebates to new or existing customers on terms as appealing as Injectafer® or other IV irons. Even if we continue to seek and eventually obtain labeling ofFeraheme in a broader population, Injectafer® will have already been available for a considerable period of time. During this period, physicians may continue to increase their use of Injectafer®, new physicians may begin to use Injectafer®, and physicians will gain increased familiarity with the product, making it more difficult for us to cause these physicians to useFeraheme in the future. In addition, manufacturers of
52
Table of Contents
Injectafer® may enter into commercial contracts with key customers or GPOs during this period, which could prevent or make it more difficult forFeraheme to retain its existing customers, gain sales to new customers and gain market share in its existing indication with customers or GPOs, and may make entry into the non-CKD market difficult if we were to continue to seek and receive approval for the broader patient population in the future. We face similar challenges outside of the U.S., where our recent SmPC and label changes in the EU and Canada could causeFeraheme to lose market share to competitors such as FerinjectTM (the trade name of Injectafer® in the EU), causingFeraheme to be commercially unviable for us outside of the U.S. If we are not able to differentiateFeraheme from other marketed IV iron products, including Injectafer®, or convince physicians and other customers ofFeraheme's safe and effective use, our ability to maintain a premium price, our ability to generate revenues and maintain profitability, our ability to pursue and support any commercialization efforts outside the U.S., and our long-term business prospects could be adversely affected.
Feraheme's ability to maintain its current market share, or gain wider market acceptance in the future, depends on a number of other factors, including but not limited to the following:
- •
- Our ability to demonstrate to healthcare providers, particularly hematologists, oncologists, hospitals, nephrologists, and others who may purchase or prescribeFeraheme, the clinical efficacy and safety ofFeraheme as an alternative to currently marketed IV iron products which treat IDA in CKD patients;
- •
- Our ability to convince physicians and other healthcare providers to use IV iron, andFeraheme in particular, rather than oral iron, which is the current treatment of choice of most physicians for treating IDA in CKD patients;
- •
- The actual or perceived safety and efficacy profile ofFeraheme as compared to alternative iron replacement therapeutic agents;
- •
- The relative price and level of reimbursement forFeraheme from payers, including government payers, such as Medicare and Medicaid, and private payers as compared to the price and level of reimbursement for alternative IV iron products;
- •
- The actual or perceived convenience and ease of administration ofFeraheme as compared to alternative iron replacement therapeutic agents, including iron administered orally, in light of recent or potential changes to the methods of administration;
- •
- Our ability to execute on our contracting strategy and offer competitive discounts, rebates and other incentives, which can result in increasing the rebates we are required to pay under the Medicaid Drug Rebate program and the discounts we are required to offer under the 340B drug pricing program;
- •
- Current and future limitations on the approved indications and patient populations forFeraheme;
- •
- The introduction of generic versions of ferumoxytol, which may occur in the near-term given the FDA's December 2012 draft guidance containing product-specific bioequivalence recommendations for drug products containing ferumoxytol; and
- •
- The effectiveness of our commercial organization and distribution networks in marketing, selling and supplyingFeraheme.
53
Table of Contents
The key component of our U.S. commercialization strategy forFeraheme is to market and sellFeraheme for use in non-dialysis adult CKD patients. The current U.S. non-dialysis CKD market is comprised primarily of three sites of care where a substantial number of CKD patients are treated: hospitals, hematology and oncology centers, and nephrology clinics. Competition in these practices is intense and competitors such as Injectafer® are gaining market share, particularly in hematology practices. IV iron therapeutic products are not currently widely used by certain physicians who treat non-dialysis CKD patients in the U.S., particularly nephrologists, due to safety concerns and the inconvenience and often impracticability of administering IV iron therapeutic products in their offices. It is often difficult to change physicians' existing treatment paradigms even when supportive clinical data are available. In addition, our ability to effectively market and sellFeraheme in the U.S. hospital market depends in part upon our ability to achieve acceptance ofFeraheme onto hospital formularies. Since many hospitals and hematology, oncology and nephrology practices are members of GPOs, which leverage the purchasing power of a group of entities to obtain discounts based on the collective bargaining power of the group, our ability to attract customers in these sites of care also depends in part on our ability to effectively promoteFeraheme to and enter into pricing agreements with GPOs. The GPOs can also offer opportunities for competitors toFeraheme that provide the ability to quickly gain market share by offering a more attractive price or contracts and incentives that encourage the members of the GPO to use or switch to the competing product. If we are not successful in capturing a significant share of the U.S. non-dialysis CKD market or if we are not successful in securing and maintaining formulary coverage forFeraheme, or if we cannot maintain strong relationships and offer competitive contracts to key customers and GPOs, our profitability as well as our long-term business prospects could be adversely affected.
We derive a substantial amount of our Feraheme revenue from a limited number of customers and the loss of one or more of these customers, a change in their fee structure, or a decline in revenue from one or more of these customers could have an adverse impact on our results of operations and financial condition.
In the U.S., we sellFeraheme primarily to wholesalers and specialty distributors and therefore a significant portion of our revenues is generated by a small number of customers. The loss of any of our customers, including if a customer viewsFeraheme as having a higher risk profile as compared to other IV iron products, especially in light of recent regulatory developments, could have a materially adverse impact on our results of operations. Four customers accounted for 81% of our totalFeraheme revenues during the year ended December 31, 2014, and two customers accounted for 57% of ourFeraheme accounts receivable balance as of December 31, 2014. We pay these wholesalers and specialty distributors a fee for the services that they provide to us. Because our business is concentrated with such a small number of wholesalers and specialty distributors, we could be forced to accept increases in their fees in order to maintain the current distribution networks through whichFeraheme is sold. Any increase in fees could have a negative impact on our current and future sales ofFeraheme in the U.S. and could have a negative impact on the reimbursement rate an individual physician, hospital or clinic would realize upon usingFeraheme.
In addition, a significant portion of our U.S.Feraheme sales are generated through a small number of contracts with GPOs. For example, approximately 26% of ourFeraheme end-user demand during the year ended December 31, 2014 was generated by members of a single GPO with which we have contracted. As a result of the significant percentage of our end-user demand being generated by a single GPO, we may be at a disadvantage in future contract or price negotiations with such GPO and that GPO may be able to influence the demand forFeraheme from its members in a particular quarter through communications they make to their customers. In addition, competitors ofFeraheme may be able to quickly gain market share if they are able to offer GPOs a more attractive price or contracts and incentives that encourage the members of the GPO to use or switch to the competing product, especially if such competing drug can be administered to a broader patient population. The loss of some or all of this demand to a competitor, a material reduction in sales volume, or a significant
54
Table of Contents
adverse change in our relationship with any of our key wholesalers, distributors or GPOs could have a material adverse effect on our revenue and results of operations.
We have no experience commercializing Feraheme outside of the U.S. and we may be unable to undertake such efforts or find a collaboration partner to undertake such efforts, and we may be unsuccessful even if such efforts are undertaken.
Historically, Takeda Pharmaceutical Company Limited ("Takeda") had commercializedFeraheme in the EU, Canada and Switzerland (Feraheme is marketed asRienso outside of the U.S. and Canada), but we have agreed to terminate our license arrangement with Takeda and are in the process of transitioning the product rights back to us, including the marketing authorizations for the EU and Canada. Sales ofFeraheme outside of the U.S. do not and are not expected to materially contribute to our revenues even after we regain worldwide rights. For example, net sales ofRienso by Takeda in the EU were less than $0.5 million in 2014. A number of considerations influence our analysis of our commercialization opportunities outside of the U.S., including (i) regulatory developments and the potential cost of post-approval clinical trial commitments and post-marketing obligations required by regulatory authorities outside of the U.S., (ii) the product's commercial viability (sales potential relative to the cost of maintaining the product on the market) in light of the current CKD label, the possible impact of future label changes, including any impact in the U.S., and the competitive landscape, and (iii) possible approaches in different geographies, which may include seeking a licensing or distribution partner or commercializing the product ourselves. Based on these considerations, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada. In order to continue commercialization efforts, we would have to assume the full cost of any post-marketing obligations required by the ex-U.S. regulatory authorities, both currently and in the future, some of which might have been Takeda's responsibility under a cost-sharing arrangement. Our U.S. sales could be negatively affected if patients or health-care providers in the U.S. perceive withdrawal from other markets as being the result of safety or efficacy, rather than commercial, reasons.
In the future, we may decide to seek to obtain a new marketing authorization forferumoxytol in the EU, particularly if we generate additional clinical data to support potential approval in the broader IDA indication and we may decide to continue to pursue commercial efforts in Canada. If we do pursue commercialization efforts outside of the U.S., we may not have the resources, or be able to find a suitable collaboration partner, to undertake such activities on our behalf. If we choose to commercializeFeraheme outside of the U.S. ourselves, building the internal infrastructure would be costly and time-consuming, and may be distracting to management, and we may not be successful in our efforts. Our ability to commercializeFeraheme outside of the U.S. is also dependent upon the successful transition of the product and related materials back to us and we are relying on Takeda to perform certain services and make certain payments for our benefit in connection with such transition. If Takeda, for any reason, fails to provide such transition services or does not make the expected payments, our ability to commercializeFeraheme outside of the U.S. will be significantly handicapped. In addition, and in light of the termination of the Takeda licensing arrangement, recent or future changes toFeraheme's product label or product insert, withdrawal of the marketing authorization and the Type II Variation forRienso and the fact thatRienso is approved for a narrower patient population than many of its competitors in the EU, could negatively impact the product's commercial potential outside of the U.S., and may lead us to withdraw the product or marketing authorization in additional geographics because it is not commercially viable. If we are not successful in commercializing or partnering, or choose not to commercialize,Feraheme outside of the U.S., our business may suffer, especially if we expend significant time and money pursuing such activities.
55
Table of Contents
Regulatory Risks
In the U.S. there have been, and we expect there will continue to be, a number of federal and state legislative initiatives implemented to reform the healthcare system in ways that could adversely impact our business and our ability to sell our products profitably.
In the U.S., there have been, and we expect there will continue to be, a number of legislative and regulatory proposals aimed at changing the U.S. healthcare system. For example, the Healthcare Reform Act contains a number of provisions that significantly impact the pharmaceutical industry and may negatively affect our business, including potential revenues. Changes that may affect our business include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under the health insurance exchanges, expansion of the 340B program, and fraud and abuse enforcement. For example, the percentage ofFeraheme sold to 340B institutions has grown from 11% in 2011 to 17% in 2014. Since these institutions are granted lower prices than those offered to our other customers, any further growth in our 340B business may have a negative impact on our sales price per gram and operating margins. These changes will impact existing government healthcare programs and will result in the development of new programs, including Medicare payment for performance initiatives and improvements to the physician quality reporting system and feedback program. Substantial new provisions affecting compliance have also been added, which may require us to modify our business practices with healthcare providers and potentially incur additional costs. While we are continuing to evaluate this legislation and its potential impact on our business, this legislation may adversely affect the demand forFeraheme andMakena in the U.S. or cause us to incur additional expenses and therefore adversely affect our financial position and results of operations. Please see our discussion above under the heading, "Pharmaceutical Pricing and Reimbursement" in Item 1. Business for a more detailed discussion of such changes.
Further, although the Healthcare Reform Act exempts "orphan drugs," such asMakena, from 340B ceiling price requirements for the covered entity types added to the program by the Healthcare Reform Act, on July 21, 2014, the Health Resources and Services Administration ("HRSA"), which administers the 340B program, issued an interpretive rule to implement the orphan drug exception which interprets the orphan drug exception narrowly. It exempts orphan drugs from the ceiling price requirements for the newly added covered entity types, namely certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, only when the orphan drug is used for its orphan indication. The newly added entities are entitled to purchase orphan drugs at the ceiling price when the orphan drug is not used for its orphan indication. A manufacturer trade group has filed a lawsuit challenging the interpretive rule as inconsistent with the statutory language. That challenge remains ongoing. The uncertainty regarding how the statutory orphan drug exception will be applied will increase the complexity of compliance, will make compliance more time-consuming, and could negatively impact our results of operations. If HRSA's narrow interpretation of the scope of the orphan drug exemption prevails, it could potentially negatively impact the price we are paid for ourMakena product by certain entities and increase the complexity of compliance with the 340B program.
In order to be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we participate in the Department of Veterans Affairs ("VA") Federal Supply Schedule ("FSS") pricing program. To participate, we are required to enter into an FSS contract with the VA, under which we must make our innovator "covered drugs" available to the "Big Four" federal agencies—the VA, the Department of Defense, or DoD, the Public Health Service, and the Coast Guard—at pricing that is capped pursuant to a statutory federal ceiling price ("FCP") formula set forth in Section 603 of the Veterans Health Care Act of 1992 ("VHCA"). The FCP is based on a weighted non-federal average manufacturer price, or Non-FAMP, which manufacturers are required to report on a quarterly and annual basis to the VA. In addition, pursuant to regulations issued by the DoD TRICARE Management Activity, now the Defense Health Agency, to implement Section 703 of the National Defense Authorization Act for
56
Table of Contents
Fiscal Year 2008, we are required to pay quarterly rebates on covered drug prescriptions dispensed to TRICARE beneficiaries by TRICARE network retail pharmacies. The formula for determining the rebate is established in the regulations and is based on the difference between the annual Non-FAMP and the FCP (as described above, these price points are required to be calculated by us under the VHCA).
If we overcharge the government in connection with our FSS contract or underpay our TRICARE rebates, whether due to a misstated FCP or otherwise, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act ("FCA") and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We expect that the Healthcare Reform Act, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future, could have a material adverse effect on our industry generally and on our ability to maintain or increase our product sales. In addition, various healthcare reform proposals have emerged at the state level in the U.S. We cannot predict the impact that newly enacted laws or any future legislation or regulation will have on us. We expect that there will continue to be a number of U.S. federal and state proposals to implement governmental pricing controls and limit the growth of healthcare costs. These efforts could adversely affect our business by, among other things, limiting the prices that can be charged for our products or the amount of reimbursement rates and terms available from governmental agencies or third-party payers, limiting the profitability of our products, increasing our rebate liability or limiting the commercial opportunities for our products, including its acceptance by healthcare payers.
If our products are marketed or distributed in a manner that violates federal, state or foreign healthcare fraud and abuse laws, marketing disclosure laws or other federal, state or foreign laws and regulations, we may be subject to civil or criminal penalties.
In addition to FDA and related regulatory requirements in the U.S. and abroad, our general operations, and the research, development, manufacture, sale and marketing of our products, are subject to extensive additional federal, state and foreign healthcare regulation, including the FCA, the Federal Anti-Kickback Statute, the Foreign Corrupt Practices Act, and their state analogues, and similar laws in countries outside of the U.S., laws, such as the U.K. Bribery Act of 2010 and governing sampling and distribution of products, and government price reporting laws as discussed above in Item 1. Business under the heading "Government Regulation—Fraud and Abuse Regulation."
Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws and private individuals have been active in bringing lawsuits on behalf of the government under the FCA and similar regulations in other countries. We have developed and implemented a corporate compliance program based on what we believe are current best practices in the pharmaceutical industry; however, these laws are broad in scope and there may not be regulations, guidance or court decisions that definitively interpret these laws in the context of particular industry practices. We cannot guarantee that we, our employees, our consultants or our contractors are or will be in compliance with all federal, state and foreign regulations. If we or our representatives fail to comply with any of these laws or regulations, a range of fines, penalties and/or other sanctions could be imposed on us, including, but not limited to, restrictions on how we market and sell our products, significant fines, exclusions from government healthcare programs, including Medicare and Medicaid, litigation, or other sanctions. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which could also have an adverse effect on our business, financial condition and
57
Table of Contents
results of operations. Such investigations or suits may also result in related shareholder lawsuits, which can also have an adverse effect on our business.
Further, the FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product's approved labeling. For drug products likeMakena that are approved by the FDA under the FDA's accelerated approval regulations, unless otherwise informed by the FDA, the sponsor must submit promotional materials at least 30 days prior to the intended time of initial dissemination of the promotional materials, which delays and may negatively impact our commercial team's ability to implement changes toMakena's marketing materials, thereby negatively impacting revenues. Moreover, under Subpart H, the FDA may also withdraw approval ofMakena if, among other things, the promotional materials are false or misleading, or other evidence demonstrates thatMakena is not shown to be safe or effective under its conditions of use.
The U.S. government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The government has also required companies to enter into complex corporate integrity agreements and/or non-prosecution agreements that can impose significant restrictions and other burdens on the affected companies. If we are found to have promoted such off-label uses, we may become subject to similar consequences.
In recent years, several U.S. states have enacted legislation requiring pharmaceutical companies to establish marketing and promotional compliance programs or codes of conduct and/or to file periodic reports with the state or make periodic public disclosures on sales, marketing, pricing, clinical trials, and other activities. In addition, as part of the Healthcare Reform Act, manufacturers of drugs are required to publicly report gifts and other payments or transfers of value made to U.S. physicians and teaching hospitals. Several states have also adopted laws that prohibit certain marketing-related activities, including the provision of gifts, meals or other items to certain healthcare providers. Many of these requirements are new and uncertain, and the likely extent of penalties for failure to comply with these requirements is unclear; however, compliance with these laws is difficult, time consuming and costly, and if we are found to not be in full compliance with these laws, we may face enforcement actions, fines and other penalties, and we could receive adverse publicity which could have an adverse effect on our business, financial condition and results of operations.
If we fail to comply with any federal, state or foreign laws or regulations governing our industry, we could be subject to a range of regulatory actions that could adversely affect our ability to commercialize our products, harm or prevent sales of our products, or substantially increase the costs and expenses of commercializing and marketing our products, all of which could have a material adverse effect on our business, financial condition and results of operations.
In addition, incentives exist under applicable U.S. law that encourage employees and physicians to report violations of rules governing promotional activities for pharmaceutical products. These incentives could lead to so-called whistleblower lawsuits as part of which such persons seek to collect a portion of moneys allegedly overbilled to government agencies as a result of, for example, promotion of pharmaceutical products beyond labeled claims. Such lawsuits, whether with or without merit, are typically time-consuming and costly to defend. Such suits may also result in related shareholder lawsuits, which are also costly to defend.
58
Table of Contents
If we fail to comply with our reporting and payment obligations under U.S. governmental pricing programs, we could be required to reimburse government programs for underpayments and could pay penalties, sanctions and fines which could have a material adverse effect on our business, financial condition and results of operations.
As a condition of reimbursement by various U.S. federal and state healthcare programs, we are required to calculate and report certain pricing information to U.S. federal and state healthcare agencies. For example, we participate in and have certain price reporting obligations to the Medicaid Drug Rebate program, and we have obligations to report average sales price ("ASP") for the Medicare program. Under the Medicaid Drug Rebate program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data reported by us on a monthly and quarterly basis to the Centers for Medicare and Medicaid Services ("CMS"), the federal agency that administers the Medicare and Medicaid programs. These data include the average manufacturer price and, in the case of innovator products such asFeraheme andMakena, the best price for each drug.
The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current average manufacturer prices and best prices for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed 12 quarters from the quarter in which the data originally were due. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid program. Any corrections to our rebate calculations could result in an overage or underage in our rebate liability for past quarters, depending on the nature of the correction. Price recalculations also may affect the 340B "ceiling price." The 340B pricing program requires participating manufacturers to agree to charge statutorily defined covered entities, such as safety-net providers, no more than the 340B ceiling price for the manufacturer's covered outpatient drugs. The 340B ceiling price is calculated using a statutory formula, which is based on the average manufacturer price and rebate amount for the covered outpatient drug as calculated under the Medicaid Drug Rebate program.
Federal law also requires that a company that participates in the Medicaid program report ASP information to CMS for certain categories of drugs that are paid under Part B of the Medicare program, such asFeraheme andMakena. This ASP information forms the basis for reimbursement for the majority of our currentFeraheme business, and to a lesser extent, for theMakena business. Manufacturers calculate ASP based on a statutorily defined formula and interpretations of the statute by CMS as to what should or should not be considered in computing ASP. An ASP for each National Drug Code for a product that is subject to the ASP reporting requirement must be submitted to CMS no later than 30 days after the end of each calendar quarter. CMS uses these submissions to determine payment rates for drugs under Medicare Part B. Changes affecting the calculation of ASP could affect the ASP calculations for our products and the resulting Medicare payment rate, and could negatively impact our results of operations.
Price reporting and payment obligations are highly complex and vary among products and programs. The calculations are complex and are often subject to interpretation by us, governmental or regulatory agencies and the courts. The calculations of average manufacturer price, best price, and ASP include a number of inputs from our contracts with wholesalers, specialty distributors, GPOs and other customers. The calculations also require us to make an assessment of whether these agreements are deemed to be forbona fide services and whether the fees we pay for any bona fide services represent fair market value in our industry and for our products. These calculations are very complex and could involve the need for us to unbundle or reallocate discounts or rebates offered over multiple quarters or across multiple products. Our processes for estimating amounts due under these governmental pricing
59
Table of Contents
programs involve subjective decisions and estimates. For example, almost half ofMakena sales are reimbursed through state Medicaid programs and are subject to the statutory Medicaid rebate, and in some cases, supplemental rebates offered by us. Often, state Medicaid programs may be slow to invoice pharmaceutical companies for these rebates resulting in a significant lag between the time a sale is recorded and the time the rebate is paid. This results in us having to carry a significant liability on our balance sheet for the estimate of rebate claims expected for Medicaid patients. If actual claims are higher than current estimates, our financial position and results of operations could be adversely affected. The unbundling of discounts and rebates across multiple reporting periods can also result in a restatement of government price reports and changes to the reimbursement rates for various customers covered under federal programs, such as Medicare, Medicaid or the 340B program.
If we have to restate our calculation of government price reports, we may be forced to refund certain monies back to payers to comply with federal pricing agreements. Such a restatement of our government price reports would also adversely impact our reported financial results of operations in the period of such restatement. As a result, our price reporting calculations remain subject to the risk of errors and our methodologies for calculating these prices could be challenged under the FCA or other laws. In addition, the Healthcare Reform Act modified the rules related to certain price reports, among other things. Presently, uncertainty exists as many of the specific determinations necessary to implement this new legislation have yet to be decided and communicated to industry participants. This uncertainty in the interpretation of the legislation increases the chances of an error in price reporting, which could in turn lead to a legal challenge, restatement or investigation. If we become subject to investigations, restatements, or other inquiries concerning our compliance with price reporting laws and regulations, we could be required to pay or be subject to additional reimbursements, penalties, sanctions or fines, which could have a material adverse effect on our business, financial condition and results of operations.
We are liable for errors associated with our submission of pricing data. In addition to retroactive adjustments to rebate amounts and the potential for 340B program refunds, if we are found to have knowingly submitted false average manufacturer price, average sales price, or best price information to the government, we may be liable for civil monetary penalties in the amount of $100,000 per item of false information. If we are found to have made a misrepresentation in the reporting of our ASP, the Medicare statute provides for civil monetary penalties of up to $10,000 for each misrepresentation for each day in which the misrepresentation was applied. Our failure to submit monthly/quarterly average manufacturer price, average sales price, and best price data on a timely basis could result in a civil monetary penalty of $10,000 per day for each day the information is late beyond the due date. Such failure also could be grounds for CMS to terminate our Medicaid drug rebate agreement, pursuant to which we participate in the Medicaid Drug Rebate program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs.
If we overcharge the government, we are required to refund the difference to the government. Failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the FCA and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, would be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
60
Table of Contents
We are subject to ongoing U.S. and foreign regulatory obligations and oversight of our products, and any failure by us to maintain compliance with applicable regulations may result in several adverse consequences including the suspension of the manufacturing, marketing and sale of our respective products, the incurrence of significant additional expense and other limitations on our ability to commercialize our respective products.
We are subject to ongoing regulatory requirements and review, including by periodic audits, both in the U.S. and in foreign jurisdictions pertaining to the development, manufacture, labeling, packaging, adverse event reporting, distribution, storage, marketing, promotion, record keeping and export of our respective products. Failure to comply with such regulatory requirements or the later discovery of previously unknown problems with our products or our third-party contract manufacturing facilities or processes by which we manufacture our products may result in restrictions on our ability to manufacture, market, distribute or sell our products, including potential withdrawal from the market. Any such restrictions could result in a decrease in our product sales, damage to our reputation or the initiation of lawsuits against us and/or our third-party contract manufacturers. We may also be subject to additional sanctions, including but not limited to:
- •
- Warning letters;
- •
- Civil or criminal penalties;
- •
- Variation, suspension or withdrawal of regulatory approvals;
- •
- Changes to the package insert of our products, such as additional warnings regarding potential side effects or potential limitations on the current dosage and administration;
- •
- Requirements to communicate with physicians and other customers about concerns related to actual or potential safety, efficacy, or other issues involving our products;
- •
- Implementation of risk mitigation programs and post-marketing obligations;
- •
- Restrictions on our continued manufacturing, marketing, distribution or sale of our products;
- •
- Temporary or permanent closing of the facilities of our third-party contract manufacturers;
- •
- Interruption of clinical trials; or
- •
- Recalls or a refusal by regulators to consider or approve applications for additional indications.
Any of the above sanctions could have a material adverse impact on our revenue generation and profitability and cause us to incur significant additional expenses.
Additionally, Lumara Health, as our wholly owned subsidiary following consummation of the acquisition, is subject to certain continuing obligations under a Consent Decree of Permanent Injunction ("Consent Decree") between the FDA, Lumara Health's predecessor company, K-V Pharmaceutical Company ("K-V Pharmaceutical") and certain former officers and affiliates of K-V Pharmaceutical. In particular, Lumara Health is bound by a number of provisions and requirements in the Consent Decree including, but not limited to, inspection of Lumara Health's places of business by the FDA without prior notice, and notification of FDA of particular actions and events. If Lumara Health fails to comply with applicable provisions in the Consent Decree, the FDC, or the FDC's implementing regulations, the FDA may impose specific sanctions including, but not limited to, the requirement to cease any of Lumara Health's manufacturing operations, the imposition of substantial financial penalties and the requirement to implement additional corrective actions.
61
Table of Contents
Regulators could determine that our clinical trials and/or our manufacturing processes, or those of our third parties, were not properly designed or are not properly operated, which could cause significant costs or setbacks for our commercialization activities.
We are obligated to conduct, and are in the process of conducting, certain post-approval clinical trials, and we may be required to conduct additional clinical trials, including if we pursue approval of additional indications, seek commercialization in other jurisdictions, or in support of our current indications. We may also determine to conduct additional clinical trials, including if we pursue new formulations or methods of administration for our products. The FDA could determine that our clinical trials and/or our manufacturing processes were not properly designed, did not include enough patients or appropriate administration, were not conducted in accordance with applicable laws and regulations, or were otherwise not properly managed. In addition, according to current good clinical practices regulations ("cGCP") we are responsible for conducting, recording and reporting the results of clinical trials to ensure that the data and results are credible and accurate and that the trial participants are adequately protected. The FDA may conduct inspections of clinical investigator sites which are involved in our clinical development programs to ensure their compliance with cGCP regulations. If the FDA determines that we, our CROs or our study sites fail to comply with applicable cGCP regulations, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may disqualify certain data generated from those sites or require us to perform additional clinical trials. Our clinical trials and manufacturing processes are subject to similar risks and uncertainties outside of the U.S. Any such deficiency in the design, implementation or oversight of our clinical development programs or post-approval clinical studies could cause us to incur significant additional costs, experience further delays or prevent us from commercializingMakena andFeraheme in their current indications, or obtaining marketing approval for additional indications, including the approval for use ofFeraheme for the broad IDA indication, if such approval is pursued.
Further, our third-party contract manufacturing facilities are subject to cGMP regulations enforced by the FDA and equivalent foreign regulations and regulatory agencies through periodic inspections to confirm such compliance. Contract manufacturers must continually expend time, money and effort in production, record-keeping and quality assurance and control to ensure that these manufacturing facilities meet applicable regulatory requirements. Failure to maintain ongoing compliance with cGMP or similar foreign regulations and other applicable manufacturing requirements of various U.S. or foreign regulatory agencies could result in, among other things, the issuance of warning letters, fines, the withdrawal or recall of our products from the marketplace, total or partial suspension of product production, the loss of inventory, suspension of the review of our current or future sNDAs or equivalent foreign filings, enforcement actions, injunctions or criminal prosecution and suspension of manufacturing authorizations. A government-mandated recall or a voluntary recall could divert managerial and financial resources, could be difficult and costly to correct, could result in the suspension of sales of our products and could have a severe adverse impact on our profitability and the future prospects of our business. If any regulatory agency inspects any of these manufacturing facilities and determines that they are not in compliance with cGMP or similar regulations or our contract manufacturers otherwise determine that they are not in compliance with these regulations, as applicable, such contract manufacturers could experience an inability to manufacture sufficient quantities of product to meet demand or incur unanticipated compliance expenditures.
We have also established certain testing and release specifications with the FDA and other foreign regulatory agencies. This release testing must be performed in order to allow finished product to be used for commercial sale. If a finished product does not meet these release specifications or if the release testing is variable, we may not be able to supply product to meet our projected demand. We monitor annual batches of our finished product for ongoing stability after it has been released for commercial sale. If a particular batch of finished drug product exhibits variations in its stability or begins to generate test results that demonstrate an adverse trend against our specifications, we may
62
Table of Contents
need to conduct an investigation into the test results, quarantine the product to prevent further use, destroy existing inventory no longer acceptable for commercial sale, or recall the batch or batches. If we are unable to develop, validate, transfer or gain regulatory approval for the new release test, our ability to supply product to the EU will be adversely affected. Such setbacks could have an adverse impact on our revenues, our profitability and the future prospects of our business.
Risks Related to Our Business Generally
With our acquisition of Lumara Health we have significantly expanded the size of our organization and we may experience difficulties in managing this or future expansion.
With the acquisition of Lumara Health, we increased our headcount by 108 full time employees. Management, personnel, systems and facilities that we currently have in place may not be adequate to support this recent growth, and we may not be able to retain or recruit qualified personnel in the future in this competitive environment to adequately support our new organization. To manage any future growth effectively, we may be required to continue to manage and expand the sales and marketing efforts for our existing products while continuing to identify and acquire attractive additions to our product portfolio, enhance our operational, financial and management controls, reporting systems and procedures and establish and increase our access to commercial supplies of our products, which will be challenging and for which we might not be successful, especially given our newly-expanded organization. We will be required to expand and maintain our facilities and equipment and manage our internal development efforts effectively while complying with our contractual obligations to licensors, licensees, contractors, collaborators, distributors and other third parties. In addition, management may have to divert a disproportionate amount of its attention away from day-to-day activities and towards managing these growth activities, which could be disruptive to our business. Our future financial performance and our ability to execute on our business plan will depend, in part, on our ability to effectively manage our recent and any future growth. If we experience difficulties or are unsuccessful in managing our expansion, our results of operations and business prospects will be negatively impacted.
Our level of indebtedness and the terms of the Term Loan Facility (including the financial covenants) and Convertible Notes could adversely affect our operations and limit our ability to plan for or respond to changes in our business or acquire additional products for our portfolio. If we are unable to comply with restrictions in the Term Loan Facility or cannot repay or refinance the Convertible Notes, the indebtedness under the Term Loan Facility could be accelerated.
We entered into the Term Loan Facility, which provided us with $340.0 million to finance our acquisition of Lumara Health. We also incurred significant indebtedness in the amount of $200.0 million in aggregate principal with additional accrued interest under our Convertible Notes (as defined below). Our level of indebtedness could adversely affect our business by, among other things:
- •
- requiring us to dedicate a substantial portion of our cash flow from operations to payments on our indebtedness, thereby reducing the availability of our cash flow for other purposes, including further diversification of our product portfolio and expansion of sales ofFeraheme in the current or broader indications;
- •
- limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate, thereby placing us at a competitive disadvantage compared to our competitors that may have less debt; and
- •
- increasing our vulnerability to adverse economic and industry conditions.
Our ability to make scheduled payments of the principal of, to pay interest on or to refinance our indebtedness, including the Term Loan Facility and the Convertible Notes, depends on our future
63
Table of Contents
performance, which is subject to economic, financial, competitive and other factors that may be beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt and support our growth strategies. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations, including under the Term Loan Facility or the Convertible Notes.
The Term Loan Facility requires us to make certain payments of principal and interest over time and contains a number of other restrictive covenants, including a financial covenant based on the total amount of debt we have as a multiple of our cash flow, as defined in the Facility, and a requirement that we reduce our indebtedness over time. The Term Loan Facility also contains covenants and terms limiting our ability to enter into new acquisitions, licenses, mergers, foreign investments, to take on new debt and sell assets, and requiring us to pay penalties in the event we want to prepay the Term Loan Facility early. The maturity date of the Term Loan Facility could also be accelerated in certain circumstances, including if we are not able to repay or refinance our Convertible Notes or in the event of an uncured event of default as outlined in the Term Loan Facility. The Term Loan Facility has a floating interest rate based on the prevailing London Interbank Offered Rate ("LIBOR") rate, making interest payments subject to adjustment depending on the interest rate environment. These and other terms in the Term Loan Facility have to be monitored closely for compliance and could restrict our ability to grow our business or enter into transactions that we believe will be beneficial to our business.
Further, holders of the Convertible Notes have the right to require us to repurchase their notes upon the occurrence of a fundamental change at a repurchase price equal to 100% of the principal amount of the notes to be repurchased, plus accrued and unpaid interest. Upon conversion of the Convertible Notes (which are currently convertible), unless we elect to deliver solely shares of our common stock to settle such conversion (other than paying cash in lieu of delivering any fractional share), we will be required to make cash payments in respect of the notes being converted. We may not have enough available cash or be able to obtain financing at the time we are required to make repurchases of Convertible Notes surrendered therefor or at the time Convertible Notes are being converted. Our failure to repurchase Convertible Notes at a time when the repurchase is required by the indenture or to pay any cash payable on future conversions of the Convertible Notes would constitute an event of default. If the repayment of any indebtedness were to be accelerated because of such event of default (whether under the Convertible Notes, our Term Loan Facility or otherwise), we may not have sufficient funds to repay the indebtedness and repurchase the Convertible Notes or make cash payments upon conversions thereof. Moreover, if our stock price increases, the parties with whom we entered into warrant transactions in connection with the pricing of the Convertible Notes (the "Warrants") could exercise such warrants, thereby causing substantial dilution to our stockholders.
We cannot make any assurances that our future operating results will be sufficient to ensure compliance with the covenants in these arrangements or to remedy any such default. In the event of an acceleration of this indebtedness, we may not have or be able to obtain sufficient funds to make any accelerated payments. Any of the factors discussed above could materially and adversely affect our business, financial condition and results of operations. In addition, if we incur additional indebtedness, the risks related to our business and our ability to service or repay our indebtedness would increase.
64
Table of Contents
We may need additional capital to achieve our business objectives and to service our debt obligations, including the Term Loan Facility, our Convertible Notes and contingent payments that may become due under the Lumara Agreement, which could cause significant dilution to our stockholders.
We estimate that our cash resources as of December 31, 2014, combined with cash we currently expect to receive from product sales and earnings on our investments will be sufficient to finance our currently planned operations for at least the next 12 months. We may require additional funds or need to establish additional alternative strategic arrangements to execute a business development transaction. We may at any time seek funding through additional arrangements with collaborators through public or private equity or debt financings, subject to the covenants in our Term Loan Facility. We may not be able to obtain financing or to secure alternative strategic arrangements on acceptable terms or within an acceptable timeframe, if at all, which would limit our ability to execute on our strategic plan. Moreover, the condition of the credit markets can be unpredictable and we may experience a reduction in value or loss of liquidity with respect to our investments, which would put further strain on our cash resources.
Further, in November 2014, we completed the acquisition of Lumara Health, which required us to pay $600.0 in upfront cash consideration and approximately 3.2 million shares of newly issued common stock. We used a combination of cash on hand and the $340.0 million Term Loan Facility to pay the upfront cash consideration. In addition to the consideration paid at closing, our definitive merger agreement with Lumara Health (the "Lumara Agreement") provides for contingent consideration of up to an additional $350.0 million based on the achievement of various sales milestones forMakena, which could be paid in all cash. We also incurred substantial costs and expenses associated with the transaction. As a result, our current level of cash on hand may limit our ability to take advantage of attractive business development opportunities and execute on our strategic plan. In addition, our cash on hand may not be sufficient to service the principal and interest payments under the Term Loan Facility, our existing Convertible Notes or any cash milestone payments to the former Lumara Health security holders upon the achievement of sales milestones. Our ability to make these required payments could be adversely affected if we do not achieve expected revenue and cash flow forecasts or if we are unable to find other sources of cash in the future. If we therefore need to pay the former Lumara Health security holders in stock upon the achievement of sales milestones, it will result in dilution to our stockholders.
Our long-term capital requirements will depend on many other factors, including, but not limited to the commercial success of our products and efforts we make in connection with commercialization and development, our ability to realize synergies and opportunities in connection with our acquisitions and portfolio expansion, the outcome of and costs associated with any material litigation or patent challenges to which we are or may become a party, the timing and magnitude of costs associated with qualifying additional manufacturing capacities and alternative suppliers, and our ability to raise additional capital on terms and within a timeframe acceptable to us, if necessary.
The $200.0 million of 2.5% convertible senior notes due February 15, 2019 (the "Convertible Notes") are, the Warrants may be, and any additional equity or equity-linked financings or alternative strategic arrangements would be, dilutive to our stockholders. In addition, the terms of our current debt instruments or any additional debt financing could greatly restrict our ability to raise additional capital and may provide rights and preferences to the investors in any such financing which are senior to those of, and not available to, current stockholders, impose restrictions on our day-to-day operations or place limitations on our ability to enter into combination transactions with other entities. Our inability to raise additional capital on terms and within a timeframe acceptable to us when needed could force us to dramatically reduce our expenses and delay, scale back or eliminate certain of our activities and operations, including our commercialization and development activities, any of which would have a material adverse effect on our business, financial condition and future business prospects.
65
Table of Contents
Our ability to use net operating loss carryforwards and tax credit carryforwards is dependent on generating future taxable income and may be limited, including as a result of future transactions involving our common stock.
In general, under Section 382 of the Internal Revenue Code of 1986, as amended, a corporation that undergoes an "ownership change" is subject to limitations on its ability to utilize its pre-change net operating losses and certain other tax assets to offset future taxable income. In general, an ownership change occurs if the aggregate stock ownership of certain stockholders increases by more than 50 percentage points over such stockholders' lowest percentage ownership during the testing period, which is generally three years. An ownership change could limit our ability to utilize our net operating loss and tax credit carryforwards for taxable years including or following such "ownership change" by allowing us to utilize only a portion of the net operating losses and tax credits that would otherwise be available but for such ownership change. Limitations imposed on the ability to use net operating losses and tax credits to offset future taxable income or the failure to generate sufficient taxable income could require us to pay more U.S. federal income taxes than we have estimated and could cause such net operating losses and tax credits to expire unused, in each case reducing or eliminating the benefit of such net operating losses and tax credits and potentially adversely affecting our financial position, including our after-tax net income. Similar rules and limitations may apply for state income tax purposes.
In September 2014, we adopted an amendment to our shareholder rights plan to help preserve our tax assets by deterring certain stockholders from increasing their percentage ownership in our stock; however, such amendment is merely a deterrent that does not actually prevent Section 382 ownership limitations and there can be no assurance that we will not undergo an ownership change. Even minor accumulations by certain of our stockholders could result in triggering an ownership change under Section 382. If such an ownership change were to occur, we expect that our net operating losses could become limited; however, the amount of the limitation would depend on a number of factors including our market value at the time of the ownership change. For a discussion of the amendment to our shareholder rights plan, see the discussion in Note O, "Stockholders' Equity," to our consolidated financial statements included in this Annual Report on Form 10-K.
In addition, we have recorded deferred tax assets based on our assessment that we will be able to realize the benefits of our net operating losses and other favorable tax attributes. Realization of deferred tax assets involve significant judgments and estimates which are subject to change and ultimately depends on generating sufficient taxable income of the appropriate character during the appropriate periods. Changes in circumstances may affect the likelihood of such realization, which in turn may trigger a write-down of our deferred tax assets, the amount of which would depend on a number of factors. A write-down would reduce our reported net income, which may adversely impact our financial condition or results of operations or cash flows. In addition, we are potentially subject to ongoing and periodic tax examinations and audits in various jurisdictions, including with respect to the amount of our net operating losses and any limitation thereon. An adjustment to such net operating loss carryforwards, including an adjustment from a taxing authority, could result in higher tax costs, penalties and interest, thereby adversely impacting our financial condition, results of operations or cash flows.
An adverse determination in any current or future lawsuits in which we are a defendant could have a material adverse effect on us.
The administration of our products to, or the use of our products by, humans may expose us to liability claims, whether or not our products are actually at fault for causing an injury. AsFeraheme is used over longer periods of time by a wider group of patients taking numerous other medicines or by patients with additional underlying health problems, the likelihood of adverse drug reactions or unintended side effects, including death, may increase. While these adverse events are rare, all
66
Table of Contents
IV irons, includingFeraheme, can cause patients to experience serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and/or fatal.Makena is a prescription hormone medicine (progestin) used to lower the risk of preterm birth in women who are pregnant and who have previously delivered preterm in the past. It is not known ifMakena is safe and effective in women who have other risk factors for preterm birth. In one clinical study, certain complications or events associated with pregnancy occurred more often in women who receivedMakena. These included miscarriage (pregnancy loss before 20 weeks of pregnancy), hospital admission for preterm labor, preeclampsia, gestational hypertension and gestational diabetes. In addition, other hormones administered during pregnancy have in the past been shown to cross the placenta and have negative effects on the offspring. Although we maintain product liability insurance coverage for claims arising from the use of our products in clinical trials and commercial use, coverage is expensive, and we may not be able to maintain sufficient insurance at a reasonable cost, if at all. Product liability claims, whether or not they have merit, could also decrease demand for our products, subject us to product recalls or harm our reputation, cause us to incur substantial costs, and divert management's time and attention.
As discussed below in Item 3. Legal Proceedings, we were the target of a purported class action complaint filed in March 2010 entitledSilverstrand Investments et. al. v. AMAG Pharm., Inc., et. al., which alleged certain securities laws violations. Although we settled theSilverstrand case in January 2015, we may also be the target of claims asserting violations of securities and fraud and abuse laws and derivative actions or other litigation in the future. Any future litigation could result in substantial costs and divert our management's attention and resources, which could cause serious harm to our business, operating results and financial condition. Further, we may not be successful in defending ourselves in the litigation and, as a result, our business could be materially harmed. These lawsuits may result in large judgments or settlements against us, any of which could have a negative effect on our financial condition and business if in excess of our insurance coverage. Though we maintain liability insurance, if any costs or expenses associated with litigation exceed our insurance coverage, we may be forced to bear some or all of these costs and expenses directly, which could be substantial.
Our success depends on our ability to attract and retain key employees, and any failure to do so may be disruptive to our operations.
We are a pharmaceutical company focused on marketing commercial products and we plan to expand our product portfolio with additional commercial-stage products through acquisitions and in-licensing; thus, the range of skills of our executive officers needs to be broad and deep. If we are not able to hire and retain talent to drive commercialization and expansion of our product portfolio, we will be unlikely to be profitable. For example, in October 2014, our Senior Vice President and Chief Development and Regulatory Officer resigned from the Company to pursue other opportunities, which may be disruptive to our regulatory discussions with the FDA or other regulators. Further, because of the specialized nature of our business, our success depends to a significant extent on our ability to continue to attract, retain and motivate qualified sales, technical operations, managerial, scientific, regulatory compliance and medical personnel of all levels. The loss of key personnel or our inability to hire and retain personnel who have such sales, technical operations, managerial, scientific, regulatory compliance and medical backgrounds could materially adversely affect our research and development efforts and our business.
Our operating results will likely fluctuate, including as a result of wholesaler, distributor and customer buying patterns, so you should not rely on the results of any single quarter to predict how we will perform over time.
Our future operating results will likely vary from quarter to quarter depending on a number of factors, including the factors described in these Risk Factors, many of which we cannot control, as well as the timing and magnitude of:
- •
- The timing and magnitude of product revenues;
- •
- The loss of a key customer or GPO;
67
Table of Contents
- •
- Costs and liabilities incurred in connection with business development activities or business development transactions into which we may enter;
- •
- Costs associated with the commercialization of our products in the U.S., including costs associated with pursuing a broader indication ofFeraheme or theMakena lifecycle management program;
- •
- Makena milestone payments we may be required to pay to the former shareholders of Lumara Health pursuant to the Lumara Agreement;
- •
- The timing and magnitude of tax payments and of principal and interest payments in connection with the Term Loan Facility and our Convertible Notes;
- •
- Costs associated with the manufacture of our products, including costs of raw and other materials and costs associated with maintaining commercial inventory and qualifying additional manufacturing capacities and alternative suppliers;
- •
- The recognition of deferred tax assets during periods in which we generate taxable income and our ability to preserve our net operating loss carryforwards and other tax assets;
- •
- Costs associated with our ongoing and planned clinical studies ofFeraheme in connection with our pediatric program, our current or future post-marketing commitments for the EMA and other regulatory agencies, our pursuit, if any, of additional indications and our development ofFeraheme in countries outside of the U.S;
- •
- Costs associated with the ongoing and planned clinical studies ofMakena in connection with current or future post-marketing commitments, and our pursuit, if any, of additional indications or lifecycle management program;
- •
- Any changes to the mix of our business;
- •
- Costs associated with manufacturing batch failures or inventory write-offs due to out-of-specification release testing or ongoing stability testing that results in a batch no longer meeting specifications;
- •
- Changes in accounting estimates related to reserves on revenue, returns, contingent consideration, impairment of long-lived or intangible assets or other accruals or changes in the timing and availability of government or customer discounts, rebates and incentives;
- •
- The implementation of new or revised accounting or tax rules or policies; and
- •
- Costs associated with the implementation of new or revised regulations of the Public Company Accounting Oversight Board, NASDAQ Global Select Market ("NASDAQ"), the U.S. Securities and Exchange Commission ("SEC") and similar entities.
Our results of operations, including, in particular, product sales, may also vary from period to period due to the buying patterns of our wholesalers, distributors, pharmacies, clinics or hospitals and specialty pharmacies. Further, our contracts with GPOs often require certain performance from the members of the GPOs on an individual account level or group level such as growth over prior periods or certain market share attainment goals in order to qualify for discounts off the list price of our products, and a GPO may be able to influence the demand for our products from its members in a particular quarter through communications they make to their members. In the event wholesalers, distributors, pharmacies, clinics or hospitals with whom we do business determine to limit their purchases of our products, our product sales could be adversely affected. Also, in the event wholesalers, distributors, pharmacies, clinics or hospitals purchase increased quantities of our products to take advantage of volume discounts or similar benefits, our quarterly results will fluctuate as re-orders become less frequent, and our overall net pricing may decrease as a result of such discounts and similar
68
Table of Contents
benefits. In addition, these contracts are often cancellable at any time by our customers, often without notice, and are non-exclusive agreements within theFeraheme orMakena markets. While these contracts are intended to support the use of our products, our competitors could offer better pricing, incentives, higher rebates or exclusive relationships. BecauseFeraheme is not indicated for the broad IDA population, the incentives in our contracts for a particular site of care are capped based on our estimate of their patients covered by our current CKD label. Because some of our competitors' products have the broad IDA label, they may provide additional incentives for all of a customer's IV iron usage, essentially becoming an exclusive provider to that particular customer.
Our contracting strategy can also have an impact on the timing of certain purchases causing product sales to vary from quarter to quarter. For example, in advance of an anticipated price increase, following the publication of our quarterly ASP, which affects the rate at whichFeraheme is reimbursed, or a reduction in expected rebates or discounts forFeraheme, customers may orderFeraheme in larger than normal quantities which could causeFeraheme sales to be lower in subsequent quarters than they would have been otherwise. Further, any changes in purchasing patterns or inventory levels, changes to our contracting strategy, increases in product returns, delays in purchasing products or delays in payment for products by one of our distributors or GPOs could also have a negative impact on our revenue and results of operations.
As a result of these and other factors, our quarterly operating results could fluctuate, and this fluctuation could cause the market price of our common stock to decline. Results from one quarter should not be used as an indication of future performance.
If the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial statements and/or our projected guidance prove inaccurate, our actual results may vary from those reflected in our projections and accruals.
Our consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us, and the related disclosure of contingent assets and liabilities. On an ongoing basis, our management evaluates our critical and other significant estimates and judgments, including among others those associated with revenue recognition related to product sales and collaboration agreements, product sales allowances and accruals, assessing investments for potential other-than-temporary impairment and determining the fair values of our investments, the fair value of our debt obligations, the fair value of assets acquired in a business combination, contingent consideration, the impairment of long-lived assets, including intangible assets, accrued expenses, income tax and equity-based compensation expense. We base our estimates on market data, our observance of trends in our industry, and various other assumptions that we believe to be reasonable under the circumstances. If actual results differ from these estimates, there could be a material adverse effect on our financial results and the performance of our stock.
Further, in January 2015, we issued financial guidance, including expected 2015 total revenues andFeraheme andMakena net sales, which is likewise based on estimates and the judgment of management. If, for any reason, we are unable to realize our projected 2015 revenue, we may not realize our publicly announced financial guidance. If we fail to realize or if we change or update any element of our publicly disclosed financial guidance or other expectations about our business, our stock price could decline in value.
As part of our revenue recognition policy, our estimates of product returns, rebates and chargebacks, fees and other discounts require subjective and complex judgments due to the need to make estimates about matters that are inherently uncertain. The same estimates will need to be made forMakena sales since much of Lumara Health's business provides for discounts, fees, rebates and
69
Table of Contents
chargebacks, in particular, since a significant amount ofMakena sales are to Medicaid patients. Any significant differences between our actual results and our estimates could materially affect our financial position and results of operations.
In addition, to determine the required quantities ofFeraheme andMakena and their related manufacturing schedules, we also need to make significant judgments and estimates based on inventory levels, current market trends, anticipated sales, forecasts and other factors. Because of the inherent nature of estimates, there could be significant differences between our estimates and the actual amount of product need. For example, the level of our access to wholesaler and distributor inventory levels and sales data in the U.S., which varies based on the wholesaler, distributor, clinic or hospital, affects our ability to accurately estimate certain reserves included in our financial statements. Any difference between our estimates and the actual amount of product demand could result in unmet demand or excess inventory, each of which would adversely impact our financial results and results of operations.
Our stock price has been and may continue to be volatile, and your investment in our stock could decline in value or fluctuate significantly, including as a result of analysts' activities.
The market price of our common stock has been, and may continue to be, volatile, and your investment in our stock could decline in value or fluctuate significantly. Our stock price has ranged between $16.49 and $48.50 in the fifty-two week period through February 4, 2015. The stock market has from time to time experienced extreme price and volume fluctuations, particularly in the biotechnology and pharmaceuticals sectors, which have often been unrelated to the operating performance of particular companies. Various factors and events, including the factors and events described in these Risk Factors, many of which are beyond our control, may have a significant impact on the market price of our common stock. Our stock price could also be subject to fluctuations as a result of general market conditions, shareholder activism and attempts to disrupt our strategy by activist investors or sales of large blocks of our common stock or the dilutive effect of our Convertible Notes or any other equity or equity-linked financings or alternative strategic arrangements.
In addition, the trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us and our business. As of February 4, 2015, five financial analysts publish reports about us and our business. We do not control these or any other analysts. Furthermore, there are many large, well-established, publicly traded companies active in our industry and market, which may mean that it is less likely that we will receive widespread analyst coverage. In addition, our future operating results are subject to substantial uncertainty, and our stock price could decline significantly if we fail to meet or exceed analysts' forecasts and expectations. If any of the analysts who cover us downgrade our stock, lower their price target or issue commentary or observations about us or our stock that are perceived by the market as negative, our stock price would likely decline rapidly. In addition, if these analysts cease coverage of our company, we could lose visibility in the market, which in turn could also cause our stock price to decline.
There is a potential market overhang that could depress the value of our common stock, and future sales of our common stock could put a downward pressure on the price of our shares and could have a material adverse effect on the price of our shares.
In connection with the Lumara Agreement, we issued approximately 3.2 million shares of newly issued common stock to former Lumara Health security holders. Upon the demand of a certain number or percentage of such former Lumara Health security holders, we have agreed to file a registration statement to register the disposition of the shares of our common stock issued to such shareholders. In accordance with the Lumara Agreement, on February 10, 2015, we filed a registration statement on Form S-3 to register the resale of approximately 1.6 million shares of such common stock. Furthermore, upon the expiration of a contractual lock-up period, the remainder (approximately 1.6 million shares) of the common stock issued to former Lumara Health security holders can be sold.
70
Table of Contents
If these shares are sold, or if other existing stockholders or our officers or directors sell, or indicate an intention to sell (which may include sales pursuant to written plans for trading shares in reliance on Rule 10b5-1 under the Securities Act of 1933), substantial amounts of our common stock in the public market, the market price of our common stock could decline.
If we identify a material weakness in our internal controls over financial reporting, our ability to meet our reporting obligations and the trading price of our stock could be negatively affected.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Accordingly, a material weakness increases the risk that the financial information we report contains material errors.
We regularly review and update our internal controls, disclosure controls and procedures, and corporate governance policies. In addition, we are required under the Sarbanes-Oxley Act of 2002 to report annually on our internal control over financial reporting. Any system of internal controls, however well designed and operated, is based in part on certain assumptions and can provide only reasonable, not absolute, assurances that the objectives of the system are met. If we, or our independent registered public accounting firm, determine that our internal controls over our financial reporting are not effective, or we discover areas that need improvement in the future, or we experience high turnover of our personnel in our financial reporting functions, these shortcomings could have an adverse effect on our business and financial results, and the price of our common stock could be negatively affected.
If we cannot conclude that we have effective internal control over our financial reporting, or if our independent registered public accounting firm is unable to provide an unqualified opinion regarding the effectiveness of our internal control over financial reporting, investors could lose confidence in the reliability of our financial statements, which could lead to a decline in our stock price. Because Lumara Health had been a private company, it has not been required to comply with the Sarbanes-Oxley Act of 2002. As such, we are in the process of integrating Lumara Health related controls into our current control environment. However, our 2014 assessment did not include evaluating the effectiveness of internal control over financial reporting of Lumara Health its subsidiaries, the consolidated results of which are included in our fiscal year 2014 consolidated financial statements. Failure to comply with reporting requirements could subject us to sanctions and/or investigations by the SEC, NASDAQ or other regulatory authorities.
Our shareholder rights plan, certain provisions in our charter and by-laws, certain provisions of our Convertible Notes, certain contractual relationships and certain Delaware law provisions could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove the current members of our Board.
We have a shareholder rights plan, the provisions of which are intended to protect our net operating loss and tax credit carryforwards and could function to deter a hostile takeover by making any proposed hostile acquisition of us more expensive and less desirable to a potential acquirer by enabling our stockholders (other than the potential hostile acquiror) to purchase significant amounts of additional shares of our common stock at dilutive prices. The rights issued pursuant to our shareholder rights plan, as amended in September 2014, become exercisable generally upon the earlier of 10 days after a person or group acquires 4.99% or more of our outstanding common stock or 10 business days after the announcement by a person or group of an intention to acquire 4.99% of our outstanding common stock via tender offer or similar transaction. The shareholder rights plan could delay or discourage transactions involving an actual or potential change in control of us or our management, including transactions in which stockholders might otherwise receive a premium for their shares over then-current prices.
71
Table of Contents
In addition, certain provisions in our certificate of incorporation and our by-laws may discourage, delay or prevent a change of control or takeover attempt of our company by a third-party as well as substantially impede the ability of our stockholders to benefit from a change of control or effect a change in management and our Board. These provisions include:
- •
- the ability of our Board to increase or decrease the size of the Board without stockholder approval;
- •
- advance notice requirements for the nomination of candidates for election to our Board and for proposals to be brought before our annual meeting of stockholders;
- •
- the authority of our Board to designate the terms of and issue new series of preferred stock without stockholder approval;
- •
- non-cumulative voting for directors; and
- •
- limitations on the ability of our stockholders to call special meetings of stockholders.
As a Delaware corporation, we are subject to the provisions of Section 203 of the Delaware General Corporation Law ("Section 203"), which prevents us from engaging in any business combination with any "interested stockholder," which is defined generally as a person that acquires 15% or more of a corporation's outstanding voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in the manner prescribed in Section 203. These provisions could have the effect of delaying or preventing a change of control, whether or not it is desired by, or beneficial to, our stockholders.
In addition to the above factors, an acquisition of our company could be made more difficult by employment agreements we have in place with our executive officers, as well as a company-wide change of control policy, which provide for severance benefits as well as the full acceleration of vesting of any outstanding options or restricted stock units in the event of a change of control and subsequent termination of employment. Further, our Third Amended and Restated 2007 Equity Incentive Plan generally permits our Board to provide for the acceleration of vesting of options granted under that plan in the event of certain transactions that result in a change of control.
Furthermore, holders of the Convertible Notes have the right to require us to repurchase their notes at a price equal to 100% of the principal amount thereof and the conversion rate for the Convertible Notes may be increased as described in the indenture, in each case, upon the occurrence of certain change of control transactions. Additionally, upon certain change of control transactions, the offsetting convertible bond hedge and warrant transactions that we entered into at the time we issued the Convertible Notes may be exercised and/or terminated early. Upon any such exercise and/or early termination, the proceeds we receive upon the exercise of the convertible bond hedge transactions may prove to be significantly lower than the amounts we would be required to pay upon termination of the warrant transactions. These features of the Convertible Notes and the convertible bond hedge and warrants, including the financial implications of any renegotiation of the above-mentioned provisions, could have the effect of preventing a change of control, whether or not it is desired by, or beneficial to, our stockholders, or may result in the acquisition of us being on terms less favorable to our stockholders than it would otherwise be.
ITEM 1B. UNRESOLVED STAFF COMMENTS:
None.
ITEM 2. PROPERTIES:
In June 2013, we entered into a lease agreement with BP Bay Colony LLC (the "Waltham Landlord") for the lease of certain real property located at 1100 Winter Street, Waltham,
72
Table of Contents
Massachusetts (the "Waltham Premises") for use as our principal executive offices. Beginning in September 2013, the initial term of the lease is five years and two months with one five-year extension term at our option. During the extension period, the base rent will be an amount agreed upon by us and the Waltham Landlord. In addition to base rent, we are also required to pay a proportionate share of the Waltham Landlord's operating costs.
The Waltham Landlord agreed to pay for certain agreed-upon improvements and we agreed to pay for any increased costs due to changes by us to the agreed-upon plans. We record all tenant improvements paid by us as leasehold improvements and amortize these improvements over the shorter of the estimated useful life of the improvement or the remaining life of the initial lease term. Amortization of leasehold improvements is included in depreciation expense.
In addition, in connection with our lease for the Waltham Premises, in June 2013 we delivered to the Waltham Landlord a security deposit of $0.4 million in the form of an irrevocable letter of credit. This security deposit will be reduced to $0.3 million on the second anniversary of the date the lease commenced. The cash securing this letter of credit is classified on our balance sheet as of December 31, 2014 as a long-term asset and is restricted in its use.
In June 2013, we also entered into an Assignment and Assumption of Lease (the "Assignment Agreement") with Shire Human Genetic Therapies, Inc. ("Shire") effecting the assignment to Shire of the right to occupy our former office space located at 100 Hayden Avenue, Lexington, Massachusetts (the "Prior Space"). Under the Assignment Agreement, the assignment to Shire became effective on September 21, 2013, the date of our departure from the Prior Space, and Shire assumed all of our obligations as the tenant of the Prior Space. The Assignment Agreement also provided for the conveyance of furniture and other personal property by us to Shire.
In connection with our acquisition of Lumara Health, we have assumed the lease of certain real property located at 16640 Chesterfield Grove Road, Chesterfield, Missouri (the "St. Louis Premises"), which we are currently using as temporary office space for Lumara Health employees as they relocate to the Waltham Premises. Beginning in September 2013, the initial term of the lease is five years and two months. In addition to base rent, we are also required to pay a proportionate share of the Chesterfield Landlord's operating costs. We are attempting to sublease the St. Louis Premises and if successful, future operating lease commitments will be partially offset by proceeds received from the sublease.
The above leases for the Waltham, Massachusetts and Chesterfield, Missouri properties requires us to pay base rent during the initial term as follows (in thousands):
| | | | |
Period | | Minimum Lease
Payments | |
|---|
Year Ended December 31, 2015 | | $ | 1,451 | |
Year Ended December 31, 2016 | | | 1,456 | |
Year Ended December 31, 2017 | | | 1,462 | |
Year Ended December 31, 2018 | | | 1,174 | |
| | | | | |
Total | | $ | 5,543 | |
| | | | | |
| | | | | |
| | | | | |
ITEM 3. LEGAL PROCEEDINGS:
We accrue a liability for legal contingencies when we believe that it is both probable that a liability has been incurred and that we can reasonably estimate the amount of the loss. We review these accruals and adjust them to reflect ongoing negotiations, settlements, rulings, advice of legal counsel and other relevant information. To the extent new information is obtained and our views on the probable outcomes of claims, suits, assessments, investigations or legal proceedings change, changes in
73
Table of Contents
our accrued liabilities would be recorded in the period in which such determination is made. For the matters referenced below, the liability is not probable or the amount cannot be reasonably estimated and, therefore, accruals have not been made. In addition, in accordance with the relevant authoritative guidance, for any matters in which the likelihood of material loss is at least reasonably possible, we will provide disclosure of the possible loss or range of loss. If a reasonable estimate cannot be made, however, we will provide disclosure to that effect.
Silverstrand Class Action
A purported class action complaint was originally filed on March 18, 2010 in the U.S. District Court for the District of Massachusetts, entitled Silverstrand Investments et. al. v. AMAG Pharm., Inc., et. al., Civil Action No. 1:10-CV-10470-NMG, and was amended on September 15, 2010 and on December 17, 2010. The second amended complaint, filed on December 17, 2010 alleged that we and our former President and Chief Executive Officer, former Chief Financial Officer, the then-members of our Board, and certain underwriters in our January 2010 offering of common stock violated certain federal securities laws, specifically Sections 11 and 12(a)(2) of the Securities Act of 1933, as amended, and that our former President and Chief Executive Officer and former Chief Financial Officer violated Section 15 of such Act, respectively, by making certain alleged omissions in a registration statement filed in January 2010. The plaintiffs sought unspecified damages on behalf of a purported class of purchasers of our common stock pursuant to our common stock offering on or about January 21, 2010. After litigating the class action lawsuit for several years, on September 12, 2014, we and the other defendants entered into a stipulation of settlement with the lead plaintiffs (on behalf of themselves and each of the class members) to resolve the class action securities lawsuit. Pursuant to the stipulation of settlement, and in exchange for a release of all claims by the class members and certain other persons, and dismissal of the lawsuit with prejudice, we agreed to cause our insurer to pay eligible class members and their attorneys a total of $3.75 million. On October 2, 2014, the U.S. District Court preliminarily approved the settlement, and potential class members were notified of the proposed settlement and the procedures by which they could seek to recover from the settlement fund, object to the settlement or request to be excluded from the settlement class and on January 30, 2015, the stipulation of settlement was approved by the U.S. District Court. The U.S. District Court entered final judgment on February 2, 2015. Any appeals of the settlement are due by March 4, 2015. We have recorded the $3.75 million settlement amount in prepaid and other current assets and a corresponding amount in accrued expenses on our consolidated balance sheet as of December 31, 2014, as the settlement amount will be fully covered by our insurance carrier. There was no impact to our consolidated statement of operations for the year ended December 31, 2014.
Makena Securities Litigation
On October 19, 2011, plaintiff Frank Julianello filed a complaint against Lumara Health (then-named K-V Pharmaceutical Company ("K-V Pharmaceutical")) and certain individual defendants, in the United States District Court for the Eastern District of Missouri (the "Court"), alleging violations of the anti-fraud provisions of the federal securities laws on behalf of all purchasers of the publicly traded securities of Lumara Health between February 14, 2011 and April 4, 2011. The complaint alleges class members were damaged by paying artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On October 31, 2011, plaintiff Ramakrishna Mukku filed a complaint against Lumara Health, in the United States District Court for the Eastern District of Missouri, alleging violations of the anti-fraud provisions of the federal securities laws on behalf of all purchasers of the publicly traded securities of Lumara Health between February 14, 2011 and April 4, 2011. The complaint alleges class members were damaged by paying artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On November 2, 2011, plaintiff Hoichi Cheong filed a complaint against Lumara Health, in the United States District Court for the Eastern District of
74
Table of Contents
Missouri, on behalf of purchasers of the securities of Lumara Health, who purchased or otherwise acquired K-V Pharmaceutical securities between February 14, 2011 and April 4, 2011, seeking to pursue remedies under the Exchange Act. The complaint alleges class members were damaged by purchasing artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On March 8, 2012, the Julianello, Mukku and Cheong cases were consolidated and the consolidated action is now styled In Re K-V Pharmaceutical Company Securities Litigation, Case No. 4:11-CV-1816. On May 4, 2012, the Court appointed Lori Anderson as Lead Plaintiff in the matter. On April 22, 2013, the individual defendants moved to dismiss the complaint and oral argument was held before the Court on November 26, 2013. Lumara Health joined in the motion to dismiss on February 10, 2014. On March 27, 2014, the Court entered an order granting Lumara Health's motion to dismiss the class action complaint without prejudice to the Plaintiffs' ability to file a second amended complaint with respect to a limited issue of whether Lumara Health's statements about Lumara Health's financial assistance program forMakena were materially false or misleading. On April 16, 2014, the Plaintiff's filed a motion to reconsider asking the Court to reconsider its order restricting the scope of Plaintiff's ability to amend its complaint. The Court denied Plaintiff's motion to reconsider and entered a judgment granting Lumara Health's motion to dismiss on June 6, 2014. On July 1, 2014, Plaintiffs filed a Notice of Appeal with the Eighth Circuit Court of Appeals and briefs have been submitted to the Court. The Court of Appeals has set March 12, 2015 as the date for oral argument.
European Patent Organization Appeal
In July 2010, Sandoz GmbH ("Sandoz") filed with the European Patent Office ("EPO") an opposition to a previously issued patent which covers ferumoxytol in EU jurisdictions. In October 2012, at an oral hearing, the Opposition Division of the EPO revoked this patent. In December 2012, our notice of appeal of that decision was recorded with the EPO, which also suspended the revocation of our patent. On May 13, 2013, we filed a statement of grounds of appeal and on September 27, 2013, Sandoz filed a response to that statement. In the event that we withdraw our appeal or we do not experience a successful outcome from the appeals process, under EU regulations ferumoxytol would still be entitled to eight years of data protection and ten years of market exclusivity from the date of approval, which we believe would create barriers to entry for any generic version of ferumoxytol into the EU market until sometime between 2020 and 2022. This decision had no impact on our revenues for the year ended December 31, 2014. However, any future unfavorable outcome in this matter could negatively affect the magnitude and timing of future revenues. We do not expect to incur any related liability regardless of the outcome of the appeal and therefore have not recorded any liability as of December 31, 2014. We continue to believe the patent is valid and intend to vigorously appeal the decision.
We may periodically become subject to legal proceedings and claims arising in connection with ongoing business activities, including claims or disputes related to patents that have been issued or that are pending in the field of research on which we are focused. Other than the above actions, we are not aware of any material claims against us as of December 31, 2014.
ITEM 4. MINE SAFETY DISCLOSURES
Not Applicable.
75
Table of Contents
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES:
Market Information
Our common stock trades on the NASDAQ Global Select Market ("NASDAQ") under the trading symbol "AMAG." On February 4, 2015, the closing price of our common stock, as reported on the NASDAQ, was $41.50 per share. The following table sets forth, for the periods indicated, the high and low sale prices per share for our common stock as reported on the NASDAQ.
| | | | | | | |
| | High | | Low | |
|---|
Year Ended December 31, 2014 | | | | | | | |
First quarter | | $ | 24.93 | | $ | 18.52 | |
Second quarter | | $ | 20.88 | | $ | 16.49 | |
Third quarter | | $ | 33.57 | | $ | 17.79 | |
Fourth quarter | | $ | 44.81 | | $ | 29.76 | |
Year Ended December 31, 2013 | | |
| | |
| |
First quarter | | $ | 23.98 | | $ | 15.00 | |
Second quarter | | $ | 25.67 | | $ | 18.46 | |
Third quarter | | $ | 27.00 | | $ | 20.35 | |
Fourth quarter | | $ | 28.42 | | $ | 18.94 | |
Stockholders
On February 4, 2015, we had approximately 156 stockholders of record of our common stock, and we believe that the number of beneficial holders of our common stock was approximately 8,776 based on responses from brokers to a search conducted by Broadridge Financial Solutions, Inc. on our behalf.
Dividends
We have never declared or paid a cash dividend on our common stock. We currently anticipate that we will retain all of our earnings for use in the development of our business and do not anticipate paying any cash dividends in the foreseeable future.
76
Table of Contents
Repurchases of Equity Securities
The following table provides certain information with respect to our purchases of shares of our stock during the fourth quarter of 2014.
| | | | | | | | | | | | | |
Period | | Total Number
of Shares
Purchased(1) | | Average Price
Paid per
Share | | Total Number of
Shares Purchased
as Part of Publicly Announced
Plans or Programs(2) | | Maximum Number of
Shares that May Yet Be
Purchased Under the
Plans or Programs(2) | |
|---|
October 1, 2014 through October 31, 2014 | | | — | | | — | | | — | | | — | |
November 1, 2014 through November 30, 2014 | | |
— | | |
— | | |
— | | |
— | |
December 1, 2014 through December 31, 2014 | | |
25,498 | |
$ |
40.31 | | |
— | | |
— | |
| | | | | | | | | | | | | | |
Total | | |
25,498 | |
$ |
40.31 | | |
— | | |
— | |
- (1)
- Represents shares of our common stock withheld by us to satisfy the minimum tax withholding obligations in connection with the vesting of restricted stock units held by our employees.
- (2)
- We do not currently have any publicly announced repurchase programs or plans.
Securities Authorized for Issuance Under Equity Compensation Plans
See Part III, Item 12 for information regarding securities authorized for issuance under our equity compensation plans. Such information is incorporated by reference to our definitive proxy statement pursuant to Regulation 14A, which we intend to file with the U.S. Securities and Exchange Commission (the "SEC") not later than 120 days after the close of our year ended December 31, 2014.
77
Table of Contents
Five-Year Comparative Stock Performance
The following graph compares the yearly percentage change in the cumulative total stockholder return on our common stock with the cumulative total return on the NASDAQ Global Market Composite Index and NASDAQ Biotechnology Index over the past five years. The comparisons assume $100 was invested on December 31, 2009 in our common stock, in the NASDAQ Global Market and the NASDAQ Biotechnology Index, and assumes reinvestment of dividends, if any.
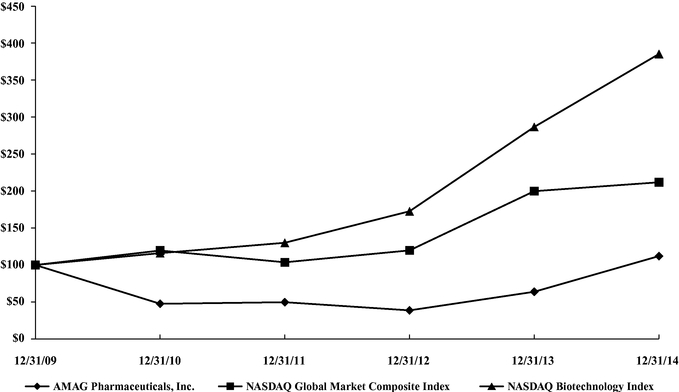
| | | | | | | | | | | | | | | | | | | |
| | 12/31/2009 | | 12/31/2010 | | 12/31/2011 | | 12/31/2012 | | 12/31/2013 | | 12/31/2014 | |
|---|
AMAG Pharmaceuticals, Inc. | | | 100.00 | | | 47.59 | | | 49.72 | | | 38.68 | | | 63.84 | | | 112.07 | |
NASDAQ Global Market Composite Index | | | 100.00 | | | 119.59 | | | 103.67 | | | 119.76 | | | 199.82 | | | 211.84 | |
NASDAQ Biotechnology Index | | | 100.00 | | | 116.06 | | | 130.08 | | | 172.67 | | | 286.67 | | | 385.29 | |
The stock price performance shown in this performance graph is not necessarily indicative of future price performance. Information used in the graph was obtained from Zach's Investment Research, Inc., a source we believe is reliable. However, we are not responsible for any errors or omissions in such information.
The material in this section captionedFive-Year Comparative and Stock Performance is being furnished and shall not be deemed "filed" with the SEC for purposes of Section 18 of the Exchange Act or otherwise subject to the liability of that section, nor shall the material in this section be deemed to be incorporated by reference in any registration statement or other document filed with the SEC under the Securities Act of 1933, except to the extent we specifically and expressly incorporate it by reference into such filing.
78
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA:
The following table sets forth selected financial data as of and for the years ended December 31, 2014, 2013, 2012, 2011 and 2010. The selected financial data set forth below has been derived from our audited financial statements. This information should be read in conjunction with the financial statements and the related notes thereto included in Part II, Item 8 of this Annual Report on Form 10-K and Management's Discussion and Analysis of Financial Condition and Results of Operations included in Part II, Item 7 of this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014(1) | | 2013 | | 2012 | | 2011 | | 2010 | |
|---|
| | (in thousands, except per share data)
| |
|---|
Statement of Operations Data | | | | | | | | | | | | | | | | |
Revenues: | | | | | | | | | | | | | | | | |
U.S. product sales, net | | $ | 108,795 | | $ | 71,362 | | $ | 58,287 | | $ | 52,097 | | $ | 59,339 | |
License fee and other collaboration revenues | | | 10,886 | | | 8,385 | | | 26,475 | | | 8,321 | | | 6,132 | |
Other product sales and royalties | | | 4,703 | | | 1,109 | | | 616 | | | 831 | | | 774 | |
| | | | | | | | | | | | | | | | | |
Total revenues | | | 124,384 | | | 80,856 | | | 85,378 | | | 61,249 | | | 66,245 | |
| | | | | | | | | | | | | | | | | |
Costs and expenses: | | | | | | | | | | | | | | | | |
Cost of product sales(2) | | | 20,306 | | | 11,960 | | | 14,220 | | | 10,531 | | | 7,606 | |
Research and development expenses | | | 24,160 | | | 20,564 | | | 33,296 | | | 58,140 | | | 54,462 | |
Selling, general and administrative expenses | | | 72,254 | | | 59,167 | | | 53,071 | | | 68,863 | | | 84,939 | |
Acquisition-related costs | | | 9,478 | | | 782 | | | — | | | — | | | — | |
Restructuring expenses | | | 2,023 | | | — | | | 2,215 | | | 3,508 | | | 2,224 | |
| | | | | | | | | | | | | | | | | |
Total costs and expenses | | | 128,221 | | | 92,473 | | | 102,802 | | | 141,042 | | | 149,231 | |
| | | | | | | | | | | | | | | | | |
Operating loss | | | (3,837 | ) | | (11,617 | ) | | (17,424 | ) | | (79,793 | ) | | (82,986 | ) |
| | | | | | | | | | | | | | | | | |
Other income (expense): | | | | | | | | | | | | | | | | |
Interest expense | | | (14,697 | ) | | — | | | — | | | — | | | — | |
Interest and dividend income, net | | | 975 | | | 1,051 | | | 1,286 | | | 1,747 | | | 1,741 | |
Gains on sales of assets | | | 103 | | | 924 | | | — | | | — | | | — | |
Gains (losses) on investments, net | | | 114 | | | 40 | | | (1,466 | ) | | (193 | ) | | 408 | |
Fair value adjustment of settlement rights | | | — | | | — | | | — | | | — | | | (788 | ) |
| | | | | | | | | | | | | | | | | |
Total other income (expense) | | | (13,505 | ) | | 2,015 | | | (180 | ) | | 1,554 | | | 1,361 | |
| | | | | | | | | | | | | | | | | |
Net income (loss) before income taxes | | | (17,342 | ) | | (9,602 | ) | | (17,604 | ) | | (78,239 | ) | | (81,625 | ) |
Income tax benefit(3) | | | 153,159 | | | — | | | 854 | | | 1,170 | | | 472 | |
| | | | | | | | | | | | | | | | | |
Net income (loss) | | $ | 135,817 | | $ | (9,602 | ) | $ | (16,750 | ) | $ | (77,069 | ) | $ | (81,153 | ) |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
Net income (loss) per share: | | | | | | | | | | | | | | | | |
Basic | | $ | 6.06 | | $ | (0.44 | ) | $ | (0.78 | ) | $ | (3.64 | ) | $ | (3.90 | ) |
Diluted | | $ | 5.45 | | $ | (0.44 | ) | $ | (0.78 | ) | $ | (3.64 | ) | $ | (3.90 | ) |
Weighted average shares outstanding used to compute net income (loss) per share: | | |
| | |
| | |
| | |
| | |
| |
Basic | | | 22,416 | | | 21,703 | | | 21,392 | | | 21,189 | | | 20,806 | |
Diluted | | | 25,225 | | | 21,703 | | | 21,392 | | | 21,189 | | | 20,806 | |
| | | | | | | | | | | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | | 2011 | | 2010 | |
|---|
| | (in thousands)
| |
|---|
Balance Sheet Data | | | | | | | | | | | | | | | | |
Working capital (current assets less current liabilities) | | $ | 107,548 | | $ | 211,284 | | $ | 221,423 | | $ | 201,037 | | $ | 254,073 | |
Total assets | | $ | 1,388,933 | | $ | 265,459 | | $ | 258,137 | | $ | 267,224 | | $ | 336,076 | |
Long-term liabilities | | $ | 762,492 | | $ | 59,930 | | $ | 52,383 | | $ | 47,634 | | $ | 54,079 | |
Stockholders' equity | | $ | 459,953 | | $ | 172,408 | | $ | 172,797 | | $ | 180,596 | | $ | 245,286 | |
- (1)
- Includes the results of operations of Lumara Health during the post-acquisition period from November 12, 2014 through December 31, 2014. See Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information regarding the November 2014 acquisition of Lumara Health.
- (2)
- Cost of product sales in 2014 includes approximately $6.1 million of non-cash expense related to the amortization of the step-up of Lumara's inventories and intangible assets to fair value at the acquisition date. See Note C, "Business
79
Table of Contents
Combinations," and Note I, "Goodwill and Intangible Assets, Net," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information.
- (3)
- The $153.2 million income tax benefit in 2014 reflects a $132.3 million decrease in our valuation allowance due to taxable temporary differences available as a source of income to realize the benefit of certain of our pre-existing deferred tax assets as a result of the acquisition of Lumara Health. See Note K, "Income Taxes," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information.
80
Table of Contents
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS:
Overview
AMAG Pharmaceuticals, Inc., a Delaware corporation, was founded in 1981. We are a specialty pharmaceutical company with a focus on maternal health, anemia and cancer supportive care. We currently market Makena® (hydroxyprogesterone caproate injection), Feraheme® (ferumoxytol) Injection for Intravenous ("IV") use and MuGard® Mucoadhesive Oral Wound Rinse. The primary goal of our company is to bring to market therapies that provide clear benefits and improve patients' lives.
Currently, our two primary sources of revenue are from the sale ofMakena andFeraheme. On November 12, 2014, we acquired Lumara Health Inc. ("Lumara Health"), a privately held pharmaceutical company specializing in women's health, for approximately $600.0 million in upfront cash consideration (subject to finalization of certain adjustments related to Lumara Health's financial position at the time of closing, including adjustments related to working capital, net debt and transaction expenses as set forth in the definitive agreement with Lumara Health (the "Lumara Agreement")) and approximately 3.2 million shares of our common stock having a fair value of approximately $112.0 million at the time of closing. The Lumara Agreement includes future contingent payments of up to $350.0 million in cash (or upon mutual agreement between us and the former Lumara Health security holders, future contingent payments may also be made in common stock or some combination thereof) payable by us to the former Lumara Health security holders based upon the achievement of certain sales milestones through calendar year 2019. In connection with the acquisition of Lumara Health, we acquiredMakena, a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. We sellMakena to specialty pharmacies and distributors, who, in turn sellMakena to healthcare providers, hospitals, government agencies and integrated delivery systems. Additional details regarding the Lumara Agreement can be found in Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K.
Feraheme was approved for marketing in the U.S. in June 2009 by the U.S. Food and Drug Administration (the "FDA") for use as an IV iron replacement therapy for the treatment of iron deficiency anemia ("IDA") in adult patients with chronic kidney disease ("CKD"). We began sellingFeraheme in the U.S. in July 2009 through our commercial organization, including a specialty sales force. We sellFeraheme to authorized wholesalers and specialty distributors, who in turn, sellFeraheme to healthcare providers who administerFeraheme primarily within hospitals, hematology and oncology centers, and nephrology clinics.
In addition to continuing to pursue opportunities to make new advancements in patients' health and to enhance treatment accessibility, we intend to continue to expand and diversify our portfolio through the in-license or purchase of additional pharmaceutical products or companies. We are seeking complementary products that will leverage our corporate infrastructure, sales force call points and commercial expertise, with a particular focus on maternal health specialists, hematology and oncology centers, nephrology clinics and hospitals. We are evaluating and plan to pursue commercial products as well as late-stage development assets. In addition, we are contemplating transactions that allow us to realize cost synergies to increase cash flows, as well as transactions that potentially optimize after-tax cash flows.
In June 2013, we entered into the License Agreement with PlasmaTech Biopharmaceuticals, Inc. (formerly known as Access Pharmaceuticals, Inc.), under which we acquired the U.S. commercial rights toMuGard for the management of oral mucositis (the "MuGard License Agreement"). Under the
81
Table of Contents
MuGard License Agreement, we obtained an exclusive, royalty-bearing license, with the right to grant sublicenses, to certain intellectual property rights, including know-how, patents and trademarks, to use, import, offer for sale, sell, manufacture and commercializeMuGard in the U.S. and its territories for the management of all diseases or conditions of the oropharyngeal cavity, including mucositis ("the "MuGard Rights"). See Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information on the MuGard License Agreement.
In June 2014, we proposed changes to the FDA related to our current U.S. label ofFeraheme based on a review of global post-marketing data to strengthen the warnings and precautions section of the label and mitigate the risk of serious hypersensitivity reactions, including anaphylaxis, in order to enhance patient safety. After considering our June 2014 submission and other information, in January 2015, the FDA notified us that it believes new safety information should be included in the labeling forFeraheme, including, among other things, a boxed warning to highlight the risks of serious hypersensitivity/anaphylaxis reactions and revisions thatFeraheme should only be administered through an IV infusion (i.e., not by IV injection) and should be contraindicated for patients with any known history of drug allergy. The FDA's recommended label changes go beyond what we proposed in June 2014. We plan to work with the FDA to finalize an updated U.S.Feraheme label.
In December 2012, we submitted a supplemental new drug application ("sNDA") to the FDA seeking approval forFeraheme for the treatment of IDA in adult patients who had failed or could not use oral iron. In January 2014, we received a complete response letter from the FDA for the sNDA informing us that our sNDA could not be approved in its present form and stating that we have not provided sufficient information to permit labeling ofFeraheme for safe and effective use for the proposed broader indication. The FDA indicated that its decision was based on the cumulative ferumoxytol data, including the global Phase III IDA program and global post-marketing safety reports for the currently indicated CKD patient population. The FDA suggested, among other things, that we submit additional clinical trial data in the proposed broad IDA patient population with a primary composite safety endpoint of serious hypersensitivity/anaphylaxis, cardiovascular events and death, events that are included in the labels ofFeraheme and other IV irons and that have been reported in the post-marketing environment forFeraheme. Additionally, the FDA proposed potentially evaluating alternative dosing and/or administration ofFeraheme as well as potential changes to labeling that would be intended to reduce the risk of serious hypersensitivity reactions associated withFeraheme. In June 2014, we met with the FDA to discuss our proposed approach to resolving the points that were raised in the complete response letter. Based on the FDA's feedback, we submitted a revised proposal that includes the design of a potential clinical trial, a safety endpoint for such trial and alternative methods of administration ofFeraheme. We expect to receive feedback from the FDA during 2015 and expect thereafter to be able to assess and determine the path forward, if any, forFeraheme in the broad IDA patient population in the U.S., including the related timing and cost of any clinical trials.
Further, in October 2014, we filed with the FDA a prior approval supplement to the originalMakena New Drug Application seeking approval of a 1 mL preservative-free vial ofMakena and we are seeking to expandMakena's formulations and drug delivery technologies as part of the product's lifecycle management program. We expect a decision in the second quarter of 2015.
Outside of the U.S., ferumoxytol has been granted marketing approval in the European Union ("EU"), Canada and Switzerland for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD. In March 2010, we entered into a License, Development and Commercialization Agreement (the "Takeda Agreement"), which was amended in June 2012 (the "Amended Takeda Agreement") with Takeda Pharmaceutical Company Limited ("Takeda"). On December 29, 2014, we entered into an agreement with Takeda to terminate the Amended Takeda
82
Table of Contents
Agreement and we will regain all worldwide development and commercialization rights forFeraheme following the transfer of marketing authorizations from Takeda to us (the "Takeda Termination Agreement"). Under the Amended Takeda Agreement, Takeda had an exclusive license to market and sell ferumoxytol in the EU, Canada, and Switzerland, as well as certain other geographic territories. The EU marketing authorization forRienso is valid in the 28 EU Member States as well as in Iceland, Liechtenstein and Norway. The trade name for ferumoxytol in Canada isFeraheme and outside of the U.S. and Canada the trade name isRienso. Additional details regarding the Takeda Termination Agreement can be found in Note R, "Collaborative Agreements," to our consolidated financial statements included in this Annual Report on Form 10-K.
Sales ofFeraheme/Rienso outside of the U.S. do not and are not expected to materially contribute to our revenues. As such, and in light of the Takeda Termination Agreement, we have been assessing various commercialization strategies forRienso in the EU and Switzerland andFeraheme in Canada. A number of considerations influence our analysis of our commercialization opportunities outside of the U.S., including (i) regulatory developments and the potential cost of post-approval clinical trial commitments and post-marketing obligations required by regulatory authorities outside of the U.S., (ii) the product's commercial viability (sales potential relative to the cost of maintaining the product on the market) in light of the current CKD label, the possible impact of future label changes, including any impact in the U.S., and the competitive landscape, and (iii) possible approaches in different geographies, which may include seeking a licensing or distribution partner or commercializing the product ourselves. Based on these considerations, we have come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
In the future, we may decide to seek to obtain a new marketing authorization for ferumoxytol in the EU, particularly if we generate additional clinical data to support potential approval in the broader IDA indication. There can be no assurance that we will be able to develop an approach that would be economically viable for us or a commercialization partner.
In February 2014 we issued $200.0 million of 2.5% convertible senior notes due February 15, 2019 (the "Convertible Notes"). Interest is payable semi-annually in arrears on February 15 and August 15 of each year, beginning on August 15, 2014. The initial conversion rate is 36.9079 shares of our common stock per $1,000 principal amount of the Convertible Notes, which corresponds to an initial conversion price of approximately $27.09 per share of our common stock and represents a conversion premium of approximately 35% based on the last reported sale price of our common stock of $20.07 on February 11, 2014, the date the Convertible Notes offering was priced. In addition, in connection with the pricing of the Convertible Notes and in order to reduce the potential dilution to our common stock and/or offset cash payments due upon conversion of the Convertible Notes, we also entered into convertible bond hedge and warrant transactions in February 2014. The initial exercise price of the warrants is $34.12 per share, subject to adjustment upon certain events, which is 70% above the last reported sale price of our common stock of $20.07 on February 11, 2014.
On November 12, 2014, in connection with the acquisition of Lumara Health, we entered into the Term Loan Facility, which provides for term loans in the aggregate principal amount of $340.0 million (the "Term Loan Facility"). We used $327.5 million of the Term Loan Facility proceeds to partially finance the $600.0 million cash portion of the Lumara Health acquisition. The Term Loan Facility bears interest, at our option, at either the Eurodollar rate plus a margin of 6.25% or the prime rate plus a margin of 5.25%. The Eurodollar rate is subject to a 1.00% floor and the prime rate is subject to a 2.00% floor. As of December 31, 2014 the stated interest rate was 7.25%. We must repay the Term Loan Facility in installments of (a) $8.5 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2015 through the quarter ending December 31, 2015, and
83
Table of Contents
(b) $12.8 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2016 through the quarter ending September 30, 2020, with the balance due in a final installment on November 12, 2020. The Term Loan Facility matures on November 12, 2020, except that the Term Loan Facility will mature on September 30, 2018 if:
- (a)
- more than $25.0 million in aggregate principal amount of our Convertible Notes remain outstanding and not converted to common stock or refinanced and replaced with debt that matures following, and has no amortization prior to, the date that is six and one half years following the closing date; and
- (b)
- the aggregate principal amount of the Term Loan Facility (including all undrawn incremental commitments) is greater than $50.0 million on and as of such date.
See Note S, "Debt," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information regarding the Convertible Notes, the bond hedge and warrant transactions, as well as the Term Loan Facility.
Our common stock trades on the NASDAQ Global Select Market ("NASDAQ") under the trading symbol "AMAG."
Critical Accounting Policies
Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. The preparation of these financial statements requires management to make certain estimates and assumptions that affect the reported amount of assets, liabilities, revenues and expenses, and the related disclosure of contingent assets and liabilities. The most significant estimates and assumptions are used to determine amounts and values of, but are not limited to:, revenue related to product sales and collaboration agreements, product sales allowances and accruals, potential other-than-temporary impairment of investments; acquisition date fair value and subsequent fair value estimates used to assess impairment of long-lived assets, including goodwill, in-process research and development ("IPR&D") and other intangible assets; contingent consideration; debt obligations; accrued expenses, income taxes and equity-based compensation expense. Actual results could differ materially from those estimates. In making these estimates and assumptions, management employs critical accounting policies. Our critical accounting policies include revenue recognition and related sales allowances and accruals, valuation of investments, equity-based compensation, business combinations, including goodwill, intangible assets and acquisition-related contingent consideration, and income taxes.
1. Revenue Recognition and Related Sales Allowances and Accruals
We recognize revenue from the sale of our products as well as license fee and other collaboration revenues, including milestone payments, other product sale revenues, and royalties we receive from our licensees. We recognize revenue in accordance with current accounting guidance related to the recognition, presentation and disclosure of revenue in financial statements, which outlines the basic criteria that must be met to recognize revenue and provides guidance for disclosure of revenue in financial statements. We recognize revenue when:
- •
- Persuasive evidence of an arrangement exists;
- •
- Delivery of product has occurred or services have been rendered;
- •
- The sales price charged is fixed or determinable; and
- •
- Collection is reasonably assured.
84
Table of Contents
U.S. Product Sales, Net
We record product sales allowances and accruals related to prompt payment discounts, chargebacks, government and other rebates, distributor, wholesaler and group purchasing organization ("GPO") fees, and product returns as a reduction of revenue in our consolidated statement of operations at the time product sales are recorded. Calculating these gross to net sales adjustments involves estimates and judgments based primarily on actual product sales data, forecasted customer buying patterns, and market research data related to utilization rates by various end-users. In addition, we also monitor our distribution channel to determine whether additional allowances or accruals are required based on inventory in our sales channel.
Classification of U.S. Product Sales Allowances and Accruals
Product sales allowances and accruals are primarily comprised of both direct and indirect fees, discounts and rebates, and provisions for estimated product returns. Direct fees, discounts and rebates are contractual fees and price adjustments payable to wholesalers, specialty distributors and other customers that purchase products directly from us. Indirect fees, discounts and rebates are contractual price adjustments payable to healthcare providers and organizations, such as certain physicians, clinics, hospitals, GPOs, and dialysis organizations that typically do not purchase products directly from us but rather from wholesalers and specialty distributors. In accordance with guidance related to accounting for fees and consideration given by a vendor to a customer, including a reseller of a vendor's products, these fees, discounts and rebates are presumed to be a reduction of the selling price. Product sales allowances and accruals are based on definitive contractual agreements or legal requirements (such as Medicaid laws and regulations) related to the purchase and/or utilization of the product by these entities and are recorded in the same period that the related revenue is recognized. We estimate product sales allowances and accruals using either historical, actual and/or other data, including estimated patient usage, applicable contractual rebate rates, contract performance by the benefit providers, other current contractual and statutory requirements, historical market data based upon experience of our products and other products similar to them, specific known market events and trends such as competitive pricing and new product introductions, current and forecasted customer buying patterns and inventory levels, and the shelf life of our products. As part of this evaluation, we also review changes to federal and other legislation, changes to rebate contracts, changes in the level of discounts, and changes in product sales trends. Although allowances and accruals are recorded at the time of product sale, certain rebates are typically paid out, on average, up to three months or longer after the sale.
Allowances against receivable balances primarily relate to prompt payment discounts, provider chargebacks and certain government agency rebates and are recorded at the time of sale, resulting in a reduction in product sales revenue and the reporting of product sales receivables net of allowances. Accruals related to Medicaid and provider volume rebates, wholesaler and distributor fees, GPO fees, other discounts to healthcare providers and product returns are recorded at the time of sale, resulting in a reduction in product sales revenue and the recording of an increase in accrued expenses.
We typically offer a 2% prompt payment discount to our customers as an incentive to remit payment in accordance with the stated terms of the invoice, generally thirty days. Because we anticipate that those customers who are offered this discount will take advantage of the discount, we accrue 100% of the prompt payment discount, at the time of sale, based on the gross amount of each invoice. We adjust the accrual quarterly to reflect actual experience.
85
Table of Contents
Chargeback reserves represent our estimated obligations resulting from the difference between the prices at which we sell our products to wholesalers and the sales price ultimately paid to wholesalers under fixed price contracts by third-party payers, including governmental agencies. We determine our chargeback estimates based on actual product sales data and forecasted customer buying patterns. Actual chargeback amounts are determined at the time of resale to the qualified healthcare provider, and we generally issue credits for such amounts within several weeks of receiving notification from the wholesaler. Estimated chargeback amounts are recorded at the time of sale, and we adjust the allowance quarterly to reflect actual experience.
Government and other rebate reserves relate to our reimbursement arrangements with state Medicaid programs, and contractual or performance rebate agreements with certain classes of trade. We determine our estimates for Medicaid rebates, if applicable, based on actual product sales data and our historical product claims experience. In estimating these reserves, we provide for a Medicaid rebate associated with both those expected instances where Medicaid will act as the primary insurer as well as in those instances where we expect Medicaid will act as the secondary insurer. For rebates associated with reaching defined performance goals, we determine our estimates using actual product sales data and forecasted customer buying and utilization patterns. Rebate amounts generally are invoiced quarterly and are paid in arrears, and we expect to pay such amounts within several weeks of notification by the Medicaid or provider entity. Estimated government and other rebates are recorded at the time of sale and, with the exception of Medicaid as discussed below, we adjust the accrual quarterly to reflect actual experience.
During 2013 and 2012, we revised our estimatedFeraheme Medicaid reserve rate based on actual product-specific rebate claims received since the 2009 launch ofFeraheme, our expectations of state level activities, and estimated rebate claims not yet submitted, which resulted in a reduction of our then estimated Medicaid rebate reserve related to prior periodFeraheme sales of $0.6 million in each of the respective years. These changes in estimates were reflected as an increase in our net product sales for 2013 and 2012 and resulted in reductions to our gross to net percentages in those periods. The reduction of our estimated Medicaid rebate reserve had an impact of $0.03 per basic and diluted share for each of the respective years. We regularly assess our Medicaid reserve balance and the rate at which we accrue for claims against product sales. If we determine in future periods that our actual rebate experience is not indicative of expected claims, or if actual claims experience changes, or if other factors affect estimated claims rates, we may be required to adjust our current Medicaid accumulated reserve estimate, which would affect net product sales in the period of the adjustment and could be significant.
A 1% increase in our estimate of our Medicaid utilization rate for 2014 would have resulted in approximately a $0.4 million decrease in net product sales.
Fees under our arrangements with distributors and wholesalers are usually based upon units of product purchased during the prior month or quarter and are usually paid by us within several weeks of our receipt of an invoice from the wholesaler or distributor, as the case may be. Fees under our arrangements with GPOs are usually based upon member purchases during the prior quarter and are generally billed by the GPO within 30 days after period end. Current accounting standards related to consideration given by a vendor to a customer, including a reseller of a vendor's products, specify that cash consideration given by a vendor to a customer is presumed to be a reduction of the selling price of the vendor's products or services and therefore should be characterized as a reduction of revenue.
86
Table of Contents
Consideration should be characterized as a cost incurred if we receive, or will receive, an identifiable benefit (goods or services) in exchange for the consideration and we can reasonably estimate the fair value of the benefit received. Because the fees we pay to wholesalers do not meet the foregoing conditions to be characterized as a cost, we have characterized these fees as a reduction of revenue. We generally pay such amounts within several weeks of our receipt of an invoice from the distributor, wholesaler GPO. Accordingly, we accrue the estimated fee due at the time of sale, based on the contracted price invoiced to the customer. We adjust the accrual quarterly to reflect actual experience.
Consistent with industry practice, we generally offer our wholesalers, specialty distributors and other customers a limited right to return our products based on the product's expiration date. Currently, the expiration dates forFeraheme in the U.S.,Makena andMuGard are five years, three years and three years, respectively. We estimate product returns based on the historical return patterns and known or expected changes in the marketplace. We track actual returns by individual production lots. Returns on lots eligible for credits under our returned goods policy are monitored and compared with historical return trends and rates.
We expect that wholesalers and healthcare providers will not stock significant inventory due to the cost of the product, the expense to store our products and/or that our products are readily available for distribution. We record an estimate of returns at the time of sale. If necessary, our estimated rate of returns may be adjusted for actual return experience as it becomes available and for known or expected changes in the marketplace. During 2014, we reduced our reserve forFeraheme product returns by approximately $1.8 million, primarily as a result of a lower than expected rate of product returns. We did not significantly adjust our reserve for product returns during 2013. During 2012, we reduced our reserve forFeraheme product returns by approximately $2.2 million, primarily as a result of a lower than expected rate of product returns as well as the lapse of the product return period on certain manufacturedFeraheme lots. The reduction of our estimated product returns reserve had a positive impact of $0.12 and $0.14 per basic and diluted share, respectively, in 2014 and $0.10 per basic and diluted share in 2012. To date, returns ofFeraheme have been relatively limited; however, returns experience may change over time. As we continue to gain more historical experience with actual returns forFeraheme and gain additional experience with return rates forMakena, we may be required to make a future adjustment to our product returns estimate, which would result in a corresponding change to our net product sales in the period of adjustment and could be significant. A 1% increase in our returns as a percentage of gross sales for the year ended December 31, 2014, would have resulted in approximately a $1.8 million decrease in net product sales.
Other product sales and royalties primarily includedFeraheme product sales to Takeda and royalties from Takeda as well as net product sales ofMuGard. Prior to the Takeda Termination Agreement, as discussed and defined below, we recorded all product sales ofFeraheme sold to Takeda in deferred revenues in our consolidated balance sheet. We recognized these deferred product revenues, and the associated cost of product sales, in our consolidated statement of operations at the time Takeda reported to us that sales had been made to its customers. At December 31, 2014, as the result of the termination of the Amended Takeda Agreement, we recognized these remaining balances of deferred product revenues and associated cost of product sales.
From time to time, we may enter into collaborative license and development agreements with biotechnology and pharmaceutical companies for the development and commercialization of our products. The terms of the agreements may include non-refundable license fees, payments based on the
87
Table of Contents
achievement of certain milestones and performance goals, reimbursement of certain out-of-pocket costs, payments for manufacturing services, and royalties on product sales.
We evaluate revenue from arrangements that have multiple elements to determine whether the components of the arrangement represent separate units of accounting as defined in the accounting guidance related to revenue arrangements with multiple deliverables. Under current accounting guidance, which governs any agreements that contain multiple elements that are either entered into or materially modified subsequent to January 1, 2011, companies are required to establish the fair value of undelivered products and services based on a separate revenue recognition process using management's best estimate of the selling price for an undelivered item when there is no vendor-specific objective evidence or third-party evidence to determine the fair value of that undelivered item. Agreements entered into prior to January 1, 2011, that have not been materially modified are accounted for under previous accounting guidance, which provides that an element of a contract can be accounted for separately if the delivered elements have standalone value and the fair value of all undelivered elements is determinable. If an element is considered to have standalone value but the fair value of any of the undelivered items cannot be determined, all elements of the arrangement are recognized as revenue as a single unit of accounting over the period of performance for such undelivered items or services. Significant management judgment is required in determining what elements constitute deliverables and what deliverables or combination of deliverables should be considered units of accounting.
When multiple deliverables are combined and accounted for as a single unit of accounting, we base our revenue recognition pattern on the last to be delivered element. Revenue is recognized using either a proportional performance or straight-line method, depending on whether we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and whether such performance obligations are provided on a best-efforts basis. To the extent we cannot reasonably estimate our performance obligations, we recognize revenue on a straight-line basis over the period we expect to complete our performance obligations. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement. We may have to revise our estimates based on changes in the expected level of effort or the period we expect to complete our performance obligations.
Our collaboration agreements may entitle us to additional payments upon the achievement of performance-based milestones. If a milestone involves substantive effort on our part and its achievement is not considered probable at the inception of the collaboration, we recognize the milestone consideration as revenue in the period in which the milestone is achieved only if it meets the following additional criteria:
- •
- The milestone consideration received is commensurate with either the level of effort required to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from our performance to achieve the milestone;
- •
- The milestone is related solely to our past performance; and
- •
- The milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement.
There is significant judgment involved in determining whether a milestone meets all of these criteria. For milestones that do not meet the above criteria and are therefore not considered substantive milestones, we recognize that portion of the milestone payment equal to the percentage of the performance period completed at the time the milestone is achieved and the above conditions are met. The remaining portion of the milestone will be recognized over the remaining performance period using a proportional performance or straight-line method.
88
Table of Contents
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in our consolidated balance sheets. Amounts not expected to be recognized within the next 12 months are classified as long-term deferred revenue.
See Note R,"Collaborative Agreements" to our consolidated financial statements included in this Annual Report on Form 10-K for additional information regarding our prior collaboration agreement with Takeda.
2. Valuation of investments
We account for and classify our investments as either "available-for-sale," "trading," or "held-to-maturity," in accordance with current guidance related to the accounting and classification of certain investments in debt and equity securities. The determination of the appropriate classification by us is based on a variety of factors, including management's intent at the time of purchase. As of December 31, 2014 and 2013, all of our investments were classified as available-for-sale securities.
Available-for-sale securities are those securities which we view as available for use in current operations, if needed. We generally classify our available-for-sale securities as short-term investments, even though the stated maturity date may be one year or more beyond the current balance sheet date. Available-for-sale investments are stated at fair value with their unrealized gains and losses included as a separate component of stockholders' equity entitled "Accumulated other comprehensive loss," until such gains and losses are realized or until an unrealized loss is considered other-than-temporary.
We recognize and report other-than-temporary impairments of our debt securities in accordance with current accounting guidance, which requires that for debt securities with a decline in fair value below amortized cost basis, an other-than-temporary impairment exists if (a) we have the intent to sell the security or (b) it is more likely than not that we will be required to sell the security prior to recovery of its amortized cost basis. If either of these conditions is met, we recognize the difference between the amortized cost of the security and its fair value at the impairment measurement date in our consolidated statement of operations. If neither of these conditions is met, we must perform additional analyses to evaluate whether the unrealized loss is associated with the creditworthiness of the security rather than other factors, such as interest rates or market factors. These factors include evaluation of the security, issuer and other factors such as the duration of the period that, and extent to which, the fair value was less than cost basis, the financial health of and business outlook for the issuer, including industry and sector performance, operational and financing cash flow factors, overall market conditions and trends, underlying collateral, whether we have a favorable history in redeeming similar securities at prices at or above fair value, and credit ratings with respect to our investments provided by investments ratings agencies. If we determine from this analysis that we do not expect to receive cash flows sufficient to recover the entire amortized cost of the security, a credit loss exists. In this situation, the impairment is considered other-than-temporary and is recognized in our consolidated statement of operations.
We also analyze when the volume and level of activity for an asset or liability have significantly decreased and when circumstances indicate that a transaction may not be considered orderly. In order to determine whether the volume and level of activity for an asset or liability have significantly decreased, we assess current activity as compared to normal market activity for the asset or liability. We rely on many factors such as trading volume, trading frequency, the levels at which market participants indicate their willingness to buy and sell our securities, as reported by market participants, and current market conditions. Using professional judgment and experience, we evaluate and weigh the relevance and significance of all applicable factors to determine if there has been a significant decrease in the volume and level of activity for an asset, group of similar assets or liabilities. Similarly, in order to identify transactions that are not orderly, we take into consideration the activity in the market which can influence the determination and occurrence of an orderly transaction. Also, we inquire as to
89
Table of Contents
whether there may have been restrictions on the marketing of the security to a single or limited number of participants. Where possible, we assess the financial condition of the seller to determine whether observed transactions may have been forced. If there is a significant disparity between the trading price for a security held by us as compared to the trading prices of similar recent transactions, we consider whether this disparity is an indicator of a disorderly trade. Using professional judgment and experience, we evaluate and weigh the relevance and significance of all applicable factors to determine if the evidence suggests that a transaction or group of similar transactions is not orderly. Based upon these procedures, we determined that market activity for our assets appeared normal and that transactions did not appear disorderly as of December 31, 2014 and 2013. Please see Note D, "Investments," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information regarding our investments.
3. Equity-Based Compensation
Under the fair value recognition guidance of equity-based compensation accounting rules, equity-based compensation cost is generally required to be measured at the grant date (based upon an estimate of the fair value of the compensation granted) and recorded to expense over the requisite service period, which generally is the vesting period. Because equity-based compensation expense is based on awards ultimately expected to vest, we must make certain judgments about whether employees and directors will complete the requisite service period. Accordingly, we have reduced the compensation expense being recognized for estimated forfeitures. Under current accounting guidance, forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Forfeitures are estimated based upon historical experience, adjusted for unusual events such as corporate restructurings, which can result in higher than expected turnover and forfeitures. If factors change and we employ different assumptions in future periods, the compensation expense that we record in the future may differ significantly from what we have recorded in the current period.
We estimate the fair value of equity-based compensation involving stock options based on the Black-Scholes option pricing model. We estimate the fair value of our restricted stock units ("RSUs") whose vesting is contingent upon market conditions using the Monte-Carlo simulation model. These models require the input of several factors such as the expected option term, the expected risk-free interest rate over the expected option term, the expected volatility of our stock price over the expected option term, and the expected dividend yield over the expected option term and are subject to various assumptions. The fair value of awards whose fair values are calculated using the Black-Scholes option pricing model is generally being amortized on a straight-line basis over the requisite service period and is recognized based on the proportionate amount of the requisite service period that has been rendered during each reporting period. The fair value of awards with market conditions is being amortized based upon the estimated derived service period. We believe our valuation methodologies are appropriate for estimating the fair value of the equity awards we grant to our employees and directors. Our equity award valuations are estimates and thus may not be reflective of actual future results or amounts ultimately realized by recipients of these grants. These amounts, and the amounts applicable to future quarters, are also subject to future quarterly adjustments based upon a variety of factors, which include, but are not limited to, changes in estimated forfeiture rates and the issuance of new equity-based awards. The fair value of RSUs granted to our employees and directors is determined based upon the quoted closing market price per share on the date of grant, adjusted for estimated forfeitures.
4. Business Combinations
We account for acquired businesses using the acquisition method of accounting, under which the total purchase price of an acquisition is allocated to the net tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values as of the date of acquisition.
90
Table of Contents
Acquisition-related costs are expensed as incurred. Any excess of the consideration transferred over the estimated fair values of the net assets acquired is recorded as goodwill. See Note C, "Business Combinations," and Note I, "Goodwill and Intangible Assets, Net," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information.
The purchase price allocations are initially prepared on a preliminary basis and are subject to change as additional information becomes available concerning the fair value and tax basis of the assets acquired and liabilities assumed. Any adjustments to the purchase price allocations are made as soon as practicable but no later than one year from the acquisition date.
Acquired inventory is recorded at its fair value, which generally requires a step-up adjustment to recognize the inventory at its expected net realizable value. The inventory step-up is recorded to cost of sales in our consolidated statements of operations as the related inventory is sold.
Goodwill represents the excess purchase price paid in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized, but is reviewed for impairment on an annual basis or more frequently if indicators of impairment are present. We determine whether goodwill may be impaired by comparing the carrying value of the reporting unit, including goodwill, to the fair value of the reporting unit. If the fair value is less than the carrying amount, a more detailed analysis is performed to determine whether goodwill is impaired. The impairment loss, if any, is measured as the excess of the carrying value of the goodwill over the implied value of the goodwill and is recorded in our consolidated statements of operations.
Finite-lived intangible assets are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. When such facts and circumstances exist, management compares the projected undiscounted future cash flows associated with the asset over its estimated useful life against the carrying amount. The impairment loss, if any, is measured as the excess of the carrying amount of the asset over its fair value.
Acquired IPR&D represents the fair value assigned to research and development assets that we acquire that have not been completed at the date of acquisition. The fair value of IPR&D acquired in a business combination is capitalized on our consolidated balance sheets at the acquisition-date fair value and is determined by estimating the costs to develop the technology into commercially viable products, estimating the resulting revenue from the projects, and discounting the net cash flows to present value. IPR&D is not amortized, but is reviewed for impairment on an annual basis or more frequently if indicators of impairment are present, until completion or abandonment of the projects. If we determine that IPR&D becomes impaired or is abandoned, the carrying value of the IPR&D is written down to its fair value with the related impairment charge recognized in our consolidated statement of operations in the period in which the impairment occurs. Upon successful completion of each project and launch of the product, we will make a separate determination of the estimated useful life of the IPR&D intangible asset and the related amortization will be recorded as an expense prospectively over its estimated useful life.
The projected discounted cash flow models used to estimate our IPR&D reflect significant assumptions regarding the estimates a market participant would make in order to evaluate a drug development asset including the following:
- •
- Probability of successfully completing clinical trials and obtaining regulatory approval;
- •
- Market size, market growth projections, and market share;
91
Table of Contents
- •
- Estimates regarding the timing of and the expected costs to advance our clinical programs to commercialization;
- •
- Estimates of future cash flows from potential product sales; and
- •
- A discount rate.
Contingent consideration arising from a business combination is included as part of the purchase price and is recognized at fair value as of the acquisition date. Subsequent to the acquisition date, we measure contingent consideration arrangements at fair value for each period until the contingency is resolved. These changes in fair value are recognized in our consolidated statements of operations. Changes in fair values reflect new information about the likelihood of the payment of the contingent consideration and the passage of time.
5. Income Taxes
We use the asset and liability method of accounting for deferred income taxes. Under this method, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. A deferred tax asset is established for the expected future benefit of net operating loss ("NOL") and credit carryforwards. Deferred tax assets and liabilities are measured using enacted rates in effect for the year in which those temporary differences are expected to be recovered or settled. A valuation allowance against net deferred tax assets is required if, based on available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. Significant judgments, estimates and assumptions regarding future events, such as the amount, timing and character of income, deductions and tax credits, are required in the determination of our provision for income taxes and whether valuation allowances are required against deferred tax assets. In evaluating our ability to recover our deferred tax assets, we consider all available evidence, both positive and negative, including the existence of taxable temporary differences, our past operating results, the existence of cumulative income in the most recent fiscal years, changes in the business in which we operate and our forecast of future taxable income. In determining future taxable income, we are responsible for assumptions utilized including the amount of state and federal operating income, the reversal of temporary differences and the implementation of feasible and prudent tax planning strategies. These assumptions require significant judgment about the forecasts of future taxable income and are consistent with the plans and estimates that we are using to manage the underlying businesses. As of December 31, 2014, we maintained a partial valuation allowance on the net deferred tax assets as we benefitted only those deferred tax assets to the extent we had existing taxable temporary differences of the appropriate character turning within the carryforward period of the existing deferred tax assets.
We account for uncertain tax positions using a "more-likely-than-not" threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. We evaluate uncertain tax positions on a quarterly basis and adjust the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. Any changes to these estimates, based on the actual results obtained and/or a change in assumptions, could impact our income tax provision in future periods. Interest and penalty charges, if any, related to unrecognized tax benefits would be classified as a provision for income tax in our consolidated statement of operations. See Note K, "Income Taxes," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information.
92
Table of Contents
Impact of Recently Issued and Proposed Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board or other standard setting bodies that are adopted by us as of the specified effective date. Unless otherwise discussed, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
Results of Operations—2014 as compared to 2013
Revenues
Total revenues for 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | | |
| | Years Ended December 31, | | 2014 to 2013 change | |
|---|
| | 2014 | | 2013 | | $ Change | | % Change | |
|---|
Makena sales, net | | $ | 22,513 | | $ | — | | $ | 22,513 | | | N/A | |
U.S.Feraheme sales, net | | | 86,282 | | | 71,362 | | | 14,920 | | | 21% | |
License fee and other collaboration revenues | | | 10,886 | | | 8,385 | | | 2,501 | | | 30% | |
Other product sales and royalties | | | 4,703 | | | 1,109 | | | 3,594 | | | >100% | |
| | | | | | | | | | | | | | |
Total | | $ | 124,384 | | $ | 80,856 | | $ | 43,528 | | | 54% | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Our total revenues in 2014 increased by $43.5 million, or 54%, as compared to 2013, primarily as the result of $22.5 million ofMakena net product sales following our November 2014 acquisition of Lumara Health, as discussed above, and a $14.9 million increase in U.S.Feraheme net product sales. In addition and as discussed below, included in our net product sales for 2014 and 2013 was a $1.8 million reduction in our reserves forFeraheme product returns and a $0.6 million reduction of our estimated Medicaid rebate reserve related to priorFeraheme sales, respectively.
The following table sets forth customers who represented 10% or more of our total revenues for 2014 and 2013:
| | | | | | | |
| | Years Ended
December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
AmerisourceBergen Drug Corporation | | | 34% | | | 41% | |
McKesson Corporation | | | 21% | | | 24% | |
Cardinal Health, Inc. | | | 15% | | | 16% | |
Takeda Pharmaceuticals Company Limited | | | 11% | | | 11% | |
93
Table of Contents
Makena Product Sales, Net
Makena product sales and product sales allowances and accruals from November 12, 2014 through December 31, 2014 consisted of the following (in thousands):
| | | | | | | |
| | Year Ended December 31, 2014 | | Percent of gross Makena product sales | |
|---|
GrossMakena product sales | | $ | 35,718 | | | | |
Less provision for product sales allowances and accruals: | | | | | | | |
Discounts and chargebacks | | | 3,451 | | | 10% | |
Government and other rebates | | | 9,665 | | | 27% | |
Returns | | | 89 | | | 0% | |
| | | | | | | | |
Total | | | 13,205 | | | 37% | |
| | | | | | | | |
NetMakena product sales | | $ | 22,513 | | | | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
U.S. Feraheme Product Sales, Net
U.S.Feraheme product sales and product sales allowances and accruals for 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | |
| |
| |
|---|
| | 2014 | | Percent of
gross U.S.
Feraheme
product sales | | 2013 | | Percent of
gross U.S.
Feraheme
product sales | | $ Change | | % Change | |
|---|
Gross U.S.Feraheme product sales | | $ | 152,428 | | | | | $ | 119,712 | | | | | $ | 32,716 | | | 27% | |
Less provision for product sales allowances and accruals: | | | | | | | | | | | | | | | | | | | |
Discounts and chargebacks | | | 51,969 | | | 34% | | | 37,098 | | | 31% | | | | | | | |
Government and other rebates and other fees | | | 15,426 | | | 10% | | | 10,868 | | | 9% | | | | | | | |
Medicaid rebate reserve adjustment | | | — | | | 0% | | | (568 | ) | | 0% | | | | | | | |
Returns | | | (1,249 | ) | | –1% | | | 952 | | | 1% | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Total | | | 66,146 | | | 43% | | | 48,350 | | | 40% | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Net U.S.Feraheme product sales | | $ | 86,282 | | | | | $ | 71,362 | | | | | $ | 14,920 | | | 21% | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Our grossFeraheme U.S. product sales increased by $32.7 million, or 27%, during 2014 as compared to 2013. Of the $32.7 million increase in gross U.S.Feraheme sales, $17.4 million was due to price increases and $15.3 million was due to increased units sold. This increase was partially offset by $19.0 million of additional allowances and accruals in 2014, excluding a $1.8 million reduction in our reserves forFeraheme product returns in 2014 and a $0.6 million reduction of our estimated Medicaid rebate reserve related to priorFeraheme sales in 2013. As a result, total net U.S.Feraheme product sales increased by $14.9 million, or 21%, during 2014 as compared to 2013.
94
Table of Contents
TotalFeraheme discounts and chargebacks for 2014 were $52.0 million, or 34% of total gross U.S.Feraheme product sales, as compared to $37.1 million, or 31%, in 2013. The increase in total discounts and chargebacks as a percentage of total gross U.S.Feraheme product sales was related primarily to a change in our customer mix.
TotalFeraheme government and other rebates (excluding any changes in estimates related to Medicaid rebate reserves) were $15.4 million, or 10% of total gross U.S.Feraheme product sales, in 2014 as compared to $10.9 million, or 9%, in 2013. The increase in total government and other rebates as a percentage of gross U.S.Feraheme product sales was related primarily to increased sales to clinics and hospitals that had volume or market share contracts with us during 2014 as compared to 2013 and changes in the structure of our performance-based rebate programs.
We are subject to reimbursement arrangements with state Medicaid programs for which we estimate and record rebate reserves. We determine our estimates from Medicaid rebates based on actual product sales data and our historical product claims experience. During 2013, we reduced our estimated Medicaid rebate reserve related to priorFeraheme sales by approximately $0.6 million based on actual product-specific rebate claims received since the July 2009 launch ofFeraheme, our expectations of state level activity, and estimated rebate claims not yet submitted. The $0.6 million Medicaid rebate reserves adjustment resulted in an increase to product sales during that period.
We generally offer our wholesalers, specialty distributors and other customers a limited right to return our products purchased directly from us, principally based on the product's expiration date which, once packaged is currently five years in the U.S forFeraheme and three years forMakena. Reserves for product returns are recorded in the period the related revenue is recognized, resulting in a reduction to product sales. We evaluate our estimated product returns rate each period based on the historical return patterns and known or expected changes in the marketplace. During 2014, we reduced our reserve forFeraheme product returns by approximately $1.8 million, primarily as the result of a lower than expected rate of product returns. As a result, the product returns provision applied to gross product sales for 2014 was a credit of $1.2 million, resulting in an increase to product sales. There were no significant adjustments to our reserve for product returns in 2013.
We regularly assess our Medicaid and product return reserve balances and accrual rates. If we determine in future periods that our actual rebate or returns experience is not indicative of expected claims or returns, if our actual claims or returns experience changes, or if other factors affect estimated claims or returns rates, we may be required to change our reserve or reserve estimates and/or the current rates at which we estimate Medicaid reserves or returns, which would affect our earnings in the period of the change and could be significant.
95
Table of Contents
An analysis of the amount ofMakena product reserves for 2014 and the amount and change inFeraheme product reserves for 2014 and 2013 is as follows (in thousands):
| | | | | | | | | | | | | |
| | Discounts and
Chargebacks | | Government
and Other
Rebates | | Returns | | Total | |
|---|
Balance at January 1, 2013 | | $ | 1,771 | | $ | 2,430 | | $ | 1,018 | | $ | 5,219 | |
| | | | | | | | | | | | | | |
Current provisions relating to sales in current year | | | 37,098 | | | 10,868 | | | 952 | | | 48,918 | |
Adjustments relating to sales in prior years | | | — | | | (568 | ) | | — | | | (568 | ) |
Payments/returns relating to sales in current year | | | (34,538 | ) | | (8,194 | ) | | — | | | (42,732 | ) |
Payments/returns relating to sales in prior years | | | (1,648 | ) | | (1,699 | ) | | (8 | ) | | (3,355 | ) |
| | | | | | | | | | | | | | |
Balance at December 31, 2013 | | $ | 2,683 | | $ | 2,837 | | $ | 1,962 | | $ | 7,482 | |
| | | | | | | | | | | | | | |
Current provisions relating to sales in current year | | | 59,372 | | | 52,468 | | | 1,391 | | | 113,231 | |
Adjustments relating to sales in prior years | | | — | | | — | | | (1,780 | ) | | (1,780 | ) |
Payments/returns relating to sales in current year | | | (47,729 | ) | | (10,771 | ) | | — | | | (58,500 | ) |
Payments/returns relating to sales in prior years | | | (2,851 | ) | | (2,126 | ) | | (229 | ) | | (5,206 | ) |
| | | | | | | | | | | | | | |
Balance at December 31, 2014 | | $ | 11,475 | | $ | 42,408 | | $ | 1,344 | | $ | 55,227 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
During 2014 and 2013, we implemented gross price increases forFeraheme, some portion of which were discounted back to customers under volume or market share based contracts. When portions of price increases are discounted back to customers, it can have the effect of widening the gross to net adjustment percentage while still resulting in a greater net price per gram.
In 2015, we expect discounts, chargebacks and government and other rebates to continue to increase as a percentage of gross sales due to our contracting and discounting strategy and the mix of business for our products and increasing competitive pressure forFeraheme.
The Health Care and Education Reconciliation Act of 2010 (the "Healthcare Reform Act") was enacted in the U.S. in March 2010 and includes certain cost containment measures including an increase to the minimum rebates for products covered by Medicaid programs and the extension of such rebates to drugs dispensed to Medicaid beneficiaries enrolled in Medicaid managed care organizations as well as the expansion of the 340B Drug Discount Program under the Public Health Service Act. This legislation contains provisions that can affect the operational results of companies in the pharmaceutical industry and healthcare related industries, including us, by imposing on them additional costs.
The Healthcare Reform Act also requires pharmaceutical manufacturers to pay a prorated share of the overall Branded Drug Fee, based on the dollar value of its branded prescription drug sales to certain federal programs identified in the legislation. The amount of our annual share of the Branded Drug Fee for each of the 2014 and 2013 annual periods was less than $0.1 million and these payments were non-deductible for income tax purposes. We have included these amounts in selling, general and administrative expense in our consolidated statements of operations. The amount of this annual payment could increase in future years due to both higher eligibleFeraheme sales and the increasing amount of the overall fee assessed across manufacturers, but any such increases are not expected to be material to our results of operations or financial condition. In addition, although the Healthcare Reform Act exempts "orphan drugs" such asMakena from 340B ceiling price requirements, on July 21, 2014, the Health Resources and Services Administration, which administers the 340B program, issued an interpretive rule to implement the orphan drug exception which interprets the orphan drug exception narrowly. It exempts orphan drugs from the ceiling price requirements for certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community
96
Table of Contents
hospitals only when the orphan drug is used for its orphan indication. The entities are entitled to purchase orphan drugs at the ceiling price when the orphan drug is not used for its orphan indication. There is ongoing litigation challenging the interpretive rule as inconsistent with the statutory language and it is unclear at this time what, if any, impact the final outcome will have on our futureMakena sales.
In addition, the number of 340B institutions, which provide drugs at reduced rates, was expanded by the Healthcare Reform Act to include additional hospitals. As a result, the volume ofFeraheme business sold to 340B eligible entities has increased since the implementation of the Healthcare Reform Act.Feraheme sold to 340B eligible entities comprised approximately 17% and 15% of our totalFeraheme sales in grams for 2014 and 2013, respectively. Because these institutions are eligible for federal pricing discounts, the revenue realized per unit ofFeraheme sold to 340B institutions is lower than from some of our other customers.
Further, under the sequestration required by the Budget Control Act of 2011, as amended by the American Taxpayer Relief Act of 2012, Medicare payments for all items and services under Parts A and B incurred on or after April 1, 2013 have been reduced by up to 2%. Therefore, after adjustment for deductible and co-insurance, the reimbursement rate for physician-administered drugs, includingFeraheme, under Medicare Part B has been reduced from average selling price ("ASP") plus 6% to ASP plus 4.3%. Beginning in April 2013, we amended certain of ourFeraheme customer contracts to try to partially address the impact of sequestration on our customers and their patients. These amendments have led to increased discounts and rebates in 2014 as compared to 2013.
We were not materially impacted by healthcare reform legislation during 2014 or 2013. Presently, we have not identified any provisions that could materially impact our business but we continue to monitor ongoing legislative developments and we are assessing what impact recent healthcare reform legislation will have on our business following the consummation of our acquisition of Lumara Health.
License fee and other collaboration revenues for the 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2014 to 2013 change |
|---|
| | 2014 | | 2013 | | $ Change | | % Change |
|---|
Deferred license fee revenues recognized from Takeda | | $ | 8,217 | | $ | 7,896 | | $ | 321 | | | 4% |
Reimbursement revenues from Takeda | | | 1,669 | | | 489 | | | 1,180 | | | >100% |
Deferred revenues recognized from 3SBio termination | | | 1,000 | | | — | | | 1,000 | | | N/A |
| | | | | | | | | | | | | |
Total | | $ | 10,886 | | $ | 8,385 | | $ | 2,501 | | | 30% |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Our license fee and other collaboration revenues in 2014 increased by $2.5 million as compared to 2013 primarily as the result of the reimbursement of $1.2 million of certain out-of pocket development costs received from Takeda in connection with the Takeda Termination Agreement and the accelerated recognition of $0.3 million of deferred revenues associated with upfront and milestone payments received to date from Takeda and previously deferred. We expect to recognize the remaining $44.4 million balance of the deferred revenue within the next twelve months. In addition, during 2014 we recognized $1.0 million of previously deferred revenue from our former partnership with 3SBio, Inc. ("3SBio") as the result of the termination of our license agreement in January 2014. We have no further obligations under the agreement with 3SBio.
We expect that our license fee and other collaborative revenues will increase in 2015 as compared to 2014 due to the recognition of the remaining $44.4 million of deferred revenues as a result of the December 2014 termination of the Amended Takeda Agreement.
97
Table of Contents
Other product sales and royalties primarily includedFeraheme product sales to Takeda and royalties from Takeda as well as net product sales ofMuGard. Other product sales and royalties increased by $3.6 million for 2014 as compared to 2013 due primarily to $3.0 million related to the termination of the Amended Takeda Agreement, including the recognition of the remaining $2.5 million balance of previously deferred product sales to Takeda. In addition, the increase in other product sales and royalties reflects a $0.9 million increase inMuGard sales.
We expect other product sales and royalties to decrease in 2015 as compared to 2014 due to the absence ofFeraheme product sales to Takeda in 2015 as a result of the December 2014 termination of the Amended Takeda Agreement.
Costs and Expenses
Cost of product sales for 2014 and 2013 were as follows (in thousands):
| | | | | | | | | | | | | |
| | Years Ended
December 31, | | 2014 to 2013 change | |
|---|
| | 2014 | | 2013 | | $ Change | | % Change | |
|---|
Cost of Product Sales | | $ | 20,306 | | $ | 11,960 | | $ | 8,346 | | | 70 | % |
Percentage of Net Product Sales and Royalties | | | 18% | | | 17% | | | | | | | |
Our cost of product sales are primarily comprised of manufacturing costs, costs of managing our contract manufacturers, and costs for quality assurance and quality control associated with our U.S. product sales, sales ofFeraheme to Takeda and the amortization of product related intangible assets and inventory step-up related to the November 2014 acquisition of Lumara Health. The $8.3 million increase in our cost of product sales for 2014 as compared to 2013 was attributable to the following factors:
- •
- $6.1 million increase related to the amortization of the Lumara Health intangible asset andMakena inventory step-up;
- •
- $2.6 million increase in costs related to sales ofFeraheme to Takeda, including the accelerated recognition of product costs previously deferred as a result of the Takeda Termination Agreement; and
- •
- $2.2 million decrease due to a lower average cost per vial ofFeraheme sold, partially offset by a $1.7 million increase due to a higher volume ofFeraheme vials sold in 2014.
We expect our cost of product sales as a percentage of net product sales and royalties, excluding any impact from the amortization of intangible assets on theMakena marketed product and MuGard Rights and the amortization of inventory step-up onMakena inventory, to decrease in 2015 as compared to 2014 due to the addition ofMakena sales to our product portfolio and the resulting lower blended cost to produce our products.
Research and development expenses include external expenses, such as costs of clinical trials, contract research and development expenses, certain manufacturing research and development costs, regulatory filing fees, consulting and professional fees and expenses, and internal expenses, such as compensation of employees engaged in research and development activities, the manufacture of product needed to support research and development efforts, related costs of facilities, and other general costs related to research and development. Where possible, we track our external costs by
98
Table of Contents
major project. To the extent that external costs are not attributable to a specific project or activity, they are included in other external costs. Prior to the initial regulatory approval of our products or development of new manufacturing processes, costs associated with manufacturing process development and the manufacture of drug product are recorded as research and development expenses. Subsequent to initial regulatory approval, costs associated with the manufacture of our products for commercial sale are capitalized in inventory and recorded as cost of product sales when sold.
Research and development expenses for 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2014 to 2013 change |
|---|
| | 2014 | | 2013 | | $ Change | | % Change |
|---|
External Research and Development Expenses | | | | | | | | | | | | |
Feraheme to treat IDA in CKD patients | | $ | 8,374 | | $ | 4,280 | | $ | 4,094 | | | 96% |
Feraheme manufacturing process development and materials | | | 2,214 | | | 2,690 | | | (476 | ) | | –18% |
Makenaclinical trial costs | | | 775 | | | — | | | 775 | | | N/A |
Makena manufacturing process development and materials | | | 928 | | | — | | | 928 | | | N/A |
Other external costs | | | 980 | | | 2,026 | | | (1,046 | ) | | –52% |
| | | | | | | | | | | | | |
Total | | $ | 13,271 | | $ | 8,996 | | $ | 4,275 | | | 48% |
| | | | | | | | | | | | | |
Internal Research and Development Expenses | | | | | | | | | | | | |
Compensation, payroll taxes, benefits and other | | | 9,293 | | | 9,419 | | | (126 | ) | | –1% |
Equity-based compensation | | | 1,596 | | | 2,149 | | | (553 | ) | | –26% |
| | | | | | | | | | | | | |
Total | | $ | 10,889 | | $ | 11,568 | | $ | (679 | ) | | –6% |
| | | | | | | | | | | | | |
Total Research and Development Expenses | | $ | 24,160 | | $ | 20,564 | | $ | 3,596 | | | 17% |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Total research and development expenses incurred in 2014 increased by $3.6 million, or 17%, as compared to 2013. The increase was primarily due to a $4.3 million increase in external research and development costs pertaining to our CKD-related trials during 2014 as well as new costs related toMakena clinical trials and related manufacturing costs. This increase was partially offset by reduced internal research and development costs of $0.7 million.
We expect research and development expenses to increase in 2015 due to the timing of expenses related to our pediatric clinical studies and clinical trial to determine the safety and efficacy of repeat doses ofFeraheme for the treatment of IDA in patients with hemodialysis dependent CKD as well as current clinical trials related toMakena's post approval commitments and its lifecycle management program. In addition, research and development expenses could increase further and significantly depending on the outcome of discussions with the FDA on the regulatory path forward forFeraheme in the broad indication and any resulting clinical trials or development efforts that we may undertake.
We track our external costs on a major project basis, in most cases through the later of the completion of the last trial in the project or the last submission of a regulatory filing to the FDA or applicable foreign regulatory body. We do not track our internal costs by project since our research and development personnel work on a number of projects concurrently and much of our fixed costs benefit multiple projects or our operations in general. The following major research and development projects were ongoing as of December 31, 2014:
- •
- Feraheme to treat IDA in CKD patients. This project currently includes the following (a) a completed clinical study evaluatingFeraheme treatment as compared to treatment to another IV iron to support the 2010 marketing authorization application ("MAA") submission; (b) a pediatric study that is being conducted as part of our post-approval Pediatric Research Equity
99
Table of Contents
Act requirement to support pediatric CKD labeling ofFeraheme; (c) two additional pediatric studies to be completed in accordance with our approved pediatric investigation plan to support the MAA submission; and (d) an ongoing multi-center clinical trial to be conducted to determine the safety and efficacy of repeat doses ofFeraheme for the treatment of IDA in patients with hemodialysis dependent CKD, including a treatment arm with iron sucrose using an magnetic resonance imaging sub-analysis to evaluate the potential for iron to accumulate in the body following repeated IV iron administration.
- •
- Makena: This project currently includes studies conducted as part of the post-approval commitments under the provisions of the FDA's "Subpart H" Accelerated Approval regulations, including (a) an ongoing efficacy and safety clinical study of Makena; (b) an ongoing follow-up study of the babies born to mothers from the efficacy and safety clinical study; and (c) a completed pharmacokinetic trial of women taking Makena.
Through December 31, 2014, we have incurred aggregate external research and development expenses of approximately $36.6 million related to our current program for the development ofFeraheme to treat IDA in CKD patients, described above. We currently estimate that the total remaining external costs associated with this development project will be in the range of approximately $20.0 million to $30.0 million over the next several years, not including any potential costs related to any clinical trials or development efforts that we may undertake as an outcome of discussions with the FDA on the regulatory path forward forFeraheme in the broad indication.
From November 12, 2014 through December 31, 2014, we have incurred aggregate external research and development expenses of approximately $0.8 million related to our current program forMakena, described above. We currently estimate that the total remaining external costs associated with this development project will be in the range of approximately $20.0 million to $30.0 million over the next several years.
In accordance with our policy of tracking external research and development costs through the later of the completion of the last trial in a project or the last submission of a regulatory filing to the FDA, we discontinued tracking our expenses related toFeraheme to treat IDA regardless of the underlying cause in the third quarter of 2013, at which point we had incurred $57.8 million of external research and development expenses. In January 2014, we received a complete response letter from the FDA in response to our sNDA submission forFeraheme for the treatment of IDA in adult patients who had failed or could not use oral iron. We are currently unable to estimate with any certainty the future costs we will incur, if any, related to our project forFeraheme to treat IDA regardless of the cause. In future periods, we may resume the disclosure of such expected future costs as the facts and circumstances warrant.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses include costs related to our commercial personnel, including our specialty sales force, medical education professionals, pharmacovigilance and safety monitoring and commercial support personnel, costs related to our administrative personnel, including our legal, finance, business development and executive personnel, external and facilities costs required to support the marketing and sale of our products and other costs associated with our corporate activities.
100
Table of Contents
Selling, general and administrative expenses for 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2014 to 2013 change |
|---|
| | 2014 | | 2013 | | $ Change | | % Change |
|---|
Compensation, payroll taxes and benefits | | $ | 31,261 | | $ | 22,819 | | $ | 8,442 | | | 37% |
Sales and marketing consulting, professional fees, and other | | | 16,702 | | | 13,407 | | | 3,295 | | | 25% |
General and administrative consulting, professional fees and other expenses | | | 18,065 | | | 16,133 | | | 1,932 | | | 12% |
Fair value of contingent consideration liability | | | (681 | ) | | 1,074 | | | (1,755 | ) | | –163% |
Equity-based compensation expense | | | 6,907 | | | 5,734 | | | 1,173 | | | 20% |
| | | | | | | | | | | | | |
Total | | $ | 72,254 | | $ | 59,167 | | $ | 13,087 | | | 22% |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Total selling, general and administrative expenses incurred in 2014 increased by $12.3 million, or 21%, as compared to 2013 for the following reasons:
- •
- $8.4 million increase in compensation, payroll taxes and benefits primarily due to increased costs associated with additional personnel in our commercial functions, including the Lumara Health sales force acquired in connection with the November 2014 acquisition of Lumara Health, and increased costs associated with certain of our general and administrative functions, including the addition of Lumara Health employees;
- •
- $3.3 million increase in sales and marketing consulting, professional fees, and other expenses primarily due to costs related toMakena marketing activities since the November 2014 acquisition and increased consulting costs related to the commercialization ofMuGard;
- •
- $1.9 increase in general and administrative consulting, professional fees and other expenses primarily due to increased costs associated with business development, consulting and other legal-related activities in support of our product portfolio expansion as well as costs associated with Lumara Health after the November 2014 acquisition. These increased costs were offset by a number of non-recurring costs in 2013, including $1.9 million of accelerated depreciation expense recognized related to our prior corporate headquarters, $0.6 million of costs related to the closure of our Cambridge, Massachusetts manufacturing facility and $0.6 million of costs associated with the relocation of our corporate headquarters;
- •
- $1.8 million decrease to the contingent consideration liability due to a $3.4 million reduction of theMuGard-related contingent consideration primarily resulting from a 2014 revision of our total projectedMuGard sales, partially offset by a $1.6 million increase to the Lumara Health-related contingent consideration; and
- •
- $1.2 million increase in equity-based compensation expense due primarily to one-time charges associated with the departure of our former Senior Vice President of Business Development and Chief Business Officer in June 2014 as well as the expense associated with equity awards to new and existing employees.
We expect that total selling, general and administrative expenses will increase in 2015 as compared to 2014 as a result of the increased headcount following the November 2014 acquisition of Lumara Health and other costs associated withMakena related commercial activities.
Acquisition-related costs
We incurred approximately $9.5 million of acquisition-related costs in 2014 related to our acquisition of Lumara Health, which primarily represented financial advisory fees, legal fees, due diligence and other costs and expenses. During 2013, in connection with the acquisition of the MuGard Rights we incurred approximately $0.8 million of expenses primarily related to professional and legal fees.
101
Table of Contents
Restructuring Expense
In connection with the November 2014 Lumara Health acquisition, we initiated a restructuring program in the fourth quarter of 2014, which included severance benefits primarily related to former Lumara Health employees. As a result of the restructuring, we recorded charges of approximately $2.0 million in 2014. We expect to pay substantially all of these restructuring costs during 2015.
Other Income (Expense)
Other income (expense) for 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2014 to 2013 change |
|---|
| | 2014 | | 2013 | | $ Change | | % Change |
|---|
Interest expense | | $ | (14,697 | ) | $ | — | | | (14,697 | ) | | N/A |
Interest and dividend income, net | | | 975 | | | 1,051 | | | (76 | ) | | –7% |
Gains on sale of asset | | | 103 | | | 924 | | | (821 | ) | | N/A |
Gains (losses) on investments, net | | | 114 | | | 40 | | | 74 | | | >100% |
| | | | | | | | | | | | | |
Total | | $ | (13,505 | ) | $ | 2,015 | | $ | (15,520 | ) | | <(100%) |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Other income (expense) for 2014 decreased by $15.5 million as compared to 2013 primarily as the result of the recognition of $14.7 million of interest expense, which was comprised of the amortization of debt discount, contractual interest expense and amortization of debt issuance costs in connection with the issuance of the $200.0 million of 2.5% convertible senior notes due February 15, 2019 (the "Convertible Notes") and our November 2014 $340.0 million Term Loan Facility. In addition, the decrease in other income (expense) reflects 2013 non-recurring gains of $0.5 million in connection with the sale of Combidex®, a legacy product of the Company, and $0.4 million in connection with the sale of fixed assets related to our previously owned Cambridge, Massachusetts manufacturing facility.
We expect our net expense to increase in 2015 as compared to 2014 as a result of recording a full year of interest expense related to our debt obligations in 2015.
Income Tax Benefit
The $153.2 million income tax benefit for 2014 reflects a $132.9 million decrease in our valuation allowance due to taxable temporary differences available as a source of income to realize the benefit of certain of our pre-existing deferred tax assets as a result of the November 2014 acquisition of Lumara Health. As of December 31, 2014, we had approximately $542.3 million of federal NOL carryforwards and up to $242.2 million of state operating loss carryforwards, which expire on various dates through the year 2034. These loss carryforwards may be available to reduce future taxable income, if any, and are subject to review and possible adjustment by the applicable taxing authorities. The available loss carryforwards that may be utilized in any future period may be subject to limitation based on historical changes in the ownership of our stock. We have a remaining valuation allowance of $33.6 million on certain of our deferred tax assets, which was recorded based on the uncertainty surrounding utilization of these deferred tax assets. It should be noted that the allocation of the purchase price related to the Lumara Health transaction is subject to adjustment upon finalization of fair valuation procedures and therefore the impact of the tax benefit associated with the valuation allowance release and deferred tax assets and liabilities (including uncertain tax positions) are subject to change.
Net Income (Loss)
For the reasons stated above, we have earned net income of $135.8 million, or $6.06 per basic share and $5.45 per diluted share, for 2014 as compared to a net loss of $9.6 million, or $0.44 per basic
102
Table of Contents
and diluted share for 2013. Included in the $135.8 million net income during 2014, is a tax benefit of $153.2 million reflecting a $132.9 million decrease in our valuation allowance due to taxable temporary differences related to the acquisition of Lumara Health, as discussed above.
Results of Operations—2013 as compared to 2012
Revenues
Our total revenues for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change |
|---|
| | 2013 | | 2012 | | $ Change | | % Change |
|---|
U.S.Feraheme product sales, net | | $ | 71,362 | | $ | 58,287 | | $ | 13,075 | | | 22% |
License fee and other collaboration revenues | | | 8,385 | | | 26,475 | | | (18,090 | ) | | –68% |
Other product sales and royalties | | | 1,109 | | | 616 | | | 493 | | | 80% |
| | | | | | | | | | | | | |
Total | | $ | 80,856 | | $ | 85,378 | | $ | (4,522 | ) | | –5% |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Our total revenues in 2013 decreased by $4.5 million as compared to 2012, primarily as the result of our recognition of approximately $20.0 million in 2012 related to milestone payments we received from Takeda in 2012 as compared to $1.8 million recognized in 2013. The net decrease was partially offset by a $13.1 million increase in U.S. netFeraheme product sales and a $0.5 million increase in other product sales and royalties. Our net product sales for each of 2013 and 2012 included a $0.6 million reduction of our estimated Medicaid rebate reserve related to priorFeraheme sales, as discussed below. In addition, our net product sales for 2012 included a $2.2 million reduction of our estimated product return reserve, as discussed below.
The following table sets forth customers who represented 10% or more of our total revenues for 2013 and 2012:
| | | | | | | |
| | Years Ended
December 31, | |
|---|
| | 2013 | | 2012 | |
|---|
AmerisourceBergen Drug Corporation | | | 41% | | | 34% | |
McKesson Corporation | | | 24% | | | 17% | |
Cardinal Health, Inc. | | | 16% | | | 12% | |
Takeda Pharmaceuticals Company Limited | | | 11% | | | 31% | |
103
Table of Contents
U.S. Feraheme Product Sales, Net
U.S.Feraheme product sales and product sales allowances and accruals for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | |
| |
| |
|---|
| | 2013 | | Percent of
gross U.S.
Feraheme
product
sales | | 2012 | | Percent of
gross U.S.
Feraheme
product
sales | | $ Change | | % Change | |
|---|
Gross U.S.Feraheme product sales | | $ | 119,712 | | | | | $ | 88,725 | | | | | $ | 30,987 | | | 35% | |
Less provision for product sales allowances and accruals: | | | | | | | | | | | | | | | | | | | |
Discounts and chargebacks | | | 37,098 | | | 31% | | | 26,517 | | | 30% | | | | | | | |
Government and other rebates | | | 10,868 | | | 9% | | | 6,058 | | | 7% | | | | | | | |
Medicaid rebate reserve adjustment | | | (568 | ) | | 0% | | | (621 | ) | | –1% | | | | | | | |
Returns | | | 952 | | | 1% | | | (1,516 | ) | | –2% | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Total | | | 48,350 | | | 40% | | | 30,438 | | | 34% | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Net U.S.Feraheme product sales | | $ | 71,362 | | | | | $ | 58,287 | | | | | $ | 13,075 | | | 22% | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
Our gross U.S.Feraheme product sales increased by $31.0 million, or 35%, during 2013 as compared to 2012. Of the $31.0 million increase, $21.5 million was due to increased units sold and $9.5 million was due to price increases. This increase was partially offset by $15.7 million of additional allowances and accruals in 2013, excluding a $2.2 million reduction of our estimated product return reserves in 2012 and a $0.6 million reduction of our estimated Medicaid rebate reserve related to priorFeraheme sales for each of 2013 and 2012, as described below. As a result of these factors, total net U.S.Feraheme product sales increased by $13.1 million, or 22%, during 2013 as compared to 2012.
Total discounts and chargebacks for 2013 were $37.1 million, or 31% of total gross product sales, as compared to $26.5 million, or 30%, in 2012. The 1% increase in total discounts and chargebacks as a percentage of total gross U.S.Feraheme product sales was related primarily to a change in our customer mix.
Total government and other rebates (excluding any changes in estimates related to Medicaid rebate reserves) were $10.9 million, or 9% of total gross U.S.Feraheme product sales, in 2013 as compared to $6.1 million, or 7%, in 2012. The 2% increase in total government and other rebates as a percentage of gross U.S.Feraheme product sales was related primarily to higher prices charged forFeraheme in 2013 as compared to 2012 and increased sales to clinics and hospitals that had volume or market share contracts with us during 2013 as compared to 2012.
During each of 2013 and 2012, we reduced our estimated Medicaid reserve related to priorFeraheme sales by approximately $0.6 million based on actual product-specific rebate claims received since the 2009 launch ofFeraheme, our expectations of state level activity, and estimated rebate claims not yet submitted. These changes in estimates were reflected as an increase of $0.6 million in our net product sales for 2013 and 2012 and resulted in reductions to our gross to net percentage in these respective periods.
During 2012, we reduced our reserve for product returns by approximately $2.2 million, primarily as a result of a lower than expected rate of product returns as well as the lapse of the product return period on certain manufacturedFeraheme lots that carried a two year expiration. As a result, the product returns provision applied to gross product sales for 2012 was a credit of $1.5 million, resulting
104
Table of Contents
in an increase to product sales, as compared to a $1.0 million charge in 2013, resulting in a decrease to product sales. There was no significant adjustment of our reserve for product returns in 2013.
An analysis of the amount of, and change in, reserves for 2013 and 2012 is as follows (in thousands):
| | | | | | | | | | | | | |
| | Discounts and
Chargebacks | | Government
and Other
Rebates | | Returns | | Total | |
|---|
Balance at January 1, 2012 | | $ | 1,822 | | $ | 3,101 | | $ | 2,842 | | $ | 7,765 | |
| | | | | | | | | | | | | | |
Current provisions relating to sales in current year | | | 26,517 | | | 6,152 | | | 577 | | | 33,246 | |
Adjustments relating to sales in prior years | | | — | | | (715 | ) | | (2,093 | ) | | (2,808 | ) |
Payments/returns relating to sales in current year | | | (24,709 | ) | | (4,511 | ) | | — | | | (29,220 | ) |
Payments/returns relating to sales in prior years | | | (1,859 | ) | | (1,597 | ) | | (308 | ) | | (3,764 | ) |
| | | | | | | | | | | | | | |
Balance at December 31, 2012 | | $ | 1,771 | | $ | 2,430 | | $ | 1,018 | | $ | 5,219 | |
| | | | | | | | | | | | | | |
Current provisions relating to sales in current year | | | 37,098 | | | 10,868 | | | 952 | | | 48,918 | |
Adjustments relating to sales in prior years | | | — | | | (568 | ) | | — | | | (568 | ) |
Payments/returns relating to sales in current year | | | (34,538 | ) | | (8,194 | ) | | — | | | (42,732 | ) |
Payments/returns relating to sales in prior years | | | (1,648 | ) | | (1,699 | ) | | (8 | ) | | (3,355 | ) |
| | | | | | | | | | | | | | |
Balance at December 31, 2013 | | $ | 2,683 | | $ | 2,837 | | $ | 1,962 | | $ | 7,482 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
During 2013 and 2012, we decreased our product sales allowances and accruals by approximately $0.6 million and $2.8 million, respectively, for changes in estimates relating to sales in prior years, as discussed above.
License fee and other collaboration revenues for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change |
|---|
| | 2013 | | 2012 | | $ Change | | % Change |
|---|
Milestone revenues recognized from Takeda | | $ | 1,800 | | $ | 19,950 | | $ | (18,150 | ) | | –91% |
Deferred license fee revenues recognized from Takeda | | | 6,096 | | | 6,096 | | | — | | | 0% |
Reimbursement revenues from Takeda | | | 489 | | | 429 | | | 60 | | | 14% |
| | | | | | | | | | | | | |
Total | | $ | 8,385 | | $ | 26,475 | | $ | (18,090 | ) | | –68% |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Our license fee and other collaboration revenues in 2013 decreased by $18.1 million as compared to 2012 primarily due to milestones received in 2012. Our milestone revenues in 2012 included a $15.0 million milestone payment from Takeda associated with the regulatory approval ofRienso in the EU, which we deemed a substantive milestone and recorded in its entirety. In addition, our 2012 milestone revenues included the recognition of a portion of an aggregate of $18.0 million of milestone payments related to the commercial launches ofFeraheme/Rienso in the EU and Canada, which we deemed non-substantive milestones and are amortizing into revenue on a cumulative catch up basis using the proportional performance method extended over the original life of the Takeda Agreement. We did not receive any milestone payments in 2013. In 2013 and 2012, we also recorded $7.9 million and $26.1 million, respectively, of revenues associated with the amortization of the upfront payments and the milestone payments we have received since the inception of our agreement with Takeda. As of December 31, 2013, we had approximately $49.3 million remaining in deferred revenues related to the
105
Table of Contents
$61.0 million in upfront payments and the $18.0 million in non-substantive milestone payments received from Takeda.
During 2013 and 2012, we recorded $0.5 million and $0.4 million, respectively, of revenues associated with certain out-of pocket development costs in connection with the Amended Takeda Agreement.
As of December 31, 2013, we had approximately $2.4 million in deferred revenue related to product shipped to Takeda, but not yet sold through to Takeda's customers, of which $0.3 million was classified as short-term and $2.1 million was classified as long-term. In addition, we had $2.3 million in deferred cost of product sales, of which $0.3 million was classified as short-term and $2.0 million was classified as long-term. These deferred revenue and deferred cost of product sales are recorded in our consolidated balance sheet as of December 31, 2013.
Costs and Expenses
Cost of product sales for 2013 and 2012 were as follows (in thousands):
| | | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change | |
|---|
| | 2013 | | 2012 | | $ Change | | % Change | |
|---|
Cost of Product Sales | | $ | 11,960 | | $ | 14,220 | | $ | (2,260 | ) | | –16 | % |
Percentage of Net Product Sales and Royalties | | | 17% | | | 24% | | | | | | | |
The $2.3 million decrease in our cost of product sales for 2013 as compared to 2012 was attributable to the following factors:
- •
- $3.6 million decrease due to costs related to the 2012 closure of our Cambridge, Massachusetts manufacturing facility, including $2.3 million in accelerated depreciation and impairment costs related to the 2012 impairment of our manufacturing facility and other related production costs;
- •
- $1.5 million decrease due to a lower average cost per vial sold, partially offset by $1.2 million increase due to a higher volume ofFeraheme vials sold in 2013;
- •
- $0.8 million increase due to the sale of pre-approval validation lots in 2012, which in accordance with our capitalization policy, excluded costs that had been expensed prior to FDA approval of the manufacturing process;
- •
- $0.5 million increase due to a write-off of inventory that was affected by a voluntary recall of a specific batch ofRienso from the Swiss market in May 2013; and
- •
- $0.3 million increase related to sales ofMuGard and sales to our partners.
106
Table of Contents
Research and development expenses for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change | |
|---|
| | 2013 | | 2012 | | $ Change | | % Change | |
|---|
External Research and Development Expenses | | | | | | | | | | | | | |
Feraheme to treat IDA in CKD patients | | $ | 4,280 | | $ | 3,226 | | $ | 1,054 | | | 33% | |
Feraheme to treat IDA regardless of the underlying cause | | | 86 | | | 12,357 | | | (12,271 | ) | | –99% | |
Feraheme as a therapeutic agent, general | | | 1,615 | | | 1,033 | | | 582 | | | 56% | |
Feraheme manufacturing process development and materials | | | 2,690 | | | 2,297 | | | 393 | | | 17% | |
Other external costs | | | 325 | | | 152 | | | 173 | | | >100% | |
| | | | | | | | | | | | | | |
Total | | $ | 8,996 | | $ | 19,065 | | $ | (10,069 | ) | | –53% | |
| | | | | | | | | | | | | | |
Internal Research and Development Expenses | | | | | | | | | | | | | |
Compensation, payroll taxes, benefits and other expenses | | | 9,419 | | | 12,237 | | | (2,818 | ) | | –23% | |
Equity-based compensation expense | | | 2,149 | | | 1,994 | | | 155 | | | 8% | |
| | | | | | | | | | | | | | |
Total | | $ | 11,568 | | $ | 14,231 | | $ | (2,663 | ) | | –19% | |
| | | | | | | | | | | | | | |
Total Research and Development Expenses | | $ | 20,564 | | $ | 33,296 | | $ | (12,732 | ) | | –38% | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Total research and development expenses incurred in 2013 decreased by $12.7 million, or 38%, as compared to 2012. The decrease was primarily due to reduced external research and development costs of $10.1 million in 2013. In addition, 2013 internal research and development costs decreased by $2.7 million as compared to 2012.
The $10.1 million, or 53%, decrease in our external research and development expenses was due to a $12.3 million decrease in costs incurred in connection with our Phase III clinical development program forFeraheme to treat IDA regardless of the underlying cause, which was completed in 2012, partially offset by a $1.1 million increase in our costs associated with our CKD-related trials and a $0.4 million increase in manufacturing process development and materials-related costs.
The $2.7 million, or 19%, decrease in our internal research and development expenses was primarily attributable to the decrease in compensation and related benefit costs in 2013 following our 2012 and 2011 corporate restructurings, which resulted in lower headcount in our research and development departments.
107
Table of Contents
Selling, General and Administrative Expenses
Selling, general and administrative expenses for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change | |
|---|
| | 2013 | | 2012 | | $ Change | | % Change | |
|---|
Compensation, payroll taxes and benefits | | $ | 22,819 | | $ | 23,273 | | $ | (454 | ) | | –2% | |
Sales and marketing consulting, professional fees, and other expenses | | | 13,407 | | | 12,133 | | | 1,274 | | | 11% | |
General and administrative consulting, professional fees and other expenses expenses | | | 17,989 | | | 12,860 | | | 5,129 | | | 40% | |
Equity-based compensation expense | | | 5,734 | | | 4,805 | | | 929 | | | 19% | |
| | | | | | | | | | | | | | |
Total | | $ | 59,949 | | $ | 53,071 | | $ | 6,878 | | | 13% | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Total selling, general and administrative expenses incurred in 2013 increased by $6.9 million, or 13%, as compared to 2012 for the following reasons:
- •
- $0.5 million decrease in compensation, payroll taxes and benefits due to a $0.9 million decrease in one-time retention payments made in 2012, partially offset by an increase of $0.4 million in 2013 due to increased headcount;
- •
- $1.3 million increase in sales and marketing consulting, professional fees, and other expenses primarily due to increased consulting costs related to the acquisition and commercialization ofMuGard;
- •
- $5.1 million increase in general and administrative consulting, professional fees and other expenses primarily due to $1.9 million of accelerated depreciation expense related to certain leasehold improvements and furniture and fixtures associated with our prior office facility, $1.4 million of increased costs associated with consulting, business development and other legal-related activities, $1.1 million adjustment to the fair value of our contingent consideration liability related to the MuGard Rights, $0.8 million of transaction and other costs related to the acquisition of the MuGard Rights (these $0.8 million costs have been reclassified to acquisition-related costs in our 2014 financial statements), $0.4 million of costs associated with the relocation of our corporate headquarters, and $0.3 million of costs related to the closure of our Cambridge, Massachusetts manufacturing facility. These increased costs in 2013 were partially offset by $1.6 million in termination fees which we paid in 2012 to ourGastroMARK licensees in connection with the termination our license agreements with them; and
- •
- $0.9 million increase in equity-based compensation expense due primarily to the expense associated with equity awards to new and existing employees.
Restructuring Expense
During 2012, we initiated corporate restructurings including a workforce reduction plan. The majority of the workforce reduction plan was associated with our manufacturing and development infrastructure, including our decision to divest our Cambridge, Massachusetts manufacturing facility. The workforce reduction was substantially completed by the end of 2012, and the majority of the related expenses were paid by the end of 2012.
108
Table of Contents
Other Income (Expense)
Other income (expense) for 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | | | |
| | Years Ended
December 31, | | 2013 to 2012 change |
|---|
| | 2013 | | 2012 | | $ Change | | % Change |
|---|
Interest and dividend income, net | | $ | 1,051 | | $ | 1,286 | | $ | (235 | ) | | –18% |
Gains on sale of asset | | | 924 | | | — | | | 924 | | | N/A |
Gains (losses) on investments, net | | | 40 | | | (1,466 | ) | | 1,506 | | | <(100%) |
| | | | | | | | | | | | | |
Total | | $ | 2,015 | | $ | (180 | ) | $ | 2,195 | | | <(100%) |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Other income (expense) for 2013 increased by $2.2 million as compared to 2012 primarily as the result of the non-recurring nature of the June 2012 $1.5 million loss realized on the sale of our then-remaining auction rate securities. Additionally, during 2013, we recognized $0.5 million of gains in connection with the sale of Combidex®, a molecular imaging agent which we were not actively pursuing development, and a $0.4 million gains on the sale of fixed assets related to our Cambridge, Massachusetts manufacturing facility. These increases were partially offset by a decrease in interest and dividend income as the result of lower average cash balances during 2013 as compared to 2012.
Income Tax Benefit
We did not recognize any income tax benefit during 2013. We recognized an income tax benefit of $0.9 million during 2012 as the result of our recognition of a corresponding income tax expense associated with the increase in value of certain securities as a result of their redemption at prices higher than the fair market value at which they were recorded. This income tax expense was recorded in other comprehensive loss.
Net Loss
For the reasons stated above, we incurred a net loss of $9.6 million and $16.8 million, or $0.44 and $0.78 per basic and diluted share, for 2013 and 2012, respectively.
Liquidity and Capital Resources
General
We currently finance our operations primarily from the sale of our products, cash generated from our investing activities and the sale of our securities. We expect to continue to incur significant expenses as we continue to market, sell and contract for the manufacture ofMakena andFeraheme and as we market and sellMuGard, and as and if we further develop and seek regulatory approval forFeraheme for the treatment of IDA in a broad range of patients in the U.S. For a detailed discussion regarding the risks and uncertainties related to our liquidity and capital resources, please refer to our Risk Factor, "We may need additional capital to achieve our business objectives and to service our debt obligations, including the Term Loan Facility, our Convertible Notes and contingent payments that may become due under the Lumara Agreement, which could cause significant dilution to our stockholders."
109
Table of Contents
Cash, cash equivalents, investments and certain financial obligations as of December 31, 2014 and 2013 consisted of the following (in thousands):
| | | | | | | | | | | | | |
| | December 31, | |
| |
| |
|---|
| | 2014 | | 2013 | | $ Change | | % Change | |
|---|
Cash and cash equivalents | | $ | 119,296 | | $ | 26,986 | | $ | 92,310 | | | >100% | |
Investments | | | 24,890 | | | 186,803 | | | (161,913 | ) | | –87% | |
| | | | | | | | | | | | | | |
Total | | $ | 144,186 | | $ | 213,789 | | $ | (69,603 | ) | | –33% | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Outstanding principal on convertible notes | | $ | 200,000 | | $ | — | | $ | 200,000 | | | N/A | |
Outstanding principal on term loan | | | 340,000 | | | — | | | 340,000 | | | N/A | |
| | | | | | | | | | | | | | |
| | $ | 540,000 | | $ | — | | $ | 540,000 | | | N/A | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
As of December 31, 2014, our investments consisted solely of corporate debt securities. We place our cash in instruments that meet high credit quality and diversification standards, as specified in our investment policy. Our investment policy also limits the amount of our credit exposure to any one issue or issuer, excluding U.S. government entities and money market funds, and seeks to manage these assets to achieve our goals of preserving principal, maintaining adequate liquidity at all times, and maximizing returns.
The $69.6 million decrease in cash, cash equivalents and investments as of December 31, 2014, as compared to December 31, 2013, was primarily due to the $272.5 million cash payment used to partially fund the acquisition of Lumara Health in November 2014, including the liquidation of approximately $170.4 million of our investments, partially offset by net proceeds of $179.1 million received during 2014 in connection with the issuance of $200.0 million aggregate principal amount of the Convertible Notes. We issued the Convertible Notes to help facilitate our corporate, clinical and commercial activities, and which, along with the convertible bond hedge transactions, are discussed in greater detail in Note S, "Debt," to our consolidated financial statements including in this Annual Report on Form 10-K. In addition, the increase in cash was partially offset by net cash expended to fund our operations and working capital.
In November 2014, we completed our acquisition of Lumara Health for approximately $600.0 million in upfront cash consideration (subject to finalization of certain adjustments related to Lumara Health's financial position at the time of closing, including adjustments related to working capital, net debt and transaction expenses as set forth in the Lumara Agreement) and approximately 3.2 million shares of our common stock having a fair value of approximately $112.0 million at the time of closing. The Lumara Agreement includes future contingent payments of up to $350.0 million in cash (or upon mutual agreement between us and the former Lumara Health security holders, future contingent payments may also be made in common stock or some combination thereof) payable by us to the former Lumara Health security holders based upon the achievement of certain sales milestones through calendar year 2019. See Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K for additional information.
In November 2014, we financed the $600.0 million cash portion of the Lumara Health acquisition through $327.5 million of net proceeds from borrowings under a new $340.0 million term loan (the "Term Loan Facility"), as discussed in more detail in Note S, "Debt," to our consolidated financial statements included in this Annual Report on Form 10-K, and $272.50 million of existing cash on hand.
110
Table of Contents
The Term Loan Facility imposes restrictive covenants on us, including a requirement that we reduce our leverage over time, and obligates us to make certain payments of principal and interest over time.
In addition, on February 14, 2014, we issued $200.0 million aggregate principal amount of Convertible Notes, as discussed in more detail in Note S, "Debt," to our consolidated financial statements included in this Annual Report on Form 10-K. We received net proceeds of $193.3 million from the sale of the Convertible Notes, after deducting fees and expenses of $6.7 million. We used $14.1 million of the net proceeds from the sale of the Convertible Notes to pay the cost of the convertible bond hedges (after such cost was partially offset by the proceeds to us from the sale of warrants in the warrant transactions). In connection with the issuance of the Convertible Notes, we incurred approximately $6.7 million of debt issuance costs, which primarily consisted of underwriting, legal and other professional fees, and allocated these costs to the liability and equity components based on the allocation of the proceeds.
The Convertible Notes are senior unsecured obligations and bear interest at a rate of 2.5% per year, payable semi-annually in arrears on February 15 and August 15 of each year, beginning on August 15, 2014. The Convertible Notes will mature on February 15, 2019, unless earlier repurchased or converted. The Convertible Notes will be convertible into cash, shares of our common stock, or a combination thereof, at our election (subject to certain limitations in the Term Loan Facility), at an initial conversion rate of approximately 36.9079 shares of common stock per $1,000 principal amount of the Convertible Notes, which corresponds to an initial conversion price of approximately $27.09 per share of our common stock and represents a conversion premium of approximately 35% based on the last reported sale price of our common stock of $20.07 on February 11, 2014, the date the notes offering was priced.
We expect that our cash, cash equivalents and investments balances, in the aggregate, will increase due to increased sales fromMakena andFeraheme during 2015, partially offset by debt-related payments. Our expectation assumes our continued investment in the development and commercialization of our products and the continued pursuit of business development transactions. We believe that our cash, cash equivalents and investments as of December 31, 2014, and the cash we currently expect to receive from sales of our products, earnings on our investments, will be sufficient to satisfy our cash flow needs for at least the next twelve months.
Year Ended December 31, 2014
Cash flows from operating activities
During 2014, our $11.4 million of cash provided by operations was attributable principally to our net operating income of approximately $135.8 million, adjusted for the following:
- •
- Non-cash operating items of ($128.2) million, including deferred income taxes, equity-based compensation expense, a write-down of inventory, amortization of debt discount and debt issuance costs, amortization of premium/discount on purchased securities, change in fair value of contingent consideration, depreciation and amortization, and other non-cash items;
- •
- $0.3 million of cash provided by operating activities due to increases in accounts receivable, inventories and prepaid assets;
- •
- $10.7 million of cash provided by operating activities due to increases in accounts payable and accrued expenses;
- •
- $9.2 million of cash used in operating activities due to decreases in deferred revenues and other long-term liabilities; and
- •
- $2.0 million of cash provided by operating activities due to decreases in other long-term assets.
111
Table of Contents
Our net income of $135.8 million was primarily the result of the recognition of a $153.2 million income tax benefit resulting from our merger with Lumara Health, partially offset by our costs to operate our business, including compensation to employees, commercialization expenses, such as marketing and promotion costs, costs to manufacture our products, research and development costs, including costs associated with our clinical trials, general and administrative costs, and interest from our debt obligations, partially offset by net product sales and collaboration revenues.
Cash flows from investing activities
Cash used in investing activities in 2014 was $432.9 million and was primarily attributable to the $595.6 million net cash used to fund the acquisition of Lumara Health, partially offset by proceeds from the sales and maturities of our investments, including the liquidation of $170.4 million to partially fund the acquisition of Lumara Health as well as a $2.9 million change in restricted cash following the return of escrowed funds related to a 2013 business development transaction that we did not complete.
Cash flows from financing activities
Cash provided by financing activities in 2014 was $513.5 million and was primarily attributable to the $327.5 million proceeds from the Term Loan Facility, which were used to partially fund the acquisition of Lumara Health and $177.8 million in net proceeds received from the issuance of the Convertible Notes in February 2014. In addition, we received $8.5 million in proceeds from the exercise of stock options.
Year Ended December 31, 2013
Cash flows from operating activities
During 2013 our use of $6.8 million of cash in operations was attributable principally to our net loss of approximately $9.6 million, adjusted for the following:
- •
- Non-cash operating items of $15.9 million including equity-based compensation expense, depreciation and amortization, amortization of premium/discount on purchased securities, change in fair value of contingent consideration, gains on the sale of assets, a write-down of inventory, and other non-cash items;
- •
- An aggregate decrease in deferred revenues and other long-term liabilities of $6.9 million;
- •
- An aggregate decrease of $5.7 million in accounts payable and accrued expenses;
- •
- An aggregate decrease of $1.5 million in accounts receivable, prepaid assets and inventories; and
- •
- An increase of $2.0 million in other long-term assets.
Our net loss of $9.6 million was primarily the result of compensation to employees, commercialization expenses, including marketing and promotion costs, research and development costs, including costs associated with our clinical trials, and general and administrative costs, partially offset by net product sales and collaboration revenues.
Cash flows from investing activities
Cash used in investing activities in 2013 was $13.9 million and was primarily attributable to the purchases of investments, partially offset by proceeds from the sales and maturities of our investments. In addition, we used $3.4 million of available cash and cash equivalents to purchase the MuGard Rights and related inventory, $2.9 million was held in an escrow account related to a business development transaction that we did not complete, and approximately $1.6 million to purchase leasehold improvements and furniture and fixtures for our new corporate headquarters. We also
112
Table of Contents
received $2.5 million from the sale of our Cambridge, Massachusetts manufacturing facility and related fixtures and equipment and $0.5 million from the sale of Combidex®, a molecular imaging agent which we were not actively pursuing development.
Cash flows from financing activities
Cash provided by financing activities in 2013 was $1.4 million and was primarily attributable to the proceeds from the exercise of stock options.
Contractual Obligations
Our long-term contractual obligations include commitments and estimated purchase obligations entered into in the normal course of business. These include commitments related to our facility leases, purchases of inventory and other purchases related to our products, debt obligations, non-cancellable operating leases, and other purchase obligations. Future lease obligations and purchase commitments, as of December 31, 2014, are as follows (in thousands):
| | | | | | | | | | | | | | | | |
| | Payment due by period | |
|---|
| | Total | | Less than 1 year | | 1-3 years | | 3-5 years | | More than 5 years | |
|---|
Facility lease obligations | | $ | 5,543 | | $ | 1,451 | | $ | 2,918 | | $ | 1,174 | | $ | — | |
Purchase commitments | | | 4,780 | | | 4,780 | | | — | | | — | | | — | |
Term loan | | | 433,973 | | | 61,213 | | | 140,110 | | | 125,238 | | | 107,413 | |
Convertible 2.5% senior notes | | | 222,500 | | | 5,000 | | | 10,000 | | | 207,500 | | | — | |
Operating lease obligations, excluding facility lease | | | 1,514 | | | 642 | | | 872 | | | — | | | — | |
| | | | | | | | | | | | | | | | | |
Total | | $ | 668,308 | | $ | 73,085 | | $ | 153,899 | | $ | 333,912 | | $ | 107,413 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
We have entered into certain operating leases, including certain office equipment and automobile leases, which expire through 2017.
In June 2013, we entered into a lease agreement with BP Bay Colony LLC (the "Landlord") for the lease of certain real property located at 1100 Winter Street, Waltham, Massachusetts (the "Waltham Premises") for use as our principal executive offices. Beginning in September 2013, the initial term of the lease is five years and two months with one five-year extension term at our option.
In June 2013, we also entered into an Assignment and Assumption of Lease (the "Assignment Agreement") with Shire Human Genetic Therapies, Inc. ("Shire") effecting the assignment to Shire of the right to occupy our former office space located at 100 Hayden Avenue, Lexington, Massachusetts (the "Prior Space"). Under the Assignment Agreement, the assignment to Shire became effective on September 21, 2013, the date of our departure from the Prior Space, and Shire assumed all of our obligations as the tenant of the Prior Space. The Assignment Agreement also provided for the conveyance of furniture and other personal property by us to Shire. As a result, our former lease obligations related to our prior office space are no longer shown in the table above.
In connection with our acquisition of Lumara Health, we have assumed the lease of certain real property located at 16640 Chesterfield Grove Road, Chesterfield, Missouri (the "St. Louis Premises"), which we are currently using as temporary office space for Lumara Health employees as they relocate to the Waltham premises. Beginning in September 2013, the initial term of the lease is five years and two months. In addition to base rent, we are also required to pay a proportionate share of the Landlord's operating costs. We are attempting to sublease the St. Louis Premises and if successful, future operating lease commitments will be partially offset by proceeds received from the sublease.
113
Table of Contents
During 2014, we entered into various agreements with third parties for which we had remaining purchase commitments of approximately $4.8 million as of December 31, 2014. These agreements principally related to certain purchase orders for the production ofFeraheme, certain outsourced commercial activities, manufacturing commitments, our information technology infrastructure and other operational activities.
Debt Obligations
Our long-term debt obligations reflect our obligations under the Convertible Notes and Term Loan Facility to pay interest on the $540.0 million aggregate principal amount and to make scheduled principal payments on the Term Loan Facility and principal payments at maturity or upon conversion, in the case of the Convertible Notes.
In connection with our acquisition of Lumara Health, we agreed to pay up to an additional $350.0 million to the former Lumara Health security holders based on the achievement of certain sales milestones. Due to the contingent nature of these milestone payments, we cannot predict the amount or timing of such payments and have therefore not included them in the table above. See Note C, "Business Combinations," for more information on the Lumara Health acquisition and related milestone payments.
As of December 31, 2014, we had several ongoing clinical studies in various clinical trial stages. Our most significant clinical trial expenditures were to clinical research organizations ("CROs"). The contracts with CROs are generally cancellable, with notice, at our option. We have recorded accrued expenses in our consolidated balance sheet of approximately $1.9 million representing expenses incurred with these organizations as of December 31, 2014, net of any amounts prepaid to these CROs. As a result of our cancellation rights, we have not included these CRO contracts in the contractual obligations table above.
We have entered into employment agreements or other arrangements with our executive officers and certain other employees, which provide for salary continuation payments and, in certain instances, the acceleration of the vesting of certain equity awards to such individuals in the event that the individual is terminated other than for cause, as defined in the applicable employment agreements or arrangements.
In the course of operating our business, we have entered into a number of indemnification arrangements under which we may be required to make payments to or on behalf of certain third parties including our directors, officers, and certain employees as well as certain other third parties with whom we enter into agreements. For further discussion of how this may affect our business, see Note Q, "Commitments and Contingencies," to our consolidated financial statements included in this Annual Report on Form 10-K.
114
Table of Contents
Legal Proceedings
For detailed information on our legal proceedings, see Note Q, "Commitments and Contingencies," to our consolidated financial statements included in this Annual Report on Form 10-K.
Off-Balance Sheet Arrangements
As of December 31, 2014, we did not have any off-balance sheet arrangements as defined in Regulation S-K, Item 303(a)(4)(ii).
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK:
Interest Rate Risk
As of December 31, 2014 and 2013, our investments equaled $24.9 million and $186.8 million, respectively, and were invested in corporate debt securities, commercial paper and U.S. treasury and government agency securities. Our investments meet high credit quality and diversification standards, as specified in our investment policy. Our investment policy also limits the amount of our credit exposure to any one issue or issuer, excluding U.S. government entities, and seeks to manage these assets to achieve our goals of preserving principal, maintaining adequate liquidity at all times, and maximizing returns. These investments are subject to interest rate risk. The modeling technique used measures the change in fair values arising from an immediate hypothetical shift in market interest rates and assumes that ending fair values include principal plus accrued interest. If market interest rates for comparable investments were to increase immediately and uniformly by 50 basis points, or one-half of a percentage point, from levels as of December 31, 2014 and 2013, this would have resulted in a hypothetical decline in fair value of our investments of approximately $0.1 million and $1.3 million, respectively, and if market interest rates for comparable investments were to decrease immediately and uniformly by 50 basis points, or one-half of a percentage point, from levels as of December 31, 2014 and 2013, this would have resulted in a hypothetical increase in fair value of our investments of approximately $0.1 million and $1.2 million, respectively. These amounts are determined by considering the impact of the hypothetical interest rate movements on our available-for-sale investment portfolios. This analysis does not consider the effect of credit risk as a result of the changes in overall economic activity that could exist in such an environment.
Equity Price Risk
Our Convertible Notes include conversion and settlement provisions that are based on the price of our common stock at conversion or at maturity of the notes. The amount of cash we may be required to pay is determined by the price of our common stock. The fair values of our Convertible Notes are dependent on the price and volatility of our common stock and will generally increase or decrease as the market price of our common stock changes.
On February 14, 2014, we issued $200.0 million aggregate principal amount of Convertible Notes. The Convertible Notes are senior unsecured obligations and bear interest at a rate of 2.5% per year, payable semi-annually in arrears on February 15 and August 15 of each year, beginning on August 15, 2014. The Convertible Notes will mature on February 15, 2019, unless earlier repurchased or converted. The Convertible Notes (which are currently convertible) will be convertible into cash, shares of our common stock, or a combination thereof, at our election (subject to certain limitations in the Term Loan Facility), at an initial conversion rate of approximately 36.9079 shares of common stock per $1,000 principal amount of the Convertible Notes, which corresponds to an initial conversion price of approximately $27.09 per share of our common stock and represents a conversion premium of approximately 35% based on the last reported sale price of our common stock of $20.07 on
115
Table of Contents
February 11, 2014, the date the notes offering was priced. As of December 31, 2014, the fair value of the Convertible Notes was $332.0 million.
In order to reduce the potential dilution to our common stock and/or offset cash payments due upon conversion of the Convertible Notes, in February 2014 we entered into convertible bond hedge transactions covering approximately 7.4 million shares of our common stock underlying the $200.0 million aggregate principal amount of the Convertible Notes. The convertible bond hedges have an exercise price of approximately $27.09 per share, subject to adjustment upon certain events, and are exercisable when and if the Convertible Notes are converted. If upon conversion of the Convertible Notes, the price of our common stock is above the exercise price of the convertible bond hedges, each of JPMorgan Chase Bank, National Association, London Branch, Morgan Stanley & Co. International plc and Royal Bank of Canada will deliver shares of our common stock and/or cash with an aggregate value approximately equal to the difference between the price of our common stock at the conversion date and the exercise price, multiplied by the number of shares of our common stock related to the convertible bond hedges being exercised.
In February 2014, we also entered into separate warrant transactions relating to, in the aggregate, approximately 7.4 million shares of our common stock underlying the $200.0 million aggregate principal amount of the Convertible Notes. The initial exercise price of the warrants is $34.12 per share, subject to adjustment upon certain events, which is 70% above the last reported sale price of our common stock of $20.07 on February 11, 2014. The warrants would separately have a dilutive effect to the extent that the market value per share of our common stock, as measured under the terms of the warrants, exceeds the applicable exercise price of the warrants.
116
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA:
Index To Consolidated Financial Statements
| | | |
Management's Annual Report on Internal Control over Financial Reporting | | 118 | |
Report of Independent Registered Public Accounting Firm | |
119 | |
Consolidated Balance Sheets—as of December 31, 2014 and 2013 | |
121 | |
Consolidated Statements of Operations—for the years ended December 31, 2014, 2013 and 2012 | |
122 | |
Consolidated Statements of Comprehensive Income (Loss)—for the years ended December 31, 2014, 2013 and 2012 | |
123 | |
Consolidated Statements of Stockholders' Equity—as of December 31, 2014, 2013 and 2012 | |
124 | |
Consolidated Statements of Cash Flows—for the years ended December 31, 2014, 2013 and 2012 | |
125 | |
Notes to Consolidated Financial Statements | |
126 | |
117
Table of Contents
MANAGEMENT'S ANNUAL REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Management is responsible for establishing and maintaining adequate internal controls over financial reporting, as such term is defined in Rule 13a-15(f) under the Securities and Exchange Act of 1934, as amended. Our internal control over financial reporting is a process designed under the supervision of our principal executive officer and principal financial officer to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external reporting purposes in accordance with U.S. generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management, including our principal executive officer and principal financial officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2014 based on the framework inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013. Our assessment did not include evaluating the effectiveness of internal control over financial reporting of recently acquired Lumara Health Inc. or Lumara Health Inc.'s subsidiaries, the consolidated results of which are included in our fiscal year 2014 consolidated financial statements and constituted 7% of total assets as of December 31, 2014 and 18% of total revenue for the year then ended. Based on this assessment, management concluded that our internal control over financial reporting was effective as of December 31, 2014.
The effectiveness of our internal control over financial reporting as of December 31, 2014, has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report which is included below.
118
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of AMAG Pharmaceuticals, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, comprehensive loss, stockholders' equity, and cash flows present fairly, in all material respects, the financial position of AMAG Pharmaceuticals, Inc. and its subsidiaries at December 31, 2014, and 2013, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2014 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedules listed in the accompanying index present fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013. The Company's management is responsible for these financial statements and financial statement schedules, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Annual Report on Internal Control over Financial Reporting. Our responsibility is to express opinions on these financial statements, on the financial statement schedules, and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
As described in Management's Annual Report on Internal Control over Financial Reporting, management has excluded Lumara Health Inc. from its assessment of internal control over financial
119
Table of Contents
reporting as of December 31, 2014 because it was acquired by the Company in a purchase business combination during 2014. We have also excluded Lumara Health Inc. from our audit of internal control over financial reporting. Lumara Health Inc. is a wholly-owned subsidiary whose total assets and total revenues represent 7% and 18%, respectively, of the related consolidated financial statement amounts as of and for the year ended December 31, 2014.
Boston, Massachusetts
February 17, 2015
120
Table of Contents
AMAG PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(IN THOUSANDS, EXCEPT SHARE AND PER SHARE DATA)
| | | | | | | |
| | As of December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
ASSETS | | | | | | | |
Current assets: | | | | | | | |
Cash and cash equivalents | | $ | 119,296 | | $ | 26,986 | |
Investments | | | 24,890 | | | 186,803 | |
Accounts receivable, net | | | 38,172 | | | 6,842 | |
Inventories | | | 40,610 | | | 17,217 | |
Receivable from collaboration | | | 4,518 | | | 278 | |
Deferred tax assets | | | 32,094 | | | — | |
Prepaid and other current assets | | | 14,456 | | | 3,396 | |
Restricted cash | | | — | | | 2,883 | |
| | | | | | | | |
Total current assets | | | 274,036 | | | 244,405 | |
Property and equipment, net | | | 1,519 | | | 1,846 | |
Goodwill | | | 205,824 | | | — | |
Intangible assets, net | | | 887,908 | | | 16,844 | |
Restricted cash | | | 2,397 | | | 400 | |
Other long-term assets | | | 17,249 | | | 1,964 | |
| | | | | | | | |
Total assets | | $ | 1,388,933 | | $ | 265,459 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
LIABILITIES AND STOCKHOLDERS' EQUITY | | | | | | | |
Current liabilities: | | | | | | | |
Accounts payable | | $ | 7,301 | | $ | 2,629 | |
Accrued expenses | | | 80,811 | | | 22,266 | |
Current portion of long-term debt | | | 34,000 | | | — | |
Deferred revenues | | | 44,376 | | | 8,226 | |
| | | | | | | | |
Total current liabilities | | | 166,488 | | | 33,121 | |
Long-term liabilities: | | | | | | | |
Long-term debt, net | | | 293,905 | | | — | |
Convertible 2.5% senior notes, net | | | 167,441 | | | — | |
Acquisition-related contingent consideration | | | 217,984 | | | 13,609 | |
Deferred income tax liability | | | 77,619 | | | — | |
Deferred revenues | | | — | | | 44,534 | |
Other long-term liabilities | | | 5,543 | | | 1,787 | |
| | | | | | | | |
Total liabilities | | | 928,980 | | | 93,051 | |
Commitments and contingencies | | | | | | | |
Stockholders' equity: | | | | | | | |
Preferred stock, par value $0.01 per share, 2,000,000 shares authorized; none issued | | | — | | | — | |
Common stock, par value $0.01 per share, 58,750,000 shares authorized; 25,599,550 and 21,772,571 shares issued and outstanding at December 31, 2014 and 2013, respectively | | | 256 | | | 218 | |
Additional paid-in capital | | | 793,757 | | | 641,941 | |
Accumulated other comprehensive loss | | | (3,617 | ) | | (3,491 | ) |
Accumulated deficit | | | (330,443 | ) | | (466,260 | ) |
| | | | | | | | |
Total stockholders' equity | | | 459,953 | | | 172,408 | |
| | | | | | | | |
Total liabilities and stockholders' equity | | $ | 1,388,933 | | $ | 265,459 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
121
Table of Contents
AMAG PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(IN THOUSANDS, EXCEPT PER SHARE DATA)
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Revenues: | | | | | | | | | | |
U.S. product sales, net | | $ | 108,795 | | $ | 71,362 | | $ | 58,287 | |
License fee and other collaboration revenues | | | 10,886 | | | 8,385 | | | 26,475 | |
Other product sales and royalties | | | 4,703 | | | 1,109 | | | 616 | |
| | | | | | | | | | | |
Total revenues | | | 124,384 | | | 80,856 | | | 85,378 | |
| | | | | | | | | | | |
Costs and expenses: | | | | | | | | | | |
Cost of product sales | | | 20,306 | | | 11,960 | | | 14,220 | |
Research and development expenses | | | 24,160 | | | 20,564 | | | 33,296 | |
Selling, general and administrative expenses | | | 72,254 | | | 59,167 | | | 53,071 | |
Acquistion-related costs | | | 9,478 | | | 782 | | | — | |
Restructuring expenses | | | 2,023 | | | — | | | 2,215 | |
| | | | | | | | | | | |
Total costs and expenses | | | 128,221 | | | 92,473 | | | 102,802 | |
| | | | | | | | | | | |
Operating loss | | | (3,837 | ) | | (11,617 | ) | | (17,424 | ) |
| | | | | | | | | | | |
Other income (expense): | | | | | | | | | | |
Interest expense | | | (14,697 | ) | | — | | | — | |
Interest and dividend income, net | | | 975 | | | 1,051 | | | 1,286 | |
Gains on sale of assets | | | 103 | | | 924 | | | — | |
Gains (losses) on investments, net | | | 114 | | | 40 | | | (1,466 | ) |
| | | | | | | | | | | |
Total other income (expense) | | | (13,505 | ) | | 2,015 | | | (180 | ) |
| | | | | | | | | | | |
Net income (loss) before income taxes | | | (17,342 | ) | | (9,602 | ) | | (17,604 | ) |
Income tax benefit | | | 153,159 | | | — | | | 854 | |
| | | | | | | | | | | |
Net income (loss) | | $ | 135,817 | | $ | (9,602 | ) | $ | (16,750 | ) |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Net income (loss) per share: | | | | | | | | | | |
Basic | | $ | 6.06 | | $ | (0.44 | ) | $ | (0.78 | ) |
Diluted | | $ | 5.45 | | $ | (0.44 | ) | $ | (0.78 | ) |
Weighted average shares outstanding used to compute net income (loss) per share: | | | | | | | | | | |
Basic | | | 22,416 | | | 21,703 | | | 21,392 | |
Diluted | | | 25,225 | | | 21,703 | | | 21,392 | |
The accompanying notes are an integral part of these consolidated financial statements.
122
Table of Contents
AMAG PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(IN THOUSANDS)
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Net income (loss) | | $ | 135,817 | | $ | (9,602 | ) | $ | (16,750 | ) |
| | | | | | | | | | | |
Other comprehensive income (loss): | | | | | | | | | | |
Unrealized gains (losses) on securities: | | | | | | | | | | |
Holding gains (losses) arising during period, net of tax | | | (191 | ) | | (268 | ) | | 129 | |
Reclassification adjustment for (gains) losses included in net income (loss) | | | 65 | | | 24 | | | 1,466 | |
| | | | | | | | | | | |
Net unrealized gains (losses) on securities | | | (126 | ) | | (244 | ) | | 1,595 | |
| | | | | | | | | | | |
Total comprehensive income (loss) | | $ | 135,691 | | $ | (9,846 | ) | $ | (15,155 | ) |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
123
Table of Contents
AMAG PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(IN THOUSANDS)
| | | | | | | | | | | | | | | | | | | |
| | Common Stock | |
| | Accumulated
Other
Comprehensive
Income (Loss) | |
| |
| |
|---|
| | Additional
Paid-in
Capital | | Accumulated
Deficit | | Total
Stockholders'
Equity | |
|---|
| | Shares | | Amount | |
|---|
Balance at December 31, 2011 | | | 21,306 | | | 213 | | | 625,133 | | | (4,842 | ) | | (439,908 | ) | | 180,596 | |
| | | | | | | | | | | | | | | | | | | | |
Net shares issued in connection with the exercise of stock options and restricted stock units | | | 178 | | | 2 | | | 98 | | | — | | | — | | | 100 | |
Shares issued in connection with employee stock purchase plan | | | 23 | | | — | | | 270 | | | — | | | — | | | 270 | |
Non-cash equity-based compensation | | | — | | | — | | | 6,986 | | | — | | | — | | | 6,986 | |
Unrealized gains on securities, net of tax of $0.9 million | | | — | | | — | | | — | | | 1,595 | | | — | | | 1,595 | |
Net loss | | | — | | | — | | | — | | | — | | | (16,750 | ) | | (16,750 | ) |
| | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | | | 21,507 | | | 215 | | | 632,487 | | | (3,247 | ) | | (456,658 | ) | | 172,797 | |
| | | | | | | | | | | | | | | | | | | | |
Net shares issued in connection with the exercise of stock options and restricted stock units | | | 252 | | | 3 | | | 1,274 | | | — | | | — | | | 1,277 | |
Shares issued in connection with employee stock purchase plan | | | 14 | | | — | | | 176 | | | — | | | — | | | 176 | |
Non-cash equity-based compensation | | | — | | | — | | | 8,004 | | | — | | | — | | | 8,004 | |
Unrealized losses on securities | | | — | | | — | | | — | | | (244 | ) | | — | | | (244 | ) |
Net loss | | | — | | | — | | | — | | | — | | | (9,602 | ) | | (9,602 | ) |
| | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2013 | | | 21,773 | | | 218 | | | 641,941 | | | (3,491 | ) | | (466,260 | ) | | 172,408 | |
| | | | | | | | | | | | | | | | | | | | |
Equity component of Convertible Notes, net of issuance costs | | | — | | | — | | | 36,907 | | | — | | | — | | | 36,907 | |
Purchase of convertible bond hedges, net of tax | | | — | | | — | | | (39,760 | ) | | — | | | — | | | (39,760 | ) |
Sale of warrants | | | — | | | — | | | 25,620 | | | — | | | — | | | 25,620 | |
Net shares issued in connection with the acquisition of Lumara Health | | |
3,210 | | |
32 | | |
111,932 | | |
— | | |
— | | |
111,964 | |
Net shares issued in connection with the exercise of stock options and vesting of restricted stock units | | | 617 | | | 6 | | | 8,492 | | | — | | | — | | | 8,498 | |
Non-cash equity-based compensation | | | — | | | — | | | 8,625 | | | — | | | — | | | 8,625 | |
Unrealized losses on securities | | | — | | | — | | | — | | | (126 | ) | | — | | | (126 | ) |
Net income | | | — | | | — | | | — | | | — | | | 135,817 | | | 135,817 | |
| | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2014 | | | 25,600 | | $ | 256 | | $ | 793,757 | | $ | (3,617 | ) | $ | (330,443 | ) | $ | 459,953 | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
124
Table of Contents
AMAG PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(IN THOUSANDS)
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Cash flows from operating activities: | | | | | | | | | | |
Net income (loss) | | $ | 135,817 | | $ | (9,602 | ) | $ | (16,750 | ) |
Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | | |
| | |
| | |
| |
Depreciation and amortization | | | 6,984 | | | 3,085 | | | 3,084 | |
Impairment loss on assets held for sale | | | — | | | — | | | 1,100 | |
Amortization of premium/discount on purchased securities | | | 2,080 | | | 2,758 | | | 2,808 | |
Write-down of inventory to net realizable value | | | 1,309 | | | 2,175 | | | 1,822 | |
Non-cash equity-based compensation expense | | | 8,625 | | | 8,004 | | | 7,024 | |
Amortization of debt discount and debt issuance costs | | | 6,870 | | | — | | | — | |
Non-cash income tax benefit | | | — | | | — | | | (854 | ) |
Gains on sale of assets | | | (103 | ) | | (924 | ) | | — | |
(Gains) losses on investments, net | | | (114 | ) | | (40 | ) | | 1,466 | |
Change in fair value of contingent consideration | | | (681 | ) | | 1,074 | | | — | |
Deferred income taxes | | | (153,159 | ) | | — | | | — | |
Changes in operating assets and liabilities: | | | | | | | | | | |
Accounts receivable, net | | | 3,588 | | | (432 | ) | | (478 | ) |
Inventories | | | (1,360 | ) | | (1,040 | ) | | 4,069 | |
Receivable from collaboration | | | (4,239 | ) | | (15 | ) | | 165 | |
Prepaid and other current assets | | | 2,331 | | | 2,817 | | | 75 | |
Other long-term assets | | | 1,964 | | | (1,964 | ) | | — | |
Accounts payable and accrued expenses | | | 10,694 | | | (5,730 | ) | | (12,195 | ) |
Deferred revenues | | | (8,384 | ) | | (6,694 | ) | | 7,912 | |
Other long-term liabilities | | | (808 | ) | | (246 | ) | | (405 | ) |
| | | | | | | | | | | |
Total adjustments | | | (124,403 | ) | | 2,828 | | | 15,593 | |
| | | | | | | | | | | |
Net cash provided by (used in) operating activities | | | 11,414 | | | (6,774 | ) | | (1,157 | ) |
| | | | | | | | | | | |
Cash flows from investing activities: | | | | | | | | | | |
Acquisition of Lumara Health, net of acquired cash | | | (595,602 | ) | | — | | | — | |
Proceeds from sales or maturities of investments | | | 223,568 | | | 106,030 | | | 133,061 | |
Purchase of investments | | | (63,747 | ) | | (115,046 | ) | | (149,406 | ) |
Acquisition of MuGard Rights and inventory | | | — | | | (3,434 | ) | | — | |
Proceeds from sale of assets | | | 103 | | | 2,970 | | | — | |
Change in restricted cash | | | 2,883 | | | (2,823 | ) | | — | |
Capital expenditures | | | (147 | ) | | (1,632 | ) | | (47 | ) |
| | | | | | | | | | | |
Net cash (used in) investing activities | | | (432,942 | ) | | (13,935 | ) | | (16,392 | ) |
| | | | | | | | | | | |
Cash flows from financing activities: | | | | | | | | | | |
Payment of contingent consideration | | | (270 | ) | | (51 | ) | | — | |
Proceeds from term loan | | | 327,509 | | | — | | | — | |
Proceeds from issuance of convertible 2.5% senior notes | | | 200,000 | | | — | | | — | |
Payment of debt issuance costs | | | (7,760 | ) | | — | | | — | |
Proceeds from issuance of warrants | | | 25,620 | | | — | | | — | |
Purchase of convertible bond hedges | | | (39,760 | ) | | — | | | — | |
Proceeds from the exercise of stock options | | | 8,499 | | | 1,277 | | | 98 | |
Proceeds from the issuance of common stock under ESPP | | | — | | | 176 | | | 270 | |
| | | | | | | | | | | |
Net cash provided by financing activities | | | 513,838 | | | 1,402 | | | 368 | |
| | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents | | | 92,310 | | | (19,307 | ) | | (17,181 | ) |
Cash and cash equivalents at beginning of the year | | | 26,986 | | | 46,293 | | | 63,474 | |
| | | | | | | | | | | |
Cash and cash equivalents at end of the year | | $ | 119,296 | | $ | 26,986 | | $ | 46,293 | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
Supplemental data of cash flow information: | | | | | | | | | | |
Interest paid on convertible 2.5% senior notes | | $ | 2,500 | | $ | — | | $ | — | |
Non-cash investing activities: | | |
| | |
| | |
| |
Accrued construction in progress | | $ | — | | $ | — | | $ | 228 | |
Fair value of acquisition-related contingent consideration | | $ | 205,000 | | $ | 13,700 | | $ | — | |
Fair value of common stock issued in connection with the Lumara Health acquisition | | $ | 111,964 | | $ | — | | $ | — | |
The accompanying notes are an integral part of these consolidated financial statements.
125
Table of Contents
AMAG PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
A. DESCRIPTION OF BUSINESS
AMAG Pharmaceuticals, Inc., a Delaware corporation, was founded in 1981. We are a specialty pharmaceutical company that markets Makena® (hydroxyprogesterone caproate injection), Feraheme® (ferumoxytol) Injection for Intravenous ("IV") use and MuGard® Mucoadhesive Oral Wound Rinse.
On November 12, 2014, we acquired Lumara Health Inc. ("Lumara Health"), a privately held pharmaceutical company specializing in women's health, for approximately $600.0 million in upfront cash consideration (subject to finalization of certain adjustments related to Lumara Health's financial position at the time of closing, including adjustments related to net working capital, net debt and transaction expenses) and approximately 3.2 million shares of our common stock having a fair value of approximately $112.0 million at the time of closing (the "Lumara Agreement"). The Lumara Agreement includes future contingent payments of up to $350.0 million in cash (or upon mutual agreement between us and the former Lumara Health security holders, future contingent payments may also be made in common stock or some combination thereof) payable by us to the former Lumara Health security holders based upon the achievement of certain sales milestones through calendar year 2019. In connection with the acquisition of Lumara Health, we acquiredMakena, a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth. We sellMakena to specialty pharmacies and distributors, who, in turn sellMakena to healthcare providers, hospitals, government agencies and integrated delivery systems. Additional details regarding the acquisition of Lumara Health can be found in Note C, "Business Combinations," to our consolidated financial statements included in this Annual Report on Form 10-K.
We also market and sellFeraheme, which was approved for marketing in the U.S. in June 2009 by the U.S. Food and Drug Administration ("FDA") for use as an IV iron replacement therapy for the treatment of iron deficiency anemia ("IDA") in adult patients with chronic kidney disease ("CKD"). We began sellingFeraheme in the U.S. in July 2009 through our commercial organization, including a specialty sales force. We sellFeraheme to authorized wholesalers and specialty distributors, who in turn, sellFeraheme to healthcare providers who administerFeraheme primarily within hospitals, hematology and oncology centers, and nephrology clinics.
Outside of the U.S., ferumoxytol has been granted marketing approval in the European Union ("EU"), Canada and Switzerland for use as an IV iron replacement therapy for the treatment of IDA in adult patients with CKD. In March 2010, we entered into a License, Development and Commercialization Agreement (the "Takeda Agreement"), which was amended in June 2012 (the "Amended Takeda Agreement") with Takeda Pharmaceutical Company Limited ("Takeda"). On December 29, 2014, we entered into an agreement with Takeda to terminate the Amended Takeda Agreement and we will regain all worldwide development and commercialization rights forFeraheme following the transfer of marketing authorizations (the "Takeda Termination Agreement"). Additional details regarding the Takeda Termination Agreement can be found in Note R, "Collaborative Agreements". Under the Amended Takeda Agreement, Takeda had an exclusive license to market and sell ferumoxytol in the EU, Canada and Switzerland, as well as certain other geographic territories. The EU marketing authorization forRienso is valid in the 28 EU Member States as well as in Iceland, Liechtenstein and Norway. We have recently come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada. The trade name for ferumoxytol in Canada isFeraheme and outside of the U.S. and Canada the trade name isRienso.
We are subject to risks common to companies in the pharmaceutical industry including, but not limited to (as such risks pertain to our business) our dependence on the success of our product
126
Table of Contents
portfolio and maintaining commercialization of our products, includingMakena andFeraheme; intense competition, including from generic products; maintaining the proprietary nature of our technology; our dependence upon third party manufacturers; our reliance on other third parties in our business, including to conduct our clinical trials and undertake our product distribution; our reliance on and the extent of reimbursement from third parties for the use of our products, includingMakena's high Medicaid reimbursement concentration; the impact ofMakena's loss of orphan drug exclusivity in February 2018; competition from compounded pharmacies; our ability to implementMakena's lifecycle management program; perceptions related to pricing and access forMakena; post-marketing commitments forMakena; limitations onFeraheme sales given its narrow CKD indication and the potential impact on sales of any actual or perceived safety problems; our ability to receive regulatory approval forFeraheme in the broader IDA indication andFeraheme's ability to compete in such market even if regulatory approval is pursued and received; our customer concentration, especially with regard toFeraheme; the impact of the termination of our license arrangement with Takeda and our commercialization efforts, if any and including cessation thereof, forFeraheme outside of the U.S., including the impact on U.S. sales; uncertainties regarding federal and state legislative initiatives; potential inability to obtain raw or other materials; our potential inadvertent failure to comply with federal, state or foreign healthcare fraud and abuse laws, marketing disclosure laws or other federal, state or foreign laws and regulations; uncertainties regarding reporting and payment obligations under government pricing programs and our level of indebtedness, our access to sufficient capital, the availability of net operating loss carryforwards and other tax assets, employee retention, our ability to be profitable in the future, the potential fluctuation of our operating results, potential differences between actual future results and the estimates or assumptions used by us in preparation of our consolidated financial statements, the volatility of our stock price, potential litigation, including securities and product liability suits and the impact of market overhang on our stock price.
Throughout this Annual Report on Form 10-K, AMAG Pharmaceuticals, Inc. and our consolidated subsidiaries are collectively referred to as "the Company," "AMAG," "we," "us," or "our." Unless the context suggests otherwise, references to "Feraheme" refer to bothFeraheme (the trade name for ferumoxytol in the U.S. and Canada) andRienso (the trade name for ferumoxytol in the EU and Switzerland).
B. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates and Assumptions
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the U.S. ("GAAP") requires management to make certain estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses, and the related disclosure of contingent assets and liabilities. The most significant estimates and assumptions are used to determine amounts and values of, but are not limited to: revenue related to product sales and collaboration agreements; product sales allowances and accruals; potential other-than-temporary impairment of investments; acquisition date fair value and subsequent fair value estimates used to assess impairment of long-lived assets, including goodwill, in-process research and development ("IPR&D") and other intangible assets; contingent consideration; debt obligations; accrued expenses; income taxes and equity-based compensation expense. Actual results could differ materially from those estimates.
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements have been prepared in conformity with GAAP and include the accounts of our wholly owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation. As of November 12, 2014 (the "Lumara Acquisition Date"), the
127
Table of Contents
operating results of Lumara Health have been consolidated with ours. See Note C, "Business Combinations," for additional information.
Cash and Cash Equivalents
Cash and cash equivalents consist principally of cash held in commercial bank accounts, money market funds and U.S. Treasury securities having an original maturity of less than three months. We consider all highly liquid investments with a maturity of three months or less as of the acquisition date to be cash equivalents. At December 31, 2014, substantially all of our cash and cash equivalents were held in either commercial bank accounts or money market funds.
Investments
We account for and classify our investments as either "available-for-sale," "trading," or "held-to-maturity," in accordance with current guidance related to the accounting and classification of certain investments in debt and equity securities. The determination of the appropriate classification by us is based on a variety of factors, including management's intent at the time of purchase. As of December 31, 2014 and 2013, all of our investments were classified as available-for-sale securities.
Available-for-sale securities are those securities which we view as available for use in current operations, if needed. We generally classify our available-for-sale securities as short-term investments, even though the stated maturity date may be one year or more beyond the current balance sheet date. Available-for-sale investments are stated at fair value with their unrealized gains and losses included as a separate component of stockholders' equity entitled "Accumulated other comprehensive loss," until such gains and losses are realized or until an unrealized loss is considered other-than-temporary.
We recognize and report other-than-temporary impairments of our debt securities in accordance with current accounting guidance, which requires that for debt securities with a decline in fair value below amortized cost basis, an other-than-temporary impairment exists if (a) we have the intent to sell the security or (b) it is more likely than not that we will be required to sell the security prior to recovery of its amortized cost basis. If either of these conditions is met, we recognize the difference between the amortized cost of the security and its fair value at the impairment measurement date in our consolidated statement of operations. If neither of these conditions is met, we must perform additional analyses to evaluate whether the unrealized loss is associated with the creditworthiness of the security rather than other factors, such as interest rates or market factors. These factors include evaluation of the security, issuer and other factors such as the duration of the period that, and extent to which, the fair value was less than cost basis, the financial health of and business outlook for the issuer, including industry and sector performance, operational and financing cash flow factors, overall market conditions and trends, underlying collateral, whether we have a favorable history in redeeming similar securities at prices at or above fair value, and credit ratings with respect to our investments provided by investments ratings agencies. If we determine from this analysis that we do not expect to receive cash flows sufficient to recover the entire amortized cost of the security, a credit loss exists. In this situation, the impairment is considered other-than-temporary and is recognized in our consolidated statement of operations.
Fair Value Measurements
Under current accounting standards, fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs.
128
Table of Contents
Current accounting guidance establishes a hierarchy used to categorize how fair value is measured and which is based on three levels of inputs, of which the first two are considered observable and the third unobservable, as follows:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities, quoted prices in markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
We hold certain assets and liabilities that are required to be measured at fair value on a recurring basis, including our cash equivalents, investments, and acquisition-related contingent consideration.
We also analyze when the volume and level of activity for an asset or liability have significantly decreased and when circumstances indicate that a transaction may not be considered orderly. In order to determine whether the volume and level of activity for an asset or liability have significantly decreased, we assess current activity as compared to normal market activity for the asset or liability. We rely on many factors such as trading volume, trading frequency, the levels at which market participants indicate their willingness to buy and sell our securities, as reported by market participants, and current market conditions. Using professional judgment and experience, we evaluate and weigh the relevance and significance of all applicable factors to determine if there has been a significant decrease in the volume and level of activity for an asset, group of similar assets or liabilities. Similarly, in order to identify transactions that are not orderly, we take into consideration the activity in the market which can influence the determination and occurrence of an orderly transaction. Also, we inquire as to whether there may have been restrictions on the marketing of the security to a single or limited number of participants. Where possible, we assess the financial condition of the seller to determine whether observed transactions may have been forced. If there is a significant disparity between the trading price for a security held by us as compared to the trading prices of similar recent transactions, we consider whether this disparity is an indicator of a disorderly trade. Using professional judgment and experience, we evaluate and weigh the relevance and significance of all applicable factors to determine if the evidence suggests that a transaction or group of similar transactions is not orderly. Based upon these procedures, we determined that market activity for our assets appeared normal and that transactions did not appear disorderly as of December 31, 2014 and 2013.
Inventory
Inventory is stated at the lower of cost or market (net realizable value), with approximate cost being determined on a first-in, first-out basis. Prior to initial approval from the FDA or other regulatory agencies, we expense costs relating to the production of inventory in the period incurred. After such time as the product receives initial regulatory approval, we begin to capitalize the inventory costs related to the product. We continue to expense costs associated with clinical trial material as research and development expense.
On a quarterly basis, we analyze our inventory levels to determine whether we have any obsolete, expired, or excess inventory. If any inventory is expected to expire prior to being sold, has a cost basis in excess of its net realizable value, is in excess of expected sales requirements as determined by internal sales forecasts, or fails to meet commercial sale specifications, the inventory is written-down through a charge to cost of product sales. The determination of whether inventory costs will be realizable requires estimates by management of future expected inventory requirements, based on sales forecasts. Once packaged,Feraheme currently has a shelf-life of five years in the U.S. and between two
129
Table of Contents
and three years outside of the U.S. and Makena has a shelf-life of three years. As a result of comparison to internal sales forecasts, we expect to fully realize the carrying value of our currentFeraheme andMakena finished goods inventory. If actual market conditions are less favorable than those projected by management, inventory write-downs may be required. Charges for inventory write-downs are not reversed if it is later determined that the product is saleable.
Restricted Cash
As of December 31, 2014 and 2013, we classified $2.4 million and $3.3 million as restricted cash, respectively. The $2.4 million in our December 31, 2014 restricted cash balances includes $2.0 million held in a restricted fund previously established by Lumara Health in connection with its Chapter 11 plan of reorganization to pay potential claims against its former directors and officers and a $0.4 million security deposit delivered to the landlord of our Waltham, Massachusetts headquarters in the form of an irrevocable letter of credit. Included in the $3.3 million restricted cash balance as of December 31, 2013 was a $2.9 million escrow payment related to a business development transaction that we did not complete as well as the $0.4 million security deposit related to our Waltham, Massachusetts headquarters described above. The escrow payment was returned to us in January 2014 and as such was classified as short-term as of December 31, 2013.
Property and Equipment
Property and equipment are recorded at cost and depreciated when placed into service using the straight-line method based on their estimated useful lives. Our laboratory and production equipment and furniture and fixtures are being depreciated over five years. Furniture, fixtures, and leasehold improvements associated with our facility lease are being depreciated over the shorter of their useful lives or the remaining life of the original lease (excluding optional lease renewal terms).
Costs for capital assets not yet placed in service are capitalized on our balance sheets and will be depreciated in accordance with the above guidelines once placed into service. Costs for maintenance and repairs are expensed as incurred. Upon sale or other disposition of property and equipment, the cost and related depreciation are removed from the accounts and any resulting gain or loss is charged to our consolidated statement of operations. Long-lived assets to be held and used are evaluated for impairment whenever events or changes in circumstances indicate that the carrying value of the asset may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset (asset group) and its eventual disposition. In the event such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values. Assets classified as held for sale are no longer subject to depreciation and are recorded at the lower of carrying value or estimated net realizable value.
Business Combinations
We account for acquired businesses using the acquisition method of accounting, which requires that assets acquired and liabilities assumed be recognized at their estimated fair values as of the acquisition date. Acquisition-related costs are expensed as incurred. Any excess of the consideration transferred over the estimated fair values of the net assets acquired is recorded as goodwill.
Acquisition-Related Contingent Consideration
Contingent consideration arising from a business combination is included as part of the purchase price and is recognized at fair value as of the acquisition date. Subsequent to the acquisition date, we measure contingent consideration arrangements at fair value for each period until the contingency is resolved. These changes in fair value are recognized in our consolidated statements of operations.
130
Table of Contents
Changes in fair values reflect new information about the likelihood of the payment of the contingent consideration and the passage of time.
Goodwill and Intangible Assets
Goodwill represents the excess purchase price paid in a business combination over the fair value of identifiable net assets acquired. Goodwill is not amortized, but is reviewed for impairment on an annual basis or more frequently if indicators of impairment are present. We determine whether goodwill may be impaired by comparing the carrying value of the reporting unit, including goodwill, to the fair value of the reporting unit. If the fair value is less than the carrying amount, a more detailed analysis is performed to determine whether goodwill is impaired. The impairment loss, if any, is measured as the excess of the carrying value of the goodwill over the implied value of the goodwill and is recorded in our consolidated statements of operations.
Finite-lived intangible assets are amortized to their estimated residual values over their estimated useful lives and reviewed for impairment if certain events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. When such facts and circumstances exist, management compares the projected undiscounted future cash flows associated with the asset over its estimated useful life against the carrying amount. The impairment loss, if any, is measured as the excess of the carrying amount of the asset over its fair value.
Acquired IPR&D represents the fair value assigned to research and development assets that we acquire that have not been completed at the date of acquisition. The fair value of IPR&D acquired in a business combination is capitalized on our consolidated balance sheet at the acquisition-date fair value. IPR&D is not amortized, but is reviewed for impairment on an annual basis or more frequently if indicators of impairment are present, until completion or abandonment of the projects. If we determine that IPR&D becomes impaired or is abandoned, the carrying value of the IPR&D is written down to its fair value with the related impairment charge recognized in our consolidated statement of operations in the period in which the impairment occurs. Upon successful completion of each project and launch of the product, we will make a separate determination of the estimated useful life of the IPR&D intangible asset and the related amortization will be recorded as an expense prospectively over its estimated useful life.
The projected discounted cash flow models used to estimate our IPR&D reflect significant assumptions regarding the estimates a market participant would make in order to evaluate a drug development asset including the following:
- •
- Probability of successfully completing clinical trials and obtaining regulatory approval;
- •
- Market size, market growth projections, and market share;
- •
- Estimates regarding the timing of and the expected costs to advance our clinical programs to commercialization;
- •
- Estimates of future cash flows from potential product sales; and
- •
- a discount rate.
Patents
We expense all patent-related costs as incurred.
Revenue Recognition and Related Sales Allowances and Accruals
We recognize revenue from the sale of our products as well as license fee and other collaboration revenues, including milestone payments, other product sale revenues, and royalties we receive from our
131
Table of Contents
licensees. We recognize revenue in accordance with current accounting guidance related to the recognition, presentation and disclosure of revenue in financial statements, which outlines the basic criteria that must be met to recognize revenue and provides guidance for disclosure of revenue in financial statements. We recognize revenue when:
- •
- Persuasive evidence of an arrangement exists;
- •
- Delivery of product has occurred or services have been rendered;
- •
- The sales price charged is fixed or determinable; and
- •
- Collection is reasonably assured.
U.S. Product Sales, Net
We record product sales allowances and accruals related to prompt payment discounts, chargebacks, government and other rebates, distributor, wholesaler and group purchasing organization ("GPO") fees, and product returns as a reduction of revenue in our consolidated statement of operations at the time product sales are recorded. Calculating these gross to net sales adjustments involves estimates and judgments based primarily on actual product sales data, forecasted customer buying patterns, and market research data related to utilization rates by various end-users. In addition, we also monitor our distribution channel to determine whether additional allowances or accruals are required based on inventory in our sales channel.
An analysis of our U.S. product sales allowances and accruals for the years ended December 31, 2014, 2013 and 2012 is as follows (in thousands):
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Provision for U.S. product sales allowances and accruals | | | | | | | | | | |
Discounts and chargebacks | | $ | 55,420 | | $ | 37,098 | | $ | 26,517 | |
Government and other rebates | | | 25,091 | | | 10,868 | | | 6,058 | |
Medicaid rebate reserve adjustment | | | — | | | (568 | ) | | (621 | ) |
Returns | | | (1,160 | ) | | 952 | | | (1,516 | ) |
| | | | | | | | | | | |
Total provision for U.S. product sales allowances and accruals | | $ | 79,351 | | $ | 48,350 | | $ | 30,438 | |
Total gross U.S. product sales | | $ | 188,146 | | $ | 119,712 | | $ | 88,725 | |
Total provision for U.S. product sales allowances and accruals as a percent of total gross U.S. product sales | | | 42% | | | 40% | | | 34%
| |
The increases in discounts and chargebacks and government and other rebates primarily reflects the addition of theMakena product to our portfolio as a result of the November 2014 acquisition of Lumara Health. In addition, as discussed below, we reduced our reserve forFeraheme product returns by approximately $1.8 million and $2.2 million during 2014 and 2012, respectively.
Classification of U.S. Product Sales Allowances and Accruals
Product sales allowances and accruals are primarily comprised of both direct and indirect fees, discounts and rebates and provisions for estimated product returns. Direct fees, discounts and rebates are contractual fees and price adjustments payable to wholesalers, specialty distributors and other customers that purchase products directly from us. Indirect fees, discounts and rebates are contractual price adjustments payable to healthcare providers and organizations, such as certain physicians, clinics, hospitals, GPOs, and dialysis organizations that typically do not purchase products directly from us but rather from wholesalers and specialty distributors. In accordance with guidance related to accounting for fees and consideration given by a vendor to a customer, including a reseller of a vendor's products,
132
Table of Contents
these fees, discounts and rebates are presumed to be a reduction of the selling price. Product sales allowances and accruals are based on definitive contractual agreements or legal requirements (such as Medicaid laws and regulations) related to the purchase and/or utilization of the product by these entities and are recorded in the same period that the related revenue is recognized. We estimate product sales allowances and accruals using either historical, actual and/or other data, including estimated patient usage, applicable contractual rebate rates, contract performance by the benefit providers, other current contractual and statutory requirements, historical market data based upon experience of our products and other products similar to them, specific known market events and trends such as competitive pricing and new product introductions, current and forecasted customer buying patterns and inventory levels, and the shelf life of our products. As part of this evaluation, we also review changes to federal and other legislation, changes to rebate contracts, changes in the level of discounts, and changes in product sales trends. Although allowances and accruals are recorded at the time of product sale, certain rebates are typically paid out, on average, up to three months or longer after the sale.
Allowances against receivable balances primarily relate to prompt payment discounts, provider chargebacks and certain government agency rebates and are recorded at the time of sale, resulting in a reduction in product sales revenue and the reporting of product sales receivables net of allowances. Accruals related to Medicaid and provider volume rebates, wholesaler and distributor fees, GPO fees, other discounts to healthcare providers and product returns are recorded at the time of sale, resulting in a reduction in product sales revenue and the recording of an increase in accrued expenses.
Discounts
We typically offer a 2% prompt payment discount to our customers as an incentive to remit payment in accordance with the stated terms of the invoice, generally thirty days. Because we anticipate that those customers who are offered this discount will take advantage of the discount, we accrue 100% of the prompt payment discount, at the time of sale, based on the gross amount of each invoice. We adjust the accrual quarterly to reflect actual experience.
Chargebacks
Chargeback reserves represent our estimated obligations resulting from the difference between the prices at which we sell our products to wholesalers and the sales price ultimately paid to wholesalers under fixed price contracts by third-party payers, including governmental agencies. We determine our chargeback estimates based on actual product sales data and forecasted customer buying patterns. Actual chargeback amounts are determined at the time of resale to the qualified healthcare provider, and we generally issue credits for such amounts within several weeks of receiving notification from the wholesaler. Estimated chargeback amounts are recorded at the time of sale, and we adjust the allowance quarterly to reflect actual experience.
Government and Other Rebates
Government and other rebate reserves relate to our reimbursement arrangements with state Medicaid programs, and contractual or performance rebate agreements with certain classes of trade. We determine our estimates for Medicaid rebates, if applicable, based on actual product sales data and our historical product claims experience. In estimating these reserves, we provide for a Medicaid rebate associated with both those expected instances where Medicaid will act as the primary insurer as well as in those instances where we expect Medicaid will act as the secondary insurer. For rebates associated with reaching defined performance goals, we determine our estimates using actual product sales data and forecasted customer buying and utilization patterns. Rebate amounts generally are invoiced quarterly and are paid in arrears, and we expect to pay such amounts within several weeks of notification by the Medicaid or provider entity. Estimated government and other rebates are recorded at the time of sale and, with the exception of Medicaid as discussed below, we adjust the accrual quarterly to reflect actual experience.
133
Table of Contents
During 2013 and 2012, we revised our estimatedFeraheme Medicaid reserve rate based on actual product-specific rebate claims received since the 2009 launch ofFeraheme, our expectations of state level activities, and estimated rebate claims not yet submitted, which resulted in a reduction of our then estimated Medicaid rebate reserve related to prior periodFeraheme sales of $0.6 million in each of the respective years. These changes in estimates were reflected as an increase in our net product sales for 2013 and 2012 and resulted in reductions to our gross to net percentages in those periods. The reduction of our estimated Medicaid rebate reserve had an impact of $0.03 per basic and diluted share for each of the respective years. We regularly assess our Medicaid reserve balance and the rate at which we accrue for claims against product sales. If we determine in future periods that our actual rebate experience is not indicative of expected claims, or if actual claims experience changes, or if other factors affect estimated claims rates, we may be required to adjust our current Medicaid accumulated reserve estimate, which would affect net product sales in the period of the adjustment and could be significant.
Distributor/Wholesaler and Group Purchasing Organization Fees
Fees under our arrangements with distributors and wholesalers are usually based upon units of product purchased during the prior month or quarter and are usually paid by us within several weeks of our receipt of an invoice from the wholesaler or distributor, as the case may be. Fees under our arrangements with GPOs are usually based upon member purchases during the prior quarter and are generally billed by the GPO within 30 days after period end. Current accounting standards related to consideration given by a vendor to a customer, including a reseller of a vendor's products, specify that cash consideration given by a vendor to a customer is presumed to be a reduction of the selling price of the vendor's products or services and therefore should be characterized as a reduction of revenue. Consideration should be characterized as a cost incurred if we receive, or will receive, an identifiable benefit (goods or services) in exchange for the consideration and we can reasonably estimate the fair value of the benefit received. Because the fees we pay to wholesalers do not meet the foregoing conditions to be characterized as a cost, we have characterized these fees as a reduction of revenue and have included them in government and other rebates in the table above. We generally pay such amounts within several weeks of our receipt of an invoice from the distributor, wholesaler or GPO. Accordingly, we accrue the estimated fee due at the time of sale, based on the contracted price invoiced to the customer. We adjust the accrual quarterly to reflect actual experience.
Product Returns
Consistent with industry practice, we generally offer our wholesalers, specialty distributors and other customers a limited right to return our products based on the product's expiration date. Currently the expiration dates forFeraheme in the U.S.,Makena andMuGard are five years, three years and three years, respectively. We estimate product returns based on the historical return patterns and known or expected changes in the marketplace. We track actual returns by individual production lots. Returns on lots eligible for credits under our returned goods policy are monitored and compared with historical return trends and rates.
We expect that wholesalers and healthcare providers will not stock significant inventory due to the cost of the product, the expense to store our products, and/or that our products are readily available for distribution. We record an estimate of returns at the time of sale. If necessary, our estimated rate of returns may be adjusted for actual return experience as it becomes available and for known or expected changes in the marketplace. During 2014, we reduced our reserve forFeraheme product returns by approximately $1.8 million, primarily as a result of a lower than expected rate of product returns. We did not significantly adjust our reserve for product returns during 2013. During 2012, we reduced our reserve forFeraheme product returns by approximately $2.2 million, primarily as a result of a lower than expected rate of product returns as well as the lapse of the product return period on
134
Table of Contents
certain manufacturedFeraheme lots. The reduction of our reserve had an impact of increasing our 2014 net income by $0.12 and $0.14 per basic and diluted share, respectively, and by $0.10 per basic and diluted share in 2012. To date, returns ofFeraheme have been relatively limited; however, returns experience may change over time. As we continue to gain more historical experience with actual returns and continue to gain additional experience with return rates forMakena, we may be required to make a future adjustment to our product returns estimate, which would result in a corresponding change to our net product sales in the period of adjustment and could be significant.
Other Product Sales and Royalties
Other product sales and royalties primarily includedFeraheme product sales to Takeda and royalties from Takeda as well as net product sales ofMuGard. Prior to the Takeda Termination Agreement, we recorded all product sales ofFeraheme sold to Takeda in deferred revenues in our consolidated balance sheet. We recognized these deferred revenues, and the associated cost of product sales, in our consolidated statement of operations at the time Takeda reported to us that sales had been made to its customers. At December 31, 2014, as the result of terminating the Amended Takeda Agreement, we recognized these remaining balances of deferred revenues and associated cost of product sales.
License Fee and Other Collaboration Revenues
The terms of product development and commercialization agreements entered into between us and our collaborative licensees may include non-refundable license fees, payments based on the achievement of certain milestones and performance goals, reimbursement of certain out-of-pocket costs, payment for manufacturing services, and royalties on product sales. We recognize license fee and research and development revenue under collaborative arrangements over the term of the applicable agreements using a proportional performance model, if practical. Otherwise, we recognize such revenue on a straight-line basis. Under this model, revenue is generally recognized in an amount equal to the lesser of the amount due under the agreements or an amount based on the proportional performance to date. In cases where project costs or other performance metrics are not estimable but there is an established contract period, revenues are recognized on a straight-line basis over the term of the relevant agreement. In cases where we are reimbursed for certain research and development costs associated with our collaboration agreements and where we are acting as the principal in carrying out these services, any reimbursement payments are recorded in license fee and other collaboration revenues in our consolidated statement of operations to match the costs that we incur during the period in which we perform those services. Nonrefundable payments and fees are recorded as deferred revenue upon receipt and may require deferral of revenue recognition to future periods.
Multiple Element Arrangements and Milestone Payments
We evaluate revenue from arrangements that have multiple elements to determine whether the components of the arrangement represent separate units of accounting as defined in the accounting guidance related to revenue arrangements with multiple deliverables. Under current accounting guidance, which governs any agreements that contain multiple elements that are either entered into or materially modified subsequent to January 1, 2011, companies are required to establish the fair value of undelivered products and services based on a separate revenue recognition process using management's best estimate of the selling price for an undelivered item when there is no vendor-specific objective evidence or third-party evidence to determine the fair value of that undelivered item. Agreements entered into prior to January 1, 2011, that have not been materially modified are accounted for under previous accounting guidance, which provides that an element of a contract can be accounted for separately if the delivered elements have standalone value and the fair value of all undelivered elements is determinable. If an element is considered to have standalone value but the fair value of any
135
Table of Contents
of the undelivered items cannot be determined, all elements of the arrangement are recognized as revenue as a single unit of accounting over the period of performance for such undelivered items or services. Significant management judgment is required in determining what elements constitute deliverables and what deliverables or combination of deliverables should be considered units of accounting.
When multiple deliverables are combined and accounted for as a single unit of accounting, we base our revenue recognition pattern on the last to be delivered element. Revenue is recognized using either a proportional performance or straight-line method, depending on whether we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and whether such performance obligations are provided on a best-efforts basis. To the extent we cannot reasonably estimate our performance obligations, we recognize revenue on a straight-line basis over the period we expect to complete our performance obligations. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement. We may have to revise our estimates based on changes in the expected level of effort or the period we expect to complete our performance obligations.
Our collaboration agreements may entitle us to additional payments upon the achievement of performance-based milestones. If a milestone involves substantive effort on our part and its achievement is not considered probable at the inception of the collaboration, we recognize the milestone consideration as revenue in the period in which the milestone is achieved only if it meets the following additional criteria:
- •
- The milestone consideration received is commensurate with either the level of effort required to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from our performance to achieve the milestone;
- •
- The milestone is related solely to our past performance; and
- •
- The milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement.
There is significant judgment involved in determining whether a milestone meets all of these criteria. For milestones that do not meet the above criteria and are therefore not considered substantive milestones, we recognize that portion of the milestone payment equal to the percentage of the performance period completed at the time the milestone is achieved and the above conditions are met. The remaining portion of the milestone will be recognized over the remaining performance period using a proportional performance or straight-line method.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in our consolidated balance sheets. Amounts not expected to be recognized within the next 12 months are classified as long-term deferred revenue.
Research and Development Expenses
Research and development expenses include external expenses, such as costs of clinical trials, contract research and development expenses, certain manufacturing research and development costs, regulatory filing fees, consulting and professional fees and expenses, and internal expenses, such as compensation of employees engaged in research and development activities, the manufacture of product needed to support research and development efforts, related costs of facilities, and other general costs related to research and development. Manufacturing costs are generally expensed as incurred until a product has received the necessary initial regulatory approval.
136
Table of Contents
Advertising Costs
Advertising costs are expensed as incurred and are included in selling, general and administrative expenses in our consolidated statements of operations. Advertising costs, including promotional expenses and costs related to trade shows were $2.1 million, $1.9 million and $1.8 million for the years ended December 31, 2014, 2013 and 2012, respectively.
Shipping and Handling Costs
We utilize two third-party logistics providers, both of which are subsidiaries of one of our distribution customers, to provide us with various shipping and handling services related to sales of our products. As we receive an identifiable benefit and we can reasonably estimate the fair value of this benefit, we have recorded $0.3 million, $0.3 million and $0.2 million as a selling, general and administrative expense during 2014, 2013 and 2012, respectively.
Equity-Based Compensation
Under the fair value recognition guidance of equity-based compensation accounting rules, equity-based compensation cost is generally required to be measured at the grant date (based upon an estimate of the fair value of the compensation granted) and recorded to expense over the requisite service period, which generally is the vesting period. Because equity-based compensation expense is based on awards ultimately expected to vest, we must make certain judgments about whether employees and directors will complete the requisite service period. Accordingly, we have reduced the compensation expense being recognized for estimated forfeitures. Under current accounting guidance, forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Forfeitures are estimated based upon historical experience and adjusted for unusual events such as corporate restructurings, which can result in higher than expected turnover and forfeitures. If factors change and we employ different forfeiture rate assumptions in future periods, the compensation expense that we record in the future may differ significantly from what we have recorded in the current period.
We estimate the fair value of equity-based compensation involving stock options based on the Black-Scholes option pricing model. We estimate the fair value of our restricted stock units ("RSUs") whose vesting is contingent upon market conditions using the Monte-Carlo simulation model. These models require the input of several factors such as the expected option term, the expected risk-free interest rate over the expected option term, the expected volatility of our stock price over the expected option term, and the expected dividend yield over the expected option term and are subject to various assumptions. The fair value of awards whose fair values are calculated using the Black-Scholes option pricing model is generally being amortized on a straight-line basis over the requisite service period and is recognized based on the proportionate amount of the requisite service period that has been rendered during each reporting period. The fair value of awards with market conditions is being amortized based upon the estimated derived service period, even if the market condition is never achieved. The fair value of awards with performance conditions is being amortized over the requisite service period if we determine that it is probable that the performance condition will be achieved. We believe our valuation methodologies are appropriate for estimating the fair value of the equity awards we grant to our employees and directors. Our equity award valuations are estimates and thus may not be reflective of actual future results or amounts ultimately realized by recipients of these grants. These amounts, and the amounts applicable to future quarters, are also subject to future quarterly adjustments based upon a variety of factors, which include, but are not limited to, changes in estimated forfeiture rates and the issuance of new equity-based awards. The fair value of service-based RSUs granted to our employees and directors is determined based upon the quoted closing market price per share on the date of grant, adjusted for estimated forfeitures.
137
Table of Contents
Income Taxes
Deferred tax assets and deferred tax liabilities are recognized based on temporary differences between the financial reporting and tax basis of assets and liabilities using future enacted rates. A valuation allowance is recorded against deferred tax assets if it is more likely than not that some or all of our deferred tax assets will not be realized.
We account for uncertain tax positions using a "more-likely-than-not" threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. We evaluate uncertain tax positions on a quarterly basis and adjust the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. Any changes to these estimates, based on the actual results obtained and/or a change in assumptions, could impact our income tax provision in future periods. Interest and penalty charges, if any, related to unrecognized tax benefits would be classified as a provision for income tax in our consolidated statement of operations.
Concentrations and Significant Customer Information
Financial instruments which potentially subject us to concentrations of credit risk consist principally of cash and cash equivalents, investments, and accounts receivable. As of December 31, 2014, our cash, cash equivalents and investments amounted to approximately $144.2 million. We currently invest our excess cash primarily in corporate debt securities. As of December 31, 2014, we had approximately $77.3 million of our total $119.3 million cash and cash equivalents balance invested in institutional money market funds, of which $60.3 million was invested in a single fund.
Our operations are located solely within the U.S. We are focused principally on developing, manufacturing, and commercializingMakena andFeraheme and commercializingMuGard. We perform ongoing credit evaluations of our customers and generally do not require collateral. The following table sets forth customers who represented 10% or more of our total revenues for 2014, 2013 and 2012:
| | | | | | | | | | |
| | Years Ended
December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
AmerisourceBergen Drug Corporation | | | 34% | | | 41% | | | 34% | |
McKesson Corporation | | | 21% | | | 24% | | | 17% | |
Cardinal Health, Inc. | | | 15% | | | 16% | | | 12% | |
Takeda Pharmaceuticals Company Limited | | | 11% | | | 11% | | | 31% | |
In addition, approximately 26%, 30% and 32% of ourFeraheme end-user demand in 2014, 2013 and 2012, respectively, was generated by members of a single GPO with which we have contracted. Revenues from customers outside of the U.S. amounted to approximately 12%, 11% and 32% of our total revenues for 2014, 2013 and 2012, respectively, and were principally related to collaboration revenue recognized in connection with the Amended Takeda Agreement with Takeda, which is headquartered in Japan.
We are currently solely dependent on a single supply chain forFeraheme drug substance and drug product and a single supply chain forMakena drug product. We are exposed to a significant loss of revenue from the sale ofFeraheme andMakena if our suppliers and/or manufacturers cannot fulfill demand for any reason.
138
Table of Contents
Comprehensive Income (Loss)
The current accounting guidance related to comprehensive income (loss) requires us to display comprehensive income (loss) and its components as part of our consolidated financial statements. Our comprehensive income (loss) consists of net income (loss) and other comprehensive income (loss). Other comprehensive income (loss) includes changes in equity that are excluded from net income (loss), which for all periods presented in these financial statements related to unrealized holding gains and losses on available-for-sale investments, net of tax.
Basic and Diluted Net Income (Loss) per Share
We compute basic net income (loss) per share by dividing net income (loss) by the weighted average number of common shares outstanding during the relevant period. Diluted net income (loss) per common share has been computed by dividing net income (loss) by the diluted number of shares outstanding during the period. Except where the result would be antidilutive to net income (loss), diluted net income (loss) per common share would be computed assuming the impact of the conversion of the $200.0 million of 2.5% convertible senior notes due February 15, 2019 (the "Convertible Notes"), the exercise of outstanding stock options, and the vesting of RSUs.
We have a choice to settle the conversion obligation under the Convertible Notes in cash, shares or any combination of the two. Pursuant to certain covenants in our Term Loan Facility (as defined in Note S, "Debt" below), which we entered into to partially fund the acquisition of Lumara Health, we may be restricted from settling conversion in whole or in part with cash unless certain conditions in the Term Loan Facility are satisfied, including a first lien leverage ratio. Therefore, after November 12, 2014 we utilized the if-converted method, which assumes the conversion of the Convertible Notes and reflects the elimination of the interest expense recorded from November 12, 2014 through December 31, 2014. The conversion premium is reflected in the calculation of diluted earnings per share as if it were a freestanding written call option on our shares. The impact of the conversion premium has been considered in the calculation of diluted net income per share by applying the weighted average of the closing price of our common stock, over a certain number of days pursuant to the terms of the Convertible Notes, to calculate the number of shares issuable under the conversion premium. In addition, in February 2014, in connection with the issuance of the Convertible Notes, we entered into convertible bond hedges. The convertible bond hedges are not included for purposes of calculating the number of diluted shares outstanding, as their effect would be anti-dilutive. The convertible bond hedges are generally expected, but not guaranteed, to reduce the potential dilution and/or offset the cash payments we are required to make upon conversion of the Convertible Notes. See Note S, "Debt," for additional information.
The dilutive effect of the stock options and RSUs has been calculated using the treasury stock method.
The components of basic and diluted net income (loss) per share were as follows (in thousands, except per share data):
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Net income (loss) | | $ | 135,817 | | $ | (9,602 | ) | $ | (16,750 | ) |
Weighted average common shares outstanding (basic) | | | 22,416 | | | 21,703 | | | 21,392 | |
Effect of dilutive securities (in shares): | | | | | | | | | | |
Stock options and restricted stock units | | | 520 | | | — | | | — | |
Convertible 2.5% senior notes | | | 2,289 | | | — | | | — | |
| | | | | | | | | | | |
Shares used in calculating dilutive net income (loss) per share | | | 25,225 | | | 21,703 | | | 21,392 | |
Net income (loss) per share: | | |
| | |
| | |
| |
Basic | | $ | 6.06 | | $ | (0.44 | ) | $ | (0.78 | ) |
Diluted | | $ | 5.45 | | $ | (0.44 | ) | $ | (0.78 | ) |
139
Table of Contents
The following table sets forth the potential common shares issuable upon the exercise of outstanding options, the vesting of RSUs and warrants (prior to consideration of the treasury stock method), which were excluded from our computation of diluted net income (loss) per share because their inclusion would have been anti-dilutive (in thousands):
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Options to purchase shares of common stock | | | 2,708 | | | 2,820 | | | 2,190 | |
Shares of common stock issuable upon the vesting of restricted stock units | | | 322 | | | 465 | | | 374 | |
Warrants | | | 7,382 | | | — | | | — | |
| | | | | | | | | | | |
Total | | | 10,412 | | | 3,285 | | | 2,564 | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
During 2014, the average common stock price was below the exercise price of the warrants.
Reclassifications
Certain amounts in prior periods have been reclassified in order to conform to the current period presentation.
C. BUSINESS COMBINATIONS
As part of our strategy to expand our portfolio with additional commercial-stage products, in November 2014, we acquired Lumara Health and its productMakena. In addition, in June 2013, we entered into the MuGard License Agreement pursuant to which we obtained an exclusive, royalty-bearing license, with the right to grant sublicenses, to certain intellectual property rights, including know-how, patents and trademarks, to use, import, offer for sale, sell, manufacture and commercializeMuGard in the U.S. and its territories (the "U.S. Territory") for the management of all diseases or conditions of the oropharyngeal cavity, including mucositis.
Lumara Health
On the Lumara Acquisition Date, we completed our acquisition of 100% of the equity ownership of Lumara Health, excluding the assets and liabilities of the Women's Health Division and certain other assets and liabilities, pursuant to the Lumara Agreement upon which time Lumara Health became our wholly owned subsidiary. In connection with the acquisition of Lumara Health, we acquiredMakena, a progestin indicated to reduce the risk of preterm birth in women with a singleton pregnancy who have a history of singleton spontaneous preterm birth.
Upon the closing of the Lumara Health acquisition (the "Closing"), we paid approximately $600.0 million in cash (subject to finalization of certain adjustments related to Lumara Health's financial position at the time of closing, including adjustments related to net working capital, net debt and transaction expenses) (the "Cash Consideration") and issued approximately 3.2 million shares of our common stock, par value $0.01, having a value of approximately $112.0 million at the time of the Closing, to the holders of Lumara Health common stock, stock options, and RSUs.
We have agreed to pay additional merger consideration, up to a maximum of $350.0 million, based on the achievement of certain net sales milestones ofMakena for the period from December 1, 2014 through December 19, 2019 as follows:
- •
- A one-time payment of $100.0 million payable upon achievement of $300.0 million in aggregate net sales in any consecutive 12-month period commencing in the month following the Lumara Acquisition Date ("the First Milestone"); plus
140
Table of Contents
- •
- A one-time payment of $100.0 million payable upon achievement of $400.0 million in aggregate net sales in any consecutive 12-month period commencing in the month following the last month in the First Milestone period (the "Second Milestone"); if the Third Milestone payment (described below) has been or is required to be made prior to achieving the Second Milestone, the Second Milestone payment shall be reduced from $100.0 million to $50.0 million; plus
- •
- A one-time payment of $50.0 million payable if aggregate net sales equal or exceed $700.0 million in any consecutive 24-calendar month period (which may include the First Milestone period) (the "Third Milestone"); however, no Third Milestone payment will be made if the Second Milestone payment has been or is required to be made in the full amount of $100.0 million; plus
- •
- A one-time payment of $100.0 million payable upon achievement of $500.0 million in aggregate net sales in any consecutive 12-month period commencing in the month following the last month in the Second Milestone period (the "Fourth Milestone"); plus
- •
- A one-time payment of $50.0 million payable upon achievement of $200.0 million in aggregate net sales in each of the five consecutive calendar years from and including the 2015 calendar year to the 2019 calendar year (the "Fifth Milestone").
In the event that the conditions to more than one contingent payment are met in any calendar year, any portion of the total amount of contingent payment due in such calendar year in excess of $100.0 million shall be deferred until the next calendar year in which less than $100.0 million in contingent payments is due.
The following table summarizes the components of the estimated total purchase price at fair value, subject to adjustment upon finalization of Lumara Health's net working capital, net debt and transaction expenses as of the Lumara Acquisition Date (in thousands):
| | | | |
| | Total Acquisition Date
Fair Value | |
|---|
Cash consideration | | $ | 600,000 | |
Fair value of 3.2 million shares of AMAG common stock | | | 111,964 | |
Fair value of contingent milestone payments | | | 205,000 | |
Estimated working capital and other adjustments | | | 821 | |
| | | | | |
Purchase price paid at closing | | | 917,785 | |
Less: | | | | |
Due from sellers | | | (5,119 | ) |
Cash acquired from Lumara Health | | | (5,219 | ) |
| | | | | |
Total purchase price | | $ | 907,447 | |
| | | | | |
| | | | | |
| | | | | |
We financed the $600.0 million upfront cash portion of the acquisition through $327.5 million of net proceeds from borrowings under a new $340.0 million term loan (the "Term Loan Facility"), as discussed in more detail in Note S, "Debt", and $272.5 million of existing cash on hand.
The fair value of the 3.2 million shares of AMAG common stock was determined based on the closing price of our common stock on the NASDAQ Global Select Market of $34.88 per share on November 11, 2014, the closing price immediately prior to the closing of the transaction.
The fair value of the contingent milestone payments was determined based on our probability-adjusted discounted cash flows estimated to be realized from the net sales ofMakena from December 1, 2014 through December 31, 2019. The cash flows were discounted at a rate of 5%, which we believe is reasonable given the level of certainty of the pay-out.
141
Table of Contents
The net working capital and other adjustments were estimated to be $0.8 million which we paid at the Closing. Subsequent to the Closing, we estimate that the net working capital and other adjustments will result in a reduction to the cash consideration of approximately $5.1 million. Accordingly, we recorded a $5.1 million receivable in prepaid and other current assets in the consolidated balance sheet at December 31, 2014. The net working capital and other adjustments are subject to change upon finalization of certain adjustments related to Lumara Health's financial position at the time of closing.
At the Closing, $7.0 million of the Cash Consideration was contributed into an escrow fund to secure any Lumara Health security holders' payment obligations with respect to the working capital, net debt and transaction expenses adjustments, which escrow will be released upon the final determination of the Cash Consideration. Also at the Closing, $35.0 million of the Cash Consideration was contributed to a separate escrow fund (the "Indemnification Escrow") to secure the former Lumara Health security holders' obligations to indemnify us for certain matters, including breaches of representations and warranties, covenants included in the Lumara Agreement, payments made by us to dissenting stockholders, specified tax claims, excess parachute claims, and certain claims related to the Women's Health division of Lumara Health, which was divested by Lumara Health prior to the Closing. The portion of the Indemnification Escrow that has not been reduced by any claims by us and is not subject to any unresolved claims will be released to the former Lumara Health security holders at the earlier of (a) March 15, 2016 and (b) five days after the date on which our audited financial statements for our fiscal year ending December 31, 2015 are filed with the Securities and Exchange Commission.
We accounted for the acquisition of Lumara Health as a business combination using the acquisition method of accounting. Under the acquisition method of accounting, the total purchase price of an acquisition is allocated to the net tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair values as of the date of acquisition. We have made a preliminary allocation of the purchase price to the net tangible and intangible assets acquired and liabilities assumed, based on available information and various assumptions we believe are reasonable, with the remaining purchase price recorded as goodwill. Due to the close proximity of the Lumara Acquisition Date to the fiscal 2014 year-end, we were unable to complete our analysis of fair value.
The following table summarizes the preliminary fair values assigned to the assets acquired and the liabilities assumed by us at the Lumara Acquisition Date (in thousands):
| | | | |
Accounts receivable | | $ | 34,918 | |
Inventories | | | 30,300 | |
Prepaid and other current assets | | | 3,322 | |
Deferred income tax assets | | | 94,965 | |
Property and equipment | | | 60 | |
Makena marketed product | | | 797,100 | |
IPR&D | | | 79,100 | |
Restricted cash | | | 1,997 | |
Other long-term assets | | | 3,412 | |
Accounts payable | | | (3,807 | ) |
Accrued expenses | | | (41,532 | ) |
Deferred income tax liabilities | | | (293,649 | ) |
Other long-term liabilities | | | (4,563 | ) |
| | | | | |
Total estimated identifiable net assets | | | 701,623 | |
Goodwill | | | 205,824 | |
| | | | | |
Total | | $ | 907,447 | |
| | | | | |
| | | | | |
| | | | | |
142
Table of Contents
The preliminary values assigned to accounts receivable, prepaid and other current assets, other long-term assets, accounts payable, accrued expenses, deferred income taxes, other long-term liabilities and goodwill presented in the table above are subject to change as additional information becomes available concerning the fair value and tax basis of the assets acquired and liabilities assumed. Any adjustments to the preliminary fair value of these acquired assets and liabilities assumed will be made as soon as practicable but not later than one year from the Lumara Acquisition Date.
The gross contractual amount of accounts receivable at the Lumara Acquisition Date was $40.5 million. The $30.3 million fair value of inventories included a fair value step-up adjustment of $26.1 million, which will be amortized and recognized as cost of product sales in our consolidated statements of operations as the related inventories are sold. We recognized $1.3 million of the fair value adjustment as cost of product sales during the year ended December 31, 2014. The remaining $24.8 million is estimated to be recognized as follows: $11.1 million in fiscal 2015, $3.5 million in fiscal 2016, $4.0 million in fiscal 2017, $3.5 million in fiscal 2018 and $2.7 million in fiscal 2019.
The fair value amounts for theMakena marketed product (the "Marketed Product") and IPR&D were determined based on assumptions that market participants would use in pricing an asset, based on the most advantageous market for the assets (i.e., its highest and best use). We determined the fair value of the Marketed Product and the IPR&D using the income approach, which is a valuation technique that provides an estimate of the fair value of an asset based on market participant expectations of the cash flows an asset would generate over its remaining useful life. Some of the more significant assumptions used in the income approach from the perspective of a market participant include the estimated net cash flows for each year for each project or product (including net revenues, cost of product sales, research and development costs, selling and marketing costs and working capital/asset contributory asset charges), the discount rate that measures the risk inherent in each future cash flow stream, the assessment of each asset's life cycle, competitive trends impacting the asset and each cash flow stream as well as other factors, including the major risks and uncertainties associated with the timely and successful completion of the IPR&D projects, such as legal risk and regulatory risk.
The fair value of the acquired IPR&D asset represents the value assigned to acquired research and development projects that, as of the Lumara Acquisition Date, had not established technological feasibility and had no alternative future use, including certain programs associated with theMakena lifecycle management program to extend the brand franchise beyond the February 2018 exclusivity date, such as new routes of administration, the use of new delivery technologies, as well as reformulation technologies. We believe the fair values assigned to the Marketed Product and IPR&D assets are based upon reasonable estimates and assumptions given available facts and circumstances as of the Lumara Acquisition Date. If these assets are not successful or successfully developed, sales and profitability may be adversely affected in future periods, and as a result, the value of the assets may become impaired.
The acquisition of Lumara Health is expected to result in carryover basis for all tax attributes. Both AMAG and Lumara Health have deferred tax assets for which full valuation allowances were provided in the pre-acquisition financial statements. However, we have considered certain of the deferred tax liabilities recorded in acquisition accounting as sources of income to support realization of Lumara Health's deferred tax assets at December 31, 2014. Based on the preliminary fair value adjustments primarily related to inventories and identifiable intangible assets acquired, we recorded a net deferred tax liability of $198.7 million in our consolidated balance sheet as of December 31, 2014 using a combined federal and state statutory income tax rate of 38.8%. The net deferred tax liability represents the $293.7 million of deferred tax liabilities recorded in acquisition accounting (primarily related to the fair value adjustments to Lumara Health's inventories and identifiable intangible assets) offset by $95.0 million of deferred tax assets acquired from Lumara Health which we have determined, on a preliminary basis, are 'more likely than not' to be realized. See Note K, "Income Taxes," for additional information.
143
Table of Contents
These tax estimates are preliminary and subject to change based on, among other things, management's final determination of the fair values of the tangible and identifiable intangible assets acquired and liabilities assumed by jurisdiction, the deductibility of acquisition-related costs and other costs deducted by Lumara pre-acquisition, and management's assessment of the combined company's ability to utilize the future benefits from acquired and legacy deferred tax assets.
Goodwill is calculated as the difference between the acquisition date fair value of the consideration transferred and the fair values of the net assets acquired and liabilities assumed. The $205.8 million of goodwill resulting from the acquisition was primarily due to the net deferred tax liabilities recorded on the fair value adjustments to Lumara Health's inventories and identifiable intangible assets. The goodwill is not deductible for income tax purposes.
Acquisition-related costs are not included as a component of consideration transferred and are expensed as incurred. We incurred approximately $9.5 million of acquisition-related costs in 2014 related to the merger with Lumara Health. These costs primarily represented financial advisory fees, legal fees, due diligence and other costs and expenses.
During the post-acquisition period in fiscal 2014, Lumara Health generated $22.5 million of revenue from sales ofMakena. We determined that separate disclosure of Lumara Health's earnings for the post-acquisition period in fiscal 2014 is not practicable due to the integration of Lumara Health's operations into our business upon acquisition.
The following table presents our revenue and net income (loss) on a pro forma combined basis, assuming that the merger occurred on January 1, 2013 and does not include any expected cost savings or restructuring actions which may be achievable or which may occur subsequent to the acquisition of Lumara Health or the impact of any non-recurring activity. For purposes of preparing the following pro forma information, certain items recorded in 2014, such as the $153.2 million tax benefit and the $9.5 million of acquisition-related costs are reflected in 2013 as if the acquisition occurred on January 1, 2013. In addition, the pro forma combined net income (loss) in fiscal 2013 does not give effect to the elimination of approximately $385.9 million of non-recurring reorganization gains, net of losses and expenses, realized in connection with Lumara Health's exit from bankruptcy in September 2013 as such amounts are not directly related to the acquisition of Lumara Health (in thousands):
| | | | | | | |
| | Year Ended
December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Pro forma combined revenues | | $ | 267,705 | | $ | 179,561 | |
Pro forma combined net income (loss) | | $ | (23,942 | ) | $ | 463,522 | |
This pro forma financial information is not necessarily indicative of our consolidated operating results that would have been reported had the transactions been completed as described herein, nor is such information necessarily indicative of our consolidated results for any future period.
MuGard
MuGard was launched in the U.S. by PlasmaTech Biopharmaceuticals, Inc. (formerly known as Access Pharmaceuticals, Inc. ("PlasmaTech") in 2010 after receiving 510(k) clearance from the FDA and is indicated for the management of oral mucositis/stomatitis (that may be caused by radiotherapy and/or chemotherapy) and all types of oral wounds (mouth sores and injuries), including aphthous ulcers/canker sores and traumatic ulcers, such as those caused by oral surgery or ill-fitting dentures or braces.
PlasmaTech remains responsible for the manufacture ofMuGard and we have entered into a quality agreement and a supply agreement with PlasmaTech under which we purchaseMuGard inventory from PlasmaTech. Our inventory purchases are at the price actually paid by PlasmaTech to purchase it from a third-party plus a mark-up to cover administration, handling and overhead.
144
Table of Contents
In consideration for the license, we paid PlasmaTech an upfront payment of $3.3 million on June 6, 2013 (the "MuGard License Date"). We are required to pay royalties to PlasmaTech on future net sales ofMuGard until the later of (a) the expiration of the licensed patents or (b) the tenth anniversary of the first commercial sale ofMuGard under the MuGard License Agreement in the U.S. Territory ("the "Royalty Term"). These tiered, double-digit royalty rates decrease for any part of the Royalty Term occurring after the expiration of the licensed patents and are subject to off-set against certain of our expenses. After the expiration of the Royalty Term, the license shall become a fully paid-up, royalty-free and perpetual license in the U.S. Territory. In addition to making an upfront payment of $3.3 million, we also acquired $0.2 million of existingMuGard inventory from PlasmaTech, which was included in our consolidated balance sheet as of the MuGard License Date.
We did not assume any pre-existing liabilities related to theMuGard business, contingent or otherwise, arising prior to the MuGard License Date. We are accounting for the acquisition of the MuGard Rights as a business combination under the acquisition method of accounting since we acquired the U.S. commercial rights forMuGard and inventory, and obtained access to certain related regulatory assets and product supply, employees and other assets, including certain patent and trademark rights, contracts, and related books and records, held by PlasmaTech which are exclusively related toMuGard (inputs), including the infrastructure to sell, distribute and marketMuGard (processes) and net sales ofMuGard (outputs). In addition, during the term of the MuGard License Agreement, we will have control over sales, distribution and marketing ofMuGard in the U.S. as PlasmaTech has assigned to us all of its right, title and interest inMuGard-related internet and social media outlets and other sales, marketing and promotional materials currently owned or controlled by PlasmaTech. PlasmaTech will no longer commercialize, market, promote, sell or make public communications relating toMuGard in the U.S Territory, except as may be agreed to by us. PlasmaTech has also agreed to not, directly or indirectly, research, develop, market, sell or commercialize any medical devices that directly compete withMuGard for the treatment of any diseases or conditions of the oropharyngeal cavity in the U.S. Territory.
We estimated the fair value of the acquired MuGard Rights using the income approach, which is a valuation technique to convert future amounts to a single present amount (discounted) and is described above.
The following table summarizes the total consideration for the MuGard Rights (in thousands):
| | | | |
| | Total
Acquisition
Date Fair
Value | |
|---|
Cash | | $ | 3,434 | |
Acquisition-related contingent consideration | | | 13,700 | |
| | | | | |
Total consideration | | $ | 17,134 | |
| | | | | |
| | | | | |
| | | | | |
The $17.1 million total consideration includes the estimated fair value of the contingent consideration at the MuGard License Date. During 2013, we completed the valuation for the acquisition of the MuGard Rights and determined the fair value of the contingent consideration to be $13.7 million as of the MuGard License Date, and the fair value of the intangible asset was determined to be $16.9 million as of the MuGard License Date. The acquisition date fair value of the contingent consideration was determined based on various market factors, including an analysis of estimated sales using a discount rate of approximately 15%. As of December 31, 2014, we estimated that the undiscounted royalty amounts we could pay under the MuGard License Agreement may range from $20.0 million to $28.0 million over a ten year period, which is our best estimate of the period over which we expect the majority of the asset's cash flows to be derived.
145
Table of Contents
The following table summarizes the fair values of the assets acquired related to the business combination as of the MuGard License Date (in thousands):
| | | | |
Assets Acquired: | | | | |
MuGard intangible asset | | $ | 16,893 | |
Inventory | | | 241 | |
| | | | | |
Net identifiable assets acquired | | $ | 17,134 | |
| | | | | |
| | | | | |
| | | | | |
The acquisition date fair value of the intangible asset was determined based on various market factors, including an analysis of estimated sales using a discount rate of 19%. This measure is based on significant Level 3 inputs not observable in the market. Such valuations require significant estimates and assumptions including but not limited to: estimating future cash flows from product sales and developing appropriate discount and probability rates. We believe the estimated fair values of the MuGard Rights are based on reasonable assumptions, however, we cannot provide assurance that the underlying assumptions used to forecast the cash flows will materialize as we estimated and thus, our actual results may vary significantly from the estimated results.
Acquisition-related costs are not included as a component of consideration transferred and are expensed as incurred. We incurred approximately $0.8 million of acquisition-related costs in 2013, which were primarily related to professional and legal fees.
Pro forma results of operations would not be materially different as a result of the acquisition of the MuGard Rights and therefore are not presented.
D. INVESTMENTS
As of December 31, 2014 and 2013, our investments equaled $24.9 million and $186.8 million, respectively, and consisted of securities classified as available-for-sale in accordance with accounting standards which provide guidance related to accounting and classification of certain investments in debt and equity securities.
The following is a summary of our investments as of December 31, 2014 and 2013 (in thousands):
| | | | | | | | | | | | | |
| | December 31, 2014 | |
|---|
| | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value | |
|---|
Corporate debt securities | | | | | | | | | | | | | |
Due in one year or less | | $ | 11,656 | | $ | 3 | | $ | (4 | ) | $ | 11,655 | |
Due in one to three years | | | 13,258 | | | 10 | | | (33 | ) | | 13,235 | |
| | | | | | | | | | | | | | |
Total investments | | $ | 24,914 | | $ | 13 | | $ | (37 | ) | $ | 24,890 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
146
Table of Contents
| | | | | | | | | | | | | |
| | December 31, 2013 | |
|---|
| | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Estimated
Fair
Value | |
|---|
Corporate debt securities | | | | | | | | | | | | | |
Due in one year or less | | $ | 42,609 | | $ | 44 | | $ | (4 | ) | $ | 42,649 | |
Due in one to three years | | | 91,443 | | | 137 | | | (106 | ) | | 91,474 | |
U.S. treasury and government agency securities | | | | | | | | | | | | | |
Due in one year or less | | | 18,526 | | | 19 | | | — | | | 18,545 | |
Due in one to three years | | | 34,123 | | | 37 | | | (25 | ) | | 34,135 | |
| | | | | | | | | | | | | | |
Total investments | | $ | 186,701 | | $ | 237 | | $ | (135 | ) | $ | 186,803 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
During the year ended December 31, 2014, we liquidated $170.4 million of our investments in order to partially fund the acquisition of Lumara Health in November 2014.
Impairments and Unrealized Gains and Losses on Investments
We did not recognize any other-than-temporary impairment losses in our consolidated statements of operations related to our securities during either 2014, 2013 or 2012. We considered various factors, including the length of time that each security was in an unrealized loss position and our ability and intent to hold these securities until the recovery of their amortized cost basis occurs. As of December 31, 2014, none of our investments has been in an unrealized loss position for more than one year. Future events may occur, or additional information may become available, which may cause us to identify credit losses where we do not expect to receive cash flows sufficient to recover the entire amortized cost basis of a security and which may necessitate the recording of future realized losses on securities in our portfolio. Significant losses in the estimated fair values of our investments could have a material adverse effect on our earnings in future periods.
Realized Gains and Losses on Investments
Gains and losses are determined on the specific identification method. Net realized gains were $0.1 million during 2014 and insignificant during 2013. During 2012, we recorded realized losses of $1.5 million to our consolidated statement of operations related to the sale of our then-remaining auction rate securities portfolio.
147
Table of Contents
E. FAIR VALUE MEASUREMENTS
The following tables represent the fair value hierarchy as of December 31, 2014 and 2013, for those assets and liabilities that we measure at fair value on a recurring basis (in thousands):
| | | | | | | | | | | | | |
| | Fair Value Measurements at December 31, 2014 Using: | |
|---|
| | Total | | Quoted Prices in
Active Markets for
Identical Assets
(Level 1) | | Significant Other
Observable Inputs
(Level 2) | | Significant
Unobservable
Inputs
(Level 3) | |
|---|
Assets: | | | | | | | | | | | | | |
Money market funds | | $ | 77,254 | | $ | 77,254 | | $ | — | | $ | — | |
Corporate debt securities | | | 24,890 | | | — | | | 24,890 | | | — | |
| | | | | | | | | | | | | | |
Total Assets | | $ | 102,144 | | $ | 77,254 | | $ | 24,890 | | $ | — | |
Liabilities: | | | | | | | | | | | | | |
Contingent consideration—Lumara Health | | $ | 206,600 | | $ | — | | $ | — | | $ | 206,600 | |
Contingent consideration—MuGard | | | 12,102 | | | — | | | — | | | 12,102 | |
| | | | | | | | | | | | | | |
Total Liabilities | | $ | 218,702 | | $ | — | | $ | — | | $ | 218,702 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | Fair Value Measurements at December 31, 2013 Using: | |
|---|
| | Total | | Quoted Prices in
Active Markets for
Identical Assets
(Level 1) | | Significant Other
Observable Inputs
(Level 2) | | Significant
Unobservable
Inputs
(Level 3) | |
|---|
Assets: | | | | | | | | | | | | | |
Money market funds | | $ | 18,767 | | $ | 18,767 | | $ | — | | $ | — | |
Corporate debt securities | | | 134,123 | | | — | | | 134,123 | | | — | |
U.S. treasury and government agency securities | | | 52,680 | | | — | | | 52,680 | | | — | |
| | | | | | | | | | | | | | |
Total Assets | | $ | 205,570 | | $ | 18,767 | | $ | 186,803 | | $ | — | |
Liabilities: | | | | | | | | | | | | | |
Contingent consideration—MuGard | | $ | 14,550 | | $ | — | | $ | — | | $ | 14,550 | |
| | | | | | | | | | | | | | |
Total Liabilities | | $ | 14,550 | | $ | — | | $ | — | | $ | 14,550 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
With the exception of our money market funds and our acquisition-related contingent consideration, the fair value of our investments is primarily determined from independent pricing services. Independent pricing services normally derive security prices from recently reported trades for identical or similar securities, making adjustments based upon other significant observable market transactions. At the end of each reporting period, we perform quantitative and qualitative analyses of prices received from third parties to determine whether prices are reasonable estimates of fair value. After completing our analyses, we did not adjust or override any fair value measurements provided by our pricing services as of either December 31, 2014 or 2013. In addition, there were no transfers or reclassifications of any securities between Level 1 and Level 2 during either 2014 or 2013.
Contingent consideration
We accounted for the acquisitions of Lumara Health and the MuGard Rights as business combinations under the acquisition method of accounting. Additional details regarding the Lumara Health acquisition and the MuGard License Agreement can be found in Note C, "Business Combinations." The fair value measurements of contingent consideration obligations and the related intangible assets arising from business combinations are determined using unobservable ("Level 3")
148
Table of Contents
inputs. These inputs include (a) the estimated amount and timing of projected cash flows; (b) the probability of the achievement of the factors on which the contingency is based; and (c) the risk-adjusted discount rate used to present value the probability-weighted cash flows. Significant increases (decreases) in any of those inputs in isolation could result in a significantly lower or higher fair value measurement.
Lumara Health
The following table presents a reconciliation of contingent consideration obligations related to the acquisition of Lumara Health measured on a recurring basis using Level 3 inputs as of December 31, 2014 (in thousands):
| | | | |
Balance as of November 12, 2014 | | $ | — | |
Acquisition date fair value of contingent consideration | | | 205,000 | |
Adjustments to fair value of contingent consideration | | | 1,600 | |
| | | | | |
Balance as of December 31, 2014 | | $ | 206,600 | |
| | | | | |
| | | | | |
| | | | | |
The $1.6 million increase of the contingent consideration related to Lumara Health was due to the time value of money. This adjustment to our contingent consideration liability is included in selling, general and administrative expenses in our consolidated statements of operations. We have classified all of the Lumara Health contingent consideration as a long-term liability in our consolidated balance sheet as of December 31, 2014.
MuGard
The following table presents a reconciliation of contingent consideration obligations related to our acquisition of the MuGard Rights measured on a recurring basis using Level 3 inputs as of December 31, 2014 and 2013 (in thousands):
| | | | |
Balance as of June 6, 2013 | | $ | — | |
Acquisition date fair value of contingent consideration | | | 13,700 | |
Payments made | | | (51 | ) |
Adjustments to fair value of contingent consideration | | | 1,074 | |
Other adjustments | | | (173 | ) |
| | | | | |
Balance as of December 31, 2013 | | $ | 14,550 | |
Payments made | | | (270 | ) |
Adjustments to fair value of contingent consideration | | | (2,281 | ) |
Other adjustments | | | 103 | |
| | | | | |
Balance as of December 31, 2014 | | $ | 12,102 | |
| | | | | |
| | | | | |
| | | | | |
During 2014, we revised our forecast of total projected net sales forMuGard and reassessed the fair value of the contingent consideration liability related to the MuGard Rights. As a result, we reduced our contingent consideration liability by $2.3 million for year ended December 31, 2014. During the year ended December 31, 2013, we increased ourMuGard related contingent consideration liability by $1.1 million. These adjustments to contingent consideration liability are included in selling, general and administrative expenses in our consolidated statements of operations.
As of December 31, 2014, we estimate that the undiscounted royalty amounts we could pay under the MuGard License Agreement may range from $20.0 million to $28.0 million over a ten year period beginning on the MuGard License Date, which is our best estimate of the period over which we expect the majority of the asset's cash flows to be derived. This measure is based on significant Level 3 inputs not observable in the market. Key assumptions include a discount rate of approximately 15%. We have
149
Table of Contents
classified $0.8 million of the MuGard contingent consideration as a short-term liability, which was included in accrued expenses in our consolidated balance sheet as of December 31, 2014.
In addition, in connection with the acquisition of the MuGard Rights, we acquired an intangible asset of $16.9 million, which was originally determined based on fair value measurements. These measures were based on significant Level 3 inputs not observable in the market. Key assumptions include a discount rate of 19%. We believe the estimated fair values of the MuGard Rights are based on reasonable assumptions, however, we cannot provide assurance that the underlying assumptions used to forecast the cash flows will materialize as we estimated and thus, our actual results may vary significantly from the estimated results.
Debt
In February 2014, we issued the Convertible Notes. As of December 31, 2014, the fair value of our Convertible Notes was $332.0 million, which differs from their carrying values. The fair value of our Convertible Notes is influenced by interest rates and our stock price and stock price volatility and is determined by prices for the Convertible Notes observed in market trading, which are Level 2 inputs.
In November 2014, in connection with the acquisition of Lumara Health, we entered into the Term Loan Facility. The fair value of our outstanding borrowings under the Term Loan Facility was approximately $342.0 million at December 31, 2014, which differs from their carrying values. The fair value of our Term Loan debt is influenced by interest rates and are Level 2 inputs.
See Note S, "Debt," for additional information on our debt obligations.
F. ACCOUNTS RECEIVABLE, NET
Our net accounts receivable were $38.2 million and $6.8 million as of December 31, 2014 and 2013, respectively, and primarily represented amounts due from wholesalers, distributors and specialty pharmacies to whom we sell our products directly. The increase in accounts receivable in 2014 is due primarily to the inclusion of Lumara Health. Accounts receivable are recorded net of reserves for estimated chargeback obligations, prompt payment discounts and any allowance for doubtful accounts.
As part of our credit management policy, we perform ongoing credit evaluations of our customers, and we have not required collateral from any customer. To date, we have not experienced significant bad debts. Accordingly, we have not established an allowance for doubtful accounts at either December 31, 2014 or 2013. If the financial condition of any of our significant customers was to deteriorate and result in an impairment of its ability to make payments owed to us, an allowance for doubtful accounts may be required which could have a material effect on earnings in the period of any such adjustment.
Customers which represented greater than 10% of our accounts receivable balances as of December 31, 2014 and 2013 were as follows:
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
AmerisourceBergen Drug Corporation | | | 45% | | | 43% | |
McKesson Corporation | | | 12% | | | 29% | |
Cardinal Health, Inc. | | | <10% | | | 19% | |
150
Table of Contents
G. INVENTORIES
Our major classes of inventories were as follows as of December 31, 2014 and 2013 (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Raw materials | | $ | 14,188 | | $ | 3,157 | |
Work in process | | | 5,965 | | | 8,322 | |
Finished goods | | | 20,457 | | | 5,738 | |
| | | | | | | | |
Total | | | 40,610 | | | 17,217 | |
| | | | | | | | |
Included in other long-term assets: | | | | | | | |
Raw materials | | | 7,798 | | | — | |
| | | | | | | | |
Total inventories | | $ | 48,408 | | $ | 17,217 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
During 2014 and 2013, we expensed $0.7 million and $1.1 million ofFeraheme commercial inventory, respectively, which we determined would be solely used in development activities at our third-party suppliers and which we recorded in research and development expenses. In addition, during 2014 and 2013, we expensed $0.6 million and $1.1 million ofFeraheme commercial inventory deemed no longer saleable, which we recorded in cost of product sales.
The increase in total raw materials and finished goods for the year ended December 31, 2014 is primarily due to the inclusion ofMakena inventory acquired in connection with the merger with Lumara Health. We recorded the acquiredMakena inventory at fair value, which required a step-up adjustment to recognize the inventory at its expected net realizable value. The $30.3 million fair value ofMakena inventories included a fair value step-up adjustment of $26.1 million, which will be amortized and recognized as cost of product sales in our consolidated statements of operations as the related inventories are sold. We recognized $1.3 million of the fair value adjustment as cost of product sales during the year ended December 31, 2014. The remaining $24.8 million is estimated to be recognized as follows: $11.1 million in fiscal 2015, $3.5 million in fiscal 2016, $4.0 million in fiscal 2017, $3.5 million in fiscal 2018 and $2.7 million in fiscal 2019. In connection with the fair value step-up adjustment ofMakena inventory, we have recorded a portion of the associated raw material inventory and associated step-up adjustment in other long-term assets as we believe that the amount of inventory purchased in the acquisition exceeds our normal inventory cycle. See Note C, "Business Combinations," for additional information.
H. PROPERTY AND EQUIPMENT, NET
Property and equipment consisted of the following as of December 31, 2014 and 2013 respectively (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Furniture and fixtures | | $ | 1,574 | | $ | 1,536 | |
Leasehold improvements | | | 430 | | | 430 | |
Laboratory and production equipment | | | 493 | | | 376 | |
| | | | | | | | |
| | | 2,497 | | | 2,342 | |
Less—accumulated depreciation | | | (978 | ) | | (496 | ) |
| | | | | | | | |
Property and equipment, net | | $ | 1,519 | | $ | 1,846 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
151
Table of Contents
In September 2013, we relocated our corporate offices from Lexington, Massachusetts to Waltham, Massachusetts and recorded $1.6 million of new leasehold improvements and furniture and fixtures related to our new location.
During 2014, 2013 and 2012, we incurred $0.5 million, $3.0 million and $4.2 million of depreciation expense, respectively. The $3.0 million of depreciation expense in 2013 included $1.9 million of accelerated depreciation expense related to fixed assets at our prior office facility. The $4.2 million of depreciation expense in 2012 included $1.4 million of accelerated depreciation related to our former Cambridge, Massachusetts manufacturing facility and related assets and a $1.1 million impairment loss to decrease the carrying value of these assets to our best estimate of fair value.
I. GOODWILL AND INTANGIBLE ASSETS, NET
Goodwill
In connection with our November 2014 acquisition of Lumara Health, we recognized $205.8 million of goodwill as of December 31, 2014. See Note C, "Business Combinations," for additional information. There has been no change in the goodwill balance since the acquisition.
Intangible Assets, Net
Our identifiable intangible assets consist of license agreements, product rights and other identifiable intangible assets, which result from product and business acquisitions. As of December 31, 2014 and 2013, our identifiable intangible assets consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | | | |
| | December 31, 2014 | | December 31, 2013 | |
|---|
| | Cost | | Accumulated
Amortization | | Net | | Cost | | Accumulated
Amortization | | Net | |
|---|
Amortizable intangible assets: | | | | | | | | | | | | | | | | | | | |
Makena Marketed Product | | $ | 797,100 | | $ | 4,834 | | $ | 792,266 | | $ | — | | $ | — | | $ | — | |
MuGard Rights | | | 16,893 | | | 351 | | | 16,542 | | | 16,893 | | | 49 | | | 16,844 | |
| | | | | | | | | | | | | | | | | | | | |
| | | 813,993 | | | 5,185 | | | 808,808 | | | 16,893 | | | 49 | | | 16,844 | |
Indefinite-lived intangible assets: | | | | | | | | | | | | | | | | | | | |
IPR&D | | | 79,100 | | | — | | | 79,100 | | | — | | | — | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total intangible assets | | $ | 893,093 | | $ | 5,185 | | $ | 887,908 | | $ | 16,893 | | $ | 49 | | $ | 16,844 | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
The Marketed Product and IPR&D intangible assets were acquired in connection with our acquisition of Lumara Health in November 2014. Amortization of the Marketed Product asset is being recognized using an economic consumption model over twenty years, which we believe is an appropriate amortization period due to the estimated economic lives of the product rights and related intangibles. See Note C, "Business Combinations," for additional information.
The MuGard Rights were acquired from PlasmaTech in June 2013. Amortization of the MuGard Rights is being recognized using an economic consumption model over ten years, which represents our best estimate of the period over which we expect the majority of the asset's cash flows to be derived. We believe this is the best approximation of the period over which we will derive the majority of value of the MuGard Rights. See Note C, "Business Combinations," for additional information.
We recorded $5.1 million, less than $0.1 million and no amortization expense for the years ended December 31, 2014, 2013 and 2012, respectively. Amortization expense is recorded in cost of product
152
Table of Contents
sales in our consolidated statements of operations. We expect amortization expense related to our finite-lived intangible assets for the next five fiscal years to be as follows (in thousands):
| | | | |
Period | | Estimated
Amortization
Expense | |
|---|
Year Ended December 31, 2015 | | | 51,886 | |
Year Ended December 31, 2016 | | | 64,977 | |
Year Ended December 31, 2017 | | | 76,679 | |
Year Ended December 31, 2018 | | | 84,359 | |
Year Ended December 31, 2019 | | | 55,746 | |
| | | | | |
Total | | $ | 333,647 | |
| | | | | |
| | | | | |
| | | | | |
J. CURRENT AND LONG-TERM LIABILITIES
Accrued Expenses
Accrued expenses consisted of the following as of December 31, 2014 and 2013 (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Commercial rebates, fees and returns | | $ | 44,807 | | $ | 4,839 | |
Professional, license, and other fees and expenses | | | 14,888 | | | 1,932 | |
Salaries, bonuses, and other compensation | | | 10,176 | | | 5,419 | |
Clinical, manufacturing and regulatory consulting fees and expenses | | | 7,181 | | | 7,834 | |
Restructuring expense | | | 1,952 | | | — | |
Commercial consulting fees and expenses | | | 1,089 | | | 1,301 | |
Short-term contingent consideration | | | 718 | | | 941 | |
| | | | | | | | |
Total accrued expenses | | $ | 80,811 | | $ | 22,266 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Deferred Revenues
Deferred revenues consisted of the following as of December 31, 2014 and 2013 (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Short-term deferred revenues: | | | | | | | |
Takeda | | $ | 44,376 | | $ | 8,226 | |
| | | | | | | | |
Total | | $ | 44,376 | | $ | 8,226 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Long-term deferred revenues: | | | | | | | |
Takeda | | $ | — | | $ | 43,534 | |
3SBio | | | — | | | 1,000 | |
| | | | | | | | |
Total | | $ | — | | $ | 44,534 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Our deferred revenues related to Takeda were recorded in our consolidated balance sheets and include the following as of December 31, 2014:
- •
- $41.2 million related to the amortization of upfront payments and milestone payments recognized under the Amended Takeda Agreement. Included in the $41.2 million was the
153
Table of Contents
amortization of upfront payments we received from Takeda in 2010, which we were recognizing on a straight-line basis over a period of 10 years, which represented the then current patent life ofFeraheme and our best estimate of the period over which we were to substantially perform our obligations. In addition, included in the $41.2 million was the amortization of an aggregate of $18.0 million in milestone payments we received from Takeda in 2012 associated with the commercial launches ofFeraheme in the EU and Canada, which we were amortizing over the original life of the Takeda Agreement. During 2014 and 2013, we recorded $8.2 million and $7.9 million to license fee and other collaboration revenues in our consolidated statements of operations, including, as a result of the Takeda Termination Agreement, the accelerated recognition of $0.3 million of the remaining deferred revenue associated with upfront and milestone payments received to date from Takeda and previously deferred; and
- •
- $3.2 million related to certain agreed upon costs under the terms of the Takeda Termination Agreement.
We expect to recognize the remaining balance of the deferred revenue related to Takeda within the next 12 months. Further, as of December 31, 2014, we recognized the $2.5 million remaining balance of previously deferred product sales to Takeda and the related cost of product sales. See Note R, "Collaborative Agreements," for further information.
In consideration of the grant of the license to 3SBio Inc. ("3SBio") in 2008, we received an upfront payment of $1.0 million, the recognition of which had been deferred. In January 2014, we mutually terminated the agreement with 3SBio, effective immediately, due to the fact that, despite the best efforts of the parties, regulatory approval in China could not be obtained within the agreed upon time period, at which time we recognized the $1.0 million to income in our consolidated statement of operations.
Other Long-Term Liabilities
Other long-term liabilities at December 31, 2014 consisted of deferred rent related to the lease of our principal executive offices in Lexington, Massachusetts and after September 2013, Waltham, Massachusetts, as well as our lease obligations assumed under the lease of Lumara Health's former principal executive offices in St. Louis, Missouri. In addition, other long-term liabilities include future payments to be made to certain states in compliance with a 2011 Lumara Health Settlement Agreement with the Department of Justice, which resolved certain claims under the qui tam provisions of the False Claims Act. Other long-term liabilities at December 31, 2013 consisted solely of deferred rent related to the lease of our principal executive offices.
K. INCOME TAXES
The income tax benefit consisted of the following (in thousands):
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Current: | | | | | | | | | | |
Federal | | $ | — | | $ | — | | $ | — | |
State | | | — | | | — | | | — | |
| | | | | | | | | | | |
Total current | | $ | — | | $ | — | | $ | — | |
Deferred: | | | | | | | | | | |
Federal | | $ | (142,884 | ) | $ | — | | $ | (833 | ) |
State | | | (10,275 | ) | | — | | | (21 | ) |
| | | | | | | | | | | |
Total deferred | | $ | (153,159 | ) | $ | — | | $ | (854 | ) |
| | | | | | | | | | | |
Total income tax benefit | | $ | (153,159 | ) | $ | — | | $ | (854 | ) |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
154
Table of Contents
The reconciliation of the statutory U.S. federal income tax rate to our effective income tax rate is as follows:
| | | | | | | | | |
| | Years Ended December 31, |
|---|
| | 2014 | | 2013 | | 2012 |
|---|
Statutory U.S. federal tax rate | | | (34.0)% | | | (34.0)% | | | (34.0)% |
State taxes, net of federal benefit | | | (7.9)% | | | 2.4% | | | 4.2% |
Equity-based compensation expense | | | 10.6% | | | 9.4% | | | 42.4% |
Permanent items, net | | | 16.0% | | | 5.3% | | | 1.2% |
Tax credits | | | (3.0)% | | | 0.5% | | | 0.8% |
Valuation allowance | | | (864.9)% | | | 16.4% | | | (19.5)% |
| | | | | | | | | | |
Total tax benefit | | | (883.2)% | | | 0.0% | | | (4.9)% |
| | | | | | | | | | |
| | | | | | | | | | |
| | | | | | | | | | |
For the year ended December 31, 2014, we recognized an income tax benefit of $153.2 million, representing an effective tax rate of (883.2%). The difference between the statutory tax rate and the effective tax rate was attributable to a non-recurring benefit of $153.2 million for the release of a portion of the valuation allowance due to taxable temporary differences available as a source of income to realize the benefit of certain pre-existing AMAG deferred tax assets as a result of the Lumara Health acquisition. Excluding the impact of this item, our overall tax provision and effective tax rate would have been zero. Other factors resulting in a difference between the statutory tax rate and the effective tax rate included certain non-deductible stock compensation expenses, certain non-deductible expenses for tax purposes and tax credits. See Note C, "Business Combinations," for more information on the Lumara Health acquisition.
We did not recognize any current federal or state income tax benefit for the year ended December 31, 2013 as we were subject to a full valuation allowance. For the year ended December 31, 2012, we recognized a $0.9 million deferred tax benefit, as the result of the recognition of corresponding income tax expense associated with the decrease in the unrealized loss on our investments, primarily related to auction rate securities, which we carried at fair market value during 2012. The corresponding income tax expense was recorded in other comprehensive loss.
Deferred tax assets and deferred tax liabilities are recognized based on temporary differences between the financial reporting and tax basis of assets and liabilities using future enacted rates. A valuation allowance is recorded against deferred tax assets if it is more likely than not that some or all
155
Table of Contents
of the deferred tax assets will not be realized. The components of our deferred tax assets and liabilities are as follows (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Assets | | | | | | | |
Net operating loss carryforwards | | $ | 188,873 | | $ | 85,269 | |
Tax credit carryforwards | | | 24,574 | | | 12,396 | |
Deferred revenue | | | 17,216 | | | 20,368 | |
Equity-based compensation expense | | | 3,436 | | | 4,176 | |
Capitalized research & development | | | 32,359 | | | 39,214 | |
Intangibles | | | 272 | | | 680 | |
Debt instruments | | | 731 | | | — | |
Reserves | | | 7,782 | | | — | |
Property, plant and equipment | | | 61 | | | — | |
Other | | | 5,014 | | | 4,371 | |
Liabilities | | | | | | | |
Property, Plant, and Equipment Depreciation | | | — | | | (58 | ) |
Intangible Assets and Inventory Amortization | | | (290,491 | ) | | — | |
Other | | | (1,795 | ) | | — | |
| | | | | | | | |
| | | (11,968 | ) | | 166,416 | |
Valuation allowance | | | (33,557 | ) | | (166,416 | ) |
| | | | | | | | |
Net deferred taxes | | $ | (45,525 | ) | $ | — | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The valuation allowance decreased by approximately $132.9 million for the year ended December 31, 2014 primarily due to taxable temporary differences available as a source of income to realize the benefit of certain pre-existing AMAG deferred tax assets as a result of the Lumara Health acquisition, which provided a tax benefit of $153.2 million offset by an increase in the valuation allowance of $20.3 million primarily related to certain Lumara deferred tax assets established in purchase accounting. In determining the amount of valuation allowance release, we considered the relevant tax law ordering rules for utilization of tax assets to determine whether the acquired Lumara Health or the pre-existing AMAG deferred tax assets were realizable. As of December 31, 2014, we maintained a partial valuation allowance on the net deferred tax assets as we benefitted only those deferred tax assets to the extent that existing taxable temporary differences could be used as a source of future income.
At December 31, 2014, we had federal net operating loss ("NOL") carryforwards of approximately $542.3 million and state NOL carryforwards of up to $242.2 million of which $254.1 million and $124.7 million were acquired as part of the Lumara Health transaction. We also had federal capital loss carryforwards of $2.1 million to offset future capital gains. At December 31, 2014, $30.6 million and $5.4 million of federal and state NOLs, respectively, related to excess equity-based compensation tax deductions the benefits for which will be recorded to additional paid-in capital when recognized through a reduction of cash taxes paid. The federal NOLs and the most significant state NOLs expire at various dates through 2034. The capital loss carryforwards will expire through 2017. We have federal tax credits of approximately $21.6 million, to offset future tax liabilities of which $12.0 million were acquired as part of the Lumara Health transaction. We have state tax credits of $4.5 million to offset future tax liabilities. These federal and state tax credits will expire periodically through 2034 if not utilized.
Utilization of our NOLs and research and development ("R&D") credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations that have occurred
156
Table of Contents
previously or that could occur in the future in accordance with Section 382 of the Internal Revenue Code of 1986 ("Section 382") as well as similar state provisions. These ownership changes may limit the amount of NOL and R&D credit carryforwards that can be utilized annually to offset future taxable income and taxes, respectively. In general, an ownership change as defined by Section 382 results from transactions increasing the ownership of certain shareholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period. Since our formation, we have raised capital through the issuance of capital stock on several occasions. These financings, combined with the purchasing shareholders' subsequent disposition of those shares, may have resulted in a change of control as defined by Section 382 or could result in a change of control in the future upon subsequent disposition. We conducted an analysis under Section 382 to determine if historical changes in ownership through December 31, 2012 would limit or otherwise restrict our ability to utilize these NOL and R&D credit carryforwards. As a result of this analysis, we do not believe there are any significant limitations on our ability to utilize these carryforwards. However, future changes in ownership after December 31, 2012 could affect the limitation in future years. Any limitation may result in expiration of a portion of the NOL or R&D credit carryforwards before utilization.
At December 31, 2014 and 2013, we had no unrecognized tax benefits. We have not, as yet, conducted a study of our R&D credit carryforwards. Such a study could result in an adjustment to our R&D credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position.
We would recognize both accrued interest and penalties related to unrecognized benefits in income tax expense. We have not recorded any interest or penalties on any unrecognized benefits since inception.
The statute of limitations for assessment by the Internal Revenue Service (the "IRS") and state tax authorities is closed for tax years prior to December 31, 2011, although carryforward attributes that were generated prior to tax year 2011 may still be adjusted upon examination by the IRS or state tax authorities if they either have been or will be used in a future period. We file income tax returns in the U.S. federal and various state jurisdictions. There are currently no federal or state audits in progress.
It should be noted that the allocation of the purchase price related to the Lumara Health transaction is subject to adjustment upon finalization of fair valuation procedures and therefore the impact of the tax benefit associated with the valuation allowance release and deferred tax assets and liabilities (including uncertain tax positions) are subject to change.
L. ACCUMULATED OTHER COMPREHENSIVE INCOME (LOSS)
The table below presents information about the effects of net income (loss) of significant amounts reclassified out of accumulated other comprehensive loss ("AOCI"), net of tax, during 2014 and 2013 (in thousands):
| | | | | | | |
| | December 31, | |
|---|
| | 2014 | | 2013 | |
|---|
Beginning Balance | | $ | (3,491 | ) | $ | (3,247 | ) |
Other comprehensive income (loss) before reclassifications | | | (191 | ) | | (268 | ) |
Gain (loss) reclassified from other accumulated comprehensive loss | | | 65 | | | 24 | |
| | | | | | | | |
Ending Balance | | $ | (3,617 | ) | $ | (3,491 | ) |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The amounts reclassified from other comprehensive loss for 2014 and 2013 primarily represented realized gains on investments, which are included in our consolidated statement of operations under "Gains (losses) on investments, net."
157
Table of Contents
M. EQUITY-BASED COMPENSATION
We currently maintain three equity compensation plans, including our Third Amended and Restated 2007 Equity Incentive Plan (the "2007 Plan"), our Amended and Restated 2000 Stock Plan (the "2000 Plan") (under which we no longer grant awards) and the Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (the "Lumara Health 2013 Plan"). During 2014, 2013 and 2012, we also granted equity through inducement grants outside of these plans to certain newly hired executive officers and certain employees, including in connection with the Lumara Health acquisition.
Third Amended and Restated 2007 Equity Incentive Plan
Our 2007 Plan was originally approved by our stockholders in November 2007. In each of May 2009, May 2010, and May 2013 our stockholders approved proposals to amend and restate our 2007 Plan to, among other things, increase the number of shares authorized for issuance thereunder by 600,000, 800,000 and 1,100,000 shares, respectively. In addition, the amendment approved by our stockholders in May 2009 replaced a limitation on the number of shares in the aggregate which could be issued under the 2007 Plan with respect to RSUs, restricted stock, stock and similar equity interests in our company with a fungible share reserve whereby the number of shares available for issuance under the 2007 Plan is reduced by one share of our common stock issued pursuant to an option or stock appreciation right and by 1.5 shares for each share of our common stock issued pursuant to a RSU award or other similar equity-based award.
The 2007 Plan provides for the grant of stock options, RSUs, restricted stock, stock, and other equity interests in our company to employees, officers, directors, consultants, and advisors of our company and our subsidiaries. We generally issue common stock from previously authorized but unissued shares to satisfy option exercises and restricted stock awards. The terms and conditions of each such grant, including, but not limited to, the number of shares, the exercise price, term of the option/award and vesting requirements, are determined by our Board of Directors (the "Board") or the Compensation Committee of our Board. Our Board may award stock options in the form of nonqualified stock options or incentive stock options ("ISOs"). Stock options may be granted at an exercise price no less than fair market value of a share of our common stock on the date of grant, as determined by our Board or the Compensation Committee of our Board, subject to certain limitations.
As of December 31, 2014, we have granted options and RSUs covering 7,414,752 shares of common stock under our 2007 Plan, of which 3,112,356 stock options and 703,124 RSUs have expired or terminated, and of which 610,297 options have been exercised and 577,132 shares of common stock have been issued pursuant to RSUs that became fully vested. The number of options and RSUs outstanding under this plan as of December 31, 2014, was 2,051,017 and 360,826, respectively. The remaining number of shares available for future grants as of December 31, 2014 was 1,707,989, not including shares subject to outstanding awards under the 2000 Plan, which will be added to the total number of shares available for issuance under the 2007 Plan to the extent that such awards expire or terminate for any reason prior to exercise. All outstanding stock options granted under our 2007 Plan have an exercise price equal to the closing price of a share of our common stock on the grant date and have either a seven or ten-year term.
Amended and Restated 2000 Stock Plan
Our 2000 Plan provided for the grant of options and other equity-based awards to our directors, officers, employees and consultants. The terms and conditions of each such grant, including, but not limited to, the number of shares, the exercise price, term of the option/award and vesting requirements, were determined by our Board or the Compensation Committee of our Board. As of December 31, 2014, we have granted stock options and RSUs covering 2,182,700 shares of common stock under the 2000 Plan, of which 1,049,339 stock options and 1,500 RSUs have expired or terminated, and of which
158
Table of Contents
1,054,095 stock options have been exercised and 42,500 shares of common stock have been issued pursuant to RSUs that became fully vested. The remaining number of shares underlying outstanding stock options which were issued pursuant to our 2000 Plan as of December 31, 2014, was 35,266. There were no remaining RSUs which were issued pursuant to our 2000 Plan as of December 31, 2014. All outstanding stock options granted under the 2000 Plan have an exercise price equal to the closing price of our common stock on the grant date and have a ten year term. In November 2007, the 2000 Plan was succeeded by our 2007 Plan and, accordingly, no further grants may be made under this plan. Any shares that remained available for issuance under the 2000 Plan as of the date of adoption of the 2007 Plan are included in the number of shares that may be issued under the 2007 Plan. Any shares subject to outstanding awards granted under the 2000 Plan that expire or terminate for any reason prior to exercise will be added to the total number of shares available for issuance under the 2007 Plan.
Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan
On November 12, 2014, we assumed the Lumara Health 2013 Plan in connection with the acquisition of Lumara Health. The total number of shares issuable pursuant to awards under this plan as of the effective date of the acquisition and after taking into account any adjustments as a result of the acquisition, was 200,000 shares.
The Lumara Health 2013 Plan provides for the grant of stock options, RSUs, restricted stock, stock, stock appreciation rights and other equity interests in our company to certain of our employees, officers, directors, consultants, and advisors of our company and our subsidiaries that are newly-hired or that previously performed services for Lumara Health. We generally issue common stock from previously authorized but unissued shares to satisfy option exercises and restricted stock awards. The terms and conditions of each award assumed in the acquisition of Lumara Health were previously determined by Lumara Health prior to being assumed in connection with the acquisition, subject to applicable adjustments made in connection with such acquisition. The terms and conditions of each award made after the acquisition of Lumara Health, including, but not limited to, the number of shares, the exercise price, term of the option/award and vesting requirements, are determined by our Board or the Compensation Committee of our Board. Our Board may award stock options in the form of nonqualified stock options or ISOs. Stock options may be granted at an exercise price no less than fair market value of a share of our common stock on the date of grant, as determined by our Board or the Compensation Committee of our Board, subject to certain limitations.
As of December 31, 2014, we have granted new options and RSUs covering 64,000 shares of common stock under the Lumara Health 2013 Plan. The number of options and RSUs outstanding under this plan as of December 31, 2014, was 44,000 and 20,000, respectively. The remaining number of shares available for future grants as of December 31, 2014 was 136,000. All outstanding stock options granted under the Lumara Health 2013 Plan have an exercise price equal to the closing price of a share of our common stock on the grant date and have either a seven or ten-year term.
Other Equity Compensation Grants
In August 2014, we granted certain members of our senior management performance-based RSUs under our 2007 Plan covering a maximum of 195,000 shares of common stock, which will be earned, if at all, based on the achievement of certain (a) targets based upon the calculated value expected to be realized with respect to certain business and corporate development transactions and (b) stock price minimums, during the 30-month period ending January 2, 2017, measured as of January 4, 2016 and January 2, 2017. Fifty percent of the RSU grant that is earned through January 4, 2016 shall vest as of such date, and 100% of the RSU grant that is earned through January 2, 2017 (less the portion previously vested) shall vest as of January 2, 2017, subject to the continued employment of the grantee through each such date. In the event that the minimum conditions of these RSUs are not met as of the measurement dates, none of the RSUs will vest. The maximum total fair value of these RSUs is $6.3 million, which will be recognized to expense over a period of approximately three years from the date the vesting conditions outlined in these grants are deemed probable, net of any estimated and actual forfeitures. We recognized $0.1 million of expense related to these awards during December 31, 2014.
159
Table of Contents
In February 2013, we granted RSUs under our 2007 Plan to certain members of our senior management covering a maximum of 82,500 shares of common stock, which are subject to a performance condition tied to the price of our common stock. These RSUs vest, if at all, at the end of the three-year period ending December 31, 2015 based on the achievement of a minimum, target or maximum stock price range. In the event that the minimum stock price range is not achieved at the measurement date, none of the RSUs will vest. The maximum total fair value of these RSUs is $0.7 million, which is being recognized to expense over a period of three years from the date of grant, net of any estimated and actual forfeitures.
During 2014, 2013 and 2012, our Board granted options to purchase 165,000, 270,000 and 300,000 shares of our common stock, respectively, and 87,900, 115,000 and 100,000 RSUs, respectively, to certain new-hire employees, including members of our senior management, to induce them to accept employment with us. In addition to these inducement grants, during December 2014, we issued options covering 304,600 shares of our common stock and 22,600 RSUs to certain employees of Lumara Health in order to induce them to accept employment with us following our acquisition of Lumara Health. The options were granted at an exercise price equal to the fair market value of a share of our common stock on the respective grant dates and will be exercisable in four equal annual installments beginning on the first anniversary of the respective grant dates. The RSU grants will vest in four equal annual installments beginning on the first anniversary of the respective grant dates. The foregoing grants were made pursuant to inducement grants outside of our 2007 Plan and the Lumara Health 2013 Plan as permitted under the NASDAQ Stock Market listing rules. We assessed the terms of these awards and determined there was no possibility that we would have to settle these awards in cash and therefore, equity accounting was applied.
Since we first began issuing inducement grants outside of our plans in 2012 as permitted under the NASDAQ Stock Market listing rules, we have issued a total of 1,344,000 shares of common stock pursuant to inducement grants, of which 146,250 stock options and 67,500 RSUs have been expired or terminated and of which 28,750 options have been exercised and 75,000 shares of common stock have been issued pursuant to RSUs that became fully vested.
Equity-based compensation expense
Equity-based compensation expense for 2014, 2013 and 2012 consisted of the following (in thousands):
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
Cost of product sales | | $ | 122 | | $ | 121 | | $ | 225 | |
Research and development | | | 1,596 | | | 2,149 | | | 1,994 | |
Selling, general and administrative | | | 6,907 | | | 5,734 | | | 4,805 | |
| | | | | | | | | | | |
Total equity-based compensation expense | | $ | 8,625 | | $ | 8,004 | | $ | 7,024 | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
We reduce the compensation expense being recognized to account for estimated forfeitures, which we estimate based primarily on historical experience, adjusted for unusual events such as corporate restructurings, which may result in higher than expected turnover and forfeitures. Under current accounting guidance, forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
Due to the uncertainty surrounding the realization of the favorable tax attributes in future tax returns associated with operating losses we incurred in the past, we have not recognized any excess tax benefits from the exercise of options. Accordingly, there was no impact recorded in cash flows from financing activities or cash flows from operating activities as reported in the accompanying consolidated statements of cash flows.
160
Table of Contents
The following table summarizes the weighted average assumptions we utilized for purposes of valuing grants of options to our employees and non-employee directors:
| | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2014 | | 2013 | | 2012 | |
|---|
| | Employees | | Non-Employee
Directors | | Employees | | Non-Employee
Directors | | Employees | | Non-Employee
Directors | |
|---|
Risk free interest rate (%) | | | 1.56 | | | 1.28 | | | 0.95 | | | 0.85 | | | 0.66 | | | 0.68 | |
Expected volatility (%) | | | 47 | | | 46 | | | 59 | | | 46 | | | 57 | | | 56 | |
Expected option term (years) | | | 5.00 | | | 4.00 | | | 5.00 | | | 4.00 | | | 4.66 | | | 4.00 | |
Dividend yield | | | none | | | none | | | none | | | none | | | none | | | none | |
Risk free interest rates utilized are based upon published U.S. Treasury yields at the date of the grant for the expected option term. During 2014 and 2013, we estimated our expected stock price volatility by basing it on the historical volatility of our own common stock price over the prior period equivalent to our expected option term to better reflect expected future volatility. During 2012, we estimated our expected stock price volatility by basing it on a blend of our own common stock price and the historical volatility of other similar companies over the prior period equivalent to our expected option term to better reflect expected future volatility. To compute the expected option term, we analyze historical exercise experience as well as expected stock option exercise patterns.
The following table summarizes details regarding stock options granted under our equity incentive plans for the year ended December 31, 2014:
| | | | | | | | | | | | | |
| | December 31, 2014 | |
|---|
| | Options | | Weighted
Average
Exercise Price | | Weighted
Average
Remaining
Contractual
Term | | Aggregate
Intrinsic Value
($ in millions) | |
|---|
Outstanding at beginning of year | | | 2,819,676 | | $ | 21.31 | | | | | | | |
Granted | | | 1,391,776 | | | 25.75 | | | | | | | |
Exercised | | | (494,576 | ) | | 19.43 | | | | | | | |
Expired and/or forfeited | | | (720,493 | ) | | 25.81 | | | | | | | |
| | | | | | | | | | | | | | |
Outstanding at end of year | | | 2,996,383 | | $ | 22.60 | | | 7.7 | | $ | 60,546 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Outstanding at end of year—vested and unvested expected to vest | | | 2,688,944 | | $ | 22.60 | | | 7.6 | | $ | 54,410 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Exercisable at end of year | | | 946,787 | | $ | 22.47 | | | 6.0 | | $ | 60,459 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
The weighted average grant date fair value of stock options granted during 2014, 2013 and 2012 was $10.63, $8.60 and $6.90, respectively. A total of 668,321 stock options vested during 2014. The total grant date fair value of options that vested during 2014, 2013 and 2012 was $5.7 million, $4.5 million and $5.5 million, respectively. The aggregate intrinsic value of options exercised during 2014, 2013 and 2012, excluding purchases made pursuant to our employee stock purchase plans, measured as of the exercise date, was approximately $5.9 million, $1.0 million and $0.1 million, respectively. The intrinsic value of a stock option is the amount by which the fair market value of the underlying stock on a specific date exceeds the exercise price of the common stock option.
In 2014, we issued an aggregate of 356,626 RSUs to our employees and directors (including inducement grants). In general, these grants vest on an annual basis over a four year period. The estimated fair value of RSUs granted was determined at the grant date based upon the quoted market price per share on the date of the grant.
161
Table of Contents
The following table summarizes details regarding RSUs granted under our equity incentive plans for the year ended December 31, 2014:
| | | | | | | |
| | December 31, 2014 | |
|---|
| | Unvested
Restricted
Stock Units | | Weighted
Average Grant
Date Fair Value | |
|---|
Outstanding at beginning of year | | | 465,394 | | $ | 17.28 | |
Granted | | | 356,626 | | | 22.88 | |
Vested | | | (151,518 | ) | | 17.71 | |
Forfeited | | | (129,276 | ) | | 18.25 | |
| | | | | | | | |
Outstanding at end of year | | | 541,226 | | $ | 20.62 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
Outstanding at end of year and expected to vest | | | 465,686 | | $ | 20.59 | |
| | | | | | | | |
| | | | | | | | |
| | | | | | | | |
The weighted average grant date fair value of RSUs granted during 2014, 2013 and 2012 was $22.88, $16.31 and $15.64, respectively. The total grant date fair of RSUs that vested during 2014, 2013 and 2012 was $2.7 million, $2.8 million and $3.5 million, respectively.
At December 31, 2014, the amount of unrecorded equity-based compensation expense for both option and RSU awards, net of forfeitures, attributable to future periods was approximately $24.0 million. Of this amount, $15.9 million was associated with stock options and is expected to be amortized on a straight-line basis to expense over a weighted average period of approximately two years, and $8.1 million was associated with RSUs and is expected to be amortized to on a straight-line basis to expense over a weighted average period of approximately two years. Such amounts will be amortized primarily to research and development or selling, general and administrative expense. These future estimates are subject to change based upon a variety of future events, which include, but are not limited to, changes in estimated forfeiture rates, employee turnover, and the issuance of new stock options and other equity-based awards.
N. EMPLOYEE SAVINGS PLAN
We provide a 401(k) Plan to our employees by which they may defer compensation for income tax purposes under Section 401(k) of the Internal Revenue Code. Each employee may elect to defer a percentage of his or her salary up to a specified maximum. Our 401(k) Plan provides, among other things, for a company contribution of 3% of each employee's combined salary and certain other compensation for the plan year. Salary deferred by employees and contributions by us to the 401(k) Plan are not taxable to employees until withdrawn from the 401(k) Plan and contributions are deductible by us when made. The amount of our company contribution for the 401(k) Plan was $0.8 million, $0.7 million, and $0.8 million for 2014, 2013 and 2012, respectively.
O. STOCKHOLDERS' EQUITY
Our certificate of incorporation authorizes our Board to issue preferred stock from time to time in one or more series. The rights, preferences, restrictions, qualifications and limitations of such stock are determined by our Board. In September 2009, our Board adopted a shareholder rights plan (the "Rights Agreement").
On February 11, 2014, in connection with the pricing of the Convertible Notes, we and American Stock Transfer & Trust Company, LLC (the "Rights Agent") entered into an amendment (the "Convertible Notes Amendment") to the Rights Agreement. The Convertible Notes Amendment, among other things, provides that, notwithstanding anything in the Rights Agreement to the contrary, each of JPMorgan Chase Bank, National Association, London Branch, Morgan Stanley & Co. International plc and Royal Bank of Canada (together the "Call Spread Counterparties") shall be
162
Table of Contents
deemed not to beneficially own any common shares underlying, or synthetically owned pursuant to, any warrant held by such Call Spread Counterparty, any common shares held by such Call Spread Counterparty (or any affiliate thereof) to hedge its exposure with respect to the convertible bond hedges and warrants, any common shares underlying, or synthetically owned pursuant to, any Derivative Securities (as such term is defined in the Rights Agreement), including the Convertible Notes, held, or entered into, by such Call Spread Counterparty (or any affiliate thereof) to hedge its exposure with respect to the convertible bond hedges and warrants or any Convertible Notes held by such Call Spread Counterparty (or any affiliate thereof) in its capacity as underwriter in the notes offering.
On September 26, 2014, we adopted another amendment to our Rights Agreement to help preserve our substantial tax assets associated with NOLs and other tax benefits by deterring certain stockholders from increasing their percentage ownership in our stock (the "NOL Amendment"). The NOL Amendment shortens the expiration date of the Rights Agreement from September 17, 2019 to March 31, 2017, decreases the exercise price of the rights from $250.0 to $80.0 in connection therewith, and makes changes to the definition of "beneficial ownership," as used in the Rights Agreement, as amended, to make it consistent with how ownership is defined under Section 382 of the Internal Revenue Code of 1986, as amended. The original Rights Agreement provided for a dividend distribution of one preferred share purchase right (a "Right") for each outstanding share of our common stock, which dividend was paid on September 17, 2009. Rights will separate from the common stock and will become exercisable upon the earlier of (a) the close of business on the 10th calendar day following the first public announcement that a person or group of affiliated or associated persons has acquired beneficial ownership of 4.99% or more (which percentage had been 20% before the NOL Amendment) of the outstanding shares of common stock, other than as a result of repurchases of stock by us or certain inadvertent actions by a stockholder or (b) the close of business on the 10th business day (or such later day as the Board may determine) following the commencement of a tender offer or exchange offer that could result, upon its consummation, in a person or group becoming the beneficial owner of 4.99% or more (which percentage had been 20% before the NOL Amendment) of the outstanding shares of common stock (the earlier of such dates being herein referred to as the "Distribution Date").
The NOL Amendment provides that the Rights are not exercisable until the Distribution Date and will expire at the earliest of (a) March 31, 2017, (b) the time at which the Rights are redeemed or exchanged, (c) the effective date of the repeal of Section 382 or any successor statute if the Board determines that the NOL Rights Plan is no longer necessary or desirable for the preservation of our tax benefits, (d) the first day of our taxable year to which the Board determines that no tax benefits may be carried forward or (e) September 26, 2015 if stockholder approval of the NOL Amendment has not been obtained by or on such date.
We expect to submit the NOL Amendment to a vote of our stockholders at our 2015 annual meeting of stockholders. There can be no assurance that the NOL Amendment will result in us being able to preserve all or any of the substantial tax assets associated with NOLs and other tax benefits.
P. BUSINESS SEGMENTS
We have determined that we conduct our operations in one business segment: the manufacture, development and commercialization of products for use in treating various conditions. Long-lived assets consist entirely of property and equipment and are located in the U.S. for all periods presented.
Q. COMMITMENTS AND CONTINGENCIES
Commitments
Our long-term contractual obligations include commitments and estimated purchase obligations entered into in the normal course of business. These include commitments related to purchases of inventory of our products, research and development service agreements, operating leases and selling,
163
Table of Contents
general and administrative obligations, and milestone payments due under our licensing and acquisition agreements.
Operating and Facility Lease Obligations
We have entered into certain operating leases, including certain office equipment and automobiles, which expire through 2017. Expense associated with these operating leases, including previous leases of certain automobiles, amounted to approximately $0.2 million, $(0.3) million and, $0.9 million for 2014, 2013 and 2012, respectively. The net credit for operating lease expense in 2013 is due to the excess of the sales value of certain automobiles we previously leased over the contracted value in connection with the 2013 termination of the automobile leases and the subsequent sales of the automobiles by the leasing companies. Future minimum lease payments associated with all non-cancellable equipment, service and lease agreements, excluding facility-related leases are approximately $0.6 million for 2015.
In June 2013, we entered into a lease agreement with BP Bay Colony LLC (the "Landlord") for the lease of certain real property located at 1100 Winter Street, Waltham, Massachusetts (the "Waltham Premises") for use as our principal executive offices. Beginning in September 2013, the initial term of the lease is five years and two months with one five-year extension term at our option. During the extension period, the base rent will be an amount agreed upon by us and the Landlord. In addition to base rent, we are also required to pay a proportionate share of the Landlord's operating costs.
The Landlord agreed to pay for certain agreed-upon improvements to the Waltham Premises and we agreed to pay for any increased costs due to changes by us to the agreed-upon plans. We record all tenant improvements paid by us as leasehold improvements and amortize these improvements over the shorter of the estimated useful life of the improvement or the remaining life of the initial lease term. Amortization of leasehold improvements is included in depreciation expense.
In addition, in connection with our facility lease for the Waltham Premises, in June 2013 we delivered to the Landlord a security deposit of $0.4 million in the form of an irrevocable letter of credit. This security deposit will be reduced to $0.3 million on the second anniversary of the date the lease commenced. The cash securing this letter of credit is classified on our balance sheet as of December 31, 2014 and 2013 as a long-term asset and is restricted in its use.
In June 2013, we also entered into an Assignment and Assumption of Lease (the "Assignment Agreement") with Shire Human Genetic Therapies, Inc. ("Shire") effecting the assignment to Shire of the right to occupy our former office space located at 100 Hayden Avenue, Lexington, Massachusetts (the "Prior Space"). Under the Assignment Agreement, the assignment to Shire became effective on September 21, 2013, the date of our departure from the Prior Space, and Shire assumed all of our obligations as the tenant of the Prior Space. The Assignment Agreement also provided for the conveyance of furniture and other personal property by us to Shire.
In connection with our acquisition of Lumara Health, we have assumed the lease of certain real property located at 16640 Chesterfield Grove Road, Chesterfield, Missouri (the "St. Louis Premises"), which we are currently using as temporary office space for Lumara Health employees as they relocate to the Waltham Premises. Beginning in September 2013, the initial term of the lease is five years and two months. In addition to base rent, we are also required to pay a proportionate share of the Landlord's operating costs. We are attempting to sublease the St. Louis Premises and if successful, future operating lease commitments will be partially offset by proceeds received from the sublease.
164
Table of Contents
Future minimum payments under our non-cancelable facility-related leases as of December 31, 2014 are as follows (in thousands):
| | | | |
Period | | Minimum Lease
Payments | |
|---|
Year Ended December 31, 2015 | | $ | 1,451 | |
Year Ended December 31, 2016 | | | 1,456 | |
Year Ended December 31, 2017 | | | 1,462 | |
Year Ended December 31, 2018 | | | 1,174 | |
| | | | | |
Total | | $ | 5,543 | |
| | | | | |
| | | | | |
| | | | | |
Facility-related rent expense, net of deferred rent amortization, for the Waltham Premises and the Prior Space, as applicable, was $0.8 million, $1.5 million and $1.7 million for 2014, 2013, and 2012. Facility-related rent expense for the St. Louis Premises was less than $0.1 million from November 12, 2014 through December 31, 2014.
Debt Obligations
Our long-term debt obligations reflect our obligations under the Convertible Notes and Term Loan Facility to pay interest on the $540.0 million aggregate principal amount and to make scheduled principal payments on the Term Loan Facility and principal payments at maturity or upon conversion, in the case of the Convertible Notes.
Purchase Commitments
During 2014, we entered into various agreements with third parties for which we had remaining purchase commitments of approximately $4.8 million as of December 31, 2014. These agreements principally related to certain purchase orders for the production of our products, certain outsourced commercial activities, manufacturing commitments, our information technology infrastructure, and other operational activities.
Contingent Consideration Related to Business Combinations
In connection with our acquisition of Lumara Health in November 2014, we agreed to pay up to an additional $350.0 million based on the achievement of certain sales milestones. Due to the contingent nature of these milestone payments, we cannot predict the amount or timing of such payments. See Note C, "Business Combinations," for more information on the Lumara Health acquisition and related milestone payments.
Other Funding Commitments
As of December 31, 2014, we had several ongoing clinical studies in various clinical trial stages. Our most significant clinical trial expenditures were to clinical research organizations ("CROs"). The contracts with CROs are generally cancellable, with notice, at our option. We have recorded accrued expenses in our consolidated balance sheet of approximately $1.9 million representing expenses incurred with these organizations as of December 31, 2014, net of any amounts prepaid to these CROs.
Severance Arrangements
We have entered into employment agreements or other arrangements with most of our executive officers and certain other employees, which provide for the continuation of salary and certain benefits and, in certain instances, the acceleration of the vesting of certain equity awards to such individuals in the event that the individual is terminated other than for cause, as defined in the applicable employment agreements or arrangements.
165
Table of Contents
Indemnification Obligations
As permitted under Delaware law, pursuant to our certificate of incorporation, by-laws and agreements with all of our current directors, executive officers, and certain of our employees, we are obligated to indemnify such individuals for certain events or occurrences while the officer, director or employee is, or was, serving at our request in such capacity. The maximum potential amount of future payments we could be required to make under these indemnification obligations is not capped. Our director and officer insurance policy limits our initial exposure to $1.0 million and our policy provides significant coverage. As a result, we believe the estimated fair value of these indemnification obligations is likely to be immaterial.
We are also a party to a number of other agreements entered into in the ordinary course of business, which contain typical provisions and which obligate us to indemnify the other parties to such agreements upon the occurrence of certain events. Such indemnification obligations are usually in effect from the date of execution of the applicable agreement for a period equal to the applicable statute of limitations. Our aggregate maximum potential future liability under such indemnification provisions is uncertain. Except for expenses we incurred related to the Silverstrand class action lawsuit, described below, filed against us in March 2010, we have not incurred any expenses as a result of such indemnification provisions. Accordingly, we have determined that the estimated aggregate fair value of our potential liabilities under such indemnification provisions is not significant, and we have not recorded any liability related to such indemnification.
Contingencies
Legal Proceedings
We accrue a liability for legal contingencies when we believe that it is both probable that a liability has been incurred and that we can reasonably estimate the amount of the loss. We review these accruals and adjust them to reflect ongoing negotiations, settlements, rulings, advice of legal counsel and other relevant information. To the extent new information is obtained and our views on the probable outcomes of claims, suits, assessments, investigations or legal proceedings change, changes in our accrued liabilities would be recorded in the period in which such determination is made. For the matters referenced below, the liability is not probable or the amount cannot be reasonably estimated and, therefore, accruals have not been made. In addition, in accordance with the relevant authoritative guidance, for any matters in which the likelihood of material loss is at least reasonably possible, we will provide disclosure of the possible loss or range of loss. If a reasonable estimate cannot be made, however, we will provide disclosure to that effect.
A purported class action complaint was originally filed on March 18, 2010 in the U.S. District Court for the District of Massachusetts, entitled Silverstrand Investments et. al. v. AMAG Pharm., Inc., et. al., Civil Action No. 1:10-CV-10470-NMG, and was amended on September 15, 2010 and on December 17, 2010. The second amended complaint, filed on December 17, 2010 alleged that we and our former President and Chief Executive Officer, former Chief Financial Officer, the then-members of our Board, and certain underwriters in our January 2010 offering of common stock violated certain federal securities laws, specifically Sections 11 and 12(a)(2) of the Securities Act of 1933, as amended, and that our former President and Chief Executive Officer and former Chief Financial Officer violated Section 15 of such Act, respectively, by making certain alleged omissions in a registration statement filed in January 2010. The plaintiffs sought unspecified damages on behalf of a purported class of purchasers of our common stock pursuant to our common stock offering on or about January 21, 2010. After litigating the class action lawsuit for several years, on September 12, 2014, we and the other defendants entered into a stipulation of settlement with the lead plaintiffs (on behalf of themselves and each of the class members) to resolve the class action securities lawsuit. Pursuant to the stipulation of settlement, and in exchange for a release of all claims by the class members and certain other persons,
166
Table of Contents
and dismissal of the lawsuit with prejudice, we agreed to cause our insurer to pay eligible class members and their attorneys a total of $3.75 million. On October 2, 2014, the U.S. District Court preliminarily approved the settlement, and potential class members were notified of the proposed settlement and the procedures by which they could seek to recover from the settlement fund, object to the settlement or request to be excluded from the settlement class and on January 30, 2015, the stipulation of settlement was approved by the U.S. District Court. The U.S. District Court entered final judgment on February 2, 2015. Any appeals of the settlement are due by March 4, 2015. We have recorded the $3.75 million settlement amount in prepaid and other current assets and a corresponding amount in accrued expenses on our consolidated balance sheet as of December 31, 2014, as the settlement amount will be fully covered by our insurance carrier. There was no impact to our consolidated statement of operations for the year ended December 31, 2014.
On October 19, 2011, plaintiff Frank Julianello filed a complaint against Lumara Health (then-named K-V Pharmaceutical Company ("K-V Pharmaceutical")) and certain individual defendants, in the United States District Court for the Eastern District of Missouri (the "Court"), alleging violations of the anti-fraud provisions of the federal securities laws on behalf of all purchasers of the publicly traded securities of Lumara Health between February 14, 2011 and April 4, 2011. The complaint alleges class members were damaged by paying artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On October 31, 2011, plaintiff Ramakrishna Mukku filed a complaint against Lumara Health, in the United States District Court for the Eastern District of Missouri, alleging violations of the anti-fraud provisions of the federal securities laws on behalf of all purchasers of the publicly traded securities of Lumara Health between February 14, 2011 and April 4, 2011. The complaint alleges class members were damaged by paying artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On November 2, 2011, plaintiff Hoichi Cheong filed a complaint against Lumara Health, in the United States District Court for the Eastern District of Missouri, on behalf of purchasers of the securities of Lumara Health, who purchased or otherwise acquired K-V Pharmaceutical securities between February 14, 2011 and April 4, 2011, seeking to pursue remedies under the Exchange Act. The complaint alleges class members were damaged by purchasing artificially inflated stock prices due to Lumara Health's purportedly misleading statements regardingMakena related to access and exclusivity. On March 8, 2012, the Julianello, Mukku and Cheong cases were consolidated and the consolidated action is now styled In Re K-V Pharmaceutical Company Securities Litigation, Case No. 4:11-CV-1816-AGF. On May 4, 2012, the Court appointed Lori Anderson as Lead Plaintiff in the matter. On April 22, 2013, the individual defendants moved to dismiss the complaint and oral argument was held before the Court on November 26, 2013. Lumara Health joined in the motion to dismiss on February 10, 2014. On March 27, 2014, the Court entered an order granting Lumara Health's motion to dismiss the class action complaint without prejudice to the Plaintiffs' ability to file a second amended complaint with respect to a limited issue of whether Lumara Health's statements about Lumara Health's financial assistance program forMakena were materially false or misleading. On April 16, 2014, the Plaintiff's filed a motion to reconsider asking the Court to reconsider its order restricting the scope of Plaintiff's ability to amend its complaint. The Court denied Plaintiff's motion to reconsider and entered a judgment granting Lumara Health's motion to dismiss on June 6, 2014. On July 1, 2014, Plaintiffs filed a Notice of Appeal with the Eighth Circuit Court of Appeals and briefs have been submitted to the Court. The Court of Appeals has set March 12, 2015 as the date for oral argument.
In July 2010, Sandoz GmbH ("Sandoz") filed with the European Patent Office (the "EPO") an opposition to a previously issued patent which covers ferumoxytol in EU jurisdictions. In October 2012, at an oral hearing, the Opposition Division of the EPO revoked this patent. In December 2012, our
167
Table of Contents
notice of appeal of that decision was recorded with the EPO, which also suspended the revocation of our patent. On May 13, 2013, we filed a statement of grounds of appeal and on September 27, 2013, Sandoz filed a response to that statement. We filed a reply to that response on March 17, 2014 and oral proceedings for the appeal is scheduled for June 16, 2015. In the event that we withdraw our appeal or that do not experience a successful outcome from the appeals process, under EU regulations ferumoxytol would still be entitled to eight years of data protection and ten years of market exclusivity from the date of approval, which we believe would create barriers to entry for any generic version of ferumoxytol into the EU market until sometime between 2020 and 2022. This decision had no impact on our revenues for the year ended December 31, 2014. However, any future unfavorable outcome in this matter could negatively affect the magnitude and timing of future revenues. We do not expect to incur any related liability regardless of the outcome of the appeal and therefore have not recorded any liability as of December 31, 2014. We continue to believe the patent is valid and intend to vigorously appeal the decision.
We may periodically become subject to other legal proceedings and claims arising in connection with ongoing business activities, including claims or disputes related to patents that have been issued or that are pending in the field of research on which we are focused. Other than the above actions, we are not aware of any material claims against us at December 31, 2014. We expense legal costs as they are incurred.
R. COLLABORATIVE AGREEMENTS
Our commercial strategy includes the formation of collaborations with other pharmaceutical companies to facilitate the sale and distribution ofFeraheme, primarily outside of the U.S., as well as expanding our portfolio through the in-license or acquisition of additional pharmaceutical products or companies, including revenue-generating commercial products and late-state development assets.
Takeda
In March 2010, we entered into the Takeda Agreement, as amended in June 2012, under which we granted exclusive rights to Takeda to develop and commercializeFeraheme as a therapeutic agent in certain agreed-upon territories. In February 2014, we entered into a supply agreement with Takeda, which provides the terms under which we sellFeraheme to Takeda in order for Takeda to meet its requirements for commercial use ofFeraheme in its licensed territories (the "Supply Agreement"). On December 29, 2014, we entered into the Takeda Termination Agreement to terminate the Amended Takeda Agreement and we will regain all worldwide development and commercialization rights forFeraheme following the transfer of the outstanding marketing authorizations to us. Pursuant to the Takeda Termination Agreement, we and Takeda have agreed to effectuate the termination of the Amended Takeda Agreement on a rolling basis, whereby the termination will be effective for a particular geographic territory (e.g., countries under the regulatory jurisdictions of Health Canada, the EMA and SwissMedic) upon the earlier of effectiveness of the transfer to us or a Withdrawal (as defined below) of the marketing authorization for such territory, with the final effective termination date to be on the third such effective date ("Termination Date"). We have recently come to a mutual decision with Takeda to initiate withdrawal of the marketing authorization forRienso in the EU and Switzerland. We are currently assessing the commercial opportunity forFeraheme in Canada.
Under the Amended Takeda Agreement, except under limited circumstances, we retained the right to manufactureFeraheme and, accordingly, were responsible for supply ofFeraheme to Takeda at a fixed price per unit. We were also responsible for conducting, and bearing the costs related to, certain pre-defined clinical studies. We determined that our obligations under the Amended Takeda Agreement had not changed from those under the original Takeda Agreement and included the following four deliverables: the license, access to future know-how and improvements to theFeraheme technology, regulatory and clinical research activities, and the manufacturing and supply of product. Pursuant to the accounting guidance in effect in March 2010, when we signed the original Takeda Agreement and
168
Table of Contents
which governed revenue recognition on multiple element arrangements, we evaluated the four deliverables under the original Takeda Agreement and determined that our obligation to provide manufacturing supply of product met the criteria for separation and was therefore treated as a single unit of accounting, which we refer to as the supply unit of accounting. Further, we concluded that the license was not separable from the undelivered future know-how and technological improvements or the undelivered regulatory and clinical research activities. Accordingly, these deliverables were being combined and also treated as a single unit of accounting, which we refer to as the combined unit of accounting. With respect to the combined unit of accounting, our then obligation to provide access to our future know-how and technological improvements was the final deliverable and was an obligation which existed throughout the term of the Amended Takeda Agreement.
In connection with the execution of the original Takeda Agreement, we received a $60.0 million upfront payment from Takeda in April 2010, which we recorded as deferred revenue, as well as approximately $1.0 million reimbursed to us during 2010 for certain expenses incurred prior to entering the agreement, which we considered an additional upfront payment. Because we could not reasonably estimate the total level of effort required to complete the obligations under the combined deliverable, we were recognizing the entire $60.0 million upfront payment and the $1.0 million reimbursed to us in 2010, into revenues on a straight-line basis over a period of ten years from March 31, 2010, the date on which we originally entered the Takeda Agreement, which represented the then current patent life ofFeraheme and our best estimate of the period over which we were to substantively perform our obligations.
In June 2012, we earned a $15.0 million milestone payment from Takeda based on the European Commission marketing authorization for ferumoxytol. We deemed the $15.0 million milestone payment as a substantive milestone and therefore recognized the full amount as revenue. During 2012, we received an aggregate of $18.0 million in milestone payments from Takeda associated with the commercial launches ofFeraheme in the EU and Canada, which we deemed to be non-substantive milestone payments. Revenues related to the combined unit of accounting are recorded in license fee and other collaboration revenues in our consolidated statement of operations. In 2014, we recognized $8.2 million in revenues associated with the amortization of the upfront and milestone payments in license fees and other collaboration revenues in our consolidated statement of operations, including the acceleration of $0.3 million of upfront and milestone payments as a result of the termination of the Amended Takeda Agreement. We have classified all remaining upfront and milestone payments received to date as short-term deferred revenues at December 31, 2014, and we expect to recognize the remaining balance of the deferred revenue related to Takeda within the next 12 months. In addition, we recorded $3.2 million related to the Takeda Termination Agreement as deferred revenue at December 31, 2014.
Prior to entering into the December 2014 Takeda Termination Agreement, under the terms of the Amended Takeda Agreement, Takeda was responsible for reimbursing us for certain out-of-pocket regulatory and clinical trial supply costs associated with carrying out our regulatory and clinical research activities under the collaboration agreement. Because we were acting as the principal in carrying out these services, any reimbursement payments received from Takeda were recorded in license fee and other collaboration revenues in our consolidated statement of operations to match the costs that we incurred during the period in which we performed those services. We recorded $1.7 million, $0.5 million and $0.4 million for 2014, 2013 and 2012, respectively, associated with other reimbursement revenues received from Takeda. We have assumed any post-marketing obligations of Takeda as part of the Takeda Termination Agreement, including costs that otherwise would have been Takeda's obligation under the Amended Takeda Agreement for the ongoing pediatric studies and the initiated multi-center clinical trial to be conducted to determine the safety and efficacy of repeat doses ofFeraheme for the treatment of IDA in patients with hemodialysis dependent CKD, including a treatment arm with iron sucrose using a magnetic resonance imaging sub-analysis. In connection with our decision to withdraw the marketing authorization forRienso in the EU and Switzerland, we may modify or terminate clinical trials being conducted as part of our post-approval commitments to the EMA.
169
Table of Contents
Prior to entering into the December 2014 Takeda Termination Agreement, at the time of shipment, we deferred recognition of all revenue forFeraheme sold to Takeda in our consolidated balance sheets. We recognized revenues from product sales to Takeda, the related cost of product sales, and any royalty revenues due from Takeda, in our consolidated statement of operations at the time Takeda reported to us that sales had been made to their customers. During 2014, we recognized $3.5 million in product sales and royalty revenue and $2.8 million of cost of product sales related to the Amended Takeda Agreement, which included all amounts previously deferred prior to the termination of the Amended Takeda Agreement, and we have included in other product sales and royalties, and cost of product sales, respectively, in our consolidated statement of operations.
In connection with each Termination Date and in accordance with the terms of the Takeda Termination Agreement, Takeda is obligated, with respect to the applicable terminated territory, to transfer and assign to us all applicable regulatory materials and approvals and certain product data, unlabeled inventory, third party contracts intellectual property rights and know-how to us, and to grant us an exclusive license for certain Takeda technology used and applied to commercializeFeraheme in the applicable territory. The Takeda Termination Agreement also details the regulatory activities each party is required to perform in connection with transferring the marketing authorization from Takeda to us in each of the territories and the allocation of the costs of such activities. We and Takeda have agreed to use commercially reasonable efforts to transfer all required activities to us on a territory-by-territory basis within 60 days after the applicable Termination Date (subject to a 30-day extension upon our request and Takeda's consent). In addition, Takeda is obligated pursuant to the Takeda Termination Agreement to provide transition assistance to us, at no cost to us, for up to 180 days after each Termination Date for the applicable termination territory. With Takeda's consent (which shall not be unreasonably withheld or delayed), we may extend the transition services period for a terminated territory for a period of time reasonably necessary to complete any services that cannot be reasonably transitioned to us during the initial 180-day period, which extension will not exceed an additional 180 days. If we request, and Takeda agrees to conduct, additional transition services after the end of the applicable transition services period, as may be extended, we will reimburse Takeda's fully burdened costs for such additional services plus 5%.
The Takeda Termination Agreement also provides that if the marketing authorization for the product is suspended in a particular territory and the parties are prevented from completing the transfer of such marketing authorization to us within 120 days after such suspension due to applicable laws or any regulatory requirements or restrictions, or if we do not fulfill our obligations to initiate marketing authorization transfer by the agreed-upon, territory-specific deadline, Takeda will have the right, in Takeda's sole discretion, to withdraw such marketing authorization (a "Withdrawal").
In consideration for the early termination of the Amended Takeda Agreement and the activities to be performed by us earlier than contemplated under the Amended Takeda Agreement, and in lieu of any future cost-sharing and milestone payments contemplated by the Amended Takeda Agreement, Takeda agreed to make certain payments to us, subject to certain terms and conditions, including up to approximately $6.7 million in connection with clinical study obligations, pharmacovigilance activities, regulatory filings and support, commercialization and back-office support and distribution expenditures and a $3.0 million milestone payment payable subject to certain regulatory conditions.
Additionally, the supply agreement we entered into with Takeda in February 2014, together with the Amended Takeda Agreement, which continues in effect until the expiration or termination of the Amended Takeda Agreement, will also terminate as of the respective Termination Date in the applicable geographic territory.
170
Table of Contents
3SBio
In 2008, we entered into a Collaboration and Exclusive License Agreement (the "3SBio License Agreement") and a Supply Agreement with 3SBio Inc. for the development and commercialization ofFeraheme as an IV iron replacement therapeutic agent in China. In consideration of the grant of the license, we received an upfront payment of $1.0 million, the recognition of which had been deferred. In January 2014, we mutually terminated the agreement with 3SBio, effective immediately, due to the fact that, despite the best efforts of the parties, regulatory approval in China could not be obtained within the agreed upon time period.
PlasmaTech
Please refer to Note C, "Business Combinations," for a detailed description of the MuGard License Agreement.
S. DEBT
2.5% Convertible Notes
On February 14, 2014, we issued $200.0 million aggregate principal amount of Convertible Notes, which includes $25.0 million principal amount of Convertible Notes issued pursuant to the full exercise of an over-allotment option granted to the underwriters in the offering. We received net proceeds of $193.3 million from the sale of the Convertible Notes, after deducting fees and expenses of $6.7 million. We used $14.1 million of the net proceeds from the sale of the Convertible Notes to pay the cost of the convertible bond hedges, as described below (after such cost was partially offset by the proceeds to us from the sale of warrants in the warrant transactions described below).
The Convertible Notes are governed by the terms of an indenture between us, as issuer, and Wilmington Trust, National Association, as the Trustee. The Convertible Notes are senior unsecured obligations and bear interest at a rate of 2.5% per year, payable semi-annually in arrears on February 15 and August 15 of each year, beginning on August 15, 2014. The Convertible Notes will mature on February 15, 2019, unless earlier repurchased or converted. The Convertible Notes (which are currently convertible) will be convertible into cash, shares of our common stock, or a combination thereof, at our election (subject to certain limitations in the Term Loan Facility), at an initial conversion rate of approximately 36.9079 shares of common stock per $1,000 principal amount of the Convertible Notes, which corresponds to an initial conversion price of approximately $27.09 per share of our common stock and represents a conversion premium of approximately 35% based on the last reported sale price of our common stock of $20.07 on February 11, 2014, the date the notes offering was priced.
The conversion rate is subject to adjustment from time to time upon the occurrence of certain events, including, but not limited to, the issuance of stock dividends and payment of cash dividends. At any time prior to the close of business on the business day immediately preceding May 15, 2018, holders may convert their Convertible Notes at their option only under the following circumstances:
- (1)
- during any calendar quarter commencing after the calendar quarter ended on March 31, 2014 (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day;
- (2)
- during the five business day period after any five consecutive trading day period (the "measurement period") in which the trading price per $1,000 principal amount of the Convertible Notes for each trading day of the measurement period was less than 98% of the
171
Table of Contents
On or after May 15, 2018 until the close of business on the second scheduled trading day immediately preceding the maturity date, holders may convert all or any portion of their Convertible Notes, in multiples of $1,000 principal amount, at the option of the holder regardless of the foregoing circumstances.
If a make-whole fundamental change, as described in the indenture, occurs and a holder elects to convert its Convertible Notes in connection with such make-whole fundamental change, such holder may be entitled to an increase in the conversion rate as described in the indenture.
We may not redeem the Convertible Notes prior to the maturity date and no "sinking fund" is provided for the Convertible Notes, which means that we are not required to periodically redeem or retire the Convertible Notes. Upon the occurrence of certain fundamental changes involving us, holders of the Convertible Notes may require us to repurchase for cash all or part of their Convertible Notes at a repurchase price equal to 100% of the principal amount of the Convertible Notes to be repurchased, plus accrued and unpaid interest.
The indenture does not contain any financial or maintenance covenants or restrictions on the payments of dividends, the incurrence of indebtedness or the issuance or repurchase of securities by us or any of our subsidiaries. The indenture contains customary terms and covenants and events of default. If an event of default (other than certain events of bankruptcy, insolvency or reorganization involving us) occurs and is continuing, the Trustee by notice to us, or the holders of at least 25% in principal amount of the outstanding Convertible Notes by written notice to us and the Trustee, may declare 100% of the principal of and accrued and unpaid interest, if any, on all of the Convertible Notes to be due and payable. Upon such a declaration of acceleration, such principal and accrued and unpaid interest, if any, will be due and payable immediately. Upon the occurrence of certain events of bankruptcy, insolvency or reorganization involving us, 100% of the principal of and accrued and unpaid interest, if any, on all of the Convertible Notes will become due and payable automatically. Notwithstanding the foregoing, the indenture provides that, to the extent we elect and for up to 270 days, the sole remedy for an event of default relating to certain failures by us to comply with certain reporting covenants in the indenture consists exclusively of the right to receive additional interest on the Convertible Notes.
In accordance with accounting guidance for debt with conversion and other options, we separately account for the liability and equity components of the Convertible Notes by allocating the proceeds between the liability component and the embedded conversion option ("equity component") due to our ability to settle the Convertible Notes in cash, common stock or a combination of cash and common stock, at our option (subject to certain limitations in the Term Loan Facility). The carrying amount of the liability component was calculated by measuring the fair value of a similar liability that does not have an associated convertible feature. The allocation was performed in a manner that reflected our non-convertible debt borrowing rate for similar debt. The equity component of the Convertible Notes was recognized as a debt discount and represents the difference between the proceeds from the issuance of the Convertible Notes and the fair value of the liability of the Convertible Notes on their respective dates of issuance. The excess of the principal amount of the liability component over its carrying amount ("debt discount") is amortized to interest expense using the effective interest method over five years (the "life of the Convertible Notes"). The equity component is not remeasured as long as it continues to meet the conditions for equity classification.
172
Table of Contents
Our outstanding Convertible Note balances as December 31, 2014 consisted of the following (in thousands):
| | | | |
| | December 31, 2014 | |
|---|
Liability component: | | | | |
Principal | | $ | 200,000 | |
Less: debt discount, net | | | (32,559 | ) |
| | | | | |
Net carrying amount | | $ | 167,441 | |
| | | | | |
| | | | | |
| | | | | |
Equity component | | $ | 38,188 | |
| | | | | |
| | | | | |
| | | | | |
In connection with the issuance of the Convertible Notes, we incurred approximately $6.7 million of debt issuance costs, which primarily consisted of underwriting, legal and other professional fees, and allocated these costs to the liability and equity components based on the allocation of the proceeds. Of the total $6.7 million of debt issuance costs, $1.3 million were allocated to the equity component and recorded as a reduction to additional paid-in capital and $5.4 million were allocated to the liability component and recorded as assets on the balance sheet. The portion allocated to the liability component is amortized to interest expense over the expected life of the Convertible Notes using the effective interest method.
We determined the expected life of the debt was equal to the five year term on the Convertible Notes. As of December 31, 2014, the carrying value of the Convertible Notes was $167.4 million and the fair value of the Convertible Notes was $332.0 million. The effective interest rate on the liability component was 7.23% for the period from the date of issuance through December 31, 2014. The following table sets forth total interest expense recognized related to the Convertible Notes during the year ended December 31, 2014 (in thousands):
| | | | |
| | Year Ended
December 31, 2014 | |
|---|
Contractual interest expense | | $ | 4,375 | |
Amortization of debt issuance costs | | | 800 | |
Amortization of debt discount | | | 5,629 | |
| | | | | |
Total interest expense | | $ | 10,804 | |
| | | | | |
| | | | | |
| | | | | |
Convertible Bond Hedge and Warrant Transactions
In connection with the pricing of the Convertible Notes and in order to reduce the potential dilution to our common stock and/or offset cash payments due upon conversion of the Convertible Notes, on February 11, 2014 and February 13, 2014, we entered into convertible bond hedge transactions covering approximately 7.4 million shares of our common stock underlying the $200.0 million aggregate principal amount of the Convertible Notes, including the exercise of the over-allotment option, with the Call Spread Counterparties. The convertible bond hedges have an exercise price of approximately $27.09 per share, subject to adjustment upon certain events, and are exercisable when and if the Convertible Notes are converted. If upon conversion of the Convertible Notes, the price of our common stock is above the exercise price of the convertible bond hedges, the Call Spread Counterparties will deliver shares of our common stock and/or cash with an aggregate value approximately equal to the difference between the price of our common stock at the conversion date and the exercise price, multiplied by the number of shares of our common stock related to the convertible bond hedges being exercised. The convertible bond hedges are separate transactions entered into by us and are not part of the terms of the Convertible Notes or the warrants, discussed below. Holders of the Convertible Notes will not have any rights with respect to the convertible bond
173
Table of Contents
hedges. We paid $39.8 million for these convertible bond hedges and recorded this amount as a reduction to additional paid-in capital, net of tax, in the first quarter of 2014.
In February 2014, we also entered into separate warrant transactions with each of the Call Spread Counterparties relating to, in the aggregate, approximately 7.4 million shares of our common stock underlying the $200.0 million aggregate principal amount of the Convertible Notes, including the exercise of the over-allotment option. The initial exercise price of the warrants is $34.12 per share, subject to adjustment upon certain events, which is 70% above the last reported sale price of our common stock of $20.07 on February 11, 2014. The warrants would separately have a dilutive effect to the extent that the market value per share of our common stock, as measured under the terms of the warrants, exceeds the applicable exercise price of the warrants. The warrants were issued to the Call Spread Counterparties pursuant to the exemption from registration set forth in Section 4(a)(2) of the Securities Act of 1933, as amended. We received $25.7 million for these warrants and recorded this amount to additional paid-in capital in the first quarter of 2014.
Aside from the initial payment of a $39.8 million premium to the Call Spread Counterparties under the convertible bond hedges, which amount is partially offset by the receipt of a $25.7 million premium under the warrants, we are not required to make any cash payments to the Call Spread Counterparties under the convertible bond hedges and will not receive any proceeds if the warrants are exercised.
Term Loan Facility
On November 12, 2014 (the "Closing Date"), in connection with the acquisition of Lumara Health, we entered into the Term Loan Facility. The proceeds of the Term Loan Facility borrowed on the Closing Date were used to finance, in part, the Cash Consideration. We borrowed $340.0 million under the Term Loan Facility to fund a portion of the purchase price of Lumara Health. We realized net proceeds of $327.5 million after deducting $12.5 million of original issue discount costs and other lender fees and expenses. At December 31, 2014, the carrying value of the outstanding borrowings, net of unamortized original issue costs and other lender fees and expenses, was $327.9 million.
The Term Loan Facility provides for the aggregate principal amount of $340.0 million and allows for the incurrence of incremental term loans in an amount up to $40.0 million on the terms and subject to the conditions set forth in the Term Loan Facility. The Term Loan Facility bears interest, at our option, at either the Eurodollar rate plus a margin of 6.25% or the prime rate plus a margin of 5.25%. The Eurodollar rate is subject to a 1.00% floor and the prime rate is subject to a 2.00% floor. As of December 31, 2014 the stated interest rate was 7.25% and the effective interest rate was 8.55%.
We must repay the Term Loan Facility in installments of (a) $8.5 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2015 through the quarter ending December 31, 2015, and (b) $12.8 million per quarter due on the last day of each quarter beginning with the quarter ending March 31, 2016 through the quarter ending September 30, 2020, with the balance due in a final installment on November 12, 2020. The Term Loan Facility matures on November 12, 2020, except that the Term Loan Facility will mature on September 30, 2018 if:
- (c)
- more than $25.0 million in aggregate principal amount of our Convertible Notes remain outstanding and not converted to common stock or refinanced and replaced with debt that matures following, and has no amortization prior to, the date that is six and one half years following the Closing Date; and
- (b)
- the aggregate principal amount of all the Term Loan Facility (including all undrawn incremental commitments) is greater than $50.0 million on and as of such date (the "Maturity Date").
174
Table of Contents
Additionally, the Term Loan Facility includes an annual mandatory prepayment of the Term Loan Facility from 75% of our excess cash flow as measured on an annual basis, with step-downs to 50%, 25% and 0% of our excess cash flow if our Total Net Leverage Ratio (as defined in the Term Loan Facility), tested as of the last day of our fiscal year, is less than or equal to 2.00 to 1.00, 1.00 to 1.00 and 0.50 to 1.00, respectively. Excess cash flow is generally defined as our adjusted Earnings Before Interest, Taxes, Depreciation and Amortization ("EBITDA") less debt service costs, unfinanced capital expenditures, unfinanced acquisition expenditures, and current income taxes paid, as adjusted for changes in our working capital. Additionally, the Term Loan Facility requires mandatory prepayment of the term loan from the net cash proceeds of (i) certain debt issuances and (ii) certain asset sales outside the ordinary course of business and from proceeds of property insurance and condemnation events, in each case of this clause (ii) subject to our right to reinvest such proceeds in our business. Any voluntary prepayment or mandatory prepayment pursuant to the preceding sentence other than such prepayments not exceeding $50.0 million in the aggregate, shall be accompanied by a prepayment premium equal to (a) 2.0% of the principal amount of such prepayment, if such prepayment is made on or prior to the date that is twelve months after the Closing Date or (b) 1.0% of the principal amount of such prepayment, if such prepayment is made after the date that is twelve months after the Closing Date and on or prior to the date that is twenty-four months after the Closing Date.
The Term Loan Facility has a lien on substantially all of our assets, including a pledge of 100% of the equity interests in our domestic subsidiaries and an obligation to pledge 65% of the equity interests in our direct foreign subsidiaries.
The Term Loan Facility contains customary affirmative covenants for transactions of this type and other affirmative covenants agreed to by the parties, including, among others, the provision of annual and quarterly financial statements and compliance certificates, maintenance of property, insurance, compliance with laws and environmental matters. The Term Loan Facility contains customary negative covenants for transactions of this type and other negative covenants agreed to by the parties, including, among others, restrictions on the incurrence of indebtedness, granting of liens, making investments and acquisitions, paying dividends, repurchases of equity interests in the Company, entering into affiliate transactions and asset sales. The Term Loan Facility also provides for a number of customary events of default, including, among others, payment, bankruptcy, covenant, representation and warranty, change of control and judgment defaults. In addition, the Term Loan Facility contains certain restrictions regarding the use of our funds to pay certain debts.
The Term Loan Facility requires that we comply with a Total Net Leverage Ratio. Under the terms of the Term Loan Facility, we must maintain a Total Net Leverage Ratio that is less than or equal to 4.60 to 1.00 for the fiscal quarter ended March 31, 2015 and declining over time to a range of 1.00 to 1.00 for the fiscal quarter ending September 30, 2017 through the Maturity Date. For purposes of testing our Total Net Leverage Ratio, we are permitted to net from our outstanding total indebtedness up to $25.0 million of its domestic unrestricted cash and cash equivalents. As of December 31, 2014, we were in compliance with these covenants.
All obligations under the Term Loan Facility are unconditionally guaranteed by substantially all of our direct and indirect domestic subsidiaries. These guarantees are secured by substantially all of the present and future property and assets of the guarantors.
175
Table of Contents
Future annual principal payments on our long-term debt as of December 31, 2014 were as follows:
| | | | |
2015 | | $ | 34,000 | |
2016 | | | 51,000 | |
2017 | | | 51,000 | |
2018 | | | 51,000 | |
2019 | | | 51,000 | |
Thereafter | | | 102,000 | |
| | | | | |
Total | | $ | 340,000 | |
| | | | | |
| | | | | |
| | | | | |
T. RESTRUCTURING
In connection with the Lumara Health acquisition, we initiated a restructuring program in the fourth quarter of 2014, which included severance benefits primarily related to certain former Lumara Health employees. As a result of the restructuring, we recorded charges of approximately $2.0 million in 2014. We expect to pay substantially all of these restructuring costs during 2015.
The following table outlines the components of our restructuring expenses which were included in current liabilities for 2014 (in thousands):
| | | | |
| | December 31, 2014 | |
|---|
Accrued restructuring, beginning of period | | $ | — | |
Employee severance, benefits and related costs | | | 2,023 | |
Payments | | | (70 | ) |
| | | | | |
Accrued restructuring, end of period | | $ | 1,953 | |
| | | | | |
| | | | | |
| | | | | |
176
Table of Contents
U. CONSOLIDATED QUARTERLY FINANCIAL DATA—UNAUDITED
The following tables provide unaudited consolidated quarterly financial data for 2014 and 2013 (in thousands, except per share data):
| | | | | | | | | | | | | |
| | March 31, 2014 | | June 30, 2014 | | September 30, 2014 | | December 31, 2014 | |
|---|
U.S. product sales, net | | $ | 17,375 | | $ | 22,225 | | $ | 22,547 | | $ | 46,648 | |
License fee and other collaboration revenues | | | 3,120 | | | 2,122 | | | 2,182 | | | 3,462 | |
Other product sales and royalties | | | 340 | | | 455 | | | 765 | | | 3143 | |
| | | | | | | | | | | | | | |
Total revenues | | | 20,835 | | | 24,802 | | | 25,494 | | | 53,253 | |
Cost of product sales | | | 2,837 | | | 2,743 | | | 2,968 | | | 11,758 | |
Gross margin | | | 17,998 | | | 22,059 | | | 22,526 | | | 41,495 | |
Operating expenses | | | 23,989 | | | 20,824 | | | 18,233 | | | 44,869 | |
Interest expense | | | (1,476 | ) | | (3,051 | ) | | (3,129 | ) | | (7,041 | ) |
Interest and dividend income, net | | | 265 | | | 253 | | | 291 | | | 166 | |
Gains on sale of assets | | | 100 | | | 2 | | | — | | | 1 | |
Gains on investments, net | | | — | | | 14 | | | 3 | | | 97 | |
| | | | | | | | | | | | | | |
Net income (loss) before income taxes | | | (7,102 | ) | | (1,547 | ) | | 1,458 | | | (10,151 | ) |
Income tax benefit | | | — | | | — | | | — | | | 153,159 | |
| | | | | | | | | | | | | | |
Net income (loss) | | $ | (7,102 | ) | $ | (1,547 | ) | $ | 1,458 | | $ | 143,008 | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Net income (loss) per share—basic | | $ | (0.33 | ) | $ | (0.07 | ) | $ | 0.07 | | $ | 5.98 | |
Net income (loss) per share—diluted | | $ | (0.33 | ) | $ | (0.07 | ) | $ | 0.07 | | $ | 4.67 | |
| | | | | | | | | | | | | |
| | March 31, 2013 | | June 30, 2013 | | September 30, 2013 | | December 31, 2013 | |
|---|
U.S.Feraheme product sales, net | | $ | 15,578 | | $ | 17,456 | | $ | 19,347 | | $ | 18,981 | |
License fee and other collaboration revenues | | | 2,003 | | | 2,055 | | | 1,998 | | | 2,329 | |
Other product sales and royalties | | | 299 | | | 138 | | | 271 | | | 401 | |
| | | | | | | | | | | | | | |
Total revenues | | | 17,880 | | | 19,649 | | | 21,616 | | | 21,711 | |
Cost of product sales | | | 2,942 | | | 3,145 | | | 2,547 | | | 3,326 | |
Gross margin | | | 14,938 | | | 16,504 | | | 19,069 | | | 18,385 | |
Operating expenses | | | 19,409 | | | 19,260 | | | 19,464 | | | 22,380 | |
Interest and dividend income, net | | | 271 | | | 256 | | | 246 | | | 278 | |
Gains on assets held for sale | | | 299 | | | 566 | | | — | | | 59 | |
Gains on investments, net | | | 6 | | | 26 | | | 4 | | | 4 | |
| | | | | | | | | | | | | | |
Net loss | | $ | (3,895 | ) | $ | (1,908 | ) | $ | (145 | ) | $ | (3,654 | ) |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Net loss per share—basic and diluted | | $ | (0.18 | ) | $ | (0.09 | ) | $ | (0.01 | ) | $ | (0.17 | ) |
Quarterly loss per share totals differ from annual loss per share totals due to rounding. On November 12, 2014, we completed our acquisition of Lumara Health and recorded $22.5 million inMakena sales in 2014 and additional operating costs incurred as a result of the acquisition.
177
Table of Contents
V. VALUATION AND QUALIFYING ACCOUNTS (IN THOUSANDS)
| | | | | | | | | | | | | |
| | Balance at
Beginning
of Period | | Additions(a) | | Deductions
Charged to
Reserves | | Balance at
End of
Period | |
|---|
Year ended December 31, 2014: | | | | | | | | | | | | | |
Accounts receivable allowances(b) | | $ | 2,683 | | $ | 59,372 | | $ | (50,580 | ) | $ | 11,475 | |
Rebates, fees and returns reserves | | $ | 4,799 | | $ | 52,079 | | $ | (13,126 | ) | $ | 43,752 | |
Year ended December 31, 2013: | | |
| | |
| | |
| | |
| |
Accounts receivable allowances(b) | | $ | 1,771 | | $ | 37,098 | | $ | (36,186 | ) | $ | 5,850 | |
Rebates, fees and returns reserves | | $ | 3,448 | | $ | 11,820 | | $ | (10,469 | ) | $ | 4,799 | |
Year ended December 31, 2012: | | |
| | |
| | |
| | |
| |
Accounts receivable allowances(b) | | $ | 1,822 | | $ | 26,517 | | $ | (26,568 | ) | $ | 1,771 | |
Rebates, fees and returns reserves | | $ | 5,943 | | $ | 6,729 | | $ | (9,224 | ) | $ | 3,448 | |
- (a)
- Additions to sales discounts, rebates, fees and returns reserves are recorded as a reduction of revenues.
- (b)
- We have not recorded an allowance for doubtful accounts in any of the years presented above. These accounts receivable allowances represent discounts and other chargebacks related to the provision for our product sales.
W. RECENTLY ISSUED AND PROPOSED ACCOUNTING PRONOUNCEMENTS
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board ("FASB") or other standard setting bodies that are adopted by us as of the specified effective date. Unless otherwise discussed, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
In May 2014, the FASB issued Accounting Standards Update ("ASU") 2014-09,Revenue from Contracts with Customers, as a new Topic, Accounting Standards Codification Topic 606. The new revenue recognition standard provides a five-step analysis of transactions to determine when and how revenue is recognized. The core principle is that a company should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. This ASU is effective for us on January 1, 2017 and shall be applied retrospectively to each period presented or as a cumulative-effect adjustment as of the date of adoption. We are in the process of evaluating the effect of adopting this new accounting guidance and are uncertain at this point of the impact on our results of operations, cash flows or financial position.
In August 2014, the FASB issued ASU No. 2014-15,Presentation of Financial Statements—Going Concern: Disclosure of Uncertainties about an Entity's Ability to Continue as a Going Concern. ASU No. 2014-15 is intended to define management's responsibility to evaluate whether there is substantial doubt about an organization's ability to continue as a going concern and to provide related footnote disclosures, if required. ASU 2014-15 will be effective for annual reporting periods ending after December 15, 2016, which will be our fiscal year ending December 31, 2016, and to annual and interim periods thereafter. We are in the process of evaluating the impact of adoption of ASU 2014-15 on our consolidated financial statements and related disclosures and currently do not expect it to have a material impact our results of operations, cash flows or financial position.
178
Table of Contents
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE:
None.
ITEM 9A. CONTROLS AND PROCEDURES:
Managements' Evaluation of our Disclosure Controls and Procedures
Our principal executive officer and principal financial officer, after evaluating the effectiveness of our "disclosure controls and procedures" (as defined in the Exchange Act Rule 13a-15(e), or Rule 15d-15(e)), with the participation of our management, have each concluded that, as of December 31, 2014, the end of the period covered by this Annual Report on Form 10-K, our disclosure controls and procedures were designed and were effective to provide reasonable assurance that information we are required to disclose in the reports that we file or submit under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission's rules and forms and that such information is accumulated and communicated to management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. It should be noted that any system of controls is designed to provide reasonable, but not absolute, assurances that the system will achieve its stated goals under all reasonably foreseeable circumstances.
Management's Annual Report on Internal Control Over Financial Reporting
Management's Report on Internal Control over Financial Reporting is contained in Part II, Item 8 "Financial Statements and Supplementary Data" of this Annual Report on Form 10-K for the year ended December 31, 2014 and is incorporated herein by reference.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the fourth quarter of the year ended December 31, 2014 that materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION:
As noted elsewhere in this Annual Report on Form 10-K, sales ofRienso in the EU do not and are not expected to materially contribute to our revenues. As such, and considering our entry into the December 2014 Takeda Termination Agreement, we have been assessing the commercial opportunity forRienso and have come to a mutual decision with Takeda to initiate withdrawal ofRienso's current marketing authorizations in the EU and Switzerland. The decision to initiate withdrawal of the marketing authorizations was made solely for commercial reasons.
179
Table of Contents
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE:
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which we plan to file with the Securities and Exchange Commission (the "SEC") not later than 120 days after the close of our year ended December 31, 2014.
ITEM 11. EXECUTIVE COMPENSATION:
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which we plan to file with the SEC not later than 120 days after the close of our year ended December 31, 2014.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS:
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which we plan to file with the SEC not later than 120 days after the close of our year ended December 31, 2014.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE:
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which we plan to file with the SEC not later than 120 days after the close of our year ended December 31, 2014.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES:
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which we plan to file with the SEC not later than 120 days after the close of our year ended December 31, 2014.
180
Table of Contents
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES:
| | |
| Exhibit Number | | Description |
|---|
| 2.1 | | Agreement and Plan of Merger, dated as of September 28, 2014, by and among Lumara Health Inc., the Company, Snowbird, Inc., and Lunar Representative, LLC as the Stockholders' Representative (incorporated herein by reference to Exhibit 2.1 to the Company's Current Report on Form 8-K filed September 29, 2014, File No. 001-10865) |
| 3.1, 4.1 | | Certificate of Incorporation of the Company, as restated (incorporated herein by reference to Exhibit 3.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, File No. 0-14732) |
| 3.2, 4.2 | | By-Laws of the Company, as amended (incorporated herein by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K filed November 28, 2008, File No. 0-14732) |
| 3.3, 4.3 | | Certificate of Designation of Series A Junior Participating Preferred Stock (incorporated herein by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| 4.4 | | Specimen certificate representing the Company's Common Stock (incorporated herein by reference to Exhibit 4.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2009, File No. 0-14732) |
| 4.5 | | Rights Agreement dated as of September 4, 2009 by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.2 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| 4.6 | | Amendment to Rights Agreement, dated as of May 10, 2012, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| 4.7 | | Amendment to Rights Agreement, dated as of February 11, 2014, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.4 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 4.8 | | NOL Amendment to Rights Agreement, dated as of September 26, 2014, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed September 29, 2014, File No. 001-10865) |
| 4.9 | | Form of Rights Certificate (incorporated herein by reference to Exhibit 4.3 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| 4.10 | | Base Indenture, dated as of February 14, 2014, by and between the Company and Wilmington Trust, National Association (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 4.11 | | First Supplemental Indenture, dated as of February 14, 2014, by and between the Company and Wilmington Trust, National Association (incorporated herein by reference to Exhibit 4.2 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 4.12 | | Form of 2.5% Convertible Senior Note due 2019 (incorporated herein by reference to Exhibit 4.3 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
181
Table of Contents
| | |
| Exhibit Number | | Description |
|---|
| 4.13 | | Form of Registration Rights and Lock-up Agreement, dated as of November 12, 2014, by and between the Company and stockholders party thereto (incorporated herein by reference to Exhibit 4.9 to the Company's Registration Statement on Form S-3 filed February 10, 2015) |
| 10.1* | | Representative Form of Indemnification Agreement (incorporated herein by reference to Exhibit 10.2 to the Company's Annual Report on Form 10-K for the year ended December 31, 2009, File No. 001-10865) |
| 10.2* | | Summary of the Company's Change of Control Policy applicable to executive officers (incorporated herein by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed February 13, 2006, File No. 0-14732) |
| 10.3* | | The Company's Non-Employee Director Compensation Policy (incorporated herein by reference to Exhibit 10.4 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
| 10.4* | | The Company's Amended and Restated 2000 Stock Plan (incorporated herein by reference to Appendix A to the Company's Definitive Proxy Statement on Schedule 14A filed December 14, 2005, File No. 0-14732) |
| 10.5* | | The Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Appendix A to the Company's Definitive Proxy Statement on Schedule 14A filed April 19, 2013, File No. 001-10865) |
| 10.6*+ | | Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan |
| 10.7* | | Form of Incentive Stock Option Agreement for Company Employees under the Company's Third Amended and Restated 2007 Equity Incentive Plan and Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| 10.8* | | Form of Non-Qualified Stock Option Agreement for Company Employees under the Company's Third Amended and Restated 2017 Equity Incentive Plan and the Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| 10.9* | | Form of Restricted Stock Unit Agreement for Company Employees under the Company's Third Amended and Restated 2007 Equity Incentive Plan and the Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| 10.10* | | Form of Non-Qualified Stock Option Agreement for Non-Employee Directors under the Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.10 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| 10.11* | | Form of Restricted Stock Unit Agreement for Non-Employee Directors under the Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.11 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| 10.12* | | Form of February 2013 Performance-based Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and the Company's executive officers (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013, File No. 001-10865) |
182
Table of Contents
| | |
| Exhibit Number | | Description |
|---|
| 10.13* | | Form of November 2011 Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and the Company's executive officers (incorporated herein by reference to Exhibit 10.21 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
| 10.14* | | Form of November 2011 Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and each non-executive employee of the Company (incorporated herein by reference to Exhibit 10.22 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
| 10.15* | | Form of Non-Plan Restricted Stock Unit Agreement, by and between the Company and William K. Heiden (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| 10.16* | | Form of Non-Plan Stock Option Agreement, by and between the Company and William K. Heiden (incorporated herein by reference to Exhibit 10.3 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| 10.17* | | Form of Non-Qualified Stock Option Agreement—Non-Plan Inducement Grant (incorporated herein by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-8, filed August 7, 2013, File No. 333-190435) |
| 10.18* | | Form of Restricted Stock Unit Agreement—Non-Plan Inducement Grant (incorporated herein by reference to Exhibit 4.4 to the Company's Registration Statement on Form S-8, filed August 7, 2013, File No. 333-190435) |
| 10.19+ | | Termination Agreement, dated December 29, 2014, by and between the Company and Takeda Pharmaceutical Company Limited (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| 10.20 | | License, Development and Commercialization Agreement by and between the Company and Takeda Pharmaceutical Company Limited, dated March 31, 2010 (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, File No. 001-10865) (confidential treatment previously granted) |
| 10.21 | | Amendment to the License, Development and Commercialization Agreement, dated June 25, 2012, by and between the Company and Takeda Pharmaceutical Company Limited (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed June 29, 2012, File No. 001-10865) (confidential treatment previously granted) |
| 10.22 | | Supply Agreement, dated February 7, 2014, by and between the Company and Takeda Pharmaceuticals International, GMBH A/S, an affiliate of Takeda Pharmaceutical Company Limited (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 (confidential treatment previously granted) |
| 10.23 | | Lease Agreement, dated as of May 27, 2008, by and between the Company and Mortimer B. Zuckerman and Edward H. Linde, trustees of 92 Hayden Avenue Trust under Declaration of Trust dated August 18, 1983 This Lease Agreement was assigned in June 2013. (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed May 29, 2008, File No. 0-14732) |
183
Table of Contents
| | |
| Exhibit Number | | Description |
|---|
| 10.24 | | Assignment and Assumption of Lease, dated as of June 10, 2013, by and among the Company, Mortimer B. Zuckerman and Edward H. Linde, Trustees of 92 Hayden Avenue Trust under Declaration of Trust dated August 18, 1983 and Shire Human Genetic Therapies, Inc. (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed June 13, 2013, File No. 001-10865) |
| 10.25 | | Lease Agreement, dated as of June 10, 2013, by and between the Company and BP BAY COLONY LLC (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed June 13, 2013, File No. 001-10865) |
| 10.26 | | License Agreement between the Company and Plasmatech Biopharmaceuticals Inc. (formerly Access Pharmaceuticals, Inc.) dated as of June 6, 2013 (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) (confidential treatment previously granted) |
| 10.27 | | Commercial Supply Agreement, dated effective as of August 29, 2012, by and between the Company and Sigma-Aldrich, Inc. (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012, File No. 001-10865) (confidential treatment previously granted) |
| 10.28 | | Amendment No.1 to Commercial Supply Agreement, dated October 3, 2013, by and between the Company and Sigma-Aldrich, Inc. (incorporated herein by reference to Exhibit 10.54 to the Company's Annual Report on Form 10-K for the year ended December 31, 2013, File No. 001-10865) (confidential treatment previously granted) |
| 10.29 | | Pharmaceutical Manufacturing and Supply Agreement, dated January 8, 2010, by and between the Company and Patheon Manufacturing Services LLC (as assignee from DSM Pharmaceuticals, Inc.) (incorporated herein by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012, File No. 001-10865) (confidential treatment previously granted) |
| 10.30 | | Amendment No. 1 to Pharmaceutical Manufacturing and Supply Agreement, dated July 5, 2014, by and between the Company and Patheon Manufacturing Services LLC (as assignee from DSM Pharmaceuticals, Inc.) (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2014, File No. 001-10865) |
| 10.31+ | | Development and Supply Agreement, dated as of September 17, 2009, by and between Lumara Health Inc. (as successor in interest to Hologic, Inc.) and Hospira Worldwide, Inc. (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| 10.32+ | | First Amendment to Development and Supply Agreement, dated as of March 28, 2014, by and between Lumara Health Inc. and Hospira Worldwide, Inc. (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| 10.33 | | Underwriting Agreement, dated as of February 11, 2014, among AMAG Pharmaceuticals, Inc. and J.P. Morgan Securities LLC, on its own behalf and as representative of the several underwriters named in Schedule 1 thereto (incorporated herein by reference to Exhibit 1.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
184
Table of Contents
| | |
| Exhibit Number | | Description |
|---|
| 10.34 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.35 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.36 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.3 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.37 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.4 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.38 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.39 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.6 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.40 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.7 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.41 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.8 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.42 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.9 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.43 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.10 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.44 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.11 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.45 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.12 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| 10.46 | | Credit Agreement, dated as of November 12, 2014, by and among the Company, the financial institutions and agents listed therein, and Jefferies Finance LLC (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed November 12, 2014, File No. 001-10865) |
185
Table of Contents
| | |
| Exhibit Number | | Description |
|---|
| 10.47* | | Form of Employment Agreement between the Company and each of its executive officers (other than William Heiden) (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, File No. 001-10865) |
| 10.48* | | Employment Agreement dated as of February 7, 2014 between the Company and William Heiden (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, File No. 001-10865) |
| 21.1+ | | Subsidiaries of AMAG Pharmaceuticals, Inc. |
| 23.1+ | | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
| 24.1 | | Power of Attorney (included on the signature page(s) hereto) |
| 31.1+ | | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Exchange Act, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 31.2+ | | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Exchange Act, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| 32.1++ | | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 32.2++ | | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 101.INS+ | | XBRL Instance Document |
| 101.SCH+ | | XBRL Taxonomy Extension Schema Document |
| 101.CAL+ | | XBRL Taxonomy Extension Calculation Linkbase Document |
| 101.DEF+ | | XBRL Taxonomy Extension Definition Linkbase Document |
| 101.LAB+ | | XBRL Taxonomy Extension Label Linkbase Document |
| 101.PRE+ | | XBRL Taxonomy Extension Presentation Linkbase Document |
- +
- Exhibits marked with a plus sign ("+") are filed herewith.
- ++
- Exhibits marked with a double plus sign ("++") are furnished herewith.
- *
- Exhibits marked with a single asterisk reference management contracts, compensatory plans or arrangements, filed in response to Item 15(a)(3) of the instructions to Form 10-K.
The other exhibits listed have previously been filed with the SEC and are incorporated herein by reference, as indicated.
- (b)
- Exhibits. We hereby file or furnish as exhibits, as the case may be, to this Form 10-K those exhibits listed in Part IV, Item 15(a)(3) above.
- (c)
- Financial Statement Schedules. No financial statement schedules have been submitted because they are not required, not applicable, or because the information required is included in the financial statements or the notes thereto.
186
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | |
| | | AMAG PHARMACEUTICALS, INC. |
|
|
By: |
|
/s/ WILLIAM K. HEIDEN
William K. Heiden
President and Chief Executive Officer |
|
|
Date: |
|
February 17, 2015 |
We, the undersigned officers and directors of AMAG Pharmaceuticals, Inc., hereby severally constitute and appoint William K. Heiden and Scott A. Holmes, and each of them singly, our true and lawful attorneys, with full power to them and each of them singly, to sign for us in our names in the capacities indicated below, all amendments to this report, and generally to do all things in our names and on our behalf in such capacities to enable AMAG Pharmaceuticals, Inc. to comply with the provisions of the Securities Exchange Act of 1934, as amended, and all requirements of the Securities and Exchange Commission.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
Name | | Title | | Date |
|---|
/s/ WILLIAM K. HEIDEN
William K. Heiden | | President, Chief Executive Officer (Principal Executive Officer) and Director | | February 17, 2015 |
/s/ SCOTT A. HOLMES
Scott A. Holmes |
|
Chief Accounting Officer, Treasurer and Senior Vice President, Finance and Investor Relations (Principal Financial and Accounting Officer) |
|
February 17, 2015 |
/s/ BARBARA DEPTULA
Barbara Deptula |
|
Director |
|
February 17, 2015 |
/s/ JOHN FALLON, M.D.
John Fallon, M.D. |
|
Director |
|
February 17, 2015 |
187
Table of Contents
| | | | |
Name | | Title | | Date |
|---|
/s/ ROBERT J. PEREZ
Robert J. Perez | | Director | | February 17, 2015 |
/s/LESLEY RUSSELL, MB. CH.B., MRCP
Lesley Russell, MB. Ch.B., MRCP |
|
Director |
|
February 17, 2015 |
/s/ GINO SANTINI
Gino Santini |
|
Director |
|
February 17, 2015 |
/s/ DAVEY S. SCOON
Davey S. Scoon |
|
Director |
|
February 17, 2015 |
/s/ JAMES SULAT
James Sulat |
|
Director |
|
February 17, 2015 |
188
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 2.1 | | Agreement and Plan of Merger, dated as of September 28, 2014, by and among Lumara Health Inc., the Company, Snowbird, Inc., and Lunar Representative, LLC as the Stockholders' Representative (incorporated herein by reference to Exhibit 2.1 to the Company's Current Report on Form 8-K filed September 29, 2014, File No. 001-10865) |
| | 3.1, 4.1 | | Certificate of Incorporation of the Company, as restated (incorporated herein by reference to Exhibit 3.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, File No. 0-14732) |
| | 3.2, 4.2 | | By-Laws of the Company, as amended (incorporated herein by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K filed November 28, 2008, File No. 0-14732) |
| | 3.3, 4.3 | | Certificate of Designation of Series A Junior Participating Preferred Stock (incorporated herein by reference to Exhibit 3.1 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| | 4.4 | | Specimen certificate representing the Company's Common Stock (incorporated herein by reference to Exhibit 4.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2009, File No. 0-14732) |
| | 4.5 | | Rights Agreement dated as of September 4, 2009 by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.2 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| | 4.6 | | Amendment to Rights Agreement, dated as of May 10, 2012, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| | 4.7 | | Amendment to Rights Agreement, dated as of February 11, 2014, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.4 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 4.8 | | NOL Amendment to Rights Agreement, dated as of September 26, 2014, by and between the Company and American Stock Transfer & Trust Company, LLC (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed September 29, 2014, File No. 001-10865) |
| | 4.9 | | Form of Rights Certificate (incorporated herein by reference to Exhibit 4.3 to the Company's Current Report on Form 8-K filed September 4, 2009, File No. 0-14732) |
| | 4.10 | | Base Indenture, dated as of February 14, 2014, by and between the Company and Wilmington Trust, National Association (incorporated herein by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 4.11 | | First Supplemental Indenture, dated as of February 14, 2014, by and between the Company and Wilmington Trust, National Association (incorporated herein by reference to Exhibit 4.2 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 4.12 | | Form of 2.5% Convertible Senior Note due 2019 (incorporated herein by reference to Exhibit 4.3 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 4.13 | | Form of Registration Rights and Lock-up Agreement, dated as of November 12, 2014, by and between the Company and stockholders party thereto (incorporated herein by reference to Exhibit 4.9 to the Company's Registration Statement on Form S-3 filed February 10, 2015) |
189
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 10.1* | | Representative Form of Indemnification Agreement (incorporated herein by reference to Exhibit 10.2 to the Company's Annual Report on Form 10-K for the year ended December 31, 2009, File No. 001-10865) |
| | 10.2* | | Summary of the Company's Change of Control Policy applicable to executive officers (incorporated herein by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed February 13, 2006, File No. 0-14732) |
| | 10.3* | | The Company's Non-Employee Director Compensation Policy (incorporated herein by reference to Exhibit 10.4 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
| | 10.4* | | The Company's Amended and Restated 2000 Stock Plan (incorporated herein by reference to Appendix A to the Company's Definitive Proxy Statement on Schedule 14A filed December 14, 2005, File No. 0-14732) |
| | 10.5* | | The Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Appendix A to the Company's Definitive Proxy Statement on Schedule 14A filed April 19, 2013, File No. 001-10865) |
| | 10.6*+ | | Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan |
| | 10.7* | | Form of Incentive Stock Option Agreement for Company Employees under the Company's Third Amended and Restated 2007 Equity Incentive Plan and Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| | 10.8* | | Form of Non-Qualified Stock Option Agreement for Company Employees under the Company's Third Amended and Restated 2017 Equity Incentive Plan and the Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| | 10.9* | | Form of Restricted Stock Unit Agreement for Company Employees under the Company's Third Amended and Restated 2007 Equity Incentive Plan and the Lumara Health Inc. Amended and Restated 2013 Incentive Compensation Plan (incorporated herein by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| | 10.10* | | Form of Non-Qualified Stock Option Agreement for Non-Employee Directors under the Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.10 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| | 10.11* | | Form of Restricted Stock Unit Agreement for Non-Employee Directors under the Company's Third Amended and Restated 2007 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.11 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) |
| | 10.12* | | Form of February 2013 Performance-based Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and the Company's executive officers (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013, File No. 001-10865) |
| | 10.13* | | Form of November 2011 Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and the Company's executive officers (incorporated herein by reference to Exhibit 10.21 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
190
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 10.14* | | Form of November 2011 Restricted Stock Unit Agreement under the Company's Second Amended and Restated 2007 Equity Incentive Plan between the Company and each non-executive employee of the Company (incorporated herein by reference to Exhibit 10.22 to the Company's Annual Report on Form 10-K for the year ended December 31, 2011, File No. 001-10865) |
| | 10.15* | | Form of Non-Plan Restricted Stock Unit Agreement, by and between the Company and William K. Heiden (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| | 10.16* | | Form of Non-Plan Stock Option Agreement, by and between the Company and William K. Heiden (incorporated herein by reference to Exhibit 10.3 to the Company's Current Report on Form 8-K filed May 10, 2012, File No. 001-10865) |
| | 10.17* | | Form of Non-Qualified Stock Option Agreement—Non-Plan Inducement Grant (incorporated herein by reference to Exhibit 4.3 to the Company's Registration Statement on Form S-8, filed August 7, 2013, File No. 333-190435) |
| | 10.18* | | Form of Restricted Stock Unit Agreement—Non-Plan Inducement Grant (incorporated herein by reference to Exhibit 4.4 to the Company's Registration Statement on Form S-8, filed August 7, 2013, File No. 333-190435) |
| | 10.19+ | | Termination Agreement, dated December 29, 2014, by and between the Company and Takeda Pharmaceutical Company Limited (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| | 10.20 | | License, Development and Commercialization Agreement by and between the Company and Takeda Pharmaceutical Company Limited, dated March 31, 2010 (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, File No. 001-10865) (confidential treatment previously granted) |
| | 10.21 | | Amendment to the License, Development and Commercialization Agreement, dated June 25, 2012, by and between the Company and Takeda Pharmaceutical Company Limited (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed June 29, 2012, File No. 001-10865) (confidential treatment previously granted) |
| | 10.22 | | Supply Agreement, dated February 7, 2014, by and between the Company and Takeda Pharmaceuticals International, GMBH A/S, an affiliate of Takeda Pharmaceutical Company Limited (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 (confidential treatment previously granted) |
| | 10.23 | | Lease Agreement, dated as of May 27, 2008, by and between the Company and Mortimer B. Zuckerman and Edward H. Linde, trustees of 92 Hayden Avenue Trust under Declaration of Trust dated August 18, 1983 This Lease Agreement was assigned in June 2013. (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed May 29, 2008, File No. 0-14732) |
| | 10.24 | | Assignment and Assumption of Lease, dated as of June 10, 2013, by and among the Company, Mortimer B. Zuckerman and Edward H. Linde, Trustees of 92 Hayden Avenue Trust under Declaration of Trust dated August 18, 1983 and Shire Human Genetic Therapies, Inc. (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed June 13, 2013, File No. 001-10865) |
191
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 10.25 | | Lease Agreement, dated as of June 10, 2013, by and between the Company and BP BAY COLONY LLC (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed June 13, 2013, File No. 001-10865) |
| | 10.26 | | License Agreement between the Company and Plasmatech Biopharmaceuticals Inc. (formerly Access Pharmaceuticals, Inc.) dated as of June 6, 2013 (incorporated herein by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended June 30, 2013, File No. 001-10865) (confidential treatment previously granted) |
| | 10.27 | | Commercial Supply Agreement, dated effective as of August 29, 2012, by and between the Company and Sigma-Aldrich, Inc. (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012, File No. 001-10865) (confidential treatment previously granted) |
| | 10.28 | | Amendment No.1 to Commercial Supply Agreement, dated October 3, 2013, by and between the Company and Sigma-Aldrich, Inc. (incorporated herein by reference to Exhibit 10.54 to the Company's Annual Report on Form 10-K for the year ended December 31, 2013, File No. 001-10865) (confidential treatment previously granted) |
| | 10.29 | | Pharmaceutical Manufacturing and Supply Agreement, dated January 8, 2010, by and between the Company and Patheon Manufacturing Services LLC (as assignee from DSM Pharmaceuticals, Inc.) (incorporated herein by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2012, File No. 001-10865) (confidential treatment previously granted) |
| | 10.30 | | Amendment No. 1 to Pharmaceutical Manufacturing and Supply Agreement, dated July 5, 2014, by and between the Company and Patheon Manufacturing Services LLC (as assignee from DSM Pharmaceuticals, Inc.) (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended September 30, 2014, File No. 001-10865) |
| | 10.31+ | | Development and Supply Agreement, dated as of September 17, 2009, by and between Lumara Health Inc. (as successor in interest to Hologic, Inc.) and Hospira Worldwide, Inc. (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| | 10.32+ | | First Amendment to Development and Supply Agreement, dated as of March 28, 2014, by and between Lumara Health Inc. and Hospira Worldwide, Inc. (Certain confidential information contained in this exhibit was omitted by means of redacting a portion of the text and replacing it with [***]. This exhibit has been filed separately with the SEC without any redactions pursuant to a Confidential Treatment Request under Rule 24b-2 of the Securities and Exchange Act of 1934, as amended) |
| | 10.33 | | Underwriting Agreement, dated as of February 11, 2014, among AMAG Pharmaceuticals, Inc. and J.P. Morgan Securities LLC, on its own behalf and as representative of the several underwriters named in Schedule 1 thereto (incorporated herein by reference to Exhibit 1.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.34 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
192
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 10.35 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.36 | | Base Call Option Transaction Confirmation, dated as of February 11, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.3 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.37 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.4 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.38 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.5 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.39 | | Base Warrants Confirmation, dated as of February 11, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.6 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.40 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.7 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.41 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.8 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.42 | | Additional Call Option Transaction Confirmation, dated as of February 13, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.9 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.43 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and JPMorgan Chase Bank, National Association, London Branch (incorporated herein by reference to Exhibit 10.10 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.44 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and Royal Bank of Canada (incorporated herein by reference to Exhibit 10.11 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.45 | | Additional Warrants Confirmation, dated as of February 13, 2014, between the Company and Morgan Stanley & Co. International plc (incorporated herein by reference to Exhibit 10.12 to the Company's Current Report on Form 8-K filed February 14, 2014, File No. 001-10865) |
| | 10.46 | | Credit Agreement, dated as of November 12, 2014, by and among the Company, the financial institutions and agents listed therein, and Jefferies Finance LLC (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed November 12, 2014, File No. 001-10865) |
| | 10.47* | | Form of Employment Agreement between the Company and each of its executive officers (other than William Heiden) (incorporated herein by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, File No. 001-10865) |
193
Table of Contents
| | | |
Exhibit
Number | | Description |
|---|
| | 10.48* | | Employment Agreement dated as of February 7, 2014 between the Company and William Heiden (incorporated herein by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, File No. 001-10865) |
| | 21.1+ | | Subsidiaries of AMAG Pharmaceuticals, Inc. |
| | 23.1+ | | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
| | 24.1 | | Power of Attorney (included on the signature page(s) hereto) |
| | 31.1+ | | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Exchange Act, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| | 31.2+ | | Certification Pursuant to Rule 13a-14(a)/15d-14(a) of the Exchange Act, as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| | 32.1++ | | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| | 32.2++ | | Certification Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| | 101.INS+ | | XBRL Instance Document |
| | 101.SCH+ | | XBRL Taxonomy Extension Schema Document |
| | 101.CAL+ | | XBRL Taxonomy Extension Calculation Linkbase Document |
| | 101.DEF+ | | XBRL Taxonomy Extension Definition Linkbase Document |
| | 101.LAB+ | | XBRL Taxonomy Extension Label Linkbase Document |
| | 101.PRE+ | | XBRL Taxonomy Extension Presentation Linkbase Document |
- +
- Exhibits marked with a plus sign ("+") are filed herewith.
- ++
- Exhibits marked with a double plus sign ("++") are furnished herewith.
- *
- Exhibits marked with a single asterisk reference management contracts, compensatory plans or arrangements, filed in response to Item 15(a)(3) of the instructions to Form 10-K.
The other exhibits listed have previously been filed with the SEC and are incorporated herein by reference, as indicated.
194