Filed Pursuant to Rule 424(b)(3) and 424(c) Commission File No. 333-150580 |
Manhattan Pharmaceuticals, Inc.
33,928,571 Shares
Common Stock
This prospectus supplement supplements the prospectus dated October 15, 2008, which relates to the shares of our common stock that may be sold by the selling securityholders named therein.
This prospectus supplement should be read in connection with, and may not be delivered or utilized without, the prospectus dated October 15, 2008 and the prospectus supplements dated November 21, 2008, November 26, 2008, January 21, 2009, January 22, 2009, January 28, 2009 and February 4, 2009. This prospectus supplement is qualified by reference to the prospectus and the prospectus supplements, except to the extent that the information in this prospectus supplement updates or supersedes the information contained in the prospectus dated October 15, 2008 or the prospectus supplements dated November 21, 2008, November 26, 2008, January 21, 2009, January 22, 2009, January 28, 2009 and February 4, 2009.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus supplement. Any representation to the contrary is a criminal offense.
The date of this prospectus supplement is April 14, 2009
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
x Annual Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the fiscal year ended December 31, 2008
¨ Transition Report pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 for the transition period from ___to___
Commission File Number 1-32639
MANHATTAN PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 36-3898269 (I.R.S. Employer Identification No.) |
48 Wall Street, 11thFloor, New York, New York (Address of Principal Executive Offices) | 10005 (Zip Code) |
(212) 582-3950
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered |
| Common Stock, $0.001 par value | OTC Bulletin Board |
Securities registered pursuant to Section 12(g) of the Exchange Act: None |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. ¨ Yes x No
Indicate by check mark whether the registrant (1) filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ | Accelerated filer ¨ |
Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ¨ Yes x No
The aggregate market value of the voting and non-voting common equity of the registrant held by non-affiliates of the registrant on June 30, 2008 based on the closing price of the common stock as reported on the OTC Bulletin Board on such date was $7,778,172.
As of March 17, 2009 there were 70,624,232 outstanding shares of common stock, par value $.001 per share.
TABLE OF CONTENTS
| Page | ||
| PART I | ||
| Item 1 | Business | 3 |
| Item 1A | Risk Factors | 18 |
| Item 2 | Properties | 28 |
| Item 3 | Legal Proceedings | 28 |
| PART II | ||
| Item 5 | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 29 |
| Item 7 | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 35 |
| Item 8 | Financial Statements and Supplementary Data | 52 |
| Item 9 | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 52 |
| Item 9A(T) | Controls and Procedures | 52 |
| Item 9B | Other Information | 53 |
| PART III | ||
| Item 10 | Directors, Executive Officers and Corporate Governance | 54 |
| Item 11 | Executive Compensation | 58 |
| Item 12 | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 62 |
| Item 13 | Certain Relationships and Related Transactions, and Director Independence | 64 |
| Item 14 | Principal Accounting Fees and Services | 68 |
| PART IV | ||
| Item 15 | Exhibits and Financial Statement Schedules | 69 |
| Index to Financial Statements | F-1 |
1
References to the “Company,” the “Registrant,” “we,” “us,” or “our” or in this Annual Report on Form 10-K refer to Manhattan Pharmaceuticals, Inc., a Delaware corporation, and our consolidated subsidiaries, together taken as a whole, unless the context indicates otherwise.
Forward-Looking Statements
This annual report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Exchange Act. Any statements about our expectations, beliefs, plans, objectives, assumptions or future events or performance are not historical facts and may be forward-looking. These statements are often, but not always, made through the use of words or phrases such as “anticipate,” “estimate,” “plan,” “project,” “expect,” “may,” “intend” and similar words or phrases. Accordingly, these statements involve estimates, assumptions and uncertainties that could cause actual results to differ materially from those expressed in them. These statements are therefore subject to risks and uncertainties, known and unknown, which could cause actual results and developments to differ materially from those expressed or implied in such statements. Such risks and uncertainties relate to, among other factors:
| · | our ability to obtain adequate financing to continue our operations; |
| · | the development of our pharmaceutical product candidates; |
| · | the regulatory approval of our pharmaceutical product candidates; |
| · | our use of clinical research centers and other contractors; |
| · | our ability to find collaborative partners for research, development and commercialization of potential products; |
| · | acceptance of our products by doctors, patients or payers; |
| · | our ability to market any of our products; |
| · | our history of operating losses; |
| · | our ability to compete against other companies and research institutions; |
| · | our ability to secure adequate protection for our intellectual property; |
| · | our ability to attract and retain key personnel; |
| · | availability of reimbursement for our products; |
| · | the effect of potential strategic transactions on our business; and |
| · | the volatility of our stock price. |
Further, any forward-looking statement speaks only as of the date on which it is made, and we undertake no obligation to update any forward-looking statement or statements to reflect events or circumstances after the date on which such statement is made or to reflect the occurrence of unanticipated events. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
2
PART I
ITEM 1. BUSINESS
Overview
We are a specialty healthcare product company focused on developing and commercializing innovative treatments for underserved patient populations. We aim to acquire rights to these technologies by licensing or otherwise acquiring an ownership interest, funding their research and development and eventually either bringing the technologies to market or out-licensing. In the short term we are focusing our efforts on the commercialization of the two product candidates we currently have in development: Hedrin™, a novel, non-insecticide treatment for pediculosis (head lice), which we are developing through a joint venture, and a topical product for the treatment of psoriasis. Longer term, we intend to acquire and commercialize low risk, quick to market products, specifically products that could be marketed over-the-counter (“OTC”), treat everyday maladies, are simple to manufacture, and/or could be classified as medical devices by the FDA.
During 2007, we discontinued development of Oleoyl-estrone and Propofol Lingual Spray. In March 2009, we discontinued development of Altoderm and Altolyn. We have not received regulatory approval for, or generated commercial revenues from marketing or selling any drugs.
Our executive offices are located at 48 Wall Street, 11th floor, New York, NY 10005 USA. Our telephone number is (212) 582-3950 and our internet website address is www.manhattanpharma.com.
Corporate History – Merger Transaction(s)
We were incorporated in Delaware in 1993 under the name “Atlantic Pharmaceuticals, Inc.” and, in March 2000, we changed our name to “Atlantic Technology Ventures, Inc.” In 2003, we completed a “reverse acquisition” of privately held “Manhattan Research Development, Inc.” In connection with this transaction, we also changed our name to “Manhattan Pharmaceuticals, Inc.” From an accounting perspective, the accounting acquirer is considered to be Manhattan Research Development, Inc. and accordingly, the historical financial statements are those of Manhattan Research Development, Inc.
During 2005 we merged with Tarpan Therapeutics, Inc. (“Tarpan”). Tarpan was a privately held New York based biopharmaceutical company developing dermatological therapeutics. This transaction was accounted for as a purchase of Tarpan by the Company.
Active Research and Development Programs
Hedrin™
In June 2007, Manhattan Pharmaceuticals entered into an exclusive license agreement with Thornton & Ross Ltd. (“T&R”) and Kerris, S.A. (“Kerris”) for a product candidate called Hedrin (the “Hedrin License Agreement”). We acquired an exclusive North American license to certain patent rights and other intellectual property relating to Hedrin, a non-insecticide product candidate for the treatment of head lice. In addition, and at the same time, we also entered into a Supply Agreement with T&R pursuant to which T&R will be the Company’s exclusive supplier of Hedrin product (the “Hedrin Supply Agreement”).
3
In February 2008, Manhattan Pharmaceuticals announced that it had entered into a joint venture agreement with Nordic Biotech Advisors ApS (“Nordic”) to develop and commercialize Hedrin. The 50/50 joint venture entity, H Pharmaceuticals (“H Pharmaceuticals” or the “Hedrin JV”), now owns, is developing, and is working to secure commercialization partners for Hedrin in North America. Manhattan Pharmaceuticals manages the day-to-day operations of the Hedrin JV under a management contract with the Hedrin JV. H Pharmaceuticals is independently funded and is responsible for all costs associated with the Hedrin project, including any necessary United States (“U.S.”) clinical trials, patent costs, and future milestones owed to the original licensor, T&R.
Pediculosis (Head lice)
Head lice (Pediculus humanus capitis) are small parasitic insects that live mainly on the human scalp and neck hair. Head lice are not known to transmit disease, but they are highly contagious and are acquired by direct head-to-head contact with an infested person’s hair, and may also be transferred with shared combs, hats, and other hair accessories. They can also live on bedding or upholstered furniture for a brief period. Head lice are seen across the socioeconomic spectrum and are unrelated to personal cleanliness or hygiene. Children are more frequently infested than are adults, and Caucasians more frequently than other ethnic groups. Lice are most commonly found on the scalp, behind the ears, and near the neckline at the back of the neck. Common symptoms include a tickling feeling of something moving in the hair, itching, irritability caused by poor sleep, and sores on the head caused by scratching. According to our internal analysis, a majority of the currently available prescription and over-the-counter (“OTC”) head lice treatments are chemical insecticides.
Mechanism of Action
Hedrin is a novel, non-insecticide combination of silicones (dimethicone and cyclomethicone) that acts as a pediculicidal (lice killing) agent by disrupting the insect’s mechanism for managing fluid and breathing. In contrast with most currently available lice treatments, Hedrin contains no chemical insecticides. Because Hedrin kills lice by preventing the louse from excreting waste fluid, rather than by acting on the central nervous system, the insects cannot build up resistance to the treatment. Recent studies have indicated that resistance to chemical insecticides may be increasing and therefore contributing to insecticide treatment failure. Manhattan Pharmaceuticals believes there is significant market potential for convenient, non-insecticide treatment alternatives. Both silicones in this proprietary formulation of Hedrin are used extensively in cosmetics and toiletries.
Product Development
To date, Hedrin has been clinically studied in 362 subjects and is currently marketed as a medical device in Western Europe and as a pharmaceutical in the United Kingdom (“U.K.”).
In a randomized, controlled, equivalence, clinical study (conducted in Europe), Hedrin was administered to 253 adult and child subjects with head lice infestation. The study results, published in the British Medical Journal in June 2005, demonstrated Hedrin’s equivalence when compared to the insecticide treatment, phenothrin, the most widely used pediculicide in the U.K. In addition, according to the same study, the Hedrin treated subjects experienced significantly less irritation (2%) than those treated with phenothrin (9%).
A clinical study published in the November 2007 issue of PLoS One, an international, peer-reviewed journal published by the Public Library of Science (PLoS), demonstrated Hedrin’s superior efficacy compared to a U.K. formulation of malathion, a widely used insecticide treatment in both Europe and North America. In this randomized, controlled, assessor blinded, parallel group clinical trial, 73 adult and child subjects with head lice infestations were treated with Hedrin or malathion liquid. Using intent-to-treat analysis, Hedrin achieved a statistically significant cure rate of 70% compared to 33% with malathion liquid. Using the per-protocol analysis Hedrin achieved a highly statistically significant cure rate of 77% compared to 35% with malathion. In Europe, it has been widely documented that head lice has become resistant to malathion, and we believe this resistance may have influenced the study results. To date, there have been no reports of malathion resistance in the U.S. Additionally, Hedrin treated subjects experienced no irritant reactions, and Hedrin showed clinical equivalence to malathion in its ability to inhibit egg hatching. Overall, investigators and study subjects rated Hedrin as less odorous, easier to apply, and easier to wash out, and 97% of Hedrin treated subjects stated they were significantly more inclined to use the product again versus 31% of those using malathion.
4
Two new, unpublished Hedrin studies were completed by T&R in 2008. In the first, Hedrin achieved a 100% kill rate in vitro, including in malathion resistant head lice. In the other, a clinical field study conducted in Manisa province, a rural area of Western Turkey, Hedrin was administered to 36 adult and child subjects with confirmed head lice infestations. Using per protocol analysis, Hedrin achieved a 97% cure rate. Using intent-to-treat analysis, Hedrin achieved a 92% cure rate since 2 subjects were eliminated due to protocol violations. No subjects reported any adverse events.
In the U.S., Manhattan Pharmaceuticals, through the Hedrin JV, is pursuing the development of Hedrin as a medical device. In January 2009, the U.S. Food and Drug Administration (“FDA”) Center for Devices and Radiological Health (“CDRH”) notified H Pharmaceuticals that Hedrin had been classified as a Class III medical device. A Class III designation means that a Premarket Approval (“PMA”) Application will need to be obtained before Hedrin can be marketed in the U.S. The Company expects to be required to complete at least one clinical trial as part of that PMA Application.
Market and Competition
In Europe, Hedrin has been launched in 27 countries and, according to T&R, has achieved 2008 annual sales through its licensees of approximately $48 million (€35 million) at in-market public prices, garnering approximately 23% market share across Europe. It is the market leader in the U.K. with $10 million in sales (25% market share) and France with a 25% market share.
According to the American Academy of Pediatrics an estimated 6-12 million Americans are infested with head lice each year, with pre-school and elementary children and their families affected most often. The total U.S. head lice market is estimated to be over $200 million with prescription and over-the-counter (OTC) therapies comprising approximately 50% of that market. The remaining 50% of the market is comprised of alternative therapies such as tea tree oils, mineral oils, and “nit picking”, or physical combing to remove lice. We believe there is significant market potential for a convenient, non-insecticide treatment for head lice.
The prescription and OTC segment of the market is dominated by 4-5 name brand products and numerous, low cost generics and store brand equivalents. The active ingredients in these pharmacological therapies are chemical insecticides. The most frequently prescribed insecticide treatments are Kwell (lindane) and Ovide (malathion), and the most frequently purchased OTC brands are Rid (pyrethrin), Nix (permethrin), and Pronto (pyrethrin). Lindane has been banned in 52 countries worldwide and has now been banned in the state of California due to its toxicity. European formulations of Malathion have experienced widespread resistance. Resistance to U.S. formulations of malathion have not been widely reported, but experts believe it may eventually develop with continued use. Head lice resistance to pyrethrin and permethrin has been reported in the U.S. and treatment failures are common.
See also “Management’s Discussion and Analysis of Financial Condition and Results of Operations- Liquidity and Capital Resources- Research and Development Projects- Hedrin.”
5
Topical Psoriasis Product
Since 2005, Manhattan Pharmaceuticals has been developing a topical formulation of parathyroid hormone (1-34) (“Topical PTH (1-34)”) for the prescription treatment of mild to moderate psoriasis. In July 2008, we announced Phase 2a study results where PTH (1-34) failed to demonstrate a statistically significant or clinically meaningful improvement in psoriasis versus a vehicle (placebo). In this study, the vehicle (placebo) was a topical gel (“GEL”) that had been developed by the Company and was identical to the Topical PTH (1-34) product minus the active ingredient, PTH (1-34). Both the active and vehicle (placebo) arms of the study showed similar improvements in study subjects’ psoriasis plaques. In summary, the study results indicated that PTH (1-34) was not effective for the treatment of psoriasis since it did not achieve superiority or a statistical separation in efficacy over the GEL. The GEL placebo may be effective enough to market as an OTC product. Due to these study results, the Company has decided to discontinue development of PTH (1-34) as a prescription topical pharmaceutical candidate, and instead, we are exploring the possibility of developing the GEL as an OTC product for mild psoriasis.
We currently hold an exclusive, worldwide license to develop and commercialize Topical PTH (1-34) for the treatment of psoriasis. This license was obtained as a result of our merger with Tarpan Therapeutics in 2005, and Tarpan had acquired the exclusive, worldwide rights pursuant to a 2004 license agreement with IGI, Inc (“IGI”). We intend to return the rights to PTH (1-34) to IGI under the terms of the license agreement.
Psoriasis
Psoriasis is a common, chronic, immune-mediated disease that results in the over-production of skin cells. In healthy skin, immature skin cells migrate from the lowest layer of the epidermis to the skin’s surface over a period of 28-30 days. In psoriasis, these cells reproduce at an extremely accelerated rate and advance to the surface in only 7 days. This results in a build up of excess, poorly differentiated skin cells that accumulate in dry, thick patches known as plaques. These plaques can appear anywhere on the body resulting in skin irritation and disability.
PTH (1-34) Product Development History
In April 2006, we encountered a stability issue with the original topical PTH (1-34) product which utilized IGI’s Novosome® formulation technology. In order to resolve that stability issue we created a new topical gel delivery system to be used with peptides, including PTH (1-34), and other types of molecules. In 2007 we filed new patent applications in the U.S. for this new proprietary GEL formulation.
In September 2007, the U.S. FDA accepted our Investigational New Drug (“IND”) application for this new gel formulation of Topical PTH (1-34), and in October 2007, we initiated and began dosing subjects in a Phase 2a clinical study of Topical PTH (1-34) for the treatment of psoriasis. This U.S., multi-center, randomized, double-blind, vehicle-controlled, parallel group study was designed to evaluate safety and preliminary efficacy of Topical PTH (1-34) in patients with mild to moderate psoriasis. Approximately 54 subjects were enrolled and randomized to receive one of two dose levels of Topical PTH (1-34), or the GEL vehicle (placebo), for an 8 week treatment period. In this study the vehicle was the topical GEL without the active ingredient, PTH (1-34). In July 2008, we announced the results of the Phase 2a study where Topical PTH (1-34) failed to demonstrate a statistically significant or clinically meaningful improvement in psoriasis. We have conducted no further clinical activities with Topical PTH (1-34) and we intend to return the product to IGI under the terms of the license agreement.
6
Topical GEL for Psoriasis
The GEL vehicle (placebo) used in the above-mentioned study is the Company’s proprietary topical GEL showed evidence of psoriasis improving properties. In this Phase 2a study, when it was utilized as the vehicle (placebo), 15% of study subjects achieved a clear or almost clear state at the end of week 2. At the end of week 4, 20% of subjects treated with the GEL had achieved a clear or almost clear state, and at the end of week 8, 25% of subjects treated with the GEL had achieved a clear or almost clear state. The Company owns worldwide rights to this topical GEL and is exploring the possibility of developing it as an OTC product for mild psoriasis.
Market and Competition
According to the National Psoriasis Foundation approximately 125 million people worldwide, including approximately 6 million Americans, suffers from psoriasis. Of these, approximately 65% (4.4 million) have mild psoriasis and maybe likely to be treated with an OTC product. According to Datamonitor, only an estimated 55% of psoriasis sufferers have been formally diagnosed by a physician, so the OTC market could potentially be much larger.
There are a number of treatments available today for psoriasis, including numerous OTC creams and ointments that help to reduce inflammation, stop itching, and soothe skin. Products such as Psoriasin, CortAid, Dermarest, and Cortizone 10 are the most common, but none are viewed as particularly effective for psoriasis. Steroids are also prescribed as an adjunct prescription therapy for pain and anti-inflammation.
See also “Management’s Discussion and Analysis of Financial Condition and Results of Operations - Liquidity and Capital Resources - Research and Development Projects – Topical Psoriasis Product.”
Discontinued Research and Development Programs
Altoderm™
In April 2007 we entered into a license agreement with T&R, pursuant to which we acquired exclusive rights to develop and commercialize Altoderm in North America. Altoderm is a novel, proprietary formulation of topical cromolyn sodium and is designed to enhance the absorption of cromolyn sodium into the skin in order to treat pruritus (itch) associated with dermatologic conditions including atopic dermatitis (eczema).
Atopic Dermatitis (Eczema)
Atopic dermatitis, also know as eczema, is a chronic disease of the skin that is believed to be caused by a combination of hereditary and environmental factors. The main symptoms of atopic dermatitis include dry, itchy skin leading to rashes on the face, hands, feet, along with inside the elbows and behind the knees. Scratching results in redness, swelling, cracking, “weeping” clear fluid, and crusting or scaling.
Product Development
In a Phase 3, randomized, double-blind, placebo-controlled, parallel-group, clinical study (conducted in Europe by T&R.) the compound was administered for 12 weeks to 114 subjects with moderately severe atopic dermatitis. The placebo (vehicle) used in this study was the Altoderm product without the active ingredient. In the study results, published in the British Journal of Dermatology in February 2005, Altoderm demonstrated a statistically significant reduction (36%) in atopic dermatitis symptoms. During the study, subjects were permitted to continue with their existing treatment, in most cases this consisted of emollients and topical steroids. A positive secondary outcome of the study was a 35% reduction in the use of topical steroids for the Altoderm treated subjects. Further analysis of the clinical data, performed by Manhattan Pharmaceuticals, showed that Altoderm treated subjects also experienced a 57% reduction in pruritus.
7
On March 6, 2008, Manhattan Pharmaceuticals announced it had successfully completed a pre-IND meeting with the FDA. Based on a review of the submitted package for Altoderm, including data from the two previously reported Phase 3 clinical studies, the FDA determined that following completion of certain nonclinical studies, and the acceptance of an IND, Phase 2 clinical studies may be initiated in the U.S. The FDA also concurred that the proposed indication of pruritus associated with dermatologic conditions including atopic dermatitis can be pursued.
In a second, Phase 3, randomized, double-blind, vehicle-controlled clinical study (also conducted in Europe by T&R) Altoderm was safe and well tolerated, and showed a trend toward improvement in pruritus, but the efficacy results were inconclusive. Altoderm treated subjects and vehicle only treated subjects experienced a similar improvement (each greater than 30%), and therefore, the study did not achieve statistical significance.
As a result of the inconclusive European study data and a lack of sufficient funds to develop Altoderm, in March 2009 the Company discontinued development and returned the project to T&R under the terms of the license agreement.
See also “Management’s Discussion and Analysis of Financial Condition and Results of Operations- Liquidity and Capital Resources- Research and Development Projects- Altoderm.”
Altolyn™
In April 2007 we entered into a license agreement with T&R, pursuant to which we acquired exclusive rights to develop and commercialize Altolyn in North America. Altolyn is a novel, proprietary oral tablet formulation of cromolyn sodium designed to treat mastocytosis and possibly other gastrointestinal disorders such as food allergy and symptoms of irritable bowel syndrome.
Mastocytosis
Mastocytosis is a rare disorder that occurs in both children and adults. It is caused by the presence of too many mast cells in the body. Mast cells are found in skin, linings of the stomach and intestine, and connective tissue (such as cartilage and tendons). Mast cells play an important role in helping the immune systems defend these tissues from disease. They release chemical “alarms” such as histamine and cytokines to attract other key players of the immune defense system to sites in the body where they might be needed. People with mastocytosis experience abdominal discomfort, nausea and vomiting, ulcers, diarrhea, and skin lesions.
Product Development
On March 6, 2008, Manhattan Pharmaceuticals announced it had successfully completed a pre-IND meeting with the FDA. Based on a review of the submitted package for Altolyn, the FDA concurred that the proposed indication of mastocytosis can be pursued and that the 505(b)(2) NDA would be an acceptable approach provided a clinical bridge is established between Altolyn and Gastrocrom®, the oral liquid formulation of cromolyn sodium currently approved in the U.S. to treat mastocytosis. Section 505(b)(2) of the Food, Drug and Cosmetic Act allows the FDA to approve a follow-on drug on the basis of data in the scientific literature or data used by FDA in the approval of other drugs.The FDA also affirmed that a single, Phase 3 study demonstrating the efficacy of Altolyn over placebo, may be sufficient to support a product approval in the U.S. In addition, the FDA also concurs that no additional nonclinical studies will be required to support an IND application. The Company is working with T&R and the current U.K. manufacturer of Altolyn to develop a Good Manufacturing Process (“cGMP”) compliant manufacturing process.
8
Due to small market opportunity and lack of sufficient funds to develop Altolyn, in March 2009 the Company discontinued development and returned the project to T&R under the terms of the license agreement.
See also “Management’s Discussion and Analysis of Financial Condition and Results of Operations- Liquidity and Capital Resources- Research and Development Projects- Altolyn.”
Oleoyl-estrone
On July 9, 2007 the Company announced the results of its two Phase 2a clinical trials of oral Oleoyl-estrone (“OE”). The results of both randomized, double-blind, placebo controlled studies, one in common obesity and the other in morbid obesity, demonstrated no statistically or clinically meaningful placebo adjusted weight loss for any of the treatment arms evaluated. Based on these results, the Company discontinued its OE programs in both common obesity and morbid obesity.
Propofol Lingual Spray
On July 9, 2007 the Company announced that it discontinued development of Propofol Lingual Spray for pre-procedural sedation.
Intellectual Property and License Agreements
Our goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and elsewhere in the world.
We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors. This knowledge and experience we call “know-how”. To help protect our proprietary know-how which is not patentable, and for inventions for which patents may be difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. To this end, we require all employees, consultants, advisors and other contractors to enter into confidentiality agreements which prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Hedrin
On June 26, 2007, the Company entered into an exclusive license the Hedrin agreement with T&R and Kerris. Pursuant to the Hedrin License Agreement, the Company has acquired an exclusive North American license to certain patent rights and other intellectual property relating to HedrinTM, a non-insecticide product candidate for the treatment of pediculosis (“head lice”):
9
| 1. | U.S. Patent Application No. 2007/0142330, entitled, “Method and composition for the control of arthropods.” Jayne Ansell, Inventor. Application filed February 12, 2007. This application is a divisional of U.S. application Ser. No. 10/097,615, filed Mar. 15, 2002, which is a continuation of International Application No. PCT/GB00/03540, which designated the United States and was filed on Sep. 14, 2000. This application has not yet issued as a patent. Any patent that issues will expire on September 14, 2020. |
This patent application has numerous, detailed and specific claims related to the use of Hedrin (novel formulation of silicon derivatives) in controlling and repelling arthropods such as insects and arachnids, and in particular control and eradication of head lice and their ova.
In addition, on June 26, 2007, the Company entered into the Hedrin Supply Agreement with T&R pursuant to which T&R will be the Company’s exclusive supplier of the Hedrin product.
In consideration for the license, the Company issued to T&R and Kerris (jointly, the “Licensor”) a combined total of 150,000 shares of its common stock valued at $120,000. In addition, the Company also made a cash payment of $600,000 to the Licensor. Further, the Company agreed to make future milestone payments to the Licensor comprised of various combinations of cash and common stock in respective aggregate amounts of $2,500,000 upon the achievement of various clinical and regulatory milestones as follows: $250,000 upon acceptance by the FDA of an IND; $1,000,000 upon the achievement of a successful outcome of a Phase 3 clinical trial; $700,000 upon the final approval of a New Drug Application (“NDA”), or its equivalent, by the FDA; $300,000 upon the issuance of a U.S. patent on Hedrin: and $250,000 upon receipt of marketing authorization in Canada.
Through December 31, 2008, none of the milestones have been reached and sales have not commenced, therefore, we have not paid any such milestones or royalties.
The Company also agreed to pay royalties to the Licensor of 8% (or, under certain circumstances, 4%) on net sales of licensed products. The Company’s exclusivity under the Hedrin Agreement is subject to an annual minimum royalty payment of $1,000,000 (or, under certain circumstances, $500,000) in each of the third through seventh years following the first commercial sale of Hedrin. The Company may sublicense its rights under the Hedrin Agreement with the consent of Licensor and the proceeds resulting from such sublicenses will be shared with the Licensor.
Pursuant to the Hedrin Supply Agreement, the Company has agreed that it and its sublicensees will purchase their respective requirements of the Hedrin product from T&R at agreed upon prices. Under certain circumstances where T&R is unable to supply Hedrin products in accordance with the terms and conditions of the Supply Agreement, the Company may obtain product from an alternative supplier subject to certain conditions. The term of the Supply Agreement ends upon termination of the Hedrin Agreement.
On February 25, 2008 the Company assigned and transferred its rights in Hedrin to the Hedrin JV. The Hedrin JV is now responsible for all of the Company’s obligations under the Hedrin License Agreement and the Hedrin Supply Agreement.
10
Topical PTH (1-34) License Agreement.
In connection with our April 2005 acquisition of Tarpan Therapeutics, Inc., we acquired Tarpan’s rights under an April 2004 Sublicense Agreement with IGI, Inc. (the “IGI Agreement”). Pursuant to this agreement we now have worldwide, exclusive license rights to the U.S. and foreign patents and patent applications for all topical uses of Topical PTH(1-34) for the treatment of hyperproliferative skin disorders including psoriasis:
| 1. | U.S. Patent No. 5,527,772, entitled “Regulation of cell proliferation and differentiation using peptides.” M.F. Holick, Inventor. Application filed July, 28, 1994. Patent issued June 18, 1996. This patent expires June 18, 2013. |
| 2. | U.S. Patent No. 5,840,690, entitled “Regulation of cell proliferation and differentation using peptides.” M.F. Holick, Inventor. Application filed June 6, 1995. Patent issued November 24, 1998. This patent expires June 18, 2013. |
| 3. | U.S. Provisional application No. US60/940,509, entitled “Topical Compositions comprising a macromolecule and methods of using same.” Application was filed on May 29, 2007. |
These patents have numerous, detailed and specific claims relating to the topical use of Topical PTH (1-34)
The IGI sublicense agreement requires us to make certain milestone payments as follows: $300,000 payable upon the commencement of a Phase 2 clinical trial; $500,000 upon the commencement of a Phase 3 clinical trial; $1,500,000 upon the acceptance of an Investigational New Drug Application (“NDA”) by the FDA; $2,400,000 upon the approval of an NDA by the FDA; $500,000 upon the commencement of a Phase 3 clinical trial for an indication other than psoriasis; $1,500,000 upon the acceptance of and NDA application for an indication other than psoriasis by the FDA; and $2,400,000 upon the approval of an NDA for an indication other than psoriasis by the FDA.
During 2007 we achieved the milestone of the commencement of a Phase 2 clinical trial. As a result $300,000 became payable to IGI. This $300,000 is included in research and development expense for the year ended December 31, 2007. Payment was made to IGI in February 2008.
In addition, we are obligated to pay IGI, Inc. an annual royalty of 6% on annual net sales up to $200,000,000. In any calendar year in which net sales exceed $200,000,000, we are obligated to pay IGI, Inc. an annual royalty of 9% annual net sales. Through December 31, 2008 sales have not commenced, therefore we have not paid any such royalties.
IGI, Inc. may terminate the agreement (i) upon 60 days’ notice if we fail to make any required milestone or royalty payments, or (ii) if we become bankrupt or if a petition in bankruptcy is filed, or if we are placed in the hands of a receiver or trustee for the benefit of creditors. IGI, Inc. may terminate the agreement upon 60 days’ written notice and an opportunity to cure in the event we commit a material breach or default. We may terminate the agreement in whole or as to any portion of the PTH patent rights upon 90 days’ notice to IGI, Inc.
11
Altoderm
On April 3, 2007, the Company entered into a license agreement for Altoderm (the “Altoderm Agreement”) with T&R. Pursuant to the Altoderm Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altoderm, a topical skin lotion product candidate with the active ingredient cromolyn sodium (also known as sodium cromoglicate) for the treatment of pruritis (itch) associated with dermatologic conditions including atopic dermatitis:
| 1. | U.S. Patent No. 7,109,246, entitled “Pharmaceutical compositions comprising an amphoteric surfactant an alkoxylated cetyl alcohol and a polar drug.” Brian Hawtin, Inventor. Application filed May 20, 1999. Patent issued September 19, 2006. This patent expires on May 20, 2019. |
| 2. | U.S. Application Publication No. 2007/0036860, entitled “Treatment of allergic conditions.” Alexander James Wigmore, Inventor. Any patent that issues will expire on November 9, 2019. This patent covers both Altoderm and Altolyn. |
These patents have numerous, detailed and specific claims related to the use of Altoderm (composition of topically administered cromolyn sodium) for treating atopic dermatitis (eczema).
In accordance with the terms of the Altoderm Agreement, the Company issued 125,000 shares of its common stock, valued at $112,500, and made a cash payment of $475,000 to T&R upon the execution of the agreement. Further, the Company agreed to make future milestone payments to T&R comprised of various combinations of cash and common stock in respective aggregate amounts of $5,675,000 and 875,000 shares of our common stock upon the achievement of various clinical and regulatory milestones. as follows: $450,000 upon acceptance by FDA of an IND; 125,000 shares of our common stock upon the first dosing of a patient in the first Phase 2 clinical trial; 250,000 shares of our common stock and $625,000 upon the first dosing of a patient in the first Phase 3 clinical trial; $1,000,000 upon the achievement of a successful outcome of a Phase 3 clinical trial; $1,100,000 upon the acceptance for filing of a NDA application by the FDA; 500,000 shares of our common stock and $2,000,000 upon the final approval of an NDA by the FDA; and $500,000 upon receipt of marketing authorization in Canada.
In addition, we are obligated to pay T&R an annual royalty of 10% on annual net sales of up to $100,000,000; 15% of the amount of annual net sales in excess of $100,000,000 and 20% of annual net sales in excess of $200,000,000. There is a minimum royalty of $1,000,000 per year. There is a one-time success fee of $10,000,000 upon the achievement of cumulative net sales of $100,000,000. Through December 31, 2008, none of the milestones have been reached and sales have not commenced, therefore, we have not paid any such milestones or royalties.
Altolyn
On April 3, 2007, the Company and T&R also entered into a license agreement for Altolyn (the “Altolyn Agreement”). Pursuant to the Altolyn Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altolyn, an oral tablet formulation product candidate using sodium cromolyn for the treatment of mastocytosis, food allergies, and inflammatory bowel disorder.
| 1. | U.S. Patent No. 7,258,872, entitled “Chromone enteric release formulation.” Alexander James Wigmore, Inventor. Application filed November 9, 1999, claiming the benefit of a GB application filed November 11, 1998. Patent issued August 21, 2007. The expected date of expiration, which was November 9, 2019, has been extended by 793 days (expiration date Jan 10, 2022). |
12
| 2. | U.S. Application Publication No. 2007/0036860, entitled “Treatment of allergic conditions.” Alexander James Wigmore, Inventor. Application filed October 13, 2006, claiming the benefit of a prior U.S. application, which claimed the benefit of a PCT application filed November 9, 1999. This application has not yet issued as a patent. Any patent that issues is expected to expire on November 9, 2019. This patent covers both Altoderm and Altolyn. |
These patents have numerous, detailed and specific claims related to Altolyn (as an oral tablet drug delivery composition), and the pending application discloses and may be used to claim the use of Altolyn (composition of orally administered sodium cromolyn) for the treatment of allergic conditions, specifically food allergies.
In accordance with the terms of the Altolyn Agreement, the Company made a cash payment of $475,000 to T&R upon the execution of the agreement. Further, the Company agreed to make future milestone payments to T&R comprised of various combinations of cash and common stock in respective aggregate amounts of $5,675,000 upon the achievement of various clinical and regulatory milestones. as follows: $450,000 upon acceptance filing by the FDA of an IND; $625,000 upon the first dosing of a patient in the first Phase 3 clinical trial; $1,000,000 upon the achievement of a successful outcome of a Phase 3 clinical trial; $1,100,000 upon the acceptance for filing of a NDA application by the FDA; $2,000,000 upon the final approval of an NDA by the FDA; and $500,000 upon receipt of marketing authorization in Canada.
In addition, we are obligated to pay T&R an annual royalty of 10% on annual net sales of up to $100,000,000; 15% of the amount of annual net sales in excess of $100,000,000 and 20% of annual net sales in excess of $200,000,000. There is a minimum royalty of $1,000,000 per year. There is a one-time success fee of $10,000,000 upon the achievement of cumulative net sales of $100,000,000. Through December 31, 2008, none of the milestones have been reached and sales have not commenced, therefore, we have not paid any such milestones or royalties.
Oleoyl-estrone
On July 9, 2007 the Company announced the results of its two Phase 2a clinical trials of oral OE. The results of both randomized, double-blind, placebo controlled studies, one in common obesity and the other in morbid obesity, demonstrated no statistically or clinically meaningful placebo adjusted weight loss for any of the treatment arms evaluated. Based on these results, the Company discontinued its OE programs in both common obesity and morbid obesity.
Propofol Lingual Spray
On July 9, 2007 the Company announced that it discontinued development of Propofol Lingual Spray for pre-procedural sedation.
Manufacturing
We do not have any manufacturing capabilities. We are in contact with several contract cGMP manufacturers for the supply of Topical PTH(1-34), the topical GEL for psoriasis and, on behalf of the Hedrin JV, Hedrin that will be necessary to conduct human clinical trials, if needed.
13
Government Regulation
The research, development, testing, manufacture, labeling, promotion, advertising, distribution, and marketing, among other things, of our products are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, and/or criminal prosecution.
Drug Approval Process. None of our drugs may be marketed in the U.S. until the drug has received FDA approval. The steps required before a drug may be marketed in the U.S. include:
| · | nonclinical laboratory tests, animal studies, and formulation studies, |
| · | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin, |
| · | adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication, |
| · | submission to the FDA of an NDA, |
| · | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practices, or cGMPs, and |
| · | FDA review and approval of the NDA. |
Nonclinical tests include laboratory evaluation of product chemistry, toxicity, and formulation, as well as animal studies. The conduct of the nonclinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the nonclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. We cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap. The study protocol and informed consent information for study subjects in clinical trials must also be approved by an Institutional Review Board for each institution where the trials will be conducted. Study subjects must sign an informed consent form before participating in a clinical trial. Phase 1 usually involves the initial introduction of the investigational drug into people to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. Phase 2 usually involves trials in a limited patient population to (i) evaluate dosage tolerance and appropriate dosage; (ii) identify possible adverse effects and safety risks; and (iii) preliminarily evaluate the efficacy of the drug for specific indications. Phase 3 trials usually further evaluate clinical efficacy and test further for safety by using the drug in its final form in an expanded patient population. There can be no assurance that Phase 1, Phase 2, or Phase 3 testing will be completed successfully within any specified period of time, if at all. Furthermore, we or the FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
14
The FDCA permits FDA and the IND sponsor to agree in writing on the design and size of clinical studies intended to form the primary basis of an effectiveness claim in an NDA application. This process is known as Special Protocol Assessment, or SPA. These agreements may not be changed after the clinical studies begin, except in limited circumstances.
Assuming successful completion of the required clinical testing, the results of the nonclinical and clinical studies, together with other detailed information, including information on the manufacture and composition of the drug, are submitted to the FDA in the form of a NDA requesting approval to market the product for one or more indications. The testing and approval process requires substantial time, effort, and financial resources. The agencies review the application and may deem it to be inadequate to support the registration and we cannot be sure that any approval will be granted on a timely basis, if at all. The FDA may also refer the application to the appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee.
The FDA has various programs, including fast track, priority review, and accelerated approval, that are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis surrogate endpoints. Generally, drugs that may be eligible for one or more of these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that provide meaningful benefit over existing treatments. We cannot be sure that any of our drugs will qualify for any of these programs, or that, if a drug does qualify, that the review time will be reduced.
Section 505(b)(2) of the FDCA allows the FDA to approve a follow-on drug on the basis of data in the scientific literature or data used by FDA in the approval of other drugs. This procedure potentially makes it easier for generic drug manufacturers to obtain rapid approval of new forms of drugs based on proprietary data of the original drug manufacturer. We intend to rely on Section 505(b)(2) to obtain approval for Altolyn.
Before approving an NDA, the FDA usually will inspect the facility or the facilities at which the drug is manufactured, and will not approve the product unless cGMP compliance is satisfactory. If the FDA evaluates the NDA and the manufacturing facilities as acceptable, the FDA may issue an approval letter, or in some cases, an approvable letter followed by an approval letter. Both letters usually contain a number of conditions that must be met in order to secure final approval of the NDA. When and if those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. The approval letter authorizes commercial marketing of the drug for specific indications. As a condition of NDA approval, the FDA may require post marketing testing and surveillance to monitor the drug’s safety or efficacy, or impose other conditions.
After approval, certain changes to the approved product, such as adding new indications, making certain manufacturing changes, or making certain additional labeling claims, are subject to further FDA review and approval. Before we can market our product candidates for additional indications, we must obtain additional approvals from FDA. Obtaining approval for a new indication generally requires that additional clinical studies be conducted. We cannot be sure that any additional approval for new indications for any product candidate will be approved on a timely basis, or at all.
15
Post-Approval Requirements. Often times, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. If such post-approval conditions are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA are required to: (i) report certain adverse reactions to the FDA, (ii) comply with certain requirements concerning advertising and promotional labeling for their products, and (iii) continue to have quality control and manufacturing procedures conform to cGMP after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. We intend to use third party manufacturers to produce our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including withdrawal of the product from the market.
Orphan Drug. The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which generally is a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting an NDA. If the FDA grants orphan drug designation, which it may not, the identity of the therapeutic agent and its potential orphan use are publicly disclosed by the FDA. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process. If a product that has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the FDA may not approve any other applications to market the same drug for the same indication, except in certain very limited circumstances, for a period of seven years. Orphan drug designation does not prevent competitors from developing or marketing different drugs for that indication.
Non-United States Regulation. Before our products can be marketed outside of the United States, they are subject to regulatory approval similar to that required in the United States, although the requirements governing the conduct of clinical trials, including additional clinical trials that may be required, product licensing, pricing and reimbursement vary widely from country to country. No action can be taken to market any product in a country until an appropriate application has been approved by the regulatory authorities in that country. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for FDA approval. In certain countries, the sales price of a product must also be approved. The pricing review period often begins after market approval is granted. Even if a product is approved by a regulatory authority, satisfactory prices may not be approved for such product.
In Europe, marketing authorizations may be submitted at a centralized, a decentralized or national level. The centralized procedure is mandatory for the approval of biotechnology products and provides for the grant of a single marketing authorization that is valid in all European Union (“EU”) members states. As of January 1995, a mutual recognition procedure is available at the request of the applicant for all medicinal products that are not subject to the centralized procedure. There can be no assurance that the chosen regulatory strategy will secure regulatory approvals on a timely basis or at all.
16
Employees
We currently have 1 part time and 3 full time employees, including: our Chief Executive Officer, our Chief Operating and Financial Officer and 2 persons in business development, administration and finance. None of our employees is covered by a collective bargaining unit. We believe our relations with our employees are satisfactory.
17
ITEM 1A. RISK FACTORS
An investment in our securities is speculative in nature, involves a high degree of risk, and should not be made by an investor who cannot bear the economic risk of its investment for an indefinite period of time and who cannot afford the loss of its entire investment. You should carefully consider the following risk factors and the other information contained elsewhere in this Annual Report before making an investment in our securities.
Risks Related to Our Business
We currently have no product revenues and will need to raise additional funds in the future. If we are unable to obtain the funds necessary to continue our operations, we will be required to delay, scale back or eliminate one or both of our remaining drug development programs and may not continue as a going concern.
We have generated no product revenues to date and will not until, and if, we receive approval from the FDA and other regulatory authorities for our two product candidates. We have already spent substantial funds developing our potential products and business, however, and we expect to continue to have negative cash flow from our operations for at least the next several years. As of December 31, 2008, we had $106,023 of cash and cash equivalents. We received additional funding of approximately $0.5 million from a joint venture agreement in February 2009 and $0.4 million from the sale of Secured 12% Notes in February 2009. We will still have to raise substantial additional funds to complete the development of our product candidates and to bring them to market. Beyond the capital requirements mentioned above, our future capital requirements will depend on numerous factors, including:
| · | the results of any clinical trials; |
| · | the scope and results of our research and development programs; |
| · | the time required to obtain regulatory approvals; |
| · | our ability to establish and maintain marketing alliances and collaborative agreements; and |
| · | the cost of our internal marketing activities. |
Swiss Pharma has received an arbitration award against us, in the Swiss Chamber of Commerce, and has initiated proceedings in New York State courts to confirm the arbitration award and enter a judgment on Swiss Pharma's behalf against us. We do not have sufficient cash or other current available assets to satisfy the arbitrators award. If Swiss Pharma is successful in entering a judgment against us, it could take actions against us that would significantly impair our ability to continue as a going concern. See "Item 3 - Legal Proceedings."
It is difficult for companies like ours to raise funds in the current economic conditions and additional financing may not be available on acceptable terms, if at all. If adequate funds are not available, we will be required to delay, scale back or eliminate one or more of our drug development programs or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain of our technologies or products that we would not otherwise relinquish. Our auditors have concluded that our net losses, negative cash flow, accumulated deficit and negative working capital as of December 31, 2008, raise substantial doubt about our ability to continue as a going concern.
18
We are not currently profitable and may never become profitable.
We have a history of losses and expect to incur substantial losses and negative operating cash flow for the foreseeable future, and we may never achieve or maintain profitability. We have incurred losses in every period since our inception on August 6, 2001. For the year ended December 31, 2008 and for the period from August 6, 2001 (inception) through December 31, 2008, we incurred net losses applicable to common shares of $4,268,858, and $59,267,928 respectively. Even if we succeed in developing and commercializing one or more of our product candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
| · | continue to undertake nonclinical development and clinical trials for our product candidates; |
| · | seek regulatory approvals for our product candidates; |
| · | implement additional internal systems and infrastructure; |
| · | lease additional or alternative office facilities; and |
| · | hire additional personnel. |
We also expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. . Our auditors have concluded that our net losses, negative cash flow, accumulated deficit and negative working capital as of December 31, 2008, raise substantial doubt about our ability to continue as a going concern.
Our failure to achieve or maintain profitability could negatively impact the value of our common stock.
We have a limited operating history upon which to base an investment decision.
We are a development-stage company and have not yet demonstrated any ability to perform the functions necessary for the successful commercialization of any product candidates. The successful commercialization of our product candidates will require us to perform a variety of functions, including:
| · | continuing to undertake nonclinical development and clinical trials; |
| · | participating in regulatory approval processes; |
| · | formulating and manufacturing products; and |
| · | conducting sales and marketing activities. |
Since inception as Manhattan Research Development, Inc., our operations have been limited to organizing and staffing, and acquiring, developing and securing our proprietary technology and undertaking nonclinical and clinical trials of principal product candidates. These operations provide a limited basis for you to assess our ability to commercialize our product candidates and the advisability of investing in our securities.
19
We depend greatly on the intellectual capabilities and experience of our key executives, and the loss of any of them could affect our ability to develop our remaining products.
We had only three full-time and one part-time employee as of March 30, 2009. The loss of Douglas Abel, our President and Chief Executive Officer, or Michael G. McGuinness, our Chief Operating and Financial Officer, could harm us. The current terms of our employment agreements with Messrs. Abel and McGuinness expire in April 2009 and July 2009, respectively. We cannot predict our success in hiring or retaining the personnel we require for continued operations.
We may not obtain the necessary U.S. or worldwide regulatory approvals to commercialize our product candidates.
We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any of our product candidates, we must first submit to the FDA an IND, which will set forth our plans for clinical testing of our product candidates. In September 2007, the FDA accepted our IND for Topical PTH(1-34). Our remaining two products, Hedrin and the Topical GEL for psoriasis are currently considered pre-clinical. We are unable to estimate the size and timing of the clinical and non clinical trials required to bring our two product candidates to market and, accordingly, cannot estimate the time when development of these product candidates will be completed.
When the clinical testing for our product candidates is complete, we will submit to the FDA a NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as nonclinical studies, as well as human tests, which are referred to as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for indicated uses. The FDA has substantial discretion in the drug approval process and may require us to conduct additional nonclinical and clinical testing or to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may:
| · | delay commercialization of, and our ability to derive product revenues from, our product candidates; |
| · | impose costly procedures on us; and |
| · | diminish any competitive advantages that we may otherwise enjoy. |
Even if we comply with all FDA requests, the FDA may ultimately reject any or all of our future NDAs. We cannot be sure that we will ever obtain regulatory clearance for any of our product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by reducing our number of salable products and, therefore, corresponding product revenues.
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize our drugs. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. We have not yet made any determination as to which foreign jurisdictions we may seek approval and have not undertaken any steps to obtain approvals in any foreign jurisdiction.
20
Clinical trials are very expensive, time consuming and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time consuming. We estimate that clinical trials of our product candidates will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
| · | unforeseen safety issues; |
| · | determination of dosing issues; |
| · | lack of effectiveness during clinical trials; |
| · | slower than expected rates of patient recruitment; |
| · | inability to monitor patients adequately during or after treatment; and |
| · | inability or unwillingness of medical investigators to follow our clinical protocols. |
In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our IND submissions or the conduct of these trials.
The results of our clinical trials may not support our product candidate claims.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims. Success in nonclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and nonclinical testing. The clinical trial process may fail to demonstrate that our product candidates are safe for humans or effective for indicated uses. This failure would cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. In addition, we anticipate that our clinical trials will involve only a small patient population. Accordingly, the results of such trials may not be indicative of future results over a larger patient population.
Physicians and patients may not accept and use our drugs.
Even if the FDA approves our product candidates, physicians and patients may not accept and use them. Acceptance and use of our product will depend upon a number of factors including:
| · | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drugs; |
| · | cost-effectiveness of our product relative to competing products; |
21
| · | availability of reimbursement for our products from government or other healthcare payers; and |
| · | effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any. |
Because we expect sales of our current product candidates, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
Our drug-development program depends upon third-party researchers who are outside our control.
We currently are collaborating with several third-party researchers, for the development of our product candidates. Accordingly, the successful development of our product candidates will depend on the performance of these third parties. These collaborators will not be our employees, however, and we cannot control the amount or timing of resources that they will devote to our programs. Our collaborators may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such programs ourselves. If outside collaborators fail to devote sufficient time and resources to our drug-development programs, or if their performance is substandard, the approval of our FDA applications, if any, and our introduction of new drugs, if any, will be delayed. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators assist our competitors at our expense, our competitive position would be harmed.
We rely exclusively on third parties to formulate and manufacture our product candidates.
We have no experience in drug formulation or manufacturing and do not intend to establish our own manufacturing facilities. We lack the resources and expertise to formulate or manufacture our own product candidates. We intend to contract with one or more manufacturers to manufacture, supply, store and distribute drug supplies for our clinical trials. If any of our product candidates receive FDA approval, we will rely on one or more third-party contractors to manufacture our drugs. Our anticipated future reliance on a limited number of third-party manufacturers, exposes us to the following risks:
| · | We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any. |
| · | Our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical needs and commercial needs, if any. |
| · | Our future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products. |
| · | Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Agency, and corresponding state agencies to ensure strict compliance with good manufacturing practice and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards. |
22
| · | If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation. |
We have no experience selling, marketing or distributing products and no internal capability to do so.
We currently have no sales, marketing or distribution capabilities. We do not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of our proposed products. Our future success depends, in part, on our ability to enter into and maintain such collaborative relationships, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products, however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that they will have effective sales forces. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. There can also be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to market and sell our product in the United States or overseas.
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our product candidates is characterized by intense competition and rapid technological advances. If our product candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors have product candidates that will compete with ours already approved or in development. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do, as well as significantly greater experience in:
| · | developing drugs; |
| · | undertaking nonclinical testing and human clinical trials; |
| · | obtaining FDA and other regulatory approvals of drugs; |
| · | formulating and manufacturing drugs; and |
| · | launching, marketing and selling drugs. |
23
Developments by competitors may render our products or technologies obsolete or non-competitive.
Many of the organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, longer drug development history in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to attract qualified personnel, parties for acquisitions, joint ventures or other collaborations.
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish.
Our success, competitive position and future revenues will depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties.
We currently do not directly own the rights to any issued patents. See “Business – Intellectual Property and License Agreements.”.
However, with regard to the patents covered by our license agreements and any future patents issued to which we will have rights, we cannot predict:
| · | the degree and range of protection any patents will afford us against competitors including whether third parties will find ways to invalidate or otherwise circumvent our patents; |
| · | if and when patents will issue; |
| · | whether or not others will obtain patents claiming aspects similar to those covered by our patents and patent applications; or |
| · | whether we will need to initiate litigation or administrative proceedings which may be costly whether we win or lose. |
Our success also depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our licensors and contractors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, we require all of our employees, consultants, advisors and contractors to enter into agreements which prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation.
Our business is substantially dependent on the intellectual property on which our product candidates are based. To date, we have not received any threats or claims that we may be infringing on another’s patents or other intellectual property rights. If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
24
| · | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| · | redesign our products or processes to avoid infringement; |
| · | stop using the subject matter claimed in the patents held by others; |
| · | pay damages; or |
| · | defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our valuable management resources. |
Our ability to generate product revenues will be diminished if our drugs sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our drugs, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
| · | government and health administration authorities; |
| · | private health maintenance organizations and health insurers; and |
| · | other healthcare payers. |
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payers increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if our product candidates are approved by the FDA, insurance coverage may not be available, and reimbursement levels may be inadequate, to cover our drugs. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for any of our products, once approved, market acceptance of our products could be reduced.
We may not successfully manage our growth.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our management and on our administrative, operational and financial resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business may suffer.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
We will need to hire additional qualified personnel with expertise in nonclinical testing, clinical research and testing, government regulation, formulation and manufacturing and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
25
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. We currently carry clinical trial insurance in an amount up to $5,000,000, which may be inadequate to protect against potential product liability claims or may inhibit the commercialization of pharmaceutical products we develop, alone or with corporate collaborators. Although we intend to maintain clinical trial insurance during any clinical trials, this may be inadequate to protect us against any potential claims. Even if our agreements with any future corporate collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
We are controlled by current officers, directors and principal stockholders.
Our directors, executive officers and principal stockholders beneficially own approximately 29 percent of our outstanding voting stock and, including shares underlying outstanding options and warrants. In addition, Nordin Biotech Venture Fund has the right to acquire up to 66,666,666 shares of our common stock (which would represent approximately 49% of our common stock as of March 17, 2009), resulting in Nordic owning approximately 49% of our common stock. Through its stock ownership, its right to acquire additional shares, its substantial control over the management of the Hedrin JV (which includes the ability to terminate our management contract with the Hedrin JV), and various covenants contained in our registration rights agreement with Nordic, Nordic has the ability to exert substantial influence over the election of our Board of Directors, the outcome of issues submitted to our stockholders, the development of Hedrin and our ability, as a company, to benefit from the successful development of Hedrin. Even without the exercise of its rights to acquire additional shares of our common stock, our directors, officers and principal stockholders, taken as a whole, have the ability to exert substantial influence over the election of our Board of Directors and the outcome of issues submitted to our stockholders.
Risks Related to Our Securities
Our stock price is, and we expect it to remain, volatile, which could limit investors’ ability to sell stock at a profit.
During the last two fiscal years, our stock price has traded at a low of $0.007 in the fourth quarter of 2008 to a high of $1.10 in the second quarter of 2007. The volatile price of our stock makes it difficult for investors to predict the value of their investment, to sell shares at a profit at any given time, or to plan purchases and sales in advance. A variety of factors may affect the market price of our common stock. These include, but are not limited to:
| · | The continuing global economic crisis, which has affected stock prices of many companies, and particularly many small pharmaceutical companies like ours; |
| · | publicity regarding actual or potential clinical results relating to products under development by our competitors or us; |
| · | delay or failure in initiating, completing or analyzing nonclinical or clinical trials or the unsatisfactory design or results of these trials; |
26
| · | achievement or rejection of regulatory approvals by our competitors or us; |
| · | announcements of technological innovations or new commercial products by our competitors or us; |
| · | developments concerning proprietary rights, including patents; |
| · | developments concerning our collaborations; |
| · | regulatory developments in the United States and foreign countries; |
| · | economic or other crises and other external factors; |
| · | period-to-period fluctuations in our revenues and other results of operations; |
| · | changes in financial estimates by securities analysts; and |
| · | sales of our common stock. |
We will not be able to control many of these factors, and we believe that period-to-period comparisons of our financial results will not necessarily be indicative of our future performance.
In addition, the stock market in general, and the market for biotechnology companies in particular, has experienced extreme price and volume fluctuations that may have been unrelated or disproportionate to the operating performance of individual companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
We have delisted from the American Stock Exchange.
As a result of our delisting, the liquidity of our common stock has been and may continue to be reduced, not only in terms of the number of shares that can be bought and sold at a given price, but also through delays in the timing of transactions and reduction in security analysts’ and the media’s coverage of us. This may result in lower prices for our common stock than might otherwise be obtained and could also result in a larger spread between the bid and asked prices for our common stock.
We have never paid dividends.
We have never paid dividends on our common stock and do not anticipate paying any dividends for the foreseeable future. You should not rely on an investment in our stock if you require dividend income. Further, you will only realize income on an investment in our stock in the event you sell or otherwise dispose of your shares at a price higher than the price you paid for your shares. Such a gain would result only from an increase in the market price of our common stock, which is uncertain and unpredictable.
27
ITEM 2. PROPERTIES
Our executive offices are located at 48 Wall Street, New York, New York 10005. We currently occupy this space pursuant to a written lease that expires on September 30, 2009 under which we pay rent of approximately $7,000 per month.
We believe that our existing facilities are adequate to meet our current requirements. We do not own any real property.
ITEM 3. LEGAL PROCEEDINGS
Swiss Pharma Contract LTD, or Swiss Pharma, a clinical site that we used in one of our obesity trials, gave notice to us that Swiss Pharma believed it was entitled to receive an additional payment of $322,776 for services in connection with that clinical trial. The contract between us and Swiss Pharma provided for arbitration in the event of a dispute, such as this claim for an additional payment. On March 10, 2008, Swiss Pharma filed for arbitration with the Swiss Chamber of Commerce. As we did not believe that Swiss Pharma was entitled to additional payments, we defended our position in arbitration. On April 2, 2008, we filed our statement of defense and counterclaim for recovery of costs incurred by us as a result of Swiss Pharma’s failure to meet agreed upon deadlines under our contract. On June 3, 2008, a hearing was held before the arbitrator under the auspices of the Swiss Chamber of Commerce. On September 5, 2008, the arbitrator rendered an award in favor of Swiss Pharma, awarding to Swiss Pharma a total of $646,000 which amount includes a $323,000 contract penalty, a final services invoice of $48,000, reimbursement of certain of Swiss Pharma’s legal and other expenses incurred in the arbitration process of $245,000, reimbursement of arbitration costs of $13,000 and interest through September 5, 2008 of $17,000. Further, the arbitrator ruled that we must pay interest at the rate of 5% per annum on $371,000, the sum of the $323,000 contract penalty and the final services invoice of $48,000, from October 12, 2007 until paid. We had previously recognized a liability to Swiss Pharma in the amount of $104,000 for the final services invoice. The remainder of the award was expensed in 2008. We have recognized research and development expense of $267,000, general and administrative expense of $257,000 and interest expense of $23,000 during the year ended December 31, 2008. On January 22, 2009, we received notice that Swiss Pharma submitted a petition to the Supreme Court of the State of New York, County of New York seeking to confirm and to enter a judgment on the Arbitration Award. On February 17, 2009, we filed an answer to Swiss Pharma's petition. A hearing has not yet been scheduled. We will continue to accrue interest at the rate of 5% per annum on the $371,000 until such amount has been settled. We do not have sufficient cash or other current available assets to satisfy the arbitrator's award.
28
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Market for Common Stock
Our common stock traded on the American Stock Exchange “AMEX”under the symbol “MHA” during the year ended December 31, 2007 and for the period from January 1, 2008 to March 26, 2008. On March 26, 2008 our common stock was voluntarily delisted from the AMEX and began trading on the Over the Counter Bulletin Board (“OCTBB”) under the symbol “MHAN”. The following table lists the high and low price for our common stock as quoted, in U.S. dollars, on the American Stock Exchange or the Over the Counter Bulletin Board for the periods indicated:
| Price Range | ||||||||||||||||
| 2008 | 2007 | |||||||||||||||
| Quarter Ended | High | Low | High | Low | ||||||||||||
| March 31 | $ | 0.230 | $ | 0.110 | $ | 0.960 | $ | 0.700 | ||||||||
| June 30 | 0.180 | 0.100 | 1.100 | 0.690 | ||||||||||||
| September 30 | 0.200 | 0.100 | 0.780 | 0.220 | ||||||||||||
| December 31 | 0.090 | 0.007 | 0.230 | 0.090 | ||||||||||||
Stock Chart
Comparison to NASDAQ Biotechnology Index.
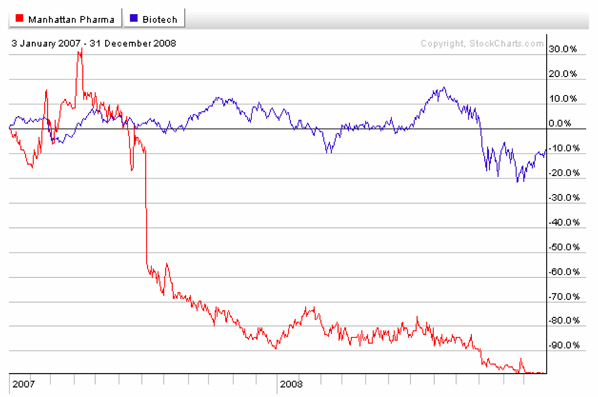
29
Record Holders
The number of holders of record of our common stock as of March 20, 2009 was 437.
Dividends
We have not paid or declared any dividends on our common stock and we do not anticipate paying dividends on our common stock in the foreseeable future, but intend instead to retain earnings, if any, for use in our business operations. The payment of dividends in the future, if any, will be at the sole discretion of our board of directors and will depend upon our debt and equity structure, earnings and financial condition, need for capital in connection with possible future acquisitions and other factors, including economic conditions, regulatory restrictions and tax considerations. We cannot guarantee that we will pay dividends or, if we pay dividends, the amount or frequency of these dividends.
Stock Repurchases
We did not make any repurchases of our common stock during 2008.
Securities authorized for issuance under equity compensation plans
| Equity Compensation Plan Information | ||||||||||||
| Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted average exercise price of outstanding options, warrants and rights | Number of securities remaing available for future issuance under equity compenstaion plans (excluding securities reflected in column (a)) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders | 10,661,612 | $ | 0.9340 | 875,628 | ||||||||
| Equity compensation plans not approved by security holders | ||||||||||||
| Total | 10,661,612 | $ | 0.934 | 875,628 | ||||||||
Recent Sales of Unregistered Securities
In connection with the Registrant’s merger of its wholly-owned subsidiary Tarpan Acquisition Corp., with Tarpan Therapeutics, Inc. (“Tarpan”), effective as of April 1, 2005, it issued an aggregate of 10,731,052 shares of its common stock to the former stockholders of Tarpan in exchange for their shares of Tarpan common stock. The Registrant relied on the exemption from federal registration under Section 4(2) of the Securities Act, based on its belief that the issuance of such securities did not involve a public offering, as there were fewer than 35 “non-accredited” investors, all of whom, either alone or through a purchaser representative, had such knowledge and experience in financial and business matters so that each was capable of evaluating the risks of the investment.
30
In August 2005, the Registrant sold in a private placement offering to accredited investors units of its securities consisting of shares of common stock and warrants to purchase additional shares of common stock. The private placement was completed in two separate closings held on August 26, 2005 and August 30, 2005. In the August 26 closing, the Registrant sold a total of 10,763,926 shares of common stock and five-year warrants to purchase 2,152,758 shares for total gross proceeds of approximately $11.95 million. The warrants issued at the August 26 closing are exercisable at a price of $1.44 per share, which represented approximately 110% of the average closing price of the Registrant’s common stock during the five trading days preceding such closing date. On August 30, 2005, the Registrant sold an additional 1,108,709 shares of common stock and five-year warrants to purchase 221,741 shares of common stock, which resulted in gross proceeds of approximately $1.28 million. The warrants issued in connection with the August 30 closing are exercisable at a price of $1.49 per share, which represented approximately 110% of the average closing price of the Company's common stock during the five trading days preceding such closing date. The total gross proceeds resulting from this offering was approximately $13.22 million, before deducting selling commissions and expenses. The Registrant paid total cash commissions of approximately $925,000 to selling agents engaged in connection with the offering and issued 5-year warrants to purchase an aggregate of 593,196 shares of common stock, of which warrants to purchase 538,196 shares are exercisable at a price of $1.44 per share and the remaining are exercisable at a price of $1.49 per share. In connection with this offering, the Registrant relied on the exemption from federal registration under Section 4(2) of the Securities Act and/or Rule 506 promulgated thereunder, based on its belief that the offer and sale of the shares and warrants did not involve a public offering as each investor was “accredited” and no general solicitation was involved in the offering.
On March 30, 2007, the Registrant entered into a series of subscription agreements with various institutional and other accredited investors for the issuance and sale in a private placement of an aggregate of 10,185,502 shares of the Registrant’s common stock for total gross proceeds of approximately $8.56 million. Of the total amount of shares issued, 10,129,947 were sold at a per share price of $0.84, and an additional 55,555 shares were sold to an entity affiliated with Neil Herskowitz, a director of Manhattan, at a per share price of $0.90, the closing sale price of the Registrant’s common stock on March 29, 2007. Pursuant to the subscription agreements, the Registrant also issued to the investors 5-year warrants to purchase an aggregate of 3,564,897 shares of the Registrant’s common stock at an exercise price of $1.00 per share. The warrants are exercisable during the period commencing September 30, 2007 and ending March 30, 2012. Pursuant to the subscription agreements, the Registrant agreed to file a registration statement with the Securities and Exchange Commission on or before May 14, 2007 covering the resale of the shares issued in the private placement, including the shares issuable upon exercise of the investor warrants. The Registrant engaged Paramount BioCapital, Inc., as its placement agent in connection with the private placement. In consideration for its services, the Registrant paid aggregate cash commissions of approximately $600,000 and issued to Paramount a 5-year warrant to purchase an aggregate of 509,275 shares at an exercise price of $1.00 per share. The sale of the shares and warrants was not registered under the Securities Act of 1933. Rather, the offer and sale of such securities was made in reliance on the exemption from registration requirements provided by Section 4(2) of the Securities Act and Regulation D promulgated thereunder. Each of the investors was “accredited” (as defined under Regulation D) and no general solicitation was used in connection with the offer and sale of such securities.
In April 2007, in partial consideration for entering into a license agreement, the Registrant issued to Thornton & Ross Ltd., the licensor, a total of 125,000 shares of the Registrant’s common stock in accordance with the terms thereof. The issuance of such common stock was considered to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, or Regulation D promulgated thereunder, as a transaction by an issuer not involving a public offering. The recipient of such common stock represented their intention to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the share certificates issued in this transaction. All recipients either received adequate information about us or had access to such information.
31
In June 2007, in consideration for entering into a license agreement, the Registrant issued to each of Thornton & Ross, Ltd. and Kerra, S.A., each a licensor thereunder, 75,000 shares of the Registrant’s common stock in accordance with the terms thereof. The issuances of such common stock were considered to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, or Regulation D promulgated thereunder, as transactions by an issuer not involving a public offering. The recipients of such common stock represented their intention to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the share certificates issued in these transactions. All recipients either received adequate information about the Registrant or had access to such information.
In January 2008, the Registrant and Nordic Biotech Venture Fund II K/S ("Nordic"), entered into a joint venture agreement the Registrant, as amended on February 18, 2008 and June 9, 2008 (the "Joint Venture Agreement"), pursuant to which in February 2008, (i) Nordic contributed cash in the amount of $2.5 million to H Pharmaceuticals K/S (formerly Hedrin Pharmaceuticals K/S), a newly formed Danish limited partnership(the "Hedrin JV") in exchange for 50% of the equity interests in the Hedrin JV, and (ii) the Registrant contributed certain assets to North American rights (under license) to our Hedrin product to the Hedrin JV in exchange for $2.0 million in cash and 50% of the equity interests in the Hedrin JV. On or around June 30, 2008, in accordance with the terms of the Joint Venture Agreement, Nordic contributed an additional $1.25 million in cash to the Hedrin JV, $1.0 million of which was distributed to us and equity in the Hedrin JV was distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%. Pursuant to the Joint Venture Agreement, upon the classification by the U.S. Food and Drug Administration, or the FDA, of Hedrin as a Class II or Class III medical device, Nordic will be required to contribute to the Hedrin JV an additional $1.25 million in cash, $0.5 million of which will be distributed to us and equity in the Hedrin JV will be distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%. Upon classification by the FDA of Hedrin as a Class II or Class III medical device, the Hedrin JV will have received a total of $1.5 million cash to be applied toward the development and commercialization of Hedrin in North America. If classification of Hedrin by the FDA as a Class II or Class III medical device is not received by June 30, 2009, then Nordic will not be obligated to make the final payment of $1.25 million and Nordic will receive an additional 20% ownership of the joint venture and enhanced control over the joint venture's operations and other important decision-making. Pursuant to the terms of the Joint Venture Agreement, Nordic has the right to nominate one person for election or appointment to our board of directors.
Pursuant to the Joint Venture Agreement, Nordic has the right to put all or a portion of its interest in the Hedrin JV in exchange for such number of shares of common stock equal to the amount of Nordic’s investment in the Hedrin JV divided by $0.14, as adjusted from time to time for stock splits and other specified events, multiplied by a conversion factor, which is (i) 1.00 for so long as Nordic's distributions from the Hedrin JV are less than the amount of its investment, (ii) 1.25 for so long as Nordic's distributions from the Hedrin JV are less than two times the amount of its investment but greater than or equal to the amount of its investment amount, (iii) 1.50 for so long as Nordic's distributions from the Hedrin JV are less than three times the amount of its investment but greater than or equal to two times the amount of its investment amount, (iv) 2.00 for so long as Nordic's distributions from the Hedrin JV are less than four times the amount of its investment but greater than or equal to three times the amount of its investment amount and (v) 3.00 for so long as Nordic’s distributions from Hedrin JV are greater than or equal to four times the amount of its investment. The put right expires upon the earlier to occur of (i) February 25, 2018 and (ii) 30 days after the date when Nordic's distributions from the Hedrin JV exceed five times the amount Nordic has invested in the Hedrin JV (or 10 days after such date if the Registrant has provided Nordic notice thereof). Pursuant to the Joint Venture Agreement, the Registrant has the right to call all or a portion of Nordic's equity interest in the Hedrin JV in exchange for such number of shares of common stock equal to the portion of Nordic's investment in the Hedrin JV that the Registrant calls by the dollar amount of Nordic's investment as of such date in the Hedrin JV, divided by $0.14, as adjusted from time to time for stock splits and other specified events. The call right is only exercisable by the Registrant if the price of common stock has closed at or above $1.40 per share for 30 consecutive trading days. During the first 30 consecutive trading days in which the common stock closes at or above $1.40 per share, the Registrant may exercise up to 25% of the call right. During the second 30 consecutive trading days in which the common stock closes at or above $1.40 per share, the Registrant may exercise up to 50% of the call right on a cumulative basis. During the third consecutive 30 trading days in which the common stock closes at or above $1.40 per share, the Registrant may exercise up to 75% of the call right on a cumulative basis. During the fourth consecutive 30 days in which the common stock closes at or above $1.40 per share, the Registrant may exercise up to 100% of the call right on a cumulative basis. Nordic may refuse the call, either by paying $1.5 million multiplied by the percentage of Nordic's investment being called or forfeiting an equivalent portion of the put right, calculated on a pro rata basis for the percentage of the Nordic equity interest called by us. The call right expires on February 25, 2013.
32
In connection with the Joint Venture Agreement, on February 25, 2008, Nordic paid the Registrant a non-refundable fee of $150,000 in exchange for the right to receive a warrant to purchase up to 7,142,857 shares of common stock at $0.14 per share, as adjusted from time to time for stock splits and other specified events, if Nordic did not exercise all or part of its put right on or before April 30, 3008. As of April 30, 2008, Nordic had not exercised all or any portion of its put right and the Registrant issued the warrant to Nordic.
The offering and sale of the securities under the Joint Venture Agreement were considered to be exempt from registration under the Securities Act, by virtue of Section 4(2) thereof and the provisions of Regulation D promulgated thereunder. Nordic has represented to the Registrant that it is an "accredited investor," as that term is defined in Rule 501(a) of Regulation D under the Securities Act.
On September 11, 2008, the Registrant entered into a series of 10% secured promissory notes with certain of its directors and officers and an employee of the Registrant (the “Note Holders”) for aggregate of $70,000. Principal and interest on the notes shall be paid in cash on March 10, 2009 unless paid earlier by the Registrant. In connection with the issuance of the notes, the Registrant also issued to the Note Holders 5-year warrants to purchase an aggregate of 140,000 shares of the Registrant's common stock at an exercise price of $0.20 per share. The Registrant granted to the Note Holders a continuing security interest in certain specific refunds, deposits and repayments due to the Registrant and expected to be repaid to the Registrant in the next several months. . The issuance of such securities was considered to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, or Regulation D promulgated thereunder, as a transaction by an issuer not involving a public offering. The recipient of such securities represented their intention to acquire the securities for investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the warrant certificates issued in this transaction. All recipients either received adequate information about us or had access to such information.
On February 3, 2009, the Registrant completed a private placement (the "2009 Private Placement") of 345 units, with each unit consisting of a 12% senior secured note promissory note in the principal amount of $5,000 and a warrant to purchase up to 166,667 shares of common stock at an exercise price of $.09 per share which expires on December 31, 2013, for aggregate gross proceeds of $1,780,500. The private placement was completed in three closings which occurred on November 19, 2008 with respect to 207 units, December 23, 2008 with respect to 56 units and February 3, 2009 with respect to 82 units.
33
On November 19, 2008, the Registrant completed the sale of 207 units in the first closing of the 2009 Private placement. The Registrant issued a warrant to purchase 5,175,010 shares of common stock at an exercise price of $.09 per share to the placement agent as partial compensation for its services. Te Registrant granted the placement agent the right to nominate a member of its Board of Directors and such director shall receive all compensation and benefits provided to its other directors. Additionally, upon such director’s appointment to the Board of Directors, he or she shall be issued a warrant to purchase 1,000,000 shares of common stock at a per share exercise price equal to the greater of (i) the fair market value on the date of issuance or (ii) $.09.
On December 23, 2008, the Registrant completed a second closing of the 2009 Private Placement under the terms of the Securities Purchase Agreement. At the second closing, the Registrant sold an additional 56 units to investors. In connection with the second closing, the Registrant issued to the placement agent a warrant to purchase 1,400,003 shares of common stock at an exercise price of $.09 per share as additional compensation for its services.
On February 3, 2009, the Registrant completed a third closing of the 2009 Private Placement under the terms of the Securities Purchase Agreement. At the third closing, the Registrant sold an additional 82 units to investors. In connection with the third closing, the Registrant issued to the placement agent a warrant to purchase 2,050,004 shares of common stock at an exercise price of $.09 per share as additional compensation for its services.
All of the investors represented in the 2009 Private Placement represented that they were “accredited investors,” as that term is defined in Rule 501(a) of Regulation D under the Securities Act, and the sale of the units was made in reliance on exemptions provided by Regulation D and Section 4(2) of the Securities Act of 1933, as amended.
34
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
Overview
We were incorporated in Delaware in 1993 under the name “Atlantic Pharmaceuticals, Inc.” and, in March 2000, we changed our name to “Atlantic Technology Ventures, Inc.” In 2003, we completed a “reverse acquisition” of privately held “Manhattan Research Development, Inc”. In connection with this transaction, we also changed our name to “Manhattan Pharmaceuticals, Inc.” From an accounting perspective, the accounting acquirer is considered to be Manhattan Research Development, Inc. and accordingly, the historical financial statements are those of Manhattan Research Development, Inc.
During 2005 we merged with Tarpan Therapeutics, Inc. (“Tarpan”). Tarpan was a privately held New York based biopharmaceutical company developing dermatological therapeutics. Through the merger, we acquired Tarpan’s primary product candidate, Topical PTH (1-34) for the treatment of psoriasis. In consideration for their shares of Tarpan’s capital stock, the stockholders of Tarpan received an aggregate of approximately 10,731,000 shares of our common stock, representing approximately 20% of our then outstanding common shares. This transaction was accounted for as a purchase of Tarpan by the Company.
We are a specialty healthcare product company focused on developing and commercializing pharmaceutical treatments for underserved patient populations. We aim to acquire rights to these technologies by licensing or otherwise acquiring an ownership interest, funding their research and development and eventually either bringing the technologies to market or out-licensing. In the short term we are focusing our efforts on the commercialization of the two product candidates we currently have in development: HedrinTM, through the Hedrin JV, a novel, non-insecticide treatment of pediculitis (head lice) and a topical product for the treatment of psoriasis. Longer term we intend to acquire and commercialize low risk, quick to market products, specifically products that could be marketed over-the-counter (“OTC”), treat everyday maladies, are simple to manufacture, and/or could be classified as medical devices by the FDA.
You should read the following discussion of our results of operations and financial condition in conjunction with the financial statements and notes thereto appearing elsewhere in this Form 10-K. This discussion includes “forward-looking” statements that reflect our current views with respect to future events and financial performance. We use words such as we “expect,” “anticipate,” “believe,” and “intend” and similar expressions to identify forward-looking statements. Investors should be aware that actual results may differ materially from our expressed expectations because of risks and uncertainties inherent in future events, particularly those risks identified under the heading “Risk Factors” following Item 1 in this Annual Report, and should not unduly rely on these forward looking statements. All share and per share information in this discussion has been adjusted for the 1-for-5 combination of our common stock effected on September 25, 2003.
Results Of Operations
2008 versus 2007
During each of the years ended December 31, 2008 and 2007, we had no revenues, and are considered a development stage company. We do not expect to have revenues relating to our products prior to December 31, 2009.
35
| Years ended December 31, | ||||||||||||||||
| 2008 | 2007 | Increase (decrease) | % Increase (decrease) | |||||||||||||
| Costs and expenses: | ||||||||||||||||
| Research and development: | ||||||||||||||||
| Share-based compensation | $ | 122,000 | $ | 539,000 | $ | (417,000 | ) | -77.37 | % | |||||||
| In-license, milestone and related fees | - | 2,245,000 | (2,245,000 | ) | -100.00 | % | ||||||||||
| Other research and development expenses | 1,681,000 | 5,752,000 | (4,071,000 | ) | -70.78 | % | ||||||||||
| Total research and development expenses | 1,803,000 | 8,536,000 | (6,733,000 | ) | -78.88 | % | ||||||||||
| General and administrative: | ||||||||||||||||
| Share-based compensation | 342,000 | 902,000 | (560,000 | ) | -62.08 | % | ||||||||||
| Other general and administrative expenses | 2,268,000 | 2,706,000 | (438,000 | ) | -16.19 | % | ||||||||||
| Total general and administrative expenses | 2,610,000 | 3,608,000 | (998,000 | ) | -27.66 | % | ||||||||||
| Other income | 144,000 | 112,000 | 32,000 | 28.57 | % | |||||||||||
| Net loss | $ | 4,269,000 | $ | 12,032,000 | $ | (7,763,000 | ) | -64.52 | % | |||||||
For the year ended December 31, 2008 research and development expense was $1,803,000 as compared to $8,536,000 for the year ended December 31, 2007. This decrease of $6,733,000, or 78.9%, is primarily comprised of a decrease in in-license, milestone and related fees of $2,245,000, a decrease in other research and development expenses of $4,071,000 and a decrease in stock based compensation of $417,000.
For the year ended December 31, 2008 general and administrative expense was $2,610,000 as compared to $3,608,000 for the year ended December 31, 2007. This decrease of $998,000, or 27.7%, is primarily comprised of a decrease in share-based compensation of $560,000 and a decrease in other general and administrative expenses of $438,000.
For the year ended December 31, 2008 other income was $144,000 as compared to $112,000 for the year ended December 31, 2007. This increase of $32,000, or 28.6%, is primarily due to increases in management fee revenue from the Hedrin JV of $447,000 and in other income of $7,000 offset by equity in losses of the Hedrin JV of $250,000, a decrease in interest income of $108,000 and an increase in interest expense of $64,000.
Net loss for the year ended December 31, 2008 was $4,269,000 as compared to $12,032,000 for the year ended December 31, 2007. This decrease of $7,763,000, or 64.5%, is primarily due to a decrease in research and development expenses of $6,733,000, a decrease in general and administrative expense of $998,000 and an increase in other income of $32,000.
Liquidity and Capital Resources
From inception to December 31, 2008, we incurred a deficit during the development stage of $59,268,000 primarily as a result of our net losses, and we expect to continue to incur additional losses through at least December 31, 2009 and for the foreseeable future. These losses have been incurred through a combination of research and development activities related to the various technologies under our control and expenses supporting those activities.
We have financed our operations since inception primarily through equity and debt financings and a joint venture transaction. During the year ended December 31, 2008, we had a net decrease in cash and cash equivalents of $544,000. This decrease resulted largely from net cash used in operating activities of $4,444,000 partially offset by net cash provided by financing activities of $3,909,000. Total liquid resources as of December 31, 2008 were $106,000 compared to $650,000 at December 31, 2007.
36
Our current liabilities as of December 31, 2008 were $1,486,000 compared to $1,872,000 at December 31, 2007, a decrease of $386,000. As of December 31, 2008, we had working capital deficit of $612,000 compared to working capital deficit of $1,006,000 at December 31, 2007.
The Company received approximately $1.8 million in February 2008 and approximately $0.9 million in June 2008 from a joint venture agreement. The Company also received $70,000 in Secured 10% Notes in September 2008 and net proceeds of $1.0 million from the sale of Secured 12% Notes in November and December 2008.
Our available working capital and capital requirements will depend upon numerous factors, including progress of our research and development programs, our progress in and the cost of ongoing and planned nonclinical and clinical testing, the timing and cost of obtaining regulatory approvals, the cost of filing, prosecuting, defending, and enforcing patent claims and other intellectual property rights, in-licensing activities, competing technological and market developments, changes in our existing collaborative and licensing relationships, the resources that we devote to developing manufacturing and commercializing capabilities, the status of our competitors, our ability to establish collaborative arrangements with other organizations and our need to purchase additional capital equipment.
Our continued operations will depend on whether we are able to raise additional funds through various potential sources, such as equity and debt financing, other collaborative agreements, strategic alliances, and our ability to realize the full potential of our technology in development. Such additional funds may not become available on acceptable terms and there can be no assurance that any additional funding that we do obtain will be sufficient to meet our needs in the long term. Through December 31, 2008, a significant portion of our financing has been through private placements of common stock and warrants. Unless our operations generate significant revenues and cash flows from operating activities, we will continue to fund operations from cash on hand and through the similar sources of capital previously described. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs. We believe that we will continue to incur net losses and negative cash flows from operating activities for the foreseeable future.
Based on the resources of the Company available at December 31, 2008, the net proceeds of $500,000 received in February 2009 from a joint venture agreement and net proceeds of $360,000 received from the sale of additional Secured 12% Notes in February 2009, management believes that the Company has sufficient capital to fund its operations through 2009. Management believes that the Company will need additional equity or debt financing or will need to generate positive cash flow from the Hedrin joint venture, or generate revenues through licensing of its products or entering into strategic alliances to be able to sustain its operations into 2010. Furthermore, the Company will need additional financing thereafter to complete development and commercialization of its products. There can be no assurances that we can successfully complete development and commercialization of our products.
These matters raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We have reported net losses of $4,269,000 and $12,032,000 for the years ended December 31, 2008 and 2007, respectively. The net loss attributable to common shares from date of inception, including preferred stock dividends, August 6, 2001 to December 31, 2008, amounts to $59,268,000. Management believes that we will continue to incur net losses through at least December 31, 2009.
37
Joint Venture Agreement
We and Nordic Biotech Venture Fund II K/S, or Nordic, entered into a joint venture agreement on January 31, 2008, which was amended on February 18, 2008 and on June 9, 2008. Pursuant to the joint venture agreement, in February 2008, (i) Nordic contributed cash in the amount of $2.5 million to H Pharmaceuticals K/S (formerly Hedrin Pharmaceuticals K/S), a newly formed Danish limited partnership, or the Hedrin JV, in exchange for 50% of the equity interests in the Hedrin JV, and (ii) we contributed certain assets to North American rights (under license) to our Hedrin product to the Hedrin JV in exchange for $2.0 million in cash and 50% of the equity interests in the Hedrin JV. On or around June 30, 2008, in accordance with the terms of the joint venture agreement, Nordic contributed an additional $1.25 million in cash to the Hedrin JV, $1.0 million of which was distributed to us and equity in the Hedrin JV was distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%.
Pursuant to the joint venture agreement, upon the classification by the U.S. Food and Drug Administration, or the FDA, of Hedrin as a Class II or Class III medical device, Nordic was required to contribute to the Hedrin JV an additional $1.25 million in cash, $0.5 million of which was to be distributed to us and equity in the Hedrin JV was to be distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%. The FDA notified the Hedrin JV that Hedrin has been classified as a Class III medical device and in February 2009, Nordic made the $1.25 million investment in the Hedrin JV, the Hedrin JV made the $0.5 million milestone payment to us and equity in the Hedrin JV was distributed to us and Nordic sufficient to maintain our respective ownership interests at 50%. In accordance with the terms of the joint venture agreement, as of December 31, 2008 the Hedrin JV had received a total of $1.75 million cash to be applied toward the development and commercialization of Hedrin in North America.
The Hedrin JV is responsible for the development and commercialization of Hedrin for the North American market and all associated costs including clinical trials, if required, regulatory costs, patent costs, and future milestone payments owed to Thornton & Ross Ltd., or T&R, the licensor of Hedrin. The Hedrin JV has engaged us to provide management services to the Hedrin JV in exchange for an annualized management fee, which for 2008, on an annualized basis, is approximately $527,000. As of December 31, 2008, we had recognized approximately $447,000 of other income from management fees earned from the Hedrin JV.
The profits of the Hedrin JV will be shared by us and Nordic in accordance with our respective equity interests in the Hedrin JV, of which we each currently hold 50%, except that Nordic is entitled to receive a minimum return each year from the Hedrin JV equal to 6% on Hedrin sales, as adjusted for any change in Nordic’s equity interest in the Hedrin JV, before any distribution is made to us. If the Hedrin JV realizes a profit in excess of the Nordic minimum return in any year, then such excess shall first be distributed to us until our distribution and the Nordic minimum return are in the same ratio as our respective equity interests in the Hedrin JV and then the remainder, if any, is distributed to Nordic and us in the same ratio as our respective equity interests. However, in the event of a liquidation of the Hedrin JV, Nordic’s distribution in liquidation must equal the amount Nordic invested in the Hedrin JV ($5 million) plus 10% per year, less the cumulative distributions received by Nordic from the Hedrin JV before any distribution is made to us. If the Hedrin JV’s assets in liquidation exceed the Nordic liquidation preference amount, then any excess shall first be distributed to us until our distribution and the Nordic liquidation preference amount are in the same ratio as our respective equity interests in the Hedrin JV and then the remainder, if any, is distributed to Nordic and us in the same ratio as our respective equity interests. Further, in no event shall Nordic’s distribution in liquidation be greater than assets available for distribution in liquidation.
Pursuant to the terms of the joint venture agreement, Nordic has the right to nominate one person for election or appointment to our board of directors. The Hedrin JV's board of directors consists of four members, two members appointed by us and two members appointed by Nordic. Nordic has the right to appoint one of the directors as chairman of the board. The chairman has certain tie breaking powers.
38
Pursuant to the joint venture agreement, Nordic has the right to put all or a portion of its interest in the Hedrin JV in exchange for such number of shares of our common stock equal to the amount of Nordic’s investment in the Hedrin JV divided by $0.09, as adjusted for the sale of the Secured 12% Notes in the fourth quarter of 2008, and as further adjusted from time to time for stock splits and other specified events, multiplied by a conversion factor, which is (i) 1.00 for so long as Nordic's distributions from the Hedrin JV are less than the amount of its investment, (ii) 1.25 for so long as Nordic's distributions from the Hedrin JV are less than two times the amount of its investment but greater than or equal to the amount of its investment amount, (iii) 1.50 for so long as Nordic's distributions from the Hedrin JV are less than three times the amount of its investment but greater than or equal to two times the amount of its investment amount, (iv) 2.00 for so long as Nordic's distributions from the Hedrin JV are less than four times the amount of its investment but greater than or equal to three times the amount of its investment amount and (v) 3.00 for so long as Nordic’s distributions from Hedrin JV are greater than or equal to four times the amount of its investment. The put right expires upon the earlier to occur of (i) February 25, 2018 and (ii) 30 days after the date when Nordic's distributions from the Hedrin JV exceed five times the amount Nordic has invested in the Hedrin JV (or 10 days after such date if we have provided Nordic notice thereof).
Pursuant to the joint venture agreement, we have the right to call all or a portion of Nordic's equity interest in the Hedrin JV in exchange for such number of shares of our common stock equal to the portion of Nordic's investment in the Hedrin JV that we call by the dollar amount of Nordic's investment as of such date in the Hedrin JV, divided by $0.09, as adjusted for the sale of the Secured 12% Notes in the fourth quarter of 2008, and as further adjusted from time to time for stock splits and other specified events. The call right is only exercisable by us if the price of our common stock has closed at or above $1.40 per share for 30 consecutive trading days. During the first 30 consecutive trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 25% of the call right. During the second 30 consecutive trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 50% of the call right on a cumulative basis. During the third consecutive 30 trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 75% of the call right on a cumulative basis. During the fourth consecutive 30 days in which our common stock closes at or above $1.40 per share, we may exercise up to 100% of the call right on a cumulative basis. Nordic may refuse the call, either by paying $1.5 million multiplied by the percentage of Nordic's investment being called or forfeiting an equivalent portion of the put right, calculated on a pro rata basis for the percentage of the Nordic equity interest called by us. The call right expires on February 25, 2013. For purposes of Nordic’s right to put, and our right to call, all or a portion of Nordic’s equity interest in the Hedrin JV, the amount of Nordic’s investment is currently $5,000,000.
In connection with our joint venture agreement, on February 25, 2008, Nordic paid us a non-refundable fee of $150,000 in exchange for the right to receive a warrant to purchase up to 11,111,111 shares of our common stock at $0.09 per share, as adjusted for the sale of the Secured 12% Notes in the fourth quarter of 2008, and as further adjusted from time to time for stock splits and other specified events, if Nordic did not exercise all or part of its put right on or before April 30, 3008. As of April 30, 2008, Nordic had not exercised all or any portion of its put right and we issued the warrant to Nordic.
In connection with the joint venture agreement, we and Nordic entered into a registration rights agreement, on February 25, 2008, as modified pursuant to a letter agreement, dated September 17, 2008, pursuant to which we agreed to file with the Securities and Exchange Commission, or the SEC, by no later than 10 calendar days following the date on which our Annual Report on Form 10-K for the year ended December 31, 2007 is required to be filed with the SEC, which was subsequently waived by Nordic until May 1, 2008, an initial registration statement registering the resale by Nordic of any shares of our common stock issuable to Nordic through the exercise of the warrant or the put right. We filed an initial registration statement on May 1, 2008, which was declared effective on October 15, 2008.
39
We also have agreed to file with the SEC any additional registration statements which may be required no later than 45 days after the date we first know such additional registration statement is required; provided, however, that (i) in the case of the classification by the FDA of Hedrin as a Class II or Class III medical device described above and the payment in full by Nordic of the related final milestone payment of $1.25 million, the registration statement with respect to the additional shares of our common stock relating to such additional investment must be filed within 45 days after achievement of such classification; and (ii) in the event we provide Nordic with notice of exercise of our right to call all or a portion of Nordic's equity interest in the Hedrin JV, a registration statement with respect to the shares of our common stock payable to Nordic in connection with such call right (after giving effect to any reduction in the number of such shares resulting from Nordic's refusal of all or a portion of such call in accordance with the terms of our joint venture agreement) must be filed within 16 days after delivery of such notice to Nordic. If we fail to file a registration statement on time or if a registration statement is not declared effective by the SEC within 105 days of the required filing date, or otherwise fail to diligently pursue registration with the SEC in accordance with the terms of the registration rights agreement, we will be required to pay as partial liquidated damages and not as a penalty, to Nordic or its assigns, an amount equal to 0.5% of the amount invested in the Hedrin JV by Nordic pursuant to the joint venture agreement per month until the registration rights agreement is declared effective by the SEC; provided, however, that in no event shall the aggregate amount payable by us exceed 9% of the amount invested in the Hedrin JV by Nordic under the joint venture agreement.
Secured 10% Notes Payable
On September 11, 2008, we issued secured 10% promissory notes to certain of our directors and officers and an employee for aggregate principal amount of $70,000. Principal and interest on the notes are payable in cash on March 10, 2009 unless paid earlier by the Company. In connection with the issuance of the notes, the Company issued to the noteholders 5-year warrants to purchase an aggregate of 140,000 shares of our common stock at an exercise price of $0.20 per share. We granted to the noteholders a continuing security interest in certain specific refunds, deposits and repayments due to us and expected to be repaid to us in the next several months. The secured 10% notes were repaid in February 2009 along with interest thereon.
Secured 12% Notes Payable
On February 3, 2009, we completed a private placement of 345 units, with each unit consisting of Secured 12% Notes in the principal amount of $5,000 and a warrant to purchase up to 166,667 shares of our common stock at an exercise price of $.09 per share which expires on December 31, 2013, for aggregate gross proceeds of $1,725,000. The private placement was completed in three closings which occurred on November 19, 2008 with respect to 207 units, December 23, 2008 with respect to 56 units and February 3, 2009 with respect to 82 units.
To secure our obligations under the notes, we entered into a security agreement and a default agreement with the investors. The security agreement provides that the notes will be secured by a pledge of our assets other than (i) our interest in the Hedrin joint venture, including, without limitation, our interest in H Pharmaceuticals K/S and H Pharmaceuticals General Partner ApS, (ii) our rent deposit for our former office space, (iii) our refund of a prepayment and (iv) our tax refund for the 2007 fiscal year from the State of New York and City of New York. In addition, to provide additional security for our obligations under the notes, we entered into a default agreement, which provides that upon an event of default under the notes, we shall, at the request of the holders of the notes, use our reasonable commercial efforts to either (i) sell a part or all of our interests in the Hedrin joint venture or (ii) transfer all or part of our interest in the Hedrin JV to the holders of the notes, as necessary, in order to fulfill our obligations under the notes, to the extent required and to the extent permitted by the applicable Hedrin joint venture agreements.
40
On November 19, 2008, we completed the sale of 207 units in our first closing of our private placement. In connection with the first closing, we issued a warrant to purchase 5,175,010 shares of common stock at an exercise price of $.09 per share to the placement agent as partial compensation for its services. Further, we granted the placement agent the right to nominate a member of our Board of Directors and such director shall receive all compensation and benefits provided to our other directors. Additionally, upon such director’s appointment to the Board of Directors, he or she shall be issued a warrant to purchase 1,000,000 shares of our common stock at a per share exercise price equal to the greater of (i) the fair market value on the date of issuance or (ii) $.09.
On December 23, 2008, we completed a second closing of our private placement. At the second closing, we sold an additional 56 units to certain investors. In connection with the second closing, we issued to the placement agent a warrant to purchase 1,400,003 shares of our common stock at an exercise price of $.09 per share as additional compensation for its services.
On February 3, 2009, we completed a third closing of our private placement. At the third closing, we sold an additional 82 units to certain investors. In connection with the third closing, we issued to the placement agent a warrant to purchase 2,050,004 shares of our common stock at an exercise price of $.09 per share as additional compensation for its services.
In connection with the private placement, we, the placement agent and the investors entered into a registration rights agreement. Pursuant to the registration rights agreement, we agreed to file a registration statement to register the resale of the shares of our common stock issuable upon exercise of the warrants issued to the investors in the private placement, within 20 days of the final closing date and to cause the registration statement to be declared effective within 90 days (or 120 days upon full review by the SEC). We filed the registration statement on February 23, 2009. On March 11, 2009 we received a comment letter from the SEC. We are addressing those comments and plan to refile the registration statement once those comments have been addressed.
American Stock Exchange
In September 2007, we received notice from the staff of AMEX, indicating that we were not in compliance with certain continued listing standards set forth in the American Stock Exchange Company guide. Specifically, the American Stock Exchange notice cited our failure to comply, as of June 30, 2007, with section 1003(a)(ii) of the AMEX Company Guide as we had less than $4,000,000 of stockholders’ equity and had losses from continuing operations and /or net losses in three or four of our most recent fiscal years and with section 1003(a)(iii) which requires us to maintain $6,000,000 of stockholders’ equity if we have experienced losses from continuing operations and /or net losses in its five most recent fiscal years.
In order to maintain our AMEX listing, we were required to submit a plan to AMEX advising the exchange of the actions we have taken, or will take, that would bring us into compliance with all the continued listing standards by April 16, 2008. We submitted such a plan in October 2007. If we were not in compliance with the continued listing standards at the end of the plan period, or if we did not make progress consistent with the plan during the period, AMEX staff could initiate delisting proceedings.
Under the terms of the Joint Venture Agreement, the number of potentially issuable shares represented by the put and call features of the Hedrin agreement, and the warrant issuable to Nordic, would exceed 19.9% of our total outstanding shares and would be issued at a price below the greater of book or market value. As a result, under AMEX regulations, we would not have been able to complete the transaction without first receiving either stockholder approval for the transaction, or a formal “financial viability” exception from AMEX’s stockholder approval requirement. We estimated that obtaining stockholder approval to comply with AMEX regulations would take a minimum of 45 days to complete. We discussed the financial viability exception with AMEX for several weeks and had neither received the exception nor been denied the exception. We determined that our financial condition required us to complete the transaction immediately, and that the Company’s financial viability depended on its completion of the transaction without further delay.
41
Accordingly, to maintain the Company’s financial viability, on February 28, 2008 we announced that we had formally notified AMEX that we intended to voluntarily delist our common stock from AMEX. The delisting became effective on March 26, 2008.
Our common stock now trades on the Over the Counter Bulletin Board (“OCTBB”) under the symbol “MHAN”. We intend to maintain corporate governance, disclosure and reporting procedures consistent with applicable law.
Commitments
General
We often contract with third parties to facilitate, coordinate and perform agreed upon research and development of our product candidates. To ensure that research and development costs are expensed as incurred, we record monthly accruals for clinical trials and nonclinical testing costs based on the work performed under the contracts.
These contracts typically call for the payment of fees for services at the initiation of the contract and/or upon the achievement of certain milestones. This method of payment often does not match the related expense recognition resulting in either a prepayment, when the amounts paid are greater than the related research and development costs recognized, or an accrued liability, when the amounts paid are less than the related research and development costs recognized.
Expenses associated with the clinical trials conducted during 2007 and 2008 were recognized on this activity based basis. At December 31, 2007 we recognized accrued expenses of $74,000 related to these clinical trials. At December 31, 2008 all clinical trials had been concluded and there was no remaining financial commitments.
Swiss Pharma Contract LTD, or Swiss Pharma, a clinical site that we used in one of our obesity trials, gave notice to us that Swiss Pharma believed it was entitled to receive an additional payment of $322,776 for services in connection with that clinical trial. The contract between us and Swiss Pharma provided for arbitration in the event of a dispute, such as this claim for an additional payment. On March 10, 2008, Swiss Pharma filed for arbitration with the Swiss Chamber of Commerce. As we did not believe that Swiss Pharma was entitled to additional payments, we defended our position in arbitration. On April 2, 2008, we filed our statement of defense and counterclaim for recovery of costs incurred by us as a result of Swiss Pharma’s failure to meet agreed upon deadlines under our contract. On June 3, 2008, a hearing was held before the arbitrator. On September 5, 2008, the arbitrator rendered an award in favor of Swiss Pharma, awarding to Swiss Pharma a total of approximately $646,000 which amount includes a contract penalty of approximately $323,000, a final services invoice of approximately $48,000, reimbursement of certain of Swiss Pharma’s legal and other expenses incurred in the arbitration process of approximately $245,000, reimbursement of arbitration costs of approximately $13,000 and interest through September 5, 2008 of approximately $17,000. Further, the arbitrator ruled that we must pay interest at the rate of 5% per annum on approximately $371,000, the sum of the contract penalty of approximately $323,000 and the final services invoice of approximately $48,000, from October 12, 2007 until paid. We had previously recognized a liability to Swiss Pharma in the amount of $104,000 for the final services invoice. The remainder of the award was expensed in 2008. We have recognized research and development expense of approximately $267,000, general and administrative expense of approximately $257,000 and interest expense of approximately $23,000 during the year ended December 31, 2008. On January 22, 2009, we received notice that Swiss Pharma submitted a petition to the Supreme Court of the State of New York, County of New York seeking to confirm and to enter a judgment on the Arbitration Award. On February 17, 2009, we filed an answer to Swiss Pharma's petition. A hearing has not yet been scheduled. We will continue to accrue interest at the rate of 5% per annum on the approximate $371,000 amount until such amount has been settled. We do not have sufficient cash or other current available assets to satisfy the arbitrator's award.
42
In February 2007, a former employee of the Company alleged an ownership interest in two of the Company’s provisional patent applications covering our discontinued product development program for Oleoyl-estrone. Also, without articulating precise legal claims, the former employee contends that the Company wrongfully characterized the former employee’s separation from employment as a resignation instead of a dismissal in an effort to harm the former employee’s immigration sponsorship efforts, and, further, to wrongfully deprive the former employee of the former employee’s alleged rights in two of the Company’s provisional patent applications. The former employee is seeking an unspecified amount in damages. The Company refutes the former employee’s contentions and intends to vigorously defend itself should the former employee file claims against the Company. There have been no further developments with respect to these contentions.
Development Commitments
At present the Company has no development commitments.
Hedrin
During 2008, the Company entered into the Hedrin JV agreement. The Hedrin JV is responsible for the development and commercialization of Hedrin for the North American market and all associated costs including clinical trials, if required, regulatory costs, patent costs, and future milestone payments owed to Thornton & Ross Ltd., or T&R, the licensor of Hedrin.
Topical PTH (1-34)
In July 2008 the Company announced the results of a phase 2a trial conducted with PTH 1-34 to evaluate the safety and preliminary efficacy of PTH 1-34 in the treatment of mild to moderate chronic plaque psoriasis. In the clinical trial, PTH 1-34 failed to show statistically or clinically meaningful improvements in the disease. The Company has conducted no further clinical activities with the product and is considering the next steps in the program, including returning the project to IGI under the terms of our license agreement.
Through our April 2005 acquisition of Tarpan Therapeutics, Inc., we acquired a Sublicense Agreement with IGI, Inc. dated April 14, 2004. Under the IGI sublicense agreement we hold the exclusive, world-wide, royalty bearing sublicense to develop and commercialize the licensed technology. Under the terms of the IGI sublicense agreement, we are responsible for the cost of the nonclinical and clinical development of the project, including research and development, manufacturing, laboratory and clinical testing and trials and marketing of licensed products.
43
The IGI sublicense agreement requires us to make certain milestone payments as follows: $300,000 payable upon the commencement of a Phase 2 clinical trial; $500,000 upon the commencement of a Phase 3 clinical trial; $1,500,000 upon the acceptance of an NDA application by the FDA; $2,400,000 upon the approval of an NDA by the FDA; $500,000 upon the commencement of a Phase 3 clinical trial for an indication other than psoriasis; $1,500,000 upon the acceptance of and NDA application for an indication other than psoriasis by the FDA; and $2,400,000 upon the approval of an NDA for an indication other than psoriasis by the FDA.
During 2007, we achieved the milestone of the commencement of Phase 2 clinical trial. As a result $300,000 became payable to IGI. This $300,000 is included in research and development expense for the year ended December 31, 2007. Payment was made to IGI in February 2008. In addition, we are obligated to pay IGI, Inc. an annual royalty of 6% on annual net sales up to $200,000,000. In any calendar year in which net sales exceed $200,000,000, we are obligated to pay IGI, Inc. an annual royalty of 9% on such excess. Through December 31, 2008, sales have not commenced, therefore, we have not paid any such royalties.
IGI, Inc. may terminate the agreement (i) upon 60 days’ notice if we fail to make any required milestone or royalty payments, or (ii) if we become bankrupt or if a petition in bankruptcy is filed, or if we are placed in the hands of a receiver or trustee for the benefit of creditors. IGI, Inc. may terminate the agreement upon 60 days’ written notice and an opportunity to cure in the event we commit a material breach or default. Eighteen months from the date of the IGI sublicense agreement, we may terminate the agreement in whole or as to any portion of the PTH patent rights upon 90 days’ notice to IGI, Inc.
Altoderm
On April 3, 2007, the Company entered into a license agreement for “Altoderm” (the “Altoderm Agreement”) with T&R. Pursuant to the Altoderm Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altoderm, a topical skin lotion product candidate using sodium cromoglicate for the treatment of atopic dermatitis. In accordance with the terms of the Altoderm Agreement, the Company issued 125,000 shares of its common stock, valued at $112,500, and made a cash payment of $475,000 to T&R upon the execution of the agreement. These amounts have been included in research and development expense. Further, the Company agreed to make future milestone payments to T&R comprised of various combinations of cash and common stock in respective aggregate amounts of $5,675,000 and 875,000 shares of common stock upon the achievement of various clinical and regulatory milestones. The Company also agreed to pay royalties on net sales of products using the licensed patent rights at rates ranging from 10% to 20%, depending on the level of annual net sales, and subject to an annual minimum royalty payment of $1 million in each year following the first commercial sale of Altoderm. The Company may sublicense the patent rights. The Company agreed to pay T&R 30% of the royalties received by the Company under such sublicense agreements.
Subsequent to December 31, 2008 the Company terminated the Altoderm Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altoderm Agreement.
44
Altolyn
On April 3, 2007, the Company and T&R also entered into a license agreement for “Altolyn” (the “Altolyn Agreement”). Pursuant to the Altolyn Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altolyn, an oral formulation product candidate using sodium cromoglicate for the treatment of mastocytosis, food allergies, and inflammatory bowel disorder. In accordance with the terms of the Altolyn Agreement, the Company made a cash payment of $475,000 to T&R upon the execution of the agreement. This amount is included in research and development expense. Further, the Company agreed to make future cash milestone payments to T&R in an aggregate amount of $5,675,000 upon the achievement of various clinical and regulatory milestones. The Company also agreed to pay royalties on net sales of products using the licensed patent rights at rates ranging from 10% to 20%, depending on the level of annual net sales, and subject to an annual minimum royalty payment of $1 million in each year following the first commercial sale of Altolyn. The Company may sublicense the patent rights. The Company agreed to pay T&R 30% of the royalties received by the Company under such sublicense agreements.
Subsequent to December 31, 2008 the Company terminated the Altolyn Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altolyn Agreement.
Oleoyl-estrone
On July 9, 2007, the Company announced the results of its two Phase 2a clinical trials of oral OE. The results of both randomized, double-blind, placebo controlled studies, one in common obesity and the other in morbid obesity, demonstrated no statistically or clinically meaningful placebo adjusted weight loss for any of the treatment arms evaluated. Based on these results, the Company discontinued its Oleoyl-estrone programs in both common obesity and morbid obesity during 2007.
Propofol Lingual Spray
On July 9, 2007, the Company announced that it is discontinued development of Propofol Lingual Spray for pre-procedural sedation.
Research and Development Projects
Hedrin
In collaboration with Nordic and through the Hedrin JV we are developing Hedrin for the treatment of pediculosis (head lice). To date, Hedrin has been clinically studied in 326 subjects and is currently marketed as a device in Western Europe and as a pharmaceutical in the United Kingdom (U.K.).
In a randomized, controlled, equivalence clinical study conducted in Europe by T&R, Hedrin was administered to 253 adult and child subjects with head louse infestation. The study results, published in the British Medical Journal in June 2005, demonstrated Hedrin’s equivalence when compared to the insecticide treatment, phenothrin, the most widely used pediculicide in the U.K. In addition, according to the same study, the Hedrin-treated subjects experienced significantly less irritation (2%) than those treated with phenothrin (9%).
An additional clinical study published in the November 2007 issue of PLoS One, an international, peer-reviewed journal published by the Public Library of Science (PLoS), demonstrated Hedrin’s superior efficacy compared to a U.K. formulation of malathion, a widely used insecticide treatment in both Europe and North America. In this randomized, controlled, assessor blinded, parallel group clinical trial, 73 adult and child subjects with head lice infestations were treated with Hedrin or malathion liquid. Using intent-to-treat analysis, Hedrin achieved a statistically significant cure rate of 70% compared to 33% with malathion liquid. Using the per-protocol analysis Hedrin achieved a highly statistically significant cure rate of 77% compared to 35% with malathion. In Europe it has been widely documented that head lice had become resistant to European formulations of malathion, and we believe this resistance had influenced these study results. To date, there have been no reports of resistance to U.S. formulations of malathion. Additionally, Hedrin treated subjects experienced no irritant reactions, and Hedrin showed clinical equivalence to malathion in its ability to inhibit egg hatching. Overall, investigators and study subjects rated Hedrin as less odorous, easier to apply, and easier to wash out, and 97% of Hedrin treated subjects stated they were significantly more inclined to use the product again versus 31% of those using malathion.
45
Two new, unpublished Hedrin studies were completed by T&R in 2008. In the first, Hedrin achieved a 100% kill rate in vitro, including in malathion resistant head lice. In the other, a clinical field study conducted in Manisa province, a rural area of Western Turkey, Hedrin was administered to 36 adult and child subjects with confirmed head lice infestations. Using per protocol analysis, Hedrin achieved a 97% cure rate. Using intent-to-treat analysis, Hedrin achieved a 92% cure rate since 2 subjects were eliminated due to protocol violations. No subjects reported any adverse events.
In the U.S., Manhattan Pharmaceuticals, through the Hedrin JV, is pursuing the development of Hedrin as a medical device. In January 2009, the U.S. Food and Drug Administration (“FDA”) Center for Devices and Radiological Health (“CDRH”) notified H Pharmaceuticals that Hedrin had been classified as a Class III medical device. A Class III designation means that a Premarket Approval (“PMA”) Application will need to be obtained before Hedrin can be marketed in the U.S. The Company expects to be required to complete at least one clinical trial as part of that PMA Application.
To date, we have incurred $1,084,000 of project costs for the development of Hedrin. During 2008, $14,000 of these costs were incurred. We do not expect to incur any future costs as the Hedrin JV is now responsible for all costs associated with Hedrin.
Topical PTH (1-34).
As a result of our merger with Tarpan Therapeutics in 2005, we hold an exclusive, worldwide license to develop and commercialize Topical PTH (1-34) for the treatment of psoriasis. Tarpan acquired the exclusive, worldwide rights pursuant to a 2004 license agreement with IGI, Inc (“IGI”).
In April 2006, we encountered a stability issue with the original topical PTH (1-34) product which utilized IGI’s Novosome® formulation technology. In order to resolve that stability issue we created a new topical gel version of PTH (1-34) and filed new patent applications in the U.S. for this new proprietary formulation.
In September 2007, the U.S. FDA accepted our Investigational New Drug (“IND”) application for this new gel formulation of Topical PTH (1-34), and in October 2007, we initiated and began dosing subjects in a Phase 2a clinical study of Topical PTH (1-34) for the treatment of psoriasis. This U.S., multi-center, randomized, double-blind, vehicle-controlled, parallel group study was designed to evaluate safety and preliminary efficacy of Topical PTH (1-34) in patients with mild to moderate psoriasis. Approximately 54 subjects were enrolled and randomized to receive one of two dose levels of Topical PTH (1-34), or the gel vehicle (placebo), for an 8 week treatment period. In this study the vehicle was the topical gel (“GEL”) without the active ingredient, PTH (1-34). In July 2008, we announced the results of the Phase 2a study where Topical PTH (1-34) failed to demonstrate a statistically significant or clinically meaningful improvement in psoriasis.
In July 2008 we announced the results of a Phase 2a clinical study where PTH (1-34) failed to show statistically or clinically meaningful improvements in psoriasis as compared to the vehicle (placebo). The Company has conducted no further clinical activities with PTH (1-34) and intends to return the project to IGI under the terms of the license agreement.
The gel vehicle (placebo) used in the above-mentioned study is the Company’s proprietary topical GEL and it unexpectedly showed evidence of psoriasis improving properties. At the end of week 2, 15% of study subjects treated with the GEL achieved a clear or almost clear state. At the end of week 4, 20% of subjects treated with the GEL had achieved a clear or almost clear state, and at the end of week 8, 25% of subjects had achieved a clear or almost clear state. The Company owns worldwide rights to this topical GEL and is exploring the possibility of developing it as an OTC product for mild psoriasis.
46
To date, we have incurred $6,504,000 of project costs related to our development of Topical PTH (1-34). These project costs have been incurred since April 1, 2005, the date of the Tarpan Therapeutics acquisition. During 2008, $1,382,000 of these costs were incurred.
Altoderm
In April 2007 we entered into a license agreement with T&R, pursuant to which we acquired exclusive rights to develop and commercialize Altoderm in North America. Altoderm is a novel, proprietary formulation of topical cromolyn sodium and is designed to enhance the absorption of cromolyn sodium into the skin in order to treat pruritus (itch) associated with dermatologic conditions including atopic dermatitis (eczema).
Atopic Dermatitis (Eczema)
Atopic dermatitis, also know as eczema, is a chronic disease of the skin that is believed to be caused by a combination of hereditary and environmental factors. The main symptoms of atopic dermatitis include dry, itchy skin leading to rashes on the face, hands, feet, along with inside the elbows and behind the knees. Scratching results in redness, swelling, cracking, “weeping” clear fluid, and crusting or scaling.
Product Development
In a Phase 3, randomized, double-blind, placebo-controlled, parallel-group, clinical study (conducted in Europe by T&R.) the compound was administered for 12 weeks to 114 subjects with moderately severe atopic dermatitis. The placebo (vehicle) used in this study was the Altoderm product without the active ingredient. In the study results, published in the British Journal of Dermatology in February 2005, Altoderm demonstrated a statistically significant reduction (36%) in atopic dermatitis symptoms. During the study, subjects were permitted to continue with their existing treatment, in most cases this consisted of emollients and topical steroids. A positive secondary outcome of the study was a 35% reduction in the use of topical steroids for the Altoderm treated subjects. Further analysis of the clinical data, performed by Manhattan Pharmaceuticals, showed that Altoderm treated subjects also experienced a 57% reduction in pruritus.
On March 6, 2008, Manhattan Pharmaceuticals announced it had successfully completed a pre-IND meeting with the FDA. Based on a review of the submitted package for Altoderm, including data from the two previously reported Phase 3 clinical studies, the FDA determined that following completion of certain nonclinical studies, and the acceptance of an IND, Phase 2 clinical studies may be initiated in the U.S. The FDA also concurred that the proposed indication of pruritus associated with dermatologic conditions including atopic dermatitis can be pursued.
In a second, Phase 3, randomized, double-blind, vehicle-controlled clinical study (also conducted in Europe by T&R) Altoderm was safe and well tolerated, and showed a trend toward improvement in pruritus, but the efficacy results were inconclusive. Altoderm treated subjects and vehicle only treated subjects experienced a similar improvement (each greater than 30%), and therefore, the study did not achieve statistical significance.
Subsequent to December 31, 2008 the Company terminated the Altoderm Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altoderm Agreement.
To date we have incurred $1,110,000 for the development of Altoderm. During 2008, $98,000 of these costs were incurred.
47
Altolyn
In April 2007 we entered into a license agreement with T&R, pursuant to which we acquired exclusive rights to develop and commercialize Altolyn in North America. Altolyn is a novel, proprietary oral tablet formulation of cromolyn sodium designed to treat mastocytosis and possibly other gastrointestinal disorders such as food allergy and symptoms of irritable bowel syndrome.
On March 6, 2008, Manhattan Pharmaceuticals announced it had successfully completed a pre-IND meeting with the FDA. Based on a review of the submitted package for Altolyn, the FDA concurred that the proposed indication of mastocytosis can be pursued and that the 505(b)(2) NDA would be an acceptable approach provided a clinical bridge is established between Altolyn and Gastrocrom®, the oral liquid formulation of cromolyn sodium currently approved in the U.S. to treat mastocytosis. Section 505(b)(2) of the Food, Drug and Cosmetic Act allows the FDA to approve a follow-on drug on the basis of data in the scientific literature or data used by FDA in the approval of other drugs.The FDA also affirmed that a single, Phase 3 study demonstrating the efficacy of Altolyn over placebo, may be sufficient to support a product approval in the U.S. In addition, the FDA also concurs that no additional nonclinical studies will be required to support an IND application. The Company is working with T&R and the current U.K. manufacturer of Altolyn to develop a Good Manufacturing Process (“cGMP”) compliant manufacturing process.
Subsequent to December 31, 2008 the Company terminated the Altolyn Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altolyn Agreement.
To date we have incurred $831,000 for the development of Altolyn. During 2008, $98,000 of these costs were incurred.
Oleoyl-estrone
On July 9, 2007, the Company announced the results of its two Phase 2a clinical trials of oral OE. The results of both randomized, double-blind, placebo controlled studies, one in common obesity and the other in morbid obesity, demonstrated no statistically or clinically meaningful placebo adjusted weight loss for any of the treatment arms evaluated. Based on these results, the Company discontinued its Oleoyl-estrone programs in both common obesity and morbid obesity.
To date, we have incurred $15,510,000 for the development of OE, none of which was incurred during 2008.
Propofol Lingual Spray
On July 9, 2007, the Company announced that it discontinued development of Propofol Lingual Spray for pre-procedural sedation.
To date, we have incurred $2,984,000 for the development of Propofol Lingual Spray, none of which was incurred during 2008.
48
Summary of Contractual Commitments
Employment Agreement
The Company has employment agreements with two employees for the payment of aggregate annual base salary of $675,000 as well as performance based bonuses. These agreements have a remaining term of three months for one employee and six months for the second employee and have a remaining obligation of $226,000 as of December 31, 2008. As per the terms of the Secured 12% Notes sold in the fourth quarter of 2008 and the first quarter of 2009 management, comprised of the two employees under contract, has agreed to reduce their salaries effective as of October 1, 2008. If the Company sells less than $1.5 million of Secured 12% Notes then their salaries shall be reduced by one-third. If the Company sells at least $1.5 million but less than $2 million of Secured 12% Notes then their salaries shall be reduced by 20%. The Company sold $1.725 million of Secured 12% Notes, management therefore was paid 80% of their salaries during the fourth quarter of 2008. Also as per the terms of the Secured 12% Notes the reduction in management’s salaries shall be further reduced to 10% if the Company realizes gross proceeds of $500,000 or more from other sources and shall be reduced to 0% if the Company realizes gross proceeds of $1,000,000 or more from other sources. In February 2009 the Company received a $500,000 milestone payment from the Hedrin JV; therefore management’s salaries are currently reduced by 10%.
Leases
Rent expense for the years ended December 31, 2008 and 2007 was $139,636 and $141,012, respectively. Future minimum rental payments subsequent to December 31, 2008 under an operating lease for the Company’s office facility are as follows:
| Years Ending December 31, | Commitment | |||
| 2009 | $ | 63,900 | ||
| 2010 and subsequent | $ | 0 | ||
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements.
Critical Accounting Policies
In December 2001, the SEC requested that all registrants discuss their most “critical accounting policies” in management’s discussion and analysis of financial condition and results of operations. The SEC indicated that a “critical accounting policy” is one which is both important to the portrayal of the company’s financial condition and results and requires management’s most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect certain reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
49
Research and Development Expenses
All research and development costs are expensed as incurred and include costs of consultants who conduct research and development on behalf of the Company and its subsidiaries. Costs related to the acquisition of technology rights and patents for which development work is still in process are expensed as incurred and considered a component of research and development costs.
The Company often contracts with third parties to facilitate, coordinate and perform agreed upon research and development of a new drug. To ensure that research and development costs are expensed as incurred, the Company records monthly accruals for clinical trials and preclinical testing costs based on the work performed under the contracts.
These contracts typically call for the payment of fees for services at the initiation of the contract and/or upon the achievement of certain milestones. This method of payment often does not match the related expense recognition resulting in either a prepayment, when the amounts paid are greater than the related research and development costs expensed, or an accrued liability, when the amounts paid are less than the related research and development costs expensed.
Share-Based Compensation
We have stockholder-approved stock incentive plans for employees, directors, officers and consultants. Prior to January 1, 2006, we accounted for the employee, director and officer plans using the intrinsic value method under the recognition and measurement provisions of Accounting Principles Board (“APB”) Opinion No. 25, “Accounting for Stock Issued to Employees” and related interpretations, as permitted by Statement of Financial Accounting Standards (“SFAS” or “Statement”) No. 123, “Accounting for Stock-Based Compensation.”
Effective January 1, 2006, the Company adopted SFAS No. 123(R), “Share-Based Payment,” (“Statement 123(R)”) for employee options using the modified prospective transition method. Statement 123(R) revised Statement 123 to eliminate the option to use the intrinsic value method and required the Company to expense the fair value of all employee options over the vesting period. Under the modified prospective transition method, the Company recognized compensation cost for the years ended December 31, 2008 and 2007 which includes a) period compensation cost related to share-based payments granted prior to, but not yet vested, as of January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of Statement 123; and b) period compensation cost related to share-based payments granted on or after January 1, 2006, based on the grant date fair value estimated in accordance with Statement 123(R). In accordance with the modified prospective method, the Company has not restated prior period results.
New Accounting Pronouncements
In February 2008, the FASB issued two Staff Positions on SFAS 157: (1) FASB Staff Position No. FAS 157-1 (“FAS 157-1”), “Application of FASB Statement No. 157 to FASB Statement No. 13 and Other Accounting Pronouncements That Address Fair Value Measurements for Purposes of Lease Classification or Measurement Under Statement 13,” and (2) FASB Staff Position No. FAS 157-2 (“FAS 157-2”), “Effective Date of FASB Statement No 157.” FAS 157-1 excludes FASB Statement No. 13, Accounting for Leases, as well as other accounting pronouncements that address fair value measurements on lease classification or measurement under Statement 13, from SFAS 157’s scope. FAS157-2 partially defers Statement 157’s effective date. The adoption of FAS 157-1 and FAS 157-2 did not have a material impact on its financial statements.
50
In October 2008, the FASB issued FASB Staff Position No. FAS 157-3 "Determining the Fair Value of a Financial Asset When the Market for That Asset is Not Active" ("FAS 157-3"), which is effective upon issuance for all financial statements that have not been issued. FAS 157-3 clarifies the application of SFAS 157, in a market that is not active. FAS 157-3 does not have a material impact on the Company’s financial position, financial performance or cash flows.
In March 2008, the FASB issued SFAS No. 161 "Disclosures About Derivative Instruments and Hedging Activities - an amendment of FASB Statement No. 133" ("SFAS 161"). SFAS 161 amends SFAS 133 by requiring expanded disclosures about an entity's derivative instruments and hedging activities. SFAS 161 requires qualitative disclosures about objectives and strategies for using derivatives, quantitative disclosures about fair value amounts of and gains and losses on derivative instruments, and disclosures about credit-risk-related contingent features in derivative instruments. SFAS 161 is effective for the Company as of January 1, 2009. The Company does not believe that SFAS 161 will have any impact on its financial statements.
In May 2008, the FASB issued SFAS No. 163, “Accounting for Financial Guarantee Insurance Contracts” (“SFAS 163”). SFAS 163 requires recognition of a claim liability prior to an event of default when there is evidence that credit deterioration has occurred in an insured financial obligation. SFAS 163 carifies how FAS 60 applies to financial guarantee insurance contracts, including the recognition and measurement to be used to account for premium revenue and claim liabilities. SFAS 163 also requires expanded disclosures about financial guarantee insurance contracts. SFAS 163 is effective for years beginning after December 15, 2008, and interim periods within those years, except for certain disclosure requirements which are effective for the first period (including interim periods) beginning after May 23, 2008. The Company does not believe that SFAS 163 will have any impact on its financial statements.
The FASB and the Securities and Exchange Commission had issued certain other accounting pronouncements as of December 31, 2008 that will become effective in subsequent periods; however, the Company does not believe that any of those pronouncements would have significantly affected its financial accounting measures or disclosures had they been in effect during the years ended December 31, 2008 and 2007 and for the period from August 6, 2001 (inception) to December 31, 2008 or that will have a significant effect at the time they become effective.
In December 2007, the FASB issued SFAS No. 141(R), a revised version of SFAS No. 141, “Business Combinations” (“SFAS 141R”). The revision is intended to simplify existing guidance and converge rulemaking under U.S. generally accepted accounting principles with international accounting standards. SFAS 141R applies prospectively to business combinations where the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. An entity may not apply it before that date. The Company does not expect the adoption of SFAS 141(R) to have a significant impact on our results of operations or financial position.
In June 2008, the FASB ratified EITF Issue No. 07-5, "Determining Whether an Instrument (or an Embedded Feature) Is Indexed to an Entity's Own Stock" (“EITF 07-5”). EITF 07-5 provides that an entity should use a two step approach to evaluate whether an equity-linked financial instrument (or embedded feature) is indexed to its own stock, including evaluating the instrument's contingent exercise and settlement provisions. It also clarifies the impact of foreign currency denominated strike prices and market-based employee stock option valuation instruments on the evaluation. EITF 07-5 is effective for fiscal years beginning after December 15, 2008. The Company is currently assessing the impact of EITF 07-5 on its financial position and results of operations.
51
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
For a list of the financial statements filed as part of this report, see the Index to Financial Statements beginning at Page F-1 of this Annual Report.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE.
None.
ITEM 9A(T). CONTROLS AND PROCEDURES.
Evaluation of Disclosure Controls and Procedures
As of December 31, 2008, we carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Securities Exchange Act of 1934, as amended). Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of that date in alerting them on a timely basis to material information required to be disclosed our reports to the Securities and Exchange Commission. There were no changes in our internal controls over financial reporting during the quarter ended December 31, 2008 that have materially affected, or are likely to materially affect, our internal controls over financial reporting.
Our management, including our Chief Executive Officer and its Chief Financial Officer, does not expect that disclosure controls or internal controls over financial reporting will prevent all errors or all instances of fraud, even as the same are improved to address any deficiencies. The design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected.
Because of the inherent limitation of a cost-effective control system, misstatements due to error or fraud may occur and not be detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls.
Management’s Report on Internal Control
Our management is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. As defined by the SEC, internal control over financial reporting is a process designed by, or under the supervision of our principal executive and principal financial officers and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the financial statements in accordance with U.S. generally accepted accounting principles.
52
Our internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of the financial statements in accordance with U.S. generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In connection with the preparation of our annual financial statements, management has undertaken an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2008, based on criteria established in Internal Control – Integrated framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO Framework. Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of those controls.
Based on this evaluation, management has concluded that our internal control over financial reporting is effective as of December 31, 2008.
This annual report does not include an attestation report of the Company’s independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the Company’s independent registered public accounting firm pursuant to temporary rules of the SEC that permit the Company to provide only management’s report on internal control in this report.
ITEM 9B. OTHER INFORMATION
On March 28, 2008 the employment agreement between the Company and Douglas Abel, the Company’s president and chief executive officer, was extended by mutual agreement for a period of one year, through April 1, 2009.
53
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
Directors
The name and age of each of our six directors as of March 27, 2009, his position with us, his principal occupation, and the period during which such person has served as a director of our company are set forth below. All directors hold office until the next annual meeting of shareholders or until their respective successors are elected and qualified.
| Name | Age | Position(s) Held | Director Since | ||||
| Douglas Abel | 47 | President, Chief Executive Officer and Director | 2005 | ||||
| Neil Herskowitz | 52 | Director | 2004 | ||||
| Malcolm Hoenlein | 65 | Director | 2004 | ||||
| Timothy McInerney | 48 | Director | 2004 | ||||
| Richard I. Steinhart | 51 | Director | 2004 | ||||
| Michael Weiser, M.D. | 46 | Director | 2003 |
Douglas Abel has been our President and Chief Executive Officer and a director of our company since April 2005. Mr. Abel was President and CEO of Tarpan Therapeutics, Inc., a privately-held biopharmaceutical company, from November 2004 until April 2005, when Tarpan was acquired by us. Prior to becoming President and CEO of Tarpan, Mr. Abel served as Vice President of the Dermatology Business Unit at Biogen Idec where he worked from August 2000 to November 2004. While at Biogen, he led more than 100 employees to support the launch of AMEVIVE®. Before that, Mr. Abel was at Allergan Pharmaceuticals from December 1987 to August of 2000, with his most recent position being Director of BOTOX® Marketing. Mr. Abel received his A.B. in chemistry from Lafayette College and an M.B.A. from Temple University.
Neil Herskowitz was appointed to our Board of Directors in July 2004. He has served as the Managing Member of ReGen Partners LLC, an investment fund located in New York, and as the President of its affiliate, Riverside Contracting LLC since June 1998. Mr. Herskowitz currently serves as a director of Innovive Pharmaceuticals (OTCBB: IVPH) a publicly traded pharmaceutical development company. He also serves on the board of directors of Starting Point Services for Children, a not-for-profit corporation, and of Vacation Village, a 220-unit development in Sullivan County, New York. Mr. Herskowitz received a B.B.A. in Finance from Bernard M. Baruch College in 1978.
Malcolm Hoenlein was appointed to our Board of Directors in July 2004. Since January 2001, he is also a director of Keryx Biopharmaceuticals, Inc. (Nasdaq: KERX). Mr. Hoenlein currently serves as the Executive Vice Chairman of the Conference of Presidents of Major American Jewish Organizations, a position he has held since 1986. He also serves as a director of Bank Leumi. Mr. Hoenlein received his B.A. from Temple University and his M.A. from the University of Pennsylvania.
Timothy McInerney has been a director of our company since July 2004. Mr. McInerney serves as a partner at Riverbank Capital Securities, Inc., a position he has held since June 2007. Mr. McInerney currently serves on the board of directors of ZIOPHARM Oncology Inc. (NASDAQ: ZIOP). From 1992 to March 2007, Mr. McInerney was a Managing Director of Paramount BioCapital, Inc. where he oversaw the overall distribution of Paramount’s private equity product. Prior to 1992, Mr. McInerney was a research analyst focusing on the biotechnology industry at Ladenburg, Thalman & Co. Prior to that, Mr. McInerney held equity sales positions at Bear, Stearns & Co. and Shearson Lehman Brothers, Inc. Mr. McInerney also worked in sales and marketing for Bristol-Myers Squibb. He received his B.S. in pharmacy from St. John’s University at New York. He also completed a post-graduate residency at the New York University Medical Center in drug information systems.
54
Richard I. Steinhart has been a director of our company since July 2004. Since April 2006, Mr. Steinhart has served as Chief Financial Officer of Electro-Optical Sciences, Inc., a publicly-held medical device company. From May 1992 to April 2006, Mr. Steinhart was principal of Forest Street Capital, a boutique investment banking, venture capital, and management consulting firm. Prior to Forest Street Capital, from May 1991 to May 1992, he was the Vice President and Chief Financial Officer of Emisphere Technologies, Inc., a publicly held biopharmaceutical company that is working to develop and commercialize a proprietary oral drug delivery system. Prior to joining Emisphere Technologies, Mr. Steinhart spent seven years at CW Group, Inc., a venture capital firm focused on medical and healthcare investments, where he was a General Partner and Chief Financial Officer. Mr. Steinhart has previously served as a director of a number of privately-held companies, including ARRIS Pharmaceuticals, Inc., a biotechnology company involved with rational drug design; Membrex, Inc., a laboratory equipment manufacturing company; and Photest, Inc., a diagnostics company. He began his career working as a certified public accountant and continues to be a New York State Certified Public Accountant. Mr. Steinhart holds a Bachelors of Business Administration and Masters of Business Administration from Pace University.
Michael Weiser, M.D., Ph.D., has served as a director of our company since February 2003. Dr. Weiser currently serves as founder and co-chairman of Actin Biomed, a position he has held since December 2006. Previously, he served as Director of Research of Paramount BioSciences, Inc. Dr. Weiser completed his Ph.D. in Molecular Neurobiology at Cornell University Medical College and received his M.D. from New York University School of Medicine, where he also completed a Postdoctoral Fellowship in the Department of Physiology and Neuroscience. Dr. Weiser currently serves on the boards of directors of Hana Biosciences, Inc. (NASDAQ: HNAB), Chelsea Therapeutics International Ltd. (NASDAQ: CHTP), Emisphere Technologies Inc. (NASDAQ: EMIS), ZIOPHARM Oncology Inc. (NASDAQ: ZIOP), and VioQuest Pharmaceuticals Inc. (OTCBB: VQPH), as well as several other privately held biotechnology companies.
There are no family relationships among any of our executive officers, directors and key employees.
Independence of the Board of Directors
Our common stock has not been listed on a national securities exchange since we voluntarily de-listed our shares from the American Stock Exchange, or AMEX, effective March 26, 2008 and therefore, we are not subject to any corporate governance requirements regarding independence of board or committee members. However, we have chosen the definition of independence contained in the AMEX rules as a benchmark to evaluate the independence of its directors. Under the AMEX listing standards, an "independent director" of a company means a person who is not an officer or employee of the company or its subsidiaries and who the board of directors has affirmatively determined does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. After review of all relevant transactions or relationships between each director, or any of his family members, and our company, our senior management and our independent registered public accounting firm, the Board has determined that all of our directors are independent directors within the meaning of the applicable AMEX listing standard, except for Mr. Abel, our President and Chief Executive Officer.
55
Board Committees
The Board of Directors has three standing committees: an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee. The following table provides membership for each of the Board committees:
| Name of Committee | Membership | |
| Audit | Messrs. Herskowitz, Hoenlein and Steinhart (Chair) | |
| Compensation | Messrs. Herskowitz, Hoenlein, Steinhart and Weiser (Chair) | |
| Nominating and Governance | Messrs. Herskowitz, Hoenlein and Steinhart (Chair) |
Audit Committee
The Audit Committee oversees our accounting and financial reporting process. For these purposes, the Audit Committee performs several functions. For example, the Committee evaluates and assesses the qualifications of the independent registered public accounting firm; determines the engagement of the independent registered public accounting firm; determines whether to retain or terminate the existing independent registered public accounting firm; reviews and approves the retention of the independent registered public accounting firm to perform any non-audit services; reviews the financial statements to be included in our Annual Report on Form 10-K; and discusses with management and the independent registered public accounting firm the results of the annual audit and the results of our quarterly financial statements. The Board of Directors adopted a written Audit Committee Charter, a copy of which can be found on our company website at www.manhattanpharma.com ..
Our Board of Directors has reviewed the definition of independence for Audit Committee members and has determined that each member of our Audit Committee is independent (as independence for audit committee members is currently defined under applicable SEC rules and the relevant AMEX listing standards. The Board has further determined that Mr. Steinhart qualifies as an “audit committee financial expert,” as defined by applicable rules of the SEC.
Compensation Committee
The Compensation Committee of the Board of Directors oversees our compensation policies, plans and programs. The Compensation Committee reviews and approves corporate performance goals and objectives relevant to the compensation of our executive officers and other senior management; reviews and recommends to the Board the compensation and other terms of employment of our Chief Executive Officer and our other executive officers; administers our equity incentive and stock option plans; and makes recommendations to the Board concerning the issuance of awards pursuant to those plans. All current members of the Compensation Committee, except for Dr. Weiser who serves as Chair of the Compensation Committee, are independent (as independence is currently defined under applicable AMEX listing standards). The Board of Directors has adopted a written charter of the Compensation Committee, a copy of which can be found on our company website at www.manhattanpharma.com.
Nominating and Governance Committee
The Nominating and Governance Committee considers and recommends to the Board persons to be nominated for election by the stockholders as directors. In addition to nominees recommended by directors, the Nominating and Governance Committee will consider nominees recommended by stockholders if submitted in writing to our Secretary at the address of Company’s principal offices. The Board believes that any candidate for director, whether recommended by stockholders or by the Board, should be considered on the basis of all factors relevant to the needs of our company and the credentials of the candidate at the time the candidate is proposed. Such factors include relevant business and industry experience and demonstrated character and judgment. All current members of the Nominating and Corporate Governance Committee are independent (as independence is currently defined under applicable AMEX listing standards). The Board of Directors adopted a written charter of the Nominating and Governance Committee, a copy of which can be found on our company website at www.manhattanpharma.com.
56
Communication with the Board of Directors
Although we have not adopted a formal process for stockholder communications with our Board of Directors, we believe stockholders should have the ability to communicate directly with the Board so that their views can be heard by the Board or individual directors, as applicable, and that appropriate and timely responses are provided to stockholders. All communications regarding general matters should be directed to our Secretary at the address below and should prominently indicate on the outside of the envelope that it is intended for the complete Board of Directors or for any particular director(s). If no designation is made, the communication will be forwarded to the entire board. Stockholder communications to the Board should be sent to: Corporate Secretary, Attention: Board of Directors (or name(s) of particular directors), Manhattan Pharmaceuticals, Inc., 48 Wall Street, New York, NY 10005.
Code of Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all officers, directors and employees of our company. A copy of our Code of Business Conduct and Ethics is available on our company’s website at www.manhattanpharma.com. If we make any substantive amendments to the Code of Business Conduct and Ethics or grant any waiver from a provision of the code to an executive officer or director, we will promptly disclose the nature of the amendment or waiver by filing with the SEC a current report on Form 8-K.
Executive Officers
Set forth below are the names, ages and titles of all of our executive officers as of February 13, 2009. All directors hold office until the next annual meeting of stockholders or until their respective successors are elected and qualified.
| Name | Age | Position | ||
| Douglas Abel | 47 | President & Chief Executive Officer and Director | ||
| Michael G. McGuinness | 55 | Chief Operating and Financial Officer & Secretary |
The biographies of our executive officers are set forth below.
Douglas Abel has been President and Chief Executive Officer and a director of our company since April 2005. His complete biography is set forth above under the caption “Management - Directors.”
Michael G. McGuinness has been our Chief Financial Officer and Secretary since July 2006. Mr. McGuinness was appointed Chief Operating Officer on April 1, 2008. Prior to joining Manhattan, Mr. McGuinness served as chief financial officer of Vyteris Holdings (Nevada), Inc. (OTCBB: VYHN), a product-based drug delivery company, from September 2001 to April 2006, and from 1998 to 2001 he was chief financial officer of EpiGenesis Pharmaceuticals, a privately-held biotechnology company. Mr. McGuinness received a BBA in public accounting from Hofstra University.
None of our executive officers is related to any other executive officer or to any of our directors.
57
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation of Executive Officers
The following table sets forth all of the compensation awarded to, earned by or paid to (i) each individual serving as our principal executive officer during our last completed fiscal year and (ii) the two most highly compensated executive officers, other than the principal executive officer, that served as an executive officer at the conclusion of the fiscal year ended December 31, 2008 and who received total compensation in excess of $100,000 during such fiscal year (collectively, the “named executives”).
| Nonqualified | ||||||||||||||||||||||||||||||
| Non-Equity | Deferred | |||||||||||||||||||||||||||||
| Option | Incentive Plan | Compensation | All Other | |||||||||||||||||||||||||||
Name and Principal Position | Year | Salary | Bonus | Awards | Compensation | Earnings | Compensation | Total | ||||||||||||||||||||||
| Douglas Abel | 2008 | $ | 338,750 | $ | 0 | (2) | $ | 153,244 | (4) | $ | 0 | $ | 0 | $ | 34,000 | (3) | $ | 525,994 | ||||||||||||
| Chief Executive Officer and President | 2007 | $ | 345,000 | $ | 90,000 | (2) | $ | 910,224 | (4) | $ | 0 | $ | 0 | $ | 42,333 | (3) | $ | 1,387,557 | ||||||||||||
| Michael McGuinness | 2008 | $ | 263,750 | $ | 0 | (2) | $ | 199,274 | (4) | $ | 0 | $ | 0 | $ | 9,000 | (5) | $ | 472,024 | ||||||||||||
| Chief Operating and Financial Officer, Secretary | 2007 | $ | 238,333 | $ | 50,000 | (2) | $ | 95,528 | (4) | $ | 0 | $ | 0 | $ | 9,000 | (5) | $ | 392,861 | ||||||||||||
Alan G. Harris (1) | 2008 | $ | 49,167 | $ | 0 | $ | 0 | (4) | $ | 0 | $ | 0 | $ | 0 | $ | 49,167 | ||||||||||||||
| Chief Medical Officer | 2007 | $ | 288,333 | $ | 0 | $ | 292,530 | (4) | $ | 0 | $ | 0 | $ | 9,000 | (5) | $ | 589,863 | |||||||||||||
| (1) | Dr. Harris’ employment with us ended effective December 31, 2007. |
| (2) | The Company accrued $180,000 for Mr. Abel and $100,00 for Mr. McGuinness as of December 31, 2007 for such bonuses but had not paid such bonuses as of that date. Payment of such bonuses were contingent upon our raising additional financing and were to be paid as follows: (i) 50% when we have consummated a financing transaction with gross proceeds to the Company of at least $1,000,000 (net of commissions) and (ii) the remaining 50% when we have consummated a financing transaction with gross proceeds (net of commissions) to us of at least $2.5 million (cumulative, including the $1 million financing transaction referred to above). We reached the condition (i) with the initial closing of the Hedrin JV transaction and such 50% was paid. We reached the condition (ii) with the closing of Secured 12% Notes transactions, however we did not pay the second 50% as we needed to retain the cash to fund operations. Mr. Abel and Mr. McGuinness have agreed to forfeit the second 50% payment. |
| (3) | For 2008 represents a payment in the amount of $25,000, which amount represents the approximate amount of additional expense incurred by Mr. Abel relating to his commuting between Boston and New York, without a tax “gross up”, and a matching contributions by us pursuant to our company’s 401(k) retirement plan of $9,000. For 2007 represents a payment in the amount of $33,333, which represents the approximate amount of additional expense incurred by Mr. Abel relating to his commuting between Boston and New York and a tax “gross up” to cover the additional tax liability to Mr. Abel from such payment, and a matching contributions by us pursuant to our company’s 401(k) retirement plan of $9,000. |
| (4) | Represents the amount of share-based costs recognized by us during 2008 and 2007 under SFAS No. 123(R). See Note 3 to our Financial Statements included in our annual reports for 2008 and 2007 on Form 10-K for the assumptions made in the valuation. |
| (5) | Represents matching contributions by us pursuant to our company’s 401(k) retirement plan. |
58
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information regarding the unexercised options held by each of our named executive officers as of December 31, 2008.
| Option Awards | ||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | ||||||||||
| Douglas Abel | 2,923,900 | 0 | $ | 1.50 | 04/01/2015 | |||||||||
| 83,333 | 166,667 | $ | 0.95 | 04/25/2017 | ||||||||||
| 433,333 | 866,667 | $ | 0.17 | 03/25/2018 | ||||||||||
| Michael McGuinness | 146,666 | 73,334 | $ | 0.70 | 07/10/2016 | |||||||||
| 40,000 | 20,000 | $ | 1.35 | 07/10/2016 | ||||||||||
| 106,667 | 213,333 | $ | 0.95 | 04/25/2017 | ||||||||||
| 366,667 | 733,333 | $ | 0.17 | 03/25/2018 | ||||||||||
Employment Agreements
Douglas Abel. We entered into an employment agreement and an extension to that employment agreement with Mr. Abel dated April 1, 2005, whereby Mr. Abel agreed to serve as our President and Chief Executive Officer for a period of four years in exchange for (i) an annual base salary of $300,000, subject to a retroactive increase in the amount of $25,000 upon our completing a financing transaction of at least $5,000,000, (ii) a signing bonus in the amount of $200,000, which was payable in two installments during the first year of the agreement, (iii) a discretionary performance-based bonus in an amount equal to up to 50% of Mr. Abel’s base salary, and (iv) an option to purchase 2,923,900 shares of our common stock at $1.50 per share with three-year annual vesting, purchasable for a 10-year term. In accordance with the terms of his employment agreement and as a result of our private placement financing that we completed in August 2005, Mr. Abel’s salary was increased to $325,000 retroactive to April 1, 2005. On November 19, 2008, at the first closing of our Secured 12% Notes private placement, we entered an amendment to the employment agreement, which provide for a reduction of up to one-third of the salary payable to Mr. Abel until we shall have received at least $2,500,000 of gross proceeds from the sale of the units or other sales of securities or from other revenue received by us in the operation of our business or any combination of the foregoing.
The employment agreement contains customary provisions relating to confidentiality, work-product assignment, non-competition and non-solicitation. In the event Mr. Abel’s employment is terminated by us (other than for cause) during the term of the agreement, including a termination upon a change of control (as defined in the agreement), we are required to pay a severance payment ranging from between 6 and 12 month of base salary, depending upon the circumstances of such termination.
Michael G. McGuinness. Mr. McGuinness’ employment with us is governed by an employment agreement dated July 7, 2006. The agreement provides for an initial three-year term of employment ending July 2009, subject to additional one-year renewal periods upon the mutual agreement of the parties. Pursuant to the agreement, Mr. McGuinness is entitled to an annual base salary of $205,000 and an annual bonus, payable in the discretion of our Board, of up to 30 percent of his annual base salary. Mr. McGuinness is also entitled to certain other fringe benefits that are made available to our senior executives from time to time, including medical and dental insurance and participation in our 401(k) plan. On November 19, 2008, at the first closing of our Secured 12% Notes private placement, we entered an amendment to the employment agreement, which provide for a reduction of up to one-third of the salary payable to Mr. McGuinness until we shall have received at least $2,500,000 of gross proceeds from the sale of the units or other sales of securities or from other revenue received by us in the operation of our business or any combination of the foregoing.
59
In addition, in accordance with the terms of the employment agreement, we issued to Mr. McGuinness two 10-year stock options pursuant to our 2003 Stock Option Plan. The first option relates to 220,000 shares of common stock and is exercisable at a price of $0.70, the closing price of our common stock on the date of his employment agreement. The second option relates to 60,000 shares and is exercisable at a price of $1.35 per share. Both options vest in three annual installments commencing July 10, 2007. To the extent Mr. McGuinness’ employment with us is terminated prior to the end of such 10-year term, the options shall remain exercisable for a period of 90 days.
Mr. McGuinness’ employment agreement further provides that in the event we terminate his employment with us other than as a result of death, for “cause,” “disability” or upon a “change of control” (as those terms are defined in the agreement), then (1) Mr. McGuinness will continue receiving his base salary and fringe benefits for a period of six months following such termination, provided, that our obligation to pay such compensation shall be offset by any amounts received by Mr. McGuinness from subsequent employment during such 6-month period, and (2) the vesting of the stock options issued to Mr. McGuinness in accordance with the employment agreement will accelerate and be deemed vested as of the date of termination and will remain exercisable for a period of 90 days following such termination. In the event we terminate Mr. McGuinness’ employment during the term of the agreement upon a “change of control” and, if at the time of such termination, the aggregate value of our outstanding common stock is less than $80 million, then (i) Mr. McGuinness will continue receiving his base salary and fringe benefits for a period of six months following such termination and (ii) the portions of the stock options issued in accordance with the employment agreement that have vested as of the date of such termination or that are scheduled to vest in the calendar year of such termination will be deemed vested and will remain exercisable for a period of 90 days following such termination.
Compensation of Directors
Non-employee directors are eligible to participate in our Non-employee Director Compensation Arrangement, which was adopted on January 30, 2007. Under the arrangement, non-employee directors are granted an option to purchase 50,000 shares of common stock upon their initial election or appointment to the board. Thereafter on an annual basis, non-employee directors are entitled to an option to purchase 50,000 shares of common stock. Each non-employee director is entitled to a retainer of $20,000 per year, payable on a quarterly basis. In addition, each such director shall be entitled to a fee of $1,000 for each meeting of the Board attended in person, or $500 for attending a meeting by telephone or other electronic means. Each non-employee director serving on a committee of the Board is entitled to a fee of $1,000 for each meeting of such committee attended by such director in person, or $500 for attending a committee meeting by telephone or other electronic means. Each non-employee director is also entitled to reimbursement for reasonable out-of-pocket expenses incurred in connection with the performance of his service as a director, including without limitation, travel related expenses incurred in connection with attendance at Board or Board committee meetings.
Due to our need to retain funds for the Company’s operations payment of fees to our directors were suspended for all periods subsequent to March 31, 2008.
60
The following table shows the compensation earned by each of our non-employee directors for the year ended December 31, 2008:
| Name | Fees Earned or Paid in Cash | Option Awards (1) | All Other Compensation | Total | ||||||||||||
| Neil Herskowitz | (2) | $ | 6,500 | $ | 14,767 | $ | - | $ | 21,267 | |||||||
| Malcolm Hoenlein | (3) | $ | 6,000 | $ | 14,767 | $ | - | $ | 20,767 | |||||||
| Timothy McInerney | (4) | $ | 5,500 | $ | 14,767 | $ | - | $ | 20,267 | |||||||
| Richard Steinhart | (5) | $ | 6,500 | $ | 14,767 | $ | - | $ | 21,267 | |||||||
| Michael Weiser | (6) | $ | 5,500 | $ | 14,767 | $ | - | $ | 20,267 | |||||||
| (1) | Represents the amount of share-based costs recognized by us during 2008 under SFAS No. 123(R). See Note 3 to our Financial Statements included in our annual report for 2008 on Form 10-K for the assumptions made in the valuation. |
| (2) | As of March 27, 2009, Mr. Herskowitz had options to purchase an aggregate of 216,010 shares of our common stock. |
| (3) | As of March 27, 2009, Mr. Hoenlein had options to purchase an aggregate of 216,010 shares of our common stock. |
| (4) | As of March 27, 2009, Mr. McInerney had options to purchase an aggregate of 250,000 shares of our common stock. |
| (5) | As of March 27, 2009, Mr. Steinhart had options to purchase an aggregate of 216,010 shares of our common stock. |
| (6) | As of March 27, 2009, Mr. Weiser had options to purchase an aggregate of 230,000 shares of our common stock. |
Compensation Committee Interlocks and Insider Participation
There were no interlocks or other relationships with other entities among our executive officers and directors that are required to be disclosed under applicable SEC regulations relating to compensation committee interlocks and insider participation.
61
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENMENT AND RELATED STOCKHOLDER MATTERS.
The following table sets forth information regarding ownership of shares of our common stock, as of February 13, 2009:
| o | by each person known by us to be the beneficial owner of 5% or more of our common stock; |
| o | by each of our directors and executive officers; and |
| o | by all of our directors and executive officers as a group. |
Except as otherwise indicated, each person and each group shown in the table has sole voting and investment power with respect to the shares of common stock indicated. For purposes of the table below, in accordance with Rule 13d-3 under the Securities Exchange Act of 1934, as amended, a person is deemed to be the beneficial owner, of any shares of our common stock over which he or she has or shares, directly or indirectly, voting or investment power or of which he or she has the right to acquire beneficial ownership at any time within 60 days. As used in this prospectus, "voting power" is the power to vote or direct the voting of shares and "investment power" includes the power to dispose or direct the disposition of shares. Common stock beneficially owned and percentage ownership as of March 27, 2009 was based on 70,624,232 shares outstanding. Unless otherwise indicated, the address of each beneficial owner is c/o Manhattan Pharmaceuticals, Inc., 48 Wall Street, New York, NY 10005.
Number of Shares Beneficially Owned (#) | Percentage Beneficially Owned (%) | |||||||
| Name of Beneficial Owners, Officers and Directors | ||||||||
Douglas Abel (1) | 4,036,232 | 5.41 | % | |||||
Michael McGuinness (2) | 1,167,334 | 1.63 | % | |||||
Michael Weiser (3) | 2,595,985 | 3.66 | % | |||||
Timothy McInerney (4) | 1,024,191 | 1.44 | % | |||||
Neil Herskowitz (5) | 188,311 | 0.27 | % | |||||
Richard I. Steinhart (6) | 183,869 | 0.26 | % | |||||
Malcolm Hoenlien (7) | 380,462 | 0.54 | % | |||||
All directors and officers as a group (8)(7 persons) | 9,576,384 | 12.43 | % | |||||
Lester Lipschutz (9) | ||||||||
| 1650 Arch Street, Philadelphia, PA 19103 | 8,941,873 | 12.66 | % | |||||
Lindsay Rosenwald (10) | ||||||||
| 787 Seventh Avenue, New York, NY 10019 | 4,023,259 | 5.61 | % | |||||
Nordic Biotech Venture Fund II K/S(11) | ||||||||
| Ostergrade 5, 3rd floor, DK-1100 | ||||||||
| Copenhagen K, Denmark | 66,666,666 | 48.56 | % | |||||
There were no directors, officers or beneficial owners of more than 10% of any class of equity securities of the Company or any other person subject to section 16 of the Exchange Act that failed to file on a timely basis reports required by section 16(a) of the Exchange Act during the most recent fiscal year or prior fiscal years.
62
| (1) | Includes 3,957,232 shares issuable upon exercise of vested portions of options and 24,000 shares issuable upon exercise of warrants. |
| (2) | Includes 1,133,334 shares issuable upon exercise of vested portions of options and 24,000 shares issuable upon exercise of warrants. |
| (3) | Includes 196,668 shares issuable upon the exercise of vested portions of options, and 151,754 shares issuable upon exercise of warrants. |
| (4) | Includes 216,668 shares issuable upon exercise of vested portions of options; and 139,863 shares issuable upon exercise of warrants. |
| (5) | Includes 182,678 shares issuable upon exercise of vested portions of options, and 43,444 shares issuance upon exercise of warrants; 77,288 shares held by Riverside Contracting, LLC, a limited liability company of which Mr. Herskowitz is a member holding 50% ownership and 44,168 shares held by ReGen Capital II, LLC, a limited liability company of which Mr. Herskowitz is a member holding 50% ownership. |
| (6) | Includes 182,678 shares issuable upon exercise of vested portions of options. |
| (7) | Includes 182,678 shares issuable upon exercise of vested portions of options. |
| (8) | Includes 5,861,936 shares issuable upon exercise of vested portions of options; 383,061 shares issuable upon the exercise of warrants; 77,288 shares held by Riverside Contracting, LLC, a limited liability company of which Mr. Herskowitz is a member holding 50% ownership and 44,168 shares held by ReGen Capital II, LLC, a limited liability company of which Mr. Herskowitz is a member holding 50% ownership. |
| (9) | Includes 8,941,873 shares of Common Stock held by separate trusts for the benefit of Dr. Rosenwald or his family with respect to which Mr. Lipschutz is either trustee or investment manager and in either case has investment and voting power. Mr. Lipschutz disclaims beneficial ownership of these shares, except to the extent of his pecuniary interest therein, if any. The foregoing information is derived from a Schedule 13G filed on behalf of the reporting person on August 1, 2007 |
| (10) | Includes 3,183,497 shares held directly by Dr. Rosenwald, 1,040,658 shares issuable upon the exercise of warrants, 80 shares held by the Dr. Rosenwald's wife, over which Dr. Rosenwald may be deemed to have sole voting and dispositive power, although he disclaims beneficial ownership of such shares except with regard to his pecuniary interest therein, if any, and 33 shares held by Dr. Rosenwald’s children, over which Dr. Rosenwald may be deemed to have sole voting and dispositive power, although he disclaims beneficial ownership of such shares except with regard to his pecuniary interest therein, if any. The foregoing information is derived from a Schedule 13G/A filed on behalf of the reporting person on February 13, 2008. |
| (11) | Includes 55,555,555 shares issuable upon exercise of Nordic's right to put all or a portion of Nordic Biotech Venture Fund II K/S' equity interest in H Pharmaceuticals K/S (formerly Hedrin Pharmaceuticals K/S), a Danish limited partnership, of which we and Nordic are partners and 11,111,111 shares issuable upon exercise of an outstanding warrant held by Nordic. Florian Schonharting and Christian Hansen have voting and investment control over such securities. |
63
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
Oleoylestrone Developments, SL
Pursuant to the terms of a license agreement dated February 15, 2002 between us and Oleoylestrone Developments, SL, or OED, which was terminated in November 2007, we had an exclusive, worldwide license to U.S. and foreign patents and patent applications relating to certain technologies. Although we were not obligated to pay royalties to OED, the license agreement required us to make certain performance-based milestone payments. As of February 13, 2009, OED held approximately 5.6% of our outstanding common stock. Additionally, Mr. Pons, a former member of our board of directors, is the chief executive officer of OED.
We also entered into a consulting agreement with OED, which became effective in February 2002 and was terminated along with the termination of the license agreement in November 2007. Pursuant to our consulting agreement, we paid OED a fee of $6,250 per month The fees associated with the consulting agreement were expensed as incurred. Pursuant the consulting agreement, OED agreed to appoint a member to serve as a member of our Scientific Advisory Board and to render consulting and advisory services to us. Such services included research, development and clinical testing of our technology as well as the reporting of the findings of such tests, assistance in the filing of patent applications and oversight and direction of efforts in regards to personnel for clinical development. For the periods ended December 31, 2007 and 2006 and from inception, fees paid to OED were $68,750, $325,000 and $931,250, respectively.
Paramount BioCapital, Inc.
In February 2007, we engaged Paramount BioCapital, Inc., as our placement agent in connection with the private placement. In consideration for its services, we paid aggregate cash commissions of approximately $600,000 and issued to Paramount a 5-year warrant to purchase an aggregate of 509,275 shares at an exercise price of $1.00 per share. At the time of the engagement, Timothy McInerney was an employee of Paramount BioCapital, Inc. or one of its affiliates. The sole shareholder of Paramount BioCapital, Inc. is Lindsay A. Rosenwald, M.D. Dr. Rosenwald beneficially owns more than 5% of our common stock. On March 30, 2007, we entered into a series of subscription agreements with various institutional and other accredited investors for the issuance and sale in a private placement of an aggregate of 10,185,502 shares of our common stock for total gross proceeds of approximately $8.56 million. Of the total amount of shares issued, 10,129,947 were sold at a per share price of $0.84, and an additional 55,555 shares were sold to an entity affiliated with Neil Herskowitz, a director of Manhattan, at a per share price of $0.90, the closing sale price of our common stock on March 29, 2007. Pursuant to the subscription agreements, we also issued to the investors 5-year warrants to purchase an aggregate of 3,564,897 shares of our common stock at an exercise price of $1.00 per share. The warrants are exercisable during the period commencing September 30, 2007 and ending March 30, 2012.
Private Placement
As described above, on March 30, 2007, we issued and sold in a private placement transaction an aggregate of 10,185,502 shares of our common stock. Of the total amount of shares issued, 10,129,947 were sold at a per share price of $0.84, and an additional 55,555 shares were sold to an entity affiliated with Neil Herskowitz, a director of Manhattan, at a per share price of $0.90, the closing sale price of our common stock on March 29, 2007. In addition to the shares of common stock, we also issued to the investors 5-year warrants to purchase an aggregate of 3,564,897 shares of our common stock at an exercise price of $1.00 per share. The warrants are exercisable during the period commencing September 30, 2007 and ending March 30, 2012. Accordingly, we received net proceeds of $7.9 million from the sale of these shares and warrants. We engaged Paramount BioCapital, Inc., as our placement agent in connection with the private placement, as discussed above.
64
The Hedrin JV
We and Nordic Biotech Venture Fund II K/S, or Nordic, entered into a joint venture agreement on January 31, 2008, which was amended on February 18, 2008 and on June 9, 2008. Pursuant to the joint venture agreement, in February 2008, (i) Nordic contributed cash in the amount of $2.5 million to H Pharmaceuticals K/S (formerly Hedrin Pharmaceuticals K/S), a newly formed Danish limited partnership, or the Hedrin JV, in exchange for 50% of the equity interests in the Hedrin JV, and (ii) we contributed certain assets to North American rights (under license) to our Hedrin product to the Hedrin JV in exchange for $2.0 million in cash and 50% of the equity interests in the Hedrin JV. On or around June 30, 2008, in accordance with the terms of the joint venture agreement, Nordic contributed an additional $1.25 million in cash to the Hedrin JV, $1.0 million of which was distributed to us and equity in the Hedrin JV was distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%.
Pursuant to the joint venture agreement, upon the classification by the U.S. Food and Drug Administration, or the FDA, of Hedrin as a Class II or Class III medical device, Nordic was required to contribute to the Hedrin JV an additional $1.25 million in cash, $0.5 million of which was to be distributed to us and equity in the Hedrin JV was to be distributed to each of us and Nordic sufficient to maintain our respective ownership interests at 50%. The FDA notified the Hedrin JV that Hedrin has been classified as a Class III medical device and in February 2009, Nordic made the $1.25 million investment in the Hedrin JV, the Hedrin JV made the $0.5 million milestone payment to us and equity in the Hedrin JV was distributed to us and Nordic Nordic sufficient to maintain our respective ownership interests at 50%. In accordance with the terms of the joint venture agreement, the Hedrin JV has received a total of $1.5 million cash to be applied toward the development and commercialization of Hedrin in North America.
The Hedrin JV will be responsible for the development and commercialization of Hedrin for the North American market and all associated costs including clinical trials, if required, regulatory costs, patent costs, and future milestone payments owed to Thornton & Ross Ltd., or T&R, the licensor of Hedrin. The Hedrin JV will engage us to provide management services to the Hedrin JV in exchange for an annualized management fee, which for 2008, on an annualized basis, is $527,000. The profits of the Hedrin JV will be shared by us and Nordic in accordance with our respective equity interests in the Hedrin JV, of which we each currently hold 50%, except that Nordic is entitled to receive a minimum return each year from the Hedrin JV equal to 6% on Hedrin sales, as adjusted for any change in Nordic’s equity interest in the Hedrin JV, before any distribution is made to us. If the Hedrin JV realizes a profit in excess of the Nordic minimum return in any year, then such excess shall first be distributed to us until our distribution and the Nordic minimum return are in the same ratio as our respective equity interests in the Hedrin JV and then the remainder, if any, is distributed to Nordic and us in the same ratio as our respective equity interests. However, in the event of a liquidation of the Hedrin JV, Nordic’s distribution in liquidation must equal the amount Nordic invested in the Hedrin JV ($5 million) plus 10% per year, less the cumulative distributions received by Nordic from the Hedrin JV before any distribution is made to us. If the Hedrin JV’s assets in liquidation exceed the Nordic liquidation preference amount, then any excess shall first be distributed to us until our distribution and the Nordic liquidation preference amount are in the same ratio as our respective equity interests in the Hedrin JV and then the remainder, if any, is distributed to Nordic and us in the same ratio as our respective equity interests. Further, in no event shall Nordic’s distribution in liquidation be greater than assets available for distribution in liquidation.
65
Pursuant to the terms of the joint venture agreement, Nordic has the right to nominate one person for election or appointment to our board of directors. The Hedrin JV's board of directors consists of four members, two members appointed by us and two members appointed by Nordic. Nordic has the right to appoint one of the directors as chairman of the board. The chairman has certain tie breaking powers. In the event that the final payment milestone described above is not achieved by March 30, 2009, then the Hedrin JV 's board of directors will increase to five members, two appointed by us and three appointed by Nordic.
Pursuant to the joint venture agreement, Nordic has the right to put all or a portion of its interest in the Hedrin JV in exchange for such number of shares of our common stock equal to the amount of Nordic’s investment in the Hedrin JV divided by $0.14, as adjusted from time to time for stock splits and other specified events, multiplied by a conversion factor, which is (i) 1.00 for so long as Nordic's distributions from the Hedrin JV are less than the amount of its investment, (ii) 1.25 for so long as Nordic's distributions from the Hedrin JV are less than two times the amount of its investment but greater than or equal to the amount of its investment amount, (iii) 1.50 for so long as Nordic's distributions from the Hedrin JV are less than three times the amount of its investment but greater than or equal to two times the amount of its investment amount, (iv) 2.00 for so long as Nordic's distributions from the Hedrin JV are less than four times the amount of its investment but greater than or equal to three times the amount of its investment amount and (v) 3.00 for so long as Nordic’s distributions from Hedrin JV are greater than or equal to four times the amount of its investment. The put right expires upon the earlier to occur of (i) February 25, 2018 and (ii) 30 days after the date when Nordic's distributions from the Hedrin JV exceed five times the amount Nordic has invested in the Hedrin JV (or 10 days after such date if we have provided Nordic notice thereof).
Pursuant to the joint venture agreement, we have the right to call all or a portion of Nordic's equity interest in the Hedrin JV in exchange for such number of shares of our common stock equal to the portion of Nordic's investment in the Hedrin JV that we call by the dollar amount of Nordic's investment as of such date in the Hedrin JV, divided by $0.14, as adjusted from time to time for stock splits and other specified events. The call right is only exercisable by us if the price of our common stock has closed at or above $1.40 per share for 30 consecutive trading days. During the first 30 consecutive trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 25% of the call right. During the second 30 consecutive trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 50% of the call right on a cumulative basis. During the third consecutive 30 trading days in which our common stock closes at or above $1.40 per share, we may exercise up to 75% of the call right on a cumulative basis. During the fourth consecutive 30 days in which our common stock closes at or above $1.40 per share, we may exercise up to 100% of the call right on a cumulative basis. Nordic may refuse the call, either by paying $1.5 million multiplied by the percentage of Nordic's investment being called or forfeiting an equivalent portion of the put right, calculated on a pro rata basis for the percentage of the Nordic equity interest called by us. The call right expires on February 25, 2013. For purposes of Nordic’s right to put, and our right to call, all or a portion of Nordic’s equity interest in the Hedrin JV, the amount of Nordic’s investment is currently $5,000,000.
Issuance of Secured Promissory Notes and Warrants
On September 11, 2008, we issued a secured promissory note in the principal amount of $12,000 to each of Douglas Abel, our President and Chief Executive Officer and a director of our company; Michael Weiser, a director of our company; Timothy McInerny, a director of our company; Neil Herskowitz, a director of our company, and Michael McGuiness, our Chief Financial Officer and Chief Operating Officer. Principal and interest on the notes are payable in cash on March 10, 2009 unless paid earlier by us. In connection with the issuance of the notes, we issued to each noteholder a 5-year warrant to purchase 24,000 shares of our common stock at an exercise price of $0.20 per share. We granted to the noteholders a continuing security interest in certain specific refunds, deposits and repayments due to us and expected to be repaid to us in the next several months. The secured 10% notes were repaid in February 2009 along with interest thereon.
66
We believe that all of the transactions set forth above were made on terms no less favorable to us than could have been obtained from unaffiliated third parties. All such transactions have been reviewed by the audit committee of our Board of Directors and approved by them. All future transactions between us and our officers, directors and principal shareholders and their affiliates will be on terms no less favorable than could be obtained from unaffiliated third parties and will be approved by our audit committee or another independent committee of our Board of Directors.
67
ITEM 14. PRINCIPAL ACCOUNTANTING FEES AND SERVICES.
Fees Billed to the Company by Its Independent Auditors
The following is a summary of the fees billed to us by J.H. Cohn LLP, our independent registered public accounting firm for professional services rendered for fiscal years ended December 31, 2008 and 2007:
| J.H. Cohn LLP | ||||||||
| Fee Category | Fiscal 2008 Fees | Fiscal 2007 Fees | ||||||
| Audit Fees | $ | 149,818 | $ | 103,940 | ||||
Audit-Related Fees (1) | 29,403 | 11,520 | ||||||
Tax Fees (2) | 12,500 | 18,708 | ||||||
All Other Fees (3) | - | - | ||||||
| Total Fees | $ | 191,721 | $ | 134,168 | ||||
| (1) | Audit-Related Fees consist principally of assurance and related services that are reasonably related to the performance of the audit or review of the Company’s financial statements but not reported under the caption “Audit Fees.” These fees include review of registration statements. |
| (2) | Tax Fees consist of fees for tax compliance, tax advice and tax planning. |
| (3) | All Other Fees consist of aggregate fees billed for products and services provided by the independent registered public accounting firm, other than those disclosed above. |
Policy on Audit Committee Pre-Approval of Audit and Permissible Non-Audit Services of Independent Registered Public Accounting Firm
At present, our audit committee approves each engagement for audit or non-audit services before we engage our independent registered public accounting firm to provide those services. Our audit committee has not established any pre-approval policies or procedures that would allow our management to engage our independent registered public accounting firm to provide any specified services with only an obligation to notify the audit committee of the engagement for those services. None of the services provided by our independent registered public accounting firm for fiscal 2008 was obtained in reliance on the waiver of the pre-approval requirement afforded in SEC regulations.
68
ITEM 15. EXHIBITS LIST
The following documents are included or incorporated by reference in this report.
| Exhibit No. | Description | |
| 2.1 | Agreement and Plan of Merger among the Company, Manhattan Pharmaceuticals Acquisition Corp. and Manhattan Research Development, Inc. (formerly Manhattan Pharmaceuticals, Inc.) dated December 17, 2002 (incorporated by reference to Exhibit 2.1 from Form 8-K filed March 5, 2003). | |
| 2.2 | Agreement and Plan of Merger among the Registrant, Tarpan Therapeutics, Inc. and Tarpan Acquisition Corp., dated April 1, 2005 (incorporated by reference to Exhibit 2.1 of the Registrant’s Form 8-K/A filed June 15, 2005). | |
| 3.1 | Certificate of incorporation, as amended through September 25, 2003 (incorporated by reference to Exhibit 3.1 to the Registrant’s Form 10-QSB for the quarter ended September 30, 2003). | |
| 3.2 | Bylaws, as amended to date (incorporated by reference from Registrant’s registration statement on Form SB-2, as amended (File No.33-98478)). | |
| 4.1 | Specimen common stock certificate (incorporated by reference from Registrant’s registration statement on Form SB-2, as amended (File No.33-98478)). | |
| 4.2 | Form of warrant issued by Manhattan Research Development, Inc., which automatically converted into warrants to purchase shares of the Registrant’s common stock upon the merger transaction with such company (incorporated by reference to Exhibit 4.1 to the Registrant’s Form 10-QSB for the quarter ended March 31, 2003). | |
| 4.3 | Form of warrant issued to placement agents in connection with the Registrant’s November 2003 private placement of Series A Convertible Preferred Stock and the Registrant’s January 2004 private placement (incorporated by reference to Exhibit 4.18 to the Registrant’s Registration Statement on Form SB-2 filed January 13, 2004 (File No. 333-111897)). | |
| 4.4 | Form of warrant issued to investors in the Registrant’s August 2005 private placement (incorporated by reference to Exhibit 4.1 of the Registrant’s Current Report on Form 8-K filed September 1, 2005). | |
| 4.5 | Form of warrant issued to placement agents in the Registrant’s August 2005 private placement (incorporated by reference to Exhibit 4.2 of the Registrant’s Form 8-K filed September 1, 2005). | |
| 4.6 | Warrant, dated April 30, 2008, issued to Nordic Biotech Venture Fund II K/S (incorporated by reference to Exhibit 4.6 of the Registrant’s Registration Statement on Form S-1 filed on May 1, 2008 (File No. 333-150580)). | |
| 4.7 | Form of Warrant issued to Noteholders on September 11, 2008 (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed on September 15, 2008) | |
| 4.8 | Form of Warrant issued to Noteholders on November 19, 2008 (incorporated by reference to Exhibit 10.6 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.1 | 1995 Stock Option Plan, as amended (incorporated by reference to Exhibit 10.18 to the Registrant’s Form 10-QSB for the quarter ended September 30, 1996). |
69
| 10.2 | Form of Notice of Stock Option Grant issued to employees of the Registrant from April 12, 2000 to February 21, 2003 (incorporated by reference to Exhibit 99.2 of the Registrant’s Registration Statement non Form S-8 filed March 24, 1998 (File 333-48531)). | |
| 10.3 | Schedule of Notices of Stock Option Grants, the form of which is attached hereto as Exhibit 4.2. | |
| 10.4 | Form of Stock Option Agreement issued to employees of the Registrant from April 12, 2000 to February 21, 2003 (incorporated by reference to Exhibit 99.3 to the Registrant’s Registration Statement on Form S-8 filed March 24, 1998 (File 333-48531)). | |
| 10.5 | License Agreement dated on or about February 28, 2002 between Manhattan Research Development, Inc. (f/k/a Manhattan Pharmaceuticals, Inc.) and Oleoyl-Estrone Developments SL (incorporated by reference to Exhibit 10.6 to the Registrant’s Amendment No. 2 to Form 10-QSB/A for the quarter ended March 31, 2003 filed on March 12, 2004). | |
| 10.6 | License Agreement dated April 4, 2003 between the Registrant and NovaDel Pharma, Inc. (incorporated by reference to Exhibit 10.1 to the Registrant’s Amendment No. 1 to Form 10-QSB/A for the quarter ended June 30, 2003 filed on March 12, 2004).++ | |
| 10.7 | 2003 Stock Option Plan (incorporated by reference to Exhibit 4.1 to Registrant’s Registration Statement on Form S-8 filed February 17, 2004). | |
| 10.8 | Employment Agreement dated April 1, 2005, between the Registrant and Douglas Abel (incorporated by reference to Exhibit 10.1 to the Registrant’s Form 8-K/A filed June 15, 2005). | |
| 10.9 | Sublicense Agreement dated April 14, 2004 between Tarpan Therapeutics, Inc., the Registrant’s wholly-owned subsidiary, and IGI, Inc. (incorporated by reference to Exhibit 10.109 to IGI Inc.’s Form 10-Q for the quarter ended March 31, 2004 (File No. 001-08568). | |
| 10.10 | Form of subscription agreement between the Registrant and the investors in the Registrant’s August 2005 private placement (incorporated by reference as Exhibit 10.1 to the Registrant’s Form 8-K filed September 1, 2005). | |
| 10.11 | Separation Agreement between the Registrant and Alan G. Harris December 21, 2007 (incorporated by reference to Exhibit 10.11 to the Registrant's Form 10-K filed March 31, 2008.) | |
| 10.12 | Employment Agreement dated July 7, 2006 between the Registrant and Michael G. McGuinness (incorporated by reference to Exhibit 10.1 of the Registrant’s Form 8-K filed July 12, 2006.) | |
| 10.13 | Summary terms of compensation plan for Registrant’s non-employee directors (incorporated by reference to Exhibit 10.1 of Registrant’s Form 8-K filed February 5, 2007). | |
| 10.14 | Form of Stock Option Agreement issued under the Registrant’s 2003 Stock Option Plan (Incorporated by reference to Exhibit 10.15 to the Registrant's Form 10-KSB filed April 2, 2078.) | |
| 10.15 | Exclusive License Agreement for “Altoderm” between Thornton & Ross Ltd. and Manhattan Pharmaceuticals, Inc. dates April 3, 2007. (Incorporated by reference to Exhibit 10.3 of the registrant’s form 10-Q for the quarter ended June 30, 2007 filed on August 14, 2007.) | |
| 10.16 | Exclusive License Agreement for “Altolyn” between Thornton &Ross Ltd. and Manhattan Pharmaceuticals, Inc. dated April 3, 2007. (Incorporated by reference to Exhibit 10.4 of the registrant’s form 10-Q for the quarter ended June 30, 2007 filed on August 14, 2007.) |
70
| 10.17 | Exclusive License Agreement for “Hedrin” between Thornton &Ross Ltd. , Kerris, S.A. and Manhattan Pharmaceuticals, Inc. dated June 26, 2007. (Incorporated by reference to Exhibit 10.5 of the registrant’s form 10-Q for the quarter ended June 30, 2007 filed on August 14, 2007.) | |
| 10.18 | Supply Agreement for “Hedrin” between Thornton & Ross Ltd. and Manhattan Pharmaceuticals, Inc. dated June 26, 2007. (Incorporated by reference to Exhibit 10.6 of the registrant’s form 10-Q for the quarter ended June 30, 2007 filed on August 14, 2007.) | |
| 10.19 | Joint Venture Agreement between Nordic Biotech Fund II K/S and Manhattan Pharmaceuticals, Inc. to develop and commercialize “Hedrin” dated January 31, 2008. | |
| 10.20 | Amendment No. 1, dated February 25, 2008, to the Joint Venture Agreement between Nordic Biotech Fund II K/S and Manhattan Pharmaceuticals, Inc. to develop and commercialize “Hedrin” dated January 31, 2008 (Incorporated by reference to Exhibit 10.20 to the Registrant's Form 10-K filed March 31, 2008). | |
| 10.21 | Omnibus Amendment to Joint Venture Agreement and Additional Agreements, dated June 9, 2008, among Manhattan Pharmaceuticals, Inc., Hedrin Pharmaceuticals K/S, Hedrin Pharmaceuticals General Partner ApS and Nordic Biotech Venture Fund II K/S. | |
| 10.22 | Assignment and Contribution Agreement between Hedrin Pharmaceuticals K/S and Manhattan Pharmaceuticals, Inc. dated February 25, 2008. (Incorporated by reference to Exhibit 10.21 to the Registrant's Form 10-K filed March 31, 2008.) | |
| 10.23 | Registration Rights Agreement between Nordic Biotech Venture Fund II K/S and Manhattan Pharmaceuticals, Inc. dated February 25, 2008. (Incorporated by reference to Exhibit 10.22 to the Registrant's Form 10-K filed March 31, 2008.) | |
| 10.24 | Letter Agreement, dated September 17, 2008, between Nordic Biotech Venture Fund II K/S and Manhattan Pharmaceuticals, Inc. | |
| 10.25 | Amendment to Employment Agreement by and between Manhattan Pharmaceuticals, Inc. and Douglas Abel (Incorporated by reference to Exhibit 10.23 to the Registrant's Form 10-K filed March 31, 2008.) | |
| 10.26 | Form of Secured Promissory Note, dated September 11, 2008 (Incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on September 15, 2008) | |
| 10.27 | Securities Purchase Agreement, dated November 19, 2008, by and among the Registrant and the investors listed on Exhibit A-1 and A-2 thereto (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.28 | Registration Rights Agreement, dated November 19, 2008, by and among the Registrant, the Placement Agent and the investors listed on Exhibit A thereto (incorporated by reference to Exhibit 10.2 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.29 | Security Agreement, dated November 19, 2008, by and among the Registrant and each person named on Exhibit A-1 and A-2 of the Securities Purchase Agreement (incorporated by reference to Exhibit 10.3 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) |
71
| 10.30 | Default Agreement, dated November 19, 2008, by and among the Registrant and the persons and entities listed on Schedule A thereto (incorporated by reference to Exhibit 10.4 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.31 | Form of 12% Senior Secured Promissory Note (incorporated by reference to Exhibit 10.5 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.32 | Amendment No. 2 to the Employment Agreement between the Registrant and Douglas Abel, dated November 19, 2008 (incorporated by reference to Exhibit 10.7 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.33 | Amendment No. 1 to the Employment Agreement between the Registrant and Michael McGuinness, dated November 19, 2008 (incorporated by reference to Exhibit 10.8 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 10.34 | Form of Placement Agent Warrant (incorporated by reference to Exhibit 10.9 to the Registrant’s Current Report on Form 8-K filed on November 25, 2008) | |
| 23.1 | Consent of J.H. Cohn LLP | |
| 31.1 | Certification of Principal Executive Officer | |
| 31.2 | Certification of Principal Financial Officer | |
| 32.1 | Certifications pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| ++ | Confidential treatment has been granted as to certain portions of this exhibit pursuant to Rule 24b-2 of the Securities Exchange Act of 1934, as amended. |
72
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act, of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 31, 2009.
| Manhattan Pharmaceuticals, Inc. | |
| By: | /s/ Douglas Abel |
| Douglas Abel | |
| Chief Executive Officer and President | |
In accordance with the Securities Exchange Act, this report has been signed below by the following persons on behalf of Manhattan Pharmaceuticals, Inc. and in the capacities and on the dates indicated.
| Signature | Title | Date | |||
| /s/ Douglas Abel | Chief Executive Officer, President and | March 31, 2009 | |||
| Douglas Abel | Director (principal executive officer) | ||||
| /s/ Michael G. McGuinness | Secretary and Chief Financial Officer | March 31, 2009 | |||
| Michael G. McGuinness | (principal accounting and financial officer) | ||||
| /s/ Neil Herskowitz | Director | March 31, 2009 | |||
| Neil Herskowitz | |||||
| /s/ Malcolm Hoenlein | Director | March 31, 2009 | |||
| Malcolm Hoenlein | |||||
| /s/ Timothy McInerney | Director | March 31, 2009 | |||
| Timothy McInerney | |||||
| /s/ Richard Steinhart | Director | March 31, 2009 | |||
| Richard Steinhart | |||||
| /s/ Michael Weiser | Director | March 31, 2009 | |||
| Michael Weiser |
73
Index to Financial Statements
| Page | ||
| Report of Independent Registered Public Accounting Firm | F-2 | |
| Balance Sheets as of December 31, 2008 and 2007 | F-3 | |
| Statements of Operations for the Years Ended December 31, 2008 and 2007 and the cumulative period from August 6, 2001 (inception) to December 31, 2008 | F-4 | |
| Statement of Stockholders’ Equity (Deficiency) for the Years Ended December 31, 2008 and 2007 and the cumulative period from August 6, 2001 (inception) to December 31, 2008 | F-5 | |
| Statements of Cash Flows for the Years Ended December 31, 2008 and 2007 and the cumulative period from August 6, 2001 (inception) to December 31, 2008 | F-7 | |
| Notes to Financial Statements | F-8 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
Manhattan Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Manhattan Pharmaceuticals, Inc. (a development stage company) as of December 31, 2008 and 2007, and the related statements of operations, stockholders' equity (deficiency) and cash flows for the years then ended, and for the period from August 6, 2001 (date of inception) to December 31, 2008. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Manhattan Pharmaceuticals, Inc. as of December 31, 2008 and 2007, and its results of operations and cash flows for the years then ended and for the period from August 6, 2001 (date of inception) to December 31, 2008, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred net losses and negative cash flows from operating activities from its inception through December 31, 2008 and has an accumulated deficit and negative working capital as of December 31, 2008. These matters raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans regarding these matters are also described in Note 2. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ J.H. Cohn LLP
Roseland, New Jersey
March 30, 2009
F-2
MANHATTAN PHARMACEUTICALS, INC.
(A Development Stage Company)
Balance Sheets
| December 31, | December 31, | |||||||
| 2008 | 2007 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 106,023 | $ | 649,686 | ||||
| Restricted cash | 730,499 | - | ||||||
| Prepaid expenses | 37,718 | 215,852 | ||||||
| Total current assets | 874,240 | 865,538 | ||||||
| Property and equipment, net | 9,072 | 44,533 | ||||||
| Secured 12% notes payable issue costs | 330,756 | - | ||||||
| Other assets | 34,895 | 70,506 | ||||||
| Total assets | $ | 1,248,963 | $ | 980,577 | ||||
| Liabilities and Stockholders' Deficiency | ||||||||
| Current Liabilities: | ||||||||
| Secured 10% notes payable | $ | 70,000 | $ | - | ||||
| Accounts payable | 542,296 | 1,279,485 | ||||||
| Accrued expenses | 874,072 | 592,177 | ||||||
| Total current liabilities | 1,486,368 | 1,871,662 | ||||||
| Secured 12% notes payable, net | 1,174,107 | - | ||||||
| Interest payable on secured 12% notes payable | 15,237 | |||||||
| Exchange obligation | 2,949,176 | - | ||||||
| Total liabilities | 5,624,888 | 1,871,662 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ deficiency: | ||||||||
| Preferred stock, $.001 par value. Authorized 1,500,000 shares; no shares issued and outstanding at December 31, 2008 and 2007 | - | - | ||||||
| Common stock, $.001 par value. Authorized 150,000,000 shares; 70,624,232 shares issued and outstanding at December 31, 2008 and December 31, 2007 | 70,624 | 70,624 | ||||||
| Additional paid-in capital | 54,821,379 | 54,037,361 | ||||||
| Deficit accumulated during the development stage | (59,267,928 | ) | (54,999,070 | ) | ||||
| Total stockholders’ deficiency | (4,375,925 | ) | (891,085 | ) | ||||
| Total liabilities and stockholders' deficiency | $ | 1,248,963 | $ | 980,577 | ||||
See accompanying notes to consolidated financial statements.
F-3
MANHATTAN PHARMACEUTICALS, INC.
(A Development Stage Company)
Statements of Operations
| Years ended December 31, | Cumulative period from August 6, 2001 (inception) to December 31, | |||||||||||
| 2008 | 2007 | 2008 | ||||||||||
| Revenue | $ | - | $ | - | $ | - | ||||||
| Costs and expenses: | ||||||||||||
| Research and development | 1,802,792 | 8,535,687 | 28,291,835 | |||||||||
| General and administrative | 2,609,910 | 3,608,270 | 16,462,273 | |||||||||
| In-process research and development charge | - | - | 11,887,807 | |||||||||
| Impairment of intangible assets | - | - | 1,248,230 | |||||||||
| Loss on disposition of intangible assets | - | - | 1,213,878 | |||||||||
| Total operating expenses | 4,412,702 | 12,143,957 | 59,104,023 | |||||||||
| Operating loss | (4,412,702 | ) | (12,143,957 | ) | (59,104,023 | ) | ||||||
| Other (income) expense: | ||||||||||||
| Equity in losses of Hedrin JV | 250,000 | 250,000 | ||||||||||
| Interest and other income | (458,634 | ) | (112,181 | ) | (1,280,531 | ) | ||||||
| Interest expense | 64,790 | 476 | 90,824 | |||||||||
| Realized gain on sale of marketable equity securities | - | - | (76,032 | ) | ||||||||
| Total other income | (143,844 | ) | (111,705 | ) | (1,015,739 | ) | ||||||
| Net loss | (4,268,858 | ) | (12,032,252 | ) | (58,088,284 | ) | ||||||
| Preferred stock dividends (including imputed amounts) | - | - | (1,179,644 | ) | ||||||||
| Net loss applicable to common shares | $ | (4,268,858 | ) | $ | (12,032,252 | ) | $ | (59,267,928 | ) | |||
| Net loss per common share: | ||||||||||||
| Basic and diluted | $ | (0.06 | ) | $ | (0.18 | ) | ||||||
| Weighted average shares of common stock outstanding: | ||||||||||||
| Basic and diluted | 70,624,232 | 68,015,075 | ||||||||||
See accompanying notes to consolidated financial statements.
F-4
MANHATTAN PAHRMACEUTICALS, INC.
(A Development Stage Company
Statement of Stockholders' Equity (Deficiency)
Series A convertible preferred stock shares | Series A convertible preferred stock amount | Common stock shares | Common stock amount | Additional paid-in capital | Subscription receivable | Deficit accumulated during development stage | Dividends payable in Series A preferred stock | Accumulated other comprehensive income (loss) | Unearned consulting services | Total stockholders’ equity (deficiency) | ||||||||||||||||||||||||||||||||||
| Stock issued at $0.0004 per share for subscription receivable | - | $ | - | 10,167,741 | $ | 10,168 | $ | (6,168 | ) | $ | (4,000 | ) | $ | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (56,796 | ) | - | - | - | (56,796 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2001 | - | - | 10,167,741 | 10,168 | (6,168 | ) | (4,000 | ) | (56,796 | ) | - | - | - | (56,796 | ) | |||||||||||||||||||||||||||||
| Proceeds from subscription receivable | - | - | - | - | - | 4,000 | - | - | - | - | 4,000 | |||||||||||||||||||||||||||||||||
| Stock issued at $0.0004 per share for license rights | - | - | 2,541,935 | 2,542 | (1,542 | ) | - | - | - | - | - | 1,000 | ||||||||||||||||||||||||||||||||
| Stock options issued for consulting services | - | - | - | - | 60,589 | - | - | - | - | (60,589 | ) | - | ||||||||||||||||||||||||||||||||
| Amortization of unearned consulting services | - | - | - | - | - | - | - | - | - | 22,721 | 22,721 | |||||||||||||||||||||||||||||||||
| Common stock issued at $0.63 per share, net of expenses | - | - | 3,043,332 | 3,043 | 1,701,275 | - | - | - | - | - | 1,704,318 | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | (1,037,320 | ) | - | - | - | (1,037,320 | ) | ||||||||||||||||||||||||||||||||
| Balance at December 31, 2002 | - | - | 15,753,008 | 15,753 | 1,754,154 | - | (1,094,116 | ) | - | - | (37,868 | ) | 637,923 | |||||||||||||||||||||||||||||||
| Common stock issued at $0.63 per share, net of expenses | - | - | 1,321,806 | 1,322 | 742,369 | - | - | - | - | - | 743,691 | |||||||||||||||||||||||||||||||||
| Effect of reverse acquisition | - | - | 6,287,582 | 6,287 | 2,329,954 | - | - | - | - | - | 2,336,241 | |||||||||||||||||||||||||||||||||
| Amortization of unearned consulting costs | - | - | - | - | - | - | - | - | - | 37,868 | 37,868 | |||||||||||||||||||||||||||||||||
| Unrealized loss on short-term investments | - | - | - | - | - | - | - | - | (7,760 | ) | - | (7,760 | ) | |||||||||||||||||||||||||||||||
| Payment for fractional shares for stock combination | - | - | - | - | (300 | ) | - | - | - | - | - | (300 | ) | |||||||||||||||||||||||||||||||
| Preferred stock issued at $10 per share, net of expenses | 1,000,000 | 1,000 | - | - | 9,045,176 | - | - | - | - | - | 9,046,176 | |||||||||||||||||||||||||||||||||
| Imputed preferred stock dividend | 418,182 | - | (418,182 | ) | - | - | ||||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (5,960,907 | ) | - | - | - | (5,960,907 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2003 | 1,000,000 | 1,000 | 23,362,396 | 23,362 | 14,289,535 | - | (7,473,205 | ) | - | (7,760 | ) | - | 6,832,932 | |||||||||||||||||||||||||||||||
| Exercise of stock options | - | - | 27,600 | 27 | 30,073 | - | - | - | - | - | 30,100 | |||||||||||||||||||||||||||||||||
| Common stock issued at $1.10, net of expenses | - | - | 3,368,952 | 3,369 | 3,358,349 | - | - | - | - | - | 3,361,718 | |||||||||||||||||||||||||||||||||
| Preferred stock dividend accrued | - | - | - | - | - | - | (585,799 | ) | 585,799 | - | - | - | ||||||||||||||||||||||||||||||||
| Preferred stock dividends paid by issuance of shares | 24,901 | 25 | - | - | 281,073 | - | - | (282,388 | ) | - | - | (1,290 | ) | |||||||||||||||||||||||||||||||
| Conversion of preferred stock to common stock at $1.10 per share | (170,528 | ) | (171 | ) | 1,550,239 | 1,551 | (1,380 | ) | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
| Warrants issued for consulting services | - | - | - | - | 125,558 | - | - | - | - | (120,968 | ) | 4,590 | ||||||||||||||||||||||||||||||||
| Amortization of unearned consulting costs | - | - | - | - | - | - | - | - | - | 100,800 | 100,800 | |||||||||||||||||||||||||||||||||
| Unrealized gain on short-term investments and reversal of unrealized loss on short-term investments | - | - | - | - | - | - | - | - | 20,997 | - | 20,997 | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (5,896,031 | ) | - | - | - | (5,896,031 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2004 | 854,373 | 854 | 28,309,187 | 28,309 | 18,083,208 | - | (13,955,035 | ) | 303,411 | 13,237 | (20,168 | ) | 4,453,816 | |||||||||||||||||||||||||||||||
F-5
MANHATTAN PHARMACEUTICALS, INC.
(A Development Stage Company)
Statement of Stockholders' Equity (Deficiency)
Series A convertible preferred stock shares | Series A convertible preferred stock amount | Common stock shares | Common stock amount | Additional paid-in capital | Subscription receivable | Deficit accumulated during development stage | Dividends payable in Series A preferred stock | Accumulated other comprehensive income (loss) | Unearned consulting services | Total stockholders’ equity (deficiency) | ||||||||||||||||||||||||||||||||||
| Common stock issued at $1.11 and $1.15, net of expenses | - | $ | - | 11,917,680 | $ | 11,918 | $ | 12,238,291 | $ | - | $ | - | $ | - | $ | - | $ | - | $ | 12,250,209 | ||||||||||||||||||||||||
| Common stock issued to vendor at $1.11 per share in satisfaction of accounts payable | - | - | 675,675 | 676 | 749,324 | - | - | - | - | - | 750,000 | |||||||||||||||||||||||||||||||||
| Exercise of stock options | - | - | 32,400 | 33 | 32,367 | - | - | - | - | - | 32,400 | |||||||||||||||||||||||||||||||||
| Exercise of warrants | - | - | 279,845 | 279 | 68,212 | - | - | - | - | - | 68,491 | |||||||||||||||||||||||||||||||||
| Preferred stock dividend accrued | - | - | - | - | - | - | (175,663 | ) | 175,663 | - | - | - | ||||||||||||||||||||||||||||||||
| Preferred stock dividends paid by issuance of shares | 41,781 | 42 | - | - | 477,736 | - | — | (479,074 | ) | - | - | (1,296 | ) | |||||||||||||||||||||||||||||||
| Conversion of preferred stock to common stock at $1.10 per share | (896,154 | ) | (896 | ) | 8,146,858 | 8,147 | (7,251 | ) | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
| Share-based compensation | - | - | - | - | 66,971 | - | - | - | - | 20,168 | 87,139 | |||||||||||||||||||||||||||||||||
| Reversal of unrealized gain on short-term investments | - | - | - | - | - | - | - | - | (12,250 | ) | - | (12,250 | ) | |||||||||||||||||||||||||||||||
| Stock issued in connection with acquisition of Tarpan Therapeutics, Inc. | - | - | 10,731,052 | 10,731 | 11,042,253 | - | - | - | - | - | 11,052,984 | |||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (19,140,997 | ) | - | - | - | (19,140,997 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2005 | - | - | 60,092,697 | 60,093 | 42,751,111 | - | (33,271,695 | ) | - | 987 | - | 9,540,496 | ||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | - | - | 27,341 | 27 | (27 | ) | - | - | - | - | - | — | ||||||||||||||||||||||||||||||||
| Share-based compensation | - | - | - | - | 1,675,499 | - | - | - | - | - | 1,675,499 | |||||||||||||||||||||||||||||||||
| Unrealized loss on short-term investments | - | - | - | - | - | - | - | - | (987 | ) | — | (987 | ) | |||||||||||||||||||||||||||||||
| Costs associated with private placement | - | - | - | - | (15,257 | ) | - | - | - | - | - | (15,257 | ) | |||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (9,695,123 | ) | - | - | - | (9,695,123 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2006 | - | - | 60,120,038 | 60,120 | 44,411,326 | - | (42,966,818 | ) | - | - | - | 1,504,628 | ||||||||||||||||||||||||||||||||
| Common stock issued at $0.84 and $0.90 per shares, net of expenses | - | - | 10,185,502 | 10,186 | 7,841,999 | - | - | - | - | - | 7,852,185 | |||||||||||||||||||||||||||||||||
| Common stock issued to directors at $0.72 per share in satisfaction of accounts payable | - | - | 27,776 | 28 | 19,972 | - | - | - | - | - | 20,000 | |||||||||||||||||||||||||||||||||
| Common stock issued to in connection with in-licensing agreement at $0.90 per share | - | - | 125,000 | 125 | 112,375 | - | - | - | - | - | 112,500 | |||||||||||||||||||||||||||||||||
| Common stock issued to in connection with in-licensing agreement at $0.80 per share | - | - | 150,000 | 150 | 119,850 | - | - | - | - | - | 120,000 | |||||||||||||||||||||||||||||||||
| Exercise of warrants | - | - | 10,327 | 15 | 7,219 | - | - | - | - | - | 7,234 | |||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | - | - | 5,589 | - | (6 | ) | - | - | - | - | - | (6 | ) | |||||||||||||||||||||||||||||||
| Share-based compensation | - | - | - | - | 1,440,956 | - | - | - | - | - | 1,440,956 | |||||||||||||||||||||||||||||||||
| Warrants issued for consulting | 83,670 | 83,670 | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (12,032,252 | ) | - | - | - | (12,032,252 | ) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2007 | - | - | 70,624,232 | 70,624 | 54,037,361 | - | (54,999,070 | ) | - | - | - | (891,085 | ) | |||||||||||||||||||||||||||||||
| Sale of warrant | 150,000 | 150,000 | ||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation | 463,890 | 463,890 | ||||||||||||||||||||||||||||||||||||||||||
| Warrants issued with secured 12% notes | 170,128 | 170,128 | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | (4,268,858 | ) | (4,268,858 | ) | ||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2008 | - | $ | - | 70,624,232 | $ | 70,624 | $ | 54,821,379 | $ | - | $ | (59,267,928 | ) | $ | - | $ | - | $ | - | $ | (4,375,925 | ) | ||||||||||||||||||||||
See accompanying notes to financial statements.
F-6
MANHATTAN PHARMACEUTICALS, INC.
(A Development Stage Company)
Statements of Cash Flows
| Years Ended, | Cumulative period from August 6, 2001 (inception) to December 31, | |||||||||||
| 2008 | 2007 | 2008 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net loss | $ | (4,268,858 | ) | $ | (12,032,252 | ) | $ | (58,088,284 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Equity in losses of Hedrin JV | 250,000 | - | 250,000 | |||||||||
| Share-based compensation | 463,890 | 1,440,956 | 3,828,873 | |||||||||
| Interest and amortization of OID and issue costs on Secured 12% Notes | 38,574 | - | 38,574 | |||||||||
| Shares issued in connection with in-licensing agreement | - | 232,500 | 232,500 | |||||||||
| Warrants issued to consultant | - | 83,670 | 83,670 | |||||||||
| Amortization of intangible assets | - | - | 145,162 | |||||||||
| Gain on sale of marketable equity securities | - | - | (76,032 | ) | ||||||||
| Depreciation | 26,106 | 48,345 | 221,931 | |||||||||
| Non cash portion of in-process research and development charge | - | - | 11,721,623 | |||||||||
| Loss on impairment and disposition of intangible assets | - | - | 2,462,108 | |||||||||
| Other | 18,327 | - | 23,917 | |||||||||
| Changes in operating assets and liabilities, net of acquisitions: | ||||||||||||
| Increase in restricted cash | (730,499 | ) | - | (730,499 | ) | |||||||
| Decrease in prepaid expenses and other current assets | 178,134 | 48,734 | 20,527 | |||||||||
| Decrease (increase) in other assets | 35,611 | - | (49,895 | ) | ||||||||
| Decrease in accounts payable | (737,189 | ) | (93,812 | ) | 962,509 | |||||||
| Increase in accrued expenses | 281,895 | 42,148 | 333,751 | |||||||||
| Net cash used in operating activities | (4,444,009 | ) | (10,229,711 | ) | (38,619,565 | ) | ||||||
| Cash flows from investing activities: | ||||||||||||
| Purchase of property and equipment | (8,973 | ) | (9,134 | ) | (239,608 | ) | ||||||
| Cash paid in connection with acquisitions | - | - | (26,031 | ) | ||||||||
| Net cash provided from the purchase and sale of short-term investments | - | - | 435,938 | |||||||||
| Proceeds from sale of license | - | - | 200,001 | |||||||||
| Net cash (used in) provided by investing activities | (8,973 | ) | (9,134 | ) | 370,300 | |||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from the Hedrin JV agreement | 2,699,176 | 2,699,176 | ||||||||||
| Sale of warrant | 150,000 | 150,000 | ||||||||||
| Proceeds from sale of Secured 10% Notes | 70,000 | 70,000 | ||||||||||
| Proceeds from sale of Secured 12% Notes | 990,143 | 1,005,143 | ||||||||||
| Repayments of notes payable to stockholders | - | - | (884,902 | ) | ||||||||
| Proceeds (costs) related to sale of common stock, net | - | 7,852,185 | 25,896,262 | |||||||||
| Proceeds from sale of preferred stock, net | - | - | 9,046,176 | |||||||||
| Proceeds from exercise of warrants and stock options | - | 7,228 | 138,219 | |||||||||
| Other, net | - | - | 235,214 | |||||||||
| Net cash provided by (used in) financing activities | 3,909,319 | 7,859,413 | 38,355,288 | |||||||||
| Net (decrease) increase in cash and cash equivalents | (543,663 | ) | (2,379,432 | ) | 106,023 | |||||||
| Cash and cash equivalents at beginning of period | 649,686 | 3,029,118 | - | |||||||||
| Cash and cash equivalents at end of period | $ | 106,023 | $ | 649,686 | $ | 106,023 | ||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Interest paid | $ | 475 | $ | 1,665 | $ | 26,033 | ||||||
| Supplemental disclosure of noncash investing and financing activities: | ||||||||||||
| Investment in Hedrin JV | $ | 250,000 | $ | - | $ | 250,000 | ||||||
| Warrants issued with Secured 12% Notes | 170,128 | - | 170,128 | |||||||||
| Common stock issued in satisfaction of accounts payable | - | 20,000 | 770,000 | |||||||||
| Imputed and accrued preferred stock dividend | - | - | 1,179,644 | |||||||||
| Conversion of preferred stock to common stock | - | - | 1,067 | |||||||||
| Preferred stock dividends paid by issuance of shares | - | - | 759,134 | |||||||||
| Issuance of common stock for acquisitions | - | - | 13,389,226 | |||||||||
| Issuance of common stock in connection with in-licensing agreement | - | 232,500 | 232,500 | |||||||||
| Marketable equity securities received in connection with sale of license | - | - | 359,907 | |||||||||
| Warrants issued to consultant | - | 83,670 | 83,670 | |||||||||
| Net liabilities assumed over assets acquired in business combination | - | - | (675,416 | ) | ||||||||
| Cashless exercise of warrants | - | 6 | 33 | |||||||||
See accompanying notes to financial statements.
F-7
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (1) | Merger and Nature of Operations |
2003 Reverse Merger
On February 21, 2003, the Company (formerly known as “Atlantic Technology Ventures, Inc.”) completed a reverse acquisition of privately held Manhattan Research Development, Inc. (“Manhattan Research”) (formerly Manhattan Pharmaceuticals, Inc.), a Delaware corporation. At the effective time of the merger, the outstanding shares of common stock of Manhattan Research automatically converted into shares of the Company’s common stock representing 80 percent of the Company’s outstanding voting stock after giving effect to the merger. Since the stockholders of Manhattan Research received the majority of the voting shares of the Company, the merger was accounted for as a reverse acquisition whereby Manhattan Research was the accounting acquirer (legal acquiree) and the Company was the accounting acquiree (legal acquirer) under the purchase method of accounting. In connection with the merger, the Company changed its name from “Atlantic Technology Ventures, Inc.” to “Manhattan Pharmaceuticals, Inc.” The results of the combined operations have been included in the Company’s financial statements since February 2003.
As described above, the Company resulted from the February 21, 2003 reverse merger between Atlantic Technology Ventures, Inc. (“Atlantic”), which was incorporated on May 18, 1993, and privately-held Manhattan Research Development, Inc., incorporated on August 6, 2001. The Company was incorporated in the State of Delaware. In connection with the merger, the former stockholders of Manhattan Research received a number of shares of Atlantic's common stock so that following the merger they collectively owned 80 percent of the outstanding shares. Upon completion of the merger, Atlantic changed its name to Manhattan Pharmaceuticals, Inc. and thereafter adopted the business of Manhattan Research.
The Company is a biopharmaceutical company focused on developing and commercializing innovative pharmaceutical therapies for underserved patient populations. The Company acquires rights to these technologies by licensing or otherwise acquiring an ownership interest, funding their research and development and eventually either bringing the technologies to market or out-licensing. We currently have two product candidates in development: Hedrin™, a novel, non-insecticide treatment of pediculitis (head lice) which is being developed by the Hedrin JV, see note 6, and a topical product for the treatment of psoriasis. During 2007, the Company discontinued development of Oleoyl-estrone and Propofol Lingual Spray.
Acquisition of Tarpan Therapeutics, Inc.
In April 2005, the Company merged with Tarpan Therapeutics, Inc., a Delaware corporation (“Tarpan”), and Tarpan Acquisition Corp., a Delaware corporation. The acquisition of Tarpan has been accounted for by the Company under the purchase method of accounting in accordance with Statement of Financial Accounting Standards (“SFAS”) No. 141 “Business Combinations”. Under the purchase method, assets acquired and liabilities assumed by the Company are recorded at their estimated fair values and the results of operations of the acquired company are consolidated with those of the Company from the date of acquisition. The excess purchase price paid by the Company to acquire the net assets of Tarpan was allocated to acquired in-process research and development totaling $11,887,807. As required by Financial Accounting Standards Board (“FASB”) Interpretation No. 4, “Applicability of FASB Statement No. 2 to Business combinations Accounted for by the Purchase Method” (“FIN 4”), the Company recorded a charge in its consolidated statement of operations for the year ended December 31, 2005 for the in-process research and development.
F-8
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (2) | Liquidity and Basis of Presentation |
Liquidity
The Company incurred a net loss of $4,268,858 and negative cash flows from operating activities of $4,444,009 for the year ended December 31, 2008 and $12,032,252 and negative cash flows from operating activities of $10,229,711 for the year ended December 31, 2007. The net loss applicable to common shares from date of inception, August 6, 2001, to December 31, 2008 amounts to $59,267,928.
The Company received approximately $7.9 million net from a private placement of common stock and warrants in March 2007. This private placement is more fully described in Note 5.
The Company received approximately $1.8 million in February 2008 and approximately $0.9 million in June 2008 from a joint venture agreement. This joint venture agreement is more fully described in Note 8. The Company also received $70,000 in Secured 10% Notes in September 2008 and $1.0 million from the sale of Secured 12% Notes in November and December 2008. These notes are more fully described in Notes 10 and 11.
Management believes that the Company will continue to incur net losses through at least December 31, 2009 and for the foreseeable future thereafter. Based on the resources of the Company available at December 31, 2008, the net proceeds of $500,000 received in February 2009 from a joint venture agreement and net proceeds of $360,000 received from the sale of additional Secured 12% Notes in February 2009, management believes that the Company has sufficient capital to fund its operations through 2009. Management believes that the Company will need additional equity or debt financing or will need to generate positive cash flow from a joint venture agreement, see Note 6, or generate revenues through licensing of its products or entering into strategic alliances to be able to sustain its operations into 2010. Furthermore, we will need additional financing thereafter to complete development and commercialization of our products. There can be no assurances that we can successfully complete development and commercialization of our products.
The Company’s continued operations will depend on its ability to raise additional funds through various potential sources such as equity and debt financing, collaborative agreements, strategic alliances and its ability to realize the full potential of its technology in development. Additional funds may not become available on acceptable terms, and there can be no assurance that any additional funding that the Company does obtain will be sufficient to meet the Company’s needs in the long-term.
These matters raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
F-9
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (3) | Summary of Significant Accounting Policies |
Basis of Presentation
The Company has not generated any revenue from its operations and, accordingly, the financial statements have been prepared in accordance with the provisions of SFAS No. 7, “Accounting and Reporting by Development Stage Enterprises.”
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect certain reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates.
Research and Development
All research and development costs are expensed as incurred and include costs of consultants who conduct research and development on behalf of the Company and its subsidiaries. Costs related to the acquisition of technology rights and patents for which development work is still in process are expensed as incurred and considered a component of research and development costs.
The Company often contracts with third parties to facilitate, coordinate and perform agreed upon research and development of a new drug. To ensure that research and development costs are expensed as incurred, the Company records monthly accruals for clinical trials and preclinical testing costs based on the work performed under the contracts.
These contracts typically call for the payment of fees for services at the initiation of the contract and/or upon the achievement of certain milestones. This method of payment often does not match the related expense recognition resulting in either a prepayment, when the amounts paid are greater than the related research and development costs expensed, or an accrued liability, when the amounts paid are less than the related research and development costs expensed.
Acquired in-process research and development
Costs to acquire in-process research and development projects and technologies which have no alternative future use at the date of acquisition are expensed.
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between financial statement carrying amounts of existing assets and liabilities, and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized.
F-10
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Computation of Net Loss per Common Share
Basic net loss per common share is calculated by dividing net loss applicable to common shares by the weighted-average number of common shares outstanding for the period. Diluted net loss per common share is the same as basic net loss per common share, since potentially dilutive securities from stock options, stock warrants and convertible preferred stock would have an antidilutive effect because the Company incurred a net loss during each period presented. The amounts of potentially dilutive securities excluded from the calculation were 80,068,144 and 16,903,292 shares at December 31, 2008 and 2007, respectively. These amounts do not include the shares issuable upon the exercise of the put or call rights issued in connection with the Hedrin JV (see Note 8) which were subject to anti-dilution rights upon the issuance of warrants with the Secured 12% Notes (see Note 11); the 41,666,667 shares of common stock issuable upon exercise of the put or call rights and the up to 13,888,889 additional shares which may become issuable upon exercise of a conditionally issuable put or call rights.
Share-Based Compensation
The Company has stockholder-approved stock incentive plans for employees, directors, officers and consultants. Prior to January 1, 2006, the Company accounted for the employee, director and officer plans using the intrinsic value method under the recognition and measurement provisions of Accounting Principles Board (“APB”) Opinion No. 25, “Accounting for Stock Issued to Employees” and related interpretations, as permitted by Statement of Financial Accounting Standards (“SFAS” or “Statement”) No. 123, “Accounting for Stock-Based Compensation.”
Effective January 1, 2006, the Company adopted SFAS No. 123(R), “Share-Based Payment,” (“Statement 123(R)”) for employee options using the modified prospective transition method. Statement 123(R) revised Statement 123 to eliminate the option to use the intrinsic value method and required the Company to expense the fair value of all employee options over the vesting period. Under the modified prospective transition method, the Company recognized compensation cost for the years ended December 31, 2008 and 2007 which includes a) period compensation cost related to share-based payments granted prior to, but not yet vested, as of January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of Statement 123; and b) period compensation cost related to share-based payments granted on or after January 1, 2006, based on the grant date fair value estimated in accordance with Statement 123(R). In accordance with the modified prospective method, the Company has not restated prior period results.
The Company recognizes compensation expense related to stock option grants on a straight-line basis over the vesting period. For the years ended December 31, 2008 and 2007, the Company recognized share-based employee compensation cost of $462,311 and $1,447,560, respectively, in accordance with Statement 123(R). The Company did not capitalize any share-based compensation cost.
F-11
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Options granted to consultants and other non-employees are accounted for in accordance with EITF No. 96-18 "Accounting for Equity Instruments That Are Issued to Other than Employees for Acquiring, or in Conjunction with Selling, Goods or Services". Accordingly, such options are recorded at fair value at the date of grant and subsequently adjusted to fair value at the end of each reporting period until such options vest, and the fair value of the options, as adjusted, is amortized to consulting expense over the related vesting period. As a result of adjusting consultant and other non-employee options to fair value as of December 31, 2008 and 2007 respectively, net of amortization, the Company recognized an increase to general and administrative and research and development expenses of $1,579 for the year ended December 31, 2008 and a reduction to general and administrative and research and development expenses of $6,604 for the year ended December 31, 2007 The Company has allocated share-based compensation costs to general and administrative and research and development expenses as follows:
| 2008 | 2007 | |||||||
| General and administrative expense: | ||||||||
| Share-based employee compensation cost | $ | 341,706 | $ | 891,897 | ||||
| Share-based consultant and non-employee cost | 158 | 10,550 | ||||||
| 341,864 | 902,447 | |||||||
| Research and development expense | ||||||||
| Share-based employee compensation cost | 120,605 | 555,663 | ||||||
| Share-based consultant and non-employee cost | 1,421 | (17,154 | ) | |||||
| 122,026 | 538,509 | |||||||
| Total share-based cost | $ | 463,890 | $ | 1,440,956 | ||||
To compute compensation expense in 2008 and 2007 the Company estimated the fair value of each option award on the date of grant using the Black-Scholes model. The Company based the expected volatility assumption on a volatility index of peer companies as the Company did not have a sufficient number of years of historical volatility of its common stock for the application of Statement 123 (R). The expected term of options granted represents the period of time that options are expected to be outstanding. The Company estimated the expected term of stock options by the simplified method as prescribed in Staff Accounting Bulletin Nos. 107 and 110. The expected forfeiture rates are based on the historical employee forfeiture experiences. To determine the risk-free interest rate, the Company utilized the U.S. Treasury yield curve in effect at the time of grant with a term consistent with the expected term of the Company’s awards. The Company has not declared a dividend on its common stock since its inception and has no intentions of declaring a dividend in the foreseeable future and therefore used a dividend yield of zero.
The following table shows the weighted average assumptions the Company used to develop the fair value estimates for the determination of the compensation charges in 2008 and 2007:
| 2008 | 2007 | |||||||
| Expected volatility | 92% | 93% | ||||||
| Dividend yield | - | - | ||||||
| Expected term (in years) | 5 - 10 | 5 - 10 | ||||||
| Risk-free interest rate | 2.81% | 3.6% - 4.9% | ||||||
| Forfeiture rate | 2% | 7% | ||||||
F-12
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Financial Instruments
At December 31, 2008 and 2007, the fair values of cash and cash equivalents and accounts payable approximate their carrying values due to the short-term nature of these instruments. At December 31, 2008, the fair values of the secured notes payable approximate their carrying values due to the recent issuance of the notes.
Cash and Cash Equivalents
Cash equivalents consist of cash or short term investments with original maturities at the time of purchase of three months or less.
Property and Equipment
Property and equipment are stated at cost. Depreciation is provided using the straight-line method over estimated useful lives. When assets are retired or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts, and any resulting gain or loss is recognized in operations for the period. Amortization of leasehold improvements is calculated using the straight-line method over the remaining term of the lease or the life of the asset, whichever is shorter. The cost of repairs and maintenance is charged to operations as incurred; significant renewals and improvements are capitalized.
Short-term Investments
Short-term investments are carried at market value since they are marketable and considered available-for-sale. The Company did not have any short-term investments at December 31, 2008 or 2007.
Equity in Joint Venture
The Company accounts for its investment in joint venture (See Note 6) using the equity method of accounting. Under the equity method, the Company records its pro-rata share of joint venture income or losses and adjusts the basis of its investment accordingly.
New Accounting Pronouncements
In February 2008, the FASB issued two Staff Positions on SFAS 157: (1) FASB Staff Position No. FAS 157-1 (“FAS 157-1”), “Application of FASB Statement No. 157 to FASB Statement No. 13 and Other Accounting Pronouncements That Address Fair Value Measurements for Purposes of Lease Classification or Measurement Under Statement 13,” and (2) FASB Staff Position No. FAS 157-2 (“FAS 157-2”), “Effective Date of FASB Statement No 157.” FAS 157-1 excludes SFAS 13, “Accounting for Leases”, as well as other accounting pronouncements that address fair value measurements on lease classification or measurement under SFAS 13, from SFAS 157’s scope. FAS157-2 partially defers SFAS 157’s effective date. The adoption of FAS 157-1 and FAS 157-2 did not have a material impact on its financial statements.
In October 2008, the FASB issued FASB Staff Position No. FAS 157-3 "Determining the Fair Value of a Financial Asset When the Market for That Asset is Not Active" ("FAS 157-3"), which is effective upon issuance for all financial statements that have not been issued. FAS 157-3 clarifies the application of SFAS 157, in a market that is not active. FAS 157-3 does not have a material impact on the Company’s financial position, financial performance or cash flows.
F-13
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
In March 2008, the FASB issued SFAS No. 161 "Disclosures About Derivative Instruments and Hedging Activities - an amendment of FASB Statement No. 133" ("SFAS 161"). SFAS 161 amends SFAS 133 by requiring expanded disclosures about an entity's derivative instruments and hedging activities. SFAS 161 requires qualitative disclosures about objectives and strategies for using derivatives, quantitative disclosures about fair value amounts of and gains and losses on derivative instruments, and disclosures about credit-risk-related contingent features in derivative instruments. SFAS 161 is effective for the Company as of January 1, 2009. The Company does not believe that SFAS 161 will have any impact on its financial statements.
In May 2008 the FASB issued SFAS No. 163, “Accounting for Financial Guarantee Insurance Contracts” (“SFAS 163”). SFAS 163 requires recognition of a claim liability prior to an event of default when there is evidence that credit deterioration has occurred in an insured financial obligation. SFAS 163 clarifies how SFAS 60 applies to financial guarantee insurance contracts, including the recognition and measurement to be used to account for premium revenue and claim liabilities. SFAS 163 also requires expanded disclosures about financial guarantee insurance contracts. SFAS 163 is effective for years beginning after December 15, 2008, and interim periods within those years, except for certain disclosure requirements which are effective for the first period (including interim periods) beginning after May 23, 2008. The Company does not believe that SFAS 163 will have any impact on its financial statements.
In December 2007, the FASB issued SFAS No. 141(R), a revised version of SFAS No. 141, “Business Combinations” (“SFAS 141R”). The revision is intended to simplify existing guidance and converge rulemaking under U.S. generally accepted accounting principles with international accounting standards. SFAS 141R applies prospectively to business combinations where the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. An entity may not apply it before that date. The Company does not expect the adoption of SFAS 141(R) to have a significant impact on our results of operations or financial position.
In June 2008, the FASB ratified EITF Issue No. 07-5, "Determining Whether an Instrument (or an Embedded Feature) Is Indexed to an Entity's Own Stock" (“EITF 07-5”). EITF 07-5 provides that an entity should use a two step approach to evaluate whether an equity-linked financial instrument (or embedded feature) is indexed to its own stock, including evaluating the instrument's contingent exercise and settlement provisions. It also clarifies the impact of foreign currency denominated strike prices and market-based employee stock option valuation instruments on the evaluation. EITF 07-5 is effective for fiscal years beginning after December 15, 2008. The Company is currently assessing the impact of EITF 07-5 on its financial position and results of operations.
F-14
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (4) | Property and Equipment |
Property and equipment consists of the following at December 31:
| 2008 | 2007 | |||||||
| Property and equipment | $ | 35,905 | $ | 226,010 | ||||
| Less accumulated depreciation | (26,833 | ) | (181,477 | ) | ||||
| Net property and equipment | $ | 9,072 | $ | 44,533 | ||||
| (5) | Stockholders’ Equity |
As described in Note 1 the Company completed a reverse acquisition of privately held Manhattan Research Development, Inc. on February 21, 2003. In July 2003, the Board of Directors adopted a resolution authorizing an amendment to the certificate of incorporation providing for a 1-for-5 combination of the Company’s common stock. The resolution approving the 1-for-5 combination was thereafter consented to in writing by holders of a majority of the Company’s outstanding common stock and became effective in September 2003. Accordingly, all share and per share information in these financial statements has been restated to retroactively reflect the 1-for-5 combination and the effects of the Reverse Merger.
2001
During 2001, the Company issued 10,167,741 shares of its common stock to investors for subscriptions receivable of $4,000 or $0.0004 per share. During 2002, the Company received the $4,000 subscription receivable.
2002
During 2002, the Company issued 2,541,935 shares of its common stock to Oleoyl-estrone Developments, S.L. (“OED”) in conjunction with a license agreement (the OED License Agreement”), as more fully described in Note 8. We valued these shares at their then estimated fair value of $1,000.
During 2002, the Company issued options to purchase 1,292,294 shares of its common stock in conjunction with several consulting agreements. The fair value of these options was $60,589. The Company expensed $22,721 in 2002 and $37,868 in 2003.
During 2002 and 2003, the Company completed two private placements. During 2002, the Company issued 3,043,332 shares of its common stock at $0.63 per share and warrants to purchase 304,333 of its common stock in a private placement. After deducting commissions and other expenses relating to the private placement, the Company received net proceeds of $1,704,318.
2003
During 2003, the Company issued an additional 1,321,806 shares of its common stock at $0.63 per share and warrants to purchase 132,181 shares of its common stock. After deducting commissions and other expenses relating to the private placement, the Company received net proceeds of $743,691. In connection with these private placements, the Company issued to the placement agent warrants to purchase 1,658,753 shares of its common stock.
F-15
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
As described in Note 1, during 2003, the Company completed a reverse acquisition. The Company issued 6,287,582 shares of its common stock with a value of $2,336,241 in the reverse acquisition.
In November 2003, the Company issued 1,000,000 shares of its newly-designated Series A Convertible Preferred Stock (the “Convertible Preferred”) at a price of $10 per share in a private placement. After deducting commissions and other expenses relating to the private placement, the Company received net proceeds of $9,046,176. Each share of Convertible Preferred was convertible at the holder’s election into shares of the Company’s common stock at a conversion price of $1.10 per share. The conversion price of the Convertible Preferred was less than the market value of the Company’s common stock on the date of issuance. Accordingly for the year ended December 31, 2003 the Company recorded a separate charge to deficit accumulated during development stage for the beneficial conversion feature associated with the issuance of Convertible Preferred of $418,182. The Convertible Preferred had a payment-in-kind annual dividend of five percent. Maxim Group, LLC of New York, together with Paramount Capital, Inc., a related party, acted as the placement agents in connection with the private placement.
2004
During 2004, the Company issued 3,368,952 shares of its common stock at a price of $1.10 per share in a private placement. After deducting commissions and other expenses relating to the private placement, the Company received net proceeds of $3,361,718. In connection with the common stock private placement and the Convertible Preferred private placement, the Company issued to the placement agents a warrant to purchase 1,235,589 shares of its common stock.
During 2004, the Company recorded a dividend on the Convertible Preferred of $585,799. 24,901 shares of Convertible Preferred were issued in payment of $282,388 of this in-kind dividend. Also during 2004, 170,528 shares of Convertible Preferred were converted into 1,550,239 shares of the Company’s common stock at $1.10 per share.
During 2004, the Company issued 27,600 shares of common stock upon the exercise of stock options.
During 2004, the Company issued warrants to purchase 110,000 shares of its common stock in conjunction with three consulting agreements. The fair value of these warrants was $120,968. The Company expensed $100,800 in 2004 and $20,168 in 2005.
2005
In August 2005, the Company issued 11,917,680 shares of its common stock and warrants to purchase 2,383,508 shares of its common stock in a private placement at $1.11 and $1.15 per share. After deducting commissions and other expenses relating to the private placement the Company received net proceeds of $12,250,209. Paramount BioCapital, Inc. (“Paramount”), an affiliate of a significant stockholder of the Company, acted as placement agent and was paid cash commissions and expenses of $967,968 of which $121,625 was paid to certain selected dealers engaged by Paramount in the private placement. The Company also issued warrants to purchase 595,449 shares of common stock to Paramount and certain select dealers, of which Paramount received warrants to purchase 517,184 common shares. Timothy McInerney and Dr. Michael Weiser, each a director of the Company, were employees of Paramount BioCapital, Inc. at the time of the transaction.
During 2005, the Company recorded a dividend on the Convertible Preferred of $175,663. 41,781 shares of Convertible Preferred were issued in payment of this $175,663 in-kind dividend and the unpaid portion of the 2004 in-kind dividend, $303,411. Also during 2005, the remaining 896,154 shares of Convertible preferred were converted into 8,146,858 shares of the Company’s common stock.
F-16
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
During 2005, the Company issued 675,675 shares of its common stock at $1.11 per share and warrants to purchase 135,135 shares of its common stock to Cato BioVentures, an affiliate of Cato Research, Inc., in exchange for satisfaction of $750,000 of accounts payable owed by the Company to Cato Research, Inc. Since the value of the shares and warrants issued was approximately $750,000, there is no impact on the statement of operations for this transaction.
During 2005, the Company issued 312,245 shares of common stock upon the exercise of stock options and warrants.
As described in Note 1, in April 2005, the Company completed the Merger with Tarpan. In accordance with the Agreement, the stockholders of Tarpan received 10,731,052 shares of the Company’s common stock with a value of $11,052,984.
2006
During 2006, the Company issued 27,341 shares of common stock upon the exercise of warrants.
2007
On March 30, 2007, the Company entered into a series of subscription agreements with various institutional and other accredited investors for the issuance and sale in a private placement of an aggregate of 10,185,502 shares of its common stock for total net proceeds of approximately $7.85 million, after deducting commissions and other costs of the transaction. Of the total amount of shares issued, 10,129,947 were sold at a per share price of $0.84, and an additional 55,555 shares were sold to an entity affiliated with a director of the Company, at a per share price of $0.90, the closing sale price of the common stock on March 29, 2007. Pursuant to the subscription agreements, the Company also issued to the investors 5-year warrants to purchase an aggregate of 3,564,897 shares of common stock at an exercise price of $1.00 per share. The warrants are exercisable during the period commencing September 30, 2007 and ending March 30, 2012. Gross and net proceeds from the private placement were $8,559,155 and $7,852,185, respectively.
Pursuant to these subscription agreements the Company filed a registration statement on Form S-3 covering the resale of the shares issued in the private placement, including the shares issuable upon exercise of the investor warrants and the placement agent warrants, with the Securities and Exchange Commission on May 9, 2007, which was declared effective by the Securities and Exchange Commission on May 18, 2007.
The Company engaged Paramount, an affiliate of a significant stockholder of the Company, as its placement agent in connection with the private placement. In consideration for its services, the Company paid aggregate cash commissions of approximately $600,000 and issued to Paramount a 5-year warrant to purchase an aggregate of 509,275 shares at an exercise price of $1.00 per share.
F-17
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
2008
During 2008, the Company issued warrants to purchase 140,000 shares of its common stock to directors, officers and an employee in conjunction with the secured 10% notes, see note 8.
During 2008, the Company issued a PUT for the issuance of 55.6 million shares of its common stock and a warrant to purchase 11.1 million shares of its common stock in conjunction with a joint venture transaction, see notes 6 and 9.
During 2008, the Company issued warrants to purchase 50.4 million shares of its common stock to the investors and the placement agent in conjunction with the sale of the secured 12% notes, see note 10.
| (6) | Joint Venture |
In February 2008, the Company and Nordic Biotech Advisors ApS through its investment fund Nordic Biotech Venture Fund II K/S (“Nordic”) entered into a 50/50 joint venture agreement (the “Hedrin JV Agreement”) to develop and commercialize the Company's North American rights (under license) to its Hedrin product.
Pursuant to the Hedrin JV Agreement, Nordic formed a new Danish limited partnership, Hedrin Pharmaceuticals K/S, (the "Hedrin JV") and provided it with initial funding of $2.5 million and the Company assigned and transferred its North American rights in Hedrin to the Hedrin JV in return for a $2.0 million cash payment from the Hedrin JV and equity in the Hedrin JV representing 50% of the nominal equity interests in the Hedrin JV. At closing the Company recognized an investment in the Hedrin JV of $250,000 and an exchange obligation of $2,054,630. The exchange obligation represents the Company’s obligation to Nordic to issue the Company’s common stock in exchange for all or a portion of Nordic’s equity interest in the Hedrin JV upon the exercise by Nordic of the put issued to Nordic in the Hedrin JV Agreement transaction. The put is described below.
The original terms of the Hedrin JV Agreement also provided that should the Hedrin JV be successful in achieving a payment milestone, namely that by September 30, 2008, the FDA determines to treat Hedrin as a medical device, Nordic will purchase an additional $2.5 million of equity in the Hedrin JV, whereupon the Hedrin JV will pay the Company an additional $1.5 million in cash and issue additional equity in the JV valued at $2.5 million, thereby maintaining the Company’s 50% ownership interest in the Hedrin JV. These terms have been amended as described below.
In June 2008, the Hedrin JV Agreement was amended (the "Hedrin JV Amended Agreement"). Under the amended terms Nordic invested an additional $1.0 million, for a total of $3.5 million, in the Hedrin JV and made an advance of $250,000 to the Hedrin JV and the Hedrin JV made an additional $1.0 million payment, for a total of $3.0 million, to the Company. The Hedrin JV also distributed additional ownership equity sufficient for each of the Company and Nordic to maintain their ownership interest at 50%. Under the amended terms, upon classification of Hedrin by the FDA as a Class II or Class III medical device Nordic is obligated to invest an additional $1.25 million, for a total investment of $5 million, into the Hedrin JV, the Hedrin JV is obligated to pay an additional $0.5 million, for a total of $3.5 million, to the Company, the $250,000 that Nordic advanced to the Hedrin JV in June becomes an equity investment in the Hedrin JV by Nordic and the Hedrin JV is obligated to issue to the Company and Nordic additional ownership interest in the Hedrin JV, thereby maintaining each of the Company’s and Nordic’s 50% ownership interest in the Hedrin JV. Subsequent to December 31, 2008, the FDA classified Hedrin as a Class III medical device, see Note 16.
F-18
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
The Company’s exchange obligation increased by $894,546 as a result of the June 2008 closing. The $894,546 represents the gross amount paid in June 2008 by the Hedrin JV to the Company of $1,000,000 offset by the costs of the Hedrin JV Agreement transaction recognized by the Company subsequent to the February closing.
During the year ended December 31, 2008, the Company recognized $250,000 of equity in the losses of the Hedrin JV, thereby reducing the carrying value of its investment to $0. During the year ended December 31, 2008 the Hedrin JV had losses of $658,000, the Company’s share of the losses is $329,000, leaving $79,000 share of losses of the Hedrin JV that were not recognized by the Company. At December 31, 2008, the Company’s exchange obligation is $2,949,176.
Nordic has an option to put all or a portion of its equity interest in the Hedrin JV to the Company in exchange for the Company’s common stock. The shares of the Company’s common stock to be issued upon exercise of the put will be calculated by multiplying the percentage of Nordic’s equity in the Hedrin JV that Nordic decides to put to the Company multiplied by the dollar amount of Nordic’s investment in Limited Partnership divided by $0.09, as adjusted from time to time, see note 12. The put option is exercisable immediately and expires at the earlier of ten years or when Nordic’s distributions from the Limited Hedrin JV exceed five times the amount Nordic invested in the Hedrin JV.
The Company has an option to call all or a portion of Nordic’s equity interest in the Hedrin JV in exchange for the Company’s common stock. The Company cannot begin to exercise its call until the price of the Company’s common stock has closed at or above $1.40 per share for 30 consecutive trading days. During the first 30 consecutive trading day period in which the Company’s common stock closes at or above $1.40 per share the Company can exercise up to 25% of its call option. During the second 30 consecutive trading day period in which the Company’s common stock closes at or above $1.40 per share the Company can exercise up to 50% of its call option on a cumulative basis. During the third 30 consecutive trading day period in which the Company’s common stock closes at or above $1.40 per share the Company can exercise up to 75% of its call option on a cumulative basis. During the fourth 30 consecutive trading day period in which the Company’s common stock closes at or above $1.40 per share the Company can exercise up to 100% of its call option on a cumulative basis. The shares of the Company’s common stock to be issued upon exercise of the call will be calculated by multiplying the percentage of Nordic’s equity in the Limited Partnership that the Company calls, as described above, multiplied by the dollar amount of Nordic’s investment in the Hedrin JV divided by $0.09. Nordic can refuse the Company’s call by either paying the Company up to $1.5 million or forfeiting all or a portion of their put, calculated on a pro rata basis for the percentage of the Nordic equity interest called by the Company.
The Hedrin JV is responsible for the development and commercialization of Hedrin for the North American market and all associated costs including clinical trials, if required, regulatory costs, patent costs, and future milestone payments owed to T&R, the licensor of Hedrin.
The Hedrin JV has engaged the Company to provide management services to the Limited Partnership in exchange for a management fee, which for 2008, on an annualized basis, is $527,000. As of December 31, 2008, the Company has recognized $446,806 of other income from management fees earned from the Hedrin JV which is included in the Company’s statement of operations for the year ended December 31, 2008 as a component of interest and other income.
F-19
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Nordic paid to the Company a non-refundable fee of $150,000 at the closing for the right to receive a warrant covering 11.1 million shares of the Company’s common stock, as adjusted due to the 12% Notes Transaction, see note 11, exercisable for $0.09 per share, as adjusted due to the 12% Notes Transaction, see note 11. The warrant is issuable 90 days from closing, provided Nordic has not exercised all or a part of its put, as described below. The Company issued the warrant to Nordic on April 30, 2008. The per share exercise price of the warrant was initially based on the volume weighted average price of the Company’s common stock for the period prior to the signing of the Hedrin JV Agreement and has been subsequently adjusted due the 12% Notes Transaction, see note 11.
The Hedrin JV's Board consists of 4 members, 2 appointed by the Company and 2 appointed by Nordic. Nordic has the right to appoint one of the directors as chairman of the Board. The chairman has certain tie breaking powers.
After the closing, at Nordic's request, the Company will nominate a person identified by Nordic to serve on the Company’s Board of Directors. Nordic has nominated a person, however, that person has declined to stand for appointment to the Company’s Board of Directors.
The Company granted Nordic registration rights for the shares to be issued upon exercise of the warrant, the put or the call. The Company filed an initial registration statement on May 1, 2008. The registration statement was declared effective on October 15, 2008. The Company is required to file additional registration statements, if required, within 45 days of the date the Company first knows that such additional registration statement was required. The Company is required to use commercially reasonable efforts to cause the additional registration statements to be declared effective by the Securities and Exchange Commission (“SEC”) within 105 calendar days from the filing date (the "Effective Date"). If the Company fails to file a registration statement on time or if a registration statement is not declared effective by the SEC within 105 days of filing the Company will be required to pay to Nordic, or its assigns, an amount in cash, as partial liquidated damages, equal to 0.5% per month of the amount invested in the Hedrin JV by Nordic until the registration statement is declared effective by the SEC. In no event shall the aggregate amount payable by the Company exceed 9% of the amount invested in the Hedrin JV by Nordic.
The profits of the Hedrin JV will be shared by the Company and Nordic in accordance with their respective equity interests in the Limited Partnership, which are currently 50% to each, except that Nordic will get a minimum distribution from the Hedrin JV equal to 5% on Hedrin sales, as adjusted for any change in Nordic’s equity interest in the Limited Partnership. If the Hedrin JV realizes a profit equal to or greater than a 10% royalty on Hedrin sales, then profits will be shared by the Company and Nordic in accordance with their respective equity interests in the Limited Partnership. However, in the event of a liquidation of the Limited Partnership, Nordic’s distribution in liquidation will be at least equal to the amount Nordic invested in the Hedrin JV ($5 million if the payment milestone described above is met, $3.5 million if it is not met) plus 10% per year, less the cumulative distributions received by Nordic from the Hedrin JV. Further, in no event shall Nordic’s distribution in liquidation be greater than assets available for distribution in liquidation.
F-20
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (7) | American Stock Exchange |
In September 2007, the Company received notice from the staff of The American Stock Exchange, or AMEX, indicating that the Company was not in compliance with certain continued listing standards set forth in the AMEX Company Guide. Specifically, AMEX notice cited the Company’s failure to comply, as of June 30, 2007, with section 1003(a)(ii) of the AMEX Company Guide as the Company had less than $4,000,000 of stockholders’ equity and had losses from continuing operations and /or net losses in three or four of our most recent fiscal years and with section 1003(a)(iii) which requires the Company to maintain $6,000,000 of stockholders’ equity if the Company has experienced losses from continuing operations and /or net losses in its five most recent fiscal years.
In order to maintain our AMEX listing, the Company was required to submit a plan to AMEX advising the exchange of the actions the Company has taken, or will take, that would bring the Company into compliance with all the continued listing standards by April 16, 2008. The Company submitted such a plan in October 2007. If the Company is not in compliance with the continued listing standards at the end of the plan period, or if the Company has not made progress consistent with the plan during the period, AMEX staff could have initiated delisting proceedings.
Under the terms of the Hedrin JV Agreement, the number of potentially issuable shares represented by the put and call features thereof and the warrant issuable to Nordic, would exceed 19.9% of the Company’s total outstanding shares and would be issued at a price below the greater of book or market value. As a result, under AMEX regulations, the Company would not have been able to complete the transaction without first receiving either stockholder approval for the transaction, or a formal “financial viability” exception from AMEX’s stockholder approval requirement. The Company estimated that obtaining stockholder approval to comply with AMEX regulations would take a minimum of 45 days to complete. The Company discussed the financial viability exception with AMEX for several weeks and had neither received the exception nor been denied the exception. The Company determined that our financial condition required the Company to complete the transaction immediately, and that the Company’s financial viability depended on the completion of the Hedrin JV Agreement without further delay.
Accordingly, to maintain the Company’s financial viability, on February 28, 2008, the Company announced that it had formally notified AMEX that the Company intended to voluntarily delist its common stock from AMEX. The delisting became effective on March 26, 2008.
The Company’s common stock now trades on the Over the Counter Bulletin Board under the symbol “MHAN”. The Company intends to maintain corporate governance, disclosure and reporting procedures consistent with applicable law.
| (8) | Secured 10% Notes Payable |
In September 2008, Manhattan entered into a series of Secured 10% Notes (the “Secured 10% Notes”) with certain of our directors, officers and an employee (the “Secured 10% Note Holders”) for aggregate of $70,000. Principal and interest on the Secured 10% Notes shall be paid in cash on March 10, 2009 unless paid earlier by us. Pursuant to the Secured 10% Notes, we also issued to the Secured 10% Note Holders 5-year warrants to purchase an aggregate of 140,000 shares of our common stock at an exercise price of $0.20 per share. Manhattan granted to the Secured 10% Note Holders a continuing security interest in certain specific refunds, deposits and repayments due Manhattan and expected to be repaid to Manhattan in the next several months. At December 31, 2008 accrued and unpaid interest on the Secured 10% Notes amounted to $1,764 and is reflected in the accompanying balance sheet as of December 31, 2008 as a component of accrued expenses. The Secured 10% Notes plus interest were repaid on February 4, 2009.
F-21
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (9) | Secured 12% Notes Payable |
On November 19 and December 23, 2008, the Company completed the first and second closings on a financing transaction (the “12% Notes Transaction”). The Company sold $1,315,000 of 12% senior secured notes (the “Secured 12% Notes”) and issued warrants to the investors to purchase 43.8 million shares of the Company’s common stock at $0.09 per share. The warrants expire on December 31, 2013. Net proceeds of $1,048,000 were realized from the first and second closings. In addition, $58,000 of issuance costs were paid outside of the first and second closings. Per the terms of the 12% Notes Transaction the net proceeds were paid into a deposit account (the “Deposit Account”) and are to be paid out to the Company in monthly installments of $106,000 retroactive to October 1, 2008. Per the terms of the 12% Notes Transaction the monthly installments are to be used exclusively to fund the current operating expenses of the Company. The Company received $318,000 of such monthly installments during 2008. The remaining balance at December 31, 2008 in the Deposit Account of approximately $730,000 is reflected in the accompanying balance sheet as of December 31, 2008 as restricted cash.
The financing transaction has a $1,000,000 minimum amount, which has been met and a $2,500,000 maximum amount. The financing transaction was still active at December 31, 2008. The third and final closing on this financing transaction occurred in February 2009, see Note 15, that brought the cumulative total to $1,725,000.
National Securities Corporation (“National”) was the placement agent for the 12% Notes Transaction. National’s compensation for acting is placement agent is a cash fee of 10% of the gross proceeds received, a non-accountable expense allowance of 1.5% of the gross proceeds, reimbursement of certain expenses and a warrant to purchase such number of shares of the Company’s common stock equal to 15% of the shares underlying the warrants issued to the investors. For the first and second closings the Company paid National a total of $155,000 in placement agent fees, a non-accountable expense allowance and reimbursement of certain expenses. In addition, the Company issued warrants to purchase 6.6 million shares of the Company’s common stock at $0.09 per share. These warrants were valued at $22,191 and are a component of Secured 12% notes payable issue costs. The warrant expires on December 13, 2013.
The Secured 12% Notes mature two years after issuance. Interest on the Secured 12% Notes is compounded quarterly and payable at maturity. At December 31, 2008, accrued and unpaid interest on the Secured 12% Notes amounted to approximately $15,000 and is reflected in the accompanying balance sheet at December 31, 2008 as interest payable on secured 12% notes payable. The Secured 12% Notes are secured by a pledge of all of the Company’s assets except for its investment in the Hedrin JV. The asset pledge includes the cash balance in the Deposit Account. In addition, to provide additional security for the Company’s obligations under the notes, the Company entered into a default agreement, which provides that upon an event of default under the notes, the Company shall, at the request of the holders of the notes, use reasonable commercial efforts to either (i) sell a part or all of the Company’s interests in the Hedrin joint venture or (ii) transfer all or part of the Company’s interest in the Hedrin JV to the holders of the notes, as necessary, in order to fulfill the Company’s obligations under the notes, to the extent required and to the extent permitted by the applicable Hedrin joint venture agreements.
F-22
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
In connection with the private placement, the Company, the placement agent and the investors entered into a registration rights agreement. Pursuant to the registration rights agreement, we agreed to file a registration statement to register the resale of the shares of our common stock issuable upon exercise of the warrants issued to the investors in the private placement, within 20 days of the final closing date and to cause the registration statement to be declared effective within 90 days (or 120 days upon full review by the Securities and Exchange Commission). we filed the registration statement on February 23, 2009. On March 11, 2009 we received a comment letter from the SEC. We are addressing those comments and plan to refile the registration statement once those comments have been addressed.
The issuance to the investors of warrants to purchase shares of the Company’s common stock at $0.09 per share changes the number of shares represented by the Nordic Put and the number of shares and exercise price of the Nordic Warrant. The Nordic Put and Nordic Warrant were issued at a value of $0.14 per share and were issued with anti-dilution rights. The issuance of any securities at a value of less than $0.14 per share activates Nordic’s anti-dilution rights. The Nordic Put and the Nordic Warrant are now exercisable at a price of $0.09 per share. The following table shows the effect of Nordic’s anti-dilution rights.
Shares Issuable Upon Exercise of Nordic's Put | Additional Shares Issuable Upon Exercise of Nordic's Put, if Certain Conditions Are Met | Shares Issuable Upon Exercise of Nordic's Warrant | Total Shares Issuable Upon Exercise of Nordic's Put and Warrant | |||||||||||||
| Before the 12% Notes Transaction | 26,785,714 | 8,928,572 | 7,142,857 | 42,857,143 | ||||||||||||
| Antidilution shares | 14,880,953 | 4,960,317 | 3,968,254 | 23,809,524 | ||||||||||||
| After the 12% Notes Transaction | 41,666,667 | 13,888,889 | 11,111,111 | 66,666,667 | ||||||||||||
The conditions for the additional shares becoming issuable upon the exercise of Nordic’s PUT were met subsequent to December 31, 2008, see note 16.
The Company incurred a total of approximately $347,000 of costs in the issuance of the $1,315,000 of Secured 12% Notes sold in 2008. These costs were capitalized and are being amortized over the life of the Secured 12% Notes into interest expense. During the year ended December 31, 2008, the amount amortized into interest expense was approximately $16,000. The remaining unamortized balance of approximately $331,000 is reflected in the accompanying balance sheet as of December 31, 2008 as a non-current asset, secured 12% notes payable issue costs.
F-23
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
The Company recognized an original issue discount (the “OID”) of approximately $148,000 on the issuance of the Secured 12% Notes sold in the first and second closings for the value of the warrants issued to the investors. The OID is being amortized over the life of the Secured 12% Notes into interest expense. During the year ended December 31, 2008 the amount amortized into interest expense was approximately $7,000. The remaining unamortized balance of approximately $141,000 has been netted against the face amount of the Secured 12% Notes in the accompanying balance sheet as of December 31, 2008.
As per the terms of the 12% Notes Transaction the Company’s officers agreed to certain modifications of their employment agreements, more fully described in Note 14.
| (10) | Stock Options |
2003 Stock Option Plan
In December 2003, the Company established the 2003 Stock Option Plan (the “2003 Plan”), which provided for the granting of up to 5,400,000 options to officers, directors, employees and consultants for the purchase of stock. In August 2005, the Company increased the number of shares of common stock reserved for issuance under the 2003 Plan by 2,000,000 shares. In May 2007, the Company increased the number of shares of common stock reserves for issuance under the 2003 Plan by 3,000,000 shares. At December 31, 2008, 10,400,000 shares were authorized for issuance. The options have a maximum term of 10 years and vest over a period determined by the Company’s Board of Directors (generally 3 years) and are issued at an exercise price equal to or greater than the fair market value of the shares at the date of grant. The 2003 Plan expires on December 10, 2013 or when all options have been granted, whichever is sooner. Under the 2003 Plan, the Company granted employees options to purchase an aggregate of 2,967,500 shares of common stock at an exercise price of $0.17 during the year ended December 31, 2008. In addition, 27,776 shares of common stock were issued during 2007 under the 2003 Plan.
At December 31, 2008, there were 875,628 shares reserved for future grants under the 2003 Plan.
1995 Stock Option Plan
In July 1995, the Company established the 1995 Stock Option Plan (the”1995 Plan”), which provided for the granting of options to purchase up to 130,000 shares of the Company’s common stock to officers, directors, employees and consultants. The 1995 Plan was amended several times to increase the number shares reserved for stock option grants. In June 2005 the 1995 Plan expired and no further options can be granted. At December 31, 2008 options to purchase 1,137,240 shares were outstanding and no shares were reserved for future stock option grants under the 1995 Plan.
F-24
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
A summary of the status of the Company’s stock options as of December 31, 2008 and changes during the year then ended is presented below:
| 2008 | ||||||||||||||||
| Shares | Weighted average exercise price | Weighted Average Remaining Contractual Term (years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at beginning of year | 8,033,838 | $ | 1.222 | |||||||||||||
| Granted | 2,967,500 | $ | 0.170 | |||||||||||||
| Exercised | - | |||||||||||||||
| Cancelled | (367,502 | ) | $ | 0.512 | ||||||||||||
| Outstanding at end of year | 10,633,836 | $ | 0.934 | 6.940 | $ | - | ||||||||||
| Options exercisable at year-end | 8,067,186 | $ | 1.122 | 6.300 | $ | - | ||||||||||
| Weighted-average fair value of options granted during the year | $ | 0.13 | ||||||||||||||
As of December 31, 2008 and 2007, the total compensation cost related to nonvested option awards not yet recognized is $425,660 and $539,046, respectively. The weighted average period over which it is expected to be recognized is approximately 1.18 and 0.5 years, respectively.
F-25
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
The following table summarizes the information about stock options outstanding at December 31, 2008:
Exercise Price | Number of Options Outstanding | Remaining Contractual Life (years) | Number of Options Exercisable | ||||||||||
| $ | 0.12 | 50,000 | 8.98 | 16,667 | |||||||||
| 0.17 | 2,900,000 | 9.24 | 1,050,015 | ||||||||||
| 0.20 | 65,000 | 3.24 | 65,000 | ||||||||||
| 0.22 | 163,750 | 3.14 | 163,750 | ||||||||||
| 0.28 | 12,000 | 3.08 | 12,000 | ||||||||||
| 0.28 | 10,000 | 1.28 | 10,000 | ||||||||||
| 0.28 | 10,000 | 2.14 | 10,000 | ||||||||||
| 0.70 | 220,000 | 7.53 | 146,667 | ||||||||||
| 0.72 | 315,000 | 8.09 | 126,668 | ||||||||||
| 0.80 | 876,490 | 4.15 | 876,490 | ||||||||||
| 0.89 | 16,667 | 7.43 | 16,667 | ||||||||||
| 0.95 | 670,000 | 8.32 | 290,000 | ||||||||||
| 0.97 | 440,000 | 5.74 | 440,000 | ||||||||||
| 1.00 | 290,696 | 6.03 | 290,696 | ||||||||||
| 1.35 | 108,333 | 7.07 | 86,666 | ||||||||||
| 1.35 | 60,000 | 7.53 | 40,000 | ||||||||||
| 1.35 | 300,000 | 7.09 | 300,000 | ||||||||||
| 1.38 | 100,000 | 6.46 | 100,000 | ||||||||||
| 1.45 | 25,000 | 6.66 | 25,000 | ||||||||||
| 1.50 | 2,923,900 | 6.25 | 2,923,900 | ||||||||||
| 1.65 | 1,077,000 | 5.08 | 1,077,000 | ||||||||||
| Total | 10,633,836 | 8,067,186 | |||||||||||
F-26
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (11) | Stock Warrants |
The following table summarizes the information about warrants to purchase shares of our common stock outstanding at December 31, 2008:
| Exercise Price | Number of Warrants Outstanding | Remaining Contractual Life (years) | Number of Warrants Exercisable | ||||||||||
| $ | 0.09 | 50,408,434 | 5.00 | 50,408,434 | |||||||||
| 0.09 | 11,111,111 | 4.33 | 11,111,111 | ||||||||||
| 0.20 | 140,000 | 4.75 | 140,000 | ||||||||||
| 0.28 | 150,000 | 4.64 | 150,000 | ||||||||||
| 0.78 | 10,000 | 1.98 | 10,000 | ||||||||||
| 1.00 | 3,564,897 | 4.25 | 3,564,897 | ||||||||||
| 1.00 | 509,275 | 4.25 | 509,275 | ||||||||||
| 1.10 | 326,499 | 1.04 | 326,499 | ||||||||||
| 1.44 | 2,161,767 | 2.65 | 2,161,767 | ||||||||||
| 1.44 | 540,449 | 2.65 | 540,449 | ||||||||||
| 1.44 | 135,135 | 2.65 | 135,135 | ||||||||||
| 1.49 | 221,741 | 2.67 | 221,741 | ||||||||||
| 1.49 | 55,000 | 2.67 | 55,000 | ||||||||||
| 1.90 | 10,000 | 1.21 | 10,000 | ||||||||||
| 1.90 | 90,000 | 1.21 | 90,000 | ||||||||||
| 69,434,308 | 69,434,308 | ||||||||||||
| (12) | Related-Party Transactions |
Oleoylestrone Developments, SL
The Company entered into a consulting agreement with OED. The agreement became effective in February 2002, at a fee of $6,250 per month. The agreement was terminated in November 2007. The fees associated with the consulting agreement are expensed as incurred. OED currently owns approximately 5.7 percent of the Company’s outstanding common stock. Additionally, Mr. Pons, chief executive officer of OED, was a member of the Company’s board of directors until his resignation in July 2007.
Total milestone payments under the license agreement of $0, $0 and $675,000 and consulting fees of $267,241, $68,750 and $698,491 are included in the accompanying statements of operations for the years ended December 31, 2008 and 2007 and for the cumulative period from August 6, 2001 to December 31, 2008.
F-27
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Paramount BioCapital, Inc.
One member of the Company’s board of directors, Timothy McInerney, was an employee of Paramount or one of its affiliates until April 2007. Another member of the Company’s board of directors, Michael Weiser, was an employee of Paramount until December 2006. In addition, two former members of the Company’s board of directors, Joshua Kazam and David Tanen, were employed by Paramount through August 2004 and were directors of the Company until September 2005. The sole shareholder of Paramount is Lindsay A. Rosenwald, M.D. Dr. Rosenwald beneficially owns more than 5 percent of the Company’s common stock as of December 31, 2008 and various trusts established for Dr. Rosenwald’s or his family’s benefit, held in excess of 12% of the Company’s common stock as of December 31, 2008. In November 2003, the Company paid to Paramount approximately $460,000 as commissions earned in consideration for placement agent services rendered in connection with the private placement of the Company’s Series A Convertible Preferred Stock, which amount represented 7 percent of the value of the shares sold by Paramount in the offering. In addition, in January 2004, the Company paid approximately $260,000 as commissions earned in consideration for placement agent services rendered by Paramount in connection with a private placement of the Company’s common stock, which amount represented 7 percent of the value of the shares sold by Paramount in the private placement. In connection with both private placements and as a result of their employment with Paramount, Mr. Kazam, Mr. McInerney and Dr. Weiser were allocated 5-year placement agent warrants to purchase 60,174, 58,642 and 103,655 shares of the Company’s common stock, respectively, at a price of $1.10 per share.
Paramount also served as the Company’s placement agent in connection with the August 2005 private placement. As placement agent, the Company paid to Paramount total cash commissions of $839,816 relating to the August 26, 2005 closing, of which $121,625 was paid to certain selected dealers engaged by Paramount in connection with the private placement and issued five-year warrants to purchase an aggregate of 540,449 shares of common stock exercisable at a price of $1.44 per share, of which Paramount received warrants to purchase 462,184 common shares. In connection with the August 30 closing, the Company paid cash commissions to Paramount of $88,550 and issued an additional five-year warrant to purchase 55,000 common shares exercisable at a price of $1.49 per share.
Paramount also served as the Company’s placement agent in connection with the March 2007 private placement. As placement agent, the Company paid to Paramount aggregate cash commissions of approximately $600,000 and issued to Paramount a 5-year warrant to purchase an aggregate of 509,275 shares of common stock at an exercise price of $1.00 per share.
F-28
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
| (13) | Income Taxes |
There was no current or deferred income tax expense for the years ended December 31, 2008 or 2007 because of the Company’s operating losses.
The components of deferred tax assets as of December 31, 2008 and 2007 are as follows:
| 2008 | 2007 | |||||||
| Deferred tax assets: | ||||||||
| Tax loss carryforwards | $ | 23,947,000 | $ | 22,513,000 | ||||
| Research and development credit | 1,769,000 | 1,769,000 | ||||||
| In-process research and development charge | 4,850,000 | 4,850,000 | ||||||
| Share-based compensation | 1,459,000 | 1,270,000 | ||||||
| Other | - | 85,000 | ||||||
| Gross deferred tax assets | 32,025,000 | 30,487,000 | ||||||
| Less valuation allowance | (32,025,000 | ) | (30,487,000 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
The reasons for the difference between actual income tax benefit for the years ended December 31, 2008 and 2007 and the amount computed by applying the statutory Federal income tax rate to losses before income tax benefit are as follows:
| 2008 | 2007 | |||||||||||||||
| Amount | % of Pre-tax loss | Amount | % of Pre-tax loss | |||||||||||||
| Federal income tax benefit at statutory rate | $ | (1,451,000 | ) | (34.0 | )% | $ | (4,102,000 | ) | (34.0 | )% | ||||||
| State income taxes, net of federal tax | (290,000 | ) | (6.8 | )% | (820,000 | ) | (6.8 | )% | ||||||||
| Research and development credits | (130,000 | ) | (3.0 | )% | (366,000 | ) | (3.0 | )% | ||||||||
| Other | 1,000 | 0.0 | % | 1,000 | 0.0 | % | ||||||||||
| Change in valuation allowance | 1,870,000 | 43.8 | % | 5,287,000 | 43.9 | % | ||||||||||
| $ | - | 0.0 | % | $ | - | 0.0 | % | |||||||||
A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. The net change in the total valuation allowance for the years ended December 31, 2008 and 2007 was an increase of $1,870,000 and $5,287,000, respectively. The tax benefit assumed using the Federal statutory tax rate of 34% has been reduced to an actual benefit of zero due principally to the aforementioned valuation allowance.
F-29
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
At December 31, 2008, the Company had unused Federal and state net operating loss carryforwards of approximately $60,426,000 and $56,963,000, respectively. The net operating loss carryforwards expire in various amounts through 2028 for Federal and state income tax purposes. The Tax Reform Act of 1986 contains provisions which limit the ability to utilize net operating loss carryforwards in the case of certain events including significant changes in ownership interests. Accordingly, a substantial portion of the Company’s net operating loss carryforwards above will be subject to annual limitations (currently approximately $100,000) in reducing any future year’s taxable income. At December 31, 2008, the Company also had research and development credit carryforwards of approximately $1,769,000 for Federal income tax purposes which expire in various amounts through 2028.
The Company files income tax returns in the U.S. Federal, State and Local jurisdictions. With certain exceptions, the Company is no longer subject to U.S. Federal and state income tax examinations by tax authorities for years prior to 2005. However, net operating loss carryforwards and tax credits generated from those prior years could still be adjusted upon audit. The Company adopted the provisions of FIN 48, “Accounting for Uncertainty in Income Taxes – an interpretation of FASB Statement No. 109” on January 1, 2007 with no material impact to the financial statements. The Company recognizes interest and penalties to uncertain tax position in income tax expense in the statement of operations.
The Company had no unrecognized tax benefits at December 31, 2008 that would affect the annual effective tax rate. Further, the Company is unaware of any positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next twelve months.
| (14) | License and Consulting Agreements |
IGI Agreement for PTH (1-34)
On April 1, 2005, as part of the acquisition of Tarpan Therapeutics, Inc., the Company acquired a Sublicense Agreement with IGI, Inc. (the “IGI Agreement”) dated April 14, 2004. Under the IGI Agreement the Company received the exclusive, world-wide, royalty bearing sublicense to develop and commercialize the licensed technology (see Note 1). Under the terms of the IGI Agreement, the Company is responsible for the cost of the preclinical and clinical development of the project, including research and development, manufacturing, laboratory and clinical testing and trials and marketing of licensed products for which the company will be responsible.
In consideration for the Company’s rights under the IGI Agreement, a payment of $300,000 was made upon execution of the agreement, prior to the Company’s acquisition of Tarpan. In addition the IGI Agreement requires the Company to make certain milestone payments as follows: $300,000 payable upon the commencement of a Phase 2 clinical trial; $500,000 upon the commencement of a Phase 3 clinical trial; $1,500,000 upon the acceptance of an NDA application by the FDA; $2,400,000 upon the approval of an NDA by the FDA; $500,000 upon the commencement of a Phase 3 clinical trial for an indication other than psoriasis; $1,500,000 upon the acceptance of and NDA application for an indication other than psoriasis by the FDA; and $2,400,000 upon the approval of an NDA for an indication other than psoriasis by the FDA.
During 2007, we achieved the milestone of the commencement of Phase 2 clinical trial. As a result $300,000 became payable to IGI. This $300,000 is included in research and development expense for the year ended December 31, 2007. Payment was made to IGI in February 2008. At December 31, 2007 this $300,000 liability is reflected in accounts payable.
F-30
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
In addition, the Company is obligated to pay IGI, Inc. an annual royalty of 6% annual net sales on annual net sales up to $200,000,000. In any calendar year in which net sales exceed $200,000,000, the Company is obligated to pay IGI, Inc. an annual royalty of 9% annual net sales. Through December 31, 2008, the Company has not paid any such royalties.
IGI, Inc. may terminate the agreement (i) upon 60 days’ notice if the Company fails to make any required milestone or royalty payments, or (ii) if the Company becomes bankrupt or if a petition in bankruptcy is filed, or if the Company is placed in the hands of a receiver or trustee for the benefit of creditors. IGI, Inc. may terminate the agreement upon 60 days’ written notice and an opportunity to cure in the event the Company commits a material breach or default. Eighteen months from the date of the IGI Agreement, the Company may terminate the agreement in whole or as to any portion of the PTH patent rights upon 90 days’ notice to IGI, Inc.
Hedrin License Agreement
On June 26, 2007, the Company entered into an exclusive license agreement for “Hedrin” (the “Hedrin License Agreement”) with Thornton & Ross Ltd. (“T&R”) and Kerris, S.A. (“Kerris”). Pursuant to the Hedrin License Agreement, the Company has acquired an exclusive North American license to certain patent rights and other intellectual property relating to Hedrin(TM), a non-insecticide product candidate for the treatment of head lice. In addition, on June 26, 2007, the Company entered into a supply agreement with T&R pursuant to which T&R will be the Company’s exclusive supplier of Hedrin product the “Hedrin Supply Agreement”.
In consideration for the license, the Company issued to T&R and Kerris (jointly, the “Licensor”) a combined total of 150,000 shares of its common stock valued at $120,000. In addition, the Company also made a cash payment of $600,000 to the Licensor. These amounts are included in research and development expense. Further, the Company agreed to make future milestone payments to the Licensor in the aggregate amount of $2,500,000 upon the achievement of various clinical, regulatory, and patent issuance milestones, as well as up to $2,500,000 in a one-time success fee based on aggregate sales of the product by the Company and its licensees of at least $50,000,000. The Company also agreed to pay royalties of 8% (or, under certain circumstances, 4%) on net sales of licensed products. The Company’s exclusivity under the Hedrin License Agreement is subject to an annual minimum royalty payment of $1,000,000 (or, under certain circumstances, $500,000) in each of the third through seventh years following the first commercial sale of Hedrin. The Company may sublicense its rights under the Hedrin Agreement with the consent of Licensor and the proceeds resulting from such sublicenses will be shared with the Licensor.
Pursuant to the supply agreement, the Company has agreed that it and its sublicensees will purchase their respective requirements of the Hedrin product from T&R at agreed upon prices. Under certain circumstances where T&R is unable to supply Hedrin products in accordance with the terms and conditions of the Supply Agreement, the Company may obtain products from an alternative supplier subject to certain conditions. The term of the Supply Agreement ends upon termination of the Hedrin Agreement.
In February 2008 the Company assigned and transferred its rights in Hedrin to a joint venture, see note 6.
F-31
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Altoderm License Agreement
On April 3, 2007, the Company entered into a license agreement for “Altoderm” (the “Altoderm Agreement”) with T&R. Pursuant to the Altoderm Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altoderm, a topical skin lotion product candidate using sodium cromoglicate for the treatment of atopic dermatitis. In accordance with the terms of the Altoderm Agreement, the Company issued 125,000 shares of its common stock, valued at $112,500, and made a cash payment of $475,000 to T&R upon the execution of the agreement. These amounts have been included in research and development expense. Further, the Company agreed to make future milestone payments to T&R comprised of various combinations of cash and common stock in respective aggregate amounts of $5,675,000 and 875,000 shares of common stock upon the achievement of various clinical and regulatory milestones. The Company also agreed to pay royalties on net sales of products using the licensed patent rights at rates ranging from 10% to 20%, depending on the level of annual net sales, and subject to an annual minimum royalty payment of $1 million in each year following the first commercial sale of Altoderm. The Company may sublicense the patent rights. The Company agreed to pay T&R 30% of the royalties received by the Company under such sublicense agreements.
Subsequent to December 31, 2008, the Company terminated the Altoderm Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altoderm Agreement.
Altolyn License Agreement
On April 3, 2007, the Company and T&R also entered into a license agreement for “Altolyn” (the “Altolyn Agreement”). Pursuant to the Altolyn Agreement, the Company acquired an exclusive North American license to certain patent rights and other intellectual property relating to Altolyn, an oral formulation product candidate using sodium cromoglicate for the treatment of mastocytosis, food allergies, and inflammatory bowel disorder. In accordance with the terms of the Altolyn Agreement, the Company made a cash payment of $475,000 to T&R upon the execution of the agreement. This amount is included in research and development expense. Further, the Company agreed to make future cash milestone payments to T&R in an aggregate amount of $5,675,000 upon the achievement of various clinical and regulatory milestones. The Company also agreed to pay royalties on net sales of products using the licensed patent rights at rates ranging from 10% to 20%, depending on the level of annual net sales, and subject to an annual minimum royalty payment of $1 million in each year following the first commercial sale of Altolyn. The Company may sublicense the patent rights. The Company agreed to pay T&R 30% of the royalties received by the Company under such sublicense agreements.
Subsequent to December 31, 2008, the Company terminated the Altolyn Agreement for convenience. The Company has no further financial liability or commitment to T&R under the Altolyn Agreement.
OED License Agreement for Oleoyl-estrone
On February 15, 2002, the Company entered into a License Agreement (the "License Agreement") with OED. Under the terms of the License Agreement, OED granted to the Company a world-wide license to make, use, lease and sell the products incorporating the licensed technology (see Note 1). OED also granted to the Company the right to sublicense to third parties the licensed technology or aspects of the licensed technology with the prior written consent of OED. OED retains an irrevocable, nonexclusive, royalty-free right to use the licensed technology solely for its internal, noncommercial use. The License Agreement shall terminate automatically upon the date of the last to expire patent contained in the licensed technology or upon the Company's bankruptcy. OED may terminate the License Agreement in the event of a material breach by the Company that is not cured within the notice period. The Company may terminate the License Agreement for any reason upon 60 days notice. The Company terminated this agreement in November 2007.
F-32
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
In addition to the License Agreement, the Company entered into a consulting agreement with OED. The agreement became effective in February 2002, at a fee of $6,250 per month, and terminated when the License Agreement terminated. The fees associated with the consulting agreement are expensed as incurred.
Under the License Agreement, the Company agreed to pay to OED certain licensing fees which are being expensed as they are incurred. The Company paid $175,000 in up front licensing fees which is included in 2002 research and development expense. In addition, pursuant to the License Agreement, the Company issued 1,000,000 shares of its common stock to OED. The Company valued these shares at their then estimated fair value of $1,000.
In connection with the License Agreement, the Company has agreed to milestone payments to OED as follows:
(i) $250,000 upon the treatment of the first patient in a Phase I clinical trial under a Company-sponsored investigational new drug application ("IND"), which was paid in 2005; (ii) $250,000 upon the treatment of the first patient in a Phase II clinical trial under a Company-sponsored IND, which was paid in 2006; (iii) $750,000 upon the first successful completion of a Company-sponsored Phase II clinical trial under a Company-sponsored IND; (iv) $2,000,000 upon the first successful completion of a Company-sponsored Phase III clinical trial under a Company sponsored IND; and (v) $6,000,000 upon the first final approval of the first new drug application for the first licensed product by the United States Food and Drug Administration (“FDA”). Through December 31, 2007, the Company paid a total of $675,000 in licensing fees and milestone payments. The Company has no further financial liability or commitment to OED under the License Agreement.
NovaDel Agreement for Propofol Lingual Spray
In April 2003, the Company entered into a license and development agreement with NovaDel, under which the Company received certain worldwide, exclusive rights to develop and commercialize products related to NovaDel’s proprietary lingual spray technology for delivering propofol for pre-procedural sedation. Under the terms of this agreement, the Company agreed to use its commercially reasonable efforts to develop and commercialize the licensed products, to obtain necessary regulatory approvals and to thereafter exploit the licensed products. The agreement also provides that NovaDel will undertake to perform, at the Company’s expense, a substantial portion of the development activities, including, without limitation, preparation and filing of various applications with applicable regulatory authorities.
In consideration for the Company’s rights under the NovaDel license agreement, the Company paid NovaDel an initial license fee of $500,000 in 2003. In addition, the license agreement requires the Company to make certain milestone payments as follows: $1,000,000 payable following the date that the first NDA for lingual spray propofol is accepted for review by the FDA; $1,000,000 following the date that the first European Marketing Application is accepted for review by any European Union country; $2,000,000 following the date when the first filed NDA for lingual spray propofol is approved by the FDA; $2,000,000 following the date when the first filed European Marketing Application for lingual spray propofol is accepted for review; $1,000,000 following the date on which an application for commercial approval of lingual spray propofol is approved by the appropriate regulatory authority in each of Australia, Canada, Japan and South Africa; and $50,000 following the date on which an application for commercial approval for lingual spray propofol is approved in any other country (other than the U.S., a member of the European Union, Australia, Canada, Japan or South Africa).
F-33
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
In addition, the Company is obligated to pay to NovaDel an annual royalty based on a fixed rate of net sales of licensed products, or if greater, the annual royalty is based on the Company’s net profits from the sale of licensed products at a rate that is twice the net sales rate. In the event the Company sublicenses the licensed product to a third party, the Company is obligated to pay royalties based on a fixed rate of fees or royalties received from the sublicensee until such time as the Company recovers its out-of-pocket costs, and thereafter the royalty rate doubles. Because of the continuing development efforts required of NovaDel under the agreement, the royalty rates are substantially higher than customary for the industry. Through December 31, 2007, the Company has incurred, and paid a total of $500,000 under the NovaDel license agreement, the initial license fee paid in 2003. The Company terminated this agreement during 2007 and has no continuing obligations under this agreement.
| (15) | Commitments and Contingencies |
Swiss Pharma
The Company has been involved in an arbitration proceeding in Switzerland with Swiss Pharma Contract LTD (“Swiss Pharma”), a clinical site that the Company used in one of its obesity trials. On September 5, 2008, the sole arbitrator in Switzerland rendered an award in favor of Swiss Pharma, awarding to Swiss Pharma a total of approximately $646,000 which amount includes a contract penalty of approximately $323,000, a final services invoice of approximately $48,000, reimbursement of certain of Swiss Pharma’s legal and other expenses incurred in the arbitration process of approximately $245,000, reimbursement of arbitration costs of approximately $13,000 and interest through September 5, 2008 of approximately $17,000. Further, the arbitrator ruled that the Company must pay interest at the rate of 5% per annum on approximately $371,000, the sum of the contract penalty of approximately $323,000 and the final services invoice of approximately $48,000, from October 12, 2007 until paid.
The Company had previously recognized a liability to Swiss Pharma in the amount of approximately $104,000 for the final services invoice. The remainder of the award, approximately $542,000, was expensed in September 2008. The Company has recognized research and development expense of approximately $267,000, general and administrative expense of approximately $257,000 and interest expense of approximately $23,000 during the year ended December 31, 2008. The Company will continue to accrue interest at the rate of 5% per annum on the approximate $371,000 until such amount has been settled.
The Company does not have sufficient cash or other current assets to satisfy the arbitrator's award.
On January 22, 2009, the Company received notice that Swiss Pharma submitted a petition to the Supreme Court of New York State, County of New York seeking to confirm and to enter a judgment on the arbitration award. On February 17, 2009, the Company filed an answer to that complaint. A hearing has not yet been scheduled.
Contentions of a Former Employee
In February 2007, a former employee of the Company alleged an ownership interest in two of the Company’s provisional patent applications. Also, without articulating precise legal claims, the former employee contends that the Company wrongfully characterized the former employee’s separation from employment as a resignation instead of a dismissal in an effort to harm the former employee’s immigration sponsorship efforts, and, further, to wrongfully deprive the former employee of the former employee’s alleged rights in two of the Company’s provisional patent applications. The former employee is seeking an unspecified amount in damages. The Company refutes the former employee’s contentions and intends to vigorously defend itself should the former employee file claims against the Company. There have been no further developments with respect to these contentions.
F-34
MANHATTAN PHARMACEUTICALS, INC.
(a Development Stage Company)
NOTES TO FINANCIAL STATEMENTS
Employment Agreement
The Company has employment agreements with two employees for the payment of aggregate annual base salary of $675,000 as well as performance based bonuses. These agreements have a remaining term of three months for one employee and six months for the second employee and have a remaining obligation of $226,000 as of December 31, 2008. As per the terms of the 12% Notes Transaction, see Note 10, management, comprised of the two employees under contract, has agreed to reduce their salaries effective as of October 1, 2008. If the Company sells less than $1.5 million of Secured 12% Notes then their salaries shall be reduced by one-third. If the Company sells at least $1.5 million but less than $2 million of Secured 12% Notes then their salaries shall be reduced by 20%. The Company sold $1.725 million of Secured 12% Notes, management therefore was paid 80% of their salaries during the fourth quarter of 2008.
Leases
Rent expense for the years ended December 31, 2008 and 2007 was $139,636 and $141,012, respectively. Future minimum rental payments subsequent to December 31, 2008 under an operating lease for the Company’s office facility are as follows:
| Years Ending December 31, | Commitment | |||
| 2009 | $ | 63,900 | ||
| 2010 and subsequent | $ | 0 | ||
F-35
| (16) | Subsequent events |
Joint Venture
In January 2009 the FDA classified Hedrin as a Class III medical device. As a result, and as per the terms of the Hedrin JV Agreement, as amended in June 2008, the $250,000 that Nordic advanced to the Hedrin JV in June 2008 became an equity investment in the Hedrin JV by Nordic and Nordic invested an additional $1.25 million in the Hedrin JV, for a total investment of $5.0 million. Also as a result of this classification of Hedrin and as per the terms of the Hedrin JV Agreement, as amended in June 2008, the Hedrin JV made a $500,000 milestone payment to the Company in February 2009 and the Hedrin JV distributed additional ownership equity sufficient for the Company and Nordic to maintain their ownership interest at 50% each.
Secured 12% Notes Payable
In February 2009 the third and final closing occurred of the 12% Notes Transaction. In this final closing an additional $410,000 of Secured 12% Notes were sold. Net proceeds of $360,000 were realized and paid into the Deposit Account. Per the terms of the 12% Notes Transaction, $200,000 was paid to the Company out of the Deposit Account for the specific purpose of making payments on its outstanding accounts payable balances as of September 30, 2008. Also per the terms of the 12% Notes Transaction, the monthly installments to be paid to the Company from the Deposit Account were increased to $113,300 from $106,000.
The Company incurred additional issuance costs of $50,000, for a total of $375,000, and recognized additional OID of $46,000, for a total of $194,000. Included in the $50,000 of issuance costs was $47,000 paid to National, for a total of $202,000.
The Company issued five-year warrants to the investors to purchase an additional 14 million shares of the Company’s common stock at $0.09 per share, for a total of 58 million shares, and issued a five-year warrant to National to purchase an additional 2 million shares of the Company’s common stock at $0.09 per share, for a total of 8.6 million shares.
F-36
Index to Exhibits Filed with this Report
| Exhibit No. | Description | |
| 23.1 | Consent of J.H. Cohn LLP. | |
23.2 | Consent of J.H. Cohn LLP. | |
| 31.1 | Certification of Principal Executive Officer. | |
| 31.2 | Certification of Principal Financial Officer. | |
| 32.1 | Certifications pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
F-37
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Our report on our audits of the financial statements of Manhattan Pharmaceuticals, Inc. as of December 31, 2008 and 2007 and for the years then ended and on the statements of operations, changes in stockholders' equity (deficiency) and cash flows for the period from August 6, 2001 (date of inception) to December 31, 2008, which contains an explanatory paragraph relating to the Company’s ability to continue as a going concern, included in this Annual Report on Form 10-K for the year ended December 31, 2008, is dated March 30, 2009. We consent to the incorporation by reference of our report in the following registration statements previously filed by the Company with the Securities and Exchange Commission pursuant to the Securities Act of 1933: the registration statements on Forms S-3 with SEC File Nos. 333-131814 and 333-142788 and the registration statements on Forms S-8 with SEC file Nos. 333-15807, 333-112888, 333-112889, 333-143838 and 333-48531.
/s/ J.H. Cohn LLP
Roseland, New Jersey
March 30, 2009
Exhibit 23.2
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the inclusion in this prospectus supplement (File No. 333-150580) of our report, which includes an explanatory paragraph relating to Manhattan Pharmaceuticals, Inc.’s ability to continue as a going concern, dated March 30, 2009, on our audits of the financial statements of Manhattan Pharmaceuticals, Inc. as of December 31, 2008 and 2007, and for the years then ended and for the period from August 6, 2001 (inception) to December 31, 2008.
/s/ J.H. Cohn LLP
Roseland, New Jersey
April 8, 2009
EXHIBIT 31.1
CERTIFICATION
I, Douglas Abel, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Manhattan Pharmaceuticals, Inc. (the “Registrant”); |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Registrant as of, and for, the periods presented in this report; |
| 4. | The Registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d - 15(e)) for the Registrant and have: |
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the Registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the Registrant's internal control over financial reporting that occurred during the Registrant's most recent fiscal quarter (the Registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the Registrant’s internal control over financial reporting; and
| 5. | The Registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Registrant’s auditors and the audit committee of the Registrant’s board of directors (or persons performing the equivalent functions): |
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Registrant’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the Registrant’s internal control over financial reporting.
| Date: March 31, 2009 | /s/ Douglas Abel |
| Douglas Abel | |
| Chief Executive Officer |
EXHIBIT 31.2
CERTIFICATION
I, Michael G. McGuinness, certify that:
| 1. | I have reviewed this Annual Report on Form 10-K of Manhattan Pharmaceuticals, Inc. (the “Registrant”); |
| 2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
| 3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Registrant as of, and for, the periods presented in this report; |
| 4. | The Registrant's other certifying officer(s) and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d – 15(e)) for the Registrant and have: |
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the Registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the Registrant's internal control over financial reporting that occurred during the Registrant's most recent fiscal quarter (the Registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the Registrant’s internal control over financial reporting; and
| 5. | The Registrant's other certifying officer(s) and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Registrant’s auditors and the audit committee of the Registrant’s board of directors (or persons performing the equivalent functions): |
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Registrant’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the Registrant’s internal control over financial reporting.
| Date: March 31, 2009 | /s/ Michael G. McGuinness |
| Michael G. McGuinness | |
| Chief Operating and Financial Officer |
Exhibit 32.1
CERTIFICATION
Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, the undersigned officers of Manhattan Pharmaceuticals, Inc. hereby certifies that, to the best of their knowledge:
(a) the Annual Report on Form 10-K of Manhattan Pharmaceuticals, Inc. for the year ended December 31, 2008 (the “Report”) fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(b) information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of Manhattan Pharmaceuticals, Inc.
| Dated: March 31, 2009 | /s/ Douglas Abel |
| Douglas Abel | |
| President and Chief Executive Officer | |
| Dated: March 31, 2009 | /s/ Michael G. McGuinness |
| Michael G. McGuinness | |
| Chief Operating and Financial Officer |