UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended: March 31, 2024
or
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission file number: 001-41277
MODULAR MEDICAL, INC.
(Exact name of registrant as specified in its charter)
| Nevada | | 87-0620495 |
(State or Other Jurisdiction of
Incorporation or Organization) | | (I.R.S. Employer
Identification No.) |
| 10470 Thornmint Road, San Diego, California | | 92127 |
| (Address of principal executive offices) | | (Zip Code) |
Registrant’s telephone number, including area code: (858) 800-3500
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
| Common Stock, par value $0.001 per share | | MODD | | The Nasdaq Stock Market, LLC |
Securities registered pursuant to Section 12(g) of the Act:
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act.
Yes ☐ No ☒
Indicate by check mark if the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ |
| Non-accelerated Filer | ☒ | Smaller reporting company | ☒ |
| Emerging growth company | ☐ | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐ No ☒
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the Registrant, based on the closing price of the shares of common stock on the Nasdaq Stock Market on September 29, 2023 was $17,971,555.
The number of shares of the registrant’s common stock outstanding, par value $0.001 per share, as of June 17, 2024, was 32,536,700.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED MARCH 31, 2024
TABLE OF CONTENTS
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (this “Report”) contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that relate to future events or to our future operations or financial performance. Any forward-looking statement involves known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to differ materially from any future results, levels of activity, performance or achievements expressed or implied by such forward-looking statement.
Words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “forecast,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “targets,” “likely,” “will,” “would,” “could,” “should,” “continue,” “scheduled” and similar expressions or phrases, or the negative of those expressions or phrases, are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Although we believe that we have a reasonable basis for each forward- looking statement contained in this report, we caution you that these statements are based on our estimates or projections of the future that are subject to known and unknown risks and uncertainties and other important factors that may cause our actual results, level of activity, performance, experience or achievements to differ materially from those expressed or implied by any forward- looking statement. Actual results, level of activity, performance, experience or achievements may differ materially from those expressed or implied by any forward-looking statement as a result of various important factors, including our critical accounting policies and risks and uncertainties relating to:
| | ● | our strategies, prospects, plans, expectations, forecasts or objectives; |
| | ● | our ability to achieve a marketable product (i.e., our insulin pump) and the costs and timing thereof; |
| | ● | acceptance of our product by our target market and our ability to compete in such market; |
| | ● | our ability to raise additional financing when needed and the terms and timing thereof; |
| | ● | our ability to expand, protect and maintain our intellectual property rights; |
| | ● | our future operations, financial position, revenues, costs, expenses, uses of cash, capital requirements, our need for additional financing or the period for which our existing cash resources will be sufficient to meet our operating requirements; |
| | ● | our analysis of the target market for our insulin pump; |
| | ● | our ability to obtain regulatory approvals and clearances relating to our insulin pump, including those of the United States Food and Drug Administration, or FDA; |
| | ● | regulatory developments in the United States and other countries; |
| | ● | the timing and costs of our obtaining all regulatory approvals and clearances identified immediately above; |
| | ● | our compliance with all applicable laws, rules and regulations, including those of the Securities and Exchange Commission, or SEC, and the FDA; |
| | ● | our ability to compete in the diabetes marketplace with larger and more substantial medical device companies; |
| | ● | general economic, business, political and social conditions; |
| | ● | our reliance on and our ability to retain (and if necessary, timely recruit and replace) our officers, directors and key employees and their ability to timely and competently perform at levels expected of them; |
| | ● | our ability to generate significant revenues and achieve profitability; |
| | ● | our ability to manage the growth of our business; |
| | ● | our commercialization, marketing and manufacturing capabilities and strategies; |
| | ● | our ability to expand, protect and maintain our intellectual property position; |
| | ● | the success of competing third-party products; |
| | ● | our ability to comply with regulatory requirements relating to our business, and the costs of compliance with those requirements, including those on data privacy and security; |
| | ● | the specific risk factors discussed under the heading “Risk Factors” set forth in this report; and |
| | ● | various other matters, many of which are beyond our control. |
Our fiscal year ends on March 31 of each calendar year. Each reference to a fiscal year in this Annual Report on Form 10-K, refers to the fiscal year ended March 31 of the calendar year indicated (for example, fiscal 2024 refers to the fiscal year ended March 31, 2024). Unless the context requires otherwise, references to “we,” “us,” “our,” and the “Company” refer to Modular Medical, Inc. and its consolidated subsidiary.
PART I
ITEM 1. BUSINESS
Overview
Modular Medical is a pre-revenue, medical device company focused on the design, development, and commercialization of innovative insulin pumps using modernized technology to increase pump adoption in the diabetes marketplace. Through the creation of an innovative two-part patch pump, we seek to fundamentally alter the trade-offs between cost and complexity and access to the higher standards of care requiring considerable motivation that presently available insulin pumps provide. By simplifying and streamlining the user experience from the initial introduction of the patient to our product, prescription assistance, establishing insurance reimbursement, streamlined training and day-to-day use with strong clinical support, we seek to expand the wearable insulin delivery device market beyond the highly motivated “super users” to expand the category into the mass market. Our product seeks to serve both the type 1 and the rapidly growing, especially in terms of device adoption, type 2 diabetes markets for those individuals requiring insulin.
Differentiation
We believe that there are a number of shortcomings and issues with currently available insulin pumps that prevent a substantial number of people who require insulin on a daily basis from choosing an insulin pump to treat their diabetes. We believe that, by tailoring our insulin pump to address such factors, we can expand the scope and adoption rate of insulin pump usage. We believe that to achieve broader market acceptance, an insulin pump must be easier to learn to use, be less time-consuming to operate, more intuitive to both patients and physicians, and meet the standards for coverage by insurance providers so that co-payments required from patients are affordable and the hurdles to insurance coverage are significantly reduced.
Among the more prominent issues are:
| | ● | Complexity: Many existing pumps are highly complex and require significant technical expertise to use effectively. We believe such pumps were designed for “super users,” who have high levels of motivation and technical competence. The complexity of pumps can be daunting to less technically inclined, less motivated users. |
| | ● | Cumbersome: We believe that a majority of existing pumps are bulky and difficult to manage, requiring a means of carrying the pump around and up to 48 inches of tubing to the injection site to connect the catheter to a pump. The tubing and the cartridge, which holds the insulin, must be replaced every few days. This requires users to carry spare parts and other equipment adding to the difficulty of using the pump. In comparison, our product only requires a cartridge change every few days. |
| | ● | Cost: Costs associated with insulin pump therapy can be high and prohibitive, especially for those on fixed or limited incomes. These costs vary by pump and insurance coverage, but multi-thousand-dollar upfront payments, often with substantial co-payments in addition to possible additional co-payments on consumables, can easily place current pumps out of reach for patients. This leads to limited or absent reimbursement/coverage and potentially high financial hurdles for patients to gain access. |
| | | |
| | ● | Outdated style: Consumer electronics devices have evolved in both form and function. Diabetes pumps have not experienced similar progress. We believe that consumers will be more receptive of products designed with the user experience in mind and that many have low tolerance for complex, difficult procedures for use and maintenance of products. |
| | | |
| | ● | Pump mechanism limitations: Traditional pumps generally utilize a syringe and plunger mechanism to deliver insulin. We believe this design limits the ability to reduce the size of the pump, and also potentially exposes the user to the unintended delivery of the full volume of insulin within the pump, which can cause hypoglycemia or death. We believe that the fear of adverse health events due to technical malfunctions related to traditional pump mechanism limitations deters the adoption of insulin pump therapy. |
Our team has substantial knowledge of the diabetes industry and experience in developing, obtaining marketing authorization for, and bringing insulin pumps to market. Based on this experience, we believe that our innovative insulin pump, using a new and proprietary method of pumping insulin, can address most or all of these shortcomings. It provides a state-of-the-art insulin pump capable of both basal (steady flow) and bolus (mealtime dosing) insulin disbursement. It also has been designed considering a natural migration path to multi-chamber/multi-liquid pumps, potentially offering an exciting array of new therapies to patients with diabetes and other conditions.
Our goal is to become the leader in expanding access to insulin pump technology to a wider portion of diabetes sufferers and provide not just care for the super users, but “diabetes care for the rest of us.” While our initial target market is people with type 1 diabetes, we believe there is a substantial opportunity to penetrate the type 2 marketplace, whether through our initial MODD1 pump or further simplification of our pump to address the type 2 marketplace.
The MODD1 is a high-precision pump that we believe represents the best choice for new pump patients because it is easy to afford, easy to learn, easy to use, and has a revolutionary design and internal technology that enable precision with low-cost manufacture and high reproducibility.
Key features include:
| | ● | Two parts - one reusable, one disposable - snap together to form the working system; |
| | ● | One button interface, easy to learn and use; |
| | ● | 90-day reusable, 3-day disposable; |
| | ● | Disposable portion removable at any time from an adhesive-backed retainer, which remains in place; |
| | ● | No external controller required, no charging, no battery replacement; and |
| | ● | Slim profile, lighter weight. |
A proprietary survey of American healthcare payors representing 50 million covered lives (approximately 1/3 of U.S. covered lives) performed for us by industry leading survey firm ISA in 2019 has demonstrated that payors are willing to grant equivalent or preferential coverage for a product with this feature set at launch in exchange for discounts of approximately 20%.
Diabetes Classifications and Therapies
Diabetes is typically classified as either type 1 or type 2:
| | ● | Type 1 diabetes, or T1D, is an auto-immune condition characterized by the body’s nearly complete inability to produce insulin. It is frequently diagnosed during childhood or adolescence, although it can sometimes have onset in adulthood. Individuals with T1D require daily insulin therapy to survive. |
| | ● | Type 2 diabetes, or T2D, represents over 90% of all individuals diagnosed with diabetes and is characterized by the body’s inability to either properly utilize insulin or produce sufficient insulin. Initially, many people with T2D attempt to manage their condition with improvements in diet and exercise and/or the use of oral medications and/or injection of glucagon-like peptide-1 (GLP-1) drugs. However, as their diabetes advances, patients often progress to requiring insulin therapies such as once-daily long-acting insulin and ultimately to intensified mealtime rapid-acting insulin therapy. This represents an important portion of the diabetes market with an estimated 1.6 million individuals with T2D intensively treated with insulin currently in the United States. |
Glucose, the primary source of energy for cells, must be maintained at certain levels in the blood in order to permit optimal cell function and health. The brain works on pure glucose, and, when sufficient glucose is available, the brain allows insulin to be released that allows the cells to absorb glucose. In people with diabetes, blood glucose levels are not well controlled by the brain due to the shortage of insulin. Frequently, blood glucose levels become very high, a condition known as hyperglycemia, or very low, a condition called hypoglycemia. Hyperglycemia can lead to serious long-term complications, including blindness, kidney disease, nervous system disorders, occlusive vascular diseases, lower-limb amputation, stroke, cardiovascular disease, and death. Hypoglycemia can lead to confusion or loss of consciousness, often requiring a visit to the emergency room or, in certain cases, result in seizures, coma, and/or death.
All people with T1D, which is our primary market, require daily insulin. According to the Seagrove 2021 Diabetes Blue Book, approximately 18% of people with T2D in the United States, or approximately 4.7 million people, require insulin (basal alone represent 3.1 million and basal plus mealtime represent 1.6 million) to manage their diabetes. In this Report, we refer to people with T1D and people with T2D who require mealtime insulin as “insulin-requiring people with diabetes.”
Currently, there are two primary therapies available for insulin-requiring people with diabetes: multiple daily insulin injections directly into the body through syringes or insulin pens (a type of syringe), referred to as Multiple Daily Injection, or MDI therapy, or the use of an insulin pump to deliver mealtime insulin boluses to help with glucose absorption after carbohydrate consumption and a continuous subcutaneous insulin infusion, or CSII therapy, into the body. Generally, CSII therapy is considered to provide a number of advantages over MDI therapy, primarily an improvement in glycemic control, as measured by certain diabetes management tests such as hemoglobin A1c (HbA1c) measure and more recently Time in Range (TIR) where a continuous glucose measuring device is used to calculate this test. Among other clinical benefits, a study conducted by Tandem Diabetes Care, Inc., or Tandem, in 2021 demonstrated that insulin pump use can decrease glucose variability, reduce the number of hypoglycemia events, and reduce the fear of hypoglycemia.
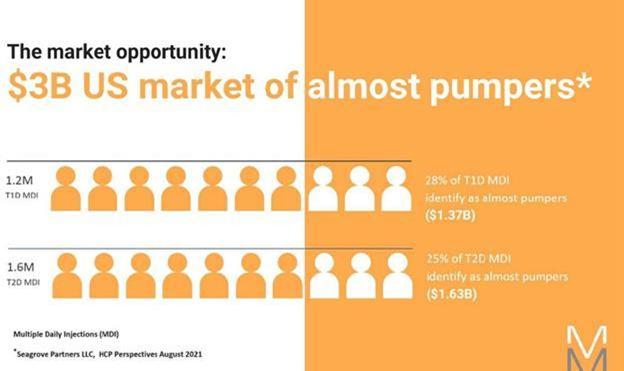
Notwithstanding these advantages, we believe the difficulty in use resulting from the complexity and cumbersome design of available insulin pumps, as well as high and often prohibitive costs for both the patient and insurance provider, has resulted not only in dissatisfaction among many existing pump users. As noted in a Seagrove Partners 2021 study, fewer than half of first-time pump users purchase a new pump after the warranty expires. We believe the cost and complexity to the user has severely limited the adoption rate of insulin pumps by a large segment of the diabetes population using MDI therapy, whom we refer to in this Report as “Almost Pumpers.”
We define Almost Pumpers as insulin-requiring people with diabetes who are aware of pumps and their potential benefits but because of past experiences, pump shortcomings, cost, complexity, and time and learning required to adopt and utilize currently available insulin pumps, continue to receive their daily insulin through MDI therapy. We undertook one-on-one interviews with over 200 of these individuals to understand their past experiences on or considering pumps, existing pump shortcomings, the cost and insurance challenges, complexity to learn and time and complexity to operate that drives them to remain on MDI. With this detailed understanding, we brought a series of prototype models to them to react to, so we could refine the design and include features that would motivate them to be able to use this technology to better care for their diabetes. Our MODD1 pump has been well received by these individuals and our clinical advisors, as applicable for this sector of the marketplace.
Our research, along with marketplace data provided by Seagrove Partners in 2023, estimates that 33% of Americans with T1D have an insulin pump and 28% of Americans with T1D (44% of those who currently utilize MDI) can be classified as having an interest in pump adoption and meeting the American Diabetes Association guidelines of glucose control if their objections to the currently available suite of products can be overcome. They do not want to closely manage their glucose levels and incur the associated time and effort involved; however, they understand, or are advised by their clinical care team, that they need to do more to achieve a reasonable level of glucose. They are the Almost Pumpers. We have developed what we believe to be the most technologically advanced delivery system overcome the objections and provide motivation for this market. We believe that there are four addressable hurdles to adoption:
| | ● | Usability: the device needs to be easy to learn and to operate; |
| | ● | Affordability: we will focus on overcoming copay and insurance hurdles rather than leaving the “insurance journey” to the clinician and patient; |
| | ● | Accessibility and Education: we will seek to engage patients to sample this new technology by supplying clinicians with free samples and simple training to allow people to see first-hand the typical barriers to adoption that have been overcome; and |
| | ● | Service and Support: where we will answer their questions and concerns during this diabetes experience. |
We believe this conversion process, engaging people to try and thereby receive the benefits of our technology will substantially increase adoption of insulin pumps among with patients with T1D and T2D who remain reliant upon multiple daily injections. Diabetes is a disease that appears randomly throughout the world. Therefore, we cannot segment the market by socioeconomics, education or level of care. We intend to create an insulin pump that appeals to all Almost Pumpers.
Market
The International Diabetes Federation estimated that, in 2021, approximately 537 million people were living with diabetes worldwide, and by 2045, this number will increase to approximately 783 million people.
An estimated 34 million people in the United States live with diabetes. Within this group, T1D accounts for approximately 1.8 million people with the remainder being T2D. All people with T1D require daily insulin. However, of the approximately 32.2 million people with T2D, about 1.6 million of them require MDI therapy to manage their diabetes. This represents a large and growing market with the effects of diabetes accounting for roughly 25% of all healthcare dollars spent annually in the United States.
According to the National Diabetes Health Care Provider Survey conducted by Seagrove Partners, LLC in 2021, approximately 25% of the 1.6 million highly insulin intensive individuals with T2D have considered going “on pump.”
Insulin pumps have been shown to provide a higher level of care for insulin-dependent people with diabetes and result in better glycemic control, fewer comorbidities, fewer trips to the emergency room, and higher overall quality of life. They also result in lower overall costs to the healthcare system, reducing typical expense per patient year from approximately $27,000 to $17,000.
Despite these benefits, only 1 in 3 (33%) of the 1.8 million Americans with T1D and very few of the 1.6 million T2D intensively treated with insulin currently use an insulin pump, for a total of approximately 670,000 current users, with only a slow increase of insulin pump use. The remaining 68% of individuals with T1D and virtually all with T2D rely on MDI therapy for glucose control. Decades of advances in technology advances have left these non-pumpers at a significant disadvantage from a control perspective versus their “pumping” counterparts.
We have identified a large segment of the market that we refer to as “Almost Pumpers.” Almost Pumpers are those insulin-requiring people with diabetes (T1D or T2D) who feel that they would adopt the pump if it were less expensive, less time consuming, less technically intimidating, and if there was no separate controller. We believe that they represent approximately 32% of the T1D market correlating to a $1.9 billion growth opportunity.
Insulin pumps on the market today require a substantial amount of time to manage the therapy, have high out-of-pocket costs that place these technologies out of reach for a large part of the population, and are feature-heavy with complex systems, which we believe have hampered adoption and intimidated many users. The most commonly used insulin pumps today require extensive training and hours of daily management. The average pump user must go through 42 steps of setup and refill process every 72 hours to “stay on track.” Our product only requires nine steps for setup and refill every 72 hours.
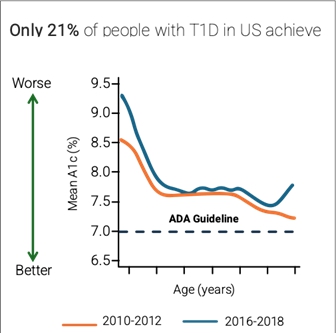
The current reluctance to adopt the insulin pump has had serious consequences on the healthcare system. In the United States, people living with T1D have struggled to attain glycemic targets. A 2019 analysis of the large T1D Exchange clinical registry found that only 21% of U.S. adults with T1D achieved the ADA A1c goal (<7.0%). Further, according to a study published in JAMA Internal Medicine, researchers found no significant improvements in diabetes care between 2005 and 2016, with persistent gaps in care related to socioeconomic status.
Another transition in the care of diabetes is the measuring of glucose from finger-stick tests to continuous glucose monitoring, or CGM, sensors, which are wearable devices. These sensors are placed under the skin and give a reading every five minutes of the user’s glucose level. While Dexcom has been a market leader in this field, the introduction and rapid adoption of the Freestyle Libre by Abbott Labs has made CGM easier and more affordable, expanded the product category, and doubled the market size. The Freestyle Libre product is a more affordable, easier to use and smaller version of the popular Dexcom, Inc. (Dexcom) CGM product. Now, for the first time, there is an easy, less painful, i.e., no more finger sticks, way for patients to have the data they need to understand more about their glucose levels and their insulin requirements. Access to such data has motivated patients to ask their diabetes clinician how they can achieve better glycemic control and made them more comfortable with using technology and wearables to treat their diabetes. Pumps offer a clear pathway to better control and better overall care. We believe that the insulin pump market is ready for a similar transition as that experienced in the CGM space. Our MODD1 pump represents a new and better offering to assist and induce a wide variety of patients to make the transition and overcome the objections to superior control by becoming a “pumper.”
We believe the present pump marketplace is approximately a $1.9 billion market, comprising 33% of T1D pumpers and a small group of T2D pumpers. Seagrove Partners surveyed clinicians, and, in its 2021 report, estimated that 28% of T1D patients and 25% of T2D patients would adopt technology that was easier to use, learn and access and eligible for insurance reimbursement. We believe this represents a total addressable market of approximately $3 billion for us, assuming cartridge revenue of approximately $4,100 per patient, per year. We expect to spend approximately 15% of our total revenue on discounts and free samples to encourage adoption of our pump product.
We are dedicated to helping all people with diabetes gain access to high quality care. We aim to help people with diabetes - especially Almost Pumpers and the historically underserved communities - gain access to insulin pump technology by making it affordable and easy to use.
Diabetes Care is at an Inflection Point
We believe that the insulin pump market stands at a crossroads as a confluence of events makes the timing for a new product introduction ideal.
2020 was a very difficult year in diabetes. Between COVID-19 and a loss of glycemic control during quarantines and isolation, deaths from diabetes rose by 17% in 2020 versus the prior year. This was sharpest among the young who saw deaths rise 29% in the 25-44 year old demographic. This became a pain point and a desire to find new and better solutions and has raised awareness among patients, caregivers, payors, and policy makers.
COVID-19 also encouraged (and required) trial and adoption of telehealth models and a great many people have found them to their liking with a high proportion of patients and of health care providers (HCPs) that want to continue to use these technologies. We expect much of this shift and newfound comfort with distance care models to persist and believes that this can provide a patient acquisition and engagement model for insulin pumps and diabetes care, especially for pumps optimized for free trial and easy learning.
At the same time, reimbursement for patch pumps has been increasingly moving to a pharmacy benefits manager (PBM) model, which simplifies reimbursement and will further aid in a “frictionless launch.” This represents a fundamental shift in the insulin pump market, making onboarding rapid and simplifying a previously complex and time-consuming “insurance journey.”
We believe these CGM device users are increasingly interested in adopting technology and wearables to manage their diabetes. We believe CGM device users are a natural market for a new type of pump, if it can meet their needs and address their objections and that the conjunction of the above trends represents a unique opportunity in the insulin pump market’s history. The CGM device provides glucose-level data, and, as necessary, the user can respond to address any issues with a simple button push on a pump to deliver their insulin versus taking out a syringe and injecting glucose.
Diabetes technology companies understand that we are at a turning point with new markets (T2D, T1D that are currently not using technologies). This can be seen with increased discussion around this topic during recent national diabetes conferences, as well as an increase in marketing promotion.
All these recent changes support the high proportion of T1D and T2D intensively treated with insulin that we consider to be Almost Pumpers, and we expect the number of Almost Pumpers to grow in the coming years and be more reachable with appropriate marketing strategies.
Our Insulin Pump
Instead of building complex, bespoke, and difficult to manufacture and maintain pumping and control systems, we began with the technology and the user in mind. Using proprietary methods of insulin measurement, we were able to eschew complex mechanisms and instead built a product candidate using only parts from high volume consumer electronics manufacturing lines, breaking the cost vs functionality curve that has existed in the insulin pump space and representing the first truly modern insulin pump design. We consider this to be a new kind of product for a new kind of patient.
In January 2024, we submitted a 510(k) premarket notification to the United States Food and Drug Administration (“FDA”) for our MODD1 insulin pump. In March 2024, we received comments from the FDA on our submission, and we are in the process of responding to those comments. A good part of our focus has shifted to managing the process of preparing to move our initial production line to our manufacturing partner, Phillips-Medisize, a large tier-one medical device manufacturer, which will manage and operate our production to produce products for human use. We believe that Phillips-Medisize will be able to rapidly scale to higher volumes at lower cost. We continue to devote substantial time and resources, including exhibiting at major diabetes conferences, to better understand the needs and preferences of Almost Pumpers and the specific patient/provider/payor requirements to motivate change from MDI therapy. By making the bolus delivery at meals simple, we believe we will drive improved health outcomes.
MODD1 has several distinguishing features:

1 - The pump has a simple button to press to deliver insulin as the patient requires it. The electronic pump uses a simple motor for rotating a cam to motivate the insulin into the patient along with a low power Bluetooth and near-field communication (NFC) chips to optionally allow the patient to communicate with their smart phone, tablet, or other mobile computing platform. Our mobile device application is included in our 510(k) submission and will be a part of our introductory product.
2 - The pump snaps together with a three-day disposable cartridge that is patient filled with insulin for delivery. It includes a simple coin cell that allows it to run through the 80-hour life of the cartridge.
3 - The infusion set contains a soft 6 mm cannula and an introducer for insertion into the skin for insulin delivery, and it automatically removes the inserted needle used to transfer insulin to the body.
MODD1 comes with a variety of methods for the patient to wear the pump. Options include: a base plate with adhesive for attaching to the body that has features for holding the pump to the patient; overwraps to hold the product to the patient; and a velcro strap with a base plate suitable for wrapping around the arm or leg of the patient.
The system will deliver a small continuous rate called a basal that will provide approximately 50% of the total daily dose required, and the user will use the on-pump button to administer boluses, typically before and after meals. The objective is to make the product simple to acquire and take home, simple to learn and most importantly, simple to use and live with, to expand the pump market, drive adoption and ultimately better clinical outcomes.
Technological Advantages
The adoption of new ultra-high volume technologies will result in far easier manufacturing scale up, as parts sourcing and assembly processes are far easier. The MODD1 was designed from the beginning for mass manufacturing, and we have partnered with a manufacturing partner, Phillips-Medisize, a Molex company, to establish processes and “lights out” or near lights out production assembly lines whereby a minimal number of workers will be required in the production facility. This advantage is compounded by the high availability and already optimized cost reduction in its components. When we achieve production scale, we believe this should result in a cost of goods for MODD1, estimated on the competitors’ announced margins and sales, of approximately 50% lower than our closest patch pump competitor.
The adoption of modern, miniaturized technologies has led to numerous other advantages, as well. For example, our MODD1 pump is smaller in overall volume than Insulet’s popular Omnipod product and has a lower profile to the skin. Despite this, the MODD1 holds a full 3 milliliter, or mL, (300 units) of insulin, in line with full sized pumps such as those offered by Tandem and Medtronic, 50% more than the 2mL reservoir in the Omnipod. We believe that this volume advantage over other patch pumps will be significant as 24% of type 1 and over 50% of the rapidly growing type 2 market require more than 2mL of insulin every three days (the expected wear time of patch pumps).
In addition, our new pumping modality will provide what we believe is the most even (and thus closest to the function of a healthy pancreas) delivery of basal insulin in the industry. We intend to demonstrate the impact of our system on glycemic control in a clinical study specifically focused on improved adherence, more bolus deliveries per day and providing the clinicians with clear data on patient use.
The technology allows the patient to simply add insulin and operate. The battery is included in each cartridge, and the device is operated without a controller. As a result, no charging is required. MODD1 has also been made push-button simple to deliver insulin to appeal to a wider audience of users.
This new technology has made the MODD1 lighter than existing offerings. Compared to the Insulet Omnipod, MODD1 weighs 20 grams (vs. 26 grams) empty and 23 grams (vs. 28 grams) fully filled (despite carrying 50% more insulin), reductions of 23% and 18%, respectively. Also, unlike existing patch pumps, the MODD1 can be removed from the needle and taken off and replaced later if the user desires. This avoids loss of insulin in a pump due to accidental dislodging of the soft canula, an issue for other patch pumps with which users have expressed considerable dissatisfaction.
Our approach to the care of diabetes can be further enhanced by leveraging the MODD1 single-pumping chamber technology and reusable pump approach to apply to dual (or more) chamber pumping solutions. We believe that such multi-chamber pumps will be integral to the realization of high time-in-range artificial pancreas solutions that require no human intervention because of the application of, for instance, drugs to raise glucose levels coupled with drugs to lower glucose. They will be the next step forward from the cumbersome and awkward solutions today that require the user to announce meals, count and input carbohydrates, and adjust delivery for exercise and sleep to prevent overdosing of insulin. Instead, if a user overdosed insulin, the user would simply pump in a drug to release sugar stores to raise it up. We believe that a pre-filled peel and stick patch pump with the ability to function in a fully autonomous closed loop system with a CGM device, which is measuring and transmitting glucose-level information, represents the next generation of diabetes care. We believe that we have demonstrated our technology and have secured, and will continue securing, intellectual property protection on our approach.
We believe this technology, especially applied in a dual chamber capacity, will open up numerous applications outside of diabetes where medication compliance of complex therapy regimes is difficult. Example applications would include weight loss, fertility, and simplifying the delivery of complex multi-drug cocktails, especially those with diverse and challenging dosing schedules.
Our Solution
Our proposed pump has been designed and developed to address the aforementioned shortcomings of the existing pump market and to appeal to: (i) the substantial group of Almost-Pumpers, who may be interested in using an insulin pump, but have not done so because of the complexity, cost or cumbersome nature of existing products and (ii) people who are using one of the currently available insulin pumps but are dissatisfied with such products. We believe that, owing to our new proprietary technology, our proposed insulin pump will be the simplest and least expensive product on the market and the easiest for providers to prescribe.
Our current pump has been built to test what we believe to be our novel approach to insulin pumps. By providing a pump that we believe will establish industry standards in terms of technology, simplicity to understand, ease of use and price, we believe our MODD 1 pump will offer the vast majority of benefits afforded by more expensive and complex pumps, but it will remain accessible to a substantially greater percentage of diabetes sufferers requiring daily insulin therapy.
We believe people generally will not use technology that intimidates them, especially for a life-sustaining therapy. In addition, we believe that physicians are hesitant to prescribe such technology due to the level of training and support required with the present pump product offerings. It is our belief that broadly-needed medical products, such as is intended with our proposed pump, must be user-friendly and affordable. We believe this approach is fundamentally different from that applied to the existing pump market today, where most pumps are continuously adding complex features appealing to super users and leaving the other people with diabetes further behind.
Our current goal is to successfully design, develop and obtain all required regulatory approvals for our proposed insulin pump, and, thereafter, commercialize the finished product. Our long-term goal is to become a leading provider of insulin pump therapy by focusing on both consumer and clinical needs.
To achieve our above stated immediate and current goals, we intend to pursue the following business strategies:
| | ● | Use of innovative proprietary technology. |
Based on the substantial experience of Paul DiPerna, our President, Chief Financial Officer, Treasurer and Chairman of our board of directors, in engineering design and innovative technology in the medical device industry and, in particular, with the invention, market vision and technical development of insulin pumps, we have generated proprietary technology that has been incorporated into our proposed insulin pump. We believe this technology allowing for a two-part, yet small enough to wear, pump product, along with simplified mechanics for pumping, has greatly assisted us in creating a simpler, user-friendly pump. We believe the completed design, engineering and technology being incorporated into our pump will make it substantially simpler and more affordable than those currently available. These features, together with the safety and reliability of our proposed pump, are designed to create the next generation of insulin pumps that will feature important and well-differentiated attributes compared to those currently available and make it available to consumers across mostly all socioeconomic groups in the United States and around the world.
| | ● | Keep costs low during our design and development process. |
To attempt to ensure that we have sufficient funds to design, develop, and obtain all required regulatory approvals for our proposed insulin pump without having to sacrifice quality and efficiency, we intend to maintain a tight budget and limit expenditures where possible. We believe this will be possible because of the extensive knowledge and experience of Mr. DiPerna, not only in the diabetes industry and more specifically in the insulin pump device market, but also his experience in designing and developing insulin pumps and other medical devices and his ability to manage a small, focused development team. We currently expect that various other expenses, such as sales and marketing costs, will not be incurred until such time as regulatory clearances are obtained.
Commercialization Strategy: Overcoming the Insurance Hurdles
Our goal is to establish MODD1 as the best option for new pump patients as we expand the market into the Almost Pumpers (Type 1 and Type 2) and newly motivated CGM users. We seek to grow the market by providing first-line insulin pump therapy that is well suited to meet the needs of both diabetes patients requiring insulin and their clinicians.
| | ● | We believe that MODD1 is approximately 50% less expensive to manufacture than Omnipod. This low cost allows us to spend more on patients and sampling. We believe that this will save money for payers because we expect to offer the pump with no upfront cost to patients. Expected benefits of MODD1 include: |
| | o | 20% discount vs Insulet will drive preferred status; |
| | o | Designed to use pharmacy benefit manager, or PBM, codes as a disposable; |
| | o | No new code needed to be reimbursed at launch because MODD1 will be able to use existing U.S. Center for Medicare and Medicaid Services, or CMS, codes; and |
| | o | Saves insurance provider an estimated $1,062 per patient per year vs Omnipod, as we will offer providers discounts from the existing reimbursement code. |
| | ● | The MODD1 will be sampled and given to patients by a doctor or diabetes nurse educator at the time of the patient visit. When a patient is motivated to make change, our starter kit will make it easy for the clinician to initiate the new therapy with training and appropriate supplies that same day. We seek to eliminate the currently challenging “insurance journey” and product acquisition timeline and significantly reduce training time for the busy clinician, which we believe are all major hurdles to pump adoption. We intend to add significant telehealth support to help the patient throughout adoption and use and to facilitate greater collaboration between patients and their physicians. |
Europe represents another large potential market for MODD1, as approximately 60 million people in Europe live with diabetes. Approximately $161 billion is spent annually on diabetes healthcare costs in Europe based on data from a Seagrove Partners 2023 study. At present, cost containment is restricting pump uptake across Europe. Current pump usage hovers between 10% and 20% in many markets. Single payor healthcare systems across Europe traditionally attempt to contain costs in the short term and seek low price technologies with moderate medical benefits. We anticipate MODD1 will offer a rebalance of this risk/reward strategy in that payors will incur only minor incremental short-term costs with the benefit of longer -term cost savings associated with reliable pump use. We intend to employ a partnership strategy across Europe following in-house managed regulatory and pricing activities in the major markets (e.g., UK) and more cost receptive markets (e.g., Nordics). We have begun the approval process for Europe and are targeting European and United Kingdom approval towards mid 2025.
Marketing
MODD1 tackles the most significant barriers to pump use-access and affordability-and makes it easier for clinicians, caregivers and individuals to manage diabetes care. We believe that MODD1 will be the only insulin pump that patients can take home immediately from the doctor’s office. Our commercialization plan will drive adoption and is designed to expand the market and is intended to do the following:
| | ● | Maximize adoption with a comprehensive frictionless launch program. We will seek to decrease the level of reimbursement effort and cost to encourage health care providers, or HCPs, to offer our pumps and encourage patient trials. Our product reduces the technical hurdles to widen appeal, encourage new starts and increase adherence. We will encourage patients utilizing MDI therapy to make the switch to the pump earlier in their treatment, ideally right at diagnosis. For those who want or need more control, a key point is that a simple button push to administer insulin is much easier than delivering an injection. |
| | ● | Leverage technology to support sales and new patient acquisition. We intend to set up technology-enabled sales teams backed with a full omnichannel program to drive awareness and trial with HCPs and patients. We will focus on educating providers that our product candidate is simple to teach and easy to support making it an ideal front line offering. |
| | ● | Facilitate patient trials. To facilitate patient trials, we intend to: |
| o | Provide a free sample pump, insurance verification, co-pay coupons and telehealth support, as may be allowed under federal and state law, to patients thereby reducing outlay of time and money; and |
| o | Partner with multiple educational and pharmacy channels to support rapid adoption and support of patients. |
| | ● | Leverage MODD1 300-unit chamber to increase adoption with Type 2 patients. We believe MODD1 has a major advantage over existing patch pumps in that the chamber carries enough insulin to meet the high doses many Type 2 patients need. We intend to promote this advantage and capture a significant share of the existing Type 2 pump users, as well as new starts. |
| | ● | Work with key organizations and policy makers to pave the way for greater access to pumps. We will promote MODD1 technology among the underserved, who are typically low users of health technology. We will identify individuals, patient organizations, professional societies, and policy and diversity and inclusion organizations that are critically important to the adoption of new technologies in the diabetes space and build relationships with these influential stakeholders. |
| | ● | Initiate a clinical study program (with key diabetes centers) We intend to provide additional clinical support for MODD1 in special patient types and clinical setting. After obtaining 510(k) clearance, we intend to conduct a soft launch and clinical research program in major markets to pave the way for the full launch, which is expected to commence in early 2025. We will work with our advisors and key diabetes associations to educate the community about the MODD1. In addition, we will conduct clinical studies to develop competitive claims and market expansion. |
| | ● | Work with major health plans to establish MODD1 as the first line pump for Type 2 patients. We believe MODD1 will be payor preferred for both Type 1 and Type 2 patients. It was designed to attain preferential reimbursement and avoid the coverage pitfalls many other pumps have experienced. |
| | o | Payors want an effective product whereby the users realize the clinical benefit. We intend to launch with a discount program for payors of 20% to drive uptake. |
| | o | Designed to use existing PBM codes as a disposable |
| | o | No new reimbursement code: Reimbursed at launch |
Tie-in with telehealth.
In recent years, telehealth has gone mainstream, and patients and providers have become comfortable with it. There are less than 4,000 patient-facing endocrinologists in the United States. The treatment of diabetes will be significantly enhanced with telehealth to drive more volume and clinical enhancements through their practices. Telemedicine is a force multiplier for a small group of doctors to better serve a large market. MODD1 was designed to be affordable enough for free sampling and trial, and simple enough for self-guided user training. We believe that by combining telehealth support with MODD1, we will decrease the burden of diabetes care and improve the lives of people with diabetes.
Soft Launch
We intend to initiate a “soft launch” following FDA clearance of the MODD1 device. Our plan is to select a group of clinicians who are well trained, experienced and have the support infrastructure to take on initial patients and monitor them carefully to provide clinical feedback on our performance to further refine our product and the support infrastructure prior to full commercial launch. Many of these clinicians will have been those who assisted in the development of the MODD1 offering.
We intend to continue to modify, refine and finalize our system to best meet:
| | ● | The general needs and preferences of our Almost Pumper target market based upon our knowledge of the diabetes industry and information available and/or obtained by us from Almost Pumpers and their caregivers; and |
| | ● | The general guidelines of third-party payors, private and public insurance companies, preferred provider organizations and other managed care providers with particular focus on the guidelines established by CMS, which administrates the United States Medicare program. To assist us in making such modifications and refinements, we have retained independent consultants to focus on ensuring that our product candidate satisfies the existing coverage and reimbursement criteria of such third-party payors. |
Manufacturing
Our pump product comprises the pump, a disposable cartridge that holds the insulin reservoir, a baseplate that affixes the pump product to the user’s body and the infusion set, which includes a cannula to infuse the insulin into the body. We intend to manufacture the pump, the cartridge and the baseplate and purchase the infusion set from third parties. Prior to shipment, our pump product will be packaged with an infusion set. In connection therewith:
| | ● | We have installed automation machines in our facility that will be capable of assembling the cartridges at a rate sufficient to supply 6,000 patients (60,000 cartridges per month), and we are in the process setting up a second line to double this capacity. |
| | ● | Product packaging will initially be performed manually by our personnel, while the cartridge automation is being refined. We expect to purchase and implement packaging automation equipment as the second phase of automation of the cartridge. |
| | ● | The infusion sets will be purchased from a third-party supplier to cost-effectively introduce our product and focus on our core expertise. |
We have commenced working with Phillips-Medisize to prepare for the transfer to its facility. We expect to transfer the cartridge automation equipment to this contract manufacturer in mid 2024 to verify and validate into its manufacturing process. Phillips Medisize would then perform all manufacturing operations to ensure compliance with FDA regulations.
FDA Clearance
The FDA requires us to meet all applicable regulations for insulin pumps, a subcategory of infusion pumps, which are generally considered Class II devices by the FDA. In January 2024, we submitted a 510(k) premarket notification to the FDA for our MODD1 insulin pump. In March 2024, we received comments from the FDA, and we are in the process of responding to those comments.
Commercialization Steps
To commercialize our product, we must successfully complete a number of material steps, including:
| | ● | Continue to ensure it meets: |
| | o | FDA requirements for 510(k) clearance, including taking such actions, if any, as may be required by the FDA as a condition to granting approval and providing 510(k) clearance for our insulin pump; |
| | o | the general needs and preferences of our Almost-Pumper target market based on our knowledge of the diabetes industry, information gathered from our soft launch and other information available and/or obtained by us from Almost Pumpers and their caregivers; and |
| o | the general guidelines of third-party payors, private and public insurance companies, preferred provider organizations and other managed care providers with particular focus on the guidelines established by the Center for Medicare and Medicaid Services, or CMS which administers the United States Medicare program, or Medicare. To assist us in making such modifications and refinements, we have retained independent consultants to focus on ensuring that our product candidate satisfies the existing coverage and reimbursement criteria of such third-party payors. |
| | ● | Transfer our manufacturing equipment and process to Phillips-Medisize prior to product launch; and |
| | ● | Hire and retain appropriate sales and marketing personnel to develop, implement and launch a promotional campaign for our insulin pump substantially focused on our target market. |
As with any medical device attempting to enter and successfully compete with existing products in an established and competitive marketplace, we will face significant hurdles to accomplish the above steps to commercialization including:
| | ● | Obtaining FDA 510(k) clearance to market and sell our insulin pump to the public; |
| | ● | Obtaining any other FDA-required authorizations with regard to our product, as required by the Federal Food, Drug, and Cosmetic Act, or FDCA, which is administered by the FDA; |
| | ● | Educating endocrinologists, physician’s assistants, nurse practitioners and nurse educators, who typically prescribe pump usage, and certified diabetes educators and dieticians, who provide education and guidance to diabetes patients, as to what we believe to be the superior qualities of our product candidate. We will continue to exhibit at the Association of Diabetes Care & Education Specialists, or ADCES, conference annually; |
| | ● | Demonstrating to select general practitioners, who have historically been skeptical of the heightened support inherent in insulin pumps, our product candidate’s ease of use and convenience; |
| | ● | Ensuring that our final product does, in fact, meet the needs of Almost Pumpers; |
| | ● | Overcoming the historic obstacles and reluctance of Almost Pumpers to using insulin pumps to treat their diabetes; and |
| | ● | Ensuring that third-party payors agree to cover all or a substantial portion of the purchase price and recurring costs of the use of our insulin pump. |
Looking Forward
Going forward, we expect to continue to evolve the MODD1 pumps and their capabilities and functionality both in response to patient needs and as part of our current platform roadmap.
| | ● | With our future MODD1+ product, we intend to seek to add phone-based control and Alternative Controller Enabled (“ACE”) and Automated Insulin Deliver (“AID”) capability to allow integration with popular continuous glucose monitors. We believe this will expand our available market to include many existing pumpers. The new model has the same modular design and low-cost components as MODD1 and provides a much desired breakthrough for patients - two-factor command authentication that allows the wearer to use an application on his/her cell phone as the controller. |
| | ● | AID control functionality is being developed and will be added via an ACE designation on the pump. |
| | ● | Any approved AID controller can drive insulin delivery in “auto” mode, when appropriate. |
| | ● | CGM integration allows the controller to potentially adjust basal insulin rate for meals and exercise with an approved algorithm. |
| | ● | With our future MODD2 product, we will seek to move to a full-featured, multi-chamber pump optimized for high time-in-range fully autonomous close loop insulin delivery utilizing the form factor and cost advantages of its pumping designs to create an affordable, easy to use drug delivery system to realize the aspiration of true “artificial pancreas” systems. We envision moving to a drug prefill model, such that cartridges can be filled with insulin or other drugs and shipped cold chain to patients, further simplifying the use process. |
Competition
Today, in the United States, only three companies are commercializing insulin pumps to T1D patients and insulin treated T2D patients and have significant market share:
| | ● | Medtronic - commercializes the durable Minimed 770G and also offers older durable pumps (670G, 630G etc.). In 2020, they held approximately 51% of the US insulin pump market. |
| | ● | Tandem - commercializes the durable t:slim X2 pump (with or without algorithms - Basal-IQ and Control-IQ). In 2020, they held approximately 28% of the US insulin pump market. |
| | ● | Insulet - commercializes the disposable Omnipod patch pump with approximately 19% of the US market in 2020. |
 |  |
| Medtronic pump and infusion set | Tandem pump and infusion set |
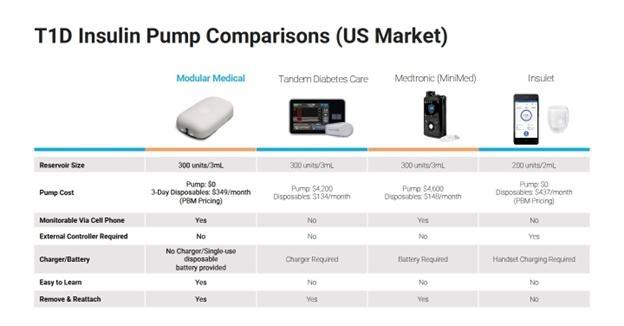
These three insulin pump offerings are vying for the attention of the most motivated and well insured in hope of converting them away from their reliance on MDI. The t:slim X2 and Minimed 770G each have a ~$5,000 list price that is covered through Durable Medical Equipment (DME) reimbursement and daily consumables, which comprise cartridge, tubing and set for each three-day period, as well. These products have controllers integrated into the pump, making them cumbersome and bulky, along with long (> 20 inch) tubing between the pump and the cannular site. The Omnipod is the third offering, a patch pump that attaches to your body for 72 hours and uses a separate controller to manage the insulin delivery process. Insurance coverage for Omnipod can be provided via DME but also via Pharmacy Benefit (PB). The Omnipod patch pump is more expensive per day and less accurate than other insulin pumps, according to a Mende 2022 study. Around 33% of people living with T1D are currently using insulin pumps; of these, the vast majority are using one of these three offerings, a statistic that has not changed significantly over the last 5+ years.

All of these pump products require extensive training to initiate and two to four hours per day to use and manage on an ongoing basis. We believe this level of sophistication and effort combined with the cost and awkwardness of these products contribute to the limited uptake.
Although there are purely mechanical pumps available to patients with a small percentage of T2D patients using the Mannkind V-Go patch pump, a fixed basal rate and a button to deliver small boluses. This pump is simple to use, though gives little performance decision to the user (e.g., no possibility to change the basal rate, no possibility to stop bolus doses, small reservoir, pump that needs to be changed every day, etc.). The last available patch pump is provided by Cequr, called Simplicity, a bolus only delivery option without basal delivery. Beta Bionics, Inc. and Deka Research and Development Corp. have received clearance for their tube-based pumps in the last 12 months, but it is too early to assess their commercial traction.
Medtronic has launched a new version of its insulin pump, the Minimed 780G, already available in some European countries with an advanced algorithm, but no obvious change in hardware. Tandem is now selling a small, no display pump called Mobi. The Mobi has a small 2mL reservoir and is controlled by a separate unit, similar to the current Omnipod product. Insulet has also launched the Omnipod 5, a similar patch pump to its current offering, that includes an AID algorithm.
Approximately 79% of the people who rely upon MDI therapy choose to not administer a shot outside of their house, which creates a poorly controlled group. MODD1 is designed to focus upon a segment of these people and mobilize them via a simple, easy to use, affordable product.
Intellectual Property
Our success depends in part on our ability to obtain patents and trademarks, maintain trade secret and know-how protection, enforce our proprietary rights against infringers, and operate without infringing on the proprietary rights of third parties. Because of the length of time and expense associated with developing new products and bringing them through the regulatory approval process, the health care industry places considerable emphasis on obtaining patent protection and maintaining trade secret protection for new technologies, products, processes, know-how, and methods.
As of March 31, 2024, we held four U.S. utility and no foreign patents, and we also held 22 pending applications in the United States and abroad. The patents and patent applications cover various aspects of our technology, including our proprietary fluid movement technology and associated features of our insulin delivery methodology. There can be no assurance that the pending patent applications will result in the issuance of patents, that patents issued to or licensed by us will not be challenged or circumvented by competitors, or that these patents will be found to be valid or sufficiently broad to protect our technology or provide us with a competitive advantage.
Government Regulation
Our operations are subject to comprehensive federal, state, and local laws and regulations in the jurisdictions in which we or our research and development partners do business. The laws and regulations governing our business and interpretations of those laws and regulations and are subject to frequent change. Our ability to operate profitably will depend in part upon our ability, and that of our research and development partners and affiliates, to operate in compliance with applicable laws and regulations. The laws and regulations relating to medical products and healthcare services that apply to our business and that of our partners and affiliates continue to evolve, and we must, therefore, devote significant resources to monitoring developments in legislation, enforcement, and regulation in such areas. As the applicable laws and regulations change, we are likely to make conforming modifications in our business processes from time to time. We cannot provide assurance that a review of our business by courts or regulatory authorities will not result in determinations that could adversely affect our operations or that the regulatory environment will not change in a way that restricts our operations.
FDA Regulation
In the United States, medical devices are strictly regulated by the FDA. Under the FDCA, a medical device is defined as “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component, part or accessory which is, among other things: intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals; or intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of any of its primary intended purposes.” This definition provides a clear distinction between a medical device and other FDA regulated products such as drugs. If the primary intended use of a medical product is achieved through chemical action or by being metabolized by the body, the product is usually a drug or biologic. If not, it is generally a medical device.
We have developed an insulin pump delivery system, which is regulated by the FDA as a medical device under the FDCA, as implemented and enforced by the FDA. The FDA regulates the development, testing, manufacturing, labeling, packaging, storage, installation, servicing, advertising, promotion, marketing, distribution, import, export, and market surveillance of medical devices.
Device Premarket Regulatory Requirements
Before being introduced into the U.S. market, each medical device must obtain marketing clearance or approval from the FDA through the premarket notification (or 510(k)) process, the de novo classification process, or the premarket approval, or PMA, process, unless they are determined to be Class I devices or to otherwise qualify for an exemption from one of these available forms of premarket review and authorization by the FDA. Under the FDCA, medical devices are classified into one of three classes - Class I, Class II or Class III - depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurance of safety and effectiveness. Classification of a device is important because the class to which a device is assigned determines, among other things, the necessity and type of FDA review required prior to marketing the device. Class I devices are those for which reasonable assurance of safety and effectiveness can be maintained through adherence to general controls which include compliance with the applicable portions of the FDA’s Quality System Regulation (the “QSR”), as well as regulations requiring facility registration and product listing, reporting of adverse medical events, and appropriate, truthful and non-misleading labeling, advertising, and promotional materials. The Class I designation also applies to devices for which there is insufficient information to determine that general controls are sufficient to provide reasonable assurance of the safety and effectiveness of the device or to establish special controls to provide such assurance, but that are not life-supporting or life-sustaining or for a use which is of substantial importance in preventing impairment of human health, and that do not present a potential, unreasonable risk of illness or injury.
Class II devices are those for which general controls alone are insufficient to provide reasonable assurance of safety and effectiveness and there is sufficient information to establish “special controls.” These special controls can include performance standards, post-market surveillance requirements, patient registries and FDA guidance documents describing device-specific special controls. While most Class I devices are exempt from the premarket notification requirement, most Class II devices require a premarket notification prior to commercialization in the United States; however, the FDA has the authority to exempt Class II devices from the premarket notification requirement under certain circumstances. As a result, manufacturers of most Class II devices must submit premarket notifications to the FDA under Section 510(k) of the FDCA (21 U.S.C. § 360(k)) in order to obtain the necessary clearance to market or commercially distribute such devices. To obtain 510(k) clearance, manufacturers must submit to the FDA adequate information demonstrating that the proposed device is “substantially equivalent” to a “predicate device” that is already on the market. A predicate device is a legally marketed device that is not subject to PMA, meaning, (i) a device that was legally marketed prior to May 28, 1976 (“pre-amendments device”) and for which a PMA is not required, (ii) a device that has been reclassified from Class III to Class II or I or (iii) a device that was found substantially equivalent through the 510(k) process. If the FDA agrees that the device is substantially equivalent to the predicate device identified by the applicant in a premarket notification submission, the agency will grant 510(k) clearance for the new device, permitting the applicant to commercialize the device. Premarket notifications are subject to user fees, unless a specific exemption applies.
If there is no adequate predicate to which a manufacturer can compare its proposed device, the proposed device is automatically classified as a Class III device. In such cases, a device manufacturer must then fulfill the more rigorous PMA requirements or can request a risk-based classification determination for its device in accordance with the de novo classification process.
Devices that are intended to be life sustaining or life supporting, devices that are implantable, devices that present a potential unreasonable risk of harm or are of substantial importance in preventing impairment of health, and devices that are not substantially equivalent to a predicate device and for which safety and effectiveness cannot be assured solely by the general controls and special controls are placed in Class III. Such devices generally require FDA approval through the PMA process, unless the device is a pre-amendments device not yet subject to a regulation requiring premarket approval. The PMA process is more demanding than the 510(k) process. For a PMA, the manufacturer must demonstrate through extensive data, including data from preclinical studies and one or more clinical trials, that the device is safe and effective for its proposed indication. The PMA must also contain a full description of the device and its components, a full description of the methods, facilities and controls used for manufacturing, and proposed labeling. Following receipt of a PMA submission, the FDA determines whether the application is sufficiently complete to permit a substantive review. If the FDA accepts the application for review, it has 180 days under the FDCA to complete its review and determine whether the proposed device can be approved for commercialization, although in practice, PMA reviews often take significantly longer, and it can take up to several years for the FDA to issue a final decision. Before approving a PMA, the FDA generally also performs an on-site inspection of manufacturing facilities for the product to ensure compliance with the QSR.
The de novo classification process allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its device to Class I or Class II, on the basis that the device presents low or moderate risk, as an alternative to following the typical Class III device pathway requiring the submission and approval of a PMA application. With our submission in January 2024, the FDA has confirmed our MODD 1 qualifies as a 510(k) eligible device and does not require a de novo classification.
Clinical trials are almost always required to support PMAs and are sometimes required to support 510(k) and de novo classification submissions. In our case, usability studies of our intended users are required and have been completed. All clinical investigations of devices to determine safety and effectiveness must be conducted in accordance with the FDA’s investigational device exemption, or IDE, regulations that govern investigational device labeling, prohibit promotion of investigational devices, and specify recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk,” as defined by the FDA, the agency requires the study sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical trials. The IDE will automatically become effective 30 days after receipt by the FDA, unless the FDA denies the application or notifies the sponsor that the investigation is on hold and may not begin until the sponsor provides supplemental information about the investigation that satisfies the agency’s concerns. If the FDA determines that there are deficiencies or other concerns with an IDE that require modification of the study, the FDA may permit a clinical trial to proceed under a conditional approval. The FDA may also notify the sponsor that the study is approved as proposed or approved with specific requested modification. Furthermore, the agency may withdraw approval of an IDE under certain circumstances. In addition, the study must be approved by, and conducted under the oversight of, an institutional review board, or IRB, for each clinical site. If the device presents a non-significant risk to the patient according to criteria established by the FDA as part of the IDE regulations, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate authorization from the FDA, but must still comply with abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements.
Post-Marketing Restrictions and Enforcement
After a device is placed on the market, numerous regulatory requirements apply. These include, but are not limited to:
| | ● | submitting and updating establishment registration and device listings with the FDA; |
| | ● | compliance with the QSR, which requires manufacturers to follow stringent design, testing, control, documentation, record maintenance, including maintenance of complaint and related investigation files, and other quality assurance controls during the manufacturing process; |
| | ● | unannounced routine or for-cause device facility inspections by the FDA, which may include our suppliers’ facilities; and |
| | ● | labeling regulations, which prohibit the promotion of products for uncleared or unapproved (or “off-label”) uses and impose other restrictions relating to promotional activities; |
| | ● | corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections or removals if undertaken to reduce a risk to health posed by a device or to remedy a violation of the FDCA that may present a risk to health; and |
| | ● | post-market surveillance regulations, which apply to certain Class II or III devices when necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
In addition, under the FDA medical device reporting, or MDR, regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or a similar device of such manufacturer were to recur. The decision to file an MDR involves a judgment by the manufacturer. If the FDA disagrees with the manufacturer’s determination, the FDA can take enforcement action.
The MDR requirements also extend to health care facilities that use medical devices in providing care to patients, or “device user facilities,” which include hospitals, ambulatory surgical facilities, nursing homes, outpatient diagnostic facilities, or outpatient treatment facilities, but not physician offices. A device user facility must report any device-related death to both the FDA and the device manufacturer, or any device-related serious injury to the manufacturer (or, if the manufacturer is unknown, to the FDA) within 10 days of the event. Device user facilities are not required to report device malfunctions that would likely cause or contribute to death or serious injury if the malfunction were to recur but may voluntarily report such malfunctions through MedWatch, the FDA’s Safety Information and Adverse Event Reporting Program.
The FDA also has the authority to require the recall of commercialized medical device products in the event of material deficiencies or defects in design or manufacture. The authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious adverse health consequences or death. Manufacturers may, under their own initiative, recall a product if any distributed devices fail to meet established specifications, are otherwise misbranded or adulterated under the FDCA, or if any other material deficiency is found. The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated.
The failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
| | ● | warning letters, fines, injunctions or civil penalties; |
| | ● | recalls, detentions or seizures of products; |
| | ● | delays in the introduction of products into the market; |
| | ● | total or partial suspension of production; |
| | ● | delay or refusal of the FDA or other regulators to grant 510(k) clearance, PMA approvals, or other marketing authorization to new products; |
| | ● | withdrawals of marketing authorizations; or |
| | ● | in the most serious cases, criminal prosecution. |
To ensure compliance with regulatory requirements, medical device manufacturers are subject to market surveillance and periodic, pre-scheduled and unannounced inspections by the FDA, and these inspections may include the manufacturing facilities of subcontractors.
Federal Trade Commission Regulatory Oversight
Our advertising for our products and services will be subject to federal truth-in-advertising laws enforced by the Federal Trade Commission (the “FTC”) as well as comparable state consumer protection laws. Under the Federal Trade Commission Act (the “FTC Act”), the FTC is empowered, among other things, to (a) prevent unfair methods of competition and unfair or deceptive acts or practices in or affecting commerce; (b) seek monetary redress and other relief for conduct injurious to consumers; and (c) gather and compile information and conduct investigations relating to the organization, business, practices, and management of entities engaged in commerce. The FTC has very broad enforcement authority, and failure to abide by the substantive requirements of the FTC Act and other consumer protection laws can result in administrative or judicial penalties, including civil penalties, injunctions affecting the manner in which we would be able to market services or products in the future, or criminal prosecution.
Healthcare Law and Regulation
United States
If our MODD1 product or our other future product candidates are approved in the United States, we will have to comply with various U.S. federal and state laws, rules and regulations pertaining to healthcare fraud and abuse, including anti-kickback laws and physician self-referral laws, rules and regulations. Violations of the fraud and abuse laws are punishable by criminal and civil sanctions, including, in some instances, exclusion from participation in federal and state healthcare programs, including Medicare and Medicaid. These laws include the following:
| | ● | the federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
| | ● | the federal False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| | ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| | ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
| | ● | the federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services; |
| | ● | the federal transparency requirements under the Physician Payments Sunshine Act require manufacturers of FDA-approved drugs, devices, biologics and medical supplies covered by Medicare or Medicaid to report, on an annual basis, to the Department of Health and Human Services information related to payments and other transfers of value to physicians, teaching hospitals, and certain advanced non-physician health care practitioners and physician ownership and investment interests; and |
| | ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by nongovernmental third-party payors, including private insurers. |
Some state laws require pharmaceutical or medical device companies to comply with the relevant industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug and device manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures.
State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. We also may be subject to, or may in the future become subject to, U.S. federal and state, and foreign laws and regulations imposing obligations on how we collect, use, disclose, store and process personal information. Our actual or perceived failure to comply with such obligations could result in liability or reputational harm and could harm our business. Ensuring compliance with such laws could also impair our efforts to maintain and expand our customer base and thereby decrease our future revenues.
The European Union approves the use of medical devices in a very different way. They have similar regulations and requirements to adhere to, however, a Notified Body, in the form of a private company, will represent their interests and is required to have sufficient expertise to review all applications and the company’s internal processes to ensure the safety of the product for which approval is being requested. We are in the process of identifying a Notified Body to represent us, and we will follow our FDA submission process with regard to preparing the materials and processes required to meet the regulations and gain clearance.
European Union
EEA
In the European Economic Area, (which is comprised of the 27 member states of the European Union plus Norway, Iceland and Liechtenstein), or EEA, manufacturers of medical devices need to comply with the Essential Requirements laid out in Annex I to the EU Medical Devices Directive (Council Directive 93/42/EEC) or with the General Safety and Performance Requirements (GSPR) of the new EU Medical Devices Regulation (EU 2017/745). Compliance with these requirements is a prerequisite to be able to affix the CE mark to medical devices, without which they cannot be marketed or sold in the EEA. To demonstrate compliance with the Essential Requirements and the GSPR and obtain the right to affix the CE Mark, manufacturers of medical devices must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low-risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements and the GSPR, a conformity assessment procedure requires the intervention of a Notified Body, which is an organization designated by a competent authority of an EEA country to conduct conformity assessments. Depending on the relevant conformity assessment procedure, the Notified Body would audit and examine the Technical File and the quality system for the manufacture, design and final inspection of the devices. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the Essential Requirements and GSPR. This Certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity. As a general rule, demonstration of conformity of medical devices and their manufacturers with the Essential Requirements and GSPR must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device are supported by suitable evidence.
All manufacturers placing medical devices into the market in the EEA must comply with the EU Medical Device Vigilance System. Under this system, incidents must be reported to the relevant authorities of the member states of the EEA, and manufacturers are required to take Field Safety Corrective Actions, or FSCAs, to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An incident is defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient or user or of other persons or to a serious deterioration in their state of health. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be communicated by the manufacturer or its legal representative to its customers and/or to the end users of the device through Field Safety Notices. Where appropriate, our products commercialized in Europe are CE marked and classified as either Class I or Class II.
In 2017, the European Parliament passed the Medical Devices Regulation, which repeals and replaces the EU Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EEA member states, the regulations would be directly applicable (i.e., without the need for adoption of EEA member State laws implementing them) in all EEA member states and are intended to eliminate current differences in the regulation of medical devices among EEA member States. The Medical Devices Regulation, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and in vitro diagnostic devices and ensure a high level of safety and health while supporting innovation.
The Medical Device Regulation became applicable on May 26, 2021. Devices lawfully placed on the market pursuant to the EU Medical Devices Directive prior to May 26, 2021 may generally continue to be made available on the market or put into service until May 26, 2026. The Medical Devices Regulation, among other things:
| | ● | strengthens the rules on placing devices on the market and reinforces surveillance once they are available; |
| | ● | establishes explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; |
| | ● | improves the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| | ● | sets up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; and |
| | ● | strengthens rules for the assessment of certain high-risk devices, such as implants, which may have to undergo an additional check by experts before they are placed on the market. |
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to such reports filed or furnished pursuant to section 13(a) or 15(d) of the Securities Exchange Act of 1934, as well as section 16 reports on Form 3, 4, or 5, are available free of charge on our website at www.modular-medical.com. as soon as it is reasonably practicable after they are filed or furnished with the SEC. Our Code of Business Conduct and Ethics and the charters for the Audit Committee, Compensation Committee and Nominating and Governance Committee are also available on our website. The Code of Business Conduct and charters are also available in print to any stockholder upon request without charge. Requests for such documents should be directed to Modular Medical, Inc., 10740 Thornmint Road, San Diego CA 92127, Attn. CFO. Our Internet website and the information contained on it or connected to it are not part of, or incorporated by, reference into this Report. Our filings with the SEC are also available on the SEC’s website at http://www.sec.gov.
Corporate Information
We are a Nevada corporation, and Quasuras, Inc., a Delaware corporation, is our only subsidiary. Our corporate headquarters and operating facilities are located at 10740 Thornmint Road, San Diego, CA 92127. Our telephone number is (858) 800-3500. We maintain a website at www.modular-medical.com.
Employees
As of March 31, 2024, we had 40 employees all of whom are located in the United States and 39 of whom are full-time, consisting of 36 in research and development and manufacturing operations and 4 in general and administrative functions.
ITEM 1A. RISK FACTORS
This Annual Report on Form 10-K contains forward-looking statements that involve risks and uncertainties, such as statements of our objectives, expectations and intentions. The cautionary statements made in this Annual Report on Form 10-K should be read as applicable to all forward-looking statements wherever they appear in this report. Our actual results could differ materially from those discussed herein. Factors that could cause or contribute to such differences include those discussed below, as well as those discussed elsewhere in this Annual Report on Form 10-K.
We might not be able to continue as a going concern.
Our consolidated financial statements as of March 31, 2024 have been prepared under the assumption that we will continue as a going concern twelve months from the date of issuance of this Report. At March 31, 2024, we had cash and cash equivalents of $9.2 million and an accumulated deficit of $65.9 million. In February 2024, we completed a public offering of common stock for net proceeds to us of approximately $10.3 million. In May 2023, we completed a public offering of common stock and warrants for net proceeds to us of approximately $9.7 million. Even with these offering proceeds, we do not believe that our cash and cash equivalents would be sufficient to fund our operations for the period of 12 months from the date of issuance of this report, and we would need to raise additional capital. As a result of our expected operating losses and cash burn for the foreseeable future and recurring losses from operations, if we are unable to raise sufficient capital through additional debt or equity arrangements, there will be uncertainty regarding our ability to maintain liquidity sufficient to operate our business effectively, which raises substantial doubt as to our ability to continue as a going concern. If we cannot continue as a viable entity, our stockholders would likely lose most or all of their investment in us.
If we are unable to generate sustainable operating profit and sufficient cash flows, then our future success will depend on our ability to raise capital. We intend to seek additional financing and evaluate financing alternatives in order to meet our cash requirements for the foreseeable future. We cannot be certain that raising additional capital, whether through selling additional debt or equity securities or obtaining a line of credit or other loan, will be available to us or, if available, will be on terms acceptable to us. If we issue additional securities to raise funds, these securities may have rights, preferences, or privileges senior to those of our common stock, and our current stockholders may experience dilution. If we are unable to obtain funds when needed or on acceptable terms, we may be required to curtail our current product development programs, cut operating costs, forego future development and other opportunities or even terminate our operations.
We are a developmental stage medical device company and have a history of significant operating losses; we expect to continue to incur operating losses, and we may never achieve or maintain profitability.
As a development-stage enterprise, we do not currently have revenues to generate cash flows to cover operating expenses. Since our inception, we have incurred operating losses in each year due to costs incurred in connection with research and development activities and general and administrative expenses associated with our operations. For the years ended March 31, 2024 and, 2023, we incurred net losses of approximately $17.5 million and $13.9 million, respectively. As a result, we need to raise additional capital in the future, which may or may not be available to us at all or only on unfavorable terms.
We expect to incur losses for the foreseeable future as we continue the development of, and seek regulatory clearance and approvals for, our insulin pump. As our MODD1 insulin pump is currently our only product, if it fails to gain regulatory approval and market acceptance, we will not be able to generate any revenue, or explore other opportunities to enhance stockholder value, such as through a sale. If we fail to generate revenue and eventually become profitable, or if we are unable to fund our continuing losses, our stockholders could lose all or a substantial part of their investment.
We will need substantial additional funding to complete subsequent phases of the development of our insulin pump product candidate and to operate our business and such funding may not be available or, if it is available, such financing is likely to substantially dilute our existing stockholders.
The discovery, development, and commercialization of new medical devices, such as our insulin pump, entails significant costs. We have completed the engineering and mechanical development of our insulin pump and cartridge, and submitted the product to the FDA for clearance. In addition, we have also implemented a production-level manufacturing process, including purchasing required equipment for low-level manufacturing. We will continue to refine our insulin pump to, among other things, meet the general needs and preferences of the Almost Pumper marketplace and the guidelines of third-party payors. To enable us to accomplish these and other related items and continue to operate our business, we will need to raise substantial additional capital and/or enter into strategic partnerships or joint ventures to enable us to:
| | ● | seek European regulatory approvals with appropriate clinical studies; |
| | ● | expand and continue to improve our manufacturing and commercialization capabilities; |
| | ● | develop, test, and, if approved, market our product candidate; |
| | ● | acquire or license additional internal systems and other infrastructure; and |
| | ● | hire and support additional management, administrative, sales and marketing, and technical personnel. |
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never achieve, we expect to finance our cash needs primarily through public or private equity offerings, debt financings or through the establishment of possible strategic alliances. We may in the future seek additional capital from public or private offerings of our capital stock or borrow additional amounts under new credit lines or from other sources. If we issue equity or debt securities to raise additional funds, our existing stockholders may experience dilution, we may incur significant financing costs, and the new equity or debt securities may have rights, preferences and privileges senior to those of our existing stockholders. In addition, if we raise additional funds through collaborations, licensing, joint ventures, strategic alliances, partnership arrangements or other similar arrangements, it may be necessary to relinquish valuable rights to the MODD1 pump or our potential future products or proprietary technologies or grant licenses on terms that are not favorable to us.
We cannot be certain that additional funding will be available on acceptable terms, or at all. If we are not able to secure additional equity funding when needed, we may have to delay, reduce the scope of, or eliminate one or more of our post-market clinical studies, development programs or future commercialization initiatives. In addition, any additional equity funding that we do obtain will dilute the ownership held by our existing equity holders. The amount of this dilution may be substantially increased if the trading price of our common stock is lower at the time of any financing. Regardless, the economic dilution to stockholders will be significant if our stock price does not increase significantly, or if the effective price of any sale is below the price paid by a particular stockholder. Any debt financing that we obtain in the future could involve substantial restrictions on activities and creditors could seek a pledge of some or all of our assets. We have not identified potential sources for such financing that we will require, and we do not have commitments from any third parties to provide any future debt financing. If we fail to obtain funding as needed, we may be forced to cease or scale back operations, and our business, prospects, results of operations, financial condition and stock price would be adversely affected.
We have a limited operating history and historical financial information upon which you may evaluate our performance.
You should consider, among other factors, our prospects for success in light of the risks and uncertainties encountered by companies that, like us, are in their early stages of development. We may not successfully address these risks and uncertainties or successfully complete our studies and/or implement our existing and new products. If we fail to do so, it could materially harm our business and impair the value of our common stock. Unanticipated problems, expenses and delays are frequently encountered in establishing a new business, conducting research, and developing new products. These include, but are not limited to, inadequate funding, failure to obtain regulatory approval, unforeseen research issues, lack of consumer, physician or third-party payor acceptance, competition, sluggish product development, and inadequate sales and marketing. The failure by us to meet any of these conditions would have a materially adverse effect upon us and may force us to reduce or curtail operations. No assurance can be given that we can or will ever operate profitably.
The amount of financing we require will depend on a number of factors, many of which are beyond our control. Our results of operations, financial condition and stock price are likely to be adversely affected if our funding requirements increase or are otherwise greater than we expect.
Our future funding requirements will depend on many factors, including, but not limited to:
| | ● | the testing costs for our insulin pump product candidate and other development activities conducted by us directly, and our ability to successfully conclude the studies and activities and achieve favorable results; |
| | ● | our ability to attract future strategic partners to pay for or share costs related to our product development efforts; |
| | ● | the costs and timing of seeking and obtaining regulatory clearance and approvals for our product candidate; |
| | ● | the costs of filing, prosecuting, maintaining and enforcing any patents and other intellectual property rights that we may have and defending against potential claims of infringement; |
| | ● | decisions to hire additional scientific, engineering or administrative personnel or consultants; |
| | ● | our ability to manage administrative and other costs of our operations; and |
| | ● | the presence or absence of adverse developments in our research program. |
If any of these factors cause our funding needs to be greater than expected, our operations, financial condition, ability to continue operations and stock price may be adversely affected.
Our future cash requirements may differ significantly from our current estimates.
Our cash requirements may differ significantly from our estimates from time to time, depending on a number of factors, including:
| | ● | the costs and results of our clinical studies regarding our insulin pump product candidate; |
| | ● | the time and costs involved in obtaining regulatory clearance and approvals; |
| | ● | whether we are able to obtain funding under future licensing agreements, strategic partnerships, or other collaborative relationships, if any; |
| | ● | the costs of compliance with laws, regulations, or judicial decisions applicable to us; and |
| | ● | the costs of general and administrative infrastructure required to manage our business and protect corporate assets and stockholder interests. |
If we fail to raise additional funds on a timely basis, we will need to scale back our business plans, which would adversely affect our business, prospects, results of operations, financial condition, and stock price, and we may even be forced to discontinue our operations and liquidate our assets.
Technological breakthroughs in diabetes monitoring, treatment or prevention could render our insulin pump obsolete.
The diabetes treatment market is subject to rapid technological change and product innovation. Our insulin pump is based on our proprietary technology, but a number of companies, medical researchers and existing pharmaceutical companies are pursuing new delivery devices, delivery technologies, sensing technologies, procedures, drugs and other therapeutics for the monitoring, treatment and/or prevention of insulin-dependent diabetes. Any technological breakthroughs in diabetes monitoring, treatment or prevention could render our insulin pump obsolete, which, since our insulin pump is our only product candidate, would have a material adverse effect on our business, our insulin pump is our only product candidate, would have a material adverse effect on our business, prospects, results of operations and financial condition and could result in stockholders losing their entire investment.
Any failure to attract and retain skilled directors, executives, employees and consultants could impair our product development and commercialization activities.
Our business depends on the skills, performance, and dedication of our directors, executive officers and key engineering, scientific and technical advisors. Many of our current engineering or scientific advisors are independent contractors and are either self-employed or employed by other organizations. As a result, they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, which may affect their ability to provide services to us in a timely manner. We will need to recruit additional directors, executive management employees, and advisers, particularly engineering, scientific and technical personnel, which will require additional financial resources. In addition, there is currently intense competition for skilled directors, executives and employees with relevant engineering, scientific and technical expertise, and this competition is likely to continue. If we are unable to attract and retain persons with sufficient engineering, scientific, technical and managerial experience, we may be forced to limit or delay our product development activities or may experience difficulties in successfully conducting our business, which would adversely affect our business, prospects, results of operations and financial condition.
Our operations are substantially dependent upon key personnel.
Our performance is substantially dependent on the continued services and performance of our senior management and certain other key personnel. The loss of services of any of our executive officers or other key employees could have a material adverse effect on our business, financial condition and results of operations. In addition, any future expansion of our business will depend on our ability to identify, attract, hire, train, retain and motivate other highly skilled managerial, marketing, customer service and manufacturing personnel, and our inability to do so could have a material adverse effect on our business, financial condition and results of operations.
We are dependent on the performance and continued engagement of our Chairman, President and Principal Financial Officer.
We are dependent on the performance and continued engagement of Paul DiPerna, our chairman, president and principal financial officer. Although we believe we will be able to engage qualified personnel for such purposes, an inability to do so could materially adversely affect our ability to market, sell, and enhance our products. While Mr. DiPerna is currently devoting his full-time working efforts to us, other employees and consultants may only be available to us on a part-time basis. The loss of one or more of our key employees, especially Mr. DiPerna, or our inability to hire and retain other qualified employees, including but not limited to research and development, sales, manufacturing, and administrative support staff, could have a material adverse effect on our business, prospects, results of operations and financial condition.
We have limited internal research and development personnel, making us dependent on consulting relationships.
We consider research and development to be an important part of the process of designing, developing, obtaining regulatory required approvals and the eventual commercialization of our insulin pump. We continue to incur increased research and development expenditures, which are primarily attributable to effort and expenses incurred in designing and developing our innovative insulin pump. We expect to continue to incur substantial costs related to research and development.
We will need to outsource and rely on third parties for various aspects relating to the development, manufacture, sales and marketing of our insulin pump as well as in connection with assisting us in the preparation and filing of our FDA submission, and our future success will be dependent on the timeliness and effectiveness of the efforts of these third parties.
We are dependent on consultants for important aspects of our product development strategy. We do not have the required financial resources and personnel to carry out independently the development of our product candidate, and do not have the capability or resources to manufacture, market or sell our current product candidate. As a result, we contract with and rely on third parties for important functions, including in connection with the development and finalization of our insulin pump, the preparation and filing of our FDA submission and eventual manufacturing and commercialization of our product candidate. We have recently entered into several agreements with third parties for such services. If problems develop in our relationships with third parties, or if such parties fail to perform as expected, it could lead to delays or lack of progress in obtaining FDA clearance, significant cost increases, changes in our strategies, and even failure of our product initiatives.
We may not be able to identify, negotiate and maintain the strategic alliances necessary to develop and commercialize our products and technologies, and we will be dependent on our corporate partners if we do.
We may seek to enter into a strategic alliance with a diabetes-related service providing company for the further development and approval of our insulin pump product candidate. At this time, we have not entered into any such strategic alliance. Strategic alliances, if entered into, could potentially provide us with additional funds, expertise, access, and other resources in exchange for exclusive or non-exclusive licenses or other rights to the product that we are currently developing or a product we may explore in the future. We cannot give any assurance that we will be able to enter into strategic relationships with a diabetes-related service providing company or others in the near future or at all. In addition, we cannot assure you that any agreements that we do reach will allow us to achieve our goals or that such grants will be on terms that prove to be economically beneficial to us. When we do enter into strategic or contractual relationships, we become dependent on the successful performance of our partners or counter-parties. If they fail to perform as expected, such failure could adversely affect our financial condition, lead to increases in our capital needs, or hinder or delay our development efforts. See “Our Business -Employees” below.
We may not receive the necessary regulatory clearance or approvals for our insulin pump, and failure to timely obtain necessary clearances and/or approvals could harm our then operations, including our ability to commercialize our product candidate.
Before we can market a new medical device, such as our insulin pump, we must first receive clearance under Section 510(k) of the Federal Food, Drug, and Cosmetic Act, or the “FDCA.” In the 510(k) clearance process, before a device may be marketed, the FDA must determine that such proposed device is “substantially equivalent” to a legally-marketed “predicate” device, which includes a device that has been previously cleared through the 510(k) process, a device that was legally marketed prior to May 28, 1976 (pre-amendments device), a device that was originally on the U.S. market pursuant to a premarket approval (PMA) and later down-classified, or a 510(k)-exempt device. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics and not raise different questions of safety or effectiveness than the predicate device. In January 2024, we submitted a 510(k) premarket notification to the United States Food and Drug Administration (“FDA”) for our MODD1 insulin pump.
Certain future modifications made to our product candidate, which we currently expect to be cleared through 510(k), may require a new 510(k) clearance. The 510(k) clearance process can be expensive, lengthy and uncertain. The FDA’s 510(k) clearance process usually takes less than 12 months, but it can last longer. Despite the time, effort and cost, a device may not be approved or cleared by the FDA. Any delay or failure to obtain necessary regulatory authorizations could harm our business, including our ability to commercialize our product candidate and our stockholders could lose their entire investment. Furthermore, even if we are granted the required regulatory authorizations, such authorizations may be subject to significant limitations on the indicated uses for the device, which may limit the market for our product candidate.
If the FDA requires us to go through a lengthier, more rigorous examination for our product candidate than we had expected, product introductions or modifications could be delayed or canceled, which could adversely affect our ability to grow our business.
The FDA can delay, limit or deny clearance or approval for our insulin pump medical device for many reasons, including, for example:
| ● | our inability to demonstrate to the satisfaction of the FDA that our product candidate is substantially equivalent to the proposed predicate device; |
| ● | the disagreement of the FDA with the design or implementation of our performance testing protocols or the interpretation of data from our performance testing; |
| ● | the data from performance testing may be insufficient to support a determination of substantial equivalence or that our device meets required special controls or applicable performance standards; |
| ● | our inability to demonstrate that the benefits of our pump outweigh the risks; |
| ● | the manufacturing process or facilities we intend to use may not meet applicable requirements; for example, we experienced issues maintaining insulin stability on an initial version of our pump product candidate, and we attributed this issue to the materials used in the production of our product; we believe we have made the necessary changes to our materials and process to address this issue and will be completing the required testing prior to our FDA submission; and |
| ● | the potential for approval policies or regulations of the FDA to change significantly in a manner rendering our data or regulatory filings insufficient for clearance or approval. |
In addition, the FDA may change its clearance and approval policies, adopt additional regulations or revise existing regulations, or take other actions, which may prevent or delay approval or clearance of our product candidate or impact our ability to modify our product candidate after clearance on a timely basis. Such policy or regulatory changes could impose additional requirements upon us that could delay our ability to obtain clearance for our pump, increase the costs of compliance or restrict our ability to maintain our current approval.
As a general rule, demonstration of conformity of medical devices and their manufacturers with the essential requirements must be based, among other things, on the evaluation of data supporting the safety and performance of the product candidates during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device are supported by suitable evidence.
Obtaining marketing authorization in the United States will not obviate the need to obtain marketing authorization in other jurisdictions We must obtain approval from foreign regulatory authorities before we can market and sell any of our product candidates in countries outside the United States. We will incur additional costs in seeking such approvals, may experience delays in obtaining such approvals and cannot be certain that such approvals will be granted.
The development, manufacture, and marketing of our product candidates outside the United States is subject to government regulation. In most foreign countries, we must complete rigorous pre-clinical testing and extensive human clinical trials that demonstrate the safety and efficacy of a product in order to apply for regulatory approval to market the product. If foreign regulatory authorities grant regulatory approval of a product, the approval may be limited to specific indications or limited with respect to its distribution. Expanded or additional indications for approved devices may not be approved, which could limit our potential revenues. Foreign regulatory authorities may refuse to grant any approval. Consequently, even if we believe that pre-clinical and clinical data are sufficient to support regulatory approval for our products, foreign regulatory authorities may not ultimately grant approval for commercial sale in any jurisdiction. If our product candidates are not approved in such jurisdictions, our ability to generate revenues will be limited and our business will be adversely affected.
Our competitors may develop products that are more effective, safer and less expensive than ours.
Existing insulin pumps are expensive, with the more popular models having purchase prices exceeding $4,000 for individuals without health insurance and often require significant patient copays. Others have daily use costs that exceed the reimbursement rates of many health insurance plans, forcing some users to spend thousands of dollars a year in copays. We believe this makes insurers hesitant to pay for any pumps and places pumps out of reach for many patients who cannot afford such out of pocket expenses.
We are engaged in the diabetes treatment sector of the healthcare marketplace, which is intensely competitive. There are current products that are quite effective at addressing the effects of diabetes, and we expect that new developments by other companies and academic institutions in the areas of diabetes treatment will continue. If approved for marketing by the FDA, depending on the approved clinical indication, our product will be competing with existing and future products related to treatments for diabetes.
Our competitors may:
| | ● | develop product candidates and market products that increase the levels of safety or efficacy that our product candidates will need to show in order to obtain regulatory approval; |
| | ● | develop product candidates and market products that are less expensive or more effective than ours; |
| | ● | commercialize competing products before we can launch any products we are working to develop; |
| | ● | hold or obtain proprietary rights that could prevent us from commercializing our products; or |
| | ● | introduce therapies or market medical products that render our potential product candidates obsolete. |
We expect to compete against large medical device companies, such as Medtronic, Inc., Tandem Diabetes Care, Inc. and Insulet Corporation, smaller companies that are collaborating with larger medical device companies, new companies, academic institutions, government agencies and other public and private research organizations. These competitors, in nearly all cases, produce similar products relative to the treatment of diabetes and have substantially greater financial resources than we do. Our competitors also have significantly greater experience in:
| | ● | developing medical device and other product candidates; |
| | ● | undertaking testing and clinical studies; |
| | ● | building relationships with key customers and opinion-leading physicians; |
| | ● | obtaining and maintaining FDA and other regulatory approvals; |
| | ● | formulating and manufacturing medical devices; |
| | ● | launching, marketing and selling medical devices; |
| ● | providing management oversight for all of the above-listed operational functions; and |
| ● | obtaining insurance coverage and reimbursement for their competitive products. |
If we fail to achieve acceptance over other existing or newly developed products, we may be unable to obtain regulatory approval or successfully commercialize our MODD1 insulin pump product candidate or any future products. If our competitors’ market medical devices that are less expensive, safer or more effective than our insulin pump, or that gain or maintain greater market acceptance, we may not be able to compete effectively, which would adversely affect our business, prospects, results of operations and financial condition. See “Business - Competition.”
We expect to rely on third-party manufacturers and will be dependent on their quality and effectiveness.
Our insulin pump requires precise, high-quality manufacturing. The failure to achieve and maintain high manufacturing standards, including failure to detect or control anticipated or unanticipated manufacturing errors or the frequent occurrence of such errors, could result in patient injury or death, discontinuance or delay of ongoing or planned clinical studies, delays or failures in product testing or delivery, cost overruns, product recalls or withdrawals and other problems that could seriously hurt our business. Contract medical device manufacturers often encounter difficulties involving production yields, quality control and quality assurance and shortages of qualified personnel. These manufacturers are subject to stringent regulatory requirements, including the FDA’s current good-manufacturing-practices regulations. If our contract manufacturers fail to maintain ongoing compliance at any time, the production of our product could be interrupted, resulting in delays or discontinuance of our clinical studies, additional costs and loss of potential revenues.
We may not be able to successfully scale-up manufacturing of our product candidate in sufficient quality and quantity, which would delay or prevent us from developing our product candidate and commercializing our product candidate.
In order to conduct larger-scale or late-stage clinical studies and for commercialization of our insulin pump, if 510(k) clearance is granted, we will need to manufacture it in larger quantities. We may not be able to successfully increase the manufacturing capacity for our product candidate in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If we are unable to successfully scale up the manufacture of our product candidate in sufficient quality and quantity, the development and testing of our product candidate and regulatory approval or commercial launch may be delayed, which could significantly harm our business.
We are dependent upon third-party suppliers to manufacture our product, and this makes us vulnerable to supply shortages and price increases; we may not be able to obtain an adequate supply of components on a timely basis or at all.
The future manufacture of our product will require the timely delivery of sufficient amounts of components from multiple suppliers in various countries. We intend to work closely with our suppliers to ensure continuity of supply, but we cannot guarantee these efforts will be successful. Due to the supply chain issues experienced by the semiconductor industry, at times, we have experienced delays obtaining integrated circuits from certain suppliers. We may need to enter into “take or pay” contracts with suppliers. We have also seen price increases for various components. We do not have supply agreements with any of our suppliers, and we make purchases based on individual purchase orders. An interruption, delay, or inability to obtain components from our third-party suppliers at acceptable prices in a timely manner, could hinder our ability to manufacture our products and have a material adverse effect on our business, prospects, financial condition and results of operations.
We may be subject to potential product liability and other claims that could materially impact our business and financial condition.
The development and sale of our insulin pump exposes us to the risk of significant damages from product liability and other claims, and the use of our product in clinical studies may result in adverse effects from liability claims. We cannot predict all the possible harms or adverse effects that may result. We intend to obtain product liability insurance to provide some protection from claims. Nonetheless, we may not have sufficient resources to pay for any liabilities resulting from a personal injury or other claim, even if it is partially covered by insurance. In addition to the possibility of direct claims, we may be required to indemnify third parties against damages and other liabilities arising out of our development, commercialization and other business activities, which would increase our liability exposure. If third parties that have agreed to indemnify us fail to do so, we may be held responsible for those damages and other liabilities as well.
Legislative, regulatory, or medical cost reimbursement changes may adversely impact our business.
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to the health care system in the U.S. and in other jurisdictions may change the nature of and regulatory requirements relating to innovations in medical devices, testing and regulatory approvals, limit or eliminate payments for medical procedures and treatments, or subject the pricing of medical devices to government control. In addition, third-party payors in the U.S. are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new products. Consequently, significant uncertainty exists as to the reimbursement status of newly approved health care products. Significant changes in the health care system in the U.S. or elsewhere, including changes resulting from adverse trends in third-party reimbursement programs, could have a material adverse effect on our projected future operating results and our ability to raise capital, commercialize products, and remain in business.
We are subject to extensive regulation by the FDA, which could restrict the sales and marketing of our insulin pump and could cause us to incur significant costs.
Our insulin pump is subject to extensive regulation by the FDA. These regulations relate to manufacturing, labeling, sale, promotion, distribution and shipping. Before a new medical device, or a new intended use of a legally marketed device, can be marketed in the United States, it must be cleared or approved by FDA through the applicable premarket review process (510(k), PMA, or de novo classification), unless an exemption applies. If we receive 510(k) clearance for our insulin pump, we may be required to obtain new 510(k) clearances for significant post-market modifications to the pump. Each premarket submission and review process can be expensive and lengthy, and entail significant user fees, unless exempt.
Medical devices may be marketed only for the indications for which they are approved or cleared. Further, 510(k) clearance can be revoked if safety or effectiveness problems develop once the device is on the market.
The current regulatory requirements to which we are subject may change in the future in a way that adversely affects us. If we fail to comply with present or future regulatory requirements that are applicable to us, we may be subject to enforcement action by the FDA, which may include any of the following sanctions:
| | ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| | ● | customer notification, or orders for repair, replacement or refunds; |
| | ● | voluntary or mandatory recall or seizure of our current or future products; |
| | ● | administrative detention by the FDA of medical devices believed to be adulterated or misbranded; |
| | ● | imposing operating restrictions, suspension or shutdown of production; |
| | ● | refusing our requests for 510(k) clearance, PMA or de-novo classification of any new products, new intended uses or modifications to our insulin pump; |
| | ● | rescinding 510(k) clearance that has already been granted; and |
The occurrence of any of these events would have a material adverse effect on our business, financial condition and results of operations and could result in stockholders losing their entire investment.
Although our insulin pump product candidate does not presently require clinical trials to apply to the FDA for clearance and even if a clinical trial is completed, the results of our clinical testing may not demonstrate the safety and efficacy of the device or may be equivocal or otherwise not be sufficient for us to obtain approval of our product candidate.
Clinical trials are almost always required to support a PMA application and may also be required to support 510(k) submissions although at this time ours does not require a PMA. If the device presents a “significant risk” to human health as defined by the FDA, the FDA requires the study sponsor to submit an investigational device exemption (“IDE”) application and obtain IDE approval prior to commencing human clinical trials. The IDE must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. An IDE will automatically become effective 30 days after receipt by the FDA, unless the FDA denies the application or notifies the sponsor that the investigation is on hold and may not begin until the sponsor provides supplemental information about the investigation that satisfies the agency’s concerns. The FDA may also notify the sponsor that the study is approved as proposed. If the FDA determines that there are deficiencies or other concerns with an IDE that require modification of the study, the FDA may permit a clinical trial to proceed under conditional approval. Furthermore, the agency may withdraw approval of an IDE under certain circumstances. Clinical trials for a significant risk device may begin once an IDE is approved by the FDA and the appropriate Institutional Review Board (“IRB”) at each clinical trial site. If the product is deemed a “non-significant risk” device, IDE approval from the FDA would not be required, but the clinical trial would need to meet other requirements including IRB approval. Our clinical trials must be conducted in accordance with FDA regulations and federal and state regulations concerning human subject protection, including informed consent and healthcare privacy. A clinical trial may be suspended by the FDA or at a specific site by the relevant IRB at any time for various reasons, including a determination that the risks to the trial participants outweigh the benefits of participation in the clinical trial. Even if a clinical trial is completed, the results of our clinical testing may not demonstrate the safety and efficacy of the device or may be equivocal or otherwise not be sufficient for us to obtain approval of our product.
Our success depends substantially upon our ability to obtain and maintain intellectual property protection relating to our product candidate and research technologies.
We have applied to the U.S. Patent and Trademark Office (the “USPTO”) and various foreign patent agencies for patents on our proprietary fluid movement technology and our insulin delivery methodology. To date, the USPTO has granted four patents to us, and we have additional applications pending and in various stages of review by the USPTO and foreign patent agencies. There can be no assurance that we will be issued additional patents by the USPTO or foreign patent agencies and that any of our patents will prevent other companies from competing with us. We will continue to attempt to patent our innovations, as appropriate, to help ensure a sustainable competitive advantage.
Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering health care product inventions, our ability to enforce our existing patents and to obtain and enforce patents that may issue from any pending or future patent applications is uncertain and involves complex legal, scientific and factual questions. To date, no consistent policy has emerged regarding the breadth of claims allowed in medical device patents. Thus, we cannot be sure that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents do issue, we cannot be sure that the claims of these patents will be held valid or enforceable by a court of law, will provide us with any significant protection against competing products, or will afford us a commercial advantage over competitive products. If, at some point in the future, one or more products resulting from our product candidates is approved for sale by the FDA and we do not have adequate intellectual property protection for those products, competitors could duplicate them for approval and sale in the United States without repeating the extensive testing required of us to obtain FDA approval.
If we are sued for infringing on third-party intellectual property rights, it will be costly and time-consuming, and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our product candidate depends on our ability to use, manufacture and sell our product candidate without infringing the patents or other proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the diabetes medical device area. There may be existing patents, unknown to us, on which our activities with our insulin pump candidate could infringe.
If a third party claims that our actions infringe on its patents or other proprietary rights, we could face a number of issues that could materially harm our competitive position, including, but not limited to:
| | ● | infringement and other intellectual property claims that, even if meritless, can be costly and time-consuming, delay the regulatory approval process and divert management’s attention from our core business operations; |
| | ● | an order that we pay substantial damages for infringement, including consequential damages for lost of profits or market share, if a court determines that our products or technologies infringe on a third party’s patent or other proprietary rights; |
| | ● | a court prohibiting us from selling or licensing our products or technologies unless the holder licenses the patent or other proprietary rights to us, which it is not required to do; and |
| | ● | even if a license is available from a holder, we may have to pay substantial royalties or grant cross-licenses to our patents or other proprietary rights. |
If any of these events occur, it could significantly harm our operations and financial condition and negatively affect our stock price.
If we are unable to protect the confidentiality of our proprietary information, the value of our technology and products could be adversely affected.
In addition to patented technology and technology for which patent protection is being sought, we rely on our unpatented technology, trade secrets and know-how. We generally seek to protect this information by confidentiality, non-disclosure and assignment of invention agreements with our officers, employees, contractors and other service providers and with parties with which we do business. These agreements may be breached, which breach may result in the misappropriation of such information, and we may not have adequate remedies for any such breach. We cannot be certain that the steps we have taken will prevent unauthorized use or reverse engineering of our technology.
Moreover, our trade secrets may be disclosed to or otherwise become known or be independently developed by competitors. To the extent that our officers, employees, contractors, other service providers, or other third parties with whom we do business use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. If, for any of the above reasons, our intellectual property is disclosed or misappropriated, it would harm our ability to protect our rights and have a material adverse effect on our business, financial condition, and results of operations.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to gain and maintain a competitive advantage. The following examples are illustrative:
| | ● | others may be able to make devices that are similar to our insulin pump but that are not covered by the claims of the patents that we own; |
| | ● | we or any collaborators might not have been the first to make the inventions covered by the issued patents or pending patent applications that we own; |
| | ● | we might not have been the first to file patent applications covering certain of our inventions; |
| | ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; |
| | ● | it is possible that our pending patent applications will not lead to issued patents; |
| | ● | issued patents that we own may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
| | ● | our competitors might conduct research and development activities in the U.S. and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; and |
| | ● | we may not develop additional proprietary technologies that are patentable. |
Healthcare reform and drug-pricing reform laws could adversely affect our product candidate and financial condition.
In the United States, there have been, and continue to be, a number of legislative initiatives to contain healthcare costs. In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (ACA), was enacted in the United States, which made a number of substantial changes in the way healthcare is financed by both governmental and private insurers. Among other ways in which it may affect our business, the ACA implemented payment system reforms, including a national pilot program on payment bundling to encourage hospitals, physicians, and other providers to improve the coordination, quality, and efficiency of certain healthcare services through bundled payment models and expanded the eligibility criteria for Medicaid programs. Since its enactment, there have been judicial, executive, and Congressional challenges to certain aspects of the ACA. It is unclear how the ACA and its implementation, as well as efforts to repeal or replace, or invalidate, the ACA, or portions thereof, will affect our insulin pump or our business. Additional legislative changes, regulatory changes, and judicial challenges related to the ACA remain possible. It is possible that the ACA, as currently enacted or as it may be amended in the future, and other healthcare reform measures that may be adopted in the future, could have an adverse effect on our industry generally and on our ability to commercialize our insulin pump and achieve profitability. We have assumed in all of our financial projections that there is not an increase in the reimbursement for our product through the pharmacy or durable medical equipment routes.
Drug pricing continues to be a subject of debate at the executive and legislative levels of U.S. government. The American Rescue Plan Act of 2021 eliminated the statutory cap on rebates that drug manufacturers pay to Medicaid beginning January 1, 2024. With the elimination of the rebate cap, manufacturers may be required to compensate states in an amount greater than what the state Medicaid programs pay for the drug. Additionally, the Inflation Reduction Act of 2022 contains substantial drug pricing reforms, including the establishment of a drug price negotiation program within the U.S. Department of Health and Human Services that would require manufacturers to charge a negotiated “maximum fair price” for certain selected drugs or pay an excise tax for noncompliance, the establishment of rebate payment requirements on manufacturers of certain drugs payable under Medicare Parts B and D to penalize price increases that outpace inflation, and requires manufacturers to provide discounts on Part D drugs. Substantial penalties can be assessed for noncompliance with the drug pricing provisions in the Inflation Reduction Act of 2022. The Inflation Reduction Act of 2022 could have the effect of reducing the prices we can charge and reimbursement we receive for our products, if approved, thereby reducing our profitability, and could have a material adverse effect on our financial condition, results of operations and growth prospects. The effect of Inflation Reduction Act of 2022 on our business and the pharmaceutical industry in general is not yet known.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We expect that additional federal, state and foreign healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in limited coverage and reimbursement and reduced demand for our products, once approved, or additional pricing pressures.
These and other healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any current product or future product candidate. Any reduction in reimbursement from Medicare or other government healthcare programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our products. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for drugs. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of any current or future product candidates, if any, may be. In addition, increased Congressional scrutiny of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Even if we are able to obtain all regulatory approvals and have completed all other steps needed to be taken to commercialize our insulin pump, if we or any contract manufacturers we select fails to comply with the FDA’s quality system regulations, the manufacturing and distribution of our product candidate could be interrupted, and our product sales and operating results could suffer.
We have established initial, low-volume manufacturing capability in our facility, and we have selected an initial, tier one contract manufacturer. We and the contract manufacturer of our insulin pump will be required to comply with the FDA’s quality system regulations, which impose a complex regulatory framework that covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of medical devices. The FDA enforces its quality system regulations through periodic unannounced inspections. We cannot assure you that, in the future, any manufacturing facilities owned by us or any contract manufacturer will pass any quality system inspection. In the event that our or any contract manufacturer’s facilities fails a quality system inspection, the manufacturing or distribution of our product candidate could be interrupted and our operations disrupted. Failure to take adequate and timely corrective action in response to an adverse quality system inspection could force a suspension or shutdown of any packaging and labeling operations or then manufacturing operations of any contract manufacturers, or a recall of our insulin pump. If any of these events were to occur, we at such time would not be able to provide our customers with the quantity of insulin pumps that they require on a timely basis, our reputation could be harmed and we could lose any customers we then have, any or all of which could have a material adverse effect on our business, financial condition and results of operations.
We may bring infringement claims or other legal proceedings against third parties, causing us to spend substantial resources on litigation and exposing our own intellectual property portfolio to challenge.
We may come to believe that third parties are infringing on our patents or other proprietary rights. To prevent infringement or unauthorized use, we may need to file infringement and/or misappropriation suits, which are very expensive and time-consuming, could result in meritorious counterclaims against us and would distract management’s attention. Also, in an infringement or misappropriation proceeding, a court may decide that one or more of our patents is invalid, unenforceable, or both, in which case third parties may be able to use our technology without paying license fees or royalties. Even if the validity of our patents is upheld, a court may refuse to stop the other party from using the technology at issue on the grounds that the other party’s activities are not covered by our patents. See “Business - Patents,” below.
We may become involved in disputes with our present or future contract partners over intellectual property ownership or other matters, which would have a significant effect on our business.
Inventions discovered in the course of performance of contracts with third parties or contractors may become jointly owned by such third-party contractors and us, in some cases, and the exclusive property of one of us, in other cases. Under some circumstances, it may be difficult to determine who owns a particular invention or whether it is jointly owned, and disputes could arise regarding ownership or use of those inventions or jointly developed improvements thereto. Other disputes may also arise relating to the performance or alleged breach of our agreements with third parties. Any disputes could be costly and time-consuming, and an unfavorable outcome could have a significant adverse effect on our business.
Assuming our insulin pump receives FDA clearance or approval, our insulin pump will still be subject to recalls, which would harm our reputation, business operations and financial results.
Even assuming we obtain FDA approval or clearance with regard to our insulin pump, the FDA has the authority to require the recall of our pump if we commence manufacturing of our insulin pump and we or any contract manufacturers we retain fail to comply with relevant regulations pertaining to manufacturing practices, labeling, advertising or promotional activities, or if new information is obtained concerning the safety or efficacy of the device. A government-mandated recall could occur if the FDA finds that there is a reasonable probability that our device would cause serious, adverse health consequences or death. A voluntary recall by us could occur as a result of manufacturing defects, labeling deficiencies, packaging defects or other failures to comply with applicable regulations. Any recall would divert management’s attention and financial resources and harm our reputation with customers. A recall involving our insulin pump would be particularly harmful to our business, financial condition and results of operations because it is currently our only product candidate.
Any disruption and/or instability in economic conditions and capital markets could adversely affect our ability to access the capital markets, and thus adversely affect our business and liquidity.
Negative economic conditions and instability or uncertainty in the financial markets could have a negative impact on our ability to access the capital markets, and thus have a negative impact on our then operations and liquidity. We face certain risks in the event of a sustained deterioration of financial market liquidity, as well as in the event of sustained deterioration in the liquidity, or failure, of our banking, cash management and custodial financial institutions. A general shortage of liquidity and credit combined with the substantial losses in worldwide equity markets could lead to an extended worldwide recession in the future. If such occurred, we would face significant challenges if conditions in the capital markets did not improve. Our ability to access the capital markets under such circumstances could be severely restricted at a time when we need to access such markets, which could have a negative impact on our business plans. Even if we are able to raise capital under such circumstances, it may not be at a price or on terms that are favorable to us. We cannot predict the occurrence of future disruptions or how long such negative conditions might continue.
Because our current insulin pump is still in the pre-clearance stage with the FDA, it does not have reimbursement and is not approved for insurance coverage. If in the future we are cleared for and are otherwise able to commercialize our insulin pump, but are unable to obtain adequate reimbursement or insurance coverage for such product candidate from third-party payors, we will be unable to generate significant revenue.
Because our current insulin pump is still in the pre-clearance stage with the FDA, it is not eligible for reimbursement and is not approved for insurance coverage. The future availability of insurance coverage and reimbursement for newly approved medical devices is highly uncertain. In the United States, patients using insulin pumps are generally reimbursed for all or part of the product cost by Medicare or other third-party payors. Any future commercial success of our insulin pump will be substantially dependent on whether third-party coverage and reimbursement is available for future customers. Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medical devices, and, as a result, they may not cover or provide adequate reimbursement for our insulin pump, assuming we are able to fully develop and obtain all regulatory approval to market it in the United States. In addition, in certain countries, no uniform policy of coverage and reimbursement for medical device products and services exists among third-party payors. Therefore, coverage and reimbursement for medical device products and services can differ significantly from payor to payor. In addition, payors continually review new technologies for possible coverage and can, without notice, deny coverage for these new products and procedures. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained or maintained, if obtained. Reimbursement systems in international markets vary significantly by country and by region within some countries, and reimbursement approvals must be obtained on a country-by-country basis. In many international markets, a product must be approved for reimbursement before it can be approved for sale in that country. Further, many international markets have government-managed healthcare systems that control reimbursement for new devices and procedures. Accordingly, unless government and other third-party payors provide coverage and reimbursement for our insulin pump, patients may not use it, which would cause investors to lose their entire investment.
Third parties might attempt to gain unauthorized access to our network or seek to compromise our insulin pump product.
Our business is dependent on the security and efficacy of our networks and computer and data management systems, and we rely on our internal computer networks for many of the systems we use to operate our business generally. From time to time, we may face attempts by others to gain unauthorized access through the Internet or otherwise or to introduce malicious software to our information technology systems. We or our products may be a target of computer hackers, organizations or malicious attackers who attempt to:
| ● | gain access to our network; |
| ● | steal proprietary information related to our business, products and employees; or |
From time to time, we may encounter attempts at gaining unauthorized access to our network, and we periodically run security checks. While we seek to detect and investigate unauthorized attempts and attacks against our network and products of which we become aware, and to prevent their recurrence where practicable through changes to our internal processes and tools and/or changes to our products, we remain potentially vulnerable to additional known or unknown threats. In addition to intentional security breaches, the integrity and confidentiality of Company and customer data and our intellectual property may be compromised as a result of human error, product defects, or technological failures. Different geographic markets may have different regulations regarding data protection, raising potential compliance risks. Further, retaliatory acts by foreign governments or terrorist organizations in response to policies of the United States government could include cyber attacks that could disrupt the economy more generally or that could also impact our operations directly or indirectly.
Any failure or perceived failure by us or our service providers to prevent information security breaches or other incidents or system disruptions, or any compromise of security that results in or is perceived or reported to result in unauthorized access to, or loss, theft, alteration, release or transfer of, our information, or any personal information, confidential information, or other data could result in loss or theft of proprietary or sensitive data and intellectual property, could harm our reputation and competitive position and could expose us to legal claims, regulatory investigations and proceedings, and fines, penalties, and other liability. Any such actual or perceived security breach, incident or system disruption could also divert the efforts of our personnel, and could require us to incur significant costs and operational consequences in connection with investigating, remediating, eliminating and putting in place additional tools, devices, policies, and other measures designed to prevent actual or perceived security breaches and other incidents and system disruptions, and in, for example, rebuilding internal systems, reduced inventory value, providing modifications to our products and services, defending against claims and litigation, responding to regulatory inquiries or actions, paying damages, or taking other remedial steps with respect to third parties. Moreover, we could be required or otherwise find it appropriate to expend significant capital and other resources to respond to, notify third parties of, and otherwise address the incident or breach and its root cause, and to notify individuals, regulatory authorities and others of security breaches involving certain types of data.
Further, we cannot assure that any limitations of liability provisions in our current or future contracts that may be applicable would be enforceable or adequate or would otherwise protect us from any liabilities or damages with respect to any particular claim relating to a security breach or other security-related matter. We also cannot be sure that any insurance coverage will continue to be available on acceptable terms or will be available in sufficient amounts to cover claims related to a security breach or incident, or that the insurer will not deny coverage as to any future claim. The successful assertion of claims against us that exceed available insurance coverage, or the occurrence of changes in our insurance policies, including premium increases or the imposition of large deductible or co-insurance requirements, could have a material adverse effect on our business, including our financial condition, operating results, and reputation.
We are subject to oversight by the SEC and other regulatory agencies. Investigations by those agencies could divert management’s focus and could have a material adverse effect on our reputation and financial condition.
We are subject to the regulation and oversight of the SEC and state regulatory agencies, in addition to the FDA. As a result, we may face legal or administrative proceedings by these agencies. We are unable to predict the effect of any investigations on our business, financial condition or reputation. In addition, publicity surrounding any investigation, even if ultimately resolved in our favor, could have a material adverse effect on our business.
We are a “smaller reporting company” and, as a result of the reduced disclosure and governance requirements applicable to smaller reporting companies, our common stock may be less attractive to investors.
We are a “smaller reporting company,” and are subject to lesser disclosure obligations in our SEC filings compared to other issuers. Specifically, “smaller reporting companies” are able to provide simplified executive compensation disclosures in their filings, are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting and have certain other decreased disclosure obligations in their SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. Decreased disclosures in our SEC filings due to our status as a “smaller reporting company” may make it harder for investors to analyze our operating results and financial prospects.
We do not expect any cash dividends to be paid on our shares of common stock for the foreseeable future.
We have never declared or paid a cash dividend and we do not anticipate declaring or paying dividends on our common stock for the foreseeable future. We expect to use future financing proceeds and earnings, if any, to fund operating expenses. Consequently, stockholders’ only opportunity to achieve a return on their investment is if the price of our stock appreciates and they sell their shares at a profit. We cannot assure stockholders of a positive return on their investment when they sell their shares or that stockholders will not lose the entire amount of their investment.
If the beneficial ownership of our common stock continues to be highly concentrated, it may prevent our stockholders from influencing significant corporate decisions.
As of March 31, 2024, our executive officers, directors and certain persons, who may be deemed affiliates, beneficially owned approximately 24% of our issued and outstanding common stock. Specifically, James Besser, our chief executive officer, and Morgan Frank, a member of our board of directors, were the beneficial owners of approximately 13% of our outstanding common stock. As a result, such persons may exercise substantial influence over the outcome of corporate actions requiring stockholder approval including, without limitation, the election of directors, certain mergers, consolidations and sales of all or substantially all of our assets or any other significant corporate transactions. Such persons may also vote against a change of control, even if such a change of control would benefit our other stockholders. Thus, investors in our common stock cannot reasonably expect to have any influence over the election of our directors or other matters submitted to a vote of our stockholders. Instead, our existing significant stockholders may exert a substantial influence on the election of our directors and any actions requiring or otherwise put to a stockholder vote, potentially in a manner that you do not support. The concentrated amount of control over our affairs held by a relatively few significant investors could serve to reduce the attractiveness or liquidity of our common stock, and thereby depress its trading price. Additionally, conflicts of interest may arise between these executive officers, directors and other affiliates, on the one hand, and us and our other stockholders, on the other hand. In resolving these conflicts of interests, these investors may favor their own interests and the interests of their affiliates, over the interests of our other stockholders, which could cause a material adverse effect on our business, prospects, financial condition and results of operations.
Future sales of our securities could adversely affect the market price of our common stock and our future capital-raising activities could involve the issuance of equity securities, which would dilute your investment and could result in a decline in the trading price of our common stock.
We may sell securities in the public or private equity markets at prices per share below the current market price of our common stock, even if we do not have an immediate need for additional capital at that time. Sales of substantial amounts of shares of our common stock, or the perception that such sales could occur, could adversely affect the prevailing market price of our shares and our ability to raise capital. We may issue additional shares of common stock in future financing transactions or as incentive compensation for our executive management and other key personnel, consultants and advisors. Issuing any equity securities would be dilutive to the equity interests represented by our then-outstanding shares of common stock. Moreover, sales of substantial amounts of shares in the public market, or the perception that such sales could occur, may adversely affect the prevailing market price of our common stock and make it more difficult for us to raise additional capital. Such resulting significant downward pressure on the price of our common stock could also encourage short sales by third parties. Such an event could place further downward pressure on the price of our common stock.
Our articles of incorporation allow for our board of directors to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our board of directors has the authority to fix and determine the relative rights and preferences of preferred stock. Currently, our board of directors has the authority to designate and issue up to 5,000,000 shares of our preferred stock without further stockholder approval. In the future, our board of directors could authorize the issuance of one or more series of preferred stock that would grant to holders, among other rights, the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of our preferred shares acquired by such persons, together with a premium, prior to the redemption of our common stock. In addition, our board of directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders.
If we fail to establish and maintain an effective system of internal controls, we may not be able to report our financial results accurately or prevent fraud. Any inability to report and file our financial results accurately and timely could harm our reputation and adversely affect the trading price of our common stock.
Effective internal controls are necessary for us to provide reliable financial reports and prevent fraud. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed. If we are unable to maintain effective internal controls, we may not have adequate, accurate or timely financial information, and we may be unable to meet our reporting obligations as a public company, including the requirements of the Sarbanes-Oxley Act of 2002 (the Sarbanes-Oxley Act). In addition, we may be unable to accurately report our financial results in future periods or report them within the timeframes required by the requirements of the SEC or the Sarbanes-Oxley Act. Failure to comply with the Sarbanes-Oxley Act, when and as applicable, could also potentially subject us to sanctions or investigations by the SEC or other regulatory authorities. Any failure to maintain or implement required new or improved controls, or any difficulties we encounter in their implementation, could result in identification of additional material weaknesses or significant deficiencies, cause us to fail to meet our reporting obligations or result in material misstatements in our financial statements.
Furthermore, Section 404 of the Sarbanes-Oxley Act and related regulations require our management to evaluate the effectiveness of our internal control over financial reporting as of the end of each fiscal year. Based on its evaluation, our management concluded that our internal controls over financial reporting were effective as of March 31, 2024. We cannot provide assurance that, in the future, a material weakness or significant deficiency will not exist or otherwise be discovered. If that were to happen, it could harm our operating results and cause stockholders to lose confidence in our reported financial information. Any such loss of confidence would have a negative effect on the trading price of our securities.
Sustained inflation could have a material adverse effect on our business, financial condition, results of operations and liquidity.
Inflation rates in the United States have remained high and may continue to rise. Inflation over the last several months has led us to experience higher costs, including, among others, labor and transportation. Some of our suppliers have raised their prices and may continue to raise prices, and, assuming we achieve FDA clearance and commence commercialization of our product, in the future, we may not be able to make corresponding price increases to obtain adequate gross margins and achieve profitability. If inflation rates continue to rise or remain elevated for a sustained period of time, they could have a material adverse effect on our business, financial condition, results of operations and liquidity.
Our board of directors is able to adopt recapitalizations through forward or reverse splits of our outstanding shares of common stock without stockholder approval.
Pursuant to our amended and restated articles of incorporation, our board of directors has the power, without obtaining stockholder approval, to effectuate recapitalizations of us through forward or reverse splits of our outstanding common stock. As a result of such provision, our board of directors can implement recapitalizations of us by effectuating a forward or reverse stock split of our outstanding common stock, which would increase or decrease each of our stockholder’s number of shares owned, and our stockholders will have no right to approve or disapprove any such action even if such actions have a material adverse effect on them.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None
Item 1C. CYBERSECURITY.
We believe a robust and proactive approach to cybersecurity risks and threats is essential to achieving our strategic business objectives and protecting our business. We may face a wide range of cybersecurity threats, such as ransomware and denial-of-service attacks. Our customers, suppliers and other business partners may also face similar cybersecurity threats, and a cybersecurity incident impacting us or any of these third parties could have a material adverse effect on our business and results of operations. Due to the risks that cybersecurity threats can pose to our business, we intend to continually evaluate best practices and methods, including cyber defense systems and training programs, to protect our business from a wide range of potential threats.
We continue to evaluate our cybersecurity control processes and procedures to address the evolving cybersecurity risks that we may face in an increasingly technically capable environment. We are implementing policies to educate and provide guidance to our personnel, including awareness programs and other related cybersecurity best practices. We plan to conduct technical risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our business practices that may affect information systems that are vulnerable to such cybersecurity threats. We also plan to conduct programmatic risk assessments, including identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks. Following these risk assessments, we will evaluate: i) whether and how to implement, and maintain reasonable safeguards to minimize identified risks, ii) how to reasonably address any identified gaps in existing safeguards; and how to regularly monitor the effectiveness of our safeguards. As we are a small pre-revenue company, we currently outsource our information technology (IT) functions to a third party. Working with the outsourced IT firm, our president will manage the risk assessment and mitigation process. Third parties will play an important role in our cybersecurity program. We intend to engage third-party service providers to conduct evaluations of our security controls, including penetration testing and consulting on best practices. The third-party services include testing both the design and operational effectiveness of security controls. This dependence exposes us, along with others who use such service providers, to the impact of a cyber-attack on their service providers. It is possible for a cyber-attack at a third-party service provider to have a significant financial, operational, or reputational impact to us. To reduce the effective impact to us of a cyber-attack on a third-party service provider, we intend to monitor the risks associated with our service providers through periodic review of these providers’ cybersecurity programs.
Our board of directors, through its audit committee, oversees our processes for identifying and mitigating risks, including cybersecurity risks. Management will periodically brief the audit committee and/or the board of directors on our cybersecurity and information security policies and plans. Our board of directors will be apprised of cybersecurity incidents deemed to have a moderate or higher business impact, and we will provide updates on management’s incident response plan for addressing and mitigating any impacts and risks associated with such an incident. We intend to develop a formal incident response plan, which sets forth the steps to be followed from incident detection and assessment to mitigation, recovery and notification and reporting within our organization and to our board of directors.
For additional information regarding whether any risks from cybersecurity threats have materially affected or are reasonably likely to materially affect us, including our business strategy, results of operations, or financial condition, please refer to Item 1A, “Risk Factors,” in this Report, including the risk factor entitled “Third parties might attempt to gain unauthorized access to our network or seek to compromise our insulin pump product.”
ITEM 2. PROPERTIES
Our principal administrative, operations, and research and development functions are located in a leased facility in San Diego, California. We currently occupy approximately 24,000 square feet of space in the San Diego facility, and the lease extends through January 2027. Under the lease, in addition to the minimum lease payments, we are responsible for property taxes, insurance and certain other operating costs. We believe that our existing facility is adequate to meet our current needs.
ITEM 3. LEGAL PROCEEDINGS
We are not a party to any material legal proceeding that we believe is likely to have a material adverse effect on our consolidated financial position or results of operations. From time to time, we may be subject to legal proceedings and claims in the ordinary course of business. These claims, even if not meritorious, could result in the expenditure of significant financial resources and diversion of management efforts. to any pending legal proceeding. To the knowledge of our management, no federal, state or local governmental agency is presently contemplating any proceeding against us. No director, executive officer or affiliate of ours or owner of record or beneficially of more than five percent of our common stock is a party adverse to us or has a material interest adverse to us in any proceeding.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information for Common Stock
Our common stock is currently listed on the Nasdaq Capital Market under the symbol “MODD.”
Authorized Capital
The Company is authorized by its Certificate of Incorporation to issue an aggregate of up to 5,000,000 shares of preferred stock, $0.001 par value per share, and 100,000,000 shares of common stock, $0.001 par value per share. As of March 31, 2024, zero and 32,463,670 shares of preferred stock and common stock, respectively, were issued and outstanding.
Holders of Record
As of March 31, 2024, we had 69 stockholders of record. The actual number of stockholders is greater than this number of stockholders of record and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees. This number of stockholders of record also does not include stockholders whose shares may be held in trust by other entities.
Securities Authorized for Issuance under Equity Compensation Plan
For information regarding securities authorized for issuance under equity compensation plans, please refer to Item 12, Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Dividend Policy
We have never declared or paid any cash dividend on our capital stock. We do not anticipate paying any cash dividends in the foreseeable future and we intend to retain all of our earnings, if any, to finance our growth and operations and to fund the expansion of our business. Payment of any dividends will be made in the discretion of our board of directors, after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs and plans for expansion. Any dividends that may be declared or paid on our common stock, must also be paid in the same consideration or manner, as the case may be, on our shares of preferred stock, if any.
Recent Sales of Unregistered Securities
Set forth below is information regarding securities issued by us within the past two years that were not registered under the Securities Act. Also included is the consideration, if any, received by us for such issuances, and information relating to the section of the Securities Act, or rule of the Securities and Exchange Commission, under which exemption from registration was claimed.
Director Compensation
On each of March 31, 2024, December 30, 2023, and June 30, 2023 we issued 6,375 shares of our common stock to four of our non-employee directors in accordance with our Outside Director Compensation Plan (the “Director Plan”). On September 30, 2023, we issued 6,265 shares of our common stock to four of our non-employee directors in accordance with the Director Plan. On March 31, 2023, December 30, 2022 and September 30, 2022, we issued 6,375 shares of our common stock to four of our non-employee directors in accordance with the Director Plan. On August 8, 2022, we issued 5,000 shares of our common stock to two of our non-employee directors in accordance with the Director Plan. On June 30, 2022, we issued 2,664 shares of our common stock to two of our non-employee directors in accordance with the Director Plan.
Service Providers
In August 2023, we issued 1,429 shares of our common stock to a service provider. In March 2023, we issued 10,000 shares of our common stock to a service provider. In March 2023, we issued 478 shares of our common stock to a service provider. In February 2023, we issued 438 shares of our common stock to a service provider. In May 2022, we issued 348 shares of our common stock to a service provider.
2022 Placement
In May 2022, we issued warrants in a private placement to purchase an aggregate of 1,438,202 shares of common stock at an exercise price of $6.60 per share. The warrants were exercisable six months from the date of issuance and have a five-year term from the date the warrants become exercisable.
ITEM 6. RESERVED
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion of our financial condition and results of operations should be read in conjunction with the financial statements and related notes included in this Annual Report on Form 10-K, or the Report. Management’s Discussion and Analysis of Financial Condition and Results of Operations may contain statements that are forward-looking. These statements are based on current expectations and assumptions that are subject to risk, uncertainties and other factors. These statements are often identified by the use of words such as “may,” “will,” “expect,” “believe,” “anticipate,” “intend,” “could,” “estimate,” or “continue,” and similar expressions or variations. Actual results could differ materially because of the factors discussed in Part I, Item 1A, These risks and uncertainties may cause actual results to differ materially from those discussed in the forward-looking statements.
Our fiscal year ends on March 31 of each calendar year. Each reference to a fiscal year in this Report, refers to the fiscal year ended March 31 of the calendar year indicated (for example, fiscal 2024 refers to the fiscal year ending March 31, 2024). Unless the context requires otherwise, references to “we,” “us,” “our,” and the “Company” refer to Modular Medical, Inc. and its consolidated subsidiary.
Company Overview
We are a pre-revenue medical device company focused on the design, development and commercialization of innovative insulin pumps using modernized technology to increase pump adoption in the diabetes marketplace. Through the creation of a novel two-part patch pump, our initial product, the MODD1, we seek to fundamentally alter the trade-offs between cost and complexity and access to the higher standards of care that presently-available insulin pumps provide. By simplifying and streamlining the user experience from introduction, prescription, reimbursement, training and day-to-day use, we seek to expand the wearable insulin delivery device market beyond the highly motivated “super users” and expand the category into the mass market. The product seeks to serve both the type 1 and the rapidly growing, especially in terms of device adoption, type 2 diabetes markets. In January 2024, we submitted a 510(k) premarket notification to the United States Food and Drug Administration (“FDA”) for our MODD1 insulin pump. In March 2024, we received comments from the FDA, and we are in the process of responding to those comments.
In February 2024, we completed a firm commitment underwritten offering and issued and sold to the underwriter 9,090,910 shares of our common stock at a price of $1.10 per share (the 2024 Offering). We received aggregate proceeds of approximately $10,000,000 before deducting underwriting discounts and commissions and other offering expenses. We also granted the underwriter a 30-day option to purchase up to an additional 1,321,989 shares of common stock to cover over allotments, if any. In March 2024, the underwriter exercised this option in full and purchased the additional securities for additional aggregate proceeds to us of approximately $1,454,000, before deducting underwriting discounts and commissions and other offering expenses.
Historically, we have financed our operations principally through private placements and public offerings of our common stock and sales of convertible promissory notes. Based on our current operating plan, substantial doubt about our ability to continue as a going concern for a period of at least one year from the date that the financial statements included in Item 8 of this Report are issued exists. Our ability to continue as a going concern depends on our ability to raise additional capital, through the sale of equity or debt securities, to support our future operations. If we are unable to secure additional capital, we will be required to curtail our research and development initiatives and take additional measures to reduce costs. We have provided additional disclosure in Note 1 to the consolidated financial statements in Item 1 of this Report and under Liquidity below.
Economic Disruptions
The global outbreak of the coronavirus disease 2019 (COVID-19) was declared a pandemic by the World Health Organization and a national emergency by the U.S. government in March 2020. This negatively affected the U.S. and global economy, disrupted global supply chains, significantly restricted travel and transportation, resulted in mandated closures and orders to “shelter-in- place” and created significant disruption of the financial markets. While the U.S. national emergency expired in May 2023 and substantially all closures and “shelter-in-place” orders have ended, there can be no assurance that the COVID-19 pandemic will not impact our operational and financial performance in the future, as the duration and spread of the pandemic and related actions taken by U.S. and foreign government agencies to prevent disease spread are uncertain, out of our control, and cannot be predicted.
Wars and acts of terrorism have led to further economic disruptions. Mounting inflationary cost pressures and recessionary fears have negatively impacted the global economy. Since mid-2022, at times, the U.S. Federal Reserve has addressed elevated inflation by increasing interest rates, as inflation remains elevated. While we were recently able to access the capital markets, in the future, we may be unable to access the capital markets, and additional capital may only be available to us on terms that could be significantly detrimental to our existing stockholders and to our business.
For additional information on risks that could impact our future results, please refer to “Risk Factors” in Part I, Item 1A of this Report.
Results of Operations
The following discussion should be read in conjunction with our consolidated financial statements and related notes included elsewhere in this Report.
Research and Development
| | | Years ended March 31, | | | Year-over-Year Change | |
| | | 2024 | | | 2023 | | | 2024 to 2023 | |
| Research and development | | $ | 12,880 | | | $ | 9,062 | | | $ | 3,818 | | | | 42.1 | % |
Our research and development, or R&D, expenses include personnel, consulting, testing, materials and supplies, depreciation and amortization and other operational costs associated with the pre-FDA clearance production of our insulin pump product. We expense R&D costs as they are incurred.
R&D expenses increased in fiscal 2024 compared with fiscal 2023 primarily due to increases in engineering and operations personnel costs of $1.6 million, consulting costs of $0.8 million, stock-based compensation expenses of $0.5 million and materials, supplies expenditures of $0.5 million and depreciation and amortization of $0.4 million. The increase in personnel costs was attributable to increased average headcount year over year, salary increases effected during fiscal 2024, payment of a bonus implemented in fiscal 2024 related to our 510(k) submission and higher payroll taxes. Our R&D employee headcount increased to 36 at March 31, 2024 from 34 at March 31, 2023. The increase in consulting costs was primarily driven by the utilization of consultants and outside testing and other firms in support of our FDA submission in January 2024. R&D expenses included stock-based compensation expenses of approximately $1.9 million and $1.4 million for fiscal 2024 and fiscal 2023, respectively. The increase in stock-based compensation costs was primarily attributable to the granting of stock options under our bonus program for our FDA submission; these options were granted in October 2023 and expensed over an expected term of four months. We expect R&D expenses will increase in fiscal 2025, as we continue to engage third parties to support our responses to the FDA on our MODD1 510(k) submission, hire additional engineering, quality assurance, and operations personnel, bring-up our manufacturing process at our medical device contract manufacturer and commence the commercialization of our product in late fiscal 2025.
General and Administrative
| | | Year ended March 31, | | | Year-over-Year Change | |
| | | 2024 | | | 2023 | | | 2024 to 2023 | |
| General and administrative | | $ | 4,649 | | | $ | 4,816 | | | $ | (167 | ) | | | (3.5 | )% |
General and administrative, or G&A, expenses consist primarily of personnel and related overhead costs for marketing, finance, human resources, facilities and general management.
G&A expenses decreased in fiscal 2024 compared with fiscal 2023 primarily as a result of reductions in stock-based compensation expense of $0.5 million, consulting fees of $0.4 million and reduced personnel costs of $0.2 million. The decreases were partially offset by increased professional services expenses of $0.4 million, which was primarily attributable to higher investor relations and financing-related costs, facilities-related expenses of $0.3 million due to our move to a larger facility in the fourth quarter of fiscal 2023, marketing expense of $0.2 million for a participant study for our product and costs incurred for initial trade show activities. G&A expenses included stock-based compensation expenses of approximately $0.8 million and $1.3 million for fiscal 2024 and fiscal 2023, respectively. We expect G&A expenses to increase in fiscal 2025, as we expect to increase headcount, as we expand our organization and implement systems to support our anticipated growth and prepare for the commercialization of our product in late fiscal 2025.
Liquidity and Capital Resources; Changes in Financial Condition
Going Concern
As a development-stage enterprise, we do not currently have revenues to generate cash flows to cover operating expenses. Since our inception, we have incurred operating losses and negative cash flows in each year due to costs incurred in connection with R&D activities and G&A expenses associated with our operations. For the years ended March 31, 2024 and 2023, we incurred net losses of approximately $17.5 million and $13.9 million, respectively. At March 31, 2024, we had a cash balance of $9.2 million and an accumulated deficit of approximately $66 million. When considered with our current operating plan, these conditions raise substantial doubt about our ability to continue as a going concern for a period of at least one year from the date that the financial statements included in Item 8 of this Report are issued. Our financial statements do not include adjustments to the amounts and classification of assets and liabilities that may be necessary should we be unable to continue as a going concern. Our operating needs include the planned costs to operate our business, including amounts required to fund continued research and development activities, working capital and capital expenditures. Our ability to continue as a going concern depends on our ability to raise additional capital, through the sale of equity or debt securities to support our future operations. Recently, during the three months ended March 31 2024, we completed the 2024 Offering for net proceeds of approximately $10.3 million. On November 22, 2023, we entered into a Sales Agreement (the “ATM Agreement”) with Leerink Partners LLC (“Leerink”) under which we may offer and sell, from time to time at our sole discretion, shares of our common stock, for aggregate gross proceeds of up to $6.5 million (subject to availability on our shelf registration statement) through an “at the market offering” program under which Leerink will act as sales agent or principal. In January 2024, we sold 153,879 shares of common stock for net proceeds of approximately $0.3 million under the ATM Agreement. We suspended sales under the ATM Agreement due to the 2024 Offering, and we may resume sales under the ATM during fiscal 2025. In addition, from December 2023 to April 2024, we received a total of approximately $0.9 million of proceeds from the exercise of common stock purchase warrants issued in a public offering we completed in May 2023. Our future capital requirements and the adequacy of our available funds will depend on many factors, including, without limitation, our ability to successfully commercialize our product, competing technological and market developments, and the need to enter into collaborations with other companies or acquire other companies or technologies to enhance or complement our product offerings. If we are unable to secure additional capital timely, we may be required to curtail R&D initiatives, reduce headcount and take additional measures to reduce costs in order to conserve our cash.
Purchase Obligations
Our primary purchase obligations include purchase orders for machinery and equipment. At March 31, 2024, we had outstanding purchase orders for machinery and equipment and related expenditures of approximately $1.1 million. In December 2023, we signed a device integration agreement with a provider of connected-care and remote monitoring diabetes technology solutions. As of March 31, 2024, we had a remaining obligation under the device integration agreement of approximately $400,000 over three years for technology license fees.
Liquidity
In fiscal 2024, we used approximately $14.0 million in operating activities, which primarily resulted from our net loss of approximately $17.5 million less changes to operating assets and liabilities of approximately $0.4 million, and as adjusted for non-cash charges and gains, which included approximately $2.7 million of stock-based compensation expenses, depreciation and amortization expenses of approximately $0.4 million, and other immaterial adjustments. The changes in operating assets and liabilities primarily related to the timing of payments to vendors. In fiscal 2023, we used approximately $11.0 million in operating activities, which primarily resulted from our net loss of approximately $13.9 million plus changes to operating assets and liabilities of approximately $0.2 million, as adjusted for non-cash charges and gains, which included stock-based compensation expenses of approximately $2.7 million, approximately $0.2 million for issuance of shares of our common stock in exchange for services, depreciation and amortization expense of approximately $0.2 million and other immaterial adjustments. The changes in operating assets and liabilities primarily related to the timing of payments to vendors.
For fiscal 2024 and fiscal 2023, cash used in investing activities of approximately $1.7 million and $1.6 million, respectively, was for the purchase of property and equipment.
Cash provided by financing activities for fiscal 2024 totaled approximately $21.1 million and was primarily attributable to proceeds of approximately $20.1 million from the sale of shares of common stock in a registered direct offering and issuance of warrants to purchase common stock in private placements that closed in May 2023 and February 2024, net of underwriter fees and issuance costs, proceeds of approximately $0.7 million for the exercise of common stock purchase warrants and proceeds of approximately $0.3 from the sale of shares under the ATM agreement. Cash provided by financing activities for fiscal 2023 totaled approximately $7.4 million and was attributable to approximately $7.4 million of net proceeds from a registered direct offering of our common stock and common stock purchase warrants in May 2022, net of placement agent fees and issuance costs.
Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in conformity with accounting principles generally accepted in the United States of America (GAAP). Note 1 to the consolidated financial statements in Item 8 of this Report describes the significant accounting policies and methods used in the preparation of our consolidated financial statements. We have identified the accounting policies below as some of the more critical to our business and the understanding of our results of operations. These policies may involve estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. Although we believe our judgments and estimates are appropriate, actual future results may differ from our estimates, and if different assumptions or conditions were to prevail, the results could be materially different from our reported results.
Use of estimates
The preparation of financial statements in conformity with GAAP requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Estimates may include those pertaining to accruals, stock-based compensation and income taxes. Actual results could materially differ from those estimates.
Stock-based compensation
We periodically issue stock options, restricted stock units and stock awards to employees and non-employees. We account for such awards based on Financial Accounting Standards Board Accounting Standards Codification (ASC) 718, whereby the value of the award is measured on the date of grant and recognized as compensation expense on a straight-line basis over the requisite service period, usually the vesting period. With respect to performance-based awards, we assess the probability of achieving the requisite performance criteria before recognizing compensation expense. We estimate the fair value of stock options on the date of grant using the Black-Scholes-Merton Option Pricing (Black Scholes) model which uses certain assumptions related to risk-free interest rates, expected volatility, expected life of the options, and future dividends. Compensation expense is recorded based upon the value derived from the Black-Scholes model. The assumptions used in the Black-Scholes model could materially affect compensation expense recorded in future periods.
Income taxes
We determine deferred tax assets and liabilities based upon the differences between the financial statement and tax bases of our assets and liabilities using tax rates in effect for the year in which we expect the differences to affect taxable income. A valuation allowance is established for any deferred tax assets for which it is more likely than not that all or a portion of the deferred tax assets will not be realized. Based on the available information and other factors, management believes it is more likely than not that our federal and state net deferred tax assets will not be fully realized, and we have recorded a full valuation allowance.
We account for uncertain tax positions in accordance with ASC Topic 740, Income Taxes. When tax returns are filed, it is likely that some positions taken would be sustained upon examination by the taxing authorities, while others are subject to uncertainty about the merits of the position taken or the amount of the position that would be ultimately sustained. The benefit of a tax position is recognized in the consolidated financial statements in the period during which, based on all available evidence, management believes it is more likely than not that the position will be sustained upon examination, including the resolution of appeals or litigation processes, if any. Tax positions taken are not offset or aggregated with other positions. Tax positions that meet the more-likely-than-not recognition threshold are measured as the largest amount of tax benefit that is more than 50 percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefits associated with tax positions taken that exceeds the amount measured as described above is reflected as a liability for unrecognized tax benefits in the accompanying consolidated balance sheets along with any associated interest and penalties that would be payable to the taxing authorities upon examination. Interest associated with unrecognized tax benefits is classified as interest expense and penalties are classified in general and administrative expenses in the consolidated statements of operations.
Leases
We account for our leases under ASC 842, Leases (ASC 842), and related ASUs, which provide supplementary guidance and clarifications. Under ASC 842, all significant lease arrangements are generally recognized at lease commencement. Operating lease right-of-use (ROU) assets and lease liabilities are recognized at the commencement date. A ROU asset and corresponding lease liability are not recorded for leases with an initial term of 12 months or less (short-term leases), and we recognize lease expense for these leases as incurred over the lease term.
ROU assets represent our right to use an underlying asset during the reasonably certain lease terms, and lease liabilities represent our obligation to make lease payments arising from the lease. Our lease terms may include options to extend or terminate the lease when it is reasonably certain that we will exercise that option. Operating lease ROU assets and liabilities are recognized at the lease commencement date based on the present value of lease payments over the lease term. We use our incremental borrowing rate, based on the information available at commencement date in determining the present value of lease payments. The operating lease ROU asset also includes any lease payments related to initial direct cost and prepayments and excludes lease incentives. Lease expense is recognized on a straight-line basis over the lease term.
Off-Balance Sheet Arrangements
We do not maintain any off-balance sheet arrangements or obligations that are reasonably likely to have a material current or future effect on our financial condition, results of operations, liquidity or capital resources.
Contractual Obligations
As a “smaller reporting company,” as defined by Item 10 of Regulation S-K, we are not required to provide the information requested by paragraph (a)(5) of this Item.
Recent Accounting Pronouncements
See Note 1 to the consolidated financial statements in Item 8 of this Report for a full description of relevant recent accounting pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8: FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - OPEN
To the Audit Committee and
Stockholders of Modular Medical, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Modular Medical, Inc. (the “Company”) as of March 31, 2024 and 2023, and the related consolidated statements of operations, stockholders’ equity, and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of March 31, 2024 and 2023, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Substantial Doubt about the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared to assume the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has incurred losses from operations and needs to raise additional funds to meet its obligations and sustain its future operations until profitability is achieved. These circumstances raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Going Concern
As described further in Note 1, the Company has incurred losses since inception, and expects to continue to incur operating losses for the foreseeable future and incur cash outflows from operations as it continues to invest in the development and subsequent commercialization of its product. The Company expects that its research and development and general and administrative expenses will continue to increase, and, as a result, the Company will need to generate significant product revenues to achieve profitability. These circumstances raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that these consolidated financial statements are issued.
We identified management’s assessment of the Company’s ability to continue as a going concern as a critical audit matter due to the inherent complexities and uncertainties related to the Company’s projections of operations.
The primary procedures we performed to address this critical audit matter included:
| - | We evaluated the reasonableness of key assumptions underlying management’s conclusion. |
| - | We evaluated that the disclosures included in the Form 10-K were complete and accurate and in accordance with accounting principles generally accepted in the United States of America. |
| - | We evaluated the impact of the Company’s existing financing arrangements and future capital needs over the next 12 months on its ability to continue as a going concern. |
Stock-Based Compensation
As discussed in Note 5, during the year ended March 31, 2024, the Company granted options to purchase shares of its common stock to employees, directors and consultants. Management is required to analyze the fair value of each option granted and amortize it over its vesting period.
We identified the valuation of stock-based compensation as a critical audit matter due to the significant judgments made by management when developing underlying assumptions regarding the fair value of the options.
The primary procedures we performed to address this critical audit matter included:
| - | We gained an understanding of Company’s processes and controls in place for determining the fair value of each granted option. |
| - | We evaluated the option price model management selected to determine the fair value, and analyzed the underlying data and assumptions used in the calculations. |
| - | We also recalculated the fair value of each option granted. |
/s/ Farber Hass Hurley LLP
PCAOB Firm ID 223
We have served as the Company’s auditor since 2018.
Chatsworth, California
June 21, 2024
Modular Medical, Inc.
Consolidated Balance Sheets
(In thousands, except par value)
| | | March 31, | |
| | | 2024 | | | 2023 | |
| ASSETS | | | | | | |
| CURRENT ASSETS | | | | | | |
| Cash and cash equivalents | | $ | 9,232 | | | $ | 3,799 | |
| Prepaid expenses and other | | | 465 | | | | 147 | |
| Security deposit | | | — | | | | 100 | |
| TOTAL CURRENT ASSETS | | | 9,697 | | | | 4,046 | |
| Property and equipment, net | | | 2,975 | | | | 1,721 | |
| Right of use assets, net | | | 1,135 | | | | 1,478 | |
| TOTAL NON-CURRENT ASSETS | | | 4,110 | | | | 3,199 | |
| TOTAL ASSETS | | $ | 13,807 | | | $ | 7,245 | |
| | | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
| | | | | | | | | |
| CURRENT LIABILITIES | | | | | | | | |
| Accounts payable | | $ | 802 | | | $ | 285 | |
| Accrued expenses | | | 280 | | | | 339 | |
| Short-term lease liabilities | | | 373 | | | | 355 | |
| TOTAL CURRENT LIABILITIES | | | 1,455 | | | | 979 | |
| Long-term lease liabilities | | | 817 | | | | 1,190 | |
| TOTAL LIABILITIES | | | 2,272 | | | | 2,169 | |
| | | | | | | | | |
| Commitments and Contingencies (Note 8) | | | | | | | | |
| | | | | | | | | |
| STOCKHOLDERS’ EQUITY | | | | | | | | |
| Preferred Stock, $0.001 par value, 5,000 shares authorized, none issued and outstanding | | | — | | | | — | |
| Common Stock, $0.001 par value, 100,000 and 50,000 shares authorized as of March 31, 2024 and 2023, respectively; 32,464 and 10,949 shares issued and outstanding as of March 31, 2024 and 2023, respectively | | | 32 | | | | 11 | |
| Additional paid-in capital | | | 77,432 | | | | 53,524 | |
| Accumulated deficit | | | (65,929 | ) | | | (48,459 | ) |
| TOTAL STOCKHOLDERS’ EQUITY | | | 11,535 | | | | 5,076 | |
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | | $ | 13,807 | | | $ | 7,245 | |
The accompanying notes are an integral part of these audited consolidated financial statements.
Modular Medical, Inc.
Consolidated Statements of Operations
(In thousands, except per-share data)
| | | Year Ended
March 31, | |
| | | 2024 | | | 2023 | |
| Operating expenses | | | | | | |
| Research and development | | $ | 12,880 | | | $ | 9,062 | |
| General and administrative | | | 4,649 | | | | 4,816 | |
| Total operating expenses | | | 17,529 | | | | 13,878 | |
| Loss from operations | | | (17,529 | ) | | | (13,878 | ) |
| Other income | | | 61 | | | | 1 | |
| | | | | | | | | |
| Loss before income taxes | | | (17,468 | ) | | | (13,877 | ) |
| Provision for income taxes | | | 2 | | | | 2 | |
| | | | | | | | | |
| Net loss | | $ | (17,470 | ) | | $ | (13,879 | ) |
| Net loss per share | | | | | | | | |
| Basic and diluted | | $ | (0.78 | ) | | $ | (1.15 | ) |
| Shares used in computing net loss per share | | | | | | | | |
| Basic and diluted | | | 22,377 | | | | 12,103 | |
The accompanying notes are an integral part of these audited consolidated financial statements.
Modular Medical, Inc.
Consolidated Statements of Stockholders’ Equity
(In thousands)
| | | | | | | | | Additional | | | | | | | |
| | | Common Stock | | | Paid-In | | | Accumulated | | | Stockholders’ | |
| | | Shares | | | Amount | | | Capital | | | Deficit | | | Equity | |
| Balance as of March 31, 2022 | | | 10,462 | | | $ | 11 | | | $ | 43,406 | | | $ | (34,580 | ) | | $ | 8,837 | |
| Issuance of common stock in registered direct offering, net of fees and issuance costs | | | 449 | | | | — | | | | 7,372 | | | | — | | | | 7,372 | |
| Shares issued for services | | | 11 | | | | — | | | | 22 | | | | — | | | | 22 | |
| Issuances under equity incentive plan | | | 27 | | | | — | | | | 86 | | | | — | | | | 86 | |
| Stock-based compensation | | | — | | | | — | | | | 2,638 | | | | — | | | | 2,638 | |
| Net loss | | | — | | | | — | | | | — | | | | (13,879 | ) | | | (13,879 | ) |
| Balance as of March 31, 2023 | | | 10,949 | | | $ | 11 | | | $ | 53,524 | | | $ | (48,459 | ) | | $ | 5,076 | |
| Issuance of common stock in public offerings, net of fees and issuance costs | | | 20,552 | | | | 20 | | | | 20,045 | | | | — | | | | 20,065 | |
| At-the-market sales of stock, net | | | 154 | | | | — | | | | 278 | | | | — | | | | 278 | |
| Exercise of warrants | | | 719 | | | | 1 | | | | 883 | | | | — | | | | 884 | |
| Shares issued for services | | | 2 | | | | — | | | | 1 | | | | — | | | | 1 | |
| Issuances under equity incentive plan | | | 88 | | | | — | | | | 37 | | | | — | | | | 37 | |
| Stock-based compensation | | | — | | | | — | | | | 2,664 | | | | — | | | | 2,664 | |
| Net loss | | | — | | | | — | | | | — | | | | (17,470 | ) | | | (17,470 | ) |
| Balance as of March 31, 2024 | | | 32,464 | | | $ | 32 | | | $ | 77,432 | | | $ | (65,929 | ) | | $ | 11,535 | |
The accompanying notes are an integral part of these audited consolidated financial statements.
Modular Medical, Inc.
Consolidated Statements of Cash Flows
(In thousands)
| | | Year ended March 31, | |
| | | 2024 | | | 2023 | |
| Cash Flows from operating activities | | | | | | |
| Net loss | | $ | (17,470 | ) | | $ | (13,879 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Stock-based compensation expense | | | 2,701 | | | | 2,724 | |
| Loss on asset disposal | | | 21 | | | | — | |
| Depreciation and amortization | | | 426 | | | | 152 | |
| Shares issued for services | | | 19 | | | | 203 | |
| Changes in assets and liabilities: | | | | | | | | |
| Prepaid expenses and other assets | | | (94 | ) | | | (14 | ) |
| Lease right-of-use assets | | | 342 | | | | 203 | |
| Accounts payable and accrued expenses | | | 458 | | | | (200 | ) |
| Change in lease liabilities | | | (355 | ) | | | (200 | ) |
| Net cash used in operating activities | | | (13,952 | ) | | | (11,011 | ) |
| | | | | | | | | |
| Cash flows from investing activities | | | | | | | | |
| Purchases of property and equipment | | | (1,700 | ) | | | (1,638 | ) |
| Net cash used in investing activities | | | (1,700 | ) | | | (1,638 | ) |
| | | | | | | | | |
| Cash flows from financing activities | | | | | | | | |
| Proceeds from at-the-market sales of common stock, net | | | 278 | | | | — | |
| Proceeds from exercise of common stock warrants | | | 742 | | | | — | |
| Proceeds from public and registered direct offerings, net | | | 20,065 | | | | 7,372 | |
| Net cash provided by financing activities | | | 21,085 | | | | 7,372 | |
| | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | | 5,433 | | | | (5,277 | ) |
| Cash and cash equivalents, at beginning of year | | | 3,799 | | | | 9,076 | |
| | | | | | | | | |
| Cash and cash equivalents, at end of year | | $ | 9,232 | | | $ | 3,799 | |
| Supplemental disclosure: | | | | | | | | |
| Noncash investing and financing activities: | | | | | | | | |
| Right-of-use asset obtained in exchange for lease liability | | $ | — | | | $ | 1,561 | |
| Receivable from transfer agent for warrant exercise proceeds | | $ | 142 | | | | — | |
| Cash paid for: | | | | | | | | |
| Income taxes | | $ | 2 | | | $ | 2 | |
The accompanying notes are an integral part of these audited consolidated financial statements.
MODULAR MEDICAL, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1 – THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Modular Medical, Inc. (the Company) was incorporated in Nevada in October 1998 under the name Bear Lake Recreation, Inc. The Company had no material business operations from 2002 until approximately 2017 when it acquired all of the issued and outstanding shares of Quasuras, Inc., a Delaware corporation (Quasuras). As the major shareholder of Quasuras retained control of both the Company and Quasuras, the share exchange was accounted for as a reverse merger. As such, the Company recognized the assets and liabilities of Quasuras, acquired in the merger, at their historical carrying amounts. Prior to the acquisition of Quasuras and, since at least 2002, the Company was a shell company, as defined in Rule 12b-2 promulgated under the Securities Exchange Act of 1934 (the Exchange Act). In June 2017, the Company changed its name from Bear Lake Recreation, Inc. to Modular Medical, Inc.
The Company is a pre-revenue, medical device company focused on the design, development and eventual commercialization of innovative insulin pumps using modernized technology to increase pump adoption in the diabetes marketplace. Through the creation of an innovative two-part patch pump, its initial product, the MODD1, the Company seeks to fundamentally alter the trade-offs between cost and complexity and access to the higher standards of care requiring considerable motivation that presently available insulin pumps provide. By simplifying and streamlining the user experience from introduction, prescription, reimbursement, training and day-to-day use, the Company seeks to expand the wearable insulin delivery device market beyond the highly motivated “super users” and expand the category into the mass market. The product seeks to serve both the type 1 and the rapidly growing, especially in terms of device adoption, type 2 diabetes markets. In January 2024, the Company submitted a 510(k) premarket notification to the United States Food and Drug Administration (FDA) for the MODD1. In March 2024, the Company received comments from the FDA on its submission, and the Company is in the process of responding to those comments.
Liquidity and Going Concern
The Company expects to continue to incur operating losses for the foreseeable future and incur cash outflows from operations as it continues to invest in the development and subsequent commercialization of its product. The Company expects that its research and development and general and administrative expenses will continue to increase, and, as a result, it will eventually need to generate significant revenue to achieve profitability. The Company’s expected operating losses and cash burn raise substantial doubt about the Company’s ability to continue as a going concern within one year after the date that these financial statements are issued. In addition, the Company’s independent registered public accounting firm, in its report on these consolidated financial statements for the year ended March 31, 2024, expressed substantial doubt about the Company’s ability to continue as a going concern. These consolidated financial statements do not include any adjustments that might result from this uncertainty. Implementation of the Company’s plans and its ability to continue as a going concern will depend upon the Company’s ability to raise additional capital, through the sale of additional equity or debt securities, to support its future operations. There can be no assurance that such additional capital, whether in the form of debt or equity financing, will be sufficient or available and, if available, that such capital will be offered on terms and conditions acceptable to the Company. In, May 2023 and February 2024, the Company completed public offerings of its common stock and warrants.
The Company’s operating needs include the planned costs to operate its business, including amounts required to fund working capital and capital expenditures. The Company’s future capital requirements and the adequacy of its available funds will depend on many factors, including the Company’s ability to successfully commercialize its product, competing technological and market developments, and the need to enter into collaborations with other companies or acquire other companies or technologies to enhance or complement its product offering. If the Company is unable to secure additional capital, it may be required to curtail its research and development initiatives and take additional measures to reduce costs in order to conserve its cash.
Basis of Presentation
The consolidated financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States of America. The Company’s fiscal year ends on March 31 of each calendar year. Each reference to a fiscal year in these notes to the consolidated financial statements refers to the fiscal year ended March 31 of the calendar year indicated (for example, fiscal 2024 refers to the fiscal year ending March 31, 2024). The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary, Quasuras. All significant intercompany transactions and balances have been eliminated in consolidation.
Use of Estimates
The preparation of the accompanying consolidated financial statements in conformity with U.S. generally accepted accounting principles (GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amount of revenues and expenses during the reporting period. Estimates may include those pertaining to accruals, stock-based compensation and income taxes. Actual results could differ from those estimates.
Reportable Segment
The Company operates in one business segment and uses one measurement of profitability for its business.
Research and Development
The Company expenses research and development expenditures as incurred.
General and Administrative
General and administrative expenses consist primarily of payroll and benefit costs, rent, stock-based compensation, legal and accounting fees, and facility and other administrative expenses.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist primarily of cash held in demand deposit accounts. The Company maintains its cash at high credit quality financial institutions within the United States, which are insured by the Federal Deposit Insurance Corporation (FDIC) up to limits of approximately $250,000. No reserve has been made in the financial statements for any possible loss due to financial institution failure.
Risks and Uncertainties
The Company is subject to risks from, among other things, competition associated with the industry in general, other risks associated with financing, liquidity requirements, rapidly changing customer requirements, limited operating history and the volatility of public markets.
Economic Disruptions
The global outbreak of the coronavirus disease 2019 (COVID-19) was declared a pandemic by the World Health Organization and a national emergency by the U.S. government in March 2020. This negatively affected the U.S. and global economy, disrupted global supply chains, significantly restricted travel and transportation, resulted in mandated closures and orders to “shelter-in- place” and created significant disruption of the financial markets. While the U.S. national emergency expired in May 2023 and substantially all closures and “shelter-in-place” orders have ended, there can be no assurance that the COVID-19 pandemic will not impact the Company’s operational and financial performance in the future, as the duration and spread of the pandemic and related actions taken by U.S. and foreign government agencies to prevent disease spread are uncertain, out of our control, and cannot be predicted.
Wars and acts of terrorism have led to further economic disruptions. Mounting inflationary cost pressures and recessionary fears have negatively impacted the global economy. Since mid-2022, at times, the U.S. Federal Reserve has addressed elevated inflation by increasing interest rates, as inflation remains elevated. While the Company was recently able to access the capital markets, in the future, the Company may be unable to access the capital markets, and additional capital may only be available to the Company on terms that could be significantly detrimental to its existing stockholders and to its business.
Cash and Cash Equivalents
Cash and cash equivalents include cash held in demand deposit and money market accounts, certificates of deposit and all highly liquid debt instruments with original maturities of three months or less.
Property and Equipment
Property and equipment are recorded at historical cost. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, generally three to five years. Depreciation is recorded in operating expenses in the consolidated statements of operations. Leasehold improvements and assets acquired through finance leases are amortized over the shorter of their estimated useful life or the lease term, and amortization is recorded in operating expenses in the consolidated statements of operations. Construction-in-process includes machinery and equipment and is stated at cost and not depreciated. Depreciation on construction-in-process commences when the assets are ready for their intended use and placed into service.
Fair Value of Financial Instruments
The Company measures the fair value of financial instruments using a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels:
| ● | Level 1 inputs to the valuation methodology are quoted prices for identical assets or liabilities in active markets. |
| ● | Level 2 inputs to the valuation methodology include quoted prices for similar assets and liabilities in active markets, and inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the financial instrument. |
| ● | Level 3 inputs to the valuation methodology are unobservable and significant to the fair value measurement. |
Due to their short-term nature, the carrying values of cash equivalents, accounts payable and accrued expenses, approximate fair value.
Leases
The Company’s right-of-use assets consist of leased assets recognized in accordance with Financial Accounting Standards Board (FASB) ASC No. 842, Leases, which requires lessees to recognize a lease liability and a corresponding lease asset for virtually all lease contracts. Right-of-use assets represent the Company’s right to use an underlying asset for the lease term and the lease liability represents the Company’s obligation to make lease payments arising from the lease, both of which are recognized based on the present value of the future minimum lease payments over the lease term at the commencement date. Leases with a lease term of 12 months or less at inception are not recorded on the consolidated balance sheets and are expensed on a straight-line basis over the lease term in the consolidated statement of operations and comprehensive loss. The Company determines the lease term by agreement with the lessor. In cases where the lease does not provide an implicit interest rate, the Company uses the Company’s incremental borrowing rate based on the information available at commencement date in determining the present value of future payments.
Stock-Based Compensation
The Company periodically issues stock options, restricted stock units and stock awards to employees and non-employees. We account for such awards based on Financial Accounting Standards Board Accounting Standards Codification (ASC) Topic 718, whereby the value of the award is measured on the date of grant and recognized as compensation expense on a straight-line basis over the requisite service period, usually the vesting period. With respect to performance-based awards, the Company assesses the probability of achieving the requisite performance criteria before recognizing compensation expense. The fair value of the Company’s stock options is estimated using the Black-Scholes-Merton Option Pricing (Black Scholes) model, which uses certain assumptions related to risk-free interest rates, expected volatility, expected life of the options, and future dividends. Compensation expense is recorded based upon the value derived from the Black-Scholes model. The assumptions used in the Black-Scholes model could materially affect compensation expense recorded in future periods.
Per-Share Amounts
Basic net loss per share is computed by dividing loss for the period by the weighted-average number of shares of common stock outstanding (WASO) during the period. In addition, the Company includes the number of shares of common stock issuable under pre-funded warrants as outstanding for purposes of the WASO calculation. Diluted net loss per share gives effect to all potentially dilutive common shares outstanding during the period. Potentially dilutive common shares consist of incremental shares of common stock issuable upon the exercise of stock options and exercise of warrants.
Prior to April 1, 2023, the Company excluded pre-funded warrants from the computation of WASO. The pre-funded warrants are now included in the computation of WASO. Prior period amounts have been conformed to the current-period presentation. The impact of the change reduced the previously reported loss per share by $0.13 and increased WASO by approximately 1,223,000 shares for the year ended March 31, 2023. The reclassification had no impact on the Company’s net loss or cash flows for the year ended March 31, 2023.
The following table sets forth securities outstanding which were excluded from the computation of diluted net loss per share as their inclusion would be anti-dilutive (in thousands):
| | | March 31, | |
| | | 2024 | | | 2023 | |
| Options to purchase common stock | | | 3,689 | | | | 2,481 | |
| Unvested restricted stock units | | | 187 | | | | — | |
| Common stock purchase warrants | | | 11,173 | | | | 6,217 | |
| Total | | | 15,049 | | | | 8,698 | |
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current period presentation. These reclassifications had no effect on the reported results of operations or cash flows.
Income Taxes
The Company determines deferred tax assets and liabilities based upon the differences between the financial statement and tax bases of the Company’s assets and liabilities using tax rates in effect for the year in which the Company expects the differences to affect taxable income. A valuation allowance is established for any deferred tax assets for which it is more likely than not that all or a portion of the deferred tax assets will not be realized. Based on the available information and other factors, management believes it is more likely than not that its federal and state net deferred tax assets will not be fully realized, and the Company has recorded a full valuation allowance.
The Company accounts for uncertain tax positions in accordance with FASB ASC Topic 740, Income Taxes. When tax returns are filed, it is likely that some positions taken would be sustained upon examination by the taxing authorities, while others are subject to uncertainty about the merits of the position taken or the amount of the position that would be ultimately sustained. The benefit of a tax position is recognized in the consolidated financial statements in the period during which, based on all available evidence, management believes it is more likely than not that the position will be sustained upon examination, including the resolution of appeals or litigation processes, if any. Tax positions taken are not offset or aggregated with other positions. Tax positions that meet the more-likely-than-not recognition threshold are measured as the largest amount of tax benefit that is more than 50 percent likely of being realized upon settlement with the applicable taxing authority. The portion of the benefits associated with tax positions taken that exceeds the amount measured as described above is reflected as a liability for unrecognized tax benefits in the accompanying consolidated balance sheets along with any associated interest and penalties that would be payable to the taxing authorities upon examination. Interest associated with unrecognized tax benefits is classified as interest expense and penalties are classified in general and administrative expenses in the consolidated statements of operations.
The Company files U.S. federal and state income tax returns in jurisdictions with varying statutes of limitations. The Company’s historical net operating loss and credit carryforwards may be adjusted by the federal and state tax authorities until the statute closes on the year in which such tax attributes are utilized.
Comprehensive Loss
Comprehensive loss represents the changes in equity of an enterprise, other than those resulting from stockholder transactions. Accordingly, comprehensive loss may include certain changes in equity that are excluded from net loss. For the years ended March 31, 2024 and 2023, the Company’s comprehensive loss was the same as its net loss.
Recently Issued Accounting Pronouncements
In November 2023, the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures, which requires disclosure of incremental segment information on an annual and interim basis. ASU No. 2023-07 is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024, and it requires retrospective application to all prior periods presented in the financial statements. The Company is currently evaluating the impact that this ASU will have on the presentation of its consolidated financial statements.
In December 2023, the FASB issued ASU No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which expands disclosures in an entity’s income tax rate reconciliation table and disclosures regarding cash taxes paid both in the U.S. and foreign jurisdictions. The update will be effective for annual periods beginning after December 15, 2024. The Company is currently evaluating the impact that this ASU will have on the presentation of its consolidated financial statements.
NOTE 2 – CONSOLIDATED BALANCE SHEET DETAIL
| | | March 31, | |
| | | 2024 | | | 2023 | |
| | | (in thousands) | |
| Prepaid and other current assets: | | | | | | | | |
| Prepaid expenses | | $ | 318 | | | $ | 142 | |
| Receivable from transfer agent for warrant exercise proceeds | | | 142 | | | | — | |
| Other receivables | | | 5 | | | | 5 | |
| | | $ | 465 | | | $ | 147 | |
| | | March 31, | |
| | | 2024 | | | 2023 | |
| | (in thousands) | |
| Property and equipment, net: | | | |
| Machinery and equipment | | $ | 3,209 | | | $ | 820 | |
| Computer equipment and software | | | 66 | | | | 66 | |
| Construction-in-process | | | 283 | | | | 1,003 | |
| Leasehold improvements | | | 33 | | | | 25 | |
| Office equipment | | | 63 | | | | 63 | |
| | | | 3,654 | | | | 1,977 | |
| Less: accumulated depreciation and amortization | | | (679 | ) | | | (256 | ) |
| | $ | 2,975 | | | $ | 1,721 | |
| | | March 31, | |
| | | 2024 | | | 2023 | |
| | | (in thousands) | |
| Accrued Expenses | | | | | | |
| Accrued wages | | $ | 243 | | | $ | 267 | |
| Other | | | 37 | | | | 72 | |
| | | $ | 280 | | | $ | 339 | |
NOTE 3 – LEASES
W. Bernardo Drive, San Diego, CA
The 39-month lease term expired on June 30, 2023, and, subsequent to expiration, the landlord refunded the Company’s security deposit.
Thornmint Road, San Diego, CA
The 48-month lease term commenced February 1, 2023, and the lease provides for an initial base monthly rent of $36,000 with annual rent increases of approximately 4%. In addition to the minimum lease payments, the Company is responsible for property taxes, insurance and other certain operating costs. A discount rate of 8%, which approximated the Company’s incremental borrowing rate, was used to measure the lease asset and liability. The Company obtained a right-of-use asset of approximately $1,560,000 in exchange for its obligations under the operating lease.
Future minimum payments under the facility operating lease, as of March 31, 2024, are listed in the table below (in thousands).
| Fiscal year ending March 31, | | | |
| 2025 | | | 452 | |
| 2026 | | | 470 | |
| 2027 | | | 405 | |
| Total future lease payments | | | 1,327 | |
| Less: Imputed interest | | | (137 | ) |
| Present value of lease liabilities | | $ | 1,190 | |
Cash paid for amounts included in the measurement of lease liabilities was approximately $476,000 and $230,000 for the years ended March 31, 2024 and 2023, respectively. Rent expense was approximately $449,000 and $237,000 for the years ended March 31, 2024 and 2023, respectively.
NOTE 4 – STOCKHOLDERS’ EQUITY
Increase in Authorized Shares
In February 2024, the Company’s stockholders approved an amendment to the Company’s Articles of Incorporation (the Amendment) to increase the number of authorized shares of common stock from 50,000,000 shares, to 100,000,000 shares. The Amendment was filed with the state of Nevada and became effective on February 15, 2024.
February 2024 Public Offering
On February 15, 2024, the Company entered into an underwriting agreement (the 2024 Underwriting Agreement) with Titan Partners Group LLC, a division of American Capital Partners, LLC (Titan), with respect to the issuance and sale 9,090,910 shares of its common stock at a price of $1.10 per share in a firm commitment underwritten offering (the 2024 Offering) by the Company. Upon the closing of the 2024 Offering, the Company received aggregate proceeds of approximately $10,000,000, before deducting underwiring discounts and commissions and other offering expenses.
Pursuant to the 2024 Underwriting Agreement, the Company granted Titan a 30-day option to purchase up to an additional 1,321,989 shares of common stock to cover over allotments, if any. On March 13, 2024, Titan exercised this option in full and purchased the additional securities for aggregate proceeds to the Company of approximately $1,454,000 before deducting underwriting discounts and commissions and other offering expenses.
Titan was paid a cash fee of 7.0% of the aggregate gross proceeds of the 2024 Offering (including the over-allotment option) and reimbursed certain out-of-pocket expenses of approximately $75,000.
ATM Offering
On November 22, 2023, the Company entered into a Sales Agreement (the ATM Agreement) with Leerink Partners LLC (Leerink) under which the Company may offer and sell, from time to time at its sole discretion, shares of its common stock, for aggregate gross proceeds of up to $6,500,000 through an “at the market offering” program under which Leerink will act as sales agent or principal. The ATM Agreement provides that Leerink will be entitled to compensation for its services equal to 3.0% of the gross proceeds from sales of any shares of common stock under the ATM Agreement. The Company has no obligation to sell any shares under the ATM Agreement and may, at any time, suspend solicitation and offers under the ATM Agreement. In January 2024, under the ATM Agreement, the Company sold 153,879 shares of common stock for net proceeds of approximately $278,000.
May 2023 Public Offering
On May 15, 2023, the Company entered into an underwriting agreement (the Underwriting Agreement) with Newbridge Securities Corporation (the Underwriter), with respect to the issuance and sale in a firm commitment underwritten offering (the 2023 Offering) by the Company of units of its securities. Upon the closing of the 2023 Offering, the Company sold 8,816,900 shares of its common stock and warrants to purchase 4,408,450 shares of its common stock for aggregate proceeds of approximately $9,390,000, before deducting underwriting discounts and commissions and other offering expenses. The securities were sold as a unit, with each unit consisting of two shares of common stock of the Company and one warrant (the 2023 Warrants) to purchase one share of common stock, at a public offering price of $2.13 per unit. The 2023 Warrants were immediately separable and exercisable, have a per share exercise price of $1.22 and expire five years from the date of issuance.
Pursuant to the Underwriting Agreement, the Company granted the Underwriter a 30-day option to purchase up to an additional 1,322,534 shares of common stock and an additional 661,267 of the 2023 Warrants to cover over-allotments, if any. On May 25, 2023, the Underwriter exercised this option in full and purchased the additional securities for aggregate gross proceeds to the Company of approximately $1,408,000, before deducting underwriting discounts and commissions and other offering expenses.
The Underwriter was paid a cash fee of 7.0% of the aggregate gross proceeds of the 2023 Offering (including the over-allotment option) and reimbursed certain out-of-pocket expenses of approximately $125,000. In addition, pursuant to the Underwriting Agreement, the Company initially issued to the Underwriter common stock purchase warrants (the UW Warrants) for a total of 709,760 shares. Subsequently, the UW Warrants were reissued to the Underwriter and its agents for a total of 604,623 shares. The UW warrants were exercisable six months from the respective issuance dates and have a four-year term and a per share exercise price of $1.32.
May 2022 Placement
On May 2, 2022, the Company entered into a securities purchase agreement (the Purchase Agreement) with an institutional investor, pursuant to which the Company sold, in a registered direct offering, which closed on May 5, 2022, an aggregate of 449,438 shares (the Shares) of the Company’s common stock, par value $0.001 per share, at a purchase price per Share of $4.45 and pre-funded warrants (the Pre-Funded Warrants) to purchase an aggregate of approximately 1,348,000 shares of common stock at a purchase price per Pre-Funded Warrant of $4.44. The Pre-Funded Warrants will be exercisable immediately on the date of issuance at an exercise price of $0.01 per share and may be exercised at any time until all of the Pre-Funded Warrants are exercised in full. In a concurrent private placement under the Purchase Agreement, the Company issued to the Investor warrants (the Private Placement Warrants) to purchase an aggregate of 1,438,202 shares of common stock at an exercise price of $6.60 per share. The Private Placement Warrants were exercisable beginning on the six-month anniversary of the date of issuance (the Initial Exercise Date) and will expire on the five-year anniversary of the Initial Exercise Date.
Issuances of Common Stock and Warrants
During the years ended March 31, 2024 and 2023, the Company issued 1,429 and 11,264 shares of common stock to service providers, respectively, with fair values of approximately $1,400 and $22,000, respectively.
As of March 31, 2024, the Company had the following warrants outstanding (share amounts in thousands):
| Type | | Number of Shares | | | Exercise Prices | | | Expiration
Dates | |
| Balance as of March 31, 2023 | | | 7,565 | | | | | | | | | |
| Issuance of common stock warrants | | | 605 | | | $ | 1.32 | | | | May 2027 | |
| Issuance of common stock warrants | | | 5,070 | | | $ | 1.22 | | | | May 2028 | |
| Common stock warrants exercised | | | (70 | ) | | $ | 1.32 | | | | | |
| Common stock warrants exercised | | | (649 | ) | | $ | 1.22 | | | | | |
| Balance as of March 31, 2024 | | | 12,521 | | | | | | | | | |
At March 31, 2024, the Company had a receivable from its transfer agent for approximately $142,000 for the proceeds from warrants exercised prior to March 31, 2024. The receivable was recorded in the prepaid and other line in the consolidated balance sheet at March 31, 2024.
As of March 31, 2023, the Company had the following warrants outstanding (share amounts in thousands):
| Type | | Number of Shares | | | Exercise Prices | | | Expiration
Dates |
| Common stock | | | 1,348 | | | $ | 0.01 | | | — |
| Common stock | | | 768 | | | $ | 6.00 | | | January - February 2027 |
| Common stock | | | 4,011 | | | $ | 6.60 | | | February 2027 |
| Common stock | | | 1,438 | | | $ | 6.60 | | | November 2027 |
| Total | | | 7,565 | | | | | | | |
NOTE 5 – STOCK-BASED COMPENSATION
Amended 2017 Equity Incentive Plan
In October 2017, the Company’s Board approved the 2017 Equity Incentive Plan (the Plan) with 1,000,000 shares of common stock reserved for issuance. In January 2020 and August 2021, the Board approved increases in the number of shares reserved for issuance under the Plan by 333,334 and 1,333,334 shares, respectively. In January 2023 and February 2024, the Company’s stockholders approved increases in the number of shares reserved for issuance under the Plan by an additional 2,000,000 and 3,000,000 shares, respectively. Under the Plan, eligible employees, directors and consultants may be granted a broad range of awards, including stock options, stock appreciation rights, restricted stock, performance-based awards and restricted stock units (RSUs). The Plan is administered by the Board or, in the alternative, a committee designated by the Board.
Stock-Based Compensation Expense
Stock options granted by the Company generally vest over 36 months and have a 10-year term. As of March 31, 2024, the unamortized compensation cost related to stock options was approximately $2,035,000 and is expected to be recognized as expense over a weighted-average period of approximately 1.3 years.
In October 2023, under its Two-Part FDA Submission and Clearance Milestone Bonus Program (the Bonus Program), the Company granted stock options for 909,533 shares, which are subject to vesting based upon the achievement of certain performance milestones by the Company and continued service by the optionees. In January 2024, options to purchase 625,326 shares (net of forfeitures), which were granted under part one of the Bonus Program, vested upon the Company’s submission to the FDA. As of March 31, 2024, the Company had not commenced expense recognition of 242,307 (net of forfeitures) of the options, which were granted under part two of the Bonus Program, based on its assessment of the probability of achievement of the applicable performance requirements.
During the year ended March 31, 2024, the Company granted options to purchase 127,500 shares that vested immediately when granted.
The weighted-average grant date fair values of stock options granted during the years ended March 31, 2024 and 2023 was $0.99 and $2.85, respectively. The following assumptions were used in the fair-value method calculations:
| | | | Year Ended March 31, | |
| | | | 2024 | | | | 2023 | |
| Risk-free interest rates | | | 3.51% - 4.72% | | | | 2.82% - 4.06% | |
| Volatility | | | 83% - 152% | | | | 83% - 223% | |
| Expected life (years) | | | 5.0 - 6.2 | | | | 5.0 – 5.7 | |
| Dividend yield | | | — | | | | — | |
The fair values of options at the grant date were estimated utilizing the Black-Scholes valuation model, which includes simplified methods to establish the fair term of options. The expected volatility is based on the historical volatility of the Company’s stock price. The risk-free interest rate was derived from the Daily Treasury Yield Curve Rates, as published by the U.S. Department of the Treasury as of the grant date for terms equal to the expected terms of the options. A dividend yield of zero was applied because the Company has never paid dividends and has no intention to pay dividends in the foreseeable future. The Company accounts for forfeitures as they occur.
The following table summarizes the activity in the shares available for grant under the Plan during the year ended March 31, 2024:
| | | | | | Options Outstanding | |
| | | Shares Available for Grant | | | Number of
Shares | | | Weighted
Average
Exercise
Price | |
| Balance at March 31, 2022 | | | 989,466 | | | | 1,650,705 | | | $ | 6.58 | |
| Additional shares authorized under the Plan | | | 2,000,000 | | | | — | | | | — | |
| Options granted | | | (1,006,074 | ) | | | 1,006,074 | | | | 3.15 | |
| Share awards | | | (26,789 | ) | | | — | | | | — | |
| Options cancelled and returned to the Plan | | | 175,689 | | | | (175,689 | ) | | | 6.48 | |
| Balance at March 31, 2023 | | | 2,132,292 | | | | 2,481,090 | | | | 5.19 | |
| Additional shares authorized under the Plan | | | 3,000,000 | | | | — | | | | — | |
| Options granted | | | (1,448,533 | ) | | | 1,448,533 | | | | 0.99 | |
| Share awards | | | (25,390 | ) | | | — | | | | — | |
| RSUs granted | | | (250,000 | ) | | | — | | | | — | |
| Options cancelled and returned to the Plan | | | 240,282 | | | | (240,282 | ) | | | 3.84 | |
| Balance at March 31, 2024 | | | 3,648,651 | | | | 3,689,341 | | | $ | 3.70 | |
No stock options were exercised during the years ended March 31, 2024 and 2023. During the years ended March 31, 2024 and 2023, the Company issued 25,390 and 26,789 shares, respectively, to its non-employee directors under the Company’s outside director compensation plan. For the years ended March 31, 2024 and 2023, the Company recorded stock-based compensation expense for these share awards of approximately $37,000 and $86,000, respectively.
A summary of RSU activity under the Plan is presented below.
| | | Number of Shares | | | Weighted Average Grant- Date Fair Value | |
| Balance at March 31, 2023 | | | — | | | | — | |
| Granted | | | 250,000 | | | $ | 0.91 | |
| Vested | | | (62,501 | ) | | | 0.91 | |
| Balance at March 31, 2024 | | $ | 187,499 | | | $ | 0.91 | |
The total intrinsic value of RSUs outstanding as of March 31, 2024 was approximately $347,000. The unamortized compensation cost at March 31, 2024 was approximately $171,000 related to RSUs and is expected to be recognized as expense over a period of approximately 2.25 years.
The following table summarizes the range of outstanding and exercisable options as of March 31, 2024:
| | | Options Outstanding | | | Options Exercisable | |
| | | | | | Weighted | | | | | | | | | | | | | |
| | | | | | Average | | | | | | | | | | | | | |
| | | | | | Remaining | | | Weighted | | | | | | Weighted | | | | |
| | | | | | Contractual | | | Average | | | | | | Average | | | Aggregate | |
| | | Number | | | Life | | | Exercise | | | Number | | | Exercise | | | Intrinsic | |
| Range of Exercise Price | | Outstanding | | | (in Years) | | | Price | | | Exercisable | | | Price | | | Value | |
| $0.93 - $2.00 | | | 2,237,066 | | | | 8.31 | | | $ | 1.48 | | | | 1,409,750 | | | $ | 1.52 | | | $ | 538,006 | |
| $3.95 - $7.51 | | | 943,145 | | | | 7.20 | | | | 5.29 | | | | 771,478 | | | | 5.52 | | | | — | |
| $8.61 - $17.70 | | | 509,130 | | | | 7.23 | | | | 10.53 | | | | 470,442 | | | | 10.53 | | | | — | |
| $0.93 - $17.70 | | | 3,689,341 | | | | 7.88 | | | $ | 3.70 | | | | 2,651,670 | | | $ | 4.28 | | | $ | 538,006 | |
The intrinsic value per share is calculated as the excess of the closing price of the common stock on the Company’s principal trading market over the exercise price of the option at March 31, 2024.
NOTE 6 – INCOME TAXES
The income tax provision consisted of the following:
| | | Year Ended March 31, | |
| | | 2024 | | | 2023 | |
| | | (in thousands) | |
| Current portion: | | | | | | |
| Federal | | $ | — | | | $ | — | |
| State | | | 2 | | | | 2 | |
| | | | 2 | | | | 2 | |
| Deferred portion: | | | | | | | | |
| Federal | | | (3,730 | ) | | | (2,933 | ) |
| State | | | (657 | ) | | | (1,467 | ) |
| | | | (4,387 | ) | | | (4,400 | ) |
| Change in valuation allowance | | | 4,387 | | | | 4,400 | |
| Provision for income taxes | | $ | 2 | | | $ | 2 | |
At March 31, 2024, the Company had net operating loss carryforwards (NOLs) of approximately $35,000,000 for federal income tax purposes and $50,400,000 for state income tax purposes. These NOLs are available to reduce future taxable income and will expire at various times from 2037 through 2045, except federal NOLs from fiscal 2018 and later, which will never expire.
The Company also had federal research and development tax credit carryforwards of approximately $2,100,000, which will begin expiring at various times from 2038 through 2044, and state research and development credits of approximately $500,000, which do not have an expiration date.
A reconciliation of income taxes provided at the federal statutory rate to the actual income tax provision is as follows:
| | | Year Ended March 31, | |
| | | 2024 | | | 2023 | |
| Federal statutory rate | | | (21 | )% | | | (21 | )% |
| State tax rate, net of federal benefit | | | (6 | )% | | | (6 | )% |
| Research and development tax credits | | | (7 | )% | | | (6 | )% |
| Change in valuation allowance | | | 32 | % | | | 29 | % |
| Other | | | 2 | % | | | 4 | % |
| Effective income tax rate | | | — | % | | | — | % |
The losses before income tax provision for the years ended March 31, 2024 and 2023 were solely attributable to US operations. Significant components of the Company’s deferred tax assets and liabilities were (in thousands):
| | | March 31, | |
| | | 2024 | | | 2023 | |
| Net operating loss carryforwards | | $ | 10,860 | | | $ | 8,742 | |
| Capitalized research and development expense | | | 3,058 | | | | 1,933 | |
| Stock-based compensation expense | | | 2,818 | | | | 2,587 | |
| Research and development tax credits | | | 2,568 | | | | 1,658 | |
| Property and equipment | | | 193 | | | | 105 | |
| Total deferred tax assets | | | 19,497 | | | | 15,025 | |
| Section 179 assets | | | (239 | ) | | | (111 | ) |
| Reserves, accruals and other | | | (49 | ) | | | (92 | ) |
| Total deferred tax liabilities | | | (288 | ) | | | (203 | ) |
| Less: valuation allowance | | | (19,209 | ) | | | (14,822 | ) |
| Deferred tax assets, net | | $ | — | | | $ | — | |
Based on the available information and other factors, management believes it is more likely than not that the net deferred tax assets at March 31, 2024 and 2023, will not be fully realizable. Accordingly, management has recorded a full valuation allowance against its net deferred tax assets at March 31, 2024 and 2023.
Management has evaluated and concluded that there were no material uncertain tax positions requiring recognition in the Company’s consolidated financial statements at March 31, 2024 and 2023. The Company does not expect any significant changes in its unrecognized tax benefits within twelve months of the reporting date.
NOTE 7 – ROYALTY AGREEMENT
In July 2017, the Company entered into a royalty agreement with its founder, then-chief executive officer, president and major shareholder (the Founder). Pursuant to the agreement, the Founder assigned and transferred all of his rights in the intellectual property of Quasuras in return for future royalty payments on the Company’s product. The Company is obligated to make royalty payments under the agreement to the Founder on any sales of the royalty product sold or otherwise commercialized by the Company equal to (a) $0.75 on each sale of a royalty product or (b) 5% of the gross sale price of the royalty product, whichever is less. The royalty payments will cease, and the agreement will terminate, at such time as the total sum of royalty payments actually paid to the Founder, pursuant to the agreement, reaches $10,000,000. The Company has the option to terminate the agreement at any time upon payment, to the Founder, of the difference between total royalty payments actually made to him to date and the sum of $10,000,000. All payments of the royalties, if due, for the preceding quarter, will be made by the Company to the Founder within 30 days after the end of each calendar quarter.
NOTE 8 – COMMITMENTS AND CONTINGENCIES
Litigations, Claims and Assessments
In the normal course of business, the Company may be involved in legal proceedings, claims and assessments arising in the ordinary course of business. The Company records legal costs associated with loss contingencies as incurred and accrues for all probable and estimable settlements.
Indemnification
In the ordinary course of business, the Company enters into contractual arrangements under which it may agree to indemnify the counterparties from any losses incurred relating to breach of representations and warranties, failure to perform certain covenants, or claims and losses arising from certain events as outlined within the particular contract, which may include, for example, losses arising from litigation or claims relating to past performance. Such indemnification clauses may not be subject to maximum loss clauses. The Company has also entered into indemnification agreements with its officers and directors. No amounts were reflected in the Company’s consolidated financial statements for the years ended March 31, 2024 and 2023 related to these indemnifications. The Company has not estimated the maximum potential amount of indemnification liability under these agreements due to the limited history of prior claims and the unique facts and circumstances applicable to each particular agreement. To date, the Company has not made any payments related to these indemnification agreements.
Purchase Obligations
The Company’s primary purchase obligations include purchase orders for machinery and equipment. At March 31, 2024, the Company had outstanding purchase orders for machinery and equipment and related expenditures of approximately $1,100,000.
In December 2023, the Company signed a device integration agreement with a provider of connected-care and remote monitoring diabetes technology solutions. As of March 31, 2024, the Company had a remaining obligation under the device integration agreement of approximately $400,000 over three years for technology license fees.
NOTE 9 – RELATED PARTY TRANSACTIONS
Manchester Management Company, LLC (MMC), as the general partner of Manchester Explorer, LP (Explorer), combined with the holdings of its affiliates, JEB Partners LP, James Besser and Morgan Frank, owned approximately 11% of the Company’s outstanding shares of common stock as of March 31, 2024. Mr. Besser is the Company’s chief executive officer and a managing member of MMC. Mr. Frank is one of our directors and serves as the portfolio manager of Explorer and as a managing member of MMC.
In February 2024, Explorer purchased 900,000 shares of common stock in the 2024 Offering at the public offering price per share of $1.10 for aggregate gross proceeds to the Company of $990,000.
The daughter of the Founder is an employee of the Company. During the years ended March 31, 2024 and 2023, the Company paid her approximately $137,000 and $201,000, respectively, which includes the aggregate grant date fair values, as determined pursuant to FASB ASC Topic 718, of stock options granted to her during each year.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are designed to ensure that information required to be disclosed in the reports filed with or furnished to the Securities and Exchange Commission, or the SEC, under the Securities Exchange Act of 1934, as amended, or the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports filed under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of our management, including our chief executive officer and our chief financial officer, we conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934 (the Exchange Act). Based on this evaluation, our management concluded that as of March 31, 2024, our disclosure controls and procedures were effective.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls. Internal control over financial reporting is the process designed by, or under the supervision of, our chief executive officer and chief financial officer, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles, and includes those policies and procedures that: (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect our transactions and dispositions of assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, cost-effective internal controls over financial reporting may not prevent or detect misstatements. All internal control systems, no matter how well designed, have inherent limitations, including the possibility of human error and the circumvention of overriding controls. Accordingly, even effective internal control over financial reporting can provide only reasonable assurance with respect to consolidated financial statement preparation. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we conducted an assessment of the effectiveness of our internal control over financial reporting as of the end of the period covered by this Annual Report on Form 10-K. In making this assessment, we used the criteria based on the framework in Internal Control—Integrated Framework (2013 Framework) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on the assessment, our management concluded that our internal control over financial reporting was effective as of March 31, 2024.
Changes in Internal Control over Financial Reporting
There were no changes in our internal controls over financial reporting during the fourth fiscal quarter of 2024 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
Not applicable.
PART III
ITEM 10: DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
The names of our directors, executive officers and certain information about each of them at March 31, 2024 are set forth below.
| Name | | Age | | Position |
| James Besser | | 48 | | Chief Executive Officer |
| Paul DiPerna | | 65 | | President, Chief Financial Officer, Treasurer and Chairman of the Board of Directors |
| Kevin Schmid | | 64 | | Chief Operating Officer |
| Duane DeSisto (1) | | 69 | | Director |
| Steven Felsher(2)(3) | | 75 | | Director |
| Morgan C. Frank | | 52 | | Director |
| Philip Sheibley(2)(3) | | 65 | | Director |
| Carmen Volkart(1)(2) | | 63 | | Director |
| Ellen O’Connor Vos | | 68 | | Director |
| (1) | Member of Compensation Committee |
| (2) | Member of Audit Committee |
| (3) | Member of Nominating and Governance Committee |
There are no family relationships among any of our directors or executive officers.
The principal occupations and positions for at least the past five years of our directors and executive officers are described below.
James “Jeb” Besser. Mr. Besser has served as our chief executive officer since February 2022 and combines over 25 years of experience in alternative investments, strategic advisory, corporate strategy and corporate governance. Since 1999, he has been a Managing Member at Manchester Management Company, LLC (“Manchester”), an investment management firm. Mr. Besser is also currently a director of River Stone Biotech, a development stage specialty bioprocessing company. He holds a B.A. in history from Brown University. We believe that Mr. Besser is qualified to serve as member of our board of directors due to his extensive prior experience conducting financial analysis of public companies (certain of which were in the development stage), including such public companies’ management teams, products, including products in the development stage, the potential markets for such products and other factors that could affect the likelihood and timing of success and market penetration of such entities’ products as well as his capital raising activities. We believe this provides us with valuable insights into the financial markets and investment criteria of institutional and other investors as well as capital raising activities.
Paul DiPerna. Mr. DiPerna has been our chairman, chief financial officer, president and treasurer since we acquired Quasuras, Inc. (“Quasuras”) in July 2017. He also served as our chief executive officer from July 2017 until August 2021, and as our Secretary from July 2017 to October 2021. In 2015, he founded Quasuras, an early-stage medical device company developing an insulin pump product, and, until its acquisition by us, he served as its chief executive officer and chairman. Prior to that, Mr. DiPerna founded Fuel Source Partners, LLC to incubate early stage medical device products and accumulate technical talent. Our current pump product was one of such proposed products and was spun-out to Quasuras in 2015. From 2012 to 2015, he served as a co-inventor at a private company with property rights in a medical device used for blood borne infection control called the Curos Cap, which was acquired by 3M Corporation. In 2003, Mr. DiPerna founded Tandem Diabetes Care, Inc. (“Tandem”) and held various positions, including as director, chief executive officer and chief technology officer and was primarily responsible for the design concept and development of Tandem’s initial insulin pump. Prior to that, he held executive and management positions at Baxter Healthcare Corporation (“Baxter”) where he was tasked with identifying synergistic opportunities in the diabetes industry. As a result, Mr. DiPerna developed substantial expertise and knowledge in the diabetes industry and led attempts by Baxter to acquire three insulin pump manufacturers. Previously, he held mechanical design engineering positions in the automated test equipment and blood separation sciences industries. Mr. DiPerna holds approximately 70 patents in medical device and microfluidic technology and has achieved numerous product clearances with the FDA. He has also achieved multiple successful exits with previous companies. Mr. DiPerna received a Masters in Engineering Management from Northeastern University and a B.S. in Mechanical Engineering from the University of Massachusetts and has spent over 35 years in the medical-device industry. We believe that Mr. DiPerna is qualified to serve as the chairman of our board of directors due to his extensive knowledge and experience in the medical-device industry generally, and, in particular, with regard to insulin pumps and the diabetes industry, as well as his management and leadership experience from holding director and senior executive positions in other public and private companies and leading project development teams of medical device companies.
Kevin Schmid. Mr. Schmid has served as our chief operating officer since July 21, 2022. He has over 19 years of experience in medical device senior management and high-volume global manufacturing operations. He served as a consultant to the Company from March 2022 until his hire date. Mr. Schmid has served as a member of the board of directors of Eitan Medical, an Israel based provider of connected infusion and wearable drug delivery solutions, since 2018. From 2018 through June 2021, he served as the Chief Executive Officer and a board member of Common Sensing, Inc., a disposable injector pen dose monitoring and reporting technology company. From 2016 to 2017, Mr. Schmid was Vice President of Drug Delivery Systems for the Stevanato Group, a provider of innovative packaging and drug delivery solutions for the pharmaceutical industry. From 2003 to 2015, Mr. Schmid was Vice President of Manufacturing, Operations, and Drug Delivery Systems for Insulet Corporation. He has a BSME degree from Clarkson University and an MBA from Sacred Heart University.
Duane DeSisto. Mr. DeSisto was appointed to our board of directors in July 2023. He has over 45 years of progressive management experience and over 25 years of experience in the medical device industry as a member of senior management and as a board member at multiple public companies. From 2001 to 2014, he served as the chief executive officer of Insulet Corporation, manufacturer of the world’s first patch insulin pump. Prior to 2001, he held executive positions with Paper Exchange, an e-business solution for the pulp and paper industry, AAI-Foster Grant, a sunglass and eyeglass provider to point-of-purchase retail, and Zoll Medical, a defibrillator manufacturer. He has an undergraduate degree from Providence College and a masters of business administration degree from Bryant University. We believe that Mr. DeSisto is qualified to serve on our board of directors because of his extensive background in operational leadership and commercialization of advanced medical devices and therapies, including insulin pumps. In addition, he has served as an executive officer and member of the board of directors at multiple public companies.
Steven Felsher. Mr. Felsher was appointed to our board of directors in November 2021. Mr. Felsher is an experienced executive with respect to finance, administration, governance and other aspects of public and private company management. He served as a member of the board of directors of Signal Hill Acquisition Corp., a special purpose acquisition company, from March 2021 to February 2023. From August 2018 to July 2020, he served as a member of the board of directors of Sito Mobile, Inc., a publicly-traded company that provided customized, data-driven solutions for brands spanning all forms of media. From January 2011 to June 2019, Mr. Felsher was a senior advisor at Quadrangle Group LLC, a private investment firm focused on the information and communications technology sectors. He spent a substantial portion of his career with Grey Global Group Inc., a global marketing services company, where he served as a senior executive from 1979 until 2007, most recently as vice chairman and chief financial officer. He holds a BA in classical Greek from Dickinson College and a J.D. from Yale University School of Law. We believe that Mr. Felsher is qualified to serve on our board of directors because of his extensive business experience with administration, governance, capital allocation and other aspects of public and private company management.
Morgan C. Frank. Mr. Frank was appointed to our board of directors in April 2017. In August 2022, he was appointed as chairman of the board of directors of SANUWAVE Health, Inc., a publicly-traded provider of wound-care products. Mr. Frank has worked with Manchester, LP since May 2002, and, prior to such time, he was a founder and managing director at First Principles Group, a boutique consultancy and principal investor specializing in corporate restructuring, restarts, intellectual property assessment and salvage, and spin outs. Prior to such time, Mr. Frank spent approximately five years as an analyst and portfolio manager at Hollis Capital, a San Francisco based hedge fund and prior thereto, Mr. Frank worked for an independent private client group at Paine Webber specializing in primary research to develop investment ideas (particularly short sale ideas) for institutional clients. Prior to his employment at Paine Webber, Mr. Frank was a currency trader for Eastern Vanguard. Mr. Frank holds a BA in Economics and in Political Science from Brown University. We believe that Mr. Frank is qualified to serve as member of our board of directors due to his extensive prior experience conducting financial analysis of public companies (certain of which were in the development stage), including such public companies’ management teams, products, including products in the development stage, the potential markets for such products and other factors that could affect the likelihood and timing of success and market penetration of such entities’ products as well as his capital raising activities. We believe this provides us with valuable insights into the financial markets and investment criteria of institutional and other investors as well as capital raising activities.
Philip Sheibley. Mr. Sheibley was appointed to our board of directors in November 2021. Mr. Sheibley is an experienced executive and venture capitalist. Since 2011, he has served as a principal at Alumni Investment Partners, a private equity firm. From 1981 to 2010, Mr. Sheibley served as a management and technology consultant with Accenture, where he focused on the life sciences area, holding a variety of leadership positions, including North American industry director for life sciences and global lead for management consulting. Mr. Sheibley holds a B.S. in industrial and systems engineering with a business minor from Lehigh University. We believe that Mr. Sheibley is qualified to serve on our board of directors because of his extensive business experience in the life sciences area and experience with venture capital investment and consulting, including financing transactions for early- stage and scale-up stage companies, assisting with scale-up strategy/execution, and participating as a board member in the medical products industry.
Carmen Volkart. Ms. Volkart was appointed to our board of directors in December 2019. Since January 2023, she has served as a member of the board of directors of Tactile Systems Technology, Inc. (Tactile Medical), a Nasdaq-listed, medical technology company developing and marketing at-home therapies for people suffering from underserved, chronic conditions. Ms. Volkart served as chief financial officer of Natureworks LLC, an advanced materials company offering a portfolio of renewably-sourced polymers, from October 2018 to September 2023. She served as a member of the board of directors, including as a member of the audit committee of Antares Pharma, Inc., a Nasdaq-listed, specialty pharmaceutical company, from October 2021 to May 2022, when it was acquired by another Nasdaq-listed company. From October 2012 to July 2018, Ms. Volkart served as chief financial officer and, for a portion of that time, as senior vice president of commercialization for NxThera, Inc., a medical device company pioneering the application of convective radiofrequency thermotherapy to treat endourological conditions. She served as global chief financial officer of Tornier N.V. from 2010 to 2012, and was chief operating and financial officer, corporate secretary, compliance officer and treasurer of Spine Wave, Inc. from 2006 to 2010. Prior to 2006, Ms. Volkart held various executive and financial positions at American Medical Systems, Inc., Medtronic, Inc. and Honeywell, Inc. She holds a B.S. in accounting from the University of North Dakota and an MBA with a concentration in strategic management from the University of Minnesota. We believe that Ms. Volkart is qualified to serve on our board of directors because of her substantial financial and public-company experience, as she has served as chief financial officer at multiple medical device and other companies.
Ellen O’Connor Vos. Ms. Vos was appointed to our board of directors in May 2021 and served as our chief executive officer from August 2021 until February 23, 2022. Ms. Vos has served as a member of VosHealth LLC since November 2020. Prior to that, she served as the president and chief executive officer of the Muscular Dystrophy Association from October 2017 to November 2020. Previously, Ms. Vos had been chief executive officer of ghg | greyhealth group from 1996 to 2017, and she has been a champion of using digital capabilities to improve the public health. Ms. Vos also serves on the board of OptimizeRX Corporation, a publicly- traded digital health company, and the Jed Foundation, a leading nonprofit dedicated to protecting the emotional health of college students, and was a founding board member of MMRF, a pioneering cancer research foundation. Ms. Vos holds a B.S. in nursing from Alfred University. We believe that Ms. Vos is qualified to serve on our board of directors because of her executive experience and extensive executive skills in digital marketing, commercialization and communications in the healthcare industry.
Family Relationships.
There are no family relationships between any of our directors or executive officers.
Involvement in Legal Proceedings
To our knowledge, none of our executive officers or our directors has, during the last ten years:
| ● | had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time; |
| ● | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity; |
| ● | been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| ● | been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| ● | been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
To our knowledge, there are no material proceedings to which any director, officer or affiliate of ours, any owner of record or beneficially of more than 5% of any class of voting securities of us, or any associate of any such director, officer, affiliate of ours, or security holder is a party adverse to us or any of our subsidiaries or has a material interest adverse to us or any of our subsidiaries.
Arrangements for Appointment of Directors and Officers
Pursuant to the Reorganization and Share Exchange Agreement, hereinafter referred to as the Share Agreement, dated as of July 24, 2017, by and among us, Quasuras, Mr. DiPerna and the other stockholders of Quasuras, until July 24, 2022, our board of directors was required to consist of no more than five and no less than two directors of which (i) Manchester Explorer, L.P. has the right to appoint two directors, pursuant to which Manchester Explorer, L.P. appointed Mr. Frank and Ms. Volkart and (ii) Mr. DiPerna, in addition to being our chairman of the board, had the right to appoint two additional directors, pursuant to which he appointed Liam Burns, who resigned from our board of directors in December 2021, and Febbo. In May 2021, the parties amended the Share Agreement and removed Manchester Explorer L.P’s and Mr. DiPerna’s rights to appoint directors. In addition, the parties agreed that Mr. DiPerna shall remain chairman of our board of directors until July 2022; provided, that in the event Mr. DiPerna resigns or is otherwise replaced as our chief executive officer, Mr. DiPerna shall remain as chairman of our board of directors for an additional period of three years. Following such amendment, our board of directors increased the size of the board to six members and, on May 18, 2021, appointed Ms. Vos as a director to our board.
Communications with our Board of Directors
Stockholders who desire to communicate with the board of directors, or a specific director, may do so by sending the communication addressed to either the board of directors or any individual director, c/o Modular Medical, Inc., 10740 Thornmint Road, San Diego, California 92127. These communications will be delivered to the board of directors, or any individual director, as specified.
Corporate Governance
Board Leadership Structure and Role in Risk Oversight
Due to our small size and early stage, we have not adopted a formal policy on whether the chairman and chief executive officer positions should be separate or combined. Since 2017, Mr. DiPerna has been serving as our chairman, and, since February 2022, Mr. Besser has been serving as our chief executive officer. Our board of directors has oversight responsibility for our risk management processes. Our board of directors receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate, regarding our assessment of risks. Our board of directors will focus on the most significant risks facing us and our general risk management strategy, and also ensure that risks undertaken by us are consistent with our appetite for risk. While our board of directors oversees our risk management processes, management is responsible for day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks facing us and that the leadership structure of our board of directors supports this approach.
We have established an audit committee, a compensation committee, and a nominating and governance committee. Each committee’s members and functions are described below.
Audit Committee
Our board of directors established the audit committee (the Audit Committee) for the purpose of overseeing the accounting and financial reporting processes and audits of our financial statements. The Audit Committee also is charged with reviewing any internal control violations under our whistleblower policy. The responsibilities of our audit committee are described in the Audit Committee Charter adopted by our board of directors, a current copy of which can be found on the investors section of our website, www.modular-medical.com.
Mr. Felsher, Mr. Sheibley and Ms. Volkart are the current members of the Audit Committee. Mr. Felsher serves as the chairperson and has been designated by the board of directors as the “audit committee financial expert,” as defined by Item 407(d)(5) of Regulation S-K under the Securities Act and the Exchange Act. That status does not impose duties, liabilities or obligations that are greater than the duties, liabilities or obligations otherwise imposed on Mr. Felsher as a member of the audit committee and the board of directors, however. Our board of directors has determined that each of our Audit Committee members satisfies the “independence” requirements of the Nasdaq listing rules and meets the independence standards under Rule 10A-3 under the Exchange Act.
Compensation Committee
Our board of directors established the compensation committee (the Compensation Committee) for the purpose of reviewing, recommending and approving our compensation policies and benefits, including the compensation of all of our executive officers and directors. Mr. DeSisto and Ms. Volkart are the current members of the compensation committee, and Mr. DeSisto serves as the chairperson. Each of our Compensation Committee members satisfies the “independence” requirements of the Nasdaq listing rules and meets the independence standards under Rule 10A-3 under the Exchange Act.
Our Compensation Committee is responsible for reviewing, recommending and approving our compensation policies and benefits, including the compensation of all of our executive officers and directors, and it also has the principal responsibility for the administration of our equity incentive plan. The responsibilities of our compensation committee are more fully described in the Compensation Committee Charter adopted by our board of directors, a current copy of which can be found on the investors section of our website, www.modular-medical.com.
Nominating and Governance Committee
Our board of directors established the nominating and governance committee (the Nominating and Governance Committee) for the purpose of (i) carrying out the responsibilities delegated by the board of directors relating to our director nominations process, (ii) developing and assessing our corporate governance policies, (iii) review our strategies, activities, and policies regarding environmental, social, and governance, or ESG, matters and (iv) provide oversight for the evaluation of the performance of the board of directors and its committees. The Nominating and Governance Committee consists of Mr. Sheibley and Mr. Felsher, and Mr. Sheibley serves as the chairperson. Each of the members of our Nominating and Governance Committee satisfies the “independence” requirements of the Nasdaq listing rules and meets the independence standards under Rule 10A-3 under the Exchange Act. The responsibilities of our Nominating and Governance committee are more fully described in the Nominating and Governance Committee Charter adopted by our board of directors, a current copy of which can be found on the investors section of our website, www.modular-medical.com.
The Nominating and Governance Committee will consider persons recommended by stockholders for inclusion as nominees for election to our board of directors if the information required by our bylaws is submitted in writing in a timely manner addressed and delivered to our secretary at the address of our executive offices. The Nominating and Governance Committee will identify and evaluate nominees for our board of directors, including nominees recommended by stockholders, based on numerous factors it considers appropriate, some of which may include strength of character, mature judgment, career specialization, relevant technical skills, diversity, and the extent to which the nominee would fill a present need on our board of directors.
Director Independence
Our board of directors has determined that each of the current directors, with the exception of Mr. DiPerna, Mr. Frank and Ms. Vos, is “independent,” as defined by the listing rules of the NASDAQ Stock Market, or Nasdaq, and the rules and regulations of the SEC. Our board of directors has standing Audit, Compensation and Nominating and Governance Committees, each of which is comprised solely of independent directors in accordance with the Nasdaq listing rules. No director qualifies as independent unless the board of directors affirmatively determines that he has no direct or indirect relationship with us that would impair his independence. We independently review the relationship of the Company to any entity employing a director or on whose board of directors such director is serving currently.
Code of Business Conduct and Ethics for Employees, Executive Officers and Directors
We have adopted a Code of Business Conduct and Ethics, or the Code of Conduct, applicable to all of our employees, executive officers and members of our board of directors. The Code of Conduct is available on our website at www.modular-medical.com. Our Nominating and Governance Committee is responsible for overseeing the Code of Conduct, and our board of directors must approve any waivers of the Code of Conduct. In addition, we intend to post on our website all disclosures that are required by law concerning any amendments to, or waivers from, any provision of the Code of Conduct.
Board Diversity
We seek diversity in experience, viewpoint, education, skill, and other individual qualities and attributes to be represented on our board of directors. We believe directors should have various qualifications, including individual character and integrity; business experience; leadership ability; strategic planning skills, ability, and experience; requisite knowledge of our industry and finance, accounting, and legal matters; communications and interpersonal skills; and the ability and willingness to devote time to our company. We also believe the skill sets, backgrounds, and qualifications of our directors, taken as a whole, should provide a significant mix of diversity in personal and professional experience, background, viewpoints, perspectives, knowledge, and abilities. Nominees are not to be discriminated against on the basis of race, religion, national origin, sex, sexual orientation, disability, or any other basis proscribed by law. The assessment of prospective directors is made in the context of the perceived needs of our board of directors from time to time.
All of our directors have held high-level positions in business or professional service firms and have experience in dealing with complex issues. We believe that all of our directors are individuals of high character and integrity, are able to work well with others, and have committed to devote sufficient time to the business and affairs of our company. In addition to these attributes, the description of each director’s background set forth above indicates the specific qualifications, skills, perspectives, and experience necessary to conclude that each individual should continue to serve as a director of ours.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors, executive officers and persons who beneficially own 10% or more of a class of securities registered under Section 12 of the Exchange Act to file reports of beneficial ownership and changes in beneficial ownership with the SEC. Directors, executive officers and greater than 10% stockholders are required by the rules and regulations of the SEC to furnish us with copies of all reports filed by them in compliance with Section 16(a).
Based solely upon a review of Forms 3 and 4 and amendments thereto furnished to us during fiscal 2024, including those reports that we filed on behalf of our directors and executive officers, no director, executive officer, beneficial owner of more than 10% of the outstanding common stock, or any other person subject to Section 16 of the Exchange Act, failed to file with the SEC on a timely basis during the fiscal year ended March 31, 2024, except that in July 2023 Mr. DeSisto failed to timely file a Form 3 to report his initial beneficial ownership and a Form 4 to report a restricted stock unit award.
ITEM 11. EXECUTIVE COMPENSATION
SUMMARY COMPENSATION TABLE
The following table sets forth compensation information for fiscal 2024 and 2023 for each of our named executive officers.
| | | | | | Salary | | | Stock Awards | | | Option Awards | | | Non-Equity Incentive Plan
Compensation | | | All Other
Compensation | | | Total | |
| Name and Principal Position | | Year | | | ($) | | | ($) | | | ($)(1) | | | ($) | | | ($) | | | ($) | |
| James E. Besser, | | | 2024 | | | | — | | | | — | | | | 130,480 | | | | — | | | | — | | | | 130,480 | |
| Chief Executive Officer (2) | | | 2023 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Paul DiPerna, | | | 2024 | | | | 300,000 | | | | — | | | | 182,792 | | | | — | | | | — | | | | 482,792 | |
| President, President Chief Financial Officer, Treasurer and Chairman | | | 2023 | | | | 300,000 | | | | — | | | | 189,413 | | | | — | | | | — | | | | 489,413 | |
| Kevin Schmid, | | | 2024 | | | | 250,000 | | | | — | | | | 160,510 | | | | — | | | | — | | | | 410,510 | |
| Chief Operating Officer (4) | | | 2023 | | | | 176,121 | | | | — | | | | 701,945 | | | | — | | | | — | | | | 878,066 | |
| (1) | Award amounts reflect the aggregate grant date fair value with respect to awards granted, as determined pursuant to Financial Accounting Standards Board (FASB) ASC Topic 718. The assumptions used to calculate the aggregate grant date fair value of option awards are set forth in the notes to the consolidated financial statements included in item 8 of this Report. These amounts do not reflect actual compensation earned or to be earned by our named executive officers. |
| (2) | Mr. Besser was appointed our chief executive officer in February 2022, and he is paid de minimis annual compensation of $1.00. |
| (4) | Mr. Schmid was appointed our chief operating officer in July 2022 at an annual base salary of $250,000. |
Outstanding Equity Awards at Fiscal Year-End
The following table shows certain information regarding outstanding equity awards held by our named executive officers as of March 31, 2024.
| Name | | Number of Securities Underlying Unexercised Options (#) Exercisable | | | Number of Securities Underlying Unexercised Options (#) Unexercisable | | | Option Exercise Price($) | | | Option Expiration Date(1) |
| James E. Besser | | | 135,136 | (2) | | | — | | | | 1.11 | | | 10/2/2033 |
| Paul DiPerna | | | 1,155 | (3) | | | — | | | | 9.48 | | | 6/1/2030 |
| | | | 1,169 | (4) | | | — | | | | 9.48 | | | 5/1/2030 |
| | | | 1,170 | (5) | | | — | | | | 9.48 | | | 4/1/2030 |
| | | | 1,660 | (6) | | | — | | | | 7.44 | | | 3/2/2030 |
| | | | 1,745 | (7) | | | — | | | | 7.44 | | | 2/1/2030 |
| | | | 1,727 | (8) | | | — | | | | 7.44 | | | 1/1/2030 |
| | | | 1,809 | (9) | | | — | | | | 6.75 | | | 12/1/2029 |
| | | | 1,811 | (10) | | | — | | | | 6.75 | | | 11/1/2029 |
| | | | 1,721 | (11) | | | — | | | | 6.75 | | | 10/1/2029 |
| | | | 1,662 | (12) | | | — | | | | 6.75 | | | 9/15/2029 |
| | | | 1,666 | (13) | | | — | | | | 6.75 | | | 8/15/2029 |
| | | | 1,660 | (14) | | | — | | | | 6.75 | | | 7/15/2029 |
| | | | 1,650 | (15) | | | — | | | | 6.75 | | | 6/15/2029 |
| | | | 1,677 | (16) | | | — | | | | 6.75 | | | 5/15/2029 |
| | | | 1,624 | (17) | | | — | | | | 6.75 | | | 4/15/2029 |
| | | | 1,694 | (18) | | | — | | | | 6.75 | | | 3/15/2029 |
| | | | 1,641 | (19) | | | — | | | | 6.75 | | | 2/15/2029 |
| | | | 1,603 | (20) | | | — | | | | 6.75 | | | 1/15/2029 |
| | | | 1,775 | (21) | | | — | | | | 6.75 | | | 12/15/2028 |
| | | | 1,775 | (22) | | | — | | | | 6.75 | | | 11/15/2028 |
| | | | 6,005 | (23) | | | — | | | | 1.98 | | | 10/15/2028 |
| | | | 6,005 | (24) | | | — | | | | 1.98 | | | 09/15/2028 |
| | | | 6,005 | (25) | | | — | | | | 1.98 | | | 08/15/2028 |
| | | | 100,000 | (26) | | | — | | | | 6.75 | | | 11/25/2029 |
| | | | 28,750 | (27) | | | 16,250 | (27) | | | 4.24 | | | 4/14/2032 |
| | | | — | | | | 50,000 | (28) | | | 1.65 | | | 4/3/2033 |
| | | | 90,091 | (29) | | | — | | | | 1.11 | | | 10/2/2033 |
| | | | — | | | | 45,046 | (30) | | | 1.11 | | | 10/2/2033 |
| Kevin Schmid | | | 97,222 | (31) | | | 77,778 | (31) | | | 4.24 | | | 7/21/2032 |
| | | | — | | | | 100,000 | (32) | | | 1.50 | | | 4/3/2033 |
| | | | 37,538 | (33) | | | — | | | | 1.11 | | | 10/2/2033 |
| | | | — | | | | 18,769 | (34) | | | 1.11 | | | 10/2/2033 |
| (1) | The standard option term is ten years, but all of the options expire automatically unless exercised within 90 days after the cessation of service as an employee, director or consultant. |
| (2) | The option was granted on October 2, 2023, and the shares subject to this option vested in January 2024 upon the Company’s 510(k) premarket submission to the FDA for its initial pump product. |
| (3) | The option was granted on June 1, 2020, and the shares subject to this option were fully vested on the grant date. |
| (4) | The option was granted on May 1, 2020, and the shares subject to this option were fully vested on the grant date. |
| (5) | The option was granted on April 1, 2020, and the shares subject to this option were fully vested on the grant date. |
| (6) | The option was granted on March 2, 2020, and the shares subject to this option were fully vested on the grant date. |
| (7) | The option was granted on February 1,2020, and the shares subject to this option were fully vested on the grant date. |
| (8) | The option was granted on January 1, 2020, and the shares subject to this option were fully vested on the grant date. |
| (9) | The option was granted on December 1, 2019, and the shares subject to this option were fully vested on the grant date. |
| (10) | The option was granted on November 1, 2019, and the shares subject to this option were fully vested on the grant date. |
| (11) | The option was granted on October 1, 2019, and the shares subject to this option were fully vested on the grant date. |
| (12) | The option was granted on September 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (13) | The option was granted on August 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (14) | The option was granted on July 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (15) | The option was granted on June 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (16) | The option was granted on May 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (17) | The option was granted on April 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (18) | The option was granted on March 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (19) | The option was granted on February 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (20) | The option was granted on January 15, 2019, and the shares subject to this option were fully vested on the grant date. |
| (21) | The option was granted on December 15, 2018, and the shares subject to this option were fully vested on the grant date. |
| (22) | The option was granted on November 15, 2018, and the shares subject to this option were fully vested on the grant date. |
| (23) | The option was granted on October 15, 2018, and the shares subject to this option were fully vested on the grant date. |
| (24) | The option was granted on September 15, 2018, and the shares subject to this option were fully vested on the grant date. |
| (25) | The option was granted on August 15, 2018, and the shares subject to this option were fully vested on the grant date. |
| (26) | The option was granted on November 25, 2019, and the shares subject to this option vested monthly over three years commencing January 1, 2020, subject to continued service as an employee, director or consultant. |
| (27) | The option was granted on April 14, 2022, and the shares subject to this option vest: i) one-third on the annual anniversary of the grant date and ii) the remaining two-thirds monthly over the next two years, subject to continued service as an employee, director or consultant |
| (28) | The option was granted on April 3, 2023, and the shares subject to this option vest: i) one-third on the annual anniversary of the grant date and ii) the remaining two-thirds monthly over the next two years subject to continued service as an employee, director or consultant. |
| (29) | The option was granted on October 2, 2023, and the shares subject to this option vested in January 2024 upon the Company’s 510(k) premarket submission to the U.S. Food and Drug Administration (“FDA”) for its initial pump product. |
| (30) | The option was granted on October 2, 2023, and the shares subject to this option vest if the Company receives notification of FDA clearance of the 510(k) premarket submission on or before August 1, 2024, subject to continued service as an employee, director or consultant. |
| (31) | The option was granted on July 21, 2022, and the shares subject to this option vest: i) one-third on the annual anniversary of the grant date and ii) the remaining two-thirds monthly over the next two years subject to continued service as an employee, director or consultant. |
| (32) | The option was granted on April 3, 2023, and the shares subject to this option vest: i) one-third on the annual anniversary of the grant date and ii) the remaining two-thirds vest over the next two years subject to continued service as an employee, director or consultant. |
| (33) | The option was granted on October 2, 2023, and the shares subject to this option vested in January 2024 upon the Company’s 510(k) premarket submission to the FDA for its initial pump product. |
| (34) | The option was granted on October 2, 2023, and the shares subject to this option vest if the Company receives notification of FDA clearance of the 510(k) premarket submission on or before August 1, 2024, subject to continued service as an employee, director or consultant. |
Employment Agreements
We have entered into our standard form of employment, confidential information and invention assignment agreement with each of our named executive officers. We also have entered into agreements to indemnify our directors and executive officers, in addition to the indemnification provided for in our articles of incorporation and bylaws. These agreements, among other things, provide for indemnification of our directors and certain executive officers for many expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by any such person in any action or proceeding, including any action by or in the right of the Company, arising out of such person’s services as a director or executive officer of ours, any subsidiary of ours or any other company or enterprise to which such person provided services at our request.
The DiPerna Employment and Related Agreements
We entered into an employment agreement dated August 1, 2018, with Mr. DiPerna pursuant to which Mr. DiPerna is employed by us as our president. Mr. DiPerna’s employment agreement had an initial two-year term and automatically renews for additional one-year terms. Pursuant to such agreement, we agreed to pay Mr. DiPerna: i) an annual salary of $200,000 in cash, ii) $100,000 per year in fully-vested stock options granted monthly at an exercise price determined by our board of directors in its sole discretion and iii) an annual bonus of $300,000, payable at the discretion of our board of directors, either in shares or in cash. If the board chooses to pay the bonus in shares, such shares will be valued at a price determined by our board of directors. Pursuant to such employment agreement (i) if (a) we terminate Mr. DiPerna’s employment without cause or he resigns with good reason, we will pay Mr. DiPerna a lump sum of $200,000, and (b) we terminate Mr. DiPerna’s employment for cause, we are not obligated to make any severance payment and Mr. DiPerna will receive only his base compensation through the last day of his employment, (ii) upon Mr. DiPerna’s death or disability, he will receive his base compensation through the last day of his employment and will remain eligible for all applicable benefits relative to death or disability pursuant to any plans that we have in place at such time, and (iii) upon a change of control (as defined in the employment agreement), Mr. DiPerna will be paid a lump sum of $100,000 within sixty days of the time at which such change of control takes place.
In May 2020, we amended our employment agreement with Mr. DiPerna to provide that in the event of a change in control:
| ● | within 60 days of the date the change in control occurs, Mr. DiPerna shall be paid by us or our successor in interest a lump sum cash payment equal to 12 months of Mr. DiPerna’s then annual Base Compensation (as defined in the employment agreement); and |
| ● | immediately prior to such change of control, any unvested stock options or other unvested securities of ours issued to Mr. DiPerna shall automatically accelerate and immediately become fully vested and exercisable. |
In June 2020, our board of directors approved an amendment to the employment agreement to provide that Mr. DiPerna’s base salary would be paid entirely in cash commencing July 1, 2020. The payment of the additional cash component of Mr. DiPerna’s annual base salary ($8,333.33 per month) was initially be deferred (the Deferred Salary) and accrue for Mr. DiPerna’s benefit until we have received $5,000,000 of cumulative gross proceeds of financing, at which time the Deferred Salary shall be paid to Mr. DiPerna and the salary deferrals will cease. The salary deferrals ceased and the Deferred Salary was paid to Mr. DiPerna in May 2021. In August 2021, Mr. DiPerna resigned as our chief executive officer, and he continues to serve as our president, chief financial officer, treasurer and chairman of our board of directors.
If a change of control occurred on March 31, 2024, under his employment agreement, Mr. DiPerna would be entitled to the following:
| ● | payment of a lump sum of $300,000 within 60 days of the time at which such change of control takes place. |
| ● | accelerated vesting of 111,296 shares of common stock under unvested stock options. The value of the shares subject to accelerated vesting is calculated as the intrinsic value per share multiplied by the number of shares that would become fully vested upon a change of control. The intrinsic value per share would be calculated as the excess of the closing price of the common stock of $1.85 on the Nasdaq Capital Market on March 28, 2024 over the exercise price of the option. As of March 31, 2024, the intrinsic value of the shares subject to accelerated vesting was approximately $67,000. |
In connection with our acquisition of Quasuras, we entered into an Intellectual Property Transfer Agreement dated as of July 24, 2017, with Quasuras and Mr. DiPerna, pursuant to which Mr. DiPerna transferred to us all intellectual property rights owned directly and/or indirectly by him related to our business. Separately, we agreed to pay Mr. DiPerna, as part of his compensation for services to be performed for us, pursuant to a royalty agreement, certain fees based upon future sales, if any, of our potential product subject to a maximum $10,000,000 cap on the aggregate amount of fees that Mr. DiPerna could earn from such arrangement.
The Schmid Offer Letter
Pursuant to an offer letter with the Company (the “Offer Letter”), Mr. Schmid shall receive an annual salary of $250,000 (the “Schmid Base Salary”) for his services as our chief operating officer. Additionally, he is eligible for an annual discretionary target incentive bonus of up to 50% of his Base Salary. In connection with his appointment, Mr. Schmid was granted a stock option to purchase 175,000 shares of our common stock. The stock option vests over a three-year period with one-third of the shares subject to the stock option vesting on the one-year anniversary of the grant date and the remaining shares vesting monthly thereafter, subject to Mr. Schmid’s continuous service with us. In the event of termination of his employment by us other than for cause or good reason (as defined in the Offer Letter), Mr. Schmid will receive an amount equal to six months of his then-current base salary as a severance payment.
James Besser
As compensation for his services as our chief executive officer, Mr. Besser is paid de minimis compensation of $1.00 per year.
Director Compensation
In the first quarter of fiscal 2022, our board of directors approved our outside (non-employee) director compensation plan (the Director Plan). Pursuant to the Director Plan, outside directors are paid the following annual retainers:
| ● | $25,000 for service as a member of the board of directors; and |
| ● | $5,000 for service as chair of a committee of the board of directors. |
The annual retainers are paid in quarterly installments in either cash, options to purchase shares of our common stock or in shares of our common stock, as directed by each director based on an annual election.
In addition, under the Director Plan, each director receives an annual service equity award of $100,000 paid in quarterly installments in either options to purchase shares of our common stock or shares of our common stock, as directed by each director based on an annual election. In July 2022, the Board amended the Director Plan to provide that a minimum price of $10.00 per share of common stock would be used to calculate the number of shares subject to options or share awards.
In addition, upon appointment to our board of directors, we award our non-employee directors an equity award under our Amended 2017 Equity Incentive Plan (the 2017 Plan), and such awards vest over three years.
The following table summarizes the compensation earned by our non-employee directors in fiscal 2024:
| | | Fee Compensation | | | Restricted
Stock Awards | | | Option
Awards | | | All Other
Compensation | | | Total | |
| Name | | ($) | | | ($)(1)(2) | | | ($)(2)(3) | | | ($)(2)(4) | | | ($) | |
| Duane DeSisto | | | 22,170 | | | | 227,500 | | | | — | | | | 12,103 | | | | 261,773 | |
| William Febbo(5) | | | 7,500 | | | | — | | | | — | | | | 2,328 | | | | 9,828 | |
| Steven Felsher | | | — | | | | — | | | | 36,312 | | | | 4,377 | | | | 40,689 | |
| Morgan Frank | | | — | | | | — | | | | 45,390 | | | | — | | | | 45,390 | |
| Philip Sheibley | | | 30,000 | | | | — | | | | — | | | | 14,550 | | | | 60,795 | |
| Carmen Volkart | | | — | | | | — | | | | 36,312 | | | | 3,658 | | | | 39,970 | |
| Ellen O’Connor Vos | | | 25,000 | | | | — | | | | 36,312 | | | | — | | | | 42,562 | |
| (1) | Mr. DeSisto was appointed to our board of directors in July 2023, and, upon his appointment, was granted a restricted stock unit (the “DeSisto RSU”) for 250,000 shares of our common stock. As of March 31, 2024, 187,499 shares of our common stock remained unvested under the DeSisto RSU. |
| (2) | Award amounts reflect the aggregate grant date fair value with respect to awards granted, as determined pursuant to FASB ASC Topic 718. The assumptions used to calculate the aggregate grant date fair value of option awards are set forth in the notes to the consolidated financial statements included in Item 8 of this Annual Report on Form 10-K. These amounts do not reflect actual compensation earned or to be earned by our directors. |
| (3) | As of March 31, 2024, our non-employee directors each held outstanding options to purchase the following number of shares of our common stock: Steven Felsher, 98,084; Morgan Frank, 177,458; Philip Sheibley, 16,667; Carmen Volkart; 150,558, Ellen O’Connor Vos, 166,020. |
| (4) | Represents stock awards under the Director Plan; we calculated the estimated fair value of the stock awards issued to Messrs. DeSisto, Felsher and Sheibley and Ms. Volkart using the closing price per share of our common stock on the day prior to the grant date in accordance with the Director Plan. |
| (5) | Mr. Febbo resigned from our board of directors in July 2023. |
ITEM 12: SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information as of May 31, 2024 concerning the ownership of our common stock by:
| ● | each stockholder known by us to be the beneficial owner of more than 5% of the outstanding shares of our common stock (currently our only class of voting securities); |
| ● | each of our executive officers; and |
| ● | all directors and executive officers as a group. |
Beneficial ownership is determined in accordance with Rule 13d-3 of the Exchange Act, and includes all shares over which the beneficial owner exercises voting or investment power. Shares that are issuable upon the exercise of options, warrants and other rights to acquire common stock that are presently exercisable or exercisable within 60 days of June 15, 2024 are reflected in a separate column in the table below. These shares are taken into account in the calculation of the total number of shares beneficially owned by a particular holder and the total number of shares outstanding for the purpose of calculating percentage ownership of the particular holder. We have relied on information supplied by our officers, directors and certain stockholders and on information contained in filings with the SEC. Except as otherwise indicated, and subject to community property laws where applicable, we believe, based on information provided by these persons, that the persons named in the table have sole voting and investment power with respect to all shares of common stock shown as beneficially owned by them. The percentage of beneficial ownership is based on 32,463,670 shares of common stock outstanding as of May 31, 2024.
Unless otherwise stated, the business address of each of our directors and executive officers listed in the table is 10740 Thornmint Road, San Diego, California 92127.
| Name and principal position | | Number of Shares
Beneficially Owned
(Excluding
Outstanding
Equity Awards and
Warrants)(1) | | | Number of
Shares Issuable on Exercise of
Outstanding
Equity Awards and
Warrants(2) | | | Percent of
Class | |
| JEB Partners, L.P. | | | 330,473 | (3) | | | — | | | | 1.02 | |
| Manchester Explorer, L.P. | | | 3,118,077 | (3) | | | 653,511 | | | | 11.36 | |
| Manchester Management Company, LLC | | | 3,448,550 | (3) | | | 653,511 | | | | 12.36 | |
| 683 Capital Management, LLC | | | 1,570,000 | (4) | | | 641,999 | | | | 6.67 | |
| Sio Capital Management, LLC | | | 689,352 | (5) | | | 2,786,516 | (5) | | | 9.84 | |
| Directors and Officers: | | | | | | | | | | | | |
| James Besser | | | 3,593,300 | (3) | | | 788,647 | | | | 13.15 | |
| Paul DiPerna | | | 2,553,586 | (6) | | | 330,436 | | | | 8.77 | |
| Kevin Schmid | | | — | | | | 195,871 | | | | * | |
| Duane DeSisto | | | 69,891 | | | | 20,833 | | | | * | |
| Steven Felsher | | | 126,177 | | | | 92,528 | | | | * | |
| Morgan C. Frank | | | 3,324,303 | (3) | | | 830,969 | | | | 12.45 | |
| Philip Sheibley | | | 51,139 | | | | 11,112 | | | | * | |
| Carmen Volkart | | | 9,585 | | | | 10,558 | | | | * | |
| Ellen O’Connor Vos | | | 18,519 | | | | 199,353 | | | | * | |
| All current directors and executive officers as a group (9 persons) | | | 6,153,200 | | | | 1,831,661 | | | | 24.77 | |
| (1) | Excludes shares subject to outstanding options, restricted stock units and warrants to acquire common stock that are exercisable within 60 days of May 31, 2024. |
| (2) | Represents the number of shares subject to outstanding options, restricted stock units and warrants to acquire common stock that are exercisable within 60 days of May 31, 2024. |
| (3) | Includes (i) 144,750 shares directly held by Mr. Besser, of which: (a) 60,277 shares were received in exchange for Mr. Besser’s shares as a result of our acquisition of Quasuras; (b) 29,630 shares purchased in a private placement in 2018 (the “2018 Placement”) and (c) 34,843 shares were purchased in a private placement in 2020 (the “2020 Placement”) and (d) 20,000 shares were purchased in the open market; (ii) 2,218,077 held by Manchester Explorer, L.P. of which: (a) 1,515,152 shares were purchased in a private placement in 2017 (the “2017 Placement”), (b) 157,037 shares were purchased in the 2018 Placement, (c) 11,614 were purchased in the 2020 Placement, (d) 300,000 shares were purchased in a public offering in February 2022, (e) 234,274 shares were acquired upon the conversion of a convertible note in February 2022 and (f) 900,000 shares were purchased in the Company’s February 2024 public offering; (iii) 330,473 shares held by JEB Partners, L.P. of which (a) 252,526 shares were purchased in the 2017 Placement, (b) 53,333 shares were purchased in the 2018 Placement and (c) 11,614 shares were purchased in the 2020 Placement; and (iv) 206,226 shares held by Mr. Frank, which shares were received in our acquisition of Quasuras in exchange for Mr. Frank’s shares of Quasuras. Mr. Besser, as the managing member, and Mr. Frank, as the portfolio manager and consultant of Manchester Management Company, LLC, (“MMC”) the general partner of Manchester Explorer, L.P. and JEB Partners, L. P., have shared voting and dispositive power over shares held by Manchester Explorer, L.P. and JEB Partners, L.P. The address for Manchester Explorer, L.P is c/o MMC, 2 Calle Candina, No. 1701, San Juan, Puerto Rico 00907. |
| (4) | Based on information reported by 683 Capital Management, LLC (“683 Management”) on Schedule 13G filed with the SEC on February 16, 2024. 683 Management, as the investment manager of 683 Capital Partners, LP (“683 Capital”), may be deemed to have beneficially owned the shares of Common Stock and warrants to purchase shares of Common Stock beneficially owned by 683 Capital. Ari Zweiman as the Managing Member of 683 Management may be deemed to have beneficially owned the shares of common stock and warrants to purchase shares of common stock beneficially owned by 683 Management. The address for 683 Management, 683 Capital and Mr. Zweiman is 1700 Broadway, Suite 4200, New York, NY 10019. |
| (5) | Based on information reported by Sio Capital Management, LLC (“Sio”) on Schedule 13G filed with the SEC on February 6, 2024. Sio and Sio GP, LLC (the “GP”) act as investment advisor and general partner, respectively, to various clients that are the record owners of the shares of our common stock reported on this Schedule 13G. Because Sio’s investment discretion with respect to such clients is subject to oversight by the GP, the GP may be deemed to be the beneficial owner of the common stock of the Issuer owned by such clients. In addition, both Sio and the GP are controlled by Michael Castor. As such, he may be deemed to control the voting and dispositive decisions with respect to, and therefore be the beneficial owner of, the shares of our common stock. The address for Sio, the GP and Mr. Castor is 600 Third Avenue, New York, New York 10016. |
| (6) | Includes (i) 2,000,000 shares directly held by the Paul DiPerna Irrevocable Trust, (ii) 333,334 shares directly held by Mr. DiPerna’s adult daughters, Kelsie DiPerna and Alaria DiPerna, which shares Mr. DiPerna has sole voting power over; (iii) 207,906 shares directly held by the Paul DiPerna Trust, of which 101,010 shares were purchased in the 2017 Placement and 23,429 shares were acquired upon the conversion off a convertible note in February 2022 and (iv) 12,346 shares held by Mr. DiPerna. The 2,000,000 shares held by the Paul DiPerna Irrevocable Trust, 333,334 shares held by Mr. DiPerna’s adult daughters and 73,480 shares held by the Paul DiPerna Trust that were issued in 2017 to Mr. DiPerna in the Control Block Acquisition and transferred to such persons in December 2020 by Mr. DiPerna. Mr. DiPerna is the chairman of our board of directors, and also serves as our president, chief financial officer and treasurer. Mr. DiPerna is the trustee of both the Paul DiPerna Irrevocable Trust and the Paul DiPerna Trust. |
Changes in Control
We are not aware of any arrangement that may result in a “changes in control,” as that term is defined by the provisions of Item 403(c) of Regulation S-K.
Equity Compensation Plan Information
The following table shows the number of securities to be issued upon exercise or vesting of outstanding equity awards under the 2017 Plan as of March 31, 2024.
| | | Number of
securities to be
issued upon
exercise or
vesting of
outstanding
equity awards
(a) | | | Weighted-
average
exercise price
of outstanding
options
(b) | | | Number of
securities
remaining available
for future issuance
under equity
compensation plans
(excluding securities
reflected in
column(a))
(c) | |
| Equity compensation plans not approved by security holders | | | 3,689,341 | | | $ | 3.70 | | | | 3,648,651 | |
ITEM 13: CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with Related Persons
Below we describe any transactions to which we have been a participant, in which the amount involved in the transaction exceeds or will exceed the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years and in which any of our directors, director nominees, executive officers, or holders of more than 5% of our capital stock, or any immediate family member of, or person sharing the household with, any of these individuals, had or will have a direct or indirect material interest since April 1, 2022.
MMC as the general partner of Manchester Explorer, L.P. (Explorer), combined with the holdings of its affiliates, JEB Partners LP, Mr. Besser and Mr. Frank, owned approximately 13% of our outstanding shares of common stock at March 31, 2024. Mr. Besser is our chief executive officer and a managing member of MMC. Mr. Frank is one of our directors, and he serves as the portfolio manager of Explorer and as a managing member of MMC. In February 2024, we closed a public offering of our common stock (the 2024 Offering), and Explorer purchased 900,000 shares in the 2024 Offering for aggregate gross proceeds to us of $990,000.
Mr. DiPerna’s daughter is an employee of ours, and, during fiscal 2024 and fiscal 2023, we paid her approximately $137,000 and $201,000, respectively, which includes the aggregate grant date fair value, as determined pursuant to FASB ASC Topic 718, of stock options granted to her.
See “Management” above for other related-party transactions involving our executive officers and directors.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following table shows the fees billed to us by Farber Hass Hurley LLP, or Farber, our independent registered public accounting firm, for the audit of our consolidated financial statements and other services provided (in thousands).
| | | Year ended March 31, | |
| | | 2024 | | | 2023 | |
| Audit fees(1) | | $ | 63 | | | $ | 53 | |
| Audit-related fees(2) | | | 38 | | | | 6 | |
| Total(3) | | $ | 101 | | | $ | 59 | |
| (1) | Audit fees consisted of fees for professional services rendered for the audit of our annual consolidated financial statements and reviews of our quarterly consolidated financial statements. |
| (2) | Audit-related fees consisted of fees for services related to our issuance of SEC registration statements and sales of our securities under registration statements. |
| (3) | Farber did not provide any non-audit or other services other than those reported under “Audit fees” and “Audit-related fees.” |
The Audit Committee meets with our independent registered public accounting firm at least four times a year. At such times, the Audit Committee reviews and approves both audit and non-audit services performed by the independent registered public accounting firm, as well as the fees charged for such services. The Audit Committee is responsible for pre-approving all auditing services and non-auditing services (other than non-audit services falling within the de minimis exception set forth in Section 10A(i)(1)(B) of the Exchange Act and non-audit services that independent auditors are prohibited from providing to us) in accordance with the following guidelines: (1) pre-approval policies and procedures must be detailed as to the particular services provided; (2) the Audit Committee must be informed about each service; and (3) the Audit Committee may delegate pre-approval authority to one or more of its members, who shall report to the full committee, but shall not delegate its pre-approval authority to management. Among other things, the Audit Committee examines the effect that performance of non-audit services may have upon the independence of the auditors.
PART IV
ITEM 15: EXHIBITS
(a) Financial Statements and Financial Statement Schedules are set forth under Part II, Item 8 of this report.
(b)
Other Schedules may omitted because they are not applicable, not required, or because the required information is included in the Consolidated Financial Statements or notes thereto.
| Exhibit | | | | Reference | | Filed or Furnished |
| Number | | Exhibit Description | | Form | | Exhibit | | Filing Date | | Herewith |
| 1.1 | | Form of Underwriting Agreement dated May 15, 2023 | | 8-K | | 1.1 | | 05/17/2023 | | |
| 1.2 | | Underwriting Agreement dated as of February 15, 2024 between the Registrant and Titan Partners Group LLC | | 8-K | | 1.1 | | 02/16/2024 | | |
| 1.3 | | Sales Agreement, dated as of November 22, 2023, between Modular Medical, Inc. and Leerink Partners LLC | | 8-K | | 1.1 | | 11/22/2023 | | |
| 2.1 | | Reorganization and Share Exchange Agreement dated as of July 24, 2017, by and among the Registrant, Quasuras, Inc., Paul DiPerna and the other stockholders of Quasuras, Inc. | | 8-K | | 2.1 | | 07/28/2017 | | |
| 2.2 | | Addendum No. 1 to Reorganization and Share Exchange Agreement dated as of July 24, 2017, by and among the Registrant, Quasuras, Inc., Paul DiPerna and the other Stockholders of Quasuras, Inc. dated May 3, 2021 | | 8-K | | 2.2 | | 05/12/2021 | | |
| 3.1 | | Third Amended and Restated Articles of Incorporation, as filed with the Secretary of State of Nevada on June 27, 2017 | | 8-K | | 3.1 | | 06/29/2017 | | |
| 3.2 | | Certificate of Amendment to the Amended and Restated Articles of Incorporation of Modular Medical, Inc., filed with the Secretary of State of the State of Nevada on November 24, 2021 | | 8-K | | 3.1 | | 12/01/2021 | | |
| 3.3 | | Certificate of Amendment to the Amended and Restated Articles of Incorporation of Modular Medical, Inc., filed with the Secretary of State of the State of Nevada on February 15, 2024 | | 8-K | | 3.1 | | 02/15/2024 | | |
| 3.4 | | Amended Bylaws | | 10-SB | | 3.2 | | 03/08/2002 | | |
| 4.1* | | 2017 Equity Incentive Plan, as amended and restated | | | | | | | | X |
| 4.2 | | Form of Warrant to Purchase Common Stock dated February 14, 2022 | | 8-K | | 4.1 | | 02/14/2022 | | |
| 4.3 | | Form of Pre-Funded Warrant to Purchase Common Stock dated May 2, 2022 | | 8-K | | 4.1 | | 05/05/2022 | | |
| 4.4 | | Form of Private Placement Warrant dated May 2, 2022 | | 8-K | | 4.2 | | 05/05/2022 | | |
| 4.5 | | Form of Warrant | | S-1/A | | 4.5 | | 05/05/2023 | | |
| 4.6 | | Form of Underwriter’s Warrant | | S-1/A | | 4.6 | | 05/05/2023 | | |
| 4.7 | | Description of Registrant’s Securities | | 10-K | | 4.7 | | 06/26/2023 | | |
| 10.1* | | Employment Agreement dated August 1, 2018, by and between the Registrant and Paul DiPerna | | S-1 | | 10.4 | | 06/27/2019 | | |
| 10.2 | | Intellectual Property Assignment Agreement dated July 24, 2017, by and between the Registrant, Quasuras, Inc. and Paul DiPerna | | 8-K | | 10.3 | | 07/28/2017 | | |
| 10.3* | | Technology Royalty Agreement dated as of July 24, 2017, by and between the Registrant, Quasuras, Inc. and Paul DiPerna | | 8-K | | 10.4 | | 07/28/2017 | | |
| 10.4 | | Standard Industrial/Commercial Agreement between the Registrant and Michael Summers dated January 5, 2023 | | S-1 | | 10.28 | | 04/24/2023 | | |
| 10.5* | | Form of Indemnification Agreement between the Registrant and each of its directors and officers used from January 23, 2020 | | 10-Q | | 10.15 | | 02/13/2020 | | |
| 10.6* | | Form of Notice of Stock Option Grant and Stock Option Agreement under the Amended 2017 Equity Incentive Plan | | 10-Q | | 10.16 | | 02/13/2020 | | |
| 10.7* | | First Amendment to the Employment Agreement between the Registrant and Paul DiPerna effective as of May 12, 2020 | | 8-K | | 10.18 | | 05/27/2020 | | |
| 10.8* | | Second Amendment to Employment Agreement between the Registrant and Paul DiPerna effective as of July 1, 2020 | | 10-Q | | 10.20 | | 08/12/2020 | | |
| 10.9 | | Form of Convertible Promissory Note issued in the 2021 Private Placement | | 8-K | | 10.21 | | 05/12/2021 | | |
| 10.10 | | Form of Common Stock Purchase Agreement dated March 2020 by and between the Registrant and the Investors named therein | | S-1 | | 10.17 | | 04/09/2020 | | |
| 10.11 | | Form of Securities Purchase Agreement for the 2021 Private Placement | | 8-K | | 10.23 | | 05/12/2021 | | |
| 10.12 | | Form of Registration Rights Agreement for the 2021 Private Placement | | 8-K | | 10.24 | | 05/12/2021 | | |
| 10.13 | | Form of Common Stock Purchase Warrant issued in the 2021 Private Placement | | 8-K | | 10.22 | | 05/12/2021 | | |
| 10.14 | | Promissory Note dated October 28, 2021 between the Registrant and Manchester Explorer, L.P. | | 8-K | | 10.27 | | 10/29/2021 | | |
| 10.15 | | Warrant Agency Agreement between the Registrant and Colonial Stock Transfer Company, Inc., dated February 14, 2022 | | 8-K | | 10.1 | | 02/14/2022 | | |
| 10.16 | | Form of Warrant Omnibus Amendment Agreement | | S-1/A | | 10.31 | | 02/07/2022 | | |
| 10.17 | | Form of Securities Purchase Agreement dated May 2, 2022 | | 8-K | | 10.1 | | 05/05/2022 | | |
| 10.18* | | Severance and Release Agreement between the Registrant and Ellen O’Connor Vos dated February 23, 2022 | | S-1 | | 10.33 | | 07/06/2022 | | |
| 10.19* | | Offer Letter Agreement between the Registrant and Kevin Schmid dated July 13, 2022 | | 8-K | | 10.1 | | 07/26/2022 | | |
| 10.20* | | Form of Notice of Grant of Restricted Stock Unit Award and Agreement under the Amended and Restated Modular Medical, Inc. 2017 Equity Incentive Plan | | 10-Q | | 4.11 | | 08/14/2023 | | |
| 10.21 | | Form of Warrant Agency Agreement | | S-1/A | | 10.29 | | 05/05/2023 | | |
| 10.22 | | Form of Common Stock Purchase Agreement dated October 28, 2021 between the Registrant and the Investors named therein | | 8-K | | 10.29 | | 10/29/2021 | | |
| 10.23 | | Modular Medical, Inc. Two-Part FDA Submission and Clearance Milestone Bonus Program | | 8-K | | 10.1 | | 10/05/2023 | | |
| 10.24* | | Third Amendment to Employment Agreement between the Company and Paul DiPerna | | 8-K | | 10.1 | | 04/10/2024 | | |
| 21.1 | | List of Subsidiaries | | | | | | | | X |
| 23.1 | | Consent of Independent Registered Public Accounting Firm | | | | | | | | X |
| 24.1 | | Power of Attorney (see signature page of this Report) | | | | | | | | X |
| 31.1 | | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | | | | | | | X |
| 31.2 | | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | | | | | | | | X |
| 32.1 | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | | | | | | | | X |
| 97.1 | | Compensation Recovery Policy | | | | | | | | X |
| 101.INS | | Inline XBRL Instance Document. | | | | | | | | X |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Linkbase Document. | | | | | | | | X |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | | | | | | | | X |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document. | | | | | | | | X |
| 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document. | | | | | | | | X |
| 101.PRE | | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | | | | | | | | X |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). | | | | | | | | X |
| * | Indicates a management contract or compensatory plan or arrangement. |
Item 16. Form 10-K Summary
Not applicable.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on the 21st day of June, 2024.
| | MODULAR MEDICAL, INC. |
| | |
| | By: | /s/ James E. Besser |
| | | James E. Besser |
| | | Chief Executive Officer, |
| | | (Principal Executive Officer) |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints James E. Besser and Paul DiPerna as her/his true and lawful attorneys-in-fact and agent, with full power of substitution and resubstitution, for her and him and in her or his name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in- fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | | Title | | Date |
| | | | | |
| /s/ James E. Besser | | Chief Executive Officer | | June 21, 2024 |
| James E. Besser | | (Principal Executive Officer) | | |
| | | | | |
| /s/ Paul DiPerna | | Chairman, President and Chief Financial Officer
(Principal Financial and Accounting Officer) | | June 21, 2024 |
| Paul DiPerna | | | | |
| | | | | |
| /s/ Duane DeSisto | | Director | | June 21, 2024 |
| Duane DeSisto | | | | |
| | | | | |
| /s/ Steven Felsher | | Director | | June 21, 2024 |
| Steven Felsher | | | | |
| | | | | |
| /s/ Morgan C. Frank | | Director | | June 21, 2024 |
| Morgan C. Frank | | | | |
| | | | | |
| /s/ Philip Sheibley | | Director | | June 21, 2024 |
| Philip Sheibley | | | | |
| | | | | |
| /s/ Carmen Volkart | | Director | | June 21, 2024 |
| Carmen Volkart | | | | |
| | | | | |
| /s/ Ellen O’Connor Vos | | Director | | June 21, 2024 |
| Ellen O’Connor Vos | | | | |
61
iso4217:USD xbrli:shares