UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K/A
Amendment No. 1
| x | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2012
| ¨ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ______to _______
Commission File Number: 000-35737
NORTHWEST BIOTHERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or Other Jurisdiction of Incorporation or Organization) | 94-3306718 (I.R.S. Employer Identification No.) |
4800 Montgomery Lane, Suite 800, Bethesda, MD 20814
(Address of principal executive offices) (Zip Code)
(240) 497-9024
(Registrant's telephone number)
N/A
(Former Name, Former Address and Former Fiscal Year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.001 par value | The NASDAQ Capital Market | |
| Warrants to purchase Common Stock | The NASDAQ Capital Market |
Securities registered pursuant to section 12(g) of the Act:None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 (the "Exchange Act") during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of "large accelerated filer," "accelerated filer," and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer ¨ (Do not check if a smaller reporting company) | Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was $18.2 million on June 29, 2012
As of April 1, 2013 the registrant had 27,140,417 shares of Common Stock outstanding.
FORM 10-K/A
EXPLANATORY NOTE
The purpose of this Amendment No. 1 on Form 10-K/A (the “Amendment”) is to amend and restate Part III, Items 10 through 14 of our previously filed Annual Report on Form 10-K for the year ended December 31, 2012, filed with the Securities and Exchange Commission on April 8, 2013 (the “Original Filing”), to include information previously omitted in reliance on General Instruction G to Form 10-K, which provides that registrants may incorporate by reference certain information from a definitive proxy statement prepared in connection with the election of directors. We have determined to include such Part III information by amendment of the Original Filing rather than by incorporation by reference to a definitive proxy statement. Accordingly, Part III of the Original Filing is hereby amended and restated as set forth below.
Except as set forth above, we have not modified or updated disclosures presented in the Original Filing to reflect events or developments that have occurred after the date of the Original Filing. Among other things, forward-looking statements made in the Original Filing have not been revised to reflect events, results or developments that have occurred or facts that have become known to us after the date of the Original Filing (other than as discussed above), and such forward-looking statements should be read in their historical context. Accordingly, this Amendment should be read in conjunction with our filings made with the SEC subsequent to the filing of the Original Filing.
NORTHWEST BIOTHERAPEUTICS, INC.
FORM 10-K
TABLE OF CONTENTS
| PART I | ||
| Item 1. | Business | 3 |
| Item 1A. | Risk Factors | 19 |
| Item 1B. | Unresolved Staff Comments | 30 |
| Item 2. | Properties | 31 |
| Item 3. | Legal Proceedings | 31 |
| Item 4. | Mine Safety Disclosures | 31 |
| PART II | ||
| Item 5. | Market for the Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 31 |
| Item 6. | Selected Financial Data | 34 |
| Item 7. | Management's Discussion and Analysis of Financial Condition And Results of Operations | 34 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 38 |
| Item 8. | Financial Statements and Supplementary Data | 38 |
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosures | 38 |
| Item 9A. | Controls and Procedures | 38 |
| Item 9B. | Other Information | 39 |
| PART III | ||
| Item 10. | Directors, Executive Officers and Corporate Governance | 39 |
| Item 11. | Executive Compensation | 39 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 39 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | 39 |
| Item 14. | Principal Accountant Fees and Services | 39 |
| PART IV | ||
| Item 15. | Exhibits and Financial Statement Schedules | 40 |
| SIGNATURES | 44 | |
| 2 |
PART I
This Report on Form 10-K for Northwest Biotherapeutics, Inc. may contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934. Such forward-looking statements are characterized by future or conditional verbs such as "may," "will," "expect," "intend," "anticipate," believe," "estimate" and "continue" or similar words. You should read statements that contain these words carefully because they discuss future expectations and plans, which contain projections of future results of operations or financial condition or state other forward-looking information. Such statements are only predictions and our actual results may differ materially from those anticipated in these forward-looking statements. We believe that it is important to communicate future expectations to investors. However, there may be events in the future that we are not able to accurately predict or control. Factors that may cause such differences include, but are not limited to, those discussed under Item 1A. Risk Factors and elsewhere in this Form 10-K for the year ended December 31, 2012, as filed with the Securities and Exchange Commission, including the uncertainties associated with product development, the risk that products that appeared promising in early clinical trials do not demonstrate safety and efficacy in larger-scale clinical trials, the risk that we will not obtain approval to market our products, the risks associated with dependence upon key personnel and the need for additional financing. We do not assume any obligation to update forward-looking statements as circumstances change.
Unless the context otherwise requires, “Northwest Biotherapeutics,” the “company,” “we,” “us,” “our” and similar names refer to Northwest Biotherapeutics, Inc. DCVax® is a registered trademark of the Company
ITEM 1. BUSINESS.
Overview
We are a development stage biotechnology company focused on developing immunotherapy products to treat cancers more effectively than current treatments, without toxicities of the kind associated with chemotherapies, and, through a proprietary batch manufacturing process, on a cost-effective affordable basis. initially in both the United States and Europe (the two largest medical markets in the world).
We have developed a platform technology, DCVax, which uses activated dendritic cells to mobilize a patient's own immune system to attack their cancer. The DCVax technology is expected to be applicable to most cancers, and is embodied in several distinct product lines. One of the product lines (DCVax-L) is designed to cover all solid tumor cancers in which the tumors can be surgically removed. Another product line (DCVax-Direct) is designed for all solid tumor cancers which are considered inoperable and cannot be surgically removed. The broad applicability of DCVax to many cancers provides multiple opportunities for commercialization and partnering.
After more than a decade of pre-clinical and clinical development, the DCVax technology has reached late stage development for two different cancers (brain and prostate), with a Phase III clinical trial in glioblastoma multiforme, or GBM, brain cancer currently under way, and a Phase III clinical trial in prostate cancer which was previously cleared to proceed by the U.S. Food and Drug Administration, or FDA, which we anticipate will proceed when we secure a partner. We have also completed a small early stage trial in metastatic ovarian cancer, and we have received clearance from the FDA for early stage trials in multiple other diverse cancers.
In the Phase I/II trials which formed the foundation for reaching these Phase III trials, the clinical results with DCVax were striking. DCVax treatment delayed disease progression and extended survival by years, rather than weeks or months as is typical with cancer drugs. In addition, DCVax was non-toxic: no serious adverse events related to the treatment were seen. These clinical results (both the efficacy and the lack of toxicity) are consistent with a large and growing body of scientific literature and clinical experience, relating to the underlying biology involved.
As of March 31, 2013, our Phase III clinical trial in GBM is being conducted at 43 sites across the United States. We are also in the process of adding further US sites.
The Phase III GBM trial is also progressing in Europe. We have accelerated and strengthened our programs in Europe by partnering with large, prominent institutions, including the Fraunhofer IZI Institute in Germany and Kings College Hospital in the U.K.
In the U.K., we received approval from the Medicines and Healthcare Products Regulatory Authority, or MHRA, on August 23, 2012, to proceed with our Phase III clinical trial in GBM in the U.K.. We have been working on manufacturing arrangements for the clinical trial in the U.K. with both Kings College London and the Fraunhofer Institute, under the oversight and management of Cognate BioServices, to obtain the necessary approvals from the German and U.K. authorities for DCVax products to be able to be manufactured in either Germany or the U.K. for the clinical trial in the U.K. These approvals were completed in February 2013. Four major medical centers in the U.K. are preparing to proceed with the trial.
We have also been working on preparations for the clinical trial in Germany. On July 25, 2012, we announced that manufacturing certification has been received from the German regulatory authorities for the clinical trial in Germany, which is the first step towards implementation of the Phase III trial in Germany. We submitted the application to the German regulatory authority (the Paul Ehrlich Insitute, or PEI) for approval of the Phase III trial. As of March 31, 2013, 24 clinical centers are in varying stages of preparations as trial sites in Germany. Also, in October, 2012, ten major hospital centers across Germany, including the key opinion leaders in brain cancer, all applied to the German healthcare system for reimbursement of DCVax-L for brain cancer.
In parallel with these developments in our Phase III brain cancer program, we have been making arrangements to launch our DCVax-Direct program. On September 20, 2012, we announced that we had obtained approval from FDA for a combined Phase I/II trial with DCVax-Direct for all solid tumor cancers. In the following months, we initiated the processes for manufacturing of the DCVax-Direct products for the clinical trial.
We also entered into collaborations with premiere institutions for the DCVax-Direct trial, as we have done for the DCVax-L trial. On November 6, 2012, we announced that we had entered into a Letter of Intent for such a collaboration with Sarah Cannon Research Institute, which specializes in oncology and has a network of more than 700 physicians in the US and UK who see more than 75,000 new cancer patients per year.
During Q1 of 2013, we have continued and accelerated the manufacturing work and the preparations for launch of the Phase I/II clinical trial with DCVax-Direct for inoperable tumors in multiple diverse cancers. The trial is expected to be launched in Q2 of this year. As is standard with Phase I/II trials, the DCVax-Direct trial will not be blinded, and the results will be visible as the trial proceeds over the course of 2013. The Phase I stage of the trial involves dose escalation and confirmation. The Phase II stage of the trial will focus on efficacy. The primary measure of efficacy will be regression (i.e., shrinkage or elimination) of the inoperable tumors. Such regression is a rapid endpoint: if it is going to occur, is anticipated to occur within a couple months of treatment.
| 3 |
Our DCVax immunotherapies are based on a platform technology involving dendritic cells, the master cells of the immune system, and are designed to reinvigorate and educate the immune system to attack cancers. The dendritic cells are able to mobilize all parts of the immune system, including T cells, B cells and antibodies, natural killer cells and many others. Mobilizing the entire immune system provides a broader attack on the cancer than mobilizing just a particular component, such as T cells alone, or a particular antibody alone. Likewise, our DCVax technology is designed to attack the full set of biomarkers, or antigens, on a patient’s cancer, rather than just a particular selected target or several targets. Clinical experience indicates that when just one or a few biomarkers on a cancer are targeted by a drug or other treatment, sooner or later the cancer usually develops a way around that drug, and the drug stops working. We believe that mobilizing all agents of the immune system, and targeting all biomarkers on the patient’s cancer, contributes to the effectiveness of DCVax.
We believe that the market potential of this technology is particularly large because the DCVax products are expected to be applicable to most or all solid tumor cancers. We believe that the market potential is also enhanced by our two-continent strategy. By conducting our Phase III clinical trial in GBM on an international basis, with trial sites in both the United States and Europe, we believe we are positioned to potentially apply for product approval in both markets.
In clinical trials to date, our DCVax treatments have been achieving what we believe to be striking results. In patients with newly diagnosed GBM, the most aggressive and lethal form of brain cancer, patients treated with full standard of care treatment today (surgery, radiation and chemotherapy), typically have recurrence of their cancer within a median of 6.9 months, and typically die within a median of 14.6 months. In contrast, our early stage clinical trials showed that patients who received DCVax in addition to standard of care typically did not experience recurrence until approximately 2 years, rather than 6.9 months, and typically lived for approximately 3 years, rather than just 14.6 months. This data, if reproducible in a larger study, such as our current Phase III trial, would demonstrate that patients with GBM can derive significant clinical benefit from DCVax treatment. Moreover, long-term follow-up data on the GBM patients treated with DCVax in prior clinical trials show that, as of the latest update, 33% of the patients have reached or exceeded 4 years’ survival, and 27% of the patients have reached or exceeded 6 years’ survival (as compared with the median survival of 14.6 months with standard of care treatment today).
Similar results (i.e., significant extension or doubling of survival time) have been obtained in patients with late stage prostate cancer, either with or without metastases, in our prostate cancer clinical trial. Encouraging early results, significantly delaying progression of the cancer, have also been seen in patients in the initial metastatic ovarian cancer clinical trial.
Nearly as important in clinical trials to date, there has been no toxicity (no serious adverse events) related to DCVax. The broad and rapidly growing body of scientific literature about dendritic cells is consistent with the DCVax clinical experience, and provides added support regarding the lack of toxicity.
We are developing and positioning DCVax as a front line therapy that could potentially become standard of care. Accordingly, we are highly sensitive to the cost and affordability of DCVax. We have spent more than a decade pioneering a unique method of single-batch manufacturing which now results in costs and pricing of DCVax lower than most cancer drugs, even though DCVax is a personalized product.
We have also worked to make DCVax an extremely simple product for both physicians and patients. DCVax is administered to patients as a simple intra-dermal injection in the arm, similar to a flu shot and does not involve any complex procedures for physicians or patients. Unlike chemical or biologic drugs, however, DCVax must remain frozen throughout the distribution and delivery process, until the time of administration to the patient, and cannot be handled at room temperature. Hospitals, pharmacies and physicians may need to adopt new requirements for handling, distribution and delivery of DCVax.
| 4 |
We have continued to focus intensively on manufacturing, as we have done for many years. In the US, due to the levels of demand for the Phase III brain cancer trial, during 2012 we arranged for doubling of the manufacturing capacity for DCVax-L. Our contract manufacturer, Cognate BioServices, undertook the necessary construction for this capacity increase.
In Europe, as part of our partnering arrangements, the Fraunhofer Institute in Germany and Kings College in the U.K. have dedicated their own “cGMP” (clean room) state-of-the-art manufacturing facilities to our programs. We thereby obtained these manufacturing facilities without capital cost to us, and without the 18-month or more lead time usually required.
These manufacturing arrangements at Fraunhofer in Germany and Kings College London in the U.K. have been developed by (including the training of all personnel) and are being supervised by Cognate BioServices, Inc., our contract manufacturer in the U.S., to ensure consistency. Adding these two manufacturing operations in Europe carries several important benefits for us: it increases capacity, it provides local operations to satisfy European regulators, and it provides important risk mitigation in case of any disruption in the U.S. manufacturing operation (In such case, we believe our DCVax product could be produced in Europe for the U.S. market).
During 2012, Fraunhofer, Cognate and we completed the 1-1/2 year long regulatory processes and the final inspections for regulatory approval and certification for the manufacture of DCVax-L for the clinical trial in Germany. In addition, Fraunhofer, Cognate, we and Kings College began the 7-month processes for regulatory approvals and institutional approvals in both the UK and Germany to enable the manufacturing in Germany to supply DCVax-L for the clinical trial in the UK as well. This German supply arrangement is in addition to the manufacturing under development in the UK. Having two manufacturing locations in Europe will provide added flexibility for capacity management as well as risk protection.
Product Information
Immune therapies for cancer
Development of effective immune therapies for cancer has long been a goal of the medical and scientific communities. The human immune system is very powerful, and also very complex: an “army” with many divisions and many different kinds of weapons. A diagram of some key agents and weapons of the immune system is set forth below:
| 5 |
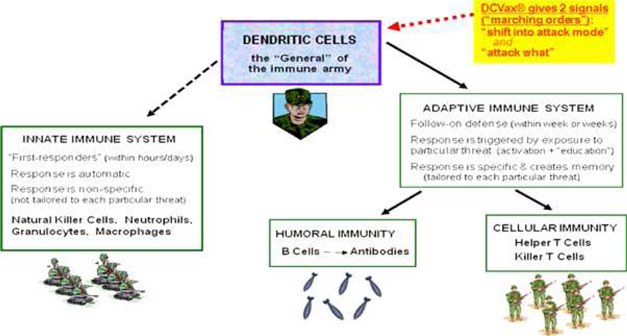
Diagram 1: The immune system “army” includes many diverse agents. Dendritic cells are the “General” of the army.
It has taken decades of research to identify the many different types of agents and weapons, to determine the relationships among them, and to determine how they work together to attack and defeat invaders such as bacteria, viruses and cancers. While the research was in process, early versions of immune therapies against cancers were tried, with mixed results and a number of failures. Over the course of the 1990s and 2000s, the first commercially successful category of immune agents to treat cancers emerged: drugs that consisted of individual antibodies, such as Avastin, Herceptin and Erbitux.
Antibodies are just one category of weapon in the overall immune “army,” and there are many, many kinds of individual antibodies within this category. Each antibody drug, such as Avastin, consists of just a single one of the many kinds of antibodies within this one category of immune weapon. These drugs do not involve the numerous other important agents in the immune army, such as T cells, NK cells, and so on.
Antibody drugs have been moderate medical successes and huge commercial successes. These drugs have delivered moderate extensions of patient survival compared with traditional chemotherapy drugs, with somewhat lesser (though still significant) toxicity. On this basis, these antibody drugs are achieving multi-billion dollars per year in sales.
Now, more broad based immune therapies are starting to come of age: “therapeutic vaccines” designed to mobilize the entire immune “army,” rather than just a single agent or single category of agents. Therapeutic vaccines are similar to preventive vaccines in that they work by mobilizing the immune system. However, therapeutic vaccines are administered to patients who already have a given disease, for the purpose of preventing or delaying recurrence or progression of the existing disease.
Several of the therapeutic vaccines that are now coming of age are focusing on dendritic cells in various ways, or on T cells. The vaccines focusing on dendritic cells offer a broader potential immune response because dendritic cells are the master cells of the immune system — the “General” of the “army.” When dendritic cells are activated against a particular pathogen (or cancer) they, in turn, mobilize all of the other agents (including T cells as well as B cells, NK cells and others) to attack that pathogen (or cancer). The process by which dendritic cells mobilize other agents takes place to a large extent in the lymph nodes.
| 6 |
A major challenge faced by immune therapies for cancer has been that, unlike in a healthy patient with an infectious disease, in cancer patients the dendritic cells fail to do their job, and the other immune agents also fail to do their job. Pathologists analyzing tumor tissue removed from cancer patients have long observed that there are often substantial numbers of immune cells in the surrounding tissue, but they are not infiltrating and attacking the tumor — as though the immune cells have made it to the doorstep of the tumor and then stopped.
The mechanisms by which cancer cells selectively suppress or block the immune system are still the subject of much research. It is known that cancer cells have many such mechanisms, including secretion of biochemical signals that jam normal immune signaling, that make tumor cells invisible to immune detection and/or that convey false messages to the immune system. Different therapeutic vaccines are taking different approaches to trying to overcome these cancer mechanisms and put the immune system back in action.
Many of the therapeutic vaccines for cancer (e.g., Cell Genesys, CancerVax) have targeted existing dendritic cellsin situ in a patient’s body, by administering various compounds or factors that are designed to attract dendritic cells to the tumor or enhance the tumor signals to the dendritic cells (in essence, making the tumor signals “louder”).
We and a few others (e.g., Dendreon) are taking a different approach, based on the belief that existing dendritic cellsin situ in a patient’s body are impaired and their ability to receive and process the necessary signals is blocked. Under this view, if the signaling is blocked, then no matter how “loud” the signal may be, it will not get through and will not achieve the activation needed.
The DCVax Technology
Our platform technology, DCVax, is a personalized immune therapy which consists of a therapeutic vaccine that uses a patient's own dendritic cells, or DCs, the master cells of the immune system, as the therapeutic agent. The patient’s DCs are obtained through a blood draw, or leukapheresis. The DCs are then activated and loaded with biomarkers (“antigens”) from the patient’s own tumor. The activation shifts the DCs into “attack mode.” The loading of biomarkers into the DCs “educates” the DCs aboutwhatto attack. The activated, educated DCs are then isolated with very high purity and constitute the DCVax personalized vaccine.
Injection of DCVax (the activated, educated dendritic cells of the patient) back into the patient, through a simple intra-dermal injection, similar to a flu shot, in the upper arm initiates a potent immune response against cancer cells, mobilizing the overall immune system and doing so in the natural way, with the numerous immune agents acting in their normal roles and in combination with each other. In short, DCVax is designed to restore the potent natural functioning of the immune system which has otherwise been impaired or blocked by the cancer.
Importantly, each activated, educated dendritic cell has a large multiplier effect, mobilizing hundreds of T cells and other immune cells. As a result, small doses of such dendritic cells can mobilize large and sustained immune responses.

Diagram 2: One Educated Dendritic Cell Activates Hundreds of Anti-Cancer Cells
| 7 |
We believe that at least three key aspects of the DCVax technology contribute to the positive results (described more fully below) seen in clinical trials to date:
| (1) | DCVax is personalized, and targets the particular biomarkers expressed onthat patient’stumor. Extensive scientific evidence has shown that there is substantial variation in tumor profiles and characteristics among patients with the “same” cancer. The degree of variation is particularly enormous in some of the most aggressive cancers, such as GBM brain cancer and pancreatic cancer. Cancer drugs are typically keyed to a single target which is believed to be found on the cancer cells’ surface or in one of the cancer cells’ signaling pathways in a substantial percentage of patients with a given type of cancer. Such drugs can be of no use in patients whose cancers do not happen to express that particular target, or cease expressing that target as the disease progresses. Most cancer drugs only achieve clinical benefits in a limited percentage of the patients with the type of cancer being targeted (e.g., 25 – 30% of the patients). In contrast, DCVax has achieved clinical benefits (i.e., longer delay in disease progression and longer extension of survival than with standard of care treatment) in over 80% of the patients who have received DCVax in clinical trials to date. Since DCVax is made with biomarkers from the patient’s own tumor, it is automatically tailored to targets that are present on that patient’s cancer. |
| (2) | DCVax is designed to target not just one but thefull set of biomarkers on the patient’s tumor. As mentioned above, cancer drugs are typically rifle shots aimed at just one target on a patient’s cancer. However, cancer is a complex and variable disease. Tumor profiles vary among patients with the “same” cancer and also vary as the disease progresses. Further, when rifle shot drugs hit individual targets on cancers, the cancers find ways around them (called “escape variants”) — and the rifle shot treatments then usually stop working. DCVax takes the opposite approach: instead of aiming at a single target, DCVax is designed at the full set of biomarkers on a patient’s cancer. Such treatment approach is expected to make it more difficult for tumors to develop escape variants. |
| (3) | DCVax is designed to mobilize theentire immune system , not just one among the many different categories of immune agents in that overall system. As described above, DCVax is comprised of activated, educated dendritic cells, and dendritic cells are the master cells of the immune system, that mobilize or help the entire immune system. Some of the prominent cancer drugs today are composed of just one type of antibody — and antibodies themselves are just one type of agent in the overall immune “army” (see Diagram 1 above). In contrast, the full immune system involves many types of antibodies, and also many other kinds of agents besides antibodies. Similarly, there have been a variety of early immune therapies that failed in the past. These, too, typically involved single agents, such as a single one among the many, many types of immune signaling molecules (e.g., a particular interferon or interleukin), or a single type of agent such as T cells alone, etc. In contrast, dendritic cells mobilize all of these different categories of agents, comprising the whole immune “army,” in combination with each other and in their natural relationships to each other. |
DCVax Product Lines
We have developed several different product lines based on the DCVax technology, to address multiple different cancers and different patient situations. There are two main components to each DCVax product: the immune cells (dendritic cells) and the cancer biomarkers (antigens).
All of our DCVax product lines are made from the patient’s own dendritic cells. The dendritic cells are freshly isolated, and newly matured and activated. We believe that the existing dendritic cells in a cancer patient have already been compromised by the cancer, and we believe that is the reason other vaccines aimed at the existing dendritic cells in patients have largely failed. However, the patient’s body continues to produce new precursors of dendritic cells, and these precursors (monocytes) circulate in the patient’s blood stream. For all DCVax products, these precursors are obtained through a blood draw, and then (through our proprietary manufacturing processes), the precursors are matured into a fresh, uncompromised batch of new dendritic cells.
The antigen (biomarker) component, which is combined with the fresh, personalized dendritic cells, varies among the DCVax product lines.
| 8 |
DCVax-L — is made with cancer antigens from tumor lysate (a protein extract from processed tumor cells) from the patient’s own tumor tissue. As such, DCVax-L incorporates thefull set of tumor antigens, making it difficult for tumors to find ways around it (“escape variants”), as described above. This is the DCVax product that has been used in our brain cancer and ovarian cancer clinical trials, and is currently in our Phase III trial. DCVax-L is expected to be used for any solid tumor cancers in situations in which the patient has their tumor surgically removed as part of standard of care.
DCVax-Direct — is designed for situations in which the tumors are inoperable — where it is not feasible or not desirable for patients to have their tumors surgically removed. This includes situations in which patients have multiple metastases, or for other reasons cannot have their tumors removed. Like DCVax-L, DCVax-Direct also incorporates thefull set of tumor antigens — but it does soin situ in the patient’s body rather than at the manufacturing facility. With DCVax Direct, the fresh, new dendritic cells are partially matured in a special (patent-protected) way so as to be ready to pick up antigens directly from tumor tissue in the patient’s body, and also communicate the information about those antigens to other agents of the immune system, such as T cells. The partially matured dendritic cells are then injected directly into the patient’s tumor(s). There, the dendritic cells pick up the antigensin situ rather than picking up the antigens from lysate in a lab dish at the manufacturing facility, as is done with DCVax-L.
DCVax-Prostate — is designed specifically for late stage, hormone independent prostate cancer. Such cancer involves the spread of micro-metastases beyond the prostate tissue. In most patients, there is no focal tumor which can be surgically removed and used to make lysate, or into which dendritic cells can be directly injected. Instead, the cancer cells are diffuse. We have developed a DCVax product line using a particular proprietary antigen — PSMA (Prostate Specific Membrane Antigen) — which is found on essentially all late stage (hormone independent) prostate cancer. The PSMA is produced through recombinant manufacturing methods, and is then combined with the fresh, personalized dendritic cells to make DCVax-Prostate.
Simplicity of DCVax for Physicians and Patients
All of the DCVax product lines are designed to be very simple for both physicians and patients, to fit within existing medical practices and procedures, and to be deliverable in virtually any clinic or doctor’s office. A number of complex, sophisticated and proprietary technologies are required for the production and frozen storage of DCVax, but these technologies are mostly deployed at the manufacturing facility and do not entail any effort or involvement by physicians or patients.
Front-end simplicity
For all DCVax product lines, the precursors (monocytes) for the fresh, new dendritic cells are obtained through a blood draw. This blood draw can be done not only at the hospital or cancer center where the patient is being treated, but at any blood center such as the Red Cross.
For DCVax-L, the collection of the patient’s tumor tissue, which is to be used to make lysate and provide the antigen component of the vaccine, involves a simple kit. The kit consists of a box with a vial which has a grinder top and is pre-loaded with a proprietary mix of enzymes. Such kits can be kept on hand like any inventory item at medical centers. In the operating room, after the tumor has been surgically removed, instead of disposing of the tissue in the medical waste, the nurse or technician chops the tissue coarsely and drops it into the vial, puts the vial back into the box, and hands the box to a courier pick-up service such as FedEx’s or UPS’ life science division, or a specialized courier such as World Courier.
For DCVax-Direct and DCVax-Prostate, there is no tumor tissue collection involved.
Back-end simplicity
For all DCVax products, administration to the patient involves a simple intra-dermal injection under the skin. All DCVax products are stored frozen in single doses. Such doses are tiny, and require less than 5 minutes to thaw. DCVax must remain frozen throughout the distribution and delivery process, until the time of administration to the patient, and cannot be handled at room temperature before that. Hospitals, pharmacies and physicians may need to adopt new requirements for handling, distribution and delivery of DCVax.
| 9 |
There are no handling steps at the point of care except thawing the frozen DCVax product to room temperature. There are also no intravenous infusions. DCVax-L and DCVax-Prostate are administered through a simple intra-dermal injection, similar to a flu shot, and are just a few drops in size. With the absence of handling steps at the point of care, and the simple intra-dermal injection, these DCVax products can be delivered to patients in virtually any clinic or doctor’s office.
The simplicity for patients also lies in the fact that DCVax is non-toxic. Patients do not have to take a second set of drugs to manage side effects of DCVax.
Clinical Programs and Clinical Trial Results
Overall Clinical Pipeline
Over the last ten years, we have built a robust clinical pipeline with DCVax products for multiple cancers, which we believe provides us with multiple opportunities for success. Our lead products, DCVax-L for GBM brain cancer and DCVax-Prostate for late stage prostate cancer, have reached late stage clinical trials. In addition to these, our DCVax-L has also been administered in an early stage trial for metastatic ovarian cancer, and other DCVax products have been cleared by the FDA to begin early stage trials in multiple other cancers.
The results seen in patients who received DCVax treatments in our Phase I/II clinical trials have been quite consistent. More than 80% of patients who received DCVax in trials to date have shown clinical benefits (longer delays in disease progression and longer extension of survival than with standard of care), compared with only 25 – 30% of patients showing clinical benefits with typical cancer drugs. Further, the clinical effects observed were largely consistent across diverse types of cancer, and diverse patient profiles (including, age, gender, physical condition, and different stages of disease). Nearly as important, in clinical trials to date, there have been no serious adverse events related to the study drug (DCVax).
Brain Cancer (GBM)
As discussed above, GBM is the most aggressive and lethal type of brain cancer. With full standard of care treatment today, including surgery, radiation and chemotherapy, the cancer recurs in a median of just 6.9 months and kills the patient in a median of just 14.6 months. There has been very little improvement in clinical outcomes for GBM patients in the last 30 years. The incidence of GBM appears to be on the rise, for unknown reasons, and there is an urgent need for new and better treatments.
Our Prior Clinical Trials
We, together with our collaborator, Dr. Linda Liau, have conducted two prior Phase I/II clinical trials at UCLA with DCVax-L for GBM brain cancer. These trials consisted of 30 patients with newly diagnosed GBM and recurrent GBM. The newly diagnosed patients who received DCVax in addition to standard of care treatment typically did not have tumor recurrence for a median of approximately 2 years, more than triple the usual time with standard of care, and patients survived for a median of approximately 3 years, approximately 2½ times the usual period attained with standard of care treatment.
Furthermore, a substantial percentage of patients who received DCVax in the prior clinical trials have continued in a “long tail” far beyond even the 3 year median survival. As of the latest long-term data update in July, 2011, 33% of the patients had reached or exceeded 4 years’ median survival and 27% had reached or exceeded 6 years’ median survival, compared with 14.6 months median survival with full standard of care treatment today.
| 10 |

Although the number of patients in our prior clinical trials for GBM has been limited, the difference in clinical outcomes with DCVax has been very large relative to outcomes with standard of care treatments. Comparisons of the patient outcomes in our trials with the outcomes of matched pools of patients treated with the same standard of care, in the same time period at the same hospital, display a high level of statistical significance rarely seen, even in clinical trials with much larger numbers of patients. The data on the results of our prior Phase I/II trials, if reproducible in a larger study, such as our current 312-patient Phase III trial, would demonstrate that patients with GBM can derive significant clinical benefit from DCVax treatment.
The measure of statistical significance, or “p value,” measures the probability that a set of clinical results are due to chance or random events. Accordingly, the smaller the “p value,” the smaller the chance that the results are random and the higher the statistical significance of the results. The FDA generally requires that the results of clinical trials reach a “p value” of .05 or less, meaning that there is a 5% or less chance that the trial results were due to chance or random events.
Comparisons of the clinical results in our two prior clinical trials with DCVax for GBM, with a small number of patients, and the clinical results in a matched pool of patients as described above, achieved the following “p values”:
| · | For the delay in time to recurrence, from 6.9 months with standard of care to approximately 2 years in patients treated with DCVax, the comparison “p value” was .00001 (i.e., a 1 in 100,000 chance that these results were random events). In general, the FDA requires a p value of 0.05 or less for product approval (i.e., a 5 in 100 chance or less that the clinical trial results were random events). |
| · | For the extension of survival time, from 14.6 months with standard of care to approximately 3 years in patients treated with DCVax, the comparison “p value” was .0003 (i.e., a 3 in 10,000 chance that these results were random events). In general, the FDA requires a p value of 0.05 or less for product approval (i.e., a 5 in 100 chance or less that the clinical trial results were random events). |
Following up on these results, in 2007 – 2008, we designed and began a 140-patient randomized, controlled Phase II trial but without a placebo and without blinding (which can only be achieved with a placebo that is indistinguishable from the new treatment being tested), as no placebo had been developed for our living cell product, DCVax. Unfortunately, without a placebo and blinding, patients who were randomized to the control group in the trial knew that this was the case — and, not surprisingly, they tended to drop out of the trial. As a result, that 140-patient Phase II trial had to be stopped and a placebo had to be developed to enable blinding, so that patients would not know whether they were receiving DCVax or a placebo.
| 11 |
Placebos to look indistinguishable from various kinds of pills can readily be made, but creating or selecting a placebo to be indistinguishable from living cells in a vial (such as the living immune cells that comprise DCVax) was a different and difficult challenge. Not only must the placebo look indistinguishable from the DCVaxvisually, it must also not have any positivefunctional action of its own that would muddy the trial results. After considerable work, we succeeded in developing such placebo arrangements and re-designing the Phase II trial to accommodate them, including nearly doubling the number of patients (from 140 to 240 patients).
We obtained a new FDA clearance and re-approvals by all the clinical sites, and commenced the new Phase II trial in early 2008. Unfortunately, we had only been underway for a short period when the economic crisis hit. We were able to keep the trial open, and continue treating the patients already enrolled in the trial, but we had to suspend new enrollment of additional patients into the trial. This suspension continued through the end of 2010, solely due to the severe economic downturn and resulting resource constraints.
In Q1 of 2011, we began the process of resuming new enrollment – obtaining renewals of institutional review board or IRB approvals and other necessary steps. We resumed the enrollment activity in Q2 of 2011. At that time, the trial was only at a dozen sites and only in the US. During 2011, we expanded the trial to 25 sites across the US.
In an amendment to the clinical trial protocol which became effective on May 3, 2012, the FDA, among other things, accepted the re-designation of this ongoing trial from a Phase II to a Phase III. In August 2012, the UK regulatory authority (the Medicines and Healthcare Products Regulatory Authority, or MHRA) also approved this trial to proceed in the UK as a Phase III trial.
Our Current Phase III Clinical Trial
During 2012, the Phase III brain cancer trial was expanded to 42 sites in the US, and nearly 30 additional sites were identified and in varying stages in the UK and Germany.
As of March 31, 2013, there are 43 clinical sites open and operating for the trial across the United States with more expected to become operational during 2013.
Inoperable Solid Tumor Cancers
Our DCVax-Direct product offers a potential new treatment option for the wide range of clinical situations in which patients' tumors are considered “inoperable” because the patient has multiple tumors, or their tumor cannot be completely removed, or the surgery would cause undue damage to the patient and impair their quality of life.
A large number of patients with a variety of cancer types (including lung, colon, pancreatic, liver, ovarian, head and neck, and others) are faced with this situation, because their tumors are already locally advanced or have begun to metastasize by the time symptoms develop and the patients seek treatment. For these patients, the outlook today is bleak and survival remains quite limited.
DCVax-Direct is administered by direct injection into a patient's tumors. It can be injected into any number of tumors, enabling patients with locally advanced disease or with metastases to be treated. DCVax-Direct can also be injected into tumors in virtually any location in the body: not only tissues at or near the surface of the body but also, with ultra-sound guidance, into interior tissues.
As described above, we have been making arrangements to launch our initial Phase I/II clinical trial with DCVax-Direct, which is approved by FDA to proceed in all solid tumor cancers. In the fall of 2012, we initiated the processes for manufacturing DCVax-Direct for the clinical trial. During Q1 of 2013, we have expanded and accelerated the manufacturing preparations, including test runs and other qualification and optimization work.
During 2012, we entered into collaborations with premiere institutions for the DCVax-Direct trial, such as the Sarah Cannon Research Institute, with its network of more than 700 physicians in the US and UK who see more than 75,000 new cancer patients per year. During Q1 of 2013, major cancer centers across the US have contacted us expressing interest in becoming sites in the DCVax-Direct clinical trial, and we have entered into discussions to expand the trial and the collaborations relating to it. We anticipate launching the trial during Q2 of 2013.
As is standard with Phase I/II trials, the DCVax-Direct trial will not be blinded, and the results will be visible as the trial proceeds over the course of 2013. The Phase I stage of the trial involves dose escalation and confirmation. The Phase II stage of the trial will focus on efficacy. The primary measure of efficacy will be regression (i.e., shrinkage or elimination) of the inoperable tumors. Such regression is a rapid endpoint: if it is going to occur, is anticipated to occur within a couple months of treatment.
| 12 |
Prostate Cancer
Prostate cancer is the most common cancer in men in the U.S., accounting for more than 25% of all cancers in men, and nearly twice as many cases per year as lung cancer in men, according to the American Cancer Society. For late stage prostate cancer, there is a pressing unmet need for new treatments. This late stage cancer includes two subsets of patients, comprising two distinct markets: (A) about 80 – 85% of patients do not yet show metastases, have a last good period of life, and typically live for about 36 months; and (B) about 15 – 20% of patients have more aggressive disease, show metastases right away, and only live for about 18 months. Nearly 100,000 men reach these late stages of prostate cancer every year in the United States alone (with similar numbers in Europe). Yet, there is no FDA approved drug specifically for the patients in group A, who comprise the vast majority of late stage prostate cancer patients.
For the patients in group B, there are a growing number of FDA approved drugs, including taxotere (docetaxel), Provenge, Zytiga and Xtandi, but they only add a few months of survival. Taxotere adds about 10 weeks of survival, in only a limited percentage of patients, and has toxic side effects. The Provenge immune therapy developed by Dendreon Corporation adds about 4 months of survival. Zytiga and Xtandi work through different mechanisms of action, and they, too, add only 4 – 5 months of survival.
We believe that DCVax-Prostate can offer a much needed treatment for late stage prostate cancer patients in group A, for whom there is no treatment specifically approved by FDA today. In addition, for patients in group B, we believe that DCVax-Prostate can potentially offer a much longer extension of survival.
| 13 |
Our Prior Clinical Trials
Clinical experience with DCVax-Prostate dates back more than a decade, and has reached the Phase III trial stage. More than one hundred patients were treated with DCVax-Prostate in an academic clinical setting in the mid and late 1990s. Based on encouraging results from those treatments, we undertook a Phase I/II clinical trial with 35 patients at two leading clinical centers: MD Anderson and UCLA. Based upon positive results from that trial, we designed a large 612-patient, Phase III clinical trial, and previously obtained FDA clearance to proceed with this trial. The details of these clinical programs are described below.
Our Phase I/II clinical trial, conducted at MD Anderson and UCLA, included both subsets of hormone independent prostate cancer patients: group A, without visible metastases, and group B, with metastases. As is standard for Phase I/II trials, in our Phase I/II trial all patients in the trial received the DCVax treatment (there was no placebo control arm). For Group A patients, the information below shows a comparison of our clinical results with the natural course of the disease in group A (for whom there is no established standard of care treatment). For group B patients, the information below shows a comparison of our clinical results with the results reported in clinical trials and clinical practice with two of the four treatments that are currently FDA approved for these patients (Taxotere and Provenge). The results of this clinical trial were as follows. Two drugs not shown below (Zytiga and Xtandi) produced clinical outcomes similar to Provenge.
Group A: Hormone Independent Prostate Cancer Patientswithout Metastases
| Natural Course of Disease | With DCVax-Prostate | |||
| Median time to disease progression (appearance of bone metastases) | 28 - 34 weeks | 59 weeks | ||
| Median survival | 36 months | >54 months and continuing** | ||
| **(more than half of these patients still alive as of 12/31/05-last data follow-up) |
Group B: Hormone Independent Prostate Cancer Patientswith Metastases
| With Standard of Care (Taxotere) | With Provenge | With DCVax-Prostate | ||||
| Median survival | 18.9 months | 25.9 months | 38.7 months | |||
| Overall survival at 3 years | 11% | 33% | 64% |
| 14 |
Thus, in the prior Phase I/II clinical trial, patients without metastases (group A) who were treated with DCVax-Prostate typically lived at least 1½ years longer than patients going through the natural course of the disease.
Patients with metastases (group B) who were treated with DCVax-Prostate lived twice as long as patients typically do with standard of care (receiving the drug taxotere), and more than a year longer than Dendreon has reported that such patients lived when treated with its Provenge immune therapy in the clinical trials upon which FDA approval of Provenge was based.
Following these positive results in both group A and group B patients, we determined to focus our Phase III clinical trial on the patients in group A, because 80-85% of late stage prostate cancer patients fall into this group, while only 15 – 20% fall into group B. In contrast, Dendreon focused its clinical trials on the group B patients, and obtained FDA approval only for that group of patients. Thus, the addressable market for our DCVax-Prostate will be at least four times the size of the addressable market for Provenge. Due to the size and anticipated cost of the Phase III trial (at least $75 million or more), we plan to proceed with that trial only in the context of partnering arrangements.
Target Markets
Since DCVax is expected, ultimately, to be applicable to most types of solid tumor cancers, we believe the potential market for DCVax can be very large. According to the American Cancer Society, 1 in 2 men, and 1 in 3 women in the U.S. will develop some form of cancer in their lifetime. There are nearly 1.5 million new cases of cancer per year in the U.S., and nearly 600,000 deaths from cancer. The statistics are similar in Europe and in much of the rest of the world.
Even focusing just on the two DCVax products which have already reached late stage clinical trials — for GBM brain cancer and for hormone independent prostate cancer, as described above — we believe that the target markets for each of these have very large (billion dollar) revenue potential.
Brain cancer
Brain cancers fall into two broad categories: primary (meaning the cancer first originates in the brain) and metastatic (meaning the cancer first appears elsewhere in the body, but subsequently metastasizes to the brain). In the U.S. alone, on an annual basis, there are some 40,000 new cases of primary brain cancer, and 160,000 new cases of metastatic brain cancer. The numbers are similar in Europe and the rest of the world.
Within the category of primary brain cancer, Grade 4 GBM is the most aggressive and lethal type. Among the 40,000 new cases of primary brain cancer per year in the U.S., at least 12,000 cases are GBM (with some estimates as high as 17,000) and the incidence is increasing.
In addition, brain cancer is a serious medical problem in children 18 years and under. It is the second most frequent type of childhood cancers (after leukemias) and, following progress in reducing death rates from leukemias, it is now the leading cause of childhood cancer deaths.
Very little has changed in the last 30 years in the treatment and clinical outcomes for GBM. With all standard of care treatment today — surgery, radiation and chemotherapy — patients still die within a median of about 14.6 months from diagnosis.
The one drug which has become the standard of care chemotherapy treatment for GBM, Temodar, achieved market saturation extremely rapidly, within two years of product launch. Temodar added 10 weeks of survival (extending survival from its historical 12 months to the 14.6 months typical today), and did so in a limited percentage of patients. Other drugs approved by FDA for GBM, such as Avastin, did not extend survival at all.
| 15 |
Against this backdrop, we believe DCVax is well positioned for this target market. Further, after seeking regulatory approval for DCVax for the GBM subset of primary brain cancers, in the future we plan to conduct clinical trials and seek approval for other (lower grade) primary brain cancers and for metastatic brain cancers.
We believe that the market potential of DCVax for brain cancer, even under conservative assumptions, is very large. For example, if one counts only GBM cases (and not other primary brain cancers nor any metastatic brain cancers), only in the U.S. and Europe (and not rest of world), and one assumes a 50% market share (compared with Temodar whose market share rapidly reached saturation), the number of cases to be treated with DCVax would be at least 12,000 per year.
Prostate cancer
We also believe that the market potential of DCVax for prostate cancer is very large, even under conservative assumptions. Prostate cancer is the most common cancer in men. More than 200,000 new cases per year are diagnosed in the U.S. alone, according to the American Cancer Society, with similar numbers in Europe. Among these, at least 100,000 new cases reach late stage prostate cancer each year in the U.S. (with similar numbers in Europe).
Among these 100,000 new late stage prostate cancer cases per year, 80 – 85% of the patients have no visible metastases, and only 15 – 20% already have visible metastases. As noted above, in prior clinical trials, patients in both groups were treated with DCVax, and both groups showed positive results (substantial extensions of survival, far beyond existing treatment options — including Provenge). We are focusing our Phase III trial on the much larger market: patients without visible metastases, comprising 80 – 85% of the 100,000 new cases per year in the U.S.
If one counts only those 80 – 85,000 late stage patients, only in the U.S. (not counting either Europe or rest of world), and one assumes only a 25% market share (compared with Taxotere, whose market share is very high despite adding only 10 weeks of survival), the number of cases to be treated with DCVax would be 20 – 21,000 cases per year.
Manufacturing of DCVax
We believe that our proprietary manufacturing process for DCVax products is a key to our favorable product economics, and we are positioning DCVax to be a potential front line therapy that can be provided to patients everywhere. We have spent more than a decade honing this manufacturing process.
We have pioneered a manufacturing model under which at least 3 years of treatments are produced in one large batch in each manufacturing cycle. In addition, we have implemented special cryopreservation methods which enable this multi-year quantity of product to be frozen, and kept frozen for years, while maintaining its potency.
Both of these technologies, the multi-year batch manufacturing and the cryopreservation, are essential elements of our manufacturing model and product economics. Together, they enable us to incur the high costs of manufacturing just one time, and then store the multi-year quantity of product, frozen, in single doses. This makes DCVax effectively an “off the shelf” product for the patient, even though it is personalized, and enables the price of DCVax to be at or below the price level of modern, non-personalized cancer drugs while still achieving reasonable profit margins. This is already the case while we are using first generation manufacturing, without automation and have not yet scaled up to obtain significant economies of scale. We believe that both automation and economies of scale will further enhance the product economics.
Our manufacturing process has been taught to, and replicated at, Kings College in England and the Fraunhofer Institute in Germany, so that the same efficiencies and quality controls will be present for the DCVax produced both in Europe and the United States.
Our manufacturing process for DCVax-L takes about 8 days, followed by quality control and sterility testing. It involves several main steps as follows:
| 16 |
Isolation of Precursors. The precursors of new dendritic cells are isolated from the patient's white blood cells, which were obtained through a blood draw and sent to the manufacturing facility.
Differentiation of Precursors into Immature Dendritic Cells. Precursors are differentiated (transformed) into immature dendritic cells through a six-day culture period, during which specific growth factors are applied in a manner that mimics the natural process in a healthy person's body.
Maturation of Dendritic Cells. Immature dendritic cells are exposed to proprietary maturation factors and methods.
Antigen Display and Activation of Dendritic Cells. Cancer-associated antigens or antigen fragments obtained from the patient’s own tumor tissue or, for prostate cancer, produced recombinantly, are added to the maturing dendritic cells. The dendritic cells ingest and process the antigen materials, and then display fragments on their outer cell surfaces (which will serve to pass along the activation signals from these dendritic cells to other agents in the immune system, such as T cells and B cells, when the dendritic cells are injected back into the patient.
Harvest. These matured and activated new dendritic cells are isolated with very high purity, and divided into single-dose vials. They are then frozen and stored until needed.
For DCVax-Direct, our manufacturing process is similar, but simpler as it does not involve full maturation of the dendritic cells and does not involve the antigen display and activation stage. The DCVax-Direct manufacturing process is partly automated with a proprietary system, and takes about 6 days.
We contract out the manufacturing of our DCVax products to Cognate BioServices. Although there are many contract manufacturers for small molecule drugs and for biologics, there are only a few major contract manufacturers in the U.S. that specialize in producing living cell products. Cognate is one of those few and appears to have the most substantial track record of clinical trial approvals from FDA for cellular products. The manufacturing of living cell products is highly specialized and entirely different than production of biologics: the physical facilities and equipment are different, the types of personnel and skill sets are different, and the processes are different.
In addition, the regulatory requirements for living cell products are exceptionally difficult to meet — particularly forpersonalized living cell products, which can vary considerably from patient to patient. We believe that among companies developing such living cell products, nearly all cases in which clinical trials have been put on clinical hold (i.e., stopped) by FDA have been because of product or manufacturing related issues.
Cognate has a leading regulatory track record. According to Cognate, the Cognate team has been responsible for the product and manufacturing aspects of more than 20 INDs (applications for approval of clinical trials) for living cells products, and all of these INDs have been approved. Moreover, the Company believes, based upon information provided by Cognate, that no client of Cognate has been put on clinical hold in connection with its product.
Cognate’s manufacturing facility for clinical-grade cell products is located in Memphis, Tennessee, near the airport. Memphis is a worldwide air shipping hub for both Federal Express and UPS. Cognate's facility is approximately 35,000 square feet and contains substantial expansion space in addition to the portions currently built out and in use. The current manufacturing facilities are sufficient to produce DCVax for at least several thousand patients per year — an amount well in excess of what is needed for the late stage clinical trial under way. There is a large amount of expansion space, which is already planned for build- -out in stages to allow for scale -up of production capacity in a modular fashion as the need increases for commercialization. This would allow Cognate's current facility to increase to a total capacity of some 5,000 patients per year. In addition, the manufacturing arrangements with Fraunhofer in Germany and Kings College London in the United Kingdom provide further manufacturing capacity and flexibility. As a comparison, Dendreon commercially launched its Provenge dendritic cell vaccine for prostate cancer with initial manufacturing capacity for only 2,000 patients per year for a cancer that occurs in at least 100,000 new cases per year In the U.S. alone.
| 17 |
Intellectual Property and Orphan Drug Designation
We have an integrated strategy for protection of our technology through both patents and other mechanisms, such as Orphan Drug status. As of December 31, 2012, we have over 180 issued and pending patents worldwide, grouped into 17 patent families. Some cover the use of dendritic cells in particular DCVax products. Others cover key processes for manufacturing and quality control for DCVax, as well as an automated system which we believe will play a major role in the scale-up of production for large numbers of patients on a cost-effective basis.
During 2012, a dozen new patents were issued to us as part of our worldwide patent portfolio. The newly issued patents covered a variety of subject matter, such as the proprietary partial maturation for DCVax-Direct, the machines and systems to manufacture DCVax-Direct, certain processes for enhancing the potency of dendritic cells in general, certain measures of product quality, and other matters.
The expiration dates of the issued patents in our portfolio range from 2015 to 2026. For some of the earlier dates, we plan to seek extensions of the patent life, and believe we have reasonable grounds for doing so.
In addition to our patent portfolio, we have obtained Orphan Drug designation for our lead product, DCVax-L for brain cancer. Such designation brings with it a variety of benefits, including potential market exclusivity for seven years in the U.S. and ten years in Europe if our product is the first of its type to reach the market.
This market exclusivity applies regardless of patents, according product exclusivity on the market even if the company that developed it has no patent coverage on the product. In addition, the time period for such market exclusivity does not begin to run until product sales begin. In contrast, the time period of a patent begins when the patent is filed and runs down during the years while the product is going through development and clinical trials.
In order to qualify for these incentives, a company must apply for designation of its product as an “Orphan Drug” and obtain approval from the FDA, or its counterpart, abroad. In addition, for the market exclusivity, a product must be either the first of its kind for a particular disease to reach the market, or clinically superior to a product currently on the market. The U.S. and the European Union each granted an Orphan Drug designation for our DCVax-L product for GBM.
Competition
The biotechnology and biopharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. Several companies, such as Dendreon, Celldex Therapeutics, Inc., Ark Therapeutics plc, Oxford Biomedica plc, Argos Therapeutics, Inc., Agenus, Inc., Prima Biomed, Ltd., Avax Technologies, Inc., Immunocellular Therapeutics, Ltd., Activartis, Bavarian Nordic, Bellicum Pharmaceuticals and others are actively involved in the research and development of immune therapies or cell-based therapies for cancer. In addition, other novel technologies for cancer are under development, such as the electro-therapy device of NovoCure. Of these companies, only one has obtained approval of such an immune therapy: Dendreon (for its Provenge treatment of prostate cancer). Additionally, several companies, such as Medarex, Inc., Amgen, Inc., Agensys, Inc., and Genentech, Inc., are actively involved in the research and development of monoclonal antibody-based cancer therapies. Currently, a substantial number of antibody-based drugs are approved for commercial sale for cancer therapy, and a large number of additional ones are under development. Many other third parties compete with us in developing alternative therapies to treat cancer, including: biopharmaceutical companies; biotechnology companies; pharmaceutical companies; academic institutions; and other research organizations, as well as some medical device companies (e.g., NovoCure and MagForce Nano Technologies AG).
| 18 |
We face extensive competition from companies developing new treatments for brain cancer. These include a variety of immune therapies, as mentioned above, as well as a variety of small molecule drugs and biologics. There are also a number of existing drugs used for the treatment of brain cancer that may compete with our product, including, Avastin® (Roche Holding AG), Gliadel® (Eisai Co. Ltd.), and Temodar® (Merck & Co., Inc.).
Most of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing and sales than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly if they enter into collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, as well as in acquiring technologies complementary to our programs, and in obtaining sites for our clinical trials and enrolling patients.
Recent Developments
In August 2012, our Board and a majority of our stockholders approved an amendment to our Amended and Restated 2007 Stock Option Plan providing that, on an ongoing basis, effective January 1 each year, the aggregate number of shares of common stock that are available for issuance under the plan shall automatically be increased in such manner as to maintain the option pool capped at twenty percent of our issued and outstanding stock.
On September 25, 2012, we effected a 1-for-16 reverse stock split of our issued and outstanding common stock. In addition, we filed an amendment to our certificate of incorporation increasing our authorized shares of preferred stock from 20,000,000 to 40,000,000.
In December, 2012, we retired $36.4 million in aggregate amount of convertible notes, notes and payables and accrued interest by entering into Conversion Agreements with our non-affiliated and affiliated note holders and creditors, including certain of our directors and executive officers. This aggregate debt amount was converted into 9.8 million common shares and warrants exercisable for 3.8 million shares of common stock. The warrants have an exercise period of five years from the date of issuance and a weighted average exercise price of $3.66 per share.
In April 2012, we announced the addition of two highly respected experts to our Board: Dr. Navid Malik and Mr. Jerry Jasinowski. Dr. Malik is Head of Life Sciences Research for Cenkos Securities Plc. in the UK, and has been one of the most influential analysts in the UK and Europe over the last decade, covering the life sciences industry worldwide. Mr. Jasinowski is a nationally recognized chief executive who headed up the largest industrial trade association in the US (the National Association of Manufacturers) for fourteen years, and has extensive board experience across a wide range of manufacturing, technology, and financial firms, including Fortune 1000 and Fortune 500 companies.
In September 2012, we added a pharma industry veteran, Dr. Guenter Rosskamp, to our management team as CEO of our German subsidiary. Dr. Rosskamp previously served for many years as Head of Central Nervous System Therapeutics, and as head of Strategic Business Development, for Schering AG (now part of Bayer AG). In those capacities, Dr. Rosskamp was responsible for the development and commercialization of multiple drugs.
Corporate Information
We were formed in 1996 and incorporated in Delaware in July 1998. Our principal executive offices are located in Bethesda, Maryland, and our telephone number is (240) 497-9024. Our website address iswww.nwbio.com. The information on our website is not part of this report. We have included our website address as a factual reference and do not intend it to be an active link to our website.
Employees
As of March 31, 2013, we had 8 full-time and 2 part-time employees. We believe our employee relations are satisfactory.
ITEM 1A. RISK FACTORS
RISK FACTORS
Our business, financial condition, operating results and prospects are subject to the following material risks. Additional risks and uncertainties not presently foreseeable to us may also impair our business operations. If any of the following risks actually occurs, our business, financial condition or operating results could be materially adversely affected. In such case, the trading price of our common stock could decline, and our stockholders may lose all or part of their investment in the shares of our Common Stock.
| 19 |
Risks Related to our Operations
We will need to raise substantial funds, on an ongoing basis, for general corporate purposes and operations, including our clinical trials. Such funding may not be available or may not be available on attractive terms.
We will need substantial additional funding, on an ongoing basis, in order to continue execution of our clinical trials, to move our product candidates towards commercialization, to continue prosecution and maintenance of our large patent portfolio, to continue development and optimization of our manufacturing and distribution arrangements, and for other corporate purposes. Any financing, if available, may include restrictive covenants and provisions that could limit our ability to take certain actions, preference provisions for the investors, and/or discounts, warrants or other incentives. Any financing will involve issuance of equity and/or debt, and such issuances will be dilutive to existing shareholders. There can be no assurance that we will be able to complete any of the financings, or that the terms for such financings will be attractive. If we are unable to obtain additional funds on a timely basis or on acceptable terms, we may be required to curtail or cease some or all of our operations at any time.
We are likely to continue to incur substantial losses, and may never achieve profitability.
As of December 31, 2012, we have an aggregate accumulated cash deficit, since inception of the Company, of $140.0 million, and accumulated non-cash (accounting measures) deficit of $179.1 million, making a combined cash and non-cash accumulated deficit of $319.1 million since the Company’s inception. We may never achieve or sustain profitability.
Our auditors have issued a “going concern” audit opinion.
Our independent auditors have indicated, in their report on our December 31, 2012 financial statements, that there is substantial doubt about our ability to continue as a going concern. A “going concern” opinion indicates that the financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets, or the amounts and classification of liabilities that may result if we do not continue as a going concern. Therefore, you should not rely on our consolidated balance sheet as an indication of the amount of proceeds that would be available to satisfy claims of creditors, and potentially be available for distribution to shareholders, in the event of liquidation.
As a development stage company with a novel technology and unproven business strategy, our limited history of operations makes an evaluation of our business and prospects difficult.
We have had a limited operating history and we are still in the process of developing our product candidates through clinical trials. Our technology is novel and involves mobilizing the immune system to fight a patient’s cancer. Immune therapies have been pursued by many parties for decades, and have experienced many failures. In addition, our technology involves personalized treatment products, a new approach to medical products that involves new product economics and business strategies, which have not yet been shown to be commercially feasible or successful. We have not yet gone through scale-up of our operations to commercial scale. This limited operating history, along with the novelty of our technology, product economics, and business strategy, and the limited scale of our operations to date, makes it difficult to assess our prospects for generating revenues commercially in the future.
We will need to expand our management and technical personnel as our operations progress, and we may not be able to recruit such additional personnel and/or retain existing personnel.
As of December 31, 2012, we employ eight (8) full-time employees and two (2) part-time employees. The rest of our personnel are retained on a consulting or contractor basis. Biotech companies would typically have a larger number of employees by the time they reach late stage clinical trials. Such trials require extensive management activities and skill sets, including scientific, medical, regulatory (for FDA and foreign regulatory counterparts), manufacturing, distribution and logistics, site management, business, financial, legal, public relations outreach to both the patient community and physician community, intellectual property, administrative, regulatory (SEC), investor relations and other.
In order to fully perform all these diverse functions, with late stage trials under way at many sites across the U.S. and soon in Europe, we will need to expand our management and technical personnel. However, the pool of such personnel with expertise and experience with living cell products, such as our DCVax immune cell product, is very limited. In addition, we are a small company with limited resources, our business prospects are uncertain and our stock price is volatile. For some or all of such reasons, we may not be able to recruit all the management and technical personnel we need, and/or we may not be able to retain all of our existing personnel. In such event, we may have to continue our operations with a smaller than usual team of personnel, and our business and financial results may suffer.
| 20 |
We rely at present on third-party contract manufacturers. As a result, we may be at risk for capacity limitations and/or supply disruptions.
We currently rely upon Cognate BioServices, Inc., or Cognate, to produce all of our DCVax product in the U.S., and to supervise the production of our DCVax product candidates outside the U.S. The majority owner of Cognate is Toucan Capital, one of our major stockholders, and its affiliates. We have an agreement in place with Cognate pursuant to which Cognate has agreed to provide manufacturing and other services for the next five years, in connection with our Phase III clinical trial of DCVax -L in brain cancer, and other programs. The agreement requires us to make certain minimum monthly payments to Cognate in order to have dedicated manufacturing capacity available for our products, irrespective of whether we actually order any DCVax products. The agreement also specifies the amounts we must pay for Cognate's actual manufacturing of DCVax for patients.
We have entered into an agreement with King’s College London to manufacture DCVax for our clinical trial and our compassionate use cases. Cognate will manage and supervise the processing in London. In addition, our partner, Fraunhofer, has received approval and certification from the regional and national regulatory agencies in Germany for the manufacture of DCVax for GBM. We anticipate that the manufacturing facilities in the U.K. and Germany will eventually obtain the necessary approvals and be able to supply DCVax products for anywhere in Europe, however this may not turn out to be feasible, for regulatory, operational and/or logistical reasons.
Problems with the manufacturing facilities or processes of Cognate or our partners in the U.K. and/or Germany could result in a failure to produce, or a delay in production, of adequate supplies of our DCVax product candidates. A number of factors could cause interruptions or delays, including the inability of a supplier to provide raw materials, equipment malfunctions or failures, damage to a facility due to natural disasters or otherwise, changes in FDA or European regulatory requirements or standards that require modifications to our manufacturing processes, action by the FDA or European regulators, or by us that results in the halting or slowdown of production of components or finished products due to regulatory issues, our manufacturers going out of business or failing to produce product as contractually required, and/or other similar factors. Because manufacturing processes for our DCVax product candidates are highly complex, require specialized facilities and personnel that are not widely available in the industry, involve equipment and training with long lead times, and are subject to lengthy regulatory approval processes, alternative qualified production capacity may not be available on a timely basis or at all. Difficulties, delays or interruptions in the manufacturing and supply of our DCVax product candidates could require us to stop enrolling additional new patients into our trial, and/or require us to stop the trial or other program, increase our costs, damage our reputation and, if our product candidates are approved for sale, cause us to lose revenue or market share if our manufacturers are unable to timely meet market demands.
The manufacturing of our product candidates will have to be greatly scaled up for commercialization, and neither we nor other parties in the industry have experience with such scale-up.
As is the case with any clinical trial, our Phase III clinical trial of DCVax-L for GBM involves a number of patients that is a small fraction of the number of potential patients for whom DCVax-L may be applicable in the commercial market. The same will be true of our other clinical programs with our other DCVax product candidates. If our DCVax-L, and/or other DCVax product candidates, are approved for commercial sale, it will be necessary to greatly scale up the volume of manufacturing, far above its level for the trials. Neither we nor our contract manufacturers have experience with such scale-up. In addition, there are virtually no consultants or advisors in the industry who have such experience and can provide guidance or assistance, because active immune therapies such as DCVax are a fundamentally new category of product in two major ways: these active immune therapy products consist of living cells, not chemical or biologic compounds, and the products are personalized. To our knowledge, no such products have successfully completed the necessary scale-up for commercialization without material difficulties. For example, Dendreon has encountered substantial difficulties trying to scale up the manufacturing of its Provenge product for commercialization.
The necessary specialized facilities, equipment and personnel may not be available or obtainable for the scale-up of manufacturing of our product candidates.
The manufacture of living cells requires specialized facilities, equipment and personnel which are entirely different than what is required for manufacturing of chemical or biologic compounds. Scaling up the manufacturing of living cell products to volume levels required for commercialization will require enormous amounts of these specialized facilities, equipment and personnel — especially where, as in the case of our DCVax product candidates, the product is personalized and must be made for each patient individually. Since living cell products are so new, and have barely begun to reach commercialization, the supply of the specialized facilities, equipment and personnel needed for them has not yet developed. It may not be possible for us or our manufacturers to obtain all of the specialized facilities, equipment and personnel needed for commercialization of our DCVax product candidates. This could delay or halt our commercialization.
| 21 |
Our technology is novel, involves complex immune system elements, and may not prove to be effective.
Data already obtained, or in the future obtained, from pre-clinical studies and clinical trials do not necessarily predict the results that will be obtained from later pre-clinical studies and clinical trials. Over the course of several decades, there have been many different immune therapy product designs — and many product failures and company failures. To our knowledge, to date, only one active immune therapy, Provenge, has been approved by the FDA. The human immune system is complex, with many diverse elements, and the state of scientific understanding of the immune system is still limited. Some immune therapies previously developed by other parties showed surprising and unexpected toxicity in clinical trials. Other immune therapies developed by other parties delivered promising results in early clinical trials, but failed in later stage clinical trials. To date, we have only completed early stage trials in limited numbers of patients. Although the results of those trials were quite positive, those results may not be achieved in our later stage clinical trials, such as the 312-patient Phase III trial we are now conducting for GBM, and our product candidates may not ultimately be found to be effective.
Clinical trials for our product candidates are expensive and time consuming, and their outcome is uncertain.
The process of obtaining and maintaining regulatory approvals for new therapeutic products is expensive, lengthy and uncertain. Costs and timing of clinical trials may vary significantly over the life of a project owing to any or all of the following non-exclusive reasons:
| • | the duration of the clinical trial; | |
| • | the number of sites included in the trials; | |
| • | the countries in which the trial is conducted; | |
| • | the length of time required and ability to enroll eligible patients; | |
| • | the number of patients that participate in the trials; | |
| • | the number of doses that patients receive; | |
| • | the drop-out or discontinuation rates of patients; | |
| • | per patient trial costs; | |
| • | third party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; | |
| • | our final product candidates having different properties in humans than in lab testing; | |
| • | the need to suspect or terminate our clinical trials; | |
| • | insufficient or inadequate supply of quality of necessary materials to conduct our trials; | |
| • | potential additional safety monitoring, or other conditions required by FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials, or other studies requested by regulatory agencies; | |
| • | problems engaging IRBs to oversee trials or in obtaining and maintaining IRB approval of studies; | |
| • | the duration of patient follow-up; | |
| • | the efficacy and safety profile of a product candidate; | |
| • | the costs and timing of obtaining regulatory approvals; and | |
| • | the costs involved in enforcing or defending patent claims or other intellectual property rights. |
Late stage clinical trials, such as our Phase III clinical trial for GBM patients, are especially expensive, typically requiring tens of millions of dollars, and take years to reach their outcomes. Such outcomes often fail to reproduce the results of earlier trials. It is often necessary to conduct multiple late stage trials (including multiple Phase III trials) in order to obtain sufficient results to support product approval, which further increases the expense. Sometimes trials are further complicated by changes in requirements while the trials are under way (for example, when the standard of care changes for the disease that is being studied in the trial). Accordingly, any of our current or future product candidates could take a significantly longer time to gain regulatory approval than we expect, or may never gain approval, either of which could delay or stop the commercialization of our DCVax product candidates.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
| 22 |
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials.
We have limited experience in conducting and managing clinical trials.
We rely on third parties to assist us, on a contract services basis, in managing and monitoring all of our clinical trials. We do not have experience conducting late stage clinical trials ourselves without third party service firms, other than our current Phase III trial, nor do we have experience in supervising such third parties in managing late stage, multi-hundred patient clinical trials, other than our current Phase III trial. Our lack of experience and/or our reliance on these third party service firms may result in delays or failure to complete these trials successfully and on time. If the third parties fail to perform, we may not be able to find sufficient alternative suppliers of those services in a reasonable time period, or on commercially reasonable terms, if at all. If we were unable to obtain alternative suppliers of such services, we might be forced to delay, suspend or stop our 312-patient Phase III clinical trial of DCVax-L for GBM.
Multiple late stage clinical trials of DCVax-L for GBM, our lead product, may be required before we can obtain regulatory approval.
Typically, companies conduct multiple late stage clinical trials of their product candidates before seeking product approval. Our current Phase III 312-patient clinical trial of DCVax-L for GBM is our first late stage trial. We may be required to conduct additional late stage trials with DCVax-L for GBM before we can obtain product approval. This would substantially delay our commercialization. There is also some possibility that changes requested by the FDA could complicate the application process for product approval. In addition, a number of products are under development for brain cancer and at least one has recently been approved in the U.S.. It is possible that the standard of care for brain cancer could change while our Phase III trial is still under way. This could necessitate further clinical trials with our DCVax-L product candidate for brain cancer.
Changes in manufacturing methods for DCVax-L could require us to conduct equivalency studies and/or additional clinical trials.
With biologics products, “the process is the product”: i.e., the manufacturing process is considered to be as integral to the product as is the composition of the product itself. If any changes are made in the manufacturing process, and such changes are considered material by the regulatory authorities, the company sponsor may be required to conduct equivalency studies to show that the product is equivalent under the changed manufacturing processes as under the original manufacturing processes, and/or the company sponsor may be required to conduct additional clinical trials. Our manufacturing processes have undergone some changes during the early clinical trials. Accordingly, we may be required to conduct equivalency studies, and/or additional clinical trials, before we can obtain product approval, unless the regulatory authorities are satisfied that the changes in processes do not affect the quality, efficacy or safety of the product.
We may not receive regulatory approvals for our product candidates or there may be a delay in obtaining such approvals.
Our products and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the European Medicines Agency, or EMA, will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval of DCVax-L.
The time required to obtain regulatory approval varies between countries. In the U.S., for products without “Fast Track” status, it can take up to eighteen (18) months after submission of an application for product approval to receive the FDA's decision. Even with Fast Track status, FDA review and decision can take up to twelve (12) months. At present, we do not have Fast Track status for our lead product, DCVax-L for GBM. We plan to apply for Fast Track status, but there can be no assurance that FDA will grant us such status for DCVax-L.
| 23 |
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including current good manufacturing practices, or cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
Regulatory approval of our product candidates may be withdrawn at any time.
After regulatory approval has been obtained for medicinal products, the product and the manufacturer are subject to continual review, including the review of adverse experiences and clinical results that are reported after our products are made available to patients, and there can be no assurance that such approval will not be withdrawn or restricted. Regulators may also subject approvals to restrictions or conditions, or impose post-approval obligations on the holders of these approvals, and the regulatory status of such products may be jeopardized if such obligations are not fulfilled. If post-approval studies are required, such studies may involve significant time and expense.
The manufacturer and manufacturing facilities we use to make any of our products will also be subject to periodic review and inspection by the FDA or EMA, as applicable. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the product or manufacturer or facility, including withdrawal of the product from the market. We will continue to be subject to the FDA or EMA requirements, as applicable, governing the labeling, packaging, storage, advertising, promotion, recordkeeping, and submission of safety and other post-market information for all of our product candidates, even those that the FDA or EMA, as applicable, had approved. If we fail to comply with applicable continuing regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
Our product candidates will require a different distribution model than conventional therapeutic products, and this may impede commercialization of our product candidates.
Our DCVax product candidates consist of living human immune cells. Such products are entirely different from chemical or biologic drugs, and require different handling, distribution and delivery than chemical or biologic drugs. One crucial difference is that our DCVax products must remain frozen throughout the distribution and delivery process, until the time of administration to the patient, and cannot be handled at room temperature until then. In addition, our DCVax product candidates are personalized and they involve ongoing treatment cycles over several years for each patient. Each product shipment for each patient must be tracked and managed individually. For all of these reasons, among others, we will not be able to simply use the distribution networks and processes that already exist for conventional drugs. It may take time for shipping companies, hospitals, pharmacies and physicians to adapt to the requirements for handling, distribution and delivery of these products, which may adversely affect our commercialization.
Our product candidates will require different marketing and sales methods and personnel than conventional therapeutic products. Also, we lack sales and marketing experience. These factors may result in significant difficulties in commercializing our product candidates.
The commercial success of any of our product candidates will depend upon the strength of our sales and marketing efforts. We do not have a marketing or sales force and have no experience in marketing or sales of products like our lead product, DCVax-L for GBM. To fully commercialize our product candidates, we will need to recruit and train marketing staff and a sales force with technical expertise and ability to manage the distribution of our DCVax-L for GBM. As an alternative, we could seek assistance from a corporate partner or a third party services firm with a large distribution system and a large direct sales force. However, since our DCVax living cell, immune therapy products are a fundamentally new and different type of product than are on the market today, we would still have to train such partner’s or such services firms’ personnel about our products, and would have to make changes in their distribution processes and systems to handle our products. We may be unable to recruit and train effective sales and marketing forces or our own, or of a partner or a services firm, and/or doing so may be more costly and difficult than anticipated. Such factors may result in significant difficulties in commercializing our product candidates, and we may be unable to generate significant revenues.
| 24 |
We may not obtain or maintain the benefits associated with orphan drug status, including market exclusivity.
Although our lead product, DCVax-L for GBM, has been granted orphan drug status in both the United States and the European Union, or EU, we may not receive the benefits associated with orphan drug designation (including the benefit providing for market exclusivity for a number of years). This may result from a failure to maintain orphan drug status, or result from a competing product reaching the market that has an orphan designation for the same disease indication. Under U.S. and EU rules for orphan drugs, if such a competing product reaches the market before ours does, the competing product could potentially obtain a scope of market exclusivity that limits or precludes our product from being sold in the U.S. for seven years or from being sold in the EU for ten years. Also, in the EU, even after orphan status has been granted, that status is re-examined shortly prior to the product receiving any regulatory approval. The EMA must be satisfied that there is evidence that the product offers a significant benefit relative to existing therapies, in order for the therapeutic product to maintain its orphan drug status. Accordingly, our product candidates will have to re-qualify for orphan drug status prior to any potential product approval in the EU.
The availability and amount of potential reimbursement for our product candidates by government and private payers is uncertain and may be delayed and/or inadequate.
The availability and extent of reimbursement by governmental and/or private payers is essential for most patients to be able to afford expensive treatments, such as cancer treatments. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services, as CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare. Private payers tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for fundamentally novel products such as ours, as there is no body of established practices and precedents for these new products. To date, we are aware of only one active immune therapy that has reached the stage of a reimbursement decision (Provenge). Although CMS approved coverage and reimbursement for Provenge, and private payers followed suit, there remain substantial questions and concerns about reimbursement for Provenge, and such questions and concerns appear to be impeding sales.
Reimbursement agencies in Europe can be even more conservative than CMS in the U.S. A number of cancer drugs which have been approved for reimbursement in the U.S. have not been approved for reimbursement in certain European countries, and/or the level of reimbursement approved in Europe is lower than in the U.S.
Various factors could increase the difficulties for our DCVax products to obtain reimbursement. Costs and/or difficulties associated with the reimbursement of Provenge could create an adverse environment for reimbursement of other immune therapies, such as our DCVax products. Approval of other competing products (drugs and/or devices) for the same disease indications could make the need for our products and the cost-benefit balance seem less compelling. The cost structure of our product is not a typical cost structure for medical products, as the majority of our costs are incurred up front, when the manufacturing of the personalized product is done. Our atypical cost structure may not be accommodated in any reimbursement for our products. If we are unable to obtain adequate levels of reimbursement, our ability to successfully market and sell our product candidates will be adversely affected.
The manner and level at which reimbursement is provided for services related to our product candidates (e.g., for administration of our product to patients) is also important. If the reimbursement for such services is inadequate, that may lead to physician resistance and adversely affect our ability to market or sell our products.
The methodology under which CMS makes coverage and reimbursement determinations is subject to change, particularly because of budgetary pressures facing the Medicare program. For example, the Medicare Prescription Drug, Improvement, and Modernization Act, or Medicare Modernization Act, enacted in 2003, provided for a change in reimbursement methodology that has reduced the Medicare reimbursement rates for many drugs, including oncology therapeutics.
In markets outside the U.S., where we plan to operate in the future, the prices of medical products are subject to direct price controls and/or to reimbursement with varying price control mechanisms, as part of national health systems. In general, the prices of medicines under such systems are substantially lower than in the U.S. Some jurisdictions operate positive and/or negative list systems under which products may only be marketed once a reimbursement price has been agreed. Other countries allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. Accordingly, in markets outside the U.S., the reimbursement for our products may be reduced compared with the U.S. and may be insufficient to generate commercially reasonable revenues and profits.
| 25 |
Competition in the biotechnology and biopharmaceutical industry is intense and most of our competitors have substantially greater resources than we do.
The biotechnology and biopharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. Several companies, such as Novartis, Amgen and Bluebird Bio, Dendreon, Celldex Therapeutics, Inc., Activartis, Oxford Biomedica plc, Argos Therapeutics, Inc., Agenus, Inc., Prima Biomed, Ltd., Avax Technologies, Inc., Immunocellular Therapeutics, Ltd., Bavarian Nordic, Bellicum Pharmaceuticals, and others are actively involved in the research and development of immune therapies or cell-based therapies for cancer. In addition, other novel technologies for cancer are under development or commercialization, such as the electro-therapy device of NovoCure. Of these companies, only two have obtained approval of such therapy: Dendreon (for its Provenge treatment of prostate cancer) and NovoCure. Additionally, several companies, such as Medarex, Inc., Amgen, Inc., Agensys, Inc., and Genentech, Inc., are actively involved in the research and development of monoclonal antibody-based cancer therapies. Currently, a substantial number of antibody-based products are approved for commercial sale for cancer therapy, and a large number of additional ones are under development. Genentech is also engaged in several Phase III clinical trials for additional antibody-based therapeutics for a variety of cancers, and several other companies are in early stage clinical trials for such products. Many other third parties compete with us in developing alternative therapies to treat cancer, including: biopharmaceutical companies; biotechnology companies; pharmaceutical companies; academic institutions; and other research organizations, as well as some medical device companies (e.g., NovoCure and MagForce Nano Technologies AG).
We face extensive competition from companies developing new treatments for brain cancer. These include a variety of immune therapies, as mentioned above, as well as a variety of small molecule drugs and biologics drugs. There are also a number of existing drugs used for the treatment of brain cancer that may compete with our product, including, Avastin® (Roche Holding AG), Gliadel® (Eisai Co. Ltd.), and Temodar® (Merck & Co., Inc.), as well as NovoCure’s electrotherapy device.
Most of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing and sales than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly if they enter into collaborative arrangements with large and established companies.
These third parties compete with us in recruiting and retaining qualified scientific and management personnel, as well as in acquiring technologies complementary to our programs, and in obtaining sites for our clinical trials and enrolling patients.
Our competitors may develop more effective or affordable products, or achieve earlier patent protection or earlier product marketing and sales. Any products developed by us may be rendered obsolete and non-competitive.
Competing generic medicinal products may be approved.
In the EU, there exists a process for approval of generic biological medicinal products once patent protection and other forms of data and market exclusivity have expired. Arrangements for approval of generic biologics products exist and are under consideration in the U.S., as well. Other jurisdictions are considering adopting legislation that would allow the approval of generic biological medicinal products. If generic medicinal products are approved, competition from such products may substantially reduce sales of our products.
We may be exposed to potential product liability claims, and our existing insurance may not cover these claims in whole or in part. In addition, insurance against such claims may not be available to us on reasonable terms in the future, if at all.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing, marketing and sale of therapeutic products. We carry insurance coverage but this insurance may not cover any claims made. In the future, insurance coverage may not be available to us on commercially reasonable terms (including acceptable cost), if at all. Insurance that we obtain may not be adequate to cover claims against us. Regardless of whether they have any merit or not, and regardless of their eventual outcome, product liability claims may result in substantially decreased demand for our product candidates, injury to our reputation, withdrawal of clinical trial participants or physicians, and/or loss of revenues. Thus, whether or not we are insured, a product liability claim or product recall may result in losses that could be material.
| 26 |
We store, handle, use and dispose of controlled hazardous, radioactive and biological materials in our business. Our current use of these materials generally is below thresholds giving rise to burdensome regulatory requirements. Our development efforts, however, may result in our becoming subject to additional requirements, and if we fail to comply with applicable requirements we could be subject to substantial fines and other sanctions, delays in research and production, and increased operating costs. In addition, if regulated materials were improperly released at our current or former facilities or at locations to which we send materials for disposal, we could be liable for substantial damages and costs, including cleanup costs and personal injury or property damages, and we could incur delays in research and production and increased operating costs.
Insurance covering certain types of claims of environmental damage or injury resulting from the use of these materials is available but can be expensive and is limited in its coverage. We have no insurance specifically covering environmental risks or personal injury from the use of these materials and if such use results in liability, our business may be seriously harmed.
Our intellectual property rights may be overturned, narrowed or blocked, and may not provide sufficient commercial protection for our product candidates, or third parties may infringe upon our intellectual property.
Patent laws afford only limited protection and may not protect our rights to the extent necessary to sustain any competitive advantage we may have. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and we may encounter significant problems in protecting our proprietary rights in those countries. Moreover patents and patent applications relating to living cell products are relatively new, involve complex factual and legal issues, and are largely untested in litigation — and as a result, are uncertain. Third parties may challenge our existing patents, and such challenges could result in overturning or narrowing some of our patents. Even if our patents are not challenged, third parties could assert that their patents block some or all of our patents
As of December 31, 2012, we have over 180 issued and pending patents worldwide relating to our product candidates and related matters such as manufacturing processes.
The issued patents expire at various dates from 2015 to 2026. Our issued patents may be challenged, and such challenges may result in reductions in scope or invalidations. Our pending patent applications may not result in issued patents. Moreover, our patents and patent applications may not be sufficiently broad to prevent others from using substantially similar technologies or from developing competing products. We also face the risk that others may independently develop similar or alternative technologies, or design around our patented technologies.
We have taken security measures (including execution of confidentiality agreements) to protect our proprietary information, especially proprietary information that is not covered by patents or patent applications. These measures, however, may not provide adequate protection for our trade secrets or other proprietary information. In addition, others may independently develop substantially equivalent proprietary information or techniques or otherwise gain access to our trade secrets.
We may be exposed to claims or lawsuits — with or without merit — on various subjects, including that our products infringe patents or other proprietary rights of other parties.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and biopharmaceutical industries generally. The patent landscape is especially uncertain in regard to cell therapy products, as it involves complex legal and factual questions for which important legal principles remain unresolved. Infringement and other intellectual property claims — with or without merit — can be expensive and time-consuming to litigate and can divert management’s attention. We have already been exposed to one frivolous patent lawsuit by a large company, which we vigorously defended and forced the large company to withdraw all of the claims made. We have also been exposed to frivolous claims (without a lawsuit) by a competitor asserting or implying inaccurately that a recent patent issued to them somehow covers our products (which it does not). In the future, we may again be exposed to claims by third parties — with or without merit — that our products infringe their intellectual property rights. Such claims or lawsuits may involve substantial costs and diversion of management attention to defend.
In addition, because patents can take many years to issue, and patent applications are not published until up to eighteen months after they are filed, there may be currently pending applications, unknown to us, which may later result in issued patents that our products may inadvertently infringe. There could also be existing patents of which we are not aware that one or more of our products may inadvertently infringe.
| 27 |
DCVax is our only technology in clinical development.
Unlike many pharmaceutical companies that have a number of products in development and which utilize many different technologies, we are dependent on the success of our DCVax platform technology. While the DCVax technology has a wide scope of potential use, and is embodied in several different product lines for different clinical situations, if the core DCVax technology is not effective or is not commercially viable, our business could fail. We do not currently have other technologies that could provide alternative support for us.
Collaborations play an important role in our business, and could be vulnerable to competition or termination.
We work with scientists and medical professionals at academic and other institutions, including UCLA, among others, some of whom have conducted research for us or have assisted in developing our research and development strategy. These scientists and medical professionals are collaborators, not our employees. They may have commitments to, or contracts with, other businesses or institutions that limit the amount of time they have available to work with us. We have little control over these individuals. We can only expect that they devote time to us and our programs as required by any license, consulting or sponsored research agreements we may have with them. In addition, these individuals may have arrangements with other companies to assist in developing technologies that may compete with our products. If these individuals do not devote sufficient time and resources to our programs, or if they provide substantial assistance to our competitors, our business could be seriously harmed.
The success of our business strategy may partially depend upon our ability to develop and maintain our collaborations and to manage them effectively. Due to concerns regarding our ability to continue our operations or the commercial feasibility of our personalized DCVax product candidates, these third parties may decide not to conduct business with us or may conduct business with us on terms that are less favorable than those customarily extended by them. If either of these events occurs, our business could suffer significantly.
We may have disputes with our collaborators, which could be costly and time consuming. Failure to successfully defend our rights could seriously harm our business, financial condition and operating results. We intend to continue to enter into collaborations in the future. However, we may be unable to successfully negotiate any additional collaboration and any of these relationships, if established, may not be scientifically or commercially successful.
Our business could be adversely affected by new legislation and/or product related issues.
Changes in applicable legislation and/or regulatory policies or discovery of problems with the product, production process, site or manufacturer may result in delays in bringing products to market, the imposition of restrictions on the product’s sale or manufacture, including the possible withdrawal of the product from the market, or may otherwise have an adverse effect on our business.
Our business could be adversely affected by animal rights activists.
Our business activities have involved animal testing, as such testing is required before new medical products can be tested in clinical trials in human patients. Animal testing has been the subject of controversy and adverse publicity. Some organizations and individuals have attempted to stop animal testing by pressing for legislation and regulation in these areas. To the extent that the activities of such groups are successful, our business could be adversely affected. Negative publicity about us, our pre-clinical trials and our product candidates could also adversely affect our business.
Risks Related to our Common Stock
The market price of our common stock may be volatile and adversely affected by several factors.
The share prices of publicly traded biotechnology and emerging pharmaceutical companies, particularly companies without consistent product revenues and earnings, can be highly volatile and are likely to remain highly volatile in the future. The price which investors may realize in sales of their shares of our common stock may be materially different than the price at which our common stock is quoted, and will be influenced by a large number of factors, some specific to us and our operations, and some unrelated to our operations. Such factors may cause the price of our stock to fluctuate frequently and substantially. Such factors may include large purchases or sales of our common stock, positive or negative events relating to other companies developing immune therapies for cancer, positive or negative events relating to healthcare and the overall pharmaceutical and biotech sector, currency fluctuations, legislative or regulatory changes, and/or general economic conditions. In the past, shareholder class action litigation has been brought against other companies that experienced volatility in the market price of their shares. Whether or not meritorious, litigation brought against a company following fluctuations in the trading price of its common stock can result in substantial costs, divert management’s attention and resources, and harm the company’s financial condition and results of operations.
| 28 |
Toucan Capital and its affiliates are the principal holders of our shares of common stock, and this concentration of ownership may have a negative effect on the market price of our common stock.
As of March 31, 2013, Toucan Capital and its affiliates (including Cognate BioServices, Toucan Partners and Linda Powers, who also serves as our Chief Executive Officer and Chairperson of the Board of Directors), collectively, owned an aggregate of 10,898,882 shares of our common stock, representing approximately 40 percent of our issued and outstanding common stock. This concentration of ownership may adversely affect the trading price of our common stock because investors may perceive disadvantages in owning stock of companies with controlling stockholders. Toucan Capital and its affiliates have the ability to exert substantial influence over all matters requiring approval by our stockholders, including the election and removal of directors and any proposed merger, consolidation or sale of all or substantially all of our assets. This influence could have the effect of delaying, deferring or preventing a change in control, or impeding a merger or consolidation, takeover or other business combination that could be favorable to investors.
The requirements of the Sarbanes-Oxley Act of 2002 and other U.S. securities laws impose substantial costs, and may drain our resources and distract our management.
We are subject to certain of the requirements of the Sarbanes-Oxley Act of 2002 in the U.S., as well as the reporting requirements under the Exchange Act. The Exchange Act requires, among other things, filing of annual reports on Form 10-K, quarterly reports on Form 10-Q and periodic reports on Form 8-K following the happening of certain material events, with respect to our business and financial condition. The Sarbanes-Oxley Act recommends among other things, that we maintain effective disclosure controls and procedures and internal controls over financial reporting. Our existing controls have some weaknesses, as described below. Meeting the requirements of the Exchange Act and the Sarbanes-Oxley Act may strain our resources and may divert management's attention from other business concerns, both of which may have a material adverse effect on our business.
Our management and our independent auditor have identified internal control deficiencies, which our management and our independent auditor believe constitute material weaknesses.
In connection with the preparation of our financial statements for the year ended December 31, 2012, and prior years, our management and our independent auditor identified certain internal control deficiencies that, in the aggregate, represent material weaknesses, including:
| • | insufficient segregation of duties and oversight of work performed in our finance and accounting function due to limited personnel, and | |
| • | lack of controls in place to ensure that all material transactions and developments impacting the financial statements are reflected. |
As part of our independent auditors’ communications with our audit committee with respect to audit procedures for the year ended December 31, 2012, our independent auditors informed the audit committee that these deficiencies constituted material weaknesses, as defined by Auditing Standard No. 5, “An Audit of Internal Control Over Financial Reporting that is Integrated with an Audit of Financial Statements and Related Independence Rule and Conforming Amendments,” established by the Public Company Accounting Oversight Board, or PCAOB. We have begun taking appropriate and reasonable steps, and will continue and complete such steps in due course, to make the necessary improvements to address these deficiencies, but the timing of such steps is uncertain and the availability of funding and resources for such steps are also uncertain. Our ability to retain qualified individuals to serve on our Board and to take on key management roles within the Company is also uncertain. Our failure to successfully complete the remedies of the existing weaknesses could lead to heightened risk for financial reporting mistakes and irregularities, and/or lead to a loss of public confidence in our internal controls that could have a negative effect on the market price of our common stock.
| 29 |
We do not intend to pay any cash dividends in the foreseeable future and, therefore, any return on your investment in our common stock must come from increases in the market price of our common stock.
We have not paid any cash dividends on our common stock to date in our history, and we do not intend to pay cash dividends on our common stock in the foreseeable future. We intend to retain future earnings, if any, for reinvestment in the development and expansion of our business. Also, any credit agreements which we may enter into with institutional lenders may restrict our ability to pay dividends. Therefore, any return on your investment in our capital stock must come from increases in the fair market value and trading price of our common stock. Such increases in the trading price of our stock may not occur.
Our certificate of incorporation and bylaws, our shareholder rights plan and Delaware law have anti-takeover provisions that could discourage, delay or prevent a change in control, which may cause our stock price to decline.
Our certificate of incorporation and bylaws and Delaware law contain provisions which could make it more difficult for a third party to acquire us, even if closing such a transaction would be beneficial to our stockholders. We are authorized to issue up to 40,000,000 shares of preferred stock. This preferred stock may be issued in one or more series, the terms of which may be determined at the time of issuance by our Board of Directors without further action by stockholders. The terms of any series of preferred stock may include voting rights (including the right to vote as a series on particular matters), preferences as to dividend, liquidation, conversion and redemption rights and sinking fund provisions. No preferred stock is currently outstanding. The issuance of any preferred stock could materially adversely affect the rights of the holders of our common stock, and therefore, reduce the value of our common stock. In particular, specific rights granted to future holders of preferred stock could be used to restrict our ability to merge with, or sell our assets to, a third party and thereby preserve control by the present management.
Provisions of our certificate of incorporation and bylaws and Delaware law also could have the effect of discouraging potential acquisition proposals or making a tender offer or delaying or preventing a change in control, including changes a stockholder might consider favorable. Such provisions may also prevent or frustrate attempts by our stockholders to replace or remove our management. In particular, the certificate of incorporation and bylaws and Delaware law, as applicable, among other things:
| · | provide the Board of Directors with the ability to alter the bylaws without stockholder approval; |
| · | establish staggered terms for board members; |
| · | place limitations on the removal of directors; and |
| · | provide that vacancies on the Board of Directors may be filled by a majority of directors in office, although less than a quorum. |
We expect to adopt a shareholder rights plan and declare a dividend distribution of one right for each outstanding share of common stock as fixed by our Board of Directors. Each right, when exercisable, will entitle the registered holder to purchase from us shares of a new series of preferred stock on the terms stated in the rights plan. The rights will generally separate from the common stock and become exercisable if any person or group acquires or announces a tender offer to acquire 15% or more of our outstanding common stock without the consent of our Board of Directors. Because the rights may substantially dilute the stock ownership of a person or group attempting to take us over without the approval of our Board of Directors, our stockholder rights plan could make it more difficult for a third party to acquire us (or a significant percentage of our outstanding capital stock) without first negotiating with our Board of Directors.
We are also subject to Section 203 of the Delaware General Corporation Law which, subject to certain exceptions, prohibits “business combinations” between a publicly-held Delaware corporation and an “interested stockholder,” which is generally defined as a stockholder who becomes a beneficial owner of 15% or more of a Delaware corporation’s voting stock for a three-year period following the date that such stockholder became an interested stockholder.
These provisions are expected to discourage certain types of coercive takeover practices and inadequate takeover bids and to encourage persons seeking to acquire control of our company to first negotiate with its Board. These provisions may delay or prevent someone from acquiring or merging with us, which may cause the market price of our common stock to decline.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None
| 30 |
ITEM 2. PROPERTIES
On July 31, 2012, we entered into a non-cancelable operating lease for 7,097 square feet of office space in Bethesda, Maryland, which expires in March 2018. As of December 31, 2012, obligations for future minimum lease payments under the lease, in aggregate over the rest of the lease term, total approximately $1.5 million.
ITEM 3. LEGAL PROCEEDINGS
On March 1, 2011, we entered into a transaction under Section 3(a)(10) of the Securities Act of 1933, as amended with Socius CG II, Ltd. (“Socius”). Pursuant to this 3(a)(10) transaction, Socius purchased certain claims for payment totaling $1,650,000 from Cognate. Thereafter, Socius elected to convert the claims into shares of our common stock. The conversion was effected through a settlement agreement between Socius and us. The settlement agreement was then the subject of a court proceeding (nominally brought by Socius against us, but handled on a cooperative basis through a Joint Stipulation by both parties) in order to obtain court approval of the settlement in accordance with the requirements of Section 3(a)(10). That Court approval was obtained on March 1, 2011. Pursuant to the settlement, the full amount of the $1,650,000 debt was converted into shares of common stock and the transaction with Socius was completed.
From time to time, we are involved in claims and suits that arise in the ordinary course of our business. At present, we are not involved in any suits.
ITEM 4. MINE SAFETY DISCLOSURES
Not Applicable
PART II
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUERS PURCHASES OF EQUITY SECURITIES
Market for Common Equity and Related Stockholder Matters
Our common stock and warrants trade on The NASDAQ Capital Market under the trading symbols “NWBO” and “NWBOW” effective December 12, 2012. Prior to listing on The NASDAQ Capital Market, our common stock was quoted on the OTCQB beginning on July 23, 2012. Previously our common stock was quoted on the Over The Counter Bulletin Board from December 23, 2002 to July 23, 2012. The table below sets forth the high and low prices for our common stock for the last two recent fiscal years. Quotations reflect inter-dealer prices, without retail mark-up, mark-down commission, and may not represent actual transactions. No assurance can be given that an active market will exist for our common stock.
| High | Low | |||||||
| Year end December 31, 2012 | ||||||||
| First Quarter | $ | 6.40 | $ | 4.64 | ||||
| Second Quarter | 5.44 | 3.04 | ||||||
| Third Quarter | 10.95 | 3.49 | ||||||
| Fourth Quarter | 8.00 | 3.03 | ||||||
| Year end December 31, 2011 | ||||||||
| First Quarter | $ | 12.64 | $ | 5.03 | ||||
| Second Quarter | 14.24 | 6.24 | ||||||
| Third Quarter | 12.00 | 5.68 | ||||||
| Fourth Quarter | 9.12 | 5.12 | ||||||
| 31 |
As of March 31, 2013, there were approximately 5,000 holders of record of our common stock. Such holders may include any broker or clearing agencies as holders of record, and in such cases exclude the individual stockholders whose shares are held by such brokers or clearing agencies.
Effective September 25, 2012, all shares of our common stock issued and outstanding were combined and reclassified on a one-for-sixteen basis.
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all future earnings, if any, to fund the ongoing development and growth of our business. We do not currently anticipate paying any cash dividends in the foreseeable future.
Securities Authorized for Issuance under Equity Compensation Plan
We have an employee stock option plan (“2007 Stock Option Plan”) under which 2,250,000 shares had been reserved for issuance as of December 31, 2012. As of January 1, 2013, the number of shares authorized for issuance under the 2007 Stock Option Plan is 5,309,166, of which 1,551,000 shares have been reserved for issuance pursuant to outstanding stock options (see below). The following table shows information with respect this plan as of December 31, 2012 (options in thousands).
Equity Compensation Plan Information
| Plan category | Number of securities to be issued upon Exercise of outstanding options, warrants and Rights (a) | Weighted-average exercise price of Outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
| Equity compensation plans approved by security holders | 1,551 | $ | 10.56 | 699 | ||||||||
| Equity compensation plans not approved by security holders | — | — | — | |||||||||
| Total | 1,551 | $ | 10.56 | 699 | ||||||||
Amended and Restated 2007 Stock Option Plan
| 32 |
Summary of the Plan
General. The Plan authorizes the grant to eligible individuals of two types of stock option awards — (1) Incentive Stock Options, within the meaning of Section 422 of the Internal Revenue Code of 1986, as amended (the “Code”); and (2) Nonstatutory Stock Options – as well as other types of securities such as shares or restricted stock units.
Stock Subject to the Plan. The 2007 Stock Option Plan became effective on June 15, 2007. In April 2008, we increased the number of shares reserved for issuance by 32,446 shares for an aggregate of 375,000 shares. In May 2010, the Board approved an increase in the number of shares reserved for issuance under the 2007 Stock Option Plan by an additional 625,000 shares. In 2011, the Board approved an increase in the shares available by 1.25 million. The increases approved by the Board in May 2010 and 2011 were not (prior to the stockholder action described) submitted to stockholders. In August 2012 as previously reported, the Board and shareholders approved amendments of the Plan under which, on an ongoing basis, effective as of January 1 each year (starting with January 1, 2013), the aggregate numbers of shares of Common Stock that are available for issuance shall automatically be increased in such manner as to maintain the option pool capped at twenty percent of our issued and outstanding stock. As of January 1, 2013, the number of shares authorized for issuance under the Plan is 5,309,166, of which 1,551,000 shares have been reserved for issuance pursuant to outstanding stock options. Pursuant to the Plan, if on any January 1st 20% of our total issued and outstanding stock is less than the number of shares of Common Stock available for issuance under the Plan, no change will be made to the aggregate number of shares of Common Stock issuable under the Plan for that year (such that the aggregate number of shares available for issuance under the Plan will never decrease).
Eligibility. Employees of, and consultants to, us or our affiliates, and members of the Board are eligible to receive stock options under the Plan. Only our employees are eligible to receive Incentive Stock Options. Employees, directors (including non-employee directors) and consultants are eligible to receive Nonstatutory Stock Options.
Purpose. The Plan is designed to: (1) encourage our employees, directors and consultants to exert maximum efforts for our success; and (2) provide such individuals with an opportunity to benefit from increases in the value of the Common Stock.
Administration. The Board has delegated to the Compensation Committee administration authority over the Plan. Subject to the provisions of the Plan, the Board and/or Compensation Committee has the power to construe and interpret the Plan and to make key determinations such as the persons to whom and the dates on which stock options or other securities will be granted, the number of shares of Common Stock to be subject to each option or other security, the time or times during the term of each option within which all or a portion of such option may be exercised, the exercise price, the type of consideration and other terms of the option.
Term. The Board may suspend or terminate the Plan without stockholder approval or ratification at any time or from time to time. Unless sooner terminated, the Plan will terminate on June 14, 2017. The Board may also amend the Plan at any time or from time to time. However, except in the case of a Capitalization Adjustment, stockholder approval shall be required, but only to the extent required by applicable law or listing requirements, for any amendment to the Plan that either (i) materially increases the number of shares of Common Stock available for issuance under the Plan, (ii) materially expands the class of individuals eligible to receive options under the Plan, (iii) materially increases the benefits accruing to participants under the Plan or materially reduces the price at which shares of Common Stock may be issued or purchased under the Plan, (iv) materially extends the term of the Plan, (v) expands the types of stock awards available for issuance under the Plan, or (vi) changes any other provision of the Plan in any other way if such modification requires stockholder approval in order to comply with Rule 16b-3 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) or satisfy the requirements of Section 422 of the Code or any securities exchange listing requirements. The Board may submit any other amendment to the Plan for stockholder approval, including, but not limited to, amendments intended to satisfy the requirements of Section 162(m) of the Code regarding the exclusion of performance-based compensation from the limitation on the deductibility of compensation paid to certain employees.
| 33 |
Recent Sales of Unregistered Securities
In October and November 2012, we issued 1.9 million shares of redeemable common stock, at a purchase price of $4.80 per share to accredited investors in separate private placement transactions and as part of conversions of debt into redeemable common stock. Total cash received amounted to $5.3 million and total conversions of debt into redeemable common stock amounted to $4.6 million. These transactions were completed pursuant to a Securities Purchase Agreement which we entered into with each investor. The agreements provide that such investors can redeem the securities for cash at a premium to the original issuance ranging from 15% to 25%. The redemption provisions occur within 12 months from the date of issuance.
The sales of the securities identified above were made pursuant to privately negotiated transactions that did not involve a public offering of securities and, accordingly, we believe that these transactions were exempt from the registration requirements of the Securities Act pursuant to Section 4(2) of the Securities Act and Rule 506 of Regulation D. The agreements executed in connection with this sale contain representations to support our reasonable belief that the security holder had access to information concerning our operations and financial condition, the security holder acquired the securities for their own account and not with a view to the distribution thereof in the absence of an effective registration statement or an applicable exemption from registration, and that the security holder is sophisticated within the meaning of Section 4(2) of the Securities Act and are “accredited investors” (as defined by Rule 501 under the Securities Act). In addition, the issuances did not involve any public offering; we made no solicitation in connection with the sale other than communications with the security holder; we obtained representations from the security holder regarding their investment intent, experience and sophistication; and the security holder either received or had access to adequate information about us in order to make an informed investment decision. All of the foregoing securities are deemed restricted securities for purposes of the Securities Act.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read this discussion together with the Financial Statements, related Notes and other financial information included elsewhere in this Form 10-K. The following discussion contains assumptions, estimates and other forward-looking statements that involve a number of risks and uncertainties, including those discussed under “Risk Factors,” and elsewhere in this Form 10-K. These risks could cause our actual results to differ materially from those anticipated in these forward-looking statements
Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in conformity with accounting principles generally accepted in the United States of America. In preparing these financial statements, we make assumptions, judgments and estimates that can have a significant impact on amounts reported in our financial statements. We base our assumptions, judgments and estimates on historical experience and various other factors that we believe to be reasonable under the circumstances. Actual results could differ materially from these estimates under different assumptions or conditions. On a regular basis we evaluate our assumptions, judgments and estimates and make changes accordingly. We believe that, of the significant accounting policies discussed in Note 3 to our consolidated financial statements, the following accounting policies require our most difficult, subjective and/or complex judgments:
Policy for Reclassified Equity Contracts
We account for potential shares that can be converted to common stock and if converted, will be in excess of authorized shares, as a liability that is recorded on the balance sheet (at fair value) only until the authorized number of shares is increased (at which time the whole liability will be re-measured, with changes in value included in other income/(expense), and then reclassified to additional paid-in capital).
Embedded Derivative Liability
We evaluate financial instruments for freestanding or embedded derivatives. Derivative instruments that have been separated from the host contract and do not qualify for hedge accounting are recorded at fair value with changes in value recognized as other income (expense) in the consolidated statements of operations in the period of change.
| 34 |
Fair Value Measurements
Fair value is defined as the price that would be received for sale of an asset or paid for transfer of a liability, in an orderly transaction between market participants at the measurement date. U.S. GAAP establishes a three-tier fair value hierarchy which prioritizes the inputs used in measuring fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). These tiers include:
| · | Level 1, defined as observable inputs such as quoted prices for identical instruments in active markets; |
| · | Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable such as quoted prices for similar instruments in active markets or quoted prices for identical or similar instruments in markets that are not active; and |
| · | Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions, such as valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable. |
Stock-based Compensation
Compensation expense for all stock-based awards is measured at the grant date based on the fair value of the award and is recognized as an expense, on a straight-line basis, over the employee's requisite service period (generally the vesting period of the equity award). The fair value of each option award is estimated on the date of grant using a Black-Scholes option valuation model. Stock-based compensation expense is recognized only for those awards that are expected to vest using an estimated forfeiture rate. Estimates of pre-vesting forfeiture are periodically revised in subsequent periods if actual forfeitures differ from those estimates. To the extent that actual results differ from our estimates, such amounts will be recorded as cumulative adjustments in the period the estimates are revised.
Redeemable Securities
We account for redeemable Common Stock in accordance with Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) ASC 480-10-S99-3A,Classification and Measurement of Redeemable Securities, which provides that securities that are optionally redeemable by the holder for cash or other assets are classified outside of permanent equity in temporary equity.
Recent Accounting Pronouncements
In September 2011, the Financial Accounting Standards Board issued ASU No. 2011-05, Comprehensive Income, or ASU 2011-05. The guidance in ASU 2011-05 revises the manner in which entities present comprehensive income in their financial statements. An entity is required to report the components of comprehensive income in either one or two consecutive financial statements:
| · | a single, continuous statement must present the components of net income and total net income, the components of other comprehensive income and total other comprehensive income, and a total for comprehensive income; or |
| · | in a two-statement approach, an entity must present the components of net income and total net income in the first statement. That statement must be immediately followed by a financial statement that presents the components of other comprehensive income, a total for other comprehensive income, and a total for comprehensive income. |
ASU 2011-05 does not change the items that must be reported in other comprehensive income. The amendments in ASU 2011-05 are effective for fiscal years beginning after December 15, 2011. We adopted this guidance, and implemented the two-statement approach.
Results of Operations
Operating costs:
Operating costs and expenses consist primarily of research and development expenses, including clinical trial expenses which increase when we are actively participating in clinical trials (and are especially high when we are in a phase III trial, as we now are), and general and administrative expenses.
| 35 |
Research and development:
Discovery and preclinical research and development expenses include scientific personnel-related salary and benefit expenses, costs of laboratory supplies used in our internal research and development projects, travel, regulatory compliance, and expenditures for preclinical and clinical trial operation and management when we are actively engaged in clinical trials.
Because we are a development stage company, we do not allocate research and development costs on a project basis. We adopted this policy, in part, due to the unreasonable cost burden associated with accounting at such a level of detail and our limited number of financial and personnel resources.
General and administrative:
General and administrative expenses include administrative personnel related salary and benefit expenses, cost of facilities, insurance, travel, legal support, property and equipment and amortization of stock options and warrants.
Twelve Months Ended December 31, 2012 and 2011
We recognized a net loss of $22.8 million in cash outlays and $44.5 million in non-cash accounting charges (i.e. inducement expense, stock-based compensation, beneficial conversion charges etc.), for a combined (cash and non-cash) total net loss of $67.3 million for the twelve months ended December 31, 2012 compared to a net loss of $32.8 million for the twelve months ended December 31, 2011.
Research and Development Expense.Research and development expense was a combined (cash and non-cash) total of $28.9 million for the twelve months ended December 31, 2012 compared to $13.5 million for the twelve months ended December 31, 2011. The increase was primarily attributable to increased number of clinical trial sites open and recruiting across the United States in our ongoing Phase III clinical trial of DCVax®-L immune therapy for GBM, as well as expansion of the trial into Europe.
As of December 31, 2012 we had 42 clinical trial sites in operation in the U.S., compared to 25 clinical trial sites at December 31, 2011. We also had established a wholly owned subsidiary in Germany and substantially expanded both our manufacturing operations and our clinical trial related activities in both the U.K. and Germany, compared to December 31, 2011.
General and Administrative Expense.General and administrative expense was a combined (cash and non-cash) $15.7 million for the twelve months ended December 31, 2012 compared to $13.3 million for the twelve months ended December 31, 2011. The increase in general and administrative expenses is as a result of the issuance of warrants for fundraising services to related parties and expenses related to investor relations and public relations.
Valuation of reclassified equity contracts. During the twelve months ended December 31, 2012, the Company recognized a non-cash gain amounting to $0.5 million from the decrease in value of reclassified equity contracts. There was a non-cash gain of $8.8 million in reclassified equity contracts during the twelve months ended December 31, 2011.
Derivative valuation gain and loss. During the twelve months ended December 31, 2012 we recognized a gain on derivative liabilities of $0.6 million due to the change in value of the financial instruments. We recognized a $0.6 million gain in the twelve months ended December 31, 2011.
Interest (Expense). Interest expense (including non-cash elements such as amortization of debt discount and accretion on redeemable securities) increased to $13.4 million for the twelve months ended December 31, 2012 from $7.7 million for the twelve months ended December 31, 2011. Interest expense increase was primarily attributable to financing transactions including the repayment of convertible notes, conversion of notes and issuance of redeemable securities.
Liquidity and Capital Resources
We have experienced recurring losses from operations. Net cash outflows from operations were $22.8 million for the twelve months ended December 31, 2012.
| 36 |
At December 31, 2012, cash and cash equivalents totaled $7.3 million, compared to $0.02 million at December 31, 2011. Working capital was a deficit of $3.3 million at December 31, 2012, compared to a deficit of $50.6 million at December 31, 2011. The working capital deficit decreased as of December 31, 2012 as compared to December 31, 2011 primarily due to the retirement of $36.4 million in convertible notes, notes and payables and accrued interest by entering into Conversion Agreements with our non-affiliated and affiliated note holders and creditors, including certain of our directors and executive officers. This $36.4 million aggregate debt amount was converted into 9.8 million common shares (of which 0.9 million are redeemable) and warrants exercisable for 3.8 million shares of common stock. The warrants have an exercise period of five years from the date of issuance and a weighted average exercise price of $3.66 per share.
Operating Activities
We used $22.8 million in cash for operating activities during the twelve months ended December 31, 2012. The increase in cash used in operating activities was primarily attributable to increased number of clinical trial sites open and recruiting across the United States, and expansion of both the trial activities and the manufacturing activities and capacity in Europe, in our ongoing Phase III clinical trial of DCVax®-L immune therapy for GBM.
As of December 31, 2012 we had 42 clinical trial sites in operation in the U.S., compared to 25 clinical trial sites at December 31, 2011. At December 31, 2012, we also had substantially expanded operations compared with December 31, 2011 including, for example, fully operational and approved manufacturing in Germany, an established wholly owned German subsidiary with a CEO who is an industry veteran from the senior management of big pharma, manufacturing activity in the U.K. and nearly 30 clinical trial sites in the U.K. and Germany in varying stages of preparation.
Investing Activities
Investing activities for the periods presented are not material.
Financing Activities
During the twelve months ended December 31, 2012, our financing activities consisted of proceeds from the issuance of convertible notes payable amounting to $13.1 million, proceeds from the issuance of common stock amounting to $13.9 million as well as the repayment of $2.2 million of notes payable, and net proceeds from the issuance of redeemable securities of $5.3 million. The increase in our financing activities was primarily attributable to raising financing to support the increased number of clinical trial sites open and recruiting across the U.S. in our ongoing Phase III clinical trial of DCVax®-L immune therapy for GBM.
In order to continue with our current activities under our DCVax®-L program, we will have to obtain substantial amounts of further funding, as described in the Risk Factors section. Our on-going funding requirements will depend on many factors, including the extent to which we realize and draw upon various sources of non-dilutive funding. One such source of non-dilutive funding is a $5.5 million German grant awarded on May 1, 2012, by the German government through its Saxony Development Bank. The grant will provide funding on matching basis for up to 50% of the costs incurred by the Company for the DCVax-L clinical trial and manufacturing in Germany. Upon the Company’s presentation of such eligible expenditures, the Company will receive funds from the grant. Our first draws under this grant are anticipated to occur in Q2 of 2013.
Other factors affecting our ongoing funding requirements include the number of staff we employ, the pace of patient enrollment in our Phase III brain cancer trial, the pace and number of sites at which we execute our Phase I/II clinical trials with DCVax-Direct, the cost of establishing clinical studies and compassionate use/named patient programs in other countries, and unanticipated developments. The extent of resources available to us will determine the pace at which we can move forward with both our DCVax-L program and our DCVax-Direct program.
| 37 |
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The full text of our audited consolidated financial statements as of December 31, 2012 and 2011 and for the fiscal years ended December 31, 2012 and 2011, begins on page F-1 of this Annual Report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our principal executive, financial and accounting officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended. Based on this evaluation, our principal executive, financial and accounting officer concluded that, as of December 31, 2012, in light of the material weaknesses described below, our disclosure controls and procedures were not effective to ensure that the information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act of 1934 is accumulated and communicated to management, including our chief executive officer, financial and accounting officer, to allow timely decisions regarding required disclosure, and that such information is recorded, processed, summarized and reported within the time periods prescribed by the SEC.
Management's Report on Internal Controls Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal controls over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our principal executive, financial and accounting officer, we conducted an evaluation of the effectiveness of our internal controls over financial reporting as of December 31, 2012. This evaluation was based on the framework in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO").
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company's annual or interim financial statements will not be prevented or detected on a timely basis.
Based on management's evaluation as of December 31, 2012, our management identified the material weaknesses set forth below in our internal control over financial reporting:
| (i) | The Company's process for internally reporting material information in a systematic manner to allow for timely filing of material information is ineffective, due to its inherent limitations from being a small company, and there exist material weaknesses in internal control over financial reporting that contribute to the weaknesses in our disclosure controls and procedures. These weaknesses include: |
| • | insufficient segregation of duties and oversight of work performed in our finance and accounting function due to limited personnel; and |
| • | lack of controls in place to ensure that all material transactions and developments impacting the financial statements are reflected. |
| 38 |
Our company's management concluded that in light of the material weaknesses described above, our company did not maintain effective internal control over financial reporting as of December 31, 2012 based on the criteria set forth in Internal Control—Integrated Framework issued by the COSO.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management's report was not subject to attestation by our public accounting firm pursuant to rules of the SEC that permit us to provide only management's report in this annual report.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting during the quarter ended December 31, 2012 that have materially affected, or are reasonable likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The following table sets forth information regarding the members of the Company’s current board of directors and its executive officers. All directors hold office until the election and qualification of their successors or their earlier removal or retirement.
| Name | Age | Position | ||
| Linda F. Powers | 56 | Class III Director, Chairperson, President and Chief Executive Officer | ||
| Alton L. Boynton, Ph.D. | 68 | Class I Director, Chief Scientific Officer | ||
| Anthony Maida, Ph.D. | 59 | Chief Operating Officer | ||
| Leslie Goldman | 66 | Senior Vice President, Business Development | ||
| Marnix Bosch, Ph.D. | 52 | Chief Technical Officer | ||
| Robert A. Farmer | 72 | Class II Director | ||
| Dr. Navid Malik | 43 | Class III Director | ||
| Jerry Jasinowski | 69 | Class II Director |
Our Board of Directors is classified into three classes, with the term of office of one class expiring each year. The term of Class I directors expires at the Company’s annual meeting of stockholders to be held in 2013, the term of Class II directors expires at the Company’s annual meeting of stockholders to be held in 2014, and the term of office of Class III directors expires at the Company’s annual meeting of stockholders to be held in 2015.
| 39 |
Executive Biographies
The principal occupations for the past five years (and, in some instances, for prior years) of each of our directors and executive officers are as follows:
Linda F. Powers. Ms. Powers has served as the Chairperson of our Board of Directors since her appointment on May 17, 2007 and Chief Executive Officer since June 8, 2011. Ms. Powers served as a managing director of Toucan Capital Fund II, a provider of venture capital, for a decade, starting in 2001. She also has over 15 years’ experience in corporate finance and restructurings, mergers and acquisitions joint ventures and intellectual property licensing. Ms. Powers is a Board member of M2GEN (an affiliate of Moffitt Cancer Center), the Trudeau Institute (a specialized research institute focused on immunology), the Chinese Biopharmaceutical Association, and the Rosalind Franklin Society. She was the Chair of the Maryland Stem Cell Research Commission for the first two years of the state’s stem cell funding program, and has served an additional 4 years on the Commission. Ms. Powers served for several years on a Steering Committee of the National Academy of Sciences, evaluating government research funding, and has been appointed to three Governors’ commissions created to determine how to build the respective states’ biotech and other high-tech industries. For six years, Ms. Powers taught an annual internal course at the National Institutes of Health for the bench scientists and technology transfer personnel on the development and commercialization of medical products. Ms. Powers serves on the Boards of six private biotechnology companies. Ms. Powers holds a B.A. from Princeton University, where she graduated magna cum laude and Phi Beta Kappa. She also earned a JD, magna cum laude, from Harvard Law School. We believe Ms. Powers’ background and experience makes her well qualified to serve as a Director.
Alton L. Boynton, Ph.D. Dr. Boynton co-founded our company, has served as our Chief Scientific Officer and a Director since our inception in 1998, was appointed our Chief Operating Officer in August 2001, was appointed President in May, 2003, and served as Chief Executive Officer from June, 2007, to June, 2011. Prior to founding our company, Dr. Boynton headed the Molecular Oncology research lab at the Pacific Northwest Research Foundation (the original foundation of Bill Hutchinson, from which the Fred Hutchinson Cancer Center was spun off). Dr. Boynton has also served as Director of the Department of Molecular Medicine of Northwest Hospital from 1995-2003 where he coordinated the establishment of a program centered on carcinogenesis. Prior to joining Northwest Hospital, Dr. Boynton was Associate Director of the Cancer Research Center of Hawaii, The University of Hawaii, where he also held the positions of Director of Molecular Oncology of the Cancer Research Center and Professor of Genetics and Molecular Biology. Dr. Boynton received his Ph.D. in Radiation Biology from the University of Iowa in 1972. As a result of Dr. Boynton’s significant years of service as a director and running a number of programs focusing on oncology and cancer-related research programs, including a Ph.D. in Radiation Biology, we concluded that Dr. Boynton is well qualified to serve as a Director.
Anthony E. Maidajoined our company in June, 2011, as Chief Operating Officer bringing more than 20 years’ experience in building oncology companies, with expertise in the business, financial, clinical and regulatory aspects of and the underlying science of oncology business. Over these two decades, Dr. Maida has held positions as Chairman, CEO, COO, CSO, CFO and VP Business Development. Among these experiences, he served as CEO of CancerVax Corporation, an early leader in cancer vaccines. In that role, he was responsible for conducting multi-hundred patient, multi-center clinical trials with the company’s cancer vaccines. Prior to joining us, Dr. Maida was serving as global head of oncology for a leading contract research organization that manages clinical trials in the U.S. and internationally.
Leslie J. Goldman joined us as Senior Vice President, Business Development, in June, 2011. Prior to joining us, Mr. Goldman was a partner at the law firm of Skadden, Arps for over 30 years, specializing in a wide array of advanced technologies and their commercialization. Mr. Goldman also serves as an advisor to a number of other technology companies. In addition, for eight years, Mr. Goldman has served as Chairman of the Board of a group of TV stations in four mid-size cities across the country.
Marnix L. Boschjoined us in 2000, and has been serving as Chief Technical Officer for a number of years. In this capacity, he plays a key role in the preparation and submission of our regulatory applications, as well as ongoing development of our product lines, and ongoing development and/or acquisition of new technologies. Dr. Bosch led the process of designing the protocols, and managed the successful preparation and submission of our Investigational New Drug (IND) applications for FDA approval to conduct clinical trials, for prostate cancer, brain cancer and multiple other cancers. He also led the processes for other regulatory submissions in both the U.S. and abroad (including the successful applications for orphan drug status in both the U.S. and Europe for DCVax-L for brain cancer). He spearheaded the development of our manufacturing and quality control processes, and is working with Cognate on next-generation further development of these processes. Prior to joining us in 2000, Dr. Bosch worked at the Dutch National Institutes of Health (RIVM) as head of the Department of Molecular Biology, as well as in academia as a professor of Pathobiology. He has authored more than 40 peer-reviewed research publications in immunology and virology, and is an inventor on several patent applications on dendritic cell product manufacturing.
Robert A. Farmerwas appointed to the Board of Directors in December 2009. Mr. Farmer served as the national treasurer of four presidential campaigns, including those for John Kerry, Bill Clinton, Michael Dukakis and John Glenn. In these roles he led fundraising of over $800 million. He served under Ron Brown as treasurer of the Democratic National Committee, and served for eight years as treasurer of the Democratic Governor’s Association. President Clinton appointed Farmer as the United States Consul General to Bermuda, where he served from 1994 to 1999. Mr. Farmer also had a successful career as an entrepreneur, including building his own publishing company, which he sold in 1983. Mr. Farmer currently serves on the Boards of Directors of International Data Group, Dale Carnegie Associates, Sober Steering Sensors, LLC, Charlesbridge Publishing, and Haute Living. Mr. Farmer is a graduate of Dartmouth College and Harvard Law School. Mr. Farmer’s experience in four presidential campaigns and his service on other Boards of Directors make him well qualified to serve as a Director.
| 40 |
Dr. Navid Malikwas appointed to the Board of Directors in April 2012. Dr. Navid Malik is the Head of Life Sciences Research at Cenkos Securities Plc. in the U.K., an independent specialist institutional securities firm. From September 2011 through January 2012, Dr. Malik was the Head of Life Sciences Research at Sanlam (Merchant Securities), a global financial services firm. Dr. Malik was Partner and Head of Life Sciences at Matrix Investment Banking Division, Matrix Group, a financial services firm in London, from December, 2008, through September, 2011. Dr. Malik was a Senior Pharmaceuticals and Biotechnology Analyst at Wimmer Financial LLP from September, 2008, through December, 2008, and was the Senior Life Sciences Analyst at Collins Stewart Plc from January, 2005, through September, 2008. In 2011, Dr. Malik was awarded two Starmine Awards (awarded each year by Thomson Reuters and the Financial Times): Number One Stock Picker in the European Pharmaceutical Sector, and Number Two Stock Picker in the UK and Ireland Healthcare Sector. Dr. Malik holds a PhD in Drug Delivery within Pharmaceutical Sciences, as well as degrees in Biomedical Sciences Research (M.Sc.) and Biochemistry and Physiology (B.Sc., joint honors). Dr. Malik also holds an MBA in finance from the City University Business School, London. We believe that Dr. Malik’s extensive experience in the life sciences fields and investment banking sector make him well qualified to serve as a Director.
Jerry Jasinowskiwas appointed to the Board of Directors in April 2012. Mr. Jasinowski currently serves on the Boards of Procurian and the Washington Tennis and Education Foundation and has held directorships in several other companies since 1990. From 2004 through 2007, Mr. Jasinowski has served as the Founder and President of the Manufacturing Institute, an organization dedicated to improving and expanding manufacturing in the United States. Mr. Jasinowski was also the President and CEO of the National Association of Manufactures, a trade association with 13,000 corporate members. Mr. Jasinowski holds an A.B. in Economics from Indiana University and an M.A in Economics from Columbia University. We concluded that Mr. Jasinowski should serve on its Board of Directors because of his extensive Board experience across a wide range of manufacturing, technology, and financial firms, including Fortune 1000 and Fortune 500 companies. We believe Mr. Jasinowski’s background and experience make him well qualified to serve as a Director.
Family Relationships
None.
Board Leadership Structure
The Board believes that Ms. Powers’ service as both Chairman of the Board and Chief Executive Officer is in our best interest and our stockholders best interests. Ms. Powers possesses detailed and in-depth knowledge of the issues, opportunities, and challenges facing us, and is thus best positioned to develop agendas that ensure that the Board’s time and attention are focused on the most critical matters. Her combined role enables decisive leadership, ensures clear accountability, and enhances our ability to communicate our message and strategy clearly and consistently to our stockholders, employees and partners.
Director Independence
Our Board of Directors has determined that a majority of the Board consists of members who are currently “independent” as that term is defined under current listing standards of NASDAQ. The Board of Directors considers Messrs. Farmer, Malik and Jasinowski to be independent.
Audit Committee
The Audit Committee has responsibility for recommending the appointment of our independent accountants, supervising our finance function (which includes, among other matters, our investment activities), reviewing our internal accounting control policies and procedures, and providing the Board such additional information and materials as it may deem necessary to make the Board aware of significant financial matters which require the attention of the Board. The Audit Committee provides the opportunity for direct contact between our independent registered public accounting firm and the Board. The Board has adopted a written charter for the Audit Committee. A copy of the charter is available on the Company’ website at www.nwbio.com.
The Audit Committee currently consists of Messrs. Farmer, Malik and Jasinowski. Our Board of Directors considers each of Messrs. Farmer, Malik and Jasinowski as “independent” as that term is defined under applicable SEC and NASDAQ rules.
Mr. Jasinowski meets the definition of an “audit committee financial expert” as defined by the SEC.
| 41 |
Compensation Committee
The Compensation Committee is responsible for determining the overall compensation levels of our executive officers and administering our stock option plans. The Board has adopted a written charter for the Compensation Committee and its current members are Messrs. Farmer, Malik and Jasinowski. A copy of the charter is available on the Company’s website at www.nwbio.com. Our Board of Directors has determined that all of the members are “independent” under the current listing standards of NASDAQ.
Corporate Governance/Nominations Committee
The Corporate Governance/Nominating Committee has responsibility for assisting the Board of Directors in, among other things, effecting Board organization, membership and function including identifying qualified Board nominees; effecting the organization, membership and function of Board committees including composition and recommendation of qualified candidates; establishment of and subsequent periodic evaluation of successor planning for the chief executive officer and other executive officers; development and evaluation of criteria for Board membership such as overall qualifications, term limits, age limits and independence; and oversight of compliance with the Corporate Governance Guidelines. The Corporate Governance/Nominating Committee shall identify and evaluate the qualifications of all candidates for nomination for election as directors. Potential nominees are identified by the Board of Directors based on the criteria, skills and qualifications that have been recognized by the Corporate Governance/Nominating Committee. While our nomination and corporate governance policy does not prescribe specific diversity standards, the Corporate Governance/Nominating Committee and its independent members seek to identify nominees that have a variety of perspectives, professional experience, education, difference in viewpoints and skills, and personal qualities that will result in a well-rounded Board of Directors.
The Corporate Governance/Nominating Committee currently consists of Messrs. Farmer, Malik and Jasinowski. The Board of Directors has determined that all of the members are “independent” under the current listing standards of NASDAQ. The Board of Directors has adopted a written charter setting forth the authority and responsibilities of the Corporate Governance/Nominating Committee. A copy of the charter is available on the Company’ website at www.nwbio.com.
Board of Directors’ Role in Risk Oversight
The Board plays an active role in risk oversight of us. The Board does not have a formal risk management committee, but administers this oversight function through various standing committees of the Board of Directors. The Audit Committee maintains responsibility for oversight of financial reporting-related risks, including those related to our accounting, auditing and financial reporting practices. The Audit Committee also reviews reports and considers any material allegations regarding potential violations of the Company’s Code of Ethics. The Compensation Committee oversees risks arising from our compensation policies and programs. This Committee has responsibility for evaluating and approving our executive compensation and benefit plans, policies and programs. The Nominations/Corporate Governance Committee oversees corporate governance risks and oversees and advises the Board with respect to our policies and practices regarding significant issues of corporate responsibility.
Code of Business Conduct and Ethics
We have adopted a formal Code of Business Conduct and Ethics applicable to all Board members, executive officers and employees. A copy of our Code of Business Conduct and Ethics will be provided free of charge upon request to: Secretary, Northwest Biotherapeutics, Inc., 4800 Montgomery Lane, Suite 800, Bethesda, Maryland, 20814.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires the Company’s directors, executive officers and persons who own more than 10% of the Company’s stock (collectively, “Reporting Persons”) to file with the SEC initial reports of ownership and changes in ownership of the Company’s Common Stock. Reporting Persons are required by SEC regulations to furnish the Company with copies of all Section 16(a) reports they file. To the Company’s knowledge, based solely on its review of the copies of such reports received or written representations from certain Reporting Persons that no other reports were required, the Company believes that during its fiscal year ended December 31, 2012 all Reporting Persons timely complied with all applicable filing requirements, except that Linda Powers made non-timely filings of Form 4 reports for certain stock gifts to family members for no consideration completed on July 24, 2012, December 12, 2012 and December 29, 2012. In addition, each of Dr. Navid Malik and Jerry Jasinowski has not filed a Form 3 in connection with their appointment to the Board of Directors.
Communication with the Board of Directors
We have established a procedure by which our stockholders may communicate directly with our Board. All communications should be in written form and directed to our corporate secretary at the following address:
Northwest Biotherapeutics, Inc.
4800 Montgomery Lane, Suite 800
Bethesda, Maryland 20814
Attention: Secretary
| 42 |
ITEM 11. EXECUTIVE COMPENSATION
Summary Compensation Table
The following table sets forth certain information concerning compensation paid or accrued to our named executive officers (the “Named Executive Officers”) during the years ended December 31, 2012 and 2011. The Option Awards shown in the table below do not constitute cash or value actually received by the named executive officer. Instead, the amounts shown are the non-cash aggregate fair values of Option Awards that were granted during the periods presented but were then subject to vesting requirements. The majority of the Options were not vested and will not vest unless certain milestones are met in the future, or certain employment period requirements are met in the future. With respect to the portion of the Options that did vest, in the case Ms. Powers and Mr. Goldman, upon vesting all of those Options became subject to an extended lock-up (until the earlier of 18 months or the Company reaching the primary endpoint of its GBM brain cancer clinical trial).
| Name and Principal Position | Year | Salary | Bonus | Option Awards | Total | |||||||||||||||
| Linda F. Powers | 2012 | $ | 360,000 | $ | — | $ | — | $ | 360,000 | |||||||||||
| Chairperson, President | 2011 | $ | 203,308 | $ | — | $ | 9,262,133 | $ | 9,465,441 | |||||||||||
| & Chief Executive Officer | ||||||||||||||||||||
| Alton L. Boynton, Ph.D.(1) | 2012 | $ | 295,000 | $ | — | $ | — | $ | 295,000 | |||||||||||
| Chief Scientific Officer and Secretary | 2011 | $ | 334,732 | $ | — | $ | 1,547,963 | $ | 1,882,695 | |||||||||||
| Anthony Maida, Ph.D. | 2012 | $ | 300,000 | $ | — | $ | — | $ | 300,000 | |||||||||||
| Chief Operating Officer | 2011 | $ | 160,384 | $ | — | $ | 814,182 | $ | 974,566 | |||||||||||
| Leslie Goldman | 2012 | $ | 348,000 | $ | — | $ | — | $ | 348,000 | |||||||||||
| Senior Vice President, | 2011 | $ | 149,092 | $ | — | $ | 1,465,527 | $ | 1,614,619 | |||||||||||
| Business Development | ||||||||||||||||||||
| Marnix L. Bosch, Ph.D., M.B.A. | 2012 | $ | 325,000 | $ | — | $ | — | $ | 325,000 | |||||||||||
| Chief Technical Officer | 2011 | $ | 339,362 | $ | 20,000 | $ | 1,192,963 | $ | 1,552,325 | |||||||||||

| (1) | Dr. Boynton resigned as Chief Executive Officer on June 8, 2011 and was appointed Chief Scientific officer on the same day. |
| 43 |
Outstanding Equity Awards at Fiscal Year-End
The following table shows outstanding stock option awards classified as exercisable and un-exercisable as of December 31, 2012.
| Number of Securities Underlying Unexercised Options | ||||||||||||||
| Name and Principal Position | Exercisable | Un-exercisable | Option Exercise Price ($) | Option Expiration Date | ||||||||||
| Linda F. Powers | 543,125 | (1) | 345,625 | $ | 10.56 | 6/21/2018 | ||||||||
| Chairperson, President & Chief Executive | ||||||||||||||
| Alton Boynton | 92,285 | (2) | 56,250 | $ | 10.56 | 6/21/2018 | ||||||||
| Chief Scientific Officer and Secretary | 330 | (6) | — | $ | 300.00 | 4/18/2014 | ||||||||
| 126 | (6) | — | $ | 21.60 | 2/18/2016 | |||||||||
| 89,428 | (7) | — | $ | 8.80 | 8/20/2022 | |||||||||
| Anthony Maida | 6,250 | (3) | 71,875 | $ | 10.56 | 6/21/2018 | ||||||||
| Chief Operating Officer | ||||||||||||||
| Leslie Goldman | 85,938 | (4) | 54,687 | $ | 10.56 | 6/21/2018 | ||||||||
| Senior Vice President, Business Development | ||||||||||||||
| Marnix Bosch | 58,221 | (5) | 56,250 | $ | 10.56 | 6/21/2018 | ||||||||
| Chief Technical Officer | 21 | (8) | — | $ | 300.00 | 9/20/2014 | ||||||||
| 52 | (8) | — | $ | 1200.00 | 1/10/2015 | |||||||||
| 200 | (8) | 9 | $ | 21.60 | 2/18/2016 | |||||||||
| 250 | (8) | 83 | $ | 28.80 | 12/1/2016 | |||||||||
| 31,770 | (9) | 21,355 | $ | 11.20 | 6/23/2022 | |||||||||
| 15,625 | (10) | — | $ | 8.80 | 8/20/2022 | |||||||||
| (1) | In conjunction with the employment agreement entered into between us and Ms. Powers on June 8, 2011, and in recognition of Ms. Powers’ service to our company while serving as Chair during the preceding four years, we granted Ms. Powers an option to purchase 888,750 shares of our stock with an exercise price of $10.56 per share. One-third of the options vested on the grant date, and upon vesting became subject to a lock-up which extends to the earlier of 18 months or our reaching the primary endpoint of its GBM brain cancer clinical trial. One-third of the options will vest in equal monthly portions over the term of the employment agreement. The remaining one-third will vest in portions tied to material milestones in multiple programs, if and to the extent those milestones are achieved. |
| (2) | In conjunction with the employment agreement entered into between us and Dr. Boynton on June 8, 2011, we issued Dr. Boynton an option to purchase 148,535 shares of our stock with an exercise price of $10.56 per share. 86,035 options vested on the grant date. 7,500 options will vest in equal monthly portions over the term of the employment agreement. The remaining 55,000 options will vest in portions tied to material milestones in multiple programs, if and to the extent those milestones are achieved. |
| (3) | In conjunction with the employment agreement entered into between us and Dr. Maida on June 8, 2011, we issued Dr. Maida an option to purchase 78,125 shares of our stock with an exercise price of $10.56 per share. No options vested on the grant date. 7,500 options will vest in equal monthly portions over the term of the employment agreement. The remainder will vest in portions tied to material milestones in multiple programs, if and to the extent those milestones are achieved. |
| (4) | In conjunction with the employment agreement entered into between us and Mr. Goldman on June 8, 2011, we issued Mr. Goldman an option to purchase 140,625 shares of our stock with an exercise price of $10.56 per share. One-third of the options vested on the grant date, and upon vesting became subject to a lock-up which extends to the earlier of 18 months or our reaching the primary endpoint of our GBM brain cancer clinical trial. One-third will vest in equal monthly portions over the term of the employment agreement. The remaining one-third will vest in portions tied to material milestones in multiple programs, if and to the extent those milestones are achieved. |
| (5) | In conjunction with the employment agreement entered into between us and Dr. Bosch on June 8, 2011, we issued Dr. Bosch an option to purchase 114,471 shares of our stock with an exercise price of $10.56 per share. 51,971 options vested on the grant date. 7,500 options will vest in equal monthly portions over the term of the employment agreement. The remaining 55,000 options will vest in portions tied to material milestones in multiple programs, if and to the extent those milestones are achieved. |
| (6) | These options were granted under the 1999 Plan, the 2001 Plan and under Dr. Boynton’s previous employment agreement. Each of these option grants vests over a four year period. One-fourth of each option grant vests on the first anniversary of the grant date and the remaining three-fourths of each grant vests in equal monthly installments over the remaining three year vesting period. |
| (7) | This option was granted under the 2007 Stock Option Plan. The options were granted August 21, 2009 and vested over a one year period and are exercisable over a 10 year period from issuance at a price of $8.80 per share. |
| (8) | These options were granted under the 1999 Plan and the 2001 Plan. Each of these option grants vests over a four year period. One-fourth of each option grant vests on the first anniversary of the grant date and the remaining three-fourths of each grant vests in equal monthly installments over the remaining three year vesting period. |
| (9) | These options were granted under the 2007 Stock Option Plan. 1,250 options vest each month until May 31, 2013. In addition, 6,250 options vest upon each of Swiss Approval, full Enrollment in Phase II Glioblastoma Multiforme clinical study and FDA approval of NDA. |
| (10) | This option was granted under the 2007 Stock Option Plan. This option grant vested over the balance of 2009 with 7,813 vesting on the grant date and the remainder vesting on December 31, 2009. |
| 44 |
Director Compensation
The following table sets forth certain information concerning compensation paid or accrued to our non-executive directors during the year ended December 31, 2012.
| Name | Year | Fees Earned or Paid in Cash | All Other Compensation(1) | Total | ||||||||||
| Robert A. Farmer | 2012 | $ | 25,000 | $ | 292,000 | $ | 317,000 | |||||||
| (1) | Robert Farmer was issued 63,364 shares in September 20012 for consulting services. |
Only non-employee directors receive director fees. Effective June 22, 2007, we were required to pay Linda F. Powers, as Chairperson and a non-executive member of the Board of Directors, approximately $100,000 per annum for her services. These payments effectively ended subsequent to Ms. Powers assuming the position of Chief Executive Officer. Also effective December 10, 2009 we were required to issue Robert A. Farmer $50,000 per annum for his services as a non-executive member of the Board of Directors. During 2011, Mr. Farmer elected to take 50% of his 2011 directors’ fee in our common stock.
Compensation Committee Interlocks and Insider Participation
None of our executive officers served during our last completed fiscal year as a director of any other entity whose executive officers served as a director on our Board or as a member of our Compensation Committee.
Employment Agreements
Linda Powers
On June 8, 2011, we entered into an employment agreement with Linda Powers, pursuant to which Ms. Powers serves as our CEO. The agreement provides for an annual compensation of $360,000 and options to purchase 888,750 shares of our common stock at an exercise price of $10.56 per share. One third of the options vested upon execution of the employment agreement, 1/3 vest monthly over the term of the agreement and the remaining 1/3 vest according to certain performance milestones. The employment agreement is for a term of two years and can be terminated at any time by either party. If the Agreement is terminated without cause, Ms. Powers is entitled to three months severance; provided however, such payment shall be conditional and shall terminate in the event Ms. Powers obtains employment prior to the expiration of the three month period. If Ms. Powers is terminated without cause, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested under certain conditions. Ms. Powers is not entitled to severance if she resigns or is terminated for Cause, as defined in the agreement. If Ms. Powers is terminated for cause, all vested options will expire thirty days from such termination and all unvested options will lapse immediately. In the event of Ms. Powers’ resignation upon at least 45 days notice, Ms. Powers shall not forfeit any issued/vested options. However, if Ms. Powers provides more than 30 days notice but less than 45 days notice then she shall forfeit all options issued/vested in the preceding 60 days, and shall forfeit all options which were issued/vested during the preceding 90 days if less than 30 days notice is provided. In the event of a change in control, as defined in the agreement, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested.
Dr. Anthony Maida
On June 21, 2011, we entered into an employment agreement with Dr. Anthony Maida pursuant to which Dr. Maida serves as our Chief Operating Officer. The agreement provides for an annual compensation of $300,000 and options to purchase 78,125 shares of common stock with an exercise price of $10.56. 313 options vest on the last day of each month during the term of the agreement with the remainder of the options vesting based on certain milestones. The employment agreement is for a term of two years and can be terminated at any time by either party. If the Agreement is terminated without cause, Dr. Maida is entitled to three months severance; provided however, such payment shall be conditional and shall terminate in the event Dr. Maida obtains employment prior to the expiration of the three month period. Dr. Maida is not entitled to severance if he resigns or is terminated for Cause, as defined in the agreement. If Dr. Maida is terminated without cause, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested under certain conditions. If Dr. Maida is terminated for cause, all issued/vested options will expire thirty days from such termination and all other options will lapse immediately. In the event of Dr. Maida’ resignation upon at least 45 days notice, Dr. Maida shall not forfeit any issued/vested options. However, if Dr. Maida provides more than 30 days notice but less than 45 days notice then he shall forfeit all options issued/vested in the preceding 60 days, and shall forfeit all options which were issued/vested during the preceding 90 days if less than 30 days notice is provided. In the event of a change in control, as defined in the agreement, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested.
| 45 |
Leslie J. Goldman
On June 8, 2011, we entered into an employment agreement with Leslie J. Goldman pursuant to which Mr. Goldman serves as Senior Vice President for Business Development. The agreement provides for an annual compensation of $264,000 and options to purchase 140,625 shares of common stock with an exercise price of $10.56. One third of the options vested upon execution of the employment agreement, 1/3 vest monthly over the term of the agreement and the remaining 1/3 vest according to certain performance milestones. The employment agreement is for a term of two years and can be terminated at any time by either party. If the Agreement is terminated without cause, Mr. Goldman is entitled to three months severance; provided however, such payment shall be conditional and shall terminate in the event Mr. Goldman obtains employment prior to the expiration of the three month period. Mr. Goldman is not entitled to severance if he resigns or is terminated for cause, as defined in the agreement. If Mr. Goldman is terminated without cause, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested under certain conditions. If Mr. Goldman is terminated for cause, all issued/vested options will expire thirty days from such termination. In the event of Mr. Goldman’s resignation upon at least 45 days notice, Mr. Goldman shall not forfeit any issued/vested options. However, if Mr. Goldman provides more than 30 days notice but less than 45 days notice then he shall forfeit all options issued/vested in the preceding 60 days, and shall forfeit all options which were issued/vested during the preceding 90 days if less than 30 days notice is provided. In the event of a change in control, as defined in the agreement, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested.
Marnix Bosch
On June 8, 2011, we entered into an employment agreement with Marnix Bosch pursuant to which Mr. Bosch serves as our Chief Technical Officer. The agreement provides for an annual compensation of $325,000 and options to purchase 62,500 shares of common stock with an exercise price of $10.56, which vests over the term based on certain milestones. In addition, the employment agreement extends the term of existing options for an additional 3 years. The employment agreement is for a term of two years and can be terminated at any time by either party. If the Agreement is terminated without cause, Mr. Bosch is entitled to three months severance; provided however, such payment shall be conditional and shall terminate in the event Mr. Bosch obtains employment prior to the expiration of the three month period. Mr. Bosch is not entitled to severance if he resigns or is terminated for cause, as defined in the agreement. If Mr. Bosch is terminated without cause, as defined in the agreement, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested under certain conditions. If Mr. Bosch is terminated for cause, all issued/vested options will expire thirty days from such termination. In the event of Mr. Bosch’s resignation upon at least 45 days notice, Mr. Bosch shall not forfeit any issued/vested options. However, if Mr. Bosch provides more than 30 days notice but less than 45 days notice then he shall forfeit all options issued/vested in the preceding 60 days, and shall forfeit all options which were issued/vested during the preceding 90 days if less than 30 days notice is provided. In the event of a change in control, as defined in the agreement, all options that may be earned under the agreement and which have not yet been issued/vested will accelerate and become fully vested.
| 46 |
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The following table presents information regarding the beneficial ownership of our common stock as of April 29, 2013 by:
| · | each person, or group of affiliated persons, who is known by us to own beneficially 5% or more of any class of our equity securities; |
| · | our directors; |
| · | each of our named executive officers, as defined in Item 402(a)(3) of Regulation S-K; and |
| · | our directors and executive officers as a group. |
Shares of common stock beneficially owned and the respective percentages of beneficial ownership of common stock assumes the exercise of all options, warrants and other securities convertible into common stock beneficially owned by such person or entity currently exercisable or exercisable within 60 days of April 29, 2013. Shares issuable pursuant to the exercise of stock options and warrants exercisable within 60 days are deemed outstanding and held by the holder of such options or warrants for computing the percentage of outstanding common stock beneficially owned by such person, but are not deemed outstanding for computing the percentage of outstanding common stock beneficially owned by any other person.
Except as indicated by the footnotes below, we believe, based on the information furnished to us, that the persons and the entities named in the table have sole voting and investment power with respect to all shares of common stock that they beneficially own, subject to applicable community property laws.
Except as otherwise noted, the address of the individuals in the following table below is c/o Northwest Biotherapeutics, Inc., 4800 Montgomery Lane, Suite 800, Bethesda, MD 20814.
| Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage(1) | ||||||
| Officers and Directors | ||||||||
| Alton L. Boynton, Ph.D.(2) | 194,358 | * | % | |||||
| Marnix L. Bosch, Ph.D., M.B.A.(3) | 115,941 | * | % | |||||
| Linda F. Powers(4) | 18,431,652 | 52.9 | % | |||||
| Robert A. Farmer(5) | 144,674 | * | % | |||||
| Leslie Goldman(6) | 544,825 | 2.0 | % | |||||
| Dr. Navid Malik | — | — | ||||||
| Anthony Maida, Ph.D.(7) | 6,250 | * | ||||||
| Jerry Jasinowski(8) | 80,357 | * | % | |||||
| All executive officer s and directors as a group (8 persons) | 19,518,057 | 55.1 | % | |||||
| 5% Security Holders | ||||||||
| Toucan Capital Fund III, L.P.(9) 4800 Montgomery Lane, Suite 801 Bethesda, MD 20814 | 1,900,772 | 6.7 | % | |||||
| Toucan Partners, LLC(10) 4800 Montgomery Lane, Suite 801 Bethesda, MD 20814 | 5,681,519 | 19.9 | % | |||||
| Cognate BioServices, Inc.(11) 4800 East Shelby Drive, Suite 108, Memphis, TN | 7,072,158 | 24.7 | % | |||||
| 47 |
| * | Less than 1%. | |
| (1) | Percentage represents beneficial ownership percentage of common stock calculated in accordance with SEC rules and does not equate to voting percentages. Based upon 27,140,417 shares of common stock issued and outstanding as of April 29, 2013. |
| (2) | Includes 182,169 shares of common stock underlying options that are currently exercisable. |
| (3) | Includes 106,139 shares of common stock underlying options that are currently exercisable. |
| (4) | Includes (i) 543,125 shares of common stock underlying currently exercisable options held by Ms. Powers; (ii) 3,234,078 shares of common stock underlying currently exercisable warrants held by Ms. Powers; (iii) 804,145 shares of common stock held by Toucan Capital Fund III, L.P.; (iv) 1,096,627 shares of common stock underlying currently exercisable warrants held by Toucan Capital; (v) 4,311,184 shares of common stock held by Toucan Partners, LLC; (vi) 1,370,335 shares of common stock underlying currently exercisable warrants held by Toucan Partners; (vii) 1,437,500 shares of common stock underlying warrants held by Cognate Bioservices, Inc.; and (vii) 5,634,658 shares of common stock held by Cognate Bioservices, Inc. Ms. Powers has voting and dispositive power over the securities owned by Toucan Capital Fund III, L.P., Toucan Partners LLC and (through Toucan Capital and Toucan Partners) Cognate Bioservices, Inc. |
| (5) | Includes 20,525 shares of common stock underlying currently exercisable warrants. |
| (6) | Includes (i) 160,139 shares of common stock underlying currently exercisable warrants; and (ii) 85,938 shares of common stock underlying currently exercisable options. |
| (7) | Consists of shares of common stock underlying currently exercisable options. |
| (8) | Includes 49,107 shares of Common Stock underlying currently exercisable warrants. |
| (9) | Consists of (i) 1,096,627 shares of common stock underlying currently exercisable warrants; and (ii) 804,145 shares of common stock. Linda Powers holds the voting and dispositive power over the shares held by Toucan Capital Fund III, L.P. |
| (10) | Includes (i) 1,370,335 shares of common stock underlying currently exercisable warrants; and (ii) 4,311,184 shares of common stock. Linda Powers holds the voting and dispositive power over the shares held by Toucan Partners, LLC. |
| (11) | Represents (i) 5,634,658 shares of common stock and (ii) 1,437,500 shares of common stock underlying a currently exercisable warrant. Linda Powers through Toucan Capital III LP and Toucan Partners LLC holds the voting and dispositive power over the shares held by Cognate BioServices, Inc. |
| 48 |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Cognate BioServices
In April, 2011, the Company entered into a new service agreement with Cognate Therapeutics, Inc. (“Cognate”), a contract manufacturing and services organization in which Toucan Capital has a majority interest. In addition, two of the principals of Toucan Capital are members of Cognate’s board of directors and Linda Powers who is a director of Cognate and managing director of Toucan Capital is Chairperson of the Company’s Board of Directors and Chief Executive Officer of the Company. This agreement replaces the agreement dated May 17, 2007 between the Company and Cognate, which had expired. Under the service agreement, the Company agreed to continue to utilize Cognate’s services, for certain consulting and manufacturing services to the Company for its ongoing DCVax®-Brain Phase III clinical trial. The scope of services is comparable to the prior agreement, and the structure and process for payments are simplified. Under the terms of the current agreement the Company pays Cognate a monthly facility fee and a fixed fee (in lieu of cost-plus charges) for each patient in the study, subject to specified minimum number of patients per month, plus charges for certain patient and product data services if such services are requested by the Company. The current service agreement will expire on March 31, 2016. During December 2012, the Company issued five year warrants for the purchase of 1,437,500 shares of common stock at an exercise price of $6.40 in connection with the conversion agreement with Cognate and as additional consideration under the service agreement. The fair value of the warrants amounting to $1,878,247 was recorded as research and development expenses and was determined using the Black-Scholes Model with the following assumptions: risk free interest rate – 0.4% volatility – 65%, expected term – five years, expected dividends – N/A.
On October 16, 2012, we entered into a conversion agreement with Cognate BioServices pursuant to which, upon the closing of the public offering in December 2012, an aggregate of $7.5 million unpaid invoiced amounts and payables were converted into equity. With respect to the unpaid invoice amounts and payables, Cognate had a right to convert on terms no less favorable than provided to any other creditor. However, Cognate agreed instead to convert on terms equal to the median of the conversion terms provided to other non-affiliated creditors over the preceding six months. Accordingly, liabilities payable to Cognate amounting to $7.5 million were converted into a total of 2.8 million shares of restricted common stock and 1.4 million warrants exercisable for common stock. The term of the warrants is five years and the exercise price is $3.20 per share. The difference between the fair value of the shares of common stock and warrants issued in excess of the carrying amount of the liabilities amounting to $3.1 was recorded as conversion inducement expense in 2012. The fair value of the common stock issued was valued after determining a 14% discount from the closing price of the Company’s common stock on the date the stock was issued for lack of marketability. The fair value of the warrants was determined using the Black-Scholes model with the following assumptions: risk free interest rate – 0.4%, volatility – 65%, expected term – 5 years, expected dividends – N/A.
In 2012, the Company issued approximately 0.5 million shares and 0.1 million warrants with an exercise price of $6.40 to an outside party in order to settle a note payable that Cognate owed to the unrelated party. The Company does not expect reimbursement from Cognate and as such, the fair value of the common stock and warrants issued to the outside party amounting to $2.2 million and was recorded as inducement expense for 2012.
During the twelve months ended December 31, 2012 and 2011, the Company recognized approximately $16.5 million and $4.7 million, respectively, of research and development costs related to these service agreements. As of December 31, 2012 and December 31 2011, the Company owed Cognate approximately $1.8 million and $0.6 million, respectively.
Toucan Capital and Toucan Partners
In 2011 the Company received proceeds of $1.4 million from Toucan in connection with issuing unsecured convertible notes. The notes were payable within one year and carried an original issue discount of 10%. The conversion price was $3.20.
In 2011 the Company converted notes payable to Toucan amounting to $0.7 million into approximately 0.2 million shares of common stock.
In 2012 the Company issued convertible notes payable of $3.1 million to Toucan. Warrants to purchase 572,402 shares of common stock at an exercise price of $6.40, and a five year term, were issued in connection with the notes. The notes were payable within one year and carried an original issue discount of 10%. The conversion price was $3.20.
On October 16, 2012, the Company entered into conversion agreements with Toucan Partners and its affiliates (with the exception of Cognate BioServices as discussed above), pursuant to which, upon the closing of the public offering in December 2012, an aggregate of $10.7 million of convertible notes and payables were converted into equity. The notes were converted substantially in accordance with their existing terms. With respect to the payables, Toucan had a right to convert on terms no less favorable than provided to any other creditor. However, Toucan agreed to instead convert on terms equal to the median of the conversion terms provided to other non-affiliated creditors over the preceding six months. The payables arose under an agreement with Toucan where the Company agreed to reimburse Toucan for certain payables, including interest, amounting to $4.6 million. The expense of $4.6 million associated with the payables was recognized in the Company’s financial statements during 2012 when the Company and Toucan concluded negotiations as to the amount owed for expenses Toucan paid on the Company’s behalf during the period of 2004 to 2012. Accordingly, the Toucan entities’ $10.7 million aggregate conversion amount was converted into a total of 3.6 million shares of restricted common stock and 1.8 million warrants exercisable for common stock. The warrants’ exercise period is five years, and the exercise price is $3.20 per share. The difference between the fair value of the shares of common stock and warrants issued in excess of the carrying amount of the liabilities amounting to$1.9was recorded as conversion inducement expense in 2012. The fair value of the common stock issued was valued after determining a 14% discount from the closing price of the Company’s common stock on the date the stock was issued for lack of marketability. The fair value of the warrants was determined using the Black-Scholes model with the following assumptions: risk free interest rate – 0.4%, volatility – 65%, expected term – 5 years, expected dividends – N/A. Total liabilities owed to Toucan as of December 31, 2012 were less than $0.01 million. Toucan continues to pay expenses on the Company’s behalf which have not been formally billed to the Company. Toucan may seek reimbursement for the expenses incurred subsequent to the settlement on October 16, 2012. Management is not able to estimate the financial statement impact of further expense reimbursements to Toucan as of the date these financial statements were issued.
| 49 |
Other Related Parties
The Company received proceeds of $0.2 million in connection with issuing an unsecured convertible note to an officer of the Company on January 3, 2012. The notes included 44,532 warrants to purchase common stock. The exercise price of the warrants is $6.40, and the exercise period is 5 years. The note was payable on demand with 7 days written notice and carried an original issue discount of 10% and a one-time interest charge of 10%. The conversion price of the note was 95% of the average of the lowest five days’ closing prices of the Company's common stock in the twenty trading days prior to conversion. This note plus accrued interest was converted into 49,500 shares of common stock in December 2012.
The Company received proceeds of $0.3 million in connection with issuing an unsecured convertible demand note to an officer of the Company on June 29, 2012. The conversion price was $5.28 and the interest rate was 10%. The notes included 43,750 warrants to purchase common stock. The exercise price is $5.60 and the exercise period of the warrants is five years. This note plus accrued interest was converted into 66,341 shares of common stock in December 2012.
During the previous three years, a Board member of the Company arranged multiple financings for the Company. The Company agreed to compensate the Board member at a rate below the usual fees of investment banks for such transactions, by paying the Board member 63,000 shares of common stock valued at $0.3 million during the third quarter of 2012.
Review, approval or ratification of transactions with related persons
Any transactions with officers, directors or 5% stockholders are on terms no less favorable to us than could be obtained from independent parties, and are approved by a majority of our independent and disinterested directors who have access to our counsel or independent legal counsel at our expense.
Board Determination of Independence
Our Board of Directors has determined that a majority of the board consists of members who are currently “independent” as that term is defined under current listing standards of NASDAQ
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES.
Audit Fees
The aggregate fees billed and unbilled for the fiscal years ended December 31, 2012 and December 31, 2011 for professional services rendered by our principal accountants for the audits of our annual financial statements, the review of our financial statements included in our quarterly reports on Form 10-Q and consultations and consents were approximately $188,495 and $158,347.
Audit-Related Fees
There were no aggregate fees billed for the fiscal year ended December 31, 2012 and 2011 for assurance and related services rendered by our principal accountants related to the performance of the audit or review of our financial statements, specifically accounting research.
Tax and Other Fees
The aggregate fees billed for the fiscal year ended December 31, 2012 and December 31, 2011 for professional services rendered by our principal accountants for tax compliance fees were $4,576 and $3,281, respectively.
Consistent with SEC policies and guidelines regarding audit independence, the Audit Committee is responsible for the pre-approval of all audit and permissible non-audit services provided by our principal accountants on a case-by-case basis. Our Audit Committee has established a policy regarding approval of all audit and permissible non-audit services provided by our principal accountants. Our Audit Committee pre-approves these services by category and service. Our Audit Committee has pre-approved all of the services provided by our principal accountants.
| 50 |
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The Exhibits listed below are identified by numbers corresponding to the Exhibit Table of Item 601 of Regulation S-K. The Exhibits designated by an asterisk (*) are management contracts or compensatory plans or arrangements required to be filed pursuant to Item 15.
EXHIBIT INDEX
| Exhibit Number | Description | |
| 3.1 | Seventh Amended and Restated Certificate of Incorporation (incorporated by reference to exhibit 3.1 filed with the Registrant’s Amendment No. 1 to the Registration Statement on Form S-1(File No. 333-134320) on July 17, 2006) | |
| 3.2 | Third Amended and Restated Bylaws of the Company (incorporated by reference to exhibit 3.1 filed with the Registrant’s Current Report on Form 8-K on June 22, 2007) | |
| 3.3 | Amendment to Seventh Amended and Restated Certificate of Incorporation (incorporated by reference to exhibit 3.2 filed with the Registrant’s Current Report on Form 8-K on June 22, 2007) | |
| 3.4 | Amendment to Seventh Amended and Restated Certificate of Incorporation (incorporated by reference to exhibit 3.4 filed with Post-Effective Amendment No. 2 to the Registrant’s Registration Statement on Form S-1 on January 28, 2008) | |
| 3.5 | Amendment to Seventh Amended and Restated Certificate of Incorporation (incorporated by reference to exhibit 3.1 filed with the Registrant’s Quarterly Report on Form 10-Q on May 21, 2012) | |
| 3.6 | Amendment to Seventh Amended and Restated Certificate of Incorporation (incorporated by reference to exhibit 3.1 filed with the Registrant’s Current Report on Form 8-K on September 26, 2012). | |
| 3.7 | Amendment to Third Amended and Restated Bylaws of the Company (incorporated by reference to exhibit 3.1 filed with the Registrant’s Current Report on Form 8-K on December 11, 2012) | |
| 4.1 | Form of common stock certificate (incorporated by reference to exhibit 4.1 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 4.2 | Form of Warrant Agency Agreement by and between Northwest Biopharmaceuticals, Inc. and Computershare Trust Company, N.A. and Form of Warrant Certificate (incorporated by reference to Exhibit 4.2 filed with the Registrant’s Form S-1 on December 4, 2012). | |
| 10.1 | Amended and Restated Loan Agreement and 10% Promissory Note dated November 14, 2005 in the principal amount of $400,000 as amended and restated on April 14, 2007 between the Company and Toucan Partners, LLC (incorporated by reference to exhibit 10.1 filed with the Registrant’s Form 10-K on April 17, 2007) | |
| 10.2 | Second Amended and Restated Loan Agreement and 10% Promissory Note originally dated December 30, 2005, and amended and restated on April 17, 2006 and April 14, 2007 in the principal amount of $250,000 between the Company and Toucan Partners, LLC (incorporated by reference to exhibit 10.2 filed with the Registrant’s Form 10-K on April 17, 2007) |
| 51 |
| 10.3 | Second Amended and Restated Loan Agreement and 10% Promissory Note originally dated March 9, 2006, and as amended and restated on April 17, 2006 and April 14, 2007 in the principal amount of $300,000 between the Company and Toucan Partners, LLC (incorporated by reference to exhibit 10.3 filed with the Registrant’s Form 10-K on April 17, 2007) | |
| 10.4 | Form of Loan Agreement and 10% Convertible, Promissory Note between the Company and Toucan Partners, LLC (incorporated by reference to exhibit 10.4 filed with the Registrant’s Form 10-K on April 17, 2007) | |
| 10.5 | Second Amended and Restated Investor Rights Agreement dated June 22, 2007 between the Company and Toucan Capital Fund II, LLP (incorporated by reference to exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K on June 22, 2007) | |
| 10.6 | Warrant to purchase securities of the Company dated July 26, 2005 issued to Toucan Capital Fund II, L.P (incorporated by reference to exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K on August 1, 2005) | |
| 10.7 | Warrant to purchase securities of the Company dated September 7, 2005 issued to Toucan Capital Fund II, L.P (incorporated by reference to exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K on September 9, 2005) | |
| 10.8 | Amended Form of Warrant to purchase securities of the Company dated November 14, 2005 and April 17, 2006, as amended April 14, 2007, issued to Toucan Partners, LLC (incorporated by reference to exhibit 10.21 filed with the Registrant’s Form 10-K on April 17, 2007) | |
| 10.9 | Form of Warrant to purchase securities of the Company dated April 14, 2007 issued to Toucan Partners, LLC (incorporated by reference to exhibit 10.22 filed with the Registrant’s Form 10-K on April 17, 2007) | |
| 10.10 | Loan Agreement and 10% Convertible Promissory Note in the principal amount of $100,000 between the Company and Toucan Partners, LLC, dated April 27, 2007 (incorporated by reference to exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K on May 3, 2007) | |
| 10.11 | Warrant to purchase securities of the Company issued to Toucan Partners, LLC, dated April 27, 2007 (incorporated by reference to exhibit 10.2 filed with the Registrant’s Current Report on Form 8-K on May 3, 2007) | |
| 10.12 | Form of Toucan Partners Loan Agreement and 10% Convertible Note, dated as of June 1, 2007 (incorporated by reference to exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K on June 7, 2007) | |
| 10.13 | Form of Toucan Partners Warrant, dated as of June 1, 2007 (incorporated by reference to exhibit 10.2 filed with the Registrant’s Current Report on Form 8-K on June 7, 2007) | |
| 10.14 | Amended and Restated Warrant to purchase Series A Preferred Stock issued to Toucan Capital Fund II, L.P., dated as of June 1, 2007 (incorporated by reference to exhibit 10.3 filed with the Registrant’s Current Report on Form 8-K on June 7, 2007) | |
| 10.15 | Warrant to purchase Series A-1 Preferred Stock issued to Toucan Capital Fund II, L.P., dated as of June 1, 2007 (incorporated by reference to exhibit 10.4 filed with the Registrant’s Current Report on Form 8-K on June 7, 2007) | |
| 10.16 | Warrant to purchase Series A-1 Preferred Stock issued to Toucan Capital Fund II, L.P., dated as of June 1, 2007 (incorporated by reference to exhibit 10.5 filed with the Registrant’s Current Report on Form 8-K on June 7, 2007) | |
| 10.17 | Northwest Biotherapeutics, Inc. $225,000 Demand Note dated June 13, 2007 (incorporated by reference to exhibit 10.1 filed with the Registrant’s Current Report on Form 8-K on June 18, 2007) | |
| 10.18 | Conversion Agreement dated June 15, 2007 and effective June 22, 2007 between the Company and Toucan Capital Fund II, LLP (incorporated by reference to exhibit 10.1filed with the Registrant’s Current Report on Form 8-K on June 22, 2007) |
| 52 |
| 10.19 | Services Agreement between Cognate BioServices, Inc. and Northwest Biotherapeutics dated April 1, 2011. Portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission pursuant to an order dated July 26, 2012 granting confidential treatment. (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on June 29, 2012) | |
| 10.20 | 1998 Stock Option Plan (incorporated by reference to exhibit 10.15 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 10.21 | 1999 Executive Stock Option Plan (incorporated by reference to exhibit 10.16 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 10.22 | 2001 Stock Option Plan (incorporated by reference to exhibit 10.17 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 10.23 | 2001 Nonemployee Director Stock Incentive Plan (incorporated by reference to exhibit 10.18 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 10.24 | Employee Stock Purchase Plan (incorporated by reference to exhibit 10.19 filed with the Registrant’s Amendment No. 3 to the Registration Statement on Form S-1 (Registration No. 333-67350) on November 14, 2001) | |
| 10.25 | 2007 Stock Option Plan (incorporated by reference to exhibit 10.5 filed with the Registrant’s Current Report on Form 8-K on June 22, 2007) | |
| 10.26 | Form of Stock Option Agreement under the 2007 Stock Option Plan (incorporated by reference to exhibit 10.2 filed with the Registrant’s Registration Statement on Form S-8 on November 21, 2007) | |
| 10.27 | Loan Agreement and Promissory Note, dated May 6, 2008 between the Company and Al Rajhi Holdings WLL (incorporated by reference to exhibit 4.5 filed with the Registrant’s Current Report on Form 8-K on May 15, 2008) | |
| 10.28 | Loan Agreement and Promissory Note, dated August 19, 2008 between the Company and Toucan Partners LLC (incorporated by reference to exhibit 10.1 filed with the Registrant’s Quarterly Report on Form 10-Q on August 19, 2008) | |
| 10.29 | Loan Agreement and Promissory Note, dated October 1, 2008 between the Company and SDS Capital Group SPC, Ltd (incorporated by reference to exhibit 10.2 filed with the Registrant’s Quarterly Report on Form 10-Q on November 19, 2008) | |
| 10.30 | Warrant, dated October 1, 2008, between the Company and SDS Capital Group SPC, Ltd (incorporated by reference to exhibit 10.3 filed with the Registrant’s Quarterly Report on Form 10-Q on November 19, 2008) | |
| 10.31 | Loan Agreement and Promissory Note, dated October 21, 2008, between the Company and SDS Capital Group SPC, Ltd (incorporated by reference to exhibit 10.4 filed with the Registrant’s Quarterly Report on Form 10-Q on November 19, 2008) | |
| 10.32 | Form of Loan Agreement and Promissory Note, between the Company and a Group of Private Investors (incorporated by reference to exhibit 10.5 filed with the Registrant’s Quarterly Report on Form 10-Q on November 19, 2008) | |
| 10.33 | Form of Warrant, between the Company and SDS Capital Group SPC. Ltd and a Group of Private Investors (incorporated by reference to exhibit 10.6 filed with the Registrant’s Quarterly Report on Form 10-Q on November 19, 2008) |
| 53 |
| 10.34 | Loan Agreement and Promissory Note, dated December 22, 2008, between the Company and Toucan Partners LLC (incorporated by reference to exhibit 10.62 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.35 | Form of Warrant, dated December 22, 2008, between the Company and Toucan Partners LLC (incorporated by reference to exhibit 10.63 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.36 | Form of Securities Purchase Agreement, by and among the Company and Al Rajhi Holdings (incorporated by reference to exhibit 10.64 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.37 | Securities Purchase Agreement, by and among the Company and a Group of Equity Investors (incorporated by reference to exhibit 10.65 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.38 | Form of Warrant, between the Company and a Group of Equity Investors (incorporated by reference to exhibit 10.66 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.39 | Form of Loan Agreement and Promissory Note, dated March 27 2009, between the Company and a Group of Private Lenders (incorporated by reference to exhibit 10.67 filed with the Registrant’s Form 10-K on April 15, 2009) | |
| 10.40 | Employment Agreement dated May 31, 2011 between the Company and Marnix Bosch (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on November 15, 2012) | |
| 10.41 | Employment Agreement dated June 8, 2011 between the Company and Alton Boynton (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on November 15, 2012) | |
| 10.42 | Employment Agreement dated May 31, 2011 between the Company and Anthony Maida (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on November 15, 2012) | |
| 10.43 | Employment Agreement dated June 8, 2011 between the Company and Leslie Goldman (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on November 15, 2012) | |
| 10.44 | Employment Agreement dated June 8, 2011 between the Company and Linda Powers (incorporated by reference to exhibit 10.19 filed with the Registrant’s Registration Statement on Form S-1 (Registration No. 333-182470 on November 15, 2012) | |
| 10.45 | Amended and Restated Northwest Biotherapeutics, Inc. 2007 Stock Option Plan (incorporated by reference to Schedule 14C filed on August 16, 2012) | |
| 21 | Subsidiary of the Registrant (Incorporated by reference to exhibit 21.1 filed with Post-Effective No. 1 to the Registrant’s Form S-1 on December 17, 2007) | |
| 23 | Consent of Peterson Sullivan LLP, Independent Registered Public Accounting Firm | |
| 31 | Certification of the Principal Executive and Principal Financial and Accounting Officer pursuant to Section 302 of the Sarbanes Oxley Act of 2002 | |
| 32 | Certification of the Principal Executive and Principal Financial and Accounting Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 54 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K/A to be signed on its behalf by the undersigned, thereunto duly authorized.
NORTHWEST BIOTHERAPEUTICS, INC. (Registrant) | |||
| Date: April 30, 2013 | By: | /s/ Linda F. Powers | |
| Linda F. Powers, | |||
| Chief Executive Officer(Principal Executive Officer | |||
| and Principal Financial and Accounting Officer) | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K/A has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
| /s/ Linda F. Powers | Chief Executive Officer (Principal | April 30, 2013 | ||
| LINDA F. POWERS | Executive Officer and Principal | |||
| Financial and Accounting Officer)) | ||||
| /s/ Alton L. Boynton | Director | April 30, 2013 | ||
| ALTON L. BOYNTON | ||||
| /s/ Robert A Farmer | Director | April 30, 2013 | ||
| ROBERT A FARMER | ||||
| /s/ Dr. Navid Malik | Director | April 30, 2013 | ||
| DR. NAVID MALIK | ||||
| /s/Jerry Jasinowski | Director | April 30, 2013 | ||
| JERRY JASINOWSKI |
| 55 |
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
INDEX TO THE CONSOLIDATED FINANCIAL STATEMENTS
| Report of Independent Registered Public Accounting Firm | F-1 | |
| Consolidated Balance Sheets as of December 31, 2012 and 2011 | F-2 | |
| Consolidated Statements of Operations for the two years ended December 31, 2012 and 2011 and March 18, 1996 (inception) to December 31, 2012 | F-3 | |
| Consolidated Statements of Comprehensive Loss for the two years ended December 31, 2012 and 2011 and March 18, 1996 (inception) to December 31, 2012 | F-4 | |
| Consolidated Statements of Changes in Stockholders’ Equity (Deficit) for the period March 18, 1996 (inception) to December 31, 2012 | F-5 | |
| Consolidated Statements of Cash Flows for the two years ended December 31, 2012 and 2011 and for the period March 18, 1996 (inception) to December 31, 2012 | F-6 | |
| Notes to the Consolidated Financial Statements | F-8 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Northwest Biotherapeutics, Inc.
Bethesda, Maryland
We have audited the accompanying consolidated balance sheets of Northwest Biotherapeutics, Inc. and Subsidiaries (a development stage company) ("the Company") as of December 31, 2012 and 2011, and the related consolidated statements of operations, comprehensive loss, stockholders' equity (deficit), and cash flows for the years then ended, and for the period from March 18, 1996 (date of inception) to December 31, 2012. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company has determined that it is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Northwest Biotherapeutics, Inc. and Subsidiaries (a development stage company) as of December 31, 2012 and 2011, and the results of their operations and their cash flows for the years then ended, and for the period from March 18, 1996 (date of inception) to December 31, 2012, in conformity with accounting principles generally accepted in the United States.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has experienced recurring losses from operations since inception, net operating cash flow deficits, and has a deficit accumulated during the development stage. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans regarding these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/S/ PETERSON SULLIVAN LLP
Seattle, Washington
April 8, 2013
| F-1 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED BALANCE SHEET
(in thousands, except per share data)
| December 31, | ||||||||
| 2012 | 2011 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 7,346 | $ | 24 | ||||
| Prepaid expenses and other current assets | 112 | 94 | ||||||
| Total current assets | 7,458 | 118 | ||||||
| Property and equipment: | ||||||||
| Laboratory equipment | 60 | 29 | ||||||
| Office furniture and other equipment | 172 | 172 | ||||||
| Less accumulated depreciation and amortization | (137 | ) | (123 | ) | ||||
| Property and equipment, net | 95 | 78 | ||||||
| Deposit and other non-current assets | 17 | 16 | ||||||
| Total assets | $ | 7,570 | $ | 212 | ||||
| LIABILITIES, REDEEMABLE COMMON STOCK AND STOCKHOLDERS' DEFICIT | ||||||||
| Current liabilities: | ||||||||
| Accounts payable (includes related party of $3,397 and $1,589 in 2012 and 2011, respectively) | $ | 8,165 | $ | 3,808 | ||||
| Accrued expenses (includes related party of $28 and $630 in 2012 and 2011, respectively) | 589 | 2,815 | ||||||
| Note payable (includes related party of $0 and $2,056 in 2012 and 2011, respectively) | 934 | 5,205 | ||||||
| Convertible notes, net (includes related party of $0 and $3,588 in 2012 and 2011, respectively) | 1,056 | 8,420 | ||||||
| Embedded derivative liability | - | 601 | ||||||
| Liability for reclassified equity contracts | - | 29,903 | ||||||
| Total current liabilities | 10,744 | 50,752 | ||||||
| Non-current liabilities: | ||||||||
| Notes payable | - | 200 | ||||||
| Convertible notes payable, net | 1,882 | 1,433 | ||||||
| Total long term liabilities | 1,882 | 1,633 | ||||||
| Total liabilities | 12,626 | 52,385 | ||||||
| Redeemable common stock ($0.001 par value) | 11,017 | - | ||||||
| Stockholders' deficit: | ||||||||
| Preferred stock ($0.001 par value); 40,000,000 and 20,000,000 shares authorized; 0 and 0 shares issued and outstanding as of December 31, 2012 and December 31, 2011, respectively | - | - | ||||||
| Common stock ($0.001 par value); 450,000,000 and 150,000,000 shares authorized; 26,545,828 and 9,334,101 shares issued and outstanding as of December 31, 2012 and December 31, 2011, respectively | 27 | 150 | ||||||
| Additional paid-in capital | 303,188 | 199,605 | ||||||
| Deficit accumulated during the development stage | (319,098 | ) | (251,778 | ) | ||||
| Cumulative translation adjustment | (190 | ) | (150 | ) | ||||
| Total stockholders' deficit | (16,073 | ) | (52,173 | ) | ||||
| Total liabilities, redeemable common stock and stockholders' deficit | $ | 7,570 | $ | 212 | ||||
See accompanying notes to the consolidated financial statements
| F-2 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| Years Ended December 31, | Period from Inception (March 18, 1996) to | |||||||||||
| 2012 | 2011 | December 31, 2012 | ||||||||||
| Revenues: | ||||||||||||
| Research material sales | $ | - | $ | 10 | $ | 580 | ||||||
| Contract research and development from related parties | - | - | 1,128 | |||||||||
| Research grants and other | 772 | - | 1,833 | |||||||||
| Total revenues | 772 | 10 | 3,541 | |||||||||
| Operating cost and expenses: | ||||||||||||
| Cost of research material sales | - | - | 382 | |||||||||
| Research and development | 28,908 | 13,452 | 119,172 | |||||||||
| General and administration | 15,675 | 13,335 | 90,999 | |||||||||
| Depreciation and amortization | 14 | 10 | 2,377 | |||||||||
| Loss on facility sublease | - | - | 895 | |||||||||
| Asset impairment loss | - | - | 2,445 | |||||||||
| Total operating costs and expenses | 44,597 | 26,797 | 216,270 | |||||||||
| Loss from operations | (43,825 | ) | (26,787 | ) | (212,729 | ) | ||||||
| Other income (expense): | ||||||||||||
| Valuation of reclassified equity instruments | 491 | 8,821 | 16,071 | |||||||||
| Conversion inducement expense | (9,103 | ) | (7,944 | ) | (27,337 | ) | ||||||
| Accretion of redeemable securities | (2,042 | ) | - | (2,042 | ) | |||||||
| Derivative valuation gain/(loss) | 601 | 728 | 1,383 | |||||||||
| Gain on sale of intellectual property and property and equipment | - | - | 3,664 | |||||||||
| Interest expense | (13,442 | ) | (7,648 | ) | (55,006 | ) | ||||||
| Interest income and other | - | - | 1,707 | |||||||||
| Net loss | (67,320 | ) | (32,830 | ) | (274,289 | ) | ||||||
| Issuance of common stock in connection with elimination of Series A and Series A-1 preferred stock preferences | - | - | (12,349 | ) | ||||||||
| Modification of Series A preferred stock warrants | - | - | (2,306 | ) | ||||||||
| Modification of Series A-1 preferred stock warrants | - | - | (16,393 | ) | ||||||||
| Series A preferred stock dividends | - | - | (334 | ) | ||||||||
| Series A-1 preferred stock dividends | - | - | (917 | ) | ||||||||
| Warrants issued on Series A and Series A-1 preferred stock dividends | - | - | (4,664 | ) | ||||||||
| Accretion of Series A preferred stock mandatory redemption obligation | - | - | (1,872 | ) | ||||||||
| Series A preferred stock redemption fee | - | - | (1,700 | ) | ||||||||
| Beneficial conversion feature of Series D preferred stock | - | �� | - | (4,274 | ) | |||||||
| Net loss applicable to common stockholders | $ | (67,320 | ) | $ | (32,830 | ) | $ | (319,098 | ) | |||
| Net loss per share applicable to common stockholders - basic | $ | (5.72 | ) | $ | (5.58 | ) | ||||||
| Weighted average shares used computing basic loss per share | 11,759 | 5,887 | ||||||||||
See accompanying notes to the consolidated financial statements
| F-3 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(in thousands)
| Years Ended December 31, | Period from Inception (March 18, 1996) to | |||||||||||
| 2012 | 2011 | December 31, 2012 | ||||||||||
| Net loss | $ | (67,320 | ) | $ | (32,830 | ) | $ | (274,289 | ) | |||
| Other comprehensive loss | ||||||||||||
| Foreign currency translation adjustment | (40 | ) | 3 | (190 | ) | |||||||
| Total comprehensive loss | $ | (67,360 | ) | $ | (32,827 | ) | $ | (274,479 | ) | |||
See accompanying notes to the consolidated financial statements
| F-4 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF CHANGE IN STOCKHOLDERS’ EQUITY
(in thousands)
| Years Ended December 31, | Period from Inception (March 18, 1996) to | |||||||||||
| 2012 | 2011 | December 31, 2012 | ||||||||||
| Common stock: | ||||||||||||
| Balance, beginning of the period | $ | 150 | $ | 73 | $ | - | ||||||
| Conversion of common stock at par related to reverse stock splits | (140 | ) | - | (201 | ) | |||||||
| Conversion of notes payable due to management to common stock | - | - | 5 | |||||||||
| Conversion of Series A and A-1 preferred stock into common stock | - | - | 25 | |||||||||
| Exercise of stock options and warrants | - | - | 5 | |||||||||
| Loan conversion and conversion inducement | 12 | 62 | 91 | |||||||||
| Stock and warrants issued for cash | 5 | 7 | 80 | |||||||||
| Stock and warrants issued for preferred stock conversion | - | - | 7 | |||||||||
| Stock and warrants issued for services | - | 8 | 15 | |||||||||
| Balance, end of the period | $ | 27 | $ | 150 | $ | 27 | ||||||
| Additional paid-in-capital: | ||||||||||||
| Balance, beginning of the period | $ | 199,605 | $ | 191,344 | $ | - | ||||||
| Adjustment of par value related to reverse stock splits | 140 | - | 201 | |||||||||
| Amortization of deferred compensation, net | - | - | 1,779 | |||||||||
| Beneficial conversion feature of Series D preferred stock | - | - | 4,274 | |||||||||
| Beneficial conversion feature remeasurement | - | - | 1,198 | |||||||||
| Cancellation of employee restricted stock grants | - | - | (412 | ) | ||||||||
| Cancellation of employee stock options | - | - | (437 | ) | ||||||||
| Change in warrant liability | - | - | 4,863 | |||||||||
| Common stock - ESOP | - | - | 6 | |||||||||
| Common Stock warrant liability | - | - | (7,127 | ) | ||||||||
| Conversion of notes payable due to management to common stock | - | - | 266 | |||||||||
| Conversion of notes payable due to Toucan Capital to Series A-1 preferred stock | - | - | 7,702 | |||||||||
| Conversion of Series A and A-1 preferred stock into common stock | - | - | 31,592 | |||||||||
| Debt discount related to beneficial conversion and warrants | 7,885 | 4,267 | 21,972 | |||||||||
| Exercise of stock options and warrants | - | - | 436 | |||||||||
| Loan conversion and conversion inducement | 34,315 | 21,712 | 69,865 | |||||||||
| Reclassification of embedded derviatives liability | - | 900 | 900 | |||||||||
| Re-classification of equity instruments, net | 29,412 | (38,723 | ) | (9,311 | ) | |||||||
| Revaluation of preferred stock and warrants | - | - | 18,699 | |||||||||
| Stock and warrants issued for cash | 13,917 | 5,494 | 84,923 | |||||||||
| Stock and warrants issuances for accounts payable and accrued expenses | 11,026 | - | 11,476 | |||||||||
| Stock and warrants issued for dividends | - | - | 4,664 | |||||||||
| Stock and warrants issued for preferred stock conversion | - | - | 12,342 | |||||||||
| Stock and warrants issued for services | 3,685 | 6,166 | 20,255 | |||||||||
| Stock compensation expense | 3,203 | 8,445 | 23,062 | |||||||||
| Balance, end of the period | $ | 303,188 | $ | 199,605 | $ | 303,188 | ||||||
| Deficit accumulated during development stage: | ||||||||||||
| Balance, beginning of the period | $ | (251,778 | ) | $ | (218,948 | ) | $ | - | ||||
| Net loss | (67,320 | ) | (32,830 | ) | (319,098 | ) | ||||||
| Balance, end of the period | $ | (319,098 | ) | $ | (251,778 | ) | $ | (319,098 | ) | |||
| Cumulative translation adjustment: | ||||||||||||
| Balance, beginning of the period | $ | (150 | ) | $ | (153 | ) | $ | - | ||||
| Foreign currency translation adjustment | (40 | ) | 3 | (190 | ) | |||||||
| Balance, end of the period | $ | (190 | ) | $ | (150 | ) | $ | (190 | ) | |||
| Total stockholders' deficit: | $ | (16,073 | ) | $ | (52,173 | ) | $ | (16,073 | ) | |||
See accompanying notes to the consolidated financial statements
| F-5 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | Period from Inception (March 18, 1996) to | |||||||||||
| 2012 | 2011 | December 31, 2012 | ||||||||||
| Cash Flows from Operating Activities: | ||||||||||||
| Net Loss | $ | (67,320 | ) | $ | (32,830 | ) | $ | (274,289 | ) | |||
| Reconciliation of net loss to net cash used in operating activities: | ||||||||||||
| Depreciation and amortization | 14 | 10 | 2,377 | |||||||||
| Amortization of deferred financing costs | - | - | 320 | |||||||||
| Amortization debt discount and accretion on redeemable securities | 14,020 | 6,016 | 44,245 | |||||||||
| Derivative valuation gain | (601 | ) | (728 | ) | (1,383 | ) | ||||||
| Accrued interest converted to stock | - | - | 260 | |||||||||
| Accreted interest on convertible promissory note | - | - | 1,484 | |||||||||
| Stock-based compensation costs | 3,203 | 8,445 | 23,062 | |||||||||
| Stock and warrants issued for services and other expenses | 3,685 | 10,934 | 23,954 | |||||||||
| Loan and accounts payable conversion inducement | 9,103 | 125 | 19,518 | |||||||||
| Valuation of reclassified equity contracts | (491 | ) | (8,820 | ) | (16,070 | ) | ||||||
| Asset impairment loss and loss (gain) on sale of properties | - | - | (936 | ) | ||||||||
| Loss on facility sublease | - | - | 895 | |||||||||
| Increase (decrease) in cash resulting from changes in assets and liabilities: | ||||||||||||
| Prepaid expenses and other current assets | (18 | ) | (8 | ) | 598 | |||||||
| Accounts payable and accrued expenses | 577 | 1,356 | 7,723 | |||||||||
| Related party accounts payable and accrued expenses | 15,075 | - | 28,144 | |||||||||
| Deposits and other non-current assets | (1 | ) | 793 | (1 | ) | |||||||
| Accrued loss on sublease | - | - | (265 | ) | ||||||||
| Deferred rent | - | - | 410 | |||||||||
| Net Cash used in Operating Activities | (22,754 | ) | (14,707 | ) | (139,954 | ) | ||||||
| Cash Flows from Investing Activities: | ||||||||||||
| Purchase of property and equipment, net | (31 | ) | (49 | ) | (5,124 | ) | ||||||
| Proceeds from sale of property and equipment | - | - | 258 | |||||||||
| Proceeds from sale of intellectual property | - | - | 1,816 | |||||||||
| Proceeds from sale of marketable securities | - | - | 2,000 | |||||||||
| Refund of security deposit | - | - | (3 | ) | ||||||||
| Transfer of restricted cash | - | - | (1,035 | ) | ||||||||
| Net Cash used in Investing Activities | (31 | ) | (49 | ) | (2,088 | ) | ||||||
| Cash Flows from Financing Activities: | ||||||||||||
| Proceeds from issuance of redeemable securities | 5,302 | - | 5,302 | |||||||||
| Proceeds from issuance of notes payable | - | 2,130 | 7,980 | |||||||||
| Proceeds from issuance of convertible notes payable | 13,181 | 7,242 | 38,414 | |||||||||
| Proceeds from issuance of note payable to related parties | - | 600 | 11,250 | |||||||||
| Repayment of note payable to related parties | - | (450 | ) | (8,050 | ) | |||||||
| Repayment of convertible promissory note | (2,193 | ) | (399 | ) | (3,262 | ) | ||||||
| Borrowing under line of credit, Northwest Hospital | - | - | 2,834 | |||||||||
| Repayment of line of credit, Northwest Hospital | - | - | (2,834 | ) | ||||||||
| Payment on capital lease obligations | - | - | (323 | ) | ||||||||
| Payments on note payable | - | - | (420 | ) | ||||||||
| Proceeds from issuance preferred stock, net | - | - | 28,708 | |||||||||
| Proceeds from exercise of stock options and warrants | - | - | 228 | |||||||||
| Proceeds from issuance common stock, net | 13,857 | 5,501 | 72,932 | |||||||||
| Proceeds from sale of stock warrant | - | - | 90 | |||||||||
| Payment of preferred stock dividends | - | - | (1,251 | ) | ||||||||
| Series A preferred stock redemption fee | - | - | (1,700 | ) | ||||||||
| Deferred financing costs | - | - | (320 | ) | ||||||||
| Net Cash provided by Financing Activities | 30,147 | 14,624 | 149,578 | |||||||||
| Effect of exchange rates on cash and cash equivalents | (40 | ) | 3 | (190 | ) | |||||||
| Net increase (decrease) in cash and cash equivalents | 7,322 | (129 | ) | 7,346 | ||||||||
| Cash and cash equivalents at beginning of period | 24 | 153 | - | |||||||||
| Cash and cash equivalent at end of period | $ | 7,346 | $ | 24 | $ | 7,346 | ||||||
See accompanying notes to the consolidated financial statements
| F-6 |
NORTHWEST BIOTHERAPEUTICS, INC.
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Years Ended December 31, | Period from Inception (March 18, 1996) to | |||||||||||
| 2012 | 2011 | December 31, 2012 | ||||||||||
| Supplemental disclosure of cash flow information - Cash paid during the period for interest | $ | - | $ | - | $ | 1,879 | ||||||
| Supplemental schedule of non-cash financing activities: | ||||||||||||
| Equipment acquired through capital leases | - | - | 285 | |||||||||
| Issuance of common stock in connection with elimination of Series A and Series A-1 preferred stock preferences | - | - | 12,349 | |||||||||
| Issuance of common stock in connection with conversion of liabilities | 40,127 | 15,769 | 59,182 | |||||||||
| Issuance of redeemable common stock in connection with conversion of liabilities | 3,673 | 3,673 | ||||||||||
| Warrants issued on Series A and Series A-1 preferred stock dividends | - | - | 4,664 | |||||||||
| Liability for reclassified equity contracts | 29,412 | - | 41,253 | |||||||||
| Accretion of mandatorily redeemable Series A preferred stock redemption obligation | - | - | 1,872 | |||||||||
| Debt discount on promissory notes | 7,913 | 5,410 | 27,414 | |||||||||
| Issuance of Series C preferred stock warrants in connection with lease agreement | - | - | 43 | |||||||||
| Issuance of common stock to settle accounts payable | - | - | 4 | |||||||||
| Liability for and issuance of common stock and warrants to Medarex | - | - | 840 | |||||||||
| Issuance of common stock to landlord | - | - | 35 | |||||||||
| Deferred compensation on issuance of stock options and restricted stock grants | - | - | 759 | |||||||||
| Cancellation of options and restricted stock | - | - | 849 | |||||||||
| Financing of prepaid insurance through note payable | - | - | 491 | |||||||||
| Stock subscription receivable | - | - | 480 | |||||||||
| Modification of Series A preferred stock warrants | - | - | 2,306 | |||||||||
| Modification of Series A-1 preferred stock warrants | - | - | 16,393 | |||||||||
| Conversion of convertible promissory notes and accrued interest to Series A-1 preferred stock | - | - | 7,707 | |||||||||
| Conversion of convertible promissory notes and accrued interest to Series D preferred stock | - | - | 5,324 | |||||||||
| Conversion of debt to accounts payable | 1,428 | - | 1,428 | |||||||||
| Conversion of convertible promissory notes and accrued interest to stock | - | - | 269 | |||||||||
See accompanying notes to the consolidated financial statements
| F-7 |
| 1. | Organization and Description of Business and Basis of Presentation |
Northwest Biotherapeutics, Inc. and its majority owned subsidiaries NW Bio Europe S.A.R.L and NW Bio GmBh (collectively, the “Company”, “we”, “us” and “our”) was organized to discover and develop innovative diagnostics and immunotherapies for prostate and brain cancer. During 1998, the Company incorporated as a Delaware corporation. Prior to 1998, the Company was a limited liability company, which was formed on March 18, 1996. The Company is a development stage company, has yet to generate significant revenues from its intended business purpose and has no assurance of future revenues. While in the development stage, the Company’s principal activities have included defining and conducting research programs, conducting clinical trials, raising capital and recruiting scientific and management personnel.
The accompanying consolidated financial statements include the accounts of Northwest Biotherapeutics, Inc. and its subsidiaries, NW Bio Europe S.A.R.L. and NW Bio GmBh (the “Company”).
Effective September 25, 2012, all shares of the Company’s common stock issued and outstanding were combined and reclassified on a one-for-sixteen basis. The effect of this reverse stock split has been retroactively applied to all periods presented.
In December 2012, the Company retired $36.4 million of convertible notes, notes and payables and accrued interest by entering into Conversion Agreements with our non-affiliated and affiliated noteholders and creditors, including certain of our directors and executive officers. This aggregate debt amount was converted into 9.8 million common shares (of which 0.9 million are redeemable) and warrants exercisable for 3.8 million shares of common stock. The warrants have an exercise period of five years from the date of issuance and a weighted average exercise price of $3.66 per share.
| 2. | Liquidity and Going Concern |
The Company has experienced recurring losses from operations. Net cash outflows from operations were $22.8 million for the twelve months ended December 31, 2012. The Company had a working capital deficit of $3.3 million at December 31, 2012 (excluding redeemable common stock amounting to $11.0 million).
The Company had a deficit accumulated during the development stage of $319.1 million.
Since 2004, Toucan Capital Fund II, L.P. (“Toucan Capital”), Toucan Partners LLC (“Toucan Partners”), entities controlled by Ms. Linda Powers, the Company’s CEO and the managing director of Toucan Capital and managing member of Toucan Partners, and Ms. Linda Powers (collectively “Toucan”) have provided substantial funding to the Company. From 2004 through December 31, 2012, Toucan loaned the Company approximately $5.3 million net of repayments and conversions. In addition, Toucan provided other financing to the Company through the purchase of common stock and preferred stock. As of the date of this report, Toucan (other then Cognate BioServices) holds approximately 20% of common stock outstanding as of December 31, 2012.
On October 16, 2012, the Company entered into conversion agreements with Toucan Partners and its affiliates (other than Cognate BioServices), pursuant to which, upon the closing of the public offering in December 2012, an aggregate of $10.7 million of convertible notes and payables was to be converted into equity. The notes were converted substantially in accordance with their existing terms. With respect to the payables, Toucan had a right to convert on terms no less favorable than provided to any other creditor. However, Toucan agreed to instead convert on terms equal to the median of the conversion terms provided to other non-affiliated creditors over the preceding six months. Accordingly, the Toucan entities’ $10.7 million aggregate conversion amounted to 3.6 million shares of restricted common stock and 1.8 million warrants exercisable for common stock. The warrants’ exercise period are five years, and the exercise price is $3.20 per share. Total liabilities owed to Toucan as of December 31, 2012 were less than $0.01 million.
In addition to financing obtained from Toucan and related entities, the Company has raised additional capital by issuing common stock and debt securities. As of December 31, 2012 the Company had approximately $7.3 million of cash and cash equivalents and had current liabilities of $10.7 million and redeemable common stock of $11.0 million. The Company will need to raise additional capital in the near future to continue to fund its clinical trials and other operating activities and there can be no assurance that its efforts to seek such funding will be successful. The Company may seek funding from Toucan Capital or Toucan Partners or their affiliates or other third parties. Such parties are under no obligation to provide the Company with any additional funds, and any such funding may be dilutive to stockholders and may contain restrictive covenants. The Company is currently exploring additional financings with several other parties; however, there can be no assurance that the Company will be able to complete any such financings or that the terms of such financings will be attractive to the Company. If the Company’s capital raising efforts are unsuccessful, its inability to obtain additional cash as needed could have a material adverse effect on the Company’s financial position, results of operations and the Company’s ability to continue its existence.
| F-8 |
The independent registered public accounting firm’s report on the financial statements for the fiscal year ended December 31, 2012 states that because of recurring operating losses, net operating cash flow deficits, and a deficit accumulated during the development stage, there is substantial doubt about the Company’s ability to continue as a going concern. A “going concern” opinion indicates that the financial statements have been prepared assuming the Company will continue as a going concern and do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
| 3. | Summary of Significant Accounting Policies |
| a. | Principles of Consolidation and Basis of Presentation |
The accompanying consolidated financial statements of the Company were prepared in accordance with U.S. GAAP and include the assets, liabilities, revenues and expenses of the Company’s majority-owned subsidiaries over which the Company exercises control. Intercompany transactions and balances were eliminated in consolidation. The first of the Company's European subsidiaries was established in Switzerland during the third quarter of 2007 and the second subsidiary was established in Germany during the fourth quarter of 2011. The German subsidiary is wholly-owned. The Company contributed 95% of the initial share capital in the Swiss subsidiary and Cognate, a related party to the Company, contributed the remaining 5%. Non-controlling interest is not material for all periods presented.
| b. | Foreign Currency Translation |
For operations outside the U.S. that prepare financial statements in currencies other than U.S. dollars, we translate the financial statements into U.S. dollars. Results of operations and cash flows are translated at average exchange rates during the period, and assets and liabilities are translated at end of period exchange rates, except for equity transactions and advances not expected to be repaid in the foreseeable future, which are translated at historical cost. The effects of exchange rate fluctuations on translating foreign currency assets and liabilities into U.S. dollars are accumulated as a separate component in other comprehensive income (loss).
| c. | Use of Estimates in Preparation of Financial Statements |
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
| d. | Embedded Derivative Liability |
The Company evaluates financial instruments for freestanding or embedded derivatives. Derivative instruments that have been separated from the host contract and do not qualify for hedge accounting are recorded at fair value with changes in value recognized as other income (expense) in the consolidated statements of operations in the period of change.
| e. | Fair Value of Financial Instruments |
The fair value of financial instruments other than liabilities payable to related parties approximate the recorded value based on the short term nature of these financial instruments. The fair value of liabilities payable to related parties is presently undeterminable due to the related party nature of the obligations. The fair value of derivative liabilities is measured using a binomial model or Monte Carlo simulation depending on the complexity of the derivative. SeeNote 4.
| f. | Cash |
Cash consists of checking accounts. While cash held by financial institutions may at times exceed federally insured limits, management believes that no material credit or market risk exposure exists due to the high quality of the institutions. The Company has not experienced any losses on such accounts.
| F-9 |
| g. | Property and Equipment |
Property and equipment are stated at cost, as adjusted for any prior impairment charges. Property and equipment are depreciated on a straight-line basis over the estimated useful lives which range from three to seven years.
Expenditures for maintenance and repairs are expensed as incurred. Gains and losses from disposal representing the difference between any proceeds received from the sale of property and equipment and the recorded values of the asset disposed are recorded in total operating costs and expenses.
| h. | Operating Leases |
The Company recognizes lease expense on a straight-line basis over the initial lease term. For leases that contain rent holidays or escalation clauses, the Company recognizes rent expense on a straight-line basis and records the difference between the rent expense and rental amount payable as deferred rent. As of December 31, 2012 and 2011 deferred rent was $0 and $19,004, respectively.
| i. | Revenue Recognition |
In various situations, the Company receives certain payments from patients. These payments are generally non-refundable, and are not dependent on the Company’s ongoing future performance. The Company has adopted a policy of recognizing these payments as revenue when received.
| j. | Research and Development Expenses |
Research and development costs are expensed as incurred. These costs include, but are not limited to, contract manufacturing costs, personnel costs, lab supplies, depreciation, amortization and other indirect costs directly related to the Company’s research and development activities.
| k. | Income Taxes |
We recognize income taxes on an accrual basis based on tax positions taken or expected to be taken in our tax returns. A tax position is defined as a position in a previously filed tax return or a position expected to be taken in a future tax filing that is reflected in measuring current or deferred income tax assets and liabilities. Tax positions are recognized only when it is more likely than not (i.e., likelihood of greater than 50%), based on technical merits, that the position would be sustained upon examination by taxing authorities. Tax positions that meet the more likely than not threshold are measured using a probability-weighted approach as the largest amount of tax benefit that is greater than 50% likely of being realized upon settlement. Income taxes are accounted for using an asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in our financial statements or tax returns. A valuation allowance is established to reduce deferred tax assets if all, or some portion, of such assets will more than likely not be realized. Should they occur, our policy is to classify interest and penalties related to tax positions as income tax expense. Since our inception, no such interest or penalties have been incurred, however prior to 1998 the Company was a limited liability company and the Company’s tax losses and credits generally flowed directly to the members.
| l. | Stock-based Compensation |
Compensation expense for all stock-based awards is measured on the grant date based on the fair value of the award and is recognized as an expense, on a straight-line basis, over the employee's requisite service period (generally the vesting period of the equity award). The fair value of each option award is estimated on the grant date using a Black-Scholes option valuation model. Stock-based compensation expense is recognized only for those awards that are expected to vest using an estimated forfeiture rate. We estimate pre-vesting option forfeitures at the time of grant and reflect the impact of estimated pre-vesting option forfeitures in compensation expense recognized. For options and warrants issued to non-employees, the Company recognizes stock compensation costs utilizing the fair value methodology over the related period of benefit.
| F-10 |
Stock-based compensation expense was as follows for the twelve months ended December 31, 2012 and 2011 (in thousands):
| Year Ended December 31, | ||||||||
| 2012 | 2011 | |||||||
| Research and development | $ | 460 | $ | 2,355 | ||||
| General and administrative | 2,743 | 6,090 | ||||||
| Total stock- based compensation expense | $ | 3,203 | $ | 8,445 | ||||
The assumptions used to estimate the fair value of awards granted for the periods presented are noted as follows. Expected volatility is based on the separate historical volatility of the market prices of our common stock over the most recent period commensurate with the estimated expected life of the award. The risk-free rate for the expected term of the option is based on the U.S. Treasury yield curve in effect at the time of grant. In 2011, the Company granted stock options and valued such options using the following assumptions: risk free interest rate – 2.27%, volatility – 193.5%, expected term – 7 years, expected dividends – N/A.
| m. | Loss per Share |
Basic loss per share is computed on the basis of the weighted average number of shares outstanding for the reporting period. Diluted loss per share is computed on the basis of the weighted average number of common shares (including redeemable shares) plus dilutive potential common shares outstanding using the treasury stock method. Any potentially dilutive securities are anti-dilutive due to the Company’s net losses. For the years presented, there is no difference between the basic and diluted net loss per share.
| n. | Operating Segments |
The Company is principally engaged in the discovery and development of innovative immunotherapies for cancer and has a single operating segment as management reviews all financial information together for the purposes of making decisions and assessing the financial performance of the Company.
Operating costs. Operating costs and expenses consist primarily of research and development expenses, including clinical trial expenses which arise when we are actively participating in clinical trials, and general and administrative expenses.
Research and development. Discovery and preclinical research and development expenses include scientific personnel related salary and benefit expenses, stock-based compensation, costs of laboratory supplies used in our internal research and development projects, travel, regulatory compliance, and expenditures for preclinical and clinical trial operation and management when we are actively engaged in clinical trials.
Because the Company is a development stage company, it does not allocate research and development costs on a project basis. The Company adopted this policy, in part, due to the unreasonable cost burden associated with accounting at such a level of detail and its limited number of financial and personnel resources. The Company’s business judgment continues to be that there is little value associated with evaluating expenditures at the project level since the Company is focusing primarily on its lead clinical trial programs as most of the Company’s expenditures relate to those programs.
General and administration. General and administration expenses include administrative personnel related salary and benefit expenses, cost of facilities, insurance, travel, legal support, property and equipment depreciation, stock-based compensation, and amortization of debt discounts, inducement expenses and beneficial conversion costs associated with the Company’s debt financing.
| o. | Reclassifications |
Certain reclassifications have been made to prior period financial statements and footnotes in order to conform to the current period's presentation.
| p. | Recent and Adopted Accounting Pronouncements |
In September 2011, the Financial Accounting Standards Board issued Accounting Standards Update (“ASU”) No. 2011-05,Comprehensive Income, or ASU 2011-05. The guidance in ASU 2011-05 revises the manner in which entities present comprehensive income in their financial statements. An entity is required to report the components of comprehensive income in either one or two consecutive financial statements:
| F-11 |
| · | A single, continuous statement must present the components of net income and total net income, the components of other comprehensive income and total other comprehensive income, and a total for comprehensive income; or |
| · | In a two-statement approach, an entity must present the components of net income and total net income in the first statement. That statement must be immediately followed by a financial statement that presents the components of other comprehensive income, a total for other comprehensive income, and a total for comprehensive income. |
ASU 2011-05 does not change the items that must be reported in other comprehensive income. The amendments in ASU 2011-05 are effective for fiscal years beginning after December 15, 2011. The Company adopted this guidance, and implemented the two-statement approach.
| q. | Reclassified Equity Contracts |
The Company accounts for potential shares that can be converted to common stock and if converted, will be in excess of authorized shares, as a liability that is recorded on the balance sheet (at fair value) only until the authorized number of shares is increased (at which time the whole liability will be re-measured, with changes in value included in other income/(expense), and then reclassified to additional paid-in capital). The value of the liability was computed by valuing the securities that management believed were most likely to be converted. This liability is revalued at each reporting date with any change in value included in other income / (expense) until such time as enough shares are authorized to cover all potentially convertible instruments.
| r. | Redeemable Securities |
The Company accounts for redeemable common stock in accordance with Accounting Standards Codification (“ASC”) 480-10-S99-3A,Classification and Measurement of Redeemable Securities, which provides that securities that are optionally redeemable by the holder for cash or other assets are classified outside of permanent equity in temporary equity.
| 4. | Fair Value Measurements |
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. U.S. GAAP establishes a three tier fair value hierarchy which prioritizes the inputs used in measuring fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 inputs) and the lowest priority to unobservable inputs (Level 3 inputs). Level 1, 2, and 3 inputs are defined as follows:
| · | Level 1 inputs, are observable inputs such as quoted prices for identical instruments in active markets; |
| · | Level 2 inputs, are inputs other than quoted prices in active markets that are either directly or indirectly observable such as quoted prices for similar instruments in active markets or quoted prices for identical or similar instruments in markets that are not active; and |
| · | Level 3 inputs, are unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions, such as valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable. |
The Company's liability for reclassified equity contracts was measured using significant unobservable (Level 3) inputs.
In 2011, as a result of the Company entering into convertible promissory notes and issuing stock options and warrants to purchase common stock, the Company's total potential outstanding common stock exceeded the Company’s authorized shares by approximately 6.8 million shares as of December 31, 2011, as also discussed inNote 9. As a result, the Company was required to value a number of shares equal to the excess issuable on exercise of warrants and options and on conversion of convertible notes and recognize the value as a liability. Effective February 6, 2012, the Company’s stockholders approved an increase in the number of authorized shares sufficient to cover the excess. At that time, the liability was re-measured, with changes in value included in other income/(expense), and then reclassified to additional paid-in capital (thereby removing it from the Company’s liabilities).
The Company concluded that certain conversion features and warrant agreements included down-round provisions and were not indexed to the Company’s stock (and are therefore recorded as derivative liabilities). The Company recognizes the derivative liabilities at their respective fair values using a binomial model adjusted for the probability of issuance using a Monte Carlo simulation. Changes in the fair value are recorded in derivative valuation gain (loss) in the consolidated statements of operations. Key assumptions for determining fair values during the periods presented included expected terms ranging from 3 to 15 months, volatility ranging from 95% to 190% and risk-free interest rate of 0.18%.
| F-12 |
The Company's embedded derivative liability was measured using significant unobservable (Level 3) inputs. The following table represents the Company’s embedded derivative liability activity (in thousands) for the twelve months ended December 31, 2012 and 2011:
| Year Ended December 31, | ||||||||
| 2012 | 2011 | |||||||
| Beginning balance | $ | 601 | $ | 839 | ||||
| Reclassification to stockholders' equity upon conversion and expiration of derivative | - | (899 | ) | |||||
| Embedded derivative liability recognized | - | 1,389 | ||||||
| Net change in fair value of embedded derivative liabilities | (601 | ) | (728 | ) | ||||
| Ending balance | $ | - | $ | 601 | ||||
The following table represents the activity for the Company's liability for reclassified equity contracts (in thousands) for the twelve months December 31, 2012 and 2011:
| Year Ended December 31, | ||||||||
| 2012 | 2011 | |||||||
| Beginning Balance | $ | 29,903 | $ | - | ||||
| Liabilities reclassified | 693 | 38,724 | ||||||
| Change in value of liabilities reclassified | (491 | ) | (8,821 | ) | ||||
| Liabilities reclassified to equity | (30,105 | ) | - | |||||
| Ending Balance | $ | - | $ | 29,903 | ||||
| 5. | Stock-based Compensation |
Stock Option Plans
The Company’s stock option plans are administered by the Board of Directors, which determines the terms and conditions of the options granted, including exercise price, number of options granted and vesting period of such options.
Stock Option Activity
A summary of stock option activity for 2012 is as follows (shares in thousands):
| Number of Options | Weighted Average Exercise Price | Weighted Average Grant Date Fair Value | Average Remaining Contractual Life | Average Intrinsic Value | ||||||||||||||||
| Outstanding at December 31, 2011 | 1,551 | $ | 10.56 | $ | 10.56 | 6.6 | $ | - | ||||||||||||
| Granted | - | - | - | - | - | |||||||||||||||
| Expired | - | - | - | - | - | |||||||||||||||
| Forfeited | - | - | - | - | - | |||||||||||||||
| Outstanding at December 31, 2012 | 1,551 | $ | 10.56 | $ | 10.56 | 5.6 | $ | - | ||||||||||||
| Exercisable as of December 31, 2012 | 1027 | 8.02 | ||||||||||||||||||
| F-13 |
Additional information regarding stock options outstanding and exercisable at December 31, 2012 is as follows (in thousands, except option price and weighted average exercise price):
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Range of Exercise Prices | Number Outstanding | Weighted Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | Number Exercisable | Weighted Average Exercise Price | |||||||||||||||
| $ 8.80 - 9.60 | 105 | 6.64 | $ | 8.80 | 60 | $ | 8.96 | |||||||||||||
| $ 9.76 - 33.60 | 1,446 | 5.50 | 10.72 | 967 | 10.72 | |||||||||||||||
| Total | 1,551 | 8.02 | $ | 10.56 | 1027 | $ | 10.72 | |||||||||||||
Options granted under the plans are generally priced at or above the estimated fair market value of the Company’s common stock on the date of grant and generally vest between four and nine years. Compensation expense, if any, is charged over the period of vesting. All options, if not previously exercised or canceled, expire ten years from the date of grant, or the expiration date specified in the individual option agreement, if earlier.
During 2011, the Company granted options to purchase 1,383,000 shares of common stock. The Company granted no options in 2012. The weighted average exercise price and grant date fair value of options granted in 2011 was $10.56. Stock compensation expense amounted to $3.2 million and $8.4 million during 2012 and 2011, respectively.
There were no exercises of options in either 2012 or 2011. Our policy, in the event of exercise, is to issue new shares to fulfill the requirements for options that are exercised.
The aggregate fair value of options vested during 2012 and 2011 was $0.1 million and $7.0 million, respectively.
As of December 31, 2012 the total unrecognized compensation expense related to unvested stock option awards was $4.1 million which is expected to be recognized over a weighted average term of approximately 2 years.
| 6. | Notes Payable |
The Company regularly issues notes and the proceeds from the notes are used to finance operations. The notes may contain conversion features and may be issued along with warrants to purchase common stock. For convertible notes, the Company allocates the proceeds received between convertible notes payable and warrants on a relative fair value basis, if applicable. The resulting discount for warrants is amortized using the effective interest method over the life of the debt instrument. After allocating a portion of the proceeds to the warrants, the effective conversion price of the convertible note payable can be determined. If the effective conversion price is lower than the market price of the Company's common stock on the date of issuance, a beneficial conversion feature is recorded as an additional discount to the convertible notes payable. The beneficial conversion feature discount is also amortized using the effective interest method over the life of the debt instrument. The amortization is recorded as interest expense on the consolidated statement of operations. During the twelve months ended December 31, 2012 and December 31, 2011, the Company received proceeds from the issuance of convertible notes of $13.2 million and $7.2 million, respectively. The notes are payable on various dates through September 2014 and have interest rates between 0% and 12%. During the twelve months ended December 31, 2012 and December 31, 2011, the Company recorded a debt discount related to the beneficial conversion feature for convertible notes and detachable warrants of $7.9 million and $5.4 million, respectively. There were approximately 1.6 million common stock purchase warrants with a weighted average exercise price of $7.30 also issued in connection with the notes during 2012. The conversion prices of the notes, with fixed terms, range between $3.20 and $20.00. The beneficial conversion feature recorded on the notes payable for 2012 and 2011 was estimated based on the effective conversion price of convertible notes after allocating debt proceeds between the note instruments and warrants on a relative fair value basis.
| F-14 |
During 2012, the Company converted notes payable of $27.8 million into approximately 7.5 million shares of common stock. The Company also issued approximately 4.7 million warrants to purchase common stock in connection with the conversion of notes payable during 2012. Certain notes were converted under inducement agreements during 2012 and as such, the difference between the fair value of the 10.1 million shares of common stock and 3.8 million warrants issued in excess of the fair value of securities issuable pursuant to original conversion terms amounted to $9.1 million and was recorded as conversion inducement expense in 2012. The fair value of the common stock issued in connection with the inducement agreements was valued after determining a 14% discount from the closing price of the Company’s common stock on the date the stock was issued for lack of marketability. The fair value of the warrants was determined using the Black-Scholes model with the following assumptions: risk free interest rate – 0.4%, volatility – 65%, expected term – 5 years, expected dividends – N/A. The principal and accrued interest payable converted to common stock during 2011 amounted to $4.8 million.
During 2012 and 2011, the Company repaid $2.4 million and $0.4 million of notes payable, respectively. The Company is currently negotiating the repayment terms of notes payable outstanding as of December 31, 2012, which have due dates prior to December 31, 2012.
Notes payable consist of the following at December 31, 2012 and December 31, 2011 (in thousands):
| 2012 | 2011 | |||||||
| Notes payable - current | ||||||||
| 12% unsecured originally due July 2011 | $ | 934 | $ | 935 | ||||
| 6% unsecured (net of discount of $0 in 2012 and $236 in 2011) | - | 1,764 | ||||||
| 12% unsecured | - | 450 | ||||||
| 934 | 3,149 | |||||||
| Notes payable related parties - current | ||||||||
| 12% unsecured (net of discount of $0 in 2012 and $21 in 2011) | - | 2,056 | ||||||
| Convertible notes payable, net - current | ||||||||
| 6% unsecured (net of discount of $0 in 2012 and $34 in 2011) | 435 | 2,676 | ||||||
| 10% unsecured (net of discount of $0 in 2012 and $38 in 2011) | - | 150 | ||||||
| 8% unsecured due April 2013 (net of discount of $4 in 2012) | 71 | - | ||||||
| 10% unsecured convertible note originally due November 2012 (net of discount of $0 in 2012 and $1,833 in 2011) | 500 | 1,167 | ||||||
| 6% unsecured originally due June 2012 (net of discount of $0 in 2012 and $182 in 2011) | - | 839 | ||||||
| 1,006 | 4,832 | |||||||
| Convertible Notes payable related party, net - current | ||||||||
| 10% unsecured (net of discount of $0 in 2012) | - | - | ||||||
| 12% unsecured | - | 2,430 | ||||||
| 6% due on demand (net of discount of $0 in 2012 and $92 in 2011) | 50 | 1,158 | ||||||
| 50 | 3,588 | |||||||
| Long term notes payable | ||||||||
| 6% unsecured note due October 2012 | - | 200 | ||||||
| Long term convertible notes, net | ||||||||
| 4% unsecured (net of discount of $0 in 2012 and $42 in 2011) | - | 402 | ||||||
| 0% unsecured | 53 | - | ||||||
| 4% unsecured (net of discount of $9 in 2011) | - | 67 | ||||||
| 4% unsecured due September 2014 (net of discount of $415 in 2012) | 419 | - | ||||||
| 11% unsecured convertible note due December 2013 (net of discount of $0 in 2012 and $321 in 2011) | - | 964 | ||||||
| 8% unsecured due 2014 (net of discount of $114 in 2012) | 1,410 | - | ||||||
| 1,882 | 1,433 | |||||||
| Total notes payable, net | $ | 3,872 | $ | 15,258 | ||||
Principal amounting to $2.0 million is payable during 2013 and $2.4 million is payable during 2014.
| F-15 |
| 7. | Reclassified Equity Contracts |
The Company accounts for potential shares that can be converted to common stock and that if converted, will be in excess of authorized shares, as a liability that is recorded on the balance sheet (at fair value) only until the authorized number of shares is increased (at which time the whole liability will be remeasured, with changes in fair value included in other income (expense), and then reclassified to additional paid-in capital). The fair value of the liability was computed by valuing the securities that management believed were most likely to be converted. This liability is revalued at each reporting date with any change in fair value included in other income (expense) until such time as enough shares are authorized to cover all potentially convertible instruments.
In 2011, as a result of the Company entering into convertible promissory notes and issuing stock options, and warrants to purchase common stock, the Company's total potential outstanding common stock exceeded the Company's authorized shares by approximately 6.8 million shares at December 31, 2011. During 2012, the number of potential shares in excess of authorized shares increased to approximately 7.0 million. Effective February 6, 2012, the number of authorized common shares was increased and the liability for potential shares in excess of total authorized shares was revalued at that date. This valuation resulted in non-cash gain of approximately $0.5 million during the twelve months ended December 31, 2012. The liability that was reclassified to additional paid-in capital (and thereby removed from the Company’s liabilities) during the twelve months ended December 31, 2012 amounted to approximately $30.1 million.
| 8. | Net Loss Per Share Applicable to Common Stockholders |
Options, warrants, and convertible debt outstanding were all considered anti-dilutive for the twelve months ended December 31, 2012, and for the twelve months ended December 31, 2011, due to net losses.
The following securities were not included in the diluted net income (loss) per share calculation because their effect was anti-dilutive as of the periods presented (in thousands):
| Year Ended December 31, | ||||||||
| 2012 | 2011 | |||||||
| Common stock options | 1,551 | 1,551 | ||||||
| Common stock warrants | 12,087 | 3,568 | ||||||
| Convertible notes | 765 | 1,978 | ||||||
| Excluded potentially dilutive securities | 14,403 | 7,097 | ||||||
| 9. | Related Party Transactions |
| a. | Cognate BioServices |
In April, 2011, the Company entered into a new service agreement with Cognate Therapeutics, Inc. (“Cognate”), a contract manufacturing and services organization in which Toucan Capital has a majority interest. In addition, two of the principals of Toucan Capital are members of Cognate’s board of directors and Linda Powers who is a director of Cognate and managing director of Toucan Capital is Chairperson of the Company’s Board of Directors and Chief Executive Officer of the Company. This agreement replaces the agreement dated May 17, 2007 between the Company and Cognate, which had expired. Under the service agreement, the Company agreed to continue to utilize Cognate’s services, for certain consulting and manufacturing services to the Company for its ongoing DCVax®-Brain Phase III clinical trial. The scope of services is comparable to the prior agreement, and the structure and process for payments are simplified. Under the terms of the current agreement the Company pays Cognate a monthly facility fee and a fixed fee (in lieu of cost-plus charges) for each patient in the study, subject to specified minimum number of patients per month, plus charges for certain patient and product data services if such services are requested by the Company. The current service agreement will expire on March 31, 2016. During December 2012, the Company issued five year warrants for the purchase of 1,437,500 shares of common stock at an exercise price of $6.40 in connection with the conversion agreement with Cognate and as additional consideration under the service agreement. The fair value of the warrants amounting to $1,878,247 was recorded as research and development expenses and was determined using the Black-Scholes Model with the following assumptions: risk free interest rate – 0.4% volatility – 65%, expected term – five years, expected dividends – N/A.
| F-16 |
On October 16, 2012, we entered into a conversion agreement with Cognate BioServices pursuant to which, upon the closing of the public offering in December 2012, an aggregate of $7.5 million unpaid invoiced amounts and payables were converted into equity. With respect to the unpaid invoice amounts and payables, Cognate had a right to convert on terms no less favorable than provided to any other creditor. However, Cognate agreed instead to convert on terms equal to the median of the conversion terms provided to other non-affiliated creditors over the preceding six months. Accordingly, liabilities payable to Cognate amounting to $7.5 million were converted into a total of 2.8 million shares of restricted common stock and 1.4 million warrants exercisable for common stock. The term of the warrants is five years and the exercise price is $3.20 per share. The difference between the fair value of the shares of common stock and warrants issued in excess of the carrying amount of the liabilities amounting to $3.1 was recorded as conversion inducement expense in 2012. The fair value of the common stock issued was valued after determining a 14% discount from the closing price of the Company’s common stock on the date the stock was issued for lack of marketability. The fair value of the warrants was determined using the Black-Scholes model with the following assumptions: risk free interest rate – 0.4%, volatility – 65%, expected term – 5 years, expected dividends – N/A.
In 2012, the Company issued approximately 0.5 million shares and 0.1 million warrants with an exercise price of $6.40 to an outside party in order to settle a note payable that Cognate owed to the unrelated party. The Company does not expect reimbursement from Cognate and as such, the fair value of the common stock and warrants issued to the outside party amounting to $2.2 million and was recorded as inducement expense for 2012.
During the twelve months ended December 31, 2012 and 2011, the Company recognized approximately $16.5 million and $4.7 million, respectively, of research and development costs related to these service agreements. As of December 31, 2012 and December 31 2011, the Company owed Cognate approximately $1.8 million and $0.6 million, respectively.
| b. | Toucan Capital and Toucan Partners |
In 2011 the Company received proceeds of $1.4 million from Toucan in connection with issuing unsecured convertible notes. The notes were payable within one year and carried an original issue discount of 10%. The conversion price was $3.20.
In 2011 the Company converted notes payable to Toucan amounting to $0.7 million into approximately 0.2 million shares of common stock.
In 2012 the Company issued convertible notes payable of $3.1 million to Toucan. Warrants to purchase 572,402 shares of common stock at an exercise price of $6.40, and a five year term, were issued in connection with the notes. The notes were payable within one year and carried an original issue discount of 10%. The conversion price was $3.20.
| F-17 |
On October 16, 2012, the Company entered into conversion agreements with Toucan Partners and its affiliates (with the exception of Cognate BioServices as discussed above), pursuant to which, upon the closing of the public offering in December 2012, an aggregate of $10.7 million of convertible notes and payables were converted into equity. The notes were converted substantially in accordance with their existing terms. With respect to the payables, Toucan had a right to convert on terms no less favorable than provided to any other creditor. However, Toucan agreed to instead convert on terms equal to the median of the conversion terms provided to other non-affiliated creditors over the preceding six months. The payables arose under an agreement with Toucan where the Company agreed to reimburse Toucan for certain payables, including interest, amounting to $4.6 million. The expense of $4.6 million associated with the payables was recognized in the Company’s financial statements during 2012 when the Company and Toucan concluded negotiations as to the amount owed for expenses Toucan paid on the Company’s behalf during the period of 2004 to 2012. Accordingly, the Toucan entities’ $10.7 million aggregate conversion amount was converted into a total of 3.6 million shares of restricted common stock and 1.8 million warrants exercisable for common stock. The warrants’ exercise period is five years, and the exercise price is $3.20 per share. The difference between the fair value of the shares of common stock and warrants issued in excess of the carrying amount of the liabilities amounting to$1.9was recorded as conversion inducement expense in 2012. The fair value of the common stock issued was valued after determining a 14% discount from the closing price of the Company’s common stock on the date the stock was issued for lack of marketability. The fair value of the warrants was determined using the Black-Scholes model with the following assumptions: risk free interest rate – 0.4%, volatility – 65%, expected term – 5 years, expected dividends – N/A. Total liabilities owed to Toucan as of December 31, 2012 were less than $0.01 million. Toucan continues to pay expenses on the Company’s behalf which have not been formally billed to the Company. Toucan may seek reimbursement for the expenses incurred subsequent to the settlement on October 16, 2012. Management is not able to estimate the financial statement impact of further expense reimbursements to Toucan as of the date these financial statements were issued.
| c. | Other Related Parties |
The Company received proceeds of $0.2 million in connection with issuing an unsecured convertible note to an officer of the Company on January 3, 2012. The notes included 44,532 warrants to purchase common stock. The exercise price of the warrants is $6.40, and the exercise period is 5 years. The note was payable on demand with 7 days written notice and carried an original issue discount of 10% and a one-time interest charge of 10%. The conversion price of the note was 95% of the average of the lowest five days’ closing prices of the Company's common stock in the twenty trading days prior to conversion. This note plus accrued interest was converted into 49,500 shares of common stock in December 2012.
The Company received proceeds of $0.3 million in connection with issuing an unsecured convertible demand note to an officer of the Company on June 29, 2012. The conversion price was $5.28 and the interest rate was 10%. The notes included 43,750 warrants to purchase common stock. The exercise price is $5.60 and the exercise period of the warrants is five years. This note plus accrued interest was converted into 66,341 shares of common stock in December 2012.
During the previous three years, a Board member of the Company arranged multiple financings for the Company. The Company agreed to compensate the Board member at a rate below the usual fees of investment banks for such transactions, by paying the Board member 63,000 shares of common stock valued at $0.3 million during the third quarter of 2012.
| F-18 |
| 10. | Redeemable Common Stock |
In October and November 2012, the Company issued 1.9 million shares of redeemable common stock, at a purchase price of $4.80 per share to accredited investors (collectively, the “Investors”) in separate private placement transactions and as part of conversions of debt into redeemable common stock. Total cash received amounted to $5.3 million and total conversions of debt into redeemable common stock amounted to $4.6 million. These transactions were completed pursuant to a Securities Purchase Agreement (the “Agreement”) which the Company entered into with each of the respective investor. The Agreements provide that such investors can redeem the securities for cash at a premium to the original issuance ranging from 15% to 25%. The redemption provisions occur within 12 months from the date of issuance.
The Company first assessed the redeemable common stock to determine if the instrument should be accounted for as a liability in accordance withASC 480. As the put option is optionally redeemable by the holder, the common stock was not required to be accounted for as a liability. Next, the Company assessed the put option within the redeemable common stock as a potential embedded derivative pursuant to the provisions of ASC 815,Derivatives and Hedging, and concluded that the put option did not meet the net settlement criteria within the definition of a derivative. Therefore, the Company has accounted for the common stock issued pursuant to the Agreement in accordance with ASC 480-10-S99-3A,Classification and Measurement of Redeemable Securities, which provides that securities that are optionally redeemable by the holder for cash or other assets are classified outside of permanent equity in temporary equity. The 1.9 million shares of common stock issued pursuant to the Agreement were recorded as redeemable common stock at an initial carrying value of $9.9 million. The Company elected to record the common stock at its redemption value of $11.0 million, as if it were currently redeemable, immediately and accordingly recorded accretion of $2.2 million to accretion of redeemable securities expense during the twelve months ended December 31, 2012.
| 11. | Stockholders’ Deficit |
| a. | Preferred Stock Issuances |
During 2005 the Company issued 32,500,000 of Series A Preferred Stock for $1,276,000 and 4,817,000 of Series B Preferred Stock for no consideration. Such shares were converted into 938,250 shares of common stock in 2007.
| b. | Common Stock Issuances |
Issuances of common stock during the twelve months ended December 31, 2011 and 2012 were as follows (shares and dollars in thousands):
| Shares issued | Purchase/ Conversion Price | Fair Value/ Proceeds/ Debt Conversion | ||||||||||
| Issuance of shares to private investors | 18 | $ | 11.89 | $ | 214 | |||||||
| Conversion of accounts payable | 346 | 10.32 | 3,572 | |||||||||
| Conversion of notes payable | 110 | 7.98 | 878 | |||||||||
| Shares issued for consulting services | 73 | 9.97 | 728 | |||||||||
| Total 1st Quarter 2011 | 547 | 9.86 | 5,392 | |||||||||
| �� | ||||||||||||
| Issuance of shares to private investors | 478 | 10.73 | 5,130 | |||||||||
| Conversion of notes payable | 202 | 6.54 | 1,322 | |||||||||
| Shares issued for consulting services | 42 | 10.48 | 440 | |||||||||
| Total 2nd Quarter 2011 | 722 | 9.55 | 6,892 | |||||||||
| Conversion of accounts payable | 42 | 11.90 | 500 | |||||||||
| Conversion of notes payable | 189 | 4.68 | 885 | |||||||||
| Total 3rd Quarter 2011 | 231 | 16.59 | 1,385 | |||||||||
| Issuance of shares to private investors | 16 | 9.38 | 150 | |||||||||
| Conversion of accounts payable | 2,875 | 5.92 | 17,020 | |||||||||
| Shares issued for consulting services | 373 | 4.40 | 1,640 | |||||||||
| Total 4th Quarter 2011 | 3,264 | 5.76 | 18,810 | |||||||||
| Total Twelve Months Ended December 31, 2011 | 4,764 | $ | 6.82 | $ | 32,479 | |||||||
| F-19 |
| Shares issued | Purchase/ Conversion Price | Fair Value/ Proceeds/Debt Conversion | ||||||||||
| Issuance of shares to private investors | 14 | $ | 5.00 | $ | 140 | |||||||
| Conversion of notes payable | 590 | 6.30 | 3,700 | |||||||||
| Conversion of accounts payable | 3 | 5.11 | 15 | |||||||||
| Total 1st Quarter 2012 | 607 | 6.24 | 3,855 | |||||||||
| Issuance of shares to private investors | 56 | 3.68 | 207 | |||||||||
| Conversion of notes payable | 338 | 3.76 | 932 | |||||||||
| Shares issued for consulting services | 1 | 4.00 | 3 | |||||||||
| Total 2nd Quarter 2012 | 395 | 2.89 | 1,142 | |||||||||
| Issuance of shares to private investors | 222 | 5.11 | 1,135 | |||||||||
| Conversion of notes payable | 1,080 | 2.94 | 3,172 | |||||||||
| Shares issued for consulting services | 190 | 4.76 | 905 | |||||||||
| Total 3rd Quarter 2012 | 1,492 | 3.49 | 5,212 | |||||||||
| Issuance of shares to public and private investors(A) | 4,575 | 3.86 | 17,675 | |||||||||
| Conversion of notes payable and payables | 10,127 | 3.17 | 32,152 | |||||||||
| Shares issued for consulting services | 15 | 5.75 | 92 | |||||||||
| Total 4th Quarter 2012 | 14,717 | 3.39 | 49,919 | |||||||||
| Total Twelve Months Ended December 31, 2012 | 17,211 | $ | 3.49 | $ | 60,063 | |||||||
| (A) includes redeemable shares. | ||||||||||||
| c. | Stock Purchase Warrants |
Through December 31, 2012, the Company has issued warrants to strategic partners, consultants and investors with exercise prices ranging from $2.40 to $51.84 and exercise periods ranging from three to five years. Each warrant is exercisable into one share of common stock. The following is a summary of warrant activity for the twelve months ended December 31, 2012:
| Number of Warrants | Weighted Average Exercise Price | |||||||
| Outstanding as of December 31, 2010 | 2,884,887 | $ | 9.83 | |||||
| Issued in 2011 | 845,283 | 10.16 | ||||||
| Expired in 2011 | (161,862 | ) | 30.69 | |||||
| Outstanding as of December 31, 2011 | 3,568,308 | 8.96 | ||||||
| Issued in 2012 | 8,784,974 | 5.08 | ||||||
| Expired in 2012 | (266,781 | ) | 7.35 | |||||
| Outstanding as of December 31, 2012 | 12,086,501 | $ | 6.18 | |||||
| F-20 |
| 12. | Commitments and Contingencies |
On July 31, 2012, the Company entered into a non-cancelable operating lease for 7,097 square feet of office space in Bethesda, Maryland, which expires in March 2018. Rent expense for 2012 and 2011 amounted to $0.2 million and $0.2 million, respectively.
The Company’s future minimum lease payments are as follows as of December 31, 2012 (in thousands of dollars):
| Office Leases | ||||
| 2013 | $ | 213 | ||
| 2014 | 234 | |||
| 2015 | 300 | |||
| 2016 | 308 | |||
| 2017 | 318 | |||
| Thereafter | 81 | |||
| Total | $ | 1,454 | ||
| 13. | Income Taxes |
There was no income tax benefit attributable to net losses for 2012 and 2011. The difference between actual tax provisions and taxes computed by applying the corporate rate of 40% and 34% in 2012 and 2011, respectively, is primarily the result of establishing a valuation allowance on the Company’s deferred tax assets arising primarily from tax loss carry forwards.
The tax effects of temporary differences and tax loss and credit carry forwards that give rise to significant portions of deferred tax assets and liabilities at December 31, 2012 and 2011 are comprised of the following (in thousands):
| Year Ended December 31, | ||||||||
| 2012 | 2011 | |||||||
| Net operating loss carry forwards | $ | 72,044 | $ | 48,184 | ||||
| Research and development credit carry forwards | 3,570 | 3,270 | ||||||
| Stock-based compensation and other | 12,004 | 24 | ||||||
| Gross deferred tax assets | 87,618 | 51,478 | ||||||
| Less valuation allowance | (87,618 | ) | (51,478 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
The increase in the valuation allowance for deferred tax assets for 2012 and 2011 of $36.1 million and $4.6 million, respectively, was primarily due to the inability to utilize net operating losses. The increase in the valuation allowance for 2012 reflects the adjustment of the corporate rate during 2012.
At December 31, 2012, the Company had net operating loss carry forwards for income tax purposes of approximately $180 million and unused research and development tax credits of approximately $3.6 million available to offset future taxable income and income taxes, respectively, expiring in 2019 through 2032. The Company’s ability to utilize net operating loss and credit carry forwards is limited pursuant to the Tax Reform Act of 1986, due to cumulative changes in stock ownership in excess of 50% such that some net operating losses may never be utilized. The tax years 2009 through 2012 remain open to examination by federal agencies and other jurisdictions in which the Company operates.
| 14. | Subsequent Events |
During the first quarter of 2013, the Company converted $0.9 million of notes payable into 0.4 million shares of common stock and issued 0.1 million shares of common stock as compensation for services.
The Company received short-term loans of $0.4 million from two executive officers in March 2013. The repayment terms were not finalized as of the date of this report.
| F-21 |