Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
From the transition period from to
Commission file number: 000-50633
CYTOKINETICS, INCORPORATED
(Exact name of registrant as specified in its charter)
| Delaware | 94-3291317 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
280 East Grand Avenue South San Francisco, CA | 94080 | |
| (Address of principal executive offices) | (Zip Code) |
(650) 624-3000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.001 par value | The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the Registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of the Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ | Accelerated filer þ | Non-accelerated filer ¨ | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of the voting and non-voting common equity held by non-affiliates was $331.2 million, computed by reference to the last sales price of $11.57 as reported by the NASDAQ Market as of the last business day of the Registrant’s most recently completed second fiscal quarter, June 28, 2013. This calculation does not reflect a determination that certain persons are affiliates of the Registrant for any other purpose. The number of shares of common stock held by non-affiliates excluded 58,501 shares of common stock held by directors, officers and affiliates of directors. The number of shares owned by affiliates of directors was determined based upon information supplied by such persons and upon Schedules 13D and 13G, if any, filed with the SEC. Exclusion of shares held by any person should not be construed to indicate that such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the Registrant, that such person is controlled by or under common control with the Registrant, or that such persons are affiliates for any other purpose.
The number of shares outstanding of the Registrant’s common stock on February 28, 2014 was 36,078,367 shares.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for its 2014 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission, no later than 120 days after the end of the fiscal year, are incorporated by reference to Part III of this Annual Report on Form 10-K.
Table of Contents
CYTOKINETICS, INCORPORATED
FORM 10-K
Year Ended December 31, 2013
Table of Contents
PART I
This report contains forward-looking statements indicating expectations about future performance and other forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the Private Securities Litigation Reform Act of 1995, that involve risks and uncertainties. We intend that such statements be protected by the safe harbor created thereby. Forward-looking statements involve risks and uncertainties and our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Examples of such forward-looking statements include, but are not limited to, statements about or relating to:
| • | guidance concerning revenues, research and development expenses and general and administrative expenses for 2014; |
| • | the sufficiency of existing resources to fund our operations for at least the next 12 months; |
| • | our capital requirements and needs for additional financing; |
| • | the initiation, design, conduct, enrollment, progress, timing and scope of clinical trials and development activities for our drug candidates conducted by ourselves or our partners, Amgen Inc. and Astellas Pharma Inc. (“Astellas”), including the anticipated timing for initiation of clinical trials, anticipated rates of enrollment for clinical trials and anticipated timing of results becoming available or being announced from clinical trials; |
| • | the results from the clinical trials and non-clinical and preclinical studies of our drug candidates and other compounds, and the significance and utility of such results; |
| • | the ability of our amendments to the protocol of our BENEFIT-ALS clinical trial to maintain the originally intended statistical power of the trial; |
| • | our and our partners’ plans or ability to conduct the continued research and development of our drug candidates and other compounds; |
| • | our expected roles in research, development or commercialization under our strategic alliances with Amgen and Astellas; |
| • | the properties and potential benefits of, and the potential market opportunities for, our drug candidates and other compounds, including the potential indications for which they may be developed; |
| • | the sufficiency of the clinical trials conducted with our drug candidates to demonstrate that they are safe and efficacious; |
| • | our receipt of milestone payments, royalties, reimbursements and other funds from current or future partners under strategic alliances, such as with Amgen or Astellas; |
| • | our ability to continue to identify additional potential drug candidates that may be suitable for clinical development; |
| • | our plans or ability to commercialize drugs with or without a partner, including our intention to develop sales and marketing capabilities; |
| • | the focus, scope and size of our research and development activities and programs; |
| • | the utility of our focus on the biology of muscle function, and our ability to leverage our experience in muscle contractility to other muscle functions; |
| • | our ability to protect our intellectual property and to avoid infringing the intellectual property rights of others; |
| • | expected future sources of revenue and capital; |
| • | losses, costs, expenses and expenditures; |
1
Table of Contents
| • | future payments under loan and lease obligations; |
| • | potential competitors and competitive products; |
| • | retaining key personnel and recruiting additional key personnel; |
| • | expected timing for recognition of compensation cost related to unvested stock options; and |
| • | the potential impact of recent accounting pronouncements on our financial position or results of operations. |
Such forward-looking statements involve risks and uncertainties, including, but not limited to:
| • | our ability to acquire the funding necessary to conduct the one or more confirmatory Phase III clinical trials for tirasemtiv in patients with amyotrophic lateral sclerosis (also known as ALS or Lou Gehrig’s disease), if supported by the results from our BENEFIT-ALS clinical trial, that we expect will be required to obtain marketing approval for tirasemtiv for the treatment of ALS; |
| • | Amgen’s decisions with respect to the timing, design and conduct of research and development activities for omecamtiv mecarbil and other related compounds, including decisions to postpone or discontinue research or development activities relating to omecamtiv mecarbil and other related compounds; |
| • | Astellas’ decisions with respect to the timing, design and conduct of research and development activities for CK-2127107 and other skeletal muscle activators, including decisions to postpone or discontinue research or development activities relating to CK-2127107 and other skeletal muscle activators; |
| • | our ability to enter into strategic partnership agreements for any of our programs on acceptable terms and conditions or in accordance with our planned timelines; |
| • | our ability to obtain additional financing on acceptable terms, if at all; |
| • | our receipt of funds and access to other resources under our current or future strategic alliances; |
| • | difficulties or delays in the development, testing, manufacturing or commercialization of our drug candidates; |
| • | difficulties or delays, or slower than anticipated patient enrollment, in our or partners’ clinical trials; |
| • | difficulties or delays in the manufacture and supply of clinical trial materials; |
| • | failure by our contract research organizations, contract manufacturing organizations and other vendors to properly fulfill their obligations or otherwise perform as expected; |
| • | results from non-clinical studies that may adversely impact the timing or the further development of our drug candidates and other compounds; |
| • | the possibility that the U.S. Food and Drug Administration (“FDA”) or foreign regulatory agencies may delay or limit our or our partners’ ability to conduct clinical trials or may delay or withhold approvals for the manufacture and sale of our products; |
| • | changing standards of care and the introduction of products by competitors or alternative therapies for the treatment of indications we target that may limit the commercial potential of our drug candidates; |
| • | difficulties or delays in achieving market access and reimbursement for our drug candidates and the potential impacts of health care reform; |
| • | changes in laws and regulations applicable to drug development, commercialization or reimbursement; |
| • | the uncertainty of protection for our intellectual property, whether in the form of patents, trade secrets or otherwise; |
| • | potential infringement or misuse by us of the intellectual property rights of third parties; |
| • | activities and decisions of, and market conditions affecting, current and future strategic partners; |
| • | accrual information provided by our contract research organizations and other vendors; |
2
Table of Contents
| • | potential ownership changes under Internal Revenue Code Section 382; and |
| • | the timeliness and accuracy of information filed with the U.S. Securities and Exchange Commission (the “SEC”) by third parties. |
In addition such statements are subject to the risks and uncertainties discussed in the “Risk Factors” section and elsewhere in this document. Such statements speak only as of the date on which they are made, and, except as required by law, we undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. New factors emerge from time to time, and it is not possible for us to predict which factors will arise. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements.
Overview
We were incorporated in Delaware in August 1997 as Cytokinetics, Incorporated. We are a clinical stage biopharmaceutical company focused on the discovery and development of novel small molecule therapeutics that modulate muscle function for the potential treatment of serious diseases and medical conditions. Our research and development activities relating to the biology of muscle function have evolved from our knowledge and expertise regarding the cytoskeleton, a complex biological infrastructure that plays a fundamental role within every human cell. Our most advanced research and development programs relate to the biology of muscle function and are directed to small molecule modulators of the contractility of skeletal or cardiac muscle. We are also conducting earlier-stage research directed to other compounds with the potential to modulate muscle contractility and other muscle functions, such as growth, energetics and metabolism.
Our lead drug candidate from our skeletal muscle contractility program, tirasemtiv (formerly known as CK-2017357), is a fast skeletal muscle troponin activator. Cytokinetics holds the rights to tirasemtiv and is independently developing this drug candidate for the potential treatment of ALS and other neuromuscular disorders. We are currently conducting a Phase II clinical trials program for tirasemtiv, including an ongoing Phase IIb clinical trial in patients with ALS, known as BENEFIT-ALS (Blinded Evaluation of Neuromuscular Effects and Functional Improvement with Tirasemtiv in ALS). We have concluded enrollment in BENEFIT-ALS and we anticipate reporting results from this clinical trial in April 2014. Tirasemtiv has been granted orphan drug designation and fast track status by the FDA and orphan medicinal product designation by the European Medicines Agency, in each case for the potential treatment of ALS. We anticipate that we will need to conduct at least one confirmatory Phase III clinical trial of tirasemtiv in patients with ALS, if supported by the results of BENEFIT-ALS, to gain marketing approval.
We are also developing CK-2127107, a structurally distinct, fast skeletal muscle troponin activator, under a strategic alliance with Astellas established in June 2013. CK-2127107 is being evaluated in Phase I clinical trials for potential non-neuromuscular indications associated with muscle weakness. We also are conducting joint research with Astellas directed to next-generation skeletal muscle activators. Further details regarding our strategic alliance with Astellas can be found below in Item 1 of this report under “Research and Development Programs — Skeletal Muscle Contractility Program — CK-2127107 and Other Skeletal Muscle Activators — Astellas Strategic Alliance.”
Our lead drug candidate from our cardiac muscle contractility program, omecamtiv mecarbil (formerly known as CK-1827452), is a novel cardiac muscle myosin activator that is being developed under a strategic alliance with Amgen. In June 2013, we expanded the collaboration to include Japan. As a result, Amgen holds an exclusive license to develop and commercialize omecamtiv mecarbil worldwide, subject to our development and commercialization participation rights. An intravenous formulation of omecamtiv mecarbil was studied in a Phase IIb clinical trial known as ATOMIC-AHF (Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure), which was designed to evaluate the safety and efficacy of omecamtiv mecarbil in patients with left ventricular systolic dysfunction who are hospitalized with acute heart failure.
3
Table of Contents
Another Phase II clinical trial, known as COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure), is currently being conducted. The primary objectives of COSMIC-HF are selecting an oral modified release formulation and doses of omecamtiv mecarbil for chronic twice-daily dosing in patients with heart failure and left ventricular systolic dysfunction, and characterizing its pharmacokinetics during 20 weeks of treatment.
We also are conducting joint research with Amgen directed to next-generation compounds in our cardiac muscle contractility program. Further details regarding our strategic alliance with Amgen can be found below in Item 1 of this report under “Research and Development Programs — Cardiac Muscle Contractility Program — Amgen Strategic Alliance.”
Two of our drug candidates have demonstrated pharmacodynamic activity in patients: tirasemtiv in patients with ALS, in patients with myasthenia gravis and in patients with claudication associated with peripheral artery disease; and omecamtiv mecarbil in patients with heart failure. Our drug candidate CK-2127107 has demonstrated pharmacological activity in preclinical models. In 2014, we expect to continue to focus on translating the observed pharmacodynamic activity of these compounds into potentially meaningful clinical benefits for patients.
Following is a summary of the planned clinical development activities for our drug candidates:
Drug Candidate (Mechanism of Action) | Mode of Administration | Potential Indication(s) | Development Status and Planned Development Activities | |||
| Skeletal Muscle Contractility Program | ||||||
Tirasemtiv1 (fast skeletal muscle troponin activator) | oral | neuromuscular diseases and conditions associated with muscle weakness | • We anticipate reporting results from BENEFIT-ALS at the American Academy of Neurology Annual Meeting in Philadelphia, PA in April 2014. | |||
CK-21271072 (fast skeletal muscle troponin activator) | oral | non-neuromuscular indications associated with muscle weakness | • We anticipate conducting additional Phase I studies and certain Phase II readiness activities in 2014 pursuant to our collaboration agreement with Astellas. | |||
| Cardiac Muscle Contractility Program | ||||||
Omecamtiv mecarbil3 (cardiac muscle myosin activator) | oral | heart failure | • We anticipate the commencement of patient enrollment in the expansion phase of COSMIC-HF to occur in the first half of 2014 following regulatory authorities’ review of responses relating to information requests provided in connection with the protocol submitted for the trial.
• We anticipate the enrollment of patients in the expansion phase of COSMIC-HF to be completed in 2014. | |||
Omecamtiv mecarbil3 (cardiac muscle myosin activator) | IV/oral | heart failure | • We anticipate commencement of patient enrollment in CY 1211 to occur in the first half of 2014 following regulatory authorities’ review of responses relating to information requests provided in connection with the protocol submitted for the trial.
• We anticipate the conduct of CY 1211 to be completed in 2014. | |||
| 1 | Independent development by Cytokinetics |
| 2 | Development pursuant to our strategic alliance with Astellas |
| 3 | Development pursuant to our strategic alliance with Amgen |
4
Table of Contents
All of our drug candidates have arisen from our cytoskeletal research activities. Our focus on the biology of the cytoskeleton distinguishes us from other biopharmaceutical companies, and potentially positions us to discover and develop novel therapeutics that may be useful for the treatment of severe diseases and medical conditions. We believe that this focus and the resulting knowledge and expertise that we have developed, especially with our proprietary technologies that permit us to evaluate the function of cytoskeletal proteins in high information content biological assays, has allowed us to increase the efficiency of our drug discovery activities. Our research and development activities since our inception in 1997 have produced multiple drug candidates that have progressed into clinical testing. Each of these drug candidates has a novel mechanism of action compared to currently marketed drugs, which we believe validates our focus on the cytoskeleton as a productive area for drug discovery. We intend to leverage our experience in muscle contractility in order to expand our current pipeline, and expect to identify additional potential drug candidates that may be suitable for clinical development.
Our Corporate Strategy
Our goal is to discover, develop and commercialize novel drug products that modulate muscle function in ways that may benefit patients with serious diseases or medical conditions, with the intent of establishing a fully integrated biopharmaceutical company. We intend to achieve this by:
| • | Focusing on drug discovery and development activities relating to the biology of muscle function. We intend to capitalize on the knowledge and expertise we have acquired in each of our muscle contractility research and development programs. In these programs, we are investigating potential treatments for diseases or medical conditions where impaired regulation of the contractile function of muscle plays a key role and such diseases or conditions may be amenable to treatment by modulation of muscle contractility, such as heart failure, and medical conditions associated with skeletal muscle weakness or wasting. |
| • | Leveraging our cytoskeletal expertise and proprietary technologies to increase the speed, efficiency and yield of our drug discovery and development processes. We believe that our unique understanding of the cytoskeleton and our proprietary research technologies should enable us to discover and potentially to develop drug candidates with novel mechanisms of action that may offer potential benefits not provided by existing drugs and which may have application across a broad array of diseases and medical conditions. We expect that we may be able to leverage our expertise in muscle contractility to expand programs related to other areas of muscle function and which may extend to the potential treatment of other serious medical diseases and conditions. Progressing related programs in parallel may afford us an opportunity to build a broader business that could benefit from multiple products that serve related clinical and commercial needs associated with impaired muscle function, muscle weakness and fatigue. In addition, this strategy may enable us to diversify certain technical, financial and operating risks by advancing several drug candidates. |
| • | Focusing on comprehensive development programs that may enhance the success of our activities directed to potential registration. We believe that by focusing on disease areas with well-organized physician-investigator groups, significant clinical unmet need, and strong patient and disease advocacy, we may enhance our effectiveness in enrolling and conducting clinical trials that may answer important questions about the dosing, tolerability, pharmacokinetics and pharmacodynamics as well as the potential safety and efficacy of our drug candidates. We believe that our considered clinical trial designs and well-executed development programs can improve our ability to realize value from our clinical development activities. We believe that our investing in these activities may result in more successful later-stage clinical development activities that may increase the likelihood of our achieving our objectives to develop effective therapeutics that may address the needs of patients with grievous diseases and conditions. |
| • | Building development and commercialization capabilities directed at concentrated and growing markets. We focus our drug discovery and development activities on disease areas for which there are serious unmet medical needs. In particular, we direct our activities to potential commercial opportunities |
5
Table of Contents
in concentrated and tractable customer segments, such as hospital specialists and disease-specific centers of excellence, which may be addressed by a smaller, targeted sales force. In preparing for the potential commercialization of our drug candidates directed to these markets, we are focusing our activities on a broad range of issues facing patients and payors, including the principal drivers of clinical and economic burdens associated with these diseases. We also seek to focus on opportunities that the multiple constituencies and stakeholders for these markets may recognize as creating value. Accordingly, targeting unmet medical needs in these areas may provide us competitive opportunities and support development of a franchise in diseases involving muscle weakness, wasting and fatigue. In these markets, we believe that a company with limited resources may be able to compete effectively against larger, more established companies with greater financial and commercial resources. For these opportunities, we intend to develop clinical development and sales and marketing capabilities with the goal of becoming a fully-integrated biopharmaceutical company. |
| • | Establishing select strategic alliances to support our drug development programs while preserving significant development and commercialization rights. We believe that such alliances may allow us to obtain financial support and to capitalize on the therapeutic area expertise and resources of our partners that can potentially accelerate the development and commercialization of our drug candidates. Where we deem appropriate, we plan to retain certain rights to participate in the development of drug candidates and commercialization of potential drugs arising from our programs and alliances, so that we can expand and capitalize on our own internal development capabilities and build our commercialization capabilities. |
Research and Development Programs
Our long-standing interest in the cytoskeleton has led us to focus our research and development activities on the biology of muscle function, and in particular, small molecule modulation of muscle contractility. We believe that our expertise in the modulation of muscle contractility is an important differentiator for us. Our preclinical and clinical experience in muscle contractility may position us to discover and develop additional novel therapies that have the potential to improve the health of patients with severe and debilitating diseases or medical conditions.
Small molecules that affect muscle contractility may have several applications for a variety of serious diseases and medical conditions. For example, certain diseases and medical conditions associated with muscle weakness may be amenable to treatment by enhancing the contractility of skeletal muscle, and heart failure is a disease often characterized by impaired cardiac muscle contractility which may be treated by modulating the contractility of cardiac muscle.
Because the modulation of the contractility of different types of muscle, such as cardiac muscle and skeletal muscle, may be relevant to multiple diseases or medical conditions, we believe we can leverage our expertise in these areas to more efficiently discover and develop as potential drugs compounds that modulate the applicable muscle type for multiple indications.
We are currently developing a number of small molecule compounds arising from our muscle contractility programs. Tirasemtiv is our lead drug candidate from our skeletal muscle contractility program. We are conducting a Phase II clinical trials program for tirasemtiv, including an ongoing Phase IIb clinical trial. Potential indications for which this drug candidate may be useful include skeletal muscle weakness associated with neuromuscular diseases, such as ALS. CK-2127107, another drug candidate from this program, is partnered with Astellas world-wide, and is in Phase I clinical development for the potential treatment of non-neuromuscular indications associated with muscle weakness. Omecamtiv mecarbil, a novel cardiac muscle myosin activator, is partnered with Amgen world-wide. An intravenous formulation of omecamtiv mecarbil has been studied in a Phase IIb clinical trial in patients with acute heart failure, and oral formulations of omecamtiv mecarbil are being studied in a Phase II clinical trial in patients with heart failure. We are also planning a Phase I study comparing the pharmacokinetics of omecamtiv mecarbil between healthy Japanese and Caucasian volunteers.
6
Table of Contents
We are continuing to conduct discovery, characterization and lead optimization activities for other compounds with the potential to modulate muscle contractility and other muscle functions, such as growth, energetics and metabolism.
Research and Development Expense. Our research and development expenses were $49.5 million, $35.6 million and $37.2 million for 2013, 2012 and 2011, respectively.
Skeletal Muscle Contractility Program
Overview. Our skeletal muscle contractility program is focused on the activation of the skeletal sarcomere, the basic unit of skeletal muscle contraction. The skeletal sarcomere is a highly ordered cytoskeletal structure composed of skeletal muscle myosin, actin, and a set of regulatory proteins, which include the troponins and tropomyosin. This program leverages our expertise developed in our ongoing discovery and development of cardiac sarcomere activators, including the cardiac muscle myosin activator omecamtiv mecarbil.
We believe that our skeletal sarcomere activators may lead to new therapeutic options for diseases and medical conditions associated with aging, muscle weakness and wasting and neuromuscular dysfunction. The clinical effects of muscle weakness and wasting, fatigue and loss of mobility can range from decreased quality of life to, in some instances, life-threatening complications. By directly improving skeletal muscle function, a small molecule activator of the skeletal sarcomere potentially could enhance functional performance and quality of life in patients suffering from diseases or medical conditions characterized or complicated by muscle weakness or wasting. These may include diseases and medical conditions associated with skeletal muscle weakness or wasting, such as ALS, claudication, myasthenia gravis, sarcopenia (general frailty associated with aging), post-surgical rehabilitation and cachexia in connection with heart failure or cancer.
Tirasemtiv is the lead drug candidate from this program, and is in Phase II clinical development. Cytokinetics holds the rights to tirasemtiv. We are also developing another drug candidate from this program, CK-2127107, which is being evaluated in Phase I clinical trials in collaboration with Astellas for potential non-neuromuscular indications associated with muscle weakness. Tirasemtiv and CK-2127107 are structurally distinct and selective small molecules that activate the fast skeletal muscle troponin complex in the sarcomere by increasing its sensitivity to calcium, leading to an increase in skeletal muscle contractility. Each of tirasemtiv and CK-2127107 has demonstrated pharmacological activity in preclinical models and tirasemtiv has demonstrated evidence of potentially clinically relevant pharmacodynamic effects in humans. We are evaluating potential indications for which tirasemtiv and CK-2127107 may be useful.
Tirasemtiv. Tirasemtiv, a fast skeletal troponin activator, is the lead drug candidate from our skeletal muscle contractility program. We have conducted three “evidence of effect” Phase IIa clinical trials of tirasemtiv. These evidence of effect clinical trials were randomized, double-blind, placebo-controlled, three-period cross-over studies of single doses of tirasemtiv administered to patients with impaired muscle function. These studies were intended to translate the mechanism of action of tirasemtiv into potentially clinically relevant pharmacodynamic effects, which may then form the basis for larger clinical trials designed to demonstrate proof of concept and possibly even to support registration. The first of these trials was conducted in patients with ALS, a chronic and progressive disease in which the motor neurons die, thus denervating skeletal muscles and causing them to atrophy. This leads to weakness, fatigue, and eventually complete paralysis and death, primarily from respiratory complications. The second of these trials was conducted in patients with myasthenia gravis, a chronic, autoimmune, neuromuscular disease which is the most common primary disorder of neuromuscular transmission. The third of these trials was conducted in patients with symptoms of claudication, which is pain or cramping in the leg muscles due to inadequate blood flow during exercise, associated with peripheral artery disease. Evidence of potentially clinically relevant pharmacodynamic effects was observed in each of these trials. We are now conducting BENEFIT-ALS (Blinded Evaluation of Neuromuscular Effects and Functional Improvement with Tirasemtiv in ALS), a Phase IIb clinical trial of tirasemtiv in patients with ALS. We anticipate that we will need to conduct at least one confirmatory Phase III clinical trial of tirasemtiv in patients with ALS, if supported by the results of BENEFIT-ALS, to gain marketing approval.
7
Table of Contents
Tirasemtiv Clinical Development
BENEFIT-ALS (Blinded Evaluation of Neuromuscular Effects and Functional Improvement with Tirasemtiv in ALS). In October 2012, we initiated BENEFIT-ALS, a multi-national, double-blind, randomized, placebo-controlled trial originally planned to enroll at least 400 patients and subsequently increased to enroll up to 500 patients. All patients began treatment with open-label tirasemtiv at 125 mg twice daily. Patients who completed a week of open-label tirasemtiv at this starting dose were randomized 1-to-1 to receive 12 weeks of double-blind treatment with tirasemtiv or placebo. Clinical assessments take place monthly during double-blinded treatment. Randomized patients also participate in follow-up evaluations at both 7 and 28 days after their final dose of double-blind study drug. The primary analysis of BENEFIT-ALS will compare the mean change from baseline in the ALS Functional Rating Scale in its revised form, or ALSFRS-R (a clinically validated instrument designed to measure disease progression and changes in functional status), in patients receiving tirasemtiv versus those receiving placebo. Secondary endpoints will include maximum voluntary ventilation, or MVV (a clinical assessment of pulmonary function and endurance), and measures of skeletal muscle function. Patients will receive tirasemtiv or placebo dosed twice daily. Patients taking riluzole (an approved treatment for ALS) at the time of enrollment who were randomized to receive double-blind tirasemtiv received riluzole at a reduced dose of 50 mg daily, in a blinded manner. We are conducting BENEFIT-ALS at over 70 sites across the United States, Canada and several European countries.
In July 2013, we were informed by our data management vendor that a programming error in the electronic data capture system controlling study drug assignment caused 58 patients initially randomized to and treated with tirasemtiv to receive placebo instead at a certain study visit and for the remainder of the study. No patients randomized to placebo were dispensed incorrect treatment. Cytokinetics and all clinical trial site personnel remain blinded to the specific patients affected by the error. Following detection of the error, we took steps to ensure that no further incorrect study drug assignments occurred and to correct the programming error in the electronic data capture system controlling study drug assignment. In addition, we convened an ad hoc meeting of the Data Safety Monitoring Board (DSMB) for BENEFIT-ALS to assess whether the error in dispensing study drug had impacted the safety of the 58 affected patients. After review of the then-available safety data from BENEFIT-ALS, the DSMB reported no concerns regarding patient safety. Following interactions with regulatory authorities, we amended the protocol for BENEFIT-ALS to enable increased enrollment to approximately 680 patients and to update the statistical methods section, in both cases with the objective to maintain the originally intended statistical power of the trial. We have concluded enrollment in BENEFIT-ALS with 711 patients of which over 400 have completed 12 weeks of treatment, and anticipate reporting the results from this clinical trial at the American Academy of Neurology (AAN) Annual Meeting in Philadelphia, PA in April 2014.
Prior ALS Clinical Trials. In June 2012, we announced the publication of our Phase IIa evidence of effect clinical trial of tirasemtiv (CY 4021) in the online edition of the journal Amyotrophic Lateral Sclerosis. In that trial, the single doses of tirasemtiv evaluated appeared generally well-tolerated, with dizziness and general fatigue being the most frequent adverse events. In addition, both patients and investigators perceived a positive change in the patients’ overall status, in a dose-dependent fashion, at 6 hours after dosing with tirasemtiv, based on a global assessment in which the patient and the investigator each independently assessed patients’ status compared to prior to dosing. There was a clear relationship between improvements in global assessments and the plasma concentrations of tirasemtiv. Also at this 6-hour time point, there was a trend towards decreased muscle fatigability, as evidenced by data from a test of sub-maximal hand-grip endurance. Data from that clinical trial also demonstrated a statistically significant increase in MVV at both 6 and 24 hours after 500 mg of tirasemtiv, and small but statistically significant increases in maximum strength of certain muscle groups tested.
In April 2012, at the AAN 64th Annual Meeting, results were presented from CY 4024, a Phase II, two-part, randomized, double-blind, placebo-controlled, multiple-dose, safety, tolerability, pharmacokinetic and pharmacodynamic clinical trial of tirasemtiv in patients with ALS. Patients in Part A of this trial were not taking riluzole; patients in Part B received riluzole at the reduced dose of 50 mg daily. In this trial, tirasemtiv appeared to be generally safe and well-tolerated when dosed daily at 125 mg, 250 mg and 375 mg once daily for two weeks. This trial was not designed or powered to evaluate statistically the effects of tirasemtiv on the various
8
Table of Contents
outcome measures that were assessed during the study. However, encouraging dose-related trends were observed in measurements of ALSFRS-R and in MVV. Plasma concentrations of tirasemtiv were unaffected by co-administration with riluzole, while riluzole plasma levels were greater when co-administered with tirasemtiv than when not co-administered with tirasemtiv. Adverse events and clinical assessments during treatment with tirasemtiv appeared similar, with or without co-administration of riluzole. Dizziness, the most commonly reported adverse event, was mostly mild and generally began and resolved early after initiating treatment. The incidence and persistence of dizziness appeared dose-related but was mild in severity in all patients who completed study drug treatment. Most reports of dizziness began early after initiating treatment and resolved spontaneously within the first week of treatment in all but one patient who nevertheless completed the trial. No serious adverse events were reported.
Also in April 2012 at the AAN 64th Annual Meeting, results were presented from a Phase II, randomized, double-blind, placebo-controlled, multiple-dose, clinical trial of tirasemtiv in patients with ALS receiving riluzole at the reduced dose of 50 mg daily (CY 4025). The authors concluded that the twice-daily dose titration regimen evaluated in the trial appeared generally safe and well-tolerated, and that the majority of patients could be titrated successfully to a tirasemtiv dose level of 250 mg twice daily. This trial was not designed or powered to evaluate statistically the effects of tirasemtiv on the various outcome measures that were assessed during the study. However, encouraging trends toward functional improvements were observed in patients receiving tirasemtiv versus those receiving placebo. In this trial, tirasemtiv treatment was associated with increases in measurements of ALSFRS-R that were similar in direction, and in MVV that were similar in both direction and magnitude, to those observed in CY 4024.
Background on ALS Market. Limited options exist for the treatment of patients with ALS, which affects as many as 30,000 Americans, with an estimated 5,600 new cases diagnosed each year in the U.S. Based on our proprietary research, the per capita prevalence and incidence appears similar in the major European markets. ALS is 20% more common in men than women; however, with increasing age, the prevalence becomes more equal between men and women. The life expectancy of an ALS patient averages two to five years from the time of diagnosis with 90 to 95% of those diagnosed with ALS having the sporadic form. Of the remaining ALS patient population, 5 to 10% have a family history of the disease (familial ALS). In cases of familial ALS, there is an approximately 50% chance each offspring will develop the disease. Based on our proprietary research, the majority of patients with ALS in the U.S. and Europe receive treatment at multidisciplinary centers that specialize in the unique needs of these patients. In the U.S., there are approximately 90 ALS centers of excellence, according to either the ALS Association or the Muscular Dystrophy Association. For most patients with ALS, death is usually due to respiratory failure because of diminished strength in the skeletal muscles responsible for breathing. We are evaluating other market opportunities for our skeletal muscle sarcomere activators.
CK-2127107 and Other Skeletal Muscle Activators
Astellas Strategic Alliance. In June 2013, we entered into a collaboration and license agreement with Astellas (the “Astellas Agreement”). Under the Astellas Agreement, we granted Astellas an exclusive license to co-develop and jointly commercialize CK-2127107 for potential application in non-neuromuscular indications associated with skeletal muscle weakness worldwide. CK-2127107 is being developed jointly by Cytokinetics and Astellas. Cytokinetics is primarily responsible for the conduct of Phase I clinical trials and certain Phase II readiness activities for CK-2127107. Astellas will be primarily responsible for the conduct of subsequent development and commercialization activities for CK-2127107.
The companies are jointly conducting research to identify next-generation skeletal muscle activators to be nominated as potential drug candidates, at Astellas’ expense, over a two-year term, which may be extended by the companies’ mutual consent. Astellas has the exclusive rights to develop and commercialize fast skeletal troponin activators from this research program in non-neuromuscular indications and to develop and commercialize other novel mechanism skeletal muscle activators from this research program in all indications,
9
Table of Contents
subject to certain Cytokinetics’ co-development and co-promotion rights. Astellas will be responsible for the costs associated with the development of all collaboration products, including CK-2127107.
Under the Astellas Agreement, we retain an option to conduct early-stage development for certain agreed indications at our initial expense, subject to reimbursement if development continues under the collaboration. We also retain an option to co-promote collaboration products in the United States and Canada. Astellas will reimburse us for certain expenses associated with our co-promotion activities.
In July 2013, we received an upfront payment of $16 million in connection with the execution of the Astellas Agreement, and we are eligible to potentially receive over $24 million in reimbursement of sponsored research and development activities during the initial two years of the collaboration. Based on the achievement of pre-specified criteria, we may receive over $250 million in milestone payments relating to the development and commercial launch of collaboration products, including up to $112 million in development and commercial launch milestones for CK-2127107. We may also receive up to $200 million in payments for achievement of pre-specified sales milestones related to net sales of all collaboration products under the Astellas Agreement. If Astellas commercializes any collaboration products, we will also receive royalties on sales of such collaboration products, including royalties ranging from the high single digits to the high teens on sales of products containing CK-2127107. In addition to these development, commercial launch and sales milestones, we may also receive payments for the achievement of pre-specified milestones relating to the joint research program.
CK-2127107 Clinical Development
CY 5011. In April 2013, we announced the initiation of a first-time-in-humans Phase I clinical trial of CK-2127107 in healthy male volunteers, known as CY 5011. CY 5011 was a double-blind, randomized, placebo-controlled study designed to assess the safety, tolerability, and pharmacokinetics of single ascending oral doses of CK-2127107 administered in a three-period crossover design. We announced results from this trial in December 2013 at the 7th International Conference of the Society on Sarcopenia, Cachexia and Wasting Disorders in Kobe, Japan. Planned single doses of CK-2127107 up to 4000 mg, the highest dose administered in this trial, were well-tolerated; therefore, a maximum tolerated dose was not defined. The pharmacokinetic profile of CK-2127107 was linear and dose-proportional across the dose range studied, with a mean terminal half-life compatible with once or twice daily dosing.
CY 5014. During the fourth quarter of 2013, Cytokinetics completed dosing in CY 5014, a Phase I clinical trial of CK-2127107 in healthy male volunteers. CY 5014 is a randomized, open-label, two-period crossover study designed to assess the relative oral bioavailability, pharmacokinetics, safety and tolerability of two oral formulations of CK-2127107.
In 2014, we expect to conduct additional Phase I studies and certain Phase II readiness activities pursuant to our collaboration agreement with Astellas.
Ongoing Research in Skeletal Muscle Activators. Our research on the direct activation of skeletal muscle continues in two areas. We are conducting translational research in preclinical models of disease and muscle function with fast skeletal muscle troponin activators to explore the potential clinical applications of this novel mechanism in diseases or conditions associated with skeletal muscle dysfunction. We also intend to conduct preclinical research on other chemically and pharmacologically distinct mechanisms to activate the skeletal sarcomere. We are conducting a joint research program with Astellas directed to the discovery of next-generation skeletal muscle activators. Under the Astellas Agreement, Astellas will reimburse us for certain research activities we perform.
Cardiac Muscle Contractility Program
Overview. Our cardiac muscle contractility program is focused on the cardiac sarcomere, the basic unit of muscle contraction in the heart. The cardiac sarcomere is a highly ordered cytoskeletal structure composed of
10
Table of Contents
cardiac muscle myosin, actin and a set of regulatory proteins. This program is currently directed towards the discovery and development of small molecule cardiac muscle myosin activators with the goal of developing novel drugs to treat acute and chronic heart failure. Cardiac muscle myosin is the cytoskeletal motor protein in the cardiac muscle cell. It is directly responsible for converting chemical energy into the mechanical force, resulting in cardiac muscle contraction. This program is based on the hypothesis that activators of cardiac muscle myosin may address certain adverse properties of existing positive inotropic agents. Current positive inotropic agents, such as beta-adrenergic receptor agonists or inhibitors of phosphodiesterase activity, increase the concentration of intracellular calcium, thereby increasing cardiac sarcomere contractility. The effect on calcium levels, however, also has been linked to potentially life-threatening side effects. In contrast, our novel cardiac muscle myosin activators work by a mechanism that directly stimulates the activity of the cardiac muscle myosin motor protein, without increasing the intracellular calcium concentration. They accelerate the rate-limiting step of the myosin enzymatic cycle and shift it in favor of the force-producing state. Rather than increasing the velocity of cardiac contraction, this mechanism instead lengthens the systolic ejection time, which results in increased cardiac function in a potentially more oxygen-efficient manner.
Amgen Strategic Alliance. In December 2006, we entered into a collaboration and option agreement with Amgen to discover, develop and commercialize novel small molecule therapeutics, including omecamtiv mecarbil, that activate cardiac muscle contractility for potential applications in the treatment of heart failure (the “Amgen Agreement”). The agreement granted Amgen an option to obtain an exclusive license worldwide, except Japan, to develop and commercialize omecamtiv mecarbil and other drug candidates arising from the collaboration. In May 2009, Amgen exercised its option. As a result, Amgen became responsible for the development and commercialization of omecamtiv mecarbil and related compounds at its expense worldwide (excluding Japan), subject to our development and commercialization participation rights. Amgen will reimburse us for certain research and development activities we perform under the collaboration.
In June 2013, Cytokinetics and Amgen executed an amendment to the Amgen Agreement to include Japan, resulting in a worldwide collaboration (the “Amgen Agreement Amendment”). Under the terms of the Amgen Agreement Amendment, we received a non-refundable upfront license fee of $15 million in June 2013. Under the Amgen Agreement Amendment, we plan to conduct a Phase I pharmacokinetic study intended to support inclusion of Japan in a potential Phase III clinical development program and potential global registration dossier for omecamtiv mecarbil. Amgen will reimburse us for the costs of this study. In addition, we are eligible to receive additional pre-commercialization milestone payments relating to the development of omecamtiv mecarbil in Japan of up to $50 million, and royalties on sales of omecamtiv mecarbil in Japan. In conjunction with the Amgen Agreement Amendment, we also entered into a common stock purchase agreement which provided for the sale of 1,404,100 shares of our common stock to Amgen at a price per share of $7.12 and an aggregate purchase price of $10.0 million which was received in June 2013. Pursuant to this agreement, Amgen has agreed to certain trading and other restrictions with respect to our common stock.
Under the Amgen Agreement, as amended, we are eligible for potential pre-commercialization and commercialization milestone payments of up to $650 million in the aggregate on omecamtiv mecarbil and other potential products arising from research under the collaboration, and royalties that escalate based on increasing levels of annual net sales of products commercialized under the agreement. The Amgen Agreement also provides for us to receive increased royalties by co-funding Phase III development costs of omecamtiv mecarbil and other drug candidates under the collaboration. If we elect to co-fund such costs, we would be entitled to co-promote the co-funded drug in North America and participate in agreed commercialization activities in institutional care settings, at Amgen’s expense.
In July 2013, Amgen announced that it had granted an option to commercialize omecamtiv mecarbil in Europe to Servier.
Omecamtiv Mecarbil. Our lead drug candidate from this program is omecamtiv mecarbil, a novel cardiac muscle myosin activator.
11
Table of Contents
We expect omecamtiv mecarbil to be developed as a potential treatment across the continuum of care in heart failure both as an intravenous formulation for use in the hospital setting and as an oral formulation for use in the outpatient setting.
Omecamtiv Mecarbil Clinical Development
Current Clinical Trials. In March 2013, we announced the initiation of dosing of patients in COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure). COSMIC-HF is a Phase II, double-blind, randomized, placebo-controlled, multicenter, dose escalation study designed to evaluate several modified-release oral formulations of omecamtiv mecarbil in patients with heart failure and left ventricular systolic dysfunction. COSMIC-HF is being conducted by Amgen in collaboration with Cytokinetics. The primary objectives of this trial are to select an oral modified release formulation and doses of omecamtiv mecarbil for chronic twice-daily dosing in patients with heart failure and left ventricular systolic dysfunction and to characterize its pharmacokinetics during 20 weeks of treatment. The secondary objective is to evaluate the safety and tolerability of oral omecamtiv mecarbil. In addition, we will have an opportunity to evaluate the potential for sustained pharmacodynamic effects and their relationship to the pharmacokinetics of this drug candidate. During the third quarter of 2013, the second cohort of the dose escalation phase of COSMIC-HF completed enrollment. We and Amgen recently reviewed results from COSMIC-HF and selected an oral formulation of omecamtiv mecarbil for evaluation in the planned expansion phase of the trial. We and Amgen have agreed to amend the protocol to evaluate a plasma concentration-guided dose titration strategy in the expansion phase of COSMIC-HF. The size of the expansion phase has been increased with the objective to provide greater statistical power for the planned evaluation of several pharmacodynamic parameters during oral dosing with omecamtiv mecarbil. This trial is being conducted by Amgen in collaboration with Cytokinetics.
Recently, Cytokinetics and Amgen agreed on the protocol and budget for the planned Phase I pharmacokinetic study, CY 1211, in healthy volunteers of both Japanese and non-Japanese ethnicity. The trial will be conducted by Cytokinetics in collaboration with Amgen. The costs of the trial will be reimbursed by Amgen.
We are collaborating with Amgen to respond to information requests received from regulatory authorities relating to their ongoing review of the protocols submitted for COSMIC-HF and CY 1211. We anticipate commencement of patient enrollment in both the expansion phase of COSMIC-HF and in CY 1211 to occur in the first half of 2014 following regulatory authorities’ review of responses relating to these information requests. We expect both the enrollment of patients in the expansion phase of COSMIC-HF and the conduct of CY 1211 to be completed in 2014.
ATOMIC-AHF. In September 2013, results from ATOMIC-AHF (Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) were presented at the European Society of Cardiology Congress and the Heart Failure Society of America Annual Scientific Meeting. ATOMIC-AHF was an international, randomized, double-blind, placebo-controlled, Phase IIb clinical trial of intravenous omecamtiv mecarbil in patients with left ventricular systolic dysfunction hospitalized with acutely decompensated heart failure. ATOMIC-AHF was conducted by Amgen in collaboration with Cytokinetics. This clinical trial enrolled over 600 patients in three sequential, ascending-dose cohorts. In each cohort, patients were randomized to receive omecamtiv mecarbil or placebo. The primary efficacy objective of this trial was to evaluate the effect of 48 hours of intravenous omecamtiv mecarbil compared to placebo on dyspnea (shortness of breath). The secondary objectives were to assess the safety and tolerability of three dose levels of intravenous omecamtiv mecarbil compared with placebo and to evaluate the effects of 48 hours of treatment with intravenous omecamtiv mecarbil on additional measures of dyspnea, patients’ global assessments, change in N-terminal pro brain-type natriuretic peptide (a biomarker associated with the severity of heart failure) and short-term clinical outcomes in these patients. In addition, the trial evaluated the relationship between plasma concentrations of omecamtiv mecarbil and echocardiographic parameters in patients with acute heart failure.
12
Table of Contents
The omecamtiv mecarbil treatment groups were not statistically different in their 7-point Likert scale dyspnea symptom response rates compared to the pooled placebo group (p=0.33); therefore, the primary endpoint was not met. Omecamtiv mecarbil demonstrated favorable dose- and concentration-related trends (nominal p=0.025 and nominal p=0.007, respectively) on dyspnea response. Improvement in dyspnea was observed in the highest omecamtiv mecarbil dose group when compared against its paired placebo group in the third cohort (dyspnea symptom response in 51 percent of subjects on omecamtiv mecarbil versus 37 percent on placebo, nominal p=0.03). The incidence of worsening heart failure within seven days of initiating treatment was 17 percent in the pooled placebo group and was 13 percent, 8 percent and 9 percent on omecamtiv mecarbil in the first, second and third cohorts, respectively. Systolic ejection time, the echocardiographic signature of omecamtiv mecarbil, increased in a concentration-dependent manner similar to that previously reported in healthy volunteers and stable heart failure patients.
Rates of adverse events (AEs), serious AEs, adjudicated deaths and hospitalizations were similar between omecamtiv mecarbil and placebo groups. There were seven post-randomization myocardial infarctions in the treatment groups receiving omecamtiv mecarbil compared with three in the placebo groups (2.3 percent vs. 1.0 percent, respectively). However, there was no relationship between the maximum increase from the baseline troponin (a biomarker specific for cardiac muscle damage) and increasing plasma concentrations of omecamtiv mecarbil. Four of the myocardial infarctions occurred more than seven days following termination of the 48-hour drug infusion. The estimated plasma concentrations near the time of these events were zero. Three of these events occurred in patients who received omecamtiv mecarbil and one occurred in a patient who received placebo. One myocardial infarction occurred subsequent to a percutaneous coronary intervention in a patient who received omecamtiv mecarbil. One myocardial infarction occurred in a patient with sepsis who received placebo. Omecamtiv mecarbil was not associated with an increased incidence of tachyarrhythmias nor were heart rate or blood pressure adversely affected.
Prior Clinical Experience with Omecamtiv Mecarbil. Prior to Amgen’s exercise of its option, Cytokinetics conducted a clinical trials program for omecamtiv mecarbil comprised of multiple Phase I and Phase IIa clinical trials designed to evaluate the safety, tolerability, pharmacodynamic and pharmacokinetic profiles of both intravenous and oral formulations in a diversity of patients, including patients with stable heart failure and patients with ischemic cardiomyopathy. In these trials, omecamtiv mecarbil exhibited generally linear, dose-proportional pharmacokinetics across the dose ranges studied. The adverse effects observed at intolerable doses in humans appeared similar to the adverse findings which occurred in preclinical safety studies at similar plasma concentrations. These effects are believed to be related to the mechanism of action of this drug candidate which, at intolerable doses, resulted in an excessive prolongation of the systolic ejection time (i.e., the time in which the heart is contracting). However, these effects resolved promptly with discontinuation of the infusions of omecamtiv mecarbil.
Phase IIa stable heart failure (safety, tolerability, pharmacokinetics and pharmacodynamics). In 2009, we presented final results from our Phase IIa clinical trial evaluating omecamtiv mecarbil administered intravenously to patients with stable heart failure. The final results showed statistically significant increases in systolic ejection time, and in stroke volume, cardiac output, fractional shortening and ejection fraction (all measures of cardiac function), that occurred across the patient population in a concentration-dependent manner. In addition, the results demonstrated statistically significant correlations between increasing omecamtiv mecarbil plasma concentrations and decreases in left ventricular end-systolic volume, left ventricular end-diastolic volume and heart rate. Omecamtiv mecarbil appeared to be generally well-tolerated in stable heart failure patients over a range of plasma concentrations during continuous intravenous administration. Patients with reduced stroke volumes (<50 ml) at baseline had generally greater pharmacodynamic responses to omecamtiv mecarbil than those in patients with greater stroke volumes at baseline, demonstrating robust pharmacodynamic activity in this more severely affected sub-population of patients from the trial.
Phase IIa ischemic cardiomyopathy and angina (safety and tolerability). In 2009, we presented results from a double-blind, randomized, placebo-controlled Phase IIa clinical trial evaluating the effect of omecamtiv
13
Table of Contents
mecarbil on symptom-limited exercise tolerance in heart failure patients with ischemic cardiomyopathy and angina. The primary safety endpoint of this clinical trial was stopping an exercise treadmill test due to angina at a stage earlier than the shorter of two baseline exercise treadmill tests. This endpoint occurred in one patient receiving placebo and in no patients receiving either the lower or higher of two dose levels of omecamtiv mecarbil. In heart failure patients with ischemic cardiomyopathy and angina, who theoretically could be most vulnerable to the possible deleterious consequences of systolic ejection time prolongation, treatment with omecamtiv mecarbil, at doses producing plasma concentrations previously demonstrated in other trials to increase cardiac function, did not appear to deleteriously affect a broad range of safety assessments in the setting of exercise.
Phase I Clinical Trials. Seven Phase I clinical trials of omecamtiv mecarbil have been conducted in healthy subjects: five conducted by Cytokinetics and two conducted by Amgen in collaboration with Cytokinetics. Results from these trials were reported previously.
Ongoing Research in Cardiac Muscle Contractility. In 2013, we agreed with Amgen to additional research activities intended to be conducted through 2014 under the research plan directed to next-generation compounds in our cardiac muscle contractility program. Under the Amgen Agreement, Amgen will reimburse us for certain research activities we perform.
Background on Heart Failure Market. Heart failure is a widespread and debilitating syndrome affecting millions of people in the United States. The high and rapidly growing prevalence of heart failure translates into significant hospitalization rates and associated societal costs. About 5.8 million people in the United States have heart failure, resulting in nearly one million hospital discharges with the primary diagnosis of heart failure and approximately 300,000 deaths each year. For people over 65 years of age, heart failure incidences approach 10 per 1000 and approximately 50% of people diagnosed with heart failure will die within 5 years of diagnosis. These numbers are increasing due to the aging of the U.S. population and an increased likelihood of survival following acute myocardial infarctions. The costs to society attributable to the prevalence of heart failure are high, especially as many chronic heart failure patients suffer repeated acute episodes. Despite currently available therapies, readmission rates for heart failure patients remain high. In general, the mortality following hospitalization for patients with heart failure is 10.4% at 30 days, 22% at one year and 42.3% at 5 years, despite the availability of therapeutic alternatives for treatment of these patients. These poor outcomes in the setting of current therapies points to the need for novel therapeutics that may offer further reductions in morbidity and mortality. The annual cost of heart failure to the U.S. health care system is estimated to be $32 billion and is predicted to grow 120% to almost $70 billion by the year 2030. Today, a portion of that cost is attributable to drugs used to treat each of chronic and acute heart failure. Approximately 70% of those costs are due to hospitalization, home health and physician care. In the U.S., Medicare is one of the largest payors for heart failure related costs. Approximately 50% of Medicare beneficiaries with heart failure are concentrated in the top 20% of the hospital referral regions in the U.S, which generally include 5 to 10 hospitals in a geographic area. New drug therapies that could reduce the number of hospitalizations could decrease the cost to the health care system.
Beyond Muscle Contractility
We have developed preclinical expertise in the mechanics of skeletal, cardiac and smooth muscle that extends from proteins to tissues to intact animal models. Our translational research in muscle contractility has enabled us to better understand the potential impact of small molecule compounds that increase skeletal or cardiac muscle contractility and to apply those findings to the further evaluation of our drug candidates in clinical populations. In addition to contractility, the other major functions of muscle include metabolism, growth and energetics, with each of these functions playing a role in certain diseases that could benefit from novel mechanism treatments. Accordingly, our knowledge of muscle contractility may serve as an entry point to the discovery of novel treatments for disorders involving muscle functions other than muscle contractility. We are leveraging our current understandings of muscle biology to investigate new ways of modulating these other
14
Table of Contents
aspects of muscle function for other potential therapeutic applications. For example, we are conducting research with compounds that affect muscle growth and that may have applications for serious diseases and medical conditions such as cachexia. Cachexia is a condition that can be associated with cancer, heart failure, chronic obstructive pulmonary disease or other conditions. This syndrome is characterized by the loss of muscle mass and may lead to weakness and disability. We are performing research on compounds that may increase muscle mass and which may impact patient functionality or potentially alter the course of diseases associated with muscle wasting.
Our Intellectual Property
Our policy is to seek patent protection for the technologies, inventions and improvements that we develop that we consider important to the advancement of our business. As of December 31, 2013, we owned or controlled 119 issued U.S. patents and over 150 additional pending U.S. and foreign patent applications. We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. Our commercial success will depend on obtaining and maintaining patent protection and trade secret protection for our drug candidates and technologies and our successfully defending these patents against third-party challenges. We will only be able to protect our technologies from unauthorized use by third parties to the extent that valid and enforceable patents cover them or we maintain them as trade secrets.
With regard to our drug candidates directed to muscle biology targets, we have a U.S. patent covering omecamtiv mecarbil and a U.S. patent covering our skeletal muscle sarcomere activators including, but not limited to, tirasemtiv, each of which will expire in 2027 unless extended. We also have additional U.S. and foreign patent applications pending for each of our drug candidates. It is not known or determinable whether other patents will issue from any of our other pending applications or what the expiration dates would be for any other patents that do issue.
All of our drug candidates are still in clinical development and have not yet been approved by the FDA. If any of these drug candidates is approved, then pursuant to federal law, we may apply for an extension of the U.S. patent term for one patent covering the approved drug, which could extend the term of the applicable patent by up to a maximum of five additional years.
The degree of future protection of our proprietary rights is uncertain because legal means may not adequately protect our rights or permit us to gain or keep our competitive advantage. Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the claim scope of these patents, our ability to enforce our existing patents and to obtain and enforce patents that may issue from any pending or future patent applications is uncertain and involves complex legal, scientific and factual questions. The standards that the U.S. Patent and Trademark Office and its foreign counterparts use to grant patents are not always applied predictably or uniformly and are subject to change. To date, no consistent policy has emerged regarding the breadth of claims allowed in biotechnology and pharmaceutical patents. Thus, we cannot be sure that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents do issue, we cannot be sure that the claims of these patents will be held valid or enforceable by a court of law, will provide us with any significant protection against competitive products, or will afford us a commercial advantage over competitive products. For example:
| • | we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| • | we or our licensors might not have been the first to file patent applications for the inventions covered by our pending patent applications and issued patents; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| • | some or all of our or our licensors’ pending patent applications may not result in issued patents or the claims that issue may be narrow in scope and not provide us with competitive advantages; |
15
Table of Contents
| • | our and our licensors’ issued patents may not provide a basis for commercially viable drugs or therapies or may be challenged and invalidated by third parties; |
| • | our or our licensors’ patent applications or patents may be subject to interference, opposition or similar administrative proceedings that may result in a reduction in their scope or their loss altogether; |
| • | we may not develop additional proprietary technologies or drug candidates that are patentable; or |
| • | the patents of others may prevent us or our partners from discovering, developing or commercializing our drug candidates. |
The defense and prosecution of intellectual property infringement suits, interferences, oppositions and related legal and administrative proceedings are costly, time-consuming to pursue and result in diversion of resources. The outcome of these types of proceedings is uncertain and could significantly harm our business.
Our ability to commercialize drugs depends on our ability to use, manufacture and sell those drugs without infringing the patents or other proprietary rights of third parties. U.S. and foreign issued patents and pending patent applications owned by third parties exist that may be relevant to the therapeutic areas and chemical compositions of our drug candidates. While we are aware of certain relevant patents and patent applications owned by third parties, there may be issued patents or pending applications of which we are not aware that could cover our drug candidates. Because patent applications are often not published immediately after filing, there may be currently pending applications, unknown to us, which could later result in issued patents that our activities with our drug candidates could infringe.
The development of our drug candidates and the commercialization of any resulting drugs may be impacted by patents of companies engaged in competitive programs with significantly greater resources. This could result in the expenditure of significant legal fees and management resources.
We also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are often difficult to protect, especially outside of the United States. While we believe that we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, partners and other advisors may unintentionally or willfully disclose our trade secrets to competitors. Enforcing a claim that a third party illegally obtained and is using our trade secrets would be expensive and time-consuming, and the outcome would be unpredictable. Even if we are able to maintain our trade secrets as confidential, our competitors may independently develop information that is equivalent or similar to our trade secrets.
We seek to protect our intellectual property by requiring our employees, consultants, contractors and other advisors to execute nondisclosure and invention assignment agreements upon commencement of their employment or engagement, through which we seek to protect our intellectual property. Agreements with our employees also preclude them from bringing the proprietary information or materials of third parties to us. We also require confidentiality agreements or material transfer agreements from third parties that receive our confidential information or materials.
For further details on the risks relating to our intellectual property, please see the risk factors under Item 1A of this report, including, but not limited to, the risk factors entitled “Our success depends substantially upon our ability to obtain and maintain intellectual property protection relating to our drug candidates and research technologies” and “If we are sued for infringing third party intellectual property rights, it will be costly and time-consuming, and an unfavorable outcome would have a significant adverse effect on our business.”
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, manufacture, marketing and distribution of
16
Table of Contents
drugs. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, labeling, storage, record keeping, approval, advertising and promotion of our drug candidates and drugs.
In the United States, the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act and implementing regulations. The process required by the FDA before our drug candidates may be marketed in the United States generally involves the following:
| • | completion of extensive preclinical laboratory tests, preclinical animal studies and formulation studies, all performed in accordance with the FDA’s good laboratory practice regulations; |
| • | submission to the FDA of an IND, which must become effective before clinical trials may begin; |
| • | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the drug candidate for each proposed indication in accordance with good clinical practices; |
| • | submission of a new drug application (“NDA”) to the FDA, which must usually be accompanied by payment of a substantial user fee; |
| • | satisfactory completion of an FDA preapproval inspection of the manufacturing facilities at which the product is produced to assess compliance with current good manufacturing practice (“cGMP”) regulations and FDA audits of select clinical investigator sites to assess compliance with good clinical practices (“GCP”); and |
| • | FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug. |
Similar regulatory procedures generally apply in countries outside of the United States. This testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our drug candidates will be granted on a timely basis, if at all.
Nonclinical tests include laboratory evaluation of product chemistry, formulation and stability, and studies to evaluate toxicity and pharmacokinetics in animals. The results of nonclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects may be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Our submission of an IND or a foreign equivalent, or those of our collaborators, may not result in authorization from the FDA or its foreign equivalent to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board (“IRB”) or its foreign equivalent for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the clinical trial until completed. The FDA, the IRB or their foreign equivalents, or the clinical trial sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Clinical Trials. For purposes of an NDA or equivalent submission and approval, clinical trials are typically conducted in the following three sequential phases, which may overlap:
| • | Phase I: Phase I includes the initial introduction of a drug candidate into humans. These studies are closely monitored and may be conducted in patients, but are usually conducted in healthy volunteer subjects. These studies are designed to determine the metabolic and pharmacologic actions of the drug candidate in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. During Phase I, sufficient information about the drug candidate’s pharmacokinetics and pharmacological effects should be obtained to permit the design of well-controlled, scientifically valid, Phase II studies. |
17
Table of Contents
| • | Phase II: Phase II includes the early controlled clinical studies conducted to obtain some preliminary data on the effectiveness of the drug for a particular indication or indications in patients with the disease or condition. This phase of testing also helps determine the common short-term side effects and risks associated with the drug candidate. These clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to make an initial determination of potential efficacy of the drug candidate for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase III clinical trials. Phase IIa clinical trials generally are designed to study the pharmacokinetic or pharmacodynamic properties and to conduct a preliminary assessment of safety of the drug candidate over a measured dose response range. In some cases, a sponsor may decide to conduct a Phase IIb clinical trial, which is a second, typically larger, confirmatory Phase II trial that could, if positive and accepted by a regulatory authority, serve as a pivotal clinical trial in the approval of a drug candidate. |
| • | Phase III: These clinical trials are commonly referred to as pivotal clinical trials. If the Phase II clinical trials demonstrate that a dose range of the drug candidate is potentially effective and has an acceptable safety profile, Phase III clinical trials are then undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites. Phase III trials are also intended to provide an adequate basis for extrapolating the results to the general population and transmitting that information in the drug labeling. Phase III studies usually include several hundred to several thousand people. |
At any time during the conduct of a clinical trial, the FDA or a foreign equivalent can impose a clinical hold on the trial if it believes the trial is unsafe or that the protocol is clearly deficient in design in meeting its stated objectives, which requires the conduct of the trial to cease until the clinical hold is removed. In some cases, the FDA or foreign equivalent may condition approval of marketing approval for a drug candidate on the sponsor’s agreement to conduct additional clinical trials to further assess the drug’s safety and effectiveness after marketing approval, known as Phase IV clinical trials.
The clinical trials we conduct for our drug candidates, both before and after approval, and the results of those trials, are generally required to be included in a clinical trials registry database that is available and accessible to the public via the internet. A failure by us to properly participate in the clinical trial database registry could subject us to significant civil monetary penalties.
Health care providers in the United States, including research institutions from which we or our partners obtain patient information, are subject to privacy rules under the Health Insurance Portability and Accountability Act of 1996 and state and local privacy laws. In the European Union, these entities are subject to the Directive 95/46-EC of the European Parliament on the protection of individuals with regard to the processing of personal data and individual European Union member states implementing additional legislation. Other countries have similar privacy legislation. We could face substantial penalties if we knowingly receive individually identifiable health information from a health care provider that has not satisfied the applicable privacy laws. In addition, certain privacy laws and genetic testing laws may apply directly to our operations and/or those of our partners and may impose restrictions on the use and dissemination of individuals’ health information and use of biological samples.
New Drug/Marketing Approval Application. The results of drug candidate development, preclinical testing and clinical trials are submitted to the FDA as part of an NDA. The NDA also must contain extensive manufacturing information. In addition, the FDA may require that a proposed Risk Evaluation and Mitigation Strategy, also known as a REMS, be submitted as part of the NDA if the FDA determines that it is necessary to ensure that the benefits of the drug outweigh its risks. Similar, and in some cases additional, requirements apply in foreign jurisdictions for marketing approval applications for drugs in those jurisdictions. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application
18
Table of Contents
should be approved. The FDA often, but not always, follows the advisory committee’s recommendations. The FDA may deny approval of an NDA by issuance of a complete response letter if the applicable regulatory criteria are not satisfied, or it may require additional clinical data, including data in a pediatric population, or an additional pivotal Phase III clinical trial or impose other conditions that must be met in order to secure final approval for an NDA. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our partners do. Once issued, the FDA or foreign equivalent may withdraw a drug approval if ongoing regulatory requirements are not met or if safety problems occur after the drug reaches the market. In addition, the FDA or its foreign counterparts may require further testing, including Phase IV clinical trials, and surveillance or restrictive distribution programs to monitor the effect of approved drugs which have been commercialized. The FDA and its foreign counterparts have the power to prevent or limit further marketing of a drug based on the results of these post-marketing programs. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to a drug, including changes in indications, labeling or manufacturing processes or facilities, we may be required to submit and obtain prior FDA approval of a new NDA or NDA supplement, or the foreign equivalent, which may require us to develop additional data or conduct additional preclinical studies and clinical trials.
Satisfaction of FDA regulations and requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years. The actual time required may vary substantially based upon the type, complexity and novelty of the drug candidate or disease. Typically, if a drug candidate is intended to treat a chronic disease, as is the case with some of our drug candidates, safety and efficacy data must be gathered over an extended period of time. Government regulation may delay or prevent marketing of drug candidates for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our drug candidates on a timely basis, if at all. Even if a drug candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages or restrictive distribution programs. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a drug may result in restrictions on the drug or even complete withdrawal of the drug from the market. Delays in obtaining, or failures to obtain, regulatory approvals for any of our drug candidates would harm our business. In addition, we cannot predict what future U.S. or foreign governmental regulations may be implemented.
Orphan Drug Designation. Some jurisdictions, including the United States, may designate drugs for relatively small patient populations as orphan drugs. The FDA grants orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. For example, the FDA has granted tirasemtiv an orphan drug designation for the treatment of ALS. In addition, the European Medicines Agency has granted tirasemtiv orphan medicinal product status for the treatment of ALS.
An FDA orphan drug designation does not shorten the duration of the regulatory review and approval process. If a drug candidate which has an orphan drug designation receives the first FDA marketing approval for the indication for which the designation was granted, then the approved drug is entitled to orphan drug exclusivity. This means that the FDA may not approve another company’s application to market the same drug for the same indication for a period of seven years, except in certain circumstances, such as a showing of clinical superiority to the drug with orphan exclusivity or if the holder of the orphan drug designation cannot assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the designation was granted. Competitors may receive approval of different drugs or biologics for the indications for which the orphan drug has exclusivity.
Fast Track Designation. Fast track is a process designed by the FDA to facilitate the development and expedite the review of drugs to treat serious diseases and fill an unmet medical need. Tirasemtiv has been granted fast track designation by the FDA for the treatment of ALS. Although fast track designation does not affect the standards for approval, the benefits of this designation include scheduled meetings to seek FDA input into
19
Table of Contents
development plans, the option of submitting an NDA in sections rather than all components simultaneously, and the option of requesting evaluation of studies using surrogate endpoints, which are laboratory measurements or physical signs used as an indirect or substitute measurement representing a clinically meaningful outcome.
Other Regulatory Requirements. Any drugs manufactured or distributed by us or our partners pursuant to FDA approvals or their foreign counterparts are subject to continuing regulation by the applicable regulatory authority, including recordkeeping requirements and reporting of adverse experiences associated with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and other applicable regulatory authorities, and are subject to periodic unannounced inspections by these regulatory authorities for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA and other regulatory requirements. If our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA or its foreign counterparts may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug.
For further details on the risks relating to government regulation of our business, please see the risk factors under Item 1A of this report, including, but not limited to, the risk factor entitled “The regulatory approval process is expensive, time-consuming and uncertain and may prevent our partners or us from obtaining approvals to commercialize some or all of our drug candidates.”
Competition
We compete in the segments of the pharmaceutical, biotechnology and other related markets that address neuromuscular and cardiovascular diseases and other diseases relating to muscle dysfunction, each of which is highly competitive. We face significant competition from most pharmaceutical companies and biotechnology companies that are also researching and selling products designed to address cardiovascular diseases and diseases and medical conditions associated with skeletal muscle weakness and wasting. Many of our competitors have significantly greater financial, manufacturing, marketing and drug development resources than we do. Large pharmaceutical companies in particular have extensive experience in clinical testing and in obtaining regulatory approvals for drugs. These companies also have significantly greater research capabilities than we do. In addition, many universities and private and public research institutes are active in research of neuromuscular and cardiovascular diseases and other diseases where there is muscle dysfunction, some in direct competition with us.
We believe that our ability to successfully compete will depend on, among other things:
| • | our drug candidates’ efficacy, safety and tolerability; |
| • | the speed and cost-effectiveness with which we develop our drug candidates; |
| • | the selection of suitable indications for which to develop our drug candidates; |
| • | the successful completion of clinical development and laboratory testing of our drug candidates; |
| • | the timing and scope of any regulatory approvals we or our partners obtain for our drug candidates; |
| • | our or our partners’ ability to manufacture and sell commercial quantities of our approved drugs to meet market demand; |
| • | acceptance of our drugs by physicians and other health care providers; |
| • | the willingness of third party payors to provide reimbursement for the use of our drugs; |
| • | our ability to protect our intellectual property and avoid infringing the intellectual property of others; |
20
Table of Contents
| • | the quality and breadth of our technology; |
| • | our employees’ skills and our ability to recruit and retain skilled employees; |
| • | our cash flows under existing and potential future arrangements with licensees, partners and other parties; and |
| • | the availability of substantial capital resources to fund development and commercialization activities. |
Our competitors may develop drug candidates and market drugs that are less expensive and more effective than our future drugs or that may render our drugs obsolete. Our current or future competitors may also commercialize competing drugs before we or our partners can launch any drugs developed from our drug candidates. These organizations also compete with us to attract qualified personnel and potential parties for acquisitions, joint ventures or other strategic alliances.
If tirasemtiv is approved for marketing by the FDA or other regulatory authorities for the treatment of ALS, it may then compete with other potential new therapies for ALS that are currently being developed by companies such as Mitsubishi Tanabe Pharma Corporation, Eisai Inc., Trophos SA, Neuraltus Pharmaceuticals, Inc., Isis Pharmaceuticals, Inc. and GlaxoSmithKline plc. In addition, BrainStorm Cell Therapeutics and Neuralstem, Inc. are each conducting clinical development of stem cell therapies for the potential treatment of ALS. If CK-2127107 is approved by the FDA for the potential treatment of non-neuromuscular indications associated with muscle weakness, potential competitors include Ligand Pharmaceuticals, Inc., which is developing LGD-4033, a selective androgen receptor modulator, for muscle wasting; and GTx, Inc., which is developing ostarine, a selective androgen receptor modulator, for cancer cachexia and potentially other indications. Novartis (in collaboration with Morphosys AG), is conducting clinical development with an activin type-IIB receptor antagonist, bimagrumab, to evaluate its ability to treat diseases involving the loss of muscle mass, strength and function. Drugs that could compete with CK-2127107 could also compete against tirasemtiv in ALS or other neuromuscular diseases, should the appropriate clinical trials be conducted.
If omecamtiv mecarbil is approved for marketing by the FDA or other regulatory authorities for the treatment of heart failure, it would compete against other drugs used for the treatment of acute and chronic heart failure. These include generic drugs, such as milrinone, dobutamine or digoxin and branded drugs such as Natrecor (nesiritide) and Procoralan (ivabradine). Omecamtiv mecarbil could also potentially compete against other novel drug candidates and therapies in development, such as bucindolol, which is being developed by ARCA biopharma, Inc.; Reasanz (serelaxin) and LCZ-696, which are being developed by Novartis; cenderitide (CD-NP), which is being developed by Carpicor Therapeutics, Inc., TRV-027, which is being developed by Trevena; ularitide, which is being developed by Cardiorentis Ltd.; aladorian, which is being developed by ARMGO Pharma, Inc; certain cardioprotectants which are being developed by Cardioxyl Pharmaceuticals, Inc.; glial growth factor (GGF-2) which is being developed by Acorda Therapeutics, Inc.; Neurocardin, which is being developed by Zensun Sci & Tech, Ltd; Mydicar, a genetically-targeted enzyme replacement therapy for advanced heart failure which is being developed by Celladon Corporation; and levosimendan, which was acquired for development by Oxygen Biotherapeutics, Inc. In addition, there are a number of medical devices being developed for the potential treatment of heart failure.
For further details on the risks relating to our competitors, please see the risk factors under Item 1A of this report, including, but not limited to, the risk factor entitled “Our competitors may develop drugs that are less expensive, safer or more effective than ours, which may diminish or eliminate the commercial success of any drugs that we may commercialize.”
Employees
As of December 31, 2013, our workforce consisted of 85 full-time employees, 28 of whom hold Ph.D. or M.D. degrees, or both, and 16 of whom hold other advanced degrees. Of our total full-time employees, 64 are engaged in research and development and 21 are engaged in business development, finance and administration functions.
21
Table of Contents
In October 2011, we announced a restructuring plan intended to align our workforce and operations in connection with our commitment to focus resources primarily on our later-stage development programs for tirasemtiv and omecamtiv mecarbil, and on our follow-on skeletal muscle troponin activator program and joint research with Amgen directed to next-generation compounds in our cardiac muscle contractility program. As a result, we reduced our workforce by approximately 18%, or 18 employees, to 83 employees. We provided severance, employee benefit continuation and career transition assistance to the employees directly affected by the restructuring.
We have no collective bargaining agreements with our employees, and we have not experienced any work stoppages. We believe that our relations with our employees are good.
Investor Information
We file electronically with the SEC our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13 or 15(d) of the Exchange Act. The public may read or copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of that site is www.sec.gov.
You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports on the day of filing with the SEC on our website at www.cytokinetics.com or by contacting the Investor Relations Department at our corporate offices by calling 650-624-3060. The information found on our website is not part of this or any other report filed with or furnished to the SEC.
In evaluating our business, you should carefully consider the following risks in addition to the other information in this report. Any of the following risks could materially and adversely affect our business, results of operations, financial condition or your investment in our securities, and many are beyond our control. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also adversely affect our business.
Risks Related To Our Business
We have a history of significant losses and may not achieve or sustain profitability and, as a result, you may lose all or part of your investment.
We have generally incurred operating losses in each year since our inception in 1997, due to costs incurred in connection with our research and development activities and general and administrative costs associated with our operations. Our drug candidates are all in early and mid-stage clinical testing, and we and our partners must conduct significant additional clinical trials before we and our partners can seek the regulatory approvals necessary to begin commercial sales of our drugs. We expect to incur increasing losses for at least several more years, as we continue our research activities and conduct development of, and seek regulatory approvals for, our drug candidates, and commercialize any approved drugs. If our drug candidates fail or do not gain regulatory approval, or if our drugs do not achieve market acceptance, we will not be profitable. If we fail to become and remain profitable, or if we are unable to fund our continuing losses, you could lose all or part of your investment.
22
Table of Contents
We will need substantial additional capital in the future to sufficiently fund our operations.
We have consumed substantial amounts of capital to date, and our operating expenditures will increase over the next several years if we expand our research and development activities. We have funded all of our operations and capital expenditures with proceeds from private and public sales of our equity securities, strategic alliances with Amgen, Astellas and others, equipment financings, interest on investments and government grants. We believe that our existing cash and cash equivalents, short-term investments and interest earned on investments should be sufficient to meet our projected operating requirements for at least the next 12 months. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development of our drug candidates and other research and development activities, including risks and uncertainties that could impact the rate of progress of our development activities, we are unable to estimate with certainty the amounts of capital outlays and operating expenditures associated with these activities.
For the foreseeable future, our operations will require significant additional funding, in large part due to our research and development expenses and the absence of any revenues from product sales. For example, we anticipate that we will need to conduct at least one Phase III clinical trial for tirasemtiv following the BENEFIT-ALS trial, if supported by the results, in order to obtain marketing approval for tirasemtiv for the potential treatment of ALS. We will require significant additional funding to enable us to conduct any such Phase III clinical trials. Until we can generate a sufficient amount of product revenue, we expect to raise future capital through strategic alliance and licensing arrangements, public or private equity offerings and debt financings. We do not currently have any commitments for future funding other than reimbursements, milestone and royalty payments that we may receive under our collaboration agreements with Amgen and Astellas. We may not receive any further funds under those agreements. Our ability to raise funds may be adversely impacted by current economic conditions. As a result of these and other factors, we do not know whether additional financing will be available when needed, or that, if available, such financing would be on terms favorable to our stockholders or us.
To the extent that we raise additional funds through strategic alliances or licensing and other arrangements with third parties, we will likely have to relinquish valuable rights to our technologies, research programs or drug candidates, or grant licenses on terms that may not be favorable to us. To the extent that we raise additional funds by issuing equity securities, our stockholders will experience additional dilution and our share price may decline. To the extent that we raise additional funds through debt financing, the financing may involve covenants that restrict our business activities. In addition, funding from any of these sources, if needed, may not be available to us on favorable terms, or at all, or in accordance with our planned timelines.
If we cannot raise the funds we need to operate our business, we will need to delay or discontinue certain research and development activities. For example, if we cannot raise the funds necessary to enable the conduct of the one or more Phase III clinical trials for tirasemtiv for the potential treatment of ALS, if supported by the results of BENEFIT-ALS, that we anticipate will be required for marketing approval, our ability to complete the development of tirasemtiv will be delayed or suspended. If we delay or discontinue research and development activities, our stock price may be negatively affected.
We have never generated, and may never generate, revenues from commercial sales of our drugs and we will not have drugs to market for at least several years, if ever.
We currently have no drugs for sale and we cannot guarantee that we will ever develop or obtain approval to market any drugs. To receive marketing approval for any drug candidate, we must demonstrate that the drug candidate satisfies rigorous standards of safety and efficacy to the FDA in the United States and other regulatory authorities abroad. We and our partners will need to conduct significant additional research and preclinical and clinical testing before we or our partners can file applications with the FDA or other regulatory authorities for approval of any of our drug candidates. In addition, to compete effectively, our drugs must be easy to use, cost-effective and economical to manufacture on a commercial scale, compared to other therapies available for the treatment of the same conditions. We may not achieve any of these objectives. Currently, our only drug
23
Table of Contents
candidates in clinical development are omecamtiv mecarbil for the potential treatment of heart failure, tirasemtiv for the potential treatment of ALS and other neuromuscular disorders and CK-2127107 for the potential treatment of non-neuromuscular indications associated with muscle weakness. We cannot be certain that the clinical development of these or any future drug candidates will be successful, that they will receive the regulatory approvals required to commercialize them, that they will ultimately be accepted by prescribers or reimbursed by insurers or that any of our other research programs will yield a drug candidate suitable for clinical testing or commercialization. Our commercial revenues, if any, will be derived from sales of drugs that we do not expect to be commercially marketed for at least several years, if at all. The development of any one or all of these drug candidates may be discontinued at any stage of our clinical trials programs and we may not generate revenue from any of these drug candidates.
Clinical trials may fail to demonstrate the desired safety and efficacy of our drug candidates, which could prevent or significantly delay completion of clinical development and regulatory approval.
Prior to receiving approval to commercialize any of our drug candidates, we or our partners must adequately demonstrate to the satisfaction of FDA and foreign regulatory authorities that the drug candidate is sufficiently safe and effective with substantial evidence from well-controlled clinical trials. We or our partners will need to demonstrate efficacy in clinical trials for the treatment of specific indications and monitor safety throughout the clinical development process and following approval. None of our drug candidates have yet been demonstrated to be safe and effective in clinical trials and they may never be. In addition, for each of our preclinical compounds, we or our partners must adequately demonstrate satisfactory chemistry, formulation, stability and toxicity in order to submit an investigational new drug application (“IND”) to the FDA, or an equivalent application in foreign jurisdictions, that would allow us to advance that compound into clinical trials. Furthermore, we or our partners may need to submit separate INDs (or foreign equivalent) to different divisions within the FDA (or foreign regulatory authorities) in order to pursue clinical trials in different therapeutic areas. Each new IND (or foreign equivalent) must be reviewed by the new division before the clinical trial under its jurisdiction can proceed, entailing all the risks of delay inherent to regulatory review. If our or our partners’ current or future preclinical studies or clinical trials are unsuccessful, our business will be significantly harmed and our stock price could be negatively affected.
All of our drug candidates are prone to the risks of failure inherent in drug development. Preclinical studies may not yield results that would adequately support the filing of an IND (or a foreign equivalent) with respect to our potential drug candidates. Even if the results of preclinical studies for a drug candidate are sufficient to support such a filing, the results of preclinical studies do not necessarily predict the results of clinical trials. As an example, because the physiology of animal species used in preclinical studies may vary substantially from other animal species and from humans, it may be difficult to assess with certainty whether a finding from a study in a particular animal species will result in similar findings in other animal species or in humans. For any of our drug candidates, the results from Phase I clinical trials in healthy volunteers and clinical results from Phase I and II trials in patients are not necessarily indicative of the results of larger Phase III clinical trials that are necessary to establish whether the drug candidate is safe and effective for the applicable indication. Likewise, interim results from a clinical trial may not be indicative of the final results from that trial.
In addition, while the clinical trials of our drug candidates are designed based on the available relevant information, such information may not accurately predict what actually occurs during the course of the trial itself, which may have consequences for the conduct of an ongoing clinical trial or for the eventual results of that trial. For example, the number of patients planned to be enrolled in a placebo-controlled clinical trial is determined in part by estimates relating to expected treatment effect and variability about the primary endpoint. These estimates are based upon earlier nonclinical and clinical studies of the drug candidate itself and clinical trials of other drugs thought to have similar effects in a similar patient population. If information gained during the conduct of the trial shows these estimates to be inaccurate, we may elect to adjust the enrollment accordingly, which may cause delays in completing the trial, additional expense or a statistical penalty to apply to the evaluation of the trial results.
24
Table of Contents
Furthermore, in view of the uncertainties inherent in drug development, such clinical trials may not be designed with focus on indications, patient populations, dosing regimens, safety, efficacy or pharmacokinetic parameters or other variables that will provide the necessary safety or efficacy data to support regulatory approval to commercialize the resulting drugs. Clinical trials of our drug candidates are designed based on guidance or advice from regulatory agencies, which is subject to change during the development of the drug candidate at any time. Such a change in a regulatory agency’s guidance or advice may cause that agency to deem results from trials to be insufficient to support approval of the drug candidate and require further clinical trials of that drug candidate to be conducted. In addition, individual patient responses to the dose administered of a drug may vary in a manner that is difficult to predict. Also, the methods we select to assess particular safety, efficacy or pharmacokinetic parameters may not yield the same statistical precision in estimating our drug candidates’ effects as may other methodologies. Even if we believe the data collected from clinical trials of our drug candidates are promising, these data may not be sufficient to support approval by the FDA or foreign regulatory authorities. Preclinical and clinical data can be interpreted in different ways. Accordingly, the FDA or foreign regulatory authorities could interpret these data in different ways from us or our partners, which could delay, limit or prevent regulatory approval.
Administering any of our drug candidates or potential drug candidates may produce undesirable side effects, also known as adverse effects. Toxicities and adverse effects observed in preclinical studies for some compounds in a particular research and development program may also occur in preclinical studies or clinical trials of other compounds from the same program. Potential toxicity issues may arise from the effects of the active pharmaceutical ingredient itself or from impurities or degradants that are present in the active pharmaceutical ingredient or could form over time in the formulated drug candidate or the active pharmaceutical ingredient. These toxicities or adverse effects could delay or prevent the filing of an IND (or a foreign equivalent) with respect to our drug candidates or potential drug candidates or cause us, our partners or the FDA or foreign regulatory authorities to modify, suspend or terminate clinical trials with respect to any drug candidate at any time during the development program. Further, the administration of two or more drugs contemporaneously can lead to interactions between them, and our drug candidates may interact with other drugs that trial subjects are taking. For example, in a Phase I drug-drug interaction study of tirasemtiv administered orally to healthy volunteers, co-administration of tirasemtiv and riluzole (an approved treatment for ALS) approximately doubled the average maximum riluzole plasma level, although it also appeared to reduce the variability of the riluzole plasma levels of the study subjects. If the adverse effects are severe or frequent enough to outweigh the potential efficacy of a drug candidate, the FDA or other regulatory authorities could deny approval of that drug candidate for any or all targeted indications. Even if one or more of our drug candidates were approved for sale as drugs, the occurrence of even a limited number of toxicities or adverse effects when used in large populations may cause the FDA or foreign regulatory authorities to impose restrictions on, or stop, the further marketing of those drugs. Indications of potential adverse effects or toxicities which do not seem significant during the course of clinical trials may later turn out to actually constitute serious adverse effects or toxicities when a drug is used in large populations or for extended periods of time.
We have observed certain adverse effects in the clinical trials conducted with our drug candidates. For example, in Phase II clinical trials of tirasemtiv, adverse events of dizziness, fatigue, headache, somnolence (sleepiness), euphoric mood, muscle spasms, gait disturbance, pain in extremity, feeling drunk, blurred vision, muscular weakness, nausea, balance disorder, asthenia (loss of strength and energy), abnormal coordination and dysarthria (difficulty speaking) occurred more frequently during treatment with tirasemtiv than with placebo, with a possible trend for their frequencies to increase with increasing doses of tirasemtiv. In clinical trials of omecamtiv mecarbil, dose-limiting effects were associated with complaints of chest discomfort, palpitations, dizziness and feeling hot, increases in heart rate, declines in blood pressure, electrocardiographic changes consistent with acute myocardial ischemia and transient rises in the MB fraction of creatine kinase and cardiac troponins I and T, which are indicative of myocardial infarction.
In addition, clinical trials of tirasemtiv and omecamtiv mecarbil enroll patients who typically suffer from serious diseases which put them at increased risk of death. These patients may die while receiving our drug
25
Table of Contents
candidates. In such circumstances, it may not be possible to exclude with certainty a causal relationship to our drug candidate, even though the responsible clinical investigator may view such an event as not study drug-related.
Any failure or significant delay in completing preclinical studies or clinical trials for our drug candidates, or in receiving and maintaining regulatory approval for the sale of any resulting drugs, may significantly harm our business and negatively affect our stock price.
Clinical trials are expensive, time-consuming and subject to delay.
Clinical trials are subject to rigorous regulatory requirements and are very expensive, difficult and time-consuming to design and implement. The length of time and number of trial sites and patients required for clinical trials vary substantially based on the type, complexity, novelty, intended use of the drug candidate and safety concerns. We estimate that the clinical trials of our current drug candidates will each continue for several more years. However, the clinical trials for all or any of our drug candidates may take significantly longer to complete. The commencement and completion of our clinical trials could be delayed or prevented by many factors, including, but not limited to:
| • | delays in obtaining, or inability to obtain, regulatory or other approvals to commence and conduct clinical trials in the manner we or our partners deem necessary for the appropriate and timely development of our drug candidates and commercialization of any resulting drugs; |
| • | delays in identifying and reaching agreement, or inability to identify and reach agreement, on acceptable terms, with prospective clinical trial sites and other entities involved in the conduct of our clinical trials; |
| • | delays or additional costs in developing, or inability to develop, appropriate formulations of our drug candidates for clinical trial use, including an appropriate modified release oral formulation for omecamtiv mecarbil; |
| • | slower than expected rates of patient recruitment and enrollment, including as a result of competition for patients with other clinical trials; limited numbers of patients that meet the enrollment criteria; patients’, investigators’ or trial sites’ reluctance to agree to the requirements of a protocol; or the introduction of alternative therapies or drugs by others; |
| • | for those drug candidates that are the subject of a strategic alliance, delays in reaching agreement with our partner as to appropriate development strategies; |
| • | a regulatory authority may require changes to a protocol for a clinical trial that then may require approval from regulatory agencies in other jurisdictions where the trial is being conducted; |
| • | an institutional review board (“IRB”) or its foreign equivalent may require changes to a protocol that then require approval from regulatory agencies and other IRBs and their foreign equivalents, or regulatory authorities may require changes to a protocol that then require approval from the IRBs or their foreign equivalents; |
| • | for clinical trials conducted in foreign countries, the time and resources required to identify, interpret and comply with foreign regulatory requirements or changes in those requirements, and political instability or natural disasters occurring in those countries; |
| • | lack of effectiveness of our drug candidates during clinical trials; |
| • | unforeseen safety issues; |
| • | inadequate supply, or delays in the manufacture or supply, of clinical trial materials; |
| • | uncertain dosing issues; |
| • | failure by us, our partners, or clinical research organizations, investigators or site personnel engaged by us or our partners to comply with good clinical practices and other applicable laws and regulations, including those concerning informed consent; |
26
Table of Contents
| • | inability or unwillingness of investigators or their staffs to follow clinical protocols; |
| • | failure by our clinical research organizations, clinical manufacturing organizations and other third parties supporting our clinical trials to fulfill their obligations; |
| • | inability to monitor patients adequately during or after treatment; |
| • | introduction of new therapies or changes in standards of practice or regulatory guidance that render our drug candidates or their clinical trial endpoints obsolete; and |
| • | results from non-clinical studies that may adversely impact the timing or further development of our drug candidates. |
We do not know whether planned clinical trials will begin on time, or whether planned or currently ongoing clinical trials will need to be restructured or will be completed on schedule, if at all. Significant delays in clinical trials will impede our ability to commercialize our drug candidates and generate revenue and could significantly increase our development costs.
We depend on Amgen for the conduct, completion and funding of the development and commercialization of omecamtiv mecarbil.
Under our strategic alliance, Amgen holds an exclusive license to our drug candidate omecamtiv mecarbil worldwide. As a result, Amgen is responsible for the clinical development and obtaining and maintaining regulatory approval of omecamtiv mecarbil for the potential treatment of heart failure worldwide.
We do not control the development activities being conducted or that may be conducted in the future by Amgen, including, but not limited to, the timing of initiation, termination or completion of clinical trials, the analysis of data arising out of those clinical trials or the timing of release of data concerning those clinical trials, which may impact our ability to report on Amgen’s results. Amgen may conduct these activities more slowly or in a different manner than we would if we controlled the development of omecamtiv mecarbil. Amgen is responsible for filing future applications with the FDA or other regulatory authorities for approval of omecamtiv mecarbil and will be the owner of any marketing approvals issued by the FDA or other regulatory authorities for omecamtiv mecarbil. If the FDA or other regulatory authorities approve omecamtiv mecarbil, Amgen will also be responsible for the marketing and sale of the resulting drug, subject to our right to co-promote omecamtiv mecarbil in North America if we exercise our option to co-fund Phase III development costs of omecamtiv mecarbil under the collaboration. However, we cannot control whether Amgen will devote sufficient attention and resources to the development of omecamtiv mecarbil or will proceed in an expeditious manner, even if we do exercise our option to co-fund the development of omecamtiv mecarbil. Even if the FDA or other regulatory agencies approve omecamtiv mecarbil, Amgen may elect not to proceed with the commercialization of the resulting drug in one or more countries.
If the results of one or more clinical trials with omecamtiv mecarbil do not meet Amgen’s expectations at any time, Amgen may elect to terminate further development of omecamtiv mecarbil or certain of the potential clinical trials for omecamtiv mecarbil, even if the actual number of patients treated at that time is relatively small. In addition, Amgen generally has discretion to elect whether to pursue or abandon the development of omecamtiv mecarbil and may terminate our strategic alliance for any reason upon six months prior notice. If Amgen abandons omecamtiv mecarbil, it would result in a delay in or could prevent us from commercializing omecamtiv mecarbil, and would delay and could prevent us from obtaining revenues for this drug candidate. Disputes may arise between us and Amgen, which may delay or cause the termination of any omecamtiv mecarbil clinical trials, result in significant litigation or cause Amgen to act in a manner that is not in our best interest. If development of omecamtiv mecarbil does not progress for these or any other reasons, we would not receive further milestone payments or royalties on product sales from Amgen with respect to omecamtiv mecarbil. If Amgen abandons development of omecamtiv mecarbil prior to regulatory approval or if it elects not to proceed with commercialization of the resulting drug following regulatory approval, we would have to seek a
27
Table of Contents
new partner for development or commercialization, curtail or abandon that development or commercialization, or undertake and fund the development of omecamtiv mecarbil or commercialization of the resulting drug ourselves. If we seek a new partner but are unable to do so on acceptable terms, or at all, or do not have sufficient funds to conduct the development or commercialization of omecamtiv mecarbil ourselves, we would have to curtail or abandon that development or commercialization, which could harm our business.
We will depend on Astellas for the conduct, completion and funding of the development and commercialization of CK-2127107.
In June 2013, we entered into a strategic alliance with Astellas focused on the research, development and commercialization of skeletal muscle activators, other than tirasemtiv and certain related compounds. The primary objective of the strategic alliance is to advance novel therapies for indications associated with muscle weakness.
As part of the strategic alliance, we granted Astellas an exclusive license to co-develop and commercialize CK-2127107 for potential application in non-neuromuscular indications worldwide. Following Cytokinetics’ conduct of Phase I clinical trials and certain Phase II readiness activities for CK-2127107, Astellas will be primarily responsible for the conduct of subsequent development and commercialization activities for CK-2127107. Astellas may elect not to continue development of CK-2127107 following Cytokinetics’ completion of these activities. In such event, we would need significant additional funding to continue the development of CK-2127107 on our own, which may not be available on attractive or acceptable terms, if at all, and we would be limited in the indications that we could pursue with this drug candidate.
We do not control the development activities that may be conducted by Astellas, including, but not limited to, the timing of initiation, termination or completion of clinical trials, the analysis of data arising out of those clinical trials or the timing of release of data concerning those clinical trials, which may impact our ability to report on Astellas’ results. Astellas may conduct these activities more slowly or in a different manner than we would. Astellas is responsible for filing future applications with the FDA or other regulatory authorities for approval of CK-2127107 and will be the owner of any marketing approvals issued by the FDA or other regulatory authorities for CK-2127107. If the FDA or other regulatory authorities approve CK-2127107, Astellas will also be responsible for the marketing and sale of the resulting drug, subject to our right to co-promote the drug in the United States and Canada. However, we cannot control whether Astellas will devote sufficient attention and resources to the development of CK-2127107 or will proceed in an expeditious manner. Even if the FDA or other regulatory agencies approve CK-2127107, Astellas may elect not to proceed with the commercialization of the resulting drug in one or more countries.
If the results of one or more clinical trials with CK-2127107 do not meet Astellas’ expectations at any time, Astellas may elect to terminate further development of CK-2127107 or certain of the potential clinical trials for CK-2127107, even if the actual number of patients treated at that time is relatively small. In addition, Astellas generally has discretion to elect whether to pursue or abandon the development of CK-2127107. Astellas may terminate our strategic alliance in whole or in part for any reason upon six months prior notice at any time following expiration of the strategic alliance’s two-year research term. If Astellas abandons CK-2127107, it would result in a delay in or could prevent us from further developing or commercializing CK-2127107, and would delay and could prevent us from obtaining revenues for this drug candidate. Disputes may arise between us and Astellas, which may delay or cause the termination of any CK-2127107 clinical trials, result in significant litigation or cause Astellas to act in a manner that is not in our best interest. If development of CK-2127107 does not progress for these or any other reasons, we would not receive further milestone payments or royalties on product sales from Astellas with respect to CK-2127107. If Astellas abandons development of CK-2127107 prior to regulatory approval or if it elects not to proceed with commercialization of the resulting drug following regulatory approval, we would have to seek a new partner for development or commercialization, curtail or abandon that development or commercialization, or undertake and fund the development of CK-2127107 or commercialization of the resulting drug ourselves. If we seek a new partner but are unable to do so on acceptable terms, or at all, or do not have sufficient funds to conduct the development or commercialization of CK-2127107 ourselves, we would have to curtail or abandon that development or commercialization, which could harm our business.
28
Table of Contents
If we do not enter into strategic alliances for our unpartnered drug candidates or research and development programs or fail to successfully maintain our current or future strategic alliances, we may have to reduce, delay or discontinue our advancement of our drug candidates and programs or expand our research and development capabilities and increase our expenditures.
Drug development is complicated and expensive. We currently have limited financial and operational resources to carry out drug development. Our strategy for developing, manufacturing and commercializing our drug candidates currently requires us to enter into and successfully maintain strategic alliances with pharmaceutical companies or other industry participants to advance our programs and reduce our expenditures on each program. Accordingly, the success of our development activities depends in large part on our current and future strategic partners’ performance, over which we have little or no control.
We have retained the rights to develop and commercialize tirasemtiv. We currently do not have a strategic partner for this drug candidate. We may seek one or more strategic partners or other arrangements with third parties to support Phase III clinical development, if supported by the results of BENEFIT-ALS, and commercialization of tirasemtiv. However, we may not be able to negotiate and enter into such strategic alliances or arrangements on acceptable terms, if at all, or in accordance with our planned timelines. If we are unable to enter into a strategic alliance for tirasemtiv, we will be unable to conduct the one or more Phase III clinical trials we believe will be necessary to obtain marketing approval for tirasemtiv for the potential treatment of ALS unless we are able to acquire the funding to do so from another source.
Our ability to commercialize drugs that we develop with our partners and that generate royalties from product sales depends on our partners’ abilities to assist us in establishing the safety and efficacy of our drug candidates, obtaining and maintaining regulatory approvals and achieving market acceptance of the drugs once commercialized. Our partners may elect to delay or terminate development of one or more drug candidates, independently develop drugs that could compete with ours or fail to commit sufficient resources to the marketing and distribution of drugs developed through their strategic alliances with us. Our partners may not proceed with the development and commercialization of our drug candidates with the same degree of urgency as we would because of other priorities they face. In addition, new business combinations or changes in a partner’s business strategy may adversely affect its willingness or ability to carry out its obligations under a strategic alliance.
If we are not able to successfully maintain our existing strategic alliances or establish and successfully maintain additional strategic alliances, we will have to limit the size or scope of, or delay or discontinue, one or more of our drug development programs or research programs, or undertake and fund these programs ourselves. Alternatively, if we elect to continue to conduct any of these drug development programs or research programs on our own, we will need to expand our capability to conduct clinical development by bringing additional skills, technical expertise and resources into our organization. This would require significant additional funding, which may not be available to us on acceptable terms, or at all.
To the extent we elect to fund the development of a drug candidate, such as omecamtiv mecarbil, tirasemtiv or CK-2127107, or the commercialization of a drug at our expense, we will need substantial additional funding.
The discovery, development and commercialization of new drugs is costly. As a result, to the extent we elect to fund the development of a drug candidate, such as omecamtiv mecarbil, tirasemtiv or CK-2127107, or the commercialization of a drug, we will need to raise additional capital to:
| • | fund clinical trials and seek regulatory approvals; |
| • | expand our development capabilities; |
| • | engage third party manufacturers for such drug candidate or drug; |
| • | build or access commercialization capabilities; |
| • | implement additional internal systems and infrastructure; |
29
Table of Contents
| • | maintain, defend and expand the scope of our intellectual property; and |
| • | hire and support additional management and scientific personnel. |
Our future funding requirements will depend on many factors, including, but not limited to:
| • | the rate of progress and costs of our clinical trials and other research and development activities; |
| • | the costs and timing of seeking and obtaining regulatory approvals; |
| • | the costs associated with establishing manufacturing and commercialization capabilities; |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | the costs of acquiring or investing in businesses, products and technologies; |
| • | the effect of competing technological and market developments; and |
| • | the status of, payment and other terms, and timing of any strategic alliance, licensing or other arrangements that we have entered into or may establish. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to continue to finance our future cash needs primarily through strategic alliances, public or private equity offerings and debt financings. We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or future commercialization initiatives.
We depend on contract research organizations to conduct our clinical trials and have limited control over their performance.
We have used and intend to continue to use contract research organizations (“CROs”) within and outside of the United States to conduct clinical trials of our drug candidates, such as tirasemtiv, CK-2127107 and omecamtiv mecarbil, and related activities. We do not have control over many aspects of our CROs’ activities, and cannot fully control the amount, timing or quality of resources that they devote to our programs. CROs may not assign as high a priority to our programs or pursue them as diligently as we would if we were undertaking these programs ourselves. The activities conducted by our CROs therefore may not be completed on schedule or in a satisfactory manner. CROs may also give higher priority to relationships with our competitors and potential competitors than to their relationships with us. Outside of the United States, we are particularly dependent on our CROs’ expertise in communicating with clinical trial sites and regulatory authorities and ensuring that our clinical trials and related activities and regulatory filings comply with applicable laws.
Our CROs’ failure to carry out development activities on our behalf as agreed and in accordance with our and the FDA’s or other regulatory agencies’ requirements and applicable U.S. and foreign laws, or our failure to properly coordinate and manage these activities, could increase the cost of our operations and delay or prevent the development, approval and commercialization of our drug candidates. For example, in June 2013 we learned from our data management vendor for our BENEFIT-ALS clinical trial that a programming error in the electronic data capture system controlling study drug assignment caused 58 patients initially randomized to and treated with tirasemtiv to receive placebo instead at a certain trial visit and for the remainder of the trial. In order to maintain the originally intended statistical power of the trial, we amended the protocol to permit enrollment of approximately 680 patients, or 180 patients in addition to the 500 patients allowed under the existing protocol. This protocol amendment will result in additional costs and delays in conducting BENEFIT-ALS. In addition, if a CRO fails to perform as agreed, our ability to collect damages may be contractually limited. If we fail to effectively manage the CROs carrying out the development of our drug candidates or if our CROs fail to perform as agreed, the commercialization of our drug candidates will be delayed or prevented.
30
Table of Contents
We have no manufacturing capacity and depend on our strategic partners and contract manufacturers to produce our clinical trial materials, including our drug candidates, and anticipate continued reliance on contract manufacturers for the development and commercialization of our potential drugs.
We do not currently operate manufacturing facilities for clinical or commercial production of our drug candidates. We have limited experience in drug formulation and manufacturing, and we lack the resources and the capabilities to manufacture any of our drug candidates on a clinical or commercial scale. Amgen has assumed responsibility to conduct these activities for the ongoing clinical development of omecamtiv mecarbil worldwide. Following our conduct of the Phase I clinical trials for CK-2127107, Astellas will assume responsibility to conduct these activities for the ongoing clinical development of CK-2127107 worldwide. For tirasemtiv, we rely on a limited number of contract manufacturers. In particular, we rely on single-source contract manufacturers for the active pharmaceutical ingredient and the drug product supply for our clinical trials, as well as other materials required to conduct our clinical trials. We expect to rely on contract manufacturers to supply all future drug candidates for which we conduct clinical development, as well as other materials required to conduct our clinical trials. If any of our existing or future contract manufacturers fail to perform satisfactorily, it could delay clinical development or regulatory approval of our drug candidates or commercialization of our drugs, producing additional losses and depriving us of potential product revenues. In addition, if a contract manufacturer fails to perform as agreed, our ability to collect damages may be contractually limited.
Our drug candidates require precise high-quality manufacturing. The failure to achieve and maintain high manufacturing standards, including failure to detect or control anticipated or unanticipated manufacturing errors or the frequent occurrence of such errors, could result in patient injury or death, discontinuance or delay of ongoing or planned clinical trials, delays or failures in product testing or delivery, cost overruns, product recalls or withdrawals and other problems that could seriously hurt our business. Contract drug manufacturers often encounter difficulties involving production yields, quality control and quality assurance and shortages of qualified personnel. These manufacturers are subject to stringent regulatory requirements, including the FDA’s current good manufacturing practices regulations and similar foreign laws and standards. Each contract manufacturer must pass a pre-approval inspection before we can obtain marketing approval for any of our drug candidates and following approval will be subject to ongoing periodic unannounced inspections by the FDA, the U.S. Drug Enforcement Agency and other regulatory agencies, to ensure strict compliance with current good manufacturing practices and other applicable government regulations and corresponding foreign laws and standards. We seek to ensure that our contract manufacturers comply fully with all applicable regulations, laws and standards. However, we do not have control over our contract manufacturers’ compliance with these regulations, laws and standards. If one of our contract manufacturers fails to pass its pre-approval inspection or maintain ongoing compliance at any time, the production of our drug candidates could be interrupted, resulting in delays or discontinuance of our clinical trials, additional costs and potentially lost revenues. In addition, failure of any third party manufacturers or us to comply with applicable regulations, including pre-or post-approval inspections and the current good manufacturing practice requirements of the FDA or other comparable regulatory agencies, could result in sanctions being imposed on us. These sanctions could include fines, injunctions, civil penalties, failure of regulatory authorities to grant marketing approval of our products, delay, suspension or withdrawal of approvals, license revocation, product seizures or recalls, operational restrictions and criminal prosecutions, any of which could significantly and adversely affect our business.
In addition, our existing and future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to successfully produce, store and distribute our drug candidates. If a natural disaster, business failure, strike or other difficulty occurs, we may be unable to replace these contract manufacturers in a timely or cost-effective manner and the production of our drug candidates would be interrupted, resulting in delays and additional costs.
Switching manufacturers or manufacturing sites would be difficult and time-consuming because the number of potential manufacturers is limited. In addition, before a drug from any replacement manufacturer or manufacturing site can be commercialized, the FDA and, in some cases, foreign regulatory agencies, must approve that site. These approvals would require regulatory testing and compliance inspections. A new
31
Table of Contents
manufacturer or manufacturing site also would have to be educated in, or develop substantially equivalent processes for, production of our drugs and drug candidates. It may be difficult or impossible to transfer certain elements of a manufacturing process to a new manufacturer or for us to find a replacement manufacturer on acceptable terms quickly, or at all, either of which would delay or prevent our ability to develop drug candidates and commercialize any resulting drugs.
We may not be able to successfully scale-up manufacturing of our drug candidates in sufficient quality and quantity, which would delay or prevent us from developing our drug candidates and commercializing resulting approved drugs, if any.
To date, our drug candidates have been manufactured in small quantities for preclinical studies and early and mid-stage clinical trials. In order to conduct larger scale or late-stage clinical trials for a drug candidate and for commercialization of the resulting drug if that drug candidate is approved for sale, we will need to manufacture it in larger quantities. We may not be able to successfully increase the manufacturing capacity for any of our drug candidates, whether in collaboration with third-party manufacturers or on our own, in a timely or cost-effective manner or at all. If a contract manufacturer makes improvements in the manufacturing process for our drug candidates, we may not own, or may have to share, the intellectual property rights to those improvements. Significant scale-up of manufacturing may require additional validation studies, which are costly and which the FDA must review and approve. In addition, quality issues may arise during those scale-up activities because of the inherent properties of a drug candidate itself or of a drug candidate in combination with other components added during the manufacturing and packaging process, or during shipping and storage of the finished product or active pharmaceutical ingredients. If we are unable to successfully scale-up manufacture of any of our drug candidates in sufficient quality and quantity, the development of that drug candidate and regulatory approval or commercial launch for any resulting drugs may be delayed or there may be a shortage in supply, which could significantly harm our business.
The mechanisms of action of our drug candidates are unproven, and we do not know whether we will be able to develop any drug of commercial value.
We have discovered and are currently developing drug candidates that have what we believe are novel mechanisms of action directed against cytoskeletal targets, and intend to continue to do so. Because no currently approved drugs appear to operate via the same biochemical mechanisms as our compounds, we cannot be certain that our drug candidates will result in commercially viable drugs that safely and effectively treat the indications for which we intend to develop them. The results we have seen for our compounds in preclinical models may not translate into similar results in humans, and results of early clinical trials in humans may not be predictive of the results of larger clinical trials that may later be conducted with our drug candidates. Even if we are successful in developing and receiving regulatory approval for a drug candidate for the treatment of a particular disease, we cannot be certain that it will be accepted by prescribers or be reimbursed by insurers or that we will also be able to develop and receive regulatory approval for that or other drug candidates for the treatment of other diseases. If we or our partners are unable to successfully develop and commercialize our drug candidates, our business will be materially harmed.
Our success depends substantially upon our ability to obtain and maintain intellectual property protection relating to our drug candidates, compounds and research technologies.
We own, or hold exclusive licenses to, a number of U.S. and foreign patents and patent applications directed to our drug candidates, compounds and research technologies. Our success depends on our ability to obtain patent protection both in the United States and in other countries for our drug candidates, their methods of manufacture and use, and our technologies. Our ability to protect our drug candidates, compounds and technologies from unauthorized or infringing use by third parties depends substantially on our ability to obtain and enforce our patents. If our issued patents and patent applications, if granted, do not adequately describe, enable or otherwise provide coverage of our technologies and drug candidates, including omecamtiv mecarbil,
32
Table of Contents
tirasemtiv and CK-2127107, we or our licensees would not be able to exclude others from developing or commercializing these drug candidates. Furthermore, the degree of future protection of our proprietary rights is uncertain because legal means may not adequately protect our rights or permit us to gain or keep our competitive advantage.
Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the claim scope of these patents, our ability to enforce our existing patents and to obtain and enforce patents that may issue from any pending or future patent applications is uncertain and involves complex legal, scientific and factual questions. The standards which the U.S. Patent and Trademark Office and its foreign counterparts use to grant patents are not always applied predictably or uniformly and are subject to change. To date, no consistent policy has emerged regarding the breadth of claims allowed in biotechnology and pharmaceutical patents. Thus, we cannot be sure that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents do issue, we cannot be sure that the claims of these patents will be held valid or enforceable by a court of law, will provide us with any significant protection against competitive products, or will afford us a commercial advantage over competitive products. In particular:
| • | we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| • | we or our licensors might not have been the first to file patent applications for the inventions covered by our pending patent applications and issued patents; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| • | some or all of our or our licensors’ pending patent applications may not result in issued patents or the claims that issue may be narrow in scope and not provide us with competitive advantages; |
| • | our and our licensors’ issued patents may not provide a basis for commercially viable drugs or therapies or may be challenged and invalidated by third parties; |
| • | our or our licensors’ patent applications or patents may be subject to interference, opposition or similar administrative proceedings that may result in a reduction in their scope or their loss altogether; |
| • | we may not develop additional proprietary technologies or drug candidates that are patentable; or |
| • | the patents of others may prevent us or our partners from discovering, developing or commercializing our drug candidates. |
Patent protection is afforded on a country-by-country basis. Some foreign jurisdictions do not protect intellectual property rights to the same extent as in the United States. Many companies have encountered significant difficulties in protecting and defending intellectual property rights in foreign jurisdictions. Some of our development efforts are performed in countries outside of the United States through third party contractors. We may not be able to effectively monitor and assess intellectual property developed by these contractors. We therefore may not be able to effectively protect this intellectual property and could lose potentially valuable intellectual property rights. In addition, the legal protection afforded to inventors and owners of intellectual property in countries outside of the United States may not be as protective of intellectual property rights as in the United States. Therefore, we may be unable to acquire and protect intellectual property developed by these contractors to the same extent as if these development activities were being conducted in the United States. If we encounter difficulties in protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
We rely on intellectual property assignment agreements with our corporate partners, employees, consultants, scientific advisors and other collaborators to grant us ownership of new intellectual property that is developed. These agreements may not result in the effective assignment to us of that intellectual property. As a result, our ownership of key intellectual property could be compromised.
33
Table of Contents
Changes in either the patent laws or their interpretation in the United States or other countries may diminish the value of our intellectual property or our ability to obtain patents. For example, the America Invents Act of 2011 may affect the scope, strength and enforceability of our patent rights in the United States or the nature of proceedings which may be brought by us related to our patent rights in the United States.
If one or more products resulting from our drug candidates is approved for sale by the FDA and we do not have adequate intellectual property protection for those products, competitors could duplicate them for approval and sale in the United States without repeating the extensive testing required of us or our partners to obtain FDA approval. Regardless of any patent protection, under current law, an application for a generic version of a new chemical entity cannot be approved until at least five years after the FDA has approved the original product. When that period expires, or if that period is altered, the FDA could approve a generic version of our product regardless of our patent protection. An applicant for a generic version of our product may only be required to conduct a relatively inexpensive study to show that its product is bioequivalent to our product, and may not have to repeat the lengthy and expensive clinical trials that we or our partners conducted to demonstrate that the product is safe and effective. In the absence of adequate patent protection for our products in other countries, competitors may similarly be able to obtain regulatory approval in those countries of generic versions of our products.
We also rely on trade secrets to protect our technology, particularly where we believe patent protection is not appropriate or obtainable. However, trade secrets are often difficult to protect, especially outside of the United States. While we endeavor to use reasonable efforts to protect our trade secrets, our or our partners’ employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our information to competitors. In addition, confidentiality agreements, if any, executed by those individuals may not be enforceable or provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure. Pursuing a claim that a third party had illegally obtained and was using our trade secrets would be expensive and time-consuming, and the outcome would be unpredictable. Even if we are able to maintain our trade secrets as confidential, if our competitors independently develop information equivalent or similar to our trade secrets, our business could be harmed.
If we are not able to defend the patent or trade secret protection position of our technologies and drug candidates, then we will not be able to exclude competitors from developing or marketing competing drugs, and we may not generate enough revenue from product sales to justify the cost of development of our drugs or to achieve or maintain profitability.
If we are sued for infringing third party intellectual property rights, it will be costly and time-consuming, and an unfavorable outcome could have a significant adverse effect on our business.
Our ability to commercialize drugs depends on our ability to use, manufacture and sell those drugs without infringing the patents or other proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the therapeutic areas in which we are developing drug candidates and seeking new potential drug candidates. In addition, because patent applications can take several years to issue, there may be currently pending applications, unknown to us, which could later result in issued patents that our activities with our drug candidates could infringe. There may also be existing patents, unknown to us, that our activities with our drug candidates could infringe.
Other future products of ours may be impacted by patents of companies engaged in competitive programs with significantly greater resources. Further development of these products could be impacted by these patents and result in significant legal fees.
If a third party claims that our actions infringe its patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including, but not limited to:
| • | infringement and other intellectual property claims that, even if meritless, can be costly and time-consuming to litigate, delay the regulatory approval process and divert management’s attention from our core business operations; |
34
Table of Contents
| • | substantial damages for past infringement which we may have to pay if a court determines that our drugs or technologies infringe a third party’s patent or other proprietary rights; |
| • | a court prohibiting us from selling or licensing our drugs or technologies unless the holder licenses the patent or other proprietary rights to us, which it is not required to do; and |
| • | if a license is available from a holder, we may have to pay substantial royalties or grant cross-licenses to our patents or other proprietary rights. |
If any of these events occur, it could significantly harm our business and negatively affect our stock price.
We may undertake infringement or other legal proceedings against third parties, causing us to spend substantial resources on litigation and exposing our own intellectual property portfolio to challenge.
Third parties may infringe our patents. To prevent infringement or unauthorized use, we may need to file infringement suits, which are expensive and time-consuming. In an infringement proceeding, a court may decide that one or more of our patents is invalid, unenforceable, or both. In this case, third parties may be able to use our technology without paying licensing fees or royalties. Even if the validity of our patents is upheld, a court may refuse to stop the other party from using the technology at issue on the ground that the other party’s activities are not covered by our patents. Policing unauthorized use of our intellectual property is difficult, and we may not be able to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States. In addition, third parties may affirmatively challenge our rights to, or the scope or validity of, our patent rights.
We may become involved in disputes with our strategic partners over intellectual property ownership, and publications by our research collaborators and clinical investigators could impair our ability to obtain patent protection or protect our proprietary information, either of which would have a significant impact on our business.
Inventions discovered under our current or future strategic alliance agreements may become jointly owned by our strategic partners and us in some cases, and the exclusive property of one of us in other cases. Under some circumstances, it may be difficult to determine who owns a particular invention or whether it is jointly owned, and disputes could arise regarding ownership or use of those inventions. These disputes could be costly and time-consuming, and an unfavorable outcome could have a significant adverse effect on our business if we were not able to protect or license rights to these inventions. In addition, our research collaborators and clinical investigators generally have contractual rights to publish data arising from their work. Publications by our research collaborators and clinical investigators relating to our research and development programs, either with or without our consent, could benefit our current or potential competitors and may impair our ability to obtain patent protection or protect our proprietary information, which could significantly harm our business.
We may be subject to claims that we or our employees have wrongfully used or disclosed trade secrets of their former employers.
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending these claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper or prevent our ability to develop and commercialize certain potential drugs, which could significantly harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and distract management.
35
Table of Contents
Our competitors may develop drugs that are less expensive, safer or more effective than ours, which may diminish or eliminate the commercial success of any drugs that we may commercialize.
We compete with companies that have developed drugs or are developing drug candidates for cardiovascular diseases, diseases and conditions associated with muscle weakness or wasting and other diseases for which our drug candidates may be useful treatments. For example, if tirasemtiv is approved for marketing by the FDA or other regulatory authorities for the treatment of ALS, it may then compete with other potential new therapies for ALS that are currently being developed by companies such as Mitsubishi Tanabe Pharma Corporation, Eisai Inc., Trophos SA, Neuraltus Pharmaceuticals, Inc., Isis Pharmaceuticals, Inc. and GlaxoSmithKline plc. In addition, BrainStorm Cell Therapeutics and Neuralstem, Inc. are each conducting clinical development of stem cell therapies for the potential treatment of ALS. If CK-2127107 is approved by the FDA for the potential treatment of non-neuromuscular indications associated with muscle weakness, potential competitors include Ligand Pharmaceuticals, Inc., which is developing LGD-4033, a selective androgen receptor modulator, for muscle wasting; and GTx, Inc., which is developing ostarine, a selective androgen receptor modulator, for cancer cachexia and potentially other indications. Novartis (in collaboration with Morphosys AG), is conducting clinical development with an activin type-IIB receptor antagonist, bimagrumab, to evaluate its ability to treat diseases involving the loss of muscle mass, strength and function. Drugs that could compete with CK-2127107 could also compete against tirasemtiv in ALS or other neuromuscular diseases, should the appropriate clinical trials be conducted.
If omecamtiv mecarbil is approved for marketing by the FDA for heart failure, it would compete against other drugs used for the treatment of acute and chronic heart failure. These include generic drugs, such as milrinone, dobutamine or digoxin and branded drugs such as Natrecor (nesiritide). Omecamtiv mecarbil could also potentially compete against other novel drug candidates and therapies in development, such as bucindolol, which is being developed by ARCA biopharma, Inc.; Reasanz (serelaxin) and LCZ-696, which are being developed by Novartis; cenderitide (CD-NP), which is being developed by Carpicor Therapeutics, Inc., TRV-027, which is being developed by Trevena; ularitide, which is being developed by Cardiorentis Ltd.; aladorian, which is being developed by Armgo Pharma, Inc; certain cardioprotectants which are being developed by Cardioxyl Pharmaceuticals, Inc.; glial growth factor (GGF-2) which is being developed by Acorda Therapeutics, Inc.; Neurocardin, which is being developed by Zensun Sci & Tech, Ltd; Mydicar, a genetically-targeted enzyme replacement therapy for advanced heart failure which is being developed by Celladon Corporation; and levosimendan, which was recently acquired for development by Oxygen Biotherapeutics, Inc. In addition, there are a number of medical devices being developed for the potential treatment of heart failure.
Our competitors may:
| • | develop drug candidates and market drugs that are less expensive or more effective than our future drugs; |
| • | commercialize competing drugs before we or our partners can launch any drugs developed from our drug candidates; |
| • | hold or obtain proprietary rights that could prevent us from commercializing our products; |
| • | initiate or withstand substantial price competition more successfully than we can; |
| • | more successfully recruit skilled scientific workers and management from the limited pool of available talent; |
| • | more effectively negotiate third-party licenses and strategic alliances; |
| • | take advantage of acquisition or other opportunities more readily than we can; |
| • | develop drug candidates and market drugs that increase the levels of safety or efficacy that our drug candidates will need to show in order to obtain regulatory approval; or |
| • | introduce therapies or market drugs that render the market opportunity for our potential drugs obsolete. |
36
Table of Contents
We will compete for market share against large pharmaceutical and biotechnology companies and smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their partners, may develop new drug candidates that will compete with ours. Many of these competitors have larger research and development programs or substantially greater financial resources than we do. Our competitors may also have significantly greater experience in:
| • | developing drug candidates; |
| • | undertaking preclinical testing and clinical trials; |
| • | building relationships with key customers and opinion-leading physicians; |
| • | obtaining and maintaining FDA and other regulatory approvals of drug candidates; |
| • | formulating and manufacturing drugs; and |
| • | launching, marketing and selling drugs. |
If our competitors market drugs that are less expensive, safer or more efficacious than our potential drugs, or that reach the market sooner than our potential drugs, we may not achieve commercial success. In addition, the life sciences industry is characterized by rapid technological change. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Our competitors may render our technologies obsolete by improving existing technological approaches or developing new or different approaches, potentially eliminating the advantages in our drug discovery process that we believe we derive from our research approach and proprietary technologies.
Our failure to attract and retain skilled personnel could impair our drug development and commercialization activities.
Our business depends on the performance of our senior management and key scientific and technical personnel. The loss of the services of any member of our senior management or key scientific or technical staff may significantly delay or prevent the achievement of drug development and other business objectives by diverting management’s attention to transition matters and identifying suitable replacements. We also rely on consultants and advisors to assist us in formulating our research and development strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us. In addition, if and as our business grows, we will need to recruit additional executive management and scientific and technical personnel. There is intense competition for skilled executives and employees with relevant scientific and technical expertise, and this competition is likely to continue. Our inability to attract and retain sufficient scientific, technical and managerial personnel could limit or delay our product development activities, which would adversely affect the development of our drug candidates and commercialization of our potential drugs and growth of our business.
Any future workforce and expense reductions may have an adverse impact on our internal programs and our ability to hire and retain skilled personnel.
Our future success will depend in large part upon our ability to attract and retain highly skilled personnel. In light of our continued need for funding and cost control, we may be required to implement future workforce and expense reductions, which could further limit our research and development activities. For example, in October 2011, we reduced our workforce by approximately 18% in order to reduce expenses and to focus resources primarily on our later-stage development programs for tirasemtiv and omecamtiv mecarbil and certain other research and development programs also directed to muscle biology. These headcount reductions and the cost control measures we have implemented may negatively affect our productivity and limit our research and development activities. We may have difficulty retaining and attracting such personnel as a result of a perceived
37
Table of Contents
risk of future workforce reductions. In addition, the implementation of any additional workforce or expense reduction programs may divert the efforts of our management team and other key employees, which could adversely affect our business.
We may expand our development and clinical research capabilities and, as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We may have growth in our expenditures, the number of our employees and the scope of our operations, in particular with respect to those drug candidates that we elect to develop or commercialize independently or together with a partner. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We currently have no sales or marketing capabilities and, if we are unable to enter into or maintain strategic alliances with marketing partners or to develop our own sales and marketing capabilities, we may not be successful in commercializing our potential drugs.
We currently have no sales, marketing or distribution capabilities. We plan to commercialize drugs that can be effectively marketed and sold in concentrated markets that do not require a large sales force to be competitive. To achieve this goal, we will need to establish our own specialized sales force and marketing organization with technical expertise and supporting distribution capabilities. Developing such an organization is expensive and time-consuming and could delay a product launch. In addition, we may not be able to develop this capacity efficiently, cost-effectively or at all, which could make us unable to commercialize our drugs. If we determine not to market our drugs on our own, we will depend on strategic alliances with third parties, such as Amgen and Astellas, which have established distribution systems and direct sales forces to commercialize them. If we are unable to enter into such arrangements on acceptable terms, we may not be able to successfully commercialize these drugs. To the extent that we are not successful in commercializing any drugs ourselves or through a strategic alliance, our product revenues and business will suffer and our stock price would decrease.
Risks Related To Our Industry
The regulatory approval process is expensive, time-consuming and uncertain and may prevent our partners or us from obtaining approvals to commercialize some or all of our drug candidates.
The research, testing, manufacturing, selling and marketing of drugs are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, and regulations differ from country to country. Neither we nor our partners are permitted to market our potential drugs in the United States until we receive approval of a new drug application (“NDA”) from the FDA. Neither we nor our partners have received NDA or other marketing approval for any of Cytokinetics’ drug candidates.
Obtaining NDA approval is a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable foreign and U.S. regulatory requirements may subject us to administrative or judicially imposed sanctions. These include warning letters, civil and criminal penalties, injunctions, product seizure or detention, product recalls, total or partial suspension of production, and refusal to approve pending NDAs or supplements to approved NDAs.
Regulatory approval of an NDA or NDA supplement is never guaranteed, and the approval process typically takes several years and is extremely expensive. The FDA and foreign regulatory agencies also have substantial discretion in the drug approval process, and the guidance and advice issued by such agencies is subject to change at any time. Despite the time and efforts exerted, failure can occur at any stage, and we may encounter problems
38
Table of Contents
that cause us to abandon clinical trials or to repeat or perform additional preclinical testing and clinical trials. The number and focus of preclinical studies and clinical trials that will be required for approval by the FDA and foreign regulatory agencies varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. In addition, the FDA may require that a proposed Risk Evaluation and Mitigation Strategy, also known as a REMS, be submitted as part of an NDA if the FDA determines that it is necessary to ensure that the benefits of the drug outweigh its risks. The FDA and foreign regulatory agencies can delay, limit or deny approval of a drug candidate for many reasons, including, but not limited to:
| • | they might determine that a drug candidate is not safe or effective; |
| • | they might not find the data from preclinical testing and clinical trials sufficient and could request that additional trials be performed; |
| • | they might not approve our, our partner’s or the contract manufacturer’s processes or facilities; or |
| • | they might change their approval policies or adopt new regulations. |
Even if we receive regulatory approval to manufacture and sell a drug in a particular regulatory jurisdiction, other jurisdictions’ regulatory authorities may not approve that drug for manufacture and sale. If we or our partners fail to receive and maintain regulatory approval for the sale of any drugs resulting from our drug candidates, it would significantly harm our business and negatively affect our stock price.
If we or our partners receive regulatory approval for our drug candidates, we or they will be subject to ongoing obligations to and continued regulatory review by the FDA and foreign regulatory agencies, and may be subject to additional post-marketing obligations, all of which may result in significant expense and limit commercialization of our potential drugs.
Any regulatory approvals that we or our partners receive for our drug candidates may be subject to limitations on the indicated uses for which the drug may be marketed or require potentially costly post-marketing follow-up studies or compliance with a REMS. In addition, if the FDA or foreign regulatory agencies approves any of our drug candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record-keeping for the drug will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the drug, including adverse events of unanticipated severity or frequency, or the discovery that adverse effects or toxicities observed in preclinical research or clinical trials that were believed to be minor actually constitute much more serious problems, may result in restrictions on the marketing of the drug or withdrawal of the drug from the market.
The FDA and foreign regulatory agencies may change their policies and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our drugs and our business would suffer.
If physicians and patients do not accept our drugs, we may be unable to generate significant revenue, if any.
Even if our drug candidates obtain regulatory approval, the resulting drugs, if any, may not gain market acceptance among physicians, healthcare payors, patients and the medical community. Even if the clinical safety and efficacy of drugs developed from our drug candidates are established for purposes of approval, physicians may elect not to recommend these drugs for a variety of reasons including, but not limited to:
| • | introduction of competitive drugs to the market; |
| • | clinical safety and efficacy of alternative drugs or treatments; |
| • | cost-effectiveness; |
39
Table of Contents
| • | availability of coverage and reimbursement from health maintenance organizations and other third-party payors; |
| • | convenience and ease of administration; |
| • | prevalence and severity of adverse side effects; |
| • | other potential disadvantages relative to alternative treatment methods; or |
| • | insufficient marketing and distribution support. |
If our drugs fail to achieve market acceptance, we may not be able to generate significant revenue and our business would suffer.
The coverage and reimbursement status of newly approved drugs is uncertain and failure to obtain adequate coverage and reimbursement could limit our ability to market any drugs we may develop and decrease our ability to generate revenue.
Even if one or more of our drug candidates is approved for sale, the commercial success of our drugs in both domestic and international markets will be substantially dependent on whether third-party coverage and reimbursement is available for our drugs by the medical profession for use by their patients, which is highly uncertain. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs. As a result, they may not cover or provide adequate payment for our drugs. They may not view our drugs as cost-effective and reimbursement may not be available to consumers or may be insufficient to allow our drugs to be marketed on a competitive basis. If we are unable to obtain adequate coverage and reimbursement for our drugs, our ability to generate revenue will be adversely affected. Likewise, current and future legislative or regulatory efforts to control or reduce healthcare costs or reform government healthcare programs, such as the Patient Protection Affordable Care Act and the Health Care and Education Reconciliation Act of 2010, could result in lower prices or rejection of coverage and reimbursement for our potential drugs. Changes in coverage and reimbursement policies or healthcare cost containment initiatives that limit or restrict reimbursement for any of our drug candidates that are approved could cause our potential future revenues to decline.
We may be subject to costly product liability or other liability claims and may not be able to obtain adequate insurance.
The use of our drug candidates in clinical trials may result in adverse effects. We cannot predict all the possible harms or adverse effects that may result from our clinical trials. We currently maintain limited product liability insurance. We may not have sufficient resources to pay for any liabilities resulting from a personal injury or other claim excluded from, or beyond the limit of, our insurance coverage. Our insurance does not cover third parties’ negligence or malpractice, and our clinical investigators and sites may have inadequate insurance or none at all. In addition, in order to conduct clinical trials or otherwise carry out our business, we may have to contractually assume liabilities for which we may not be insured. If we are unable to look to our own or a third party’s insurance to pay claims against us, we may have to pay any arising costs and damages ourselves, which may be substantial.
In addition, if we commercially launch drugs based on our drug candidates, we will face even greater exposure to product liability claims. This risk exists even with respect to those drugs that are approved for commercial sale by the FDA and foreign regulatory agencies and manufactured in licensed and regulated facilities. We intend to secure additional limited product liability insurance coverage for drugs that we commercialize, but may not be able to obtain such insurance on acceptable terms with adequate coverage, or at reasonable costs. Even if we are ultimately successful in product liability litigation, the litigation would consume substantial amounts of our financial and managerial resources and may create adverse publicity, all of which would impair our ability to generate sales of the affected product and our other potential drugs. Moreover,
40
Table of Contents
product recalls may be issued at our discretion or at the direction of the FDA and foreign regulatory agencies, other governmental agencies or other companies having regulatory control for drug sales. Product recalls are generally expensive and often have an adverse effect on the reputation of the drugs being recalled and of the drug’s developer or manufacturer.
We may be required to indemnify third parties against damages and other liabilities arising out of our development, commercialization and other business activities, which could be costly and time-consuming and distract management. If third parties that have agreed to indemnify us against damages and other liabilities arising from their activities do not fulfill their obligations, then we may be held responsible for those damages and other liabilities.
Our employees or contractors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, anti-fraud and abuse laws, anti-bribery laws and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees or contractors. Such misconduct could include failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with anti-bribery laws (such as the Foreign Corrupt Practice Act) or healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, marketing and promotion, sales commission, incentive programs and other business arrangements and practices. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent these activities may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Responding to any claims relating to improper handling, storage or disposal of the hazardous chemicals and radioactive and biological materials we use in our business could be time-consuming and costly.
Our research and development processes involve the controlled use of hazardous materials, including chemicals and radioactive and biological materials. Our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resultant injury from those materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. We may be sued for any injury or contamination that results from our or third parties’ use of these materials. Compliance with environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production activities.
Our facilities in California are located near an earthquake fault, and an earthquake or other types of natural disasters, catastrophic events or resource shortages could disrupt our operations and adversely affect our results.
All of our facilities and our important documents and records, such as hard copies of our laboratory books and records for our drug candidates and compounds and our electronic business records, are located in our corporate headquarters at a single location in South San Francisco, California near active earthquake zones. If a natural disaster, such as an earthquake or flood, a catastrophic event such as a disease pandemic or terrorist attack, or a localized extended outage of critical utilities or transportation systems occurs, we could experience a
41
Table of Contents
significant business interruption. Our partners and other third parties on which we rely may also be subject to business interruptions from such events. In addition, California from time to time has experienced shortages of water, electric power and natural gas. Future shortages and conservation measures could disrupt our operations and cause expense, thus adversely affecting our business and financial results.
Risks Related To an Investment in Our Securities
We expect that our stock price will fluctuate significantly, and you may not be able to resell your shares at or at or above your investment price.
The stock market, particularly in recent years, has experienced significant volatility, particularly with respect to pharmaceutical, biotechnology and other life sciences company stocks, which often does not relate to the operating performance of the companies represented by the stock. Factors that could cause volatility in the market price of our common stock include, but are not limited to:
| • | announcements concerning any of the clinical trials for our drug candidates, such as tirasemtiv for the potential treatment of ALS, CK-2127107 for the potential treatment of non-neuromuscular indications associated with muscle weakness and omecamtiv mecarbil for the potential treatment of heart failure (including, but not limited to, the timing of initiation or completion of such trials and the results of such trials, and delays or discontinuations of such trials, including delays resulting from slower than expected or suspended patient enrollment or discontinuations resulting from a failure to meet pre-defined clinical end points); |
| • | announcements concerning our strategic alliance with Amgen or Astellas or future strategic alliances; |
| • | failure or delays in entering additional drug candidates into clinical trials; |
| • | failure or discontinuation of any of our research programs; |
| • | issuance of new or changed securities analysts’ reports or recommendations; |
| • | failure or delay in establishing new strategic alliances, or the terms of those alliances; |
| • | market conditions in the pharmaceutical, biotechnology and other healthcare-related sectors; |
| • | actual or anticipated fluctuations in our quarterly financial and operating results; |
| • | developments or disputes concerning our intellectual property or other proprietary rights; |
| • | introduction of technological innovations or new products by us or our competitors; |
| • | issues in manufacturing our drug candidates or drugs; |
| • | market acceptance of our drugs; |
| • | third-party healthcare coverage and reimbursement policies; |
| • | FDA or other U.S. or foreign regulatory actions affecting us or our industry; |
| • | litigation or public concern about the safety of our drug candidates or drugs; |
| • | additions or departures of key personnel; |
| • | substantial sales of our common stock by our existing stockholders, whether or not related to our performance; |
| • | automated trading activity by algorithmic and high-frequency trading programs; and |
| • | volatility in the stock prices of other companies in our industry or in the stock market generally. |
These and other external factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may
42
Table of Contents
otherwise negatively affect the liquidity of our common stock. In addition, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert our management’s time and attention.
If the ownership of our common stock continues to be highly concentrated, it may prevent you and other stockholders from influencing significant corporate decisions and may result in conflicts of interest that could cause our stock price to decline.
As of February 28, 2014, our executive officers, directors and their affiliates beneficially owned or controlled approximately 5.6% of the outstanding shares of our common stock (after giving effect to the exercise of all outstanding vested and unvested options, restricted stock units and warrants). Accordingly, these executive officers, directors and their affiliates, acting as a group, will have substantial influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transactions. These stockholders may also delay or prevent a change of control of us, even if such a change of control would benefit our other stockholders. The significant concentration of stock ownership may adversely affect the trading price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
Volatility in the stock prices of other companies may contribute to volatility in our stock price.
The stock market in general, and the NASDAQ stock exchanges and the market for technology companies in particular, have experienced significant price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Further, there has been particular volatility in the market prices of securities of early stage and clinical stage life sciences companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class action litigation has often been instituted. A securities class action suit against us could result in substantial costs, potential liabilities and the diversion of management’s attention and resources, and could harm our reputation and business.
Our common stock is thinly traded and there may not be an active, liquid trading market for our common stock.
There is no guarantee that an active trading market for our common stock will be maintained on NASDAQ, or that the volume of trading will be sufficient to allow for timely trades. Investors may not be able to sell their shares quickly or at the latest market price if trading in our stock is not active or if trading volume is limited. In addition, if trading volume in our common stock is limited, trades of relatively small numbers of shares may have a disproportionate effect on the market price of our common stock.
Our stockholders will experience substantial additional dilution if outstanding options or warrants are exercised for common stock.
As of February 28, 2014, there were 7,691,096 shares of common stock issuable upon the exercise of warrants, having a weighted average exercise price of $5.95 per share, and 1,699,603 shares of common stock issuable upon the exercise of stock options outstanding, having a weighted average exercise price of $18.96 per share. The exercise of outstanding options or warrants for common stock would be substantially dilutive to the outstanding shares of common stock. Any dilution or potential dilution may cause our stockholders to sell their shares, which would contribute to a downward movement in the stock price of our common stock.
Ownership changes may limit our ability to use our net operating losses and tax credits in the future.
In general, under Section 382 of the Internal Revenue Code (“Section 382”), a corporation that undergoes an ‘ownership change’ is subject to limitations on its ability to utilize its pre-change net operating losses and tax
43
Table of Contents
credits to offset future taxable income. We have performed a Section 382 analysis and do not believe that we have experienced an ownership change since 2006. A portion of our existing net operating losses and tax credits are subject to limitations arising from previous ownership changes. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 and result in additional limitations. We intend to continue to monitor public filings made by third parties with the SEC to assess whether an ownership change under Section 382 has occurred. Our ability to accurately assess any such ownership change is limited by the timeliness and accuracy of these public filings.
Evolving regulation of corporate governance and public disclosure may result in additional expenses, use of resources and continuing uncertainty.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010 and new SEC regulations and NASDAQ Stock Market LLC rules create uncertainty for public companies. We regularly evaluate and monitor developments with respect to new and proposed laws, regulations and standards. We cannot accurately predict or estimate the amount of the additional costs we may incur in connection with complying with such laws, regulations and standards or the timing of these costs. For example, compliance with the internal control requirements of Section 404 of the Sarbanes-Oxley Act has to date required us to commit significant resources to document and test the adequacy of our internal control over financial reporting. We can provide no assurance as to conclusions of management or by our independent registered public accounting firm with respect to the effectiveness of our internal control over financial reporting in the future. In addition, the SEC has adopted regulations that require us to file corporate financial statement information in an interactive data format known as XBRL. We may incur significant costs and need to invest considerable resources to remain in compliance with these regulations.
These new or changed laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We intend to maintain high standards of corporate governance and public disclosure. As a result, we intend to invest the resources necessary to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies, due to ambiguities related to practice or otherwise, regulatory authorities may initiate legal proceedings against us, which could be costly and time-consuming, and our reputation and business may be harmed.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our businesses. In addition, the terms of existing or any future debts may preclude us from paying these dividends.
Item 1B. Unresolved Staff Comments
None.
Our facilities consist of approximately 81,587 square feet of research and office space. We lease 50,195 square feet located at 280 East Grand Avenue, and 31,392 square feet at 256 East Grand Avenue, in South San Francisco, California until 2018 with an option to renew the lease for an additional three years. We believe that these facilities are suitable and adequate for our current needs.
44
Table of Contents
We are not a party to any material legal proceedings.
Item 4. Mine Safety Disclosures
Not applicable.
45
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases ofEquity Securities |
Prior to our initial public offering on April 29, 2004, there was no public market for our common stock. Our common stock was quoted under the symbol “CYTK” on the NASDAQ Global Market from the date of our initial public offering through December 19, 2012, and has since been quoted on the NASDAQ Capital Market. The following table sets forth the high and low closing sales price per share of our common stock as reported on the NASDAQ Global Market or NASDAQ Capital Market, as applicable, for the periods indicated (as adjusted for the one-for-six reverse split of our common stock which became effective June 24, 2013).
| Closing Sale Price | ||||||||
| High | Low | |||||||
2012: | ||||||||
First Quarter | $ | 7.26 | $ | 5.88 | ||||
Second Quarter | $ | 7.20 | $ | 3.60 | ||||
Third Quarter | $ | 5.52 | $ | 3.72 | ||||
Fourth Quarter | $ | 5.20 | $ | 3.60 | ||||
2013: | ||||||||
First Quarter | $ | 7.14 | $ | 4.02 | ||||
Second Quarter | $ | 12.96 | $ | 6.42 | ||||
Third Quarter | $ | 13.82 | $ | 7.57 | ||||
Fourth Quarter | $ | 7.47 | $ | 6.01 | ||||
On February 28, 2014, the last reported sale price for our common stock on the NASDAQ Capital Market was $9.85 per share. We currently expect to retain future earnings, if any, for use in the operation and expansion of our business and have not paid and do not in the foreseeable future anticipate paying any cash dividends. As of February 28, 2014, there were 73 holders of record of our common stock.
46
Table of Contents
Equity Compensation Information
Information regarding our equity compensation plans and the securities authorized for issuance thereunder is set forth in Part III, Item 12.
Comparison of Historical Cumulative Total Return Among Cytokinetics, Incorporated, the NASDAQ Stock Market (U.S.) Index and the NASDAQ Biotechnology Index(*)
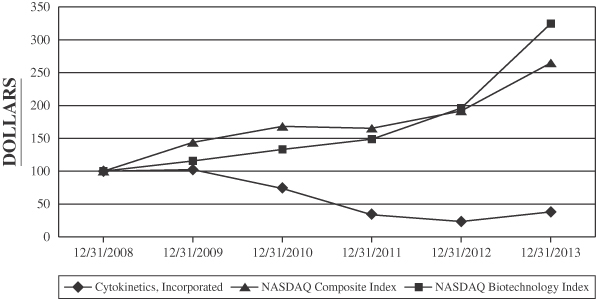
| (*) | The above graph shows the cumulative total stockholder return of an investment of $100 in cash from December 31, 2009 through December 31, 2013 for: (i) our common stock; (ii) the NASDAQ Stock Market (U.S.) Index; and (iii) the NASDAQ Biotechnology Index. All values assume reinvestment of the full amount of all dividends. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns. |
| 12/31/08 | 12/31/09 | 12/31/10 | 12/31/11 | 12/31/12 | 12/31/13 | |||||||||||||||||||
Cytokinetics, Incorporated | $ | 100.00 | $ | 102.11 | $ | 73.33 | $ | 33.68 | $ | 23.16 | $ | 38.01 | ||||||||||||
NASDAQ Composite Index | $ | 100.00 | $ | 143.89 | $ | 168.22 | $ | 165.19 | $ | 191.47 | $ | 264.84 | ||||||||||||
NASDAQ Biotechnology Index | $ | 100.00 | $ | 115.63 | $ | 132.98 | $ | 148.69 | $ | 196.12 | $ | 324.80 | ||||||||||||
The information contained under this caption “Comparison of Historical Cumulative Total Return Among Cytokinetics, Incorporated, the NASDAQ Stock Market (U.S.) Index and the NASDAQ Biotechnology Index” shall not be deemed to be soliciting material or to be filed with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act or the Exchange Act, except to the extent that we specifically incorporate it by reference into such filing.
Sales of Unregistered Securities
On June 14, 2013, we sold 1,404,100 shares of our common stock at a price per share of $7.12 and an aggregate purchase price of $10.0 million to Amgen.
We relied on the exemption from registration contained in Section 4(2) of the Securities Act, and Regulation D, Rule 506 thereunder, in connection with the issuance and sale of the common stock to Amgen.
47
Table of Contents
Item 6. Selected Financial Data
The following selected financial data should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Item 8, “Financial Statements and Supplemental Data” of this report on Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| 2013 | 2012 | 2011 | 2010 | 2009 | ||||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
Statement of Operations Data: | ||||||||||||||||||||
Revenues: | ||||||||||||||||||||
Research and development revenues from related parties(1) | $ | 2,019 | $ | 4,177 | $ | 2,054 | $ | 1,487 | $ | 7,171 | ||||||||||
Research and development, grant and other revenues | 7,547 | 3,382 | 1,946 | 1,090 | — | |||||||||||||||
License revenues from related parties(1) | 17,230 | — | — | — | 74,367 | |||||||||||||||
License revenues | 3,852 | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total revenues | 30,648 | 7,559 | 4,000 | 2,577 | 81,538 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Operating expenses: | ||||||||||||||||||||
Research and development | 49,450 | 35,643 | 37,182 | 38,013 | 39,840 | |||||||||||||||
General and administrative | 15,092 | 12,429 | 13,590 | 14,199 | 15,626 | |||||||||||||||
Restructuring charges (reversals) | — | (56 | ) | 1,192 | — | (23 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total operating expenses | 64,542 | 48,016 | 51,964 | 52,212 | 55,443 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Operating income (loss) | (33,894 | ) | (40,457 | ) | (47,964 | ) | (49,635 | ) | 26,095 | |||||||||||
Interest and other, net(2) | 177 | 87 | 104 | 172 | (1,401 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Income (loss) before income taxes | (33,717 | ) | (40,370 | ) | (47,860 | ) | (49,463 | ) | 24,694 | |||||||||||
Income tax provision (benefit) | — | — | — | (176 | ) | 150 | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net income (loss) | (33,717 | ) | (40,370 | ) | (47,860 | ) | (49,287 | ) | 24,544 | |||||||||||
Deemed dividend related to beneficial conversion feature of convertible preferred stock | — | (1,307 | ) | (2,857 | ) | — | — | |||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net income (loss) allocable to common stockholders: | $ | (33,717 | ) | $ | (41,677 | ) | $ | (50,717 | ) | $ | (49,287 | ) | $ | 24,544 | ||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Net income (loss) per share allocable to common stockholders:(3) | ||||||||||||||||||||
Basic | $ | (1.24 | ) | $ | (2.30 | ) | $ | (4.30 | ) | $ | (4.61 | ) | $ | 2.57 | ||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Diluted | $ | (1.24 | ) | $ | (2.30 | ) | $ | (4.30 | ) | $ | (4.61 | ) | $ | 2.54 | ||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Weighted average shares used in computing net income (loss) per share allocable to common stockholders:(4) | ||||||||||||||||||||
Basic | 27,275 | 18,107 | 11,800 | 10,694 | 9,565 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Diluted | 27,275 | 18,107 | 11,800 | 10,694 | 9,660 | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
48
Table of Contents
| As of December 31, | ||||||||||||||||||||
| 2013 | 2012 | 2011 | 2010 | 2009 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
Balance Sheet Data: | ||||||||||||||||||||
Cash and cash equivalents, investments, auction rate securities (“ARS”) and investment put option related to ARS | $ | 80,230 | $ | 74,000 | $ | 49,023 | $ | 72,845 | $ | 114,727 | ||||||||||
Restricted cash | — | — | 196 | 788 | 1,674 | |||||||||||||||
Working capital | 52,634 | 69,322 | 46,548 | 66,174 | 96,735 | |||||||||||||||
Total assets | 83,188 | 77,551 | 52,773 | 77,992 | 122,599 | |||||||||||||||
Long-term portion of equipment financing lines | — | — | — | 152 | 985 | |||||||||||||||
Accumulated deficit | (482,597 | ) | (448,880 | ) | (408,510 | ) | (360,650 | ) | (311,363 | ) | ||||||||||
Total stockholders’ equity(3) | 54,442 | 70,085 | 48,178 | 70,516 | 101,428 | |||||||||||||||
| (1) | Revenues from related parties consisted of revenues recognized under our research and development arrangements with related parties, including Amgen. See Note 7 in the Notes to Financial Statements for further details. |
| (2) | Interest and Other, net consisted of interest income/expense and other income/expense. For the years ended December 31, 2010, and 2009, it also included unrealized gains (losses) on ARS and investment put option related to the Series C-2 ARS Rights issued to us by UBS AG. For the year ended December 31, 2009, it also included warrant expense. See Note 15 in the Notes to Financial Statements for further details. |
| (3) | On June 24, 2013, we effected a one-for-six reverse stock split of our common stock through an amendment to our amended and restated certificate of incorporation (the “COI Amendment”). As of the effective time of the reverse stock split, every six shares of our issued and outstanding common stock were converted into one issued and outstanding share of common stock, without any change in par value per share. The reverse stock split affected all shares of our common stock outstanding immediately prior to the effective time of the reverse stock split, as well as the number of shares of common stock available for issuance under equity incentive plans. In addition, the reverse stock split effected a reduction in the number of shares of common stock issuable upon the conversion of shares of preferred stock or upon the exercise of stock options or warrants outstanding immediately prior to the effectiveness of the reverse stock split. No fractional shares were issued as a result of the reverse stock split. Stockholders who would otherwise have been entitled to receive a fractional share received cash payments in lieu thereof. In addition, the COI Amendment reduced the number of authorized shares of common stock to 81.5 million. |
All references to shares of common stock and per share data for all periods presented in the accompanying selected financial data have been adjusted to reflect the reverse stock split on a retroactive basis.
| (4) | In 2009, we sold 599,455 shares of common stock to Kingsbridge Capital Limited (“Kingsbridge”) pursuant to the 2007 committed equity financing facility for net proceeds of $6.9 million. In May 2009, we sold 1,184,433 shares of common stock in a registered direct offering for net proceeds of approximately $12.9 million. In 2010, we sold 889,970 shares of common stock to Kingsbridge pursuant to the 2007 committed equity financing facility for net proceeds of $14.0 million. In April 2011, we sold 883,333 shares of common stock, 8,070 shares of Series A convertible preferred stock and warrants to purchase 1,114,168 shares of common stock to Deerfield Private Design Fund II, L.P., Deerfield Private Design International II, L.P., Deerfield Special Situations Fund, L.P., and Deerfield Special Situations Fund International Limited for net proceeds of approximately $19.9 million. In the fourth quarter of 2011, we sold 429,868 shares of common stock through McNicoll, Lewis & Vlak LLC (“MLV”) for net proceeds of $2.4 million. In June 2012, we issued to various investors (i) 9,320,176 shares of common stock for a purchase price of $4.56 per share, (ii) 23,026 shares of Series B convertible preferred stock for a purchase price of $760.00 per share, and (iii) warrants to purchase 7,894,704 shares of the Company’s common stock at an exercise price of $5.28 per share, for aggregate gross proceeds of approximately $60.0 million. In 2012, we sold 432,724 shares of common stock through MLV for net proceeds of $2.8 million. In June 2013, we sold 1,404,100 |
49
Table of Contents
shares of common stock to Amgen at a price per share of $7.12 and an aggregate purchase price of $10.0 million, pursuant to the Amgen Agreement Amendment. In 2013, we sold 1,170,583 shares of common stock through MLV for net proceeds of $7.5 million. See Note 13, “Stockholders’ Equity (Deficit)” in the Notes to Financial Statements for further details. |
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with our financial statements and accompanying notes included elsewhere in this report. Operating results are not necessarily indicative of results that may occur in future periods.
We were incorporated in Delaware in August 1997 as Cytokinetics, Incorporated. We are a clinical stage biopharmaceutical company focused on the discovery and development of novel small molecule therapeutics that modulate muscle function for the potential treatment of serious diseases and medical conditions. Our research and development activities relating to the biology of muscle function have evolved from our knowledge and expertise regarding the cytoskeleton, a complex biological infrastructure that plays a fundamental role within every human cell. Our most advanced research and development programs relate to the biology of muscle function and are directed to small molecule modulators of the contractility of skeletal or cardiac muscle. We are also conducting or may conduct earlier-stage research directed to other compounds with the potential to modulate muscle contractility and other muscle functions, such as growth, energetics and metabolism.
Our drug candidates currently in clinical development are our skeletal muscle activators tirasemtiv and CK-2127107, and our cardiac muscle activator omecamtiv mecarbil. Tirasemtiv is being evaluated for the potential treatment of ALS and other neuromuscular disorders. CK-2127107 is being evaluated for the potential treatment of non-neuromuscular indications associated with skeletal muscle weakness. Omecamtiv mecarbil is being evaluated for the potential treatment of heart failure.
Muscle Contractility Programs
Skeletal Muscle Contractility Program
Tirasemtiv is the lead drug candidate from this program, and is in Phase II clinical development. Cytokinetics holds the rights to tirasemtiv. We are also developing another drug candidate from this program, CK-2127107, which is being evaluated in Phase I clinical trials in collaboration with Astellas Pharma Inc. (“Astellas”) for potential non-neuromuscular indications associated with muscle weakness. Tirasemtiv and CK-2127107 are structurally distinct and selective small molecules that activate the fast skeletal muscle troponin complex in the sarcomere by increasing its sensitivity to calcium, leading to an increase in skeletal muscle contractility. Each of tirasemtiv and CK-2127107 has demonstrated pharmacological activity in preclinical models and tirasemtiv has demonstrated evidence of potentially clinically relevant pharmacodynamic effects in humans. We are evaluating potential indications for which tirasemtiv and CK-2127107 may be useful.
Tirasemtiv. We have conducted three “evidence of effect” Phase IIa clinical trials of tirasemtiv. These evidence of effect clinical trials were randomized, double-blind, placebo-controlled, three-period cross-over studies of single doses of tirasemtiv administered to patients with impaired muscle function. These studies were intended to translate the mechanism of action of tirasemtiv into potentially clinically relevant pharmacodynamic effects, which may then form the basis for larger clinical trials designed to demonstrate proof of concept and possibly even to support registration. The first of these trials was conducted in patients with amyotrophic lateral sclerosis, also known as ALS or Lou Gehrig’s disease, a chronic and progressive disease in which the motor neurons die, thus denervating skeletal muscles and causing them to atrophy. This leads to weakness, fatigue, and eventually complete paralysis and death, primarily from respiratory complications. The second of these trials was conducted in patients with myasthenia gravis, a chronic, autoimmune, neuromuscular disease which is the most common primary disorder of neuromuscular transmission. The third of these trials was conducted in patients with symptoms of claudication, which is pain or cramping in the leg muscles due to inadequate blood flow during
50
Table of Contents
exercise, associated with peripheral artery disease. Evidence of potentially clinically relevant pharmacodynamic effects was observed in each of these trials. We are now conducting BENEFIT-ALS (Blinded Evaluation of Neuromuscular Effects and Functional Improvement with Tirasemtiv in ALS), a Phase IIb clinical trial of tirasemtiv in patients with ALS. We anticipate that we will need to conduct at least one confirmatory Phase III clinical trial of tirasemtiv in patients with ALS, if supported by the results of BENEFIT-ALS, to gain marketing approval.
In 2010, the National Institute of Neurological Disorders and Stroke (“NINDS”) awarded us a grant of $2.8 million under the American Recovery and Reinvestment Act of 2009, which was intended to support three years of research and development of tirasemtiv for the potential treatment of myasthenia gravis. In September 2012, the NINDS awarded us an additional $0.5 million for this program under a separate grant. We recognized revenue under this grant in 2013, 2012 and 2011 of $0.1 million, $1.3 million and $1.7 million, respectively, which we recorded as research and development grant and other revenues. The project period for both of these grants ended June 30, 2013, and no additional funds are available to us under these grants.
Tirasemtiv Clinical Development
ALS
BENEFIT-ALS (Blinded Evaluation of Neuromuscular Effects and Functional Improvement with Tirasemtiv in ALS): In October 2012, we initiated BENEFIT-ALS, a multi-national, double-blind, randomized, placebo-controlled trial originally planned to enroll at least 400 patients and subsequently increased to enroll up to 500 patients. All patients began treatment with open-label tirasemtiv at 125 mg twice daily. Patients who completed a week of open-label tirasemtiv at this starting dose were randomized 1-to-1 to receive 12 weeks of double-blind treatment with tirasemtiv or placebo. Clinical assessments take place monthly during double-blinded treatment. Randomized patients also participate in follow-up evaluations at both 7 and 28 days after their final dose of double-blind study drug. The primary analysis of BENEFIT-ALS will compare the mean change from baseline in the ALS Functional Rating Scale in its revised form, or ALSFRS-R (a clinically validated instrument designed to measure disease progression and changes in functional status), in patients receiving tirasemtiv versus those receiving placebo. Secondary endpoints will include maximum voluntary ventilation, or MVV (a clinical assessment of pulmonary function and endurance), and measures of skeletal muscle function. Patients will receive tirasemtiv or placebo dosed twice daily. Patients taking riluzole at the time of enrollment who are randomized to receive double-blind tirasemtiv will receive riluzole at a reduced dose of 50 mg daily, in a blinded manner. We are conducting BENEFIT-ALS at over 70 sites across the United States, Canada and several European countries.
In July 2013, we were informed by our data management vendor that a programming error in the electronic data capture system controlling study drug assignment caused 58 patients initially randomized to and treated with tirasemtiv to receive placebo instead at a certain study visit and for the remainder of the study. No patients randomized to placebo were dispensed incorrect treatment. Cytokinetics and all clinical trial site personnel remain blinded to the specific patients affected by the error. Following detection of the error, we took steps to ensure that no further incorrect study drug assignments occurred and to correct the programming error in the electronic data capture system controlling study drug assignment. In addition, we convened an ad hoc meeting of the Data Safety Monitoring Board (DSMB) for BENEFIT-ALS to assess whether the error in dispensing study drug had impacted the safety of the 58 affected patients. After review of the then-available safety data from BENEFIT-ALS, the DSMB reported no concerns regarding patient safety. Following interactions with regulatory authorities, we amended the protocol for BENEFIT-ALS to enable increased enrollment to approximately 680 patients and to update the statistical methods section, in both cases with the objective to maintain the originally intended statistical power of the trial. We have completed enrollment in BENEFIT-ALS with 711 patients, of which over 400 have completed 12 weeks of treatment, and anticipate reporting the results from this clinical trial at the American Academy of Neurology (AAN) Annual Meeting in Philadelphia, PA in April 2014.
Prior ALS Clinical Trials. In June 2012, we announced the publication of our Phase IIa evidence of effect clinical trial of tirasemtiv (CY 4021) in the online edition of the journal Amyotrophic Lateral Sclerosis. In that
51
Table of Contents
trial, the single doses of tirasemtiv evaluated appeared generally well-tolerated, with dizziness and general fatigue being the most frequent adverse events. In addition, both patients and investigators perceived a positive change in the patients’ overall status, in a dose-dependent fashion, at 6 hours after dosing with tirasemtiv, based on a global assessment in which the patient and the investigator each independently assessed patients’ status compared to prior to dosing. There was a clear relationship between improvements in global assessments and the plasma concentrations of tirasemtiv. Also at this 6-hour time point, there was a trend towards decreased muscle fatigability, as evidenced by data from a test of sub-maximal hand-grip endurance. Data from that clinical trial also demonstrated a statistically significant increase in MVV at both 6 and 24 hours after 500 mg of tirasemtiv, and small but statistically significant increases in maximum strength of certain muscle groups tested.
In April 2012, at the AAN 64th Annual Meeting, results were presented from CY 4024, a Phase II, two-part, randomized, double-blind, placebo-controlled, multiple-dose, safety, tolerability, pharmacokinetic and pharmacodynamic clinical trial of tirasemtiv in patients with ALS. Patients in Part A of this trial were not taking riluzole; patients in Part B received riluzole at the reduced dose of 50 mg daily. In this trial, tirasemtiv appeared to be generally safe and well-tolerated when dosed daily at 125 mg, 250 mg and 375 mg once daily for two weeks. This trial was not designed or powered to evaluate statistically the effects of tirasemtiv on the various outcome measures that were assessed during the study. However, encouraging dose-related trends were observed in measurements of ALSFRS-R and in MVV. Plasma concentrations of tirasemtiv were unaffected by co-administration with riluzole, while riluzole plasma levels were greater when co-administered with tirasemtiv than when not co-administered with tirasemtiv. Adverse events and clinical assessments during treatment with tirasemtiv appeared similar, with or without co-administration of riluzole. Dizziness, the most commonly reported adverse event, was mostly mild and generally began and resolved early after initiating treatment. The incidence and persistence of dizziness appeared dose-related but was mild in severity in all patients who completed study drug treatment. Most reports of dizziness began early after initiating treatment and resolved spontaneously within the first week of treatment in all but one patient who nevertheless completed the trial. No serious adverse events were reported.
Also in April 2012 at the AAN 64th Annual Meeting, results were presented from a Phase II, randomized, double-blind, placebo-controlled, multiple-dose, clinical trial of tirasemtiv in patients with ALS receiving riluzole at the reduced dose of 50 mg daily (CY 4025). The authors concluded that the twice-daily dose titration regimen evaluated in the trial appeared generally safe and well-tolerated, and that the majority of patients could be titrated successfully to a tirasemtiv dose level of 250 mg twice daily. This trial was not designed or powered to evaluate statistically the effects of tirasemtiv on the various outcome measures that were assessed during the study. However, encouraging trends toward functional improvements were observed in patients receiving tirasemtiv versus those receiving placebo. In this trial, tirasemtiv treatment was associated with increases in measurements of ALSFRS-R that were similar in direction, and in MVV that were similar in both direction and magnitude, to those observed in CY 4024.
CK-2127107 and Other Skeletal Muscle Activators
Astellas Strategic Alliance. In June 2013, we entered into a collaboration and license agreement with Astellas (the “Astellas Agreement”). Under the Astellas Agreement, we granted Astellas an exclusive license to co-develop and jointly commercialize CK-2127107 for potential application in non-neuromuscular indications associated with skeletal muscle weakness worldwide. CK-2127107 is being developed jointly by Cytokinetics and Astellas. Cytokinetics is primarily responsible for the conduct of Phase I clinical trials and certain Phase II readiness activities for CK-2127107. Astellas will be primarily responsible for the conduct of subsequent development and commercialization activities for CK-2127107.
The companies are jointly conducting research to identify next-generation skeletal muscle activators to be nominated as potential drug candidates, at Astellas’ expense. Astellas has the exclusive rights to develop and commercialize fast skeletal troponin activators from this research program in non-neuromuscular indications and to develop and commercialize other novel mechanism skeletal muscle activators from this research program in
52
Table of Contents
all indications, subject to certain Cytokinetics’ co-development and co-promotion rights. Astellas will be responsible for the costs associated with the development of all collaboration products, including CK-2127107.
Under the Astellas Agreement, we retain an option to conduct early-stage development for certain agreed indications at our initial expense, subject to reimbursement if development continues under the collaboration. We also retain an option to co-promote collaboration products in the United States and Canada. Astellas will reimburse us for certain expenses associated with our co-promotion activities.
In July 2013, we received an upfront payment of $16 million in connection with the execution of the Astellas Agreement, and we are eligible to potentially receive over $24 million in reimbursement of sponsored research and development activities during the initial two years of the collaboration. Based on the achievement of pre-specified criteria, we may receive over $250 million in milestone payments relating to the development and commercial launch of collaboration products, including up to $112 million in development and commercial launch milestones for CK-2127107. We may also receive up to $200 million in payments for achievement of pre-specified sales milestones related to net sales of all collaboration products under the Astellas Agreement. If Astellas commercializes any collaboration products, we will also receive royalties on sales of such collaboration products, including royalties ranging from the high single digits to the high teens on sales of products containing CK-2127107. In addition to these development, commercial launch and sales milestones, we may also receive payments for the achievement of pre-specified milestones relating to the joint research program.
CK-2127107 Clinical Development
CY 5011. In April 2013, we announced the initiation of a first-time-in-humans Phase I clinical trial of CK-2127107 in healthy male volunteers, known as CY 5011. CY 5011 was a double-blind, randomized, placebo-controlled study designed to assess the safety, tolerability, and pharmacokinetics of single ascending oral doses of CK-2127107 administered in a three-period crossover design. We announced results from this trial in December 2013 at the 7th International Conference of the Society on Sarcopenia, Cachexia and Wasting Disorders in Kobe, Japan. Planned single doses of CK-2127107 up to 4000 mg, the highest dose administered in this trial, were well-tolerated; therefore, a maximum tolerated dose was not defined. The pharmacokinetic profile of CK-2127107 was linear and dose-proportional across the dose range studied, with a mean terminal half-life compatible with once or twice daily dosing.
CY 5014. During the fourth quarter of 2013, Cytokinetics completed dosing in CY 5014, a Phase I clinical trial of CK-2127107 in healthy male volunteers. CY 5014 is a randomized, open-label, two-period crossover study designed to assess the relative oral bioavailability, pharmacokinetics, safety and tolerability of two oral formulations of CK-2127107.
Other Development. We expect to conduct additional Phase I studies and certain Phase II readiness activities pursuant to our collaboration agreement with Astellas.
Ongoing Research in Skeletal Muscle Activators. Our research on the direct activation of skeletal muscle continues in two areas. We are conducting translational research in preclinical models of disease and muscle function with fast skeletal muscle troponin activators to explore the potential clinical applications of this novel mechanism in diseases or conditions associated with skeletal muscle dysfunction. We also intend to conduct preclinical research on other chemically and pharmacologically distinct mechanisms to activate the skeletal sarcomere. We are conducting a joint research program with Astellas directed to the discovery of next-generation skeletal muscle activators. Under the Astellas Agreement, Astellas will reimburse us for certain research activities we perform.
The clinical trials programs for each of tirasemtiv and CK-2127107 may proceed for several years, and we will not be in a position to generate any revenues or material net cash flows from sales of this drug candidate until the program is successfully completed, regulatory approval is achieved, and the drug is commercialized.
53
Table of Contents
Tirasemtiv and CK-2127107 are each at too early a stage of development for us to predict if or when this may occur. Our expenditures will increase if and as we move tirasemtiv into later development. Our expenditures will also increase if Astellas terminates development of CK-2127107 or related compounds and we elect to develop them independently, or if we conduct early-stage development for certain agreed indications at our initial expense, subject to reimbursement if development continues under the collaboration.
We recorded research and development expenses for activities relating to our skeletal muscle contractility program of approximately $40.8 million, $24.9 million and $24.0 million in the years ended December 31, 2013, 2012 and 2011 respectively. We recognized research and development revenue from Astellas of $6.4 million in 2013, consisting of reimbursements of full-time employee equivalents (“FTEs”) and other expenses. We anticipate that our expenditures relating to the research and development of compounds in our skeletal muscle contractility program will increase significantly if and as we advance tirasemtiv, CK-2127107 or other compounds from this program into and through development.
Cardiac Muscle Contractility Program
Our lead drug candidate from this program is omecamtiv mecarbil, a novel cardiac muscle myosin activator. We expect omecamtiv mecarbil to be developed as a potential treatment across the continuum of care in heart failure both as an intravenous formulation for use in the hospital setting and as an oral formulation for use in the outpatient setting.
Amgen Agreement. In December 2006, we entered into a collaboration and option agreement with Amgen Inc. to discover, develop and commercialize novel small molecule therapeutics, including omecamtiv mecarbil, that activate cardiac muscle contractility for potential applications in the treatment of heart failure (the “Amgen Agreement”). The agreement granted Amgen an option to obtain an exclusive license worldwide, except Japan, to develop and commercialize omecamtiv mecarbil and other drug candidates arising from the collaboration. In May 2009, Amgen exercised its option. As a result, Amgen became responsible for the development and commercialization of omecamtiv mecarbil and related compounds at its expense worldwide (excluding Japan), subject to our development and commercialization participation rights. Amgen will reimburse us for certain research and development activities we perform under the collaboration.
In June 2013, Cytokinetics and Amgen executed an amendment to the Amgen Agreement to include Japan, resulting in a worldwide collaboration (the “Amgen Agreement Amendment”). Under the terms of the Amgen Agreement Amendment, we received a non-refundable upfront license fee of $15 million in June 2013. Under the Amgen Agreement Amendment, we plan to conduct a Phase I pharmacokinetic study intended to support inclusion of Japan in a potential Phase III clinical development program and potential global registration dossier for omecamtiv mecarbil. Amgen will reimburse us for the costs of this study. In addition, we are eligible to receive additional pre-commercialization milestone payments relating to the development of omecamtiv mecarbil in Japan of up to $50 million, and royalties on sales of omecamtiv mecarbil in Japan. In conjunction with the Amgen Agreement Amendment, we also entered into a common stock purchase agreement which provided for the sale of 1,404,100 shares of our common stock to Amgen at a price per share of $7.12 and an aggregate purchase price of $10.0 million which was received in June 2013. Pursuant to this agreement, Amgen has agreed to certain trading and other restrictions with respect to our common stock.
Under the Amgen Agreement, as amended, we are eligible for potential pre-commercialization and commercialization milestone payments of up to $650 million in the aggregate on omecamtiv mecarbil and other potential products arising from research under the collaboration, and royalties that escalate based on increasing levels of annual net sales of products commercialized under the agreement. The Amgen Agreement also provides for us to receive increased royalties by co-funding Phase III development costs of omecamtiv mecarbil and other drug candidates under the collaboration. If we elect to co-fund such costs, we would be entitled to co-promote the co-funded drug in North America and participate in agreed commercialization activities in institutional care settings, at Amgen’s expense.
54
Table of Contents
In July 2013, Amgen announced that it had granted an option to commercialize omecamtiv mecarbil in Europe to Servier.
Omecamtiv Mecarbil Clinical Development
Current Clinical Trials. In March 2013, we announced the initiation of dosing of patients in COSMIC-HF (Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure). COSMIC-HF is a Phase II, double-blind, randomized, placebo-controlled, multicenter, dose escalation study designed to evaluate several modified-release oral formulations of omecamtiv mecarbil in patients with heart failure and left ventricular systolic dysfunction. COSMIC-HF is being conducted by Amgen in collaboration with Cytokinetics. The primary objectives of this trial are to select an oral modified release formulation and doses of omecamtiv mecarbil for chronic twice-daily dosing in patients with heart failure and left ventricular systolic dysfunction and to characterize its pharmacokinetics during 20 weeks of treatment. The secondary objective is to evaluate the safety and tolerability of oral omecamtiv mecarbil. In addition, we will have an opportunity to evaluate the potential for sustained pharmacodynamic effects and their relationship to the pharmacokinetics of this drug candidate. During the third quarter of 2013, the second cohort of the dose escalation phase of COSMIC-HF completed enrollment. We and Amgen recently reviewed results from COSMIC-HF and selected an oral formulation of omecamtiv mecarbil for evaluation in the planned expansion phase of the trial. We and Amgen have agreed to amend the protocol to evaluate a plasma concentration-guided dose titration strategy in the expansion phase of COSMIC-HF. The size of the expansion phase has been increased with the objective to provide greater statistical power for the planned evaluation of several pharmacodynamic parameters during oral dosing with omecamtiv mecarbil. This trial is being conducted by Amgen in collaboration with Cytokinetics.
Recently, Cytokinetics and Amgen agreed on the protocol and budget for the planned Phase I pharmacokinetic study, CY 1211, in healthy volunteers of both Japanese and non-Japanese ethnicity. The trial will be conducted by Cytokinetics in collaboration with Amgen. The costs of the trial will be reimbursed by Amgen.
We are collaborating with Amgen to respond to information requests received from regulatory authorities relating to their ongoing review of the protocols submitted for COSMIC-HF and CY 1211. We anticipate commencement of patient enrollment in both the expansion phase of COSMIC-HF and in CY 1211 to occur in the first half of 2014 following regulatory authorities’ review of responses relating to these information requests. We expect both the enrollment of patients in the expansion phase of COSMIC-HF and the conduct of CY 1211 to be completed in 2014.
ATOMIC-AHF. In September 2013, results from ATOMIC-AHF (Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure) were presented at the European Society of Cardiology Congress and the Heart Failure Society of America Annual Scientific Meeting. ATOMIC-AHF was an international, randomized, double-blind, placebo-controlled, Phase IIb clinical trial of intravenous omecamtiv mecarbil in patients with left ventricular systolic dysfunction hospitalized with acutely decompensated heart failure. ATOMIC-AHF was conducted by Amgen in collaboration with Cytokinetics. This clinical trial enrolled over 600 patients in three sequential, ascending-dose cohorts. In each cohort, patients were randomized to receive omecamtiv mecarbil or placebo. The primary efficacy objective of this trial was to evaluate the effect of 48 hours of intravenous omecamtiv mecarbil compared to placebo on dyspnea (shortness of breath). The secondary objectives were to assess the safety and tolerability of three dose levels of intravenous omecamtiv mecarbil compared with placebo and to evaluate the effects of 48 hours of treatment with intravenous omecamtiv mecarbil on additional measures of dyspnea, patients’ global assessments, change in N-terminal pro brain-type natriuretic peptide (a biomarker associated with the severity of heart failure) and short-term clinical outcomes in these patients. In addition, the trial evaluated the relationship between plasma concentrations of omecamtiv mecarbil and echocardiographic parameters in patients with acute heart failure.
The omecamtiv mecarbil treatment groups were not statistically different in their 7-point Likert scale dyspnea symptom response rates compared to the pooled placebo group (p=0.33); therefore, the primary endpoint
55
Table of Contents
was not met. Omecamtiv mecarbil demonstrated favorable dose- and concentration-related trends (nominal p=0.025 and nominal p=0.007, respectively) on dyspnea response. Improvement in dyspnea was observed in the highest omecamtiv mecarbil dose group when compared against its paired placebo group in the third cohort (dyspnea symptom response in 51 percent of subjects on omecamtiv mecarbil versus 37 percent on placebo, nominal p=0.03). The incidence of worsening heart failure within seven days of initiating treatment was 17 percent in the pooled placebo group and was 13 percent, 8 percent and 9 percent on omecamtiv mecarbil in the first, second and third cohorts, respectively. Systolic ejection time, the echocardiographic signature of omecamtiv mecarbil, increased in a concentration-dependent manner similar to that previously reported in healthy volunteers and stable heart failure patients.
Rates of adverse events (AEs), serious AEs, adjudicated deaths and hospitalizations were similar between omecamtiv mecarbil and placebo groups. There were seven post-randomization myocardial infarctions in the treatment groups receiving omecamtiv mecarbil compared with three in the placebo groups (2.3 percent vs. 1.0 percent, respectively). However, there was no relationship between the maximum increase from the baseline troponin (a biomarker specific for cardiac muscle damage) and increasing plasma concentrations of omecamtiv mecarbil. Four of the myocardial infarctions occurred more than seven days following termination of the 48-hour drug infusion. The estimated plasma concentrations near the time of these events were zero. Three of these events occurred in patients who received omecamtiv mecarbil and one occurred in a patient who received placebo. One myocardial infarction occurred subsequent to a percutaneous coronary intervention in a patient who received omecamtiv mecarbil. One myocardial infarction occurred in a patient with sepsis who received placebo. Omecamtiv mecarbil was not associated with an increased incidence of tachyarrhythmias nor were heart rate or blood pressure adversely affected.
Prior Clinical Experience with Omecamtiv Mecarbil. Prior to Amgen’s exercise of its option, Cytokinetics conducted a clinical trials program for omecamtiv mecarbil comprised of multiple Phase I and Phase IIa clinical trials designed to evaluate the safety, tolerability, pharmacodynamic and pharmacokinetic profiles of both intravenous and oral formulations in a diversity of patients, including patients with stable heart failure and patients with ischemic cardiomyopathy. In these trials, omecamtiv mecarbil exhibited generally linear, dose-proportional pharmacokinetics across the dose ranges studied. The adverse effects observed at intolerable doses in humans appeared similar to the adverse findings which occurred in preclinical safety studies at similar plasma concentrations. These effects are believed to be related to the mechanism of action of this drug candidate which, at intolerable doses, resulted in an excessive prolongation of the systolic ejection time (i.e., the time in which the heart is contracting). However, these effects resolved promptly with discontinuation of the infusions of omecamtiv mecarbil.
Phase IIa stable heart failure (safety, tolerability, pharmacokinetics and pharmacodynamics). In 2009, we presented final results from our Phase IIa clinical trial evaluating omecamtiv mecarbil administered intravenously to patients with stable heart failure. The final results showed statistically significant increases in systolic ejection time, and in stroke volume, cardiac output, fractional shortening and ejection fraction (all measures of cardiac function), that occurred across the patient population in a concentration-dependent manner. In addition, the data demonstrated statistically significant correlations between increasing omecamtiv mecarbil plasma concentrations and decreases in left ventricular end-systolic volume, left ventricular end-diastolic volume and heart rate. Omecamtiv mecarbil appeared to be generally well-tolerated in stable heart failure patients over a range of plasma concentrations during continuous intravenous administration. Patients with reduced stroke volumes (<50 ml) at baseline had generally greater pharmacodynamic responses to omecamtiv mecarbil than those in patients with greater stroke volumes at baseline, demonstrating robust pharmacodynamic activity in this more severely affected sub-population of patients from the trial.
Phase IIa ischemic cardiomyopathy and angina (safety and tolerability). In 2009, we presented results from a double-blind, randomized, placebo-controlled Phase IIa clinical trial evaluating the effect of omecamtiv mecarbil on symptom-limited exercise tolerance in heart failure patients with ischemic cardiomyopathy and angina. The primary safety endpoint of this clinical trial was stopping an exercise treadmill test due to angina at a
56
Table of Contents
stage earlier than the shorter of two baseline exercise treadmill tests. This endpoint occurred in one patient receiving placebo and in no patients receiving either the lower or higher of two dose levels of omecamtiv mecarbil. In heart failure patients with ischemic cardiomyopathy and angina, who theoretically could be most vulnerable to the possible deleterious consequences of systolic ejection time prolongation, treatment with omecamtiv mecarbil, at doses producing plasma concentrations previously demonstrated in other trials to increase cardiac function, did not appear to deleteriously affect a broad range of safety assessments in the setting of exercise.
Phase I Clinical Trials. Seven Phase I clinical trials of omecamtiv mecarbil have been conducted in healthy subjects: five conducted by Cytokinetics and two conducted by Amgen in collaboration with Cytokinetics. Results from these trials were reported previously.
Ongoing Research in Cardiac Muscle Contractility. In 2013, we agreed with Amgen to additional research activities intended to be conducted through 2014 under the research plan directed to next-generation compounds in our cardiac muscle contractility program. Under the Amgen Agreement, Amgen will reimburse us for certain activities we perform.
The clinical trials program for omecamtiv mecarbil may proceed for several years, and we will not be in a position to generate any revenues or material net cash flows from sales of this drug candidate until the program is successfully completed, regulatory approval is achieved, and the drug is commercialized. Omecamtiv mecarbil is at too early a stage of development for us to predict if or when this may occur. We funded all research and development costs associated with this program prior to Amgen’s option exercise in May 2009. We recorded research and development expenses for activities relating to our cardiac muscle contractility program of approximately $3.4 million, $4.5 million and $2.8 million in the years ended December 31, 2013, 2012 and 2011, respectively. We recognized research and development revenue from Amgen of $2.0 million in 2013, $4.2 million in 2012 and $2.1 million in 2011, consisting of reimbursements of FTEs and other expenses.
We anticipate that our expenditures relating to the research and development of compounds in our cardiac muscle contractility program will increase if we participate in the future advancement of omecamtiv mecarbil through clinical development. Our expenditures will also increase if Amgen terminates development of omecamtiv mecarbil or related compounds and we elect to develop them independently, or if we elect to co-fund later-stage development of omecamtiv mecarbil or other compounds in our cardiac muscle contractility program under our collaboration and option agreement with Amgen.
Beyond Muscle Contractility
We have developed preclinical expertise in the mechanics of skeletal, cardiac and smooth muscle that extends from proteins to tissues to intact animal models. Our translational research in muscle contractility has enabled us to better understand the potential impact of small molecule compounds that increase skeletal or cardiac muscle contractility and to apply those findings to the further evaluation of our drug candidates in clinical populations. In addition to contractility, the other major functions of muscle include metabolism, growth and energetics, with each of these functions playing a role in certain diseases that could benefit from novel mechanism treatments. Accordingly, our knowledge of muscle contractility may serve as an entry point to the discovery of novel treatments for disorders involving muscle functions other than muscle contractility. We are leveraging our current understandings of muscle biology to investigate new ways of modulating these other aspects of muscle function for other potential therapeutic applications. For example, we are conducting research with compounds that affect muscle growth and that may have applications for serious diseases and medical conditions such as cachexia. Cachexia is a condition that can be associated with cancer, heart failure, chronic obstructive pulmonary disease or other conditions. This syndrome is characterized by the loss of muscle mass and may lead to weakness and disability. We are performing research on compounds that may increase muscle mass and which may impact patient functionality or potentially alter the course of diseases associated with muscle wasting.
57
Table of Contents
Development Risks
The successful development of any of our drug candidates is highly uncertain. We cannot estimate with certainty or know the exact nature, timing and costs of the activities necessary to complete the development of any of our drug candidates or the date of completion of these development activities due to numerous risks and uncertainties, including, but not limited to:
| • | decisions made by Amgen with respect to the development of omecamtiv mecarbil and by Astellas with respect to the development of CK-2127107; |
| • | our potential inability to obtain the additional funding necessary for us to conduct the one or more confirmatory Phase III clinical trials for tirasemtiv in patients with ALS, if supported by the results of BENEFIT-ALS, that we anticipate will be required to obtain marketing approval for this indication; |
| • | the uncertainty of the timing of the initiation and completion of patient enrollment and treatment in our or our partners’ clinical trials; |
| • | the possibility of delays in the collection of clinical trial data and the uncertainty of the timing of the analyses of our clinical trial data after these trials have been initiated and completed; |
| • | our potential inability to obtain additional funding and resources for our development activities on acceptable terms, if at all, including, but not limited to, our potential inability to obtain or retain partners to assist in the design, management, conduct and funding of clinical trials; |
| • | failure by our clinical trial sites, clinical research organizations, clinical manufacturing organizations and other third parties supporting our or our partners’ clinical trials to fulfill their obligations or otherwise perform as expected. |
| • | delays or additional costs in manufacturing of our drug candidates for clinical trial use, including developing appropriate formulations of our drug candidates; |
| • | the uncertainty of clinical trial results, including variability in patient response; |
| • | the uncertainty of obtaining FDA or other foreign regulatory agency approval required for the clinical investigation of our drug candidates; |
| • | the uncertainty related to the development of commercial scale manufacturing processes and qualification of a commercial scale manufacturing facility; |
| • | the possibility that results from non-clinical studies may adversely impact the timing or further development of our drug candidates; and |
| • | possible delays in the characterization, formulation and manufacture of drug candidates and other compounds. |
If we fail to complete the development of any of our drug candidates in a timely manner, it could have a material adverse effect on our operations, financial position and liquidity. In addition, any failure by us or our partners to obtain, or any delay in obtaining, regulatory approvals for our drug candidates could have a material adverse effect on our results of operations. A further discussion of the risks and uncertainties associated with completing our programs as planned, or at all, and certain consequences of failing to do so are discussed further in the risk factors entitled “We will need substantial additional capital in the future to sufficiently fund our operations,” “We have never generated, and may never generate, revenues from commercial sales of our drugs and we may not have drugs to market for at least several years, if ever,” “Clinical trials may fail to demonstrate the desired safety and efficacy of our drug candidates, which could prevent or significantly delay completion of clinical development and regulatory approval” and “Clinical trials are expensive, time-consuming and subject to delay,” and other risk factors.
58
Table of Contents
Revenues
Our current revenue sources are limited, and we do not expect to generate any revenue from product sales for several years, if at all. We have recognized revenues from our strategic alliances with Amgen, Astellas, Global Blood Therapeutics, Inc., formerly known as Global Blood Targeting, Inc. (“Global Blood”) and MyoKardia, Inc. (“MyoKardia”) and grant revenues from NINDS.
In June 2013, we and Amgen executed an amendment to the Amgen Agreement to include Japan, resulting in a worldwide collaboration. (See Note 7 to financial statements.) Under the terms of the Amgen Agreement Amendment, we received a non-refundable upfront license fee of $15 million in June 2013. Under the Amgen Agreement Amendment, we plan to conduct a Phase I pharmacokinetic study intended to support inclusion of Japan in a potential Phase III clinical development program and potential global registration dossier for omecamtiv mecarbil. Amgen will reimburse us for the costs of this study. In addition, we are eligible to receive additional pre-commercialization milestone payments relating to the development of omecamtiv mecarbil in Japan of up to $50 million, and royalties on sales of omecamtiv mecarbil in Japan. In the fourth quarter of 2013, we determined that all conditions necessary for revenue recognition of the non-refundable upfront license fee under Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 605-10 had been met and accordingly, in the fourth quarter of 2013, we recognized a total of $17.2 million in license revenue attributable to the Amgen Agreement Amendment.
We have received reimbursements from Amgen for certain research and development activities, which we recorded as revenue as the related expenses were incurred. We may be eligible to receive further reimbursements from Amgen for certain research and development activities, which we will record as revenue if and when the related expenses are incurred. We record amounts received in advance of performance as deferred revenue. Revenues related to the reimbursement of FTEs were based on negotiated rates intended to approximate the costs for our FTEs.
In July 2013, we received an upfront payment of $16 million in connection with the execution of the Astellas Agreement, and we are eligible to potentially receive over $24 million in reimbursement of sponsored research and development activities during the initial two years of the collaboration. Based on the achievement of pre-specified criteria, we may receive over $250 million in milestone payments relating to the development and commercial launch of collaboration products, including up to $112 million in development and commercial launch milestones for CK-2127107. We may also receive up to $200 million in payments for achievement of pre-specified sales milestones related to net sales of all collaboration products under the Agreement. If Astellas commercializes any collaboration products, we will also receive royalties on sales of such collaboration products, including royalties ranging from the high single digits to the high teens on sales of products containing CK-2127107. In addition to these development, commercial launch and sales milestones, we may also receive payments for the achievement of pre-specified milestones relating to the joint research program. We determined the license and the research and development services relating to the Astellas Agreement are a single unit of accounting as the license was determined to not have stand-alone value. Accordingly, we are recognizing this revenue using the proportional performance model. In 2013, we recognized $3.9 million of the $16 million upfront license fee as license revenue and as of December 31, 2013, we deferred the remaining $12.1 million. We also recognized $6.4 million in revenue in 2013 for reimbursement of sponsored research and development activities under the Astellas Agreement.
Because a substantial portion of our revenues for the foreseeable future will depend on achieving development and other pre-commercialization milestones under our strategic alliances with Amgen and Astellas, our results of operations may vary substantially from year to year.
If one or more of our drug candidates is approved for sale as a drug, we expect that our future revenues will most likely be derived from royalties on sales from drugs licensed to Amgen and Astellas under our respective strategic alliances and from those licensed to future partners, and from direct sales of our drugs. We retain a
59
Table of Contents
product-by-product option to co-fund certain Phase III development activities under our strategic alliance with Amgen, thereby potentially increasing our royalties and affording us co-promotion rights in North America. If we exercise our co-promotion rights under the Amgen strategic alliance, we are entitled to receive reimbursement for certain sales force costs we incur in support of our commercial activities. Under our strategic alliance with Astellas, we retain an option to co-promote collaboration products in the United States and Canada. Astellas will reimburse us for certain expenses associated with our co-promotion activities.
As part of an initiative to seek certain smaller collaborations intended to allow us to offset our research costs, during 2011 and 2012, we entered into collaborative research agreements with two early-stage biopharmaceutical companies. In October 2011, we entered into an agreement with Global Blood. Under an agreed research plan, scientists from Global Blood and our FTEs conducted research focused on small molecule therapeutics that target the blood. We provided to Global Blood access to certain research facilities, FTEs and other resources at agreed reimbursement rates that approximated our costs. We were the primary obligor in the collaboration arrangement, and accordingly, we recorded expense reimbursements from Global Blood as research and development revenue. In April 2012, we extended this agreement through December 2012. Research and development revenue of $14,000 in 2013 from Global Blood was pursuant to a separate agreement.
In August 2012, we entered into a collaboration agreement with MyoKardia. Under an agreed research plan, scientists from MyoKardia and our FTEs conduct research focused on small molecule therapeutics that inhibit cardiac sarcomere proteins. We provided to MyoKardia access to certain research facilities, and continue to provide FTEs and other resources at agreed reimbursement rates that approximate our costs. We are the primary obligor in the collaboration arrangement, and accordingly, we record expense reimbursements from MyoKardia as research and development revenue. The research plan ended as planned in August 2013.
In July 2010 and in September 2012, the NINDS awarded us grants to support research and development of tirasemtiv directed to the potential treatment for myasthenia gravis for a period of up to three years. The grants were completed in June of 2013.
Research and Development
We incur research and development expenses associated with both partnered and unpartnered research activities. We expect to incur research and development expenses for the clinical development of tirasemtiv and CK-2127107 and pre-clinical research of other skeletal sarcomere activators for the potential treatment of diseases and medical conditions associated with muscle weakness or wasting. We expect to incur research and development expenses for omecamtiv mecarbil for the potential treatment of heart failure in accordance with agreed upon research and development plans with Amgen.
Research and development expenses related to any development and commercialization activities we elect to fund consist primarily of employee compensation, supplies and materials, costs for consultants and contract research and manufacturing, facilities costs and depreciation of equipment.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and administrative functions, including, but not limited to, finance, human resources, legal, business and commercial development and strategic planning. Other significant costs include facilities costs and professional fees for accounting and legal services, including legal services associated with obtaining and maintaining patents and regulatory compliance.
Restructuring
In October 2011, we announced a restructuring plan to realign our workforce and operations in line with our continued commitment to focus primarily on the development of our key later-stage development programs for
60
Table of Contents
tirasemtiv and omecamtiv mecarbil and on our follow-on skeletal muscle troponin activator program and joint research with Amgen directed to next-generation compounds in our cardiac muscle contractility program. As a result, we reduced our workforce by 18 employees, or approximately 18%, to 83 employees. We provided severance, employee benefit continuation and career transition assistance to the employees directly affected by the restructuring. We incurred restructuring charges of $1.2 million in the fourth quarter of 2011, primarily personnel-related termination costs. We completed all restructuring activities and recognized all anticipated restructuring charges by December 31, 2012.
Stock Compensation
The following table summarizes stock-based compensation related to stock options, restricted stock awards, restricted stock units, and employee stock purchases for 2013, 2012 and 2011 (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Research and development | $ | 1,538 | $ | 1,801 | $ | 1,331 | ||||||
General and administrative | 2,059 | 1,982 | 1,738 | |||||||||
|
|
|
|
|
| |||||||
Stock-based compensation included in operating expenses | $ | 3,597 | $ | 3,783 | $ | 3,069 | ||||||
|
|
|
|
|
| |||||||
As of December 31, 2013, there was $3.2 million of unrecognized compensation cost related to unvested stock options, which is expected to be recognized over a weighted-average period of 2.54 years and $149,000 of unrecognized compensation cost related to unvested restricted stock units, which is expected to be recognized over a weighted-average period of 1.18 years.
Income Taxes
We account for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce the deferred tax assets to the amounts expected to be realized. We did not record an income tax provision in the years ended December 31, 2013, 2012 or 2011 because we had a net taxable loss in these periods.
Based upon the weight of available evidence, which includes our historical operating performance, reported cumulative net losses since inception and difficulty in accurately forecasting our future results, we maintained a full valuation allowance on the net deferred tax assets as of December 31, 2013, 2012 and 2011. The valuation allowance was determined pursuant to the accounting guidance for income taxes, which requires an assessment of both positive and negative evidence when determining whether it is more likely than not that deferred tax assets are recoverable. We intend to maintain a full valuation allowance on the U.S. deferred tax assets until sufficient positive evidence exists to support reversal of the valuation allowance. The valuation allowance increased by $13.7 million in 2013, $21.1 million in 2012 and $18.5 million in 2011.
We also follow the accounting guidance that defines the threshold for recognizing the benefits of tax return positions in the financial statements as “more-likely-than-not” to be sustained by the taxing authorities based solely on the technical merits of the position. If the recognition threshold is met, the tax benefit is measured and recognized as the largest amount of tax benefit that, in our judgment, is greater than 50% likely to be realized. Historically, we have filed income tax returns with the federal Internal Revenue Service (“IRS”) and the state of California. For jurisdictions in which tax filings are made, we are subject to income tax examination for all fiscal years since inception. In general, the statute of limitations for tax liabilities for these years remains open for the purpose of adjusting the amounts of the losses and credits carried forward from those years.
61
Table of Contents
Interest accrued related to unrecognized tax benefits and penalties was zero for 2013, 2012 and 2011. We account for interest related to unrecognized tax benefits and penalties by classifying both as income tax expense in the financial statements in accordance with the accounting guidance for uncertainty in income taxes. We do not expect our unrecognized tax benefits to change materially over the next twelve months.
In general, under Section 382 of the Internal Revenue Code (“Section 382), a corporation that undergoes an ‘ownership change’ is subject to limitations on its ability to utilize its pre-change net operating losses (“NOLs”) and tax credits to offset future taxable income. We have performed a Section 382 analysis and do not believe that we have experienced an ownership change since 2006. A portion of our existing NOLs and tax credits are subject to limitations arising from previous ownership changes. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 and result in additional limitations.
Results of Operations
Years ended December 31, 2013, 2012 and 2011
Revenues
| Years Ended December 31, | Increase (Decrease) | |||||||||||||||||||
| 2013 | 2012 | 2011 | 2013 | 2012 | ||||||||||||||||
| (In millions) | ||||||||||||||||||||
Research and development revenues from related parties | $ | 2.0 | $ | 4.2 | $ | 2.1 | $ | (2.2 | ) | $ | 2.1 | |||||||||
Research and development, grant and other revenues | 7.5 | 3.4 | 1.9 | 4.1 | 1.5 | |||||||||||||||
License revenues from related parties | 17.2 | — | — | 17.2 | — | |||||||||||||||
License revenues | 3.9 | — | — | 3.9 | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total revenues | $ | 30.6 | $ | 7.6 | $ | 4.0 | $ | 23.0 | $ | 3.6 | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Research and development revenues from related parties refers to research and development revenues from our strategic alliance with Amgen. Revenues from Amgen were $2.0 million, $4.2 million and $2.1 million in 2013, 2012 and 2011, respectively. Revenues from Amgen in 2013 and 2012 consisted of reimbursement of FTE expenses. Revenues from Amgen in 2011 consisted of $2.0 million for reimbursement of FTE expenses and $0.1 million for other research and development expenses.
Research and development, grant and other revenues in 2013 included $6.4 million of research program reimbursement revenues from our collaboration with Astellas. Research and development, grant and other revenues in 2013, 2012 and 2011 also included grant revenue from the NINDS, revenue from Global Blood and revenue from MyoKardia. In July 2010 and in September 2012, the NINDS awarded us grants to support research and development of tirasemtiv directed to the potential treatment for myasthenia gravis for a period of up to three years. We recognized grant revenue of $69,000, $1.3 million and $1.7 million under this grant arrangement in 2013, 2012 and 2011, respectively. As part of an initiative to seek certain smaller collaborations intended to allow us to offset our research costs, during 2011 and 2012, we entered into collaborative research agreements with two early-stage biopharmaceutical companies. We recognized revenue from Global Blood of $14,000 in 2013, $1.5 million in 2012 and $0.3 million in 2011. We recognized revenue from Myokardia of $42,000 in 2013 and $0.6 million in 2012.
License revenues from related parties refers to license revenues from our strategic alliance with Amgen. Revenues from Amgen of $17.2 million in 2013 included recognition of an upfront license fee of $15 million, along with additional license revenues of $2.2 million, resulting from the allocation of a portion of the excess of the cash received over the fair value of the common stock issued to Amgen as part of that transaction. In conjunction with the Amgen Amendment Agreement, we sold 1,404,100 shares of our common stock to Amgen
62
Table of Contents
for $10.0 million, which was received in June 2013. We determined the fair value of the stock issued to Amgen to be $7.5 million. A portion of the excess of cash received over fair value of $2.5 million was also allocated to the services performed and was deferred and will be recognized as revenue as services are performed. At December 31, 2013, deferred revenue relating to the Amgen Amendment Agreement was approximately $300,000.
License revenues in 2013 refers to license revenues from our collaboration with Astellas. Revenues included $3.9 million of the $16 million upfront license fee received from Astellas in July 2013 in connection with the execution of the Astellas Agreement. We are recognizing this revenue over time using the proportional performance model.
Research and development expenses
| Years Ended December 31, | Increase (Decrease) | |||||||||||||||||||
| 2013 | 2012 | 2011 | 2013 | 2012 | ||||||||||||||||
| (In millions) | ||||||||||||||||||||
Research and development expenses | $ | 49.5 | $ | 35.6 | $ | 37.2 | $ | 13.9 | $ | (1.6 | ) | |||||||||
The increase in research and development expenses in 2013 was primarily due to increased spending for outsourced clinical costs and laboratory costs totaling $14.0 million, partially offset by decreased spending for outsourced preclinical expense. The decrease in 2012 was primarily due to decreases of $2.1 million in laboratory expenses and $1.0 million in personnel-related costs, partially offset by an increase of $1.3 million in outsourced clinical and pre-clinical costs and $0.2 million in facilities costs.
From a program perspective, the $13.9 million increase in research and development spending in 2013 compared to 2012 was primarily due to increased spending of $15.9 million for our skeletal muscle contractility program. The $1.6 million decline in research and development spending in 2012 compared to 2011 was primarily due to decreases of $3.8 million for our smooth muscle contractility program and $0.4 million for our other research programs, partially offset by increased spending of $0.9 million for our skeletal muscle contractility program and $1.7 million for our cardiac muscle contractility program.
| Years Ended December 31, | Increase (Decrease) | |||||||||||||||||||
| 2013 | 2012 | 2011 | 2013 | 2012 | ||||||||||||||||
| (In millions) | ||||||||||||||||||||
Cardiac muscle contractility | $ | 3.4 | $ | 4.5 | $ | 2.8 | $ | (1.1 | ) | $ | 1.7 | |||||||||
Skeletal muscle contractility | 40.8 | 24.9 | 24.0 | 15.9 | 0.9 | |||||||||||||||
Smooth muscle contractility | 0.2 | 1.8 | 5.6 | (1.6 | ) | (3.8 | ) | |||||||||||||
All other research programs | 5.1 | 4.4 | 4.8 | 0.7 | (0.4 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Total research and development expenses | $ | 49.5 | $ | 35.6 | $ | 37.2 | $ | 13.9 | $ | (1.6 | ) | |||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Clinical development timelines, likelihood of success and total completion costs vary significantly for each drug candidate and are difficult to estimate. We anticipate that we will determine on an ongoing basis which research and development programs to pursue and how much funding to direct to each program, taking into account the scientific and clinical success of each drug candidate. The lengthy process of seeking regulatory approvals and subsequent compliance with applicable regulations requires the expenditure of substantial resources. Any failure by us to obtain and maintain, or any delay in obtaining, regulatory approvals could cause our research and development expenditures to increase and, in turn, could have a material adverse effect on our results of operations.
We expect our research and development expenditures to increase in 2014 compared to 2013 and that they will be in the range of $52 million to $55 million. We expect to continue development of our drug candidate
63
Table of Contents
tirasemtiv for the potential treatment of neuromuscular diseases and medical conditions associated with muscle weakness or wasting. As part of our strategic alliance with Astellas, we expect to continue development of our drug candidate CK-2127107 for the potential treatment of non-neuromuscular diseases and medical conditions associated with muscle weakness or wasting. As part of our strategic alliance with Amgen, we expect to continue development of our drug candidate omecamtiv mecarbil for the potential treatment of heart failure. Non-cash expenses such as stock-based compensation and depreciation of approximately $1.5 million are included in our estimate of 2014 research and development expenses.
General and administrative expenses
| Years Ended December 31, | Increase (Decrease) | |||||||||||||||||||
| 2013 | 2012 | 2011 | 2013 | 2012 | ||||||||||||||||
| (In millions) | ||||||||||||||||||||
General and administrative expenses | $ | 15.1 | $ | 12.4 | $ | 13.6 | $ | 2.7 | $ | (1.2 | ) | |||||||||
General and administrative expenses increased in 2013 compared to 2012 and decreased in 2012 compared to 2011. The increase in 2013 was primarily due to increased spending of $1.7 million for personnel-related costs, $1.4 million for outside services and $0.2 million for legal expenses. The decrease in 2012 compared to 2011 was primarily due to decreases of $0.4 million in financial services costs, $0.3 million in personnel expenses, and $0.5 million in facilities costs. We expect that general and administrative expenses in 2014 will increase compared to 2013 and will be in the range of $16 million to $18 million. Non-cash expenses such as stock-based compensation and depreciation of approximately $1.5 million are included in our estimate of 2014 general and administrative expenses.
Interest and Other, net
Components of Interest and Other, net are as follows:
| Years Ended December 31, | Increase (Decrease) | |||||||||||||||||||
| 2013 | 2012 | 2011 | 2013 | 2012 | ||||||||||||||||
| (In millions) | ||||||||||||||||||||
Interest income and other income | $ | 0.1 | $ | 0.1 | $ | 0.2 | $ | — | $ | (0.1 | ) | |||||||||
Interest expense and other expense | 0.1 | — | (0.1 | ) | 0.1 | 0.1 | ||||||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Interest and Other, net | $ | 0.2 | $ | 0.1 | $ | 0.1 | $ | 0.1 | $ | — | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
Interest income and other income consisted primarily of interest income generated from our cash, cash equivalents and investments. Interest income and other income was $0.1 million in both 2013 and 2012 as the effect upon interest income of higher average cash balances was offset by lower average interest rates. Interest income and other income decreased in 2012 compared to 2011 primarily due to lower average effective interest rates and lower average invested balances.
Interest expense and other expense in 2013 consisted solely of net gains realized upon disposal of equipment. Interest expense and other expense in 2011 primarily consisted of interest expense on borrowings under our equipment financing lines. We repaid the remaining outstanding equipment financing debt in March 2012.
Liquidity and Capital Resources
From August 5, 1997, our date of inception, through December 31, 2013, we funded our operations through the sale of equity securities, non-equity payments from collaborators, equipment financings, government grants and interest income.
64
Table of Contents
We have received net proceeds from the sale of equity securities of $449.2 million from August 5, 1997, the date of our inception, through December 31, 2013, excluding sales of equity to GlaxoSmithKline (“GSK”) and Amgen. Included in these proceeds are $94.0 million received upon closing of the initial public offering of our common stock in May 2004. In connection with execution of our collaboration and license agreement in 2001, GSK made a $14.0 million equity investment in Cytokinetics. GSK made additional equity investments in Cytokinetics in 2003 and 2004 of $3.0 million and $7.0 million, respectively. In January 2007, in connection with the execution of our collaboration agreement with Amgen, we received net proceeds of $32.9 million from a stock purchase agreement with Amgen. In June 2013, in conjunction with the Amgen Agreement Amendment, we sold 1,404,100 shares of common stock to Amgen for an aggregate purchase price of $10.0 million, which we received in June 2013.
On a cumulative basis through December 31, 2013, we have received $126.1 million in non-equity payments from Amgen and $54.5 million in non-equity payments from GSK.
Amgen Agreement Amendment
In June 2013, we entered into the Amgen Agreement Amendment, which expanded our strategic alliance to include Japan (see Note 7, “Related Party Transactions” in the Notes to Financial Statements). Under the terms of the Amgen Agreement Amendment, we received an upfront, non-refundable license payment of $15 million in June 2013. In conjunction with the Amgen Agreement Amendment, we also entered into a common stock purchase agreement pursuant to which we sold 1,404,100 shares common stock to Amgen at a price per share of $7.12. The aggregate purchase price of $10.0 million was received in June 2013. We determined the fair value of the stock issued to Amgen to be $7.5 million. The excess of cash received over fair value of $2.5 million was deferred and will be recognized as revenue as services are performed over approximately 12 months.
Astellas Agreement
In June 2013, we entered into the Astellas Agreement (see Note 8 to Financial Statements). In July 2013, we received an upfront, non-refundable license payment of $16 million in connection with the execution of the Astellas Agreement. We are eligible to potentially receive over $24 million in reimbursement of sponsored research and development activities during the initial two years of the collaboration. In addition, we may also receive payments for the achievement of pre-specified milestones relating to the joint research program.
April 2011 Private Offering
In April 2011, we entered into a securities purchase agreement with Deerfield Private Design Fund II, L.P., Deerfield Private Design International II, L.P., Deerfield Special Situations Fund, L.P., and Deerfield Special Situations Fund International Limited (collectively, “Deerfield”). In April 2011, pursuant to the agreement, we issued to Deerfield (i) 833,333 shares of common stock for a purchase price of $1.50 per share, (ii) 8,070 shares of Series A convertible preferred stock (the “Series A Preferred Stock”) for a purchase price of $1,500.00 per share, and (iii) warrants to purchase 1,114,168 shares of our common stock at an initial exercise price of $9.90 per share, for aggregate gross proceeds of approximately $20.1 million. After issuance costs of approximately $0.2 million, the net proceeds were approximately $19.9 million. The offering was made pursuant to a shelf registration statement that we filed with the SEC on November 10, 2008, which became effective on November 19, 2008 (File No. 333-155259).
On September 26, 2012, 8,070 shares of Series A Preferred Stock were converted into 1,345,000 shares of our common stock. The conversion was in accordance with the terms of the agreement with Deerfield under which the Series A Preferred Stock was issued in 2011.
65
Table of Contents
MLV
In June 2011, we entered into an At-The-Market Issuance Sales Agreement (the “MLV Agreement”) with McNicoll, Lewis & Vlak LLC (“MLV”), pursuant to which we sold, through January 2014, an aggregate of 2,397,278 shares of common stock through MLV for net proceeds of approximately $15.1 million. No shares remain available to us for sale through the MLV Agreement. (See Note 17, “Subsequent Events” in the Notes to Financial Statements.)
June 2012 Public Offerings
On June 20, 2012, we entered into underwriting agreements for two separate, concurrent offerings of our securities (the “June 2012 Public Offerings”). On June 25, 2012, pursuant to the underwriting agreements, in aggregate we issued to various investors (i) 9,320,176 shares of common stock for a purchase price of $4.56 per share, (ii) 23,026 shares of Series B convertible preferred stock (the “Series B Preferred Stock”) for a purchase price of $760.00 per share, and (iii) warrants to purchase 7,894,704 shares of our common stock at an exercise price of $5.28 per share, for aggregate gross proceeds of approximately $60.0 million. After issuance costs of approximately $4.0 million, the net proceeds from the June 2012 Public Offerings were approximately $56.0 million.
The warrants issued in the June 2012 Public Offerings became exercisable upon issuance and will remain exercisable for five years until June 25, 2017. The warrant holders are prohibited from exercising the warrants and obtaining shares of common stock if, as a result of such exercise, the holder and its affiliates would own more than 9.98% of the total number of shares of our common stock then issued and outstanding. We valued the warrants as of the date of issuance at $16.2 million using the Black-Scholes option pricing model and the following assumptions: a contractual term of five years, a risk-free interest rate of 0.73%, volatility of 76%, and the fair value of our common stock on the issuance date of $3.78. In February 2013, warrants to purchase 1,000 shares of our common stock at an exercise price of $5.28 per share were exercised in accordance with the June 2012 Public Offerings underwriting agreements. In April 2013, we issued 358,460 shares of common stock related to cashless exercise of warrants. As of December 31, 2013, warrants to purchase 6,577,928 shares of our common stock were outstanding and exercisable.
In the first quarter of 2013, 4,000 shares of Series B convertible preferred stock were converted into 666,667 shares of our common stock. In the second quarter of 2013, 15,026 shares of Series B convertible preferred stock were converted into 2,504,333 shares of our common stock. In July, 2013, 4,000 shares of Series B convertible preferred stock, which represented all remaining shares of Series B convertible preferred stock, were converted into 666,681 shares of our common stock. The conversions were in accordance with the June 2012 Public Offerings underwriting agreements.
The June 2012 Public Offerings were made pursuant to a shelf registration statement that we filed with the SEC on November 25, 2011, which became effective on December 8, 2011 (File No. 333-178189) and a supplemental shelf registration statement on Form S-3MEF that we filed with the SEC on June 20, 2012, which became effective on June 20, 2012 (File No. 333-182226). The closing of the June 2012 Public Offerings took place on June 25, 2012.
The fair value of the common stock into which the Series B Preferred Stock was convertible exceeded the allocated purchase price of the Series B Preferred Stock by $1.3 million on the date of issuance, resulting in a beneficial conversion feature. We recognized the beneficial conversion feature as a one-time, non-cash, deemed dividend to the holders of Series B Preferred Stock on the date of issuance, which is the date the stock first became convertible.
February 2014 Public Offering
On February 25, 2014, the Company closed an underwritten public offering for the issuance and sale of 5,031,250 shares of its common stock. The gross public offering proceeds were approximately $40.3 million. The
66
Table of Contents
net proceeds from the sale of the shares were approximately $37.4 million, after deducting the underwriting discount and estimated offering expenses. (See Note 17, “Subsequent Events” in the Notes to Financial Statements.)
Sources and Uses of Cash
Our cash, cash equivalents and investments totaled $80.2 million at December 31, 2013, up from $74.0 million at December 31, 2012. The increase of $6.2 million in 2013 was primarily due to upfront license fees received of $31.0 million and $14.5 million in net proceeds from equity issuances, partially offset by cash used to fund operations.
Net cash used in operating activities in 2013 was $7.7 million and resulted principally from the net loss of $33.7 million less $16.2 million of deferred revenue, $3.6 million of non-cash stock compensation expense and increased payables and accruals of $5.1 million. Net cash used in operating activities in 2012 was $33.4 million and primarily resulted from the net loss of $40.4 million. Net cash used in operating activities in 2011 was $45.6 million and primarily resulted from our net loss of $47.9 million.
Net cash used in investing activities was $1.5 million in 2013 and primarily consisted of cash used to purchase investments, net of proceeds from the maturity of investments, of $1.0 million and equipment purchases of $0.5 million. Net cash used in investing activities was $28.8 million in 2012 and primarily consisted of cash used to purchase investments, net of proceeds from the maturity of investments, of $28.9 million. Net cash provided by investing activities in 2011 was $25.3 million and primarily consisted of proceeds from maturities of investments, net of cash used to purchase investments, of $25.1 million.
Net cash provided by financing activities was $14.5 million in 2013 and primarily consisted of the purchase of common stock by Amgen totaling $7.5 million (See Note 7, “Related Party Transactions” in the Notes to Financial Statements) and common stock sold pursuant to the MLV agreement totaling $7.5 million. Repurchases of common stock in 2013 to satisfy employee withholding obligations totaled $0.6 million. Net cash provided by financing activities was $58.3 million in 2012 and primarily consisted of net proceeds of $56.0 million from the sale of 55,921,054 shares of common stock and 23,026 shares of Series B Preferred Stock in the June 2012 Public Offerings and net proceeds of $2.8 million from our sale of 2,596,341 shares of common stock through MLV. We repaid the remaining balance of our equipment financing line debt in March 2012 and no further funds are available to us under this line. Net cash provided by financing activities was $21.6 million in 2011 and primarily consisted of net proceeds of $19.9 million from our financing with Deerfield and $2.4 million from sales of our common stock through MLV.
Shelf Registration Statements. In November 2011, we filed a shelf registration statement with the SEC, which was declared effective in December 2011 (the “December 2011 Shelf”). The December 2011 Shelf allowed us to issue shares of our common stock from time to time for an aggregate offering price of up to $100.0 million. In June 2012, we filed a supplemental shelf registration statement with the SEC, which was declared effective in June 2012 (the “Supplemental Shelf”). The Supplemental Shelf allows us to issue additional securities from time to time for an aggregate offering price of up to $20.0 million, and for a total aggregate offering price under the December 2011 Shelf and the Supplemental Shelf of up to $120.0 million. As of February 28, 2014, $18.3 million remains available to us under this shelf registration statement. The specific terms of offerings, if any, under the shelf registration statement would be established at the time of such offerings.
In November 2013 we filed a shelf registration statement with the SEC, which was declared effective in December 2013 (the “December 2013 Shelf”). The December 2013 Shelf allows us to issue common stock and preferred stock, and/or warrants to purchase any of such securities with a total value of up to $150.0 million. As of February 28, 2014, $109.8 million remains available to us under the December 2013 Shelf. The specific terms of offerings, if any, under the December 2013 Shelf would be established at the time of such offerings.
67
Table of Contents
As of December 31, 2013, future minimum payments under our lease obligations were as follows (in thousands):
| Within One Year | One to Three Years | Three to Five Years | After Five Years | Total | ||||||||||||||||
Operating lease(1) | $ | 3,357 | $ | 7,057 | $ | 5,619 | $ | — | $ | 16,033 | ||||||||||
|
|
|
|
|
|
|
|
|
| |||||||||||
| (1) | Our long-term commitment under operating lease relates to payments under our facility lease in South San Francisco, California, which expires in 2018. |
In future periods, we expect to incur substantial costs as we continue to expand our research programs and related research and development activities. We expect to continue development of our drug candidate tirasemtiv for the potential treatment of neuromuscular diseases and medical conditions associated with muscle weakness or wasting. As part of our strategic alliance with Astellas, we expect to continue development of our drug candidate CK-2127107 for the potential treatment of non-neuromuscular diseases and medical conditions associated with muscle weakness or wasting. As part of our strategic alliance with Amgen, we expect to continue development of our drug candidate omecamtiv mecarbil for the potential treatment of heart failure.
Our future capital uses and requirements depend on numerous factors. These factors include, but are not limited to, the following:
| • | the initiation, progress, timing, scope and completion of preclinical research, non-clinical development and clinical trials for our drug candidates and other compounds; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | delays that may be caused by requirements of regulatory agencies; |
| • | Amgen’s decisions with regard to funding of development and commercialization of omecamtiv mecarbil or other compounds for the potential treatment of heart failure under our collaboration; |
| • | Astellas’ decisions with regard to funding of development and commercialization of CK-2127107 and other skeletal muscle activators; |
| • | our level of funding for the development of current or future drug candidates; |
| • | the number of drug candidates we pursue; |
| • | the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; |
| • | our ability to establish and maintain selected strategic alliances required for the development of drug candidates and commercialization of our potential drugs; |
| • | our plans or ability to expand our drug development capabilities, including our capabilities to conduct clinical trials for our drug candidates; |
| • | our plans or ability to engage third party manufacturers for our drug candidates and potential drugs; |
| • | our plans or ability to build or access sales and marketing capabilities and to achieve market acceptance for potential drugs; |
| • | the expansion and advancement of our research programs; |
| • | the hiring of additional employees and consultants; |
| • | the expansion of our facilities; |
| • | the acquisition of technologies, products and other business opportunities that require financial commitments; and |
| • | our revenues, if any, from successful development of our drug candidates and commercialization of potential drugs. |
68
Table of Contents
We have incurred an accumulated deficit of $482.6 million since inception and there can be no assurance that we will attain profitability. We are subject to risks common to clinical stage companies including, but not limited to, development of new drug candidates, dependence on key personnel, and the ability to obtain additional capital as needed to fund our future plans. Our liquidity will be impaired if sufficient additional capital is not available on terms acceptable to us, if at all. To date, we have funded our operations primarily through sales of our common stock and convertible preferred stock, contract payments under our collaboration agreements, debt financing arrangements, government grants and interest income. Until we achieve profitable operations, we intend to continue to fund operations through payments from strategic collaborations, additional sales of equity securities, government grants and debt financings. We have never generated revenues from commercial sales of our drugs and may not have drugs to market for at least several years, if ever. Our success is dependent on our ability to obtain additional capital by entering into new strategic collaborations and/or through equity or debt financings, and ultimately on our and our collaborators’ ability to successfully develop and market one or more of our drug candidates. We cannot be certain that sufficient funds will be available from such collaborators or financings when needed or on satisfactory terms. Additionally, there can be no assurance that any of drugs based on our drug candidates will be accepted in the marketplace or that any future products can be developed or manufactured at an acceptable cost. These factors could have a material adverse effect on our future financial results, financial position and cash flows.
Based on the current status of our development plans, we believe that our existing cash and cash equivalents, investments and interest earned on investments will be sufficient to meet our projected operating requirements for at least the next 12 months. If, at any time, our prospects for internally financing our research and development programs decline, we may decide to reduce research and development expenses by delaying, discontinuing or reducing our funding of development of one or more of our drug candidates or potential drug candidates or of other research and development programs. Alternatively, we might raise funds through strategic relationships, public or private financings or other arrangements. There can be no assurance that funding, if needed, will be available on attractive terms, or at all, or in accordance with our planned timelines. Furthermore, financing obtained through future strategic relationships may require us to forego certain commercialization and other rights to our drug candidates. Similarly, any additional equity financing may be dilutive to stockholders and debt financing, if available, may involve restrictive covenants. Our failure to raise capital as and when needed could have a negative impact on our financial condition and our ability to pursue our business strategy.
Off-balance Sheet Arrangements
We are not party to any off-balance sheet arrangements that have, or are reasonably likely to have, a material current or future effect on our financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our financial statements included in this Form 10-K, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
69
Table of Contents
Investments
Available-for-sale investments. Our investments consist of municipal and government agency bonds, commercial paper, U.S. Treasury securities, and money market funds. We designate all investments as available-for-sale. Therefore, they are reported at fair value, with unrealized gains and losses recorded in accumulated other comprehensive income. See Note 3, “Cash Equivalents and Investments” in the Notes to Financial Statements for further detailed discussion. Investments with original maturities greater than three months and remaining maturities less than one year are classified as short-term investments. Investments with remaining maturities greater than one year are classified as long-term investments.
Other-than-temporary impairment. All of our available-for-sale investments are subject to a periodic impairment review. We recognize an impairment charge when a decline in the fair value of our investments below the cost basis is judged to be other-than-temporary. Factors considered by management in assessing whether an other-than-temporary impairment has occurred include: the nature of the investment; whether the decline in fair value is attributable to specific adverse conditions affecting the investment; the financial condition of the investee; the severity and the duration of the impairment; and whether we have the intent and ability to hold the investment to maturity. When we determine that an other-than-temporary impairment has occurred, the investment is written down to its market value at the end of the period in which we determine that an other-than-temporary decline occurred. The amortized cost of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income. Realized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income or expense. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in Interest and Other, net.
Revenue Recognition
We recognize revenue when the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the fee is fixed or determinable; and collectability is reasonably assured. Determination of whether persuasive evidence of an arrangement exists and whether delivery has occurred or services have been rendered are based on management’s judgments regarding the fixed nature of the fee charged for research performed and milestones met, and the collectability of those fees. Should changes in conditions cause management to determine these criteria are not met for certain future transactions, revenue recognized for any reporting period could be adversely affected.
Revenue under our license and collaboration arrangements is recognized based on the performance requirements of the contract. Research and development revenues, which are earned under agreements with third parties for agreed research and development activities, may include non-refundable license fees, research and development funding, cost reimbursements and contingent milestones and royalties. Our collaborations prior to January 1, 2011 with multiple elements were evaluated and divided into separate units of accounting if certain criteria were met, including whether the delivered element had stand-alone value to the customer and whether there were vendor-specific objective and reliable evidence (“VSOE”) of the fair value of the undelivered items. The consideration we received was allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria were applied to each of the separate units. The consideration we received was combined and recognized as a single unit of accounting when criteria for separation were not met.
On January 1, 2011, ASC Topic 605-25, Revenue Recognition — Multiple-Element Arrangements (“ASC 605-25”) on the recognition of revenues for agreements with multiple deliverables became effective and applies to any agreements we entered into on or after January 1, 2011. Under this updated guidance, revenue will be allocated to each element using a selling price hierarchy, where the selling price for an element is based on VSOE if available; third-party evidence (“TPE”), if available and VSOE is not available; or the best estimate of selling price, if neither VSOE nor TPE is available.
70
Table of Contents
Upfront, non-refundable licensing payments are assessed to determine whether or not the licensee is able to obtain stand-alone value from the license. Where stand-alone value is indicated, revenue from the license is recognized upon delivery once all terms of the arrangement are finalized. Where this is not the case, we do not consider the license deliverable to be a separate unit of accounting, and we defer revenue with revenue recognition for the license fee being recognized in conjunction with the other deliverables that constitute the combined unit of accounting over the estimated performance period for noncontingent deliverables, following the pattern of performance.
Also on January 1, 2011, ASC Topic 605-28,Revenue Recognition — Milestone Method (“ASC 605-28”) became effective and established the milestone method as an acceptable method of revenue recognition for certain contingent event-based payments under research and development arrangements. Under the milestone method, a payment that is contingent upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. A milestone is an event (i) that can be achieved based in whole or in part on either our performance or on the occurrence of a specific outcome resulting from our performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved, and (iii) that would result in additional payments being due to us. The determination that a milestone is substantive is based on management’s judgment and is made at the inception of the arrangement. Milestones are considered substantive when the consideration earned from the achievement of the milestone is (i) commensurate with either our performance to achieve the milestone or the enhancement of value of the item delivered as a result of a specific outcome resulting from our performance to achieve the milestone, (ii) relates solely to past performance and (iii) is reasonable relative to all deliverables and payment terms in the arrangement.
Other contingent event-based payments received for which payment is either contingent solely upon the passage of time or the results of a collaborative partner’s performance are not considered milestones under ASC 605-28. In accordance with ASC 605-25, such payments will be recognized as revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; price is fixed or determinable; and collectability is reasonably assured.
For our collaborations entered into prior to January 1, 2011, we recognized and will continue to recognize milestone payments as revenue upon achievement of the milestone, provided the milestone payment was non-refundable, substantive effort and risk was involved in achieving the milestone and the amount of the milestone was reasonable in relation to the effort expended or risk associated with the achievement of the milestone. If these conditions were not met, we deferred the milestone payment and recognized it as revenue over the estimated period of performance under the contract as we completed our performance obligations. We have concluded that all of the future contingent milestone payments pursuant to our research and development arrangements entered into prior to January 1, 2011 are not considered substantive as they are the results of a collaborative partner’s performance. Therefore, they are not considered milestones under ASC 605-28.
With respect to milestones related to our collaboration and license agreement entered into with Astellas in 2013, we believe the milestones related to research and early development are substantive as there is uncertainty that the milestones will be met, the milestones can only be achieved with our past and current performance and the achievement of the milestone will result in additional payments to us. Therefore, they are considered milestones under ASC 605-28. We believe that the milestones related to later development and commercialization are not substantive as they are primarily the result of the collaborative partner’s performance and therefore will be recognized as we complete our performance obligations under the agreement, if any.
Research and development revenues and cost reimbursements are based upon negotiated rates for our FTEs and actual out-of-pocket costs. FTE rates are negotiated rates that are based upon our costs, and which we believe approximate fair value. Any amounts received in advance of performance are recorded as deferred revenue. None of the revenues recognized to date are refundable if the relevant research effort is not successful. In revenue arrangements in which both parties make payments to each other, we evaluate the payments to determine whether
71
Table of Contents
payments made by us will be recognized as a reduction of revenue or as expense. Revenue we recognize may be reduced by payments made to the other party under the arrangement unless we receive a separate and identifiable benefit in exchange for the payments and we can reasonably estimate the fair value of the benefit received.
Funds received from third parties under grant arrangements are recorded as revenue if we are deemed to be the principal participant in the grant arrangement as the activities under the grant are part of our development programs. If we are not the principal participant, the grant funds are recorded as a reduction to research and development expense. Grant funds received are not refundable and are recognized when the related qualified research and development costs are incurred and when there is reasonable assurance that the funds will be received. Funds received in advance are recorded as deferred revenue.
Preclinical Study and Clinical Trial Accruals
A substantial portion of our preclinical studies and all of our clinical trials have been performed utilizing third-party contract research organizations (“CROs”) and other vendors. For preclinical studies, the significant factors used in estimating accruals include the percentage of work completed to date and contract milestones achieved. For clinical trial expenses, the significant factors used in estimating accruals include the number of patients enrolled, duration of enrollment and percentage of work completed to date. We monitor patient enrollment levels and related activities to the extent possible through internal reviews, correspondence and status meetings with CROs and review of contractual terms. Our estimates are dependent on the timeliness and accuracy of data provided by our CROs and other vendors. If we have incomplete or inaccurate data, we may under- or overestimate activity levels associated with various studies or clinical trials at a given point in time. In this event, we could record adjustments to research and development expenses in future periods when the actual activity levels become known. No material adjustments to preclinical study and clinical trial expenses have been recognized to date.
Stock-Based Compensation
We apply the accounting guidance for stock compensation, which establishes the accounting for share-based payment awards made to employees and directors, including employee stock options and employee stock purchases. Under this guidance, stock-based compensation cost is measured at the grant date based on the calculated fair value of the award, and is recognized as an expense on a straight-line basis over the employee’s requisite service period, generally the vesting period of the award.
Under the guidance for stock compensation for non-employees, we measure the fair value of the award each period until the award is fully vested.
As required under the accounting rules, we review our valuation assumptions at each grant date and, as a result, from time to time we will likely change the valuation assumptions we use to value stock based awards granted in future periods. The assumptions used in calculating the fair value of share-based payment awards represent management’s best estimates at the time, but these estimates involve inherent uncertainties and the application of management judgment. As a result, if conditions change and we use different assumptions, our stock-based compensation expense could be materially different in the future. In addition, we are required to estimate the expected forfeiture rate and recognize expense only for those shares expected to vest. If our actual forfeiture rate is materially different from our estimate, the stock-based compensation expense could be significantly different from what we have recorded in the current period.
Income Taxes
We account for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable
72
Table of Contents
income. Valuation allowances are established when necessary to reduce the deferred tax assets to the amounts expected to be realized. We did not record an income tax provision in the years ended December 31, 2012 and 2011 because we had a net taxable loss in these periods.
Based upon the weight of available evidence, which includes our historical operating performance, reported cumulative net losses since inception and difficulty in accurately forecasting our future results, we maintained a full valuation allowance on the net deferred tax assets as of December 31, 2013, 2012 and 2011. The valuation allowance was determined pursuant to the accounting guidance for income taxes, which requires an assessment of both positive and negative evidence when determining whether it is more likely than not that deferred tax assets are recoverable. We intend to maintain a full valuation allowance on the U.S. deferred tax assets until sufficient positive evidence exists to support reversal of the valuation allowance. The valuation allowance increased by $13.7 million in 2013, $21.1 million in 2012 and $18.5 million in 2011.
We also follow the accounting guidance that defines the threshold for recognizing the benefits of tax return positions in the financial statements as “more-likely-than-not” to be sustained by the taxing authorities based solely on the technical merits of the position. If the recognition threshold is met, the tax benefit is measured and recognized as the largest amount of tax benefit that, in our judgment, is greater than 50% likely to be realized. Historically, we have filed income tax returns with the IRS and the state of California. For jurisdictions in which tax filings are made, we are subject to income tax examination for all fiscal years since inception. The IRS’s Large Business and International Division concluded its audit of the 2009 tax year with no material adjustments. However, in general, the statute of limitations for tax liabilities for these years remains open for the purpose of adjusting the amounts of the losses and credits carried forward from those years.
Interest accrued related to unrecognized tax benefits and penalties were zero for 2013, 2012 and 2011. We account for interest related to unrecognized tax benefits and penalties by classifying both as income tax expense in the financial statements in accordance with the accounting guidance for uncertainty in income taxes. We do not expect our unrecognized tax benefits to change materially over the next twelve months.
In general, under Section 382 a corporation that undergoes an ‘ownership change’ is subject to limitations on its ability to utilize its pre-change NOLs and tax credits to offset future taxable income. We have performed a Section 382 analysis and do not believe that we have experienced an ownership change since 2006. A portion of our existing NOLs and tax credits are subject to limitations arising from previous ownership changes. Future changes in our stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 and result in additional limitations.
Recent Accounting Pronouncements
See “Recent Accounting Pronouncements” in Note 1, “Organization and Significant Accounting Policies” in the Notes to Financial Statements for a discussion of recently adopted accounting pronouncements and accounting pronouncements not yet adopted, and their expected impact on our financial position and results of operations.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Interest Rate and Market Risk
Our exposure to market risk is limited to interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because the majority of our investments are in short-term debt securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. We are exposed to the impact of interest rate changes and changes in the market values of our investments. Our interest income is sensitive to changes in the general level of U.S. interest rates. Our exposure to market rate risk for changes in interest rates relates primarily to our investment portfolio. We have not used derivative financial instruments in our investment portfolio. We invest
73
Table of Contents
the majority of our excess cash in U.S. Treasuries and, by policy, limit the amount of credit exposure in any one issuer and investment class, with the exception of obligations of the U.S. Treasury and federal agencies, for which there are no such limits. We protect and preserve our invested funds by attempting to limit default, market and reinvestment risk. Investments in both fixed-rate and floating-rate interest-earning instruments carry a degree of interest rate risk. Fixed-rate securities may have their fair market value adversely impacted due to a rise in interest rates, while floating-rate securities may produce less income than expected if interest rates fall. Due in part to these factors, our future investment income may fall short of expectations due to changes in interest rates. To minimize risk, we maintain our portfolio of cash and cash equivalents and short- and long-term investments in a variety of interest-bearing instruments, including U.S. government and agency securities, high grade municipal and U.S. bonds and money market funds. Our investment portfolio of short- and long-term investments is subject to interest rate risk, and will fall in value if market interest rates increase.
Our cash and cash equivalents are invested in highly liquid securities with maturities of three months or less at the time of purchase. Consequently, we do not consider our cash and cash equivalents to be subject to significant interest rate risk and have therefore excluded them from the table below. We do not have any foreign currency or derivative financial instruments.
The table below presents the principal amounts and weighted average interest rates by year of maturity for our investment portfolio (dollars in thousands):
| 2014 | 2015 | Total | Fair Value at December 31, 2013 | |||||||||||||
Assets: | ||||||||||||||||
Investments | $ | 57,570 | $ | 2,502 | $ | 60,072 | $ | 60,072 | ||||||||
Average interest rate | 0.12 | % | 0.20 | % | 0.13 | % | ||||||||||
74
Table of Contents
Item 8. Financial Statements and Supplementary Data
CYTOKINETICS, INCORPORATED
INDEX TO FINANCIAL STATEMENTS
| Page | ||||
| 76 | ||||
| 77 | ||||
| 78 | ||||
| 79 | ||||
| 81 | ||||
| 82 | ||||
75
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Cytokinetics, Incorporated:
In our opinion, the accompanying balance sheets and the related statements of comprehensive loss, of stockholders’ equity (deficit) and of cash flows present fairly, in all material respects, the financial position of Cytokinetics, Incorporated at December 31, 2013 and December 31, 2012, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2013 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2013, based on criteria established inInternal Control — Integrated Framework 1992 issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PRICEWATERHOUSECOOPERS LLP
San Jose, CA
March 7, 2014
76
Table of Contents
BALANCE SHEETS
| December 31, | ||||||||
| 2013 | 2012 | |||||||
| (In thousands, except | ||||||||
| share and per share data) | ||||||||
| ASSETS | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 20,158 | $ | 14,907 | ||||
Short-term investments | 57,570 | 59,093 | ||||||
Related party accounts receivable | 5 | 4 | ||||||
Prepaid and other current assets | 1,605 | 2,423 | ||||||
|
|
|
| |||||
Total current assets | 79,338 | 76,427 | ||||||
Property and equipment, net | 1,221 | 997 | ||||||
Long-term investments | 2,502 | — | ||||||
Other assets | 127 | 127 | ||||||
|
|
|
| |||||
Total assets | $ | 83,188 | $ | 77,551 | ||||
|
|
|
| |||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
Current liabilities: | ||||||||
Accounts payable | $ | 3,709 | $ | 2,002 | ||||
Accrued liabilities | 8,272 | 4,877 | ||||||
Deferred revenue, current | 14,701 | — | ||||||
Related party payables and accrued liabilities | — | 150 | ||||||
Short-term portion of deferred rent | 22 | 76 | ||||||
|
|
|
| |||||
Total current liabilities | 26,704 | 7,105 | ||||||
Deferred revenue, non-current | 1,500 | — | ||||||
Long-term portion of deferred rent | 542 | 361 | ||||||
|
|
|
| |||||
Total liabilities | 28,746 | 7,466 | ||||||
|
|
|
| |||||
Commitments and contingencies (Note 11) | ||||||||
Stockholders’ equity: | ||||||||
Preferred stock, $0.001 par value: | — | — | ||||||
Authorized: 10,000,000 shares ; Issued and outstanding: | ||||||||
Series A Convertible Preferred Stock — zero shares at December 31, 2013 and December 31, 2012 | ||||||||
Series B Convertible Preferred Stock — zero shares at December 31, 2013 and 23,026 shares at December 31, 2012 | ||||||||
Common stock, $0.001 par value: | ||||||||
Authorized: 81,500,000 shares; | ||||||||
Issued and outstanding: 30,681,624 shares at December 31, 2013 and 23,742,911 shares at December 31, 2012 | 31 | 24 | ||||||
Additional paid-in capital | 537,001 | 518,923 | ||||||
Accumulated other comprehensive income | 7 | 18 | ||||||
Accumulated deficit | (482,597 | ) | (448,880 | ) | ||||
|
|
|
| |||||
Total stockholders’ equity | 54,442 | 70,085 | ||||||
|
|
|
| |||||
Total liabilities and stockholders’ equity | $ | 83,188 | $ | 77,551 | ||||
|
|
|
| |||||
The accompanying notes are an integral part of these financial statements.
77
Table of Contents
STATEMENTS OF COMPREHENSIVE LOSS
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (In thousands, except per share data) | ||||||||||||
Revenues: | ||||||||||||
Research and development revenues from related parties | $ | 2,019 | $ | 4,177 | $ | 2,054 | ||||||
Research and development, grant and other revenues | 7,547 | 3,382 | 1,946 | |||||||||
License revenues from related parties | 17,230 | — | — | |||||||||
License revenues | 3,852 | — | — | |||||||||
|
|
|
|
|
| |||||||
Total revenues | 30,648 | 7,559 | 4,000 | |||||||||
|
|
|
|
|
| |||||||
Operating expenses: | ||||||||||||
Research and development | 49,450 | 35,643 | 37,182 | |||||||||
General and administrative | 15,092 | 12,429 | 13,590 | |||||||||
Restructuring charges (reversals) | — | (56 | ) | 1,192 | ||||||||
|
|
|
|
|
| |||||||
Total operating expenses | 64,542 | 48,016 | 51,964 | |||||||||
|
|
|
|
|
| |||||||
Operating loss | (33,894 | ) | (40,457 | ) | (47,964 | ) | ||||||
Interest and other, net | 177 | 87 | 104 | |||||||||
|
|
|
|
|
| |||||||
Loss before income taxes | (33,717 | ) | (40,370 | ) | (47,860 | ) | ||||||
Income tax benefit | — | — | — | |||||||||
|
|
|
|
|
| |||||||
Net loss | (33,717 | ) | (40,370 | ) | (47,860 | ) | ||||||
Deemed dividend related to beneficial conversion feature of convertible preferred stock | — | (1,307 | ) | (2,857 | ) | |||||||
|
|
|
|
|
| |||||||
Net loss allocable to common stockholders | $ | (33,717 | ) | $ | (41,677 | ) | $ | (50,717 | ) | |||
|
|
|
|
|
| |||||||
Net loss per share allocable to common stockholders — basic and diluted | $ | (1.24 | ) | $ | (2.30 | ) | $ | (4.30 | ) | |||
|
|
|
|
|
| |||||||
Weighted-average number of shares used in computing net loss per share allocable to common stockholders — basic and diluted | 27,275 | 18,107 | 11,800 | |||||||||
|
|
|
|
|
| |||||||
Comprehensive loss | $ | (33,728 | ) | $ | (40,355 | ) | $ | (47,853 | ) | |||
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these financial statements.
78
Table of Contents
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
Common Stock |
Preferred Stock | Additional Paid-In Capital | Deferred Stock-Based Compensation | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||||||||||||||||||
Balance, December 31, 2010 | 11,151,267 | $ | 11 | — | $ | — | $ | 431,159 | $ | — | $ | (4 | ) | $ | (360,650 | ) | $ | 70,516 | ||||||||||||||||||
Issuance of common stock upon exercise of stock options for cash at $6.00-$7.20 per share | 2,667 | — | — | — | 17 | — | — | — | 17 | |||||||||||||||||||||||||||
Issuance of common stock pursuant to ESPP at a weighted price of $6.66 per share | 18,822 | — | — | — | 125 | — | — | — | 125 | |||||||||||||||||||||||||||
Issuance of common stock to Deerfield at $9.00 per share, net of issuance costs of $53 | 883,333 | 1 | — | — | 6,126 | — | — | — | 6,127 | |||||||||||||||||||||||||||
Issuance of Series A convertible preferred stock to Deerfield at $1,500 per share, net of issuance costs of $81 | — | — | 8,070 | — | 9,329 | — | — | — | 9,329 | |||||||||||||||||||||||||||
Issuance of warrants to Deerfield, net of issuance costs of $38 | — | — | — | — | 4,427 | — | — | — | 4,427 | |||||||||||||||||||||||||||
Issuance of common stock to MLV at $6.00-$6.12 per share, net of commission and issuance costs of $160 | 429,868 | 1 | — | 2,420 | — | — | — | 2,421 | ||||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | 3,069 | — | — | — | 3,069 | |||||||||||||||||||||||||||
Other comprehensive income | — | — | — | — | — | — | 7 | — | 7 | |||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (47,860 | ) | (47,860 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
Balance, December 31, 2011 | 12,485,957 | 13 | 8,070 | — | 456,672 | — | 3 | (408,510 | ) | 48,178 | ||||||||||||||||||||||||||
Issuance of common stock pursuant to ESPP at a weighted price of $4.32 per share | 15,010 | — | — | — | 65 | — | — | — | 65 | |||||||||||||||||||||||||||
Issuance of common stock upon exercise of restricted stock units | 144,045 | — | — | — | (401 | ) | — | — | — | (401 | ) | |||||||||||||||||||||||||
Issuance of common stock pursuant to June 2012 public offerings at $4.56 per share, net of issuance costs of $2,139 | 9,320,176 | 9 | — | — | 29,907 | — | — | — | 29,916 | |||||||||||||||||||||||||||
Issuance of Series B convertible preferred stock pursuant to June 2012 public offerings at $760 per share, net of issuance costs of $881 | — | — | 23,026 | — | 12,318 | — | — | — | 12,318 | |||||||||||||||||||||||||||
Issuance of warrants pursuant to June 2012 public offerings, net of issuance costs of $984 | — | — | — | — | 13,761 | — | — | — | 13,761 | |||||||||||||||||||||||||||
Issuance of common stock to MLV at $6.30-$7.20 per share, net of commission and issuance costs of $89 | 432,724 | 1 | — | — | 2,819 | — | — | — | 2,820 | |||||||||||||||||||||||||||
Conversion of Series A convertible preferred stock to common stock at $1,000 per share | 1,345,000 | 1 | (8,070 | ) | — | (1 | ) | — | — | — | — | |||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | 3,783 | — | — | — | 3,783 | |||||||||||||||||||||||||||
Other comprehensive income | — | — | — | — | — | — | 15 | — | 15 | |||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (40,370 | ) | (40,370 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
79
Table of Contents
CYTOKINETICS, INCORPORATED
STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT) — (Continued)
Common Stock |
Preferred Stock | Additional Paid-In Capital | Deferred Stock-Based Compensation | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||||||||||||||||||
Balance, December 31, 2012 | 23,742,912 | 24 | 23,026 | — | $ | 518,923 | — | 18 | (448,880 | ) | 70,085 | |||||||||||||||||||||||||
Issuance of common stock upon exercise of stock options for cash at $4.02-$11.10 per share | 21,397 | — | — | — | 114 | — | — | — | 114 | |||||||||||||||||||||||||||
Issuance of common stock pursuant to ESPP at a weighted price of $3.66 per share | 14,985 | — | — | — | 55 | — | — | — | 55 | |||||||||||||||||||||||||||
Issuance of common stock upon exercise of restricted stock units | 130,534 | — | — | — | (623 | ) | — | — | — | (623 | ) | |||||||||||||||||||||||||
Issuance of common stock to related party for $7.12 per share, net of issuance costs of $21 | 1,404,100 | 2 | — | — | 7,448 | — | — | — | 7,450 | |||||||||||||||||||||||||||
Issuance of common stock upon exercise of warrants | 359,460 | — | — | — | 5 | — | — | — | 5 | |||||||||||||||||||||||||||
Conversion of Series B convertible preferred stock to common stock at $1,000 per share | 3,837,681 | 4 | (23,026 | ) | — | (4 | ) | — | — | — | — | |||||||||||||||||||||||||
Fractional shares settlement pursuant to reverse stock split | (28 | ) | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||
Issuance of common stock to MLV at $6.50-$6.79 per share, net of commission and issuance costs of $232 | 1,170,583 | 1 | — | — | 7,486 | — | — | — | 7,487 | |||||||||||||||||||||||||||
Stock-based compensation | — | — | — | — | 3,597 | — | — | — | 3,597 | |||||||||||||||||||||||||||
Other comprehensive loss | — | — | — | — | — | — | (11 | ) | — | (11 | ) | |||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (33,717 | ) | (33,717 | ) | |||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
Balance, December 31, 2013 | 30,681,624 | $ | 31 | — | $ | — | $ | 537,001 | $ | — | $ | 7 | $ | (482,597 | ) | $ | 54,442 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
80
Table of Contents
STATEMENTS OF CASH FLOWS
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
| (In thousands) | ||||||||||||
Cash flows from operating activities: | ||||||||||||
Net loss | $ | (33,717 | ) | $ | (40,370 | ) | $ | (47,860 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Depreciation and amortization of property and equipment | 433 | 591 | 1,297 | |||||||||
(Gain) loss on disposal of equipment | (79 | ) | (2 | ) | 3 | |||||||
Non-cash restructuring expenses, net of reversals | — | (56 | ) | 194 | ||||||||
Stock-based compensation | 3,597 | 3,783 | 3,069 | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Related party accounts receivable | — | 10 | 32 | |||||||||
Prepaid and other assets | 818 | (320 | ) | (238 | ) | |||||||
Accounts payable | 1,656 | 690 | 162 | |||||||||
Accrued and other liabilities | 3,523 | 2,098 | (2,266 | ) | ||||||||
Related party payables and accrued liabilities | (150 | ) | 138 | 12 | ||||||||
Deferred revenue | 16,201 | — | — | |||||||||
|
|
|
|
|
| |||||||
Net cash used in operating activities | (7,718 | ) | (33,438 | ) | (45,595 | ) | ||||||
|
|
|
|
|
| |||||||
Cash flows from investing activities: | ||||||||||||
Purchases of investments | (79,434 | ) | (92,788 | ) | (48,025 | ) | ||||||
Proceeds from sales and maturities of investments | 78,444 | 63,900 | 73,174 | |||||||||
Purchases of property and equipment | (542 | ) | (125 | ) | (443 | ) | ||||||
Proceeds from sales of property and equipment | 13 | 2 | 3 | |||||||||
Decrease in restricted cash | — | 196 | 592 | |||||||||
|
|
|
|
|
| |||||||
Net cash provided by (used in) investing activities | (1,519 | ) | (28,815 | ) | 25,301 | |||||||
|
|
|
|
|
| |||||||
Cash flows from financing activities: | ||||||||||||
Proceeds from initial public offering, sale of common stock to related party, and public offerings, net of issuance costs | 7,450 | 43,677 | — | |||||||||
Proceeds from draw down of committed equity financing facilities and at-the-market facility, net of commission and issuance costs | — | 2,820 | 2,421 | |||||||||
Proceeds from other issuances of common stock and warrants, net | 7,038 | (336 | ) | 10,696 | ||||||||
Proceeds from issuance of preferred stock, net of issuance costs | — | 12,318 | 9,329 | |||||||||
Repayment of equipment financing lines | — | (152 | ) | (833 | ) | |||||||
|
|
|
|
|
| |||||||
Net cash provided by financing activities | 14,488 | 58,327 | 21,613 | |||||||||
|
|
|
|
|
| |||||||
Net increase (decrease) in cash and cash equivalents | 5,251 | (3,926 | ) | 1,319 | ||||||||
Cash and cash equivalents, beginning of period | 14,907 | 18,833 | 17,514 | |||||||||
|
|
|
|
|
| |||||||
Cash and cash equivalents, end of period | $ | 20,158 | $ | 14,907 | $ | 18,833 | ||||||
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these financial statements.
81
Table of Contents
NOTES TO FINANCIAL STATEMENTS
Note 1 — Organization and Significant Accounting Policies
Organization
Cytokinetics, Incorporated (the “Company”, “we” or “our”) was incorporated under the laws of the state of Delaware on August 5, 1997. The Company is a clinical stage biopharmaceutical company focused on the discovery and development of novel small molecule therapeutics that modulate muscle function for the potential treatment of serious diseases and medical conditions.
The Company was in the development stage at December 31, 2012, as defined in Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 915, “Development Stage Entities.” During the year ended December 31, 2013, the Company exited the development stage with the execution of the Amgen Agreement Amendment and the Astellas Agreement (See Note 7 and Note 8), from which the Company received significant revenues from its principal operations, indicative that the Company was no longer in the development stage.
The Company’s financial statements contemplate the conduct of the Company’s operations in the normal course of business. The Company has incurred an accumulated deficit of $482.6 million since inception and there can be no assurance that the Company will attain profitability. The Company had a net loss of $33.7 million and net cash used in operations of $7.8 million for the year ended December 31, 2013. Cash, cash equivalents and investments increased to $80.2 million at December 31, 2013 from $74.0 million at December 31, 2012. The Company anticipates that it will continue to have operating losses and net cash outflows in future periods.
The Company is subject to risks common to clinical stage biopharmaceutical companies including, but not limited to, development of new drug candidates, dependence on key personnel, and the ability to obtain additional capital as needed to fund its future plans. The Company’s liquidity will be impaired if sufficient additional capital is not available on terms acceptable to the Company. To date, the Company has funded its operations primarily through sales of its common stock and convertible preferred stock, contract payments under its collaboration agreements, debt financing arrangements, government grants and interest income. Until it achieves profitable operations, the Company intends to continue to fund operations through payments from strategic collaborations, additional sales of equity securities, government grants and debt financings. The Company has never generated revenues from commercial sales of its drugs and may not have drugs to market for at least several years, if ever. The Company’s success is dependent on its ability to enter into new strategic collaborations and/or raise additional capital and to successfully develop and market one or more of its drug candidates. As a result, the Company may choose to raise additional capital through equity or debt financings to continue to fund its operations in the future. The Company cannot be certain that sufficient funds will be available from such a financing or through a collaborator when required or on satisfactory terms. Additionally, there can be no assurance that the Company’s drug candidates will be accepted in the marketplace or that any future products can be developed or manufactured at an acceptable cost. These factors could have a material adverse effect on the Company’s future financial results, financial position and cash flows.
Based on the current status of its development plans, the Company believes that its existing cash, cash equivalents and investments at December 31, 2013 will be sufficient to fund its cash requirements for at least the next 12 months. If, at any time, the Company’s prospects for financing its research and development programs decline, the Company may decide to reduce research and development expenses by delaying, discontinuing or reducing its funding of one or more of its research or development programs. Alternatively, the Company might raise funds through strategic collaborations, public or private financings or other arrangements. Such funding, if needed, may not be available on favorable terms, or at all.
The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
82
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Basis of Presentation
The financial statements include all adjustments (consisting only of normal recurring adjustments) that management believes are necessary for the fair presentation of the balances and results for the periods presented.
Concentration of Credit Risk and Other Risks and Uncertainties
Financial instruments that potentially subject the Company to concentrations of risk consist principally of cash and cash equivalents, investments and accounts receivable. The Company’s cash, cash equivalents and investments are invested in deposits with three major financial institutions in the U.S. Deposits in these banks may exceed the amount of insurance provided on such deposits. The Company has not experienced any realized losses on its deposits of cash, cash equivalents or investments.
The economic turmoil in the United States in recent years, the extraordinary volatility in the stock markets and other current negative macroeconomic indicators could negatively impact the Company’s ability to raise the funds necessary to support its business and may materially adversely affect its business, operating results and financial condition.
The Company performs an ongoing credit evaluation of its strategic partners’ financial conditions and generally does not require collateral to secure accounts receivable from its strategic partners. The Company’s exposure to credit risk associated with non-payment will be affected principally by conditions or occurrences within Amgen Inc. (“Amgen”) and Astellas Pharma Inc. (“Astellas”), its strategic partners. Approximately 63%, 55% and 51% of total revenues for the years ended December 31, 2013, 2012 and 2011, respectively, were derived from Amgen. Accounts receivable due from Amgen were zero at December 31, 2013 and 2012. See also Note 7, “Related Party Transactions,” regarding the collaboration agreement with Amgen. Approximately 34% of total revenues for the year ended December 31, 2013 was derived from Astellas. Accounts receivable due from Astellas were zero at December 31, 2013. See also Note 8, “Other Research and Development Revenue Arrangements” regarding collaboration agreement with Astellas.
Drug candidates developed by the Company may require approvals or clearances from the U.S. Food and Drug Administration (“FDA”) or international regulatory agencies prior to commercialized sales. There can be no assurance that the Company’s drug candidates will receive any of the required approvals or clearances. If the Company was to be denied approval or clearance or any such approval or clearance was to be delayed, it would have a material adverse impact on the Company.
The Company’s operations and employees are located in the United States. In the year ended December 31, 2013, 66% of the Company’s revenues were received from entities located in the United States and 34% were received from a Japanese entity. In the years ended 2012 and 2011, all of the Company’s revenues were received from entities located in the United States or from United States affiliates of foreign corporations.
Cash and Cash Equivalents
The Company considers all highly liquid investments with a maturity of three months or less at the time of purchase to be cash equivalents.
83
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Investments
Available-for-sale investments. The Company’s investments consist of U.S. Treasury securities, money market funds, U.S. municipal and government agency bonds, and commercial paper. The Company designates all investments as available-for-sale and therefore reports them at fair value, based on quoted marked prices, with unrealized gains and losses recorded in accumulated other comprehensive loss. The cost of securities sold is based on the specific-identification method. Investments with original maturities greater than three months and remaining maturities of one year or less are classified as short-term investments. Investments with remaining maturities greater than one year are classified as long-term investments.
Other-than-temporary impairment. All of the Company’s available-for-sale investments are subject to a periodic impairment review. The Company recognizes an impairment charge when a decline in the fair value of its investments below the cost basis is judged to be other-than-temporary. Factors considered by management in assessing whether an other-than-temporary impairment has occurred include: the nature of the investment; whether the decline in fair value is attributable to specific adverse conditions affecting the investment; the financial condition of the investee; the severity and the duration of the impairment; and whether the Company has the intent and ability to hold the investment to maturity. When the Company determines that an other-than-temporary impairment has occurred, the investment is written down to its market value at the end of the period in which it is determined that an other-than-temporary decline has occurred. The amortized cost of debt securities in this category is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization is included in interest income. Recognized gains and losses and declines in value judged to be other-than-temporary, if any, on available-for-sale securities are included in other income or expense. The cost of securities sold is based on the specific identification method. Interest and dividends on securities classified as available-for-sale are included in Interest and other, net.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and are depreciated on a straight-line basis over the estimated useful lives of the related assets, which are generally three years for computer equipment and software, five years for laboratory equipment and office equipment, and seven years for furniture and fixtures. Amortization of leasehold improvements is computed using the straight-line method over the shorter of the remaining lease term or the estimated useful life of the related assets, typically ranging from three to seven years. Upon sale or retirement of assets, the costs and related accumulated depreciation and amortization are removed from the balance sheet and the resulting gain or loss is reflected in operations. Maintenance and repairs are charged to operations as incurred.
Impairment of Long-lived Assets
In accordance with the accounting guidance for the impairment or disposal of long-lived assets, the Company reviews long-lived assets, including property and equipment, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. Under the accounting guidance, an impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Impairment, if any, is measured as the amount by which the carrying amount of a long-lived asset exceeds its fair value.
Revenue Recognition
The accounting guidance for revenue recognition requires that certain criteria must be met before revenue can be recognized: persuasive evidence of an arrangement exists; delivery has occurred or services have been
84
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
rendered; the fee is fixed or determinable; and collectability is reasonably assured. Determination of whether persuasive evidence of an arrangement exists and whether delivery has occurred or services have been rendered are based on management’s judgments regarding the fixed nature of the fee charged for research performed and milestones met, and the collectability of those fees. Should changes in conditions cause management to determine these criteria are not met for certain future transactions, revenue recognized for any reporting period could be adversely affected.
Revenue under our license and collaboration arrangements is recognized based on the performance requirements of the contract. Research and development revenues, which are earned under agreements with third parties for agreed research and development activities, may include non-refundable license fees, research and development funding, cost reimbursements and contingent milestones and royalties. The Company’s collaborations prior to January 1, 2011 with multiple elements were evaluated and divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there was vendor-specific objective and reliable evidence (“VSOE”) of the fair value of the undelivered items. The consideration the Company received was allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria were applied to each of the separate units. The consideration the Company received was combined and recognized as a single unit of accounting when criteria for separation were not met. On January 1, 2011, ASC Topic 605-25, Revenue Recognition — Multiple-Element Arrangements (“ASC 605-25”) on the recognition of revenues for agreements with multiple deliverables became effective and applies to any agreements the Company entered into on or after January 1, 2011. Under this updated guidance, revenue is allocated to each element using a selling price hierarchy, where the selling price for an element is based on VSOE if available; third-party evidence (“TPE”), if available and VSOE is not available; or the best estimate of selling price, if neither VSOE nor TPE is available.
Non-refundable license fees are recognized as revenue as the Company performs under the applicable agreement. Where the level of effort is relatively consistent over the performance period, the Company recognizes total fixed or determined revenue on a straight-line basis over the estimated period of expected performance.
ASC Topic 605-28, Revenue Recognition — Milestone Method (“ASC 605-28”), established the milestone method as an acceptable method of revenue recognition for certain contingent event-based payments under research and development arrangements. Under the milestone method, a payment that is contingent upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. A milestone is an event (i) that can be achieved based in whole or in part on either the Company’s performance or on the occurrence of a specific outcome resulting from the Company’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved, and (iii) that would result in additional payments being due to the Company. The determination that a milestone is substantive is judgmental and is made at the inception of the arrangement. Milestones are considered substantive when the consideration earned from the achievement of the milestone is (i) commensurate with either the Company’s performance to achieve the milestone or the enhancement of value of the item delivered as a result of a specific outcome resulting from the Company’s performance to achieve the milestone, (ii) relates solely to past performance and (iii) is reasonable relative to all deliverables and payment terms in the arrangement.
Other contingent event-based payments received for which payment is either contingent solely upon the passage of time or the results of a collaborative partner’s performance are not considered milestones under ASC 605-28. In accordance with ASC 605-25, such payments will be recognized as revenue when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; price is fixed or determinable; and collectability is reasonably assured.
85
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Prior to January 1, 2011, the Company recognized and will continue to recognize milestone payments as revenue upon achievement of the milestone, provided the milestone payment was non-refundable, substantive effort and risk were involved in achieving the milestone and the amount of the milestone was reasonable in relation to the effort expended or risk associated with the achievement of the milestone. If these conditions were not met, the Company deferred the milestone payment and recognized it as revenue over the estimated period of performance under the contract as the Company completed its performance obligations. The Company has concluded that all of the future contingent milestone payments pursuant to its research and development arrangements entered into prior to January 1, 2011 are not considered substantive as they are the results of a collaborative partner’s performance. Therefore, they are not considered milestones under ASC605-28.
With respect to milestones related to the collaboration and license agreement entered into in 2013 with Astellas, Inc., the Company believes the milestones related to research and early development are substantive as there is uncertainty that the milestones will be met, the milestones can only be achieved with our past and current performance and the achievement of the milestone will result in additional payments to us. Therefore, they are considered milestones under ASC 605-28. The Company believes the milestones related to later development and commercialization are not substantive as they are primarily the result of the collaborative partner’s performance and therefore will be recognized as the Company completes its performance obligations under the agreement, if any.
Research and development revenues and cost reimbursements are based upon negotiated rates for the Company’s full-time employee equivalents (“FTE”) and actual out-of-pocket costs. FTE rates are negotiated rates that are based upon the Company’s costs, and which the Company believes approximate fair value. Any amounts received in advance of performance are recorded as deferred revenue. None of the revenues recognized to date are refundable if the relevant research effort is not successful. In revenue arrangements in which both parties make payments to each other, the Company evaluates the payments in accordance with the accounting guidance for arrangements under which consideration is given by a vendor to a customer, including a reseller of the vendor’s products, to determine whether payments made by us will be recognized as a reduction of revenue or as expense. In accordance with this guidance, revenue recognized by the Company may be reduced by payments made to the other party under the arrangement unless the Company receives a separate and identifiable benefit in exchange for the payments and the Company can reasonably estimate the fair value of the benefit received. The application of the accounting guidance for consideration given to a customer has had no material impact to the Company.
Funds received from third parties under grant arrangements are recorded as revenue if the Company is deemed to be the principal participant in the grant arrangement as the activities under the grant are part of the Company’s development program. If the Company is not the principal participant, the grant funds are recorded as a reduction to research and development expense. Grant funds received are not refundable and are recognized when the related qualified research and development costs are incurred and when there is reasonable assurance that the funds will be received. Funds received in advance are recorded as deferred revenue.
Preclinical Studies and Clinical Trial Accruals
A substantial portion of the Company’s preclinical studies and all of the Company’s clinical trials have been performed by third-party contract research organizations (“CROs”) and other vendors. For preclinical studies, the significant factors used in estimating accruals include the percentage of work completed to date and contract milestones achieved. For clinical trial expenses, the significant factors used in estimating accruals include the number of patients enrolled, duration of enrollment and percentage of work completed to date. The Company monitors patient enrollment levels and related activities to the extent practicable through internal reviews, correspondence and status meetings with CROs, and review of contractual terms. The Company’s estimates are
86
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
dependent on the timeliness and accuracy of data provided by its CROs and other vendors. If the Company has incomplete or inaccurate data, it may under- or overestimate activity levels associated with various studies or trials at a given point in time. In this event, it could record adjustments to research and development expenses in future periods when the actual activity level becomes known. No material adjustments to preclinical study and clinical trial expenses have been recognized to date.
Research and Development Expenditures
Research and development costs are charged to operations as incurred.
Retirement Plan
The Company sponsors a 401(k) defined contribution plan covering all employees. There were no employer contributions to the plan from inception through December 31, 2013.
Income Taxes
The Company accounts for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amounts expected to be realized.
The Company also follows the accounting guidance that defines the threshold for recognizing the benefits of tax return positions in the financial statements as “more-likely-than-not” to be sustained by the taxing authorities based solely on the technical merits of the position. If the recognition threshold is met, the tax benefit is measured and recognized as the largest amount of tax benefit that, in the Company’s judgment, is greater than 50% likely to be realized.
Comprehensive Loss
The Company follows the accounting standards for the reporting and presentation of comprehensive income (loss) and its components. In June 2011, the Financial Accounting Standards Board (“FASB”) issued new accounting guidance that revised the manner in which entities present comprehensive income in their financial statements. The new guidance requires entities to present comprehensive income either in a continuous statement of comprehensive income, which replaces the statement of operations, or in two separate, consecutive statements. The new guidance does not change the items that must be reported in other comprehensive income, nor does it require new disclosures. On January 1, 2012, the Company adopted new accounting guidance and presents comprehensive income (loss) in a continuous statement of comprehensive income (loss) which replaced the statement of operations. Comprehensive loss includes all changes in stockholders’ equity during a period from non-owner sources. Comprehensive loss for each of the years ended December 31, 2013, 2012, and 2011 was equal to net loss adjusted for unrealized gains and losses on investments.
Segment Reporting
The Company has determined that it operates in only one segment.
Reverse Stock Split
On June 24, 2013, the Company effected a one-for-six reverse stock split of its common stock through an amendment to its amended and restated certificate of incorporation (the “COI Amendment”). As of the
87
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
effective time of the reverse stock split, every six shares of the Company’s issued and outstanding common stock were converted into one issued and outstanding share of common stock, without any change in par value per share. The reverse stock split affected all shares of the Company’s common stock outstanding immediately prior to the effective time of the reverse stock split, as well as the number of shares of common stock available for issuance under the Company’s equity incentive plans. In addition, the reverse stock split effected a reduction in the number of shares of common stock issuable upon the conversion of shares of preferred stock or upon the exercise of stock options or warrants outstanding immediately prior to the effectiveness of the reverse stock split. No fractional shares were issued as a result of the reverse stock split. Stockholders who would otherwise have been entitled to receive a fractional share received cash payments in lieu thereof. In addition, the COI Amendment reduced the number of authorized shares of common stock to 81.5 million.
As the par value per share of the Company’s common stock remained unchanged at $0.001 per share, a total of $139,000 was reclassified from common stock to additional paid-in capital. All references to shares of common stock and per share data for all periods presented in the accompanying financial statements and notes thereto have been adjusted to reflect the reverse stock split on a retroactive basis.
Recent Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In February 2013, the FASB issued Accounting Statement Update (“ASU”) 2013-02,Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income. This update requires entities to disclose items reclassified out of accumulated other comprehensive income and into net income in a single location within the financial statements. On January 1, 2013, the Company adopted this new accounting guidance and discloses reclassifications out of accumulated other comprehensive income and into net income in the footnotes to the financial statements.
Accounting Pronouncements Not Yet Adopted
In July 2013, the FASB issued ASU 2013-11,Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists. ASU 2013-11 amends accounting guidance on the presentation of an unrecognized tax benefit when a net operating loss carryforward, a similar tax loss, or tax credit carryforward exists. This new guidance requires entities, if certain criteria are met, to present an unrecognized tax benefit, or portion of an unrecognized tax benefit, in the financial statements as a reduction to a deferred tax asset for a net operating loss carryforward, a similar tax loss, or a tax credit carryforward when such items exist in the same taxing jurisdiction. The provisions of ASU 2013-11 are effective for fiscal years and interim periods beginning after December 15, 2013, which corresponds to the Company’s first quarter of fiscal year 2014. The Company does not expect the adoption of ASU 2013-11 will have a material effect on its financial statements.
88
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Note 2 — Net Loss Per Share
Basic net loss per share allocable to common stockholders is computed by dividing net loss allocable to common stockholders by the weighted average number of vested common shares outstanding during the period. Diluted net income loss per share allocable to common stockholders is computed by giving effect to all potentially dilutive common shares, including outstanding stock options, unvested restricted stock, warrants, convertible preferred stock and shares issuable under the Company’s Employee Stock Purchase Plan (“ESPP”), by applying the treasury stock method. The following is the calculation of basic and diluted net loss per share allocable to common stockholders (in thousands except per share data):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Net loss | $ | (33,717 | ) | $ | (40,370 | ) | $ | (47,860 | ) | |||
Deemed dividend related to beneficial conversion feature of convertible preferred stock | — | (1,307 | ) | (2,857 | ) | |||||||
|
|
|
|
|
| |||||||
Net loss allocable to common stockholders | $ | (33,717 | ) | $ | (41,677 | ) | $ | (50,717 | ) | |||
|
|
|
|
|
| |||||||
Weighted-average shares used in computing net loss per share allocable to common stockholders — basic and diluted | 27,275 | 18,107 | 11,800 | |||||||||
|
|
|
|
|
| |||||||
Net loss per share allocable to common stockholders — basic and diluted | $ | (1.24 | ) | $ | (2.30 | ) | $ | (4.30 | ) | |||
The following instruments were excluded from the computation of diluted net loss per common share allocable to common stockholders for the periods presented because their effect would have been antidilutive (in thousands):
| December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Options to purchase common stock | 2,449 | 1,791 | 1,599 | |||||||||
Warrants to purchase common stock | 7,692 | 9,009 | 1,114 | |||||||||
Series A convertible preferred stock (as converted to common stock) | — | — | 1,345 | |||||||||
Series B convertible preferred stock (as converted to common stock) | — | 3,838 | — | |||||||||
Restricted stock units | 42 | 217 | 518 | |||||||||
Shares issuable related to the ESPP | 14 | 11 | 8 | |||||||||
|
|
|
|
|
| |||||||
Total shares | 10,200 | 14,866 | 4,584 | |||||||||
|
|
|
|
|
| |||||||
Note 3 — Supplementary Cash Flow Data
Supplemental cash flow information was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Cash paid for interest | $ | — | $ | 3 | $ | 41 | ||||||
Cash paid for income taxes | 1 | 1 | 1 | |||||||||
Significant non-cash investing and financing activities: | ||||||||||||
Purchases of property and equipment through accounts payable | 193 | 116 | 13 | |||||||||
Purchases of property and equipment through accrued liabilities | (2 | ) | 37 | — | ||||||||
Purchases of property and equipment through trade in value of disposed property and equipment | 81 | — | — | |||||||||
89
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Note 4 — Cash Equivalents and Investments
Cash Equivalents and Available for Sale Investments
The amortized cost and fair value of cash equivalents and available for sale investments at December 31, 2013 and 2012 were as follows (in thousands):
| December 31, 2013 | ||||||||||||||||||||
| Amortized Cost | Unrealized Gains | Unrealized Losses | Fair Value | Maturity Dates | ||||||||||||||||
Cash equivalents — money market funds | $ | 15,858 | $ | — | $ | — | $ | 15,858 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||||||
Short-term investments — U.S. Treasury securities | $ | 57,564 | $ | 7 | $ | (1 | ) | $ | 57,570 | 1/2014 — 12/2014 | ||||||||||
|
|
|
|
|
|
|
| |||||||||||||
Long-term investments — U.S. Treasury securities | $ | 2,502 | $ | — | $ | — | $ | 2,502 | 1/2015 | |||||||||||
|
|
|
|
|
|
|
| |||||||||||||
| December 31, 2012 | ||||||||||||||||||||
| Amortized Cost | Unrealized Gains | Unrealized Losses | Fair Value | Maturity Dates | ||||||||||||||||
Cash equivalents — money market funds | $ | 10,655 | $ | — | $ | — | $ | 10,655 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||||||
Short-term investments — U.S. Treasury securities | $ | 59,075 | $ | 18 | $ | — | $ | 59,093 | 1/2013 — 11/2013 | |||||||||||
|
|
|
|
|
|
|
| |||||||||||||
As of December 31, 2013 and December 31, 2012, the Company’s U.S. Treasury securities classified as short-term investments had unrealized losses of approximately $1 and zero, respectively. The unrealized losses in 2013 were primarily caused by slight increases in short-term interest rates subsequent to the purchase date of the related securities. The Company collected the contractual cash flows on its U.S. Treasury securities that matured from January 1, 2014 through February 28, 2014 and expects to be able to collect all contractual cash flows on the remaining maturities of its U.S. Treasury securities.
Interest income was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Interest income | $ | 792 | $ | 83 | $ | 132 | ||||||
Note 5 — Fair Value Measurements
The Company adopted the fair value accounting guidance to value its financial assets and liabilities. Fair value is defined as the price that would be received for assets when sold or paid to transfer a liability in an orderly transaction between market participants at the measurement date (exit price). The Company utilizes market data or assumptions that the Company believes market participants would use in pricing the asset or liability, including assumptions about risk and the risks inherent in the inputs to the valuation technique. These inputs can be readily observable, market corroborated or generally unobservable.
The Company primarily applies the market approach for recurring fair value measurements and endeavors to utilize the best information reasonably available. Accordingly, the Company utilizes valuation techniques that
90
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible, and considers the security issuers’ and the third-party insurers’ credit risk in its assessment of fair value.
The Company classifies the determined fair value based on the observability of those inputs. Fair value accounting guidance establishes a fair value hierarchy that prioritizes the inputs used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurement) and the lowest priority to unobservable inputs (Level 3 measurement). The three defined levels of the fair value hierarchy are as follows:
Level 1 — Observable inputs, such as quoted prices in active markets for identical assets or liabilities;
Level 2 — Inputs, other than the quoted prices in active markets, that are observable either directly or through corroboration with observable market data; and
Level 3 — Unobservable inputs, for which there is little or no market data for the assets or liabilities, such as internally-developed valuation models.
Financial assets measured at fair value on a recurring basis as of December 31, 2013 and 2012 are classified in the table below in one of the three categories described above (in thousands):
| December 31, 2013 | ||||||||||||||||
| Fair Value Measurements Using | Assets At Fair Value | |||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
Money market funds | $ | 15,858 | $ | — | $ | — | $ | 15,858 | ||||||||
U.S. Treasury securities | 60,072 | — | — | 60,072 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | 75,930 | $ | — | $ | — | $ | 75,930 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Amounts included in: | ||||||||||||||||
Cash and cash equivalents | $ | 15,858 | $ | — | $ | — | $ | 15,858 | ||||||||
Short-term investments | 57,570 | — | — | 57,570 | ||||||||||||
Long-term investments | 2,502 | — | — | 2,502 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | 75,930 | $ | — | $ | — | $ | 75,930 | ||||||||
|
|
|
|
|
|
|
| |||||||||
| December 31, 2012 | ||||||||||||||||
| Fair Value Measurements Using | Assets At Fair Value | |||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
Money market funds | $ | 10,655 | $ | — | $ | — | $ | 10,655 | ||||||||
U.S. Treasury securities | 59,093 | — | — | 59,093 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | 69,748 | $ | — | $ | — | $ | 69,748 | ||||||||
|
|
|
|
|
|
|
| |||||||||
Amounts included in: | ||||||||||||||||
Cash and cash equivalents | $ | 10,655 | $ | — | $ | — | $ | 10,655 | ||||||||
Short-term investments | 59,093 | — | — | 59,093 | ||||||||||||
|
|
|
|
|
|
|
| |||||||||
Total | $ | 69,748 | $ | — | $ | — | $ | 69,748 | ||||||||
|
|
|
|
|
|
|
| |||||||||
The valuation technique used to measure fair value for the Company’s Level 1 assets is a market approach, using prices and other relevant information generated by market transactions involving identical assets. As of December 31, 2013 and 2012, the Company had no financial assets measured at fair value on a recurring basis using significant Level 2 or Level 3 inputs.
91
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
The carrying amount of the Company’s accounts receivable and accounts payable approximates fair value due to the short-term nature of these instruments.
Note 6 — Balance Sheet Components
Property and equipment balances were as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
Property and equipment, net: | ||||||||
Laboratory equipment | $ | 15,317 | $ | 17,064 | ||||
Computer equipment and software | 2,549 | 3,190 | ||||||
Office equipment, furniture and fixtures | 1,050 | 638 | ||||||
Leasehold improvements | 3,297 | 3,393 | ||||||
|
|
|
| |||||
| 22,213 | 24,285 | |||||||
Less: Accumulated depreciation and amortization | (20,992 | ) | (23,288 | ) | ||||
|
|
|
| |||||
| $ | 1,221 | $ | 997 | |||||
|
|
|
| |||||
Depreciation expense was $0.4 million, $0.6 million and $1.3 million for the years ended December 31, 2013, 2012 and 2011 respectively.
Accrued liabilities were as follows (in thousands):
| December 31, | ||||||||
| 2013 | 2012 | |||||||
Accrued liabilities: | ||||||||
Clinical and preclinical costs | $ | 4,975 | $ | 2,170 | ||||
Consulting and professional fees | 697 | 312 | ||||||
Bonus | 1,614 | 1,355 | ||||||
Vacation pay | 778 | 696 | ||||||
Other payroll related | 93 | 87 | ||||||
Other accrued expenses | 115 | 257 | ||||||
|
|
|
| |||||
| $ | 8,272 | $ | 4,877 | |||||
|
|
|
| |||||
Interest receivable on cash equivalents and investments of $215,000 and $187,000 is included in prepaid and other current assets at December 31, 2013 and 2012, respectively.
Note 7 — Related Party Transactions
Research and Development Arrangements
Amgen
In December 2006, the Company entered into a collaboration and option agreement with Amgen to discover, develop and commercialize novel small molecule therapeutics, including omecamtiv mecarbil, that activate cardiac muscle contractility for potential applications in the treatment of heart failure (the “Amgen
92
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Agreement”). The agreement granted Amgen an option to obtain an exclusive license worldwide, except Japan, to develop and commercialize omecamtiv mecarbil and other drug candidates arising from the collaboration. In May 2009, Amgen exercised its option. As a result, Amgen became responsible for the development and commercialization of omecamtiv mecarbil and related compounds at its expense worldwide (excluding Japan), subject to the Company’s development and commercialization participation rights. Amgen will reimburse the Company for certain research and development activities it performs under the collaboration.
In June 2013, Cytokinetics and Amgen announced an amendment to the Amgen Agreement to include Japan, resulting in a worldwide collaboration (the “Amgen Agreement Amendment”). Under the terms of the Amgen Agreement Amendment, the Company received a non-refundable upfront license fee of $15 million in June 2013. Under the Amgen Agreement Amendment, the Company plans to conduct a Phase I pharmacokinetic study intended to support inclusion of Japan in a potential Phase III clinical development program and potential global registration dossier for omecamtiv mecarbil. Amgen will reimburse the Company for the costs of this study. In addition, the Company is eligible to receive additional pre-commercialization milestone payments relating to the development of omecamtiv mecarbil in Japan of up to $50 million, and royalties on sales of omecamtiv mecarbil in Japan.
In conjunction with the Amgen Agreement Amendment, the Company also entered into a common stock purchase agreement which provided for the sale of 1,404,100 shares of its common stock to Amgen at a price per share of $7.12 and an aggregate purchase price of $10.0 million which was received in June 2013. The Company determined the fair value of the stock issued to Amgen to be $7.5 million. The excess of cash received over fair value of $2.5 million was deferred and is being allocated between the license and services based on their relative selling prices using best estimate of selling price. Allocated consideration will be recognized as revenue as revenue criteria is satisfied, or as services are performed over approximately 12 months. Pursuant to this agreement, Amgen has agreed to certain trading and other restrictions with respect to the Company’s common stock.
Under the Amgen Agreement, as amended, the Company is eligible for potential pre-commercialization and commercialization milestone payments of up to $650 million in the aggregate on omecamtiv mecarbil and other potential products arising from research under the collaboration, and royalties that escalate based on increasing levels of annual net sales of products commercialized under the agreement. None of the future contingent milestone payments pursuant to this arrangement as of January 1, 2011 are considered substantive as they are the results of Amgen’s performance. Therefore, they are not considered milestones under ASC 605-28. The Amgen Agreement also provides for the Company to receive increased royalties by co-funding Phase III development costs of omecamtiv mecarbil and other drug candidates under the collaboration. If the Company elects to co-fund such costs, it would be entitled to co-promote the co-funded drug in North America and participate in agreed commercialization activities in institutional care settings, at Amgen’s expense.
In October 2013, the Company determined that all conditions necessary for revenue recognition under ASC 605-10 had been satisfied and accordingly, recognized a total of $17.2 million in license revenue attributable to the Amgen Agreement Amendment in the fourth quarter of 2013.
At December 31, 2013, the Company had $300,000 of deferred revenue under the Amgen Agreement Amendment.
Pursuant to the Amgen Agreement, the Company has recognized research and development revenue from Amgen for reimbursements of its costs of certain full-time employee equivalents (“FTEs”) supporting a collaborative research program directed to the discovery of next-generation cardiac sarcomere activator
93
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
compounds and of other costs related to that research program. These reimbursements were recorded as research and development revenues from related parties. Revenue from Amgen was as follows (in thousands):
Revenue from Amgen was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
License revenues | $ | 17,230 | $ | — | $ | — | ||||||
FTE reimbursements | 2,019 | 4,174 | 1,988 | |||||||||
Reimbursements of other costs | — | 3 | 66 | |||||||||
|
|
|
|
|
| |||||||
Total research and development revenues from Amgen | $ | 19,249 | $ | 4,177 | $ | 2,054 | ||||||
|
|
|
|
|
| |||||||
Note 8 — Other Research and Development Revenue Arrangements
Astellas Pharma Inc. (“Astellas”)
In June 2013, the Company entered into a collaboration and license agreement (the “Astellas Agreement”) with Astellas. The primary objective of the collaboration to be conducted under the Astellas Agreement is to advance novel therapies for diseases and medical conditions associated with muscle weakness.
Under the Astellas Agreement, the Company granted Astellas an exclusive license to co-develop and jointly commercialize CK-2127107, a fast skeletal troponin activator, for potential application in non-neuromuscular indications worldwide. CK-2127107, which is currently in Phase I clinical development, is being developed jointly by the Company and Astellas. The Company is primarily responsible for the conduct of Phase I clinical trials and certain Phase II readiness activities for CK-2127107 and Astellas will be primarily responsible for the conduct of subsequent development and commercialization activities for CK-2127107.
The companies are jointly conducting research to identify next-generation skeletal muscle activators to be nominated as potential drug candidates, at Astellas’ expense, over a two-year term, which may be extended by the companies’ mutual consent. Astellas has the exclusive rights to develop and commercialize fast skeletal troponin activators from this research program in non-neuromuscular indications and to develop and commercialize other novel mechanism skeletal muscle activators from this research program in all indications, subject to certain co-development and co-promotion rights of the Company under the Astellas Agreement. Astellas will be responsible for the costs associated with the development of all collaboration products, including CK-2127107.
The Company retains an option to conduct early-stage development for certain agreed upon indications at its initial expense, subject to reimbursement if development continues under the collaboration. The Company also retains an option to co-promote collaboration products in the United States and Canada. Astellas will reimburse the Company for certain expenses associated with its co-promotion activities.
In July 2013, the Company received an upfront, non-refundable license fee of $16 million in connection with the execution of the Astellas Agreement, and is eligible to potentially receive over $24 million in reimbursement of sponsored research and development activities during the initial two years of the collaboration. Based on the achievement of pre-specified criteria, the Company may receive over $250 million in milestone payments relating to the development and commercial launch of collaboration products, including up to $112 million in development and commercial launch milestones for CK-2127107. The Company may also receive up to $200 million in payments for achievement of pre-specified sales milestones related to net sales of all collaboration products under the Astellas Agreement. In the event Astellas commercializes any collaboration
94
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
products, the Company will also receive royalties on sales of such collaboration products, including royalties ranging from the high single digits to the high teens on sales of products containing CK-2127107. In addition to the foregoing development, commercial launch and sales milestones, the Company may also receive payments for the achievement of pre-specified milestones relating to the joint research program.
The Company retains the exclusive right to develop and commercialize tirasemtiv for the potential treatment of amyotrophic lateral sclerosis and other neuromuscular disorders independently from the Astellas Agreement.
As of June 30, 2013, the Company deferred revenue related to the Astellas Agreement in accordance with ASC 605-25. The Company evaluated whether the delivered elements under the arrangement have value on a stand-alone basis. Upfront, non-refundable licensing payments are assessed to determine whether or not the licensee is able to obtain stand-alone value from the license. Where this is not the case, the Company does not consider the license deliverable to be a separate unit of accounting, and the revenue is deferred with revenue recognition for the license fee being recognized in conjunction with the other deliverables that constitute the combined unit of accounting.
The Company determined that the license and the research and development services are a single unit of accounting as the license was determined to not have stand-alone value. Accordingly, the Company is recognizing this revenue using the proportional performance model. As of December 31, 2013, the Company has recognized $3.9 million of the $16 million upfront license fee as license revenue and deferred the remaining $12.1 million.
The Company recognizes milestone payments utilizing the milestone method of revenue recognition. The Company believes the milestones related to research and early development under the Astellas Agreement are substantive as there is uncertainty that the milestones will be met, the milestone can only be achieved with the Company’s past and current performance and the achievement of the milestone will result in additional payment to the Company. The Company believes that the milestones related to later development and commercialization are not substantive as they are primarily the result of the collaborative partner’s performance and therefore will be recognized as the Company completes its performance obligations under the agreement, if any. To date, the Company has not recognized any milestone revenue from its collaboration with Astellas.
Research and development revenue from Astellas was as follows (in thousands):
| Year Ended December 31, 2013 | ||||
License revenues | $ | 3,852 | ||
FTE reimbursements | 3,285 | |||
Reimbursements of other costs | 3,130 | |||
|
| |||
Total revenue from Astellas | $ | 10,267 | ||
|
| |||
At December 31, 2013, the Company had $15.9 million of deferred revenue under the Astellas Agreement, reflecting the unrecognized portion of the license revenue and prepayment on expenses expected to be incurred in the first quarter of 2014.
Grants
In 2010, the National Institute of Neurological Disorders and Strokes (“NINDS”) awarded to the Company a $2.8 million grant to support research and development of tirasemtiv directed to the potential treatment for
95
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
myasthenia gravis for a period of up to three years. In September 2012, the NINDS awarded to us an additional $0.5 million under a separate grant. Management determined that the Company was the principal participant in the grant arrangements, and, accordingly, the Company recorded amounts earned under the arrangements as revenue. The grants were completed in June of 2013.
In November 2010, the Company was notified by the U.S. Department of the Treasury that it would receive total cash grants of $0.7 million based on its applications for certain investments in qualified therapeutic discovery projects under Section 48D of the Internal Revenue Code. The grants related to certain research and development costs the Company incurred in 2009 in connection with its cardiac, skeletal and smooth muscle contractility programs.
Total grant revenues were as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
NINDS myasthenia gravis | $ | 69 | $ | 1,308 | $ | 1,680 | ||||||
U.S. Department of the Treasury | 25 | — | — | |||||||||
|
|
|
|
|
| |||||||
Total grant revenue | $ | 94 | $ | 1,308 | $ | 1,680 | ||||||
|
|
|
|
|
| |||||||
Global Blood Therapeutics, Inc. (“Global Blood”)
In October 2011, the Company entered into a collaboration agreement with Global Blood Therapeutics, Inc. (formerly called Global Blood Targeting, Inc.) (“Global Blood”). Under an agreed research plan, scientists from Global Blood and our FTEs conducted research focused on small molecule therapeutics that target the blood. The Company provided to Global Blood access to certain research facilities, FTEs and other resources at agreed reimbursement rates that approximated our costs. In April 2012, the Company extended this agreement through December 2012. The Company was the primary obligor in the collaboration arrangement, and accordingly, the Company recorded expense reimbursements from Global Blood as research and development revenue.
Research and development revenue from Global Blood was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Expense reimbursements from Global Blood | $ | 14 | $ | 1,479 | $ | 266 | ||||||
|
|
|
|
|
| |||||||
MyoKardia, Inc.
In August 2012, the Company entered into a collaboration agreement with MyoKardia, Inc. Under an agreed research plan, scientists from MyoKardia and our FTEs conduct research focused on small molecule therapeutics that inhibit cardiac sarcomere proteins. The Company provided to MyoKardia access to certain research facilities, and continues to provide FTEs and other resources at agreed reimbursement rates that approximate our costs. The Company is the primary obligor in the collaboration arrangement, and accordingly, the Company records expense reimbursements from MyoKardia as research and development revenue. The research plan terminated as planned in August 2013.
96
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Research and development revenue from MyoKardia was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Expense reimbursements from MyoKardia | $ | 1,024 | $ | 595 | $ | — | ||||||
|
|
|
|
|
| |||||||
Note 9 — Debt
In April 2006, the Company entered into an equipment financing agreement with GE Capital. As of December 31, 2011, the balance of equipment loans outstanding under this line was $152,000. The Company repaid the remaining outstanding equipment financing debt in March 2012 and no further funds are available under this line of credit.
Interest Expense
Total interest expense incurred by the Company was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Interest expense | $ | — | $ | 3 | $ | 35 | ||||||
Note 10 — Restructuring
In October 2011, the Company announced a restructuring plan to realign its workforce and operations in line with its continued commitment to focus primarily on the development of its key later-stage development programs for tirasemtiv and omecamtiv mecarbil and on its follow-on skeletal muscle troponin activator program and joint research with Amgen directed to next-generation compounds in its cardiac muscle contractility program. As a result, the Company reduced its workforce by 18 employees, or approximately 18%, to 83 employees. The Company provided severance, employee benefit continuation and career transition assistance to the employees directly affected by the restructuring. The Company incurred restructuring charges of $1.2 million in the fourth quarter of 2011, primarily personnel-related termination costs.
The following table summarizes the activity for the restructuring plan (in thousands):
| Employee Severance and Related Benefits | ||||
Restructuring liability at December 31, 2011 | $ | 194 | ||
2012 reversals | (58 | ) | ||
Cash payments | (136 | ) | ||
|
| |||
Restructuring liability at December 31, 2012 | $ | — | ||
|
| |||
There was no activity in the restructuring liability subsequent to December 31, 2012 and the balance of the restructuring liability at December 31, 2013 was zero.
97
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Note 11 — Commitments and Contingencies
Leases
The Company leases office space and equipment under a non-cancelable operating lease that expires in 2018, with an option to extend the lease for an additional three-year period. The lease terms provide for rental payments on a graduated scale and the Company’s payment of certain operating expenses. The Company recognizes rent expense on a straight-line basis over the lease period.
Rent expense was as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Rent expense | $ | 3,306 | $ | 3,375 | $ | 2,990 | ||||||
|
|
|
|
|
| |||||||
As of December 31, 2013, future minimum lease payments under noncancelable operating leases were as follows (in thousands):
2014 | $ | 3,357 | ||
2015 | 3,470 | |||
2016 | 3,587 | |||
2017 | 3,713 | |||
2018 | 1,906 | |||
Thereafter | — | |||
|
| |||
Total | $ | 16,033 | ||
|
|
In the ordinary course of business, the Company may provide indemnifications of varying scope and terms to vendors, lessors, business partners and other parties with respect to certain matters, including, but not limited to, losses arising out of the Company’s breach of such agreements, services to be provided by or on behalf of the Company, or from intellectual property infringement claims made by third parties. In addition, the Company has entered into indemnification agreements with its directors and certain of its officers and employees that will require the Company, among other things, to indemnify them against certain liabilities that may arise by reason of their status or service as directors, officers or employees. The Company maintains director and officer insurance, which may cover certain liabilities arising from its obligation to indemnify its directors and certain of its officers and employees, and former officers and directors in certain circumstances. The Company maintains product liability insurance and comprehensive general liability insurance, which may cover certain liabilities arising from its indemnification obligations. It is not possible to determine the maximum potential amount of exposure under these indemnification obligations due to the limited history of prior indemnification claims and the unique facts and circumstances involved in each particular indemnification obligation. Such indemnification obligations may not be subject to maximum loss clauses.
Note 12 — Convertible Preferred Stock
As of December 31, 2010 there were 10,000,000 shares of preferred stock authorized and no shares outstanding.
On April 18, 2011, the Company entered into a securities purchase agreement (the “Deerfield Agreement”) with Deerfield Private Design Fund II, L.P., Deerfield Private Design International II, L.P., Deerfield Special Situations Fund, L.P., and Deerfield Special Situations Fund International Limited (collectively, “Deerfield”). On
98
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
April 20, 2011, pursuant to the Deerfield Agreement, the Company issued to Deerfield 8,070 shares of Series A convertible preferred stock (the “Series A Preferred Stock”) for a purchase price of $1,500.00 per share for net proceeds of approximately $9.3 million, as well as common stock and warrants that are discussed in Note 13 — Stockholders’ Equity (Deficit).
The fair value of the common stock into which the Series A Preferred Stock was convertible exceeded the allocated purchase price of the Series A Preferred Stock by $2.9 million on the date of issuance, resulting in a beneficial conversion feature. The Company recognized the beneficial conversion feature as a one-time, non-cash, deemed dividend to the holders of Series A Preferred Stock on the date of issuance, which is the date the stock first became convertible.
On September 26, 2012, all 8,070 shares of Series A Preferred Stock were converted into 1,345,000 shares of our common stock. The conversion was in accordance with the terms of the agreement with Deerfield under which the Series A Preferred Stock was issued in 2011.
On June 20, 2012, the Company entered into underwriting agreements for two separate, concurrent public offerings of the Company’s securities (the “June 2012 Public Offerings”). On June 25, 2012, pursuant to the underwriting agreements, in aggregate the Company issued to certain investors 23,026 shares of Series B convertible preferred stock (the “Series B Preferred Stock”) for a purchase price of $760.00 per share, for net proceeds of approximately $12.3 million.
Each share of Series B Preferred Stock was convertible into common stock at any time at the holder’s option. However, the holder was prohibited from converting the Series B Preferred Stock into shares of common stock if, as a result of such conversion, the holder and its affiliates would own more than 9.98% of the total number of shares of common stock then issued and outstanding. In the event of the Company’s liquidation, dissolution, or winding up, holders of Series B Preferred Stock would receive a payment equal to $0.001 per share before any proceeds are distributed to the common stockholders. Shares of Series B Preferred Stock generally have no voting rights, except as required by law and except that the consent of holders of a majority of the outstanding Series B Preferred Stock is required to amend the terms of the Series B Preferred Stock. Holders of Series B Preferred Stock were not entitled to receive any dividends, unless and until specifically declared by the Company’s board of directors. The Series B Preferred Stock ranked senior to the Company’s common stock as to distributions of assets upon the Company’s liquidation, dissolution or winding up, whether voluntarily or involuntarily. The Series B Preferred Stock may have ranked senior to, on parity with or junior to any class or series of the Company’s capital stock created in the future depending upon the specific terms of such future stock issuance. As a result of the one-for-six reverse stock split effected in June 2013, the conversion ratio for Series B convertible preferred stock changed from 1,000 shares of common stock per share of Series B convertible preferred stock to 166.67 shares of common stock per share of Series B convertible preferred stock.
The fair value of the common stock into which the Series B Preferred Stock is convertible exceeded the allocated purchase price of the Series B Preferred Stock by $1.3 million on the date of issuance, resulting in a beneficial conversion feature. The Company recognized the beneficial conversion feature as a one-time, non-cash, deemed dividend to the holders of Series B Preferred Stock on the date of issuance, which is the date the stock first became convertible.
In the first quarter of 2013, 4,000 shares of Series B convertible preferred stock were converted into 666,667 shares of common stock. In the second quarter of 2013, 15,026 shares of Series B convertible preferred stock were converted into 2,504,334 shares of common stock. On July 2, 2013, 4,000 shares of Series B convertible preferred stock, which represented all remaining shares of Series B convertible preferred stock, were converted into 666,681 shares of common stock. The conversions were in accordance with the terms of the original agreement under which the Series B convertible preferred stock was issued in 2012.
99
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Note 13 — Stockholders’ Equity (Deficit)
Accumulated Other Comprehensive Loss
In 2013, the Company reclassified insignificant amounts of unrealized gains (losses) in investments out of accumulated other comprehensive income into net loss.
Authorized Shares
On May 18, 2011, the stockholders approved an increase in the number of authorized shares of common stock from 170,000,000 to 245,000,000. The increase became effective in August 2011, when the Company filed the Certificate of Amendment of Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware. In June 2013, upon the stockholder approval of the one-for-six reverse stock split and the amendment to the Company’s amended and restated certificate of incorporation, the number of authorized shares of common stock was reduced to 81,500,000 (See Note 1).
Common Stock Outstanding
On April 20, 2011, pursuant to the Deerfield Agreement, the Company issued to Deerfield (i) 883,333 shares of common stock for a purchase price of $9.00 per share, (ii) 8,070 shares of Series A convertible preferred stock (the “Series A Preferred Stock”) for a purchase price of $1,500.00 per share, and (iii) warrants to purchase 1,114,168 shares of the Company’s common stock at an initial exercise price of $9.90 per share, for aggregate gross proceeds of approximately $20.1 million. After issuance costs of approximately $0.2 million, the net proceeds were approximately $19.9 million.
The offering was made pursuant to a shelf registration statement that the Company filed with the SEC on November 10, 2008, which became effective on November 19, 2008 (File No. 333-155259). The closing of the offering took place on April 20, 2011.
In accordance with the accounting guidance for valuing stock and warrants when preferred stock, common stock and warrants are issued in a single transaction and all are to be accounted for as equity, the Company allocated the gross purchase proceeds using the relative fair value method. The fair value of the common stock issued to Deerfield was calculated based on the closing price of the stock on the commitment date as quoted on The NASDAQ Global Market. The Series A Preferred Stock was valued based on the fair value of the Company’s common stock on the commitment date times the conversion ratio of one share of preferred to one thousand shares of common stock. The fair value of the Series A Preferred Stock was determined to be essentially equivalent to the fair value of the common stock into which it is convertible, based on the preferred holders’ ability to immediately convert the Series A Preferred Stock to common stock and the fact that the liquidation preference of the Series A Preferred Stock is only $0.001 per share. The fair value of the warrants was determined using the Black-Scholes pricing model, as discussed above. The relative fair value ratio of each of the instruments issued was then applied to the total gross proceeds of $20.1 million, resulting in allocated purchase prices of $6.2 million for the common stock, $9.4 million for the Series A Preferred Stock and $4.5 million for the warrants.
On September 26, 2012, all 8,070 shares of Series A Convertible Preferred Stock were converted into 1,345,000 shares of our common stock. The conversion was in accordance with the terms of the original agreement under which the Series A Convertible Preferred Stock was issued in 2011.
In June 2011, the Company entered into an At-The-Market Issuance Sales Agreement (the “MLV Agreement”) with McNicoll, Lewis & Vlak LLC (“MLV”), pursuant to which the Company sold, through December 31, 2013, 2,033,175 shares of common stock through MLV for net proceeds of approximately $12.7 million. (See Note 17.)
100
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
On June 25, 2012, pursuant to the June 2012 Public Offerings, in aggregate the Company issued to various investors (i) 9,320,176 shares of common stock for a purchase price of $4.56 per share, (ii) 23,026 shares of the Series B Preferred Stock for a purchase price of $760.00 per share, and (iii) warrants to purchase 7,894,704 shares of the Company’s common stock at an exercise price of $5.28 per share, for aggregate gross proceeds of approximately $60.0 million. After issuance costs of approximately $4.0 million, the net proceeds from the June 2012 Public Offerings were approximately $56.0 million.
The offerings were made pursuant to a shelf registration statement that the Company filed with the SEC on November 25, 2011, which became effective on December 8, 2011 (File No. 333-178189) and a supplemental shelf registration statement on Form S-3MEF that the Company filed with the SEC on June 20, 2012, which became effective on June 20, 2012 (File No. 333-182226). The closing of the offerings took place on June 25, 2012.
In accordance with the accounting guidance for valuing stock and warrants when stock is issued in conjunction with other securities, and the stock and other securities are to be accounted for as equity, the Company allocated the gross purchase proceeds using the relative fair value method. For accounting purposes, the June 2012 Public Offerings were considered to be one transaction. The fair value of the common stock issued in the June 2012 Public Offerings was calculated based on the closing price of the stock on the commitment date as quoted on The NASDAQ Global Market. The Series B Preferred Stock was valued based on the fair value of the Company’s common stock on the commitment date times the conversion ratio of one share of preferred stock to one thousand shares of common stock. The fair value of the Series B Preferred Stock was determined to be essentially equivalent to the fair value of the common stock into which it is convertible, based on the preferred holders’ ability to immediately convert the Series B Preferred Stock to common stock and the fact that the liquidation preference of the Series B Preferred Stock is only $0.001 per share. The fair value of the warrants was determined using the Black-Scholes pricing model, as discussed above. The relative fair value ratio of each of the instruments issued was then applied to the total gross proceeds of $60.0 million, resulting in allocated purchase prices of $32.1 million for the common stock, $13.2 million for the Series B Preferred Stock, and $14.7 million for the warrants.
In conjunction with the Amgen Agreement Amendment (see Note 7), in June 2013, Amgen purchased 1,404,100 shares of the Company’s common stock at a price per share of $7.12 and an aggregate purchase price of $10.0 million which was received in June 2013. Under the terms of this agreement, Amgen agreed to certain trading and other restrictions with respect to the Company’s common stock. The Company determined the fair value of the stock issued to Amgen to be $7.5 million. The excess of cash received over fair value of $2.5 million was deferred and is being allocated between the license and services based on their relative selling prices using best estimate of selling price.
In February 2014, the Company closed an underwritten public offering for the issuance and sale of 5,031,250 shares of its common stock. The gross public offering proceeds were approximately $40.3 million. The net proceeds from the sale of the shares were approximately $37.4 million, after deducting the underwriting discount and estimated offering expenses. (See Note 17.)
Warrants
On April 20, 2011, pursuant to the Deerfield Agreement, the Company issued to Deerfield warrants to purchase 1,114,168 shares of the Company’s common stock at an initial exercise price of $9.90 per share, for aggregate gross proceeds of approximately $4.5 million. After issuance costs of approximately $0.1 million, the net proceeds were approximately $4.4 million. The warrants issued to Deerfield became exercisable on October 20, 2011 and will remain exercisable until April 20, 2015. The warrant holders are prohibited from exercising the warrants and obtaining shares of common stock if, as a result of such exercise, the holder and its
101
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
affiliates would own more than 9.98% of the total number of shares of the Company’s common stock then issued and outstanding. The Company valued the warrants as of the date of issuance at $5.8 million using the Black-Scholes option pricing model and the following assumptions: a contractual term of four years, a risk-free interest rate of 1.66%, volatility of 80%, and the fair value of the Company’s common stock on the issuance date of $9.12 ($1.52 before adjustment for the Company’s 2013 reverse stock split, see Note 1).
On June 25, 2012, pursuant to the June 2012 Public Offerings, the Company issued warrants to purchase 7,894,704 shares of the Company’s common stock at an exercise price of $5.28 per share, for an aggregate gross proceeds of approximately $14.7 million. The warrant holders are prohibited from exercising the warrants and obtaining shares of common stock if, as a result of such exercise, the holder and its affiliates would own more than 9.98% of the total number of shares of the Company’s common stock then issued and outstanding. The Company valued the warrants as of the date of issuance at $16.2 million using the Black-Scholes option pricing model and the following assumptions: a contractual term of five years, a risk-free interest rate of 0.73%, volatility of 76%, and the fair value of the Company’s common stock on the issuance date of $3.78 ($0.63 before adjustment for the Company’s 2013 reverse stock split, see Note 1).
In February 2013, warrants to purchase 1,000 shares of our common stock at an exercise price of $5.28 per share were cash exercised in accordance with the June 2012 Public Offerings underwriting agreements.
In April 2013, the Company issued 358,460 shares of common stock related to cashless exercises of warrants in accordance with the June 2012 Public Offerings.
Outstanding warrants as of December 31, 2013 were as follows:
| Number of Shares | Exercise Price | Expiration Date | ||||||||||
Issued 4/20/2011 | 1,114,168 | $ | 9.90 | 04/20/15 | ||||||||
Issued 6/25/2012 | 6,577,928 | $ | 5.28 | 06/25/17 | ||||||||
Stock Option Plans
2004 Plan
In January 2004, the Board of Directors adopted the 2004 Equity Incentive Plan (the “2004 Plan”), which was approved by the stockholders in February 2004. The 2004 Plan provides for the granting of incentive stock options, nonstatutory stock options, restricted stock, stock appreciation rights, stock performance units and stock performance shares to employees, directors and consultants. Under the 2004 Plan, options may be granted at prices not lower than 100% of the fair market value of the common stock on the date of grant for nonstatutory stock options and incentive stock options and may be granted for terms of up to ten years from the date of grant. Options granted to new employees generally vest 25% after one year and monthly thereafter over a period of four years. Options granted to existing employees generally vest monthly over a period of four years. At the May 2013 Annual Meeting of Stockholders, the number of shares of common stock authorized for issuance under the 2004 Plan was increased by 2,000,000. As of December 31, 2013, there were 2,161,829 shares of common stock reserved for issuance under the 2004 Plan.
1997 Plan
In 1997, the Company adopted the 1997 Stock Option/Stock Issuance Plan (the “1997 Plan”). The Plan provides for the granting of stock options to employees and consultants of the Company. Options granted under the 1997 Plan may be either incentive stock options or nonstatutory stock options. Incentive stock options may be
102
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
granted only to Company employees (including officers and directors who are also employees). Nonstatutory stock options may be granted to Company employees and consultants. Options under the Plan may be granted for terms of up to ten years from the date of grant as determined by the Board of Directors, provided, however, that (i) the exercise price of an incentive stock option and nonstatutory stock option shall not be less than 100% and 85% of the estimated fair market value of the shares on the date of grant, respectively, and (ii) with respect to any 10% stockholder, the exercise price of an incentive stock option or nonstatutory stock option shall not be less than 110% of the estimated fair market value of the shares on the date of grant and the term of the grant shall not exceed five years. Options may be exercisable immediately and are subject to repurchase options held by the Company which lapse over a maximum period of ten years at such times and under such conditions as determined by the Board of Directors. Options granted under the 1997 Plan generally vested over four or five years (generally 25% after one year and monthly thereafter). As of December 31, 2013, the Company had reserved 11,504 shares of common stock for issuance related to options outstanding under the 1997 Plan, and there were no shares available for future grants under the 1997 Plan.
Activity under the two stock option plans was as follows:
| Shares Available for Grant of Option or Award | Stock Options Outstanding | Weighted Average Exercise Price per Share - Stock Options | Weighted Average remaining contractual Life | Aggregate Intrinsic Value | ||||||||||||||||
Balance at December 31, 2010 | 854,862 | 1,349,223 | $ | 25.89 | ||||||||||||||||
Increase in authorized shares | 500,000 | — | — | |||||||||||||||||
Options granted | (425,420 | ) | 425,420 | 9.56 | ||||||||||||||||
Restricted stock units granted | (531,701 | ) | — | — | ||||||||||||||||
Options exercised | — | (2,667 | ) | 6.56 | ||||||||||||||||
Options forfeited/expired | 173,569 | (173,568 | ) | 22.63 | ||||||||||||||||
Restricted stock units forfeited | 14,166 | — | — | |||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2011 | 585,476 | 1,598,408 | 21.93 | |||||||||||||||||
Increase in authorized shares | 416,667 | — | — | |||||||||||||||||
Options granted | (403,108 | ) | 403,108 | 6.14 | ||||||||||||||||
Options forfeited/expired | 210,989 | (210,989 | ) | 17.00 | ||||||||||||||||
Restricted stock units forfeited | 68,702 | — | — | |||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2012 | 878,726 | 1,790,527 | 18.96 | |||||||||||||||||
Increase in authorized shares | 2,000,000 | — | — | |||||||||||||||||
Options granted | (797,629 | ) | 797,629 | 5.95 | ||||||||||||||||
Restricted stock units granted | (41,661 | ) | — | — | ||||||||||||||||
Options exercised | — | (21,397 | ) | 5.32 | ||||||||||||||||
Options forfeited/expired | 117,394 | (117,394 | ) | 12.55 | ||||||||||||||||
Restricted stock units forfeited | 4,999 | — | — | |||||||||||||||||
|
|
|
| |||||||||||||||||
Balance at December 31, 2013 | 2,161,829 | 2,449,365 | $ | 15.15 | 6.67 | $ | 589,077 | |||||||||||||
|
|
|
| |||||||||||||||||
Exercisable at December 31, 2013 | 1,622,205 | $ | 19.49 | 5.60 | $ | 288,844 | ||||||||||||||
Vested and expected to vest as of December 31, 2013 | 2,428,631 | $ | 15.22 | 6.65 | $ | 580,858 | ||||||||||||||
Total intrinsic value of stock options exercised was $107,000, zero and $8,000 during the years ended December 31, 2013, 2012 and 2011, respectively. The intrinsic value is calculated as the difference between the market value as of December 31, 2013 and the exercise price of shares. The market value as of December 31, 2013 was $6.50 per share as reported by NASDAQ. The weighted average grant date fair value of stock options granted was $3.85, $3.89 and $6.25 per share during the years ended December 31, 2013, 2012 and 2011,
103
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
respectively. The number of option shares vested was 937,994, 245,946 and 225,622 in 2013, 2012 and 2011, respectively. The fair value of option shares vested was $3.5 million, $1.5 million and $2.0 million in 2013, 2012 and 2011, respectively.
Restricted stock unit activity was as follows:
| Number of Shares | Weighted Average Award Date Fair Value per Share | |||||||
Restricted stock units outstanding at December 31, 2010 | — | $ | — | |||||
Restricted stock units granted | 531,701 | 6.78 | ||||||
Restricted stock units forfeited | (14,166 | ) | 6.78 | |||||
|
| |||||||
Restricted stock units outstanding at December 31, 2011 | 517,535 | 6.78 | ||||||
Restricted stock units vested | (231,935 | ) | 6.78 | |||||
Restricted stock units forfeited | (68,702 | ) | 6.78 | |||||
|
| |||||||
Restricted stock units outstanding at December 31, 2012 | 216,898 | 6.78 | ||||||
Restricted stock units granted | 41,661 | 6.00 | ||||||
Restricted stock units vested | (211,897 | ) | 6.78 | |||||
Restricted stock units forfeited | (4,999 | ) | 6.78 | |||||
|
| |||||||
Unvested restricted stock units outstanding at December 31, 2013 | 41,663 | $ | 6.00 | |||||
|
| |||||||
The total fair value of restricted stock units vested during the years ended December 31, 2013, 2012 and 2010 was $1.4 million, $1.6 million and zero, respectively. The Company measures compensation expense for restricted stock units at fair value on the grant date and recognizes the expense over the expected vesting period. The fair value for restricted stock units is based on the closing price of the Company’s common stock on the grant date. Unvested restricted stock awards are subject to repurchase at no cost to the Company.
Stock-Based Compensation
The Company applies the accounting guidance for stock compensation, which establishes accounting for share-based payment awards made to employees and directors, including employee stock options and employee stock purchases. Under this guidance, stock-based compensation cost is measured at the grant date based on the calculated fair value of the award, and is recognized as an expense on a straight-line basis over the employee’s requisite service period, generally the vesting period of the award.
The following table summarizes stock-based compensation related to stock options, restricted stock awards, restricted stock unit, and employee stock purchases (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Research and development | $ | 1,538 | $ | 1,801 | $ | 1,331 | ||||||
General and administrative | 2,059 | 1,982 | 1,738 | |||||||||
|
|
|
|
|
| |||||||
Stock-based compensation included in operating expenses | $ | 3,597 | $ | 3,783 | $ | 3,069 | ||||||
|
|
|
|
|
| |||||||
The Company uses the Black-Scholes option pricing model to determine the fair value of stock options and employee stock purchase plan shares. The key input assumptions used to estimate fair value of these awards
104
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
include the exercise price of the award, the expected option term, the expected volatility of the Company’s stock over the option’s expected term, the risk-free interest rate over the option’s expected term, and the Company’s expected dividend yield, if any.
The fair value of share-based payments was estimated on the date of grant using the Black-Scholes option pricing model based on the following weighted average assumptions:
| Year Ended December 31, 2013 | Year Ended December 31, 2012 | Year Ended December 31, 2011 | ||||||||||||||||||||||
| Employee Stock Options | ESPP | Employee Stock Options | ESPP | Employee Stock Options | ESPP | |||||||||||||||||||
Risk-free interest rate | 1.1 | % | 0.2 | % | 1.1 | % | 0.2 | % | 2.4 | % | 0.3 | % | ||||||||||||
Volatility | 73.2 | % | 74.6 | % | 71.1 | % | 72.0 | % | 72.0 | % | 72.0 | % | ||||||||||||
Expected term in years | 6.20 | 1.25 | 6.13 | 1.25 | 6.10 | 1.25 | ||||||||||||||||||
Expected dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % | 0.0 | % | ||||||||||||
The risk-free interest rate that the Company uses in the option pricing model is based on the U.S. Treasury zero-coupon issues with remaining terms similar to the expected terms of the options. The Company does not anticipate paying dividends in the foreseeable future and therefore uses an expected dividend yield of zero in the option pricing model. The Company is required to estimate forfeitures at the time of grant and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. Historical data is used to estimate pre-vesting option forfeitures and record stock-based compensation expense only on those awards that are expected to vest.
The Company uses its own historical exercise activity and extrapolates the life cycle of options outstanding to arrive at its estimated expected term for new option grants. The Company uses its own volatility history based on its stock’s trading history for the period subsequent to the Company’s initial public offering in April 2004. The Company measures compensation expense for awards of restricted stock and restricted stock units at fair value on the date of grant and recognizes the expense over the expected vesting period. The fair value for restricted stock and restricted stock unit awards is based on the closing price of the Company’s common stock on the date of grant.
As of December 31, 2013, there was $3.2 million of unrecognized compensation cost related to unvested stock options, which is expected to be recognized over a weighted-average period of 2.54 years and $149,000 of unrecognized compensation cost related to unvested restricted stock units, which is expected to be recognized over a weighted-average period of 1.18 years.
Non-employee Stock-Based Compensation
The Company records stock option grants to non-employees at their fair value on the measurement date. The measurement of stock-based compensation is subject to adjustment as the underlying equity instruments vest.
There were no stock option grants to non-employees in the years ended December 31, 2013, 2012 or 2011. When terminating, if employees continue to provide service to the Company as consultants and their grants are permitted to continue to vest, the expense associated with the continued vesting of the related stock options is classified as non-employee stock compensation expense after the status change.
In connection with services rendered by non-employees, the Company recorded stock-based compensation expense of $104,000, $56,000, and $18,000 in 2013, 2012 and 2011, respectively.
105
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
ESPP
In January 2004, the Board of Directors adopted the ESPP, which was approved by the stockholders in February 2004. Under the ESPP, statutory employees may purchase common stock of the Company up to a specified maximum amount through payroll deductions. The stock is purchased semi-annually at a price equal to 85% of the fair market value at certain plan-defined dates. The Company issued 14,985, 15,010 and 18,822 shares of common stock during 2013, 2012 and 2011, respectively, pursuant to the ESPP at an average price of $3.66, $4.32 and $6.66 per share, in 2013, 2012 and 2011, respectively. At December 31, 2013 the Company had 189,896 shares of common stock reserved for issuance under the ESPP.
Note 14 — Income Taxes
The Company accounts for income taxes under the liability method. Under this method, deferred tax assets and liabilities are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce the deferred tax assets to the amounts expected to be realized. The Company did not record an income tax provision in the years ended December 31, 2013, 2012, or 2011 because the Company had a net taxable loss in the period.
For financial statement purposes, income before taxes includes the following components (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
United States | $ | (33,717 | ) | $ | (40,370 | ) | $ | (47,860 | ) | |||
Foreign | — | — | — | |||||||||
|
|
|
|
|
| |||||||
Total | $ | (33,717 | ) | $ | (40,370 | ) | $ | (47,860 | ) | |||
|
|
|
|
|
| |||||||
The Company recorded the following income tax provision as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Current: | ||||||||||||
Federal | $ | — | $ | — | $ | — | ||||||
State | — | — | — | |||||||||
|
|
|
|
|
| |||||||
Total | $ | — | $ | — | $ | — | ||||||
|
|
|
|
|
| |||||||
Deferred: | ||||||||||||
Federal | $ | — | $ | — | $ | — | ||||||
State | — | — | — | |||||||||
|
|
|
|
|
| |||||||
Total | $ | — | $ | — | $ | — | ||||||
|
|
|
|
|
| |||||||
106
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Deferred income taxes reflect the net tax effect of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The significant components of the Company’s deferred tax assets and liabilities were as follows (in thousands):
| As of December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Deferred tax assets: | ||||||||||||
Depreciation and amortization | $ | 918 | $ | 1,024 | $ | 997 | ||||||
Capitalized R&D | 20,702 | 4,932 | 6,582 | |||||||||
Reserves and accruals | 4,946 | 5,101 | 3,524 | |||||||||
Net operating losses | 144,254 | 153,193 | 141,226 | |||||||||
Tax credits | 33,043 | 25,943 | 16,778 | |||||||||
|
|
|
|
|
| |||||||
Total deferred tax assets | 203,863 | 190,193 | 169,107 | |||||||||
Less: Valuation allowance | (203,863 | ) | (190,193 | ) | (169,107 | ) | ||||||
|
|
|
|
|
| |||||||
Net deferred tax assets | $ | — | $ | — | $ | — | ||||||
|
|
|
|
|
| |||||||
Based upon the weight of available evidence, which includes the Company’s historical operating performance, reported cumulative net losses since inception and difficulty in accurately forecasting the Company’s future results, the Company maintained a full valuation allowance on the net deferred tax assets as of December 31, 2013, 2012 and 2011. The valuation allowance was determined pursuant to the accounting guidance for income taxes, which requires an assessment of both positive and negative evidence when determining whether it is more likely than not that deferred tax assets are recoverable. The Company intends to maintain a full valuation allowance on the U.S. deferred tax assets until sufficient positive evidence exists to support reversal of the valuation allowance. The valuation allowance increased by $13.7 million in 2013, $21.1 million in 2012, and $18.5 million in 2011.
As a result of certain realization requirements of accounting guidance for stock compensation, the table of deferred tax assets and liabilities shown above does not include certain deferred tax assets at December 31, 2013, 2012 and 2011 that arose directly from tax deductions related to equity compensation in excess of compensation recognized for financial reporting. Equity will be increased by $1.8 million if and when such benefits are ultimately realized and reduce taxes payable.
The following are the Company’s valuation and qualifying accounts (in thousands):
| Balance at Beginning of Period | Charged to Expenses | Charged to Other Accounts | Deductions | Balance at End of Period | ||||||||||||||||
Year Ended December 31, 2011: | ||||||||||||||||||||
Deferred tax valuation allowance | $ | 150,635 | $ | 18,472 | — | — | $ | 169,107 | ||||||||||||
Year Ended December 31, 2012: | ||||||||||||||||||||
Deferred tax valuation allowance | $ | 169,107 | $ | 21,086 | — | — | $ | 190,193 | ||||||||||||
Year Ended December 31, 2013: | ||||||||||||||||||||
Deferred tax valuation allowance | $ | 190,193 | $ | 13,670 | — | — | $ | 203,863 | ||||||||||||
107
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
The following is a reconciliation of the statutory federal income tax rate to the Company’s effective tax rate:
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Tax at federal statutory tax rate | (34 | )% | (34 | )% | (34 | )% | ||||||
State income tax, net of federal tax benefit | (4 | )% | (6 | )% | (6 | )% | ||||||
State Apportionment | 7 | % | 0 | % | 0 | % | ||||||
Tax credits (net) | (14 | )% | (19 | )% | (1 | )% | ||||||
Deferred tax assets (utilized) not benefited | 41 | % | 56 | % | 39 | % | ||||||
Stock-based compensation | 2 | % | 0 | % | 1 | % | ||||||
NOL Expiration | 1 | % | 2 | % | 0 | % | ||||||
Other | 1 | % | 1 | % | 1 | % | ||||||
|
|
|
|
|
| |||||||
Total | 0 | % | 0 | % | 0 | % | ||||||
|
|
|
|
|
| |||||||
The Company had federal net operating loss carryforwards of approximately $375.2 million and apportioned state net operating loss carryforwards of approximately $286.0 million before federal benefit at December 31, 2013. If not utilized, the federal and state operating loss carryforwards will begin to expire in various amounts beginning 2020 and 2014, respectively. The net operating loss carryforwards include deductions for stock options.
The Company had general business credit of approximately $29.1 million and $13.6 million for federal and California state income tax purposes, respectively, at December 31, 2013. Amounts are comprised of Research and Development Credits and Orphan Drug Credits. If not utilized, the federal carryforwards will expire in various amounts beginning in 2021. The California state credit can be carried forward indefinitely. Since its filing of its 2011 tax return, the Company has claimed the orphan drug credit. For qualifying expenses, the orphan drug credit offers an increased benefit relative to the research and development credit taken in years prior.
As required by California state law, the Company apportions income to California based on a “market-based” sourcing approach. Accordingly, the Company’s California apportionment formula is sensitive to changes in the source of the Company’s mix of revenue. As a result of agreements in place in 2013, the Company adjusted deferred tax assets to reflect these changes.
In general, under Section 382 of the Internal Revenue Code (“Section 382”), a corporation that undergoes an ‘ownership change’ is subject to limitations on its ability to utilize its pre-change net operating losses and tax credits to offset future taxable income. The Company has performed a section 382 analysis for the year ended December 31, 2013 and has not experienced an ownership change since 2006. A portion of the Company’s existing net operating losses and tax credits are subject to limitations arising from previous ownership changes. Future changes in the Company’s stock ownership, some of which are outside of our control, could result in an ownership change under Section 382 and result in additional limitations.
Section 59(e) of the Internal Revenue Code allows a Company to capitalize R&D expenses. The Company elected to capitalize R&D expenses on its 2012 tax returns after completing a reverse stock split in the second quarter of 2013. The Company anticipates doing the same in its 2013 tax return.
The Company follows the accounting guidance that prescribes a comprehensive model for how companies should recognize, measure, present, and disclose in their financial statements uncertain tax positions taken or expected to be taken on a tax return. Tax positions are initially recognized in the financial statements when it is more likely than not that the position will be sustained upon examination by the tax authorities. Such tax
108
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
positions are initially and subsequently measured as the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate settlement with the tax authority assuming full knowledge of the position and relevant facts.
The Company historically files income tax returns for federal and California purposes. In general, the statute of limitations for tax liabilities for all years remains open for the purpose of adjusting the amounts of the losses and credits carried forward from those years.
The following is a tabular reconciliation of the total amounts of unrecognized tax benefits (“UTBs”) (in thousands):
| Federal and State Tax | Federal Tax Benefit of State Income Tax UTBs | Unrecognized Income Tax Benefits - Net of Federal Benefit of State UTBs | ||||||||||
Unrecognized tax benefits balance at December 31, 2011 | $ | 5,491 | $ | 1,174 | $ | 4,317 | ||||||
Addition for tax positions of prior years | 547 | — | 547 | |||||||||
Reduction for tax positions of prior years | (1,059 | ) | (16 | ) | (1,043 | ) | ||||||
Addition for tax positions related to the current year | 361 | — | 361 | |||||||||
|
|
|
|
|
| |||||||
Unrecognized tax benefits balance at December 31, 2012 | 5,340 | 1,158 | 4,182 | |||||||||
Addition for tax positions of prior years | 24 | 8 | 16 | |||||||||
Addition for tax positions related to the current year | 807 | 43 | 764 | |||||||||
|
|
|
|
|
| |||||||
Unrecognized tax benefits balance at December 31, 2013 | $ | 6,171 | $ | 1,209 | $ | 4,962 | ||||||
|
|
|
|
|
| |||||||
Included in the balance of unrecognized tax benefits as of December 31, 2013, 2012 and 2011 are $5.0 million, $4.2 million and $4.3 million of tax benefits, respectively, that, if recognized, would result in adjustments to other tax accounts, primarily deferred taxes.
The Company recognizes interest accrued related to unrecognized tax benefits and penalties as income tax expense. Related to the unrecognized tax benefits noted above, the Company did not accrue any penalties or interest during 2013, 2012 or 2011. The Company does not expect its unrecognized tax benefit to change materially over the next twelve months.
Note 15 — Interest and Other, Net
Components of Interest and Other, Net were as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2013 | 2012 | 2011 | ||||||||||
Warrant expense | $ | — | $ | — | $ | — | ||||||
Interest income and other income | 98 | 89 | 143 | |||||||||
Interest expense and other expense | 79 | (2 | ) | (39 | ) | |||||||
|
|
|
|
|
| |||||||
Interest and Other, net | $ | 177 | $ | 87 | $ | 104 | ||||||
|
|
|
|
|
| |||||||
Warrant expense for the period from inception to December 31, 2013 was related to the change in the fair value of the warrant liability that was recorded in connection with the Company’s registered direct equity offering in May 2009.
109
Table of Contents
CYTOKINETICS, INCORPORATED
NOTES TO FINANCIAL STATEMENTS — (Continued)
Interest income and other income primarily consisted of interest income generated from the Company’s cash, cash equivalents and investments.
Interest expense and other expense in 2013 consisted solely of net gains realized upon disposal of equipment. Interest expense and other expense in 2011 primarily consisted of interest expense on borrowings under equipment financing lines.
Note 16 — Quarterly Financial Data (Unaudited)
Quarterly results were as follows (in thousands, except per share data):
| First Quarter | Second Quarter | Third Quarter | Fourth Quarter | |||||||||||||
2013 | ||||||||||||||||
Total revenues | $ | 821 | $ | 1,009 | $ | 4,469 | $ | 24,349 | ||||||||
Net income (loss) | (12,619 | ) | (15,041 | ) | (12,588 | ) | 6,531 | |||||||||
Net income (loss) allocable to common stockholders | (12,619 | ) | (15,041 | ) | (12,588 | ) | 6,531 | |||||||||
Net income (loss) per share allocable to common stockholders — basic | $ | (0.53 | ) | $ | (0.58 | ) | $ | (0.43 | ) | $ | 0.22 | |||||
Net income (loss) per share allocable to common stockholders — diluted | $ | (0.53 | ) | $ | (0.58 | ) | $ | (0.43 | ) | $ | 0.21 | |||||
2012 | ||||||||||||||||
Total revenues | $ | 1,820 | $ | 1,841 | $ | 1,714 | $ | 2,184 | ||||||||
Net loss | (9,928 | ) | (8,943 | ) | (10,044 | ) | (11,455 | ) | ||||||||
Net loss allocable to common stockholders | (9,928 | ) | (10,250 | ) | (10,044 | ) | (11,455 | ) | ||||||||
Net loss per share allocable to common stockholders — basic and diluted | $ | (0.78 | ) | $ | (0.76 | ) | $ | (0.45 | ) | $ | (0.48 | ) | ||||
Note 17 — Subsequent Events
In January 2014, the Company sold 364,103 shares of common stock through MLV for net proceeds of approximately $2.4 million. As of January 8, 2014, the Company had sold 2,397,278 shares, the maximum allowable under the MLV Agreement and therefore, no shares remain available to the Company for sale through MLV.
On February 25, 2014, the Company closed an underwritten public offering for the issuance and sale of 5,031,250 shares of its common stock. The gross public offering proceeds were approximately $40.3 million. The net proceeds from the sale of the shares were approximately $37.4 million, after deducting the underwriting discount and estimated offering expenses.
110
Table of Contents
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures. Our management evaluated, with the participation of our Chief Executive Officer and our Chief Financial Officer, the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that the Company’s disclosure controls and procedures are effective.
Management’s Report on Internal Control over Financial Reporting. Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act). Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2013. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework. Our management has concluded that, as of December 31, 2013, our internal control over financial reporting is effective based on these criteria.
Our independent registered public accounting firm, PricewaterhouseCoopers LLP, has audited the effectiveness of our internal control over financial reporting as of December 31, 2013, as stated in their report, which is included herein.
Changes in Internal Control over Financial Reporting. There was no change in our internal control over financial reporting that occurred during the quarter ended December 31, 2013 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations on Effectiveness of Controls. Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal controls, will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within Cytokinetics have been detected.
None.
111
Table of Contents
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The information regarding our directors and executive officers, our director nominating process and our audit committee is incorporated by reference from our definitive Proxy Statement for our 2014 Annual Meeting of Stockholders, where it appears under the headings “Board of Directors” and “Executive Officers.”
Section 16(a) Beneficial Ownership Reporting Compliance
The information regarding our Section 16 beneficial ownership reporting compliance is incorporated by reference from our definitive Proxy Statement described above, where it appears under the headings “Section 16(a) Beneficial Ownership Reporting Compliance.”
Code of Ethics
We have adopted a Code of Ethics that applies to all directors, officers and employees of the Company. We publicize the Code of Ethics through posting the policy on our website, www.cytokinetics.com. We will disclose on our website any waivers of, or amendments to, our Code of Ethics within four business days following the date of such amendment or waiver.
Item 11. Executive Compensation
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above, where it appears under the headings “Executive Compensation” and “Compensation Committee Interlocks and Insider Participation.”
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item regarding security ownership of certain beneficial owners and management is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above, where it appears under the heading “Security Ownership of Certain Beneficial Owners and Management.”
The following table summarizes the securities authorized for issuance under our equity compensation plans as of December 31, 2013:
Plan Category | Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | Weighted Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||||||
Equity compensation plans approved by stockholders | 1,622,205 | $ | 19.49 | 2,351,725 | (1) | |||||||
Equity compensation plans not approved by stockholders | — | — | — | |||||||||
|
|
|
|
|
| |||||||
Total | 1,622,205 | $ | 19.49 | 2,351,725 | ||||||||
|
|
|
|
|
| |||||||
| (1) | Includes 226,321 shares of common stock reserved for issuance under the Employee Stock Purchase Plan. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the headings “Certain Business Relationships and Related Party Transactions” and “Board of Directors.”
Item 14. Principal Accounting Fees and Services
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above, where it appears under the heading “Principal Accountant Fees and Services.”
112
Table of Contents
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) The following documents are filed as part of this Form 10-K:
(1) Financial Statements (included in Part II of this report):
| • | Report of Independent Registered Public Accounting Firm |
| • | Balance Sheets |
| • | Statements of Comprehensive Loss |
| • | Statements of Stockholders’ Equity (Deficit) |
| • | Statements of Cash Flows |
| • | Notes to Financial Statements |
(2) Financial Statement Schedules:
None — All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
(3) Exhibits:
Exhibit | Description | |
| 3.1 | Amended and Restated Certificate of Incorporation.(1) | |
| 3.2 | Certificate of Amendment of Amended and Restated Certificate of Incorporation.(18) | |
| 3.3 | Certificate of Amendment of Amended and Restated Certificate of Incorporation.(21) | |
| 3.4 | Amended and Restated Bylaws.(2) | |
| 3.5 | Certificate of Designation of Preferences, Rights and Limitations of Series A Convertible Preferred Stock.(3) | |
| 3.6 | Certificate of Designation of Preferences, Rights and Limitations of Series B Convertible Preferred Stock.(17) | |
| 4.1 | Specimen Common Stock Certificate.(4) | |
| 4.2 | Registration Rights Agreement, dated as of December 29, 2006, by and between the Company and Amgen Inc.(5) | |
| 4.3 | Form of Warrant to Purchase Common Stock of Cytokinetics, Inc.(3) | |
| 4.4 | Form of Common Stock Warrant Agreement(15) | |
| 4.5 | Form of Preferred Stock Warrant Agreement(15) | |
| 4.6 | Form of Warrant(10) | |
| 4.7 | Form of Common Stock Warrant and Warrant Certificate(22) | |
| 4.8 | Form of Preferred Stock Warrant and Warrant Certificate(22) | |
| 10.1 | 1997 Stock Option/Stock Issuance Plan.(2) | |
| 10.2 | 2004 Equity Incentive Plan, as amended.(14) | |
| 10.3 | 2004 Employee Stock Purchase Plan.(14) | |
| 10.4 | Build-to-Suit Lease, dated May 27, 1997, by and between Britannia Pointe Grand Limited Partnership and Metaxen, LLC.(2) | |
113
Table of Contents
Exhibit | Description | |
| 10.5 | First Amendment to Lease, dated April 13, 1998, by and between Britannia Pointe Grand Limited Partnership and Metaxen, LLC.(2) | |
| 10.6 | Sublease Agreement, dated May 1, 1998, by and between the Company and Metaxen, LLC.(2) | |
| 10.7 | Sublease Agreement, dated March 1, 1999, by and between Metaxen, LLC and Exelixis Pharmaceuticals, Inc.(2) | |
| 10.8 | Assignment and Assumption Agreement and Consent, dated July 11, 1999, by and among Exelixis Pharmaceuticals, Metaxen, LLC, Xenova Group PLC and Britannia Pointe Grande Limited Partnership.(2) | |
| 10.9 | Second Amendment to Lease, dated July 11, 1999, by and between Britannia Pointe Grand Limited Partnership and Exelixis Pharmaceuticals, Inc.(2) | |
| 10.10 | First Amendment to Sublease Agreement, dated July 20, 1999, by and between the Company and Metaxen, LLC.(2) | |
| 10.11 | Agreement and Consent, dated July 20, 1999, by and among Exelixis Pharmaceuticals, Inc., the Company and Britannia Pointe Grand Limited Partnership.(2) | |
| 10.12 | Amendment to Agreement and Consent, dated July 31, 2000, by and between the Company, Exelixis, Inc., and Britannia Pointe Grande Limited Partnership.(2) | |
| 10.13 | Assignment and Assumption of Lease, dated September 28, 2000, by and between the Company and Exelixis, Inc.(2) | |
| 10.14 | Sublease Agreement, dated September 28, 2000, by and between the Company and Exelixis, Inc.(2) | |
| *10.15 | Collaboration and Option Agreement, dated as of December 29, 2006, by and between the Company and Amgen Inc.(11) | |
| 10.16 | Common Stock Purchase Agreement, dated October 15, 2007, by and between the Company and Kingsbridge Capital Limited.(8) | |
| 10.17 | Form of Indemnification Agreement between the Company and each of its directors and executive officers.(9) | |
| *10.18 | Scientific Advisory Board Consulting Agreement, dated April 1, 2008, by and between the Company and James. H. Sabry.(12) | |
| 10.19 | Amended and Restated Executive Employment Agreement, dated May 21, 2007, by and between the Company and Robert Blum.(9) | |
| 10.20 | Form of Executive Employment Agreement between the Company and its executive officers.(9) | |
| *10.21 | Amendment No. 1, dated June 17, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.22 | Amendment No. 2, dated September 30, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.23 | Amendment No. 3, dated October 31, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.24 | Amendment No. 4, dated February 20, 2009, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| 10.25 | Form of Amendment No. 1 to Amended and Restated Executive Employment Agreements.(13) | |
| 10.26 | Third Amendment to Lease, dated December 10, 2010, by and between the Company and Britannia Pointe Grand Limited Partnership.(16) | |
114
Table of Contents
Exhibit | Description | |
| *10.27 | Amendment No. 5, dated November 1, 2010, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(16) | |
| 10.28 | Securities Purchase Agreement, dated April 18, 2011, between the Company and Deerfield Private Design Fund II, L.P., Deerfield Private Design International II, L.P., Deerfield Special Situations Fund, L.P., and Deerfield Special Situations Fund International Limited.(3) | |
| 10.29 | At the Market Issuance Sales Agreement, dated June 10, 2011, between the Company and McNicoll, Lewis & Vlak LLC.(6) | |
| *10.30 | Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(24) | |
| *10.31 | Amendment No. 1, dated May 1, 2012, to Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(19) | |
| *10.32 | Amendment No. 2, dated October 30, 2012 to Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(20) | |
| 10.33 | Compensation Information for the Company’s Named Executive Officers.(23) | |
| 10.34 | Form of Option Agreement.(20) | |
| 10.35 | Form of Restricted Stock Unit Award Agreement.(20) | |
| 10.36 | Common Stock Purchase Agreement dated June 11, 2013, by and between the Company and Amgen, Inc.(25) | |
| *10.37 | Amendment No. 6, dated June 11, 2013, to the Collaboration and Option Agreement by and between the Company and Amgen, Inc.(14) | |
| *10.38 | Collaboration and License Agreement, dated June 21, 2013, by and between the Company and Astellas Pharma, Inc.(14) | |
| 10.39 | Form of Executive Employment Agreement between the Company and its executive officers. | |
| 23.1 | Consent of Independent registered public accounting firm. | |
| 24.1 | Power of Attorney (included in the signature page to this report). | |
| 31.1 | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certifications of the Principal Executive Officer and the Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (18 U.S.C. Section 1350). | |
| 101.INS | XBRL Instance Document. | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | |
| (1) | Incorporated by reference from our registration statement on Form S-3, registration number 333-174869, filed with the Securities and Exchange Commission on June 13, 2011. |
115
Table of Contents
| (2) | Incorporated by reference from our registration statement on Form S-1, registration number 333-112261, declared effective by the Securities and Exchange Commission on April 29, 2004. |
| (3) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on April 18, 2011. |
| (4) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Security and Exchange Commission on May 9, 2007. |
| (5) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 3, 2007. |
| (6) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 13, 2011. |
| (7) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 20, 2006. |
| (8) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on October 15, 2007. |
| (9) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 5, 2008. |
| (10) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 6, 2012. |
| (11) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 12, 2007. |
| (12) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on April 2, 2008. |
| (13) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 12, 2009. |
| (14) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 7, 2013. |
| (15) | Incorporated by reference from our registration statement on Form S-3, registration number 333-178189, filed with the Securities and Exchange Commission on November 25, 2011. |
| (16) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 11, 2011. |
| (17) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 20, 2012. |
| (18) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 4, 2011. |
| (19) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on May 4, 2012. |
| (20) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 15, 2013. |
| (21) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 25, 2013. |
| (22) | Incorporated by reference from our registration statement on Form S-3, registration number 333-192125, filed with the Securities and Exchange Commission on November 6, 2013. |
116
Table of Contents
| (23) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on February 27, 2014. |
| (24) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 13, 2012. |
| (25) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 12, 2013. |
| * | Pursuant to a request for confidential treatment, portions of this Exhibit have been redacted from the publicly filed document and have been furnished separately to the Securities and Exchange Commission as required by Rule 406 under the Securities Act or Rule 24b-2 under the Exchange Act, as applicable. |
(b) Exhibits |
The exhibits listed under Item 15(a)(3) hereof are filed as part of this Form 10-K, other than Exhibit 32.1 which shall be deemed furnished.
(c) Financial Statement Schedules |
None — All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
117
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| CYTOKINETICS, INCORPORATED | ||
By: | /s/ ROBERT I. BLUM | |
Robert I. Blum President, Chief Executive Officer and Director | ||
Dated: March 7, 2014
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Robert I. Blum and Sharon A. Barbari, and each of them, his true and lawful attorneys-in-fact, each with full power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities and Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ ROBERT I. BLUM Robert I. Blum | President, Chief Executive Officer and Director (Principal Executive Officer) | March 7, 2014 | ||
/s/ SHARON A. BARBARI Sharon A. Barbari | Executive Vice President, Finance and Chief Financial Officer (Principal Financial and Accounting Executive) | March 7, 2014 | ||
/s/ L. PATRICK GAGE, PH.D. L. Patrick Gage, Ph.D. | Chairman of the Board of Directors | March 7, 2014 | ||
/s/ SANTO J. COSTA Santo J. Costa | Director | March 7, 2014 | ||
/s/ DENISE M. GILBERT, PH.D. Denise M. Gilbert, Ph.D. | Director | March 7, 2014 | ||
/s/ JOHN T. HENDERSON, M.B. CH.B. John T. Henderson, M.B. Ch.B. | Director | March 7, 2014 | ||
/s/ B. LYNNE PARSHALL, ESQ. B. Lynne Parshall, Esq. | Director | March 7, 2014 | ||
/s/ SANDFORD D. SMITH Sandford D. Smith | Director | March 7, 2014 | ||
/s/ WENDELL WIERENGA, PH.D. Wendell Wierenga, Ph.D. | Director | March 7, 2014 | ||
118
Table of Contents
Exhibit Number | Description | |
| 3.1 | Amended and Restated Certificate of Incorporation.(1) | |
| 3.2 | Certificate of Amendment of Amended and Restated Certificate of Incorporation.(18) | |
| 3.3 | Certificate of Amendment of Amended and Restated Certificate of Incorporation.(21) | |
| 3.4 | Amended and Restated Bylaws.(2) | |
| 3.5 | Certificate of Designation of Preferences, Rights and Limitations of Series A Convertible Preferred Stock.(3) | |
| 3.6 | Certificate of Designation of Preferences, Rights and Limitations of Series B Convertible Preferred Stock.(17) | |
| 4.1 | Specimen Common Stock Certificate.(4) | |
| 4.2 | Registration Rights Agreement, dated as of December 29, 2006, by and between the Company and Amgen Inc.(5) | |
| 4.3 | Form of Warrant to Purchase Common Stock of Cytokinetics, Inc.(3) | |
| 4.4 | Form of Common Stock Warrant Agreement(15) | |
| 4.5 | Form of Preferred Stock Warrant Agreement(15) | |
| 4.6 | Form of Warrant(10) | |
| 4.7 | Form of Common Stock Warrant and Warrant Certificate(22) | |
| 4.8 | Form of Preferred Stock Warrant and Warrant Certificate(22) | |
| 10.1 | 1997 Stock Option/Stock Issuance Plan.(2) | |
| 10.2 | 2004 Equity Incentive Plan, as amended.(14) | |
| 10.3 | 2004 Employee Stock Purchase Plan.(14) | |
| 10.4 | Build-to-Suit Lease, dated May 27, 1997, by and between Britannia Pointe Grand Limited Partnership and Metaxen, LLC.(2) | |
| 10.5 | First Amendment to Lease, dated April 13, 1998, by and between Britannia Pointe Grand Limited Partnership and Metaxen, LLC.(2) | |
| 10.6 | Sublease Agreement, dated May 1, 1998, by and between the Company and Metaxen, LLC.(2) | |
| 10.7 | Sublease Agreement, dated March 1, 1999, by and between Metaxen, LLC and Exelixis Pharmaceuticals, Inc.(2) | |
| 10.8 | Assignment and Assumption Agreement and Consent, dated July 11, 1999, by and among Exelixis Pharmaceuticals, Metaxen, LLC, Xenova Group PLC and Britannia Pointe Grande Limited Partnership.(2) | |
| 10.9 | Second Amendment to Lease, dated July 11, 1999, by and between Britannia Pointe Grand Limited Partnership and Exelixis Pharmaceuticals, Inc.(2) | |
| 10.10 | First Amendment to Sublease Agreement, dated July 20, 1999, by and between the Company and Metaxen, LLC.(2) | |
| 10.11 | Agreement and Consent, dated July 20, 1999, by and among Exelixis Pharmaceuticals, Inc., the Company and Britannia Pointe Grand Limited Partnership.(2) | |
| 10.12 | Amendment to Agreement and Consent, dated July 31, 2000, by and between the Company, Exelixis, Inc., and Britannia Pointe Grande Limited Partnership.(2) | |
| 10.13 | Assignment and Assumption of Lease, dated September 28, 2000, by and between the Company and Exelixis, Inc.(2) | |
119
Table of Contents
Exhibit Number | Description | |
| 10.14 | Sublease Agreement, dated September 28, 2000, by and between the Company and Exelixis, Inc.(2) | |
| *10.15 | Collaboration and Option Agreement, dated as of December 29, 2006, by and between the Company and Amgen Inc.(11) | |
| 10.16 | Common Stock Purchase Agreement, dated October 15, 2007, by and between the Company and Kingsbridge Capital Limited.(8) | |
| 10.17 | Form of Indemnification Agreement between the Company and each of its directors and executive officers.(9) | |
| *10.18 | Scientific Advisory Board Consulting Agreement, dated April 1, 2008, by and between the Company and James. H. Sabry.(12) | |
| 10.19 | Amended and Restated Executive Employment Agreement, dated May 21, 2007, by and between the Company and Robert Blum.(9) | |
| 10.20 | Form of Executive Employment Agreement between the Company and its executive officers.(9) | |
| *10.21 | Amendment No. 1, dated June 17, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.22 | Amendment No. 2, dated September 30, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.23 | Amendment No. 3, dated October 31, 2008, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| *10.24 | Amendment No. 4, dated February 20, 2009, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(13) | |
| 10.25 | Form of Amendment No. 1 to Amended and Restated Executive Employment Agreements.(13) | |
| 10.26 | Third Amendment to Lease, dated December 10, 2010, by and between the Company and Britannia Pointe Grand Limited Partnership.(16) | |
| *10.27 | Amendment No. 5, dated November 1, 2010, to the Collaboration and Option Agreement by and between the Company and Amgen Inc.(16) | |
| 10.28 | Securities Purchase Agreement, dated April 18, 2011, between the Company and Deerfield Private Design Fund II, L.P., Deerfield Private Design International II, L.P., Deerfield Special Situations Fund, L.P., and Deerfield Special Situations Fund International Limited.(3) | |
| 10.29 | At the Market Issuance Sales Agreement, dated June 10, 2011, between the Company and McNicoll, Lewis & Vlak LLC.(6) | |
| *10.30 | Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(24) | |
| *10.31 | Amendment No. 1, dated May 1, 2012, to Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(19) | |
| *10.32 | Amendment No. 2, dated October 30, 2012 to Consulting Agreement between the Company and David J. Morgans, dated November 1, 2011.(20) | |
| 10.33 | Compensation Information for the Company’s Named Executive Officers.(23) | |
| 10.34 | Form of Option Agreement.(20) | |
| 10.35 | Form of Restricted Stock Unit Award Agreement.(20) | |
120
Table of Contents
Exhibit Number | Description | |
| 10.36 | Common Stock Purchase Agreement dated June 11, 2013, by and between the Company and Amgen, Inc.(25) | |
| *10.37 | Amendment No. 6, dated June 11, 2013, to the Collaboration and Option Agreement by and between the Company and Amgen, Inc.(14) | |
| *10.38 | Collaboration and License Agreement, dated June 21, 2013, by and between the Company and Astellas Pharma, Inc.(14) | |
| 10.39 | Form of Executive Employment Agreement between the Company and its executive officers. | |
| 23.1 | Consent of Independent registered public accounting firm. | |
| 24.1 | Power of Attorney (included in the signature page to this report). | |
| 31.1 | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certifications of the Principal Executive Officer and the Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (18 U.S.C. Section 1350). | |
| 101.INS | XBRL Instance Document. | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | |
| (1) | Incorporated by reference from our registration statement on Form S-3, registration number 333-174869, filed with the Securities and Exchange Commission on June 13, 2011. |
| (2) | Incorporated by reference from our registration statement on Form S-1, registration number 333-112261, declared effective by the Securities and Exchange Commission on April 29, 2004. |
| (3) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on April 18, 2011. |
| (4) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Security and Exchange Commission on May 9, 2007. |
| (5) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 3, 2007. |
| (6) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 13, 2011. |
| (7) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 20, 2006. |
| (8) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on October 15, 2007. |
| (9) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 5, 2008 |
121
Table of Contents
| (10) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 6, 2012. |
| (11) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 12, 2007. |
| (12) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on April 2, 2008. |
| (13) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 12, 2009. |
| (14) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 7, 2013. |
| (15) | Incorporated by reference from our registration statement on Form S-3, registration number 333-178189, filed with the Securities and Exchange Commission on November 25, 2011. |
| (16) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 11, 2011. |
| (17) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 20, 2012. |
| (18) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on August 4, 2011. |
| (19) | Incorporated by reference from our Quarterly Report on Form 10-Q, filed with the Securities and Exchange Commission on May 4, 2012. |
| (20) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 15, 2013. |
| (21) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 25, 2013. |
| (22) | Incorporated by reference from our registration statement on Form S-3, registration number 333-192125, filed with the Securities and Exchange Commission on November 6, 2013. |
| (23) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on February 27, 2014. |
| (24) | Incorporated by reference from our Annual Report on Form 10-K, filed with the Securities and Exchange Commission on March 13, 2012. |
| (25) | Incorporated by reference from our Current Report on Form 8-K, filed with the Securities and Exchange Commission on June 12, 2013. |
| * | Pursuant to a request for confidential treatment, portions of this Exhibit have been redacted from the publicly filed document and have been furnished separately to the Securities and Exchange Commission as required by Rule 406 under the Securities Act or Rule 24b-2 under the Exchange Act, as applicable. |
(b) Exhibits |
The exhibits listed under Item 15(a)(3) hereof are filed as part of this Form 10-K, other than Exhibit 32.1 which shall be deemed furnished.
122
Table of Contents
(c) Financial Statement Schedules |
None — All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
123