UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2013
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from __________________ to __________________
Commission File Number: 001-34236
| BIOSPECIFICS TECHNOLOGIES CORP. |
(Exact name of registrant as specified in its charter)
| Delaware | 11-3054851 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
| 35 Wilbur Street, Lynbrook, NY | 11563 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: 516.593.7000
Securities registered under Section 12(b) of the Exchange Act:
Title of each class | Name of each exchange on which registered |
| Common Stock | The Nasdaq Global Market |
Securities registered under Section 12(g) of the Exchange Act: NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
¨Yes þNo
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
¨Yes þNo
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Exchange Act from their obligations under those Sections.
Indicate by check mark whether the registrant (1) filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
þYes ¨No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.
¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
i
Large accelerated filer ¨ | Accelerated filer þ | |
Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
¨Yes þNo
The aggregate market value of voting and non-voting common stock held by non-affiliates of the Registrant as of June 28, 2013, the last business day of the registrant’s most recently completed second fiscal quarter, was approximately $77.0 million.
Note – If a determination as to whether a particular person or entity is an affiliate cannot be made without involving unreasonable effort and expense, the aggregate market value of the common stock held by non-affiliates may be calculated on the basis of assumptions reasonable under the circumstances, provided that the assumptions are set forth in this Form.
The number of shares outstanding of the registrant’s common stock as of March 3, 2014 is 6,388,468.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for its 2014 Annual Meeting of Stockholders scheduled to be held on June , 24, 2014, which is expected to be filed with the Securities and Exchange Commission not later than 120 days after the end of the registrant’s fiscal year ended December 31, 2013, are incorporated by reference into Part III of this annual report on Form 10-K. With the exception of the portions of the registrant’s definitive proxy statement for its 2014 Annual Meeting of Stockholders that are expressly incorporated by reference into this annual report on Form 10-K, such proxy statement shall not be deemed filed as part of this annual report on Form 10-K.
Page | |||
| PART I | 3 | ||
| Item 1. | 3 | ||
| Item 1A. | 19 | ||
| Item IB. | 30 | ||
| Item 2. | 30 | ||
| Item 3. | 30 | ||
| Item 4. | 30 | ||
| PART II | 31 | ||
| Item 5. | 31 | ||
| Item 6. | 34 | ||
| Item 7. | 34 | ||
| Item 7A. | 52 | ||
| Item 8. | 52 | ||
| Item 9. | 52 | ||
| Item 9A. | 52 | ||
| Item 9B. | 53 | ||
| PART III | 53 | ||
| Item 10 | 53 | ||
| Item 11. | 53 | ||
| Item 12. | 53 | ||
| Item 13. | 53 | ||
| Item 14. | 54 | ||
| PART IV | 54 | ||
| Item 15. | 54 | ||
Introductory Comments – Terminology
Throughout this annual report on Form 10-K (this “Report”), the terms “BioSpecifics,” “Company,” “we,” “our,” and “us” refer to BioSpecifics Technologies Corp. and its subsidiary, Advance Biofactures Corporation (“ABC-NY”).
Introductory Comments – Forward-Looking Statements
This Report includes “forward-looking statements” within the meaning of, and made pursuant to the safe harbor provisions of, the Private Securities Litigation Reform Act of 1995. All statements other than statements of historical fact, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management, expected revenue growth, and the assumptions underlying or relating to such statements, are “forward-looking statements”. The forward-looking statements in this Report include statements concerning, among other things, the market potential for the use of XIAFLEX to treat Dupuytren’s contracture and Peyronie’s disease and the likelihood of success of Auxilium’s plans for marketing and sales for those indications; the potential approval by the FDA of the sBLA filed by Auxilium to expand the label for Dupuytren’s contracture for multiple injections; the timing for Swedish Orphan Biovitrum AB to submit to the EMA a MAA for Peyronie’s disease; the timing of the release by Auxilium of top-line data from its phase IIb study of collagenase clostridium histolyticum (“CCH”) as a treatment for frozen shoulder; the timing of the release by Auxilium of top-line data from its phase IIa study of CCH for cellulite; the expectation of XIAFLEX to be the first and only pharmaceutical therapy for frozen shoulder and cellulite; the timing for BioSpecifics to submit to Auxilium the Chien-804 final study report for canine lipoma and the likelihood that Auxilium will exercise its opt in rights for such indication; the timing for BioSpecifics’ to initiate and complete enrollment of a placebo-controlled trial for human lipoma; the timing of the release by BioSpecifics of top-line data from its pre-clinical study in uterine fibroids and the potential success of, and commercial market for, such indication; the expected life of patents; the expected marketing exclusivity for XIAFLEX for Dupuytren’s contracture in Europe; the timing for receiving the final earn-out payment under our agreement with DFB Biotech, Inc.; and the projected receipt of payments from Auxilium. In some cases, these statements can be identified by forward-looking words such as “believe,” “expect,” “anticipate,” “plan,” “estimate,” “likely,” “may,” “will,” “could,” “continue,” “project,” “predict,” “goal,” the negative or plural of these words, and other similar expressions. These forward-looking statements are predictions based on BioSpecifics’ current expectations and its projections about future events. There are a number of important factors that could cause BioSpecifics’ actual results to differ materially from those indicated by such forward-looking statements, including the timing of regulatory filings and action; the ability of BioSpecifics’ partner, Auxilium Pharmaceuticals, Inc., and its partners, Asahi Kasei Pharma Corporation, Actelion Pharmaceuticals Ltd. and Swedish Orphan Biovitrum AB, to achieve their objectives for XIAFLEX in their applicable territories; the market for XIAFLEX in, and timing, initiation and outcome of clinical trials for, additional indications including frozen shoulder, cellulite, human lipoma and canine lipoma and uterine fibroids, all of which will determine the amount of milestone, royalty, mark-up on cost of goods sold and sublicense income BioSpecifics may receive; the potential of XIAFLEX to be used in additional indications; and other risk factors set forth in Part I, Item 1A of this Report under the heading “Risk Factors”. All forward-looking statements included in this Report are made as of the date hereof, and we assume no obligation to update these forward-looking statements.
PART I
Overview
We are a biopharmaceutical company involved in the development of an injectable collagenase for multiple indications. We have a development and license agreement with Auxilium Pharmaceuticals, Inc. (“Auxilium”) for injectable collagenase (which Auxilium has named XIAFLEX®) for marketed indications and collagenase clostridium histolyticum (“CCH”) for indications in development. Auxilium has an option to acquire additional indications that we may pursue, including human and canine lipoma. Auxilium is currently selling XIAFLEX in the U.S. for the treatment of Dupuytren’s contracture and Peyronie’s disease. Following the termination of the agreement between Auxilium and Pfizer, Inc. (“Pfizer”), Auxilium entered into an agreement with Swedish Orphan Biovitrum AB (“Sobi”) pursuant to which Sobi has marketing rights for XIAPEX® (the EU trade name for collagenase clostridium histolyticum) for Dupuytren’s contracture and Peyronie’s disease in Europe and certain Eurasian countries. Sobi is currently selling XIAPEX in Europe for the treatment of Dupuytren’s contracture. In addition, Auxilium has an agreement with Asahi Kasei Pharma Corporation (“Asahi”) pursuant to which Asahi has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Japan. Auxilium also has an agreement with Actelion Pharmaceuticals Ltd. (“Actelion”) pursuant to which Actelion has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Canada, Australia, Brazil and Mexico.
Operational Highlights
Peyronie’s Disease. In December 2013, the U.S. Food and Drug Administration (“FDA”) approved Auxilium’s supplemental Biologics License Application (“sBLA”) for XIAFLEX for the treatment of Peyronie's disease. As a result, we recognized a $2.0 million milestone payment from Auxilium. This is the first and only FDA-approved biologic therapy indicated for the treatment of Peyronie's disease in men with a palpable plaque and a curvature of 30 degrees or greater at the start of therapy.
Dupuytren’s Contracture. In the fourth quarter of 2013, Auxilium presented results from Year 4 of the Collagenase Optimal Reduction of Dupuytren’s Long-term Evaluations of Success Study (“CORDLESS”). CORDLESS is a five-year observational study designed to assess the rates of recurrence following treatment with XIAFLEX, as well as long-term safety and progression of disease in patients from earlier Auxilium studies. Also in the fourth quarter of 2013, Auxilium announced positive results from the open label, phase IIIb MULTICORD (Multiple Treatment Investigation of Collagenase Optimizing the Resolution of Dupuytren's) study evaluating XIAFLEX for the concurrent treatment of adult Dupuytren's contracture patients with multiple palpable cords. The study demonstrated that two concurrent injections of XIAFLEX in patients with multiple Dupuytren's contractures resulted in comparable improvement in joint contracture and range of motion to those seen in previous studies when XIAFLEX was administered as single injections, 30 days apart. Adverse event (AE) rates were also comparable to single injection administration 30 days apart. Based on the results, Auxilium submitted a sBLA to the FDA in the fourth quarter 2013 seeking expansion of labeling for the concurrent treatment of multiple palpable cords and hopes to receive approval by the end of 2014. On February 24, 2014, Auxilium reported that the FDA had accepted the submission with a PDUFA date of October 20, 2014.
Cellulite. Auxilium expanded the field of its license for injectable collagenase to include the potential treatment of cellulite by exercising, in January 2013, its exclusive option under our development and license agreement. In October 2013, Auxilium dosed the first patient in its phase IIa clinical trial of collagenase clostridium histolyticum (“CCH”) for the treatment of cellulite. Auxilium anticipates top-line results from the study in the first quarter of 2015. No FDA-approved pharmaceutical therapies are currently available for the treatment of cellulite.
Frozen Shoulder. Auxilium reported positive top-line data in the first quarter of 2013 from its phase IIa clinical trial of XIAFLEX for the potential treatment of frozen shoulder (adhesive capsulitis). In December 2013, Auxilium dosed the first patient in its phase IIb study of CCH for the treatment of frozen shoulder. Auxilium anticipates top-line results from the study in the first quarter of 2015. No FDA-approved pharmaceutical therapies are currently available for the treatment of frozen shoulder.
Human Lipoma. In the first quarter 2014, we announced top-line data from the phase II dose escalation clinical trial of CCH for the treatment of human lipoma. The primary efficacy outcome of active reduction of the visible surface area of the lipoma as measured by caliper was met, combining all patients (p<0.0001). There were no serious adverse events reported during the trial.
Canine Lipoma. In fourth quarter 2013, we announced top-line data from Chien-804, the placebo-controlled, double-blind, randomized phase II trial evaluating the efficacy of CCH in canines with benign subcutaneous lipomas. The trial did not meet its primary endpoint of a statistically significant post-treatment difference in the mean percent change in lipoma volume by CT scan; however, in the responder analysis there was a statistically significant reduction in lipoma surface area among dogs treated with CCH (p=0.0084). Auxilium has the option to exclusively license development and marketing rights to the canine lipoma indication, which would trigger an opt-in payment and potential future milestone and royalty payments from Auxilium. We anticipate submitting a final study report to Auxilium in the first quarter of 2014, which will trigger the 120 day opt-in period. If Auxilium does not opt-in, then the rights will revert back to us.
Uterine Fibroids. In third quarter 2013, we announced that a poster titled, “Biomechanical Evaluation of Human Uterine Fibroids after Exposure to Purified Clostridial Collagenase” was presented at the Society for the Study of Reproduction 46th Annual Meeting in Montreal, Quebec, Canada. The poster provided data which show that highly purified collagenase can reduce the stiffness of human uterine fibroid tissue in laboratory experiments. Increased tissue rigidity has been implicated as a cause of the morbidity associated with uterine fibroids. We anticipate releasing top-line data from a pre-clinical study in the second quarter of 2014.
Research and Development of Injectable Collagenase for Multiple Indications
On June 3, 2004, we entered into, and later amended, a development and license agreement with Auxilium pursuant to which we granted to Auxilium an exclusive worldwide license to develop, market and sell products containing our injectable collagenase for the treatment of Dupuytren’s contracture, Peyronie's disease, frozen shoulder and cellulite, as well as an exclusive option to develop and license the technology for use in additional indications, such as human and canine lipoma and uterine fibroids, other than dermal formulations labeled for topical administration. We have amended and restated that agreement twice, once on December 11, 2008 in connection with the Development, Commercialization and Supply Agreement, dated December 17, 2008 between an Auxilium subsidiary and Pfizer, and more recently on August 31, 2011 (the “Auxilium Agreement”). The Auxilium Agreement and other licensing agreements are discussed more fully throughout this Item 1, in particular under the section titled “Licensing and Marketing Agreements.”
Background on Collagenase
Collagenase is the only protease that can hydrolyze the triple helical region of collagen under physiological conditions. The specific substrate collagen comprises approximately one-third of the total protein in mammalian organisms, and it is the main constituent of skin, tendon, and cartilage, as well as the organic component of teeth and bone. The body relies on endogenous collagenase production to remove dead tissue, and collagenase production is an essential biological mechanism, which regulates matrix remodeling and the normal turnover of tissue. The Clostridial collagenase produced by us has a broad specificity towards all types of collagen and is acknowledged as much more efficient than mammalian collagenases. Clostridial collagenase cleaves the collagen molecule at multiple sites along the triple helix whereas the mammalian collagenase is only able to cleave the molecule at a single site along the triple helix.
Collagenase is widely used for cell dispersion for tissue disassociation and cell culture because it does not damage the cell membrane. Since the main component of scar tissue is collagen, collagenase has been used in a variety of clinical investigations to remove scar tissue without surgery. Histological and biochemical studies have shown that the tissue responsible for the deformities associated with Dupuytren’s contracture and Peyronie’s disease is primarily composed of collagen. Surgical removal of scar tissue has the potential to result in complications including increased scar formation. Due to the highly specific nature of the Clostridial collagenase enzyme, we consider its use to be more desirable for the removal of unwanted tissue than the application of general proteolytic enzymes. Treatment with injectable collagenase for removal of excessive scar tissue represents a first in class minimally-invasive approach to this unmet medical need. The lead indications involving our injectable collagenase are Dupuytren’s contracture, Peyronie’s disease, frozen shoulder, cellulite, human lipoma and canine lipoma and uterine fibroids. New clinical indications involving the therapeutic application of Clostridial collagenase to supplement the body’s own natural enzymes are continuously being proposed to us by specialists in the medical community.
Collagenase for Treatment of Dupuytren’s Contracture
Dupuytren’s contracture is a deforming condition of the hand in which one or more fingers contract toward the palm, often resulting in physical disability. The onset of Dupuytren’s contracture is characterized by the formation of nodules in the palm that are composed primarily of collagen. As the disease progresses, the collagen nodules begin to form a cord causing the patient’s finger(s) to contract, making it impossible to open the hand fully. Patients often complain about the inability to wash their hands, wear gloves, or grasp some objects. Dupuytren’s contracture has a genetic basis and is most prevalent in individuals of northern European ancestry. Well-known individuals with Dupuytren’s contracture include President Ronald Reagan, President George Bush, and Prime Minister Margaret Thatcher.
XIAFLEX is the only drug approved by the FDA and the EMA for the treatment of Dupuytren’s contracture. Prior to FDA approval of XIAFLEX, the only proven treatment for Dupuytren’s contracture was surgery.
Commercialization of XIAFLEX for Dupuytren’s Contracture in the United States
Auxilium has been marketing XIAFLEX for the treatment of adult Dupuytren's contracture patients with a palpable cord since it became available by prescription in March 2010, following Auxilium’s receipt of marketing approval from the FDA. The prescribing information for XIAFLEX made available by Auxilium lists “tendon rupture or other serious injury to the injected extremity,” as well as “pulley rupture, ligament injury, complex regional pain syndrome, and sensory abnormality of the hand,” and one “anaphylactic reaction reported in a post-marketing clinical study in a patient who had previous exposure to XIAFLEX for the treatment of Dupuytren’s contracture” as reported serious adverse reactions to XIAFLEX. The prescribing information for XIAFLEX also states that the most frequently reported adverse drug reactions in XIAFLEX clinical trials included swelling of the injected hand, contusion, injection site reaction, injection site hemorrhage, and pain in the treated extremity. The prescribing information notes that adverse reaction rates observed in clinical trials of a drug may not reflect those observed in practice because such trials “are conducted under widely varying conditions.” As a condition of its approval of XIAFLEX, the FDA and Auxilium agreed upon a risk evaluation and mitigation strategy (“REMS”) program for XIAFLEX, which consists of a communication plan and a medication guide. This REMS program is designed (1) to evaluate and mitigate known and potential risks and serious adverse events; (2) to inform healthcare providers about how to properly inject XIAFLEX and perform finger extension procedures; and (3) to inform patients about the serious risks associated with XIAFLEX.
In the fourth quarter of 2013, Auxilium presented results from Year 4 of the Collagenase Optimal Reduction of Dupuytren’s Long-term Evaluations of Success Study (“CORDLESS”). CORDLESS is a five-year observational study designed to assess the rates of recurrence following treatment with XIAFLEX, as well as long-term safety and progression of disease in patients from earlier Auxilium studies. These data indicated that 57.9 percent of patients previously successfully treated with XIAFLEX did not experience disease recurrence based on the study's definition of recurrence, which is a 20 degree change of contracture with a palpable cord, or the joint undergoing medical or surgical intervention. Of the 623 joints assessed, only 12.8 percent of those joints received medical or surgical intervention through Year 4 and of these patients, most were retreated with XIAFLEX. The data also reveal no new long-term adverse events. Of the 86 serious AEs reported through four years of follow-up, only one was considered related to XIAFLEX (decrease in ring finger circumference due to Dupuytren's contracture resolution).
Also in the fourth quarter of 2013, Auxilium announced positive results from the open label, phase IIIb, MULTICORD (Multiple Treatment Investigation of Collagenase Optimizing the Resolution of Dupuytren's) study evaluating XIAFLEX for the concurrent treatment of adult Dupuytren's contracture patients with multiple palpable cords. The study demonstrated that two concurrent injections of XIAFLEX in patients with multiple Dupuytren's contractures resulted in comparable improvement in joint contracture and range of motion to those seen in previous studies when XIAFLEX was administered as single injections, 30 days apart. Adverse event (AE) rates were also comparable to single injection administration 30 days apart. The MULTICORD study found that concurrent injections of XIAFLEX reduced total fixed flexion contracture (FFC) by an average of 74.4 percent and improved the total range of motion by a combined average 66.6 degrees. Hand functionality as measured by the URAM ( U nité R humatologique des A ffections de la M ain) scale, a 9-item validated scale developed to assess functional outcome of patients suffering from Dupuytren’s disease, improved an average of 12.3 points. The estimated clinically important change of the URAM scale is 2.9 points.(i) The timing of the finger extension procedure was also examined in this study. XIAFLEX injection is currently followed by the finger extension procedure at 24 hours when needed. In MULTICORD, finger extension was performed at 24, 48 or 72 hours. There was no difference in the efficacy or safety profile based upon finger extension times. Based on the results, Auxilium submitted a sBLA to the FDA in the fourth quarter 2014 seeking expansion of labeling for the concurrent treatment of multiple palpable cords and expects to receive approval by the end of 2014. On February 24, 2014, Auxilium reported that the FDA had accepted its submission with a PDUFA date of October 20, 2014.
In its Corporate Overview - 4Q and FY 2013 Financial Results presented on February 28, 2014 (the “Auxilium Presentation”), Auxilium stated that the number of procedures in 2013 were up 10.6 % from 2012 and the XIAFLEX market share of procedures reached 27.0% in 2013. Total XIAFLEX revenues increased by 22% from 2012 to 2013 with an increase from $65.8 million dollars in 2012 to $80.1 million dollars in 2013.
Status of Regulatory Approval of XIAFLEX for Dupuytren’s Contracture Outside of the United States
Sobi has exclusive rights to commercialize XIAPEX for Dupuytren's contracture and Peyronie's disease, subject to applicable regulatory approvals, in 28 EU member countries, Switzerland, Norway, Iceland, 18 Central Eastern Europe/Commonwealth of Independent countries, including Russia and Turkey, and 22 Middle Eastern & North African countries. Sobi, via its Partner Products business unit, is primarily responsible for the applicable regulatory, clinical and commercialization activities for XIAPEX in Dupuytren's contracture and Peyronie's disease in these countries. As Auxilium reported in its 10K, “XIAPEX is now available in Austria, Belgium, Czech Republic, Denmark, Finland, Hungary, Iceland, Ireland, Italy, Luxembourg, Netherlands, Norway, Poland, Portugal, Romania, Spain, Sweden, Switzerland and the United Kingdom.”
XIAFLEX for the treatment of Dupuytren’s contracture has also been approved for sale in Canada and Australia.
Collagenase for Treatment of Peyronie’s Disease
Peyronie’s disease is characterized by the presence of a collagen plaque on the shaft of the penis, which can distort an erection and make intercourse difficult or impossible in advanced cases. In some mild cases, the plaque can resolve spontaneously without medical intervention. In severe cases, the penis can be bent at a 90-degree angle during erection. Significant psychological distress has been noted in patients with Peyronie’s disease who are sexually active. Frequent patient complaints include increased pain, painful erections, palpable plaque, penile deformity, and erectile dysfunction. Patients with Peyronie’s disease have been reported to have an increased likelihood of having Dupuytren’s contracture, frozen shoulder, plantar fibromatosis, knuckle pads, hypertension and diabetes. Peyronie’s disease typically affects males in the range of 40-70 years. The cause of Peyronie’s disease is unknown, although some investigators have proposed that it may be due to trauma or an autoimmune component. A number of researchers have suggested that the incidence of Peyronie’s disease has increased due to the use of erectile dysfunction drugs. Although the estimated prevalence of Peyronie’s disease in adult men has been reported to be approximately 5% (See Bella A. Peyronie’s Disease J Sex Med 2007; 4:1527-1538), the disease is thought to be underdiagnosed and undertreated. (See L.A. Levine Peyronie’s Disease: A Guide to Clinical Management. Humana Press: 10-17, 2007). According to Auxilium, based on U.S. historical medical claims data, it is estimated that between 65,000 and 120,000 patients are diagnosed with Peyronie’s disease every year, but only 5,000 to 6,500 Peyronie’s disease patients are treated with injectables or surgery annually.
Approval by the FDA
As announced on December 6, 2013, the FDA approved the sBLA submitted by Auxilium for XIAFLEX, an in-office, biologic for this treatment of Peyronie's disease. This is the first and only FDA-approved biologic therapy indicated for the treatment of Peyronie's disease in men with a palpable plaque and a curvature of 30 degrees or greater at the start of therapy. XIAFLEX is already approved in the U.S., EU, Canada and Australia for the treatment of adult Dupuytren's contracture patients with a palpable cord in the palm.
The approval by the FDA of Auxilium's sBLA for XIAFLEX for the treatment of Peyronie's disease is based on safety and efficacy data from Auxilium's Phase III clinical trials and other controlled and open label clinical studies in which over 1,000 patients with Peyronie's disease were enrolled and received over 7,400 injections of XIAFLEX. In the two identical Phase III double-blind placebo-controlled studies, XIAFLEX demonstrated statistically significant improvement in the co-primary endpoints of penile curvature deformity and patient-reported bother versus placebo. The approved dose of XIAFLEX for the treatment of Peyronie's disease is 0.58 mg per injection administered into a Peyronie's plaque. Up to eight injections (four treatment cycles) may be administered in the course of treatment. Also, a penile modeling procedure is recommended after every treatment cycle of two injections in an effort to further disrupt the plaque. If more than one plaque is present, it should be injected into the plaque causing the curvature deformity.
Auxilium has created Auxilium Advantage™ to support access to XIAFLEX and provide a single point of contact for health care providers and patients for help accessing the product. A risk evaluation and mitigation strategy (REMS) for XIAFLEX went into effect after the product first received FDA approval in February 2010 for adults with Dupuytren's contracture with a palpable cord, and Auxilium has further collaborated with the FDA to update the REMS with an Elements to Assure Safe Use (ETASU) for XIAFLEX for the treatment of Peyronie's disease in men with a palpable plaque and curvature deformity of 30 degrees or greater at the start of therapy. The goal of the XIAFLEX REMS with an ETASU for Peyronie's disease is to certify that the appropriate physicians and practice sites are trained in the use of XIAFLEX and to attempt to mitigate the serious risk of penile fracture (corporal rupture) and other serious injuries to the penis such as hematoma. These serious risks are highlighted in the Boxed Warning within the Full Prescribing Information (the label).
Commercialization of Peyronie’s Diseases in the United States
The Auxilium Presentation noted that the initial patient focus if 5,000-6,000 invasive treatment patients per year and the initial launch focus is on 400 physicians who perform 90% of all surgeries, with 225 in the first phase of outreach. To date, 452 physicians and 249 sites have been certified. With respect to reimbursement, 827 patients have submitted requests for reimbursement and 84% of the 32% for whom decisions have been made received reimbursement.
Frozen shoulder is a clinical syndrome of pain and decreased motion in the shoulder joint. It is estimated to affect 20 to 50 million people worldwide with a slightly higher incidence in women. It is estimated that 700,000 patients visit doctors annually in the U.S. in connection with frozen shoulder. It typically occurs between the ages of 40-70. Individuals with insulin dependent diabetes have been reported to have a 36% higher incidence rate and are more likely to have bilateral symptoms. No FDA-approved pharmaceutical therapies are currently available for the treatment of frozen shoulder. The most common treatments for frozen shoulder syndrome are extensive physical therapy, corticosteroids and/or arthroscopy, and some drugs are used to manage pain.
Phase II
In the first quarter of 2013, reported the top-line results of its phase IIa study. The phase IIa study was an open-label, controlled dose-ranging study designed to assess the safety and efficacy of CCH for the treatment of Stage 2 unilateral idiopathic frozen shoulder in comparison to an exercise-only control group. The study involved 50 adult men and women at 11 sites throughout the U.S. Four cohorts of 10 patients each received up to three ultrasound-guided extraarticular injections of varying doses of CCH (ranging from 0.29 mg to 0.58 mg in three different volumes; 0.5, 1.0, or 2.0 mL), separated by a minimum of 21 days. All patients were instructed to perform home shoulder exercises. The fifth cohort of ten patients received no CCH injections and only performed home shoulder exercises. The study’s primary endpoint was the change (in degrees) from baseline to the day 92 follow-up in active forward flexion in the affected shoulder compared to the exercise-only cohort. Safety assessments were made during all study visits and immunogenicity testing was performed at screening and day 92.
Both the 0.58mg(1mL) and 0.58mg(2mL) dosing arms showed positive, statistically significant improvement from baseline in forward flexion vs. the exercise-only group. The 0.58mg(1mL) dosing arm also showed statistically significant improvement from baseline in shoulder abduction vs. the exercise-only group. Positive trends with improvement in degrees were also seen in other active range of motion (“AROM”) assessments vs. the exercise-only group. Twenty-nine study patients (72.5%) received three CCH injections with 5 subjects receiving two injections and 6 subjects receiving one injection only.
AROM Assessment Results from Phase IIa FSS Study
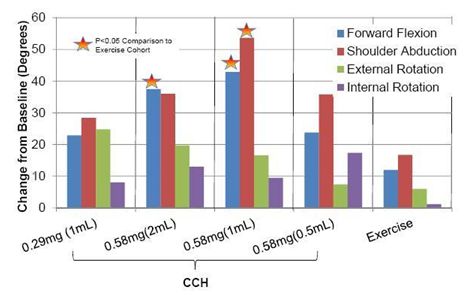
Patients were also assessed using the American Shoulder and Elbow Surgeons (“ASES”) Scale for function and pain. Both the 0.58 mg(1mL) and 0.58 mg(2mL) cohort demonstrated statistically significant improvement in pain and function over baseline scores vs. the exercise-only group (p<0.05).
Treatment-related adverse events with CCH were mostly localized bruising, injection site pain and swelling, hematoma, and musculoskeletal pain. All such events resolved without intervention, and are consistent with XIAFLEX/CCH use in other approved and potential indications. No subjects discontinued the study due to an adverse event. A shoulder MRI was performed on all patients at screening and day 92. Screening MRIs were performed to exclude the presence of other clinically significant conditions such as concomitant rotator cuff injury. Day 92 MRI evaluations indicated there were no rotator cuff injuries. There were no drug-related serious adverse events reported.
In the fourth quarter of 2013, Auxilium reported that it had initiated a phase IIb double-blind, placebo-controlled study of the safety and efficacy of CCH for the treatment of Stage 2 unilateral idiopathic frozen shoulder. The study will enroll approximately 300 adult men and women at approximately 35 sites in the U.S. and Australia. Subjects will be randomized 3:1 to receive CCH or placebo and will receive up to three ultrasound-guided injections of study drug. Each injection will be separated by a minimum of 21 days. All subjects will also perform home shoulder exercises after the first injection.
The primary endpoint of the phase IIb study will be change in degrees from baseline to the Day 95 follow-up visit in active forward flexion in the affected shoulder compared to placebo. Patients will also be assessed using the ASES Scale for function and pain as well as additional patient reported outcome measures. Safety assessments will be made during all study visits and immunogenicity testing will be performed at screening and at the end of the study.
Collagenase For Treatment of Cellulite (Edematous Fibrosclerotic Panniculopathy)
Edematous fibrosclerotic panniculopathy, commonly known as cellulite, describes a condition, in which lobules of subcutaneous adipose tissue extend into the dermal layer. Cellulite can involve the loss of elasticity or shrinking of collagen cords, called septae, that attach the skin to lower layers of muscle. When fat in cellulite prone areas swells and expands, the septae tether the skin, which causes surface dimpling characteristic of cellulite. These changes can visibly affect the shape of the epidermis and resemble an orange peel-like dimpling of the skin. (See Avram, Cellulite: a review of its physiology and treatment, Journal of Cosmetic Laser Therapy 2004; 6: 181–185). XIAFLEX treatment is intended to target and lyse, or break, those collagen tethers with the goal of releasing the skin dimpling and potentially resulting in smoothing of the skin.
Cellulite has been reported to occur in 85-98% of post-pubertal females and rarely in men, and it is believed to be prevalent in women of all races. (See Avram, Cellulite: a review of its physiology and treatment, Journal of Cosmetic Laser Therapy 2004; 6: 181–185; Khan MH et al. Treatment of cellulite: Part I. Pathophysiology. J Am Acad Dermatol. 2010 Mar;62(3):361-70). As Auxilium reported in its 10 K, “[c]urrent treatments for cellulite include massage devices, creams, unapproved injectables, laser-based procedures or liposuction. There are no drugs currently approved by the FDA to treat cellulite, and devices cleared by the FDA to treat the condition have varying degrees of success in eliminating cellulite.”
In December 2012, Auxilium announced top-line 30-day data from its phase Ib, single site, open-label dose escalation study of CCH for the treatment of cellulite. The study enrolled 99 women between 21 and 60 years of age. Study participants were assigned to one of 11 arms, each of which varied in treatment dose, injection concentration and volume, to receive a single injection of CCH, divided into 10 aliquots over a pre-defined 8x10cm template around a target dimple. The objectives of the study are to assess the safety and effectiveness of a single injection of CCH for the treatment of cellulite at 30, 60 and 90 days across multiple dosing arms. Pharmacokinetic evaluations were made as well. Across all dosing arms, 60 patients (63%) who were treated experienced some improvement in the volume of their target cellulite dimple at Day 30. Overall, 17% of patients had a greater than or equal to 30% improvement in their target dimple at Day 30; however, multiple CCH dosing arms had more than 40 percent of patients experience an improvement greater than or equal to 30% in their target dimple at Day 30. Treatment-related adverse events with CCH were mostly localized bruising, injection site discomfort and swelling, and all such events resolved without intervention, which are all consistent with CCH use in other indications.
In January 2013, Auxilium exercised its exclusive option under the Auxilium Agreement to expand the field of its license for injectable collagenase to include the potential treatment of adult patients with cellulite. As a result, we received a one-time license fee payment of $500,000, a portion of which we paid to the Research Foundation of the State University of New York at Stony Brook pursuant to the terms of our in-licensing agreement described below in the “In-Licensing and Royalty Agreements” section under the heading “Cellulite”. Auxilium’s exclusive, worldwide license has now been expanded, subject to the terms of the Auxilium Agreement, to include all research, development, use, commercialization, marketing, sales and distribution rights for injectable collagenase for the potential treatment of cellulite.
In October 2013, Auxilium announced the initiation of its phase IIa study of CCH for the treatment of cellulite. The phase IIa study is a randomized, double-blind multiple-dose study expected to enroll approximately 144 women between the ages of 18 and 45 in the U.S. Patients will be evaluated for treatment efficacy by investigator and patient assessments, as well as 3-D photographic imaging techniques. The study will be conducted in two stages and safety will be evaluated through the collection of adverse events. If the safety and local tolerability profile from the first stage has been found to be acceptable, subjects will be enrolled in stage 2.
To qualify for the study, participants must have cellulite in the posterolateral thighs and/or buttocks for at least 12 months prior to a screening visit. Eligible study participants will be assigned to one of four groups that vary in treatment dose (low, medium, high, and placebo) and will be randomized to low-dose CCH, mid-dose CCH, high-dose CCH, or placebo in a 5:5:5:3 ratio. Total treatment doses per treatment session include doses both lower and higher than the dose used in Dupuytren’s contracture with a palpable cord. Each subject may receive up to three treatment sessions of study drug according to randomization and each treatment session will be approximately 21 days apart. In this study, only the dimples treated on Day 1 may be retreated on Day 22 (Treatment Session 2) and Day 43 (Treatment Session 3) if, in the opinion of the investigator, the dimple continues to be evident. A variable number of dimples may be treated within one treatment quadrant. Topline results from the study are expected in the first quarter of 2015.
Additional Clinical Indications For Collagenase
Human Lipoma
Lipomas are benign fatty tumors that occur as bulges under the skin and affect humans and canines. It is estimated that lipomas are the primary diagnosis in 575,000 patients in the U.S. annually. The only proven therapy for lipoma treatment is surgery, which is often not practical for patients with multiple lipomas. Based on observations made during preclinical studies that a collagenase injection decreased the size of fat pads in animals, we initiated, monitored and supplied the requisite study drug for a phase I open label clinical trial for the treatment of human lipomas with a single injection of collagenase. Favorable initial results (10 out of 12 patients had a 50-90% reduction in the size of the lipomas) from this trial for the treatment of human lipomas were presented at a meeting of the American Society of Plastic Surgeons.
In January 2014, we announced the top-line data from the phase II dose escalation clinical trial of CCH for the treatment of human lipoma. This phase II open-label single-center dose escalation study assessed the safety and efficacy of CCH in 14 patients with lipoma, divided into four dose cohorts. Each patient received a single injection of CCH in one of four ascending doses based on the current commercial dose of CCH in marketed indications, ranging from 0.058mg (10% of commercial dose) to 0.44mg (75% of commercial dose). The primary efficacy outcome was reduction in lipoma visible surface area as measured by caliper. Data showed patients in the highest dose group (75% of commercial dose) achieved the best efficacy results with an average of 67% reduction of lipoma visible surface area as measured by caliper at six months post-treatment. Additionally, data demonstrated that 75% of patients in the highest dose group achieved reduction of 50% or more in lipoma visible surface area. We anticipate initiating a placebo-controlled trial in the first half of 2014.
There were no drug-related serious adverse events reported during the trial. The most frequent treatment-related adverse events were localized to the injection site and included bruising, injection site swelling and injection site pain. These adverse events are consistent with those seen previously in clinical experience.
Canine Lipoma
Based on the encouraging results reported in the clinical investigations in human lipoma, we began clinical trials in canine lipoma. Lipomas are found in 2.3% of canines, and there may be as many as 1.7 million canines affected with skin lipomas in the U.S. Lipomas in older canines are very common, and lipomas that restrict motion in older canines are a serious problem. The only proven therapy for this condition is surgical excision of the lipoma, which necessarily involves the use of general anesthesia. It has been estimated that up to 2% of sick canines die as a complication of general anesthesia (See Brodbelt Vet J 2009 Dec; 182 (3): 375-6). We surveyed 77 veterinarians which included participants from the academic field and others that are in private practice. The participants indicated that on average they perform 25 lipoma excision surgeries per year at an average cost of $530 for the surgical procedure. It is conservatively estimated that 47,000 veterinarians are in active practice in the U.S.
Chien-804
In December 2013, we announced top-line data from Chien-804, the placebo-controlled, double-blind, randomized phase II trial evaluating the efficacy of CCH in canines with benign subcutaneous lipomas. The Chien-804 trial enrolled 37 dogs in a single injection study randomized 1:1 CCH to placebo with lipoma volume being measured by CT scan and lipoma surface area being measured by caliper at baseline, one month and 90 days. The data at 90 days show a post-treatment difference in the mean percent change in lipoma volume by CT scan between the CCH and placebo-treated groups of -11.58% (p=0.52), which was not statistically significant. The percent change at 90 days in mean visible surface area measured by caliper showed a difference of -24.18% (p=0.09), which approached statistical significance. Among those dogs whose lipomas decreased by 50% or more, the results achieved statistical significance and showed that the visible surface area as measured by caliper decreased by 50% or more in 47.4% of CCH-treated dogs (9 out of 19) versus 5.9% of placebo-treated dogs (1 out of 17), with a p-value of 0.0084. A questionnaire administered to pet owners, while blinded to the study, showed 84.2% satisfaction with the results of CCH treatment versus 33.4% satisfaction with the placebo results (p=0.005). We anticipate providing Auxilium with the Chien-804 final study report in the first quarter of 2014.
There were no drug-related serious adverse events reported during the trial. The most frequent treatment-related adverse events were local injection site reactions including bruising, injection site swelling, injection site pain and injection site edema. These adverse events are consistent with those seen previously in clinical experience in humans.
Uterine Fibroids
In July 2013, we announced that a poster titled, "Biomechanical Evaluation of Human Uterine Fibroids after Exposure to Purified Clostridial Collagenase" was presented at the Society for the Study of Reproduction 46th Annual Meeting in Montreal, Quebec, Canada. The poster provided data which show that highly purified collagenase can reduce the stiffness of human uterine fibroid tissue in laboratory experiments. Increased tissue rigidity has been implicated as a cause of the morbidity associated with uterine fibroids.
The results of this ex vivo study showed that treatment of fibroids with determined doses of purified collagenase caused a statistically significant decrease in the stiffness of the tissue. This hypothesis was tested in fibroid tissue obtained after hysterectomy or myomectomy surgery from patients.
Tissues were injected with collagenase and compared to control-injected tissue. The stiffness in the fibroid tissue was reduced in a time and dose dependent manner with a p-value ≤ 0.001.
The study is being led by Dr. Phyllis Leppert, a Professor of Obstetrics and Gynecology and Professor of Pathology and her colleague, Dr. Friederike Jayes at Duke Medicine with our support. We anticipate reporting top-line data from the pre-clinical study in the first half of 2014.
Other Clinical Indications
Other clinical indications for which our collagenase injection has been tested include keloids, hypertrophic scars, scarred tendons, glaucoma, herniated intervertebral discs, and as an adjunct to vitrectomy.
LICENSING AND MARKETING AGREEMENTS
Auxilium Agreement
Under the Auxilium Agreement, we granted to Auxilium exclusive worldwide rights to develop, market and sell certain products containing our injectable collagenase. Currently its licensed rights cover the indications of Dupuytren’s contracture, Peyronie’s disease, frozen shoulder and cellulite. Auxilium may further expand the Auxilium Agreement, at its exclusive option, to develop and license our injectable collagenase for use in additional indications.
Auxilium’s existing agreement with Pfizer terminated as of April 24, 2013. Pursuant to a transition services agreement, Pfizer continued support of the supply of XIAPEX until February 28, 2014. Currently, Sobi has exclusive rights to commercialize XIAPEX for Dupuytren's contracture and Peyronie's disease, subject to applicable regulatory approvals, in 28 EU member countries, Switzerland, Norway, Iceland, 18 Central Eastern Europe/Commonwealth of Independent countries, including Russia and Turkey, and 22 Middle Eastern & North African countries.
Sobi, via its Partner Products business unit, is primarily responsible for the applicable regulatory, clinical and commercialization activities for XIAPEX in Dupuytren's contracture and Peyronie's disease in these countries. We will receive a certain percentage of milestone payments that Sobi pays to Auxilium. We will also receive royalties from net sales and payments on costs of goods sold in Sobi territories from Auxilium, which will be a specified percentage of what Auxilium receives from Sobi.
Auxilium has granted to Asahi the exclusive right to develop and commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Japan. Auxilium has granted to Actelion the exclusive right to develop and commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Canada, Australia, Brazil and Mexico. XIAFLEX has been approved for sale for Dupuytren’s contracture in Canada and Australia.
Through December 31, 2013, Auxilium has paid us up-front licensing and sublicensing fees and milestone payments under the Auxilium Agreement of $26.4 million, including amounts in connection with Auxilium’s agreements with Pfizer, Asahi and Actelion. In addition to the payments already received by us and to be received by us with respect to the Dupuytren’s contracture indication, Auxilium will be obligated to make contingent milestone payments to us, with respect to each of frozen shoulder and cellulite indications, upon the acceptance of the regulatory filing and upon receipt by Auxilium, its affiliate or sublicensee of regulatory approval. The remaining contingent milestone payments that may be received, in the aggregate, from Auxilium in respect of frozen shoulder and cellulite are $3.0 million. To the extent there is sub-licensing income as defined in the Auxilium Agreement, Auxilium will also be obligated to make sublicense fee payments to us if it out-licenses to third parties the right to market and sell XIAFLEX for the treatment of frozen shoulder or cellulite. Additional milestone obligations will be due if Auxilium exercises its option to develop and license XIAFLEX for additional indications, such as human and canine lipoma. In the first quarter 2014, we anticipate we will present the opt-in study for canine lipoma to Auxilium and Auxilium will have 120 days to exercise its option and pay the associated option amount.
We will receive a certain percentage of milestone payments that Sobi pays to Auxilium. We will also receive royalties from net sales and payments on costs of goods sold in Sobi territories from Auxilium, which will be a specified percentage of what Auxilium receives from Sobi. To the extent Auxilium enters into an agreement or agreements related to other territories, the percentage of sublicense income that Auxilium would pay us will depend on the stage of development and approval of XIAFLEX for the particular indication at the time such other agreement or agreements are executed.
Auxilium must pay us on a country-by-country and product-by-product basis a low double digit royalty as a percentage of net sales for products covered by the Auxilium Agreement and sold in the United States, Europe and certain Eurasian countries and Japan. In the case of products covered by the Auxilium Agreement and sold in other countries (the “Rest of the World”), Auxilium must pay us on a country-by-country and product-by-product basis a specified percentage of the royalties it is entitled to receive from a partner or partners with whom it has contracted for such countries (a “Rest of the World Partner”), which in the case of Canada, Australia, Brazil and Mexico is Actelion. The royalty rate is independent of sales volume and clinical indication in the United States, Europe and certain Eurasian countries and Japan, but is subject to set-off in those countries and the Rest of the World for certain expenses we owe to Auxilium relating to certain development and patent costs. In addition, the royalty percentage may be reduced if (i) market share of a competing product exceeds a specified threshold; or (ii) Auxilium is required to obtain a license from a third party in order to practice our patents without infringing such third party’s patent rights, although Auxilium has confirmed to us that no license from a third party is required. In addition, if Auxilium out-licenses to a third party, then we will receive a specified percentage of certain payments made to Auxilium in consideration of such out-licenses.
These royalty obligations extend, on a country-by-country and product-by-product basis, for the longer of the patent life (including pending patents), the expiration of any regulatory exclusivity period based on orphan drug designation or foreign equivalent thereof or June 3, 2016. Auxilium may terminate the Auxilium Agreement upon 90 days prior written notice. If Auxilium terminates the Auxilium Agreement other than because of an uncured, material breach by us, all rights revert to us. Pursuant to our August 31, 2011 settlement agreement with Auxilium, we are now co-owners and are or will be co-inventors of U.S. Patent No. 7,811,560 and any continuations and divisionals thereof. Auxilium expects this patent will expire in July 2028.
On top of the payments set forth above, Auxilium must pay to us an amount equal to a specified mark-up of the cost of goods sold for products sold in the United States, Europe and certain Eurasian countries or Japan. For products sold in the Rest of the World, Auxilium must pay to us a specified percentage of the mark-up of the cost of goods sold it is entitled to receive from a Rest of the World Partner, including Actelion, without regard to any set-offs that the Rest of the World Partner may have with respect to Auxilium.
Auxilium is generally responsible, at its own cost and expense, for developing the formulation and finished dosage form of products and arranging for the clinical supply of products. Auxilium is generally responsible for all clinical development and regulatory costs for Peyronie’s disease, Dupuytren’s contracture, frozen shoulder, cellulite and all additional indications for which it exercises its options.
A redacted copy of the Auxilium Agreement was filed on Form 8-K with the SEC on September 1, 2011. The foregoing descriptions of the Auxilium Agreement do not purport to be complete and are qualified in their entirety by reference to the full text of the Auxilium Agreement.
DFB
In connection with a March 2006 agreement (the “DFB Agreement”), pursuant to which we sold our topical collagenase business to DFB Biotech, Inc. and its affiliates (“DFB”), we expect to receive in March 2014 the final earn out payment of $3.5 million which was recognized as income in 2013.
In-Licensing and Royalty Agreements
We have entered into several in-licensing and royalty agreements with various investigators, universities and other entities throughout the years.
Dupuytren’s Contracture
On November 21, 2006, we entered into a license agreement (the “Dupuytren’s License Agreement”) with the Research Foundation of the State University of New York at Stony Brook (the “Research Foundation”), pursuant to which the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the development, manufacture, use or sale of (i) the collagenase enzyme obtained by a fermentation and purification process (the “Enzyme”), and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment and prevention of Dupuytren’s contracture.
In consideration of the license granted under the Dupuytren’s License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme or injectable collagenase for the treatment and prevention of Dupuytren’s contracture (each a “Dupuytren’s Licensed Product”).
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Dupuytren’s Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of such Dupuytren’s Licensed Product on a country-by-country basis. The royalty rate is 0.5% of net sales. Our obligation to pay royalties to the Research Foundation will continue until the later of (i) the expiration of the last valid claim of a patent pertaining to the Dupuytren’s Licensed Product; (ii) the expiration of the regulatory exclusivity period conveyed by the FDA’s Office of Orphan Products Development (“OOPD”) with respect to the Dupuytren’s Licensed Product; or (iii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Dupuytren’s License Agreement and the licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Dupuytren’s License Agreement will become fully paid, irrevocable exclusive licenses.
A redacted copy of the Dupuytren’s License Agreement was filed on Form 8-K with the SEC on November 28, 2006. The foregoing descriptions of the Dupuytren’s License Agreement do not purport to be complete and are qualified in their entirety by reference to the full text of the Dupuytren’s License Agreement.
Peyronie’s Disease
On August 27, 2008, we entered into an agreement with Dr. Martin K. Gelbard to improve the deal terms related to our future royalty obligations for Peyronie’s disease by buying down our future royalty obligations with a one-time cash payment. A redacted copy of the agreement was filed on Form 8-K with the SEC on September 5, 2008. On March 31, 2012, we entered into an amendment to this agreement, which enables us to buy down a portion of our future royalty obligations in exchange for an initial cash payment and five additional cash payments. A redacted copy of the amendment was filed on Form 8-K/A with the SEC on August 8, 2012. The foregoing descriptions of the agreement with Dr. Gelbard and the amendment to that agreement do not comport to be complete and are qualified in their entirety by reference to the full text of that agreement, as amended.
Frozen Shoulder
On November 21, 2006, we entered into a license agreement (the “Frozen Shoulder License Agreement”) with the Research Foundation, pursuant to which the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the development, manufacture, use or sale of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment and prevention of frozen shoulder.
Additionally, the Research Foundation granted to us an exclusive license to the patent applications in respect of frozen shoulder. The license granted to us under the Frozen Shoulder License Agreement is subject to the non-exclusive license (with right to sublicense) granted to the U.S. government by the Research Foundation in connection with the U.S. government’s funding of the initial research.
In consideration of the license granted under the Frozen Shoulder License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme or injectable collagenase for the treatment and prevention of frozen shoulder (each a “Frozen Shoulder Licensed Product”). In addition, we and the Research Foundation will share in any milestone payments and sublicense income received by us in respect of the rights licensed under the Frozen Shoulder License Agreement.
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Frozen Shoulder Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Frozen Shoulder Licensed Product. Our obligation to pay royalties to the Research Foundation will continue until, the later of (i) the expiration of the last valid claim of a patent pertaining to a Frozen Shoulder Licensed Product or (ii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Frozen Shoulder License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Frozen Shoulder License Agreement will become fully paid, irrevocable exclusive licenses.
A redacted copy of the Frozen Shoulder License Agreement was filed on Form 8-K with the SEC on November 28, 2006. The foregoing descriptions of the Frozen Shoulder License Agreement do not purport to be complete and are qualified in their entirety by reference to the full text of the Frozen Shoulder License Agreement.
In connection with the execution of the Dupuytren’s License Agreement and the Frozen Shoulder License Agreement, we made certain up-front payments to the Research Foundation and the clinical investigators working on the Dupuytren’s contracture and frozen shoulder indications for the Enzyme.
Cellulite
We have two in-licensing and royalty agreements related to cellulite. One is a license agreement (the “Cellulite License Agreement”) with the Research Foundation that we entered into on August 23, 2007. Pursuant to the Cellulite License Agreement, the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the manufacture, preparation, formulation, use or development of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme, which are made, used and sold for the prevention or treatment of cellulite. Additionally, the Research Foundation granted to us an exclusive license to the patent applications in respect of cellulite. The license granted to us under the Cellulite License Agreement is subject to the non-exclusive license (with right to sublicense) granted to the U.S. government by the Research Foundation in connection with the U.S. government’s funding of the initial research.
In consideration of the license granted under the Cellulite License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme, which are made, used and sold for the prevention or treatment of cellulite (each a “Cellulite Licensed Product”). In addition, we and the Research Foundation will share in any milestone payments and sublicense income received by us in respect of the rights licensed under the Cellulite License Agreement. We paid a portion of the $500,000 milestone payment we received from Auxilium in respect of its exercise of cellulite as an addition indication under the Auxilium Agreement, subject to certain credits for certain up-front payments we made to the Research Foundation.
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Cellulite Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Cellulite Licensed Product. Our obligation to pay royalties to the Research Foundation will continue until, the later of (i) the expiration of the last valid claim of a patent pertaining to a Cellulite Licensed Product or (ii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Cellulite License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Cellulite License Agreement will become fully paid, irrevocable exclusive licenses.
The other in-licensing and royalty agreement we have related to cellulite is a license agreement with Dr. Zachary Gerut that we entered into on March 27, 2010 (the “Gerut License Agreement”). Pursuant to the Gerut License Agreement, Dr. Gerut granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties to know-how owned by Dr. Gerut related to the manufacture, preparation, formulation, use or development of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment of fat. As the in-license granted in the Gerut License Agreement pertains to the treatment of fat, this in-license also relates to human lipoma and canine lipoma.
In consideration of the license granted under the Gerut License Agreement, we agreed to pay to Dr. Gerut certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme which are made, used and sold for the removal or treatment of fat in humans or animals (each a “Gerut Licensed Product”). In addition, in the event the FDA approves a Gerut Licensed Product, we have agreed to make a one-time stock option grant to Dr. Gerut with a strike price equal to the closing trading price on the day before the date of such grant.
Our obligation to pay royalties to Dr. Gerut with respect to sales by us, our affiliates or any sublicensee of any Gerut Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Gerut Licensed Product. Our obligation to pay royalties to Dr. Gerut will continue until June 3, 2016 or such longer period as we continue to receive royalties for such Gerut Licensed Product.
Unless terminated earlier in accordance with its termination provisions, the Gerut License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Gerut License Agreement will become fully paid, irrevocable exclusive licenses.
Redacted copies of the Cellulite License Agreement and the Gerut License Agreement were filed on our Form 10-K filed with the SEC March 15, 2013. The foregoing descriptions of the Cellulite License Agreement and the Gerut License Agreement do not purport to be complete and are qualified in their entirety by reference to the full text of these agreements.
Other Indications
We have or may enter into certain other license and royalty agreements with respect to other indications that we may elect to pursue.
COMPETITION
We and our licensees face worldwide competition from larger pharmaceutical companies, specialty pharmaceutical companies and biotechnology firms, universities and other research institutions and government agencies that are developing and commercializing pharmaceutical products. Many of our competitors have substantially greater financial, technical and human resources than we have and may subsequently develop products that are more effective, safer or less costly than any products that we have developed, are developing or will develop, or that are generic products. Our success will depend on our ability to acquire, develop and commercialize products and our ability to establish and maintain markets for our products that receive marketing approval.
COST OF RESEARCH AND DEVELOPMENT ACTIVITIES
During fiscal years 2013 and 2012, the Company invested $1.5 million dollars and $1.2 million dollars, respectively, in research and development activities.
GOVERNMENT REGULATION
Any product labeled for use in humans requires regulatory approval by government agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical trials to demonstrate safety and efficacy and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal, state, local, and foreign statutes and regulations also govern testing, manufacturing, labeling, distribution, storage, and record-keeping related to such products and their promotion and marketing. The process of obtaining these approvals and the compliance with federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. In addition, the current political environment and the current regulatory environment at the FDA could lead to increased testing and data requirements which could impact regulatory timelines and costs.
Clinical trials involve the administration of the investigational product candidate or approved products to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study and the parameters to be used in assessing the safety and the effectiveness of the drug. Typically, clinical evaluation involves a time-consuming and costly three-phase sequential process, but the phases may overlap. Each trial must be reviewed, approved and conducted under the auspices of an independent institutional review board, and each trial must include the patient’s informed consent.
Clinical testing may not be completed successfully within any specified time period, if at all. The FDA monitors the progress of all clinical trials that are conducted in the U.S. and may, at its discretion, reevaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA can also provide specific guidance on the acceptability of protocol design for clinical trials. The FDA, we or our partners may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional clinical trials be conducted as a condition to product approval. During all clinical trials, physicians monitor the patients to determine effectiveness and/or to observe and report any reactions or other safety risks that may result from use of the drug candidate.
Assuming successful completion of the required clinical trials, drug developers submit the results of preclinical studies and clinical trials, together with other detailed information including information on the chemistry, manufacture and control of the product, to the FDA, in the form of an NDA or BLA, requesting approval to market the product for one or more indications. In most cases, the NDA/BLA must be accompanied by a substantial user fee. The FDA reviews an NDA/BLA to determine, among other things, whether a product is safe and effective for its intended use.
Before approving an NDA or BLA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve the NDA or BLA unless cGMP compliance is satisfactory. The FDA will issue an approval letter if it determines that the NDA or BLA, manufacturing process and manufacturing facilities are acceptable. If the FDA determines that the NDA or BLA, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the NDA or BLA does not satisfy the regulatory criteria for approval and refuse to approve the NDA or BLA by issuing a “not approvable” letter.
The testing and approval process requires substantial time, effort and financial resources, which may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We or our partners may encounter difficulties or unanticipated costs in our or their efforts to secure necessary governmental approvals, which could delay or preclude us or them from marketing our products. Furthermore, the FDA may prevent a drug developer from marketing a product under a label for its desired indications or place other conditions, including restrictive labeling, on distribution as a condition of any approvals, which may impair commercialization of the product. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
If the FDA approves the NDA or BLA, the drug can be marketed to physicians to prescribe in the U.S. After approval, the drug developer must comply with a number of post-approval requirements, including delivering periodic reports to the FDA (i.e., annual reports), submitting descriptions of any adverse reactions reported, biological product deviation reporting, and complying with drug sampling and distribution requirements and any other requirements set forth in the FDA’s approval (such as the REMS program, which the FDA has required for XIAFLEX and consists of a communication plan and a medication guide). The holder of an approved NDA/BLA is required to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. Drug manufacturers and their subcontractors are required to register their facilities and are subject to periodic unannounced inspections by the FDA to assess compliance with cGMP which impose procedural and documentation requirements relating to manufacturing, quality assurance and quality control. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other regulatory requirements. The FDA may require post-market testing and surveillance to monitor the product’s safety or efficacy, including additional studies to evaluate long-term effects.
In addition to studies requested by the FDA after approval, a drug developer may conduct other trials and studies to explore use of the approved drug for treatment of new indications, which require submission of a supplemental or new NDA/BLA and FDA approval of the new labeling claims. The purpose of these trials and studies is to broaden the application and use of the drug and its acceptance in the medical community.
We use, and will continue to use, third party manufacturers to produce our products in clinical quantities. Future FDA inspections may identify compliance issues at our facilities, at the facilities of our contract manufacturers or at those of our partners that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product or the failure to comply with requirements may result in restrictions on a product, manufacturer or holder of an approved NDA/BLA, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications. Also, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
INTELLECTUAL PROPERTY AND RIGHTS
Our success will depend in part on our ability to protect our existing products and the products we acquire or in-license by obtaining and maintaining a strong proprietary position both in the U.S. and in other countries. To develop and maintain such a position, we intend to continue relying upon patent protection, trade secrets, know-how, continuing technological innovations and licensing opportunities. In addition, we intend to seek patent protection whenever available for any products or product candidates and related technology we develop or acquire in the future.
Patents
We are the assignee or licensee of various U.S. patents, which have received patent protection in various foreign countries. Pursuant to our August 31, 2011 settlement agreement with Auxilium, we are now co-owners and either have been or will be added as co-inventors of U.S. Patent No. 7,811,560 and any continuations and divisionals thereof. Auxilium expects this patent will expire in July 2028. In addition, we have licenses to other pending patent applications. Although we believe these patent applications, if they issue as patents, will provide a competitive advantage, the scope of the patent positions of pharmaceutical firms involves complex legal, scientific and factual questions and, as such, is generally uncertain. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued. Consequently, we do not know whether any of our current patent applications, or the products or product candidates we develop, acquire or license will result in the issuance of patents or, if any patents are issued, whether they will provide significant proprietary protection, will be of any value to us or will be challenged, circumvented or invalidated by our competitors or otherwise.
While we attempt to ensure that our product candidates and the methods we employ to manufacture them do not infringe other parties’ patents and proprietary rights, competitors or other parties may assert that we infringe their proprietary rights. Because patent applications in the U.S. and some other jurisdictions can proceed in secrecy until patents issue, third parties may obtain other patents without our knowledge prior to the issuance of patents relating to our product candidates, which they could attempt to assert against us. Also, since publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain of the priority of inventions covered by pending patent applications. Moreover, we may have to participate in interference proceedings declared by the U.S. Patent and Trademark Office (the “USPTO”) to determine priority of invention, or in opposition proceedings in the USPTO, either of which could result in substantial cost to us, even if the eventual outcome is favorable to us. In the U.S., issued patents may be broadened, narrowed or even canceled as a result of post-issuance procedures instituted by us or third parties, including reissue, ex parte reexamination, and the new inter partes review, post grant review, and supplemental examination procedures enacted as part of the Leahy-Smith America Invents Act. There can be no assurance that the patents, if issued and challenged in a court of competent jurisdiction, would be found valid or enforceable. An adverse outcome could subject us to significant liabilities to third parties, require disputed rights to be licensed from third parties or require us to cease using such technology.
Although we believe that our product candidates, production methods and other activities do not currently infringe the intellectual property rights of third parties, we cannot be certain that a third party will not challenge our position in the future. If a third party alleges that we are infringing its intellectual property rights, we may need to obtain a license from that third party, but there can be no assurance that any such license will be available on acceptable terms or at all. Any infringement claim that results in litigation could result in substantial cost to us and the diversion of management’s attention from our core business. To enforce patents issued, assigned or licensed to us or to determine the scope and validity of other parties’ proprietary rights, we may also become involved in litigation or in interference proceedings declared by the USPTO, which could result in substantial costs to us or an adverse decision as to the priority of our inventions. We may be involved in interference and/or opposition proceedings in the future. We believe there will continue to be litigation in our industry regarding patent and other intellectual property rights.
We licensed to Auxilium our injectable collagenase for the treatment of Dupuytren’s contracture, Peyronie’s disease, frozen shoulder and cellulite. We have two use patents in the U.S. covering the enzyme underlying our injectable collagenase, one for the treatment of Dupuytren’s contracture, which issued from a reissue proceeding in December 2007, and one for the treatment of Peyronie’s disease. The Dupuytren’s patent expires in 2014, and the Peyronie’s patent expires in 2019. Both the Dupuytren’s and Peyronie’s patents are limited to the use of the enzyme for the treatment of Dupuytren’s contracture and Peyronie’s disease within certain dose ranges. An application to extend the term of the Dupuytren’s patent to August 22, 2019 based upon regulatory delay in granting approval to sell XIAFLEX was filed in the USPTO on April 1, 2010. A letter was issued by the Food and Drug Administration on March 11, 2013, indicating that XIAFLEX was subject to a regulatory review period before its commercial marketing or use, and that submission of the application was timely. However, the USPTO has not taken any action on the request for extension, and we cannot be certain how much of an extension, if any, will be granted by the USPTO.
Orphan Drug Designations
Two indications, Dupuytren’s contracture and Peyronie’s disease, have received orphan drug designation from the OOPD. These indications did not receive the European equivalent of orphan drug designation.
The OOPD administers the major provisions of the Orphan Drug Act, an innovative program that provides incentives for sponsors to develop products for rare diseases. The incentives for products that qualify under the Orphan Drug Act include seven-year exclusive marketing rights post-FDA approval (which means, with respect to Dupuytren’s contracture, exclusivity until February 2, 2017 and Peyronie’s Disease until December 6, 2020), tax credits for expenses associated with clinical trials including a 20 year tax carry-forward, availability of FDA grants, and advice on design of the clinical development plan.
The orphan drug provisions of the Federal Food, Drug, and Cosmetic Act also provide incentives to drug and biologics suppliers to develop and supply drugs for the treatment of rare diseases, currently defined as diseases that affect fewer than 200,000 individuals in the U.S. or, for a disease that affects more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for such disease or condition will be recovered from its sales in the U.S. Under these provisions, a supplier of a designated orphan product can seek tax benefits, and the holder of the first FDA approval of a designated orphan product will be granted a seven-year period of marketing exclusivity for that product for the orphan indication. It would not prevent other drugs from being approved for the same indication.
Patient Protection and Affordable Care Act
As Auxilium reported in its 10 K, “the Patient Protection and Affordable Care Act (“PPACA”, which was enacted in 2010, includes provisions covering biological product exclusivity periods and a specific reimbursement methodology for biosimilars. As a new biological product, we expect that XIAFLEX will be eligible for 12 years of marketing exclusivity from the date of its approval by the FDA (although this could change as the regulations are enacted) which was February 2, 2010. PPACA also establishes an abbreviated licensure pathway for products that are biosimilar to or interchangeable with FDA-approved biological products, such as XIAFLEX. As a result, we could face competition from other pharmaceutical companies that develop biosimilar versions of XIAFLEX that do not infringe our patents or other proprietary rights. Similar legislation has been adopted in the EU.”
Trade Secrets
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technology or that we can meaningfully protect our trade secrets.
It is our policy to require certain employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees, the agreements provide that all inventions conceived by the individual shall be our exclusive property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
EMPLOYEES
The Company currently has five employees, who are all full-time employees.
CORPORATE INFORMATION
BioSpecifics Technologies Corp. was incorporated in Delaware in 1990. ABC-NY was incorporated in New York in 1957. Our telephone number is 516-593-7000. Our corporate headquarters are currently located at 35 Wilbur St., Lynbrook, NY 11563, as further described in this Report under “Item 2 - Description of Property”.
AVAILABLE INFORMATION
We file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy any document we file with the SEC at the SEC's public reference room at 100 F. Street, N.E., Washington, DC 20549, at prescribed rates. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. You may also obtain our SEC filings free of charge from the SEC's Internet website at www.sec.gov.
Our Internet website address is www.biospecifics.com. We make available free of charge through our Internet website's “Investors Relations” page most of our filings with the SEC, including our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and other information. These reports and information are available as soon as reasonably practicable after such material is electronically filed with, or furnished to, the SEC.
In addition to the other information included in this Report, the following factors should be considered in evaluating our business and future prospects. Any of the following risks, either alone or taken together, could materially and adversely affect our business, financial position or results of operations. If one or more of these or other risks or uncertainties materialize or if our underlying assumptions prove to be incorrect, our actual results may vary materially from what we projected. There may be additional risks that we do not presently know or that we currently believe are immaterial which could also impair our business or financial position.
Risks Related to Our Limited Sources of Revenue
Our future revenue is primarily dependent upon option, milestone and contingent royalty payments from Auxilium.
Our primary sources of revenues are from option, milestone, mark-up on cost of goods sold and contingent royalty payments from Auxilium under the Auxilium Agreement. In 2013, we recognized the final earnout payment of $3.5 million from DFB, which will be received in March 2014.
Auxilium
As described in Item 1 above, under the Auxilium Agreement, in exchange for the right to receive royalties and other payments, we granted to Auxilium the right to develop, manufacture, market and sell worldwide products (other than dermal formulations for topical administration) that contain collagenase for the treatment of Dupuytren’s contracture, Peyronie’s disease, frozen shoulder and cellulite. However, we have no control over Auxilium’s ability to successfully market, sell and manufacture candidate products for the treatment of Dupuytren’s contracture, or, in the case of Peyronie’s disease, frozen shoulder and cellulite, to pursue commercialization, and we may receive limited, if any, royalty payments from Auxilium. We have received in the past, and are entitled to receive in the future, certain milestone payments from Auxilium in respect of its efforts to commercialize candidate products, but we have no control over Auxilium’s ability to achieve the milestones. As also described in Item 1 above, Auxilium has sublicensed to third parties some of the development and commercialization rights it licenses from us. We have received in the past a percentage of sublicense income that Auxilium receives from these third parties based on the achievement of certain regulatory and sales related milestones. There is no guarantee that these third parties will continue to pursue development and commercialization of XIAFLEX. As in the case with Pfizer, if any third party stops pursuing such development and commercialization, sublicense income would no longer be payable to Auxilium or us.
Even if Auxilium or its sublicensees pursues development and commercialization, there is no guarantee that the FDA or equivalent foreign regulatory body will approve XIAFLEX for a given indication or that commercialization will be successful, if the FDA or equivalent foreign regulatory body does approve XIAFLEX for a given indication. Moreover, under the Auxilium Agreement, royalty payments are subject to set-off for certain expenses we owe Auxilium related to development and patent costs. We anticipate that the amount of royalties due to us will exceed the amount of any set-offs on a going forward basis.
In addition, we have granted to Auxilium an option to expand its license and development rights to one or more additional indications for injectable collagenase not currently licensed to Auxilium, including for the treatment of human and canine lipoma. If Auxilium exercises its option with respect to an additional indication, we are entitled to receive a one-time license fee for the rights to, as well as potential milestone, royalty and other payments with respect to, such new indication. If Auxilium does not exercise its option as to any additional indication, we may offer to any third party such development rights with regard to products in the Auxilium Territory (as defined in the Auxilium Agreement), provided that we first offer the same terms to Auxilium, or develop the product ourselves. Auxilium has no obligation to exercise its option with respect to any such additional indication, and its decision to do so is in its complete discretion. Clinical trials can be expensive and the results are subject to different interpretations, therefore, there is no assurance that after conducting phase II clinical trials on any additional indication, and incurring the associated expenses, Auxilium will exercise its option or we will receive any revenue from it. Under the Auxilium Agreement, we may only offer to a third party development rights with regard to products in the Auxilium Territory and not in Europe and certain Eurasian countries. Even if Auxilium exercises its option as to any additional indication, its obligations to develop the product for such indication are limited to initiating Stage II Development (as defined in the Auxilium Agreement) for such additional indication within one year of exercising the option as to such indication. Auxilium may decide to allocate its resources other than to the development of XIAFLEX, and we have no control over that decision. For instance, Auxilium has acquired Actient Holdings LLC, a private urology specialty therapeutics company, for $585 million in upfront cash plus certain contingent consideration and warrants to purchase Auxilium common stock. Any such non-XIAFLEX related acquisition may result in Auxilium reallocating its priorities away from XIAFLEX.
DFB
As part of the sale of our topical collagenase business to DFB, we were entitled to receive earn out payments in respect of sales of certain products developed and manufactured by DFB that contain collagenase for topical administration through the end of August 2013. A final payment of $3.5 million was recognized in 2013, but will be received in March 2014.
Our dependence upon revenue from Auxilium make us subject to the commercialization and other risk factors affecting Auxilium over which we have limited or no control.
Auxilium has disclosed in its securities filings a number of risk factors to consider when evaluating its business and future prospects. Given our dependence upon revenue from Auxilium, Auxilium’s operating success or failure has a significant impact on our potential royalty stream and other payment rights. As such, we refer you to the full text of Auxilium’s disclosed risk factors in its securities filings, which were most recently included in the Auxilium 10-K.
If we are unable to obtain option, milestone, mark-up on cost of goods sold and royalty payments from Auxilium or meet our needs for additional funding from other sources, we may be required to limit, scale back or cease our operations.
Our business strategy contains elements that we will not be able to implement if we do not receive the anticipated option, milestone, royalty or earn out payments from Auxilium, or secure additional funding from other sources. While we anticipate being profitable on an ongoing, annual basis, our future funding requirements will depend on many factors, including:
| • | Auxilium’s ability to manufacture and commercialize XIAFLEX for which we would receive milestone, mark-up on cost of goods sold and royalty payments; |
| • | Sobi’s ability to commercialize XIAPEX in its territory and Actelion’s ability to commercialize XIAFLEX in Canada or Australia; |
| • | the amount actually owed to Auxilium for certain patent costs; |
| • | the scope, rate of progress, cost and results of our clinical trials on additional indications, including human lipoma, for which Auxilium could exercise its option to acquire rights to them, and whether Auxilium exercises the option for canine lipoma; |
| • | the terms and timing of any future collaborative, licensing, co-promotion and other arrangements that we may establish; |
| • | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights or defending against any other litigation; |
| • | the extent to which Auxilium’s acquisition of Actient and in-licensing of STENDRA™ results in Auxilium’s reallocation of priority away from XIAFLEX; and |
| • | the extent to which Auxilium focuses on men’s health and away from orthopedic or dermatology (Dupuytren’s contracture, frozen shoulder, cellulite). |
These factors could result in variations from our currently projected operating requirements. If our existing resources are insufficient to satisfy our operating requirements, we may need to limit, scale back or cease operations or, in the alternative, borrow money. Given our operations and history, we may not be able to borrow money on commercially reasonable terms, if at all. If we issue any equity or debt securities, the terms of such issuance may not be acceptable to us and would likely result in substantial dilution of our stockholders’ investment. If we do not receive revenues from Auxilium or DFB, and are unable to secure additional financing, we may be required to cease operations.
In order to finance and to secure the rights to conduct clinical trials for products we have licensed to Auxilium, we have granted to third parties significant rights to share in royalty payments received by us.
To finance and secure the rights to conduct clinical trials for products we have licensed to Auxilium, we have granted to third parties certain rights to share in royalty payments received by us from Auxilium under the Auxilium Agreement. Consequently, we will be required to share a significant portion of the payments due to us from Auxilium under the Auxilium Agreement.
If we breach our agreements with third parties, our business could be materially harmed.
Our agreements with third parties impose on us various obligations, such as those related to intellectual property rights, non-competition, and development of products, as described throughout this Item 1A of this Report. If we fail to comply with such obligations, or a counterparty to our agreements believes that we have failed to comply with such obligations, we may be sued and the costs of the resulting litigation could materially harm our business.
Risks Related to Clinical Trials and Development of Drug Candidates
Our ability to conduct clinical trials may be limited by the Auxilium Agreement.
Under the Auxilium Agreement, we have the right to conduct trials, studies or development work for, among other things, indications in canine lipomas and human lipomas, and, upon approval by the parties’ joint development committee (“JDC”), additional indications. Auxilium has pre-approved our protocols for canine lipomas and human lipomas. However, certain material changes to the protocols must be approved by the JDC, and the JDC may decide not to approve such changes if the JDC has reasonable safety concerns. In addition, the JDC has the right to stop a study or trial in canine lipomas and human lipomas if the rate of serious adverse events exceeds certain thresholds. If the JDC fails to approve changes to our protocols for canine lipomas and human lipomas or if the JDC stops our studies or trials in canine lipomas and human lipomas due to safety concerns, our ability to obtain option, milestone and royalty payments with respect to those indications would be limited. We may only conduct in vivo trials, studies or development work for additional indications beyond the pre-approved indications upon submission to and approval by the JDC of our development plan which includes in vivo studies of uterine fibroids. In the case of indications in keloids, capsular contraction after breast augmentation, arthrofibrosis following total joint replacement in humans and equine suspensory ligament desmitis, the JDC may reject our submission only for reasonable safety concerns. The JDC may reject our submission for any other additional indications for safety or commercial concerns. If the JDC rejects our submissions in any additional indications, our ability to obtain option, milestone and royalty payments with respect to those additional indications would be limited.
We are dependent on Auxilium for access to XIAFLEX, which may limit our ability to conduct clinical trials and to obtain the associated option, milestone and contingent royalty payments under the Auxilium Agreement.
Under the Auxilium Agreement, we have agreed to buy at cost plus a mark-up XIAFLEX from Auxilium for conducting our trials, studies and development work. If Auxilium does not supply XIAFLEX to us, our ability to conduct clinical trials using XIAFLEX would be limited because we do not have the right to make XIAFLEX or to purchase it from third parties. Moreover, our ability to use our own clinical material may be limited both by lack of availability and by certain potential regulatory restrictions. Without adequate supply of clinical material our ability to obtain additional option, milestone and royalty payments under the Auxilium Agreement would be limited.
If clinical trials in humans or veterinarian trials for our potential new indications are delayed, we may not be able to obtain option, milestone or royalty payments under the Auxilium Agreement for new indications.
Clinical trials that we or our investigators may conduct may not begin on time or may need to be restructured or temporarily suspended after they have begun. Clinical trials can be delayed or may need to be restructured for a variety of reasons, including delays or restructuring related to:
| · | changes to the regulatory approval process for product candidates; |
| · | obtaining regulatory approval to commence a clinical trial; |
| · | timing of responses required from regulatory authorities; |
| · | negotiating acceptable clinical trial agreement terms with prospective investigators or trial sites; |
| · | obtaining institutional review board, or equivalent, approval to conduct a clinical trial at a prospective site; |
| · | recruiting subjects to participate in a clinical trial; |
| · | competition in recruiting clinical investigators; |
| · | shortage or lack of availability of clinical trial supplies from external and internal sources; |
| · | the need to repeat clinical trials as a result of inconclusive results or poorly executed testing; |
| · | failure to validate a patient-reported outcome questionnaire; |
| · | the placement of a clinical hold on a study; |
| · | the failure of third parties conducting and overseeing the operations of our clinical trials to perform their contractual or regulatory obligations in a timely fashion; and |
| · | exposure of clinical trial subjects to unexpected and unacceptable health risks or noncompliance with regulatory requirements, which may result in suspension of the trial. |
The process of conducting clinical trials and developing product candidates involves a high degree of risk, may take several years, and may ultimately not be successful.
Product candidates that appear promising in the early phases of development may fail to reach the market for several reasons, including:
| • | clinical trials may show product candidates to be ineffective or not as effective as anticipated or to have harmful side effects or any unforeseen result; |
| • | product candidates may fail to receive regulatory approvals required to bring the products to market; |
| • | manufacturing costs, the inability to scale up to produce supplies for clinical trials or other factors may make our product candidates uneconomical; and |
| • | the proprietary rights of others and their competing products and technologies may prevent product candidates from being effectively commercialized or from obtaining exclusivity. |
Success in preclinical and early clinical trials does not ensure that large-scale clinical trials will be successful. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. The length of time necessary to complete clinical trials and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly and may be difficult to predict. Any changes to the U.S. regulatory approval process could significantly increase the timing or cost of regulatory approval for product candidates making further development uneconomical or impossible. In addition, once Auxilium exercises its option with respect to an additional indication, further clinical trials, development, manufacturing, marketing and selling of such product are out of our control. Our interest is limited to receiving option, milestone and royalty payments.
Successful development of drug candidates is inherently difficult and uncertain, and our long-term prospects depend upon our ability and the ability of our partners, particularly with respect to XIAFLEX, to continue to successfully commercialize these drug candidates.
Successful development of drugs is inherently difficult and uncertain. Our business requires investments in research and development over many years, often for drug candidates that may fail during the research and development process. Even if the Company is able to successfully complete the development of our drug candidates, our long-term prospects depend upon our ability and the ability of our partners, particularly with respect to XIAFLEX, to continue to successfully commercialize these drug candidates.
There is significant uncertainty regarding our ability to successfully develop drug candidates in other indications. These risks include the uncertainty of:
| · | the nature, timing and estimated costs of the efforts necessary to complete the development of our drug candidate projects; |
| · | the anticipated completion dates for our drug candidate projects; |
| · | the scope, rate of progress and cost of our clinical trials that we are currently running or may commence in the future with respect to our drug candidate projects; |
| · | the scope, rate of progress of our preclinical studies and other research and development activities related to our drug candidate projects; |
| · | clinical trial results for our drug candidate projects; |
| · | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights relating to our drug candidate projects; |
| · | the terms and timing of any strategic alliance, licensing and other arrangements that we have or may establish in the future relating to our drug candidate projects; |
| · | the cost and timing of regulatory approvals with respect to our drug candidate projects; and |
| · | the cost of establishing clinical supplies for our drug candidate projects. |
Risks Related to Our Agreements with Auxilium and DFB
Our ability to conduct clinical trials and develop products for injectable administration of collagenase is limited by the Auxilium Agreement.
Under the Auxilium Agreement, we have licensed or granted options to certain of our rights to conduct clinical trials and develop products for injectable administration of collagenase. We agreed, for example, to certain non-competition provisions, which may limit our clinical development activities.
Our ability to conduct clinical trials and develop products for topical administration of collagenase is limited by the agreement we have signed with DFB.
Under the DFB Agreement, we have sold, licensed, or granted options to certain of our rights to conduct clinical trials and develop products for topical administration of collagenase. Under the terms of the DFB Agreement, we have agreed to certain non-competition provisions, which may limit our clinical development activities.
Risks Related to Regulatory Requirements
We are subject to numerous complex regulatory requirements and failure to comply with these regulations, or the cost of compliance with these regulations, may harm our business.
Conducting clinical trials for human drugs and, in certain circumstances, veterinarian trials for animal drugs, and the testing, development and manufacturing and distribution of product candidates are subject to regulation by numerous governmental authorities in the U.S. and other jurisdictions, if we desire to export the resulting products to such other jurisdictions. These regulations govern or affect the testing, manufacture, safety, labeling, storage, record-keeping, approval, distribution, advertising and promotion of product candidates, as well as safe working conditions. Noncompliance with any applicable regulatory requirements can result in suspension or termination of any ongoing clinical trials of a product candidate or refusal of the government to approve a product candidate for commercialization, criminal prosecution and fines, recall or seizure of products, total or partial suspension of production, prohibitions or limitations on the commercial sale of products or refusal to allow the entering into of federal and state supply contracts. The FDA and comparable governmental authorities have the authority to suspend or terminate any ongoing clinical trials of a product candidate or withdraw product approvals that have been previously granted. Even after a product candidate has been approved, the FDA and comparable governmental authorities subject such product to continuing review and regulatory requirements including, for example, requiring the conducting and reporting of the results of certain clinical studies or trials and commitments to voluntarily conduct additional clinical trials. In addition, regulatory approval could impose limitations on the indicated or intended uses for which product candidates may be marketed. With respect to its approval of XIAFLEX for the treatment of adult Dupuytren’s contracture patients with a palpable cord, for example, the FDA and Auxilium agreed upon a REMS program consisting of a communication plan and a medication guide. With respect to its approval of XIAFLEX for Peyronie’s disease, Auxilium has further collaborated with the FDA to update the REMS with an Elements to Assure Safe Use (“ETASU”) for XIAFLEX for the treatment of Peyronie's disease in men with a palpable plaque and curvature deformity of 30 degrees or greater at the start of therapy. The goal of the XIAFLEX REMS with an ETASU for Peyronie’s disease is to certify that the appropriate physicians and practice sites are trained in the use of XIAFLEX and to attempt to mitigate the serious risk of penile fracture (corporal rupture) and other serious injuries to the penis such as hematoma. Currently, there is a substantial amount of congressional and administrative review of the FDA and the regulatory approval process for drug candidates in the U.S. As a result, there may be significant changes made to the regulatory approval process in the U.S. In addition, the regulatory requirements relating to the development, manufacturing, testing, promotion, marketing and distribution of product candidates may change in the U.S. Such changes may increase our costs and adversely affect our operations.
Additionally, failure to comply with, or changes to applicable regulatory requirements may result in a variety of consequences, including the following:
| • | restrictions on our products or manufacturing processes; |
| • | warning letters; |
| • | withdrawal of a product from the market; |
| • | voluntary or mandatory recall of a product; |
| • | fines; |
| • | suspension or withdrawal of regulatory approvals for a product; |
| • | refusal to permit the import or export of our products; |
| • | refusal to approve pending applications or supplements to approved applications that we submit; |
| • | denial of permission to file an application or supplement in a jurisdiction; |
| • | product seizure; and |
| • | injunctions or the imposition of civil or criminal penalties against us. |
Our corporate compliance program cannot guarantee that we are in compliance with all potentially applicable laws and regulations and we have incurred and will continue to incur costs relating to compliance with applicable laws and regulations.
We are a small company and we rely heavily on third parties and outside consultants to conduct many important functions. As a biopharmaceutical company, we are subject to a large body of legal and regulatory requirements. In addition, as a publicly traded company we are subject to significant regulations, including the Sarbanes-Oxley Act of 2002 (“SOX”), some of which have only recently been revised or adopted. We cannot assure you that we are or will be in compliance with all potentially applicable laws and regulations. Failure to comply with all potentially applicable laws and regulations could lead to the imposition of fines, cause the value of our common stock to decline, impede our ability to raise capital or list our securities on certain securities exchanges. New rules could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees and as executive officers. We cannot predict or estimate the amount of the additional costs we may incur or the timing of such costs to comply with these rules and regulations.
We may fail to maintain effective internal controls over external financial reporting or such controls may fail or be circumvented.
SOX requires us to report annually on our internal controls over financial reporting, and our business and financial results could be adversely effected if we, or our independent registered public accounting firm, determine that these controls are not effective. In addition, any failure or circumvention of our internal controls and procedures or failure to comply with regulations concerning controls and procedures could have a material effect on our business, results of operation and financial condition. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees and as executive officers.
Risks Related to Growth and Employees
Adverse events or lack of efficacy in clinical trials may force us and/or our partners upon whom we are wholly dependent to stop development of our product candidates or prevent regulatory approval of our product candidates or significant safety issues could arise after regulatory approval of our products, any of which could materially harm our business.
The prescribing information for XIAFLEX for Dupuytren’s contracture made available by Auxilium lists “tendon ruptures or other serious injury to the injected extremity” and one “anaphylactic reaction reported in a post-marketing clinical study in a patient who had previous exposure to XIAFLEX for the treatment of Dupuytren’s contracture” as a reported serious adverse reaction to XIAFLEX and states that the most frequently reported adverse drug reactions in XIAFLEX clinical trials included swelling of the injected hand, contusion, injection site reaction, injection site hemorrhage, and pain in the treated extremity. The prescribing information notes that adverse reaction rates observed in clinical trials of a drug may not reflect those observed in practice because such trials “are conducted under widely varying conditions.”
In the case of Peyronie’s disease, the serious risks include penile fracture (corporal rupture) and other serious injuries to the penis such as hematoma. These serious risks are highlighted in the Boxed Warning within the Full Prescribing Information (the label).
Adverse events or lack of efficacy may force us to stop development of our product candidates or prevent regulatory approval of our product candidates, which could materially harm our business. In addition, any adverse events or lack of efficacy may force Auxilium to stop development of the products we have licensed to it or prevent regulatory approval of such products, which could materially impair all or a material part of the future revenue we hope to receive from Auxilium. Even if our product candidates receive regulatory approval, new safety issues may be reported and we or our partners may be required to amend the conditions of use for a product.
We and our licensees face competition in our product development and marketing efforts from pharmaceutical and biotechnology companies, universities and other not-for-profit institutions.
We and our licensees face competition in our product development and marketing efforts from entities that have substantially greater research and product development capabilities and greater financial, scientific, marketing and human resources. These entities include pharmaceutical and biotechnology companies, as well as universities and not-for-profit institutions. Our and our licensees’ competitors may succeed in developing products or intellectual property earlier than we or our licensees do, entering into successful collaborations before us or our licensees, obtaining approvals from the FDA or other regulatory agencies for such products before us or our licensees, or developing or marketing products that are more effective than those we or our licensees could develop or market. The success of any one competitor in these or other respects will have a material adverse effect on our business, our ability to receive option payments from Auxilium or our ability to generate revenues from third party arrangements with respect to additional indications for which Auxilium does not exercise its option.
Because of the specialized nature of our business, the termination of relationships with key management, consulting and scientific personnel or the inability to recruit and retain additional personnel could prevent us from developing our technologies, conducting clinical trials and/or obtaining financing.
The competition for qualified personnel in the biotechnology field is intense, and we rely heavily on our ability to attract and contract with qualified independent scientific and medical investigators, and technical and managerial personnel. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. To the extent we are unable to attract and retain any of these individuals on favorable terms our business may be adversely affected.
If product liability lawsuits are brought against us, we may incur substantial liabilities.
We continue to have product liability exposure for topical product sold by us prior to the sale of our topical business to DFB. In addition, under the Auxilium Agreement, we are obligated to indemnify Auxilium and its affiliates for any harm or losses they suffer relating to any personal injury and other product liability resulting from our development, manufacture or commercialization of any injectable collagenase product. In addition, the clinical testing and, if approved, commercialization of our product candidates involves significant exposure to product liability claims. We have clinical trial and product liability insurance in the aggregate amount of $5.0 million dollars that we believe is adequate in both scope and amount and has been placed with what we believe are reputable insurers. We may not be able to maintain our clinical trial and product liability insurance at an acceptable cost, if at all, and this insurance may not provide adequate coverage against potential claims or losses. If losses from product liability claims exceed our insurance coverage, we may incur substantial liabilities that exceed our financial resources, and our business and results of operations may be harmed. Whether or not we are ultimately successful in product liability litigation, such litigation could consume substantial amounts of our financial and managerial resources, and might result in adverse publicity, all of which could impair our business.
Risks Related to Intellectual Property Rights
If we breach any of the agreements under which we license rights to products or technology from others, we could lose license rights that are critical to our business and our business could be harmed.
We are a party to a number of license agreements by which we have acquired rights to use the intellectual property of third parties that are necessary for us to operate our business. If any of the parties terminates its agreement, whether by its terms or due to our breach, our right to use the party’s intellectual property may negatively affect our licenses to Auxilium, and, in turn, their obligation to make option, milestone, contingent royalty or other payments to us.
Our ability and the ability of our licensors, licensees and collaborators to develop and license products based on our patents may be impaired by the intellectual property of third parties.
Auxilium’s, and our commercial success in developing and manufacturing collagenase products based on our patents is dependent on these products not infringing the patents or proprietary rights of third parties. While we currently believe that we, our licensees, licensors and collaborators have freedom to operate in the collagenase market, others may challenge that position in the future. There has been, and we believe that there will continue to be, significant litigation in the pharmaceutical industry regarding patent and other intellectual property rights.
Third parties could bring legal actions against us, our licensees, licensors or collaborators claiming damages and seeking to enjoin clinical testing, manufacturing and marketing of the affected product or products. A third party might request a court to rule that the patents we in-licensed or licensed to others, or those we may in-license in the future, are invalid or unenforceable. In such a case, even if the validity or enforceability of those patents were upheld, a court might hold that the third party’s actions do not infringe the patent we in-license or license to others, which could, in effect, limit the scope of our patent rights and those of our licensees, licensors or collaborators. Our agreements with Auxilium require us to indemnify them against any claims for infringement based on the use of our technology. If we become involved in any litigation, it could consume a substantial portion of our resources, regardless of the outcome of the litigation. If Auxilium becomes involved in such litigation, it could also consume a substantial portion of their resources, regardless of the outcome of the litigation, thereby jeopardizing their ability to commercialize candidate products and/or their ability to make option, milestone or royalty payments to us. If any of these actions is successful, in addition to any potential liability for damages, we could be required to obtain a license to permit ourselves, our licensees, licensors or our collaborators to conduct clinical trials, manufacture or market the affected product, in which case we may be required to pay substantial royalties or grant cross-licenses to our patents. However, there can be no assurance that any such license will be available on acceptable terms or at all. Ultimately, we, our licensees, licensors or collaborators could be prevented from commercializing a product, or forced to cease some aspect of their or our business, as a result of patent infringement claims, which could harm our business or right to receive option, milestone and contingent royalty payments.
Risks Related to our Common Stock
Future sales of our common stock could negatively affect our stock price.
If our common stockholders sell substantial amounts of common stock in the public market, or the market perceives that such sales may occur, the market price of our common stock could decline. In addition, we may need to raise additional capital in the future to fund our operations. If we raise additional funds by issuing equity securities, our stock price may decline and our existing stockholders may experience dilution of their interests. Because we historically have not declared dividends, stockholders must rely on an increase in the stock price for any return on their investment in us.
Our stock price has, in the past, been volatile, and the market price of our common stock may drop below the current price.
Our stock price has, at times, been volatile. Currently, our common stock is traded on The Nasdaq Global Market (“Nasdaq”) and is thinly traded.
Market prices for securities of pharmaceutical, biotechnology and specialty pharmaceutical companies have been particularly volatile. Some of the factors that may cause the market price of our common stock to fluctuate include:
| • | results of our clinical trials; |
| • | failure of any product candidates we have licensed to Auxilium to achieve commercial success; |
| • | failure of Auxilium to exercise any opt in rights to new indications including canine lipoma; |
| • | regulatory developments in the U.S. and foreign countries; |
| • | developments or disputes concerning patents or other proprietary rights; |
| • | litigation involving us or our general industry, or both; |
| • | future sales of our common stock by the estate of our former Chairman and CEO, directors,officers, or others; |
| • | changes in the structure of healthcare payment systems, including developments in price control legislation; |
| • | departure of key personnel; |
| • | termination of agreements with our licensees or their sublicensees; |
| • | announcements of material events by those companies that are our competitors or perceived to be similar to us; |
| • | changes in estimates of our financial results; |
| • | investors’ general perception of us; |
| • | general economic, industry and market conditions; and |
| • | the reallocation by Auxilium of its priorities away from XIAFLEX or orthopedic or dermatological indications. |
If any of these risks occurs, or continues to occur, it could cause our stock price to fall and may expose us to class action lawsuits that, even if unsuccessful, could be costly to defend and a distraction to management. In addition, purchases of our common stock pursuant to our stock repurchase program may, depending on the timing and volume of such repurchases, result in our stock price being higher than it would be in the absence of such repurchases. If repurchases pursuant to the program are discontinued, our stock price may fall.
We may become subject to stockholder activism efforts that could cause material disruption to our business.
Certain influential institutional investors, hedge funds and other stockholders have taken steps to involve themselves in the governance and strategic direction of certain companies due to governance or strategic related disagreements between such companies and such stockholders. If we become subject to such stockholder activism efforts, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business and adversely affect the market price of our common stock.
We have no current plan to pay dividends on our common stock and investors may lose the entire amount of their investment.
We have no current plans to pay dividends on our common stock. Therefore, investors will not receive any funds absent a sale of their shares. We cannot assure investors of a positive return on their investment when they sell their shares nor can we assure that investors will not lose the entire amount of their investment.
Our outstanding options to purchase shares of common stock could have a possible dilutive effect.
As of December 31, 2013, options to purchase 1,167,000 shares of common stock were outstanding. In addition, as of December 31, 2013 a total of 239,098 options were available for grant under our stock option plans. The issuance of common stock upon the exercise of these options could adversely affect the market price of the common stock or result in substantial dilution to our existing stockholders.
If securities analysts do not publish research reports about our business or if they downgrade us or our sector, the price of our common stock could decline.
The trading market for our common stock will depend in part on research reports that industry or financial analysts publish about us or our business. If analysts downgrade us or any of our licensees, or other research analysts downgrade the industry in which we operate or the stock of any of our competitors or licensees, the price of our common stock will probably decline.
Provisions in our certificate of incorporation and bylaws may prevent or frustrate a change in control.
Provisions of our certificate of incorporation and bylaws may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions:
| • | provide for a classified board of directors; |
| • | give our Board the ability to designate the terms of and issue new series of preferred stock without stockholder approval, commonly referred to as “blank check” preferred stock, with rights senior to those of our common stock; |
| • | limit the ability of the stockholders to call special meetings; and |
| • | impose advance notice requirements on stockholders concerning the election of directors and other proposals to be presented at stockholder meetings. |
In addition, during May 2002, the Board implemented a rights agreement (commonly known as a “Poison Pill”), which effectively discourages or prevents acquisitions of more than 15% of our common stock in transactions (mergers, consolidations, tender offer, etc.) that have not been approved by our Board. The Board amended the Poison Pill in February 2011 to increase the threshold from 15% to 18% and extended the expiration date of the Poison Pill for an additional two years, to May 31, 2014. In February 2014, the Board amended the Poison Pill again to extend the term for an additional two years, to May 31, 2016. These provisions could make it more difficult for common stockholders to replace members of the Board. Because our Board is responsible for appointing the members of our management team, these provisions could in turn affect any attempt to replace the current management team.
If our principal stockholders, executive officers and directors choose to act together, they may be able to control our operations, acting in their own best interests and not necessarily those of other stockholders.
As of March 3, 2014 our executive officers, directors and their affiliates, in the aggregate, beneficially owned shares representing approximately 28.5% of our common stock. Beneficial ownership includes shares over which an individual or entity has investment or voting power and includes shares that could be issued upon the exercise of options within 60 days. As a result, if these stockholders were to choose to act together, they may be able to control all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these individuals, if they chose to act together, could control the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of ownership could have the effect of delaying, deferring or preventing a change in control or impeding a merger or consolidation, takeover or other business combination that could be favorable to other stockholders.
This significant concentration of share ownership may adversely affect the trading price for our common stock because investors often perceive disadvantages in owning stock in companies with controlling stockholders.
None.
Our corporate headquarters are currently located at 35 Wilbur St., Lynbrook, NY 11563. On November 21, 2013, the Company entered into an Agreement of Lease (the “New Lease”) with 35 Wilbur Street Associates, LLC (“New Landlord”) for the Company’s corporate headquarters located at 35 Wilbur Street, Lynbrook, New York 11563 (the “Premises”). Neither the Company nor its affiliates have a material relationship or affiliation with the New Landlord. As previously reported, the Company formerly leased the Premises from Wilbur St. Corp. (“WSC”). On November 21, 2013, WSC sold the Premises to the New Landlord, and the Company entered into the New Lease with the New Landlord and simultaneously terminated the existing lease. The term of the New Lease is twenty-four months, provided, however, that the Company has the option to cancel the New Lease after the first year by giving three months’ notice, which may be given before the expiration of the first year. Pursuant to the New Lease, the Company’s monthly base rent is $12,000.00. The Company is required to pay as additional rent an amount equal to the increase in taxes over a specified base year.
None.
Not Applicable.
| Item 5. |
Market Information
Our common stock currently trades under the symbol BSTC on Nasdaq.
The table below sets forth the high and low closing sale prices for our common stock for each of the quarterly periods in 2013 and 2012 as reported by and as quoted by Nasdaq, as applicable:
2013 | HIGH | LOW | ||||||
| Fourth Quarter | $ | 22.94 | $ | 18.42 | ||||
| Third Quarter | $ | 19.47 | $ | 15.98 | ||||
| Second Quarter | $ | 17.29 | $ | 15.17 | ||||
| First Quarter | $ | 17.20 | $ | 14.64 | ||||
2012 | HIGH | LOW | ||||||
| Fourth Quarter | $ | 19.43 | $ | 12.75 | ||||
| Third Quarter | $ | 20.27 | $ | 17.53 | ||||
| Second Quarter | $ | 19.06 | $ | 13.70 | ||||
| First Quarter | $ | 21.10 | $ | 15.76 | ||||
These quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission and may not represent actual transactions.
Holders of Record
As of February 26, 2014, there were approximately 71 holders of record of our common stock. Because many of such shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividends
It has been our policy to retain potential earnings to finance the growth and development of our business and not pay dividends, and we have no current plans to pay dividends. Any payment of cash dividends in the future will depend upon our financial condition, capital requirements and earnings as well as such other factors as our board of directors may deem relevant.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information as of December 31, 2013 with respect to the shares of our common stock that may be issued under our existing equity compensation plans:
| Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted-average exercise price of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
Equity compensation plans approved by security holders(1) | 1,167,000 | $ | 9.03 | 239,098 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 1,167,000 | $ | 9.03 | 239,098 | ||||||||
(1) Please see Note 10, “Stockholders’ Equity,” of the notes to the consolidated financial statements for a description of the material features of each of our plans.
Recent Sales of Unregistered Securities
For the year ended December 31, 2013, we did not issue any unregistered shares of securities.
Issuer Purchases of Equity Securities (1)
On December 10, 2013, our Board of Directors reauthorized the repurchase of up to $2.0 million of our common stock under the stock repurchase program.
The following table presents a summary of share repurchases made by us during the quarter ended December 31, 2013.
Month | Total Number of Shares Purchased (2) | Average Price Paid Per Share (3) | Total Cumulative Number of Shares Purchased as Part of Publicly Announced Plan | Maximum Dollar Value of Shares that may yet be Purchased under the Plan | ||||||||||||
| October 1, 2013 to October 31, 2013 | 3,539 | $ | 18.88 | 169,472 | $ | 997,271 | ||||||||||
| November 1, 2013 – November 30, 2013 | - | - | - | - | ||||||||||||
| December 1, 2013 to December 31, 2013 | - | - | - | $ | 2,000,000 | (5) | ||||||||||
| Total | 3,539 | |||||||||||||||
| (1) | On June 4, 2010, we announced that our board of directors authorized a stock repurchase program under Rule 10b-18 of the Exchange Act of up to $2.0 million of our outstanding common stock over a period of 12 months. On June 20, 2011, we announced that our Board of Directors had reauthorized this stock repurchase program. On November 15, 2012, we announced that our Board of Directors had reauthorized this stock repurchase program. |
| (2) | The purchases were made in open-market transactions. |
| (3) | Includes commissions paid, if any, related to the stock repurchase transactions. |
| (4) | On November 15, 2012, we announced that our Board of Directors had reauthorized the repurchase of up to $2.0 million of our common stock under the stock repurchase program. |
| (5) | On December 10, 2013, we announced that our Board of Directors had reauthorized the repurchase of up to $2.0 million of our common stock under the stock repurchase program. |
Performance Graph
The graph below compares the cumulative total stockholder return on our common stock with the cumulative total stockholder return of (i) the NASDAQ Biotechnology Index, and (ii) the NASDAQ Composite Index, assuming an investment of $100 on December 31, 2008, in each of our common stock; the stocks comprising the NASDAQ Composite Index; and the stocks comprising the NASDAQ Biotechnology Index.
Comparison of Cumulative Total Return* Among BioSpecifics Technologies Corp, the NASDAQ Biotechnology Index and the NASDAQ Composite Index

| 12/31/2008 | 12/31/2009 | 12/31/2010 | 12/31/2011 | 12/31/2012 | 12/31/2013 | |||||||||||||||||||
| Biospecifics Technologies Corp | $ | 100.00 | $ | 137.15 | $ | 119.63 | $ | 77.66 | $ | 69.86 | $ | 101.26 | ||||||||||||
| Nasdaq Biotechnology Index | $ | 100.00 | $ | 115.60 | $ | 132.98 | $ | 148.69 | $ | 196.12 | $ | 324.80 | ||||||||||||
| Nasdaq Composite Index | $ | 100.00 | $ | 143.89 | $ | 168.22 | $ | 165.19 | $ | 191.47 | $ | 264.84 | ||||||||||||
*Total return assumes $100 invested on December 31, 2008 in our common stock, the NASDAQ Composite Index, and the NASDAQ Biotechnology Index and reinvestment of dividends through fiscal year ended December 31, 2013.
The following selected consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes appearing elsewhere in this Report. The consolidated statements of operations data for the years ended December 31, 2013, 2012 and 2011 and the consolidated balance sheet data as of December 31, 2013 and 2012 have been derived from our audited consolidated financial statements and related notes, which are included elsewhere in this Report. The consolidated statement of operations data for the years ended December 31, 2010 and 2009 and the consolidated balance sheet data as of December 31, 2011, 2010 and 2009 have been derived from audited financial statements which do not appear in this Report. The historical results presented are not necessarily indicative of results to be expected in any future period.
| Years Ended December 31, | ||||||||||||||||||||
| Consolidated Statement of Operations Data | 2013 | 2012 | 2011 | 2010 | 2009 | |||||||||||||||
| Net revenues | $ | 14,467,240 | $ | 11,145,078 | $ | 11,395,726 | $ | 5,661,348 | $ | 3,155,757 | ||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 1,484,416 | 1,249,755 | 972,078 | 1,223,931 | 488,646 | |||||||||||||||
| General and administrative | 5,038,363 | 4,774,828 | 5,231,881 | 6,470,449 | 4,832,019 | |||||||||||||||
| Total operating expenses | 6,522,779 | 6,024,583 | 6,203,959 | 7,694,380 | 5,320,665 | |||||||||||||||
| Operating income (loss) | 7,944,461 | 5,120,495 | 5,191,767 | (2,033,032 | ) | (2,164,908 | ) | |||||||||||||
| Other income (expense): | ||||||||||||||||||||
| Interest income | 26,202 | 34,634 | 55,780 | 86,310 | 55,693 | |||||||||||||||
| Interest expense | - | - | - | - | (39 | ) | ||||||||||||||
| Other | - | - | 15,823 | 13,130 | (8,863 | ) | ||||||||||||||
| Qualifying Therapeutic Credit | - | - | - | 426,403 | - | |||||||||||||||
| 26,202 | 34,634 | 71,603 | 525,843 | 46,791 | ||||||||||||||||
| Income (loss) before income tax | 7,970,663 | 5,155,129 | 5,263,370 | (1,507,189 | ) | (2,118,117 | ) | |||||||||||||
| Income tax benefit (expense) | (2,684,816 | ) | (2,174,054 | ) | 1,338,256 | (1,351 | ) | 161,574 | ||||||||||||
| Net income (loss) | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | $ | (1,508,540 | ) | $ | (1,956,543 | ) | ||||||||
| Basic net income (loss) per share | $ | 0.83 | $ | 0.47 | $ | 1.04 | $ | (0.24 | ) | $ | (0.32 | ) | ||||||||
| Diluted net income (loss) per share | $ | 0.76 | $ | 0.43 | $ | 0.95 | $ | (0.24 | ) | $ | (0.32 | ) | ||||||||
| Shares used in computation of basic net income (loss) per share | 6,345,615 | 6,351,245 | 6,340,648 | 6,261,214 | 6,065,939 | |||||||||||||||
| Shares used in computation of diluted net income (loss) per share | 6,922,274 | 6,981,527 | 6,952,386 | 6,261,214 | 6,065,939 | |||||||||||||||
| Years Ended December 31, | ||||||||||||||||||||
| Consolidated Balance Sheet Data: | 2013 | 2012 | 2011 | 2010 | 2009 | |||||||||||||||
| Cash and cash equivalents | $ | 5,624,860 | $ | 3,383,737 | $ | 3,196,831 | $ | 2,470,852 | $ | 3,950,389 | ||||||||||
| Short-term investments | 6,966,964 | 5,120,000 | 5,000,000 | 5,360,970 | 4,548,541 | |||||||||||||||
| Total assets | 23,252,244 | 18,390,264 | 16,265,073 | 11,518,701 | 11,748,478 | |||||||||||||||
| Total stockholders’ equity | 22,322,439 | 17,458,346 | 14,872,314 | 6,700,723 | 6,092,107 | |||||||||||||||
| Item 7. |
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes appearing at the end of this Report. Some of the information contained in this discussion and analysis or set forth elsewhere in this Report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” section of this Report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a biopharmaceutical company involved in the development of an injectable collagenase for multiple indications. We have a development and license agreement with Auxilium Pharmaceuticals, Inc. (“Auxilium”) for injectable collagenase (which Auxilium has named XIAFLEX®) for marketed indications and collagenase clostridium histolyticum (“CCH”) for indications in development. Auxilium has an option to acquire additional indications that we may pursue, including human and canine lipoma. Auxilium is currently selling XIAFLEX in the U.S. for the treatment of Dupuytren’s contracture and Peyronie’s disease. Following the termination of the agreement between Auxilium and Pfizer, Inc. (“Pfizer”), Auxilium entered into an agreement with Swedish Orphan Biovitrum AB (“Sobi”) pursuant to which Sobi has marketing rights for XIAPEX® (the EU trade name for collagenase clostridium histolyticum) for Dupuytren’s contracture and Peyronie’s disease in Europe and certain Eurasian countries. Sobi is currently selling XIAPEX in Europe for the treatment of Dupuytren’s contracture. In addition, Auxilium has an agreement with Asahi Kasei Pharma Corporation (“Asahi”) pursuant to which Asahi has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Japan. Auxilium also has an agreement with Actelion Pharmaceuticals Ltd. (“Actelion”) pursuant to which Actelion has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Canada, Australia, Brazil and Mexico.
Peyronie’s Disease. In December 2013, the U.S. Food and Drug Administration (“FDA”) approved Auxilium’s supplemental Biologics License Application (“sBLA”) for XIAFLEX for the treatment of Peyronie's disease. As a result, we recognized a $2.0 million milestone payment from Auxilium. This is the first and only FDA-approved biologic therapy indicated for the treatment of Peyronie's disease in men with a palpable plaque and a curvature of 30 degrees or greater at the start of therapy.
Dupuytren’s Contracture. In the fourth quarter of 2013, Auxilium presented results from Year 4 of the Collagenase Optimal Reduction of Dupuytren’s Long-term Evaluations of Success Study (“CORDLESS”). CORDLESS is a five-year observational study designed to assess the rates of recurrence following treatment with XIAFLEX, as well as long-term safety and progression of disease in patients from earlier Auxilium studies. Also in the fourth quarter of 2013, Auxilium announced positive results from the open label, phase IIIb MULTICORD (Multiple Treatment Investigation of Collagenase Optimizing the Resolution of Dupuytren's) study evaluating XIAFLEX for the concurrent treatment of adult Dupuytren's contracture patients with multiple palpable cords. The study demonstrated that two concurrent injections of XIAFLEX in patients with multiple Dupuytren's contractures resulted in comparable improvement in joint contracture and range of motion to those seen in previous studies when XIAFLEX was administered as single injections, 30 days apart. Adverse event (AE) rates were also comparable to single injection administration 30 days apart. Based on the results, Auxilium submitted a sBLA to the FDA in the fourth quarter 2013 seeking expansion of labeling for the concurrent treatment of multiple palpable cords and hopes to receive approval by the end of 2014. On February 24, 2014, Auxilium reported that the FDA had accepted the submission with a PDUFA date of October 20, 2014.
Cellulite. Auxilium expanded the field of its license for injectable collagenase to include the potential treatment of cellulite by exercising, in January 2013, its exclusive option under our development and license agreement. In October 2013, Auxilium dosed the first patient in its phase IIa clinical trial of collagenase clostridium histolyticum (“CCH”) for the treatment of cellulite. Auxilium anticipates top-line results from the study in the first quarter of 2015. No FDA-approved pharmaceutical therapies are currently available for the treatment of cellulite.
Frozen Shoulder. Auxilium reported positive top-line data in the first quarter of 2013 from its phase IIa clinical trial of XIAFLEX for the potential treatment of frozen shoulder (adhesive capsulitis). In December 2013, Auxilium dosed the first patient in its phase IIb study of CCH for the treatment of frozen shoulder. Auxilium anticipates top-line results from the study in the first quarter of 2015. No FDA-approved pharmaceutical therapies are currently available for the treatment of frozen shoulder.
Human Lipoma. In the first quarter 2014, we announced top-line data from the phase II dose escalation clinical trial of CCH for the treatment of human lipoma. The primary efficacy outcome of active reduction of the visible surface area of the lipoma as measured by caliper was met, combining all patients (p<0.0001). There were no serious adverse events reported during the trial.
Canine Lipoma. In fourth quarter 2013, we announced top-line data from Chien-804, the placebo-controlled, double-blind, randomized phase II trial evaluating the efficacy of CCH in canines with benign subcutaneous lipomas. The trial did not meet its primary endpoint of a statistically significant post-treatment difference in the mean percent change in lipoma volume by CT scan; however, in the responder analysis there was a statistically significant reduction in lipoma surface area among dogs treated with CCH (p=0.0084). Auxilium has the option to exclusively license development and marketing rights to the canine lipoma indication, which would trigger an opt-in payment and potential future milestone and royalty payments from Auxilium. We anticipate submitting a final study report to Auxilium in the first quarter of 2014, which will trigger the 120 day opt-in period. If Auxilium does not opt-in, then the rights will revert back to us.
Uterine Fibroids. In third quarter 2013, we announced that a poster titled, “Biomechanical Evaluation of Human Uterine Fibroids after Exposure to Purified Clostridial Collagenase” was presented at the Society for the Study of Reproduction 46th Annual Meeting in Montreal, Quebec, Canada. The poster provided data which show that highly purified collagenase can reduce the stiffness of human uterine fibroid tissue in laboratory experiments. Increased tissue rigidity has been implicated as a cause of the morbidity associated with uterine fibroids. We anticipate releasing top-line data from a pre-clinical study in the second quarter of 2014.
Outlook
We generated revenue from two primary sources: in connection with the Auxilium Agreement, we receive license, sublicense income, royalties, milestones and mark-up on cost of goods sold payments related to the sale and approval of XIAFLEX/XIAPEX as described below in Part II, Item 7, "Auxilium Agreement". Under the DFB Agreement, pursuant to which we sold our topical collagenase business to DFB, we received earn out payments based on the sales of certain products, but our right to receive these payments expired at the end of August 2013. We expect to receive the final of these earn out payments in March 2014.
Beginning in the fourth quarter of 2013, we expect to generate revenue from one primary source: in connection with the Auxilium Agreement.
Auxilium Agreement
Under the Auxilium Agreement, we granted to Auxilium exclusive worldwide rights to develop, market and sell certain products containing our injectable collagenase. Currently its licensed rights cover the indications of Dupuytren’s contracture, Peyronie’s disease, frozen shoulder and cellulite. Auxilium may further expand the Auxilium Agreement, at its exclusive option, to develop and license our injectable collagenase for use in additional indications.
Auxilium’s existing agreement with Pfizer terminated as of April 24, 2013. Pursuant to a transition services agreement, Pfizer continued support of the supply of XIAPEX until February 28, 2014. Currently, Sobi has exclusive rights to commercialize XIAPEX for Dupuytren's contracture and Peyronie's disease, subject to applicable regulatory approvals, in 28 EU member countries, Switzerland, Norway, Iceland, 18 Central Eastern Europe/Commonwealth of Independent countries, including Russia and Turkey, and 22 Middle Eastern & North African countries. As Auxilium reported in its 10K, “XIAPEX is now available in Austria, Belgium, Czech Republic, Denmark, Finland, Hungary, Iceland, Ireland, Italy, Luxembourg, Netherlands, Norway, Poland, Portugal, Romania, Spain, Sweden, Switzerland and the United Kingdom.”
Sobi, via its Partner Products business unit, is primarily responsible for the applicable regulatory, clinical and commercialization activities for XIAPEX in Dupuytren's contracture and Peyronie's disease in these countries. We will receive a certain percentage of milestone payments that Sobi pays to Auxilium. We will also receive royalties from net sales and payments on costs of goods sold in Sobi territories from Auxilium, which will be a specified percentage of what Auxilium receives from Sobi.
Auxilium has granted to Asahi the exclusive right to develop and commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Japan. Auxilium has granted to Actelion the exclusive right to develop and commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Canada, Australia, Brazil and Mexico. XIAFLEX has been approved for sale for Dupuytren’s contracture in Canada and Australia.
Through December 31, 2013, Auxilium has paid us up-front licensing and sublicensing fees and milestone payments under the Auxilium Agreement of $26.4 million, including amounts in connection with Auxilium’s agreements with Pfizer, Asahi and Actelion. In addition to the payments already received by us and to be received by us with respect to the Dupuytren’s contracture indication, Auxilium will be obligated to make contingent milestone payments to us, with respect to each of frozen shoulder and cellulite indications, upon the acceptance of the regulatory filing and upon receipt by Auxilium, its affiliate or sublicensee of regulatory approval. The remaining contingent milestone payments that may be received, in the aggregate, from Auxilium in respect of frozen shoulder and cellulite are $3.0 million. To the extent there is sub-licensing income as defined in the Auxilium Agreement, Auxilium will also be obligated to make sublicense fee payments to us if it out-licenses to third parties the right to market and sell XIAFLEX for the treatment of frozen shoulder or cellulite. Additional milestone obligations will be due if Auxilium exercises its option to develop and license XIAFLEX for additional indications, such as human and canine lipoma. In the first quarter 2014, we anticipate we will present the opt-in study for canine lipoma to Auxilium and Auxilium will have 120 days to exercise its option and pay the associated option amount.
We will receive a certain percentage of milestone payments that Sobi pays to Auxilium. We will also receive royalties from net sales and payments on costs of goods sold in Sobi territories from Auxilium, which will be a specified percentage of what Auxilium receives from Sobi. To the extent Auxilium enters into an agreement or agreements related to other territories, the percentage of sublicense income that Auxilium would pay us will depend on the stage of development and approval of XIAFLEX for the particular indication at the time such other agreement or agreements are executed.
Auxilium must pay us on a country-by-country and product-by-product basis a low double digit royalty as a percentage of net sales for products covered by the Auxilium Agreement and sold in the United States, Europe and certain Eurasian countries and Japan. In the case of products covered by the Auxilium Agreement and sold in other countries (the “Rest of the World”), Auxilium must pay us on a country-by-country and product-by-product basis a specified percentage of the royalties it is entitled to receive from a partner or partners with whom it has contracted for such countries (a “Rest of the World Partner”), which in the case of Canada, Australia, Brazil and Mexico is Actelion. The royalty rate is independent of sales volume and clinical indication in the United States, Europe and certain Eurasian countries and Japan, but is subject to set-off in those countries and the Rest of the World for certain expenses we owe to Auxilium relating to certain development and patent costs. In addition, the royalty percentage may be reduced if (i) market share of a competing product exceeds a specified threshold; or (ii) Auxilium is required to obtain a license from a third party in order to practice our patents without infringing such third party’s patent rights, although Auxilium has confirmed to us that no license from a third party is required. In addition, if Auxilium out-licenses to a third party, then we will receive a specified percentage of certain payments made to Auxilium in consideration of such out-licenses.
These royalty obligations extend, on a country-by-country and product-by-product basis, for the longer of the patent life (including pending patents), the expiration of any regulatory exclusivity period based on orphan drug designation or foreign equivalent thereof or June 3, 2016. Auxilium may terminate the Auxilium Agreement upon 90 days prior written notice. If Auxilium terminates the Auxilium Agreement other than because of an uncured, material breach by us, all rights revert to us. Pursuant to our August 31, 2011 settlement agreement with Auxilium, we are now co-owners and are or will be co-inventors of U.S. Patent No. 7,811,560 and any continuations and divisionals thereof. Auxilium expects this patent will expire in July 2028.
On top of the payments set forth above, Auxilium must pay to us an amount equal to a specified mark-up of the cost of goods sold for products sold in the United States, Europe and certain Eurasian countries or Japan. For products sold in the Rest of the World, Auxilium must pay to us a specified percentage of the mark-up of the cost of goods sold it is entitled to receive from a Rest of the World Partner, including Actelion, without regard to any set-offs that the Rest of the World Partner may have with respect to Auxilium.
Auxilium is generally responsible, at its own cost and expense, for developing the formulation and finished dosage form of products and arranging for the clinical supply of products. Auxilium is generally responsible for all clinical development and regulatory costs for Peyronie’s disease, Dupuytren’s contracture, frozen shoulder, cellulite and all additional indications for which it exercises its options.
In connection with a March 2006 agreement (the “DFB Agreement”), pursuant to which we sold our topical collagenase business to DFB Biotech, Inc. and its affiliates (“DFB”), we expect to receive in March 2014 the final earn out payment of $3.5 million which was recognized as income in 2013.
In-Licensing and Royalty Agreements
We have entered into several in-licensing and royalty agreements with various investigators, universities and other entities throughout the years.
Dupuytren’s Contracture
On November 21, 2006, we entered into a license agreement (the “Dupuytren’s License Agreement”) with the Research Foundation of the State University of New York at Stony Brook (the “Research Foundation”), pursuant to which the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the development, manufacture, use or sale of (i) the collagenase enzyme obtained by a fermentation and purification process (the “Enzyme”), and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment and prevention of Dupuytren’s contracture.
In consideration of the license granted under the Dupuytren’s License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme or injectable collagenase for the treatment and prevention of Dupuytren’s contracture (each a “Dupuytren’s Licensed Product”).
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Dupuytren’s Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of such Dupuytren’s Licensed Product on a country-by-country basis. The royalty rate is 0.5% of net sales. Our obligation to pay royalties to the Research Foundation will continue until the later of (i) the expiration of the last valid claim of a patent pertaining to the Dupuytren’s Licensed Product; (ii) the expiration of the regulatory exclusivity period conveyed by the FDA’s Office of Orphan Products Development (“OOPD”) with respect to the Dupuytren’s Licensed Product; or (iii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Dupuytren’s License Agreement and the licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Dupuytren’s License Agreement will become fully paid, irrevocable exclusive licenses.
Peyronie’s Disease
On August 27, 2008, we entered into an agreement with Dr. Martin K. Gelbard to improve the deal terms related to our future royalty obligations for Peyronie’s disease by buying down our future royalty obligations with a one-time cash payment. A redacted copy of the agreement was filed on Form 8-K with the SEC on September 5, 2008. On March 31, 2012, we entered into an amendment to this agreement, which enables us to buy down a portion of our future royalty obligations in exchange for an initial cash payment and five additional cash payments. A redacted copy of the amendment was filed on Form 8-K/A with the SEC on August 8, 2012. The foregoing descriptions of the agreement with Dr. Gelbard and the amendment to that agreement do not comport to be complete and are qualified in their entirety by reference to the full text of that agreement, as amended.
Frozen Shoulder
On November 21, 2006, we entered into a license agreement (the “Frozen Shoulder License Agreement”) with the Research Foundation, pursuant to which the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the development, manufacture, use or sale of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment and prevention of frozen shoulder. Additionally, the Research Foundation granted to us an exclusive license to the patent applications in respect of frozen shoulder. The license granted to us under the Frozen Shoulder License Agreement is subject to the non-exclusive license (with right to sublicense) granted to the U.S. government by the Research Foundation in connection with the U.S. government’s funding of the initial research.
In consideration of the license granted under the Frozen Shoulder License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme or injectable collagenase for the treatment and prevention of frozen shoulder (each a “Frozen Shoulder Licensed Product”). In addition, we and the Research Foundation will share in any milestone payments and sublicense income received by us in respect of the rights licensed under the Frozen Shoulder License Agreement.
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Frozen Shoulder Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Frozen Shoulder Licensed Product. Our obligation to pay royalties to the Research Foundation will continue until, the later of (i) the expiration of the last valid claim of a patent pertaining to a Frozen Shoulder Licensed Product or (ii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Frozen Shoulder License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Frozen Shoulder License Agreement will become fully paid, irrevocable exclusive licenses.
In connection with the execution of the Dupuytren’s License Agreement and the Frozen Shoulder License Agreement, we made certain up-front payments to the Research Foundation and the clinical investigators working on the Dupuytren’s contracture and frozen shoulder indications for the Enzyme.
Cellulite
We have two in-licensing and royalty agreements related to cellulite. One is a license agreement (the “Cellulite License Agreement”) with the Research Foundation that we entered into on August 23, 2007. Pursuant to the Cellulite License Agreement, the Research Foundation granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties, to know-how owned by the Research Foundation related to the manufacture, preparation, formulation, use or development of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme, which are made, used and sold for the prevention or treatment of cellulite. Additionally, the Research Foundation granted to us an exclusive license to the patent applications in respect of cellulite. The license granted to us under the Cellulite License Agreement is subject to the non-exclusive license (with right to sublicense) granted to the U.S. government by the Research Foundation in connection with the U.S. government’s funding of the initial research.
In consideration of the license granted under the Cellulite License Agreement, we agreed to pay to the Research Foundation certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme, which are made, used and sold for the prevention or treatment of cellulite (each a “Cellulite Licensed Product”). In addition, we and the Research Foundation will share in any milestone payments and sublicense income received by us in respect of the rights licensed under the Cellulite License Agreement. We paid a portion of the $500,000 milestone payment we received from Auxilium in respect of its exercise of cellulite as an addition indication under the Auxilium Agreement, subject to certain credits for certain up-front payments we made to the Research Foundation.
Our obligation to pay royalties to the Research Foundation with respect to sales by us, our affiliates or any sublicensee of any Cellulite Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Cellulite Licensed Product. Our obligation to pay royalties to the Research Foundation will continue until, the later of (i) the expiration of the last valid claim of a patent pertaining to a Cellulite Licensed Product or (ii) June 3, 2016.
Unless terminated earlier in accordance with its termination provisions, the Cellulite License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Cellulite License Agreement will become fully paid, irrevocable exclusive licenses.
The other in-licensing and royalty agreement we have related to cellulite is a license agreement with Dr. Zachary Gerut that we entered into on March 27, 2010 (the “Gerut License Agreement”). Pursuant to the Gerut License Agreement, Dr. Gerut granted to us and our affiliates an exclusive worldwide license, with the right to sublicense to certain third parties to know-how owned by Dr. Gerut related to the manufacture, preparation, formulation, use or development of (i) the Enzyme and (ii) all pharmaceutical products containing the Enzyme or injectable collagenase, in each case to the extent it pertains to the treatment of fat. As the in-license granted in the Gerut License Agreement pertains to the treatment of fat, this in-license also relates to human lipoma and canine lipoma.
In consideration of the license granted under the Gerut License Agreement, we agreed to pay to Dr. Gerut certain royalties on net sales (if any) of pharmaceutical products containing the Enzyme which are made, used and sold for the removal or treatment of fat in humans or animals (each a “Gerut Licensed Product”). In addition, in the event the FDA approves a Gerut Licensed Product, we have agreed to make a one-time stock option grant to Dr. Gerut with a strike price equal to the closing trading price on the day before the date of such grant.
Our obligation to pay royalties to Dr. Gerut with respect to sales by us, our affiliates or any sublicensee of any Gerut Licensed Product in any country (including the U.S.) arises only upon the first commercial sale of a Gerut Licensed Product. Our obligation to pay royalties to Dr. Gerut will continue until June 3, 2016 or such longer period as we continue to receive royalties for such Gerut Licensed Product.
Unless terminated earlier in accordance with its termination provisions, the Gerut License Agreement and licenses granted under that agreement will continue in effect until the termination of our royalty obligations. After that, all licenses granted to us under the Gerut License Agreement will become fully paid, irrevocable exclusive licenses.
Other Indications
We have or may enter into certain other license and royalty agreements with respect to other indications that we may elect to pursue.
Significant Risks
We are dependent to a significant extent on third parties, and our principal licensee, Auxilium, may not be able to continue successfully commercializing XIAFLEX for Dupuytren’s contracture and Peyronie’s Disease, successfully develop CCH for additional indications, obtain required regulatory approvals, manufacture XIAFLEX at an acceptable cost, in a timely manner and with appropriate quality, or successfully market products or maintain desired margins for products sold, and as a result we may not achieve sustained profitable operations.
Critical Accounting Policies, Estimates and Assumptions
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. These estimates are based on historical experience and on various other assumptions that we believe are reasonable under the circumstances. Actual results could differ from those estimates. While our significant accounting policies are described in more detail in the notes to our consolidated financial statements, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Cash, Cash Equivalents and Short-term Investments. Cash, cash equivalents and short-term investments are stated at market value. Cash equivalents include only securities having a maturity of three months or less at the time of purchase. The Company limits its credit risk associated with cash, cash equivalents and short-term investments by placing its investments with banks it believes are highly creditworthy and with highly rated money market funds, U.S. government securities, or short-term commercial paper which are held to maturity.
Fair Value Measurements. Management believes that the carrying amounts of the Company’s financial instruments, including cash, cash equivalents, short-term investments, accounts receivable, accounts payable and accrued expenses approximate fair value due to the short-term nature of those instruments.
Concentration of Credit Risk and Major Customers. The Company maintains bank account balances, which, at times, may exceed insured limits. The Company has not experienced any losses with these accounts and believes that it is not exposed to any significant credit risk on cash. The Company maintains its investment in FDIC insured certificates of deposits with several banks.
At December 31, 2013, the accounts receivable balance of $5.0 million was primarily from two customers, comprised of $3.5 million (70% of total) from DFB and $1.5 million (30% of total) from Auxilium. The Company has been dependent in each year on two customers who generate almost all its revenues. (With the expiration of right to receive payments on Santyl sales in August 2013, the primary source of our revenues is Auxilium Pharmaceutical, Inc.)
Treasury Stock. The Company accounts for treasury stock under the cost method and includes treasury stock as a component of stockholders’ equity.
Revenue Recognition. We recognize revenues from product sales when there is persuasive evidence that an arrangement exists, title passes, the price is fixed and determinable, and payment is reasonably assured. We currently recognize revenues resulting from the licensing, sublicensing and use of our technology and from services we sometimes perform in connection with the licensed technology.
We enter into product development licenses and collaboration agreements that may contain multiple elements, such as upfront license and sublicense fees, milestones related to the achievement of particular stages in product development and royalties. As a result, significant contract interpretation is sometimes required to determine the appropriate accounting, including whether the deliverables specified in a multiple-element arrangement should be treated as separate units of accounting for revenue recognition purposes, and if so, how the aggregate contract value should be allocated among the deliverable elements and when to recognize revenue for each element.
We recognize revenue for delivered elements only when the fair values of undelivered elements are known, when the associated earnings process is complete and, to the extent the milestone amount relates to our performance obligation, when our licensee confirms that we have met the requirements under the terms of the agreement, and when payment is reasonably assured. Changes in the allocation of the contract value between various deliverable elements might impact the timing of revenue recognition, but in any event, would not change the total revenue recognized on the contract. For example, nonrefundable upfront product license fees, for product candidates for which we are providing continuing services related to product development, are deferred and recognized as revenue over the development period.
Milestones, in the form of additional license fees, typically represent nonrefundable payments to be received in conjunction with the achievement of a specific event identified in a contract, such as completion of specified clinical development activities and/or regulatory submissions and/or approvals. We believe that a milestone represents the culmination of a distinct earnings process when it is not associated with ongoing research, development or other performance on our part. We recognize such milestones as revenue when they become due and payment is reasonably assured. When a milestone does not represent the culmination of a distinct earnings process, we recognize revenue in a manner similar to that of an upfront product license fee.
Royalty/ Mark-up on Cost of Goods Sold / Earn-Out Revenue. For those arrangements for which royalty, mark-up on cost of goods sold or earn-out payment information becomes available and collectability is reasonably assured, we recognize revenue during the applicable period earned. For interim quarterly reporting purposes, when collectability is reasonably assured but a reasonable estimate of royalty, mark-up on cost of goods sold or earn-out payment revenues cannot be made, the royalty, mark-up on cost of goods sold or earn-out payment revenues are generally recognized in the quarter that the applicable licensee provides the written report and related information to us.
Under the Auxilium Agreement, we do not participate in the selling, marketing or manufacturing of products for which we receive royalties and a mark-up of the cost of goods sold revenues. The royalty and mark-up on cost of goods sold revenues will generally be recognized in the quarter that Auxilium provides the written reports and related information to us, that is, royalty and mark-up on cost of goods sold revenues are generally recognized one quarter following the quarter in which the underlying sales by Auxilium occurred. The royalties payable by Auxilium to us are subject to set-off for certain patent costs.
Under the DFB Agreement, pursuant to which we sold our topical collagenase business to DFB, we had the right to receive earn-out payments based on sales of certain products. This right to receive payments on Santyl sales expired in August 2013. Generally, under the DFB Agreement we would receive payments and a report within ninety (90) days from the end of each calendar year after DFB has sold the royalty-bearing product. DFB has provided us earn-out reports on a quarterly basis. BioSpecifics has now recognized all income from the Santyl sales under the DFB agreement, and expects to receive the corresponding cash payment, the income recognized in 2013, in March 2014.
Consulting and Technical Assistance Services. We recognize revenues from consulting and technical assistance contracts primarily as a result of the DFB Agreement. Consulting revenues are recognized ratably over the term of the contract. The consulting and technical assistance obligations to DFB expired during March 2011.
Reimbursable Third Party Development Costs. We accrued patent expenses for research and development that are reimbursable by us under the Auxilium Agreement. We capitalize certain patent costs related to estimated third party development costs that are reimbursable under the Auxilium Agreement. In August 2011, through the amendment and restatement of our development and license agreement with Auxilium, we have clarified the rights and responsibilities of the joint development of collagenase clostridium histolyticum (“CCH”). We resolved what had been an on-going dispute with Auxilium concerning the appropriate amount of creditable third party development expenses related to the lyophilization of the injection formulation and certain patent expenses for research and development costs that are reimbursable under the Auxilium Agreement. We agreed and have reimbursed Auxilium by offsetting future royalties payable for the amount invoiced us for third party development costs related to the development of the lyophilization of the injection formulation. We do not expect any additional third party development cost related to the lyophilization of the injection formulation.
As of December 31, 2013 our net reimbursable third party patent expense accrual was approximately $60,000.
Receivables and Deferred Revenue. Accounts receivable as of December 31, 2013 is approximately $5.0 million, which consists of approximately $3.5 million due from DFB in accordance with the earn-out under the DFB Agreement and approximately $1.5 million in royalties and mark-up on costs of goods sold due from Auxilium in accordance with the terms of the Auxilium Agreement. Deferred revenue of $0.2 million consist of licensing fees related to the cash payments received under the Auxilium Agreement in prior years and amortized over the expected development period of certain indications for CCH.
Third-Party Royalties. We have entered into licensing and royalty agreements with third parties and agreed to pay certain royalties on net sales of products for specific indications. We accrue third-party royalty expenses on net sales reported to us by Auxilium. Third-party royalty expense is generally expensed in the quarter that Auxilium provides the written reports and related information to us, that is, generally one quarter following the quarter in which the underlying sales by Auxilium occurred. We expect our third party royalty expense under General and Administrative expenses will continue to increase as net sales by Auxilium for XIAFLEX increase and potential new indications for CCH are approved.
Royalty Buy-Down. On March 31, 2012, we entered into an amendment to our existing agreement with Dr. Gelbard, dated August 27, 2008, related to our future royalty obligations for Peyronie’s disease. The amendment enables us to buy down a portion of our future royalty obligations in exchange for an initial cash payment and five additional cash payments, one of which was paid in December 2013.
As of December 31, 2013, we have capitalized $3.35 million related to this agreement which will be amortized over approximately five years beginning on the date in which we receive our quarterly royalty report from Auxilium reporting the first commercial sale of XIAFLEX for the treatment of Peyronie’s disease, which represents the period estimated to be benefited using the straight-line method. In accordance with Accounting Standards Codification 350, Intangibles, Goodwill and Other, the Company amortizes intangible assets with finite lives in a manner that reflects the pattern in which the economic benefits of the assets are consumed or otherwise used up. If that pattern cannot be reliably determined, the assets are amortized using the straight-line method. We perform an evaluation of the recoverability of the carrying value of our intangible assets to determine if facts and circumstances indicate that the carrying value of intangible assets may be impaired and if any adjustment is warranted. Based on our evaluation as of December 31, 2013, no impairment existed for intangible assets.
Stock Based Compensation. Under ASC 718, we estimate the fair value of our employee stock awards at the date of grant using the Black-Scholes option-pricing model, which requires the use of certain subjective assumptions. The most significant assumptions are our estimates of the expected volatility of the market price of our stock and the expected term of an award. Expected volatility is based on the historical volatility of our common stock. When establishing an estimate of the expected term of an award, we consider the vesting period for the award, our historical experience of employee stock option exercises (including forfeitures) and the expected volatility. As required under the accounting rules, we review our valuation assumptions at each grant date and, as a result, we are likely to change our valuation assumptions used to value future employee stock-based awards granted, to the extent any such awards are granted.
Further, ASC 718 requires that employee stock-based compensation costs to be recognized over the requisite service period, or the vesting period, in a manner similar to all other forms of compensation paid to employees. The allocation of employee stock-based compensation costs to each operating expense line are estimated based on specific employee headcount information at each grant date and estimated stock option forfeiture rates and revised, if necessary, in future periods if actual employee headcount information or forfeitures differ materially from those estimates. As a result, the amount of employee stock-based compensation costs we recognize in each operating expense category in future periods may differ significantly from what we have recorded in the current period.
RESULTS OF OPERATIONS
YEAR ENDED DECEMBER 31, 2013 COMPARED WITH YEAR ENDED DECEMBER 31, 2012
Net revenues
Net revenues for the two years ended December 31, 2013 and 2012 comprise the following:
| Year Ended December 31 | ||||||||||||||||
| 2013 | 2012 | Change | % Change | |||||||||||||
| Net sales | $ | 37,458 | $ | 18,219 | 19,239 | 106 | % | |||||||||
| Royalties | 11,767,758 | 9,155,654 | 2,612,104 | 29 | % | |||||||||||
| Licensing revenue | 2,662,024 | 1,971,205 | 690,819 | 35 | % | |||||||||||
| Total net revenues | $ | 14,467,240 | $ | 11,145,078 | 3,322,162 | 30 | % | |||||||||
Product Revenues, net
Product revenues include the sales of the collagenase for laboratory use recognized at the time it is shipped to customers. We had a small amount of revenue from the sale of collagenase for laboratory use. For the calendar years ended 2013 and 2012 product revenues were $37,458 and $18,219, respectively. This increase of $19,239, or 106%, was primarily related to the amount of material required to perform testing and additional research by our customers.
Royalties
Royalties consist of royalties and the mark-up on cost of goods sold under the Auxilium Agreement and earn-out revenues associated with the DFB Agreement. Total royalty, mark-up on cost of goods sold and earn-out revenues for year ended December 31, 2013 were $11.8 million as compared to $9.2 million in the 2012 period, an increase of $2.6 million, or 29%. Royalty and the mark-up on cost of goods sold revenues recognized under the Auxilium Agreement were $8.2 million for the 2013 period and $6.3 million for the 2012 period. The increase of $1.9 million, or 32%, was due to increased net sales of XIAFLEX during 2013 reported to us by Auxilium.
We received earn-out revenues from DFB under the earn-out payment provision of the DFB Agreement after certain net sales levels are achieved. Revenues recognized under the DFB Agreement were $3.5 million for the year ended December 31, 2013 and $2.9 million for the same period in 2012. This increase of $0.6 million, or 22%, is mainly related to the increase in net sales during the 2013 period reported to us by DFB. We expect to receive the final earn-out payment for revenue recognized during 2013 in March 2014.
Licensing Revenue
Licensing revenue consists of licensing fees, sublicensing fees and milestones. For the years ended December 31, 2013 and 2012, we recognized total licensing and milestone revenue of $2.7 million and $2.0 million, respectively, an increase of $0.7 million, or 35%. Certain licensing revenues recognized are related to the cash payments received under the Auxilium Agreement in prior years and amortized over the expected development period. Licensing revenue recognized for the years ended December 31, 2013 and 2012 were $0.6 million and $0.4 million, respectively. The increase of $0.2 million was mainly due to licensing fees recognized of $0.5 million related to the exercise by Auxilium of its exclusive option to expand the field of its license for injectable collagenase to include the potential treatment of adult patients with edematous fibrosclerotic panniculopathy, commonly known as cellulite, partially offset by lower license fees recognized related to development of $0.1 million as compared to $0.4 million in the comparable period of 2012. Sublicensing fees recognized in 2013 were zero compared to $0.6 million in the same period in 2012. In 2012, we recognized $0.6 million in sublicensing fees, which were related to the $10.0 million paid to Auxilium by Actelion for the rights to develop and commercialize XIAFLEX for the treatment of Dupuytren's contracture and Peyronie's disease in Canada, Australia, Brazil and Mexico.
Milestone revenue recognized for the years ended December 31, 2013 and 2012 were $2.0 million and $1.0 million, respectively. In 2013, we recognized a $2.0 million milestone related to the FDA’s approval of XIAFLEX for the treatment of Peyronie's disease. In addition, a $28,500 milestone was recognized in the 2013 period related to product approval for XIAFLEX for the treatment of Dupuytren's contracture in adults with a palpable cord in Australia granted to Actelion. In the 2012 period, we recognized a $1.0 million milestone related to the FDA’s December 2012 acceptance of Auxilium’s sBLA for XIAFLEX for the potential treatment of Peyronie's disease. We also, recognized a milestone of $28,500 related to the Notice of Compliance (approval) by Health Canada for XIAFLEX for the treatment of Dupuytren's contracture in adults with a palpable cord in Canada granted to Auxilium.
Under current accounting guidance, nonrefundable upfront license fees for product candidates for which we are providing continuing services related to product development are deferred and recognized as revenue over the development period. The remaining balance will be recognized over the respective development periods or when we determine that we have no ongoing performance obligations.
Research and Development Activities
Research and development expenses include, but are not limited to, internal costs, such as salaries and benefits, costs of materials, lab expense, facility costs and overhead. Research and development expenses also consist of third party costs, such as medical professional fees, product costs used in clinical trials, consulting fees and costs associated with clinical study arrangements. Research and development expenses were $1.5 million and $1.2 million, respectively, for calendar years 2013 and 2012, representing an increase in 2013 of $0.3 million, or 19%. This increase in research and development expenses was primarily due to expenses related to our clinical development programs partially offset by lower stock-based compensation.
We are currently working to develop CCH for the treatment of human and canine lipoma and have begun a pre-clinical study in uterine fibroids.
Human Lipoma
Lipomas are benign fatty tumors that occur as bulges under the skin and affect humans and canines. It is estimated that lipomas are the primary diagnosis in 575,000 patients in the U.S. annually. The only proven therapy for lipoma treatment is surgery, which is often not practical for patients with multiple lipomas. Based on observations made during preclinical studies that a collagenase injection decreased the size of fat pads in animals, we initiated, monitored and supplied the requisite study drug for a phase I open label clinical trial for the treatment of human lipomas with a single injection of collagenase. Favorable initial results (10 out of 12 patients had a 50-90% reduction in the size of the lipomas) from this trial for the treatment of human lipomas were presented at a meeting of the American Society of Plastic Surgeons.
In January 2014, we announced the top-line data from the phase II dose escalation clinical trial of CCH for the treatment of human lipoma. This phase II open-label single-center dose escalation study assessed the safety and efficacy of CCH in 14 patients with lipoma, divided into four dose cohorts. Each patient received a single injection of CCH in one of four ascending doses based on the current commercial dose of CCH in marketed indications, ranging from 0.058mg (10% of commercial dose) to 0.44mg (75% of commercial dose). The primary efficacy outcome was reduction in lipoma visible surface area as measured by caliper. Data showed patients in the highest dose group (75% of commercial dose) achieved the best efficacy results with an average of 67% reduction of lipoma visible surface area as measured by caliper at six months post-treatment. Additionally, data demonstrated that 75% of patients in the highest dose group achieved reduction of 50% or more in lipoma visible surface area. We anticipate initiating a placebo-controlled trial in the first half of 2014.
There were no drug-related serious adverse events reported during the trial. The most frequent treatment-related adverse events were localized to the injection site and included bruising, injection site swelling and injection site pain. These adverse events are consistent with those seen previously in clinical experience.
Canine Lipoma
Based on the encouraging results reported in the clinical investigations in human lipoma, we began clinical trials in canine lipoma. Lipomas are found in 2.3% of canines, and there may be as many as 1.7 million canines affected with skin lipomas in the U.S. Lipomas in older canines are very common, and lipomas that restrict motion in older canines are a serious problem. The only proven therapy for this condition is surgical excision of the lipoma, which necessarily involves the use of general anesthesia. It has been estimated that up to 2% of sick canines die as a complication of general anesthesia (See Brodbelt Vet J 2009 Dec; 182 (3): 375-6). We surveyed 77 veterinarians which included participants from the academic field and others that are in private practice. The participants indicated that on average they perform 25 lipoma excision surgeries per year at an average cost of $530 for the surgical procedure. It is conservatively estimated that 47,000 veterinarians are in active practice in the U.S.
Chien-804
In December 2013, we announced top-line data from Chien-804, the placebo-controlled, double-blind, randomized phase II trial evaluating the efficacy of CCH in canines with benign subcutaneous lipomas. The Chien-804 trial enrolled 37 dogs in a single injection study randomized 1:1 CCH to placebo with lipoma volume being measured by CT scan and lipoma surface area being measured by caliper at baseline, one month and 90 days. The data at 90 days show a post-treatment difference in the mean percent change in lipoma volume by CT scan between the CCH and placebo-treated groups of -11.58% (p=0.52), which was not statistically significant. The percent change at 90 days in mean visible surface area measured by caliper showed a difference of -24.18% (p=0.09), which approached statistical significance. Among those dogs whose lipomas decreased by 50% or more, the results achieved statistical significance and showed that the visible surface area as measured by caliper decreased by 50% or more in 47.4% of CCH-treated dogs (9 out of 19) versus 5.9% of placebo-treated dogs (1 out of 17), with a p-value of 0.0084. A questionnaire administered to pet owners, while blinded to the study, showed 84.2% satisfaction with the results of CCH treatment versus 33.4% satisfaction with the placebo results (p=0.005). We anticipate providing Auxilium with the Chien-804 final study report in the first quarter of 2014.
There were no drug-related serious adverse events reported during the trial. The most frequent treatment-related adverse events were local injection site reactions including bruising, injection site swelling, injection site pain and injection site edema. These adverse events are consistent with those seen previously in clinical experience in humans.
Uterine Fibroids
In July 2013, we announced that a poster titled, "Biomechanical Evaluation of Human Uterine Fibroids after Exposure to Purified Clostridial Collagenase" was presented at the Society for the Study of Reproduction 46th Annual Meeting in Montreal, Quebec, Canada. The poster provided data which showed that highly purified collagenase can reduce the stiffness of human uterine fibroid tissue in laboratory experiments. Increased tissue rigidity has been implicated as a cause of the morbidity associated with uterine fibroids.
The results of this ex vivo study showed that treatment of fibroids with determined doses of purified collagenase caused a statistically significant decrease in the stiffness of the tissue. This hypothesis was tested in fibroid tissue obtained after hysterectomy or myomectomy surgery from patients. Tissues were injected with collagenase and compared to control-injected tissue. The stiffness in the fibroid tissue was reduced in a time and dose dependent manner with a p-value ≤ 0.001.
The study is being led by Dr. Phyllis Leppert, a Professor of Obstetrics and Gynecology and Professor of Pathology and her colleague, Dr. Friederike Jayes at Duke Medicine with our support. We anticipate reporting top-line data from the pre-clinical study in the first half of 2014.
The following table summarizes our research and development expenses related to our pre-clinical and clinical development programs:
Year Ended December 31, 2013 | Year Ended December 31, 2012 | Accumulated Expenses Since January 1, 2010 | ||||||||||
Program | ||||||||||||
| Canine Lipoma | $ | 463,208 | $ | 442,741 | $ | 1,436,196 | ||||||
| Human Lipoma | 333,230 | 168,879 | 738,099 | |||||||||
| Uterine Fibroids | 157,630 | - | 157,630 | |||||||||
Successful development of drugs is inherently difficult and uncertain. Our business requires investments in research and development over many years, often for drug candidates that may fail during the research and development process. Even if the Company is able to successfully complete the development of our drug candidates, our long-term prospects depend upon our ability and the ability of our partners, particularly with respect to XIAFLEX and CCH, to continue to successfully commercialize these drug candidates.
There is significant uncertainty regarding our ability to successfully develop drug candidates in other indications. These risks include the uncertainty of:
| · | the nature, timing and estimated costs of the efforts necessary to complete the development of our drug candidate projects; |
| · | the anticipated completion dates for our drug candidate projects; |
| · | the scope, rate of progress and cost of our clinical trials that we are currently running or may commence in the future with respect to our drug candidate projects; |
| · | the scope, rate of progress of our preclinical studies and other research and development activities related to our drug candidate projects; |
| · | clinical trial results for our drug candidate projects; |
| · | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights relating to our drug candidate projects; |
| · | the terms and timing of any strategic alliance, licensing and other arrangements that we have or may establish in the future relating to our drug candidate projects; |
| · | the cost and timing of regulatory approvals with respect to our drug candidate projects; and |
| · | the cost of establishing clinical supplies for our drug candidate projects. |
Our current resources and liquidity are sufficient to advance our significant current research and development projects and, Auxilium will have the option to exclusively license the canine and human lipoma indications upon completion of the appropriate opt-in study.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs for personnel, consultant costs, legal fees, investor relations, professional fees and overhead costs. General and administrative expenses were $5.0 million and $4.8 million for calendar years 2013 and 2012, respectively, an increase of $0.2 million or 6%, from 2012. The increase in general and administrative expenses was mainly due to increased third party licensing and royalty fees, investor relations, professional fees, consulting services partially offset by lower legal fees, stock-based compensation and director fees.
Other Income and expense
Other income for calendar year 2013 was $26,202 compared to $34,634 for calendar year 2012. For calendar year 2013 and 2012, other income consisted of interest earned on our investments.
Income Taxes
Our deferred tax liabilities, deferred tax assets and related valuation allowances are impacted by events and transactions arising in the ordinary course of business, research and development activities, vesting of nonqualified options, deferred revenues and other items. Deferred tax assets are affected by the valuation allowance which is dependent upon several factors, including estimates of the realization of deferred income tax assets, and the impact of estimated future taxable income. Significant judgment is required to determine the estimated amount of valuation allowance to record. Changes in the estimate of the valuation allowance could materially increase or decrease our provision for income taxes in future periods.
The provision for income taxes and corresponding taxes payable in 2013 was $2.7 million as compared to $2.2 million in 2012. In 2013, we utilized tax assets of $0.1 million related to deferred licensing revenue and stock based compensation and a $17,000 research and development credit to reduce our taxes payable which was partially offset by an increase to our deferred taxes for employee based compensation. The amount of refundable federal income taxes as of December 31, 2013 is approximately $0.2 million.
In 2012, we used $1.0 million of our Orphan Drug tax credit to reduce our federal income tax payable. We recognized the tax effect of $0.8 million related to the exercise of nonqualified options in our financial statements, which lowered our taxes payable by $0.3 million, reduced our tax assets related to non-qualified stock options by $32,000 and increased additional paid in capital by $0.3 million. Additionally, we utilized tax assets from our federal and state net operating loss carryforwards of $16,000 and deferred licensing revenue of $0.1 million to reduce our taxes payable. Because our state net operating losses of $4.2 million exceeded our federal net operating losses of $47,000 we set up a valuation allowance of $0.3 million against our tax asset of our state net operating loss carryforwards.
YEAR ENDED DECEMBER 31, 2012 COMPARED WITH YEAR ENDED DECEMBER 31, 2011
Net revenues
Net revenues for the two years ended December 31, 2012 and 2011 comprise the following:
| Year Ended December 31 | ||||||||||||||||
| 2012 | 2011 | Change | % Change | |||||||||||||
| Net sales | $ | 18,219 | $ | 21,998 | (3,779 | ) | (17 | )% | ||||||||
| Royalties | 9,155,654 | 6,314,959 | 2,840,695 | 45 | % | |||||||||||
| Licensing revenue | 1,971,205 | 5,012,102 | (3,040,897 | ) | (61 | )% | ||||||||||
| Consulting fees | - | 46,667 | (46,667 | ) | (100 | )% | ||||||||||
| Total net revenues | $ | 11,145,078 | $ | 11,395,726 | $ | (250,648 | ) | (2 | )% | |||||||
Product Revenues, net
Product revenues include the sales of the collagenase for laboratory use recognized at the time it is shipped to customers. We had a small amount of revenue from the sale of collagenase for laboratory use. For the calendar years ended 2012 and 2011 product revenues were $18,219 and $21,998, respectively. This decrease of $3,779, or 17%, was primarily related to the amount of material required to perform testing and additional research by our customers.
Royalties
Royalties consist of royalties and the mark-up on cost of goods sold under the Auxilium Agreement and earn-out revenues associated with the DFB Agreement. Total royalty, mark-up on cost of goods sold and earn-out revenues for year ended December 31, 2012 were $9.2 million as compared to $6.3 million in the 2011 period, an increase of $2.9 million, or 45%. Royalty and the mark-up on cost of goods sold revenues recognized under the Auxilium Agreement were $6.3 million for the 2012 period and $4.0 million for the 2011 period. The increase of $2.3 million, or 55%, was due to increased net sales of XIAFLEX during 2012 reported to us by Auxilium.
We receive earn-out revenues from DFB under the earn-out payment provision of the DFB Agreement after certain net sales levels are achieved. Revenues recognized under the DFB Agreement were $2.9 million for the year ended December 31, 2012 and $2.3 million for the same period in 2011. This increase of $0.6 million, or 27%, is mainly related to the increase in net sales during the 2012 period reported to us by DFB.
Licensing Revenue
Licensing revenue consists of licensing fees, sublicensing fees and milestones. For the years ended December 31, 2012 and 2011, we recognized total licensing and milestone revenue of $2.0 million and $5.0 million, respectively, a decrease of $3.0 million, or 61%. Certain licensing revenues recognized are related to the cash payments received under the Auxilium Agreement in prior years and amortized over the expected development period. Licensing revenue recognized for the years ended December 31, 2012 and 2011 were $372,705 and $437,102, respectively. The decrease of $64,397, or 15%, was mainly due to the completed recognition of licensing revenue associated with the Dupuytren’s contracture indication during the first quarter of 2010 and a slight change in the development timeline associated with the Peyronie’s indication. Sublicensing fees recognized in 2012 were $0.6 million compared to $0.8 million in the same period in 2011. In 2012, we recognized $0.6 million in sublicensing fees, which were related to the $10.0 million paid to Auxilium by Actelion for the rights to develop and commercialize XIAFLEX for the treatment of Dupuytren's contracture and Peyronie's disease in Canada, Australia, Brazil and Mexico. In the 2011 period, we recognized $0.8 million of the $15.0 million paid to Auxilium by Asahi for the rights to commercialize XIAFLEX for the treatment of Dupuytren's contracture and Peyronie's disease in Japan.
Milestone revenue recognized for the years ended December 31, 2012 and 2011 were $1.0 million and $3.8 million, respectively. In the 2012 period, we recognized a $1.0 million milestone related to the FDA’s December 2012 acceptance of Auxilium’s sBLA for XIAFLEX for the potential treatment of Peyronie's disease. We also, recognized a milestone of $28,500 related to the Notice of Compliance (approval) by Health Canada for XIAFLEX for the treatment of Dupuytren's contracture in adults with a palpable cord in Canada granted to Auxilium. In the 2011 period, we recognized a $2.6 million milestone related to the first sale of XIAFLEX in Europe, a $0.6 million milestone related to the first sale of XIAFLEX in Germany, and a $0.6 million milestone related to the first sale of XIAFLEX in Spain.
Under current accounting guidance, nonrefundable upfront license fees for product candidates for which we are providing continuing services related to product development are deferred and recognized as revenue over the development period. The remaining balance will be recognized over the respective development periods or when we determine that we have no ongoing performance obligations.
Consulting Services
We recognize revenues from consulting and technical assistance contracts primarily as a result of the DFB Agreement. We recognize consulting revenues ratably over the term of the contract. For calendar years 2012 and 2011 we recognized zero and $46,667 respectively. The decrease in revenues resulting from consulting and technical assistance contracts is due to the expiration in March 2011 of our consulting obligations under the DFB Agreement.
Research and Development Activities
Research and development expenses include, but are not limited to, internal costs, such as salaries and benefits, costs of materials, lab expense, facility costs and overhead. Research and development expenses also consist of third party costs, such as medical professional fees, product costs used in clinical trials, consulting fees and costs associated with clinical study arrangements. Research and development expenses were $1.2 million and $1.0 million, respectively, for calendar years 2012 and 2011, representing an increase in 2012 of $0.2 million, or 29%. This increase in research and development expenses was primarily due to expenses related to our clinical development programs.
We were working to develop CCH for the treatment of human and canine lipoma. We initiated a placebo controlled randomized study to evaluate the efficacy of CCH for the treatment of subcutaneous benign lipomas in canines. The treatment was a single injection of CCH or placebo. The primary efficacy endpoint was the relative change in lipoma volume from baseline to 3 months, as determined by CT scan. We completed this study, and top-line data were released in December 2013.
Also, we initiated a 14-patient, single center dose escalation, phase II clinical trial of CCH for the treatment of human lipomas. The study was a single injection, open-label trial, and CCH was being administered in four ascending doses (0.058 mg to 0.44 mg). The primary efficacy endpoint was a change in the visible surface area of the target lipoma, as determined at six months post-injection. In January 2014, we announced the top-line data from the phase II dose escalation clinical trial of CCH for the treatment of human lipoma.
The following table summarizes our research and development expenses related to our clinical development programs:
Year Ended December 31, 2012 | Year Ended December 31, 2011 | Accumulated Expenses Since January 1, 2010 | ||||||||||
Program | ||||||||||||
| Canine Lipoma | $ | 442,741 | $ | 332,217 | $ | 972,988 | ||||||
| Human Lipoma | 168,879 | 110,800 | 404,869 | |||||||||
Successful development of drugs is inherently difficult and uncertain. Our business requires investments in research and development over many years, often for drug candidates that may fail during the research and development process. Even if the Company is able to successfully complete the development of our drug candidates, our long-term prospects depend upon our ability and the ability of our partners, particularly with respect to XIAFLEX and CCH, to continue to successfully commercialize these drug candidates.
There is significant uncertainty regarding our ability to successfully develop drug candidates in other indications. These risks include the uncertainty of:
| · | the nature, timing and estimated costs of the efforts necessary to complete the development of our drug candidate projects; |
| · | the anticipated completion dates for our drug candidate projects; |
| · | the scope, rate of progress and cost of our clinical trials that we are currently running or may commence in the future with respect to our drug candidate projects; |
| · | the scope, rate of progress of our preclinical studies and other research and development activities related to our drug candidate projects; |
| · | clinical trial results for our drug candidate projects; |
| · | the cost of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights relating to our drug candidate projects; |
| · | the terms and timing of any strategic alliance, licensing and other arrangements that we have or may establish in the future relating to our drug candidate projects; |
| · | the cost and timing of regulatory approvals with respect to our drug candidate projects; and |
| · | the cost of establishing clinical supplies for our drug candidate projects. |
Our current resources and liquidity are sufficient to advance our significant current research and development projects and, Auxilium will have the option to exclusively license the canine and human lipoma indications upon completion of the appropriate opt-in study.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs for personnel, consultant costs, legal fees, investor relations, professional fees and overhead costs. General and administrative expenses were $4.8 million and $5.2 million for calendar years 2012 and 2011, respectively, a decrease of $0.5 million or 9%, from 2011. The decrease in general and administrative expenses was due to lower general legal fees and stock based compensation partially offset by third party royalty fees and consulting fees.
Other Income and expense
Other income for calendar year 2012 was $34,634 compared to $71,603 for calendar year 2011. For calendar year 2012, other income consisted of interest earned on our investments of $34,634. For calendar year 2011, other income consisted of interest earned on our investments of $55,780 and a reversal of accrued tax penalties associated with our delinquent tax filings in previous years of $15,823.
Income Taxes
Our deferred tax liabilities, deferred tax assets and related valuation allowances are impacted by events and transactions arising in the ordinary course of business, research and development activities, vesting of nonqualified options, deferred revenues and other items. Deferred tax assets are affected by the valuation allowance which is dependent upon several factors, including estimates of the realization of deferred income tax assets, and the impact of estimated future taxable income. Significant judgment is required to determine the estimated amount of valuation allowance to record. Changes in the estimate of the valuation allowance could materially increase or decrease our provision for income taxes in future periods.
The provision for income taxes and corresponding taxes payable in 2012 was $2.2 million as compared to a tax benefit of $1.3 million in 2011. In 2012, we used $1.0 million of our Orphan Drug tax credit to reduce our federal income tax payable. We recognized the tax effect of $0.8 million related to the exercise of nonqualified options in our financial statements, which lowered our taxes payable by $0.3 million, reduced our tax assets related to non-qualified stock options by $32,000 and increased additional paid in capital by $0.3 million. Additionally, we utilized tax assets from our federal and state net operating loss carryforwards of $16,000 and deferred licensing revenue of $0.1 million to reduce our taxes payable. Because our state net operating losses of $4.2 million exceeded our federal net operating losses of $47,000 we set up a valuation allowance of $0.3 million against our tax asset of our state net operating loss carryforwards.
In the 2011 period, the $1.3 million in income tax benefit consisted of an income tax expense of $2.3 million which was offset by a one-time $3.6 million tax benefit related to our deferred tax asset valuation allowance and the recording of our deferred tax assets as we believe that our tax assets are more likely than not to be realized as we achieved sustained profitability on an on-going annual basis. In making such determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax planning strategies and recent financial operations. In 2011, we recognized the tax effect of $4.6 million in disqualifying disposition of options and the exercise of nonqualified options in our financial statements. The exercise of nonqualified options and disposition of disqualified options lowered our taxes payable by $1.9 million, reduced our tax assets related to non-qualified options by $0.2 million and increased additional paid in capital and provision for income tax benefits by $1.7 million and $30,239, respectively. Additionally, we utilized tax assets from our federal and state net operating loss carryforwards of $0.3 million and deferred licensing revenue of $0.2 million to reduce our taxes payable. Because our state net operating losses exceeded our federal net operating losses we set up a valuation allowance of $0.2 million against our tax asset for our state net operating loss carryforwards.
Liquidity and Capital Resources
To date, we have financed our operations primarily through product sales, debt instruments, licensing revenues and royalties under agreements with third parties and sales of our common stock. At December 31, 2013, 2012 and 2011, we had cash and cash equivalents and short-term investments in the aggregate of approximately $12.6 million, $8.5 million and $8.2 million, respectively.
Sources and Uses of Cash
Net cash provided by (used in) operating activities was $5.1 million, $2.4 million and $(0.7) million for 2013, 2012 and 2011. Net cash provided by operating activities for 2013 was primarily due to our net income, net of stock compensation expenses and other non-cash charges. Net cash provided by operations for 2012 was primarily due to our net income, net of stock compensation expenses and other non-cash charges. Net cash used in operations for 2011 was mainly due to a decrease in accrued expenses, increases in deferred tax assets and accounts receivable, net of stock compensation expense and other non-cash charges
The majority of our cash expenditures in 2013, 2012, and 2011 were to fund research and development, our business activities and our stock repurchase program.
Net cash used in investing activities was $2.4 million in 2013 and $1.6 million in 2012, as compared to net cash provided by investing activities of $0.4 million in 2011. The net cash used in investing activities in the 2013 period reflects the maturing of investments of $9.7 million and reinvestment of $11.6 million in marketable securities and a cash payment related to our future royalty obligations for Peyronie's disease of $0.6 million. The net cash used in investing activities in the 2012 period reflects the maturing of investments of $5.1 million and reinvestment of $5.2 million in marketable securities and a one-time cash payment related to our future royalty obligations for Peyronie's disease of $1.5 million. The net cash provided by investing activities in the 2011 period reflects the maturing of investments of $5.4 million and reinvestment of $5.0 million in marketable securities.
Net cash used in financing activities was $0.4 million in 2013 and $0.6 million in 2012, as compared to net cash provided by financing activities of $1.1 million for 2011. In 2013, net cash used in financing activities of was mainly related to the repurchase of our common stock under our 2010 Stock Repurchase Program of $0.7 million partially offset by excess tax benefits related to share-based payments and stock option proceeds of $0.2 million. In 2012, net cash used in financing activities was mainly related to the repurchase of our common stock under our 2010 Stock Repurchase Program of $1.0 million partially offset by excess tax benefits related to share-based payments and stock option proceeds of $0.4 million. In 2011, net cash provided by financing activities was due to excess tax benefits related to share-based payments and stock option proceeds partly offset by the repurchase of our common stock under our 2010 Stock Repurchase Program.
Contractual Commitments
We are involved with licensing of products which are generally associated with payments to third parties from whom we have licensed the product. Such payments may take the form of an up-front payment; milestone payments which are paid when certain parts of the overall development program are accomplished; payments upon certain regulatory events, such as the filing of an IND, an NDA or BLA, approval of an NDA or BLA, or the equivalents in other countries; and payments based on a percentage of sales.
We may also out-license products, for which we hold the rights, to other companies for commercialization in other territories, or at times, for other uses. When this happens, the payments to us would also take the same form as described above.
Operating Leases
Our operating leases are principally for facilities and equipment. We currently lease approximately 15,000 square feet of space at our headquarters in Lynbrook, New York. Additionally, we lease certain vehicle and certain office equipment which generally expire in 2014 and 2017, respectively.
Operating lease expenses amounted to approximately $143,000 for calendar years 2013, 2012 and 2011, respectively.
Future minimum annual payments required under non-cancelable operating leases are approximated as follows:
Year ending December 31,
| 2014 | $ | 137,000 | ||
| 2015 | $ | 4,000 | ||
| 2016 | $ | 4,000 | ||
| 2017 | $ | 2,000 |
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
We do not use derivative financial instruments or derivative commodity instruments for trading purposes. Our financial instruments consist of cash, cash equivalents, short-term investments, trade accounts receivable, accounts payable and long-term obligations. We consider investments that, when purchased, have a remaining maturity of 90 days or less to be cash equivalents.
Our investment portfolio is subject to interest rate risk, although limited given the nature of the investments, and will fall in value in the event market interest rates increase. All our cash and cash equivalents at December 31, 2013, amounting to approximately $5.6 million, were maintained in bank demand accounts and money market accounts. Our short-term investments of $7.0 million were maintained in certificates of deposit. We do not hedge our interest rate risks, as we believe reasonably possible near-term changes in interest rates would not materially affect our results of operations, financial position or cash flows.
For the discussion of Item 8, “Financial Statements” please see the Consolidated Financial Statements, beginning on page F-1 of this Report.
None.
| Item 9A. | CONTROLS AND PROCEDURES. |
The Company, under the supervision and with the participation of Thomas L. Wegman, the Company’s President, Principal Executive Officer, Principal Financial Officer and Principal Accounting Officer, evaluated the effectiveness of its disclosure controls and procedures as of the end of the period covered by this Report. Based on that evaluation, management has concluded that the Company’s disclosure controls and procedures are effective to ensure that information required to be disclosed in reports filed under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC, and that such information is accumulated and communicated to the Company’s management, to allow timely decisions regarding required disclosure. Because of the inherent limitations in all control systems, any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Furthermore, our controls and procedures can be circumvented by the individual acts of some persons, by collusion of two or more people or by management override of the control, and misstatements due to error or fraud may occur and not be detected on a timely basis.
Management’s Annual Report on Internal Control Over Financial Reporting
Management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting for the Company as defined in Rule 13a-15(f) under the Exchange Act. The Company’s internal control over financial reporting is designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements and the reliability of financial reporting.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2013. In making this assessment, management used the 1992 criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control – Integrated Framework. We believe that, as of December 31, 2013, the Company’s internal control over financial reporting was effective based on this criteria.
Tabriztchi & Co, the independent registered public accounting firm that audited our Consolidated Financial Statements included in this Report, audited the effectiveness of our internal control over financial reporting as of December 31, 2013, as stated in their report which is included in Part IV, Item 15 of this Report.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting identified in connection with the evaluation of our controls performed during the year ended December 31, 2013 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Not applicable.
The information required by this item is incorporated herein by reference to the sections captioned “Directors and Executive Officers,” “Committees of the Board of Directors,” and “Section 16(a) Beneficial Ownership Reporting Compliance” in our definitive Proxy Statement relating to our 2014 Annual Meeting of Stockholders.
The information required by this item is incorporated herein by reference to the section captioned “Executive Compensation” in our definitive Proxy Statement relating to our 2014 Annual Meeting of Stockholders.
| Item 12. |
The information required by this item is incorporated herein by reference to the sections captioned “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in our definitive Proxy Statement relating to our 2014 Annual Meeting of Stockholders.
The information required by this item is incorporated herein by reference to the section captioned “Certain Relationships and Related Transactions” in our definitive Proxy Statement relating to our 2014 Annual Meeting of Stockholders.
The information required by this item is incorporated herein by reference to the section captioned “Ratification of Selection of Independent Registered Public Accounting Firm” in our definitive Proxy Statement relating to our 2014 Annual Meeting of Stockholders.
| (a) | The following documents are filed as part of this Report: |
| (1) | Consolidated Financial Statements (See Index to Consolidated Financial Statements on page F-1) |
| (2) | Financial Statement Schedules |
All schedules to the consolidated financial statements are omitted as the required information is either inapplicable or presented in the consolidated financial statements
| (3) | Exhibits |
The information required by this Item is set forth in the Exhibit Index hereto which is incorporated herein by reference.
| (b) | Exhibits |
The information required by this Item is set forth in the Exhibit Index hereto which is incorporated herein by reference.
BIOSPECIFICS TECHNOLOGIES CORP.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS FOR THE YEAR
ENDED DECEMBER 31, 2013
| Page | |
Reports of Independent Registered Public Accounting Firm | F-2 |
| Consolidated Balance Sheets | F-4 |
| Consolidated Statements of Operations | F-5 |
| Consolidated Statements of Cash Flows | F-6 |
| Consolidated Statements of Stockholders’ Deficit | F-7 |
| Notes to Consolidated Financial Statements | F-8 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of
BioSpecifics Technologies Corp.
We have audited the accompanying consolidated balance sheets of BioSpecifics Technologies Corp. (the Company) as of December 31, 2013 and 2012, and the related consolidated statements of operations, comprehensive income, stockholders' equity and cash flows for each of the years in the three-year period ended December 31, 2013. BioSpecifics Technologies Corp.'s management is responsible for these financial statements. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of BioSpecifics Technologies Corp. as of December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2012 in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), BioSpecifics Technologies Corp.'s internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) (1992 framework), and our report dated March 6, 2014 expressed an unqualified opinion thereon.
/s/ Tabriztchi & Co., CPA, P.C. | |
| Garden City, NY | |
March 6, 2014 | |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of
BioSpecifics Technologies Corp.
We have audited BioSpecifics Technologies Corp.'s internal control over financial reporting as of December 31, 2013 based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) (the COSO criteria). BioSpecifics Technologies Corp's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, BioSpecifics Technologies Corp. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2013, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of BioSpecifics Technologies Corp. , as of December 31, 2013 and 2012, and the related statements of income, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2013 of and our report dated March 6, 2014 expressed an unqualified opinion.
/s/ Tabriztchi & Co., CPA, P.C. | |
| Garden City, NY | |
March 6, 2014 | |
BioSpecifics Technologies Corp.
Consolidated Balance Sheet
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 5,624,860 | $ | 3,383,737 | ||||
| Short term investments | 6,966,964 | 5,120,000 | ||||||
| Accounts receivable, net | 5,004,418 | 5,082,360 | ||||||
| Income tax receivable | 255,708 | 51,070 | ||||||
| Deferred tax asset | 94,992 | 88,910 | ||||||
| Prepaid expenses and other current assets | 326,519 | 149,724 | ||||||
| Total current assets | 18,273,461 | 13,875,801 | ||||||
| Deferred royalty buy-down | 3,350,000 | 2,750,000 | ||||||
| Deferred tax assets –long term | 1,412,784 | 1,484,141 | ||||||
| Patent costs, net | 215,999 | 280,322 | ||||||
| Total assets | 23,252,244 | 18,390,264 | ||||||
| Liabilities and Stockholders' Equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | 634,277 | 512,866 | ||||||
| Deferred revenue | 69,130 | 133,524 | ||||||
| Accrued liabilities of discontinued operations | 78,138 | 78,138 | ||||||
| Total current liabilities | 781,545 | 724,528 | ||||||
| Deferred revenue - license fees | 138,260 | 207,390 | ||||||
| Stockholders' equity: | ||||||||
| Series A Preferred stock, $.50 par value, 700,000 shares authorized; none outstanding | - | - | ||||||
| Common stock, $.001 par value; 10,000,000 shares authorized; 6,655,168 and 6,625,168 shares issued at December 31, 2013 and 2012, respectively | 6,655 | 6,625 | ||||||
| Additional paid-in capital | 20,951,796 | 20,688,706 | ||||||
| Retained earnings (accumulated deficit) | 4,975,018 | (310,829 | ) | |||||
| Treasury stock, 300,739 and 260,632 shares at cost as of December 31, 2013 and 2012 | (3,601,030 | ) | (2,926,156 | ) | ||||
| Total stockholders' equity | 22,332,439 | 17,458,346 | ||||||
| Total liabilities and stockholders’ equity | $ | 23,252,244 | $ | 18,390,264 | ||||
See accompanying notes to consolidated financial statements
BioSpecifics Technologies Corp.
Consolidated Statements of Operations
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Revenues: | ||||||||||||
| Net sales | $ | 37,458 | $ | 18,219 | $ | 21,998 | ||||||
| Royalties | 11,767,758 | 9,155,654 | 6,314,959 | |||||||||
| Licensing revenue | 2,662,024 | 1,971,205 | 5,012,102 | |||||||||
| Consulting fees | - | - | 46,667 | |||||||||
| Total revenues | 14,467,240 | 11,145,078 | 11,395,726 | |||||||||
| Costs and expenses: | ||||||||||||
| Research and development | 1,484,416 | 1,249,755 | 972,078 | |||||||||
| General and administrative | 5,038,363 | 4,774,828 | 5,231,881 | |||||||||
| Total costs and expenses | 6,522,779 | 6,024,583 | 6,203,959 | |||||||||
| Operating income | 7,944,461 | 5,120,495 | 5,191,767 | |||||||||
| Other income (expense): | ||||||||||||
| Interest income | 26,202 | 34,634 | 55,780 | |||||||||
| Other | - | - | 15,823 | |||||||||
| 26,202 | 34,634 | 71,603 | ||||||||||
| Income before benefit (expense) for income tax | 7,970,663 | 5,155,129 | 5,263,370 | |||||||||
| Income tax benefit (expense) | (2,684,816 | ) | (2,174,054 | ) | 1,338,256 | |||||||
| Net income | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | ||||||
| Basic net income per share | $ | 0.83 | $ | 0.47 | $ | 1.04 | ||||||
| Diluted net income per share | $ | 0.76 | $ | 0.43 | $ | 0.95 | ||||||
| Shares used in computation of basic net income per share | 6,345,615 | 6,351,245 | 6,340,648 | |||||||||
| Shares used in computation of diluted net income per share | 6,922,274 | 6,981,527 | 6,952,386 | |||||||||
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Net income | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | ||||||
| Other comprehensive income | - | - | - | |||||||||
| Comprehensive income | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | ||||||
See accompanying notes to consolidated financial statements
BioSpecifics Technologies Corp.
Consolidated Statements of Cash Flows
| Years Ended December 31, | ||||||||||||
| Cash flows from operating activities: | 2013 | 2012 | 2011 | |||||||||
| Net income | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | ||||||
| Adjustments to reconcile net income to net cash used in operating activities: | ||||||||||||
| Depreciation and amortization | 64,323 | 64,190 | 50,685 | |||||||||
| Stock-based compensation expense | 111,636 | 228,485 | 517,367 | |||||||||
| Deferred income tax | (10,653 | ) | 1,474,904 | (3,047,955 | ) | |||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accounts receivable | 77,942 | (1,845,443 | ) | (1,250,792 | ) | |||||||
| Prepaid expenses and other current assets | (381,433 | ) | 142,160 | (65,642 | ) | |||||||
| Accounts payable and accrued expenses | 121,411 | (242,233 | ) | (3,009,109 | ) | |||||||
| Deferred revenue | (133,524 | ) | (372,705 | ) | (483,769 | ) | ||||||
| Net cash provided by (used in) operating activities from operations | 5,135,549 | 2,430,433 | (687,589 | ) | ||||||||
| Cash flows from investing activities: | ||||||||||||
| Maturities of marketable securities | 9,710,000 | 5,070,000 | 5,360,970 | |||||||||
| Purchases of marketable securities | (11,556,964 | ) | (5,190,000 | ) | (5,000,000 | ) | ||||||
| Payment for royalty buy down | (600,000 | ) | (1,500,000 | ) | - | |||||||
| Net cash provided by (used in) investing activities from operations | (2,446,964 | ) | (1,620,000 | ) | 360,970 | |||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from stock option exercises | 30,000 | 148,425 | 82,450 | |||||||||
| Repurchases of common stock | (674,874 | ) | (1,034,647 | ) | (739,551 | ) | ||||||
| Excess tax benefits from share-based payment arrangements | 197,412 | 262,695 | 1,709,699 | |||||||||
| Net cash provided by (used in) financing activities from operations | (447,462 | ) | (623,527 | ) | 1,052,598 | |||||||
| Increase in cash and cash equivalents | 2,241,123 | 186,906 | 725,979 | |||||||||
| Cash and cash equivalents at beginning of year | 3,383,737 | 3,196,831 | 2,470,852 | |||||||||
| Cash and cash equivalents at end of year | $ | 5,624,860 | $ | 3,383,737 | $ | 3,196,831 | ||||||
| Supplemental disclosures of cash flow information: | ||||||||||||
| Cash paid during the year for: | ||||||||||||
| Interest | $ | - | $ | - | $ | - | ||||||
| Taxes | $ | 2,713,500 | $ | 232,000 | $ | 190,000 | ||||||
Supplemental disclosures of non-cash transactions:
Under our agreement with Auxilium certain patent costs and expenses paid by Auxilium on behalf of the Company are creditable against future royalties. During the year ended December 31, 2013 we accrued $60,000 related to patent expense which was offset against our royalties’ receivable from Auxilium. The amortization of patent costs was $64,323, $64,190 and $50,685 in the 2013, 2012 and 2011 periods, respectively.
Our deferred tax assets and additional paid in capital decreased by approximately $75,000 as a result of the cancelation of 15,000 stock options.
See accompanying notes to consolidated financial statements
BioSpecifics Technologies Corp.
Consolidated Statement of Stockholders' Equity (Deficit)
| Shares | Amount | Additional Paid in Capital | Retained Earnings (Accumulated Deficit) | |||||||||||||
| Balances - December 31, 20010 | 6,445,743 | $ | 6,446 | $ | 17,739,765 | $ | (9,893,530 | ) | ||||||||
| Issuance of common stock under stock option plans | 85,000 | 85 | 82,365 | - | ||||||||||||
| Stock compensation expense | - | - | 517,367 | - | ||||||||||||
| Repurchases of common stock | - | - | - | - | ||||||||||||
| Excess tax benefits from share-based payment arrangements | - | - | 1,709,699 | - | ||||||||||||
| Net profit | - | - | - | 6,601,626 | ||||||||||||
| Balances - December 31, 2011 | 6,530,743 | $ | 6,531 | $ | 20,049,196 | $ | (3,291,904 | ) | ||||||||
| Issuance of common stock under stock option plans | 94,425 | 94 | 148,330 | - | ||||||||||||
| Stock compensation expense | - | - | 228,485 | - | ||||||||||||
| Repurchases of common stock | - | - | - | - | ||||||||||||
| Excess tax benefits from share-based payment arrangements | - | - | 262,695 | - | ||||||||||||
| Net profit | - | - | - | 2,981,075 | ||||||||||||
| Balances - December 31, 2012 | 6,625,168 | $ | 6,625 | $ | 20,688,706 | $ | (310,829 | ) | ||||||||
| Issuance of common stock under stock option plans | 30,000 | 30 | 29,970 | - | ||||||||||||
| Effect of expiration of stock options | - | - | (75,928 | ) | - | |||||||||||
| Stock compensation expense | - | - | 111,636 | - | ||||||||||||
| Repurchases of common stock | - | - | - | - | ||||||||||||
| Excess tax benefits from share-based payment arrangements | - | - | 197,412 | - | ||||||||||||
| Net profit | - | - | - | 5,285,847 | ||||||||||||
| Balances - December 31, 2013 | 6,655,168 | 6,655 | $ | 20,951,796 | $ | 4,975,018 | ||||||||||
Treasury Stock | Shareholder Equity Total | |||||||
| Balances - December 31, 2010 | $ | (1,151,958 | ) | $ | 6,700,723 | |||
| Issuance of common stock under stock option plans | - | 82,450 | ||||||
| Stock compensation expense | - | 517,367 | ||||||
| Repurchases of common stock | (739,551 | ) | (739,551 | ) | ||||
| Excess tax benefits from share-based payment arrangements | - | 1,709,699 | ||||||
| Net profit | - | 6,601,626 | ||||||
| Balances - December 31, 2011 | $ | (1,891,509 | ) | $ | 14,872,314 | |||
| Issuance of common stock under stock option plans | - | 148,424 | ||||||
| Stock compensation expense | - | 228,485 | ||||||
| Repurchases of common stock | (1,034,647 | ) | (1,034,647 | ) | ||||
| Excess tax benefits from share-based payment arrangements | - | 262,695 | ||||||
| Net profit | - | 2,981,075 | ||||||
| Balances - December 31, 2012 | $ | (2,926,156 | ) | $ | 17,458,346 | |||
| Issuance of common stock under stock option plans | - | 30,000 | ||||||
| Effect of expiration of stock options | - | (75,928 | ) | |||||
| Stock compensation expense | - | 111,636 | ||||||
| Repurchases of common stock | (674,874 | ) | (674,874 | ) | ||||
| Excess tax benefits from share-based payment arrangements | - | 197,412 | ||||||
| Net profit | - | 5,285,847 | ||||||
| Balances - December 31, 2013 | $ | (3,601,030 | ) | $ | 22,332,439 | |||
BIOSPECIFICS TECHNOLOGIES CORP.
Notes to Consolidated Financial Statements
December 31, 2013, 2012 and 2011
1. ORGANIZATION AND DESCRIPTION OF BUSINESS
We are a biopharmaceutical company involved in the development of an injectable collagenase for multiple indications. We have a development and license agreement with Auxilium Pharmaceuticals, Inc. (“Auxilium”) for injectable collagenase (which Auxilium has named XIAFLEX®) for marketed indications and collagenase clostridium histolyticum (“CCH”) for indications in development. Auxilium has an option to acquire additional indications that we may pursue, including human and canine lipoma. Auxilium is currently selling XIAFLEX in the U.S. for the treatment of Dupuytren’s contracture and Peyronie’s disease. Following the termination of the agreement between Auxilium and Pfizer, Inc. (“Pfizer”), Auxilium entered into an agreement with Swedish Orphan Biovitrum AB (“Sobi”) pursuant to which Sobi has marketing rights for XIAPEX® (the EU trade name for collagenase clostridium histolyticum) for Dupuytren’s contracture and Peyronie’s disease in Europe and certain Eurasian countries. Sobi is currently selling XIAPEX in Europe for the treatment of Dupuytren’s contracture. In addition, Auxilium has an agreement with Asahi Kasei Pharma Corporation (“Asahi”) pursuant to which Asahi has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Japan. Auxilium also has an agreement with Actelion Pharmaceuticals Ltd. (“Actelion”) pursuant to which Actelion has the right to commercialize XIAFLEX for the treatment of Dupuytren’s contracture and Peyronie’s disease in Canada, Australia, Brazil and Mexico.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Principles of Consolidation
The audited consolidated financial statements include the accounts of the Company and its subsidiary, Advance Biofactures Corp. (“ABC-NY”).
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires the use of management’s estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Cash, Cash Equivalents and Short-term Investments
Cash, cash equivalents and short-term investments are stated at market value. Cash equivalents include only securities having a maturity of three months or less at the time of purchase. The Company limits its credit risk associated with cash, cash equivalents and short-term investments by placing its investments with banks it believes are highly creditworthy and with highly rated money market funds, U.S. government securities, or short-term commercial paper which are held to maturity.
Fair Value Measurements
Management believes that the carrying amounts of the Company’s financial instruments, including cash, cash equivalents, short-term investments, accounts receivable, accounts payable and accrued expenses approximate fair value due to the short-term nature of those instruments.
Concentration of Credit Risk and Major Customers
The Company maintains bank account balances, which, at times, may exceed insured limits. The Company has not experienced any losses with these accounts and believes that it is not exposed to any significant credit risk on cash. The Company maintains its investment in FDIC insured certificates of deposits with several banks.
At December 31, 2013, the accounts receivable balance of $5.0 million was primarily from two customers, comprising of $3.5 million (70% of total) from DFB Biotech, Inc. and $1.5 million (30% of total) from Auxilium Pharmaceutical, Inc.
The Company has been dependent in each year on a two customers who generate almost all its revenues. In the year ended December 31, 2013, the licensing and royalty revenues from Auxilium Pharmaceutical, Inc. were $8.2 million (70% of total) and royalties and consulting revenues from DFB Biotech, Inc. were $3.5 million (30% of total). (With the expiration of right to receive payments on Santyl sales in August 2013, the primary source of our revenues is Auxilium Pharmaceutical, Inc.)
Revenue Recognition
We currently recognize revenues resulting from product sales, the licensing and sublicensing of the use of our technology and from services we sometimes perform in connection with the licensed technology under the guidance of Accounting Standards Codification 605, Revenue Recognition (“ASC 605”).
If we determine that separate elements exist in a revenue arrangement under ASC 605, we recognize revenue for delivered elements only when the fair values of undelivered elements are known, when the associated earnings process is complete, when payment is reasonably assured and, to the extent the milestone amount relates to our performance obligation, when our customer confirms that we have met the requirements under the terms of the agreement.
Revenues, and their respective treatment for financial reporting purposes, are as follows:
Product Sales
We recognize revenue from product sales when there is persuasive evidence that an arrangement exists, title passes, the price is fixed or determinable and collectability is reasonably assured. No right of return exists for our products except in the case of damaged goods. To date, we have not experienced any significant returns of our products.
Net sales include the sales of the collagenase for laboratory use that are recognized at the time the product is shipped to customers for laboratory use.
Royalty/Mark-Up on Cost of Goods Sold / Earn-Out Revenue
For those arrangements for which royalty, mark-up on cost of goods sold or earn-out payment information becomes available and collectability is reasonably assured, we recognize revenue during the applicable period earned. For interim quarterly reporting purposes, when collectability is reasonably assured but a reasonable estimate of royalty, mark-up on cost of goods sold or earn-out payment revenues cannot be made, the royalty, mark-up on cost of goods sold or earn-out payment revenues are generally recognized in the quarter that the applicable licensee provides the written report and related information to us.
Under the Auxilium Agreement, we do not participate in the selling, marketing or manufacturing of products for which we receive royalties and a mark-up on the cost of goods sold revenues. The royalty and mark-up on cost of goods sold revenues will generally be recognized in the quarter that Auxilium provides the written reports and related information to us, that is, royalty and mark-up on cost of goods sold revenues are generally recognized one quarter following the quarter in which the underlying sales by Auxilium occurred. The royalties payable by Auxilium to us are subject to set-off for certain patent costs.
Under the DFB Agreement, pursuant to which we sold our topical collagenase business to DFB, we had the right to receive earn-out payments based on sales of certain products. This right to receive payments on Santyl sales expired in August 2013. Generally, under the DFB Agreement we would receive payments and a report within ninety (90) days from the end of each calendar year after DFB has sold the royalty-bearing product. DFB has provided us earn-out reports on a quarterly basis. BioSpecifics has now recognized all income from the Santyl sales under the DFB agreement, and expects to receive the corresponding cash payment, the income recognized in 2013, in March 2014.
Licensing Revenue
We include revenue recognized from upfront licensing, sublicensing and milestone payments in “License Revenues” in our consolidated statements of operations in this Report.
Upfront License and Sublicensing Fees
We generally recognize revenue from upfront licensing and sublicensing fees when the agreement is signed, we have completed the earnings process and we have no ongoing performance obligation with respect to the arrangement. Nonrefundable upfront technology license for product candidates for which we are providing continuing services related to product development are deferred and recognized as revenue over the development period.
Milestones
Milestones, in the form of additional license fees, typically represent nonrefundable payments to be received in conjunction with the achievement of a specific event identified in the contract, such as completion of specified development activities and/or regulatory submissions and/or approvals. We believe that a milestone represents the culmination of a distinct earnings process when it is not associated with ongoing research, development or other performance on our part. We recognize such milestones as revenue when they become due and collection is reasonably assured. When a milestone does not represent the culmination of a distinct earnings process, we recognize revenue in a manner similar to that of an upfront license fee.
The timing and amount of revenue that we recognize from licenses of technology, either from upfront fees or milestones where we are providing continuing services related to product development, is primarily dependent upon our estimates of the development period. We define the development period as the point from which research activities commence up to regulatory approval of either our, or our partners’ submission assuming no further research is necessary. As product candidates move through the development process, it is necessary to revise these estimates to consider changes to the product development cycle, such as changes in the clinical development plan, regulatory requirements, or various other factors, many of which may be outside of our control. Should the U.S. Food and Drug Administration or other regulatory agencies require additional data or information, we would adjust our development period estimates accordingly. The impact on revenue of changes in our estimates and the timing thereof is recognized prospectively over the remaining estimated product development period.
Consulting and Technical Assistance Services
We recognized revenues from consulting and technical assistance contracts primarily as a result of our DFB Agreement and Auxilium Agreement. Consulting revenues are recognized ratably over the term of the contract. The consulting and technical assistance obligations to DFB expired in March 2011.
Treasury Stock
The Company accounts for treasury stock under the cost method and includes treasury stock as a component of stockholders’ equity.
Receivables, Deferred Revenue and Allowance for Doubtful Accounts
Trade accounts receivable are stated at the amount the Company expects to collect. We maintain allowances for doubtful accounts for estimated losses resulting from the inability of its customers to make required payments. We consider the following factors when determining the collectability of specific customer accounts: customer credit-worthiness, past transaction history with the customer, current economic industry trends, and changes in customer payment terms. Our accounts receivable balance is typically due from its two large pharmaceutical customers. These companies have historically paid timely and have been financially stable organizations. Due to the nature of the accounts receivable balance, we believe the risk of doubtful accounts is minimal. If the financial condition of our customers were to deteriorate, adversely affecting their ability to make payments, additional allowances would be required. We provide for estimated uncollectible amounts through a charge to earnings and a credit to a valuation allowance. Balances that remain outstanding after we have used reasonable collection efforts are written off through a charge to the valuation allowance and a credit to accounts receivable.
Accounts receivable as of December 31, 2013 is approximately $5.0 million, which consists of approximately $3.5 million due from DFB in accordance with the expired earn-out under the DFB Agreement and approximately $1.5 million in royalties and mark-up on costs of goods sold due from Auxilium in accordance with the terms of the Auxilium Agreement. Deferred revenue of $0.2 million consist of licensing fees related to the cash payments received under the Auxilium Agreement in prior years and amortized over the expected development period of certain indications for CCH. We recorded no material bad debt expense in each of the last three years. The allowance for doubtful accounts balance was $30,095, at December 31, 2013 and 2012.
Reimbursable Third Party Development Costs
We accrued patent expenses for research and development that are reimbursable by us under the Auxilium Agreement. We capitalize certain patent costs related to estimated third party development costs that are reimbursable under the Auxilium Agreement. In August 2011, through the amendment and restatement of our development and license agreement with Auxilium, we have clarified the rights and responsibilities of the joint development of XIAFLEX and CCH. We resolved what had been an on-going dispute with Auxilium concerning the appropriate amount of creditable third party development expenses related to the lyophilization of the injection formulation and certain patent expenses for research and development costs that are reimbursable under the Auxilium Agreement. We agreed and have reimbursed Auxilium by offsetting future royalties payable for the amount invoiced us for third party development costs related to the development of the lyophilization of the injection formulation. We do not expect any additional third party development cost related to the lyophilization of the injection formulation.
As of December 31, 2013 our net reimbursable third party patent expense accrual was approximately $60,000.
Third-Party Royalties
We have entered into licensing and royalty agreements with third parties and agreed to pay certain royalties on net sales of products for specific indications. We accrue third-party royalty expenses on net sales reported to us by Auxilium. Third-party royalty expense is generally expensed in the quarter that Auxilium provides the written reports and related information to us, that is, generally one quarter following the quarter in which the underlying sales by Auxilium occurred. We expect our third party royalty expense under General and Administrative expenses will continue to increase as net sales by Auxilium for XIAFLEX increase and potential new indications for CCH are approved.
Royalty Buy-Down
On March 31, 2012, we entered into an amendment to our existing agreement, dated August 27, 2008, related to our future royalty obligations for Peyronie’s disease. The amendment enables us to buy down a portion of our future royalty obligations in exchange for an initial cash payment of $1.5 million and five additional cash payments, one of which was paid in December 2013.
As of December 31, 2013, we have capitalized $3.35 million related to this agreement which will be amortized over approximately five years beginning on the date of the first commercial sale of XIAFLEX for the treatment of Peyronie’s disease, which represents the period estimated to be benefited using the straight-line method. In accordance with Accounting Standards Codification 350, Intangibles, Goodwill and Other, the Company amortizes intangible assets with finite lives in a manner that reflects the pattern in which the economic benefits of the assets are consumed or otherwise used up. If that pattern cannot be reliably determined, the assets are amortized using the straight-line method. We perform an evaluation of the recoverability of the carrying value of our intangible assets to determine if facts and circumstances indicate that the carrying value of intangible assets may be impaired and if any adjustment is warranted. Based on our evaluation as of December 31, 2013, no impairment existed for intangible assets.
Research and Development Expenses
Research and development expenses include, but are not limited to, internal costs, such as salaries and benefits, costs of materials, lab expense, facility costs and overhead. Research and development (“R&D”) expenses also consist of third party costs, such as medical professional fees, product costs used in clinical trials, consulting fees and costs associated with clinical study arrangements. We may fund R&D at medical research institutions under agreements that are generally cancelable. All of these costs are charged to R&D as incurred, which may be measured by percentage of completion, contract milestones, patient enrollment, or the passage of time.
Clinical Trial Expenses
Our cost accruals for clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with various clinical trial centers and clinical research organizations. In the normal course of business we contract with third parties to perform various clinical trial activities in the ongoing development of potential drugs. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Payments under the contracts depend on factors such as the achievement of certain events, the successful enrollment of patients, the completion of portions of the clinical trial, or similar conditions. The objective of our accrual policy is to match the recording of expenses in our financial statements to the actual cost of services received and efforts expended. As such, expenses related to each patient enrolled in a clinical trial are recognized ratably beginning upon entry into the trial and over the course of the patient’s continued participation in the trial. In the event of early termination of a clinical trial, we accrue an amount based on our estimate of the remaining non-cancelable obligations associated with the winding down of the clinical trial. Our estimates and assumptions could differ significantly from the amounts that may actually be incurred.
Income Taxes
Deferred tax assets and liabilities are recognized based on the expected future tax consequences, using current tax rates, of temporary differences between the financial statement carrying amounts and the income tax basis of assets and liabilities. A valuation allowance is applied against any net deferred tax asset if, based on the weighted available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
We use the asset and liability method of accounting for income taxes, as set forth in Accounting Standards Codification 740-10-25-2. Under this method, deferred income taxes, when required, are provided on the basis of the difference between the financial reporting and income tax basis of assets and liabilities at the statutory rates enacted for future periods. In accordance with Accounting Standards Codification 740-10-45-25, Income Statement Classification of Interest and Penalties, we classify interest associated with income taxes under interest expense and tax penalties under other.
Stock Based Compensation
The Company has two stock-based compensation plans in effect which are described more fully in Note 10. Accounting Standards Codification 718, Compensation - Stock Compensation (“ASC 718”) requires the recognition of compensation expense, using a fair-value based method, for costs related to all share-based awards including stock options and common stock issued to our employees and directors under our stock plans. It requires companies to estimate the fair value of share-based awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the requisite service periods in our Consolidated Statements of Operations.
Under the ASC 718, we estimate the fair value of our employee stock awards at the date of grant using the Black-Scholes option-pricing model, which requires the use of certain subjective assumptions. The most significant of these assumptions are our estimates of the expected volatility of the market price of our stock and the expected term of an award. When establishing an estimate of the expected term of an award, we consider the vesting period for the award, our recent historical experience of employee stock option exercises (including forfeitures) and the expected volatility. When there is uncertainty in the factors used to determine the expected term of an award, we use the simplified method. As required under the accounting rules, we review our valuation assumptions at each grant date and, as a result, our valuation assumptions used to value employee stock-based awards granted in future periods may change. The company granted 30,000, 15,000 and zero stock options in 2013, 2012 and 2011, respectively.
Further, ASC 718 requires that employee stock-based compensation costs to be recognized over the requisite service period, or the vesting period, in a manner similar to all other forms of compensation paid to employees. The allocation of employee stock-based compensation costs to each operating expense line are estimated based on specific employee headcount information at each grant date and estimated stock option forfeiture rates and revised, if necessary, in future periods if actual employee headcount information or forfeitures differ materially from those estimates. As a result, the amount of employee stock-based compensation costs we recognize in each operating expense category in future periods may differ significantly from what we have recorded in the current period.
Stock-based compensation expense recognized under ASC 718 was as follows:
| December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Research and development | $ | 92,249 | $ | 171,217 | $ | 96,849 | ||||||
| General and administrative | 19,387 | 57,268 | 420,518 | |||||||||
| Total stock-based compensation expense | $ | 111,636 | $ | 228,485 | $ | 517,367 | ||||||
We account for stock options granted to persons other than employees or directors at fair value using the Black-Scholes option-pricing model in accordance with Accounting Standards Codification 505-50, Equity Based Payments to Non-Employees (“ASC 505-50”). Stock options granted to such persons and stock options that are modified and continue to vest when an employee has a change in employment status are subject to periodic revaluation over their vesting terms. We recognize the resulting stock-based compensation expense during the service period over which the non-employee provides services to us. The stock-based compensation expense related to non-employees for the years ended December 31, 2013, 2012 and 2011 was $79,049, $109,479, and zero, respectively.
Property, Plant and Equipment
Property, plant and equipment are stated at cost, less accumulated depreciation. Machinery and equipment, furniture and fixtures, and autos are depreciated on the straight-line basis over their estimated useful lives of 5 to 10 years.
Patent Costs
We amortize intangible assets with definite lives on a straight-line basis over their estimated useful lives, ranging from 1 to 13 years, and review for impairment on a quarterly basis and when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable.
As of December 31, 2013, the Company’s capitalized costs related to certain patents paid by Auxilium on behalf of the Company and are reimbursable to Auxilium under the Auxilium Agreement. These patent costs are creditable against future royalty revenues. At December 31, net patent costs consisted of:
| 2013 | 2012 | 2011 | ||||||||||
| Patents, net | $ | 215,999 | $ | 280,322 | $ | 190,416 | ||||||
The amortization expense for patents was $64,323, $64,190and $50,685, for the years ended December 31, 2013, 2012 and 2011. The estimated aggregate amortization expense for each of the next five years is as follows:
| 2014 | $ | 53,000 | ||
| 2015 | 25,000 | |||
| 2016 | 20,000 | |||
| 2017 | 20,000 | |||
| 2018 | 20,000 |
Income Taxes
In accordance with Accounting Standards Codification 740-10-45-25, Income Statement Classification of Interest and Penalties (“ASC 740-10-45-25”) we classify interest associated with income taxes under interest expense and tax penalties under other.
| 3. | FAIR VALUE MEASUREMENTS |
The authoritative literature for fair value measurements established a three-tier fair value hierarchy, which prioritizes the inputs in measuring fair value. These tiers are as follows: Level 1, defined as observable inputs such as quoted market prices in active markets; Level 2, defined as inputs other than the quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as significant unobservable inputs (entity developed assumptions) in which little or no market data exists.
As of December 31, 2013, the Company held certain investments that are required to be measured at fair value on a recurring basis. The following tables present the Company’s fair value hierarchy for these financial assets as of December 31, 2013, 2012 and 2011:
December 31, 2013 | Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||
| Cash and cash equivalents | $ | 5,624,860 | $ | 5,624,860 | - | - | ||||||||||
| Certificates of Deposit | 6,966,964 | 6,966,964 | - | - | ||||||||||||
December 31, 2012 | Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||
| Cash and cash equivalents | $ | 3,383,737 | $ | 3,383,737 | - | - | ||||||||||
| Certificates of Deposit | 5,120,000 | 5,120,000 | - | - | ||||||||||||
December 31, 2011 | Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||
| Cash and cash equivalents | $ | 3,196,831 | $ | 3,196,831 | - | - | ||||||||||
| Certificates of Deposit | 5,000,000 | 5,000,000 | - | - | ||||||||||||
4. EARNINGS PER SHARE
Basic earnings per share is computed by dividing net income by the weighted-average number of common shares outstanding during the period. Diluted earnings per share is computed by dividing net income by the weighted-average number of common shares outstanding during the period increased to include all additional common shares that would have been outstanding assuming potentially dilutive common shares, resulting from option exercises, had been issued and any proceeds thereof used to repurchase common stock at the average market price during the period.
2013 | 2012 | 2011 | ||||||||||
| Net income for diluted computation | $ | 5,285,847 | $ | 2,981,075 | $ | 6,601,626 | ||||||
| Weighted average shares: | ||||||||||||
| Basic | 6,345,615 | 6,351,245 | 6,340,648 | |||||||||
| Effect of dilutive securities: | ||||||||||||
| Stock options | 576,659 | 630,282 | 611,738 | |||||||||
| Diluted | 6,922,274 | 6,981,527 | 6,952,386 | |||||||||
| Net income per share: | ||||||||||||
| Basic | $ | 0.83 | $ | 0.47 | $ | 1.04 | ||||||
| Diluted | $ | 0.76 | $ | 0.43 | $ | 0.95 | ||||||
5. INVENTORIES, NET
None.
6. PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment from continuing operations consist of:
| December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Machinery and equipment | $ | 562,610 | $ | 562,610 | $ | 562,610 | ||||||
| Furniture and fixtures | 91,928 | 91,928 | 91,928 | |||||||||
| Leasehold improvements | 1,185,059 | 1,185,059 | 1,185,059 | |||||||||
| 1,839,597 | 1,839,597 | 1,839,597 | ||||||||||
| Less accumulated depreciation and amortization | (1,839,597 | ) | (1,839,597 | ) | (1,839,597 | ) | ||||||
| $ | - | $ | - | $ | - | |||||||
Total depreciation expense amounted to zero for each calendar year 2013, 2012 and 2011, respectively.
7. COMPREHENSIVE INCOME
For the years ended 2013, 2012, 2011, we had no components of other comprehensive income other than net income itself.
8. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
Accounts payable and accrued liabilities consist of the following:
| December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| Trade accounts payable and accrued expenses | $ | 409,617 | $ | 304,635 | $ | 407,954 | ||||||
| Accrued legal and other professional fees | 61,538 | 61,147 | 50,000 | |||||||||
| Accrued payroll and related costs | 163,122 | 147,084 | 143,048 | |||||||||
| $ | 634,277 | $ | 512,866 | $ | 601,002 | |||||||
9. INCOME TAXES
The provision for income taxes consists of the following:
Year ended December 31,
| 2013 | 2012 | 2011 | ||||||||||
Current taxes: | ||||||||||||
| Federal | $ | 2,724,597 | $ | 686,968 | $ | - | ||||||
| State | 25,491 | 12,182 | 3,525 | |||||||||
| Total current taxes | 2,750,088 | 699,150 | 3,525 | |||||||||
Deferred taxes: | ||||||||||||
| Federal | (68,298 | ) | 1,134,532 | (1,219,190 | ) | |||||||
| State | 3,023 | 340,372 | (122,593 | ) | ||||||||
| Total deferred taxes | (65,274 | ) | 1,474,904 | (1,341,783 | ) | |||||||
| Total provision for income taxes | $ | 2,684,814 | $ | 2,174,054 | $ | (1,338,258 | ) | |||||
The effective income tax rate of the Company differs from the federal statutory tax rate of 34% due to the following items:
Year ended December 31,
| 2013 | 2012 | 2011 | ||||||||||
| Statutory rate | 34.00 | % | 34.00 | % | 34.0 | % | ||||||
| State income taxes, net of federal income tax benefit | 0.21 | % | 0.16 | % | 7.1 | % | ||||||
| Stock-based compensation | 0.11 | % | 1.51 | % | 4.0 | % | ||||||
| Change in effective state tax rate | 0.02 | % | 6.59 | % | - | |||||||
| Other, net | (0.66 | )% | (0.08 | )% | (5.9 | )% | ||||||
| Increase (decrease) in valuation allowance | - | - | (64.6 | )% | ||||||||
| Effective tax rate (benefit) | 33.68 | % | 42.18 | % | (25.4 | %) | ||||||
The effective rate reconciliation includes the permanent differences and changes in valuation allowance for windfalls, stock-based compensation, and net operating loss.
Deferred income taxes reflect the tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The components of deferred income tax assets and liabilities are as follows:
Year ended December 31,
2013 | 2012 | 2011 | ||||||||||
| Tax credit carry forward | $ | - | $ | - | $ | 1,027,633 | ||||||
| Deferred revenues | 71,062 | 132,514 | 293,297 | |||||||||
| Other | 71,304 | 27,322 | 17,253 | |||||||||
| Options | 1,365,409 | 1,413,214 | 1,687,780 | |||||||||
| Net operating loss carry forward | - | - | 21,992 | |||||||||
| Net deferred tax assets before valuation allowance | 1,507,776 | 1,573,050 | 3,047,955 | |||||||||
| Valuation allowance | - | - | - | |||||||||
| Net deferred tax asset | $ | 1,507,776 | $ | 1,573,050 | $ | 3,047,955 | ||||||
Company considers all available information, including operating results, ongoing tax planning, and forecasts of future taxable income. Based on the results of our operations in 2011, the growth in the market for XIAFLEX, and the trend in actual and anticipated royalty income, we had determined that it was more likely than not that the benefit of our deferred tax assets would be realized. Consequently, in 2011, we eliminated the valuation allowance of $3.6 million.
Stock-based compensation, recorded in the Company's financial statements is non-deductible for tax purposes and increases the Company's effective tax rate. Deferred tax assets, including those associated with stock based compensation, are reviewed and adjusted for apportionment and potential tax rates changes in various jurisdictions. In 2012, our tax assets related to stock-based compensation decreased by $0.3 million, due to a reduction in our estimated state tax apportionment rate.
We recognized $0.6 million, $0.8 million and $0.7 million of tax deductible expenses from the exercise of non-qualified or a disqualified disposition of incentive stock options, in 2013, 2012 and 2011 respectively. The windfall tax benefits of $0.2 million, $0.3 million and $1.7 million realized upon exercise of stock-based awards were classified as additional paid in capital and recorded under cash flows from financing activities, in 2013, 2012 and 2011, respectively.
The provision for income taxes and corresponding taxes payable in 2013 was $2.7 million. We utilized tax assets of $0.1 million related to deferred licensing revenue and stock based compensation and a $17,000 research and development credit to reduce our taxes payable which was partially offset by an increase to our deferred taxes for employee based compensation. The amount of refundable federal income taxes as of December 31, 2013 is approximately $0.2 million.
In 2012, we used $1.0 million of our Orphan Drug tax credit to reduce our federal income tax payable. We recognized the tax effect of $0.8 million related to the exercise of nonqualified options in our financial statements, which lowered our taxes payable by $0.3 million, reduced our tax assets related to non-qualified stock options by $32,000 and increased additional paid in capital by $0.3 million. Additionally, we utilized tax assets from our federal and state net operating loss carryforwards of $16,000 and deferred licensing revenue of $0.1 million to reduce our taxes payable. Because our state net operating losses of $4.2 million exceeded our federal net operating losses of $47,000 we set up a valuation allowance of $0.3 million against our tax asset of our state net operating loss carryforwards.
As of December 31, 2013, the Company believes that there are no significant uncertain tax positions, and no amounts have been recorded for interest and penalties. The Company does not expect that it would be required to record a liability related to an uncertain tax position. The tax periods open to examination by the major taxing jurisdictions to which the Company is subject include fiscal years 2010 through 2012.
10. STOCKHOLDERS’ EQUITY
Stock Option Plans
In July 1997, the Company's stockholders approved a stock option plan (the “1997 Plan”) for eligible key employees, directors, independent agents, and consultants who make a significant contribution toward the Company's success and development and to attract and retain qualified employees which expired in July 2007. Under the 1997 Plan, qualified incentive stock options and non-qualified stock options may be granted to purchase up to an aggregate of 500,000 shares of the Company's common stock, subject to certain anti-dilution provisions. The exercise price per share of common stock may not be less than 100% (110% for qualified incentive stock options granted to stockholders owning at least 10% of common shares) of the fair market value of the Company's common stock on the date of grant. In general, the options vest and become exercisable in four equal annual installments following the date of grant, although the Company’s board of directors, at its discretion, may provide for different vesting schedules. The options expire ten years (five years for qualified incentive stock options granted to stockholders owning at least 10% of common shares) after such date. The Company filed with the Securities and Exchange Commission a Registration Statement on Form S-8 for the 1997 Plan on September 26, 1997 to register these securities. In accordance with terms of the 1997 Plan, no options were granted ten years after the effective date of the 1997 Plan, or July 2007. In July 2007, approximately 231,000 stock options expired unissued, and there are no shares available for grant remaining under the 1997 Plan. As of December 31, 2012 there were zero options outstanding under the 1997 Plan.
In August 2001, the Company's stockholders approved a stock option plan (the “2001 Plan”), with terms similar to the 1997 Plan. The 2001 Plan authorizes the granting of awards of up to an aggregate of 750,000 shares of the Company's common stock, subject to certain anti-dilution provisions. On December 16, 2003, stockholders approved an amendment to the 2001 Plan, which increased the number of shares authorized for grant from 750,000 shares to 1,750,000 shares, an increase of 1,000,000 shares. On June 17, 2009, our stockholders approved an amendment to the 2001 Plan to extend the term of the 2001 Plan from April 6, 2011 to April 23, 2019 and to authorize an additional 300,000 shares of our common stock for issuance under the 2001 Plan. A total of 2,050,000 shares of common stock are now authorized for issuance under the amended 2001 Plan. The Company filed with the Securities and Exchange Commission a Registration Statement on Form S-8 for the 2001 Plan on October 5, 2007 and on July 15, 2009 as amended to register these securities. As of December 31, 2013 options to purchase 1,167,000 shares of common stock were outstanding under the 1997 Plan and 2001 Plan, and a total of 239,098 shares remain available for grant under the 2001 Plan.
The following table presents the assumptions used to estimate the fair values of the stock options granted in the periods presented:
September 2013 | April 2013 | May 2012 | ||||||||||
| Risk-free interest rate | 1.73 | % | 0.68 | % | 0.69 | % | ||||||
| Expected volatility | 33 | % | 37 | % | 54 | % | ||||||
| Expected life (in years) | 5 | 5 | 5 | |||||||||
| Dividend yield | - | - | - | |||||||||
| Weighted-average estimated fair value of options granted during the year | $ | 85,000 | $ | 79,000 | $ | 110,000 | ||||||
The summary of the stock options activity is as follows for year ended:
Shares | Weighted Average Exercise Price | |||||||
| Outstanding at December 31, 2012 | 1,182,000 | $ | 8.90 | |||||
December 31, 2013 | ||||||||
| Options granted | 30,000 | $ | 16.88 | |||||
| Options exercised | (30,000 | ) | 1.00 | |||||
| Options canceled or expired | 15,000 | 30.79 | ||||||
| Outstanding at end of year | 1,167,000 | 9.03 | ||||||
| Options exercisable at year end | 1,132,000 | 8.55 | ||||||
| Shares available for future grant | 239,098 | -- | ||||||
The following table summarizes information relating to stock options by exercise price at December 31, 2013:
| Outstanding Shares | Exercisable Shares | |||||||||||||||||||||
Option Exercise Price | Number of Shares | Weighted Average Life (years) | Weighted Average Exercise Price | Number of Shares | Weighted Average Option Price | |||||||||||||||||
| $ | 0.83 - 2.00 | 467,500 | 2.10 | $ | 1.01 | 467,500 | $ | 1.01 | ||||||||||||||
| 4.00 - 6.00 | 242,000 | 3.41 | 4.69 | 242,000 | 4.69 | |||||||||||||||||
| 13.00 - 16.00 | 155,000 | 5.24 | 13.96 | 155,000 | 13.96 | |||||||||||||||||
17.00 - 19.00 | 85,000 | 5.77 | 17.73 | 70,000 | 17.69 | |||||||||||||||||
| 20.00 - 21.00 | 112,500 | 4.75 | 20.57 | 112,500 | 20.57 | |||||||||||||||||
26.00 - 30.00 | 105,000 | 5.82 | 28.02 | 85,000 | 27.74 | |||||||||||||||||
| 1,167,000 | 3.72 | $ | 9.03 | 1,132,000 | $ | 8.55 | ||||||||||||||||
We granted 30,000 stock options during 2013. The weighted-average grant-date fair value for options granted during 2013 was $16.88 per share. During the 2013, 2012 and 2011, $30,000, $148,425and $82,450 were received from stock options exercised by employees, respectively. The aggregate intrinsic value of options outstanding and exercisable as of December 31, 2013 was approximately $14.8 million. Aggregate intrinsic value represents the total pre-tax intrinsic value, based on the closing price of our common stock of $21.67 on December 31, 2013, which would have been received by the option holders had all option holders exercised their options as of that date. Total unrecognized compensation cost related to non-vested stock options outstanding as of December 31, 2013 was approximately $79,000 which we expect to recognize over a weighted-average period of 3.75 years.
11. COMMITMENTS AND CONTINGENCIES
Lease Agreements
Our corporate headquarters are currently located at 35 Wilbur St., Lynbrook, NY 11563. On November 21, 2013, the Company entered into an Agreement of Lease (the “New Lease”) with 35 Wilbur Street Associates, LLC (“New Landlord”) for the Company’s administrative headquarters located at 35 Wilbur Street, Lynbrook, New York 11563 (the “Premises”). Neither the Company nor its affiliates have a material relationship or affiliation with the New Landlord. As previously reported, the Company formerly leased the Premises from Wilbur St. Corp. (“WSC”). On November 21, 2013, WSC sold the Premises to the New Landlord, and the Company entered into the New Lease with the New Landlord and simultaneously terminated the existing lease. The term of the New Lease is twenty-four months, provided, however, that the Company has the option to cancel the New Lease after the first year by giving three months’ notice, which may be given before the expiration of the first year. Pursuant to the New Lease, the Company’s monthly base rent is $12,000.00. The Company is required to pay as additional rent an amount equal to the increase in taxes over a specified base year.
The Company's operations are principally conducted on leased premises. Future minimum annual rental payments required under non-cancelable operating leases are $132,000.
Rent expense under all operating leases amounted to approximately $135,000 for calendar years 2013, 2012 and 2011, respectively.
12. RELATED PARTY TRANSACTIONS
As described above in Note 11, the Tenant and the Landlord were parties to the Lease Agreement. The rent expense, under the lease agreement, were $120,000, $135,000 and $135,000 for the years ended December 31, 2013, 2012 and 2011, respectively. As of December 31, 2013 there were no remaining related party transactions.
13. EMPLOYEE BENEFIT PLANS
ABC-NY has a 401(k) Profit Sharing Plan for employees who meet minimum age and service requirements. Contributions to the plan by ABC-NY are discretionary and subject to certain vesting provisions. The Company made no contributions to this plan for calendar years 2013, 2012 or 2011.
14. SUBSEQUENT EVENTS
We have evaluated subsequent events for recognition or disclosure through the time of filing these consolidated financial statements on Form 10-K with the U.S. Securities and Exchange Commission on March 7, 2014.
15. SELECTED QUARTERLY DATA (Unaudited)
The following table sets forth certain unaudited quarterly data for each of the four quarters in the years ended December 31, 2013 and 2012. The data has been derived from the Company's unaudited consolidated financial statements that, in management's opinion, include all adjustments (consisting of normal recurring adjustments) necessary for a fair presentation of such information when read in conjunction with the Consolidated Financial Statements and Notes thereto. The results of operations for any quarter are not necessarily indicative of the results of operations for any future period.
| First | Second | �� | Third | Fourth | ||||||||||||
| Quarter | Quarter | Quarter | Quarter | |||||||||||||
| Year ended December 31, 2013 | ||||||||||||||||
| Net revenues | $ | 3,980,024 | $ | 3,269,983 | $ | 3,145,123 | $ | 4,072,110 | ||||||||
| Operating profit | 2,066,668 | 1,563,685 | 1,738,501 | 2,575,607 | ||||||||||||
| Net income | 1,353,084 | 1,028,186 | 1,178,775 | 1,725,802 | ||||||||||||
| Basic earnings per share | $ | 0.21 | $ | 0.16 | $ | 0.19 | $ | 0.27 | ||||||||
| Diluted earnings per share | $ | 0.19 | $ | 0.15 | $ | 0.17 | $ | 0.25 | ||||||||
| First | Second | Third | Fourth | |||||||||||||
| Quarter | Quarter | Quarter | Quarter | |||||||||||||
| Year ended December 31, 2012 | ||||||||||||||||
| Net revenues | $ | 2,586,748 | $ | 2,601,834 | $ | 2,448,225 | $ | 3,508,271 | ||||||||
| Operating profit | 1,248,474 | 1,047,562 | 779,527 | 2,044,932 | ||||||||||||
| Net income | 742,390 | 666,682 | 471,047 | 1,100,956 | ||||||||||||
| Basic earnings per share | $ | 0.12 | $ | 0.11 | $ | 0.07 | $ | 0.17 | ||||||||
| Diluted earnings per share | $ | 0.11 | $ | 0.10 | $ | 0.07 | $ | 0.16 | ||||||||
EXHIBIT INDEX
The documents listed below are being filed or have previously been filed on behalf of the Company and are incorporated herein by reference from the documents indicated and made a part hereof. Exhibits not identified as previously filed are filed herewith:
Exhibit Number | Description |
| 3.1 | Registrant’s Certificate of Incorporation, as amended (incorporated by reference to Exhibit 3.1 of the Registrant’s Form 10-KSB filed with the Commission on March 2, 2007) |
| Registrant’s Amended and Restated By-laws as amended February 25, 2014* | |
| 3.3 | Amendment to Amended and Restated By-laws (incorporated by reference to Exhibit 3.1 of the Registrant’s Form 8-K filed with the Commission on February 26, 2014) |
| 4.1 | Rights Agreement dated as of May 14, 2002 (incorporated by reference as Exhibit 1 to the Registrant’s Form 8-A filed with the Commission on May 30, 2002) |
| 4.2 | Amendment No. 1 to Rights Agreement, dated June 19, 2003 (incorporated by reference to Exhibit 10.19 of the Registrant’s Form 10-KSB filed with the Commission on March 2, 2007) |
| 4.3 | Amendment No. 2 to Rights Agreement, dated as of February 3, 2011 (incorporated by reference to Exhibit 4.1 to the Registrant’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 4, 2011) |
| Amendment No.3 Rights Agreement, dated as of March 5, 2014* | |
| Agreement of Lease, dated as of November 21, 2013, between the Company, ABC-NY and 35 Wilbur Street Associates, LLC* | |
| Lease Termination Agreement, dated as of November 21, 2013, between the Company, ABC-NY and Wilbur St. Corp.* | |
| 10.3 | Asset Purchase Agreement between the Company, ABC-NY and DFB dated March 3, 2006 (incorporated by reference to Exhibit 2.1 of the Registrant’s Form 8-K filed with the Commission on March 9, 2006) |
| 10.4 | Amendment to Asset Purchase Agreement between the Company, ABC-NY and DFB dated January 8, 2007 (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-K filed with the Commission on January 12, 2007) |
| 10.5 | Dupuytren’s License Agreement dated November 21, 2006 between the Company and the Research Foundation (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-K filed with the Commission on November 28, 2006) |
| 10.6 | Frozen Shoulder License Agreement dated November 21, 2006 between the Company and the Research Foundation (incorporated by reference to Exhibit 10.2 of the Registrant’s Form 8-K filed with the Commission on November 28, 2006) |
| 10.7 | Cellulite License Agreement dated August 23, 2007 between the Company and the Research Foundation (incorporated by reference as Exhibit 10.7 of the Registrant’s Form 10-KSB filed with the Commission on March 15, 2013) |
| 10.8 | License Agreement dated March 27, 2010 between the Company and Zachary Gerut, M.D. (incorporated by reference as Exhibit 10.8 of the Registrant’s Form 10-KSB filed with the Commission on March 15, 2013) |
| 10.9 | Form of 1997 Stock Option Plan of Registrant (incorporated by reference as Exhibit 4.1 of the Registrant’s Form S-8 filed with the Commission on September 26, 1997) |
| 10.10 | Amended and Restated 2001 Stock Option Plan of Registrant (incorporated by reference to Appendix D of the Registrant’s Schedule14A filed with the Commission on April 30, 2009) |
| 10.11 | Change of Control Agreement, dated June 18, 2007 between the Company and Henry Morgan (incorporated by reference to Exhibit 10.21 of the Registrant’s Form 10-KSB filed with the Commission on September 26, 2007) |
| 10.12 | Change of Control Agreement, dated June 18, 2007 between the Company and Michael Schamroth (incorporated by reference to Exhibit 10.22 of the Registrant’s Form 10-KSB filed with the Commission on September 26, 2007) |
| 10.13 | Change of Control Agreement, dated June 18, 2007 between the Company and Dr. Paul Gitman (incorporated by reference to Exhibit 10.23 of the Registrant’s Form 10-KSB filed with the Commission on September 26, 2007) |
| 10.14 | Amendment and Restated Agreement between the Company and Dr. Marty Gelbard dated March 31, 2012 between the Company and Marty Gelbard (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-KA filed with the Commission on August 8, 2012) |
| 10.15 | Amended and Restated Development and License Agreement dated December 11, 2008 and effective December 17, 2008 between the Company and Auxilium Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-K filed with the Commission on December 19, 2008) |
| 10.16 | Executive Employment Agreement, dated August 5, 2008 between the Company and Thomas L. Wegman (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-K filed with the Commission on August 8, 2008) |
| 10.17 | Change of Control Agreement, dated October 1, 2008 between the Company and Dr. Matthew Geller (incorporated by reference to Exhibit 10.23 of the Registrant’s Form 10-K filed with the Commission on March 31, 2009) |
| Change of Control Agreement, dated as of September 17, 2013, between the Company and George Gould* | |
| 10.19 | Second Amended and Restated Development and License Agreement, dated as of August 31, 2011, by and between BioSpecifics Technologies Corp. and Auxilium Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.1 of the Registrant's Form 8-K filed with the SEC on September 1, 2011) |
| 10.20 | Settlement Agreement, dated as of August 31, 2011, by and between BioSpecifics Technologies Corp. and Auxilium Pharmaceuticals, Inc. (incorporated by reference to Exhibit 10.2 of the Registrant's Form 8-K filed with the SEC on September 1, 2011) |
| 14 | Amended and Restated Code of Business Conduct and Ethics (incorporated by reference to Exhibit 14.1 of the Registrant’s Form 10-KSB filed with the Commission on March 2, 2007) |
| 21 | Subsidiaries of the Registrant (incorporated by reference to Exhibit 21.1 of the Registrant’s Form 10-KSB filed with the Commission on March 2, 2007) |
| Consent of Tabriztchi & Co. CPA, P.C.* | |
| Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | |
| Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002* |
| * | filed herewith |
SIGNATURES
In accordance with section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant caused this Report on Form 10-K to be signed on its behalf by the undersigned, thereto duly authorized individual.
| Date: March 7, 2014 | |||
BIOSPECIFICS TECHNOLOGIES CORP. | |||
| By: | /s/ Thomas L. Wegman | ||
| Name: | Thomas L. Wegman | ||
| Title: | President | ||
In accordance with the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| SIGNATURE | TITLE | |
| /s/ Thomas L. Wegman | President, Director, and Principal Executive, Financial | |
| Name: Thomas L. Wegman | and Accounting Officer | |
| Date: March 7, 2014 | ||
| /s/ Paul Gitman | Director | |
| Name: Dr. Paul Gitman | ||
| Date: March 7, 2014 | ||
| /s/ George Gould | Director | |
| Name: George Gould | ||
| Date: March 7, 2014 | ||
| /s/ Henry G. Morgan | Director | |
| Name: Henry G. Morgan | ||
| Date: March 7, 2014 | ||
| /s/ Michael Schamroth | Director | |
| Name: Michael Schamroth | ||
| Date: March 7, 2014 | ||
| /s/ Dr. Mark Wegman | Director | |
| Name: Dr. Mark Wegman | ||
| Date March 7, 2014 | ||
| /s/ Toby Wegman | Director | |
| Name: Toby Wegman | ||
| Date: March 7, 2014 |