UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): December 10, 2022
SANGAMO THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| | | | | | | | | | | | | | |
| | | | |
| Delaware | | 000-30171 | | 68-0359556 |
(State or other jurisdiction of
incorporation) | | (Commission
File Number) | | (IRS Employer
ID Number) |
7000 Marina Blvd., Brisbane, California 94005
(Address of principal executive offices) (Zip Code)
(510) 970-6000
(Registrant’s telephone number, including area code)
Not Applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| | | | | |
| ☐ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| | | | | |
| ☐ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| | | | | |
| ☐ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| | | | | |
| ☐ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act: | | | | | | | | | | | | | | |
| | | | |
| Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
| Common Stock, $0.01 par value per share | | SGMO | | Nasdaq Global Select Market |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§ 230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§ 240.12b-2 of this chapter).
Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Item 8.01 Other Events.
On December 10, 2022, Sangamo Therapeutics, Inc., or Company, presented updated preliminary proof-of-concept clinical data from the Phase 1/2 PRECIZN-1 study of BIVV003, a zinc finger nuclease gene-edited autologous cell therapy product candidate for the treatment of sickle cell disease, or SCD, at the 64th American Society for Hematology Annual Meeting and Exposition, or ASH. Also on December 11, 2022, the Company presented at ASH updated follow-up data from the Phase 1/2 Alta study of giroctocogene fitelparvovec, an investigational gene therapy for patients with moderately severe to severe hemophilia A in development with Pfizer Inc. Summaries of the data presented are below.
Summary of Updated Preliminary Safety, Tolerability and Efficacy Results from the Phase 1/2 PRECIZN-1 Study of BIVV003 Presented at ASH
•PRECIZN-1 is an ongoing, first-in-human, open label, single arm, multi-site study evaluating the safety, tolerability and efficacy of BIVV003 in patients with severe SCD (n=8; aged 18-40 years).
•Eligible patients underwent mobilization and apheresis with plerixafor. Autologous hematopoietic stem and progenitor cells, or HSPCs, were transfected ex vivo with zinc finger nuclease messenger ribonucleic acid to manufacture BIVV003. A single intravenous infusion was administered at least 72 hours after pre-conditioning with busulfan.
•Patients were monitored for stem cell engraftment and hematopoietic recovery, adverse events, clinical and laboratory hemolysis markers, total hemoglobin, or Hb, and fetal hemoglobin, or HbF, percentage of F cells and SCD-related events post‑BIVV003 infusion.
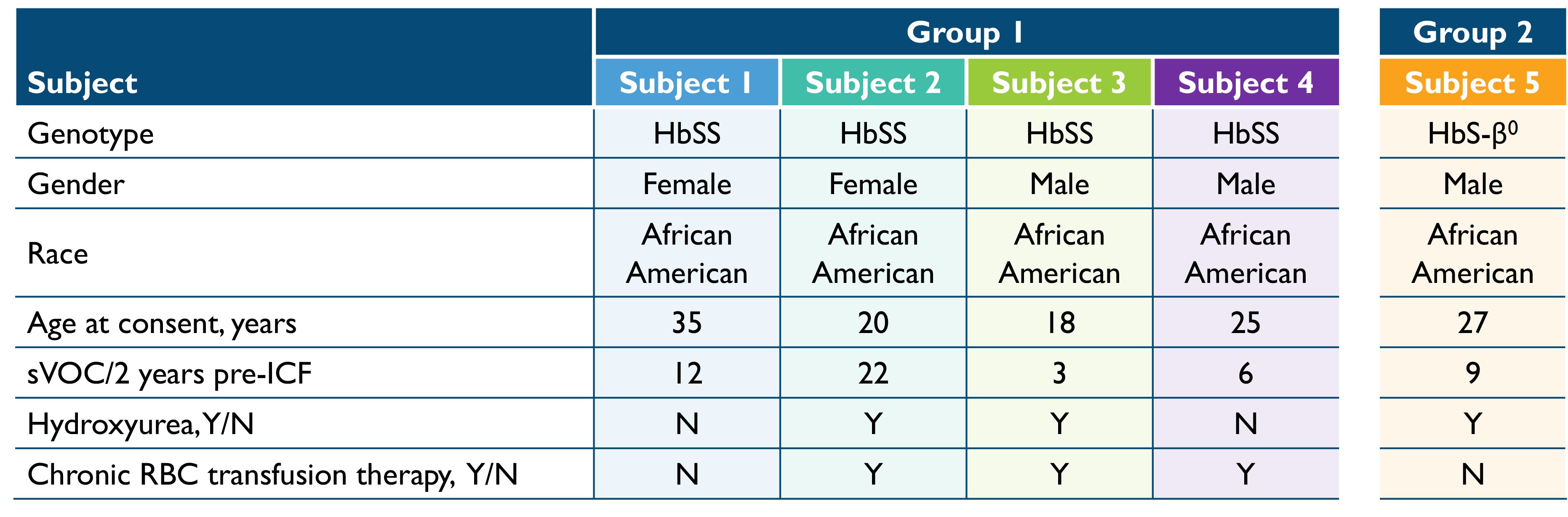
•Six patients achieved successful target yields of HSPCs. Five of the six patients achieving successful target yields of HSPCs had been infused with BIVV003 as of the September 30, 2022 cutoff date. Baseline characteristics of these five patients are in Table 1 below. The first four patients dosed received BIVV003 produced using the initial manufacturing process and are referred to in Table 1 below as Group 1. Group 1 patients have been followed for up to 30 months post-infusion. The patient referred to in Table 1 below as Group 2 received BIVV003 manufactured using improved methods that have been shown in internal experiments to increase the number of long-term progenitor cells in the final product. As of the cutoff date, the first patient treated in Group 2, or Patient 5, had been followed for 5 months. A second patient in Group 2 was dosed after the cutoff date.
•Four of five patients in aggregate across both Groups 1 and 2 improved clinically since BIVV003 infusion through the September 30, 2022, cutoff date.
In Group 1:
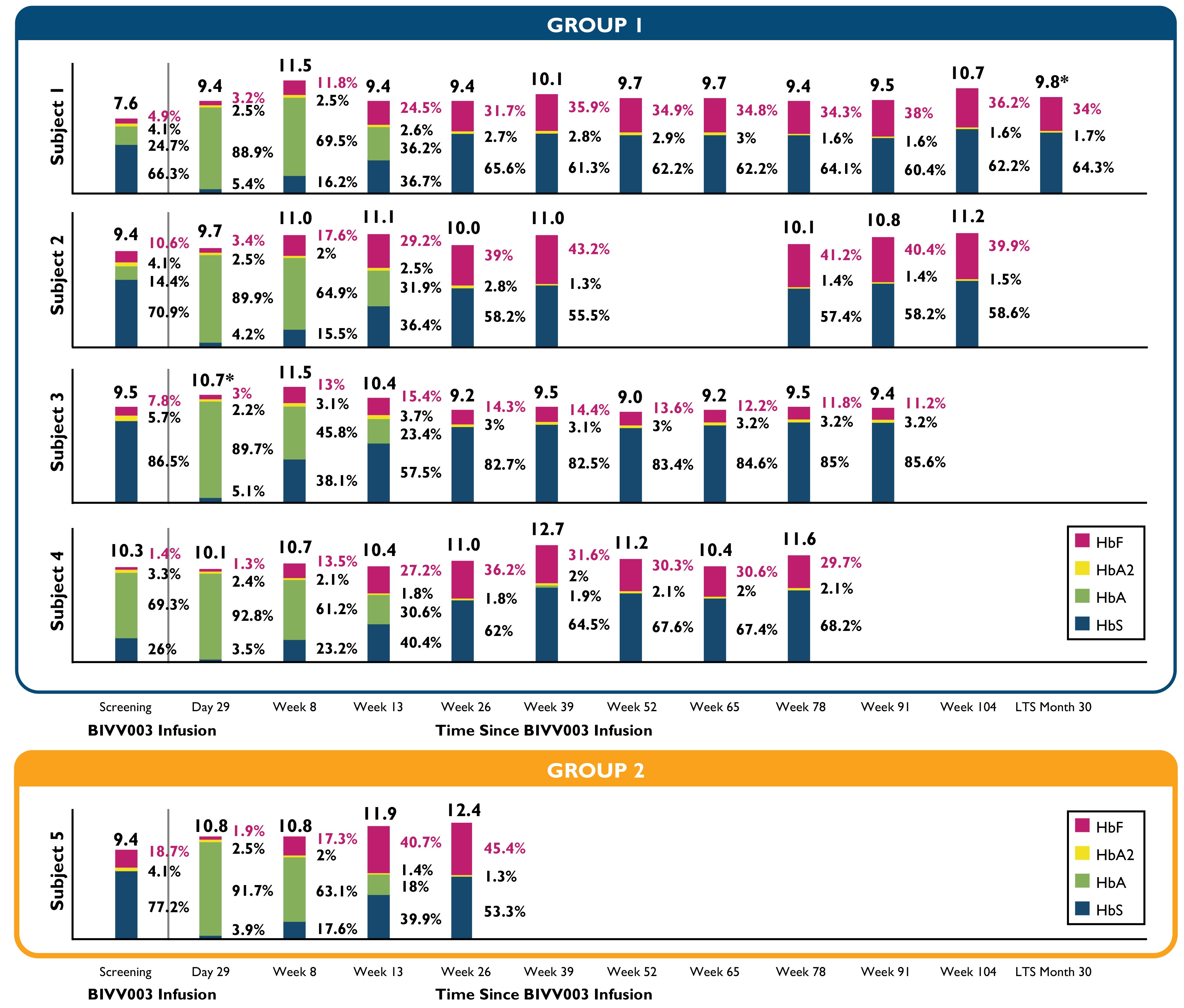
•The effects of BIVV003 infusion on total Hb and HbF levels were maintained up to 30 months.
•Three of the four patients had stable engraftment of zinc finger nuclease-modified HSPCs, resulting in sustained elevated HbF levels greater than 30% and an absence of severe vaso-occlusive crisis, or VOCs, post-BIVV003 administration.
In Group 2:
•Patient 5 received BIVV003 manufactured using improved methods that have been shown in internal experiments to increase the number of long-term progenitor cells in the final product.
•The HbF level of 45% and total Hb of 12.4 g/dL at week 26 post-infusion for Patient 5 in the latest sample collected post data cutoff were greater than the levels observed in Group 1 at week 26.
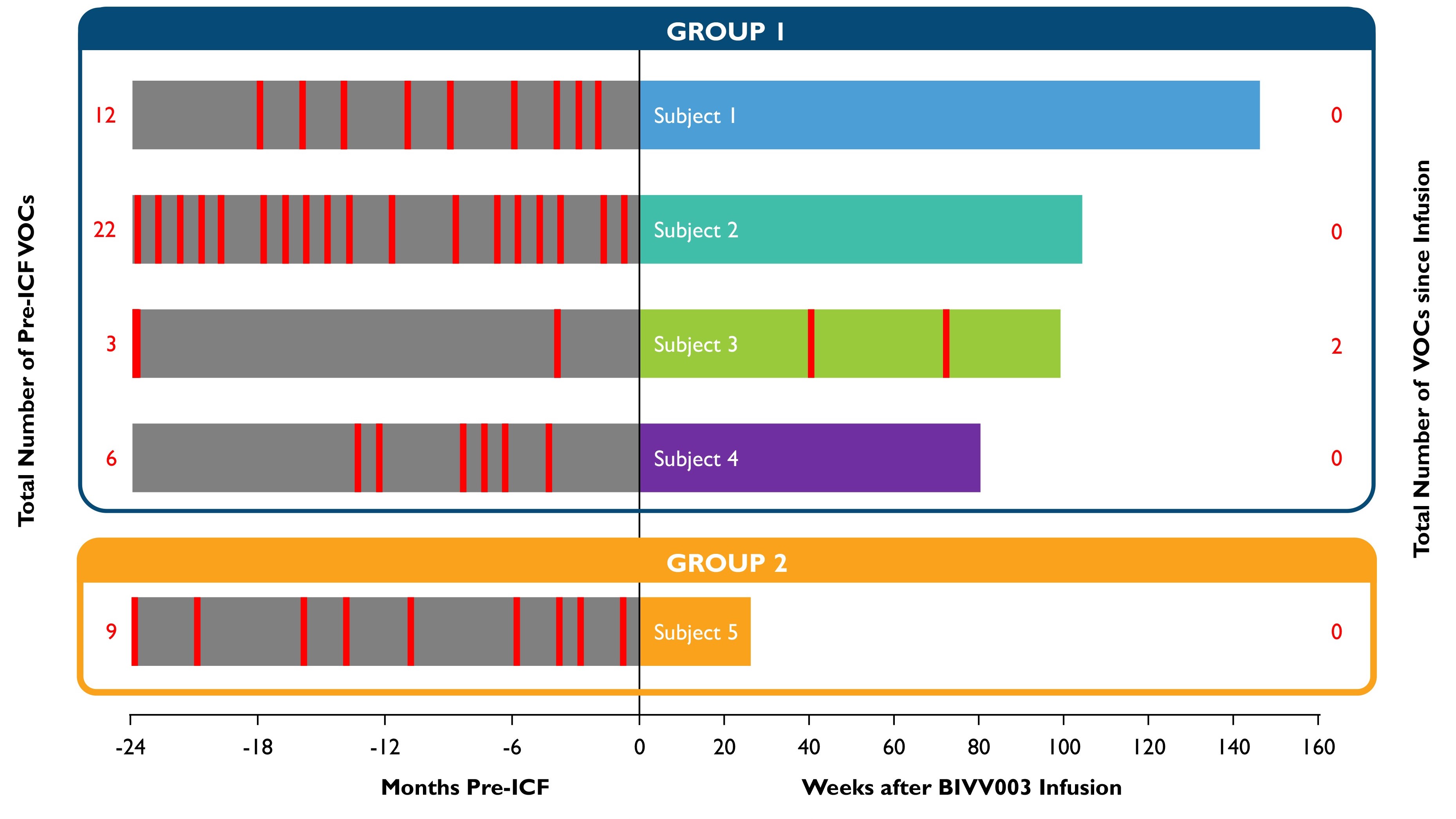
•As of the September 30, 2022 cutoff date, BIVV003 was generally well tolerated, and most adverse events reported in the screening, mobilization, apheresis and conditioning periods were SCD-related events. The investigator reported two serious adverse events of sickle cell anemia with a VOC as related to plerixafor, and one serious adverse event of nausea as related to busulfan. Nearly half of the adverse events reported after infusion of BIVV003 were related to busulfan. The investigator reported two serious adverse events of sickle cell anemia with a VOC nine months and 16 months after infusion in the one patient in Group 1 who had low HbF levels (11-14%). No other SCD-related serious adverse events were reported after infusion. No adverse events related to BIVV003 were reported by the investigator or sponsor. See Figure 2 below for VOCs reported before and after infusion of BIVV003.
Table 1: Baseline Characteristics of First 5 Subjects Dosed
VOC = vaso-occlusive crisis, ICF= informed consent form, RBC = red blood cell
Group 1 includes the first four patients dosed, who received BIVV003 produced using the initial manufacturing process.
Group 2 includes patient 5 who received BIVV003 manufactured using improved methods that have been shown in internal experiments to increase the number of long-term progenitor cells in the final product.
Table 2: Total Hb and Hb Fractionation
Total hemoglobin and hemoglobin fractionation at screening and post-BIVV003 infusion over time.
HbA = adult hemoglobin, HbA2 = variant adult hemoglobin, HbF = fetal hemoglobin, HbS = sickle hemoglobin
LTS= Long-term follow-up study
(*) indicates the Hb value from local lab, since the central lab value was not collected
Group 1 includes the first four patients dosed, who received BIVV003 produced using the initial manufacturing process.
Group 2 includes patient 5 who received BIVV003 manufactured using improved methods that have been shown in internal experiments to increase the number of long-term progenitor cells in the final product.
Table 3: Absence of VOC After BIVV003 Infusion
Number of severe vaso-occlusive crises (VOCs) reported in the 24 months before signing the study informed consent form (ICF) and in the post-BIVV003 infusion period.
Red lines represent severe VOCs; 2 severe VOCs occurring in the same month appear as one red line
Group 1 includes the first four patients dosed, who received BIVV003 produced using the initial manufacturing process.
Group 2 includes patient 5 who received BIVV003 manufactured using improved methods that have been shown in internal experiments to increase the number of long-term progenitor cells in the final product.
Summary of Updated Follow-up Data from the Phase 1/2 Alta Study of Giroctocogene Fitelparvovec Presented at ASH
•Alta is a Phase 1/2 single-dose multicenter dose-ranging study to assess the safety and tolerability of giroctocogene fitelparvovec in adults with severe hemophilia A.
•Patients were monitored for adverse events, change in circulating Factor VIII, or FVIII, activity, change from baseline in use of FVIII replacement therapy, change in frequency and severity of bleeding episodes, measurement of FVIII inhibitor levels and vector shedding in bodily fluids.
•Maintenance of FVIII activity in the mild range (>5%) or greater improves outcomes for patients with severe hemophilia A.
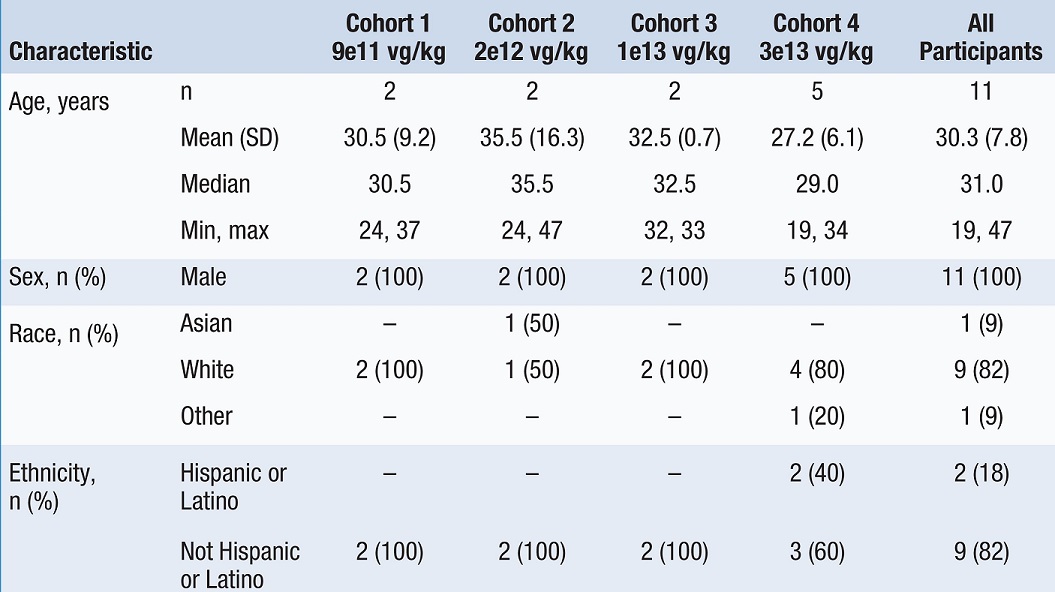
•Eleven male patients participated in the study overall, with five patients in the 3e13-vg/kg highest dose cohort. See Table 1 below for baseline patient demographics. As of the September 6, 2022 cutoff date, all patients had been followed for 153 to 263 weeks and all participants have completed at least 35 months of follow-up.
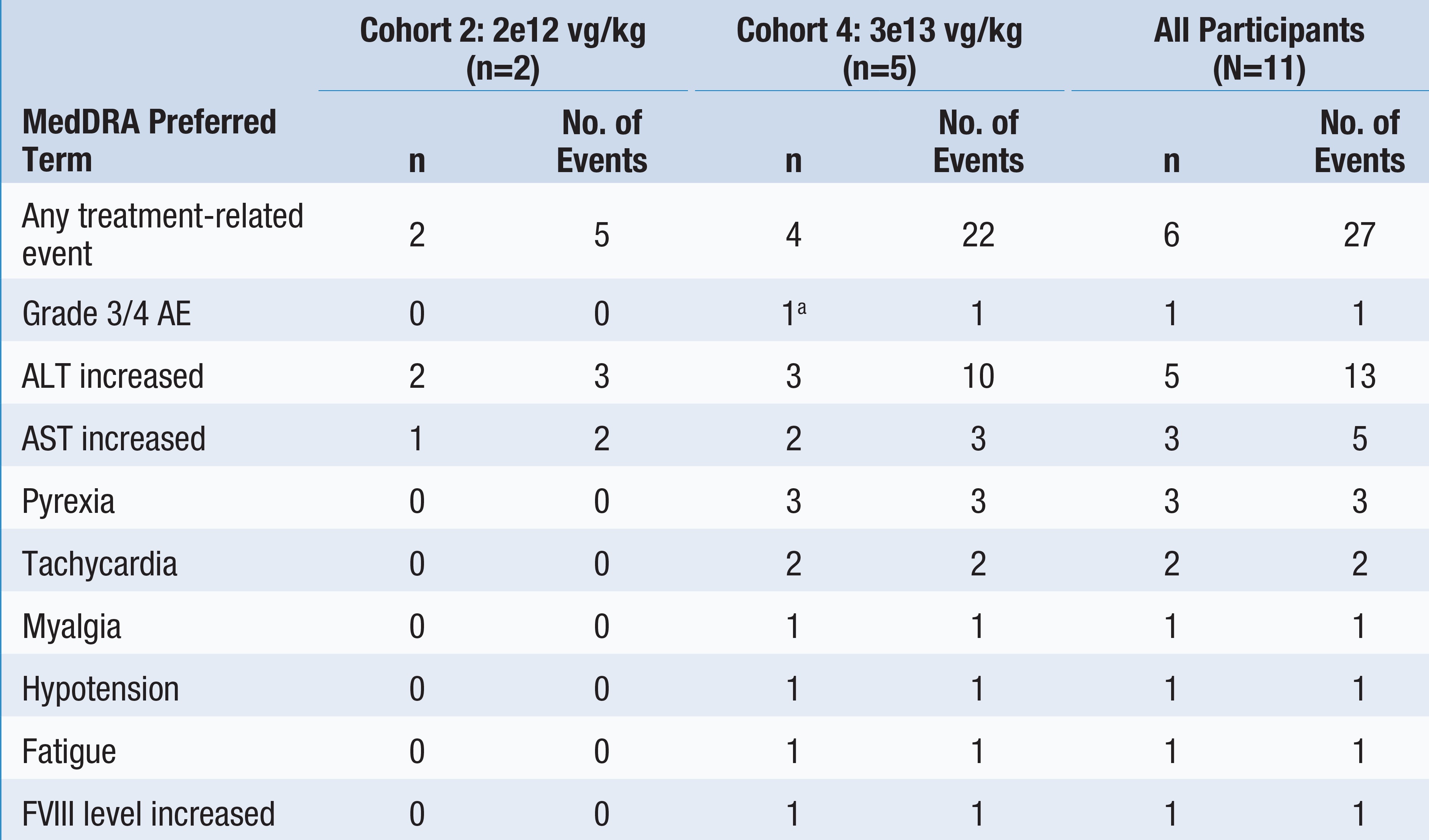
•As of the September 6, 2022 cutoff date, six of the eleven patients had experienced treatment-related adverse events, including four of the five patients in the highest dose cohort. The most commonly reported treatment-related adverse events included elevated liver enzymes and infusion-related reactions: increased alanine aminotransferase, or ALT (5/11 (45.5%) overall; 3/5 (60.0%) in the highest dose cohort), increased aspartate aminotransferase, or AST (3/11 (27.3%) overall; 2/5 (40.0%) in the highest dose cohort), pyrexia (3/11 (27.3%) overall; 3/5 (60.0%) in the highest dose cohort), and tachycardia (2/11 (18.2%) overall; 2/5 (40.0%) in the highest dose cohort).
•Treatment-related serious adverse events were reported in one patient in the highest dose cohort who experienced grade 3 hypotension and fever with onset approximately six hours after giroctocogene fitelparvovec infusion; the events fully resolved with treatment and did not delay post-infusion discharge the next day. See Table 2 below for more details on treatment-related adverse events.
•As of the September 6, 2022 cutoff date, no confirmed FVIII inhibitor development occurred, and no thrombotic events, neoplastic events, abnormal alfa-fetoprotein and/or liver masses were reported.
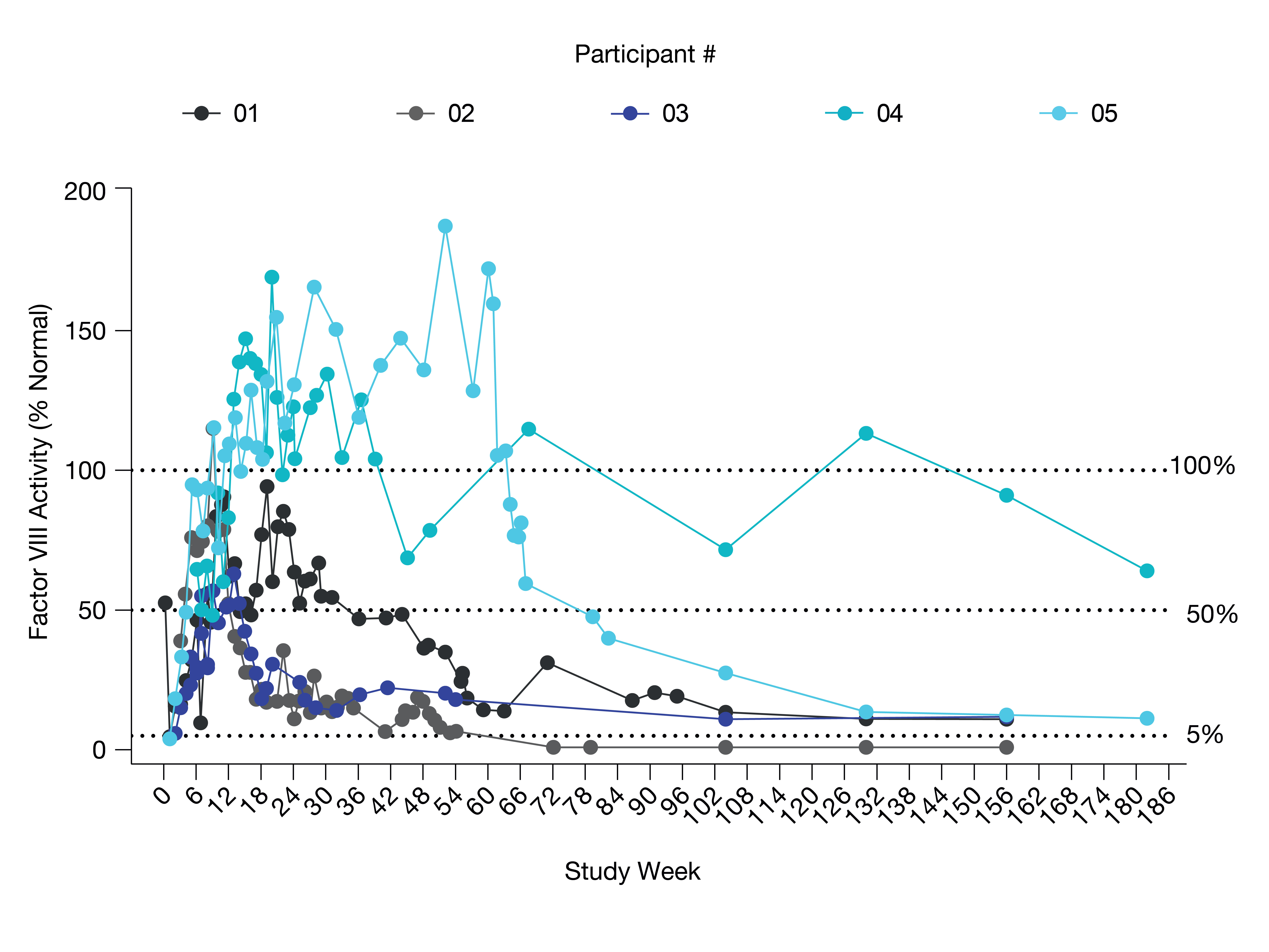
•All five patients in the highest dose cohort demonstrated FVIII activity as shown in Table 3 below through week 156. Mean FVIII activity at week 156 was 25.5% of normal as measured by chromogenic clotting assay at the central laboratory. In this highest dose cohort, the annualized bleeding rate, meaning the number of all bleeding episodes starting three weeks after infusion divided by the observation period in years, was zero for the first year post-infusion and the mean overall annual bleeding rate throughout the total duration of follow-up was 1.2 as of the September 6, 2022 cutoff date.
•In this highest dose cohort, two patients experienced bleeding events necessitating treatment with exogenous FVIII: one patient experienced 17 bleeding events (8 traumatic, 5 spontaneous, 4 unknown), and one patient experienced one bleeding event in a target joint, circumstances unknown. No participants in this highest dose cohort have resumed prophylaxis as of the cutoff date.
•Additional follow-up is required to assess durability of therapeutic effect and other long-term effects of giroctocogene fitelparvovec, such as impact on overall patient liver health.
Table 1: Baseline Patient Demographics by Giroctocogene Fitelparvovec Dose Cohort
Data cut: September 6, 2022
Max = maximum, Min = minimum, SD = standard deviation, vg = vector genomes
Table 2: Treatment-Related Adverse Events by Giroctocogene Fitelparvovec Dose Cohort
Data cut: September 6, 2022
(a) One patient experienced grade 3 hypotension that was considered related to study drug and resolved with treatment
AE = adverse event, ALT = alanine transaminase, AST = aspartate aminotransferase, vg = vector genomes
Table 3: FVIII Activity Levels (Measured with Chromogenic Assay) for Individuals in the Giroctocogene Fitelparvovec Highest Dose Cohort (3e13 vg/kg, Cohort 4)
Latest available FVIII values from September 6, 2022 data cut
FVIII = Factor VIII, vg = vector genomes
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| | | | | | | | | | | | | | | | | | | | |
| | | | | | |
| | | | SANGAMO THERAPEUTICS, INC. |
| | | |
| Dated: December 12, 2022 | | | | By: | | /s/ SCOTT B. WILLOUGHBY |
| | | | Name: | | Scott B. Willoughby |
| | | | Title: | | Senior Vice President, General Counsel and Corporate Secretary |