Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington D.C. 20549
Form 10-K
| (Mark One) | ||
þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the fiscal year ended December 31, 2006 | ||
| or | ||
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
| For the transition period from to . |
Commission File Number: 0-28402
Aradigm Corporation
(Exact Name of Registrant as Specified in Its Charter)
| California | 94-3133088 | |||
| (State or Other Jurisdiction of | (I.R.S. Employer | |||
| Incorporation or Organization) | Identification No. | ) |
3929 Point Eden Way, Hayward, CA 94545
(Address of Principal Executive Offices)
Registrant’s telephone number, including area code:
(510) 265-9000
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, no par value
Indicate by check mark whether the registrant is a well-know seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes o No þ
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and larger accelerated filer” inRule 12b-2 of the Exchange Act. (check one):
Large accelerated filer o Accelerated filer o Non-accelerated filer þ
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Exchange Act). Yes o No þ
The aggregate market value of registrant’s common stock held by non-affiliates of the registrant, based upon the closing price of a share of the registrant’s common stock on June 30, 2006 was: $19,056,180
The number of shares of the registrant’s common stock outstanding as of February 28, 2007 was: 53,946,300
TABLE OF CONTENTS
Table of Contents
Forward Looking Statements
This Annual Report onForm 10-K contains forward-looking statements. When used in this Annual Report the words “anticipate,” “objective,” “may,” “might,” “should,” “could,” “can,” “intend,” “expect,” “believe,” “estimate,” “predict,” “potential,” “plan” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about: our expectations regarding our future expenses, sales and operations; our anticipated cash needs and our estimates regarding our capital requirements and our need for additional financing; the expected development path and timing of our product candidates; our expectations regarding the use of Section 505(b)(2) of the United States Food, Drug and Cosmetic Act and an expedited development and regulatory process; our ability to obtain and derive benefits from orphan drug designation; our ability to anticipate the future needs of our customers; our plans for future products and enhancements of existing products; our growth strategy elements; the anticipated trends and challenges in the markets in which we operate; and our ability to attract customers.
These statements reflect our current views with respect to uncertain future events and are based on imprecise estimates and assumptions and subject to risk and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. While we believe our plans, intentions and expectations reflected in those forward-looking statements are reasonable, these plans, intentions or expectations may not be achieved. Our actual results, performance or achievements could differ materially from those contemplated, expressed or implied by the forward-looking statements contained in this Annual Report for a variety of reasons, including those under the heading “Risk Factors.”
All forward-looking statements attributable to us or persons acting on our behalf are expressly qualified in their entirety by the risk factors and other cautionary statements set forth in this Annual Report. Other than as required by applicable securities laws, we are under no obligation, and we do not intend, to update any forward-looking statement, whether as result of new information, future events or otherwise.
PART I
| Item 1. | Business |
Overview
We are an emerging specialty pharmaceutical company focused on the development and commercialization of a portfolio of drugs delivered by inhalation for the treatment of severe respiratory diseases by pulmonologists. Local delivery of drugs to the respiratory tract by inhalation for the treatment of respiratory disease has been shown to be safe and efficacious and to provide a rapid onset of action in conditions such as asthma, chronic bronchitis and cystic fibrosis. We have developed a significant amount of expertise and intellectual property in pulmonary drug delivery for respiratory and systemic diseases over the last decade. We have demonstrated in our laboratory research and clinical trials that our hand-held AERx pulmonary drug delivery system, with a product candidate currently in Phase 3 clinical trials, is particularly suitable for drugs where highly efficient and precise delivery to the respiratory tract is advantageous or essential.
We currently have three respiratory product candidates in development: innovative treatments for cystic fibrosis, pulmonary arterial hypertension, and inhalation anthrax. In selecting our development programs, we seek drugs approved by the United States Food and Drug Administration, or the FDA, that can be reformulated for both existing and new indications in respiratory disease. Our intent is to use our pulmonary delivery methods and formulations to improve their safety, efficacy and convenience to patients. We believe that this strategy will allow us to reduce cost, development time and risk of failure, when compared to the discovery and development of new chemical entities. We intend to commercialize our respiratory product candidates with our own focused sales and marketing force addressing pulmonary specialty doctors in the United States, where we believe that a proprietary sales force will enhance the return to our shareholders. Where our products can benefit a broader population of patients in the United States or in other countries, we may enter into co-development, co-promotion or other marketing arrangements with collaborators, thereby reducing costs and increasing revenues through license fees, milestone payments and royalties.
1
Table of Contents
Pulmonary delivery by inhalation is already a widely used, well accepted method of administration of a variety of drugs for the treatment of respiratory diseases. Compared to other routes of administration, inhalation provides local delivery of the drug to the respiratory tract, offering a number of potential advantages, including rapid onset of action, less drug required to achieve the desired therapeutic effect, and reduced side effects because the rest of the body has lower exposure to the drug. We believe that there still are significant unmet medical needs in the respiratory disease market, both to replace existing therapies that over prolonged use in patients demonstrate reduced efficacy or increased side effects, as well as to provide novel treatments to patient populations and for disease conditions that are inadequately treated. Based on our analysis of market data from Business Insights and Wolters Kluwer PHAST, we believe that we could potentially address a market opportunity currently estimated at approximately $20 billion, and growing at over 10% per year, for inhaled treatments of chronic respiratory diseases.
In addition to its use in the treatment of respiratory diseases, there is also an increasing awareness of the value of the inhalation route of delivery to administer drugs via the lung for the systemic treatment of disease elsewhere in the body. For many drugs, the large and highly absorptive area of the lung enables bioavailability via pulmonary delivery that could otherwise only be obtained by injection. We believe that the features of our AERx delivery system make it more attractive for many systemic drug applications than alternative methods. The most advanced product candidate based on the AERx delivery system is in Phase 3 clinical trials being conducted by our licensee, Novo Nordisk A/S, to deliver insulin systemically via the lungs for the treatment of diabetes. We believe particular opportunities exist for the use of our pulmonary delivery technology for the delivery of biologics, including proteins, antibodies and peptides, that today must be delivered by injection, as well as small molecule drugs, where rapid absorption is desirable. We intend to pursue selected opportunities for systemic delivery via inhalation by seeking collaborations that will fund development and commercialization.
We believe that our proprietary formulation and delivery technologies and our experience in the development and management of pulmonary clinical programs uniquely position us to benefit from the opportunities in the respiratory disease market as well as other pharmaceutical markets that would benefit from the efficient, non-invasive inhalation delivery of drugs.
Our Strategy
We are transitioning our business model toward a specialty pharmaceutical company focused on development and commercialization of a portfolio of drugs delivered by inhalation for the treatment of respiratory diseases. We have chosen to focus on respiratory diseases based on the expertise of our management team and the history of our company. We have significant experience in the treatment of respiratory diseases and specifically in the development of inhalation products that are uniquely suited for their treatment. We have a portfolio of proprietary technologies that may potentially address significant unmet medical needs for better products in the global respiratory market, which showed over 10% growth overall in 2005 with higher growth rates in the areas of innovative products, based on our analysis of market data from Business Insights and Wolters Kluwer PHAST. There are five key elements of our strategy:
| • | Develop a proprietary portfolio of products for the treatment of respiratory diseases. We believe our expertise in the development of pulmonary pharmaceutical products should enable us to advance and commercialize respiratory products for a variety of indications. We will continue to evaluate appropriate drugs and biologics for inclusion in our proprietary pipeline. We will do so in consideration of the expected market opportunity, cost, time and potential returns and the resources needed to advance our self-initiated programs and programs with collaborators. We select for development those products that can benefit from our experience in pulmonary delivery and that we believe are likely to provide a superior therapeutic profile or other valuable benefits to patients when compared to existing products. A key component of our strategy will be to continue to actively seek product opportunities where we can pursue either a new indication or route of administration for drugs already approved by the FDA. In each case, we will then combine the drug with the most appropriate pulmonary delivery system and formulation to create a proprietary product candidate with an attractive therapeutic profile and that is safe, effective and convenient for patients to use. | |
| • | Accelerate the regulatory approval process. We believe our management team’s regulatory expertise in pharmaceutical inhalation products, new indications and reformulations of existing drugs will enable us to |
2
Table of Contents
| pursue the most appropriate regulatory pathway for our product candidates. Because many of our product candidates incorporate FDA-approved drugs, we believe that the most expedient review and approval pathway for many of these product candidates in the United States will be under Section 505(b)(2) of the Food, Drug and Cosmetic Act, or the FDCA. Section 505(b)(2) permits the FDA to rely on scientific literature or on the FDA’s prior findings of safetyand/or effectiveness for approved drug products. By choosing to develop new applications or reformulations of FDA-approved drugs, we believe that we can substantially reduce or potentially eliminate the significant time, expenditure and risks associated with preclinical testing of new chemical entities and biologics, as well as utilize knowledge of these approved drugs to reduce the risk, time and cost of the clinical trials needed to obtain drug approval. In addressing niche market opportunities, we intend to pursue orphan drug designation for our products when appropriate. Orphan drug designation may be granted to drugs and biologics that treat rare life-threatening diseases that affect fewer than 200,000 persons in the United States. Such designation provides a company with the possibility of market exclusivity for up to seven years as well as regulatory assistance, reduced filing fees and possible tax credits. |
| • | Develop our own sales and marketing capacity for products in niche markets. We intend to develop our own targeted sales and marketing force for those of our products prescribed primarily by the approximately 11,000 pulmonologists, or their subspecialty segments, in the United States. We expect to begin establishing a sales force as we approach commercialization of the first of such products. We believe that by developing a small sales group dedicated to interacting with disease-specific physicians in the respiratory field, we can create greater value from our products for our shareholders. For markets where maximizing sales of the product would depend on marketing to primary healthcare providers that are only addressable with a large sales force, we plan to enter into co-marketing arrangements. We also intend to establish collaborative relationships to commercialize our products in cases where we cannot meet these goals with a small sales force or when we need collaborators with relevant expertise and capabilities, such as the ability to address international markets. Through such collaborations, we may also utilize our collaborators’ resources and expertise to conduct large late-stage clinical development. | |
| • | Exploit the broad applicability of our delivery technology through opportunistic collaborations. We continue to believe that companies can benefit by collaborating with us when our proprietary delivery technologies can create new pharmaceutical and biologics product opportunities. We intend to continue to exploit the broad applicability of our delivery technologies for systemic applications of our validated technologies in collaborations with companies that will fund development and commercialization. We intend to continue to out-license technologies and product opportunities that we have already developed to a certain stage and that are outside of our core strategic focus. Collaborations and out-licensing may generate additional revenues while we progress towards the development and potential launch of our own proprietary products. | |
| • | Outsource manufacturing activities. We intend to to outsource the late stage clinical and commercial scale manufacturing of our products to conserve our capital for product development. We believe that the manufacturing processes for our AERx delivery systems are now sufficiently advanced that the required late stage clinical and commercial manufacturing capacity can be obtained from contract manufacturers. We are also utilizing contract manufacturers to make our liposomal formulations. With this approach, we seek manufacturers whose expertise should allow us to reduce risk and costs normally incurred if we were to build, operate and maintain large-scale production facilities ourselves. |
Product Candidates
Product candidates in development include both our own proprietary products and products under development with collaborators. They consist of approved drugs combined with our controlled inhalation deliveryand/or
3
Table of Contents
formulation technologies. The following table shows the disease indication and stage of development for each product candidate in our portfolio.
Product Candidate | Indication | Stage of Development | ||
Proprietary Programs Under Development | ||||
| ARD-3100 (Liposomal ciprofloxacin) | Cystic Fibrosis | Preclinical | ||
| ARD-1100 (Liposomal ciprofloxacin) | Inhalation Anthrax | Preclinical | ||
Collaborative Programs Under Development | ||||
| AERx iDMS (Insulin) | Type 1 and Type 2 Diabetes | Phase 3 | ||
| ARD-1300 (Hydroxychloroquine) | Asthma | Phase 2(1) | ||
| ARD-1500 (Liposomal treprostinil) | Pulmonary Arterial Hypertension | Preclinical |
| (1) | A Phase 2a clinical study did not meet pre-specified clinical endpoints. The program is currently under review by APT, and in our view is unlikely to continue. |
In addition to these programs, we are continually evaluating opportunities for product development where we can apply our expertise and intellectual property to produce better therapies and where we believe the investment could provide significant value to our shareholders.
Liposomal Ciprofloxacin
Ciprofloxacin is approved by the FDA as an anti-infective agent and is widely used for the treatment of a variety of bacterial infections. Today ciprofloxacin is delivered by oral or intravenous administration. We believe that delivering this potent antibiotic directly to the lung may improve its safety and efficacy in the treatment of pulmonary infections. We believe that our novel sustained release formulation of ciprofloxacin may be able to maintain therapeutic concentrations of the antibiotic within infected lung tissues, while reducing systemic exposure and the resulting side effects seen with currently marketed ciprofloxacin products. To achieve this sustained release, we employ liposomes, which are lipid-based nanoparticles dispersed in water that encapsulate the drug during storage and release the drug slowly upon contact with fluid covering the airways and the lung. In an animal experiment, ciprofloxacin delivered to the lung of mice appeared to be rapidly absorbed into the bloodstream, with no drug detectable four hours after administration. In contrast, the liposomal formulation of ciprofloxacin produced significantly higher levels of ciprofloxacin in the lung at all time points and was still detectable at 12 hours. We also believe that for certain respiratory disease indications it may be possible that a liposomal formulation enables better interaction of the drug with the disease target, leading to improved effectiveness over other therapies. We have at present two target indications with distinct delivery systems for this formulation that share much of the laboratory and production development efforts, as well as a common safety data base.
ARD-3100 — Liposomal Ciprofloxacin for the Treatment of Cystic Fibrosis
One of our liposomal ciprofloxacin programs is a proprietary program using our liposomal formulation of ciprofloxacin for the treatment and control of respiratory infections common to patients with cystic fibrosis, or CF. CF is a genetic disease that causes thick, sticky mucus to form in the lungs, pancreas and other organs. In the lungs, the mucus tends to block the airways, causing lung damage and making these patients highly susceptible to lung infections. According to the Cystic Fibrosis Foundation, CF affects roughly 30,000 children and adults in the United States and roughly 70,000 children and adults worldwide. According to the American Lung Association, the direct medical care costs for an individual with CF are currently estimated to be in excess of $40,000 per year.
The inhalation route affords direct administration of the drug to the infected part of the lung, maximizing the dose to the affected site and minimizing the wasteful exposure to the rest of the body where it could cause side effects. Therefore, treatment of CF-related lung infections by direct administration of antibiotics to the lung may improve both the safety and efficacy of treatment compared to systemic administration by other routes, as well as improving patient convenience as compared to injections. Oral and injectable forms of ciprofloxacin are approved
4
Table of Contents
for the treatment ofPseudomonas aeruginosa,a lung infection to which CF patients are vulnerable. Currently, there is only one inhalation antibiotic approved for the treatment of this infection. We believe that local lung delivery via inhalation of ciprofloxacin in a sustained release formulation could provide a convenient, effective and safe treatment of the debilitating and often life-threatening lung infections that afflict patients with CF.
Our liposomal ciprofloxacin CF program represents the first program in which we intend to retain full ownership and development rights. We believe we have the preclinical development, clinical and regulatory knowledge to advance this product through development in the most efficient manner. We intend to commercialize this program on our own.
Development
We have received orphan drug designations from the FDA for this product for the management of CF, and for the treatment of respiratory infections associated with non-CF bronchiectasis — a chronic pulmonary disease with symptoms similar to cystic fibrosis affecting over 100,000 patients in USA. As a designated orphan drug, liposomal ciprofloxacin is eligible for tax credits based upon its clinical development costs, as well as assistance from the FDA to coordinate study design. The designation also provides the opportunity to obtain market exclusivity for seven years from the date of New Drug Application, or NDA, approval.
We initiated preclinical studies for liposomal ciprofloxacin in 2006 and expect to initiate human clinical studies in the first half of 2007. We expect to incur expenses of approximately $20 million in 2007 to complete preclinical studies and fund early stage clinical trials and related manufacturing requirements for ARD-3100. In order to reach commercialization of ARD-3100, we estimate we will need to spend an additional $15 million to $20 million. In order to expedite anticipated time to market and increase market acceptance, we have elected to deliver ciprofloxacin via nebulizer, as most CF patients already own a nebulizer and are familiar with this method of drug delivery. We intend to examine the potential for delivery of ciprofloxacin via our AERx delivery system. We share the formulation and manufacturing development as well as the safety data developed for our inhalation anthrax program discussed below in the development of this CF opportunity. We also intend to explore the utility of liposomal ciprofloxacin for the treatment of other serious respiratory infections associated with other respiratory diseases.
ARD-1100 — Liposomal Ciprofloxacin for the Treatment of Inhalation Anthrax
The second of our liposomal ciprofloxacin programs is for the prevention and treatment of pulmonary anthrax infections. Anthrax spores are naturally occurring in soil throughout the world. Anthrax infections are most commonly acquired through skin contact with infected animals and animal products or, less frequently, by inhalation or ingestion of spores. With inhalation anthrax, once symptoms appear, fatality rates are high even with the initiation of antibiotic and supportive therapy. Further, a portion of the anthrax spores, once inhaled, may remain dormant in the lung for several months and germinate. Anthrax has been identified by the Centers for Disease Control as a likely potential agent of bioterrorism. In the fall of 2001, when anthrax-contaminated mail was deliberately sent through the United States Postal Service to government officials and members of the media, five people died and many more became sick. These attacks highlighted the concern that inhalation anthrax as a bioterror agent represents a real and current threat.
Ciprofloxacin has been approved by the FDA for use orally and via injection for the treatment of inhalation anthrax (post-exposure) since 2000. This ARD-1100 research and development program has been funded by Defence Research and Development Canada, or DRDC, a division of the Canadian Department of National Defence. We believe that this product candidate may potentially be able to deliver a long acting formulation of ciprofloxacin directly into the lung and could have fewer side effects and be more effective to prevent and treat inhalation anthrax than currently available therapies.
Development
We began our research into liposomal ciprofloxacin under a technology demonstration program funded by DRDC as part of their interest to develop products to counter bioterrorism. DRDC had already demonstrated the feasibility of inhaled liposomal ciprofloxacin for post-exposure prophylaxis ofFrancisella tularensis,a potential
5
Table of Contents
bioterrorism agent similar to anthrax. Mice were exposed to a lethal dose ofF. tularensisand then 24 hours later were exposed via inhalation to a single dose of free ciprofloxacin, liposomal ciprofloxacin or saline. All the mice in the control group and the free ciprofloxacin group were dead within 11 days post-infection; in contrast, all the mice in the liposomal ciprofloxacin group were alive 14 days post-infection. The same results were obtained when the mice received the single inhaled treatment as late as 48 or 72 hours post-infection. The DRDC has funded our development efforts to date and additional development of this program is dependent on negotiating for and obtaining continued funding from DRDC or on identifying other collaborators or sources of funding. We plan to use our preclinical and clinical safety data from our CF program to supplement the data needed to have this product candidate considered for approval for use in treating inhalation anthrax and possibly other inhaled life-threatening bioterrorism infections.
If we can obtain sufficient additional funding, we would anticipate developing this drug for approval under FDA regulations relating to the approval of new drugs or biologics for potentially fatal diseases where human studies cannot be conducted ethically or practically. Unlike most drugs, which require large, well controlled Phase 3 clinical trials in patients with the disease or condition being targeted, these regulations allow for a drug to be evaluated and approved by the FDA on the basis of demonstrated safety in humans combined with studies in animal models to show effectiveness.
AERx iDMS — Inhaled Insulin for the Treatment of Diabetes
AERx iDMS is being developed to control blood glucose levels in patients with diabetes. This product is currently in Phase 3 clinical trials, and our licensee, Novo Nordisk, is responsible for all remaining development, manufacturing and commercialization. We are entitled to receive royalties under our license agreement that will rise to an average of five percent or higher by the fifth year after commercialization, from any sales of this product as well as from future enhancements or generations of this technology. According to 2005 statistics from the American Diabetes Association, approximately 20.8 million Americans suffer from either Type 1 or Type 2 diabetes. Over 90% of these Americans have Type 2 diabetes, the prevalence of which is increasing dramatically due to lifestyle factors such as inappropriate diet and lack of physical activity. Patients with Type 1 diabetes do not have the ability to produce their own insulin and must administer insulin injections to survive. Patients with Type 2 diabetes are insulin resistant and unable to efficiently use the insulin that their bodies produce. While many Type 2 patients can initially maintain adequate control over blood glucose through diet, exercise and oral medications, most Type 2 patients progress within three years to where they cannot maintain adequate control over their glucose levels and insulin therapy is needed. However, given the less acute nature of Type 2 diabetes, many of these patients are reluctant to take insulin by injection despite the risks. Inadequate regulation of glucose levels in diabetes patients is associated with a variety of short and long-term effects, including blindness, kidney disease, heart disease, amputation resulting from chronic or extended periods of reduced blood circulation to body tissue and other circulatory disorders. The global market for diabetes therapies in 2005 was in excess of $18 billion, according to Business Insights. The majority of this amount was from sales of oral antidiabetics, while insulin and insulin analogues accounted for $7.3 billion, a 17% increase over the prior year. Sales of insulin and insulin analogues are forecast to grow to $9.8 billion in 2011. Type 2 patients consume the majority of insulin used in the United States. We believe that when patients are provided a non-invasive delivery alternative to injection, they will be more likely to self-administer insulin as often as needed to keep tight control over their blood-glucose levels.
We believe that AERx iDMS possesses features that will benefit diabetes patients and will provide an advantage over competitive pulmonary insulin products or can be used as a replacement for or adjunct to currently available therapies. Our patented breath control methods and technologies guide patients into the optimal breathing pattern for effective insulin deposition in and absorption from the lung. An optimal breathing pattern for insulin delivery depends on several elements: actuation of drug delivery at the early part of inspiration, control of inspiratory flow rate, and the state of inflation of the lung after the insulin is deposited, with the fully inflated lung providing the most desirable absorption profile. We believe a patient’s ability to breathe reproducibly will be required to assure adequate safety and efficacy of inhaled insulin for the treatment of Type 1 and Type 2 diabetes. Our system also allows patients to adjust dosage in single unit increments, which is key to proper glucose control in diabetes. AERx iDMS offers the ability for patients and physicians to monitor and review a patient’s dosing
6
Table of Contents
regimen. We believe the combination of breath control, high efficiency of delivery to the lung and single unit adjustable dosing in an inhalation device will make AERx iDMS a competitively attractive product.
Development
Over a decade ago, we initiated research and development into the inhalation delivery of insulin to meet a major unmet medical need in the treatment of Type 1 and Type 2 diabetes: a system that could provide similar levels of safety and efficacy as injected insulin but with the added benefit of a non-invasive route of delivery. We successfully completed a Phase 1 clinical study and filed an Investigational New Drug application, or IND, relating to our AERx iDMS program in 1998. After our initial work, we entered into a collaboration for our AERx iDMS in June 1998 with a world leader in the treatment of diabetes, Novo Nordisk. From 1998 to January 2005, we received an aggregate of $150.1 million from Novo Nordisk to fund development of the AERx delivery system for delivering insulin, production for preclinical and clinical testing and process development and scale up. In January 2005, we transferred the partially completed initial commercial production facility and associated personnel to Novo Nordisk for $55.3 million, and Novo Nordisk assumed responsibility for continuing production and bringing the facility up to its planned capacity of 750 million dosage forms per year.
AERx iDMS is currently undergoing testing in Phase 3 clinical trials, begun in May 2006 by Novo Nordisk. These trials follow significant prior clinical work that showed AERx iDMS to be comparable to injectable insulin in the overall management of Type 1 and Type 2 diabetes. Past clinical testing has shown:
| • | HbAlc levels, a key marker of blood glucose control, are statistically the same in patients using AERx iDMS and patients using subcutaneous insulin injections. | |
| • | The onset of action of inhaled insulin via AERx iDMS is not significantly different from subcutaneous injection of rapid-acting insulin, but significantly faster than subcutaneous injection of human regular insulin. | |
| • | The duration of action of inhaled insulin via AERx iDMS is not significantly different from subcutaneous injection of human regular insulin, but significantly longer than subcutaneous injection of rapid-acting insulin. | |
| • | Although small declines were seen on some pulmonary function parameters following12-24 months of dosing on AERx iDMS, these declines were not considered to be of clinical significance, and the findings are not expected to have an impact on overall pulmonary safety of the product. |
The Phase 3 clinical trials are expected to include a total of approximately 3,400 Type 1 and Type 2 diabetes patients. The trials include treatment comparisons with other antidiabetics. The longest trial is expected to last 27 months. Novo Nordisk announced in October 2006 that it expects the commercial launch of the product in 2010. As with any clinical program, there are many factors that could delay the launch or could result in AERx iDMS not receiving or maintaining regulatory approval.
In January 2005 and in July 2006, we announced restructurings of the AERx iDMS program. Under the new arrangements, Novo Nordisk is responsible for all further clinical, manufacturing and commercial development, while we and Novo Nordisk continue to cooperate and share in technology development, as well as intellectual property development and defense. We are entitled to receive royalty payments on any commercial sales.
ARD-1300 — Hydroxychloroquine for the Treatment of Asthma
The ARD-1300 program is investigating a novel aerosolized formulation of hydroxychloroquine, or HCQ, as a treatment for asthma under a collaboration with APT Pharmaceuticals, a privately held biotechnology company. Asthma is a common chronic disorder of the lungs characterized by airway inflammation, airway hyperresponsiveness or airway narrowing due to certain stimuli. Despite several treatment options, asthma remains a major medical problem associated with high morbidity and large economic costs to the society. According to the American Lung Association, asthma accounts for $11.5 billion in direct healthcare costs annually in the United States, of which the largest single expenditure, at $5 billion, was prescription drugs. Primary symptoms of asthma include coughing, wheezing, shortness of breath and tightness of the chest with symptoms varying in frequency and degree. According
7
Table of Contents
to Datamonitor, asthma affected 41.5 million people in developed countries in 2005, with 9.5 million of those affected being children. The highest prevalence of asthma occurs in the United States and the United Kingdom.
The most common treatment for the inflammatory condition causing chronic asthma is inhaled steroid therapy via metered dose inhalers, dry powder inhalers or nebulizers. While steroids are effective, they have side effects, including oral thrush, throat irritation, hoarseness and growth retardation in children, particularly at high doses and with prolonged use. HCQ is an FDA-approved drug that has been used for over 20 years in oral formulations as an alternative to steroid therapy for treatment for lupus and rheumatoid arthritis. Data from studies in which HCQ was orally administered to humans suggested that HCQ could be effective in the treatment of asthma. APT and Aradigm hypothesized that targeted delivery of HCQ to the airways may enhance the effectiveness of the treatment of asthma relative to systemic delivery of HCQ while reducing side effects by decreasing exposure of the drug to other parts of the body.
Development
APT has funded all activities in the development of this program to date. The ARD-1300 program advanced into Phase 2 clinical trials following positive preclinical testing and Phase 1 clinical results. The Phase 2a clinical trial, begun in March 2006, was a randomized, double-blind, placebo-controlled, multi-dose study in patients with asthma. The trial enrolled 100 patients with moderate-persistent asthma who were randomized to one of two treatments groups: either aerosolized placebo or aerosolized HCQ given once daily for 21 consecutive days. Both treatment groups were administered the drug via our AERx delivery system with efficacy, safety and tolerability assessments being performed throughout the study. The dosing of patients in the trial was completed in August 2006 and the results of the study were announced in November.
The results of the Phase 2a clinical study of inhaled HCQ as a treatment for patients with moderate-persistent asthma did not meet the pre-specified clinical efficacy endpoints. No serious adverse effects were noted or associated with the aerosolized HCQ or with the AERx system. While APT and Aradigm are currently analyzing the data from this study in order to determine whether additional studies of inhaled HCQ are warranted, it is unlikely that the current development path for HCQ for the treatment of asthma can be advanced without further research. This will delay development of this product candidate. Moreover, APT may choose not to conduct such research, in which case this collaborative program would be terminated. While APT has not yet indicated its intention, we believe it is unlikely that this program will continue.
ARD-1500 — Treprostinil for the Treatment of Pulmonary Arterial Hypertension
The ARD-1500 program is a part of a collaboration with United Therapeutics and is investigating a sustained-release liposomal formulation of a prostacyclin analogue for administration using our AERx delivery system for the treatment of pulmonary arterial hypertension, or PAH. PAH is a rare disease that results in the progressive narrowing of the arteries of the lungs, causing continuous high blood pressure in the pulmonary artery and eventually leading to heart failure. According to Decision Resources, in 2003, the more than 130,000 people worldwide affected by PAH purchased $600 million of PAH-related medical treatments and sales are expected to reach $1.2 billion per year by 2013.
Prostacyclin analogues are an important class of drugs used for the treatment of pulmonary arterial hypertension. However, the current methods of administration of these drugs are burdensome on patients. Treprostinil is marketed by United Therapeutics under the name Remodulin and is administered by intravenous or subcutaneous infusion. CoTherix, (acquired in 2007 by Actelion Pharmaceuticals Ltd.), markets in the United States another prostacyclin analogue, iloprost, under the name Ventavis that is administered six to nine times per day using a nebulizer, with each treatment lasting four to six minutes. We believe administration of liposomal treprostinil by inhalation using our AERx delivery system may be able to deliver an adequate dose for the treatment of PAH in a small number of breaths. We also believe that our sustained release formulation may lead to a reduction in the number of daily administrations that are needed to be effective when compared to existing therapies. We believe that our ARD-1500 product candidate potentially could offer a non-invasive, more direct and patient-friendly approach to treatment to replace or complement currently available treatments.
8
Table of Contents
Development
United Therapeutics has funded our activities in this program to date. We have completed initial preclinical (“in vitro”) testing of selected formulations. We are currently in negotiations with United Therapeutics on further development of the ARD-1500 program.
Additional Potential Product Applications
We have demonstrated in human clinical trials to date effective deposition and, where required, systemic absorption of a wide variety of drugs, including small molecules, peptides and proteins, using our AERx delivery system. We intend to identify additional pharmaceutical product opportunities that could potentially utilize our proprietary delivery systems for the pulmonary delivery of various drug types, including proteins, peptides, oligonucleotides, gene products and small molecules. We have demonstrated in the past our ability to successfully enter into collaborative arrangements for our programs, and we believe additional opportunities for collaborative arrangements exist outside of our core respiratory disease focus, for some of which we have data as well as intellectual property positions. The following are descriptions of two potential opportunities:
| • | Smoking Cessation. Based on internal work and work funded under grants from the National Institutes of Health, we are developing intellectual property in the area of smoking cessation. To date, we have two issued United States patents containing claims directed towards the use of titrated nicotine replacement therapy for smoking cessation. | |
| • | Pain Management System. Based on our internal work and a currently dormant collaboration with GlaxoSmithKline, we have developed a significant body of preclinical and Phase 1 clinical data on the use of inhaled morphine and fentanyl, and Phase 2 clinical data on inhaled morphine, with our proprietary AERx delivery system for the treatment of breakthrough pain in cancer and post-surgical patients. |
We are currently examining our previously conducted preclinical and clinical programs to identify molecules that may be suitable for further development consistent with our current business strategy. In most cases, we have previously demonstrated the feasibility of delivering these compounds via our proprietary AERx delivery system but we have not been able to continue development due to a variety of reasons, most notably the lack of funding from collaborators. If we identify any such programs during this review, we will consider continuing the development of such compounds on our own.
Pulmonary Drug Delivery Background
Pulmonary delivery describes the delivery of drugs by oral inhalation and is a common method of treatment of many respiratory diseases, including asthma, chronic bronchitis and CF. The current global market for inhalation products includes delivery through metered-dose inhalers, dry powder inhalers and nebulizers. The advantage of inhalation delivery for the diagnosis, prevention and treatment of lung disease is that the active agent is delivered in high concentration directly to the desired targets in the respiratory tract while keeping the body’s exposure to the rest of the drug, and resulting side effects, at minimum. Over the last two decades, there has also been increased interest in the use of the inhalation route for systemic delivery of drugs throughout the body, either for the purpose of rapid onset of action or to enable noninvasive delivery of drugs that are not orally bioavailable.
The efficacy, safety and efficient delivery of any inhaled drug depend on delivering the dose of the drug to the specified area of the respiratory tract. To achieve reproducible delivery of the dose, it is essential to control three factors:
| • | emitted dose; | |
| • | particle size distribution; and | |
| • | breathing maneuver. |
Breathing maneuver includes synchronization of the dose administration with the inhalation, inspiratory flow rate and the amount of air that the patient inhales at the time of dose — the “lung volume.”
9
Table of Contents
Lack of control of any of these factors may impair patient safety and therapeutic benefits. Further, the efficiency of delivery has economic implications, especially for drugs whose inherent production costs are high, such as biologics.
Traditional inhalation delivery systems, such as inhalers, have been designed and used primarily for delivery of drugs to the respiratory airways, not to the deep lung. While these systems have been useful in the treatment of certain diseases such as asthma, they generate a wide range of particle sizes, only a portion of which can reach the deep lung tissues. In order for an aerosol to be delivered to the deep lung where there is a large absorptive area suitable for effective systemic absorption, the medication needs to be delivered into the airstream early during inhalation. This is best achieved with systems that are breath-actuated,i.e., the dose delivery is automatically started at the beginning of inhalation. Further, the drug formulation must be transformed into very fine particles or droplets (typically one to three microns in diameter). In addition, the velocity of these particles must be low as they pass through the airways into the deep lung. The particle velocity is determined by the particle generator and the inspiratory flow rate of the patient. Large or fast-moving particles typically get deposited in the mouth and upper airways, where they may not be absorbed and could cause side effects. Most of the traditional drug inhalation delivery systems have difficulty in generating appropriate drug particle sizes or consistent emitted doses, and they also rely heavily on proper patient breathing technique to ensure adequate and reproducible lung delivery. To achieve appropriate drug particle sizes and consistent emitted doses, most traditional inhalation systems require the use of various additives such as powder carrier materials, detergents, lubricants, propellants, stabilizers and solvents, which may potentially cause toxicity or allergic reactions. It is also well documented that the typical patient frequently strays from proper inhalation technique after training and may not be able to maintain a consistent approach over even moderate periods of time. Since precise and reproducible dosing with medications is necessary to ensure safety and therapeutic efficacy, any variability in breathing technique among patients or from dose to dose may negatively impact the therapeutic benefits to the patient. We believe high efficiency and reproducibility of lung delivery will be required in order for inhalation to successfully replace certain injectable products.
The rate of absorption of drug molecules such as insulin from the lung has been shown to depend also on the lung volume following the deposition of the drug in the lung. In order to achieve safety and efficacy comparable to injections, this absorption step also needs to be highly reproducible. We therefore believe that an inhalation system that will coach the patient to breathe reproducibly to the same lung volume will be required to assure adequate safety and reproducibility of delivery of certain drugs delivered systemically via the lung.
The AERx Delivery Technology
The AERx delivery technology provides an efficient and reproducible means of targeting drugs to the diseased parts of the lung, or to the lung for systemic absorption, through a combination of fine mist generation technology and breath control mechanisms. Similar to nebulizers, the AERx delivery technology is capable of generating aerosols from simple liquid drug formulations, avoiding the need to develop complex dry powder or other formulations. However, in contrast to nebulizers, AERx is a hand-held unit that can deliver the required dosage typically in one or two breaths due to its enhanced efficiency, compared to nebulization treatments, which commonly last about 15 minutes. We believe the ability to make small micron- size droplets from a hand-held device that incorporates breath control will be the preferred method of delivery for many medications.
We have demonstrated in the laboratory and in many human clinical trials that our AERx delivery system enables pulmonary delivery of a wide range of pharmaceuticals in liquid formulations for local or systemic effects. Our proprietary technologies focus principally on delivering liquid medications through small particle aerosol generation and controlling patient inhalation technique for efficient and reproducible delivery of the aerosol drug to the deep lung. We have developed these proprietary technologies through an integrated approach that combines expertise in physics, engineering and pharmaceutical sciences. The key features of the AERx delivery system include the following:
| • | Liquid Formulation. Most drugs being considered by us for pulmonary delivery, especially biologics, are currently marketed in stable water formulations. The AERx delivery system takes advantage of existing liquid-drug formulations, reducing the time, cost and risk of formulation development compared to dry-powder-based technologies. The formulation technology of the AERx delivery system allows us to use |
10
Table of Contents
| conventional, sterile pharmaceutical manufacturing techniques. We believe that this approach will result in lower cost production methods than those used in dry powder systems because we are able to bypass much of the complex formulation and manufacturing processes required for those systems. Moreover, the liquid drug formulations used in the AERx delivery system are expected to have the same stability profile as the currently marketed versions of the same drugs. Because of the nature of liquid formulations, the additives we use are standard and therefore minimize safety concerns. |
| • | Efficient, Precise Aerosol Generation. Our proprietary technology produces the low-velocity, small-particle aerosols necessary for efficient deposition of a drug in the deep lung. The AERx delivery system aerosolizes liquid drug formulations from pre-packaged, single-use, disposable packets. Each disposable packet comprises a small blister package of the drug and an adjacent aerosolization nozzle. The AERx device compresses the packet to push the drug through the nozzle and thereby creates the aerosol. No propellants are required since mechanical pressure is used to generate the aerosol. Each packet is used only once to avoid plugging or wearing that could degenerate aerosol quality if reused. Through this technology, we believe we can achieve highly efficient and reproducible aerosols. The AERx device also has the ability to deliver a range of patient-selected doses, making it ideal for applications where the dose must be changed between uses or over time. | |
| • | Breath-Control Technology and Automated Breath-Controlled Delivery. Studies have shown that even well trained patients tend to develop improper inhalation technique over time, resulting in less effective therapy. The typical problems are associated with the inability to coordinate the start of inhalation with the activation of the dose delivery, inappropriate inspiratory flow rate and inhaled volume of air with the medication. The AERx delivery system employs breath control methods and technologies to guide the patient into the proper breathing maneuver. As a result, a consistent dose of medication is delivered each time the product is used. The characteristics of the breath control can be customized for different patient groups, such as young children or other patients with small lung volumes. |
The AERx delivery system offers additional patented features that we believe provide an advantage over competitive pulmonary products for certain important indications. For example, we believe our system for insulin delivery is unique in allowing the patients to adjust dosage in single insulin unit increments. This adjustable dosing feature may provide an advantage in certain other types of disease management where precise dosing adjustment is critical. The electronic version of the AERx delivery system can also be designed to incorporate the ability for a physician to monitor and download a patient’s dosing regimen, which we believe will aid in patient care and assist physicians in addressing potential issues of non-compliance. We have also developed a lockout feature for the AERx delivery system, which can be used to prevent use of the system by anyone other than the prescribed patient, and to prevent excessive dosing in any given time frame. These features of our AERx delivery system are protected by our strong intellectual property estate that includes patent claims directed toward the design, manufacture and testing of the AERx dosage forms and the various AERx pulmonary drug delivery systems.
We currently have two versions of the first-generation hand-held AERx delivery system in clinical use: AERx iDMS, which has been customized for the delivery of insulin, and the AERx Single Dose Platform, which is designed for general clinical use. We are also in the final stages of development of a next-generation system, the AERx Essence, which retains the key features of breath control and aerosol quality, but which is a much smaller, palm-sized device.
Formulation Technologies
We have a number of formulation technologies for drugs delivered by inhalation. We have proprietary knowledge and trade secrets relating to the formulation of drugs to achieve products with adequate stability and safety, and the manufacture and testing of inhaled drug formulations. We have been exploring the use of liposomal formulations of drugs that may be used for the prevention and treatment of respiratory diseases. Liposomes are lipid-based nanoparticles dispersed in water that encapsulate the drug during storage and release the drug slowly upon contact with fluid covering the airways and the lung. We are developing liposomal formulations particularly for those drugs that currently need to be dosed several times a day, or when the slow release of the drug is likely to improve the efficacy and safety profile. We believe a liposomal formulation will provide extended duration of
11
Table of Contents
protection and treatment against lung infection, greater convenience for the patient and reduced systemic levels of the drug. The formulation may also enable better interaction of the drug with the disease target, potentially leading to greater efficacy. We have applied this technology to ciprofloxacin and treprostinil. We are also examining other potential applications of this formulation technology for respiratory therapies.
Intellectual Property and Other Proprietary Rights
Our success will depend to a significant extent on our ability to obtain, expand and protect our intellectual property estate, enforce patents, maintain trade secret protection and operate without infringing the proprietary rights of other parties. As of February 28, 2007, we had 71 issued United States patents, with 20 additional United States patent applications pending. In addition, we had 72 issued foreign patents and additional 71 foreign patent applications pending. The bulk of our patents and patent applications contain claims directed toward our proprietary delivery technologies, including methods for aerosol generation, devices used to generate aerosols, breath control, compliance monitoring, certain pharmaceutical formulations, design of dosage forms and their manufacturing and testing methods. In addition, we have purchased three United States patents containing claims that are relevant to our inhalation technologies. The bulk of our patents, including fundamental patents directed toward our proprietary AERx delivery technology, expire between 2013 and 2023. For certain of our formulation technologies we have in-licensed some technology and will seek to supplement such intellectual property rights with complementary proprietary processes, methods and formulation technologies, including through patent applications and trade secret protection. Because patent positions can be highly uncertain and frequently involve complex legal and factual questions, the breadth of claims obtained in any application or the enforceability of our patents cannot be predicted.
In connection with the further restructuring of our collaboration with Novo Nordisk in July 2006, we transferred to Novo Nordisk the ownership of 23 issued United States patents and their correspondingnon-United States counterparts, if any. These transferred patents are especially important for the AERx iDMS program. We retain exclusive, royalty-free control of these patents outside the field of glucose control and will continue to be entitled to royalties under our license agreement that will rise to an average of five percent or higher by the fifth year after commercialization in respect of any inhaled insulin products marketed or licensed by Novo Nordisk.
In December 2004, as part of our research and development efforts funded by DRDC for the development of liposomal ciprofloxacin for the treatment of biological terrorism-related inhalation anthrax, we obtained worldwide exclusive rights to a patented liposomal formulation technology for the pulmonary delivery of ciprofloxacin, and may have the ability to expand the exclusive license to other fields.
We seek to protect our proprietary position by protecting inventions that we determine are or may be important to our business. We do this when we are able through the filing of patent applications with claims directed toward the devices, methods and technologies we develop. Our ability to compete effectively will depend to a significant extent on our ability and the ability of our collaborators to obtain and enforce patents and maintain trade secret protection over our proprietary technologies. The coverage claimed in a patent application typically is significantly reduced before a patent is issued, either in the United States or abroad. Consequently, any of our pending or future patent applications may not result in the issuance of patents or, to the extent patents have been issued or will be issued, these patents may be subjected to further proceedings limiting their scope and may in any event not contain claims broad enough to provide meaningful protection. Patents that are issued to us or our collaborators may not provide significant proprietary protection or competitive advantage, and may be circumvented or invalidated.
We also rely on our trade secrets and the know-how of our employees, officers, consultants and other service providers. Our policy is to require our officers, employees, consultants and advisors to execute proprietary information and invention assignment agreements upon commencement of their relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the relationship shall be kept confidential except in specified circumstances. These agreements also provide that all inventions developed by the individual on behalf of us shall be assigned to us and that the individual will cooperate with us in connection with securing patent protection for the invention if we wish to pursue such protection. These agreements may not provide meaningful protection for our inventions, trade secrets or other proprietary information in the event of unauthorized use or disclosure of such information.
12
Table of Contents
We also execute confidentiality agreements with outside collaborators and consultants. However, disputes may arise as to the ownership of proprietary rights to the extent that outside collaborators or consultants apply technological information developed independently by them or others to our projects, or apply our technology or proprietary information to other projects, and any such disputes may not be resolved in our favor. Even if resolved in our favor, such disputes could result in substantial expense and diversion of management attention.
In addition to protecting our own intellectual property rights, we must be able to develop products without infringing the proprietary rights of other parties. Because the markets in which we operate involve established competitors with significant patent portfolios, including patents relating to compositions of matter, methods of use and methods of delivery and products in those markets, it may be difficult for us to develop products without infringing the proprietary rights of others.
We would incur substantial costs if we are required to defend ourselves in suits, regardless of their merit. These legal actions could seek damages and seek to enjoin development, testing, manufacturing and marketing of the allegedly infringing product. In addition to potential liability for significant damages, we could be required to obtain a license to continue to manufacture or market the allegedly infringing product and any license required under any such patent may not be available to us on acceptable terms, if at all.
We may determine that litigation is necessary to enforce our proprietary rights against others. Such litigation could result in substantial expense and diversion of management attention, regardless of its outcome and any litigation may not be resolved in our favor.
Competition
We are in a highly competitive industry. We are in competition with pharmaceutical and biotechnology companies, hospitals, research organizations, individual scientists and nonprofit organizations engaged in the development of drugs and other therapies for the respiratory disease indications we are targeting. Our competitors may succeed, and many have already succeeded, in developing competing products, obtaining FDA approval for products or gaining patient and physician acceptance of products before us for the same markets and indications that we are targeting. Many of these companies, and large pharmaceutical companies in particular, have greater research and development, regulatory, manufacturing, marketing, financial and managerial resources and experience than we have and many of these companies may have products and product candidates that are in a more advanced stage of development than our product candidates. If we are not “first to market” for a particular indication, it may be more difficult for us or our collaborators to enter markets unless we can demonstrate our products are clearly superior to existing therapies.
Examples of competitive therapies include:
| • | ARD-3100. Currently marketed products include Tobi marketed by Novartis, Pulmozyme marketed by Genentech, Zithromax marketed by Pfizer and Cipro marketed by Bayer. CF products under development include inhaled aztreonam under development by Gilead, inhaled amikacin under development by Transave, Doripenam under development by Johnson & Johnson and inhaled ciprofloxacin under development by Bayer. | |
| • | ARD-1100. Current anthrax treatment products include various oral generic and proprietary antibiotics, such as Cipro marketed by Bayer. | |
| • | AERx iDMS. Currently marketed diabetes products include insulin products marketed by companies such as Novo Nordisk, Eli Lilly and sanofi-aventis. Pfizer, in collaboration with Nektar Therapeutics, has recently obtained FDA approval for Exubera, an inhaled form of insulin. Eli Lilly, in collaboration with Alkermes Pharmaceuticals, and Mannkind Corporation have announced they have inhaled insulin products in development. | |
| • | ARD-1300. Currently marketed products include Advair marketed by GlaxoSmithKline, Xolair marketed by Novartis in collaboration with Genentech, Singulair marketed by Merck, Asmanex marketed by Schering-Plough and Pulmicort marketed by AstraZeneca International. Similar asthma products under |
13
Table of Contents
| development include Symbicort under development by AstraZeneca and Alvesco under development by sanofi-aventis. |
| • | ARD-1500. Currently marketed products include intravenous delivery and subcutaneous infusion of prostacyclins, such as Remodulin marketed by United Therapeutics, and inhaled prostacyclins, such as Ventavis, marketed by Schering AG and CoTherix, (acquired by Actelion Pharmaceuticals Ltd. In 2007). |
Many of these products have substantial current sales and long histories of effective and safe use. In addition, we believe there are a number of additional drug candidates in various stages of development that, if approved, would compete with any future products we may develop. Moreover, one or more of our competitors that have developed or are developing pulmonary drug delivery technologies, such as Alkermes, Nektar, Mannkind or Alexza Pharmaceuticals, or other competitors with alternative drug delivery methods, may negatively impact our potential competitive position.
We believe that our respiratory expertise and pulmonary delivery and formulation technologies provide us with an important competitive advantage for our potential products. We intend to compete by developing products that are safer, more efficacious, more convenient, less costly, earlier to market, marketed with smaller sales forces or cheaper to develop than existing products, or any combination of the foregoing.
Government Regulation
United States
The research, development, testing, manufacturing, labeling, advertising, promotion, distribution, marketing, and export, among other things, of any products we develop are subject to extensive regulation by governmental authorities in the United States and other countries. The FDA regulates drugs in the United States under the FDCA and implementing regulations thereunder.
If we or our product development collaborators fail to comply with the FDCA or FDA regulations, we, our collaborators, and our products could be subject to regulatory actions. These may include delay in approval or refusal by the FDA to approve pending applications, injunctions ordering us to stop sale of any products we develop, seizure of our products, warning letters, imposition of civil penalties or other monetary payments, criminal prosecution, and recall of our products. Any such events would harm our reputation and our results of operations.
Before one of our drugs may be marketed in the United States, it must be approved by the FDA. None of our product candidates has received such approval. We believe that our products currently in development will be regulated by FDA as drugs.
The steps required before a drug may be approved for marketing in the United States generally include:
| • | preclinical laboratory and animal tests, and formulation studies; | |
| • | the submission to the FDA of an Investigational New Drug application, or IND, for human clinical testing that must become effective before human clinical trials may begin; | |
| • | adequate and well controlled human clinical trials to establish the safety and efficacy of the product candidate for each indication for which approval is sought; | |
| • | the submission to the FDA of a New Drug Application, or NDA, and FDA’s acceptance of the NDA for filing; | |
| • | satisfactory completion of an FDA inspection of the manufacturing facilities at which the product is to be produced to assess compliance with the FDA’s Good Manufacturing Practices, or GMP; and | |
| • | FDA review and approval of the NDA. |
Preclinical Testing
The testing and approval process requires very substantial time, effort, and financial resources, and the receipt and timing of approval, if any, is highly uncertain. Preclinical tests include laboratory evaluations of product chemistry, toxicity, and formulation, as well as animal studies. The results of the preclinical studies, together with
14
Table of Contents
manufacturing information and analytical data, are submitted to the FDA as part of the IND. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the proposed clinical trials as outlined in the IND prior to that time. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. Submission of an IND may not result in FDA authorization to commence clinical trials. Once an IND is in effect, the protocol for each clinical trial to be conducted under the IND must be submitted to the FDA, which may or may not allow the trial to proceed.
Clinical Trials
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified investigators and healthcare personnel. Clinical trials are conducted under protocols detailing, for example, the parameters to be used in monitoring patient safety and the safety and effectiveness criteria, or end points, to be evaluated. Clinical trials are typically conducted in three defined phases, but the phases may overlap or be combined. Each trial must be reviewed and approved by an independent institutional review board overseeing the institution conducting the trial before it can begin.
These phases generally include the following:
| • | Phase 1. Phase 1 clinical trials usually involve the initial introduction of the drug into human subjects, frequently healthy volunteers. In Phase 1, the drug is usually evaluated for safety, including adverse effects, dosage tolerance, absorption, distribution, metabolism, excretion and pharmacodynamics. | |
| • | Phase 2. Phase 2 usually involves studies in a limited patient population with the disease or condition for which the drug is being developed to (1) preliminarily evaluate the efficacy of the drug for specific, targeted indications; (2) determine dosage tolerance and appropriate dosage; and (3) identify possible adverse effects and safety risks. | |
| • | Phase 3. If a drug is found to be potentially effective and to have an acceptable safety profile in Phase 2 studies, the clinical trial program will be expanded, usually to further evaluate clinical efficacy and safety by administering the drug in its final form to an expanded patient population at geographically dispersed clinical trial sites. Phase 3 studies usually include several hundred to several thousand patients. |
Phase 1, Phase 2, or Phase 3 clinical trials may not be completed successfully within any specified period of time, if at all. Further, we, our product development collaborators, or the FDA may suspend clinical trials at any time on various grounds, including a finding that the patients are being exposed to an unacceptable health risk. For example, in 2004, Novo Nordisk, our collaborator in the AERx iDMS program, amended the protocols of a Phase 3 clinical program, which resulted in a significant delay in the development of AERx iDMS.
Assuming successful completion of the required clinical testing, the results of preclinical studies and clinical trials, together with detailed information on the manufacture and composition of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. Before approving an application, the FDA usually will inspect the facility or facilities at which the product is manufactured, and will not approve the product unless continuing GMP compliance is satisfactory. If the FDA determines the NDA is not acceptable, the FDA may outline the deficiencies in the NDA and often will request additional information or additional clinical trials. Notwithstanding the submission of any requested additional testing or information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If regulatory approval of a product is granted, such approval will usually entail limitations on the indicated uses for which such product may be marketed. Once approved, the FDA may withdraw the product approval if compliance with pre and post-marketing regulatory requirements and conditions of approvals are not maintained, if GMP compliance is not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies, referred to as Phase 4 studies, to monitor the effect of approved products and may limit further marketing of the product based on the results of these post-marketing studies.
After approval, certain changes to the approved product, such as adding new indications, certain manufacturing changes, or additional labeling claims are subject to further FDA review and approval. Post-approval
15
Table of Contents
marketing of products can lead to new findings about the safety or efficacy of the products. This information can lead to a product sponsor making, or the FDA requiring, changes in the labeling of the product or even the withdrawal of the product from the market.
Section 505(b)(2) Applications
Some of our product candidates may be eligible for submission of applications for approval under the FDA’s Section 505(b)(2) approval process, which requires less information than the NDAs described above. Section 505(b)(2) applications may be submitted for drug products that represent a modification (e.g., a new indication or new dosage form) of an eligible approved drug and for which investigations other than bioavailability or bioequivalence studies are essential to the drug’s approval. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the listed drug, scientific literature, and information obtained by the 505(b)(2) applicant needed to support the modification of the listed drug. For this reason, preparing Section 505(b)(2) applications is generally less costly and time-consuming than preparing an NDA based entirely on new data and information from a full set of clinical trials. The law governing Section 505(b)(2) or FDA’s current policies may change in such a way as to adversely affect our applications for approval that seek to utilize the Section 505(b)(2) approach. Such changes could result in additional costs associated with additional studies or clinical trials and delays.
The FDCA provides that reviewsand/or approvals of applications submitted under Section 505(b)(2) will be delayed in various circumstances. For example, the holder of the NDA for the listed drug may be entitled to a period of market exclusivity, during which the FDA will not approve, and may not even review a Section 505(b)(2) application from other sponsors. If the listed drug is claimed by patent that the NDA holder has listed with the FDA, the Section 505(b)(2) applicant must submit a patent certification. If the 505(b)(2) applicant certifies that the patent is invalid, unenforceable, or not infringed by the product that is the subject of the Section 505(b)(2), and the 505(b)(2) applicant is sued within 45 days of its notice to the entity that holds the approval for the listed drug and the patent holder, the FDA will not approve the Section 505(b)(2) application until the earlier of a court decision favorable to the Section 505(b)(2) applicant or the expiration of 30 months. The regulations governing marketing exclusivity and patent protection are complex, and it is often unclear how they will be applied in particular circumstances.
In addition, both before and after approval is sought, we and our collaborators are required to comply with a number of FDA requirements. For example, we are required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain limitations and other requirements concerning advertising and promotion for our products. Also, quality control and manufacturing procedures must continue to conform to continuing GMP after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with continuing GMP. In addition, discovery of problems such as safety problems may result in changes in labeling or restrictions on a product manufacturer or NDA holder, including removal of the product from the market.
Orphan Drug Designation
The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition” which generally is a disease or condition that affects fewer than 200,000 individuals in the United States. A sponsor may request orphan drug designation of a previously unapproved drug, or of a new indication for an already marketed drug. Orphan drug designation must be requested before an NDA is submitted. If the FDA grants orphan drug designation, which it may not, the identity of the therapeutic agent and its potential orphan status are publicly disclosed by the FDA. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process. If a drug which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the drug is entitled to orphan exclusivity, meaning that the FDA may not approve any other applications to market the same drug for the same indication for a period of seven years, unless the subsequent application is able to demonstrate clinical superiority in efficacy or safety. Orphan drug designation does not prevent competitors from developing or marketing different drugs for that indication, or the same drug for other indications.
16
Table of Contents
We have obtained orphan drug designation from the FDA for inhaled liposomal ciprofloxacin for the management of cystic fibrosis. We may seek orphan drug designation for other eligible product candidates we develop. However, our liposomal ciprofloxacin may not receive orphan drug marketing exclusivity. Also, it is possible that our competitors could obtain approval, and attendant orphan drug designation or exclusivity, for products that would preclude us from marketing our liposomal ciprofloxacin for this indication for some time.
International Regulation
We are also subject to foreign regulatory requirements governing clinical trials, product manufacturing, marketing and product sales. Our ability to market and sell our products in countries outside the United States will depend upon receiving marketing authorization(s) from appropriate regulatory authorities. We will only be permitted to commercialize our products in a foreign country if the appropriate regulatory authority is satisfied that we have presented adequate evidence of safety, quality and efficacy. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing of the product in those countries. Approval of a product by the FDA does not assure approval by foreign regulators. Regulatory requirements, and the approval process, vary widely from country to country, and the time, cost and data needed to secure approval may be longer or shorter than that required for FDA approval. The regulatory approval and oversight process in other countries includes all of the risks associated with the FDA process described above.
Scientific Advisory Board
We have assembled a scientific advisory board comprised of scientific and product development advisors who provide expertise, on a consulting basis from time to time, in the areas of respiratory diseases, allergy and immunology, hormonal and metabolic disorders, pharmaceutical development and drug delivery, including pulmonary delivery, but are employed elsewhere on a full-time basis. As a result, they can only spend a limited amount of time on our affairs. We access scientific and medical experts in academia, as needed, to support our scientific advisory board. The scientific advisory board assists us on issues related to potential product applications, product development and clinical testing. Its members, and their affiliations and areas of expertise, include:
Name | Affiliation | Area of Expertise | ||
| Peter R. Byron, Ph.D. | Medical College of Virginia, Virginia Commonwealth University | Aerosol Science/Pharmaceutics | ||
| Peter S. Creticos, M.D. | The Johns Hopkins University School of Medicine | Allergy/Immunology/Asthma | ||
| Stephen J. Farr, Ph.D. | Zogenix, Inc. | Pulmonary Delivery/ Pharmaceutics | ||
| Michael Powell, Ph.D. | Sofinnova Ventures | Drug Development | ||
| Robert E. Ratner, M.D. | MedStar Research Institute | Endocrinology | ||
| Adam Wanner, M.D. | University of Miami | Chronic Obstructive Pulmonary Disease (COPD) | ||
| Martin Wasserman, Ph.D. | Roche, AtheroGenics (retired) | Asthma |
In addition to our scientific advisory board, for certain indications and programs we assemble groups of experts to assist us on issues specific to such indications and programs.
Employees
As of December 31, 2006, we had 54 employees, 15 of whom have advanced degrees. Of these, 36 are involved in research and development, product development and commercialization; and 18 are involved in business development, finance and administration. Our employees are not represented by any collective bargaining agreement.
17
Table of Contents
Corporate History and Website Information
We were incorporated in California in 1991. Our principal executive offices are located at 3929 Point Eden Way, Hayward, California 94545, and our main telephone number is(510) 265-9000. Investors can obtain access to this annual report onForm 10-K, our quarterly reports onForm 10-Q, our current reports onForm 8-K and all amendments to these reports, free of charge, on our website at http://www.aradigm.com as soon as reasonably practicable after such filings are electronically filed with the Securities and Exchange Commission (“SEC”). The public may read and copy any material we file with the SEC at the SEC’s Public Reference Room at 450 Fifth Street, N.W., Washington, D.C., 20549. The public may obtain information on the operations of the Public Reference Room by calling the SEC at1-800-SEC-0330. The SEC maintains an Internet site, http://www.sec.gov that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
We have adopted a code of ethics, which is part of our Code of Business Conduct and Ethics that applies to all of our employees, including our principal executive officer, our principal financial officer and our principal accounting officer. This code of ethics is posted on our website.
Executive Officers and Directors
Our directors and executive officers and their ages as of February 28, 2007 are as follows:
Name | Age | Position | ||||
| Igor Gonda, Ph.D. | 59 | President, Chief Executive Officer and Director | ||||
| Thomas C. Chesterman | 47 | Senior Vice President and Chief Financial Officer | ||||
| Babatunde A. Otulana, M.D. | 50 | Senior Vice President, Development and Chief Medical Officer | ||||
| Frank H. Barker(1)(3) | 76 | Director | ||||
| Stephen O. Jaeger(1)(2) | 62 | Director | ||||
| John M. Siebert, Ph.D.(2)(3) | 66 | Director | ||||
| Virgil D. Thompson(1)(2)(3) | 67 | Chairman of the Board | ||||
| (1) | Member of the Audit Committee. | |
| (2) | Member of the Compensation Committee. | |
| (3) | Member of the Nominating and Corporate Governance Committee. |
Igor Gonda, Ph.D. has served as our President and Chief Executive Officer since August 2006, and as a director since September 2001. From December 2001 to August 2006, Dr. Gonda was the Chief Executive Officer and Managing Director of Acrux Limited, a publicly traded specialty pharmaceutical company located in Melbourne, Australia. From July 2001 to December 2001, Dr. Gonda was our Chief Scientific Officer and, from October 1995 to July 2001, was our Vice President, Research and Development. From February 1992 to September 1995, Dr. Gonda was a Senior Scientist and Group Leader at Genentech, Inc. His key responsibilities at Genentech were the development of the inhalation delivery of rhDNase (Pulmozyme) for the treatment of cystic fibrosis and non-parenteral methods of delivery of biologics. Prior to that, Dr. Gonda held academic positions at the University of Aston in Birmingham, United Kingdom, and the University of Sydney, Australia. Dr. Gonda holds a B.Sc. in Chemistry and a Ph.D. in Physical Chemistry from Leeds University, United Kingdom. Dr. Gonda was the Chairman of our Scientific Advisory Board until August 2006.
Thomas C. Chestermanhas served as our Senior Vice President and Chief Financial Officer since August 2002. From March 1996 to December 2001, Mr. Chesterman was Vice President and Chief Financial Officer at Bio-Rad Laboratories, Inc., a life-science research products and clinical diagnostics company. From 1993 to 1996, Mr. Chesterman was Vice President of Strategy and Chief Financial Officer of Europolitan AB, a telecommunications company. Mr. Chesterman holds a B.A. from Harvard University and an M.B.A. in Finance and Accounting from the University of California at Davis.
Babatunde A. Otulana, M.D. has served as our Senior Vice President, Development, since August 2006. Prior to that, Dr. Otulana served as our Vice President, Clinical and Regulatory Affairs since October 1997. From 1991 to
18
Table of Contents
September 1997, Dr. Otulana was a Medical Reviewer in the Division of Pulmonary Drug Products at the Center for Drug Evaluation and Research of the United States Food and Drug Administration. Dr. Otulana currently serves as an Associate Clinical Professor in Pulmonary Medicine at the School of Medicine, University of California at Davis. Dr. Otulana holds an M.D. from the University of Ibadan, Nigeria, and completed Pulmonary Fellowships at Papworth Hospital, University of Cambridge, United Kingdom, and at Howard University Hospital, Washington, D.C.
Frank H. Barkerhas been a director since May 1999. From January 1980 to January 1994, Mr. Barker served as a company group chairman of Johnson & Johnson, Inc., a diversified health care company, and was Corporate Vice President from January 1989 to January 1996. Mr. Barker retired from Johnson & Johnson, Inc. in January 1996. Mr. Barker holds a B.A. in Business Administration from Rollins College, Winter Park, Florida. Mr. Barker is a director of Jenex Corporation, a Canadian medical devices company.
Stephen O. Jaegerhas been a director since March 2004. Mr. Jaeger was Chairman, President and Chief Executive Officer of eBT International, Inc. a privately held software products and services company, from 1999 to December 2005. Prior to joining eBT, Mr. Jaeger was the Executive Vice President and Chief Financial Officer of Clinical Communications Group, Inc., a provider of educational marketing services to the pharmaceutical and biotech industries, from 1997 to 1998. From 1995 to 1997, Mr. Jaeger served as Vice President, Chief Financial Officer and Treasurer of Applera Corp., formerly known as Perkin Elmer Corporation, an analytical instrument and systems company with a focus on life science and genetic discovery. Prior to 1995, Mr. Jaeger was Chief Financial Officer and a director of Houghton Mifflin Company and held various financial positions with BP, Weeks Petroleum Limited and Ernst & Young LLP. Mr. Jaeger holds a B.A. in Psychology from Fairfield University and an M.B.A. in Accounting from Rutgers University and is a Certified Public Accountant. Mr. Jaeger is the Chairman of the Board of Savient Pharmaceuticals Inc., a publicly traded specialty pharmaceutical company, and a director of Arlington Tankers, Ltd., a publicly traded shipping company. Mr. Jaeger is the Chairman of and the designated “audit committee financial expert” on our, Savient Pharmaceuticals’ and Arlington Tankers’ audit committees.
John M. Siebert Ph.D. has been a director since November 2006. Since May 2003, Dr. Siebert has been the Chairman and Chief Executive Officer of CyDex, Inc., a privately held specialty pharmaceutical company. From September 1995 to April 2003, he was President and Chief Executive Officer of CIMA Labs Inc., a publicly traded drug delivery company, and from July 1995 to September 1995 he was President and Chief Operating Officer of CIMA Labs. From 1992 to 1995, Dr. Siebert was Vice President, Technical Affairs at Dey Laboratories, Inc., a privately held pharmaceutical company. From 1988 to 1992, he worked at Bayer Corporation. Prior to that, Dr. Siebert was employed by E.R. Squibb & Sons, Inc., G.D. Searle & Co. and The Procter & Gamble Company. Dr Siebert holds a B.S. in Chemistry from Illinois Benedictine University, an M.S. in Organic Chemistry from Wichita State University and a Ph.D. in Organic Chemistry from the University of Missouri.
Virgil D. Thompsonhas been a director since June 1995 and has been Chairman of the Board since January 2005. Since November 2002, Mr. Thompson has been President and Chief Executive Officer of Angstrom Pharmaceuticals, Inc., a privately held pharmaceutical company. From September 2000 to November 2002, Mr. Thompson was President, Chief Executive Officer and a director of Chimeric Therapies, Inc., a privately held biotechnology company. From May 1999 until September 2000, Mr. Thompson was the President, Chief Operating Officer and a director of Savient Pharmaceuticals, a publicly traded specialty pharmaceutical company. From January 1996 to April 1999, Mr. Thompson was the President and Chief Executive Officer and a director of Cytel Corporation, a publicly traded biopharmaceutical company that was subsequently acquired by IDM Pharma, Inc. From 1994 to 1996, Mr. Thompson was President and Chief Executive Officer of Cibus Pharmaceuticals, Inc., a privately held drug delivery device company. From 1991 to 1993, Mr. Thompson was President of Syntex Laboratories, Inc., a U.S. subsidiary of Syntex Corporation, a publicly traded pharmaceutical company. Mr. Thompson holds a B.S. in Pharmacy from Kansas University and a J.D. from The George Washington University Law School. Mr. Thompson is a director of Questcor Pharmaceuticals, Inc., a publicly traded pharmaceutical company, and Savient Pharmaceuticals.
19
Table of Contents
| Item 1A. | Risk Factors |
Except for historical information contained herein, the discussion in this Report onForm 10-K contains forward-looking statements, including, without limitation, statements regarding timing and results of clinical trials, the establishment of corporate partnering arrangements, the anticipated commercial introduction of our products and the timing of our cash requirements. These forward-looking statements involve certain risks and uncertainties that could cause actual results to differ materially from those in such forward-looking statements. Potential risks and uncertainties include, without limitation, those mentioned in this report and in particular the factors described below.
Risks Related to Our Business
We are an early-stage company.
You must evaluate us in light of the uncertainties and complexities present in an early-stage company. All of our potential products are in an early stage of research or development. Our potential drug delivery products require extensive research, development and pre-clinical and clinical testing. Our potential products also may involve lengthy regulatory reviews before they can be sold. Because none of our product candidates has yet received approval by the FDA, we cannot assure you that our research and development efforts will be successful, any of our potential products will be proven safe and effective or regulatory clearance or approval to sell any of our potential products will be obtained. We cannot assure you that any of our potential products can be manufactured in commercial quantities or at an acceptable cost or marketed successfully. We may abandon the development of some or all of our product candidates at any time and without prior notice. We must incur substantial up-front expenses to develop and commercialize products and failure to achieve commercial feasibility, demonstrate safety, achieve clinical efficacy, obtain regulatory approval or successfully manufacture and market products will negatively impact our business.
We recently changed our product development strategy, and if we do not successfully implement this new strategy our business and reputation will be damaged.
Since our inception in 1991 we have focused on developing drug delivery technologies. In May, 2006, we transitioned our business focus from the development of delivery technologies to the application of our pulmonary drug delivery technologies and expertise to the development of novel drug products to treat respiratory diseases. As part of this transition we have implemented workforce reductions in an effort to reduce our expenses and improve our cash flows. We have not yet implemented or are only in the early stages of implementing various aspects of our new strategy, and we may not be successful in implementing our new strategy. Even if we are able to implement the various aspects of our new strategy, it may not be successful.
We will need additional capital, and we may not be able to obtain it.
Our operations to date have consumed substantial amounts of cash and have generated no product revenues. While our refocused development strategy will reduce capital expenditures, we expect negative operating cash flows to continue for at least the foreseeable future. Even though we do not plan to engage in drug discovery, we will nevertheless need to commit substantial funds to develop our product candidates and we may not be able to obtain sufficient funds on acceptable terms or at all. Our future capital requirements will depend on many factors, including:
| • | our progress in the application of our delivery and formulation technologies, which may require further refinement of these technologies; | |
| • | the number of product development programs we pursue and the pace of each program; | |
| • | our progress with formulation development; | |
| • | the scope, rate of progress, results and costs of preclinical testing and clinical trials; | |
| • | the time and costs associated with seeking regulatory approvals; |
20
Table of Contents
| • | our ability to outsource the manufacture of our product candidates and the costs of doing so; | |
| • | the time and costs associated with establishing in-house resources to market and sell certain of our products; | |
| • | our ability to establish and maintain collaborative arrangements with others and the terms of those arrangements; | |
| • | the costs of preparing, filing, prosecuting, maintaining and enforcing patent claims, and | |
| • | our need to acquire licenses or other rights for our product candidates. |
Since inception, we have financed our operations primarily through private placements and public offerings of our capital stock, proceeds from equipment lease financings, contract research funding and interest earned on investments. We believe that our existing cash and cash equivalent balances at December 31, 2006, interest earned on our investments and net proceeds from our public offering that closed on January 30, 2007, should be sufficient to meet our needs for at least the next 24 months. We will need to obtain substantial additional funds before we would be able to bring any of our product candidates to market. Our estimates of future capital use are uncertain, and changing circumstances, including those related to implementation of our new development strategy or further changes to our development strategy, could cause us to consume capital significantly faster than currently expected, and our expected sources of funding may not be sufficient. If adequate funds are not available, we will be required to delay, reduce the scope of, or eliminate one or more of our product development programs and reduce personnel-related costs, or to obtain funds through arrangements with collaborators or other sources that may require us to relinquish rights to certain of our technologies or products that we would not otherwise relinquish. If we are able to obtain funds through the issuance of debt securities or borrowing, the terms may restrict our operations. If we are able to obtain funds through the issuance of equity securities, your interest will be diluted and our stock price may drop as a result.
We have a history of losses, we expect to incur losses for at least the foreseeable future, and we may never attain or maintain profitability.
We have never been profitable and have incurred significant losses in each year since our inception. As of December 31, 2006, we have an accumulated deficit of $287.9 million. We have not had any product sales and do not anticipate receiving any revenues from product sales for at least the next few years, if ever. While our recent shift in development strategy may result in reduced capital expenditures, we expect to continue to incur substantial losses over at least the next several years as we:
| • | expand drug product development efforts; | |
| • | conduct preclinical testing and clinical trials; | |
| • | pursue additional applications for our existing delivery technologies; | |
| • | outsource the commercial-scale production of our products; and | |
| • | establish a sales and marketing force to commercialize certain of our proprietary products if these products obtain regulatory approval. |
To achieve and sustain profitability, we must, alone or with others, successfully develop, obtain regulatory approval for, manufacture, market and sell our products. We expect to incur substantial expenses in our efforts to develop and commercialize products and we may never generate sufficient product or contract research revenues to become profitable or to sustain profitability.
Our dependence on collaborators may delay or prevent the progress of certain of our programs.
Our commercialization strategy for certain of our product candidates depends on our ability to enter into agreements with collaborators to obtain assistance and funding for the development and potential commercialization of our product candidates. Collaborations may involve greater uncertainty for us, as we have less control over certain aspects of our collaborative programs than we do over our proprietary development and commercialization programs. We may determine that continuing a collaboration under the terms provided is not in our best
21
Table of Contents
interest, and we may terminate the collaboration. Our existing collaborators could delay or terminate their agreements, and our products subject to collaborative arrangements may never be successfully commercialized. For example, Novo Nordisk has control over and responsibility for development and commercialization of AERx iDMS. The development and commercialization of AERx iDMS could be delayed further or terminated if Novo Nordisk fails to conduct these activities in a timely manner or at all. In 2004, Novo Nordisk amended the protocols of a Phase 3 clinical program, which resulted in a significant delay of the development of the product. If, due to delays or otherwise, we do not receive development funds or achieve milestones set forth in the agreements governing our collaborations, or if any of our collaborators breach or terminate their collaborative agreements or do not devote sufficient resources or priority to our programs, our business prospects and potential to receive revenues would be hurt.
Further, our existing or future collaborators may pursue alternative technologies or develop alternative products either on their own or in collaboration with others, including our competitors, and the priorities or focus of our collaborators may shift such that our programs receive less attention or resources than we would like. Any such actions by our collaborators may adversely affect our business prospects and ability to earn revenues. In addition, we could have disputes with our existing or future collaborators regarding, for example, the interpretation of terms in our agreements. Any such disagreements could lead to delays in the development or commercialization of any potential products or could result in time-consuming and expensive litigation or arbitration, which may not be resolved in our favor.
Even with respect to certain other programs that we intend to commercialize ourselves, we may enter into agreements with collaborators to share in the burden of conducting clinical trials, manufacturing and marketing our product candidates or products. In addition, our ability to apply our proprietary technologies to develop proprietary drugs will depend on our ability to establish and maintain licensing arrangements or other collaborative arrangements with the holders of proprietary rights to such drugs. We may not be able to establish such arrangements on favorable terms or at all, and our existing or future collaborative arrangements may not be successful.
The results of later stage clinical trials of our product candidates may not be as favorable as earlier trials and that could result in additional costs and delay or prevent commercialization of our products.
Although we believe the limited and preliminary data we have regarding our potential products is encouraging, the results of initial preclinical testing and clinical trials do not necessarily predict the results that we will get from subsequent or more extensive preclinical testing and clinical trials. Clinical trials of our product candidates may not demonstrate that they are safe and effective to the extent necessary to obtain regulatory approvals. Many companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after receiving promising results in earlier trials. If we cannot adequately demonstrate through the clinical trial process that a therapeutic product we are developing is safe and effective, regulatory approval of that product would be delayed or prevented, which would impair our reputation, increase our costs and prevent us from earning revenues.
If our clinical trials are delayed because of patient enrollment or other problems, we would incur additional cost and postpone the potential receipt of revenues.
Before we or our collaborators can file for regulatory approval for the commercial sale of our potential products, the FDA will require extensive preclinical safety testing and clinical trials to demonstrate their safety and efficacy. Completing clinical trials in a timely manner depends on, among other factors, the timely enrollment of patients. Our collaborators’ and our ability to recruit patients depends on a number of factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the study and the existence of competing clinical trials. Delays in planned patient enrollment in our current or future clinical trials may result in increased costs, program delays or both, and the loss of potential revenues.
22
Table of Contents
We are subject to extensive regulation, including the requirement of approval before any of our product candidates can be marketed. We may not obtain regulatory approval for our product candidates on a timely basis, or at all.
We, our collaborators and our products are subject to extensive and rigorous regulation by the federal government, principally the FDA, and by state and local government agencies. Both before and after regulatory approval, the development, testing, manufacture, quality control, labeling, storage, approval, advertising, promotion, sale, distribution and export of our potential products are subject to regulation. Pharmaceutical products that are marketed abroad are also subject to regulation by foreign governments. Our products cannot be marketed in the United States without FDA approval. The process for obtaining FDA approval for drug products is generally lengthy, expensive and uncertain. To date, we have not sought or received approval from the FDA or any corresponding foreign authority for any of our product candidates.
Even though we intend to apply for approval of most of our products in the United States under Section 505(b)(2) of the United States Food, Drug and Cosmetic Act, which applies to reformulations of approved drugs and that may require smaller and shorter safety and efficacy testing than that for entirely new drugs, the approval process will still be costly, time-consuming and uncertain. We or our collaborators may not be able to obtain necessary regulatory approvals on a timely basis, if at all, for any of our potential products. Even if granted, regulatory approvals may include significant limitations on the uses for which products may be marketed. Failure to comply with applicable regulatory requirements can, among other things, result in warning letters, imposition of civil penalties or other monetary payments, delay in approving or refusal to approve a product candidate, suspension or withdrawal of regulatory approval, product recall or seizure, operating restrictions, interruption of clinical trials or manufacturing, injunctions and criminal prosecution.
Regulatory authorities may not approve our product candidates even if the product candidates meet safety and efficacy endpoints in clinical trials or the approvals may be too limited for us to earn sufficient revenues.
The FDA and other foreign regulatory agencies can delay approval of or refuse to approve our product candidates for a variety of reasons, including failure to meet safety and efficacy endpoints in our clinical trials. Our product candidates may not be approved even if they achieve their endpoints in clinical trials. Regulatory agencies, including the FDA, may disagree with our trial design and our interpretations of data from preclinical studies and clinical trials. Even if a product candidate is approved, it may be approved for fewer or more limited indications than requested or the approval may be subject to the performance of significant post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates. Any limitation, condition or denial of approval would have an adverse affect on our business, reputation and results of operations.
Even if we are granted initial FDA approval for any of our product candidates, we may not be able to maintain such approval, which would reduce our revenues.
Even if we are granted initial regulatory approval for a product candidate, the FDA and similar foreign regulatory agencies can limit or withdraw product approvals for a variety of reasons, including failure to comply with regulatory requirements, changes in regulatory requirements, problems with manufacturing facilities or processes or the occurrence of unforeseen problems, such as the discovery of previously undiscovered side effects. If we are able to obtain any product approvals, they may be limited or withdrawn or we may be unable to remain in compliance with regulatory requirements. Both before and after approval we, our collaborators and our products are subject to a number of additional requirements. For example, certain changes to the approved product, such as adding new indications, certain manufacturing changes and additional labeling claims are subject to additional FDA review and approval. Advertising and other promotional material must comply with FDA requirements and established requirements applicable to drug samples. We, our collaborators and our manufacturers will be subject to continuing review and periodic inspections by the FDA and other authorities where applicable and must comply with ongoing requirements, including the FDA’s Good Manufacturing Practices, or GMP, requirements. Once the FDA approves a product, a manufacturer must provide certain updated safety and efficacy information, submit copies of promotional materials to the FDA and make certain other required reports. Product approvals may be
23
Table of Contents
withdrawn if regulatory requirements are not complied with or if problems concerning safety or efficacy of the product occur following approval. Any limitation or withdrawal of approval of any of our products could delay or prevent sales of our products, which would adversely affect our revenues. Further continuing regulatory requirements involve expensive ongoing monitoring and testing requirements.
Since one of our key proprietary programs, the ARD-3100 liposomal ciprofloxacin program, relies on the FDA’s granting of orphan drug designation for potential market exclusivity, the product may not be able to obtain market exclusivity and could be barred from the market for up to seven years.
The FDA has granted orphan drug designation for our proprietary liposomal ciprofloxacin for the management of cystic fibrosis. Orphan drug designation is intended to encourage research and development of new therapies for diseases that affect fewer than 200,000 patients in the United States. The designation provides the opportunity to obtain market exclusivity for seven years from the date of the FDA’s approval of a new drug application, or NDA. However, the market exclusivity is granted only to the first chemical entity to be approved by the FDA for a given indication. Therefore, if another inhaled ciprofloxacin product were to be approved by the FDA for a cystic fibrosis indication before our product, then we may be blocked from launching our product in the United States for seven years, unless we are able to demonstrate to the FDA clinical superiority of our product on the basis of safety or efficacy. We may seek to develop additional products that incorporate drugs that have received orphan drug designations for specific indications. In each case, if our product is not the first to be approved by the FDA for a given indication, we will be unable to access the target market in the United States, which would adversely affect our ability to earn revenues.
We have limited manufacturing capacity and will have to depend on contract manufacturers and collaborators; if they do not perform as expected, our revenues and customer relations will suffer.
We have limited capacity to manufacture our requirements for the development and commercialization of our product candidates. We intend to use contract manufacturers to produce key components, assemblies and subassemblies in the clinical and commercial manufacturing of our products. We may not be able to enter into or maintain satisfactory contract manufacturing arrangements. Specifically, an affiliate of Novo Nordisk has agreed to supply devices and dosage forms to us for use in the development of our products that incorporate our proprietary AERx technology through January 27, 2008. We may not be able to extend this agreement at satisfactory terms, if at all, and we may not be able to find a replacement contract manufacturer at satisfactory terms.
We may decide to invest in additional clinical manufacturing facilities in order to internally produce critical components of our product candidates and to handle critical aspects of the production process, such as assembly of the disposable unit-dose packets and filling of the unit-dose packets. If we decide to produce components of any of our product candidates in-house, rather than use contract manufacturers, it will be costly and we may not be able to do so in a timely or cost-effective manner or in compliance with regulatory requirements.
With respect to some of our product development programs targeted at large markets, either our collaborators or we will have to invest significant amounts to attempt to provide for the high-volume manufacturing required to take advantage of these product markets, and much of this spending may occur before a product is approved by the FDA for commercialization. Any such effort will entail many significant risks. For example, the design requirements of our products may make it too costly or otherwise infeasible for us to develop them at a commercial scale, or manufacturing and quality control problems may arise as we attempt to expand production. Failure to address these issues could delay or prevent late-stage clinical testing and commercialization of any products that may receive FDA approval.
Further, we, our contract manufacturers and our collaborators are required to comply with the FDA’s GMP requirements that relate to product testing, quality assurance, manufacturing and maintaining records and documentation. We, our contract manufacturers or our collaborators may not be able to comply with the applicable GMP and other FDA regulatory requirements for manufacturing, which could result in an enforcement or other action, prevent commercialization of our product candidates and impair our reputation and results of operations.
24
Table of Contents
We rely on a small number of vendors and contract manufacturers to supply us with specialized equipment, tools and components; if they do not perform as we need them to, we will not be able to develop or commercialize products.
We rely on a small number of vendors and contract manufacturers to supply us and our collaborators with specialized equipment, tools and components for use in development and manufacturing processes. These vendors may not continue to supply such specialized equipment, tools and components, and we may not be able to find alternative sources for such specialized equipment and tools. Any inability to acquire or any delay in our ability to acquire necessary equipment, tools and components would increase our expenses and could delay or prevent our development of products.
In order to market our proprietary products, we are likely to establish our own sales, marketing and distribution capabilities. We have no experience in these areas, and if we have problems establishing these capabilities, the commercialization of our products would be impaired.
We intend to establish our own sales, marketing and distribution capabilities to market products to concentrated, easily addressable prescriber markets. We have no experience in these areas, and developing these capabilities will require significant expenditures on personnel and infrastructure. While we intend to market products that are aimed at a small patient population, we may not be able to create an effective sales force around even a niche market. In addition, some of our product development programs will require a large sales force to call on, educate and support physicians and patients. While we intend to enter into collaborations with one or more pharmaceutical companies to sell, market and distribute such products, we may not be able to enter into any such arrangement on acceptable terms, if at all. Any collaborations we do enter into may not be effective in generating meaningful product royalties or other revenues for us.
If any products that we or our collaborators may develop do not attain adequate market acceptance by healthcare professionals and patients, our business prospects and results of operations will suffer.
Even if we or our collaborators successfully develop one or more products, such products may not be commercially acceptable to healthcare professionals and patients, who will have to choose our products over alternative products for the same disease indications, and many of these alternative products will be more established than ours. For our products to be commercially viable, we will need to demonstrate to healthcare professionals and patients that our products afford benefits to the patient that are cost-effective as compared to the benefits of alternative therapies. Our ability to demonstrate this depends on a variety of factors, including:
| • | the demonstration of efficacy and safety in clinical trials; | |
| • | the existence, prevalence and severity of any side effects; | |
| • | the potential or perceived advantages or disadvantages compared to alternative treatments; | |
| • | the timing of market entry relative to competitive treatments; | |
| • | the relative cost, convenience, product dependability and ease of administration; | |
| • | the strength of marketing and distribution support; | |
| • | the sufficiency of coverage and reimbursement of our product candidates by governmental and other third-party payors; and | |
| • | the product labeling or product insert required by the FDA or regulatory authorities in other countries. |
Our product revenues will be adversely affected if, due to these or other factors, the products we or our collaborators are able to commercialize do not gain significant market acceptance.
25
Table of Contents
We depend upon our proprietary technologies, and we may not be able to protect our potential competitive proprietary advantage.
Our business and competitive position is dependent upon our and our collaborators’ ability to protect our proprietary technologies related to various aspects of pulmonary drug delivery and drug formulation. While our intellectual property rights may not provide a significant commercial advantage for us, our patents and know-how are intended to provide protection for important aspects of our technology, including methods for aerosol generation, devices used to generate aerosols, breath control, compliance monitoring, certain pharmaceutical formulations, design of dosage forms and their manufacturing and testing methods. In addition, we are maintaining as non-patented trade secrets some of the key elements of our manufacturing technologies, for example, those associated with production of disposable unit-dose packets for our AERx delivery system.
Our ability to compete effectively will also depend to a significant extent on our and our collaborators’ ability to obtain and enforce patents and maintain trade secret protection over our proprietary technologies. The coverage claimed in a patent application typically is significantly reduced before a patent is issued, either in the United States or abroad. Consequently, any of our pending or future patent applications may not result in the issuance of patents and any patents issued may be subjected to further proceedings limiting their scope and may in any event not contain claims broad enough to provide meaningful protection. Any patents that are issued to us or our collaborators may not provide significant proprietary protection or competitive advantage, and may be circumvented or invalidated. In addition, unpatented proprietary rights, including trade secrets and know-how, can be difficult to protect and may lose their value if they are independently developed by a third party or if their secrecy is lost. Further, because development and commercialization of pharmaceutical products can be subject to substantial delays, patents may expire early and provide only a short period of protection, if any, following commercialization of products.
In July 2006, we assigned 23 issued United States patents to Novo Nordisk along with correspondingnon-United States counterparts and certain related pending applications. In August 2006, Novo Nordisk brought suit against Pfizer, Inc. claiming infringement of certain claims in one of the assigned United States patents. In December 2006, Novo Nordisk’s motion for a preliminary injunction in this case was denied. That patent is placed at risk in connection with this infringement lawsuit. Other patents assigned to Novo Nordisk may become the subject of future litigation. If all or any of the patents assigned to Novo Nordisk are invalidated, it may reduce Novo Nordisk’s commitment to move forward with AERx iDMS and would adversely affect any royalties or other compensation which we might potentially otherwise receive based directly on such patents. Further, the patents assigned to Novo Nordisk encompass, in some instances, technology beyond inhaled insulin and, if all or any of these patents are invalidated, it could harm our ability to obtain market exclusivity with respect to other product candidates.
We may infringe on the intellectual property rights of others, and any litigation could force us to stop developing or selling potential products and could be costly, divert management attention and harm our business.
We must be able to develop products without infringing the proprietary rights of other parties. Because the markets in which we operate involve established competitors with significant patent portfolios, including patents relating to compositions of matter, methods of use and methods of drug delivery, it could be difficult for us to use our technologies or develop products without infringing the proprietary rights of others. We may not be able to design around the patented technologies or inventions of others and we may not be able to obtain licenses to use patented technologies on acceptable terms, or at all. If we cannot operate without infringing the proprietary rights of others, we will not earn product revenues.
If we are required to defend ourselves in a lawsuit, we could incur substantial costs and the lawsuit could divert management attention, regardless of the lawsuit’s merit or outcome. These legal actions could seek damages and seek to enjoin testing, manufacturing and marketing of the accused product or process. In addition to potential liability for significant damages, we could be required to obtain a license to continue to manufacture or market the accused product or process and any license required under any such patent may not be made available to us on acceptable terms, if at all.
26
Table of Contents
Periodically, we review publicly available information regarding the development efforts of others in order to determine whether these efforts may violate our proprietary rights. We may determine that litigation is necessary to enforce our proprietary rights against others. Such litigation could result in substantial expense, regardless of its outcome, and may not be resolved in our favor.
Furthermore, patents already issued to us or our pending patent applications may become subject to dispute, and any disputes could be resolved against us. For example, Eli Lilly and Company brought an action against us seeking to have one or more employees of Eli Lilly named as co-inventors on one of our patents. This case was determined in our favor in 2004, but we may face other similar claims in the future and we may lose or settle cases at significant loss to us. In addition, because patent applications in the United States are currently maintained in secrecy for a period of time prior to issuance, and patent applications in certain other countries generally are not published until more than 18 months after they are first filed, and because publication of discoveries in scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first creator of inventions covered by our pending patent applications or that we were the first to file patent applications on such inventions.
We are in a highly competitive market, and our competitors have developed or may develop alternative therapies for our target indications, which would limit the revenue potential of any product we may develop.
We are in competition with pharmaceutical, biotechnology and drug delivery companies, hospitals, research organizations, individual scientists and nonprofit organizations engaged in the development of drugs and therapies for the disease indications we are targeting. Our competitors may succeed before we can, and many already have succeeded, in developing competing technologies for the same disease indications, obtaining FDA approval for products or gaining acceptance for the same markets that we are targeting. If we are not “first to market,” it may be more difficult for us and our collaborators to enter markets as second or subsequent competitors and become commercially successful. We are aware of a number of companies that are developing or have developed therapies to address indications we are targeting, including major pharmaceutical companies such as Bayer, Eli Lilly, Genentech, Gilead Sciences, Merck & Co., Novartis and Pfizer. Certain of these companies are addressing these target markets with pulmonary products that are similar to ours. These companies and many other potential competitors have greater research and development, manufacturing, marketing, sales, distribution, financial and managerial resources and experience than we have and many of these companies may have products and product candidates that are on the market or in a more advanced stage of development than our product candidates. Our ability to earn product revenues and our market share would be substantially harmed if any existing or potential competitors brought a product to market before we or our collaborators were able to, or if a competitor introduced at any time a product superior to or more cost-effective than ours.
If we do not continue to attract and retain key employees, our product development efforts will be delayed and impaired.
We depend on a small number of key management and technical personnel. Our success also depends on our ability to attract and retain additional highly qualified marketing, management, manufacturing, engineering and development personnel. There is a shortage of skilled personnel in our industry, we face intense competition in our recruiting activities, and we may not be able to attract or retain qualified personnel. Losing any of our key employees, particularly our new President and Chief Executive Officer, Dr. Igor Gonda, who plays a central role in our strategy shift to a specialty pharmaceutical company, could impair our product development efforts and otherwise harm our business. Any of our employees may terminate their employment with us at will.
Acquisition of complementary businesses or technologies could result in operating difficulties and harm our results of operations.
While we have not identified any definitive targets, we may acquire products, businesses or technologies that we believe are complementary to our business strategy. The process of investigating, acquiring and integrating any business or technology into our business and operations is risky and we may not be able to accurately predict or
27
Table of Contents
derive the benefits of any such acquisition. The process of acquiring and integrating any business or technology may create operating difficulties and unexpected expenditures, such as:
| • | diversion of our management from the development and commercialization of our pipeline product candidates; | |
| • | difficulty in assimilating and efficiently using the acquired assets or personnel; and | |
| • | inability to retain key personnel. |
In addition to the factors set forth above, we may encounter other unforeseen problems with acquisitions that we may not be able to overcome. Any future acquisitions may require us to issue shares of our stock or other securities that dilute the ownership interests of our other shareholders, expend cash, incur debt, assume liabilities, including contingent or unknown liabilities, or incur additional expenses related to write-offs or amortization of intangible assets, any of which could materially adversely affect our operating results.
If we market our products in other countries, we will be subject to different laws and we may not be able to adapt to those laws, which could increase our costs while reducing our revenues.
If we market any approved products in foreign countries, we will be subject to different laws, particularly with respect to intellectual property rights and regulatory approval. To maintain a proprietary market position in foreign countries, we may seek to protect some of our proprietary inventions through foreign counterpart patent applications. Statutory differences in patentable subject matter may limit the protection we can obtain on some of our inventions outside of the United States. The diversity of patent laws may make our expenses associated with the development and maintenance of intellectual property in foreign jurisdictions more expensive than we anticipate. We probably will not obtain the same patent protection in every market in which we may otherwise be able to potentially generate revenues. In addition, in order to market our products in foreign jurisdictions, we and our collaborators must obtain required regulatory approvals from foreign regulatory agencies and comply with extensive regulations regarding safety and quality. We may not be able to obtain regulatory approvals in such jurisdictions and we may have to incur significant costs in obtaining or maintaining any foreign regulatory approvals. If approvals to market our products are delayed, if we fail to receive these approvals, or if we lose previously received approvals, our business would be impaired as we could not earn revenues from sales in those countries.
We may be exposed to product liability claims, which would hurt our reputation, market position and operating results.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates in humans and will face an even greater risk upon commercialization of any products. These claims may be made directly by consumers or by pharmaceutical companies or others selling such products. We may be held liable if any product we develop causes injury or is found otherwise unsuitable during product testing, manufacturing or sale. Regardless of merit or eventual outcome, liability claims would likely result in negative publicity, decreased demand for any products that we may develop, injury to our reputation and suspension or withdrawal of clinical trials. Any such claim will be very costly to defend and also may result in substantial monetary awards to clinical trial participants or customers, loss of revenues and the inability to commercialize products that we develop. Although we currently have product liability insurance, we may not be able to maintain such insurance or obtain additional insurance on acceptable terms, in amounts sufficient to protect our business, or at all. A successful claim brought against us in excess of our insurance coverage would have a material adverse effect on our results of operations.
If we cannot arrange for adequate third-party reimbursement for our products, our revenues will suffer.
In both domestic and foreign markets, sales of our potential products will depend in substantial part on the availability of adequate reimbursement from third-party payors such as government health administration authorities, private health insurers and other organizations. Third-party payors often challenge the price and cost-effectiveness of medical products and services. Significant uncertainty exists as to the adequate reimbursement
28
Table of Contents
status of newly approved health care products. Any products we are able to successfully develop may not be reimbursable by third-party payors. In addition, our products may not be considered cost-effective and adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize a profit. Legislation and regulations affecting the pricing of pharmaceuticals may change before our products are approved for marketing and any such changes could further limit reimbursement. If any products we develop do not receive adequate reimbursement, our revenues will be severely limited.
Our use of hazardous materials could subject us to liabilities, fines and sanctions.
Our laboratory and clinical testing sometimes involve use of hazardous and toxic materials. We are subject to federal, state and local laws and regulations governing how we use, manufacture, handle, store and dispose of these materials. Although we believe that our safety procedures for handling and disposing of such materials comply in all material respects with all federal, state and local regulations and standards, there is always the risk of accidental contamination or injury from these materials. In the event of an accident, we could be held liable for any damages that result and such liability could exceed our financial resources. Compliance with environmental and other laws may be expensive and current or future regulations may impair our development or commercialization efforts.
If we are unable to effectively implement or maintain a system of internal controls over financial reporting, we may not be able to accurately or timely report our financial results and our stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us to evaluate the effectiveness of our internal controls over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal controls over financial reporting in our annual report onForm 10-K for that fiscal year. Section 404 also requires our independent registered public accounting firm, beginning with our fiscal year ending December 31, 2008, to attest to, and report on, management’s assessment of our internal controls over financial reporting. Additionally, if our market capitalization increases significantly and we become an accelerated filer, our auditors may need to issue such a report for calendar year 2007. Our ability to comply with the annual internal control report requirements will depend on the effectiveness of our financial reporting and data systems and controls across our company. We expect these systems and controls to involve significant expenditures and to become increasingly complex as our business grows and to the extent that we make and integrate acquisitions. To effectively manage this complexity, we will need to continue to improve our operational, financial and management controls and our reporting systems and procedures. Any failure to implement required new or improved controls, or difficulties encountered in the implementation or operation of these controls, could harm our operating results and cause us to fail to meet our financial reporting obligations, which could adversely affect our business and reduce our stock price.
Risks Related to Our Common Stock
Our stock price is likely to remain volatile.
The market prices for securities of many companies in the drug delivery and pharmaceutical industries, including ours, have historically been highly volatile, and the market from time to time has experienced significant price and volume fluctuations unrelated to the operating performance of particular companies. Prices for our common stock may be influenced by many factors, including:
| • | investor perception of us; | |
| • | research analyst recommendations and our ability to meet or exceed quarterly performance expectations of analysts or investors; | |
| • | fluctuations in our operating results; | |
| • | market conditions relating to our segment of the industry or the securities markets in general; | |
| • | announcements of technological innovations or new commercial products by us or our competitors; |
29
Table of Contents
| • | publicity regarding actual or potential developments relating to products under development by us or our competitors; | |
| • | failure to maintain existing or establish new collaborative relationships; | |
| • | developments or disputes concerning patents or proprietary rights; | |
| • | delays in the development or approval of our product candidates; | |
| • | regulatory developments in both the United States and foreign countries; | |
| • | concern of the public or the medical community as to the safety or efficacy of our products, or products deemed to have similar safety risk factors or other similar characteristics to our products; | |
| • | period-to-period fluctuations in financial results; | |
| • | future sales or expected sales of substantial amounts of common stock by shareholders; | |
| • | our ability to raise financing; and | |
| • | economic and other external factors. |
In the past, class action securities litigation has often been instituted against companies promptly following volatility in the market price of their securities. Any such litigation instigated against us would, regardless of its merit, result in substantial costs and a diversion of management’s attention and resources.
Our common stock was delisted from the Nasdaq Capital Market; this delisting may reduce the liquidity of our common stock and the price may decline.
On November 10, 2006, our common stock was delisted from the Nasdaq Capital Market due to non-compliance with Nasdaq’s continued listing standards. Our common stock is currently quoted on the OTC Bulletin Board. This delisting may reduce the liquidity of our common stock, may cause investors not to trade in our stock and may result in a lower stock price. In addition, investors may find it more difficult to obtain accurate quotations of the share price of our common stock.
We have implemented certain anti-takeover provisions, which make it less likely that we would be acquired and you would receive a premium price for your shares.
Certain provisions of our articles of incorporation and the California Corporations Code could discourage a party from acquiring, or make it more difficult for a party to acquire, control of our company without approval of our board of directors. These provisions could also limit the price that certain investors might be willing to pay in the future for shares of our common stock. Certain provisions allow our board of directors to authorize the issuance, without shareholder approval, of preferred stock with rights superior to those of the common stock. We are also subject to the provisions of Section 1203 of the California Corporations Code, which requires us to provide a fairness opinion to our shareholders in connection with their consideration of any proposed “interested party” reorganization transaction.
We have adopted a shareholder rights plan, commonly known as a “poison pill.” We have also adopted an Executive Officer Severance Plan and a Form of Change of Control Agreement, both of which may provide for the payment of benefits to our officers in connection with an acquisition. The provisions of our articles of incorporation, our poison pill, our severance plan and our change of control agreements, and provisions of the California Corporations Code may discourage, delay or prevent another party from acquiring us or reduce the price that a buyer is willing to pay for our common stock.
We have never paid dividends on our capital stock, and we do not anticipate paying cash dividends for at least the foreseeable future.
We have never declared or paid cash dividends on our capital stock. We do not anticipate paying any cash dividends on our common stock for at least the foreseeable future. We currently intend to retain all available funds
30
Table of Contents
and future earnings, if any, to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be your sole source of potential gain for at least the foreseeable future.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
At December 31, 2006, we leased one building with an aggregate of 72,000 square feet of office and laboratory facilities at 3929 Point Eden Way, Hayward, California. The aggregate lease payment in 2006 was approximately $1.9 million. Minimum payments under this lease, net of sublease payments, will be approximately $2.4 million in 2007 and an aggregate of $19.0 million for the period 2008 through 2016. As a result of our recent restructuring activities, we have consolidated our operations to a portion of the space at our current address and we are actively investigating sublease opportunities for the vacated space. The lease expires in June 2016, subject to our option to extend the term for six months to December 2016.
| Item 3. | Legal Proceedings |
We are not currently a party to any pending legal proceedings.
| Item 4. | Submission of Matters to a Vote of Security Holders |
There were no submissions of matters to a vote of security holders in the quarter ended December 31, 2006.
PART II
| Item 5. | Market for the Registrant’s Common Stock Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Since December 21, 2006, our common stock has been quoted on the OTC Bulletin Board, an electronic quotation service for securities tradedover-the-counter, under the symbol “ARDM”. Between June 20, 1996 and May 1, 2006 our common stock was listed on the Nasdaq Global Market (formerly the Nasdaq National Market), at which time, not being in compliance with continued listing requirements, we voluntarily moved our listing to the Nasdaq Capital Market (formerly the Nasdaq SmallCap Market); between May 2, 2006 and November 9, 2006 our common stock was listed on the Nasdaq Capital Market (formerly the Nasdaq SmallCap Market), at which time, not being in compliance with continued listing requirements, we were delisted; and between November 10, 2006 and December 20, 2006 our common stock was quoted on the Pink Sheets, until such time as our quotation on the OTC Bulletin Board was initiated.
31
Table of Contents
The following table sets forth the high and low closing sale prices of our common stock for the periods indicated.
| High | Low | |||||||
2004 | ||||||||
| First Quarter | $ | 13.70 | $ | 9.05 | ||||
| Second Quarter | 11.35 | 4.20 | ||||||
| Third Quarter | 6.40 | 3.30 | ||||||
| Fourth Quarter | 10.00 | 5.90 | ||||||
2005 | ||||||||
| First Quarter | $ | 8.65 | $ | 5.60 | ||||
| Second Quarter | 5.90 | 5.00 | ||||||
| Third Quarter | 6.05 | 4.95 | ||||||
| Fourth Quarter | 5.25 | 3.45 | ||||||
2006 | ||||||||
| First Quarter | $ | 5.04 | $ | 3.03 | ||||
| Second Quarter | 3.32 | 1.29 | ||||||
| Third Quarter | 2.24 | 1.40 | ||||||
| Fourth Quarter | 1.69 | 0.81 | ||||||
2007 | ||||||||
| First Quarter (through February 28, 2007) | 1.46 | 0.90 | ||||||
On February 28, 2007, there were approximately 225 stockholders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our capital stock and we do not currently intend to pay any cash dividends on our capital stock for at least the foreseeable future. We expect to retain future earnings, if any, to fund the development and growth of our business. Any future determination to pay dividends on our capital stock will be, subject to applicable law, at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements and contractual restrictions in loan agreements or other agreements.
32
Table of Contents
Stock Performance Graph
The following graph shows the total shareholder return of an investment of $100 in cash on December 31, 2001 for (i) our common stock, (ii) the Nasdaq composite Market Index (the “Nasdaq Index”) and (iii) the Nasdaq Pharmaceutical Index (the “Nasdaq-Pharmaceutical”). The total return for our stock and for each index assumes the reinvestment of dividends, although dividends have never been declared on our stock, and is based on the returns of the component companies weighted according to their capitalizations as of the end of each quarterly period. The Nasdaq Index tracks the aggregate price performance of equity securities traded on the Nasdaq. The Nasdaq-Pharmaceutical tracks the aggregate price performance of equity securities of pharmaceutical companies traded on the Nasdaq Index. Our common stock is quoted on the OTC Bulletin Board, an electronic quotation service for securities tradedover-the-counter.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among Aradigm Corporation, The NASDAQ Composite Index
And The NASDAQ Pharmaceutical Index
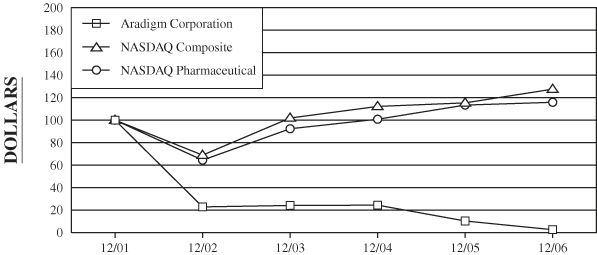
| Cumulative Total Return | ||||||||||||||||||||||||||||||
| 12/01 | 12/02 | 12/03 | 12/04 | 12/05 | 12/06 | |||||||||||||||||||||||||
Aradigm Corporation | 100.00 | 22.82 | 24.08 | 24.37 | 10.28 | 2.54 | ||||||||||||||||||||||||
NASDAQ Composite | 100.00 | 68.85 | 101.86 | 112.16 | 115.32 | 127.52 | ||||||||||||||||||||||||
NASDAQ Pharmaceutical | 100.00 | 64.40 | 92.31 | 100.78 | 113.36 | 115.84 | ||||||||||||||||||||||||
| * | This section is not “soliciting material,” is not deemed “filed” with the SEC, and is not to be incorporated by reference into any filing of the Company under the 1933 Act or 1934 Act, whether made before or after the date hereof and irrespective of any general incorporation language contained in such filing. |
33
Table of Contents
| Item 6. | Selected Financial Data |
The following selected financial data should be read in conjunction with the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements and notes thereto included in this Report onForm 10-K.
| Years Ended December 31, | ||||||||||||||||||||
| 2006(1) | 2005 | 2004 | 2003 | 2002 | ||||||||||||||||
Statements of operations data: | ||||||||||||||||||||
| Contract and license revenues | $ | 4,814 | $ | 10,507 | $ | 28,045 | $ | 33,857 | $ | 28,967 | ||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 22,1998 | 30,174 | 46,477 | 49,636 | 54,680 | |||||||||||||||
| General and administrative | 10,717 | 10,895 | 11,934 | 10,391 | 10,394 | |||||||||||||||
| Restructuring and asset impairment | 6,003 | — | — | — | — | |||||||||||||||
| Total operating expenses | 38,918 | 41,069 | 58,411 | 60,027 | 65,074 | |||||||||||||||
| Loss from operations | (34,104 | ) | (30,562 | ) | (30,366 | ) | (26,170 | ) | (36,107 | ) | ||||||||||
| Gain on sale of patent and royalty interest (to related party) | 20,000 | — | — | — | — | |||||||||||||||
| Interest income | 1,251 | 1,317 | 194 | 338 | 818 | |||||||||||||||
| Interest expense | (197 | ) | (6 | ) | (16 | ) | (138 | ) | (499 | ) | ||||||||||
| Other income (expense) | 23 | 36 | (1 | ) | — | (143 | ) | |||||||||||||
| Net Loss | $ | (13,027 | ) | $ | (29,215 | ) | $ | (30,189 | ) | $ | (25,970 | ) | $ | (35,931 | ) | |||||
| Basic and diluted net loss per share | $ | (0.89 | ) | $ | (2.01 | ) | $ | (2.37 | ) | $ | (2.59 | ) | $ | (5.94 | ) | |||||
| Shares used in computing basic and diluted net loss per share | 14,642 | 14,513 | 12,741 | 10,039 | 6,052 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||||||
| Pro Forma | ||||||||||||||||||||||||
| 2006(2) | 2006(1) | 2005 | 2004 | 2003 | 2002 | |||||||||||||||||||
Balance sheet data: | ||||||||||||||||||||||||
| Cash, cash equivalents and short-term investments | $ | 61,403 | $ | 27,514 | $ | 27,694 | $ | 16,763 | $ | 29,770 | $ | 31,443 | ||||||||||||
| Working capital | 59,295 | 25,406 | 21,087 | 4,122 | 19,708 | 16,039 | ||||||||||||||||||
| Total assets | 66,115 | 32,226 | 39,497 | 79,741 | 95,218 | 97,129 | ||||||||||||||||||
| Non-current portion of notes payable and capital lease obligations | 7,686 | 7,686 | — | — | — | 497 | ||||||||||||||||||
| Convertible preferred stock | — | 23,669 | 23,669 | 23,669 | 23,669 | 30,665 | ||||||||||||||||||
| Accumulated deficit | (287,865 | ) | (287,865 | ) | (274,838 | ) | (245,623 | ) | (215,436 | ) | (189,443 | ) | ||||||||||||
| Total shareholders’ equity (deficit) | 53,611 | (3,947 | ) | 7,171 | 35,754 | 52,970 | 41,410 | |||||||||||||||||
| (1) | On July 3, 2006, we further restructured our relationship with Novo Nordisk through an intellectual property assignment, a royalty prepayment and an eight-year promissory note with Novo Nordisk. The promissory note was secured by the royalty payments on any AERx iDMS sales by Novo Nordisk under the license with us. The key features of this restructuring included: |
| • | our transfer to Novo Nordisk of the ownership of 23 issued United States patents and their correspondingnon-United States counterparts, if any, as well as related pending applications, in exchange for $12.0 million paid to us in cash. We retained exclusive, royalty-free control of these patents outside the field of glucose control and will continue to be entitled to royalties under our license agreement that will rise to an average of |
34
Table of Contents
| five percent or higher by the fifth year after commercialization with respect to any inhaled insulin products marketed or licensed by Novo Nordisk. |
| • | our receipt of a royalty prepayment of $8.0 million in exchange for a one percent reduction on our average royalty rate for the commercialized AERx iDMS product. As a result, we are entitled to receive royalty rates under our license agreement with Novo Nordisk that will commence at a minimum of 3.25% on launch, and that we estimate will average 5% over the life of the product. | |
| • | our issuance of an eight-year promissory note to Novo Nordisk in connection with our receipt from Novo Nordisk of a loan in the principal amount of $7.5 million with interest accruing at 5% per year. The principal and interest are payable to Novo Nordisk in three equal payments of $3.5 million on July 2, 2012, July 1, 2013 and June 30, 2014. Our obligations under the note are secured by royalty payments upon any commercialization of the AERx iDMS product. |
| (2) | Pro forma data reflects the issuance of 37,950,000 shares of common stock in an underwritten public offering that closed on January 30, 2007 and resulted in net proceeds, after underwriting discount and expenses, of approximately $33.3 million. This public offering triggered the automatic conversion of all outstanding shares of Series A convertible preferred stock to common stock and eliminated the Series A liquidation preference of $41.9 million, equal to the original issue price plus all accrued and unpaid dividends (as adjusted for any stock dividends, combinations, splits, recapitalizations and other similar events). |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The discussion below contains forward-looking statements that are based on the beliefs of management, as well as assumptions made by, and information currently available to, management. Our future results, performance or achievements could differ materially from those expressed in, or implied by, any such forward-looking statements as a result of certain factors, including, but not limited to, those discussed in this section as well as in the section entitled “Risk Factors” and elsewhere in this report. This discussion should be read in conjunction with the financial statements and notes to the financial statements contained in this report.
Overview
We are an emerging specialty pharmaceutical company focused on the development and commercialization of a portfolio of drugs delivered by inhalation for the treatment of severe respiratory diseases by pulmonologists. Over the last decade, we have invested a large amount of capital to develop drug delivery technologies, and in doing so we have developed a significant amount of expertise in pulmonary drug delivery. We have also invested considerable effort into the generation of a large volume of laboratory and clinical data demonstrating the performance of our AERx pulmonary drug delivery platform. We have not been profitable since inception and expect to incur additional operating losses over at least the next several years as we expand product development efforts, preclinical testing and clinical trial activities and possible sales and marketing efforts and as we secure production capabilities from outside contract manufacturers. To date, we have not had any significant product sales, and we do not anticipate receiving any revenues from the sale of products for at least the next several years. As of December 31, 2006, we had an accumulated deficit of $287.9 million. Historically we have funded our operations primarily through private placements and public offerings of our capital stock, proceeds from equipment lease financings, license fees and milestone payments from collaborators, proceeds from our restructuring transaction with Novo Nordisk and interest earned on investment.
We have performed initial feasibility work and conducted early stage clinical work on a number of potential products and have been compensated for expenses incurred while performing this work in several cases pursuant to feasibility study agreements and other collaborative arrangements. We will seek to develop certain potential products ourselves, including those that can benefit from our experience in pulmonary delivery, and that have markets we can address with a targeted sales and marketing force and that we believe are likely to provide a superior therapeutic profile or other valuable benefits to patients when compared to existing products. For other potential products with larger or less concentrated markets we may seek to enter into development and commercialization agreements with collaborators.
35
Table of Contents
In 2004, we executed a development agreement with Defence Research and Development Canada, a division of the Canadian Department of National Defence, for the development of liposomal ciprofloxacin for the treatment of biological terrorism-related inhalation anthrax. We are also exploring the use of liposomal ciprofloxacin to treat other indications. We have received orphan drug designation for this formulation from the United States Food and Drug Administration, or the FDA, for the management of cystic fibrosis, or CF. We initiated preclinical studies for our ARD-3100 product candidate in 2006 and expect to initiate human clinical studies for the CF indication in the first half of 2007. We anticipate using safety data from these studies to support our expected application for approval of the ARD-1100 product candidate for the prevention and treatment of inhalation anthrax and possibly other inhaled life-threatening bioterrorism infections as well.
The AERx insulin Diabetes Management System, or AERx iDMS, which we initially developed and is now licensed to Novo Nordisk, is being developed to control blood glucose levels in patients with diabetes. Following the restructuring of our collaborative arrangement in January 2005, all responsibility for funding and conducting the remaining development and commercialization of this product, including manufacturing, clinical trials, regulatory filings, marketing and sales, has been transferred to Novo Nordisk. We have the right to receive royalties on any sales of AERx iDMS. AERx iDMS is currently undergoing testing in a Phase 3 clinical program, which began in May 2006. This program follows significant prior clinical work which provided preliminary evidence that AERx iDMS is comparable to regular injectable insulin in the overall management of Type 1 and Type 2 diabetes. The Phase 3 clinical program is expected to include a total of approximately 3,400 Type 1 and Type 2 diabetes patients and is taking place worldwide with primary focus on Europe and the United States. The program includes treatment comparisons with other antidiabetics. The longest of these trials is expected to last 27 months. Novo Nordisk announced in October 2006 that it expects commercial launch of the product in 2010. As with any clinical program, there are many factors that could delay the launch or could result in AERx iDMS not receiving or maintaining regulatory approval.
We have other ongoing collaborator-funded and proprietary programs under development. In 2007, we expect self-initiated research and development expenses to remain unchanged from 2006; however, the extent of and costs associated with future research and development efforts are uncertain and difficult to predict due to the early stage of development of our programs.
Restructured Relationship with Novo Nordisk
During 2005, our collaborative agreement with Novo Nordisk and its subsidiary, Novo Nordisk Delivery Technologies, or NNDT, contributed approximately 76% of our total contract revenues. From the inception of our collaboration in June 1998 through December 31, 2005, we have received from Novo Nordisk $150.1 million in product development and milestone payments and $35.0 million from the purchase of our common stock by Novo Nordisk and its affiliates. All product development and milestone payments received to date have been recognized as revenue.
As of January 26, 2005, we restructured the AERx iDMS program, pursuant to a restructuring agreement entered into with Novo Nordisk and NNDT in September 2004. Under the terms of the restructuring agreement, we sold certain equipment, leasehold improvements and other tangible assets used in the AERx iDMS program to NNDT, for a cash payment of $55.3 million (before refund of cost advances made by Novo Nordisk). Our expenses related to this transaction for legal and other consulting costs were $1.1 million. In connection with the restructuring transaction, we entered into various related agreements with Novo Nordisk and NNDT, including the following:
| • | an amended and restated license agreement amending the development and license agreement previously in place with Novo Nordisk, expanding Novo Nordisk’s development and manufacturing rights to the AERx iDMS program and providing for royalties to us on future AERx iDMS net sales in lieu of a percentage interest in the gross profits from the commercialization of AERx iDMS, which royalties run until the later of last patent expiry or last use of our intellectual property and which apply to future enhancements or generations of our AERx delivery technology; | |
| • | a three-year agreement under which NNDT agreed to perform contract manufacturing of AERx iDMS-identical devices and dosage forms filled with compounds provided by us in support of preclinical and initial clinical development of other products that incorporate our AERx delivery system; and |
36
Table of Contents
| • | an amendment of the common stock purchase agreement in place with Novo Nordisk prior to the closing of the restructuring transaction, (i) deleting the provisions whereby we can require Novo Nordisk to purchase certain additional amounts of common stock, (ii) imposing certain restrictions on the ability of Novo Nordisk to sell shares of our common stock and (iii) providing Novo Nordisk with certain registration and information rights with respect to these shares. |
As a result of this transaction, we recorded our final project development revenues from Novo Nordisk in the first quarter of 2005, and, as we were no longer obligated to continue work related to the non-refundable milestone payment from Novo Nordisk in connection with the commercialization of AERx, we recognized the remaining balance of the deferred revenue associated with the milestone of $5.2 million as revenue in the first quarter of 2005. In 2005, we recorded revenues of approximately $727,000 from NNDT related to transition and support agreements. As a result of this transaction, we were released from our contractual obligations relating to future operating lease payments for two buildings assigned to NNDT and accordingly reversed the deferred rent liability related to the two buildings of $1.4 million, resulting in a reduction of operating expenses in 2005. In addition, pursuant to the restructuring agreement, we terminated a manufacturing and supply agreement and a patent cooperation agreement, each previously in place with Novo Nordisk and dated October 22, 2001.
On July 3, 2006, we further restructured our relationship with Novo Nordisk through an intellectual property assignment, a royalty prepayment and an eight-year promissory note with Novo Nordisk. The promissory note was secured by the royalty payments on any AERx iDMS sales by Novo Nordisk under the license with us. The key features of this restructuring included:
| • | our transfer to Novo Nordisk of the ownership of 23 issued United States patents and their correspondingnon-United States counterparts, if any, as well as related pending applications, in exchange for $12.0 million paid to us in cash. We retained exclusive, royalty-free control of these patents outside the field of glucose control and will continue to be entitled to royalties that will rise to an average of five percent or higher by the fifth year after commercialization with respect to any inhaled insulin products marketed or licensed by Novo Nordisk. | |
| • | our receipt of a royalty prepayment of $8.0 million in exchange for a one percent reduction on our average royalty rate for the commercialized AERx iDMS product. As a result, we are entitled to receive royalty rates under our license agreement with Novo Nordisk that will commence at a minimum of 3.25% on launch, and that we estimate will average 5% over the life of the product. | |
| • | our issuance of an eight-year promissory note to Novo Nordisk in connection with our receipt from Novo Nordisk of a loan in the principal amount of $7.5 million with interest accruing at 5% per year. The principal and interest are payable to Novo Nordisk in three equal payments of $3.5 million on July 2, 2012, July 1, 2013 and June 30, 2014. Our obligations under the note are secured by royalty payments upon any commercialization of the AERx iDMS product. |
We and Novo Nordisk continue to cooperate and share in technology development, as well as intellectual property development and defense. Both we and Novo Nordisk have access to any developments or improvements the other might make to the AERx delivery system, within their respective fields of use. As of December 31, 2006, Novo Nordisk was a substantial holder of our common stock and is restricted from disposing of any of our common stock until January 1, 2009 or the earlier occurrence of certain specified events. Pursuant to the Company’s public offering completed on January 30, 2007, Novo Nordisk owned approximately 3.0% of the Company’s stock on an as-converted basis.
In August 2006, Novo Nordisk announced that it had filed a lawsuit against Pfizer claiming that Exubera, an inhaled insulin product that Pfizer has been developing with Nektar Therapeutics, infringes a patent originally owned by us and now owned by Novo Nordisk with rights retained by us outside the field of glucose control. In December 2006, Novo Nordisk’s motion for a preliminary injunction in this case was denied. While the outcome of this lawsuit is highly uncertain, we are entitled to a portion of any proceeds, net of litigation costs, that may be received by Novo Nordisk from a favorable outcome.
37
Table of Contents
Purchase and Sale of Intraject Technology
In May 2003, we acquired select assets from the Weston Medical Group, a company based in the United Kingdom, including the Intraject needle-free delivery technology, related manufacturing equipment and intellectual property and associated transfer costs, for a total of $2.9 million. The purchase price and additional costs were allocated to the major pieces of purchased commercial equipment for the production of Intraject and were recorded in property and equipment as construction in progress. No costs or expenses were allocated to intellectual property or in-process research and development on a pro-rata basis, because of the lack of market information, the early stage of development and the immateriality of any allocation to intellectual property or in-process research and development based on the substantial value of the tangible assets acquired.
In October 2004, we announced positive results from the clinical performance verification trial of the Intraject needle-free delivery system. Following the results from the configuration trial, we initiated a pilot pharmacokinetic study comparing Intraject with sumatriptan, a treatment for migraines, to the currently marketed needle-injected product. In June 2005, we announced results from this study, which showed that Intraject sumatriptan was bioequivalent to the marketed injectable product and that patients were able to self-administer using Intraject.
In August 2006, we sold all of our assets related to the Intraject technology platform and products, including 12 United States patents along with any foreign counterparts corresponding to those United States patents, to Zogenix, Inc., a newly created private company that has some officers who were former officers of our company. Zogenix is responsible for further development and commercialization efforts of Intraject. We received a $4.0 million initial payment and we will be entitled to a milestone payment upon initial commercialization and royalty payments upon any commercialization of products that may be developed and sold using the Intraject technology. Our potential royalty payments will be affected by the ability of Zogenix to maintain and, if necessary, enforce the patents we assigned to it.
Critical Accounting Policies and Estimates
We consider certain accounting policies related to revenue recognition, stock-based compensation and impairment of long-lived assets to be critical accounting policies that require the use of significant judgments and estimates relating to matters that are inherently uncertain and may result in materially different results under different assumptions and conditions. The preparation of financial statements in conformity with United States generally accepted accounting principles requires us to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes to the financial statements. These estimates include useful lives for property and equipment and related depreciation calculations, estimated amortization periods for payments received from product development and license agreements as they relate to the revenue recognition of deferred revenue and assumptions for valuing options, warrants and other stock-based compensation. Our actual results could differ from these estimates.
Revenue Recognition
Contract revenues consist of revenues from collaboration agreements and feasibility studies. We recognize revenues under the provisions of the Securities and Exchange Commission issued Staff Accounting Bulletin, or SAB, No. 104, “Revenue Recognition.” Under collaboration agreements, revenues are recognized as costs are incurred. Deferred revenue represents the portion of all refundable and nonrefundable research payments received that have not been earned. In accordance with contract terms, milestone payments from collaborative research agreements are considered reimbursements for costs incurred under the agreements and, accordingly, are generally recognized as revenues either upon the completion of the milestone effort when payments are contingent upon completion of the effort or are based on actual efforts expended over the remaining term of the agreements when payments precede the required efforts. Costs of contract revenues are approximate to or are greater than such revenues and are included in research and development expenses when incurred. Refundable development and license fee payments are deferred until the specified performance criteria are achieved. Refundable development and license fee payments are generally not refundable once the specific performance criteria are achieved and accepted.
38
Table of Contents
Impairment of Long-Lived Assets
We review for impairment whenever events or changes in circumstances indicate that the carrying amount of property and equipment may not be recoverable in accordance with Statement of Financial Accounting Standard, or SFAS, No. 144, “Accounting for the Impairment or Disposal of Long-Lived Assets.” Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. Future cash flows that are contingent in nature are generally not recognized. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values and the loss is recognized in the statements of operations. We recorded a non-cash impairment charge of $4.0 million in 2006, related to our estimate of the net realizable value of the Intraject-related assets, based on the expected sale of those assets.
Stock-Based Compensation Expense
Effective January 1, 2006, we adopted the fair value recognition provisions of SFAS No. 123 (revised 2004), “Share-Based Payment,” or SFAS No. 123R, using the modified prospective transition method and, therefore, have not restated prior periods’ results. Under this method, we recognize compensation expense, net of estimated forfeitures, for all stock-based payments granted after January 1, 2006 and all stock-based payments granted prior to but not vested as of January 1, 2006.
Under the provisions of SFAS No. 123R, stock-based compensation cost is estimated at the grant date based on the award’s fair value and is recognized as expense, net of estimated forfeitures, ratably over the requisite vesting period. We have elected to calculate an award’s fair value based on the Black-Scholes option-pricing model. The Black-Scholes model requires various assumptions including expected option life and expected stock price volatility. If any of the assumptions used in the Black-Scholes model or the estimated forfeiture rate change significantly, stock-based compensation expense may differ materially in the future from that recorded in the current period.
On August 10, 2006, we agreed to grant Dr Gonda, our President and Chief Executive Officer, a stock bonus of up to 100,000 shares of common stock in two 50,000 share tranches to be earned based on achievement of minimum share price appreciation objectives after each of the first two years from the employment start date. We valued Dr. Gonda’s stock bonus on a Monte-Carlo simulation due to the path-dependency of the award. We believe the Monte-Carlo simulation provides a more precise estimate for the grant date fair value of a market-based equity award as the Monte Carlo simulation allows for vesting throughout the vesting period.
Under SFAS No. 123R, we recognized compensation expense for stock-based compensation of $1.6 million for the year ended December 31, 2006. As of December 31, 2006, the total unrecorded stock based compensation balance for unvested shares, net of expected forfeitures, was $2.8 million which is expected to be amortized over a weighted average period of 1.41 years.
Results of Operations
Years Ended December 31, 2006, 2005 and 2004
Revenues
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2004 to 2005 | 2005 to 2006 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Related parties | $ | 59 | $ | 8,013 | $ | 26,999 | (99 | )% | (70 | )% | ||||||||||
| Unrelated parties | 4,755 | 2,494 | 1,046 | 91 | % | 138 | % | |||||||||||||
| Total revenues | $ | 4,814 | $ | 10,507 | $ | 28,045 | (54 | ) | (63 | )% | ||||||||||
We reported revenues from collaborative contracts of $4.8 million in 2006, compared to $10.5 million in 2005 and $28.0 million in 2004. The decrease in revenue in 2006 compared to 2005 is due primarily to the decreases in partner-funded project development revenue from Novo Nordisk, which was $59,000 in 2006 compared to
39
Table of Contents
$8.0 million in 2005 and offset by contract revenue from other partner-funded programs, which totaled $4.8 million in 2006 and $2.5 million in 2005. Revenue in 2004 consisted of $27.0 million from partner-funded project development revenue from Novo Nordisk and $1.0 million from other partner-funded project development programs. The reduction in partner revenues in 2005 from 2004 is primarily due to the sale of AERx iDMS program related assets to Novo Nordisk pursuant to the restructuring transaction completed in January 2005. Costs associated with contract research revenue are included in research and development expenses.
As a result of the sale of the Intraject platform to Zogenix, which was completed in August 2006, we recorded revenues of $869,000 related to transition and support agreements entered into in connection with the sale transaction. Milestone revenues from our ARD-1300 development program were $484,000 in 2006 and $162,000 in 2005.
Research and Development Expenses
Research and development expenses decreased in 2006 compared to 2005 and 2004. These expenses were $22.2 million in 2006 compared to $30.2 million in 2005 and $46.5 million in 2004.
Spending for collaborative and self-initiated research and development projects was as follows (in millions of dollars):
Research and Development
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2005 to 2006 | 2004 to 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Research and development expenses: | ||||||||||||||||||||
| Collaborative | $ | 4,440 | $ | 5,996 | $ | 28,164 | (26 | )% | (79 | )% | ||||||||||
| Self-initiated | 17,758 | 24,178 | 18,313 | (27 | )% | 32 | % | |||||||||||||
| Total research and development expenses | $ | 22,198 | $ | 30,174 | $ | 46,477 | (26 | )% | (35 | )% | ||||||||||
These expenses represent proprietary research expenses as well as the costs related to contract research revenue and include salaries and benefits of scientific and development personnel, laboratory supplies, consulting services and the expenses associated with the development of manufacturing processes.
Research and development expenses in 2006 decreased by $8.0 million, or 26%, compared to 2005. The decrease in research and development expense was due primarily to the completion of our Intraject clinical batch registration lot activities being substantially completed at year-end 2005 and finalized in early 2006. In addition, on May 15, 2006, we announced the implementation of a strategic restructuring of our business operations to focus resources on advancing the current product pipeline and initiated a reduction in force to better align our cost structure with our new focus. Research and development expenses in 2005 decreased by $16.3 million, or 35%, compared to 2004. The decrease in research and development expenses is primarily due to a reduction in headcount and facility costs associated with the restructuring transaction with Novo Nordisk and cost reduction programs. As a result of the restructuring of the AERx iDMS program, which was completed on January 26, 2005, our development agreement with Novo Nordisk ended in 2005. This was offset by $5.9 million increase from 2004 to 2005 in self-initiated development efforts primarily relating to Intraject. In August 2006, for milestone payments and royalties, we sold all of our assets related to the Intraject technology platform to Zogenix, a newly created private company that is responsible for further development and commercialization efforts of Intraject.
We have other on-going partner-funded and self-initiated programs under development. In 2007, we expect research and development expense to increase from 2006, however, future research and development efforts for our partner-funded programs are difficult to predict at this time due to their early stage of development.
Stock based compensation expense charged to research and development in 2006 was $882,000 due to the adoption of SFAS No. 123R effective January 1, 2006.
40
Table of Contents
General and Administrative Expenses
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2005 to 2006 | 2004 to 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| General and administrative expenses | $ | 10,717 | $ | 10,895 | $ | 11,934 | (2 | )% | (9 | )% | ||||||||||
General and administrative expenses were $10.7 million in 2006 compared to $10.9 million in 2005 and $11.9 million in 2004. General and administrative expenses decreased in 2006 over 2005 by $178,000, or 2.0%, and decreased in 2005 over 2004 by $1.0 million, or 9.0%, resulting primarily from legal and consulting costs incurred in 2004 associated with the restructuring transaction with Novo Nordisk, which closed on January 26, 2005. Other than the restructuring transaction, there were no significant corporate transactions in 2006, 2005 and 2004. In 2007, we expect general and administrative expenses to decrease, compared with 2006.
Stock based compensation expense charged to general and administration in 2006 was $740,000 due to the adoption of SFAS No. 123R effective January 1, 2006.
Restructuring and Asset Impairment
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2005 to 2006 | 2004 to 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Restructuring and asset impairment expenses | $ | 6,003 | — | — | 100 | % | — | |||||||||||||
Restructuring and asset impairment expenses are comprised of severance related expenses including payroll, health insurance payments, outplacement expenses and Intraject related asset impairment expenses. Severance-related expense for the year ended December 31, 2006 was $2.0 million and is primarily related to the reduction in workforce announced on May 15, 2006. We expect to pay the severance-related balance in full by the end of 2007. The asset impairment charge of $4.0 million in 2006 is related to our estimate of the net realizable value of the Intraject-related assets, based on the sale of those assets in August 2006.
Gain on sale of patents and royalty interest
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2005 to 2006 | 2004 to 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Gain on sale of patent and royalty interest | $ | 20,000 | — | — | 100 | % | — | |||||||||||||
On July 3, 2006, we entered into a Second Amended and Restated License Agreement with Novo Nordisk A/S to reflect: (i) the transfer by us of certain intellectual property, including all right, title and interest to its patents that contain claims that pertain generally to breath control or specifically to the pulmonary delivery of monomeric insulin and monomeric insulin analogs, together with interrelated patents, which are linked via terminal disclaimers, as well as certain pending patent applications and continuations thereof for a cash payment to us of $12.0 million, with the Company retaining exclusive, royalty-free control of these patents outside the field of glucose control; (ii) a reduction by 100 basis points of each royalty rate payable by Novo Nordisk to us for a cash payment to us of $8.0 million; and (iii) a loan to us in the principal amount of $7.5 million, secured by a pledge of the net royalty stream payable to us by Novo Nordisk pursuant to the License Agreement.
The $12.0 million and the $8.0 million payments are included in gain on sale of patent and royalty interest line item for the year ended December 31, 2006. There were no similar transactions in prior years.
41
Table of Contents
Interest Income, Interest Expense and Other Income (Expense)
| Years Ended December 31, | % Increase (Decrease) | |||||||||||||||||||
| 2006 | 2005 | 2004 | 2005 to 2006 | 2004 to 2005 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Interest income, interest expense and other income (expense): | ||||||||||||||||||||
| Interest income | $ | 1,251 | $ | 1,317 | $ | 194 | (5 | )% | 579 | % | ||||||||||
| Interest expense | (197 | ) | (6 | ) | (16 | ) | 3,183 | % | (63 | )% | ||||||||||
| Other income (expense) | 23 | 36 | (1 | ) | (36 | )% | (3,700 | )% | ||||||||||||
| Total interest income, interest expense and other income (expense) | $ | 1,077 | $ | 1,347 | $ | 177 | (20 | )% | 661 | % | ||||||||||
Interest income was $1.3 million in 2006 and 2005 compared to $194,000 in 2004. The average cash and investment balances in 2006, included the receipts of proceeds of approximately $20.0 million from the sale of patents and royalty interest to Novo Nordisk in July 2006, proceeds from a $7.5 million promissory note issued to Novo Nordisk and the sale of Intraject related assets to Zogenix for $4.0 million in August 2006. The average cash and investment balances in 2005 included the receipt of net proceeds of approximately $11.7 million from a private placement of common stock in December 2004 and net proceeds of approximately $51.1 million from the closing of the restructuring transaction with Novo Nordisk in January 2005. The increase in interest income in 2005 compared to 2004 of $1.1 million was due to an increase in interest rates earned and higher average invested balances in 2005.
Interest expense was $197,000 in 2006, $6,000 in 2005 and $16,000 in 2004. Interest expense in 2006 primarily reflects the interest expense on the $7.5 million note issued to Novo Nordisk in July 2006. The note accrues interest at 5% per annum. We expect to recognize $384,000 in interest expense in 2007 related to this note.
Other income (expense) was approximately $23,000 in 2006 compared to $36,000 in 2005 and ($1,000) in 2004. The increase in 2006 over 2005 was due to a $41,000 gain in foreign currency exchange translation offset by a $13,000 net loss on sale of assets and $4,000 in accounts payable adjustments and state tax expense. The increase in other income (expenses) in 2005 compared to 2004 is due primarily to a $49,000 net gain on the sale of assets offset by a $12,000 loss on foreign currency exchange translation and $1,000 in state tax expense.
Liquidity and Capital Resources
As of December 31, 2006, we had cash, cash equivalents and short-term investments of approximately $27.5 million. On January 30, 2007 we closed our public offering of 37,950,000 shares of common stock in an underwritten public offering that resulted in proceeds of approximately $33.9 million and net proceeds, after underwriting discount and expenses, of approximately $33.3 million. This public offering triggered the automatic conversion of all outstanding shares of Series A convertible preferred stock to common stock and eliminated the Series A liquidation preference of $41.9 million.
Net cash used in operating activities in 2006 was $30.3 million compared to $34.6 million in 2005 and $23.1 million in 2004. The $4.3 million decrease in net cash used in 2006 compared to 2005 was the result of a lower net loss in 2006 compared to 2005, down $16.2 million, due primarily from proceeds of $20.0 million from the sale of patents and royalty rights to Novo Nordisk, offset by non-cash charges, including stock-based compensation expense under SFAS 123R of $1.6 million, asset impairment charge on property and equipment of $4.0 million and lower depreciation expense incurred in the 2006, down $477,000 from 2005, primarily as a result of the sale of Intraject-related assets to Zogenix. Cash was used to pay for invoices outstanding primarily related to the Intraject program, an increase of $2.4 million from 2005, to pay for severance-related expenses accrued for the reduction in workforce, an increase of $2.8 million and to fund accounts receivable, primarily related to partnered programs. The change in prepaid of $856,000 between years is primarily due to the capitalization of financing related expense. In 2006, we recognized less deferred revenue due to the sale of AERx iDMS program to Novo Nordisk completed in early 2005. The change in accrued liabilities of $687,000 is due primarily to clinical trial expenses related to our ARD-3100 program and the change in deferred rent of $1.6 million is primarily as a result of the Novo Nordisk restructuring completed in January 2005.
42
Table of Contents
The increase in net cash used in operating activities in 2005 compared to 2004 was due primarily to the result of a slightly lower net loss in 2005 compared to 2004, down $974,000, offset by lower depreciation expense incurred in the 2005, down $2.4 million from 2004 and changes in deferred revenue activity of $5.8 million and deferred rent activity of $1.8 million. The reduction in depreciation expense, the change in deferred revenue and the change in deferred rent were primarily due to the restructuring with Novo Nordisk. In 2005, we recognized less depreciation expense due to the sale of AERx iDMS program-related assets to Novo Nordisk pursuant to the restructuring transaction, recognized AERx iDMS program related funding and milestone payments received in prior years as revenue and recorded the reversal of deferred rent liability for lease obligations associated with the AERx iDMS program to the statement of operations as a credit to rent expense. Additionally, 2005 activity in accounts payable and accrued liabilities reflects a $2.0 million decrease in operating cash used when compared to 2004 activity. The smaller increase in accounts payable results primarily from liabilities for expenses associated with the private placement of equity in December 2004, the restructuring transaction with Novo Nordisk, and capital expenditures recorded at 2004 year-end.
Net cash provided by investing activities in 2006 was $21.7 million compared to $47.4 million in 2005 and $6.5 million in 2004. The lower balance in 2006 compared to 2005 is primarily due to $20.0 million in proceeds from the sale of patents and royalty rights to Novo Nordisk and $4.0 million in proceeds from the sale of Intraject related assets to Zogenix in 2006, compared to $50.3 million provided from the sale of assets to NNDT in connection with the restructuring transaction with Novo Nordisk that closed in January 2005. This was offset by a $3.5 million decrease in capital expenditures primarily due to a decrease in our Intraject clinical batch activities that were finalized in early 2006 and a $2.9 million decrease in proceeds from sales and maturities ofavailable-for-sale investments, net of purchases. The 2005 increase in cash provided from investing activities compared to 2004 is primarily due to $50.3 million provided from the sale of assets to NNDT in connection with the restructuring transaction with Novo Nordisk that closed in January 2005, off-set by $3.0 million increase in capital expenditures primarily related to Intraject and $6.6 million decrease in proceeds from sales and maturities ofavailable-for-sale investments, net of purchases.
Net cash provided by financing activities in 2006 was $7.9 million compared to $592,000 in 2005 and $12.6 million in 2004. Cash provided by financing activities in 2006 primarily represents proceeds from the $7.5 million promissory note issued to Novo Nordisk, $160,000 repayment of notes receivable from officers and $288,000 in net cash provided by purchases under our employee stock plans. Cash provided by financing activities in 2005 was from the issuance of common stock upon exercise of stock options and purchase of common stock under the employee stock purchase plan of $500,000 and repayment of notes receivable from officers and employees of $92,000. Financing activities in 2004 included the sale of common stock through private placements in December 2004, which raised net proceeds of approximately $11.7 million. In addition, net proceeds received from the issuance of common stock upon exercise of stock options and purchase of common stock under the employee stock purchase plan was $911,000, offset by $427,000 of cash used for capital lease obligations.
Our research and development efforts have and will continue to require a commitment of substantial funds to conduct the costly and time-consuming research and preclinical and clinical testing activities necessary to develop and refine our technology and proposed products and to bring any such products to market. Our future capital requirements will depend on many factors, including continued progress and the results of the research and development of our technology and drug delivery systems, our ability to establish and maintain favorable collaborative arrangements with others, progress with preclinical studies and clinical trials and the results thereof, the time and costs involved in obtaining regulatory approvals, the cost of development and the rate ofscale-up of our production technologies, the cost involved in preparing, filing, prosecuting, maintaining and enforcing patent claims, and the need to acquire licenses or other rights to new technology.
Since inception, we have financed our operations primarily through private placements and public offerings of our capital stock, proceeds from equipment lease financings, contract research funding, proceeds from the sale of assets to Novo Nordisk in connection with the restructuring transaction, including sale of patents and royalty interest and interest earned on investments.
We continue to review our planned operations through the end of 2007, and beyond. We particularly focus on capital spending requirements to ensure that capital outlays are not expended sooner than necessary. If we make
43
Table of Contents
satisfactory progress in our development programs, we would expect our cash requirements for capital spending and operations to increase in future periods. We currently expect our total capital outlays for 2007 will be approximately $3.0 million. The majority of these outlays will be associated with completing the clinical trials of our lead candidate, ARD-3100.
We have incurred significant losses and negative cash flows from operations since our inception. At December 31, 2006, we have an accumulated deficit of $287.9 million, working capital of $25.4 million, and shareholders’ deficit of $3.9 million. Management believes that cash and cash equivalents on hand at December 31, 2006 together with approximately $33.3 million in net proceeds from our completed public offering in January 30, 2007 will be sufficient to enable us to meet our obligations for the next 24 months.
Off-Balance Sheet Financings and Liabilities
Other than contractual obligations incurred in the normal course of business, we do not have any off-balance sheet financing arrangements or liabilities, guarantee contracts, retained or contingent interests in transferred assets or any obligation arising out of a material variable interest in an unconsolidated entity. We do not have any majority-owned subsidiaries.
Contractual Obligations
Our contractual obligations and future minimum lease payments that are non-cancelable at December 31, 2006 are disclosed in the following table.
| Payment Due by Period | ||||||||||||||||||||
| Less Than | After | |||||||||||||||||||
| Total | 1 Year | 1-3 Years | 3-5 Years | 5 Years | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Operating lease obligations | $ | 21,361 | $ | 2,366 | $ | 4,810 | $ | 4,245 | $ | 9,940 | ||||||||||
| Unconditional capital purchase obligations | 485 | 485 | — | — | — | |||||||||||||||
| Unconditional purchase obligations | 1,595 | 1,595 | — | — | — | |||||||||||||||
| Total contractual commitments | $ | 23,441 | $ | 4,446 | $ | 4,810 | $ | 4,245 | $ | 9,940 | ||||||||||
Recent Accounting Pronouncements
In June 2006, the Financial Accounting Standards Board issued Interpretation No. 48, Accounting for Uncertainty in Income Taxes, an interpretation of FAS 109,Accounting for Income Taxes(FIN 48), to create a single model to address accounting for uncertainty in tax positions. FIN 48 clarifies the accounting for income taxes, by prescribing a minimum recognition threshold a tax position is required to meet before being recognized in the financial statements. FIN 48 also provides guidance on derecognition, measurement, classification, interest and penalties, accounting in interim periods, disclosure and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. We will adopt FIN 48 as of January 1, 2007, as required. We are currently evaluating the effect, if any, that the adoption of FIN 48 will have on our financial statements.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements (“SFAS No. 157”). Among other requirements, SFAS No. 157 defines fair value and establishes a framework for measuring fair value and also expands disclosure about the use of fair value to measure assets and liabilities. SFAS No. 157 is effective beginning the first fiscal year that begins after November 15, 2007. We are currently evaluating SFAS No. 157 and expect to adopt this guidance beginning on January 1, 2008.
Item 7A. Quantitative and Qualitative Disclosure About Market Risk
In the normal course of business, our financial position is routinely subject to a variety of risks, including market risk associated with interest rate movement. We regularly assess these risks and have established policies and business practices intended to protect against these and other exposures. As a result, we do not anticipate material potential losses in these areas.
44
Table of Contents
As of December 31, 2006, we had cash, cash equivalents and short-term investments of $27.5 million, consisting of cash, cash equivalents and highly liquid short-term investments. Our short-term investments will likely decline by an immaterial amount if market interest rates increase and, therefore, we believe our exposure to interest rate changes is immaterial. Declines of interest rates over time will, however, reduce our interest income from short-term investments.
45
Table of Contents
| Item 8. | Financial Statements and Supplementary Data |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Shareholders of Aradigm Corporation
We have audited the accompanying balance sheets of Aradigm Corporation as of December 31, 2006 and 2005, and the related statements of operations, convertible preferred stock and shareholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2006. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Aradigm Corporation at December 31, 2006 and 2005, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2006, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the financial statements, in 2006 Aradigm Corporation changed its method of accounting for stock — based compensation in accordance with guidance provided in Statement of Financial Accounting Standards No. 123(R), “Share-Based Payment”.
/s/ Ernst & Young LLP
Palo Alto, California
March 2, 2007
46
Table of Contents
ARADIGM CORPORATION
| December 31, | ||||||||||||
| Pro Forma | ||||||||||||
| (Note 13) | ||||||||||||
| 2006 | 2006 | 2005 | ||||||||||
| (In thousands, except share data) | ||||||||||||
| (Unaudited) | ||||||||||||
ASSETS | ||||||||||||
| Current assets: | ||||||||||||
| Cash and cash equivalents | $ | 60,902 | $ | 27,013 | $ | 27,694 | ||||||
| Short-term investments | 501 | 501 | — | |||||||||
| Receivables | 643 | 643 | 400 | |||||||||
| Current portion of notes receivable from officers and employees | — | — | 62 | |||||||||
| Prepaid and other current assets | 1,002 | 1,002 | 874 | |||||||||
| Total current assets | 63,048 | 29,159 | 29,030 | |||||||||
| Property and equipment, net | 2,592 | 2,592 | 9,875 | |||||||||
| Non-current portion of notes receivable from officers and employees | 31 | 31 | 129 | |||||||||
| Other assets | 444 | 444 | 463 | |||||||||
| Total assets | $ | 66,115 | $ | 32,226 | $ | 39,497 | ||||||
LIABILITIES, CONVERTIBLE PREFERRED STOCK AND SHAREHOLDERS’ EQUITY (DEFICIT) | ||||||||||||
| Current liabilities: | ||||||||||||
| Accounts payable | $ | 1,151 | 1,151 | $ | 3,034 | |||||||
| Accrued clinical and cost of other studies | 278 | 278 | 398 | |||||||||
| Accrued compensation | 1,814 | 1,814 | 3,814 | |||||||||
| Deferred revenue | — | — | 222 | |||||||||
| Other accrued liabilities | 511 | 511 | 475 | |||||||||
| Total current liabilities | 3,754 | 3,754 | 7,943 | |||||||||
| Non-current portion of deferred rent | 1,035 | 1,035 | 714 | |||||||||
| Non-current portion of capital lease | 29 | 29 | — | |||||||||
| Note payable and accrued interest to related party | 7,686 | 7,686 | — | |||||||||
| Commitments and contingencies | ||||||||||||
| Convertible preferred stock, no par value; 2,050,000 shares authorized; issued and outstanding shares: 1,544,626 at December 31, 2006 and 2005; liquidation preference of $41,866 at December 31, 2006 and 2005; none outstanding on an unaudited pro forma basis | — | 23,669 | 23,669 | |||||||||
| Shareholders’ equity (deficit): | ||||||||||||
| Preferred stock, 2,950,000 shares authorized but none outstanding | ||||||||||||
| Common stock, no par value; authorized shares: 100,000,000 at December 31, 2006; 150,000,000 at December 31, 2005; issued and outstanding shares: 14,765,474 at December 31, 2006; 14,562,809 at December 31, 2005; and 53,951,175 on an unaudited pro forma basis | 341,472 | 283,914 | 282,004 | |||||||||
| Accumulated other comprehensive income | 4 | 4 | 5 | |||||||||
| Accumulated deficit | (287,865 | ) | (287,865 | ) | (274,838 | ) | ||||||
| Total shareholders’ equity (deficit) | 53,611 | (3,947 | ) | 7,171 | ||||||||
| Total liabilities, convertible preferred stock and shareholders’ equity (deficit) | $ | 66,115 | 32,226 | $ | 39,497 | |||||||
See accompanying Notes to Financial Statements.
47
Table of Contents
ARADIGM CORPORATION
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| (In thousands, except per share data) | ||||||||||||
| Contract and license revenues: | ||||||||||||
| Related parties | $ | 59 | $ | 8,013 | $ | 26,999 | ||||||
| Unrelated parties | 4,755 | 2,494 | 1,046 | |||||||||
| Total revenues | 4,814 | 10,507 | 28,045 | |||||||||
| Operating expenses: | ||||||||||||
| Research and development | 22,198 | 30,174 | 46,477 | |||||||||
| General and administrative | 10,717 | 10,895 | 11,934 | |||||||||
| Restructuring and asset impairment | 6,003 | — | — | |||||||||
| Total expenses | 38,918 | 41,069 | 58,411 | |||||||||
| Loss from operations | (34,104 | ) | (30,562 | ) | (30,366 | ) | ||||||
| Gain on sale of patent and royalty interest (to related party) | 20,000 | — | — | |||||||||
| Interest income | 1,251 | 1,317 | 194 | |||||||||
| Interest expense | (197 | ) | (6 | ) | (16 | ) | ||||||
| Other income (expense) | 23 | 36 | (1 | ) | ||||||||
| Net loss | $ | (13,027 | ) | $ | (29,215 | ) | $ | (30,189 | ) | |||
| Basic and diluted net loss per common share | $ | (0.89 | ) | $ | (2.01 | ) | $ | (2.37 | ) | |||
| Shares used in computing basic and diluted net loss per common share | 14,642 | 14,513 | 12,741 | |||||||||
See accompanying Notes to Financial Statements.
48
Table of Contents
ARADIGM CORPORATION
AND SHAREHOLDERS’ EQUITY (DEFICIT)
| Accumulated | Total | |||||||||||||||||||||||||||||||
| Convertible | Other | Shareholders’ | ||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Deferred | Comprehensive | Accumulated | Equity | |||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Compensation | Income (Loss) | Deficit | (Deficit) | |||||||||||||||||||||||||
| (In thousands, except share data) | ||||||||||||||||||||||||||||||||
| Balances at December 31, 2003 | 1,544,626 | $ | 23,669 | 12,550,239 | $ | 268,406 | $ | — | $ | (2 | ) | $ | (215,434 | ) | $ | 52,970 | ||||||||||||||||
| Issuance of common stock for cash, net of issuance costs of $817, including warrants valued at $2,278 | — | — | 1,666,679 | 11,683 | — | — | — | 11,683 | ||||||||||||||||||||||||
| Issuance of common stock under the employee stock purchase plan | — | — | 167,946 | 911 | — | — | — | 911 | ||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options | — | — | 81 | 1 | — | — | — | 1 | ||||||||||||||||||||||||
| Issuance of common stock upon exercise of warrants | — | — | 74,200 | 304 | — | — | — | 304 | ||||||||||||||||||||||||
| Issuance of options and warrants to purchase common stock for services | — | — | — | 82 | — | — | — | 82 | ||||||||||||||||||||||||
| Comprehensive loss: | ||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | (30,189 | ) | (30,189 | ) | ||||||||||||||||||||||
Net change in unrealized loss onavailable-for-sale investments | — | — | — | — | — | (8 | ) | — | (8 | ) | ||||||||||||||||||||||
| Total comprehensive loss | (30,197 | ) | ||||||||||||||||||||||||||||||
| Balances at December 31, 2004 | 1,544,626 | 23,669 | 14,459,145 | 281,387 | — | (10 | ) | (245,623 | ) | 35,754 | ||||||||||||||||||||||
| Issuance of common stock under the employee stock purchase plan | — | — | 93,662 | 458 | — | — | — | 458 | ||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options | — | — | 10,077 | 42 | — | — | — | 42 | ||||||||||||||||||||||||
| Adjustment to common stock shares for rounding of partial shares from the reverse stock split | — | — | (75 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Warrant revaluation | — | — | — | 90 | — | — | — | 90 | ||||||||||||||||||||||||
| Issuance of options for services | — | — | — | 27 | (21 | ) | — | — | 6 | |||||||||||||||||||||||
| Amortization of deferred compensation | — | — | — | — | 21 | — | — | 21 | ||||||||||||||||||||||||
| Comprehensive loss: | ||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | (29,215 | ) | (29,215 | ) | ||||||||||||||||||||||
Net change in unrealized gain (loss) onavailable-for-sale investments | — | — | — | — | — | 15 | — | 15 | ||||||||||||||||||||||||
| Total comprehensive loss | (29,200 | ) | ||||||||||||||||||||||||||||||
| Balances at December 31, 2005 | 1,544,626 | 23,669 | 14,562,809 | 282,004 | — | 5 | (274,838 | ) | 7,171 | |||||||||||||||||||||||
| Issuance of common stock under the employee stock purchase plan | — | — | 111,553 | 286 | — | — | — | 286 | ||||||||||||||||||||||||
| Issuance of common stock under the restricted stock award plan | — | — | 145,500 | — | — | — | — | — | ||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options | — | — | 645 | 2 | — | — | — | 2 | ||||||||||||||||||||||||
| Stock-based compensation related to issuance of stock option grants | — | — | — | 1,622 | — | — | — | 1,622 | ||||||||||||||||||||||||
| Reversal of restricted stock award due to forfeiture | — | — | (55,033 | ) | — | — | — | — | — | |||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | (13,027 | ) | (13,027 | ) | ||||||||||||||||||||||
Net change in unrealized gain (loss) onavailable-for-sale investments | — | — | — | — | — | (1 | ) | — | (1 | ) | ||||||||||||||||||||||
| Total comprehensive loss | (13,028 | ) | ||||||||||||||||||||||||||||||
| Balances at December 31, 2006 | 1,544,626 | $ | 23,669 | 14,765,474 | $ | 283,914 | $ | — | $ | 4 | $ | (287,865 | ) | $ | (3,947 | ) | ||||||||||||||||
See accompanying Notes to Financial Statements.
49
Table of Contents
ARADIGM CORPORATION
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| (In thousands) | ||||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net loss | $ | (13,027 | ) | $ | (29,215 | ) | $ | (30,189 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Non-cash asset impairment on property and equipment | 4,014 | — | — | |||||||||
| Amortization and accretion of investments | — | 50 | 166 | |||||||||
| Depreciation and amortization | 934 | 1,412 | 3,813 | |||||||||
| Stock-based compensation expense related to employee stock options and employee stock purchases | 1,622 | — | — | |||||||||
| Loss on retirement and sale of property and equipment | 164 | 268 | 544 | |||||||||
| Cost of warrants and common stock options for services | — | 117 | 82 | |||||||||
| Amortization and accretion of investments | — | 50 | — | |||||||||
| Gain on sale of patent and royalty interest | (20,000 | ) | — | — | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Receivables | (243 | ) | (301 | ) | 41 | |||||||
| Prepaid and other current assets | (128 | ) | 728 | 308 | ||||||||
| Other assets | 19 | (24 | ) | (51 | ) | |||||||
| Accounts payable | (1,883 | ) | 565 | 1,584 | ||||||||
| Accrued compensation | (2,000 | ) | 830 | 963 | ||||||||
| Accrued liabilities | 129 | (558 | ) | 439 | ||||||||
| Deferred rent | 321 | (1,229 | ) | 620 | ||||||||
| Deferred revenue | (222 | ) | (7,250 | ) | (1,440 | ) | ||||||
| Net cash used in operating activities | (30,298 | ) | (34,607 | ) | (23,120 | ) | ||||||
| Cash flows from investing activities: | ||||||||||||
| Capital expenditures | (1,829 | ) | (5,311 | ) | (2,300 | ) | ||||||
| Sales of property and equipment | 4,000 | 50,292 | — | |||||||||
Purchases ofavailable-for-sale investments | (502 | ) | (5,330 | ) | (6,376 | ) | ||||||
Proceeds from maturities ofavailable-for-sale investments | — | 7,750 | 15,190 | |||||||||
| Proceeds from sale of patents and royalty interest | 20,000 | — | — | |||||||||
| Net cash provided by investing activities | 21,669 | 47,401 | 6,514 | |||||||||
| Cash flows from financing activities: | ||||||||||||
| Proceeds from the issuance of note payable to related party | 7,500 | — | — | |||||||||
| Proceeds from issuance of common stock | 288 | 500 | 12,899 | |||||||||
| Payments received on notes receivable from officers and employees | 160 | — | — | |||||||||
| Forgiveness of notes receivable from officers and employees | — | 92 | 115 | |||||||||
| Payments on capital lease obligations and equipment loans | — | — | (427 | ) | ||||||||
| Net cash provided by financing activities | 7,948 | 592 | 12,587 | |||||||||
| Net increase (decrease) in cash and cash equivalents | (681 | ) | 13,386 | (4,019 | ) | |||||||
| Cash and cash equivalents at beginning of year | 27,694 | 14,308 | 18,327 | |||||||||
| Cash and cash equivalents at end of year | $ | 27,013 | $ | 27,694 | $ | 14,308 | ||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Cash paid for interest | $ | 11 | $ | 6 | $ | 16 | ||||||
| Non-cash investing and financing activities: | ||||||||||||
| Issuance of options and warrants to purchase common stock for services | — | 117 | 82 | |||||||||
| Issuance of warrants with private placement of common stock | — | — | 2,278 | |||||||||
See accompanying Notes to Financial Statements.
50
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS
| 1. | Organization and Summary of Significant Accounting Policies |
Organization
Aradigm Corporation (the “Company”) is a California corporation focused on the development and commercialization of a portfolio of drugs delivered by inhalation for the treatment of severe respiratory diseases by pulmonologists. The Company’s principal activities to date have included obtaining financing, recruiting management and technical personnel, securing operating facilities, conducting research and development, and expanding commercial production capabilities. The Company does not anticipate receiving any revenues from the sale of products in the upcoming year. The Company operates as a single operating segment.
Liquidity and Financial Condition
The Company has incurred significant losses and negative cash flows from operations since its inception. At December 31, 2006, the Company had an accumulated deficit of $287.9 million and working capital of $25.4 million and shareholders’ deficit of $3.9 million. Management believes that cash, cash equivalents and short-term investments on hand at December 31, 2006, together with proceeds from the public offering completed on January 30, 2007, (See Note 13) will be sufficient to enable the Company meet its obligations for the next 24 months. Management plans to continue to fund the Company with funds obtained through collaborative arrangements, equity issuancesand/or debt arrangements.
Use of Estimates
The preparation of financial statements in conformity with United States generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. These estimates include useful lives for property and equipment and related depreciation calculations, estimated amortization period for payments received from product development and license agreements as they relate to the revenue recognition of deferred revenue and assumptions for valuing options and warrants. Actual results could differ from these estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with a maturity of three months or less from purchase date to be cash equivalents. The Company places its cash and cash equivalents in money market funds, commercial paper and corporate notes.
Investments
Management determines the appropriate classification of the Company’s marketable securities, which consist solely of debt securities, at the time of purchase. All marketable securities are classified asavailable-for-sale, carried at estimated fair value and reported in either cash equivalents or short-term investments. Unrealized gains and losses onavailable-for-sale securities are excluded from earnings and reported as a separate component in the statement of convertible preferred stock and shareholders’ equity until realized. Fair values of investments are based on quoted market prices where available. Interest income is recognized when earned and includes interest, dividends, amortization of purchase premiums and discounts, and realized gains and losses on sales of securities. The cost of securities sold is based on the specific identification method. The Company regularly reviews all of its investments forother-than-temporary declines in fair value. When the Company determines that the decline in fair value of an investment below the Company’s accounting basis isother-than-temporary, the Company reduces the carrying value of the securities held and records a loss in the amount of any such decline. No such reductions have been required during any of the periods presented.
51
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Notes Receivable
Notes receivable are related to advances granted to employees for relocation. All amounts classified as current are due within 12 months. All amounts classified as long-term are due no later than April 2008. All balances were forgivenand/or paid as of December 31, 2006.
Property and Equipment
The Company records property and equipment at cost and calculates depreciation using the straight-line method over the estimated useful lives of the respective assets. Machinery and equipment includes external costs incurred for validation of the equipment. The Company does not capitalize internal validation expense. Computer equipment and software includes capitalized computer software. All of the Company’s capitalized software is purchased; the Company has not internally developed computer software. Leasehold improvements are depreciated over the shorter of the term of the lease or useful life of the improvement.
The estimated useful lives of property and equipment are as follows:
| Machinery and equipment | 5 to 7 years | |||
| Furniture and fixtures | 5 to 7 years | |||
| Lab equipment | 5 to 7 years | |||
| Computer equipment and software | 3 to 5 years | |||
| Leasehold improvements | 5 to 17 years |
Impairment of Long-Lived Assets
In accordance with Statement of Financial Accounting Standards (“SFAS”) No. 144, “Accounting for the Impairment or Disposal of Long-Lived Assets,” the Company reviews for impairment whenever events or changes in circumstances indicate that the carrying amount of property and equipment may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values and the loss is recognized in the Statements of Operations. The Company recorded an impairment charge of $4.0 million in 2006 related to the anticipated sale of Intraject related assets (see Note 11).
Revenue Recognition
Contract revenues consist of revenues from grants, collaboration agreements and feasibility studies. The Company recognizes revenue under the provisions of the Securities and Exchange Commission issued Staff Accounting Bulletin No. 104, “Revenue Recognition.” Under the agreements, revenue is recognized once costs are incurred and collectibility is reasonably assured. Under some agreements the Company’s collaborators have the right to withhold reimbursement of costs incurred until the work performed under the agreement is mutually agreed upon. For these agreements revenue is recognized upon confirmation from the collaborator of acceptance of work performed and payment amount. Deferred revenue represents the portion of all refundable and nonrefundable research payments received that have not been earned. In accordance with contract terms, milestone payments from collaborative research agreements are considered reimbursements for costs incurred under the agreements and, accordingly, are generally recognized as revenue either upon the completion of the milestone effort, when payments are contingent upon completion of the effort, or are based on actual efforts expended over the remaining term of the agreements when payments precede the required efforts. Costs of contract revenues are approximate to or are greater than such revenues and are included in research and development expenses. Refundable development and license fee payments are deferred until the specified performance criteria are achieved. Refundable development and license fee payments are generally not refundable once the specific performance criteria are achieved and accepted.
52
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Research and Development
Research and development expenses consist of costs incurred for company-sponsored, collaborative and contracted research and development activities. These costs include direct and research-related overhead expenses. Research and development expenses under collaborative and government grants approximate the revenue recognized under such agreements. The Company expenses research and development costs as such costs are incurred.
Advertising
Advertising costs are charged to general and administrative expense as incurred. Advertising expenses for the years ended December 31, 2006, 2005 and 2004 were $39,000, $265,000 and $233,000, respectively.
Stock-Based Compensation
Prior to January 1, 2006, the Company had elected to follow Accounting Principles Board (“APB”) Opinion No. 25, “Accounting for Stock Issued to Employees” (“APB 25”), and related interpretations in accounting for its employee stock options. Compensation expense was based on the difference, if any, between the fair value of the Company’s common stock and the exercise price of the option or share right on the measurement date, which is typically the date of grant. In accordance with SFAS 123, “Accounting for Stock-Based Compensation,” (“SFAS 123”) as amended by SFAS 148, “Accounting for Stock-Based Compensation — Transition and Disclosure,” the Company has provided below the pro forma disclosures of the effect on net loss and loss per share as if SFAS 123 had been applied in measuring compensation expense for all periods presented (in thousands, except per share data).
| Years Ended December 31, | ||||||||
| 2005 | 2004 | |||||||
| Net loss — as reported | $ | (29,215 | ) | $ | (30,189 | ) | ||
| Add: | ||||||||
| Stock-based employee compensation expense included in reported net loss | 21 | — | ||||||
| Less: | ||||||||
| Total stock-based employee compensation expense determined under fair value based method for all awards | (3,066 | ) | (4,585 | ) | ||||
| Pro forma net loss | $ | (32,260 | ) | $ | (34,774 | ) | ||
| Basic and diluted net loss per common share: | ||||||||
| As reported | $ | (2.01 | ) | $ | (2.37 | ) | ||
| Pro forma | $ | (2.22 | ) | $ | (2.73 | ) | ||
Valuation assumptions
Pro forma information regarding net loss and basic and diluted net loss per common share prepared in accordance with SFAS 123, as amended, has been determined as if the Company had accounted for its employee and non-employee director stock options granted using the fair value method prescribed by this statement. The fair
53
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
value of options was estimated at the date of grant using the Black-Scholes option pricing model with the following assumptions:
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
Employee Stock Options | ||||||||||||
| Dividend yield | 0 | .0% | 0 | .0% | 0 | .0% | ||||||
| Volatility factor | 86 | .6% | 97 | .6% | 98 | .0% | ||||||
| Risk-free interest rate | 4 | .9% | 3 | .8% | 3 | .1% | ||||||
| Expected life (years) | 4 | .2 | 4 | .0 | 4 | .0 | ||||||
| Weighted-average fair value of options granted during the periods | $1 | .40 | $4 | .10 | $5 | .50 | ||||||
The Company accounts for options and warrants issued to non-employees under SFAS 123 and Emerging Issues Task Force Issue No. (“EITF”)96-18, “Accounting for Equity Instruments that are Issued to Other than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,” using the Black-Scholes option pricing model. The value of such non-employee options and warrants are periodically re-measured over their vesting terms. The fair value of options and warrants was remeasured at period-end using the Black-Scholes option pricing model with the following assumptions: a risk-free interest rate of 2.0% to 4.9%, using applicable United States Treasury rates; a dividend yield of 0.0%; an annual volatility factor of 87% to 98%; and an average expected life based on the terms of the option grant or contractual term of the warrant of 1 to 4.2 years. Expense recognized related to options and warrants issued to non-employees was $130, $117,000 and $82,000 during the years ended December 31, 2006, 2005 and 2004, respectively.
Adoption of SFAS No. 123R
The Company adopted the fair value recognition provisions of SFAS 123(R) (revised 2004), “Share-based Payment,” (“SFAS 123(R)”) effective January 1, 2006. Stock-based compensation expense is based on the fair value of that portion of employee stock options that are ultimately expected to vest during the period. Stock-based compensation expense recognized in our statement of operations during 2006 included compensation expense for stock-based awards granted prior to, but not yet vested as of, December 31, 2005, based on the grant date fair value estimated in accordance with the pro forma provisions of SFAS 123, and share-based payment awards granted subsequent to December 31, 2005 based on the grant date fair value estimated in accordance with SFAS 123(R). For stock options granted after January 1, 2006, the fair value of each award is amortized using the straight-line single-option method. For share awards granted prior to 2006, the fair value of each award is amortized using the accelerated multiple-option valuation method prescribed by SFAS 123. Stock-based compensation expense is based on awards ultimately expected to vest, therefore, it has been reduced for estimated forfeitures. SFAS 123(R)requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company estimated forfeitures based on historical experience. In the information required under SFAS 123 for the periods prior to 2006, the Company accounted for forfeitures as they occurred.
In November 2005, the FASB issued FASB Staff Position No.FAS123(R)-3, “Transition Election Related to Accounting for the Tax Effects ofShare-Based Payment Awards”. We have adopted the simplified method to calculate the beginning balance of the additionalpaid-in-capital (“APIC”) pool of the excess tax benefit, and to determine the subsequent impact on the APIC pool and our Statements of Cash Flows of the tax effects of employeestock-based compensation awards that were outstanding upon our adoption of SFAS 123(R).
54
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
The following table shows the effect of SFAS 123(R) on stock-based employee compensation expense included in the statement of operations for the year ended December 31, 2006 (in thousands except per share amount):
| December 31, | ||||
| 2006 | ||||
| Costs and expenses: | ||||
| Research and development | $ | 882 | ||
| General and administrative | 740 | |||
| Total stock-based compensation expense | $ | 1,622 | ||
| Impact on basic and diluted net loss per common share | $ | (0.11 | ) | |
There was no capitalized stock-based employee compensation cost as of December 31, 2006. Since the Company incurred net losses in 2006, there was no recognized tax benefit as of December 31, 2006 associated with stock-based compensation expense. Total compensation expense for restricted stock awards recognized by the Company under SFAS No. 123R was $85,000 for the year ended December 31, 2006 and 69,849 shares subject to restricted share awards are issued and outstanding. As of December 31, 2006, $241,000 of total unrecognized compensation costs, net of forfeitures, related to non-vested awards is expected to be recognized over a weighted average period of 1.64 years.
For restricted common stock issued at discounted prices, the Company recognizes compensation expense over the vesting period for the difference between the exercise or purchase price and the fair market value on the measurement date.
During the year ended December 31, 2006, the Company granted options to purchase approximately 2,498,000 shares of common stock, with an estimated weighted-average fair value of $1.39 per share, respectively, on the date of grant.
Total compensation expense for options recognized by the Company under SFAS No. 123(R), was $1.4 million for the year ended December 31, 2006. The weighted-average period over which compensation expense related to options outstanding at December 31, 2006 is expected to be recognized is 1.41 years.
Income Taxes
The Company uses the liability method to account for income taxes as required by SFAS 109, “Accounting for Income Taxes.” Under this method, deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
Net Loss Per Common Share
Basic net loss per common share on a historical basis is computed using the weighted-average number of shares of common stock outstanding less the weighted-average number of shares subject to repurchase. There were no shares subject to repurchase in the years ended December 31, 2006, 2005 and 2004. Stock options, warrants, unvested restricted stock and shares to be issued upon conversion of the convertible preferred stock were not included in the net loss per share calculation for the years ended December 31, 2006, 2005 and 2004 because the inclusion of such shares would have had an anti-dilutive effect.
55
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Potentially dilutive securities include the following (in thousands):
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| Outstanding stock options | 3,064 | 1,730 | 1,883 | |||||||||
| Unvested restricted stock | 70 | — | — | |||||||||
| Warrants to purchase common stock | 1,255 | 2,120 | 2,566 | |||||||||
| Convertible preferred stock | 1,236 | 1,236 | 1,236 | |||||||||
Significant Concentrations
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash, cash equivalents and short-term investments. Risks associated with these instruments are mitigated by banking with and only purchasing commercial paper from creditworthy institutions. The maximum amount of loss due to credit risk associated with these financial instruments is their respective fair values as stated in the balance sheet.
The Company has development arrangements with various collaborators. For the years ended December 31, 2005 and 2004, the Novo Nordisk AERx iDMS program contributed approximately 76% and 96% of total contract revenues, respectively and $59,000 in 2006. In January 2005, the Company completed the restructuring of the AERx iDMS program, pursuant to the Restructuring Agreement entered into with Novo Nordisk A/S (“Novo Nordisk”) and Novo Nordisk Delivery Technologies, Inc. (“NNDT”) in September 2004. Under the current agreements between Novo Nordisk and the Company, completed on July 3, 2006, Novo Nordisk has assumed responsibility for the completion of development, manufacturing and commercialization of the AERx iDMS insulin product. The Company will be entitled to receive royalties that will rise to an average of five percent or higher by the fifth year after commercialization on any future sales of the commercialized product. Novo Nordisk, a company publicly traded in Denmark, is considered to be a related party due to its ownership interest in the Company. Novo Nordisk owned approximately 9.8% of the Company’s common stock on an as-converted basis as of December 31, 2006. Pursuant to the Company’s public offering completed on January 30, 2007, Novo Nordisk owned approximately 3.0% of the Company’s stock on an as-converted basis.
Comprehensive Income (Loss)
SFAS 130, “Reporting Comprehensive Income,” requires unrealized gains or losses on the Company’savailable-for-sales securities to be recorded in other comprehensive income (loss). Total comprehensive loss has been disclosed on the statement of redeemable convertible preferred stock and shareholders’ equity (deficit).
Recently Issued Accounting Pronouncements
In June 2006, the Financial Accounting Standards Board issued Interpretation No. 48, Accounting for Uncertainty in Income Taxes, an interpretation of FAS 109,Accounting for Income Taxes(FIN 48), to create a single model to address accounting for uncertainty in tax positions. FIN 48 clarifies the accounting for income taxes, by prescribing a minimum recognition threshold a tax position is required to meet before being recognized in the financial statements. FIN 48 also provides guidance on de-recognition, measurement, classification, interest and penalties, accounting in interim periods, disclosure and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006. The Company will adopt FIN 48 as of January 1, 2007, as required. Management currently evaluating the effect, if any, that the adoption of FIN 48 will have on our financial statements
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements (“SFAS No. 157”). Among other requirements, SFAS No. 157 defines fair value and establishes a framework for measuring fair value and also expands disclosure about the use of fair value to measure assets and liabilities. SFAS No. 157 is effective beginning the first fiscal year that begins after November 15, 2007. We are evaluating the impact of SFAS No. 157 on our financial position, results of operations and cash flows.
56
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 2. | Cash and Cash Equivalents and Investments |
The following summarizes the fair value of cash and cash equivalents and investments (amounts in thousands):
| December 31, | ||||||||
| 2006 | 2005 | |||||||
| Cash equivalents: | ||||||||
| Money market fund | $ | 1,248 | $ | 1,321 | ||||
| Commercial paper | 25,765 | 26,373 | ||||||
| $ | 27,013 | $ | 27,694 | |||||
| Short-term investments: | ||||||||
| Corporate and government notes | $ | 501 | — | |||||
The Company places its cash and cash equivalents in money market funds, commercial paper and corporate and government notes. All short-term investments at December 31, 2006 and 2005 mature in less than one year. Unrealized holding gains and losses on securities classified asavailable-for-sale are recorded in accumulated other comprehensive income. As of December 31, 2006 and 2005 the difference between the fair value and amortized cost ofavailable-for-sale securities were gains of $4,000 and $5,000 respectively. The individual gross unrealized gains and individual gross unrealized losses for 2006 and 2005 were not material.
| 3. | Property and Equipment |
Property and equipment consist of the following (amounts in thousands):
| December 31, | ||||||||
| 2006 | 2005 | |||||||
| Machinery and equipment | $ | 3,905 | $ | 4,505 | ||||
| Furniture and fixtures | 1,142 | 1,150 | ||||||
| Lab equipment | 2,658 | 2,539 | ||||||
| Computer equipment and software | 3,798 | 3,790 | ||||||
| Leasehold improvements | 1,068 | 1,564 | ||||||
| Property and equipment at cost | 12,571 | 13,548 | ||||||
| Less accumulated depreciation and amortization | (10,204 | ) | (10,201 | ) | ||||
| Net depreciable assets | 2,367 | 3,347 | ||||||
| Construction in progress | 225 | 6,528 | ||||||
| Property and equipment, net | $ | 2,592 | $ | 9,875 | ||||
Depreciation expense was $934,000, $1.4 million and $3.8 million in 2006, 2005 and 2004, respectively.
| 4. | Leases, Commitments and Contingencies |
The Company has a lease for a building containing office, laboratory and manufacturing facilities, which expires in 2016. A minor portion of this lease expense was offset by a sublease to NNDT of $10,000 per month through December 2006. Additionally, the Company entered into a new copier lease agreement in July 2005 for
57
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
$5,030 per month for 60 months. Future minimum lease payments non-cancelable at December 31, 2006 for the remaining lease agreements are as follows (amounts in thousands):
| Operating | ||||
| Leases | ||||
| Year ending December 31: | ||||
| 2007 | $ | 2,366 | ||
| 2008 | 2,432 | |||
| 2009 | 2,378 | |||
| 2010 | 2,253 | |||
| 2011 | 1,992 | |||
| 2012 and thereafter | 9,940 | |||
| Total minimum lease payments | $ | 21,361 | ||
The Company’s operating lease has a rent escalation clause and, accordingly, the Company recognizes rent expense on a straight-line basis. At December 31, 2006 and 2005, the Company had $1.0 million and $714,000 of deferred rent, respectively. A portion of the lease commitment for 2006 is offset by a sublease to NNDT of $10,000 per month through December 2006 and a sublease to Zogenix of $7,000 per month starting in September through December 2006. As a result of the restructuring activities, the Company has consolidated its operations to a portion of the space at its current address and are actively investigating sublease opportunities for the vacated space.
For the years ended December 31, 2006, 2005 and 2004, building rent expense, net of sublease income, under operating leases totaled $1.9 million, $1.3 million and $5.5 million, respectively.
At December 31, 2006, the Company had contractual non-cancelable purchase commitments for capital equipment purchases of $485,000 and for services of $1.6 million.
Indemnification
The Company from time to time enters into contracts that contingently require the Company to indemnify parties against third party claims. These contracts primarily relate to: (i) real estate leases, under which the Company may be required to indemnify property owners for environmental and other liabilities, and other claims arising from the Company’s use of the applicable premises, and (ii) agreements with the Company’s officers, directors and employees, under which the Company may be required to indemnify such persons from certain liabilities arising out of such persons’ relationships with the Company. To date, the Company has made no payments related to such indemnifications and no liabilities have been recorded for these obligations on the balance sheets at December 31, 2006 or 2005.
Legal Matters
From time to time, the Company is involved in litigation arising out of the ordinary course of its business. Currently there are no known claims or pending litigation expected to have a material effect on the Company’s overall financial position, results of operations, or liquidity.
| 5. | Convertible Preferred Stock and Common Stock Warrants |
The Company completed a $48.4 million preferred stock financing in December 2001. Under the terms of the financing the Company sold to a group of investors 2,001,236 shares of Series A convertible preferred stock (“preferred stock”) at a purchase price of $24.20 per share. Each share of preferred stock, together with accrued and unpaid dividends, is convertible at the option of the holder into 0.8 shares of common stock. Each share of outstanding preferred stock will automatically convert into common stock upon either the closing of a registered
58
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
underwritten public offering covering the offer and sale of common stock with gross proceeds (before underwriting discounts, commissions and fees) to the Company exceeding $25.0 million or the date on which the common stock closing bid price has been above $52.9375 per share for at least 20 consecutive trading days. The Company also issued warrants to the investors to purchase approximately 1,040,642 shares of common stock at an exercise price of $34.85 per share. Issuance costs of approximately $3.0 million were accounted for as a reduction to proceeds from the preferred stock financing. The warrants were exercisable through December 2006. In March, June and July of 2003, certain holders of shares of the Company’s preferred stock elected to convert an aggregate of 456,610 shares of preferred stock to common stock. The Company issued 365,288 shares of common stock in connection with those conversions. There were no dividends declared as of December 31, 2006, 2005 or 2004.
Pursuant to the completion of the Company’s public offering on January 30, 2007, the remainder of the preferred stock was automatically converted to shares of common stock at a conversion ratio of 0.8 shares of common stock for each share of preferred stock.
| 6. | Shareholders’ Equity |
In a private placement in December 2004, the Company issued 1,666,679 shares of common stock at a price of $7.50 per share and warrants to purchase 416,669 shares of common stock at $10.50 per share, for aggregate consideration of approximately $12.5 million. The warrants are exercisable at the election of the warrant holders for a four-year term. The Company valued the warrants as of December 2004, the date of financing, using the Black-Scholes option pricing model using the following assumptions: estimated volatility of 88%, risk-free interest rate of 3.6%, no dividend yield, and an expected life of four years, and recorded approximately $2.3 million as issuance costs related to the private placement. These warrants are exercisable through December 2008.
In November 2003, the Company issued 1,556,110 shares of common stock at $9.00 per share and warrants to purchase 389,027 shares of common stock at $12.50 per share to certain investors for an aggregate purchase price of approximately $14.0 million in a private placement. The warrants are exercisable at the election of the warrant holders for a four-year term. The Company valued the warrants as of November 2003, the date of financing, using the Black-Scholes option pricing model using the following assumptions: estimated volatility of 88%, risk-free interest rate of 2.5%, no dividend yield, and an expected life of four years, and recorded approximately $2.6 million as issuance costs related to the private placement. These warrants are exercisable through November 2007.
In March 2003, the Company issued 3,798,478 shares of common stock at $3.95 per share and warrants to purchase 854,654 shares of common stock at $5.35 per share to certain investors for an aggregate purchase price of approximately $15.0 million in a private placement. The warrants are exercisable at the election of the warrant holders for a four-year term. The Company valued the warrants as of March 2003, the date of financing, using the Black-Scholes option pricing model using the following assumptions: estimated volatility of 84%, risk-free interest rate of 2.5%, no dividend yield, and an expected life of four years, and recorded approximately $1.9 million as issuance costs related to the private placement. In addition, in connection with this private placement and as an inducement for investors to purchase shares of common stock, the Company issued warrants (“replacement warrants”) to purchase an aggregate of 803,205 shares of its common stock at $5.60 per share to certain of the investors in the private placement in exchange for the cancellation of an equal number of warrants to purchase shares of the common stock at $34.85 per share, held by the same investors. The Company valued the replacement warrants as of March 2003, the date of the replacement, using the Black-Scholes option pricing model using the following assumptions: estimated volatility of 84%, risk-free interest rate of 2.5%, no dividend yield, and an expected life of 3.8 years, and recorded an additional $1.1 million as issuance costs related to the private placement. As of December 31, 2006, 418,896 of these warrants are exercisable through March 2007.
In June 2004, the Company filed a Certificate of Amendment to the Company’s Amended and Restated Articles of Incorporation with the Secretary of State of the State of California to increase the Company’s authorized number of shares of common stock from 100,000,000 to 150,000,000 shares. The additional shares of common stock authorized by the amendment have rights identical to the common stock of the Company outstanding
59
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
immediately before the filing of the amendment. Issuances of common stock from the additional authorized shares do not affect the rights of the holders of the Company’s common stock and preferred stock outstanding immediately before the filing of the amendment, except for effects that may be incidental to increasing the number of shares of the Company’s common stock outstanding, such as dilution of any earnings per share and voting rights of holders of other common stock.
In January 2006, the Company filed a Certificate of Amendment to the Company’s Amended and Restated Articles of Incorporation with the Secretary of State of the State of California to decrease the Company’s authorized number of shares of common stock from 150,000,000 to 100,000,000 shares.
Reverse Stock Split
On January 4, 2006, the Company filed a Certificate of Amendment to the Company’s Amended and Restated Articles of Incorporation with the Secretary of State of the State of California effecting a1-for-5 reverse split of the Company’s common stock. All share and per share amounts have been retroactively restated in the financial statements and these accompanying notes for all periods presented.
Reserved Shares
At December 31, 2006, the Company had 1,394,002 shares of our common stock reserved for issuance of new grants, 1,254,592 shares of common stock reserved for issuance upon exercise of common stock warrants, 3,063,981 shares reserved for issuance upon exercise of options under all plans, 1,235,701 shares reserved for issuance upon conversion of preferred stock, 100,000 shares reserved for issuance upon vesting of Igor Gonda’s performance bonus and 340,847 available authorized shares under the Employee Stock Purchase Plan. Pursuant to the completion of public offering in January 30, 2007, the automatic conversion of all outstanding preferred stock resulted in the issuance of 1,235,701 common stock.
Other Common Stock Warrants
In January 2004, the Company amended the payment terms of the operating lease for its primary offices. In consideration for the amended lease agreement, Aradigm replaced common stock warrants to purchase 27,000 shares of common stock at $50.80 — $108.60 per share with new common stock warrants with an exercise price equal to $8.55 per share. The $88,000 incremental fair value of the replacement warrants, as defined as the fair value of the new warrant less the fair value of the old warrant on date of replacement, is being amortized to operating expenses on a straight-line basis over the remaining life of the lease. The fair value of the warrants was measured as of January 2004, the date of the amendment, using the Black-Scholes option pricing model with the following assumptions: risk-free interest rates between 1.3% and 2.4%; a dividend yield of 0.0%; annual volatility factor of 88%; and a weighted average expected life based on the contractual term of the warrants from 1 to 3.5 years. As of December 31, 2006, 5,000 of these warrants are exercisable through July 2007.
In October 2002, the Company issued warrants in connection with a financial relations service agreement that entitles the holder to purchase 15,000 shares of common stock, 5,000 of which are exercisable at $9.95 per share, 5,000 shares of which are exercisable at $11.95 per share and 5,000 shares of which are exercisable at $13.95 per share. At the execution of the agreement 3,000 shares immediately vested and the remaining shares shall vest based on the achievement of various performance benchmarks set forth in the agreement: all benchmarks were achieved as of March 2004. The Company valued the warrants as of October 2002, the date of agreement, using the Black-Scholes option pricing model using the following assumptions: estimated volatility of 88%, risk-free interest rate of 2.0%, no dividend yield, and an expected life of four years. The fair value of these warrants is re-measured as the underlying warrants vest and is being expensed over the vesting period of the warrants. For the year ended December 31, 2003 the Company recognized $77,000 of expense associated with these warrants. In the year ended December 31, 2004, due to all benchmarks being achieved during the year, the Company reversed previously recognized expense of $48,000. The warrants are exercisable through October 2007.
60
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
1996 Equity Incentive Plan, 2005 Equity Incentive Plan and 1996 Non-Employee Directors’ Plan
The 1996 Equity Incentive Plan (the “1996 Plan”) and the 2005 Equity Incentive Plan (the “2005 Plan”), which amended, restated and retitled the 1996 Plan, were adopted to provide a means by which officers, non-employee directors, scientific advisory board members and employees of and consultants to the Company and its affiliates could be given an opportunity to acquire an equity interest in the Company. All officers, non-employee directors, scientific advisory board members and employees of and consultants to the Company are eligible to participate in the 2005 Plan.
In April 1996, the Company’s Board of Directors adopted and the Company’s shareholders approved the 1996 Plan, which amended and restated an earlier stock option plan. The 1996 Plan reserved 960,000 shares for future grants. During May 2001, the Company’s shareholders approved an amendment to the Plan to include an evergreen provision. In 2003, the 1996 Plan was amended, to increase the maximum number of shares available for issuance under the evergreen feature of the 1996 Plan by 400,000 shares to 2,000,000 shares. The evergreen provision automatically increased the number of shares reserved under the 1996 Plan, subject to certain limitations, by 6% of the issued and outstanding shares of common stock of the Company or such lesser number of shares as determined by the board of directors on the date of the annual meeting of shareholders of each fiscal year beginning 2001 and ending 2005.
Options granted under the 1996 Plan may be immediately exercisable if permitted in the specific grant approved by the Company’s board of directors and, if exercised early, the issued shares may be subject to repurchase provisions. The shares acquired generally vest over a period of four years from the date of grant. The 1996 Plan also provides for a transition from employee to consultant status without termination of the vesting period as a result of such transition. Any unvested stock issued is subject to repurchase agreements whereby the Company has the option to repurchase unvested shares upon termination of employment at the original issue price. The common stock subject to repurchase has voting rights but does not have resale rights prior to vesting. The Company has repurchased a total of 7,658 shares in accordance with these agreements through December 31, 1998. Subsequently, no grants with early exercise provisions have been made under the 1996 Plan and no shares have been repurchased. During 2005, the Company granted options to purchase 279,420 shares of common stock under the 1996 Plan. As of December 31, 2005, the Company had 1,662,883 options outstanding under the 1996 Plan.
In March 2005, the Company’s board of directors adopted and in May 2005 the Company’s shareholders approved the 2005 Plan, which amended, restated and retitled the 1996 Plan. All outstanding awards granted under the 1996 Plan remain subject to the terms of the 1996 Plan. All stock awards granted on or after the adoption date are subject to the terms of the 2005 Plan. No shares were added to the share reserve under the 2005 Plan other than the shares available for future issuance under the 1996 Plan. Pursuant to the 2005 Plan, the Company had 2,918,638 shares of common stock authorized for issuance. Options (net of canceled or expired options) covering an aggregate of 1,999,252 shares of the Company’s Common Stock had been granted under the 1996 Plan, and 919,386 shares became available for future grant under the 2005 Plan. In March 2006 the Company’s board of directors amended and in May 2006 the Company’s shareholders approved the amendment to the 2005 Plan, increasing the shares of common stock authorized for issuance by 2,000,000. As of December 31, 2006, 1,394,002 shares remained available for future grant.
Options granted under the 2005 Plan expire no later than 10 years from the date of grant. Options granted under the 2005 Plan may be either incentive or non-statutory stock options. For incentive and non-statutory stock option grants, the option price shall be at least 100% and 85%, respectively, of the fair value on the date of grant, as determined by the Company’s board of directors. If at any time the Company grants an option, and the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of stock of the Company, the option price shall be at least 110% of the fair value and shall not be exercisable more than five years after the date of grant.
61
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Options granted under the 2005 Plan may be immediately exercisable if permitted in the specific grant approved by the board of directors and, if exercised early may be subject to repurchase provisions. The shares acquired generally vest over a period of four years from the date of grant. The 2005 Plan also provides for a transition from employee to consultant status without termination of the vesting period as a result of such transition. Under the 2005 Plan, employees may exercise options in exchange for a note payable to the Company, if permitted under the applicable grant. As of December 31, 2006 there were no outstanding notes receivable from shareholders. Any unvested stock issued is subject to repurchase agreements whereby the Company has the option to repurchase unvested shares upon termination of employment at the original issue price. The common stock subject to repurchase has voting rights but cannot be resold prior to vesting. No grants with early exercise provisions have been made under the 2005 Plan and no shares have been repurchased. During 2005, the Company granted options to purchase 46,040 shares of common stock and 2,498,000 shares in 2006, under the 2005 Plan.
The 1996 Non-Employee Directors’ Stock Option Plan (the “Directors’ Plan”) had 45,000 shares of common stock authorized for issuance. Options granted under the Directors’ Plan expire no later than 10 years from date of grant. The option price shall be at 100% of the fair value on the date of grant as determined by the board of directors. The options generally vest quarterly over a period of one year. During 2000, the board of directors approved the termination of the Directors’ Plan. No more options can be granted under the plan after its termination. The termination of the Directors’ Plan had no effect on the options already outstanding. There was no activity in the Directors’ Plan during the year ended December 31, 2006 and, as of December 31, 2005 and 2006, 21,186 outstanding options with exercise prices ranging from $41.25 — $120.63 remained with no additional shares available for grant.
62
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
The following is a summary of activity under the 1996 Plan, the 2005 Plan and the Directors’ Plan as of December 31, 2006:
| Options Outstanding | ||||||||||||||||||||||||
| Weighted | ||||||||||||||||||||||||
| Shares Available | Average | |||||||||||||||||||||||
| for Grant of | Number of | Exercise | ||||||||||||||||||||||
| Option | Shares | Price per Share | Price | |||||||||||||||||||||
| Balance at December 31, 2003 | 599,704 | 1,296,765 | $ | 1.85 | - | $ | 120.65 | $ | 31.15 | |||||||||||||||
| Options authorized | 762,774 | — | — | — | ||||||||||||||||||||
| Options granted | (697,760 | ) | 697,760 | $ | 3.80 | - | $ | 12.00 | $ | 7.75 | ||||||||||||||
| Options exercised | — | (81 | ) | $ | 4.75 | - | $ | 7.10 | $ | 6.20 | ||||||||||||||
| Options cancelled | 111,274 | (111,274 | ) | $ | 1.85 | - | $ | 117.85 | $ | 31.60 | ||||||||||||||
| Balance at December 31, 2004 | 775,992 | 1,883,170 | $ | 2.15 | - | $ | 120.65 | $ | 22.20 | |||||||||||||||
| Options authorized | — | — | — | — | ||||||||||||||||||||
| Options granted | (325,460 | ) | 325,460 | $ | 4.30 | - | $ | 7.95 | $ | 5.97 | ||||||||||||||
| Options exercised | — | (10,077 | ) | $ | 2.17 | - | $ | 4.75 | $ | 4.39 | ||||||||||||||
| Adjustment for rounding for reverse stock split | — | 10 | — | — | ||||||||||||||||||||
| Options cancelled | 468,854 | (468,854 | ) | $ | 2.83 | - | $ | 120.63 | $ | 21.84 | ||||||||||||||
| Balance at December 31, 2005 | 919,386 | 1,729,709 | $ | 2.83 | - | $ | 120.63 | $ | 19.47 | |||||||||||||||
| Options authorized | 2,000,000 | — | — | — | ||||||||||||||||||||
| Options granted | (2,498,000 | ) | 2,498,000 | $ | 1.02 | - | $ | 3.77 | $ | 2.11 | ||||||||||||||
| Options exercised | — | (645 | ) | $ | 2.83 | $ | 2.83 | |||||||||||||||||
| Restricted stock awards granted | (145,500 | ) | — | — | — | |||||||||||||||||||
| Performance Bonus stock award granted | (100,000 | ) | — | — | — | |||||||||||||||||||
| Options cancelled | 1,163,083 | (1,163,083 | ) | $ | 1.64 | - | $ | 115.00 | $ | 10.03 | ||||||||||||||
| Restricted share awards cancelled | 55,033 | — | — | — | ||||||||||||||||||||
| Balance at December 31, 2006 | 1,394,002 | 3,063,981 | $ | 1.02 | - | $ | 120.63 | $ | 8.90 | |||||||||||||||
63
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
The following table summarizes information about stock options outstanding and exercisable as of December 31, 2006:
| Options Outstanding | ||||||||||||||||||||
| Weighted | Options Exercisable | |||||||||||||||||||
| Average | Weighted | Weighted | ||||||||||||||||||
| Number | Remaining | Average | Average | |||||||||||||||||
| of | Contractual | Exercise | Number of | Exercise | ||||||||||||||||
Exercise Price Range | Shares | Life (In Years) | Price | Shares | Price | |||||||||||||||
| $ 1.02 - $ 1.29 | 348,000 | 9.54 | $ | 1.26 | 37,642 | $ | 1.29 | |||||||||||||
| $ 1.52 - $ 1.70 | 397,500 | 9.50 | 1.68 | 48,687 | 1.67 | |||||||||||||||
| $ 1.80 - $ 1.80 | 603,287 | 9.66 | 1.80 | 162,385 | 1.80 | |||||||||||||||
| $ 1.87 - $ 1.87 | 500,000 | 9.61 | 1.87 | — | — | |||||||||||||||
| $ 3.14 - $ 5.30 | 384,494 | 8.09 | 4.37 | 179,257 | 4.95 | |||||||||||||||
| $ 5.35 - $ 12.00 | 348,813 | 7.54 | 8.33 | 270,530 | 8.55 | |||||||||||||||
| $ 13.00 - $ 35.00 | 313,659 | 4.91 | 22.49 | 313,659 | 22.49 | |||||||||||||||
| $ 41.25 - $112.50 | 161,056 | 2.14 | 72.32 | 161,056 | 72.32 | |||||||||||||||
| $115.00 - $115.00 | 400 | 3.83 | 115.00 | 400 | 115.00 | |||||||||||||||
| $120.63 - $120.63 | 6,772 | 3.11 | 120.63 | 6,772 | 120.63 | |||||||||||||||
| 3,063,981 | 8.28 | $ | 8.90 | 1,180,388 | $ | 19.65 | ||||||||||||||
Performance Stock Award
In October 2006, as provided in his employment offer letter, the Company agreed to pay to Dr. Gonda, our President and Chief Executive Officer, a stock bonus of up to 100,000 shares of its common stock to be earned based on the common stock price reaching certain price targets after each of the first two years of his employment. The Company valued Dr. Gonda’s stock bonus on a Monte-Carlo simulation due to the path-dependency of the award. The Company believes that the Monte-Carlo simulation provides a more precise estimate for the grant date fair value of a market-based equity award as the simulation allows for vesting throughout the vesting period. The fair value of the performance stock award is $94,000.
Employee Stock Purchase Plan
Employees generally are eligible to participate in the Employee Stock Purchase Plan (the “Purchase Plan”) if they have been continuously employed by the Company for at least 10 days prior to the first day of the offering period and are customarily employed at least 20 hours per week and at least five months per calendar year and are not a 5% or greater shareholder. Shares may be purchased under the Purchase Plan at 85% of the lesser of the fair market value of the common stock on the grant date or purchase date. Employee contributions, through payroll deductions, are limited to the lesser of 15% of earnings or $25,000.
As of December 31, 2006, a total of 709,153 shares have been issued under the Purchase Plan, leaving a balance of 340,847 available authorized shares. Compensation expense was $111,000 in 2006. Under SFAS No. 123(R), stock-based compensation cost is reported for the fair value of the employees’ purchase rights, which was estimated using the Black-Scholes model and the following assumptions for 2006: expected volatility of 86.37%; risk-free interest rates of 4.90%; an average expected life of 0.49 years and a dividend yield of 0.0%. The weighted-average fair value of the purchase rights granted was $0.60 per share in 2006 and $2.90 per share in 2005 and in 2004. Pro forma compensation expense was $485,000 and $623,000 for the years ended December 31, 2005 and 2004 respectively, under the Employee Stock Purchase Plan.
64
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 7. | Employee Benefit Plans |
The Company has a 401(k) Plan which stipulates that all full-time employees with at least 30 days of employment can elect to contribute to the 401(k) Plan, subject to certain limitations, up to $14,000 annually on a pretax basis. Subject to a maximum dollar match contribution of $7,000 per year, the Company will match 50% of the first 6% of the employee’s contribution on a pretax basis. The Company expensed total employer matching contributions of $194,000, $283,000 and $461,000 in 2006, 2005 and 2004, respectively.
| 8. | Related Party Transactions |
Novo Nordisk and its affiliate, Novo Nordisk Pharmaceuticals, Inc., are considered related parties and at December 31, 2006 owned 1,573,674 shares of the Company’s common stock, representing 10.6% of the Company’s total outstanding common stock (9.8% on an as-converted basis). Pursuant to the Company’s public offering completed on January 30, 2007, Novo Nordisk owned approximately 3.0% of the Company’s stock on an as-converted basis.
Development and License Agreement
In June 1998, the Company executed a development and commercialization agreement with Novo Nordisk to jointly develop a pulmonary delivery system for administering insulin by inhalation. Under the terms of the agreement, Novo Nordisk has been granted exclusive rights to worldwide sales and marketing rights for any products developed under the terms of the agreement. Through December 31, 2006, the Company received from Novo Nordisk $150.1 million in product development and milestone payments and, of this amount, the Company has recognized all of these funds as contract revenues. Under the terms of the development agreement in effect at December 31, 2005 between the Company and Novo Nordisk, prior to completion of the restructuring transaction noted below, Novo Nordisk was to fund all product development costs incurred by the Company under the terms of the agreement, and the Company was to be the initial manufacturer of the product and was to receive a share of the overall gross profits resulting from Novo Nordisk’s sales of the product while Novo Nordisk and the Company agreed to co-fund final development of the AERx device.
January 2005 Restructuring
On January 26, 2005, the Company completed a restructuring of its AERx iDMS program, pursuant to a restructuring agreement entered into with Novo Nordisk and NNDT, a newly created wholly owned subsidiary of Novo Nordisk. Under the terms of the restructuring agreement the Company sold certain equipment, leasehold improvements and other tangible assets currently utilized in the AERx iDMS program to NNDT for $55.3 million, of which the Company received net proceeds of $51.3 million after applying a refund of cost advances of $4.0 million previously made by Novo Nordisk. The Company’s expenses related to this transaction for legal and other consulting costs were $1.1 million. In connection with the restructuring transaction, the Company entered into various related agreements with Novo Nordisk and NNDT, effective January 26, 2005, including the following:
| • | an amended and restated license agreement amending the Development and License Agreement previously in place with Novo Nordisk, expanding Novo Nordisk’s development and manufacturing rights to the AERx iDMS program and providing for royalties that will rise to an average of five percent or higher by the fifth year after commercialization to the Company on future AERx iDMS net sales; and | |
| • | a three-year agreement under which NNDT agreed to perform contract manufacturing of AERx iDMS-identical devices and dosage forms filled with compounds provided by the Company in support of preclinical and initial clinical development by the Company of other AERx products. |
As a result of the restructuring transaction, contract revenue from the Company’s development agreement with Novo Nordisk ceased in January 2005. The Company received $59,000 related to its iDMS consulting arrangement
65
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
with Novo Nordisk in 2006. For the years ended December 31, 2005 and 2004, the Company recognized contract revenues of $8.0 million and $27.0 million, respectively.
Significant payments from collaborators, contract and milestone revenues and deferred revenue are as follows (amounts in thousands):
| December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| Deferred revenue — beginning balance | $ | 222 | $ | 11,491 | $ | 12,931 | ||||||
| Payments: | ||||||||||||
| Novo Nordisk | 59 | 727 | 25,373 | |||||||||
| Other collaborator-funded programs | 4,524 | 2,530 | 1,232 | |||||||||
| Total payments | 4,583 | 3,257 | 26,605 | |||||||||
| Contract revenues recognized: | ||||||||||||
| Novo Nordisk | 59 | 8,013 | 26,999 | |||||||||
| Other collaborator-funded programs | 4,746 | 2,494 | 1,046 | |||||||||
| Total contract revenues recognized | 4,805 | 10,507 | 28,045 | |||||||||
| Deferred revenue at December 31, 2004 recognized on January 26, 2005 as payment for assets pursuant to the restructuring agreement with Novo Nordisk | — | 4,019 | — | |||||||||
| Deferred revenue — ending balance | — | 222 | 11,491 | |||||||||
| Less: non-current portion of deferred revenue | — | — | (3,966 | ) | ||||||||
| Current portion of deferred revenue | $ | — | $ | 222 | $ | 7,525 | ||||||
The Company receives revenues from other collaborator-funded programs. These programs are generally early-stage feasibility programs and may not necessarily develop into long-term development agreements with the collaborators.
July 2006 Restructuring
On July 3, 2006, the Company and Novo Nordisk A/ S entered into a Second Amended and Restated License Agreement (the “License Agreement”) to reflect: (i) the transfer by the Company of certain intellectual property, including all right, title and interest to its patents that contain claims that pertain generally to breath control or specifically to the pulmonary delivery of monomeric insulin and monomeric insulin analogs, together with interrelated patents, which are linked via terminal disclaimers, as well as certain pending patent applications and continuations thereof by the Company for a cash payment to the Company of $12.0 million, with the Company retaining exclusive, royalty-free control of these patents outside the field of glucose control; (ii) a reduction by 100 basis points of each royalty rate payable by Novo Nordisk to the Company for a cash payment to the Company of $8.0 million; and (iii) a loan to the Company in the principal amount of $7.5 million.
The $12.0 million and the $8.0 million are included in gain on sale of patent and royalty interest line item in the accompanying statements of operations for the year ended December 31, 2006. The loan bears interest accruing at 5% per annum and the principal along with the accrued interest is payable in three equal payments of $3.5 million at July 2, 2012, July 1, 2013 and June 30, 2014. The loan is secured by a pledge of the net royalty stream payable to the Company by Novo Nordisk pursuant to the License Agreement.
66
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Securities Purchase Agreements
In 1998, the Company raised $5.0 million through the sale of common stock to Novo Nordisk at a 25% premium to the market price. In June 2001, the Company raised an additional $5.0 million through the sale of common stock to Novo Nordisk at the market price. In October 2001, the Company entered into a new common stock purchase agreement with Novo Nordisk Pharmaceuticals. Under the new agreement, Novo Nordisk Pharmaceuticals committed to purchase up to $45.0 million of the Company’s common stock at fair market value specified in the agreement, of which $20.0 million was invested initially. In July 2002, the Company raised $5.0 million through the sale of common stock to Novo Nordisk Pharmaceuticals under the terms of the agreement. Since the inception of the collaboration in June 1998 through December 31, 2006, the Company raised $35.0 million through the sale of common stock to Novo Nordisk.
In connection with the July 2002 restructuring transaction, the Company entered into an amendment of the common stock purchase agreement in place with Novo Nordisk, deleting the provisions whereby the Company can require Novo Nordisk to purchase certain amounts of common stock and imposing certain restriction on the ability of Novo Nordisk to sell shares of the Company’s common stock that it holds.
| 9. | Income Taxes |
There is no provision for income taxes because the Company has incurred operating losses. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for tax purposes.
Significant components of the Company’s deferred tax assets are as follows (amounts in thousands):
| December 31, | ||||||||
| 2006 | 2005 | |||||||
| Net operating loss carry forward | $ | 96,000 | $ | 91,800 | ||||
| Deferred revenue | — | 100 | ||||||
| Research and development credits | 19,600 | 14,400 | ||||||
| Capitalized research and development | 3,900 | 3,100 | ||||||
| Other | 1,100 | 1,300 | ||||||
| Total deferred tax assets | 120,600 | 110,700 | ||||||
| Valuation allowance | (120,600 | ) | (110,700 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
Management believes that, based on a number of factors, it is not determinable that it is more likely than not that the deferred tax asset will be realized. Accordingly, a full valuation allowance has been recorded for all deferred tax assets at December 31, 2006 and 2005. The valuation allowance increased for each of the years ended December 31 by $9.9 million for 2006, $8.9 million for 2005, and $13.2 million for 2004.
As of December 31, 2006, the Company had federal net operating loss carry forwards of approximately $250.3 million and federal research and development tax credits of approximately $13.4 million, which expire in the years 2006 through 2026.
As of December 31, 2006, the Company had California net operating loss carry forwards of approximately $146.6 million, which expire in the years 2007 through 2016, and California research and development tax credits of approximately $8.7 million, which do not expire, and California Manufacturer’s Investment Credit of approximately $904,000, which expire in the years 2006 through 2013.
67
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
Approximately $42,000 of the federal and state net operating loss carryforwards represents the stock option deduction arising from activity under the Company’s stock option plan, the benefit of which will increase additional paid in capital when realized.
Utilization of net operating loss and tax credit carryforwards are subject to certain limitations under Section 382 of the Internal Revenue Code of 1986 (“Section 382”), as amended, in the event of a change in the Company’s ownership, as defined in Section 382. The Company has not performed this ownership analysis; however, it is possible that there has been a “Section 382” change in ownership, which would limit the amount of net operating loss available to be used in future years.
The Tax Reform Act of 1986 limits the annual use of net operating loss and tax credit carry forwards in certain situations where changes occur in stock ownership of a company. In the event the Company has a change in ownership, as defined, the annual utilization of such carry forwards could be limited.
| 10. | Restructuring and Asset Impairment |
On May 15, 2006, the Company announced the implementation of a strategic restructuring of its business operations to focus resources on advancing the current product pipeline and developing products focused on respiratory disease, leveraging the Company’s core expertise and intellectual property. The Company recorded an initial charge of $1.3 million. The Company accounted for the restructuring activity in accordance with FAS No. 146,Accounting for Costs Associated with Exit or Disposal. The restructuring included a reduction in force of 36 employees, the majority of which were research personnel. On August 25, 2006, the Company recorded an additional restructuring charge of $566,000 in severance expense related to the termination of employment of V. Bryan Lawlis, the Company’s former President and Chief Executive Officer, and Bobba Venkatadri, the Company’s former Senior Vice President of Operations, offset by a reduction of previously recognized severance costs of $233,000 related to the departure of employees in connection with the sale of Intraject-related assets to Zogenix. On October 20, 2006 the Company identified additional redundant positions affecting five employees and recorded severance costs of $268,000. The Company recorded net restructuring charges of $1.9 million for the year ended December 31, 2006 which is included in the restructuring and asset impairment expense line item on the accompanying statement of operations. The Company expects to pay the severance-related expenses in full by the end of 2007. The accrual for employee related expenses is included in accrued compensation in the accompanying balance sheet as of December 31, 2006.
The Company recorded a non-cash impairment charge of $4.0 million which was incurred to write down its Intraject-related assets to their net realizable value. The net realizable value did not include any potential future contingent milestones or royalties. The Company sold these assets to Zogenix in 2006 for an initial payment of $4.0 million and recorded an additional impairment charge of $14,000 (See note 12). The following table summarizes the Company’s restructuring and asset impairment expenses in 2006 (in thousands):
| Balance at | ||||||||||||||||||||
| Non-Cash | Restructuring | December 31, | ||||||||||||||||||
Type of Liability | Impairment | Charges | Adjustments | Payments | 2006 | |||||||||||||||
Year ended December 31, 2006 | ||||||||||||||||||||
| Severance and related benefits | $ | — | $ | 2,134 | $ | (233 | ) | $ | (1,185 | ) | $ | 716 | ||||||||
| Out-placement services | — | 88 | — | (52 | ) | 36 | ||||||||||||||
| Impairment on Intraject-related assets | 4,014 | — | — | — | — | |||||||||||||||
| $ | 4,014 | $ | 2,222 | $ | (233 | ) | $ | (1,237 | ) | $ | 752 | |||||||||
68
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
| 11. | Sale of Intraject-Related Assets |
In August 2006, the Company sold all of its assets related to the Intraject technology platform and products, including 12 United States patents along with any foreign counterparts corresponding to those United States patents, to Zogenix, Inc., a newly created private company that has some officers who were former officers of Aradigm. Zogenix is responsible for further development and commercialization efforts of Intraject. The Company recorded a non-cash impairment charge of $4.0 million in 2006, which was incurred to write down its Intraject-related assets to their net realizable value. The Company sold these assets for a $4.0 million initial payment and will be entitled to a milestone payment upon initial commercialization and royalty payments upon any commercialization of products that may be developed and sold using the Intraject technology. The net book value of these assets at the time of sale was $4.0 million. The net realizable value did not include any potential future contingent milestones or royalties.
In connection with the sale of its Intraject platform, the Company entered into an agreement with Zogenix for transitional support through December 31, 2006. The Company is to be reimbursed for the provision of consulting services, information technology and document control support and office facilities. The Company recorded revenues of $869,000 from Zogenix in 2006.
| 12. | Quarterly Results of Operations (unaudited) |
Following is a summary of the quarterly results of operations for the years ended December 31, 2006 and 2005 (amounts in thousands, except per share amounts):
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| 2006 | 2006 | 2006 | 2006 | |||||||||||||
| Contract and license revenues | $ | 1,073 | $ | 1,807 | $ | 1,126 | $ | 807 | ||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 6,740 | 6,357 | 4,547 | 4,554 | ||||||||||||
| General and administrative | 2,853 | 2,685 | 3,514 | 1,665 | ||||||||||||
| Restructuring and asset impairment | — | 5,370 | 347 | 286 | ||||||||||||
| Total expenses | 9,593 | 14,412 | 8,408 | 6,505 | ||||||||||||
| Loss from operations | (8,520 | ) | (12,605 | ) | (7,282 | ) | (5,698 | ) | ||||||||
| Gain on sale of patent and royalty interest (to related parties) | — | — | 20,000 | — | ||||||||||||
| Interest income | 245 | 135 | 459 | 412 | ||||||||||||
| Interest expense | (3 | ) | (3 | ) | (95 | ) | (96 | ) | ||||||||
| Other income (expense) | (7 | ) | 40 | (7 | ) | (3 | ) | |||||||||
| Net loss | $ | (8,285 | ) | $ | (12,433 | ) | $ | 13,075 | $ | (5,385 | ) | |||||
| Basic net income (loss) per common share | $ | (0.57 | ) | $ | (0.85 | ) | $ | 0.89 | $ | (0.37 | ) | |||||
| Diluted net income (loss) per common share | $ | (0.57 | ) | $ | (0.85 | ) | $ | 0.82 | $ | (0.37 | ) | |||||
| Shares used in computing basic net income (loss) per common share | 14,563 | 14,656 | 14,660 | 14,531 | ||||||||||||
| Shares used in computing diluted net income (loss) per common share | 14,563 | 14,656 | 15,982 | 14,531 | ||||||||||||
69
Table of Contents
ARADIGM CORPORATION
NOTES TO FINANCIAL STATEMENTS — (Continued)
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| 2005 | 2005 | 2005 | 2005 | |||||||||||||
| Contract and license revenues | $ | 7,714 | $ | 1,212 | $ | 719 | $ | 862 | ||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 7,070 | 7,317 | 6,471 | 9,316 | ||||||||||||
| General and administrative | 3,235 | 2,713 | 2,326 | 2,621 | ||||||||||||
| Total expenses | 10,305 | 10,030 | 8,797 | 11,937 | ||||||||||||
| Loss from operations | (2,591 | ) | (8,818 | ) | (8,078 | ) | (11,075 | ) | ||||||||
| Interest income | 288 | 350 | 342 | 337 | ||||||||||||
| Interest expense | — | — | — | (6 | ) | |||||||||||
| Other income (expense) | (37 | ) | (8 | ) | 8 | 73 | ||||||||||
| Net loss | $ | (2,340 | ) | $ | (8,476 | ) | $ | (7,728 | ) | $ | (10,671 | ) | ||||
| Basic and diluted net loss per common share | $ | (0.16 | ) | $ | (0.58 | ) | $ | (0.53 | ) | $ | (0.73 | ) | ||||
| Shares used in computing basic and diluted net loss per common share | 14,459 | 14,512 | 14,518 | 14,563 | ||||||||||||
| 13. | Subsequent Events |
Public Offering of Common Shares
On January 30, 2007, the Company received $33.9 million from the closing of its public offering of 37,950,000 shares of common stock in an underwritten public offering with net proceeds, after underwriting discount and expenses, of approximately $33.3 million. This public offering triggered the automatic conversion of all outstanding shares of Series A convertible preferred stock to common stock and eliminated the Series A liquidation preference of $41.9 million, equal to the original issue price plus all accrued and unpaid dividends (as adjusted for any stock dividends, combinations, splits, recapitalizations and other similar events). Following the offering, the 1,544,626 shares of Series A convertible preferred stock has been converted to 1,235,701 shares of common stock, and no liquidation preference or other preferential rights remain.
The unaudited pro forma balance sheet information in the accompanying balance sheet is provided as a result of the significant changes in the Company’s capital structure subsequent to the balance sheet date and assumes the transactions noted above that were completed subsequent to December 31, 2006 had occurred as of December 31, 2006.
70
Table of Contents
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A. | Controls and Procedures |
Evaluation of disclosure controls and procedures: Based on their evaluation of our disclosure controls and procedures (as defined in the rules promulgated under the Securities Exchange Act of 1934, as amended), our chief executive officer and our chief financial officer have concluded that our disclosure controls and procedures were effective as of the end of the period covered by this report.
We believe that a controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives, and our chief executive officer and our chief financial officer have concluded that these controls and procedures are effective at the “reasonable assurance” level.
Changes in internal controls: There were no significant changes in our internal controls over financial reporting during the period covered by this report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
Identification of Directors and Executive Officers
The information required by this Item concerning our directors and executive officers is set forth in Part I of this Annual Report.
Audit Committee Information
The Audit Committee of the Board oversees our corporate accounting and financial reporting process. For this purpose, the Audit Committee performs several functions. The Audit Committee evaluates the performance of and assesses the qualifications of the independent auditors; determines and approves the engagement of the independent auditors; determines whether to retain or terminate the existing independent auditors or to appoint and engage new independent auditors; reviews and approves the retention of the independent auditors to perform any proposed permissible non-audit services; monitors the rotation of partners of the independent auditors on our audit engagement team as required by law; confers with management and the independent auditors regarding the effectiveness of internal controls over financial reporting; establishes procedures, as required under applicable law, for the receipt, retention and treatment of complaints received by us regarding accounting, internal accounting controls or auditing matters and the confidential and anonymous submission by employees of concerns regarding questionable accounting or auditing matters; reviews the financial statements to be included in our Annual Report onForm 10-K and our quarterly financial statements; and discusses with management and the independent auditors the results of the annual audit. The Audit Committee held seven meetings during the fiscal year. Currently, three directors comprise the Audit Committee: Messrs. Barker, Jaeger and V. Thompson. The Audit Committee has adopted a written Audit Committee Charter.
The Board annually reviews the SEC listing standards definition of independence for Audit Committee members and has determined that Mr. Jaeger qualifies as an “audit committee financial expert,” as defined in applicable rules of the SEC. The Board made a qualitative assessment of Mr. Jaeger’s level of knowledge and
71
Table of Contents
experience based on a number of factors, including his formal education and experience as a Chief Financial Officer for public reporting companies.
Section 16(a) Compliance
Section 16(a) of the 1934 Act requires our directors and executive officers, and persons who own more than ten percent of a registered class of our equity securities, to file with the SEC initial reports of ownership and reports of changes in ownership of our common stock and other equity securities. Officers, directors and greater than ten percent shareholders are required by SEC regulation to furnish us with copies of all Section 16(a) forms they file.
To our knowledge, based solely on a review of the copies of such reports furnished to us and, if applicable, written representations that no other reports were required, during the fiscal year ended December 31, 2006, all Section 16(a) filing requirements applicable to our officers, directors and greater than ten percent beneficial owners were complied with, other than two Forms 4 that were filed late (one for Mr. Siebert and one for Dr. Bryan Lawlis, our former Chief Executive Officer).
Code of Conduct and Ethics
We have adopted the Aradigm Corporation Code of Business Conduct and Ethics that applies to all of our officers, directors and employees. The Code of Business Conduct and Ethics is available on our website at www.aradigm.com. If we make any substantive amendments to the Code of Business Conduct and Ethics or grant any waiver from a provision of the Code to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website.
| Item 11. | Executive Compensation |
The policies of the Compensation Committee, or the Committee, with respect to the compensation of executive officers, including the Chief Executive Officer, or CEO, are designed to provide compensation sufficient to attract, motivate and retain executives of outstanding ability and potential and to establish an appropriate relationship between executive compensation and the creation of shareholder value. To meet these goals, the Committee recommends executive compensation packages to our board of directors that are based on a mix of salary, bonus and equity awards. Although the Committee has not adopted any formal guidelines for allocating total compensation between equity compensation and cash compensation, our executives’ compensation packages have more recently reflected an increased focus on performance and equity-based compensation, as we believe it is important to maintain a strong link between executive incentives and the creation of shareholder value. We believe that performance and equity-based compensation are the most important component of the total executive compensation package for maximizing shareholder value while, at the same time, attracting, motivating and retaining high-quality executives.
Overall, we seek to provide total compensation packages that are competitive in terms of total potential value to our executives, and that are tailored to the unique characteristics of our company in order to create an executive compensation program that will adequately reward our executives for their roles in creating value for our shareholders. We intend to be competitive with other similarly situated companies in our industry.
Benchmarking of Cash and Equity Compensation
The Committee believes it is important when making its compensation-related decisions to be informed as to current practices of similarly situated publicly held companies in the life sciences industry. In early 2006 the Committee commissioned a study conducted by an outside consulting firm that specializes in executive compensation. This study reviewed the cash and equity compensation practices of 19 publicly held companies in the life sciences industry. These companies were chosen for inclusion in the study based on certain business characteristics similar to ours, including revenues, stage of development, employee headcount and market capitalization. In addition to benchmarking studies, the Committee has historically taken into account input from other sources, including input from other independent members of the board of directors and publicly available data relating to the compensation practices and policies of other companies within and outside of our industry. While benchmarking may not always be appropriate as a stand-alone tool for setting compensation due to the aspects of our business and
72
Table of Contents
objectives that may be unique to us, we generally believe that gathering this information is an important part of our compensation-related decision-making process.
The Committee intends to retain the services of third-party executive compensation specialists from time to time, as the Committee sees fit, in connection with the establishment of cash and equity compensation and related policies.
Compensation Components
Base Salary. Generally, we believe that executive base salaries should be set near the median of the range of salaries for executives in similar positions and with similar responsibilities at comparable companies. We believe that maintaining base salary amounts at or near the industry median minimizes competitive disadvantage while avoiding paying amounts in excess of what we believe to be necessary to motivate executives to meet corporate goals. Base salaries are generally reviewed annually, and the Committee and board will seek to adjust base salary amounts to realign such salaries with median market levels after taking into account individual responsibilities, performance and experience.
For 2006, the average increase in the salaries of the executive officers, including the CEO, from 2005 salaries was 4%. For 2007, base salaries for executives will not be increased from their 2006 levels.
Annual Executive Bonus Plan. In addition to base salaries, we believe that performance-based cash bonuses play an important role in providing incentives to our executives to achieve defined annual corporate goals. Near the beginning of each year, the board, upon the recommendation of the Committee and subject to any applicable employment agreement, determines a target bonus amount for each executive. The target percentages are set at levels that, upon achievement of 100% of the target percentage, are likely to result in bonus payments that the Committee believes to be at or near the median for target bonus amounts for comparable companies and that, upon achievement beyond target, can result in bonuses of up to 150% of that amount. The Committee then reviews a detailed set of overall corporate performance goals prepared by management that are intended to apply to the executives’ bonus awards and, with some distinctions, to the bonus awards for all of our other employees. The Committee then works with management to develop final corporate performance goals that are set at a level the Committee believes management can reasonably achieve with hard work over the next year.
At the end of each year, the board, upon the recommendation of the Committee, determines the level of achievement for each corporate goal and awards credit for the achievement of goals as a percentage of the target bonus. Final determinations as to bonus levels are then based on the achievement of these corporate goals, which are the same for all executives, as well as our assessment as to the overall success of our company and the development of our business. Actual bonuses are paid to the executives at the end of each fiscal year and may be above or below target bonus levels, at the discretion of the board. Bonus payments under our annual bonus plan are contingent on continued employment with the company at the end of the year.
In March 2006, the board, upon recommendation of the Committee, established target bonus awards (as a percentage of base salary) of 50% for Dr. V. Bryan Lawlis, our former CEO, and 40% for Mr. Chesterman, Dr. Otulana, Dr. Stephen J. Farr, our former Senior Vice President and Chief Scientific Officer, and Mr. Bobba Venkatadri, our former Senior Vice President of Operations. Upon Dr. Gonda’s appointment as our CEO in August 2006, pursuant to the terms of his employment offer letter, the board set his target bonus at 33% of his prorated 2006 salary. In 2006, the corporate goals identified by the Committee and the board included meeting various objectives relating generally to the development and progression of existing product candidates and collaborations, establishing new collaborative arrangements, developing our supply chain and successfully securing funding and managing expense levels.
In December 2006, the Committee and board determined that applicable corporate performance goals that were achieved in 2006 merited a bonus award of 79% of the target bonus award for executive officers. However, based on the overall development of our business in 2006, the Committee and board decided to apply a discretionary discount and awarded the executive officers reduced bonuses of $22,364 to Dr. Gonda, $48,202 to Mr. Chesterman and $45,352 to Dr. Otulana. The bonus awards were 20%, 16% and 16% of the 2006 salary paid to Dr. Gonda, Mr. Chesterman and Dr. Otulana, respectively. The board also indicated to management that it would consider
73
Table of Contents
awarding additional discretionary bonuses to the executive officers upon successful completion of our follow-on public offering. In February 2007, the board, upon recommendation of the Committee, approved one-time discretionary bonuses for our executives in the following amounts: $50,000 for Dr. Gonda and $78,000 for each of Mr. Chesterman and Dr. Otulana. Dr. Gonda’s bonus award is based on the prorated salary for the portion of 2006 during which Dr. Gonda served as our CEO. Drs. Lawlis and Farr and Mr. Venkatadri did not receive 2006 bonuses as they were not serving as officers at the end of 2006.
In February 2007, the board, upon recommendation of the Committee, established 2007 target bonus awards (as a percentage of base salary) of 50% for Dr. Gonda and 40% for Mr. Chesterman and Dr. Otulana. 2007 bonus awards will again be capped at 150% of the target award, based on maximum goal achievement. The board, upon recommendation of the Committee, also approved performance goals with respect to bonuses under the 2007 Executive Bonus Plan. In 2007, half of the executives’ bonus awards will be earned based on the achievement of specified corporate performance goals, including meeting various objectives relating generally to the development and progression of existing product candidates and collaborations, establishing new programs and collaborative arrangements and managing expense levels. The remaining half of the executives’ bonus awards will be earned based on our common stock achieving specified price targets by the end of the year. If our common stock closes at between $1.19 and $1.42 per share (equal to approximately 125% and 150% of the offering price in our recently completed follow-on public offering), each executive officer will earn his target bonus with respect to that portion of the bonus, and if our common stock closes at or above $1.43 per share (equal to approximately 150% of the offering price in our recently completed follow-on public offering), each executive officer will earn 150% of his target bonus with respect to that portion of the bonus. Actual bonus payments under the 2007 Executive Bonus Plan will be paid at the end of the year and may be above or below the target bonus levels, at the discretion of the board and the Committee.
Equity Awards. We believe that providing a significant portion of our executives’ total compensation package in stock options and other equity awards aligns the incentives of our executives with the interests of our shareholders and with our long-term success. The Committee and board develop their equity award determinations based on their judgments as to whether the complete compensation packages provided to our executives, including prior equity awards, are sufficient to retain, motivate and adequately award the executives. This judgment is based in part on information provided by benchmarking studies.
We grant equity awards through our 2005 Equity Incentive Plan, which was adopted by our board and shareholders to permit the grant of stock options, stock appreciation rights, restricted shares, restricted stock units, performance shares and other stock-based awards to our officers, directors, scientific advisory board members, employees and consultants. All of our employees, directors, scientific advisory board members and consultants are eligible to participate in the 2005 Equity Incentive Plan. The material terms of the 2005 Equity Incentive Plan are further described in note 6 to our financial statements included elsewhere in this Annual Report.
In March 2006, we issued to Dr. Lawlis, Mr. Chesterman, Dr. Otulana, Dr. Farr and Mr. Venkatadri options to purchase up to 150,000, 80,000, 60,000, 60,000 and 40,000 shares of our common stock, respectively. In June 2006, we issued to each of Mr. Chesterman and Dr. Otulana an option to purchase up to 300,000 shares of our common stock. Upon Dr. Gonda’s appointment as our CEO in August 2006, we granted to Dr. Gonda an option to purchase up to 500,000 shares of our common stock. All options we granted have an exercise price equal to the fair market of our common stock on the date of grant.
In addition, in October 2006, as provided in Dr. Gonda’s employment offer letter, we agreed to pay to Dr. Gonda a stock bonus of up to 100,000 shares of our common stock to be earned based on our common stock price reaching certain price targets after each of the first two years of his employment as more fully described below in the section entitled “2006 Grants of Plan-based Awards.” We believe this award structure is consistent with our approach of providing significant equity-based compensation to our executives in order to align our executives’ interests with those of our shareholders.
For 2007, the Committee has yet to consider whether to grant additional equity awards to our executives.
Severance Benefits. We have adopted an Executive Officer Severance Plan and have entered into change of control agreements with each of our executive officers, the terms of which are more described below in the section
74
Table of Contents
entitled “Potential Payments Upon Termination or Change in Control.” We believe these severance and change in control benefits are an essential element of our executive compensation package and assist us in recruiting and retaining talented individuals.
Other Compensation. All of our executives are eligible to participate in our employee benefit plans, including medical, dental, life insurance and 401(k) plans. These plans are available to all salaried employees and do not discriminate in favor of executive officers. It is generally our policy to not extend significant perquisites to our executives that are not available to our employees generally. We have no current plans to make changes to levels of benefits and perquisites provided to executives.
The following table sets forth information regarding compensation earned in 2006 by our CEO, our Chief Financial Officer, our Chief Medical Officer, our former CEO and two other former executive officers who would have been among the five most highly compensated executives if they had been employed at the end of the fiscal year (these individuals are collectively referred to as our “named executive officers”):
| Non-Equity | ||||||||||||||||||||||||||||||||
| Stock | Option | Incentive Plan | All Other | |||||||||||||||||||||||||||||
| Salary | Bonus | Awards | Awards(1) | Compensation(2) | Compensation | Total | ||||||||||||||||||||||||||
| Year | ($) | ($) | ($) | ($) | ($) | ($) | ($) | |||||||||||||||||||||||||
| Igor Gonda, Ph.D.(3) | 2006 | 113,230 | 100,000 | 20,340 | 68,942 | 22,363 | 50,966 | 375,841 | ||||||||||||||||||||||||
| President and Chief Executive Officer | ||||||||||||||||||||||||||||||||
| Thomas C. Chesterman | 2006 | 305,076 | — | — | 202,471 | 48,202 | 28,596 | 584,291 | ||||||||||||||||||||||||
| Senior Vice President and Chief Financial Officer | ||||||||||||||||||||||||||||||||
| Babatunde A. Otulana, M.D. | 2006 | 287,038 | — | — | 183,319 | 45,352 | 22,980 | 538,689 | ||||||||||||||||||||||||
| Senior Vice President of Development and Chief Medical Officer | ||||||||||||||||||||||||||||||||
Former Executive Officers | ||||||||||||||||||||||||||||||||
| V. Bryan Lawlis, Jr., Ph.D.(4) | 2006 | 274,139 | — | — | 259,936 | — | 452,393 | 986,468 | ||||||||||||||||||||||||
| Former President and Chief Executive Officer | ||||||||||||||||||||||||||||||||
| Stephen J. Farr, Ph.D.(5) | 2006 | 210,000 | — | — | 163,357 | — | 167,456 | 540,813 | ||||||||||||||||||||||||
| Former Senior Vice President and Chief Scientific Officer | ||||||||||||||||||||||||||||||||
| Bobba Venkatadri(6) | 2006 | 260,089 | — | — | 141,237 | — | 379,339 | 780,665 | ||||||||||||||||||||||||
| Former Senior Vice President of Operations | ||||||||||||||||||||||||||||||||
| (1) | The method of and assumptions used to calculate the value of the options granted to our named executive officers is discussed in note 1 to our financial statements included elsewhere in this prospectus. | |
| (2) | Each executive officer employed at the end of 2006 received a cash bonus for achievement of certain corporate and personal goals pursuant to our 2006 Executive Bonus Plan. | |
| (3) | Dr. Gonda commenced his employment with us on August 10, 2006. Dr. Gonda’s bonus reflects a $100,000 signing bonus for accepting his offer of employment. Dr. Gonda also received a stock bonus award for up to 100,000 shares of our common stock. We valued Dr. Gonda’s stock bonus on a Monte-Carlo simulation due to the path-dependency of the award. We believe that the Monte-Carlo simulation provides a more precise estimate for the grant date fair value of a market-based equity award as the simulation allows for vesting throughout the vesting period. In accordance with Securities and Exchange Commission regulations, the cost reflected in the table above only includes the portion of the award’s value that was amortized for 2006. Dr. Gonda’s compensation includes $21,000 in director fees and options expense of $2,485 that were both attributable to his services as a director prior to his appointment as our CEO in August 2006. Dr. Gonda has not received any additional compensation for his services as a director since he was appointed CEO in August 2006. | |
| (4) | Dr. Lawlis ceased serving as our CEO on August 10, 2006. |
75
Table of Contents
| (5) | Dr. Farr ceased serving as an executive officer on June 16, 2006. | |
| (6) | Mr. Venkatadri ceased serving as an executive officer on September 15, 2006. |
All Other Compensation in the summary compensation table above includes the following components:
| Life | 401(k) | Employee | ||||||||||||||||||||||||||||||||||||||
| Club | Health Care | Moving | Insurance | Matching | Stock | Director | Severance | |||||||||||||||||||||||||||||||||
| Memberships | Contribution | Allowance | Premiums | Contributions | Purchase | Fees | Benefits | Total | ||||||||||||||||||||||||||||||||
Name | Year | ($) | ($) | ($) | ($) | ($) | ($) | ($) | ($) | ($) | ||||||||||||||||||||||||||||||
| Igor Gonda, Ph.D. | 2006 | 350 | 3,386 | 23,304 | 595 | 2,331 | — | 21,000 | — | 50,966 | ||||||||||||||||||||||||||||||
| Thomas C. Chesterman | 2006 | 350 | 14,855 | — | 1,707 | 9,065 | 2,618 | — | — | 28,596 | ||||||||||||||||||||||||||||||
| Babatunde A. Otulana, M.D. | 2006 | 350 | 14,855 | — | 1,613 | 6,163 | — | — | — | 22,980 | ||||||||||||||||||||||||||||||
Former Executive Officers | ||||||||||||||||||||||||||||||||||||||||
| V. Bryan Lawlis, Jr., Ph.D. | 2006 | — | 6,585 | — | 1,172 | 7,000 | — | — | 437,636 | 452,393 | ||||||||||||||||||||||||||||||
| Stephen J. Farr, Ph.D. | 2006 | 300 | 7,427 | — | 872 | 4,643 | — | — | 154,214 | 167,456 | ||||||||||||||||||||||||||||||
| Bobba Venkatadri | 2006 | 300 | 7,230 | — | 1,184 | 5,344 | — | — | 365,281 | 379,339 | ||||||||||||||||||||||||||||||
2006 Grants of Plan-Based Awards
The following table sets forth information regarding plan-based awards to our named executive officers in 2006:
| All Other | ||||||||||||||||||||||||||||||||||||
| Option | Grant | |||||||||||||||||||||||||||||||||||
| Estimated Future | Estimated Future | Awards: | Exercise | Date Fair | ||||||||||||||||||||||||||||||||
| Payouts Under Non- | Payouts Under | Number of | or Base | Value of | ||||||||||||||||||||||||||||||||
| Equity Incentive | Equity Incentive | Securities | Price of | Stock and | ||||||||||||||||||||||||||||||||
| Plan Awards(1) | Plan Awards(2) | Underlying | Option | Option | ||||||||||||||||||||||||||||||||
| Grant | Approval | Target | Maximum | Target | Maximum | Options | Awards | Awards(3) | ||||||||||||||||||||||||||||
Name | Date | Date | ($) | ($) | (#) | (#) | (#) | ($/sh) | ($) | |||||||||||||||||||||||||||
| Igor Gonda, Ph.D. | 5/18/2006 | 5/18/2006 | — | — | — | — | 4,000 | 1.52 | 3,996 | |||||||||||||||||||||||||||
| 8/10/2006 | 8/8/2006 | 37,743 | 56,615 | — | — | — | — | — | ||||||||||||||||||||||||||||
| 8/10/2006 | 8/8/2006 | — | — | — | — | 500,000 | 1.87 | 621,500 | ||||||||||||||||||||||||||||
| 10/4/2006 | 10/4/2006 | — | — | 100,000 | 100,000 | — | — | 94,000 | ||||||||||||||||||||||||||||
| Thomas C. Chesterman | 3/7/2006 | 3/2/2006 | 122,031 | 183,046 | — | — | — | — | — | |||||||||||||||||||||||||||
| 3/7/2006 | 3/2/2006 | — | — | — | — | 80,000 | 3.77 | 198,032 | ||||||||||||||||||||||||||||
| 6/27/2006 | 6/27/2006 | — | — | — | — | 300,000 | 1.29 | 255,660 | ||||||||||||||||||||||||||||
| Babatunde A. Otulana, M.D. | 3/7/2006 | 3/2/2006 | 114,815 | 172,223 | — | — | — | — | — | |||||||||||||||||||||||||||
| 3/7/2006 | 3/2/2006 | — | — | — | — | 60,000 | 3.77 | 148,524 | ||||||||||||||||||||||||||||
| 6/8/2006 | 6/8/2006 | — | — | — | — | 300,000 | 1.70 | 334,650 | ||||||||||||||||||||||||||||
Former Executive Officers | ||||||||||||||||||||||||||||||||||||
| V. Bryan Lawlis, Jr., Ph.D. | 3/7/2006 | 3/2/2006 | 157,500 | 236,250 | — | — | — | — | — | |||||||||||||||||||||||||||
| 3/7/2006 | 3/2/2006 | — | — | — | — | 150,000 | 3.77 | 371,310 | ||||||||||||||||||||||||||||
| Stephen J. Farr, Ph.D. | 3/7/2006 | 3/2/2006 | 119,524 | 179,286 | — | — | — | — | — | |||||||||||||||||||||||||||
| 3/7/2006 | 3/2/2006 | — | — | — | — | 60,000 | 3.77 | 148,524 | ||||||||||||||||||||||||||||
| Bobba Venkatadri | 3/7/2006 | 3/2/2006 | 112,692 | 169,038 | — | — | — | — | — | |||||||||||||||||||||||||||
| 3/7/2006 | 3/2/2006 | — | — | — | — | 40,000 | 3.77 | 99,016 | ||||||||||||||||||||||||||||
76
Table of Contents
| (1) | Reflects each executive officer’s participation in our 2006 Executive Bonus Plan. The amount of bonus actually paid under the plan is reflected in the summary compensation table above. | |
| (2) | Reflects a stock bonus award for up to 100,000 shares of our common stock granted to Dr. Gonda. Dr. Gonda will earn half of the shares underlying his award if the average closing price of our common stock between June 9, 2007 and August 9, 2007 is 120% of the average closing price between June 9, 2006 and August 9, 2006 and he will earn the other half of the shares underlying his award if the average closing price of our common stock between June 9, 2008 and August 9, 2008 is 125% of the greater of (i) the average closing price between June 9, 2006 and August 9, 2006 or (ii) the average closing price between June 9, 2007 and August 9, 2007. In addition, if the average closing price of our common stock between June 9, 2008 and August 9, 2008 is 150% of the average closing price between June 9, 2006 and August 9, 2006, Dr. Gonda will be entitled to receive the full award of 100,000 share notwithstanding either of the first two price targets being achieved. If we undergo a change in control on or prior to August 9, 2008, Dr. Gonda, will earn the full 100,000 shares underlying the award if our shareholders receive consideration in the transaction that reflects at least a 15% return per annum from the average closing price of our common stock between June 9, 2006 and August 9, 2006. If we terminate Dr. Gonda’s employment without cause between August 10, 2007 and August 9, 2008 and Dr. Gonda had previously received the first half of his bonus award, he will be entitled to receive the second half of the bonus award upon his termination. In no event will the value of the shares awarded to Dr. Gonda under the bonus award exceed $1,000,000. | |
| (3) | The method of and assumptions used to calculate the value of options granted to our named executive officers is discussed in note 1 to our Financial Statements included elsewhere in this prospectus. We valued Dr. Gonda’s stock bonus on a Monte-Carlo simulation due to the path-dependency of the award. We believe that the Monte-Carlo simulation provides a more precise estimate for the grant date fair value of a market-based equity award as the simulation allows for vesting throughout the vesting period. |
77
Table of Contents
Outstanding Equity Awards At December 31, 2006
The following table provides information regarding each unexercised stock option held by each of our named executive officers as of December 31, 2006:
| Option Awards | Stock Awards | |||||||||||||||||||||||||||
| Market or | ||||||||||||||||||||||||||||
| Number of | Payout Value | |||||||||||||||||||||||||||
| Unearned | of Unearned | |||||||||||||||||||||||||||
| Shares, Units | Shares, Units | |||||||||||||||||||||||||||
| Number of Securities | or Other | or Other | ||||||||||||||||||||||||||
| Underlying Unexercised | Option | Rights That | Rights That | |||||||||||||||||||||||||
| Options | Exercise | Option | Have Not | Have Not | ||||||||||||||||||||||||
| Exercisable | Unexercisable | Price(1) | Expiration | Vested | Vested | |||||||||||||||||||||||
Name | (#) | (#) | ($) | Date | (#) | ($) | ||||||||||||||||||||||
| Igor Gonda, Ph.D. | (2 | ) | — | — | — | — | 100,000 | 94,000 | ||||||||||||||||||||
| (3 | ) | — | 500,000 | 1.87 | 08/10/2016 | — | — | |||||||||||||||||||||
| (4 | ) | 2,000 | 2,000 | 1.52 | 05/18/2016 | — | — | |||||||||||||||||||||
| 4,000 | — | 5.30 | 05/19/2015 | — | — | |||||||||||||||||||||||
| 4,000 | — | 5.30 | 05/20/2014 | — | — | |||||||||||||||||||||||
| 2,000 | — | 5.30 | 05/13/2014 | — | — | |||||||||||||||||||||||
| 4,000 | — | 6.50 | 05/15/2013 | — | — | |||||||||||||||||||||||
| 4,000 | — | 4.75 | 02/19/2013 | — | — | |||||||||||||||||||||||
| 2,000 | — | 17.25 | 05/21/2012 | — | — | |||||||||||||||||||||||
| 4,000 | — | 17.15 | 05/17/2012 | — | — | |||||||||||||||||||||||
| 3,000 | — | 24.10 | 02/11/2012 | — | — | |||||||||||||||||||||||
| Thomas C. Chesterman | (5 | ) | 37,500 | 262,500 | 1.29 | 06/27/2016 | — | — | ||||||||||||||||||||
| (6 | ) | — | 80,000 | 3.77 | 03/07/2016 | — | — | |||||||||||||||||||||
| (6 | ) | 15,312 | 19,688 | 5.90 | 02/22/2015 | — | — | |||||||||||||||||||||
| (6 | ) | 13,061 | 5,939 | 12.00 | 02/27/2014 | — | — | |||||||||||||||||||||
| (5 | ) | 6,562 | 438 | 4.75 | 02/19/2013 | — | — | |||||||||||||||||||||
| 30,000 | — | 13.00 | 09/05/2012 | — | — | |||||||||||||||||||||||
| Babatunde A. Otulana, M.D. | (5 | ) | 37,500 | 262,500 | 1.70 | 06/08/2016 | — | — | ||||||||||||||||||||
| (6 | ) | — | 60,000 | 3.77 | 03/07/2016 | — | — | |||||||||||||||||||||
| (6 | ) | 10,936 | 14,064 | 5.90 | 02/22/2015 | — | — | |||||||||||||||||||||
| (6 | ) | 15,123 | 6,877 | 12.00 | 02/27/2014 | — | — | |||||||||||||||||||||
| (5 | ) | 9,375 | 625 | 4.75 | 02/19/2013 | — | — | |||||||||||||||||||||
| 26,000 | — | 24.10 | 02/11/2012 | — | — | |||||||||||||||||||||||
| 8,000 | — | 17.20 | 09/20/2011 | — | — | |||||||||||||||||||||||
| 12,000 | — | 30.00 | 03/15/2011 | — | — | |||||||||||||||||||||||
| 8,000 | — | 112.50 | 02/15/2010 | — | — | |||||||||||||||||||||||
| 3,000 | — | 35.00 | 05/21/2009 | — | — | |||||||||||||||||||||||
| 3,000 | — | 60.00 | 02/02/2009 | — | — | |||||||||||||||||||||||
| 2,000 | — | 61.25 | 03/30/2008 | — | — | |||||||||||||||||||||||
| 11,000 | — | 64.38 | 10/21/2007 | — | — | |||||||||||||||||||||||
| V. Bryan Lawlis, Jr., Ph.D. | 9,420 | — | 5.90 | 02/22/2015 | — | — | ||||||||||||||||||||||
| 23,437 | — | 12.00 | 02/27/2014 | — | — | |||||||||||||||||||||||
| Stephen J. Farr, Ph.D. | — | — | — | — | — | — | ||||||||||||||||||||||
| Bobba Venkatadri | — | — | — | — | — | — | ||||||||||||||||||||||
78
Table of Contents
| (1) | Represents the fair market value of a share of our common stock on the grant date of the option. | |
| (2) | The stock bonus award vests as described in the section above entitled “2006 Grants of Plan-based Awards.” | |
| (3) | The option vests over four years with 1/4 of the shares of underlying common stock vesting on the first anniversary of the grant date and 1/48 of the shares of underlying common stock vesting each month thereafter. | |
| (4) | The option vests over one year with 1/4 of the shares of underlying common stock vesting every three months from the grant date. | |
| (5) | The option vests over four years with 1/16 of the shares of underlying common stock vesting every three months from the grant date. | |
| (6) | The option vests over four years with 1/4 of the shares of underlying common stock vesting on the first anniversary of the grant date and 1/16 of the shares of underlying common stock vesting every three months thereafter. |
2006 Option Exercises and Stock Vested
None of our named executive officers exercised options or had shares of stock vest in 2006.
Pension Benefits
None of our named executive officers participate in or have account balances in qualified or non-qualified defined benefit plans sponsored by us. The Committee may elect to adopt qualified or non-qualified defined benefit plans in the future if the Committee determines that doing so is in our best interests.
Nonqualified Deferred Compensation
None of our named executive officers participate in or have account balances in nonqualified defined contribution plans or other nonqualified deferred compensation plans maintained by us. The Committee may elect to provide our officers and other employees with non-qualified defined contribution or other nonqualified deferred compensation benefits in the future if the Committee determines that doing so is in our best interests.
79
Table of Contents
Potential Payments Upon Termination or Change in Control
The following table and summary set forth potential payments payable to our current executive officers upon termination of employment or a change in control. The Committee may in its discretion revise, amend or add to the benefits if it deems advisable. The table below reflects amounts payable to our executive officers assuming their employment was terminated on December 31, 2006:
| Termination Without | ||||||||||||||
| Cause or | ||||||||||||||
| Constructive | ||||||||||||||
| Termination Without | Termination | |||||||||||||
| Cause Prior to a | Change in | Following a Change | ||||||||||||
| Change in Control | Control | in Control | ||||||||||||
Name | Benefit | ($) | ($) | ($) | ||||||||||
| Igor Gonda, Ph.D. | Salary | 320,000 | — | 640,000 | ||||||||||
| Bonus | 114,032 | — | 228,064 | |||||||||||
| Option acceleration | — | — | 562,180 | |||||||||||
| Stock bonus acceleration(1) | — | 90,000 | 90,000 | |||||||||||
| Benefits continuation | 12,539 | — | 25,078 | |||||||||||
| Career transition assistance | — | — | 20,000 | |||||||||||
| Total value: | 446,571 | 90,000 | 1,565,322 | |||||||||||
| Thomas C. Chesterman | Salary | 306,000 | — | 459,000 | ||||||||||
| Bonus | 87,234 | — | 130,851 | |||||||||||
| Option acceleration | — | — | 482,388 | |||||||||||
| Benefits continuation | 18,906 | — | 28,360 | |||||||||||
| Career transition assistance | — | — | 10,000 | |||||||||||
| Total value: | 412,140 | — | 1,110,599 | |||||||||||
| Babatunde A. Otulana, M.D. | Salary | 289,000 | — | 433,500 | ||||||||||
| Bonus | 82,388 | — | 123,582 | |||||||||||
| Option acceleration | — | — | 497,583 | |||||||||||
| Benefits continuation | 18,906 | — | 28,360 | |||||||||||
| Career transition assistance | — | — | 10,000 | |||||||||||
| Total value: | 390,294 | — | 1,093,025 | |||||||||||
| (1) | The value of Dr. Gonda’s stock bonus is calculated using a value of $0.90 per share of common stock, which was the last reported closing sale price of our common stock prior to December 31, 2006. Dr. Gonda is only entitled to acceleration of his stock bonus upon a change in control if our shareholders receive consideration in the transaction that reflects at least a 15% return per annum from the average closing price of our common stock between June 9, 2006 and August 9, 2006. |
Termination without cause prior to a change in control. If any of our executives is terminated by us without cause prior to a change in control, upon executing a general release and waiver, such executive is entitled to receive (less applicable withholding taxes) in a lump sum payment or in installments, at our discretion:
| • | an amount equal to such executive’s annual base salary; | |
| • | an amount equal to such executive’s current target bonus multiplied by the average annual percentage achievement of corporate goals for the three complete fiscal years preceding the termination date; and | |
| • | continuation of such executive’s health insurance benefits for 12 months. |
In addition, if we terminate Dr. Gonda’s employment without cause between August 10, 2007 and August 9, 2008 and he received the initial 50,000 shares of his stock bonus award on August 9, 2007, he will be entitled to receive the remaining 50,000 unvested shares of his stock bonus award.
80
Table of Contents
Termination without cause or constructive termination following a change in control. If any of our executives is terminated by us without cause or constructively terminated (which includes a material reduction in title or duties, a material reduction in salary or benefits or a relocation of 50 miles or more) during the18-month period following a change in control, upon executing a general release and waiver, such executive is entitled to receive (less applicable withholding taxes):
| • | a lump sum payment equal to such executive’s annual base salary multiplied by two, in the case of Dr. Gonda, and one and one-half, in the case of Mr. Chesterman and Dr. Otulana; | |
| • | a lump sum payment equal to such executive’s current target bonus multiplied by (i) the average annual percentage achievement of corporate goals for the three complete fiscal years preceding the termination date and (ii) two, in the case of Dr. Gonda, and one and one-half, in the case of Mr. Chesterman and Dr. Otulana; | |
| • | continuation of such executive’s health insurance benefits for 24 months, in the case of Dr. Gonda, and 18 months, in the case of Mr. Chesterman and Dr. Otulana; | |
| • | reimbursement of actual career transition assistance (outplacement services) incurred by such executive within six months of termination in an amount up to $20,000, in the case of Dr. Gonda, and $10,000, in the case of Mr. Chesterman and Dr. Otulana; and | |
| • | acceleration of vesting of any stock options or restricted stock awards that remain unvested as of the date of such executive’s termination. |
Compensation Committee Interlocks and Insider Participation
During the 2006 fiscal year, the Committee initially consisted of Messrs. Jaeger and V. Thompson and Dr. Gonda. Dr. Gonda had previously served as an officer from October 1995 until December 2001. Dr. Gonda stepped down from the Compensation Committee after his appointment as our CEO and was replaced by Mr. Barker. No interlocking relationship exists between our board or the Committee and the board of directors or the compensation committee of any other company, nor has any such interlocking relationship existed in the past.
Report of the Compensation Committee
The Committee has reviewed and discussed with management the Compensation Discussion and Analysis required by Item 402(b) ofRegulation S-K, which contained in this Annual Report onForm 10-K. Based on this review and discussion, the Compensation Committee has recommended to the Board of Directors that the Compensation Discussion and Analysis be included in this Annual Report onForm 10-K.
Members of the Aradigm Corporation
Compensation Committee:
Frank H. Barker
Stephen O. Jaeger
Virgil D. Thompson
In February, 2007, John M. Siebert was appointed to the Compensation Committee, replacing Frank H. Barker.
81
Table of Contents
Non-Employee Director Compensation
The following table sets forth a summary of the compensation we paid to our non-employee directors in 2006:
| Fees | ||||||||||||
| Earned or | ||||||||||||
| Paid in | Option | |||||||||||
| Cash | Awards(1) | Total | ||||||||||
Name | ($) | ($) | ($) | |||||||||
| Frank H. Barker(2) | 39,500 | 12,266 | 51,766 | |||||||||
| Igor Gonda(3) | 21,000 | 2,485 | 23,485 | |||||||||
| Stephen O. Jaeger(4) | 45,750 | 12,266 | 58,016 | |||||||||
| John Nehra(5) | 8,500 | 5,626 | 14,126 | |||||||||
| Wayne Roe(6) | 10,000 | 5,626 | 15,626 | |||||||||
| John M. Siebert(7) | 4,810 | 3,165 | 7,975 | |||||||||
| Richard Thompson(8) | 10,000 | 213,333 | 223,333 | |||||||||
| Virgil D. Thompson(9) | 68,250 | 20,889 | 89,139 | |||||||||
| (1) | The method of and assumptions used to calculate the value of the options granted to our directors is discussed in note 1 to our financial statements included elsewhere in this prospectus. | |
| (2) | Mr. Barker owns options to purchase up to 54,043 shares of our common stock as of December 31, 2006, of which 36,043 shares are vested as of December 31, 2006. | |
| (3) | Represents compensation received by Dr. Gonda in 2006 for services as a director prior to his appointment as our CEO in August 2006. Dr. Gonda did not receive compensation for his services as a director after he was appointed as our CEO. Dr. Gonda owns options to purchase up to 551,809 shares of our common stock as of December 31, 2006, of which 49,809 shares are vested as of December 31, 2006. | |
| (4) | Mr. Jaeger owns options to purchase up to 32,000 shares of our common stock as of December 31, 2006, of which 14,000 shares are vested as of December 31, 2006. | |
| (5) | Mr. Nehra did not stand for re-election to our board of directors at the 2006 Annual Meeting of Shareholders. Mr. Nehra did not own any outstanding options as of December 31, 2006. | |
| (6) | Mr. Roe did not stand for re-election to our board of directors at the 2006 Annual Meeting of Shareholders. Mr. Roe did not own any outstanding options as of December 31, 2006. | |
| (7) | Dr. Siebert joined our board in November 2006. The fees paid to him represent a pro-rated portion of his retainer for the period during which he served as a director. Dr. Siebert owns options to purchase up to 30,000 shares of our common stock as of December 31, 2006, of which no shares are vested as of December 31, 2006. | |
| (8) | Mr. R. Thompson did not stand for re-election to our board of directors at the 2006 Annual Meeting of Shareholders. Mr. R. Thompson’s option awards includes option expense related to the options that were granted when he previously served as our CEO. Mr. R. Thompson did not own any outstanding options as of December 31, 2006. | |
| (9) | Mr. V. Thompson owns options to purchase up to 87,600 shares of our common stock as of December 31, 2006, of which 54,600 shares are vested as of December 31, 2006. |
In 2007, the Chairman of the Board will receive an annual retainer of $50,000 and all other non-employee directors will receive an annual cash retainer of $30,000. Board members also receive additional annual retainers for serving on board committees. The additional annual retainer for the Chairman of the Audit Committee will be $15,000 and the additional annual retainer for all other members of the Audit Committee will be $5,000. The additional annual retainer for the Chairman of the Compensation Committee and the Chairman of the Nominating and Corporate Governance Committee will be $10,000 and the additional annual retainer for all other members will be $5,000. The board retainer covers six meetings in a year and, if exceeded, the Chairman of the Board will receive $1,500 for each additional meeting and the other board members will receive $1,000 for each additional meeting. If the number of meetings in a year for any given committee exceeds four, the chairman of the committee will receive $1,500 for each additional meeting and the other committee members will receive $1,000 for each additional
82
Table of Contents
meeting. Our directors are also entitled to receive reimbursement of reasonableout-of-pocket expenses incurred by them to attend board meetings.
In addition to the cash compensation, each non-employee director will be granted an annual stock option award. Each non-employee director will automatically receive an option to purchase up to 30,000 shares of our common stock upon election to the board. The Chairman of the Board will be granted automatically an option to purchase up to 35,000 shares of our common stock upon re-election to the board and the other members of the board will be granted automatically an option to purchase up to 20,000 shares of our common stock upon re-election to the board.
Limitation of Liability of Officers and Directors and Indemnification
Our articles of incorporation and bylaws include provisions to (i) eliminate the personal liability of our directors for monetary damages resulting from breaches of their fiduciary duty, to the extent permitted by California law and (ii) permit us to indemnify our directors and officers, employees and other agents to the fullest extent permitted by the California Corporations Code. Pursuant to Section 317 of the California Corporations Code, a corporation generally has the power to indemnify its present and former directors, officers, employees and agents against any expenses incurred by them in connection with any suit to which they are, or are threatened to be made, a party by reason of their serving in such positions so long as they acted in good faith and in a manner they reasonably believed to be in, or not opposed to, the best interests of a corporation and, with respect to any criminal action, they had no reasonable cause to believe their conduct was unlawful. We believe that these provisions are necessary to attract and retain qualified persons as directors and officers. These provisions do not eliminate liability for breach of the directors’ duty of loyalty to us or our shareholders, for acts or omissions not in good faith or involving intentional misconduct or knowing violations of law, for any transaction from which the director derived an improper personal benefit or for any willful or negligent payment of any unlawful dividend.
We have entered into indemnification agreements with certain officers, including each of our named executive officers, and each of our directors that provide, among other things, that we will indemnify such officer or director, under the circumstances and to the extent provided for therein, for expenses, damages, judgments, fines and settlements such officer or director may be required to pay in actions or proceedings to which such officer or director is or may be made a party by reason of such officer’s or director’s position as an officer, director or other agent of us, and otherwise to the full extent permitted under California law and our bylaws.
83
Table of Contents
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The following table sets forth certain information regarding the ownership of our common stock as of March 1, 2007 by: (i) each director and nominee for director; (ii) each of the executive officers named in the Summary Compensation Table (provided above); (iii) all of our executive officers and directors as a group; and (iv) all those known by us to be beneficial owners of more than five percent of our common stock.
| Beneficial Ownership | ||||||||
| Common | ||||||||
| Number of | Percent of | |||||||
Beneficial Owner | Shares | Total (%) | ||||||
| Wellington Management Company LLP(1) | 6,441,400 | 11.9 | ||||||
| 75 State Street Boston, MA 02109 | ||||||||
| RA Capital Management, LLC(2) | 6,355,000 | 11.8 | ||||||
| 111 Huntington Avenue, Suite 610 Boston, MA 02199 | ||||||||
| Kevin C. Tang(3) | 6,262,500 | 11.6 | ||||||
| 4401 Eastgate Mall San Diego, CA 92121 | ||||||||
| Highbridge International LLC(4) | 4,330,000 | 8.0 | ||||||
c/o Harmonic Fund Services The Cayman Corporate Centre, 4th Floor 27 Hospital Road Grand Cayman, Cayman Islands, B.W.I. | ||||||||
| Deerfield Capital, L.P.(5) | 3,303,100 | 6.1 | ||||||
780 3rd Avenue, 37th Floor New York, NY 10017 | ||||||||
| Igor Gonda(6) | 79,626 | * | ||||||
| Thomas C. Chesterman(7) | 155,001 | * | ||||||
| Babatunde A. Otulana(8) | 191,266 | * | ||||||
| Virgil D. Thompson(9) | 75,000 | * | ||||||
| Frank H. Barker(10) | 45,043 | * | ||||||
| Stephen O. Jaeger(11) | 23,000 | * | ||||||
| John M. Siebert(12) | 11,500 | * | ||||||
| V. Bryan Lawlis(13) | 43,869 | * | ||||||
| Stephen J. Farr(14) | 10,470 | * | ||||||
| All executive officers and directors as a group (7 persons)(15) | 580,436 | 1.1 | ||||||
| (1) | Based upon information contained in a Schedule 13G as filed with the Securities and Exchange Commission (“SEC”) on February 12, 2007 and a letter to us dated March 16, 2007, Wellington Management Company LLP (“Wellington”), in its capacity as investment adviser, may be deemed to beneficially own 6,441,400 shares of the Issuer which are held of record by clients of Wellington. Those clients have the right to receive, or the power to direct the receipt of, dividends from, or the proceeds from the sale of, such securities. No such client is known by Wellington to have such right or power with respect to more than five percent of our securities. Wellington disclaims beneficial ownership and any pecuniary interest in these shares. | |
| (2) | Based upon information contained in a Schedule 13G filed with the SEC on February 5, 2007, Mr. Richard H. Aldrich and Mr. Peter Kolchinsky (together, the “Managers”) are the managers of RA Capital Management, LLC (“Capital”), which is the sole general partner of RA Capital Biotech Fund, L.P. (the “Fund”). In the aggregate, the Reporting Persons beneficially own 6,355,000 shares of common stock (the “RA Shares”). Each Reporting Person beneficially own the RA Shares. The Fund has the power to vote and dispose of the shares beneficially owned by it (as described above). Capital, as the sole general partner of the Fund, has the sole authority to vote and dispose of the RA Shares. The Manager, by virtue of his position as manager of Capital, has the shared authority to vote and dispose of all of the RA Shares. |
84
Table of Contents
| (3) | Based upon information contained in a Schedule 13G filed with the SEC on February 2, 2007, Kevin C Tang may be deemed to beneficially own 6,262,500 shares of our common stock, which includes 5,310,000 shares owned of record by Tang Capital Partners, for which Tang Capital Management, of which Mr. Tang is manager, serves as general partner. Mr. Tang shares voting and dispositive power over such shares with Tang Capital Management and Tang Capital Partners. With respect to the remaining 952,500 shares that Mr. Tang may be deemed to beneficially own, Mr. Tang has shared dispositive power and no voting power over 210,000 shares and sole voting and dispositive power over 742,500 shares. Mr. Tang disclaims beneficial ownership of all such shares except to the extent of his pecuniary interest therein. | |
| (4) | Based upon information contained in a Schedule 13G filed with the SEC on February 1, 2007, each reporting person therein may be deemed the beneficial owner of 4,330,000 shares of common stock owned by Highbridge International LLC. Highbridge International LLC is a subsidiary of Highbridge Master L.P. Highbridge Capital Corporation and Highbridge Capital L.P. are limited partners of Highbridge Master L.P. Highbridge GP, Ltd. is the General Partner of Highbridge Master L.P. Highbridge GP, LLC is the General Partner of Highbridge Capital L.P. Highbridge Capital Management, LLC is the trading manager of Highbridge Capital Corporation, Highbridge Capital L.P., Highbridge International LLC and Highbridge Master L.P. Glenn Dubin is a Co-Chief Executive Officer of Highbridge Capital Management, LLC. Henry Swieca is a Co-Chief Executive Officer of Highbridge Capital Management, LLC. The foregoing should not be construed in and of itself as an admission by any reporting person as to beneficial ownership of shares of common stock owned by another reporting person. In addition, each of Highbridge Master L.P., Highbridge Capital Corporation, Highbridge Capital L.P., Highbridge GP, Ltd., Highbridge GP, LLC, Highbridge Capital Management, LLC, Glenn Dubin and Henry Swieca disclaims beneficial ownership of shares of common stock owned by Highbridge International LLC. | |
| (5) | Based upon information contained in a Schedule 13G filed with the SEC on February 2, 2007, and information provided directly to us, Deerfield Capital, L.P., Deerfield Special Situations Fund, L.P., Deerfield Management Company, L.P., Deerfield Special Situations Fund International Limited and James E. Flynn share voting and dispositive power over the 3,303,100 shares reflected as beneficially owned. | |
| (6) | Includes 30,000 shares of common stock subject to options exercisable within 60 days of March 1, 2007. | |
| (7) | Includes 144,998 shares of common stock subject to options and 570 shares of common stock subject to outstanding warrants that are each exercisable within 60 days of March 1, 2007. | |
| (8) | Includes 183,247 shares of common stock subject to options exercisable within 60 days of March 1, 2007. | |
| (9) | Includes 71,100 shares of common stock subject to options exercisable within 60 days of March 1, 2007. |
| (10) | Includes 45,043 shares of common stock subject to options exercisable within 60 days of March 1, 2007. | |
| (11) | Includes 23,000 shares of common stock subject to options exercisable within 60 days of March 1, 2007. | |
| (12) | Includes 7,500 shares of common stock subject to options exercisable within 60 days of March 1, 2007. | |
| (13) | Includes 32,857 shares of common stock subject to options and 1,709 shares of common stock subject to outstanding warrants that are each exercisable within 60 days of March 1, 2007. Dr. Lawlis ceased serving as our President and Chief Executive Officer as of August 10, 2006. | |
| (14) | Dr. Farr ceased serving as an executive officer on June 16, 2006. | |
| (15) | See footnotes (6) through (12) above. |
85
Table of Contents
Equity Compensation Plan Information
The following table summarizes our equity compensation plan information as of December 31, 2006. Information is included for the equity compensation plans approved by our stockholders. There are no equity compensation plans not approved by our stockholders.
| Common Stock | ||||||||||||
| Available for | ||||||||||||
| Future Issuance | ||||||||||||
| Common Stock to | Under Equity | |||||||||||
| be Issued Upon | Weighted-Average | Compensation | ||||||||||
| Exercise of | Exercise Price of | Plans (Excluding | ||||||||||
| Outstanding Options | Outstanding Options | Securities Reflected | ||||||||||
Plan Category | and Rights(1) | and Rights | in Column (a))(1) | |||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by Aradigm stockholders | 3,163,981 | (1) | $ | 8.90 | 1,834,849 | (2) | ||||||
| Equity compensation plans not approved by Aradigm stockholders | — | — | — | |||||||||
| (1) | Includes 100,000 shares that we agreed to grant to our President and Chief Executive Officer, in two 50,000 share tranches, to be earned based on achievement of minimum share price appreciation objectives after each of the first two years from the employment start date of August 10, 2006. | |
| (2) | Issued pursuant to the Company’s 1996 Equity Incentive Plan, the 1996 Non-Employee Directors’ Plan, and the 2005 Equity Incentive Plan and shares available for future issuance includes 340,847 shares reserved under ESPP. (See Note 6 of the Financial Statements.) |
| Item 13. | Certain Relationships and Related Transactions and Director Independence |
Independence of Directors
Our board of directors has determined that Messrs. Barker, Jaeger and V. Thompson and Dr. Siebert are each independent under the standards set forth in Nasdaq Stock Market, or Nasdaq, Marketplace Rule 4200(a)(15). We intend to maintain at least two directors on the board that meet these independence standards. Our board has also determined that each member of our Compensation Committee and Nominating and Corporate Governance Committee is independent under Nasdaq Marketplace Rule 4200(a)(15) and each member of our Audit Committee is independent under the standards set forth in Nasdaq Marketplace Rules 4350(d)(2)(A)(i) and (ii).
Review, Approval or Ratification of Transactions with Related Persons
Our policy is to require that any transaction with a related party required to be reported under applicable Securities and Exchange Commission rules, other than compensation-related matters and waivers of our code of business conduct and ethics, be reviewed and approved or ratified by a majority of independent, disinterested directors. We have not adopted procedures for review of, or standards for approval of, these transactions, but instead review such transactions on a case by case basis. Our policy is to require that all compensation-related matters be recommended for board approval by the Compensation Committee and that any waiver of our code of business conduct and ethics be reviewed and approved by the Nominating and Corporate Governance Committee and be reported under applicable SEC rules.
Transactions with Novo Nordisk
As of January 26, 2005, we restructured the AERx iDMS program, pursuant to a restructuring agreement entered into with Novo Nordisk and its subsidiary, Novo Nordisk Delivery Technologies, or NNDT, in September 2004. Under the terms of the restructuring agreement, we sold certain equipment, leasehold improvements and other tangible assets used in the AERx iDMS program to NNDT, for a cash payment of $55.3 million (before refund of cost advances made by Novo Nordisk). Our expenses related to this transaction for legal and other consulting
86
Table of Contents
costs were $1.1 million. In connection with the restructuring transaction, we entered into various related agreements with Novo Nordisk and NNDT, including the following:
| • | an amended and restated license agreement amending the development and license agreement previously in place with Novo Nordisk, expanding Novo Nordisk’s development and manufacturing rights to the AERx iDMS program and providing for royalties to us on future AERx iDMS net sales in lieu of a percentage interest in the gross profits from the commercialization of AERx iDMS, which royalties run until the later of last patent expiry or last use of our intellectual property and which apply to future enhancements or generations of our AERx delivery technology; | |
| • | a three-year agreement under which NNDT agreed to perform contract manufacturing of AERxiDMS-identical devices and dosage forms filled with compounds provided by us in support of preclinical and initial clinical development of other products that incorporate our AERx delivery system; and | |
| • | an amendment of the common stock purchase agreement in place with Novo Nordisk prior to the closing of the restructuring transaction, (i) deleting the provisions whereby we can require Novo Nordisk to purchase certain additional amounts of common stock, (ii) imposing certain restrictions on the ability of Novo Nordisk to sell shares of our common stock and (iii) providing Novo Nordisk with certain registration and information rights with respect to these shares. |
As a result of this transaction, we were no longer obligated to continue work related to the non-refundable milestone payment from Novo Nordisk in connection with the commercialization of AERx iDMS. We also entered into transition and support agreements with NNDT and we were released from our contractual obligations relating to future operating lease payments for two buildings assigned to NNDT. Pursuant to the restructuring agreement, we terminated a manufacturing and supply agreement and a patent cooperation agreement, each previously in place with Novo Nordisk and dated October 22, 2001. As part of the restructuring, one of our officers and many of our employees became employees of NNDT.
On July 3, 2006, we further restructured our relationship with Novo Nordisk through an intellectual property assignment, a royalty prepayment and an eight-year promissory note with Novo Nordisk. The promissory note was secured by the royalty payments on any AERx iDMS sales by Novo Nordisk under the license with us. The key features of this restructuring included:
| • | our transfer to Novo Nordisk of the ownership of 23 issued United States patents and their correspondingnon-United States counterparts, if any, as well as related pending applications, in exchange for $12.0 million paid to us in cash. We retained exclusive, royalty-free control of these patents outside the field of glucose control and will continue to be entitled to royalties that will rise to an average of five percent or higher by the fifth year after commercialization with respect to any inhaled insulin products marketed or licensed by Novo Nordisk. | |
| • | our receipt of a royalty prepayment of $8.0 million in exchange for a one percent reduction on our average royalty rate for the commercialized AERx iDMS product. As a result, we will receive royalty rates under our license agreement with Novo Nordisk that will commence at a minimum of 3.25% on launch, and that we estimate will average 5% over the life of the product. | |
| • | our issuance of an eight-year promissory note to Novo Nordisk in connection with our receipt from Novo Nordisk of a loan in the principal amount of $7.5 million with interest accruing at 5% per year. The principal and accrued interest will be payable to Novo Nordisk in three equal payments of $3.5 million on July 2, 2012, July 1, 2013 and June 30, 2014, commencing in six years at a five percent annual interest rate. Our obligations under the note are secured by royalty payments upon any commercialization of the AERx iDMS product. |
We and Novo Nordisk continue to cooperate and share in technology development, as well as intellectual property development and defense. Both we and Novo Nordisk have access to any developments or improvements the other might make to the AERx delivery system, within their respective fields of use. In August 2006, Novo Nordisk announced that it had filed a lawsuit against Pfizer claiming that Exubera, an inhaled insulin product that Pfizer has been developing with Nektar Therapeutics, infringes a patent originally developed by us and now
87
Table of Contents
owned by Novo Nordisk with rights retained by us outside the field of glucose control. In December 2006, Novo Nordisk’s motion for a preliminary injunction in this case was denied. While the outcome of this lawsuit is highly uncertain, we are entitled to a portion of any proceeds, net of litigation costs, that may be received by Novo Nordisk from a favorable outcome
| Item 14. | Principal Accountant Fees and Services |
The following table represents aggregate fees billed to us for fiscal years ended December 31, 2006 and December 31, 2005, by Ernst & Young LLP, our independent registered public accounting firm. All services described below were pre-approved by the Audit Committee.
| Fiscal Year Ended | ||||||||
| December 31, | ||||||||
| 2006 | 2005 | |||||||
| (In thousands) | ||||||||
| Audit Fees(1) | $ | 505 | $ | 377 | ||||
| Audit-related Fees | — | 30 | ||||||
| Tax Fees | — | — | ||||||
| All Other Fees | — | — | ||||||
| Total Fees | $ | 505 | $ | 407 | ||||
| (1) | Audit fees represent fees for professional services related to the performance of the audit of our annual financial statements, review of our quarterly financial statements and consents on SEC filings. |
The Audit Committee pre-approves audit services, audit-related services and non-audit services provided by our independent registered public accounting firm, Ernst & Young, LLP, and will not approve services that the Audit Committee determines are outside the bounds of applicable laws and regulations. Pre-approval may also be given as part of the Audit Committee’s approval of the scope of the engagement of the independent registered public accounting firm or on an individual explicitcase-by-case basis before the independent registered public accounting firm is engaged to provide each service. The pre-approval of services may be delegated to one or more of the Audit Committee’s members, but the decision must be reported to the full Audit Committee at its next scheduled meeting.
The Audit Committee has determined that the rendering of the services, other than audit services, by Ernst & Young LLP is compatible with maintaining the principal accountant’s independence.
PART IV
| Item 15. | Exhibits and Financial Statements Schedules |
(a)(1) Financial Statements.
Included in Part II of this Report:
| Page in | ||||
| Form 10-K | ||||
| 46 | ||||
| 47 | ||||
| 48 | ||||
| 49 | ||||
| 50 | ||||
| 51 | ||||
88
Table of Contents
(2) Financial Statement Schedules.
All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
(3) Exhibits.
Exhibit No. | Description | |||
| 3 | .1(1) | Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .2(2) | Bylaws of the Company, as amended. | ||
| 3 | .3(3) | Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .4(4) | Amended and Restated Certificate of Determination of Preferences of Series A Convertible Preferred Stock. | ||
| 3 | .5(3) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .6(3) | Certificate of Amendment of Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .7(5) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .8(5) | Certificate of Amendment of Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .9(6) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 4 | .1 | Reference is made to Exhibits 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8 and 3.9. | ||
| 4 | .2(1) | Specimen common stock certificate. | ||
| 10 | .1(1)+ | Form of Indemnity Agreement between the Registrant and each of its directors and officers. | ||
| 10 | .2(7)+ | 2005 Equity Incentive Plan, as amended. | ||
| 10 | .3(1)+ | Form of the Company’s Incentive Stock Option Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .4(1)+ | Form of the Company’s Non-statutory Stock Option Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .5(1)+ | 1996 Non-Employee Directors’ Stock Option Plan. | ||
| 10 | .6(1)+ | Form of the Company’s Non-statutory Stock Option Agreement under the 1996 Non-Employee Directors’ Stock Option Plan. | ||
| 10 | .7(7)+ | Employee Stock Purchase Plan, as amended. | ||
| 10 | .8(1)+ | Form of the Company’s Employee Stock Purchase Plan Offering Document. | ||
| 10 | .9(8) | Lease Agreement for the property located in Phase V of the Britannia Point Eden Business Park in Hayward, California, dated January 28, 1998, between the Company and Britannia Point Eden, LLC. | ||
| 10 | .10(9) | Rights Agreement, dated as of August 31, 1998, between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10a(3) | Amendment to Rights Agreement, dated as of October 22, 2001, by and between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10b(3) | Amendment to Rights Agreement, dated as of December 6, 2001, by and between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10c(10) | Amendment No. 3 to Rights Agreement, dated as of January 24, 2007, by and between the Company and Computershare Trust Company, N.A. | ||
| 10 | .11(11) | Securities Purchase Agreement, dated as of November 7, 2003, by and among the Company and the purchasers named therein. | ||
| 10 | .12(12) | Securities Purchase Agreement, dated as of November 14, 2003, by and among the Company and the purchaser named therein. | ||
| 10 | .13(13)# | Restructuring Agreement, dated as of September 28, 2004, by and among the Company, Novo Nordisk A/ S and Novo Nordisk Delivery Technologies, Inc. | ||
| 10 | .14(14) | Securities Purchase Agreement, dated as of December 17, 2004, by and among the Company and the purchasers named therein. | ||
| 10 | .15(7) | Amended and Restated Stock Purchase Agreement, dated as of January 26, 2005, by and among the Company, Novo Nordisk A/ S and Novo Nordisk Pharmaceuticals, Inc. | ||
| 10 | .16(7)+ | Form of Change of Control Agreement entered into between the Company and certain of the Company’s senior officers. | ||
| 10 | .17(15)+ | Executive Officer Severance Benefit Plan. | ||
89
Table of Contents
Exhibit No. | Description | |||
| 10 | .18(6)+ | Form of the Company’s Restricted Stock Bonus Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .19(16)# | Second Amended and Restated License Agreement, dated as of July 3, 2006, by and between the Company and Novo Nordisk A/ S. | ||
| 10 | .20(7) | Promissory Note and Security Agreement, dated July 3, 2006, by and between the Company and Novo Nordisk A/ S. | ||
| 10 | .21(7)# | Asset Purchase Agreement, dated as of August 25, 2006, by and between the Company and Zogenix, Inc. | ||
| 10 | .22(7)+ | Employment Agreement, dated as of August 10, 2006, with Dr. Igor Gonda. | ||
| 23 | .1 | Consent of Independent Registered Public Accounting Firm. | ||
| 24 | .1 | Power of Attorney. Reference is made to the signature page. | ||
| 31 | .1 | Section 302 Certification of the Chief Executive Officer. | ||
| 31 | .2 | Section 302 Certification of the Chief Financial Officer. | ||
| 32 | .1 | Section 906 Certification of the Chief Executive Officer and the Chief Financial Officer. | ||
| + | Represents a management contract or compensatory plan or arrangement. | |
| # | The Commission has granted the Company’s request for confidential treatment with respect to portions of this exhibit. | |
| (1) | Incorporated by reference to the Company’sForm S-1(No. 333-4236) filed on April 30, 1996, as amended. | |
| (2) | Incorporated by reference to the Company’sForm 10-Q filed on August 14, 1998. | |
| (3) | Incorporated by reference to the Company’sForm 10-K filed on March 29, 2002. | |
| (4) | Incorporated by reference to the Company’sForm S-3(No. 333-76584) filed on January 11, 2002, as amended. | |
| (5) | Incorporated by reference to the Company’sForm 10-Q filed on August 13, 2004. | |
| (6) | Incorporated by reference to the Company’sForm 10-K filed on March 31, 2006. | |
| (7) | Incorporated by reference to the Company’sForm S-1(No. 333-138169) filed on October 24, 2006, as amended. | |
| (8) | Incorporated by reference to the Company’sForm 10-K filed on March 24, 1998, as amended. | |
| (9) | Incorporated by reference to the Company’sForm 8-K filed on September 2, 1998. | |
| (10) | Incorporated by reference to the Company’sForm 8-K filed on January 30, 2007. | |
| (11) | Incorporated by reference to the Company’sForm 8-K filed on November 12, 2003. | |
| (12) | Incorporated by reference to the Company’sForm 8-K filed on November 20, 2003. | |
| (13) | Incorporated by reference to the Company’sForm 8-K filed on December 23, 2004. | |
| (14) | Incorporated by reference to the Company’sForm 10-Q filed on August 14, 2006. | |
| (15) | Incorporated by reference to the Company’sForm 10-Q filed on November 15, 2004. | |
| (16) | Incorporated by reference to the Company’sForm 8-K filed on October 13, 2005. |
| (b) | Index to Exhibits. |
See Exhibits listed under Item 15(a) (3).
| (c) | Financial Statement Schedules. |
All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
90
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Hayward, State of California, on the 30th day of March 2007.
ARADIGM CORPORATION
| By: | /s/ Igor Gonda |
Igor Gonda
President and Chief Executive Officer
KNOWN ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints, jointly and severally, Igor Gonda and Thomas C. Chesterman, and each one of them,attorneys-in-fact for the undersigned, each with power of substitution, for the undersigned in any and all capacities, to sign any and all amendments to this Report onForm 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of saidattorneys-in-fact, or their substitutes, may do or cause to be done by virtue hereof.
IN WITNESS WHEREOF, each of the undersigned has executed this Power of Attorney as of the date indicated opposite his name.
Pursuant to the requirements of the Securities and Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||||
/s/ Igor Gonda Igor Gonda | President, Chief Executive Officer and Director (Principal Executive Officer) | March 30, 2007 | ||||
/s/ Thomas C. Chesterman Thomas C. Chesterman | Sr. VP and Chief Financial Officer (Principal Financial and Accounting Officer) | March 30, 2007 | ||||
/s/ Virgil D. Thompson Virgil D. Thompson | Chairman of the Board and Director | March 30, 2007 | ||||
/s/ Frank H. Barker Frank H. Barker | Director | March 30, 2007 | ||||
/s/ Stephen O. Jaeger Stephen O. Jaeger | Director | March 30, 2007 | ||||
/s/ John M. Siebert John M. Siebert | Director | March 30, 2007 | ||||
91
Table of Contents
EXHIBIT INDEX
Exhibit No. | Description | |||
| 3 | .1(1) | Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .2(2) | Bylaws of the Company, as amended. | ||
| 3 | .3(3) | Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .4(4) | Amended and Restated Certificate of Determination of Preferences of Series A Convertible Preferred Stock. | ||
| 3 | .5(3) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .6(3) | Certificate of Amendment of Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .7(5) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 3 | .8(5) | Certificate of Amendment of Certificate of Determination of Series A Junior Participating Preferred Stock. | ||
| 3 | .9(6) | Certificate of Amendment of Amended and Restated Articles of Incorporation of the Company. | ||
| 4 | .1 | Reference is made to Exhibits 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8 and 3.9. | ||
| 4 | .2(1) | Specimen common stock certificate. | ||
| 10 | .1(1)+ | Form of Indemnity Agreement between the Registrant and each of its directors and officers. | ||
| 10 | .2(7)+ | 2005 Equity Incentive Plan, as amended. | ||
| 10 | .3(1)+ | Form of the Company’s Incentive Stock Option Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .4(1)+ | Form of the Company’s Non-statutory Stock Option Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .5(1)+ | 1996 Non-Employee Directors’ Stock Option Plan. | ||
| 10 | .6(1)+ | Form of the Company’s Non-statutory Stock Option Agreement under the 1996 Non-Employee Directors’ Stock Option Plan. | ||
| 10 | .7(7)+ | Employee Stock Purchase Plan, as amended. | ||
| 10 | .8(1)+ | Form of the Company’s Employee Stock Purchase Plan Offering Document. | ||
| 10 | .9(8) | Lease Agreement for the property located in Phase V of the Britannia Point Eden Business Park in Hayward, California, dated January 28, 1998, between the Company and Britannia Point Eden, LLC. | ||
| 10 | .10(9) | Rights Agreement, dated as of August 31, 1998, between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10a(3) | Amendment to Rights Agreement, dated as of October 22, 2001, by and between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10b(3) | Amendment to Rights Agreement, dated as of December 6, 2001, by and between the Company and ComputerShare Trust Company, N.A. | ||
| 10 | .10c(10) | Amendment No. 3 to Rights Agreement, dated as of January 24, 2007, by and between the Company and Computershare Trust Company, N.A. | ||
| 10 | .11(11) | Securities Purchase Agreement, dated as of November 7, 2003, by and among the Company and the purchasers named therein. | ||
| 10 | .12(12) | Securities Purchase Agreement, dated as of November 14, 2003, by and among the Company and the purchaser named therein. | ||
| 10 | .13(13)# | Restructuring Agreement, dated as of September 28, 2004, by and among the Company, Novo Nordisk A/ S and Novo Nordisk Delivery Technologies, Inc. | ||
| 10 | .14(14) | Securities Purchase Agreement, dated as of December 17, 2004, by and among the Company and the purchasers named therein. | ||
| 10 | .15(7) | Amended and Restated Stock Purchase Agreement, dated as of January 26, 2005, by and among the Company, Novo Nordisk A/ S and Novo Nordisk Pharmaceuticals, Inc. | ||
| 10 | .16(7)+ | Form of Change of Control Agreement entered into between the Company and certain of the Company’s senior officers. | ||
92
Table of Contents
Exhibit No. | Description | |||
| 10 | .17(15)+ | Executive Officer Severance Benefit Plan. | ||
| 10 | .18(6)+ | Form of the Company’s Restricted Stock Bonus Agreement under the 2005 Equity Incentive Plan. | ||
| 10 | .19(16)# | Second Amended and Restated License Agreement, dated as of July 3, 2006, by and between the Company and Novo Nordisk A/ S. | ||
| 10 | .20(7) | Promissory Note and Security Agreement, dated July 3, 2006, by and between the Company and Novo Nordisk A/ S. | ||
| 10 | .21(7)# | Asset Purchase Agreement, dated as of August 25, 2006, by and between the Company and Zogenix, Inc. | ||
| 10 | .22(7)+ | Employment Agreement, dated as of August 10, 2006, with Dr. Igor Gonda. | ||
| 23 | .1 | Consent of Independent Registered Public Accounting Firm. | ||
| 24 | .1 | Power of Attorney. Reference is made to the signature page. | ||
| 31 | .1 | Section 302 Certification of the Chief Executive Officer. | ||
| 31 | .2 | Section 302 Certification of the Chief Financial Officer. | ||
| 32 | .1 | Section 906 Certification of the Chief Executive Officer and the Chief Financial Officer. | ||
| + | Represents a management contract or compensatory plan or arrangement. | |
| # | The Commission has granted the Company’s request for confidential treatment with respect to portions of this exhibit. | |
| (1) | Incorporated by reference to the Company’sForm S-1(No. 333-4236) filed on April 30, 1996, as amended. | |
| (2) | Incorporated by reference to the Company’sForm 10-Q filed on August 14, 1998. | |
| (3) | Incorporated by reference to the Company’sForm 10-K filed on March 29, 2002. | |
| (4) | Incorporated by reference to the Company’sForm S-3(No. 333-76584) filed on January 11, 2002, as amended. | |
| (5) | Incorporated by reference to the Company’sForm 10-Q filed on August 13, 2004. | |
| (6) | Incorporated by reference to the Company’sForm 10-K filed on March 31, 2006. | |
| (7) | Incorporated by reference to the Company’sForm S-1(No. 333-138169) filed on October 24, 2006, as amended. | |
| (8) | Incorporated by reference to the Company’sForm 10-K filed on March 24, 1998, as amended. | |
| (9) | Incorporated by reference to the Company’sForm 8-K filed on September 2, 1998. | |
| (10) | Incorporated by reference to the Company’sForm 8-K filed on January 30, 2007. | |
| (11) | Incorporated by reference to the Company’sForm 8-K filed on November 12, 2003. | |
| (12) | Incorporated by reference to the Company’sForm 8-K filed on November 20, 2003. | |
| (13) | Incorporated by reference to the Company’sForm 8-K filed on December 23, 2004. | |
| (14) | Incorporated by reference to the Company’sForm 10-Q filed on August 14, 2006. | |
| (15) | Incorporated by reference to the Company’sForm 10-Q filed on November 15, 2004. | |
| (16) | Incorporated by reference to the Company’sForm 8-K filed on October 13, 2005. |
93