UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended September 30, 2013
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 1-15803
Avanir Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 33-0314804 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
| |
| 20 Enterprise Suite 200, Aliso Viejo, California | | 92656 |
| (Address of principal executive offices) | | (Zip Code) |
(949) 389-6700
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15 (d) of the Act. YES ¨ NO þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YES þ NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES þ NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer | | ¨ | | Accelerated filer | | þ |
| | | |
| Non-accelerated filer | | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YES ¨ NO þ
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of March 31, 2013 was approximately $270.8 million, based upon the closing price on the NASDAQ Global Market reported for such date. Shares of common stock held by each officer and director and by each person who is known to own 10% or more of the outstanding Common Stock have been excluded in that such persons may be deemed to be affiliates of the Company. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of December 2, 2013, the registrant had 152,106,667 shares of common stock issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be disclosed in Part III of this report is incorporated by reference from the registrant’s definitive Proxy Statement for the 2014 Annual Meeting of Stockholders, which will be held on February 12, 2014 and which proxy statement will be filed not later than 120 days after the end of the fiscal year covered by this report.
Table of Contents
PART I
Except for the historical information contained herein, the matters set forth in this Annual Report on Form10-K, including statements regarding the Company’s plans, potential opportunities, financial or other expectations, projections, goals objectives, milestones, strategies, market growth, timelines, legal matters, product pipeline, clinical studies, product development and the potential benefits of its commercialized products and products under development are forward-looking statements within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from the anticipated or estimated future results, including the risks and uncertainties associated with Avanir’s future operating performance and financial position, developments in Avanir’s ongoing NUEDEXTA patent litigation, the market demand for and acceptance of Avanir’s products domestically and internationally, research, development and commercialization of new products domestically and internationally, obtaining and maintaining regulatory approvals domestically and internationally, including, but not limited to potential regulatory delays or rejections, risks associated with meeting the objectives of clinical studies, including, but not limited to, delays or failures in enrollment, and the occurrence of adverse safety events, and other risks set forth below under Item 1A, “Risk Factors” and other documents subsequently filed with or furnished to the Securities and Exchange Commission. These forward-looking statements are based on current information that may change and you are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. All forward-looking statements are qualified in their entirety by this cautionary statement, and the Company undertakes no obligation to revise or update any forward-looking statement to reflect events or circumstances after the date hereof.
References in this report to Avanir, the Company, we, our and us refer to Avanir Pharmaceuticals, Inc. and its subsidiaries, on a consolidated basis. “Avanir” and “NUEDEXTA” are registered trademarks of Avanir Pharmaceuticals, Inc. or its subsidiaries in the U.S. and/or other countries. Other trademarks or service marks appearing in this report may be trademarks or service marks of other owners.
EXECUTIVE OVERVIEW
Avanir is a biopharmaceutical company focused on acquiring, developing and commercializing novel therapeutic products for the treatment of central nervous system disorders. Our lead product NUEDEXTA® (referred to as AVP-923 during clinical development) is a first-in-class dual N-methyl-D-aspartate (“NMDA”) receptor antagonist and sigma-1 agonist. NUEDEXTA, 20/10 mg, was approved in the United States in October 2010 for the treatment of pseudobulbar affect (“PBA”). It is also approved for the treatment of PBA in the European Union in two dose strengths, NUEDEXTA 20/10 mg and NUEDEXTA 30/10 mg. We commercially launched NUEDEXTA in the United States in February 2011 and we are currently assessing plans regarding the potential commercialization of NUEDEXTA in the European Union.
We are studying the clinical utility of AVP-923 in other mood/behavior disorders, movement disorders and pain.We have two ongoing Phase II clinical trials exploring the potential treatment of agitation in patients with Alzheimer’s disease and potential treatment of levodopa-induced dyskinesia in Parkinson’s disease (the “LID study”). The LID study is supported by a grant from the Michael J. Fox Foundation. In December 2013, we completed a Phase II clinical trial of AVP-923 for the potential treatment of central neuropathic pain in patients with multiple sclerosis (“MS”). The data from this Phase II clinical trial did not meet the primary efficacy endpoint. In the study, AVP-923 treated patients experienced levels of pain reduction commensurate with those observed in similar studies and the improvement in pain scores from baseline reached statistical significance; however, there was no difference between the treatment arms and placebo. We believe that a higher than expected placebo response negatively impacted the study results. AVP-923 was generally safe and well tolerated in the study.
We are also developing AVP-786, a next generation drug product containing deuterium-modified dextromethorphan and quinidine for the treatment of neurologic and psychiatric disorders. We completed a pharmacokinetic study with AVP-786, and based on this data, we believe that we have identified a formulation of
3
AVP-786 with a comparable pharmacokinetic profile, and likely a comparable, safety and tolerability profile to AVP-923. This AVP-786 formulation contains significantly less quinidine than used in AVP-923.
We are also developing a novel Breath Powered™ intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, AVP-825. If approved, this product would be the first and only fast-acting dry-powder nasal delivery form of sumatriptan. AVP-825 is licensed from Optinose AS (“OptiNose”). Under the terms of the agreement, we assumed responsibility for regulatory, manufacturing, supply-chain and commercialization activities for the investigational product. Both parties will work together on the remaining activities in support of the New Drug Application (“NDA”) submission. We have begun preparing the NDA and expect to file the application with the U.S. Food and Drug Administration (“FDA”) in the first quarter of calendar 2014.
We entered into a multi-year agreement with Merck Sharp & Dohme Corp. (“Merck”) to co-promote Merck’s type 2 diabetes therapies JANUVIA® (sitagliptin) and the sitagliptin family of products in the long-term care institutional setting in the United States beginning October 1, 2013. The term of the agreement will continue for three years following the launch date of the co-promotion activities. Under the terms of the agreement, the Company will be compensated via a (i) fixed monthly fee and (ii) performance fee based on the amount of the products sold by the Company above a predetermined baseline. A significant majority of the fee is performance based. Over the three years of the agreement, Avanir could receive up to a maximum of $60.0 million in compensation.
In addition to our focus on products for the central nervous system, we also have partnered programs in other therapeutic areas that may generate future revenue for us. Our first commercialized product, docosanol 10% cream (sold in the United States and Canada as Abreva® by our marketing partner GlaxoSmithKline Consumer Healthcare), is the only over-the-counter treatment for cold sores that has been approved by the FDA.
Avanir was incorporated in California in August 1988 and was reincorporated in Delaware in March 2009.
MARKETED PRODUCTS AND DRUG CANDIDATES
The following chart illustrates the status of research and development activities for our products and product candidates that are commercialized or under development.

In addition to the research and development programs identified above, Avanir has provided unrestricted research grants to support several investigator initiated studies with AVP-923. Current studies planned or ongoing include potential treatment of behavioral symptoms of adults with autism spectrum disorder, treatment of bulbar function (impaired speech, swallowing, and saliva control) associated with amyotrophic lateral sclerosis (ALS), and treatment of refractory depression. For additional information regarding these studies please see http://clinicaltrials.gov.
NUEDEXTA for the Treatment of Pseudobulbar Affect
NUEDEXTA is the first and only FDA-approved treatment for PBA. PBA occurs secondary to a variety of otherwise unrelated neurological conditions, and is characterized by involuntary, sudden, and frequent episodes of
4
laughing and/or crying. PBA episodes typically occur out of proportion or incongruent to the patient’s underlying emotional state.
NUEDEXTA is an innovative combination of two well-characterized components: dextromethorphan hydrobromide (20 mg), the ingredient that is pharmacologically active in the central nervous system, and quinidine sulfate (10 mg), a metabolic inhibitor enabling dextromethorphan to reach therapeutic plasma concentrations. NUEDEXTA acts on sigma-1 and NMDA receptors in the brain, although the mechanism by which NUEDEXTA exerts therapeutic effects in patients with PBA is unknown.
Studies to support the effectiveness of NUEDEXTA were performed in patients with PBA and underlying amyotrophic lateral sclerosis (“ALS”) and MS. The primary outcome measure, the number of laughing and crying episodes, was significantly lower in the NUEDEXTA cohort compared with placebo. The secondary outcome measure, the Center for Neurologic Studies Lability Scale (“CNS-LS”), demonstrated a significantly greater mean decrease in CNS-LS score from baseline for the NUEDEXTA cohort compared with placebo. NUEDEXTA has not been studied in other types of emotional lability that can commonly occur, for example, in Alzheimer’s disease and other dementias.
NUEDEXTA safety information
For a description of the NUEDEXTA safety information, a copy has been filed as Exhibit 99.1 on this Annual Report on Form 10-K for the period ended September 30, 2013.
PBA indication and market
PBA is a distinct neurologic syndrome that is characterized by a loss of control of emotional expression, involving episodes of involuntary crying or laughing that are contrary or exaggerated relative to the patient’s inner feelings.
There are an estimated 18 to 20 million people in the United States who suffer from the underlying neurologic conditions that can give rise to PBA. These underlying neurologic conditions include but are not limited to ALS, MS, Alzheimer’s disease, Parkinson’s disease, stroke and traumatic brain injury. Based on the epidemiologic medical literature, physician estimates, market research, an Avanir-sponsored patient survey of 2,464 neurologic patients and their caregivers, and the PRISM PBA registry which enrolled 5,290 neurologic patients across 173 investigator sites in the U.S., we estimate that approximately 10% of people in the United States who suffer from neurological disease or injury also suffer from moderate to severe PBA symptoms, with many more suffering from mild PBA symptoms. Initial research indicates a similar rate of prevalence of PBA in the European Union.
Other than NUEDEXTA, there are no FDA-approved therapies or European Medicines Agency (“EMA”)-approved therapies indicated to treat PBA. Some physicians treat PBA using a range of drugs off-label, including: selective serotonin reuptake inhibitors/serotonin-norepinephrine reuptake inhibitors (SSRIs/SNRIs), antidepressants and atypical antipsychotics. According to our market research, physicians are generally only moderately satisfied with these off-label therapies as a treatment for PBA. We periodically conduct this market research through an Internet-based survey of approximately 240 physicians, consisting of Neurologists, Internal Medicine/Geriatrics, Psychiatrists and long-term care affiliated physicians.
We believe that NUEDEXTA represents a more attractive treatment option for patients suffering from PBA. In our Phase III STAR trial that was completed in 2009, patients treated with NUEDEXTA reported an average 82% reduction in PBA episodes at the end of the 12-week study compared to baseline, with an average 44% reduction in episodes after the first week of treatment. Over the course of the 12-week study, patients receiving NUEDEXTA experienced significantly lower PBA episode rates versus placebo (P<0.0001). Over the final two weeks of the STAR trial, 51% of patients treated with NUEDEXTA achieved episode-free remission.
NUEDEXTA commercial strategy
We currently market NUEDEXTA in the U.S. to approximately 10,000 physicians and other healthcare providers who specialize in psychiatry, neurology, internal medicine or geriatric medicine and practice in outpatient
5
or long-term care settings in the U.S. Our U.S. commercial strategy for NUEDEXTA includes the following key objectives:
| | • | | Maximize sales force reach in retail and institutional channels — We have two separate sales teams in the retail and institutional channels which has expanded our sales force reach and increased our efficiency by allowing each team to focus their specialized skill set and expertise towards each distinct channel. |
| | • | | Increase awareness and diagnosis of PBA — Since PBA is under-diagnosed and often not treated, we use a range of tools to educate physicians and nurses about the prevalence of PBA, the burden of the disease and encourage the utilization of screening tools to diagnose PBA in patients with underlying neurological disorders. |
| | • | | Expand adoption of NUEDEXTA — We are focused on driving awareness of NUEDEXTA and educating health care providers on the risks and benefits of the drug. These interactions take place in a wide range of forums including physician offices, long term care facilities, speaker programs and medical meetings and conferences. Our medical affairs team is active in publishing results from NUEDEXTA studies that are disseminated through appropriate scientific channels. |
| | • | | Minimize barriers to patient access — We work actively with major insurance plans and pharmacy benefit managers to ensure that NUEDEXTA receives the appropriate coverage and patients with PBA have access to the drug. Additionally, we have a range of patient programs to help defray some of the costs of the medication including co-pay cards, out-of-pocket patient assistance program for Medicare Part D patients and patient assistance programs for patients lacking insurance. |
| | • | | Motivate patients and caregivers to request and adhere to NUEDEXTA therapy — Our market research has shown that many neurological patients are not aware that PBA is a distinct medical condition and do not discuss symptoms with their physicians. Research also shows that when they engage with a physician, they are likely to receive a medication like NUEDEXTA. Based on this data, we have engaged in a series of programs directly communicating with patients and their caregivers to provide educational material on PBA and NUEDEXTA where appropriate. These programs include direct database marketing, Internet advertising and direct response television advertising. |
We received approval in June 2013 to market NUEDEXTA in the European Union and are currently evaluating our commercial strategy for the European Union.
AVP-923 for the Potential Treatment of Neuropathic Pain
AVP-923 for the treatment central neuropathic pain in multiple sclerosis
Multiple Sclerosis is one of the most frequent chronic neurologic diseases causing significant disability in young adults. Among the many neurological complications of MS, chronic pain has a significant impact on the daily life of patients with this disease.
Several distinct pain conditions associated with MS have been characterized in various literature, including optic neuritis, headache, musculoskeletal pain (which includes painful tonic spasms, back pain and muscle spasms) and central neuropathic pain (which includes extremity pain, trigeminal neuralgia and Lhermitte’s sign). While the specific pain mechanisms associated with each condition have not been fully identified, it is believed that neuropathic pain results from nerve damage caused by MS. Approximately 30% of MS patients experience central neuropathic pain during the course of the disease.
In November 2011, we initiated a Phase II study, known as Pain Research In Multiple sclErosis (“PRIME”), for the treatment of central neuropathic pain in MS. The objectives of PRIME were to evaluate the safety, tolerability, and efficacy of three dose levels of AVP-923 capsules for the treatment of central neuropathic pain in a population of patients with MS. The trial was a multicenter, randomized, double-blind, placebo-controlled, 4-arm parallel group study. Eligible patients were randomized to receive one of the three dose levels of AVP-923 (45/10 mg, 30/10 mg, or 20/10 mg) or placebo daily for 12 weeks. The primary efficacy measure was the Pain Rating Scale obtained from patient diaries. The primary efficacy endpoint looked at the plasma levels of DM at different doses versus the pain relief observed. Secondary endpoints included measure of fatigue, impact of MS on
6
daily life, sleep quality, cognition and depression. Safety was assessed by monitoring adverse events, clinical laboratory tests, electrocardiograms, physical examinations, and vital signs. In February 2013, based on the successful interim Phase I results from the AVP-786 study, we modified the target study enrollment from approximately 400 to 200 patients both in the U.S. and internationally. The enrollment target was modified in order to accelerate completion of the study so that data would be available to help plan further development of our next generation compound AVP-786. In December 2013, we completed the PRIME study. The data from the PRIME study did not meet the primary efficacy endpoint. In the study, AVP-923 treated patients experienced levels of pain reduction commensurate with those observed in similar studies and the improvement in pain scores from baseline reached statistical significance; however, there was no difference between the treatment arms and placebo. Avanir believes that a higher than expected placebo response negatively impacted the study results. AVP-923 was generally safe and well tolerated in the study.
AVP-923 for the treatment of diabetic neuropathic pain
Diabetic peripheral neuropathic pain (“DPN pain”), which arises from nerve injury, can result in a chronic and debilitating form of pain that has historically been poorly diagnosed and treated. It is often described as burning, tingling, stabbing, or pins and needles in the feet, legs, hands or arms. An estimated 3.5 million people in the United States experience DPN pain according to the American Diabetes Association. DPN pain currently is most commonly treated with antidepressants, anticonvulsants, opioid analgesics and local anesthetics. Most of these treatments have limited effectiveness or undesirable side effects resulting in a high degree of unmet medical need. The global neuropathic pain market was approximately $2.4 billion in 2010 and is expected to grow to $3.6 billion by 2020 among the seven largest markets, consisting of the United States, Japan, France, Germany, Italy, Spain and the United Kingdom.
Avanir has completed a Phase III clinical trial for AVP-923 in the treatment of patients with DPN pain. In April 2007, we announced positive top-line data from our first Phase III clinical trial of AVP-923 for the treatment of patients with DPN pain. The most commonly reported adverse events from this Phase III study were dizziness, nausea, diarrhea, fatigue and somnolence, which were mild to moderate in nature. We are continuing to evaluate our options for this program, including the use of AVP-786 in the advancement of this program.
Other Programs
AVP-923 for the treatment of agitation in patients with Alzheimer’s disease
Alzheimer’s disease is generally characterized by cognitive decline, impaired performance of daily activities, and behavioral disturbances. Behavioral and psychiatric symptoms develop in as many as 60% of community-dwelling dementia patients and in more than 80% of patients with dementia living in nursing homes; as the disease progresses the risk of such complications approaches 100%. Dementia-related behavioral symptoms, including agitation, can be extremely distressing to the individual, the family, and caregivers. These behavioral disturbances have been associated with more rapid cognitive decline, institutionalization, and increased caregiver burden.
The objectives of this proof-of-concept study are to evaluate the safety, tolerability, and efficacy of AVP-923 for the treatment of agitation in Alzheimer’s patients. The trial is a multicenter, randomized, double-blind, placebo-controlled study that is expected to enroll up to 200 Alzheimer’s patients in the United States. Eligible patients will be randomized to receive either AVP-923 or placebo for 10 weeks. The primary efficacy measure is the agitation/aggression domain of the Neuropsychiatric Inventory or NPI. Secondary outcome measures include assessments of disease severity, behavioral abnormalities, cognition, activities of daily living, quality of life and caregiver strain. Standard safety assessments will also be conducted.
AVP-923 for the treatment of levodopa-induced-dyskinesia
Levodopa-induced-dyskinesia (“LID”) occurs in most patients with Parkinson’s disease (“PD”) after several years of treatment, generally in association with other motor response complications, such as wearing-off or “on-off” fluctuations. Dyskinesia may be as disabling as the parkinsonism itself, and current treatment options are limited and are not always effective.
7
This proof-of-concept, double blind, randomized, crossover study will compare AVP-923 (45mg DM/10mg Q) with placebo for treatment of LID. The study will enroll approximately 16 PD patients across three study centers in the U.S. and Canada. Study participants will receive, in a random order, a 2-week treatment with AVP-923 and a2-week placebo treatment, separated by a 2-week break. At the end of each 2-week treatment period, patients will receive a 2-hour levodopa infusion to test the drug effect on dyskinesia. Patients will be carefully monitored throughout the 6-week study for side effects, Parkinson’s symptoms and general health status. The results of this study will help inform future development of AVP-923 for LID. This study is being funded through a grant awarded by the Michael J. Fox Foundation.
AVP-786 for the treatment of neurologic and psychiatric disorders
AVP-786 is a novel investigational drug product consisting of a combination of deuterium-modified dextromethorphan (a new chemical entity or NCE) and the metabolic inhibitor quinidine. The compound was developed through incorporation of deuterium into molecular positions of dextromethorphan, resulting in strengthened molecular bonds which reduce susceptibility to enzyme cleavage. Based on interim data, we believe that we have identified a formulation of AVP-786 with a comparable pharmacokinetic profile, and likely a comparable safety and tolerability profile to AVP-923. This AVP-786 formulation contains significantly less quinidine than used in AVP-923. In June 2013, the FDA agreed to an expedited development pathway for AVP-786, requiring only a limited non-clinical package as part of the Investigational New Drug application. Upon completion of these non-clinical studies, the Company intends to proceed directly into human clinical trials. The Company currently intends to study AVP-786 in neuropathic pain, agitation in patients with Alzheimer’s disease and treatment resistant depression.
AVP-825 for the treatment of acute migraine
AVP-825 is an investigational drug-device combination product consisting of low-dose sumatriptan powder to treat acute migraine. The powder is delivered intranasally utilizing a novel Breath Powered delivery technology. If approved, AVP-825 would be the first and only fast-acting dry-powder intranasal form of sumatriptan. We have begun preparing the NDA and expect to file the application with the FDA in the first quarter of calendar 2014.
Migraine represents an area of significant unmet medical need. According to the Centers for Disease Control and Prevention, over 37 million Americans suffer from migraine headaches. The triptan class of medications is generally considered the standard of care with over 13 million prescriptions written annually. Sumatriptan is the class leader with a market share of over 50%, making it the most commonly prescribed migraine drug in the U.S. An online survey of over 2,500 frequent migraine sufferers revealed that 66% were dissatisfied with their treatments. As a result, many migraine sufferers are seeking fast-acting, well tolerated treatment options.
Docosanol 10% cream — cold sore treatment
Docosanol 10% cream is a topical treatment for cold sores. In 2000, we received FDA approval for marketing docosanol 10% cream as an over-the-counter product. Since that time, docosanol 10% cream has been approved by regulatory agencies in Asia, North America, and Europe. In March 2000, we granted a subsidiary of GlaxoSmithKline, SB Pharmco Puerto Rico, Inc. (“GSK”), the exclusive rights under a license to market docosanol 10% cream in the United States and Canada (“GSK License Agreement”). GSK markets the product under the name Abreva® in these markets. Under the terms of the GSK License Agreement, GSK is responsible for all sales and marketing activities and the manufacturing and distribution of docosanol 10% cream. Under the GSK license agreement, we received a total of $25 million in milestone payments from GSK and we were entitled to receive an 8% royalty on net sales of Abreva by GSK.
In November 2002, we sold to Drug Royalty USA an undivided interest in our right to receive future Abreva royalties under the GSK License Agreement for $24.1 million (the “Drug Royalty Agreement”). Under the Drug Royalty Agreement, Drug Royalty USA has the right to receive royalties from GSK on sales of Abreva until the expiration of the patent for Abreva on April 30, 2014. We retained the right to receive 50% of all royalties (a net of 4%) under the GSK License Agreement for annual net sales of Abreva in the U.S. and Canada in excess of $62.0 million. From the effective date of the GSK License Agreement up to the 2002 sale of our royalty rights to
8
Drug Royalty USA, Inc., we received a total of approximately $5.9 million in royalty payments from GSK attributed to the 8% royalty on net sales by GSK.
Under the terms of our docosanol license agreements, our partners are generally responsible for all regulatory approvals, sales and marketing activities, and manufacturing and distribution of the product in the licensed territories. The terms of the license agreements typically provide for us to receive a data transfer fee, potential milestone payments and royalties on product sales. We purchase the active pharmaceutical ingredient (“API”), docosanol, from a large supplier in Western Europe and have, on occasion, sold material to our licensees. We currently store our API in the United States. Any material disruption in manufacturing could cause a delay in shipments and possible loss of sales.
Competition
The pharmaceutical industry is characterized by rapidly evolving technology and intense competition. A large number of companies of all sizes, including major pharmaceutical companies and specialized biotechnology companies, engage in activities similar to our activities. Many of our competitors have substantially greater financial and other resources available to them. In addition, colleges, universities, governmental agencies and other public and private research organizations continue to conduct research and are becoming more active in seeking patent protection and licensing arrangements to collect royalties for use of technologies that they have developed. Some of our competitors’ current or future products and technologies may be in direct competition with ours. We also must compete with these institutions in recruiting highly qualified personnel.
NUEDEXTA for Pseudobulbar Affect. Although NUEDEXTA is the first product to be marketed for the treatment of PBA, we are aware that physicians may prescribe other products in an off-label manner for the treatment of this disorder. For example, NUEDEXTA may face competition from the following products:
| | • | | Antidepressants, including Prozac®, Celexa®, Zoloft®, Paxil®, Elavil® and Pamelor® and others; |
| | • | | Atypical antipsychotic agents, including Zyprexa®, Risperdal®, Seroquel, Abilify®, Geodon® and others; and |
| | • | | Miscellaneous agents, including Symmetrel®, Lithium and others. |
While it is also possible that compounding pharmacies could combine the components of NUEDEXTA in an unauthorized fashion that is in violation of our patents, it is inconsistent with the policies of the Pharmacy Compounding Accreditation Board.
Manufacturing
We currently have no manufacturing or production facilities and, accordingly, rely on third parties for clinical production of our products and product candidates. We obtain the API for NUEDEXTA from a sole supplier who is one of several available commercial suppliers. (See Item 1A, “Risk Factors”). Further, we licensed to various pharmaceutical companies the exclusive rights to manufacture and distribute docosanol 10% cream.
Intellectual Property Rights
Patents
We own and have licensed a number of our patents relating to our products, which in the aggregate are believed to be of material importance to us in the operation of our business. See Item 1A, “Risk Factors.”
Trademarks and Other Intellectual Property Rights
We have made a practice of selling our products under trademarks and of obtaining protection for these trademarks by all available means. These trademarks are protected under the common law and/or by registration in the United States and other countries. We consider these trademarks in the aggregate to be of material importance in the operation of our business.
Our intellectual property also includes copyrights, confidential and trade secret information.
9
Government Regulations
The FDA and comparable regulatory agencies in foreign countries extensively regulate the manufacture and sale of the pharmaceutical products that we have developed or are currently developing. The FDA has established guidelines and safety standards that are applicable to the nonclinical evaluation and clinical investigation of therapeutic products and stringent regulations that govern the manufacture and sale of these products. The process of obtaining regulatory approval for a new therapeutic product usually requires a significant amount of time and substantial resources. The steps typically required before a product can be tested in humans include:
| | • | | Animal pharmacology studies to obtain preliminary information on the safety and efficacy of a drug; and |
| | • | | Non-clinical evaluationin vitroandin vivoincluding extensive toxicology studies. |
The results of these non-clinical studies may be submitted to the FDA as part of an IND application. The sponsor of an IND application may commence human testing of the compound 30 days after submission of the IND, unless notified to the contrary by the FDA.
The clinical testing program for a new drug typically involves three phases:
| | • | | Phase I investigations are generally conducted in healthy subjects. In certain instances, subjects with a life-threatening disease, such as cancer, may participate in Phase I studies that determine the maximum tolerated dose and initial safety of the product; |
| | • | | Phase II studies are conducted in limited numbers of subjects with the disease or condition to be treated and are aimed at determining the most effective dose and schedule of administration, evaluating both safety and whether the product demonstrates therapeutic effectiveness against the disease; and |
| | • | | Phase III studies involve large, well-controlled investigations in diseased subjects and are aimed at verifying the safety and effectiveness of the drug. |
Data from all clinical studies, as well as all nonclinical studies and evidence of product quality, typically are submitted to the FDA in a NDA. Although the FDA’s requirements for clinical trials are well established and we believe that we have planned and conducted our clinical trials in accordance with the FDA’s applicable regulations and guidelines, these requirements, including requirements relating to testing the safety of drug candidates, may be subject to change or new interpretation. Additionally, we could be required to conduct additional trials beyond what we had planned due to the FDA’s safety and/or efficacy concerns or due to differing interpretations of the meaning of our clinical data. (See Item 1A, “Risk Factors”).
The FDA’s Center for Drug Evaluation and Research must approve a NDA for a drug before it may be marketed in the U.S. If we begin to market our proposed products for commercial sale in the U.S., any manufacturing operations that may be established in or outside the U.S. will also be subject to rigorous regulation, including compliance with current good manufacturing practices. We also may be subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substance Control Act, the Export Control Act and other present and future laws of general application.
Regulatory obligations continue post-approval, and include the reporting of adverse events when a drug is utilized in the broader commercial population. Promotion and marketing of drugs is also strictly regulated, with penalties imposed for violations of FDA regulations, the Lanham Act (trademark statute), and other federal and state laws, including the federal anti-kickback statute.
We currently intend to continue to seek, directly or through our partners, approval to market our products and product candidates in foreign countries, which may have regulatory processes that differ materially from those of the FDA. We anticipate that we will rely upon pharmaceutical or biotechnology companies to license our proposed products or independent consultants to seek approvals to market our proposed products in foreign countries. We cannot assure you that approvals to market any of our proposed products will be obtained in any country. Approval to market a product in any one foreign country does not necessarily indicate that approval can be obtained in other countries.
10
Product Liability Insurance
We maintain product liability insurance on our products and clinical trials that provides coverage in the amount of $20 million per incident and $20 million in aggregate.
Executive Officers and Key Employees of the Registrant
Information concerning our executive officers and key employees, including their names, ages and certain biographical information can be found in Part III, Item 10 under the caption, “Executive Officers and Key Employees of the Registrant.” This information is incorporated by reference into Part I of this report.
Employees
As of December 2, 2013, we employed 267 persons, including 34 engaged in research, development, regulatory and medical affairs activities, 202 in selling and marketing, and 31 in general and administrative functions.
Financial Information about Segments
We operate in a single accounting segment — the development and commercialization of novel treatments that target the central nervous system. Refer to Note 14, “Segment Information” in the Notes to Consolidated Financial Statements.
General Information
Our principal executive offices are located at 20 Enterprise, Suite 200, Aliso Viejo, California 92656. Our telephone number is (949) 389-6700 and our e-mail address isinfo@avanir.com. Our Internet website address iswww.avanir.com.No portion of our website is incorporated by reference into this Annual Report on Form 10-K.
You are advised to read this Annual Report on Form 10-K in conjunction with other reports and documents that we file from time to time with the Securities and Exchange Commission (“SEC”). In particular, please read our definitive proxy statement, which will be filed with the SEC in connection with our 2014 Annual Meeting of Stockholders, our Quarterly Reports on Form 10-Q and any Current Reports on Form 8-K that we may file from time to time. You may obtain copies of these reports after the date of this annual report directly from us or from the SEC at the SEC’s Public Reference Room at 100 F Street, N.E. Washington, D.C. 20549. In addition, the SEC maintains information for electronic filers (including Avanir) at its website at www.sec.gov. The public may obtain information regarding the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. We make our periodic and current reports available on our internet website, free of charge, as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC.
This Annual Report on Form 10-K contains forward-looking information based on our current expectations. Because our actual results may differ materially from any forward-looking statements that we make or that are made on our behalf, this section includes a discussion of important factors that could affect our actual future results, including, but not limited to, our product sales, capital resources, commercial market estimates, safety of NUEDEXTA, future development efforts, patent protection, effects of healthcare reform, reliance on third parties, and other risks set forth below. We disclaim any intent to update forward-looking statements to reflect subsequent developments or actual results.
11
Risks Relating to Our Business
Our near-term prospects depend on reaching profitability from the commercialization of NUEDEXTA in the United States. If we are unable to continue to increase NUEDEXTA revenues, including through raising PBA awareness among patients and physicians, driving higher rates of physician adoption and obtaining reimbursement and third party payer coverage, our ability to generate significant revenue or achieve profitability will be adversely affected.
Although NUEDEXTA has been approved for marketing, our ability to generate significant revenue in the near term is entirely dependent upon our ability to continue the successful commercialization of NUEDEXTA. To continue to be successful we must:
| | • | | maintain successful sales, marketing and educational programs for our targeted physicians and other health care providers; |
| | • | | raise patient and physician awareness of PBA and encourage physicians to screen patients for the condition; |
| | • | | obtain adequate reimbursement for NUEDEXTA from a broad range of payers; and |
| | • | | maintain and defend our patent protection and maintain regulatory exclusivity for NUEDEXTA. |
Supplying the market for NUEDEXTA requires us to manage relationships with an increasing number of collaborative partners, suppliers and third-party contractors. If we are unable to successfully maintain the required sales and marketing infrastructure, as well as successfully manage an increasing number of relationships, including with suppliers, manufacturers, distributors, insurance carriers and prescribers, we will have difficulty growing our business. In addition, pharmacies, institutions and prescribers may rely on third-party medical information systems to interpret the NUEDEXTA approved product label and guide utilization of NUEDEXTA. If these information systems load incorrect information or misinterpret the approved product label, it may result in lower adoption or utilization than expected. For example, because NUEDEXTA contains quinidine, which is a known pro-arrhythmic drug at antiarrhythmic doses exceeding 600 mg per day, it is possible that medical information systems may incorrectly identify NUEDEXTA as contraindicated or otherwise inappropriate for a patient, even in situations where the risks are substantially less than perceived.
In addition, we may enter into co-promotion or out-licensing arrangements with other pharmaceutical or biotechnology partners for NUEDEXTA where necessary to reach customers in domestic or foreign market segments and when deemed strategically and economically advantageous. To the extent that we enter into co-promotion or other licensing arrangements, our product revenues may be lower than if we directly marketed and sold NUEDEXTA, and some or all of the revenues we receive will depend upon the efforts of third parties, which may not be successful. If we are unable to accomplish any of these key objectives, we may not be able to generate significant product revenue or become profitable.
We have a history of net losses and an accumulated deficit, and we may be unable to generate sufficient revenue to achieve or maintain profitability in the future.
We have experienced significant net losses and negative cash flows from operations and we expect our negative cash flow from operations to continue until we are able to generate significant revenues from sales of NUEDEXTA. As of September 30, 2013, we had an accumulated deficit of approximately $500.5 million. We have incurred these losses principally from costs incurred in funding the research, development and clinical testing of our drug candidates, from our general and administrative expenses and from our commercialization activities for NUEDEXTA. We may continue incurring net losses for the foreseeable future and we expect our operating losses to continue for the foreseeable future as we continue to grow NUEDEXTA sales, invest in the development of AVP-923 and AVP-786, seek to commercialize NUEDEXTA in the European Union (“EU”), and seek FDA approval and subsequently commercialize AVP-825.
Our ability to generate revenue and achieve profitability in the near term is dependent on our ability, alone or with partners, to successfully manufacture and market NUEDEXTA for the treatment of patients with PBA in the United States. We expect to continue to spend substantial amounts on the ongoing marketing of NUEDEXTA domestically for the treatment of PBA, invest in Europe to commercialize NUEDEXTA, and seek regulatory
12
approvals for use of NUEDEXTA in other geographic markets and indications. As a result, we may be unable to generate sufficient revenue from product sales to become profitable or generate positive cash flows.
Certain of our key issued patents covering NUEDEXTA are currently being challenged and our pending patent applications may be denied. An adverse outcome in either case would adversely affect our ability to generate significant product revenue or become profitable.
We have invested in an extensive patent portfolio and we rely substantially on the protection of our intellectual property through our ownership or control of issued patents and patent applications. The degree of patent protection that will ultimately be afforded to us in the U.S. and in other important markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts and lawmakers in these countries. If we cannot prevent others from exploiting claims in our patent portfolio, we will not derive the benefit from it that we currently expect. Further, we may incur substantial expense from litigation to protect our patent portfolio.
The validity, enforceability and scope of our core patents covering NUEDEXTA are currently being challenged as a result of abbreviated new drug application (“ANDA”) filings from generic drug companies. An adverse outcome in the current or any future challenges to the validity, enforceability or scope of our patent portfolio could significantly reduce revenues from NUEDEXTA and any future products. More broadly, investors should be aware that the pharmaceutical industry is highly competitive. Our ability to compete in this space involves various risks relating to our intellectual property, including:
| | • | | our patents covering NUEDEXTA may be found to be invalid and unenforceable or insufficiently broad to block the introduction of a generic form of NUEDEXTA; |
| | • | | the claims in any of our pending patent applications may not be allowed and/or our patent applications may not be granted; |
| | • | | competitors may develop similar or superior technologies independently, duplicate our technologies, or design around the patented aspects of our technologies; |
| | • | | any of our issued patents may not provide us with significant competitive advantages; and |
| | • | | we may not be able to secure additional worldwide intellectual property protection for our NUEDEXTA patent portfolio. |
An adverse outcome with respect to any of these risks could adversely affect our ability to generate significant product revenue or become profitable.
We have received notices of ANDA filings for NUEDEXTA submitted by five generic drug companies. These ANDA filings assert that a generic form of NUEDEXTA would not infringe our FDA Orange Book listed patents and/or those patents are invalid. As a result of these filings, we have commenced litigation to defend our patent rights, and have subsequently settled three of these cases. The litigation has been costly and time consuming and, depending on the outcome of the litigation, we may face competition from lower cost generic or follow-on products in the near term.
NUEDEXTA is approved under the provisions of the Federal Food, Drug and Cosmetic Act (“FDCA”), which renders it susceptible to potential competition from generic manufacturers via the Hatch-Waxman Act and ANDA process. Generic manufacturers pursuing ANDA approval are not required to conduct costly and time-consuming clinical trials to establish the safety and efficacy of their products; rather, they are permitted to rely on the innovator’s data regarding safety and efficacy. Additionally, generic drug companies generally do not expend significant sums on sales and marketing activities, instead relying on physicians or payers to substitute the generic form of a drug for the branded form. Thus, generic manufacturers can sell their products at prices much lower than those charged by the innovative pharmaceutical or biotechnology companies who have incurred substantial expenses associated with the research and development of the drug product and who must spend significant sums marketing a new drug.
The ANDA procedure includes provisions allowing generic manufacturers to challenge the innovator’s patent protection by submitting “Paragraph IV” certifications to the FDA in which the generic manufacturer claims that the innovator’s patent is invalid, unenforceable and/or will not be infringed by the manufacture, use, or sale of the
13
generic product. A patent owner who receives a Paragraph IV certification may choose to sue the generic applicant for patent infringement. In recent years, generic manufacturers have used Paragraph IV certifications extensively to challenge the applicability of patents listed in the FDA’s Approved Drug Products List with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book, on a wide array of innovative therapeutic products. We expect this trend to continue and to affect drug products with even relatively modest revenues.
We have received Paragraph IV certification notices from five separate companies contending that our patents listed in the Orange Book (U.S. Patents 7,659,282, 8,227,484 and RE 38,115, which expire in August 2026, July 2023 and January 2016, respectively) are invalid, unenforceable and/or will not be infringed by the manufacture, use, or sale of a generic form of NUEDEXTA. In response to these notices, we have filed suit against all of the generic drug companies to defend our patent rights. We have entered into settlement agreements with three of the companies to resolve pending patent litigation in response to their ANDAs seeking approval to market generic versions of NUEDEXTA capsules. The settlement agreements grant the three companies the right to begin selling a generic version of NUEDEXTA on July 30, 2026, or earlier under certain circumstances. The parties also filed stipulations and orders of dismissal with the United States District Court for the District of Delaware which conclude the litigation with respect to the three companies. The settlements do not end our ongoing litigation against the other two ANDA filers.
As a result of the 30-month stay associated with the filing of the ANDA actions, the FDA cannot grant final approval to any ANDA before December 30, 2013, unless there is an earlier court decision holding that the Orange Book-listed ’282, ’484 and ’115 patents are not infringed and/or are invalid.
Although we intend to vigorously enforce our intellectual property rights relating to NUEDEXTA, there can be no assurance that we will prevail in our defense of our patent rights. Our existing patents could be invalidated, found unenforceable or found not to cover a generic form of NUEDEXTA. If an ANDA filer were to receive FDA approval to sell a generic version of NUEDEXTA and/or prevail in any patent litigation, NUEDEXTA would become subject to increased competition and our revenue would be adversely affected.
PBA is a new market and estimates vary significantly over the potential market size and our anticipated revenues over the near and long term.
NUEDEXTA is being made available to patients to treat PBA, an indication for which there was no previously established pharmaceutical market. Industry sources and equity research analysts have a wide divergence of estimates for the near- and long-term market potential of our product. A variety of assumptions directly impact the estimates for our drug’s market potential, including estimates of underlying neurologic condition prevalence, severity of PBA prevalence among these conditions, rates of physician adoption of our drug for treatment of PBA among these populations, health plan reimbursement rates, and patient adherence and compliance rates within each underlying neurological condition. Small differences in these assumptions can lead to widely divergent estimates of the market potential of our product. Additionally, although our approved product label is indicated to treat PBA, without regard to the underlying neurological condition, it is possible that physicians, the FDA’s Office of Prescription Drug Promotion (OPDP), payers or others may interpret the label more narrowly than the FDA’s Division of Neurology Products approval for a broad PBA label and believe that PBA secondary to certain conditions, such as Alzheimer’s disease, is not an indicated use. If such misinterpretations are widespread, the actual market size may be smaller than we have estimated. Accordingly, investors are cautioned not to place undue reliance on any particular estimates of equity research analysts or industry sources.
Significant safety or drug interaction problems could arise with respect to NUEDEXTA, which could result in restrictions in NUEDEXTA’s label, recalls, withdrawal of NUEDEXTA from the market, an adverse impact on potential sales of NUEDEXTA, or cause us to alter or terminate current or future NUEDEXTA clinical development programs, any of which would adversely impact our future business prospects.
Discovery of previously unknown safety or drug interaction problems with an approved product may result in a variety of adverse regulatory actions. This risk may be increased as physicians, at their own discretion, may prescribe NUEDEXTA for off-label uses, which may result in unknown safety or drug interactions. Under the Food and Drug Administration Amendments Act of 2007, the FDA has broad authority to force drug manufacturers to
14
take any number of actions if previously unknown safety or drug interaction problems arise, including, but not limited to: (i) requiring manufacturers to conduct post-approval clinical studies to assess known risks or signals of serious risks, or to identify unexpected serious risks; (ii) mandating labeling changes to a product based on new safety information; or (iii) requiring manufacturers to implement a Risk Evaluation Mitigation Strategy, or REMS, where necessary to assure safe use of the drug. In addition, previously unknown safety or drug interaction problems could result in product recalls, restrictions on the product’s permissible uses, or withdrawal of the product from the market.
The combination of dextromethorphan and quinidine has never been marketed for the treatment of any condition until the approval of NUEDEXTA for the treatment of PBA. NUEDEXTA has only been studied in a limited number of patients in clinical trials. The data submitted to the FDA and the European Medicines Agency (“EMA”) were obtained in controlled clinical trials of limited duration. In connection with the approval of NUEDEXTA, the FDA and EMA have required that we conduct certain post-approval studies, which include clinical and non-clinical studies. New safety or drug interaction issues may arise from these studies or as NUEDEXTA is used over longer periods of time by a wider group of patients. For example, elderly patients may be more prone to have multiple risk factors for adverse events such as certain cardiac conditions, hepatic or renal insufficiency, or multi-drug treatment regimens. In addition, as we conduct other clinical trials for AVP-923 in other indications, new safety or drug interaction problems may be identified which could negatively impact both our ability to successfully complete these studies and the use and/or regulatory status of NUEDEXTA for the treatment of PBA. New safety or drug interaction issues may result in product liability lawsuits and may require us to, among other things, provide additional warnings and/or restrictions on the NUEDEXTA prescribing information, including a boxed warning, directly alert healthcare providers of new safety information, narrow our approved indications, or alter or terminate current or planned trials for additional uses of AVP-923, any of which could have a significant adverse impact on potential sales of NUEDEXTA and our ability to achieve or maintain profitability.
In addition, if we are required to conduct additional post-approval clinical studies, implement a REMS, or take other similar actions, such requirements or restrictions could have a material adverse impact on our ability to generate revenues from sales of NUEDEXTA, and/or require us to expend significant additional funds.
We have limited capital resources and may need to raise additional funds to support our operations.
We have experienced significant operating losses due to costs associated with funding the research, development, clinical testing and commercialization of NUEDEXTA and our drug candidates. We expect to continue to incur substantial operating losses for the foreseeable future as we continue to expand our commercialization efforts for NUEDEXTA in the U.S. and in European markets, continue to develop AVP-923 and AVP-786, and seek FDA approval and subsequently commercialize AVP-825. Although we had approximately $57.5 million in cash and cash equivalents, restricted cash and cash equivalents, and restricted investments as of September 30, 2013, we currently do not have sufficient revenue from NUEDEXTA or other sources of recurring revenue or cash flow from operations to sustain our operations and it is possible that we may not be able to achieve profitability with our current capital resources.
In light of our substantial long-term capital needs, we may need to partner our rights to NUEDEXTA (either in the U.S. or outside the U.S.) or raise additional capital in the future to finance our long-term operations, until we are able to generate sufficient revenue from product sales to fund our operations. Based on our current loss rate and existing capital resources as of the date of this report, we estimate that we have sufficient funds to sustain our operations at their current and anticipated levels over the next 12 months, which includes the costs associated with the ongoing commercialization of NUEDEXTA for the treatment of PBA in the U.S. and European markets, seeking FDA approval and subsequently commercialize AVP-825. Although we expect to be able to raise additional capital if needed, there can be no assurance that we will be able to do so or that the available terms of any financing would be acceptable to us. If we are unable to raise additional capital to fund future operations, we may experience significant delays or cutbacks in the commercialization of NUEDEXTA and may be forced to further curtail our operations.
If we raise additional capital, we may do so through various financing alternatives, including licensing or sales of our technologies, drugs and/or drug candidates, selling shares of common or preferred stock, through the
15
acquisition of other companies, or through the issuance of debt securities. Each of these financing alternatives carries certain risks.
Raising capital through the issuance of common stock may depress the market price of our stock. Any such financing will dilute our existing stockholders and, if our stock price is relatively depressed at the time of any such offering, the levels of dilution would be greater.
In addition, debt financing may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as encumbering our assets, making capital expenditures or entering into certain licensing transactions.
Our Loan Agreement with Oxford and SVB contains certain covenants that could adversely affect our operations and, if an event of default were to occur, we could be forced to repay the outstanding indebtedness sooner than planned and possibly at a time when we do not have sufficient capital to meet this obligation. The occurrence of any of these events could cause a significant adverse impact on our business, prospects and stock price.
Pursuant to our Loan Agreement with Oxford and SVB, we have pledged all of our assets, other than our patents and other intellectual property rights, and have agreed that we may not sell or assign rights to our patents and other intellectual property without the prior consent of Oxford and SVB. The Loan Agreement also requires us to maintain a minimum sales level relative to projected NUEDEXTA revenues, measured on a trailing three-month basis, or maintain cash and cash equivalents in accounts subject to control agreements in favor of the collateral agent equal to at least 1.5 times the outstanding amount of obligations under the Loan Agreement. The failure to satisfy both of these tests would result in an event of default, which could accelerate our repayment obligations. Additionally, the Loan Agreement contains affirmative and negative covenants that, among other things, restrict our ability to:
| | • | | incur additional indebtedness or guarantees; |
| | • | | make investments, loans and acquisitions; |
| | • | | sell assets, including capital stock of subsidiaries; |
| | • | | alter the business of the Company; |
| | • | | engage in transactions with affiliates; and |
| | • | | enter into agreements limiting dividends and distributions of certain subsidiaries. |
The Loan Agreement also includes events of default, including, among other things, payment defaults; breaches of representations, warranties or covenants; certain bankruptcy events; the occurrence of certain material adverse changes; and a commercial, generic version of NUEDEXTA (for the treatment of PBA) becoming available. Upon the occurrence of an event of default and following any cure periods (if applicable), a default interest rate of an additional 5.0% per annum may be applied to the outstanding loan balances, and the lenders may declare all outstanding obligations immediately due and payable and take such other actions as set forth in the Loan Agreement.
These terms of the Loan Agreement could prevent us from taking certain actions without the consent of our lenders and, if an event of default should occur, we could be required to immediately repay the outstanding indebtedness. If we are unable to repay this debt, the lenders would be able to foreclose on the secured collateral, including our cash accounts, and take other remedies permitted under the security agreement. Even if we are able to repay the indebtedness on an event of default, the repayment of these sums may significantly reduce our working capital and impair our ability to operate as planned. The occurrence of any of these events could cause a significant adverse impact on our business, prospects and stock price.
16
We recently entered into a co-promotion agreement for our institutional sales force that could have negative business implications.
We recently entered into a multi-year agreement involving our institutional sales team. This co-promote agreement involves certain financial and operating risks, including:
| | • | | promotional efforts being diverted to the co-promote product which could result in a negative impact on NUEDEXTA revenues; |
| | • | | increased compliance risk which could harm our reputation; and |
| | • | | if we are unable to generate significant revenue or achieve profitability for the co-promote activity. |
If any of these risks occurred, it could adversely affect our business, financial condition and operating results.
There can be no assurance that the FDA will approve AVP-825 for the treatment of acute migraine.
A Phase II and Phase III clinical trial of AVP-825 for acute treatment of migraine have been completed and will form the basis for a 505(b)(2) NDA filing with the FDA together with previously completed studies with the reference drug, sumatriptan. The FDA and other regulatory authorities will have substantial discretion in evaluating the results of the Phase III clinical trial and our NDA filing.
It is possible that the FDA may require us to conduct additional non-clinical, clinical or chemical manufacturing control-related studies before we gain approval for AVP-825. Prior to approving a new drug, the FDA generally requires that the efficacy of the drug be demonstrated in two adequate and well-controlled clinical trials. In some situations, the FDA approves drugs on the basis of a single well-controlled clinical trial and / or on the basis of referencing data generated previously with the reference drug under the 505(b)(2) application process. If the FDA determines that the clinical trials already conducted do not demonstrate a clinically meaningful benefit and an acceptable safety profile, or do not reflect an acceptable risk-benefit profile or if the FDA requires us to conduct additional clinical trials in order to gain approval, we may incur significant additional development costs and commercialization of AVP-825 would be prevented or delayed and our business would be adversely affected.AVP-825 is classified as a new drug-device combination which requires additional conditions to be satisfied for FDA approval beyond what is required for other drug products.
In addition, this Breath Powered intranasal device has not been previously reviewed or approved by the FDA and therefore, it is possible that other issues may arise during the review process which could delay or preclude the approval and require additional capital investment.
We established a joint steering committee, a joint intellectual property committee and joint development committee which will give OptiNose input on matters related to development of AVP-825 and intellectual property related to the product. As a result, our success depends partially on the success of OptiNose in performing its responsibilities and enforcing their intellectual property rights.
There can be no assurance that we will be able to successfully manufacture, distribute and commercialize AVP-825, including adequate sales, marketing, distribution and manufacturing capabilities. If we are unable to successfully commercialize AVP-825, our ability to generate significant revenue and achieve product launch timelines may be adversely affected.
We are primarily responsible for the manufacturing and distribution of AVP-825. We will utilize third parties to manufacture, package and distribute AVP-825. We may encounter challenges in the purchase of manufacturing equipment or in entering into manufacturing and supply agreements. We have no experience in manufacturing AVP-825 in commercial quantities. Any delays or difficulties in obtaining API or in the manufacturing, packaging or distribution of AVP-825 could negatively affect our sales revenues as well as delay FDA approval.
If AVP-825 is approved by the FDA, our ability to generate significant revenue is entirely dependent upon our ability to commercialize AVP-825. Our future results could be impacted by important factors which include, but are not limited to, commercial market estimates, reliance on market research, competition in the migraine segment, effect of healthcare reform, ability to secure reasonable pricing and patent protection. If we are unable to generate revenues from AVP-825, including through raising awareness among patients and physicians of the benefits of
17
using the device for the treatment of acute migraine, driving higher rates of physician adoption and obtaining reimbursement and third party payer coverage, our ability to generate significant revenue or achieve profitability will be adversely affected.
We may not be able to adequately build or maintain necessary sales, marketing, supply chain management or reimbursement capabilities on our own or enter into arrangements with third parties to perform these functions in a timely manner or on acceptable terms. Additionally, maintaining sales, marketing and distribution capabilities may be more expensive than we anticipate, requiring us to divert capital from other intended purposes or preventing us from building our sales, marketing and distribution capabilities to the desired levels. To be successful we must:
| | • | | recruit and retain adequate numbers of effective sales personnel; |
| | • | | effectively train our sales personnel on AVP-825; |
| | • | | reach an adequate number of health care providers which treat acute migraine; |
| | • | | manage geographically dispersed sales and marketing operations; and |
| | • | | rely on OptiNose to maintain and defend the patent protection and maintain regulatory exclusivity forAVP-825. |
The commercialization of AVP-825 requires us to manage relationships with an increasing number of collaborative partners, suppliers and third-party contractors. If we are unable to successfully establish and maintain the required infrastructure, either internally or through third parties, and successfully manage an increasing number of relationships, we will have difficulty growing our business. In addition, we may enter into co-promotion or out-licensing arrangements with other pharmaceutical or biotechnology partners where necessary to reach customers when deemed strategically and economically advisable. To the extent that we enter into co-promotion or other licensing arrangements, our product revenues are likely to be lower than if we directly marketed and sold AVP-825, and some or all of the revenues we receive will depend upon the efforts of third parties, which may not be successful. If we are unable to develop and maintain adequate sales, marketing and distribution capabilities, independently or with others, we may not be able to generate significant product revenue or become profitable.
We recently entered into a license agreement with obligations that could require significant capital infusions and could involve many financial and operating risks.
In July 2013, we entered into an exclusive license agreement for the development and commercialization of a Breath Powered intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, now named AVP-825. The licensed territories are the United States, Canada and Mexico. Our obligations pursuant to this license agreement could require significant capital infusions and could involve many financial and operating risks, including, but not limited to, the following:
| | • | | we may have to issue debt or equity securities to meet our obligations under this license agreement, which would dilute our stockholders and could adversely affect the market price of our common stock, and we may issue securities or rights with contingent payment obligations, which could have variable accounting treatment and negative accounting consequences; |
| | • | | our obligations pursuant to this license agreement may result in a negative impact on our results of operations and, as such, delay profitability; |
| | • | | we may encounter difficulties in assimilating and integrating AVP-825 into our existing business, including related technologies, personnel or operations; |
| | • | | our obligations pursuant to this license agreement may require significant capital infusions and AVP-825 may not generate sufficient value to justify the acquisition cost; |
| | • | | focus on integrating AVP-825 into our existing business may disrupt our ongoing business, divert resources, increase our expenses and distract our management; and |
| | • | | we have little or no prior experience in the migraine market and our assumptions surrounding the market, including revenue forecasts, may not be accurate. |
18
If any of these risks occurred, it could adversely affect our business, financial condition and operating results.
Our co-promotion agreement with Merck may be terminated with or without cause, which could negatively impact our business.
The co-promotion agreement with Merck may be terminated with cause at any time or without cause upon 90 days written notice at any time after the first year anniversary of the launch date. If the agreement were to be terminated, with or without cause, our business could be negatively impacted, including failure to achieve profitability for the co-promote activity and damage to our reputation.
We are seeking partners to market NUEDEXTA in the EU, and we will be substantially dependent on any such marketing partners in those countries to successfully commercialize NUEDEXTA. We may not be successful in establishing a partnership, but even if we are successful in establishing a partnership or decide to launch ourselves, we may be unable to generate NUEDEXTA revenue in the European market, including through raising PBA awareness among patients and physicians, driving higher rates of physician adoption and obtaining reimbursement and third party payer coverage, our ability to generate significant revenue or achieve profitability in the European market will be adversely affected.
NUEDEXTA has been approved for marketing in the EU and our ability to generate significant revenue in the EU is entirely dependent upon our ability to successfully commercialize NUEDEXTA. To be successful in the EU we must:
| | • | | maintain successful sales, marketing and educational programs for our targeted physicians and other health care providers; |
| | • | | raise patient and physician awareness of PBA and encourage physicians to screen patients for the condition; |
| | • | | obtain adequate reimbursement for NUEDEXTA from a broad range of payers; and |
| | • | | maintain and defend our patent protection and maintain regulatory exclusivity for NUEDEXTA. |
Our prospects to successfully commercialize NUEDEXTA in the EU will depend, among other things, on our ability to establish successful arrangements with international distribution and marketing partners. Consummation of NUEDEXTA partnering arrangements is subject to the negotiation of complex contractual relationships and we may not be able to negotiate such agreements on a timely basis, if at all, or on terms acceptable to us. Where we are successful in entering into these third party arrangements, our revenues from NUEDEXTA sales will be lower than if we commercialized directly, as we will be required to share the revenues with our licensing, commercialization and development partners. If our commercialization efforts with our partners are unsuccessful or we are unable to launch NUEDEXTA in certain countries, we may realize little or no revenue from sales in the EU despite having received marketing approval. In the event that we are unsuccessful in obtaining a partner, we may establish a NUEDEXTA sales and marketing sales infrastructure in the EU.
We are developing AVP-786, a next generation drug product containing deuterium-modified dextromethorphan. It is possible that studies of AVP-786 may not produce favorable results and future studies utilizing AVP-786 carry certain risks.
We have licensed exclusive, worldwide rights to develop and commercialize deuterium-modified dextromethorphan compounds for the potential treatment of neurologic and psychiatric disorders, as well as certain rights to other deuterium-modified dextromethorphan compounds. The goal of the AVP-786 program is to deliver therapeutically effective levels of deuterium-modified dextromethorphan, but with a reduction in the need for an enzyme inhibitor such as quinidine. Although we believe that a drug product containing deuterium-modified dextromethorphan will allow us to significantly reduce the level of quinidine, we are not certain that AVP-786 will have a similar efficacy profile to AVP-923 and, if approved by the FDA, that the reduction in quinidine will result in improved safety language in the package insert.
We completed a pharmacokinetic study with AVP-786 and, based on this data, we believe that we have identified a formulation of AVP-786 with a comparable pharmacokinetic profile, and likely a comparable safety and tolerability profile toAVP-923. In June 2013, the FDA agreed to an expedited development pathway for AVP-786,
19
requiring only a limited non-clinical package as part of the Investigational New Drug application. Although the FDA has agreed to allow us to reference the extensive data generated during the AVP-923 development programs in support of the AVP-786 development programs and regulatory filings, there can be no assurance that we will be successful developing this investigational drug or that we will obtain regulatory approval domestically or internationally. In addition, our initial discussions regarding the development of AVP-786 have been with the FDA’s Division of Neurology. There can be no assurance that other divisions at the FDA will agree with the expedited development plan discussed with the Division of Neurology.
Additionally, we established a joint steering committee and a joint patent committee which will give Concert input on development and patent prosecution for a period of time. As a result, our success depends partially on the success of Concert in performing its responsibilities.
We have licensed or sold most of our non-core drug development programs and related assets and these and other possible future dispositions carry certain risks.
We have entered into agreements for the licensing or sale of our non-core assets, including FazaClo, our anthrax antibody program, and other antibodies in our infectious disease program, as well as docosanol in the U.S. and other markets worldwide. From time to time, we have been and will continue to be engaged in discussions with potential licensing or development partners for NUEDEXTA for the treatment of PBA and/or AVP-923/AVP-786 for other indications and we may choose to pursue a partnership or license involving NUEDEXTA and/orAVP-923/AVP-786 if the terms are attractive. Additionally, we may seek to acquire rights to other drugs or technologies. However, all of these transactions involve numerous risks, including:
| | • | | diversion of management’s attention from normal daily operations of the business; |
| | • | | disputes over earn-outs, working capital adjustments or contingent payment obligations; |
| | • | | inability to effectively integrate an acquired business or asset or to achieve the efficiencies or synergies we anticipate; |
| | • | | insufficient proceeds to offset expenses associated with the transactions; and |
| | • | | the potential loss of key employees following such a transaction. |
Transactions such as these carry significant risks where a large portion of the total consideration is contingent upon post-closing events, such as regulatory, commercialization or sales milestones. We may not have control over whether these milestones are met and, if they are not met, then a potentially large portion of the value of the transaction may not be realized. Disputes may also develop over these and other terms, such as representations and warranties, indemnities, earn-outs, and other provisions in the transaction agreements. If disputes are resolved unfavorably, our financial condition and results of operations may be adversely affected and we may not realize the anticipated benefits from the transactions.
Disputes relating to these transactions can lead to expensive and time-consuming litigation and may subject us to unanticipated liabilities or risks, disrupt our operations, divert management’s attention from day-to-day operations, and increase our operating expenses. See Item 3. “Legal Proceedings” for further discussion relating to a lawsuit filed by Neal R. Cutler, M.D., founder of Alamo Pharmaceuticals, LLC.
The FDA’s safety concerns regarding our prior formulation of NUEDEXTA, known as AVP-923, for the treatment of PBA extend to other clinical indications that we have been pursuing, including DPN pain. Due to these concerns, any future development of AVP-923 or AVP-786 for other indications, including central neuropathic pain in patients with MS, is expected to use an alternative lower-dose quinidine formulation, which may negatively affect efficacy.
We have completed a single Phase III trial for AVP-923 in the treatment of DPN pain. In communications regarding the continued development of AVP-923 for this indication, the FDA has stated that certain safety concerns and questions raised in the PBA approvable letter issued in 2006 necessitate the testing of a low-dose quinidine formulation for the DPN pain indication. Additionally, based on feedback we have received from the FDA on the proposed continued development of AVP-923 for DPN pain, we believe it is likely that two additional large well-controlled Phase III trials would be needed to support a supplemental NDA filing for this indication. Due to our limited
20
capital resources, we do not expect that we will be able to conduct the trials needed for this indication without additional capital, a development partner for AVP-923 for DPN pain, or a commercialization partner for NUEDEXTA. Moreover, although we achieved positive results in our initial Phase III trial, an alternative low-dose quinidine formulation may not yield the same levels of efficacy as seen in the earlier trials or as predicted based on our subsequent pharmacokinetic study. Any decrease in efficacy may be so great that the drug does not demonstrate a statistically significant improvement over placebo or an active comparator. Additionally, any alternative low-dose quinidine formulation that we develop may not sufficiently satisfy the FDA’s safety concerns. If this were to happen, we may not be able to pursue the development of AVP-923 or AVP-786 for DPN pain or other indications, including central neuropathic pain in patients with MS and symptoms of agitation in patients with Alzheimer’s disease, or may need to undertake significant additional clinical trials, which would be costly and cause potentially substantial delays.
If our products infringe the intellectual property rights of others, we may incur damages and be required to incur the expense of obtaining a license.
Even if we successfully secure our intellectual property rights, third parties, including other biotechnology or pharmaceutical companies, may allege that our technology infringes on their rights. Intellectual property litigation is costly, and even if we were to prevail in such a dispute, the cost of litigation could adversely affect our business, financial condition, and results of operations. Litigation is also time consuming and could divert management’s attention and resources away from our operations and other activities. If we were to lose any litigation, in addition to any damages we would have to pay, we could be required to stop the infringing activity or obtain a license. Any required license might not be available to us on acceptable terms, or at all. Some licenses might be non-exclusive, and our competitors could have access to the same technology licensed to us. If we were to fail to obtain a required license or were unable to design around a competitor’s patent, we would be unable to sell or continue to develop some of our products, which would have a material adverse effect on our business, financial condition and results of operations.
We rely on market research to evaluate the commercial acceptance of NUEDEXTA and AVP-825.
Based on the results of our market research, we believe that physicians are likely to continue to support the use and adoption of NUEDEXTA for the treatment of PBA. In addition, we believe that physicians are likely to support and adopt the use of AVP-825 for the treatment of acute migraine, if approved by the FDA. We conduct market research in accordance with Good Marketing Research Practices; however, research findings may not be indicative of the response we might receive from a broader sample of physicians. Moreover, these results are based on physicians’ impressions formed from a description of the product or their actual experience from having prescribed the product, which could result in different impressions or intended behaviors compared to other physicians in our target audience. If the actual use and adoption rates of NUEDEXTA and AVP-825 (if approved by the FDA) are significantly lower than market research or other data suggest, our financial condition and results of operations could be adversely affected.
It is unclear whether we would be eligible for patent term extension in the U.S. and supplementary protection certificates in Europe and we therefore do not know whether our patent term can be extended.
Market exclusivity provisions under the FDCA may also delay the submission or the approval of certain applications for competing product candidates. The FDCA provides three years of non-patent marketing exclusivity for an NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving abbreviated NDAs (ANDA) for drugs containing the original active agent.
Once the three-year FDCA exclusivity period has passed and after the patents (including the patent extension term, if any) that cover NUEDEXTA expire or have been invalidated, generic drug companies would be able to introduce competing versions of the drug. If we are unsuccessful in defending our patents against generic competition, our long-term revenues from NUEDEXTA sales may be significantly less than expected, we may have greater difficulty finding a development partner or licensee for NUEDEXTA and the costs to defend the patents would be significant.
21
In Europe, based on the European Commission’s Article 14(11) of Regulation (EC) No. 726/2004, NUEDEXTA qualifies for ten years of regulatory data protection. Similar to the U.S., market exclusivity provisions provide for a maximum five-year extension for certain patents through the granting of Supplementary Protection Certificates. Although all countries in the European Union are required to provide Supplementary Protection Certificates that come into force after expiry of the patent upon which they are based, no unified cross-recognition exists. Applications for Supplementary Protection Certificates must be filed with each country’s patent office and approved on a country-by-country basis. Although we believe that NUEDEXTA will qualify for this extension and we plan to apply for Supplementary Protection Certificates, we cannot assure you that NUEDEXTA will be granted any Supplementary Protection Certificates nor, if a Supplementary Protection Certificate is granted, that the term of the extension will be five years.
We may be unable to protect our unpatented proprietary technology and information.
In addition to our patented intellectual property, we also rely on trade secrets and confidential information. We may be unable to effectively protect our rights to such proprietary technology or information. Other parties may independently develop or gain access to equivalent technologies or information and disclose it for others to use. Disputes may arise about inventorship and corresponding rights to know-how and inventions resulting from the joint creation or use of intellectual property by us and our corporate partners, licensees, scientific and academic collaborators and consultants. In addition, confidentiality agreements and material transfer agreements we have entered into with these parties and with employees and advisors may not provide effective protection of our proprietary technology or information or, in the event of unauthorized use or disclosure, may not provide adequate remedies. If we fail to protect our trade secrets and confidential information, our business and results of operations could be adversely affected.
We face challenges recruiting and retaining members of management and other key personnel.
The industry in which we compete has a high level of employee mobility and aggressive recruiting of skilled employees. This type of environment creates intense competition for qualified personnel, particularly in commercial, clinical and regulatory affairs, research and development and accounting and finance. Because we have a relatively small management team, the loss of any executive officers, including the Chief Executive Officer, key members of senior management or other key employees, could adversely affect our operations.
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our principal operations are located in Aliso Viejo, California. We depend on our facilities and on our partners, contractors and vendors for the continued operation of our business. Natural disasters or other catastrophic events, interruptions in the supply of natural resources, political and governmental changes, wildfires and other fires, floods, explosions, actions of animal rights activists, earthquakes and civil unrest could disrupt our operations or those of our partners, contractors and vendors. Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors’ insurance policies or for which we or our contractors do not have coverage. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. However, we have a disaster recovery plan in place for our information technology infrastructure that generally allows us to have our critical systems operational in as little as four hours of triggering the disaster recovery plan, depending on the severity of the disaster. Moreover, any such event could adversely impact the commercialization of NUEDEXTA and our research and development programs.
Risks Relating to Our Industry
The pharmaceutical industry is highly competitive and most of our competitors have larger operations and greater resources. As a result, we face significant competitive hurdles.
The pharmaceutical and biotechnology industries are highly competitive and subject to significant and rapid technological change. We compete with hundreds of companies that develop and market products and technologies in areas similar to those in which we are performing our research. For example, we expect that NUEDEXTA may face competition from off-label use of other agents in the treatment of PBA, even though none of these agents has
22
proven to be safe and effective for the treatment of PBA. Additionally, NUEDEXTA may face direct competition from a generic form of NUEDEXTA, if approved, as described above.
Our competitors may have specific expertise and development technologies that are better than ours and many of these companies, which include large pharmaceutical companies, either alone or together with their research partners, have substantially greater financial resources, larger research and development capabilities and substantially greater experience than we do. Accordingly, our competitors may successfully develop competing products. We are also competing with other companies and their products with respect to manufacturing efficiencies and marketing capabilities, areas where we have limited or no direct experience.
Further, AVP-825 will have to compete with existing and any newly developed migraine products or therapies. There are also likely to be numerous competitors developing new products to treat migraine, which could render AVP-825 obsolete or non-competitive.
If we fail to comply with regulatory requirements, regulatory agencies may take action against us, which could significantly harm our business.
Marketed products, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for these products, are subject to continual requirements and review by the FDA, the EMA and other regulatory bodies. In addition, regulatory authorities subject a marketed product, its manufacturer and the manufacturing facilities to ongoing review and periodic inspections. We are subject to ongoing FDA requirements, including required submissions of safety and other post-market information and reports, registration requirements, current Good Manufacturing Practices (“cGMP”) regulations, requirements regarding the distribution of samples to physicians and recordkeeping requirements.
The cGMP regulations also include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. We rely on the compliance by our contract manufacturers with cGMP regulations and other regulatory requirements relating to the manufacture of products. We are also subject to state laws and registration requirements covering the distribution of our products. If we fail to comply with any of these requirements, we may be subject to action by regulatory agencies, which could negatively affect our business.
Regulatory agencies may also change existing requirements or adopt new requirements or policies. We may be slow to adapt or may not be able to adapt to these changes or new requirements.
There are a number of difficulties and risks associated with clinical trials and our trials may not yield the expected results.
There are a number of difficulties and risks associated with conducting clinical trials. For instance, we may discover that a product candidate does not exhibit the expected therapeutic results, may cause harmful side effects or have other unexpected characteristics that may delay or preclude regulatory approval or limit commercial use if approved. It typically takes several years to complete a late-stage clinical trial and a clinical trial can fail at any stage of testing. If clinical trial difficulties or failures arise, our product candidates may never be approved for sale or become commercially viable.
In addition, the possibility exists that:
| | • | | the results from earlier clinical trials may not be predictive of results that will be obtained from subsequent clinical trials, particularly larger trials; |
| | • | | institutional review boards or regulators, including the FDA, may hold, suspend or terminate our clinical research or the clinical trials of our product candidates for various reasons, including noncompliance with regulatory requirements or if, in their opinion, the participating subjects are being exposed to unacceptable health risks; |
| | • | | subjects may drop out of our clinical trials; |
| | • | | our non-clinical studies or clinical trials may produce negative, inconsistent or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical studies or clinical trials; |
23
| | • | | trial results derived from top-line data, which is based on a preliminary analysis of efficacy and safety data related to primary and secondary endpoints, may change following a more comprehensive review of the complete data set derived from a particular clinical trial or may change due to FDA requests to analyze the data differently; |
| | • | | the cost of our clinical trials may be greater than we currently anticipate and clinical trials may take longer than expected to enroll patients and complete, particularly for progressive diseases such as MS where our drug candidates are primarily aimed at treating associated symptoms and not the underlying disease itself; and |
| | • | | there could be a delay in initiating our clinical trials. |
It is possible that earlier clinical and non-clinical trial results may not be predictive of the results of subsequent clinical trials. If earlier clinical and/or non-clinical trial results cannot be replicated or are inconsistent with subsequent results, our development programs may be cancelled or deferred. In addition, the results of these prior clinical trials may not be acceptable to the FDA or similar foreign regulatory authorities because the data may be incomplete, outdated or not otherwise acceptable for inclusion in our submissions for regulatory approval.
Additionally, the FDA has substantial discretion in the approval process and may reject our data, disagree with our interpretations of regulations, draw different conclusions from our clinical trial data or ask for additional information at any time during their review.
Although we would work to be able to fully address any such FDA concerns, we may not be able to resolve all such matters favorably, if at all. Disputes that are not resolved favorably could result in one or more of the following:
| | • | | delays in our ability to submit an NDA; |
| | • | | the refusal by the FDA to accept for filing any NDA we may submit; |
| | • | | requests for additional studies or data; |
| | • | | delays in obtaining an approval; |
| | • | | the rejection of an application; or |
| | • | | the approval of the drug, but with restrictive labeling that could adversely affect the commercial market. |
If we do not receive regulatory approval to sell our product candidates or cannot successfully commercialize our product candidates, we may not be able to generate sufficient revenues or achieve or maintain profitability.
We face uncertainty related to healthcare reform, pricing and reimbursement, which could reduce our revenue.
In the United States, President Obama signed in March 2010 the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, “PPACA”), which is expected to substantially change the way health care is financed by both governmental and private payers. PPACA provides for changes to extend medical benefits to those who currently lack insurance coverage, encourages improvements in the quality of health care items and services, and significantly impacts the U.S. pharmaceutical industry in a number of ways, further listed below. By extending coverage to a larger population, PPACA may substantially change the structure of the health insurance system and the methodology for reimbursing medical services, drugs and devices. These structural changes, as well as other changes that may be made as part of deficit and debt reduction efforts in Congress, could entail modifications to the existing system of private payers and government programs, such as Medicare, Medicaid and State Children’s Health Insurance Program, as well as the creation of a government-sponsored healthcare insurance source, or some combination of both. Such restructuring of the coverage of medical care in the United States could impact the extent of reimbursement for prescribed drugs, including our product candidates, biopharmaceuticals, and medical devices. While the constitutionality of key provisions of the Healthcare Reform Act were recently upheld by the Supreme Court, legislative changes to it remain possible. We expect that the Healthcare Reform Act, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future could have a material adverse effect on our industry in general and on our ability to maintain or increase our product sales, successfully commercialize
24
our product candidates or could limit or eliminate our future spending on development projects. Some of the specific PPACA provisions, among other things:
| | • | | Establish annual, non-deductible fees on any entity that manufactures or imports certain branded prescription drugs and biologics; |
| | • | | Increase minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program; |
| | • | | Extend manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations or extension of statutory rebates to a broader patient population; |
| | • | | Establish a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research; |
| | • | | Require manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50 percent point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and |
| | • | | Revised the definition of average manufacturer price by changing the classes of purchasers included in the calculation, and expanded the entities eligible for discounted 340B pricing. |
A majority of sales of NUEDEXTA come from institutional settings. Any adverse change in reimbursement policy affecting patients, providers and payers in institutional settings could have a material and adverse impact on our business.
If future reimbursement for NUEDEXTA or any other approved product candidates, if any, is substantially less than we project, or rebate obligations associated with them are substantially increased, our business could be materially and adversely impacted.
Sales of NUEDEXTA for treatment of patients with PBA will depend in part on the availability of coverage and reimbursement from third-party payers such as government insurance programs, including Medicare and Medicaid, private health insurers, health maintenance organizations and other health care related organizations. Accordingly, coverage and reimbursement may be uncertain. Adoption of NUEDEXTA by the medical community may be limited if third-party payers will not offer coverage. Cost control initiatives may decrease coverage and payment levels for NUEDEXTA and, in turn, the price that we will be able to charge. We are unable to predict all changes to the coverage or reimbursement methodologies that will be applied by private or government payers to NUEDEXTA. Any denial of private or government payer coverage of NUEDEXTA could harm our business and reduce our revenue.
In addition, both the federal and state governments in the United States and foreign governments continue to propose and pass new legislation affecting coverage and reimbursement policies, which are designed to contain or reduce the cost of health care, as well as hold public hearings on these matters, which has resulted in certain private companies dropping the prices of their drugs. Further federal and state proposals and healthcare reforms are likely, which could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. There may be future changes that result in reductions in current coverage and reimbursement levels for our products, if commercialized, and we cannot predict the scope of any future changes or the impact that those changes would have on our operations.
If we fail to obtain regulatory approval in foreign jurisdictions, we would not be able to market our products abroad and our revenue prospects would be limited.
We are seeking to have our products or product candidates marketed outside the United States. In order to market our products in the EU and many other foreign jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and jurisdictions and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval processes may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all, and may not qualify or be accepted for accelerated review in foreign countries. For example, our development partner in Japan encountered
25
significant difficulty in seeking approval of docosanol in that country and was forced to abandon efforts to seek approval in that country. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
We rely on insurance companies to mitigate our exposure for business activities, including developing and marketing pharmaceutical products for human use.
The conduct of our business, including the testing, marketing and sale of pharmaceutical products, involves the risk of liability claims by consumers, stockholders, and other third parties including products in which we co-promote. Although we maintain various types of insurance, including product liability and director and officer liability, claims can be high and our insurance may not sufficiently cover our actual liabilities. If liability claims were made against us, it is possible that our insurance carriers may deny, or attempt to deny, coverage in certain instances. If a lawsuit against us is successful, then the lack or insufficiency of insurance coverage could materially and adversely affect our business and financial condition. Furthermore, various distributors of pharmaceutical products require minimum product liability insurance coverage before their purchase or acceptance of products for distribution. Failure to satisfy these insurance requirements could impede our ability to achieve broad distribution of our products and the imposition of higher insurance requirements could impose additional costs on us. Additionally, we are potentially at risk if our insurance carriers become insolvent. Although we have historically obtained coverage through highly rated and capitalized firms, there can be no assurance that we will be able to maintain coverage under existing policies at the current rates or purchase insurance under new policies at reasonable rates.
If we market products in a manner that violates health care fraud and abuse laws, we may be subject to civil and criminal penalties, which may adversely affect our business, financial condition and results of operations.
We are subject to healthcare fraud and abuse regulation and enforcement by both the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include:
| | • | | the federal healthcare programs’ Anti-Kickback Law (as amended by the PPACA, which modified the intent requirement of the Anti-Kickback Law so that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it), which prohibits, among other things, persons from soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
| | • | | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent and, under the PPACA, the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes; |
| | • | | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; and |
| | • | | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payer, including commercial insurers. |
In addition, the compliance environment is changing as some states mandate implementation of commercial compliance programs to ensure compliance with these laws. The PPACA also imposes new reporting and disclosure requirements on drug manufacturers for any “transfer of value” made or distributed to prescribers and other healthcare providers and such information will be made publicly available in a searchable format. Drug manufacturers are required to begin collecting required information on August 1, 2013 and to submit reports disclosing any investment interests held by physicians and their immediate family members from August through December of 2013 by March 31, 2014. Failure to submit required information may result in civil monetary penalties
26
of up to an aggregate of $150,000 per year (and up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests not reported in an annual submission. PPACA also requires pharmaceutical manufacturers and distributors to provide the U.S. Department of Health and Human Services with an annual report on the drug samples they provide to physicians. There has also been a recent trend of increased federal and state regulation of payments made to physicians for marketing, including the tracking and reporting of gifts, compensation, and other remuneration to physicians. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements increases the possibility that a healthcare company may run afoul of one or more of the requirements.
If our operations are found to be in violation of any of the laws described above or any other domestic or foreign laws or governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, exclusion from governmental health care programs, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Even if we are not found to be in violation of any of the laws described above or any other domestic or foreign laws or governmental regulations that apply to us, any action against us for violation of these laws could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
We may incur significant liability if it is determined that we are promoting the “off-label” use of drugs.
We have taken numerous steps to ensure compliance is a high priority throughout the organization and we believe that our communications regarding our products are in compliance with the relevant regulatory requirements. However, the FDA or another regulatory authority may disagree.
Responding to government investigations, defending any claims raised, and any resulting fines, restitution, damages and penalties, settlement payments or administrative actions, as well as any related actions brought by stockholders or other third parties, could have a material impact on our reputation, business and financial condition and divert the attention of our management from operating our business. Our distribution and contracting partners may also be the subject of regulatory investigations involving, or remedies or sanctions for, off-label promotion of products we have licensed to them, or for which they provide vendor support services, which may have an adverse impact on sales of such licensed products, or indemnification obligations, which may, in turn, have a material adverse effect on our business, financial condition and results of operations and could cause the market value of our common shares to decline. The risks of a regulatory investigation are increased by the practice of some stock market participants to publicly issue reports alleging compliance violations. A report of this nature was recently issued regarding the Company’s practices.
Companies may not promote drugs for “off-label” uses — that is, uses that are not described in the product’s labeling and that differ from those approved by the FDA or other applicable regulatory agencies. Physicians may prescribe drug products for off-label uses, and such off-label uses are common across medical specialties. Although the FDA and other regulatory agencies do not regulate a physician’s choice of treatments for a given medical condition, the FDA and other regulatory agencies do restrict communications by pharmaceutical companies and their sales representatives regarding dissemination of information concerning off-label use. The FDA and other regulatory agencies actively enforce regulations prohibiting promotion of product for off-label uses and the promotion of products for which marketing authorization has not been obtained. A company that is found to have promoted product for off-label uses may be subject to significant liability, including civil and administrative remedies as well as criminal sanctions. Notwithstanding the regulatory restrictions on off-label promotion, the FDA and other regulatory authorities allow companies to engage in truthful, non-misleading, and non-promotional scientific exchange with health care professionals concerning their products.
27
Risks Related to Reliance on Third Parties
We depend on third parties to manufacture, package and distribute compounds for our drugs and drug candidates. The failure of these third parties to perform successfully could harm our business.
We have utilized, and intend to continue utilizing, third parties to manufacture, package and distribute NUEDEXTA and the active pharmaceutical ingredient (“API”) for docosanol 10% cream and to provide clinical supplies of our drug candidates. We will also utilize third parties to manufacture, package and distribute AVP-825. We have no experience in manufacturing and do not have any manufacturing facilities. Currently, we have sole suppliers for the API for docosanol and NUEDEXTA, and a sole manufacturer for the finished form of NUEDEXTA. In addition, these materials are custom-made and available from only a limited number of sources. In particular, there may be a limited supply source for APIs in NUEDEXTA. Although we maintain a significant supply of APIs on hand, any sustained disruption in this supply could adversely affect our operations and revenues. Any material disruption in manufacturing could cause a delay in shipments and possible loss of sales. We do not have any long-term agreements in place with our current docosanol supplier or NUEDEXTA API suppliers. If we are required to change manufacturers, we may experience delays associated with finding an alternate manufacturer that is properly qualified to produce supplies of our products and product candidates in accordance with FDA requirements and our specifications. Any delays or difficulties in obtaining APIs or in manufacturing, packaging or distributing NUEDEXTA could negatively affect our sales revenues, as well as delay our clinical trials of AVP-923 for DPN pain, MS-related pain or other potential indications. The third parties we rely on for manufacturing and packaging are also subject to regulatory review, and any regulatory compliance problems with these third parties could significantly delay or disrupt our commercialization activities. Additionally, macro-economic conditions may adversely affect these third parties, causing them to suffer liquidity or operational problems. If a key third party vendor becomes insolvent or is forced to lay off workers assisting with our projects, our results and development timing could suffer.
Because we depend on clinical research centers and other contractors for clinical testing and for certain research and development activities, the results of our clinical trials and such research activities are, to a certain extent, beyond our control.
The nature of clinical trials and our business strategy of outsourcing a substantial portion of our research require that we rely on clinical research centers and other contractors to assist us with research and development, clinical testing activities, patient enrollment and regulatory submissions to the FDA. As a result, our success depends partially on the success of these third parties in performing their responsibilities. Although we pre-qualify our contractors and we believe that they are fully capable of performing their contractual obligations, we cannot directly control the adequacy and timeliness of the resources and expertise that they apply to these activities. Additionally, macro-economic conditions may affect our development partners and vendors, which could adversely affect their ability to timely perform their tasks. If our contractors do not perform their obligations in an adequate and timely manner, the pace of clinical development, regulatory approval and commercialization of our drug candidates could be significantly delayed and our prospects could be adversely affected.
We generally do not control the development of compounds licensed to third parties and, as a result, we may not realize a significant portion of the potential value of any such license arrangements.
Under our typical out-license arrangement we have no direct control over the development of drug candidates and have only limited, if any, input on the direction of development efforts. These development efforts are made by our licensing partner, and if the results of their development efforts are negative or inconclusive, it is possible that our licensing partner could elect to defer or abandon further development of these programs. We similarly rely on licensing partners to obtain regulatory approval for docosanol in foreign jurisdictions. Because much of the potential value of these license arrangements is contingent upon the successful development and commercialization of the licensed technology, the ultimate value of these licenses will depend on the efforts of licensing partners. If our licensing partners do not succeed in developing the licensed technology for whatever reason, or elect to discontinue the development of these programs, we may be unable to realize the potential value of these arrangements. If we were to license NUEDEXTA to a third party or a development partner, it is likely that much of the long-term success of that drug will similarly depend on the efforts of the licensee.
28
We expect to rely entirely on third parties for international registration, sales and marketing efforts.
In the event that we attempt to enter into international markets, in some instances we expect to rely on collaborative partners to obtain regulatory approvals and to market and sell our product(s) in those markets. We have not yet entered into any collaborative arrangement with respect to marketing or selling NUEDEXTA, with the exception of one such agreement relating to Israel. We may be unable to enter into any other arrangements on terms favorable to us, or at all, and even if we are able to enter into sales and marketing arrangements with collaborative partners, we cannot assure you that their sales and marketing efforts will be successful. If we are unable to enter into favorable collaborative arrangements with respect to marketing or selling NUEDEXTA in international markets, or if our collaborators’ efforts are unsuccessful, our ability to generate revenues from international product sales will suffer.
Risks Relating to Our Stock
Our stock price has historically been volatile and we expect that this volatility will continue for the foreseeable future.
The market price of our common stock has been, and is likely to continue to be, highly volatile. This volatility can be attributed to many factors independent of our operating results, including the following:
| | • | | comments made by securities analysts, including changes in their recommendations; |
| | • | | short selling activity by certain investors, including any failures to timely settle short sale transactions; |
| | • | | announcements by us of financing transactions and/or future sales of equity or debt securities; |
| | • | | sales of our common stock by our directors, officers or significant stockholders, including sales effected pursuant to predetermined trading plans adopted under the safe-harbor afforded by Rule 10b5-1; |
| | • | | negative opinions that are misleading and inaccurate regarding our business, management or future prospects published by certain market participants intent on putting downward pressure on our stock price; |
| | • | | regulatory developments in the U.S. and foreign countries, including the passage of laws, rules or regulations relating to healthcare and reimbursement or the public announcement of inquiries relating to these subjects; |
| | • | | lack of volume of stock trading leading to low liquidity; and |
| | • | | market and economic conditions. |
If a substantial number of shares are sold into the market at any given time, particularly following any significant announcements or large swings in our stock price (whether sales are under our existing “shelf” registration statements, from an existing stockholder, or the result of warrant or stock options exercised), there may not be sufficient demand in the market to purchase the shares without a decline in the market price for our common stock. Moreover, continuous sales into the market of a number of shares in excess of the typical trading volume for our common stock, or even the availability of such a large number of shares, could depress the trading market for our common stock over an extended period of time.
Additionally, our stock price has been volatile as a result of announcements of regulatory actions and decisions relating to NUEDEXTA, and periodic variations in our operating results. We expect that our operating results will continue to vary from quarter to quarter, particularly as we continue to market NUEDEXTA and report sales results. Our operating results and prospects may also vary depending on the status of our partnering arrangements.
As a result of these factors, we expect that our stock price may continue to be volatile and investors may be unable to sell their shares at a price equal to, or above, the price paid. Additionally, any significant drops in our stock price could give rise to stockholder lawsuits, which are costly and time consuming to defend against and which may adversely affect our ability to raise capital while the suits are pending, even if the suits are ultimately resolved in our favor. We have, from time to time, been a party to such suits and although none have been material to date, there can be no assurance that any such suit will not have an adverse effect on us.
29
If our internal controls over financial reporting are not considered effective, our business and stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us to evaluate the effectiveness of our internal controls over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal controls over financial reporting in our annual report on Form 10-K for that fiscal year. Section 404 also requires our independent registered public accounting firm to attest to, and report on, management’s assessment of our internal controls over financial reporting.
Our management, including our chief executive officer and principal financial officer, does not expect that our internal controls over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud involving a company have been, or will be, detected. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and we cannot assure you that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become ineffective because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. We cannot assure you that we or our independent registered public accounting firm will not identify a material weakness in our internal controls in the future. A material weakness in our internal controls over financial reporting would require management and our independent registered public accounting firm to consider our internal controls as ineffective. If our internal controls over financial reporting are not considered effective, we may experience a loss of public confidence, which could have an adverse effect on our business and on the market price of our common stock.
Because we do not expect to pay dividends in the foreseeable future, you must rely on stock appreciation for any return on your investment.
We have paid no cash dividends on our common stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, we do not expect to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. In addition, under the terms of our Loan Agreement, we are precluded from paying cash dividends without the prior written consent of the lenders. Accordingly, the success of your investment in our common stock will likely depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value or even maintain the price at which you purchased your shares, and you may not realize a return on your investment in our common stock.
Our corporate governance documents and Delaware law may delay or prevent an acquisition of us that stockholders may consider favorable, which could decrease the value of our common stock.
Our certificate of incorporation and bylaws and Delaware law contain provisions that could make it more difficult for a third party to acquire us without the consent of our board of directors. These provisions include restrictions on the ability of our stockholders to remove directors and supermajority voting requirements for stockholders to amend our organizational documents and a classified board of directors. In addition, our board of directors has the right to issue preferred stock without stockholder approval, which could be used to dilute the stock ownership of a potential hostile acquirer. Delaware law, for instance, also imposes some restrictions on mergers and other business combinations between any holder of 15% or more of our outstanding common stock and us. Although we believe these provisions protect our stockholders from coercive or otherwise unfair takeover tactics and thereby provide for an opportunity to receive a higher bid by requiring potential acquirers to negotiate with our board of directors, these provisions apply even if the offer may be considered beneficial by some stockholders.
30
| Item 1B. | Unresolved Staff Comments |
None.
We recently entered into a lease for approximately 61,000 square feet in Aliso Viejo, California, which will become our headquarters, commercial and administrative offices. This lease expires in December 2020.
Currently our headquarters, commercial and administrative offices are also located in Aliso Viejo, California, where we occupy approximately 30,000 square feet. Additionally, we have approximately 8,000 square feet of unoccupied space for future expansion. This Aliso Viejo office lease expires in April 2018. Once we have relocated to our new offices, our intention is to sublet the 30,000 square feet of our current offices.
NUEDEXTA ANDA Litigation
In fiscal 2011 and 2012, we received Paragraph IV certification notices from five separate companies contending that certain of our patents listed in the FDA’s publication, “Approved Drug Products with Therapeutic Equivalence Evaluation” (“FDA Orange Book”) (U.S. Patents 7,659,282 (“’282 Patent”), 8,227,484 (“’484 Patent”) and RE 38,115 (“’115 Patent”), which expire in August 2026, July 2023 and January 2016, respectively) are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of NUEDEXTA as described in those companies’ ANDAs. The FDA Orange Book provides potential competitors, including generic drug companies, with a list of issued patents covering approved drugs. In August 2011 and March 2012, we filed lawsuits in the U.S. District Court for the District of Delaware against Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc. (collectively “Par”), Actavis South Atlantic LLC and Actavis, Inc. (collectively “Actavis”), Wockhardt USA, LLC and Wockhardt, Ltd. (collectively, “Wockhardt”), Impax Laboratories, Inc. (“Impax”) and Watson Pharmaceuticals, Inc., Watson Laboratories, Inc. and Watson Pharma, Inc. (collectively “Watson”) (Par, Actavis, Wockhardt, Impax and Watson, collectively the “Defendants”). In March 2012, we also filed a protective suit in the U.S. District Court for the District of New Jersey against Watson. The New Jersey suit was voluntarily dismissed by us in May 2012. In September and October 2012, we filed lawsuits in the U.S. District Court for the District of Delaware against the Defendants. All lawsuits (collectively, the “ANDA Actions”) were filed on the basis that the Defendants’ submissions of their respective ANDAs to obtain approval to manufacture, use, sell, or offer for sale generic versions of NUEDEXTA prior to the expiration of the ’282 Patent, the ’484 Patent and the ’115 Patent listed in the FDA Orange Book constitute infringement of one or more claims of those patents. On October 31, 2012, Watson announced the divestiture of its ANDA for a generic form of NUEDEXTA to Sandoz, Inc. As a result of Sandoz’ acquisition and maintenance of said ANDA, on May 30, 2013, we filed suit in the U.S. District Court for the District of Delaware against Sandoz. This suit was filed on the basis that Sandoz’ ANDA to obtain approval to manufacture, use, sell, or offer for sale generic versions of NUEDEXTA prior to the expiration of the ’282 Patent, the ’484 Patent and the ’115 Patent listed in the FDA Orange Book constitutes infringement of one or more claims of those patents.
On December 3, 2012, we received a Memorandum Opinion and Order (the “Order”) issued by Judge Leonard P. Stark of the United States District Court for the District of Delaware (the “Court”) related to the Markman hearing held October 5, 2012 in our ongoing patent infringement case against the Defendants. The Order establishes the meaning of patent claim terms in dispute between the parties. After comprehensive briefing and oral argument, Judge Stark’s Order adopted our proposed or stipulated patent term constructions.
A bench trial was held in September 2013 and concluded on October 15, 2013. We are currently awaiting Judge Stark’s decision.
On August 9, August 30, and September 6, 2013, we entered into settlement agreements with Sandoz, Actavis and Wockhardt, respectively, to resolve pending patent litigation in response to their ANDAs seeking approval to market generic versions of NUEDEXTA capsules. The settlement agreements grant Sandoz, Actavis and Wockhardt the right to begin selling a generic version of NUEDEXTA on July 30, 2026, or earlier under certain circumstances. The parties also filed stipulations and orders of dismissal with the United States District Court for
31
the District of Delaware which conclude the litigation with respect to Sandoz, Actavis and Wockhardt. The settlements do not end our ongoing litigation against the other two ANDA filers.
As a result of the 30-month stay associated with the filing of the ANDA Actions, the FDA cannot grant final approval to any ANDA before December 30, 2013, unless there is an earlier court decision holding that the Orange Book-listed ’282, ’484 and ’115 patents are not infringed and/or are invalid. We intend to vigorously enforce our intellectual property rights relating to NUEDEXTA, but we cannot predict the outcome of these matters.
Alamo and Azur Litigation
In October 2011, Neal R. Cutler, M.D., the founder of Alamo Pharmaceuticals, LLC (“Alamo”), filed a lawsuit in California Superior Court against Azur Pharma International III Limited and Azur Pharma Limited (collectively, “Azur”) and Avanir (the “Cutler Action”). We purchased Alamo in 2006 to acquire rights to FazaClo, an approved anti-psychotic drug. In connection with this acquisition of Alamo, we agreed to provide the Alamo equity holders, including Dr. Cutler, with milestone payments tied to the aggregate net revenues of FazaClo. In 2007, we sold FazaClo and its related assets and operations to Azur. In connection with this sale, Azur agreed to assume the milestone payment obligations due to Dr. Cutler under the Alamo purchase agreement, although we may remain liable for these payments if Azur defaults on these obligations. In the Cutler Action, Dr. Cutler alleges a breach of contract and a breach of implied covenant of good faith and fair dealing. Dr. Cutler alleges that Azur has failed to make certain sales information available to Dr. Cutler and has withheld payments to which Dr. Cutler is entitled. Dr. Cutler alleges that Avanir has acquiesced in this conduct, and Dr. Cutler seeks to hold both Azur and Avanir liable for these actions. Azur Ltd. and Jazz Pharmaceuticals, Inc. entered into a business combination creating Jazz Pharmaceuticals, plc (“Jazz”). As successor in interest to Azur Ltd., Jazz agreed to indemnify Avanir in full for the claims asserted in the Cutler Action.
General and Other
In the ordinary course of business, we may face various claims brought by third parties and we may, from time to time, make claims or take legal actions to assert our rights, including intellectual property rights as well as claims relating to employment matters and the safety or efficacy of our products. Any of these claims could subject us to costly litigation and, while we generally believe that we have adequate insurance to cover many different types of liabilities, our insurance carriers may deny coverage, may be inadequately capitalized to pay on valid claims, or our policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on our consolidated operations, cash flows and financial position. Additionally, any such claims, whether or not successful, could damage our reputation and business. Management believes the outcome of currently pending claims or lawsuits will not likely have a material effect on our operations or financial position.
| Item 4. | Mine Safety Disclosures |
Not applicable.
32
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
The following table sets forth the high and low prices for our common stock in each of the quarters over the past two fiscal years, as quoted on the NASDAQ Global Market.
| | | | | | | | | | | | | | | | |
| | | Common Stock Price | |
| | | Fiscal 2013 | | | Fiscal 2012 | |
| | | High | | | Low | | | High | | | Low | |
First Quarter | | $ | 3.57 | | | $ | 2.07 | | | $ | 3.25 | | | $ | 1.77 | |
Second Quarter | | $ | 3.32 | | | $ | 2.65 | | | $ | 3.56 | | | $ | 2.00 | |
Third Quarter | | $ | 4.78 | | | $ | 2.60 | | | $ | 4.05 | | | $ | 2.65 | |
Fourth Quarter | | $ | 6.00 | | | $ | 3.98 | | | $ | 4.05 | | | $ | 2.76 | |
On December 2, 2013, the closing sales price of our common stock was $4.46 per share.
As of December 2, 2013, we had approximately 23,701 stockholders, including 376 holders of record and an estimated 23,325 beneficial owners. We have not paid any dividends on our common stock since our inception and do not expect to pay dividends on our common stock in the foreseeable future.
The following graph compares the cumulative 5-year return provided to a hypothetical stockholder in Avanir Pharmaceuticals, Inc.’s common stock relative to the cumulative total returns of the NASDAQ Composite index and the NASDAQ Biotechnology index. An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our common stock and in each of the indexes on October 1, 2008 and its relative performance is tracked through September 30, 2013.
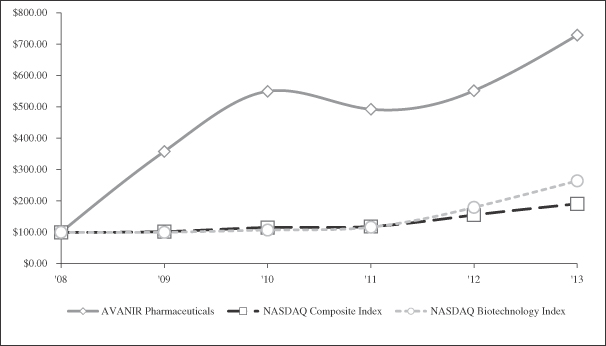
33
Information About Our Equity Compensation Plans
Information regarding our equity compensation plans is incorporated by reference to Item 12, “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” of Part III of this annual report on Form 10-K.
| Item 6. | Selected Financial Data |
The selected consolidated financial data set forth below at September 30, 2013 and 2012, and for the fiscal years ended September 30, 2013, 2012, and 2011, are derived from our audited consolidated financial statements included elsewhere in this report. This information should be read in conjunction with those consolidated financial statements, the notes thereto, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The selected consolidated financial data set forth below at September 30, 2011, 2010 and 2009, and for the years ended September 30, 2010 and 2009, are derived from our audited consolidated financial statements that are contained in reports previously filed with the SEC, not included herein. The quarterly consolidated financial data are derived from unaudited consolidated financial statements included in our Quarterly Reports on Form 10-Q.
The following tables include selected consolidated financial data for each of our last five fiscal years and quarterly data for the last two fiscal years. Operating expense includes cost of product sales, cost of research and development services, and cost of government research grant services for all periods presented.
Summary Financial Information
| | | | | | | | | | | | | | | | | | | | |
| | | Fiscal Years Ended September 30, | |
Statement of operations data: | | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
Total revenues | | $ | 75,365,884 | | | $ | 41,275,073 | | | $ | 10,495,895 | | | $ | 2,895,474 | | | $ | 4,176,509 | |
Operating expenses(1) | | $ | 146,792,584 | | | $ | 99,677,254 | | | $ | 71,125,310 | | | $ | 29,992,198 | | | $ | 26,101,174 | |
Loss from operations | | $ | (71,426,700 | ) | | $ | (58,402,181 | ) | | $ | (60,629,415 | ) | | $ | (27,096,724 | ) | | $ | (21,924,665 | ) |
Net loss | | $ | (75,475,863 | ) | | $ | (59,743,827 | ) | | $ | (60,631,563 | ) | | $ | (26,694,148 | ) | | $ | (21,996,016 | ) |
Basic and diluted net loss per share: | | $ | (0.53 | ) | | $ | (0.45 | ) | | $ | (0.51 | ) | | $ | (0.30 | ) | | $ | (0.28 | ) |
Basic and diluted weighted average number of shares of common stock outstanding | | | 142,269,399 | | | | 133,358,571 | | | | 119,405,230 | | | | 87,614,420 | | | | 78,844,251 | |
| (1) | Includes an up-front payment of $20.0 million for the in-licensing of AVP-825. |
| | | | | | | | | | | | | | | | | | | | |
| | | September 30, | |
Balance sheet data: | | 2013 | | | 2012 | | | 2011 | | | 2010 | | | 2009 | |
Cash and cash equivalents | | $ | 55,259,073 | | | $ | 69,778,406 | | | $ | 79,542,564 | | | $ | 38,771,469 | | | $ | 31,486,012 | |
Restricted cash and cash equivalents and restricted investments | | $ | 2,269,924 | | | $ | 2,356,599 | | | $ | 2,252,939 | | | $ | 601,550 | | | $ | 468,475 | |
Total cash, cash equivalents, restricted cash and cash equivalents, and restricted investments | | $ | 57,528,997 | | | $ | 72,135,005 | | | $ | 81,795,503 | | | $ | 39,373,019 | | | $ | 31,954,487 | |
Working capital | | $ | 37,051,425 | | | $ | 60,596,300 | | | $ | 70,200,594 | | | $ | 32,967,188 | | | $ | 26,685,567 | |
Total assets | | $ | 76,079,007 | | | $ | 86,011,796 | | | $ | 89,648,994 | | | $ | 42,141,198 | | | $ | 34,068,072 | |
Deferred revenues | | $ | 1,288,514 | | | $ | 4,049,318 | | | $ | 7,791,416 | | | $ | 8,476,831 | | | $ | 9,912,367 | |
Notes payable, net of debt discount | | $ | 29,365,108 | | | $ | 28,860,526 | | | $ | — | | | $ | — | | | $ | — | |
Total liabilities | | $ | 57,607,453 | | | $ | 49,174,689 | | | $ | 18,309,330 | | | $ | 14,134,642 | | | $ | 14,126,337 | |
Stockholders’ equity | | $ | 18,471,554 | | | $ | 36,837,107 | | | $ | 71,339,664 | | | $ | 28,006,556 | | | $ | 19,941,735 | |
34
| | | | | | | | | | | | | | | | |
Quarterly statement of operations data for fiscal 2013 (Unaudited): | | Fiscal Quarters Ended | |
| | December 31,
2012 | | | March 31, 2013 | | | June 30, 2013 | | | September 30,
2013 | |
Total revenues | | $ | 16,520,272 | | | $ | 17,434,188 | | | $ | 19,758,662 | | | $ | 21,652,762 | |
Operating expenses(1) | | $ | 27,556,440 | | | $ | 32,916,906 | | | $ | 30,168,785 | | | $ | 56,150,453 | |
Loss from operations | | $ | (11,036,168 | ) | | $ | (15,482,718 | ) | | $ | (10,410,123 | ) | | $ | (34,497,691 | ) |
Net loss | | $ | (12,076,082 | ) | | $ | (16,526,701 | ) | | $ | (11,424,630 | ) | | $ | (35,448,450 | ) |
Basic and diluted net loss per share | | $ | (0.09 | ) | | $ | (0.12 | ) | | $ | (0.08 | ) | | $ | (0.24 | ) |
Basic and diluted weighted average number of common shares outstanding | | | 136,772,557 | | | | 139,173,746 | | | | 145,244,796 | | | | 147,851,544 | |
| (1) | Includes an up-front payment of $20.0 million for the in-licensing of AVP-825. |
| | | | | | | | | | | | | | | | |
Quarterly statement of operations data for fiscal 2012 (Unaudited): | | Fiscal Quarters Ended | |
| | December 31,
2011 | | | March 31, 2012 | | | June 30, 2012 | | | September 30,
2012 | |
Total revenues | | $ | 7,164,883 | | | $ | 10,040,388 | | | $ | 10,537,996 | | | $ | 13,531,806 | |
Operating expenses | | $ | 23,129,337 | | | $ | 27,090,504 | | | $ | 25,250,002 | | | $ | 24,207,411 | |
Loss from operations | | $ | (15,964,454 | ) | | $ | (17,050,116 | ) | | $ | (14,712,006 | ) | | $ | (10,675,605 | ) |
Net loss | | $ | (15,946,157 | ) | | $ | (17,042,159 | ) | | $ | (15,030,214 | ) | | $ | (11,725,297 | ) |
Basic and diluted net loss per share | | $ | (0.12 | ) | | $ | (0.13 | ) | | $ | (0.11 | ) | | $ | (0.09 | ) |
Basic and diluted weighted average number of common shares outstanding | | | 127,934,257 | | | | 133,463,300 | | | | 135,825,005 | | | | 136,239,668 | |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
Except for the historical information contained herein, the matters set forth in this Annual Report on Form 10-K, including statements regarding the Company’s plans, potential opportunities, financial or other expectations, projections, goals objectives, milestones, strategies, market growth, timelines, legal matters, product pipeline, clinical studies, product development and the potential benefits of its commercialized products and products under development are forward-looking statements within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from the anticipated or estimated future results, including the risks and uncertainties associated with Avanir’s future operating performance and financial position, developments in Avanir’s ongoing NUEDEXTA patent litigation, the market demand for and acceptance of Avanir’s products domestically and internationally, research, development and commercialization of new products domestically and internationally, obtaining and maintaining regulatory approvals domestically and internationally, including, but not limited to potential regulatory delays or rejections in the filing or acceptance of the Marketing Authorization Application, uncertainty regarding use of the data package which served as the basis for the U.S. FDA approval, risks associated with meeting the objectives of clinical studies, including, but not limited to, delays or failures in enrollment, and the occurrence of adverse safety events, and other risks set forth below under Item 1A, “Risk Factors” and other documents subsequently filed with or furnished to the Securities and Exchange Commission. These forward-looking statements are based on current information that may change and you are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K. All forward-looking statements are qualified in their entirety by this cautionary statement, and the Company undertakes no obligation to revise or update any forward-looking statement to reflect events or circumstances after the date hereof. Except as otherwise indicated herein, all dates referred to in this report represent periods or dates fixed with reference to the calendar year, rather than our fiscal year ending September 30.
Executive Overview
We are a biopharmaceutical company focused on acquiring, developing and commercializing novel therapeutic products for the treatment of central nervous system disorders. Our lead product NUEDEXTA® (referred to as
35
AVP-923 during clinical development) is a first-in-class dual NMDA receptor antagonist and sigma-1 agonist. NUEDEXTA, 20/10 mg, is approved in the United States for the treatment of pseudobulbar affect (“PBA”). It is also approved for PBA in the European Union in two dose strengths, NUEDEXTA 20/10 mg and NUEDEXTA 30/10 mg.
The table below shows quarterly gross product sales and dispensed prescription data for NUEDEXTA over the past two years.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Three Months Ended | |
| | | December 31,
2011 | | | March 31,
2012(1) | | | June 30,
2012 | | | September 30,
2012 | | | December 31,
2012 | | | March 31,
2013 | | | June 30,
2013 | | | September 30,
2013 | |
Gross product sales | | $ | 6,285,631 | | | $ | 11,185,298 | | | $ | 12,205,811 | | | $ | 15,431,249 | | | $ | 18,373,831 | | | $ | 20,849,581 | | | $ | 24,315,422 | | | $ | 27,666,348 | |
Total dispensed units (capsules) | | | 746,343 | | | | 1,012,777 | | | | 1,332,982 | | | | 1,584,263 | | | | 1,727,356 | | | | 1,839,842 | | | | 2,334,898 | | | | 2,606,289 | |
Percentage growth over previous quarter | | | 45 | % | | | 36 | % | | | 32 | % | | | 19 | % | | | 9 | % | | | 7 | % | | | 27 | % | | | 12 | % |
| (1) | Includes a one-time adjustment to gross product sales of approximately $1.9 million as a result of a change in accounting estimate. |
We are studying the clinical utility of AVP-923 in other mood/behavior disorders, movement disorders and pain.We have two ongoing Phase II clinical trials exploring the potential treatment of agitation in patients with Alzheimer’s disease and potential treatment of levodopa-induced dyskinesia in Parkinson’s disease (the “LID study”). The LID study is supported by a grant from the Michael J. Fox Foundation. In December 2013, we completed a Phase II clinical trial of AVP-923 for the potential treatment of central neuropathic pain in patients with multiple sclerosis (“MS”). The data from this Phase II clinical trial did not meet the primary efficacy endpoint. In the study, AVP-923 treated patients experienced levels of pain reduction commensurate with those observed in similar studies and the improvement in pain scores from baseline reached statistical significance; however, there was no difference between the treatment arms and placebo. We believe that a higher than expected placebo response negatively impacted the study results. AVP-923 was generally safe and well tolerated in the study.
AVP-923 has also completed a Phase III trial for the treatment of patients with diabetic peripheral neuropathic pain (“DPN pain”) with positive results. Additional Phase III trials would need to be completed for approval of this indication.
We are also developing AVP-786, a next generation drug product containing deuterium-modified dextromethorphan and quinidine for the treatment of neurologic and psychiatric disorders. We completed a pharmacokinetic study with AVP-786, and based on this data, we believe that we have identified a formulation of AVP-786 with a comparable pharmacokinetic profile, and likely a comparable safety and tolerability profile to AVP-923. This AVP-786 formulation contains significantly less quinidine than used in AVP-923.
We are also developing a novel Breath Powered intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, AVP-825. If approved, this product would be the first and only fast-acting dry-powder nasal delivery form of sumatriptan. AVP-825 is licensed from OptiNose. Under the terms of the agreement, we assumed responsibility for regulatory, manufacturing, supply-chain and commercialization activities for the investigational product, now named AVP-825. Both parties will work together on the remaining activities in support of the New Drug Application (“NDA”) submission. We have begun preparing the NDA and expect to file the application with the FDA in the first quarter of calendar 2014.
We entered into a multi-year agreement with Merck to co-promote Merck’s type 2 diabetes therapies JANUVIA® (sitagliptin) and the sitagliptin family of products in the long-term care institutional setting in the United States beginning October 1, 2013. The term of the agreement will continue for three years following the launch date of the co-promotion activities. Under the terms of the agreement, the Company will be compensated via a (i) fixed monthly fee and (ii) performance fee based on the amount of the products sold by the Company above a predetermined baseline. A significant majority of the fee is performance based. Over the three years of the agreement, Avanir could receive up to a maximum of $60.0 million in compensation.
36
In addition to our focus on products for the central nervous system, we also have partnered programs in other therapeutic areas which may generate future revenue for us. Our first commercialized product, docosanol 10% cream, (sold in the United States and Canada as Abreva® by our marketing partner GlaxoSmithKline Consumer Healthcare) is the only over-the-counter treatment for cold sores that has been approved by the FDA.
Avanir was incorporated in California in August 1988 and was reincorporated in Delaware in March 2009.
In addition to the summary above, additional accomplishments in fiscal 2013 and subsequent to the end of fiscal 2013 through the date of this filing are:
| | • | | In June 2013, the European Commission approved NUEDEXTA in the European Union for the treatment of PBA, irrespective of underlying neurological disease or injury. |
| | • | | In August 2013, we published the findings from the PRISM patient registry, the largest registry ever conducted to further understand prevalence and impact of PBA in the United States. |
| | • | | In the fourth quarter of fiscal 2013, we initiated a sales force expansion adding approximately 25 representatives to focus on the institutional care setting and currently have approximately 80 representatives focused in the institutional care setting and approximately 72 representatives focused in the retail setting. |
| | • | | In the fourth quarter of fiscal 2013, we entered into settlement agreements with three of five filers to resolve pending patent litigation in response to their abbreviated new drug applications seeking approval to market generic versions of NUEDEXTA capsules. |
| | • | | In fiscal 2013, we sold 12,965,465 shares of our common stock under an at-the-market sales agreement. Gross proceeds were $50.0 million, before deducting underwriting discounts, commissions and offering expenses. |
| | • | | In October 2013, we enrolled the first patient in our Phase II study investigating the use of AVP-923 for the treatment of levodopa-induced dyskinesia in patients with Parkinson’s disease. |
Our principal focus is on the commercialization of NUEDEXTA for the PBA indication. We believe that cash and cash equivalents, restricted cash and cash equivalents, and restricted investments of approximately $57.5 million at September 30, 2013, together with funds generated from sales of NUEDEXTA will be sufficient to fund our operations for at least the next 12 months. For additional information about the risks and uncertainties that may affect our business and prospects, please see Item 1A, “Risk Factors.”
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our consolidated financial statements prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these consolidated financial statements requires us to make a number of assumptions and estimates that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reported periods. Significant estimates and assumptions made by management include, among others, sales returns, discounts and allowances, provisions for uncollectible receivables, realizability of inventories, valuation of investments, recoverability of long-lived assets, recognition of deferred revenue, the fair value of stock options, warrants and shares issued for non-cash consideration, determination of expenses in outsourced contracts, and realization of deferred tax assets. We base our estimates on historical experience and various other assumptions that are available at the time of the estimate and that we believe to be reasonable under the circumstances. Some of these judgments can be subjective and complex. For any given individual estimate or assumption made by us, there may also be other estimates or assumptions that are reasonable. These items are monitored and analyzed by management for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are recorded in the period in which they become known. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
37
Revenue Recognition
We have historically generated revenues from product sales, collaborative research and development arrangements, and other commercial arrangements such as royalties, the sale of royalty rights and sales of technology rights. Payments received under such arrangements may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements designated in the agreements, royalties on sales of products resulting from collaborative arrangements, and payments for the sale of rights to future royalties.
We recognize revenue when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) our price to the buyer is fixed or determinable; and (4) collectability is reasonably assured. In addition, certain product sales are subject to rights of return. For products sold where the buyer has the right to return the product, we recognize revenue at the time of sale only if (1) our price to the buyer is substantially fixed or determinable at the date of sale, (2) the buyer has paid us, or is obligated to pay us and the obligation is not contingent on resale of the product, (3) the buyer’s obligation to us would not be changed in the event of theft or physical destruction or damage of the product, (4) the buyer acquiring the product for resale has economic substance apart from that provided by us, (5) we do not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (6) the amount of future returns can be reasonably estimated. We recognize such product revenues when we have met all the above criteria, including the ability to reasonably estimate future returns, or when we can reasonably estimate that the return privilege has substantially expired, whichever occurs first.
Product Sales — NUEDEXTA. NUEDEXTA is sold primarily to third-party wholesalers that, in turn, sell the product to retail pharmacies, hospitals, and other dispensing organizations. We have entered into agreements with wholesale customers, certain medical institutions and third-party payers throughout the United States. These agreements frequently contain commercial terms, which may include favorable product pricing and discounts and rebates payable upon dispensing the product to patients. Additionally, these agreements customarily provide the customer with rights to return the product, subject to the terms of each contract. Consistent with pharmaceutical industry practice, wholesale customers can return purchased product during an 18-month period that begins six months prior to the product’s expiration date and ends 12 months after the expiration date.
Our net product sales represent gross product sales less allowances for customer credits, including estimated discounts, rebates, chargebacks and co-pay assistance. These allowances provided by us to a customer are presumed to be a reduction of the selling prices of our products or services and, therefore, are characterized as a reduction of revenue when recognized in our statement of operations. Allowances for discounts, rebates, chargebacks and co-pay assistance are estimated based on contractual terms with customers and sell-through data purchased from third parties. We believe the assumptions used to estimate these allowances are reasonable considering known facts and circumstances. However, actual rebates and chargebacks could differ materially from estimated amounts because of, among other factors, unanticipated changes in prescription trends and any change in assumptions affecting sell-through data purchased from third parties. Product shipping and handling costs are included in cost of product sales.
Prior to the second quarter of fiscal 2012, we were unable to reasonably estimate future returns due to the lack of sufficient historical return data for NUEDEXTA. Accordingly, we invoiced the wholesaler, recorded deferred revenue at gross invoice sales price less estimated cash discounts and distribution fees, and classified the inventory shipped as finished goods. We previously deferred recognition of revenue and the related cost of product sales on shipments of NUEDEXTA until the right of return no longer existed, i.e. when we received evidence that the products had been dispensed to patients. We estimated patient prescriptions dispensed using an analysis of third-party information.
Product Sales — Active Pharmaceutical Ingredient docosanol (“docosanol”). Revenue from our sales of docosanol is recorded when title and risk of loss have passed to the buyer provided the criteria for revenue recognition has been met. We sell docosanol to various licensees upon receipt of a written order for the materials. Shipments generally occur fewer than three times per year. Our contracts for sales of docosanol include buyer acceptance provisions that give buyers the right of replacement if the delivered product does not meet specified criteria. That right requires that they give us notice within 30 days after receipt of the product. We have the option to refund or replace any such defective materials; however, we have historically demonstrated that the materials
38
shipped from the same pre-inspected lot have consistently met the specified criteria and no buyer has rejected any of our shipments from the same pre-inspected lot to date. Therefore, we recognize revenue at the time of delivery without providing any returns reserve. We recognize revenue upon shipment of NUEDEXTA to its wholesalers and other customers.
Multiple Element Arrangements. We have, in the past, entered into arrangements whereby we deliver to the customer multiple elements including technology and/or services. Such arrangements have included some combination of the following: antibody generation services; licensed rights to technology, patented products, compounds, data and other intellectual property; and research and development services. At the inception of each such arrangement, we analyze the multiple elements contained within the arrangement to determine whether the elements can be separated. If a product or service is not separable, the combined deliverables will be accounted for as a single unit of accounting.
A delivered element can be separated from other elements when it meets both of the following criteria: (1) the delivered item has value to the customer on a standalone basis; and (2) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in our control. If an element can be separated, we allocate amounts based upon the selling price of each element. We determine the selling price of a separate deliverable using the price we charge other customers when we sell that product or service separately; however, if we do not sell the product or service separately, we use third-party evidence of selling price of a similar product or service to a similarly situated customer. We consider licensed rights or technology to have standalone value to our customers if we or others have sold such rights or technology separately or our customers can sell such rights or technology separately without the need for our continuing involvement. We have not entered into any multiple element arrangements that have required us to estimate selling prices during fiscal 2013, 2012 or 2011.
License Arrangements. License arrangements may consist of non-refundable up-front license fees, data transfer fees, research reimbursement payments, exclusive licensed rights to patented or patent pending compounds, technology access fees, and various performance or sales milestones. These arrangements are often multiple element arrangements.
Non-refundable, up-front fees that are not contingent on any future performance by us, and require no consequential continuing involvement on our part, are recognized as revenue when the license term commences and the licensed data, technology and/or compound is delivered. Such deliverables may include physical quantities of compounds, design of the compounds and structure-activity relationships, the conceptual framework and mechanism of action, and rights to the patents or patents pending for such compounds. We defer recognition of non-refundable up-front fees if we have continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of our performance under the other elements of the arrangement. In addition, if we have required continuing involvement through research and development services that are related to our proprietary know-how and expertise of the delivered technology, or can only be performed by us, then such up-front fees are deferred and recognized over the period of continuing involvement.
Payments related to substantive, performance-based milestones in a research and development arrangement are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements when they represent the culmination of the earnings process.
Royalty Arrangements. We recognize royalty revenues from licensed products when earned in accordance with the terms of the license agreements. Net sales amounts generally required to be used for calculating royalties include deductions for returned product, pricing allowances, cash discounts, freight and warehousing. These arrangements are often multiple element arrangements.
Certain royalty arrangements provide that royalties are earned only if a sales threshold is exceeded. Under these types of arrangements, the threshold is typically based on annual sales. For royalty revenue generated from the license agreement with GSK, we recognize royalty revenue in the period in which the threshold is exceeded.
When we sell our rights to future royalties under license agreements and also maintain continuing involvement in earning such royalties, we defer recognition of any up-front payments and recognize them as revenues over the
39
life of the license agreement. We recognize revenues for the sale of an undivided interest of our Abreva® license agreement to Drug Royalty USA under the “units-of-revenue method.” Under this method, the amount of deferred revenues to be recognized in each period is calculated by multiplying the ratio of the royalty payments due to Drug Royalty USA by GSK for the period to the total remaining royalties we expect GSK will pay Drug Royalty USA over the remaining term of the agreement.
Cost of Product Sales
Cost of product sales includes third-party royalties and direct and indirect costs to manufacture product sold, including packaging, storage, shipping and handling costs and the write-off of obsolete inventory.
Recognition of Expenses in Outsourced Contracts
Pursuant to our assessment of the services that have been performed on clinical trials and other contracts, we recognize expense as the services are provided. Our assessments include, but are not limited to: (1) an evaluation by the project manager of the work that has been completed during the period, (2) measurement of progress prepared internally and/or provided by the third-party service provider, (3) analyses of data that justify the progress, and (4) management’s judgment.
Research and Development Expenses
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outsourced contracts. Research and development expenses are charged to operations as they are incurred. Up-front payments to collaborators made in exchange for the avoidance of potential future milestone and royalty payments on licensed technology are also charged to research and development expense when the drug is still in the development stage, has not been approved by the FDA for commercialization and has no alternative uses.
We assess our obligations to make milestone payments that may become due under licensed or acquired technology to determine whether the payments should be expensed or capitalized. We charge milestone payments to research and development expense when:
| | • | | The technology is in the early stage of development and has no alternative uses; |
| | • | | There is substantial uncertainty regarding the future success of the technology or product; |
| | • | | There will be difficulty in completing the remaining development; and |
| | • | | There is substantial cost to complete the work. |
Acquired contractual rights. Payments to acquire contractual rights to a licensed technology or drug candidate are expensed as incurred when there is uncertainty in receiving future economic benefits from the acquired contractual rights. We consider the future economic benefits from the acquired contractual rights to a drug candidate to be uncertain until such drug candidate is approved by the FDA or when other significant risk factors are abated.
Share-Based Compensation
We grant options, restricted stock units and restricted stock awards to purchase our common stock to our employees, directors and consultants under our stock option plans. The benefits provided under these plans are share-based payments that we account for using the fair value method.
The fair value of each option award is estimated on the date of grant using a Black-Scholes-Merton option pricing model (“Black-Scholes model”) that uses assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility, actual and projected employee stock option exercise behaviors, risk-free interest rates and expected dividends. Expected volatilities are based on historical volatility of our common stock and other factors. The expected terms of options granted are based on analyses of historical employee termination rates and option exercises. The expected risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant. Since we do not expect to pay dividends on our common stock in the foreseeable future, we estimated the dividend yield to be 0%.
40
If factors change and we employ different assumptions in calculating the fair value in future periods, the compensation expense that we record may differ significantly from what we have recorded in the current period. There is a high degree of subjectivity involved when using option pricing models to estimate share-based compensation. Because changes in the subjective input assumptions can materially affect our estimates of fair value of our share-based compensation, in our opinion, existing valuation models, including the Black-Scholes model and lattice binomial models, may not provide reliable measures of the fair value of our share-based compensation. Consequently, there is a risk that our estimates of the fair values of our share-based compensation awards on the grant dates may bear little resemblance to the actual values realized upon the exercise, early termination or forfeiture of those share-based payments in the future. Certain share-based payments, such as employee stock options, may expire worthless or otherwise result in zero intrinsic value as compared to the fair value originally estimated on the grant dates and reported in our financial statements.
Alternatively, values may be realized from these instruments that are significantly in excess of the fair value originally estimated on the grant date and reported in our financial statements. There is no current market-based mechanism or other practical application to verify the reliability and accuracy of the estimates stemming from these valuation models, nor is there a means to compare and adjust the estimates to actual values. Although the fair value of employee share-based awards is determined using an option-pricing model, the value derived from that model may not be indicative of the value observed in a willing buyer/willing seller market transaction.
The application of the fair value method of accounting for share-based compensation may be subject to further interpretation and refinement over time. There are significant differences among valuation models, and there is a possibility that we will adopt different valuation models in the future. This may result in a lack of consistency in future periods and materially affect the fair value estimate of share-based payments. It may also result in a lack of comparability with other companies that use different models, methods and assumptions.
Theoretical valuation models and market-based methods are evolving and may result in lower or higher fair value estimates for share-based compensation. The timing, readiness, adoption, general acceptance, reliability and testing of these methods is uncertain. Sophisticated mathematical models may require voluminous historical information, modeling expertise, financial analyses, correlation analyses, integrated software and databases, consulting fees, customization and testing for adequacy of internal controls. Market-based methods are emerging that, if employed by us, may dilute our earnings per share and involve significant transaction fees and ongoing administrative expenses. The uncertainties and costs of these extensive valuation efforts may outweigh the benefits to investors.
Share-based compensation expense recognized during a period is based on the value of the portion of share-based payment awards that is ultimately expected to vest and is amortized using the straight-line attribution method. As share-based compensation expense recognized in the consolidated statements of operations for fiscal 2013, 2012, and 2011 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. The fair value method requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We estimate forfeitures based on our historical experience. Changes to the estimated forfeiture rate are accounted for as a cumulative effect of change in the period the change occurred.
Effects of Inflation
We believe the impact of inflation and changing prices on net revenues and on operations has been minimal during the past three years.
Results of Operations
We operate our business on the basis of a single reportable segment, which is the business of development, acquisition and commercialization of novel therapeutics for chronic diseases. Our chief operating decision-maker is the Chief Executive Officer, who evaluates our company as a single operating segment.
We categorize revenues by type of revenue in five different categories: 1) product sales, 2) royalties and royalty rights, 3) licensing, 4) government research grant services and 5) research and development services.
41
All long-lived assets for fiscal 2013 and 2012 were located in the United States.
Comparison of Fiscal 2013 and 2012
Revenues
| | | | | | | | | | | | | | | | |
| | | Year ended September 30, | | | | | | | |
| | | 2013 | | | 2012 | | | $ Change | | | % Change | |
REVENUES | | | | | | | | | | | | | | | | |
Gross product sales | | $ | 91,205,182 | | | $ | 45,107,989 | | | $ | 46,097,193 | | | | 102 | % |
Less: discounts and allowances | | | 20,513,155 | | | | 8,033,422 | | | | 12,479,733 | | | | 155 | % |
| | | | | | | | | | | | | | | | |
Net product sales | | | 70,692,027 | | | | 37,074,567 | | | | 33,617,460 | | | | 91 | % |
Revenues from royalties | | | 4,453,707 | | | | 4,200,506 | | | | 253,201 | | | | 6 | % |
Revenues from research grant services | | | 220,150 | | | | — | | | | 220,150 | | | | — | |
| | | | | | | | | | | | | | | | |
Total revenues | | $ | 75,365,884 | | | $ | 41,275,073 | | | $ | 34,090,811 | | | | 83 | % |
| | | | | | | | | | | | | | | | |
Total revenues were $75.4 million for the fiscal year ended September 30, 2013 compared to $41.3 million for the fiscal year ended September 30, 2012. The increase in total revenues of approximately $34.1 million was primarily attributed to an increase of 86% in volume for NUEDEXTA net product sales when compared to the same period of the prior year, as well as price increases implemented over the last twelve months.
Discounts and allowances increased approximately $12.5 million for the fiscal year ended September 30, 2013 when compared to the same period in fiscal 2012. Discounts and allowances were 22.5% of gross product sales in fiscal 2013 compared to 17.8% of gross product sales in fiscal 2012. The increase in the discount and allowances percentage of gross product sales is primarily due to new contracts entered into with managed care entities during fiscal 2013 and increases in government mandated rebates.
Revenue from royalties related to GSK was approximately $4.5 million and $4.2 million in fiscal 2013 and 2012, respectively. We expect to recognize revenue from royalties from GSK through April 2014. Revenue from royalties from Azur was approximately $492,000 for fiscal 2012. The maximum royalty payments from the Azur license agreement was reached in fiscal 2012 and we did not recognize any additional revenue from royalties under this agreement in fiscal 2013.
Potential revenue-generating contracts that remained active as of September 30, 2013 include licensing revenue from our agreement with GSK and co-promote revenues from our co-promotion agreement with Merck and modest potential revenue generated from various other licensing agreements. Partnering, licensing and research collaborations have been, and may continue to be, an important part of our business development strategy. We may continue to seek partnerships with pharmaceutical companies that can help fund our operations in exchange for sharing in the success of any licensed compounds or technologies. See Note 11, “Research, License, Supply and other Agreements,” and Note 14, “Segment Information,” in the Notes to Consolidated Financial Statements.
Operating Expenses
| | | | | | | | | | | | | | | | |
| | | Year Ended September 30, | | | | | | | |
| | | 2013 | | | 2012 | | | $ Change | | | % Change | |
OPERATING EXPENSES | | | | | | | | | | | | | | | | |
Cost of product sales | | $ | 4,002,303 | | | $ | 2,120,221 | | | $ | 1,882,082 | | | | 89 | % |
Cost of research grant services | | | 147,431 | | | | — | | | | 147,431 | | | | — | |
Research and development | | | 49,506,199 | | | | 23,066,037 | | | | 26,440,162 | | | | 115 | % |
Selling and marketing | | | 63,201,776 | | | | 52,463,409 | | | | 10,738,367 | | | | 20 | % |
General and administrative | | | 29,934,875 | | | | 22,027,587 | | | | 7,907,288 | | | | 36 | % |
| | | | | | | | | | | | | | | | |
Total operating expenses | | $ | 146,792,584 | | | $ | 99,677,254 | | | $ | 47,115,330 | | | | 47 | % |
| | | | | | | | | | | | | | | | |
42
Cost of Product Sales
In fiscal 2013, cost of product sales was $4.0 million compared to $2.1 million in fiscal 2012. The increase in cost of product sales is attributable to an increase of 86% in the volume of NUEDEXTA net product sales as compared to the same period of the prior year.
Research and Development Expenses
Research and development expenses increased by approximately $26.4 million for the fiscal year ended September 30, 2013 compared to the fiscal year ended September 30, 2012. The increase is primarily due to the $20.0 million up-front payment for the in-licensing of AVP-825, increased clinical study costs of approximately $5.9 million and increased personnel costs of approximately $0.5 million to support the increased number of studies.
Selling and Marketing Expenses
Selling and marketing expenses increased by approximately $10.7 million for the fiscal year ended September 30, 2013 compared to the fiscal year ended September 30, 2012. The increase is primarily attributed to increased personnel costs of approximately $8.4 million resulting from sales force expansions which increased our sales force by 30% and increased marketing and other costs of approximately $2.3 million.
General and Administrative Expenses
General and administrative expenses increased by approximately $7.9 million for the fiscal year ended September 30, 2013, compared to the fiscal year ended September 30, 2012. The increase is primarily attributed to increased legal costs of approximately $5.6 million associated with the enforcement of our intellectual property rights related to NUEDEXTA, increased personnel costs of approximately $1.1 million and increased other costs of approximately $1.2 million.
Share-Based Compensation
Total compensation expense for our share-based payments in the fiscal years ended September 30, 2013 and 2012 was approximately $5.8 million and $4.9 million, respectively. Research and development expense in the fiscal years ended September 30, 2013 and 2012 includes share-based compensation expense of approximately $1.1 million and $1.0 million, respectively. Selling and marketing expense in the fiscal years ended September 30, 2013 and 2012 includes share-based compensation expense of approximately $1.6 million and $0.9 million, respectively. General and administrative expense in the fiscal years ended September 30, 2013 and 2012 includes share-based compensation expense of approximately $3.1 million and $3.0 million, respectively. As of September 30, 2013, approximately $11.3 million of total unrecognized compensation costs related to unvested awards is expected to be recognized over a weighted average period of 2.6 years. See Note 10, “Stockholders’ Equity – Employee Equity Incentive Plans,” in the Notes to Consolidated Financial Statements for further discussion.
Interest Expense
For the fiscal year ended September 30, 2013, interest expense was approximately $4.1 million compared to interest expense of $1.4 million for the prior fiscal year. The increase in interest expense is due to a financing arrangement entered into in June 2012, resulting in fiscal 2013 having a full year of interest expense compared to only four months of interest expense for the same period in the prior year.
Net Loss
Net loss was approximately $75.5 million, or $0.53 per share, for the fiscal year ended September 30, 2013, compared to a net loss of approximately $59.7 million, or $0.45 per share for the fiscal year ended September 30, 2012. The increase in net loss is primarily attributed to an increase in operating expenses and interest expense, partially offset by increased NUEDEXTA net product sales.
43
Comparison of Fiscal 2012 and 2011
Revenues
| | | | | | | | | | | | | | | | |
| | | Year ended September 30, | | | | | | | |
| | | 2012 | | | 2011 | | | $ Change | | | % Change | |
REVENUES | | | | | | | | | | | | | | | | |
Gross product sales | | $ | 45,107,989 | | | $ | 7,014,778 | | | $ | 38,093,211 | | | | 543 | % |
Less: discounts and allowances | | | 8,033,422 | | | | 925,472 | | | | 7,107,950 | | | | 768 | % |
| | | | | | | | | | | | | | | | |
Net product sales | | | 37,074,567 | | | | 6,089,306 | | | | 30,985,261 | | | | 509 | % |
Revenues from royalties | | | 4,200,506 | | | | 4,406,589 | | | | (206,083 | ) | | | -5 | % |
| | | | | | | | | | | | | | | | |
Total revenues | | $ | 41,275,073 | | | $ | 10,495,895 | | | $ | 30,779,178 | | | | 293 | % |
| | | | | | | | | | | | | | | | |
Total revenues were $41.3 million for the fiscal year ended September 30, 2012 compared to $10.5 million for the fiscal year ended September 30, 2011. The increase in total revenues of approximately $30.8 million was primarily attributed to an increase in volume for NUEDEXTA net product sales which we began promoting commercially in February 2011.
Discounts and allowances increased approximately $7.1 million for the fiscal year ended September 30, 2012 when compared to the same period in fiscal 2011. Discounts and allowances were 17.8% in fiscal 2012 compared to 13.2% in fiscal 2011. The increase in the discount and allowances percentages is primarily due to new contracts entered into with managed care entities during fiscal 2012.
Revenue from royalties related to GSK was approximately $3.7 million and $3.9 million in fiscal 2012 and 2011, respectively. We expect to recognize revenue from royalties from GSK through April 2014. Revenue from royalties from Azur was approximately $0.5 million for each of the fiscal years 2012 and 2011.
Operating Expenses
| | | | | | | | | | | | | | | | |
| | | Year ended September 30, | | | | | | | |
| | | 2012 | | | 2011 | | | $ Change | | | % Change | |
OPERATING EXPENSES | | | | | | | | | | | | | | | | |
Cost of product sales | | $ | 2,120,221 | | | $ | 445,980 | | | $ | 1,674,241 | | | | 375 | % |
Research and development | | | 23,066,037 | | | | 15,253,739 | | | | 7,812,298 | | | | 51 | % |
Selling and marketing | | | 52,463,409 | | | | 38,140,558 | | | | 14,322,851 | | | | 38 | % |
General and administrative | | | 22,027,587 | | | | 17,285,033 | | | | 4,742,554 | | | | 27 | % |
| | | | | | | | | | | | | | | | |
Total operating expenses | | $ | 99,677,254 | | | $ | 71,125,310 | | | $ | 28,551,944 | | | | 40 | % |
| | | | | | | | | | | | | | | | |
Cost of Product Sales
In fiscal 2012, cost of product sales was $2.1 million compared to $0.4 million in fiscal 2011. The increase in cost of product sales is attributable to the increase in product sales of NUEDEXTA which we began promoting commercially in February 2011.
Research and Development Expenses
Research and development expenses increased by approximately $7.8 million for the fiscal year ended September 30, 2012 compared to the fiscal year ended September 30, 2011. The increase is primarily due to costs of approximately $3.5 million associated with PRIME, a Phase II clinical trial of AVP-923 for the treatment of central neuropathic pain in patients with MS, for which we enrolled our first patient in November 2011, costs of approximately $2.5 million for the d-DM development program which began in the second quarter of fiscal 2012, and approximately $0.7 million of increased regulatory costs, primarily to support pursuing approval for NUEDEXTA in Europe.
44
Selling and Marketing Expenses
Selling and marketing expenses increased by approximately $14.3 million for the fiscal year ended September 30, 2012, compared to the fiscal year ended September 30, 2011. The increase is primarily attributed to increased personnel costs of approximately $11.1 million resulting from sales force expansion and other key corporate and commercial headcount resulting in an increase of over 40 employees; increased marketing and market research costs of approximately $2.9 million; and increased other costs of approximately $0.3 million.
General and Administrative Expenses
General and administrative expenses increased by approximately $4.7 million for the fiscal year ended September 30, 2012, compared to the fiscal year ended September 30, 2011. The increase is primarily attributed to increased personnel costs of approximately $1.8 million resulting from the hiring of key corporate headcount to support the commercialization of NUEDEXTA; increased legal expense of approximately $2.3 million primarily due to the enforcement of our intellectual property rights; and increased other corporate costs of approximately $0.6 million.
Share-Based Compensation
Total compensation expense for our share-based payments in the fiscal years ended September 30, 2012 and 2011 was approximately $4.9 million and $3.8 million, respectively. Research and development expense in the fiscal years ended September 30, 2012 and 2011 includes share-based compensation expense of approximately $1.0 million and $0.6 million, respectively. Selling and marketing expense in the fiscal years ended September 30, 2012 and 2011 includes share-based compensation expense of approximately $0.9 million and $0.6 million, respectively. General and administrative expense in the fiscal years ended September 30, 2012 and 2011 includes share-based compensation expense of approximately $3.0 million and $2.5 million, respectively.
Interest Expense
For the fiscal year ended September 30, 2012, interest expense was approximately $1.4 million and there was no interest expense for the fiscal year ended September 30, 2011. Interest expense in fiscal 2012 is related to the Oxford and SVB financing arrangement we entered into in June 2012.
Net Loss
Net loss was approximately $59.7 million, or $0.45 per share, for the fiscal year ended September 30, 2012, compared to a net loss of approximately $60.6 million, or $0.51 per share for the fiscal year ended September 30, 2011. The decrease in net loss is primarily attributed to increased NUEDEXTA product sales, partially offset by an increase in operating expenses.
Liquidity and Capital Resources
We assess our liquidity based on our ability to generate cash to fund future operations. Key factors in the management of our liquidity are: cash required to fund operating activities including expected operating losses and the levels of inventories, accounts payable and capital expenditures; the timing and extent of cash received from or cash paid for milestone payments under license agreements; adequate credit facilities; and financial flexibility to attract long-term equity capital on favorable terms. Historically, cash required to fund on-going business operations has been provided by financing activities and has been used to fund operations, working capital requirements and investing activities.
45
Cash, cash equivalents, restricted cash and cash equivalents and restricted investments, as well as net cash provided by or used in operating, investing and financing activities, are summarized in the table below.
| | | | | | | | | | | | | | | | | | | | |
| | | September 30,
2013 | | | Decrease
During
Period | | | September 30,
2012 | | | Decrease
During
Period | | | September 30,
2011 | |
Cash, cash equivalents, restricted cash and cash equivalents, and restricted investments | | $ | 57,528,997 | | | $ | (14,606,008 | ) | | $ | 72,135,005 | | | $ | (9,660,498 | ) | | $ | 81,795,503 | |
Cash and cash equivalents | | $ | 55,259,073 | | | $ | (14,519,333 | ) | | $ | 69,778,406 | | | $ | (9,764,158 | ) | | $ | 79,542,564 | |
Net working capital | | $ | 37,051,425 | | | $ | (23,544,875 | ) | | $ | 60,596,300 | | | $ | (9,604,294 | ) | | $ | 70,200,594 | |
| | | | | |
| | | Year Ended
September 30,
2013 | | | Change
Between
Periods | | | Year Ended
September 30,
2012 | | | Change
Between
Periods | | | Year Ended
September 30,
2011 | |
Net cash used in operating activities | | $ | (65,422,227 | ) | | $ | (7,765,607 | ) | | $ | (57,656,620 | ) | | $ | (1,135,331 | ) | | $ | (56,521,289 | ) |
Net cash used in investing activities | | | (361,623 | ) | | | 555,040 | | | | (916,663 | ) | | | 2,558,081 | | | | (3,474,744 | ) |
Net cash provided by financing activities | | | 51,264,517 | | | | 2,455,392 | | | | 48,809,125 | | | | (51,958,003 | ) | | | 100,767,128 | |
| | | | | | | | | | | | | | | | | | | | |
Net change in cash and cash equivalents | | $ | (14,519,333 | ) | | $ | (4,755,175 | ) | | $ | (9,764,158 | ) | | $ | (50,535,253 | ) | | $ | 40,771,095 | |
| | | | | | | | | | | | | | | | | | | | |
Operating activities. Net cash used in operating activities was approximately $65.4 million in fiscal 2013 compared to approximately $57.7 million in fiscal 2012. The increase is primarily due to increased operating expenses of $47.1 million, including a $20.0 million up-front payment for the in-licensing of AVP-825, and an increase in interest expense of approximately $2.7 million, partially offset by increased net product sales of approximately $33.6 million and an increase in non-cash charges and changes in operating assets and liabilities of approximately $8.0 million. Net cash used in operating activities was approximately $56.5 million in fiscal 2011. The increase in fiscal 2012 when compared to fiscal 2011 is primarily due to expenses related to NUEDEXTA sales and marketing expenses and our clinical and regulatory activities, partially offset by increased revenues.
Investing activities. Net cash used in investing activities was approximately $0.4 million in fiscal 2013, compared to approximately $0.9 million in fiscal 2012. The decrease is primarily related to a decrease in purchases of property and equipment. Net cash used in investing activities in fiscal 2011 was approximately $3.5 million. The decrease in fiscal 2012 when compared to fiscal 2011 is primarily related to a decrease in purchases of property and equipment which were high in fiscal 2011 in support of our commercial launch of NUEDEXTA and reduced purchases of investments in securities.
Financing activities. Net cash provided by financing activities was approximately $51.3 million in fiscal 2013 compared to approximately $48.8 million in fiscal 2012. In fiscal 2013, we raised approximately $50.0 million in gross proceeds (approximately $48.8 million net of offering costs and commissions) from the sale of common stock and approximately $2.5 million from the exercise of warrants and stock options. In fiscal 2012, we received proceeds from debt, net of issuance costs of approximately $29.6 million. In addition, we raised approximately $10.3 million in gross proceeds (approximately $10.1 million net of offering costs and commissions) from the sale of common stock and approximately $9.1 million from the exercise of warrants and stock options. In fiscal 2011, we raised approximately $96.7 million in gross proceeds (approximately $91.4 million net of offering costs and commissions) from the sale of common stock and approximately $9.3 million from the exercise of warrants and stock options.
In August 2012, we filed with the SEC a shelf registration statement on Form S-3 to sell an aggregate of up to $100.0 million in common stock, preferred stock, debt securities and warrants. Included in this shelf registration on
46
Form S-3 is a prospectus relating to a financing facility with Cowen and Company, LLC (“Cowen”), providing for the sale of up to $25.0 million worth of shares of our common stock from time to time in the open market at prevailing prices in accordance with the terms of a sales agreement entered into with Cowen on August 8, 2012. As of September 30, 2013, 7,935,395 shares of common stock had been sold under this facility for total gross proceeds of approximately $25.0 million. In December 2013, we amended our sales agreement with Cowen, providing for the sale of up to an additional $30.0 million worth of shares of our common stock from time to time in the open market at prevailing prices. As of September 30, 2013, approximately $75.0 million remains available under this shelf registration statement.
In July 2013, we filed with the SEC a shelf registration statement on Form S-3 to sell an aggregate of up to $150.0 million in common stock, preferred stock, debt securities and warrants. Included in this shelf registration on Form S-3 is a prospectus relating to a financing facility with Cowen, providing for the sale of up to $25.0 million worth of shares of our common stock from time to time in the open market at prevailing prices in accordance with the terms of a sales agreement entered into on August 8, 2012 and amended in July 2013. As of September 30, 2013, 5,030,070 shares of common stock had been sold under this facility for total gross proceeds of approximately $25.0 million. As of September 30, 2013, approximately $125.0 million remains available under this shelf registration statement.
As of September 30, 2013, we have contractual obligations for trade expenses and operating lease obligations, as summarized in the table that follows. We have no off-balance sheet arrangements.
| | | | | | | | | | | | | | | | | | | | |
| | | Payments Due by Period | |
| | | Total | | | Less Than
1 Year | | | 1-3 Years | | | 3-5 Years | | | More than
5 Years | |
Debt obligation | | $ | 36,363,829 | | | $ | 10,749,024 | | | $ | 25,614,805 | | | $ | — | | | $ | — | |
Operating lease obligations(1) | | | 19,294,515 | | | | 2,615,863 | | | | 5,982,087 | | | | 5,876,404 | | | | 4,820,161 | |
Purchase obligations(2) | | | 11,915,169 | | | | 11,915,169 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total | | $ | 67,573,513 | | | $ | 25,280,056 | | | $ | 31,596,892 | | | $ | 5,876,404 | | | $ | 4,820,161 | |
| | | | | | | | | | | | | | | | | | | | |
| (1) | Includes a lease entered into in November 2013 for approximately 61,000 square feet of office space. See Note 15, “Subsequent Events.” |
| (2) | Purchase obligations consist of the total of trade accounts payable and trade related accrued expenses at September 30, 2013, which approximates our contractual commitments for goods and services supported by purchase obligations in the normal course of our business. |
NUEDEXTA License Milestone Payments. We hold the exclusive worldwide marketing rights to NUEDEXTA for certain indications pursuant to an exclusive license agreement with the Center for Neurologic Study (“CNS”).
We paid to CNS a $75,000 milestone upon FDA approval of NUEDEXTA for the treatment of PBA in fiscal 2011. In addition, we are obligated to pay CNS a royalty ranging from approximately 5% to 8% of net U.S. GAAP revenue generated by sales of NUEDEXTA. During fiscal 2013, 2012 and 2011 respectively, we recorded royalties of approximately $3.5 million, $1.8 million and $309,000 related to NUEDEXTA product shipments as cost of product sales in the consolidated statements of operations. Under certain circumstances, we may have the obligation to pay CNS a portion of net revenues received if we sublicense NUEDEXTA to a third party.
Under the agreement with CNS, we are required to make payments on achievements of up to a maximum of ten milestones, based upon five specific clinical indications. Maximum payments for these milestone payments could total approximately $1.1 million if we pursued the development of NUEDEXTA for all five of the licensed indications. In general, individual milestones range from $75,000 to $125,000 for each accepted new drug application (“NDA”) and a similar amount for each approved NDA in addition to the royalty discussed above on net U.S. GAAP revenues. We are not obligated to develop additional indications under the CNS license agreement.
47
Deuterium-Modified Dextromethorphan License Milestone Payments. We hold the exclusive worldwide marketing rights to develop and commercialize deuterium-modified dextromethorphan compounds for the potential treatment of neurological and psychiatric disorders, as well as certain rights to other deuterium-modified dextromethorphan compounds pursuant to a license agreement with Concert.
Under the agreement with Concert, we are obligated to make milestone and royalty payments to Concert based on successful advancement of deuterium-modified dextromethorphan products for one or more indications in the United States, Europe, and Japan. Individual milestone payments range from $2.0 — $6.0 million, $1.5 — $15.0 million, and $25.0 — $60.0 million for clinical, regulatory and commercial targets respectively, and in aggregate could total over $200.0 million. Royalty payments are tiered, beginning in the single-digits and increasing to the low double-digits for worldwide net sales of deuterium-modified dextromethorphan products exceeding $1 billion annually. As of September 30, 2013, we have paid $2.0 million for milestones that have been achieved pursuant to this agreement.
AVP-825 License Milestone Payments. In July 2013, we entered into an exclusive license agreement for the development and commercialization of OptiNose’s novel Breath Powered intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine. The licensed territories are the United States, Canada and Mexico. If approved, this product would be the first and only fast-acting dry-powder nasal delivery form of sumatriptan.
Under the terms of the agreement, OptiNose received an up-front cash payment of $20.0 million and is eligible to receive reimbursement for certain shared development costs and up to an additional $90.0 million in total payments resulting from the achievement of future clinical, regulatory and commercial milestones. In addition, if approved, we will make tiered royalty payments of a low double-digit percentage of net sales in the United States, Canada and Mexico.
Management Outlook
We believe that cash, cash equivalents, restricted cash and cash equivalents, and restricted investments of approximately $57.5 million at September 30, 2013, together with funds expected to be generated from sales of NUEDEXTA, will be sufficient to sustain our planned level of operations for at least the next 12 months. However, we cannot provide assurances that our plans will not change, or that changed circumstances will not result in the depletion of capital resources more rapidly than anticipated.
For information regarding the risks associated with our need to raise capital to fund our ongoing and planned operations, please see Item 1A, “Risk Factors.”
Recent Authoritative Guidance
See Note 2, “Summary of Significant Accounting Policies” in the Notes to Consolidated Financial Statements for a discussion of recently issued authoritative guidance and its effect, if any, on us.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
As described below, we are exposed to market risks related to changes in interest rates. Because substantially all of our revenue, expenses, and capital purchasing activities are transacted in U.S. dollars, our exposure to foreign currency exchange rates is immaterial. However, in the future we could face increasing exposure to foreign currency exchange rates if we obtain EMA approval to market NUEDEXTA and distribute internationally, expand international distribution of docosanol 10% cream and purchase additional services from outside the U.S. Until such time as we are faced with material amounts of foreign currency exchange rate risks, we do not plan to use derivative financial instruments, which can be used to hedge such risks. We will evaluate the use of derivative financial instruments to hedge our exposure as the needs and risks should arise.
48
Interest Rate Sensitivity
Our investment portfolio consists primarily of cash equivalent fixed income instruments invested in government money market funds. The primary objective of our investments in these securities is to preserve principal. We classify our restricted investments, which are primarily certificates of deposit, as of September 30, 2013 as held-to-maturity. These held-to-maturity securities are subject to interest rate risk. Based on our current low yield, any decrease in interest rates is not likely to have a material effect on interest income.
We maintain cash balances at major financial institutions in the United States that are insured by the Federal Deposit Insurance Corporation (“FDIC”) up to $250,000 per owner. At times, deposits held with these financial institutions may exceed the amount of insurance provided by the FDIC. Generally, these deposits may be redeemed upon demand and, therefore, bear minimal risk.
Financial instruments that potentially subject us to concentrations of credit risk consist principally of cash and cash equivalents and trade receivables. Our cash and cash equivalents are placed at various money market mutual funds and financial institutions of high credit standing.
We extend credit to our customers in the form of trade receivables. We perform ongoing credit evaluations of our customers’ financial conditions and may limit the amount of credit extended if deemed necessary but usually we have required no collateral.
| Item 8. | Financial Statements and Supplementary Data |
Our consolidated financial statements are annexed to this report beginning on page F-1.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Vice President, Finance, of the effectiveness of our “disclosure controls and procedures” as of the end of the period covered by this report, pursuant to Rules 13a-15(b) and 15d-15(b) under the Securities Exchange Act of 1934, as amended.
In connection with that evaluation, our CEO and Vice President, Finance concluded that our disclosure controls and procedures were effective and designed to provide reasonable assurance that the information required to be disclosed is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission rules and forms as of September 30, 2013. For the purpose of this review, disclosure controls and procedures means controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. These disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit is accumulated and communicated to management, including our principal executive officer, principal financial officer and principal accounting officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Under the supervision and with
49
the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework inInternal Control — Integrated Framework(1992) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) and related COSO guidance. Based on our evaluation under this framework, our management concluded that our internal control over financial reporting was effective as of September 30, 2013.
Our assessment of the effectiveness of our internal control over financial reporting as of September 30, 2013 has been audited by KMJ Corbin & Company LLP, an independent registered public accounting firm, as stated in their report, which is set forth at page F-3.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) during our fourth fiscal quarter ended September 30, 2013, that has materially affected, or is reasonably likely to materially affect our internal control over financial reporting.
| Item 9B. | Other Information |
None.
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
The information relating to our directors that is required by this item is incorporated by reference from the information under the captions “Election of Directors,” “Corporate Governance,” “Board of Directors and Committees” and “Executive Officers” contained in our definitive proxy statement (the “Proxy Statement”), which will be filed with the Securities and Exchange Commission in connection with our 2014 Annual Meeting of Stockholders.
Additionally, information relating to reporting of insider transactions in Company securities is incorporated by reference from the information under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in the Proxy Statement.
Executive Officers and Key Employees of the Registrant
The names of our executive officers and other key employees and their ages as of December 1, 2013 are set forth below. Officers are elected annually by the Board of Directors and hold office until their respective successors are qualified and appointed or until their resignation, removal or disqualification.
| | | | | | |
Name | | Age | | | Position |
Keith A. Katkin | | | 42 | | | President and Chief Executive Officer |
Gregory J. Flesher | | | 43 | | | Senior Vice President, Corporate Development and Chief Business Officer |
Randall E. Kaye, M.D. | | | 51 | | | Senior Vice President, Medical Affairs and Chief Medical Officer |
Rohan Palekar | | | 48 | | | Senior Vice President and Chief Commercial Officer |
Joao Siffert, M.D. | | | 49 | | | Senior Vice President, Research and Development, Chief Scientific Officer |
Christine G. Ocampo, CPA | | | 41 | | | Vice President, Finance and Secretary |
Keith Katkin. Mr. Katkin was appointed President and Chief Executive Officer of Avanir and was elected as a member of the Board of Directors in March of 2007. From July 2005 until March 2007, Mr. Katkin served as Senior Vice President of Sales and Marketing. Prior to joining Avanir, Mr. Katkin previously served as Vice President, Commercial Development for Peninsula Pharmaceuticals, playing a key role in the management and
50
ultimate sale of the company to Johnson & Johnson in 2005. Additionally, Mr. Katkin’s employment experience includes leadership roles at InterMune, Amgen and Abbott Laboratories. Mr. Katkin also served as strategic advisor to Cerexa, a pharmaceutical company that was sold to Forest Laboratories in 2007. Mr. Katkin received a B.S. degree in Business and Accounting from Indiana University and an M.B.A. degree in Finance from the Anderson School of Management at UCLA, graduating with honors. Mr. Katkin became a licensed Certified Public Accountant in 1995.
Gregory J. Flesher. Mr. Flesher was appointed Senior Vice President, Corporate Development and Chief Business Officer in February 2011. From August 2007 to February 2011, he served as Vice President of Business Development. From June 2006 to August 2007, he served as Senior Director of Commercial Strategy and, in November 2006, assumed the additional responsibility for Business Development and Portfolio Planning. Prior to joining Avanir, Mr. Flesher held positions as Director of Sales — Hepatology (from 2004 to 2006) and Director of Marketing — Pulmonary at InterMune, Inc. (from 2002 to 2004). Prior to his tenure at InterMune, Mr. Flesher held both oncology and nephrology marketing positions with Amgen Inc., a global biotechnology company, from 1999 to 2002. Mr. Flesher also has global marketing and clinical development experience from Eli Lilly and Company, where he worked from 1995 to 1998. Mr. Flesher graduated from Purdue University with a Bachelor of Science in Biology.
Randall E. Kaye, M.D. Dr. Kaye was appointed Senior Vice President, Medical Affairs and Chief Medical Officer in March 2007. From November 2006 until March 2007, he served as Vice President Clinical and Medical Affairs, and from January 2006 to November 2006 as Vice President of Medical Affairs. Immediately prior to joining Avanir, Dr. Kaye was the Vice President of Medical Affairs for Scios Inc., a division of Johnson & Johnson, from 2004 to 2006. From 2002 to 2004, Dr. Kaye recruited and managed the Medical Affairs department for InterMune Inc. Previously, Dr. Kaye served for nearly a decade in a variety of Medical Affairs and Marketing positions for Pfizer Inc. Dr. Kaye earned his Doctor of Medicine, Masters in Public Health and Bachelor of Science degrees at George Washington University in Washington, D.C. and was a Research Fellow in Allergy and Immunology at Harvard Medical School.
Rohan Palekar. Mr. Palekar joined Avanir in March 2012 as Senior Vice President and Chief Commercial Officer. Mr. Palekar has over 20 years of experience in the biopharmaceutical industry and has worked on significant brands including Remicade® and Stelara® as well as the investigational therapy MDV3100. Mr. Palekar’s most recent commercial leadership role was as Chief Commercial Officer for Medivation, a biopharmaceutical company, where he was responsible for all commercial activities, chemistry, manufacturing and controls, medical affairs and public relations functions. Prior to Medivation, Mr. Palekar spent over 16 years at Johnson & Johnson from July 1991 to January 2008 in various senior commercial and strategic management roles, most recently as Vice President of Sales & Marketing at Centocor, a subsidiary of Johnson & Johnson, where he successfully launched two new indications for Remicade. Prior to that, Mr. Palekar was the worldwide Vice President of Immunology and held marketing leadership roles at McNeil Consumer and Specialty Pharmaceuticals. Mr. Palekar earned his M.B.A. degree from the Amos Tuck School of Business Administration Dartmouth College, his B.Com. in Accounting from the University of Bombay and his L.L.B. in Law from the University of Bombay.
Joao Siffert, M.D. Dr. Siffert joined Avanir in August 2011 as Senior Vice President, Research and Development and became Chief Scientific Officer in December 2012. Dr. Siffert previously served as Vice President and Chief Medical Officer at Ceregene, Inc., a biotechnology company focused on the development of neurotrophic gene therapies for Alzheimer’s and Parkinson’s diseases, from September 2007 to August 2011. Prior to his work at Ceregene, Dr. Siffert served as the Chief Medical Officer at Avera Pharmaceuticals, a CNS specialty pharmaceutical company, from May 2005 to September 2007. Prior to joining Avera, Dr. Siffert held positions with Pfizer (from February 2002 to May 2005) first as a medical director for Relpax and subsequently as the worldwide medical team leader of Lyrica and Neurontin focusing in areas of pain and epilepsy. Prior to Pfizer, Dr. Siffert held academic positions at Beth Israel Medical Center, where he served as director of the Adult Neuro-Oncology program, and Albert Einstein College of Medicine, where he was assistant professor of neurology. Dr. Siffert completed residencies in pediatrics at New York University School of Medicine and in neurology at Harvard Medical School. Dr. Siffert was certified by the American Board of Neurology and Psychiatry in 1996. He holds an M.D. degree from the University of Sao Paulo School of Medicine as well as an M.B.A. degree from Columbia University Business School.
51
Christine G. Ocampo, CPA. Ms. Ocampo was appointed Vice President, Finance in February 2008. Ms. Ocampo previously served as Avanir’s Controller since March 2007. Prior to joining Avanir, Ms. Ocampo served as Senior Vice President, Chief Financial Officer, Chief Accounting Officer, Treasurer and Secretary of Cardiogenesis Corporation, a medical device company, from November 2003 until April 2006. From 2001 to November 2003, Ms. Ocampo served in the role of Vice President and Corporate Controller at Cardiogenesis. Prior to joining Cardiogenesis in April 1997, Ms. Ocampo held a management position in Finance at Mills-Peninsula Health Systems in Burlingame, California, and spent three years as an auditor for Ernst & Young LLP. Ms. Ocampo graduated with a Bachelor of Science in Accounting from Seattle University and became a licensed Certified Public Accountant in 1996.
Code of Ethics
We have adopted a code of Business Conduct and Ethics for directors, officers (including our principal executive officer, principal financial officer, and principal accounting officer and controller), and employees. This code is available on our website at www.avanir.com. Any waivers from or amendments to the code of ethics will be posted to our website. You may also request a printed copy of our code of ethics, without charge, by writing to us at 20 Enterprise, Suite 200, Aliso Viejo, California 92656, Attn: Investor Relations.
| Item 11. | Executive Compensation |
The information required by this item is incorporated by reference to the information under the captions “Executive Compensation” and “Compensation Committee Interlocks and Insider Participation” and “Director Compensation” contained in the Proxy Statement.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item is incorporated by reference to the information under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” contained in the Proxy Statement.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
The information required by this item is incorporated by reference to the information under the captions “Certain Relationships and Related Party Transactions,” “Director Independence” and “Board Committees” contained in the Proxy Statement.
| Item 14. | Principal Accounting Fees and Services |
The information required by this item is incorporated by reference to the information under the caption “Fees for Independent Registered Public Accounting Firm” contained in the Proxy Statement.
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
| | (99) | Financial Statements and Schedules |
See index to the financial statements on page F-1.
52
(b) Exhibits
The following exhibits are incorporated by reference or filed as part of this report.
| | | | | | |
Exhibit Number | | | | Incorporated by Reference Herein |
| | Description | | Form | | Date |
| | | |
| 3.1 | | Certificate of Incorporation of the Registrant | | Current Report on Form 8-K, as Exhibit 3.1 | | March 25, 2009 |
| | | |
| 3.2 | | Bylaws of the Registrant | | Current Report on Form 8-K, as Exhibit 3.2 | | March 25, 2009 |
| | | |
| 3.3 | | Certificate of Ownership and Merger merging Avanir Pharmaceuticals, a California corporation, with and into Avanir Pharmaceuticals, Inc., a Delaware corporation | | Current Report on Form 8-K, as Exhibit 3.3 | | March 25, 2009 |
| | | |
| 4.1 | | Form of Common Stock Certificate | | Current Report on Form 8-K, as Exhibit 4.1 | | March 25, 2009 |
| | | |
| 4.7 | | Form of Common Stock Warrant, issued in connection with the Loan and Security Agreement dated May 7, 2012 | | Quarterly Report on Form 10-Q for the quarter ended June 30, 2012, as Exhibit 4.1 | | August 9, 2012 |
| | | |
| 10.1 | | License Agreement, dated as of March 31, 2000, by and between Avanir Pharmaceuticals and SB Pharmco Puerto Rico, a Puerto Rico Corporation | | Current Report on Form 8-K, as Exhibit 10.1 | | May 4, 2000 |
| | | |
| 10.2 | | License Purchase Agreement, dated as of November 22, 2002, by and between Avanir Pharmaceuticals and Drug Royalty USA, Inc. | | Current Report on Form 8-K, as Exhibit 99.1 | | January 7, 2003 |
| | | |
| 10.3 | | License Agreement, dated as of August 1, 2000, by and between Avanir Pharmaceuticals and IriSys Research & Development, LLC* | | Quarterly Report on Form 10-Q for the quarter ended June 30, 2000, as Exhibit 10.2 | | August 14, 2000 |
| | | |
| 10.4 | | Exclusive Patent License Agreement, dated as of April 2, 1997, by and between IriSys Research & Development, LLC and the Center for Neurologic Study | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2005, as Exhibit 10.1 | | May 13, 2005 |
| | | |
| 10.5 | | Amendment to Exclusive Patent License Agreement, dated as of April 11, 2000, by and between IriSys Research & Development, LLC and the Center for Neurologic Study | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2005, as Exhibit 10.2 | | May 13, 2005 |
| | | |
| 10.6 | | Manufacturing Services Agreement, dated as of January 4, 2006, by and between Patheon Inc. and Avanir Pharmaceuticals* | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2006, as Exhibit 10.1 | | May 10, 2006 |
53
| | | | | | |
Exhibit Number | | | | Incorporated by Reference Herein |
| | Description | | Form | | Date |
| | | |
| 10.7 | | Amended and Restated 1998 Stock Option Plan | | Annual Report on Form 10-K for the fiscal year ended September 30, 2001, as Exhibit 10.2 | | December 21, 2001 |
| | | |
| 10.8 | | Amended and Restated 1994 Stock Option Plan | | Annual Report on Form 10-K for the fiscal year ended September 30, 2001, as Exhibit 10.4 | | December 21, 2001 |
| | | |
| 10.9 | | Amended and Restated 2000 Stock Option Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.1 | | May 14, 2003 |
| | | |
| 10.10 | | Form of Restricted Stock Grant Notice for use with Amended and Restated 2000 Stock Option Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.2 | | May 14, 2003 |
| | | |
| 10.11 | | 2003 Equity Incentive Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.3 | | May 14, 2003 |
| | | |
| 10.12 | | Form of Non-Qualified Stock Option Award Notice for use with 2003 Equity Incentive Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.4 | | May 14, 2003 |
| | | |
| 10.13 | | Form of Restricted Stock Grant for use with 2003 Equity Incentive Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.5 | | May 14, 2003 |
| | | |
| 10.14 | | Form of Restricted Stock Grant Notice (cash consideration) for use with 2003 Equity Incentive Plan | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2003, as Exhibit 10.6 | | May 14, 2003 |
| | | |
| 10.15 | | 2005 Equity Incentive Plan | | Filed herewith | | |
| | | |
| 10.16 | | Form of Stock Option Agreement for use with 2005 Equity Incentive Plan | | Current Report on Form 8-K, as Exhibit 10.1 | | March 23, 2005 |
| | | |
| 10.17 | | Form of Restricted Stock Unit Grant Agreement for use with 2005 Equity Incentive Plan and 2003 Equity Incentive Plan | | Annual Report on Form 10-K for the fiscal year ended September 30, 2010, as Exhibit 10.21 | | December 8, 2010 |
| | | |
| 10.18 | | Form of Restricted Stock Unit Director Grant Agreement for use with 2005 Equity Incentive Plan and 2003 Equity Incentive Plan | | Annual Report on Form 10-K for the fiscal year ended September 30, 2010, as Exhibit 10.22 | | December 8, 2010 |
| | | |
| 10.19 | | Form of Restricted Stock Purchase Agreement for use with 2005 Equity Incentive Plan | | Annual Report on Form 10-K for the fiscal year ended September 30, 2006, as Exhibit 10.35 | | December 18,
2006 |
| | | |
| 10.20 | | Form of Indemnification Agreement with certain Directors and Executive Officers of the Registrant | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2012, as Exhibit 10.1 | | May 9, 2012 |
54
| | | | | | |
Exhibit Number | | | | Incorporated by Reference Herein |
| | Description | | Form | | Date |
| | | |
| 10.21 | | Form of Change of Control Agreement | | Annual Report on Form 10-K for the fiscal year ended September 30, 2011, as Exhibit 10.24 | | December 13, 2011 |
| | | |
| 10.22 | | Employment Agreement with Randall Kaye, dated as of December 23, 2005 | | Quarterly Report on Form 10-Q for the quarter ended December 31, 2005, as Exhibit 10.1 | | February 9, 2006 |
| | | |
| 10.23 | | Employment Agreement with Keith Katkin, dated as of March 13, 2007 | | Annual Report on Form 10-K for the fiscal year ended September 30, 2007, as Exhibit 10.44 | | December 21, 2007 |
| | | |
| 10.24 | | Asset Purchase and License Agreement, dated as of March 6, 2008, by and among Avanir Pharmaceuticals, Xenerex Biosciences and Emergent Biosolutions, Inc.* | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2008, as Exhibit 10.1 | | May 14, 2008 |
| | | |
| 10.25 | | Amendment #1 to Manufacturing Services Agreement, dated as of January 19, 2010, by and between Avanir Pharmaceuticals, Inc. and Patheon Inc. | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, as Exhibit 10.1 | | May 3, 2010 |
| | | |
| 10.26 | | Quality Agreement, dated as of January 19, 2010, by and between Avanir Pharmaceuticals, Inc. and Patheon Inc.* | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2010, as Exhibit 10.2 | | May 3, 2010 |
| | | |
| 10.27 | | First Amendment to Offer of Employment, dated as of December 31, 2008, by and between Keith A. Katkin and Avanir Pharmaceuticals, Inc. | | Annual Report on Form 10-K for the fiscal year ended September 30, 2010, as Exhibit 10.37 | | December 8, 2010 |
| | | |
| 10.28 | | Summit Office Lease, dated as of February 1, 2011, by and between Aliso Viejo RO-V1, LLC and Avanir Pharmaceuticals, Inc. | | Quarterly Report on Form 10-Q for the quarter ended March 31, 2011, as Exhibit 10.1 | | May 10, 2011 |
| | | |
| 10.29 | | First Amendment to Summit Office Lease, dated September 27, 2013, by and between Aliso Viejo RP-V1, LLC and Avanir Pharmaceuticals Inc. | | Filed herewith | | |
| | | |
| 10.30 | | Loan and Security Agreement, dated as of May 7, 2012, among the Registrant, Oxford Finance LLC, Silicon Valley Bank, Avanir Acquisition Corp., Avanir Holding Company and Xenerex Biosciences.* | | Quarterly Report on Form 10-Q for the quarter ended June 30, 2012, as Exhibit 10.1 | | August 9, 2012 |
55
| | | | | | |
Exhibit Number | | | | Incorporated by Reference Herein |
| | Description | | Form | | Date |
| | | |
| 10.31 | | Sales Agreement dated as of August 8, 2012, by and between Avanir Pharmaceuticals, Inc. and Cowen and Company, LLC | | Registration Statement on Form S-3 (File No. 333-183153), as Exhibit 1.2 | | August 8, 2012 |
| | | |
| 10.32 | | Amendment No. 1 to Sales Agreement dated as of July 5, 2013, by and between Avanir Pharmaceuticals, Inc. and Cowen and Company, LLC | | Registration Statement on Form S-3 (File No. 333-189831), as Exhibit 1.3 | | July 5, 2013 |
| | | |
| 10.33 | | License Agreement dated July 1, 2013, by and between Avanir Pharmaceuticals, Inc. and OptiNose AS** | | Filed herewith | | |
| | | |
| 10.34 | | Sublease Agreement dated November 8, 2013, by and between Valeant Pharmaceuticals International, Inc. and Avanir Pharmaceuticals, Inc. | | Filed herewith | | |
| | | |
| 21.1 | | List of Subsidiaries | | Annual Report on Form 10-K for the fiscal year ended September 30, 2010, as Exhibit 21.1 | | December 8, 2010 |
| | | |
| 23.1 | | Consent of Independent Registered Public Accounting Firm | | Filed herewith | | |
| | | |
| 31.1 | | Certification of Chief Executive Officer pursuant to Section 302 of Sarbanes-Oxley Act of 2002 | | Filed herewith | | |
| | | |
| 31.2 | | Certification of Chief Financial Officer pursuant to Section 302 of Sarbanes-Oxley Act of 2002 | | Filed herewith | | |
| | | |
| 32.1 | | Certification of Chief Executive Officer pursuant to Section 906 of Sarbanes-Oxley Act of 2002 | | Filed herewith | | |
| | | |
| 32.2 | | Certification of Chief Financial Officer pursuant to Section 906 of Sarbanes-Oxley Act of 2002 | | Filed herewith | | |
| | | |
| 99.1 | | NUEDEXTA Safety Information | | Filed herewith | | |
| | | |
| 101.INS | | XBRL Instance Document | | Filed herewith | | |
| | | |
| 101.SCH | | XBRL Taxonomy Extension Schema Document | | Filed herewith | | |
| | | |
| 101.CAL | | XBRL Taxonomy Calculation Linkbase Document | | Filed herewith | | |
| | | |
| 101.DEF | | XBRL Taxonomy Definition Linkbase Document | | Filed herewith | | |
56
| | | | | | |
Exhibit Number | | | | Incorporated by Reference Herein |
| | Description | | Form | | Date |
| | | |
| 101.LAB | | XBRL Taxonomy Extension Labels Linkbase Document | | Filed herewith | | |
| | | |
| 101.PRE | | XBRL Taxonomy Extension Presentation Linkbase Document | | Filed herewith | | |
| * | Confidential treatment has been granted with respect to portions of this exhibit pursuant to an application requesting confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934. A complete copy of this exhibit, including the redacted terms, has been separately filed with the Securities and Exchange Commission. |
| ** | Confidential treatment has been requested with respect to portions of this exhibit pursuant to an application requesting confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934. A complete copy of this exhibit, including the redacted terms, has been separately filed with the Securities and Exchange Commission. |
57
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | |
| AVANIR PHARMACEUTICALS, INC. |
| |
| By: | | /S/ KEITH A. KATKIN |
| | Keith A. Katkin |
| | President and Chief Executive Officer |
Date: December 10, 2013
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the date indicated.
| | | | |
Signature | | Title | | Date |
| | |
/S/ KEITH A. KATKIN Keith A. Katkin | | President and Chief Executive Officer (Principal Executive Officer) | | December 10, 2013 |
| | |
/S/ CHRISTINE G. OCAMPO, CPA Christine G. Ocampo, CPA | | Vice President, Finance (Principal Financial and Accounting Officer) | | December 10, 2013 |
| | |
/S/ CRAIG A. WHEELER Craig A. Wheeler | | Director, Chairman of the Board | | December 10, 2013 |
| | |
/S/ HANS E. BISHOP Hans E. Bishop | | Director | | December 10, 2013 |
| | |
/S/ DAVID J. MAZZO, PH.D. David J. Mazzo, Ph.D. | | Director | | December 10, 2013 |
| | |
/S/ CORINNE NEVINNY Corinne Nevinny | | Director | | December 10, 2013 |
| | |
/S/ DENNIS G. PODLESAK Dennis G. Podlesak | | Director | | December 10, 2013 |
| | |
/S/ SCOTT M. WHITCUP, M.D. Scott M. Whitcup, M.D. | | Director | | December 10, 2013 |
58
Avanir Pharmaceuticals, Inc.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Avanir Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Avanir Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of September 30, 2013 and 2012, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended September 30, 2013. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall consolidated financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Avanir Pharmaceuticals, Inc. and subsidiaries as of September 30, 2013 and 2012, and the results of their operations and their cash flows for each of the three years in the period ended September 30, 2013, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of September 30, 2013, based on the criteria established inInternal Control — Integrated Framework(1992) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated December 10, 2013 expressed an unqualified opinion on the Company’s internal control over financial reporting.
/s/ KMJ Corbin & Company LLP
Costa Mesa, California
December 10, 2013
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
ON INTERNAL CONTROL OVER FINANCIAL REPORTING
To the Board of Directors and Stockholders of
Avanir Pharmaceuticals, Inc.
We have audited the internal control over financial reporting of Avanir Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of September 30, 2013, based on criteria established in Internal Control — Integrated Framework (1992) issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting included in Item 9A. Our responsibility is to express an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness of the internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Avanir Pharmaceuticals, Inc. and subsidiaries maintained, in all material respects, effective internal control over financial reporting as of September 30, 2013, based on the criteria established inInternal Control — Integrated Framework (1992) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Avanir Pharmaceuticals, Inc. and subsidiaries as of September 30, 2013 and 2012, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended September 30, 2013 and our report dated December 10, 2013 expressed an unqualified opinion on those consolidated financial statements.
/s/ KMJ Corbin & Company LLP
Costa Mesa, California
December 10, 2013
F-3
Avanir Pharmaceuticals, Inc.
CONSOLIDATED BALANCE SHEETS
| | | | | | | | |
| | | September 30, | |
| | | 2013 | | | 2012 | |
| ASSETS | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 55,259,073 | | | $ | 69,778,406 | |
Restricted cash and cash equivalents | | | 965,986 | | | | 652,913 | |
Trade receivables, net | | | 12,525,992 | | | | 7,231,759 | |
Inventories, net | | | 710,179 | | | | 415,475 | |
Prepaid expenses | | | 1,391,210 | | | | 1,569,255 | |
Other current assets | | | 991,200 | | | | 865,335 | |
Restricted short-term investments | | | — | | | | 401,550 | |
| | | | | | | | |
Total current assets | | | 71,843,640 | | | | 80,914,693 | |
Restricted long-term investments | | | 1,303,938 | | | | 1,302,136 | |
Property and equipment, net | | | 1,592,791 | | | | 1,808,594 | |
Non-current inventories, net | | | 784,186 | | | | 908,364 | |
Other assets | | | 554,452 | | | | 1,078,009 | |
| | | | | | | | |
Total assets | | $ | 76,079,007 | | | $ | 86,011,796 | |
| | | | | | | | |
|
| LIABILITIES AND STOCKHOLDERS’ EQUITY | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 5,876,425 | | | $ | 2,932,961 | |
Accrued expenses | | | 11,908,570 | | | | 7,062,159 | |
Accrued compensation and payroll taxes | | | 7,775,761 | | | | 5,603,546 | |
Current portion of deferred royalty revenues | | | 1,288,514 | | | | 2,557,464 | |
Current portion of notes payable, net of debt discount | | | 7,942,945 | | | | 2,162,263 | |
| | | | | | | | |
Total current liabilities | | | 34,792,215 | | | | 20,318,393 | |
Other liabilities | | | 1,393,075 | | | | 666,179 | |
Notes payable, net of current portion and debt discount | | | 21,422,163 | | | | 26,698,263 | |
Deferred royalty revenues, net of current portion | | | — | | | | 1,491,854 | |
| | | | | | | | |
Total liabilities | | | 57,607,453 | | | | 49,174,689 | |
| | | | | | | | |
Commitments and contingencies | | | | | | | | |
Stockholders’ equity: | | | | | | | | |
Preferred stock — $0.0001 par value, 10,000,000 shares authorized, no shares issued | | | — | | | | — | |
Common stock — $0.0001 par value, 200,000,000 shares authorized; 152,063,621 and 136,435,492 shares issued and outstanding as of September 30, 2013 and 2012, respectively | | | 15,206 | | | | 13,643 | |
Additional paid-in capital | | | 518,992,237 | | | | 461,883,490 | |
Accumulated deficit | | | (500,535,889 | ) | | | (425,060,026 | ) |
| | | | | | | | |
Total stockholders’ equity | | | 18,471,554 | | | | 36,837,107 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 76,079,007 | | | $ | 86,011,796 | |
| | | | | | | | |
See notes to consolidated financial statements.
F-4
Avanir Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | | | | | | | | | | |
| | | Years Ended September 30, | |
| | | 2013 | | | 2012 | | | 2011 | |
Revenues: | | | | | | | | | | | | |
Net product sales | | $ | 70,692,027 | | | $ | 37,074,567 | | | $ | 6,089,306 | |
Revenues from royalties | | | 4,453,707 | | | | 4,200,506 | | | | 4,406,589 | |
Revenues from research grant services | | | 220,150 | | | | — | | | | — | |
| | | | | | | | | | | | |
Total revenues | | | 75,365,884 | | | | 41,275,073 | | | | 10,495,895 | |
| | | | | | | | | | | | |
Operating expenses: | | | | | | | | | | | | |
Cost of product sales | | | 4,002,303 | | | | 2,120,221 | | | | 445,980 | |
Cost of research grant services | | | 147,431 | | | | — | | | | — | |
Research and development | | | 49,506,199 | | | | 23,066,037 | | | | 15,253,739 | |
Selling and marketing | | | 63,201,776 | | | | 52,463,409 | | | | 38,140,558 | |
General and administrative | | | 29,934,875 | | | | 22,027,587 | | | | 17,285,033 | |
| | | | | | | | | | | | |
Total operating expenses | | | 146,792,584 | | | | 99,677,254 | | | | 71,125,310 | |
| | | | | | | | | | | | |
Loss from operations | | | (71,426,700 | ) | | | (58,402,181 | ) | | | (60,629,415 | ) |
Other income (expense): | | | | | | | | | | | | |
Interest income | | | 51,914 | | | | 42,815 | | | | 38,785 | |
Interest expense | | | (4,097,846 | ) | | | (1,385,342 | ) | | | — | |
Other, net | | | (31 | ) | | | 4,081 | | | | (37,733 | ) |
| | | | | | | | | | | | |
Loss before provision for income taxes | | | (75,472,663 | ) | | | (59,740,627 | ) | | | (60,628,363 | ) |
Provision for income taxes | | | 3,200 | | | | 3,200 | | | | 3,200 | |
| | | | | | | | | | | | |
Net loss | | $ | (75,475,863 | ) | | $ | (59,743,827 | ) | | $ | (60,631,563 | ) |
| | | | | | | | | | | | |
Basic and diluted net loss per share | | $ | (0.53 | ) | | $ | (0.45 | ) | | $ | (0.51 | ) |
| | | | | | | | | | | | |
Basic and diluted weighted average number of common shares outstanding | | | 142,269,399 | | | | 133,358,571 | | | | 119,405,230 | |
| | | | | | | | | | | | |
See notes to consolidated financial statements.
F-5
Avanir Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | | Common Stock
Subscribed but
Unissued | | | Additional
Paid-in
Capital | | | Accumulated
Deficit | | | Total
Stockholders’
Equity | |
| | | Shares | | | Amount | | | Shares | | | Amount | | | | |
BALANCE AT SEPTEMBER 30, 2010 | | | 95,965,572 | | | $ | 9,597 | | | | 180,000 | | | $ | 580,910 | | | $ | 332,100,685 | | | $ | (304,684,636 | ) | | $ | 28,006,556 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (60,631,563 | ) | | | (60,631,563 | ) |
Issuance of common stock in connection with: | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | | 814,454 | | | | 81 | | | | — | | | | — | | | | 637,995 | | | | — | | | | 638,076 | |
Exercise of warrants | | | 6,085,267 | | | | 608 | | | | — | | | | — | | | | 8,701,323 | | | | — | | | | 8,701,931 | |
Sale of stock, net of offering costs | | | 22,327,000 | | | | 2,233 | | | | (180,000 | ) | | | (580,910 | ) | | | 91,430,553 | | | | — | | | | 90,851,876 | |
Vesting of restricted stock units | | | 252,941 | | | | 25 | | | | — | | | | — | | | | (25 | ) | | | — | | | | — | |
Common stock surrendered | | | (1,446 | ) | | | — | | | | — | | | | — | | | | (5,665 | ) | | | — | | | | (5,665 | ) |
Share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 3,778,453 | | | | — | | | | 3,778,453 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
BALANCE AT SEPTEMBER 30, 2011 | | | 125,443,788 | | | | 12,544 | | | | — | | | | — | | | | 436,643,319 | | | | (365,316,199 | ) | | | 71,339,664 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (59,743,827 | ) | | | (59,743,827 | ) |
Issuance of common stock in connection with: | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | | 1,673,811 | | | | 167 | | | | — | | | | — | | | | 1,344,961 | | | | — | | | | 1,345,128 | |
Exercise of warrants | | | 5,445,061 | | | | 545 | | | | — | | | | — | | | | 7,785,892 | | | | — | | | | 7,786,437 | |
Sale of stock, net of offering costs | | | 3,668,656 | | | | 367 | | | | — | | | | — | | | | 10,062,629 | | | | — | | | | 10,062,996 | |
Vesting of restricted stock units | | | 204,176 | | | | 20 | | | | — | | | | — | | | | (20 | ) | | | — | | | | — | |
Issuance of warrants to purchase common stock in connection with notes payable | | | — | | | | — | | | | — | | | | — | | | | 1,169,227 | | | | — | | | | 1,169,227 | |
Share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 4,877,482 | | | | — | | | | 4,877,482 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
BALANCE AT SEPTEMBER 30, 2012 | | | 136,435,492 | | | | 13,643 | | | | — | | | | — | | | | 461,883,490 | | | | (425,060,026 | ) | | | 36,837,107 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | (75,475,863 | ) | | | (75,475,863 | ) |
Issuance of common stock in connection with: | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | | 1,033,833 | | | | 103 | | | | — | | | | — | | | | 1,496,151 | | | | — | | | | 1,496,254 | |
Exercise of warrants | | | 782,294 | | | | 78 | | | | — | | | | — | | | | 1,015,379 | | | | — | | | | 1,015,457 | |
Sale of stock, net of offering costs | | | 12,965,465 | | | | 1,297 | | | | — | | | | — | | | | 48,751,509 | | | | — | | | | 48,752,806 | |
Vesting of restricted stock units | | | 846,537 | | | | 85 | | | | — | | | | — | | | | (85 | ) | | | — | | | | — | |
Share-based compensation expense | | | — | | | | — | | | | — | | | | — | | | | 5,845,793 | | | | — | | | | 5,845,793 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
BALANCE AT SEPTEMBER 30, 2013 | | | 152,063,621 | | | $ | 15,206 | | | | — | | | $ | — | | | $ | 518,992,237 | | | $ | (500,535,889 | ) | | $ | 18,471,554 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See notes to consolidated financial statements.
F-6
Avanir Pharmaceuticals, Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | | | | | | | | | | |
| | | Years Ended September 30, | |
| | | 2013 | | | 2012 | | | 2011 | |
Cash flows from operating activities: | | | | | | | | | | | | |
Net loss | | $ | (75,475,863 | ) | | $ | (59,743,827 | ) | | $ | (60,631,563 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation and amortization | | | 826,482 | | | | 699,738 | | | | 437,845 | |
Allowance for doubtful accounts | | | 207,828 | | | | — | | | | — | |
Amortization of debt discount and debt issuance costs | | | 596,096 | | | | 213,319 | | | | — | |
Share-based compensation expense | | | 5,845,793 | | | | 4,877,482 | | | | 3,778,453 | |
Loss on disposal of assets | | | 279 | | | | — | | | | 9,271 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Trade receivables, net | | | (5,502,061 | ) | | | (5,220,594 | ) | | | (2,011,165 | ) |
Inventories, net | | | (170,526 | ) | | | (278,662 | ) | | | (164,342 | ) |
Prepaid expenses and other assets | | | 484,223 | | | | (208,909 | ) | | | (2,245,098 | ) |
Accounts payable | | | 2,888,227 | | | | (327,627 | ) | | | 1,285,100 | |
Accrued expenses and other liabilities | | | 5,465,884 | | | | 4,722,878 | | | | 1,601,130 | |
Accrued compensation and payroll taxes | | | 2,172,215 | | | | 1,351,680 | | | | 2,104,495 | |
Deferred product revenues, net | | | — | | | | (1,652,788 | ) | | | 1,652,788 | |
Deferred royalty revenues | | | (2,760,804 | ) | | | (2,089,310 | ) | | | (2,338,203 | ) |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (65,422,227 | ) | | | (57,656,620 | ) | | | (56,521,289 | ) |
| | | | | | | | | | | | |
Cash flows from investing activities: | | | | | | | | | | | | |
Purchases of property and equipment | | | (448,298 | ) | | | (813,003 | ) | | | (1,823,355 | ) |
Purchases of restricted investments and restricted cash and cash equivalents | | | (314,875 | ) | | | (103,660 | ) | | | (1,651,389 | ) |
Proceeds from maturities of restricted short-term investments | | | 401,550 | | | | — | | | | — | |
| | | | | | | | | | | | |
Net cash used in investing activities | | | (361,623 | ) | | | (916,663 | ) | | | (3,474,744 | ) |
| | | | | | | | | | | | |
Cash flows from finaning activities: | | | | | | | | | | | | |
Proceeds from debt, net of issuance costs | | | — | | | | 29,614,564 | | | | — | |
Proceeds from issuances of common stock, net of commissions and offering costs | | | 48,752,806 | | | | 10,062,996 | | | | 90,851,876 | |
Proceeds from exercise of stock options and warrants | | | 2,511,711 | | | | 9,131,565 | | | | 9,340,007 | |
Collection of stock subscriptions receivable | | | — | | | | — | | | | 580,910 | |
Shares surrendered to pay for tax withholding | | | — | | | | — | | | | (5,665 | ) |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 51,264,517 | | | | 48,809,125 | | | | 100,767,128 | |
| | | | | | | | | | | | |
Net change in cash and cash equivalents | | | (14,519,333 | ) | | | (9,764,158 | ) | | | 40,771,095 | |
Cash and cash equivalents at beginning of year | | | 69,778,406 | | | | 79,542,564 | | | | 38,771,469 | |
| | | | | | | | | | | | |
Cash and cash equivalents at end of year | | $ | 55,259,073 | | | $ | 69,778,406 | | | $ | 79,542,564 | |
| | | | | | | | | | | | |
Supplemental disclosures of cash flow information: | | | | | | | | | | | | |
Interest paid | | $ | 2,685,000 | | | $ | 648,875 | | | $ | — | |
Income taxes paid | | $ | 3,200 | | | $ | 3,200 | | | $ | 3,200 | |
Supplemental disclosures of non-cash investing and financing activities: | | | | | | | | | | | | |
Purchases of property and equipment in accounts payable and accrued expenses | | $ | 162,660 | | | $ | — | | | $ | — | |
Issuance of warrants for common stock in connection with notes payable | | $ | — | | | $ | 1,169,227 | | | $ | — | |
See notes to consolidated financial statements.
F-7
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1. | Description of Business and Basis of Presentation |
Description of Business
Avanir Pharmaceuticals, Inc. and subsidiaries (“Avanir”, the “Company” or “we”) is a biopharmaceutical company focused on acquiring, developing and commercializing novel therapeutic products for the treatment of central nervous system disorders. The Company’s lead product NUEDEXTA® (referred to as AVP-923 during clinical development) is a first-in-class dual N-methyl-D-aspartate (“NMDA”) receptor antagonist and sigma-1 agonist. NUEDEXTA, 20/10 mg, was approved in the United States in October 2010 for the treatment of pseudobulbar affect (“PBA”). It is also approved for the treatment of PBA in the European Union in two dose strengths, NUEDEXTA 20/10 mg and NUEDEXTA 30/10 mg.
The Company is studying the clinical utility of AVP-923 in other mood/behavior disorders, movement disorders and pain.The Company has two ongoing Phase II clinical trials exploring the potential treatment of agitation in patients with Alzheimer’s disease and potential treatment of levodopa-induced dyskinesia in Parkinson’s disease (the “LID study”). The LID study is supported by a grant from the Michael J. Fox Foundation. In December 2013, the Company completed a Phase II clinical trial of AVP-923 for the potential treatment of central neuropathic pain in patients with multiple sclerosis (“MS”). The data from this Phase II clinical trial did not meet the primary efficacy endpoint. In the study, AVP-923 treated patients experienced levels of pain reduction commensurate with those observed in similar studies and the improvement in pain scores from baseline reached statistical significance; however, there was no difference between the treatment arms and placebo. The Company believes that a higher than expected placebo response negatively impacted the study results. AVP-923 was generally safe and well tolerated in the study.
The Company is also developing AVP-786, a next generation drug product containing deuterium-modified dextromethorphan and quinidine for the treatment of neurologic and psychiatric disorders. The Company completed a pharmacokinetic study with AVP-786, and based on this data, the Company believes that it has identified a formulation of AVP-786 with a comparable pharmacokinetic profile, and likely a comparable safety and tolerability profile to AVP-923. ThisAVP-786 formulation contains significantly less quinidine than used in AVP-923.
The Company is also developing a novel Breath Powered™ intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, AVP-825. If approved, this product would be the first and only fast-acting dry-powder nasal delivery form of sumatriptan. AVP-825 is licensed from OptiNose AS (“OptiNose”). Under the terms of the agreement, the Company assumed responsibility for regulatory, manufacturing, supply-chain and commercialization activities for the investigational product. Both parties will work together on the remaining activities in support of the New Drug Application (“NDA”) submission. The Company has begun preparing the NDA and expects to file the application with the FDA in the first quarter of calendar 2014.
We entered into a multi-year agreement with Merck Sharp & Dohme Corp. (“Merck”) to co-promote Merck’s type 2 diabetes therapies JANUVIA® (sitagliptin) and the sitagliptin family of products in the long-term care institutional setting in the United States beginning October 1, 2013. The term of the Agreement will continue for three years following the launch date of the co-promotion activities. Under the terms of the Agreement, the Company will be compensated via a (i) fixed monthly fee and (ii) performance fee based on the amount of the Products sold by the Company above a predetermined baseline. A significant majority of the fee is performance based. Over the three years of the agreement, Avanir could receive up to a maximum of $60.0 million in compensation. See Note 11, “Research, License, Supply and other Agreements.”
In addition to the Company’s focus on products for the central nervous system, the Company also has partnered programs in other therapeutic areas which may generate future revenue. The Company’s first commercialized product, docosanol 10% cream, (sold in the United States and Canada as Abreva® by our marketing partner GlaxoSmithKline Consumer Healthcare) is the only over-the-counter treatment for cold sores that has been approved by the FDA.
F-8
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Company’s operations are subject to certain risks and uncertainties frequently encountered by companies in the early stages of operations, particularly in the evolving market for small biotech and specialty pharmaceutical companies. Such risks and uncertainties include, but are not limited to, the occurrence of adverse safety events with NUEDEXTA; that NUEDEXTA may not gain broader acceptance by the medical field or that the indicated use may not be clearly understood; the Company’s dependence on third parties for manufacturing and distribution of NUEDEXTA; that the Company may not adequately build or maintain the necessary sales, marketing, supply chain management and reimbursement capabilities on its own or enter into arrangements with third parties to perform these functions in a timely manner or on acceptable terms; the ability to successfully protect and enforce its intellectual property rights relating to NUEDEXTA; timing and uncertainty of achieving milestones in clinical trials; obtaining approvals by the FDA, European Medicines Agency (“EMA”) and regulatory agencies in other countries; and potential regulatory delays or rejections in the filing or acceptance of an NDA. The Company’s ability to generate revenues in the future may depend on market acceptance of NUEDEXTA for the treatment of PBA, pricing and reimbursement in European countries, the ability to obtain a partner to market NUEDEXTA in the EU, commercial market estimates and related revenue projections for AVP-825 and the timing and success of reaching clinical development milestones and obtaining regulatory approvals for other formulations and indications for AVP-923 and AVP-786. The Company’s cash expenditures depend substantially on the level of expenditures for the ongoing marketing of NUEDEXTA, clinical development activities for AVP-923 and AVP-786, milestone payments related to AVP-786 and AVP-825 and regulatory activities for AVP-825.
Avanir was incorporated in California in August 1988 and was reincorporated in Delaware in March 2009.
Basis of presentation
The consolidated financial statements include the accounts of Avanir Pharmaceuticals, Inc. and its wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated. The Company’s fiscal year ends on September 30 of each year. The years ended September 30, 2013, 2012, and 2011 are herein referred to as fiscal 2013, fiscal 2012 and fiscal 2011, respectively.
The Company has evaluated subsequent events through the filing date of this Form 10-K, and determined that no subsequent events have occurred that would require recognition in the consolidated financial statements or disclosure in the notes thereto other than as discussed in the accompanying notes. See Note 15, “Subsequent Events” and Note 9, “Commitments and Contingencies – Legal Contingencies.”
Reclassifications
Sales, general and administrative expenses for fiscal 2012 and 2011 are now reported under two separate captions, sales and marketing and general and administrative expenses, in the accompanying consolidated statement of operations in order to conform to the current period presentation.
| 2. | Summary of Significant Accounting Policies |
Management estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates. Significant estimates made by management include, among others, sales returns, discounts and allowances, provisions for uncollectible receivables, realizability of inventories, valuation of investments, recoverability of long-lived assets, recognition of deferred revenue, the fair value of stock options, warrants and shares issued for non-cash consideration, determination of expenses in outsourced contracts, and realization of deferred tax assets.
Cash and cash equivalents
Cash and cash equivalents consist of cash and highly liquid investments with maturities of three months or less at the date of acquisition.
F-9
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Concentration of credit risk and sources of supply
Financial assets that potentially subject the Company to concentrations of credit risk consist of cash and cash equivalents and trade receivables. The Company’s cash and cash equivalents are placed in various money market mutual funds and at financial institutions of high credit standing. At times, deposits held with these financial institutions may exceed the amount of insurance provided by the Federal Deposit Insurance Corporation (FDIC). Generally, these deposits may be redeemed upon demand and, therefore, bear minimal risk. The Company performs ongoing credit evaluations of customers’ financial condition and may limit the amount of credit extended if necessary; however, the Company has historically required no collateral from its customers.
The Company currently has sole suppliers for the active pharmaceutical ingredients (“APIs”) for NUEDEXTA and a sole manufacturer for the finished form of NUEDEXTA. In addition, these materials are custom and available from only a limited number of sources. Any material disruption in manufacturing could cause a delay in shipments and possible loss of revenue. If the Company is required to change manufacturers, the Company may experience delays associated with finding an alternative manufacturer that is properly qualified to produce NUEDEXTA in accordance with FDA requirements and the Company’s specifications.
Inventories
Inventories are stated at the lower of cost or market. Cost is determined on a first-in, first-out (“FIFO”) basis. The Company evaluates the carrying value of inventories on a regular basis, based on the price expected to be obtained for products in their respective markets compared with historical cost. Write-downs of inventories are considered to be permanent reductions in the cost basis of inventories.
The Company also regularly evaluates its inventories for excess quantities and obsolescence (expiration), taking into account such factors as historical and anticipated future sales or use in production compared to quantities on hand and the remaining shelf life of products and active pharmaceutical ingredients on hand. The Company establishes reserves for excess and obsolete inventories as required based on its analyses.
Property and equipment
Property and equipment, net, is stated at cost less accumulated depreciation and amortization. Depreciation and amortization is computed using the straight-line method over the estimated useful life of the asset. Computer equipment and related software are depreciated over three to five years. Office equipment, furniture and fixtures are depreciated over five years. Manufacturing equipment is depreciated over eight years. Leasehold improvements are amortized over the estimated useful life or remaining lease term, whichever is shorter.
Valuation of long-lived assets
Long-lived assets are evaluated for impairment whenever events or changes in circumstances indicate that their carrying value may not be recoverable. If the review indicates that a long-lived asset is not recoverable (i.e. the carrying amount is less than the future projected undiscounted cash flows), its carrying amount would be reduced to fair value. Factors management considers important that could trigger an impairment review include the following:
| | • | | A significant underperformance relative to expected historical or projected future operating results; |
| | • | | A significant change in the manner of the Company’s use of the acquired asset or the strategy for its overall business; and/or |
| | • | | A significant negative industry or economic trend. |
Based on its analysis, the Company’s management believes that no impairment of the carrying value of its long-lived assets existed at September 30, 2013 and 2012.
F-10
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Deferred rent
The Company accounts for rent expense related to operating leases by determining total minimum rent payments on the leases over their respective periods and recognizing the rent expense on a straight-line basis. The difference between the actual amount paid and the amount recorded as rent expense in each fiscal year is recorded as an adjustment to deferred rent. Deferred rent as of September 30, 2013 and 2012 was approximately $367,000 and $434,000, respectively, and is included in accrued expenses and other liabilities in the accompanying consolidated balance sheets.
Fair value of financial instruments
The Company measures the fair value of certain of its financial assets on a recurring basis. A fair value hierarchy is used to rank the quality and reliability of the information used to determine fair values. Financial assets and liabilities carried at fair value will be classified and disclosed in one of the following three categories:
| | • | | Level 1-Quoted prices (unadjusted) in active markets for identical assets and liabilities. |
| | • | | Level 2-Inputs other than Level 1 that are observable, either directly or indirectly, such as unadjusted quoted prices for similar assets and liabilities, unadjusted quoted prices in the markets that are not active, or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| | • | | Level 3-Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
At September 30, 2013 and 2012, the Company’s financial instruments include cash and cash equivalents, restricted cash and cash equivalents, trade receivables, restricted investments, accounts payable, accrued expenses and other liabilities, accrued compensation and payroll taxes, and notes payable. The carrying amount of cash and cash equivalents, restricted cash and cash equivalents, trade receivables, accounts payable, accrued expenses and other liabilities, and accrued compensation and payroll taxes approximates fair value due to the short-term maturities of these instruments. The Company’s restricted investments are carried at amortized cost which approximates fair value. Based on borrowing rates currently available to the Company, the carrying value of notes payable approximates fair value.
Restricted cash and cash equivalents and restricted investments
Restricted cash and cash equivalents and restricted investments consist of certificates of deposit, which are classified as held-to-maturity.
Restricted cash and cash equivalents consist of a certificate of deposit relating to the Company’s corporate credit card agreement and automatically renews every three months. Restricted short-term investment consists of a certificate of deposit related to an irrevocable standby letter of credit connected to the short-term portion of an office lease which expired in February 2013.
Long-term restricted investments consist of two certificates of deposit related to irrevocable standby letters of credit connected to fleet rentals and an office lease with an expiration date in 2018. The certificates of deposit automatically renew annually.
Debt issuance costs and debt discount
Debt issuance costs are stated at cost, net of accumulated amortization in other assets in the consolidated balance sheets. Debt discount is recorded within notes payable in the consolidated balance sheets. Amortization expense of debt issuance costs and the debt discount is calculated using the interest method over the term of the debt and is recorded in interest expense in the accompanying consolidated statements of operations.
F-11
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Revenue recognition
The Company has historically generated revenues from product sales, collaborative research and development arrangements, and other commercial arrangements such as royalties, the sale of royalty rights and sales of technology rights. Payments received under such arrangements may include non-refundable fees at the inception of the arrangements, milestone payments for specific achievements designated in the agreements, royalties on sales of products resulting from collaborative arrangements, and payments for the sale of rights to future royalties.
The Company recognizes revenue when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the Company’s price to the buyer is fixed or determinable; and (4) collectability is reasonably assured. In addition, certain product sales are subject to rights of return. For products sold where the buyer has the right to return the product, the Company recognizes revenue at the time of sale only if (1) the Company’s price to the buyer is substantially fixed or determinable at the date of sale, (2) the buyer has paid the Company, or is obligated to pay the Company and the obligation is not contingent on resale of the product, (3) the buyer’s obligation to the Company would not be changed in the event of theft or physical destruction or damage of the product, (4) the buyer acquiring the product for resale has economic substance apart from that provided by the seller, (5) the Company does not have significant obligations for future performance to directly bring about resale of the product by the buyer, and (6) the amount of future returns can be reasonably estimated. The Company recognizes such product revenues when either it has met all the above criteria, including the ability to reasonably estimate future returns, or when it can reasonably estimate that the return privilege has substantially expired, whichever occurs first.
Product Sales — NUEDEXTA. NUEDEXTA is sold primarily to third-party wholesalers that, in turn, sell this product to retail pharmacies, hospitals, and other dispensing organizations. The Company has entered into agreements with wholesale customers, certain medical institutions and third-party payers throughout the United States. These agreements frequently contain commercial terms, which may include favorable product pricing and discounts and rebates payable upon dispensing the product to patients. Additionally, these agreements customarily provide the customer with rights to return the product, subject to the terms of each contract. Consistent with pharmaceutical industry practice, wholesale customers can return purchased product during an 18-month period that begins six months prior to the product’s expiration date and ends 12 months after the expiration date.
The Company’s net product sales represent gross product sales less allowances for customer credits, including estimated discounts, rebates, chargebacks and co-pay assistance. These allowances provided by the Company to a customer are presumed to be a reduction of the selling prices of the Company’s products or services and, therefore, are characterized as a reduction of revenue when recognized in the Company’s consolidated statement of operations. Allowances for discounts, rebates, chargebacks and co-pay assistance are estimated based on contractual terms with customers and sell-through data purchased from third parties. The Company believes the assumptions used to estimate these allowances are reasonable considering known facts and circumstances. However, actual rebates and chargebacks could differ materially from estimated amounts because of, among other factors, unanticipated changes in prescription trends and any change in assumptions affecting sell-through data purchased from third parties. Product shipping and handling costs are included in cost of product sales.
Prior to the second quarter of fiscal 2012, the Company was unable to reasonably estimate future returns due to the lack of sufficient historical return data for NUEDEXTA. Accordingly, the Company invoiced the wholesaler, recorded deferred revenue at gross invoice sales price less estimated cash discounts and distribution fees, and classified the inventory shipped as finished goods. The Company previously deferred recognition of revenue and the related cost of product sales on shipments of NUEDEXTA until the right of return no longer existed, i.e. when the Company received evidence that the products had been dispensed to patients. The Company estimated patient prescriptions dispensed using an analysis of third-party information.
Product Sales — Active Pharmaceutical Ingredient docosanol (“docosanol”). Revenue from sales of the Company’s docosanol is recorded when title and risk of loss have passed to the buyer, provided the criteria for
F-12
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
revenue recognition have been met. The Company sells the docosanol to various licensees upon receipt of a written order for the materials. Shipments generally occur fewer than three times per year. The Company’s contracts for sales of the docosanol include buyer acceptance provisions that give the Company’s buyers the right of replacement if the delivered product does not meet specified criteria. That right requires that they give the Company notice within 30 days after receipt of the product. The Company has the option to refund or replace any such defective materials; however, it has historically demonstrated that the materials shipped from the same pre-inspected lot have consistently met the specified criteria and no buyer has rejected any of the Company’s shipments from the same pre-inspected lot to date. Therefore, the Company recognizes revenue at the time of delivery without providing any returns reserve.
Multiple Element Arrangements. The Company has, in the past, entered into arrangements whereby it delivers to the customer multiple elements including technology and/or services. Such arrangements have included some combination of the following: antibody generation services; licensed rights to technology, patented products, compounds, data and other intellectual property; and research and development services. At the inception of each such arrangement, the Company analyzes the multiple elements contained within the arrangement to determine whether the elements can be separated. If a product or service is not separable, the combined deliverables will be accounted for as a single unit of accounting.
A delivered element can be separated from other elements when it meets both of the following criteria: (1) the delivered item has value to the customer on a standalone basis; and (2) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the Company’s control. If an element can be separated, the Company allocates amounts based upon the selling price of each element. The Company determines the selling price of a separate deliverable using the price it charges other customers when it sells that product or service separately; however, if the Company does not sell the product or service separately, it uses third-party evidence of selling price of a similar product or service to a similarly situated customer. The Company considers licensed rights or technology to have standalone value to its customers if it or others have sold such rights or technology separately or its customers can sell such rights or technology separately without the need for the Company’s continuing involvement. The Company has not entered into any multiple element arrangements which have required the Company to estimate selling prices during fiscal 2013, 2012 and 2011.
License Arrangements. License arrangements may consist of non-refundable up-front license fees, data transfer fees, research reimbursement payments, exclusive licensed rights to patented or patent pending compounds, technology access fees, and various performance or sales milestones. These arrangements are often multiple element arrangements.
Non-refundable, up-front fees that are not contingent on any future performance by the Company, and require no consequential continuing involvement on its part, are recognized as revenue when the license term commences and the licensed data, technology and/or compound is delivered. Such deliverables may include physical quantities of compounds, design of the compounds and structure-activity relationships, the conceptual framework and mechanism of action, and rights to the patents or patents pending for such compounds. The Company defers recognition of non-refundable up-front fees if it has continuing performance obligations without which the technology, right, product or service conveyed in conjunction with the non-refundable fee has no utility to the licensee that is separate and independent of the Company’s performance under the other elements of the arrangement. In addition, if the Company has required continuing involvement through research and development services that are related to its proprietary know-how and expertise of the delivered technology, or can only be performed by the Company, then such up-front fees are deferred and recognized over the period of continuing involvement.
Payments related to substantive, performance-based milestones in a research and development arrangement are recognized as revenues upon the achievement of the milestones as specified in the underlying agreements when they represent the culmination of the earnings process.
F-13
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Royalty Arrangements. The Company recognizes royalty revenues from licensed products when earned in accordance with the terms of the license agreements. Net sales amounts generally required to be used for calculating royalties include deductions for returned product, pricing allowances, cash discounts, freight and warehousing. These arrangements are often multiple element arrangements.
Certain royalty arrangements provide that royalties are earned only if a sales threshold is exceeded. Under these types of arrangements, the threshold is typically based on annual sales. For royalty revenue generated from the license agreement with GlaxoSmithKline (“GSK”), the Company recognizes royalty revenue in the period in which the threshold is exceeded. For royalty revenue generated from the license agreement with Azur Pharma (“Azur”), the Company recognizes revenue when it has determined that the threshold has been exceeded.
When the Company sells its rights to future royalties under license agreements and also maintains continuing involvement in earning such royalties, it defers recognition of any up-front payments and recognizes them as revenues over the life of the license agreement. The Company recognizes revenues for the sale of an undivided interest of its Abreva® license agreement to Drug Royalty USA under the “units-of-revenue method.” Under this method, the amount of deferred revenues to be recognized in each period is calculated by multiplying the ratio of the royalty payments due to Drug Royalty USA by GSK for the period to the total remaining royalties the Company expects GSK will pay Drug Royalty USA over the remaining term of the agreement.
Cost of product sales
Cost of product sales includes third-party royalties and direct and indirect costs to manufacture product sold, including packaging, storage, shipping and handling costs and the write-off of obsolete inventory.
Recognition of expenses in outsourced contracts
Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, the Company recognizes expense as the services are provided. Such management assessments include, but are not limited to: (1) an evaluation by the project manager of the work that has been completed during the period, (2) measurement of progress prepared internally and/or provided by the third-party service provider, (3) analyses of data that justify the progress, and (4) management’s judgment. Several of the Company’s contracts extend across multiple reporting periods.
Research and development expenses
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outsourced contracts. Research and development expenses are charged to operations as they are incurred. Up-front payments to collaborators made in exchange for the avoidance of potential future milestone and royalty payments on licensed technology are also charged to research and development expense when the drug is still in the development stage, has not been approved by the FDA for commercialization and has no alternative uses.
The Company assesses its obligations to make milestone payments that may become due under licensed or acquired technology to determine whether the payments should be expensed or capitalized. The Company charges milestone payments to research and development expense when:
| | • | | The technology is in the early stage of development and has no alternative uses; |
| | • | | There is substantial uncertainty regarding the future success of the technology or product; |
| | • | | There will be difficulty in completing the remaining development; and |
| | • | | There is substantial cost to complete the work. |
F-14
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Acquired contractual rights. Payments to acquire contractual rights to a licensed technology or drug candidate are expensed as incurred when there is uncertainty in receiving future economic benefits from the acquired contractual rights. The Company considers the future economic benefits from the acquired contractual rights to a drug candidate to be uncertain until such drug candidate is approved by the FDA or when other significant risk factors are abated.
Share-based compensation
The Company grants options, restricted stock units and restricted stock awards to purchase the Company’s common stock to employees, directors and consultants under stock option plans. The benefits provided under these plans are share-based payments that the Company accounts for using the fair value method.
The fair value of each option award is estimated on the date of grant using a Black-Scholes-Merton option pricing model (“Black-Scholes model”) that uses assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, expected stock price volatility, actual and projected employee stock option exercise behaviors, risk-free interest rate and expected dividends. Expected volatilities are based on the historical volatility of the Company’s common stock and other factors. The expected terms of options granted are based on analyses of historical employee termination rates and option exercises. The risk-free interest rate is based on the U.S. Treasury yield in effect at the time of the grant. Since the Company does not expect to pay dividends on common stock in the foreseeable future, it estimated the dividend yield to be 0%.
Share-based compensation expense recognized during a period is based on the value of the portion of share-based payment awards that is ultimately expected to vest and is amortized under the straight-line attribution method. As share-based compensation expense recognized in the accompanying consolidated statements of operations for fiscal 2013, 2012, and 2011 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. The fair value method requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company estimates forfeitures based on historical experience. Changes to the estimated forfeiture rate are accounted for as a cumulative effect of change in the period the change occurred.
Total compensation expense related to all of the Company’s share-based awards for fiscal 2013, 2012 and 2011 was comprised of the following:
| | | | | | | | | | | | |
| | | Years Ended September 30, | |
| | | 2013 | | | 2012 | | | 2011 | |
Share-based compensation classified as: | | | | | | | | | | | | |
Research and development expense | | $ | 1,071,932 | | | $ | 964,876 | | | $ | 644,057 | |
Selling and marketing expense | | | 1,650,256 | | | | 908,299 | | | | 600,023 | |
General and administrative expense | | | 3,123,605 | | | | 3,004,307 | | | | 2,534,373 | |
| | | | | | | | | | | | |
Total | | $ | 5,845,793 | | | $ | 4,877,482 | | | $ | 3,778,453 | |
| | | | | | | | | | | | |
| | | | | | | | | | | | |
| | | Years Ended September 30, | |
| | | 2013 | | | 2012 | | | 2011 | |
Share-based compensation expense from: | | | | | | | | | | | | |
Stock options | | $ | 4,233,133 | | | $ | 3,643,210 | | | $ | 2,580,836 | |
Restricted stock units | | | 1,612,660 | | | | 1,234,272 | | | | 1,197,617 | |
| | | | | | | | | | | | |
Total | | $ | 5,845,793 | | | $ | 4,877,482 | | | $ | 3,778,453 | |
| | | | | | | | | | | | |
F-15
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Since the Company has a net operating loss carry-forward as of September 30, 2013, 2012, and 2011, no excess tax benefits for tax deductions related to share-based awards were recognized in the accompanying consolidated statements of operations. Additionally, no incremental tax benefits were recognized from stock options exercised in fiscal 2013, 2012, and 2011 that would have resulted in a reclassification from cash flows from operating activities to cash flows from financing activities.
Advertising expenses
Advertising costs are expensed as incurred, and these costs are included in both research and development expenses as related to clinical trials, and selling, general and administrative expenses, which include promotional materials for physicians. Advertising costs were approximately $8.9 million, $7.3 million and $5.7 million for fiscal 2013, 2012, and 2011, respectively.
Income taxes
The Company accounts for income taxes and the related accounts under the liability method. Deferred tax assets and liabilities are determined based on the differences between the consolidated financial statement carrying amounts and the income tax bases of assets and liabilities. A valuation allowance is applied against any net deferred tax asset if, based on available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company recognizes any uncertain income tax positions on income tax returns at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
The total unrecognized tax benefit resulting in a decrease in deferred tax assets and corresponding decrease in the valuation allowance at September 30, 2013 is $3.6 million. There are no unrecognized tax benefits included in the consolidated balance sheet that would, if recognized, affect the effective tax rate.
The Company’s policy is to recognize interest and/or penalties related to income tax matters in income tax expense. The Company had $0 accrued for interest and penalties on the Company’s consolidated balance sheets at September 30, 2013 and 2012.
The Company is subject to taxation in the U.S. and various state jurisdictions. The Company’s tax years for 1995 and forward for federal purposes and 1989 and forward for California purposes are subject to examination by the U.S. and California tax authorities due to the carryforward of unutilized net operating losses and research and development credits.
The Company does not foresee material changes to its gross uncertain income tax position liability within the next twelve months.
Recent authoritative guidance
In July 2013, the FASB issued Accounting Standards Update (“ASU”) No. 2013-11, “Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists.” ASU 2013-11 provides explicit guidance on the financial statement presentation of an unrecognized tax benefit when a net operating loss carryforward, a similar tax loss, or a tax credit carryforward exists. The guidance is effective prospectively for fiscal years, and interim periods within those years, beginning after December 15, 2013, with an option for early adoption. The Company intends to adopt this guidance at the beginning of its first quarter of fiscal year 2015, and is currently evaluating the impact on its consolidated financial statements and disclosures.
Proposed Amendments to Current Accounting Standards. The Financial Accounting Standard Board (“FASB”) is currently working on amendments to existing accounting standards governing a number of areas including, but not limited to, revenue recognition and lease accounting.
F-16
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
In June 2010, the FASB issued an exposure draft,Revenue from Contracts with Customers, which would supersede most of the existing guidance on revenue recognition in Accounting Standards Codification (“ASC”) Topic 605,Revenue Recognition. In November 2011, the FASB re-exposed this draft and it expects a final standard to be issued during the first quarter of calendar 2014. As the standard-setting process is still ongoing, the Company is unable to determine the impact this proposed change in accounting will have in the Company’s consolidated financial statements at this time.
In August 2010, the FASB issued an exposure draft,Leases, which would result in significant changes to the accounting requirements for both lessees and lessors in ASC Topic 840,Leases. In May 2013, the FASB re-exposed this draft and the comment period closed in September 2013. As the standard-setting process is still ongoing, the Company is unable to determine the impact this proposed change in accounting will have in the Company’s consolidated financial statements at this time.
Inventories are comprised of NUEDEXTA finished goods and the active pharmaceutical ingredients of NUEDEXTA, dextromethorphan (“DM”) and quinidine (“Q”), as well as the active pharmaceutical ingredient docosanol.
The composition of inventories as of September 30, 2013 and 2012 is as follows:
| | | | | | | | |
| | | September 30, | |
| | | 2013 | | | 2012 | |
Raw materials | | $ | 936,856 | | | $ | 1,026,250 | |
Work in progress | | | 36,453 | | | | 43,828 | |
Finished goods | | | 521,056 | | | | 253,761 | |
| | | | | | | | |
Total inventory | | | 1,494,365 | | | | 1,323,839 | |
Less: current portion | | | (710,179 | ) | | | (415,475 | ) |
| | | | | | | | |
Non-current portion | | $ | 784,186 | | | $ | 908,364 | |
| | | | | | | | |
The amount classified as non-current inventories is comprised of the raw material components for NUEDEXTA, DM and Q, which will be used in the manufacture of NUEDEXTA capsules beyond our one year operating cycle.
The following table presents the activity in inventory reserves for the last three fiscal years:
| | | | | | | | | | | | | | | | |
| | | Balance at
September 30,
2012 | | | Additional
Reserves | | | Usage | | | Balance at
September 30,
2013 | |
Reserve for excess and obsolete inventory | | | | | | | | | | | | | | | | |
Reserve for NUEDEXTA | | $ | 57,080 | | | $ | — | | | $ | (16,249 | ) | | $ | 40,831 | |
Reserve for docosanol | | | 316,400 | | | | — | | | | — | | | | 316,400 | |
Reserve for DM and Q | | | 378,829 | | | | — | | | | — | | | | 378,829 | |
| | | | | | | | | | | | | | | | |
Total | | $ | 752,309 | | | $ | — | | | $ | (16,249 | ) | | $ | 736,060 | |
| | | | | | | | | | | | | | | | |
F-17
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| | | | | | | | | | | | | | | | |
| | | Balance at
September 30,
2011 | | | Additional
Reserves | | | Usage | | | Balance at
September 30,
2012 | |
Reserve for excess and obsolete inventory | | | | | | | | | | | | | | | | |
Reserve for NUEDEXTA | | $ | 82,363 | | | $ | — | | | $ | (25,283 | ) | | $ | 57,080 | |
Reserve for docosanol | | | 316,400 | | | | — | | | | — | | | | 316,400 | |
Reserve for DM and Q | | | 378,829 | | | | — | | | | — | | | | 378,829 | |
| | | | | | | | | | | | | | | | |
Total | | $ | 777,592 | | | $ | — | | | $ | (25,283 | ) | | $ | 752,309 | |
| | | | | | | | | | | | | | | | |
| | | | |
| | | Balance at
September 30,
2010 | | | Additional
Reserves | | | Usage | | | Balance at
September 30,
2011 | |
Reserve for excess and obsolete inventory | | | | | | | | | | | | | | | | |
Reserve for NUEDEXTA | | $ | — | | | $ | 82,363 | | | $ | — | | | $ | 82,363 | |
Reserve for docosanol | | | 316,400 | | | | — | | | | — | | | | 316,400 | |
Reserve for DM and Q | | | 383,000 | | | | — | | | | (4,171 | ) | | | 378,829 | |
| | | | | | | | | | | | | | | | |
Total | | $ | 699,400 | | | $ | 82,363 | | | $ | (4,171 | ) | | $ | 777,592 | |
| | | | | | | | | | | | | | | | |
Property and equipment as of September 30, 2013 and 2012 consist of the following:
| | | | | | | | |
| | | September 30, | |
| | | 2013 | | | 2012 | |
Computer equipment and related software | | $ | 2,987,917 | | | $ | 2,534,289 | |
Leasehold improvements | | | 308,532 | | | | 299,032 | |
Office equipment, furniture and fixtures | | | 1,460,304 | | | | 1,426,647 | |
Manufacturing equipment | | | 232,423 | | | | 125,000 | |
| | | | | | | | |
| | | 4,989,176 | | | | 4,384,968 | |
Accumulated depreciation | | | (3,396,385 | ) | | | (2,576,374 | ) |
| | | | | | | | |
| | $ | 1,592,791 | | | $ | 1,808,594 | |
| | | | | | | | |
Depreciation and amortization expense associated with property and equipment was approximately $826,000, $700,000, and $438,000 for fiscal 2013, 2012, and 2011, respectively. In fiscal 2011, approximately $574,000 of office equipment was disposed of as it was determined to no longer be in use. The loss on disposal related to the net book value of approximately $9,000 is included in “other, net” on the accompanying consolidated statements of operations.
F-18
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Accrued expenses at September 30, 2013 and 2012 are as follows:
| | | | | | | | |
| | | September 30, | |
| | | 2013 | | | 2012 | |
Accrued royalties, rebates, chargebacks, and distribution fees(1) | | $ | 5,525,103 | | | $ | 2,851,140 | |
Accrued research and development expenses | | | 2,146,210 | | | | 1,222,140 | |
Accrued selling, general and administrative expenses | | | 3,892,395 | | | | 2,170,142 | |
Other current liabilities | | | 344,862 | | | | 818,737 | |
| | | | | | | | |
Total accrued expenses | | $ | 11,908,570 | | | $ | 7,062,159 | |
| | | | | | | | |
| (1) | Accrued royalties, rebates, chargebacks and distribution fees are directly impacted by product revenue and will fluctuate over time in relation to the change in product revenue. |
The following table sets forth as of September 30, 2013 and 2012 the net deferred revenue balances for NUEDEXTA product shipments and the Company’s sale of future Abreva® royalty rights to Drug Royalty USA.
| | | | | | | | | | | | |
| | | NUEDEXTA
Product
Shipments, Net | | | Drug Royalty
USA Agreement | | | Total | |
Net deferred revenues as of October 1, 2012 | | $ | — | | | $ | 4,049,318 | | | $ | 4,049,318 | |
Shipments, net | | | — | | | | — | | | | — | |
Recognized as revenues during period | | | — | | | | (2,760,804 | ) | | | (2,760,804 | ) |
| | | | | | | | | | | | |
Net deferred revenues as of September 30, 2013 | | $ | — | | | $ | 1,288,514 | | | $ | 1,288,514 | |
| | | | | | | | | | | | |
Classified and reported as: | | | | | | | | | | | | |
Current portion of deferred revenues | | $ | — | | | $ | 1,288,514 | | | $ | 1,288,514 | |
Deferred revenues, net of current portion | | | — | | | | — | | | | — | |
| | | | | | | | | | | | |
Total deferred revenues | | $ | — | | | $ | 1,288,514 | | | $ | 1,288,514 | |
| | | | | | | | | | | | |
Net deferred revenues as of October 1, 2011 | | $ | 1,652,788 | | | $ | 6,138,628 | | | $ | 7,791,416 | |
Shipments, net | | | 5,968,371 | | | | — | | | | 5,968,371 | |
Recognized as revenues during period | | | (5,766,315 | ) | | | (2,089,310 | ) | | | (7,855,625 | ) |
Recognized as a result of a change in accounting estimate(1) | | | (1,854,844 | ) | | | — | | | | (1,854,844 | ) |
| | | | | | | | | | | | |
Net deferred revenues as of September 30, 2012 | | $ | — | | | $ | 4,049,318 | | | $ | 4,049,318 | |
| | | | | | | | | | | | |
Classified and reported as: | | | | | | | | | | | | |
Current portion of deferred revenues | | $ | — | | | $ | 2,557,464 | | | $ | 2,557,464 | |
Deferred revenues, net of current portion | | | — | | | | 1,491,854 | | | | 1,491,854 | |
| | | | | | | | | | | | |
Total deferred revenues | | $ | — | | | $ | 4,049,318 | | | $ | 4,049,318 | |
| | | | | | | | | | | | |
| (1) | The amount ultimately recognized as net product sales was approximately $1.7 million due to other discounts and allowances, including, but not limited to, rebates, chargebacks and co-pay assistance totaling approximately $191,000. |
F-19
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
NUEDEXTA Product Shipments, net — The amount of deferred revenue from NUEDEXTA product shipments is shown net of estimated prompt-pay discounts and wholesaler fees based on wholesaler purchases. The amount that ultimately will be recognized as net product sales in the Company’s consolidated financial statements may be different due to other discounts and allowances, including, but not limited to, wholesaler fees based on wholesaler shipments, rebates, chargebacks and co-pay assistance. See Note 2, “Summary of Significant Accounting Policies – Revenue recognition – Product Sales – NUEDEXTA,” for further discussion.
Drug Royalty USA Agreement — In November 2002, the Company sold to Drug Royalty USA an undivided interest in the Company’s rights to receive future Abreva royalties under the license agreement with GSK for $24.1 million (the “Drug Royalty Agreement” and the “GSK License Agreement,” respectively). Under the Drug Royalty Agreement, Drug Royalty USA has the right to receive royalties from GSK on sales of Abreva until the expiration of the patent for Abreva on April 30, 2014. The Company retained the right to receive 50% of all royalties (a net of 4%) under the GSK License Agreement for annual net sales of Abreva in the U.S. and Canada in excess of $62.0 million. In fiscal 2013, 2012, and 2011, the Company recognized royalties related to the annual net Abreva sales in excess of $62.0 million in the amount of approximately $1.7 million, $1.6 million and $1.5 million, respectively, which is included in the accompanying consolidated statements of operations as revenues from royalties.
Revenues are recognized when earned, collection is reasonably assured and no additional performance of services is required. The Company classified the proceeds received from Drug Royalty USA as deferred revenue, and is recognizing the revenue over the life of the license agreement because of the Company’s continuing involvement over the term of the Drug Royalty Agreement. Such continuing involvement includes overseeing the performance of GSK and its compliance with the covenants in the GSK License Agreement, monitoring patent infringement, adverse claims or litigation involving Abreva, and undertaking to find a new license partner in the event that GSK terminates the agreement. The Drug Royalty Agreement contains both covenants and events of default that require such performance on the Company’s part. Therefore, nonperformance on the Company’s part could result in default of the arrangement, and could give rise to additional rights in favor of Drug Royalty USA under a separate security agreement with Drug Royalty USA, which could result in loss of the Company’s rights to share in future Abreva royalties if wholesale sales by GSK exceed $62.0 million a year. The deferred revenue is being recognized as revenue using the “units-of-revenue method” over the life of the license agreement. Based on a review of the Company’s continuing involvement, the Company concluded that the sale proceeds did not meet any of the rebuttable presumptions that would require classification of the proceeds as debt.
In May 2012, the Company entered into a Loan and Security Agreement (the “Loan Agreement”) with Oxford Finance LLC and Silicon Valley Bank. The Loan Agreement provides for a term loan of $30.0 million which was funded upon closing of the transaction in June 2012. Under the terms of the Loan Agreement, interest accrues on the outstanding balance at a rate of 8.95% per annum. In the third fiscal quarter of 2013, the Company met the criteria to extend the interest only payment for six months. Therefore, the Company will make monthly payments of interest only until January 1, 2014 (the “Amortization Date”). Beginning on the Amortization Date, the outstanding loan balance will be repaid in thirty equal monthly payments of principal and interest. In addition to the original principal, a final payment equal to 7% of the original principal amount of the loan will be due thirty months from the Amortization Date. The final payment is being accreted as interest expense over the term of the debt using the interest method and the related liability of approximately $1.1 million and $299,000 as of September 30, 2013 and 2012, respectively, is included in other liabilities in the accompanying consolidated balance sheets.
In accordance with the terms of the Loan Agreement, the Company issued to the lenders warrants to purchase shares of the Company’s common stock equal to 4.55% of the original principal at a price per share equal to the lower of the 10-day average share price prior to closing or the price per share on the day of funding. Accordingly,
F-20
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
the Company issued to the lenders warrants to purchase 491,007 shares of the Company’s common stock at an exercise price of $2.78 per share. The relative fair value of the warrants was approximately $1.2 million and was estimated using the Black-Scholes model with the following assumptions: fair value of the Company’s common stock at issuance of $2.80 per share; ten year contractual term; 96.7% volatility; 0% dividend rate; and a risk-free interest rate of 1.8%. The relative fair value of the warrants was recorded as a debt discount, decreasing notes payable and increasing additional paid-in capital on the accompanying consolidated balance sheets. The debt discount is being amortized to interest expense over the term of the debt using the interest method. For the years ended September 30, 2013 and 2012 debt discount amortization was approximately $505,000 and $180,000, respectively.
The loan is secured by a first priority security interest in all of the Company’s assets, other than its intellectual property and its rights under license agreements granting it rights to intellectual property.
The Loan Agreement contains standard affirmative and restrictive covenants. The affirmative covenants include, among other items, that the Company maintain a minimum sales level relative to projected NUEDEXTA revenues, measured on a trailing three-month basis, or maintain cash and cash equivalents in accounts subject to control agreements in favor of the collateral agent equal to at least 1.5 times the outstanding amount of obligations under the Loan Agreement. Additionally, the affirmative and restrictive covenants, among other items, restrict the Company’s ability to incur additional indebtedness or guarantees; incur liens; make investments, loans and acquisitions; consolidate or merge; sell assets, including capital stock of subsidiaries; alter the business of the Company; engage in transactions with affiliates; and enter into agreements limiting dividends and distributions of certain subsidiaries. The Loan Agreement also includes events of default, including, among other things, payment defaults, breaches of representations, warranties or covenants, certain bankruptcy events, the occurrence of certain material adverse changes, and a commercial, generic version of NUEDEXTA (for the treatment of PBA) becoming available. Upon the occurrence of an event of default and following any cure periods (if applicable), a default interest rate of an additional 5.0% per annum may be applied to the outstanding loan balances, and the lenders may declare all outstanding obligations immediately due and payable and take such other actions as set forth in the Loan Agreement. As of September 30, 2013, the Company was in compliance with all covenants in the Loan Agreement.
Future payments as of September 30, 2013 are as follows:
| | | | |
Year Ending September 30, | | | |
2014 | | $ | 10,749,024 | |
2015 | | | 13,437,031 | |
2016 | | | 12,177,774 | |
| | | | |
Total minimum payments | | | 36,363,829 | |
Less amount representing interest | | | 6,363,829 | |
| | | | |
Notes payable, gross | | | 30,000,000 | |
Unamortized discount on notes payable | | | (634,892 | ) |
| | | | |
Notes payable balance | | | 29,365,108 | |
Current portion of notes payable, net of debt discount | | | 7,942,945 | |
| | | | |
Notes payable, net of current portion and debt discount | | $ | 21,422,163 | |
| | | | |
| 8. | Computation of Net Loss Per Share |
Basic net loss per common share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is computed by dividing net loss
F-21
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
by the weighted-average number of common and dilutive common equivalent shares outstanding during the period. In the loss periods, the shares of common stock issuable upon exercise of stock options and warrants or upon vesting of restricted stock units are excluded from the computation of diluted net loss per share, as their effect is anti-dilutive.
For fiscal 2013, 2012, and 2011, the following options and warrants to purchase shares of common stock and restricted stock units were excluded from the computation of diluted net loss per share, as the inclusion of such shares would be anti-dilutive:
| | | | | | | | | | | | |
| | | Fiscal 2013 | | | Fiscal 2012 | | | Fiscal 2011 | |
Stock options | | | 8,823,041 | | | | 8,142,468 | | | | 8,083,896 | |
Stock warrants | | | 53,957 | | | | 1,201,116 | | | | 6,155,170 | |
Restricted stock units(1) | | | 2,904,423 | | | | 2,617,188 | | | | 1,798,213 | |
| (1) | Includes 1,267,215, 1,600,564, and 1,413,481 shares of restricted stock in fiscal 2013, 2012, and 2011, respectively, awarded to directors that have vested but are still restricted until the directors resign. |
| 9. | Commitments and Contingencies |
Operating lease commitments. The Company leases approximately 38,000 square feet of office space in Aliso Viejo, California. The lease has scheduled rent increases each year and expires in April 2018. As of September 30, 2013, the financial commitment for the remainder of the term of this lease is approximately $5.0 million.
In November 2013, the Company entered into a lease for approximately 61,000 square feet of office space commencing in November 2013 for an initial lease term of 86 months. The lease has scheduled rent increases each year. The aggregate minimum payments over the initial term of the lease are expected to be approximately $14.3 million.
Rent expense, excluding common area charges and other costs, was approximately $1.1 million, $2.0 million and $1.9 million in fiscal 2013, 2012, and 2011, respectively. Sublease rent income totaled approximately $294,000, $997,000 and $961,000 in fiscal 2013, 2012 and 2011, respectively. Future minimum rental payments under non-cancelable operating lease commitments as of September 30, 2013 and including the lease the Company entered into November 2013 are as follows:
| | | | |
Year Ending September 30, | | Minimum
Payments | |
2014 | | $ | 2,615,863 | |
2015 | | | 2,946,688 | |
2016 | | | 3,035,399 | |
2017 | | | 3,141,553 | |
2018 | | | 2,734,851 | |
Thereafter | | | 4,820,161 | |
| | | | |
Total | | $ | 19,294,515 | |
| | | | |
Legal contingencies.
NUEDEXTA ANDA Litigation
In fiscal 2011 and 2012, the Company received Paragraph IV certification notices from five separate companies contending that certain of its patents listed in the FDA’s publication, “Approved Drug Products with
F-22
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Therapeutic Equivalence Evaluation” (“FDA Orange Book”) (U.S. Patents 7,659,282 (“’282 Patent”), 8,227,484 (“’484 Patent”) and RE 38,115 (“’115 Patent”), which expire in August 2026, July 2023 and January 2016, respectively) are invalid, unenforceable and/or will not be infringed by the manufacture, use, sale or offer for sale of a generic form of NUEDEXTA as described in those companies’ abbreviated new drug application (“ANDA”). The FDA Orange Book provides potential competitors, including generic drug companies, with a list of issued patents covering approved drugs. In August 2011 and March 2012, the Company filed lawsuits in the U.S District Court for the District of Delaware against Par Pharmaceutical, Inc. and Par Pharmaceutical Companies, Inc. (collectively “Par”), Actavis South Atlantic LLC and Actavis, Inc. (collectively “Actavis”), Wockhardt USA, LLC and Wockhardt, Ltd. (collectively, “Wockhardt”), Impax Laboratories, Inc. (“Impax”) and Watson Pharmaceuticals, Inc., Watson Laboratories, Inc. and Watson Pharma, Inc. (collectively “Watson”) (Par, Actavis, Wockhardt, Impax and Watson, collectively the “Defendants”). In September and October 2012, the Company filed lawsuits in the U.S. District Court for the District of Delaware against the Defendants. All lawsuits (collectively, the “ANDA Actions”) were filed on the basis that the Defendants’ submissions of their respective ANDAs to obtain approval to manufacture, use, sell, or offer for sale generic versions of NUEDEXTA prior to the expiration of the ‘282 Patent, the ‘484 Patent and the ‘115 Patent listed in the FDA Orange Book constitute infringement of one or more claims of those patents. On October 31, 2012, Watson announced the divestiture of its ANDA for a generic form of NUEDEXTA to Sandoz, Inc. (“Sandoz”). As a result of Sandoz’ acquisition and maintenance of said ANDA, on May 30, 2013, the Company filed suit in the U.S. District Court for the District of Delaware against Sandoz. This suit was filed on the basis that Sandoz’ ANDA to obtain approval to manufacture, use, sell, or offer for sale generic versions of NUEDEXTA prior to the expiration of the ’282 Patent, the ’484 Patent and the ’115 Patent listed in the FDA Orange Book constitutes infringement of one or more claims of those patents.
On December 3, 2012, the Company received a Memorandum Opinion and Order (the “Order”) issued by Judge Leonard P. Stark of the United States District Court for the District of Delaware (the “Court”) related to the Markman hearing held October 5, 2012 in the Company’s ongoing patent infringement case against the Defendants. The Order establishes the meaning of patent claim terms in dispute between the parties. After comprehensive briefing and oral argument, Judge Stark’s Order adopted the Company’s proposed or stipulated patent term constructions.
A bench trial was held in September 2013 and concluded on October 15, 2013. We are currently awaiting Judge Stark’s decision.
On August 9, August 30, and September 6, 2013, Avanir entered into settlement agreements with Sandoz, Actavis and Wockhardt, respectively, to resolve pending patent litigation in response to their ANDAs seeking approval to market generic versions of NUEDEXTA capsules. The settlement agreements grant Sandoz, Actavis and Wockhardt the right to begin selling a generic version of NUEDEXTA on July 30, 2026, or earlier under certain circumstances. The parties also filed stipulations and orders of dismissal with the United States District Court for the District of Delaware which conclude the litigation with respect to Sandoz, Actavis and Wockhardt. The settlements do not end the Company’s ongoing litigation against the other two ANDA filers.
As a result of the 30-month stay associated with the filing of the ANDA Actions, the FDA cannot grant final approval to any ANDA before December 30, 2013, unless there is an earlier court decision holding that the Orange Book-listed ’282, ‘484 and ‘115 patents are not infringed and/or are invalid. The Company intends to vigorously enforce its intellectual property rights relating to NUEDEXTA, but the Company cannot predict the outcome of these matters.
Alamo and Azur Litigation
In October 2011, Neal R. Cutler, M.D., the founder of Alamo Pharmaceuticals, LLC (“Alamo”), filed a lawsuit in California Superior Court against Azur Pharma International III Limited and Azur Pharma Limited (collectively, “Azur”) and Avanir (the “Cutler Action”). The Company purchased Alamo in 2006 to acquire rights to FazaClo, an
F-23
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
approved anti-psychotic drug. In connection with this acquisition of Alamo, the Company agreed to provide the Alamo equity holders, including Dr. Cutler, with milestone payments tied to the aggregate net revenues of FazaClo. In 2007, the Company sold FazaClo and its related assets and operations to Azur. In connection with this sale, Azur agreed to assume the milestone payment obligations due to Dr. Cutler under the Alamo purchase agreement, although the Company could remain liable for these payments if Azur defaults on these obligations. In the Cutler Action, Dr. Cutler alleges a breach of contract and a breach of implied covenant of good faith and fair dealing. Dr. Cutler alleges that Azur has failed to make certain sales information available to Dr. Cutler and has withheld payments to which Dr. Cutler is entitled. Dr. Cutler alleges that Avanir has acquiesced in this conduct, and Dr. Cutler seeks to hold both Azur and Avanir liable for these actions. Azur Ltd. and Jazz Pharmaceuticals, Inc. entered into a business combination creating Jazz Pharmaceuticals, plc (“Jazz”). As successor in interest to Azur Ltd., Jazz agreed to indemnify Avanir in full for the claims asserted in the Cutler Action.
General and Other
In the ordinary course of business, the Company may face various claims brought by third parties and the Company may, from time to time, make claims or take legal actions to assert the Company’s rights, including intellectual property rights as well as claims relating to employment and the safety or efficacy of products. Any of these claims could subject the Company to costly litigation and, while the Company generally believes that it has adequate insurance to cover many different types of liabilities, the Company’s insurance carriers may deny coverage or policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on the Company’s consolidated operations, cash flows and financial position. Additionally, any such claims, whether or not successful, could damage the Company’s reputation and business. Management believes the outcomes of currently pending claims and lawsuits will not likely have a material effect on the Company’s consolidated operations or financial position.
In addition, it is possible that the Company could incur termination fees and penalties if it elected to terminate contracts with certain vendors, including clinical research organizations.
Guarantees and Indemnities. The Company indemnifies its directors, officers and certain executives to the maximum extent permitted under the laws of the State of Delaware, and various lessors in connection with facility leases for certain claims arising from such facilities or leases. Additionally, the Company periodically enters into contracts that contain indemnification obligations, including contracts for the purchase and sale of assets, wholesale distribution agreements, clinical trials, pre-clinical development work and securities offerings. These indemnification obligations provide the contracting parties with the contractual right to have Avanir pay for the costs associated with the defense and settlement of claims, typically in circumstances where Avanir has failed to meet its contractual performance obligations in some fashion.
The maximum amount of potential future payments under such indemnifications is not determinable. The Company has not incurred significant costs related to these guarantees and indemnifications, and no liability has been recorded in the consolidated financial statements for guarantees and indemnifications as of September 30, 2013 and 2012.
Center for Neurologic Study (“CNS”) —The Company holds the exclusive worldwide marketing rights to NUEDEXTA for certain indications pursuant to an exclusive license agreement with CNS.
The Company paid a $75,000 milestone upon FDA approval of NUEDEXTA for the treatment of PBA in fiscal 2011. In addition, the Company is obligated to pay CNS a royalty ranging from approximately 5% to 8% of net U.S. GAAP revenue generated by sales of NUEDEXTA. Under certain circumstances, the Company may have the obligation to pay CNS a portion of net revenues received if the Company sublicenses NUEDEXTA to a third party.
Under the agreement with CNS, the Company is required to make payments on achievements of up to a maximum of ten milestones, based upon five specific clinical indications. Maximum payments for these milestone
F-24
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
payments could total approximately $1.1 million if the Company pursued the development of NUEDEXTA for all five of the licensed indications. In general, individual milestones range from $75,000 to $125,000 for each accepted new drug application (“NDA”) and a similar amount for each approved NDA in addition to the royalty discussed above on net U.S. GAAP revenues. The Company does not have the obligation to develop additional indications under the CNS license agreement.
Concert Pharmaceuticals, Inc. –The Company holds the exclusive worldwide marketing rights to develop and commercialize Concert’s d-DM compounds for the potential treatment of neurological and psychiatric disorders, as well as certain rights to other deuterium-modified dextromethorphan compounds pursuant to a license agreement with Concert.
Under the agreement with Concert, the Company is obligated to make milestone and royalty payments to Concert based on successful advancement of d-DM products for one or more indications in the United States, Europe, and Japan. Individual milestone payments range from $2.0 — $6.0 million, $1.5 — $15.0 million, and $25.0 — $60.0 million for clinical, regulatory and commercial targets respectively, and in aggregate could total over $200 million. Royalty payments are tiered, beginning in the single-digits and increasing to the low double-digits for worldwide net sales of d-DM products exceeding $1.0 billion annually. As of September 30, 2013, the Company has paid $2.0 million for milestones that have been achieved pursuant to this agreement, which were recorded to research and development expense in the accompanying consolidated statement of operations.
OptiNose AS — In July 2013, the Company entered into an exclusive license agreement for the development and commercialization of a novel Breath Powered intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, AVP-825.
AVP-825 is licensed from OptiNose. Under the terms of the agreement, OptiNose received an up-front cash payment of $20.0 million, which was recorded to research and development expense in the accompanying consolidated statement of operations. The Company and OptiNose will share certain development costs. OptiNose is eligible to receive up to an additional $90.0 million in aggregate milestone payments resulting from the achievement of future clinical, regulatory and commercial milestones. In addition, following product approval, Avanir will be required to make tiered royalty payments to OptiNose of a low double-digits percentage of net sales in the United States, Canada and Mexico.
Preferred stock
During fiscal 2013, the Company had a stockholder rights plan (“the Plan”). In November 2013, the Plan was amended such that it expired in November 2013.
The Plan provided for the issuance of Series A junior participating preferred stock to each stockholder of record under certain circumstances. None of the Series A junior participating preferred stock were outstanding on September 30, 2013 and 2012. The Plan provided for a dividend distribution of one preferred share purchase right (the “Right”) on each outstanding share of common stock, payable on shares outstanding as of March 20, 2009 (the “Record Date”). All shares of common stock issued by the Company after the Record Date have been issued with such Rights attached. Subject to limited exceptions, the Rights would become exercisable if a person or group acquired 20% or more of the Company’s common stock or announced a tender offer for 20% or more of the Company’s common stock (a “Trigger Event”).
If and when the Rights become exercisable, each Right would entitle stockholders, excluding the person or group causing the Trigger Event (an “Acquiring Person”), to buy a fraction of a share of Series A junior participating preferred stock at a fixed price. In certain circumstances following a Trigger Event, each Right would entitle its owner, who is not an Acquiring Person, to purchase at the Right’s then current exercise price, a number of
F-25
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
shares of common stock having a market value equal to twice the Right’s exercise price. Rights held by any Acquiring Person would become void and not be exercisable to purchase shares at the discounted purchase price.
The Board of Directors may redeem the Rights at $0.0001 per Right at any time before a person has acquired 20% or more of the outstanding common stock. The Rights would expire on March 20, 2019, subject to a periodic review of the Plan by a committee of independent directors.
Common stock
Fiscal 2013. In August 2012, the Company filed with the SEC a shelf registration statement on Form S-3 to sell an aggregate of up to $100.0 million in common stock, preferred stock, debt securities and warrants. Included in this shelf registration on Form S-3 is a prospectus relating to a financing facility with Cowen and Company, LLC (“Cowen”), providing for the sale of up to $25.0 million worth of shares of our common stock from time to time into the open market at prevailing prices in accordance with the terms of a sales agreement entered into with Cowen in August 2012. During fiscal 2013, the Company issued 7,935,395 shares of common stock under the sales agreement at an average price of $3.15 per share raising proceeds of approximately $25.0 million ($24.4 million after offering expenses, including commissions).
In July 2013, the Company filed with the SEC a shelf registration statement on Form S-3 to sell an aggregate of up to $150.0 million in common stock, preferred stock, debt securities and warrants. Included in this shelf registration on Form S-3 is a prospectus relating to a financing facility with Cowen, providing for the sale of up to an additional $25.0 million worth of shares of our common stock from time to time into the open market at prevailing prices in accordance with the terms of a sales agreement entered into with Cowen in August 2012 and amended in July 2013. During fiscal 2013, the Company issued 5,030,070 shares of common stock under the sales agreement at an average price of $4.97 per share raising proceeds of approximately $25.0 million ($24.4 million after offering expenses, including commissions).
During fiscal 2013, the Company issued 510,188 shares of common stock in connection with restricted stock units which were awarded to directors and vested at September 30, 2012, but were restricted until the resignation of the directors, 336,349 shares of common stock in connection with the vesting of restricted stock units and 1,033,833 shares of common stock in connection with the exercise of stock options resulting in proceeds of approximately $1.5 million.
During fiscal 2013, restricted stock unit awards for a total of 176,839 shares awarded to directors vested, but the issuance and delivery of these shares are deferred until the director resigns.
Fiscal 2012. On July 30, 2009, the Company entered into a Controlled Equity Offering Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co. (“Cantor”), providing for the sale of up to 12,500,000 shares of common stock from time to time in the open market at prevailing prices. Pursuant to the Sales Agreement, sales of common stock are made in such quantities and on such minimum price terms as the Company may set from time to time. During fiscal 2012, 3,668,656 shares of common stock were sold under the Sales Agreement at an average price of $2.81 per share raising proceeds of approximately $10.3 million ($10.1 million after offering expenses, including commissions). As of September 30, 2012, a total of 12,355,166 shares of common stock were sold under the Sales Agreement at an average price of $2.83 per share raising gross proceeds of approximately $35.0 million ($33.8 million after offering expenses, including commissions).
During fiscal 2012, the Company received proceeds of approximately $7.8 million from the exercise of warrants to purchase 5,445,061 shares of the Company’s common stock. The warrants had been issued in connection with the Company’s registered securities offering in April 2008 at an exercise price of $1.43 per share.
During fiscal 2012, the Company issued 204,176 shares of common stock in connection with the vesting of restricted stock units and 1,673,811 shares of the Company’s common stock upon the exercise of outstanding options resulting in proceeds of approximately $1.3 million.
F-26
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
During fiscal 2012, restricted stock unit awards for a total of 187,083 shares awarded to directors vested, but the issuance and delivery of these shares are deferred until the director resigns.
Fiscal 2011. In November 2010, the Company completed a common stock offering raising $88.0 million in gross proceeds and approximately $83.0 million in net proceeds to the Company after deducting underwriting discounts, commissions and offering expenses. In connection with the offering, 20 million shares of common stock were sold at a public offering price of $4.40 per share.
During fiscal 2011, 2,147,000 shares of common stock were sold under the Sales Agreement at an average price of $3.78 per share raising proceeds of $8.1 million ($7.9 million after offering expenses, including commissions).
During fiscal 2011, the Company issued 252,941 shares of common stock in connection with the vesting of restricted stock units. In connection with the vesting, an officer exercised his option to pay for minimum required withholding taxes associated with the vesting of restricted stock by surrendering 1,446 shares of common stock at an average market price of $3.92 per share.
Also during fiscal 2011, the Company issued 814,454 shares of common stock upon the exercise of outstanding options resulting in proceeds of approximately $638,000.
Warrants
During fiscal 2013, the Company received proceeds of approximately $1.0 million from the exercise of warrants to purchase 710,109 shares of the Company’s common stock. The warrants had been issued in connection with the Company’s registered securities offering in April 2008 at an exercise price of $1.43 per share.
During fiscal 2012, the Company issued warrants to purchase 491,007 shares of the Company’s common stock at an exercise price of $2.78 per share in connection with the financing transaction in May 2012. (See Note 7, “Notes Payable.”) In fiscal 2013, 437,050 of these warrants were exercised in a cashless transaction resulting in the issuance of 72,185 shares of the Company’s common stock. As of September 30, 2013, 53,957 warrants remain outstanding and exercisable. The warrants expire in May 2022.
During fiscal 2012, the Company received proceeds of approximately $7.8 million from the exercise of warrants to purchase 5,445,061 shares of the Company’s common stock. The warrants had been issued in connection with the Company’s registered securities offering in April 2008 at an exercise price of $1.43 per share.
A following table summarizes all warrant activity for fiscal 2013 and 2012:
| | | | | | | | | | | | |
| | | Shares of
Common Stock
Purchasable Upon
Exercise of Warrants | | | Weighted
Average
Exercise Price
per Share | | | Range of
Exercise Prices | |
Outstanding at September 30, 2011 | | | 6,155,170 | | | $ | 1.43 | | | $ | 1.43 | |
Issued | | | 491,007 | | | $ | 2.78 | | | $ | 2.78 | |
Exercised | | | (5,445,061 | ) | | $ | 1.43 | | | $ | 1.43 | |
| | | | | | | | | | | | |
Outstanding at September 30, 2012 | | | 1,201,116 | | | $ | 1.98 | | | $ | 1.43-$2.78 | |
Exercised | | | (1,147,159 | ) | | $ | 1.94 | | | $ | 1.43-$2.78 | |
| | | | | | | | | | | | |
Outstanding at September 30, 2013 | | | 53,957 | | | $ | 2.78 | | | $ | 2.78 | |
| | | | | | | | | | | | |
F-27
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Employee equity incentive plans
The Company currently has one equity incentive plan, which is the 2005 Equity Incentive Plan (the “2005 Plan”). The Amended and Restated 1994 Stock Option Plan (the “1994 Plan”), the Amended and Restated 1998 Stock Option Plan (the “1998 Plan”), the 2000 Stock Option Plan (the “2000 Plan”) and the 2003 Equity Incentive Plan (the “2003 Plan” and, together with the 1994 Plan, 1998 Plan, 2000 Plan and 2005 Plan, the “Plans”) are expired and the Company no longer grants share-based awards from these plans. All of the Plans were approved by the stockholders, except for the 2003 Plan, which was approved solely by the Board of Directors. Share-based awards are subject to terms and conditions established by the Compensation Committee of the Company’s Board of Directors. The Company’s policy is to issue new common shares upon the exercise of stock options, conversion of share units or purchase of restricted stock.
During fiscal 2013, 2012, and 2011, the Company granted share-based awards under both the 2003 Plan and the 2005 Plan. Under the 2003 Plan and 2005 Plan, options to purchase shares, restricted stock units, restricted stock and other share-based awards may be granted to the Company’s directors, employees and consultants. Effective March 2013, the Company is no longer able to issue grants from the 2003 Plan. Under the Plans, as of September 30, 2013, the Company had an aggregate of 12,439,448 shares of common stock reserved for issuance. Of those shares, 11,505,566 were subject to outstanding options and other awards and 933,882 shares were available for future grants of share-based awards. The Company may also issue share-based awards outside of the Plans. As of September 30, 2013, there were 221,900 options to purchase shares of common stock that were issued outside of the Plans (inducement option grants). None of the share-based awards are classified as a liability as of September 30, 2013.
2005 Equity Incentive Plan. On March 17, 2005, the Company’s stockholders approved the adoption of the 2005 Plan that initially provided for the issuance of up to 500,000 shares of common stock, plus an annual increase beginning in fiscal 2006 equal to the lesser of (a) 1% of the shares of common stock outstanding on the last day of the immediately preceding fiscal year, (b) 325,000 shares of common stock, or (c) such lesser number of shares of common stock as the Board of Directors shall determine. Pursuant to the provisions of annual increases, the number of authorized shares of common stock for issuance under the 2005 Plan increased by 273,417 shares effective November 16, 2005, 317,084 shares effective November 30, 2006, and an additional 325,000 shares effective for each date of December 4, 2007, November 6, 2008, November 11, 2009, November 10, 2010, November 10, 2011 and November 15, 2012 to a total of 3,040,501 shares. In February 2006, the Company’s stockholders eliminated the limitation on the number of shares of common stock that may be issued as restricted stock under the 2005 Plan. The 2005 Plan allows the Company to grant options, restricted stock awards and stock appreciation rights to the Company’s directors, officers, employees and consultants. As of September 30, 2013, 933,882 shares of common stock remained available for issuance under the 2005 Plan.
Stock Options. Stock options are granted with an exercise price equal to the current market price of the Company’s common stock at the grant date and have 10-year contractual terms. For option awards, typically 25% of the option shares vest and become exercisable on the first anniversary of the grant date and the remaining 75% of
F-28
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
the option shares vest and become exercisable quarterly in equal installments thereafter over three years. Certain option awards provide for accelerated vesting if there is a change in control (as defined in the Plans). Summaries of stock options outstanding and changes during fiscal 2013 are presented below.
| | | | | | | | | | | | | | | | |
| | | Number of
Shares | | | Weighted Average
Exercise Price Per
Share | | | Weighted Average
Remaining
Contractual Term
(In Years) | | | Aggregate
Intrinsic
Value | |
Outstanding at September 30, 2012 | | | 8,142,468 | | | $ | 2.67 | | | | | | | | | |
Granted | | | 2,151,507 | | | $ | 2.89 | | | | | | | | | |
Exercised | | | (1,033,833 | ) | | $ | 1.45 | | | | | | | | | |
Forfeited | | | (437,101 | ) | | $ | 3.08 | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Outstanding at September 30, 2013 | | | 8,823,041 | | | $ | 2.85 | | | | 7.4 | | | $ | 14,106,009 | |
| | | | | | | | | | | | | | | | |
Vested and expected to vest in the future at September 30, 2013 | | | 8,566,637 | | | $ | 2.85 | | | | 7.3 | | | $ | 13,748,940 | |
| | | | | | | | | | | | | | | | |
Exercisable at September 30, 2013 | | | 4,621,266 | | | $ | 2.83 | | | | 6.3 | | | $ | 8,232,487 | |
| | | | | | | | | | | | | | | | |
The weighted average grant-date fair values of options granted during fiscal 2013, 2012, and 2011 were $1.95, $1.89 and $3.18 per share, respectively. The total intrinsic value of options exercised during fiscal 2013, 2012 and 2011 was $2.2 million, $3.7 million and $2.5 million, respectively, based on the differences in market prices on the dates of exercise and the option exercise prices. As of September 30, 2013, the total unrecognized compensation cost related to options was approximately $8.1 million, which is expected to be recognized over a weighted-average period of 2.5 years, based on the vesting schedules.
The fair value of each option award is estimated on the date of grant using the Black-Scholes model that uses the assumptions noted in the following table. Expected volatilities are based on historical volatility of the Company’s common stock and other factors. The expected term of options granted is based on analyses of historical employee termination rates and option exercises. The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant.
Assumptions used in the Black-Scholes model for options granted during fiscal 2013, 2012, and 2011 were as follows:
| | | | | | |
| | | 2013 | | 2012 | | 2011 |
Expected volatility | | 82.7% — 84.1% | | 103.7% — 108.7% | | 103.7% — 108.0% |
Weighted-average volatility. | | 83.7% | | 107.9% | | 107.7% |
Average expected term in years | | 5.4 | | 5.8 | | 5.8 |
Risk-free interest rate (zero coupon U.S. Treasury Note) . | | 0.8% — 1.7% | | 0.9% — 1.3% | | 1.1% — 2.5% |
Expected dividend yield | | 0% | | 0% | | 0% |
Restricted stock units (“RSU”). RSUs granted to employees generally vest based on three or four years of continuous service from the date of grant. RSUs granted to non-employee directors generally vest over the term of one year from the grant date and are not released until the awardee’s termination of service. Vesting for non-employee director grants allow for accelerated vesting of RSUs in the case of a non-employee director’s resignation where either: (i) he/she has served for at least four years as a member of the Board and is in good standing at the time of resignation, or (ii) he/she resigns for reasons related to health or family matters and is otherwise in good standing at the time of resignation. The following table summarizes the RSU activities for fiscal 2013.
F-29
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| | | | | | | | |
| | | Number of Shares | | | Weighted Average
Grant Date
Fair Value | |
Unvested at September 30, 2012 | | | 1,016,624 | | | $ | 2.23 | |
Granted | | | 1,166,303 | | | $ | 2.65 | |
Vested | | | (513,188 | ) | | $ | 2.64 | |
Forfeited | | | (32,529 | ) | | $ | 2.23 | |
| | | | | | | | |
Unvested at September 30, 2013 | | | 1,637,210 | | | $ | 2.40 | |
| | | | | | | | |
The weighted average grant-date fair value of RSUs granted during fiscal 2013, 2012, and 2011 was $2.65, $2.15 and $4.12 per unit, respectively. The fair value of RSUs vested during fiscal 2013, 2012, and 2011 was approximately $1.4 million, $1.2 million and $1.1 million, respectively. As of September 30, 2013, the total unrecognized compensation cost related to unvested stock units was approximately $3.1 million, which is expected to be recognized over a weighted-average period of 2.6 years, based on the vesting schedules and assuming no forfeitures.
At September 30, 2013, there were 1,267,215 shares of restricted stock with a weighted-average grant date fair value of $2.08 per share awarded to directors that have vested but are still restricted until the directors resign. In fiscal 2013, 2012, and 2011, 176,839, 187,083 and 255,343 shares of restricted stock, respectively, vested but remained restricted.
During fiscal 2013, the Company granted performance-based RSUs to purchase 325,436 shares of common stock from the 2003 Stock Option Plan. These performance-based RSUs are included in the above unvested RSU table. The RSUs have a performance goal related to fiscal 2013 revenue that determines when vesting begins and the actual number of shares to be awarded ranging from 0% to 100% of target. Vesting is over three years beginning on the date the performance goal is achieved (“Achievement Date”), with 50% of the RSU shares vesting on the first anniversary of the Achievement Date and the remaining 50% of the RSU shares vesting annually in equal installments thereafter over two years. At September 30, 2013, the performance goal had been met and 321,436 performance RSUs are outstanding and have begun to vest.
During fiscal 2012, the Company granted performance-based RSUs to purchase 30,000 shares of common stock from the 2003 Stock Option Plan. These performance-based RSUs are included in the above unvested RSU table. The RSUs have a performance goal related to revenue that determines when vesting begins and the actual number of shares to be awarded ranging from 0% to 100% of target. Vesting is over 4 years beginning on the Achievement Date, with 25% of the RSU shares vesting on the first anniversary of the Achievement Date and the remaining 75% of the RSU shares vesting quarterly in equal installments thereafter over three years. At September 30, 2013, the performance goal had not been met and all 30,000 performance-based RSUs were outstanding.
During fiscal 2010, the Company awarded an RSU representing the right to acquire a total of 120,000 shares of common stock to a non-employee. The grant date fair value of this award was $2.08 per share. The restricted stock units vested on October 15, 2011, and were re-measured at each balance sheet date and again on the date vested at $3.22 per share.
| 11. | Research, License, Supply and other Agreements |
Center for Neurologic Study. The Company holds the exclusive worldwide marketing rights to NUEDEXTA for certain indications pursuant to an exclusive license agreement with CNS.
The Company paid a $75,000 milestone upon FDA approval of NUEDEXTA for the treatment of PBA in fiscal 2011. In addition, the Company is obligated to pay CNS a royalty ranging from approximately 5% to 8% of
F-30
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
net U.S. GAAP revenue generated by sales of NUEDEXTA. During fiscal 2013, 2012 and 2011, royalties of approximately $3.5 million, $1.8 million and $309,000, respectively, are recorded to cost of product sales in the accompanying consolidated statements of operations. Under certain circumstances, the Company may have the obligation to pay CNS a portion of net revenues received if the Company sublicenses NUEDEXTA to a third party.
Under the agreement with CNS, the Company is required to make payments on achievements of up to a maximum of ten milestones, based upon five specific clinical indications. Maximum payments for these milestone payments could total approximately $1.1 million if the Company pursued the development of NUEDEXTA for all five of the licensed indications. In general, individual milestones range from $75,000 to $125,000 for each accepted NDA and a similar amount for each approved NDA in addition to the royalty discussed above on net U.S. GAAP revenues. The Company does not have the obligation to develop additional indications under the CNS license agreement.
Concert Pharmaceuticals, Inc.— The Company holds the exclusive worldwide marketing rights to develop and commercialize Concert’s d-DM compounds for the potential treatment of neurological and psychiatric disorders, as well as certain rights to other deuterium-modified dextromethorphan compounds pursuant to a license agreement with Concert.
Under the agreement with Concert, the Company is obligated to make milestone and royalty payments to Concert based on successful advancement of d-DM products for one or more indications in the United States, Europe, and Japan. Individual milestone payments range from $2.0 — $6.0 million, $1.5 — $15.0 million, and $25.0 — $60.0 million for clinical, regulatory and commercial targets respectively, and in aggregate could total over $200 million. Royalty payments are tiered, beginning in the single-digits and increasing to the low double-digits for worldwide net sales of d-DM products exceeding $1.0 billion annually. As of September 30, 2013, the Company has paid $2.0 million for milestones that have been achieved pursuant to this agreement, which were recorded to research and development expense in the accompanying consolidated statement of operations.
OptiNose AS. The Company holds an exclusive license agreement for the development and commercialization of a novel Breath Powered intranasal delivery system containing low-dose sumatriptan powder to treat acute migraine, AVP-825. The licensed territories are the United States, Canada and Mexico.
Under the terms of the agreement, OptiNose received an up-front cash payment of $20.0 million, which was recorded to research and development expense in the accompanying consolidated statement of operations. The Company and OptiNose will share certain shared development costs. OptiNose is eligible to receive up to an additional $90.0 million in total payments resulting from the achievement of future clinical, regulatory and commercial milestones. In addition, if approved, we will make tiered royalty payments to OptiNose of a low double-digit percentage of net sales in the United States, Canada and Mexico.
Merck. In August 2013, the Company entered into a multi-year agreement with Merck to co-promote Merck’s type 2 diabetes therapies JANUVIA® (sitagliptin) and the sitagliptin family of products in the long-term care institutional setting in the United States beginning October 1, 2013. The term of the Agreement will continue for three years following the launch date of the co-promotion activities.
Under the terms of the Agreement, the Company will be compensated via a (i) fixed monthly fee and (ii) performance fee based on the amount of the products sold by the Company above a predetermined baseline. A significant majority of the fee is performance based. Over the three years of the agreement, the Company could receive up to a maximum of $60.0 million in compensation.
The parties will jointly develop a product marketing and promotion plan. The Company will lead sales implementation with its institutional sales force. Merck will be required to provide a specific level of marketing and promotional support, and the Company is required to provide predetermined level of sales efforts. Merck will continue to be responsible for all other aspects of research, manufacturing, supply, regulatory, medical, managed care and marketing activities for the products.
F-31
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Agreement may be terminated without cause by either party upon 90 days written notice, at any time after the first anniversary of the launch date. If after the first anniversary, Merck terminates the Agreement without cause, Merck will be required to continue paying a portion of the performance based fee that would otherwise be due for a period of twelve months (though not beyond the expiration of three years following the launch date.) The Agreement may be terminated with cause by either party based on certain events, including a material uncured breach by either party, a failure to achieve certain sales thresholds by the Company, an assignment for the benefit of creditors, or upon mutual agreement. Additionally, Merck may terminate the Agreement (a) upon change of control of the Company; (b) if the Company adds promotional responsibilities for additional products to its sales representatives promoting the products; or (c) upon a violation of applicable law.
GlaxoSmithKline Subsidiary, SB Pharmco Puerto Rico, Inc. On March 31, 2000, the Company signed an exclusive license agreement with GSK for rights to manufacture and sell Abreva (docosanol 10% cream) as an over-the-counter product in the United States and Canada as a treatment for cold sores. Under the terms of the license agreement, GSK Consumer Healthcare is responsible for all sales and marketing activities and the manufacturing and distribution of Abreva in the U.S. and Canada. The terms of the license agreement provide for the Company to earn royalties on product sales. In October 2000 and August 2005, GSK launched Abreva in the United States and Canada, respectively. All milestones under the agreement were earned and paid prior to fiscal 2003. During fiscal 2003, the Company sold an undivided interest in the GSK license agreement to Drug Royalty with a term until the later of December 13, 2013 or until the expiration of the patent for Abreva on April 30, 2014. (See Note 6, “Deferred Revenues.”)
Azur Pharma. In August 2007, the Company entered into a license agreement with Azur, where the Company could receive up to $2.0 million in royalties, based on 3% of annualized net product revenues in excess of $17.0 million. During fiscal 2012 and 2011, the Company recorded royalty revenues of approximately $492,000 and $527,000 in connection with this agreement. As of September 30, 2012, the Company had received $2.0 million in royalties related to annual revenue in excess of $17.0 million and no further royalties will be recognized related under this license agreement.
Components of the income tax provision are as follows for the fiscal years ended September 30, 2013, 2012 and 2011:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | 2011 | |
Current: | | | | | | | | | | | | |
State | | $ | 3,200 | | | $ | 3,200 | | | $ | 3,200 | |
| | | | | | | | | | | | |
Deferred: | | | | | | | | | | | | |
Federal | | | (23,055,582 | ) | | | (15,486,906 | ) | | | (18,297,582 | ) |
State | | | (4,938,691 | ) | | | (2,731,249 | ) | | | (2,020,800 | ) |
| | | | | | | | | | | | |
| | | (27,994,273 | ) | | | (18,218,155 | ) | | | (20,318,382 | ) |
| | | | | | | | | | | | |
Increase in deferred income tax asset valuation allowance | | | 27,994,273 | | | | 18,218,155 | | | | 20,318,382 | |
| | | | | | | | | | | | |
Total income tax provision | | $ | 3,200 | | | $ | 3,200 | | | $ | 3,200 | |
| | | | | | | | | | | | |
F-32
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Deferred income taxes reflect the income tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred income tax balance are as follows:
| | | | | | | | | | | | |
| | | September 30, | |
| | | 2013 | | | 2012 | | | 2011 | |
Net operating loss carryforwards | | $ | 155,960,858 | | | $ | 135,430,308 | | | $ | 117,027,433 | |
Deferred revenue | | | 504,143 | | | | 1,570,370 | | | | 2,960,454 | |
Research credit carryforwards | | | 9,185,508 | | | | 11,202,848 | | | | 11,560,603 | |
Capitalized license fees and patents | | | 10,176,119 | | | | 2,017,029 | | | | 2,342,589 | |
Capitalized research and development costs | | | 29,705 | | | | 99,671 | | | | 263,942 | |
Share-based compensation and options | | | 8,452,457 | | | | 6,979,532 | | | | 5,586,697 | |
Foreign tax credits | | | — | | | | — | | | | 595,912 | |
Other | | | 3,404,764 | | | | 2,579,478 | | | | 1,480,613 | |
| | | | | | | | | | | | |
Deferred income tax assets | | | 187,713,554 | | | | 159,879,236 | | | | 141,818,243 | |
| | | | | | | | | | | | |
Deferred tax liabilities: | | | | | | | | | | | | |
Other | | | (162,824 | ) | | | (322,779 | ) | | | (479,941 | ) |
| | | | | | | | | | | | |
Deferred tax liabilities | | | (162,824 | ) | | | (322,779 | ) | | | (479,941 | ) |
| | | | | | | | | | | | |
Less valuation allowance for net deferred income tax assets | | | (187,550,730 | ) | | | (159,556,457 | ) | | | (141,338,302 | ) |
| | | | | | | | | | | | |
Net deferred tax assets / (liabilities) | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
The Company has provided a full valuation allowance against the net deferred income tax assets recorded as of September 30, 2013, 2012 and 2011 as the Company concluded that they are unlikely to be realized. As of September 30, 2013 the Company had federal and state net operating loss carryforwards of $407.4 million and $356.1 million, respectively. As of September 30, 2013 the Company had federal and California research and development credits of $7.8 million and $6.8 million, respectively. The net operating loss and research credit carryforwards will expire on various dates through 2029, unless previously utilized. The Company also has no foreign tax credit carryforwards at September 30, 2013. In the event of certain ownership changes, the Tax Reform Act of 1986 imposes certain restrictions on the amount of net operating loss and credit carryforwards that the Company may use in any year.
Pursuant to Internal Revenue Code (“IRC”) Sections 382 and 383, annual use of the Company’s net operating loss and research and development credit carryforwards may be limited in the event of a cumulative change in ownership of more than 50% within a three-year period. The Company has completed an analysis under IRC Sections 382 and 383 through September 30, 2013, and has determined that it has not experienced an ownership change as defined by Section 382. The Company continues to monitor changes in our ownership as any future ownership changes may significantly reduce the utilization of the net operating loss carryforwards and research tax credits before they expire.
F-33
Avanir Pharmaceuticals, Inc.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
A reconciliation of the federal statutory income tax rate and the effective income tax rate is as follows for the fiscal years ended September 30:
| | | | | | | | | | | | |
| | | 2013 | | | 2012 | | | 2011 | |
Federal statutory rate | | | (34 | )% | | | (34 | )% | | | (34 | )% |
Increase in deferred income tax asset valuation allowance | | | 37 | | | | 31 | | | | 34 | |
State income taxes, net of federal effect | | | (5 | ) | | | (5 | ) | | | (4 | ) |
Research and development credits | | | 3 | | | | 1 | | | | 0 | |
Expired net operating loss and other credits | | | — | | | | 6 | | | | 4 | |
Other | | | (1 | ) | | | 1 | | | | 0 | |
| | | | | | | | | | | | |
Effective income tax rate | | | 0 | % | | | 0 | % | | | 0 | % |
| | | | | | | | | | | | |
The Company has established an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code. The plan allows participating employees to deposit into tax deferred investment accounts up to 50% of their salary, subject to annual limits. The Company is not required to make matching contributions under the plan. However, the Company voluntarily contributed approximately $401,000, $412,000 and $204,000 in fiscal 2013, 2012, and 2011, respectively, to the plan.
The Company operates the business on the basis of a single reportable segment, which is the business of discovery, development and commercialization of novel therapeutics for chronic diseases. The Company’s chief operating decision-maker is the Chief Executive Officer, who evaluates the company as a single operating segment.
The Company categorizes revenues by geographic area based on selling location. All operations are currently located in the United States; therefore, total revenues for fiscal 2013, 2012, and 2011 are attributed to the United States. All long-lived assets at September 30, 2013 and 2012 are located in the United States.
The Company sells NUEDEXTA to a limited number of wholesalers. Three wholesalers accounted for 88%, 89% and 91% of gross product sales in fiscal 2013, 2012 and fiscal 2011, respectively. In addition, the three wholesalers accounted for 91% of trade receivables at September 30, 2013 and 2012.
Less than 10% of the Company’s total revenues in fiscal 2013 and 2012 were derived from a license agreement with GSK and the sale of rights to royalties under that agreement. Royalties from GSK were approximately 37% of total revenues in fiscal 2011.
In November 2013, the Company entered into a lease for approximately 61,000 square feet of office space commencing in November 2013 for an initial lease term of 86 months. The lease has scheduled rent increases each year. The aggregate minimum payments over the initial term of the lease are expected to be approximately $14.3 million.
* * * * *
F-34