UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | ||
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended December 31, 2009 | ||
or | ||
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to | ||
Commission File No. 0-30319
THERAVANCE, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 94-3265960 (I.R.S. Employer Identification No.) | |
901 Gateway Boulevard, South San Francisco, California (Address of principal executive offices) | 94080 (Zip Code) | |
Registrant's telephone number, including area code:650-808-6000 | ||
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
| Title of Each Class | Name of Each Exchange On Which Registered | |
|---|---|---|
| Common Stock $0.01 Par Value | Nasdaq Global Market |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT:NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 205 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer. See definition of "accelerated filer and large accelerated filer" in Rule 12b-2 of the Exchange Act (Check One):
| Large accelerated filer o | Accelerated filer ý | Non-accelerated filer o (Do not check if a smaller reporting company) | Smaller reporting companyo |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
The aggregate market value of the voting and non-voting common equity (consisting of Common Stock, $0.01 par value and Class A Common Stock, $0.01 par value) held by non-affiliates of the registrant based upon the closing price of the Common Stock on the Nasdaq Global Market on June 30, 2009 was $576,528,588.
On February 16, 2010, there were 54,830,359 shares of the registrant's Common Stock and 9,401,499 shares of the registrant's Class A Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant's definitive Proxy Statement to be issued in conjunction with the registrant's 2010 Annual Meeting of Stockholders, which is expected to be filed not later than 120 days after the registrant's fiscal year ended December 31, 2009, are incorporated by reference into Part III of this Annual Report. Except as expressly incorporated by reference, the registrant's Proxy Statement shall not be deemed to be a part of this Annual Report on Form 10-K.
2009 Form 10-K Annual Report
Table of Contents
PART I | ||||
Item 1. | Business | 3 | ||
Item 1A. | Risk Factors | 15 | ||
Item 1B. | Unresolved Staff Comments | 33 | ||
Item 2. | Properties | 33 | ||
Item 3. | Legal Proceedings | 33 | ||
Item 4. | Submission of Matters to a Vote of Security Holders | 33 | ||
PART II | ||||
Item 5. | Market for the Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 34 | ||
Item 6. | Selected Financial Data | 37 | ||
Item 7. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 39 | ||
Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 50 | ||
Item 8. | Financial Statements and Supplementary Data | 51 | ||
Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | 81 | ||
Item 9A. | Controls and Procedures | 81 | ||
Item 9B. | Other Information | 84 | ||
PART III | ||||
Item 10. | Directors, Executive Officers and Corporate Governance | 84 | ||
Item 11. | Executive Compensation | 84 | ||
Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 84 | ||
Item 13. | Certain Relationships and Related Transactions, and Director Independence | 84 | ||
Item 14. | Principal Accounting Fees and Services | 84 | ||
PART IV | ||||
Item 15. | Exhibits and Financial Statement Schedules | 85 | ||
Signatures | 89 | |||
Exhibits | ||||
2
Special Note regarding Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Such forward-looking statements involve substantial risks, uncertainties and assumptions. All statements in this Annual Report on Form 10-K, other than statements of historical facts, including statements regarding our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans, intentions, expectations and objectives could be forward-looking statements. The words "anticipates," "believes," "designed," "estimates," "expects," "goal," "intends," "may," "plans," "projects," "pursuing," "will," "would" and similar expressions (including the negatives thereof) are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. We may not actually achieve the plans, intentions, expectations or objectives disclosed in our forward-looking statements and the assumptions underlying our forward-looking statements may prove incorrect. Therefore, you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions, expectations and objectives disclosed in the forward-looking statements that we make. Factors that we believe could cause actual results or events to differ materially from our forward-looking statements include, but are not limited to, those discussed below in "Risk Factors" in Item 1A, "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Item 7 and elsewhere in this Annual Report on Form 10-K. Our forward-looking statements in this Annual Report on Form 10-K are based on current expectations and we do not assume any obligation to update any forward-looking statements.
Overview
Theravance is a biopharmaceutical company with a pipeline of internally discovered product candidates. We are focused on the discovery, development and commercialization of small molecule medicines across a number of therapeutic areas including respiratory disease, bacterial infections and gastrointestinal motility dysfunction. Our key programs include: VIBATIV™ (telavancin) with Astellas Pharma Inc. (Astellas) and our RELOVAIR™ program (formerly referred to as Horizon) and the Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA) program with GlaxoSmithKline plc (GSK). By leveraging our proprietary insight of multivalency to drug discovery focused primarily on validated targets, we are pursuing a next generation strategy designed to discover superior medicines in areas of significant unmet medical need. Our headquarters are located at 901 Gateway Boulevard, South San Francisco, California 94080. Theravance was incorporated in Delaware in November 1996 under the name Advanced Medicine, Inc. and began operations in May 1997. The Company changed its name to Theravance, Inc. in April 2002.
Our strategy focuses on the discovery, development and commercialization of medicines with superior efficacy, convenience, tolerability and/or safety. By primarily focusing on biological targets that have been clinically validated either by existing medicines or by potential medicines in late-stage clinical studies, we can leverage years of available knowledge regarding a target's activity and the animal models used to test potential medicines against such targets. We move a product candidate into development after it demonstrates the potential to be superior to existing medicines or drug candidates in animal models that we believe correlate to human clinical experience. This strategy of developing the next generation of existing medicines or potential medicines is designed to reduce technical risk and increase productivity. We believe that we can enhance the probability of successfully developing and commercializing medicines by identifying at least two structurally different product candidates, whenever practicable, in each therapeutic program. In total, our research and development expenses,
3
including stock-based compensation expense, incurred for all of our therapeutic programs in 2009, 2008 and 2007 were $77.5 million, $82.0 million and $155.3 million, respectively.
We have entered into collaboration arrangements with GSK and Astellas for the development and commercialization of our product candidates. In November 2002 we entered into our long-acting beta2 agonist (LABA) collaboration with GSK to develop and commercialize a once-daily LABA product candidate both as a single-agent new medicine for the treatment of chronic obstructive pulmonary disease (COPD) and as part of a new combination medicine with an inhaled corticosteroid (ICS) for the treatment of asthma and/or a long-acting muscarinic antagonist (LAMA) for COPD. This collaboration is now known as the RELOVAIR™ program. In March 2004 we entered into a strategic alliance agreement with GSK under which GSK received an option to license exclusive development and commercialization rights to product candidates from all of our full drug discovery programs initiated prior to September 1, 2007, on pre-determined terms and on an exclusive, worldwide basis. Our 2005 collaboration arrangement with Astellas covers the development and commercialization of VIBATIV™, a bactericidal, once-daily injectable antibiotic developed by us for the treatment of Gram-positive infections, including methicillin-resistantStaphylococcus aureus. The U.S. Food and Drug Administration has approved VIBATIV™ for the treatment of adult patients with complicated skin and skin structure infections (cSSSI) caused by susceptible Gram-positive bacteria, includingStaphylococcus aureus, both methicillin-resistant (MRSA) and methicillin-susceptible (MSSA) strains. VIBATIV™ is also approved in Canada for the treatment of adult patients with cSSSI.
4
Our Programs
Our drug discovery efforts are based on the principles of multivalency. Multivalency involves the simultaneous attachment of a single molecule to multiple binding sites on one or more biological targets. We have applied our expertise in multivalency to discover product candidates and lead compounds in a wide variety of therapeutic areas. We have conducted extensive research in both relevant laboratory and animal models to demonstrate that by applying the design principles of multivalency, we can achieve significantly stronger and more selective attachment of our compounds to a variety of intended biological targets. We believe that medicines that attach more strongly and selectively to their targets will be superior to many medicines by substantially improving potency, duration of action and/or safety. The table below summarizes the status of our most advanced product candidates for internal development or co-development. Prior to entering into human clinical studies, a product candidate undergoes preclinical studies which include formulation development or safety testing in animal models.
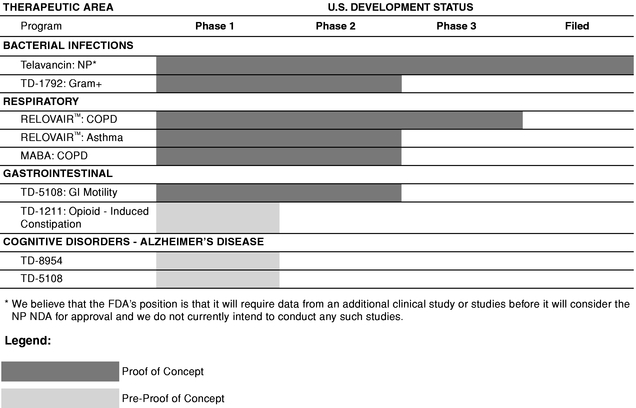
In the table above:
- •
- Development Status indicates the most advanced stage of development that has been completed or is in process.
- •
- Phase 1 indicates initial clinical safety testing in healthy volunteers, or studies directed toward understanding the mechanisms of action of the drug.
- •
- Phase 2 indicates further clinical safety testing and preliminary efficacy testing in a limited patient population.
- •
- Phase 3 indicates evaluation of clinical efficacy and safety within an expanded patient population.
- •
- Filed indicates that a New Drug Application (NDA) or Market Authorization Application (MAA) has been submitted to and accepted for filing by the FDA or the European Medicines Agency (EMEA), respectively.
- •
- We consider programs in which at least one compound has successfully completed a Phase 2a study showing efficacy and tolerability as having achieved Proof of Concept.
5
Our Relationship with Astellas
2005 License, Development and Commercialization Agreement
In November 2005, we entered into a collaboration arrangement with Astellas for the development and commercialization of telavancin. In July 2006, Japan was added to our telavancin collaboration, thereby giving Astellas worldwide rights to this medicine. Through December 31, 2009, we have received $190.0 million in upfront, milestone and other fees from Astellas and we are eligible to receive up to an additional $30.0 million in remaining milestone payments related to regulatory filings and approvals in various regions of the world. Additionally, certain costs related to the collaboration are reimbursable by Astellas.
In 2009 the FDA approved VIBATIVTM for the treatment of adult patients with cSSSI caused by susceptible Gram-positive bacteria, includingStaphylococcus aureus, both MRSA and MSSA strains. VIBATIVTM also was approved in Canada in 2009 for the treatment of adult patients with cSSSI. We are entitled to receive royalties on global net sales of VIBATIVTM that, on a percentage basis, range from the high teens to the upper twenties depending on sales volume. We were responsible for substantially all costs to develop and obtain U.S. regulatory approval for VIBATIVTM and Astellas is responsible for substantially all costs associated with commercialization of VIBATIVTM.
Our Relationship with GlaxoSmithKline
RELOVAIRTM Program
In November 2002, we entered into our long-acting beta2 agonist (LABA) collaboration with GSK to develop and commercialize a once-daily LABA product candidate both as a single-agent new medicine for the treatment of COPD and as part of a new combination medicine with an ICS for the treatment of asthma and/or a LAMA for COPD. These programs, now known collectively as the RELOVAIRTM program, are aimed at developing next generation respiratory products to replace GSK's Seretide and Advair franchise, which reported 2009 sales of approximately $8.0 billion. Each company contributed four LABA product candidates to the collaboration.
In connection with the RELOVAIRTM program, in 2002 we received from GSK an upfront payment of $10.0 million and sold to an affiliate of GSK shares of our Series E preferred stock for an aggregate purchase price of $40.0 million. In addition, we were eligible to receive up to $495.0 million in development, approval, launch, and sales milestones and royalties on the sales of any product resulting from this program. Through December 31, 2009, we have received a total of $60.0 million in upfront and development milestone payments. GSK has determined to focus the collaboration's resources on the development of the lead LABA, GW642444 ('444), a GSK-discovered compound, together with GSK's ICS, fluticasone furoate (FF). Accordingly, we do not expect to receive any further milestone payments from the RELOVAIRTM program. In the event that a LABA product candidate discovered by GSK is successfully developed and commercialized, we will be obligated to make milestone payments to GSK which could total as much as $220.0 million if both a single-agent and a combination product were launched in multiple regions of the world. Based on available information, we do not estimate that a significant portion of these potential milestone payments to GSK are likely to be made in the next two years. Moreover, we are entitled to receive the same royalties on sales of medicines from the RELOVAIRTM program, regardless of whether the product candidate originated with Theravance or with GSK. Theravance is entitled to annual royalties of 15% on the first $3.0 billion of annual global net sales and 5% for all annual global net sales above $3.0 billion. Sales of single-agent LABA medicines and combination medicines would be combined for the purposes of this royalty calculation. For other products combined with a LABA from the RELOVAIRTM program, such as a combination LABA/LAMA medicine, which are launched after a LABA/ICS combination medicine, royalties are upward tiering and range from the mid-single digits to 10%. However, if GSK is
6
not selling a LABA/ICS combination product at the time that the first other LABA combination is launched, then the royalties described above for the LABA/ICS combination medicine are applicable.
2004 Strategic Alliance
In March 2004, we entered into our strategic alliance with GSK. Under this alliance, GSK received an option to license exclusive development and commercialization rights to product candidates from all of our full drug discovery programs initiated prior to September 1, 2007, on pre-determined terms and on an exclusive, worldwide basis. Pursuant to the terms of the strategic alliance agreement, we initiated three new full discovery programs between May 2004 and August 2007. These three programs are (i) our peripheral Opioid-Induced Bowel Constipation (PUMA) program, (ii) our AT1 Receptor—Neprilysin Inhibitor (ARNI) program for cardiovascular disease and (iii) our MonoAmine Reuptake Inhibitor (MARIN) program for chronic pain. GSK has the right to license product candidates from these three programs, and must exercise this right no later than sixty days subsequent to the "proof-of-concept" stage (generally defined as the successful completion of a Phase 2a clinical study showing efficacy and tolerability if the biological target for the drug has been clinically validated by an existing medicine, and successful completion of a Phase 2b clinical study showing efficacy and tolerability if the biological target for the drug has not been clinically validated by an existing medicine). Under the terms of the strategic alliance, GSK has only one opportunity to license each of our programs. Upon its decision to license a program, GSK is responsible for funding all future development, manufacturing and commercialization activities for product candidates in that program. In addition, GSK is obligated to use diligent efforts to develop and commercialize product candidates from any program that it licenses. Consistent with our strategy, we are obligated at our sole cost to discover two structurally different product candidates for any programs that are licensed by GSK under the alliance. If these programs are successfully advanced through development by GSK, we are entitled to receive clinical, regulatory and commercial milestone payments and royalties on any sales of medicines developed from these programs. For product candidates licensed to date under this agreement, the royalty structure for a product containing one of our compounds as a single active ingredient would result in an average percentage royalty rate in the low double digits. If a product is successfully commercialized, in addition to any royalty revenue that we receive, the total upfront and milestone payments that we could receive in any given program that GSK licenses range from $130.0 million to $162.0 million for programs with single-agent medicines and up to $252.0 million for programs with both a single-agent and a combination medicine. If GSK chooses not to license a program, we retain all rights to the program and may continue the program alone or with a third party. To date, GSK has licensed our two COPD programs: LAMA and MABA. We received a $5.0 million payment from GSK in connection with its license of each of our LAMA and MABA programs in August 2004 and March 2005, respectively. However, in 2009, GSK returned the LAMA program to us because the formulation of the lead product candidate was incompatible with GSK's proprietary inhaler device. GSK has chosen not to license our bacterial infections program, our anesthesia program and our 5-HT4 program. There can be no assurance that GSK will license any of the remaining programs under the alliance agreement, which could have an adverse effect on our business and financial condition.
In connection with the strategic alliance with GSK, we received from GSK a payment of $20.0 million. In May 2004, GSK purchased through an affiliate 6,387,096 shares of our Class A common stock for an aggregate purchase price of $108.9 million. Through December 31, 2009, we have received $46.0 million in upfront and milestone payments from GSK relating to the strategic alliance agreement. In addition, pursuant to a partial exercise of its rights under the governance agreement, upon the closing of our initial public offering on October 8, 2004, GSK purchased through an affiliate an additional 433,757 shares of Class A common stock. GSK's ownership position of our outstanding stock was approximately 14.6% as of February 16, 2010.
7
Development Programs
Respiratory Programs
RELOVAIRTM
In December 2008, we announced positive results from a Phase 2b study evaluating the dose-response, safety, and efficacy of five doses of the lead LABA compound, '444, in patients with moderate-to-severe COPD, and in February 2009 we announced positive results from three separate Phase 2b clinical studies assessing the safety and efficacy of GSK's ICS, FF across a range of eight doses in over 1,800 patients with mild, moderate and severe asthma.
In late October 2009, we and GSK announced that the first patient commenced treatment in the Phase 3 program in COPD. The program comprises a broad range of large-scale Phase 3 clinical studies to evaluate the once-a-day LABA, '444, in combination with the once-a-day ICS, FF, for the treatment of COPD. The overall registrational program, which will study more than 6,000 patients, includes two 12-month exacerbation studies, two six-month efficacy and safety studies and a detailed lung function profile study. In addition, other studies are planned to assess the potential for superiority of the fixed combination of '444 and FF versus other treatments for COPD. GSK is responsible for funding the aforementioned studies.
In addition to the COPD development program, we and GSK remain committed to the progression of the RELOVAIRTM program for the treatment of asthma, details of which are expected to be announced later in 2010.
Inhaled Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA) Program
In our MABA program, we are developing with GSK a bifunctional long-acting inhaled bronchodilator. By combining bifunctional activity and high lung selectivity, we intend to develop a medicine with greater efficacy than single mechanism bronchodilators (such as tiotropium or salmeterol) and with equal or better tolerability. In our MABA program in COPD, we are currently waiting for the completion and review of Phase 2b enabling studies before determining whether to commence the next stage of clinical development. All clinical studies in this program are fully funded and paid for by GSK.
Bacterial Infections Program
Telavancin
In October 2009, Astellas and we announced that Astellas Pharma Europe B.V. submitted a MAA to the EMEA for telavancin for the treatment of NP, including ventilator-associated pneumonia, and complicated skin and soft tissue infections in adults (cSSTI). The EMEA has since completed the Validation Phase for the MAA and initiated the scientific review of the application.
On November 27, 2009 we announced that we received a Complete Response letter from the FDA relating to our telavancin NDA for NP, which was filed in January 2009. The Complete Response instructed us that submission of additional data and analyses for the NP patient population to support an evaluation of all-cause mortality as the primary efficacy endpoint was necessary to demonstrate the safety and efficacy of telavancin. The Phase 3 NP clinical program included clinical response as the primary efficacy endpoint, consistent with current draft FDA guidelines for antibacterial clinical trial design in NP, and all-cause mortality as a secondary endpoint. The Complete Response did not specify the time point at which the FDA will measure the all-cause mortality data, nor did it indicate the populations in which these analyses will be considered. The Complete Response letter also requested a scientific rationale for pooling the all-cause mortality data from the two studies as they may individually
8
be of insufficient size and statistical power to support the evaluation of all-cause mortality as the primary efficacy endpoint.
We responded to the Complete Response letter in December 2009. The key elements of our response included a rationale for pooling the two Phase 3 NP studies to evaluate all-cause mortality as the primary efficacy endpoint and all available all-cause mortality data that was analyzed using Kaplan-Meier survival estimates. In January 2010 the FDA sent us a letter notifying us that it considered our response "incomplete," and stating that even if pooling of the two studies is acceptable for analyzing mortality, the two pooled studies would then equate to only one adequate and well-controlled trial and therefore would not constitute the substantial evidence of efficacy required for approval. In addition, the FDA noted that the adequacy and similarity of populations across the studies for the purposes of pooling had not yet been determined, and is still a review issue. Finally, the FDA also noted several design criteria that should be taken into account in the design of new clinical trials. These design criteria do not include a specific primary endpoint for the evaluation of efficacy, the size or number of studies required, or what the appropriate statistical analysis might be. As a result, the design, size and scope of any additional studies required by the FDA are unclear at this time. With regard to our telavancin NP NDA, we believe that the FDA's position is that it will require data from an additional clinical study or studies before it will consider the NP NDA for approval and we do not currently intend to conduct any such studies.
Other Pipeline Programs
In addition to telavancin, RELOVAIRTM and MABA, we have a number of other clinical-stage programs for bacterial infections, gastrointestinal motility and cognitive disorders.
TD-1792 is our investigational heterodimer antibiotic that combines the antibacterial activities of a glycopeptide and a beta-lactam in one molecule. The goal of our program with TD-1792 is to develop a next-generation antibiotic for the treatment of serious infections caused by Gram-positive bacteria. During the third quarter of 2009, we began a Phase 1 bronchoalveolar lavage (BAL) study that will provide data on the penetration of TD-1792 into lung tissue and lung fluids in order to evaluate the potential of this compound as a treatment for NP.
Our Gastrointestinal (GI) Motility Dysfunction program is dedicated to finding new medicines for GI motility disorders such as chronic idiopathic constipation (CIC) and other disorders related to reduced gastrointestinal motility. Our lead compound in this area is TD-5108, a highly selective 5-HT4 receptor agonist that has successfully completed a 400 patient Phase 2 study in CIC.
We are also developing TD-1211, an oral peripheral Mu-opioid antagonist (PUMA) for the treatment of opioid-induced bowel constipation. We completed a successful single-ascending dose Phase 1 study with TD-1211 and recently progressed the compound into a multiple-ascending dose Phase 1 study.
In cognitive disorders, we are currently evaluating compounds TD-5108 and TD-8954 as potential treatments for Alzheimer's disease. In the second quarter of 2009, we announced that TD-8954 successfully completed a single-ascending dose Phase 1 study. Recently we began multiple-ascending dose Phase 1 studies with each of TD-5108 and TD-8954 to evaluate their penetration into the central nervous system.
In our MARIN program for the treatment of neuropathic pain, we have completed IND-enabling studies with compound TD-9855 and anticipate commencing Phase 1 studies later in 2010.
Multivalency
Our proprietary approach combines chemistry and biology to efficiently discover new product candidates using our expertise in multivalency. Multivalency refers to the simultaneous attachment of a
9
single molecule to multiple binding sites on one or more biological targets. When compared to monovalency, whereby a molecule attaches to only one binding site, multivalency can significantly increase a compound's potency, duration of action and/or selectivity. Multivalent compounds generally consist of several individual small molecules, at least one of which is biologically active when bound to its target, joined by linking components.
Our approach is based on an integration of the following insights:
- •
- many targets have multiple binding sites and/or exist in clusters with similar or different targets;
- •
- biological targets with multiple binding sites and/or those that exist in clusters lend themselves to multivalent drug design;
- •
- molecules that simultaneously attach to multiple binding sites can exhibit considerably greater potency, duration of action and/or selectivity than molecules that attach to only one binding site; and
- •
- greater potency, duration of action and/or selectivity provides the basis for superior therapeutic effects, including enhanced convenience, tolerability and/or safety compared to conventional drugs.
Our Strategy
Our objective is to discover, develop and commercialize new medicines with superior efficacy, convenience, tolerability and/or safety. The key elements of our strategy are to:
Apply our expertise in multivalency primarily to validated targets to efficiently discover and develop superior medicines in areas of significant unmet medical need. We intend to continue to concentrate our efforts on discovering and developing product candidates where:
- •
- existing drugs have levels of efficacy, convenience, tolerability and/or safety that are insufficient to meet an important medical need;
- •
- we believe our expertise in multivalency can be applied to create superior product candidates that are more potent, longer acting and/or more selective than currently available medicines;
- •
- there are established animal models that can be used to provide us with evidence as to whether our product candidates have the potential to provide superior therapeutic benefits relative to current medicines; and
- •
- there is a relatively large commercial opportunity.
Identify two structurally different product candidates in each therapeutic program whenever practicable. We believe that we can increase the likelihood of successfully bringing superior medicines to market by identifying, whenever practicable, two product candidates for development in each program. Our second product candidates are typically in a different structural class from the first product candidate. Applying this strategy can reduce our dependence on any one product candidate and provide us with the potential opportunity to commercialize two compounds in a given area.
Partner with global pharmaceutical companies. Our strategy is to seek collaborations with leading global pharmaceutical companies to accelerate development and commercialization of our product candidates at the strategically appropriate time. The RELOVAIRTM program and our strategic alliance with GSK, and our telavancin collaboration with Astellas, are examples of these types of partnerships.
Leverage the extensive experience of our people. We have an experienced senior management team with many years of experience discovering, developing and commercializing new medicines with
10
companies such as Bristol-Myers Squibb Company, Merck & Co., Gilead Sciences, Pfizer and ICOS Corporation.
Improve, expand and protect our technical capabilities. We have created a substantial body of know-how and trade secrets in the application of our multivalent approach to drug discovery. We believe this is a significant asset that distinguishes us from our competitors. We expect to continue to make substantial investments in drug discovery using multivalency and other technologies to maintain what we believe are our competitive advantages.
Manufacturing
We primarily rely on a number of third parties, including contract manufacturing organizations and our collaborative partners, to produce our active pharmaceutical ingredient and drug product. Manufacturing of compounds in our RELOVAIRTM and MABA programs is handled by GSK. Additionally, GSK will be responsible for the manufacturing of any additional product candidates associated with the programs that it licenses under the strategic alliance agreement.
We believe that we have in-house expertise to manage a network of third-party manufacturers. We believe that we will be able to continue to negotiate third party manufacturing arrangements on commercially reasonable terms and that it will not be necessary for us to develop internal manufacturing capacity in order to commercialize our products. However, if we are unable to obtain contract manufacturing or obtain such manufacturing on commercially reasonable terms, or if manufacturing is interrupted at one of our suppliers, whether due to regulatory or other reasons, we may not be able to develop or commercialize our products as planned.
Government Regulation
The development and commercialization of our product candidates and our ongoing research are subject to extensive regulation by governmental authorities in the United States and other countries. Before marketing in the United States, any medicine we develop must undergo rigorous preclinical studies and clinical studies and an extensive regulatory approval process implemented by the FDA under the Federal Food, Drug, and Cosmetic Act. Outside the United States, our ability to market a product depends upon receiving a marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical studies, marketing authorization, pricing and reimbursement vary widely from country to country. In any country, however, we will be permitted to commercialize our medicines only if the appropriate regulatory authority is satisfied that we have presented adequate evidence of the safety, quality and efficacy of our medicines.
Before commencing clinical studies in humans in the United States, we must submit to the FDA an Investigational New Drug application that includes, among other things, the results of preclinical studies. If the FDA accepts the Investigational New Drug submission, clinical studies are usually conducted in three phases and under FDA oversight. These phases generally include the following:
Phase 1. The product candidate is introduced into healthy human volunteers and is tested for safety, dose tolerance and pharmacokinetics.
Phase 2. The product candidate is introduced into a limited patient population to assess the efficacy of the drug in specific, targeted indications, assess dosage tolerance and optimal dosage, and identify possible adverse effects and safety risks.
Phase 3. If a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 evaluations, the clinical study will be expanded to further demonstrate clinical efficacy, optimal dosage and safety within an expanded patient population.
11
The results of product development, preclinical studies and clinical studies must be submitted to the FDA as part of a new drug application, or NDA. The NDA also must contain extensive manufacturing information. NDAs for new chemical entities are subject to performance goals defined in the Prescription Drug User Fee Act (PDUFA) which suggests a goal for FDA action within 6 months for applications that are granted priority review and 10 months for applications that receive standard review. For a product candidate no active ingredient of which has been previously approved by the FDA, the FDA must either refer the product candidate to an advisory committee for review or provide in the action letter on the application for the product candidate a summary of the reasons why the product candidate was not referred to an advisory committee prior to approval. In addition, under the 2008 Food and Drug Administration Amendments Act, the FDA has authority to require submission of a formal Risk Evaluation and Management Strategy (REMS) to ensure safe use of the product. At the end of the review period, the FDA communicates an approval of the NDA or issues a complete response listing the application's deficiencies.
Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if safety or quality issues are identified after the product reaches the marketplace. In addition, the FDA may require post-marketing studies, referred to as Phase 4 studies, to monitor the effect of approved products, and may limit further marketing of the product based on the results of these post-marketing studies. The FDA has broad post-market regulatory and enforcement powers, including the ability to suspend or delay issuance of approvals, seize or recall products, withdraw approvals, enjoin violations, and institute criminal prosecution.
If we obtain regulatory approval for a medicine, this clearance to market the product will be limited to those diseases and conditions for which the medicine is effective, as demonstrated through clinical studies and included in the medicine's labeling. Even if this regulatory approval is obtained, a marketed medicine, its manufacturer and its manufacturing facilities are subject to continual review and periodic inspections by the FDA. The FDA ensures the quality of approved medicines by carefully monitoring manufacturers' compliance with its current Good Manufacturing Practice (cGMP) regulations. The cGMP regulations for drugs contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a medicine. The regulations make sure that a medicine is safe for use, and that it has the ingredients and strength it claims to have. Discovery of previously unknown problems with a medicine, manufacturer or facility may result in restrictions on the medicine or manufacturer, including costly recalls or withdrawal of the medicine from the market.
We are also subject to various laws and regulations regarding laboratory practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances in connection with our research. In each of these areas, as above, the FDA and other regulatory authorities have broad regulatory and enforcement powers, including the ability to suspend or delay issuance of approvals, seize or recall products, withdraw approvals, enjoin violations, and institute criminal prosecution, any one or more of which could have a material adverse effect upon our business, financial condition and results of operations.
Outside the United States our ability to market our products will also depend on receiving marketing authorizations from the appropriate regulatory authorities. Risks similar to those associated with FDA approval described above exist with the regulatory approval processes in other countries.
Patents and Proprietary Rights
We will be able to protect our technology from unauthorized use by third parties only to the extent that our technology is covered by valid and enforceable patents or is effectively maintained as trade secrets. Our success in the future will depend in part on obtaining patent protection for our product candidates. Accordingly, patents and other proprietary rights are essential elements of our business.
12
Our policy is to seek in the United States and selected foreign countries patent protection for novel technologies and compositions of matter that are commercially important to the development of our business. For proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our drug discovery process that involve proprietary know-how and technology that is not covered by patent applications, we rely on trade secret protection and confidentiality agreements to protect our interests. We require all of our employees, consultants and advisors to enter into confidentiality agreements. Where it is necessary to share our proprietary information or data with outside parties, our policy is to make available only that information and data required to accomplish the desired purpose and only pursuant to a duty of confidentiality on the part of those parties.
As of December 31, 2009, we own 183 issued United States patents and 765 granted foreign patents. In addition, we have 122 United States patent applications pending and 740 foreign patent applications pending. The claims in these various patents and patent applications are directed to compositions of matter, including claims covering product candidates, lead compounds and key intermediates, pharmaceutical compositions, methods of use and processes for making our compounds along with methods of design, synthesis, selection and use relevant to multivalency in general and to our research and development programs in particular.
United States issued patents and foreign patents generally expire 20 years after filing. The patent rights relating to telavancin owned by us and licensed to Astellas currently consist of United States patents that expire between 2019 and 2024, additional pending United States patent applications and counterpart patents and patent applications in a number of jurisdictions, including Europe. Nevertheless, issued patents can be challenged, narrowed, invalidated or circumvented, which could limit our ability to stop competitors from marketing similar products and threaten our ability to commercialize our product candidates. Our patent position, similar to other companies in our industry, is generally uncertain and involves complex legal and factual questions. To maintain our proprietary position we will need to obtain effective claims and enforce these claims once granted. It is possible that, before any of our products can be commercialized, any related patent may expire or remain in force only for a short period following commercialization, thereby reducing any advantage of the patent. Also, we do not know whether any of our patent applications will result in any issued patents or, if issued, whether the scope of the issued claims will be sufficient to protect our proprietary position.
We have entered into a License Agreement with Janssen Pharmaceutica pursuant to which we have licensed rights under certain patents owned by Janssen covering an excipient used in the formulation of telavancin. We believe that the general and financial terms of the agreement with Janssen are ordinary course terms. Pursuant to the terms of this license agreement, we are obligated to pay royalties and milestone payments to Janssen based on any commercial sales of telavancin. Astellas has agreed to assume responsibility for these payments under the terms of our license agreement with them. The license is terminable by us upon prior written notice to Janssen or upon an uncured breach or a liquidation event of one of the parties.
Competition
Our objective is to discover, develop and commercialize new medicines with superior efficacy, convenience, tolerability and/or safety. To the extent that we are able to develop medicines, they are likely to compete with existing drugs that have long histories of effective and safe use and with new therapeutic agents. We expect that any medicines that we commercialize with our collaborative partners or on our own will compete with existing, market-leading medicines.
Many of our potential competitors have substantially greater financial, technical and personnel resources than we have. In addition, many of these competitors have significantly greater commercial
13
infrastructures than we have. Our ability to compete successfully will depend largely on our ability to leverage our experience in drug discovery and development to:
- •
- discover and develop medicines that are superior to other products in the market;
- •
- attract qualified scientific, product development and commercial personnel;
- •
- obtain patent and/or other proprietary protection for our medicines and technologies;
- •
- obtain required regulatory approvals; and
- •
- successfully collaborate with pharmaceutical companies in the discovery, development and commercialization of new medicines.
VIBATIVTM (telavancin). VIBATIV™ competes with vancomycin, a generic drug that is manufactured by a variety of companies, as well as other drugs targeted at Gram-positive bacterial infections. Currently marketed products include but are not limited to daptomycin (marketed by Cubist Pharmaceuticals), linezolid (marketed by Pfizer) and tigecycline (marketed by Wyeth). To effectively compete with these medicines, and in particular with the relatively inexpensive generic option of vancomycin, we and our partner Astellas will need to demonstrate to physicians that, based on experience, clinical data, side-effect profiles and other factors, VIBATIV™ is preferable to vancomycin and other existing or subsequently-developed anti-infective drugs in certain clinical situations.
RELOVAIRTM Program with GSK. We anticipate that, if approved, any product from our RELOVAIRTM program with GSK will compete with a number of approved bronchodilator drugs and drug candidates under development that are designed to treat asthma and COPD. These include but are not limited to salmeterol and fluticasone (marketed by GSK), formoterol (marketed by a number of companies) and formoterol and budesonide as a combination (marketed by AstraZeneca), and tiotropium (marketed by Boehringer Ingelheim and Pfizer). Indacaterol is being developed as a single-agent by Novartis and, in combination with an ICS (mometasone). In addition, indacaterol combined with a muscarinic antagonist is being developed by Novartis. New combinations of formoterol with fluticasone or mometasone are being developed by Abbott (with SkyePharma), and Merck respectively. Boehringer-Ingelheim is developing a combination product with tiotropium and the long-acting beta agonist BI-1744 for the treatment of COPD. In addition, several firms are reported to be developing new formulations of salmeterol-fluticasone and formoterol-budesonide which may be marketed as generics or branded generics relative to the innovator products from GSK and AstraZeneca respectively. However, the ability of such generics to achieve fully substitutable status is uncertain as there is no well established regulatory pathway for demonstration of bioequivalence for inhaled medicines. All of these efforts represent potential competition for any product from our RELOVAIRTM program.
In addition, as the principles of multivalent medicine design become more widely known and appreciated based on patent and scientific publications and regulatory filings, we expect the field to become highly competitive. Pharmaceutical companies, biotechnology companies and academic and research institutions may seek to develop product candidates based upon the principles underlying our multivalent technologies.
Employees
As of December 31, 2009, we had 194 employees, 146 of which were primarily engaged in research and development activities. None of our employees are represented by a labor union. We consider our employee relations to be good.
14
Available Information
Our Internet address iswww.theravance.com. Our investor relations website is located athttp://ir.theravance.com. We make available free of charge on our investors relations website under "SEC Filings" our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, our directors' and officers' Section 16 Reports and any amendments to those reports as soon as reasonably practicable after filing or furnishing such materials to the U.S. Securities and Exchange Commission (SEC). The information found on our website is not part of this or any other report that we file with or furnish to the SEC.
In addition to the other information in this Annual Report on Form 10-K, the following risk factors should be considered carefully in evaluating our business and us.
Risks Related to our Business
If the RELOVAIR™ program does not progress into Phase 3 asthma studies, or if the Phase 3 program in asthma or chronic obstructive pulmonary disease (COPD) does not demonstrate safety and efficacy, the RELOVAIRTM program will be significantly delayed, our business will be harmed, and the price of our securities could fall.
In late 2008 and early 2009, we announced results from multiple RELOVAIRTM program Phase 2b asthma studies and a COPD study, and the Phase 3 program for COPD commenced in October 2009. Any adverse developments or results or perceived adverse developments or results with respect to the RELOVAIRTM program will significantly harm our business and could cause the price of our securities to fall. Examples of such adverse developments include, but are not limited to:
- •
- the U.S. Food and Drug Administration (FDA) determining that any of the Phase 2b asthma studies failed to meet study endpoints or raised safety concerns, or that additional clinical studies are required prior to commencing Phase 3 asthma studies;
- •
- the FDA concluding that any of the Phase 3 enabling studies or other clinical or preclinical studies currently underway raise safety or other concerns;
- •
- the FDA, after being presented with data from the Phase 2b studies as well as additional studies, requiring further evidence that the long-acting beta2 agonist (LABA) is a once-daily medication;
- •
- the Phase 3 program in asthma or COPD raising safety concerns or not demonstrating efficacy; or
- •
- any change in FDA policy or guidance regarding the use of LABAs to treat asthma or COPD.
With regard to changes in FDA policy or guidance concerning LABAs, on March 10-11, 2010, the FDA has scheduled an Advisory Committee to discuss the design of medical research studies (known as "clinical trial design") to evaluate serious asthma outcomes (such as hospitalizations, a procedure using a breathing tube known as intubation, or death) with the use of LABAs in the treatment of asthma in adults, adolescents, and children.
In addition, on February 18, 2010 the FDA announced that LABAs should not be used alone in the treatment of asthma, and will require manufacturers to include this warning in the product labels of these drugs, along with taking other steps to reduce the overall use of these medicines. The FDA will now require that the product labels for LABA medicines reflect, among other things, that the use of LABAs is contraindicated without the use of an asthma controller medication such as an inhaled corticosteroid, that LABAs should only be used long-term in patients whose asthma cannot be adequately controlled on asthma controller medications, and LABAs should be used for the shortest
15
duration of time required to achieve control of asthma symptoms and discontinued, if possible, once asthma control is achieved.
It is unknown at this time what, if any, effect these recent or future FDA actions will have on the development of the RELOVAIRTM program.
With regard to our telavancin NP NDA, we believe that the FDA's position is that it will require data from an additional clinical study or studies before it will consider the NP NDA for approval and we do not currently intend to conduct any such studies.
Our first New Drug Application (NDA) for telavancin was submitted in late 2006 and on September 11, 2009 the FDA approved VIBATIV™ (telavancin) for the treatment of adults with complicated skin and skin structure infections (cSSSI) caused by susceptible Gram-positive bacteria. In January 2009 we submitted a second telavancin NDA to the FDA for the NP indication and we received a Complete Response letter from the FDA in late November 2009. The Complete Response instructed us that submission of additional data and analyses for the NP patient population to support an evaluation of all-cause mortality as the primary efficacy endpoint is necessary to demonstrate the safety and efficacy of telavancin. The Phase 3 NP clinical program included clinical response as the primary efficacy endpoint, consistent with current draft FDA guidelines for antibacterial clinical trial design in NP, and all-cause mortality as a secondary endpoint. The Complete Response did not specify the time point at which the FDA will measure the all-cause mortality data, nor did it indicate the populations in which these analyses will be considered. The Complete Response letter also requested a scientific rationale for pooling the all-cause mortality data from the two studies as they may individually be of insufficient size and statistical power to support the evaluation of all-cause mortality as the primary efficacy endpoint.
We responded to the Complete Response letter in December 2009. The key elements of our response included a rationale for pooling the two Phase 3 NP studies to evaluate all-cause mortality as the primary efficacy endpoint and all available all-cause mortality data which was analyzed using Kaplan-Meier survival estimates. In January 2010 the FDA sent us a letter notifying us that it considered our response "incomplete," and stating that even if pooling of the two studies is acceptable for analyzing mortality, the two pooled studies would then equate to only one adequate and well-controlled trial and therefore would not constitute the substantial evidence of efficacy required for approval. In addition, the FDA noted that the adequacy and similarity of populations across the studies for the purposes of pooling had not yet been determined, and is still a review issue. Finally, the FDA also suggested several design criteria that should be taken into account in the design of new clinical trials. These design criteria do not include a specific primary endpoint for the evaluation of efficacy, the size or number of studies required, or what the appropriate statistical analysis might be. As a result, the design, size and scope of any additional studies required by the FDA are unclear at this time. With regard to our telavancin NP NDA, we believe that the FDA's position is that it will require data from an additional clinical study or studies before it will consider the NP NDA for approval and we do not currently intend to conduct any such studies. Any further adverse developments or perceived adverse developments with respect to telavancin for the NP indication, could harm our business and cause the price of our securities to fall.
If telavancin is not approved by the European Medicines Agency (EMEA) or if the EMEA requires data from additional clinical studies of telavancin, our business will be adversely affected and the price of our securities could fall.
On October 28, 2009, Astellas Pharma Europe B.V., a subsidiary of our telavancin partner, Astellas Pharma Inc. (Astellas), announced that it submitted a new European marketing authorization application (MAA) for telavancin to the European Medicines Agency (EMEA) for the treatment of
16
complicated skin and soft tissue infections (cSSTI) and NP and on November 30, 2009 we announced that the EMEA had completed the validation phase for the MAA and the EMEA's scientific review process had begun. In October 2008, we announced that Astellas Pharma Europe B.V. voluntarily withdrew a previously filed MAA for telavancin for the treatment of cSSTI from the EMEA based on communications from the Committee for Medicinal Products for Human Use (CHMP) of the EMEA that the data provided were not sufficient to allow the CHMP to conclude a positive benefit-risk balance for telavancin for the sole indication of cSSTI at that time.
If the EMEA does not approve our application, requires data from additional clinical studies regarding telavancin, or if telavancin is ultimately approved by the EMEA but with restrictions, including labeling that may limit the targeted patient population, our business will be harmed and the price of our securities could fall.
If our product candidates, in particular the lead compounds in the RELOVAIRTM program with GSK that recently commenced a Phase 3 clinical program in COPD, and telavancin for the treatment of NP are determined to be unsafe or ineffective in humans, our business will be adversely affected and the price of our securities could fall.
Although our first approved product, VIBATIV™, was commercially launched in the U.S. by our partner Astellas in November 2009, we have not yet commercialized any of our other product candidates. We are uncertain whether any of our other product candidates will prove effective and safe in humans or meet applicable regulatory standards. In addition, our approach to applying our expertise in multivalency to drug discovery may not result in the creation of successful medicines. The risk of failure for our product candidates is high. For example, in late 2005, we discontinued our overactive bladder program based upon the results of our Phase 1 studies with compound TD-6301, and GSK discontinued development of TD-5742, the first LAMA compound licensed from us, after completing initial Phase 1 studies. To date, the data supporting our drug discovery and development programs is derived solely from laboratory experiments, preclinical studies and clinical studies. A number of other compounds remain in the lead identification, lead optimization, preclinical testing or early clinical testing stages.
Several well-publicized approvable and Complete Response letters issued by the FDA and safety-related product withdrawals, suspensions, post-approval labeling revisions to include boxed warnings and changes in approved indications over the last few years, as well as growing public and governmental scrutiny of safety issues, have created an increasingly conservative regulatory environment. The implementation of new laws and regulations, and revisions to FDA clinical trial design guidelines, have increased uncertainty regarding the approvability of a new drug. In addition, there are additional requirements for approval of new drugs, including advisory committee meetings for new chemical entities, and formal risk evaluation and mitigation strategy (REMS) at the FDA's discretion. These new laws, regulations, additional requirements and changes in interpretation could cause non-approval or further delays in the FDA's review and approval of our product candidates.
With regard to all of our programs, any delay in commencing or completing clinical studies for product candidates, as we are currently experiencing in our Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA) program with GSK, and any adverse results from clinical or preclinical studies or regulatory obstacles product candidates may face, would harm our business and could cause the price of our securities to fall.
Each of our product candidates must undergo extensive preclinical and clinical studies as a condition to regulatory approval. Preclinical and clinical studies are expensive, take many years to complete and study results may lead to delays in further studies or decisions to terminate programs. For example, we had planned to commence Phase 2b clinical studies in our MABA Program with GSK in 2009, but we are awaiting the completion and review of data from several preclinical studies. These
17
key studies, which we have also referred to as "Phase 2b enabling studies," will likely determine whether or not Phase 2b clinical studies in this program proceed as planned. If the analysis of the results of these studies lead to a decision not to proceed, GSK may need to conduct additional work which could significantly delay the MABA Program, or GSK may decide to terminate the entire program.
The commencement and completion of clinical studies for our product candidates may be delayed by many factors, including:
- •
- lack of effectiveness of product candidates during clinical studies;
- •
- adverse events, safety issues or side effects relating to the product candidates or their formulation into medicines;
- •
- inability to raise additional capital in sufficient amounts to continue our development programs, which are very expensive;
- •
- the need to sequence clinical studies as opposed to conducting them concomitantly in order to conserve resources;
- •
- our inability to enter into partnering arrangements relating to the development and commercialization of our programs and product candidates;
- •
- our inability or the inability of our collaborators or licensees to manufacture or obtain from third parties materials sufficient for use in preclinical and clinical studies;
- •
- governmental or regulatory delays and changes in regulatory requirements, policy and guidelines;
- •
- failure of our partners to advance our product candidates through clinical development;
- •
- delays in patient enrollment, which we experienced in our Phase 3 NP program for telavancin, and variability in the number and types of patients available for clinical studies;
- •
- difficulty in maintaining contact with patients after treatment, resulting in incomplete data;
- •
- a regional disturbance where we or our collaborative partners are enrolling patients in our clinical trials, such as a pandemic, terrorist activities or war, or a natural disaster; and
- •
- varying interpretations of data by the FDA and similar foreign regulatory agencies.
If our product candidates that we develop on our own or through collaborative partners are not approved by regulatory agencies, including the FDA, we will be unable to commercialize them.
The FDA must approve any new medicine before it can be marketed and sold in the United States. We must provide the FDA and similar foreign regulatory authorities with data from preclinical and clinical studies that demonstrate that our product candidates are safe and effective for a defined indication before they can be approved for commercial distribution. We will not obtain this approval for a product candidate unless and until the FDA approves a NDA. The processes by which regulatory approvals are obtained from the FDA to market and sell a new product are complex, require a number of years and involve the expenditure of substantial resources. In order to market our medicines in foreign jurisdictions, we must obtain separate regulatory approvals in each country. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. Conversely, failure to obtain approval in one or more jurisdictions may make approval in other jurisdictions more difficult.
18
Clinical studies involving our product candidates may reveal that those candidates are ineffective, inferior to existing approved medicines, unacceptably toxic, or that they have other unacceptable side effects. In addition, the results of preclinical studies do not necessarily predict clinical success, and larger and later-stage clinical studies may not produce the same results as earlier-stage clinical studies.
Frequently, product candidates that have shown promising results in early preclinical or clinical studies have subsequently suffered significant setbacks or failed in later clinical studies. In addition, clinical studies of potential products often reveal that it is not possible or practical to continue development efforts for these product candidates. If our clinical studies are substantially delayed or fail to prove the safety and effectiveness of our product candidates in development, we may not receive regulatory approval of any of these product candidates and our business and financial condition will be materially harmed.
VIBATIV™ may not be accepted by physicians, patients, third party payors, or the medical community in general.
The commercial success of VIBATIV™ will depend upon its acceptance by physicians, patients, third party payors and the medical community in general. We cannot be sure that VIBATIV™ will be accepted by these parties. VIBATIV™ competes with vancomycin, a relatively inexpensive generic drug that is manufactured by a variety of companies, a number of existing anti-infectives manufactured and marketed by major pharmaceutical companies and others, and potentially against new anti-infectives that are not yet on the market. Even if the medical community accepts that VIBATIV™ is safe and efficacious for its indicated use, physicians may choose to restrict the use of VIBATIV™. If we and our partner, Astellas, are unable to demonstrate to physicians that, based on experience, clinical data, side-effect profiles and other factors, VIBATIV™ is preferable to vancomycin and other existing or subsequently-developed anti-infective drugs, we may never generate meaningful revenue from VIBATIV™. The degree of market acceptance of VIBATIV™ depends on a number of factors, including, but not limited to:
- •
- the demonstration of the clinical efficacy and safety of VIBATIV™;
- •
- the approved labeling for VIBATIV™;
- •
- the advantages and disadvantages of VIBATIV™ compared to alternative therapies;
- •
- potential negative perceptions, if any, of physicians related to delays with our NP NDA;
- •
- our and Astellas' ability to educate the medical community about the safety and effectiveness of VIBATIV™;
- •
- the reimbursement policies of government and third party payors; and
- •
- the market price of VIBATIV™ relative to competing therapies.
We commenced a workforce restructuring in April 2008 to focus our efforts on our key research and exploratory development programs and to reduce our overall cash burn rate. Even after giving effect to this restructuring, we do not have sufficient cash to fully develop and commercialize our un-partnered product candidates, and the restructuring may impact our ability to execute our business plan.
In April 2008, we commenced a significant workforce restructuring involving the elimination of approximately 40% of our positions through layoffs from all departments throughout our organization, including senior management. Our objective with the restructuring was to reduce our overall cash burn rate and focus on our key clinical programs while maintaining core research and exploratory development capability. However, the restructuring has adversely affected the pace and breadth of our research and development efforts. We may in the future decide to restructure operations and reduce expenses further by taking such measures as additional reductions in our workforce and program
19
spending. There can be no assurance that following this restructuring, or any future restructuring, we will have sufficient cash resources to allow us to fund our operations as planned.
Even if our product candidates receive regulatory approval, such as VIBATIV™, commercialization of such products may be adversely affected by regulatory actions and oversight.
Even if we receive regulatory approval for our product candidates, this approval may include limitations on the indicated uses for which we can market our medicines or the patient population that may utilize our medicines, which may limit the market for our medicines or put us at a competitive disadvantage relative to alternative therapies. For example, VIBATIV™'s labeling contains a boxed warning regarding the risks of use of VIBATIV™ during pregnancy. Products with boxed warnings are subject to more restrictive advertising regulations than products without such warnings. These restrictions could make it more difficult to market VIBATIV™ effectively. Further, now that VIBATIV™ is approved, we remain subject to continuing regulatory obligations, such as safety reporting requirements and additional post-marketing obligations, including regulatory oversight of promotion and marketing. In addition, the labeling, packaging, adverse event reporting, advertising, promotion and recordkeeping for the approved product remain subject to extensive and ongoing regulatory requirements. If we become aware of previously unknown problems with an approved product in the U.S. or overseas or at our contract manufacturers' facilities, a regulatory agency may impose restrictions on the product, our contract manufacturers or on us, including requiring us to reformulate the product, conduct additional clinical studies, change the labeling of the product, withdraw the product from the market or require our contract manufacturer to implement changes to its facilities. In addition, we may experience a significant drop in the sales of the product, our royalties on product revenues and reputation in the marketplace may suffer, and we could face lawsuits.
We are also subject to regulation by regional, national, state and local agencies, including the Department of Justice, the Federal Trade Commission, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies with respect to VIBATIV™, as well as governmental authorities in those foreign countries in which any of our product candidates are approved for commercialization. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal and state statutes and regulations govern to varying degrees the research, development, manufacturing and commercial activities relating to prescription pharmaceutical products, including preclinical and clinical testing, approval, production, labeling, sale, distribution, import, export, post-market surveillance, advertising, dissemination of information and promotion. If we or any third parties that provide these services for us are unable to comply, we may be subject to regulatory or civil actions or penalties that could significantly and adversely affect our business. Any failure to maintain regulatory approval will limit our ability to commercialize our product candidates, which would materially and adversely affect our business and financial condition.
We have incurred operating losses in each year since our inception and expect to continue to incur substantial losses for the foreseeable future.
We have been engaged in discovering and developing compounds and product candidates since mid-1997. Our first approved product, VIBATIV™, was launched by our partner Astellas in the U.S. in November 2009, and we expect modest revenues and royalties during its launch phase. We may never generate sufficient revenue from selling medicines to achieve profitability. As of December 31, 2009, we had an accumulated deficit of approximately $1.1 billion.
We expect to incur substantial expenses as we continue our drug discovery and development efforts, particularly to the extent we advance our product candidates into and through clinical studies, which are very expensive. As a result, we expect to continue to incur substantial losses for the foreseeable future. We are uncertain when or if we will be able to achieve or sustain profitability.
20
Failure to become and remain profitable would adversely affect the price of our securities and our ability to raise capital and continue operations.
If we fail to obtain the capital necessary to fund our operations, we may be unable to develop our product candidates and we could be forced to share our rights to commercialize our product candidates with third parties on terms that may not be favorable to us.
We need large amounts of capital to support our research and development efforts. If we are unable to secure capital to fund our operations we will not be able to continue our discovery and development efforts and we might have to enter into strategic collaborations that could require us to share commercial rights to our medicines to a greater extent than we currently intend. Based on our current operating plans, milestone forecasts and spending assumptions, we believe that our cash and cash equivalents and marketable securities will be sufficient to meet our anticipated operating needs for at least the next twelve months. We are likely to require additional capital to fund operating needs thereafter. If we were to conduct additional studies to support the telavancin NP NDA and we were required to fund such studies, our capital needs could increase substantially. In addition, under our RELOVAIR™ program with GSK, in the event that a LABA product candidate discovered by GSK is successfully developed and commercialized, we will be obligated to pay GSK milestone payments which could total as much as $220.0 million if both a single-agent and a combination product were launched in multiple regions of the world. The current lead LABA candidate, GW642444, is a GSK-discovered compound and GSK has determined to focus the collaboration's LABA development resources on the development of this compound only. If this GSK-discovered compound, which recently commenced a Phase 3 program in COPD, is advanced through regulatory approval and commercialization, we would not be entitled to receive any further milestone payments from GSK with regard to the RELOVAIR™ program and we would have to pay GSK the milestones noted above. We cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Even if we are able to raise additional capital, such financing may result in significant dilution to existing security holders. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have to make additional reductions in our workforce and may be prevented from continuing our discovery and development efforts and exploiting other corporate opportunities. This could harm our business, prospects and financial condition and cause the price of our securities to fall.
Global financial and economic conditions have had an impact on our industry, may adversely affect our business and financial condition in ways that we currently cannot predict, and may limit our ability to raise additional funds.
Global financial conditions and general economic conditions, including the decreased availability of credit, have had an impact on our industry, and may adversely affect our business and our financial condition. Our ability to access the capital or debt markets and raise funds required for our operations may be severely restricted at a time when we would like, or need, to do so, which would have an adverse effect on our ability to fund our operations as planned. In addition, many biotechnology and biopharmaceutical companies with limited funds have been unable to raise capital during the recent period of financial and economic uncertainty and volatility, and they are left with limited alternatives including merging with other companies or out-licensing their assets. The large number of companies in this situation has led to an increase in supply of biotechnology and biopharmaceutical assets available for license or sale, which disadvantages companies like us that intend to partner certain of their assets.
If our partners do not satisfy their obligations under our agreements with them, or if they terminate our partnership with them, we will be unable to develop our partnered product candidates as planned.
We entered into our collaboration agreement for the RELOVAIR™ program with GSK in November 2002, our strategic alliance agreement with GSK in March 2004, and our telavancin
21
development and commercialization agreement with Astellas in November 2005. In connection with these agreements, we have granted to these parties certain rights regarding the use of our patents and technology with respect to compounds in our development programs, including development and marketing rights. Under our GSK agreements, GSK has full responsibility for development and commercialization of any product candidates in the programs that it has in-licensed, including RELOVAIR™ and MABA. Any future milestone payments or royalties to us from these programs will depend on the extent to which GSK advances the product candidate through development and commercial launch. In connection with our license, development and commercialization agreement with Astellas, Astellas is responsible for the commercialization of VIBATIV™ and any royalties to us from net sales of VIBATIV™ will depend upon Astellas' ability to commercialize the medicine.
Our partners might not fulfill all of their obligations under these agreements, and, in certain circumstances, they may terminate our partnership with them. In either event, we may be unable to assume the development and commercialization of the product candidates covered by the agreements or enter into alternative arrangements with a third party to develop and commercialize such product candidates. In addition, with the exception of product candidates in our RELOVAIR™ program, our partners generally are not restricted from developing and commercializing their own products and product candidates that compete with those licensed from us. If a partner elected to promote its own products and product candidates in preference to those licensed from us, future payments to us could be reduced and our business and financial condition would be materially and adversely affected. Accordingly, our ability to receive any revenue from the product candidates covered by these agreements is dependent on the efforts of the partner. We could also become involved in disputes with a partner, which could lead to delays in or termination of our development and commercialization programs and time-consuming and expensive litigation or arbitration.
If a partner terminates or breaches its agreements with us, or otherwise fails to complete its obligations in a timely manner, the chances of successfully developing or commercializing our product candidates would be materially and adversely affected. For example, under the terms of our telavancin license, development and commercialization agreement, Astellas has the right to terminate the agreement since VIBATIV™ was not approved by December 31, 2008. If Astellas chooses to terminate the agreement, the further commercialization of VIBATIV™ would be delayed.
In addition, while our strategic alliance with GSK sets forth pre-agreed upfront payments, development obligations, milestone payments and royalty rates under which GSK may obtain exclusive rights to develop and commercialize certain of our product candidates, GSK may in the future seek to negotiate more favorable terms on a project-by-project basis. To date, GSK has licensed our LAMA program and our MABA program under the terms of the strategic alliance agreement and has chosen not to license our bacterial infections program, our anesthesia program and our 5-HT4 program. In February 2009, GSK returned the LAMA program to us because the current formulation of the lead product candidate is incompatible with GSK's proprietary inhaler device. There can be no assurance that GSK will license any other development program under the terms of the strategic alliance agreement, or at all. GSK's failure to license our development programs or its return of programs to us could adversely affect the perceived prospects of the product candidates that are the subject of these development programs, which could negatively affect both our ability to enter into collaborations for these product candidates with third parties and the price of our securities.
We rely on a limited number of manufacturers for our product candidates, and our business will be harmed if these manufacturers are not able to satisfy our demand and alternative sources are not available.
We have limited in-house active pharmaceutical ingredient (API) production capabilities and depend primarily on a number of third-party API and drug product manufacturers. We may not have long-term agreements with these third parties and our agreements with these parties may be terminable
22
at will by either party at any time. If, for any reason, these third parties are unable or unwilling to perform, or if their performance does not meet regulatory requirements, we may not be able to locate alternative manufacturers or enter into favorable agreements with them. Any inability to acquire sufficient quantities of API and drug product in a timely manner from these third parties could delay clinical studies, prevent us from developing our product candidates in a cost-effective manner or on a timely basis and adversely affect the commercial introduction of any approved products. In addition, manufacturers of our API and drug product are subject to the FDA's cGMP regulations and similar foreign standards and we do not have control over compliance with these regulations by our manufacturers.
We have had manufactured sufficient telavancin API and drug product for the anticipated six-month commercial launch supply of VIBATIV™ and this inventory has been delivered to our collaboration partner. All further manufacture of VIBATIV™ API and drug product is now our collaboration partner's responsibility. For the foreseeable future, we anticipate that our collaboration partner will rely on third parties. If, for any reason, these third parties are unable or unwilling to perform, or if their performance does not meet regulatory requirements, including maintaining cGMP compliance, our collaboration partner may not be able to locate alternative manufacturers or enter into favorable agreements with them. Any inability to acquire sufficient quantities of API and drug product in a timely manner from these third parties could delay further telavancin studies and development, and adversely affect the commercialization of VIBATIV™ and any other telavancin products, if approved.
Our manufacturing strategy presents the following additional risks:
- •
- because of the complex nature of our compounds, our manufacturers may not be able to successfully manufacture our APIs and/or drug products in a cost effective and/or timely manner and changing manufacturers for our APIs or drug products could involve lengthy technology transfer and validation activities for the new manufacturer;
- •
- the processes required to manufacture certain of our APIs and drug products are specialized and available only from a limited number of third-party manufacturers;
- •
- some of the manufacturing processes for our APIs and drug products have not been scaled to quantities needed for continued clinical studies or commercial sales, and delays in scale-up to commercial quantities could delay clinical studies, regulatory submissions and commercialization of our product candidates; and
- •
- because some of the third-party manufacturers are located outside of the U.S., there may be difficulties in importing our APIs and drug products or their components into the U.S. as a result of, among other things, FDA import inspections, incomplete or inaccurate import documentation or defective packaging.
Our relationship with GSK may have a negative effect on our ability to enter into relationships with third parties.
As of February 16, 2010, GSK beneficially owned approximately 14.6% of our outstanding capital stock. Pursuant to our strategic alliance with GSK, GSK has the right to license exclusive development and commercialization rights to our product candidates arising from (i) our oral peripheral opioid-induced bowel constipation (PUMA) program, (ii) our AT1 Receptor—Neprilysin Inhibitor (ARNI) program for cardiovascular disease and (iii) our MonoAmine Reuptake Inhibitor (MARIN) program for chronic pain. Because GSK may license these three development programs at any time prior to successful completion of a Phase 2 proof-of-concept study, we may be unable to collaborate with other partners with respect to these programs until we have expended substantial resources to advance them through clinical studies. We may not have sufficient funds to pursue such programs in the event GSK
23
does not license them at an early stage. Pharmaceutical companies other than GSK that may be interested in developing products with us may be less inclined to do so because of our relationship with GSK, or because of the perception that development programs that GSK does not license, or returns to us, pursuant to our strategic alliance agreement are not promising programs. If our ability to work with present or future strategic partners or collaborators is adversely affected as a result of our strategic alliance with GSK, our business prospects may be limited and our financial condition may be adversely affected.
If we are unable to enter into future collaboration arrangements or if any such collaborations with third parties are unsuccessful, we will be unable to fully develop and commercialize our product candidates and our business will be adversely affected.
We have active collaborations with GSK for the RELOVAIR™ and MABA programs and with Astellas for telavancin, and we have licensed our anesthesia compound to AstraZeneca AB (AstraZeneca). Additional collaborations will be needed to fund later-stage development of our product candidates that have not been licensed to a collaborator, and to commercialize these product candidates if approved by the necessary regulatory agencies. Each of TD-5108, our lead 5-HT4 compound, and TD-1792, our investigational antibiotic, has successfully completed a Phase 2 proof-of-concept study, and TD-4208, our LAMA compound that GSK returned to us in February 2009 under the terms of the strategic alliance agreement, has completed a Phase 1 study. We currently intend to pursue collaboration arrangements for the development and commercialization of these compounds. Collaborations with third parties regarding these programs or our other programs may require us to relinquish material rights, including revenue from commercialization of our medicines, on terms that are less attractive than our current arrangements or to assume material ongoing development obligations that we would have to fund. These collaboration arrangements are complex and time-consuming to negotiate, and if we are unable to reach agreements with third-party collaborators, we may fail to meet our business objectives and our financial condition may be adversely affected. We face significant competition in seeking third-party collaborators, especially in the current weak economy which is driving many biotechnology and biopharmaceutical companies to seek to sell or license their assets, and we may be unable to find third parties to pursue product collaborations on a timely basis or on acceptable terms. Furthermore, for any collaboration, we may not be able to control the amount of time and resources that our partners devote to our product candidates and our partners may choose to pursue alternative products. Our inability to successfully collaborate with third parties would increase our development costs and would limit the likelihood of successful commercialization of our product candidates.
We depend on third parties in the conduct of our clinical studies for our product candidates.
We depend on independent clinical investigators, contract research organizations and other third party service providers in the conduct of our preclinical and clinical studies for our product candidates. We rely heavily on these parties for execution of our preclinical and clinical studies, and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that our clinical studies are conducted in accordance with good clinical practices (GCPs) and other regulations as required by the FDA and foreign regulatory agencies, and the applicable protocol. Failure by these parties to comply with applicable regulations, GCPs and protocols in conducting studies of our product candidates can result in a delay in our development programs or non-approval of our product candidates by regulatory authorities.
The FDA enforces good clinical practices and other regulations through periodic inspections of trial sponsors, clinical research organizations (CROs), principal investigators and trial sites. For example, in connection with the FDA's review of our telavancin NDAs, the FDA conducted inspections of Theravance and certain of our study sites, clinical investigators and CROs. If we or any of the third
24
parties on which we have relied to conduct our clinical studies are determined to have failed to comply with GCPs, the study protocol or applicable regulations, the clinical data generated in our studies may be deemed unreliable. This could result in non-approval of our product candidates by the FDA, or we or the FDA may decide to conduct additional audits or require additional clinical studies, which would delay our development programs and could result in significant additional costs.
We face substantial competition from companies with more resources and experience than we have, which may result in others discovering, developing, receiving approval for or commercializing products before or more successfully than we do.
Our ability to succeed in the future depends on our ability to demonstrate and maintain a competitive advantage with respect to our approach to the discovery and development of medicines. Our objective is to discover, develop and commercialize new small molecule medicines with superior efficacy, convenience, tolerability and/or safety. Because our strategy is to develop new product candidates primarily for biological targets that have been validated by existing medicines or potential medicines in late stage clinical studies, to the extent that we are able to develop medicines, they are likely to compete with existing drugs that have long histories of effective and safe use. We expect that any medicines that we commercialize with our collaborative partners will compete with existing or future market-leading medicines.
Many of our potential competitors have substantially greater financial, technical and personnel resources than we have. In addition, many of these competitors have significantly greater commercial infrastructures than we have. Our ability to compete successfully will depend largely on our ability to leverage our experience in drug discovery and development to:
- •
- discover and develop medicines that are superior to other products in the market;
- •
- attract and retain qualified personnel;
- •
- obtain patent and/or other proprietary protection for our medicines and technologies;
- •
- obtain required regulatory approvals; and
- •
- successfully collaborate with pharmaceutical companies in the discovery, development and commercialization of new medicines.
Established pharmaceutical companies may invest heavily to quickly discover and develop or in-license novel compounds that could make our product candidates obsolete. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA approval or discovering, developing and commercializing medicines before we do. Other companies are engaged in the discovery of medicines that would compete with the product candidates that we are developing.
Any new medicine that competes with a generic or proprietary market leading medicine must demonstrate compelling advantages in efficacy, convenience, tolerability and/or safety in order to overcome severe price competition and be commercially successful. VIBATIV™ must demonstrate these advantages, as it competes with vancomycin, a relatively inexpensive generic drug that is manufactured by a number of companies, and a number of existing anti-infectives marketed by major and other pharmaceutical companies. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
As the principles of multivalency become more widely known, we expect to face increasing competition from companies and other organizations that pursue the same or similar approaches. Novel therapies, such as gene therapy or effective vaccines for infectious diseases, may emerge that will make both conventional and multivalent medicine discovery efforts obsolete or less competitive.
25
We have no experience selling or distributing products and no internal capability to do so.
Generally, our strategy is to engage pharmaceutical or other healthcare companies with an existing sales and marketing organization and distribution system to market, sell and distribute our products. We may not be able to establish these sales and distribution relationships on acceptable terms, or at all. If we receive regulatory approval to commence commercial sales of any of our product candidates that are not covered by our current agreements with GSK, Astellas or AstraZeneca, we will need a partner in order to commercialize such products unless we establish a sales and marketing organization with appropriate technical expertise and supporting distribution capability. At present, we have no sales personnel and a limited number of marketing personnel. Factors that may inhibit our efforts to commercialize our products without strategic partners or licensees include:
- •
- our inability to recruit and retain adequate numbers of effective sales and marketing personnel;
- •
- the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe our products;
- •
- the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
- •
- unforeseen costs and expenses associated with creating an independent sales and marketing organization.
If we are not able to partner with a third party and are not successful in recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty commercializing our product candidates, which would adversely affect our business and financial condition.
If we lose key management or scientific personnel, or if we fail to retain our key employees, our ability to discover and develop our product candidates will be impaired.
We are highly dependent on principal members of our management team and scientific staff to operate our business. We have become even more dependent on existing personnel since the significant workforce restructuring announced in April 2008, which involved the elimination of approximately 40% of our positions through layoffs from all departments throughout our organization, including senior management. While we planned our restructuring with the purpose of focusing on our key clinical programs while maintaining core research and exploratory development capability, the restructuring has adversely affected the pace and breadth of our research and development efforts. While the remaining scientific team has expertise in many different aspects of drug discovery and exploratory development, there is less depth to the team and we are more susceptible to remaining team members voluntarily leaving employment with us. Our company is located in northern California, which is headquarters to many other biotechnology and biopharmaceutical companies and many academic and research institutions. As a result, competition for certain skilled personnel in our market remains intense. None of our employees have employment commitments for any fixed period of time and may leave our employment at will.
If we fail to retain our remaining qualified personnel or replace them when they leave, we may be unable to continue our development and commercialization activities.
Our business and operations would suffer in the event of system failures.
Although we have security measures in place, our internal computer systems and those of our CROs and other service providers are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. We have not experienced any such system failure, accident or security breach to date, but if such an event were to
26
occur, it could result in a material disruption to our business. For example, the loss of clinical trial data from completed or ongoing clinical trials of our product candidates could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. If a disruption or security breach results in a loss of or damage to our data or regulatory applications, or inadvertent disclosure of confidential or proprietary information, we could incur liability and the further development of our product candidates could be delayed.
Our principal facility is located near known earthquake fault zones, and the occurrence of an earthquake, extremist attack or other catastrophic disaster could cause damage to our facilities and equipment, which could require us to cease or curtail operations.
Our principal facility is located in the San Francisco Bay Area near known earthquake fault zones and therefore is vulnerable to damage from earthquakes. In October 1989, a major earthquake struck this area and caused significant property damage and a number of fatalities. We are also vulnerable to damage from other types of disasters, including power loss, attacks from extremist organizations, fire, floods, communications failures and similar events. If any disaster were to occur, our ability to operate our business could be seriously impaired. In addition, the unique nature of our research activities and of much of our equipment could make it difficult for us to recover from this type of disaster. We may not have adequate insurance to cover our losses resulting from disasters or other similar significant business interruptions and we do not plan to purchase additional insurance to cover such losses due to the cost of obtaining such coverage. Any significant losses that are not recoverable under our insurance policies could seriously impair our business and financial condition.
Risks Related to our Alliance with GSK
GSK's ownership of a significant percentage of our stock and its ability to acquire additional shares of our stock may create conflicts of interest, and may inhibit our management's ability to continue to operate our business in the manner in which it is currently being operated.
As of February 16, 2010, GSK beneficially owned approximately 14.6% of our outstanding capital stock, and GSK has the right to maintain its percentage ownership of our capital stock. GSK could have substantial influence in the election of our directors, delay or prevent a transaction in which stockholders might receive a premium over the prevailing market price for their shares and have significant control over certain changes in our business.
In addition, GSK may make an offer to our stockholders to acquire outstanding voting stock that would bring GSK's percentage ownership of our voting stock to no greater than 60%, provided that:
- •
- the offer includes no condition as to financing;
- •
- the offer is approved by a majority of our independent directors;
- •
- the offer includes a condition that the holders of a majority of the shares of the voting stock not owned by GSK accept the offer by tendering their shares in the offer; and
- •
- the shares purchased will be subject to the provisions of the governance agreement on the same basis as the shares of GSK's Class A common stock.
Further, pursuant to our certificate of incorporation, we renounce our interest in and waive any claim that a corporate or business opportunity taken by GSK constitutes a corporate opportunity of ours unless such corporate or business opportunity is expressly offered to one of our directors who is a director, officer or employee of GSK, primarily in his or her capacity as one of our directors.
27
GSK's rights under the strategic alliance and governance agreements may deter or prevent efforts by other companies to acquire us, which could prevent our stockholders from realizing a control premium.
Our governance agreement with GSK requires us to exempt GSK from our stockholder rights plan, affords GSK certain rights to offer to acquire us in the event third parties seek to acquire our stock and contains other provisions that could deter or prevent another company from seeking to acquire us. For example, GSK may offer to acquire 100% of our outstanding stock from stockholders in certain circumstances, such as if we are faced with a hostile acquisition offer or if our board of directors acts in a manner to facilitate a change in control of us with a party other than GSK. In addition, pursuant to our strategic alliance agreement with GSK, GSK has the right to license (i) our PUMA program, (ii) our ARNI program and (iii) our MARIN program. As a result of these rights, other companies may be less inclined to pursue an acquisition of us and therefore we may not have the opportunity to be acquired in a transaction that stockholders might otherwise deem favorable, including transactions in which our stockholders might realize a substantial premium for their shares.
GSK could sell or transfer a substantial number of shares of our common stock, which could depress the price of our securities or result in a change in control of our company.
GSK may sell or transfer our common stock either pursuant to a public offering registered under the Securities Act of 1933, as amended (the "1933 Act"), or pursuant to Rule 144 of the 1933 Act. In addition, beginning in September 2012, GSK will have no restrictions on its ability to sell or transfer our common stock on the open market, in privately negotiated transactions or otherwise, and these sales or transfers could create substantial declines in the price of our securities or, if these sales or transfers were made to a single buyer or group of buyers, could contribute to a transfer of control of our company to a third party.
Risks Related to Legal and Regulatory Uncertainty
If our efforts to protect the proprietary nature of the intellectual property related to our technologies are not adequate, we may not be able to compete effectively in our market.
We rely upon a combination of patents, patent applications, trade secret protection and confidentiality agreements to protect the intellectual property related to our technologies. Any involuntary disclosure to or misappropriation by third parties of this proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market. The status of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and is very uncertain. As of December 31, 2009, we owned 183 issued United States patents and 765 granted foreign patents, as well as additional pending United States and foreign patent applications. Our patent applications may be challenged or fail to result in issued patents and our existing or future patents may be invalidated or be too narrow to prevent third parties from developing or designing around these patents. If the sufficiency of the breadth or strength of protection provided by our patents with respect to a product candidate is threatened, it could dissuade companies from collaborating with us to develop, and threaten our ability to commercialize, the product candidate. Further, if we encounter delays in our clinical trials or in obtaining regulatory approval of our product candidates, the patent lives of the related product candidates would be reduced.
In addition, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, for processes for which patents are difficult to enforce and for any other elements of our drug discovery and development processes that involve proprietary know-how, information and technology that is not covered by patent applications. Although we require our employees, consultants, advisors and any third parties who have access to our proprietary
28
know-how, information and technology to enter into confidentiality agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Further, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or, if established, maintain a competitive advantage in our market, which could materially adversely affect our business, financial condition and results of operations.
Litigation or third-party claims of intellectual property infringement would require us to divert resources and may prevent or delay our drug discovery and development efforts.
Our commercial success depends in part on us and our partners not infringing the patents and proprietary rights of third parties. Third parties may assert that we or our partners are using their proprietary rights without authorization. There are third party patents that may cover materials or methods for treatment related to our product candidates. At present, we are not aware of any patent claims with merit that would adversely and materially affect our ability to develop our product candidates, but nevertheless the possibility of third party allegations cannot be ruled out. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Furthermore, parties making claims against us or our partners may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates. For example, an action has been filed in the United States Patent and Trademark office opposing registration of the trademark VIBATIV™. Failure to register this trademark may have an adverse impact on sales of VIBATIV™, which could adversely affect our business. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties or pay royalties. In addition, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize one or more of our product candidates, which could harm our business significantly. In addition, in the future we could be required to initiate litigation to enforce our proprietary rights against infringement by third parties. Prosecution of these claims to enforce our rights against others would involve substantial litigation expenses and divert substantial employee resources from our business. If we fail to effectively enforce our proprietary rights against others, our business will be harmed.
Product liability lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our medicines.
The risk that we may be sued on product liability claims is inherent in the development and commercialization of pharmaceutical products. Side effects of, or manufacturing defects in, products that we or our partners develop or commercialize could result in the deterioration of a patient's condition, injury or even death. Once a product is approved for sale and commercialized, the likelihood of product liability lawsuits tends to increase. Our partner Astellas launched VIBATIV™, our first approved product, in the U.S. in November 2009. Claims may be brought by individuals seeking relief for themselves or by individuals or groups seeking to represent a class. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are
29
held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forgo further commercialization of the applicable products.
Although we maintain general liability and product liability insurance, this insurance may not fully cover potential liabilities. In addition, inability to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercial production and sale of our products, which could adversely affect our business. Product liability claims could also harm our reputation, which may adversely affect our and our partners' ability to commercialize our products successfully.
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability to generate revenues.
The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care costs to contain or reduce costs of health care may adversely affect one or more of the following:
- •
- our or our collaborators' ability to set a price we believe is fair for our products, if approved;
- •
- our ability to generate revenues and achieve profitability; and
- •
- the availability of capital.
Legislative proposals to reform healthcare and government insurance programs, the current Presidential administration and its focus on health care reform, along with the trend toward managed healthcare in the United States could influence the purchase of healthcare products and reduce demand and prices for our products, if approved. This could harm our or our collaborators' ability to market our potential medicines and generate revenues. Cost containment measures that health care payors and providers are instituting and the effect of probable further health care reform could significantly reduce potential revenues from the sale of any product candidates approved in the future. In addition, in certain foreign markets, the pricing of prescription drugs is subject to government control and reimbursement may in some cases be unavailable. We believe that pricing pressures at the state and federal level, as well as internationally, will continue and may increase, which may make it difficult for us to sell our potential medicines that may be approved in the future at a price acceptable to us or our collaborators.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including chemical, biological and radioactive materials. In addition, our operations produce hazardous waste products. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of hazardous materials. We may incur significant additional costs to comply with these and other applicable laws in the future. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials and we may incur liability as a result of any such contamination or injury. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. We do not have any insurance for liabilities arising from hazardous materials. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research, development and production efforts, which could harm our business.
30
General Company Related Risks
The price of our securities has been extremely volatile and may continue to be so, and purchasers of our securities could incur substantial losses.
The price of our securities has been extremely volatile and may continue to be so. The stock market in general and the market for biotechnology and biopharmaceutical companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies, in particular during the last few years. The following factors, in addition to the other risk factors described in this section, may also have a significant impact on the market price of our securities:
- •
- any further adverse developments or perceived adverse developments with respect to the FDA's review of the telavancin NP NDA, which could include, without limitation, non-approval of the NDA;
- •
- any adverse developments or perceived adverse developments with respect to the commercial launch of VIBATIV™;
- •
- any adverse developments or results or perceived adverse developments or results with respect to the RELOVAIR™ program with GSK, including without limitation any difficulties or delays encountered with regard to the regulatory path for the RELOVAIR™ program;
- •
- any adverse developments or perceived adverse developments with respect to regulatory matters concerning telavancin in any foreign jurisdiction, in particular the MAA that our partner Astellas submitted to the EMEA in October 2009 and of which the EMEA commenced scientific review in November 2009;
- •
- any adverse developments or results or perceived adverse developments or results with respect to the MABA program with GSK, including without limitation the possibility that the analysis of results from key preclinical studies may lead to significant delay of the MABA program or perhaps a decision to terminate the entire program;
- •
- any adverse developments or perceived adverse developments in the field of LABAs, including any change in FDA policy or guidance (such as the recent pronouncement warning that LABAs should not be used alone in the treatment of asthma or the outcome of the upcoming FDA Advisory Committee on LABAs);
- •
- any announcements of developments with, or comments by, the FDA with respect to products we or our partners have under development or have commercialized;
- •
- our workforce restructuring commenced in April 2008 and uncertainties or perceived uncertainties related to the restructuring, including without limitation concerns regarding our ability to retain key employees and the possibility that we will have to implement further workforce reductions;
- •
- the extent to which GSK advances (or does not advance) our product candidates through development into commercialization;
- •
- any adverse developments or perceived adverse developments with respect to our relationship with GSK;
- •
- any adverse developments or perceived adverse developments with respect to our relationship with Astellas, including without limitation, disagreements that may arise between us and Astellas concerning regulatory strategy or further development of telavancin, or Astellas' termination of our telavancin license, development and commercialization agreement, which it now has the right to do;
31
- •
- any adverse developments or perceived adverse developments with respect to our partnering efforts with our 5-HT4 program, TD-1792 or TD-4208, the LAMA product candidate that GSK returned to us in February 2009 under the terms of the strategic alliance agreement;
- •
- announcements regarding GSK's decisions whether or not to license any of our development programs or to return to us any previously licensed program, such as our experience with our LAMA program licensed from us by GSK in 2004 under the strategic alliance agreement and then returned to us by GSK in February 2009;
- •
- announcements regarding GSK or Astellas generally;
- •
- announcements of patent issuances or denials, technological innovations or new commercial products by us or our competitors;
- •
- developments concerning any collaboration we may undertake with companies other than GSK or Astellas;
- •
- publicity regarding actual or potential study results or the outcome of regulatory review relating to products under development by us, our partners or our competitors;
- •
- regulatory developments in the United States and foreign countries;
- •
- economic and other external factors beyond our control;
- •
- sales of stock by us or by our stockholders, including sales by certain of our employees and directors whether or not pursuant to written pre-determined selling plans under Rule 10b5-1 of the Securities Exchange Act of 1934, some of which plans are currently in effect, such as plans adopted by our employees to sell shares to cover taxes due upon the quarterly vesting of restricted stock units, and other plans which may be entered into; and
- •
- potential sales or purchases of our capital stock by GSK.
Concentration of ownership will limit your ability to influence corporate matters.
As of February 16, 2010, GSK beneficially owned approximately 14.6% of our outstanding capital stock and our directors, executive officers and investors affiliated with these individuals beneficially owned approximately 14.1% of our outstanding capital stock. Based on our review of publicly available filings as of February 16, 2010, our six largest stockholders other than GSK collectively owned approximately 52.0% of our outstanding capital stock. These stockholders could control the outcome of actions taken by us that require stockholder approval, including a transaction in which stockholders might receive a premium over the prevailing market price for their shares.
Anti-takeover provisions in our charter and bylaws, in our rights agreement and in Delaware law could prevent or delay a change in control of our company.
Provisions of our certificate of incorporation and bylaws may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions include:
- •
- requiring supermajority stockholder voting to effect certain amendments to our certificate of incorporation and bylaws;
- •
- restricting the ability of stockholders to call special meetings of stockholders;
- •
- prohibiting stockholder action by written consent; and
- •
- establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings.
32
In addition, our board of directors has adopted a rights agreement that may prevent or delay a change in control of us. Further, some provisions of Delaware law may also discourage, delay or prevent someone from acquiring us or merging with us.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None
Our headquarters are located in South San Francisco, CA, and consist of two leased buildings of approximately 110,000 and 60,000 square feet, respectively. The leases expire in March 2012 and may be extended for two additional five-year periods. The current annual rental expense under these leases is approximately $6.6 million, subject to annual increases.
We are not a party to any material legal proceedings.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS
No matters were submitted to a vote of stockholders during the fourth quarter of the fiscal year covered by this report.
33
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock has been traded on the Nasdaq Global Market under the symbol "THRX" since October 5, 2004. The following table sets forth the high and low closing prices of our common stock on a per share basis for the periods indicated and as reported on the Nasdaq Global Market:
Calendar Quarter | High | Low | |||||
|---|---|---|---|---|---|---|---|
2009 | |||||||
First Quarter | $ | 18.48 | $ | 10.94 | |||
Second Quarter | $ | 17.60 | $ | 12.94 | |||
Third Quarter | $ | 18.38 | $ | 13.13 | |||
Fourth Quarter | $ | 15.40 | $ | 13.00 | |||
2008 | |||||||
First Quarter | $ | 22.21 | $ | 9.40 | |||
Second Quarter | $ | 14.23 | $ | 11.16 | |||
Third Quarter | $ | 16.82 | $ | 12.16 | |||
Fourth Quarter | $ | 12.40 | $ | 5.77 | |||
As of February 16, 2010, there were 216 stockholders of record of our common stock. There is no established public trading market for our Class A common stock, all of which is owned by GSK. We did not make any unregistered sales of equity securities during the fourth quarter of 2009.
Dividend Policy
We currently intend to retain any future earnings to finance our research and development efforts. We have never declared or paid cash dividends and do not intend to declare or pay cash dividends on our common stock or Class A common stock in the foreseeable future.
Equity Compensation Plans
The following table provides certain information with respect to all of our equity compensation plans in effect as of December 31, 2009:
| | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted-average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
Plan Category | (a) | (b) | (c) | |||||||
Equity compensation plans approved by security holders | 9,934,936 | (1) | $ | 16.95 | (3) | 2,020,047 | (4) | |||
Equity compensation plans not approved by security holders | 521,032 | (2) | 11.63 | (3) | 178,116 | |||||
Total | 10,455,968 | (1)(2) | $ | 16.63 | (3) | 2,198,163 | (4) | |||
- (1)
- Includes 7,914,869 shares issuable upon exercise of outstanding options and 2,020,067 shares issuable upon vesting of outstanding restricted stock units.
34
- (2)
- Includes 499,000 shares issuable upon exercise of outstanding options and 22,032 shares issuable upon vesting of outstanding restricted stock units.
- (3)
- Does not take into account outstanding restricted stock units as these awards have no exercise price.
- (4)
- Includes 478,619 shares of common stock available under our Employee Stock Purchase Plan.
The Theravance, Inc. 2008 New Employee Equity Incentive Plan (the 2008 EIP) is a non-stockholder approved plan, which was adopted by the board of directors on January 29, 2008 and is intended to satisfy the requirements of Nasdaq Marketplace Rule 5635(c)(4). Non-statutory options, restricted stock units, and restricted stock awards may be granted under the 2008 EIP to our employees. The Board authorized 500,000 shares of Common Stock for issuance under the 2008 EIP upon its adoption in 2008 and the Compensation Committee of the Board authorized an additional 200,000 shares for issuance under the 2008 EIP in July 2009. All option grants will have an exercise price per share of no less than 100% of the fair market value per share of Common Stock on the grant date. Each stock option, restricted stock unit and restricted stock award will vest in installments over the holder's period of service. Additional features of the 2008 EIP are outlined in Note 11 to the Consolidated Financial Statements.
Stock Performance Graph
The graph set forth below compares the cumulative total stockholder return on our common stock for the period commencing on October 5, 2004 and ending on December 31, 2009, with the cumulative total return of (i) the Nasdaq Composite Index and (ii) the AMEX Biotechnology Index, over the same period. This graph assumes the investment of $100.00 on October 5, 2004 in our common stock and $100.00 on September 30, 2004 in the Nasdaq Composite Index and the AMEX Biotechnology Index, and assumes the reinvestment of dividends, if any, although dividends have never been declared on our common stock.
The comparisons shown in the graph below are based upon historical data. We caution that the stock price performance shown in the graph below is not necessarily indicative of, nor is it intended to forecast, the potential future performance of our common stock. Information used in the graph was obtained from Research Data Group, Inc., a source believed to be reliable, but we are not responsible for any errors or omissions in such information.
Notwithstanding anything to the contrary set forth in any of our previous or future filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, that might incorporate this Annual Report on Form 10-K or future filings made by us under those statutes, this Stock Performance Graph section shall not be deemed filed with the United States Securities and Exchange Commission and shall not be deemed incorporated by reference into any of those prior filings or into any future filings made by us under those statutes.
35
COMPARISON OF 5 MONTH CUMULATIVE TOTAL RETURN*
Among Theravance, Inc., The NASDAQ Composite Index
And The NYSE Arca Biotechnology Index

*100 invested on 12/31/04 in stock or index, including reinvestment of dividends.
Fiscal year ending December 31.
36
ITEM 6. SELECTED FINANCIAL DATA
The following tables reflect selected consolidated summary financial data for each of the last five fiscal years and are derived from our audited financial statements. This data should be read in conjunction with Item 8, "Financial Statements and Supplementary Data", and with Item 7, "Management's Discussion and Analysis of Financial Condition and Results of Operations" in this Annual Report on Form 10-K.
| | Year Ended December 31, | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | 2006 | 2005 | ||||||||||||
| | (in thousands, except per share data) | ||||||||||||||||
CONSOLIDATED STATEMENT OF OPERATIONS DATA: | |||||||||||||||||
Revenue | $ | 24,374 | $ | 23,096 | $ | 22,002 | $ | 19,587 | $ | 12,054 | |||||||
Operating expenses: | |||||||||||||||||
Research and development | 77,524 | 82,020 | 155,254 | 166,564 | 137,936 | ||||||||||||
General and administrative | 27,066 | 28,861 | 35,313 | 32,193 | 23,674 | ||||||||||||
Restructuring charges | 1,145 | 5,419 | — | — | — | ||||||||||||
Total operating expenses (1) | 105,735 | 116,300 | 190,567 | 198,757 | 161,610 | ||||||||||||
Loss from operations | (81,361 | ) | (93,204 | ) | (168,565 | ) | (179,170 | ) | (149,556 | ) | |||||||
Interest and other income | 2,111 | 5,242 | 8,661 | 13,319 | 6,687 | ||||||||||||
Interest expense | (6,052 | ) | (5,681 | ) | (93 | ) | (193 | ) | (295 | ) | |||||||
Net loss | $ | (85,302 | ) | $ | (93,643 | ) | $ | (159,997 | ) | $ | (166,044 | ) | $ | (143,164 | ) | ||
Basic and diluted net loss per share | $ | (1.35 | ) | $ | (1.53 | ) | $ | (2.64 | ) | $ | (2.81 | ) | $ | (2.69 | ) | ||
Shares used in computing basic and net loss per share (2) | 63,027 | 61,390 | 60,498 | 59,013 | 53,270 | ||||||||||||
| | As of December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||
CONSOLIDATED BALANCE SHEET DATA: | ||||||||||||||||
Cash, cash equivalents and marketable securities | $ | 155,390 | $ | 200,605 | $ | 129,272 | $ | 235,570 | $ | 200,009 | ||||||
Working capital | 123,096 | 166,006 | 78,554 | 147,582 | 118,677 | |||||||||||
Total assets | 181,393 | 236,156 | 161,983 | 262,424 | 224,835 | |||||||||||
Long-term liabilities(3)(4) | 331,441 | 327,150 | 172,714 | 139,505 | 117,078 | |||||||||||
Accumulated deficit | (1,116,754 | ) | (1,031,452 | ) | (937,809 | ) | (777,812 | ) | (611,768 | ) | ||||||
Total stockholders' equity (net capital deficiency) | (188,994 | ) | (134,949 | ) | (66,264 | ) | 63,310 | 59,584 | ||||||||
- (1)
- The following table discloses the allocation of stock-based compensation expense included in total operating expenses:
| | Year Ended December 31, | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||
Research and development | $ | 11,542 | $ | 10,264 | $ | 13,133 | $ | 12,635 | $ | 3,259 | ||||||
General and administrative | 8,458 | 7,755 | 9,361 | 9,196 | 2,364 | |||||||||||
Total stock-based compensation | $ | 20,000 | $ | 18,019 | $ | 22,494 | $ | 21,831 | $ | 5,623 | ||||||
37
- (2)
- In February 2006, we completed a secondary offering with the sale of 5,200,000 shares of common stock. The financing raised proceeds, net of issuance costs, of $139.9 million.
- (3)
- Long-term liabilities include the long-term portion of deferred revenue as follows:
(in thousands) | 2009 | 2008 | 2007 | 2006 | 2005 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Deferred revenue | $ | 157,426 | $ | 152,771 | $ | 166,136 | $ | 134,383 | $ | 111,251 | ||||||
- (4)
- In January 2008, we closed an underwritten public offering of $172.5 million aggregate principal amount of unsecured convertible subordinated notes which will mature on January 15, 2015. The financing raised proceeds, net of issuance costs, of $166.7 million.
38
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management's Discussion and Analysis (MD&A) is intended to facilitate an understanding of our business and results of operations. You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and related notes included in Item 8, "Financial Statements and Supplementary Data" in this Annual Report on Form 10-K. The information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to our plans and strategy for our business, includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Such statements are based upon current expectations that involve risks and uncertainties. You should review the section entitled "Risk Factors" in Item 1A of Part I above for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Executive Summary
Theravance is a biopharmaceutical company with a pipeline of internally discovered product candidates. We are focused on the discovery, development and commercialization of small molecule medicines across a number of therapeutic areas including respiratory disease, bacterial infections and gastrointestinal motility dysfunction. Our key programs include: VIBATIVTM (telavancin) with Astellas Pharma Inc. (Astellas) and our RELOVAIRTM (formerly referred to as Horizon) program and the Bifunctional Muscarinic Antagonist-beta2 Agonist program with GlaxoSmithKline plc (GSK). By leveraging our proprietary insight of multivalency to drug discovery focused primarily on validated targets, we are pursuing a next generation strategy designed to discover superior medicines in areas of significant unmet medical need.
Our net loss for the year ended December 31, 2009 was $85.3 million compared to $93.6 million in 2008. This decrease was primarily due to lower research and development and restructuring expenses. Research and development expenses for the year ended December 31, 2009 decreased to $77.5 million compared to $82.0 million in 2008. This decrease was primarily driven by lower external clinical study costs as well as lower employee related costs due to the reduction in force initiated in April 2008. In 2009, we incurred restructuring expenses primarily due to charges recognized for the sublease of excess space in a portion of one of our South San Francisco, CA buildings whereas in 2008 we incurred restructuring expenses for severance and other termination benefit charges resulting from our workforce reduction announced in April 2008. Cash, cash equivalents, and short-term investments totaled $155.4 million at December 31, 2009, a decrease of $45.2 million since December 31, 2008. We expect to incur substantial losses for at least the next several years as we continue to invest in research and development.
Respiratory Programs
RELOVAIR™
In October 2009, GSK and Theravance announced that the first patient had commenced treatment in the Phase 3 program to develop a next-generation combination treatment for patients with chronic obstructive pulmonary disease (COPD). The Phase 3 program comprises a broad range of large scale Phase 3 studies to evaluate the investigational once-a-day long-acting beta agonist (LABA), 642444 ('444), in combination with the once-a-day inhaled corticosteroid (ICS), fluticasone furoate (FF), for the treatment of COPD. The overall program, which will study more than 6,000 patients, includes two 12-month exacerbation studies, two 6-month efficacy and safety studies, a detailed lung function profile
39
study, and studies to assess the potential for superiority of the fixed combination of '444 and FF versus other treatments for COPD.
Bacterial Infections Program
VIBATIV™ (telavancin)
In November 2009, Theravance and Astellas announced the commercial launch in the United States of VIBATIV™ for the treatment of adult patients with complicated skin and skin structure infections (cSSSI) caused by susceptible Gram-positive bacteria, includingStaphylococcus aureus, both methicillin-resistant (MRSA) and methicillin-susceptible (MSSA) strains. VIBATIV™, also approved by Health Canada for the treatment of adult patients with cSSSI, is targeted to launch in Canada in 2010 with our partner, Astellas.
Since the commercial launch in November and through December 31, 2009, Astellas recorded VIBATIV™ had net sales of $4.3 million, a substantial portion of which was related to the initial wholesaler stocking.
Telavancin
In November 2009, the European Medicines Agency completed the validation phase for the Marketing Authorization Application (MAA) for telavancin for the treatment of nosocomial pneumonia (NP), including ventilator-associated pneumonia, and complicated skin and soft tissue infections (cSSTI) in adults. Astellas Pharma Europe B.V., a European affiliate of Astellas, submitted the MAA in October 2009 under the Centralized Procedure and applied for marketing authorization for telavancin in the Member States of the European Union, plus Iceland, Liechtenstein and Norway.
On January 28, 2010, we announced that we received a letter from the FDA indicating our response to the November 2009 Complete Response letter for our telavancin New Drug Application for the treatment of NP due to Gram-positive organisms was incomplete. We have not met with the FDA yet to discuss this letter. It is unclear at this point what the standard for approval is for this indication. We do not have an estimated timeline for the resolution of these issues. We believe that the FDA's position is that it will require data from an additional clinical study or studies before it will consider the NP NDA for approval and we do not currently intend to conduct any such studies.
Critical Accounting Policies
This discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenue and expenses during the reporting periods. We periodically evaluate our material estimates and judgments based on the terms of underlying agreements, the expected course of development, historical experience and other factors that we believe are reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 1 to our consolidated financial statements contained in Item 8, "Financial Statements and Supplementary Data" in this Annual Report on Form 10-K, we believe that the following accounting policies relating to revenue recognition, preclinical study and clinical study expenses, stock-based compensation charges and inventory require us to make significant estimates, assumptions and judgments.
40
Revenue Recognition
In connection with our agreements with GSK and Astellas, we have determined that the deliverables under these collaboration agreements do not meet the criteria required for separate accounting units for the purposes of revenue recognition. As a result, we recognize revenue from non-refundable, upfront fees and development milestone payments ratably over the term of our performance under the agreements. These advance payments are recorded as deferred revenue pending recognition and are classified as a short- or long-term liability on the balance sheet. We periodically review the estimated performance period, which could impact the deferral period and, therefore, the timing and the amount of revenue recognized. Significant milestones in the development process typically include initiation or completion of various phases of clinical studies and approvals by regulatory agencies. We have made various changes to our performance periods under our agreements based upon updated product development timelines. It is possible that future adjustments will be made if actual conditions differ from our current plan and development assumptions.
Pursuant to our agreement with Astellas, we delivered the estimated first six months of commercial sale stock of VIBATIV™ to Astellas in October 2009. In December 2009, we expensed inventory that was no longer realizable. We recognize as revenue the net impact of transactions with our partner Astellas related to VIBATIV™ inventory including revenue specifically attributable to any sales and cost of inventory either transferred or expensed as unrealizable.
We have been reimbursed by GSK and Astellas for certain external development costs under their respective collaboration agreements. Such reimbursements have been reflected as a reduction of research and development expense and not as revenue.
We recognize royalty revenue from Astellas on their net sales of VIBATIV™ in the period the royalties are earned, based on net sales reporting provided by Astellas.
Preclinical Study and Clinical Study Expenses
A substantial portion of our preclinical studies and all of our clinical studies have been performed by third-party contract research organizations (CROs). Some CROs bill monthly for services performed, while others bill based upon milestones achieved. We review the activities performed under the significant contracts each quarter. For preclinical studies, the significant factors used in estimating accruals include the percentage of work completed to date and contract milestones achieved. For clinical study expenses, the significant factors used in estimating accruals include the number of patients enrolled and percentage of work completed to date. Vendor confirmations are obtained for contracts with longer duration when necessary to validate our estimate of expenses. Our estimates are highly dependent upon the timeliness and accuracy of the data provided by our CROs regarding the status of each program and total program spending and adjustments are made when deemed necessary. To date, we have not recorded any material adjustments as a result of changes to our estimates.
Stock-Based Compensation
We use the fair value method of accounting for stock-based compensation arrangements. Stock-based compensation arrangements currently include stock options granted, restricted shares issued and restricted stock unit awards (RSUs) granted under the 2004 Equity Incentive Plan and the 2008 New Employee Equity Incentive Plan and purchases of common stock by our employees at a discount to the market price during offering periods under our Employee Stock Purchase Plan (ESPP). The estimated fair value of stock options, restricted shares and RSUs is expensed on a straight-line basis over the expected term of the grant and the fair value of performance-contingent RSUs is expensed during the term of the award when we determine that it is probable that certain performance milestones will be met. Compensation expense for purchases under the ESPP is recognized based on the estimated fair value of the common stock during each offering period and the percentage of the purchase discount.
41
Stock-based compensation expense for stock options and RSUs has been reduced for estimated forfeitures so that compensation expense is based on options and RSUs ultimately expected to vest. We estimate annual forfeiture rates for stock options and RSUs based on our historical forfeiture experience.
Inventory
Our VIBATIV™ inventory is stated at the lower of cost or market and is included with prepaid and other current assets. Our inventory has a limited shelf life and can only be sold to our partner Astellas. If information becomes available that suggests that Astellas will not purchase our inventory due to insufficient remaining shelf life or product demand, it will not be realizable, and we will be required to expense a portion or all of the capitalized inventory costs.
Collaboration Arrangements
2005 License, Development and Commercialization Agreement with Astellas
In November 2005, we entered into a collaboration arrangement with Astellas for the development and commercialization of telavancin. In July 2006, Japan was added to the collaboration, thereby giving Astellas worldwide rights to this medicine. Through December 31, 2009, we have received $190.0 million in upfront, milestone and other fees from Astellas. We are eligible to receive up to an additional $30.0 million in remaining milestone payments related to regulatory filings and approvals in various regions of the world. We record these payments as deferred revenue and are amortizing them ratably over our estimated period of performance (development and commercialization period). We recognized $10.8 million and $10.3 million in amortization of deferred revenue under this agreement in 2008 and 2007, respectively.
We are entitled to receive royalties on global net sales of VIBATIV™ by Astellas that, on a percentage basis, range from the high teens to the upper twenties depending on sales volume. Under this arrangement, we are responsible for substantially all costs to develop and obtain U.S. regulatory approval for telavancin for cSSSI and NP, and Astellas is responsible for substantially all other costs associated with commercialization and further development of telavancin.
Pursuant to the collaboration arrangement, we delivered the estimated first six months of commercial sale stock of VIBATIV™ to Astellas out of our capitalized inventory in October 2009. In December 2009, we expensed inventory that was no longer realizable. In 2009, our revenue from this collaboration agreement was composed of:
(in thousands) | Year Ended December 31, 2009 | |||
|---|---|---|---|---|
Amortization of deferred revenue | $ | 11,338 | ||
Royalties from net sales of VIBATIV™ | 766 | |||
Cost of estimated first six months of commercial sale stock of VIBATIV™ | (1,629 | ) | ||
Cost of expensed unrealizable VIBATIV™ | (1,175 | ) | ||
Net Astellas collaboration revenue | $ | 9,300 | ||
RELOVAIR™ Program with GSK
In November 2002, we entered into our long-acting beta2 agonist (LABA) collaboration with GSK to develop and commercialize a LABA product candidate both as a single-agent new medicine for the treatment of COPD and as part of a new combination medicine with an ICS for the treatment of asthma and/or a long-acting muscarinic antagonist (LAMA) for COPD.
42
In connection with the RELOVAIR™ program, in 2002 we received from GSK an upfront payment of $10.0 million and sold to an affiliate of GSK shares of our Series E Preferred Stock for an aggregate purchase price of $40.0 million. In addition, we were eligible to receive up to $495.0 million in development, approval, launch and sales milestones and royalties on the sales of any product resulting from this program. Through December 31, 2009, we have received a total of $60.0 million in upfront and development milestone payments. GSK has determined to focus the collaboration's resources on the development of the lead LABA, GW642444, a GSK-discovered compound, together with GSK's ICS, fluticasone furoate. Accordingly, we do not expect to receive any further milestone payments from the RELOVAIR™ program. In the event that a LABA product candidate discovered by GSK is successfully developed and commercialized, we would be obligated to make milestone payments to GSK which could total as much as $220.0 million if both a single-agent and a combination product were launched in multiple regions of the world. Based on available information, we do not estimate that a significant portion of these potential milestone payments to GSK are likely to be made in the next two years. Moreover, we are entitled to receive the same royalties on sales of medicines from the RELOVAIR™ program, regardless of whether the product candidate originated with Theravance or with GSK. We are entitled to receive royalties of 15% on the first $3.0 billion of annual global net sales, and 5% on annual global net sales above $3.0 billion, for approved single-agent LABA and combination LABA-ICS medicines. Sales of single-agent LABA medicines and combination medicines would be combined for the purposes of this royalty calculation. For other products combined with a LABA from the RELOVAIR™ program, such as a combination LABA/LAMA medicine, which are launched after a LABA/ICS combination medicine, royalties are upward tiering and range from the mid-single digits to 10%. However, if GSK is not selling a LABA/ICS combination product at the time that the first other LABA combination is launched, then the royalties described above for the LABA/ICS combination medicine are applicable.
We recorded the initial cash payment and subsequent milestone payments as deferred revenue and are amortizing them ratably over our estimated period of performance (the product development period). Collaboration revenue from GSK under this agreement was $5.1 million, $6.8 million and $6.8 million in 2009, 2008 and 2007, respectively.
2004 Strategic Alliance with GSK
In March 2004, we entered into our strategic alliance with GSK. Under this alliance, GSK received an option to license exclusive development and commercialization rights to product candidates from our entire full drug discovery programs initiated prior to September 1, 2007, on pre-determined terms and on an exclusive, worldwide basis. Under the terms of the strategic alliance, GSK has only one opportunity to license each of our programs. Upon GSK's decision to license a program, GSK is responsible for funding all future development, manufacturing and commercialization activities for product candidates in that program. In addition, GSK is obligated to use diligent efforts to develop and commercialize product candidates from any program that it licenses. Consistent with our strategy, we are obligated at our sole cost to discover two structurally different product candidates for any programs that are licensed by GSK under the alliance. If these programs are successfully advanced through development by GSK, we are entitled to receive clinical, regulatory and commercial milestone payments and royalties on any sales of medicines developed from these programs. For product candidates licensed to date under this agreement, the royalty structure for a product containing one of our compounds as a single active ingredient would result in an average percentage royalty rate in the low double digits. If a product is successfully commercialized, in addition to any royalty revenue that we receive, the total upfront and milestone payments that we could receive in any given program that GSK licenses range from $130.0 million to $162.0 million for programs with single-agent medicines and up to $252.0 million for programs with both a single-agent and a combination medicine. If GSK chooses not to license a program, we retain all rights to the program and may continue the program alone or with a third party. To date, GSK has licensed our two COPD programs: long-acting muscarinic antagonist
43
(LAMA) and Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA). We received $5.0 million payments from GSK in connection with our license of each of our LAMA and MABA programs in August 2004 and March 2005, respectively. GSK has chosen not to license our bacterial infections program, anesthesia program or 5-HT4 program.
In connection with the strategic alliance with GSK, we received from GSK a payment of $20.0 million. This payment is being amortized over the initial performance period during which GSK may exercise its right to license certain of our programs under the agreement. In connection with the strategic alliance, we recognized $2.7 million in revenue for each of the years ended December 31, 2009, 2008 and 2007. In addition, in May 2004, GSK purchased through an affiliate 6,387,096 shares of our Class A common stock for an aggregate purchase price of $108.9 million.
Through December 31, 2009, we have received $46.0 million in upfront and milestone payments from GSK relating to the strategic alliance agreement. In addition, pursuant to a partial exercise of its rights under the governance agreement, upon the closing of our initial public offering on October 8, 2004, GSK purchased through an affiliate an additional 433,757 shares of Class A common stock for $6.9 million.
In August 2004, GSK exercised its right to license our LAMA program pursuant to the terms of the strategic alliance. We received a $5.0 million payment from GSK in connection with its licensing of our LAMA program. Through December 31, 2009, we received a milestone payment from GSK of $3.0 million related to clinical progress of our product candidate. These payments were amortized ratably over the estimated period of performance (the product development period) until 2009, when we recognized the remaining $4.2 million of deferred revenue related to the LAMA program as a result of the program being returned to us from GSK. We recognized $4.2 million, $0.8 million and $0.8 million in revenue related to the LAMA program in 2009, 2008 and 2007, respectively.
In March 2005, GSK exercised its right to license our MABA program pursuant to the terms of the strategic alliance. We received a $5.0 million payment from GSK in connection with the license of our MABA program. Through December 31, 2009, we received milestone payments from GSK of $13.0 million related to clinical progress of our candidate. These payments are being amortized ratably over the estimated period of performance (the product development period). In connection with the MABA program, we recognized $3.0 million, $2.0 million and $1.0 million in revenue in 2009, 2008 and 2007, respectively.
Results of Operations
Revenue
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
Revenue | $ | 24.4 | $ | 23.1 | $ | 22.0 | $ | 1.3 | 6 | % | $ | 1.1 | 5 | % | ||||||||
From GSK, we recognize revenue from the amortization of upfront and milestone payments related to our RELOVAIR™ program and strategic alliance agreements. From Astellas, we recognize revenue from the amortization of upfront and milestone payments related to our telavancin collaboration, royalties from net sales of VIBATIV™ and the impact of VIBATIV™ inventory transfers or dispositions. The table below reflects the upfront and milestone payments received from GSK under
44
the RELOVAIR™ program and strategic alliance agreements and from Astellas under the telavancin collaboration through December 31, 2009 (in millions).
Agreements/Programs | Signed Agreement/Licensed Program | Upfront, Milestone and Other Payments | |||||||
|---|---|---|---|---|---|---|---|---|---|
GSK Collaborations | |||||||||
RELOVAIR™ program | 2002 | $ | 60.0 | ||||||
Strategic Alliance agreement execution | 2004 | 20.0 | |||||||
Strategic Alliance—LAMA license | 2004 | 8.0 | |||||||
Strategic Alliance—MABA license | 2005 | 18.0 | |||||||
Astellas License agreement | 2005 | 190.0 | |||||||
Total | $ | 296.0 | |||||||
Upfront fees and milestone payments received have been deferred and are being amortized ratably into revenue over the applicable estimated performance period with end dates ranging between 2011 and 2021. Future revenue will include the ongoing amortization of upfront and milestone payments earned, royalties from Astellas on net sales of VIBATIV™ and proceeds from Astellas for transfers of inventory offset by our cost for manufacturing that inventory. We periodically review and if necessary revise the estimated performance periods of our contracts.
Research & Development
Research and development expenses, as compared to the prior years, were as follows:
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
External research and development | $ | 13.8 | $ | 17.9 | $ | 68.3 | $ | (4.1 | ) | (23 | )% | $ | (50.4 | ) | (74 | )% | ||||||
Employee-related | 29.3 | 30.9 | 49.4 | (1.6 | ) | (5 | )% | (18.5 | ) | (37 | )% | |||||||||||
Stock-based compensation | 11.5 | 10.3 | 13.1 | 1.2 | 12 | % | (2.8 | ) | (21 | )% | ||||||||||||
Facilities, depreciation and other allocated | 22.9 | 22.9 | 24.5 | — | — | % | (1.6 | ) | (7 | )% | ||||||||||||
Total research and development expenses | $ | 77.5 | $ | 82.0 | $ | 155.3 | $ | (4.5 | ) | (5 | )% | $ | (73.3 | ) | (47 | )% | ||||||
Research and development expenses decreased in 2009 compared to 2008 primarily due to lower external costs related to the regulatory process for telavancin. Employee-related expenses decreased in 2009 compared to 2008 primarily due to our reduction in force announced in April 2008. Stock-based compensation increased in 2009 compared to 2008 primarily due to credits taken in 2008 as a result of our reduction in force announced in April 2008.
External research and development costs decreased in 2008 compared to 2007 primarily due to the completion, during 2007, of our Phase 2 clinical studies for TD-5108, our lead GI Motility Dysfunction compound, and TD-1792, our investigational antibiotic and completion of our Phase 3 NP program for telavancin. Employee-related expenses decreased in 2008 compared to 2007 primarily due to our reduction in force announced in April 2008, as well as the costs related to our long-term bonus program having been fully accrued in 2007. Stock-based compensation expenses decreased in 2008 compared to 2007 primarily due to our reduction in force announced in April 2008. Facilities, depreciation and other allocated expenses decreased in 2008 compared to 2007 primarily due to lower supplies and facilities administration costs in 2008.
Research and development expenses for 2010 are expected to be driven largely by employee related expenses, costs associated with our continued development efforts in our oral peripheral
45
Mu-opioid-antagonist, or PUMA, program for opioid-induced bowel constipation with TD-1211, our 5-HT4 program with TD-5108 and TD-8954, our MonoAmine Reuptake Inhibitor, or MARIN, program for chronic pain with TD-9855, ongoing efforts to obtain FDA approval of telavancin for NP and costs associated with new drug discovery programs.
Under our agreement with Astellas, we are responsible for completion of the telavancin Phase 3 programs, publication of the results of these studies and preparation and submission of an NDA to the FDA for the cSSSI and NP indications. The FDA approved VIBATIV™ for the treatment of cSSSI in September 2009. In April 2009, the FDA accepted for review our telavancin NDA for nosocomial pneumonia and in November 2009, the FDA issued a Complete Response letter with regard to the application. We responded to the Complete Response letter in December 2009 and in January 2010, the FDA sent us a letter notifying us that it considered our response "incomplete." We anticipate that our aggregate external costs, net of amounts reimbursed to us by Astellas, associated with our obligations with regard to telavancin described above, assuming that no additional clinical studies will be conducted, will be towards the upper end of the range of $160.0 million to $170.0 million.
We have not provided program costs in detail because we do not track, and have not tracked, all of the individual components (specifically the internal cost components) of our research and development expenses on a program basis. We do not have the systems and processes in place to accurately capture these costs on a program basis.
General and administrative
General and administrative expenses, as compared to the prior years, were as follows:
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
General and administrative | $ | 27.1 | $ | 28.9 | $ | 35.3 | $ | (1.8 | ) | (6 | )% | $ | (6.4 | ) | (18 | )% | ||||||
General and administrative expenses decreased in 2009 compared to 2008 primarily due to lower external expenses related to telavancin marketing preparations and lower facilities related expenses.
General and administrative expenses decreased in 2008 compared to 2007 primarily due to lower employee related expenses due to our reduction in force announced in April 2008.
We anticipate general and administrative expenses in 2010 to be at similar levels to 2009.
Restructuring charges
Restructuring charges, as compared to the prior years, were as follows:
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
Restructuring charges | $ | 1.1 | $ | 5.4 | $ | — | $ | (4.3 | ) | (80 | )% | $ | 5.4 | NA | ||||||||
Restructuring charges decreased in 2009 compared to 2008. The expenses in 2009 were primarily due to restructuring charges recognized for the sublease of excess space in a portion of one of our South San Francisco, CA buildings whereas the expenses in 2008 were due to restructuring charges recognized for severance and other termination benefit charges resulting from our workforce reduction announced in April 2008.
We do not anticipate incurring additional restructuring charges from the actions taken in 2009 and 2008.
46
Interest and other income
Interest and other income, as compared to the prior years, were as follows:
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
Interest and other income | $ | 2.1 | $ | 5.2 | $ | 8.7 | $ | (3.1 | ) | (60 | )% | $ | (3.5 | ) | (40 | )% | ||||||
Interest and other income decreased in 2009 compared to 2008 and in 2008 compared to 2007 primarily due to a trend of lower interest income earned on our investments.
Interest expense
Interest expense, as compared to the prior years, was as follows:
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions, except percentages) | 2009 | 2008 | 2007 | $ | % | $ | % | |||||||||||||||
Interest expense | $ | 6.1 | $ | 5.7 | $ | 0.1 | $ | 0.4 | 7 | % | $ | 5.6 | 5600 | % | ||||||||
Interest expense increased in 2009 compared to 2008 primarily due to a full year of interest expense and amortization of debt issuance costs on our convertible subordinated notes issued in January 2008. Interest expense increased in 2008 compared to 2007 primarily due to interest expense and amortization of debt issuance costs on our convertible subordinated notes issued in January 2008.
Income Taxes
At December 31, 2009, we had net operating loss carryforwards for federal income taxes of $825.2 million and federal research and development tax credit carryforwards of $36.5 million. We recorded a valuation allowance to offset in full the benefit related to our deferred tax assets because realization of these benefits is uncertain.
Since January 1, 2007, we have increased our unrecognized tax benefits by $12.9 million. We had unrecognized tax benefits of $36.2 million and $39.6 million as of January 1, 2009 and December 31, 2009, respectively. If we eventually are able to recognize these uncertain positions, most of the $39.6 million of the unrecognized benefit would reduce the effective tax rate, except for excess tax benefits related to stock-based payments.
Utilization of net operating loss and tax credit carryforwards may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. We conducted an analysis through 2009 to determine whether an ownership change had occurred since inception. The analysis indicated that two ownership changes occurred in prior years. However, notwithstanding the applicable annual limitations, we estimate that no portion of the net operating loss or credit carryforwards will expire before becoming available to reduce federal and state income tax liabilities. Annual limitations may result in expiration of net operating loss and tax credit carryforwards before some or all of such amounts have been utilized.
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily through private placements and public offerings of equity and debt securities and payments received under corporate collaboration agreements. As of December 31, 2009, we had $155.4 million in cash, cash equivalents and marketable securities, excluding $1.3 million in restricted cash that was pledged as collateral for certain of our leases.
47
We expect to incur substantial expenses as we continue our discovery and development efforts; particularly to the extent we advance our product candidates into clinical studies, which are very expensive. We believe that our cash, cash equivalents and marketable securities will be sufficient to meet our anticipated operating needs for at least the next twelve months based upon current operating plans, milestone and royalty forecasts and spending assumptions. We will require additional capital to fund operating needs thereafter. If our current operating plans, milestone and royalty forecasts or spending assumptions change, we may require additional funding sooner in the form of public or private equity offerings or debt financings. Furthermore, if favorable financing opportunities arise, we may seek additional funding sooner. However, future financing may not be available in amounts or on terms acceptable to us, if at all. This could leave us without adequate financial resources to fund our operations as presently conducted.
Cash Flows
| | Year Ended December 31, | Change 2009/2008 | Change 2008/2007 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in millions) | 2009 | 2008 | 2007 | $ | $ | |||||||||||
Net cash used in operating activities | $ | (58.1 | ) | $ | (99.9 | ) | $ | (104.4 | ) | $ | 41.8 | $ | 4.5 | |||
Net cash provided by (used in) investing activities | $ | 1.7 | $ | (67.4 | ) | $ | 110.6 | $ | 69.1 | $ | (178.0 | ) | ||||
Net cash provided by financing activities | $ | 11.6 | $ | 173.1 | $ | 7.8 | $ | (161.5 | ) | $ | 165.3 | |||||
The decrease in cash used in operations in 2009 compared to 2008 was primarily due to higher milestone payments received from our collaboration partners, lower expenses and lower uses of cash for other operating assets and liabilities in 2009. The decrease in cash used in operations in 2008 compared to 2007 was primarily due to a lower expenses in 2008, partially offset by lower milestone payments received from our collaboration partners in 2008 and higher uses of cash for other operating assets and liabilities during 2008.
Investing activities provided cash in 2009 and 2007 and used cash in 2008. The usage of cash in 2008 resulted primarily from purchases of marketable securities as a result of investing the proceeds of our convertible subordinated notes offering which closed in January 2008.
The decrease in cash provided by financing activities in 2009 compared to 2008 and the increase in cash provided by financing activities in 2008 compared to 2007 were primarily due to net proceeds of $166.7 million received in January 2008 from our convertible subordinated notes offering.
Contractual Obligations and Commitments
Our major outstanding contractual obligations relate to our convertible subordinated notes, a note payable, a capital lease, operating leases and outstanding purchase commitments primarily for services under contract research, development and clinical supply agreements. These contractual obligations as of December 31, 2009 are as follows:
(in millions) | Less than 1 year | 1-3 years | 4-5 years | After 5 years | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Convertible subordinated notes | $ | 5.2 | $ | 10.4 | $ | 10.4 | $ | 175.0 | $ | 201.0 | ||||||
Note payable | 0.1 | 0.2 | — | — | 0.3 | |||||||||||
Capital lease | 0.0 | 0.1 | — | — | 0.1 | |||||||||||
Operating leases | 6.4 | 8.3 | — | — | 14.7 | |||||||||||
Purchase obligations | 4.3 | 0.5 | 0.1 | — | 4.9 | |||||||||||
Total | $ | 16.0 | $ | 19.5 | $ | 10.5 | $ | 175.0 | $ | 221.0 | ||||||
48
In January 2008, we closed an underwritten public offering of $172.5 million aggregate principal amount of unsecured convertible subordinated notes that will mature on January 15, 2015. The financing raised proceeds, net of issuance costs, of $166.7 million which is being used for general corporate purposes. The notes bear interest at the rate of 3.0% per year, which is payable semi-annually in arrears in cash on January 15 and July 15 of each year, beginning on July 15, 2008. The notes are convertible, at the option of the holder, into shares of our common stock at an initial conversion rate of 38.6548 shares per $1,000 principal amount of the notes, subject to adjustment in certain circumstances, which represents an initial conversion price of approximately $25.87 per share.
In addition to our debt commitment mentioned above, our other outstanding contractual obligations relate to operating leases, fixed purchase commitments under contract research, development and clinical supply agreements, a capital lease and a note payable. As security for performance of certain obligations under the operating leases for our headquarters, we have issued letters of credit in the aggregate of approximately $1.3 million, collateralized by an equal amount of restricted cash. The terms of the facilities leases require us to maintain an unrestricted cash and marketable securities balance of at least $50.0 million on the last day of each calendar quarter.
Pursuant to our RELOVAIR™ program with GSK, in the event that a LABA product candidate discovered by GSK is successfully developed and commercialized, we will be obligated to make milestone payments to GSK which could total as much as $220.0 million if both a single-agent and a combination product were launched in multiple regions of the world. The current lead LABA candidate, GW642444, is a GSK-discovered compound. Based on available information, we do not estimate that any significant portion of these potential milestone payments to GSK is likely to be made in the next two years.
Recent Accounting Pronouncements
In November 2007, the Financial Accounting Standards Board (FASB) ratified a consensus which requires participants in a collaboration to make separate disclosures regarding the nature and purpose of an arrangement, their rights and obligations under the arrangement, the accounting policy for the arrangement and the income statement classification and amounts arising from the arrangement between participants for each period an income statement is presented. This consensus is effective for us beginning in the first quarter of fiscal year 2009. We have determined that the adoption of this consensus will have no material impact on our financial position, results of operations and cash flows.
In June 2009, the FASB approved the FASB Accounting Standards Codification (Codification), as the single source of authoritative GAAP for all non-governmental entities, with the exception of the SEC and its staff. The Codification changes the referencing and organization of accounting guidance and is effective for interim and annual periods ending after September 15, 2009. Since it is not intended to change or alter existing GAAP, we have determined that the Codification had no material impact on our financial position, results of operations and cash flows.
In September 2009, the FASB ratified guidance to address criteria for separating consideration received in multiple-element revenue arrangements. Companies will be required to allocate the overall consideration to each deliverable by using a best estimate of the selling price of individual deliverables in the arrangement in the absence of vendor-specific objective evidence or other third-party evidence of the selling price. This guidance will be effective in fiscal years beginning on or after June 15, 2010 and early adoption will be permitted. Companies may elect to adopt this guidance prospectively for all revenue arrangements entered into or materially modified after the date of adoption or retrospectively for all periods presented. We are currently evaluating the potential impact, if any, of the adoption of this guidance on our financial position, results of operations and cash flows.
49
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risk, including changes to interest rates which are confined to our cash, cash equivalents, restricted cash and marketable securities. We have invested primarily in money market funds, federal agency notes, corporate debt securities and U.S. treasury notes. To reduce the volatility relating to these exposures, we have put investment and risk management policies and procedures in place. The securities in our investment portfolio are not leveraged, are classified as available-for-sale and, due to their very short-term nature, are subject to minimal interest rate risk. We currently do not engage in hedging activities. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have any significant negative impact on the realized value of our investment portfolio. Our outstanding note payable has a fixed interest rate and therefore, we have no exposure to interest rate fluctuations.
Most of our transactions are conducted in U.S. dollars, although we do conduct some preclinical activities and manufacture some active pharmaceutical ingredients with vendors located outside the United States. Some of these expenses are paid in U.S. dollars, and some are paid in the local foreign currency. If the exchange rate undergoes a change of 10%, we do not believe that it would have a material impact on our results of operations or cash flows.
50
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Consolidated Balance Sheets as of December 31, 2009 and December 31, 2008 | 52 | |
Consolidated Statements of Operations for each of the three years in the period ended December 31, 2009 | 53 | |
Consolidated Statements of Stockholders' Equity (net capital deficiency) for each of the three years in the period ended December 31, 2009 | 54 | |
Consolidated Statements of Cash Flows for each of the three years in the period ended December 31, 2009 | 55 | |
Notes to Consolidated Financial Statements | 56 | |
Report of Independent Registered Public Accounting Firm | 80 |
51
THERAVANCE, INC.
Consolidated Balance Sheets
(in thousands, except per share data)
| | December 31, | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | |||||||
Assets | |||||||||
Current assets: | |||||||||
Cash and cash equivalents | $ | 47,544 | $ | 92,280 | |||||
Marketable securities | 107,846 | 108,325 | |||||||
Receivable from related party | 274 | 287 | |||||||
Notes receivable | 144 | 266 | |||||||
Prepaid and other current assets | 6,234 | 8,803 | |||||||
Total current assets | 162,042 | 209,961 | |||||||
Restricted cash | 1,310 | 3,810 | |||||||
Property and equipment, net | 12,927 | 16,206 | |||||||
Notes receivable | 947 | 1,185 | |||||||
Other long-term assets | 4,167 | 4,994 | |||||||
Total assets | $ | 181,393 | $ | 236,156 | |||||
Liabilities and stockholders' net capital deficiency | |||||||||
Current liabilities: | |||||||||
Accounts payable | $ | 1,792 | $ | 3,277 | |||||
Accrued personnel-related expenses | 6,314 | 8,932 | |||||||
Accrued clinical and development expenses | 1,805 | 3,434 | |||||||
Other accrued liabilities | 5,129 | 4,407 | |||||||
Current portion of note payable and capital lease | 184 | 117 | |||||||
Current portion of deferred revenue | 23,722 | 23,788 | |||||||
Total current liabilities | 38,946 | 43,955 | |||||||
Convertible subordinated notes | 172,500 | 172,500 | |||||||
Deferred rent | 851 | 1,560 | |||||||
Note payable and capital lease | 275 | 319 | |||||||
Deferred revenue | 157,426 | 152,771 | |||||||
Other long-term liabilities | 389 | — | |||||||
Commitments and contingencies (Notes 3, 9 and 10) | |||||||||
Stockholders' net capital deficiency: | |||||||||
Preferred stock, $0.01 par value, 230 shares authorized, no shares issued and outstanding | — | — | |||||||
Common stock, $0.01 par value; 200,000 shares authorized, issuable in series; 54,830 and 52,576 shares issued and outstanding at December 31, 2009 and December 31, 2008, respectively | 549 | 525 | |||||||
Class A Common Stock, $0.01 par value, 30,000 shares authorized, 9,402 issued and outstanding at December 31, 2009 and December 31, 2008 | 94 | 94 | |||||||
Additional paid-in capital | 927,082 | 895,383 | |||||||
Accumulated other comprehensive income | 35 | 501 | |||||||
Accumulated deficit | (1,116,754 | ) | (1,031,452 | ) | |||||
Total stockholders' net capital deficiency | (188,994 | ) | (134,949 | ) | |||||
Total liabilities and stockholders' net capital deficiency | $ | 181,393 | $ | 236,156 | |||||
See accompanying notes to consolidated financial statements.
52
THERAVANCE, INC.
Consolidated Statements of Operations
(in thousands, except per share data)
| | Year Ended December 31, | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | ||||||||
Revenue (includes amounts from GSK, a related party, of $15,073, $12,303 and $11,297 in 2009, 2008 and 2007, respectively) | $ | 24,374 | $ | 23,096 | $ | 22,002 | |||||
Operating expenses: | |||||||||||
Research and development | 77,524 | 82,020 | 155,254 | ||||||||
General and administrative | 27,066 | 28,861 | 35,313 | ||||||||
Restructuring charges | 1,145 | 5,419 | — | ||||||||
Total operating expenses | 105,735 | 116,300 | 190,567 | ||||||||
Loss from operations | (81,361 | ) | (93,204 | ) | (168,565 | ) | |||||
Interest and other income | 2,111 | 5,242 | 8,661 | ||||||||
Interest expense | (6,052 | ) | (5,681 | ) | (93 | ) | |||||
Net loss | $ | (85,302 | ) | $ | (93,643 | ) | $ | (159,997 | ) | ||
Basic and diluted net loss per share | $ | (1.35 | ) | $ | (1.53 | ) | $ | (2.64 | ) | ||
Shares used in computing basic and diluted net loss per share | 63,027 | 61,390 | 60,498 | ||||||||
See accompanying notes to consolidated financial statements.
53
THERAVANCE, INC.
Consolidated Statements of Stockholders' Equity (net capital deficiency)
(in thousands)
| | | | Class A Common Stock | | | | | | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Common Stock | | Notes Receivable from Stockholders | Accumulated Other Comprehensive Income | | Total Stockholders' Equity (net capital deficiency) | |||||||||||||||||||||||
| | Additional Paid-In Capital | Accumulated Deficit | |||||||||||||||||||||||||||
| | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
Balance at December 31, 2006 | 50,746 | $ | 507 | 9,402 | $ | 94 | $ | 840,498 | $ | (3 | ) | $ | 26 | $ | (777,812 | ) | $ | 63,310 | |||||||||||
Common stock issuances from employee stock option and purchase plan, net of repurchases, restricted stock awards and early exercised stock vested | 938 | 9 | — | — | 7,924 | — | — | — | 7,933 | ||||||||||||||||||||
Stock-based compensation | — | — | — | — | 22,494 | — | — | — | 22,494 | ||||||||||||||||||||
Forgiveness and repayments of notes receivable | — | — | — | — | (38 | ) | 3 | — | — | (35 | ) | ||||||||||||||||||
Comprehensive loss: | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (159,997 | ) | (159,997 | ) | ||||||||||||||||||
Net unrealized gain on marketable securities | — | — | — | — | — | — | 31 | — | 31 | ||||||||||||||||||||
Total comprehensive loss | (159,966 | ) | |||||||||||||||||||||||||||
Balance at December 31, 2007 | 51,684 | 516 | 9,402 | 94 | 870,878 | — | 57 | (937,809 | ) | (66,264 | ) | ||||||||||||||||||
Common stock issuances from employee stock option and purchase plan | 892 | 9 | — | — | 6,485 | — | — | — | 6,494 | ||||||||||||||||||||
Stock-based compensation | — | — | — | — | 18,019 | — | — | — | 18,019 | ||||||||||||||||||||
Forgiveness and repayments of notes receivable | — | — | — | — | 1 | — | — | — | 1 | ||||||||||||||||||||
Comprehensive loss: | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (93,643 | ) | (93,643 | ) | ||||||||||||||||||
Net unrealized gain on marketable securities | — | — | — | — | — | — | 444 | — | 444 | ||||||||||||||||||||
Total comprehensive loss | (93,199 | ) | |||||||||||||||||||||||||||
Balance at December 31, 2008 | 52,576 | 525 | 9,402 | 94 | 895,383 | — | 501 | (1,031,452 | ) | (134,949 | ) | ||||||||||||||||||
Common stock issuances from employee stock option, purchase plan and restricted stock units | 2,254 | 24 | — | — | 11,699 | — | — | — | 11,723 | ||||||||||||||||||||
Stock-based compensation | — | — | — | — | 20,000 | — | — | — | 20,000 | ||||||||||||||||||||
Comprehensive loss: | |||||||||||||||||||||||||||||
Net loss | — | — | — | — | — | — | — | (85,302 | ) | (85,302 | ) | ||||||||||||||||||
Net unrealized loss on marketable securities | — | — | — | — | — | — | (466 | ) | — | (466 | ) | ||||||||||||||||||
Total comprehensive loss | (85,768 | ) | |||||||||||||||||||||||||||
Balance at December 31, 2009 | 54,830 | $ | 549 | 9,402 | $ | 94 | $ | 927,082 | $ | — | $ | 35 | $ | (1,116,754 | ) | $ | (188,994 | ) | |||||||||||
See accompanying notes to consolidated financial statements.
54
THERAVANCE, INC.
Consolidated Statements of Cash Flows
(in thousands)
| | Year Ended December 31, | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | |||||||||
Cash flows from operating activities | ||||||||||||
Net loss | $ | (85,302 | ) | $ | (93,643 | ) | $ | (159,997 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Depreciation and amortization | 5,541 | 6,962 | 4,058 | |||||||||
Stock-based compensation | 20,000 | 18,019 | 22,494 | |||||||||
Other-than-temporary impairment loss on marketable securities | — | 20 | 1,140 | |||||||||
Loss on sale of equipment | — | 42 | — | |||||||||
Notes receivable | (13 | ) | 15 | 3 | ||||||||
Changes in operating assets and liabilities: | ||||||||||||
Receivables, prepaid and other assets | 2,755 | (3,523 | ) | (787 | ) | |||||||
Accounts payable and accrued liabilities | (2,696 | ) | (8,217 | ) | (11,383 | ) | ||||||
Accrued personnel-related expenses | (2,618 | ) | (2,908 | ) | 3,525 | |||||||
Deferred rent | (709 | ) | (443 | ) | (295 | ) | ||||||
Deferred revenue | 4,589 | (12,096 | ) | 34,999 | ||||||||
Other liabilities | 389 | (4,139 | ) | 1,871 | ||||||||
Net cash used in operating activities | (58,064 | ) | (99,911 | ) | (104,372 | ) | ||||||
Cash flows from investing activities | ||||||||||||
Purchases of property and equipment | (744 | ) | (1,031 | ) | (9,818 | ) | ||||||
Purchases of marketable securities | (123,460 | ) | (371,625 | ) | (93,329 | ) | ||||||
Maturities of marketable securities | 118,065 | 286,177 | 121,804 | |||||||||
Sales of marketable securities | 5,000 | 18,729 | 90,760 | |||||||||
Sale of equipment | — | 103 | — | |||||||||
Release of restricted cash | 2,500 | — | 50 | |||||||||
Additions to notes receivable | — | (100 | ) | (250 | ) | |||||||
Decrease in notes receivable | 375 | 381 | 1,375 | |||||||||
Net cash provided by (used in) investing activities | 1,736 | (67,366 | ) | 110,592 | ||||||||
Cash flows from financing activities | ||||||||||||
Payments on notes payable and capital leases | (131 | ) | (101 | ) | (88 | ) | ||||||
Net proceeds from issuances of common stock | 11,723 | 6,493 | 7,913 | |||||||||
Net proceeds from issuance of convertible subordinated notes | — | 166,732 | — | |||||||||
Net cash provided by financing activities | 11,592 | 173,124 | 7,825 | |||||||||
Net increase (decrease) in cash and cash equivalents | (44,736 | ) | 5,847 | 14,045 | ||||||||
Cash and cash equivalents at beginning of period | 92,280 | 86,433 | 72,388 | |||||||||
Cash and cash equivalents at end of period | $ | 47,544 | $ | 92,280 | $ | 86,433 | ||||||
Supplemental Disclosure of Cash Flow Information | ||||||||||||
Cash paid for interest | $ | 5,229 | $ | 2,535 | $ | 86 | ||||||
Supplemental Disclosure of Non-Cash Investing Activity | ||||||||||||
Acquisition cost of property and equipment under capital lease | $ | 154 | $ | — | $ | — | ||||||
See accompanying notes to consolidated financial statements.
55
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Summary of Significant Accounting Policies
Description of Operations and Principles of Consolidation
Theravance, Inc. (the Company or Theravance) is a biopharmaceutical company with a pipeline of internally discovered product candidates. Theravance is focused on the discovery, development and commercialization of small molecule medicines across a number of therapeutic areas including respiratory disease, bacterial infections and gastrointestinal motility dysfunction. The Company's key programs include: VIBATIVTM (telavancin) with Astellas Pharma Inc. (Astellas) and the RELOVAIRTM program and Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA) program with GlaxoSmithKline plc (GSK). By leveraging the Company's proprietary insight of multivalency toward drug discovery focused primarily on validated targets, Theravance is pursuing a next generation strategy designed to discover superior medicines in areas of significant unmet medical need. The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Use of Management's Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with a maturity of three months or less on the date of purchase to be cash equivalents. Cash equivalents are carried at cost, which approximates fair value.
Under certain lease agreements and letters of credit, the Company has pledged cash and cash equivalents as collateral. There was $1.3 million and $3.8 million of restricted cash related to such agreements at December 31, 2009 and 2008, respectively.
Marketable Securities
The Company classifies its marketable securities as available-for-sale and has the ability and the intent of holding these securities for a period of time sufficient to allow for any anticipated recovery in market value. Available-for-sale securities are carried at estimated fair value, with the unrealized gains and losses reported in stockholders' equity (net capital deficiency) and included in accumulated other comprehensive income. The cost of securities in this category is adjusted for amortization of premiums and accretion of discounts from the date of purchase to maturity. Such amortization is included in interest and other income. Realized gains and losses and declines in value judged to be other than temporary on marketable securities are also included in interest and other income. The cost of securities sold is based on the specific-identification method.
Other-than-Temporary Impairment Assessment
The Company reviews its investment portfolio to identify and evaluate investments that have indications of possible impairment. Factors considered in determining whether a loss is other-than-temporary include the length of time and extent to which fair value has been less than the
56
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
1. Summary of Significant Accounting Policies (Continued)
cost basis, the financial condition and near-term prospects of the investee, credit quality and the Company's conclusion that it does not intend to sell an impaired investment and is not more likely than not to be required to sell the security before it recovers its amortized cost basis. If the Company determines that the impairment of an investment is other-than-temporary, the investment is written down with a charge recorded in interest and other income, net.
Fair Value of Financial Instruments
Financial instruments include cash equivalents, marketable securities, receivables from related party, accounts payable, accrued liabilities and convertible subordinated notes. Marketable securities are carried at fair value. The carrying value of cash equivalents, receivables from related party, accounts payable and accrued liabilities approximate their fair value due to the relatively short nature of these instruments. Convertible subordinated notes are described in Note 7.
Inventory
Inventory is stated at the lower of cost or market and is included with prepaid and other current assets. Inventory consisted of $3.4 million and $5.6 million of VIBATIV™ finished goods, active pharmaceutical ingredient, or other commercial launch supplies as of December 31, 2009 and 2008, respectively. Under the Company's 2005 License, Development and Commercialization Agreement with Astellas, the Company was responsible to deliver to Astellas approximately six months of first commercial sale stock (as defined) in preparation for the commercialization of VIBATIV™ in the United States. In October 2009, the Company delivered and expensed on an average cost basis the estimated first six months of commercial sale stock out of its capitalized inventory. In December 2009, the Company expensed on an average cost basis $1.2 million of inventory that was no longer realizable. If Astellas decides not to purchase any of the remaining VIBATIV™ inventory, the Company may be required to expense a portion or all of the remaining capitalized inventory.
Revenue Recognition
The Company recognizes revenue in accordance with the criteria outlined in Staff Accounting Bulletin No. 101 (SAB 101) "Revenue Recognition in Financial Statements", as amended by SAB 104 and Financial Accounting Standards Board Accounting Standards Codification (ASC) 605, "Revenue Recognition" and ASC 808, "Collaborative Arrangements." The Company has determined that the deliverables under its various collaboration agreements do not meet the criteria required for separate accounting units for the purposes of revenue recognition.
In connection with the Company's agreements with GSK and Astellas, the Company has determined that the deliverables under these collaboration agreements do not meet the criteria required for separate accounting units for the purposes of revenue recognition. As a result, the Company recognizes revenue from non-refundable, upfront fees and development milestone payments ratably over the term of its performance under the agreements. These upfront or milestone payments received, pending recognition as revenue, are recorded as deferred revenue and are classified as a short-term or long-term liability on the balance sheet to be amortized over the period of deferral. Deferred revenue that is classified as short-term or long-term liabilities is amortized to revenue and is not settled with cash. The Company periodically reviews the estimated performance periods of its contracts based on the progress of its programs. Pursuant to the Company's agreement with Astellas,
57
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
1. Summary of Significant Accounting Policies (Continued)
the Company delivered the estimated first six months of commercial sale stock of VIBATIV™ to Astellas in October 2009. In December 2009, the Company expensed inventory that was no longer realizable. The Company recognizes as revenue the net impact of transactions with Astellas related to VIBATIV™ inventory including revenue specifically attributable to any sales and cost of inventory either transferred or expensed as unrealizable. The Company has been reimbursed by GSK and Astellas for certain external development costs under their respective collaboration agreements. Such reimbursements have been reflected as a reduction of research and development expense and not as revenue.
The Company recognizes royalty revenue from Astellas on their net sales of VIBATIV™ in the period in which the royalties are earned based on net sales reporting provided by Astellas.
Property and Equipment
Property, equipment and leasehold improvements are stated at cost and depreciated using the straight-line method as follows:
Leasehold improvements | Shorter of remaining lease terms or useful life | |
Equipment, furniture and fixtures | 5-7 years | |
Software and computer equipment | 3 years |
Capitalized Software
The Company capitalizes certain costs related to direct material and service costs for software obtained for internal use. Capitalized software costs are depreciated over 3 years.
Impairment of Long-Lived Assets
Long-lived assets include property and equipment. The carrying value of long-lived assets is reviewed for impairment whenever events or changes in circumstances indicate that the asset may not be recoverable. An impairment loss is recognized when the total of estimated future cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount.
Concentration of Credit Risks
The Company invests in a variety of financial instruments and, by its policy, limits the amount of credit exposure with any one issuer, industry or geographic area for investments other than instruments backed by the U.S. federal government.
Related Parties
Transactions with GSK are described in Note 3.
Robert V. Gunderson, Jr. is a director of the Company. The Company has engaged Gunderson Dettmer Stough Villeneuve Franklin & Hachigian, LLP, of which Mr. Gunderson is a partner, as its primary legal counsel. Fees are incurred in the ordinary course of business, and were $0.4 million, $0.4 million and $0.6 million for the years ended December 31, 2009, 2008 and 2007, respectively.
58
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
1. Summary of Significant Accounting Policies (Continued)
Notes Receivable
The Company has provided loans to its employees primarily to assist them with the purchase of a primary residence, which collateralizes the resulting loans. Interest receivable related to the loans was $12,000 and $10,000 at December 31, 2009 and 2008, respectively, and is included in other current assets. The Company accrues interest on the loans at rates ranging up to 8%. The outstanding loans have maturity dates ranging from January 2010 through January 2013.
Bonus Accruals
The Company has short-term bonus programs for eligible employees. Bonuses are determined based on various criteria, including the achievement of corporate, departmental and individual goals. Bonus accruals are estimated based on various factors, including target bonus percentages per level of employee and probability of achieving the goals upon which bonuses are based. The Company's management periodically reviews the progress made towards the goals under the bonus programs. As bonus accruals are dependent upon management's judgments of the likelihood of achieving the various goals, it is possible for bonus expense to vary significantly in future periods if changes occur in those management estimates.
Deferred Rent
Deferred rent consists of the difference between cash payments and the recognition of rent expense on a straight-line basis for the buildings the Company occupies. Rent expense is being recognized ratably over the life of the leases. Because the Company's operating leases provide for rent increases over the terms of the leases, average annual rent expense during the first 5.5 years of the leases exceeded the Company's actual cash rent payments.
Research and Development Costs
Research and development costs are expensed as incurred. Research and development costs consist of salaries and benefits, laboratory supplies and facility costs, as well as fees paid to third parties that conduct certain research and development activities on behalf of the Company, net of certain external development costs reimbursed by GSK and Astellas.
Preclinical Study and Clinical Study Expenses
Most of the Company's preclinical studies and all of its clinical studies have been performed by third-party contract research organizations (CROs). Some CROs bill monthly for services performed, while others bill based upon milestones achieved. The Company reviews the activities performed under the significant contracts each quarter. For preclinical studies, the significant factors used in estimating accruals include the percentage of work completed to date and contract milestones achieved. For clinical study expenses, the significant factors used in estimating accruals include the number of patients enrolled and percentage of work completed to date. Vendor confirmations are obtained for contracts with longer duration when necessary to validate the Company's estimate of expenses. The Company's estimates are highly dependent upon the timeliness and accuracy of the data provided by its CROs regarding the status of each program and total program spending and adjustments are made when deemed necessary.
59
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
1. Summary of Significant Accounting Policies (Continued)
Fair Value of Stock-based Compensation Awards
The Company uses the fair value method of accounting for stock-based compensation arrangements. Stock-based compensation arrangements currently include stock options granted, restricted shares issued, restricted stock unit awards (RSUs) granted and performance-contingent RSUs granted under the 2004 Equity Incentive Plan and the 2008 New Employee Equity Incentive Plan and purchases of common stock by the Company's employees at a discount to the market price during offering periods under the Company's Employee Stock Purchase Plan (ESPP). The estimated fair value of stock options, restricted shares and RSUs is expensed on a straight-line basis over the expected term of the grant and the fair value of performance-contingent RSUs is expensed during the term of the award when the Company determines that it is probable that certain performance milestones will be met. Compensation expense for purchases under the ESPP is recognized based on the estimated fair value of the common stock during each offering period and the percentage of the purchase discount.
Stock-based compensation expense for stock options and RSUs has been reduced for estimated forfeitures so that compensation expense is based on options and RSUs ultimately expected to vest. The Company's estimated annual forfeiture rates for stock options and RSUs are based on its historical forfeiture experience.
Segment Reporting
The Company has determined that it operates in only one segment which is the research and development of human therapeutics. Revenues are primarily generated from the Company's collaborations with GSK and Astellas, located in the United Kingdom and Japan, respectively. All long-lived assets are maintained in the United States.
Comprehensive Loss
Comprehensive loss is comprised of net loss and other comprehensive income (loss). Other comprehensive income (loss) consists of changes in unrealized gains and losses on the Company's available-for-sale securities. Comprehensive income or loss for the years ended December 31, 2009, 2008 and 2007 has been presented in the Company's Consolidated Statements of Stockholders' Equity (net capital deficiency).
Income Taxes
The Company utilizes the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax basis of assets and liabilities and are measured using enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
Recent Accounting Pronouncements
In November 2007, the Financial Accounting Standards Board (FASB) ratified a consensus which requires participants in a collaboration to make separate disclosures regarding the nature and purpose of an arrangement, their rights and obligations under the arrangement, the accounting policy for the arrangement and the income statement classification and amounts arising from the arrangement
60
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
1. Summary of Significant Accounting Policies (Continued)
between participants for each period an income statement is presented. This consensus is effective for the Company beginning in the first quarter of fiscal year 2009. The Company has determined that the adoption of this consensus will have no material impact on its financial position, results of operations and cash flows.
In June 2009, the FASB approved the FASB Accounting Standards Codification (Codification), as the single source of authoritative GAAP for all non-governmental entities, with the exception of the SEC and its staff. The Codification changes the referencing and organization of accounting guidance and is effective for interim and annual periods ending after September 15, 2009. Since it is not intended to change or alter existing GAAP, the Company has determined the Codification had no material impact on its financial position, results of operations and cash flows.
In September 2009, the FASB ratified guidance to address criteria for separating consideration received in multiple-element revenue arrangements. Companies will be required to allocate the overall consideration to each deliverable by using a best estimate of the selling price of individual deliverables in the arrangement in the absence of vendor-specific objective evidence or other third-party evidence of the selling price. This guidance will be effective in fiscal years beginning on or after June 15, 2010 and early adoption will be permitted. Companies may elect to adopt this guidance prospectively for all revenue arrangements entered into or materially modified after the date of adoption or retrospectively for all periods presented. The Company is currently evaluating the potential impact, if any, of the adoption of this guidance on its financial position, results of operations and cash flows.
2. Net Loss per Share
Basic net loss per share (basic EPS) is computed by dividing net loss by the weighted-average number of common shares outstanding during the period, less shares subject to repurchase. Diluted net loss per share (diluted EPS) is computed by dividing net loss by the weighted-average number of common shares outstanding during the period, less shares subject to repurchase, plus dilutive potential common shares. Diluted EPS is identical to basic EPS for all periods presented since potential common shares are excluded from the calculation, as their effect is anti-dilutive.
Using the treasury stock method, potential common shares that were excluded from the calculation are as follows:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | 2007 | |||||||
Shares issuable upon the exercise of stock options | 2,092 | 2,167 | 5,202 | |||||||
Shares issuable under restricted stock unit and restricted stock awards | 378 | 134 | 33 | |||||||
Shares issuable upon the conversion of convertible debt | 6,668 | 6,249 | — | |||||||
61
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
2. Net Loss per Share (Continued)
The calculation of basic and diluted EPS is as follows:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | |||||||
| | (in thousands, except for per share data) | |||||||||
Basic and diluted: | ||||||||||
Net loss | $ | (85,302 | ) | $ | (93,643 | ) | $ | (159,997 | ) | |
Weighted average shares of common stock outstanding | 63,084 | 61,466 | 60,642 | |||||||
Less: weighted average shares subject to repurchase | (57 | ) | (76 | ) | (144 | ) | ||||
Weighted average shares used in computing basic and diluted net loss per share | 63,027 | 61,390 | 60,498 | |||||||
Basic and diluted net loss per share | $ | (1.35 | ) | $ | (1.53 | ) | $ | (2.64 | ) | |
3. Collaboration Agreements
2005 License, Development and Commercialization Agreement with Astellas
In November 2005, the Company entered into a collaboration arrangement with Astellas for the development and commercialization of telavancin. In July 2006, Japan was added to the collaboration, thereby giving Astellas worldwide rights to this medicine. Through December 31, 2009, the Company has received $190.0 million in upfront, milestone and other fees from Astellas. The Company is eligible to receive up to an additional $30.0 million in remaining milestone payments related to regulatory filings and approvals in various regions of the world. The Company records these payments as deferred revenue and is amortizing them ratably over its estimated period of performance (development and commercialization period). The Company recognized $10.8 million and $10.3 million in amortization of deferred revenue under this agreement in 2008 and 2007, respectively.
The Company is entitled to receive royalties on global net sales of VIBATIV™ by Astellas that, on a percentage basis, range from the high teens to the upper twenties depending on sales volume. Under this arrangement, the Company is responsible for substantially all costs to develop and obtain U.S. regulatory approval for telavancin for complicated skin and skin structure infections (cSSSI) and nosocomial pneumonia (NP) and Astellas is responsible for substantially all other costs associated with commercialization and further development of telavancin.
Pursuant to the collaboration arrangement, the Company delivered the estimated first six months of commercial sale stock of VIBATIV™ to Astellas out of its capitalized inventory in October 2009. In
62
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Collaboration Agreements (Continued)
December 2009, the Company expensed inventory that was no longer realizable. In 2009, the Company's revenue from this collaboration agreement was composed of:
(in thousands) | Year Ended December 31, 2009 | |||
|---|---|---|---|---|
Amortization of deferred revenue | $ | 11,338 | ||
Royalties from net sales of VIBATIV™ | 766 | |||
Cost of estimated first six months of commercial sale stock of VIBATIV™ | (1,629 | ) | ||
Cost of expensed unrealizable VIBATIV™ | (1,175 | ) | ||
Net Astellas collaboration revenue | $ | 9,300 | ||
RELOVAIR™ Program with GSK
In November 2002, the Company entered into its long-acting beta2 agonist (LABA) collaboration with GSK to develop and commercialize a long-acting beta2 agonist (LABA) product candidate both as a single-agent new medicine for the treatment of chronic obstructive pulmonary disease (COPD) and as part of a new combination medicine with an inhaled corticosteroid (ICS) for the treatment of asthma and/or a long-acting muscarinic antagonist (LAMA) for COPD.
In connection with the RELOVAIR™ program, in 2002 the Company received from GSK an upfront payment of $10.0 million and sold to an affiliate of GSK shares of the Company's Series E Preferred Stock for an aggregate purchase price of $40.0 million. In addition, the Company was eligible to receive up to $495.0 million in development, approval, launch and sales milestones and royalties on the sales of any product resulting from this program. Through December 31, 2009, the Company has received a total of $60.0 million in upfront and development milestone payments. GSK has determined to focus the collaboration's resources on the development of the lead LABA, GW642444, a GSK-discovered compound, together with GSK's ICS, fluticasone furoate. Accordingly, the Company does not expect to receive any further milestone payments from the RELOVAIR™ program. In the event that a LABA product candidate discovered by GSK is successfully developed and commercialized, the Company would be obligated to make milestone payments to GSK which could total as much as $220.0 million if both a single-agent and a combination product were launched in multiple regions of the world. Based on available information, the Company does not estimate that a significant portion of these potential milestone payments to GSK are likely to be made in the next two years. Moreover, the Company is entitled to receive the same royalties on sales of medicines from the RELOVAIR™ program, regardless of whether the product candidate originated with Theravance or with GSK. The Company is entitled to receive royalties of 15% on the first $3.0 billion of annual global net sales, and 5% on annual global net sales above $3.0 billion, for approved single-agent LABA and combination LABA-ICS medicines. Sales of single-agent LABA medicines and combination medicines would be combined for the purposes of this royalty calculation. For other products combined with a LABA from the RELOVAIR™ program, such as a combination LABA/LAMA medicine, which are launched after a LABA/ICS combination medicine, royalties are upward tiering and range from the mid-single digits to 10%. However, if GSK is not selling a LABA/ICS combination product at the time that the first other LABA combination is launched, then the royalties described above for the LABA/ICS combination medicine are applicable.
63
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Collaboration Agreements (Continued)
The Company recorded the initial cash payment and subsequent milestone payments as deferred revenue and is amortizing them ratably over its estimated period of performance (the product development period). Collaboration revenue from GSK under this agreement was $5.1 million, $6.8 million and $6.8 million in 2009, 2008 and 2007, respectively.
2004 Strategic Alliance with GSK
In March 2004, the Company entered into its strategic alliance with GSK. Under this alliance, GSK received an option to license exclusive development and commercialization rights to product candidates from all of the Company's full drug discovery programs initiated prior to September 1, 2007, on pre-determined terms and on an exclusive, worldwide basis. Under the terms of the strategic alliance, GSK has only one opportunity to license each of the Company's programs. Upon GSK's decision to license a program, GSK is responsible for funding all future development, manufacturing and commercialization activities for product candidates in that program. In addition, GSK is obligated to use diligent efforts to develop and commercialize product candidates from any program that it licenses. Consistent with the Company's strategy, it is obligated at its sole cost to discover two structurally different product candidates for any programs that are licensed by GSK under the alliance. If these programs are successfully advanced through development by GSK, the Company is entitled to receive clinical, regulatory and commercial milestone payments and royalties on any sales of medicines developed from these programs. For product candidates licensed to date under this agreement, the royalty structure for a product containing one of its compounds as a single active ingredient would result in an average percentage royalty rate in the low double digits. If a product is successfully commercialized, in addition to any royalty revenue that the Company receives, the total upfront and milestone payments that it could receive in any given program that GSK licenses range from $130.0 million to $162.0 million for programs with single-agent medicines and up to $252.0 million for programs with both a single-agent and a combination medicine. If GSK chooses not to license a program, the Company retains all rights to the program and may continue the program alone or with a third party. To date, GSK has licensed the Company's two COPD programs: long-acting muscarinic antagonist (LAMA) and Bifunctional Muscarinic Antagonist-beta2 Agonist (MABA). The Company received $5.0 million payments from GSK in connection with its license of each of the Company's LAMA and MABA programs in August 2004 and March 2005, respectively. GSK has chosen not to license the Company's bacterial infections program, anesthesia program or 5-HT4 program.
In connection with the strategic alliance with GSK, the Company received from GSK a payment of $20.0 million. This payment is being amortized over the initial performance period during which GSK may exercise its right to license certain of the Company's programs under the agreement. In connection with the strategic alliance, the Company recognized $2.7 million in revenue for each of the years ended December 31, 2009, 2008 and 2007. In addition, in May 2004, GSK purchased through an affiliate 6,387,096 shares of the Company's Class A common stock for an aggregate purchase price of $108.9 million.
Through December 31, 2009, the Company has received $46.0 million in upfront and milestone payments from GSK relating to the strategic alliance agreement. In addition, pursuant to a partial exercise of its rights under the governance agreement, upon the closing of the Company's initial public offering on October 8, 2004, GSK purchased through an affiliate an additional 433,757 shares of Class A common stock for $6.9 million.
64
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
3. Collaboration Agreements (Continued)
In August 2004, GSK exercised its right to license the Company's LAMA program pursuant to the terms of the strategic alliance. The Company received a $5.0 million payment from GSK in connection with its licensing of the Company's LAMA program. Through December 31, 2009, the Company received a milestone payment from GSK of $3.0 million related to clinical progress of the Company's product candidate. These payments were amortized ratably over the estimated period of performance (the product development period) until 2009, when the Company recognized the remaining $4.2 million of deferred revenue related to the LAMA program as a result of the program being returned to the Company from GSK. The recognition of the remaining deferred revenue related to the LAMA program had a favorable impact to basic net loss per share of $0.07 for 2009. The Company recognized $4.2 million, $0.8 million and $0.8 million in revenue related to the LAMA program in 2009, 2008 and 2007, respectively.
In March 2005, GSK exercised its right to license the Company's MABA program pursuant to the terms of the strategic alliance. The Company received a $5.0 million payment from GSK in connection with the license of the Company's MABA program. Through December 31, 2009, the Company received milestone payments from GSK of $13.0 million related to clinical progress of its candidate. These payments are being amortized ratably over the estimated period of performance (the product development period). In connection with the MABA program, the Company recognized $3.0 million, $2.0 million and $1.0 million in revenue in 2009, 2008 and 2007, respectively.
4. Marketable Securities
The Company manages, monitors and measures its investments in highly liquid investment-grade securities by major security type. The following is a summary of the Company's cash, cash equivalents, marketable securities and restricted cash by major security type:
| | December 31, 2009 | December 31, 2008 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | |||||||||||||||||
U.S. government securities | $ | 45,123 | $ | 27 | $ | (5 | ) | $ | 45,145 | $ | 39,483 | $ | 149 | $ | — | $ | 39,632 | ||||||||
U.S. government agencies | 18,032 | 10 | — | 18,042 | 28,785 | 284 | — | 29,069 | |||||||||||||||||
U.S. corporate notes | 11,181 | 8 | (5 | ) | 11,184 | 19,635 | 55 | (13 | ) | 19,677 | |||||||||||||||
U.S. commercial paper | 43,473 | 1 | — | 43,474 | 24,916 | 26 | — | 24,942 | |||||||||||||||||
Certificates of deposit | — | — | — | — | 60 | — | — | 60 | |||||||||||||||||
Money market funds | 35,425 | — | — | 35,425 | 86,876 | — | — | 86,876 | |||||||||||||||||
Total | 153,234 | 46 | (10 | ) | 153,270 | 199,755 | 514 | (13 | ) | 200,256 | |||||||||||||||
Less amounts classified as cash equivalents | (44,114 | ) | — | — | (44,114 | ) | (88,121 | ) | — | — | (88,121 | ) | |||||||||||||
Less amounts classified as restricted cash | (1,310 | ) | — | — | (1,310 | ) | (3,810 | ) | — | — | (3,810 | ) | |||||||||||||
Amounts classified as marketable securities | $ | 107,810 | $ | 46 | $ | (10 | ) | $ | 107,846 | $ | 107,824 | $ | 514 | $ | (13 | ) | $ | 108,325 | |||||||
65
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
4. Marketable Securities (Continued)
The estimated fair value amounts were determined using available market information. At December 31, 2009, 100% of marketable securities have contractual maturities within twelve months and the average duration of marketable securities was approximately five months.
The following table provides the net realized gains (losses) on marketable securities for the periods presented:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | 2007 | |||||||
Realized gains | $ | — | $ | 28 | $ | 224 | ||||
Realized losses | — | (20 | ) | (1,188 | ) | |||||
Net realized gains (losses) | $ | — | $ | 8 | $ | (964 | ) | |||
In the year ended December 31, 2009, the Company realized no gains or losses that were previously classified as unrealized gains and losses in accumulated other comprehensive income at December 31, 2008.
In the year ended December 31, 2008, the Company realized $18,000 in gains and no losses that were previously classified as unrealized gains and losses in accumulated other comprehensive income at December 31, 2007.
The following table provides the breakdown of the marketable securities with unrealized losses at December 31, 2009:
| | In loss position for less than 12 months | In loss position for more than 12 months | Total | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | Fair Value | Gross Unrealized losses | Fair Value | Gross Unrealized Losses | Fair Value | Gross Unrealized Losses | |||||||||||||
U.S. corporate notes | $ | 7,495 | $ | (5 | ) | $ | — | $ | — | $ | 7,495 | $ | (5 | ) | |||||
U.S. government securities | 15,004 | (5 | ) | — | — | 15,004 | (5 | ) | |||||||||||
Total | $ | 22,499 | $ | (10 | ) | $ | — | $ | — | $ | 22,499 | $ | (10 | ) | |||||
The Company does not intend to sell the investments which are in an unrealized loss position and it is not more likely than not the Company will be required to sell the investments before recovery of their amortized cost basis, which may be maturity. The Company has determined that the gross unrealized losses on its marketable securities at December 31, 2009 were temporary in nature.
5. Fair Value Measurements
The Company defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date.
66
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
5. Fair Value Measurements (Continued)
The Company's valuation techniques are based on observable and unobservable inputs. Observable inputs reflect readily obtainable data from independent sources, while unobservable inputs reflect the Company's market assumptions. The Company classifies these inputs into the following hierarchy:
Level 1 Inputs—Quoted prices for identical instruments in active markets.
Level 2 Inputs—Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations whose inputs are observable or whose significant value drivers are observable.
Level 3 Inputs—Unobservable inputs and little, if any, market activity for the assets.
The fair value of the Company's financial assets were as follows:
| | Fair Value Measurements at Reporting Date Using | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | | |||||||||
December 31, 2009 (in thousands) | Level 1 | Level 2 | Level 3 | Total | |||||||||
U.S. government securities | $ | 45,145 | $ | — | $ | — | $ | 45,145 | |||||
U.S. government agency securities | 18,042 | — | — | 18,042 | |||||||||
U.S. corporate notes | 1,020 | 10,164 | — | 11,184 | |||||||||
U.S. commercial paper | — | 43,474 | — | 43,474 | |||||||||
Money market funds | 35,425 | — | — | 35,425 | |||||||||
Total | $ | 99,632 | $ | 53,638 | $ | — | $ | 153,270 | |||||
| | Fair Value Measurements at Reporting Date Using | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | | |||||||||
December 31, 2008 (in thousands) | Level 1 | Level 2 | Level 3 | Total | |||||||||
U.S. government securities | $ | 39,632 | $ | — | $ | — | $ | 39,632 | |||||
U.S. government agency securities | 28,103 | 966 | — | 29,069 | |||||||||
U.S. corporate notes | 9,712 | 9,965 | — | 19,677 | |||||||||
U.S. commercial paper | — | 24,942 | — | 24,942 | |||||||||
Certificates of deposit | 60 | — | — | 60 | |||||||||
Money market funds | 86,876 | — | — | 86,876 | |||||||||
Total | $ | 164,383 | 35,873 | $ | — | $ | 200,256 | ||||||
67
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
6. Property and Equipment
Property and equipment consists of the following:
| | December 31, | ||||||
|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | |||||
Computer equipment | $ | 3,454 | $ | 3,194 | |||
Software | 4,546 | 4,546 | |||||
Furniture and fixtures | 3,577 | 3,423 | |||||
Laboratory equipment | 27,234 | 26,621 | |||||
Leasehold improvements | 15,381 | 15,381 | |||||
| 54,192 | 53,165 | ||||||
Less accumulated depreciation and amortization | (41,265 | ) | (36,959 | ) | |||
Property and equipment, net | $ | 12,927 | $ | 16,206 | |||
Depreciation expense was $4.3 million, $4.5 million and $4.1 million for the years ended December 31, 2009, 2008 and 2007, respectively. The change in accumulated depreciation is net of asset retirements.
7. Long-Term Obligations
Long-term obligations are as follows:
| | December 31, 2009 | December 31, 2008 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | Carrying value | Estimated fair value | Carrying value | Estimated fair value | |||||||||
Convertible subordinated notes | $ | 172,500 | $ | 137,784 | $ | 172,500 | $ | 103,931 | |||||
Note payable to lessor | 329 | 329 | 436 | 436 | |||||||||
Convertible Subordinated Notes
In January 2008, the Company closed an underwritten public offering of $172.5 million aggregate principal amount of unsecured convertible subordinated notes which will mature on January 15, 2015. The financing raised proceeds, net of issuance costs, of $166.7 million. The notes bear interest at the rate of 3.0% per year, which is payable semi-annually in arrears in cash on January 15 and July 15 of each year, beginning on July 15, 2008. The notes are convertible, at the option of the holder, into shares of the Company's common stock at an initial conversion rate of 38.6548 shares per $1,000 principal amount of the notes, subject to adjustment in certain circumstances, which represents an initial conversion price of approximately $25.87 per share. The debt issuance costs, which are included in other long-term assets, are being amortized on a straight-line basis over the life of the notes. Unamortized debt issuance costs totaled $4.2 million as of December 31, 2009.
Holders of the notes will be able to require the Company to repurchase some or all of their notes upon the occurrence of a fundamental change (as defined) at 100% of the principal amount of the notes being repurchased plus accrued and unpaid interest. The Company may not redeem the notes prior to January 15, 2012. On or after January 15, 2012 and prior to the maturity date, the Company, upon notice of redemption, may redeem for cash all or part of the notes if the last reported sale price of its common stock has been greater than or equal to 130% of the conversion price then in effect for
68
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
7. Long-Term Obligations (Continued)
at least 20 trading days during any 30 consecutive trading day period prior to the date on which it provides notice of redemption. The redemption price will equal 100% of the principal amount of the notes to be redeemed, plus accrued and unpaid interest up to but excluding the redemption date.
Note Payable
In connection with the Company's lease agreement for its 60,000 square foot facility in South San Francisco, California (see Note 9), the Company received approximately $0.9 million in July 2002 under a tenant improvement loan from the lessor, which is payable in monthly installments through 2012, bears interest at 14.5% per annum and is secured by the underlying leasehold improvements.
The aggregate maturities of the note payable for each of the remaining three years are as follows: $0.1 million in 2010, $0.2 million in 2011 and $42,000 in 2012.
Capital Lease
The Company entered into a capital lease agreement for communications equipment during 2009 and has accounted for the arrangement as follows:
(in thousands) | Year Ended December 31, 2009 | |||
|---|---|---|---|---|
Initiation of lease arrangement | $ | 154 | ||
2009 minimum lease payments less interest | (24 | ) | ||
Present value of future payments | 130 | |||
Less current portion | (51 | ) | ||
Long-term portion | $ | 79 | ||
The equipment under the capital lease arrangement is included in property and equipment and the related amortization is included in depreciation expense in the consolidated statements of operations and cash flows. The cost of equipment financed under capital leases was $0.2 million and the related accumulated amortization was $10,000 as of December 31, 2009.
8. Restructuring charges
In response to the completion of its Phase 3 development activities and to reduce its overall cash burn rate, the Company announced a plan to reduce its workforce by approximately 40% through layoffs from all departments throughout the organization in April 2008.
In February 2009, the Company entered into a sublease agreement with a third party to sublease excess space in a portion of one of its South San Francisco, CA buildings. The sublease has a 37 month term that began March 2009. For the year ended December 31, 2009, the Company recorded a restructuring charge of $1.3 million of which $1.1 million represents the estimated fair value of the Company's lease payments and expenses less sublease income through March 2012.
69
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
8. Restructuring charges (Continued)
The following table summarizes the accrual balance and utilization by cost type for the restructuring for the years ended December 31, 2009 and 2008:
(in thousands) | Employee Severance and Benefits | Excess Facilities | |||||
|---|---|---|---|---|---|---|---|
Restructuring charges accrued | $ | 5,533 | $ | — | |||
Cash payments | (4,874 | ) | — | ||||
Adjustments | (157 | ) | — | ||||
Balance as of December 31, 2008 | 502 | — | |||||
Restructuring charges accrued | 85 | 1,264 | |||||
Cash payments | (443 | ) | (570 | )* | |||
Adjustments | (28 | ) | — | ||||
Balance as of December 31, 2009 | $ | 116 | $ | 694 | |||
- *
- Includes fair value of cash payments less sublease payments received
The restructuring accrual related to employee severance and benefits is recorded within accrued personnel-related expenses and the restructuring accrual related to excess facilities is recorded within other accrued liabilities and other long-term liabilities on the Company's consolidated balance sheets.
9. Operating Leases and Subleases
The Company leases an 110,000 square foot facility and an adjacent 60,000 square foot facility in South San Francisco, California. Both of the leases expire in 2012 and have two renewal options of five years each. As security for performance of its future obligations under these leases, the Company has letters of credit for an aggregate $1.3 million, collateralized by an equal amount of restricted cash. If the Company's unrestricted cash and marketable securities balance is less than $50.0 million during the terms of the leases, then the letters of credit must be increased by an aggregate of $1.0 million.
At December 31, 2009, the Company's future minimum commitments under noncancelable operating leases are as follows:
(in thousands) | Minimum Lease Commitments | ||||
|---|---|---|---|---|---|
Year ending December 31: | |||||
2010 | $ | 6,435 | |||
2011 | 6,596 | ||||
2012 | 1,659 | ||||
Total | $ | 14,690 | |||
70
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
9. Operating Leases and Subleases (Continued)
Expenses and income associated with operating leases were as follows:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | 2007 | |||||||
Rent expense | $ | 6,559 | $ | 6,873 | $ | 6,958 | ||||
Sublease income, net | (580 | ) | — | (128 | ) | |||||
As of December 31, 2009, the Company expects to receive up to $1.9 million of minimum rentals through the end of a noncancelable sublease in March 2012.
10. Commitments and Contingencies
Indemnifications
The Company indemnifies its officers and directors for certain events or occurrences, subject to certain limits. The Company may be subject to contingencies that may arise from matters such as product liability claims, legal proceedings, shareholder suits and tax matters, as such, the Company is unable to estimate the potential exposure related to these indemnification agreements. The Company has not recognized any liabilities relating to these agreements as of December 31, 2009.
Purchase Obligations
At December 31, 2009, the Company had outstanding purchase obligations on commercially reasonable terms, primarily for services under contract research, development and clinical supply agreements totaling $4.9 million.
11. Stock-Based Compensation
Determining Fair Value of Stock-Based Compensation
The Company uses the Black-Scholes valuation model for stock-based payment awards granted. The Company's determination of the fair value of stock-based payment awards on the grant date using the Black-Scholes option valuation model requires the input of highly subjective assumptions, including the expected stock price volatility and the expected life of the award. As the Company has been operating as a public company for a period of time that is shorter than its estimated expected option life, the Company is unable to use actual price volatility or option life data as input assumptions within its Black-Scholes valuation model when determining the fair value of its stock options. As a result, the Company uses the "simplified" method as described in Staff Accounting Bulletin No. 107 for expected option life and peer company price volatility.
71
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Stock-Based Compensation (Continued)
The weighted-average assumptions used to value employee stock-based compensation for stock options granted and employee stock purchase plan issuances were as follows:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| | 2009 | 2008 | 2007 | |||||||
Employee stock options | ||||||||||
Risk-free interest rate | 1.55% - 2.98 | % | 1.50% - 3.50 | % | 3.48% - 5.03 | % | ||||
Expected life (in years) | 5 - 6 | 6 | 5 - 6 | |||||||
Volatility | 0.48 - 0.57 | 0.49 - 0.57 | 0.46 - 0.49 | |||||||
Dividend yield | — % | — % | — % | |||||||
Weighted average estimated fair value of stock options granted | $7.48 | $6.19 | $16.47 | |||||||
Employee stock purchase plan issuances | ||||||||||
Risk-free interest rate | 0.17% - 0.88 | % | 0.25% - 2.80 | % | 3.23% - 4.98 | % | ||||
Expected life (in years) | 0.5 - 2 | 0.5 - 2 | 0.5 - 2 | |||||||
Volatility | 0.50 - 0.84 | 0.45 - 0.92 | 0.26 - 0.41 | |||||||
Dividend yield | — | % | — | % | — | % | ||||
Weighted average estimated fair value of ESPP issuances | $6.42 | $4.10 | $8.17 | |||||||
Total stock-based compensation expense recognized for the year ended December 31, 2009 was $20.0 million, which consisted of $19.0 million related to employee stock options, RSUs and employee stock purchases, $0.5 million related to options and RSUs issued to non-employees for services rendered and $0.5 million related to shares of restricted stock. As of December 31, 2009, there was $11.8 million, $18.9 million and $0.8 million of total unrecognized compensation cost related to unvested stock options, RSUs and restricted stock, respectively. This cost is expected to be recognized over a weighted-average period of approximately 2.18 years, 2.69 years and 2.29 years for stock options, RSUs and restricted stock, respectively. Total stock-based compensation expense recognized for the year ended December 31, 2008 was $18.0 million, which consisted of $16.9 million related to employee stock awards and employee stock purchases, $0.6 million related to the value of options and RSUs issued to non-employees for services rendered and $0.5 million related to the value of shares of restricted stock. The Company has not recognized, and does not expect to recognize in the near future, any tax benefit related to employee stock-based compensation costs as a result of the full valuation allowance on the Company's net deferred tax assets including deferred tax assets related to its net operating loss carryforwards.
The following table discloses the allocation of stock-based compensation expense included in the consolidated statements of operations:
| | Year Ended December 31, | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | 2007 | |||||||
Research and development | $ | 11,542 | $ | 10,264 | $ | 13,133 | ||||
General and administrative | 8,458 | 7,755 | 9,361 | |||||||
Total stock-based compensation expense | $ | 20,000 | $ | 18,019 | $ | 22,494 | ||||
72
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Stock-Based Compensation (Continued)
The Company does not currently pay dividends and does not intend to declare or pay cash dividends on its common stock in the foreseeable future.
Equity Incentive Plans
The Company issues stock options, restricted stock awards and RSUs under the 2004 Equity Incentive Plan, as amended (the 2004 Plan) and the 2008 New Employee Equity Incentive Plan, as amended (the 2008 Plan).
2008 New Employee Equity Incentive Plan
The Company authorized 500,000 shares of Common Stock for issuance under the 2008 Plan upon its adoption in 2008 and authorized an additional 200,000 shares for issuance under the 2008 Plan in July 2009. The 2008 Plan provides for the granting of non-qualified stock options, restricted stock awards and RSUs to newly hired employees. Stock options may be granted with an exercise price not less than 100% of the fair market value of the common stock on the date of grant. Stock options are generally granted with terms of up to ten years and vest over a period of four years. For the year ended December 31, 2009, the Company granted stock options to purchase 314,250 shares at a weighted average exercise price of $14.88 and granted 18,000 RSUs with a weighted-average fair value of $14.50 per share under the 2008 Plan. For the year ended December 31, 2008, the Company granted stock options to purchase 192,250 shares at a weighted average exercise price of $6.31 and granted 5,376 RSUs with a weighted-average fair value of $6.15 per share under the 2008 Plan. As of December 31, 2009, there were 178,116 shares remaining available for issuance under the 2008 Plan.
2004 Equity Incentive Plan
The 2004 Plan provides for the granting of stock options, restricted stock awards, stock appreciation rights and RSUs to employees, officers, directors and consultants of the Company. Stock options may be granted with an exercise price not less than 100% of the fair market value of the common stock on the date of grant. Stock options are generally granted with terms of up to ten years and vest over a period of four years. During the years ended December 31, 2009, 2008 and 2007, the Company granted stock options to purchase 42,000, 191,500 and 2,127,256 shares, respectively, at weighted average stock prices of $14.98, $18.08 and $32.06, respectively, under the 2004 Plan. During the years ended December 31, 2009 and 2008, the Company granted 931,636 and 1,042,113 RSUs, respectively, with a weighted-average fair value of $14.66 and $16.33 per share, respectively, under the 2004 Plan. As of December 31, 2009, total shares remaining available for issuance under the 2004 Plan were 1,541,428.
For the years ended December 31, 2009, 2008 and 2007, the Company granted zero, 113,636 and 2,061,206 performance-contingent RSUs, respectively, to employees. These performance-contingent RSUs have dual triggers of vesting based upon the successful achievement of certain corporate operating milestones during 2009, as well as a requirement for continued employment through 2009 and 2010. In 2008, 25% of the performance-contingent RSUs granted to senior management in 2007 was cancelled and the performance-contingent RSUs held by non-executive employees were amended such that half of the RSUs would vest over time and the other half will remain subject to certain performance targets. As none of the performance milestones were achieved on December 31, 2009,
73
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Stock-Based Compensation (Continued)
433,491 of these RSUs were forfeited pursuant to their terms and the Company anticipates that the remaining 544,410 will be forfeited in April 2010 pursuant to their terms.
The following table summarizes equity award activity under the 2008 Plan and the 2004 Plan, and related information:
| | Number of Shares Available for Grant | Number of Shares Subject to Outstanding Options | Weighted-Average Exercise Price of Outstanding Options | Number of Shares Subject to Outstanding RSUs | Weighted-Average Fair Value per Share at Grant | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | (in thousands, except per share data) | ||||||||||||||||
Balance at December 31, 2006 | 1,070 | 10,390 | $ | 12.92 | — | $ | — | ||||||||||
Additional shares authorized | 3,500 | — | $ | — | — | $ | — | ||||||||||
Granted | (4,259 | ) | 2,127 | $ | 32.06 | 2,061 | $ | 32.45 | |||||||||
Exercised | — | (815 | ) | $ | 6.93 | — | $ | — | |||||||||
Forfeited | 282 | (266 | ) | $ | 24.61 | (16 | ) | $ | 33.25 | ||||||||
Balance at December 31, 2007 | 593 | 11,436 | $ | 16.63 | 2,045 | $ | 32.44 | ||||||||||
Additional shares authorized | 500 | — | — | — | — | ||||||||||||
Granted | (1,431 | ) | 384 | $ | 12.18 | 1,047 | $ | 16.28 | |||||||||
Exercised | — | (692 | ) | $ | 6.76 | — | $ | — | |||||||||
Forfeited | 2,007 | (1,175 | ) | $ | 26.30 | (832 | ) | $ | 30.56 | ||||||||
Balance at December 31, 2008 | 1,669 | 9,953 | $ | 16.01 | 2,260 | $ | 21.51 | ||||||||||
Additional shares authorized | 200 | — | — | — | — | ||||||||||||
Granted | (1,306 | ) | 356 | $ | 14.90 | 950 | $ | 14.66 | |||||||||
Exercised | — | (1,333 | ) | $ | 7.77 | — | $ | — | |||||||||
Released | — | — | — | (603 | ) | $ | 14.62 | ||||||||||
Forfeited | 1,127 | (562 | ) | $ | 25.43 | (565 | ) | $ | 29.78 | ||||||||
Shares withheld for taxes | 29 | — | — | — | $ | 14.65 | |||||||||||
Balance at December 31, 2009 | 1,719 | 8,414 | $ | 16.63 | 2,042 | $ | 14.15 | ||||||||||
74
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Stock-Based Compensation (Continued)
As of December 31, 2009, all outstanding options to purchase common stock of the Company are summarized in the following table (in thousands, except years and per share data):
| | Options Outstanding | Options Exercisable | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Range of Exercise Prices | Number Outstanding | Weighted Average Remaining Contractual Life in Years | Weighted Average Exercise Prices | Aggregate Intrinsic Value | Options Exercisable | Weighted Average Remaining Contractual Life in Years | Weighted Average Exercise Price | Aggregate Intrinsic Value | |||||||||||||||||
$3.10 | 744 | 3.2 | $ | 3.10 | 744 | 3.2 | $ | 3.10 | |||||||||||||||||
$6.15 - $6.70 | 187 | 8.9 | $ | 6.21 | 47 | 8.9 | $ | 6.22 | |||||||||||||||||
$8.53 | 1,566 | 1.8 | $ | 8.53 | 1,566 | 1.8 | $ | 8.53 | |||||||||||||||||
$9.69 | 1,404 | 4.0 | $ | 9.69 | 1,404 | 4.0 | $ | 9.69 | |||||||||||||||||
$12.40 - $18.25 | 1,391 | 6.2 | $ | 15.89 | 1,005 | 5.0 | $ | 16.20 | |||||||||||||||||
$18.26 - $21.70 | 715 | 5.4 | $ | 19.12 | 663 | 5.3 | $ | 19.06 | |||||||||||||||||
$21.71 - $29.65 | 1,260 | 6.2 | $ | 27.97 | 1,084 | 6.0 | $ | 28.25 | |||||||||||||||||
$29.66 - $35.46 | 1,147 | 6.8 | $ | 33.57 | 826 | 6.8 | $ | 33.59 | |||||||||||||||||
Total | 8,414 | 4.8 | $ | 16.63 | $ | 20,652 | 7,339 | 4.3 | $ | 15.92 | $ | 19,694 | |||||||||||||
As of December 31, 2009, the aggregate intrinsic value of the options outstanding and the options exercisable was $20.7 million and $19.7 million, respectively.
The total intrinsic value of the options exercised during the years ended December 31, 2009, 2008 and 2007 was $10.0 million, $4.9 million and $19.0 million, respectively. The total fair value of options vested for the years ended December 31, 2009, 2008 and 2007 was $15.7 million, $20.4 million and $32.5 million, respectively.
Employee Stock Purchase Plan
Under the 2004 Employee Stock Purchase Plan (ESPP), the Company's non-officer employees may purchase common stock through payroll deductions at a price equal to 85 percent of the lower of the fair market value of the stock at the beginning of the offering period or at the end of each applicable purchase period. The ESPP provides for consecutive and overlapping offering periods of 24 months in duration, with each offering period composed of four consecutive six-month purchase periods. The purchase periods end on either May 15th or November 15th. ESPP contributions are limited to a maximum of 15 percent of an employee's eligible compensation.
The Company's ESPP plan also includes a feature that provides for a new offering period to begin when the fair market value of the Company's common stock on any purchase date during an offering period falls below the fair market value of the Company's common stock on the first day of such offering period. This feature is called a reset. The Company had resets for new twenty-four month offering periods starting on November 16, 2007, May 16, 2008 and November 16, 2008. The Company applied modification accounting to determine the incremental fair value associated with the ESPP resets and recognized the related incremental stock-based compensation expense. For the years ended December 31, 2009, 2008 and 2007, the Company recognized $0.4 million, $0.4 million and $0.1 million, respectively, in incremental fair value for the ESPP resets. Including the incremental fair value for the ESPP resets, the total stock-based compensation expense recognized relating to the ESPP for the years ended December 31, 2009, 2008 and 2007 was $1.3 million, $0.9 million and $1.4 million, respectively.
75
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
11. Stock-Based Compensation (Continued)
As of December 31, 2009, a total of 1,475,000 shares of common stock were approved and authorized for issuance under the ESPP. Through December 31, 2009, the Company issued 996,381 shares under the ESPP at an average price of $10.88 per share.
Restricted Stock
The Company's board of directors approved the grant of 50,000 shares of restricted stock in 2005 and 71,000 shares of restricted stock in 2007 to members of the Company's management. These restricted shares of common stock vest based on continued service, with pre-determined vesting percentages and anniversary dates. The Company valued the awards based on the closing market price of the Company's common stock on the date of the respective awards. The 50,000 share award from 2005 was valued at $0.9 million, a 50,000 share award from 2007 was valued at $1.3 million and a 21,000 award from 2007 was valued at $0.5 million. The fair value of restricted stock that vested for the years ended December 31, 2009, 2008 and 2007 was $0.4 million for each year. The total intrinsic value of unvested restricted stock at December 31, 2009, 2008 and 2007 was $0.7 million, $0.9 million and $1.9 million, respectively. The Company recognized stock-based compensation expense of $0.5 million, $0.5 million and $0.4 million related to these awards for the years ended December 31, 2009, 2008 and 2007, respectively.
Director Compensation Program
Pursuant to the Company's director compensation program, each independent director receives an annual retainer plus a fee for attending each board and committee meeting. The Chairman of the Board receives a flat rate per year for his service. Also under this program, each independent director who first becomes a director after January 1, 2008 is automatically granted an initial RSU award for 12,000 shares of common stock that vest monthly over the first two years of service. In addition, at each annual stockholder meeting, each independent director is automatically granted an RSU award for 6,000 shares of common stock and an option covering 6,000 shares of common stock, both of which vest monthly over one year.
12. Income Taxes
Due to ongoing operating losses and the inability to recognize any income tax benefit, there is no provision for income taxes.
76
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. Income Taxes (Continued)
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company's deferred tax assets are as follows:
| | December 31, | |||||||
|---|---|---|---|---|---|---|---|---|
(in thousands) | 2009 | 2008 | ||||||
Deferred tax assets: | ||||||||
Net operating loss carryforwards | $ | 274,000 | $ | 244,000 | ||||
Deferred revenues | 72,000 | 70,000 | ||||||
Capitalized research and development expenditures | 34,000 | 34,000 | ||||||
Research and development tax credit carryforwards | 31,000 | 28,000 | ||||||
Other | 22,000 | 25,000 | ||||||
Valuation allowance | (433,000 | ) | (401,000 | ) | ||||
Net deferred tax assets | $ | — | $ | — | ||||
Realization of deferred tax assets is dependent on future taxable income, if any, the timing and the amount of which are uncertain. Accordingly, the deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $32.0 million, $38.0 million and $63.0 million for the years ended December 31, 2009, 2008 and 2007, respectively.
As of December 31, 2009, the Company had federal net operating loss carryforwards of approximately $825.2 million, which will expire from 2011 through 2029 and federal research and development tax credit carryforwards of approximately $36.5 million, which will expire from 2018 through 2029. The Company also had state net operating loss carryforwards of approximately $198.7 million expiring in the years 2010 through 2029 and state research tax credits of approximately $39.3 million, which carry forward indefinitely.
The net operating loss deferred tax asset balances at December 31, 2009 and 2008 do not include excess tax benefits from stock option exercises. Stockholders' net capital deficiency will be increased if and when such excess tax benefits are ultimately realized.
Utilization of net operating loss and tax credit carryforwards may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. The Company conducted an analysis through 2009 to determine whether an ownership change had occurred since inception. The analysis indicated that two ownership changes occurred in prior years. However, notwithstanding the applicable annual limitations, no portion of the net operating loss or credit carryforwards are expected to expire before becoming available to reduce federal and state income tax liabilities. Annual limitations may result in expiration of net operating loss and tax credit carryforwards before some or all of such amounts have been utilized.
77
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
12. Income Taxes (Continued)
Uncertain Tax Positions
A reconciliation of the beginning and ending balances of the total amounts of gross unrecognized tax benefits is as follows (in thousands):
Gross unrecognized tax benefits at January 1, 2007 | $ | 26,700 | ||
Gross increase in tax positions for current year | 6,500 | |||
Unrecognized tax benefits at December 31, 2007 | 33,200 | |||
Gross increase in tax positions for current year | 3,000 | |||
Unrecognized tax benefits at December 31, 2008 | 36,200 | |||
Gross decrease for tax positions for prior years | (100 | ) | ||
Gross increase in tax positions for current year | 3,500 | |||
Unrecognized tax benefits at December 31, 2009 | $ | 39,600 | ||
If the Company eventually is able to recognize these uncertain positions, most of the $39.6 million of the unrecognized benefit would reduce the effective tax rate, except for excess tax benefits related to stock-based payments. The Company currently has a full valuation allowance against its deferred tax asset which would impact the timing of the effective tax rate benefit should any of these uncertain positions be favorably settled in the future. The Company does not believe it is reasonably possible that its unrecognized tax benefits will significantly change within the next twelve months.
The Company is subject to taxation in the U.S. and various state jurisdictions. The tax years 1996 and forward remain open to examination by the federal and most state tax authorities due to net operating loss and overall credit carryforward positions.
13. Quarterly Consolidated Results of Operations (Unaudited)
The following table presents certain unaudited consolidated quarterly financial information for the eight quarters in the period ended December 31, 2009. This information has been prepared on the same basis as the audited Consolidated Financial Statements and includes all adjustments (consisting
78
THERAVANCE, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS (Continued)
13. Quarterly Consolidated Results of Operations (Unaudited) (Continued)
only of normal recurring adjustments) necessary to present fairly the unaudited quarterly results of operations set forth herein.
| | March 31 | June 30 | September 30 | December 31 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | (in thousands except per share data) | |||||||||||||
2009: | ||||||||||||||
Revenue | $ | 9,544 | $ | 5,493 | $ | 5,515 | $ | 3,822 | ||||||
Operating expenses | (27,892 | ) | (26,846 | ) | (26,596 | ) | (24,401 | ) | ||||||
Loss from operations | (18,348 | ) | (21,353 | ) | (21,081 | ) | (20,579 | ) | ||||||
Net loss | (19,217 | ) | (21,692 | ) | (22,183 | ) | (22,210 | ) | ||||||
Basic and diluted net loss per share | $ | (0.31 | ) | $ | (0.35 | ) | $ | (0.35 | ) | $ | (0.35 | ) | ||
2008: | ||||||||||||||
Revenue | $ | 5,645 | $ | 5,505 | $ | 5,999 | $ | 5,947 | ||||||
Operating expenses | (35,945 | ) | (32,315 | ) | (26,619 | ) | (21,421 | ) | ||||||
Loss from operations | (30,300 | ) | (26,810 | ) | (20,620 | ) | (15,474 | ) | ||||||
Net loss | (29,764 | ) | (27,026 | ) | (20,928 | ) | (15,925 | ) | ||||||
Basic and diluted net loss per share | $ | (0.49 | ) | $ | (0.44 | ) | $ | (0.34 | ) | $ | (0.26 | ) | ||
79
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Theravance, Inc.
We have audited the accompanying consolidated balance sheets of Theravance, Inc. (the "Company") as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders' equity (net capital deficiency), and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Theravance, Inc. at December 31, 2009 and 2008, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2009, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Theravance, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 26, 2010 expressed an unqualified opinion thereon.
/s/ERNST & YOUNG LLP
Palo Alto, California
February 26, 2010
80
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures.
We conducted an evaluation as of December 31, 2009, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures, which are defined under SEC rules as controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files under the Securities Exchange Act of 1934 (Exchange Act) is recorded, processed, summarized and reported within required time periods. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective.
Management's Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) of the Exchange Act. Internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in theInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management's assessment included evaluation of such elements as the design and operating effectiveness of key financial reporting controls, process documentation, accounting policies, and our overall control environment. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2009.
Our independent registered public accounting firm, Ernst & Young LLP, has audited our internal control over financial reporting as of December 31, 2009. The report on the audit of internal control over financial reporting is included below.
Limitations on the Effectiveness of Controls
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent all error and all fraud. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefit of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within Theravance have been detected. Also, projections of any evaluation of
81
effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act) identified in connection with the evaluation required by paragraph (d) of Rule 13a-15 of the Exchange Act, which occurred during the fourth fiscal quarter of the year ended December 31, 2009 which has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
82
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Theravance, Inc.
We have audited Theravance, Inc.'s internal control over financial reporting as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Theravance, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Theravance, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Theravance, Inc. as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders' equity (net capital deficiency), and cash flows for each of the three years in the period ended December 31, 2009 of Theravance, Inc. and our report dated February 26, 2010 expressed an unqualified opinion thereon.
/s/ERNST & YOUNG LLP
Palo Alto, California
February 26, 2010
83
On February 24, 2010, the Company and Dr. P. Roy Vagelos, Chairman of the Board of Directors of the Company, agreed that Dr. Vagelos will not stand for reelection at the Company's annual stockholders meeting on April 27, 2010. In addition, Dr. Vagelos and the Company entered into a consulting agreement to be effective on April 27, 2010 for services to commence upon Dr. Vagelos' departure from the board of directors following the 2010 annual stockholders meeting. Pursuant to this agreement, Dr. Vagelos has agreed to provide consulting services for the period from April 27, 2010 through December 31, 2011 for the primary purpose of assisting the Company's Chief Executive Officer and the Senior Vice President—Research and Early Clinical Development with questions relating to the business of Theravance. As consideration for these services, Dr. Vagelos will receive a consulting fee of $4,000 per month and his currently outstanding stock options and restricted stock unit awards will continue to vest. In addition, all of Dr. Vagelos' outstanding stock options will be amended to provide that such options will remain exercisable with respect to any option shares that are vested as of the termination of his service for the length of the option term. The Company's Board of Directors also (i) appointed Rick E Winningham, its Chief Executive Officer, as Chairman of the Board of Directors, such appointment to be effective April 27, 2010, and (ii) reconfirmed William H. Waltrip as the Lead Independent Director of the Board.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
For the information required by this Item, see "Questions and Answers About this Proxy Material and Voting", "Election of Directors", "Nominees", "Meetings of the Board of Directors", "Executive Officers", "Section 16(a) Beneficial Ownership Reporting Compliance", "Audit Committee" and "Code of Business Conduct" in the Proxy Statement to be filed with the SEC, which sections are incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
For the information required by this Item, see "2009 Director Compensation", "Compensation of Executive Officers", "Compensation Committee Report" and "Compensation Committee Interlocks and Insider Participation" in the Proxy Statement to be filed with the SEC, which sections are incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
For the information required by this Item, see "Security Ownership of Certain Beneficial Owners and Management" and "Securities Authorized for Issuance Under Equity Compensation Plans" in the Proxy Statement to be filed with the SEC, which sections are incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
For the information required by this Item, see "Independence of the Board of Directors" and "Review, Approval or Ratification of Transactions with Related Persons" in the Proxy Statement to be filed with the SEC, which sections are incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
For the information required by this Item, see "Independent Registered Public Accounting Firm's Fees" and "Pre-Approval Policies and Procedures" in the Proxy Statement to be filed with the SEC, which sections are incorporated herein by reference.
84
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
- (a)
- The following documents are filed as part of this Annual Report on Form 10-K:
- 1.
- Financial Statements:
The following financial statements and schedules of the Registrant are contained in Item 8 of this Annual Report on Form 10-K:
Consolidated Balance Sheets as of December 31, 2009 and 2008 | 52 | |
| 53 | ||
| 54 | ||
| 55 | ||
| 56 | ||
| 80 |
- 2.
- Financial Statement Schedules:
All schedules are omitted because they are either not applicable or the required information is shown in the Consolidated Financial Statements or notes thereto.
- 3.
- Exhibits
The representations and warranties made by the parties to the agreements listed below were made solely for purposes of the agreements and to allocate risk between the parties. You should not rely on the representations, warranties or covenants in these agreements.
| Exhibit Number | Description | Form | Filing Date/ Period End Date | ||||
|---|---|---|---|---|---|---|---|
| 3.3 | Amended and Restated Certificate of Incorporation | S-1 | 7/26/04 | ||||
| 3.4 | Certificate of Amendment of Restated Certificate of Incorporation | 10-Q | 3/31/07 | ||||
| 3.5 | Amended and Restated Bylaws (as amended by the board of directors April 25, 2007) | 10-Q | 9/30/08 | ||||
| 4.1 | Specimen certificate representing the common stock of the registrant | 10-K | 12/31/06 | ||||
| 4.2 | Amended and Restated Rights Agreement between the registrant and The Bank of New York, as Rights Agent, dated as of June 22, 2007 | 10-Q | 6/30/07 | ||||
| 4.3 | Indenture dated as of January 23, 2008 by and between Theravance, Inc. and The Bank of New York Trust Company, N.A., as trustee | 8-K | 1/23/08 | ||||
| 4.4 | Form of 3.0% Convertible Subordinated Note Due 2015 (included in Exhibit 4.3) | ||||||
| 4.5 | Amendment to Amended and Restated Rights Agreement between the registrant and The Bank of New York Mellon Corporation, as Rights Agent, dated November 21, 2008 | 8-K | 11/25/08 | ||||
| 10.1 | + | 1997 Stock Plan | S-1 | 6/10/04 | |||
| 10.2 | + | Long-Term Stock Option Plan | S-1 | 6/10/04 | |||
85
| Exhibit Number | Description | Form | Filing Date/ Period End Date | ||||
|---|---|---|---|---|---|---|---|
| 10.3 | + | 2004 Equity Incentive Plan, as amended December 6, 2006 | 10-K | 12/31/06 | |||
| 10.4 | Employee Stock Purchase Plan, as amended December 10, 2008 | 10-Q | 3/31/08 | ||||
| 10.5 | + | Change in Control Severance Plan, as amended and restated on July 27, 2007 | 10-Q | 6/30/08 | |||
| 10.8 | Amended and Restated Lease Agreement, 951 Gateway Boulevard, between the registrant and HMS Gateway Office L.P., dated January 1, 2001 | S-1 | 6/10/04 | ||||
| 10.9 | Lease Agreement, 901 Gateway Boulevard, between the registrant and HMS Gateway Office L.P., dated January 1, 2001 | S-1 | 6/10/04 | ||||
| 10.10 | * | Collaboration Agreement between the registrant and Glaxo Group Limited, dated as of November 14, 2002 | S-1 | 9/29/04 | |||
| 10.11 | + | Form of Indemnification Agreement for directors and officers of the registrant | S-1 | 6/10/04 | |||
| 10.12 | Class A Common Stock Purchase Agreement between the registrant and SmithKline Beecham Corporation, dated as of March 30, 2004 | S-1 | 6/10/04 | ||||
| 10.13 | Amended and Restated Investors' Rights Agreement by and among the registrant and the parties listed therein, dated as of May 11, 2004 | S-1 | 6/10/04 | ||||
| 10.14 | Amended and Restated Governance Agreement by and among the registrant, SmithKline Beecham Corporation and GlaxoSmithKline dated as of June 4, 2004 | S-1 | 7/26/04 | ||||
| 10.15 | * | Strategic Alliance Agreement between the registrant and Glaxo Group Limited, dated as of March 30, 2004 | S-1 | 9/30/04 | |||
| 10.16 | * | License Agreement between the registrant and Janssen Pharmaceutica, dated as of May 14, 2002 | S-1 | 9/29/04 | |||
| 10.17 | + | Offer Letter with Rick E Winningham dated August 23, 2001 | S-1 | 6/10/04 | |||
| 10.18 | Form of Class A Common Stock Purchase Agreement between the registrant and GSK | S-1 | 9/29/04 | ||||
| 10.19 | + | Offer Letter with Michael W. Aguiar dated as of January 31, 2005 | 10-K | 12/31/04 | |||
| 10.20 | + | Form of Notice of Grant and Stock Option Agreement under 2004 Equity Incentive Plan | 10-K | 12/31/04 | |||
| 10.21 | + | Form of Notice of Restricted Stock Award and Restricted Stock Agreement under 2004 Equity Incentive Plan | 10-Q | 6/30/07 | |||
| 10.22 | + | Description of Cash Bonus Program, as amended | |||||
| 10.23 | * | License, Development and Commercialization Agreement between the registrant and Astellas Pharma Inc. dated November 7, 2005 | S-3 | 1/30/06 | |||
| 10.24 | + | Form of Notice of Stock Option Grant and Stock Option Agreement between the registrant and P. Roy Vagelos | 8-K | 5/2/06 | |||
| 10.25 | * | Amendment to License, Development and Commercialization Agreement between the registrant and Astellas Pharma Inc. dated as of July 18, 2006 | 10-Q | 9/30/06 | |||
86
| Exhibit Number | Description | Form | Filing Date/ Period End Date | ||||
|---|---|---|---|---|---|---|---|
| 10.26 | + | Form of Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect from 2007) | 10-Q | 6/30/07 | |||
| 10.27 | + | Form of Non-Employee Director Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect through 2006) | 10-Q | 6/30/07 | |||
| 10.28 | + | Form of Non-Employee Director Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect from 2007) | 10-Q | 6/30/07 | |||
| 10.29 | + | Form of Performance-Contingent Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 6/30/07 | |||
| 10.30 | + | Offer letter with Leonard Blum dated July 27, 2007 | 10-Q | 9/30/07 | |||
| 10.31 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-K | 12/31/07 | |||
| 10.32 | + | 2008 New Employee Equity Incentive Plan, as of December 16, 2009 | 10-K | 12/31/07 | |||
| 10.33 | + | Form of Notice of Grant and Stock Option Agreement under 2008 New Employee Equity Incentive Plan | 10-K | 12/31/07 | |||
| 10.34 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan between the registrant and P. Roy Vagelos | 10-Q | 3/31/08 | |||
| 10.35 | + | Form of Non-Employee Director Time-Based Vesting Notice of Initial Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 3/31/08 | |||
| 10.36 | + | Form of Non-Employee Director Time-Based Vesting Notice of Annual Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 3/31/08 | |||
| 10.37 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (sales plan applicable to more than one award) | 10-Q | 6/30/08 | |||
| 10.38 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (sales plan applicable to one award) | 10-Q | 6/30/08 | |||
| 10.39 | + | Separation Agreement between Michael Kitt and the registrant dated June 22, 2008 | 10-Q | 6/30/08 | |||
| 10.40 | + | Form of Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2008 New Employee Equity Incentive Plan | 10-Q | 9/30/08 | |||
| 10.41 | + | Amendment to Offer Letter between the registrant and Leonard Blum dated July 23, 2008 | 10-K | 12/31/08 | |||
| 10.42 | + | Amendment to Offer Letter between the registrant and Rick E Winningham dated December 23, 2008 | 10-K | 12/31/08 | |||
87
| Exhibit Number | Description | Form | Filing Date/ Period End Date | ||||
|---|---|---|---|---|---|---|---|
| 10.43 | + | Description of long-term cash bonus arrangement with Mathai Mammen | 10-K | 12/31/08 | |||
| 10.44 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (executive officer replenishment 2009) | 10-K | 12/31/08 | |||
| 10.45 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (employee replenishment 2009) | 10-K | 12/31/08 | |||
| 10.46 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan between the registrant and P. Roy Vagelos (2009) | 10-Q | 3/31/09 | |||
| 10.47 | + | Amendment to Change in Control Severance Plan effective December 31, 2009 | |||||
| 10.48 | + | 2009 Change in Control Severance Plan adopted December 31, 2009 | |||||
| 21.1 | List of Subsidiaries | 10-K | 12/31/05 | ||||
| 23.1 | Consent of Independent Registered Public Accounting Firm | ||||||
| 24.1 | Power of Attorney (see signature page to this Annual Report on Form 10-K) | ||||||
| 31.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14 under the Securities Exchange Act of 1934 | ||||||
| 31.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14 under the Securities Exchange Act of 1934 | ||||||
| 32 | Certifications Pursuant to 18 U.S.C. Section 1350 | ||||||
- +
- Management contract or compensatory plan or arrangement required to be filed pursuant to Item 15(b) of Form 10-K.
- *
- Confidential treatment has been requested for certain portions which are omitted in the copy of the exhibit electronically filed with the Securities and Exchange Commission. The omitted information has been filed separately with the Securities and Exchange Commission pursuant to Theravance Inc.'s application for confidential treatment.
88
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| THERAVANCE, INC. | ||||
Date: February 26, 2010 | By: | /s/ RICK E WINNINGHAM Rick E Winningham Chief Executive Officer | ||
KNOWN ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Rick E Winningham and Michael W. Aguiar, each of whom may act without joinder of the other, as their true and lawful attorneys-in-fact and agents, each with full power of substitution and resubstitution, for such person and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to the annual report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he or she could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or their substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||
|---|---|---|---|---|
| /s/ P. ROY VAGELOS, M.D. P. Roy Vagelos, M.D | Chairman of the Board and Directors | February 26, 2010 | ||
/s/ RICK E WINNINGHAM Rick E Winningham | Chief Executive Officer and Director (Principal Executive Officer) | February 26, 2010 | ||
/s/ MICHAEL W. AGUIAR Michael W. Aguiar | Senior Vice President, Finance and Chief Financial Officer (Principal Financial and Accounting Officer) | February 26, 2010 | ||
/s/ JEFFREY M. DRAZAN Jeffrey M. Drazan | Director | February 26, 2010 |
89
Signature | Title | Date | ||
|---|---|---|---|---|
| /s/ ROBERT V. GUNDERSON, JR. Robert V. Gunderson, Jr. | Director | February 26, 2010 | ||
/s/ ARNOLD J. LEVINE, PH.D. Arnold J. Levine, Ph.D | Director | February 26, 2010 | ||
/s/ BURTON G. MALKIEL, PH.D. Burton G. Malkiel, Ph.D | Director | February 26, 2010 | ||
/s/ WILLIAM H. WALTRIP William H. Waltrip | Director | February 26, 2010 | ||
/s/ GEORGE M. WHITESIDES, PH.D. George M. Whitesides, Ph.D | Director | February 26, 2010 | ||
/s/ WILLIAM D. YOUNG William D. Young | Director | February 26, 2010 |
90
| | | Incorporated by Reference | |||||
|---|---|---|---|---|---|---|---|
| Exhibit Number | Description | Form | Filing Date/Period End Date | ||||
| 3.3 | Amended and Restated Certificate of Incorporation | S-1 | 7/26/04 | ||||
| 3.4 | Certificate of Amendment of Restated Certificate of Incorporation | 10-Q | 3/31/07 | ||||
| 3.5 | Amended and Restated Bylaws (as amended by the board of directors April 25, 2007) | 10-Q | 9/30/08 | ||||
| 4.1 | Specimen certificate representing the common stock of the registrant | 10-K | 12/31/06 | ||||
| 4.2 | Amended and Restated Rights Agreement between the registrant and The Bank of New York, as Rights Agent, dated as of June 22, 2007 | 10-Q | 6/30/07 | ||||
| 4.3 | Indenture dated as of January 23, 2008 by and between Theravance, Inc. and The Bank of New York Trust Company, N.A., as trustee | 8-K | 1/23/08 | ||||
| 4.4 | Form of 3.0% Convertible Subordinated Note Due 2015 (included in Exhibit 4.3) | ||||||
| 4.5 | Amendment to Amended and Restated Rights Agreement between the registrant and The Bank of New York Mellon Corporation, as Rights Agent, dated November 21, 2008 | 8-K | 11/25/08 | ||||
| 10.1 | + | 1997 Stock Plan | S-1 | 6/10/04 | |||
| 10.2 | + | Long-Term Stock Option Plan | S-1 | 6/10/04 | |||
| 10.3 | + | 2004 Equity Incentive Plan, as amended December 6, 2006 | 10-K | 12/31/06 | |||
| 10.4 | Employee Stock Purchase Plan, as amended December 10, 2008 | 10-Q | 3/31/08 | ||||
| 10.5 | + | Change in Control Severance Plan, as amended and restated on July 27, 2007 | 10-Q | 6/30/08 | |||
| 10.8 | Amended and Restated Lease Agreement, 951 Gateway Boulevard, between the registrant and HMS Gateway Office L.P., dated January 1, 2001 | S-1 | 6/10/04 | ||||
| 10.9 | Lease Agreement, 901 Gateway Boulevard, between the registrant and HMS Gateway Office L.P., dated January 1, 2001 | S-1 | 6/10/04 | ||||
| 10.10 | * | Collaboration Agreement between the registrant and Glaxo Group Limited, dated as of November 14, 2002 | S-1 | 9/29/04 | |||
| 10.11 | + | Form of Indemnification Agreement for directors and officers of the registrant | S-1 | 6/10/04 | |||
| 10.12 | Class A Common Stock Purchase Agreement between the registrant and SmithKline Beecham Corporation, dated as of March 30, 2004 | S-1 | 6/10/04 | ||||
| 10.13 | Amended and Restated Investors' Rights Agreement by and among the registrant and the parties listed therein, dated as of May 11, 2004 | S-1 | 6/10/04 | ||||
| 10.14 | Amended and Restated Governance Agreement by and among the registrant, SmithKline Beecham Corporation and GlaxoSmithKline dated as of June 4, 2004 | S-1 | 7/26/04 | ||||
91
| | | Incorporated by Reference | |||||
|---|---|---|---|---|---|---|---|
| Exhibit Number | Description | Form | Filing Date/Period End Date | ||||
| 10.15 | * | Strategic Alliance Agreement between the registrant and Glaxo Group Limited, dated as of March 30, 2004 | S-1 | 9/30/04 | |||
| 10.16 | * | License Agreement between the registrant and Janssen Pharmaceutica, dated as of May 14, 2002 | S-1 | 9/29/04 | |||
| 10.17 | + | Offer Letter with Rick E Winningham dated August 23, 2001 | S-1 | 6/10/04 | |||
| 10.18 | Form of Class A Common Stock Purchase Agreement between the registrant and GSK | S-1 | 9/29/04 | ||||
| 10.19 | + | Offer Letter with Michael W. Aguiar dated as of January 31, 2005 | 10-K | 12/31/04 | |||
| 10.20 | + | Form of Notice of Grant and Stock Option Agreement under 2004 Equity Incentive Plan | 10-K | 12/31/04 | |||
| 10.21 | + | Form of Notice of Restricted Stock Award and Restricted Stock Agreement under 2004 Equity Incentive Plan | 10-Q | 6/30/07 | |||
| 10.22 | + | Description of Cash Bonus Program, as amended | |||||
| 10.23 | * | License, Development and Commercialization Agreement between the registrant and Astellas Pharma Inc. dated November 7, 2005 | S-3 | 1/30/06 | |||
| 10.24 | + | Form of Notice of Stock Option Grant and Stock Option Agreement between the registrant and P. Roy Vagelos | 8-K | 5/2/06 | |||
| 10.25 | * | Amendment to License, Development and Commercialization Agreement between the registrant and Astellas Pharma Inc. dated as of July 18, 2006 | 10-Q | 9/30/06 | |||
| 10.26 | + | Form of Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect from 2007) | 10-Q | 6/30/07 | |||
| 10.27 | + | Form of Non-Employee Director Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect through 2006) | 10-Q | 6/30/07 | |||
| 10.28 | + | Form of Non-Employee Director Notice of Stock Option Grant and Stock Option Agreement under 2004 Equity Incentive Plan (form in effect from 2007) | 10-Q | 6/30/07 | |||
| 10.29 | + | Form of Performance-Contingent Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 6/30/07 | |||
| 10.30 | + | Offer letter with Leonard Blum dated July 27, 2007 | 10-Q | 9/30/07 | |||
| 10.31 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-K | 12/31/07 | |||
| 10.32 | + | 2008 New Employee Equity Incentive Plan, as of December 16, 2009 | 10-K | 12/31/07 | |||
| 10.33 | + | Form of Notice of Grant and Stock Option Agreement under 2008 New Employee Equity Incentive Plan | 10-K | 12/31/07 | |||
92
| | | Incorporated by Reference | |||||
|---|---|---|---|---|---|---|---|
| Exhibit Number | Description | Form | Filing Date/Period End Date | ||||
| 10.34 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan between the registrant and P. Roy Vagelos | 10-Q | 3/31/08 | |||
| 10.35 | + | Form of Non-Employee Director Time-Based Vesting Notice of Initial Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 3/31/08 | |||
| 10.36 | + | Form of Non-Employee Director Time-Based Vesting Notice of Annual Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan | 10-Q | 3/31/08 | |||
| 10.37 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (sales plan applicable to more than one award) | 10-Q | 6/30/08 | |||
| 10.38 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (sales plan applicable to one award) | 10-Q | 6/30/08 | |||
| 10.39 | + | Separation Agreement between Michael Kitt and the registrant dated June 22, 2008 | 10-Q | 6/30/08 | |||
| 10.40 | + | Form of Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2008 New Employee Equity Incentive Plan | 10-Q | 9/30/08 | |||
| 10.41 | + | Amendment to Offer Letter between the registrant and Leonard Blum dated July 23, 2008 | 10-K | 12/31/08 | |||
| 10.42 | + | Amendment to Offer Letter between the registrant and Rick E Winningham dated December 23, 2008 | 10-K | 12/31/08 | |||
| 10.43 | + | Description of long-term cash bonus arrangement with Mathai Mammen | |||||
| 10.44 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (executive officer replenishment 2009) | 10-K | 12/31/08 | |||
| 10.45 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan (employee replenishment 2009) | 10-K | 12/31/08 | |||
| 10.46 | + | Form of Time-Based Vesting Notice of Restricted Stock Unit Award and Restricted Stock Unit Agreement under 2004 Equity Incentive Plan between the registrant and P. Roy Vagelos (2009) | 10-Q | 3/31/09 | |||
| 10.47 | + | Amendment to Change in Control Severance Plan effective December 31, 2009 | |||||
| 10.48 | + | 2009 Change in Control Severance Plan adopted December 31, 2009 | |||||
| 21.1 | List of Subsidiaries | 10-K | 12/31/05 | ||||
| 23.1 | Consent of Independent Registered Public Accounting Firm | ||||||
| 24.1 | Power of Attorney (see signature page to this Annual Report on Form 10-K) | ||||||
93
| | | Incorporated by Reference | |||||
|---|---|---|---|---|---|---|---|
| Exhibit Number | Description | Form | Filing Date/Period End Date | ||||
| 31.1 | Certification of Chief Executive Officer Pursuant to Rule 13a-14 under the Securities Exchange Act of 1934 | ||||||
| 31.2 | Certification of Chief Financial Officer Pursuant to Rule 13a-14 under the Securities Exchange Act of 1934 | ||||||
| 32 | Certifications Pursuant to 18 U.S.C. Section 1350 | ||||||
- +
- Management contract or compensatory plan or arrangement required to be filed pursuant to Item 15(b) of Form 10-K.
- *
- Confidential treatment has been requested for certain portions which are omitted in the copy of the exhibit electronically filed with the Securities and Exchange Commission. The omitted information has been filed separately with the Securities and Exchange Commission pursuant to Theravance Inc.'s application for confidential treatment.
94