QuickLinks -- Click here to rapidly navigate through this document
As filed with the United States Securities and Exchange Commission on May 13, 2008
Registration No. 333-149663
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 1
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
CYDEX PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 74-2807105 | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification No.) |
10513 W. 84th Terrace
Lenexa, KS 66214
(913) 685-8850
(Address, including zip code, and telephone number, including area code, of registrant's principal executive offices)
JOHN M. SIEBERT, Ph.D.
CHAIRMAN AND CHIEF EXECUTIVE OFFICER
CYDEX PHARMACEUTICALS, INC.
10513 W. 84th TERRACE
LENEXA, KS 66214
(913) 685-8850
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
| Brett D. White, Esq. Eric H. Anderson, Esq. Cooley Godward KronishLLP Five Palo Alto Square 3000 El Camino Real Palo Alto, CA 94306 (650) 843-5000 | William C. Davisson, III, Esq. Goodwin ProcterLLP 135 Commonwealth Drive Menlo Park, CA 94025 (650) 752-3100 |
Approximate date of commencement of proposed sale to the public:As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filero | Accelerated filero | |
| Non-accelerated filerý (Do not check if a smaller reporting company) | Smaller reporting company o |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment that specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED MAY 13, 2008
Shares
| CyDex Pharmaceuticals, Inc. |  |
Common Stock
$ per share
- •
- We are offering shares of common stock.
- •
- This is our initial public offering and no public market currently exists for our shares.
- •
- We anticipate the initial public offering price will be between $ and $ per share.
- •
- We have applied to have the common stock approved for listing on the Nasdaq Global Market under the symbol CYDX.
Investing in our common stock involves risks. See "Risk Factors" beginning on page 10.
Per Share | Total | |||||
|---|---|---|---|---|---|---|
| Initial public offering price | $ | $ | ||||
| Underwriting discount | $ | $ | ||||
| Proceeds, before expenses, to CyDex Pharmaceuticals, Inc. | $ | $ | ||||
We have granted the underwriters a 30-day option to purchase up to additional shares of common stock from us to cover over-allotments, if any.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares to purchasers on or about , 2008.
Pacific Growth Equities, LLC
JMP Securities
Fortis Securities LLC
, 2008
You should rely only on the information contained in this prospectus. We have not authorized anyone to provide you with different information. We are not making an offer of these securities in any state where the offer is not permitted. You should not assume that the information contained in this prospectus is accurate as of any date other than the date on the front of this prospectus.
| | Page | |
|---|---|---|
| Prospectus Summary | 1 | |
| Risk Factors | 10 | |
| Forward-Looking Statements and Industry Data | 30 | |
| Use of Proceeds | 32 | |
| Dividend Policy | 32 | |
| Capitalization | 33 | |
| Dilution | 35 | |
| Selected Consolidated Financial Data | 37 | |
| Management's Discussion and Analysis of Financial Condition and Results of Operations | 39 | |
| Business | 57 | |
| Management | 85 | |
| Certain Relationships and Related Persons Transactions | 112 | |
| Principal Stockholders | 114 | |
| Description of Capital Stock | 117 | |
| Certain United States Federal Tax Consequences to Non-United States Holders | 121 | |
| Shares Eligible for Future Sale | 124 | |
| Underwriting | 126 | |
| Legal Matters | 129 | |
| Experts | 129 | |
| Where You Can Find Additional Information | 130 | |
| Index to Consolidated Financial Statements | F-1 |
The items in the following summary are described in more detail later in this prospectus. This summary provides an overview of selected information and does not contain all of the information you should consider. After you read the following summary, you should read and consider carefully the more detailed information and financial statements and related notes that we include in this prospectus. If you invest in our common stock, you are assuming a high degree of risk. See the section of this prospectus entitled "Risk Factors."
Overview
We are a specialty pharmaceutical company focused on the development and commercialization of drugs specifically designed to address limitations of current therapies in selected established markets. We have developed a broad portfolio of 12 product candidates utilizing our drug formulation technology using Captisol® cyclodextrins. Captisol cyclodextrins are a patent protected, specifically modified family of cyclodextrins designed to improve solubility, stability, bioavailability, safety and/or dosing of a number of active pharmaceutical ingredients, or APIs. Captisol cyclodextrins are ring-shaped molecules comprised of sugar molecules. A number of different types of APIs that are not otherwise readily soluble in water can interact with Captisol cyclodextrins, resulting in compounds that are more water-soluble than the API would be standing alone. By applying our Captisol technology to APIs with established safety and efficacy characteristics, we believe that we can create differentiated products with significant commercial opportunities, while simultaneously reducing development risk, costs and timelines as compared to the development of new chemical entities.
We generate revenue through a number of activities, including outlicensing our drug formulation technology to established pharmaceutical companies. Our technology has been validated by the launch of four Captisol-Enabled® products commercialized by Pfizer and Bristol-Myers Squibb, or BMS. Our outlicensing effort has allowed us to advance our Captisol technology, generate cash flow for our company and expand our strategy to capitalize on the growth opportunities and business lines enabled by Captisol. We intend to leverage the formulation, development, regulatory and intellectual property expertise garnered from our experience to facilitate the development and commercialization of our own product candidates for use in the acute care hospital setting. We refer to these product candidates as our retained product candidates. For those product candidates that will likely require more extensive development and commercialization efforts, which we refer to as our partner product candidates, we partner with established pharmaceutical or specialty pharmaceutical companies. All of our retained product candidates and partner product candidates are currently in development and have not yet been commercialized. We plan to continue to outlicense our Captisol technology to third parties for use in the development of their own products. The following table presents information
1
describing our seven leading product candidates, as well as the four Captisol-Enabled products currently marketed by Pfizer and BMS:
| Captisol-Enabled Product Candidate or Product | Brand Name of Approved Third- Party Product Containing Chemically Equivalent API | Target Indication | Stage of Development | Partner or Outlicensee | ||||
|---|---|---|---|---|---|---|---|---|
| Retained Product Candidates | ||||||||
Captisol-Enabled Fosphenytoin IV and IM | Cerebyx | Status Epilepticus | NDA Submission Anticipated in Late 2008 | Retained by CyDex | ||||
Captisol-Enabled Melphalan IV | Alkeran | Multiple Myeloma | Preclinical | Retained by CyDex | ||||
Captisol-Enabled Topiramate IV | Topamax | Epilepsy | Pre-IND Meeting Held in May 2008 | Retained by CyDex | ||||
Partner Product Candidates | ||||||||
Captisol-Enabled Amiodarone IV | Cordarone | Arrhythmia | NDA Submitted in 2008 | Prism | ||||
Captisol-Enabled Clopidogrel IV | Plavix | Atherothrombosis | NDA Submission Anticipated in 2010 | Prism | ||||
Captisol-Enabled Budesonide solution for inhalation | Pulmicort Respules | Asthma | Phase 2 Clinical Trials Completed in the U.K. | AstraZeneca | ||||
Captisol-Enabled Budesonide nasal spray | Rhinocort | Nasal Allergy | Phase 2 Clinical Trial Completed in Canada | Under discussion | ||||
Marketed Products | ||||||||
Geodon IM | — | Bipolar Disorder and Schizophrenia | Market | Pfizer | ||||
Vfend IV | — | Antifungal | Market | Pfizer | ||||
Abilify Injection IM | — | Bipolar Disorder and Schizophrenia | Market | BMS | ||||
Cerenia | — | Canine Motion Sickness | Market | Pfizer | ||||
Issued patents relating to our product candidates and the Captisol-Enabled products marketed by Pfizer and BMS expire between 2010 and 2022, as more fully described in the section entitled "Business—Intellectual Property."
All of our product candidates are formulations of APIs used in commercially available drug products that have been approved by the Food and Drug Administration, or the FDA, for which we can potentially improve delivery efficiency. We believe we will be able to leverage the known safety and efficacy characteristics of these APIs to rapidly develop and commercialize product candidates on a Section 505(b)(2) regulatory pathway, under which the FDA may approve new drug applications, in part, on the basis of the FDA's previous findings of safety and efficacy for other drug products. This means that, upon a showing of bioequivalence to an approved drug product, one or more of our new drug applications could be approved on the basis of literature or FDA findings of safety and efficacy
2
related to the currently marketed APIs used in our formulations. While the FDA has discretion to request additional data to support approval, we believe that in some cases this path to registration should only necessitate the completion of a single or a small number of clinical trials demonstrating bioequivalence to the existing, previously approved formulation.
Our Products and Product Candidates
Retained Product Candidates
We are developing six product candidates for use in the acute care hospital setting, and we intend to build a focused U.S.-based specialty sales force to market these product candidates. The following describes our three leading retained product candidates:
Captisol-Enabled Fosphenytoin IV and IM. Fosphenytoin is an approved injectable therapy marketed by Pfizer under the brand name Cerebyx for the acute treatment of status epilepticus. The current formulation of Cerebyx requires refrigeration, making it difficult to utilize in emergency settings that do not have access to a refrigeration unit, such as many ambulances or helicopters. Our product candidate is a Captisol-Enabled formulation of fosphenytoin for either intravenous, or IV, or intramuscular, or IM, injection that we have designed to not require refrigeration. We anticipate filing a new drug application, or NDA, for Captisol-Enabled Fosphenytoin IV and IM in late 2008.
Captisol-Enabled Melphalan IV. Melphalan, marketed by Celgene Corporation under the name Alkeran, is an injectable, palliative treatment for patients with multiple myeloma for whom oral therapy is not appropriate. The current formulation of melphalan is presented as two separate vials that must be reconstituted immediately prior to use and is stable for only a period of up to two hours. As the actual administration of each dose of Alkeran is done over a minimum of 15 minutes, the prepared product must be administered immediately after mixing. We have developed Captisol-Enabled Melphalan IV, which is a one-vial system that we believe will have a longer use time. Assuming we are able to demonstrate bioequivalence to Alkeran in anticipated clinical trials, we plan to submit an NDA for Captisol-Enabled Melphalan IV in late 2009.
Captisol-Enabled Topiramate IV. Topiramate is an orally administered drug marketed by Ortho-McNeil Neurologics under the brand name Topamax for the prevention of migraines and the treatment of epilepsy. Topiramate is an oral solid dosage form, which may take up to two hours to reach peak blood levels, making it a suboptimal treatment option for the acute treatment of either a migraine or a seizure. Additionally, many patients in the acute setting are unable to take oral medications and require IV formulated products. We are developing Captisol-Enabled Topiramate IV to be used for the treatment of epilepsy, which we believe will provide significantly faster therapeutic onset compared to the oral formulation. A pre-investigational new drug, or pre-IND, meeting was held with the FDA in May 2008.
Partner Product Candidates
We are developing six product candidates that we have either collaborated with, outlicensed or intend to outlicense to partners for further development and commercialization. To date, we have entered into agreements with respect to Captisol-Enabled Amiodarone IV, Captisol-Enabled Clopidogrel IV and Captisol-Enabled Budesonide solution for inhalation. We believe that these agreements validate our technology and our ability to identify, develop and subsequently outlicense these opportunities. The following describes our leading partner product candidates:
Captisol-Enabled Amiodarone IV. Amiodarone is currently marketed under the brand name Cordarone for the treatment of life-threatening recurrent ventricular arrhythmia when patients have
3
not responded to other available antiarrhythmics or when alternative agents could not be tolerated. Due to the poor water-solubility of this API, the injectable formulation utilizes a solvent system that is associated with a potential major adverse side effect, a rapid decrease in blood pressure. We have developed Captisol-Enabled Amiodarone IV, which does not contain the solvent system found in the current formulation. We believe this will offer a potentially safer therapeutic option for patients. We have outlicensed Captisol-Enabled Amiodarone IV to Prism Pharmaceuticals, Inc., or Prism, and Prism is developing this product candidate. Based on our communications with Prism, we understand that Prism submitted an NDA for this product candidate in 2008.
Captisol-Enabled Clopidogrel IV. Clopidogrel, marketed as Plavix, is available as a solid oral dose to reduce the incidence of clot formation associated with cardiovascular surgery and cardiovascular complications. Clopidogrel is highly insoluble in water, and there are currently no IV or oral liquid dosage forms of this API available. As a result, physicians must wait a number of hours prior to surgery in order to allow the active drug in the oral dose to reach maximum blood levels. We believe that Captisol-Enabled Clopidogrel IV has the potential to reach peak blood levels within 30 minutes of administration. Furthermore, we also believe that our formulation may enable physicians to administer a lower dose of Clopidogrel relative to Plavix while achieving equivalent levels of bioactivity. We have engaged in a collaboration with Prism to develop Captisol-Enabled Clopidogrel IV. Based on our communications with Prism, we believe that Prism could submit an NDA for this product candidate in 2010.
Captisol-Enabled Budesonide. Budesonide is a steroid currently approved for the treatment of multiple indications, including asthma, hay fever and other allergies. However, current formulations require a relatively high dose and can require a relatively long time to administer. We are currently developing three formulations of Captisol-Enabled Budesonide: an inhalation solution for the treatment of asthma; a nasal spray for the treatment of nasal allergies; and an ophthalmic solution for the treatment of allergies. When tested on human volunteers, we observed that the Captisol-Enabled Budesonide solution for inhalation produced similar plasma levels in approximately one-fourth of the time relative to a budesonide suspension prepared from the commercially available Pulmicort Respules. As compliance is a serious concern with asthma patients, especially pediatric patients, we believe that this improved administration profile could be a distinct commercial advantage. We have also observed that when using a more efficient nebulizer, we may be able to use a significantly lower concentration of budesonide to achieve the same bioactivity as Pulmicort Respules, thereby potentially reducing the amount of steroid delivered. In 2006, we entered into a license agreement with Verus Pharmaceuticals Inc., or Verus, for the North American rights to the Captisol-Enabled Budesonide solution for inhalation, which Verus recently assigned to AstraZeneca AB, or AstraZeneca, through its subsidiary Tika Läkemedel AB. In 2008, we entered into an option agreement with AstraZeneca pursuant to which we granted AstraZeneca an exclusive, irrevocable option to expand the territory covered by the North American license agreement and related supply agreement to include all the countries in the world. We and our partners have already carried out three Phase 1 and two Phase 2 proof-of-concept clinical trials with Captisol-Enabled Budesonide.
Captisol Outlicensing Pipeline
We have outlicensed our Captisol technology to a number of organizations, including Critical Therapeutics, Daiichi Asubio, Proteolix, Sunesis, Taisho and TargeGen, and we have over 30 agreements currently in place. We currently receive royalties from Pfizer and BMS on the sales of Captisol-Enabled Geodon IM, Vfend IV, Abilify Injection IM and Cerenia. In 2007, we earned $3.8 million in royalty revenue from these outlicensees.
4
Product Candidate Selection and Development
In order for us to initiate a clinical development program, a drug compound must have a strong technical fit with our Captisol technology and also address an important unmet medical need in a significant patient population. We believe that combining our Captisol technology with APIs that have known safety and efficacy characteristics will enable us to move our retained product candidates from initial screening through submission of an NDA in 18 to 36 months. If our clinical trials are successful, our goal is to submit one or more NDAs per year beginning in 2008 for the foreseeable future for our retained product candidates, as our resources permit. Simultaneously, we will continue to outlicense or enter into collaborations for non-core product candidates and outlicense our Captisol technology.
Our Strategy
Our goal is to become a leading integrated specialty pharmaceutical company. To accomplish this goal we intend to:
- •
- Focus our internal development efforts on low risk product opportunities targeting the acute care hospital setting. We believe that utilizing APIs that have known biological activity and established safety and efficacy profiles will enable us to bring our retained product candidates to market on a relatively shortened development and regulatory timeline and with lower risk than new chemical entities. We believe that a small specialty sales force can effectively market our products to hospitals. However, if we are unable to bring our retained product candidates to market on a
shortened timeline or if a more extensive sales force than anticipated is required, this strategy may not be successful.
- •
- Establish strategic partnerships for product opportunities that do not fit into our core strategy. We believe that this strategy will enable the commercialization of partner product candidates that require longer timelines and greater financial outlay than our retained product candidates, without requiring us to incur the full cost of development and the financial risk of commercialization. However, if we are not able to establish additional partnering arrangements or if our partnering arrangements require substantial resources on our part, impose unfavorable restrictions or are terminated, we may not realize the benefits of this strategy.
- •
- Utilize APIs that have known safety and efficacy characteristics to gain rapid market acceptance. We believe that physicians will be more likely to use APIs that have known safety and efficacy characteristics than new chemical entities. Nevertheless, perceptions of the safety and efficacy characteristics of APIs we utilize may subsequently change, which could make it more difficult for us to gain market acceptance.
- •
- Continue to build intellectual property around our product candidates and our Captisol technology. We continue to pursue intellectual property protection for our product candidates. We are also pursuing additional intellectual property protection to address the expiration of the Captisol composition of matter patents in the U.S. in 2010, and expiration of the Captisol composition of matter patents outside the U.S. between 2011 and 2013, with one expiring in 2016. However, if we are unsuccessful in obtaining additional intellectual property protection or if our intellectual property protection is inadequate to protect against generic competition, this strategy may be unsuccessful.
- •
- Maintain our technology outlicensing business. We plan to continue to outlicense Captisol to organizations on an opportunistic basis to provide us with revenue to support our
5
development efforts relating to our own product candidates. If our licensees terminate or reduce their development programs or if we are unable to successfully license our technology in the future, this could limit our ability to support our development efforts relating to our product candidates.
Risks Related to Our Business
We are subject to many risks that you should be aware of before you decide to buy our common stock. These risks could adversely affect our business, offset or eliminate any advantages of our approach or prevent us from successfully implementing our business strategy, and are discussed more fully in the section entitled "Risk Factors." For example:
- •
- We have a history of net losses, and although we achieved profitability in 2006, we may not be able to return to or maintain profitability.
- •
- We rely on our platform technology, Captisol, for the continuation of our business, and if any adverse events were to occur relating to our Captisol technology, our business would be seriously harmed.
- •
- The basic composition of matter patents relating to Captisol expire in 2010 in the U.S. and between 2011 and 2013 outside the U.S., with one expiring in 2016, which may enable competitors to sell generic versions of Captisol, which would substantially reduce our revenue from our technology outlicensing business and could result in generic competition for our products and product candidates.
- •
- There is a risk that our product candidates will not successfully complete clinical trials and will not advance to the regulatory approval stage.
- •
- Even if we or our partners complete clinical trials of product candidates, we or our partners may never succeed in obtaining regulatory approval for any product candidates. Without regulatory approval, we or our partners will be unable to commercialize product candidates, and our growth prospects will be materially impaired.
Company Information
Our business began as CyDex, L.C., a Kansas limited liability company. The members of CyDex, L.C. entered into member contribution agreements with CyDex, Inc., a Kansas corporation, whereby the members of CyDex, L.C. exchanged their membership interests for shares of stock in CyDex, Inc. in December 1996, leaving CyDex, Inc. as the sole member of CyDex, L.C. In connection with this exchange, CyDex, L.C. transferred its assets to CyDex, Inc. CyDex, Inc. reincorporated in Delaware on July 19, 2000 by merging the Kansas corporation into CyDex, Inc., a Delaware corporation, which was then a subsidiary of the Kansas corporation. In November 2007, we changed our name to CyDex Pharmaceuticals, Inc. Our principal executive offices are located at 10513 W. 84th Terrace, Lenexa, KS 66214, and our telephone number is (913) 685-8850. Our website address is www.cydexpharma.com. The information contained in, or that can be accessed through, our website is not part of this prospectus. Unless the context indicates otherwise, as used in this prospectus, the terms "CyDex," "we," "us" and "our" refer to CyDex Pharmaceuticals, Inc., a Delaware corporation.
CAPTISOL,Captisol-Enabled and our CD logo are registered trademarks of our company in the United States. We use CAPTISOL,Captisol-Enabled and the CyDex logo as trademarks in the United States and other countries. We have also filed trademark applications in the United States for CYDEX, CYDEX PHARMA, and CYDEX PHARMACEUTICALS. All references to "Captisol" in this prospectus refer to CAPTISOL® cyclodextrins, and all references to "Captisol-Enabled" in this prospectus refer toCaptisol-Enabled® products or product candidates. All other trademarks and tradenames mentioned in this prospectus are the property of their respective owners.
6
| Common stock offered by us | shares | |
Common stock to be outstanding after this offering | shares | |
Use of proceeds | We intend to use the net proceeds from this offering to support our clinical trials and further development and eventual commercialization of our retained product candidates, to further develop our partner product candidates to advance them to Phase 2 clinical trials, to build our internal infrastructure and capabilities, and for working capital and other general corporate purposes. | |
Proposed Nasdaq Global Market symbol | CYDX |
The foregoing information regarding the number of shares of our common stock to be outstanding after this offering is based on shares outstanding as of December 31, 2007 and excludes:
- •
- 7,695,668 shares of common stock issuable upon the exercise of outstanding options under our Amended and Restated 1997 Equity Compensation Plan with a weighted average exercise price of $0.77 per share;
- •
- 4,460,966 shares of common stock issuable upon the exercise of outstanding warrants with an exercise price of $0.01 per share; and
- •
- shares of common stock reserved for future issuance under our 2008 Equity Incentive Plan, our 2008 Employee Stock Purchase Plan and our 2008 Non-Employee Directors' Stock Option Plan, as well as automatic increases in the number of shares of our common stock reserved for future issuance under these benefit plans, each of which will become effective immediately upon the signing of the underwriting agreement for this offering.
Except as otherwise indicated, all information in this prospectus assumes:
- •
- a -for- reverse split of our common stock to be effected immediately prior to the effectiveness of this offering;
- •
- the conversion of all outstanding shares of our preferred stock into shares of common stock, immediately prior to the closing of this offering;
- •
- the approval of our 2008 Equity Incentive Plan, 2008 Employee Stock Purchase Plan and 2008 Non-Employee Directors' Stock Option Plan by our stockholders;
- •
- the filing of our amended and restated certificate of incorporation immediately following the closing of this offering; and
- •
- no exercise of the underwriters' over-allotment option.
7
Summary Consolidated Financial Data
The following tables summarize our historical consolidated financial data for the last three years. The consolidated statement of operations data for the years ended December 31, 2005, 2006 and 2007 and the balance sheet data as of December 31, 2007 are derived from our audited consolidated financial statements included elsewhere in this prospectus. You should read this data together with the consolidated financial statements and related notes included elsewhere in this prospectus and the information under "Selected Consolidated Financial Data" and "Management's Discussion and Analysis of Financial Condition and Results of Operations." Our historical results are not necessarily indicative of our operating results or financial position to be expected in the future.
| | Years Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||||||||
| | (in thousands, except share and per share data) | |||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||
Revenue: | ||||||||||||
| Milestone and license | $ | 1,947 | $ | 1,982 | $ | 1,172 | ||||||
| Material sales | 4,164 | 5,671 | 6,819 | |||||||||
| Royalties | 2,511 | 2,989 | 3,829 | |||||||||
| Contract research | 158 | 845 | 683 | |||||||||
| Other | 39 | 93 | 242 | |||||||||
| Total revenue | 8,819 | 11,580 | 12,745 | |||||||||
Operating expenses: | ||||||||||||
| Cost of sales(1) | 2,566 | 2,482 | 2,508 | |||||||||
| Research and development(1) | 4,419 | 4,435 | 9,078 | |||||||||
| Selling, general and administrative(1) | 2,488 | 2,681 | 4,328 | |||||||||
| Total operating expenses | 9,473 | 9,598 | 15,914 | |||||||||
| Operating income (loss) | (654 | ) | 1,982 | (3,169 | ) | |||||||
Other income (expense): | ||||||||||||
| Interest income | 223 | 428 | 582 | |||||||||
| Other, net | (7 | ) | (2 | ) | — | |||||||
| Total other income | 216 | 426 | 582 | |||||||||
| Income (loss) before income taxes | (438 | ) | 2,408 | (2,587 | ) | |||||||
Provision for income taxes | — | 53 | — | |||||||||
| Net income (loss) | (438 | ) | 2,355 | (2,587 | ) | |||||||
| Preferred dividends and accretion | 4,441 | 4,455 | 4,471 | |||||||||
| Net loss attributable to common stockholders | $ | (4,879 | ) | $ | (2,100 | ) | $ | (7,058 | ) | |||
| Basic and diluted net loss per common share(2) | $ | (0.87 | ) | $ | (0.35 | ) | $ | (1.04 | ) | |||
| Shares used to compute basic and diluted net loss per common share(2) | 5,617,270 | 6,082,357 | 6,798,681 | |||||||||
- (1)
- Effective January 1, 2006, we adopted Statement of Financial Accounting Standards, or SFAS, No. 123 (revised 2004),Share-Based Payment, or SFAS No. 123R. SFAS No. 123R superseded SFAS No. 123,Accounting for Stock-Based Compensation,
footnotes continued on following page
8
or SFAS No. 123, and Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees, or APB No. 25. SFAS No. 123R requires that all stock-based compensation be recognized as an expense in the financial statements and that such cost be measured at the fair value of the award. As we previously used the minimum value method, as defined by SFAS No. 123, for purposes of measuring the fair value of stock options, we adopted SFAS No. 123R using the prospective method of application, which requires us to recognize compensation cost based on fair value for any awards granted after January 1, 2006, and any existing awards modified after that date. Under the prospective method, we recorded stock-based compensation expense for awards granted prior to January 1, 2006 using the intrinsic value method of APB No. 25.
The periods presented above include employee stock-based compensation in the following amounts:
| | Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||||||
| | (in thousands) | |||||||||
| Cost of sales | $ | 4 | $ | 2 | $ | 5 | ||||
| Research and development | 39 | 15 | 21 | |||||||
| Selling, general and administrative | 6 | 23 | 177 | |||||||
| Total | $ | 49 | $ | 40 | $ | 203 | ||||
- (2)
- See Notes 2(r) and 12 of the notes to our consolidated financial statements for an explanation of the method used to calculate basic and diluted net loss per common share.
| | As of December 31, 2007 | ||||
|---|---|---|---|---|---|
| | Actual | Pro Forma As Adjusted(1) | |||
| | (in thousands) | ||||
| Consolidated Balance Sheet Data: | |||||
| Cash, cash equivalents and short-term investments | $ | 8,109 | |||
| Working capital | 10,821 | ||||
| Total assets | 18,432 | ||||
| Redeemable convertible preferred stock | 34,600 | ||||
| Accumulated deficit | (21,927 | ) | |||
| Total stockholders' equity (deficit) | (19,032 | ) | |||
- (1)
- On a pro forma as adjusted basis to give effect to the conversion of all outstanding shares of our preferred stock into an aggregate of shares of common stock immediately prior to the closing of this offering and the sale of shares of our common stock we are offering at an assumed initial public offering price of $ per share, after deducting estimated underwriting discounts and estimated offering expenses payable by us. A $1.00 increase (decrease) in the assumed public offering price of $ per share would increase (decrease) each of cash, cash equivalents and short-term investments, working capital, total assets and stockholders' equity (deficit) by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and estimated offering expenses payable by us. The pro forma as adjusted information presented above is illustrative only and will change based on the actual initial public offering price and other terms of this offering determined at pricing.
9
An investment in our common stock involves a high degree of risk. You should consider carefully the risks described below, together with the other information contained in this prospectus, including in our consolidated financial statements and the related notes appearing at the end of this prospectus, before you decide whether to buy shares of our common stock. If any of the following risks actually occur, our business, results of operations and financial condition could suffer significantly. In any of these cases, the market price of our common stock could decline, and you may lose all or part of your investment in our common stock.
Risks Related to Our Business
We have a history of net losses, and although we achieved profitability in 2006, we may not be able to return to or maintain profitability.
We have incurred significant net losses since the inception of our business through 2005. As of December 31, 2007, we had an accumulated deficit of $21.9 million. Although we had net income of $2.4 million in 2006, we had net losses of $2.6 million in 2007 and $438,000 in 2005. Our historical losses have resulted primarily from investing in our research and development programs. We expect to continue to make significant investments in our research and development programs, and our selling, general and administrative expenses have been and will continue to be a significant component of our cost structure. For example, we anticipate that in 2008 we will invest up to $8.0 million in research and development and will incur up to $5.0 million in general and administrative expenses. We expect to continue to incur significant and increasing expenses for the foreseeable future as we continue our research activities, conduct development of or seek regulatory approvals for our product candidates, incur the additional costs of operating as a public company, and establish a specialty sales and marketing organization to commercialize our products. As a result of these and other activities, we do not anticipate being profitable in 2008, and there is no guarantee that we will be able to return to profitability in the future. We also expect that expenses from these activities will exceed any revenue generated by the increased spending for at least the next two years. Even if we successfully develop and commercialize one or more of our product candidates, we may not be able to maintain profitability. Even if we do maintain profitability, we may not be able to increase profitability on a quarterly or annual basis. If we are unable to maintain or increase profitability, the market value of our common stock may decline and you could lose all or part of your investment.
We rely on our platform technology, Captisol, for the continuation of our business, and if any adverse events were to occur relating to our Captisol technology, our business would be seriously harmed.
All of our products and product candidates, as well as our technology that we outlicense, are based on Captisol. In addition, we or our partners are attempting to develop some product candidates that may contain significantly higher levels of Captisol than in any currently-approved product and at levels at which the FDA has previously expressed concerns regarding potential renal toxicity. If products or product candidates incorporating our Captisol technology were to cause any unexpected adverse events, whether in preclinical studies, clinical trials or as commercialized products, whether as a result of Captisol or otherwise, the perception of Captisol safety could be seriously harmed. If this were to occur, we may not be able to market our products unless and until we are able to demonstrate that the adverse event was unrelated to Captisol, which we may not be able to do. Further, whether or not the adverse event was a result of Captisol, we could be required by the FDA to submit to additional regulatory reviews or approvals, including extensive safety testing or clinical testing of products using Captisol, which would be expensive and, even if we were to demonstrate that the adverse event was unrelated to Captisol, would delay our marketing of Captisol-Enabled products and receipt of revenue related to those products.
10
The basic composition of matter patents relating to Captisol expire in 2010 in the U.S. and between 2011 and 2013 outside the U.S., with one expiring in 2016, which may enable competitors to sell generic versions of Captisol, which would substantially reduce our revenue from our technology outlicensing business and could result in generic competition for our products and product candidates.
The basic composition of matter patents relating to Captisol expire in 2010 in the U.S. and between 2011 and 2013 outside the U.S., with one expiring in 2016. We have obtained patent protection on a number of combinations of APIs and Captisol through three combination patents in the U.S., and we have applied for six additional combination patents in the U.S. relating to the combination of Captisol with specific APIs. Our U.S. combination patent relating to Fosphenytoin expires June 12, 2018 and our U.S. combination patent relating to Amiodarone expires May 4, 2022. Our U.S. combination patent relating to one of our early-stage product candidates expires March 19, 2022. There is no guarantee that these patents will be sufficient to prevent competitors from using Captisol after 2010 and competing against us, or from developing combination patents for products that will prevent us from developing products using those APIs. In addition, most of the agreements in our Captisol outlicensing business, including our agreements with Pfizer relating to Geodon IM, Vfend IV and Cerenia, provide that once the relevant patent expires, the amount of royalties we receive will be reduced or eliminated.
There is a risk that our product candidates will not successfully complete clinical trials and will not advance to the regulatory approval stage.
We or our partners will only receive regulatory approval to commercialize a product candidate if we or our partners can demonstrate to the satisfaction of the FDA, the European Medicines Agency, or EMEA, or other applicable regulatory authorities, in well designed and properly conducted clinical trials, that the product candidate is safe and effective and otherwise meets the appropriate standards required for approval for a particular indication. If lengthy or complex trials are required to support approval, we would have to decide whether to cease pursuit of regulatory approval of the product candidate. Factors we expect to consider in this decision would include the anticipated time, complexity and cost associated with continuing to pursue regulatory approval measured against the perceived benefit to be derived from successful completion. Historically, favorable results from preclinical studies and early clinical trials have often not been confirmed in later clinical trials. Many companies in the pharmaceutical industry have experienced significant setbacks in advanced clinical trials or during the regulatory approval process, despite promising initial results. The clinical effects of our product candidates may be different than expected or may include undesirable side effects that delay, extend or preclude regulatory approval or limit their commercial use if approved.
A number of events or factors, including any of the following, could delay the completion of our and our partners' ongoing and planned clinical trials and negatively impact the ability to obtain regulatory approval for, and to market and sell, our products and product candidates:
- •
- conditions imposed on us or our partners by the FDA, the EMEA or other applicable regulatory authorities, regarding the scope or design of clinical trials;
- •
- the results of clinical trials may not meet the level of statistical significance required for approval by the FDA, the EMEA or other applicable regulatory authorities;
- •
- the FDA, the EMEA or other applicable regulatory authorities may require additional or expanded clinical trials;
11
- •
- delays in obtaining, or the inability to obtain or maintain, required approvals from institutional review boards, or IRBs, or other reviewing entities at clinical sites selected for participation in clinical trials;
- •
- insufficient supply or deficient quality of product candidates or other materials necessary to conduct clinical trials;
- •
- difficulties in manufacturing;
- •
- difficulties enrolling subjects and high drop-out rates of subjects in clinical trials;
- •
- negative or inconclusive results from clinical trials, or results that are inconsistent with earlier results, that necessitate additional clinical trials;
- •
- serious or unexpected drug-related side effects experienced by subjects in clinical trials;
- •
- failure of our third-party contractors or our investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner; or
- •
- lack of resources or expertise to fund, manage or monitor the clinical trial or third-party contractors.
Clinical trials may not begin as planned, may need to be redesigned, and may not be completed on schedule, if at all. In addition, standards enunciated by regulatory agencies are constantly subject to change as a result of factors outside of our control. For example, we have designed the clinical trials for our lead product candidates to meet FDA requirements; however, the FDA could change its requirements before we seek or obtain regulatory approval. Delays in clinical trials may result in increased development costs for our product candidates, which would cause the market price of our shares to decline and could require us to obtain additional financing, which may not be available on favorable terms or at all. In addition, if one or more clinical trials are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of our product candidates could be significantly reduced.
Even if we or our partners complete clinical trials of product candidates, we or our partners may never succeed in obtaining regulatory approval for product candidates. Without regulatory approval, we or our partners will be unable to commercialize product candidates, and our growth prospects will be materially impaired.
All of our product candidates must successfully complete development and gain regulatory approval before we or our partners can market them. If we or our partners do not demonstrate the safety and efficacy of product candidates, we or our partners will not obtain the required regulatory approvals to commercialize these product candidates. Any product candidate that we or our partners seek to commercialize is subject to extensive regulation by the FDA, EMEA and other applicable regulatory authorities relating to research, development, testing, manufacture, quality control, safety, efficacy, record-keeping, labeling, packaging, storage, approval, advertising, marketing, promotion, sale and distribution and import and export. Satisfaction of these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays. In addition, approval of our product candidates may be delayed in some circumstances due to market exclusivity granted to other parties by the FDA. Our growth prospects may be materially impaired as a result of any delay in, or failure to receive, required regulatory approval for some or all of our product candidates.
12
We intend to seek FDA approval of our retained product candidates and partner product candidates utilizing Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act. 505(b)(2) allows the submission of an NDA that relies in part on data in the public domain or on the FDA's prior findings regarding the safety and effectiveness of approved drugs, which could expedite the development program for our product candidates by potentially decreasing the overall scope of work. If we or our partners are unable to utilize 505(b)(2), the development program for our retained product candidates and partner product candidates would be materially longer than we expect, and we or our partners would also have to conduct significantly more costly trials than we anticipate, which would harm our business. While we believe that in some cases the 505(b)(2) path to registration should only necessitate the completion of a single or a small number of clinical trials demonstrating bioequivalence to the existing, previously approved formulation, the FDA has discretion to request additional data to support approval. In addition, 505(b)(2) NDAs are subject to special requirements designed to protect the property rights of sponsors of previously approved drugs that are referenced in a 505(b)(2) NDA. These requirements may give rise to patent litigation and mandatory delays in approval of our NDAs for up to 30 months or longer depending on the outcome of any litigation. Moreover, even if we or our partners are able to utilize 505(b)(2), there is no guarantee that this would ultimately lead to faster drug development or to approval. None of the Captisol-Enabled products currently marketed by our outlicensees were approved under 505(b)(2).
We are a small company with limited experience in taking product candidates through the regulatory process and commercializing product candidates. If we are unable to hire additional experienced personnel and manage the regulatory process in an efficient manner, we may not be able to commercialize our product candidates.
During our existence as a privately-held company, we have operated with a very small number of employees, and have not taken any of our product candidates through the regulatory process with the FDA. In order to successfully commercialize our product candidates, we will need to significantly expand our internal resources and capabilities to improve and manage our regulatory compliance process. Attracting highly qualified scientific and other personnel to manage the regulatory process may be difficult, as competition exists among other companies and research and academic institutions for qualified personnel, and personnel with the skills that we require are difficult to find in the Kansas City area. If we cannot attract and retain sufficiently qualified personnel on acceptable terms to manage the regulatory process, we may experience significant delay in carrying out the regulatory compliance process or may not be able to develop and commercialize competitive products.
In addition, our growth and success depend on our ability to attract and retain additional highly qualified scientific, technical and sales personnel. Intense competition exists among other companies and research and academic institutions for qualified personnel. If we cannot attract and retain sufficiently qualified employees on acceptable terms, we may not be able to develop and commercialize competitive products.
We obtain Captisol from a sole source supplier, and if this supplier were to cease to be able to supply Captisol to us, or decline to supply Captisol to us, we would be unable to continue to derive revenue or continue to develop our product candidates until we obtained an alternative source, which could take a considerable length of time.
We currently have one supplier of Captisol, Hovione FarmaCiencia SA, or Hovione, through its agent Hovione LLC. Hovione is a major supplier of APIs and API intermediates located in Lisbon, Portugal. Hovione has a second production site in Macau, China, but that site is not yet qualified to make Captisol. If a major disaster were to happen at Hovione or Hovione were to suffer major production problems or were to fail to deliver Captisol to us for any other reason, there could be a significant interruption of our Captisol supply. While we carry a significant inventory of Captisol for this type of
13
occurrence, which should permit us to satisfy our existing supply obligations through mid-2010 under current and anticipated demand conditions, an unusually large order or two could rapidly deplete that inventory and cause significant problems with our licensees and disrupt our business. In addition, if we fail to supply Captisol under our supply agreements, our customers could obtain the right to have Captisol manufactured by other suppliers, which would significantly harm our business.
We depend on our senior management to manage our business, which dependence will become even greater as a public company, and if we are not able to retain our senior management, or attract and retain other senior management, our business would be harmed.
During our existence as a privately-held company, we have operated with a very small number of employees. However, given our transition to a public company, as well as our expected growth and the change in emphasis of our business model, our internal management needs will change. We will need to significantly expand our management team, which will require the hiring of significant additional personnel as well as the expansion of responsibilities by our current management team. We expect that we will need to hire a vice president of business development, a vice president of sales and marketing, a head of regulatory function and additional managerial and finance personnel. High demand exists for senior management and other key personnel in the pharmaceutical industry, and personnel with the skills that we require are difficult to find in the Kansas City area. The loss of any of our current management personnel, or failure to hire the additional management personnel that we will need, may negatively impact our ability to manage our company effectively and to carry out our business plan. Of the members of our senior management team, only our chief executive officer, Dr. Siebert, has an employment agreement. Notwithstanding his employment agreement, Dr. Siebert could resign at any time.
We have material weaknesses in our internal control over financial reporting, and these material weaknesses create a reasonable possibility that a material misstatement of our interim or annual financial statements will not be prevented or detected in a timely manner.
As a publicly-traded company, we must maintain effective disclosure controls and procedures and internal control over financial reporting. Our independent registered public accounting firm determined in February 2008 that we have five material weaknesses as follows:
- •
- our control environment is ineffective due to the operating style of our senior management, which does not consistently emphasize effective internal control over financial reporting or the importance of the financial reporting process;
- •
- we do not have an effective process for identifying financial reporting risks and evaluating whether adequate policies, procedures and control activities are implemented on a timely basis, particularly for complex or non-recurring transactions;
- •
- we are not able to effectively monitor whether our internal control over financial reporting is functioning properly because documentation of the operation of control activities and internal control policies and procedures is insufficient to assess performance, and responsibilities for our personnel involved in internal control over financial reporting are not clearly delineated;
- •
- we did not appropriately segregate duties, or implement alternative controls, relative to executing and accounting for new revenue arrangements; and
14
- •
- we do not have personnel with the necessary experience and training to ensure appropriate revenue recognition for key revenue streams; specifically, we lack employees with the expertise to analyze multiple element arrangements for appropriate revenue recognition.
While we have a number of material weaknesses that we are in the process of remediating, we do not have material weaknesses related to the policies and procedures that:
- •
- pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and disposition of assets;
- •
- provide reasonable assurance that our receipts and expenditures are being made in accordance with authorizations of our management and directors; or
- •
- provide reasonable assurance that we will prevent or timely detect unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
As a result of these material weaknesses in internal control over financial reporting, material misstatements existed with respect to the accounting for revenue recognition, preferred stock and warrants and other items that resulted in a recent restatement of our consolidated financial statements for the years ended December 31, 2004, 2005 and 2006. Additionally, material misstatements were identified in our revenue recognition, research and development costs, stock-based compensation and other items for the year ended December 31, 2007, which were corrected prior to the issuance of the consolidated financial statements.
In order to improve and to maintain the effectiveness of our internal control over financial reporting and disclosure controls and procedures, significant resources and management oversight will be required. We have begun the process of remediating these material weaknesses, but this process will take time, and we will not be able to assert that we have remediated these material weaknesses until the procedures that we put in place have been working for a sufficient period of time for us to determine that they are effective. As a result of this and similar activities, management's attention may be diverted from other business concerns, which could have a material adverse effect on our business, financial condition and results of operations. If we are unable to remediate these material weaknesses, or in the future report one or more additional material weaknesses, there is a reasonable possibility that a material misstatement of our interim or annual financial statements will not be prevented or detected on a timely basis. This could result in a restatement of our financial statements or impact our ability to accurately report financial information on a timely basis, which could adversely affect our stock price.
We rely on contract manufacturers for the manufacture of our products and product candidates, and if these contract manufacturers fail to perform as we expect, we will incur delays in our ability to generate revenue and substantial additional expenses in obtaining new contract manufacturers.
We do not manufacture our products or product candidates, but rather contract with contract manufacturers for the manufacture of our products and product candidates. With respect to any specific product or product candidate, we only contract with one contract manufacturer due to the high cost of compliance with good manufacturing practices prior to the contract manufacturer being permitted to manufacture the product or product candidate for use in humans. If a contract manufacturer is unable or unwilling to continue to manufacture for us in the future, we would be required to contract with a new contract manufacturer for the specific product or product candidate. In the case of products, this would cause us to lose revenue during the qualification process, and in the
15
case of product candidates, this could cause a delay in the commercialization of the product candidate. In addition, in either case we would incur substantial additional expenses as a result of the new contract manufacturer becoming qualified. Further, if a contract manufacturer were to experience a delay in producing products or product candidates due to a failure to meet strict FDA manufacturing requirements or otherwise, we would also experience a delay in development and commercialization of the product candidate or, in the case of products, sales of the product. This risk is exacerbated in the case of manufacture of injectables, which require heightened sterility and other conditions as well as specialized facilities for preparation.
Our revenue is currently dependent upon our Captisol outlicensing business. If any or all of our current outlicensees were to fail in commercialization of their products, or if we fail to establish new licensing arrangements, our business and growth prospects would be materially harmed.
Revenue from our Captisol outlicensing business accounted for approximately 95%, 78% and 87% of our total revenue in 2005, 2006 and 2007, respectively. In particular, our revenue is currently dependent on royalties from two of our outlicensees, Pfizer and Bristol-Myers Squibb, or BMS. Pfizer accounted for 27% of our total revenue in 2006 and 40% of our total revenue in 2007, and BMS accounted for 9% of our total revenue in 2006 and 4% of our total revenue in 2007. We currently depend on our arrangements with our outlicensees to sell products using our Captisol technology. If our outlicensees discontinue sales of products using our Captisol technology, fail to obtain regulatory approval for their products using our Captisol technology, fail to satisfy their obligations under their agreements with us, or if we are unable to establish new licensing and marketing relationships, our financial results and growth prospects would be materially affected. Further, under most of our Captisol outlicenses, the amount of royalties we receive will be reduced or will cease when the relevant patent expires. The basic composition of matter patents relating to Captisol expire in 2010 in the U.S. and between 2011 and 2013 outside the U.S., with one expiring in 2016, and if our other intellectual property rights are not sufficient to prevent a generic form of Captisol from coming to market, the source of the vast majority of our revenue may cease to exist.
If we are not successful in establishing and maintaining our partnering relationships, we will not be able to grow our business.
An element of our business strategy is to establish relationships with third parties to accelerate the development of our partner product candidates. The process of establishing new partnering relationships is difficult, time-consuming and involves significant uncertainty. We face, and will continue to face, significant competition in seeking appropriate partners. Our obligations under these arrangements can include performance of development activities, such as feasibility studies, early-stage development activities, formulation optimization, stability testing and scale-up of the manufacturing process, supply of the product to the partner for clinical testing, assistance in the preparation of regulatory filings by our partner and supply of the product for sale by our partner. If we fail to meet any of these obligations, we may lose our rights to future development fees and future royalty and milestone payments, and our partners may have the right to terminate the agreement. In addition, our agreements sometimes allow for our partner to terminate the agreement with limited notice and without penalty or in the event the partner reasonably determines that the product candidate does not justify continued development or commercialization.
In addition, our partners may fail to fulfill their responsibilities under these arrangements or may seek to renegotiate or terminate their relationships with us due to unsatisfactory clinical results, a change in business strategy, a change in control or other reasons. If we are unable to establish and maintain development arrangements on acceptable terms, we may have to delay or discontinue further development of one or more of our partner product candidates, seek regulatory approval or undertake
16
commercialization activities at our own expense or find alternative sources of funding, and our growth prospects will be materially harmed.
We currently have no internal sales, marketing and distribution capabilities. If we are unable to develop our sales, marketing and distribution capabilities, we will not be successful in commercializing our products.
We do not currently plan to hire a vice president of sales and marketing to initiate the development of our internal sales, marketing and distribution capabilities until 2009. If Captisol-Enabled Fosphenytoin IV and IM, one of our lead retained product candidates, is approved in 2009, we intend to commercialize it in the United States by establishing our own sales force or contracting with a specialty sales and marketing organization with technical experience. We also expect to support distribution capabilities. We do not anticipate making significant expenditures to establish the organization to sell any potential product (including Captisol-Enabled Fosphenytoin IV and IM) in advance of knowing whether it has received FDA approval. Failure or delay in the establishment of a sales force or contracting with a specialty sales and marketing organization could delay any product launch.
We currently do not intend to sell any of our products directly outside the United States. Therefore, we must successfully enter into arrangements with third parties to register our products and perform these services outside the United States.
If we do not establish sales and distribution capabilities successfully, either on our own or in collaboration with third parties, we may not successfully commercialize any future products, and our future product revenue could suffer.
Development of pharmaceutical products is expensive, time-consuming and subject to uncertainties, and we may not realize a return on our investment in product development for a significant period of time, if at all.
Developing pharmaceutical products is expensive, and there is typically a significant amount of time prior to realizing a return on an investment in product development, if a return is realized at all. In 2005, 2006 and the 2007, our research and development expenses were $4.4 million, $4.4 million and $9.1 million, respectively, or approximately 50%, 38% and 71%, respectively, of our revenue. The increase in our research and development expenses in 2007 as compared to 2006 was primarily due to the timing of clinical trials. Our future plans include significant investments in research and development, including clinical trials and related product opportunities. We believe that we must continue to dedicate a significant amount of financial and operational resources to our research and development efforts to grow our business and maintain our competitive position. However, whether we or our partners will obtain regulatory approval for our product candidates is uncertain, and if we or our partners are not able to do so, we will have expended significant resources and received no benefit. Even if we or our partners are successful in obtaining regulatory approval, we do not expect that we will receive significant revenue from these investments for several years. If our cash flows and financial resources are not sufficient to fund the development of our products and product candidates, we would be required to raise additional capital to continue to develop our products and product candidates, which we may not be able to do on favorable financial terms, or at all. In addition, if we do raise equity capital, it could be dilutive to our stockholders.
17
We rely on third parties to conduct, supervise and monitor our clinical trials, and those third parties may perform in an unsatisfactory manner, such as by failing to meet established deadlines for the completion of these trials.
We rely on third parties such as contract research organizations, or CROs, medical institutions and clinical investigators to enroll qualified patients and conduct, supervise and monitor our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Our reliance on these third parties, however, does not relieve us of our regulatory responsibilities, including ensuring that our clinical trials are conducted in accordance with good clinical practices, or GCP, and the investigational plan and protocols contained in the relevant regulatory application, such as the investigational new drug application. In addition, the CROs with which we contract may not complete activities on schedule, or may not conduct our preclinical studies or clinical trials in accordance with regulatory requirements or our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for, and to commercialize, our product candidates may be delayed or prevented.
We may not be able to patent some formulations of our product candidates in development and may not be able to obtain adequate protection under the Hatch-Waxman Act to prevent generics from copying our product candidates.
Our product candidates utilize APIs that we believe are in or will be in the public domain. While we are working to obtain patent protection for our Captisol-Enabled formulations of these APIs, as well as manufacturing processes and uses of Captisol, there is no guarantee that we will be able to do so. In cases where no patent protection can be obtained, limited regulatory exclusivity providing protection against generic competition may in some circumstances be obtained under the Hatch-Waxman Act. There is no guarantee that we will receive exclusivity for any particular product candidate. If, as we believe, our retained product candidates only require bioequivalence trials as the basis for approval, they would not qualify for Hatch-Waxman exclusivity. Following the expiration of the basic Captisol composition of matter patents in the U.S. in 2010, biotechnology or pharmaceutical companies, which may have greater financial and personnel resources than we do, may be able to obtain regulatory approval to market one or more of our Captisol-Enabled formulations of these APIs before we are able to obtain regulatory approval. Such approval for our competitors' products could block our ability to obtain approval for up to three years. Failure to obtain patent protection or regulatory exclusivity will adversely impact our ability to commercialize our products and realize a positive return on our investment.
Even if our product candidates receive regulatory approval, they may not become commercially viable products.
Even if our product candidates are approved for commercialization, they may not become commercially viable products. For example, even if we or our partners receive regulatory approval to market a commercial product, any approval may be subject to limitations on the indicated uses for which we or our partners may market the product. A product or product candidate may not result in commercial success for other reasons, including:
- •
- lack of perceived differentiation of our Captisol-Enabled products from the pioneer drug or generic form of the API;
- •
- difficulty in large-scale manufacturing;
- •
- low market acceptance by physicians, healthcare payers, patients and the medical community as a result of lower demonstrated clinical safety or efficacy compared to other
18
- •
- insufficient or unfavorable levels of reimbursement from government or third-party payers;
- •
- infringement on proprietary rights of others for which we have not received licenses;
- •
- incompatibility with other drugs;
- •
- other potential advantages of alternative treatment methods;
- •
- ineffective marketing and distribution support;
- •
- lack of cost-effectiveness; or
- •
- timing of market introduction of competitive products.
products, prevalence and severity of adverse side effects, or other potential disadvantages relative to alternative treatment methods;
If our product candidates do not receive commercial acceptance, our business, results of operations and financial condition will be adversely affected.
Our product candidates, if they receive regulatory approval for marketing, remain subject to ongoing regulatory requirements. If we or our partners fail to comply with these requirements, regulatory authorities could withdraw these approvals and the sales of any approved commercial products could be suspended.
After receipt of regulatory marketing approval, if any, each of our products remains subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event and manufacturing problem reporting, storage, advertising, promotion, distribution and record-keeping. Furthermore, the approval may be subject to limitations on the uses for which the product may be marketed or the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, which could reduce our revenue, increase our expenses and render the approved product candidate not commercially viable. In addition, as clinical experience with a drug expands after approval because it is typically used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed that were not seen or anticipated during pre-approval clinical trials or other studies. Any adverse effects observed after the approval and marketing of a product candidate could result in detrimental labeling changes, limitations on the use of the approved product or its withdrawal from the marketplace. Absence of long-term safety data may also limit the approved uses of our products, if any. If we or our partners fail to comply with the regulatory requirements of the FDA, the EMEA and other applicable regulatory authorities, or if previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including:
- •
- restrictions on the products, manufacturers or manufacturing processes;
- •
- warning letters and untitled letters;
- •
- civil penalties and criminal prosecutions and penalties;
- •
- fines;
19
- •
- injunctions;
- •
- product seizures or detentions;
- •
- import or export bans or restrictions;
- •
- company-initiated or FDA-requested product recalls and related publicity requirements;
- •
- suspension or withdrawal of regulatory approvals;
- •
- total or partial suspension of production; and
- •
- refusal to approve pending applications for marketing approval of new products or of supplements to approved applications.
If we or our partners are slow or unable to adapt to changes in existing regulatory requirements or the promulgation of new regulatory requirements or policies, we or our partners may lose marketing approval for our products, resulting in decreased revenue from milestone payments, product sales or royalties.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our products for unapproved or "off-label" uses.
If our product candidates receive FDA clearance or approval, our promotional materials and training methods regarding physicians need to comply with FDA and other applicable laws and regulations. If the FDA determines that our promotional materials or training constitute promotion for an unapproved use, it could request that we modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and adoption of our products would be impaired.
New legal and regulatory requirements could make it more difficult for us or our partners to obtain approvals for our product candidates and could limit or make more burdensome our or our partners' ability to commercialize any approved products.
New federal legislation was recently enacted known as the FDA Amendments Act of 2007, which grants the FDA new authority to impose post-approval clinical study and clinical trial requirements, require safety-related changes to product labeling and require the adoption of risk management plans, referred to in the legislation as risk evaluation and mitigation strategies, or REMS. The REMS may include requirements for special labeling or medication guides for patients, special communication plans to healthcare professionals, and restrictions on distribution and use. For example, if the FDA makes the requisite findings, it might require that a new product be used only by physicians with specified specialized training, only in specified designated healthcare settings, or only in conjunction with special patient testing and monitoring. The legislation also includes the following: requirements for providing the public information on ongoing clinical trials through a clinical trial registry and for disclosing clinical trial results to the public through a clinical trial database; renewed requirements for conducting trials to generate information on the use of products in pediatric patients; and new penalties, for example for false or misleading consumer advertisements. Other proposals have been made to impose additional requirements on drug approvals, further expand post-approval requirements, and restrict sales and promotional activities. The new legislation, and the additional
20
proposals if enacted, may make it more difficult or burdensome for us or our partners to obtain approval of our product candidates, any approvals we or our partners receive may be more restrictive or be subject to onerous post-approval requirements, our or our partners' ability to successfully commercialize approved products may be hindered, and our business may be harmed as a result.
Rapid technological change could make our products, product candidates or technologies obsolete.
Pharmaceutical technologies and products are subject to rapid and significant technological change. We expect our competitors may develop new technologies and products that may render our products, product candidates or drug formulation technologies uncompetitive or obsolete. The products and technologies of our competitors may be more effective than the products, product candidates and drug formulation technologies developed by us. As a result, our products and product candidates may become obsolete before we recover expenses incurred in connection with their development from sales revenue from any commercialized product. We are aware of other pharmaceutical companies that are developing competing technologies for at least two of our retained product candidates and two of our partner product candidates.
Our competitors may develop products that are less expensive, safer or more effective than our products, which would reduce the commercial potential of our products that we or our partners may bring to market.
We face intense competition from pharmaceutical and biotechnology companies, including other specialty pharmaceutical companies, CROs, academic institutions and government agencies. Some of these competitors are also our partners.
Our competitors may be able to use alternative technologies to formulate the active materials in our product candidates. They may, therefore, bring to market products that are able to compete with our product candidates or products that we may develop in the future. If successful, products derived from alternative technologies will compete against our and our partners' products and product candidates. Competing technologies include hydroxypropyl beta cyclodextrin marketed by Janssen Pharmaceutica, natural cyclodextrins, liposome technologies, emulsifier technologies and nanoparticle technologies. The products derived from these technologies may be safer or more effective than our and our partners' products and product candidates.
Certain of our product candidates, as well as the Captisol-Enabled products currently marketed by Pfizer and BMS, compete with products that utilize competitive technologies. For example, Vfend IV, which is marketed by Pfizer and uses our Captisol technology, competes with antifungal products such as Sporanox and AmBisome. Sporanox is formulated using hydroxypropyl beta cyclodextrin technology and is marketed by Johnson & Johnson, and AmBisome is formulated using liposomal technology and is marketed by Astellas. In addition, our Captisol-Enabled Budesonide solution for inhalation may compete with a budesonide solution for inhalation developed by MAP Pharmaceuticals that uses nanoparticle technology.
Potential products being tested in the United States and Europe of which we are not currently aware may also compete with our product candidates. Generic products may enter the market or may not infringe our granted patents and any patent applications that issue as patents. Our partners could choose a competing drug formulation system to use with their drugs instead of Captisol. Our partners could also develop or license products using competing technology that may displace or replace product candidates we intend to license to them or product candidates we have already licensed to them. There may be competitive products that beat us to the marketplace ahead of the retained specialized injectable product candidates we are developing. In addition, our partners themselves face competition from other major pharmaceutical companies for products using our Captisol technology,
21
which could adversely impact the potential for our technologies and partner product candidates, as well as our royalty revenue and business and financial condition.
Many of our competitors have greater capital resources, manufacturing and marketing experience, research and development resources and production facilities than we have. Many of them also have more experience than we do in preclinical studies and clinical trials of new drugs and in obtaining FDA, EMEA and other applicable regulatory approvals.
Patent protection for our products and product candidates is important and uncertain. If we are unable to obtain patent protection, we will experience intense competition.
Our success will depend on our ability and on the ability of our partners to obtain patent protection for our products and product candidates. We have filed and plan to continue to file for patents in the U.S., Europe, Japan and other jurisdictions. Because the patent position of specialty pharmaceutical companies involves complex legal and factual questions, we cannot predict the validity and enforceability of our patents with certainty. Our issued patents may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties. In addition, our patents may not prevent our competitors from developing similar products using different processes that are not covered by our patents. Our pending patent applications, those we may file in the future or those we may license from third parties may not result in patents being issued or receipt of the claims we desire. If these patents are issued, they may not provide us with protection or competitive advantages against competitors with similar technology. Our patents may not adequately protect our rights or permit us to gain or maintain a competitive advantage.
Patent rights are territorial, which means that the patent protection we do have will only extend to those countries in which we have issued patents. In addition, the laws of some countries do not protect intellectual property rights to the same extent as do the laws of the United States and various European countries. Competitors may successfully challenge our patents, produce similar drugs or products that do not infringe our patents, or produce drugs in countries where we have not applied for patent protection or that do not respect our patents. Additionally, the nature of claims contained in unpublished patent filings around the world is unknown to us and it is not possible to know whether known patent filings may be extended to other countries. Furthermore, it is not possible to know the scope of claims that will ultimately be allowed in published applications and it is also not possible to know which claims of granted patents, if any, will be deemed enforceable in a court of law. If any of our patents are invalidated or if we do not obtain additional patents in the future, we may experience intense competition and may lose royalties and sales revenue, which would have an adverse effect on our financial performance.
If we are unable to protect the confidentiality of our trade secrets or know-how, our proprietary information may be used by others to compete against us.
In addition to patent protection, we rely on a combination of trade secrets and know-how to maintain our competitive position. We generally try to protect trade secrets and know-how by entering into confidentiality or non-disclosure agreements with parties that have access to it, such as our partners, licensees, employees and consultants. Any of these parties may breach the confidentiality agreements and willfully or unintentionally disclose our confidential information, or our competitors might learn of the information in some other way. The disclosure to, or independent development by, a competitor of any trade secret or know-how not protected by a patent could materially adversely affect any competitive advantage we may have over the competitor.
22
Legal proceedings or third-party claims of intellectual property infringement may require us to spend substantial time and money and could prevent us or our partners from developing or commercializing products.
The manufacture, use, offer for sale, sale or importation of Captisol, our products or product candidates might infringe on the claims of third-party patents. A party might file an infringement action against us. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of litigation or proceedings more effectively because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation or defense of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant financial and human resources. We are aware of a third-party patent potentially relating to Captisol, although to date no claim has been made against us relating to this patent. Although we believe that the manufacture, use, offer for sale, sale, or importation of Captisol should not be held to infringe any valid claim of this patent, the patent could nevertheless be asserted against us. In addition, we are aware of third-party patents potentially relating to some of our current product candidates, and are further investigating the potential relevance of these patents to such product candidates. Should Captisol or any Captisol-Enabled product candidate be held to infringe a claim of a third-party patent, it could have a material adverse effect on our business.
Furthermore, some of our license agreements with partners require us to indemnify our partner for the infringement of third-party intellectual property rights. In the event that one of our partners' Captisol-Enabled products is deemed to infringe a third-party intellectual property right, such as a third-party patent, we may be obligated to indemnify our partner for the alleged infringement. The cost of defending and/or resolving a claim of patent infringement could be substantial. Furthermore, if we are unable to successfully defend or settle an action, the potential damages and potential loss of royalties from the sale of our partner's product may have a material adverse effect on our business.
We are exposed to political, economic and other risks that arise from operating a business that contracts with companies outside the United States.
We derive revenue from several different countries. For the years ended December 31, 2005, 2006 and 2007, approximately 37%, 22% and 29%, respectively, of our revenue was derived from sources outside the United States. The 29% of revenue derived outside the United States in 2007 was comprised of approximately 15% from the United Kingdom, 6% from Sweden, 3% from France, 1% from Belgium and the remainder from all other countries. We are therefore exposed to risks inherent in international operations. These risks include, but are not limited to:
- •
- changes in general economic, social and political conditions;
- •
- adverse tax consequences;
- •
- the difficulty of enforcing agreements and collecting receivables through legal systems outside the United States;
- •
- inadequate protection of intellectual property;
- •
- required compliance with a variety of laws and regulations of jurisdictions outside the United States, including labor and tax laws;
- •
- customers outside the United States may have longer payment cycles;
- •
- changes in laws and regulations of jurisdictions outside the United States; and
23
- •
- terrorist acts and natural disasters.
Our business success depends in part on our ability to anticipate and effectively manage these and other regulatory, economic, social and political risks inherent in contracting with businesses outside the United States. We cannot provide assurance that we will be able to effectively manage these risks or that they will not have a material adverse effect on our business.
Uncertainty in the credit market may negatively affect our ability to sell the auction-rate preferred securities owned by us and the value of these securities.
Our short-term investments as of March 31, 2008 include various auction-rate preferred securities, or ARPS, with a par value of $5.6 million and an estimated fair value of $5.2 million. ARPS are variable-rate debt securities. While the underlying security has a long-term maturity, the interest rate is reset through Dutch auctions that are held at pre-determined intervals, usually every 7 to 28 days. In the past, liquidity for ARPS was provided by this auction process that allowed investors to roll over their holdings or obtain immediate liquidity by selling the ARPS at market. Recent uncertainty in the global credit and capital markets has negatively affected these auctions and beginning in February 2008, many of these auctions have not had sufficient bidders to allow investors to complete a sale of ARPS. As a result, the ARPS we hold have experienced multiple failed auctions, and we have recorded an impairment charge of $410,000 during the first quarter of 2008. We will not be able to liquidate these ARPS until a future auction is successful or until we decide and are able to sell the ARPS in a secondary market. Consequently, we have also reclassified our investments in ARPS to long-term investments, since we may not be able to liquidate them in the near term. If we need to liquidate these ARPS to fund our business efforts and are not able to do so, our business could be harmed. In addition, failed auctions in the future may further affect the fair value of our ARPS and may result in additional impairment charges.
Currency exchange rate fluctuations may have a negative effect on our financial condition.
We are exposed to fluctuations in currency from purchases of goods, services and equipment and investments in other countries and funding denominated in the currencies of other countries. In particular, we are exposed to the fluctuations in the exchange rate between the U.S. dollar and the Euro. Currently, our source of Captisol is located in Portugal, which is a Euro currency country. Fluctuations in currency exchange rates may affect our results of operations, which in turn may adversely affect reported earnings and the comparability of period-to-period results of operations. Due to the constantly changing currency exposures and the potential substantial volatility of currency exchange rates, we cannot predict the effect of exchange rate fluctuations on our future results. If our current exposure to exchange rate fluctuations increases, adverse movements in exchange rates could have a material adverse effect on our financial condition and results of operations.
If plaintiffs bring product liability lawsuits against us, we may incur substantial liabilities and may be required to limit commercialization of our approved products and product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates in clinical trials and face an even greater risk for commercialized products. Although we are not currently a party to product liability litigation, if we are sued, we may be held liable if any product or product candidate we develop causes injury or is found otherwise unsuitable during product testing, manufacturing, marketing or sale. Regardless of merit or eventual outcome, liability claims may result in decreased demand for any product candidates or products that we may develop, injury to our reputation, discontinuation of clinical trials, costs to defend litigation, substantial monetary awards to clinical trial participants or patients, loss of revenue and the inability to commercialize any products that we develop. We have product liability insurance that covers our clinical trials up to a $5.0 million annual limit. We intend to expand product liability insurance coverage to include the sale of
24
commercial products if we obtain marketing approval for any products that we may develop. However, this insurance may be prohibitively expensive, or may not fully cover our potential liabilities. Inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or delay the commercialization of our product candidates. If we are sued for any injury caused by our product candidates or any future products, our liability could exceed our total assets.
We use hazardous chemicals in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research and development processes involve the controlled use of hazardous materials, and our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge or injury from these materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these materials. We could be subject to civil damages in the event of an improper or unauthorized release of, or exposure of individuals to, hazardous materials. In addition, claimants may sue us for injury or contamination that results from our use or the use by third parties of these materials and our liability may exceed our total assets. We maintain insurance for the use of hazardous materials in the aggregate amount of $2.0 million, which may not be adequate to cover any claims. Compliance with environmental and other laws and regulations may be expensive, and current or future regulations may impair our research, development or production efforts.
In the future, we may develop product candidates containing drug substances which are regulated by the U.S. Drug Enforcement Administration. Failure to comply with applicable regulations could harm our business.
The Controlled Substances Act imposes various registration, recordkeeping and reporting requirements, procurement and manufacturing quotas, labeling and packaging requirements, security controls and a restriction on prescription refills on certain prescription drugs regulated as controlled substances. A principal factor in determining the particular requirements, if any, applicable to a product is its actual or potential abuse profile. The U.S. Drug Enforcement Administration, or DEA, regulates drug substances as Schedule I, II, III, IV or V substances, with Schedule I and Schedule II substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. We may develop product candidates which are subject to DEA regulations relating to manufacture, storage, distribution and dispensing, and for which the DEA regulates the amount of the scheduled substance that is available for clinical trials and commercial distribution. These products are subject to stringent controls, including quotas on the amount of product that can be manufactured as well as a prohibition on the refilling of prescriptions without a new prescription from the physician. The DEA periodically inspects facilities for compliance with its rules and regulations. Failure to comply with current and future regulations of the DEA could lead to a variety of sanctions, including revocation, or denial of renewal, or of DEA registrations, injunctions, or civil or criminal penalties and could harm our business and financial condition. Most states also regulate controlled substances and impose independent licensing, recordkeeping and security requirements.
We will need to implement additional finance and accounting systems, procedures and controls in the future as we grow and to satisfy new reporting requirements.
The laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002, or Sarbanes-Oxley, and rules enacted and proposed by the U.S. Securities and Exchange Commission, or SEC, and by the Nasdaq Global Market, will result in increased costs to us as we undertake efforts to comply with rules and respond to the requirements applicable to public companies. The rules make it more difficult and costly for us to obtain some types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits
25
and coverage or incur substantially higher costs to obtain the same or similar coverage as compared to the policies previously available to public companies. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or our board committees or as executive officers.
As a public company, we need to comply with Sarbanes-Oxley and the related rules and regulations of the SEC, including expanded disclosure, accelerated reporting requirements and more complex accounting rules. Compliance with Section 404 of Sarbanes-Oxley and other requirements will increase our costs and require additional management resources. The rules and regulations of the SEC will require among other things, that we file annual, quarterly and current reports with respect to our business and financial condition. The requirements of these rules and regulations will make some activities more difficult, time-consuming or costly and may also place undue strain on our personnel, systems and resources. We have been upgrading our finance and accounting systems, procedures and controls and will need to continue to implement additional finance and accounting systems, procedures and controls as we grow to satisfy new reporting requirements. We currently do not have an internal audit group. In addition, we will need to hire additional legal and accounting staff with appropriate experience and technical knowledge, and we cannot provide assurance that we will be able to hire appropriate personnel in a timely fashion. If we are unable to complete the required assessment as to the effectiveness of our internal control over financial reporting or if we continue to have material weaknesses in internal control over financial reporting or develop additional material weaknesses in internal control over financial reporting in the future, investors could lose confidence in the reliability of our internal control over financial reporting, which could adversely affect our stock price.
Our facilities are located in a tornado zone, and the occurrence of a tornado or other catastrophic disaster could damage our facilities and equipment, which could cause us to curtail or cease operations.
Our facilities are located outside of Kansas City, Kansas, which is in a tornado zone. We are therefore vulnerable to damage from tornados. We are also vulnerable to damage from other types of disasters, such as power loss, fire, floods and similar events. If any disaster were to occur, our ability to operate our business could be seriously impaired. We are insured against up to $2.6 million in damages resulting from natural disasters, including tornados. We currently may not have adequate insurance to cover our losses resulting from disasters or other similar significant business interruptions, and we do not plan to purchase additional insurance to cover such losses due to the cost of obtaining such coverage. Any significant losses that are not recoverable under our insurance policies could seriously impair our business, financial condition and prospects.
Risks Related to Ownership of our Common Stock and this Offering
As our common stock has not been publicly traded, we expect that the price of our common stock may fluctuate substantially.
Before this offering, there has been no public market for our common stock. An active public trading market may not develop after completion of this offering or, if developed, may not be sustained. The price of our common stock sold in this offering will not necessarily reflect the market price of our common stock after this offering. The market price for our common stock after this offering may be affected by a number of factors, including:
- •
- quarterly variations in our operating results, or the operating results of our competitors;
- •
- the timing of revenue recognition;
- •
- the announcement of new products by us or our competitors;
26
- •
- announcements related to litigation;
- •
- changes in earnings estimates, investors' perceptions, recommendations by securities analysts or our failure to achieve analysts' earning estimates;
- •
- the depth and liquidity of the market for our common stock;
- •
- changing legal or regulatory requirements;
- •
- developments in our industry; and
- •
- general market conditions and other factors unrelated to our operating performance or the operating performance of our competitors.
In addition, the stock market in general, and the Nasdaq Global Market in particular, have experienced substantial price and volume volatility that is often seemingly unrelated to the operating performance of particular companies. These broad market fluctuations may cause the trading price of our common stock to decline. In the past, securities class action litigation has often been brought against a company after a period of volatility in the market price of its common stock. We may become involved in this type of litigation in the future. Any securities litigation claims brought against us could result in substantial expense and the diversion of our management's attention from our business.
New investors in our common stock will experience immediate and substantial dilution after this offering.
The assumed initial public offering price is substantially higher than the book value per share of our outstanding common stock. If you purchase common stock in this offering, you will incur immediate dilution of $ per share based on the assumed initial offering price of $ per share. This dilution is due in large part to earlier investors in our company having paid substantially less than the assumed initial public offering price when they purchased their shares. Investors who purchase shares of common stock in this offering will contribute approximately % of the total amount we have raised to fund our operations, but will own only approximately % of our common stock, based upon the number of shares outstanding as of December 31, 2007. The exercise of outstanding options and warrants and other future equity issuances, including future public offerings or private placements of equity securities and any additional shares issued in connection with acquisitions, may result in further economic dilution to investors. For a further description of dilution that you will experience immediately after this offering, see the section of this prospectus entitled "Dilution."
A sale of a substantial number of shares of our common stock may cause the price of our common stock to decline.
If our stockholders sell substantial amounts of our common stock in the public market after this offering or the public market perceives that existing stockholders might sell shares of common stock, the market price of our common stock could decline. After this offering, we will have shares of common stock outstanding based on the number of shares outstanding as of December 31, 2007. All of the shares offered under this prospectus will be freely tradable without restriction or further registration under the federal securities laws, unless purchased by our "affiliates" as that term is defined in Rule 144 under the Securities Act of 1933. Of the remaining shares outstanding upon the closing of this offering, shares may be sold pursuant to Rules 144, 144(k) and 701 upon the expiration of lock-up agreements that expire 180 days after the date of this prospectus unless otherwise extended or waived as described in "Shares Eligible for Future Sale."
27
Following this offering, existing stockholders holding an aggregate of shares of common stock will have rights, subject to some conditions, that permit them to require us to file a registration statement with the SEC or include their shares in registration statements that we may file for ourselves or other stockholders. If we register their shares of common stock following the expiration of the lock-up agreements, they can sell those shares in the public market. Promptly following this offering, we intend to register shares of common stock for issuance under our stock option plans. As of December 31, 2007, 7,695,668 shares were subject to outstanding options, with a weighted average exercise price of $0.77 per share, of which 4,174,584 were vested. Once we register these shares, they can be freely sold in the public market upon issuance, subject to the lock-up agreements referred to above and the restrictions imposed on our affiliates under Rule 144.
Our directors, officers and principal stockholders have significant voting power and may take actions that may not be in the best interests of our other stockholders.
After this offering, our officers, directors and principal stockholders each holding more than five percent of our common stock, collectively will control approximately % of our outstanding common stock, without giving effect to the purchase of shares by any such persons in this offering. As a result, these stockholders, if they act together, will be able to control our management and affairs and most matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control and might adversely affect the market price of our common stock. This concentration of ownership may not be in the best interests of our other stockholders.
We have broad discretion in the use of proceeds of this offering.
Our management will have broad discretion over the use and investment of the net proceeds of this offering, and accordingly investors in this offering will need to rely upon the judgment of our management with respect to the use of proceeds, with only limited information concerning management's specific intentions. Our management may spend a portion or all of the net proceeds from this offering in ways that our stockholders may not desire or that may not yield a favorable return. The failure of our management to apply the net proceeds of this offering effectively could harm our business, financial condition and results of operations.
Anti-takeover provisions in our amended and restated certificate of incorporation and amended and restated bylaws, and Delaware law, contain provisions that could discourage a takeover.
In addition to the effect that the concentration of ownership by our officers, directors and significant stockholders may have, our amended and restated certificate of incorporation and our amended and restated bylaws contain provisions that may enable our management to resist a change in control. These provisions may discourage, delay or prevent a change in our ownership or a change in our management. In addition, these provisions could limit the price that investors would be willing to pay in the future for shares of our common stock. Such provisions, to be set forth in our amended and restated certificate of incorporation or amended and restated bylaws effective upon the completion of this offering, include:
- •
- advance notice requirements for stockholders for nominations to serve on our board of directors or for proposals that can be acted upon at stockholder meetings;
- •
- stockholder action by written consent will be prohibited;
- •
- special meetings of the stockholders will be permitted to be called only by a majority of our board of directors, the chairman of our board of directors or our chief executive officer;
28
- •
- our board of directors will be classified, consisting of three classes of directors with staggered three-year terms, with each class consisting, as nearly as possible, of one-third of the total number of directors;
- •
- stockholders will not be permitted to cumulate their votes for the election of directors;
- •
- stockholders will not be permitted to change the authorized number of directors constituting our board of directors;
- •
- newly created directorships resulting from an increase in the authorized number of directors or vacancies on our board of directors will be filled only by majority vote of the remaining directors, even though less than a quorum is then in office;
- •
- our board of directors will be expressly authorized to modify, alter or repeal our amended and restated bylaws; and
- •
- stockholders will be permitted to amend our amended and restated bylaws only upon receiving at least a majority of the votes entitled to be cast by holders of all outstanding shares then entitled to vote generally in the election of directors, voting together as a single class.
We are also subject to the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit some business combinations with stockholders owning 15% or more of our outstanding voting stock. These and other provisions in our amended and restated certificate of incorporation, our amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our board of directors or initiate actions that are opposed by our then-current board of directors, including delaying or impeding a merger, tender offer or proxy contest involving us. Any delay or prevention of a change in control transaction or changes in our board of directors could cause the market price of our common stock to decline.
We have not paid dividends and do not intend to pay dividends in the future.
We have not previously declared or paid any cash dividends on our capital stock, and at present we do not intend to do so in the forseeable future. See also "Dividend Policy." You should not rely on an investment in our common stock if you require dividend income. Income derived from an investment in our common stock would only come from a rise in the market price of our common stock and you would have to sell some or all of our common stock you purchased to realize this gain. Any increase in the market price of our common stock will be uncertain and unpredictable.
29
FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
This prospectus contains forward-looking statements that are based on our management's beliefs and assumptions and on information currently available to our management. The forward-looking statements are contained principally in the sections entitled "Prospectus Summary," "Risk Factors," "Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Business." Forward-looking statements include, but are not limited to, statements about:
- •
- expected timing of regulatory filings, such as the filing of an NDA for Captisol-Enabled Fosphenytoin IV and IM;
- •
- expectations regarding the limited number, scope and duration of clinical trials necessary to receive regulatory approval of our product candidates;
- •
- expectations of future operating results or financial performance;
- •
- introduction of new products;
- •
- plans for growth and future operations;
- •
- our plans to develop and commercialize our product candidates;
- •
- our use of proceeds from this offering;
- •
- the success and timing of our preclinical studies and clinical trials;
- •
- the ability to obtain and maintain regulatory approval for our product candidates;
- •
- the size and growth potential of possible markets for our product candidates and our ability to serve those markets;
- •
- regulatory developments in the United States and foreign countries;
- •
- the rate and degree of market acceptance of any future products;
- •
- the accuracy of our estimates regarding expenses, future revenue, capital requirements and needs for additional financing and our ability to obtain additional financing;
- •
- our ability to attract strategic partners with development, regulatory and commercialization expertise;
- •
- our ability to obtain and maintain intellectual property protection for our products and product candidates;
- •
- the development of our marketing capabilities;
- •
- the success of competing drugs that are or may become available; and
- •
- the performance of third-party manufacturers who supply the components included in our products and product candidates.
30
In some cases, you can identify forward-looking statements by terms such as "anticipates," "believes," "can," "continue," "could," "estimates," "expects," "intend," "may," "plan," "potential," "predict," "project," "should," "will," "would" and similar expressions intended to identify forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance time frames or achievements to be materially different from any future results, performance, time frames or achievements expressed or implied by the forward-looking statements. We discuss many of these risks, uncertainties and other factors in this prospectus in greater detail in the section of this prospectus entitled "Risk Factors." While we are responsible for these forward-looking statements, given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this prospectus. You should read this prospectus and the documents that we have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We hereby qualify all of our forward-looking statements by these cautionary statements.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
This prospectus also contains statistical data and estimates that we obtained from industry reports, publications and information generated by the Multiple Myeloma Research Foundation,American Family Physician, the Health Care Cost and Utilization Project and IMS Health, or IMS. The information from IMS is an estimate derived from the use of information under license from the following IMS Health information service: IMS National Sales Perspectives™ database. Although we believe that the reports, publications and information are reliable, we have not independently verified the data.
31
We estimate that we will receive approximately $ million in net proceeds from this offering, or $ million if the underwriters' over-allotment option is exercised in full, based upon an assumed initial public offering price of $ per share, after deducting estimated underwriting discounts and estimated offering expenses payable by us. A $1.00 increase (decrease) in the assumed public offering price of $ per share would increase (decrease) our net proceeds by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and estimated offering expenses payable by us. We do not expect that a change in the initial offering price by these amounts would have a material effect on our uses of the net proceeds from this offering.
We expect to use our net proceeds from this offering as follows:
- •
- approximately $ million for our clinical trials and further development and eventual commercialization of our retained product candidates;
- •
- approximately $ million for further development of our partner product candidates to advance them to Phase 2 clinical trials;
- •
- approximately $ million for building our internal infrastructure and capabilities; and
- •
- the balance for working capital and general corporate expenses.
The actual costs and timing of clinical trials are highly uncertain, subject to risk and may change depending upon the clinical indication targeted, the development strategy pursued and the results of preclinical studies and earlier clinical trials. The amounts and timing of other expenditures will depend upon numerous factors, including the status of our product development and commercialization efforts, the amount of proceeds actually raised in this offering, competition, manufacturing, activities and any strategic partnering arrangements we may enter into.
The amounts we actually expend in these areas may vary significantly from our current intentions and will depend on a number of factors, including FDA approval of our product candidates, future sales growth, success of research and product development efforts, cash generated from future operations and actual expenses to operate our business. We will retain broad discretion in the allocation of a substantial portion of the net proceeds of this offering. We may also use a portion of the net proceeds to acquire or invest in complementary businesses, technologies, services or products. We have no current plans, agreements or commitments with respect to any such acquisition or investment, and we are not currently engaged in any negotiations with respect to any such transaction. Pending such uses, we will invest the net proceeds of this offering in short-term, interest-bearing, investment-grade securities.
We have not declared or paid any cash dividends on our capital stock, and all dividends automatically accrued to date by the holders of our preferred stock will be terminated without cash payment in connection with this offering. We currently intend to retain any future earnings to finance the growth and development of our business and therefore do not anticipate paying any cash dividends in the foreseeable future. Any future determination to pay cash dividends will be at the discretion of our board of directors and will depend upon our financial condition, operating results, capital requirements, any contractual restrictions and such other factors that our board of directors deems appropriate.
32
The following table sets forth our capitalization as of December 31, 2007:
- •
- on an actual basis; and
- •
- on a pro forma as adjusted basis to give effect to the conversion of all outstanding shares of our preferred stock into an aggregate of shares of common stock immediately prior to the closing of this offering and the sale of shares of our common stock we are offering at an assumed initial public offering price of $ per share, after deducting estimated underwriting discounts and estimated offering expenses payable by us.
You should read this table together with our consolidated financial statements and related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations" appearing elsewhere in this prospectus.
| | December 31, 2007 | ||||
|---|---|---|---|---|---|
| | Actual | Pro Forma As Adjusted(1) | |||
| | (in thousands, except share data) | ||||
| Consolidated Balance Sheet Data: | |||||
| Cash, cash equivalents and short-term investments | $ | 8,109 | |||
| Redeemable convertible preferred stock, $0.01 par value per share; 79,665,642 shares authorized, 79,665,637 issued and outstanding, actual; no shares authorized, issued and outstanding, pro forma as adjusted | $ | 34,600 | |||
| Stockholders' equity (deficit): | |||||
| Convertible preferred stock, $0.01 par value per share; 2,000,000 shares authorized, issued and outstanding, actual; no shares authorized, issued and outstanding, pro forma as adjusted | 20 | ||||
| Common stock, $0.01 par value per share; 103,000,000 shares authorized, 7,380,438 issued and 7,280,438 outstanding, actual; 100,000,000 shares authorized, issued and outstanding, pro forma as adjusted | 74 | ||||
| Additional paid-in capital | 3,001 | ||||
| Accumulated deficit | (21,927 | ) | |||
| Treasury stock, at cost | (200 | ) | |||
| Total stockholders' equity (deficit) | (19,032 | ) | |||
| Total capitalization | $ | 15,568 | |||
- (1)
- A $1.00 increase (decrease) in the assumed public offering price of $ per share would increase (decrease) each of cash, cash equivalents and short-term investments, additional paid-in capital, total stockholders' equity (deficit) and total capitalization by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and estimated offering expenses payable by us.
33
The foregoing information regarding the number of shares of our common stock to be outstanding immediately after this offering is based on shares outstanding as of December 31, 2007 and excludes:
- •
- 7,695,668 shares of common stock issuable upon the exercise of outstanding options under our Amended and Restated 1997 Equity Compensation Plan with a weighted average exercise price of $0.77 per share;
- •
- 4,460,966 shares of common stock issuable upon the exercise of outstanding warrants with an exercise price of $0.01 per share; and
- •
- shares of common stock reserved for future issuance under our 2008 Equity Incentive Plan, our 2008 Employee Stock Purchase Plan and our 2008 Non-Employee Directors' Stock Option Plan, as well as automatic increases in the number of shares of our common stock reserved for future issuance under these benefit plans, each of which will become effective immediately upon the signing of the underwriting agreement for this offering.
34
If you invest in our common stock in this offering, your interest will be diluted to the extent of the difference between the initial public offering price per share of our common stock in this offering and the pro forma as adjusted net tangible book value per share of common stock immediately after this offering.
Net tangible book value per share represents total tangible assets less total liabilities, divided by the number of shares of common stock outstanding. Our pro forma net tangible book value as of December 31, 2007 was approximately $ million, or approximately $ per share of common stock, after giving effect to the conversion of our preferred stock into common stock. After further giving effect to the sale by us of shares of common stock in this offering at an assumed initial public offering price of $ per share, after deducting estimated underwriting discounts and estimated offering expenses payable by us, our pro forma as adjusted net tangible book value as of December 31, 2007 would have been approximately $ million, or approximately $ per share of common stock. This represents an immediate increase in net tangible book value of $ per share to existing stockholders and an immediate dilution of $ per share to new investors.
The following table illustrates this dilution on a per share basis, without giving effect to the over-allotment option granted to the underwriters:
| Assumed initial public offering price per share | $ | |||||
| Historical net tangible book value per share as of December 31, 2007 | $ | |||||
| Increase in pro forma as adjusted net tangible book value per share attributable to this offering | ||||||
| Pro forma as adjusted net tangible book value per share after this offering | ||||||
| Dilution per share to new investors participating in this offering | $ | |||||
A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) our pro forma as adjusted net tangible book value by $ million, or $ per share, and the dilution to new investors participating in this offering by $ per share, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and estimated offering expenses payable by us.
If the underwriters exercise their over-allotment option to purchase additional shares from us, our pro forma as adjusted net tangible book value per share as of December 31, 2007 would have been $ per share, the increase in the pro forma as adjusted net tangible book value per share attributable to this offering would be $ and the dilution to new investors participating in this offering would be $ per share.
The following table summarizes, as of December 31, 2007, on the pro forma as adjusted basis described above, the number of shares of our common stock purchased from us, the total consideration paid to us and the average price per share paid to us by existing stockholders and to be paid by new investors purchasing shares of our common stock in this offering. The table assumes an
35
initial public offering price of $ per share, after deducting estimated underwriting discounts and estimated offering expenses payable by us.
| | Shares Purchased | Total Consideration | | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average Price Per Share | ||||||||||||
| | Number | Percent | Amount | Percent | |||||||||
| Existing stockholders | % | $ | % | $ | |||||||||
| New investors | |||||||||||||
| Total | 100 | % | $ | 100 | % | ||||||||
A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) total consideration paid by new investors by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same, and after deducting the estimated underwriting discounts and estimated offering expenses payable by us.
If the underwriters' over-allotment option to purchase additional shares from us in this offering is exercised in full, the following will occur:
- •
- the percentage of shares of common stock held by existing stockholders after the completion of this offering will be approximately % of the total number of shares of our common stock outstanding after this offering; and
- •
- the number of shares held by new investors after the completion of this offering will be , or approximately % of the total number of shares of our common stock outstanding after this offering.
The foregoing information as to the number of shares of our common stock to be outstanding immediately after this offering is based on shares outstanding as of December 31, 2007 and excludes:
- •
- 7,695,668 shares of common stock issuable upon the exercise of outstanding options under our Amended and Restated 1997 Equity Compensation Plan with a weighted average exercise price of $0.77 per share;
- •
- 4,460,966 shares of common stock issuable upon the exercise of outstanding warrants with an exercise price of $0.01 per share; and
- •
- shares of common stock reserved for future issuance under our 2008 Equity Incentive Plan, our 2008 Employee Stock Purchase Plan and our 2008 Non-Employee Directors' Stock Option Plan, as well as automatic increases in the number of shares of our common stock reserved for future issuance under these benefit plans, each of which will become effective immediately upon the signing of the underwriting agreement for this offering.
Assuming the exercise in full of all of our outstanding options and warrants at December 31, 2007, historical net tangible book value before this offering at December 31, 2007 would be $ per share, representing an immediate dilution of $ per share to our existing stockholders and, after giving effect to the sale of shares of common stock in this offering, there would be an immediate dilution of per share to purchasers of our common stock in this offering.
36
SELECTED CONSOLIDATED FINANCIAL DATA
The following tables summarize our historical consolidated financial data. The consolidated statement of operations data for the years ended December 31, 2005, 2006 and 2007 and the consolidated balance sheet data as of December 31, 2006 and 2007 are derived from our consolidated financial statements audited by KPMG LLP, independent registered public accounting firm, included elsewhere in this prospectus. The consolidated statement of operations data for the year ended December 31, 2004 and consolidated balance sheet data as of December 31, 2005 are derived from our audited consolidated financial statements not included in this prospectus. The consolidated statement of operations data for the year ended December 31, 2003 and the consolidated balance sheet data as of December 31, 2003 and 2004 are derived from our unaudited consolidated financial statements not included in this prospectus. The following selected consolidated financial data should be read in conjunction with the consolidated financial statements and related notes and "Management's Discussion and Analysis of Financial Condition and Results of Operations." The unaudited consolidated financial statements include, in the opinion of management, all adjustments, consisting only of normal recurring adjustments, which management considers necessary for a fair statement of the results of those periods. Our historical results are not necessarily indicative of our operating results or financial position to be expected in the future.
| | Years Ended December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2003 | 2004 | 2005 | 2006 | 2007 | |||||||||||||
| | (in thousands, except share and per share data) | |||||||||||||||||
| Consolidated Statement of Operations Data: | ||||||||||||||||||
Revenue: | ||||||||||||||||||
| Milestone and license | $ | 1,920 | $ | 1,008 | $ | 1,947 | $ | 1,982 | $ | 1,172 | ||||||||
| Material sales | 915 | 1,398 | 4,164 | 5,671 | 6,819 | |||||||||||||
| Royalties | 1,737 | 2,249 | 2,511 | 2,989 | 3,829 | |||||||||||||
| Contract research | — | 15 | 158 | 845 | 683 | |||||||||||||
| Other | 96 | 59 | 39 | 93 | 242 | |||||||||||||
| Total revenue | 4,668 | 4,729 | 8,819 | 11,580 | 12,745 | |||||||||||||
| Operating expenses: | ||||||||||||||||||
| Cost of sales(1) | 1,228 | 1,183 | 2,566 | 2,482 | 2,508 | |||||||||||||
| Research and development(1) | 3,299 | 3,993 | 4,419 | 4,435 | 9,078 | |||||||||||||
| Selling, general and administrative(1) | 3,235 | 2,422 | 2,488 | 2,681 | 4,328 | |||||||||||||
| Total operating expenses | 7,762 | 7,598 | 9,473 | 9,598 | 15,914 | |||||||||||||
| Operating income (loss) | (3,094 | ) | (2,869 | ) | (654 | ) | 1,982 | (3,169 | ) | |||||||||
| Other income (expense): | ||||||||||||||||||
| Interest income (expense), net | (94 | ) | (76 | ) | 223 | 428 | 582 | |||||||||||
| Warrant income (expense) | (830 | ) | 42 | — | — | — | ||||||||||||
| Other, net | 94 | 87 | (7 | ) | (2 | ) | — | |||||||||||
| Total other income (expense) | (830 | ) | 53 | 216 | 426 | 582 | ||||||||||||
| Income (loss) before income taxes | (3,924 | ) | (2,816 | ) | (438 | ) | 2,408 | (2,587 | ) | |||||||||
| Provision for income taxes | — | — | — | 53 | — | |||||||||||||
| Net income (loss) | (3,924 | ) | (2,816 | ) | (438 | ) | 2,355 | (2,587 | ) | |||||||||
| Preferred dividends and accretion | 1,326 | 2,418 | 4,441 | 4,455 | 4,471 | |||||||||||||
| Net loss attributable to common stockholders | $ | (5,250 | ) | $ | (5,234 | ) | $ | (4,879 | ) | $ | (2,100 | ) | $ | (7,058 | ) | |||
| Basic and diluted net loss per common share(2) | $ | (1.25 | ) | $ | (1.11 | ) | $ | (0.87 | ) | $ | (0.35 | ) | $ | (1.04 | ) | |||
| Shares used to compute basic and diluted net loss per common share(2) | 4,200,516 | 4,725,717 | 5,617,270 | 6,082,357 | 6,798,681 | |||||||||||||
- (1)
- Effective January 1, 2006, we adopted Statement of Financial Accounting Standards, or SFAS, No. 123 (revised 2004),Share-Based Payment, or SFAS No. 123R. SFAS No. 123R superseded SFAS No. 123,Accounting for Stock-Based Compensation, or SFAS No. 123, and Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees, or APB No. 25. SFAS No. 123R requires that all stock-based compensation be recognized as an expense in the financial statements and that
footnotes continued on following page
37
such cost be measured at the fair value of the award. As we previously used the minimum value method, as defined by SFAS No. 123, for purposes of measuring the fair value of stock options, we adopted SFAS No. 123R using the prospective method of application, which requires us to recognize compensation cost based on fair value for any awards granted after January 1, 2006, and any existing awards modified after that date. Under the prospective method, we recorded stock-based compensation expense for awards granted prior to January 1, 2006 using the intrinsic value method of APB No. 25.
The periods presented above include employee stock-based compensation in the following amounts:
| | Years Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2003 | 2004 | 2005 | 2006 | 2007 | |||||||||||
| | (in thousands) | |||||||||||||||
| Cost of sales | $ | 26 | $ | — | $ | 4 | $ | 2 | $ | 5 | ||||||
| Research and development | 173 | 104 | 39 | 15 | 21 | |||||||||||
| Selling, general and administrative | 410 | 167 | 6 | 23 | 177 | |||||||||||
| Total | $ | 609 | $ | 271 | $ | 49 | $ | 40 | $ | 203 | ||||||
- (2)
- See Notes 2(r) and 12 of the notes to our consolidated financial statements for an explanation of the method used to calculate basic and diluted net loss per common share.
| | December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2003 | 2004 | 2005 | 2006 | 2007 | |||||||||||
| | (in thousands) | |||||||||||||||
| Consolidated Balance Sheet Data: | ||||||||||||||||
Cash, cash equivalents and short-term investments | $ | 2,380 | $ | 7,822 | $ | 7,533 | $ | 11,041 | $ | 8,109 | ||||||
| Working capital | (484 | ) | 8,725 | 10,444 | 13,557 | 10,821 | ||||||||||
| Total current assets | 4,747 | 10,044 | 11,441 | 14,592 | 13,629 | |||||||||||
| Total assets | 6,111 | 16,997 | 16,354 | 18,884 | 18,432 | |||||||||||
| Total current liabilities | 5,231 | 1,319 | 997 | 1,035 | 2,808 | |||||||||||
| Total liabilities | 5,243 | 1,328 | 1,021 | 1,052 | 2,864 | |||||||||||
| Redeemable convertible preferred stock | 14,304 | 21,652 | 25,954 | 30,269 | 34,600 | |||||||||||
| Accumulated deficit | (18,441 | ) | (21,257 | ) | (21,695 | ) | (19,340 | ) | (21,927 | ) | ||||||
| Total stockholders' deficit | (13,436 | ) | (5,983 | ) | (10,621 | ) | (12,437 | ) | (19,032 | ) | ||||||
38
MANAGEMENT'S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read together with our financial statements and related notes appearing elsewhere in this prospectus and the information under "Selected Financial Data." This discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. These forward-looking statements fall within the meaning of the federal securities laws that relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as "may," "will," "should," "expect," "plan," "anticipate," "believe," "estimate," "predict," "intend," "potential" or "continue" or the negative of these terms or other comparable terminology. Forward-looking statements involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those set forth under "Risk Factors," "Forward-Looking Statements" and elsewhere in this prospectus.
Overview
We are a specialty pharmaceutical company focused on the development and commercialization of drugs specifically designed to address limitations of current therapies in selected established markets. We have developed a broad portfolio of 12 product candidates utilizing our drug formulation technology using Captisol cyclodextrins. Captisol cyclodextrins are a patent protected, specifically modified family of cyclodextrins designed to improve solubility, stability, bioavailability, safety and/or dosing of a number of active pharmaceutical ingredients, or APIs. Captisol cyclodextrins are ring-shaped molecules comprised of sugar molecules. A number of different APIs that are not otherwise readily soluble in water can interact with Captisol cyclodextrins, resulting in compounds that are more water-soluble than the API would be standing alone. In order to best utilize our internal resources, experience and technology, we are focusing on the development and commercialization of product candidates for use in the acute care hospital setting. For our product candidates that will likely require more extensive development and commercialization efforts, we partner with established pharmaceutical or specialty pharmaceutical companies. We also outlicense our Captisol technology to third parties for use in the development of their own products.
Historically, we have focused on the outlicense of our Captisol technology to established pharmaceutical companies for the application to, and improvement of, product candidates in their pipelines. Our technology has been validated by the launch of four Captisol-Enabled products commercialized by two of our outlicensees, Pfizer and Bristol-Myers Squibb, or BMS. We originally focused on this strategy in order to accelerate the commercialization of our Captisol technology and create cash flow for our company. The gross profits generated by this outlicensing business have allowed us to expand our strategy to capitalize on the growth opportunities and business lines enabled by Captisol. More recently, we have been focusing on the development of our retained product candidates for use in the acute care hospital setting, as well as our partner product candidates which we have collaborated with, outlicensed or intend to outlicense to established pharmaceutical companies for development and commercialization. All of our retained product candidates and partner product candidates are currently in development and have not yet been commercialized.
As a result of our historic focus on Captisol outlicensing, revenue derived from Captisol outlicensing accounted for 95%, 78% and 87% of total revenue during 2005, 2006 and 2007, respectively. However, with the execution of three agreements for our partner product candidates in 2006 and 2007, we expect to derive a significant portion of our future revenue from our partner product candidates. We do not expect to have any revenue from retained products sales until at least 2010. We expect our revenue over the next several years to be derived primarily from milestone payments,
39
license fees, material sales and royalties on product sales. These payments will be conditional upon our partners' continued development and commercialization of products using Captisol and the signing of new agreements.
In 2004, we implemented a general program of providing a limited clinical use agreement for companies early in the development process, which simplifies processing within pharmaceutical companies developing new compounds utilizing Captisol. Pursuant to this type of agreement, the licensee is granted a limited license to use Captisol in small Phase 1 human clinical trials and we agree to supply Captisol to the licensee for such clinical trials. This change in our Captisol outlicensing effort has resulted in many new development efforts, and is a primary contributor to the significant increase in material sales since 2004.
Material Weaknesses
Our independent registered public accounting firm determined that we had five material weaknesses as of December 31, 2007:
- •
- our control environment is ineffective due to the operating style of our senior management, which does not consistently emphasize effective internal control over financial reporting or the importance of the financial reporting process;
- •
- we do not have an effective process for identifying financial reporting risks and evaluating whether adequate policies, procedures and control activities are implemented on a timely basis, particularly for complex or non-recurring transactions;
- •
- we are not able to effectively monitor whether our internal control over financial reporting is functioning properly because documentation of the operation of control activities and internal control policies and procedures is insufficient to assess performance, and responsibilities for our personnel involved in internal control over financial reporting are not clearly delineated;
- •
- we did not appropriately segregate duties, or implement alternative controls, relative to executing and accounting for new revenue arrangements; and
- •
- we do not have personnel with the necessary experience and training to ensure appropriate revenue recognition for key revenue streams; specifically, we lack employees with the expertise to analyze multiple element arrangements for appropriate revenue recognition.
As a result of these material weaknesses in internal control over financial reporting, material misstatements existed with respect to the accounting for revenue recognition, preferred stock and warrants and other items that resulted in a recent restatement of our consolidated financial statements for the years ended December 31, 2004, 2005 and 2006. Additionally, material misstatements were identified in our revenue recognition, research and development costs, stock-based compensation and other items for the year ended December 31, 2007, which were corrected prior to the issuance of the consolidated financial statements. We have begun a process of remediating these material weaknesses, but this process will take time, and we will not be able to assert that we have remediated any of these material weaknesses until the procedures that we put in place operate for a sufficient period of time for us to determine that they are effective.
Our management team recognizes the significance of the reported material weaknesses in internal controls, and has made remediation a high priority. In addition to addressing the matters specifically
40
raised by our independent registered public accounting firm, we have initiated an aggressive "top down" approach to increase the visibility, awareness and importance of internal controls and compliance across our organization. Remedial actions we have taken to address the material weaknesses include the following:
- •
- Our board of directors adopted a revised audit committee charter that strengthened the role of the audit committee. Specifically, it indicates that the primary purpose of the committee is to act on behalf of the board of directors in fulfilling the board of directors' oversight responsibilities for our corporate accounting and financial reporting processes, systems of internal control over financial reporting, and audits of financial statements, as well as the qualifications, independence and performance of our independent registered public accounting firm. Our audit committee and the chairman of our audit committee are committed to open communication among the committee, our independent registered public accounting firm, our financial management, our senior management and third-party consultants hired to assist us in evaluating our finance organization.
- •
- We have strengthened our corporate accounting department by increasing the number of corporate accounting personnel. In October 2007, we hired a corporate controller, who is responsible for the periodic financial statement close process, accounting for new revenue arrangements and revenue recognition for key revenue streams. Currently, we are reevaluating roles, reporting relationships, processes, and training needs in the accounting area. This examination will likely result in hiring additional personnel with relevant experience and may involve a realignment of existing responsibilities, reporting relationships and additional training of current personnel.
- •
- In May 2008, Theron E. Odlaugh, Ph.D. accepted an offer to join our company as president and chief operating officer, which we believe will allow us to better segregate duties. In addition, we are actively searching for a vice president of business development. We believe filling these positions will allow our chief executive officer and chief financial officer to focus more of their efforts on developing and maintaining programs to improve the overall control environment of the organization. These additions are expected to remediate the material weakness related to segregation of duties.
We have engaged an independent consultant and an independent third-party risk consulting and internal audit services firm to aid us in evaluating the necessary personnel, resources and processes specifically related to identifying financial reporting risks and identifying whether adequate policies, procedures and control activities are implemented as well as revenue recognition documentation, controls and policy application. Through these efforts, we have developed and continue to refine a comprehensive remediation plan to address the material weaknesses in internal control identified by our independent registered public accounting firm.
Critical Accounting Policies
This discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in conformity with U.S. generally accepting accounting principles. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenue, expenses and related disclosures. On an ongoing basis, we evaluate our estimates and judgments, including those described below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making assumptions about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results could differ
41
from those estimates. We believe the following accounting policies require significant judgment and estimates by management in the preparation of our consolidated financial statements.
Revenue Recognition
We enter into license agreements with pharmaceutical companies for the use of Captisol in their product development activities and for commercialization of Captisol-Enabled products. The terms of the agreements, whether with respect to our partner products or our Captisol outlicensing program, may include nonrefundable license fees, nonrefundable payments based on the achievement of specified milestones and royalties on sales of products containing Captisol. We have also entered into exclusive supply agreements with many of our customers that include pricing for the supply of customers' requirements of Captisol by us. The pricing of Captisol frequently reflects purchase volumes of individual customers. However, there are no minimum purchase commitments by the customer.
The majority of our revenue arrangements involve the bundling of a license with the option to purchase manufactured product. We analyze our agreements to determine whether there are multiple elements in the agreements that should be separated and accounted for individually or as a single unit of accounting. We believe that our licenses have stand-alone value at the outset of an agreement because the customer obtains the right to use Captisol in its formulations without any additional input by us, and in a hypothetical stand-alone transaction, the customer would be able to procure inventory from another manufacturer in the absence of contractual provisions for exclusive supply by us. We evaluate the pricing of manufactured product in these agreements to determine if such agreements are priced at fair value or include a significant potential incremental discount. If we determine that a significant potential incremental discount exists, we recognize a portion of the nonrefundable license fee over the lesser of the term of the agreement or the period remaining through 2012, which is the earliest date we would anticipate a possible generic competitor. We recognize nonrefundable license fees, for which we have no future development obligations and for which we provide no significant potential incremental discount on the pricing of manufactured products, upon the execution of the agreement if all other conditions for revenue recognition have been met. We recognize milestone payments, which are nonrefundable, when earned if we determine that no significant potential incremental discount exists related to the pricing of manufactured product. If we determine that a significant potential incremental discount exists these milestone payments are deferred at a percentage equal to the deferred percentage of the remaining nonrefundable license fee. We recognize license revenue in our multiple-element revenue agreements using the residual method. Under the residual method, we may allocate revenue to a significant potential incremental discount for the customer's option to purchase manufactured product, and we recognize the residual amount of license revenue after all other general conditions for revenue recognition have been met. To the extent that revenue agreements contain a research and development element that has stand-alone value, we determine fair value of the research and development element by considering competitor pricing for comparable services. We recognize research and development revenue, including any payments received in advance of performance, on a straight-line basis over the life of the contract, with the cumulative amount of revenue recognized on a contract limited to the payments received or amounts currently due and payable as of that point in time. We do not use the substantive milestone method to recognize research and development revenue due to the lack of historical documentation to support the substantive efforts involved in the achievement of milestones, as well as the relatively short time period covered by such arrangements.
We recognize material sales revenue upon transfer of title which normally passes to buyer upon shipment to the customer. We recognize sales royalties as earned. We recognize contract research revenue on a straight-line basis over the life of the contract, with the cumulative amount of revenue recognized on a contract at a point in time not to exceed the payments earned as of that point in time.
42
We defer payments, if any, received in advance of performance under an agreement and recognize these payments as revenue when earned. Other revenue consists primarily of third-party patent and development expenses we incur that are reimbursed to us from two of our customers.
Stock-Based Compensation
We provide equity incentive compensation to our employees, officers, directors and consultants by granting stock options to them under our equity incentive plans. Prior to 2006, we applied the intrinsic value method as prescribed in Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees, or APB No. 25, and related interpretations, in accounting for stock options granted under our Amended and Restated 1997 Equity Compensation Plan. Under the intrinsic value method, we did not recognize compensation cost when the exercise price of employee stock options was equal to or greater than the fair value of the underlying stock on the date of grant. For options granted between December 1997 and August 2004, we determined that the exercise price was below the fair value of the underlying common stock on the date of grant. We recorded stock compensation expense using the straight-line method over the vesting period for the difference between the exercise price and the estimated fair value of the common stock at the date of grant. Options granted between September 2004 and December 2005 had an exercise price equal to the estimated fair value of the underlying common stock and therefore no stock compensation expense was recognized.
In the absence of a public trading market for our common stock, our board of directors, with input from management, considered numerous objective and subjective factors in estimating and determining the fair value of our common stock at each option grant date, including the factors described below:
- •
- progress and milestones attained in our business;
- •
- the common stock underlying the option involved illiquid securities in a private company;
- •
- the price of our convertible preferred stock issued by us primarily to outside investors in arms-length transactions, and the rights, preferences and privileges of the convertible preferred stock relative to the common stock;
- •
- our performance and the status of our partners' research and product development efforts;
- •
- our stage of development and business strategy, including our review status with regulatory authorities; and
- •
- the likelihood of achieving a liquidity event for the shares of common stock underlying these stock options, such as an initial public offering, or IPO, merger or sale, given prevailing market conditions.
Effective January 1, 2006, we adopted Statement of Financial Accounting Standards, or SFAS, No. 123 (revised 2004),Share-Based Payment, or SFAS No. 123R. This statement supersedes SFAS No. 123,Accounting for Stock-Based Compensation, or SFAS No. 123, and APB No. 25. SFAS No. 123R requires that all stock-based compensation be recognized as an expense in the financial statements and that such cost be measured at the fair value of the award. As we previously used the minimum value method, as defined by SFAS No. 123, for purposes of measuring the fair value of stock options, we adopted SFAS No. 123R using the prospective method of application, which requires us to recognize stock compensation for all awards granted after January 1, 2006 and any existing awards modified after that date. Under this method, we did not record stock-based compensation expense for awards granted prior to, but not yet vested as of, January 1, 2006, using the fair value amounts estimated
43
under SFAS No. 123. For stock-based awards granted after January 1, 2006, we recognize compensation expense based on estimated grant date fair value using the Black-Scholes option-pricing model. We recognized stock-based compensation determined under APB No. 25 for awards granted prior to January 1, 2006, and in-the-money at the date of grant, as these awards continued to vest in 2006 and 2007. Stock-based compensation reflected in net income (loss) was $48,786, $39,797 and $202,803 during 2005, 2006 and 2007, respectively.
As of December 31, 2007, total compensation related to unvested options not yet recognized in the consolidated financial statements was $312,481 and the weighted average period over which we expect it to be recognized is approximately three years. We have not recognized, and do not expect to recognize in the near future, any tax benefit related to employee stock-based compensation costs as a result of the full valuation allowance on our net deferred tax assets and our net operating loss carryforwards.
To assist us in making valuation determinations, we retained American Appraisal Associates, Inc., an independent valuation firm, to perform valuation analyses of our common stock and options. However, American Appraisal does not audit nor does it independently verify the accuracy or completeness of financial data, projections or other information provided to it in connection with its valuation services. Our board of directors considered these analyses in determining the fair value of our common stock. We have received valuations from American Appraisal as of June 7, 2005, March 3, 2006 and September 5, 2007. Prior to 2007, we did not obtain a contemporaneous independent valuation of our common stock due to capital resources constraints and because we believed that our board was capable of determining the fair market value of our common stock. In April 2007, American Appraisal performed retrospective valuations for 2005 and 2006 using the current value method.
The current value method is deemed to be appropriate when an enterprise is at such an early stage of its development that (a) no material progress has been made on the enterprise's business plan, (b) no significant common equity value has been created in the business above the liquidation preference on the preferred shares and (c) there is no reasonable basis for estimating the amount and timing of any common equity value above the liquidation preference that might be created in the future. While we have been in operation since 1993, there have been significant changes in our business in recent years, such that we were still considered to be in the early stages of development as of June 7, 2005 and March 3, 2006.
In the September 5, 2007 valuation, American Appraisal considered two methods in valuing our common stock: the current value method and the probability weighted expected return method. We determined that the probability weighted expected return method was the more appropriate method under the circumstances. Using that method, American Appraisal arrived at a fair value of our common stock of $0.35 per share on September 5, 2007, the date that we issued options to purchase 1,712,500 shares of our common stock at an exercise price of $0.35 per share. We issued no other stock options in 2007.
In connection with preparing for our initial public offering and the preparation of our consolidated financial statements included in this prospectus, we asked American Appraisal to conduct a retrospective evaluation of the fair value of our common stock based upon assumptions revised in January 2008. In this evaluation, American Appraisal re-calculated the fair value of our common stock using the probability weighted expected return method and applying specific assumptions as directed
44
by us, including assumptions as to expected pre-money valuation, discount rate and probability of occurrence of the following events:
- •
- initial public offering (assuming the conversion of each share of preferred stock into one share of common stock) (10% probability and 38% discount rate);
- •
- initial public offering (assuming the conversion of each share of preferred stock into more than one share of common stock) (50% probability and 38% discount rate);
- •
- sale or merger (30% probability and 38% discount rate);
- •
- dissolution (5% probability and 22% discount rate); and
- •
- continuation as a private company (5% probability and 38% discount rate).
We believe this retrospective evaluation provides a more appropriate measure of the fair value of our common stock for accounting purposes, given our efforts toward a public offering of our common stock and the additional information now available to us. Based on this reassessment, we determined the fair value of a share of common stock to be $0.48 at September 5, 2007, which is the value that we used for purposes of calculating the stock-based compensation related to the stock option grants made on September 5, 2007.
Current Value Method
The current value method utilizes financial projections to determine the fair value of the total equity. To arrive at the indicated value of the common stock, American Appraisal deducted the liquidation values of the three preferred equity classes from the fair value of the total equity. Due to the multiple differences between the preferred equity classes, which have unique sets of other features and rights, such as redemption, voting rights, board representation and dividend rights, and the common stock, a discount for the lack of marketability was applied to the indicated value of the common stock to arrive at the fair value of the common stock.
Probability Weighted Expected Return Method
The probability weighted expected return method considers the probability of different future events occurring, including an initial public offering, merger or sale, dissolution and remaining a private company, to determine the fair value of the common stock. American Appraisal deducted from the expected pre-money valuations the liquidation values of the three preferred equity classes to determine the value to be allocated to the common shares.
Following this offering, we will continue to use the Black-Scholes pricing model to determine the fair value of stock options and we will estimate our volatility assumption considering peer company data until we have a sufficient share price history to calculate historical volatility. Stock-based compensation expense includes the fair value of each option to purchase shares of our common stock on the date of grant and is amortized over the vesting period of the underlying option, generally three or four years, using the straight-line method. There are significant judgments and estimates inherent in the determination of fair value. For this and other reasons, the fair value used to compute stock-based compensation expense may not be reflective of the fair market value that would result from the application of other valuation methods, including accepted valuation methods for tax purposes.
45
Intrinsic Value of Stock Options
Based on an assumed initial public offering price of $ per share, the intrinsic value of the stock options outstanding at December 31, 2007 was $ million, of which $ million related to vested options and $ million related to unvested options.
Accounting for Income Taxes
We account for income taxes in accordance with SFAS No. 109,Accounting for Income Taxes. SFAS No. 109 requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences and carryforwards are expected to be recovered or settled.
As of December 31, 2007, we had federal and state net operating loss carryforwards of $14.6 million. If not utilized, the net operating loss carryforwards will begin expiring in 2019 for federal purposes and in 2009 for Kansas state tax purposes. Additionally, we have a federal alternative minimum tax credit carryforward of $52,841, which does not expire. Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Internal Revenue Code, substantial changes in our ownership may limit the amount of net operating loss carryforwards that could be utilized annually in the future to offset taxable income. Any such annual limitation may significantly reduce the utilization of net operating losses before they expire. We have determined that we have not experienced a substantial ownership change, as defined by Section 382 of the Internal Revenue Code, during the period of January 1, 1997 through the date of this offering and we do not anticipate that this offering will result in a substantial change in ownership for purposes of Section 382 of the Internal Revenue Code. In each period since inception, we have recorded a valuation allowance for the full amount of our deferred tax asset, as the realization of the deferred tax asset cannot be currently assessed as more likely than not. As a result, we have not recorded any federal or state income tax benefit in our consolidated statement of operations.
Deferred Charge and Inventory
We outsource the production of Captisol to Hovione FarmaCiencia SA, or Hovione, through its agent Hovione LLC. Hovione is a major supplier of APIs and API intermediates. Pursuant to our production agreement that ends in 2014, we have paid Hovione LLC $1.0 million, which we have recorded as a deferred charge. We have ongoing minimum annual purchase commitments under the agreement and are required to purchase a total of $15.0 million of Captisol over the term of the agreement. Under the agreement, the amount we pay for Captisol over the second half of the term of the agreement will be proportionately reduced to allow for the recovery of this deferred charge. Our ability to realize this deferred charge is based upon purchase forecasts during the term of the agreement, and if the actual purchases are less than those forecasts, we may not be able to realize this asset. The pricing for the future inventory purchases is based upon purchasing levels and is subject to adjustment for changes in the exchange rate between the United States dollar and the Euro, which we and Hovione will share evenly, and to consumer price index adjustments.
We record inventory at the lower of cost or market using the first-in, first-out method. We analyze our inventory levels periodically and write down inventory to its net realizable value if it has become obsolete, has a cost basis in excess of its expected net realizable value or is in excess of expected requirements.
46
Results of Operations
The following table sets forth our financial results, as a percentage of revenue, for the years ended December 31, 2005, 2006 and 2007:
| | Years Ended December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||||
| Revenue | 100.0 | % | 100.0 | % | 100.0 | % | ||
Cost of sales | 29.1 | 21.4 | 19.7 | |||||
| Research and development | 50.1 | 38.3 | 71.2 | |||||
| Selling, general and administrative | 28.2 | 23.2 | 34.0 | |||||
| Total operating expenses | 107.4 | 82.9 | 124.9 | |||||
| Operating income (loss) | (7.4 | ) | 17.1 | (24.9 | ) | |||
| Other income (expense): | ||||||||
| Interest income | 2.5 | 3.7 | 4.6 | |||||
| Other, net | (0.1 | ) | — | — | ||||
| Income (loss) before income taxes | (5.0 | ) | 20.8 | (20.3 | ) | |||
| Provision for income taxes | — | 0.5 | — | |||||
| Net income (loss) | (5.0 | )% | 20.3 | % | (20.3 | )% | ||
Comparison of Years Ended December 31, 2005, 2006 and 2007
Revenue
| | Years Ended December 31, | Increase from Prior Period | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2006 | 2007 | |||||||||||
| | (dollars in thousands) | |||||||||||||||
| Revenue | $ | 8,819 | $ | 11,580 | $ | 12,745 | $ | 2,761 | $ | 1,165 | ||||||
| Percentage change | 31.3 | % | 10.1 | % | ||||||||||||
Our revenue consists of milestone payments, license fees, material sales, royalty revenue, contract research revenue and other revenue. Milestone payments are due based on notification of development and regulatory milestone achievements. License fees are nonrefundable upfront license fees, for which we have no future development obligations, which are due upon signing of the license agreement. Material sales consist of revenue from the sale of Captisol. Customers typically enter into material supply agreements for Captisol for use in clinical trials and commercialized products. Royalties are based on product sales of approved drugs that contain Captisol. Contract research revenue consists of development services provided by us to our licensees. Other revenue consists primarily of third-party patent and development expenses incurred by us and reimbursed by two of our customers. See "Critical Accounting Policies — Revenue Recognition" above for a description of how we recognize revenue.
Milestone and license revenue decreased from $2.0 million in 2006 to $1.2 million in 2007, a 41% decrease. The decrease was due to the achievement of a significant milestone from BMS in 2006 for the new drug application, or NDA, approval of Abilify Injection IM in the United States. This was offset in part by the signing of a greater number of agreements and agreements with larger upfront fees in 2007, including an agreement with AstraZeneca for Captisol-Enabled Budesonide solution for
47
inhalation, and the receipt of a milestone payment from BMS for the registration of Abilify Injection IM in the European Union. Milestone and license revenue increased from $1.9 million in 2005 to $2.0 million in 2006, a 2% increase. In 2006, milestone and license revenue was due to the signing of license agreements with Verus Pharmaceuticals, or Verus, and Prism Pharmaceuticals, or Prism, signing six new limited clinical use agreements and the achievement of a milestone from BMS for the NDA approval of Abilify Injection IM in the United States. In 2005, milestone and license revenue was due to the signing of three new technology license agreements, signing a new limited clinical use agreement and the achievement of a milestone from BMS for the NDA submission of Abilify Injection IM in the United States.
Material sales increased from $5.7 million in 2006 to $6.8 million in 2007, a 20% increase. The increase was due to the development timeline of four of our major licensees' clinical programs, which resulted in five major material purchases totaling $3.6 million in 2007 by these major customers for use in their clinical programs. Material sales increased from $4.2 million in 2005 to $5.7 million in 2006, a 36% increase, due to the signing of new license agreements with Verus and Prism, five more limited clinical use agreements than were signed in 2005, and additional Captisol licensees continuing development of their Captisol-Enabled formulations, all of which required additional purchases of material.
Royalties increased from $3.0 million in 2006 to $3.8 million in 2007, a 28% increase. The increase was due to increased product sales of Pfizer's Vfend IV drug in Japan and Europe and the launch of Pfizer's animal health product, Cerenia. Royalties increased from $2.5 million in 2005 to $3.0 million in 2006, a 19% increase. The increase from 2005 to 2006 was due to the first full year of product sales in Japan and increasing sales in the European market for Pfizer's Vfend IV drug.
Contract research revenue decreased from $845,000 in 2006 to $683,000 in 2007, a 19% decrease. The decrease is due to the timing of licensees' requests for and completion of formulation development services. Contract research revenue increased from $158,000 in 2005 to $845,000 in 2006, a 435% increase. The increase from 2005 to 2006 was due to the agreement with Verus, which provided for a recurring annual minimum amount for contract research, until such time as the initial NDA for the first licensed product is accepted, to perform development services, as well as the timing of other customer requests and completion of development services.
Other revenue increased from $93,000 in 2006 to $242,000 in 2007, a 160% increase. The increase is due to increased reimbursable third-party development expenses of $136,000 and increased reimbursable patent prosecution and maintenance expenses of $10,000. Other revenue increased from $39,000 in 2005 to $93,000 in 2006, a 138% increase. The increase is due to increased reimbursable third-party development expenses of $15,000 and increased reimbursable patent prosecution and maintenance expenses of $38,000.
48
| | Years Ended December 31, | Increase (Decrease) from Prior Period | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2006 | 2007 | |||||||||||
| | (dollars in thousands) | |||||||||||||||
| Cost of sales | $ | 2,566 | $ | 2,482 | $ | 2,508 | $ | (84 | ) | $ | 26 | |||||
| Percentage change | (3.3 | )% | 1.0 | % | ||||||||||||
Cost of sales consists of inventory and operational costs related to inventory management, quality assurance, quality control and amortization of product and license rights.
Cost of sales increased from 2006 to 2007 due to the corresponding increase in revenue from material sales. Cost of sales as a percentage of revenue decreased from 21% in 2006 to 20% in 2007 due to a larger percentage of our material sales consisting of clinical materials sales, which carry higher margins.
Cost of sales decreased from 2005 to 2006 due to the lower cost of acquiring material as a result of a change in supplier. Cost of sales as a percentage of revenue decreased from 29% in 2005 to 21% in 2006 due to the lower cost of acquiring material, as well as a larger percentage of our material sales consisting of clinical materials sales, which carry higher margins.
We expect that, for the next several quarters, cost of sales as a percentage of revenue will remain relatively constant with that experienced in 2007.
Research and development
Most of our operating expenses to date have been research and development expenses associated with either the development of our technology or the development of product candidates using our technology, including expenses relating to preclinical development, safety and toxicology, as well as Phase 1 and Phase 2 clinical trials. We have also incurred research and development expenses relating to production, stability, analytical and large-scale bioanalytical testing. We expense all research and development costs as they are incurred.
As described in more detail in the section entitled "Business," our Captisol-Enabled product candidates are at various stages of development. For example, we anticipate submitting an NDA for Captisol- Enabled Fosphenytoin IV and IM in late 2008, and are preparing for clinical trials for our Captisol-Enabled Melphalan IV and Captisol-Enabled Topiramate IV product candidates.
We are dependent on the FDA and its determinations with respect to each of our product candidates. In addition, clinical trials vary widely in cost, success rate and timing. As a result, we are unable to estimate the total costs we will incur and the timing of these costs with respect to any particular product candidate. In addition, we are unable to determine, with any precision, the anticipated completion dates for our research and development programs or the period in which material cash flows will commence.
49
The table below sets forth our research and development expense for the years ended December 31, 2007.
| | Years Ended December 31, | Increase from Prior Period | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2006 | 2007 | |||||||||||
| | (dollars in thousands) | |||||||||||||||
| Research and development | $ | 4,419 | $ | 4,435 | $ | 9,078 | $ | 16 | $ | 4,643 | ||||||
| Percentage change | 0.4 | % | 104.7 | % | ||||||||||||
The expenses we incur for research and development consist primarily of salaries and related employee benefits, costs associated with preparation for potential commercial manufacturing, validation of analytical and quality assurance methods, stability testing, clinical trial expenses managed by our contract research organizations, or CROs, and costs associated with regulatory activities. Our most significant costs are clinical trial and manufacturing scale-up and validation expenses. Clinical trial expenses include payments to vendors such as CROs and investigators, and related expenses. Clinical supplies are generally prepared by our contract manufacturers once validated methods and a process are developed. We charge all research and development expenses to operations as incurred because the underlying development efforts and technology associated with these expenses relate to our research and development efforts and have no alternative future use.
Research and development expenses increased from 2006 to 2007 primarily due to increased payments to our CROs of $3.7 million, increased intellectual property activities of $419,000 and increased wage expense of $120,000 related to the hiring of additional personnel, all to support the development of our Captisol-Enabled formulations. These increases were partially offset by a decrease in payments related to regulatory activities of $120,000, due to the status of our programs.
Research and development expenses remained relatively flat from 2005 to 2006 due to completion of a number of obligated support services in 2005 for two of our licensees resulting in payments totaling $954,000, offset by additional product concept technology and evaluation of new technology activities of $485,000, additional regulatory activities to support the development of our Captisol-Enabled formulations of $212,000, an increase in payments to our CROs of $169,000 and additional intellectual property activities of $88,000.
We expect research and development expenses to increase as a result of our expected continued clinical trial and manufacturing scale up, including increased payments to vendors such as CROs and investigators, and related expenses, as well as increased materials expenses.
Selling, general and administrative
| | Years Ended December 31, | Increase from Prior Year | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2006 | 2007 | |||||||||||
| | (dollars in thousands) | |||||||||||||||
| Selling, general and administrative | $ | 2,488 | $ | 2,681 | $ | 4,328 | $ | 193 | $ | 1,647 | ||||||
| Percentage change | 7.8 | % | 61.4 | % | ||||||||||||
Our selling expenses consist of market research costs and a portion of business development and administrative costs. Our general and administrative expenses consist of salaries, benefits and professional fees related to our administrative finance, human resources, legal, business development,
50
insurance and facility costs, and internal support systems. Legal costs are a large percentage of these costs as we enter into many confidentiality agreements, limited clinical use agreements, license agreements, license and supply agreements, supply agreements, collaborative agreements, development agreements and strategic alliances.
Selling, general and administrative expenses increased from 2006 to 2007 primarily due to increased legal costs of $233,000 to support the increased number of our licensing related agreements and general corporate matters, additional market research spending of $175,000, increased stock option expense of $150,000, an increase in accounting fees of $963,000, increased wage expense of $113,000 related to the hiring of additional personnel and competitive intelligence spending of $66,000 to support our Captisol-Enabled formulations. These increases were partially offset by a decrease in payments for trade show and exhibitor sponsorships of $34,000.
Selling, general and administrative expenses increased from 2005 to 2006 due to an increase in wage and benefit expense of $354,000, primarily related to the hiring of additional personnel, increased legal costs of $115,000 related mostly to support the increased number of our Captisol outlicensing agreements and an increase in other professional fees of $51,000 primarily related to increased accounting and valuation services fees. These increases were partially offset by the decrease in payments for severance expense of $273,000 and a decrease of $26,000 related to marketing research fees related to our acute care hospital product candidates.
We anticipate increasing selling expenses once we complete the submission of our NDA for Captisol-Enabled Fosphenytoin IV and IM. We also contemplate the addition of key sales force management in the future. We have not made a decision as to whether we will use a contract sales force or hire our own sales force. Sales force expenses will increase as we submit NDAs for our lead product candidates. We expect to incur increased salary and administrative expenses as we add personnel, comply with the reporting obligations applicable to public companies, and continue to build our support structures that will facilitate our continued development and preparation for commercialization of our acute care hospital product candidates.
Interest income
| | Years Ended December 31, | Increase from Prior Year | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2006 | 2007 | |||||||||||
| | (dollars in thousands) | |||||||||||||||
| Interest income | $ | 223 | $ | 428 | $ | 582 | $ | 205 | $ | 154 | ||||||
| Percentage change | 91.9 | % | 36.0 | % | ||||||||||||
Interest income consists of interest earned on our cash, cash equivalents and short-term investments. Interest income increased from 2006 to 2007 due to higher interest rates as well as a higher average cash, cash equivalents and short-term investments balance. Interest income increased from 2005 to 2006 due to higher interest rates as well as a higher average cash, cash equivalents and short-term investments balance.
Provision for income taxes
Income tax expense was $0, $52,841 and $0 in the years ended December 31, 2005, 2006 and 2007, respectively. The entire amount in 2006 was federal income tax expense due to Alternative Minimum Tax requirements.
51
Liquidity and Capital Resources
Since inception, we have financed our operations primarily through revenue and the private placement of equity securities. Through December 31, 2007, we have received net proceeds of $31.0 million from sales of shares of our preferred and common stock.
As of December 31, 2007, we had $8.1 million in cash, cash equivalents and short-term investments, compared to $11.0 million at December 31, 2006. Our cash, cash equivalents and short-term investment balances are held in a variety of interest bearing instruments, including certificates of deposit, corporate bonds and auction-rate preferred securities, or ARPS. Cash in excess of immediate requirements is invested with a view toward liquidity and capital preservation.
In April 2006, we entered into an agreement and related promissory note with First National Bank of Kansas for a $3.0 million revolving line of credit. In April 2008, we entered into a new promissory note. The interest rate is variable and is initially set at 5.25% per annum subject to periodic adjustments based on the First National Bank of Kansas prime rate. This line of credit is secured by a pledge of all inventory, chattel paper, accounts receivable, equipment and general intangibles except intellectual property rights. Although we have made no borrowings against the line of credit, if we were to make borrowings we would be required to repay all outstanding principal and all accrued unpaid interest on April 29, 2009. The credit available under this revolving line of credit as of April 30, 2008 was $3.0 million.
Our operating activities provided (used) net cash in the amount of $(2.0) million in 2007, $3.7 million in 2006 and $(298,000) in 2005. In 2007, cash used in operating activities was primarily related to funding research and development activities and other professional-related expenses. We progressed from cash used in operating activities in 2005 to cash provided by operating activities in 2006 primarily as a result of the growth of our business and our efforts to contain our cash expenses.
Our investing activities provided (used) net cash in the amount of $(83,000) in 2007, $(1.7) million in 2006 and $172,000 in 2005.
Our financing activities provided (used) net cash in the amount of $(0.7) million in 2007, $104,000 in 2006 and $52,000 in 2005. In 2007, cash used in financing activities was primarily related to costs associated with our initial public offering. In 2006 and 2005, cash provided by financing activities consisted solely of proceeds from the exercise of common stock options.
We hold certain ARPS, which are marketable securities with long-term stated maturities for which the interest rates are reset through a Dutch auction every 7 to 28 days. The auctions historically have provided a liquid market for these securities as investors historically could readily sell their investments at auction. With the liquidity issues experienced in the global credit and capital markets, beginning in February 2008 many of these auctions have not had sufficient bidders to allow investors to complete a sale of ARPS as the amount of securities submitted for sale has exceeded the amount of purchase orders. As a result, ARPS held by us have experienced multiple failed auctions. In the case of a failed auction, the dividends continue to be paid at the applicable "failure" rate for each security until an auction can establish a market clearing rate. Generally, for most of the securities we own, the specified "failure" rate ranges from the then-current applicable LIBOR rate to 150% of such rate. All of the ARPS held by us continue to carry AAA ratings and have not experienced any payment defaults. Until the auction process is reinstated or these securities are redeemed by the issuers, we will not be able to liquidate the ARPS we hold. Issuers of many of the closed-end funds who have issued ARPS have made public announcements of their intent to work toward redeeming the securities. At March 31, 2008, we estimate the fair value of our investments in ARPS to be $5.2 million. This estimate of fair
52
value is based on a discounted cash flow model that uses significant unobservable inputs. We have recorded an other-than-temporary impairment charge of $410,000 as of March 31, 2008 to write these investments down to their estimated fair value. We also reclassified the investments which we may not be able to liquidate in the near term to long-term investments as of March 31, 2008, as the investments are not generally available to support our current operations.
We have prepared a near-term liquidity forecast assuming the long-term portion of these investments are not converted to cash over the next 12 months. Based on our present forecast of operating cash flows together with amounts available on our credit line, currently $3.0 million, we expect to continue to be able to meet our obligations in the normal course and do not expect to reduce our planned research and development efforts. However, this cash flow forecast anticipates certain milestone payments on contracts which are expected but not assured. If we are unable to achieve these milestones or if revenue does not meet our expectations, we may need to reduce our research and development efforts or reduce our administrative costs until we are able to liquidate our ARPS.
We believe that the net proceeds from this offering and interest earned thereon, net of cash flows used in operations, and our current cash, cash equivalents and short-term investments, will be sufficient to satisfy our anticipated cash needs for working capital and capital expenditures through at least the next 24 months. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available financial resources sooner than we currently expect. Our forecast of the period of time that our financial resources will be adequate to support operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed in "Risk Factors." In light of the numerous risks and uncertainties associated with the development and commercialization of our product candidates and the extent to which we enter into collaborations with third parties to participate in their development and commercialization, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated activities. Our future funding requirements will depend on many factors, including:
- •
- the scope, rate of progress, results and costs of our preclinical testing, clinical trials and other research and development activities;
- •
- the terms and timing of any distribution, collaborative or licensing agreements that we may establish;
- •
- the cost, timing and outcomes of regulatory approvals;
- •
- the number and characteristics of product candidates that we pursue;
- •
- the cost and timing of establishing manufacturing, marketing and sales capabilities;
- •
- the cost of establishing clinical and commercial supplies of our product candidates;
- •
- the cost of preparing, filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and
- •
- the extent to which we acquire or invest in businesses, products or technologies, although we currently have no commitments or agreements relating to any of these types of transactions.
53
We may need to raise additional funds to support our operations, and such funding may not be available to us on acceptable terms, or at all. If we are unable to raise additional funds when needed, we may not be able to continue development of our product candidates or we could be required to delay, scale back or eliminate some or all of our development programs and other operations. We may seek to raise additional funds through public or private financing, strategic partnerships or other arrangements. Any additional equity financing would be dilutive to stockholders and debt financing, if available, may involve restrictive covenants. If we raise funds through collaborative or licensing arrangements, we may be required to relinquish, on terms that are not favorable to us, rights to some of our intellectual property or product candidates that we would otherwise seek to develop or commercialize ourselves. Our failure to raise capital when needed may harm our business and operating results.
Contractual Obligations and Contingent Liabilities and Commitments
We lease office space and some equipment under non-cancelable operating lease agreements. In addition, we have an annual minimum purchase obligation to Hovione for the purchase of Captisol. The following are contractual commitments at December 31, 2007 associated with lease obligations and contractual commitments (in thousands):
| | Payments due by period | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Total | Less than 1 Year | 1-3 Years | 4-5 Years | Over 5 Years | |||||||||||
| Operating lease obligations | $ | 243 | $ | 224 | $ | 19 | $ | — | $ | — | ||||||
| Contractual commitments obligations | 9,385 | 1,620 | 4,860 | 2,905 | — | |||||||||||
| Total | $ | 9,628 | $ | 1,844 | $ | 4,879 | $ | 2,905 | $ | — | ||||||
As of December 31, 2007, we had no debt obligations or capital lease obligations. In January 2008, we entered into an amendment with Hovione extending the term of the agreement to 2014. Since December 31, 2007, our operating lease and contractual commitment obligations have not otherwise changed materially.
Recent Developments
During the first quarter of 2008, we received a royalty payment from a customer for previously unreported royalties. The amount of royalties received in excess of amounts previously reported was $709,000. This particular customer self reports royalties due to us. We recorded this amount as revenue in the first quarter resulting from a change in estimate.
Off-balance Sheet Transactions
We are not party to any off-balance sheet transactions.
Recent Accounting Pronouncements
In September 2006, the SEC issued Staff Accounting Bulletin No. 108,Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements, or SAB No. 108, which addresses how uncorrected errors in previous years should be considered when quantifying errors in current year financial statements. The SEC staff believes registrants must quantify errors using both a balance sheet and a statement of operations approach and evaluate whether either approach results in quantifying a misstatement that, when all relevant quantitative and qualitative
54
factors are considered, is material. We adopted SAB No. 108 as of December 31, 2006, retroactively to January 1, 2006, with no impact on our operating results or financial position.
In June 2006, the Financial Accounting Standards Board, or FASB, issued FASB Interpretation No 48,Accounting for Uncertainty in Income Taxes, or FIN 48. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an entity's financial statements and prescribes a threshold of more-likely-than-not for recognition of tax benefits of specified tax positions taken or expected to be taken in a tax return. FIN 48 also provides related guidance on measurement, derecognition, classification, interest and penalties, and disclosure. On January 1, 2007, we adopted FIN 48, which did not have any impact on our consolidated financial statements.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements, or SFAS No. 157. SFAS No. 157 defines fair value as used in numerous accounting pronouncements, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosure related to the use of fair value measures in financial statements. SFAS No. 157 does not expand the use of fair value measures in financial statements. SFAS No. 157 emphasizes that fair value is a market-based measurement and not an entity-specific measurement, based on an exchange transaction in which the entity sells an asset or transfers a liability. SFAS No. 157 establishes a fair value hierarchy from observable market data at the highest level, to fair value based on an entity's own fair value assumptions, at the lowest level. SFAS No. 157 became effective for us as of January 1, 2008. Per FASB Staff Position 157-2,Effective Date of FASB Statement No. 157, implementation of SFAS No. 157 to certain nonfinancial assets and nonfinancial liabilities will be deferred until annual reporting periods beginning after November 15, 2008 and interim periods within those annual reporting periods. We are currently evaluating the impact, if any, that the adoption of SFAS No. 157 will have on our financial position and results of operations.
In February 2007, the FASB issued SFAS No. 159,The Fair Value Option for Financial Assets and Financial Liabilities, or SFAS No. 159. SFAS No. 159 permits, but does not require, entities to measure specified financial instruments and other assets and liabilities at fair value on an instrument-by-instrument basis. Unrealized gains and losses on items for which the fair value option has been elected should be recognized in earnings at each subsequent reporting date. SFAS No. 159 became effective for us as of January 1, 2008 and cannot be adopted early unless SFAS No. 157 is also adopted. We are currently evaluating the impact, if any, the adoption of SFAS No. 159 may have on our financial position and results of operations.
In June 2007, the FASB issued Emerging Issues Task Force, or EITF, Issue No. 07-3,Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities, or EITF Issue No. 07-3, which applies to companies involved in research and development activities that make nonrefundable advance payments for goods that will be used or for services that will be performed on future research and development activities. EITF Issue No. 07-3 requires that nonrefundable advance payments for future research and development activities be deferred and recognized as an expense as goods are delivered or the related services are performed. EITF Issue No. 07-3 is effective for financial statements issued for fiscal years beginning after December 15, 2007 and interim periods within those fiscal years. We do not expect the adoption of EITF Issue No. 07-3 to have a material impact on our financial position and results of operations.
In December 2007, the FASB issued EITF Issue No. 07-1,Accounting for Collaborative Arrangements, or EITF Issue No. 07-1, which applies to collaborative arrangements that are conducted by the participants without the creation of a separate legal entity for the arrangements and clarifies, among other things, how to determine whether a collaborative agreement is within the scope of this issue. EITF Issue No. 07-1 is effective for financial statements issued for fiscal years beginning after
55
December 15, 2008. We do not expect the adoption of EITF Issue No. 07-1 to have a material impact on our financial position and results of operations.
Quantitative and Qualitative Disclosures About Market Risk
Interest rate risk represents the risk of changes in value of a financial instrument caused by fluctuations in interest rates. We did not have any indebtedness for borrowed money as of December 31, 2007. The annual interest rate on our credit facility is currently 5.25% per annum, subject to periodic adjustment. If and when we do enter into future borrowing arrangements or borrow under our existing revolving credit facility, we may seek to manage exposure to interest rate changes by using a mix of debt maturities and variable- and fixed-rate debt, together with interest rate swaps where appropriate, to fix or lower our borrowing costs.
Our exposure to market risks for changes in interest rates relates primarily to our investment portfolio. As of December 31, 2007, our cash equivalents and short-term investments consisted primarily of money market funds, ARPS and certificates of deposit. Due to the short-term nature of our investment portfolio, we do not believe that an immediate 10% change in interest rates would have a material effect on the fair market value of our portfolio. We do not expect our operating results or cash flows to be materially affected to any significant degree by a sudden change in market interest rates on our investment portfolio. We do not hold any of these instruments for trading purposes.
We do not make material sales or purchases in currencies other than in U.S. dollars. However, our contract with Hovione, our supplier of Captisol, requires that we adjust the price we pay for Captisol as a result of fluctuations in the Euro against the U.S. dollar that exceed a fixed percent. As a result, changes in the exchange rate between the U.S. dollar and Euro expose us to foreign currency exchange risk. However, we do not believe that a 10% change in exchange rates would have a material adverse effect on our business. For example, a weakening of the dollar would increase the costs to us but would also have an offsetting effect in terms of royalties from sales in foreign countries. We have not entered into any financial instruments to hedge against this foreign currency risk exposure.
56
Overview
We are a specialty pharmaceutical company focused on the development and commercialization of drugs specifically designed to address limitations of current therapies in selected established markets. We have developed a broad portfolio of 12 product candidates utilizing our drug formulation technology using Captisol cyclodextrins. Captisol cyclodextrins are a patent protected, specifically modified family of cyclodextrins designed to improve solubility, stability, bioavailability, safety and/or dosing of a number of active pharmaceutical ingredients, or APIs. Captisol cyclodextrins are ring shaped molecules comprised of sugar molecules. A number of different APIs that are not otherwise readily soluble in water can interact with Captisol cyclodextrins, resulting in compounds that are more water soluble than the API standing alone. In order to best utilize our internal resources, experience and technology, we are focusing on the development and commercialization of product candidates for use in the acute care hospital setting. For our product candidates that will likely require more extensive development and commercialization efforts, we partner with established pharmaceutical or specialty pharmaceutical companies. All of our product candidates and our partner product candidates are currently in development and have not yet been commercialized. We also outlicense our Captisol technology to third parties for use in the development of their own products.
We generate revenue through a number of activities, including outlicensing our drug formulation technology to established pharmaceutical companies. Our technology has been validated by the launch of four Captisol-Enabled products commercialized by Pfizer and Bristol-Myers Squibb, or BMS. Our outlicensing effort has allowed us to advance our Captisol technology, generate cash flow for our company and expand our strategy to capitalize on the growth opportunities and business lines enabled by Captisol. We intend to leverage the formulation, development, regulatory and intellectual property expertise garnered from our experience to facilitate the development and commercialization of our own product candidates for use in the acute care hospital setting.
We develop product candidates using APIs that are in Food and Drug Administration, or FDA, approved and commercially available drug products, for which we can potentially improve delivery efficiency. By applying our Captisol technology to APIs with known safety and efficacy characteristics, we believe that we can create differentiated products with significant commercial opportunities, while simultaneously reducing development risk, costs and timelines as compared to the development of new chemical entities. Specifically, we believe we will be able to leverage the known safety and efficacy characteristics of these APIs to rapidly develop and commercialize product candidates on a Section 505(b)(2) regulatory pathway, under which the FDA may approve new drug applications, in part, on the basis of the FDA's prior findings of safety and efficacy for other drug products. This means that, upon a showing of bioequivalence to an approved drug product, one or more of our new drug applications could be approved on the basis of literature or FDA findings of safety and efficacy related to the currently marketed APIs used in our formulations. While the FDA has discretion to request additional data to support approval, we believe that in some cases this path to registration should only necessitate the completion of a single or a small number of clinical trials demonstrating bioequivalence to the existing, previously approved formulation.
In order for us to initiate a clinical development program, a drug compound must have a strong technical fit with our Captisol technology and also address an important unmet medical need in a significant patient population. We believe that the solubility, stability, bioavailability, safety and/or dose sparing properties of our Captisol technology will enable us to move a compound from initial screening through submission of a new drug application, or NDA, in 18 to 36 months. If our clinical trials are successful, our goal is to submit one or more NDAs per year beginning in 2008 for the foreseeable future for our retained product candidates, as our resources permit. Simultaneously, we will
57
continue to outlicense or enter into collaborations for non-core product candidates and outlicense our Captisol technology. We have established and intend to continue to broaden our intellectual property estate to protect both Captisol and our Captisol-Enabled product candidates.
Our three business lines are:
- •
- Retained Product Candidates. We are developing six product candidates for use in the acute care hospital setting, and we intend to build a focused U.S.-based, specialty sales force to market these product candidates. The following table presents information describing our three leading retained product candidates:
| | Captisol-Enabled Product Candidate | Brand Name of Approved Third- Party Product Containing Chemically Equivalent API | Target Indication | Stage of Development | |||
|---|---|---|---|---|---|---|---|
| Captisol-Enabled Fosphenytoin IV and IM | Cerebyx | Status Epilepticus | NDA Submission Anticipated in Late 2008 | ||||
| Captisol-Enabled Melphalan IV | Alkeran | Multiple Myeloma | Preclinical | ||||
| Captisol-Enabled Topiramate IV | Topamax | Epilepsy | Pre-IND Meeting Held in May 2008 |
We have an additional three retained product candidates in preclinical development. We have also received an issued patent for one of our retained product candidates.
- •
- Partner Product Candidates. We are developing six product candidates that we have either collaborated with, outlicensed or intend to outlicense to partners for further development and commercialization. These product candidates typically require significant financial resources or extensive development to commercialize. The following table presents information describing our four leading partner product candidates:
| | Captisol-Enabled Product Candidate | Brand Name of Approved Third- Party Product Containing Chemically Equivalent API | Target Indication | Stage of Development | Partner | ||||
|---|---|---|---|---|---|---|---|---|---|
| Captisol-Enabled Amiodarone IV | Cordarone | Arrhythmia | NDA Submitted in 2008 | Prism | |||||
| Captisol-Enabled Clopidogrel IV | Plavix | Atherothrombosis | NDA Submission Anticipated in 2010 | Prism | |||||
| Captisol-Enabled Budesonide solution for inhalation | Pulmicort Respules | Asthma | Phase 2 Clinical Trials Completed in the U.K. | AstraZeneca | |||||
| Captisol-Enabled Budesonide nasal spray | Rhinocort | Nasal Allergy | Phase 2 Clinical Trial Completed in Canada | Under discussion |
- Our other two partner product candidates are scheduled to begin Phase 2 proof-of-concept clinical trials in 2008.
- •
- Captisol Outlicensing. We outlicense our Captisol technology to pharmaceutical companies for implementation in their own product lines. This strategy was the basis for our agreements with Pfizer and BMS and has resulted in four marketed products to date. In
58
addition, we have licensed Captisol technology to 18 pharmaceutical, specialty pharmaceutical and biotech companies to conduct early human clinical testing.
Market Opportunity
Acute medical conditions are characterized by a rapid onset of symptoms that are temporary and severe, and that occur at irregular intervals. Most of the approved drugs for the treatment of acute conditions are typically delivered either in tablets, in an inhaled format or by injection. We believe that convenience, cost effectiveness, rapid onset of action, consistent bioavailability and dose delivery, and dose purity are the keys for product acceptance in the hospital.
Though oral administration is often the most convenient and painless way to deliver drugs for the patient, oral drugs are not optimal to deliver high concentrations of the drug to the target site in a rapid fashion due to difficulty of administration, poor absorption qualities or metabolism by the liver prior to reaching the target site. A traditional method of improving onset of activity and bioavailability is to change the route of administration from oral to intravenous or inhaled. Intravenous, or IV, injections are currently the most common form of administration of drugs used in the hospital for acute conditions as they provide a rapid onset of action for a wide range of indications and can be used to provide rapid changes in effect. However, not all APIs are amenable to IV formulation due to issues such as water insolubility. In cases where an API can be formulated as an injectable product, efficacy can be limited by toxicity or instability.
Many APIs, though shown to be biologically active in cellular assays, do not possess the chemical properties that make them amenable to administration in humans. One of the initial challenges in formulating drugs so that they are effective in humans is water solubility. A drug needs to be water soluble to be readily delivered to a cellular membrane, but needs to be relatively water insoluble, or hydrophobic, to cross the membrane. According to Drug Discovery World Summer 2005, almost half of new molecular entities identified by pharmaceutical industry screening programs have failed to be developed because of poor water solubility. Formulation systems used to make drugs more soluble include the following:
- •
- Cyclodextrins. Cyclodextrins are circular carbohydrates that are generally used to formulate oral pharmaceuticals. There are six cyclodextrins currently available for use in pharmaceutical products. However, some of these have demonstrated limited aqueous solubility and/or have shown toxicity when given by injection.
- •
- pH modifications. The use of low and high pH buffers to solubilize amines and acids, respectively, is a traditional method utilized by many medicinal chemists. pH modifications are also used to optimize stability. However, the injection of these formulations into the body can cause significant pain and injection site irritation.
- •
- Emulsification. Combinations of organic chemicals can be used to emulsify many hydrophobic APIs. However, these products are often associated with increased injection site irritation, pain and toxicity.
- •
- Solvent systems. Specific combinations of solvents, such as glycols and alcohol, can also be used to effectively solubilize some hydrophobic APIs. However, in some cases, solvent systems require mixing solvents and the API shortly before administration. These products are often associated with increased injection site irritation and toxicity and may require a complicated administration protocol.
59
- •
- Liposomes. The use of artificial vesicles known as liposomes to deliver microscopic amounts of drugs to cells has been known for several decades and in recent years has been applied to a number of products. However, liposome technology is often limited by the high cost of production, low particle stability and toxicity.
- •
- Micronization. Micronization of APIs is another technique utilized to address formulation issues by reducing the size of hydrophobic molecules for oral drug delivery. However, micronization is generally insufficient for injectables. In order to overcome this limitation, some organizations have gone a step further in the reduction of particle size, utilizing nanoparticle technology. However, nanoparticle technology is often limited by technological challenges, high cost and low particle stability.
As a result of the limitations of oral tablets and current formulations of marketed inhaled and IV therapeutics, we believe there is a significant unmet medical and patient need for products for the treatment of acute conditions that can provide safe and rapid therapeutic onset, can be efficiently solubilized and can be easily stored and made ready for use.
Our Solution: Captisol-Enabled Products
We have developed our Captisol technology platform as a potential solution to the problems of solubility, stability, bioavailability and safety faced by many APIs. Captisol is a patent protected family of derivatives of natural cyclodextrins. Natural cyclodextrins are known to improve the aqueous solubility of hydrophobic drugs. Captisol is the trade name for our modifiedb-cyclodextrin known as SBE7-b-CD. We selected this derivative based on its improved safety profile and drug carrier properties relative to other cyclodextrins. Captisol can provide up to a 40-fold improvement in water solubility over the naturalb-cyclodextrin. Captisol has an established safety record as demonstrated by its use in four FDA-approved products to date. Captisol is currently being used in or is planned for use in an additional 27 product candidates in clinical trials.
The following diagram illustrates how Captisol facilitates the administration of hydrophobic APIs:
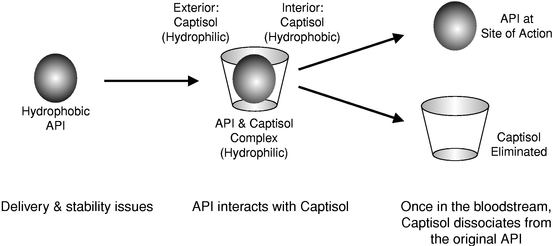
Captisol is a ring structure that surrounds or otherwise interacts with the water-insoluble, or hydrophobic, API. This structure is hydrophilic, or water soluble, on the exterior and hydrophobic on the interior, thereby increasing solubility of the API during preparation, storage and administration. Following administration, the API separates from the Captisol molecule in the bloodstream and is delivered to the site of action. The Captisol molecule, which is inert, is then eliminated from the body.
60
We believe that our Captisol technology has the potential to enable improved formulation, delivery characteristics and overall product profiles as compared to other methods of solubilization and formulation:
- •
- Improved solubility. It is generally difficult to formulate water-insoluble APIs in all dosage forms, and in particular, as injectables or inhalation products. This presents particular challenges when attempting to deliver high levels of API to reach specific dosing targets. We believe that Captisol may provide an innovative solution to these problems by potentially enabling an optimized formulation of many different otherwise insoluble APIs.
- •
- Rapid onset. Captisol has been shown to enable the aqueous formulation of many water insoluble APIs as injectable or inhaled medications. We believe that our Captisol technology has the potential to be used to enable faster acting versions of many currently approved oral products.
- •
- Improved bioavailability and delivery efficiency. Captisol may provide better bioavailability than methods of solubilization utilizing nanoparticles and solvent systems. Additionally, in some cases Captisol may permit lower dosing of API. For example, when dosed in an air jet nebulizer, data suggests that our Captisol-Enabled Budesonide solution provides equivalent systemic levels of budesonide with a significantly lower dosage of the API compared to the reference suspension product.
- •
- Improved safety. To date, Captisol has demonstrated acceptable safety at doses planned for most of our retained product candidates and partner product candidates when administered by injection and has not exhibited the toxic effects sometimes associated with naturalb-cyclodextrin in preclinical studies. The first product utilizing Captisol was approved in 2001 and, to date, we are not aware of any significant adverse reactions attributed to Captisol use in humans, including with any of the commercialized products.
- •
- Administration convenience. Captisol may simplify a multi-step preparation process and eliminate the need for refrigeration required for some current commercial products.
- •
- Improved stability. Because the Captisol molecule surrounds or otherwise interacts with the API molecule, the API may be protected from some degradation effects. As an example, stability data suggests that Captisol-Enabled Budesonide provides greater UV light protection for budesonide than the budesonide in solution as part of the currently marketed suspension formulation.
Our Captisol technology has the potential to be used across a broad spectrum of molecules including both water soluble and insoluble compounds, small molecules, peptides and protein macromolecules. We have screened over 200 APIs and approximately 40% of those screened have exhibited drug characteristics that we believe make them candidates for combination with our Captisol technology.
Our Strategy
Our goal is to become a leading integrated specialty pharmaceutical company. To accomplish this goal we intend to:
- •
- Focus our internal development efforts on low risk product opportunities targeting the acute care hospital setting. We are focusing on developing a portfolio of acute care hospital product candidates using APIs that already have extensive historical use in humans, a well-defined safety profile and known biological activity. We believe that this
61
- •
- Establish strategic partnerships for product opportunities that do not fit into our core strategy. We intend to partner with organizations to develop and commercialize our product candidates that have higher risk because they may require significant resources to conduct clinical trials, or larger sales forces to market products once approved. We believe that this strategy will enable the commercialization of our partner product candidates, without requiring us to incur the full cost of development and the financial risk of commercialization.
- •
- Utilize APIs that have known safety and efficacy characteristics to gain rapid market acceptance. Many of the APIs used in our product candidates are well known to physicians. We believe that introducing an improved version of a product with which physicians are already familiar may mitigate the delay and commercial risk often associated with the introduction of new chemical entities.
- •
- Continue to build intellectual property around our product candidates and our Captisol technology. We continue to pursue composition of matter patents for the formulation of APIs in combination with Captisol. Additionally, we are developing enhanced versions of Captisol and have filed patent applications on these versions, and are also developing improved processes for the manufacturing of Captisol.
- •
- Maintain our technology outlicensing business. We currently generate revenue from material sales, royalties on products our licensees commercialize, license fees and milestones, and fees related to our efforts to help licensees take advantage of our Captisol technology. We plan to continue to outlicense Captisol to organizations on an opportunistic basis to provide us with revenue to support our development efforts relating to our own product candidates.
strategy will enable us to bring our product candidates to market on a relatively shortened development timeline and with lower risk than new chemical entities. We intend to utilize a 505(b)(2) pathway to registration which may require only a single or a small number of clinical trials demonstrating bioequivalence to the current formulation of the API. We believe that a small specialty sales force can effectively market our products to hospitals.
Products and Product Candidates
We combine small and large molecule APIs with our Captisol technology to create product candidates. We believe that the existing APIs for which we are currently developing Captisol-Enabled formulations either will go off patent soon or are no longer eligible for patent protection, based on the Orange Book listing for such APIs. Our portfolio is comprised of product candidates that we intend to commercialize ourselves and product candidates that we have either collaborated with, outlicensed or intend to outlicense to partners. In addition, we outlicense our Captisol technology to other companies for their products.
62
The following table lists our three leading retained product candidates targeting an indication in the acute care hospital setting.
| Captisol-Enabled Product Candidate | Brand Name of Approved Third- Party Product Containing Chemically Equivalent API | Target Indication | Stage of Development | |||
|---|---|---|---|---|---|---|
| Captisol-Enabled Fosphenytoin IV and IM | Cerebyx | Status Epilepticus | NDA Submission Anticipated in Late 2008 | |||
Captisol-Enabled Melphalan IV | Alkeran | Multiple Myeloma | Preclinical | |||
Captisol-Enabled Topiramate IV | Topamax | Epilepsy | Pre-IND Meeting Held in May 2008 |
Captisol-Enabled Fosphenytoin IV and IM
Status epilepticus is a life threatening condition in which the brain is in a state of persistent seizure. According toAmerican Family Physician, August 2003, an estimated 152,000 cases occur each year in the U.S., resulting in 42,000 deaths and an inpatient cost of $3.8 billion to $7.0 billion per year. The injectable prescription market for the acute treatment of these episodes includes Cerebyx (fosphenytoin IV), marketed by Pfizer, and Ativan (lorazepam IV), marketed by Biovail. According to a published report, Cerebyx generated $71 million in hospital-based revenue in 2006.
Fosphenytoin is converted in the body to its active form, phenytoin, after administration. Phenytoin itself is insoluble in water. In the current formulation of fosphenytoin, there is a small amount of fosphenytoin that is converted to phenytoin in the vial during storage. This conversion creates a precipitate. A vial with precipitate is not acceptable for use as an injectable product due to potential safety reasons. To prevent any fosphenytoin from converting to phenytoin during storage, the current formulation of fosphenytoin has a pH of approximately 8.6 to 9.0, which is significantly more basic than human blood, which has a pH of 7.4. As a result, this injection is occasionally associated with injection site irritation. Furthermore, the vial must be refrigerated in order to prevent further precipitation. This requirement makes it difficult to utilize in emergency settings that do not have access to a refrigeration unit, such as many ambulances or helicopters. We believe that fosphenytoin's requirement for refrigeration may limit its overall usage and that the introduction of a more stable and biocompatible product into the marketplace could provide a significant commercial opportunity.
We are developing a Captisol-Enabled formulation of fosphenytoin for intramuscular, or IM, and intravenous, or IV, administration for the treatment of status epilepticus. We believe our Captisol- Enabled formulations can be stored and administered at room temperature either in vials or in a pre-filled bag. This would allow for ease of use and quick administration with limited preparation time while also potentially reducing the possibility of medical error.
Development Status
We recently completed two pivotal clinical trials comparing our Captisol-Enabled Fosphenytoin formulations directly to Cerebyx in healthy volunteers. One trial examined the bioequivalence of our IM formulation while the other examined our IV formulation. Each trial was set up as a double blind crossover study, and we enrolled 38 subjects in the IV trial and 52 subjects in the IM trial. In both trials, key pharmacokinetic parameters were analyzed and compared to Cerebyx. In addition to general safety monitoring in both trials, the FDA has required that potential renal effects be closely evaluated through the analysis of urine samples pre- and post-dosing to further evaluate the safety of Captisol in
63
these Captisol-Enabled formulations. We have completed the clinical testing portion of these trials and are awaiting the final report.
We are also developing a pre-filled bag formulation of Captisol-Enabled Fosphenytoin IV. The pre-filled bag will contain the same basic formulation as our IV fosphenytoin product, thereby increasing the convenience of our product by eliminating the need to transfer the vial solution into an IV bag for administration. We believe that bioequivalence testing may not be necessary for this dosage form because the planned bioequivalency study will use the same vial formulation diluted into a bag, resulting in the same final formulation. As such, we believe we should be able to use the data from our IV bioequivalence study in order to complete our registration package. Under this assumption, we believe we will only have to complete stability testing and Chemical Manufacturing Control, or CMC, documentation in order to file for regulatory approval for this product candidate once our initial IV formulation receives approval, if any. We submitted additional information to the FDA in early 2008 and, assuming the FDA confirms that it will not require bioequivalence testing for this dosage form, we intend to submit an NDA in the fourth quarter of 2008.
Captisol-Enabled Melphalan IV
According to the Multiple Myeloma Research Foundation, over 50,000 patients in the United States suffer from multiple myeloma with approximately 16,000 new cases being diagnosed annually. Traditionally, when first diagnosed with multiple myeloma, patients either undergo a stem cell transplant or are initiated on a treatment regimen that may include chemotherapy and radiation, corticosteroids, biologics, bisphosphonates or angiogenesis inhibitors such as thalidomide.
Melphalan is currently marketed by Celgene under the name Alkeran as either an oral or injectable palliative therapy for patients with multiple myeloma or ovarian cancer. Alkeran is a chemotherapeutic agent that competes with other chemotherapeutics in cancer treatment, such as cyclophosphamide (Cytoxan), doxorubicin (Adriamycin) and vincristine (Oncovin). According to documents filed by Celgene with the SEC, Alkeran generated approximately $50 million in revenue in 2006, 95% of which came from the sale of the injectable formulation. Much of this is attributed to the off-label use of melphalan as a conditioning regimen prior to stem cell transplants. We currently have no plans to seek approval for this regimen and therefore could not market this regimen with our Captisol-Enabled Melphalan IV product candidate. The FDA may require that our labeling, if any, discuss safety concerns with Captisol-Enabled Melphalan IV at higher doses such as the doses used off-label.
We believe that our Captisol technology has the potential to improve the delivery and stability of the current formulation of melphalan. Alkeran is currently packaged as two separate vials that must be combined prior to use. Once mixed, the formulation is stable for a period of up to two hours. As the actual administration of each dose of Alkeran is done over a minimum of 15 minutes, the prepared product must be administered immediately after mixing. We believe that the mixing process and strict timing required to prepare Alkeran for administration provides an attractive commercial opportunity for a potentially more streamlined and stable product offering.
Our Captisol-Enabled Melphalan IV product candidate is a one vial formulation of melphalan that we believe will have a longer use time after mixing with saline. If achieved, this increased stability would provide for ease of use in administration while maintaining therapeutic effect.
Development Status
We are currently finalizing the formulation of Captisol-Enabled Melphalan IV. Once completed, we intend to initiate registration batch production, submit an investigational new drug application, or IND, and prepare for a single bioequivalence study which we believe, along with our stability data, will be sufficient for regulatory submission. We anticipate that this pivotal bioequivalence trial will be a multi-center, randomized, blinded two period crossover trial comparing our Captisol-Enabled
64
Melphalan IV to intravenous administration of Alkeran. We will make an effort to measure Captisol-complexed Melphalan in the plasma as requested by the FDA in our pre-IND meeting. We believe we will not be required to complete any additional non-clinical studies. Assuming FDA and Institutional Review Board, or IRB, concurrence, we anticipate initiating the bioequivalence study by the end of 2008 and, if the study is successful, submitting our NDA in late 2009.
Captisol-Enabled Topiramate IV
Topiramate is marketed by Ortho-McNeil Neurologics under the brand name Topamax as an orally administered drug for the prevention of migraines as well as for the treatment of chronic epilepsy. According to the Epilepsy Foundation, there are approximately 2.7 million epilepsy patients in the U.S. who use a range of therapeutic options, including Depakote, Gabapentin, Lamictal and Keppra. According to the Health Care Cost and Utilization Project, in 2005 the total hospital costs for epilepsy and convulsion patients was nearly $1.8 billion.
The treatment paradigm for neurological disorders typically involves some trial and error, with physicians initiating multiple different types of therapy before identifying a preferred medication that is found to be both tolerable and efficacious for the patient. We believe that there is a preference among many prescribing physicians to use the same therapeutic agent utilized to treat patients on a chronic, prophylactic basis for acute epileptic episodes. However, the current formulation of Topamax is not optimal for use in the acute setting because it is an oral dosage form and requires up to two hours to reach peak blood levels. Additionally, many patients in the acute setting are unable to take oral medications and require IV formulated products. Acute use is not currently approved for Topamax and may require one or more additional clinical trials for approval.
We are developing an IV formulation of topiramate in conjunction with the University of Minnesota to be used for the treatment of epilepsy. We believe our Captisol-Enabled Topiramate IV will provide for significantly faster onset of drug compared to the oral formulation while providing administration to a wider audience, including those currently on Topamax for chronic treatment of epilepsy.
Development Status
We held a pre-IND meeting with the FDA to discuss the development of Captisol-Enabled Topiramate IV in May 2008. We are also assisting in the preparation of clinical grade material for use in separate pharmacokinetic studies to be initiated by the University of Minnesota. Based on our communication with the University of Minnesota, we expect an initial bioavailability study conducted by the University of Minnesota to begin in mid-2008 in order to examine Captisol-Enabled Topiramate IV in 24 patients at lower doses but at the same rate of administration we are targeting. Data from this study will be used to finalize our IV formulation and dosing strategy for our pivotal studies. Based on our pre-IND meeting, we may need to conduct additional preclinical studies to demonstrate the safety of rapid administration of high doses of intravenous Captisol prior to initiating clinical trials at our planned dose and administration schedule for Captisol-Enabled Topiramate IV. In addition, we may be required to conduct our bioequivalence studies of Captisol-Enabled Topiramate IV in patients with epilepsy, rather than healthy normal volunteers, to monitor for potential toxic effects of Captisol on renal tubular function at very high rates of administration. Finally, we may be required to assess seizure control and various renal safety parameters as well as bioequivalence in our clinical trials.
We are evaluating the use of topiramate for additional therapeutic areas in which Topamax may have clinical benefits, including potential acute use for the treatment of epilepsy. In addition, some premature infants experience cardiovascular problems that require surgery. During this surgery, there is the possibility that they will develop hypoxia, or low levels of oxygen in the blood, which may have a negative effect on their future mental development. We believe our intravenous topiramate product candidate may also have potential utility in the treatment of neonatal seizure and hypoxia. These indications will likely require a full preclinical and clinical development effort. We believe the
65
indications qualify for Orphan Drug status, defined as a drug treating diseases affecting fewer than 200,000 people in the United States. This status could provide us with marketing exclusivity for up to seven years for the Orphan Drug designated indication and specified tax credits. We intend to request Orphan Drug designation if we pursue development for these indications.
Other Retained Product Candidates
We are developing three additional product candidates that are currently in preclinical testing. These product candidates are being developed to address central nervous system and other indications. We believe these product candidates indicate the potential for a significant difference relative to currently marketed products and for which we believe that we may be able to obtain intellectual property protection. These product candidates will be the subject of a pre-IND meeting and, if the results of the meeting are positive, will be eligible to enter full product development.
Partner Product Candidates
We are developing six partner product candidates that we have either collaborated with, outlicensed or intend to outlicense to partners for further development and commercialization. To date, we have entered into a collaboration agreement with respect to Captisol-Enabled Clopidogrel IV and have outlicensed Captisol-Enabled Amiodarone IV and Captisol-Enabled Budesonide solution for inhalation. We believe that our agreements to date validate both our technology as well as our ability to identify, develop and subsequently outlicense these opportunities. Our four leading partner product candidates are listed below:
| Captisol-Enabled Product Candidate | Brand Name of Approved Third- Party Product Containing Chemically Equivalent API | Target Indication | Stage of Development | Partner | ||||
|---|---|---|---|---|---|---|---|---|
| Captisol-Enabled Amiodarone IV | Cordarone | Arrhythmia | NDA Submitted in 2008 | Prism | ||||
| Captisol-Enabled Clopidogrel IV | Plavix | Atherothrombosis | NDA Submission Anticipated in 2010 | Prism | ||||
| Captisol-Enabled Budesonide solution for inhalation | Pulmicort Respules | Asthma | Phase 2 Clinical Trials Completed in the U.K. | AstraZeneca | ||||
| Captisol-Enabled Budesonide nasal spray | Rhinocort | Nasal Allergy | Phase 2 Clinical Trial Completed in Canada | Under discussion |
Captisol-Enabled Amiodarone IV
Amiodarone is currently marketed under the brand name Cordarone for the treatment of life-threatening recurrent ventricular arrhythmia when patients have not responded to other available antiarrhythmics or when alternative agents could not be tolerated. Amiodarone is currently available in an injectable formulation as well as an oral tablet. Due to the poor water- solubility of this API, the injectable formulation utilizes a solvent system for solubilization, which is associated with a potential major adverse side effect, a rapid decrease in blood pressure. We have developed Captisol-Enabled Amiodarone IV, which does not contain the solvent system found in the current formulation. We believe this will offer a potentially safer therapeutic option for patients.
We have outlicensed an injectable formulation combining amiodarone and Captisol to Prism Pharmaceuticals, Inc., or Prism. Under the terms of this agreement, Prism is responsible, under an exclusive worldwide license from us, for all development and commercialization of Captisol-Enabled Amiodarone IV, at its sole expense. We are supplying Captisol to Prism for use in accordance with the
66
terms of the license agreement under a separate supply agreement. Under the license agreement, Prism will pay us milestone payments and royalties on sales of Captisol-Enabled Amiodarone IV. Prism may terminate the license agreement with or without cause, and either party may terminate the agreement for the other party's uncured material breach, bankruptcy, or criminal conviction in connection with the business associated with the licensed products. We may terminate the license agreement if Prism fails to meet a diligence milestone. Either party may terminate the supply agreement for the other party's uncured material breach, bankruptcy or other specified events. Prism may terminate the supply agreement if a regulatory authority prohibits or significantly restricts the manufacture, sale or use of Captisol or Captisol-Enabled Amiodarone IV. Prism is conducting development of this product candidate and based on communications with Prism, we understand that Prism submitted an NDA for this product candidate in 2008.
Captisol-Enabled Clopidogrel IV
Clopidogrel, marketed as Plavix, is available as a tablet to reduce the incidence of clot formation associated with cardiovascular surgery and cardiovascular complications. Clopidogrel is highly insoluble in water, and there are currently no IV or oral liquid dosage forms of this API available. Currently, many patients receive 600 mg of oral Plavix prior to surgeries such as stent placement in order to reduce the chances of clot formation. However, with this administration, physicians must wait for a number of hours prior to initiating the procedure in order to allow the active drug to reach maximum blood levels. We and our partner plan to target hospitals where there is a pretreatment use of Plavix for some cardiovascular procedures. We believe that our injectable formulation has the potential to reach peak blood levels within 30 minutes of administration, allowing physicians to operate sooner after administration. Furthermore, we also believe that our formulation could enable administration of less clopidogrel relative to Plavix while achieving equivalent levels of bioactivity.
In June 2006, we entered into a joint product due diligence agreement with Prism, followed in October 2007 by a Development Collaboration Agreement for the development of clopidogrel formulations employing Captisol. Under the terms of this agreement, Prism will conduct clinical testing and file an NDA for Captisol-Enabled Clopidogrel IV. The agreement will terminate either one year after FDA approval of Captisol-Enabled Clopidogrel IV or by a party giving written notice of termination to the other party. Based on our communications with Prism, we believe that Prism could submit an NDA for this product candidate in 2010. If Captisol-Enabled Clopidogrel IV is not outlicensed to a third party, we intend to enter into a commercialization agreement with Prism. As of the date of this prospectus, we are currently collaborating with Prism in the early stages of clinical development of Captisol-Enabled Clopidogrel IV.
Additionally, we and Prism are in discussions with other pharmaceutical organizations for potential strategic relationships relating to this product candidate.
Captisol-Enabled Budesonide
Budesonide is a glucocorticoid steroid currently approved for the treatment of asthma, non-infectious rhinitis, including hay fever and other allergies, and inflammatory bowel disease and for treatment and prevention of abnormal lesions in the nasal passages, or nasal polyposis. There are multiple formulations of the product, including a nasal inhalation suspension (Rhinocort, AstraZeneca), an oral inhalation suspension (Pulmicort Respules, AstraZeneca), an enema (Entocort enema, AstraZeneca) and a modified-release capsule (Entocort EC, Prometheus Labs). AstraZeneca also sells a combination of budesonide and formoterol (Oxis) as a single inhaler for the treatment of asthma under the brand Symbicort. We are currently developing three formulations of budesonide in combination with Captisol: an inhalation solution for the treatment of asthma; a nasal spray for the treatment of nasal allergies; and an ophthalmic solution for the treatment of allergic conjunctivitis. The first two of these are described below.
67
Captisol-Enabled Budesonide Solution for Inhalation
We entered into a license agreement with Verus Pharmaceuticals Inc., or Verus, in 2006 for the North American rights to the Captisol-Enabled Budesonide solution for inhalation. Verus assigned the license to AstraZeneca AB, or AstraZeneca, in 2007 through its subsidiary Tika Läkemedel AB. In 2008, we entered into an option agreement with AstraZeneca pursuant to which we granted AstraZeneca an exclusive, irrevocable option to expand the territory covered by the North American license agreement and related supply agreement to include all the countries in the world. AstraZeneca currently markets budesonide under the brand name Pulmicort Respules. According to the IMS National Sales Perspectives database, U.S. sales of Pulmicort Respules were over $700 million in 2006.
Pulmicort Respules are a suspension formulation of budesonide marketed by AstraZeneca for the treatment of asthma in children and adults. Pulmicort Respules is a 500-microgram, two milliliter volume that requires five to sixteen minutes to be delivered by air jet nebulizer. We are developing a Captisol-Enabled solution of budesonide, which when tested on human volunteers preliminarily indicated it could be delivered in about one-fourth of the time compared to Pulmicort Respules. As compliance is a serious concern with asthma patients, especially pediatric patients, we believe that this improved administration profile will be a distinct commercial advantage.
We have also observed that when using a more efficient nebulizer, we can use a significantly lower dosage of budesonide to achieve the same bioavailability as Pulmicort Respules, with shorter administration times. As the chronic administration of steroids is associated with a number of adverse side effects in addition to restricted use in children, we believe that the introduction of a product with less overall steroid exposure may be well received and may provide a therapeutic option for individuals, such as some children and adults, who cannot tolerate the current formulations of budesonide.
For a description of these agreements, see "Licensing, Collaboration and Marketing Agreements—License Agreement, Supply Agreement and Option Agreement with AstraZeneca" below. Captisol-Enabled Budesonide solution for inhalation has already undergone three Phase 1 and two Phase 2 proof-of-concept clinical trials.
Captisol-Enabled Budesonide Nasal Spray
We are developing a Captisol-Enabled solution formulation of budesonide nasal spray. Rhinocort is currently marketed by AstraZeneca for the treatment of nasal allergies. We have conducted a double-blinded, crossover efficacy trial against the original product and a saline control in an environmental chamber in which 65 patients were exposed to ragweed pollen. In this trial, we observed a trend toward faster onset of activity compared to Rhinocort in total nasal symptoms and a significantly faster onset and better scores in non-nasal symptoms.
Other Captisol-Enabled Budesonide Product Candidates
We are also developing a formulation of the Captisol-Enabled Budesonide solution with a currently marketed antihistamine. We believe that this combination product could build on the clinical potential of the nasal spray solution by utilizing multiple mechanisms of action to address disease. A disease state such as chronic sinusitis, or inflammation of the nasal passages, requires a multiple mechanism of action product. We are in discussions with several potential partners to outlicense this technology.
Captisol Outlicensing Pipeline
We have outlicensed our Captisol technology to a number of organizations and we have over 30 agreements currently in place. We currently receive royalties from Pfizer and BMS on sales of Captisol-Enabled Geodon IM, Vfend IV, Abilify Injection IM (launched December 2006) and Cerenia (launched in 2007). There is no minimum royalty payment due to us from BMS.
68
Below is a list of the Captisol-Enabled marketed products.
| Licensee (Approved Third-Party Brand Name) | Indication | Royalty Term Expires | ||
|---|---|---|---|---|
| Pfizer (Geodon) | Bipolar Disorder and Schizophrenia | During 2010 (in the U.S.) Beginning 2011 (outside the U.S.) | ||
Pfizer (Vfend) | Antifungal | During 2010 (in the U.S.) Beginning 2011 (outside the U.S.) | ||
BMS (Abilify Injection IM) | Bipolar Disorder and Schizophrenia | During 2010 (in the U.S.) Beginning 2011 (outside the U.S.) | ||
Pfizer (Cerenia) | Canine Motion Sickness | During 2010 (in the U.S.) Beginning 2011 (outside the U.S.) |
In addition to the products listed in the table above, we have licensed our Captisol technology to a number of companies for a variety of uses. Our outlicensees include Critical Therapeutics, Daiichi Asubio, Proteolix, Sunesis, Taisho and TargeGen.
Using Captisol early in the development process can increase the number of candidates that can be evaluated, decrease development time and increase the likelihood of a candidate's potential success. In fact, many of our Captisol outlicensing agreements are the result of the use of Captisol in the early drug discovery process. Additionally, since adopting this approach to licensing, we have significantly increased our sales of clinical grade Captisol.
Research and Development
As of December 31, 2007, we had nine full time employees in research and development, five of whom hold Ph.D. or M.D. degrees. Our research and development employees have been responsible for all aspects of product development, including product conceptualization, formulation development, product engineering, clinical trials and regulatory compliance.
Our research group has extensive experience in preformulation, formulation, analysis, pharmacokinetics and drug delivery. In addition to preclinical research and development work, the group is also responsible for generating patent applications and patent execution, with the assistance of external patent counsel, conceiving new product ideas, and evaluating potential in-license technologies and products. The group currently develops all our technologies and intellectual property.
We have chosen to focus the majority of our development effort on injectable pharmaceuticals because we have historical expertise in this area. For example, we have an extensive Type 5 Drug Master File, or DMF, as well as a Type 4 DMF on file with the FDA, both of which are focused mainly on injectable products. The DMFs we have assembled contain basic safety and toxicity testing as well as specifications and analytical methodology, thereby permitting the FDA's access to the information and reducing the regulatory submission burden for our partners who can cross-reference our DMFs in their regulatory submissions. In addition, the FDA's experience with this information and the technology may reduce the risk of other potential issues.
During 2005, 2006 and 2007, we have spent $4.4 million, $4.4 million and $9.1 million, respectively, on research and development activities.
Sales and Marketing
In connection with the commercial launch of any retained products that receive FDA approval, we intend to build a commercial organization focused on promoting our products principally in the acute care hospital setting in the United States. We believe that we can efficiently market our products
69
through a small specialized sales force, as the number of institutions comprising the hospital marketplace is relatively limited. In addition, we believe a small number of these institutions account for a substantial portion of the prescribing activity. The concentrated nature of this market also creates the opportunity to leverage our sales force across multiple therapeutic categories in the hospital. Outside the United States, we intend to establish strategic partnerships for the commercialization of our products. Our partner product candidates will be marketed and sold by our partners. See Note 14 of our consolidated financial statements for revenue by geographic area.
Manufacturing and Supply
Hovione Agreement
We currently outsource the production of Captisol to Hovione FarmaCiencia SA, or Hovione, a major supplier of APIs and API intermediates located in Lisbon, Portugal. In December 2002, we entered into a Captisol supply agreement with Hovione, under which Hovione is our exclusive supplier of Captisol and is restricted from supplying Captisol to third parties, so long as specified conditions are met. In addition to its main manufacturing site in Loures, Portugal, Hovione will qualify a second site in Macau if our forecast requirements for Captisol exceed the capabilities of the Loures site. We have ongoing minimum purchase commitments under the agreement and are required to pay Hovione an aggregate minimum amount during the agreement term. Hovione must supply amounts exceeding our forecasts by a fixed percent. In January 2008, we entered into an amendment to the supply agreement, under which we and Hovione agreed to reduce our minimum annual purchase requirement of Captisol and to extend the term of the agreement.
We pay Hovione unit prices, in U.S. dollars, for all Captisol supplied after the commercial production date, which prices may be adjusted based on the following:
- •
- fluctuation in currency exchange rates;
- •
- change in raw material prices;
- •
- change in the Portuguese consumer price index; and
- •
- our requested changes to the Captisol manufacturing process or specifications.
In the January 2008 amendment to the supply agreement, we and Hovione agreed to clarify how the exchange rate between the dollar and the Euro would be determined and applied.
In the event of a Captisol supply interruption, we are permitted to designate and, with Hovione's assistance, qualify one or more alternate suppliers. If the supply interruption continues beyond a designated period, we may terminate the agreement. In addition, if Hovione cannot supply our requirements of Captisol due to an uncured force majeure event or if the unit price of Captisol exceeds a set figure, we may obtain Captisol from a third party. In the January 2008 amendment to the supply agreement, we and Hovione agreed to remove the obligation of Hovione to hold additional quantities of Captisol inventory on our behalf.
Unless terminated earlier, the agreement will continue until the expiration of the initial term in March 2014. The term will automatically continue after the initial term for successive two year renewal terms, unless either party gives written notice of its intention to terminate the agreement no less than two years prior to the expiration of the initial term or renewal term. In addition, either party may terminate the agreement for the uncured material breach or bankruptcy of the other party or an extended force majeure event. We may terminate the agreement for extended supply interruption, regulatory action related to Captisol or other specified events.
70
Under the agreement, there are two relationship management committees. The first committee is a technical committee that is responsible for resolving technical issues relating to qualification of facilities, change control, and regulatory compliance of the manufacture of Captisol. The second committee is a management committee that is responsible for managing the overall relationship between the parties. We have designated one employee to represent us on each of the two committees.
Manufacture of Injectable Pharmaceuticals
Injectables are traditionally more difficult to develop than other formulations of drugs because of the expense of injectable manufacturing facilities, additional regulatory scrutiny and the stringent contamination hurdles associated with the production of sterile products. For example, manufacturing of injectables is more demanding relative to other formulations as the FDA requires that injectables be produced in sterile facilities because they are injected directly into the body. Because of the stringencies around the manufacture of injectable pharmaceuticals and, subsequently, a decrease in relative competition, there is often less price attrition from branded versions to generic versions of these drugs.
Competition
The pharmaceutical industry is subject to intense competition and characterized by extensive research efforts, intensive intellectual property development and litigation, and rapid technological progress. Competition in our industry occurs on a variety of fronts, including discovery efforts, developing and bringing new products to market before others, developing new products to provide the same benefits as existing products at lower cost and developing new products to provide benefits superior to those of existing products. There are many companies, including generic manufacturers as well as established pharmaceutical companies, that have significantly greater financial and other resources than we do, as well as academic and other research institutions that are engaged in research and development efforts for the indications targeted by our product candidates. It is possible that formulations of the APIs that we are developing could be formulated effectively using more traditional formulation approaches such as solvent systems, emulsification or nanoparticles. If tested and registered, these formulations may become competitive to our approach. When any product candidate is approved for marketing, we will compete with the existing API, based on the increased effectiveness and difference in price of our Captisol-Enabled product.
Certain of our product candidates, as well as the Captisol-Enabled products currently marketed by Pfizer and BMS, compete with products that utilize competitive technologies. For example, Vfend IV, which is marketed by Pfizer and uses our Captisol technology, competes with antifungal products such as Sporanox and AmBisome. Sporanox is formulated using hydroxypropyl beta cyclodextrin technology and is marketed by Johnson & Johnson, and AmBisome is formulated using liposomal technology and is marketed by Astellas. In addition, our Captisol-Enabled Budesonide solution for inhalation may compete with a budesonide solution for inhalation developed by MAP Pharmaceuticals that uses nanoparticle technology.
Licensing, Collaboration and Marketing Agreements
Option Agreement, Exclusive License Agreement, and Nonexclusive License Agreement With Pfizer
In September 1993, we obtained from the University of Kansas, or KU, an exclusive, worldwide license, with the right to sublicense, under the original Captisol patents, as well as intellectual property covering the results generated during specified CyDex-sponsored research activities at KU. We are responsible for maintaining the DMF for Captisol. KU retained a research license to the technology for noncommercial educational and research purposes, and agreed to assign to us its then-pending license agreements with Pfizer relating to Captisol.
In August 2004, we amended the KU license agreement to replace all future payment terms under the agreement, including royalties, with a concurrent lump sum payment and issuance of stock to KU. KU
71
also granted us a right of first refusal to acquire exclusive, worldwide rights to any future improvements to Captisol, including any next generation formulations of Captisol, that are developed by KU or by third parties pursuant to research sublicenses granted by KU. KU is obligated to disclose to us in writing any such improvement, and upon receipt of such information, we may exercise our right of first refusal to obtain such an exclusive license upon terms and conditions not materially different than those described in the original KU license agreement. To date, we have not exercised our right of first refusal to acquire any such improvements. The license agreement with KU will remain in force until the expiration of all licensed patents, the last of which will expire in 2010 in the United States and between 2011 and 2013 outside the U.S., with one expiring in 2016.
In December 1993, as contemplated by our license agreement with KU, KU entered into an option agreement with Pfizer, simultaneously transferring to us all of KU's rights and obligations under that agreement. Under the option agreement, we granted Pfizer a research license under the Captisol technology, as well as options to obtain an exclusive license to manufacture, use, and sell antifungal products, and a nonexclusive license to manufacture, use and sell non-antifungal products. Pfizer exercised both options in June 1996. Pfizer has paid all required option payments and exercise fees required under the option agreement.
Under the option agreement, Pfizer is obligated to disclose to us all results and improvements it generates related to Captisol, and granted us a worldwide, royalty-free, perpetual nonexclusive license, with the right to grant sublicenses, under all improvements it makes to Captisol (and any patent claims relating to Captisol) to make, use, and sell Captisol, including as incorporated into commercial products. All of Pfizer's chemistry, pharmaceutical, and toxicology data generated under the option agreement are included in our Captisol DMF. We are also obligated to disclose our (or our licensees') improvements to Captisol to Pfizer, which Pfizer may use consistent with the other rights granted.
Pfizer reimburses us for patent prosecution and maintenance fees for all licensed patents and patent applications that Pfizer requests we file, prosecute and maintain. Pfizer has the first right, and if not exercised we have a backup right, to prosecute or prevent any infringement of the licensed rights by pharmaceutical products prescribed for the same indication as a Pfizer product. We have the first right, and Pfizer has a backup right, to prosecute or prevent all other infringement of the licensed rights.
Under the exclusive license, Pfizer is commercializing its antifungal product Vfend IV, and pays us royalties on sales in all countries in which we have Captisol patents. Regardless of Pfizer's sales, we receive a minimum quarterly royalty, unless Pfizer decides to convert the exclusive license to a nonexclusive license. Pfizer has already paid us all product development milestones under the exclusive license agreement.
Pfizer's antipsychotic product, Geodon IM, and its animal health product, Cerenia, are both covered by the nonexclusive license agreement. In 2001, we amended the nonexclusive license to provide for separate royalty payments for human and animal health products. Pfizer pays royalties on sales of licensed products in all countries in which we have Captisol patents. Pfizer has made all payments for animal health development milestones under the nonexclusive agreement.
Absent early termination, the option agreement will expire upon the expiration of all licensed patents. The nonexclusive license and exclusive licenses will expire, on a country-by-country basis, upon expiration of the last-to-expire patent in such country. Pfizer may terminate any of the agreements with or without cause, and either party may terminate any agreement for the other party's uncured material breach of that agreement. Termination of one agreement does not affect the other agreements, and the research license to Pfizer and Pfizer's license to us will survive termination of the option agreement.
72
License Agreement, Supply Agreement and Option Agreement with AstraZeneca
In January 2006, we entered into a license agreement with Verus, subsequently amended and restated in November 2006, pursuant to which we granted Verus an exclusive license under our patents claiming or covering Captisol-Enabled Budesonide and, subject to rights granted to Pfizer, Captisol, as well as know-how and trademarks related to Captisol-Enabled Budesonide, to exploit Captisol-Enabled Budesonide for use with a nebulizer in North America. In March 2006, we entered into a supply agreement with Verus to supply Captisol for Verus's use in accordance with the license agreement covering North America. Verus and AstraZeneca AB, or AstraZeneca, informed us that Verus and AstraZeneca, through its subsidiary Tika Läkemedel AB, had executed an agreement in May 2007 under which AstraZeneca acquired all of Verus's rights, title, and interest in and to our agreements with Verus. In August 2007, following the assignment of the license agreement and the supply agreement to AstraZeneca, we and AstraZeneca amended the license agreement to reflect AstraZeneca's development and commercialization of Captisol-Enabled Budesonide.
Under the amended license agreement, AstraZeneca is responsible for the development, regulatory, and commercialization activities of licensed products, including all costs. At AstraZeneca's request, but subject to a minimum requirement, we will provide requested development services to AstraZeneca in exchange for payment. AstraZeneca owns all technology developed under the agreement. AstraZeneca also has a right of first negotiation to specified other products we develop or acquire, outside of specified indications. In the future, the parties will discuss potential development of additional specified compounds. Until at least the expiration of all licensed patents, we may not compete with AstraZeneca with respect to the licensed products and other products containing compounds specified in the agreement, except that we may develop such products for particular indications. Upon our request, we and AstraZeneca will negotiate in good faith a waiver of specified restrictions, or the terms of a license under AstraZeneca's intellectual property, in either case, for us to commercialize products for limited indications.
AstraZeneca owes us upfront, annual and development milestone fees, royalties on sales of licensed products, and a portion of revenue received from its sublicensees.
Under the amended license agreement, we are responsible for prosecuting and maintaining all licensed Captisol patents and all patents outside the licensed territory that claim Captisol-Enabled Budesonide, and AstraZeneca is responsible for such patents inside the licensed territory. Each party pays all costs of the patents for which it is responsible. AstraZeneca has the sole right to institute, prosecute and control litigation with respect to a Paragraph IV certification, which is a certification that a new product will not infringe an already approved product's Orange Book listed patents or that such patents are invalid, and has the first right to enforce the licensed patents against any other infringement or misappropriation by a third party.
Under the supply agreement, we supply AstraZeneca with Captisol pursuant to forecasts and purchase orders submitted by AstraZeneca, using Hovione as our third-party manufacturer. In the event of a supply failure, AstraZeneca may obtain Captisol from a generic supplier or, if no generic supplier exists, an alternate manufacturer we select with AstraZeneca. If we cannot agree on an alternate supplier, AstraZeneca may select an alternate manufacturer of its choice. We will provide necessary technology and assistance to any alternate manufacturer.
73
The license agreement will remain in force until terminated by AstraZeneca for any reason or by either party for the other party's uncured material breach. The supply agreement will remain in force until terminated by AstraZeneca upon termination of the license agreement or by either party for the other party's uncured material breach.
In 2008, we entered into an option agreement with AstraZeneca pursuant to which we granted AstraZeneca an exclusive, irrevocable option to expand the territory and proprietary rights currently covered by the North American license agreement and related supply agreement to include all the countries in the world, as well as specified additional proprietary rights. AstraZeneca may exercise the option beginning on a negotiated date and ending no later than December 15, 2008 unless otherwise agreed to by the parties to extend such option period. If AstraZeneca exercises its option, the license agreement and related supply agreement assigned by Verus to AstraZeneca will be automatically amended and restated to expand the license granted by us to AstraZeneca to include all countries in the world. If specified conditions are satisfied, AstraZeneca will pay us a fee in connection with the exercise of the option. All other terms of the North American license agreement and related supply agreement would be substantially similar to the terms of the worldwide license agreement and related supply agreement between us and AstraZeneca. Under the worldwide license agreement, AstraZeneca will continue to owe us a recurring annual minimum amount to perform development services, until such time as the initial NDA for the first licensed product is accepted, as well as development milestone fees and royalties for sales of licensed products in the expanded territory. The option agreement will terminate upon the earlier of AstraZeneca's exercise of the option or the expiration of the unexercised option period. AstraZeneca may terminate the option at any time without cause.
Other License and Supply Agreements
We have entered into license and supply agreements with various parties interested in development and commercialization of Captisol-Enabled APIs, pursuant to which we grant exclusive licenses for the development and commercialization of the specific Captisol-Enabled API. The license agreements typically include milestone payments and royalty obligations. Under the supply agreements, we agree to supply clinical and commercial grade Captisol for use in accordance with the license granted. To date, we have entered into commercial license and supply agreements with ten different parties, in addition to the relationships with Pfizer and AstraZeneca described above.
Limited Clinical Use Agreements
We also enter into standard limited clinical use agreements with third parties, pursuant to which we grant a limited right to use Captisol in small Phase 1 human clinical trials of Captisol-Enabled forms of the third party's API. These agreements also include the supply of limited quantities of clinical grade Captisol. To date, we have entered into 18 limited clinical use agreements.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, to obtain and maintain proprietary protection for our technology and know-how, to operate without infringing on the proprietary rights of others and to prevent others from infringing on our proprietary rights. We actively seek to patent the technologies, inventions and improvements we consider important to the development of our business. In addition, we rely upon trade secrets and contractual arrangements to protect our proprietary information.
As of the date of this prospectus, we own or have exclusive rights, subject to certain third party rights, to seven issued U.S. patents and 13 pending or allowed U.S. patent applications. We also have rights, subject to certain third party rights, to 99 corresponding foreign patents (including five European
74
patents) and 111 corresponding foreign patent applications (including 12 European applications and applications in other key foreign markets such as Japan (13 applications) and Canada (11 applications)) pending, as well as six pending PCT International patent applications that have not yet entered the 30-month national stage. These issued patents and patent applications reflect our efforts to aggressively pursue patent protection in the major pharmaceutical markets.
We have rights under two patent families, which include U.S. 5,134,127 and U.S. 5,376,645 and corresponding foreign patents and patent applications, through an exclusive, irrevocable, worldwide, fully-paid license from KU. These basic composition of matter patents relate to Captisol's underlying sulfoalkyl ether cyclodextrin, or SAE-CD, platform technology and compositions containing SAE-CD complexed with a drug. Following an opposition filed against the European patent corresponding to the '645 U.S. patent, the claims that will issue in the European patent are directed to oral formulations only. The '127 and '645 U.S. patents will expire in 2010, and the corresponding foreign patents will expire between 2011 and 2013, with one expiring in 2016. To date, we have relied primarily on these two patent families for the protection of our technology. Following the expiration of these patents, we will rely on the intellectual property rights discussed below for the protection of our technology. In connection with our arrangement with KU and as a result of our relationship with Pfizer, we have also obtained a non-exclusive, irrevocable, worldwide, royalty-free license under Pfizer's U.S. 6,153,746 patent and its corresponding foreign patents or application. These patents relate to the current manufacturing process for Captisol and other SAE-CD's, and the U.S. patent will expire in 2018.
As of the date of this prospectus, we own three issued U.S. patents and their corresponding foreign patents and applications directed to combinations of SAE-CD with specific drugs or specific classes of drugs. Our first issued U.S. patent for combinations of SAE-CD with specific drugs is U.S. 6,133,248. The '248 patent relates to our Captisol-Enabled Fosphenytoin IV and IM product candidates and will expire in 2018. In addition, we have obtained an issued patent U.S. 6,869,939 relating to our Captisol-Enabled Amiodarone IV product candidate, which will expire in 2022. In addition to patents relating to our current product candidates, we have also obtained an issued patent U.S. 7,034,013 relating to Captisol-Enabled Propofol liquid formulations, which will expire in 2022. We have also filed a number of corresponding foreign patent applications relating to these U.S. patents.
As of the date of this prospectus, we have filed four U.S. patent applications that are currently pending, as well as 40 currently pending PCT International and foreign patent applications relating to our Captisol-Enabled Budesonide technology. If issued, the projected expiration date for the U.S. patents in this patent family would be 2024. We have filed two PCT International patent applications relating to our Captisol-Enabled Clopidogrel IV product candidate. Upon filing of U.S. national phase patent applications, if issued, the projected expiration date for these patents would be 2028.
In addition to the above patents and patent applications relating to our current product candidates, we have additional patent families, including U.S. 6,046,177 and U.S. 5,874,418 and corresponding foreign patents and applications, which relate to compositions and pharmaceutical formulations wherein SAE-CD is present as a physical mixture with a drug. These patents are complementary to our '127 and '645 patent families, as they relate to products in which a majority of the drug is not complexed with SAE-CD. This technology is intended mainly for use in solid dosage forms such as powders and compressed dosage forms. These U.S. patents will expire in 2017.
Given that the composition of matter patents for SAE-CD will expire in 2010 in the U.S., we have developed strategies for obtaining additional patent protection for our technology. Our strategies include developing patents relating to:
- •
- combinations of SAE-CD with specific drugs or specific classes of drugs;
75
- •
- SAE-CD compositions having specified physical properties;
- •
- the use of SAE-CD as a preservative in pharmaceutical formulations;
- •
- capsules containing an aqueous-fill composition including SAE-CD;
- •
- formulations containing two or more compositions of SAE-CD; and
- •
- manufacturing processes and techniques.
In addition to filing patent applications for our product candidates, we also enter into license arrangements from time to time as part of our intellectual property strategy. In August 2007, we entered into a license agreement with Dr. Falk Pharma GmbH, under which Dr. Falk Pharma granted us a nonexclusive, worldwide license under three patents in respect of products for specified types of indications under patents issued in the U.S., Canada and Japan. We also have an option to obtain a license to related improvements under additional patents Dr. Falk Pharma may obtain. We have paid a one-time fee under the agreement and are obligated to pay royalties on future sales, including sales by our sublicensees. There is no minimum royalty obligation under our agreement with Dr. Falk Pharma. We sublicensed our rights under this agreement to AstraZeneca in the amendment to AstraZeneca's license agreement. See "License Agreement, Supply Agreement and Option Agreement with AstraZeneca" under "Licensing, Collaboration and Marketing Agreements" above for more information regarding our agreements with AstraZeneca. Our agreement with Dr. Falk Pharma will remain in force until at least the expiration of the last-to-expire of the licensed patents, unless terminated earlier by either party. Either party may terminate the agreement for the other party's uncured material breach or bankruptcy, and Dr. Falk Pharma may also terminate the agreement if we challenge the validity of the licensed patents.
In the course of our business, we may receive notices of claims of infringement, misappropriation or misuse of other parties' proprietary rights. Some of these claims may lead to litigation. We cannot provide any assurance that we will prevail in these actions, or that other future actions alleging misappropriation or misuse by us of third-party trade secrets, patents or trademarks, or challenging the validity of patents issued to us will not be asserted or prosecuted against us, or that any claims will not adversely affect our business, financial condition and results of operations. We are aware of a third-party patent potentially relating to Captisol. Although we believe that the manufacture, use, offer for sale, sale, or importation of Captisol should not be held to infringe any valid claim of this patent, the patent could nevertheless be asserted against us. In addition, we are aware of third-party patents potentially relating to some of our current product candidates, and are further investigating the potential relevance of these patents to such product candidates. Should Captisol or any Captisol-Enabled product candidate be held to infringe a claim of a third-party patent, it could have a material adverse effect on our business.
An adverse determination in litigation or interference proceedings to which we may become a party relating to any patents issued to us or any patents owned by third parties could subject us to significant liabilities to third parties or require us to seek licenses from third parties. Furthermore, if we are found to willfully infringe third-party patents, we could, in addition to other penalties, be required to pay triple damages. Regardless of the outcome, costs associated with litigation and settlements may be substantial and could include ongoing royalties. We may be unable to obtain necessary licenses on satisfactory or commercially feasible terms, if at all.
Furthermore, some of our license agreements with partners require us to indemnify our partner for the infringement of third-party intellectual property rights. In the event that one of our partners' Captisol-
76
Enabled products is deemed to infringe a third-party intellectual property right, such as a third-party patent, we may be obligated to indemnify our partner for the alleged infringement. The cost of defending and/or resolving a claim of patent infringement could be substantial. Furthermore, if we are unable to successfully defend or settle an action, the potential damages and potential loss of royalties from the sale of our partner's product may have a material adverse effect on our business.
All employees and technical consultants working for us are required to execute confidentiality agreements in connection with their employment and consulting relationships with us. Our confidentiality agreements provide that all confidential information developed or made known to others during the course of the employment, consulting or business relationship shall be kept confidential except in specified circumstances. Agreements with employees provide that all inventions conceived by the individual while employed by us are our exclusive technology. We cannot provide any assurance that employees and consultants will abide by the confidentiality or assignment terms of these agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our technology or obtain and use information that we regard as proprietary.
Government Regulation
The testing, manufacturing, labeling, advertising, promotion, distribution, export and marketing of our product candidates are subject to extensive regulation by governmental authorities and federal, state, and local statutes and regulations in the United States and other countries.
FDA regulation of development and approval
Our product candidates consist of drug compounds combined with Captisol and are considered "new drugs" in the United States. The U.S. Food and Drug Administration, or the FDA, under the Federal Food, Drug, and Cosmetic Act, or FDCA, regulates drugs in the United States, including the research, development, testing, manufacture, quality control, storage, labeling, packaging, promotion and advertising, distribution, marketing, and export and import of drug products and their components. Our drugs must be approved by the FDA through the new drug application, or NDA, process before they may be legally marketed in the U.S. The steps required before a drug may be approved for marketing in the United States generally include:
- •
- completion of preclinical laboratory studies, animal studies, and formulation studies according to Good Laboratory Practices;
- •
- the submission to the FDA of an investigational new drug application, or IND, for human clinical testing, which must become effective before human clinical trials commence;
- •
- approval of proposed human clinical trials by Institutional Review Boards, or IRBs, governing study sites;
- •
- performance of adequate and well-controlled human clinical trials according to Good Clinical Practices to establish the safety and efficacy of the product; or the performance of a clinical bioavailability/bioequivalence study to establish bioequivalence or similarity to reference approved products;
- •
- the submission to the FDA of an NDA and the FDA's acceptance of the NDA for filing;
- •
- satisfactory completion of an FDA inspection of the manufacturing facilities at which the product is made to assess compliance with current Good Manufacturing Practices, or cGMP, and to ensure that the facilities, methods and controls are adequate to preserve the
77
- •
- FDA review, including review by an FDA advisory committee, if applicable; and
- •
- obtaining FDA approval of the NDA.
drug's identity, strength, quality, and purity. The FDA may also audit clinical trial sites that generated the data in support of the NDA and business office sites to inspect original documentation and effective Standard Operating Procedures for compliance with regulations for sponsors regarding surveillance and marketing procedures;
The testing and approval process requires substantial time, effort, and financial resources, and the receipt and timing of approval is uncertain.
Preclinical studies include laboratory evaluations of the product candidate's chemistry and formulation, as well as animal studies to assess, among other things, the potential toxicity of the product candidate. The results of the preclinical studies, together with drug product and manufacturing information, data and information on any previous human use and other analytical data, are submitted to the FDA as part of the IND, which must become effective before clinical trials may be commenced. The IND becomes effective automatically 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the trials as outlined in the IND prior to that time. In that case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed.
Clinical trials involve the administration of the product candidates to healthy volunteers or patients under the supervision of a qualified principal investigator. Further, each clinical trial protocol must be reviewed and approved by an IRB, at or servicing each institution at which the clinical trial will be conducted. The IRB will consider, among other things, whether the risks to trial subjects are minimized and are reasonable in relation to the potential benefits. The IRB also approves the informed consent form that must be explained to each subject or his or her legal representative in the process of obtaining their consent, and considers the possible liability of the institution. Each new clinical trial protocol must be presented to the IRB for approval and submitted to the FDA for review. Protocols detail, among other things, subject selection and exclusion criteria, the objectives of the study, dosing procedures, stopping rules and parameters for monitoring subject safety, and data analysis.
Clinical trials typically are conducted in three sequential phases prior to approval, but the phases may overlap. A fourth, or post-approval, phase may include additional clinical trials. These phases generally include the following:
- •
- Phase 1. Phase 1 clinical trials involve the initial introduction of the drug into human subjects, frequently healthy volunteers. These studies are designed to determine the metabolism and pharmacologic actions of the drug in humans, the adverse effects associated with increasing doses and, if possible, to gain early evidence of effectiveness. In Phase 1, the drug is usually tested for safety, including adverse effects, dosage tolerance, absorption, distribution, metabolism, excretion and pharmacodynamics.
- •
- Phase 2. Phase 2 clinical trials usually involve studies in a limited patient population to (1) evaluate the efficacy of the drug for specific, targeted indications; (2) determine dosage tolerance and optimal dosage; and (3) identify possible adverse effects and safety risks.
- •
- Phase 3, or Pivotal Trials. If a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 studies, the clinical trial program will be expanded to further demonstrate clinical efficacy, optimal dosage and safety within an expanded
78
- •
- Phase 4. Phase 4 clinical trials are studies required of, or agreed to, by a sponsor that are conducted after the FDA has approved a product for marketing. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to further document safety and clinical benefit in the case of drugs approved under accelerated approval regulations. Failure to promptly conduct Phase 4 clinical trials could result in withdrawal of approval for products approved under accelerated approval regulations.
patient population at geographically dispersed clinical trial sites. Phase 3 studies usually include several hundred to several thousand patients.
Progress reports on clinical trials must be submitted to FDA at least annually, and safety reports must be submitted for serious and unexpected adverse events or any finding from animal testing that suggests a significant risk for human subjects. FDA or the sponsor may suspend a clinical trial at any time for various reasons, including a finding that there is an unacceptable health risk to subjects, and an IRB may suspend or terminate its approval if the clinical trial is not conducted according to the IRB's requirements or if the drug has been associated with an unexpected serious harm to patients.
The results of preclinical studies and clinical trials, together with detailed information on the manufacture and composition of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product. Generally, regulatory approval of a new drug by the FDA may follow one of three routes. The most traditional of these routes is the submission of a full NDA under 505(b)(1) of the FDCA. Another route is the submission of an Abbreviated NDA, or ANDA, for products that are the same as, and are shown to be bioequivalent to, previously approved drug products under 505(j) of the FDCA. A final route, which is possible where an applicant chooses to rely in part on the FDA's conclusion about the safety and effectiveness of previously approved drugs in seeking approval of a modified version of the drug, is to submit an NDA under 505(b)(2) of the FDCA. For our product candidates that are currently undergoing clinical trials, we are currently pursuing the 505(b)(2) application route. We believe that 505(b)(2) is the development pathway that will maximize the commercial opportunities for these product candidates and our other product candidates. As such, we intend to engage in discussions with the FDA to determine which, if any, portions of our development program can be modified, based on previous FDA findings of a drug's safety and effectiveness.
Applications submitted under either 505(b)(1) or 505(b)(2) are required by the FDA to contain full reports of investigations of safety and effectiveness. However, in contrast to a traditional NDA submitted pursuant to 505(b)(1) in which the applicant submits all of the data demonstrating safety and effectiveness, a 505(b)(2) application is one in which one or more of the investigations relied upon for approval were not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Instead, an application submitted pursuant to 505(b)(2) may rely upon published literature, and/or the finding by the FDA that an approved drug containing the same or a related API as the drug that is the subject of the 505(b)(2) application is safe and effective. The 505(b)(2) applicant must submit information, typically including a bioavailability/bioequivalency study, and potentially including other clinical and non-clinical data, to support changes to the approved drug. Because large scale clinical trials potentially may not be required, the preclinical and clinical development programs leading to the submission of an NDA under 505(b)(2) may be less expensive to carry out and may be concluded in a shorter period of time than programs required for a 505(b)(1) application. In its review of any NDA submissions, however, the FDA has broad discretion to require an applicant to generate additional data related to safety and efficacy and it is impossible to predict the number or nature of the studies that may be required before the FDA will grant approval. Notwithstanding the approval of many products by the FDA pursuant to 505(b)(2) over the last few
79
years, many for brand name companies, some brand-name pharmaceutical companies and others have objected to the FDA's interpretation under 505(b)(2) that the FDA may rely upon prior findings of safety and effectiveness for an approved drug in the review of a competitor's 505(b)(2) NDA. If the FDA changes its interpretation of 505(b)(2) or a court disagrees with the FDA, this could delay or even prevent the FDA from approving any 505(b)(2) NDA that we submit.
The FDA reviews all NDAs submitted before it accepts them for filing and may refuse to file an NDA that is not sufficiently complete to permit review. In addition to the review, a user must pay a fee for process of the NDA. As of the date hereof, fees are approximately $1.2 million for applications that require clinical data (excluding bioequivalency or bioavailability data) and $589,000 for applications that do not require clinical data for support, such as some 505(b)(2) applications.
Once the NDA submission has been accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA currently has ten months in which to complete its initial review of a standard NDA, including 505(b)(2) NDAs, and respond to the applicant, and six months for a priority NDA for a drug that represents a significant improvement in treatment, prevention, or diagnosis of disease. However, the FDA announced in January 2008 that it may miss these goals by up to two months due to staffing shortages and other priorities. In addition, drugs for serious and life-threatening diseases that demonstrate the potential to address unmet medical needs for such a condition may receive "fast track" designation and may be approved on the basis of adequate and well-controlled studies that establish an effect on a clinical endpoint or on a surrogate endpoint that is reasonably likely to predict clinical benefit. Approval of fast track products is often conditioned on conduct of post-market, or Phase 4, studies to validate the surrogate endpoint or confirm the clinical endpoint and is subject to expedited withdrawal of approval for various reasons, including a failure to validate or confirm the endpoints used. FDA may delay approval of an NDA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product. FDA approval of any NDA submitted by us will be at a time the FDA chooses. Also, if regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory requirements and conditions of approvals are not maintained or if problems occur after the product reaches the marketplace. In addition, the FDA may require post-marketing studies to monitor the effect of approved products and may limit further marketing of the product based on the results of these post-marketing studies.
Under the Pediatric Research Equity Act, or the PREA, NDAs for new active ingredients, new indications, new dosage forms, and new routes of administration must include data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals or partial waivers of these requirements. Depending on whether PREA assessments were conducted on the approved drugs that we intend to combine with Captisol, and whether such combination triggers additional PREA assessments, we may have to submit data to comply with the PREA. Pediatric studies conducted in response to a written request from FDA may result in a six-month extension of any existing marketing or patent exclusivity we may have.
Before approving an NDA, the FDA will conduct pre-approval inspections at facilities where the product is manufactured, whether ours or our third-party manufacturers', and will not approve the product unless the manufacturing facility complies with cGMP and the facilities, controls, and methods used ensure the identity, strength, purity and quality of the drug product.
80
If we obtain regulatory approval for a product, the approval will be limited to those diseases and conditions for which the product is effective, as demonstrated through clinical trials. Changes to an approved drug's indications for use, formulation, dosing, labeling, manufacturing, or other aspects of the drug product are subject to FDA review and all changes except some minor changes require supplemental approval. Even after regulatory approval is obtained, a marketed product, and all its manufacturing facilities, whether owned by the applicant or a third-party contractor, are subject to continual review and periodic inspections by the FDA, and in our case the State of Kansas, to ensure compliance with cGMP and for other reasons. Such inspections may identify compliance issues that may disrupt production or distribution and require substantial resources to correct.
Other post-market requirements include complying with record-keeping and specified electronic record and signature requirements, adverse experience and production problem reporting and annual reporting requirements, drug sampling and distribution requirements, and complying with FDA's labeling and advertising requirements. FDA strictly regulates labeling, advertising and promotion, and other information regarding marketed and investigational products. Marketed drugs may only be promoted for the approved indications and in accordance with the approved product label.
Discovery of previously unknown problems with a medicine, device, manufacturer or facility may result in restrictions on the marketing or manufacturing of an approved product, including costly recalls or withdrawal of the product from the market. The FDA has broad regulatory and enforcement powers, including suspension or delaying issuance of approvals or withdrawing approvals, product seizures and injunctions on production, distribution, or marketing of products, recalls, warning letters, civil and criminal penalties, restitution and disgorgement, and refusals of government contracts. Any agency or judicial enforcement could have a material adverse effect on us.
From time to time legislation is passed by Congress that significantly affects the statutory provisions covering the FDA and the products and firms it regulates. In addition, the FDA from time to time promulgates new rules or guidance, or revises or reinterprets its regulations and guidance. Such changes are difficult to predict, and could significantly affect our business and products.
The Hatch-Waxman Act
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant's product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
The ANDA or 505(b)(2) NDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA's Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product's Orange Book listed patents or that such patents are invalid is called a Paragraph IV certification. If an applicant does not challenge the listed patents, the ANDA or 505(b)(2) NDA will not be approved until all the listed patents claiming the referenced product have expired.
If an applicant makes a Paragraph IV certification, it must also send notice of the Paragraph IV certification to the patent and NDA holders once the ANDA or 505(b)(2) NDA has been submitted to
81
the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. An infringement suit brought within 45 days of the receipt of a Paragraph IV certification could result in costly litigation and automatically prevents the FDA from approving the ANDA or 505(b)(2) NDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA or 505(b)(2) NDA applicant.
An ANDA or 505(b)(2) NDA also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active ingredients, during which ANDAs or 505(b)(2) NDAs for versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original product approval.
As a result of these requirements, to the extent our NDAs relate to APIs contained in existing drug products, we will be prevented from submitting or receiving approval of a 505(b)(2) or ANDA during the exclusivity period applicable to the new chemical entity. Although this exclusivity will not prevent us from pursuing a separate 505(b)(1) application for a new chemical entity, we do not intend to pursue 505(b)(1) NDAs for our current product candidates. We do not anticipate that we will be entitled to new chemical entity exclusivity for any of our current product candidates. In contrast, a 505(b)(2) application we submit for a drug product utilizing an API contained in a previously approved drug might itself be eligible for three years of marketing exclusivity, provided new clinical investigations (not including bioavailability or bioequivalence studies) that we conduct or sponsor are essential to the FDA's approval of the application. This three-year market exclusivity may block the approval of generic or 505(b)(2) competitors, but may not have substantial value if there is a generic version of the pioneer drug. In addition, because we only plan to conduct bioequivalence studies for Captisol-Enabled Fosphenytoin IV and IM and our other retained product candidates, those product candidates will not be eligible for three-year market exclusivity.
In addition, in any NDA we file, we must submit for listing in the FDA's Orange Book all of our patents claiming the drug product, drug substance or a method of using the drug with respect to which a claim of infringement could reasonably be asserted if a person not licensed by the patent owner manufactured, used, or sold the drug. We must also certify as to the status of the listed patents relating to the existing approved drug product or must challenge the validity or applicability of those patents. Similarly, in order for generic or 505(b)(2) applicants to rely on the FDA's approval of any NDA we submit, such generic applicants would have to certify to the status of our listed patents and would be subject to marketing or patent exclusivity, if any, covering our approved drug product. If a generic or 505(b)(2) entrant challenged the validity of our patents or their applicability to the product that is the subject of the ANDA or 505(b)(2) application, we would likely be involved in costly litigation to defend our patent from such competitors. In addition, neither market exclusivity nor patent exclusivity will block submission or approval of an NDA for a product similar to ours that is submitted as a new chemical entity under 505(b)(1).
Additional Regulations
In addition to regulation by the FDA and some state regulatory agencies, the United States Drug Enforcement Administration, or DEA, imposes various registration, recordkeeping and reporting requirements, procurement and manufacturing quotas, labeling and packaging requirements, security controls and a restriction on prescription refills on controlled prescription products under the Controlled Substances Act. A principal factor in determining the particular requirements, if any,
82
applicable to a product is its actual or potential abuse profile. The DEA regulates drug substances as Schedule I, II, III, IV or V substances, with Schedule I and II substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. The DEA regulations related to manufacturing, storage, distribution and dispensing and the DEA also regulates the amount of the scheduled substance that would be available for clinical trials and commercial distribution. The DEA periodically inspects facilities for compliance with its rules and regulations. Failure to comply with current and future regulations of the DEA could lead to a variety of sanctions, including revocation, or denial of renewal of DEA registrations, injunctions, or civil or criminal penalties, and could harm our business and financial condition. Most states also regulate controlled substances and impose independent licensing, recordkeeping and security requirements.
We also will be subject to a variety of foreign regulations governing clinical trials and the marketing of any future products. Outside the United States, our ability to market a product depends upon receiving a marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. In any country, however, we will only be permitted to commercialize our products if the appropriate regulatory authority is satisfied that we have presented adequate evidence of safety, quality and efficacy. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing of the product in those countries. The time needed to secure approval may be longer or shorter than that required for FDA approval. The regulatory approval and oversight process in other countries includes all of the risks associated with the FDA process described above.
Pharmaceutical Pricing and Reimbursement
In both domestic and foreign markets, our ability to commercialize successfully and attract strategic partners for our product candidates depends in significant part on the availability of adequate coverage and reimbursement from third-party payors, including, in the United States, governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Third-party payors are increasingly challenging prices charged for medical products and services and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of any future products. Even with studies, our product candidates may be considered less safe, less effective or less cost effective than existing products, and third-party payors therefore may not provide coverage and reimbursement for our product candidates, in whole or in part.
Political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental changes. There have been, and we expect there will continue to be, a number of legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. We anticipate that Congress, state legislatures and the private sector will continue to consider and may adopt healthcare policies intended to curb rising healthcare costs. These cost containment measures include:
- •
- controls on government funded reimbursement for medical products and services;
- •
- controls on healthcare providers;
- •
- challenges to the pricing of medical products and services or limits or prohibitions on reimbursement for specific products and therapies through other means;
- •
- reform of drug importation laws; and
83
- •
- expansion of use of managed care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person.
We are unable to predict what additional legislation, regulations or policies, if any, relating to the healthcare industry or third-party coverage and reimbursement may be enacted in the future or what effect such legislation, regulations or policies would have on our business. Any cost containment measures, including those listed above, or other healthcare system reforms that are adopted could have a material adverse effect on our ability to operate profitably.
Employees
As of December 31, 2007, we had 25 full time employees, nine of whom held Ph.D. or M.D. degrees and nine of whom were engaged in full time research and development activities. We plan to continue to expand our product candidate development programs and hire additional staff to facilitate this growth. We continue to search for qualified individuals with interdisciplinary training to address the various aspects and applications of our development candidates and our technologies. None of our employees is represented by a labor union, and we consider our employee relations to be good.
Facilities
Our principal offices and facilities are currently located in one leased building totaling approximately 19,880 square feet in Lenexa, Kansas. A portion of this space is used for research and development, and the balance is used for office space for general and administrative functions. The initial term of the lease expires on January 31, 2009. We have an option to renew the lease for an additional five-year term, beginning on February 1, 2009 and expiring on January 31, 2014. While we believe that the current space under the lease is adequate to meet our needs for the foreseeable future, the lease also provides us with a contingent right to lease additional adjoining space during the lease term.
Legal Proceedings
We are not currently party to any legal proceedings.
84
Executive Officers, Directors and Key Employees
Our current executive officers, directors and key employees, and their respective ages and positions as of March 31, 2008 are:
| Name | Age | Position | ||
|---|---|---|---|---|
| John M. Siebert, Ph.D. | 68 | Chairman and Chief Executive Officer | ||
| Allen K. Roberson, CPA | 41 | Chief Financial Officer | ||
| Gerold Mosher, Ph.D. | 51 | Director, Product Development | ||
| J.D. Pipkin, Ph.D. | 56 | Director, Product Development | ||
| Vincent Antle, Ph.D. | 39 | Director, Technical Operations | ||
| Steven D. Cosler | 52 | Director | ||
| James C. Gale | 58 | Director | ||
| Elliot F. Hahn, Ph.D. | 63 | Director | ||
| David Poltack | 36 | Director | ||
| Philip L. Smith, Ph.D. | 58 | Director |
John M. Siebert, Ph.D. has served on our board of directors since January 2004. Dr. Siebert joined CyDex as a consultant in May 2003 and became chairman and chief executive officer in January 2004. Prior to joining CyDex, Dr. Siebert was president and chief executive officer of CIMA Labs, a drug delivery technology company, from 1995 to 2003. Prior to joining CIMA, Dr. Siebert served in scientific, quality assurance and product development roles with G. D. Searle, E.R.Squibb, Bayer and Dey Laboratories, as well as Procter & Gamble. Dr. Siebert serves on the board of directors of Aradigm Corporation, a public pharmaceutical company, and Primus Pharmaceuticals. Dr. Siebert holds a bachelor's degree in chemistry from Illinois Benedictine University, a master's degree in organic chemistry from Wichita State University, and a doctorate in organic chemistry from the University of Missouri.
Allen K. Roberson, CPA has served as our chief financial officer since September 2007. Prior to becoming chief financial officer, Mr. Roberson served as our vice president of finance and accounting. Mr. Roberson joined CyDex in February 2003. During his ten years with Sanofi-Aventis and its predecessor companies in the pharmaceutical industry, Mr. Roberson's responsibilities included global accounting, sales regional controller, operations finance and contract management. From 1988 to 1991, Mr. Roberson served at Deloitte & Touche, where he managed audits and financial reviews across multiple industries. Mr. Roberson received his bachelor's degree in accounting from Drake University.
Gerold Mosher, Ph.D. joined CyDex in 1997 and has served as our Director, Product Development since August 2000. Dr. Mosher holds a bachelor's degree in pharmacy, a master's degree in pharmaceutical chemistry and a doctorate in pharmaceutical chemistry, all from the University of Kansas.
J.D. Pipkin, Ph.D. joined CyDex in March 2001 and has served as our Director, Product Development since that time. Dr. Pipkin holds a bachelor's degree in mathematics and chemistry, a master's degree in pharmaceutical chemistry and a doctorate in pharmaceutical chemistry, all from the University of Kansas.
Vincent Antle, Ph.D. has served as our Director, Technical Operations since June 2006. Prior to becoming a director, Dr. Antle served as our manager of technical operations. Dr. Antle joined CyDex
85
in September 2005. Prior to joining CyDex, Dr. Antle was manager of technical operations at EaglePicher Pharmaceutical Services from 1999 to 2005, where his responsibilities included project oversight, client interactions, proposal preparation, product development, setting production time-lines and project budget assessments. Dr. Antle holds a bachelor's degree in chemistry from the University of Minnesota — Morris and a doctorate in medicinal chemistry from the University of Cincinnati.
Steven D. Cosler has served on our board of directors since January 2005. Mr. Cosler has done consulting work for CyDex and is currently a consultant for various companies. Mr. Cosler was president and chief executive officer of Priority Healthcare Corporation from July 1996 to October 2005. Mr. Cosler joined Priority, at that time a subsidiary of Bindley Western Industries, in 1996 and held positions of increasing responsibility until he was named a director in 2000, president in 2001 and chief executive officer in 2002. His earlier positions included senior vice president and general manager, executive vice president — pharmacy services, executive vice president and chief operating officer. Mr. Cosler is a director of SXC Health Solutions, Inc., a public company, and serves on the Board of Trustees of two funds of Claymore Advisors, LLC. Mr. Cosler holds a bachelor's degree in industrial management from the Krannert School of Management at Purdue University.
James C. Gale has served on our board of directors since August 2004 and is chairman of the nominating and corporate governance committee. Mr. Gale is a managing director of Sanders Morris Harris, a Houston-based investment firm. Since October 1998, he has served as managing partner of LOF Partners LLC, which manages the firm's three life sciences private equity funds. Mr. Gale serves on the boards of directors of Indevus Pharmaceuticals and several private companies. He is the chairman of the board of directors of Alpex Pharma S.A. From February 2004 to present, Mr. Gale was head of investment banking for Gruntal & Co., LLC. Earlier, he served with Home Insurance Co., E.F. Hutton & Co. and Adam Cohen Securities, Inc., where he was involved in making principal investments. Mr. Gale received his master's degree in business administration from The University of Chicago. Messrs. Gale and Smith both serve as directors of Avantium Technologies, B.V., or Avantium. CyDex Pharmaceuticals, Inc. does not have any relationship with Avantium.
Elliot F. Hahn, Ph.D. has served on our board of directors since August 2004. Dr. Hahn has been chairman and chief executive officer of SoLapharm Inc., a privately held pharmaceutical company, since December 2007 and was President of Accu-Break Pharmaceuticals, Inc., a privately held company, from October 2004 to December 2007. Dr. Hahn currently serves as the Chairman of Accu-Break Phamaceuticals. He also co-founded Andrx Corporation, a specialty pharmaceutical company acquired by Watson Pharmaceuticals, Inc. in November 2006. Dr. Hahn was president of Andrx from 1993 until 2003. He served as chairman of the board from 2002 to June 2003, and as interim chief executive officer from 2001 to June 2002. From June 1989 to February 1993, Dr. Hahn served as vice president for scientific affairs of IVAX, a manufacturer and distributor of generic pharmaceutical products acquired by Teva Pharmaceutical Industries Ltd. in January 2006, where he was involved in the evaluation and licensing of product opportunities, and was responsible for maintaining IVAX's intellectual property. Prior to that, he was vice president of research at the pharmaceutical subsidiary of IVAX. Before joining IVAX, Dr. Hahn was an associate professor at The Rockefeller University, an assistant professor at Albert Einstein College of Medicine, and a member of the Institute for Steroid Research at Montefiore Hospital. In addition to CyDex, Dr. Hahn serves as a director of Pharmasset, Inc., a pharmaceutical company, and NationsHealth, Inc., a provider of diabetes supplies. He holds a bachelor's degree from City College of New York and a doctorate in organic chemistry from Cornell University.
David Poltack has served on our board of directors since May 2007 and is chairman of our audit committee. Mr. Poltack currently serves as general partner for TVM Capital, a venture capital firm, which he joined in September 2000. Prior to joining TVM Capital, he worked for more than six years
86
at PricewaterhouseCoopers LLP, where he was a Manager in the venture capital and healthcare practices. Mr. Poltack is a Certified Public Accountant and holds a bachelor of science in accountancy from Boston College and a master's degree in business administration with honors from Suffolk University.
Philip L. Smith, Ph.D. has served on our board of directors since August 2004 and is chairman of our compensation committee. Dr. Smith is a general partner at SR One, a venture capital fund that is wholly owned by GlaxoSmithKline, which he joined in June 2002 and has been a general partner since January 2003. From 1985 to June 2002, Dr. Smith held positions of increasing responsibility in the Pharmaceutical Development Group at SmithKline Beecham/GlaxoSmithKline, ultimately heading up a transnational group responsible for identifying and recommending internal development of drug delivery technologies for product development. From 1981 to 1985, Dr. Smith was an assistant professor at the University of Kansas Medical Center. Dr. Smith serves on the board of directors of OctoPlus, a drug delivery company, and RedPoint Bio, a biotechnology company, as well as several private companies. Dr. Smith received a master's degree and doctorate in medicinal chemistry/pharmacology from Northeastern University and a bachelor's degree in chemistry from the University of Maine. Messrs. Gale and Smith both serve as directors of Avantium Technologies, B.V., or Avantium. CyDex Pharmaceuticals, Inc. does not have any relationship with Avantium.
Executive Officers
Our executive officers are elected by, and serve at the discretion of, our board of directors. There are no family relationships among our directors and executive officers. In addition to the two executive officers listed above, Theron E. Odlaug, Ph.D. has accepted an offer to join CyDex as president and chief operating officer, and is expected to start in May 2008. Dr. Odlaug is a global healthcare industry executive and has had a career with Baxter International, Inc., Bayer AG, Fujisawa and Astellas Pharma U.S., Inc. Presently, he is Managing Director of EIR Healthcare Advisors LLC and a member of the board of directors of Advanced Life Sciences Holdings, Inc. Dr. Odlaug has been a member of the board of the privately held Wahl Clipper Corporation and its compensation committee since 1999. He was a member of the board of the Illinois Biotechnology Industry Association (iBIO) from 2003 to 2006. He earned his bachelor's and master's degrees in biology from the University of Missouri and his doctorate in public health from the University of Minnesota.
Board Composition
Our board of directors currently consists of six members and one vacancy. Effective upon the closing of this offering, we will divide our board of directors into three classes, as follows:
- •
- Class I, which will consist of Mr. Gale and Mr. Poltack, whose term will expire at our annual meeting of stockholders to be held in 2008;
- •
- Class II, which will consist of Dr. Hahn and Dr. Smith, whose term will expire at our annual meeting of stockholders to be held in 2009; and
- •
- Class III, which will consist of Mr. Cosler and Dr. Siebert, whose term will expire at our annual meeting of stockholders to be held in 2010.
At each annual meeting of stockholders to be held after the initial classification, the successors to directors whose terms then expire will serve until the third annual meeting following their election and until their successors are duly elected and qualified. The authorized number of directors may be changed only by resolution of the board of directors. Any additional directorships resulting from an increase in the number of directors will be distributed between the three classes so that, as nearly as
87
possible, each class will consist of one-third of the directors. This classification of the board of directors may have the effect of delaying or preventing changes in our control or management. Under Delaware law, our directors may be removed for cause by the affirmative vote of the holders of a majority of our voting stock.
Our current directors were elected pursuant to a shareholders' agreement that we entered into with some holders of our common and convertible preferred stock, as well as the provisions of our certificate of incorporation. The relevant provisions of the shareholders' agreement will terminate upon the closing of this offering and the certificate of incorporation will be amended and restated effective upon the closing of this offering, and there will be no further contractual obligations regarding the election of our directors, except that in connection with our offer to Dr. Odlaug to become our president and chief operating officer, we have agreed to recommend to the board of directors that Dr. Odlaug be elected as a member of the board of directors six months after the commencement of his employment.
Lead Independent Director
If at any time the chairman of our board of directors is not an independent director, the board of directors may designate one of the independent directors then on our board of directors to serve as our lead independent director. The responsibilities of our lead independent director include:
- •
- consulting with and acting as a liaison between the board of directors and our chairman;
- •
- establish the agendas for meetings of the board of directors and serve as chairman of such meetings in the absence of our chairman;
- •
- establish the agenda and preside over meetings of the independent members of the board of directors;
- •
- coordinate with committee chairs regarding committee meeting agendas and informational requirements;
- •
- preside over any portions of meetings of our board of directors at which the evaluation or compensation of our chief executive officer or the performance of our board of directors is presented or discussed;
- •
- coordinate the activities of the other independent directors and perform such other duties as may be established or delegated by our chairman; and
- •
- otherwise consult with our chairman and management on matters relating to corporate governance and board of directors performance matters.
In performing the duties described above, the lead independent director is expected to consult with the chair of any committees of the board of directors and solicit their participation in order to avoid diluting the authority or responsibilities of such committee. If our chairman is an independent director, then he or she shall perform the functions otherwise assigned to our lead independent director. Our current chairman, Dr. Siebert, is not an independent director, and the board of directors has designated as our lead independent director.
88
Committees of the Board of Directors
Our board of directors currently has an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and responsibilities described below.
Audit Committee
Our audit committee is comprised of Messrs. Cosler, Gale and Poltack, each of whom is a non-employee member of our board of directors. Mr. Poltack is the chairman of the audit committee. The board of directors has determined that Mr. Poltack is an "audit committee financial expert" as defined under SEC rules and regulations. We believe that the composition of our audit committee meets the requirements for independence and financial sophistication under the current requirements of the Nasdaq Global Market and SEC rules and regulations. Messrs. Gale and Poltack each beneficially own in excess of 10% of our stock and, as a result, own in excess of the amount that would automatically qualify them as being independent under SEC rules. This does not, however, create a presumption that they are not independent. In addition, our audit committee has the specific responsibilities and authority necessary to comply with the current requirements of the Nasdaq Global Market and SEC rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
Our audit committee is responsible for, among other things:
- •
- overseeing the accounting and financial reporting processes and audits of our financial statements;
- •
- appointing an independent registered public accounting firm to audit our financial statements;
- •
- overseeing and monitoring:
- •
- the integrity of our financial statements;
- •
- our compliance with legal and regulatory requirements as they relate to financial statements or accounting matters;
- •
- our independent registered public accounting firm's qualifications, independence and performance; and
- •
- our internal accounting and financial controls;
- •
- preparing the report that SEC rules require be included in our annual proxy statement;
- •
- providing the board of directors with the results of its monitoring and recommendations; and
- •
- providing to the board of directors additional information and materials as it deems necessary to make the board of directors aware of significant financial matters that require the attention of the board of directors.
Our independent registered public accounting firm and internal financial personnel have unrestricted access to our audit committee and meet privately with our audit committee on a regular basis.
89
Compensation Committee
Our compensation committee is comprised of Messrs. Cosler and Poltack and Dr. Smith, each of whom is a non-employee member of our board of directors. Dr. Smith is the chairman of the compensation committee. Each member of our compensation committee is an "outside director" as that term is defined in Section 162(m) of the Internal Revenue Code and a "non-employee director" within the meaning of Rule 16b-3 of the rules promulgated under the Securities Exchange Act of 1934, as amended. We believe that the composition of our compensation committee meets the requirements for independence under the current requirements of the Nasdaq Global Market and SEC rules and regulations. We intend to comply with future requirements to the extent they become applicable to us.
Our compensation committee is responsible for, among other things:
- •
- reviewing and approving for our chief executive officer and other executive officers:
- •
- annual base salary;
- •
- annual incentive bonus, including the specific goals and amount;
- •
- equity compensation;
- •
- employment agreements, severance arrangements and change in control agreements/provisions;
- •
- any other benefits, compensations, compensation policies or arrangements;
- •
- the criteria on which compensation paid to our chief executive officer for the last completed fiscal year is based;
- •
- the relationship of such compensation to our performance;
- •
- the committee's executive compensation policies applicable to executive officers; and
- •
- acting as administrator of our current benefit plans;
- •
- reviewing and making recommendations to the board of directors regarding general compensation goals and guidelines for employees and the criteria by which bonuses to employees are determined; and
- •
- preparing a report to be included in our annual proxy statement.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee is comprised of Mr. Gale and Dr. Hahn. Mr. Gale and Dr. Hahn are non-employee members of our board of directors and Mr. Gale is the chairman of the nominating and corporate governance committee. We believe that the composition of our nominating and corporate governance committee meets the requirements for independence under the current requirements of the Nasdaq Global Market.
90
Our nominating and corporate governance committee is responsible for, among other things:
- •
- reviewing board structure, composition and practices, and making recommendations on these matters to the board of directors;
- •
- reviewing, soliciting and making recommendations to the board of directors and stockholders with respect to candidates for election to the board; and
- •
- overseeing compliance by employees with our Code of Business Conduct and Ethics.
Compensation Committee Interlocks and Insider Participation
During the last fiscal year, none of the members of our compensation committee was one of our officers or employees. None of our executive officers serves, or has served in the past year, as a member of the board of directors or compensation committee of any entity that has one or more executive officers who have served on our board of directors or compensation committee.
Compensation Discussion and Analysis
The following discussion and analysis of compensation arrangements of our named executive officers for 2007 should be read together with the compensation tables and related disclosures set forth below.
Overview of Compensation Program
Our board of directors has delegated the responsibility for assessing and recommending executive compensation to the compensation committee. It is the policy of the compensation committee, as delegated to it by our board of directors, to seek to maintain an overall compensation structure designed to attract, retain and motivate management and other employees by providing appropriate levels of risk and reward, assessed on a relative basis at all levels within our company and in proportion to individual contribution and performance, and to seek to establish appropriate incentives for management and employees at all levels to further our long-term strategic plan and long-term value as a going concern and to avoid undue emphasis on short-term market value. Once the compensation committee makes its determinations regarding executive compensation, it then recommends these determinations to our board of directors, which has the ultimate authority to approve or modify these recommendations.
Generally, the types of compensation and benefits provided to our executive officers are similar to those provided to executive officers of other similarly-situated companies. The discussion below focuses on the compensation relating to the individuals included in the Summary of Executive Compensation Table under the caption "Summary of Executive Compensation" below, who are referred to as our "named executive officers."
Compensation Philosophy and Objectives
The compensation committee's objective in establishing our executive compensation program is to provide executive compensation that is both successful in attracting and retaining individuals with the skills necessary for us to achieve our long-term business plan, as well as motivate and reward those individuals who perform at or above the levels that we expect. In order to do so, the compensation committee establishes:
- •
- base cash compensation necessary as compensation for services rendered;
91
- •
- a significant cash incentive component which will only be paid upon the achievement of company-specific annual, long-term and strategic goals and which will increase with performance exceeding those goals and, in the case of our chief executive officer, personal goals established by the compensation committee; and
- •
- equity incentives to further align executive officers' interests with those of the stockholders.
Because we face strong competition for individuals with the skills necessary to make our company successful, the compensation committee reviews compensation surveys and takes industry trends into account with respect to each of these components, and believes that it is necessary for our executive compensation to be at or above the 25th percentile, with the potential to be at the 75th percentile, of companies with which we compete for executive talent, in order for us to meet our hiring and retention goals. As part of its review, the compensation committee examines aggregated survey data of companies participating in surveys prepared by various consulting groups, which consist of in excess of 50 companies.
Role of Executive Officers in Compensation Decisions
The compensation committee recommends to our board of directors all compensation decisions for the named executive officers, and our board of directors then either approves the compensation recommendations as made or modifies these recommendations. Historically, our board of directors has accepted the recommendations of the compensation committee without modification. Our chief executive officer and chief financial officer assess their performance, as well as the performance of any other executive officers, on an annual basis and reach a consensus as to the recommendations they will make to the compensation committee, based upon numerous industry compensation surveys, company performance, individual performance and any other information they believe to be relevant. Our chief executive officer and chief financial officer then present their recommendations to the compensation committee, including with respect to salary adjustments, annual cash bonus plans and equity grants, along with the basis for their recommendations. The compensation committee reviews and considers these recommendations in making their compensation decisions. These recommendations are just one factor that the compensation committee takes into account in making its compensation decisions. Other factors are discussed below.
Setting Executive Compensation
In 2007, the compensation committee did not engage an executive compensation consulting firm, but rather relied on industry compensation surveys, its own experience, as well as our needs as a company, in setting executive compensation.
In making compensation decisions, the compensation committee compares each element of total compensation against norms taken from the industry compensation surveys referenced above. The compensation committee pays particular attention to surveys in which the companies surveyed reflect the types of companies against which we compete for talent.
We compete with many larger companies as well as companies of similar size for top executive-level talent. Accordingly, the compensation committee generally targets base compensation for executive officers between the 25th and 75th percentiles of salaries paid to similarly situated executive officers, based on numerous industry compensation surveys, and designs cash bonus compensation plans to deliver cash compensation between the 25th and 75th percentiles. The decisions of the compensation committee may vary within these ranges, and may occasionally be outside of these ranges as the compensation committee deems appropriate, based upon the experience level of the individual,
92
individual performance and relative contributions of the executive officer, ability to affect our performance, and market factors. For example, the compensation committee targeted Dr. Siebert's compensation above the 50th percentile range primarily due to his experience and ability to affect our performance, and Mr. Roberson's compensation below the 50th percentile range due to his having a lesser amount of experience than Dr. Siebert.
The compensation committee recommends stock option grants in order to assist us in:
- •
- aligning the link between the creation of stockholder value and long-term executive incentive compensation;
- •
- providing an opportunity for increased equity ownership by executive officers; and
- •
- maintaining competitive levels of total compensation.
In addressing equity compensation, the compensation committee evaluates the survey information from industry compensation surveys as well as the experience members have had in their careers and in other board positions they hold.
The compensation committee allocates a significant percentage of total compensation to bonus and equity incentives as a result of the philosophy mentioned above. There is no pre-established policy or target for the allocation between either cash and non-cash or short-term and long-term incentive compensation. Our executive officers realize income from cash incentive compensation as a result of our performance compared to established goals, their personal performance towards company long-term goals, and other factors, and realize equity compensation as a result of our performance.
Executive Compensation Components
Base Salary
In determining base salaries for 2007, the compensation committee analyzed the base salary and performance-based compensation of each executive officer as compared to the surveys described above.
Performance-Based Incentive Compensation
Annual target performance-based compensation for 2007 for each named executive officer was a percentage of the named executive officer's base salary. The compensation committee established the target performance-based compensation levels using the principles described above. Applying these principles, the compensation committee determined to set Dr. Siebert's bonus percentage at 50% of base salary, and Mr. Roberson's bonus percentage at 30% of base salary. In order to earn the target bonus, the compensation committee set company-specific performance goals as follows:
93
For Dr. Siebert, the compensation committee and our board of directors determined the performance goals and relative weighting, as well as subsequent percentage achievement, as follows:
| Goals | Relative Weight of Goal | Percent of Goal Achieved | Weighted Average Portion of Bonus Earned and Paid | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Financial Objectives | 10 | % | 100 | % | 10 | % | ||||
| a. | Achieve 2007 revenue of $11.7 million. EBITDA loss not to exceed $2.2 million. | ||||||||||
2. | Implement Intellectual Property Plan | 15 | % | 93 | % | 14 | % | ||||
| a. | File at least one new Captisol patent application, of a material nature, to extend intellectual property, by June 30, 2007 | ||||||||||
| b. | File at least one newg-SBECD patent application, of a material nature, by October 31, 2007 | ||||||||||
| c. | File at least one new alkyl-SBECD patent application, of a material nature, by November 30, 2007 | ||||||||||
| d. | File at least two new substituted cyclodextrin/product patent applications, of a material nature, by December 31, 2007. This objective is in addition to b and c above. | ||||||||||
3. | Current Products Activities | 15 | % | 93 | % | 14 | % | ||||
| a. | Complete and review bioequivalence, or BE, results for fosphenytoin BE study by October 1, 2007 | ||||||||||
| b. | Complete a specified product candidate pre-IND meeting with the FDA by March 15, 2007 | ||||||||||
| c. | Initiate first patient enrolled proof-of-concept human clinical trial for a specified product candidate by July 31, 2007 | ||||||||||
| d. | Initiate Phase 3 clinical trials for a specified product candidate or complete outlicense agreement by June 30, 2007 | ||||||||||
4. | New Products Activities | 15 | % | 53 | % | 8 | % | ||||
| a. | Initiate a Phase 1 clinical trial (Pharmacokinetic, Proof of Concept, or efficacy) of a new product by August 31, 2007 | ||||||||||
| b. | Complete a development plan or outlicense agreement for a specified product candidate by July 31, 2007 | ||||||||||
5. | Technology Licensing Activities | 5 | % | 100 | % | 5 | % | ||||
| a. | Complete two new technology license and supply agreements by November 30, 2007 | ||||||||||
| b. | Complete four new limited clinical use agreements by November 30, 2007 | ||||||||||
94
6. | Product Licensing Activities | 5 | % | 100 | % | 5 | % | ||||
| a. | Complete one new license agreement by November 30, 2007 | ||||||||||
7. | Exit Strategy Activities | 35 | % | 0 | % | 0 | % | ||||
| a. | Complete a strategic combination of the Company or complete an initial public offering with a minimum established pre-money valuation by December 31, 2007 | 56 | % | ||||||||
Our board of directors had determined that the most important activity for Dr. Siebert in 2007 was to identify the best possible method for delivering liquidity to our stockholders, and established this as the major focus of Dr. Siebert's activity in 2007. The compensation committee and the board of directors determined that Dr. Siebert had achieved the goals at the levels set forth above.
For Mr. Roberson, the compensation committee and our board of directors determined the performance goals and relative weighting, with the elements of each of the following goals being identical with the corresponding goals for Dr. Siebert, as well as subsequent percentage achievement, as follows:
| Goals | Relative Weight of Goal | Percent of Goal Achieved | Weighted Average Portion of Bonus Earned and Paid | ||||
|---|---|---|---|---|---|---|---|
| 1. Financial Objectives | 20 | % | 100 | % | 20 | % | |
| 2. Implement Intellectual Property Plan | 25 | % | 80 | % | 20 | % | |
| 3. Current Products Activities | 20 | % | 60 | % | 12 | % | |
| 4. New Products Activities | 15 | % | 53 | % | 8 | % | |
| 5. Technology Licensing Activities | 10 | % | 100 | % | 10 | % | |
| 6. Product Licensing Activities | 10 | % | 100 | % | 10 | % | |
| 80 | % | ||||||
The focus for Mr. Roberson was on initiating development of new products deemed to be important to our future valuation, as well as achieving financial objectives, especially revenue and EBITDA. Our board of directors determined that this focus was particularly important from the viewpoint that an unusually high amount of development spending was being allocated to further develop product candidates already in the pipeline. The compensation committee and the board of directors determined that Mr. Roberson had achieved the goals at the levels set forth above.
Our board of directors and compensation committee determined that development of new product candidates was of particular importance for our future and that less emphasis should be placed on Captisol outlicensing activities. This determination is reflected in the relative weighting of the goals reflected above. The compensation committee reviewed the accomplishments against goals from the viewpoint that these two officers were responsible for achieving the goals to increase the valuation of our company in the overall interest of the stockholders.
95
Stock Option Program
The only equity awards that we have granted to executive officers to date have been stock options. We have not granted any restricted stock or restricted stock units. We have limited equity awards to stock options to date because we believe they are effective in aligning executive officers' interests with the interests of our stockholders, and in encouraging such officers to remain with CyDex. In the future, we may grant restricted stock or restricted stock units, but we have not determined to do so to date. Generally, the compensation committee recommends significant stock option grants when an executive officer commences employment. The initial grants are made as negotiated with the executive officer. In negotiating the stock option grants, the compensation committee takes into account the position the executive officer will have, the level of responsibility, and other relevant factors particular to the circumstances. The relative weight given to each of these factors will vary from individual to individual at the compensation committee's discretion. The compensation committee makes adjustments as it deems reasonable to attract candidates in the competitive environment in which we operate.
The compensation committee recommends subsequent stock option grants at varying times and in varying amounts at the discretion of the compensation committee. Historically, these grants have been made in the first or second board of directors meeting each year. The compensation committee assesses each executive officer's performance during the prior year during the performance appraisal process, and also considers corporate performance in recommending subsequent stock option grants. The compensation committee also reviews grant practices by reviewing industry surveys for comparative purposes. The compensation committee looks at averages of similar sized companies by employee position/skill level to assess appropriate levels. The compensation committee also takes into account unvested existing stock options in determining the size of subsequent grants. The compensation committee determines the vesting schedule and the number of shares granted to ensure that the executive officer has meaningful incentives to remain in our company's employ. The stock option will provide a return to the executive officer only if he remains in our employ, and will only provide a benefit if the price of our common stock is greater than the exercise price of the option at the time of exercise. Based on these factors and the number of shares available for grant, our compensation committee recommends the levels of subsequent stock option grants.
The exercise price of stock options is determined by our board of directors on the date of grant. Most of the stock options granted have a ten-year maximum term and vest at a rate of 25% following one year of employment, and then annually thereafter over three additional years, for a total of four years. Vesting ceases on termination of employment and exercise rights cease either 90 days or five years after termination of employment, as the case may be, except in the case of death (subject to an 18 month limitation) or disability (subject to a 12-month limitation). Prior to the exercise of a stock option, the holder has no rights as a stockholder with respect to the shares subject to the stock option, including voting rights and the right to receive dividends or dividend equivalents.
The compensation committee believes that stock option grants currently provide a significant incentive to our employees and executive officers.
In addition to the option grants made in the ordinary course as described above, in March 2006, our board of directors granted a stock option to Dr. Siebert to purchase 1,800,000 shares of our common stock with an exercise price equal to the fair value of our common stock on the date of grant, which grant would vest five years from the date of grant. The board provided that the vesting of 20% of the shares would accelerate upon either BMS or Pfizer entering into an agreement with us to extend the royalty term with respect to marketed products until the last to expire applicable claim in a Captisol patent. These conditions to acceleration were met and the shares vested in September 2006. The board also provided that the vesting of this grant would accelerate with respect to 80% of the shares if there was a liquidation event, such as a sale of our company or an initial public offering of our common
96
stock, by September 30, 2007. The right to acceleration with respect to 80% of the shares lapsed on September 30, 2007. The board made this grant to provide Dr. Siebert with significant additional incentive to cause an event to occur that would provide our holders of preferred stock liquidity with respect to their investment, and secondarily to provide Dr. Siebert with significant additional incentive to preserve our ability to fund the development of our product candidates with royalties received from marketed products.
Retirement and Other Benefits
401(k) Plan. We maintain a deferred savings retirement plan for our U.S. employees. The deferred savings retirement plan is intended to qualify as a tax-qualified plan under Section 401 of the Internal Revenue Code. The deferred savings retirement plan permits us to make discretionary contributions, subject to established limits and a vesting schedule. In 2006 and 2007, the board approved a match of up to 2% of the employee's salary for our 401(k) plan participants.
Perquisites and Other Personal Benefits. Our compensation committee has approved, and we have obtained and pay the premiums on, a term life insurance policy on Dr. Siebert with Dr. Siebert's wife as the beneficiary. The compensation committee approved this policy in recognition of the tremendous physical and mental effort Dr. Siebert was making to increase shareholder value and to recognize the risk involved in the great amount of travel undertaken by Dr. Siebert in that pursuit. Our executive officers participate in the same group insurance and employee benefit plans as our other salaried employees. At this time we do not provide any material special benefits or other perquisites to our executive officers other than as described above.
Summary of Executive Compensation
The following table sets forth the total compensation awarded or paid to, or earned by, our chairman and chief executive officer and our chief financial officer for the year ended December 31, 2007. We refer to these individuals as our named executive officers. We did not have any other executive officers during 2007.
Summary of Executive Compensation Table
| Name and Principal Position | Year | Salary ($) | Option Awards ($)(1) | Non-Equity Incentive Plan Compensation ($)(2) | All Other Compensation ($) | Total ($) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| John M. Siebert, Ph.D. Chairman and Chief Executive Officer | 2007 | 346,100 | 18,946 | 96,908 | 13,695 | (3) | 475,649 | |||||
Allen K. Roberson, CPA Chief Financial Officer(4) | 2007 | 160,869 | 8,406 | 38,609 | 3,185 | (5) | 211,069 |
- (1)
- The dollar amount in this column represents the compensation cost for the year ended December 31, 2007 of stock option awards granted in and prior to 2007. These amounts have been calculated in accordance with SFAS No. 123R ignoring the estimates of forfeiture and using the Black-Scholes option-pricing model.
- (2)
- Paid pursuant to our performance-based compensation plan for 2007 as described in the section entitled "Compensation Discussion and Analysis" above.
- (3)
- Includes the value of life insurance premiums and tax adjustments of $8,560 and matching contributions to the 401(k) plan of $5,135 for the benefit of Dr. Siebert.
- (4)
- Mr. Roberson became our chief financial officer in September 2007. Mr. Roberson previously served as our vice president, finance and accounting.
- (5)
- Includes the value of matching contributions to the 401(k) plan for the benefit of Mr. Roberson.
97
Grants of Plan-Based Awards
The following table shows information regarding grants of plan-based awards to the named executive officers for the year ended December 31, 2007:
Grants of Plan-Based Awards in Fiscal 2007
| | | Estimated Future Payouts Under Non-Equity Incentive Plan Awards | All Other Option Awards: Number of Securities Underlying Options (#)(2) | | | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | Grant Date Fair Value of Stock and Option Awards ($)(4) | |||||||||||
| | | Exercise or Base Price of Option Awards ($/Sh)(3) | ||||||||||||
| Name | Grant Date | Threshold ($) | Target ($)(1) | Maximum ($) | ||||||||||
| John M. Siebert, Ph.D. | September 5, 2007 | 0 | 173,050 | 173,050 | 250,000 | 0.35 | 64,834 | |||||||
Allen K. Roberson, CPA | September 5, 2007 | 0 | 48,261 | 48,261 | 150,000 | 0.35 | 38,900 | |||||||
- (1)
- This column sets forth the target amount of each named executive officer's annual non-equity incentive plan award for the year ended December 31, 2007 under our annual cash bonus award program. The amounts set forth in this column do not represent additional compensation earned by the named executive officers for the year ended December 31, 2007. For a description of our annual cash bonus award program, see "Compensation Discussion and Analysis."
- (2)
- Unless otherwise noted, stock options were granted pursuant to the 1997 Equity Compensation Plan.
- (3)
- Represents the per share fair value of our common stock, as determined in good faith by our board of directors on the grant date.
- (4)
- Represents the grant date fair value of such option award as determined in accordance with SFAS No. 123R. These amounts have been calculated in accordance with SFAS No. 123R using the Black-Scholes valuation model.
A description of our 2007 management incentive compensation arrangements, as well as a description of Dr. Siebert's special stock option grant of 1,800,000 shares, is set forth in "Compensation Discussion and Analysis" above.
Outstanding Equity Awards at Year-End
The following table shows information regarding outstanding equity awards at year end for the named executive officers for the year ended December 31, 2007.
Outstanding Equity Awards At December 31, 2007
| | Option Awards | |||||||
|---|---|---|---|---|---|---|---|---|
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($)(1) | Option Expiration Date | ||||
| John M. Siebert, Ph.D., Chairman and Chief Executive Officer | 316,666 360,000 125,000 0 | 0 1,440,000 375,000 250,000 | (2) (3) (4) (5) | 0.15 0.15 0.15 0.35 | 11/02/14 12/05/15 05/24/16 09/05/17 | |||
Allen K. Roberson, CPA, Chief Financial Officer | 5,000 300,000 225,000 0 | 0 0 375,000 150,000 | (6) (7) (8) | 2.50 0.15 0.15 0.35 | 03/03/13 11/02/14 03/22/16 09/05/17 | |||
- (1)
- Represents the per share fair market value of our common stock, as determined in good faith by our board of directors on the grant date.
- (2)
- Original grant of option to purchase 950,000 shares. One-third of the shares subject to the option award vest on the first anniversary of the vesting commencement date of November 2, 2004 and one-third of the shares subject to the option award vest annually thereafter.
98
- (3)
- Original grant of option to purchase 1,800,000 shares. All of the shares subject to the option award vest on the fifth anniversary of the vesting commencement date of December 5, 2005 unless certain performance based milestones are achieved in which case vesting may be accelerated.
- (4)
- Original grant of option to purchase 500,000 shares. One-fourth of the shares subject to the option award vest on the first anniversary of the vesting commencement date of May 24, 2006 and one-fourth of the shares subject to the option award vest annually thereafter.
- (5)
- Original grant of option to purchase 250,000 shares. One-third of the shares subject to the option award vest on the first anniversary of the vesting commencement date of September 5, 2007 and one-third of the shares subject to the option award vest annually thereafter.
- (6)
- Original grant of option to purchase 300,000 shares. One-third of the shares subject to the option award vest on the first anniversary of the vesting commencement date of November 2, 2004 and one-third of the shares subject to the option award vest annually thereafter.
- (7)
- Two grants of options to purchase 100,000 and 500,000 shares. Grant of option to purchase 100,000 shares not subject to vesting. Grant of option to purchase 500,000 shares subject to vesting as follows: one-fourth of the shares subject to the option award vest on the first anniversary of the vesting commencement date of March 22, 2006 and one-fourth of the shares subject to the option award vest annually thereafter.
- (8)
- Original grant of option to purchase 150,000 shares. One-third of the shares subject to the option award vest on the first anniversary of the vesting commencement date of September 5, 2007 and one-third of the shares subject to the option award vest annually thereafter
Option Exercises
The following table sets forth certain information regarding option awards exercised by our named executive officers during 2007:
| | Option Awards | ||||
|---|---|---|---|---|---|
| Name | Number of Shares Acquired on Exercise | Value Realized on Exercise(1) | |||
| John M. Siebert, Ph.D. | 316,667 | $ | |||
- (1)
- Calculated assuming a price per share of $ , which is the mid-point of the range reflected on the cover page of this prospectus.
Nonqualified Deferred Compensation
We do not currently maintain non-qualified defined contribution plans or other deferred compensation plans.
Pension Plans
We do not currently maintain any pension plans.
Employment Agreement, Severance Agreements and Potential Payments Upon Termination Or Change In Control
John M. Siebert, Ph.D., Chairman and Chief Executive Officer
In June 2006, we entered into an employment agreement with Dr. Siebert, our chairman and chief executive officer. The agreement provides that Dr. Siebert will initially receive an annual base salary of $336,000, subject to annual increases approved by the board of directors, and will be eligible to receive an annual performance bonus of up to 50% of his annual base salary. Dr. Siebert is also eligible to participate in our general employee benefit plans in accordance with the terms and conditions of such plans. The agreement provides that Dr. Siebert is employed "at-will," and his employment may be terminated at any time by us or by Dr. Siebert.
99
Under the employment agreement with Dr. Siebert, if Dr. Siebert is involuntarily terminated without cause, then Dr. Siebert will receive 12 months base salary. This payment will be delayed for a period of six months after termination if, at the time of his termination, Dr. Siebert is a "specified employee" within the meaning of Section 409A of the Internal Revenue Code. Moreover, if Dr. Siebert's employment is terminated due to a permanent disability he will receive term life insurance for a period of up to five years, with a benefit of $1 million, provided that the annual premium for such policy does not exceed $10,000. The payment of such premiums will be delayed for a period of six months after termination if Dr. Siebert is a "specified employee" at the time of such termination. In addition, Dr. Siebert is entitled to full acceleration of his unvested stock options if involuntarily terminated without cause, or if he terminates his employment for good reason, in either case within 13 months of a change in control.
Dr. Siebert is also a party to a severance agreement with us, entered into in January 2004, and amended in February 2008. Under the severance agreement, Dr. Siebert will be entitled to receive severance payments upon (i) an involuntary termination that occurs within six months prior to a change in control if the termination is caused by the change in control, (ii) an involuntary termination other than for cause within two years following a change in control or (iii) a voluntary termination for good reason within two years following a change in control. Dr. Siebert will be entitled to receive one year of base salary, plus one-twelfth of base salary for each year of Dr. Siebert's employment with us, up to an aggregate of one times his base salary. Dr Siebert will also be entitled to receive any bonuses paid by us to Dr. Siebert for the fiscal year ending immediately prior to the change in control and additional amounts to equalize excise tax payments under Section 4999 of the Internal Revenue Code. In addition, Dr. Siebert will be eligible to receive a continuation for 12 months of health, disability, and group life insurance plans or any retirement, pension or profit sharing plans, selected by him immediately prior to the change in control. Lump sum severance payments and various continuing coverage payments will be delayed for a period of six months if, at the time of termination, Dr. Siebert is a "specified employee" within the meaning of Section 409A of the Internal Revenue Code.
The following table describes the potential payments to Dr. Siebert upon his termination without good cause, or resignation for good reason, in connection with a change in control, or upon termination without cause not in connection with a change in control:
| Event | Salary(1) | Cash payments(2) | Equity Acceleration(3) | Benefits(4) | Tax Gross Ups | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Termination without good cause, or resignation for good reason, within 24 months following a change in control. | $ | 346,100 | $ | 259,299 | $ | 5,836 | $ | 280,428 | ||||||||
| Termination without cause, no change in control. | $ | 346,100 | — | — | — | |||||||||||
- (1)
- Represents 12 months of continued salary.
- (2)
- Calculated based on a change in control taking place as of December 31, 2007. Cash payments of $259,299 representing one-twelfth of annual salary for each year of employment with the company and bonuses paid by the company for the fiscal year ending immediately prior to the change in control.
- (3)
- Calculated based on a change in control taking place as of December 31, 2007 and assuming a price per share of $ , which is the mid-point of the range reflected on the cover page of this prospectus. Represents the full acceleration of unvested stock options held by Dr. Siebert if terminated within 13 months of a change in control as specified in Dr. Siebert's employment agreement.
- (4)
- Represents 12 months of COBRA health benefits and group life insurance.
100
Theron E. Odlaug, Ph.D., President and Chief Operating Officer
Dr. Odlaug has accepted an offer to join CyDex as president and chief operating officer, and we anticipate he will start in May 2008. The offer letter with Dr. Odlaug provides that Dr. Odlaug will initially receive an annual base salary of $290,000, subject to increases approved by the board of directors, and will be eligible to receive an annual performance bonus of up to 40% of his annual base salary which will be prorated from the date of hire for 2008. In connection with his employment, and subject to board of directors approval, Dr. Odlaug will receive an option to purchase 1,000,000 shares of our common stock. Dr. Odlaug is also eligible to participate in our general employee benefit plans in accordance with the terms and conditions of such plans. The offer letter also contemplates that we will recommend to the board of directors that Dr. Odlaug be elected as a member of the board of directors six months after the commencement of his employment. The offer letter provides that Dr. Odlaug is employed "at-will," and his employment may be terminated at any time by us or by Dr. Odlaug.
Under the offer letter with Dr. Odlaug, if Dr. Odlaug is involuntary terminated without cause during the first 18 months of his employment, then Dr. Odlaug will receive nine months base salary, payable no later than March 15 following the year of such involuntary termination period. In connection with our offer to Dr. Odlaug, we also expect to enter into a severance agreement with Dr. Odlaug on substantially the same terms as Dr. Siebert's severance agreement described above.
Allen K. Roberson, CPA, Chief Financial Officer
Mr. Roberson is a party to a severance agreement with us, entered into in November 2003, and amended in February 2008. Under the severance agreement, Mr. Roberson will be entitled to receive severance payments upon (i) an involuntary termination that is within six months prior to a change in control if the termination is caused by the change in control, (ii) an involuntary termination other than for cause within two years following a change in control or (iii) a voluntary termination for good reason within two years following a change in control. Mr. Roberson will be entitled to receive one year of base salary, plus one-twelfth of base salary for each year of Mr. Roberson's employment with us, up to an aggregate of one times his base salary. Mr. Roberson will also be entitled to receive any bonuses paid by us to Mr. Roberson for the fiscal year ending immediately prior to the change in control and additional amounts to equalize excise tax payments under Section 4999 of the Internal Revenue Code. In addition, Mr. Roberson will be eligible to receive a continuation for 12 months of health, disability, and group life insurance plans or any retirement, pension or profit sharing plans, selected by him immediately prior to the change in control. Lump sum severance payments and various continuing coverage payments will be delayed for a period of six months if, at the time of termination, Mr. Roberson is a "specified employee" within the meaning of Section 409A of the Internal Revenue Code.
The following table describes the potential payments to Mr. Roberson upon his termination without good cause, or resignation for good reason, within 24 months following a change in control or upon involuntary termination within six months prior to a change in control, assuming a change in control had occurred on December 31, 2007:
| Salary(1) | Cash payments(2) | Benefits(3) | Tax Gross ups | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| $175,000 | $ | 101,201 | $ | 5,872 | $ | 129,412 | $ | 411,485 | ||||
- (1)
- Represents 12 months of continued salary.
- (2)
- Cash payments of $101,201 representing one-twelfth of annual salary for each year of employment with the company and bonuses paid by the company for the fiscal year ending immediately prior to the change in control.
- (3)
- Represents 12 months of COBRA health benefits and group health life insurance.
101
Summary of Non-Employee Director Compensation
The following table sets forth the total compensation awarded or paid to, or earned by, our non-employee directors for the year ended December 31, 2007.
Non-Employee Director Compensation Table
| Name | Fees Earned or Cash Paid ($) | Option Awards ($)(1)(2)(3) | Total ($) | |||
|---|---|---|---|---|---|---|
| Steven D. Cosler | 10,500 | 25,882 | 36,382 | |||
| James C. Gale | — | 25,882 | 25,882 | |||
| Elliot F. Hahn, Ph.D. | 7,500 | 25,882 | 33,382 | |||
| Stephen Hoffman(4) | — | — | — | |||
| David Poltack(5) | — | 25,882 | 25,882 | |||
| Peter Thornton(6) | — | — | — | |||
| Philip L. Smith, Ph.D. | — | 25,882 | 25,882 |
- (1)
- The amounts shown in this column represent the dollar amounts recognized for financial statement reporting purposes for the fiscal year ended December 31, 2007 in accordance with SFAS No. 123R, excluding an estimate of forfeitures related to service-based vesting conditions, and thus include amounts from awards granted in and prior to 2007. Assumptions used in the calculation of these amounts are described in Note 11 to our audited consolidated financial statements for the year ended December 31, 2007, included elsewhere in this prospectus.
- (2)
- The following options were outstanding as of December 31, 2007: Mr. Cosler —180,000; Mr. Gale —160,000; Dr. Hahn —50,000; Mr. Hoffman —5,000; Mr. Poltack —80,000; Mr. Thornton —0; and Dr. Smith —160,000.
- (3)
- On September 5, 2007, each of the applicable directors received a grant of options to purchase 80,000 shares of common stock with an exercise price of $0.35 per share.
- (4)
- Mr. Hoffman resigned from our board of directors in May 2007.
- (5)
- Mr. Poltack joined our board of directors in May 2007.
- (6)
- Mr. Thornton resigned from our board of directors in July 2007.
Under our current director compensation policy, each independent director receives an option to purchase 200,000 shares of our common stock upon becoming a member of the board of directors, subject to vesting restrictions, and an annual grant of an option to purchase 40,000 shares of common stock per year thereafter. Each independent member of the board of directors receives as compensation for their service as directors an annual retainer of $12,000 paid quarterly, $3,000 plus expenses for attendance at board meetings, $500 for attendance at telephonic meetings, $5,000 per year for service as the chair of the audit committee of the board of directors and $4,000 per year for service as the chair of each of the compensation committee and nominating and corporate governance committees, respectively.
In addition, under the terms of our 2008 Non-Employee Directors' Stock Option Plan, our non-employee directors will be entitled to receive automatic grants of stock options once we are a publicly-traded company. See the description of our 2008 Non-Employee Directors' Stock Option Plan in the section immediately below.
Employee Benefit Plans
Amended and Restated 1997 Equity Compensation Plan
Historically we have granted stock options under our Amended and Restated 1997 Equity Compensation Plan, or 1997 plan, which our board of directors adopted in January 1997 and our
102
stockholders subsequently approved. An aggregate of 580,248 shares of common stock is reserved for issuance under the 1997 plan.
The 1997 plan provides for the grant of incentive stock options and nonqualified stock options. As of December 31, 2007, options to purchase 7,695,668 shares of common stock at a weighted average exercise price per share of $0.77 remained outstanding and 580,248 shares remained available for future issuance.
Our board of directors has the authority to construe and interpret the terms of the 1997 plan and the awards granted under it. Upon the signing of the underwriting agreement for this offering, no further awards will be granted under the 1997 plan. Although the 1997 plan will terminate, all outstanding awards will continue to be governed by their existing terms.
Stock Options. The 1997 plan provides for the grant of incentive stock options under federal tax laws and nonqualified stock options. Only employees may receive incentive stock options, but nonqualified stock options may be granted to employees, directors, and consultants. The exercise price of incentive stock options may not be less than 100% of the fair market value of our common stock on the date of grant. Shares subject to options under the 1997 plan generally vest in a series of installments over an optionee's period of service.
In general, the maximum term of options granted under the 1997 plan is ten years. Unless the terms of an optionee's option agreement provide otherwise, if an optionee's service relationship with us, or any of our affiliates, ceases for any reason other than disability, death, or cause, the optionee may exercise the vested portion of any options for three months after the date of such termination. If an optionee's service relationship with us, or any of our affiliates, ceases due to disability or death, the optionee or a beneficiary may exercise any vested options for a period of 12 months. If an optionee's service relationship is terminated for cause, the option will expire immediately. In no event, however, may an option be exercised beyond the expiration of its term.
Acceptable consideration for the purchase of common stock issued upon the exercise of a stock option may include (a) cash or cash equivalents, (b) the tender of common stock previously owned by the optionee, (c) a net exercise of the option, and (d) other legal consideration.
Changes in Control. In the event of a specified change in control transaction, outstanding options may be assumed, continued, or substituted for by any surviving or acquiring corporation. If the surviving or acquiring corporation elects not to assume, continue, or substitute for outstanding options, then such options will become fully exercisable and terminate to the extent not exercised prior to the change in control transaction.
2008 Equity Incentive Plan
Our board of directors adopted the 2008 Equity Incentive Plan, or 2008 incentive plan, in 2008 and our stockholders approved the 2008 incentive plan in 2008. The 2008 incentive plan will become effective immediately upon the signing of the underwriting agreement for this offering. The 2008 incentive plan will terminate in December 2018, unless sooner terminated by our board of directors.
Stock Awards. The 2008 incentive plan provides for the grant of incentive stock options, nonstatutory stock options, restricted stock awards, restricted stock unit awards, stock appreciation rights, performance stock awards, and other forms of equity compensation, or stock awards, which may be granted to employees, including officers, non-employee directors, and consultants.
103
Share Reserve. Following this offering, the aggregate number of shares of common stock that may be issued initially pursuant to stock awards under the 2008 incentive plan is shares. Such share reserve consists of: (a) the shares remaining available for future issuance under the 1997 plan, including shares subject to outstanding stock options under the 1997 plan, plus (b) an additional shares reserved for issuance under the 2008 incentive plan. The number of shares of common stock reserved for issuance will automatically increase on January 1st, from January 1, 2009 through January 1, 2018, by the lesser of (a) % of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, or (b) a lesser number of shares of common stock determined by our board of directors prior to the start of a year for which an increase applies. The maximum number of shares that may be issued pursuant to the exercise of incentive stock options over the term of the 2008 incentive plan is shares.
No person may be granted stock options or stock appreciation rights covering more than shares of common stock under the 2008 incentive plan during any calendar year. Such limitation is designed to help assure that any deductions to which we would otherwise be entitled upon the exercise of such stock options and stock appreciation rights or upon the subsequent sale of shares acquired under such awards, will not be subject to the $1 million limitation on the income tax deductibility of compensation paid to specified executive officers imposed by Section 162(m) of the Internal Revenue Code.
If a stock award granted under the 2008 incentive plan or the 1997 plan expires or otherwise terminates without being exercised in full, the shares of common stock not acquired pursuant to the stock award again become available for subsequent issuance under the 2008 incentive plan. In addition, the following types of shares under the 2008 incentive plan or the 1997 plan will become available for the grant of new stock awards under the 2008 incentive plan: (a) shares that are forfeited or repurchased by us prior to becoming fully vested; (b) shares subject to stock awards that are settled in cash; (c) shares withheld to satisfy income and employment withholding taxes; (d) shares used to pay the exercise price of an option in a net exercise arrangement; (e) shares tendered to us to pay the exercise price of an option; and (f) shares that are cancelled pursuant to an exchange or repricing program. Shares issued under the 2008 incentive plan may be previously unissued shares or reacquired shares, including shares bought on the open market. As of the date of this prospectus, no shares of common stock have been issued under the 2008 incentive plan.
Administration. Our board of directors has delegated its authority to administer the 2008 incentive plan to our compensation committee. Subject to the terms of the 2008 incentive plan, our board of directors or an authorized committee, referred to as the plan administrator, determines recipients, dates of grant, the numbers and types of stock awards to be granted, and the terms and conditions of the stock awards, including the period of their exercisability and vesting. Subject to the limitations set forth below, the plan administrator will also determine the exercise price of options granted, the consideration to be paid for restricted stock awards, and the strike price of stock appreciation rights.
The plan administrator has the authority to:
- •
- reduce the exercise price of any outstanding option or the strike price of any outstanding stock appreciation right;
- •
- cancel any outstanding option or stock appreciation right and to grant in exchange one or more of the following:
- •
- new options or stock appreciation rights covering the same or a different number of shares of common stock,
104
- •
- new stock awards,
- •
- cash, and/or
- •
- other valuable consideration; or
- •
- engage in any action that is treated as a repricing under generally accepted accounting principles.
Stock Options. Incentive and nonstatutory stock options are granted pursuant to incentive and nonstatutory stock option agreements adopted by the plan administrator. The plan administrator determines the exercise price for a stock option provided that the exercise price of an incentive stock option and nonstatutory stock option cannot be less than 100% of the fair market value of our common stock on the date of grant. Options granted under the 2008 incentive plan vest at the rate specified by the plan administrator.
Generally, the plan administrator determines the term of stock options granted under the 2008 incentive plan, up to a maximum of ten years (except in the case of some incentive stock options, as described below). Unless the terms of an optionee's stock option agreement provide otherwise, if an optionee's relationship with us, or any of our affiliates, ceases for any reason other than disability or death, the optionee may exercise any vested options for a period of three months following the cessation of service. If an optionee's service relationship with us, or any of our affiliates, ceases due to disability or death (or an optionee dies within a specified period following cessation of service), the optionee or a beneficiary may exercise any vested options for a period of 12 months in the event of disability, and 18 months in the event of death. The option term may be extended in the event that exercise of the option following termination of service is prohibited by applicable securities laws. In no event, however, may an option be exercised beyond the expiration of its term.
Acceptable consideration for the purchase of common stock issued upon the exercise of a stock option will be determined by the plan administrator and may include (a) cash or cash equivalents, (b) a broker-assisted cashless exercise, (c) the tender of common stock previously owned by the optionee, (d) a net exercise of the option, and (e) other legal consideration approved by the plan administrator.
Unless the plan administrator provides otherwise, options generally are not transferable except by will, the laws of descent and distribution, or pursuant to a domestic relations order. An optionee may designate a beneficiary, however, who may exercise the option following the optionee's death.
Tax Limitations on Incentive Stock Options. Incentive stock options may be granted only to our employees. The aggregate fair market value, determined at the time of grant, of shares of our common stock with respect to incentive stock options that are exercisable for the first time by an optionee during any calendar year under all of our stock plans may not exceed $100,000. No incentive stock option may be granted to any person who, at the time of the grant, owns or is deemed to own stock possessing more than 10% of our total combined voting power or that of any of our affiliates unless (a) the option exercise price is at least 110% of the fair market value of the stock subject to the option on the date of grant, and (b) the term of the incentive stock option does not exceed five years from the date of grant.
Restricted Stock Awards. Restricted stock awards are granted pursuant to restricted stock award agreements adopted by the plan administrator. Restricted stock awards may be granted in consideration for (a) cash or cash equivalents, (b) past or future services rendered to us or our affiliates, or (c) any other form of legal consideration approved by the plan administrator. Shares of
105
common stock acquired under a restricted stock award may, but need not, be subject to forfeiture to us in accordance with a vesting schedule to be determined by the plan administrator. Rights to acquire shares under a restricted stock award may be transferred only upon such terms and conditions as set by the plan administrator.
Restricted Stock Unit Awards. Restricted stock unit awards are granted pursuant to restricted stock unit award agreements adopted by the plan administrator. Restricted stock unit awards may be granted in consideration for any form of legal consideration approved by the plan administrator. A restricted stock unit award may be settled by cash, delivery of stock, a combination of cash and stock as deemed appropriate by the plan administrator, or in any other form of consideration set forth in the restricted stock unit award agreement. Additionally, dividend equivalents may be credited in respect of shares covered by a restricted stock unit award. Except as otherwise provided in the applicable award agreement, restricted stock units that have not vested will be forfeited upon the participant's cessation of continuous service for any reason.
Stock Appreciation Rights. Stock appreciation rights are granted pursuant to stock appreciation rights agreements adopted by the plan administrator. The plan administrator determines the strike price for a stock appreciation right which cannot be less than 100% of the fair market value of the common stock on the date of grant. Upon the exercise of a stock appreciation right, we will pay the participant an amount equal to the product of (a) the excess of the per share fair market value of our common stock on the date of exercise over the strike price, multiplied by (b) the number of shares of common stock with respect to which the stock appreciation right is exercised. A stock appreciation right granted under the 2008 incentive plan vests at the rate specified in the stock appreciation right agreement.
The plan administrator determines the term of stock appreciation rights granted under the 2008 incentive plan up to a maximum of ten years. If a participant's service relationship with us, or any of our affiliates, ceases, then the participant, or the participant's beneficiary, may exercise any vested stock appreciation right for three months (or such longer or shorter period specified in the stock appreciation right agreement) after the date such service relationship ends. In no event, however, may a stock appreciation right be exercised beyond the expiration of its term.
Performance Stock Awards. The 2008 incentive plan permits the grant of performance stock awards that may qualify as performance-based compensation that is not subject to the $1 million limitation on the income tax deductibility of compensation paid to specified executive officers imposed by Section 162(m) of the Internal Revenue Code. To assure that the compensation attributable to one or more performance stock awards will so qualify, our compensation committee can structure one or more such awards so that stock will be issued or paid pursuant to such award only upon the achievement of specified pre-established performance goals during a designated performance period. The maximum benefit to be received by a participant in any calendar year attributable to performance stock awards may not exceed 1,000,000 shares of common stock.
Other Stock Awards. The plan administrator may grant other awards based in whole or in part by reference to our common stock. The plan administrator will set the number of shares under the award and all other terms and conditions of such awards.
Changes to Capital Structure. In the event that there is a specified type of change in our capital structure, such as a stock split, appropriate adjustments will be made to (a) the number of shares reserved under the 2008 incentive plan, (b) the maximum number of shares that may be issued pursuant to the exercise of incentive stock options, (c) the maximum number of stock options, stock appreciation rights, and performance stock awards for which any one person may be granted per
106
calendar year, and (d) the number of shares and exercise price or strike price, if applicable, of all outstanding stock awards.
Corporate Transactions. In the event of specified significant corporate transactions, outstanding stock awards under the 2008 incentive plan may be assumed, continued or substituted for by any surviving or acquiring entity (or its parent company). If the surviving or acquiring entity (or its parent company) elects not to assume, continue or substitute for such stock awards, then (a) with respect to any such stock awards that are held by individuals whose service with us or our affiliates has not terminated prior to the effective date of the corporate transaction, the vesting and exercisability provisions of such stock awards will be accelerated in full and such awards will be terminated if not exercised prior to the effective date of the corporate transaction, and (b) all other outstanding stock awards will terminate if not exercised prior to the effective date of the corporate transaction. Our board of directors may also provide that the holder of an outstanding stock award not assumed in the corporate transaction will surrender such stock award in exchange for a payment equal to the excess of (a) the value of the property that the stock award would have received upon exercise of the stock award, over (b) the exercise price otherwise payable in connection with the stock award.
Changes in Control. Our board of directors has the discretion to provide that a stock award under the 2008 incentive plan will immediately vest as to all or any portion of the shares subject to the stock award (a) immediately upon the occurrence of specified specified change in control transactions, whether or not such stock award is assumed, continued, or substituted by a surviving or acquiring entity in the transaction, or (b) in the event a participant's service with us or a successor entity is terminated actually or constructively in connection with or within a designated period following the occurrence of specified specified change in control transactions. Stock awards held by participants under the 2008 incentive plan will not vest on such an accelerated basis unless specifically provided by the participant's applicable award agreement.
2008 Employee Stock Purchase Plan
Our board of directors adopted our 2008 Employee Stock Purchase Plan, or 2008 purchase plan, in 2008 and our stockholders approved the 2008 purchase plan in 2008. The 2008 purchase plan will become effective immediately upon the signing of the underwriting agreement for this offering although we have no current plans to commence an offering under the 2008 purchase plan.
Share Reserve. Following this offering, the 2008 purchase plan authorizes the issuance of shares of common stock pursuant to purchase rights granted to our employees or to employees of any of our designated affiliates. The number of shares of common stock reserved for issuance will automatically increase on January 1st, from January 1, 2009 through January 1, 2018, by the lesser of (a) % of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, or (b) a lesser number of shares of common stock determined by our board of directors prior to the start of a year for which an increase applies. The maximum number of shares that may be issued pursuant to the exercise of purchase rights over the term of the 2008 purchase plan is shares. The 2008 purchase plan is intended to qualify as an "employee stock purchase plan" within the meaning of Section 423 of the Internal Revenue Code. As of the date hereof, no shares of common stock have been purchased under the 2008 purchase plan.
Administration. Our board of directors has delegated its authority to administer the 2008 purchase plan to our compensation committee. The 2008 purchase plan is implemented through a series of offerings of purchase rights to eligible employees. Under the 2008 purchase plan, we may specify offerings with a duration of not more than 27 months, and may specify shorter purchase periods
107
within each offering. Each offering will have one or more purchase dates on which shares of common stock will be purchased for employees participating in the offering.
Payroll Deductions. Generally, all regular employees, including executive officers, employed by us or by any of our affiliates may participate in the 2008 purchase plan and may contribute, normally through payroll deductions, a percentage of their earnings for the purchase of common stock under the 2008 purchase plan. Unless otherwise determined by our board of directors, common stock will be purchased for accounts of employees participating in the 2008 purchase plan at a price per share equal to the lower of (a) 85% of the fair market value of a share of our common stock on the first date of an offering, or (b) 85% of the fair market value of a share of our common stock on the date of purchase.
Reset Feature. Our board of directors may specify that if the fair market value of a share of our common stock on any purchase date within a particular offering period is less than the fair market value on the start date of that offering period, then the employees in that offering period will automatically be transferred and enrolled in a new offering period which will begin on the next day following such a purchase date.
Limitations. Employees may have to satisfy one or more of the following service requirements before participating in the 2008 purchase plan, as determined by our board of directors: (a) customarily employed for more than 20 hours per week, (b) customarily employed for more than five months per calendar year, or (c) continuous employment with us or one of our affiliates for a period of time not to exceed two years. No employee may purchase shares under the 2008 purchase plan at a rate in excess of $25,000 worth of our common stock valued based on the fair market value per share of our common stock at the beginning of an offering for each year such a purchase right is outstanding. Finally, no employee will be eligible for the grant of any purchase rights under the 2008 purchase plan if immediately after such rights are granted, such employee has voting power over 5% or more of our outstanding capital stock measured by vote or value.
Changes to Capital Structure. In the event that there is a specified type of change in our capital structure, such as a stock split, appropriate adjustments will be made to (a) the number of shares reserved under the 2008 purchase plan, (b) the maximum number of shares that may be issued pursuant to the exercise of purchase rights over the term of the 2008 purchase plan, and (c) the number of shares and purchase price in effect for all outstanding purchase rights.
Corporate Transactions. In the event of specified significant corporate transactions, any then-outstanding rights to purchase our stock under the 2008 purchase plan will be assumed, continued or substituted for by any surviving or acquiring entity (or its parent company). If the surviving or acquiring entity (or its parent company) elects not to assume, continue or substitute for such purchase rights, then the participants' accumulated contributions will be used to purchase shares of our common stock within ten business days prior to such corporate transaction, and such purchase rights will terminate immediately thereafter.
2008 Non-Employee Directors' Stock Option Plan
Our board of directors adopted our 2008 Non-Employee Directors' Stock Option Plan, or 2008 directors' plan, in 2008 and our stockholders approved the 2008 directors' plan in 2008. The 2008 directors' plan will become effective immediately upon the signing of the underwriting agreement for this offering. The 2008 directors' plan provides for the automatic grant of nonstatutory stock options to purchase shares of common stock to our non-employee directors over their period of service on our board.
108
Share Reserve. Following this offering, the aggregate number of shares of common stock that may be issued initially under the 2008 directors' plan is shares. The number of shares of common stock reserved for issuance will automatically increase on January 1st, from January 1, 2009 through January 1, 2018, by the excess of (a) the number of shares of common stock subject to options granted during the preceding calendar year, over (b) the number of shares added back to the share reserve during the preceding calendar year. If any option expires or terminates for any reason, in whole or in part, without having been exercised in full, the shares of common stock not acquired under such option will become available for future issuance under the 2008 directors' plan. The following types of shares issued under the 2008 directors' plan may again become available for the grant of new options: (a) any shares withheld to satisfy withholding taxes, (b) any shares used to pay the exercise price of an option in a net exercise arrangement, and (c) shares tendered to us to pay the exercise price of an option. As of the date of this prospectus, no shares of common stock have been issued under the 2008 directors' plan.
Administration. All options granted under the 2008 directors' plan are made in strict compliance with its express provisions. Subject to the provisions of the 2008 directors' plan, our board of directors has the authority to construe and interpret the 2008 directors' plan and the stock options granted under it, and to establish rules for its administration.
Initial Option. Pursuant to the terms of the 2008 directors' plan, each individual who first becomes a non-employee director after the completion of this offering will automatically be granted an option to purchase shares of common stock on such date. The shares subject to each such initial option vest with respect to (a) one-third of the shares subject to the initial grant upon completion of one year of continuous service, and (b) the balance of the shares in a series of 24 successive equal monthly installments measured from the first anniversary of the date of grant.
Annual Option. Pursuant to the terms of the 2008 directors' plan, each individual who is serving as a non-employee director following the close of the annual meeting each year, beginning in 2008, will automatically be granted an option to purchase shares of common stock on such date. The shares subject to each such annual option vest in a series of 12 successive equal monthly installments measured from the date of grant.
Terms of All Options. The exercise price of each option granted under the 2008 directors' plan will be equal to 100% of the fair market value of our common stock on the date of grant. The maximum term of options granted under the 2008 directors' plan is ten years. If a non-employee director's service relationship with us, or any of our affiliates, whether as a non-employee director or subsequently as an employee, director or consultant of ours or an affiliate, ceases for any reason other than disability, death, or within 12 months following a change in control, the optionee may exercise any vested options for a period of three months following the cessation of service. If such an optionee's service relationship with us, or any of our affiliates, ceases due to disability or death (or an optionee dies within a specified period following cessation of service), the optionee or a beneficiary may exercise the option for a period of 12 months in the event of disability, and 18 months in the event of death. If such an optionee's service terminates within 12 months following a specified change in control transaction, the optionee may exercise the option for a period of 12 months following the effective date of such a transaction. The option term may be extended in the event that exercise of the option following termination of service is prohibited by applicable securities laws. In no event, however, may an option be exercised beyond the expiration of its term.
Transferability of Options. Options granted under the 2008 directors' plan are generally not transferable except by will, the laws of descent and distribution, or pursuant to a domestic relations order. However, an option may be transferred for no consideration upon written consent of our board
109
of directors if (a) at the time of transfer, a Form S-8 registration statement under the Securities Act is available for the issuance of shares upon the exercise of such transferred option, or (b) the transfer is to the optionee's employer or its affiliate at the time of transfer.
Changes to Capital Structure. In the event that there is a specified type of change in our capital structure, such as a stock split, appropriate adjustments will be made to (a) the number of shares reserved under the 2008 directors' plan, (b) the number of shares for which options are to be granted subsequently to new and continuing non-employee directors and (c) the number of shares and exercise price of all outstanding options.
Corporate Transactions. In the event of specified significant corporate transactions, all outstanding options under the 2008 directors' plan may be assumed, continued or substituted for by any surviving or acquiring entity (or its parent company). If the surviving or acquiring entity (or its parent company) elects not to assume, continue or substitute for such options, then (a) with respect to any such options that are held by optionees then performing services for us or our affiliates, the vesting and exercisability of such options will be accelerated in full and such options will be terminated if not exercised prior to the effective date of the corporate transaction, and (b) all other outstanding options will terminate if not exercised prior to the effective date of the corporate transaction. Our board of directors may also provide that the holder of an outstanding option not assumed in the corporate transaction will surrender such option in exchange for a payment equal to the excess of (a) the value of the property that the optionee would have received upon exercise of the option, over (b) the exercise price otherwise payable in connection with the option.
Changes in Control. The vesting and exercisability of options held by non-employee directors who are either (a) required to resign their position in connection with a specified change in control transaction, or (b) removed from their position in connection with such a change in control will be accelerated in full.
Limitations of Liability and Indemnification of Officers and Directors
Our amended and restated certificate of incorporation, which will become effective immediately following the completion of this offering, includes provisions that limit the liability of our directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties except for:
- •
- any breach of the director's duty of loyalty to us or to our stockholders;
- •
- acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law;
- •
- unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or
- •
- any transaction from which a director derived an improper personal benefit.
Our amended and restated bylaws provide that we must indemnify our directors and executive officers and may indemnify our officers, employees and other agents to the fullest extent permitted by law. Our amended and restated bylaws also permit us to advance expenses, as incurred by an indemnified party in connection with the defense of any action or proceeding arising out of his or her status or
110
service as a director, officer, employee or other agent of us upon an undertaking by him or her to repay any advances if it is ultimately determined that he or she is not entitled to indemnification.
We have entered into separate indemnification agreements with our directors and executive officers. These agreements require us to, among other things, indemnify the director or executive officer against expenses, including attorney's fees, judgments, fines and settlements paid by the individual in connection with any action, suit or proceeding arising out of the individual's status or service as our director or executive officer, other than liabilities arising from willful misconduct or conduct that is knowingly fraudulent or deliberately dishonest, and to advance expenses incurred by the individual in connection with any proceeding against him or her individually with respect to which he or she may be entitled to indemnification by us. We believe that the provisions of our amended and restated certificate of incorporation and amended and restated bylaws and the indemnification agreements are necessary to attract and retain qualified persons as directors and executive officers. We also maintain directors' and officers' liability insurance, including coverage for public securities matters.
As of the date of this prospectus, we are not aware of any pending litigation or proceeding involving any of our directors, officers, employees or agents where indemnification will be required or permitted. Furthermore, we are not aware of any threatened litigation or proceeding that might result in a claim for indemnification.
111
CERTAIN RELATIONSHIPS AND RELATED PERSONS TRANSACTIONS
Related Person Transactions Policy
Our audit committee has authority to review and approve all related person transactions as set forth in our audit committee charter. We have a simple organizational structure in which potential transactions with related persons are generally easily recognizable by the members of our board of directors and management in the ordinary course of business. In addition, each member of our board of directors and management is aware that all related person transactions must be approved by our audit committee. As such, we rely on each member of our board of directors and management to report any potential related person transactions to our chief executive officer, who would then present the potential related person transaction to the audit committee for approval. In addition, we require our directors and officers to complete director and officer questionnaires on at least an annual basis identifying any transactions with us in which the director or executive officer or his or her family members have an interest.
We review related person transactions due to the potential for a conflict of interest. In addition, our nominating and corporate governance committee determines, on an annual basis, which members of our board of directors are independent (as independence is currently defined in Rule 4200(a)(15) of the Nasdaq listing standards). Our nominating and corporate governance committee reviews and discusses any relationships with directors that would potentially interfere with the director's exercise of independent judgment in carrying out the responsibilities of a director. In cases where the nominating and corporate governance committee determines that it is likely a particular board member or board members will have a conflict of interest concerning an aspect of our business, it may recommend the establishment of a special committee of the board of directors comprised solely of non-conflicted directors to act with respect to such business. Finally, our Code of Business Conduct and Ethics establishes the standards of behavior for all employees, officers, and directors.
Sales of Securities
From January 1, 2005 through January 31, 2008, the following executive officers, directors and holders of 5% or more of our outstanding stock have purchased securities in the amounts and as of the dates set forth below.
| Purchaser | Shares of Common Stock | |
|---|---|---|
| Steve Cosler(1) | 140,000 | |
| Elliot Hahn(2) | 270,000 | |
| John Siebert(3) | 983,334 | |
| Peter Thornton(4) | 190,000 |
- (1)
- Consists of shares acquired pursuant to the exercise of options to purchase a total of 140,000 shares of common stock for $0.15 per share on June 26, 2006.
- (2)
- Consists of shares acquired pursuant to the exercise of options to purchase a total of 190,000 shares of common stock for $0.15 per share on February 21, 2006 and August 8, 2007 and an option to purchase 80,000 shares of common stock for $0.35 per share on November 16, 2007.
- (3)
- Consists of shares acquired pursuant to the exercise of options to purchase a total of 983,334 shares of common stock for $0.15 per share on April 25, 2005, October 16, 2006 and June 11, 2007.
- (4)
- Consists of shares acquired pursuant to the exercise of options to purchase a total of 190,000 shares of common stock for $0.15 per share on October 5, 2007.
112
Registration Rights Agreement
We have entered into a registration rights agreement with the purchasers of our outstanding preferred stock, including our chief executive officer and entities with which some of our directors are affiliated. Under the registration rights agreement, specified existing stockholders may require us to file a registration statement under the Securities Act to register the sale of shares of our common stock, subject to specified limitations. The registration rights agreement also grants "piggyback" registration rights in connection with most registered offerings of common stock that we initiate, either for our own account or for the account of another stockholder. We will pay all expenses relating to these registrations, other than underwriting discounts and commissions. As of December 31, 2007, the holders of shares of our common stock, including the shares of common stock issuable upon the automatic conversion of our preferred stock, and warrants to purchase up to shares of our common stock, are entitled to rights with respect to the registration of their shares under the Securities Act following this offering. See "Description of Capital Stock — Registration Rights."
Shareholders' Agreement
We have entered into a shareholders' agreement with the purchasers of our outstanding preferred stock, including our chief executive officer and entities with which some of our directors are affiliated, relating to, among other things, information rights, rights of first refusal and the obligation of each party to vote or consent to maintain the size of our board of directors and to elect the nominees of specified parties to the board of directors. Upon the closing of this offering, these rights will terminate and none of our stockholders will have any special rights regarding the election or designation of members of our board of directors.
Employment Agreement and Severance Agreement with John M. Siebert, Ph.D.
We have entered into an employment agreement with Dr. Siebert. Dr. Siebert is also entitled to specified severance and change in control benefits. See "Management — Employment Agreement, Severance Agreements and Potential Payments Upon Termination Or Change In Control" above.
Severance Agreement with Mr. Allen Roberson
Mr. Roberson is entitled to specified severance and change in control benefits. See "Management — Employment Agreement, Severance Agreements and Potential Payments Upon Termination Or Change In Control" above.
Director and Officer Indemnification
Our amended and restated certificate of incorporation contains provisions limiting the liability of directors. In addition, we have entered into agreements to indemnify our directors and executive officers to the fullest extent permitted under Delaware law and have entered into indemnification agreements with each of our directors and executive officers. See "Management — Limitations of Liability and Indemnification of Officers and Directors."
All future transactions between us and our officers, directors, principal stockholders and their affiliates will be approved by a majority of our board of directors, including a majority of the independent and disinterested directors in these transactions.
113
The following table sets forth, as of December 31, 2007, information regarding beneficial ownership of our capital stock by the following:
- •
- each person or entity who beneficially owns more than 5% of our common stock;
- •
- each of our directors;
- •
- all of our directors and executive officers as a group; and
- •
- each of our other named executive officers.
Beneficial ownership is determined according to the rules of the SEC and generally means that a person possesses sole or shared voting or investment power of that security, and includes shares underlying options and warrants that are currently exercisable or exercisable within 60 days. Information with respect to beneficial ownership has been furnished to us by each director, executive officer or 5% or more stockholder, as the case may be. Except as otherwise indicated, we believe that the beneficial owners of the common stock listed below, based on the information each of them has given to us, have sole investment and voting power with respect to their shares, except where community property laws may apply.
Unless otherwise indicated, shares underlying options and warrants to purchase shares of our common stock that are exercisable within 60 days of December 31, 2007 are deemed to be beneficially owned by the persons holding these options and warrants for the purpose of computing percentage ownership of that person, but are not treated as outstanding for the purpose of computing any other person's ownership percentage.
This table lists applicable percentage ownership based on shares of common stock outstanding as of December 31, 2007, including shares of preferred stock, on an as-converted basis, assuming an initial public offering price of $ per share, and also lists applicable percentage ownership based on shares of common stock outstanding after the closing of this offering.
Unless otherwise indicated, the address for each of the stockholders in the table below is c/o CyDex Pharmaceuticals, Inc., 10513 W. 84th Terrace, Lenexa, Kansas 66214.
| | | Percent | ||||
|---|---|---|---|---|---|---|
| Name and Address of Beneficial Owner | Shares Beneficially Owned | Before Offering | After Offering | |||
| 5% or Greater Stockholders: | ||||||
| Entities affiliated with TVM Capital Corporation(1) | ||||||
| RiverVest Venture Fund I, L.P.(2) | ||||||
| Entities affiliated with Life Sciences Opportunities Funds(3) | ||||||
| Private Equity Direct Finance(4) | ||||||
| Entities affiliated with S.R. One Ltd.(5) | ||||||
| Eastman Chemical Company Investments, Inc.(6) | ||||||
Named Executive Officers and Directors: | ||||||
| John M. Siebert, Ph.D.(7) | ||||||
| Allen K. Roberson(8) | ||||||
| Steven D. Cosler(9) | ||||||
| James C. Gale(10) | ||||||
| Elliot F. Hahn, Ph.D.(11) | ||||||
| Stephen Hoffman(12) | ||||||
| Peter Thornton(13) | ||||||
| David Poltack(14) | ||||||
| Philip L. Smith, Ph.D.(15) | ||||||
| All directors and executive officers as a group (9 persons) | ||||||
footnotes on following page
114
- **Less
- than 1%
- (1)
- Consists of (a) shares of common stock held by TVM IV GmbH & Co. KG, (b) warrants to purchase shares of common stock held by TVM IV GmbH & Co. KG that are exercisable within 60 days of December 31, 2007, and (c) shares of common stock held by TVM V Life Science Ventures GmbH & Co. KG. TVM IV Management GmbH & Co. KG is the Managing Limited Partner of TVM IV GmbH & Co. KG. TVM V Life Science Ventures Management GmbH & Co. KG is the Managing Limited Partner of TVM V Life Science Ventures GmbH & Co. KG. Certain investment committees, composed of certain Managing Limited Partners of TVM, have voting and dispositive authority over the shares held by each of these entities and therefore beneficially own such shares. Decisions of the investment committees are made by a majority vote of their members and, as a result, no single member of the investment committees has voting or dispositive authority over the shares. Hubert Birner, Gert Caspritz, Mark Cipriano, John J. DiBello, Alexandra Goll, Stefan Fischer, David Poltack, Helmut Schuhsler and Bernd Siebel are the members of the investment committee of TVM V Life Science Ventures Management GmbH & Co. KG. John J. DiBello, Friedrich Bornikoel, Helmut Schuhsler, Alexandra Goll, Christian Claussen, Hubert Birner, Bernd Siebel, Gert Caspritz, and Hans Schreck are the members of the investment committee of TVM IV Management GmbH & Co. KG. David Poltack, one of our directors, is the Secretary of TVM Capital Corporation. See note 14. The address for TVM Capital Corporation and its related entities is 101 Arch St., Suite 1950, Boston, MA 02110.
- (2)
- Consists of (a) shares of common stock held by RiverVest Venture Fund I, L.P. and (b) warrants to purchase shares of common stock held by RiverVest Venture Fund I, L.P. RiverVest Venture Partners I, LLC is the general partner of RiverVest Venture Fund I, L.P. The investment committee of RiverVest Venture Partners I, LLC exercises voting and dispositive power over the shares held by RiverVest Venture Fund I, L.P. Decisions by the investment committee are made by vote and as a result no single member of the investment committee exercises voting or dispositive power over the shares. Shares may be voted pursuant to a majority vote of the investment committee and shares may be disposed of pursuant to a unanimous vote of the investment committee. Thomas Melzer, Jay A. Schmelter and Andrew B. Craig III are the members of the investment committee. The address for RiverVest Venture Fund I, L.P. and its related entities is 733 Forsyth Blvd., Suite 1650, St. Louis, MO 63105.
- (3)
- Consists of (a) shares of common stock held by Life Sciences Opportunities Fund (Institutional) II, LP, (b) shares held by Life Sciences Opportunities Fund II, LP, and (c) shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Mr. James Gale. Mr. Gale, one of our directors, is a managing partner of Signet Healthcare Partners, LLC, which manages Life Sciences Opportunities Fund II, LP and Life Sciences Opportunities Fund (Institutional) II, LP, and therefore may be deemed to beneficially own these shares. See note 10. The address for these entities is c/o Sanders Morris Harris, 527 Madison Avenue, New York, NY 10022.
- (4)
- Consists of shares of common stock held by Private Equity Direct Finance. Andrew Tyson, Gwen McLaughlin and Richard Gorter have sole voting, dispositive and investment control over the securities held by Private Equity Direct Finance and, therefore, may be deemed to beneficially own these shares. The address for Private Equity Direct Finance is P.O. Box 847, One Capital Place, 4th Floor, Grand Cayman, KYI-1103, Cayman Islands.
- (5)
- Consists of shares of common stock held by S.R. One Ltd. and shares of common stock issuable upon the exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Philip L. Smith for the benefit of S.R. One Ltd. Philip L. Smith, one of our directors, is a general partner of S.R. One Ltd. and exercises shared voting and investment control over the securities held by S.R. One Ltd. See note 15. The address for S.R. One Ltd. is 161 Washington Street, Suite 500, Conshohocken, PA 19428-2077.
- (6)
- Consists of shares of common stock held by Eastman Chemical Company Investments, Inc. The board of directors of Eastman Chemical Company Investments, Inc., or Eastman, exercises voting and dispositive power over the shares held by Eastman. Decisions by the Eastman board of directors are made by majority vote and as a result no single member of the Eastman board of directors exercises voting or dispositive power over the shares. Harold Kalbach, Kari Johnson and Amaryl Isenberg are the members of the Eastman board of directors. The address for Eastman Chemical Company Investments, Inc. is 103 Foulk Rd., Suite 205-0, Wilmington, DE 19803.
- (7)
- Consists of (a) shares of common stock held by John M. and Michelle Siebert as joint tenants, (b) shares of common stock held by John M. Siebert, and (c) shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by John M. Siebert.
- (8)
- Includes shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Allen K. Roberson.
- (9)
- Consists of (a) shares of common stock and (b) shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Steven D. Cosler.
115
- (10)
- Consists of shares held by entities affiliated with Life Sciences Opportunities Funds (see note 3), and shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by James C. Gale. Mr. Gale is a managing partner of Signet Healthcare Partners, LLC, which manages Life Sciences Opportunities Fund II, LP and Life Sciences Opportunities Fund (Institutional) II, LP.
- (11)
- Consists of (a) shares of common stock held by Elliot Hahn, and (b) shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Elliot Hahn.
- (12)
- Consists of shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007 held by Stephen Hoffman. Mr. Hoffman resigned as a director in May 2007.
- (13)
- Mr. Thornton resigned as a director in July 2007.
- (14)
- Consists of shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007, and shares held by entities and persons affiliated with TVM Capital Corporation, over which Mr. Poltack exercises shared voting and investment power. Mr. Poltack is the Secretary of TVM Capital Corporation.
- (15)
- Consists of shares of common stock issuable upon exercise of stock options that are exercisable within 60 days of December 31, 2007, and shares held by entities and persons affiliated with S.R. One Ltd., a wholly owned subsidiary of GlaxoSmithKline plc. Dr. Smith is a general partner of S.R. One Ltd. and exercises shared voting and investment control over the securities held by S.R. One Ltd. See note 5.
116
Upon the closing of this offering and the filing of our amended and restated certificate of incorporation, our authorized capital stock will consist of 100,000,000 shares of common stock, $0.01 par value per share. The following description of our capital stock does not purport to be complete and is subject to, and qualified in its entirety by, our amended and restated certificate of incorporation and bylaws, which are exhibits to the registration statement of which this prospectus forms a part.
Common Stock
As of December 31, 2007, shares of our common stock were outstanding and held of record by 98 stockholders. This amount assumes the conversion of all outstanding shares of our preferred stock into common stock, which will occur immediately prior to the closing of this offering. In addition, as of December 31, 2007, 7,695,668 shares of our common stock were subject to outstanding options and 4,460,966 shares of our common stock were subject to outstanding warrants. Upon the closing of this offering, shares of our common stock will be outstanding assuming no exercise of outstanding stock options or warrants or the underwriters' over-allotment option.
Each share of our common stock entitles its holder to one vote on all matters to be voted upon by our stockholders. Subject to preferences that may apply to any of our outstanding preferred stock, holders of our common stock will receive ratably any dividends our board of directors declares out of funds legally available for that purpose. If we liquidate, dissolve or wind up, the holders of common stock are entitled to share ratably in all assets remaining after payment of liabilities and any liquidation preference of any of our outstanding preferred stock. Our common stock has no preemptive rights, conversion rights or other subscription rights or redemption or sinking fund provisions. The shares of our common stock to be issued upon the closing of this offering will be fully paid and non-assessable.
Preferred Stock
Upon the closing of this offering, all outstanding shares of preferred stock will have been converted into shares of common stock. See Note 9 to our consolidated financial statements for a description of the currently outstanding preferred stock. Following this offering, our amended and restated certificate of incorporation will be amended and restated to delete all references to shares of preferred stock.
Warrants
As of December 31, 2007, warrants exercisable for up to an aggregate of 4,460,966 shares of our common stock were outstanding. These warrants were issued in connection with the conversion of outstanding indebtedness under a bridge financing arrangement into Series B convertible preferred stock. These warrants are immediately exercisable at an exercise price of $0.01 per share and will expire in August 2010. These warrants have a net exercise provision under which the holder may, in lieu of payment of the exercise price in cash, surrender the warrant and receive a net amount of shares of common stock based on the fair market value of our common stock at the time of exercise of the warrant after deduction of the aggregate exercise price. These warrants also contain provisions for the adjustment of the exercise price and the aggregate number of shares issuable upon the exercise of the warrant in the event of stock dividends, stock splits or stock combinations, reclassifications, combinations or exchanges.
Registration Rights
Commencing six months following the effective date of the registration statement of which this prospectus is a part, the holders of shares of our common stock or permitted transferees, including shares of common stock issuable upon the exercise of outstanding warrants, will
117
be entitled to require us to register these shares under the Securities Act, subject to limitations and restrictions. Also, if at any time, we propose to register any of our securities under the Securities Act, either for our own account or for the account of other securities holders, the holders of these shares will be entitled to notice of the registration and, subject to specified exceptions, will be entitled to include, at our expense, their shares of our common stock in the registration statement. In addition, the holders of these shares may require us, at our expense and on not more than two occasions in any twelve-month period, to file a registration statement on Form S-3 under the Securities Act, subject to specified conditions and limitations, covering their shares of our common stock. If we are required to file a registration statement, we must use our best efforts to have the registration statement declared effective. These registration rights are subject to conditions and limitations, including the right of the underwriters to limit the number of shares of our common stock included in the registration statement. These registration rights will continue in effect following this offering.
Anti-Takeover Provisions of Delaware Law and Charter Provisions
Upon the closing of this offering we will be subject to Section 203 of the Delaware General Corporation Law. In general, the statute prohibits a publicly-held Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless:
- •
- prior to that date, the board of directors approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder;
- •
- upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding those shares owned by persons who are directors and also officers, and by employee stock plans in which shares held subject to the plan will be tendered in a tender or exchange offer; or
- •
- on or subsequent to that date, the business combination is approved by the board of directors and is authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least two-thirds of the outstanding voting stock not owned by the interested stockholder.
Section 203 defines business combination to include:
- •
- any merger or consolidation involving the corporation and the interested stockholder;
- •
- any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation;
- •
- subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder;
- •
- any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; and
- •
- the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
118
In general, Section 203 defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
On the closing of this offering, our amended and restated certificate of incorporation and bylaws will include a number of provisions that may deter or impede hostile takeovers or changes of control or management. These provisions include:
- •
- Classified Board. Our amended and restated certificate of incorporation provides for a classified board of directors consisting of three classes of directors, each serving a staggered three-year term. Commencing in 2008, a portion of our board of directors will be elected each year for three-year terms. At the time of this offering:
- •
- Mr. Gale and Mr. Poltack are designated Class I directors whose term will expire at the 2008 annual meeting of stockholders;
- •
- Dr. Hahn and Dr. Smith are designated Class II directors whose term will expire at the 2009 annual meeting of stockholders;
- •
- Mr. Cosler and Dr. Siebert are designated Class III directors whose term will expire at the 2010 annual meeting of stockholders.
- •
- Board of directors vacancies. Our restated certificate of incorporation and amended and restated bylaws authorize only our board of directors to fill vacant directorships. In addition, the number of directors constituting our board of directors may be set only by resolution adopted by a majority vote of our entire board of directors. These provisions prevent a stockholder from increasing the size of our board of directors and gaining control of our board of directors by filling the resulting vacancies with its own nominees.
- •
- Stockholder action; special meetings of stockholders. Our amended and restated certificate of incorporation provides that our stockholders may not take action by written consent, but may only take action at annual or special meetings of our stockholders. Stockholders will not be permitted to cumulate their votes for the election of directors. Our amended and restated bylaws further provide that special meetings of our stockholders may be called only by a majority of our board of directors, the chairman of our board of directors, our chief executive officer or our president.
- •
- Advance notice requirements for stockholder proposals and director nominations. Our amended and restated bylaws provide advance notice procedures for stockholders seeking to bring business before our annual meeting of stockholders, or to nominate candidates for election as directors at our annual meeting of stockholders. Our amended and restated bylaws also specify requirements as to the form and content of a stockholder's notice. These provisions may preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our annual meeting of stockholders.
These provisions may have the effect of delaying or preventing a change in control.
119
Transfer Agent and Registrar
The transfer agent and registrar for the common stock is . The transfer agent and registrar's address is .
Nasdaq Global Market Listing
We have applied to list our common stock on the Nasdaq Global Market under the symbol CYDX.
120
CERTAIN UNITED STATES FEDERAL TAX CONSEQUENCES
TO NON-UNITED STATES HOLDERS
The following is a general discussion of certain United States federal income and estate tax consequences of the ownership and disposition of our common stock to a non-United States holder. For purposes of this discussion, a non-United States holder is any beneficial owner that for United States federal income tax purposes is not a United States person. The term United States person means:
- •
- an individual citizen or resident of the United States;
- •
- a corporation or other entity taxable as a corporation or a partnership or entity taxable as a partnership created or organized in the United States or under the laws of the United States or any political subdivision thereof;
- •
- an estate whose income is subject to United States federal income tax regardless of its source; or
- •
- a trust (i) whose administration is subject to the primary supervision of a United States court and which has one or more United States persons who have the authority to control all substantial decisions of the trust; or (ii) which has made an election to be treated as a United States person.
If a partnership or other pass-through entity holds common stock, the tax treatment of a partner or member in the partnership or other entity will generally depend on the status of the partner or member and upon the activities of the partnership or other entity. Accordingly, we urge partnerships or other pass-through entities which hold our common stock and partners or members in these partnerships or other entities to consult their tax advisors.
This discussion assumes that non-United States holders will hold our common stock issued pursuant to this offering as a capital asset (generally, property held for investment). This discussion does not address all aspects of United States federal income taxation that may be relevant in light of a non-United States holder's special tax status or special tax situations. United States expatriates, life insurance companies, tax-exempt organizations, dealers in securities or currency, banks or other financial institutions, pension funds and investors that hold common stock as part of a hedge, straddle or conversion transaction are among those categories of potential investors that are subject to special rules not covered in this discussion. This discussion does not address any non-income tax consequences arising under the laws of any state, local or non-United States taxing jurisdiction. Furthermore, the following discussion is based on current provisions of the Internal Revenue Code, and Treasury Regulations and administrative and judicial interpretations thereof, all as in effect on the date of this prospectus, and all of which are subject to change, possibly with retroactive effect. Accordingly, we urge each non-United States holder to consult a tax advisor regarding the United States federal, state, local and non-United States income and other tax consequences of acquiring, holding and disposing of shares of our common stock. Additionally, we have not sought any ruling from the Internal Revenue Service, or IRS, with respect to statements made and conclusions reached in this discussion, and there can be no assurance that the IRS will agree with these statements and conclusions.
Dividends
We have not paid any dividends on our common stock, and all dividends accrued to date by the holders of our preferred stock will be terminated without cash payment in connection with this offering. In addition, we do not plan to pay any dividends for the foreseeable future. However, if we
121
do pay dividends on our common stock, those payments will constitute dividends for United States tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under United States federal income tax principles. To the extent those dividends exceed our current and accumulated earnings and profits, the dividends will constitute a return of capital and will first reduce a holder's basis, but not below zero, and then will be treated as gain from the sale of stock.
Any dividend (out of earnings and profits) paid to a non-United States holder of common stock generally will be subject to United States withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable tax treaty. In order to receive a reduced treaty rate, a non-United States holder must provide us with an IRS Form W-8BEN certifying qualification for the reduced rate.
Dividends received by a non-United States holder that are effectively connected with a United States trade or business conducted by the non-United States holder (and dividends attributable to a non-United States holder's permanent establishment in the United States if a tax treaty applies) are exempt from this withholding tax. In order to obtain this exemption, a non-United States holder must provide us with an IRS Form W-8ECI properly certifying this exemption. Effectively connected dividends (and dividends attributable to a permanent establishment), although not subject to withholding tax, are taxed at the same graduated rates applicable to United States persons, net of certain deductions and credits. In addition, dividends received by a corporate non-United States holder that are effectively connected with a United States trade or business of the corporate non-United States holder (and dividends attributable to a corporate non-United States holder's permanent establishment in the United States if a tax treaty applies) may also be subject to a branch profits tax at a rate of 30% (or such lower rate as may be specified in a tax treaty).
A non-United States holder of common stock that is eligible for a reduced rate of withholding tax pursuant to a tax treaty may obtain a refund of any excess amounts currently withheld if an appropriate claim for refund is filed with the IRS.
Gain on Disposition of Common Stock
A non-United States holder generally will not be subject to United States federal income tax on gain realized upon the sale or other disposition of our common stock unless:
- •
- the gain is effectively connected with a United States trade or business of the non-United States holder (or attributable to a permanent establishment in the United States if a tax treaty applies), which gain, in the case of a corporate non-United States holder, must also be taken into account for branch profits tax purposes;
- •
- the non-United States holder is an individual who holds his or her common stock as a capital asset (generally, an asset held for investment purposes) and who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met; or
- •
- our common stock constitutes a United States real property interest by reason of our status as a "United States real property holding corporation" for United States federal income tax purposes at any time within the shorter of the five-year period preceding the disposition or the holder's holding period for our common stock. We believe that we are not currently, and that we will not become, a "United States real property holding corporation" for United States federal income tax purposes.
122
Backup Withholding and Information Reporting
Generally, we must report annually to the IRS the amount of dividends paid, the name and address of the recipient, and the amount, if any, of tax withheld. A similar report is sent to the holder. Pursuant to tax treaties or other agreements, the IRS may make its reports available to tax authorities in the recipient's country of residence.
Payments of dividends or of proceeds on the disposition of stock made to a non-United States holder may be subject to backup withholding (currently at a rate of 28%) unless the non-United States holder establishes an exemption, for example, by properly certifying its non-United States status on a Form W-8BEN or another appropriate version of Form W-8. Notwithstanding the foregoing, backup withholding may apply if either we or our paying agent has actual knowledge, or reason to know, that the holder is a United States person.
Backup withholding is not an additional tax. Rather, the United States income tax liability of persons subject to backup withholding will be reduced by the amount of tax withheld. If withholding results in an overpayment of taxes, a refund may be obtained, provided that the required information is furnished to the IRS.
Federal Estate Tax
An individual non-United States holder who is treated as the owner, or has made certain lifetime transfers, of an interest in our common stock will be required to include the value thereof in his or her gross estate for United States federal estate tax purposes, and may be subject to United States federal estate tax unless an applicable estate tax treaty provides otherwise.
123
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, no public market existed for our common stock. Market sales of shares of our common stock after this offering and from time to time, and the availability of shares for future sale, may reduce the market price of our common stock. Sales of substantial amounts of our common stock, or the perception that these sales could occur, could adversely affect prevailing market prices for our common stock and could impair our future ability to obtain capital, especially through an offering of equity securities.
Based on shares outstanding on December 31, 2007, upon the closing of this offering, shares of common stock will be outstanding, assuming no outstanding options or warrants are exercised prior to the closing of this offering. Of these outstanding shares, the shares sold in this offering will be freely tradable without restrictions or further registration under the Securities Act, unless the shares are purchased by our affiliates as that term is defined under Rule 144 under the Securities Act.
The remaining shares of common stock outstanding upon the closing of this offering are restricted securities as defined under Rule 144 of the Securities Act. Restricted securities may be sold in the U.S. public market only if registered or if they qualify for an exemption from registration, including by reason of Rules 144, 144(k) or 701 under the Securities Act, which rules are summarized below. These remaining shares will be available for sale as follows:
- •
- shares of common stock will be immediately eligible for sale in the public market without restriction;
- •
- shares of common stock will be eligible for sale in the public market under Rule 144 or Rule 701, beginning 90 days after the effective date of the registration statement of which this prospectus is a part, subject to the volume, manner of sale and other limitations under those rules; and
- •
- the remaining shares of common stock will become eligible under Rule 144 for sale in the public market from time to time after the 180-day anniversary of the effective date of the registration statement of which this prospectus is a part, upon expiration of their respective holding periods.
The above list does not take into consideration the effect of the lock-up agreements described below.
Additionally, of the 12,156,634 shares of common stock issuable upon exercise of options and warrants outstanding as of December 31, 2007, approximately shares will be vested and eligible for sale 180 days after the date of this prospectus.
Rule 144
In general, under Rule 144 as currently in effect, any person who is not our affiliate, has not been our affiliate for 90 days prior to the proposed sale, and has held their shares for at least one year including the holding period of any prior owner other than one of our affiliates if permitted under Rule 144, may sell shares without restriction. In addition, beginning 90 days after the effective date of this offering, a person who has beneficially owned restricted securities for at least six months including the holding period of any prior owner other than one of our affiliates if permitted under Rule 144, may sell shares without restriction, provided the current public information requirements of Rule 144 continue to be satisfied.
124
Rule 144 also provides that affiliates relying on Rule 144 to sell shares of our common stock held for at least six months are entitled to sell a number of shares within any three-month period that does not exceed the greater of:
- •
- one percent of the number of shares of our common stock then outstanding, which will equal approximately shares immediately after this offering; and
- •
- the average weekly trading volume of our common stock on the Nasdaq Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Sales of restricted shares under Rule 144 held by our affiliate are also subject to the Rule 144 requirements regarding the manner of sale, notice, and availability of public information about us.
Rule 701
Under Rule 701, shares of our common stock acquired upon the exercise of currently outstanding options or pursuant to other rights granted under our stock plans may be resold, by:
- •
- persons other than affiliates, beginning 90 days after the effective date of this offering, subject only to the manner-of-sale provisions of Rule 144; and
- •
- our affiliates, beginning 90 days after the effective date of this offering, subject to the manner-of-sale, current public information, and filing requirements of Rule 144, in each case, without compliance with the six-month holding period requirement of Rule 144.
As of December 31, 2007, options to purchase a total of 7,695,668 shares of common stock were outstanding, of which 4,174,584 were vested. Of the total number of shares of our common stock issuable under these options, all are subject to contractual lock-up agreements with us or the underwriters.
Form S-8 Registration Statements
We intend to file one or more registration statements on Form S-8 under the Securities Act after the closing of this offering to register the shares of our common stock that are issuable pursuant to our benefit plans. These registration statements are expected to become effective upon filing. Shares covered by these registration statements will then be eligible for sale in the public markets, subject to any applicable lock-up agreements and to Rule 144 limitations applicable to affiliates.
Lock-up Agreements
Our officers and directors and holders of substantially all of our outstanding securities have agreed, subject to customary exceptions, not to, among other things, sell or otherwise transfer the economic benefit of, directly or indirectly, any shares of our common stock, or any security convertible into or exchangeable or exercisable for our common stock, without the prior written consent of Pacific Growth Equities, LLC for a period of 180 days after the date of this prospectus. The lock-up agreements signed by our security holders generally permit them, among other customary exceptions, to makebona fide gifts to their immediate family, to transfer securities to trusts for their or their immediate family's benefit and, if the security holder is a partnership, limited liability company or corporation, to transfer securities to its partners, members or stockholders. However, the recipients of these transfers must agree to be bound by the lock-up agreement for the remainder of the 180 days. Pacific Growth Equities, LLC may, in its sole discretion, at any time, and without notice, release for sale in the public market all or any portion of the shares subject to the lock-up agreements. Substantially all of the shares that are not subject to the underwriters' lock-up agreements are subject to similar contractual lock-up restrictions with us.
125
The underwriters named below have agreed to buy, subject to the terms of the purchase agreement, the number of shares listed opposite their names below. Pacific Growth Equities, LLC is acting as sole book running manager for this offering and, together with JMP Securities LLC and Fortis Securities LLC, are acting as representatives of the underwriters. The underwriters are committed to purchase and pay for all of the shares if any are purchased, other than those shares covered by the over-allotment option described below.
| Underwriter | Number of Shares | ||
|---|---|---|---|
| Pacific Growth Equities, LLC | |||
| JMP Securities LLC | |||
| Fortis Securities LLC | |||
| Total | |||
The underwriters have advised us that they propose to offer the shares to the public at $ per share. The underwriters propose to offer the shares to certain dealers at the same price less a concession of not more than $ per share. The underwriters may allow and the dealers may re-allow a concession of not more than $ per share on sales to certain other brokers and dealers. After the offering, these figures may be changed by the underwriters.
We have granted to the underwriters an over-allotment option to purchase up to an additional s hares of common stock from us at the same price to the public, and with the same underwriting discount, as set forth above. The underwriters may exercise this option any time during the 30-day period after the date of this prospectus, but only to cover over-allotments, if any. To the extent the underwriters exercise the over-allotment option, each underwriter will become obligated, subject to certain conditions, to purchase approximately the same percentage of the additional shares as it was obligated to purchase under the purchase agreement.
We estimate that the total fees and expenses payable by us, excluding underwriting discounts and commissions, will be approximately $ . The following table shows the underwriting fees to be paid to the underwriters by us in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters' over-allotment option.
| | No Exercise | Full Exercise | ||||
|---|---|---|---|---|---|---|
| Per Share | $ | $ | ||||
| Total | $ | $ | ||||
We have agreed to indemnify the underwriters against certain liabilities, including civil liabilities under the Securities Act, or to contribute to payments that the underwriters may be required to make in respect of those liabilities.
Except for sales of common stock to the underwriters in accordance with the terms of the underwriting agreement, we, all of our executive officers and directors, and certain other security holders, holding in the aggregate % of our outstanding common stock, have agreed not to sell or otherwise dispose of any shares of common stock, whether now owned or hereafter acquired, for a period of 180 days after the date of this prospectus, subject to extensions in certain cases, without the prior written consent of Pacific Growth Equities, LLC on behalf of the underwriters. Upon the expiration of these lock-up agreements, additional shares will be available for sale in the public
126
market. The agreements provide exceptions for, among other transactions, our sales in connection with the exercise of options granted and the granting of options to purchase shares of our common stock under our existing stock option plans.
Prior to the offering, there has been no established trading market for our common stock. The initial public offering price for the shares of common stock offered by this prospectus will be negotiated by us and the underwriters. The factors to be considered in determining the initial public offering price include:
- •
- the history of and the prospects for the industry in which we compete;
- •
- our past and present operations;
- •
- our historical results of operations;
- •
- our prospects for future earnings;
- •
- the recent market prices of securities of generally comparable companies; and
- •
- the general condition of the securities markets at the time of the offering and other relevant factors.
There can be no assurance that the initial public offering price of our common stock will correspond to the price at which our common stock will trade in the public market subsequent to this offering or that an active public market for our common stock will develop and continue after this offering.
To facilitate the offering, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the price of our common stock during and after the offering. Specifically, the underwriters may over-allot or otherwise create a short position in the common stock for their own account by selling more shares of common stock than have been sold to them by us. The underwriters may elect to cover any such short position by purchasing shares of common stock in the open market or by exercising the over-allotment option granted to the underwriters. In addition, the underwriters may stabilize or maintain the price of the common stock by bidding for or purchasing shares of common stock in the open market and may impose penalty bids. If penalty bids are imposed, selling concessions allowed to syndicate members or other broker dealers participating in the offering are reclaimed if shares of common stock previously distributed in the offering are repurchased, whether in connection with stabilization transactions or otherwise. The effect of these transactions may be to stabilize or maintain the market price of the common stock at a level above that which might otherwise prevail in the open market. The imposition of a penalty bid may also affect the price of the common stock to the extent that it discourages resales of the common stock. The magnitude or effect of any stabilization or other transactions is uncertain.
These transactions may be effected on the Nasdaq Global Market or otherwise and, if commenced, may be discontinued at any time. In connection with this offering, some underwriters (and selling group members) may also engage in passive market making transactions in our common stock on the Nasdaq Global Market. Passive market making consists of displaying bids on the Nasdaq Global Market limited by the prices of independent market makers and effecting purchases limited by those prices in response to order flow. Rule 103 of Regulation M promulgated by the SEC limits the amount of net purchases that each passive market maker may make and the displayed size of each bid. Passive market making may stabilize the market price of our common stock at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
127
From time to time in the ordinary course of their respective business, certain of the underwriters and their affiliates may in the future engage in commercial banking or investment banking transactions with us and our affiliates.
In relation to each member state of the European Economic Area which has implemented the Prospectus Directive (each, a relevant member state), each underwriter has represented and agreed that with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state, or the relevant implementation date, it has not made and will not make an offer of shares of our common stock to the public in this offering in that relevant member state prior to the publication of a prospectus in relation to such shares which has been approved by the competent authority in that relevant member state or, where appropriate, approved in another relevant member state and notified to the competent authority in that relevant member state, all in accordance with the Prospectus Directive. However, with effect from and including the relevant implementation date, it may make an offer of shares of our common stock to the public in that relevant member state at any time:
- •
- to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities;
- •
- to any legal entity which has two or more of (i) an average of at least 250 employees during the last financial year, (ii) a total balance sheet of more than €43,000,000 and (iii) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts;
- •
- to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive) subject to obtaining the prior consent of the underwriters; or
- •
- in any other circumstances which do not require the publication by the issuer of a prospectus pursuant to Article 3 of the Prospectus Directive,
provided that no such offer of shares shall result in a requirement for the publication of a prospectus pursuant to Article 3 of the Prospectus Directive or any measure implementing the Prospectus Directive in a relevant member state and each person who initially acquires any share or to whom any offer is made under this offering will be deemed to have represented, acknowledged and agreed that it is a "qualified investor" within the meaning of Article 2 (1)(e) of the Prospectus Directive. For the purposes of this provision, the expression an "offer of shares of our common stock to the public" in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the shares to be offered so as to enable an investor to decide to purchase or subscribe such shares, as may be varied in that relevant member state by any measure implementing the Prospectus Directive in that member state, and the expression "Prospectus Directive" means Directive 2003/71/ EC and includes any relevant implementing measure in each relevant member state.
The shares have not been and will not be offered to the public within the meaning of the German Sales Prospectus Act (Verkaufsprospektgesetz) or the German Investment Act (Investmentgesetz). The shares have not been and will not be listed on a German exchange. No sales prospectus pursuant to the German Sales Prospectus Act has been or will be published or circulated in Germany or filed with the German Federal Financial Supervisory Authority (Bundesanstalt für Finanzdienstleistungsaufsicht) or any other governmental or regulatory authority in Germany. This prospectus does not constitute an offer to the public in Germany and it does not serve for public distribution of the shares in Germany. Neither this prospectus, nor any other document issued in connection with this offering, may be issued
128
or distributed to any person in Germany except under circumstances which do not constitute an offer to the public within the meaning of the German Sales Prospectus Act or the German Investment Act.
Each underwriter has represented, warranted and agreed that: (i) it has not offered or sold and, prior to the expiry of a period of six months from the closing date, will not offer or sell any shares to persons in the United Kingdom except to persons whose ordinary activities involve them in acquiring, holding, managing or disposing of investments (as principal or agent) for the purposes of their businesses or otherwise in circumstances which have not resulted and will not result in an offer to the public in the United Kingdom within the meaning of the Public Offers of Securities Regulations 1995; (ii) it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000, or FSMA) received by it in connection with the issue or sale of any shares in circumstances in which section 21(1) of the FSMA does not apply to our company and (iii) it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares in, from or otherwise involving the United Kingdom.
The shares offered pursuant to this prospectus will not be offered, directly or indirectly, to the public in Switzerland and this prospectus does not constitute a public offering prospectus as that term is understood pursuant to Article 652a or Article 1156 of the Swiss Federal Code of Obligations. We have not applied for a listing of the shares being offered pursuant to this prospectus on the SWX Swiss Exchange or on any other regulated securities market, and consequently, the information presented in this prospectus does not necessarily comply with the information standards set out in the relevant listing rules. The shares being offered pursuant to this prospectus have not been registered with the Swiss Federal Banking Commission as foreign investment funds, and the investor protection afforded to acquirers of investment fund certificates does not extend to acquirers of securities.
If you purchase shares of common stock offered in this prospectus, you may be required to pay stamp taxes and other charges under the laws and practices of the country of purchase, in addition to the offering price listed on the cover page of this prospectus.
The validity of the issuance of the shares of common stock offered by this prospectus will be passed upon for us by our counsel, Cooley Godward Kronish LLP, Palo Alto, California. Certain legal matters will be passed upon for the underwriters by Goodwin Procter LLP, Menlo Park, California.
The consolidated financial statements of CyDex Pharmaceuticals, Inc. as of December 31, 2006 and 2007 and for each of the years in the three-year period ended December 31, 2007, have been included herein in reliance upon the report of KPMG LLP, independent registered public accounting firm, appearing elsewhere herein, and upon the authority of said firm as experts in accounting and auditing. The audit report covering the December 31, 2007 consolidated financial statements refers to the adoption of Statement of Financial Accounting Standards No. 123 (revised 2004),Share-Based Payment.
129
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1, including all amendments and supplements thereto, under the Securities Act that registers the shares of our common stock to be sold in this offering. The registration statement, including the attached exhibits and schedules, contains additional relevant information about us and our capital stock. The rules and regulations of the SEC allow us to omit from this prospectus certain information included in the registration statement. For further information about us and our common stock, you should refer to the registration statement and the exhibits and schedules filed with the registration statement. With respect to the statements contained in this prospectus regarding the contents of any agreement or any other document, in each instance, the statement is qualified in all respects by the complete text of the agreement or document, a copy of which has been filed as an exhibit to the registration statement. In addition, upon the closing of this offering, we will file reports, proxy statements and other information with the SEC under the Securities Exchange Act of 1934, as amended. You may obtain copies of this information by mail from the Public Reference Section of the SEC, 100 F Street, NE, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy statements and other information about issuers that file electronically with the SEC. The address of that site is www.sec.gov.
We intend to provide our stockholders with annual reports containing financial statements that have been examined and reported on, with an opinion expressed by an independent registered public accounting firm, and to file with the SEC quarterly reports containing unaudited financial data for the first three quarters of each year.
130
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | Page | |
|---|---|---|
| Report of Independent Registered Public Accounting Firm | F-2 | |
Consolidated Balance Sheets as of December 31, 2006 and 2007 | F-3 | |
Consolidated Statements of Operations for the years ended December 31, 2005, 2006 and 2007 | F-4 | |
Consolidated Statements of Stockholders' Deficit for the years ended December 31, 2005, 2006 and 2007 | F-5 | |
Consolidated Statements of Cash Flows for the years ended December 31, 2005, 2006 and 2007 | F-6 | |
Notes to Consolidated Financial Statements | F-7 |
F-1
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
CyDex Pharmaceuticals, Inc.:
We have audited the accompanying consolidated balance sheets of CyDex Pharmaceuticals, Inc. and subsidiary as of December 31, 2006 and 2007, and the related consolidated statements of operations, stockholders' deficit, and cash flows for each of the years in the three-year period ended December 31, 2007. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of CyDex Pharmaceuticals, Inc. and subsidiary as of December 31, 2006 and 2007, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Notes 2 and 11 to the consolidated financial statements, effective January 1, 2006, the Company adopted Statement of Financial Accounting Standards No. 123 (revised 2004),Share-Based Payment.
/s/ KPMG LLP
Kansas City, Missouri
March 11, 2008, except as
to Note 15, which is as of
May 12, 2008.
F-2
CYDEX PHARMACEUTICALS, INC.
Consolidated Balance Sheets
(in thousands, except share and per share amounts)
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2006 | 2007 | |||||
| Assets | |||||||
Current assets: | |||||||
| Cash and cash equivalents | $ | 3,596 | $ | 809 | |||
| Short-term investments | 7,445 | 7,300 | |||||
| Accounts receivable, net | 1,466 | 2,234 | |||||
| Inventory | 1,986 | 2,691 | |||||
| Other current assets | 99 | 595 | |||||
| Total current assets | 14,592 | 13,629 | |||||
| Property and equipment, net | 549 | 542 | |||||
| Product and license rights, net | 2,728 | 2,036 | |||||
| Other noncurrent assets | 1,015 | 2,225 | |||||
| Total assets | $ | 18,884 | $ | 18,432 | |||
Liabilities and Stockholders' Deficit | |||||||
Current liabilities: | |||||||
| Accounts payable | $ | 337 | $ | 1,498 | |||
| Accrued liabilities | 638 | 1,276 | |||||
| Deferred revenue | 7 | 34 | |||||
| Income tax payable | 53 | — | |||||
| Total current liabilities | 1,035 | 2,808 | |||||
| Deferred revenue | 17 | 56 | |||||
| Total liabilities | 1,052 | 2,864 | |||||
| Commitments and contingent liabilities (Note 13) | |||||||
Series A convertible preferred stock, $0.01 par value: authorized 16,468,622 shares; issued and outstanding 16,468,618 shares; net of unaccreted issuance costs of $250 and $173 at December 31, 2006 and 2007, respectively, and unaccreted beneficial conversion feature of $6,077 and $4,045 at December 31, 2006 and 2007, respectively; liquidation preference of $17,354 and $18,194 at December 31, 2006 and 2007, respectively | 11,027 | 13,976 | |||||
| Series B convertible preferred stock, $0.01 par value: authorized 63,197,020 shares; issued and outstanding 63,197,019 shares; net of unaccreted issuance costs of $625 and $433 at December 31, 2006 and 2007, respectively; liquidation preference of $28,368 and $29,558 at December 31, 2006 and 2007, respectively | 19,242 | 20,624 | |||||
| Stockholders' deficit: | |||||||
| Series A-1 convertible preferred stock, $0.01 par value: authorized, issued and outstanding 2,000,000 shares; liquidation preference of $2,337 and $2,477 at December 31, 2006 and 2007, respectively | 20 | 20 | |||||
Common stock, $0.01 par value: authorized 103,000,000 shares; issued 6,424,211 shares and 7,380,438 shares and outstanding 6,324,211 shares and 7,280,438 shares at December 31, 2006 and 2007, respectively | 64 | 74 | |||||
| Additional paid-in capital | 7,019 | 3,001 | |||||
| Accumulated deficit | (19,340 | ) | (21,927 | ) | |||
| Treasury stock, at cost (100,000 shares of common stock) | (200 | ) | (200 | ) | |||
| Total stockholders' deficit | (12,437 | ) | (19,032 | ) | |||
| Total liabilities and stockholders' deficit | $ | 18,884 | $ | 18,432 | |||
See accompanying notes to consolidated financial statements.
F-3
CYDEX PHARMACEUTICALS, INC.
Consolidated Statements of Operations
(in thousands, except share and per share amounts)
| | Years Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | ||||||||
| Revenue: | |||||||||||
| Milestone and license | $ | 1,947 | $ | 1,982 | $ | 1,172 | |||||
| Material sales | 4,164 | 5,671 | 6,819 | ||||||||
| Royalties | 2,511 | 2,989 | 3,829 | ||||||||
| Contract research | 158 | 845 | 683 | ||||||||
| Other | 39 | 93 | 242 | ||||||||
| Total revenue | 8,819 | 11,580 | 12,745 | ||||||||
| Operating expenses: | |||||||||||
| Cost of sales | 2,566 | 2,482 | 2,508 | ||||||||
| Research and development | 4,419 | 4,435 | 9,078 | ||||||||
| Selling, general and administrative | 2,488 | 2,681 | 4,328 | ||||||||
| Total operating expenses | 9,473 | 9,598 | 15,914 | ||||||||
| Operating income (loss) | (654 | ) | 1,982 | (3,169 | ) | ||||||
| Other income (expense): | |||||||||||
| Interest income | 223 | 428 | 582 | ||||||||
| Other, net | (7 | ) | (2 | ) | — | ||||||
| Total other income | 216 | 426 | 582 | ||||||||
| Income (loss) before income taxes | (438 | ) | 2,408 | (2,587 | ) | ||||||
| Provision for income taxes | — | 53 | — | ||||||||
| Net income (loss) | (438 | ) | 2,355 | (2,587 | ) | ||||||
| Preferred dividends and accretion | 4,441 | 4,455 | 4,471 | ||||||||
| Net loss attributable to common stockholders | $ | (4,879 | ) | $ | (2,100 | ) | $ | (7,058 | ) | ||
| Basic and diluted net loss per common share | $ | (0.87 | ) | $ | (0.35 | ) | $ | (1.04 | ) | ||
| Shares used to compute basic and diluted net loss per common share | 5,617,270 | 6,082,357 | 6,798,681 | ||||||||
See accompanying notes to consolidated financial statements.
F-4
CYDEX PHARMACEUTICALS, INC.
Consolidated Statements of Stockholders' Deficit
(in thousands)
| | Series A-1 convertible preferred stock | Common stock | Additional paid-in capital | Accumulated deficit | Treasury stock | Total stockholders' deficit | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Balance at January 1, 2005 | $ | 20 | $ | 54 | $ | 15,400 | $ | (21,257 | ) | $ | (200 | ) | $ | (5,983 | ) | ||||
| Issuance of common stock | — | 3 | 49 | — | — | 52 | |||||||||||||
| Accretion of Series A convertible preferred stock issuance costs and beneficial conversion feature | — | — | (2,109 | ) | — | — | (2,109 | ) | |||||||||||
| Accretion of Series B convertible preferred stock issuance costs | — | — | (162 | ) | — | — | (162 | ) | |||||||||||
| Accrued dividends on Series A convertible preferred stock | — | — | (840 | ) | — | — | (840 | ) | |||||||||||
| Accrued dividends on Series B convertible preferred stock | — | — | (1,190 | ) | — | — | (1,190 | ) | |||||||||||
| Stock-based compensation | — | — | 49 | — | — | 49 | |||||||||||||
| Net loss | — | — | — | (438 | ) | — | (438 | ) | |||||||||||
| Balance at December 31, 2005 | 20 | 57 | 11,197 | (21,695 | ) | (200 | ) | (10,621 | ) | ||||||||||
| Issuance of common stock | — | 7 | 97 | — | — | 104 | |||||||||||||
| Accretion of Series A convertible preferred stock issuance costs and beneficial conversion feature | — | — | (2,109 | ) | — | — | (2,109 | ) | |||||||||||
| Accretion of Series B convertible preferred stock issuance costs | — | — | (176 | ) | — | — | (176 | ) | |||||||||||
| Accrued dividends on Series A convertible preferred stock | — | — | (840 | ) | — | — | (840 | ) | |||||||||||
| Accrued dividends on Series B convertible preferred stock | — | — | (1,190 | ) | — | — | (1,190 | ) | |||||||||||
| Stock-based compensation | — | — | 40 | — | — | 40 | |||||||||||||
| Net income | — | — | — | 2,355 | — | 2,355 | |||||||||||||
| Balance at December 31, 2006 | 20 | 64 | 7,019 | (19,340 | ) | (200 | ) | (12,437 | ) | ||||||||||
| Issuance of common stock | — | 10 | 110 | — | — | 120 | |||||||||||||
| Accretion of Series A convertible preferred stock issuance costs and beneficial conversion feature | — | — | (2,109 | ) | — | — | (2,109 | ) | |||||||||||
| Accretion of Series B convertible preferred stock issuance costs | — | — | (192 | ) | — | — | (192 | ) | |||||||||||
| Accrued dividends on Series A convertible preferred stock | — | — | (840 | ) | — | — | (840 | ) | |||||||||||
| Accrued dividends on Series B convertible preferred stock | — | — | (1,190 | ) | — | — | (1,190 | ) | |||||||||||
| Stock-based compensation | — | — | 203 | — | — | 203 | |||||||||||||
| Net loss | — | — | — | (2,587 | ) | — | (2,587 | ) | |||||||||||
| Balance at December 31, 2007 | $ | 20 | $ | 74 | $ | 3,001 | $ | (21,927 | ) | $ | (200 | ) | $ | (19,032 | ) | ||||
See accompanying notes to consolidated financial statements.
F-5
CYDEX PHARMACEUTICALS, INC.
Consolidated Statements of Cash Flows
(in thousands)
| | Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||||||
| Cash flows from operating activities: | ||||||||||
| Net income (loss) | $ | (438 | ) | $ | 2,355 | $ | (2,587 | ) | ||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||
| Depreciation and amortization of property and equipment | 170 | 207 | 234 | |||||||
| Loss on disposal of property and equipment | — | 3 | 1 | |||||||
| Amortization of intangible assets | 692 | 692 | 692 | |||||||
| Stock-based compensation | 49 | 40 | 203 | |||||||
| Changes in assets and liabilities: | ||||||||||
| Accounts receivable | (1,325 | ) | 709 | (768 | ) | |||||
| Inventory | (431 | ) | (329 | ) | (705 | ) | ||||
| Other current assets | 1,331 | (23 | ) | (496 | ) | |||||
| Accounts payable | (26 | ) | 319 | 853 | ||||||
| Accrued liabilities | (425 | ) | (208 | ) | 513 | |||||
| Deferred revenue | 105 | (93 | ) | 66 | ||||||
| Income tax payable | — | 53 | (53 | ) | ||||||
| Net cash provided by (used in) operating activities | (298 | ) | 3,725 | (2,047 | ) | |||||
| Cash flows from investing activities: | ||||||||||
| Additions to property and equipment | (43 | ) | (321 | ) | (228 | ) | ||||
| Purchase of short-term investments | (8,871 | ) | (8,270 | ) | (10,394 | ) | ||||
| Proceeds from sale or maturity of short-term investments | 9,086 | 6,910 | 10,539 | |||||||
| Net cash provided by (used in) investing activities | 172 | (1,681 | ) | (83 | ) | |||||
| Cash flows from financing activities: | ||||||||||
| Initial public offering costs | — | — | (777 | ) | ||||||
| Proceeds from exercise of common stock options | 52 | 104 | 120 | |||||||
| Net cash provided by (used in) financing activities | 52 | 104 | (657 | ) | ||||||
| Net increase (decrease) in cash and cash equivalents | (74 | ) | 2,148 | (2,787 | ) | |||||
| Cash and cash equivalents at beginning of year | 1,522 | 1,448 | 3,596 | |||||||
| Cash and cash equivalents at end of year | $ | 1,448 | $ | 3,596 | $ | 809 | ||||
| Supplemental disclosure of noncash investing and financing activities | ||||||||||
| Change in accrued liabilities for additions to property and equipment | $ | (40 | ) | $ | 40 | $ | — | |||
| Change in accounts payable and accrued liabilities for initial public offering costs incurred | $ | — | $ | — | $ | 433 | ||||
See accompanying notes to consolidated financial statements.
F-6
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements
(1) Business
CyDex Pharmaceuticals, Inc., a Delaware corporation, and subsidiary (collectively, the Company) is a specialty pharmaceutical company focused on the development and commercialization of drugs specifically designed to address limitations of current therapies in selected established markets. The Company has developed a broad portfolio of product candidates utilizing its drug formulation technology using Captisol® cyclodextrins. Captisol cyclodextrins are a patent protected, specifically modified family of cyclodextrins designed to improve solubility, stability, bioavailability, safety and dosing of a number of active pharmaceutical ingredients (APIs).
(2) Summary of Significant Accounting Policies
(a) Consolidation Policy
The consolidated financial statements include the accounts of CyDex Pharmaceuticals, Inc. and its wholly-owned subsidiary, CyDex, L.C., a Kansas limited liability company. All intercompany balances and transactions have been eliminated.
(b) Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and the accompanying notes. Actual results could differ from those estimates.
(c) Cash and Cash Equivalents
Cash and cash equivalents at December 31, 2006 and 2007 consist of cash and short-term securities with maturity dates of three months or less at the time of purchase.
(d) Short-Term Investments
Investments with remaining maturities of twelve months or less are classified as short-term investments. The Company classifies its marketable securities as available-for-sale in accordance with Statement of Financial Accounting Standards (SFAS) No. 115,Accounting for Certain Investments in Debt and Equity Securities. The Company's short-term investments consist of certificates of deposit, corporate bonds and auction-rate preferred securities, which are debt instruments having longer-dated (in most cases, many years) legal maturities, but with interest rates that are generally reset every seven days. Because these securities are frequently repriced, they often trade in the market on a par-in, par-out basis. Because the Company regularly liquidates its investments in these securities for reasons including, among others, changes in market interest rates and changes in the availability of and the yield on alternative investments, the Company has classified these securities as available-for-sale securities. These investments are carried at cost, which approximates fair value. There have been insignificant realized and unrealized gains or losses on these investments.
(e) Concentration Risk
The Company invests its excess cash and short-term investments in accordance with its investment policy, with an objective to ensure both liquidity and safety of principal. The policy limits investments to certain types of instruments issued by the U.S. government and institutions with strong investment
F-7
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
grade credit ratings, and places restrictions on their terms and concentrations by type and issuer to reduce the Company's credit risk.
Accounts receivable from two pharmaceutical companies were approximately 58% and 29% of total accounts receivable at December 31, 2006 and 45% and 20% of total accounts receivable at December 31, 2007.
Revenue from two pharmaceutical companies amounted to 37% and 13% of total revenue for the year ended December 31, 2005. Revenue from three pharmaceutical companies amounted to 27%, 19% and 13% of total revenue for the year ended December 31, 2006. Revenue from two pharmaceutical companies amounted to 40% and 15% of total revenue for the year ended December 31, 2007.
The Company obtains Captisol from a sole source supplier. If this supplier is not able to supply the requested amounts of Captisol, the Company would be unable to continue to derive revenue from the sale of Captisol until it obtained an alternative source, which may take a considerable length of time.
(f) Allowance for Doubtful Accounts
The Company maintains an allowance for doubtful accounts based on the best estimate of the amount of probable losses in the Company's existing accounts receivable. When determining the allowance for doubtful accounts, several factors are taken into consideration, including historical write-off experience and review of specific customer accounts for collectibility. Account balances are charged off against the allowance after collection efforts have been exhausted and the potential for recovery is considered remote. The allowance for doubtful accounts was immaterial as of December 31, 2006 and 2007.
(g) Inventory
Inventory is stated at the lower of cost or market. The Company determines cost using the first-in, first-out method. The Company analyzes its inventory levels periodically and writes down inventory to its net realizable value if it has become obsolete, has a cost basis in excess of its expected net realizable value or is in excess of expected requirements. For the periods presented, the Company does not consider any of its inventory to be impaired.
(h) Other Current Assets
Other current assets include prepaid inventory, nonrefundable advance payments for research and development activities, interest receivable and other advance payments. At December 31, 2006 and 2007, prepaid research and development costs were $0 and $374,853, respectively. Such amounts will be recognized as expense when the related services are performed.
(i) Property and Equipment
Property and equipment are recorded at cost and are depreciated on a straight-line basis over their estimated useful lives. Leasehold improvements are amortized over the shorter of the lease term or the estimated useful life of the related asset. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is recorded in the consolidated statement of operations. Repair and maintenance costs are charged to expense as incurred.
F-8
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(j) Product and License Rights
Product and license rights consist of amounts paid to the University of Kansas (the University) under a license agreement entered into in 1993 and amended in 2004. The original amount paid to the University of $450,000 is being amortized on a straight-line basis over 16 years, which was the remaining life of the patent at the time of the original license agreement. In 2004, the Company paid the University $4,424,500 in cash and issued to the University 2,000,000 shares of Series A-1 convertible preferred stock (Series A-1 Stock) having an estimated fair value of $370,000 in consideration for, among other things, the elimination of the Company's obligation to make future royalty payments as required under the license agreement. Of the total consideration paid of $4,794,500, $534,500 was recorded as a reduction of previously accrued royalty costs, and the additional license consideration of $4,260,000 was capitalized and is being amortized on a straight-line basis over 77 months, which was the remaining life of the patent at the time of the amendment.
(k) Other Noncurrent Assets
The Company outsources the production of Captisol to Hovione FarmaCiencia SA (Hovione) through its agent Hovione LLC. Hovione is a major supplier of APIs and API intermediates. Other assets include a $1,000,000 deferred charge for an amount paid to Hovione LLC related to a production agreement, as amended, for Captisol that ends in 2014. The Company has ongoing minimum annual purchase commitments under the agreement and is required to purchase a total of $15,000,000 of Captisol over the term of the agreement. Per the agreement, the amount paid for Captisol over the second half of the total commitment will be proportionately reduced to allow for the recovery of the deferred charge. The Company's ability to realize this deferred charge is based upon its purchase forecasts during the term of the agreement, and if the actual purchases are less than those forecasts, the Company may not be able to realize this asset. The pricing for the future inventory purchases is based upon purchasing levels and is subject to adjustment for changes in the exchange rate between the United States dollar and the Euro, which the Company and Hovione will share evenly, and to certain consumer price index adjustments.
(l) Income Taxes
The Company accounts for income taxes in accordance with SFAS No. 109,Accounting for Income Taxes (SFAS No. 109). SFAS No. 109 requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences and carryforwards are expected to be recovered or settled. A valuation allowance is recognized to reduce deferred tax assets to the amount that will more likely than not be realized. In assessing the need for a valuation allowance, the Company considers all available evidence including past operating results, estimates of future taxable income and the feasibility of ongoing tax strategies.
(m) Financial Instruments
The fair values of cash and cash equivalents, short-term investments, accounts receivable, accounts payable and accrued liabilities are estimated to approximate their carrying values because of the short maturity of these financial instruments.
F-9
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(n) Revenue Recognition
The Company enters into license agreements with pharmaceutical companies for the use of Captisol in their product development activities and for commercialization of Captisol-Enabled products. The terms of the agreements may include nonrefundable license fees, nonrefundable payments based on the achievement of certain milestones, and royalties on any sales of products containing Captisol. The Company generally also enters into supply agreements with many of its customers that include pricing for the exclusive supply of customers' requirements of Captisol by the Company. The pricing of Captisol frequently gives consideration to purchase volumes of individual customers. However, there are no minimum purchase commitments by the customer. Revenue is recognized when all four of the following general criteria are met: (1) the Company has persuasive evidence that an arrangement exists, (2) the fees are fixed and determinable, (3) shipment has occurred (including passage of title and risk of loss) and (4) collection is reasonably assured. The Company's credit and exchange policy includes provisions for return of its product when it (1) does not meet specifications or (2) is damaged in shipment (in limited circumstances where title does not transfer until delivery). Sales returns have been insignificant in 2005, 2006 and 2007.
The Company also analyzes its arrangements to determine whether there are multiple elements in the agreement that should be separated and accounted for individually or as a single unit of accounting in accordance with Emerging Issues Task Force (EITF) Issue No. 00-21,Revenue Arrangements with Multiple Deliverables.
The majority of the Company's revenue arrangements involve the bundling of a license with the option to purchase manufactured product. Licenses are granted to pharmaceutical companies for the use of Captisol in the development of pharmaceutical compounds. The licenses may be granted for the use of the Captisol product for all phases of clinical trials and through commercial availability of the host drug or may be limited to certain phases of the clinical trial process. The Company believes that its licenses have stand-alone value at the outset of an arrangement because the customer obtains the right to use Captisol in its formulations without any additional input by the Company, and in a hypothetical stand-alone transaction, the customer would be able to procure inventory from another manufacturer in the absence of contractual provisions for exclusive supply by the Company. The Company evaluates the pricing of manufactured product in its arrangements to determine whether such arrangements are priced at fair value or include a significant potential incremental discount. If a significant potential incremental discount has been determined to exist, a portion of the nonrefundable license fee is recognized over the lesser of the term of the agreement or the period remaining through 2012, which is the earliest date the Company would anticipate a possible generic competitor. Nonrefundable license fees, for which there are no future development obligations and for which the Company provides no significant potential incremental discount on the pricing of manufactured products, are recognized on the execution of the agreement if all other general conditions for revenue recognition have been met. Milestone payments, which are nonrefundable, on license agreements are recognized when earned if no significant potential incremental discount has been determined to exist related to the pricing of manufactured product. If a significant potential incremental discount has been determined to exist, these milestone payments are deferred at a percentage equal to the deferred percentage of the remaining nonrefundable license fee. The Company recognizes license revenue in its multiple-element revenue arrangements using the residual method. Under the residual method, revenue may be allocated to a significant potential incremental discount for the customer's option to purchase manufactured product, and the residual amount of license revenue is recognized after all other general conditions for revenue recognition have been met. To the extent that revenue arrangements contain a research and
F-10
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
development element that has stand-alone value, the Company determines fair value of the research and development element by considering competitor pricing for comparable services. The Company recognizes research and development revenue, including any payments received in advance of performance, on a straight-line basis over the life of the contract, with the cumulative amount of revenue recognized on a contract limited to the payments received or amounts currently due and payable as of that point in time. The Company does not use the substantive milestone method to recognize research and development revenue due to the lack of historical documentation to support the substantive efforts involved in the achievement of milestones, as well as the relatively short time period covered by such arrangements.
Material sales revenue is recognized upon transfer of title, which normally passes to the buyer upon shipment to the customer. Sales royalties are recognized as earned. Contract research revenue is recognized on a straight-line basis over the life of the contract, with the cumulative amount of revenue recognized on a contract at a point in time not to exceed the payments earned as of that point in time. Payments, if any, received in advance of performance under an agreement are deferred and recognized as revenue when earned. Other revenue consists of reimbursements received from pharmaceutical companies for patent expenses, research and development and shipping costs related to material sales.
(o) Research and Development
Research and development expenses are recorded in the statements of operations as incurred and include direct costs of research scientists and equipment, contracted costs, and an allocation of certain overhead expenses.
(p) Stock-based Compensation
Prior to 2006, the Company applied the intrinsic value method as prescribed in Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees (APB No. 25), and related interpretations, in accounting for stock options granted under the Company's 1997 Equity Compensation Plan. Under the intrinsic value method, no stock compensation is recognized if the exercise price of the Company's employee stock options was equal to or greater than the fair value of the underlying stock on the date of the grant.
Effective January 1, 2006, the Company adopted SFAS No. 123 (revised 2004),Share-Based Payment (SFAS No. 123R). This statement supersedes SFAS No. 123,Accounting for Stock-Based Compensation (SFAS No. 123), and APB No. 25. SFAS No. 123R requires that all stock-based compensation be recognized as an expense in the consolidated financial statements and that such cost be measured at the fair value of the award on the date of grant if classified as an equity award. As the Company previously used the minimum value method, as defined by SFAS No. 123, for purposes of measuring the fair value of stock options, SFAS No. 123R was adopted using the prospective method of application, which requires the Company to recognize stock compensation for all awards granted after January 1, 2006 and any existing awards modified after that date. For stock options granted after January 1, 2006, the Company recognizes stock compensation based on the estimated grant date fair value using the Black-Scholes option-pricing model.
(q) Net Loss Per Share
Basic net loss per common share (EPS) is calculated by dividing net loss attributable to common stockholders by the weighted average shares of common stock outstanding during the period. Diluted
F-11
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
EPS reflects the potential dilution of securities that could share in the net loss attributable to common stockholders, including the effect of dilution to net income of convertible securities, stock options and warrants.
The Company incurred a net loss attributable to common stockholders for the years ended December 31, 2005, 2006 and 2007. Potential dilutive common shares included both shares issuable upon the assumed exercise of outstanding employee stock options and warrants and the assumed conversion of outstanding convertible preferred stock. Such shares were not included in the net loss per common share calculation, as their inclusion would have been antidilutive.
(r) Segment Information
SFAS No. 131,Disclosures about Segments of an Enterprise and Related Information, establishes standards for reporting information about operating segments in annual financial statements and in interim financial reports issued to stockholders. Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated on a regular basis by the chief operating decision maker in deciding how to allocate resources to an individual segment and in assessing performance of the segment. The Company evaluates performance and makes decisions based on one segment.
(s) Recently Issued Accounting Standards
In September 2006, the Securities and Exchange Commission (SEC) issued Staff Accounting Bulletin No. 108,Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements (SAB No. 108), which addresses how uncorrected errors in previous years should be considered when quantifying errors in current year financial statements. The SEC staff believes registrants must quantify errors using both a balance sheet and a statement of operations approach and evaluate whether either approach results in quantifying a misstatement that, when all relevant quantitative and qualitative factors are considered, is material. The Company adopted SAB No. 108 as of December 31, 2006, retroactive to January 1, 2006, with no impact on its operating results or financial position.
In June 2006, the Financial Accounting Standards Board (FASB) issued FASB Interpretation No. 48,Accounting for Uncertainty in Income Taxes (FIN 48). FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an entity's financial statements and prescribes a threshold of more-likely-than-not for recognition of tax benefits of certain tax positions taken or expected to be taken in a tax return. FIN 48 also provides related guidance on measurement, derecognition, classification, interest and penalties, and disclosure. On January 1, 2007, the Company adopted FIN 48, which did not have any impact on its consolidated financial statements.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements (SFAS No. 157). SFAS No. 157 defines fair value as used in numerous accounting pronouncements, establishes a framework for measuring fair value in U.S. generally accepted accounting principles and expands disclosure related to the use of fair value measures in financial statements. SFAS No. 157 does not expand the use of fair value measures in financial statements. SFAS No. 157 emphasizes that fair value is a market-based measurement and not an entity-specific measurement, based on an exchange transaction in which the entity sells an asset or transfers a liability. SFAS No. 157 establishes a fair value hierarchy from observable market data at the highest level, to fair value based on an entity's own fair value assumptions, at the lowest level. SFAS No. 157 became effective for the Company as of January 1,
F-12
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
2008. Per FASB Staff Position 157-2,Effective Date of FASB Statement No. 157, implementation of SFAS No. 157 to certain nonfinancial assets and nonfinancial liabilities will be deferred until annual reporting periods beginning after November 15, 2008 and interim periods within those annual reporting periods. The Company is currently evaluating the impact, if any, that the adoption of SFAS No. 157 will have on its financial position and results of operations.
In February 2007, the FASB issued SFAS No. 159,The Fair Value Option for Financial Assets and Financial Liabilities (SFAS No. 159). SFAS No. 159 permits, but does not require, entities to measure certain financial instruments and other assets and liabilities at fair value on an instrument-by-instrument basis. Unrealized gains and losses on items for which the fair value option has been elected should be recognized in earnings at each subsequent reporting date. SFAS No. 159 is effective for the Company as of January 1, 2008 and cannot be adopted early unless SFAS No. 157 is also adopted. The Company is currently evaluating the impact, if any, that the adoption of SFAS No. 159 may have on its financial position and results of operations.
In June 2007, the FASB issued EITF Issue No. 07-3,Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities (EITF Issue No. 07-3), which applies to companies involved in research and development activities that make nonrefundable advance payments for goods that will be used or for services that will be performed on future research and development activities. EITF Issue No. 07-3 requires that nonrefundable advance payments for future research and development activities be deferred and recognized as an expense as goods are delivered or the related services are performed. EITF Issue No. 07-3 is effective for financial statements issued for fiscal years beginning after December 15, 2007 and interim periods within those fiscal years. The Company does not expect the adoption of EITF Issue No. 07-3 to have a material impact on its financial position and results of operations.
In December 2007, the FASB issued EITF Issue No. 07-1,Accounting for Collaborative Arrangements (EITF Issue No. 07-1), which applies to collaborative arrangements that are conducted by the participants without the creation of a separate legal entity for the arrangements and clarifies, among other things, how to determine whether a collaborative agreement is within the scope of this issue. EITF Issue No. 07-1 is effective for financial statements issued for fiscal years beginning after December 15, 2008. The Company does not expect the adoption of EITF Issue No. 07-1 to have a material impact on its financial position and results of operations.
(3) Short-Term Investments
Short-term investments classified as available-for-sale are reported at cost. At December 31, 2006 and 2007, the estimated fair value of short-term investments approximated their cost. Short-term investments consist of the following at December 31, 2006 and 2007 (in thousands):
| | December 31, | |||||
|---|---|---|---|---|---|---|
| | 2006 | 2007 | ||||
| Certificate of deposits | $ | 2,845 | $ | 1,500 | ||
| Auction-rate preferred securities | 4,600 | 5,800 | ||||
| $ | 7,445 | $ | 7,300 | |||
F-13
Notes to Consolidated Financial Statements (Continued)
(4) Property and Equipment
Property and equipment consist of the following at December 31, 2006 and 2007 (in thousands):
| | | December 31, | |||||||
|---|---|---|---|---|---|---|---|---|---|
| | Estimated Useful Life (Years) | ||||||||
| | 2006 | 2007 | |||||||
| Furniture and equipment | 3 to 7 | $ | 206 | $ | 210 | ||||
| Information technology equipment | 3 to 7 | 247 | 250 | ||||||
| Laboratory equipment | 5 to 7 | 722 | 907 | ||||||
| Leasehold improvements | 2 to 5 | 309 | 312 | ||||||
| 1,484 | 1,679 | ||||||||
| Less: accumulated depreciation and amortization | (935 | ) | (1,137 | ) | |||||
| $ | 549 | $ | 542 | ||||||
(5) Accrued Liabilities
Accrued liabilities consist of the following at December 31, 2006 and 2007 (in thousands):
| | December 31, | |||||
|---|---|---|---|---|---|---|
| | 2006 | 2007 | ||||
| Accrued employment costs | $ | 483 | $ | 403 | ||
| Accrued professional fees | 33 | 406 | ||||
| Accrued facilities | 40 | 22 | ||||
| Accrued payables | 82 | 445 | ||||
| $ | 638 | $ | 1,276 | |||
(6) Severance Payable
Severance expense for the years ended December 31, 2005, 2006 and 2007 was $431,589, $158,786 and $98,369, respectively. A summary of the severance payable at December 31, 2006 and 2007 is presented below (in thousands):
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
| | 2006 | 2007 | |||||
| Balance, beginning of period | $ | 182 | $ | — | |||
| Severance expense | 159 | 98 | |||||
| Cash payments | (341 | ) | (98 | ) | |||
| Balance, end of period | $ | — | $ | — | |||
(7) Income Taxes
The provision for income taxes for the years ended December 31, 2005, 2006 and 2007 was $0, $52,841 and $0, respectively. The amount in 2006 is for federal income tax expense related to
F-14
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
alternative minimum tax, or AMT, requirements. The provision for income taxes differed from the amounts computed by applying the U.S. federal income tax rate of 34% to pretax income (loss) as a result of the following (in thousands):
| | Years Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||||||
| Provision (benefit) for federal income taxes | $ | (149 | ) | $ | 819 | $ | (880 | ) | ||
| State and local taxes | (21 | ) | 118 | (122 | ) | |||||
| Gross change in valuation allowance | 164 | (894 | ) | 978 | ||||||
| Other | 6 | 10 | 24 | |||||||
| Provision for income taxes | $ | — | $ | 53 | $ | — | ||||
The tax effects of temporary differences and carryforwards that give rise to significant portions of deferred tax assets and deferred tax liabilities at December 31, 2006 and 2007 are as follows (in thousands):
| | December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
| | 2006 | 2007 | ||||||
| Deferred tax assets: | ||||||||
| Nonqualified stock options | $ | 1,509 | $ | 787 | ||||
| Federal and state net operating loss carryforwards | 4,998 | 5,664 | ||||||
| AMT credit carryforwards | 53 | 53 | ||||||
| Intangible asset amortization | 328 | 580 | ||||||
| Depreciation of property and equipment | — | 1 | ||||||
| Accrued compensation | 32 | 23 | ||||||
| Other | 5 | — | ||||||
| Gross deferred tax assets | 6,925 | 7,108 | ||||||
| Valuation allowance | (6,909 | ) | (7,108 | ) | ||||
| Net deferred tax assets | 16 | — | ||||||
| Deferred tax liabilities: | ||||||||
| Depreciation of property and equipment | (16 | ) | — | |||||
| Total deferred tax liabilities | (16 | ) | — | |||||
| Total net deferred tax assets | $ | — | $ | — | ||||
Deferred tax assets of $14,932, $192 and $778,970 were reversed in 2005, 2006 and 2007, respectively, along with the corresponding valuation allowance, upon the expiration of unexercised nonqualified stock options.
A valuation allowance of $6,909,300 and $7,108,400 at December 31, 2006 and 2007, respectively, was recognized because the likelihood of realization of net deferred tax assets cannot be currently assessed as more-likely-than-not.
F-15
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
At December 31, 2007, the Company had net operating loss carryforwards for federal and state purposes of $14,579,123. The state and federal net operating loss carryforwards begin to expire in 2009 and 2019, respectively. Additionally, the Company has a federal AMT credit carryforward of $52,841, which does not expire.
On January 1, 2007, the Company adopted FIN 48, which did not have any impact on its consolidated financial statements. As of December 31, 2007, the Company has not recorded any amounts for unrecognized tax benefits related to the implementation of FIN 48. The Company's policy is to record estimated interest and penalties related to the underpayment of income taxes as a component of its income tax provision. In addition, the Company is not currently subject to any ongoing federal or state audits.
(8) Product and License Rights
In September 1993, the Company entered into a license agreement pursuant to which the University granted to the Company an exclusive worldwide license to commercialize (including the right to grant sublicenses) the patent rights to certain cyclodextrin derivatives, including sulfobutyl ether beta cyclodextrin (Captisol) that was designed as a solubility and stability enhancer for drug compounds. In addition, the University assigned to the Company a pending agreement with a pharmaceutical company, which was ultimately executed. In exchange for the license, the Company issued to the University equity ownership in the Company (representing a 45% interest in the Company at such time). The Company valued the license at $450,000, which represented the estimated fair value of the membership units issued at the transaction date. The cost of the original license is being amortized and charged to expense based upon a 16-year life. Amortization expense related to the original agreement is $28,125 per year. In addition, pursuant to the terms of the license agreement between the University and the Company, the Company was obligated to pay the University royalties on monies received from commercialization of the patent rights.
On August 4, 2004, the parties entered into the second amendment to the license agreement pursuant to which the Company issued to the University of Kansas Center for Research, Inc., the University's designee, separate payments of $424,500 and $4,000,000 in cash and 2,000,000 shares of the Company's Series A-1 Stock, having an estimated fair value of $370,000, in consideration for the purchase by the Company of the then outstanding and all future payment obligations to the University owed by the Company under the license agreement. As further consideration for the payments by the Company, the University granted to the Company, subject to the University's retained research rights, an irrevocable, fully paid, worldwide, exclusive (even as to the University) license under the patents originally licensed under the license agreement and certain patent rights and intellectual property rights related to certain cyclodextrin derivatives generated through September 30, 2004 from the sponsored research performed by the University on the Company's behalf, as well as an exclusive option of the Company to acquire rights to any patented inventions claiming certain improvements to the Captisol technology from the University under research sublicenses granted by the University. Of the total consideration paid of $4,794,500, $534,500 was recorded as a reduction of previously accrued royalty costs and $4,260,000 was recorded as additional license cost, which will be amortized over the remaining life of the patent at the time of the amendment, resulting in additional annual amortization expense of $663,859. After August 4, 2004, the Company was no longer obligated to pay royalties.
F-16
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
Intangible assets consist of the following at December 31, 2006 and 2007 (dollars in thousands):
| | | | December 31, | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | 2006 | 2007 | |||||||||||||
| | Estimated Useful Lives (Months) | Cost | Accumulated Amortization | Net Carrying Amount | Accumulated Amortization | Net Carrying Amount | |||||||||||
| Captisol patent | 192 | $ | 450 | $ | 377 | $ | 73 | $ | 405 | $ | 45 | ||||||
| Captisol license | 77 | 4,260 | 1,605 | 2,655 | 2,269 | 1,991 | |||||||||||
| $ | 4,710 | $ | 1,982 | $ | 2,728 | $ | 2,674 | $ | 2,036 | ||||||||
Intangible assets are amortized over their estimated useful economic life using the straight-line method. Amortization expense for each of the years in the three-year period ended December 31, 2007 was $691,984. Estimated amortization expense of intangible assets for each of the next four years is $691,984 for 2008, $680,265 for 2009, $663,860 for 2010 and $0 thereafter.
(9) Preferred Stock
During 2000, the Company issued 2,313,223 shares of Series A convertible preferred stock (Series A Stock), in exchange for proceeds of $12,000,000. The Series A Stock was initially recorded in the consolidated financial statements net of issuance costs of $1,014,705. The difference between the carrying amount and the redemption price was accreted using the effective interest method. During 2004, a stock split of the Series A Stock occurred due to the issuance of the Series B convertible preferred stock (Series B Stock), pursuant to existing antidilution provisions. The stock split of 7.11934 for one resulted in the issuance of 14,155,395 additional shares of Series A Stock. A beneficial conversion feature of $10,985,295 was recorded, resulting in a reduction of the carrying value of the Series A Stock to $575,785. The difference between the new carrying value of $575,785 and the redemption value of $12,000,000 was comprised of the beneficial conversion feature of $10,985,295 and the remaining unaccreted issuance costs of $438,920 at the time of the stock split. Such difference is being accreted using the straight-line method. For each of the years in the three-year period ended December 31, 2007, $2,109,086 of accretion of issuance costs and beneficial conversion feature were recorded as a charge to additional paid-in capital.
During 2004, the Company issued 63,197,019 shares of Series B Stock, in exchange for cash of $14,000,000 and conversion of $2,950,000 of outstanding indebtedness issued pursuant to a bridge financing arrangement entered into in 2003 and $50,000 of accrued interest on that bridge loan. No additional shares of Series B Stock were issued for the remaining $207,490 of the accrued interest, and such amount was reclassified to additional paid-in capital. The Series B Stock was initially recorded in the consolidated financial statements net of issuance costs of $1,026,809. The difference between the carrying amount and the redemption price is being accreted using the effective interest method. For the years ended December 31, 2005, 2006 and 2007, $161,848, $175,963 and $191,312, respectively, of issuance costs were accreted and recorded as a charge to additional paid-in capital.
During 2004, the Company also issued 2,000,000 shares of Series A-1 Stock, having an estimated fair value of $370,000, in connection with the second amendment to the license agreement between the Company and the University.
F-17
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
Certain designations and preferences accompany the Series A Stock, Series A-1 Stock and Series B Stock (referred to collectively as Preferred Stock).
Liquidation Rights. The holders of Preferred Stock are entitled to certain liquidation rights in the event of any liquidation, dissolution, or winding up of the affairs of the Company. Generally, the holders of Series B Stock are entitled to be paid first, before any payments are made to holders of Series A Stock, Series A-1 Stock or common stock, out of the assets of the Company an amount equal to 1.5 times the original price of the stock purchased, plus accrued or declared but unpaid dividends; next, the holders of Series A Stock are entitled to be paid, before any payments are made to holders of Series A-1 Stock or common stock, out of the assets of the Company an amount equal to the original price of the stock purchased (as adjusted for the stock split), plus accrued or declared but unpaid dividends; next, the holders of Series A-1 Stock are entitled to be paid, before any payments are made to holders of common stock, out of the assets of the Company an amount equal to $1.00 per share, plus declared but unpaid dividends. After such preferential payment has been made, the holders of Preferred Stock will share in the distribution of the remaining assets with the holders of common stock on an as-converted basis.
Conversion. The holders of all Preferred Stock have conversion rights whereby each share of Preferred Stock is convertible, at the option of the holder, at any time, into a certain number of shares of common stock on a one for one basis. The number of shares of common stock to be received upon conversion of a share of Preferred Stock is subject to adjustment to avoid dilution of the interest of the holders of Preferred Stock. The shares of Preferred Stock, excluding accrued or declared and unpaid dividends, shall be automatically converted into shares of common stock in the event of an initial public offering of the Company in which the price per share is at least six times the Series B Stock original issue price and the aggregate gross proceeds to the Company are at least $50,000,000, or upon the written election of the holders of at least 60% of the shares of the Series A Stock and Series B Stock, voting together on an as-converted basis.
Voting Rights. Generally, holders of Preferred Stock are entitled to vote together with the holders of common stock on any matters submitted to the stockholders for a vote. Holders of Preferred Stock have that number of votes per share equal to the number of shares of common stock into which each share of Preferred Stock could be converted at that time.
Consent Rights. With certain exceptions, the affirmative vote of the holders of at least 70% of the Series B Stock is required before the Company may, among other things, (i) effect any liquidation, dissolution, or winding up of, or any consolidation or merger involving the Company; (ii) sell or encumber a substantial portion of the assets of the Company; (iii) increase or decrease the number of authorized shares of Preferred Stock or change the powers, preferences, or special rights of the Preferred Stock, or authorize or create any class of stock having preference, priority, or parity with the Preferred Stock, or amend the certificate of incorporation or bylaws in any way that adversely affects the Preferred Stock; (iv) increase or decrease the authorized number of members of the board of directors; (v) cause the redemption, repurchase, payment of dividends or other distributions with respect to the class or series of stock junior to the Series B Stock; (vi) make any capital expenditure or purchase of assets exceeding $500,000; (vii) incur indebtedness in excess of $500,000; or (viii) increase the Company's stock option pool over 10,000,000 shares.
Representation on the Board of Directors and Committees. The holders of the Series B Stock have the exclusive right, separately from the holders of the Series A Stock, Series A-1 Stock and common
F-18
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
stock, to elect two directors of the Company if at least 25% of the shares of Series B Stock remain outstanding, and the holders of the Series A Stock have the exclusive right, separately from the holders of the Series B Stock, Series A-1 Stock and common stock, to elect one director of the Company if at least 25% of the shares of Series A Stock remain outstanding.
Dividends. Cumulative dividends of 7% shall accrue on the Preferred Stock annually and be payable when and as declared, upon liquidation or dissolution, and upon redemption. No dividends shall be payable on common stock without payment of dividends on Preferred Stock equal to what would be paid to holders of Preferred Stock if it were converted to common stock. The Series A Stock and Series B Stock have been increased for total undeclared and unpaid dividends of $8,221,806 and $10,252,201 at December 31, 2006 and 2007, respectively, with the offset recorded as a reduction to additional paid-in capital. Series A-1 Stock has undeclared and unpaid dividends of $337,406 and $477,406 at December 31, 2006 and 2007, respectively.
Redemption. Periodically, the holders of Series A Stock and Series B Stock having an aggregate liquidation preference equal to at least $6,000,000 may unconditionally require the Company, to the extent funds are available and subject to applicable law, to redeem from all holders of Series A Stock and Series B Stock, with respect to each such holder, the number of shares of Series A Stock and Series B Stock as specified by such holder. The redemption price for the Series A Stock and the Series B Stock shall be the respective original purchase price per share of the stock, as adjusted, to account for any stock dividend, stock split, combination of shares, reclassification, or other similar event with respect to the Preferred Stock, plus any accrued unpaid dividends. If holders were to elect to redeem all Series A Stock and Series B Stock at the earliest redemption date, payments would aggregate $29,000,000 in 2009, plus accrued and unpaid dividends.
(10) Warrants
In 2000, the Company granted warrants for the purchase of 46,265 shares of common stock, at an exercise price of $5.19 per share, to a private placement agent for services provided. The warrants were immediately exercisable and 38,554 expired unexercised in August 2005 and 7,711 expired unexercised in October 2005.
In 2003, the Company granted warrants for the purchase of shares of subsequently to be issued preferred stock in a qualified financing under a bridge financing arrangement. The number of shares of the subsequently to be issued preferred stock that could be purchased pursuant to the warrants was to be calculated as 35% of the principal amount of the bridge financing notes divided by the price per share of the subsequently to be issued preferred stock. The exercise price was to be the lesser of 80% of the price per share of the subsequently to be issued preferred stock or $4.15. The warrants were exercisable on the earlier of (a) the closing of a qualified financing, (b) a mandatory redemption, (c) an optional conversion or (d) immediately prior to a change of control, and were to expire in June and July 2010. As of December 31, 2003, the warrants were liability-classified and had a carrying value of $829,595, based on a fair value of $2.44 per share.
In May 2004, the warrants were amended to reflect the following terms. The number of shares of the subsequently to be issued preferred stock that could be purchased pursuant to the warrants was to be calculated as 50% of the principal amount of the bridge financing notes divided by the price per share of the subsequently to be issued preferred stock sold by the Company in a qualified financing. The exercise price was to be the lesser of 80% of the price per share of the subsequently to be issued
F-19
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
preferred stock or $0.90. In May 2004, a mark-to-market adjustment of $292,970 was recorded as non-operating expense, based on a fair value of $0.86 per share.
In August 2004, the warrants were further amended to be warrants for the purchase of 5,483,270 shares of common stock, at an exercise price of $0.01 per share, in connection with the conversion of outstanding indebtedness under a bridge financing arrangement into Series B Stock. These warrants were immediately exercisable and expire in August 2010. In 2004, 743,494 of these warrants were exercised. No warrants were exercised in 2005 or 2006, and 278,810 warrants were exercised in 2007. As a result of the amendment, in August 2004, a mark-to-market adjustment of $334,697 was recorded as a reduction to non-operating expense for the decline in the estimated fair value of the warrants, based on a fair value of $0.19 per share. The warrants were also reclassified from a liability- classified award to an equity-classified award in August 2004.
The fair value of the warrants was calculated using the Black-Scholes model taking into account the current value of the shares of stock that underlie the warrants, the exercise price of the warrants, the contractual term, the expected volatility of the underlying stock, the expected dividends on the underlying stock and the risk-free interest rate over the contractual term for the warrants.
(11) Stock Options
The Company's 1997 Equity Compensation Plan provides for grants of stock options to employees, officers, directors and consultants, with the number of shares of common stock authorized for option grants of 10,000,000 shares. For the years ended December 31, 2005, 2006 and 2007, the Company issued options to purchase 1,068,000, 3,290,000 and 1,712,500 shares of common stock, respectively, which generally vest ratably over periods between three and five years and have a contractual term of ten years. Of these, stock option awards for 903,750 shares granted in 2007 vested immediately upon grant. Newly issued common stock will be issued upon exercise of stock options. At December 31, 2007, there were 580,248 additional shares available to be granted under the 1997 Equity Compensation Plan.
In March 2006, the Company's board of directors granted an option to purchase 1,800,000 shares of common stock to an employee, which is included in the option grants described above, with an exercise price equal to the fair value of the Company's common stock on the date of grant. This grant cliff vests five years from the date of grant. The vesting of 20% of the shares accelerated in September 2006 upon the execution of an agreement which extended the royalty term with respect to marketed products until the last to expire of all applicable Captisol patents. The board also provided that 80% of the shares would accelerate and vest upon specified liquidation events. This acceleration provision lapsed on September 30, 2007.
Total stock-based compensation for the years ended December 31, 2005, 2006 and 2007 was $48,786, $39,797 and $202,803, respectively. For option grants prior to 2004, the Company recorded stock compensation over the vesting period for the difference between the exercise price of $2.50 and the estimated fair value of the common stock at the date of grant of $5.00. Options granted in 2004 and 2005 had an exercise price equal to the estimated fair value of the common stock at the date of grant, and therefore no stock compensation was recognized for those options. Stock-based compensation for the years ended December 31, 2005, 2006 and 2007 was $48,786, $19,741 and $1,276, respectively, for options granted prior to 2004.
F-20
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
For stock options granted to employees during 2006 and 2007, stock compensation was recorded based upon the weighted average grant date fair value of $0.02 and $0.32 per share, respectively, in accordance with SFAS No. 123R. Stock-based compensation for these awards of $20,056 and $201,527 was reflected in net income (loss) for the years ended December 31, 2006 and 2007, respectively. The fair value of each option award was estimated on the date of grant using the Black-Scholes option-pricing model using the weighted average assumptions in the following table. The risk-free rate for the expected life of the option is based on the U.S. Treasury yield curve in effect at the time of grant, with maturities matching the expected term of the options. Expected volatility is based on comparable companies' common stock, the contractual term of the options, and the available trading history of the comparable companies' common stock. No dividends are anticipated to be paid over the expected term of the options based on current expectations of management. The Company used the following assumptions for calculating the fair value of stock options using the Black-Scholes option-pricing model:
| | 2006 | 2007 | |||
|---|---|---|---|---|---|
| Valuation assumptions: | |||||
| Expected dividend yield | 0 | % | 0 | % | |
| Expected volatility | 71 | % | 64 | % | |
| Expected term (years) | 6.11 | 5.73 | |||
| Risk-free interest rate | 4.71 | % | 4.23 | % |
A summary of the status of the Company's 1997 Equity Compensation Plan at December 31, 2005, 2006 and 2007 is presented below:
| Options | Number of shares | Weighted average exercise price | Weighted average remaining contractual term (years) | Aggregate intrinsic value (in thousands) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Outstanding at December 31, 2004 | 6,094,251 | $ | 1.14 | ||||||
| Granted | 1,043,000 | 0.15 | |||||||
| Forfeited | (1,063,925 | ) | 0.19 | ||||||
| Expired | (61,875 | ) | 1.05 | ||||||
| Exercised | (350,000 | ) | 0.15 | — | |||||
| Outstanding at December 31, 2005 | 5,661,451 | $ | 1.19 | ||||||
| Granted | 3,290,000 | 0.15 | |||||||
| Forfeited | (500,250 | ) | 0.15 | ||||||
| Expired | (123,950 | ) | 2.27 | ||||||
| Exercised | (696,667 | ) | 0.15 | — | |||||
| Outstanding at December 31, 2006 | 7,630,584 | $ | 0.89 | ||||||
| Granted | 1,712,500 | 0.35 | |||||||
| Forfeited | (147,250 | ) | 0.22 | ||||||
| Expired | (822,749 | ) | 1.63 | ||||||
| Exercised | (677,417 | ) | 0.17 | 208 | |||||
| Outstanding at December 31, 2007 | 7,695,668 | $ | 0.77 | 7.90 | 1,832 | ||||
| Options exercisable at December 31, 2007 | 4,174,584 | $ | 1.23 | 6.71 | 910 | ||||
F-21
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
Based on a retrospective appraisal of the Company's common stock for 2005 and 2006, certain awards exercised in 2005 and 2006 occurred out-of-the-money.
The following table summarizes information about stock options at December 31, 2007:
| Exercise prices | Outstanding | Weighted average remaining vesting life (years) | Exercisable | |||
|---|---|---|---|---|---|---|
| $0.15 | 4,927,166 | 2 | 2,604,957 | |||
| 0.35 | 1,582,500 | 2 | 383,750 | |||
| 2.50 | 821,577 | — | 821,452 | |||
| 6.00 | 121,200 | — | 121,200 | |||
| 7.50 | 243,225 | — | 243,225 | |||
| 7,695,668 | — | 4,174,584 | ||||
As of December 31, 2007, total compensation cost not yet recognized in the consolidated financial statements related to unvested options was $312,481 and the weighted average period over which the Company expects it to be recognized is approximately three years. The Company has not recognized, and does not expect to recognize in the near future, any tax benefit related to employee stock-based compensation costs as a result of the full valuation allowance on its net deferred tax assets and its net operating loss carryforwards.
On September 5, 2007, the Company issued 60,000 stock options with an exercise price of $0.35 to a nonemployee. These options were outstanding at December 31, 2007. Of these options, 40,000 vested immediately and 20,000 cliff vest on May 31, 2008 if the recipient provides service through such vesting date. For the year ended December 31, 2007, stock compensation related to these options was $16,399, and compensation expense for the nonvested options will be re-measured at each balance sheet date until they vest.
(12) Earnings per Share
The following table summarizes the securities outstanding at the end of each year with the potential to become common stock that have been excluded from the computation of diluted net loss per common share, as their effect would have been antidilutive.
| | December 31, | |||||
|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | |||
| Convertible preferred stock | 81,665,637 | 81,665,637 | 81,665,637 | |||
| Stock options and warrants | 10,401,227 | 12,370,360 | 12,156,634 | |||
| Total | 92,066,864 | 94,035,997 | 93,822,271 | |||
(13) Commitments and Contingent Liabilities
The Company leases office space and certain equipment under noncancelable operating lease agreements. Lease expense related to office space and equipment was $208,292 for each of the years in the three-year period ended December 31, 2007. Certain of the Company's operating leases contain scheduled rent increases over the term of the lease. Lease expense is recognized on a straight-line basis,
F-22
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
which results in initial expense recognition in advance of actual payments. As of December 31, 2006 and 2007, the Company had recorded $39,760 and $21,537, respectively, of cumulative expense in excess of payments made and has reflected those amounts as a liability in the Company's consolidated balance sheet.
Generally, in the normal course of business, leases will be renewed or replaced as they expire. Future minimum lease payments for noncancelable leases are as follows (in thousands):
| Years Ending December 31, | Minimum Lease Payments | ||
|---|---|---|---|
| 2008 | $ | 224 | |
| 2009 | 19 | ||
| 2010 | — | ||
The Company periodically issues letters of credit for certain lease obligations. At December 31, 2006 and 2007, the Company had issued letters of credit for $141,087 and $97,754, respectively, for those purposes.
At December 31, 2006 and 2007, the Company had an agreement with First National Bank of Kansas for a $3,000,000 revolving line of credit of which the entire $3,000,000 was available to the Company. The interest rate to be applied to the unpaid principal is at a fixed amount of 8.25% per annum. This line of credit is secured by a pledge of all inventory, chattel paper, accounts, equipment and general intangibles except intellectual property rights. Although the Company has made no borrowings against the line of credit, if it were to make borrowings, all outstanding principal and accrued unpaid interest would be due in one payment on April 3, 2008.
The Company is committed to purchase a total of $15,000,000 of Captisol over the term of a supply agreement. At December 31, 2007, the Company had purchased $5,615,182 toward its $15,000,000 commitment. This agreement expires in 2014.
(14) Geographic Information
Revenue by geographic area for the years ended December 31, 2005, 2006 and 2007 is as follows (in thousands):
| | December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | ||||||
| United States | $ | 5,591 | $ | 9,005 | $ | 9,109 | |||
| United Kingdom | 844 | 2,156 | 1,900 | ||||||
| Belgium | 1,089 | — | 167 | ||||||
| Other | 1,295 | 419 | 1,569 | ||||||
| Total revenue | $ | 8,819 | $ | 11,580 | $ | 12,745 | |||
(15) Subsequent Event
At December 31, 2007, the Company held $5,800,000 of AAA rated auction-rate preferred securities at cost, which approximates fair value. While the underlying security has a long-term maturity, the interest rate is reset through Dutch auctions that are held at pre-determined intervals, usually every 7 to 28 days. Disruptions in the global credit and capital markets experienced in 2008 have not
F-23
CYDEX PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
permitted the Company to liquidate certain of these securities. The Company has experienced a number of failed auctions since late March 2008, since many of these auctions have not had sufficient bidders to allow investors to complete a sale of ARPS. Prior to this time, these auctions had historically provided a liquidity source for these investments. The Company does not know if or when the credit markets will improve and allow it to liquidate these investments. Due to this situation, the Company has developed a model to estimate an illiquidity discount. The model uses significant unobservable inputs. The estimated fair value resulting from this model is less than cost. The Company has recorded an other-than-temporary impairment charge of $410,000 as of March 31, 2008 to write these investments down to their estimated fair value. The Company also reclassified the investments which it may not be able to liquidate in the near term to long-term investments as of March 31, 2008, as they are not generally available to support the Company's current operations. At December 31, 2007, these investments were classified as a current asset.
F-24
Shares
CyDex Pharmaceuticals, Inc.
Common Stock
PROSPECTUS
Until , 2008 (25 days after the date of this prospectus), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers' obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
Pacific Growth Equities, LLC
JMP Securities
Fortis Securities LLC
, 2008
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth all expenses, other than the underwriting discounts and commissions, payable by the registrant in connection with the sale of the common stock being registered. All the amounts shown are estimates except the registration fee, the FINRA filing fee and the Nasdaq Global Market initial listing fee. We intend to pay all expenses of registration, issuance and distribution.
| | Total | |||
|---|---|---|---|---|
| SEC registration fee | $ | 1,965 | ||
| FINRA filing fee | 5,500 | |||
| Nasdaq Global Market initial listing fee | 125,000 | |||
| Blue sky qualification fees and expenses | 10,000 | |||
| Printing and engraving expenses | 175,000 | |||
| Legal fees and expenses | 1,500,000 | |||
| Accountant's fees and expenses | * | |||
| Transfer agent and registrar fees | 10,000 | |||
| Miscellaneous | * | |||
| Total | $ | * | ||
*To be completed by amendment.
Item 14. Indemnification of Officers and Directors
The registrant's amended and restated certificate of incorporation provides that a director will not be personally liable to the registrant or to its stockholders for monetary damages for any breach of fiduciary duty as a director to the fullest extent permitted by Section 102 of the Delaware General Corporation Law.
As permitted by Section 145 of the Delaware General Corporation Law, the bylaws of the registrant provide that (i) the registrant is required to indemnify its directors and officers to the fullest extent not prohibited by the Delaware General Corporation Law, (ii) the registrant may, in its discretion, indemnify its other employees and agents as set forth in the Delaware General Corporation Law, (iii) the registrant is required to advance all expenses incurred by its directors and officers in connection with certain legal proceedings, (iv) the rights conferred in the bylaws are not exclusive, and (v) the registrant is authorized to enter into indemnification agreements with its directors, officers, employees and agents.
The registrant has entered into agreements with its directors and officers that require the registrant to indemnify such persons against expenses, judgments, fines, settlements, and other amounts that any such person becomes legally obligated to pay (including with respect to a derivative action) in connection with any proceeding, whether actual or threatened, to which such person may be made a party by reason of the fact that such person is or was a director or officer of the registrant or any of its affiliates. The indemnification agreements also set forth certain procedures that will apply in the event of a claim for indemnification thereunder. At present, no litigation or proceeding is pending that involves a director or officer of the registrant regarding which indemnification is sought, nor is the registrant aware of any threatened litigation that may result in claims for indemnification.
II-1
The form of underwriting agreement filed as exhibit 1.1 to this registration statement provides for indemnification under certain circumstances by the underwriters of the registrant, its directors, certain of its officers and its controlling persons for certain liabilities arising under the Securities Act, or otherwise.
The registrant maintains a directors' and officers' insurance and registrant reimbursement policy. The policy (i) insures directors and officers against losses for which the registrant does not indemnify and which losses arise from certain wrongful acts in the indemnified parties' capacities as directors and officers and (ii) reimburses the registrant for those losses for which the registrant has lawfully indemnified the directors and officers. The policy contains various exclusions, none of which apply to this offering.
The Amended and Restated Investor Rights Agreement between the registrant and certain investors provides for cross-indemnification in connection with registration of the registrant's common stock on behalf of such investors.
Item 15. Recent Sales of Unregistered Securities
The following list sets forth information regarding all securities sold by us from January 1, 2005 through January 31, 2008.
- 1.
- Between January 1, 2005 and December 31, 2007, we granted stock options to purchase 6,045,500 shares of common stock to employees, consultants and directors pursuant to our Amended and Restated 1997 Equity Compensation Plan. Of these options, 642,250 shares have been cancelled without being exercised and 450,750 have been exercised, 0 shares of which have been repurchased and 4,952,500 remain outstanding.
- 2.
- In October 2007, we sold and issued 278,810 shares of common stock for $2,788 in cash, upon the exercise of an outstanding warrant that was originally issued in 2003. These shares remain outstanding.
The issuances described in above in this Item 15 were deemed exempt from registration under the Securities Act in reliance on either (a) Rule 701 promulgated under the Securities Act as offers and sale of securities pursuant to certain compensatory benefit plans and contracts relating to compensation in compliance with Rule 701 or (b) Section 4(2) of the Securities Act, as transactions by an issuer not involving any public offering. The recipients of securities in each of these transactions represented their intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the share certificates and instruments issued in such transactions. All recipients had adequate access, through their relationships with us, to information about us.
Item 16. Exhibits and Financial Statement Schedules
- (a)
- Exhibits.
| Exhibit Number | Description of Document | |
|---|---|---|
| 1.1 | Form of Underwriting Agreement | |
| 3.1† | Amended and Restated Certificate of Incorporation currently in effect | |
| 3.2† | Certificate of Amendment to Amended and Restated Certificate of Incorporation | |
| 3.3* | Certificate of Amendment to Amended and Restated Certificate of Incorporation | |
| 3.4* | Form of Amended and Restated Certificate of Incorporation to be effective upon completion of this offering | |
| 3.5† | Bylaws currently in effect |
II-2
| 3.6 | Form of Amended and Restated Bylaws to be effective upon completion of this offering | |
| 4.1 | Reference is made to exhibits 3.1 through 3.6 | |
| 4.2* | Specimen Common Stock Certificate | |
| 4.3† | Form of Amended and Restated Warrant to purchase shares of Common Stock | |
| 4.4† | Amended and Restated Registration Rights Agreement, dated August 4, 2004, by and between the Registrant and certain of its stockholders | |
| 5.1* | Opinion of Cooley Godward Kronish LLP | |
| 10.1+ | Form of Indemnity Agreement between the Registrant and its officers and directors | |
| 10.2+† | Amended and Restated 1997 Equity Compensation Plan and forms of related agreements | |
| 10.3*+ | 2008 Equity Incentive Plan and forms of related agreements | |
| 10.4*+ | 2008 Employee Stock Purchase Plan | |
| 10.5*+ | 2008 Non-Employee Directors' Stock Option Plan and forms of related agreements | |
| 10.6+† | Employment Agreement, dated June 21, 2006, with John M. Siebert | |
| 10.6.1+† | Amendment to Employment Agreement of June 21, 2006, with John M. Siebert, dated February 24, 2008 | |
| 10.7+† | Severance Compensation Agreement Following Change in Control, dated January 1, 2004, with John M. Siebert | |
| 10.7.1+† | Amendment to Severance Compensation Agreement Following Change in Control, dated January 1, 2004, with John M. Siebert, dated February 24, 2008 | |
| 10.8+† | Severance Compensation Agreement Following Change in Control, dated November 1, 2003, with Allen K. Roberson | |
| 10.8.1+† | Amendment to Severance Compensation Agreement Following Change in Control, dated November 1, 2003, with Allen K. Roberson, dated February 23, 2008 | |
| 10.9*# | Supply Agreement, dated December 20, 2002, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.1*# | First Amendment to the Supply Agreement of December 20, 2002, dated July 29, 2005, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.2* | 2nd Amendment to the Supply Agreement of December 20, 2002 and amended July 29, 2005, dated March 1, 2007, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.3*# | 3rd Amendment to the Supply Agreement of December 20, 2002 and amended July 29, 2005 and March 1, 2007, dated January 25, 2008, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.10*# | License Agreement, dated September 3, 1993, between the Registrant and The University of Kansas | |
| 10.10.1*# | Second Amendment to the License Agreement of September 3, 1993, dated August 4, 2004, between the Registrant and The University of Kansas | |
| 10.11*# | Option Agreement, dated December 3, 1993, between Pfizer, Inc. and The University of Kansas (Assigned to the Registrant by Option Agreement Assignment, dated November 5, 1993) | |
| 10.11.1*# | Option Agreement Assignment by The University of Kansas to the Registrant, dated November 5, 1993 | |
| 10.12*# | Exclusive License Agreement, dated June 4, 1996, between Pfizer, Inc. and the Registrant | |
| 10.13*# | Nonexclusive License Agreement, dated June 4, 1996, between Pfizer, Inc. and the Registrant | |
| 10.13.1*# | Addendum to Nonexclusive License Agreement of June 4, 1996, dated December 11, 2001, between the Registrant and Pfizer, Inc. | |
| 10.14* | Acknowledgement agreement, dated March 3, 2008, between the Registrant and The University of Kansas | |
| 10.15*# | Supply Agreement, dated March 23, 2006, between the Registrant and Verus Pharmaceuticals, Inc. | |
| 10.16*# | Amended and Restated License Agreement, dated November 21, 2006, between the Registrant and Verus Pharmaceuticals, Inc. |
II-3
| 10.16.1*# | First Amendment to the Amended and Restated License Agreement of November 21, 2006, dated August 24, 2007, between the Registrant and Tika Läkemedel AB | |
| 10.17*# | Option Agreement, dated February 1, 2008, between the Registrant and Tika Läkemedel AB | |
| 10.18*# | Patent License Agreement, dated August 1, 2007, between the Registrant and Dr. Falk Pharma GmbH | |
| 10.19† | Pine Ridge Business Park Lease, dated October 17, 2003, between the Registrant and PERG Buildings, LLC | |
| 10.20 | Business Loan Agreement (Asset Based), dated April 6, 2006, between the Registrant and First National Bank of Kansas | |
| 10.20.1 | Promissory Note, dated April 30, 2008, between the Registrant and First National Bank of Kansas | |
| 10.21+ | Offer Letter, dated May 6, 2008, with Theron E. Odlaug | |
| 23.1 | Consent of KPMG LLP | |
| 23.2* | Consent of Cooley Godward Kronish LLP. Reference is made to Exhibit 5.1 | |
| 23.3 | Consent of American Appraisal Associates, Inc. | |
| 24.1† | Power of Attorney |
*To be filed by amendment.
+Management contract or compensatory plan.
#Confidential treatment has been requested with respect to portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission.
†Previously filed.
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers, and controlling persons of the registrant pursuant to the foregoing provisions or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant undertakes that:
(1) for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; and
(2) for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and this offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-4
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Amendment No. 1 to registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Lenexa, State of Kansas, on the 12th day of May, 2008.
| CYDEX PHARMACEUTICALS, INC. | |||
By: | /s/ JOHN M. SIEBERT John M. Siebert, Ph.D. Chairman and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Act of 1933, this Amendment No. 1 to registration statement has been signed by the following persons in the capacities and on the dates indicated.
| Signatures | Title | Date | ||
|---|---|---|---|---|
| /s/ JOHN M. SIEBERT JOHN M. SIEBERT, PH.D. | Chairman and Chief Executive Officer (Principal Executive Officer) | May 12, 2008 | ||
/s/ ALLEN K. ROBERSON ALLEN K. ROBERSON, CPA | Chief Financial Officer (Principal Financial and Accounting Officer) | May 12, 2008 | ||
/s/ STEVEN D. COSLER* STEVEN D. COSLER | Director | May 12, 2008 | ||
/s/ JAMES C. GALE* JAMES C. GALE | Director | May 12, 2008 | ||
/s/ ELLIOT F. HAHN* ELLIOT F. HAHN, PH.D. | Director | �� | May 12, 2008 | |
/s/ DAVID POLTACK* DAVID POLTACK | Director | May 12, 2008 | ||
/s/ PHILIP L. SMITH* PHILIP L. SMITH, PH.D. | Director | May 12, 2008 |
*By: | /s/ ALLEN K. ROBERSON Allen K. Roberson Attorney-in-fact |
II-5
| Exhibit Number | Description of Document | |
|---|---|---|
| 1.1 | Form of Underwriting Agreement | |
| 3.1† | Amended and Restated Certificate of Incorporation currently in effect | |
| 3.2† | Certificate of Amendment to Amended and Restated Certificate of Incorporation | |
| 3.3* | Certificate of Amendment to Amended and Restated Certificate of Incorporation | |
| 3.4* | Form of Amended and Restated Certificate of Incorporation to be effective upon completion of this offering | |
| 3.5† | Bylaws currently in effect | |
| 3.6 | Form of Amended and Restated Bylaws to be effective upon completion of this offering | |
| 4.1 | Reference is made to exhibits 3.1 through 3.6 | |
| 4.2* | Specimen Common Stock Certificate | |
| 4.3† | Form of Amended and Restated Warrant to purchase shares of Common Stock | |
| 4.4† | Amended and Restated Registration Rights Agreement, dated August 4, 2004, by and between the Registrant and certain of its stockholders | |
| 5.1* | Opinion of Cooley Godward Kronish LLP | |
| 10.1+ | Form of Indemnity Agreement between the Registrant and its officers and directors | |
| 10.2+† | Amended and Restated 1997 Equity Compensation Plan and forms of related agreements | |
| 10.3*+ | 2008 Equity Incentive Plan and forms of related agreements | |
| 10.4*+ | 2008 Employee Stock Purchase Plan | |
| 10.5*+ | 2008 Non-Employee Directors' Stock Option Plan and forms of related agreements | |
| 10.6+† | Employment Agreement, dated June 21, 2006, with John M. Siebert | |
| 10.6.1+† | Amendment to Employment Agreement of June 21, 2006, with John M. Siebert, dated February 24, 2008 | |
| 10.7+† | Severance Compensation Agreement Following Change in Control, dated January 1, 2004, with John M. Siebert | |
| 10.7.1+† | Amendment to Severance Compensation Agreement Following Change in Control, dated January 1, 2004, with John M. Siebert, dated February 24, 2008 | |
| 10.8+† | Severance Compensation Agreement Following Change in Control, dated November 1, 2003, with Allen K. Roberson | |
| 10.8.1+† | Amendment to Severance Compensation Agreement Following Change in Control, dated November 1, 2003, with Allen K. Roberson, dated February 23, 2008 | |
| 10.9*# | Supply Agreement, dated December 20, 2002, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.1*# | First Amendment to the Supply Agreement of December 20, 2002, dated July 29, 2005, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.2* | 2nd Amendment to the Supply Agreement of December 20, 2002 and amended July 29, 2005, dated March 1, 2007, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.9.3*# | 3rd Amendment to the Supply Agreement of December 20, 2002 and amended July 29, 2005 and March 1, 2007, dated January 25, 2008, between the Registrant and Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | |
| 10.10*# | License Agreement, dated September 3, 1993, between the Registrant and The University of Kansas | |
| 10.10.1*# | Second Amendment to the License Agreement of September 3, 1993, dated August 4, 2004, between the Registrant and The University of Kansas |
| 10.11*# | Option Agreement, dated December 3, 1993, between Pfizer, Inc. and The University of Kansas (Assigned to the Registrant by Option Agreement Assignment, dated November 5, 1993) | |
| 10.11.1*# | Option Agreement Assignment by The University of Kansas to the Registrant, dated November 5, 1993 | |
| 10.12*# | Exclusive License Agreement, dated June 4, 1996, between Pfizer, Inc. and the Registrant | |
| 10.13*# | Nonexclusive License Agreement, dated June 4, 1996, between Pfizer, Inc. and the Registrant | |
| 10.13.1*# | Addendum to Nonexclusive License Agreement of June 4, 1996, dated December 11, 2001, between the Registrant and Pfizer, Inc. | |
| 10.14* | Acknowledgement agreement, dated March 3, 2008, between the Registrant and The University of Kansas | |
| 10.15*# | Supply Agreement, dated March 23, 2006, between the Registrant and Verus Pharmaceuticals, Inc. | |
| 10.16*# | Amended and Restated License Agreement, dated November 21, 2006, between the Registrant and Verus Pharmaceuticals, Inc. | |
| 10.16.1*# | First Amendment to the Amended and Restated License Agreement of November 21, 2006, dated August 24, 2007, between the Registrant and Tika Läkemedel AB | |
| 10.17*# | Option Agreement, dated February 1, 2008, between the Registrant and Tika Läkemedel AB | |
| 10.18*# | Patent License Agreement, dated August 1, 2007, between the Registrant and Dr. Falk Pharma GmbH | |
| 10.19† | Pine Ridge Business Park Lease, dated October 17, 2003, between the Registrant and PERG Buildings, LLC | |
| 10.20 | Business Loan Agreement (Asset Based), dated April 6, 2006, between the Registrant and First National Bank of Kansas | |
| 10.20.1 | Promissory Note, dated April 30, 2008, between the Registrant and First National Bank of Kansas | |
| 10.21+ | Offer Letter, dated May 6, 2008, with Theron E. Odlaug | |
| 23.1 | Consent of KPMG LLP | |
| 23.2* | Consent of Cooley Godward Kronish LLP. Reference is made to Exhibit 5.1 | |
| 23.3 | Consent of American Appraisal Associates, Inc. | |
| 24.1† | Power of Attorney |
*To be filed by amendment.
+Management contract or compensatory plan.
#Confidential treatment has been requested with respect to portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission.
†Previously filed.
TABLE OF CONTENTS
PROSPECTUS SUMMARY
RISK FACTORS
FORWARD-LOOKING STATEMENTS AND INDUSTRY DATA
USE OF PROCEEDS
DIVIDEND POLICY
CAPITALIZATION
DILUTION
SELECTED CONSOLIDATED FINANCIAL DATA
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
BUSINESS
MANAGEMENT
Grants of Plan-Based Awards in Fiscal 2007
Outstanding Equity Awards At December 31, 2007
Non-Employee Director Compensation Table
CERTAIN RELATIONSHIPS AND RELATED PERSONS TRANSACTIONS
PRINCIPAL STOCKHOLDERS
DESCRIPTION OF CAPITAL STOCK
CERTAIN UNITED STATES FEDERAL TAX CONSEQUENCES TO NON-UNITED STATES HOLDERS
SHARES ELIGIBLE FOR FUTURE SALE
UNDERWRITING
LEGAL MATTERS
EXPERTS
WHERE YOU CAN FIND ADDITIONAL INFORMATION
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
CYDEX PHARMACEUTICALS, INC. Consolidated Balance Sheets (in thousands, except share and per share amounts)
CYDEX PHARMACEUTICALS, INC. Consolidated Statements of Operations (in thousands, except share and per share amounts)
CYDEX PHARMACEUTICALS, INC. Consolidated Statements of Stockholders' Deficit (in thousands)
CYDEX PHARMACEUTICALS, INC. Consolidated Statements of Cash Flows (in thousands)
CYDEX PHARMACEUTICALS, INC. Notes to Consolidated Financial Statements
PART II INFORMATION NOT REQUIRED IN PROSPECTUS
- Item 13. Other Expenses of Issuance and Distribution
Item 14. Indemnification of Officers and Directors
Item 15. Recent Sales of Unregistered Securities
Item 16. Exhibits and Financial Statement Schedules
Item 17. Undertakings
EXHIBIT INDEX