UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-Q
☒ QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended June 30, 2021
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to .
Commission File No. 000-26770
NOVAVAX, INC.
(Exact name of registrant as specified in its charter)
| | | | | | | | | | | |
| Delaware | 22-2816046 |
(State or other jurisdiction of
incorporation or organization) | (I.R.S. Employer
Identification No.) |
| |
21 Firstfield Road | Gaithersburg | MD | 20878 |
| (Address of principal executive offices) | (Zip code) |
(240) 268-2000
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | |
| Title of each class | Trading
Symbol(s) | Name of each exchange on which registered |
| Common Stock, Par Value $0.01 per share | NVAX | The Nasdaq Global Select Market |
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | |
| Large accelerated filer | x | Accelerated Filer | o |
| | | |
| Non-accelerated filer | o | Smaller reporting company | o |
| | | |
| Emerging growth company | ☐ | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No x
The number of shares outstanding of the Registrant's Common Stock, $0.01 par value, was 74,484,166 as of July 31, 2021.
NOVAVAX, INC.
TABLE OF CONTENTS
PART I. FINANCIAL INFORMATION
Item 1. Financial Statements
NOVAVAX, INC.
CONSOLIDATED BALANCE SHEETS
(in thousands, except share and per share information)
| | | | | | | | | | | |
| June 30,
2021 | | December 31,
2020 |
| (unaudited) | | |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 2,074,880 | | | $ | 553,398 | |
| Marketable securities | 0 | | | 157,649 | |
| Restricted cash | 46,398 | | | 93,880 | |
| Accounts receivable | 49,989 | | | 262,012 | |
| Unbilled services | 21,374 | | | 0 | |
| Prepaid expenses and other current assets | 162,351 | | | 181,264 | |
| Total current assets | 2,354,992 | | | 1,248,203 | |
| Restricted cash | 1,461 | | | 1,460 | |
| Property and equipment, net | 213,186 | | | 179,954 | |
| Intangible assets, net | 5,281 | | | 5,725 | |
| Goodwill | 134,294 | | | 135,379 | |
| Other non-current assets | 40,373 | | | 11,758 | |
| Total assets | $ | 2,749,587 | | | $ | 1,582,479 | |
| | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 73,995 | | | $ | 54,332 | |
| Accrued expenses | 249,178 | | | 142,468 | |
| | | |
| Deferred revenue | 1,220,073 | | | 273,228 | |
| Current portion of finance lease liabilities | 98,383 | | | 105,862 | |
| Other current liabilities | 9,887 | | | 3,782 | |
| Total current liabilities | 1,651,516 | | | 579,672 | |
| Convertible notes payable | 322,746 | | | 322,035 | |
| Non-current finance lease liabilities | 9,193 | | | 40,083 | |
| Other non-current liabilities | 20,570 | | | 13,480 | |
| Total liabilities | 2,004,025 | | | 955,270 | |
| | | |
| Commitments and contingencies | 0 | | 0 |
| | | |
| | | |
| Preferred stock, $0.01 par value, 2,000,000 shares authorized at June 30, 2021 and December 31, 2020; 0 shares issued and outstanding at June 30, 2021 and December 31, 2020 | 0 | | | 0 | |
| | | |
| Stockholders' equity: | | | |
| Common stock, $0.01 par value, 600,000,000 shares authorized at June 30, 2021 and December 31, 2020; and 74,672,351 shares issued and 74,248,279 shares outstanding at June 30, 2021 and 71,350,365 shares issued and 70,953,739 shares outstanding at December 31, 2020 | 747 | | | 714 | |
| Additional paid-in capital | 3,237,085 | | | 2,535,476 | |
| Accumulated deficit | (2,449,235) | | | (1,874,199) | |
| Treasury stock, 424,072 shares, cost basis at June 30, 2021 and 396,626 shares, cost basis at December 31, 2020 | (47,205) | | | (41,806) | |
| Accumulated other comprehensive income | 4,170 | | | 7,024 | |
| Total stockholders’ equity | 745,562 | | | 627,209 | |
| Total liabilities and stockholders’ equity | $ | 2,749,587 | | | $ | 1,582,479 | |
The accompanying notes are an integral part of these financial statements.
NOVAVAX, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share information)
(unaudited)
| | | | | | | | | | | | | | | | | | | | | | | |
| For the Three Months Ended
June 30, | | For the Six Months Ended
June 30, |
| 2021 | | 2020 | | 2021 | | 2020 |
| | | | | | | |
| Revenue: | | | | | | | |
| Government contracts | $ | 240,534 | | | $ | 0 | | | $ | 623,238 | | | $ | 0 | |
| Grant and other | 34,026 | | | 35,538 | | | 98,551 | | | 38,915 | |
| Royalties | 23,457 | | | 0 | | | 23,457 | | | 0 | |
| Total revenue | 298,017 | | | 35,538 | | | 745,246 | | | 38,915 | |
| | | | | | | |
| Expenses: | | | | | | | |
| Research and development | 570,685 | | | 34,846 | | | 1,163,356 | | | 51,741 | |
| General and administrative | 73,161 | | | 17,719 | | | 136,351 | | | 27,098 | |
| Total expenses | 643,846 | | | 52,565 | | | 1,299,707 | | | 78,839 | |
| Loss from operations | (345,829) | | | (17,027) | | | (554,461) | | | (39,924) | |
| Other income (expense): | | | | | | | |
| Investment income | 369 | | | 297 | | | 731 | | | 732 | |
| Interest expense | (5,968) | | | (3,403) | | | (10,807) | | | (6,806) | |
| Other income (expense) | 2,659 | | | 2,612 | | | (3,934) | | | 2,613 | |
| Net loss before income tax expense | $ | (348,769) | | | $ | (17,521) | | | $ | (568,471) | | | $ | (43,385) | |
| Income tax expense | 3,548 | | | 0 | | | 6,565 | | | 0 | |
| Net loss | $ | (352,317) | | | $ | (17,521) | | | $ | (575,036) | | | $ | (43,385) | |
| | | | | | | |
| Basic and diluted net loss per share | $ | (4.75) | | | $ | (0.30) | | | $ | (7.82) | | | $ | (0.84) | |
| | | | | | | |
| Basic and diluted weighted average number of common shares outstanding | 74,118 | | | 58,618 | | | 73,580 | | | 51,401 | |
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(in thousands)
(unaudited)
| | | | | | | | | | | | | | | | | | | | | | | |
| For the Three Months Ended
June 30, | | For the Six Months Ended
June 30, |
| 2021 | | 2020 | | 2021 | | 2020 |
| | | | | | | |
| Net loss | $ | (352,317) | | | $ | (17,521) | | | $ | (575,036) | | | $ | (43,385) | |
| Other comprehensive income (loss): | | | | | | | |
| Net unrealized losses on marketable securities available-for-sale, net of reclassifications | 0 | | | 176 | | | (9) | | | 44 | |
| Foreign currency translation adjustment | 4,527 | | | 1,115 | | | (2,845) | | | (731) | |
| Other comprehensive loss | 4,527 | | | 1,291 | | | (2,854) | | | (687) | |
| Comprehensive loss | $ | (347,790) | | | $ | (16,230) | | | $ | (577,890) | | | $ | (44,072) | |
The accompanying notes are an integral part of these financial statements.
NOVAVAX, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY (DEFICIT)
Three Months Ended June 30, 2021 and 2020
(in thousands, except share information)
(unaudited)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Treasury
Stock | | Other
Comprehensive
Income (Loss) | | Stockholders'
Equity
(Deficit) |
| Shares | | Amount | | | | | |
| |
| Balance at March 31, 2021 | 74,470,583 | | | $ | 745 | | | $ | 3,180,114 | | | $ | (2,096,918) | | | $ | (44,457) | | | $ | (357) | | | $ | 1,039,127 | |
| | | | | | | | | | | | | |
| Non-cash stock-based compensation | — | | | — | | | 53,123 | | | — | | | — | | | — | | | 53,123 | |
| Stock issued under incentive programs | 201,768 | | | 2 | | | 3,848 | | | — | | | (2,748) | | | — | | | 1,102 | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | — | | | 4,527 | | | 4,527 | |
| Net loss | — | | | — | | | — | | | (352,317) | | | — | | | — | | | (352,317) | |
| Balance at June 30, 2021 | 74,672,351 | | | $ | 747 | | | $ | 3,237,085 | | | $ | (2,449,235) | | | $ | (47,205) | | | $ | 4,170 | | | $ | 745,562 | |
| | | | | | | | | | | | | |
| Balance at March 31, 2020 | 53,906,322 | | | $ | 539 | | | $ | 1,450,279 | | | $ | (1,457,665) | | | $ | (2,638) | | | $ | (14,486) | | | $ | (23,971) | |
| Preferred stock beneficial conversion feature | — | | | — | | | 24,139 | | | (24,139) | | | — | | | — | | | 0 | |
| Non-cash stock-based compensation | — | | | — | | | 7,932 | | | — | | | — | | | — | | | 7,932 | |
| Stock issued under incentive programs | 316,815 | | | 3 | | | 8,947 | | | — | | | — | | | — | | | 8,950 | |
| Issuance of common stock, net of issuance costs of $2,647 | 7,039,495 | | | 70 | | | 207,775 | | | — | | | — | | | 0 | | | 207,845 | |
| Unrealized loss on marketable securities | — | | | — | | | — | | | — | | | — | | | 176 | | | 176 | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | — | | | 1,115 | | | 1,115 | |
| Net loss | — | | | — | | | — | | | (17,521) | | | — | | | — | | | (17,521) | |
| Balance at June 30, 2020 | 61,262,632 | | | $ | 612 | | | $ | 1,699,072 | | | $ | (1,499,325) | | | $ | (2,638) | | | $ | (13,195) | | | $ | 184,526 | |
The accompanying notes are an integral part of these financial statements.
NOVAVAX, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS' EQUITY (DEFICIT)
Six Months Ended June 30, 2021 and 2020
(in thousands, except share information)
(unaudited)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Treasury
Stock | | Other
Comprehensive
Income (Loss) | | Stockholders'
Equity
(Deficit) |
| Shares | | Amount | | | | | |
| |
| Balance at December 31, 2020 | 71,350,365 | | | $ | 714 | | | $ | 2,535,476 | | | $ | (1,874,199) | | | $ | (41,806) | | | $ | 7,024 | | | $ | 627,209 | |
| Non-cash stock-based compensation | — | | | — | | | 106,183 | | | — | | | | | — | | | 106,183 | |
| Stock issued under incentive programs | 743,019 | | | 7 | | | 30,593 | | | — | | | (5,399) | | | — | | | 25,201 | |
| Issuance of common stock, net of issuance costs of $7,292 | 2,578,967 | | | 26 | | | 564,833 | | | — | | | — | | | — | | | 564,859 | |
| Unrealized loss on marketable securities | — | | | — | | | — | | | — | | | — | | | (9) | | | (9) | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | — | | | (2,845) | | | (2,845) | |
| Net loss | — | | | — | | | — | | | (575,036) | | | — | | | — | | | (575,036) | |
| Balance at June 30, 2021 | 74,672,351 | | | $ | 747 | | | $ | 3,237,085 | | | $ | (2,449,235) | | | $ | (47,205) | | | $ | 4,170 | | | $ | 745,562 | |
| | | | | | | | | | | | | |
| Balance at December 31, 2019 | 32,399,352 | | | $ | 324 | | | $ | 1,260,551 | | | $ | (1,431,801) | | | $ | (2,583) | | | $ | (12,508) | | | $ | (186,017) | |
| Preferred stock beneficial conversion feature | — | | | — | | | 24,139 | | | (24,139) | | | | | — | | | 0 | |
| Non-cash stock-based compensation | — | | | — | | | 11,897 | | | — | | | — | | | — | | | 11,897 | |
| Stock issued under incentive programs | 350,054 | | | 3 | | | 9,007 | | | — | | | (55) | | | — | | | 8,955 | |
| Issuance of common stock, net of issuance costs of $5,145 | 28,513,226 | | | 285 | | | 393,478 | | | — | | | — | | | — | | | 393,763 | |
| Unrealized loss on marketable securities | — | | | — | | | — | | | — | | | — | | | 44 | | | 44 | |
| Foreign currency translation adjustment | — | | | — | | | — | | | — | | | — | | | (731) | | | (731) | |
| Net loss | — | | | — | | | — | | | (43,385) | | | — | | | — | | | (43,385) | |
| Balance at June 30, 2020 | 61,262,632 | | | $ | 612 | | | $ | 1,699,072 | | | $ | (1,499,325) | | | $ | (2,638) | | | $ | (13,195) | | | $ | 184,526 | |
The accompanying notes are an integral part of these financial statements.
NOVAVAX, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
(unaudited)
| | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 |
| | | |
| Operating Activities: | | | |
| Net loss | $ | (575,036) | | | $ | (43,385) | |
| Reconciliation of net loss to net cash used in operating activities: | | | |
| Depreciation and amortization | 4,727 | | | 1,905 | |
| | | |
| Non-cash stock-based compensation | 106,183 | | | 11,897 | |
| Right-of-use assets written off | 12,707 | | | 0 | |
| Other | 3,855 | | | (1,081) | |
| Changes in operating assets and liabilities: | | | |
| Receivables, prepaid expenses and other assets | 193,004 | | | (61,079) | |
| Accounts payable and accrued expenses | 115,212 | | | 27,094 | |
| Deferred revenue | 946,845 | | | 157,173 | |
| Net cash provided by operating activities | 807,497 | | | 92,524 | |
| | | |
| Investing Activities: | | | |
| Capital expenditures | (28,932) | | | (3,884) | |
| Acquisition of Novavax CZ, net of cash required | 0 | | | (164,204) | |
| Purchases of marketable securities | (2,167) | | | (107,608) | |
| Proceeds from maturities and sale of marketable securities | 159,807 | | | 29,750 | |
| Net cash provided by (used in) investing activities | 128,708 | | | (245,946) | |
| | | |
| Financing Activities: | | | |
| Net proceeds from sale of preferred stock | 0 | | | 199,822 | |
| Net proceeds from sales of common stock | 564,859 | | | 393,763 | |
| Net proceeds from the exercise of stock-based awards | 26,903 | | | 8,955 | |
| Finance lease payments | (53,618) | | | 0 | |
| Net cash provided by financing activities | 538,144 | | | 602,540 | |
| Effect of exchange rate on cash, cash equivalents and restricted cash | (348) | | | 276 | |
| Net increase in cash, cash equivalents and restricted cash | 1,474,001 | | | 449,394 | |
| Cash, cash equivalents and restricted cash at beginning of period | 648,738 | | | 82,180 | |
| Cash, cash equivalents and restricted cash at end of period | $ | 2,122,739 | | | $ | 531,574 | |
| | | |
| Supplemental disclosure of non-cash activities: | | | |
| | | |
| Right-of-use assets from new lease agreements | $ | 28,826 | | | $ | 0 | |
| Capital expenditures included in accounts payable and accrued expenses | $ | 11,037 | | | $ | 2,753 | |
| | | |
| Supplemental disclosure of cash flow information: | | | |
| Cash interest payments | $ | 10,046 | | | $ | 6,094 | |
| Cash paid for income taxes | $ | 3,017 | | | $ | 0 | |
The accompanying notes are an integral part of these financial statements.
NOVAVAX, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
June 30, 2021
(unaudited)
Note 1 – Organization
Novavax, Inc. (“Novavax,” and together with its wholly owned subsidiaries, including Novavax AB and Novavax CZ, the “Company”) is a biotechnology company that promotes improved health globally through the discovery, development and commercialization of innovative vaccines to prevent serious infectious diseases. The Company’s vaccine candidates, including both its coronavirus vaccine candidate, NVX-CoV2373, and its lead influenza vaccine candidate, NanoFlu™, are genetically engineered, three-dimensional nanostructures of recombinant proteins critical to disease pathogenesis and may elicit differentiated immune responses, which may be more efficacious than naturally occurring immunity or traditional vaccines. NVX-CoV2373 and NanoFlu™ include the use of the Company's proprietary Matrix-M™ adjuvant.
Note 2 – Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”) for interim financial information and the instructions to Form 10-Q and Article 10 of Regulation S-X. The consolidated balance sheet as of June 30, 2021, the consolidated statements of operations and the consolidated statements of comprehensive loss for the three and six months ended June 30, 2021 and 2020, the consolidated statements of changes in stockholders’ equity (deficit) for the three and six months ended June 30, 2021 and 2020 and the consolidated statements of cash flows for the six months ended June 30, 2021 and 2020 are unaudited, but include all adjustments (consisting of normal recurring adjustments) that the Company considers necessary for a fair presentation of the financial position, operating results, comprehensive loss, changes in stockholders’ equity (deficit) and cash flows, respectively, for the periods presented. Although the Company believes that the disclosures in these unaudited consolidated financial statements are adequate to make the information presented not misleading, certain information and footnote information normally included in consolidated financial statements prepared in accordance with U.S. GAAP have been condensed or omitted as permitted under the rules and regulations of the United States Securities and Exchange Commission (“SEC”).
The unaudited consolidated financial statements include the accounts of Novavax, Inc. and its wholly owned subsidiaries, including Novavax AB and Novavax CZ. All intercompany accounts and transactions have been eliminated in consolidation.
The accompanying unaudited consolidated financial statements are presented in U.S. dollars. The functional currency of Novavax AB, which is located in Sweden, is the local currency (Swedish Krona), and the functional currency of Novavax CZ, which is located in the Czech Republic, is the local currency (Czech Koruna). The translation of assets and liabilities of these subsidiaries to U.S. dollars is made at the exchange rate in effect at the consolidated balance sheet date, while equity accounts are translated at historical rates. The translation of the statement of operations data is made at the average exchange rate in effect for the period. The translation of operating cash flow data is made at the average exchange rate in effect for the period, and investing and financing cash flow data is translated at the exchange rate in effect at the date of the underlying transaction. Translation gains and losses are recognized as a component of accumulated other comprehensive income in the accompanying unaudited consolidated balance sheets. Accumulated other comprehensive income included a foreign currency translation balance of $4.2 million and $7.0 million as of June 30, 2021 and December 31, 2020, respectively.
The accompanying unaudited consolidated financial statements should be read in conjunction with the financial statements and notes thereto included in the Company's Annual Report on Form 10-K for the year ended December 31, 2020. Results for this or any interim period are not necessarily indicative of results for any future interim period or for the entire year. The Company operates in 1 business segment.
Use of Estimates
The preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ materially from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of highly liquid investments with maturities of three months or less from the date of purchase. Cash and cash equivalents consist of the following at (in thousands):
| | | | | | | | | | | |
| June 30,
2021 | | December 31,
2020 |
| Cash | $ | 82,563 | | | $ | 122,312 | |
| Money market funds | 238,028 | | | 96,116 | |
| Government-backed securities | 57,250 | | | 44,250 | |
| Treasury securities | 199,990 | | | 44,052 | |
| Corporate debt securities | 1,386,056 | | | 246,668 | |
| Agency securities | 110,993 | | | 0 | |
| Cash and cash equivalents | $ | 2,074,880 | | | $ | 553,398 | |
Cash equivalents are recorded at cost, which approximate fair value due to their short-term nature.
Marketable Securities
The Company invests in marketable securities that generally consist of debt securities with maturities greater than three months from the date of purchase that include commercial paper, government-backed securities, treasury securities, corporate notes and agency securities. Classification of marketable securities between current and non-current is dependent upon the maturity date at the balance sheet date taking into consideration the Company's ability and intent to hold the investment to maturity.
Interest and dividend income are recorded when earned and included in investment income in the consolidated statements of operations. Premiums and discounts, if any, on marketable securities are amortized or accreted to maturity and included in investment income in the consolidated statements of operations. The specific identification method is used in computing realized gains and losses on the sale of the Company's securities.
The Company classifies its marketable securities with readily determinable fair values as “available-for-sale.” Investments in securities that are classified as available-for-sale are measured at fair market value in the consolidated balance sheets, and unrealized gains and losses on marketable securities are reported as a separate component of stockholders' equity (deficit) until realized. Marketable securities are evaluated periodically to determine whether a decline in value is “other-than-temporary.” The term “other-than-temporary” is not intended to indicate a permanent decline in value. Rather, it means that the prospects for a near-term recovery of value are not necessarily favorable, or that there is a lack of evidence to support fair values equal to, or greater than, the carrying value of the security. Management reviews criteria, such as the magnitude and duration of the decline, as well as the Company's ability to hold the securities, including whether the Company will be required to sell a security prior to recovery of its amortized cost basis, the investment issuer's financial condition and business outlook to predict whether the loss in value is other-than-temporary. Realized gains and losses and declines in value determined to be other-than-temporary are recorded as other income (expense) in the consolidated statements of operations.
Restricted Cash
The Company’s current and non-current restricted cash includes payments received under the Coalition for Epidemic Preparedness Innovations (“CEPI”) funding agreements, payments received under the Bill & Melinda Gates Foundation (“BMGF”) grant agreements and cash collateral accounts under letters of credit that serve as security deposits for certain facility leases. The Company will utilize the CEPI and BMGF funds as it incurs expenses for services performed under these agreements.
As of June 30, 2021, the restricted cash balances (both current and non-current) consisted of $1.2 million for payments received from BMGF, $45.2 million of payments under the CEPI funding agreements and $1.5 million of security deposits. As of December 31, 2020, the restricted cash balances (both current and non-current) consisted of $1.5 million for payments received from BMGF, $92.4 million of payments under the CEPI funding agreements and $1.5 million of security deposits.
The following table provides a reconciliation of cash, cash equivalents and restricted cash reported in the consolidated balance sheets that sum to the total of the same such amounts shown in the statement of cash flows (in thousands):
| | | | | | | | | | | |
| June 30,
2021 | | December 31,
2020 |
| Cash and cash equivalents | $ | 2,074,880 | | | $ | 553,398 | |
| Restricted cash current | 46,398 | | | 93,880 | |
| Restricted cash non-current | 1,461 | | | 1,460 | |
| Cash, cash equivalents and restricted cash | $ | 2,122,739 | | | $ | 648,738 | |
Revenue Recognition
The Company has various arrangements that include a right for a third party to use the Company's intellectual property as a functional license. These licensing arrangements include sales-based royalties, as well as certain development and commercial milestone payments, and the license is deemed to be the predominant item to which the sales-based royalties or milestone payments relate. For arrangements that include a development or regulatory milestone payment, the Company evaluates whether the associated event is considered probable of achievement and estimates the amount to be included in the transaction price using the most likely amount method. Milestone payments that are not within the Company or licensee's control, such as those dependent upon receipt of regulatory approval, are not considered probable of achievement until the triggering event occurs. At the end of each reporting period, the Company reevaluates the probability of achievement of each milestone and any related constraint, and if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis and affect revenue and results of operations in the period of adjustment. For arrangements that include sales-based royalties, including milestone payments based upon the achievement of a certain level of product sales, wherein the license is deemed to be the sole or predominant item to which the payments relate, the Company recognizes revenue on the satisfaction (or partial satisfaction) of its performance obligation to which some or all of the payment has been allocated, which is normally on the occurrence of the related sales. As a practical expedient, the Company has elected not to disclose the aggregate amount of the transaction price for the variable consideration that represents a sales-based royalties under the licensing arrangements. Consideration for optional goods and/or services is excluded from the transaction price at contract inception. During the three and six months ended June 30, 2021, the Company recognized sales-based royalties of $23.5 million.
Income Taxes
The Company accounts for income taxes in accordance with ASC Topic 740, Income Taxes. Under the liability method, deferred income taxes are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and operating loss carryforwards.
The Company has historically generated significant federal, state and foreign tax net operating losses, which may be subject to limitation in future periods. Management has fully reserved the related deferred tax assets with a valuation allowance in the current reporting period as more likely than not the related benefit will not be realized. The Company is currently subject to examination in all open tax years.
During the three and six months ended June 30, 2021, the Company recognized $3.5 million and $6.6 million, respectively, of income tax expense related to foreign withholding tax on royalties.
Net Loss per Share
Net loss per share is computed using the weighted average number of shares of common stock outstanding. As of June 30, 2021 and 2020, the Company had outstanding stock options, stock appreciation rights (“SARs”) and unvested restricted stock units (“RSUs”) totaling 6,092,983 and 7,797,651, respectively.
As of June 30, 2021, the Company’s Notes (see Note 7) would have been convertible into approximately 2,385,800 shares of the Company’s common stock assuming a common stock price of $136.20 or higher. These shares, after giving effect to the add back of interest expense and unamortized debt issuance costs on the Notes and any shares due to the Company upon settlement of its capped call transactions, are excluded from the computation, as their effect is antidilutive.
Recent Accounting Pronouncements
Not Yet Adopted
In August 2020, the Financial Accounting Standards Board ("FASB") issued ASU No. 2020-06, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity (“ASU 2020-06”), which will simplify the accounting for certain financial instruments with characteristics of liabilities and equity, including certain convertible instruments and contracts in an entity’s own equity. Specifically, the new standard will remove the separation models required for convertible debt with cash conversion features and convertible instruments with beneficial conversion features. It will also remove certain settlement conditions that are currently required for equity contracts to qualify for the derivative scope exception and will simplify the diluted earnings per share calculation for convertible instruments. ASU 2020-06 will be effective January 1, 2022 for the Company and may be applied using a full or modified retrospective approach. Management has evaluated the impact of adopting ASU 2020-06 and has determined that it will not have a material impact on the Company’s consolidated financial statements.
Note 3 – Fair Value Measurements
The following table represents the Company's fair value hierarchy for its financial assets and liabilities (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Fair Value at June 30, 2021 | | Fair Value at December 31, 2020 |
| Assets | Level 1 | | Level 2 | | Level 3 | | Level 1 | | Level 2 | | Level 3 |
| Money market funds(1) | $ | 238,028 | | | $ | 0 | | | $ | 0 | | | $ | 96,116 | | | $ | 0 | | | $ | 0 | |
| Government-backed securities(1) | 0 | | | 57,250 | | | 0 | | | 0 | | | 44,250 | | | 0 | |
| Treasury securities(2) | 0 | | | 199,990 | | | 0 | | | 0 | | | 54,088 | | | 0 | |
| Corporate debt securities(3) | 0 | | | 1,386,056 | | | 0 | | | 0 | | | 373,681 | | | 0 | |
| Agency securities(4) | 0 | | | 110,993 | | | 0 | | | 0 | | | 20,600 | | | 0 | |
| Total cash equivalents and marketable securities | $ | 238,028 | | | $ | 1,754,289 | | | $ | 0 | | | $ | 96,116 | | | $ | 492,619 | | | $ | 0 | |
| Liabilities | | | | | | | | | | | |
| Convertible notes payable | $ | 0 | | | $ | 580,405 | | | $ | 0 | | | $ | 0 | | | $ | 407,238 | | | $ | 0 | |
(1)Classified as cash and cash equivalents as of June 30, 2021 and December 31, 2020, respectively, on the consolidated balance sheets.
(2)Includes $199,990 and $44,052 classified as cash and cash equivalents as of June 30, 2021 and December 31, 2020, respectively, on the consolidated balance sheets.
(3)Includes $1,386,056 and $246,668 classified as cash and cash equivalents as of June 30, 2021 and December 31, 2020, respectively, on the consolidated balance sheets.
(4)Includes $110,993 classified as cash and cash equivalents as of June 30, 2021 on the consolidated balance sheets.
Fixed-income investments categorized as Level 2 are valued at the custodian bank by a third-party pricing vendor's valuation models that use verifiable observable market data, e.g., interest rates and yield curves observable at commonly quoted intervals and credit spreads, bids provided by brokers or dealers or quoted prices of securities with similar characteristics. Pricing of the Company's Notes (see Note 7) has been estimated using other observable inputs, including the price of the Company's common stock, implied volatility, interest rates and credit spreads among others.
During the six months ended June 30, 2021 and 2020, the Company did not have any transfers between levels.
Note 4 – Marketable Securities
Marketable securities classified as available-for-sale as of June 30, 2021 and December 31, 2020 were comprised of (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| June 30, 2021 | | December 31, 2020 |
| Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value | | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value |
| Treasury securities | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 10,038 | | | $ | 0 | | | $ | (2) | | | $ | 10,036 | |
| Corporate debt securities | 0 | | | 0 | | | 0 | | | 0 | | | 127,003 | | | 13 | | | (3) | | | 127,013 | |
| Agency securities | 0 | | | 0 | | | 0 | | | 0 | | | 20,599 | | | 1 | | | 0 | | | 20,600 | |
| Total marketable securities | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 157,640 | | | $ | 14 | | | $ | (5) | | | $ | 157,649 | |
The primary objective of the Company's investment policy is the preservation of capital; thus, the Company's investment policy limits investments to certain types of instruments with high-grade credit ratings, places restrictions on maturities and concentrations in certain industries and requires the Company to maintain a certain level of liquidity. As of June 30, 2021, all of the Company's investments were in securities classified as cash and cash equivalents.
Note 5 – Goodwill and Other Intangible Assets
Goodwill
The change in the carrying amounts of goodwill for the six months ended June 30, 2021 was as follows (in thousands):
| | | | | |
| Amount |
| Balance at December 31, 2020 | $ | 135,379 | |
| Currency translation adjustments | (1,085) | |
| Balance at June 30, 2021 | $ | 134,294 | |
Identifiable Intangible Assets
Purchased intangible assets consisted of the following as of June 30, 2021 and December 31, 2020 (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| June 30, 2021 | | December 31, 2020 |
| Gross
Carrying
Amount | | Accumulated
Amortization | | Intangible
Assets, Net | | Gross
Carrying
Amount | | Accumulated
Amortization | | Intangible
Assets, Net |
| Finite-lived intangible assets: | | | | | | | | | | | |
| Proprietary adjuvant technology | $ | 8,741 | | | $ | (3,460) | | | $ | 5,281 | | | $ | 9,099 | | | $ | (3,374) | | | $ | 5,725 | |
| Collaboration agreements | 3,947 | | | (3,947) | | | 0 | | | 4,109 | | | (4,109) | | | 0 | |
| Total identifiable intangible assets | $ | 12,688 | | | $ | (7,407) | | | $ | 5,281 | | | $ | 13,208 | | | $ | (7,483) | | | $ | 5,725 | |
Amortization expense for the six months ended June 30, 2021 and 2020 was $0.2 million and $0.3 million, respectively.
Estimated amortization expense for existing intangible assets for the remainder of 2021 and for each of the five succeeding years ending December 31 will be as follows (in thousands):
| | | | | | | | |
| Year | | Amount |
| 2021 (remainder) | | $ | 219 | |
| 2022 | | 437 | |
| 2023 | | 437 | |
| 2024 | | 437 | |
| 2025 | | 437 | |
| 2026 | | 437 | |
Note 6 - Leases
During the second quarter of 2021, the Company evaluated the impact of changes in facts and circumstances on its Contract Manufacturing Organizations and Contract Development and Manufacturing Organizations agreements that had previously been determined to represent embedded lease arrangements. The Company concluded that the impact resulted in the modification of existing leases and, in accordance with its policy, the Company remeasured and reallocated the remaining consideration in the contracts and reassessed the lease classification as of the effective date of the modification. As a result, the Company recognized a Right-Of-Use ("ROU") asset and a corresponding long-term operating lease liability of $11.4 million on the remeasurement of one of its long-term supply agreements using an incremental borrowing rate of 6.5%. The Company expensed the ROU asset since it relates to research and development activities for the development of NVX-CoV2373 for which the Company does not have an alternative future use. Modifications to leases with a lease term of 12 months or less at the commencement date did not result in a change in lease classification and, in accordance with the Company's election to apply the practical expedient in ASC Topic 842, Leases (“ASC 842”), it did not recognize a ROU asset or lease liability but instead, lease payments are recognized as an expense on a straight-line basis over the modified lease term and variable lease payments that do not depend on an index or rate, are recognized as an expense in the period in which the variable lease costs are incurred based on performance or usage in accordance with contractual agreements.
During the three and six months ended June 30, 2021, the Company recognized a short-term lease expense of $86.6 million and $214.2 million, respectively, related to its embedded leases, including a new lease that commenced during the first quarter of 2021. The Company did 0t recognize a short-term lease expense related to embedded leases during the three and six months ended June 30, 2020.
During the three and six months ended June 30, 2021, the Company recognized $1.8 million and $4.0 of interest expenses, respectively, on its finance lease liabilities. The Company did 0t recognize any interest expense related to finance lease liabilities during the three and six months ended June 30, 2020.
During the second quarter of 2021, the Company extended the term of certain of its existing research and development facility and offices leases by two years to five years, giving rise to additional ROU assets and related long-term operating lease liabilities of approximately $7.2 million.
Note 7 – Long-Term Debt
Convertible Notes
The Company incurred approximately $10.0 million of debt issuance costs during the first quarter of 2016 relating to the issuance of $325 million aggregate principal amount of convertible senior unsecured notes that will mature on February 1, 2023 (the “Notes”), which were recorded as a reduction to the Notes on the consolidated balance sheet. The $10.0 million of debt issuance costs is being amortized and recognized as additional interest expense over the seven year contractual term of the Notes on a straight-line basis, which approximates the effective interest rate method.
Total convertible notes payable consisted of the following at (in thousands):
| | | | | | | | | | | |
| June 30,
2021 | | December 31,
2020 |
| Principal amount of Notes | $ | 325,000 | | | $ | 325,000 | |
| Unamortized debt issuance costs | (2,254) | | | (2,965) | |
| Total convertible notes payable | $ | 322,746 | | | $ | 322,035 | |
The interest expense incurred in connection with the Notes consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended
June 30, | | Six Months Ended
June 30, | | |
| 2021 | | 2020 | | 2021 | | 2020 | | | | |
| Coupon interest at 3.75% | $ | 3,047 | | | $ | 3,047 | | | $ | 6,094 | | | $ | 6,094 | | | | | |
| Amortization of debt issuance costs | 356 | | | 356 | | | 712 | | | 712 | | | | | |
| Total interest expense on Notes | $ | 3,403 | | | $ | 3,403 | | | $ | 6,806 | | | $ | 6,806 | | | | | |
Note 8 – Stockholders' Equity
During the six months ended June 30, 2021 and 2020, the Company sold 2.6 million and 28.5 million, respectively, of shares of its common stock resulting in net proceeds of approximately $565 million and $392 million, respectively, under its various At Market Issuance Sales agreements.
In June 2021, the Company entered into an At Market Issuance Sales Agreement (the "June 2021 Sales Agreement"), which allows it to issue and sell up to $500 million in gross proceeds of shares of its common stock, and terminated its existing At Market Issuance Sales agreement. As of June 30, 2021, 0 shares had been sold under the June 2021 Sales Agreement.
Note 9 – Stock-Based Compensation
Equity Plans
The 2015 Stock Incentive Plan, as amended (“2015 Plan”), was approved at the Company's annual meeting of stockholders in June 2015. Under the 2015 Plan, equity awards may be granted to officers, directors, employees and consultants of and advisors to the Company and any present or future subsidiary.
The 2015 Plan authorizes the issuance of up to 12.4 million shares of common stock under equity awards granted under the 2015 Plan, including an increase of 1.5 million shares approved for issuance under the 2015 Plan at the Company's 2021 annual meeting of stockholders. All such shares authorized for issuance under the 2015 Plan have been reserved. The 2015 Plan will expire on March 4, 2025.
The Amended and Restated 2005 Stock Incentive Plan (“2005 Plan”) expired in February 2015 and no new awards may be made under such plan, although awards will continue to be outstanding in accordance with their terms.
The 2015 Plan permits and the 2005 Plan permitted the grant of stock options (including incentive stock options), restricted stock, stock appreciation rights and restricted stock units. In addition, under the 2015 Plan, unrestricted stock, stock units and performance awards may be granted. Stock options and stock appreciation rights generally have a maximum term of ten years and may be or were granted with an exercise price that is no less than 100% of the fair market value of the Company's common stock at the time of grant. Grants of stock options are generally subject to vesting over periods ranging from one to four years.
Stock Options and Stock Appreciation Rights
The following is a summary of stock options and SARs activity under the 2015 Plan and 2005 Plan for the six months ended June 30, 2021:
| | | | | | | | | | | | | | | | | | | | | | | |
| 2015 Plan | | 2005 Plan |
| Stock
Options and SARs | | Weighted-Average
Exercise
Price | | Stock
Options | | Weighted-Average
Exercise
Price |
| Outstanding at January 1, 2021 | 5,420,463 | | | $ | 38.05 | | | 214,186 | | | $ | 88.11 | |
| Granted | 47,458 | | | $ | 148.49 | | | 0 | | | $ | 0 | |
| Exercised | (598,441) | | | $ | 44.14 | | | (35,401) | | | $ | 106.49 | |
| Canceled | (50,881) | | | $ | 121.33 | | | 0 | | | $ | 0 | |
| Outstanding at June 30, 2021 | 4,818,599 | | | $ | 37.50 | | | 178,785 | | | $ | 84.47 | |
| Shares exercisable at June 30, 2021 | 692,187 | | | $ | 70.17 | | | 178,785 | | | $ | 84.47 | |
| Shares available for grant at June 30, 2021 | 2,360,263 | | | | | | | |
The fair value of stock options granted under the 2015 Plan was estimated at the date of grant using the Black-Scholes option-pricing model with the following assumptions:
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended
June 30, | Six Months Ended
June 30, | |
| 2021 | | 2020 | | 2021 | | 2020 | | | | |
| Weighted average Black-Scholes fair value of stock options granted | $166.66 | | $77.41 | | $131.66 | | $76.99 | | | | |
| Risk-free interest rate | 0.6%-1.1% | | 0.3%-0.6% | | 0.5%-1.1% | | 0.3%-1.5% | | | | |
| Dividend yield | 0% | | 0% | | 0% | | 0% | | | | |
| Volatility | 126.2%-142.0% | | 116.0%-151.5% | | 124.7%-142.0% | | 116.0%-151.5% | | | | |
| Expected term (in years) | 4.1-6.1 | | 3.9-7.6 | | 4.1-6.1 | | 3.9-7.6 | | | | |
The total aggregate intrinsic value and weighted-average remaining contractual term of stock options and SARs outstanding under the 2015 Plan and 2005 Plan as of June 30, 2021 was approximately $865 million and 8.1 years, respectively. The total aggregate intrinsic value and weighted-average remaining contractual term of stock options and SARs exercisable under the 2015 Plan and 2005 Plan as of June 30, 2021 was approximately $121 million and 5.5 years, respectively. The aggregate intrinsic value represents the total intrinsic value (the difference between the Company's closing stock price on the last trading day of the period and the exercise price, multiplied by the number of in-the-money stock options and SARs) that would have been received by the holders had all stock option and SAR holders exercised their stock options and stock appreciation rights on June 30, 2021. This amount is subject to change based on changes to the closing price of the Company's common stock. The aggregate intrinsic value of stock options and vesting of restricted stock awards for the six months ended June 30, 2021 and 2020 was approximately $115 million and $8 million, respectively.
Employee Stock Purchase Plan
The Employee Stock Purchase Plan, as amended (the “ESPP”), was approved at the Company's annual meeting of stockholders in June 2013. The ESPP currently authorizes an aggregate of 600,000 shares of common stock to be purchased. The ESPP allows employees to purchase shares of common stock of the Company at each purchase date through payroll deductions of up to a maximum of 15% of their compensation, at 85% of the lesser of the market price of the shares at the time of purchase or the market price on the beginning date of an option period (or, if later, the date during the option period when the employee was first eligible to participate). As of June 30, 2021, there were 212,876 shares available for issuance under the ESPP.
The ESPP is considered compensatory for financial reporting purposes. As such, the fair value of ESPP shares was estimated at the date of grant using the Black-Scholes option-pricing model with the following assumptions:
| | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended
June 30, | | Six Months Ended
June 30, | | |
| 2021 | | 2020 | | 2021 | | 2020 | | | | |
| Range of Black-Scholes fair values of ESPP shares granted | $128.70-$238.85 | | $2.87-$21.80 | | $128.70-$238.85 | | $2.57-$35.00 | | | | |
| Risk-free interest rate | 0.1% | | 1.5%-2.6% | | 0.1% | | 1.5%-2.6% | | | | |
| Dividend yield | 0% | | 0% | | 0% | | 0% | | | | |
| Volatility | 120.4%-159.4% | | 66.6%-150.9% | | 120.4%-159.4% | | 66.6%-154.4% | | | | |
| Expected term (in years) | 0.5-2.0 | | 0.5-2.0 | | 0.5-2.0 | | 0.5-2.0 | | | | |
Restricted Stock Units
The following is a summary of restricted stock units activity for the six months ended June 30, 2021:
| | | | | | | | | | | |
| Number of
Shares | | Per Share
Weighted-
Average
Fair Value |
| Outstanding and Unvested at January 1, 2020 | 1,044,980 | | | $ | 72.59 | |
| Restricted stock units granted | 134,099 | | | $ | 178.71 | |
| Restricted stock units vested | (66,457) | | | $ | 79.20 | |
| Restricted stock units forfeited | (17,023) | | | $ | 128.59 | |
| Outstanding and Unvested at June 30, 2021 | 1,095,599 | | | $ | 84.30 | |
The Company recorded all stock-based compensation expense in the consolidated statements of operations as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended
June 30, | | Six Months Ended
June 30, | |
| 2021 | | 2020 | | 2021 | | 2020 | | | |
| Research and development | $ | 24,779 | | | $ | 4,098 | | | $ | 48,569 | | | $ | 6,005 | | | | |
| General and administrative | 28,344 | | | 3,834 | | | 57,614 | | | 5,892 | | | | |
| Total stock-based compensation expense | $ | 53,123 | | | $ | 7,932 | | | $ | 106,183 | | | $ | 11,897 | | | | |
As of June 30, 2021, there was approximately $230 million of total unrecognized compensation expense related to unvested stock options, SARs, restricted stock units and the ESPP. The increase in unrecognized compensation expense is primarily due to the significant increase in the Company's common stock price starting in 2020. This unrecognized non-cash compensation expense is expected to be recognized over a weighted-average period of one year, and will be allocated between research and development and general and administrative expenses accordingly. This estimate does not include the impact of other possible stock-based awards that may be made during future periods and awards that require approval by the stockholders.
Note 10 –Contingencies
In February 2021, a Novavax stockholder filed a derivative complaint against certain members of the Company's board of directors and certain members of senior management in the Delaware Court of Chancery with Novavax as a nominal defendant. The plaintiff challenges two sets of equity awards made in April 2020 and in June 2020 on the ground that they were “spring-loaded,” that is, made at a time when certain board members or members of senior management allegedly possessed undisclosed positive material information concerning the Company. The complaint asserts claims for breach of fiduciary duty, waste, and unjust enrichment. The plaintiff seeks an award of damages to the Company, an order rescinding the April 2020 and June 2020 awards or requiring disgorgement, and an award of attorneys’ fees incurred in connection with the litigation. On May 10, 2021, the defendants moved to dismiss the complaint in its entirety. On June 17, 2021, the Company’s stockholders voted FOR ratification of the April 2020 awards and ratification of the June 2020 awards. Details of the ratification proposals are set forth in the Company’s Definitive Proxy Statement filed with the SEC on May 3, 2021. The results of the vote were disclosed in the Company’s Current Report on Form 8-K filed with the SEC on June 24, 2021. Should the plaintiff elect to move forward with his claims, the defendants intend to move for summary judgment on ratification grounds while continuing to pursue dismissal. As such, the Company is not expecting any material estimable financial impact of the plaintiff's claim.
Note 11 – Revenue
During the three and six months ended June 30, 2021 and 2020, the Company performed research and development under government contracts and grant, license and clinical development agreements. The Company's revenue primarily
consisted of funding under U.S. government contracts and the Company's funding arrangement with CEPI to advance the clinical development and manufacturing of NVX-CoV2373.
The Company recorded revenue as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | |
| Three Months Ended
June 30, | | Six Months Ended
June 30, |
| 2021 | | 2020 | | 2021 | | 2020 |
| Government contracts | | | | | | | |
| OWS | $ | 239,493 | | | $ | 0 | | | $ | 603,053 | | | $ | 0 | |
| DoD | 1,041 | | | 311 | | | 20,185 | | | 311 | |
| Grants and other | | | | | | | |
| CEPI | 31,955 | | | 34,246 | | | 93,516 | | | 36,504 | |
| BMGF | 0 | | | 159 | | | 2,628 | | | 414 | |
| Other | 2,071 | | | 822 | | | 2,407 | | | 1,686 | |
| Royalties | 23,457 | | | 0 | | | 23,457 | | | 0 | |
| Total | $ | 298,017 | | | $ | 35,538 | | | $ | 745,246 | | | $ | 38,915 | |
Government Contracts and Grants
The Company’s U.S. government contracts comprise an agreement with Advanced Technology International (“ATI”), the Consortium Management Firm acting on behalf of the Medical CBRN Defense Consortium in connection with the partnership formerly known as Operation Warp Speed (“OWS”) and a contract with the U.S. Department of Defense (the “DoD”). As of June 30, 2021, the Company's OWS agreement was fully funded up to $1.75 billion to support certain activities related to the development of NVX-CoV2373 and the manufacture and delivery of 100 million doses of the vaccine candidate to the U.S. government. The U.S. government has recently instructed the Company to prioritize alignment with the U.S. Food and Drug Administration on the Company's analytic methods before conducting additional U.S. manufacturing and further indicated that the U.S. government will not fund additional U.S. manufacturing until such agreement has been made. The U.S. government also instructed the Company to proceed with work under the OWS Agreement related to all other activities, including ongoing clinical trials and nonclinical studies, regulatory interactions, analytics/assays and characterization of manufactured vaccine and project management. The Company’s revenue from CEPI comprises grant and forgivable loan funding. The latter is repayable if the proceeds from the sales of NVX-CoV2373 to one or more third parties cover the Company’s costs of manufacturing the vaccine, not including manufacturing costs funded by CEPI.
Collaboration and License Agreements
In February 2021, the Company finalized an expanded collaboration and license agreement with SK bioscience to manufacture and commercialize NVX-CoV2373 for sale to the government of Korea. Concurrently, SK bioscience finalized an advance purchase agreement with the Korean government to supply 40 million doses of NVX-CoV2373 to the Republic of Korea beginning in 2021. The agreement is in addition to the Company's existing manufacturing arrangement with SK bioscience entered into in August 2020. Under the collaboration agreement, SK bioscience was granted an exclusive license to develop, manufacture and commercialize NVX-CoV2373 in the Republic of Korea. SK bioscience expanded its capacity to manufacture the antigen component of NVX-CoV2373 for use in the final drug product globally, including product distributed by the COVAX Facility. SK bioscience will also purchase a certain quantity of NVX-CoV2373 directly from the Company, subject to approval by relevant regulatory authority, and sufficient doses of Matrix-M™ adjuvant to manufacture the remainder of the 40 million doses of NVX-CoV2373 it expects to sell to the Korean government. SK bioscience will pay the Company a tiered royalty in the low to middle double-digit range on the sale of NVX-CoV2373. The Company recognized royalties of $23.5 million during the three and six months ended June 30, 2021 related to SK bioscience's sale of the antigen component of NVX-CoV2373 to the Korean government. In May 2021, the Company entered a non-binding Memorandum of Understanding ("MOU") with the Ministry of Health and Welfare of Korea and SK bioscience to explore further cooperation in the development and manufacturing of vaccines, including NVX-CoV2373. Under the MOU, the Company agreed to potentially explore the development of new vaccine products with SK bioscience, including COVID-19 variant vaccines, and/or an influenza/COVID-19 combination vaccine.
In February 2021, the Company finalized a collaboration agreement previously announced in August 2020, with Takeda Pharmaceutical Company Limited (“Takeda”) for the exclusive development, manufacturing and commercialization of NVX-CoV2373 in Japan. Under the agreement, the Company will transfer technology and supply the Matrix-M™ adjuvant to Takeda, which will manufacture the antigen component of NVX-CoV2373. Takeda will receive funding from the Government
of Japan’s Ministry of Health, Labour and Welfare to support the technology transfer, establishment of infrastructure and scale-up of manufacturing. The Company will be entitled to receive royalties based on the achievement of certain development and commercial milestones, as well as on a portion of net profits from the sale of the vaccine.
Vaccine Supply Agreements
During the six months ended June 30, 2021, the Company entered into various Advanced Purchase Agreements ("APAs"), including an agreement with Her Majesty the Queen in Right of Canada as represented by the Minister of Public Works and Government Services to supply 52 million doses of NVX-CoV2373. The Company will submit an application for regulatory approval in Canada following its first submission for regulatory approval in another priority market and the Canada authority will provide reasonable assistance to the Company with obtaining such regulatory approval. As part of the agreement, Canada will have the option to purchase up to an additional 24 million doses of NVX-CoV2373. In February 2021, the Company reached a MOU with the Canadian government to produce NVX-CoV2373 in Canada. The Company plans to produce NVX-CoV2373 at the National Research Council’s Biologics Manufacturing Centre in Montreal once both the vaccine candidate and the facility receive Health Canada approvals.
In May 2021, the Company finalized an APA with Gavi, the Vaccine Alliance ("Gavi") building upon its MOU previously announced in February 2021. Under the terms of the agreement, 1.1 billion doses of NVX-CoV2373 are to be made available to countries participating in the COVAX Facility, which was established to allocate and distribute vaccines equitably to participating countries and economies. The Company expects to manufacture and distribute 350 million doses of NVX-CoV2373 to countries participating under the COVAX Facility. Under a separate purchase agreement with Gavi, Serum Institute of India Private Limited ("SIIPL") is expected to manufacture and deliver the balance of the 1.1 billion doses of NVX-CoV2373 for low- and middle-income countries participating in the COVAX Facility. The Company expects to deliver doses with antigen and adjuvant manufactured at facilities directly funded under the Company's funding agreement with CEPI. The Company expects to supply significant doses that Gavi would allocate to low-, middle- and high-income countries, subject to certain limitations, utilizing a tiered pricing schedule and Gavi may prioritize such doses to low- and middle- income countries, at lower prices. Additionally, the Company may provide additional doses of NVX-CoV2373, to the extent available from CEPI funded manufacturing facilities, in the event that SIIPL cannot materially deliver expected vaccine doses to the COVAX Facility. Together with SIIPL, the Company expects to initiate delivery of doses following receipt of appropriate regulatory authorizations. Under the agreement, the Company received an upfront payment from Gavi of $350 million during the second quarter of 2021 and expects to receive an additional payment of $350 million if the Company secures emergency use listing for NVX-CoV2373 by the World Health Organization ("WHO").
During the six months ended June 30, 2021, changes in the Company's accounts receivables, unbilled services and deferred revenue balances were as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| | December 31, 2020 | | Additions | | Deductions | | June 30, 2021 |
| Accounts receivable | | $ | 262,012 | | | $ | 1,309,924 | | | $ | (1,521,947) | | | $ | 49,989 | |
| Unbilled services | | 0 | | | 499,239 | | | (477,865) | | | 21,374 | |
| Deferred revenue | | 273,228 | | | 1,192,483 | | | (245,638) | | | 1,220,073 | |
As of June 30, 2021, the deferred revenue of $1.2 billion primarily comprised of approximately $1.1 billion related to upfront payments under APAs.
The aggregate amount of the transaction price allocated to performance obligations that were unsatisfied (or partially unsatisfied) was $7.0 billion as at June 30, 2021. The Company expects to fulfil its unsatisfied performance obligations within 12 months.
Note 12 – Subsequent Events
In August 2021, the Company announced that it finalized the terms of an APA with the European Commission, which the parties expect to execute during the third quarter of 2021, for the purchase of up to 100 million initial doses of NVX-CoV2373, with the option of the European Commission to purchase an additional 100 million doses through 2023.
In August 2021, the Company filed regulatory submissions in partnership with SIIPL for emergency use authorization in multiple markets. Regulatory submissions were filed with the Drugs Controller General of India, as well as regulatory agencies in Indonesia and the Philippines. In addition to these filings, the Company expects to file a submission to the WHO for emergency use listing in August of 2021.
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Any statements in the discussion below and elsewhere in this Quarterly Report about expectations, beliefs, plans, objectives, assumptions or future events or performance of Novavax, Inc. (“Novavax,” together with its wholly owned subsidiaries Novavax AB and Novavax CZ, the “Company,” “we” or “us”) are not historical facts and are forward-looking statements. Such forward-looking statements include, without limitation, statements about our capabilities, goals, expectations regarding future revenue and expense levels and capital raising activities; our operating plans and prospects; potential market sizes and demand for our product candidates; the efficacy, safety and intended utilization of our product candidates; the development of our clinical-stage product candidates and our recombinant vaccine and adjuvant technologies; the development of our preclinical product candidates; the conduct, timing and potential results from clinical trials and other preclinical studies; plans for and potential timing of regulatory filings; our expectation of manufacturing capacity, timing, production and delivery for NVX-CoV2373; our expectations with respect to the anticipated ongoing development and potential commercialization or licensure of NVX-CoV2373 and NanoFlu™; the expected timing and content of regulatory actions; funding from the U.S. government partnership formerly known as Operation Warp Speed (“OWS”), the U.S. Department of Defense (“DoD”) and the Coalition for Epidemic Preparedness Innovations (“CEPI”), and payments from the Bill & Melinda Gates Foundation (“BMGF”); funding under our advance purchase agreements and supply agreements; our available cash resources and usage and the availability of financing generally; plans regarding partnering activities and business development initiatives; and other matters referenced herein. Generally, forward-looking statements can be identified through the use of words or phrases such as “believe,” “may,” “could,” “will,” “would,” “possible,” “can,” “estimate,” “continue,” “ongoing,” “consider,” “anticipate,” “intend,” “seek,” “plan,” “project,” “expect,” “should,” “would,” “aim,” or “assume,” the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words.
Forward-looking statements are neither historical facts nor assurances of future performance. Instead, they are based only on our current beliefs and expectations about the future of our business, future plans and strategies, projections, anticipated events and trends, the economy and other future conditions. Forward-looking statements involve estimates, assumptions, risks and uncertainties that could cause actual results or outcomes to differ materially from those expressed or implied in any forward-looking statements, and, therefore, you should not place considerable reliance on any such forward-looking statements. Such risks and uncertainties include, without limitation, challenges satisfying, alone or together with partners, various safety, efficacy, and product characterization requirements, including those related to process qualification and assay validation, necessary to satisfy each applicable regulatory authority, like the U.S. Food and Drug Administration (“FDA”), World Health Organization (“WHO”), UK Medicines and Healthcare Products Regulatory Agency (“MHRA”), the European Medicines Agency (“EMA”), the Republic of Korea’s Ministry of Food and Drug Safety (“MFDS”), or Japan’s Ministry of Health, Labour and Welfare (“MHLW”); difficulty obtaining scarce raw materials; resource, including human capital and manufacturing capacity, constraints on our ability to pursue these regulatory pathways, alone or with partners, in multiple jurisdictions simultaneously, leading to staggering of regulatory filings and potential regulatory actions; challenges meeting contractual requirements under agreements with multiple commercial, governmental, and other entities; and other risks and uncertainties identified in Part II, Item 1A “Risk Factors” of this Quarterly Report and in Part I, Item 1A “Risk Factors” of our Annual Report on Form 10-K, which may be detailed and modified or updated in other documents filed with the United States Securities and Exchange Commission (“SEC”) from time to time, and are available at www.sec.gov and at www.novavax.com. You are encouraged to read these filings as they are made.
We cannot guarantee future results, events, level of activity, performance or achievement. Any or all of our forward-looking statements in this Quarterly Report may turn out to be inaccurate or materially different from actual results. Further, any forward-looking statement speaks only as of the date when it is made, and we undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, unless required by law. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Overview
Novavax, Inc., together with our wholly-owned subsidiaries, Novavax AB and Novavax CZ, is a biotechnology company promoting improved global health through the discovery, development and commercialization of innovative vaccines to prevent serious infectious diseases and address urgent, global health needs. Our vaccine candidates, including both our coronavirus vaccine candidate (“NVX-CoV2373”) and our seasonal quadrivalent influenza vaccine candidate (“NanoFlu™”), are genetically engineered, three-dimensional nanostructures of recombinant proteins critical to disease pathogenesis. We believe that our protein-subunit-based candidates elicit differentiated immune responses that may be more efficacious than naturally occurring immunity or other vaccine approaches. Additionally, our Matrix-M™ adjuvant has been shown to enhance functional immune responses and has been well-tolerated in multiple clinical trials. To date, we have formulated many of the vaccine candidates in our pipeline with our Matrix-M™ adjuvant, including NVX-CoV2373 and NanoFlu™.
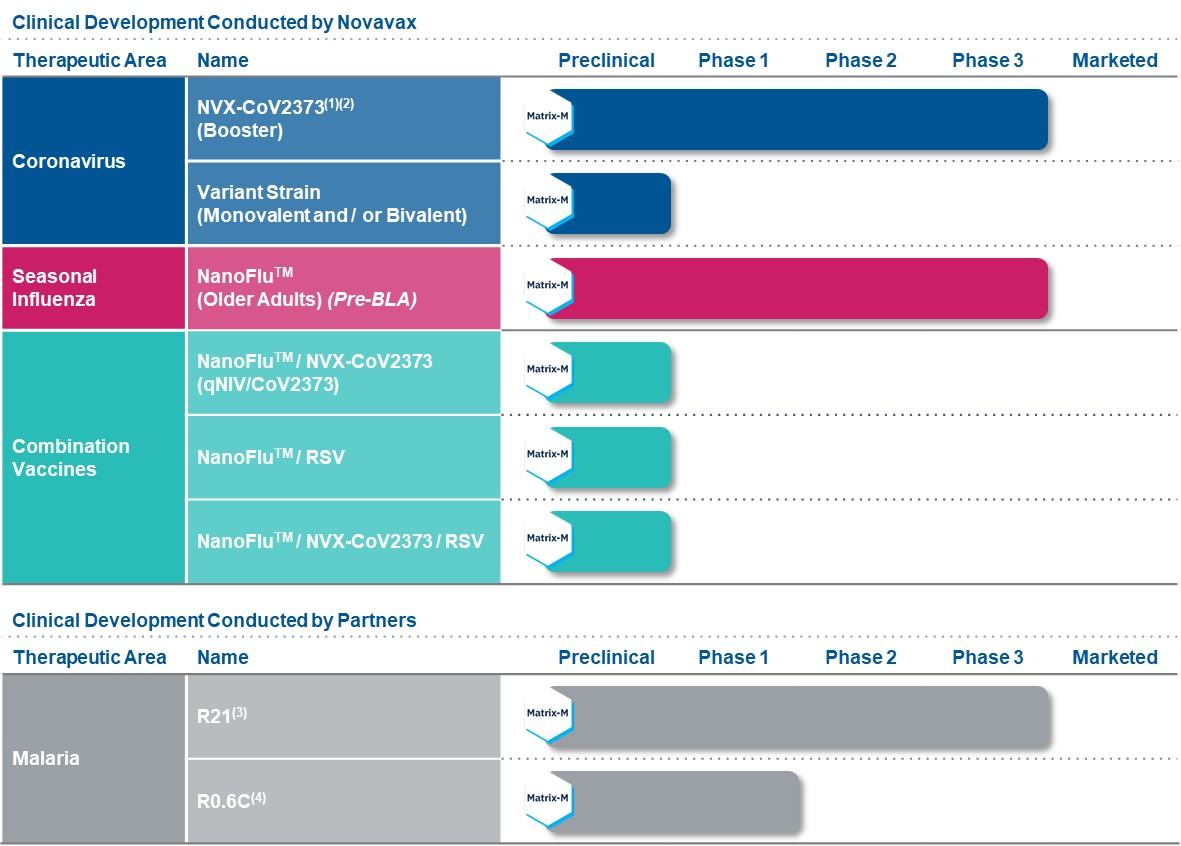
Near-term Clinical Development Pipeline
Our development pipeline encompasses vaccine candidates addressing therapeutic areas including coronavirus, seasonal influenza, respiratory syncytial virus (“RSV”) and other emerging infectious diseases. At the forefront of our pipeline is NVX-CoV2373. We have advanced NVX-CoV2373 through two Phase 3 clinical trials, which have demonstrated high efficacy against the original COVID-19 strain and commonly circulating variants of COVID-19 with a favorable safety profile. We have also advanced our NanoFlu™ vaccine program through a Phase 3 clinical trial, which demonstrated positive top-line results and achieved statistical significance in key secondary endpoints. We continue to evaluate the viability of certain combination vaccines, including combinations of our NanoFlu™, NVX-CoV2373 and respiratory syncytial virus fusion (F) protein nanoparticle vaccine candidate (“RSV F Vaccine”).
We remain focused on bringing our NVX-CoV2373 vaccine candidate to market following global regulatory approvals. Through ongoing crossover and booster studies in our clinical trials, as well as the development of our COVID-19 variant strain vaccine candidates, we continue to collect data to characterize and optimize vaccine performance. We expect to leverage these clinical insights to advance our booster strategy given the evolving COVID-19 pandemic.
In addition to our COVID-19 clinical development, NanoFlu™ continues to be a priority for our team, especially as it relates to development of our NanoFlu™ / NVX-CoV2373 combination vaccine candidate (“qNIV/CoV2373”).
Although NVX-CoV2373 and NanoFlu™ are our near-term priorities, we remain optimistic that the additional programs in our pipeline, including our vaccine candidates for RSV and other emerging infectious diseases, present viable opportunities for future development.
The pipeline chart below summarizes the clinical and preclinical development programs that we are focusing on in the near-term.
(1)Supported by funding from OWS, DoD, CEPI and BMGF
(2)Ongoing PREVENT-19, a Phase 3 clinical trial in the U.S. and Mexico; Ongoing PREVENT-19 pediatric expansion in the U.S.; Ongoing Phase 3 clinical trial in the UK; Ongoing Phase 2b clinical trial in South Africa
(3)Reflects malaria vaccine candidate (“R21”) created by the University of Oxford and formulated with Matrix-M™
adjuvant; Ongoing Phase 3 clinical trial in Africa; R21 is licensed to Serum Institute of India Private Limited ("SIIPL"); Novavax will have commercial rights to sell and distribute R21 in certain countries, primarily in travelers' and military vaccine markets
(4)Reflects malaria transmission blocking vaccine candidate (“R0.6C”) manufactured by Statens Serum Institut; Ongoing Phase 1 clinical trial in the Netherlands to evaluate R0.6C, Matrix-M™ adjuvant and common aluminum hydroxide adjuvant
Matrix-M™ Adjuvant
Our proprietary Matrix-M™ adjuvant has demonstrated potent and well-tolerated efficacy by stimulating the entry of antigen presenting cells (“APCs”) into the injection site and enhancing antigen presentation in local lymph nodes, which in turn activates T-cell, B-cell, and APC populations, thereby boosting immune response. Our Matrix-M™ adjuvant has been shown to increase neutralizing antibodies and induces long-lasting memory B cells, which enhances B-cell immunity and recruits and increases the frequency of CD4+ and CD8+ T cells to enhance T-cell immunity in preclinical models. The potent immune-stimulating mechanism of action enables a lower dose of antigen required to achieve the desired immune response, ultimately contributing to increased supply and manufacturing capacity. These immune-boosting and dose-sparing capabilities contribute to the adjuvant’s highly unique profile. Our Matrix-M™ adjuvant is being evaluated in combination with several other vaccine antigens produced by other manufacturers.
Coronavirus
NVX-CoV2373 Clinical Development
We have evaluated NVX-CoV2373 in various preclinical and clinical trials, including two Phase 3 trials, one Phase 2b trial, and one Phase 1/2 trial. Through our clinical development program to date, we confirmed the use of a 5-microgram dose of NVX-CoV2373 with Matrix-M™ adjuvant for late-stage development. We have also collected data that indicates a reassuring safety profile and high levels of efficacy for NVX-CoV2373 against original COVID-19 strain and commonly circulating variants. A summary and status of our clinical development of NVX-CoV2373 follows:
PREVENT-19 Pediatric Expansion
In June 2021, we completed enrollment of the pediatric expansion of our PREVENT-19 Phase 3 trial in the U.S. that was initiated in April 2021. The pediatric expansion is a placebo-controlled study to evaluate efficacy, safety, and immunogenicity of NVX-CoV2373 in 2,248 adolescent participants aged 12 to 17 across up to 75 sites in the U.S. Participants will randomly receive either the vaccine candidate or placebo in two doses, administered 21 days apart. Two-thirds of participants will receive intramuscular injections of the vaccine and one-third will receive placebo. All primary doses have been administered and a blinded crossover is planned to take place in August 2021 to ensure that all trial participants receive active vaccine before the start of the 2021-2022 school year. Participants will be monitored for safety for up to two years following the final administered dose.
PREVENT-19 Phase 3 U.S. and Mexico
In June 2021, we announced results from the final analysis of our PREVENT-19 Phase 3 trial in the U.S. and Mexico initiated in December 2020. The final analysis was conducted on events accrued prior to participants receiving crossover vaccine. In the final analysis, NVX-CoV2373 achieved its primary efficacy endpoint with an overall efficacy of 90.4% despite over half the illness cases being caused by Variants of Interest ("VoI") and Variants of Concern ("VoC"). Additionally, NVX-CoV2373 demonstrated 100% protection against moderate and severe disease, including those caused by variants.
PREVENT-19 was a randomized, placebo-controlled, observer-blinded trial to evaluate the efficacy, safety, and immunogenicity of NVX-CoV2373 in 29,960 participants aged 18 years or older across 119 sites in the U.S. and Mexico. Enrollment for PREVENT-19 emphasized recruiting high-risk groups most impacted by COVID-19. The participant trial population was composed of the following: 22% Latin American, 12% African American, 7% Native American, 4% Asian American, 37% with underlying medical co-morbidities and 13% older adults aged 65 years and older. The trial design was harmonized to align with other Phase 3 trials conducted under the auspices of OWS, including the use of a single external, independent Data and Safety Monitoring Board to evaluate safety. The trial’s primary endpoint was the first occurrence of PCR-confirmed, symptomatic (mild, moderate or severe) COVID-19 with onset at least seven days after the second study
vaccination in participants who had not been previously infected with SARS-CoV-2. Two-thirds of the participants were assigned to randomly receive two intramuscular injections of NVX-CoV2373 comprising 5 micrograms of antigen with 50 micrograms of Matrix-M™ adjuvant, administered 21 days apart, while one-third of the trial participants received placebo. The primary efficacy analysis was event-driven, based on the number of participants with symptomatic mild, moderate, or severe COVID-19 disease.
Efficacy endpoints were accrued from January 25, 2021, through April 30, 2021, at a time when the Alpha (B.1.1.7) variant strain became the predominant strain in the U.S. Other strains, including VoI and VoC, were also on the rise during the PREVENT-19 endpoint accrual window. PREVENT-19 met a key secondary endpoint, demonstrating 100% efficacy against variants not considered VoC/VoI. NVX-CoV2373 also demonstrated 92.6% efficacy against VoC/VoI, achieving a key exploratory endpoint of the study. Among high-risk populations (participants over age 65, under age 65 with certain comorbidities, or having life circumstances with frequent COVID-19 exposure), NVX-CoV2373 demonstrated 91.0% efficacy. Preliminary safety data showed NVX-CoV2373 to be generally well-tolerated, with serious and severe adverse events low in number and balanced between vaccine and placebo groups. Participants will be followed for 24 months following the second injection. PREVENT-19 was conducted with support and funding from OWS.
Phase 3 United Kingdom (“UK”)
In June 2021, the final analysis of our Phase 3 UK trial was published in the New England Journal of Medicine. The publication of the final analysis highlights the robust safety and efficacy data for NVX-CoV2373. The final analysis confirmed 89.7% overall efficacy, with over 60% of the cases caused by the Alpha (B.1.1.7) variant strain. The analysis also confirmed 96.4% efficacy against non-Alpha (non-B.1.1.7) variant strains, which represents strains most similar to the original COVID-19 virus. The final analysis also demonstrated that initial vaccine side effects were mostly mild and transient, and that no imbalance was seen in more serious adverse events compared with the placebo arm. The final analysis builds upon the interim analysis conducted in January 2021 and updated analysis announced in March 2021. The trial was a randomized, observer-blinded, placebo-controlled study that enrolled 15,203 adults aged 18 to 84. Half of the trial participants received two intramuscular injections of NVX-CoV2373 comprising 5 micrograms of antigen with 50 micrograms of Matrix-M™ adjuvant, administered 21 days apart, while the other half of the trial participants received placebo. The trial was conducted in partnership with the UK government Vaccines Taskforce and led by researchers at St George’s, University of London and St George’s Hospital, London.
Phase 3 UK Co-Administration Sub-Study
In June 2021, we announced data from the co-administration sub-study of NVX-CoV2373 and an approved influenza vaccine, conducted as part of our Phase 3 UK trial. In this study, 431 participants enrolled in our Phase 3 UK trial received an approved seasonal influenza vaccine (Seqirus, adjuvanted, trivalent seasonal influenza vaccine or a cell-based quadrivalent seasonal influenza vaccine). Approximately half of the participants in the study were also co-vaccinated with NVX-CoV2373, while the remainder received placebo. Results demonstrated a robust immune response and a favorable safety and reactogenicity profile. Immunogenicity of the influenza vaccine was preserved with concomitant administration, while a modest decrease in the immunogenicity of NVX-CoV2373 was found. There was an adequate number of participants aged 18 to 64 to confirm an efficacy trend of 87.5% against COVID-19. The majority of local and systemic reactogenicity events were absent or mild, with small increases in pain and tenderness at the injection site and muscle aches being elevated amongst participants who were co-vaccinated with NVX-CoV2373 and influenza vaccine. Rates of severe adverse events ("SAEs") were low in all groups, and there were no additional early safety concerns associated with co-administration. Rates of unsolicited adverse events (“AEs”), medically attended AEs, and SAEs were low and balanced between the groups. Data from this study have been submitted for peer review and are available ahead of publication via the preprint server on medRxiv. The co-administration sub-study represents the first study of a SARS-CoV-2 vaccine candidate and an approved influenza vaccine, and was led by researchers at St George’s, University of London and St George’s Hospital, London.
Phase 2b South Africa
In May 2021, results from the initial primary analysis of our Phase 2b South Africa trial were published in the New England Journal of Medicine. The publication of the initial primary analysis, which was previously announced in January 2021, highlights NVX-CoV2373’s cross-protection against the Beta (B.1.351) variant strain prevalent in South Africa during the study.
The Phase 2b South Africa trial was a randomized, observer-blinded, placebo-controlled study that enrolled 4,404 participants. Half of the trial participants received two intramuscular injections of NVX-CoV2373 comprising 5 micrograms of antigen with 50 micrograms of Matrix-M™ adjuvant, administered 21 days apart, while the other half of the trial participants received placebo.
In the complete analysis announced in March 2021, NVX-CoV2373 demonstrated 55.4% efficacy for the prevention of mild, moderate and severe COVID-19 disease in the 95% of the trial population that was HIV-negative. Overall efficacy, including both HIV-positive and HIV-negative participants, was 48.6% predominantly against the Beta (B.1.351) variant strain, with the complete analysis showing that NVX-CoV2373 achieved its primary efficacy endpoint in the overall trial population. During the efficacy analysis, the Beta (B.1.351) variant strain circulating in South Africa accounted for approximately 93% of sequenced cases in our Phase 2b trial. There were no cases of severe disease in the NVX-CoV2373 group, and all hospitalization and death occurred in the placebo group. This trial also showed that the vaccine is well-tolerated, with low levels of SAEs through day 35, balanced between vaccine and placebo groups.
While the interim analysis announced in January 2021 reported that prior infection with the original COVID-19 strain may not completely protect against subsequent infection by the variant predominantly circulating in South Africa, the complete analysis indicated that there may be a late protective effect of prior exposure with the original COVID-19 strain. In placebo recipients, at 90 days the illness rate was 8.0% in baseline seronegative participants, with a rate of 5.9% in baseline seropositive participants. CEPI funded the manufacturing of doses of NVX-CoV2373 for this Phase 2b clinical trial, which was also supported in part by a $15.0 million grant from BMGF.
NVX-CoV2373 Booster and Crossover Studies
Novavax-Led Crossover Studies
In April 2021, we initiated crossover arms in our Phase 3 UK, Phase 2b South Africa and PREVENT-19 Phase 3 trials. In June 2021, we completed crossover arms in our Phase 3 UK trial. As of August 2021, our Phase 2b South Africa and PREVENT-19 crossover arms are ongoing.
In our Phase 3 UK and PREVENT-19 trials, participants were offered the opportunity to receive an additional round of injections. Participants who elected to do so received an additional two-dose regimen of either vaccine for those who originally received placebo or placebo for those who originally received vaccine. In our Phase 2b South Africa trial, participants will receive either active vaccine for those who initially received placebo or a booster dose of active vaccine for those who initially received active vaccine. Throughout our crossover arms, participants in all of our trials will remain blinded to their courses of treatment to preserve the ability to assess clinical data in an unbiased manner.
Novavax-Led Booster Study
In August 2021, we announced data from our six-month booster study in the Phase 2 portion of our U.S. and Australia Phase 1/2 trial, which we initiated in March 2021. In this booster study, select participants in the 5 microgram dose cohort from the Phase 2 portion of the Phase 1/2 trial received a third 5 microgram dose (a booster dose) at six months to examine the functional immune response of our vaccine candidate. Results from this study showed that twenty-eight days following boosting, anti-spike IgG increased approximately 4.6-fold compared to the peak response seen after the second dose (Day 217 GMEU = 200,408 (95% CI: 159,796; 251,342)). This boosted value represents a 3.7 to 4.4-fold increase in anti-spike IgG values that were associated with protection in our PREVENT-19 and Phase 3 UK trials. Similarly, wild-type neutralization responses increased approximately 4.3-fold compared to the peak response seen after Dose 2 (IC50 neutralization titers = 6,231 (95% CI: 4,738; 8,195)). This boosted value represents a 4.6 to 5.5-fold increase over the neutralization response associated with protection in our PREVENT-19 and Phase 3 UK trials. The data showed that functional hACE-2 inhibitory antibodies were induced with primary vaccination against Alpha (B.1.1.7), Beta (B.1.351) and Delta and that these levels increased 6- to 10-fold following a single 6-month boost with prototype vaccine. Additionally, functional ACE-2 binding inhibition antibodies cross-reactive with the Delta (B.1.617.2) variant strain were more than 6-fold higher than the primary vaccination series. The administration of booster doses was generally well-tolerated. Local and systemic reactogenicity increased between dose one, dose two, and dose three, with 90% of symptoms rated as mild or moderate after the third dose.
Mix-and-Match COVID-19 Vaccine Booster Trial ("Cov-Boost")
In July 2021, enrollment was completed in Cov-Boost, a mix-and-match (vaccine interchangeability) COVID-19 vaccine booster trial, for which we announced our participation in May 2021. The trial, which was initiated in June 2021, enrolled 2,886 participants aged 30 years and older. NVX-CoV2373 is one of seven COVID-19 vaccines that is being studied to evaluate the potential for providing a booster dose from different manufacturers to individuals who have previously received two doses of an authorized vaccine. Of the 2,886 participants enrolled, 230 participants received a full dose of NVX-CoV2373, while 216 participants received a half volume dose of NVX-CoV2373. The trial assesses the safety and immune response against COVID-19 provided by the various vaccine schedules. The trial is led by the University Hospital Southampton NHS
Foundation Trust and other UK National Institute for Health Research sites. Cov-Boost is receiving support from the UK government Vaccines Taskforce and Department of Health and Social Care.
Comparing COVID-19 Vaccine Schedule Combinations - Stage 2 ("Com-COV2")
In May 2021, enrollment was completed in Com-COV2, a newly expanded investigator-initiated Phase 2 clinical trial, for which we announced our participation in April 2021. The trial enrolled 1,072 adults aged 50 years and older who received their first vaccination during the previous 8-12 weeks. NVX-CoV2373 is one of four COVID-19 vaccines that is being studied to evaluate the potential for combined regimens that mix vaccines from different manufacturers to achieve immune protection against COVID-19. Participants received one of four different vaccines as a second dose, 359 of whom were administered NVX-CoV2373. The trial compares the immune system responses from those who receive a heterologous regimen to those who receive a homologous regimen. Participants in this non-inferiority study are followed for reactogenicity (safety) and immune responses. The trial is conducted by the University of Oxford and supported by the UK government Vaccines Taskforce. The MHRA and Joint Committee on Vaccination and Immunisation formally assesses the safety and efficacy of any new vaccination regiment before it is made available to the public.
NVX-CoV2373 Clinical Development Conducted by Partners
Phase 2/3 India
In March 2021, SIIPL initiated a Phase 2/3 clinical trial of NVX-CoV2373 in India. The initial cohort was fully enrolled in April 2021 and the total study includes approximately 1,600 participants aged 18 to 65 years.
Phase 1/2 Japan
In March 2021, Takeda Pharmaceutical Company Limited (“Takeda”) completed enrollment of a Phase 1/2 clinical trial of NVX-CoV2373 in Japan. This placebo-controlled trial will evaluate the immunogenicity and safety of NVX-CoV2373 in 200 participants aged 20 years and older.
Variant Strain (Monovalent and/or Bivalent) Vaccine Development
Our nanoparticle vaccine technology is purpose-built to rapidly address evolving infectious disease threats. In January 2021, we initiated development of new constructs against the emerging strains of COVID-19, and in February 2021, we selected variant strain vaccines for preclinical evaluation. We are currently evaluating multiple variant strain vaccine candidates, including a recombinant spike protein antigen based on the Beta (B.1.351) virus lineage (“rS-B.1.351”).
COVID-19 Beta (B.1.351) Variant Strain Vaccine ("rS-B.1.351")
In June 2021, we announced data from three complementary non-clinical studies of our rS-B.1.351 variant strain vaccine. The studies compared rS-B.1.351 to NVX-CoV2373 as standalone, in combination, and as a heterologous prime boost vaccine. The data showed that rS-B.1.351 demonstrated strong immunogenicity and protection against the Alpha (B.1.1.7) variant strain, Beta (B.1.351) variant strain and the original COVID-19 virus in animal studies. The findings also showed a broad array of cellular and humoral responses in animal models against all virus strains evaluated. Data from these studies have been submitted for peer review and are available ahead of publication via the preprint server on bioRxiv. We expect to initiate clinical evaluation of rS-B.1.351 in the fall of 2021.
In one study, thirty randomly selected human serum samples from Phase 2 clinical trial participants after their second dose of NVX-CoV2373 were assayed and analyzed for their ability to neutralize Alpha (B.1.1.7) and Beta (B.1.351) variant strains. The trial participants’ sera demonstrated a neutralizing capacity of the Alpha (B.1.1.7) variant strain equal to NVX-CoV2373, with a modest reduction in neutralizing capacity again the Beta (B.1.351) variant strain. These findings support the initial development and evaluation of a B.1.351-directed vaccine as a booster or in a combination vaccine approach.
In an additional preclinical study, mice were immunized with NVX-CoV2373 or rS-B.1.351 alone, in combination, or as a heterologous prime boost. Preclinical data from this study demonstrated that rS-B.1.351 was highly immunogenic and produced neutralizing antibodies.
In another preclinical study, non-human primates (“NHPs”) that were originally immunized with NVX-CoV2373 were boosted approximately one year later with one or two doses of rS-B.1.351. Preclinical data from this study showed that NHPs
boosted with rS-B.1.351 induced strong neutralizing immune responses to the original COVID-19 strain, as well as Alpha (B.1.1.7) and Beta (B.1.351) variant strains. Data from this preclinical study builds upon initial data announced in May 2021.
NVX-CoV2373 Regulatory and Licensure
As of August 2021, we continue to work to complete various Chemistry, Manufacturing and Controls ("CMC") requirements, which ensure that our manufacturing processes are in accordance with regulatory standards (see further discussion of our manufacturing activities below under the heading "NVX-CoV2373 Manufacturing and Supply"). Below is a summary and status of our regulatory processes through the date of filing this Form 10-Q.
In August 2021, we filed regulatory submissions in partnership with SIIPL for emergency use authorization ("EUA") in multiple markets. Regulatory submissions were filed with the Drugs Controller General of India ("DCGI"), as well as regulatory agencies in Indonesia and the Philippines. In addition to these filings, we expect to file a submission to the WHO for emergency use listing ("EUL") in August of 2021.
In April 2021, SK bioscience Ltd. ("SK bioscience") initiated the regulatory submission process in collaboration with Novavax to the MFDS for authorization of NVX-CoV2373.
We expect to file for authorization with the MHRA in the third quarter of 2021. We expect to complete additional filings for authorization within weeks of filing with the MHRA, including with EMA, Australian Therapeutic Goods Administration, Health Canada, and New Zealand Medsafe.
We are actively engaged in communications with the FDA through submissions to our open investigational new drug application (“IND”) and discussions on various aspects of the program required to support the regulatory approval process. We plan to file submissions for EUA with the FDA in the fourth quarter of 2021.
COVID-19 Vaccine Funding
We have secured critical funding throughout 2020 and into 2021 to support the development of NVX-CoV2373. Through the date of filing this Form 10-Q, funding for NVX-CoV2373 encompasses over $2 billion from sources including the BMGF, CEPI, the DoD, and OWS.
In April 2021, our Base Agreement and a Project Agreement (together, as amended and supplemented, the “OWS Agreement”) entered into with Advanced Technology International, Inc., the Consortium Management Firm acting on behalf of the Medical CBRN Defense Consortium in connection with OWS, was amended to fully fund the agreement up to $1.75 billion to support certain activities related to the development of NVX-CoV2373. This includes the manufacture and delivery of 100 million doses of NVX-CoV2373 to the U.S. government. We expect this funding will assist in rapidly developing our large-scale manufacturing capacity and transitioning into ongoing production, including the capability to stockpile and distribute large quantities of NVX-CoV2373 for use in clinical trials and potentially for commercial sale, if authorized for emergency use or licensed. The OWS Agreement is funding the late-stage clinical studies necessary to determine the safety and efficacy of NVX-CoV2373, including PREVENT-19. Funding under the OWS Agreement is expected to support our plans to file submissions for EUA and licensure with the FDA. Accepted analytical methods that we can use to demonstrate our vaccine’s purity, potency and consistent lot manufacturing are critical to attaining licensure in all the territories we intend to sell our vaccine. In the U.S., these analytical methods will be reviewed and approved by the FDA. The U.S. government has recently instructed us to prioritize alignment with the FDA on our analytic methods before conducting additional U.S. manufacturing and further indicated that the U.S. government will not fund additional U.S. manufacturing until such agreement has been made. The U.S. government also instructed us to proceed with work under the OWS Agreement related to all other activities including ongoing clinical trials and nonclinical studies, regulatory interactions, analytics/assays and characterization of manufactured vaccine and project management.
A summary and status of our historical COVID-19 funding developments follows:
NVX-CoV2373 Manufacturing and Supply
We have established a global manufacturing and supply chain to support the commercialization of NVX-CoV2373. With significant progress made throughout 2020 and through the second quarter of 2021, our global supply chain spans over ten countries and includes Novavax-owned facilities in the Czech Republic and Sweden, as well as partnerships with contract manufacturing organizations around the world. In the second quarter of 2021, we remained focused on readying our global supply chain for commercialization in order to ensure we promptly deliver NXV-CoV2373 upon anticipated regulatory authorizations.
We expect our global manufacturing capacity of NVX-CoV2373 to be approximately 100 million doses per month by the end of the third quarter of 2021. We anticipate the remainder of our manufacturing capacity will be ready by the end of the fourth quarter of 2021, which we expect will support total global manufacturing capacity of approximately 150 million doses per month. Of this anticipated capacity, approximately one billion annualized doses will be manufactured by SIIPL.
A summary and status of key manufacturing and supply developments during the second quarter of 2021 follows:
In June 2021, we announced the completion of construction of the National Research Council of Canada’s Biologics Manufacturing Centre and ongoing technology transfer for the production of NVX-CoV2373. Technology transfer to establish a step-by-step process of producing NVX-CoV2373 at the Biologics Manufacturing Centre began in April 2021 following a collaboration agreement entered into with the National Research Council of Canada in March 2021. These developments build upon a Memorandum of Understanding (“MOU”) entered into in February 2021 with the Canadian government to produce
NVX-CoV2373 at the Biologics Manufacturing Centre in Canada. Large-scale GMP production is expected to begin once the facility has received Health Canada approval.
In May 2021, we entered a non-binding MOU with the MOHW and SK bioscience to explore further cooperation in the development and manufacturing of vaccines, including NVX-CoV2373. Under the agreement, we agreed to potentially explore the development of new vaccine products with SK bioscience, including COVID-19 variant vaccines, and/or an influenza/COVID-19 combination vaccine. We will continue to collaborate with SK bioscience in manufacturing of the vaccines utilizing SK bioscience’s facility, with support from the Korean government. The MOU builds upon previous agreements entered with SK bioscience, including a development and supply agreement entered into in August 2020 and a collaboration and license agreement entered into in February 2021, whereby we granted SK bioscience an exclusive license to develop, manufacture and commercialize NVX-CoV2373 in the Republic of Korea.
In May 2021, the MHLW announced it was in contract discussions to potentially order 150 million doses of our COVID-19 vaccine candidate from Takeda. This development follows the exclusive license agreement we entered into with Takeda in February 2021 for the development, manufacturing and commercialization of NVX-CoV2373 in Japan. We anticipate Takeda will have a manufacturing capacity of 250 million doses per year. Takeda is in process of establishing vaccine procurement based on technology transfer by Novavax of NVX-CoV2373. Distribution of NVX-CoV2373 in Japan by Takeda is expected to begin in late 2021 or early 2022. Takeda is responsible for regulatory submission to Japan’s Pharmaceutical and Medical Devices Agency (“PMDA”).
NVX-CoV2373 Supply Agreements
We expect our global supply chain will enable us to deliver upon our supply commitments around the world. We have entered into advance purchase agreements (referred to as "APAs" or "supply agreements" throughout this Form 10-Q), as well as multiple supply and license agreements with strategic partners. The APAs typically contain terms that include upfront payments intended to assist us in funding investments related to building out and operating our manufacturing and distribution network, among other expenses, in support of our global supply commitment. Such upfront payments generally become non-refundable upon our achievement of certain development and commercial milestones.
A summary and status of key supply agreements executed since the beginning of the second quarter of 2021 and through the date of filing this Form 10-Q follows:
In May 2021, we finalized an APA with Gavi, the Vaccine Alliance ("Gavi"), building upon our MOU previously announced in February 2021. Under the terms of the agreement, 1.1 billion doses of NVX-CoV2373 are to be made available to countries participating in the COVAX Facility, which was established to allocate and distribute vaccines equitably to participating countries and economies. We expect to manufacture and distribute 350 million doses of NVX-CoV2373 to countries participating under the COVAX Facility. Under a separate purchase agreement with Gavi, SIIPL is expected to manufacture and deliver the balance of the 1.1 billion doses of NVX-CoV2373 for low- and middle-income countries participating in the COVAX Facility. We expect to deliver doses with antigen and adjuvant manufactured at facilities directly funded by the investments previously received from CEPI (the "CEPI Funding Agreement"). We expect to supply significant doses that Gavi would allocate to low-, middle- and high-income countries, subject to certain limitations, utilizing a tiered pricing schedule and Gavi may prioritize such doses to low- and middle-income countries at lower prices. Additionally, we may provide additional doses of NVX-CoV2373, to the extent available from CEPI-funded manufacturing facilities, in the event that SIIPL cannot materially deliver expected vaccine doses to the COVAX Facility. Together with SIIPL, we expect to initiate delivery of doses following receipt of appropriate regulatory authorizations. Under the agreement, we received an upfront payment from Gavi of $350 million during the second quarter of 2021 and expect to receive an additional payment of $350 million if we secure EUL for NVX-CoV2373 by the WHO.
We expect to sign additional APAs or supply agreements that are currently in active discussions and negotiations. For example, in August 2021, we announced that we finalized the terms of an APA with the European Commission, which the parties expect to execute during the third quarter of 2021, for the purchase of up to 100 million initial doses of NVX-CoV2373, with the option of the European Commission to purchase an additional 100 million doses through 2023.
In addition to our supply agreements, we have committed 110 million doses of NVX-CoV2373 to the U.S. government under the OWS Agreement and our contract with the DoD (the "DoD Contract").
Seasonal Influenza
NanoFlu™ Vaccine Program (Older Adults)
To date, we have advanced NanoFlu™ through a Phase 3 clinical trial in which NanoFlu™ achieved all primary endpoints and achieved statistical significance in key secondary endpoints. In 2020, we took steps to ensure the continued advancement of NanoFlu™ in parallel with our COVID-19 activities and formed a leadership team solely dedicated to our NanoFlu™ vaccine program. This NanoFlu™ vaccine development unit benefits from joint shared services with key cross-functional departments and builds on the Company’s established knowledge base in the discovery and development of innovative vaccines. Our NanoFlu™ vaccine team remains focused on advancing our combination NanoFlu™ / NVX-CoV2373 vaccine candidate, qNIV/CoV2373, to clinical development.
Combination Vaccines
With the ongoing development of NanoFlu™, NVX-CoV2373 and our RSV F Vaccine, a strong rationale exists for developing combination respiratory vaccines designed to protect susceptible populations against these diseases. Although testing is at an early stage, we believe that the combination of influenza with COVID-19, influenza with RSV, and influenza with both RSV and COVID-19 may be achievable, as these vaccines all use our recombinant nanoparticle technology and include our proprietary Matrix-M™ adjuvant.
NanoFlu™/NVX-CoV2373 Combination Vaccine ("qNIV/CoV2373")
In May 2021, we announced data from a preclinical study of qNIV/CoV2373 to assess its immunogenicity and protective efficacy in animal models. Preclinical data from this study showed that qNIV/CoV2373 induced functional influenza and COVID-19 antibody responses, with hemagglutination inhibition and ACE2 receptor-inhibiting titers that were comparable between immunization with the combination vaccine and with its respective component vaccines. qNIV/CoV2373 also induced elevated levels of SARS-CoV-2 anti-S IgG two weeks after the first immunization, which increased significantly after the second dose, with levels comparable to animals that received NVX-CoV2373 alone. Human ACE2 receptor inhibiting antibody levels responded similarly. qNIV/CoV2373 also induced antibodies against SARS-CoV-2 neutralizing epitopes that are common between the original COVID-19 strain and the Beta (B.1.351) variant strain. When challenged with SARS-CoV-2, examination of viral load in the upper and lower respiratory tract showed little or no virus was detected four days after infection in animals immunized with either qNIV/CoV2373 or with NVX-CoV2373 alone. Data from this study are available ahead of publication via the preprint server for biology on bioRxiv. We expect to initiate clinical evaluation of qNIV/CoV2373 later in 2021.
Malaria
R21 - Malaria Vaccine
R21 is a malaria vaccine candidate created by the Jenner Institute, University of Oxford, and formulated with our Matrix-M™ adjuvant. The University of Oxford has partnered with SIIPL for commercial development of R21 and has granted them a license for the vaccine. We expect to manufacture and supply the Matrix-M™ adjuvant component of R21 to SIIPL. SIIPL has committed to manufacture at least 200 million doses per year of R21 after licensure. Additionally, SIIPL has rights to use Matrix-M™ adjuvant in the vaccine in regions where the disease is endemic and will pay royalties to us on its market sales of the vaccine. We will have commercial rights to sell and distribute the SIIPL-manufactured vaccine in certain countries, primarily in the travelers’ and military vaccine markets.
R21 Clinical Development
In May 2021, the first doses were administered in a Phase 3 licensure trial of R21 to assess the efficacy and safety of the malaria vaccine candidate. This double-blinded, randomized, controlled Phase 3 trial includes 4,800 participants aged five to 36 months across five sites in Burkina Faso, Kenya, Mali and Tanzania. Participants will receive three vaccinations four weeks apart and a booster vaccination one year later.
In April 2021, we announced the pre-print publication of data in Preprints with The Lancet from a Phase 2b clinical trial evaluating R21. The Phase 2b randomized, controlled, double-blind trial was conducted at the Clinical Research Unit of Nanoro / Institut de Recherche en Sciences de la Santé in Africa and recruited 450 participants aged five to 17 months from the catchment area of Nanoro, Burkina Faso. In three study arms, participants received 5 micrograms of R21 with either 25 micrograms of Matrix-M™ adjuvant, 50 micrograms of Matrix-M™ adjuvant, or a rabies vaccine as a control. R21 demonstrated
77% efficacy in the higher adjuvant dose group and 71% efficacy in the lower adjuvant dose group. Both adjuvant dose levels were well tolerated in young children, with no severe reactions to the vaccine reported.
R0.6C - Malaria Transmission Blocking Vaccine
R0.6C is a novel malaria transmission blocking vaccine candidate manufactured by Statens Serum Institut that has been formulated in a preclinical study with our Matrix-M™ adjuvant. R0.6C is a vaccine candidate based on the protective epitopes of the Pfs48/45 protein. Development of R0.6C is supported by grants from the European Union’s Horizon 2020 research and innovation program and funded by PATH’s Malaria Vaccine Initiative.
R0.6C Clinical Development
In June 2021, a Phase 1 clinical trial for R0.6C was initiated at Radboud University Medical Center in the Netherlands. The trial enrolled healthy, adult volunteers and will assess the safety and immunogenicity of R0.6C. The trial will also evaluate our Matrix-M™ adjuvant and the common aluminum hydroxide adjuvant. This Phase 1 trial follows a preclinical study of R0.6C formulated with our Matrix-M™ adjuvant, which showed greater than 80% reduction of transmission of the parasite that causes malaria.
Sales of Common Stock
During the six months ended June 30, 2021 and 2020, we sold 2.6 million and 28.5 million, respectively, of shares of our common stock resulting in net proceeds of approximately $565 million and $392 million, respectively, under our various At Market Issuance Sales agreements.
In June 2021, we entered into an At Market Issuance Sales Agreement (the "June 2021 Sales Agreement"), which allows us to issue and sell up to $500 million in gross proceeds of shares of our common stock, and terminated our existing At Market Issuance Sales agreement. As of June 30, 2021, no shares had been sold under the June 2021 Sales Agreement.
Critical Accounting Policies and Use of Estimates
There are no material changes to our critical accounting policies as described in Item 7 of our Annual Report on Form 10-K for the fiscal year ended December 31, 2020, as filed with the SEC.
Recent Accounting Pronouncements Not Yet Adopted
See “Note 2―Summary of Significant Accounting Policies” included in our Notes to Consolidated Financial Statements (under the caption “Recent Accounting Pronouncements”).
Results of Operations
The following is a discussion of the historical financial condition and results of the Company’s operations that should be read in conjunction with the unaudited consolidated financial statements and notes set forth in this Quarterly Report.
Three Months Ended June 30, 2021 and 2020
Revenue:
| | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Revenue (in thousands): | | | | | |
| Government contracts | $ | 240,534 | | | $ | — | | | $ | 240,534 | |
| Grants and other | 34,026 | | | 35,538 | | | (1,512) | |
| Royalties | 23,457 | | | — | | | 23,457 | |
| Total revenue | $ | 298,017 | | | $ | 35,538 | | | $ | 262,479 | |
Revenue for the three months ended June 30, 2021 was $298.0 million as compared to $35.5 million for the same period in 2020, an increase of $262.5 million. Revenue for the three months ended June 30, 2021 was primarily comprised of
revenue for services performed under the OWS Agreement and the CEPI Funding Agreement. Revenue for the three months ended June 30, 2020 was primarily comprised of revenue for services performed under the CEPI Funding Agreement. During the three month ended June 30, 2021, we recognized royalties of $23.5 million under our licensing arrangements. The significant increase in revenue was due to increased development activities relating to NVX-CoV2373 under the OWS Agreement and the CEPI Funding Agreement.
We expect revenue in 2021 to significantly increase as compared with 2020 due to our NVX-CoV2373 program, which we anticipate will continue to be funded by OWS and CEPI and/or other revenue sources. Further, we anticipate bringing our NVXCoV2373 vaccine candidate to market following global regulatory approvals which, if achieved, should significantly increase revenue. In anticipation, we have entered into various APA, as well as multiple supply and license agreements with strategic partners to supply NVX-CoV2373 in their specified territories under which we are entitled to receive royalties from the sale of NVX-CoV2373 by such partners.
Expenses:
| | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Expenses (in thousands): | | | | | |
| Research and development | $ | 570,685 | | | $ | 34,846 | | | $ | 535,839 | |
| General and administrative | 73,161 | | | 17,719 | | | 55,442 | |
| Total expenses | $ | 643,846 | | | $ | 52,565 | | | $ | 591,281 | |
Research and Development Expenses
In the three months ended June 30, 2021, our research and development activities were primarily focused on the development of NVX-CoV2373 and included direct external research and development expenses related to NVX-CoV2373 of $497.2 million, primarily comprised of costs related to the following:
•expenses incurred under agreements with contract research organization ("CROs") that conduct our clinical trials and third-party consultants related to the development of NVX-CoV2373;
•expenses incurred on developing and manufacturing the antigen drug substance and Matrix-M™ components of NVX-CoV2373 under agreements that we established with third-party contract manufacturing organizations ("CMOs") and contract manufacturing and development organizations ("CDMOs");
•expenses incurred for the procurement of raw materials, laboratory supplies and equipment; and
•other costs related to preclinical studies and regulatory consulting, as well as related program management activities to support our growing global operations.
Research and development expenses increased to $570.7 million for three months ended June 30, 2021 as compared to $34.8 million for three months ended June 30, 2020, an increase of $535.8 million primarily due to research and development of NVX-CoV2373, as summarized in the table below (in millions):
| | | | | | | | | | | |
| Three Months Ended June 30, |
| 2021 | | 2020 |
| NVX-CoV2373 | $ | 497,196 | | | $ | 17,293 | |
NanoFlu™ | 3,168 | | | 4,611 | |
| Other vaccine development programs | 210 | | | 738 | |
| Total direct external research and development expense | 500,574 | | | 22,642 | |
| Employee expenses | 24,556 | | | 4,794 | |
| Stock-based compensation expense | 24,779 | | | 4,098 | |
| Facility expenses | 3,410 | | | 902 | |
| Other expenses | 17,366 | | | 2,410 | |
| Total research and development expenses | $ | 570,685 | | | $ | 34,846 | |
For 2021, we expect research and development expenses to significantly increase over 2020 expenses due to our continued development activities for our NVX-CoV2373 program and increases in employee-related costs. Following a potential regulatory approval of NVX-CoV2373, we expect products sales will result in certain types of costs that have been previously recorded as research and development in our Consolidated Statement of Operations to be capitalized as inventory and expensed as cost of goods sold when product is delivered. Cost of goods sold expenses could be significant in 2021 depending on our commercial shipment levels for those shipments in which the costs were recorded into inventory.
We do not provide forward-looking estimates of costs and time to complete our research programs due to the many uncertainties associated with vaccine development. As we obtain data from preclinical studies and clinical trials, we may elect to discontinue or delay clinical trials in order to focus our resources on more promising vaccine candidates. Completion of clinical trials may take several years or more, but the length of time can vary substantially depending upon the phase, size of clinical trial, primary and secondary endpoints and the intended use of the vaccine candidate. The cost of clinical trials may vary significantly over the life of a project as a result of a variety of factors, including:
•the number of participants who participate in the clinical trials;
•the number of sites included in the clinical trials;
•if clinical trial locations are domestic, international or both;
•the time to enroll participants;
•the duration of treatment and follow-up;
•the safety and efficacy profile of the vaccine candidate; and
•the cost and timing of, and the ability to secure, regulatory approvals.
As a result of these uncertainties, we are unable to determine the duration and completion costs of our research and development projects or when, and to what extent, we will generate future cash flows from our research projects.
General and Administrative Expenses
General and administrative expenses increased to $73.2 million for the three months ended June 30, 2021 from $17.7 million for the same period in 2020, an increase of $55.4 million. The increase in general and administrative expenses is primarily due to increased employee-related costs, primarily stock-based compensation expense, and an increase in professional fees in support of our NVX-CoV2373 program. As of June 30, 2021, we had 195 employees dedicated to general and administrative functions as compared with 61 employees as of June 30, 2020. For 2021, we expect general and administrative expenses to significantly increase due to increased activities related to supporting our NVX-CoV2373 program and increases in employee-related costs and professional fees.
Other Income (Expense):
| | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Other Income (Expense) (in thousands): | | | | | |
| Investment income | $ | 369 | | | $ | 297 | | | $ | 72 | |
| Interest expense | (5,968) | | | (3,403) | | | (2,565) | |
| Other income | 2,659 | | | 2,612 | | | 47 | |
| Total other income (expense), net | $ | (2,940) | | | $ | (494) | | | $ | (2,446) | |
We had total other expense, net, of $2.9 million for the three months ended June 30, 2021 as compared to $0.5 million for the same period in 2020. In the three months ended June 30, 2021, we also recorded interest expense of $1.8 million for finance leases.
Income Tax Expense:
During the three months ended June 30, 2021, we recognized $3.5 million of income tax expense related to foreign withholding tax on royalties. We did not recognize any income tax expense for the three months ended June 30, 2020.
Net Loss:
| | | | | | | | | | | | | | | | | |
| Three Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Net Loss (in thousands, except per share information): | | | | | |
| Net loss | $ | (352,317) | | | $ | (17,521) | | | $ | (334,796) | |
| Net loss per share | $ | (4.75) | | | $ | (0.30) | | | $ | (4.45) | |
| Weighted average shares outstanding | 74,118 | | | 58,618 | | | 15,500 | |
Net loss for the three months ended June 30, 2021 was $352.3 million, or $4.75 per share, as compared to $17.5 million, or $0.30 per share, for the same period in 2020. The increase in net loss was primarily due to a significant increase in development activities relating to NVX-CoV2373 and increase in employee-related costs, primarily stock-based compensation expense, partially offset by increased revenue under the OWS Agreement and CEPI Funding Agreement.
The increase in weighted average shares outstanding for the three months ended June 30, 2021 is primarily a result of sales of our common stock in 2021 and 2020.
Six Months Ended June 30, 2021 and 2020
Revenue:
| | | | | | | | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Revenue (in thousands): | | | | | |
| Government contracts | $ | 623,238 | | | $ | — | | | $ | 623,238 | |
| Grants and other | 98,551 | | | 38,915 | | | 59,636 | |
| Royalties | 23,457 | | | — | | | 23,457 | |
| Total revenue | $ | 745,246 | | | $ | 38,915 | | | $ | 706,331 | |
Revenue for the six months ended June 30, 2021 was $745.2 million as compared to $38.9 million for the same period in 2020, an increase of $706.3 million. Revenue for the six months ended June 30, 2021 was primarily comprised of revenue for services performed under the OWS Agreement and CEPI Funding Agreement. Revenue for the six months ended June 30, 2020 was primarily comprised of revenue for services performed under the CEPI Funding Agreement. The significant increase in revenue was due to increased development activities relating to NVX-CoV2373 under the OWS Agreement and CEPI Funding Agreement.
Expenses:
| | | | | | | | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Expenses (in thousands): | | | | | |
| Research and development | $ | 1,163,356 | | | $ | 51,741 | | | $ | 1,111,615 | |
| General and administrative | 136,351 | | | 27,098 | | | 109,253 | |
| Total expenses | $ | 1,299,707 | | | $ | 78,839 | | | $ | 1,220,868 | |
Research and Development Expenses
In the six months ended June 30, 2021, our research and development activities were primarily focused on the development of NVX-CoV2373 and included direct external research and development expenses related to NVX-CoV2373 of $1.0 billion, primarily comprised of costs related to the following:
•expenses incurred under agreements with CROs that conduct our clinical trials and third-party consultants related to the development of NVX-CoV2373;
•expenses incurred on developing and manufacturing the antigen drug substance and Matrix-M™ components of NVX-CoV2373 under agreements that we established with third-party CMOs and CDMOs;
•expenses incurred for the procurement of raw materials, laboratory supplies and equipment; and
•other costs related to preclinical studies and regulatory consulting, as well as related program management activities to support our growing global operations.
Research and development expenses increased to $1.2 billion for the six months ended June 30, 2021 from $51.7 million for the same period in 2020, an increase of $1.1 billion, primarily due to increased development activities relating to NVX-CoV2373, as summarized in the table below (in millions):
| | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 |
| NVX-CoV2373 | $ | 1,035,320 | | | $ | 18,851 | |
NanoFlu™ | 4,294 | | | 8,456 | |
| Other vaccine development programs | 514 | | | 1,902 | |
| Total direct external research and development expense | 1,040,128 | | | 29,209 | |
| Employee expenses | 49,511 | | | 9,363 | |
| Stock-based compensation expense | 48,569 | | | 6,005 | |
| Facility expenses | 6,405 | | | 2,090 | |
| Other expenses | 18,743 | | | 5,074 | |
| Total research and development expenses | $ | 1,163,356 | | | $ | 51,741 | |
General and Administrative Expenses
General and administrative expenses increased to $136.4 million for the six months ended June 30, 2021 from $27.1 million for the same period in 2020, an increase of $109.3 million. The increase in general and administrative expenses is primarily due to increased employee-related costs, primarily stock-based compensation expense and an increase in professional fees in support of our NVX-CoV2373 program. As of June 30, 2021, we had 195 employees dedicated to general and administrative functions versus 61 employees as of June 30, 2020.
Other Income (Expense): | | | | | | | | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Other Income (Expense) (in thousands): | | | | | |
| Investment income | $ | 731 | | | $ | 732 | | | $ | (1) | |
| Interest expense | (10,807) | | | (6,806) | | | (4,001) | |
| Other income (expense) | (3,934) | | | 2,613 | | | (6,547) | |
| Total other income (expense), net | $ | (14,010) | | | $ | (3,461) | | | $ | (10,549) | |
We had total other expense, net of $14.0 million for the six months ended June 30, 2021 as compared to $3.5 million for the same period in 2020. In the six months ended June 30, 2021, we also recorded an interest expense of $4.0 million for finance leases. In the six months ended June 30, 2021 and 2020, other income included a loss of $3.1 million and a gain of $2.7 million, respectively, on the intercompany loan with Novavax CZ due to changes in the exchange rates.
Income Tax Expense:
During the six months ended June 30, 2021, we recognized $6.6 million of income tax expense related to foreign withholding tax on royalties. We did not recognize any income tax expense for the six months ended June 30, 2020.
Net Loss:
| | | | | | | | | | | | | | | | | |
| Six Months Ended June 30, |
| 2021 | | 2020 | | Change |
| Net Loss (in thousands, except per share information): | | | | | |
| Net loss | $ | (575,036) | | | $ | (43,385) | | | $ | (531,651) | |
| Net loss per share | $ | (7.82) | | | $ | (0.84) | | | $ | (6.98) | |
| Weighted average shares outstanding | 73,580 | | | 51,401 | | | 22,179 | |
Net loss for the six months ended June 30, 2021 was $575.0 million, or $7.82 per share, as compared to $43.4 million, or $0.84 per share, for the same period in 2020. The increase in net loss was primarily due to increased development activities relating to NVX-CoV2373, increased employee-related costs, primarily stock-based compensation expense, partially offset by increased revenue under the OWS Agreement and CEPI Funding Agreement.
The increase in weighted average shares outstanding for the six months ended June 30, 2021 is primarily a result of sales of our common stock in 2021 and 2020.
Liquidity Matters and Capital Resources
Our future capital requirements depend on numerous factors including, but not limited to, our projected activities related to the development of NVX-CoV2373, including significant commitments under various CRO, CMO and CDMO agreements, the progress of preclinical studies and clinical trials, the time and costs involved in obtaining regulatory approvals, the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights and other manufacturing, sales and distribution costs. We plan to continue developing other vaccines and product candidates, such as NanoFlu™ vaccine and potential combination vaccines candidates, which are in various stages of development. We believe our operating expenses and capital requirements will fluctuate depending upon the timing of events, such as the progress of our NVX-CoV2373 clinical trials and approval for the use of NVX-CoV2373 in the U.S. and internationally, as well as the scope, initiation and progress of our preclinical studies and clinical trials related to other research and development activities.
We have entered into APAs or supply agreements with various countries globally that, if our product candidate is approved, are expected to result in the delivery of approximately 550 million doses, including Gavi, of NVX-CoV2373 throughout 2021 and into the first half of 2022. The APAs or supply agreements typically contain terms that include upfront payments intended to assist us in funding investments related to building out and operating our manufacturing and distribution network, among other expenses, in support of our global supply commitment. Such upfront payments generally become non-refundable upon our achievement of certain development and commercial milestones. We expect to sign additional APAs or supply agreements that are currently in active discussions and negotiations.
In May 2021, we finalized an APA with Gavi, building upon our MOU previously announced in February 2021. Under the terms of the agreement, 1.1 billion doses of NVX-CoV2373 are to be made available to countries participating in the COVAX Facility, which was established to allocate and distribute vaccines equitably to participating countries and economies. We expect to manufacture and distribute 350 million doses of NVX-CoV2373 to countries participating under the COVAX Facility. Under a separate purchase agreement with Gavi, SIIPL is expected to manufacture and deliver the balance of the 1.1 billion doses of NVX-CoV2373 for low- and middle-income countries participating in the COVAX Facility. We expect to deliver doses with antigen and adjuvant manufactured at facilities directly funded by the investments previously received from CEPI. We expect to supply significant doses that Gavi would allocate to low-, middle- and high-income countries, subject to certain limitations, utilizing a tiered pricing schedule and Gavi may prioritize such doses to low- and middle- income countries, at lower prices. Additionally, we may provide additional doses, to the extent available from CEPI-funded manufacturing facilities, in the event that SIIPL cannot materially deliver expected vaccine doses to the COVAX Facility. Together with SIIPL, we expect to initiate delivery of doses following receipt of appropriate regulatory authorizations. Under the agreement, we received an upfront payment of $350 million from Gavi during the second quarter of 2021 and expect to receive an additional payment of $350 million if we secure EUL for NVX-CoV2373 by the WHO.
We have also entered into supply and license agreements with strategic partners to supply NVX-CoV2373 in their specified territories under which we are entitled to receive royalties primarily from the sale of NVX-CoV2373 by our partners. During the three and six months ended June 30, 2021, we received royalties of $23.5 million under these licensing arrangements.
In the six months ended June 30, 2021, we funded our operations with cash and marketable securities on hand, upfront payments under APAs, proceeds from the sale of common stock together with revenue under the OWS Agreement and CEPI Funding Agreement that support our NVX-CoV2373 vaccine development activities. We anticipate our future operations to be funded by our cash, cash equivalents and marketable securities, upfront payments under our APAs, revenue under our OWS Agreement and CEPI Funding Agreement, and following any potential global regulatory approvals, revenue from product sales, royalties under licensing arrangements with our strategic partners and/or other potential funding sources.
As of June 30, 2021, we had $2.1 billion in cash and cash equivalents, marketable securities and restricted cash as compared to $806.4 million as of December 31, 2020. These amounts consisted of $2.1 billion in cash and cash equivalents and $47.9 million in restricted cash as of June 30, 2021 as compared to $553.4 million in cash and cash equivalents $157.6 million in marketable securities and $95.3 million in restricted cash as of December 31, 2020.
The following table summarizes cash flows for the six months ended June 30, 2021 and 2020 (in thousands):
| | | | | | | | | | | | | | | | | |
| 2021 | | 2020 | | Change |
| Net cash provided by (used in): | | | | | |
| Operating activities | $ | 807,497 | | | $ | 92,524 | | | $ | 714,973 | |
| Investing activities | 128,708 | | | (245,946) | | | 374,654 | |
| Financing activities | 538,144 | | | 602,540 | | | (64,396) | |
| Effect on exchange rate on cash, cash equivalents and restricted cash | (348) | | | 276 | | | (624) | |
| Net increase in cash, cash equivalents and restricted cash | 1,474,001 | | | 449,394 | | | 1,024,607 | |
| Cash, cash equivalents and restricted cash at beginning of period | 648,738 | | | 82,180 | | | 566,558 | |
| Cash, cash equivalents and restricted cash at end of period | $ | 2,122,739 | | | $ | 531,574 | | | $ | 1,591,165 | |
Net cash provided by operating activities increased to $807.5 million for the six months ended June 30, 2021, as compared to $92.5 million for the same period in 2020. The increase in cash provided is primarily due to payments under APAs recorded as deferred revenue and the timing of payments to third-parties.
During the six months ended June 30, 2021 and 2020, our investing activities consisted primarily of maturities and sale of marketable securities, net of purchases, our acquisition of Novavax CZ in 2020 and, to a much lesser extent, capital expenditures. Capital expenditures for the six months ended June 30, 2021 and 2020 were $28.9 million and $3.9 million, respectively, and the increase in capital expenditures was primarily due to the build out of our facilities and related capital expenditures to support NVX-CoV2373. For 2021, we expect an increase in our capital expenditures due to further development activities for our NVX-CoV2373 program, including the additional build-out of research and development and manufacturing facilities and related equipment, and the build-out of our new corporate office facility to accommodate anticipated increases in headcount.
Our financing activities consisted primarily of sales of our common stock under our At Market Issuance Sales Agreements, payments of finance lease liabilities and exercise of stock-based awards. In the six months ended June 30, 2021 and 2020, we received net proceeds of approximately $565 million and $392 million, respectively, from selling shares of common stock through our At Market Issuance Sales Agreements.
Off-Balance Sheet Arrangements
We did not have any material off-balance sheet arrangements as of June 30, 2021.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
The primary objective of our investment activities is preservation of capital, with the secondary objective of maximizing income. As of June 30, 2021, we had cash and cash equivalents of $2.1 billion, $47.9 million in restricted cash and working capital of $703.5 million.
Our exposure to market risk is primarily confined to our investment portfolio, which historically has been classified as available-for-sale. We do not believe that a change in the market rates of interest would have any significant impact on the realizable value of our investment portfolio. Changes in interest rates may affect the investment income we earn on our marketable securities when they mature and the proceeds are reinvested into new marketable securities and, therefore, could impact our cash flows and results of operations.
Interest and dividend income is recorded when earned and included in investment income. Premiums and discounts, if any, on marketable securities are amortized or accreted to maturity and included in investment income. The specific identification method is used in computing realized gains and losses on the sale of our securities.
We are headquartered in the U.S. where we conduct the vast majority of our business activities. We have two foreign consolidated subsidiaries, Novavax AB, which is located in Sweden, and Novavax CZ, which is located in the Czech Republic. A 10% decline in the exchange rate between the U.S. dollar and Swedish Krona would result in a decline of stockholders’ equity of approximately $8 million as of June 30, 2021. A 10% decline in the exchange rate between the U.S. dollar and Czech Koruna would result in a decline of stockholders’ equity of approximately $5 million as of June 30, 2021.
Our Notes have a fixed interest rate and we have no additional material debt. As such, we do not believe that we are exposed to any material interest rate risk as a result of our borrowing activities.
Item 4. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Our management, with the assistance of our chief executive officer and chief financial officer, has reviewed and evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) as of June 30, 2021. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving such control objectives. Based on the evaluation of our disclosure controls and procedures as of June 30, 2021, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting
Our management, including our chief executive officer and chief financial officer, has evaluated any changes in our internal control over financial reporting that occurred during the quarterly period ended June 30, 2021, and has concluded that there was no change that occurred during the quarterly period ended June 30, 2021 that materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s assessment of and conclusion on the effectiveness of disclosure controls and procedures and internal controls over financial reporting did not include the internal controls related to the operations acquired in the acquisition of Novavax CZ that are included in our June 30, 2021 consolidated financial statements through May 27, 2021. Our audit of internal control over financial reporting also did not include an evaluation of the internal control over financial reporting of Novavax CZ through May 27, 2021.
PART II. OTHER INFORMATION
Item 1. Legal Proceedings
On February 26, 2021, a Novavax stockholder named Thomas Golubinski filed a derivative complaint against certain members of the Novavax board of directors and certain members of senior management in the Delaware Court of Chancery. Novavax is a nominal defendant. The plaintiff challenges two sets of equity awards, made in April 2020 and in June 2020, on the ground that they were “spring-loaded,” that is, made at a time when certain board members or members of senior management allegedly possessed undisclosed positive material information concerning the Company. The complaint asserts claims for breach of fiduciary duty, waste, and unjust enrichment. The plaintiff seeks an award of damages to the Company, an order rescinding the April 2020 and June 2020 awards or requiring disgorgement, and an award of attorneys’ fees incurred in connection with the litigation. On May 10, 2021, the defendants moved to dismiss the complaint in its entirety. On June 17,
2021, the Company’s stockholders voted FOR ratification of the April 2020 awards and ratification of the June 2020 awards. Details of the ratification proposals are set forth in the Company’s Definitive Proxy Statement on Schedule 14A filed with the SEC on May 3, 2021. The results of the vote were disclosed in the Company’s Current Report on Form 8-K filed with the SEC on June 24, 2021. Should the plaintiff elect to move forward with his claims, the defendants intend to move for summary judgment on ratification grounds while continuing to pursue dismissal.
Item 1A. Risk Factors
You should carefully consider the following risk factors in evaluating our business. A number of risks could cause our actual results to differ materially from those that are indicated by forward-looking statements. Some risks relate principally to our business and the industry in which we operate. Others relate principally to the securities market and ownership of our common stock. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties of which we are unaware, or that we currently deem immaterial, also may become important factors that affect us. Any of the following risks could result in material adverse impacts on our business, financial condition or results of operations. You also should consider the other information included in this Quarterly Report on Form 10-Q as well as our other filings with the SEC.
Summary of Risk Factors
Our business is subject to numerous risks. The following is a summary of the principal risk factors described in this section:
•We have a history of losses and our future profitability is uncertain.
•We will continue to require significant funding to maintain our current level of operations and fund the further development of our vaccine candidates.
•Because our vaccine product development efforts depend on new and rapidly evolving technologies, our efforts may not succeed.
•The regulatory and commercial success of our COVID-19 vaccine candidate, NVX-CoV2373, remains uncertain. We may be unable to obtain regulatory approval or produce a successful vaccine in a timely manner, if at all.
•We are a biotechnology company and face significant risk in developing, manufacturing and commercializing our products.
•Because we depend on third-parties to conduct some of our laboratory testing and clinical trials, and a significant amount of our vaccine manufacturing and distribution, we may encounter delays in or lose some control over our efforts to develop and supply products.
•Many of our competitors have significantly greater resources and experience, which may negatively impact our commercial opportunities and those of our current and future licensees.
•There is significant competition in the development of a vaccine against COVID-19, influenza, and RSV and we may never see returns on the significant resources we are devoting to our vaccine candidates.
•We have not completed the development of vaccine products, and we may not succeed in obtaining the FDA licensure necessary to sell such vaccine products.
•The regulatory pathway for NVX-CoV2373 is continually evolving, and may result in unexpected or unforeseen challenges.
•We are conducting, and plan to conduct in the future, a number of clinical trials for NVX-CoV2373 at sites outside the United States and the FDA may not accept data from trials conducted in such locations.
•Even if regulatory approval is received for our vaccine candidates, the later discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions, including withdrawal of the product from the market.
•Our success depends on our ability to maintain the proprietary nature of our technology.
•Our business may be adversely affected if we do not successfully execute our business development initiatives.
•Servicing our 3.75% convertible senior unsecured notes due 2023 (the “Notes”) requires a significant amount of cash, and we may not have sufficient cash flow resources to pay our debt.
•Because our stock price has been and will likely continue to be highly volatile, the market price of our common stock may be lower or more volatile than expected.
•Litigation could have a material adverse impact on our results of operation and financial condition.
We or the third parties upon whom we depend may be adversely affected by natural or man-made disasters or public health emergencies, such as the COVID-19 pandemic.
Risks Related to Our Financial Condition and Capital Requirements
We have a history of losses and our future profitability is uncertain.
Our expenses have exceeded our revenue since our formation in 1987, and our accumulated deficit at June 30, 2021 was $2.4 billion. Our revenues and expenses fluctuate significantly from period to period. For most of our history our expenses have exceeded our revenues, which may occur during most periods in the foreseeable future. Our net losses for the last three fiscal years were $418.3 million in 2020, $132.7 million in 2019 and $184.7 million in 2018.
Historically, our losses have resulted predominantly from research and development expenses for our vaccine candidates, manufacturing-related expenses, expenses associated with efforts to obtain regulatory approvals, costs related to protection of our intellectual property and other general and administrative operating expenses, a significant portion of which have been noncash. Our expenses have exceeded our revenue since inception, and we believe our expenses will fluctuate over time, and may substantially increase in some years, as a result of continuing efforts to develop, test, manufacture, and make regulatory filings for our vaccine candidates, and, if our product candidates are approved, commercialization efforts.
As of the end of the second quarter of 2021, our investment in the development and manufacture of NVX-CoV2373 has been substantial, and we expect such levels of investment to continue for the rest of 2021 and beyond, although the precise magnitude of our total investment will depend on the duration of the COVID-19 pandemic, the competitive landscape, the timing and results of our applications for regulatory approvals, the availability of funding, and whether and what booster shot protocols are recommended by governments, regulatory authorities, and healthcare providers. If we are unable to timely commercialize a vaccine against COVID-19, we likely would never recoup our investments. We expect to continue to incur significant operating expenses and anticipate significant losses over time as we seek to:
•conduct additional clinical trials and seek regulatory approvals for NVX-CoV2373 and other potential vaccine candidates;
•conduct preclinical studies for other potential vaccine candidates;
•expand our global manufacturing and distribution capacity; and
•maintain, expand and protect our intellectual property portfolio.
As a result, we expect our cumulative operating losses to increase until such time, if ever, that product sales, licensing fees, royalties, milestones, contract research and other sources generate sufficient revenue to fully fund our operations. We may never achieve profitability and may not sustain profitability, if achieved.
We will continue to require significant funding to maintain our current level of operations and fund the further development of our vaccine candidates.
We do not currently generate sufficient revenue from product sales, licensing fees, royalties, milestones, contract research or other sources to fully fund our operations. We, therefore, will use our cash resources, and expect to require additional funds, to maintain our operations, continue our research and development programs, commence future preclinical studies and clinical trials, seek regulatory approvals and manufacture and market any products that are approved for commercialization.
To date, we have financed our operations primarily through the sale of equity and debt securities, government funding and grant agreements, and additional funding may not be available to us on favorable terms, or at all. Although we have entered into supply agreements for NVX-CoV2373 that include prepayments from the purchasers, until we can generate sufficient product revenue in amounts sufficient to fully fund our operations, which we may never do, we expect to finance our cash needs through a combination of additional public or private equity or debt financings, as well as existing cash, potential collaborations, strategic alliances and marketing, distribution or licensing arrangements, funding from governmental and non-governmental funding entities, and potentially other sources. While we may continue to apply for contracts or grants from academic institutions, non-profit organizations and governmental entities, we may not be successful. Adequate additional funding may not be available to us on acceptable terms, if at all. Furthermore, negative interpretations of clinical trial data or setbacks, or perceived setbacks, with respect to manufacturing ability and/or capacity or regulatory filing timelines for NVX-CoV2373 or our other vaccine candidates, as well as the competitive landscape posed by other COVID-19 vaccines, may impair our ability to raise additional financing on favorable terms, or at all. If we cannot raise the additional funds required for our anticipated operations, we may be required to delay significantly, reduce the scope of or eliminate one or more of our research or development programs, downsize our organization, or seek alternative measures to avoid insolvency, including arrangements with collaborative partners or others that may require us to relinquish rights to certain of our technologies or vaccine candidates. If we raise additional funds through future offerings of shares of our common stock or other securities, such offerings would cause dilution of current stockholders’ percentage ownership in the Company, which could be substantial. Future offerings also could have a material and adverse effect on the price of our common stock.
Economic uncertainty may adversely affect our access to capital, cost of capital and ability to execute our business plan as scheduled.
Generally, worldwide economic conditions remain uncertain, particularly due to the COVID-19 pandemic. Access to capital markets is critical to our ability to operate. Traditionally, biotechnology companies have funded their research and development expenditures by raising capital in the equity markets. Declines and uncertainties in these markets in the past have severely restricted raising new capital and have affected companies’ ability to continue to expand or fund existing development, manufacturing, regulatory and commercialization efforts. We require significant capital for our current and expected operations. The general economic and capital market conditions, both in the U.S. and worldwide, have been volatile in the past and at times have adversely affected our access to capital and increased the cost of capital. The capital and credit markets may not be available to support future capital raising activity on favorable terms. If economic conditions decline, our future cost of equity or debt capital and access to the capital markets could be adversely affected. In addition, if we are unable to access the capital markets on favorable terms, our ability to execute our business plan as contemplated would be compromised. Moreover, we rely and intend to rely on third-parties, including clinical research organizations, contract manufacturing organizations and other important vendors and consultants. Global economic conditions may result in a disruption or delay in the performance of our third-party contractors and suppliers. If such third-parties are unable to adequately satisfy their contractual commitments to us in a timely manner, our business could be adversely affected.
Our existing funding and supply agreements do not assure success of our vaccine candidates or that we will be able to fully fund our vaccine candidates.
The OWS Agreement, the DoD Agreement and the CEPI funding agreement each reimburse a portion of the expenses associated with the development and commercialization of NVX-CoV2373. To the extent funding commitments in such agreements are conditioned on our meeting certain milestones or conditions, including regulatory approval in applicable jurisdictions, we may not ultimately receive the full amount of committed funds and could be exposed to urgent need for additional funding to support our NVX-CoV2373 development, manufacturing and distribution activities. For example, in connection with the OWS Agreement, the USG has instructed us to prioritize alignment with the FDA on our analytic methods before conducting additional U.S. manufacturing and further indicated that the USG will not fund additional U.S. manufacturing until such agreement has been made. The OWS Agreement includes provisions giving the USG termination rights based on a determination that the funded project will not produce beneficial results commensurate with the expenditure of resources and that termination would be in the USG’s interest. Such a determination would result in the loss of funding under that agreement and could result in other actions by the U.S. government. The CEPI funding agreement provides CEPI certain “march-in” rights in the event of certain breaches of that agreement. We may be unable to timely obtain additional government or private funding, if at all. Additionally, we have entered into, and plan to continue entering into, supply agreements for NVX-
CoV2373 that include prepayments from the purchasers. In the event we are unable to successfully develop and commercialize NVX-CoV2373 or fail to meet certain product volume or delivery timing obligations under our supply agreements, we may be required to refund significant portions of the prepayments, which could have a material and adverse effect on our financial condition. Our inability to succeed with key clinical or development activities could jeopardize our ability to obtain licensure from the FDA or other regulatory authorities to sell NVX-CoV2373. As a result, our existing funding and supply agreements may be insufficient to fund our commercial launch.
Risks Related to Product Development and Commercialization
Because our vaccine product development efforts depend on new and rapidly evolving technologies, our efforts may not succeed.
Our vaccine development efforts depend on new, rapidly evolving technologies and on the marketability and profitability of our products. Our development efforts and, if those are successful, commercialization of NVX-CoV2373 and our other vaccines could fail for a variety of reasons, including if:
•our recombinant nanoparticle vaccine technologies, any or all of the products based on such technologies or our proprietary manufacturing process prove ineffective or unsafe;
•new strains of COVID-19 evolve, with respect to which NVX-CoV2373 proves less effective;
•we or our third-party manufacturer facilities fail to reproducibly scale-up manufacturing with sufficiently high yields at reasonable cost and on projected timelines, or such manufacturing fails to generate product that consistently satisfies purity, potency, quality, stability, and shelf-life standards necessary for obtaining regulatory approvals or achieving commercial viability;
•the products are difficult to manufacture on a large-scale or uneconomical to market;
•our in-house or third-party manufacturing facilities fail regulatory inspections;
•proprietary rights of third-parties prevent us or our collaborators from exploiting technologies, and manufacturing or marketing products; or
•third-party competitors achieve and maintain greater market share due to earlier approvals or superior marketing capabilities.
The regulatory and commercial success of our COVID-19 vaccine candidate, NVX-CoV2373, remains uncertain. We may be unable to obtain regulatory approval or produce a successful vaccine in a timely manner, if at all.
In response to the outbreak of COVID-19, we are pursuing the development and manufacture of our vaccine candidate, NVX-CoV2373. Even though we have reported positive data from Phase 1, 2 and 3 clinical trials, such results may not be sufficient to support regulatory submissions, authorizations and approvals, accelerated or otherwise, on our projected timelines, if at all.
Additionally, even if NVX-CoV2373 receives regulatory approval, successful commercialization depends on our ability to effectively scale up manufacturing capabilities at our own locations and those of our manufacturing partners and contractors. In May 2020, we acquired Novavax CZ (formerly Praha Vaccines, a.s.) including its vaccine manufacturing facility in Bohumil, Czech Republic and approximately 150 of its employees. We also are actively entering into agreements with third parties to manufacture the antigen component of NVAX-CoV2373 and our proprietary Matrix-M™ adjuvant, as well as to distribute NVX-CoV2373. Because of contractual restraints and the limited number of third-party manufacturers with the expertise, required regulatory approvals and facilities to manufacture NVX-CoV2373 and its components at commercial scale, replacement of a manufacturer may be expensive and time-consuming and may cause interruptions in production. Manufacturing of NVX-CoV2373 and its components involves a complicated process that will require significant investments of time and financial resources to implement, and our efforts to establish manufacturing capabilities may not meet expectations as to timing, scale-up, reproducibility, yields, purity, cost, potency or quality. Shortages of raw materials and supplies also negatively impact our manufacturing efforts. We may not be able to timely and effectively produce NVX-CoV2373 in adequate quantities to address global demand.
We have not previously had a commercial launch of any vaccine product, and doing so in a pandemic environment with an urgent, critical global need creates additional challenges. In addition to scaling up our manufacturing capabilities, we
need to develop global distribution channels and form partnerships with third parties worldwide, as well as hire, train and integrate additional management, administrative and sales and marketing personnel. Rapid and significant growth may strain our administrative and operational infrastructure, imposing significant additional responsibilities on our organization, and our efforts to establish these capabilities may not meet expectations as to timing, scale-up, reproducibility, yields, purity, cost, potency or quality. If we fail to successfully manage our growth and the increased complexity of our operations, our business, financial position, results of operations and prospects may be materially and adversely affected.
We are a biotechnology company and face significant risk in developing, manufacturing and commercializing our products.
We focus our research and development activities on vaccines, an area in which we believe we have particular strengths and a technology that appears promising. The outcome of any research and development program is highly uncertain. Only a small fraction of biopharmaceutical development programs ultimately result in commercial products or even product candidates and a number of events could delay our development efforts and negatively impact our ability to make regulatory submissions or obtain regulatory approval for, and to manufacture, market and sell, NVX-CoV2373 or any other vaccine on our projected timelines, if at all. Vaccine candidates that initially appear promising often fail to yield successful products, and we may not ultimately be able to demonstrate the safety, potency, purity, stability and efficacy necessary to obtain regulatory authorization to market our product candidates. In many cases, preclinical studies or clinical trials will show that a product candidate is not efficacious or that it raises safety concerns or has other side effects that outweigh its intended benefit. Success in preclinical or early clinical trials may not translate into success in large-scale clinical trials. Further, success in clinical trials often leads to increased investment, accelerating cumulative losses. Even if clinical trial results appear positive, regulatory approval may not be obtained if the FDA does not agree with our interpretation of the results, and we may face challenges when scaling-up the production process to commercial levels. Even after a product is approved and launched, general usage or post-marketing clinical trials may identify safety or other previously unknown problems with the product, or manufacturing issues may emerge, either of which may result in regulatory approvals being suspended, limited to narrow the scope of the approval, or revoked, which may otherwise prevent successful commercialization. Intense competition in the vaccine industry could also limit the successful commercialization of any products for which we receive commercial approval.
Because we depend on third-parties to conduct some of our laboratory testing and clinical trials, and a significant amount of our vaccine manufacturing and distribution, we may encounter delays in or lose some control over our efforts to develop and supply products.
We are highly dependent on third-party organizations to conduct some of our laboratory testing and clinical trials and a significant amount of our vaccine manufacturing activities and distribution. If we are unable to obtain any necessary services on acceptable terms, we may not complete our product development or commercialization efforts in a timely manner. We may lose control over these activities or become too dependent upon these parties. These third-parties may not complete testing, manufacturing or distribution activities on schedule, or in satisfaction of regulatory or commercial requirements. Certain of our facilities are also contracted for defined time frames and through association with OWS and CEPI, and we may not be able to access those facilities for sufficient periods of time to provide adequate supply.
We are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, safety and welfare of clinical trial participants are adequately protected. The FDA and foreign regulatory agencies also require us to comply with good manufacturing practices. Our reliance on third-parties does not relieve us of these responsibilities and requirements. These third-parties may not successfully carry out their contractual duties or regulatory obligations. Furthermore, if a third-party manufacturer is producing materials or products for themselves or other companies, that manufacturer is exposed to regulatory risks for the production of such materials and products. As a result, failure to meet the regulatory requirements for the production of those materials and products may generally affect the regulatory status of the third-party manufacturer’s facility, which could impact its ability to produce our materials and products. Any of our third-party service providers may need to be replaced, the quality or accuracy of the data they obtain may be compromised, the services provided to us may be delayed, or the product they manufacture may be contaminated due to the failure to adhere to our clinical and manufacturing protocols, regulatory requirements or for other reasons. In any such event, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval of, or successfully commercially manufacture on a timely basis, our vaccine candidates.
The results from the Prepare trial, including that ResVax failed to meet the primary endpoint of the trial, will likely create challenges, some of which may be significant, around further development of that vaccine.
While the Prepare results suggest that ResVax is safe and is likely efficacious in more serious manifestations of RSV disease, the trial failed to achieve its primary clinical endpoint. Not achieving the primary clinical endpoint has been viewed negatively by our investors. Although the failure to achieve the primary endpoint in the trial is not evidence that the vaccine is
ineffective, it means that regulatory agencies like the FDA and EMA are likely to require additional clinical trial data prior to licensure. This development may be viewed negatively by our potential collaborators and partners, which may make the ongoing development of ResVax, and any other RSV F Vaccine candidates, more challenging.
We may have product liability exposure.
The administration of drugs or vaccines to humans, whether in clinical trials or after marketing approval, can result in product liability claims. We maintain product liability insurance coverage for our current clinical programs, including our NVX-CoV2373 trials. If and when we obtain marketing approval for any vaccine candidate, we intend to expand our insurance coverage to include the sale of commercial products; however, we may not be able to obtain or maintain insurance coverage on commercially reasonable terms, at a reasonable cost or in sufficient amounts to protect us against losses due to liability. Furthermore, such insurance coverage and our resources may not be sufficient to satisfy all liabilities that result from product liability claims. A successful claim may prevent us from obtaining adequate product liability insurance in the future on commercially desirable terms, if at all. Even if a claim is not successful, defending such a claim would be time- consuming and expensive, may damage our reputation in the marketplace and would likely divert management’s attention.
In addition, because we are developing NVX-CoV2373 in response to the outbreak of COVID-19, a global pandemic, we may have a widely used vaccine as an investigational vaccine or a product authorized for temporary or emergency use prior to our receipt of marketing approval. Unexpected safety issues in these circumstances could lead to product liability claims and our existing insurance may not be adequate for such claims.
Regardless of merit or eventual outcome, liability claims may result in:
•decreased demand for our products;
•withdrawal of regulatory approvals;
•voluntary or mandatory recalls of our products;
•necessity for additional nonclinical or clinical studies, changes in labeling, or changes to manufacturing processes, specifications and/or facilities;
•impairment of our business reputation and negative media attention;
•withdrawal of clinical trial participants;
•costs of related litigation;
•substantial monetary awards to participants or other claimants;
•loss of revenue; and
•inability to commercialize our vaccine candidates.
In the United States, the PREP Act, provides immunity for manufacturers from all claims under state or federal law for "loss" arising out of the administration or use of a “covered countermeasure.” However, injured persons may still bring a suit for "willful misconduct" against the manufacturer under some circumstances. "Covered countermeasures" include security countermeasures and "qualified pandemic or epidemic products", including products intended to diagnose or treat pandemic or epidemic disease, such as pandemic vaccines, as well as treatments intended to address conditions caused by such products. For these immunities to apply, the Secretary of DHHS must issue a declaration in cases of public health emergency or “credible risk” of a future public health emergency. On March 17, 2020, the Secretary of DHHS issued a declaration under the PREP Act and has issued subsequent amendments thereto to provide liability immunity for activities related to certain countermeasures against the ongoing COVID-19 pandemic. While we believe our products would be covered under the provisions of the PREP Act, this cannot be assured. Also, the Secretary of the HHS may not make other declarations in the future that cover any of our other product candidates, and the U.S. Congress may reduce coverage under the PREP Act or repeal it altogether. Product liability lawsuits may result in substantial liabilities and may require us to limit commercialization of our product candidates.
If we are unable to effectively manufacture our vaccines in sufficient quantities, at sufficient yields or are unable to obtain regulatory approvals for a manufacturing facility for our vaccines, we may experience delays or an adverse impact on product development, clinical trials, regulatory approval and commercial distribution.
Completion of our clinical trials and commercialization of our vaccine candidates require access to, or development of, facilities to effectively manufacture our vaccine candidates at sufficient yields and at commercial-scale. We have limited experience manufacturing any of our vaccine candidates in the volumes necessary to support commercial sales. While we have recently increased our projected global manufacturing capacity for NVX-CoV2373, our efforts to establish manufacturing capabilities may not meet expectations as to timing, scale-up, reproducibility, yields, purity, cost, potency or quality. The antigen component of NVX-CoV2373 is currently being manufactured at Novavax CZ, as well as numerous partnered manufacturing sites, including FUJIFILM in the United States, SIIPL in India and Takeda in Japan, among others. Challenges manufacturing either the antigen component or the adjuvant, or issues in later manufacturing stages, could compromise production of NVX-CoV2373.
Manufacturing our vaccine candidates involves a complicated process with which we have limited experience. We are highly dependent on third-party organizations to conduct a significant amount of our vaccine manufacturing activities. If we and our third-party manufacturing organizations are unable to manufacture our vaccine candidates in clinical quantities or, if and when necessary, in commercial quantities and at sufficient yields and at required specifications, then commercialization will be delayed, and we will need to identify and reach supply arrangements with additional third-parties. Third-party manufacturers also must receive FDA or equivalent foreign regulatory body approval before they can produce clinical material or commercial product. Our vaccines are in competition with other products for access to these third-party facilities and may be subject to delays in manufacture if third-parties prioritize other products. We may not be able to enter into any necessary additional third-party manufacturing arrangements on acceptable terms, or on a timely basis. In addition, we have to enter into technical transfer agreements and share our know-how with the third-party manufacturers, which can be time-consuming and may result in delays.
Because of contractual restraints and the limited number of third-party manufacturers with the expertise, required regulatory approvals and facilities to manufacture our bulk vaccines at commercial-scale, replacement of a manufacturer may be expensive and time-consuming and may cause interruptions in the production of our vaccine. We and our third-party manufacturers may also encounter production challenges related to:
•costs, scale up, and yields;
•shortages of raw materials and supplies;
•quality control and assurance;
•contamination, lot consistency, potency, and purity;
•shortages of qualified personnel and other capacity constraints;
•compliance with strictly enforced and evolving federal, state and foreign regulations that vary in each country where products might be sold including nationalization or other territory restrictions placed on our owned and third-party manufacturing sites; and
•capital funding.
Delays or interruptions could have a material adverse effect on our business, financial condition, results of operations and cash flows.
We must identify vaccines for development with our technologies and establish successful third-party relationships.
The near and long-term viability of our vaccine candidates depend in part on our ability to successfully establish new strategic collaborations with pharmaceutical and biotechnology companies, non-profit organizations and government agencies. Establishing strategic collaborations and obtaining government funding is difficult and time-consuming. Potential collaborators may reject collaborations based upon their assessment of our financial, regulatory or intellectual property position or based on their internal pipelines; government agencies may reject contract or grant applications based on their assessment of public need, the public interest, our products’ ability to address these areas, or other reasons beyond our expectations or control. Collaborators also may seek to modify or terminate relationships. Past success in establishing strategic collaborations with pharmaceutical and biotechnology companies, non-profit organizations and government agencies in the past is no guarantee of future success in entering into new relationships or in performing under existing relationships. If we fail to establish a sufficient number of collaborations or government relationships on acceptable terms, or fail to perform under collaborations or
relationships to the satisfaction of counter-parties, we may not be able to commercialize our vaccine candidates or generate sufficient revenue to fund further research and development efforts.
The collaborations we have established or may establish may not result in the successful development or commercialization of any vaccine candidates for several reasons, including the fact that:
•we may not have the ability to control the activities of our partners and cannot provide assurance that they will fulfill their obligations to us, including with respect to the license, development and commercialization of vaccine candidates, in a timely manner or at all;
•such partners may not devote sufficient resources to our vaccine candidates or properly maintain or defend our intellectual property rights;
•our partners could independently develop, or develop with third parties, products that compete directly or indirectly with our vaccine candidates if such partners believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours;
•any failure on the part of our partners to perform or satisfy their obligations to us could lead to delays in the development or commercialization of our vaccine candidates and affect our ability to realize product revenue; and
•disagreements, including disputes over the ownership of technology developed with such collaborators, could result in litigation, which would be time consuming and expensive, and may delay or terminate research and development efforts, regulatory approvals and commercialization activities.
If we or our collaborators fail to maintain our existing agreements or in the event we fail to establish agreements as necessary, we could be required to undertake research, development, manufacturing and commercialization activities solely at our own expense. These activities would significantly increase our capital requirements and, given our lack of sales, marketing and distribution capabilities, significantly delay the commercialization of our vaccine candidates.
Even if licensed to market, our vaccine products may not be initially or ever profitable.
Whether we make a profit from the sale of our vaccine products is dependent on a number of variables, including the costs we incur manufacturing, testing and releasing, packaging and shipping such vaccine product. Additionally, the CEPI funding agreement necessitates that we allocate a certain number of doses of NVX-CoV2373 to certain middle and lower income countries, and the Grant Agreement with BMGF necessitates that we commit to a specific amount of sales in certain specified middle and lower income countries, which may impact negatively our ability to generate profit. We cannot predict when, if at all, our approved vaccine products will be profitable to the Company.
Even if we successfully commercialize any of our vaccine candidates, either alone or in collaboration, we face uncertainty with respect to pricing, third-party reimbursement and healthcare reform, all of which could adversely affect any commercial success of our vaccine candidates.
Our ability to collect revenue from the commercial sale of our vaccines may depend on our ability, and that of any current or potential future collaboration partners or customers, to obtain adequate levels of approval, coverage and reimbursement for such products from third-party payers such as:
•government health administration authorities such as the Advisory Committee for Immunization Practices of the Centers for Disease Control and Prevention;
•private health insurers;
•managed care organizations;
•pharmacy benefit management companies; and
•other healthcare related organizations.
Third-party payers are increasingly challenging the prices charged for medical products and may deny coverage or offer inadequate levels of reimbursement if they determine that a prescribed product has not received appropriate clearances from the FDA, or foreign equivalent, or other government regulators; is not used in accordance with cost-effective treatment
methods as determined by the third-party payer; or is experimental, unnecessary or inappropriate. Prices could also be driven down by managed care organizations that control or significantly influence utilization of healthcare products.
In both the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory proposals and initiatives to change the health care system in ways that could affect our ability to sell vaccines and could adversely affect the prices that we receive for our vaccine candidates, if approved. Some of these proposed and implemented reforms could result in reduced pharmaceutical pricing or reimbursement rates for medical products, and while we have no current vaccines available for commercial sale, the impact of such reform could nevertheless adversely affect our business strategy, operations and financial results. For example, the Affordable Care Act (“ACA”) contained several cost containment measures that could adversely affect our future revenue, including, for example, increased drug rebates under Medicaid for brand name prescription drugs, extension of Medicaid rebates to Medicaid managed care organizations, and extension of so-called 340B discounted pricing on pharmaceuticals sold to certain healthcare providers. Additional provisions of the healthcare reform laws that may negatively affect our future revenue and prospects for profitability include the assessment of an annual fee based on our proportionate share of sales of brand name prescription drugs to certain government programs, including Medicare and Medicaid. The ACA also established a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable branded on drugs (including vaccines) to eligible beneficiaries during their coverage gap period (the so-called “donut hole”), as condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Other aspects of healthcare reform, such as expanded government enforcement authority and heightened standards that could increase compliance-related costs, could also affect our business.
Further, we face uncertainties because of occasional political, legislative, and administrative efforts to substantially modify or invalidate some or all of the provisions of the ACA. For example, in 2017, the Trump administration withheld the cost-sharing subsidies paid to ACA health insurance exchange plans serving low-income enrollees. The Tax Cut and Jobs Act (“TCJA”) was also enacted at the end of 2017 and included provisions that affected healthcare insurance coverage and payment, such as the elimination of the tax penalty for individuals who do not maintain sufficient health insurance coverage beginning in 2019 (the so-called “individual mandate”).
More recently, the Biden administration, through the American Rescue Plan Act of 2021, increased subsidies for coverage purchased through ACA health insurance exchanges and extended eligibility for subsidies to higher income levels. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the TCJA, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional but remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the U.S. Supreme Court granted the petitions for writs of certiorari to review the case, and oral arguments were heard on November 10, 2020. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Separately, President Biden issued an Executive Order to initiate a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA marketplace. The Executive Order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare, including among others, reexamining Medicaid demonstration projects and waiver programs that include work requirements, and policies that create unnecessary barriers to obtaining access to health insurance coverage through Medicaid or the ACA. It is also unclear how these and other healthcare reform measures of the Biden administration or other efforts, if any, to challenge, repeal or replace the ACA, will impact our business.
Other legislative changes have been proposed and adopted since the ACA was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011 and subsequent laws, which began in 2013 and, due to subsequent legislative amendments, will stay in effect through 2030 unless additional Congressional action is taken. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws may result in additional reductions in Medicare and other healthcare funding, which may materially adversely affect customer demand and affordability for our products and, accordingly, the results of our financial operations. Additionally, the pharmaceutical industry has also been the subject of significant publicity in recent years regarding the pricing of pharmaceutical products, including publicity and pressure resulting from prices charged by pharmaceutical companies for new products as well as price increases by pharmaceutical companies on older products that some people have deemed excessive. As a result, pharmaceutical product prices have been the focus of increased scrutiny by the U.S. government, including certain state attorneys general, members of congress, presidential candidates and the United States Department of Justice. If reforms in the health care industry make reimbursement for our potential products less likely, the market for our potential products will be reduced, and we could lose potential sources of revenue. The existence or threat of cost control measures could cause our corporate collaborators to be less willing or able to pursue research and development programs related to our vaccine candidates. Further, it is also possible that additional governmental action is taken in response to the COVID-19 pandemic. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us.
Even if we receive regulatory approvals for our vaccine candidates, including NVX-CoV2373, coverage and reimbursement may be subject to unique regulatory policies. For example, under the ACA preventive care mandate, non-grandfathered group health plans and health insurance coverage offered in the individual or group market typically have at least one year before they must provide first-dollar coverage for a newly issued preventive care requirement or guideline. However, pursuant to the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”), non-grandfathered group health plans and health insurance coverage offered in the individual or group market must cover any qualifying coronavirus preventive service 15 business days after the United States Preventive Services Task Force, or Advisory Committee on Immunization Practices (ACIP) designates such service as preventive. Further, third-party reimbursement for providers administering COVID-19 vaccines may affect market acceptance of NVX-CoV2373, if we receive regulatory approval. Currently, the CARES Act and its implementing regulations state that (i) providers that participate in the U.S. Centers for Disease Control and Prevention’s COVID-19 Vaccination Program must administer a COVID-19 immunization regardless of an individual’s ability to pay or health insurance coverage status, (ii) providers may not seek any reimbursement, including through balance billing, from an immunization recipient, (iii) coverage is required, without cost-sharing, for the administration of the immunization even if a third party, such as the federal government, pays for the cost of the immunization, and (iv) private health insurance plans must cover COVID-19 immunizations and their administration even when provided by out-of-network providers for the duration of the public health emergency for COVID-19. Even if we receive regulatory approvals for NVX-CoV237, there is no guarantee payors will provide coverage and reimbursement for our product after the termination of the public health emergency, nor can we guarantee that even if coverage is provided, the reimbursement amount will be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment. We cannot predict continued prevalence of COVID-19, whether herd immunity will be achieved (which would affect the need for future administration of COVID-19 vaccines), or whether NVX-CoV2373 will be effective against continuing mutations or variants of the SARS-CoV-2 virus.
We have limited marketing capabilities, and if we are unable to enter into collaborations with marketing partners or develop our own sales and marketing capability, we may not be successful in commercializing any approved products.
Although we have initiated preliminary activities in anticipation of commercialization of our vaccine candidates, we currently have limited dedicated sales, marketing or distribution capabilities. As a result, we depend on collaborations with third-parties that have established distribution systems and sales forces, including our collaboration with SIIPL, among others. To the extent that we enter into co-promotion or other licensing arrangements, our revenue will depend upon the efforts of third-parties, over which we may have little or no control. If we are unable to reach and maintain agreements with one or more pharmaceutical companies or collaborators, we may be required to market our products directly. Developing a marketing and sales force is expensive and time-consuming and could delay a product launch. We may not be able to attract and retain qualified sales personnel or otherwise develop this capability.
Our vaccine candidates may never achieve market acceptance even if we obtain regulatory approvals.
Even if we receive regulatory approvals for the commercial sale of our vaccine candidates, the commercial success of these vaccine candidates will depend on, among other things, their acceptance by physicians, patients and third-party payers, such as health insurance companies and other members of the medical community, as a vaccine and cost-effective alternative to competing products. If our vaccine candidates fail to gain market acceptance, we may be unable to earn sufficient revenue to continue our business. Market acceptance of, and demand for, any product that we may develop and commercialize will depend on many factors, including:
•our ability to provide acceptable evidence of safety and efficacy (including against emerging COVID-19 variants);
•the prevalence and severity of adverse side effects;
•whether our vaccines are differentiated from other vaccines;
•availability, relative cost and relative efficacy of alternative and competing treatments;
•the effectiveness of our marketing and distribution strategy;
•publicity concerning our products or competing products and treatments; and
•our ability to obtain sufficient third party insurance coverage or reimbursement.
If our vaccine candidates do not become widely accepted by physicians, patients, third-party payers and other members of the medical community, our business, financial condition and results of operations could be materially and adversely affected.
We may not be able to secure sufficient supplies of a key component of our adjuvant technology.
Because an important component of our adjuvant technology is extracted from a species of soap-bark tree (Quillaja saponaria) grown in Chile, we need long term access to quillaja extract with a consistent and sufficiently high quality. We need a secure supply of raw material, as well as back-up suppliers, or our adjuvant products may be delayed and we may not be able to meet our obligations under our various collaboration and supply agreements.
Current or future regional relationships may hinder our ability to engage in larger transactions.
We have entered into regional collaborations to develop, manufacture and distribute our vaccine candidates in certain parts of the world, and we anticipate entering into additional regional collaborations. Our relationships with SIIPL, Cadila and BMGF are examples of these regional relationships. These relationships often involve the licensing of our technology to our partner or entering into a distribution agreement, frequently on an exclusive basis. Generally, exclusive agreements are restricted to certain territories. Because we have entered into exclusive license and distribution agreements, larger companies may not be interested, or able, to enter into collaborations with us on a worldwide-scale. Also, these regional relationships may make us an unattractive target for an acquisition.
Our product candidates are sensitive to shipping and storage conditions, which could subject our vaccine candidates to risk of loss or damage.
Our vaccine candidates are sensitive to storage and handling conditions. Loss in vaccine candidates could occur if the product or product intermediates are not stored or handled properly. It is possible that our vaccine candidates could be lost due to expiration prior to use. If we do not effectively maintain our supply logistics, then we may experience an unusual number of returned or out of date products. Failure to effectively maintain our supply logistics, by us or third parties, could lead to additional manufacturing costs and delays in our ability to supply required quantities for clinical trials or otherwise.
Our vaccine candidates could become subject to a product recall which could harm our reputation, business, and financial results.
The FDA and similar foreign governmental authorities have the authority to require the recall of certain vaccine candidates. Manufacturers may, under their own initiative, recall a product if any material deficiency in a product is found. A government-mandated or voluntary recall by us or our strategic collaborators could occur as a result of manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our vaccine candidates would divert managerial and financial resources and have an adverse effect on our financial condition and results of operations. A recall announcement could harm our reputation with customers and negatively affect our sales, if any.
Risks Related to Our Industry and Competition
Many of our competitors have significantly greater resources and experience, which may negatively impact our commercial opportunities and those of our current and future licensees.
The biotechnology and pharmaceutical industries are subject to intense competition and rapid and significant technological change. We have many potential competitors, including major pharmaceutical companies, specialized biotechnology firms, academic institutions, government agencies and private and public research institutions. Many of our competitors have significantly greater financial and technical resources, experience and expertise in:
•research and development;
•preclinical testing;
•designing and implementing clinical trials;
•regulatory processes and approvals;
•production and manufacturing; and
•sales and marketing of approved products.
Principal competitive factors in our industry include:
•the quality and breadth of an organization’s technology;
•management of the organization and the execution of the organization’s strategy;
•the skill and experience of an organization’s employees and its ability to recruit and retain skilled and experienced employees;
•an organization’s intellectual property portfolio;
•the range of capabilities, from target identification and validation to drug discovery and development to manufacturing and marketing; and
•the availability of substantial capital resources to fund discovery, development and commercialization activities.
Large and established companies, such as Merck & Co., Inc., GlaxoSmithKline plc, CSL Ltd, Sanofi Pasteur, SA, Pfizer Inc., Johnson & Johnson, AstraZeneca, and Moderna, among others, compete in the vaccine market. In particular, these companies have greater experience and expertise in securing government contracts and grants to support their research and development efforts, conducting testing and clinical trials, obtaining regulatory approvals to market products, manufacturing such products on a broad scale and marketing approved products.
Regardless of the disease, smaller or early-stage companies and research institutions also may prove to be significant competitors, particularly through collaborative arrangements with large and established pharmaceutical companies. As these companies develop their technologies, they may develop proprietary positions, which may prevent or limit our product development and commercialization efforts. We will also face competition from these parties in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and participant registration for clinical trials and in acquiring and in-licensing technologies and products complementary to our programs or potentially advantageous to our business. If any of our competitors succeed in obtaining approval from the FDA or other regulatory authorities for their products sooner than we do or for products that are more effective or less costly than ours, our commercial opportunity could be significantly reduced.
In order to effectively compete, we will have to make substantial investments in development, testing, manufacturing and sales and marketing or partner with one or more established companies. We may not be successful in gaining significant market share for any vaccine. Our technologies and vaccines also may be rendered obsolete or non-competitive as a result of products introduced by our competitors to the marketplace more rapidly and at a lower cost.
There is significant competition in the development of a vaccine against COVID-19, influenza, and RSV and we may never see returns on the significant resources we are devoting to our vaccine candidates.
We may be unable to produce a successful COVID-19 vaccine, and establish a competitive market share for our vaccine before the COVID-19 outbreak is contained or significantly diminished. A large number of vaccine manufacturers, academic institutions and other organizations have developed COVID-19 vaccines or are developing COVID-19 vaccine candidates. In particular, Moderna, Pfizer/BioNTech, and Johnson & Johnson have received emergency use authorizations for their COVID-19 vaccines in the U.S. and other countries, and many other companies, including AstraZeneca, Sinovac Biotech, Sinopharm, and Inovio are in various stages of developing and obtaining marketing authorization for COVID-19 vaccine candidates. Despite funding provided to us to date, many of our competitors pursuing vaccine candidates have significantly greater product candidate development, manufacturing and marketing resources than we do. Larger pharmaceutical and biotechnology companies have extensive experience in clinical testing and obtaining regulatory approval for their products and may have the resources to heavily invest to accelerate discovery and development of their vaccine candidates. The success of our COVID-19 vaccine will depend, in part, on its relative safety, efficacy (including against emerging variant strains), side effect profile, convenience, and cost. COVID-19 vaccines approved prior to our vaccine satisfy a portion of the demand for initial vaccinations, and we no longer have access to that opportunity. In addition, COVID-19 vaccines approved prior to our vaccine may develop broad market acceptance that we are challenged to overcome. Furthermore, if any competitors are successful in producing a more efficacious vaccine or other treatment for COVID-19 (including against emerging variant strains), or if any competitors are able to manufacture and distribute any such vaccines or treatments with greater efficiency there may be a diversion of potential governmental and other funding away from us and toward such other parties.
We are allocating significant financial and personnel resources to the development of NVX-CoV2373, which may cause delays in or otherwise negatively impact our other development programs. Our business could be negatively impacted by our allocation of significant resources to combating a global health threat that is unpredictable or against which our vaccine, if commercialized, may ultimately prove unsuccessful or unprofitable.
Many seasonal influenza vaccines are currently approved and marketed. Competition in the sale of these seasonal influenza vaccines is intense. Therefore, newly developed and approved products must be differentiated from existing vaccines
in order to have commercial success. In order to show differentiation in the seasonal influenza market, a product may need to be more efficacious, particularly in older adults, and/or be less expensive or quicker to manufacture. Many competitors are working on new products and new generations of current products, intended to be more efficacious than those currently marketed. Our nanoparticle seasonal influenza vaccine candidate may not prove to be more efficacious than current products or products under development by our competitors. Further, our in-house or third-party manufacturing arrangements may not provide enough savings of time or money to provide the required differentiation for commercial success.
We are also aware that there are multiple companies with active RSV vaccine programs at various stages of development. Thus, while there is no RSV vaccine currently on the market, there is likely to be significant and consistent competition as these active programs mature. Different RSV vaccines may work better for different segments of the population, so it may be difficult for a single RSV vaccine manufacturer to provide vaccines that are marketable to multiple population segments. Geographic markets are also likely to vary significantly, which may make it difficult to market a single RSV vaccine worldwide. Even if a manufacturer brings an RSV vaccine to license, it is likely that competitors will continue to work on new products that could be more efficacious and/or less expensive. Our RSV vaccine candidate may not be as far along in development as other active RSV vaccine programs about which we are not aware, nor as efficacious as products under development by competing companies. Even if our RSV vaccine candidate receives regulatory approval, it may not achieve significant sales if other, more effective vaccines under development by our competitors are also approved.
Risks Related to Regulatory and Compliance Matters
We have not completed the development of vaccine products, and we may not succeed in obtaining the FDA licensure or foreign regulatory approvals necessary to sell such vaccine products.
The development, manufacture and marketing of our pharmaceutical and biological products are subject to government regulation by the U.S. FDA and regulatory authorities in other jurisdictions, including the European Medicines Agency EMA, the State Institute for Drug Control (SUKL) with respect to our manufacturing facility in the Czech Republic and the Swedish Medical Products Agency (Läkemedelsverket, LV) with respect to our adjuvant product being developed in Sweden, as well as other country authorities into which active pharmaceutical ingredients and excipients are imported and/or manufactured by us or our sub-contracted manufacturers. In the U.S. and most foreign countries, we must complete rigorous preclinical testing and extensive clinical trials that demonstrate the safety and efficacy of a product in order to apply for regulatory approval to market the product. Additionally, we must demonstrate that our manufacturing facilities, processes and controls are adequate with respect to such product to assure safety, purity and potency. None of our vaccine candidates has yet gained regulatory approval in the U.S. or elsewhere. We also have vaccine candidates in clinical trials and preclinical laboratory or animal studies. The steps generally required by the FDA before our proposed investigational products may be marketed in the U.S. include:
•performance of preclinical (animal and laboratory) tests;
•submission to the FDA of an IND, which must become effective before clinical trials may commence;
•performance of adequate and well controlled clinical trials to establish the safety and efficacy of the investigational product in the intended target population;
•performance of a consistent and reproducible manufacturing process at commercial scale capable of passing FDA inspection;
•submission to the FDA of a BLA or a NDA; and
•FDA approval of the BLA or NDA before any commercial sale or shipment of the product.
These processes are expensive and can take many years to complete, and we may not be able to demonstrate the safety, purity, potency and efficacy of our vaccine candidates to the satisfaction of regulatory authorities. The start of clinical trials can be delayed or take longer than anticipated for many and varied reasons, many of which are out of our control. Safety concerns may emerge that could lengthen the ongoing clinical trials or require additional clinical trials to be conducted. Promising results in early clinical trials may not be replicated in subsequent clinical trials. Regulatory authorities may also require additional testing, and we may be required to demonstrate that our proposed products represent an improved form of treatment over existing therapies, which we may be unable to do without conducting further clinical trials. Moreover, if the FDA grants regulatory approval of a product, the approval may be limited to specific indications or limited with respect to its distribution. Expanded or additional indications for approved products may not be approved, which could limit our revenue. Foreign regulatory authorities may apply similar limitations or may refuse to grant any approval. Consequently, even if we believe that preclinical and clinical data are sufficient to support regulatory approval for our vaccine candidates, the FDA and foreign regulatory authorities ultimately may not grant approval for commercial sale in any jurisdiction, or may impose regulatory
requirements that make further pursuit of approval uneconomical in one or more jurisdictions. If our vaccine candidates are not approved, our ability to generate revenue will be limited, and our business will be adversely affected.
We may fail to obtain regulatory approval for our products on a timely basis or comply with our continuing regulatory obligations after approval is obtained.
Delays in obtaining regulatory approval can be extremely costly in terms of lost sales opportunities, loss of any potential marketing advantage of being early to market and increased clinical trial costs. The speed with which we begin and complete our preclinical studies necessary to begin clinical trials, clinical trials and our applications for marketing approval will depend on several factors, including the following:
•our ability to scale-up manufacturing capability that reproducibly generates consistent yields of product with required purity, potency and quality; that such scale-up occurs on a timely basis; and that we have access to sufficient quantities of materials for use in necessary preclinical studies and clinical trials;
•regulatory agency review and approval of proposed clinical trial protocols;
•approval of clinical trials protocols and informed consent forms by institutional review boards responsible for overseeing the ethical conduct of the trial;
•the rate of participant enrollment and retention, which is a function of many factors, including the size of the participant population, the proximity of participants to clinical sites, the eligibility criteria for the clinical trial and the nature of the protocol;
•unfavorable test results or side effects experienced by clinical trial participants;
•analysis of data obtained from preclinical and clinical activities, which are susceptible to varying interpretations and which interpretations could delay, limit, result in the suspension or termination of, or prevent further conduct of clinical studies or regulatory approval;
•the availability of skilled and experienced staff to conduct and monitor clinical trials and to prepare the appropriate regulatory applications; and
•changes in the policies of regulatory authorities for drug or vaccine approval during the period of product development.
We have limited experience in conducting and managing the preclinical studies and clinical trials necessary to obtain regulatory marketing approvals. We may not be permitted to continue or commence additional clinical trials. We also face the risk that the results of our clinical trials may be inconsistent with the results obtained in preclinical studies or clinical trials of similar products or that the results obtained in later phases of clinical trials may be inconsistent with those obtained in earlier phases. A number of companies in the biotechnology and product development industry have suffered significant setbacks in advanced clinical trials, even after experiencing promising results in early animal and human testing.
Regulatory agencies may require us or our collaborators to delay, restrict or discontinue clinical trials on various grounds, including a finding that the participants are being exposed to an unacceptable health risk. In addition, we or our collaborators may be unable to submit applications to regulatory agencies within the time frame we currently expect. Once submitted, applications must be approved by various regulatory agencies before we or our collaborators can commercialize the product described in the application. All statutes and regulations governing the conduct of clinical trials are subject to change in the future, which could affect the cost of such clinical trials. Any unanticipated costs or delays in our clinical trials or regulatory submissions could delay our ability to generate revenue and harm our financial condition and results of operations.
Failure to obtain regulatory approval in foreign jurisdictions would prevent us from marketing our products internationally.
We intend to have our vaccine candidates marketed outside the U.S. In furtherance of this objective, we have entered into supply agreements with various foreign governments and international distribution agreements with commercial entities. In order to market our products in the European Union, United Kingdom, India, Asia and many other non-U.S. jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing and data review. The time required to obtain foreign
regulatory approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by one regulatory agency does not ensure approval by regulatory agencies in other jurisdictions. However, a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in other jurisdictions, including approval by the FDA. The failure to obtain regulatory approval in foreign jurisdictions could harm our business.
The regulatory pathway for NVX-CoV2373 is continually evolving and may result in unexpected or unforeseen challenges.
The regulatory pathway for NVX-CoV2373 is evolving and failure by us to comply with any laws, rules and standards, some of which may not exist yet or are subject to interpretation and may be subject to change, could result in a variety of adverse consequences, including penalties, fines and delays in vaccine licensure. Efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased general and administrative expenses and a diversion of management time and attention to regulatory compliance activities. For example, the rules, regulations and standards governing OWS are uncertain and may evolve as the program progresses. Such rules or standards may adversely affect our plans to develop NVX-CoV2373 and failure by us to comply with any laws, rules or standards, some of which may not exist yet or may change, could result in a range of adverse consequences, such as penalties, fines or failure to receive funding.
The speed at which multiple stakeholders are moving to create, test and approve vaccines for COVID-19 is highly unusual and may increase the risks associated with traditional vaccine development, which typically takes between eight and ten years. Given this accelerated timeline, we and regulators, such as the FDA, the EMA, and the MHRA may make decisions more rapidly than is typical. Evolving or changing plans or priorities at the FDA or other regulatory bodies, including based on new knowledge of COVID-19 and how the disease affects the human body, and new variants of the virus, may significantly affect the regulatory pathway for NVX-CoV2373. Results from clinical testing may raise new questions and require us to redesign proposed clinical trials, including revising proposed endpoints or adding new clinical trial sites or cohorts of subjects. In addition, the FDA’s or other regulators’ analysis of clinical data may differ from our interpretation, or regulators’ requirements and expectations for vaccine authorization or approval may change over time, with the result that the FDA or other regulators may require that we conduct additional clinical trials or non-clinical studies. The evolving regulatory pathway may impede the development, commercialization and/or licensure of NVX-CoV2373.
In addition, because the path to licensure of any vaccine against COVID-19 is unclear, we may have a widely used vaccine in circulation in certain countries as an investigational vaccine or a product authorized for temporary or emergency use prior to our receipt of full marketing approval. Unexpected safety issues in these circumstances could lead to significant reputational damage for Novavax and our technology platform going forward and other issues, including delays in our other programs, the need for re-design of our clinical trials and the need for significant additional financial resources.
We are conducting, and plan to conduct in the future, a number of clinical trials for NVX-CoV2373 at sites outside the United States and the FDA may not accept data from trials conducted in such locations.
We are currently conducting several clinical trials of NVX-CoV2373 at sites outside the U.S., including a Phase 3 trial in the U.K., a Phase 2b trial in South Africa, and a Phase 1/2 trial partially in Australia. We also plan in the future to conduct (or collaborate to conduct) a Phase 2/3 trial in India, Phase 2 trial in the Czech Republic, and Phase 1/2 trial in Japan. Although the FDA may accept data from clinical trials conducted outside the U.S., acceptance of these data is subject to conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and be performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will depend on its determination that the trials also complied with all applicable U.S. laws and regulations. If the FDA does not accept the data from any trial that we conduct outside the U.S., it could result in delay pending completion of our trials conducted in the U.S. or result in the need for additional trials, which would be costly and time-consuming and could delay or permanently halt our development of NVX-CoV2373.
Even if regulatory approval is received for our vaccine candidates, the later discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions, including withdrawal of the product from the market.
Even after a product gains regulatory approval, the product and the manufacturer of the product will be subject to continuing regulatory review, including adverse event reporting requirements and the FDA’s general prohibition against promoting products for unapproved uses. Failure to comply with any post-approval requirements can, among other things, result in warning letters, product seizures, recalls, substantial fines, injunctions, suspensions or revocations of marketing licenses, operating restrictions and criminal prosecutions. Any such enforcement actions, any unanticipated changes in existing
regulatory requirements or the adoption of new requirements, or any safety issues that arise with any approved products, could adversely affect our ability to market products and generate revenue and thus adversely affect our ability to continue our business.
We also may be restricted or prohibited from marketing or manufacturing a product, even after obtaining product approval, if previously unknown problems with the product or its manufacture are subsequently discovered. We cannot provide assurance that newly discovered or developed safety issues will not arise following regulatory approval. With the use of any vaccine by a wide patient population, serious adverse events may occur from time to time that did not arise in the clinical trials of the product or that initially appeared to be unrelated to the vaccine itself and only with the collection of subsequent information were found to be causally related to the product. Any such safety issues could cause us to suspend or cease marketing of our approved products, possibly subject us to substantial liabilities, and adversely affect our ability to generate revenue and our financial condition.
Our ability to produce a successful vaccine may be curtailed by one or more government actions or interventions, which may be more likely during a global health crisis such as COVID-19.
Given the significant global impact of the COVID-19 pandemic, it is possible that one or more government entities may take actions, including under the U.S. Government under the Defense Production Act of 1950, as amended, that directly or indirectly have the effect of diminishing some of our rights or opportunities with respect to NVX-CoV2373, and the economic value of a COVID-19 vaccine to us could be limited. In addition, during a global health crisis, such as the COVID-19 pandemic, where the spread of a disease needs to be controlled, closed or heavily regulated national borders create challenges and delays in our development, production and distribution activities and may necessitate that we pursue strategies to develop, produce and distribute our vaccine candidates within self-contained national or international borders or with additional safety measures or checks in place, at potentially much greater expense and with longer timeframes for public distribution.
Inadequate funding for the FDA, the SEC and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise perform their normal functions on which the operation of our business may rely, which could negatively impact our ability to develop or commercialize new products or services, access capital markets, or otherwise operate our business.
The ability of the FDA to review and approve new products is affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of the SEC and other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA and the SEC, have had to furlough employees and stop or slow the pace of critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. Further, in our operations as a public company, future government shutdowns could impact our ability to access the public markets and obtain necessary capital in order to properly capitalize and continue our operations.
Fast Track Designation by the FDA or other regulatory acceleration options may not actually lead to a faster development or regulatory review or approval process and does not assure approval.
If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address an unmet medical need for this condition, the drug sponsor may apply for FDA Fast Track Designation or similar fast track processes with other regulatory agencies, such as conditional marketing authorizations from the EMA. However, Fast Track Designation does not ensure that the drug sponsor will receive marketing approval or that approval will be granted within any particular timeframe. The FDA granted Fast Track Designation for NVX-CoV2373 in November 2020 and for NanoFlu™, our recombinant quadrivalent seasonal influenza vaccine candidate, in January 2020. We may also seek Fast Track Designation for more of our other vaccine candidates. If we do seek Fast Track Designation for our other vaccine candidates, we may not receive it, and even if we receive Fast Track Designation, we may not experience a faster development process, review or approval compared to conventional FDA procedures. In addition, the FDA may withdraw Fast Track designation if it believes that the designation is no longer supported by data from our clinical development program. Fast Track Designation alone does not guarantee qualification for the FDA’s priority review procedures.
Obtaining a Fast Track Designation does not change the standards for product approval, but may expedite the development or approval process. Even though the FDA has granted such designation for NVX-CoV2373 and NanoFlu™, it
may not actually result in faster clinical development or regulatory review or approval. Furthermore, such a designation does not increase the likelihood that NVX-CoV2373 or NanoFlu™ will receive marketing approval in the U.S.
Because we are subject to environmental, health and safety laws, we may be unable to conduct our business in the most advantageous manner.
We are subject to various laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals, emissions and wastewater discharges, and the use and disposal of hazardous or potentially hazardous substances used in connection with our research, including infectious disease agents. We also cannot accurately predict the extent of regulations that might result from any future legislative or administrative action. Any of these laws or regulations could cause us to incur additional expense or restrict our operations.
Our facilities in Maryland are subject to various local, state and federal laws and regulations relating to safe working conditions, laboratory practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including chemicals, microorganisms and various hazardous compounds used in connection with our research and development activities. In the U.S., these laws include the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act. Similar national and local regulations govern our facilities in Sweden and the Czech Republic. We cannot eliminate the risk of accidental contamination or discharge or injury from these materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these materials. We could be subject to civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, these hazardous materials. In addition, claimants may sue us for injury or contamination that results from our use or the use by third-parties of these materials, and our liability may exceed our total assets. Compliance with environmental laws and regulations may be expensive, and current or future environmental regulations may impair our research, development or production efforts.
Although we have general liability insurance, these policies contain exclusions from insurance against claims arising from pollution from chemicals or pollution from conditions arising from our operations. Our collaborators are working with these types of hazardous materials in connection with our collaborations. In the event of a lawsuit or investigation, we could be held responsible for any injury we or our collaborators cause to persons or property by exposure to, or release of, any hazardous materials. However, we believe that we are currently in compliance with all material applicable environmental and occupational health and safety regulations.
For our product candidates, we will be subject to additional healthcare laws and our failure to comply with those laws could have a material adverse effect on our results of operations and financial conditions.
Within the U.S. (and within foreign countries), if we obtain approval for any of our product candidates and begin commercializing them, our operations may be directly, or indirectly through our arrangements with third-party payors and customers, subject to additional healthcare regulation and enforcement by the federal and state governments (or the regulatory bodies or governments of foreign countries), which may constrain the business or financial arrangements and relationships through which we sell, market and distribute our products. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, structuring and commission(s), certain customer incentive programs and other business arrangements generally. Activities subject to these laws also involve the improper use of information obtained in the course of patient recruitment for clinical trials. The applicable U.S. federal and state healthcare laws and regulations (which may be comparable to foreign laws existing in foreign countries) that may affect our ability to operate include:
•the Federal Food, Drug and Cosmetic Act, which among other things, strictly regulates drug product marketing and promotion and prohibits manufacturers from marketing such products for unapproved uses;
•the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving or providing remuneration, directly or indirectly, to induce the referral for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid;
•federal false claims laws, including the FCA, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, information or claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent;
•manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims; the FCA also permits a private individual
acting as whistleblower to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery;
•federal laws that require pharmaceutical manufacturers to report certain calculated product prices to the government or provide certain discounts or rebates to government authorities or private entities, often as a condition of reimbursement under government healthcare programs;
•the federal Physician Payment Sunshine Act and its implementing regulations, which require manufacturers of drugs, devices, biologicals, and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the DHHS information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; effective January 1, 2022, these reporting obligations will extend to include transfers of value made to certain non-physician providers such as physician assistants and nurse practitioners;
•the federal law known as HIPAA, which, in addition to privacy protections applicable to healthcare providers and other entities, prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters;
•federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers;
•state law equivalents of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state gift ban and transparency laws, many of which state laws differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts; and
•state laws restricting interactions with healthcare providers and other members of the healthcare community or requiring pharmaceutical manufacturers to implement certain compliance standards.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to, on a corporate or individual basis, penalties, including civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and even imprisonment, any of which could materially adversely affect our ability to operate our business and our financial results. In addition, the cost of implementing sufficient systems, controls, and processes to ensure compliance with all of the aforementioned laws could be significant. Any action for violation of these laws, even if successfully defended, could cause us to incur significant legal expenses and divert management’s attention from the operation of the company’s business. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, that person or entity may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Prohibitions or restrictions on sales or withdrawal of future marketed products could materially affect business in an adverse way.
It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent inappropriate conduct may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. It is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights those actions, our business may be impaired.
Risks Related to our Intellectual Property
Our success depends on our ability to maintain the proprietary nature of our technology.
Our success in large part depends on our ability to maintain the proprietary nature of our technology and other trade secrets. To do so, we must prosecute and maintain existing patents, obtain new patents and pursue trade secret and other
intellectual property protection. We also must operate without infringing the proprietary rights of third-parties or allowing third-parties to infringe our rights. We currently have or have rights to over 450 U.S. patents and corresponding foreign patents and patent applications covering our technologies. However, patent issues relating to pharmaceuticals and biologics involve complex legal, scientific and factual questions. To date, no consistent policy has emerged regarding the breadth of biotechnology patent claims that are granted by the U.S. Patent and Trademark Office (“USPTO”) or enforced by the federal courts. Therefore, we do not know whether any particular patent applications will result in the issuance of patents, or that any patents issued to us will provide us with any competitive advantage. We also cannot be sure that we will develop additional proprietary products that are patentable. Furthermore, there is a risk that others will independently develop or duplicate similar technology or products or circumvent the patents issued to us.
There is a risk that third-parties may challenge our existing patents or claim that we are infringing their patents or proprietary rights. We could incur substantial costs in defending patent infringement suits or in filing suits against others to have their patents declared invalid or claim infringement. It is also possible that we may be required to obtain licenses from third-parties to avoid infringing third-party patents or other proprietary rights. We cannot be sure that such third-party licenses would be available to us on acceptable terms, if at all. If we are unable to obtain required third-party licenses, we may be delayed in or prohibited from developing, manufacturing or selling products requiring such licenses.
Although our patent filings include claims covering various features of our vaccine candidates, including composition, methods of manufacture and use, our patents do not provide us with complete protection against the development of competing products. Some of our know-how and technology is not patentable. To protect our proprietary rights in unpatentable intellectual property and trade secrets, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. These agreements may not provide meaningful protection for our trade secrets, know-how or other proprietary information.
Third parties may claim we infringe their intellectual property rights.
Our research, development and commercialization activities, including any vaccine candidates resulting from these activities, may be found to infringe patents owned by third-parties and to which we do not hold licenses or other rights. There may be rights we are not aware of, including applications that have been filed, but not published that, when issued, could be asserted against us. These third-parties could bring claims against us, and that may cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay research, development, manufacturing or sales of the product or biologic drug candidate that is the subject of the suit.
As a result of patent infringement claims, or in order to avoid potential claims, we may choose or be required to seek a license from the third-party. These licenses may not be available on acceptable terms, or at all. Even if we are able to obtain a license, the license would likely obligate us to pay license fees or royalties or both, and the rights granted to us might be non- exclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. All of the issues described above could also impact our collaborators, which would also impact the success of the collaboration and therefore us.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries.
We may become involved in litigation to protect or enforce our patents or the patents of our collaborators or licensors, which could be expensive and time-consuming.
Competitors may infringe our patents or the patents of our collaborators or licensors. As a result, we may be required to file suit to counter infringement for unauthorized use. This can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover its technology. An adverse determination of any litigation or defense proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at the risk of not issuing.
Even if we are successful, litigation may result in substantial costs and distraction to our management. Even with a broad portfolio, we may not be able, alone or with our collaborators and licensors, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the U.S.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of litigation, there could be public announcements of the results of hearings, motions or
other interim proceedings or developments. If investors perceive these results to be negative, the market price for our common stock could be significantly harmed.
The scope, validity, and ownership of our patent claims may be challenged in various venues and, if we do not prevail, our ability to exclude competitors may be harmed, potentially reducing our ability to succeed commercially.
We may be subject to a variety of challenges from third-parties that relate to the scope of the claims or to their validity. Such challenges can be mounted in post-grant review, ex parte re-examination, and inter partes review proceedings before the USPTO, or similar adversarial proceedings in other jurisdictions. If we are unsuccessful in any such challenge, the scope of our claims could be narrowed or could be invalidated. Any such outcome could impair our ability to exclude competitors from the market in those countries, potentially impacting our commercial success.
Our patents may be subject to various challenges related to ownership and inventorship, including interference or derivation proceedings. Third-parties may assert that they are inventors on our patents or that they are owners of the patents. While we perform inventorship analyses to insure that the correct inventors are listed on our patents, we cannot be certain that a court of competent jurisdiction would arrive at the same conclusions we do. If we are unsuccessful in defending against ownership or inventorship challenges, a court may require us to list additional inventors, may invalidate the patent, or may transfer ownership of the patent to a third-party. Any of these outcomes may harm our ability to exclude competitors and potentially impact our commercial success. Further, if ownership is transferred to a third-party we may be required to seek a license to those rights to preserve our exclusive ability to practice the invention. Such a license may not be available on commercially reasonable terms, or at all. If we are unable to obtain a license, we may be required to expend time, effort, and other resources to design around the patent. Any such license may be non-exclusive and if a competitor is able to obtain a license from the third-party, our ability to exclude that competitor from the market may be negatively impacted.
Even if we are ultimately successful, defending any such challenges may cause us to incur substantial expenses and may require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
We may need to license intellectual property from third-parties and, if our right to use the intellectual property we license is affected, our ability to develop and commercialize our vaccine candidates may be harmed.
We have in the past, and we expect in the future to license intellectual property from third-parties and that these licenses will be material to our business. We will not own the patents or patent applications that underlie these licenses, and we may not control either the prosecution or the enforcement of the patents. Under such circumstances, we may be forced to rely upon our licensors to properly prosecute and file those patent applications and prevent infringement of those patents.
While many of the licenses under which we have rights provide us with rights in specified fields, the scope of our rights under these and other licenses may be subject to dispute by our licensors or third-parties. In addition, our rights to use these technologies and practice the inventions claimed in the licensed patents and patent applications are subject to our licensors abiding by the terms of those licenses and not terminating them. Any of our licenses may be terminated by the licensor if we are in breach of a term or condition of the license agreement, or in certain other circumstances.
Further, any disputes regarding obligations in licenses may require us to take expensive and time-consuming legal action to resolve, and, even if we are successful, may delay our ability to commercialize products and generate revenue. Further, if we are unable to resolve license issues that arise we may lose rights to practice intellectual property that is required to make, use, or sell products. Any such loss could compromise our development and commercialization efforts for current or future product candidates and/or may require additional effort and expense to design around.
Our vaccine candidates and potential vaccine candidates will require several components that may each be the subject of a license agreement. The cumulative license fees and royalties for these components may make the commercialization of these vaccine candidates uneconomical.
If patent laws or the interpretation of patent laws change, our competitors may be able to develop and commercialize our discoveries.
Important legal issues remain to be resolved as to the extent and scope of available patent protection for biopharmaceutical products and processes in the U.S. and other important markets outside the U.S., such as Europe and Japan. In addition, foreign markets may not provide the same level of patent protection as provided under the U.S. patent system. Litigation or administrative proceedings may be necessary to determine the validity and scope of certain of our and others’ proprietary rights. Any such litigation or proceeding may result in a significant commitment of resources in the future and could force us to do one or more of the following: cease selling or using any of our products that incorporate the challenged intellectual property, which would adversely affect our revenue; obtain a license from the holder of the intellectual property right alleged to have been infringed, which license may not be available on reasonable terms, if at all; and redesign our products
to avoid infringing the intellectual property rights of third-parties, which may be time-consuming or impossible to do. In addition, changes in, or different interpretations of, patent laws in the U.S. and other countries may result in patent laws that allow others to use our discoveries or develop and commercialize our products. We cannot provide assurance that the patents we obtain or the unpatented technology we hold will afford us significant commercial protection.
If we do not obtain patent term extension and/or patent term adjustment in the United States under the Hatch- Waxman Act and similar extensions in foreign countries, our ability to exclude competitors may be harmed.
In the United States, the patent term is 20 years from the earliest U.S. non-provisional filing date. Extensions of patent term may be available under certain circumstances. Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, we may be able to extend the term of one patent that covers a marketed product under the Drug Price Competition and Patent Term Restoration Act of 1984, (the “Hatch-Waxman Amendments”) and similar legislation in the European Union.
The Hatch-Waxman Amendments permit patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. We may not receive any extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner.
Patent term covering our products may also be extended for time spent during the prosecution of the patent application in the USPTO. This extension is referred to as Patent Term Adjustment (“PTA”). The laws and regulations governing how the USPTO calculates the PTA is subject to change and changes in the law can reduce or increase any such PTA. Further, the PTA granted by the USPTO may be challenged by a third-party. If we do not prevail under such a challenge, the PTA may be reduced or eliminated, shortening the patent term, which may negatively impact our ability to exclude competitors.
Risks Related to Employee Matters, Managing Growth and Information Technology
Our business may be adversely affected if we do not successfully execute our business development initiatives.
We anticipate growing through both internal development projects, as well as external opportunities, which include the acquisition, partnering and in-licensing of products, technologies and companies or the entry into strategic alliances and collaborations. The availability of high quality opportunities is limited, and we may fail to identify candidates that we and our stockholders consider suitable or complete transactions on terms that prove advantageous. In order to pursue such opportunities, we may require significant additional financing, which may not be available to us on favorable terms, if at all. Even if we are able to successfully identify and complete acquisitions, like our business combinations with Novavax CZ (formerly Praha Vaccines) and Novavax AB, strategic transactions involve many risks, including, among others, those related to diversion of management’s attention from other business concerns, unanticipated expenses and liabilities, and increased complexity of our operations, which could prevent us from effectively exploiting acquired facilities, successfully integrating the acquired business and personnel, or fully realizing expected synergies.
To effectively manage our current and future potential growth, we will need to continue to enhance our operational, financial and management processes and to effectively expand, train and manage our employee base. Supporting our growth initiatives will require significant expenditures and management resources, including investments in research and development, manufacturing in-house and through third-party manufacturers and other areas of our business. Furthermore, we are in process of implementing a new enterprise resource planning system, which is intended to increase efficiency, but may result in disruptions or delays to our operations during its implementation. If we do not successfully manage our growth and do not successfully execute our growth initiatives, then our business and financial results may be adversely impacted, and we may incur asset impairment or restructuring charges.
Security breaches and other disruptions could compromise our information and expose us to liability, and our failure to comply with data protection laws and regulations could lead to government enforcement actions, which would cause our business and reputation to suffer.
In the ordinary course of our business, we and many of our current and future strategic partners, vendors, contractors, and consultants collect and store sensitive data, including intellectual property, our proprietary business information and data about our clinical participants, suppliers and business partners and personally identifiable information. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack by malicious third parties with a wide range of motives and expertise, including nation-states, organized criminal groups, “hacktivists,” patient groups, disgruntled current or former employees and others. Hacker attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be
vulnerable to such attacks or may be breached due to employee error or malfeasance. In 2020, several domestic and foreign security agencies announced that government actors or government-affiliated actors were specifically targeting organizations engaging in COVID-19 vaccine development and research. Our profile as an OWS recipient and progress on NVX-CoV2373 may result in greater risk of cyber attack. Any such breach could compromise our networks, our enterprise resource planning software and the information stored there could be accessed, publicly disclosed, lost or stolen. Furthermore, if our systems become compromised, we may not promptly discover the intrusion. Like other companies in our industry, we have experienced attacks to our data and systems, including malware and computer viruses. Attacks could have a material impact on our business, operations or financial results. Any access, disclosure or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, disrupt our operations, and damage our reputation, which could adversely affect our business. In addition, privacy and data protection laws may be interpreted and applied differently from country to country and may create inconsistent or conflicting requirements, which can increase the costs incurred by us in complying with such laws. The European Union’s GDPR, which greatly increases the jurisdictional reach of European Union law and became effective in May 2018, adds a broad array of requirements for handling personal data including the public disclosure of significant data breaches, and imposes substantial penalties for non-compliance of up to the greater of €20 million or 4% of global annual revenue for the preceding financial year. Our efforts to comply with GDPR and other privacy and data protection laws may impose significant costs and challenges that are likely to increase over time, and we could incur substantial penalties or litigation related to violations of existing or future data privacy laws and regulations.
The GDPR imposes strict rules on the transfer of personal data to countries outside the European Economic Area (“EEA”), including the United States and, in response, the EU and United States agreed in 2016 to a transfer framework for data transferred from the European Union to the United States, called the EU-US Privacy Shield. On July 16, 2020, however, the Court of Justice of the European Union issued a decision that declared the Privacy Shield framework, one of the primary mechanisms U.S. companies used to import personal information from Europe, invalid, and raised questions about whether the European Commission’s Standard Contractual Clauses (“SCCs”), an alternative to the Privacy Shield, can lawfully be used for cross-border data transfers. On June 4, 2021, the European Commission adopted new SCCs under the GDPR for personal data transfers outside of the EEA. Under this legal mechanism, we may have obligations to conduct transfer impact assessments for such cross-border data transfers and implement additional security measures. As we incorporate the new SCCs into our contractual arrangements, we may be required to expend significant resources to update our contractual arrangements and to comply with such obligations. If we are unable to implement a valid compliance mechanism for cross-border personal information transfers, we may face increased exposure to regulatory actions, substantial fines and injunctions against processing or transferring personal information from Europe. Inability to import personal information from Europe to the United States may significantly and negatively impact our business operations, including by limiting our ability to conduct clinical trials activities in Europe; limiting our ability to collaborate with contract research organizations, service providers, contractors and other companies subject to the GDPR; or requiring us to increase our data processing capabilities in Europe at significant expense.
Additionally, the CCPA, which became effective January 1, 2020, substantially expands privacy obligations of many businesses. The CCPA requires new disclosures to California consumers, imposes new rules for collecting or using information about minors, and affords consumers new abilities, such as the right to know whether the data is sold or disclosed and to whom, the right to request that a company delete personal information collected, the right to opt-out of the sale of personal information and the right to non-discrimination in terms of price or service when a consumer exercises a privacy right. If we fail to comply with these regulations, we could be subject to civil sanctions, including fines and penalties for noncompliance. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Moreover, a newly passed ballot initiative, the California Privacy Rights Act (“CPRA”), which will become operational in 2023, expands on the CCPA, creating new consumer rights and protections, including the right to correct personal information, the right to opt out of the use of personal information in automated decision making, the right to opt out of “sharing” consumer’s personal information for cross-context behavioral advertising, and the right to restrict use of and disclosure of sensitive personal information, including geolocation data to third parties. We will need to evaluate and potentially update our privacy program to ensure compliance with the CPRA and may incur additional costs and expenses in our effort to comply.
Collaborations and contracts of our wholly owned subsidiaries Novavax AB and Novavax CZ, with regional partners, such as SIIPL and Cadila, as well as with international providers, expose us to additional risks associated with doing business outside the U.S.
Swedish-based Novavax AB and Czech Republic-based Novavax CZ are wholly owned subsidiaries of Novavax, Inc. We also have established a manufacturing and distribution agreement with SIIPL, formed a joint venture with Cadila in India and have entered into other agreements and arrangements with foreign governments and companies in other countries. We plan to continue to enter into collaborations or partnerships with companies, non-profit organizations and local governments in various parts of the world. Risks of conducting business outside the U.S. include negative consequences of:
•the costs associated with seeking to comply with multiple regulatory requirements that govern our ability to develop, manufacture and sell products in local markets;
•failure to comply with anti-bribery laws such as the U.S. Foreign Corrupt Practices Act and similar anti-bribery laws in other jurisdictions;
•new or changes in interpretations of existing trade protections measures, including tariffs, embargoes and import and export licensing requirements;
•difficulties in and costs of staffing, managing and operating our international operations;
•changes in environmental, health and safety laws;
•fluctuations in foreign currency exchange rates;
•new or changes in interpretations of existing tax laws;
•political instability and actual or anticipated military or potential conflicts;
•economic instability, inflation, recession and interest rate fluctuations;
•minimal or diminished protection of intellectual property in many jurisdictions; and
•possible nationalization and expropriation.
These risks, individually or in the aggregate, could have a material adverse effect on our business, financial conditions, results of operations and cash flows.
If we are unable to attract or retain key management or other personnel, our business, operating results and financial condition could be materially adversely affected.
We depend on our senior executive officers, as well as key scientific and other personnel. The loss of these individuals could harm our business and significantly delay or prevent the achievement of research, development or business objectives. Turnover in key executive positions resulting in lack of management continuity and long-term history with our Company could result in operational and administrative inefficiencies and added costs.
We may not be able to attract qualified individuals for key positions on terms acceptable to us. Competition for qualified employees is intense among pharmaceutical and biotechnology companies, and the loss of qualified employees, or an inability to attract, retain and motivate additional highly skilled employees could hinder our ability to complete clinical trials successfully and otherwise develop marketable products.
We also rely from time to time on outside advisors who assist us in formulating our research and development and clinical strategy. We may not be able to attract and retain these individuals on acceptable terms, which could delay our development efforts.
Risks Related to Our Convertible Senior Notes
Servicing our 3.75% convertible senior unsecured notes due 2023 requires a significant amount of cash, and we may not have sufficient cash flow to pay our debt.
In 2016, we issued $325 million aggregate principal amount of Notes. Our ability to make scheduled payments of the principal of, to pay interest on, or to refinance our indebtedness, including the Notes, depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. We do not expect our business to be able to generate cash flow from operations sufficient to service our debt and make necessary capital expenditures and may therefore be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness, which is non-callable and matures in 2023, will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations, and limit our flexibility in planning for and reacting to changes in our business.
We may not have the ability to raise the funds necessary to repurchase the Notes as required upon a fundamental change, and our future debt may contain limitations on our ability to repurchase the Notes.
Holders of the Notes will have the right to require us to repurchase their Notes for cash upon the occurrence of a fundamental change at a fundamental change repurchase price equal to 100% of the principal amount of the Notes to be repurchased, plus accrued and unpaid interest, if any. A fundamental change may also constitute an event of default or prepayment under, and result in the acceleration of the maturity of, our then-existing indebtedness. We cannot assure that we will have sufficient financial resources, or will be able to arrange financing, to pay the fundamental change repurchase price in cash with respect to any Notes surrendered by holders for repurchase upon a fundamental change. In addition, restrictions in our then existing credit facilities or other indebtedness, if any, may not allow us to repurchase the Notes upon a fundamental change. Our failure to repurchase the Notes upon a fundamental change when required would result in an event of default with respect to the Notes which could, in turn, constitute a default under the terms of our other indebtedness, if any. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and repurchase the Notes.
Capped call transactions entered into in connection with our Notes may affect the value of our common stock.
In connection with our Notes, we entered into capped call transactions (the “capped call transactions”) with certain financial institutions. The capped call transactions are expected to generally reduce the potential dilution upon conversion of the Notes into shares of our common stock.
In connection with establishing their initial hedges of the capped call transactions, these financial institutions or their respective affiliates entered into various derivative transactions with respect to our common stock and/or to purchase our common stock. The financial institutions, or their respective affiliates, may modify their hedge positions by entering into or unwinding various derivatives with respect to our common stock and/or purchasing or selling our common stock or other securities of ours in secondary market transactions prior to the maturity of the Notes. This activity could also cause or avoid an increase or a decrease in the market price of our common stock or the Notes, which could affect the value of our common stock.
Risks Related to Ownership of Our Common Stock
Because our stock price has been and will likely continue to be highly volatile, the market price of our common stock may be lower or more volatile than expected.
Our stock price has been highly volatile. From January 1, 2021 through July 31, 2021, the closing sale price of our common stock has been as low as $112.98 per share and as high as $319.93 per share. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may cause the market price of our common stock to be lower or more volatile than expected.
Furthermore, given the global focus on the COVID-19 pandemic and our investment in developing a COVID-19 vaccine, information in the public arena on this topic, whether or not accurate, has had and will likely continue to have an outsized impact (positive or negative) on our stock price. Information related to our development, manufacturing, regulatory and commercialization efforts with respect to NVX-CoV2373, or information regarding such efforts by competitors with respect to their COVID-19 vaccines and vaccine candidates, may meaningfully impact our stock price. As a result of this volatility, you may not be able to sell your common stock at or above your initial purchase price. The market price of our common stock may be influenced by many other factors, including:
•future announcements about us or our collaborators or competitors, including the results of testing, technological innovations or new commercial products;
•clinical trial results;
•delays in making regulatory submissions;
•depletion of our cash reserves;
•sale of equity securities or issuance of additional debt;
•announcement by us of significant strategic partnerships, collaborations, joint ventures, capital commitments or acquisitions;
•changes in government regulations;
•impact of competitor successes and in particular development success of vaccine candidates that compete with our own vaccine candidates;
•developments in our relationships with our collaboration and funding partners;
•announcements relating to health care reform and reimbursement levels for new vaccines and other matters affecting our business and results, regardless of accuracy;
•sales of substantial amounts of our stock by us or existing stockholders (including stock by insiders or 5% stockholders);
•development, spread or new announcements related to pandemic diseases;
•litigation;
•public concern as to the safety of our products;
•significant set-backs or concerns with the industry or the market as a whole;
•regulatory inquiries, reviews and potential action, including from the FDA or the SEC;
•recommendations by securities analysts or changes in earnings estimates; and
•the other factors described in this Risk Factors section.
In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation often has been instituted against that company. Such litigation, if instituted against us, could cause us to incur substantial costs to defend such claims and divert management’s attention and resources, which could seriously harm our business, financial condition, and results of operations, and prospects.
Raising additional capital by issuing securities or through collaboration and licensing arrangements may cause dilution to existing stockholders or require us to relinquish rights to our technologies or vaccine candidates.
If we are unable to partner with a third-party to advance the development of one or more of our vaccine candidates, we will need to raise money through additional debt or equity financings. To the extent that we raise additional capital by issuing equity securities, our stockholders will experience immediate dilution, which may be significant. There is also a risk that such equity issuances may cause an ownership change under the Internal Revenue Code of 1986, as amended, and similar state provisions, thus limiting our ability to use our net operating loss carryforwards and credits. To the extent that we raise additional capital through licensing arrangements or arrangements with collaborative partners, we may be required to relinquish, on terms that may not be favorable to us, rights to some of our technologies or vaccine candidates that we would otherwise seek to develop or commercialize ourselves. In addition, economic conditions may also negatively affect the desire or ability of potential collaborators to enter into transactions with us. They may also have to delay or cancel research and development projects or reduce their overall budgets.
Provisions of our Second Amended and Restated Certificate of Incorporation and Amended and Restated By-Laws and Delaware law could delay or prevent the acquisition of the Company, even if such acquisition would be beneficial to stockholders, and could impede changes in our Board.
Provisions in our organizational documents could hamper a third-party’s attempt to acquire, or discourage a third-party from attempting to acquire control of, the Company. Stockholders who wish to participate in these transactions may not have the opportunity to do so. Our organizational documents also could limit the price investors are willing to pay in the future for our securities and make it more difficult to change the composition of our Board in any one year. For example, our organizational documents provide for a staggered board with three classes of directors serving staggered three-year terms and advance notice requirements for stockholders to nominate directors and make proposals.
As a Delaware corporation, we are also afforded the protections of Section 203 of the Delaware General Corporation Law, which will prevent us from engaging in a business combination with a person who acquires at least 15% of our common
stock for a period of three years from the date such person acquired such common stock, unless advance board or stockholder approval was obtained.
Any delay or prevention of a change of control transaction or changes in our Board or management could deter potential acquirers or prevent the completion of a transaction in which our stockholders could receive a substantial premium over the then current market price for their shares.
We have never paid dividends on our capital stock, and we do not anticipate paying any such dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We currently anticipate that we will retain all of our earnings for use in the development of our business and do not anticipate paying any cash dividends in the foreseeable future. As a result, capital appreciation, if any, of our common stock would be the only source of gain for stockholders until dividends are paid, if at all.
General Risk Factors
Litigation could have a material adverse impact on our results of operation and financial condition.
In addition to intellectual property litigation, from time to time, we may be subject to other litigation. Regardless of the merits of any claims that may be brought against us, litigation could result in a diversion of management’s attention and resources and we may be required to incur significant expenses defending against these claims. If we are unable to prevail in litigation, we could incur substantial liabilities. Where we can make a reasonable estimate of the liability relating to pending litigation and determine that it is probable, we record a related liability. As additional information becomes available, we assess the potential liability and revise estimates as appropriate. However, because of uncertainties relating to litigation, the amount of our estimates could be wrong.
We or the third parties upon whom we depend may be adversely affected by natural or man-made disasters or public health emergencies, such as the COVID-19 pandemic.
Our operations, and those of our clinical research organizations, contract manufacturing organizations, vendors of materials needed in manufacturing, collaboration partners, distributors and other third parties upon whom we depend, could be subject to fires, extreme weather conditions, earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, war, political unrest, sabotage or terrorism and other natural or man-made disasters, as well as public health emergencies, such as the COVID-19 pandemic. The occurrence of any of these business disruptions could prevent us from using all or a significant portion of our facilities and it may be difficult or impossible for us to continue certain activities for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event and we may incur substantial expenses and delays as a result. Our ability to manufacture our product candidates and obtain necessary clinical supplies for our product candidates could be disrupted if the operations of our contract manufacturing organizations or suppliers are affected by a natural or man-made disaster, or a public health emergency.
The outbreak of COVID-19 may materially and adversely affect our business and our financial results.
The COVID-19 pandemic continues to present substantial global economic and public health challenges, which may materially and adversely impact our business, financial condition and results of operations. In response to COVID-19, various aspects of our business operations have been, and could continue to be, disrupted. We continue to implement a work from home policy, with our administrative employees working outside of our offices, and on-site staff restricted to only those required to execute certain laboratory and related support activities. Working remotely could increase our cybersecurity risk, create data accessibility concerns, and make us more susceptible to communication disruptions, any of which could adversely impact our business operations. In addition, as a result of state or local restrictions, our on-site staff conducting research and development may not be able to access our laboratories, and these core activities may be significantly limited or curtailed, possibly for extended periods of time. Travel restrictions and other governmental measures may also result in a disruption or delay in the performance of our third-party contractors and suppliers. If such third parties are unable to adequately satisfy their contractual commitments to us in a timely manner, our business could be adversely affected. Furthermore, while some jurisdictions have recently started to phase out restrictions imposed on commercial activities at varying degrees, a resurgence of COVID-19, coupled with a potential surge in variant strains of COVID-19, in certain geographies could result in restrictions being reinstated.
Our clinical trials, whether planned or ongoing, may be affected by the COVID-19 pandemic. Study procedures (particularly any procedures that may be deemed non-essential), site initiation, participant recruitment and enrollment, participant dosing, shipment of our product candidates, distribution of clinical trial materials, study monitoring, site inspections and data analysis may be paused or delayed due to changes in hospital or research institution policies, federal, state or local
regulations, prioritization of hospital and other medical resources toward efforts to treat or prevent COVID-19, or other reasons related to the pandemic. In addition, there could be a potential effect of COVID-19 to the operations of the FDA or other health authorities, which could result in delays of reviews and approvals, including with respect to our product candidates. Any prolongation or de-prioritization of our clinical trials or delay in regulatory review resulting from such disruptions could materially affect the development and study of our product candidates.
The trading prices for our common stock and that of other biopharmaceutical companies have been highly volatile due to the COVID-19 pandemic, especially as a result of investor concerns and uncertainty related to the impact of the outbreak on the economies of countries worldwide. These broad market and industry fluctuations, as well as general economic, political and market conditions, may negatively impact the market price of shares of our common stock.
The COVID-19 pandemic continues to rapidly evolve. The extent to which the outbreak impacts our business, preclinical studies and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the emergence of variant strains, the duration of the pandemic, travel restrictions and social distancing in the U.S. and other countries, business closures or business disruptions and the effectiveness of actions taken in the U.S. and other countries to contain and treat the disease.
The United Kingdom’s withdrawal from the European Union could result in increased regulatory and legal complexity, which may make it more difficult for us to do business in the UK and/or Europe and impose additional challenges in securing regulatory approval of our product candidates in the UK and/or Europe.
The United Kingdom’s exit from the European Union as of January 31, 2020, with a transitional period up to December 31, 2020, commonly referred to as “Brexit”, has caused political and economic uncertainty, including in the regulatory framework applicable to our operations and vaccine candidates in the United Kingdom and the European Union, and this uncertainty may persist for years. Brexit could, among other outcomes, disrupt the free movement of goods, services and people between the United Kingdom and the European Union, and result in increased legal and regulatory complexities, as well as potential higher costs of conducting business in Europe. As one of the Brexit consequences, the EMA has relocated from the United Kingdom to the Netherlands. This has led to a significant reduction of the EMA workforce, which has resulted and could further result in significant disruption and delays in its administrative procedures, such as granting clinical trial authorization or opinions for marketing authorization, disruption of importation and export of active substance and other components of new drug formulations, and disruption of the supply chain for clinical trial product and final authorized formulations. As any European Union marketing authorization for NVX-CoV2373 would be issued after January 1, 2021, if at all, it would not be grandfathered in the UK. We therefore must seek to obtain a separate marketing authorization for the UK, increasing our regulatory burden.
The cumulative effects of the disruption to the regulatory framework may add considerably to the development lead time to marketing authorization and commercialization of products in the European Union and/or the United Kingdom. It is possible that there will be increased regulatory complexities, which can disrupt the timing of our clinical trials and regulatory approvals. In addition, changes in, and legal uncertainty with regard to, national and international laws and regulations may present difficulties for our clinical and regulatory strategy. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from commercializing our product candidates in the United Kingdom and/or the European Union and restrict our ability to generate revenues and achieve and sustain profitability.
In addition, as a result of Brexit, other European countries may seek to conduct referenda with respect to their continuing membership with the European Union. Given these possibilities and others we may not anticipate, as well as the absence of comparable precedent, it is unclear what financial, regulatory and legal implications the withdrawal of the United Kingdom from the European Union will have, how such withdrawal will affect us, and the full extent to which our business could be adversely affected.
We are increasingly a target for public scrutiny, and our business may be impacted by unfavorable publicity.
Given that COVID-19 represents an unprecedented urgent public health crisis, that we are developing NVX-CoV2373 as a COVID-19 vaccine candidate, and that we have received significant funding from the U.S. and foreign governments and other sources to support the development and potential commercialization of NVX-CoV2373, we have observed and are likely to continue to face significant public attention and scrutiny over the complex decisions we have made and will be making regarding the development, testing, manufacturing, allocation and pricing of NVX-CoV2373. If we are unable to successfully manage these risks, we could face significant reputational harm, which could negatively affect our stock price. The intense public interest, including speculation by the media, in the development of NVX-CoV2373 has caused significant volatility in our stock price, which we expect to continue as data and other information from our ongoing clinical trials become publicly available. If concerns should arise about the actual or anticipated efficacy or safety of any of our product candidates, such concerns could adversely affect the market’s perception of these candidates, which could lead to a decline in investors’ expectations and a decline in the price of our common stock.
The increasing use of social media platforms presents new risks and challenges to our business.
Social media is increasingly being used to communicate about pharmaceutical companies’ research, product candidates, and the diseases such product candidates are being developed to prevent. Social media practices in the pharmaceutical industry continue to evolve and regulations relating to such use are not always clear. This evolution creates uncertainty and risk of noncompliance with regulations applicable to our business, resulting in potential regulatory actions against us. For example, subjects may use social media channels to comment on their experience in an ongoing blinded clinical trial or to report an alleged adverse event. When such events occur, there is a risk that we fail to monitor and comply with applicable adverse event reporting obligations or we may not be able to defend our business or the public’s legitimate interests in the face of the political and market pressures generated by social media due to restrictions on what we may say about our investigational product candidates. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social media or networking website. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions, or incur reputational or other harm to our business.
Item 6. Exhibits
| | | | | | | | |
3.1
| | |
| | |
| 3.2 | | |
| | |
| 3.3 | | |
| | |
| 3.4 | | |
| | |
| 10.1* | | |
| | |
| 10.2* | | |
| | |
| 10.3 | | |
| | |
| 10.4* | | |
| | |
| 10.5*± | | |
| | |
| 10.6*± | | |
| | |
| 10.7*± | | |
| | |
| 31.1* | | |
| | |
| 31.2* | | |
| | |
| 32.1* | | |
| | |
| 32.2* | | |
| | |
| 101 | | The following financial information from our Quarterly Report on Form 10-Q for the quarter ended June 30, 2021, formatted in Inline Extensible Business Reporting Language (Inline XBRL): (i) the Consolidated Balance Sheets as of June 30, 2021 and December 31, 2020, (ii) the Consolidated Statements of Operations for the three- and six-month periods ended June 30, 2021 and 2020, (iii) the Consolidated Statements of Comprehensive Loss for the three- and six-month periods ended June 30, 2021 and 2020, (iv) the Consolidated Statements of Changes in Stockholders’ Equity (Deficit) for the three- and six-month periods ended June 30, 2021 and 2020, (v) the Consolidated Statements of Cash Flows for the six-month period ended June 30, 2021 and 2020, and (vi) the Notes to Consolidated Financial Statements. |
| | |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
| | |
___________________________________
*Filed or furnished herewith.
± Certain portions of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | | | |
| NOVAVAX, INC. |
| | |
| Date: August 5, 2021 | By: | /s/ Stanley C. Erck |
| | Stanley C. Erck
President and Chief Executive Officer
(Principal Executive Officer) |
| | |
| Date: August 5, 2021 | By: | /s/ John J. Trizzino |
| | John J. Trizzino
Executive Vice President, Chief Commercial Officer, Chief Business Officer and Interim Chief Financial Officer
(Principal Financial and Accounting Officer) |