Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the fiscal year ended December 31, 2005 | ||
or | ||
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the transition period from to . | ||
Commission file number:000-50865
MannKind Corporation
(Exact name of registrant as specified in its charter)
Delaware | 13-3607736 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 28903 North Avenue Paine Valencia, California (Address of principal executive offices) | 91355 (Zip Code) |
Registrant’s telephone number, including area code
(661) 775-5300
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $0.01 per share
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer. See definition of “accelerated filer” and “large accelerated filer” inRule 12b-2 of the Act. (Check one):
Large accelerated filer o Accelerated filer þ Non-accelerated filer o
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Act). Yes o No þ
As of June 30, 2005, the aggregate market value of the voting stock held by non — affiliates of the registrant, computed by reference to the last sale price of such stock as of such date on the Nasdaq National Market, was approximately $167,548,444.
As of March 8, 2006, there were 49,510,109 shares of the registrant’s Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the registrant’s definitive Proxy Statement for the 2006 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by thisForm 10-K, are incorporated by reference in Part III,Items 10-14 of thisForm 10-K.
MANNKIND CORPORATION
Annual Report onForm 10-K
For the Fiscal Year Ended December 31, 2005
TABLE OF CONTENTS
2
Table of Contents
Forward-Looking Statements
Statements in this report that are not strictly historical in nature are forward-looking statements. These statements include, but are not limited to, statements about: the progress or success of our research, development and clinical programs, the timing of completion of enrollment in our clinical trials, the timing of the interim analyses and the timing or success of the commercialization of our Technosphere Insulin System, or any other products or therapies that we may develop; our ability to market, commercialize and achieve market acceptance for our Technosphere Insulin System, or any other products or therapies that we may develop; our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; our estimates for future performance; our estimates regarding anticipated operating losses, future revenues, capital requirements and our needs for additional financing; and scientific studies and the conclusions we draw from them. In some cases, you can identify forward-looking statements by terms such as “anticipates,” “believes,” “could,” “estimates,” “expects,” “goal,” “intends,” “may,” “plans,” “potential,” “predicts,” “projects,” “should,” “will,” “would,” and similar expressions intended to identify forward-looking statements. These statements are only predictions or conclusions based on current information and expectations and involve a number of risks and uncertainties. The underlying information and expectations are likely to change over time. Actual events or results may differ materially from those projected in the forward-looking statements due to various factors, including, but not limited to, those set forth under the caption “Risks and Uncertainties That May Affect Results” and elsewhere in this report. Except as required by law, we undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
Technosphere® and MedTone® are our registered trademark in the United States. We have also applied for or registered company trademarks in other jurisdictions, including Europe and Japan. This document also contains trademarks and service marks of other companies that are the property of their respective owners.
PART I
| Item 1. | Business |
Unless the context requires otherwise, the words “MannKind,” “we,” “company,” “us” and “our” refer to MannKind Corporation and its subsidiary.
OVERVIEW
MannKind Corporation is a biopharmaceutical company focused on the discovery, development and commercialization of therapeutic products for diseases such as diabetes and cancer. Our lead investigational product candidate, the Technosphere Insulin System, is currently in phase 3 clinical trials in the United States and Europe to study its safety and efficacy in the treatment of diabetes. This therapy consists of a proprietary dry powder formulation of insulin that is inhaled into the deep lung using our proprietary inhaler. We believe that the performance characteristics, unique kinetics, convenience and ease of use of the Technosphere Insulin System may have the potential to change the way diabetes is treated.
In particular, we have observed in our clinical trials to date that the Technosphere Insulin System produces a profile of insulin levels in the bloodstream that approximates the insulin profile normally seen in healthy individuals immediately following the beginning of a meal, but which is absent in all patients with diabetes. As a result, we believe that our Technosphere Insulin System will be beneficial not only for insulin-using diabetes patients but also for patients with type 2 diabetes who are currently using conventional therapies other than insulin. If further clinical trials support our initial observations, we believe the Technosphere Insulin System has the potential to be indicated for the treatment of type 2 diabetes after a patient has failed to respond adequately to diet and exercise. The use of insulin early in the progression of diabetes would represent a paradigm shift in the treatment of this disease.
To date, we have conducted multiple Phase 1 and Phase 2 clinical trials of our Technosphere Insulin System in Europe and the United States, which have involved more than 800 individuals and over 60,000 patient-days of home use. In a Phase 2 clinical trial conducted in the United States, the use of Technosphere Insulin at mealtimes significantly lowered blood glucose levels in patients with type 2 diabetes who previously were experiencing inadequate control of their disease. Even in patients with only mildly elevated blood glucose levels, we observed in
3
Table of Contents
this study that the typical risks of frequent or severe hypoglycemia generally associated with insulin therapy appear not to be associated with Technosphere Insulin, suggesting that our therapy may have a significant safety advantage over other currently available insulin therapies. In a European Phase 2 dosage-tolerance study, Technosphere Insulin was observed to improve glycemic control in a dose-dependent manner as measured by decreases in HbA1c levels and by reductions in glucose excursions following a meal. In addition, there was no indication of a negative effect on pulmonary function and no weight gain at any dose of Technosphere Insulin over 12 weeks of treatment.
Currently, we are enrolling patients in three pivotal Phase 3 clinical trials, including a two-year pulmonary safety study that will compare the pulmonary function of patients with type 1 or type 2 diabetes randomized to either Technosphere Insulin or standard diabetes care. Based on our discussions with the FDA, we plan to accumulate two years of controlled safety data from patients with type 1 diabetes as well as patients with type 2 diabetes before we file a new drug application for the Technosphere Insulin System. We anticipate that our entire clinical trial program will involve more than 3,200 patients. Larger populations and longer durations of exposure may be necessary depending on the safety profile of our product.
Our Technosphere Insulin System utilizes our proprietary Technosphere formulation technology, which is based on a class of organic molecules that are designed to self-assemble into small particles onto which drug molecules can be loaded. We are also developing additional Technosphere-based products for the delivery of other drugs. We plan to initiate Phase 1 clinical trials of our therapeutic cancer vaccine by the end of 2006.
We were incorporated in the State of Delaware in 1991. Our principal executive offices are located at 28903 North Avenue Paine, Valencia, California 91355, and our telephone number at that address is(661) 775-5300. Our website address is http://www.mannkindcorp.com. Our filings with the Securities and Exchange Commission, or SEC, including our annual report onForm 10-K, quarterly reports onForm 10-Q, current reports onForm 8-K and any amendments to those reports are available free of charge through our website as soon as reasonably practicable after being electronically filed with or furnished to the SEC.
OVERVIEW OF DIABETES
Diabetes is a major disease characterized by the body’s inability to properly regulate levels of blood glucose, or blood sugar. The cells of the body use glucose as fuel, which is consumed 24 hours a day. Between meals, when glucose is not being supplied from food, the liver releases glucose into the blood to sustain adequate levels. Insulin is a hormone produced by the pancreas that regulates the body’s blood glucose levels. Patients with diabetes develop abnormally high levels of glucose, a state known as hyperglycemia, either because they produce insufficient levels of insulin or because they fail to respond adequately to insulin produced by the body. Over time, poorly controlled levels of blood glucose can lead to major complications, including high blood pressure, blindness, amputations, kidney failure, heart attack, stroke and death.
According to the United States Centers for Disease Control, or CDC, as of 2005, approximately 20.8 million people in the United States, or 7% of the population, suffered from diabetes. The CDC estimated that 14.6 million cases of diabetes were diagnosed and under treatment and that 1.5 million new cases would be diagnosed in 2005. The CDC reported that diabetes was the sixth leading cause of death listed on death certificates in 2002, but that diabetes was likely to be underreported as a cause of death. Overall, the CDC found that the risk of death among people with diabetes is about twice that of people without diabetes of similar age. The American Diabetes Association estimated that, in 2002, the total cost of diabetes in the United States was $132 billion. This amount includes $12 billion of direct costs for drug treatment, of which approximately $7 billion were for insulin and delivery supplies and approximately $5 billion were for non-insulin oral medications.
There are two major forms of diabetes, type 1 and type 2. Type 1 diabetes is an autoimmune disease characterized by a complete lack of insulin secretion by the pancreas, so insulin must be supplied from outside the body in order to sustain life. In type 2 diabetes, the pancreas continues to produce insulin; however, insulin-dependent cells become resistant toward the insulin effect. Over time, the pancreas becomes increasingly unable to secrete adequate amounts of insulin to support metabolism. According to the CDC, type 2 diabetes is the more prevalent form of the disease, affecting approximately 90% to 95% of people diagnosed with diabetes.
4
Table of Contents
Challenges of treating type 2 diabetes
Typically, the treatment of type 2 diabetes starts with management of diet and exercise and progresses to treatment with various oral medications and then to treatment with insulin. Treatment with diet and exercise alone has not been an effective long-term solution for most patients with type 2 diabetes. Oral medications, which act predominantly by increasing the amount of insulin produced by the pancreas, by increasing the sensitivity of insulin-dependent cells or by reducing the glucose output of the liver, generally have significant adverse effects and are limited in their ability to manage the disease effectively.
Insulin therapy usually involves administering several subcutaneous needle injections of insulin each day. Although this treatment regimen is accepted as effective, it has limitations, most notably the fact that patients dislike injecting themselves with insulin due to the inconvenience and pain of needles. As a result, patients tend not to comply adequately with the prescribed treatment regimens and are often improperly medicated. Moreover, even when properly administered, subcutaneous injections of insulin do not replicate the natural time-action profile of insulin. In a person without diabetes, blood insulin levels rise within several minutes of the entry into the bloodstream of glucose from a meal. By contrast, injected insulin enters the bloodstream slowly, resulting in peak insulin levels in about 120 to 180 minutes for regular human insulin or 30-90 minutes for “rapid-acting” insulin analogs. The consequence is for patients with diabetes to have inadequate levels of insulin present at the initiation of a meal and to be over-insulinized between meals. This lag in insulin delivery results in high blood glucose levels early after meal onset, followed by a tendency for glucose to fall to abnormally low levels, a state known as hypoglycemia, during the period between meals. Hypoglycemia can result in loss of mental acuity, confusion, increased heart rate, hunger, sweating and faintness and, at very low glucose levels, loss of consciousness, seizures, coma and death.
The following figure illustrates the body’s need to have insulin available shortly after the start of a meal. In a 2004 article in theAmerican Journal of Physiology, Endocrinology and Metabolism, researchers modeled the response of a healthy pancreas to changes in blood glucose. The top panel illustrates the response following the rapid introduction of glucose, such as an intravenous bolus injection of a glucose solution. In this case, the pancreas responds with a sharp spike in insulin secretion followed by an extended wave of insulin secretion. The lower panel illustrates the response to a gradual elevation in glucose levels, such as might occur during ingestion of a meal. In this situation, the insulin concentration rises less briskly and remains elevated. However, as with the intravenous bolus of glucose, there is a fairly tight coupling between the rate of change in blood glucose levels and the response by the pancreas to secrete insulin.

The early insulin response following glucose ingestion is an important part of maintaining control over glucose levels during the post-meal period. It is thought that the early surge of insulin levels shuts off glucose production by
5
Table of Contents
the liver, which otherwise would continue to release glucose into the bloodstream at the same time that glucose is being absorbed from the meal. This avoids high blood sugar levels during mealtime and prevents the pancreas from having to secrete an excessive amount of insulin.
In other words, the time-course of insulin delivery to the bloodstream appears to be a significant factor in overall glucose control — a point that was made by a 1996 study reported in theJournal of Clinical Investigation. In this study, healthy subjects and patients with type 2 diabetes were given a meal-like intravenous infusion of glucose along with an intravenous infusion of insulin. In all participants, natural insulin secretion was inhibited so that only the infused insulin reached the bloodstream. The top panel shows that when the patient group was administered insulin in a manner that resembled the normal early insulin response, glucose control improved during the post-meal period compared to the effect seen when the same patients were administered insulin in a manner that resembled the typical diabetic profile, i.e., without an early insulin response. The bottom panel shows that when the healthy subjects were administered insulin in a manner that resembled the typical diabetic profile, post-meal blood glucose levels were higher and stayed elevated for a more prolonged period than when the same subjects were administered insulin in a manner resembling the normal early insulin response.
Profile of insulin release affects post-meal glucose control

Summary
In diabetes, the core defect is insulin deficiency. Even early in the progression of the disease, there is a marked reduction in the ability of patients to secrete insulin rapidly in response to the onset of a meal. Insulin has been used for more than eight decades to treat diabetes and is widely regarded as the preferred form of therapy for the disease. However, the conventional administration of insulin by subcutaneous injection is not able to deliver insulin to the bloodstream rapidly enough to approximate the early insulin secretion seen in healthy individuals following a meal. This shortcoming, combined with the pain and inconvenience of needles, is a significant obstacle to the effectiveness of conventional insulin therapy for the treatment of diabetes.
6
Table of Contents
THE MANNKIND SOLUTION
Addresses the core defect in diabetes in a manner unlike any other insulin therapy
In our clinical studies to date, we have consistently observed that the Technosphere Insulin System produces a profile of insulin levels in the bloodstream that approximates the early insulin secretion normally seen in healthy individuals following the beginning of a meal.
This performance characteristic distinguishes the Technosphere Insulin System from other insulin therapies. A 2004 review article in theBritish Journal of Diabetes and Vascular Diseasessurveyed the data published on pulmonary insulin products in development and compared their glucose-lowering activity to that of injectable rapid-acting insulin analogs. The graph below from this article shows that most pulmonary insulin formulations have comparable time-action profiles to injectable rapid-acting insulin. The one exception was the Technosphere Insulin System, which has been observed to have a much more rapid onset of action than the other insulin therapies reviewed.
Time-action profiles of inhaled insulin systems compared to a rapid-acting insulin analog

We believe the rapid action of Technosphere Insulin may be related to the unique aspects of both the carrier molecule as well as the insulin in our formulation. Our Technosphere formulation technology is centered on a class of pH-sensitive organic molecules that are designed to self-assemble into small carrier particles. Under mildly acidic conditions, these carrier particles assume a solid state, but at a more neutral pH they spontaneously dissociate into a liquid. Certain drugs, such as insulin, can be loaded onto these particles by combining mildly acidic solutions of the drug and the Technosphere material to form a mixture, which is then dried to form a powder. The structural characteristics of loaded Technosphere particles (i.e., particle size and surface topography) impart aerodynamic properties that enable them to fly deep into the lungs. Our laboratory studies indicate that Technosphere particles, unlike penetration enhancers, do not affect the tight junctions between cells, do not alter cell permeability and do not cause disruption of cell membranes. Instead, the Technosphere particles instantaneously change from a powder to a liquid upon contact with the neutral pH of the moist lung surface, allowing insulin to move rapidly down its concentration gradient into the bloodstream without any active assistance or enhancement.
Significantly, when the Technosphere particles dissociate, we currently think that the insulin that is released is in a form that can be readily used. Ordinarily, when insulin is in solution, it tends to form a hexamer, a complex of six associated insulin molecules. For example, regular human insulin is hexameric. In order to exert a pharmacological effect, the hexamer must first dissociate into three dimers — complexes of two insulin molecules — which then further dissociate into individual insulin molecules, or monomers. Only monomeric insulin exerts a physiological effect. Rapid-acting insulin analogues are designed to further the dissolution of the hexameric form, thereby reducing the time required to achieve an effect. However, none of these products can approximate the early insulin
7
Table of Contents
release that follows the beginning of a meal. The Technosphere Insulin System, on the other hand, appears to deliver monomeric insulin to the deep lung, where the individual molecules diffuse across a thin layer of cells to reach the bloodstream. No changes to the insulin molecule are required before it can start exerting its glucose-lowering effect.
Thus, we currently believe that Technosphere Insulin has the potential to address insulin deficiency in diabetes in a manner unlike any other insulin therapy, either existing or under development. We further believe that our ability to produce a better approximation of normal insulin physiology will translate into better glucose control, thereby giving our investigational product potential clinical and commercial advantages.
More natural insulin profile translates to better glucose control
When Technosphere Insulin is administered at or shortly after the beginning of a meal, we have observed that blood glucose levels after the meal are more tightly controlled than if patients attempt to control their disease with subcutaneous insulin or oral medications. These observations were made in our clinical studies of the Technosphere Insulin System, three of which are described in more detail below. We believe that this improved glucose control stems from the synchronization of Technosphere Insulin activity to meal digestion. Meal digestion is somewhat variable, but lasts approximately three hours. The figure below, taken from our clinical data, illustrates that over 80% of the glucose-lowering activity of regular subcutaneous insulin is not exerted until at least three hours after a meal. Typically, this situation causes patients to eat snacks between meals, contributing to the weight gain often associated with insulin therapy. By contrast, up to 80% of the action of Technosphere Insulin occurs within the first three hours after a meal. We believe that this phenomenon explains why we have seen almost no risk of severe hypoglycemia and no therapy-related weight gain in our clinical trials to date.

Study 003b
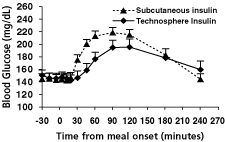
In a Phase 2 study of 16 patients with type 2 diabetes, we compared the effect of mealtime doses of Technosphere Insulin on blood glucose levels to the effect of mealtime doses of subcutaneous insulin. This study employed a cross-over design, so that patients were treated with either Technosphere Insulin or subcutaneous injections of insulin at mealtimes for approximately one week and then, after a washout period, were treated with the other treatment for a further week. At the end of each treatment period, patients were administered a standardized meal and their blood insulin and glucose levels were monitored for afour-hour period.
The graphs below show mean blood levels of insulin and glucose following administration of the meal to patients at the end of each treatment period. The panel on the left shows the characteristically rapid appearance of insulin in the bloodstream when Technosphere Insulin is inhaled as compared to the much slower increase following the subcutaneous injection of insulin. The panel on the right shows the corresponding post-meal excursions, or changes from baseline, of glucose absorbed from the meal following administration of either Technosphere Insulin or
8
Table of Contents
subcutaneous insulin. These data show that Technosphere Insulin was able to limit the excursion of blood glucose during the post-meal period in patients in this trial to a greater extent than insulin administered subcutaneously.
| Post-meal insulin levels | Post-meal glucose excursions | |
 |  |
In study 003b, we quantified the total exposure to insulin and the excursion of blood glucose levels by calculating the areas under the mean insulin and glucose curves, respectively. The results of this analysis are presented in the bar graph below. The bars on the left show that the areas under the insulin curve were virtually identical, indicating that patients in this trial received the same total exposure to insulin, whether from Technosphere Insulin or subcutaneous insulin. However, as shown by the bars on the right, when these patients inhaled Technosphere Insulin, there was a significantly decreased excursion in post-meal levels of blood glucose. Across all these patients, the mean total excursion of post-meal glucose levels following administration of Technosphere Insulin was 48% less than the mean excursion observed following administration of subcutaneous insulin.
Post-meal insulin levels and glucose excursions
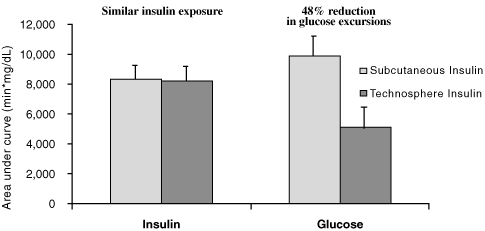
This latter observation supports our belief that delivering insulin in a manner that approximates the insulin response of healthy individuals allows patients with diabetes to achieve greater control over the glucose levels during the post-meal period.
Study 008
In a larger Phase 2 clinical trial, we studied the longer-term effects of meal-time insulin on blood glucose levels in 123 patients with type 2 diabetes whose pre-study treatment regimen consisted of either diet and exercise or one or more diabetes medications. This trial did not include patients whose diabetes had progressed to the point that they were already taking daily insulin. Patients were included in the study if their initial glycolated hemoglobin, or HbA1c, levels were between 6.5% and 10.5%, which is an indication that they were not achieving optimal glucose control on their current therapy. HbAlc levels are a measure of the average blood glucose level over the previous three to four months and an indication of how well a patient is controlling glucose levels. Patients were evaluated in two groups: those with moderate to severe elevations of HbA1c levels with a baseline of 8.0% and above and those with mild to moderate elevations of HbA1c levels with baseline levels ranging from 6.6% to 7.9%. Patients were
9
Table of Contents
randomized, in adouble-blind fashion, into either a group that inhaled Technosphere Insulin at mealtime or a control group that inhaled a placebo at mealtime. The use of a study agent at mealtime was the only variable in this study; all subjects continued their pre-study treatment regimen (diet and exercise or oral medications) and performed home blood glucose monitoring for the12-week duration of the study.
All patients were started on active or control therapy at a dose of 6 units before each meal. The physicians adjusted the inhaled study drug doses in increments of 6 or 12 units as often as weekly in order to bring blood glucose to the desired level of control. The maximum dose of mealtime Technosphere Insulin allowed in the study was 48 units. In most cases, patients did not reach their final maximum dose level until they had already completed at least 8 out of 12 weeks of treatment.
Patients with moderately severe elevations of HbA1c levels, treated with Technosphere Insulin, experienced a mean reduction in HbAlc levels of 1.37 percentage points over the limited duration of the study, compared to a mean reduction of 0.51 percentage points in the control-treated group. The difference in reduction of HbA1c levels between the Technosphere Insulin and the control groups was statistically and clinically significant in favor of Technosphere Insulin. Patients in this trial with mild to moderate elevations of HbA1c levels, treated with Technosphere Insulin, experienced a mean reduction in HbAlc levels of 0.43 percentage points, compared to a mean reduction of 0.18 percentage points in the control-treated group. This difference was also statistically and clinically significant in favor of Technosphere Insulin.
In this trial, we also conducted monthly evaluations of the response of patients to a standardized meal, during which we measured their post-meal glucose levels. We observed that the average post-meal glucose excursions fell as patients progressed to higher doses of Technosphere Insulin. At the highest average dose (30 units), the average glucose excursion was 34 mg/dl, which was approximately half the excursion observed in the control group.
Study 005
In a Phase 2b dose-ranging study, we evaluated the effect of different mealtime doses of Technosphere Insulin added to a single fixed daily injection of basal insulin glargine (Lantus) in patients with type 2 diabetes. In total, 227 patients were studied. Patients were followed on their existing therapy for four weeks, then transferred to insulin glargine, and subsequently randomized to receive, in a double blind fashion, either inhaled placebo or Technosphere Insulin at dosages of 14, 28, 42 or 56 units. Patients who received Technosphere Insulin were force-titrated weekly to their pre-assigned dose and remained on that dose for the duration of the study. HbA1c levels and post-meal glucose excursions were assessed at the initial visit, at the start of randomized treatment and at study completion. Comparisons were made between the groups receiving Technosphere Insulin and the group receiving placebo.
The study demonstrated that the addition of Technosphere Insulin to insulin glargine produced a statistically significant, dose-dependent reduction in HbA1c levels. In patients treated for eight weeks at 56 units (n=45), the mean reduction in percent HbA1c levels was 0.79 below that observed in the placebo group. We also observed a dose-dependent effect on post-meal glucose excursions. The graph below shows how each increased dose of Technosphere Insulin produced a proportional reduction in the glucose excursion following a standardized meal.
10
Table of Contents
Dose-dependent reduction of post-meal glucose excursions

At the largest dose evaluated, 56 units, the maximal post-meal glucose excursion was reduced to only 34 mg/dl, a 63% reduction compared to the control group.
Summary
We have seen in Phase 2 studies involving patients with type 2 diabetes that Technosphere Insulin can substantially reduce the excursion of glucose levels during the post-meal period and can lower average glucose levels within a few weeks of treatment. We are presently unable to assess the full glucose lowering effect of Technosphere Insulin from our Phase 2 studies because HbA1c levels are a measure of average blood glucose levels over the preceding three to four months, and in these studies patients were only dosed at their final maximum level for eight weeks or less. In the coming months, we plan to complete studies that have followed patients for a longer period of time, which we hope will give us better insight into the effect of Technosphere Insulin on HbA1c levels.
In any event, we believe the ability of Technosphere Insulin to substantially reduce post-meal glucose excursions is a significant performance characteristic. A 2005 report inClinical Diabetesreviewed evidence that links excessive post-meal glucose excursions to atherosclerosis and diabetic vascular disease, a complication of diabetes that affects the eyes, kidney and peripheral and autonomic nervous systems. The authors recommended that patients with type 2 diabetes be started on aggressive insulin therapy early in the course of the disease rather than using it as a “punishment” for failing to achieve glucose control with oral medications. We believe that the Technosphere Insulin System, if approved and commercialized, may offer physicians a means to help their patients with diabetes reduce post-meal glucose excursions without the pain and inconvenience of multiple daily insulin injections.
Favorable safety profile in clinical trials to date
To date, we have conducted clinical trials in Europe and the United States that have involved a total of more than 800 individuals and approximately 60,000 patient-days of home use. We have seen a significantly reduced number of mild, moderate and severe hypoglycemic episodes associated with Technosphere Insulin compared with the incidence reported with use of other insulin products. In some patients, we have observed mild coughing, usually limited to the period when learning to use the inhalation technique. Other adverse events reported in our clinical trials — including backache, common cold, pneumonia, anemia and diarrhea — were either found to be unrelated to the administration of Technosphere Insulin or could not be conclusively linked to its usage. We have also found no evidence of treatment-induced insulin antibodies occurring in patients treated with Technosphere Insulin.
In studies 008 and 005, we assessed pulmonary function in patients that received Technosphere Insulin over a 12 week period. In both studies, we found that there was no clinically or statistically significant difference between the baseline values and the final test results for these patients. In these studies, we also looked for evidence of weight gain in the patients that received Technosphere Insulin. The results indicated that there was no change in weight
11
Table of Contents
over the treatment period. Many of the patients from these studies have been enrolled in a long-term safety study, which should permit us to evaluate pulmonary function, weight gain and other markers over a longer period of time.
We have an ongoing program of safety surveillance and adverse event reporting for the purpose of evaluating the ongoing safety data related to the use of our Technosphere Insulin System. Our safety data are necessarily preliminary until we have completed longer-term safety studies.
Convenient and easy to use
To facilitate the delivery of Technosphere-formulated drugs to the deep lung, we developed an inhaler that utilizes single-use, disposable, plastic cartridges containing drug-loaded powder. Our MedTone inhaler is light and easy to use, and fits in the palm of the patient’s hand, which we believe facilitates patient compliance. To administer a dose, the patient opens the device, inserts a cartridge of Technosphere Insulin powder into the inhaler, inserts the mouthpiece into the mouth and takes a deep breath, thereby drawing the powder deep into the lungs. The device incorporates an airflow regulator that is designed to ensure a consistent airflow from patient to patient and from use to use, even in patients with restricted airflow capacity. In addition, the inhaler is breath actuated, which means that the patient does not need to coordinate a breath with any manipulation of the device, such as priming or pumping. In our clinical trials of our Technosphere Insulin System, patients have reported a high level of satisfaction with the MedTone inhaler.
We believe the ease of use of the MedTone inhaler complements the time-action profile of the Technosphere Insulin powder to produce a highly convenient system. Because insulin is transferred to the bloodstream rapidly with our therapy, we believe that the optimal and most convenient time for patients to take a dose of Technosphere Insulin is right at the start of a meal. In contrast, with subcutaneous regular insulin, it is recommended that the user try to time an injection 15 to 45 minutes before the expected mealtime, raising issues such as miscalculation of time or unanticipated change in meal availability, which could result in an increase in adverse events.
Higher relative bioavailability
Our clinical trials to date have indicated that a substantial percentage of the insulin formulated as Technosphere Insulin enters the bloodstream where it can exert a glucose-lowering effect. Pharmacologists use the term “bioavailability” to describe the percentage of the dosage form of a drug that enters the bloodstream and is available to exert an effect. Unless a drug is administered intravenously, its bioavailability is usually less than 100% because it must first be absorbed into the bloodstream and may be partially metabolized before it can exert an effect. The term “relative bioavailability” is used when the effect of a new formulation is compared to the effect of an established standard formulation, which, for insulin products, is regular human insulin injected subcutaneously.
Based on the results of our clinical trials and on our analysis of published reports of the performance of other pulmonary insulin systems in development, we believe that the relative bioavailability associated with our Technosphere Insulin System is up to three times greater than that reported for the other inhaled insulin platforms. We have observed that the relative bioavailability of Technosphere Insulin is as high as 28% compared to the approximately 11% relative bioavailability that has been reported by Pfizer for Exubera. In both cases, these measures are based on the amount of insulin present in the dosage form of the product, but we believe that the bioavailability is dependent on the amount that reaches the deep lung. With our Technosphere Insulin System, we know that not all of the powder contained in a cartridge makes it to the deep lung. For example, a small amount of the powder clings to the walls of the cartridge or remains in the mouthpiece. In addition, some of the powder that is discharged from our inhaler upon breath activation does not reach the bloodstream because the particles are not in the size range that we believe would reach the deep lung. Instead, these particles are likely deposited in the mouth and throat where they are swallowed and become not bioavailable. When corrected for these factors, we have calculated the relative bioavailability of Technosphere Insulin as approximately 56%.
One advantage of having a high relative bioavailability compared to other inhaled insulin systems is that the Technosphere Insulin System utilizes less insulin in order to achieve a specific glucose-lowering effect. This is likely to be safer from a clinical perspective and offers a potential cost of goods benefit from a manufacturing perspective. In addition, with a higher relative bioavailability, the response to a given dose would be expected to be less variable because a smaller portion of the insulin in the dosage form is lost before it reaches the bloodstream.
12
Table of Contents
CLINICAL DEVELOPMENT ACTIVITIES
The Technosphere Insulin System
We are currently conducting a number of studies of the safety and efficacy of the Technosphere Insulin System, including the following trials that are underway or planned for 2006:
Study 010
This ongoing trial is a three-year evaluation of the safety and tolerability of Technosphere Insulin in patients with type 2 diabetes who have participated in study 008 or study 005. The primary objective is to evaluate pulmonary function with secondary objectives to examine changes in HbA1c levels, insulin antibodies, frequency of adverse events and quality of life.
Study 030
This is a two-year, prospective, multi-site study that incorporates two design strategies. The first component is a randomized, open-label trial comparing pulmonary function in two groups of patients with diabetes. One group of approximately 625 patients is being treated with Technosphere Insulin and the other group of approximately 625 patients is being treated with existing oraland/or injectable therapies. The second component is a comparison of pulmonary function in the patients with diabetes who are not treated with Technosphere Insulin to a group of 125 subjects without any abnormalities in glucose control. Enrollment for this study began in June 2005 and is expected to be complete later in 2006.
Study 014
This study compared the efficacy of mealtime use of Technosphere Insulin plus basal insulin to mealtime use of rapid-acting, subcutaneous insulin plus basal insulin in approximately 240 patients with type 2 diabetes who had a baseline HbA1c level greater than 7.0% and less than 11.5%. The primary efficacy endpoint for this study was mean change in HbA1c levels from baseline to treatment week 24. Enrollment was completed in July 2005.
Study 101
This trial compared mealtime use of Technosphere Insulin to mealtime use of rapid-acting, subcutaneous insulin in approximately 90 patients with type 1 diabetes, who were evaluated over a12-week period. The primary efficacy endpoint was the change in blood glucose levels following a standard meal. Enrollment was completed in July 2005.
Study 009
This study is similar to Study 101, but will follow a population of approximately 500 patients with type 1 diabetes for a12-month period. Efficacy will be evaluated on the basis of changes in HbA1c levels as well as changes in blood glucose levels after a standardized mixed meal. We began enrolling patients in this study in the first quarter of 2006.
Study 102
This study will compare the efficacy of mealtime use of Technosphere Insulin to the twice-daily use of premixed insulin, a mixture of long- and short-acting insulin, in a population of approximately 500 patients with type 2 diabetes. Efficacy will be evaluated on the basis of changes in HbA1c levels as well as changes in blood glucose levels after a standardized mixed meal. We began enrolling patients in this study in the first quarter of 2006.
Study 103
This study will evaluate the efficacy of Technosphere Insulin alone and in combination with metformin, an oral medication, in approximately 500 patients with type 2 diabetes who are not achieving desired glucose control with a combination of metformin and sulphonylurea, another oral medication. Efficacy will be evaluated on the basis of
13
Table of Contents
changes in HbA1c levels after 26 weeks of treatment as well as changes in blood glucose levels after a standardized mixed meal. We plan to begin enrolling patients in this study in the second quarter of 2006.
Special population studies
We have two ongoing studies: one examining the impact of asthma and the other examining the impact of chronic smoking on the pharmacokinetics associated with Technosphere Insulin. We plan to use the results of these studies to conduct safety studies in patients with asthma and in chronic smokers that also have diabetes. We also plan to conduct a safety study in patients with both diabetes and chronic obstructive pulmonary disease. Other special population studies include a trial that will examine the elimination of Technosphere Insulin in patients with kidney disease and a trial that will examine the effect of Technosphere Insulin in patients with liver disease.
Cancer immunotherapy program
We are also developing therapies for the treatment of solid tumor cancers. The lead product candidate in this program, MKC1106, is intended for the treatment of several solid-tumor cancers, including ovarian, colorectal, pancreatic, renal, breast and prostate carcinomas. We plan to commence clinical trials for MKC1106 late in the fourth quarter of 2006.
Our cancer therapy program utilizes the body’s immune system to help eradicate tumor cells. The immune system is a network of cells and organs that defends the body against infection and abnormal cells, such as cancer. A key element of the immune system is its ability to distinguish between healthy cells and foreign or diseased cells that do not belong in the body. The immune system accomplishes this task by recognizing distinctive molecules called epitopes found on the surface of each cell as either normal or abnormal, and responding to them appropriately. Any substance capable of triggering an immune response is known as an antigen. An antigen can be all or part of a pathogenic organism or it can be a by-product of diseased cells. Certain specialized cells of the immune system sample antigens present in the body and present the epitopes associated with foreign antigens to other cells of the immune system whose function is to destroy any cell that expresses the same epitope; this is known as cell-mediated immunity. In this way, the immune system can launch a very specific response to infection or disease.
Our approach uses DNA- and peptide-based compounds that correspond to tumor-associated antigens that are expressed in a range of solid-tumor cancers. A patient is first “primed” by DNA-based compounds, or plasmids, that are injected directly into the patient’s lymph nodes. This is designed to sensitize the immune system to thetumor-associated antigens encoded by the plasmids. After a period of time, the patient’s lymph nodes are then injected with synthetic peptides that are designed to “boost” or greatly amplify the immune response to the target antigens. This prime-boost regimen is designed to provoke a potent cell-mediated immune response that destroys cancer cells along with the underlying blood supply to a tumor.
We believe that our therapeutic approach addresses several deficiencies inherent in earlier approaches to cancer immunotherapy, including:
| • | Specificity. We target cancer epitopes to which the immune system has not developed a tolerance, instead of targeting the dominant epitopes expressed by cancerous cells, many of which are tolerated by the immune system. We have developed technology designed to identify the non-tolerated epitopes on the cancer cell surface, and we have developed a method of modifying these epitopes that is designed to activate an immune response. Through this process, we believe that the body’s tolerance of the cancer cells can be broken, leading to the destruction of the cancer by the immune system. | |
| • | Administration. In contrast to the conventional subcutaneous or intramuscular route of administration, our compounds are delivered directly into the patient’s lymph nodes, where studies have shown they will have the greatest impact. We believe that the direct delivery of our compounds will bring local high concentrations of the active components of our compounds into contact with high concentrations of the cells needed to generate a potent cell-mediated immune response. | |
| • | Selectivity, potency and duration of response. We deliver our therapeutic compounds in a manner that we believe primes the immune system to respond to cancer cells expressing specific epitopes, in much the same way that a chronic infection evokes a progressively increased immune response to invading bacteria. Our |
14
Table of Contents
| administrative regimen is designed to boost the immune response over the course of a treatment cycle so that it becomes increasingly potent and long acting. |
OUR STRATEGY
Our objective is to develop products primarily in the major therapeutic areas of diabetes and cancer. Our strategy is to achieve this objective by doing the following:
| • | Commercialize our Technosphere Insulin System for the insulin-using diabetes market. We intend to advance our Technosphere Insulin System through Phase 3 clinical trials and then into commercialization, with the goal of first establishing a significant presence for Technosphere Insulin in the insulin-using diabetes market. We believe that the market for insulin products has the potential to expand as a result of the launch of Exubera, primarily due to the non-invasive nature of pulmonary insulin delivery. We believe the advantages in terms of safety, efficacy and convenience of the Technosphere Insulin System, as compared to other pulmonary insulin products, will enable us to capture a significant portion of the existing and expanded insulin-using diabetes market. | |
| • | Establish our Technosphere Insulin System as the preferred drug therapy within the broader population of people with type 2 diabetes. Our target markets also include patients with type 2 diabetes who are currently using conventional therapies other than insulin, including: |
| • | patients currently using diet and exercise therapy but who are having difficulty achieving proper blood glucose control; | |
| • | patients for whom diet and exercise therapy has failed, but who otherwise would have started non-insulin oral medications; and | |
| • | patients currently using non-insulin oral medications. |
A 2004 article in the New England Journal of Medicine reported that that aggressive insulin therapy, aimed at limiting post-meal glucose excursions, early in the course of the disease can reduce the risk of long-term cardiovascular complications. We believe that our Technosphere Insulin System, if approved, could be effective in an aggressive early insulin therapy program in two ways: it approximates the natural time-action profile of insulin in a manner that appears to provide better control over post-meal glucose excursions than injections, and it offers a more convenient way to take insulin than injections. As a result, we believe that our Technosphere Insulin System could have the potential to become a preferred drug therapy for a broad population of people with type 2 diabetes.
| • | Evaluate strategic collaborations for the development, marketing and commercialization of our Technosphere Insulin System. We are evaluating potential collaboration opportunities with large pharmaceutical companies in the United States, Europe and Japan to provide marketing, sales and financial resources to commercialize and sell our Technosphere Insulin System. We have not licensed or transferred any of our rights to this product. Our goal is to retain worldwide manufacturing rights for our Technosphere Insulin System. | |
| • | Advance our cancer immunotherapy program into clinical trials. We intend to commence clinical trials of our first cancer immunotherapy product late in the fourth quarter of 2006. Our intent is to evaluate the safety and efficacy of this program for the treatment of a range of solid-tumor cancers, including ovarian, colorectal, pancreatic, renal, breast and prostate carcinomas. | |
| • | Expand our proprietary Technosphere formulation technology for the delivery of other drugs. We are developing additional applications for our proprietary Technosphere formulation technology by formulating other drugs for pulmonary delivery. We believe our proprietary Technosphere formulation technology can also be extended to other forms of local administration, such as gastrointestinal delivery, because of its apparent ability to stabilize drugs and facilitate transport across cellular membranes without damage. |
15
Table of Contents
SALES AND MARKETING
We currently have no sales, marketing or distribution capabilities and have no experience as a company in marketing or selling pharmaceutical products. Our efforts to date have primarily been directed at developing products for a number of different markets. Assuming that we receive regulatory approval for our product candidates, we anticipate that we will have to pursue different sales and marketing strategies tailored to each particular product and market segment. In order to commercially market any of our products, we will also need either to develop a sales and marketing infrastructure ourselves or collaborate with third parties who have greater sales and marketing capabilities and have access to potentially large markets.
Although we believe that establishing our own sales and marketing organizations in North America would have substantial advantages, we recognize that this may not be practical for some of our products and that collaborating with companies with established sales and marketing capabilities in a particular market or markets may be a more effective alternative for some products. To date, we have retained worldwide commercialization rights for all of our products, including our lead product, the Technosphere Insulin System. We believe that this will give us flexibility if we enter into collaborations to provide the necessary sales and marketing support.
We are evaluating potential collaboration opportunities to assist us in the development and commercialization of our Technosphere Insulin System in the United States, Europe and Japan, and we may also create parallel in-house sales and marketing operations in certain key markets, particularly in the United States.
MANUFACTURING AND SUPPLY
We purchase human recombinant insulin under a long-term contract with Diosynth B.V., a global producer of insulin and a subsidiary of Akzo Nobel. This agreement has no specified termination date, but generally may be terminated upon two-years’ advance notice by either party. In addition, Diosynth has agreed to support our regulatory filings relating to the Technosphere Insulin System in the United States and abroad. We believe Diosynth has sufficient capacity to provide us with sufficient quantities of insulin to support our needs through the initial stages of commercialization. We must rely on our insulin supplier to maintain compliance with relevant regulatory requirements including cGMP.
We have a long-term supply agreement with Vaupell, Inc., an independent third party, for the manufacture and supply of our MedTone inhaler and the cartridges that are inserted into it. We rely on this manufacturer to comply with relevant regulatory requirements, including compliance with Quality System Regulations, or QSRs. We believe our manufacturer has the capacity to meet our Phase 3 clinical and initial commercial requirements.
Currently, we obtain our Technosphere pre-cursor raw material from Degussa AG, a major chemical manufacturer with facilities in Europe and North America. We also have the capability of manufacturing this chemical ourselves in our Danbury, Connecticut facility, which is now treated as a back up facility. We are in the process of qualifying another secondary manufacturer to supply us with commercial quantities of this raw material. Like us, our third-party manufacturers are subject to extensive governmental regulation.
We formulate and fill the Technosphere Insulin powder into plastic cartridges and blister package the cartridges in a manufacturing suite in our Danbury facility. We believe that our Danbury facility has adequate capacity to meet our currently anticipated clinical trial needs. We are continuing to increase our filling and packaging capacity through the acquisition of new equipment and the expansion of our clean rooms and other manufacturing facilities. We believe that our building improvements have been adequately validated to date and that the facility continues to substantially conform with cGMP. We have initiated the design and construction of a modular filling and packaging system that will increase our filling and packaging capacity. The new system is designed to operate at high speeds in a very small space, and capacity can be expanded by using multiple units.
INTELLECTUAL PROPERTY AND PROPRIETARY TECHNOLOGY
Our success will depend in large measure on our ability to obtain and enforce our intellectual property rights, effectively maintain our trade secrets and avoid infringing the proprietary rights of third parties. Our policy is to file patent applications on what we deem to be important technological developments that might relate to our product candidates or methods of using our product candidates and to seek intellectual property protection in the
16
Table of Contents
United States, Europe, Japan and selected other jurisdictions for all significant inventions. We have obtained, are seeking, and will continue to seek patent protection on the compositions of matter, methods and devices flowing from our research and development efforts. We have also in-licensed certain technology.
With respect to our Technosphere Insulin System, our core patents claim Technosphere particles as compositions of matter as well as methods for manufacturing Technosphere particles that incorporate drugs. The first of these patents expires in 2012, but subsequent patents provide additional coverage for the composition of matter and methods of use of the current product until 2020. We also hold patents that claim methods of using Technosphere particles for the pulmonary delivery of drugs. These patents relating to Technosphere Insulin do not expire until 2015. In addition, we are prosecuting patent applications related to the MedTone inhaler device and the capsules that contain the dry powder. We have filed and intend to continue to file additional patent applications on improvements to the Technosphere technology and its manufacture, as well as on specific compositions of matter formed using this technology in combination with drugs. To date, we have been issued 14 US and foreign Technosphere-related patents and have over 40 pending applications in different jurisdictions claiming inventions related to the Technosphere technology and the dry powder inhaler.
Our cancer immunotherapy program is built on proprietary methods for the selection, design and administration of epitopes. We have 90 pending patent applications relating to this technology, both as methods of use and compositions of matter. We are pursuing patents on the use of our administration method to induce and maintain a cell-mediated immune response. The prosecution is ongoing in many jurisdictions; however, we have been granted four patents related to this method, including two in the United States, which do not expire until 2018. We also have patent applications related to differential antigen processing and product designs. Two patents from this group have issued in the United States, which provides us with protection until 2020. In addition to applications of these broad technologies, we have filed and will continue to file patent applications on specific compounds and the protocols for administering them.
The fields of pulmonary drug delivery and cancer therapies are crowded and a substantial number of patents have been issued in these fields. In addition, because patent positions can be highly uncertain and frequently involve complex legal and factual questions, the breadth of claims obtained in any application or the enforceability of issued patents cannot be confidently predicted. Further, there can be substantial delays in commercializing pharmaceutical products, which can partially consume the statutory period of exclusivity through patents.
In addition, the coverage claimed in a patent application can be significantly reduced before a patent is issued, either in the United States or abroad. Statutory differences in patentable subject matter may limit the protection we can obtain on some of our inventions outside of the United States. For example, methods of treating humans are not patentable in many countries outside of the United States. These and other issues may limit the patent protection we will be able to secure internationally. Consequently, we do not know whether any of our pending or future patent applications will result in the issuance of patents or, to the extent patents have been issued or will be issued, whether these patents will be subjected to further proceedings limiting their scope, will provide significant proprietary protection or competitive advantage, or will be circumvented or invalidated. Furthermore, patents already issued to us or our pending applications may become subject to disputes that could be resolved against us. In addition, patent applications in the United States filed before November 29, 2000 are currently maintained in secrecy until the patent issues, although in certain countries, including the United States, for applications filed on or after November 29, 2000, applications are generally published 18 months after the application’s priority date. In any event, because publication of discoveries in scientific or patent literature often trails behind actual discoveries, we cannot be certain that we were the first creator of inventions covered by our pending patent applications or that we were the first to file patent applications on such inventions.
Although we own a number of domestic and foreign patents and patent applications relating to our Technosphere Insulin System and cancer vaccine products under development, we have identified certain third-party patents that a court may interpret to restrict our freedom to operate (that is, to cover our products) in the areas of Technosphere formulations, pulmonary insulin delivery and the treatment of cancer. Specifically, we have identified certainthird-party patents having claims relating to chemical compositions of matter and pulmonary insulin delivery that may trigger an allegation of infringement upon the commercial manufacture and sale of our Technosphere Insulin System. We have also identified third-party patents disclosing methods and compositions of matter related to
17
Table of Contents
DNA-based vaccines that also may trigger an allegation of infringement upon the commercial manufacture and sale of cancer therapy. We believe, based in part on advice of counsel, that we are not infringing any valid claims of any patent owned by a third party. However, if a court were to determine that our inhaled insulin products or cancer therapies were infringing any of these patent rights, we would have to establish with the court that these patents were invalid in order to avoid legal liability for infringement of these patents. Proving patent invalidity can be difficult because issued patents are presumed valid. Therefore, in the event that we are unable to prevail in an infringement or invalidity action we will either have to acquire the third-party patents outright or seek a royalty-bearing license. Royalty-bearing licenses effectively increase costs and therefore may materially affect product profitability. Furthermore, should the patent holder refuse to either assign or license us the infringed patents, it may be necessary to cease manufacturing the product entirelyand/or design around the patents. In either event, our business would be harmed and our profitability could be materially adversely impacted. If third parties file patent applications, or are issued patents claiming technology also claimed by us in pending applications, we may be required to participate in interference proceedings in the US Patent and Trademark Office, or USPTO, to determine priority of invention. We may be required to participate in interference proceedings involving our issued patents and pending applications.
We also rely on trade secrets and know-how, which are not protected by patents, to maintain our competitive position. We require our officers, employees, consultants and advisors to execute proprietary information and invention and assignment agreements upon commencement of their relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of our relationship must be kept confidential, except in specified circumstances. These agreements also provide that all inventions developed by the individual on behalf of us must be assigned to us and that the individual will cooperate with us in connection with securing patent protection on the invention if we wish to pursue such protection. There can be no assurance, however, that these agreements will provide meaningful protection for our inventions, trade secrets or other proprietary information in the event of unauthorized use or disclosure of such information.
We also execute confidentiality agreements with outside collaborators. However, disputes may arise as to the ownership of proprietary rights to the extent that outside collaborators apply technological information developed independently by them or others to our projects, or apply our technology to outside projects, and there can be no assurance that any such disputes would be resolved in our favor. In addition, any of these parties may breach the agreements and disclose our confidential information or our competitors might learn of the information in some other way. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business, results of operations and financial condition could be adversely affected.
COMPETITION
The pharmaceutical and biotechnology industries are intensely competitive and characterized by rapidly evolving technology and intense research and development efforts. We expect to compete with companies, including the major international pharmaceutical companies, and other institutions that have substantially greater financial, research and development, marketing and sales capabilities and have substantially greater experience in undertaking preclinical and clinical testing of products, obtaining regulatory approvals and marketing and selling biopharmaceutical products. We will face competition based on, among other things, product efficacy and safety, the timing and scope of regulatory approvals, ease of use and cost.
We believe our Technosphere Insulin System provides us with important competitive advantages in the delivery of insulin when compared with currently known alternatives. However, new drugs or further developments in alternative drug delivery methods may provide greater therapeutic benefits, or comparable benefits at lower cost, than our Technosphere Insulin System. There can be no assurance that existing or new competitors will not introduce products or processes competitive with or superior to our product candidates.
We have set forth below more detailed information about certain of our competitors. The following is based on information currently available to us.
18
Table of Contents
Other pulmonary and oral insulin delivery systems
In January 2006, the FDA approved Exubera, which was developed by Pfizer, Inc. in collaboration with Nektar Therapeutics, for the treatment of adults with type 1 and type 2 diabetes, as did the European Medicines Evaluation Agency. Eli Lilly and Company, in collaboration with Alkermes, Inc., is also developing a pulmonary insulin product. These companies initiated a Phase 3 clinical trial in July 2005, which is required to define the safety and efficacy of the Lilly/Alkermes product. Novo Nordisk A.S. has announced their intent to re-initiate Phase 3 clinical testing of their pulmonary insulin product. Kos Pharmaceuticals, Inc. is also developing a pulmonary insulin product.
There are also several companies, including Nobex Corporation, Generex Biotechnology Corporation and Emisphere Technologies, Inc., that are pursuing development of products for the oral delivery of insulin. We believe these products are currently in relatively early clinical trials.
Non-insulin oral medications
We expect that our Technosphere Insulin System will compete with currently available non-insulin oral medications for type 2 diabetes. These products include sulfonylureas, metformin and various insulin sensitizers. The sulfonylureas, which are mostly generic, act by directly stimulating insulin secretion and have been the principal non-insulin oral medication used to treat type 2 diabetes for several decades. Metformin, which is now available as a generic drug and is also marketed by Bristol-Meyers Squibb Company as Glucophage, has also been widely used for the treatment of type 2 diabetes. Insulin sensitizers, including Avandia, which is being marketed by GlaxoSmithKline PLC, and Actos, which is being marketed by Takeda Pharmaceuticals North America, Inc. and Eli Lilly & Company, are increasingly being used to treat type 2 diabetes.
Injected insulin
In the subcutaneous insulin market, our competitors have made considerable efforts to develop faster acting injectable insulin formulations. Humalog, which was developed by Eli Lilly and Company, and Insulin Aspart, or NovoLog, which was developed by Novo Nordisk A/S, are the two principal injectable insulin formulations with which we expect to compete.
Other injected diabetes medications
There is considerable interest in a new class of injectible diabetes drugs known as incretin mimetic agents. These drugs simulate or alter the activity of naturally occurring hormones that are released from the digestive system, such asGLP-1, GIP and others. These hormones modulate the metabolism of energy-containing compounds in the body, such as glucose, fat and protein. The most advanced of these drugs are Symlin and Byetta, being marketed by Amylin Pharmaceuticals, Inc. in collaboration with Eli Lilly & Company. Several other pharmaceutical companies are working on additional incretin mimetic agents or on incretin modifiers, such as DPP-IV inhibitors, which act to extend the potency ofGLP-1.
Immunotherapy
Over the last decade or so, a variety of companies have sought to develop therapeutic compounds that provide a selective immune response against cancer. Some of these companies, including Dendreon Corporation, Antigenics Inc., CancerVax Corporation, Cell Genesys Inc. and Corixa Corporation, have focused on products derived from the patients’ own cancer or other cells, or tumor cell lines, which can take the form of whole cells or cell fragments, or on tumor antigens extracted from cancerous cells. Other companies, including CancerVax Corporation, Progenics Pharmaceuticals, Inc., Therion Biologics Corporation and Vical Incorporated, are pursuing therapies designed to work across a broad spectrum of patients and tumor types.
GOVERNMENT REGULATION AND PRODUCT APPROVAL
The FDA and comparable regulatory agencies in state, local and foreign jurisdictions impose substantial requirements upon the clinical development, manufacture and marketing of medical devices and new drug products. These
19
Table of Contents
agencies, through regulations that implement the Food, Drug and Cosmetic Act, as amended, or FDCA, and other regulations, regulate research and development activities and the development, testing, manufacture, labeling, storage, shipping, approval, advertising, promotion, sale and distribution of such products. In addition, if our products are marketed abroad, they also are subject to export requirements and to regulation by foreign governments. The regulatory clearance process is generally lengthy, expensive and uncertain. Failure to comply with applicable FDA and other regulatory requirements can result in sanctions being imposed on us or the manufacturers of our products, including hold letters on clinical research, civil or criminal fines or other penalties, product recalls, or seizures, or total or partial suspension of production or injunctions, refusals to permit products to be imported into or exported out of the United States, refusals of the FDA to grant approval of drugs or to allow us to enter into government supply contracts, withdrawals of previously approved marketing applications and criminal prosecutions.
The steps typically required before an unapproved new drug product for use in humans may be marketed in the United States include:
| • | Preclinical studies that include laboratory evaluation of product chemistry and formulation, as well as animal studies to assess the potential safety and efficacy of the product. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, or requiring such studies to be replicated. In some cases, long-term preclinical studies are conducted while clinical studies are ongoing. | |
| • | Submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may commence. The results of the preclinical studies are submitted to the FDA as part of the IND. Unless the FDA objects, the IND becomes effective 30 days following receipt by the FDA. | |
| • | Approval of clinical protocols by independent investigational review boards, or IRBs, at each of the participating clinical centers conducting a study. The IRBs consider, among other things, ethical factors, the potential risks to individuals participating in the trials and the potential liability of the institution. The IRB also approves the consent form signed by the trial participants. | |
| • | Adequate and well-controlled human clinical trials to establish the safety and efficacy of the product. Clinical trials involve the administration of the drug to healthy volunteers or to patients under the supervision of a qualified medical investigator according to an approved protocol. The clinical trials are conducted in accordance with protocols that detail the objectives of the study, the parameters to be used to monitor participant safety and efficacy or other criteria to be evaluated. Each protocol is submitted to the FDA as part of the IND. Companies also must generally determine the details of properly treating pediatric patients with a drug and this can sometimes mean that specific pediatric studies must be performed. Human clinical trials are typically conducted in the following four sequential phases that may overlap or be combined: | |
| • | In Phase 1, the drug is initially introduced into a small number of individuals and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. Phase I clinical trials are often conducted in healthy human volunteers and such cases do not provide evidence of efficacy. In case of severe or life-threatening diseases, the initial human testing is often conducted in patients rather than healthy volunteers. Because these patients already have the target disease, these studies may provide initial evidence of efficacy traditionally obtained in Phase 2 clinical trials. Consequently, these types of trials are frequently referred to as Phase 1/2 clinical trials. The FDA receives reports on the progress of each phase of clinical testing and it may require the modification, suspension or termination of clinical trials if it concludes that an unwarranted risk is presented to patients or healthy volunteers. | |
| • | Phase 2 involves clinical trials in a limited patient population to further identify any possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. | |
| • | Phase 3 clinical trials are undertaken to further evaluate dosage, clinical efficacy and to further test for safety in an expanded patient population at geographically dispersed clinical study sites. Phase 3 clinical trials usually include a broader patient population so that safety and efficacy can be substantially established. |
20
Table of Contents
| Phase 3 clinical trials begin once Phase 2 evaluations demonstrate that a dosage range of the product may be effective and has an acceptable safety profile. |
| • | Phase 4 clinical trials are performed if the FDA requires, or a company pursues, additional clinical trials after a product is approved. These clinical trials may be made a condition to be satisfied after a drug receives approval. The results of Phase 4 clinical trials can confirm the effectiveness of a product candidate and can provide important safety information to augment the FDA’s voluntary adverse drug reaction reporting system. | |
| • | Concurrent with clinical trials and preclinical studies, companies also must develop information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in accordance with drug cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product and the manufacturer must develop methods for testing the quality, purity, and potency of the final products. Additionally, appropriate packaging must be selected and tested and chemistry stability studies must be conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life. | |
| • | Submission to the FDA of a new drug application, or NDA, for non-biological drugs such as insulin, based on the clinical trials. The results of pharmaceutical development, preclinical studies, and clinical trials are submitted to the FDA in the form of an NDA for approval of the marketing and commercial shipment of the product. Under the Pediatric Research Equity Act of 2003, or PREA, NDAs are required to include an assessment, generally based on clinical study data, of the safety and efficacy of drugs for all relevant pediatric populations. The statute provides for waivers or deferrals in certain situations but we can make no assurances that such situations will apply to us or our product candidates. | |
| • | The FDA reviews all NDAs submitted before it accepts them for filing. It may request additional information rather than accepting an application for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA. Also, as part of this review, the FDA may refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation. The FDA is not bound by the recommendation of an advisory committee. Manufacturing establishments often also are subject to inspections prior to NDA approval to assure compliance with cGMP and with manufacturing commitments made in the relevant marketing application. Under the Prescription Drug User Fee Act, or PDUFA, certain pharmaceutical establishment, product and application fees were established. Submission of an NDA with clinical data requires payment of a fee. For fiscal year 2005, the required fee is $672,000. Under PDUFA, the FDA assigns a review goal for standard applications of 10 months from acceptance of the application, at which time the FDA may approve the product or request additional information. It is not unusual for the FDA to ask for more information upon completion of this first review cycle. There can be no assurance that an application will be approved during the first review cycle or any subsequent review cycles or that the FDA may not extend the PDUFA deadlines. | |
| • | FDA approval of the NDA must be granted prior to any commercial sale or shipment of the drug product. The FDA may deny an NDA approval if safety, efficacy or other regulatory requirements are not satisfied. The FDA may also require additional testing or information before approving the NDA. If regulatory approval of the product is granted, such approval may require post-marketing testing and surveillance to monitor the safety of the product or may entail limitations on the indicated uses for which the product may be marketed or advertised. The FDA may require additional testing or information before approving the NDA. In addition, product approval may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following the commencement of marketing. |
Clinical trials are designed and conducted in a variety of ways. A “placebo-controlled” trial is one in which the trial tests the results of a group of patients, referred to as an “arm” of the trial, receiving the drug being tested against those of an arm that receives a placebo, which is a substance of identical appearance that is not therapeutic in a medical or chemical sense. In a “double-blind” study, neither the researcher nor the patient knows into which arm of
21
Table of Contents
the trial the patient has been placed, or whether the patient is receiving the drug or the placebo. “Randomized” means that upon enrollment, patients are placed into one arm or the other at random by computer. “Parallel control” trials generally involve a study arm with a patient population that is not exposed to the study medication (i.e., is either on placebo or standard treatment protocols) for comparison to the drug being tested and groups are assigned upon patient admission to the study and remain in those groups for the duration of the study. An “open label” study is one where the researcher and the patient know that the patient is receiving the drug. A trial is said to be “pivotal” if it is designed to meet statistical criteria with respect to pre-determined “endpoints,” or clinical objectives, that the sponsor believes, based usually on its interactions with the relevant regulatory authority, will be sufficient for regulatory approval. In most cases, at least two “pivotal” Phase 3 clinical trials are necessary for approval. Claims regarding the superiority of one drug product over another generally must be supported by so-calledhead-to-head clinical trials involving comparison of one drug to another to which superiority is claimed. Under the PREA of 2003, an NDA also must include an assessment, generally based on clinical study data, on the safety and efficacy of a drug for all relevant pediatric populations. The statute provides for waivers or deferrals in certain situations but we can make no assurances that such situations will apply to us or our product candidates.
Medical products containing a combination of new drugs, biological products, or medical devices may be regulated as “combination products” in the United States. A combination product generally is defined as a product comprised of components from two or more regulatory categories (e.g., drug/device, device/biologic, drug/biologic). Each component of a combination product is subject to the requirements established by the FDA for that type of component, whether a new drug, biologic, or device. In order to facilitate premarket review of combination products, the FDA designates one of its centers to have primary jurisdiction for the premarket review and regulation of both components. The determination whether a product is a combination product or two separate products is made by the FDA on acase-by-case basis. We have had discussions with the FDA about the status of our Technosphere Insulin System as a combination product and we have been told that the FDA considers our product a combination drug/device. There have been some indications from the FDA that the review of a future drug marketing application for our Technosphere Insulin System will involve three separate review groups of the FDA: (1) the Metabolism and Endocrine Drug Products Division; (2) the Pulmonary Drug Products Division; and (3) the Center for Devices and Radiological Health within the FDA that reviews Medical Devices. Although the FDA has not made an official final decision in this regard, we currently understand that the Metabolic and Endocrine Drug Products Division will be the lead group and obtain consulting reviews from the other two FDA groups.
The testing and approval process requires substantial time, effort and financial resources. We cannot be certain that any approval of our products will be granted on a timely basis, if at all. If any of our products are approved for marketing by the FDA, we will be subject to continuing regulation by the FDA, including record-keeping requirements, reporting of adverse experiences with the product, submitting other periodic reports, drug sampling and distribution requirements, notifying the FDA and gaining its approval of certain manufacturing or labeling changes, and complying with certain electronic records and signature requirements. Prior to and following approval, if granted, all manufacturing sites are subject to inspection by the FDA and other national regulatory bodies and must comply with cGMP, QSR and other requirements enforced by the FDA and other national regulatory bodies through their facilities inspection program. Foreign manufacturing establishments must comply with similar regulations. In addition, our drug-manufacturing facilities located in Danbury and the facilities of our insulin supplier and the supplier of our MedTone inhaler and cartridges are subject to federal registration and listing requirements and, if applicable, to state licensing requirements. Facilities are subject to inspection by the FDA and similar national agencies, as well as state and local authorities at any time. Failure, including those of our insulin and MedTone inhaler suppliers, to obtain and maintain applicable federal registrations or state licenses, or to meet the inspection criteria of the FDA or the other national regulatory bodies, would disrupt our manufacturing processes and would harm our business. In complying with standards set forth in these regulations, manufacturers must continue to expend time, money and effort in the area of production and quality control to ensure full compliance. Currently, we believe we are operating under all of the necessary guidelines and permits.
It is not yet clear to what extent we will be subject to regulations governing premarket approval or clearances of medical devices separate from approval requirements governing drugs. We currently expect that our inhaler will be approved as part of the NDA for our Technosphere Insulin System. No assurances exist that we will not be required to obtain separate device clearances or approval for use of our inhaler with our Technosphere Insulin System. This
22
Table of Contents
may result in our being subject to medical device review user fees and to other device requirements to market our inhaler and may result in significant delays in commercialization. Even if the device component is approved as part of our NDA for the Technosphere Insulin System, numerous device regulatory requirements still apply to the device part of the drug-device combination. These include:
| • | product labeling regulations; | |
| • | general prohibition against promoting products for unapproved or “off-label” uses; | |
| • | corrections and removals (e.g., recalls); | |
| • | establishment registration and device listing; | |
| • | general prohibitions against the manufacture and distribution of adulterated and misbranded devices; and | |
| • | the Medical Device Reporting regulation, which requires that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur. Failure to comply with these regulatory requirements could result in civil fines, product seizures, injunctions,and/or criminal prosecution of responsible individuals and us. Further, the company we have contracted to manufacture our MedTone inhaler and cartridges will be subject to the QSRs, which require manufacturers to follow elaborate design, testing, control, documentation and other quality assurance procedures during the manufacturing process of medical devices, among other requirements. |
Failure to adhere to regulatory requirements at any stage of development, including the preclinical and clinical testing process, the review process, or at any time afterward, including after approval, may result in various adverse consequences. These consequences include action by the FDA or other national regulatory body that has the effect of delaying approval or refusing to approve a product; suspending or withdrawing an approved product from the market; seizing or recalling a product; or imposing criminal penalties against the manufacturer. In addition, later discovery of previously unknown problems may result in restrictions on a product, its manufacturer, or the NDA holder, or market restrictions through labeling changes or product withdrawal. Also, new government requirements may be established or they may change at any time that could delay or prevent regulatory approval of our products under development. For example, in response to recent events regarding questions about the safety of certain approved prescription products, including the lack of adequate warnings, the FDA and Congress are currently considering new regulatory and legislative approaches to advertising, monitoring and assessing the safety of marketed drugs, including legislation providing the FDA with authority to mandate labeling changes for approved pharmaceutical products, particularly those related to safety. We also cannot be sure that the current Congressional and FDA initiatives pertaining to ensuring the safety of marketed drugs or other developments pertaining to the pharmaceutical industry will not adversely affect our operations. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
In addition, the FDA imposes a number of complex regulations on entities that advertise and promote drugs, which include, among other requirements, standards for and regulations ofdirect-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. Such advertising and promotional activities are also being scrutinized by the FDA and Congress as a result of recent concerns that have been raised about the safety of marketed drugs. The FDA has very broad enforcement authority under the FDCA, and failure to comply with these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from FDA standards, a requirement that future advertising and promotional materials be pre-cleared by the FDA, and state and federal civil and criminal investigations and prosecutions.
Products manufactured in the United States and marketed outside the United States are subject to certain FDA regulations, as well as regulation by the country in which the products are to be sold. We also would be subject to foreign regulatory requirements governing clinical trials and drug product sales if products are marketed abroad. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries usually must be obtained prior to the marketing of the product in those countries. The approval
23
Table of Contents
process varies from jurisdiction to jurisdiction and the time required may be longer or shorter than that required for FDA approval.
Product development and approval within this regulatory framework take a number of years, involve the expenditure of substantial resources and are uncertain. Many drug products ultimately do not reach the market because they are not found to be safe or effective or cannot meet the FDA’s other regulatory requirements. In addition, there can be no assurance that the current regulatory framework will not change or that additional regulation will not arise at any stage of our product development that may affect approval, delay the submission or review of an application or require additional expenditures by us. There can be no assurance that we will be able to obtain necessary regulatory clearances or approvals on a timely basis, if at all, for any of our product candidates under development, and delays in receipt or failure to receive such clearances or approvals, the loss of previously received clearances or approvals, or failure to comply with existing or future regulatory requirements could have a material adverse effect on our business and results of operations.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this latter procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the application and assessment report, each member state must decide whether to recognize approval. We plan to choose the appropriate route of European regulatory filing in an attempt to accomplish the most rapid regulatory approvals. However, the chosen regulatory strategy may not secure regulatory approvals or approvals of the chosen product indications. In addition, these approvals, if obtained, may take longer than anticipated.
We cannot assure you that any of our product candidates will prove to be safe or effective, will receive regulatory approvals, or will be successfully commercialized.
In addition to the foregoing, we are subject to numerous federal, state and local laws relating to such matters as laboratory practices, the experimental use of animals, the use and disposal of hazardous or potentially hazardous substances, controlled drug substances, safe working conditions, manufacturing practices, environmental protection and fire hazard control. We may incur significant costs to comply with those laws and regulations now or in the future.
Patent restoration and marketing exclusivity
The Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, permits the FDA to approve abbreviated NDAs, or ANDAs, for generic versions of innovator drugs and also provides certain patent restoration and market exclusivity protections to innovator drug manufacturers. The ANDA process permits competitor companies to obtain marketing approval for a new drug with the same active ingredient for the same uses, dosage form and strength as an innovator drug but does not require the conduct and submission of clinical studies demonstrating safety and efficacy for that product. Instead of providing completely new safety and efficacy data, therefore, a competitor could make copies of such drugs and only need to submit manufacturing information and clinical data demonstrating that the copy is bioequivalent to the innovator’s product in order to gain marketing approval from the FDA.
Another type of application allowed by the Hatch-Waxman Act, a Section 505(b)(2) application, may be permitted where a company does not own or have a right to reference all the data required for approval. Section 505(b)(2) NDAs are often submitted for drug products that contain the same active ingredient as those in first approved drug products and where additional studies are required for approval, such as for changes in routes of administration or dosage forms.
Hatch-Waxman requires a competitor that submits an ANDA or a Section 505(b)(2) application with respect to a drug to notify the innovator company of its application and potential infringement of patent rights. Hatch-Waxman places certain timing requirements on us with respect to filing an infringement action against such an applicant. The timing of competitive FDA approvals of ANDAs or Section 505(b)(2) applications is related to the patent status of the approved products, if any.
24
Table of Contents
While the Hatch-Waxman Act provides competitors the ability to market copies of approved innovator drug products with the submission of significantly less clinical data, depending on the patent status of the innovator, the Act also provides under certain circumstances for the restoration of a portion of a product’s patent term that is lost during a drug’s clinical development and NDA review by the FDA. Hatch-Waxman also provides for a statutory protection, known as market exclusivity, which prohibits the FDA’s approval or acceptance of certain competitor new drug applications. Patent term restoration can return up to five years of patent term for a patent that covers a new drug product or its use to compensate for time lost during the regulatory review process. This period is generally one-half the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application, subject to a maximum extension of five years. No extension can extend the total patent life beyond 14 years after the drug approval date. The application for patent term extension is subject to approval by the USPTO, in conjunction with the FDA. It takes at least six months to obtain approval of the application for patent term extension, and there can be no guarantee that the application will be granted.
The Hatch-Waxman Act also provides for differing periods of marketing exclusivity for new drugs approved under NDAs. Among the types of exclusivity are those for “new chemical entities” and those for innovative changes to previously approved drugs, where new clinical trials (other than bioavailability studies) are essential to the approval of the NDAs. Our lead product, the Technosphere Insulin System, is an innovative change to a previously approved drug with the same active ingredient, insulin. Marketing exclusivity for the Technosphere Insulin System, if available and granted by the FDA, would prohibit the agency for a period of three years from approving an ANDA or Section 505(b)(2) application for competitive versions of our new formulation. This three-year exclusivity, however, only covers the innovation associated with the NDA to which it attaches. Thus, the three-year exclusivity would not prohibit the FDA from approving ANDAs or Section 505(b)(2) applications for drugs containing the same active ingredient but without our new dosage formulation. Hatch-Waxman also does not prevent the approval of a full NDA containing all the safety and efficacy data and information required for approval, even where approval of the same drug would have been blocked under an ANDA or Section 505(b)(2) application.
The FDA Modernization Act of 1997 included a pediatric exclusivity provision that was extended by the Best Pharmaceuticals for Children Act of 2002. Pediatric exclusivity is designed to provide an incentive to manufacturers for conducting research about the safety and efficacy of their products in children. Pediatric exclusivity, if granted, provides an additional six months of market exclusivity in the United States for new or currently marketed drugs if certain pediatric studies requested by the FDA are completed by the applicant. To obtain this additional six months of exclusivity, it would be necessary for us to first receive a written request from the FDA to conduct pediatric studies and then to conduct the requested studies according to a previously agreed timeframe and submit the report of the study. There can be no assurances that we would receive a written request from the FDA and if so that we would complete the studies in accordance with the requirements for this six-month exclusivity. The current pediatric exclusivity provision is scheduled to end on October 1, 2007, and there can be no assurances that it will be reauthorized.
EMPLOYEES
As of December 31, 2005, we had 428 full-time employees, all of whom are employed at-will. Ninety-four of these employees were engaged in research and development, 119 in manufacturing, 128 in clinical, regulatory affairs and quality assurance and 87 in administration, finance, management, information systems, corporate development and human resources. Thirty-eight of these employees have a Ph.D. degreeand/or M.D. degree and are engaged in activities relating to research and development, manufacturing, quality assurance and business development. None of our employees is subject to a collective bargaining agreement. We believe relations with our employees are good.
SCIENTIFIC ADVISORS
We seek advice from a number of leading scientists and physicians on scientific, technical and medical matters. These advisors are leading scientists in the areas of pharmacology, chemistry, immunology and biology. Our scientific advisors are consulted regularly to assess, among other things:
| • | our research and development programs; |
25
Table of Contents
| • | the design and implementation of our clinical programs; | |
| • | our patent and publication strategies; | |
| • | market opportunities from a clinical perspective; | |
| • | new technologies relevant to our research and development programs; and | |
| • | specific scientific and technical issues relevant to our business. |
The following are our scientific advisors and their primary affiliations:
Name | Primary Affiliation | |
| Richard Bergenstal, M.D. | International Diabetes Center | |
| William Boswell, M.D., FACR | University of Southern California School of Medicine | |
| James Bryan, M.D. | Pharmaceutical Product Development, Inc. | |
| Alexander Fleming, M.D. | Kinexum Corporation | |
| Martin Kast, Ph.D. | Norris Comprehensive Cancer Center, University of Southern California | |
| Thomas Kundig, M.D. | University of Zurich | |
| Harold E. Lebovitz, M.D., FACE | State University of New York — Brooklyn | |
| Frederick Levy, Ph.D. | Ludwig Institute for Cancer Research | |
| Robert Morgan, Jr., M.D., FACP | City of Hope National Medical Center | |
| Robert Ozols, M.D., Ph.D. | Fox Chase Cancer Center | |
| Daniel Porte, M.D. | University of California, San Diego | |
| Philip Raskin, M.D., FACE, FACP | University of Texas Southwestern Medical Center | |
| Robert Rizza, M.D. | Mayo Clinic | |
| Julio Rosenstock, M.D. | Dallas Diabetes and Endocrine Center | |
| Jesse Roth, M.D. | North Shore Long Island Jewish Medical Center | |
| Jay S. Skyler, M.D. | University of Miami Diabetes Research Institute | |
| Rolf Zinkernagel, M.D., Ph.D., Nobel Laureate | University of Zurich | |
| Bernie Zinman, M.D. | Mount Sinai Hospital, Toronto |
EXECUTIVE OFFICERS
The following table sets forth our current executive officers and their ages as of December 31, 2005:
Name | Age | Position(s) | ||||
| Alfred E. Mann | 80 | Chairman of the Board of Directors and Chief Executive Officer | ||||
| Hakan S. Edstrom | 55 | President, Chief Operating Officer and Director | ||||
| Richard L. Anderson | 66 | Corporate Vice President and Chief Financial Officer | ||||
| Peter C. Richardson | 46 | Corporate Vice President and Chief Scientific Officer | ||||
| Dan R. Burns | 54 | Corporate Vice President and President, Commercial Operations and Business Development | ||||
| Juergen A. Martens, Ph.D. | 50 | Corporate Vice President, Operations | ||||
| David Thomson, Ph.D., J.D. | 39 | Corporate Vice President, General Counsel and Corporate Secretary | ||||
| Diane M. Palumbo | 52 | Corporate Vice President, Human Resources | ||||
26
Table of Contents
Alfred E. Mannhas been one of our directors since April 1999, our Chairman of the Board since December 2001 and our Chief Executive Officer since October 2003. He founded and formerly served as Chairman and Chief Executive Officer of MiniMed, Inc., a publicly traded company focused on diabetes therapy and microinfusion drug delivery that was acquired by Medtronic, Inc. in August 2001. Mr. Mann also founded and, from 1972 through 1992, served as Chief Executive Officer of Pacesetter Systems, Inc. and its successor, Siemens Pacesetter, Inc., a manufacturer of cardiac pacemakers, now the Cardiac Rhythm Management Division of St. Jude Medical Corporation. Mr. Mann founded and since 1993, has served as Chairman and Co-Chief Executive Officer of Advanced Bionics Corporation, a medical device manufacturer focused on neurostimulation to restore hearing to the deaf and to treat chronic pain and other neural deficits, that was acquired by Boston Scientific Corporation in June 2004. Mr. Mann has also founded and is non-executive Chairman of Second Sight, which is developing a visual prosthesis for the blind and Quallion, which produces batteries for medical products and for the military and aerospace industries. Mr. Mann is also non-executive Chairman of the Alfred Mann Foundation and Alfred Mann Institute at the University of Southern California, and the Alfred Mann Foundation for Biomedical Engineering, which is establishing additional institutes at other research universities. Mr. Mann holds a bachelor’s and master’s degree in Physics from the University of California at Los Angeles, honorary doctorates from Johns Hopkins University, the University of Southern California, Western University and the Technion-Israel Institute of Technology and is a member of the National Academy of Engineering.
Hakan S. Edstromhas been our President and Chief Operating Officer since April 2001 and has served as one of our directors since December 2001. Mr. Edstrom was with Bausch & Lomb, Inc., a health care product company, from January 1998 to April 2001, advancing to the position of Senior Corporate Vice President and President of Bausch & Lomb, Inc. Americas Region. From 1981 to 1997, Mr. Edstrom was with Pharmacia Corporation, where he held various executive positions, including President and Chief Executive Officer of Pharmacia Opthalmics Inc. Mr. Edstrom is currently a director of Q-Med AB, a biotechnology and medical device company, and Ixion Biotechnology, Inc., a biotechnology company. Mr. Edstrom was educated in Sweden and holds a master’s degree in business administration from the Stockholm School of Economics.
Richard L. Andersonhas been our Corporate Vice President and Chief Financial Officer since October 2002. . From January 1997 to September 2002, Mr. Anderson held various executive positions at NeoRx Corporation, a Seattle-based publicly traded biotechnology company, including President, Chief Operating Officer, Chief Financial Officer and Senior Vice President, Finance and Operations. Mr. Anderson holds a master’s degree in Management from Johns Hopkins University, a master’s degree in solid state physics from the University of Maryland and a bachelor’s degree in physics from Bucknell University
Peter C. Richardson has been our Corporate Vice President and Chief Scientific Officer since October 2005. From 1991 to October 2005, he was employed by Novartis Pharmaceuticals Corporation, which is the U.S. affiliate of Novartis AG, a world leader in healthcare, most recently as Senior Vice President, Global Head of Development Alliances. From 2003 until 2005, he was Senior Vice President and Head of Development of Novartis Pharmaceuticals KK Japan. He earlier practiced as an endocrinologist. Dr. Richardson holds a B.Med.Sci (Hons.) and a BM.BS (Hons.) from University of Nottingham Medical School; an MRCP (UK) from the Royal College of Physicians, UK; a Certificate in Pharmaceutical Medicine from Universities of Freibourg, Strasbourg and Basle; and a Diploma in Pharmaceutical Medicine from the Royal College of Physicians Faculty of Pharmaceutical Medicine.
Dan R. Burnshas been our Corporate Vice President and President, Commercial Operations and Business Development since September 2002. Prior to joining us, he served as Chief Executive Officer of HealthTalk Interactive, a pharmaceutical services firm, from 2000 to 2002. From 1998 to 1999 Mr. Burns served as Chief Executive Officer of ProScript, a biopharmaceutical company. He served as President and Chief Operating Officer of Trophix Pharmaceuticals, Inc. from 1997 to 1998. Prior to joining Trophix Pharmaceuticals, for 18 years, Mr. Burns held a number of senior executive positions both internationally and domestically with Bristol Myers Squibb. Mr. Burns holds degrees in psychology and business administration from McMaster University and Mohawk College.
Juergen A. Martens, Ph.D. has been our Corporate Vice President of Operations since September 2005. From 2000 to August 2005, he was employed by Nektar Therapeutics, Inc., most recently as Vice President of Pharmaceutical Technology Development Previously, he held technical management positions at Aerojet Fine Chemicals from
27
Table of Contents
1998 to 2000 and at FMC Corporation from 1996 to 1998. From 1987 to 1996, Dr. Martens held a variety of management positions with increased responsibility in R&D, plant management, and business process development at Lonza, in Switzerland and in the United States. Dr. Martens holds a BS in chemical engineering from the Technical College Mannheim/Germany, a BS/MS in chemistry and a doctorate in physical chemistry from the University of Marburg/Germany.
David Thomson, Ph.D., J.D. has been our Corporate Vice President, General Counsel and Corporate Secretary since January 2002. Prior to joining us, he practiced corporate/commercial and securities law at the Toronto law firm of Davies Ward Phillips & Vineberg LLP from May 1999 through December 2001, except for a period from May to December 2000, when he served as Vice President, Business Development for CTL ImmunoTherapies Corp. From March 1994 to August 1996, Dr. Thomson held a post-doctoral position at the Rockefeller University, where he conducted medical research in the Laboratory of Neurophysiology. Dr. Thomson obtained his bachelor’s degree, master’s degree and Ph.D. degree from Queens University and obtained his J.D. degree from the University of Toronto.
Diane M. Palumbohas been our Corporate Vice President of Human Resources since November 2004. From July 2003 to November 2004, she was President of her own human resources consulting company. From June 1991 to July 2003, Ms. Palumbo held various positions with Amgen, Inc., a California-based biopharmaceutical company, including Senior Director, Human Resources. In addition, Ms. Palumbo has held Human Resources positions with Unisys and Mitsui Bank Ltd. of Tokyo. She holds a master’s degree in business administration from St. John’s University, NY and a bachelor of science degree, magna cum laude, also from St. John’s University, NY.
Executive officers serve at the discretion of the Board of Directors. There are no family relationships between any of the directors and executive officers of MannKind.
Item 1A. Risk Factors
You should consider carefully the following information about the risks described below, together with the other information contained in this report, before you decide to buy or maintain an investment in our common stock. We believe the risks described below are the risks that are material to us as of the date of this annual report. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also affect our business. If any of the following risks actually occur, our business, financial condition, results of operations and future growth prospects would likely be materially and adversely affected. In these circumstances, the market price of our common stock could decline, and you may lose all or part of the money you paid to buy our common stock.
RISKS RELATED TO OUR BUSINESS
We have a history of operating losses, we expect to continue to incur losses, and we may never become profitable.
We are a development stage company with no commercial products. All of our product candidates are still being developed, and all but our Technosphere Insulin System are still in early stages of development. Our product candidates will require significant additional development, clinical trials, regulatory clearances and additional investment before they can be commercialized. We anticipate that our Technosphere Insulin System will not be commercially available for several years, if at all.
We have never been profitable, and, as of December 31, 2005, we had an accumulated deficit of $557.3 million. The accumulated deficit has resulted principally from costs incurred in our research and development programs, the write-off of goodwill and general operating expenses. We expect to make substantial expenditures and to incur increasing operating losses in the future in order to further develop and commercialize our product candidates, including costs and expenses to complete clinical trials, seek regulatory approvals and market our product candidates. This accumulated deficit may increase significantly as we expand development and clinical trial efforts.
Our losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and stockholders’ equity. Our ability to achieve and sustain profitability depends upon obtaining regulatory approvals for and successfully commercializing our Technosphere Insulin System, either alone or with third parties. We do not currently have the required approvals to market any of our product candidates, and we may not receive them. We
28
Table of Contents
may not be profitable even if we succeed in commercializing any of our product candidates. As a result, we cannot be sure when we will become profitable, if at all.
If we fail to raise additional capital, our financial condition and business would suffer.
It is costly to develop therapeutic products and conduct clinical trials for these products. Although we currently are focusing on our Technosphere Insulin System as our lead product candidate, we may in the future conduct clinical trials for a number of additional product candidates. Our future revenues may not be sufficient to support the expense of these activities.
Based upon our current expectations, we believe that our existing capital resources, including the net proceeds from our private placement in August 2005, will enable us to continue planned operations into the third quarter of 2006. However, we cannot assure you that our plans will not change or that changed circumstances will not result in the depletion of our capital resources more rapidly than we currently anticipate. Accordingly, we expect that we will need to raise additional capital, either through the sale of equityand/or debt securities, a strategic business collaboration or the establishment of other funding facilities, in order to continue the development and commercialization of our Technosphere Insulin System and other product candidates and to support our other ongoing activities. The amount of additional funds we need will depend on a number of factors, including:
| • | the rate of progress and costs of our clinical trials and research and development activities, including costs of procuring clinical materials and expanding our own manufacturing facilities; |
| • | our success in establishing strategic business collaborations and the timing and amount of any payments we might receive from any collaboration we are able to establish; |
| • | actions taken by the FDA and other regulatory authorities affecting our products and competitive products; |
| • | our degree of success in commercializing our Technosphere Insulin System or our other product candidates; |
| • | the emergence of competing technologies and products and other adverse market developments; |
| • | the timing and amount of payments we might receive from potential licensees; |
| • | the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims and other intellectual property rights or defending against claims of infringement by others; and |
| • | the costs of discontinuing projects and technologies or decommissioning existing facilities, if we undertake those activities. |
We have raised capital in the past primarily through the sale of equity securities. We may in the future pursue the sale of equityand/or debt securities, or the establishment of other funding facilities. Issuances of debt or additional equity could impact your rights as a holder of our common stock, may dilute your ownership percentage and may impose restrictions on our operations. These restrictions could include limitations on additional borrowing and specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens, pay dividends, redeem our stock or make investments.
We also may seek to raise additional capital by pursuing opportunities for the licensing, sale or divestiture of certain intellectual property and other assets, including our Technosphere technology platform. We cannot offer assurances, however, that any strategic collaborations, sales of securities or sale or license of assets will be available to us on a timely basis or on acceptable terms, if at all. We may be required to enter into relationships with third parties to develop or commercialize products or technologies that we otherwise would have sought to develop independently, and any such relationships may not be on terms as commercially favorable to us as might otherwise be the case.
In the event that sufficient additional funds are not obtained through strategic collaboration opportunities, licensing arrangements, sales of securitiesand/or asset sales on a timely basis, we may be required to reduce expenses through the delay, reduction or curtailment of our projects, including our Technosphere Insulin System development activities, or further reduction of costs for facilities and administration.
29
Table of Contents
We depend heavily on the successful development and commercialization of our lead product candidate, the Technosphere Insulin System, which is still under development, and our other product candidates, which are in preclinical development.
To date, we have not completed the development of any products through to commercialization. Only our Technosphere Insulin System is currently undergoing clinical trials, while our other product candidates are in research or preclinical development. We anticipate that in the near term our ability to generate revenues will depend solely on the successful development and commercialization of our Technosphere Insulin System.
We have expended significant time, money and effort in the development of our lead product candidate, the Technosphere Insulin System, which has not yet received regulatory approval and which may never be commercialized. Before we can market and sell our Technosphere Insulin System, we will need to advance our Technosphere Insulin System through Phase 3 clinical trials and demonstrate in these trials that our Technosphere Insulin System is safe and effective. We currently anticipate conducting several pivotal Phase 3 clinical trials as well as several special population studies involving, in total, more than 3,200 patients, which will require additional time and substantial expenditure of resources. We must also receive the necessary approvals from the FDA and similar foreign regulatory agencies before this product can be marketed in the United States or elsewhere. Even if we were to receive regulatory approval, we ultimately may be unable to gain market acceptance of our Technosphere Insulin System for a variety of reasons, including the treatment and dosage regimen, potential adverse effects, the availability of alternative treatments and cost effectiveness. If we fail to commercialize our Technosphere Insulin System, our business, financial condition and results of operations will be materially and adversely affected.
We are seeking to develop and expand our portfolio of product candidates through our internal research programs and through licensing or otherwise acquiring the rights to therapeutics in the areas of cancer and other areas. All of these product candidates will require additional research and development and significant preclinical, clinical and other testing prior to seeking regulatory approval to market them. Accordingly, these product candidates will not be commercially available for a number of years, if at all.
A significant portion of the research that we are conducting involves new and unproven compounds and technologies, including our Technosphere Insulin System, Technosphere platform technology and immunotherapy product candidates. Research programs to identify new product candidates require substantial technical, financial and human resources. Even if our research programs identify candidates that initially show promise, these candidates may fail to progress to clinical development for any number of reasons, including discovery upon further research that these candidates have adverse effects or other characteristics that indicate they are unlikely to be effective. In addition, the clinical results we obtain at one stage are not necessarily indicative of future testing results. If we fail to successfully complete the development and commercialization of our Technosphere Insulin System or develop or expand our other product candidates, or are significantly delayed in doing so, our business and results of operations will be harmed and the value of our stock could decline.
If we do not achieve our projected development goals in the timeframes we announce and expect, our business would be harmed and the market price of our common stock could decline.
For planning purposes, we estimate the timing of the accomplishment of various scientific, clinical, regulatory and other product development goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of scientific studies and clinical trials and the submission of regulatory filings. From time to time, we publicly announce the expected timing of some of these milestones. All of these milestones are based on a variety of assumptions. The actual timing of the achievement of these milestones can vary dramatically compared to our estimates — in many cases for reasons beyond our control — depending on numerous factors, including:
| • | the rate of progress, costs and results of our clinical trial and research and development activities, which will be impacted by the level of proficiency and experience of our clinical staff; | |
| • | our ability to identify and enroll patients who meet clinical trial eligibility criteria; | |
| • | our ability to access sufficient, reliable and affordable supplies of components used in the manufacture of our product candidates, including insulin and other materials for our Technosphere Insulin System; |
30
Table of Contents
| • | the costs of expanding and maintaining manufacturing operations, as necessary; | |
| • | the extent of scheduling conflicts with participating clinicians and clinical institutions; | |
| • | the receipt of approvals by our competitors and by us from the FDA and other regulatory agencies; and | |
| • | other actions by regulators. |
In addition, if we do not obtain sufficient additional funds through sales of securities, strategic collaborations or the sale or license of our assets on a timely basis, we may be required to reduce expenses by delaying, reducing or curtailing our Technosphere Insulin System or other product development activities, which would impact our ability to meet milestones. If we fail to commence or complete, or experience delays in or are forced to curtail, our proposed clinical programs or otherwise fail to adhere to our projected development goals in the timeframes we announce and expect, our business and results of operations will be harmed and the market price of our common stock may decline.
We face substantial competition in the development of our product candidates and may not be able to compete successfully, and our product candidates may be rendered obsolete by rapid technological change.
We initially are focusing on the development of the Technosphere Insulin System for the treatment of diabetes, and we face intense competition in this area. In January 2006, the FDA and the European Commission approved Exubera, developed by Pfizer, Inc. in collaboration with Nektar Therapeutics, for the treatment of adults with type 1 and type 2 diabetes. Eli Lilly and Company, in collaboration with Alkermes, Inc., initiated a Phase 3 clinical trial in July 2005, required for registration of their inhaled insulin system. Novo Nordisk A.S. has announced their intention to re-initiate Phase 3 clinical trials of their pulmonary insulin product. In addition, a number of established pharmaceutical companies have or are developing technologies for the treatment of diabetes. We also face substantial competition for the development of our other product candidates.
Many of our existing or potential competitors have, or have access to, substantially greater financial, research and development, production, and sales and marketing resources than we do and have a greater depth and number of experienced managers. As a result, our competitors may be better equipped than we are to develop, manufacture, market and sell competing products.
The rapid rate of scientific discoveries and technological changes could result in one or more of our products becoming obsolete or noncompetitive. Our competitors may develop or introduce new products that render our technology and our Technosphere Insulin System less competitive, uneconomical or obsolete. Pfizer, the first to commercialize a pulmonary insulin system, will have an advantage in being able to gain reputation and market share as well as set parameters for the pulmonary insulin market such as pricing. Our future success will depend not only on our ability to develop our products but to improve them and to keep pace with emerging industry developments. We cannot assure you that we will be able to do so.
We also expect to face increasing competition from universities and other non-profit research organizations. These institutions carry out a significant amount of research and development in the areas of diabetes and cancer. These institutions are becoming increasingly aware of the commercial value of their findings and are more active in seeking patent and other proprietary rights as well as licensing revenues.
If we fail to enter into a strategic collaboration with respect to our Technosphere Insulin System, we may not be able to execute on our business model.
We are currently evaluating potential collaborations with respect to our Technosphere Insulin System. If we are not able to enter into a collaboration on terms that are favorable to us, we could be required to undertake and fund product development, clinical trials, manufacturing and marketing activities solely at our own expense. We currently estimate that the cost to continue the development of the Technosphere Insulin System over the next 12 months would be up to $200 million. However, this estimate may change based on how the program proceeds. Failure to enter into a collaboration with respect to our Technosphere Insulin System could substantially increase our requirements for capital, which might not be available on favorable terms, if at all. Alternatively, we would have
31
Table of Contents
to substantially reduce our development efforts, which would delay or otherwise impede the commercialization of our Technosphere Insulin System.
We will face similar challenges as we seek to develop our other product candidates. Our current strategy for developing, manufacturing and commercializing our other product candidates includes evaluating the potential for collaborating with pharmaceutical and biotechnology companies at some point in the drug development process and for these collaborators to undertake the advanced clinical development and commercialization of our product candidates. It may be difficult for us to find third parties that are willing to enter into collaborations on economic terms that are favorable to us, or at all. Failure to enter into a collaboration with respect to any other product candidate could substantially increase our requirements for capital and force us to substantially reduce our development effort.
If we enter into collaborative agreements and if our third-party collaborators do not perform satisfactorily or if our collaborations fail, development or commercialization of our Technosphere Insulin System may be delayed and our business could be harmed.
We currently rely on clinical research organizations and hospitals to conduct, supervise or monitor some or all aspects of clinical trials involving our Technosphere Insulin System. Further, we may also enter into license agreements, partnerships or other collaborative arrangements to support financing, development and marketing of our Technosphere Insulin System. We may also license technology from others to enhance or supplement our technologies. These various collaborators may enter into arrangements that would make them potential competitors. These various collaborators also may breach their agreements with us and delay our progress or fail to perform under their agreements, which could harm our business.
If we enter into collaborative arrangements, we will have less control over the timing, planning and other aspects of our clinical trials, and the sale and marketing of our Technosphere Insulin System and our other product candidates. We cannot offer assurances that we will be able to enter into satisfactory arrangements with third parties as contemplated or that any of our existing or future collaborations will be successful.
Testing of our Technosphere Insulin System or another product candidate may not yield successful results, and even if it does, we may still be unable to commercialize that product candidate.
Our research and development programs are designed to test the safety and efficacy of our Technosphere Insulin System and our other product candidates through extensive preclinical and clinical testing. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of our Technosphere Insulin System or any of our other product candidates, including the following:
| • | safety and efficacy results obtained in our preclinical and initial clinical testing may be inconclusive or may not be predictive of results obtained in later-stage clinical trials or following long-term use, and we may as a result be forced to stop developing product candidates that we currently believe are important to our future; | |
| • | the data collected from clinical trials of our product candidates may not be sufficient to support FDA or other regulatory approval; | |
| • | after reviewing test results, we or any potential collaborators may abandon projects that we previously believed were promising; and | |
| • | our product candidates may not produce the desired effects or may result in adverse health effects or other characteristics that preclude regulatory approval or limit their commercial use if approved. |
We have initiated a pivotal Phase 3 safety study of our Technosphere Insulin System to evaluate pulmonary function over a period of two years. Our Technosphere Insulin System is intended for multiple uses per day. Due to the size and timeframe over which existing and planned clinical trials are conducted, the results of clinical trials, including our existing Phase 3 trials, may not be indicative of the effects of the use of our Technosphere Insulin System over longer terms. If long-term use of our Technosphere Insulin System results in adverse health effects or reduced efficacy or both, the FDA or other regulatory agencies may terminate our ability to market and sell our Technosphere Insulin System, may narrow the approved indications for use or otherwise require restrictive product
32
Table of Contents
labeling or marketing, or may require further clinical trials, which may be time-consuming and expensive, and may not produce favorable results.
As a result of any of these events, the FDA, other regulatory authorities, any collaborator or we may suspend or terminate clinical trials or marketing of our Technosphere Insulin System at any time. Any suspension or termination of our clinical trials or marketing activities may harm our business and results of operations and the market price of our common stock may decline.
If we are unable to transition successfully from an early-stage development company to a company that commercializes therapeutics, our operations would suffer.
We are reaching a critical juncture in our development, transitioning from an early-stage development company to one with multiple Phase 3 clinical trials. Phase 3 development of the Technosphere Insulin System is far more complex than the earlier phases. Overall, we plan to support a significant number of studies in the near term. We have not previously implemented the range of studies contemplated for our Phase 3 clinical program. Moreover, as a company, we have no previous experience in the Phase 3-through-new drug application, or NDA, stage of product development.
We require a well-structured plan to make this transition. In the past year, we have added a significant number of new executive personnel, particularly in clinical development, regulatory and manufacturing production, including personnel with significantPhase 3-to-commercialization experience. We have aligned our management structure to accommodate the increasing complexity of our operations, and we are implementing the following measures, among others, to accommodate our transition, complete development of our Technosphere Insulin System and successfully implement our commercialization strategy for our Technosphere Insulin System:
| • | expand our manufacturing capabilities; |
| • | develop comprehensive and detailed commercialization, clinical development and regulatory plans; and |
| • | implement standard operating procedures, including those for protocol development. |
If we are unable to accomplish these measures in a timely manner, we would be at considerable risk of failing to:
| • | complete our Phase 3 clinical trial program in a deliberate fashion, on time and within budget; and |
| • | develop through our Phase 3 trials the key clinical data needed to obtain regulatory approval and compete successfully in the marketplace. |
If our suppliers fail to deliver materials and services needed for the production of our Technosphere Insulin System in a timely and sufficient manner, or they fail to comply with applicable regulations, our business and results of operations would be harmed and the market price of our common stock could decline.
For our Technosphere Insulin System to be commercially viable, we need access to sufficient, reliable and affordable supplies of insulin, our MedTone inhaler, the related cartridges and other materials. We currently have a long-term supply agreement with Diosynth B.V., an independent supplier of insulin and a subsidiary of Akzo Nobel, which is currently our sole supplier for insulin. We are aware of at least five other suppliers of bulk insulin but to date we have not entered into a commercial relationship with any of the five. Currently we obtain our Technosphere pre-cursor raw material from Degussa AG, a major chemical manufacturer with facilities in Europe and North America. We utilize our in-house chemical manufacturing plant as a back up facility. Degussa AG has the capacity to supply our current clinical and future commercial requirements. We entered into a long-term supply agreement with Vaupell, Inc., the supplier of our MedTone inhaler and cartridges. We must rely on our suppliers to comply with relevant regulatory and other legal requirements, including the production of insulin in accordance with current drug Good Manufacturing Practices, or cGMP, and the production of MedTone inhaler and related cartridges in accordance with device Quality System Regulations, or QSR. The supply of all of these materials may be limited or the manufacturer may not meet relevant regulatory requirements, and if we are unable to obtain these materials in sufficient amounts, in a timely manner and at reasonable prices, or if we should encounter delays or difficulties in our relationships with manufacturers or suppliers, the development or manufacturing of our
33
Table of Contents
Technosphere Insulin System may be delayed. Any such events would delay the submission of our Technosphere Insulin System for regulatory approval or market introduction and subsequent sales and, if so, our business and results of operations will be harmed and the market price of our common stock may decline.
We have never manufactured our Technosphere Insulin System or any other product candidate in commercial quantities, and if we fail to develop an effective manufacturing capability for our product candidates or to engage third-party manufacturers with this capability, we may be unable to commercialize these products.
We currently obtain our Technosphere pre-cursor raw material primarily from Degussa AG. We use our Danbury, Connecticut facility to formulate Technosphere Insulin, fill plastic cartridges with Technosphere Insulin and blister package the cartridges for our clinical trials. We presently intend to increase our formulation, fill and finishing capabilities at Danbury in order to accommodate our activities through initial commercialization. This expansion will involve a number of third-party suppliers of equipment and materials as well as engineering and construction services. Our suppliers may not deliver all of the required equipment, materials and services in a timely manner or at reasonable prices. If we encounter difficulties in our relationships with these suppliers, or if a supplier becomes unable to provide us with goods or services at the agreed-upon price, our facilities expansion could be delayed or its costs increased.
We have never manufactured our Techosphere Insulin System or any other product candidate in commercial quantities. As our product candidates move through the regulatory process, we will need to either develop the capability of manufacturing on a commercial scale or engage third-party manufacturers with this capability, and we cannot offer assurances that we will be able to do either successfully. The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, especially in scaling up initial production. These problems include difficulties with production costs and yields, quality control and assurance and shortages of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. In addition, before we would be able to produce commercial quantities of Technosphere Insulin at our Danbury facility, it would have to undergo a pre-approval inspection by the FDA. The expansion process and preparation for the FDA’s pre-approval inspection for commercial production at the Danbury facility could take an additional six months or longer. If we use a third-party supplier to formulate Technosphere Insulin or produce raw material, the transition could also require significantstart-up time to qualify and implement the manufacturing process. If we engage a third-party manufacturer, our third-party manufacturer may not perform as agreed or may terminate its agreement with us.
Any of these factors could cause us to delay or suspend clinical trials, regulatory submissions, required approvals or commercialization of our product candidates, entail higher costs and result in our being unable to effectively commercialize our products. Furthermore, if we or a third-party manufacturer fail to deliver the required commercial quantities of any product on a timely basis and at commercially reasonable prices, and we were unable to promptly find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volume and on a timely basis, we would likely be unable to meet demand for such products and we would lose potential revenues.
We deal with hazardous materials and must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our research and development work involves the controlled storage and use of hazardous materials, including chemical, radioactive and biological materials. In addition, our manufacturing operations involve the use ofCBZ-lysine, which is stable and non-hazardous under normal storage conditions, but may form an explosive mixture under certain conditions. Our operations also produce hazardous waste products. We are subject to federal, state and local laws and regulations governing how we use, manufacture, store, handle and dispose of these materials. Moreover, the risk of accidental contamination or injury from hazardous materials cannot be completely eliminated, and in the event of an accident, we could be held liable for any damages that may result, and any liability could fall outside the coverage or exceed the limits of our insurance. Currently, our general liability policy provides coverage up to $1 million per occurrence and $2 million in the aggregate and is supplemented by an umbrella policy
34
Table of Contents
that provides a further $4 million of coverage; however, our insurance policy excludes pollution coverage and we do not carry a separate hazardous materials policy. In addition, we could be required to incur significant costs to comply with environmental laws and regulations in the future. Finally, current or future environmental laws and regulations may impair our research, development or production efforts.
When we purchased the facilities located in Danbury, Connecticut, in 2001 there was a soil cleanup plan in process. As part of the purchase, we obtained an indemnification from the seller related to the remediation of the soil for all known environmental conditions that existed at the time the seller acquired the property. The seller is, in turn, indemnified for these known environmental conditions by the previous owner. We estimate that the cost to complete the soil cleanup plan for industrial use is $1.5 to $3.0 million over the next 18 to 24 months. We also received an indemnification from the seller for environmental conditions created during its ownership of the property and for environmental problems unknown at the time that the seller acquired the property. These additional indemnities are limited to the purchase price that we paid for the Danbury facilities. In the event that any cleanup costs are imposed on us and we are unable to collect the full amount of these costs and expenses from the seller or the party responsible for the contamination, we may be required to pay these costs and our business and results of operations may be harmed.
If we fail to enter into collaborations with third parties, we would be required to establish our own sales, marketing and distribution capabilities, which could impact the commercialization of our products and harm our business.
A broad base of physicians and specialists treat patients with diabetes. A large sales force will be required in order to educate and support these physicians and specialists. Therefore, we plan to enter into collaborations with one or more pharmaceutical companies to sell, market and distribute our Technosphere Insulin System, if it is approved. If we fail to enter into collaborations, we would be required to establish our own direct sales, marketing and distribution capabilities. Establishing these capabilities can be time-consuming and expensive and we estimate that establishing a specialty sales force would cost more than $35 million. Because of our size, we would be at a disadvantage to our potential competitors, all of which either are or have collaborated with large pharmaceutical companies that have substantially more resources than we do. As a result, we would not initially be able to field a sales force as large as our competitors or provide the same degree of market research or marketing support. In addition, our competitors would have a greater ability to devote research resources toward expansion of the indications for their products. We cannot assure you that we will succeed in entering into acceptable collaborations, that any such collaboration will be successful or, if not, that we will successfully develop our own sales, marketing and distribution capabilities.
If any product that we may develop does not become widely accepted by physicians, patients, third-party payors and the healthcare community, we may be unable to generate significant revenue, if any.
Technosphere Insulin System and our other product candidates are new and unproven. Even if any of our product candidates obtain regulatory approvals, it may not gain market acceptance among physicians, patients, third-party payors and the healthcare community. Failure to achieve market acceptance would limit our ability to generate revenue and would adversely affect our results of operations.
The degree of market acceptance of our Technosphere Insulin System and our other product candidates will depend on many factors, including:
| • | the claims for which FDA approval can be obtained, including superiority claims; | |
| • | the perceived advantages and disadvantages of competitive products; | |
| • | the willingness and ability of patients and the healthcare community to adopt new technologies; | |
| • | the ability to manufacture the product in sufficient quantities with acceptable quality and at an acceptable cost; | |
| • | the perception of patients and the healthcare community, including third-party payors, regarding the safety, efficacy and benefits of the product compared to those of competing products or therapies; |
35
Table of Contents
| • | the convenience and ease of administration of the product relative to existing treatment methods; | |
| • | the pricing and reimbursement of the product relative to existing treatment therapeutics and methods; and | |
| • | marketing and distribution support for the product. |
Physicians will not recommend a product until clinical data or other factors demonstrate the safety and efficacy of the product as compared to other treatments. Even if the clinical safety and efficacy of our product candidates is established, physicians may elect not to recommend these product candidates for a variety of factors, including the reimbursement policies of government and third-party payors and the effectiveness of our competitors in marketing their therapies. Because of these and other factors, any product that we may develop may not gain market acceptance, which would materially harm our business, financial condition and results of operations.
If third-party payors do not reimburse customers for our products, our products might not be used or purchased, which would adversely affect our revenues.
Our future revenues and potential for profitability may be affected by the continuing efforts of governments and third-party payors to contain or reduce the costs of healthcare through various means. For example, in certain foreign markets the pricing of prescription pharmaceuticals is subject to governmental control. In the United States, there has been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental controls. We cannot be certain what legislative proposals will be adopted or what actions federal, state or private payors for healthcare goods and services may take in response to any healthcare reform proposals or legislation. Such reforms may make it difficult to complete the development and testing of our Technosphere Insulin System and our other product candidates, and therefore may limit our ability to generate revenues from sales of our product candidates and achieve profitability. Further, to the extent that such reforms have a material adverse effect on the business, financial condition and profitability of other companies that are prospective collaborators for some of our product candidates, our ability to commercialize our product candidates under development may be adversely affected.
In the United States and elsewhere, sales of prescription pharmaceuticals still depend in large part on the availability of reimbursement to the consumer from third-party payors, such as governmental and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. In addition, because each third-party payor individually approves reimbursement, obtaining these approvals is atime-consuming and costly process. We would be required to provide scientific and clinical support for the use of any product to each third-party payor separately with no assurance that approval would be obtained. This process could delay the market acceptance of any product and could have a negative effect on our future revenues and operating results. Even if we succeed in bringing one or more products to market, we cannot be certain that any such products would be considered cost-effective or that reimbursement to the consumer would be available, in which case our business and results of operations would be harmed and the market price of our common stock could decline.
If product liability claims are brought against us, we may incur significant liabilities and suffer damage to our reputation.
The testing, manufacturing, marketing and sale of our Technosphere Insulin System and our other product candidates expose us to potential product liability claims. A product liability claim may result in substantial judgments as well as consume significant financial and management resources and result in adverse publicity, decreased demand for a product, injury to our reputation, withdrawal of clinical trial volunteers and loss of revenues. We currently carry worldwide liability insurance in the amount of $5 million. We believe these limits are reasonable to cover us from potential damages arising from current and previous clinical trials of our Technosphere Insulin System. In addition, we carry local policies per trial in each country in which we conduct clinical trials that requires us to carry coverage based on local statutory requirements. We intend to obtain product liability coverage for commercial sales in the future if our Technosphere Insulin System is approved. However, we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise, and because insurance coverage in our industry can be very expensive and difficult to obtain, we cannot assure you that we will be able to obtain sufficient coverage at an acceptable cost, if at all. If losses from such claims exceed our liability insurance
36
Table of Contents
coverage, we may ourselves incur substantial liabilities. If we are required to pay a product liability claim, we may not have sufficient financial resources to complete development or commercialization of any of our product candidates and, if so, our business and results of operations would be harmed and the market price of our common stock may decline.
If we lose any key employees or scientific advisors, our operations and our ability to execute our business strategy could be materially harmed.
In order to commercialize our product candidates successfully, we will be required to expand our work force, particularly in the areas of manufacturing, clinical trials management, regulatory affairs, business development, and sales and marketing. These activities will require the addition of new personnel, including management, and the development of additional expertise by existing personnel. In October 2005, Dr. Peter Richardson joined us as Corporate Vice President and Chief Scientific Officer, and Dr. Juergen Martens, who joined us in September 2005, was appointed our Corporate Vice President of Operations in February 2006. We face intense competition for qualified employees among companies in the biotechnology and biopharmaceutical industries. Our success depends upon our ability to attract, retain and motivate highly skilled employees. We may be unable to attract and retain these individuals on acceptable terms, if at all.
The loss of the services of any principal member of our management and scientific staff could significantly delay or prevent the achievement of our scientific and business objectives. All of our employees are “at will” and we currently do not have employment agreements with any of the principal members of our management or scientific staff, and we do not have key person life insurance to cover the loss of any of these individuals. Replacing key employees may be difficult and time-consuming because of the limited number of individuals in our industry with the skills and experience required to develop, gain regulatory approval of and commercialize our product candidates successfully.
We have relationships with scientific advisors at academic and other institutions to conduct research or assist us in formulating our research, development or clinical strategy. These scientific advisors are not our employees and may have commitments to, and other obligations with, other entities that may limit their availability to us. We have limited control over the activities of these scientific advisors and can generally expect these individuals to devote only limited time to our activities. Failure of any of these persons to devote sufficient time and resources to our programs could harm our business. In addition, these advisors are not prohibited from, and may have arrangements with, other companies to assist those companies in developing technologies that may compete with our product candidates.
If our Chief Executive Officer is unable to devote sufficient time and attention to our business, our operations and our ability to execute our business strategy could be materially harmed.
Alfred Mann, our Chairman and Chief Executive Officer, is also serving as the Chairman and Co-Chief Executive Officer of Advanced Bionics Corporation, a wholly owned subsidiary of Boston Scientific Corporation. Mr. Mann is involved in many other business and charitable activities. As a result, the time and attention Mr. Mann devotes to the operation of our business varies, and he may not expend the same time or focus on our activities as other, similarly situated chief executive officers. If Mr. Mann is unable to devote the time and attention necessary to running our business, we may not be able to execute our business strategy and our business could be materially harmed.
We have been sued by our former Chief Medical Officer. As a result of this litigation, we may incur material costs and suffer other consequences, which may adversely affect us.
In May 2005, Dr. Cheatham filed a complaint against us in the California Superior Court. The complaint alleges causes of action for wrongful termination in violation of public policy, breach of contract and retaliation in connection with the termination of Dr. Cheatham’s employment. In the complaint, Dr. Cheatham seeks compensatory, punitive and exemplary damages in excess of $2.0 million as well as reimbursement of attorneys’ fees. In June 2005, we answered the complaint and also filed a cross-complaint against Dr. Cheatham, alleging claims for libel per se, trade libel, breach of contract, breach of the implied covenant of good faith and fair dealing and breach of the duty of loyalty. In July 2005, Dr. Cheatham filed a demurrer and motion to strike our cross-complaint under
37
Table of Contents
California’s anti-SLAPP statute. In September 2005, the California Superior Court overruled Dr. Cheatham’s demurrer and denied his motion to strike the Company’s cross-complaint. Dr. Cheatham then filed a notice of appeal of the Court’s ruling denying his motion to strike. Discovery as to Dr. Cheatham’s claims against us is proceeding, and this case is scheduled for trial to commence in October 2006.
The litigation will result in costs and divert management’s attention and resources, any of which could adversely affect our business, results of operations or financial position. We are also concerned that, despite the findings by an independent counsel following an investigation and despite the endorsement of the independent counsel’s report by our board of directors, investors could give undue weight to Dr. Cheatham’s allegations, resulting in damage to our reputation, or the FDA could begin an investigation, either of which could adversely affect the trading price of our common stock. To date, we have not been notified of any investigation by the FDA. If we are not successful in this litigation, we could be forced to make a significant settlement or judgment payment to Dr. Cheatham, which could adversely affect our business, results of operations or financial position.
Our facilities that are located in Southern California may be affected by natural disasters.
Our headquarters and some of our research and development activities are located in Southern California, where they are subject to an enhanced risk of natural and other disasters such as power and telecommunications failures, mudslides, fires and earthquakes. A fire, earthquake or other catastrophic loss that causes significant damage to our facilities or interruption of our business could harm our business. We do not carry insurance to cover losses caused by earthquakes, and the insurance coverage that we carry for fire damage and for business interruption may be insufficient to compensate us for any losses that we may incur.
A change in the accounting treatment of stock-based awards will adversely affect our results of operations.
In December 2004, the Financial Accounting Standards Board issued revised SFAS No. 123,Share-Based Payment, or SFAS No. 123R, which requires companies to expense employee stock options and other stock-based awards for financial reporting purposes. Under SFAS No. 123R, beginning with our first quarter of 2006, we are required to value our employee stock option grants pursuant to an option valuation model, and then amortize that value against our reported earnings over the vesting period in effect for those options. We previously accounted for stock-based awards to employees in accordance with Accounting Principles Board Opinion (“APB”) No. 25,Accounting for Stock Issued to Employees, and had adopted the disclosure-only alternative of SFAS No. 123 and SFAS No. 128, each of which has been superseded by SFAS No. 123R. The change in accounting treatment resulting from SFAS No. 123R may materially and adversely affect our reported results of operations as stock-based compensation expense is charged directly against our reported earnings. We continue to believe that expensing stock-based compensation will have an impact on our statement of operations similar to the pro forma disclosure under SFAS No. 123 (see Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation).
If our internal controls over financial reporting are not considered effective, our business and stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us to evaluate the effectiveness of our internal controls over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal controls over financial reporting in our annual report onForm 10-K for that fiscal year. Section 404 also requires our independent registered public accounting firm to attest to, and report on, management’s assessment of our internal controls over financial reporting. Our management has concluded, and our independent registered public accounting firm has attested, that our internal control over financial reporting was effective as of December 31, 2005.
Our management, including our chief executive officer and chief financial officer, does not expect that our internal controls over financial reporting will prevent all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no
38
Table of Contents
evaluation of controls can provide absolute assurance that all control issues and instances of fraud involving a company have been, or will be, detected. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and we cannot assure you that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. We cannot assure you that we or our independent registered public accounting firm will not identify a material weakness in our internal controls in the future. A material weakness in our internal controls over financial reporting would require management and our independent registered public accounting firm to evaluate our internal controls as ineffective. If our internal controls over financial reporting are not considered effective, we may experience a loss of public confidence, which could have an adverse effect on our business and on the market price of our common stock.
RISKS RELATED TO REGULATORY APPROVALS
Our product candidates must undergo rigorous preclinical and clinical testing and we must obtain regulatory approvals, which could be costly and time-consuming and subject us to unanticipated delays or prevent us from marketing any products.
Our research and development activities, as well as the manufacturing and marketing of our product candidates, including our Technosphere Insulin System, are subject to regulation, including regulation for safety, efficacy and quality, by the FDA in the United States and comparable authorities in other countries. FDA regulations are wide-ranging and govern, among other things:
| • | product design, development, manufacture and testing; | |
| • | product labeling; | |
| • | product storage and shipping; | |
| • | pre-market clearance or approval; | |
| • | advertising and promotion; and | |
| • | product sales and distribution. |
Clinical testing can be costly and take many years, and the outcome is uncertain and susceptible to varying interpretations. We expect, based on our discussions with the FDA and on our understanding of the interactions between the FDA and other pharmaceutical companies developing inhaled insulin delivery systems, that we will need safety data covering at least two years from patients treated with our Technosphere Insulin System and that we must complete a two-year carcinogenicity study of Technosphere Insulin in rodents to obtain approval, among other requirements. We cannot be certain when or under what conditions we will undertake further clinical trials. The clinical trials of our product candidates may not be completed on schedule, the FDA or foreign regulatory agencies may order us to stop or modify our research, or these agencies may not ultimately approve any of our product candidates for commercial sale. The data collected from our clinical trials may not be sufficient to support regulatory approval of our various product candidates, including our Technosphere Insulin System. Even if we believe the data collected from our clinical trials are sufficient, the FDA has substantial discretion in the approval process and may disagree with our interpretation of the data. Our failure to adequately demonstrate the safety and efficacy of any of our product candidates would delay or prevent regulatory approval of our product candidates, which could prevent us from achieving profitability.
The requirements governing the conduct of clinical trials and manufacturing and marketing of our product candidates, including our Technosphere Insulin System, outside the United States vary widely from country to country. Foreign approvals may take longer to obtain than FDA approvals and can require, among other things, additional testing and different clinical trial designs. Foreign regulatory approval processes include all of the risks associated with the FDA approval processes. Some of those agencies also must approve prices of the products. Approval of a product by the FDA does not ensure approval of the same product by the health authorities of other countries. In addition, changes in regulatory policy in the United States or in foreign countries for product approval
39
Table of Contents
during the period of product development and regulatory agency review of each submitted new application may cause delays or rejections.
The process of obtaining FDA and other required regulatory approvals, including foreign approvals, is expensive, often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. We are not aware of any precedent for the successful commercialization of products based on our technology. On January 26, 2006, the FDA approved the first pulmonary insulin product, Exubera. This may impact the development and registration of our Technosphere Insulin System in many ways, including: the approval of Exubera may increase the difficulty of enrolling patients in our clinical trials; Exubera may be viewed as standard of care by the FDA and used as a reference for the safety/efficacy evaluations of our Technosphere Insulin System; and the approval standards set for Exubera may be applied to other products that follow including our Technosphere Insulin System. The FDA has advised us that it will regulate our Technosphere Insulin System as a “combination product” because of the complex nature of the system that includes the combination of a new drug (Technosphere Insulin) and a new medical device (the MedTone inhaler used to administer the insulin). The FDA indicated that the review of a future drug marketing application for our Technosphere Insulin System will involve three separate review groups of the FDA: (1) the Metabolic and Endocrine Drug Products Division; (2) the Pulmonary Drug Products Division; and (3) the Center for Devices and Radiological Health within the FDA that reviews medical devices. We currently understand that the Metabolic and Endocrine Drug Products Division will be the lead group and will obtain consulting reviews from the other two FDA groups. The FDA has not made an official final decision in this regard, however, and we can make no assurances at this time about what impact FDA review by multiple groups will have on the review and approval of our product or whether we are correct in our understanding of how the Technosphere Insulin System will be reviewed and approved.
Also, questions that have been raised about the safety of marketed drugs generally, including pertaining to the lack of adequate labeling, may result in increased cautiousness by the FDA in reviewing new drugs based on safety, efficacy, or other regulatory considerations and may result in significant delays in obtaining regulatory approvals. Such regulatory considerations may also result in the imposition of more restrictive drug labeling or marketing requirements as conditions of approval, which may significantly affect the marketability of our drug products. FDA review of our Technosphere Insulin System as a combination product therapy may lengthen the product development and regulatory approval process, increase our development costs and delay or prevent the commercialization of our Technosphere Insulin System.
We are developing our Technosphere Insulin System as a new treatment for diabetes utilizing unique, proprietary components. As a combination product, any changes to either the MedTone inhaler, the Technosphere material or the insulin, including new suppliers, could possibly result in FDA requirements to repeat certain clinical studies. This means, for example, that switching to an alternate delivery system could require us to undertake additional clinical trials and other studies, which could significantly delay the development and commercialization of our Technosphere Insulin System. Our product candidates that are currently in development for the treatment of cancer also face similar obstacles and costs.
We currently expect that our inhaler will be reviewed for approval as part of the NDA for our Technosphere Insulin System. No assurances exist that we will not be required to obtain separate device clearances or approval for use of our inhaler with our Technosphere Insulin System. This may result in our being subject to medical device review user fees and to other device requirements to market our inhaler and may result in significant delays in commercialization. Even if the device component is approved as part of our NDA for the Technosphere Insulin System, numerous device regulatory requirements still apply to the device part of the drug-device combination.
We have only limited experience in filing and pursuing applications necessary to gain regulatory approvals, which may impede our ability to obtain timely approvals from the FDA or foreign regulatory agencies, if at all.
We will not be able to commercialize our Technosphere Insulin System and other product candidates until we have obtained regulatory approval. We have no experience as a company in late-stage regulatory filings, such as preparing and submitting NDAs, which may place us at risk of delays, overspending and human resources inefficiencies. Any delay in obtaining, or inability to obtain, regulatory approval could harm our business.
40
Table of Contents
If we do not comply with regulatory requirements at any stage, whether before or after marketing approval is obtained, we may be subject to criminal prosecution, fined or forced to remove a product from the market or experience other adverse consequences, including restrictions or delays in obtaining regulatory marketing approval.
Even if we comply with regulatory requirements, we may not be able to obtain the labeling claims necessary or desirable for product promotion. We may also be required to undertake post-marketing trials. In addition, if we or other parties identify adverse effects after any of our products are on the market, or if manufacturing problems occur, regulatory approval may be withdrawn and a reformulation of our products, additional clinical trials, changes in labeling of, or indications of use for, our productsand/or additional marketing applications may be required. If we encounter any of the foregoing problems, our business and results of operations will be harmed and the market price of our common stock may decline.
Even if we obtain regulatory approval for our product candidates, such approval may be limited and we will be subject to stringent, ongoing government regulation.
Even if regulatory authorities approve any of our product candidates, they could approve less than the full scope of uses or labeling that we seek or otherwise require special warnings or other restrictions on use or marketing. Regulatory authorities may limit the segments of the diabetes population to which we or others may market our Technosphere Insulin System or limit the target population for our other product candidates. Based on currently available clinical studies, we believe that our Technosphere Insulin System may have certain advantages over currently approved insulin products including its approximation of the natural early insulin secretion normally seen in healthy individuals following the beginning of a meal. Nonetheless, there are no assurances that these and other advantages, if any, of the Technosphere Insulin System have clinical significance or can be confirmed inhead-to-head clinical trials against appropriate approved comparator insulin drug products. Such comparative clinical trials are required to make these types of superiority claims in labeling or advertising. These aforementioned observations and others may therefore not be capable of substantiation in comparative clinical trials prior to our NDA submission, if at all, or otherwise may not be suitable for inclusion in product labeling or advertising and, as a result, our Technosphere Insulin System may not have competitive advantages when compared to other insulin products.
The manufacture, marketing and sale of these product candidates will be subject to stringent and ongoing government regulation. The FDA may also withdraw product approvals if problems concerning safety or efficacy of the product occur following approval. In response to questions that have been raised about the safety of certain approved prescription products, including the lack of adequate warnings, the FDA and Congress are currently considering new regulatory and legislative approaches to advertising, monitoring and assessing the safety of marketed drugs, including legislation providing the FDA with authority to mandate labeling changes for approved pharmaceutical products, particularly those related to safety. We also cannot be sure that the current Congressional and FDA initiatives pertaining to ensuring the safety of marketed drugs or other developments pertaining to the pharmaceutical industry will not adversely affect our operations.
We also are required to register our establishments and list our products with the FDA and certain state agencies. We and any third-party manufacturers or suppliers must continually adhere to federal regulations setting forth requirements, known as cGMP (for drugs) and QSR (for medical devices), and their foreign equivalents, which are enforced by the FDA and other national regulatory bodies through their facilities inspection programs. If our facilities, or the facilities of our manufacturers or suppliers, cannot pass a preapproval plant inspection, the FDA will not approve the marketing of our product candidates. In complying with cGMP and foreign regulatory requirements, we and any of our potential third-party manufacturers or suppliers will be obligated to expend time, money and effort in production, record-keeping and quality control to ensure that our products meet applicable specifications and other requirements. QSR requirements also impose extensive testing, control and documentation requirements. State regulatory agencies and the regulatory agencies of other countries have similar requirements. In addition, we will be required to comply with regulatory requirements of the FDA, state regulatory agencies and the regulatory agencies of other countries concerning the reporting of adverse events and device malfunctions, corrections and removals (e.g., recalls), promotion and advertising and general prohibitions against the manufacture and distribution of adulterated and misbranded devices. Failure to comply with these regulatory requirements could
41
Table of Contents
result in civil fines, product seizures, injunctionsand/or criminal prosecution of responsible individuals and us. Any such actions would have a material adverse effect on our business and results of operations.
Our insulin supplier does not yet supply human recombinant insulin for an FDA-approved product and will likely be subject to an FDA preapproval inspection before the agency will approve a future marketing application for our Technosphere Insulin System.
We can make no assurances that our insulin supplier will be acceptable to the FDA. If we were required to find a new or additional supplier of insulin, we would be required to evaluate the new supplier’s ability to provide insulin that meets our specifications and quality requirements, which would require significant time and expense and could delay the manufacturing and future commercialization of our Technosphere Insulin System. We also depend on suppliers for other materials that comprise our Technosphere Insulin System, including our MedTone inhaler and cartridges. All of our device suppliers must comply with relevant regulatory requirements including QSR. It also is likely that major suppliers will be subject to FDA preapproval inspections before the agency will approve a future marketing application for our Technosphere Insulin System. At the present time our insulin supplier is certified to the ISO9001:2000 Standard. There can be no assurance, however, that if the FDA were to conduct a preapproval inspection of our insulin supplier or other suppliers, that the agency would find that the supplier substantially comply with the QSR or cGMP requirements, where applicable. If we or any potential third-party manufacturer or supplier fails to comply with these requirements or comparable requirements in foreign countries, regulatory authorities may subject us to regulatory action, including criminal prosecutions, fines and suspension of the manufacture of our products.
Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the indicated uses for which the product candidate may be marketed or contain requirements for potentially costly post-marketingfollow-up clinical trials.
Reports of side effects or safety concerns in related technology fields or in other companies’ clinical trials could delay or prevent us from obtaining regulatory approval or negatively impact public perception of our product candidates.
At present, there are a number of clinical trials being conducted by us and other pharmaceutical companies involving insulin delivery systems. If we discover that our product is associated with a significantly increased frequency of adverse events, or if other pharmaceutical companies announce that they observed frequent adverse events in their trials involving the pulmonary delivery of insulin, we could encounter delays in the timing of our clinical trials or difficulties in obtaining the approval of our Technosphere Insulin System. As well, the public perception of our products might be adversely affected, which could harm our business and results of operations and cause the market price of our common stock to decline, even if the concern relates to another company’s product.
For example, in August 2004, an analyst reported that the United Kingdom Committee on the Safety of Medicines had expressed concern that a European application for approval of a drug for the treatment of diabetes was not licensable at the time. Earlier in 2004, sanofi-aventis, on behalf of Pfizer and Nektar, filed for regulatory approval in Europe of Exubera. Although the identity of the drug was not disclosed in the analyst’s report, the news nonetheless triggered temporary but sharp declines in the market prices of Nektar’s common stock as well as our common stock.
There are also a number of clinical trials being conducted by other pharmaceutical companies involving compounds similar to, or competitive with, our other product candidates. Adverse results reported by these other companies in their clinical trials could delay or prevent us from obtaining regulatory approval or negatively impact public perception of our product candidates, which could harm our business and results of operations and cause the market price of our common stock to decline.
42
Table of Contents
RISKS RELATED TO INTELLECTUAL PROPERTY
If we are unable to protect our proprietary rights, we may not be able to compete effectively, or operate profitably.
Our commercial success depends, in large part, on our ability to obtain and maintain intellectual property protection for our technology. Our ability to do so will depend on, among other things, complex legal and factual questions, and it should be noted that the standards regarding intellectual property rights in our fields are still evolving. We attempt to protect our proprietary technology through a combination of patents, trade secrets, know-how and confidentiality agreements. We own a number of domestic and international patents, have a number of domestic and international patent applications pending and have licenses to additional patents. We cannot assure you that our patents and licenses will successfully preclude others from using our technologies, and we could incur substantial costs in seeking enforcement of our proprietary rights against infringement. Even if issued, the patents may not give us an advantage over competitors with similar technologies.
Moreover, the issuance of a patent is not conclusive as to its validity or enforceability and it is uncertain how much protection, if any, will be afforded by our patents. A third party may challenge the validity or enforceability of a patent after its issuance by various proceedings such as oppositions in foreign jurisdictions or re-examinations in the US. If we attempt to enforce our patents, they may be challenged in court where they could be held invalid, unenforceable, or have their breadth narrowed to an extent that would destroy their value.
We also rely on unpatented technology, trade secrets, know-how and confidentiality agreements. We require our officers, employees, consultants and advisors to execute proprietary information and invention and assignment agreements upon commencement of their relationships with us. We also execute confidentiality agreements with outside collaborators. There can be no assurance, however, that these agreements will provide meaningful protection for our inventions, trade secrets or other proprietary information in the event of unauthorized use or disclosure of such information. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business, results of operations and financial condition could be adversely affected.
If we become involved in lawsuits to protect or enforce our patents or the patents of our collaborators or licensors, we would be required to devote substantial time and resources to prosecute or defend such proceedings.
Competitors may infringe our patents or the patents of our collaborators or licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings brought by the US Patent and Trademark Office, or USPTO, may be necessary to determine the priority of inventions with respect to our patent applications or those of our collaborators or licensors. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and be a distraction to our management. We may not be able, alone or with our collaborators and licensors, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States. We may not prevail in any litigation or interference proceeding in which we are involved. Even if we do prevail, these proceedings can be very expensive and distract our management.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, the market price of our common stock may decline.
43
Table of Contents
If our technologies conflict with the proprietary rights of others, we may incur substantial costs as a result of litigation or other proceedings and we could face substantial monetary damages and be precluded from commercializing our products, which would materially harm our business.
Over the past three decades the number of patents issued to biotechnology companies has expanded dramatically. As a result it is not always clear to industry participants, including us, which patents cover the multitude of biotechnology product types. Ultimately, the courts must determine the scope of coverage afforded by a patent and the courts do not always arrive at uniform conclusions.
A third party may claim that we are using inventions covered by such third party’s patents and may go to court to stop us from engaging in our normal operations and activities. These lawsuits can be expensive and would consume time and other resources. There is a risk that a court would decide that we are infringing a third party’s patents and would order us to stop the activities covered by the patents, including the commercialization of our products. In addition, there is a risk that we would have to pay the other party damages for having violated the other party’s patents (which damages may be increased, as well as attorneys’ fees ordered paid, if infringement is found to be willful), or that we will be required to obtain a license from the other party in order to continue to commercialize the affected products, or to design our products in a manner that does not infringe a valid patent. We may not prevail in any legal action, and a required license under the patent may not be available on acceptable terms or at all, requiring cessation of activities that were found to infringe a valid patent. We also may not be able to develop a non-infringing product design on commercially reasonable terms, or at all.
Although we own a number of domestic and foreign patents and patent applications relating to our Technosphere Insulin System and cancer vaccine products under development, we have identified certain third-party patents that a court may interpret to restrict our freedom to operate (that is, to cover our products) in the areas of Technosphere formulations, pulmonary insulin delivery and the treatment of cancer. Specifically, we have identified certain third-party patents having claims relating to chemical compositions of matter and pulmonary insulin delivery that may trigger an allegation of infringement upon the commercial manufacture and sale of our Technosphere Insulin System. We have also identified third-party patents disclosing methods of use and compositions of matter related to DNA-based vaccines that also may trigger an allegation of infringement upon the commercial manufacture and sale of our cancer therapy. If a court were to determine that our insulin products or cancer therapies were infringing any of these patent rights, we would have to establish with the court that these patents were invalid or unenforceable in order to avoid legal liability for infringement of these patents. However, proving patent invalidity or unenforceability can be difficult because issued patents are presumed valid. Therefore, in the event that we are unable to prevail in an infringement or invalidity action we will have to either acquire the third-party patents outright or seek a royalty-bearing license. Royalty-bearing licenses effectively increase production costs and therefore may materially affect product profitability. Furthermore, should the patent holder refuse to either assign or license us the infringed patents, it may be necessary to cease manufacturing the product entirelyand/or design around the patents, if possible. In either event, our business would be harmed and our profitability could be materially adversely impacted.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, the market price of our common stock may decline.
Patent litigation is costly and time-consuming. Among other things, such litigation may divert the attention of key personnel and we may not have sufficient resources to bring these actions to a successful conclusion. At the same time, some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Although patent and intellectual property disputes in the pharmaceutical area have often been settled for licensing or similar arrangements, associated costs may be substantial and could include ongoing royalties. An adverse determination in a judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from manufacturing and selling our products or result in substantial monetary damages, which would adversely affect our business and results of operations and cause the market price of our common stock to decline.
44
Table of Contents
We may not obtain trademark registrations for our potential trade names.
We have not selected trade names for some of our products and product candidates; therefore, we have not filed trademark registrations for our potential trade names for those products in any jurisdiction, including the United States. Although we intend to defend any opposition to our trademark registrations, no assurance can be given that any of our trademarks will be registered in the United States or elsewhere or that the use of any of our trademarks will confer a competitive advantage in the marketplace. Furthermore, even if we are successful in our trademark registrations, the FDA has its own process for drug nomenclature and its own views concerning appropriate proprietary names. It also has the power, even after granting market approval, to request a company to reconsider the name for a product because of evidence of confusion in the marketplace. We cannot assure you that the FDA or any other regulatory authority will approve of any of our trademarks or will not request reconsideration of one of our trademarks at some time in the future.
RISKS RELATED TO OUR COMMON STOCK
Our stock price is volatile.
The stock market, particularly in recent years, has experienced significant volatility particularly with respect to pharmaceutical and biotechnology stocks. Since our initial public offering in August 2004, the high and low sales price of our common stock has varied significantly, from a low of $8.42 to a high of $24.31. The volatility of pharmaceutical and biotechnology stocks often does not relate to the operating performance of the companies represented by the stock. Our business and the market price of our common stock may be influenced by a large variety of factors, including:
| • | the progress and results of our clinical trials; | |
| • | announcements by us or our competitors concerning their clinical trial results, acquisitions, strategic alliances, technological innovations and newly approved commercial products; | |
| • | the availability of critical materials used in developing and manufacturing our Technosphere Insulin System or other product candidates; | |
| • | developments concerning our patents, proprietary rights and potential infringement claims; | |
| • | developments in our litigation with our former Chief Medical Officer; | |
| • | the expense and time associated with, and the extent of our ultimate success in, securing regulatory approvals; | |
| • | changes in securities analysts’ estimates of our financial and operating performance; | |
| • | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders; and | |
| • | discussion of our Technosphere Insulin System, our other product candidates, competitors’ products, or our stock price by the financial and scientific press, the healthcare community and online investor communities such as chat rooms. |
Any of these risks, as well as other factors, could cause the market price of our common stock to decline.
If other biotechnology and biopharmaceutical companies or the securities markets in general encounter problems, the market price of our common stock could be adversely affected.
Public companies in general and companies included on The Nasdaq National Market in particular have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. There has been particular volatility in the market prices of securities of biotechnology and other life sciences companies, and the market prices of these companies have often fluctuated because of problems or successes in a given market segment or because investor interest has shifted to other segments. These broad market and industry factors may cause the market price of our common stock to decline,
45
Table of Contents
regardless of our operating performance. We have no control over this volatility and can only focus our efforts on our own operations, and even these may be affected due to the state of the capital markets.
In the past, following periods of large price declines in the public market price of a company’s securities, securities class action litigation has often been initiated against that company. Litigation of this type could result in substantial costs and diversion of management’s attention and resources, which would hurt our business. Any adverse determination in litigation could also subject us to significant liabilities.
Our Chief Executive Officer and principal stockholder can individually control our direction and policies, and his interests may be adverse to the interests of our other stockholders. After his death, his stock will be left to his funding foundations for distribution to various charities, and we cannot assure you of the manner in which those entities will manage their holdings.
Mr. Mann has been our primary source of financing to date. At December 31, 2005, Mr. Mann beneficially owned approximately 48.5% of our outstanding shares of capital stock. Members of Mr. Mann’s family beneficially owned at least an additional 2.0% of our outstanding shares of common stock, although Mr. Mann does not have voting or investment power with respect to these shares. By virtue of his holdings, Mr. Mann can and will continue to be able to effectively control the election of the members of our board of directors, our management and our affairs and prevent corporate transactions such as mergers, consolidations or the sale of all or substantially all of our assets that may be favorable from our standpoint or that of our other stockholders or cause a transaction that we or our other stockholders may view as unfavorable.
Subject to compliance with federal and state securities laws, Mr. Mann is free to sell the shares of our stock he holds at any time. Upon his death, we have been advised by Mr. Mann that his shares of our capital stock will be left to the Alfred E. Mann Medical Research Organization, or AEMMRO, and AEM Foundation for Biomedical Engineering, or AEMFBE,not-for-profit medical research foundations that serve as funding organizations for Mr. Mann’s various charities, including the Alfred Mann Foundation, or AMF, and the Alfred Mann Institute at the University of Southern California, and that may serve as funding organizations for any other charities that he may establish. The AEMMRO is a membership foundation consisting of six members, including Mr. Mann, four of his children and Dr. Joseph Schulman, the director of AMF. The AEMFBE is a membership foundation consisting of five members, including Mr. Mann and the same four of his children. Although we understand that the members of AEMMRO and AEMFBE have been advised of Mr. Mann’s objectives for these foundations, once Mr. Mann’s shares of our capital stock become the property of the foundations, we cannot assure you as to how those shares will be distributed or how they will be voted.
The future sale of our common stock could negatively affect our stock price.
As of December 31, 2005, we had approximately 50.3 million shares of common stock outstanding. Substantially all of these shares are available for public sale, subject in some cases to volume and other limitations or delivery of a prospectus. If our common stockholders sell substantial amounts of common stock in the public market, or the market perceives that such sales may occur, the market price of our common stock may decline. Furthermore, if we were to include in a company-initiated registration statement shares held by our stockholders pursuant to the exercise of their registrations rights, the sale of those shares could impair our ability to raise needed capital by depressing the price at which we could sell our common stock.
In addition, we will need to raise substantial additional capital in the future to fund our operations. If we raise additional funds by issuing equity securities, the market price of our common stock may decline and our existing stockholders may experience significant dilution.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Our amended and restated certificate of incorporation and bylaws include anti-takeover provisions, such as a prohibition on stockholder actions by written consent, the authority of our board of directors to issue preferred stock without stockholder approval, and supermajority voting requirements for specified actions. In addition, because we
46
Table of Contents
are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which generally prohibits stockholders owning 15% or more of our outstanding voting stock from merging or combining with us in certain circumstances. These provisions may delay or prevent an acquisition of us, even if the acquisition may be considered beneficial by some of our stockholders. In addition, they may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
Because we do not expect to pay dividends in the foreseeable future, you must rely on stock appreciation for any return on your investment.
We have paid no cash dividends on any of our capital stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, we do not expect to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Furthermore, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Accordingly, the success of your investment in our common stock will likely depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value after the offering or even maintain the price at which you purchased your shares, and you may not realize a return on your investment in our common stock.
| Item 1B. | Unresolved Staff Comments. |
This item is not applicable.
| Item 2. | Properties |
In 2001, we acquired a facility in Danbury, Connecticut that includes two buildings comprising approximately 190,000 square feet and currently house our research and development, administrative and manufacturing functions, primarily for Technosphere Insulin formulation, filling and packaging. We believe that our facility in Danbury has sufficient space to contain additional Technosphere Insulin manufacturing capacity necessary to satisfy potential commercial demand for the launch of our Technosphere Insulin System and the first few years thereafter for our Technosphere Insulin System and other Technosphere-related products.
We own and occupy approximately 147,000 square feet of laboratory, office and manufacturing space in Valencia, California. The facility contains our principal executive offices and houses our research and development laboratories for our cancer and other programs. We also use this facility to provide support for the development of our Technosphere programs.
We lease approximately 34,000 square feet of office space in Paramus, New Jersey pursuant to a lease that ends in January 2009.
| Item 3. | Legal Proceedings |
In May 2005, our former Chief Medical Officer filed a complaint against us in the California Superior Court, County of Los Angeles,Wayman Wendell Cheatham, M.D. v. MannKind Corporation, Case No. BC333845. The complaint alleges causes of action for wrongful termination in violation of public policy, breach of contract and retaliation in connection with our termination of Dr. Cheatham’s employment. In the complaint, Dr. Cheatham seeks compensatory, punitive and exemplary damages in excess of $2.0 million, as well as reimbursement of attorneys’ fees. In June 2005, we answered the complaint, generally denying each of Dr. Cheatham’s allegations and asserting various defenses. We believe the allegations in the complaint are without merit and intend to vigorously defend against them. We also filed a cross-complaint against Dr. Cheatham, alleging claims for libel per se, trade libel, breach of contract, breach of the implied covenant of good faith and fair dealing and breach of the duty of loyalty. The libel claims allege that Dr. Cheatham made certain false and malicious statements about us in a letter to the FDA with regard to a request by us to hold a meeting with the FDA. The remaining causes of action in the cross-complaint arise out of our allegations that Dr. Cheatham had an undisclosed consulting relationship with a
47
Table of Contents
competitor during his employment with us, in violation of our agreement. In July 2005, Dr. Cheatham filed a demurrer and motion to strike our cross-complaint under California’s anti-SLAPP statute. In September 2005, the California Superior Court overruled Dr. Cheatham’s demurrer and denied his motion to strike our cross-complaint. Dr. Cheatham then filed a notice of appeal of the Court’s ruling denying his motion to strike. Discovery as to Dr. Cheatham’s claims against us is proceeding, and this case is scheduled for trial to commence in October 2006. We believe that the ultimate resolution of this matter will not have a material impact on our financial position or results of operations.
In 2000, we issued 699,972 shares of common stock to three consultants in exchange for notes receivable aggregating approximately $10.9 million. The fixed interest bearing notes were collateralized by the underlying common stock. Thenotes-for-stock transactions were accounted for as in-substance stock option grants to non-employees. In November 2004, the consultants informed us that they had entered into an agreement in October 2001 with Mr. Mann, our Chairman, Chief Executive Officer and principal stockholder, under which Mr. Mann would purchase a portion of the consultants’ common stock, and that MannKind was to apply the proceeds to the amounts owed under the consultants’ respective notes. The consultants informed us that they believed both we and Mr. Mann were in breach of the alleged agreement, and indicated their intent to seek alleged damages arising from our failure to perform the alleged agreement. On October 19, 2005, the principal and interest on the notes aggregating $14.6 million became due and payable and we pursued collection. On November 21, 2005, the consultants filed a complaint against us in the California Superior Court, County of Los Angeles,Rollins et al. v. MannKind et al, Case No. BC343381. The complaint alleges causes of action for breach of the abovementioned agreement, among other things. On January 19, 2006, the parties mediated and settled the case. Under the settlement, MannKind repurchased 620,697 shares from the consultants in full satisfaction of the notes. MannKind also agreed to repurchase the remaining 79,275 shares held by the consultants for $1.4 million. The complaint has been dismissed in its entirety with prejudice.
| Item 4. | Submission of Matters to a Vote of Security Holders |
No matters were submitted to a vote of our security holders during the quarter ended December 31, 2005.
PART II
| Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchase of Equity Securities |
Common Stock Market Price
Our common stock has been traded on the Nasdaq National Market under the symbol “MNKD” since July 28, 2004. The following table sets forth for the quarterly periods indicated, the high and low bid prices for our common stock as reported by Nasdaq. These quotations reflect inter-dealer prices, without retail mark-up, markdown or commission, and may not represent actual transactions.
| High | Low | |||||||
| Year ended December 31, 2004 | ||||||||
| Third quarter (from July 28, 2004) | $ | 24.31 | $ | 10.71 | ||||
| Fourth quarter | $ | 20.40 | $ | 14.32 | ||||
| Year ended December 31, 2005 | ||||||||
| First quarter | $ | 16.15 | $ | 11.67 | ||||
| Second quarter | $ | 16.00 | $ | 8.58 | ||||
| Third quarter | $ | 14.48 | $ | 8.42 | ||||
| Fourth quarter | $ | 13.85 | $ | 10.60 | ||||
The closing sales price of our common stock on the Nasdaq National Market was $17.86 on March 8, 2006 and there were 230 registered holders of record as of that date.
48
Table of Contents
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently intend to retain all available funds and any future earnings for use in the operation and expansion of our business. Accordingly, we do not anticipate paying any cash dividends on our common stock in the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors.
Securities Authorized for Issuance Under Equity Compensation Plans
The information required to be disclosed by Item 201(d) ofRegulation S-K is incorporated herein by reference to the proxy Statement.
Recent Sales of Unregistered Securities
There were no sales of equity securities by us that were not registered under the Securities Act of 1933 during the fourth quarter of 2005.
Use of Proceeds
The initial public offering of our common stock, par value $0.01 per share, was effected through a Registration Statement onForm S-1 (FileNo. 333-115020) that was declared effective by the SEC on July 27, 2004, and a Registration Statement onForm S-1 (FileNo. 333-117702) that became effective upon filing with the SEC on July 28, 2004. The Registration Statements covered the offer and sale of up to 7,187,500 shares of our common stock, including an over-allotment option we granted to the underwriters to purchase up to 937,500 shares of our common stock from us, for an aggregate offering price of $100.6 million. Our initial public offering commenced on July 28, 2004. On August 2, 2004, 6,250,000 shares of our common stock were sold for an aggregate offering price of $87.5 million. The managing underwriters in the offering were UBS Investment Bank, Piper Jaffray, Wachovia Securities, Jefferies & Company, Inc. and Harris Nesbitt. The underwriters exercised 307,100 shares of the over-allotment option on August 28, 2004 and the closing occurred on September 1, 2004.
Our initial public offering resulted in aggregate net proceeds to us of approximately $83.2 million, including approximately $4.0 million in proceeds from the exercise of the underwriter’s over-allotment option. In connection with the offering, we paid $6.4 million in underwriting discounts and commissions and offering expenses of approximately $2.2 million.
No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or person owning ten percent or more of any class of our equity securities or to any other affiliates. All offering expenses were paid directly to others.
As of December 31, 2005, we estimate that we had used approximately $73.6 million of the total net proceeds of our initial public offering for operating activities and approximately $9.6 million of the net proceeds for the purchase of manufacturing equipment. The foregoing payments were direct payments made to third parties who were not our directors or officers (or their associates), persons owning ten percent or more of any class of our equity securities or any other affiliate, except that the proceeds used for working capital included regular compensation for officers and directors. The use of proceeds does not represent a material change from the use of proceeds described in the prospectus we filed pursuant to Rule 424(b) of the Securities Act with the SEC on July 28, 2004.
49
Table of Contents
| Item 6. | Selected Financial Data |
The following selected consolidated financial data should be read in conjunction with MannKind consolidated financial statements and notes thereto and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” which are included elsewhere in this report.
| Year Ended December 31, | ||||||||||||||||||||
Statement of Operations Data: | 2001 | 2002 | 2003 | 2004 | 2005 | |||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
| Revenue | $ | 326 | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 19,763 | 42,724 | 45,613 | 59,406 | 95,347 | |||||||||||||||
| General and administrative | 10,629 | 13,215 | 20,699 | 17,743 | 22,775 | |||||||||||||||
| In-process research and development costs | 19,726 | — | — | — | — | |||||||||||||||
| Goodwill impairment | — | 151,428 | — | — | — | |||||||||||||||
| Total operating expenses | 50,118 | 207,367 | 66,312 | 77,149 | 118,122 | |||||||||||||||
| Loss from operations | (49,792 | ) | (207,367 | ) | (66,312 | ) | (77,149 | ) | (118,122 | ) | ||||||||||
| Other income | 288 | 487 | 36 | 226 | 78 | |||||||||||||||
| Interest income | 1,261 | 617 | 398 | 932 | 3,707 | |||||||||||||||
| Loss before provision for income taxes | (48,243 | ) | (206,263 | ) | (65,878 | ) | (75,991 | ) | (114,337 | ) | ||||||||||
| Income tax provision | (2 | ) | (2 | ) | (1 | ) | (1 | ) | (1 | ) | ||||||||||
| Net loss | (48,245 | ) | (206,265 | ) | (65,879 | ) | (75,992 | ) | (114,338 | ) | ||||||||||
| Deemed dividends related to beneficial conversion feature of convertible preferred stock | — | (1,421 | ) | (1,017 | ) | (19,822 | ) | — | ||||||||||||
| Accretion on redeemable preferred stock | (239 | ) | (251 | ) | (253 | ) | (60 | ) | — | |||||||||||
| Net loss applicable to common stockholders | $ | (48,484 | ) | $ | (207,937 | ) | $ | (67,149 | ) | $ | (95,874 | ) | $ | (114,338 | ) | |||||
| Basic and diluted net loss per share | $ | (4.60 | ) | $ | (15.43 | ) | $ | (3.63 | ) | $ | (3.80 | ) | $ | (2.87 | ) | |||||
| Shares used to compute basic and diluted net loss per share | 10,534 | 13,472 | 18,488 | 25,221 | 39,871 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
Balance Sheet Data: | 2001 | 2002 | 2003 | 2004 | 2005 | |||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Cash, cash equivalents and marketable securities | $ | 53,730 | $ | 31,052 | $ | 55,945 | $ | 90,533 | $ | 145,634 | ||||||||||
| Working capital | 47,477 | 24,171 | 49,097 | 82,837 | 128,507 | |||||||||||||||
| Total assets | 251,487 | 104,773 | 125,876 | 163,483 | 228,371 | |||||||||||||||
| Deferred compensation and other liabilities | 231 | 207 | 404 | 76 | 29 | |||||||||||||||
| Redeemable convertible preferred stock | 4,684 | 4,935 | 5,188 | — | — | |||||||||||||||
| Deficit accumulated during the development stage | (94,827 | ) | (301,092 | ) | (366,971 | ) | (442,963 | ) | (557,301 | ) | ||||||||||
| Total stockholders’ equity | 235,017 | 90,773 | 111,577 | 150,363 | 206,977 | |||||||||||||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and notes thereto included in this report.
50
Table of Contents
OVERVIEW
We are a biopharmaceutical company focused on the discovery, development and commercialization of therapeutic products for diseases such as diabetes and cancer. We are currently in Phase 3 clinical trials in the United States and Europe of our lead product, the Technosphere Insulin System, to study its safety and efficacy in the treatment of diabetes. This therapy consists of a proprietary dry powder formulation of insulin that is inhaled into the deep lung using our proprietary inhaler. We believe that the combination of the performance characteristics, unique kinetics, convenience and ease of use of the Technosphere Insulin System may have the potential to change the way diabetes is treated. We are developing additional applications for our proprietary Technosphere platform technology by formulating other drugs for pulmonary delivery. We are also developing therapies for the treatment of solid-tumor cancers. Our other product candidates are in research and pre-clinical development.
We are a development stage enterprise and have incurred significant losses since our inception in 1991. As of December 31, 2005, we have incurred a cumulative net loss of $557.3 million. To date, we have not generated any product revenues and have funded our operations primarily through the sale of equity securities.
We do not anticipate sales of any product prior to regulatory approval and commercialization of our Technosphere Insulin System. We currently do not have the required approvals to market any of our product candidates, and we may not receive such approvals. We may not be profitable even if we succeed in commercializing any of our product candidates. We expect to make substantial and increasing expenditures and to incur additional operating losses for at least the next several years as we:
| • | continue the clinical development and commercialization of our Technosphere Insulin System for the treatment of diabetes; | |
| • | expand our manufacturing operations for our Technosphere Insulin System to meet our currently anticipated commercial production needs; | |
| • | expand our other research, discovery and development programs; | |
| • | expand our proprietary Technosphere platform technology and develop additional applications for the pulmonary delivery of other drugs; and | |
| • | enter into sales and marketing collaborations with other companies, if available on commercially reasonable terms, or develop these capabilities ourselves. |
Our business is subject to significant risks, including but not limited to the risks inherent in our ongoing clinical trials and the regulatory approval process, the results of our research and development efforts, competition from other products and technologies and uncertainties associated with obtaining and enforcing patent rights.
RESEARCH AND DEVELOPMENT EXPENSES
Our research and development expenses consist mainly of costs associated with the clinical trials of our product candidates which have not yet received regulatory approval for marketing and for which no alternative future use has been identified. This includes the salaries, benefits and stock-based compensation of research and development personnel, laboratory supplies and materials, facility costs, costs for consultants and related contract research, licensing fees, and depreciation of laboratory equipment. We track research and development costs by the type of cost incurred. We partially offset research and development expenses with the recognition of estimated amounts receivable from the State of Connecticut pursuant to a program under which we can exchange qualified research and development income tax credits for cash.
Our research and development staff conducts our internal research and development activities, which include research, product development, clinical development, manufacturing and related activities. This staff is located in our facilities in Valencia, California; Paramus, New Jersey; and Danbury, Connecticut. We expense research and development costs as we incur them.
Clinical development timelines, likelihood of success and total costs vary widely. We are focused primarily on advancing the Technosphere Insulin System through Phase 3 clinical trials and regulatory filings. We plan to commercialize our lead product as a treatment for diabetes. Based on the results of preclinical studies, we plan to
51
Table of Contents
develop additional applications of our Technosphere technology. Additionally, we anticipate that we will continue to determine which research and development projects to pursue, and how much funding to direct to each project, on an ongoing basis, in response to the scientific and clinical success of each product candidate. We cannot be certain when any revenues from the commercialization of our products will commence.
At this time, due to the risks inherent in the clinical trial process and given the early stage of development of our product candidates other than the Technosphere Insulin System, we are unable to estimate with any certainty the costs we will incur in the continued development of our product candidates for commercialization. The costs required to complete the development of our Technosphere Insulin System will be largely dependent on the scope of our clinical trials, the cost and efficiency of our manufacturing process and discussions with the FDA on its requirements. We anticipate that our research and development expenses, particularly for the Technosphere Insulin System, will increase significantly with the continuation of existing clinical trials, the initiation of new trials, the resulting manufacturing costs associated with producing clinical trial materials, and the expansion, qualification and validation of our commercial manufacturing processes and facilities. Additionally, we expect non-cash stock-based compensation expense resulting from the adoption of Statement of Financial Accounting Standards (“SFAS”) No. 123R,Share-based Payment: an Amendment of FASB Statement 123 and 95, effective as of January 1, 2006, to increase in the future. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation in the notes to our financial statements.
GENERAL AND ADMINISTRATIVE EXPENSES
Our general and administrative expenses consist primarily of salaries, benefits and stock-based compensation for administrative, finance, business development, human resources, legal and information systems support personnel. In addition, general and administrative expenses include business insurance and professional services costs.
We expect general and administrative expenses other than non-cash stock-based compensation expense to increase slightly in the future as a result of increased headcount, public company compliance and establishment of investor relations and marketing programs. We expect overall general and administrative expenses to increase significantly as a result of the adoption of SFAS No. 123R. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation in the notes to our financial statements.
CRITICAL ACCOUNTING POLICIES
We have based our discussion and analysis of our financial condition and results of operations on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making estimates of expenses such as stock option expenses and judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates under different assumptions or conditions. The significant accounting policies that are critical to the judgments and estimates used in the preparation of our financial statements are described in more detail below.
Impairment of long-lived assets
Assessing long-lived assets for impairment requires us to make assumptions and judgments regarding the carrying value of these assets. We evaluate long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. The assets are considered to be impaired if we determine that the carrying value may not be recoverable based upon our assessment of the following events or changes in circumstances:
| • | significant changes in our strategic business objectives and utilization of the assets; | |
| • | a determination that the carrying value of such assets cannot be recovered through undiscounted cash flows; |
52
Table of Contents
| • | loss of legal ownership or title to the assets; or | |
| • | the impact of significant negative industry or economic trends. |
If we believe our assets to be impaired, the impairment we recognize is the amount by which the carrying value of the assets exceeds the fair value of the assets. Any write-downs would be treated as permanent reductions in the carrying amount of the asset and an operating loss would be recognized. In addition, we base the useful lives and related amortization or depreciation expense on our estimate of the useful lives of the assets. If a change were to occur in any of the above-mentioned factors or estimates, our reported results could materially change.
To date, we have had recurring operating losses, and the recoverability of our long-lived assets is contingent upon executing our business plan. If we are unable to execute our business plan, we may be required to write down the value of our long-lived assets in future periods.
Clinical trial expenses
Our clinical trial accrual process seeks to account for expenses resulting from our obligations under contract with vendors, consultants, and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. Our objective is to reflect the appropriate trial expenses in our financial statements by matching period expenses with period services and efforts expended. We account for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. We determine accrual estimates through discussions with internal clinical personnel and outside service providers as to the progress or state of completion of trials, or the services completed. Service provider status is then compared to the contractual obligated fee to be paid for such services. During the course of a clinical trial, we adjust our rate of clinical expense recognition if actual results differ from our estimates. In the event that we do not identify certain costs that have begun to be incurred or we underestimate or overestimate the level of services performed or the costs of such services, our reported expenses for a period would be too low or too high. The date on which certain services commence, the level of services performed on or before a given date and the cost of the services are often judgmental. We make these judgments based upon the facts and circumstances known to us in accordance with generally accepted accounting principles.
Stock-based compensation
Prior to December 31, 2005, we accounted for employee stock options and the employee stock purchase plan using the intrinsic value method in accordance with APB No. 25 and adopted the disclosure only alternative of SFAS No. 123. In December 2004, the FASB issued SFAS No. 123R which requires companies to expense share-based payments to employees, including stock options, based on the fair value of the award at the grant date. SFAS No. 123R also eliminates the intrinsic value method of accounting for stock options which we followed until December 31, 2005. We are required to adopt SFAS No. 123R beginning with the first quarter of 2006. Upon adoption of SFAS No. 123R, we are required to select an adoption method and a valuation method that requires the input of highly subjective assumptions, including the expected volatility of our stock price and an expected option term. The first adoption method is a modified prospective transition method whereby a company would recognize share-based employee costs from the beginning of the fiscal period in which the recognition provisions are first applied as if the fair value accounting method had been used to account for all employee awards granted, modified, or settled after the effective date and to any awards that were not fully vested as of the effective date. Measurement and attribution of compensation cost for awards that are unvested as of the effective date of SFAS No. 123R would be based on the same estimate of the grant-date fair value and the same attribution method used previously under SFAS No. 123. The second adoption method is a modified retrospective transition method whereby a company would recognize employee compensation cost for periods presented prior to the adoption of SFAS No. 123R in accordance with the original provisions of SFAS 123; that is, an entity would recognize employee compensation costs in the amounts reported in the pro forma disclosures provided in accordance with SFAS No. 123. A company would not be permitted to make any changes to those amounts upon adoption of SFAS No. 123R unless those changes represent a correction of an error. For periods after the date of adoption of SFAS No. 123R, the modified
53
Table of Contents
prospective transition method described above would be applied. We continue to review our alternatives for adoption under this new pronouncement. We believe that the expensing of stock-based compensation will have an impact on our statement of operations similar to the pro forma disclosure under SFAS No. 123. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation.
Accounting for income taxes
We must make significant management judgments when determining our provision for income taxes, our deferred tax assets and liabilities and any valuation allowance recorded against our net deferred tax assets. At December 31, 2005, we have established a valuation allowance of $155.8 million against all of our net deferred tax asset balance, due to uncertainties related to our deferred tax assets as a result of our history of operating losses. The valuation allowance is based on our estimates of taxable income by jurisdiction in which we operate and the period over which our deferred tax assets will be recoverable. In the event that actual results differ from these estimates or we adjust these estimates in future periods, we may need to change the valuation allowance, which could materially impact our financial position and results of operations.
RESULTS OF OPERATIONS
Years ended December 31, 2005 and 2004
Revenues
No revenues were recorded for the years ended December 31, 2005 or 2004. We do not anticipate sales of any product prior to regulatory approval and commercialization of our Technosphere Insulin System.
Research and Development Expenses
The following table provides a comparison of the research and development expense categories for the years ended December 31, 2005 and 2004 (dollars in thousands):
| Year Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2005 | 2004 | $ Change | % Change | |||||||||||||
| Clinical | $ | 49,483 | $ | 23,477 | $ | 26,006 | 111 | % | ||||||||
| Manufacturing | 25,401 | 19,714 | 5,687 | 29 | % | |||||||||||
| Research | 22,449 | 17,309 | 5,140 | 30 | % | |||||||||||
| Research and development tax credit | (1,666 | ) | (4,030 | ) | 2,364 | (59 | )% | |||||||||
| Stock-based compensation expense (benefit) | (320 | ) | 2,936 | (3,256 | ) | (111 | )% | |||||||||
| Research and development expenses | $ | 95,347 | $ | 59,406 | $ | 35,941 | 61 | % | ||||||||
The increase in research and development expenses for the year ended December 31, 2005, as compared to the year ended December 31, 2004 was primarily due to ongoing expenses related to the clinical development of our Technosphere Insulin System. The expansion of our phase 3 clinical trial program for our Technosphere Insulin System and the continuation of other preclinical studies significantly increased our clinical research expenditures in 2005. This also resulted in increased Technosphere Insulin manufacturing costs to supply clinical trial materials. We continue to expand our qualification and validation of our manufacturing system. Additionally, research activities related to toxicology studies for our Technosphere Insulin System, expanding our proprietary Technosphere platform technology, developing additional applications for the pulmonary delivery of other drugs and the discovery and development of programs primarily focused on cancer therapies resulted in increased research expenditures. We anticipate that our research and development expenses associated with our Technosphere Insulin System, expanding our Technosphere platform technology and the pursuit of cancer therapies will increase significantly in 2006. Specifically, we anticipate increased expenses related to the continuation of existing and initiation of new clinical trials, and the resulting manufacturing costs associated with producing clinical trial materials.
The research and development tax credit recognized for the years ended December 31, 2005 and 2004 partially offsets our research and development expenses. The State of Connecticut provides an opportunity to exchange
54
Table of Contents
certain research and development income tax credit carryforwards for cash in exchange for forgoing the carryforward of the research and development credits. Estimated amounts receivable under the program are recorded as a reduction of research and development expenses. During the years ended December 31, 2004 and 2005, research and development expenses were offset by $4.0 million and $1.7 million, respectively, in connection with the program. The three months ended September 30, 2004 was the first period in which we were able to recognize the benefit of these credits for financial reporting purposes and accordingly the $4.0 million recognized in the year ended December 31, 2004 included amounts attributable to 2004 and prior years.
The decrease in stock-based compensation expense for the year ended December 31, 2005 compared to the year ended December 31, 2004 primarily resulted from the effect of the decrease of our stock price from December 31, 2004 to December 31, 2005. A significant portion of the compensation expense is tied to the stock options that were repriced in November 2003 as the compensation cost for all repriced options was measured on a quarterly basis until the options expired or were exercised or canceled. With the adoption of SFAS No. 123R, we expect stock-based compensation to increase significantly in the future. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation.
General and Administrative Expenses
The following table provides a comparison of the general and administrative expense categories for the years ended December 31, 2005 and 2004 (dollars in thousands):
| Year Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2005 | 2004 | $ Change | % Change | |||||||||||||
| Salaries, employee related and other general expenses | $ | 24,183 | $ | 13,869 | $ | 10,314 | 74 | % | ||||||||
| Stock-based compensation expense (benefit) | (1,408 | ) | 3,874 | (5,282 | ) | (136 | )% | |||||||||
| General and administrative expenses | $ | 22,775 | $ | 17,743 | $ | 5,032 | 28 | % | ||||||||
General and administrative expenses for the year ended December 31, 2005 increased as compared to the year ended December 31, 2004. Increased administrative services resulted in increased headcount, compensation adjustments and other employee related expenses. Additionally, litigation, public company compliance (including the Sarbanes-Oxley Act) and our establishment of a marketing function in 2005 increased both professional fees and consulting expenses. Offsetting increases to general and administrative expenses for these periods was a decrease in stock-based compensation expense resulting from the effect of the fluctuation of our stock price on the valuation of stock options that were repriced in November 2003. We expect general and administrative expenses other than non-cash stock-based compensation expense to increase slightly in the future as a result of increased headcount, public company compliance and establishment of investor relations and marketing programs. We expect overall general and administrative expenses to increase significantly as a result of the adoption of SFAS 123R. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation in the footnotes to our financial statements.
Deemed Dividend
Deemed dividend for 2004 represents the beneficial conversion charge to common stockholders related to the downward adjustment of the Series B and C preferred stock conversion price. All outstanding preferred stock automatically converted into common stock at the close of the initial public offering in the third quarter of 2004, and no further deemed dividend has been or will be recognized.
55
Table of Contents
Years ended December 31, 2004 and 2003
Revenues
No revenues were recorded for the years ended December 31, 2004 or 2003.
Research and Development Expenses
The following table provides a comparison of the research and development expense categories for the years ended December 31, 2004 and 2003 (dollars in thousands):
| Year Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2004 | 2003 | $ Change | % Change | |||||||||||||
| Clinical | $ | 23,477 | $ | 12,187 | $ | 11,290 | 93 | % | ||||||||
| Manufacturing | 19,714 | 12,246 | 7,468 | 61 | % | |||||||||||
| Research | 17,309 | 20,219 | (2,910 | ) | (14 | )% | ||||||||||
| Research and development tax credit | (4,030 | ) | — | (4,030 | ) | — | ||||||||||
| Stock-based compensation expense | 2,936 | 961 | 1,975 | 206 | % | |||||||||||
| Research and development expenses | $ | 59,406 | $ | 45,613 | $ | 13,793 | 30 | % | ||||||||
Research and development expenses increased for the year ended December 31, 2004 compared to the year ended December 31, 2003 primarily due to ongoing expenditures in 2004 related to our Technosphere Insulin System. Continuation of preclinical and clinical studies in 2004 increased research expenditures, which also resulted in increased manufacturing costs to supply clinical trial materials and to continue the validation of our manufacturing system. The overall increase in research and development costs for 2004 were offset by decreased costs resulting from the termination of AlleCure product development programs and the redesign of CTL product development programs initiated in the first quarter of 2003. Further offsetting the total increase in research and development expenses is an estimated $4.0 million benefit recognized pursuant to a program under which we can exchange qualified research and development income tax credits for cash in the State of Connecticut.
The increase in stock-based compensation expense for the year ended December 31, 2004 compared to the year ended December 31, 2003 primarily resulted from the effect of the increase of our stock price from December 31, 2003 to December 31, 2004. A significant portion of the compensation expense is tied to the stock options that were repriced in November 2003 as the compensation cost for all options repriced is measured on a quarterly basis until the options expire or are exercised or canceled.
General and administrative expenses
The following table provides a comparison of the general and administrative expense categories for the years ended December 31, 2004 and 2003 (dollars in thousands):
| Year Ended | ||||||||||||||||
| December 31, | ||||||||||||||||
| 2004 | 2003 | $ Change | % Change | |||||||||||||
| Salaries, employee related and other general expenses | $ | 13,869 | $ | 17,159 | $ | (3,290 | ) | (19 | )% | |||||||
| Stock-based compensation expense | 3,874 | 3,540 | 334 | 9 | % | |||||||||||
| General and administrative expenses | $ | 17,743 | $ | 20,699 | $ | (2,956 | ) | (14 | )% | |||||||
General and administrative expenses for the year ended December 31, 2004 decreased compared to the year ended December 31, 2003. The decrease was primarily due to transition and severance expenses resulting from the consolidation in 2003 of our California operations into our Valencia, California facility and reduction of our California workforce, offset by an increase in 2004 in non-cash stock-based compensation expense.
56
Table of Contents
Deemed dividends
Deemed dividends of $19.8 million and $1.0 million for the years ended December 31, 2004 and 2003, respectively, represent the beneficial conversion charge to common stockholders related to the downward adjustment of the Series B and C preferred stock conversion price.
LIQUIDITY AND CAPITAL RESOURCES
We have funded our operations primarily through the sale of equity securities. On August 5, 2005, we closed a $175.0 million private placement of shares of common stock and the concurrent issuance of warrants for the purchase of additional shares of common stock to accredited investors including our principal stockholder who purchased approximately $87.3 million. We sold 17,132,000 shares of our common stock in the private placement, together with warrants to purchase up to 3,426,000 shares of common stock at an exercise price of $12.228 per share. In connection with this private placement, we paid $4.5 million in commissions to our placements agents and incurred $0.3 million in other offering expenses which resulted in net proceeds of approximately $170.2 million.
During the year ended December 31, 2005, we used $101.2 million of cash for our operations compared to using $59.9 million for our operations in the year ended December 31, 2004. We had a net loss of $114.3 million for the year ended December 31, 2005, of which $5.5 million consisted of non-cash charges such as depreciation and amortization, stock-based compensation, losses on sale and abandonment or disposal of property and equipment. Deferred compensation of $1.4 million was repaid in May 2005 and $1.5 million was received in April 2005 related to the Connecticut research and development tax credit exchange. We expect our negative operating cash flow to continue at least until we obtain regulatory approval and achieve commercialization of our Technosphere Insulin System.
We used $94.4 million of cash for investing activities during the year ended December 31, 2005, compared to using $9.6 million for the year ended December 31, 2004. Cash used in investing activities was primarily from net purchases of marketable securities of $77.9 million and $17.2 million used to purchase machinery and equipment to expand our manufacturing operations and quality systems in support of our expansion of clinical trials for Technosphere Insulin System. We expect to make significant purchases of equipment in the foreseeable future.
Our financing activities provided cash of $172.7 million for the year ended December 31, 2005 compared to $101.5 million for 2004. Cash from financing activities in 2005 was primarily from the private placement in August 2005 and the exercise of stock options. For 2004, cash from financing activities was primarily from the receipt of $83.2 million in net proceeds from our initial public offering in the third quarter of 2004 and $18.2 million from the collection of stock subscriptions for 355,943 shares of Series C convertible preferred stock in the first quarter of 2004.
As of December 31, 2005, we had $145.6 million in cash, cash equivalents and marketable securities. Although we believe our existing cash resources will be sufficient to fund our anticipated cash requirements into the third quarter of 2006, we will require significant additional financing in the future to fund our operations. If adequate funds are not available, we may be required to delay, reduce or eliminate expenditures for certain of our programs, including our Technosphere Insulin System development activities. Because the majority of our anticipated expenses in the near term can be reduced or eliminated in a relatively short period, we believe that if we are unable to obtain additional capital we can continue activities, on a reduced basis, through the first quarter of 2007. Additionally, our principal stockholder has given assurances that he will provide additional funding for our operations, if necessary through the first quarter of 2007.
We intend to use our capital resources to continue the development of our Technosphere Insulin System and to develop additional applications for our proprietary Technosphere platform technology. In addition, portions of our capital resources will be devoted to expanding our other product development programs for the treatment ofsolid-tumor cancers. We anticipate that we will expend a portion of our capital to scale up our manufacturing capabilities in our Danbury facilities. We also intend to use our capital resources for general corporate purposes, which may include in-licensing or acquiring additional technologies.
If we enter into a strategic business collaboration with a pharmaceutical or biotechnology company, we would expect, as part of the transaction, to receive additional capital and reimbursements for a portion of the costs associated with the
57
Table of Contents
development, manufacture and commercialization of our Technosphere Insulin System. In addition, we expect to pursue the sale of equityand/or debt securities, or the establishment of other funding facilities. Issuances of debt or additional equity could impact the rights of our existing stockholders, dilute the ownership percentages of our existing stockholders and may impose restrictions on our operations. These restrictions could include limitations on additional borrowing, specific restrictions on the use of our assets as well as prohibitions on our ability to create liens, pay dividends, redeem our stock or make investments. We also may seek to raise additional capital by pursuing opportunities for the licensing, sale or divestiture of certain intellectual property and other assets, including our Technosphere technology platform. There can be no assurance, however, that any strategic collaboration, sale of securities or sale or license of assets will be available to us on a timely basis or on acceptable terms, if at all. If we are unable to raise additional capital, we may be required to enter into agreements with third parties to develop or commercialize products or technologies that we otherwise would have sought to develop independently, and any such agreements may not be on terms as commercially favorable to us.
However, we cannot provide assurances that our plans will not change or that changed circumstances will not result in the depletion of our capital resources more rapidly than we currently anticipate. If planned operating results are not achieved or we are not successful in raising additional equity financing or entering a business collaboration, we may be required to reduce expenses through the delay, reduction or curtailment of our projects, including our Technosphere Insulin System development activities, or further reduction of costs for facilities and administration.
Effects of Inflation
Our assets are primarily monetary, consisting of cash and cash equivalents. Because of their liquidity, these assets are not directly affected by inflation. We also believe that we have intangible assets in the value of our technology. In accordance with generally accepted accounting principles, we have not capitalized the value of this intellectual property on our consolidated balance sheet. Because we intend to retain and continue to use our equipment, furniture and fixtures and leasehold improvements, we believe that the incremental inflation related to replacement costs of such items will not materially affect our operations. However, the rate of inflation affects our expenses, such as those for employee compensation and contract services, which could increase our level of expenses and the rate at which we use our cash resources.
Off-Balance Sheet Arrangements
As of December 31, 2005, we did not have any off-balance sheet arrangements.
COMMITMENTS AND CONTINGENCIES
Our contractual obligations consist of operating leases, purchase obligations and capital lease commitments. Future payments under our operating lease obligations, including facility leases executed in March 2005 and November 2005, and open purchase commitments consisted of the following at December 31, 2005 (in thousands):
| Payments Due in | ||||||||||||||||||||
| Less than | ||||||||||||||||||||
Contractual Obligations | One Year | 1-3 Years | 3-5 Years | 5 Years | Total | |||||||||||||||
| Open purchase order commitments(1) | $ | 42,750 | $ | 44,731 | $ | — | $ | — | $ | 87,481 | ||||||||||
| Operating lease obligations | 849 | 1,454 | 61 | — | 2,364 | |||||||||||||||
| Capital lease obligations | 74 | — | — | — | 74 | |||||||||||||||
| Total contractual obligations | $ | 43,673 | $ | 46,185 | $ | 61 | $ | — | $ | 89,919 | ||||||||||
| (1) | The amounts included in open purchase order commitments are subject to performance under the purchase order by the supplier of the goods or services and do not become our obligation until such performance is rendered. The amount shown is principally for the purchase of materials for our clinical trials, the acquisition of manufacturing equipment and commitments related to our manufacturing plant expansion. |
58
Table of Contents
RELATED PARTY TRANSACTIONS
For a description of our related party transactions see Note 15 — Certain Relationships and Related Party Transactions” in the notes to our financial statements.
RECENT ACCOUNTING PRONOUNCEMENTS
In May 2005, the FASB issued SFAS No. 154,Accounting Changes and Error Corrections (“SFAS No. 154”), which replaces APB Opinion No. 20,Accounting Changes and SFAS No. 3,Reporting Accounting Changes in Interim Financial Statements — An Amendment of APB Opinion No. 28. SFAS No. 154 provides guidance on the accounting for and reporting of accounting changes and error corrections. It establishes retrospective application, or the latest practicable date, as the required method for reporting a change in accounting principle and the reporting of a correction of an error. SFAS No. 154 is effective for accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. We believe that the adoption of this statement will not have a material effect on our financial condition or results of operations.
In March 2005, the FASB issued Interpretation No. 47,Accounting for Conditional Asset Retirement Obligations (“FIN 47”), which clarifies that the term conditional asset retirement obligation as used in SFAS No. 143,Accounting for Asset Retirement Obligations, refers to a legal obligation to perform an asset retirement activity in which the timing and (or) method of settlement are conditional on a future event that may or may not be within the control of the entity. FIN 47, which went into effect in December 2005, governs the disclosures related to future environmental liabilities and affected companies must recognize future estimated cleanup costs on current financial statements. We believe the estimated costs associated with potential environmental liabilities are not material.
In December 2004, the FASB issued SFAS No. 123R which requires companies to expense share-based payments to employees, including stock options, based on the fair value of the award at the grant date. SFAS No. 123R also eliminates the intrinsic value method of accounting for stock options which we followed until December 31, 2005. We are required to adopt SFAS No. 123R beginning with the first quarter of 2006. Upon adoption of SFAS No. 123R, we are required to select an adoption method and a valuation method that requires the input of highly subjective assumptions, including the expected volatility of our stock price and an expected option term. The first adoption method is a modified prospective transition method whereby a company would recognize share-based employee costs from the beginning of the fiscal period in which the recognition provisions are first applied as if the fair value accounting method had been used to account for all employee awards granted, modified, or settled after the effective date and to any awards that were not fully vested as of the effective date. Measurement and attribution of compensation cost for awards that are unvested as of the effective date of SFAS No. 123R would be based on the same estimate of the grant-date fair value and the same attribution method used previously under SFAS No. 123. The second adoption method is a modified retrospective transition method whereby a company would recognize employee compensation cost for periods presented prior to the adoption of SFAS No. 123R in accordance with the original provisions of SFAS 123; that is, an entity would recognize employee compensation costs in the amounts reported in the pro forma disclosures provided in accordance with SFAS No. 123. A company would not be permitted to make any changes to those amounts upon adoption of SFAS No. 123R unless those changes represent a correction of an error. For periods after the date of adoption of SFAS No. 123R, the modified prospective transition method described above would be applied. We continue to review our alternatives for adoption under this new pronouncement. We believe that the expensing of stock-based compensation will have an impact on our statement of operations similar to the pro forma disclosure under SFAS No. 123. See Note 2 — Summary of Significant Accounting Policies — Stock-Based Compensation.
In March 2004, the FASB ratified the measurement and recognition guidance and certain disclosure requirements for impaired securities as described in EITF IssueNo. 03-1,The Meaning ofOther-Than-Temporary Impairment and Its Application to Certain Investments. On November 3, 2005, the FASB issued FASB Staff Position (“FSP”) Nos.FAS 115-1 andFAS 124-1 which addresses the determination as to when an investment is considered impaired, whether that impairment is other than temporary, and the measurement of an impairment loss. This FSP also includes accounting considerations subsequent to the recognition of another-than-temporary impairment and requires certain disclosures about unrealized losses that have not been recognized asother-than-temporary impairments. The guidance in this FSP amends FASB Statements No. 115,Accounting for Certain Investments
59
Table of Contents
in Debt and Equity Securities, and No. 124,Accounting for Certain Investments Held byNot-for-Profit Organizations and APB Opinion No. 18,The Equity Method of Accounting for Investments in Common Stock. The FSP is effective for reporting periods beginning after December 15, 2005. Our investment portfolio at December 31, 2005 is comprised of highly liquid short-term securities. Unrealized losses for these securities are not material.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
We have not used derivative financial instruments. However, we are exposed to market risk related to changes in interest rates. Our current policy is to maintain a highly liquid short-term investment portfolio consisting mainly of U.S. money market funds and government and investment-grade corporate debt. Our cash is deposited in and invested through highly rated financial institutions in North America. Our short-term investments are subject to interest rate risk and will fall in value if market interest rates increase. If market interest rates were to increase immediately and uniformly by ten percent from levels at December 31, 2005, we estimate that the fair value of our investment portfolio would decline by an immaterial amount.
| Item 8. | Financial Statements and Supplementary Data |
The information required by this Item is included in Items 15(a)(1) and (2) of Part IV of this report.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports filed under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Our chief executive officer and chief financial officer performed an evaluation under the supervision and with the participation of our management, of our disclosure controls and procedures (as defined inRule 13a-15(b) of the Securities Exchange Act of 1934) as of December 31, 2005. Based on that evaluation, our chief executive officer and chief financial officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange ActRule 13a-15(f). Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework set forth inInternal Control — Integrated Frameworkissued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework set forth inInternal Control — Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2005. Our management’s assessment of the effectiveness of our internal control over financial reporting as of December 31, 2005 has been audited by Deloitte & Touche LLP, an independent registered public accounting firm, as stated in their report which is included herein.
60
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of MannKind Corporation,
Valencia, California
We have audited management’s assessment, included in the accompanying Management’s Report on Internal Control Over Financial Reporting, that MannKind Corporation and subsidiaries (a development stage company) (the “Company”) maintained effective internal control over financial reporting as of December 31, 2005, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on management’s assessment and an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating management’s assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed by, or under the supervision of, the company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the company’s board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, management’s assessment that the Company maintained effective internal control over financial reporting as of December 31, 2005, is fairly stated, in all material respects, based on the criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2005, based on the criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements as of and for the year ended December 31, 2005 of the Company and our report dated March 15, 2006 expressed an unqualified opinion on those financial statements.
/s/ DELOITTE & TOUCHE LLP
Los Angeles, California
March 15, 2006
61
Table of Contents
| Item 9B. | Other Information. |
None.
PART III
Certain information required by Part III is omitted from this report because we will file a definitive proxy statement within 120 days after the end of our fiscal year pursuant to Regulation 14A for our 2006 annual meeting of stockholders, and the information included in the proxy statement is incorporated herein by reference.
| Item 10. | Directors and Executive Officers of the Registrant. |
(a) Executive Officers — For information regarding the identification and business experience of our executive officers, see “Executive Officers” in Part I, Item 1 of this report.
(b) Directors — The information required by this Item regarding the identification and business experience of our directors is contained in the section entitled “Proposal 1 — Election of Directors” in the proxy statement for the May 2006 annual meeting of stockholders to be filed with the Securities and Exchange Commission within 120 days after the end of our fiscal year ended December 31, 2005, and is incorporated herein by reference.
Additional information required by this Item is incorporated by reference to the section entitled “Section 16(a) Beneficial Ownership Reporting Compliance” in the proxy statement.
We have adopted a Code of Business Conduct and Ethics Policy that applies to our directors and employees (including our principal executive officer, principal financial officer, principal accounting officer and controller), and have posted the text of the policy on our website (www.mannkindcorp.com) in connection with “Investor Relations” materials. In addition, we intend to promptly disclose (i) the nature of any amendment to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
| Item 11. | Executive Compensation |
The information under the caption “Executive Compensation” in the proxy statement is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Executive Compensation — Securities Authorized for Issuance under Equity Compensation Plans” in the proxy statement is incorporated herein by this reference.
| Item 13. | Certain Relationships and Related Transactions |
The information under the caption “Certain Transactions” in the proxy statement is incorporated herein by reference. With the exception of the information specifically incorporated by reference from the proxy statement in this report, the proxy statement shall not be deemed to be filed as part of this report. Without limiting the foregoing, the information under the captions “Report of the Audit Committee of the Board of Directors,” “Report of the Compensation Committee of the Board of Directors” and “Performance Measurement Comparison” in the proxy statement is not incorporated by reference in this report.
| Item 14. | Principal Accounting Fees and Services |
The information under the caption “Principal Accounting Fees and Services” in the proxy statement is incorporated herein by reference.
62
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules |
(a) The following documents are filed as part of, or incorporated by reference into, this report:
(1)(2) Financial Statements and Financial Statement Schedules. The following Financial Statements of MannKind Corporation, Financial Statement Schedules and Report of Independent Registered Public Accounting Firm are included in a separate section of this report beginning onpage F-2:
| Report of Independent Registered Public Accounting Firm | F-2 | |||
| Consolidated Balance Sheets | F-3 | |||
| Statements of Operations | F-4 | |||
| Statements of Stockholders’ Equity (Deficit) | F-5 | |||
| Statements of Cash Flows | F-8 | |||
| Notes to Financial Statements | F-10 |
All financial statement schedules have been omitted because the required information is not applicable or not present in amounts sufficient to require submission of the schedule, or because the information required is included in the consolidated financial statements or the notes thereto.
(3) Exhibits. The exhibits listed under Item 15(c) hereof are filed with, or incorporated by reference into, this report. Each management contract or compensatory plan or arrangement is identified separately in Item 15(c) hereof.
(c) Exhibits. The following exhibits are filed as part of, or incorporated by reference into, this report:
Exhibit Index
| Exhibit | ||||
Number | Description of Document | |||
| 3 | .1(1) | Restated Certificate of Incorporation. | ||
| 3 | .2(1) | Amended and Restated Bylaws. | ||
| 4 | .1(1) | Form of common stock certificate. | ||
| 4 | .2(1) | Registration Rights Agreement, dated October 15, 1998 by and among CTL ImmunoTherapies Corp., Medical Research Group, LLC, McLean Watson Advisory Inc. and Alfred E. Mann, as amended. | ||
| 10 | .1(2) | Securities Purchase Agreement, dated August 2, 2005 by and among MannKind and the purchasers listed on Exhibit A thereto. | ||
| 10 | .2†(3) | Supply Agreement, dated December 31, 2004, between MannKind and Vaupell, Inc. | ||
| 10 | .3†(1) | Supply Agreement, dated January 1, 2000, between Diosynth B.V. and Pharmaceutical Discovery Corporation. | ||
| 10 | .4*(1) | Form of Indemnity Agreement entered into between MannKind and each of its directors and officers. | ||
| 10 | .5* | Description of Officers’ Incentive Program. | ||
| 10 | .6*(4) | Description of 2006 executive officer salaries. | ||
| 10 | .7*(4) | Description of 2006 non-employee director compensation. | ||
| 10 | .8*(1) | Executive Severance Agreement, dated August 1, 2003, between MannKind and Wendell Cheatham. | ||
| 10 | .9*(1) | Executive Severance Agreement, dated August 1, 2003, between MannKind and Hakan Edstrom. | ||
| 10 | .10*(1) | Executive Severance Agreement, dated August 1, 2003, between MannKind and David Thomson. | ||
| 10 | .11*(1) | Executive Severance Agreement, dated August 1, 2003, between MannKind and Dick Anderson. | ||
| 10 | .12*(1) | Executive Severance Agreement, dated August 1, 2003, between MannKind and Dan Burns. | ||
| 10 | .13*(1) | Change of Control Agreement, dated August 1, 2003, between MannKind and Wendell Cheatham. | ||
| 10 | .14*(1) | Change of Control Agreement, dated August 1, 2003, between MannKind and Hakan Edstrom. | ||
| 10 | .15*(1) | Change of Control Agreement, dated August 1, 2003, between MannKind and David Thomson. | ||
63
Table of Contents
| Exhibit | ||||
Number | Description of Document | |||
| 10 | .16*(1) | Change of Control Agreement, dated August 1, 2003, between MannKind and Dick Anderson. | ||
| 10 | .17*(1) | Change of Control Agreement, dated August 1, 2003, between MannKind and Dan Burns. | ||
| 10 | .18*(1) | 2004 Equity Incentive Plan and form of stock option agreement thereunder. | ||
| 10 | .19*(5) | Form of Phantom Stock Award Agreement under the 2004 Equity Incentive Plan. | ||
| 10 | .20* | 2004 Non-Employee Directors’ Stock Option Plan and form of stock option agreement thereunder. | ||
| 10 | .21*(1) | 2004 Employee Stock Purchase Plan and form of offering document thereunder. | ||
| 10 | .22*(1) | Pharmaceutical Discovery Corporation 1991 Stock Option Plan. | ||
| 10 | .23*(1) | Pharmaceutical Discovery Corporation 1999 Stock Plan and form of stock option plan thereunder. | ||
| 10 | .24*(1) | AlleCure Corp. 2000 Stock Option and Stock Plan. | ||
| 10 | .25*(1) | CTL Immunotherapies Corp. 2000 Stock Option and Stock Plan. | ||
| 10 | .26*(1) | 2001 Stock Awards Plan. | ||
| 31 | .1 | Certification of the Chief Executive Officer pursuant toRules 13a-14(a) and15d-14(a) of the Securities Exchange Act of 1934, as amended. | ||
| 31 | .2 | Certification of the Chief Financial Officer pursuant toRules 13a-14(a) and15d-14(a) of the Securities Exchange Act of 1934, as amended. | ||
| 32 | Certifications of the Chief Executive Officer and Chief Financial Officer pursuant toRules 13a-14(b) and15d-14(b) of the Securities Exchange Act of 1934, as amended and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. § 1350) | |||
| * | Indicates management contract or compensatory plan. | |
| † | Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the SEC. | |
| (1) | Incorporated by reference to MannKind’s registration statement onForm S-1 (FileNo. 333-115020), filed with the SEC on April 30, 2004, as amended. | |
| (2) | Incorporated by reference to MannKind’s current report onForm 8-K filed with the SEC on August 5, 2005. | |
| (3) | Incorporated by reference to MannKind’s current report onForm 8-K filed with the SEC on February 23, 2005. | |
| (4) | Incorporated by reference to MannKind’s current report onForm 8-K filed with the SEC on February 22, 2006. | |
| (5) | Incorporated by reference to MannKind’s current report onForm 8-K filed with the SEC on December 14, 2005. |
64
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Mannkind Corporation
| By: | /s/ Alfred E. Mann |
Alfred E. Mann
Chief Executive Officer
Dated: March 15, 2006
POWER OF ATTORNEY
KNOW ALL BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Hakan S. Edstrom, Richard L. Anderson and David Thomson, and each of them, as his or her true and lawfulattorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place, and stead, in any and all capacities, to sign any and all amendments to this Report, and any other documents in connection therewith, and to file the same, with all exhibits thereto, with the Securities and Exchange Commission, granting unto saidattorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that saidattorneys-in-fact and agents, or any of them or their or his substitute or substituted, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||||
| /s/ Alfred E. Mann Alfred E. Mann | Chief Executive Officer and Chairman of the Board of Directors(Principal Executive Officer) | March 15, 2006 | ||||
| /s/ Hakan S. Edstrom Hakan S. Edstrom | President, Chief Operating Officer and Director | March 15, 2006 | ||||
| /s/ Richard L. Anderson Richard L. Anderson | Corporate Vice President and Chief Financial Officer(Principal Financial and Accounting Officer) | March 15, 2006 | ||||
| /s/ Kathleen Connell, Ph.D. Kathleen Connell, Ph.D. | Director | March 15, 2006 | ||||
| /s/ Ronald J. Consiglio Ronald J. Consiglio | Director | March 15, 2006 | ||||
| /s/ Michael Friedman, M.D. Michael Friedman, M.D. | Director | March 15, 2006 | ||||
65
Table of Contents
Signature | Title | Date | ||||
| /s/ Llew Keltner M.D., Ph.D. Llew Keltner M.D., Ph.D. | Director | March 15, 2006 | ||||
| Kent Kresa | Director | March 15, 2006 | ||||
| David H. MacCallum | Director | March 15, 2006 | ||||
| /s/ Henry L. Nordhoff Henry L. Nordhoff | Director | March 15, 2006 | ||||
66
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
(A Development Stage Company)
INDEX TO FINANCIAL STATEMENTS
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-8 | ||||
| F-10 |
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of MannKind Corporation
Valencia, California
We have audited the accompanying consolidated balance sheets of MannKind Corporation and subsidiaries (a development stage company) (the “Company”) as of December 31, 2004 and 2005 and the related statements of operations, stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2005 and for the period from February 14, 1991 (date of inception) to December 31, 2005. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the financial position of MannKind Corporation and subsidiaries at December 31, 2004 and 2005 and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2005 and for the period from February 14, 1991 (date of inception) to December 31, 2005 in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of the Company’s internal control over financial reporting as of December 31, 2005, based on the criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 15, 2006 expressed an unqualified opinion on management’s assessment of the effectiveness of the Company’s internal control over financial reporting and an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
/s/ DELOITTE & TOUCHE LLP
Los Angeles, California
March 15, 2006
F-2
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
(In thousands, except share data)
| December 31, | ||||||||
| 2004 | 2005 | |||||||
Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 78,987 | $ | 56,037 | ||||
| Marketable securities | 11,546 | 89,597 | ||||||
| Restricted cash | 583 | — | ||||||
| State research and development credit exchange receivable — current | 1,500 | 1,194 | ||||||
| Prepaid expenses and other current assets | 3,265 | 3,044 | ||||||
| Total current assets | 95,881 | 149,872 | ||||||
| Property and equipment — net | 66,511 | 76,183 | ||||||
| State research and development credit exchange receivable — net of current portion | 1,030 | 2,031 | ||||||
| Other assets | 61 | 285 | ||||||
| Total | $ | 163,483 | $ | 228,371 | ||||
Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 3,039 | $ | 3,547 | ||||
| Accrued expenses and other current liabilities | 8,632 | 17,818 | ||||||
| Deferred compensation | 1,373 | — | ||||||
| Total current liabilities | 13,044 | 21,365 | ||||||
| Other liabilities | 76 | 29 | ||||||
| Total liabilities | 13,120 | 21,394 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ equity: | ||||||||
| Undesignated preferred stock, $0.01 par value — 10,000,000 shares authorized; no shares issued or outstanding at December 31, 2004 and 2005 | — | — | ||||||
| Common stock, $0.01 par value — 90,000,000 shares authorized; 32,756,237 and 50,314,108 shares issued and outstanding at December 31, 2004 and 2005, respectively | 327 | 503 | ||||||
| Additional paid-in capital | 592,999 | 763,775 | ||||||
| Deficit accumulated during the development stage | (442,963 | ) | (557,301 | ) | ||||
| Total stockholders’ equity | 150,363 | 206,977 | ||||||
| Total | $ | 163,483 | $ | 228,371 | ||||
See notes to financial statements.
F-3
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
(A Development Stage Company)
(In thousands, except per share data)
| Cumulative | ||||||||||||||||
| Period from | ||||||||||||||||
| February 14, | ||||||||||||||||
| 1991 (Date of | ||||||||||||||||
| Inception) to | ||||||||||||||||
| Year Ended December 31, | December 31, | |||||||||||||||
| 2003 | 2004 | 2005 | 2005 | |||||||||||||
| Revenue | $ | — | $ | — | $ | — | $ | 2,858 | ||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 45,613 | 59,406 | 95,347 | 298,400 | ||||||||||||
| General and administrative | 20,699 | 17,743 | 22,775 | 97,975 | ||||||||||||
| In-process research and development costs | — | — | — | 19,726 | ||||||||||||
| Goodwill impairment | — | — | — | 151,428 | ||||||||||||
| Total operating expenses | 66,312 | 77,149 | 118,122 | 567,529 | ||||||||||||
| Loss from operations | (66,312 | ) | (77,149 | ) | (118,122 | ) | (564,671 | ) | ||||||||
| Other income (expense) | 36 | 226 | 78 | (1,892 | ) | |||||||||||
| Interest income | 398 | 932 | 3,707 | 9,278 | ||||||||||||
| Loss before provision for income taxes | (65,878 | ) | (75,991 | ) | (114,337 | ) | (557,285 | ) | ||||||||
| Income taxes | (1 | ) | (1 | ) | (1 | ) | (16 | ) | ||||||||
| Net loss | (65,879 | ) | (75,992 | ) | (114,338 | ) | (557,301 | ) | ||||||||
| Deemed dividend related to beneficial conversion feature of convertible preferred stock | (1,017 | ) | (19,822 | ) | — | (22,260 | ) | |||||||||
| Accretion on redeemable preferred stock | (253 | ) | (60 | ) | — | (952 | ) | |||||||||
| Net loss applicable to common stockholders | $ | (67,149 | ) | $ | (95,874 | ) | $ | (114,338 | ) | $ | (580,513 | ) | ||||
| Net loss per share applicable to common stockholders — basic and diluted | $ | (3.63 | ) | $ | (3.80 | ) | $ | (2.87 | ) | |||||||
| Shares used to compute basic and diluted net loss per share applicable to common stockholders | 18,488 | 25,221 | 39,871 | |||||||||||||
See notes to financial statements.
F-4
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
| Series C | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series C | Convertible | Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B | Series C | Convertible | Preferred | Notes | Notes | Accumulated | ||||||||||||||||||||||||||||||||||||||||||||||
| Convertible | Convertible | Preferred | Stock | Additional | Receivable | Receivable | During the | |||||||||||||||||||||||||||||||||||||||||||||
| Preferred Stock | Preferred Stock | Stock | Subscriptions | Common Stock | Paid-In | from | from | Development | ||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Issuable | Receivable | Shares | Amount | Capital | Stockholders | Officers | Stage | Total | ||||||||||||||||||||||||||||||||||||||||
| (In thousands) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| BALANCE, FEBRUARY 14, 1991 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | — | $ | — | — | $ | — | $ | — | $ | — | 998 | $ | 10 | $ | 890 | $ | — | $ | — | $ | — | $ | 900 | |||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (911 | ) | (911 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, FEBRUARY 29, 1992 | — | — | — | — | — | — | 998 | 10 | 890 | — | — | (911 | ) | (11 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash and services | — | — | — | — | — | — | 73 | 1 | 887 | — | — | — | 888 | |||||||||||||||||||||||||||||||||||||||
| Capital contribution | — | — | — | — | — | — | — | — | 20 | — | — | — | 20 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (1,175 | ) | (1,175 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, FEBRUARY 28, 1993 | — | — | — | — | — | — | 1,071 | 11 | 1,797 | — | — | (2,086 | ) | (278 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | — | — | — | — | — | — | 11 | — | 526 | — | — | — | 526 | |||||||||||||||||||||||||||||||||||||||
| Issuance of stock for notes receivable | — | — | — | — | — | — | 8 | — | 400 | (400 | ) | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (1,156 | ) | (1,156 | ) | ||||||||||||||||||||||||||||||||||||||
| BALANCE, FEBRUARY 28, 1994 | — | — | — | — | — | — | 1,090 | 11 | 2,723 | (400 | ) | — | (3,242 | ) | (908 | ) | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash and services | — | — | — | — | — | — | 36 | — | 1,805 | — | — | — | 1,805 | |||||||||||||||||||||||||||||||||||||||
| Collection of stock subscription | — | — | — | — | — | — | — | — | — | 400 | — | — | 400 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (2,004 | ) | (2,004 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1994 | — | — | — | — | — | — | 1,126 | 11 | 4,528 | — | — | (5,246 | ) | (707 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for services | — | — | — | — | — | — | — | — | 8 | — | — | — | 8 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 1 | — | 22 | — | — | — | 22 | |||||||||||||||||||||||||||||||||||||||
| Stock compensation | — | — | — | — | — | — | — | — | 384 | — | — | — | 384 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (2,815 | ) | (2,815 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1995 | — | — | — | — | — | — | 1,127 | 11 | 4,942 | — | — | (8,061 | ) | (3,108 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash and services | — | — | — | — | — | — | 1 | — | 59 | — | — | — | 59 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 3 | — | 12 | — | — | — | 12 | |||||||||||||||||||||||||||||||||||||||
| Stock compensation | — | — | — | — | — | — | — | — | 126 | — | — | — | 126 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (2,570 | ) | (2,570 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1996 | — | — | — | — | — | — | 1,131 | 11 | 5,139 | — | — | (10,631 | ) | (5,481 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash and services | — | — | — | — | — | — | 548 | 6 | 190 | — | — | — | 196 | |||||||||||||||||||||||||||||||||||||||
| Stock compensation | — | — | — | — | — | — | — | — | 2 | — | — | — | 2 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 27 | — | 135 | — | — | — | 135 | |||||||||||||||||||||||||||||||||||||||
| Conversion of notes payable | — | — | — | — | — | — | 12 | — | 60 | — | — | — | 60 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (2,280 | ) | (2,280 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1997 | — | — | — | — | — | — | 1,718 | 17 | 5,526 | — | — | (12,911 | ) | (7,368 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash and services | — | — | — | — | — | — | 2,253 | 23 | 12,703 | — | — | — | 12,726 | |||||||||||||||||||||||||||||||||||||||
| Stock compensation | — | — | — | — | — | — | — | — | 150 | — | — | — | 150 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 68 | 1 | 24 | — | — | — | 25 | |||||||||||||||||||||||||||||||||||||||
| Conversion of notes payable | — | — | — | — | — | — | 215 | 2 | 1,200 | — | — | — | 1,202 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (3,331 | ) | (3,331 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1998 | — | — | — | — | — | — | 4,254 | 43 | 19,603 | — | — | (16,242 | ) | 3,404 | ||||||||||||||||||||||||||||||||||||||
| Issuance of common stock | — | — | — | — | — | — | 162 | 2 | 532 | — | — | — | 534 | |||||||||||||||||||||||||||||||||||||||
| Conversion of notes payable | — | — | — | — | — | — | 80 | 1 | 994 | — | — | — | 995 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (5,679 | ) | (5,679 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 1999 | — | — | — | — | — | — | 4,496 | 46 | 21,129 | — | — | (21,921 | ) | (746 | ) | |||||||||||||||||||||||||||||||||||||
| Conversion of notes payable | — | — | — | — | — | — | 63 | 1 | 1,073 | — | — | — | 1,074 | |||||||||||||||||||||||||||||||||||||||
| Issuance of Series B preferred stock for cash | 193 | 15,000 | — | — | — | — | — | — | — | — | — | — | 15,000 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash, services and notes | — | — | — | — | — | — | 4,690 | 46 | 33,945 | (2,358 | ) | — | — | 31,633 | ||||||||||||||||||||||||||||||||||||||
| Discount on notes below market rate | — | — | — | — | — | — | — | — | — | 241 | — | — | 241 | |||||||||||||||||||||||||||||||||||||||
| Accrued interest on notes | — | — | — | — | — | — | — | — | — | (117 | ) | — | — | (117 | ) | |||||||||||||||||||||||||||||||||||||
| Purchase of Series A redeemable convertible preferred stock | — | — | — | — | — | — | — | — | (993 | ) | — | — | — | (993 | ) | |||||||||||||||||||||||||||||||||||||
| Amount in excess of redemption obligation | — | — | — | — | — | — | — | — | 999 | — | — | — | 999 | |||||||||||||||||||||||||||||||||||||||
F-5
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) — (Continued)
| Series C | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series C | Convertible | Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B | Series C | Convertible | Preferred | Notes | Notes | Accumulated | ||||||||||||||||||||||||||||||||||||||||||||||
| Convertible | Convertible | Preferred | Stock | Additional | Receivable | Receivable | During the | |||||||||||||||||||||||||||||||||||||||||||||
| Preferred Stock | Preferred Stock | Stock | Subscriptions | Common Stock | Paid-In | from | from | Development | ||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Issuable | Receivable | Shares | Amount | Capital | Stockholders | Officers | Stage | Total | ||||||||||||||||||||||||||||||||||||||||
| (In thousands) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion to redemption value on Series A redeemable convertible preferred stock | — | $ | — | — | $ | — | $ | — | $ | — | — | $ | — | $ | (149 | ) | $ | — | $ | — | $ | — | $ | (149 | ) | |||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | 9,609 | — | — | — | 9,609 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (24,661 | ) | (24,661 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2000 | 193 | 15,000 | — | — | — | — | 9,249 | 93 | 65,613 | (2,234 | ) | — | (46,582 | ) | 31,890 | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | — | — | — | — | — | — | 3,052 | 30 | 78,000 | — | — | — | 78,030 | |||||||||||||||||||||||||||||||||||||||
| Cash received for common stock to be issued | — | — | — | — | — | — | — | — | 3,900 | — | — | — | 3,900 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for services | — | — | — | — | — | — | 3 | — | 60 | — | — | — | 60 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 1 | — | 13 | — | — | — | 13 | |||||||||||||||||||||||||||||||||||||||
| Accrued interest on notes | — | — | — | — | — | — | — | — | — | (189 | ) | — | — | (189 | ) | |||||||||||||||||||||||||||||||||||||
| Payments on notes receivable | — | — | — | — | — | — | — | — | — | 28 | — | — | 28 | |||||||||||||||||||||||||||||||||||||||
| Accretion to redemption value on Series A redeemable convertible preferred stock | — | — | — | — | — | — | — | — | (239 | ) | — | — | — | (239 | ) | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | 1,565 | — | — | — | 1,565 | |||||||||||||||||||||||||||||||||||||||
| Issuance of put option by stockholder | — | — | — | — | — | — | — | — | (2,949 | ) | — | — | — | (2,949 | ) | |||||||||||||||||||||||||||||||||||||
| Record merger of entities | — | — | — | — | — | — | — | — | 171,154 | — | — | — | 171,154 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (48,245 | ) | (48,245 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2001 | 193 | 15,000 | — | — | — | — | 12,305 | 123 | 317,117 | (2,395 | ) | — | (94,827 | ) | 235,018 | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | — | — | — | — | — | — | 3,922 | 40 | 58,775 | — | — | — | 58,815 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash already received | — | — | — | — | — | — | 234 | 2 | (2 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Issuance of stock award to employee | — | — | — | — | — | — | 3 | — | 84 | — | — | — | 84 | |||||||||||||||||||||||||||||||||||||||
| Cash received for common stock issuable | — | — | — | — | — | — | — | — | 98 | — | — | — | 98 | |||||||||||||||||||||||||||||||||||||||
| Accrued interest on notes | — | — | — | — | — | — | — | — | — | (229 | ) | — | — | (229 | ) | |||||||||||||||||||||||||||||||||||||
| Payments on notes receivable | — | — | — | — | — | — | — | — | — | 1,314 | — | — | 1,314 | |||||||||||||||||||||||||||||||||||||||
| Beneficial conversion feature of Series B convertible preferred stock | — | — | — | — | — | — | — | — | 1,421 | — | — | — | 1,421 | |||||||||||||||||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of Series B convertible preferred stock | — | — | — | — | — | — | — | — | (1,421 | ) | — | — | — | (1,421 | ) | |||||||||||||||||||||||||||||||||||||
| Accretion to redemption value on Series A redeemable convertible preferred stock | — | — | — | — | — | — | — | — | (251 | ) | — | — | — | (251 | ) | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | 268 | — | — | — | 268 | |||||||||||||||||||||||||||||||||||||||
| Put option redemption by stockholder | — | — | — | — | — | — | — | — | 1,921 | — | — | — | 1,921 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (206,265 | ) | (206,265 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2002 | 193 | 15,000 | — | — | — | — | 16,464 | 165 | 378,010 | (1,310 | ) | — | (301,092 | ) | 90,773 | |||||||||||||||||||||||||||||||||||||
| Issuance of Series C convertible preferred stock subscriptions | — | — | — | — | 50,000 | (50,000 | ) | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Cash collected on Series C convertible preferred stock subscriptions | — | — | — | — | — | 31,847 | — | — | — | — | — | — | 31,847 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash | — | — | — | — | — | — | 3,494 | 35 | 49,965 | — | — | — | 50,000 | |||||||||||||||||||||||||||||||||||||||
| Non-cash compensation expense of officer resulting from stockholder contribution | — | — | — | — | — | — | — | — | 70 | — | — | — | 70 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for cash already received | — | — | — | — | — | — | 17 | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Notes receivable by stockholder issued to officers | — | — | — | — | — | — | 225 | — | (225 | ) | — | — | ||||||||||||||||||||||||||||||||||||||||
| Accrued interest on notes | — | — | — | — | — | — | — | — | — | (102 | ) | (3 | ) | — | (105 | ) | ||||||||||||||||||||||||||||||||||||
| Beneficial conversion feature of Series B convertible preferred stock | — | — | — | — | — | — | — | — | 1,017 | — | — | — | 1,017 | |||||||||||||||||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of Series B convertible preferred stock | — | — | — | — | — | — | — | — | (1,017 | ) | — | — | — | (1,017 | ) | |||||||||||||||||||||||||||||||||||||
| Accretion to redemption value on Series A redeemable convertible preferred stock | — | — | — | — | — | — | — | — | (253 | ) | — | — | — | (253 | ) | |||||||||||||||||||||||||||||||||||||
F-6
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) — (Continued)
| Series C | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series C | Convertible | Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Series B | Series C | Convertible | Preferred | Notes | Notes | Accumulated | ||||||||||||||||||||||||||||||||||||||||||||||
| Convertible | Convertible | Preferred | Stock | Additional | Receivable | Receivable | During the | |||||||||||||||||||||||||||||||||||||||||||||
| Preferred Stock | Preferred Stock | Stock | Subscriptions | Common Stock | Paid-In | from | from | Development | ||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Issuable | Receivable | Shares | Amount | Capital | Stockholders | Officers | Stage | Total | ||||||||||||||||||||||||||||||||||||||||
| (In thousands) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | $ | — | — | $ | — | $ | — | $ | — | — | $ | — | $ | 4,501 | $ | — | $ | — | $ | — | $ | 4,501 | |||||||||||||||||||||||||||||
| Put shares sold to majority stockholder | — | — | — | — | — | — | — | — | 623 | — | — | — | 623 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (65,879 | ) | (65,879 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2003 | 193 | 15,000 | — | — | 50,000 | (18,153 | ) | 19,975 | 200 | 433,141 | (1,412 | ) | (228 | ) | (366,971 | ) | 111,577 | |||||||||||||||||||||||||||||||||||
| Issuance of Series C convertible preferred stock for cash | — | — | 356 | 18,153 | (18,153 | ) | 18,153 | — | — | — | — | — | — | 18,153 | ||||||||||||||||||||||||||||||||||||||
| Issuance of Series C convertible preferred stock for cash already received | — | — | 624 | 31,847 | (31,847 | ) | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 86 | — | 1,079 | — | — | — | 1,079 | |||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | 4 | — | 46 | — | — | — | 46 | |||||||||||||||||||||||||||||||||||||||
| Accrued interest on notes | — | — | — | — | — | — | — | — | — | (107 | ) | — | — | (107 | ) | |||||||||||||||||||||||||||||||||||||
| Repayment of notes receivable by stockholder issued to officers | — | — | — | — | — | — | — | — | (225 | ) | — | 228 | — | 3 | ||||||||||||||||||||||||||||||||||||||
| Repayment of stock note receivable | — | — | — | — | — | — | (90 | ) | (1 | ) | (1,518 | ) | 1,519 | — | — | — | ||||||||||||||||||||||||||||||||||||
| Conversion of Series A convertible preferred stock to common stock | — | — | — | — | — | — | 891 | 9 | 5,239 | — | — | — | 5,248 | |||||||||||||||||||||||||||||||||||||||
| Conversion of Series B convertible preferred stock to common stock | (193 | ) | (15,000 | ) | — | — | — | — | 811 | 8 | 14,992 | — | — | — | — | |||||||||||||||||||||||||||||||||||||
| Conversion of Series C convertible preferred stock to common stock | — | — | (980 | ) | (50,000 | ) | — | — | 4,464 | 45 | 49,955 | — | — | — | — | |||||||||||||||||||||||||||||||||||||
| Issuance of common shares in exchange for warrants | — | — | — | — | — | — | 22 | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Issuance of common shares under Employee Stock Purchase Plan | — | — | — | — | — | — | 36 | — | 430 | — | — | — | 430 | |||||||||||||||||||||||||||||||||||||||
| Net proceeds from initial public offering | — | — | — | — | — | — | 6,557 | 66 | 83,110 | — | — | — | 83,176 | |||||||||||||||||||||||||||||||||||||||
| Beneficial conversion feature of Series B convertible preferred stock | — | — | — | — | — | — | — | — | 19,822 | — | — | — | 19,822 | |||||||||||||||||||||||||||||||||||||||
| Deemed dividends related to beneficial conversion feature of Series B and Series C convertible preferred stock | — | — | — | — | — | — | — | — | (19,822 | ) | — | — | — | (19,822 | ) | |||||||||||||||||||||||||||||||||||||
| Accretion to redemption value on Series A redeemable convertible preferred stock | — | — | — | — | — | — | — | — | (60 | ) | — | — | — | (60 | ) | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | 6,810 | — | — | — | 6,810 | |||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (75,992 | ) | (75,992 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2004 | — | — | — | — | — | — | 32,756 | 327 | 592,999 | — | — | (442,963 | ) | 150,363 | ||||||||||||||||||||||||||||||||||||||
| Issuance of common shares in exchange for warrants | — | — | — | — | — | — | 24 | — | 245 | — | — | — | 245 | |||||||||||||||||||||||||||||||||||||||
| Issuance of common shares under Employee Stock Purchase Plan | — | — | — | — | — | — | 58 | 1 | 494 | — | — | — | 495 | |||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 304 | 3 | 1,948 | — | — | — | 1,951 | |||||||||||||||||||||||||||||||||||||||
| Issuance of stock awards to consultants | — | — | — | — | — | — | 40 | 1 | (146 | ) | — | — | — | (145 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of stock and warrants for cash | — | — | — | — | — | — | 17,132 | 171 | 170,063 | — | — | — | 170,234 | |||||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | (1,828 | ) | — | — | — | (1,828 | ) | |||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | (114,338 | ) | (114,338 | ) | |||||||||||||||||||||||||||||||||||||
| BALANCE, DECEMBER 31, 2005 | — | $ | — | — | $ | — | $ | — | $ | — | 50,314 | $ | 503 | $ | 763,775 | $ | — | $ | — | $ | (557,301 | ) | $ | 206,977 | ||||||||||||||||||||||||||||
F-7
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
(In thousands)
| Cumulative | ||||||||||||||||
| Period from | ||||||||||||||||
| February 14, | ||||||||||||||||
| 1991 (Date of | ||||||||||||||||
| Inception) to | ||||||||||||||||
| Years Ended December 31, | December 31, | |||||||||||||||
| 2003 | 2004 | 2005 | 2005 | |||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||||||
| Net loss | $ | (65,879 | ) | $ | (75,992 | ) | $ | (114,338 | ) | $ | (557,301 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||
| Depreciation and amortization | 7,657 | 7,179 | 7,391 | 30,637 | ||||||||||||
| Stock-based compensation expense (benefit) | 4,501 | 6,810 | (1,728 | ) | 22,518 | |||||||||||
| Loss on sale and abandonment/disposal of property and equipment | 2,803 | 528 | 16 | 3,367 | ||||||||||||
| Accrued interest on investments, net of amortization of premiums | — | — | (146 | ) | (146 | ) | ||||||||||
| In-process research and development | — | — | — | 19,726 | ||||||||||||
| Discount on stockholder notes below market rate | — | — | — | 241 | ||||||||||||
| Non-cash compensation expense of officer resulting from stockholder contribution | 70 | — | — | 70 | ||||||||||||
| Accrued interest expense on notes payable to stockholders | — | — | — | 1,538 | ||||||||||||
| Non-cash interest expense | — | 3 | — | 3 | ||||||||||||
| Accrued interest on notes | (105 | ) | (107 | ) | — | (747 | ) | |||||||||
| Goodwill impairment | — | — | — | 151,428 | ||||||||||||
Loss onavailable-for-sale securities | 76 | 86 | — | 229 | ||||||||||||
| Changes in assets and liabilities: | ||||||||||||||||
| State research and development credit exchange receivable | — | (2,530 | ) | (695 | ) | (3,225 | ) | |||||||||
| Prepaid expenses and other current assets | (910 | ) | (1,406 | ) | 221 | (3,044 | ) | |||||||||
| Other assets | (93 | ) | 129 | (224 | ) | (285 | ) | |||||||||
| Accounts payable | (1,172 | ) | 1,486 | 509 | 3,547 | |||||||||||
| Accrued expenses and other current liabilities | 1,147 | 4,244 | 9,185 | 17,818 | ||||||||||||
| Other liabilities | (87 | ) | (46 | ) | (47 | ) | 27 | |||||||||
| Payment of deferred compensation | (220 | ) | (271 | ) | (1,373 | ) | — | |||||||||
| Net cash used in operating activities | (52,212 | ) | (59,887 | ) | (101,229 | ) | (313,599 | ) | ||||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||||||
| Purchase of marketable securities | (51,960 | ) | (16,353 | ) | (258,150 | ) | (402,718 | ) | ||||||||
| Sales of marketable securities | 59,294 | 13,648 | 180,245 | 313,040 | ||||||||||||
| Purchase of property and equipment | (5,183 | ) | (6,895 | ) | (17,169 | ) | (110,369 | ) | ||||||||
| Proceeds from sale of property and equipment | 75 | — | 90 | 182 | ||||||||||||
| Restricted cash | (559 | ) | (24 | ) | 583 | — | ||||||||||
| Net cash (used in) provided by investing activities | 1,667 | (9,624 | ) | (94,401 | ) | (199,865 | ) | |||||||||
F-8
Table of Contents
| Cumulative | ||||||||||||||||
| Period from | ||||||||||||||||
| February 14, | ||||||||||||||||
| 1991 (Date of | ||||||||||||||||
| Inception) to | ||||||||||||||||
| Years Ended December 31, | December 31, | |||||||||||||||
| 2003 | 2004 | 2005 | 2005 | |||||||||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||||||
| Issuance of common stock and warrants for cash | 50,000 | 84,731 | 172,680 | 493,251 | ||||||||||||
| Collection of Series C convertible preferred stock subscriptions receivable | 31,847 | 18,153 | — | 50,000 | ||||||||||||
| Issuance of Series B convertible preferred stock for cash | — | — | — | 15,000 | ||||||||||||
| Cash received for common stock to be issued | — | — | — | 3,900 | ||||||||||||
| Repurchase of common stock | (1,028 | ) | — | — | (1,028 | ) | ||||||||||
| Put shares sold to majority stockholder | 623 | — | — | 623 | ||||||||||||
| Borrowings under lines of credit | — | — | — | 4,220 | ||||||||||||
| Proceeds from notes receivables | — | — | — | 1,742 | ||||||||||||
| Principal payments on notes payable | — | — | — | (1,667 | ) | |||||||||||
| Payable to stockholder | 1,406 | (1,406 | ) | — | — | |||||||||||
| Borrowings on notes payable | — | — | — | 3,460 | ||||||||||||
| Net cash provided by financing activities | 82,848 | 101,478 | 172,680 | 569,501 | ||||||||||||
| NET INCREASE (DECREASE) IN CASH AND CASH EQUIVALENTS | $ | 32,303 | $ | 31,967 | $ | (22,950 | ) | $ | 56,037 | |||||||
| CASH AND CASH EQUIVALENTS, BEGINNING OF PERIOD | 14,717 | 47,020 | 78,987 | — | ||||||||||||
| CASH AND CASH EQUIVALENTS, END OF PERIOD | $ | 47,020 | $ | 78,987 | $ | 56,037 | $ | 56,037 | ||||||||
| SUPPLEMENTAL CASH FLOWS DISCLOSURES: | ||||||||||||||||
| Cash paid for income taxes | $ | 1 | $ | 1 | $ | 1 | $ | 16 | ||||||||
| Interest paid in cash | 4 | 5 | — | 80 | ||||||||||||
| Accretion on redeemable convertible preferred stock | (253 | ) | (60 | ) | — | (952 | ) | |||||||||
| Issuance of common stock upon conversion of notes payable | — | — | — | 3,331 | ||||||||||||
| Increase in additional paid-in capital resulting from merger | — | — | — | 171,154 | ||||||||||||
| Issuance of common stock for notes receivable | — | — | — | 2,758 | ||||||||||||
| Issuance of put option by stockholder | — | — | — | (2,949 | ) | |||||||||||
| Put option redemption by stockholder | — | — | — | 1,921 | ||||||||||||
| Notes receivable by stockholder issued to officers | 225 | (225 | ) | — | — | |||||||||||
| Issuance of Series C convertible preferred stock subscriptions | 50,000 | — | — | 50,000 | ||||||||||||
| Issuance of Series A redeemable convertible preferred stock | — | — | — | 4,296 | ||||||||||||
| Conversion of Series A redeemable convertible preferred stock | — | (5,248 | ) | — | (5,248 | ) | ||||||||||
In connection with the Company’s initial public offering, all shares of Series B and Series C convertible preferred stock, in the amount of $15.0 million and $50.0 million, respectively, automatically converted into common stock in August 2004.
See notes to financial statements.
F-9
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| 1. | Description of Business and Basis of Presentation |
Business — MannKind Corporation (the “Company”) is a biopharmaceutical company focused on the development and commercialization of therapeutic products for diseases such as diabetes and cancer. The Company’s lead investigational product candidate, the Technosphere Insulin System, is currently in phase 3 clinical trials in the U.S. and Europe to study its safety and efficacy in the treatment of diabetes. The Technosphere Insulin System consists of the Company’s proprietary dry-powder Technosphere formulation of insulin that is inhaled deep into the lung using the Company’s MedTone inhaler.
Basis of Presentation — The Company is considered to be in the development stage as its primary activities since incorporation have been establishing its facilities, recruiting personnel, conducting research and development, business development, business and financial planning, and raising capital. Since its inception through December 31, 2005 the Company has reported accumulated net losses of $557.3 million, which include a goodwill impairment charge of $151.4 million (see Note 2), and negative cash flow from operations of $313.6 million. It is costly to develop therapeutic products and conduct clinical trials for these products. Based upon the Company’s current expectations, management believes the Company’s existing capital resources will enable it to continue planned operations into the third quarter of 2006. However, the Company cannot provide assurances that its plans will not change or that changed circumstances will not result in the depletion of its capital resources more rapidly than it currently anticipates. Accordingly, the Company expects that it will need to raise additional capital, either through the sale of equity and/or debt securities, a strategic business collaboration with a pharmaceutical company or the establishment of other funding facilities, in order to continue the development and commercialization of its Technosphere Insulin System and other product candidates and to support its other ongoing activities. If planned operating results are not achieved or the Company is not successful in raising additional capital, management believes that planned expenditures could be reduced or eliminated in a relatively short period to continue activities, on a reduced basis, through the first quarter of 2007. Additionally, the Company’s principal stockholder has given assurances that he will provide additional funding for the Company’s operations, if necessary, through the first quarter of 2007.
On December 12, 2001, the stockholders of AlleCure Corp. (“AlleCure”) and CTL ImmunoTherapies Corp. (“CTL”) voted to exchange their shares for shares of Pharmaceutical Discovery Corporation (“PDC”). Upon approval of the merger, PDC then changed its name to MannKind Corporation. PDC was incorporated in the State of Delaware on February 14, 1991. The stockholders of PDC did not vote on the merger. At the date of the merger, Mr. Alfred Mann owned 76% of PDC, 59% of AlleCure and 69% of CTL. Accordingly, only the minority interest of AlleCure and CTL was stepped up to fair value using the purchase method of accounting. As a result of this purchase accounting, in-process research and development of $19,726,000 and goodwill of $151,428,000 were recorded at the entity level. The historical basis of PDC and the historical basis relating to the ownership interests of Mr. Mann in AlleCure and CTL have been reflected in the financial statements. For periods prior to December 12, 2001, the results of operations have been presented on a combined basis. All references in the accompanying financial statements and notes to the financial statements to number of shares, sales price and per share amounts of the Company’s capital stock have been retroactively restated to reflect the share exchange ratios for each of the entities that participated in the merger.
For periods subsequent to December 12, 2001, the accompanying financial statements have been presented on a consolidated basis and include the wholly-owned subsidiaries, AlleCure and CTL. On December 31, 2002, AlleCure and CTL merged with and into MannKind and ceased to be separate entities.
Reclassifications — Certain reclassifications have been made to the prior years’ financial statements to conform to the 2005 presentation.
Reverse Stock Split — On May 27, 2004, the Company’s board of directors approved aone-for-three reverse stock split, which was effected on July 22, 2004. All common share and per common share amounts in the consolidated financial statements and notes to financial statements have been adjusted for all periods that are presented prior to
F-10
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
July 22, 2004 to give effect to the reverse stock split, including reclassifying an amount equal to the reduction in par value to additional paid-in capital.
Segment Information — In accordance with Statement of Financial Accounting Standards (“SFAS”) No. 131,Disclosures about Segments of an Enterprise and Related Information, operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as one segment operating entirely in the United States of America.
Initial Public Offering — On August 2, 2004, the Company completed an initial public offering of its common stock at a price to the public of $14.00 per share. The Company sold 6,250,000 shares of common stock in the offering and 307,100 shares upon the exercise of the underwriters’ option to cover over-allotments resulting in aggregate net proceeds of $83.2 million. In connection with the initial public offering, all of the outstanding shares of the Company’s preferred stock were converted into shares of its common stock. Accordingly, the automatic conversion of preferred stock on August 2, 2004 into common stock is reflected in the accompanying audited consolidated financial statements.
Private Placement — On August 5, 2005, the Company completed a $175.0 million private placement of common stock and the concurrent issuance of warrants for the purchase of additional shares of common stock to accredited investors including the Company’s principal stockholder who purchased $87.3 million of the private placement. The Company sold 17,132,000 shares of common stock in the private placement, together with warrants to purchase up to 3,426,000 shares of common stock at an exercise price of $12.228 per share. In connection with this private placement, the Company paid $4.5 million in commissions to the placement agents and incurred $300,000 in other offering expenses which resulted in net proceeds of approximately $170.2 million.
| 2. | Summary of Significant Accounting Policies |
Financial Statement Estimates — The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Cash and Cash Equivalents — The Company considers all highly liquid investments with a purchased maturity date of three months or less to be cash equivalents.
Concentration of Credit Risk — Financial instruments that potentially subject the Company to concentration of credit risk consist of cash and cash equivalents and marketable securities. Cash and cash equivalents consist primarily of interest-bearing accounts and are regularly monitored by management and held in high credit quality institutions. Marketable securities consist of highly liquid short-term investment securities such as government and investment-grade corporate debt.
Marketable Securities — The Company accounts for marketable securities as available for sale, in accordance with SFAS No. 115,Accounting for Certain Debt and Equity Securities. Unrealized holding gains and losses foravailable-for-sale securities are reported as a separate component of stockholders’ equity until realized. The Company reviews the portfolio for other than temporary impairment in accordance with Emerging Issues Task Force (“EITF”) IssueNo. 03-01,The Meaning ofOther-Than-Temporary Impairment and Its Application to Certain Investmentsand FASB Staff Position No. 115-1,The Meaning of Other-Than-Temporary Impairment and Its Application to Certain Investments.
State Research and Development Credit Exchange Receivable — The State of Connecticut provides certain companies with the opportunity to exchange certain research and development income tax credit carryforwards for
F-11
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
cash in exchange for foregoing the carryforward of the research and development credits. The program provides for an exchange of research and development income tax credits for cash equal to 65% of the value of corporation tax credit available for exchange. Estimated amounts receivable under the program are recorded as a reduction of research and development expenses.
Fair Value of Financial Instruments — The carrying amounts of financial instruments, which include cash equivalents, marketable securities, accounts payable, accrued expenses and other current liabilities and payable to stockholder, approximate their fair values due to their relatively short maturities. The carrying amounts of the notes receivable from stockholders reflect market rates of interest for similar loans of similar amounts and terms available from a third party (see Notes 7 and 8).
Goodwill and Identifiable Intangibles — As a result of the merger with AlleCure and CTL on December 12, 2001, as described in Note 1, goodwill of $151.4 million was recorded at the entity level in 2001. Upon adoption of SFAS No. 142,Goodwill and Other Intangible Assets, the Company adopted a policy of testing goodwill and intangible assets with indefinite lives for impairment at least annually, as of December 31, with any related impairment losses being recognized in earnings when identified. In December 2002 the Company concluded that the major AlleCure product development program should be terminated and that the clinical trials of the CTL product should be halted and returned to the research stage. As a result of this determination, the Company closed the CTL facility and reduced headcount for AlleCure and CTL by approximately 50%. In connection with the annual test for impairment of goodwill as of December 31, 2002, the Company determined that on the basis of the internal study, the goodwill recorded for the AlleCure and CTL units was potentially impaired. The Company performed the second step of the annual impairment test as of December 31, 2002 for each of the potentially impaired reporting units and estimated the fair value of the AlleCure and CTL programs using the expected present value of future cash flows which were expected to be negligible. Accordingly, the goodwill balance of $151.4 million, was determined to be fully impaired and an impairment loss was recorded in 2002. Subsequent to December 31, 2002, the Company had no goodwill or intangibles with indefinite lives included on its balance sheet.
Property and Equipment — Property and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the related assets. Leasehold improvements are amortized over the term of the lease or the service lives of the improvements, whichever is shorter. Assets under construction are not depreciated until placed into service.
Impairment of Long-Lived Assets — The Company evaluates long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable in accordance with SFAS No. 144,Accounting for the Impairment or Disposal of Long Lived-Assets. Assets are considered to be impaired if the carrying value may not be recoverable based upon management’s assessment of the following events or changes in circumstances:
| • | significant changes in the Company’s strategic business objectives and utilization of the assets; | |
| • | a determination that the carrying value of such assets can not be recovered through undiscounted cash flows; | |
| • | loss of legal ownership or title to the assets; or | |
| • | the impact of significant negative industry or economic trends. |
If the Company believes an asset to be impaired, the impairment recognized is the amount by which the carrying value of the assets exceeds the fair value of the assets. Any write-downs would be treated as permanent reductions in the carrying amount of the asset and an operating loss would be recognized. For the years ended December 31, 2004 and 2005, the Company did not consider any long-lived assets to be impaired based on management’s assessment.
F-12
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
Accounts Payable and Accrued Expenses — All liabilities, including accounts payable and accrued expenses, are recorded consistent with the definition of liabilities and accrual accounting as provided by Financial Accounting Standards Board (“FASB”) Statement of Financial Accounting Concepts No. 6,Elements of Financial Statements.
Income Taxes — In accordance with SFAS No. 109,Accounting for Income Taxes, deferred income tax assets and liabilities are recorded for the expected future tax consequences of temporary differences between the financial statement carrying amounts and the income tax basis of assets and liabilities. A valuation allowance is recorded to reduce net deferred income tax assets to amounts that are more likely than not to be realized.
Significant management judgment is involved in determining the provision for income taxes, deferred tax assets and liabilities and any valuation allowance recorded against net deferred tax assets. Due to uncertainties related to deferred tax assets as a result of the history of operating losses, a valuation allowance has been established against the gross deferred tax asset balance. The valuation allowance is based on management’s estimates of taxable income by jurisdiction in which the Company operates and the period over which deferred tax assets will be recoverable. In the event that actual results differ from these estimates or the Company adjusts these estimates in future periods, a change in the valuation allowance may be needed, which could materially impact the Company’s financial position and results of operations.
Contingencies — Contingencies are recorded in accordance with SFAS No. 5,Accounting for Contingencies.
Stock-Based Compensation — At December 31, 2005, the Company has four stock-based compensation plans, which are described more fully in Note 10. The Company accounts for employee stock options and the employee stock purchase plan using the intrinsic value method in accordance with Accounting Principles Board (“APB”) Opinion No. 25,Accounting for Stock Issued to Employees, and its interpretations, and has adopted the disclosure-only alternative of SFAS No. 123,Accounting for Stock-Based Compensation. Stock options issued to consultants are accounted for in accordance with the provisions of EITF IssueNo. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services, and FASB Interpretation No. 28 (“FIN 28”),Accounting for Stock Appreciation Rights and Other Variable Stock Option or Award Plans.
Under SFAS No. 123, the Company estimates the fair value of each stock option at the grant date or modification date, if any, using the Black-Scholes option valuation model with the following weighted-average assumptions:
| Year Ended December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Risk-free interest rate | 2.90 | % | 3.40 | % | 4.05 | % | ||||||
| Expected lives | 4.0 years | 4.0 years | 4.8 years | |||||||||
| Volatility | 100 | % | 100 | % | 86 | % | ||||||
F-13
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
Had compensation cost been determined under the accounting provisions of SFAS No. 123, the Company’s net loss would have been adjusted to the pro forma amounts indicated below (in thousands, except per share amounts):
| Year Ended December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Net loss — as reported | $ | (65,879 | ) | $ | (75,992 | ) | $ | (114,338 | ) | |||
| Add: Stock-based employee compensation expense (benefit) included in reported net loss | 4,224 | 5,694 | (1,892 | ) | ||||||||
| Deduct: Total stock-based employee compensation expense determined under the fair value based method for all awards | (6,586 | ) | (10,302 | ) | (13,452 | ) | ||||||
| Net loss — pro forma | (68,241 | ) | (80,600 | ) | (129,682 | ) | ||||||
| Deemed dividends related to beneficial conversion features of convertible preferred stock | (1,017 | ) | (19,822 | ) | — | |||||||
| Accretion on redeemable preferred stock | (253 | ) | (60 | ) | — | |||||||
| Net loss applicable to common stockholders — pro forma | $ | (69,511 | ) | $ | (100,482 | ) | $ | (129,682 | ) | |||
| Basic and diluted loss applicable to common stockholders per share, as reported | $ | (3.63 | ) | $ | (3.80 | ) | $ | (2.87 | ) | |||
| Basic and diluted loss applicable to common stockholders per share, pro forma | $ | (3.76 | ) | $ | (3.98 | ) | $ | (3.25 | ) | |||
Warrants — The Company has issued warrants to purchase shares of its common stock. Warrants are accounted for as equity in accordance with the provisions of EITF IssueNo. 00-19:Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock.
Research and Development Expenses — Research and development expenses consist primarily of costs associated with the clinical trials of the Company’s product candidates, manufacturing supplies and other development materials, compensation and other expenses for research and development personnel, costs for consultants and related contract research, facility costs, and depreciation. Research and development costs, which are net of any tax credit exchange recognized for the Connecticut state research and development credit exchange program, are expensed as incurred consistent with SFAS No. 2,Accounting for Research and Development Costs.
Clinical Trial Expenses — Clinical trial expenses, which are reflected in research and development expenses in the accompanying statements of operations, result from obligations under contract with vendors, consultants, and clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to the Company under such contracts. The appropriate level of trial expenses are reflected in the Company’s financial statements by matching period expenses with period services and efforts expended. These expenses are recorded according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. Clinical trial accrual estimates are determined through discussions with internal clinical personnel and outside service providers as to the progress or state of completion of trials, or the services completed. Service provider status is then compared to the contractually obligated fee to be paid for such services. During the course of a clinical trial, the Company may adjust the rate of clinical expense recognized if actual results differ from management’s estimates. The date on which certain services commence, the level of services performed on or before a given date and the cost of the services are often judgmental.
Net Loss Per Share of Common Stock — Basic net loss per share excludes dilution for potentially dilutive securities and is computed by dividing loss applicable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net loss per share reflects the potential dilution that could
F-14
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
occur if securities or other contracts to issue common stock were exercised or converted into common stock. Potentially dilutive securities are excluded from the computation of diluted net loss per share for all of the periods presented in the accompanying statements of operations because the reported net loss in each of these periods results in their inclusion being antidilutive.
Potentially dilutive securities outstanding are summarized as follows:
| December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Series A redeemable convertible preferred stock on an as converted to common stock basis | 890,706 | — | — | |||||||||
| Series B convertible preferred stock on an as converted to common stock basis | 746,408 | — | — | |||||||||
| Series C convertible preferred stock on an as converted to common stock basis | — | — | — | |||||||||
| Stock warrants to purchase common stock | 174,917 | 131,628 | 3,438,776 | |||||||||
| Restricted stock units | — | — | 164,901 | |||||||||
| Common stock options | 2,099,824 | 4,067,979 | 4,985,831 | |||||||||
Exit or Disposal Activities — SFAS No. 146,Accounting for Costs Associated with Exit or Disposal Activities, was effective for exit or disposal activities initiated after December 31, 2002. SFAS No. 146 addresses financial accounting and reporting for the costs associated with exit or disposal activities and EITF IssueNo. 94-3,Liability Recognition for Certain Employee Termination Benefits and Costs to Exit and Disposal Activity (Including Certain Costs Incurred in a Restructuring). SFAS No. 146 requires that a liability for costs associated with an exit or disposal activity be recognized when the liability is incurred and establishes that fair value is the objective for initial measurements of the liability.
Included in general and administrative expense for the year ended December 31, 2003 are approximately $2.2 million of costs related to the consolidation of the Company’s separate California facilities into the Company’s Valencia facility. The $2.2 million consists of $1.1 million in severance costs, $438,000 in office closure costs and $648,000 related to the abandonment of fixed assets. The severance and office closure costs aggregating $1.5 million were initially recorded as an accrued liability and included in accrued expenses and other current liabilities in the accompanying consolidated balance sheets. As of December 31, 2004, payments against this liability totaled $1.5 million. The remaining liability of $65,000 was paid in February 2005.
Also, as a result of the merger with AlleCure and CTL, the Company recorded a write-off of long-lived assets of approximately $2.2 million and $428,000, for the years ended December 31, 2003 and 2004, respectively.
Recently Issued Accounting Standards — In May 2005, the FASB issued SFAS No. 154,Accounting Changes and Error Corrections, which replaces APB Opinion No. 20,Accounting Changesand SFAS No. 3,Reporting Accounting Changes in Interim Financial Statements — An Amendment of APB Opinion No. 28. SFAS No. 154 provides guidance on the accounting for and reporting of accounting changes and error corrections. It establishes retrospective application, or the latest practicable date, as the required method for reporting a change in accounting principle and the reporting of a correction of an error. SFAS No. 154 is effective for accounting changes and corrections of errors made in fiscal years beginning after December 15, 2005. The Company believes that the adoption of this statement will not have a material effect on its financial condition or results of operations.
In March 2005, the FASB issued FASB Interpretation No. 47 (“FIN 47”),Accounting for Conditional Asset Retirement Obligations, which clarifies that the term conditional asset retirement obligation as used in SFAS No. 143,Accounting for Asset Retirement Obligations, refers to a legal obligation to perform an asset retirement activity in which the timing and (or) method of settlement are conditional on a future event that may or
F-15
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
may not be within the control of the entity. FIN 47, which went into effect in December 2005, governs the disclosures related to future environmental liabilities and affected companies must recognize future estimated cleanup costs on current financial statements. The Company believes the estimated costs associated with potential environmental liabilities are not material.
In December 2004, the FASB issued SFAS No. 123R,Share-based Payment: an Amendment of FASB Statements No. 123 and 95. The statement requires companies to expense share-based payments to employees, including stock options, based on the fair value of the award at the grant date. The statement also eliminates the intrinsic value method of accounting for stock options permitted by APB No. 25, which the Company followed until December 31, 2005. The Company is required to adopt SFAS No. 123R for the quarter that begins January 1, 2006. Upon adoption of SFAS No. 123R, the Company is required to select an adoption method and a valuation method that requires the input of highly subjective assumptions, including the expected volatility of its stock price and an expected option term. The first adoption method is a modified prospective transition method whereby a company would recognize share-based employee costs from the beginning of the fiscal period in which the recognition provisions are first applied as if the fair value accounting method had been used to account for all employee awards granted, modified, or settled after the effective date and to any awards that were not fully vested as of the effective date. Measurement and attribution of compensation cost for awards that are unvested as of the effective date of SFAS No. 123R would be based on the same estimate of the grant-date fair value and the same attribution method used previously under SFAS No. 123. The second adoption method is a modified retrospective transition method whereby a company would recognize employee compensation cost for periods presented prior to the adoption of SFAS No. 123R in accordance with the original provisions of SFAS 123; that is, an entity would recognize employee compensation costs in the amounts reported in the pro forma disclosures provided in accordance with SFAS No. 123. A company would not be permitted to make any changes to those amounts upon adoption of SFAS No. 123R unless those changes represent a correction of an error. For periods after the date of adoption of SFAS No. 123R, the modified prospective transition method described above would be applied. The Company continues to believe that the expensing of stock-based compensation will have an impact on the Company’s Statements of Operations similar to the pro forma disclosure under SFAS No. 123.
In March 2004, the FASB ratified the measurement and recognition guidance and certain disclosure requirements for impaired securities as described in EITF IssueNo. 03-1,The Meaning ofOther-Than-Temporary Impairment and Its Application to Certain Investments. On November 3, 2005, the FASB issued FASB Staff Position (“FSP”) Nos.FAS 115-1 andFAS 124-1 which addresses the determination as to when an investment is considered impaired, whether that impairment is other than temporary, and the measurement of an impairment loss. This FSP also includes accounting considerations subsequent to the recognition of another-than-temporary impairment and requires certain disclosures about unrealized losses that have not been recognized asother-than-temporary impairments. The guidance in this FSP amends SFAS No. 115,Accounting for Certain Investments in Debt and Equity Securities”, and SFAS No. 124,Accounting for Certain Investments Held byNot-for-Profit Organizationsand APB Opinion No. 18,The Equity Method of Accounting for Investments in Common Stock. The FSP is effective for reporting periods beginning after December 15, 2005. The Company’s investment portfolio at December 31, 2005 is comprised of highly liquid short-term securities. Unrealized losses for these securities are not material.
F-16
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 3. | Investment in securities |
The following is a summary of theavailable-for-sale securities classified as current assets (in thousands).
| December 31, | December 31, | |||||||||||||||
| 2004 | 2005 | |||||||||||||||
| Cost Basis | Fair Value | Cost Basis | Fair Value | |||||||||||||
| US government securities | $ | 1,443 | $ | 1,443 | $ | — | $ | — | ||||||||
| Corporate debt instruments | 2,353 | 2,353 | — | — | ||||||||||||
| Auction rate preferred stock | — | — | — | — | ||||||||||||
| Auction rate municipal bonds | 7,750 | 7,750 | 89,597 | 89,597 | ||||||||||||
| $ | 11,546 | $ | 11,546 | $ | 89,597 | $ | 89,597 | |||||||||
The Company’s policy is to maintain a highly liquid short-term investment portfolio. The contractual maturities for auction rate municipal bonds at December 31, 2005 are between 19 and 40 years. Despite the long-term nature of their stated contractual maturities, the Company has the ability to quickly liquidate these securities. Proceeds from the sale and maturities ofavailable-for-sale securities amounted to approximately $59.3 million, $13.6 million, and $180.2 million for the years ended December 31, 2003, 2004 and 2005, respectively. Gross realized gains and losses foravailable-for-sale securities were insignificant for the years ended December 31, 2003, 2004 and 2005. Gross realized gains and losses foravailable-for-sale securities are recorded as other income (expense). The cost of securities sold is based on the specific identification method. Unrealized gains and losses foravailable-for-sale securities for all periods presented in the table above were not material.
| 4. | State research and development credit exchange receivable |
The State of Connecticut provides certain companies with the opportunity to exchange certain research and development income tax credit carryforwards for cash in exchange for forgoing the carryforward of the research and development income tax credits. The program provides for an exchange of research and development income tax credits for cash equal to 65% of the value of corporation tax credit available for exchange. Estimated amounts receivable under the program are recorded as a reduction of research and development expenses. During the years ended December 31, 2004 and 2005, research and development expenses were offset by $4.0 million and $1.7 million, respectively, in connection with the program. The three months ended September 30, 2004 was the first period in which the Company was able to recognize the benefit of these credits for financial reporting purposes and accordingly the $4.0 million recognized in the year ended December 31, 2004 included amounts attributable to 2004 and prior years.
F-17
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 5. | Property and equipment |
Property and equipment consist of the following (dollar amounts in thousands):
| Estimated | ||||||||||||
| Useful | ||||||||||||
| Life | December 31, | |||||||||||
| (years) | 2004 | 2005 | ||||||||||
| Land | — | $ | 5,273 | $ | 5,273 | |||||||
| Buildings | 39-40 | 9,566 | 9,566 | |||||||||
| Building improvements | 5-40 | 37,397 | 39,543 | |||||||||
| Machinery and equipment | 3-10 | 18,080 | 23,087 | |||||||||
| Furniture, fixtures and office equipment | 5-10 | 2,391 | 2,573 | |||||||||
| Computer equipment and software | 3 | 3,308 | 4,808 | |||||||||
| Leasehold improvements | 627 | 5 | ||||||||||
| Construction in progress | 3,326 | 10,298 | ||||||||||
| Deposits on equipment | 5,911 | 6,411 | ||||||||||
| 85,879 | 101,564 | |||||||||||
| Less accumulated depreciation and amortization | (19,368 | ) | (25,381 | ) | ||||||||
| Property and equipment — net | $ | 66,511 | $ | 76,183 | ||||||||
Leasehold improvements are amortized over four years which is the shorter of the term of the lease or the service lives of the improvements. Depreciation and amortization expense for the years ended December 31, 2003, 2004 and 2005, and the cumulative period from February 14, 1991 (date of inception) to December 31, 2005 was $7.7 million, $7.2 million, $7.4 million and $30.6 million, respectively.
| 6. | Accrued expenses and other current liabilities |
Accrued expenses and other current liabilities are comprised of the following (in thousands):
| December 31, | ||||||||
| 2004 | 2005 | |||||||
| Salary and related expenses | $ | 2,325 | $ | 4,291 | ||||
| Research and clinical trial costs | 4,116 | 8,589 | ||||||
| Litigation settlement payable | — | 1,400 | ||||||
| Other | 2,191 | 3,538 | ||||||
| Accrued expenses and other current liabilities | $ | 8,632 | $ | 17,818 | ||||
| 7. | Notes receivable from stockholders |
During the year ended December 31, 2000, the Company issued an aggregate of 238,000 shares of common stock to an executive of CTL and a consultant of CTL in exchange for full recourse notes receivable of $1.2 million each, for an aggregate amount of $2.4 million. The notes bore interest at fixed rates and were payable in five years. The notes were prepayable at the option of the debtor. The notes were collateralized by the underlying common stock. The debtors had no obligation to provide services to the Company under the terms of the stock purchases. The notes bore fixed rates of interest that were less than market rates of interest available for similar loans of similar amounts and terms from a third party; consequently, the Company recognized compensation expense equal to the discount on the notes based on market rates of interest and the terms of the notes. The total discount on the notes of $241,000 was
F-18
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
expensed to general and administrative expense during the year ended December 31, 2000 as the debtors had no further obligation to the Company. During the year ended December 31, 2002, the executive of CTL paid his $1.2 million note in full along with $135,000 of accrued interest. As of December 31, 2003, notes receivable reflected in stockholders’ equity consisted of $1.2 million in principal and $233,000 in accrued interest. During the year ended December 31, 2004, the consultant of CTL tendered to the Company 90,025 shares of the Company’s common stock held by the consultant to satisfy in full his $1,179,000 note plus $340,000 of accrued interest.
The Company issued 110,000 shares of common stock to an executive of AlleCure in exchange for notes receivable, in the amounts of $1.2 million during the year ended December 31, 2000 and $750,000 during the year ended December 31, 2001. The notes bore interest at fixed rates and were payable in five years. The notes were pre-payable at the option of the debtor. The notes were collateralized by the underlying common stock. The executive had no further obligation to the Company under the terms of the stock purchase. During the first quarter of 2003, the executive was terminated by the Company (SeeNote 12-Commitments and Contingencies — Severance Agreements). Thenote-for-stock transactions have been accounted for as in-substance stock option grants to an employee. The in-substance stock option grants had no intrinsic value as of the transaction dates. The pre-payment feature of the notes resulted in the exercise price of the in-substance stock option being unknown until the notes were paid in full. Accordingly, the Company was required to measure the intrinsic value of the in-substance stock options on the balance sheet date of each financial reporting period. During 2001, the Company recorded approximately $815,000 of stock-based compensation expense, which was included in general and administrative expense, relating to the in-substance stock options. This amount was reversed in 2002 because the in-substance stock options had no intrinsic value as of December 31, 2002. There was no stock-based compensation expense recorded for the in-substance options in 2003 because they had no intrinsic value as of December 31, 2003. The Company recorded approximately $17,000 of stock-based compensation expense relating to the in-substance stock options during the year ended December 31, 2004, which represented the intrinsic value of the in-substance options at year end. This amount was reversed in 2005 because the in-substance stock options had no intrinsic value as of December 31, 2005. In September and October 2005, the principal and interest totaling $1.6 million on the notes issued in exchange for approximately 78,000 of the 110,000 shares of common stock issued became due and payable. The Company pursued collection and, in January 2006, the debtor tendered 143,949 shares as full repayment of the notes in default and the note issued in exchange for 32,000 shares which would become due in April 2006. These shares were valued at $2.6 million on January 31, 2006 and represented principal and interest due through that date on all notes outstanding. The Company received and cancelled 143,949 shares on January 31, 2006.
During the years ended December 31, 2000 and 2001, the Company issued an aggregate 701,333 shares of common stock to various consultants in exchange for notes receivable aggregating approximately $10.9 million. The notes bore interest at fixed rates and were payable in five years. The notes were pre-payable at the option of the debtors. The notes were collateralized by the underlying common stock. The consultants had no further obligation to the Company under the terms of the stock purchases. Thenote-for-stock transactions have been accounted for as in-substance stock option grants to non-employees. Since the in-substance stock options were 100% vested and nonforfeitable upon issuance, a measurement date was deemed to have occurred on the issuance date. Accordingly, the Company recorded stock-based compensation expense equal to the estimated fair value of the in-substance options of $8.4 million in 2000 and $15,000 in 2001. These amounts were estimated using the Black-Scholes option valuation model and the following weighted-average assumptions: volatility of 100%, term of five years, interest rate of 5.06%. Notes issued in exchange for 699,972 of the 701,333 shares aggregating $10.9 million in principal became due on October 19, 2005. The remaining note for $32,000 is due in September 2006. A total of $14.6 million in principal and interest became due and payable on October 19, 2005 and the Company pursued collection. On November 21, 2005, the consultants filed a complaint against the Company in the California Superior Court, County of Los Angeles,Rollins et al. v. MannKind et al, Case No. BC343381. The complaint alleges causes of action for breach of certain agreements. On January 19, 2006, the parties mediated and settled the case. Under the settlement,
F-19
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
the Company repurchased 620,697 shares from the consultants in full satisfaction of the principal and interest on the notes previously accounted for by the Company as in-substance stock options. These shares were tendered and cancelled on February 3, 2006. The Company also agreed to repurchase the remaining 79,275 shares held by the consultants for $1.4 million in cash. The settlement was reflected as a $1.4 million charge to general and administrative expense in the fourth quarter of the year ended December 31, 2005 with a corresponding amount payable included in accrued expenses and other current liabilities as of December 31, 2005. On February 21, 2006, the Company received and cancelled the 79,275 shares, and paid the purchase price of $1.4 million to the consultants. The complaint was dismissed in its entirety with prejudice.
| 8. | Deferred compensation |
Certain officers elected to defer part or all of their compensation from 1991 through 1998 due to Company’s cash flow difficulties in those years. The amounts due for deferred compensation were non-interest bearing with no repayment terms. The remaining balance of $1.4 million at December 31, 2004 was paid in June 2005.
In February 2003, pursuant to a settlement agreement with one of the officers, the Company agreed to pay the deferred compensation amount outstanding to the former officer in the amount of approximately $775,000 in three payments. The Company paid approximately $220,000 and $271,000 of this amount during the years ended December 31, 2003 and 2004, respectively. The remaining $284,000 was paid in April 2005. The settlement also obligated the Company to make certain severance related payments which are more fully described inNote 12-Commitment and Contingencies — Severance Agreements.
| 9. | Common and preferred stock |
Initial Public Offering — Upon closing of the initial public offering of the Company’s common stock on August 2, 2004, 6,250,000 shares of common stock were sold. The underwriters exercised a30-day option related to the initial public offering and purchased 307,100 shares of common stock on August 28, 2004.
Private Placement — On August 5, 2005, the Company closed a $175.0 million private placement of common stock and the concurrent issuance of warrants for the purchase of additional shares of common stock to accredited investors including the Company’s principal stockholder who purchased $87.3 million of the private placement. The Company sold 17,132,000 shares of common stock in the private placement, together with warrants to purchase up to 3,426,000 shares of common stock at an exercise price of $12.228 per share which became exercisable on February 1, 2006 and expire on August 5, 2010. In connection with this private placement, the Company paid $4.5 million in commissions to the placement agents and incurred $300,000 in other offering expenses which resulted in net proceeds of approximately $170.2 million.
Common Stock — The Company is authorized to issue 90,000,000 shares of common stock. As of December 31, 2005, 50,314,108 shares of common stock are issued and outstanding.
The Company had reserved shares of common stock for issuance as follows:
| December 31, | December 31, | |||||||
| 2004 | 2005 | |||||||
| Common stock options | 4,067,979 | 4,985,831 | ||||||
| Restricted stock units | — | 164,901 | ||||||
| Exercise of warrants | 131,628 | 3,438,776 | ||||||
| 4,199,607 | 8,589,508 | |||||||
Preferred Stock — The Company is authorized to issue 10,000,000 shares of preferred stock. As of December 31, 2005, no shares of preferred stock are issued and outstanding.
F-20
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
In connection with the initial public offering, all 267,212 shares of the Company’s Series A redeemable convertible preferred stock, all 192,618 shares of the Company’s Series B convertible preferred stock and all 980,392 shares of the Company’s Series C convertible preferred stock converted into 6,166,372 shares of common stock on August 2, 2004. While none of the convertible preferred stock were determined to contain beneficial conversion features on their respective offering commitment dates, pursuant to the terms of the Series B and Series C preferred stock, their conversion prices were adjusted downward through the date of their conversion resulting in beneficial conversion charges to common stockholders in the aggregate amount of $1.1 million during the year ended December 31, 2003 and $19.8 million during the year ended December 31, 2004. In addition, differences between the redemption amounts of the Series A redeemable convertible preferred stock and the net proceeds received (i.e., the costs of financing) were accreted in the amount of $253,000 during the year ended December 31, 2003 and $60,000 during the year ended December 31, 2004. These charges have been reflected by the Company as both a reduction and increase in additional paid-in capital during the respective periods. Although the charges have no net effect on stockholders’ equity, they increase the net loss applicable to the common stockholders and net loss per share.
Registration rights — As of December 31, 2005 the holders of 916,715 shares of the Company’s common stock and the holders of warrants to purchase 12,436 shares of the Company’s common stock have rights, subject to some conditions, to require the Company to file registration statements covering their shares or to include their shares in registration statements that the Company may file for itself or other stockholders.
As of December 31, 2005, the holders of 17,132,000 shares of common stock together with warrants to purchase up to 3,426,000 shares of common stock, all of which were issued in the August 2005 private placement, have rights that require the Company to keep the registration of the shares of common stock purchased in the private placement or underlying warrants continuously effective until at least August 2007.
| 10. | Stock option plans |
During 2004, the Company’s board of directors and stockholders approved the amendment and restatement of the 2001 Stock Awards Plan, now known as the 2004 Equity Incentive Plan (the “Plan”). The Plan provides for the granting of stock awards including stock options and restricted stock units, to employees, directors and consultants.
The Company’s board of directors determines eligibility, vesting schedules and exercise prices for stock awards granted under the Plan. Options and other stock awards under the Plan expire not more than ten years from the date of the grant and are exercisable upon vesting. Stock options generally vest over four years. Current stock option grants vest and become exercisable at the rate of 25% after one year and ratably on a monthly basis over a period of 36 months thereafter. Restricted stock units generally vest over four years with consideration satisfied by service to the Company.
During 2005, the Company’s board of directors granted 1,634,679 options at a weighted average exercise price of $12.03 and 165,354 restricted stock units. The restricted stock units are not included in the tables below, which describe stock option activity. As of December 31, 2005, 4,142,840 options and 164,901 restricted stock units were outstanding under the Plan and an additional 469,293 shares of common stock were available for issuance under the Plan. In February 2006, the Company’s board of directors approved an increase of 4.0 million shares in the number of shares reserved under the Plan for future issuance.
During 2004, the Company’s board of directors and stockholders also approved the 2004 Non-Employee Directors’ Stock Option Plan. The plan provides for the automatic, non-discretionary grant of options to the Company’s non-employee directors. As of December 31, 2005, 267,500 options were outstanding under the plan and an additional 532,500 shares of common stock were available for issuance under the plan.
The Company has two other stock award plans: the 1991 Stock Option Plan (the “1991 Plan”) and the 1999 Stock Plan (the “1999 Plan”). Both of these plans provide for the granting of stock options to directors, employees and consultants. As of December 31, 2005, 73,090 options were outstanding under the 1991 Plan and 179,271 options
F-21
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
were outstanding under the 1999 Plan. There were no additional shares available for issuance under the 1991 Plan or the 1999 Plan at December 31, 2005.
Prior to the merger of CTL and AlleCure into the Company, CTL had granted options to purchase shares of its common stock under its 2000 Stock Option and Stock Plan (the “CTL Plan”). Similarly, AlleCure had granted options to purchase shares of its common stock under its 2000 Stock Option and Stock Plan (the “AlleCure Plan”). Pursuant to the plans of reorganization and agreements of merger between the Company and each of CTL and AlleCure, the Company has assumed the obligation to issue shares of the Company’s common stock, at the exchange ratio agreed to in the merger agreement, upon the exercise of options granted under the CTL Plan and the AlleCure Plan. After the merger date, no further options were granted under either the CTL Plan or AlleCure Plan. As of December 31, 2005 there were an aggregate of 82,158 options outstanding under these plans. The assumption of options issued by CTL and AlleCure by the Company in connection with the merger on December 12, 2001 resulted in a new measurement date. On this date, the outstanding options had an intrinsic value of approximately $2.6 million. Approximately $632,000 of the $2.6 million related to vested options and was therefore recorded immediately as compensation expense. The remaining amount of the $2.6 million, as adjusted for subsequent cancellations, related to unvested options and was being amortized to stock-based compensation expense over the remaining vesting period. During the years ended December 31, 2002, 2003, 2004 and the cumulative period from February 14, 1991 (date of inception) to December 31, 2004, $632,000, $275,000, $214,000 and $1.8 million, respectively, was recognized as stock-based compensation expense related to these options. As of December 31, 2004, there was no further expense to recognize.
During the year ended December 31, 2002, the Company’s board of directors granted 240,972 options at an exercise price of $25.23 per share to an employee who is the principal stockholder. The options vest annually over four years. These options, which were not under any plan, were outstanding at December 31, 2005 and are included in the tables below.
In March 2004, the Company’s board of directors approved the 2004 Employee Stock Purchase Plan, which became effective upon the closing of the Company’s initial public offering. The aggregate number of shares that may be sold under the plan is 2,000,000 shares of common stock. On January 1 of each year, for a period of ten years beginning January 1, 2005, the share reserve automatically increases by the lesser of: 700,000 shares, 1% of the total number of shares of common stock outstanding on that date, or an amount as may be determined by the board of directors. However, under no event can the annual increase cause the total number of shares reserved under the purchase plan to exceed 10% of the total number of shares of capital stock outstanding on December 31 of the prior year. On January 1, 2005 and 2006, the purchase plan share reserve was increased by 327,562 and 503,141 shares, respectively. For the years ended December 31, 2004 and 2005, the Company sold 36,152 and 57,642 shares, respectively, of its common stock to employees participating in the plan.
F-22
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
The following table summarizes information about stock options outstanding:
| Year Ended December 31, | ||||||||||||||||||||||||
| 2003 | 2004 | 2005 | ||||||||||||||||||||||
| Weighted | Weighted | Weighted | ||||||||||||||||||||||
| Average | Average | Average | ||||||||||||||||||||||
| Exercise | Exercise | Exercise | ||||||||||||||||||||||
| Options | Price | Options | Price | Options | Price | |||||||||||||||||||
| Options outstanding at the beginning of the period | 1,904,603 | $ | 17.52 | 2,099,825 | $ | 10.29 | 4,067,979 | $ | 12.19 | |||||||||||||||
| Granted | 1,664,886 | 9.36 | 2,145,124 | 14.09 | 1,634,679 | 12.03 | ||||||||||||||||||
| Exercised | — | — | (86,066 | ) | 12.54 | (304,555 | ) | 6.42 | ||||||||||||||||
| Canceled | (1,469,664 | ) | 18.57 | (90,904 | ) | 13.02 | (412,272 | ) | 13.27 | |||||||||||||||
| Options outstanding at the end of the period | 2,099,825 | 10.29 | 4,067,979 | 12.19 | 4,985,831 | 12.40 | ||||||||||||||||||
| Options exercisable at the end of the period | 728,590 | 9.96 | 1,236,599 | 9.76 | 1,937,918 | 11.55 | ||||||||||||||||||
The weighted-average exercise prices and weighted-average fair values of options granted are as follows:
| Year Ended December 31, | ||||||||||||||||||||||||
| 2003 | 2004 | 2005 | ||||||||||||||||||||||
| Weighted | Weighted | Weighted | Weighted | Weighted | Weighted | |||||||||||||||||||
| Average | Average Fair | Average | Average Fair | Average | Average Fair | |||||||||||||||||||
| Exercise | Value of | Exercise | Value of | Exercise | Value of | |||||||||||||||||||
| Price | Option Granted | Price | Option Granted | Price | Option Granted | |||||||||||||||||||
| Option price equal to the fair value of common stock on grant date | $ | — | $ | — | $ | 14.63 | $ | 10.29 | $ | 12.03 | $ | 8.14 | ||||||||||||
| Option price greater than the fair value of common stock on grant date | — | — | — | — | — | — | ||||||||||||||||||
| Option price less than the fair value of common stock on grant date | 9.36 | 11.55 | 13.67 | 10.05 | — | — | ||||||||||||||||||
The following table summarizes information about stock options outstanding at December 31, 2005:
| Weighted-Average | ||||||||||||||||||||
| Remaining | ||||||||||||||||||||
| Options | Contractual Life | Weighted-Average | Options | Weighted-Average | ||||||||||||||||
Grant Price Range | Outstanding | (years) | Exercise Price | Exercisable | Exercise Price | |||||||||||||||
| $ 0.33 — $ 7.95 | 1,247,728 | 3.72 | $ | 7.47 | 1,078,422 | $ | 7.39 | |||||||||||||
| $ 9.18 — $12.75 | 1,338,701 | 9.35 | 11.46 | 123,601 | 11.36 | |||||||||||||||
| $12.99 — $15.00 | 1,883,526 | 8.66 | 13.78 | 426,545 | 13.82 | |||||||||||||||
| $17.56 — $25.23 | 515,876 | 6.88 | 21.70 | 309,350 | 22.95 | |||||||||||||||
| 4,985,831 | 7.42 | 12.40 | 1,937,918 | 11.55 | ||||||||||||||||
For employee options, the difference between the estimated fair value of the underlying stock and the option exercise price, if lower, is recognized as compensation expense over the vesting period in accordance with APB Opinion No. 25. For non-employee options, the Company recognizes stock-based compensation expense for the estimated fair value of the options, determined using the Black-Scholes option valuation model, in accordance with
F-23
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
EITFNo. 96-18 and FIN 28. During the year ended December 31, 2003, non-employee stock-based compensation expense of approximately $277,000 was recognized for outstanding non-employee options. During the years ended December 31, 2004 and 2005, non-employee stock-based compensation expense of approximately $11,000 and $65,000, respectively, was recognized for outstanding non-employee options. As of December 31, 2005, non-employee options to purchase 49,333 shares of the Company’s common stock at a weighted-average exercise price of $11.75 per share were outstanding and are reflected in the tables above. The in-substance stock options disclosed in Note 7, which resulted from the issuance of stock to certain individuals, are excluded from the tables above. In November 2004, pursuant to assignment agreements with two consultants, the Company issued 200 shares of its common stock under its 2004 Equity Incentive Plan. The Company agreed to issue 99,800 additional shares upon the achievement of certain milestones specified in consulting agreements and for the year ended December 31, 2004, the Company recorded approximately $1.1 million in stock-based compensation expense related to these agreements. In November 2005, 39,800 of the 99,800 shares were issued to the consultants and the Company decreased stock-based compensation expense by approximately $146,000 based on the fair market value of the shares when issued.
In October 2003, pursuant to a settlement agreement with the Company’s former chief executive officer, options to purchase 83,333 shares of common stock were immediately vested and remain fully exercisable through April 2005. The change in option terms resulted in a new measurement date. On the date of the agreement, the outstanding options had an intrinsic value of approximately $153,000 that was recorded immediately as compensation expense. The options were exercised in October 2004.
In November 2003, pursuant to a settlement agreement with a former employee, options to purchase 5,000 shares of common stock were immediately vested and the option exercise period was extended. These options remained fully exercisable through May 2004. The change in option terms resulted in a new measurement date. On the date of the agreement, the outstanding options had an intrinsic value of approximately $25,000 that was recorded immediately as compensation expense.
During the year ended December 31, 2003, the Company extended the exercise period for certain employee options to purchase 174,855 shares of common stock at an exercise price of $6.24 per share. Also, in February 2003, pursuant to a settlement agreement with a former executive, the Company agreed that options to purchase 124,553 shares of common stock would remain exercisable at least until April 2006 (see Note 12 — Commitments and Contingencies — Severance Agreement). The Company recorded compensation expense of approximately $2.5 million and $259,000 for the years ended December 31, 2003 and 2004, respectively. For the year ended December 31, 2005, the Company decreased compensation expense by approximately $229,000 related to these options.
On October 7, 2003, the Company’s board of directors approved a re-pricing program for certain outstanding options to purchase shares of the Company’s common stock granted under each of its stock plans. Under the re-pricing program, each holder of outstanding options granted under the stock plans who was an employee of the Company on November 5, 2003 could elect to exchange up to all of their outstanding options with an exercise price greater than $7.95 for re-priced stock options with an exercise price of $7.95 per share and a term of four years. The option re-pricing became effective on November 5, 2003. Vesting restarted immediately with 50% vesting in November 2004 and the remaining 50% vesting monthly until fully vested in November 2005. Employees who voluntarily resigned in the12-month period beginning November 5, 2003 forfeited their re-priced options. Employees who were involuntarily terminated in the12-month period beginning November 5, 2003 vested 50% upon termination. In accordance with the terms of the re-pricing program, on November 5, 2003 the Company canceled 781,572 outstanding stock options with a weighted average exercise price of $19.83 per share and issued, in exchange for the canceled options, 781,572 new options with an exercise price of $7.95 per share. Compensation cost for all options re-priced under the re-pricing program was re-measured, using the intrinsic value method proscribed by APB No. 25, on a quarterly basis. Compensation cost for these options are recognized in accordance
F-24
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
with the method prescribed by FIN 28. For the years ended December 31, 2003 and 2004, the Company recorded $595,000 and $4.4 million, respectively in stock-based compensation expense related to the re-pricing program. For the year ended December 31, 2005, the Company recorded a decrease in stock-based compensation expense of approximately $2.4 million relating to the re-pricing program. Upon adoption of SFAS No. 123R (see Note 2 — Summary of Significant Accounting Policies — Recently Issued Accounting Standards), effective January 1, 2006, the Company will no longerre-measure these options.
Total stock-based compensation expense recognized in the accompanying statements of operations is as follows (in thousands):
| Year Ended December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Employee-related | $ | 4,224 | $ | 5,694 | $ | (1,892 | ) | |||||
| Consultant-related | 277 | 1,116 | 164 | |||||||||
| Total | $ | 4,501 | $ | 6,810 | $ | (1,728 | ) | |||||
Total stock-based compensation expense recognized in the accompanying statements of operations is included in the following categories (in thousands):
| Year Ended December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Research and development | $ | 961 | $ | 2,936 | $ | (320 | ) | |||||
| General and administrative | 3,540 | 3,874 | (1,408 | ) | ||||||||
| Total | $ | 4,501 | $ | 6,810 | $ | (1,728 | ) | |||||
| 11. | Warrants |
During 1995 and 1996, the Company issued warrants to purchase shares of common stock. The warrants range in exercise price from $12.53 to $12.70 per share and expire at various dates through 2007. The warrants contain provisions for the adjustment of the exercise price and the number of shares issuable upon the exercise of the warrant in the event the Company declares any stock dividends or effects any stock split, reclassification or consolidation of its common stock. The warrants also contain a provision that provides for an adjustment to the exercise price and the number of shares issuable in the event that the Company issues securities for a per share price less than a specified price. As of December 31, 2004, warrants to purchase 131,628 shares of common stock were outstanding. During the second quarter ended June 30, 2005, warrants to purchase 110,888 shares of common stock were exchanged for 24,210 shares of common stock resulting in stock-based compensation expense of $245,000 based on a fair market value of the common stock of $10.12 per share. As of December 31, 2005, the remaining warrants to purchase 12,436 shares of common stock at a weighted average exercise price of $12.66 per share are outstanding and exercisable while warrants to purchase 8,304 shares of common stock expired during 2005.
In connection with the sale of common stock in the private placement which closed on August 5, 2005, the Company concurrently issued warrants to purchase up to 3,426,000 shares of common stock at an exercise price of $12.228 per share. See also Note 9 — Common and Preferred Stock — Private Placement. These warrants become exercisable on February 1, 2006 and expire in August 2010.
F-25
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 12. | Commitments and contingencies |
Operating Leases — The Company leases certain facilities and equipment under various operating leases, which expire at various dates through 2009. Future minimum rental payments required under operating leases are as follows at December 31, 2005:
Year Ending December 31, | ||||
| 2006 | $ | 849,000 | ||
| 2007 | 727,000 | |||
| 2008 | 727,000 | |||
| After 2008 | 61,000 | |||
| Total minimum lease payments | $ | 2,364,000 | ||
Rent expense under all operating leases for the years ended December 31, 2003, 2004 and 2005 was approximately, $1.2 million, $601,000 and $1.1 million, respectively.
Capital Leases — The Company’s capital leases were not material for the years ended December 31, 2003, 2004 and 2005.
Guarantees and Indemnifications — In the ordinary course of its business, the Company makes certain indemnities, commitments and guarantees under which it may be required to make payments in relation to certain transactions. The Company, as permitted under Delaware law and in accordance with its Bylaws, indemnifies its officers and directors for certain events or occurrences, subject to certain limits, while the officer or director is or was serving at the Company’s request in such capacity. The term of the indemnification period is for the officer’s or director’s lifetime. The maximum amount of potential future indemnification is unlimited; however, the Company has a director and officer insurance policy that may enable it to recover a portion of any future amounts paid. The Company believes the fair value of these indemnification agreements is minimal. The Company has not recorded any liability for these indemnities in the accompanying consolidated balance sheets. However, the Company accrues for losses for any known contingent liability, including those that may arise from indemnification provisions, when future payment is probable. No such losses have been recorded to date.
Litigation — The Company is involved in various legal proceedings and other matters. In accordance with SFAS No. 5,Accounting for Contingencies, the Company would record a provision for a liability when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated.
In May 2005, the Company’s former Chief Medical Officer filed a complaint against the Company in the California Superior Court, County of Los Angeles.Wayman Wendell Cheatham, M.D. v. MannKind Corporation, Case No. BC333845. The complaint alleges causes of action for wrongful termination in violation of public policy, breach of contract and retaliation, in connection with the Company’s termination of Dr. Cheatham’s employment. In the complaint, Dr. Cheatham seeks compensatory, punitive and exemplary damages in excess of $2.0 million as well as reimbursement of attorneys’ fees. In June 2005, the Company answered the complaint, generally denying each of Dr. Cheatham’s allegations and asserting various defenses. The Company believes the allegations in the complaint are without merit and intends to vigorously defend against them. The Company also filed a cross-complaint against Dr. Cheatham, alleging claims for libel per se, trade libel, breach of contract, breach of the implied covenant of good faith and fair dealing and breach of the duty of loyalty. The libel claims allege that Dr. Cheatham made certain false and malicious statements about the Company in a letter to the Food and Drug Administration (“FDA”) with regard to a request by the Company to hold a meeting with the FDA. The remaining causes of action in the cross-complaint arise out of the Company’s allegations that Dr. Cheatham had an undisclosed consulting relationship with a Company competitor during his employment with the Company, in violation of his agreement with the Company. In July 2005, Dr. Cheatham filed a demurrer and motion to strike the Company’s cross-complaint under California’s anti-SLAPP statute. In September 2005, the California Superior Court overruled Dr. Cheatham’s demurrer and
F-26
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
denied his motion to strike the Company’s cross-complaint. Dr. Cheatham then filed a notice of appeal of the Court’s ruling denying his motion to strike. Discovery as to Dr. Cheatham’s claims against the Company is proceeding, and this case is scheduled for trial to commence in October 2006. The Company believes that the ultimate resolution of this matter will not have a material impact on the Company’s financial position or results of operations.
In 2000, the Company issued 699,972 shares of common stock to three consultants in exchange for notes receivable aggregating approximately $10.9 million. The fixed interest bearing notes were collateralized by the underlying common stock. Thenotes-for-stock transactions were accounted for as in-substance stock option grants to non-employees. In November 2004, the consultants informed the Company that they had entered into an agreement in October 2001 with Alfred E. Mann, the Company’s chairman, chief executive officer and principal stockholder, under which Mr. Mann would purchase a portion of the consultants’ common stock, and that the Company was to apply the proceeds to the amounts owed under the consultants’ respective notes. The consultants informed the Company that they believed both the Company and Mr. Mann were in breach of the alleged agreement, and indicated their intent to seek alleged damages arising from the Company’s failure to perform the alleged agreement. On October 19, 2005, the principal and interest on the notes aggregating $14.6 million became due and payable and the Company pursued collection. On November 21, 2005, the consultants filed a complaint against the Company in the California Superior Court, County of Los Angeles,Rollins et al. v. MannKind et al, Case No. BC343381. The complaint alleges causes of action for breach of the abovementioned agreement, among other things. On January 19, 2006, the parties mediated and settled the case. Under the settlement, the Company repurchased 620,697 shares from the consultants in full satisfaction of the notes. These shares were tendered and cancelled on February 3, 2006. The Company also agreed to repurchase the remaining 79,275 shares held by the consultants for $1.4 million in cash. The settlement was reflected as a $1.4 million charge to general and administrative expense in the fourth quarter of the year ended December 31, 2005 with a corresponding amount payable included in accrued expenses and other current liabilities as of December 31 2005. On February 21, 2006, the Company received and cancelled the 79,275 shares, and paid the purchase price of $1.4 million to the consultants. The complaint has been dismissed in its entirety with prejudice.
In September and October 2005, the principal and interest totaling $1.6 million on notes issued in exchange for approximately 78,000 shares of common stock to a former executive of the Company became due and payable to the Company. On December 31, 2005, the 78,000 shares of common stock issued in exchange for the notes receivable had a market value of approximately $878,000. The Company pursued collection. On January 31, 2006, former executive tendered 143,949 shares as full repayment of the notes in default and an additional note due in April 2006 issued in exchange for approximately 32,000 shares of common stock. The 143,949 shares were valued at $2.6 million on January 31, 2006 and represented principal and interest due through that date on all notes. The Company received and cancelled these shares on January 31, 2006.
Severance Agreements — In February 2003, pursuant to a settlement agreement, the Company is obligated to pay a former executive approximately $1.0 million over three years, comprised of approximately $775,000 in deferred compensation (see Note 8) from prior years and the remainder comprised of other severance-related items. As of December 31, 2004 the Company paid approximately $765,000 of this amount. The remaining $284,000 was paid in April 2005. The settlement agreement further provides that the options held by the former executive to purchase up to 46,585 shares of the Company’s common stock remain fully exercisable through April 2007, and options to purchase up to 124,553 shares of the Company’s common stock remain fully exercisable until at least April 2006. The change in option terms resulted in a new measurement date. On the date of the agreement, the outstanding options had an intrinsic value of approximately $1.1 million, which was immediately expensed as part of the $2.5 million charge more fully described in Note 10.
In February 2003, pursuant to a settlement agreement, the Company became obligated to pay a former employee his base salary at the rate of approximately $22,000 per month through December 2003 and a lump sum payment of
F-27
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
$67,000, which was paid in February 2005 and was previously included in accrued expenses in the accompanying balance sheet at December 31, 2004.
In October 2003, under the terms of a severance agreement with its former chief executive officer, the Company paid a severance payment of $165,000 and agreed to pay his base salary at the rate of approximately $28,000 per month through April 2005. Amounts owed under this agreement are included in accrued expenses in the accompanying balance sheet at December 31, 2004. The agreement also provides for accelerated vesting of an option held by the former executive permitting him to purchase up to 83,333 shares of the Company’s common stock until April 7, 2005 (see Note 10). These options were exercised in October 2004.
| 13. | Employee benefit plans |
As a result of the merger in December 2001, each respective 401(k) savings retirement plans of AlleCure, CTL and PDC were converted to the 401(k) Savings Retirement Plan (the “MannKind Retirement Plan”) for the Company. For the years ended December 31, 2003, 2004 and 2005, the Company contributed $235,000, $249,000 and $372,000, respectively, to the MannKind Retirement Plan.
| 14. | Income taxes |
Deferred income taxes reflect the tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and income tax purposes. A valuation allowance is established when uncertainty exists as to whether all or a portion of the net deferred tax assets will be realized. Components of the net deferred tax asset as of December 31, 2004 and 2005 are approximately as follows (in thousands):
| December 31, | ||||||||
| 2004 | 2005 | |||||||
| Deferred Tax Assets: | ||||||||
| Net operating loss carryforwards | $ | 92,269 | $ | 137,292 | ||||
| Research and development credits | 4,566 | 5,719 | ||||||
| Accrued expenses | 5,548 | 8,100 | ||||||
| Non-qualified stock option expense | 5,027 | 3,793 | ||||||
| Depreciation | 572 | 895 | ||||||
| Total gross deferred tax assets | 107,982 | 155,799 | ||||||
| Valuation allowance | (107,982 | ) | (155,799 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
The Company’s effective income tax rate differs from the statutory federal income tax rate as follows for the years ended December 31, 2003, 2004 and 2005:
| December 31, | ||||||||||||
| 2003 | 2004 | 2005 | ||||||||||
| Federal tax benefit rate | 35.0 | % | 35.0 | % | 35.0 | % | ||||||
| State tax benefit, net of federal benefit | — | — | — | |||||||||
| Permanent items | (0.1 | ) | — | — | ||||||||
| Other | 1.6 | — | — | |||||||||
| Valuation allowance | (36.5 | ) | (35.0 | ) | (35.0 | ) | ||||||
| Effective income tax rate | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
F-28
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
As required by SFAS No. 109, management of the Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets. Management has concluded, in accordance with the applicable accounting standards, that it is more likely than not that the Company may not realize the benefit of its deferred tax assets. Accordingly, the net deferred tax assets have been fully reserved. Management reevaluates the positive and negative evidence on an annual basis. During the years ended December 31, 2003, 2004 and 2005, the change in the valuation allowance was $29.3 million, $34.1 million and $47.8 million, respectively, for income taxes.
At December 31, 2005, the Company had federal and state net operating loss carryforwards of approximately $356.0 million and $229.1 million available, respectively, to reduce future taxable income and which will expire at various dates beginning in 2006 and 2008, respectively. At December 31, 2005, the Company had research and development credits of $7.6 million that expire at various dates through 2018. Ownership changes, as defined in Section 382 of the Internal Revenue Code, such as those resulting from the issuance of common stock in connection with the Company’s initial public offering and subsequent private placement, may limit the amount of net operating loss and tax credit carryforwards that can be utilized to offset future taxable income or tax liability. The Company has not yet determined the amounts of such limitations, if any; however, they could be significant. Any such limitations that may have existed for any of the periods presented in the accompanying financial statements would not have impacted the amounts reported because the Company has provided a full valuation allowance against all of its deferred tax assets. To the extent the Company realizes taxable income in any future period, the amount of any such limitations could have a material impact on the reported financial statements.
| 15. | Related party transactions |
The Company issued 8,550,446 shares of its common stock to its principal stockholder during the year ended December 31, 2005 for proceeds of approximately $87.3 million. In connection with this issuance, the board of directors approved the issuance of warrants to purchase 1,710,091 shares of the Company’s common stock at $12.228 per share, which expire on August 2, 2010. The issuance of shares and warrants to the principal stockholder was on terms identical to the other purchasers in the private placement, as approved by the Company’s board of directors.
During the years ended December 31, 2003 and 2004, the Company paid $497,000 and $218,000, respectively, to certain universities to conduct sponsored research programs, including clinical research. Certain stockholders of the Company are employees of these universities and oversee the sponsored research programs. No sponsored research programs were conducted with and no amounts were paid to these universities in 2005.
On January 19, 2006, the Company settled a claim filed by three consultants related to certain notes-for-stock transactions (see Note 12 — Commitments and Contingencies — Litigation).
One stockholder was paid $6,000 and $48,000 for consulting services during the years ended December 31, 2003 and 2004, respectively. No services were provided or paid for the year ended December 31, 2005.
In connection with certain meetings of the Company’s board of directors and on other occasions when the Company’s business necessitated air travel for the Company’s principal stockholder and other Company employees, the Company utilized the principal stockholder’s private aircraft, and the Company paid the charter company that manages the aircraft on behalf of the Company’s majority stockholder approximately $321,000, $145,000 and $62,000, respectively, for the years ended December 31, 2003, 2004 and 2005. These payments were approved by the audit committee of the board of directors.
In 2004, the Company engaged one of its directors to provide consulting services related to seeking potential partners in the development and commercialization of the Company’s technology. The Company paid this director approximately $47,000 for consulting services rendered for the year ended December 31, 2004. No services were provided for the year ended December 31, 2005.
F-29
Table of Contents
MANNKIND CORPORATION AND SUBSIDIARIES
(A Development Stage Company)
NOTES TO FINANCIAL STATEMENTS — (Continued)
The Company has entered into indemnification agreements with each of its directors and executive officers, in addition to the indemnification provided for in its amended and restated certificate of incorporation and amended and restated bylaws (see Note 12 — Guarantees and Indemnifications).
| 16. | Selected quarterly financial data (unaudited) |
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| (In thousands, except per share data) | ||||||||||||||||
2004 | ||||||||||||||||
| Net loss | $ | (16,409 | ) | $ | (18,245 | ) | $ | (20,188 | ) | $ | (21,150 | ) | ||||
| Net loss applicable to common stockholders | $ | (17,085 | ) | $ | (18,241 | ) | $ | (39,398 | ) | $ | (21,150 | ) | ||||
| Net loss per share applicable to common stockholders — basic and diluted | $ | (0.86 | ) | $ | (0.91 | ) | $ | (1.40 | ) | $ | (0.65 | ) | ||||
| Weighted average common shares used to compute basic and diluted net loss per share applicable to common stockholders | 19,975 | 19,975 | 28,051 | 32,768 | ||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
| (In thousands, except per share data) | ||||||||||||||||
2005 | ||||||||||||||||
| Net loss | $ | (22,162 | ) | $ | (27,155 | ) | $ | (31,730 | ) | $ | (33,291 | ) | ||||
| Net loss applicable to common stockholders | $ | (22,162 | ) | $ | (27,155 | ) | $ | (31,730 | ) | $ | (33,291 | ) | ||||
| Net loss per share applicable to common stockholders — basic and diluted | $ | (0.68 | ) | $ | (0.83 | ) | $ | (0.73 | ) | $ | (0.66 | ) | ||||
| Weighted average common shares used to compute basic and diluted net loss per share applicable to common stockholders | 32,764 | 32,777 | 43,460 | 50,250 | ||||||||||||
F-30