QuickLinks -- Click here to rapidly navigate through this document
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2007 |
or |
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to |
|
COMMISSION FILE NUMBER 000-31161
ARENA PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
Delaware
(State or other jurisdiction of
incorporation or organization) | | 23-2908305
(I.R.S. Employer
Identification No.) |
6166 Nancy Ridge Drive, San Diego, CA
(Address of principal executive offices) |
|
92121
(Zip Code) |
(858) 453-7200
(Registrant's telephone number, including area code) |
Securities registered pursuant to 12(b) of the Act: |
Title of Each Class
| | Name of Each Exchange on Which Registered
|
|---|
| Common Stock, $0.0001 par value | | NASDAQ Global Market |
| Preferred Stock Purchase Rights | | NASDAQ Global Market |
Securities registered pursuant to 12(g) of the Act:None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer o | | Accelerated filer ý | | Non-accelerated filer o
(Do not check if a smaller reporting company) | | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
The approximate aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant was $659.0 million as of June 30, 2007, based on the last sale price of the registrant's common stock as reported on the NASDAQ Global Market on such date. For purposes of this calculation, shares of the registrant's common stock held by directors and officers have been excluded. This number is provided only for purposes of this Annual Report on Form 10-K and does not represent an admission that any particular person or entity is an affiliate of the registrant.
As of February 29, 2008, there were 73,759,776 shares of the registrant's common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required by Part III of this Annual Report on Form 10-K is incorporated by reference from the registrant's definitive proxy statement for the annual meeting of stockholders to be held in June 2008, which will be filed with the Securities and Exchange Commission within 120 days after the close of the registrant's fiscal year ended December 31, 2007.
ARENA PHARMACEUTICALS, INC.
TABLE OF CONTENTS
| PART I | | |
| Item 1. | | Business | | 3 |
| Item 1A. | | Risk Factors | | 22 |
| Item 1B. | | Unresolved Staff Comments | | 44 |
| Item 2. | | Properties | | 44 |
| Item 3. | | Legal Proceedings | | 45 |
| Item 4. | | Submission of Matters to a Vote of Security Holders | | 45 |
PART II |
|
|
| Item 5. | | Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | | 46 |
| Item 6. | | Selected Financial Data | | 48 |
| Item 7. | | Management's Discussion and Analysis of Financial Condition and Results of Operations | | 50 |
| Item 7A. | | Quantitative and Qualitative Disclosures About Market Risk | | 67 |
| Item 8. | | Financial Statements and Supplementary Data | | 68 |
| Item 9. | | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure | | 101 |
| Item 9A. | | Controls and Procedures | | 101 |
PART III |
|
|
| Item 10. | | Directors, Executive Officers and Corporate Governance | | 103 |
| Item 11. | | Executive Compensation | | 103 |
| Item 12. | | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | | 103 |
| Item 13. | | Certain Relationships and Related Transactions, and Director Independence | | 103 |
| Item 14. | | Principal Accountant Fees and Services | | 103 |
PART IV |
|
|
| Item 15. | | Exhibits, Financial Statement Schedules | | 104 |
2
INFORMATION RELATING TO FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K includes forward-looking statements. These forward-looking statements involve a number of risks and uncertainties. These forward-looking statements can generally be identified as such because the context of the statement will include words such as "may," "will," "intends," "plans," "believes," "anticipates," "expects," "estimates," "predicts," "potential," "continue," "likely," or "opportunity," the negative of these words or other similar words. Similarly, statements that describe our future plans, strategies, intentions, expectations, objectives, goals or prospects and other statements that are not historical facts are also forward-looking statements. For such statements, we claim the protection of the Private Securities Litigation Reform Act of 1995. Readers of this Annual Report on Form 10-K are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the time this Annual Report on Form 10-K was filed with the Securities and Exchange Commission, or SEC. These forward-looking statements are based largely on our expectations and projections about future events and future trends affecting our business, and are subject to risks and uncertainties that could cause actual results to differ materially from those anticipated in the forward-looking statements. These risks and uncertainties include, without limitation, those discussed in "Item 1A. Risk Factors" and in "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations" of this Annual Report on Form 10-K. In addition, past financial or operating performance is not necessarily a reliable indicator of future performance and you should not use our historical performance to anticipate results or future period trends. We can give no assurances that any of the events anticipated by the forward-looking statements will occur or, if any of them do, what impact they will have on our results of operations and financial condition. Except as required by law, we undertake no obligation to publicly revise our forward-looking statements to reflect events or circumstances that arise after the filing of this Annual Report on Form 10-K or documents incorporated by reference herein that include forward-looking statements.
Arena Pharmaceuticals®, Arena® and our corporate logo are registered service marks of Arena. CART™ and BRL Screening™ are unregistered service marks of Arena. All other brand names or trademarks appearing in this Annual Report on Form 10-K are the property of their respective holders.
In this Annual Report on Form 10-K, "Arena Pharmaceuticals," "Arena," "we," "us" and "our" refer to Arena Pharmaceuticals, Inc. and our wholly owned subsidiaries on a consolidated basis, unless the context otherwise provides.
PART I
Item 1. Business.
We are a clinical-stage biopharmaceutical company focused on discovering, developing and commercializing oral drugs in four major therapeutic areas: cardiovascular, central nervous system, inflammatory and metabolic diseases. Our most advanced drug candidate, lorcaserin hydrochloride, or lorcaserin, is being investigated in a Phase 3 clinical trial program for the treatment of obesity. We have a broad pipeline of novel compounds that selectively target known and orphan G protein-coupled receptors, or GPCRs, and includes compounds being developed by our partners, Ortho-McNeil Pharmaceutical, Inc., a Johnson & Johnson company, or Ortho-McNeil, and Merck & Co., Inc., or Merck.
We focus on GPCRs because they are a validated class of drug targets that mediate the majority of cell-to-cell communication in humans. A high percentage of today's prescription drugs target one or more GPCRs, and we believe that approved GPCR-based drugs target about 30% of the known non-sensory GPCRs. Selective targeting of specific GPCRs is intended to increase the likelihood of the desired pharmacology and minimize the risk of "off target" effects. We believe our GPCR-focused technologies and integrated discovery and development capabilities will allow us to continue to build our pipeline of unique and selective drug candidates.
3
In September 2006, we initiated the first of three planned Phase 3 clinical trials evaluating the efficacy and safety of lorcaserin, our lead drug candidate under investigation for the treatment of obesity. The first trial, known as BLOOM (Behavioral modification and Lorcaserin for Overweight and Obesity Management), is a double-blind, randomized and placebo-controlled trial that enrolled more than 3,100 overweight and obese patients. In September 2007, we announced the continuation of the BLOOM trial following a planned review by an independent Echocardiographic Data Safety Monitoring Board, or ESMB, of unblinded echocardiograms performed after patients completed six months of dosing in the trial. The ESMB confirmed that differences, if any, in the rates of valvulopathy, as defined by the United States Food and Drug Administration, or FDA, in patients treated with lorcaserin and in the control group did not meet their predetermined stopping criteria.
In December 2007, we initiated the second and third Phase 3 clinical trials evaluating the safety and efficacy of lorcaserin. Known as BLOSSOM (Behavioral modification and Lorcaserin Second Study for Obesity Management) and BLOOM-DM (Behavioral modification and Lorcaserin for Overweight and Obesity Management in Diabetes Mellitus), these one-year, double-blind, randomized and placebo-controlled clinical trials are expected to collectively enroll approximately 3,750 overweight and obese patients. In contrast to BLOOM, patients with FDA-defined valvulopathy are allowed to enroll in BLOSSOM and BLOOM-DM. BLOOM, BLOSSOM and BLOOM-DM comprise the entire planned Phase 3 pivotal trial program for lorcaserin.
In addition to lorcaserin, our other internal clinical programs include APD125 and APD791. In September 2007, we announced results from a Phase 2a clinical trial of APD125, an oral drug candidate that we discovered and believe has the potential to reduce insomnia symptoms and improve sleep maintenance. In the Phase 2a clinical trial, which was conducted in patients with chronic primary insomnia, APD125 significantly improved endpoints measuring improvements in sleep maintenance, including wake after sleep onset, or WASO, wake time during sleep, or WTDS, and number of awakenings and arousals. In addition, in the Phase 2a clinical trial, APD125 significantly increased time spent in deep sleep and decreased the amount of time spent in lighter sleep. During the clinical trial, treatment with APD125 was well tolerated with no observations of next day cognitive impairment. In 2008, we plan to initiate a Phase 2b clinical trial to examine subjective measures of sleep maintenance.
APD791 is an oral drug candidate that we discovered and are investigating for the treatment and prevention of arterial thromboembolic diseases such as acute coronary syndrome. We recently announced positive results from a single dose Phase 1a clinical trial of APD791 and the initiation of a multiple dose Phase 1b clinical trial to further evaluate the compound's safety, pharmacokinetics and pharmacodynamics.
In addition to internal programs, we have partnerships with pharmaceutical companies, including Ortho-McNeil and Merck. Our Ortho-McNeil partnership is focused on receptor agonists of an orphan GPCR, the Glucose-Dependent Insulinotropic Receptor, or GDIR, as treatments for diabetes and other disorders, and our Merck partnership is focused on niacin receptor agonists as treatments for atherosclerosis and other disorders. Merck recently initiated under our partnership a Phase 1 clinical trial of a second generation oral niacin receptor agonist.
We intend to commercialize our drug candidates independently and with partners. We have not received regulatory approval for, or generated commercial revenues from, marketing or selling any drugs. We were incorporated in 1997.
4
Our Research & Development Programs
We have built a broad pipeline of drug candidates that target large and attractive market opportunities in several therapeutic areas. The following table summarizes our current independent and partnered development programs and selected research programs:
Development Program (Indication)
| | Development
Status
| | Next Potential
Milestone
| | Commercial
Rights
|
|---|
| Lorcaserin (obesity) | | Phase 3 | | Month-12 ESMB review | | Arena |
| APD125 (insomnia) | | Phase 2 | | Start Phase 2b | | Arena |
| APD791 (arterial thrombosis) | | Phase 1 | | Complete Phase 1b | | Arena |
| Niacin receptor agonist (atherosclerosis and other disorders) | | Phase 1 | | Complete Phase 1 | | Merck |
| GDIR receptor agonists (diabetes) | | Preclinical | | Start Phase 1 | | Ortho-McNeil |
| APD916 (wakefulness promoter) | | Preclinical | | Start Phase 1 | | Arena |
Research Program
|
|
|
|
|
|
|
| Cardioprotection | | Research | | — | | Arena |
| Cytokine & immune cell modulators | | Research | | — | | Arena |
| Type 2 diabetes & obesity | | Research | | — | | Arena |
Note: The table above does not list all of our research programs.
We are investigating lorcaserin in a Phase 3 pivotal trial program for the treatment of obesity. Obesity affects tens of millions of adults and children in the United States and poses serious long-term threats to their health and welfare. Studies have shown that modest weight loss of as little as 5% of initial body weight can result in a meaningful reduction in the risks associated with obesity, such as diabetes. Currently, pharmaceutical treatment options for obesity are limited.
Lorcaserin is a novel and selective 5-HT2C serotonin receptor agonist. Based on our preclinical studies and clinical trial data to date, we believe that lorcaserin is unlikely to cause serotonin-mediated valvulophathy or other cardiovascular side effects. This belief is supported by the review by the independent ESMB of unblinded echocardiograms that was performed after patients completed six months of dosing in the BLOOM trial. The ESMB review confirmed that differences, if any, in the rates of FDA-defined valvulopathy in patients treated with lorcaserin and in the control group did not meet the ESMB's predetermined stopping criteria. Our belief is also supported by data from our 4- and 12-week clinical trials, in which no apparent effects of the drug were seen on heart valves or pulmonary arterial pressure, and by long-term (6-12 month) toxicity studies at high doses in animals. However, the longer-term, ongoing clinical trials of lorcaserin will be needed to confirm these results. This is a major and continuing focus of our Phase 3 pivotal trial program.
Mechanism of Action. We believe lorcaserin selectively stimulates the 5-HT2C serotonin receptor, a GPCR located in the hypothalamus. Stimulation of this hypothalamic receptor is strongly associated with feeding behavior and satiety. We conducted preclinical studies examining the activity and 5-HT receptor subtype specificity of lorcaserin. In these studies, lorcaserin demonstrated a high affinity and selectivity for the 5-HT2C receptor, with approximately 15-fold and 100-fold selectivityin vitro over the human 5-HT2A and 5-HT2B receptors, respectively, and no pharmacologic activity at other serotonin receptors except at concentrations greatly exceeding the expected therapeutic range.
Prior Clinical Development. We have completed multiple Phase 1 and Phase 2 clinical trials of lorcaserin. Our Phase 2a clinical trial included 352 obese patients dosed for 28 days, and our Phase 2b
5
clinical trial included 469 obese patients dosed for 12 weeks. Highly statistically significant, clinically meaningful and progressive weight loss was observed in both Phase 2 clinical trials, with no apparent drug effect on heart valves or pulmonary artery pressure, as assessed by serial echocardiograms. Lorcaserin was also generally well tolerated in both Phase 2 clinical trials.
The randomized, double-blind, multiple-dose, 28-day Phase 2a clinical trial of lorcaserin in obese patients compared doses of 1 mg, 5 mg and 15 mg to placebo. Over the 28-day treatment period there was a highly statistically significant (p=0.0002) mean weight loss of 2.9 pounds in patients taking the 15 mg dose of lorcaserin versus 0.9 pounds for the placebo group. Lorcaserin was generally well tolerated at all doses investigated in the trial. An assessment of follow-up echocardiograms taken at the end of dosing and approximately 90 days after patients received their first doses of lorcaserin in the Phase 2a clinical trial indicated no apparent drug effect on heart valves or pulmonary artery pressure.
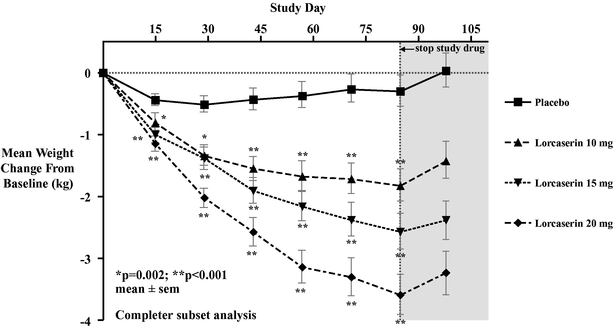
A randomized, double-blind, multiple-dose, 12-week Phase 2b clinical trial of lorcaserin in obese patients compared doses of 10 mg and 15 mg once daily and 20 mg (10 mg dosed twice daily) of lorcaserin to placebo. The primary endpoint of the trial, which excluded diet and exercise advice, was weight loss after administration of lorcaserin for 12 weeks. Patients completing the 12-week treatment period with lorcaserin achieved a highly statistically significant (p<0.001) mean weight loss of 4.0, 5.7 and 7.9 pounds at daily doses of 10 mg, 15 mg and 20 mg (10 mg dosed twice daily), respectively, compared to 0.7 pounds for the placebo group. Using an intent-to-treat, last-observation-carried-forward analysis, treatment with lorcaserin was also associated with a highly statistically significant (p<0.001) mean weight loss of 3.7, 4.8 and 6.8 pounds at daily doses of 10 mg, 15 mg and 20 mg (10 mg dosed twice daily), respectively, in patients taking lorcaserin compared to 0.4 pounds for the placebo group. The proportions of patients completing the 12-week treatment period with lorcaserin who achieved a 5% or greater weight loss from baseline were 13% (p=0.015), 20% (p<0.001) and 31% (p<0.001) at daily doses of 10 mg, 15 mg and 20 mg (10 mg dosed twice daily), respectively, compared to 2% in the placebo group. Lorcaserin was generally well tolerated at all doses investigated in the trial. Adverse events occurring in greater than 5% in any of the dosed groups were headache, nausea, dizziness, vomiting, dry mouth, nasopharyngitis, fatigue and urinary tract infection. As demonstrated by the graph below, average weight loss increased progressively at each time point measured throughout the trial for all lorcaserin dose groups, and was dose-dependent. As we expected, after patients stopped taking lorcaserin, they started to regain weight.
6
Lorcaserin Phase 2b Clinical Trial: Weight Loss by Dose and Time
An assessment of echocardiograms at baseline and day 85 indicated no apparent lorcaserin effect on heart valves or pulmonary artery pressure. No changes in valvular regurgitation greater than one category, and no significant increases in pulmonary artery pressure in any group were identified in the echocardiogram results. No significant differences in the number of patients with increased regurgitation at any value were observed between any treatment group and placebo. Valvular regurgitation, a measure of back flow or leakage of blood through heart valves due to imperfect valve closing, was scored on a five-point scale (absent, trace, mild, moderate or severe). The FDA defines significant valvulopathy as mild or greater aortic valve regurgitation or moderate or greater mitral valve regurgitation. This is one measure used in our Phase 3 program to assess potential effects of lorcaserin on heart valves. As demonstrated by the table below, the incidence of FDA-defined valvulopathy was greater in the placebo group versus the combined lorcaserin treated groups.
Lorcaserin Phase 2b Clinical Trial: Incidence of FDA-Defined Valvulopathy
| |
| | Lorcaserin
| |
| |
|---|
| | Placebo
| | 10 mg
| | 15 mg
| | 20 mg
| |
| |
|---|
| Patients (N) | | 99 | | 99A,
100M | | 96 | | 96 | | | |
| Aortic (A) Regurgitation | | 0 | | 0 | | 1 | | 0 | | | |
| Mitral (M) Regurgitation | | 2 | | 0 | | 1 | | 0 | | | |
| Percent by Dose | | 2.0 | % | 0.0 | % | 2.1 | % | 0.0 | % | | |
| |
| | 
| |
|---|
| Percent by Treatment | | 2.0 | % | | | 0.7 | % | | | | |
Phase 3 Clinical Development. In September 2006, we initiated the first of three planned Phase 3 clinical trials to evaluate the safety and efficacy of lorcaserin for the treatment of obesity. BLOOM, the first of the three clinical trials, completed enrollment in February of 2007 with more than 3,100 overweight and obese patients in approximately 100 centers in the United States.
BLOOM is a double-blind, randomized trial evaluating a 20 mg dose (10 mg dosed twice daily) of lorcaserin versus placebo over a two-year treatment period in obese patients (Body Mass Index, or
7
BMI, of 30 to 45) with or without co-morbid conditions and overweight patients (BMI of 27 to less than 30) with at least one co-morbid condition. The primary efficacy endpoint is the proportion of patients with a 5% or greater weight reduction from baseline at week 52 as compared to placebo.
Patients received echocardiograms at baseline, 6 and 12 months after initiating dosing in the trial, and will receive follow-up echocardiograms at 18 and 24 months after starting the trial. In September 2007, we announced the continuation of the BLOOM trial after the independent ESMB conducted its planned review of the unblinded echocardiograms that were performed after patients completed six months of dosing in the trial. The ESMB's review confirmed that differences, if any, in the rates of FDA-defined valvulopathy in patients treated with lorcaserin and in the control group did not meet their predetermined stopping criteria. The review also confirmed that the rate of FDA-defined valvulopathy in the trial is consistent with our statistical powering assumptions used in the design of the clinical trial program to monitor patients for any increased risk of developing valvulopathy. As with the month-6 echocardiogram analysis, the ESMB will review the month-12 echocardiographic data and, based upon its predetermined criteria, will make a second judgment as to whether it is appropriate to continue or stop the trial. We expect the month-12 ESMB review to take place in March 2008.
In December 2007, we initiated BLOSSOM and BLOOM-DM, the second and third Phase 3 clinical trials evaluating lorcaserin's efficacy and safety. These one-year, double-blind, randomized and placebo-controlled clinical trials are expected to collectively enroll approximately 3,750 overweight and obese patients. Consistent with our proposal, the FDA has allowed us to eliminate the requirement to perform echocardiographic testing prior to enrolling patients in both of these trials. As a result, patients with pre-existing FDA-defined valulopathy and other echocardiographic variants and abnormalities may be enrolled in the BLOSSOM and BLOOM-DM trials. This is different from the design of BLOOM, the initial lorcaserin pivotal study, in which echocardiography was used to screen for patients with FDA-defined valvulopathy and certain other echocardiographic abnormalities and exclude those patients from enrolling in the trial. Instead, in BLOSSOM and BLOOM-DM, there are no such echocardiographically defined exclusion criteria, although serial echocardiograms will be obtained to extend the lorcaserin safety database. BLOOM, BLOSSOM and BLOOM-DM comprise the entire planned Phase 3 clinical trial program for lorcaserin.
The BLOSSOM trial is evaluating 10 mg and 20 mg daily doses (10 mg dosed once or twice daily) of lorcaserin versus placebo over a one-year treatment period in obese patients (BMI of 30 to 45) with or without co-morbid conditions and overweight patients (BMI of 27 to less than 30) with at least one co-morbid condition at about 100 sites in the United States. The BLOOM-DM trial is evaluating 10 mg and 20 mg daily doses (10 mg dosed once or twice daily) of lorcaserin versus placebo over a one-year treatment period in obese and overweight patients with type 2 diabetes at about 45 sites in the United States.
Consistent with the BLOOM trial, diet and exercise will also be included in the BLOSSOM and BLOOM-DM trials in accordance with current FDA guidelines, and the proportion of patients with a 5% or greater weight reduction from baseline at week 52 will be the primary efficacy endpoint. Secondary endpoints include changes in serum lipids, blood pressure and quality of life; in the BLOOM-DM trial, HbA1c and other indicators of glycemic control will also be evaluated. In both of these additional trials, all patients will receive echocardiograms at baseline, at month 6, and at the end of the study to assess heart valve function over time. In contrast to the ongoing BLOOM trial, however, there will be no oversight by an independent safety monitoring board.
The complete lorcaserin Phase 3 pivotal program is planned to enroll a total of approximately 7,000 patients. In addition to the Phase 3 clinical trials, several additional smaller trials, such as drug interaction and abuse potential trials, will be conducted. Assuming we receive favorable results from the month-12 ESMB review and our clinical trials and preclinical studies, we expect to file a New Drug Application, or NDA, for lorcaserin by the end of 2009.
8
Intellectual Property. As of January 31, 2008, we owned issued patents that cover compositions of matter for lorcaserin and related compounds and methods of treatment utilizing lorcaserin and related compounds in 44 jurisdictions, including the United States, Germany, France, the United Kingdom, Italy and Spain, and had applications pending in approximately 25 other jurisdictions, including Japan, Canada and China. Based on sales statistics provided by IMS Health, the jurisdictions where lorcaserin patents have been issued accounted for more than 76% of global pharmaceutical sales in 2006, while jurisdictions where lorcaserin patents remain pending accounted for more than 20% of global pharmaceutical sales in that same year. The patent on lorcaserin issued by the United States Patent and Trademark Office is serial number US 6,953,787 and the corresponding patent granted by the European Patent Office is serial number EP 1 411 881 B1. The earliest priority date for the patents on lorcaserin is 2002. The terms of these patents are capable of continuing into 2023 in most jurisdictions without taking into account any patent term extension regimes of any country.
In September 2007, we announced positive Phase 2 clinical trial results from our lead drug candidate for the treatment of insomnia, APD125, which is a novel and selective 5-HT2A serotonin receptor inverse agonist. The National Institutes of Health estimated in 2003 that between 30 to 40% of United States adults report some level of insomnia and that insomnia is a chronic problem for about 10% of the United States population. In these cases, the lack of restful sleep impairs the person's ability to carry out their daily responsibilities because they are too tired or have trouble concentrating. However, the great majority of insomnia patients do not seek treatment. Currently approved therapies for insomnia include Ambien and Ambien CR, marketed by sanofi-aventis, Lunesta, marketed by Sepracor Inc., Sonata, marketed by King Pharmaceuticals, Inc., Rozerem, a melatonin MT1 and MT2 agonist marketed by Takeda Pharmaceuticals North America, Inc., and certain benzodiazepines. With the exception of Rozerem, these therapies work by activating the GABA-A receptor complex in the brain, causing a general suppressive effect on the central nervous system, or CNS. These GABAergic drugs are generally associated with CNS side effects, including a sensation of dullness and lethargy upon awakening, often referred to as the "hangover effect." Other potential problems associated with the GABAergic drugs include the risk of developing tolerance and drug dependency in at-risk populations. In addition, GABAergic drugs are scheduled controlled substances by the Drug Enforcement Administration of the United States Department of Justice, or DEA, due to their potential for abuse. Despite these limitations, worldwide sales estimates for insomnia medications were over $3.5 billion in 2006.
Mechanism and Preclinical Data. APD125 acts through a different mechanism than currently marketed insomnia drugs. Based on our preclinical data, we believe that by selectively targeting the 5-HT2A receptor, APD125 blocks one of several CNS-activating pathways, rather than initiating a general CNS-suppressive effect. Because of the different mechanism of action, APD125 may not have the side effects generally associated with currently approved GABAergic drugs. Through this novel mechanism, APD125 has the potential to reduce insomnia symptoms and improve sleep maintenance by decreasing the number of awakenings during the night, decreasing the amount of wake time after initial sleep onset and increasing the amount of time spent in deep sleep, or slow wave sleep (stage 3 and stage 4 sleep), the most restorative type of sleep.
Our preclinical studies have shown that, in animals, APD125 increases the total time of non-REM (absence of rapid eye movement or dreams) sleep, the most restorative phase of the sleep cycle in humans, while having no effect on REM sleep. The total increase in non-REM sleep time was manifested by fewer sleep bouts of longer duration, indicating an increase in sleep consolidation. In addition, animals treated with APD125 showed during non-REM sleep an increase in delta power, a brain wave activity associated with increased sleep intensity. The improvement in the duration of non-REM sleep observed with APD125 administration was at least as robust as that observed with a
9
prototypic GABAergic drug, Ambien. However, unlike Ambien, APD125 did not reduce REM sleep in these studies.
Prior Clinical Development. We have completed several Phase 1 clinical trials of APD125 in normal volunteers. The Phase 1 program consisted of three randomized, double-blind and placebo-controlled trials evaluating the single and multiple dose safety and pharmacokinetics of APD125 in normal volunteers. Additionally, the program evaluated the pharmacodynamics of nighttime dosing by assessing effects on sleep patterns in normal volunteers using polysomnography.
In this Phase 1 clinical trial program, APD125 was well tolerated at single doses up to 160 mg and repeated doses up to 80 mg. At 40 mg, the maximum concentration in the body, or Cmax, of APD125 plateaued; there were no significant differences in Cmax among the 40 mg, 80 mg and 160 mg doses. At 80 mg, the total overall exposure, or area under the curve, of APD125 also plateaued; the pharmacokinetics at the 160 mg dose were generally similar to the 80 mg dose. At doses from 10-40 mg, APD125 caused a robust and highly statistically significant (p=0.0002) increase in the amount of deep, or slow wave, sleep in volunteers with normal sleep/wake patterns. In addition, other statistically significant signals indicative of improved sleep maintenance were seen, including statistically significant increases in stage 3 and stage 4 sleep, reductions in stage 1 sleep, reductions in the number of awakenings and an increase in delta power, the deepest form of slow wave sleep. Adverse events were infrequent and APD125 was well tolerated. The Phase 1 results support our expectation that APD125 will not cause any limiting next-day impairment of psychomotor skills or memory.
Phase 2 Clinical Development. In March 2007, we initiated a Phase 2a clinical trial of APD125. This Phase 2a clinical trial was a randomized, double-blind, placebo-controlled study evaluating the safety and efficacy of nighttime dosing in patients with chronic insomnia. The trial evaluated standard measurements of sleep, such as WASO, WTDS, number of awakenings, number of arousals, total sleep time and latency to persistent sleep, and enrolled a total of 173 male and female patients in about 25 clinical sites in the United States. The trial employed a cross-over design, in that every patient received both active doses of APD125 (10 mg and 40 mg) and placebo in random order, for one week, separated by a seven to nine day washout period between each dosing period. Efficacy was measured objectively by averaging polysomnography values for nights one and two (N1/2) and for nights six and seven (N6/7), versus baseline values.
In the Phase 2a clinical trial, APD125 significantly improved endpoints measuring improvements in sleep maintenance, including WASO and WTDS. WTDS decreased from baseline by 45.8 and 46.4 minutes, respectively, in the 10 mg and 40 mg doses at N1/2, and by 46.1 and 46.9 minutes, respectively, at N6/7; these differences were statistically significant for both doses at N1/2 (p<0.0001 compared to placebo decrease from baseline of 32.4 minutes) and N6/7 (p=0.0009 for 10 mg, p=0.0004 for 40 mg compared to placebo decrease from baseline of 36.0 minutes). The decrease from baseline in WASO was 52.5 and 53.5 minutes, respectively, for the 10 mg and 40 mg doses at N1/2 (p<0.0001 for both compared to placebo decrease from baseline of 37.8 minutes). Improvements from baseline in WASO of 51.7 and 48.0 minutes were observed at N6/7 (p=0.0131 and p=0.1994 compared to placebo improvement from baseline of 44.0 minutes).
Significant improvements also were seen in other important measurements of sleep maintenance, including a decrease in the number of awakenings and arousals (p<0.0001 at both N1/2 and N6/7 at 10 mg and 40 mg for both variables). Changes in the number of awakenings were 0.0, -2.5 and -3.1 at N1/2 and -0.9, -2.3 and -2.5 at N6/7 for placebo, 10 mg and 40 mg, respectively. Changes in the number of arousals were +3.8, -5.8 and -8.1 on N1/2 and +2.5, -4.8 and -6.7 on N6/7 for placebo, 10 mg and 40 mg, respectively.
In the trial, APD125 also significantly increased the time spent in deep (stage 3 and 4) sleep and at the same time decreased the amount of time spent in light (stage 1) sleep (p<0.0001 at 10 mg and 40 mg for both measures), providing further evidence for the sleep maintenance properties of APD125.
10
Time in REM sleep was not reduced. As expected, based on the mechanism of APD125, no improvement in sleep onset relative to placebo was observed.
Treatment with APD125 was well tolerated in the trial, with no reports of serious adverse events and no emerging safety findings as compared to placebo. No next day impairment of cognitive function was observed.
The data from this trial indicates that APD125 is efficacious for promoting sleep maintenance in patients with chronic insomnia. The data is also consistent with the Phase 1 data and support further development of APD125 for the treatment of insomnia patients who have difficulty maintaining sleep.
While the study was not powered to demonstrate significance in the subjective endpoints, there were trends towards improvements in the quality of sleep, number of awakenings and total sleep time, with statistical significance for at least one time point and dose for each of these variables. In 2008, we intend to initiate a Phase 2b clinical trial to examine subjective measures of sleep maintenance.
Intellectual Property. As of January 31, 2008, we owned issued patents that cover compositions of matter for APD125 and related compounds and methods of treatment utilizing APD125 and related compounds in 40 jurisdictions, including Germany, France, the United Kingdom, Italy and Spain, and had applications pending in approximately 36 other jurisdictions and international patent authorities, including the United States, Japan, Canada and China. Based on sales statistics provided by IMS Health, the jurisdictions where APD125 patents have been issued accounted for more than 29% of global pharmaceutical sales in 2006, while jurisdictions where APD125 patents remain pending accounted for more than 70% of global pharmaceutical sales in that same year. The patent on APD125 issued by the European Patent Office is serial number EP 1 558 582 B1. The earliest priority date for the patents on APD125 is 2003. The terms of these patents are capable of continuing into 2024 in most jurisdictions without taking into account any patent term extension regimes of any country.
Our lead anti-thrombotic drug candidate, APD791, is currently in a Phase 1 program. APD791 is a novel, oral and selective inverse agonist of the 5-HT2A serotonin receptor intended to lower the risk of arterial thrombosis by reducing the amplification of platelet aggregation, arterial constriction and intimal hyperplasia, or thickening of the vessel wall, mediated by serotonin. Thrombosis is the formation of a clot, or thrombus, inside a blood vessel that restricts the flow of blood. The formation of a thrombus is often caused by an injury to the wall of the blood vessel. The injury to the blood vessel activates platelets, which then aggregate and adhere to one another as they start to release certain factors, including serotonin, that facilitate thrombosis. Thrombi that form in diseased atherosclerotic arteries of the heart may cause acute coronary syndrome or myocardial infarction, and thrombi that form in the vessels of the brain may cause stroke. The American Heart Association estimates that in the United States over 13.9 million people alive in 2005 had survived either a myocardial infarction or a stroke. To reduce the risk of future events, many patients receive daily anti-thrombotic therapy. Worldwide sales of Plavix, a leading anti-thrombotic marketed by Bristol-Myers Squibb and sanofi-aventis, totaled almost $6.0 billion in 2006, making it the second best selling drug in any therapeutic category.
Mechanism and Preclinical Data. APD791 is a novel, oral and selective inverse agonist of the 5-HT2A serotonin receptor. Serotonin activation of the 5-HT2A receptor on platelets and vascular smooth muscle is thought to play an important role in the events leading to thrombosis, and elevated serotonin levels have been associated with increased cardiovascular risk. Normally, when a platelet is activated by one of a number of factors such as thrombin or collagen, the platelet releases serotonin, which, based on preclinical studies, promotes platelet aggregation, vasoconstriction and intimal hyperplasia. By blocking activation of the 5-HT2A receptor on platelets and in other cardiovascular tissues, APD791 may curb platelet aggregation, vasoconstriction and intimal hyperplasia in the clinical
11
setting, thereby reducing or preventing thrombosis. We believe APD791 represents a new approach to reducing the risk of arterial thromboembolic disease.
APD791 demonstrated improved coronary artery flow in a preclinical study using the Folts model, an established model of acute coronary syndrome. In other preclinical studies, blocking activation of the 5-HT2A receptor on platelets also demonstrated an improved separation of the dose needed for inhibition of thrombosis versus the dose that increased bleeding relative to existing therapies, suggesting that APD791 has the potential for improved safety relative to existing therapies. We believe these results are consistent with blocking the role of serotonin in the thrombosis process.
Clinical Development. In July 2007, we initiated a single-ascending dose Phase 1 clinical trial evaluating APD791 in healthy adult volunteers. This Phase 1a trial was a randomized, placebo-controlled, double-blind, single-ascending dose trial in 90 healthy male and female volunteers. Doses originally intended for study ranged from 1 mg to 160 mg, but due to favorable tolerability the maximum dose was increased to 320 mg. In the Phase 1a trial, doses were generally well tolerated, without any dose related adverse events, such that a maximum tolerated dose could not be defined despite achieving high concentrations in blood. APD791 was rapidly absorbed, and exposures were generally related to dose. Terminal half-life (t1/2) of parent plus active metabolites was also related to dose, reaching approximately 11 hours at the higher doses. Dose dependent inhibition of serotonin-mediated amplification of platelet aggregation was demonstrated, supporting the preclinical data generated around APD791 and establishing initial clinical validation for APD791's novel mechanism of action.
Based on the positive Phase 1a results, we initiated a Phase 1b clinical trial in January 2008. The Phase 1b trial is a randomized, placebo-controlled, double-blind, multiple-ascending dose trial in up to 50 healthy male and female volunteers between the ages of 19 and 45 years old. In addition to evaluating APD791's safety and tolerability profile, the trial will also evaluate the pharmacokinetics and pharmacodynamics of multiple oral doses of APD791 over a period of one week. Results from the Phase 1b trial are anticipated in mid 2008.
Intellectual Property. As of January 31, 2008, we have issued patents or pending patent applications covering compositions of matter for APD791 and related methods of treatment in 57 jurisdictions, including pending applications in the United States, Japan, Canada, China and before the European Patent Office. Based on sales statistics provided by IMS Health, the jurisdictions where APD791 patents have been filed accounted for more than 99% of global pharmaceutical sales in 2006. The earliest priority date for the patents on APD791 is 2004. The terms of these patents are capable of continuing into 2025 in most jurisdictions without taking into account any patent term extension regimes of any country.
In our partnership with Ortho-McNeil, we are collaborating on the development of compounds for the treatment of type 2 diabetes and other disorders by targeting the GDIR. The GDIR is a novel receptor discovered by Arena that, in our preclinical models, demonstrated the ability to stimulate insulin production in response to increases in blood glucose. Diabetes is a major worldwide disease. The International Diabetes Federation has estimated that in 2007 there were 246 million adults with diabetes worldwide, an increase of over 20% since 2003. Approximately 90% of diabetics worldwide suffer from type 2 diabetes, which is characterized by inadequate response to insulin and/or inadequate secretion of insulin as blood glucose levels rise. Therapies for type 2 diabetes are directed toward correcting the body's inadequate response with oral or injectable medications, or directly modifying insulin levels through injection of insulin or insulin analogs.
Oral medications for type 2 diabetes include insulin releasers such as glyburide, insulin sensitizers such as Actos and Avandia, inhibitors of glucose production by the liver such as metformin, DPP-IV
12
inhibitors like Januvia, as well as Precose and Glyset, which slow the uptake of glucose from the intestine. The market for diabetes medications was nearly $12.0 billion in 2005, of which oral drugs exceeded $7.0 billion. However, a significant portion of type 2 diabetics fail oral medication and require injected insulin therapy. Current oral medications for type 2 diabetes have a number of side effects, including hypoglycemia, weight gain, edema, and perhaps an increase in cardiovascular mortality. Numerous pharmaceutical and biotechnology companies are seeking to develop insulin sensitizers, novel insulin formulations and other therapeutics to improve the treatment of diabetes.
Mechanism and Preclinical Data. We have found the GDIR to be expressed in beta cells, the cells in the pancreas responsible for producing insulin in response to increases in blood glucose. We believe the GDIR represents a novel mechanism for generating a new class of drugs for diabetes that may offer advantages over current approaches. Our preclinical results indicate that stimulating the GDIR allows beta cells to produce insulin more efficiently in response to changes in blood glucose levels. In addition, we have demonstrated in our preclinical studies that the GDIR stimulates incretin hormone release and thus may enhance glucose homeostasis by this additional mechanism. We have also found in these studies that stimulation of the GDIR leads to increased levels and activity of intracellular factors thought to be involved in the preservation of beta cells. Our preclinical studies suggest that the GDIR is amenable to oral small molecule drug development, and we have discovered potent, selective and oral small molecule agonists of the GDIR that improve glucose tolerance and lower blood glucose levels in animal models of diabetes. The GDIR mechanism is glucose dependent, so that in our animal studies our compounds only lowered blood glucose when it rose above normal levels, such as after a meal. Our preclinical results indicate these compounds do not lower normal fasting baseline glucose levels in animal models and, therefore, do not cause hypoglycemia, unlike the glucose-insensitive sulphonylureas.
Development and Partnership Status. In December 2004, we entered into a collaboration and license agreement with Ortho-McNeil to further develop GDIR agonists for the potential treatment of type 2 diabetes and other disorders. In January 2005, we received a non-refundable $17.5 million upfront payment and two milestone payments of $2.5 million each and, in February 2006, we received a $5.0 million milestone payment related to Ortho-McNeil's initiation of a Phase 1 clinical trial of APD668, a novel oral drug candidate discovered by Arena and intended to stimulate the GDIR. In January 2008, we announced that initial clinical trial results for APD668 suggest that GDIR agonists may improve glucose control in patients with type 2 diabetes.
The initial clinical trials by Ortho-McNeil evaluated healthy volunteers and patients with type 2 diabetes in randomized, double-blind, placebo-controlled trials evaluating the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple (14 day) escalating doses of APD668. Based on the data from those studies suggesting that GDIR agonists may improve glucose control in patients with type 2 diabetes, Ortho-McNeil put APD668 on hold and advanced a potentially more potent Arena-discovered GDIR agonist into preclinical development.
As of December 20, 2007, we no longer receive research funding, have significant involvement or perform services under this collaboration. From the inception of this collaboration through December 31, 2007, we have received $27.5 million from Ortho-McNeil in upfront and milestone payments and $7.2 million in research funding. We are eligible to receive a total of $295.0 million in milestone payments for each compound, as well as royalty payments associated with Ortho-McNeil's commercialization of any products discovered under the agreement. These milestones include development and approval milestone payments of up to $132.5 million for the first indication and $62.5 million for the second indication for each compound, and up to $100.0 million in sales milestone payments for each product resulting from the collaboration.
13
In our partnership with Merck, we are collaborating on three GPCRs to develop therapeutics for atherosclerosis and other disorders. We believe one or more of these GPCRs plays a role in regulating plasma lipid profiles, including HDL cholesterol, the so-called "good cholesterol," and is responsible for the HDL-raising activity of niacin. There are very successful drugs available for lowering LDL cholesterol. However, development of novel, effective therapies to increase HDL cholesterol remains a major focus of research. We believe that such therapies may reduce the risk of atherosclerotic heart disease and compete in the large dyslipidemia market.
In January 2008, Merck initiated a Phase 1 clinical trial under our partnership of a second generation oral niacin receptor agonist. From the inception of this collaboration through December 31, 2007, we have received $18.0 million from Merck in upfront and milestone payments, and equity investments totaling $8.5 million. We may receive additional milestone payments of up to $28.0 million for Merck's clinical and marketing achievements, as well as royalty payments associated with Merck's commercialization of any products discovered under the agreement. In addition, we received research funding from Merck through December 31, 2007 totaling $27.5 million. As of October 21, 2007, we no longer receive research funding, have significant involvement or perform services under this collaboration.
Cardiovascular. Acute myocardial infarction, which is commonly known as a heart attack, is often followed in survivors by heart failure. Myocardial infarction and heart failure are often a direct consequence of atherosclerosis, and both remain major causes of death. We have identified certain GPCRs that we believe play a role in the processes related to atherosclerosis and reperfusion injury, and are seeking to identify small molecules directed at these GPCR targets that we believe could provide cardioprotection following myocardial infarction.
CNS Diseases. Many GPCRs are predominately found in the brain or the CNS, and, therefore, we believe targeting GPCRs provides an opportunity to selectively treat various CNS diseases. Many approved drugs for indications ranging from insomnia and narcolepsy to depression, schizophrenia and Parkinson's disease target GPCRs. Our discovery efforts in CNS diseases are focused on indications, such as wakefulness promoters, with large market opportunities where current therapies have significant limitations.
Inflammatory Diseases. We are developing small molecule therapeutics that target GPCRs involved in the inflammatory process. We have identified GPCRs that are found in specific immune cell types. We believe these GPCRs modulate the inflammatory process, and we are applying our screening technologies to these targets to identify small molecules that could activate or inhibit these GPCRs. Some of the GPCRs we are targeting are expressed in immune cells and could be important in cell trafficking and the modulation of key cytokines, such as TNF-alpha, that mediate inflammatory processes.
Other Diabetes Programs. For metabolic diseases, we are working on a series of GPCR targets in addition to the GDIR in order to develop oral therapies to treat type 1 and type 2 diabetes. For example, we are conducting research with receptors that may act to regulate glucose uptake, glucose absorption, insulin sensitivity, insulin secretion, lipid levels and production of glucose in the liver. In order to treat general metabolic disease, we have prioritized GPCRs that have the potential to modulate blood glucose and lipid levels.
Other Obesity Programs. In addition to lorcaserin and other compounds that act on the 5-HT2C serotonin receptor, we have discovery programs focused on several different GPCRs implicated in obesity. Our drug discovery efforts are directed at identifying novel drug candidates that target GPCRs
14
in the CNS and peripheral tissues to reduce fat mass in humans. We have identified GPCRs expressed in the hypothalamus, an area of the brain known to be critical for regulating satiety and metabolism, that we believe play a role in the regulation of food intake and weight.
Our Proprietary GPCR Technologies and Programs
Our drug candidates have resulted from our GPCR-focused drug discovery technologies and capabilities, including Constitutively Activated Receptor Technology, or CART, and our Melanophore technology, and our overall approach to drug discovery and development. GPCRs are categorized as "known" when their naturally occurring, or native, ligands have been identified. Scientists have used molecular cloning in combination with the sequencing of the human genome to identify both additional receptor subtypes of known GPCRs as well as hundreds of novel GPCRs. These novel GPCRs are categorized as "orphan" GPCRs because their native ligands have not been identified. We believe both orphan and known GPCRs offer significant promise for the development of novel GPCR-based therapeutics.
Our constitutive activation technologies allow us to simultaneously identify drug leads that act as receptor activators, or agonists, which increase the detected biological response, or act as receptor inhibitors, which decrease the detected response. We can also identify inverse agonists, which inhibit ligand-independent, as well as ligand-dependent, receptor activity.
We believe that our constitutive activation technologies offer several key advantages for drug discovery, including:
- •
- eliminating the need to identify the native ligand for an orphan receptor;
- •
- enhancing the detection of, and allowing us to simultaneously identify, both receptor inhibitor and receptor activator drug leads;
- •
- allowing for the identification of drug leads that inhibit both ligand-independent and ligand-dependent activity; and
- •
- providing the ability to discover novel and improved therapeutics directed at known receptors.
We use our constitutive activation technologies in combination with our patented Melanophore technology. Our Melanophore technology is a broadly applicable high-throughput screen for GPCRs. When a GPCR is activated (either by a ligand or independent of a ligand through constitutive activation), the GPCR couples to one or more G proteins, including those belonging to the Gs, Gq, and Gi/o classes. Melanophore technology can detect GPCRs that couple to major G protein classes. We believe our Melanophore technology is, therefore, also well-suited for studies of orphan receptors whose coupling parameters are unknown. We believe Melanophore technology provides us with a robust, reproducible, high-throughput and low-cost means for identifying and optimizing GPCR agonists, antagonists and inverse agonists, and is sensitive enough to detect the constitutive activity of many GPCRs.
Our Strategy
The key elements of our scientific and business strategy are to:
- •
- Advance our lead programs. We intend to continue to advance our current drug candidates, with a partner or independently, through clinical development and, if successful, to commercialization.
- •
- Discover and develop additional small molecule drug candidates targeting GPCRs. We intend to continue to discover and develop oral, small molecule compounds for GPCRs identified or validated through our research efforts.
15
- •
- Focus on attractive market opportunities. Obesity, insomnia, diabetes, atherosclerosis and arterial thrombosis each represent large market opportunities. We intend to continue to focus on these and other markets with attractive commercial potential.
- •
- Recognize significant economic value for our drug candidates under development. We intend to maximize the value of our drug candidates through both independent development and licensing and other partnership opportunities with pharmaceutical and larger biotechnology companies.
- •
- Continue to build our capabilities. To capitalize on our discoveries, we plan to continue to improve and expand our capabilities as our drug candidates enter into, and move through, clinical trials and to commercialization.
- •
- Maintain strong discovery research capabilities. Our proprietary technologies, our drug discovery infrastructure and the integrated approach to research used by our scientists, have allowed us to identify a number of GPCR targets and novel compounds. We believe these and other discoveries will continue to fuel our pipeline.
Intellectual Property
Our success depends in large part on our ability to protect our proprietary technology, compounds and information, and to operate without infringing the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright, and trademark laws, as well as confidentiality agreements, licensing agreements and other agreements, to establish and protect our proprietary rights.
As of January 31, 2008, we owned, in part or in whole, or had exclusively licensed the following patents: 22 in the United States, 1 in Japan, 10 in Germany, 10 in France, 10 in the United Kingdom, 10 in Italy, 10 in Spain, 3 in China, and approximately 306 in other jurisdictions. In addition, as of January 31, 2008, we had approximately 1,132 patent applications before the United States Patent and Trademark Office, foreign patent offices and international patent authorities. These patents and patent applications are divided into 101 distinct families of related patents that are directed to chemical compositions of matter, methods of treatment using chemical compositions, GPCR genes, CART, Melanophore technology, or other novel screening methods. One of our patent families was exclusively in-licensed and contains a single issued patent. Ninety-three of our patent families, which include a total of about 298 patents and 1,011 patent applications, were invented solely by our employees. The remaining 7 of our patent families, which include a total of about 62 patents and 121 patent applications, were the subject of joint inventions by our employees and the employees of other entities. There is no assurance that any of our patent applications will issue, or that any of the patents will be enforceable or will cover a drug or other commercially significant product or method. Except for the US patents relating to our Melanophore technology, the term of most of our other current patents commenced, and most of our future patents, if any, will commence, on the date of issuance and terminate 20 years from the earliest effective filing date of the patent application. Since our US Melanophore patents were issued under now superseded rules that provided a patent term of 17 years from the date of issuance, the term of these patents are scheduled to end in 2012. Because the time from filing to issuance of patent applications relating to our business is often more than three years, the resulting term of our pending patent applications, if any, on our drug candidates and technologies may be substantially less than 20 years. In the United States, the European Union and some other jurisdictions, patent term extensions are available for certain delays in either patent office proceedings or marketing and regulatory approval processes. However, due to the specific requirements for obtaining these extensions, there is no assurance that our patents will be afforded extensions even if we encounter significant delays in patent office proceedings or marketing and regulatory approval.
We seek patent protection for our key inventions, including clinical candidates and drug candidates we identify, routes for chemical synthesis, CART, new receptors and new uses for receptors that we discover, as well as genetically altered receptors. It has generally been possible to obtain broad
16
composition of matter patents on novel chemical compounds. It has also generally been possible to obtain broad method patents for techniques and procedures for screening and drug-identification technologies. It has generally been more difficult to obtain broad composition of matter patents for nucleic acid and amino acid sequences. However, it has been possible to obtain patents that protect specific sequences and functional equivalents of those sequences. Furthermore, intellectual property law allows for separate and distinct patents for novel, altered genetic sequences that have improved properties over previously disclosed sequences. We believe that we can obtain patents on certain of our CART-activated receptor sequences because they are not functional equivalents of the natural version of the receptor.
In addition to patent protection, we rely on trade secrets, proprietary know-how, and continuing technological advances to develop and maintain our competitive position. To maintain the confidentiality of our trade secrets and proprietary information, all of our employees are required to enter into and adhere to an employee confidentiality and invention assignment agreement, laboratory notebook policy, and invention disclosure procedures as a condition of employment. Additionally, our employee confidentiality and invention assignment agreements require that our employees not bring to us, or use without proper authorization, any third-party proprietary technology. We also require our consultants and collaborators that have access to proprietary property and information to execute confidentiality and invention rights agreements in our favor before beginning their relationship with us. While such arrangements are intended to enable us to better control the use and disclosure of our proprietary property and provide for our ownership of proprietary technology developed on our behalf, they may not provide us with meaningful protection for such property and technology in the event of unauthorized use or disclosure.
Competition
The biotechnology and pharmaceutical industries are highly competitive and are subject to rapid and significant change. We face significant competition from organizations that are pursuing the same or similar technologies. We also face significant competition from organizations that are pursuing drugs that would compete with the drug candidates we are developing. We may not be able to compete successfully against these organizations, which include many large, well-financed and experienced pharmaceutical and biotechnology companies, as well as academic and research institutions and government agencies.
The focus of our scientific and business strategy is on GPCRs. We believe that many pharmaceutical and biotechnology companies and other organizations also have internal drug discovery programs focused on GPCRs. In addition, other companies have attempted to overcome the problems associated with traditional drug screening by embarking on a variety of alternative strategies. Developments by others may render our drug candidates or technologies obsolete or noncompetitive.
Our present competitors with respect to lorcaserin include Abbott Laboratories, which markets sibutramine under the brand name Meridia, and Hoffmann-La Roche Inc., the United States prescription drug unit of the Roche Group, which markets orlistat under the brand name Xenical. Also, GlaxoSmithKline Consumer Healthcare is marketing an over-the-counter low-dose version of orlistat under the brand name alli in the United States. Another potential competitor is sanofi-aventis, which markets rimonabant under the brand name Acomplia in Europe. Sanofi-aventis has sought and may continue to seek marketing approval for rimonabant in the United States. In addition, we believe that there are potentially competing obesity programs that may be in development at various pharmaceutical and biotechnology companies, including 5-HT2C programs.
In addition to the marketed compounds described above under the APD125 discussion, we believe sanofi-aventis, Eli Lilly and Company, and other companies are developing other potentially competing programs for insomnia, including programs targeting the 5-HT2A receptor.
17
Many of our existing and potential competitors have substantially greater drug development capabilities and financial, scientific and marketing resources than we do. Additional consolidation in the pharmaceutical industry may result in even more resources being concentrated with our competitors. As a result, our competitors may be able to devote greater resources than we can to the research, development, marketing and promotion of drug discovery techniques or therapeutic products, or to adapt more readily to technological advances than we can. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA approval, or commercializing drugs before we do.
We expect to encounter significant competition for the principal drug candidates that we are developing. Companies that complete clinical trials, obtain regulatory approvals and commence commercial sales of their drug candidates before us may achieve a significant competitive advantage. Furthermore, we may be competing against companies with substantially greater manufacturing, marketing, distribution and selling capabilities, and any drug candidate that we successfully develop may compete with existing therapies that have long histories of safe and effective use.
We may rely on our collaborators for support of development programs and for the manufacturing and marketing of drug candidates. Our collaborators may be conducting multiple drug development efforts within the same disease areas that are the subject of their agreements with us, which may negatively impact the development of drugs that they discover that are subject to our agreements. Generally, our agreements with our collaborators do not preclude them from pursuing development efforts in one or more therapeutic areas of interest in which we have internal development efforts ongoing. In addition, we face and will continue to face intense competition from other companies for such collaborative arrangements, and technological and other developments by others may make it more difficult for us to establish such relationships.
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, premarket approval, manufacture, marketing and distribution of pharmaceutical products. These agencies and other regulatory agencies regulate research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, recordkeeping, advertising and promotion of drug candidates. Failure to comply with applicable FDA or other requirements may result in civil or criminal penalties, suspension or delays in clinical development, recall or seizure of products, partial or total suspension of production or withdrawal of a product from the market.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act, or FFDCA, and its implementing regulations. The process required by the FDA before our drug candidates may be marketed in the United States generally involves the following:
- •
- completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA's current good laboratory practice (cGLP) regulations;
- •
- submission to the FDA of an investigational new drug, or IND, application, which must become effective before human clinical trials may begin;
- •
- performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug candidate for each proposed indication;
- •
- submission to the FDA of a new drug application, or NDA;
- •
- satisfactory completion of an FDA preapproval inspection of the manufacturing facilities at which the product is produced to assess compliance with cGMP regulations; and
- •
- FDA review and approval of the NDA prior to any commercial marketing or sale of the drug.
18
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our drug candidates will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation as well as cGLP studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30 day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Our IND submissions, or those of our collaborators, may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP, regulations and regulations for informed consent.
Clinical Trials. For purposes of NDA submission and approval, clinical trials are typically conducted in the following sequential phases, which may overlap:
- •
- Phase 1 Clinical Trials. Studies are initially conducted in a limited population to test the drug candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans. In some cases, a sponsor may decide to conduct what is referred to as a "Phase 1b" evaluation, which is an additional, safety-focused Phase 1 clinical trial.
- •
- Phase 2 Clinical Trials. Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted indications and to determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. In some cases, a sponsor may decide to run what is referred to as a "Phase 2b" evaluation, which is a second, confirmatory Phase 2 clinical trial.
- •
- Phase 3 Clinical Trials. These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites.
- •
- Phase 4 Clinical Trials. In some cases, the FDA may condition approval of an NDA for a drug candidate on the sponsor's agreement to conduct additional clinical trials to further assess the drug's safety and effectiveness after NDA approval. In addition, a sponsor may decide to conduct additional clinical trials after the FDA has approved an NDA. Post-approval trials are typically referred to as Phase 4 clinical trials.
New Drug Applications. The results of product development, preclinical studies and clinical trials are submitted to the FDA as part of an NDA. NDAs also must contain extensive manufacturing information. Once the submission has been accepted for filing, the FDA's goal is to review applications within 10 months or, if the application relates to a serious or life-threatening indication, six months. The review process is often significantly extended by FDA requests for additional information or
19
clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of an NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s). Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase 4 clinical trials, and surveillance programs to monitor the safety effects of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
Other Regulatory Requirements. Any products manufactured or distributed by us or our collaborators pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping and reporting requirements. Adverse event experience with the product must be reported to the FDA in a timely fashion and pharmacovigilance programs to proactively look for these adverse events may be mandated by the FDA. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as Warning Letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If we or our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in adverse publicity, Warning Letters, corrective advertising and potential civil and criminal penalties.
Physicians may prescribe legally available drugs for uses that are not described in the product's labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers' communications regarding off-label use.
In Zofingen, Switzerland, our Swiss subsidiary, Arena Pharmaceuticals GmbH, or Arena GmbH, operates a drug product manufacturing and packaging facility. In Switzerland, Swissmedic is the central Swiss supervisory authority for therapeutic products. It is a public service organization of the federal government with headquarters in Bern, Switzerland. After an inspection of our Swiss manufacturing facility by the competent regional authorities (Regionales Heilmittelinspektorat der Nordostschweiz,
20
Basel, Switzerland), acting on behalf of Swissmedic, in June and July 2007, Swissmedic issued an operation permit to Arena GmbH for the production of drugs in July 2007. This permit is valid until July 2012.
DEA Regulation. The DEA regulates drugs that are controlled substances. Controlled substances are those drugs that appear on one of the five schedules promulgated and administered by the DEA under the Controlled Substances Act, or CSA. The CSA governs, among other things, the inventory, distribution, recordkeeping, handling, security and disposal of controlled substances. If our drug candidates are scheduled by the DEA as controlled substances, we will be subject to periodic and ongoing inspections by the DEA and similar state drug enforcement authorities to assess our ongoing compliance with DEA's regulations. Any failure to comply with these regulations could lead to a variety of sanctions, including the revocation, or a denial of renewal of any DEA registration, injunctions, or civil or criminal penalties.
Sources and Availability of Raw Materials, Intermediates, and Clinical Supplies
We purchase raw materials and intermediates when necessary from commercial sources. To decrease the risk of an interruption to our supply, when reasonably possible, we source these materials from redundant suppliers so that, in general, the loss of any one source of supply would not have a material adverse effect on project timelines or inventory of clinical supplies for use in human trials. However, currently we have a primary source of supply for some key intermediates, active pharmaceutical ingredient, or API, and drug products for our lead development projects. The loss of a primary source of supply would potentially delay our lead development projects, including lorcaserin, APD125 and APD791, and potentially those of our collaborators.
In Zofingen, Switzerland, Arena GmbH operates a drug product manufacturing and packaging facility, where drugs are produced under contract for Siegfried Ltd. This facility is suitable for producing and packaging lorcaserin tablets for registration and commercial use, as well as tablets and packaging for other programs.
Compliance with Environmental Regulations
Our research and development programs involve the controlled use of hazardous materials, chemicals, biological materials and various radioactive compounds. In the United States, we are subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, the Controlled Substances Act and other present federal, state or local regulations.
With regard to Arena GmbH's drug product manufacturing and packaging facility, Arena GmbH has contracted with Siegfried Ltd to provide safety, health and environmental services and assess compliance, train personnel and oversee Arena GmbH's compliance with the applicable safety, health and environmental regulations. Arena GmbH is subject to regulation under the Environmental Protection Act (Umweltschutzgesetz, USG) and the Federal Act on the Protection of Waters (Gewässerschutzgesetz, GSchG), which refer to several ordinances such as the Ordinance on Air Pollution Control (Luftreinhalteverordnung, LRV), the Ordinance on Incentive Taxes on Volatile Organic Compounds (Verordnung über die Lenkungsabgabe auf flüchtigen organischen Verbindungen, VOCV), the Water Protection Ordinance (Gewässerschutzverordnung, GSchV), the Ordinance of the Handling of Wastes (VeVA), the Chemicals Ordinance (ChemV) and the Ordinance on Protection against Major Accidents (Störfallverordnung, StFV). The competent authority in Switzerland for the implementation of environmental regulations is BAFU (Bundesamt für Umwelt / Federal Office for the Environment), which is the Swiss agency for the environment as well as the respective authorities of the Canton of Aargau (Amt für Umwelt). Occupational health and safety is regulated by the EKAS guideline (Nr. 6508) for the evaluation of worker safety and reporting to the relevant authorities. The
21
competent authority for the implementation of occupational health and safety regulations is the Canton of Aargau (Amt für Wirtschaft und Arbeit), where exposure limits are set by SUVA (Schweizerische Unfallversicherungsanstalt), which is the Swiss Accident Insurance Fund.
We may be subject to further such regulations in the future. Although we believe that our operations comply in all material respects with the applicable environmental laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result, and the extent of that liability could exceed our resources. Our compliance with these laws and regulations has not had, and is not expected to have, a material effect upon our capital expenditures, results of operations or competitive position.
Research and Development Expenses
Research and development activities, which include personnel costs, research supplies, facility and equipment costs and clinical and preclinical study fees, are the primary source of our expenses. Such expenses related to the development and improvement of our technologies and drug candidates totaled $149.5 million for the year ended December 31, 2007, $103.4 million for the year ended December 31, 2006 and $79.7 million for the year ended December 31, 2005. Research that was sponsored by our collaborators is included in our total research and development expenses. We estimate that research expenses incurred on projects sponsored by our collaborators totaled $4.6 million for the year ended December 31, 2007, $7.7 million for the year ended December 31, 2006 and $8.7 million for the year ended December 31, 2005.
Employees
As of February 29, 2008, we had a total of 491 employees, including 419 in research, development and manufacturing and 72 in administration, which includes finance, legal, facilities, information technology and other general support areas. We consider our relationship with our employees to be good.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the "Exchange Act") are available free of charge on our website (www.arenapharm.com) as soon as reasonably practicable after they are filed with, or furnished to, the SEC.
Item 1A. Risk Factors.
Investment in our stock involves a high degree of risk. You should consider carefully the risks described below, together with other information in this Annual Report on Form 10-K and other public filings, before making investment decisions regarding our stock. If any of the following events actually occur, our business, operating results, prospects or financial condition could be materially and adversely affected. This could cause the trading price of our common stock to decline and you may lose all or part of your investment. Moreover, the risks described below are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business, operating results, prospects or financial condition.
22
Risks Relating to Our Business
We will need additional funds to conduct our planned research and development efforts, and we may not be able to obtain such funds.
Our accumulated deficit since inception has resulted in large part from the significant research and development expenditures we have made in seeking to identify and validate new drug targets and develop compounds that could become marketed drugs.
We expect that our operating expenses over the next several years will be significant and that we will continue to have significant operating losses for at least the next several years, even if we or our collaborators are successful in advancing our compounds or partnered compounds.
We do not have any commercially available drugs. It takes many years and potentially hundreds of millions of dollars to successfully develop a preclinical or early clinical compound into a marketed drug, and our efforts may not result in a marketed drug. We have substantially less money than we need to develop our compounds into marketed drugs. Additional funding may not be available to us or may not be available on terms that you or we believe are favorable. If additional funding is not available, we may have to delay, reduce the scope of or eliminate one or more of our research or development programs.
In addition, provisions of our series B-1 redeemable convertible preferred stock, or Series B-1 Preferred, require us to obtain approval of the preferred stockholders, or otherwise trigger rights of first refusal or payment provisions, to (i) offer or sell new securities, other than in specified underwritten offerings or strategic partnerships or joint venture and certain other exceptions, (ii) sell or issue common stock or securities issuable into common stock below certain prices, (iii) incur debt or allow liens on our property, other than certain permitted debt and liens, (iv) amend our certificate of incorporation so as to affect adversely any rights of the preferred stockholders, (v) authorize or create a new class of stock that will be senior or equal to the Series B-1 Preferred or our series B-2 redeemable convertible preferred stock, or Series B-2 Preferred (the "Series B-1 Preferred" and the "Series B-2 Preferred" are collectively referred to as the "Series B Preferred"), in terms of dividends, redemption or distribution of assets, or (vi) take certain other actions. These provisions may make it more difficult for us to take certain corporate actions and could delay, discourage or prevent future financings.
Our stock price could decline significantly based on the results and timing of clinical trials and preclinical studies of, and decisions affecting, our lead drug candidates.
Results of clinical trials and preclinical studies (including preclinical studies conducted after initiation of clinical trials) of our lead drug candidates may not be viewed favorably by us or third parties, including investors, analysts, potential collaborators, the academic and medical community, and regulators. The same may be true of how we design the development programs of our lead drug candidates and regulatory decisions (including by regulatory authorities) affecting those development programs. Biotechnology company stock prices have declined significantly when such results and decisions were unfavorable or perceived negatively or when a drug candidate did not otherwise meet expectations.
We have several drug programs that are currently in clinical trials. In addition to successfully completing clinical trials, in order to conduct long-term clinical trials and gain regulatory approval to commercialize drug candidates, regulatory authorities require that all drug candidates complete short- and long-term preclinical toxicity and carcinogenicity studies. These studies in animals are required to help determine the potential risk that drug candidates may be toxic or cause cancer in humans. The preclinical assessment of carcinogenic potential includes short-term in vitro and in vivo studies to look for chromosomal damage. Short-term carcinogenicity and toxicity studies have been completed for all of our clinical-stage programs. To date, we have only completed long-term preclinical toxicity studies
23
for lorcaserin, and we have not completed carcinogenicity studies for lorcaserin or any of our other clinical-stage programs. The results of our clinical trials and preclinical studies are uncertain, and the design of these trials and studies (which may change significantly and be more expensive than currently anticipated depending on our results and regulatory decisions) may also be viewed negatively by third parties. We may not be successful in advancing our programs on our projected timetable, if at all. Failure to initiate or delays in the development programs for any of our drug candidates, or unfavorable results or decisions or negative perceptions regarding any of such programs, could cause our stock price to decline significantly. This is particularly the case with respect to our most advanced drug candidate, lorcaserin, for which we have three ongoing Phase 3 clinical trials and expect a month-12 ESMB review of the echocardiographic data from one of these trials (our BLOOM trial) to occur in March 2008.
Our development of lorcaserin may be adversely impacted by cardiovascular side effects previously associated with fenfluramine and dexfenfluramine.
We have developed lorcaserin to more selectively stimulate the 5-HT2C serotonin receptor because we believe this may avoid the cardiovascular side effects associated with fenfluramine and dexfenfluramine (often used in combination with phentermine, the combination of which was commonly referred to as "fen-phen"), two serotonin-releasing agents and non-selective serotonin receptor agonists, both of which were withdrawn from the market in 1997 after reported incidences of heart valve disease and pulmonary hypertension associated with their usage. We may not be correct in this belief, however, or lorcaserin's selectivity profile may not avoid these undesired side effects. Moreover, the potential relationship between the activity of lorcaserin and the activity of fenfluramine and dexfenfluramine may result in increased United States Food and Drug Administration, or FDA, regulatory scrutiny of the safety of lorcaserin and may raise potential adverse publicity in the marketplace, which could affect clinical enrollment or ultimately sales if lorcaserin is approved for sale.
The development programs for our drug candidates are expensive, time consuming, uncertain and susceptible to change, interruption, delay or termination.
Drug development programs are very expensive, time consuming and difficult to design and implement. Our drug candidates are in various stages of development and are prone to the risks of failure inherent in drug development. We will need to complete additional clinical trials and preclinical studies before we can demonstrate that our drug candidates are safe and effective to the satisfaction of the FDA and similar non-US regulatory authorities. These trials are expensive and uncertain processes that take years to complete. Failure can occur at any stage of the process, and successful early clinical or preclinical trials do not ensure that later trials or studies will be successful. In addition, the commencement of our planned clinical trials could be substantially delayed or prevented by several factors, including:
- •
- limited number of, and competition for, suitable patients required for enrollment in our clinical trials;
- •
- limited number of, and competition for, suitable sites to conduct our clinical trials;
- •
- delay or failure to obtain FDA approval or agreement to commence a clinical trial;
- •
- delay or failure to obtain sufficient supplies of our drug candidates for our clinical trials;
- •
- delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or investigators; and
- •
- delay or failure to obtain institutional review board, or IRB, approval to conduct a clinical trial at a prospective site.
24
Even if the results of our development programs are favorable, the development programs of our most advanced drug candidates, including those being developed by our collaborators, may take significantly longer than expected to complete. In addition, the FDA, other regulatory authorities, our collaborators, or we may suspend, delay or terminate our development programs at any time for various reasons, including:
- •
- lack of effectiveness of any drug candidate during clinical trials;
- •
- side effects experienced by study participants or other safety issues;
- •
- slower than expected rates of patient recruitment and enrollment or lower than expected patient retention rates;
- •
- delays or inability to manufacture or obtain sufficient quantities of materials for use in clinical trials;
- •
- inadequacy of or changes in our manufacturing process or compound formulation;
- •
- delays in obtaining regulatory approvals to commence a study, or "clinical holds," or delays requiring suspension or termination of a study by a regulatory authority, such as the FDA, after a study is commenced;
- •
- changes in applicable regulatory policies and regulations;
- •
- delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites;
- •
- uncertainty regarding proper dosing;
- •
- unfavorable results from ongoing clinical trials and preclinical studies;
- •
- failure of our clinical research organizations to comply with all regulatory and contractual requirements or otherwise perform their services in a timely or acceptable manner;
- •
- scheduling conflicts with participating clinicians and clinical institutions;
- •
- failure to construct appropriate clinical trial protocols;
- •
- insufficient data to support regulatory approval;
- •
- termination of clinical trials by one or more clinical trial sites;
- •
- inability or unwillingness of medical investigators to follow our clinical protocols;
- •
- difficulty in maintaining contact with subjects during or after treatment, which may result in incomplete data; or
- •
- lack of sufficient funding to continue clinical trials and preclinical studies.
There is typically a high rate of attrition from the failure of drug candidates proceeding through clinical trials, and many companies have experienced significant setbacks in advanced development programs even after promising results in earlier studies or trials. We may experience similar setbacks in our development programs. If we or our collaborators abandon or are delayed in our development efforts related to lorcaserin, APD125, APD791 or any other drug candidate, we may not be able to generate sufficient revenues to continue our operations at the current level or become profitable, our reputation in the industry and in the investment community would likely be significantly damaged, additional funding may not be available to us or may not be available on terms you or we believe are favorable, and our stock price would likely decrease significantly.
25
Our drug candidates are subject to extensive regulation, and we may not receive required regulatory approvals for any of our drug candidates.
The clinical development, manufacturing, labeling, packaging, storage, recordkeeping, advertising, promotion, export, marketing and distribution, and other possible activities relating to our drug candidates are, and any resulting drugs will be, subject to extensive regulation by the FDA and other regulatory agencies in the United States. Neither our collaborators nor we are permitted to market our drug candidates in the United States until we receive regulatory approval from the FDA. Neither our collaborators nor we have received marketing approval for any of our drug candidates. Specific preclinical data, chemistry, manufacturing and controls data, a proposed clinical study protocol and other information must be submitted to the FDA as part of an investigational new drug, or IND, application, and clinical trials may commence only after the IND application becomes effective. To market a new drug in the United States, we must submit to the FDA and obtain FDA approval of a New Drug Application, or NDA. An NDA must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing and controls to demonstrate the safety and effectiveness of the drug candidate.
Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA and other applicable regulatory requirements may, either before or after product approval, if any, subject our company to administrative or judicially imposed sanctions, including:
- •
- Warning Letters;
- •
- civil and criminal penalties;
- •
- injunctions;
- •
- withdrawal of approved products;
- •
- product seizure or detention;
- •
- product recalls;
- •
- total or partial suspension of production;
- •
- imposition of restrictions on operations, including costly new manufacturing requirements; and
- •
- refusal to approve pending NDAs or supplements to approved NDAs.
Regulatory approval of an NDA or NDA supplement is not guaranteed. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional preclinical studies and clinical trials. The number of preclinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to target, and the regulations applicable to any particular drug candidate. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including:
- •
- a drug candidate may not be deemed adequately safe and effective;
- •
- FDA officials may not find the data from preclinical studies and clinical trials sufficient;
- •
- the FDA may not approve the manufacturing processes or facilities; or
- •
- the FDA may change its approval policies or adopt new regulations.
We do not expect any drugs resulting from our research and development efforts to be commercially available until 2010 or later. Our most advanced drug candidates, including lorcaserin and APD125, have not completed all preclinical studies and the large, pivotal Phase 3 clinical trials for efficacy and safety that are required for FDA approval. Also, we have not previously filed NDAs with the FDA, nor have we previously conducted Phase 3 clinical trials, which are significantly larger and
26
more complex than earlier-stage trials. This lack of corporate experience may impede our ability to successfully complete these trials and obtain FDA approval in a timely manner, if at all, for our drug candidates for which development and commercialization is our responsibility. Even if we believe that data collected from our preclinical studies and clinical trials of our drug candidates are promising and that our information and procedures regarding chemistry, manufacturing and controls are sufficient, our data may not be sufficient to support approval by the FDA or any other United States or foreign regulatory authority. As a result, we cannot predict when or whether regulatory approval will be obtained for any drug we develop. In addition, we believe that the regulatory review of NDAs for drug candidates intended for widespread use by a large proportion of the general population is becoming increasingly focused on safety. In this regard, it is possible that some of our drug candidates, including lorcaserin and APD125, will be subject to increased scrutiny to show adequate safety than would drug candidates for more acute and life-threatening diseases such as cancer. Even if approved, drug candidates may not be approved for all indications requested and such approval may be subject to limitations on the indicated uses for which the drug may be marketed. Our business and reputation may be harmed by any failure or significant delay in receiving regulatory approval for the sale of any drugs resulting from our drug candidates.
In order to market any drugs outside of the United States, we and our collaborators must comply with numerous and varying regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks associated with FDA approval as well as additional, presently unanticipated, risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects associated with regulatory approval in the United States, including the risk that our drug candidates may not be approved for all indications requested and that such approval may be subject to limitations on the indicated uses for which the drug may be marketed.
The results of preclinical studies and completed clinical trials are not necessarily predictive of future results, and our current drug candidates may not have favorable results in later studies or trials.
Preclinical studies and Phase 1 and Phase 2 clinical trials are not primarily designed to test the efficacy of a drug candidate, but rather to test safety, to study pharmacokinetics and pharmacodynamics, and to understand the drug candidate's side effects at various doses and schedules. To date, long-term safety and efficacy have not yet been demonstrated in clinical trials for any of our drug candidates. Favorable results in our early studies or trials may not be repeated in later studies or trials, including continuing preclinical studies and large-scale clinical trials, and our drug candidates in later-stage trials may fail to show desired safety and efficacy despite having progressed through earlier-stage trials. In particular, preclinical data and the limited clinical results that we have obtained for lorcaserin and APD125 may not predict results from studies in larger numbers of subjects drawn from more diverse populations treated for longer periods of time. They also may not predict the ability of lorcaserin or APD125 to achieve or sustain the desired effects in the intended population or to do so safely. Unfavorable results from ongoing preclinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials, or abandonment of a clinical program. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated, or a clinical program abandoned. In addition, we may report top-line data from time to time, which is based on a preliminary analysis of key efficacy and safety data, and is subject to change following a more comprehensive review of the data related to the applicable clinical trial.
27
Many of our research and development programs are in early stages of development, and may not result in the commencement of clinical trials.
Many of our research and development programs are in the discovery or preclinical stage of development. The process of discovering compounds with therapeutic potential is expensive, time consuming and unpredictable. Similarly, the process of conducting preclinical studies of compounds that we discover requires the commitment of a substantial amount of our technical and financial resources and personnel. We may not discover additional compounds with therapeutic potential, and any of the compounds for which we are conducting preclinical studies may not result in the commencement of clinical trials. We cannot be certain that results sufficiently favorable to justify commencement of Phase 1 clinical trials will be obtained in these preclinical investigations. If we are unable to identify and develop new drug candidates, we may not be able to maintain a clinical development pipeline or generate revenues.
Drug discovery and development is intensely competitive in the therapeutic areas on which we focus. If our competitors develop treatments that are approved faster, marketed better or demonstrated to be more effective or safer than our drug candidates, our commercial opportunities will be reduced or eliminated.
We focus our efforts on GPCRs. Because GPCRs are an important target class for drug discovery efforts, we believe that many pharmaceutical and biotechnology companies and other organizations have internal drug discovery programs focused on GPCRs. Many of the drugs that our collaborators or we are attempting to discover and develop would compete with existing therapies. In addition, many companies are pursuing the development of new drugs that target the same diseases and conditions that we target. Many of our competitors, particularly large pharmaceutical companies, have substantially greater research, development and marketing capabilities and greater financial, scientific and human resources than we do. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before we do for the same indication may achieve a significant competitive advantage, including certain patent and FDA marketing exclusivity rights. In addition, our competitors may develop drugs with fewer side effects, more desirable characteristics (such as route of administration or frequency of dosing) or greater efficacy than our drug candidates or drugs, if any, for the same indication. Any results from our research and development efforts, or from our joint efforts with our existing or any future collaborators, may not compete successfully with existing or newly discovered products or therapies.
If we do not partner one or more unpartnered programs or raise additional funds, we may have to curtail some of our activities.
Without additional capital or funding from partners, we would need to re-evaluate our strategy of moving multiple drug discovery and development programs forward while at the same time maintaining our research and discovery capabilities. Based on such evaluation, we may need to significantly curtail some of our current and planned programs and expenditures. We do not know what programs, if any, we would need to curtail, but we believe narrowing our pipeline would reduce our opportunities for success.
Our revenues depend upon the actions of our existing and potential collaborators.
We expect that, for at least the next few years, our revenues will depend upon the success of our existing collaborations, our ability to enter into new collaborations and our ability to generate revenues under our subsidiary, Arena Pharmaceuticals GmbH's, or Arena GmbH, contract manufacturing agreement with Siegfried Ltd. Our revenues of $19.3 million for the year ended December 31, 2007 were derived exclusively from our collaborations with Merck and Ortho-McNeil. Absent any new collaborator, we expect our revenues for 2008 to be derived under Arena GmbH's contract manufacturing agreement with Siegfried Ltd and, to a lesser extent, from our collaborations with Merck
28
and Ortho-McNeil. In 2008 and beyond, our revenues from our collaborations with Merck and Ortho-McNeil will depend on, in addition to patent reimbursements, milestone and royalty payments, if any. Thus, we will receive little additional revenues from our existing collaborators if our own or our collaborators' research, development or, ultimately, marketing efforts are unsuccessful.
Typically, our collaborators (and not us) control the development of partnered compounds into drugs after we have met early preclinical scientific milestones. In addition, we may not have complete access to information about the results and status of our collaborators' clinical trials and regulatory programs and strategies. We are not entitled to the more significant milestone payments under our agreements until our collaborators have advanced compounds in clinical testing. Our partners may not devote adequate resources to the development of our compounds and may not develop or implement a successful clinical or regulatory strategy. Only two of our partners, Merck and Ortho-McNeil, have advanced our drug candidates into clinical testing and paid us the applicable milestone payments. We cannot guarantee that any other development, approval or sales milestones in our existing or future collaborations will be achieved, or that we will receive any payments for the achievement of any future milestones. In addition, our existing collaborations, including our collaborations with Merck and Ortho-McNeil, may be terminated early in certain circumstances, in which case we may not receive future milestone or royalty payments or patent reimbursements.
Moreover, our ability to enter into new collaborations depends on the outcomes of our preclinical and clinical testing. We do not control these outcomes. In addition, even if our testing is successful, pharmaceutical companies may not partner with us on terms that we believe are acceptable until we have advanced our drug candidates into the clinic and, possibly, through later-stage clinical trials, if at all.
Collaborative relationships may lead to delays in drug development and commercialization and disputes.
We may have conflicts with our prospective, current or past collaborators, such as conflicts concerning the interpretation of preclinical or clinical data, the achievement of milestones, or the ownership of intellectual property. Our collaborators may stop supporting our drug candidates if they develop or obtain rights to competing drug candidates or drugs. If any conflicts arise with Ortho-McNeil, Merck or any other prospective, current or past collaborator, such collaborator may act in a manner that is adverse to our interests. Any such disagreement could result in one or more of the following, each of which could delay, or lead to termination of development or commercialization of our partnered drug candidates, and in turn prevent us from generating revenues:
- •
- unwillingness on the part of a collaborator to pay us research funding, milestone payments or royalties that we believe are due to us under a collaboration;
- •
- uncertainty regarding ownership of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations;
- •
- unwillingness on the part of a collaborator to keep us informed regarding the progress of its development and commercialization activities or to permit public disclosure of the results of those activities;
- •
- slowing or cessation of a collaborator's development or commercialization efforts with respect to our drug candidates; or
- •
- litigation or arbitration.
29
Setbacks and consolidation in the pharmaceutical and biotechnology industries, and our or our collaborators' inability to obtain third-party coverage and adequate reimbursement, could make partnering more difficult and diminish our revenues.
Setbacks in the pharmaceutical and biotechnology industries, such as those caused by safety concerns relating to high-profile drugs like Avandia, Vioxx and Celebrex, or drug candidates such as rimonabant and torcetrapib, as well as competition from generic drugs, litigation, and industry consolidation, may have an adverse effect on us. For example, pharmaceutical companies may be less willing to enter into new collaborations or continue existing collaborations if they are integrating a new operation as a result of a merger or acquisition or if their therapeutic areas of focus change following a merger. Moreover, our and our collaborators' ability to commercialize any of our drugs that may be approved will depend in part on government regulation and the availability of coverage and adequate reimbursement from third-party payers, including private health insurers and government payers, such as the Medicaid and Medicare programs, increases in government-run, single-payer health insurance plans and compulsory licenses of drugs. Government and third-party payers are increasingly attempting to contain healthcare costs by limiting coverage and reimbursement levels for new drugs. These efforts may limit our commercial opportunities by reducing the amount a potential collaborator is willing to pay to license our programs or drug candidates in the future due to a reduction in the potential revenues from drug sales. Moreover, legislation and regulations affecting the pricing of pharmaceuticals may change before regulatory agencies approve our drug candidates for marketing. Adoption of such legislation and regulations could further limit pricing approvals for, and reimbursement of, drugs. A government or third-party payor decision not to approve pricing for, or provide adequate coverage and reimbursements of, our drugs, if any, could limit market acceptance of such drugs.
We rely on third parties to conduct our clinical trials and many of our preclinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our drug candidates may not advance in a timely manner or at all.
In the course of our discovery, preclinical testing and clinical trials, we rely on third parties, including laboratories, investigators, clinical research organizations and manufacturers, to perform critical services for us. For example, we rely on third parties to conduct our clinical trials and many of our preclinical studies. Clinical research organizations are responsible for many aspects of the trials, including finding and enrolling subjects for testing and administering the trials. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner, and we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. In addition, if such third parties fail to perform their obligations in compliance with our clinical trial protocols or GCPs, our clinical trials may not meet regulatory requirements or may need to be repeated. Furthermore, we may not be able to obtain regulatory approval to commercialize the drug candidate being tested in such trials. As a result of our dependence on third parties, we may face delays or failures outside of our direct control. These risks also apply to the development activities of our collaborators, and we do not control our collaborators' research and development, clinical trials or
30
regulatory activities. We do not expect any drugs resulting from our collaborators' research and development efforts to be commercially available for many years, if ever.
We rely on third-party manufacturers and we or such third parties may encounter failures or difficulties that could delay the clinical development or regulatory approval of our drug candidates, or their ultimate commercial production if approved.
We and third parties manufacture our drug candidates. We do not have manufacturing facilities that can produce sufficient quantities of drug candidates for large-scale clinical trials. Accordingly, we must either develop such facilities, which will require substantial additional funds, or rely, at least to some extent, on third-party manufacturers for the production of drug candidates. Furthermore, should we obtain FDA approval for any of our drug candidates, we expect to rely, at least to some extent, on third-party manufacturers for commercial production. Our dependence on others for the manufacture of our drug candidates may adversely affect our ability to develop and deliver such drug candidates on a timely and competitive basis.
Any performance failure on the part of us or a third-party manufacturer could delay clinical development, regulatory approval or, ultimately, sales of our drug candidates. We or third-party manufacturers may encounter difficulties involving production yields, regulatory compliance, quality control and quality assurance, as well as shortages of qualified personnel. Approval of our drug candidates could be delayed, limited or denied if the FDA does not approve our or a third-party manufacturer's processes or facilities. Moreover, the ability to adequately and timely manufacture and supply drug candidates is dependent on the uninterrupted and efficient operation of the manufacturing facilities, which is impacted by many manufacturing variables including:
- •
- availability or contamination of raw materials and components used in the manufacturing process, particularly those for which we have no other source or supplier;
- •
- facility capacity of our facilities or those of our contract manufacturers;
- •
- facility contamination by microorganisms or viruses;
- •
- compliance with regulatory requirements;
- •
- changes in forecasts of future demand;
- •
- timing and actual number of production runs;
- •
- production success rates and bulk drug yields; and
- •
- timing and outcome of product quality testing.
In addition, we or our third-party manufacturers may encounter delays and problems in manufacturing our drug candidates or drugs for a variety of reasons, including accidents during operation, failure of equipment, delays in receiving materials, natural or other disasters, political or governmental changes, or other factors inherent in operating complex manufacturing facilities. Supply chain management is difficult. Commercially available starting materials and reagents may become scarce or more expensive to maintain, and we may not be able to obtain favorable terms in agreements with subcontractors. We or our third-party manufacturers may not be able to operate our respective manufacturing facilities in a cost-effective manner or in a time frame that is consistent with our expected future manufacturing needs. If we or our third-party manufacturers cease or interrupt production or if our third-party manufacturers and other service providers fail to supply materials, products or services to us for any reason, such interruption could delay progress on our programs, or interrupt the commercial supply, with the potential for additional costs and lost revenues. If this were to occur, we may also need to seek alternative means to fulfill our manufacturing needs.
We may not be able to enter into agreements for the manufacture of our drug candidates with manufacturers whose facilities and procedures comply with applicable law. Manufacturers are subject to
31
ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Administration of the United States Department of Justice, or DEA, and corresponding state and foreign authorities to ensure strict compliance with current Good Manufacturing Practices, or cGMPs, and other applicable government regulations and corresponding foreign standards. We do not have control over a third-party manufacturer's compliance with these regulations and standards. In addition, our Swiss subsidiary, Arena GmbH has contracted with Siegfried Ltd to provide safety, health and environmental services and assess compliance, train personnel and oversee Arena GmbH's compliance with the applicable safety, health and environmental regulations. We are, therefore, relying at least in part on Siegfried Ltd's judgment, experience and expertise. If we or one of our manufacturers fails to maintain compliance, we or they could be subject to civil or criminal penalties, the production of our drug candidates could be interrupted or suspended, or our product could be recalled or withdrawn, resulting in delays, additional costs and potentially lost revenues.
We may not be able to effectively integrate or manage our international operations and such difficulty could adversely affect our stock price, business operations, financial condition and results from operations.
In January 2008, we purchased from Siegfried Ltd certain drug product facility assets, including fixtures, equipment, other personal property and real estate assets and acquired 69 employees in Zofingen, Switzerland. There are significant risks associated with the establishment of foreign operations, including, but not limited to, compliance with local laws and regulations, the protection of our intellectual property, the ability to integrate our corporate culture with local customs and cultures, the distraction to our management and foreign currency exchange rates and the impact of shifts in the US and local economies on those rates. We will also be contract manufacturing drug products for Siegfried for at least the next several years and, therefore, be subject to liability for non-performance, product recalls and other claims against manufacturers.
We may engage in strategic transactions that could impact our liquidity, increase our expenses and present significant distractions to our management.
From time to time we consider strategic transactions, such as acquisitions of companies, asset purchases and out-licensing or in-licensing of compounds or technologies. Additional potential transactions we may consider include a variety of different business arrangements, including spin-offs, strategic partnerships, joint ventures, restructurings, divestitures, business combinations and investments. Any such transaction may require us to incur non-recurring or other charges, may increase our near- and long-term expenditures and may pose significant integration challenges, require additional expertise or disrupt our management or business, which could harm our operations and financial results.
As part of an effort to acquire a business or drug candidate or to enter into other significant transactions, we conduct business, legal and financial due diligence with the goal of identifying and evaluating material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, might not realize the intended advantages of the transaction. If we fail to realize the expected benefits from acquisitions we may consummate, whether as a result of unidentified risks, integration difficulties, regulatory setbacks or other events, our business, results of operations and financial condition could be adversely affected.
Our efforts will be seriously jeopardized if we are unable to retain and attract key employees.
Our success depends on the continued contributions of our principal management, development and scientific personnel, and the ability to hire and retain key personnel, particularly in the clinical development area as we transition more of our programs from research into drug development. We face intense competition for such personnel. The loss of services of any principal member of our management or scientific staff, particularly Jack Lief, our President, Chief Executive Officer and Chairman, and Dominic P. Behan, Ph.D., our Senior Vice President and Chief Scientific Officer, could adversely impact our operations and ability to raise additional capital. To our knowledge, neither Mr. Lief nor Dr. Behan plans to leave, retire or otherwise disassociate with us in the near future.
32
We use biological materials, hazardous materials, chemicals and radioactive compounds.
Our research and development and manufacturing activities involve the use of potentially harmful biological materials as well as materials, chemicals and various radioactive compounds that could be hazardous to human health and safety or the environment. These materials and various wastes resulting from their use are stored at our facility pending ultimate use and disposal. We cannot completely eliminate the risk of contamination, which could cause:
- •
- interruption of our research and development or manufacturing efforts;
- •
- injury to our employees and others;
- •
- environmental damage resulting in costly clean up; and
- •
- liabilities under domestic or foreign federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products.
In such an event, we may be held liable for any resulting damages, and any such liability could exceed our resources. Although we carry insurance in amounts and type that we consider commercially reasonable, we cannot be certain that the coverage or coverage limits of our insurance policies will be adequate and we do not have insurance coverage for losses relating to an interruption of our research and development efforts caused by contamination.
We may incur substantial liabilities from any product liability claims if our insurance coverage for those claims is inadequate.
We develop, test and, to a limited extent, manufacture drugs that are used by humans. We face an inherent risk of product liability exposure related to the testing of our drug candidates in clinical trials, and will face an even greater risk if we sell our own drugs commercially. An individual may bring a liability claim against us if one of our drug candidates or drugs causes, or merely appears to have caused, an injury. If we cannot successfully defend ourselves against a product liability claim, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
- •
- decreased demand for our drug;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial subjects;
- •
- costs of related litigation;
- •
- substantial monetary awards to subjects or other claimants;
- •
- loss of revenues; and
- •
- the inability to commercialize our drug candidates.
We have limited product liability insurance that covers our clinical trials. We intend to expand our insurance coverage to include the sale of drugs if marketing approval is obtained for any of our drug candidates. However, insurance coverage is increasingly expensive. We may not be able to obtain or maintain insurance coverage at a reasonable cost, and we may not have insurance coverage that will be adequate to satisfy any liability that may arise.
Damages awarded in a product liability action could be substantial and could have a negative impact on our financial condition. Whether or not we were ultimately successful in product liability litigation, such litigation would consume substantial amounts of our financial and managerial resources, and might result in adverse publicity, all of which would impair our business.
33
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our United States operations, including laboratories, offices and a chemical development facility are located in the same business park in San Diego. We also have a drug product facility that is located in Zofingen, Switzerland. We depend on our facilities and on our collaborators, contractors and vendors for the continued operation of our business. Natural disasters or other catastrophic events, including terrorist attacks, power interruptions, political and governmental changes, wildfires and other fires, actions of animal rights activists, earthquakes and wars could disrupt our operations or those of our collaborators, contractors and vendors. Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors' insurance policies or for which we or our contractors do not have coverage. For example, we are not insured against a terrorist attack. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. Moreover, any such event could delay our research and development programs.
Even if any of our drug candidates receives regulatory approval, our drug candidates will still be subject to extensive post-marketing regulation.
If we or our collaborators receive regulatory approval for our drug candidates in the United States or other jurisdictions, we will also be subject to ongoing obligations and continued regulatory review from the FDA and other applicable regulatory agencies, such as continued adverse event reporting requirements. We may also be subject to additional FDA post-marketing obligations, all of which may result in significant expense and limit our ability to commercialize such drugs in the United States or other jurisdictions.
If any of our drug candidates receive United States regulatory approval or approval in other jurisdictions, the FDA or other regulatory agencies may still impose significant restrictions on the indicated uses for which such drugs may be marketed or impose ongoing requirements for potentially costly post-approval studies. If the FDA or other regulatory agencies approve any of our drug candidates, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the drug will be subject to extensive regulatory requirements. We and the manufacturers of our products are also required to comply with cGMP regulations, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our products, and these facilities are subject to ongoing regulatory inspections. In addition, regulatory agencies subject a drug, its manufacturer and the manufacturer's facilities to continual review and inspections. The subsequent discovery of previously unknown problems with a drug, including adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured, may result in restrictions on the marketing of that drug, up to and including withdrawal of the drug from the market. In the United States, the DEA and comparable state-level agencies also heavily regulate the manufacturing, holding, processing, security recordkeeping and distribution of drugs that are considered controlled substances. If any of our drug candidates are scheduled by the DEA as controlled substances (due to abuse potential), we will become subject to the DEA's regulations. The DEA periodically inspects facilities for compliance with its rules and regulations. If our manufacturing facilities or those of our suppliers fail to comply with applicable regulatory requirements, it could result in regulatory action and additional costs to us. Failure to comply with applicable FDA and other regulatory requirements may, either before or after product approval, if any, subject our company to administrative or judicially imposed sanctions, including:
- •
- issuance of Warning Letters by the FDA or other regulatory agencies;
- •
- imposition of fines and other civil penalties;
34
- •
- criminal prosecutions;
- •
- injunctions, suspensions or revocations of regulatory approvals;
- •
- suspension of any ongoing clinical trials;
- •
- total or partial suspension of manufacturing;
- •
- delays in commercialization;
- •
- refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our collaborators;
- •
- refusals to permit drugs to be imported into or exported from the United States;
- •
- restrictions on operations, including costly new manufacturing requirements; and
- •
- product recalls or seizures.
The FDA's and other regulatory agencies' policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates or further restrict or regulate post-approval activities. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our drugs and our business could suffer.
Even if we receive regulatory approval to market our drug candidates, our ability to generate revenues from any resulting products will be subject to a variety of risks, many of which are out of our control.
Even if our drug candidates obtain regulatory approval, resulting products may not gain market acceptance among physicians, patients, healthcare payors or the medical community. We believe that the degree of market acceptance and our ability to generate revenues from such products will depend on a number of factors, including:
- •
- timing of market introduction of competitive drugs;
- •
- efficacy and safety of our drug candidates;
- •
- prevalence and severity of any side effects;
- •
- potential or perceived advantages or disadvantages over alternative treatments;
- •
- strength of sales, marketing and distribution support;
- •
- price of our future products, both in absolute terms and relative to alternative treatments;
- •
- the effect of current and future healthcare laws on our drug candidates;
- •
- availability of coverage and reimbursement from government and other third-party payors; and
- •
- product labeling or product insert requirements of the FDA or other regulatory authorities.
If our approved drugs, if any, fail to achieve market acceptance, we may not be able to generate significant revenue to achieve or sustain profitability.
Currency fluctuations may negatively affect our financial condition.
We primarily spend and generate cash in US dollars, and present our consolidated financial statements in US dollars. However, a portion of our expected and potential payments and receipts under our agreements are in foreign currencies, including Swiss francs. For example, payments and receipts under our asset purchase agreement, contract manufacturing agreement and long-term API
35
manufacturing agreement with Siegfried Ltd are required to be paid in Swiss francs. A fluctuation of the exchange rates of foreign currencies versus the US dollar may, thus, adversely affect our financial results, including cash balances, expenses and revenues. We may enter into hedging transactions to try to reduce our foreign currency exposure in the future, but there is no assurance that such transactions will occur or be successful.
Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers.
Laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002 and rules adopted by the Securities and Exchange Commission, or SEC, and by the Nasdaq Global Market, as well as the laws and regulations of foreign governments, may result in increased costs to us, particularly as we continue to develop the required capabilities in the United States and abroad to commercialize our products. These laws, rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers. We cannot estimate accurately the amount or timing of additional costs we may incur to respond to these laws, rules and regulations.
The technologies on which we rely may not result in the discovery or development of commercially viable drugs or could become obsolete.
Our GPCR technologies include technologies that allow us to discover drug-like compounds that act on receptor subtypes of known GPCRs and novel GPCRs where the native ligands have not been identified. These methods of identifying, prioritizing and screening molecular targets are unproven, and may not result in the regulatory approval and commercialization of any therapeutic products. We do not believe that there are any drugs on the market that have been discovered or developed using our proprietary technologies. If we are unable to identify additional drug candidates using our proprietary drug discovery technologies, we may not be able to maintain a clinical development pipeline or generate revenues.
Another company, organization or individual could have, or could develop, a technology targeting GPCRs to discover and develop compounds into drugs more effectively or efficiently than our screening and other technologies. Such a technology could render our technologies, in particular our constitutively activated receptor technology, or CART, and Melanophore technology, obsolete or noncompetitive.
Risks Relating to Our Intellectual Property
Our success is dependent on intellectual property rights held by us and third parties and our interest in these rights is complex and uncertain.
Our success will depend on our own and on our collaborators' abilities to obtain, secure and defend patents. In particular, the patents directed to our most advanced drug candidates and other compounds discovered using our technologies or that are otherwise part of our collaborations are important to commercializing drugs. We have numerous United States and foreign patent applications pending for our technologies. There is no assurance that any of our patent applications will issue, or that any of the patents will be enforceable or will cover a drug or other commercially significant technology or method, or that the patents will be held to be valid for their expected terms.
The procedures for obtaining a patent in the United States and in most foreign countries are complex. These procedures require an analysis of the scientific technology related to the invention and
36
many sophisticated legal issues. Obtaining patent rights outside the United States often requires the translation of highly technical documents and an improper translation may lead to the loss of, or otherwise jeopardize, the patent protection of our inventions. Ensuring adequate quality of translators and foreign patent attorneys is often very challenging. Consequently, the process for having our pending patent applications issue as patents will be difficult, complex and time consuming. Our patent position is very uncertain and we do not know when, or if, we will obtain additional patents for our technologies, or if the scope of the patents obtained will be sufficient to protect our drugs.
In addition, other entities may challenge the validity or enforceability of our patents and patent applications in litigation or administrative proceedings. Even the issuance of a patent is not conclusive as to its validity or enforceability. We cannot make assurances as to how much protection, if any, will be given to our patents if we attempt to enforce them or they are challenged. It is possible that a competitor or a generic pharmaceutical provider may successfully challenge our patents and those challenges may result in reduction or elimination of our patents' coverage.
We also rely on confidentiality agreements and trade secrets to protect our technologies. However, such information is difficult to protect. We require our employees to contractually agree not to improperly use our confidential information or disclose it to others, but we may be unable to determine if our employees have conformed or will conform to their legal obligations under these agreements. We also enter into confidentiality agreements with prospective collaborators, collaborators, service providers and consultants, but we may not be able to adequately protect our trade secrets or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of this information. Many of our employees and consultants were, and many of them may currently be, parties to confidentiality agreements with other pharmaceutical and biotechnology companies, and the use of our technologies could violate these agreements. In addition, third parties may independently discover our trade secrets or proprietary information.
Some of our academic institution licensors, research collaborators and scientific advisors have rights to publish data and information to which we have rights. We generally seek to prevent our partners from disclosing scientific discoveries before we have the opportunity to file patent applications on such discoveries. In some of our collaborations, we do not have control over our partners' ability to disclose their own discoveries under the collaboration and in some of our academic collaborations we are limited to relatively short periods to review a proposed publication and file a patent application. If we cannot maintain the confidentiality of our technologies and other confidential information in connection with our collaborations, our ability to receive patent protection or protect our proprietary information will be impaired.
The United States Patent and Trademark Office has recently tried to enact and/or proposed changes in the rules governing (i) the duties of patent applicants to disclose information that relates to their applications, (ii) the ability of patent applicants to file unlimited numbers of patent applications and patent claims that concern closely related inventions and/or different aspects of the same invention, and (iii) the manner in which the United States Patent and Trademark Office will decide whether to require patent applicants to separate closely related inventions into separate patent applications. In addition, the United States Congress is considering a change to the federal laws dealing with patents on several issues including, but not limited to: (i) what types of information can be used to determine whether an invention is not new and, therefore, not patentable, (ii) the limits on the independent administrative rulemaking authority of the United States Patent and Trademark Office, (iii) the duties of patent applicants to disclose information that relates to their applications, (iv) whether, under what circumstances, and how many times a third party will have an opportunity to challenge an issued United States patent before the United States Patent and Trademark Office, (v) whether and under what circumstances patent applicants can lose their ability to enforce their patents in the United States based on their failure to disclose certain information relating to their inventions, and (vi) how damages
37
for patent infringement may be limited and apportioned based on a number of factors including the similarity of a patented invention to pre-existing technologies.
The United States is by far the largest single market for pharmaceuticals in the world, responsible for between 40% and 50% of all such sales. Because of the critical nature of patent rights to the pharmaceutical industry, changes in United States patent rules and laws could have a profound effect on our future profits. Several of the patent rule and law changes that are being considered could significantly weaken patent protections in the United States in general. They may also have a disproportionately large negative impact on the biotechnology and pharmaceutical industries in particular, as well as tilt the balance of market control and distribution of profits between the manufacturers of patented pharmaceutical products and the manufacturers of generic pharmaceutical products towards the generics manufacturers. At present there is considerable uncertainty as to which patent rules and laws will be changed and whether changes to the patent rules will ultimately be enforced or struck down by the courts.
A dispute regarding the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be costly and result in delays or termination of our future research, development, manufacturing and sales activities.
Our commercial success also depends upon our ability to develop and manufacture our drug candidates and market and sell drugs, if any, and conduct our research and development activities without infringing or misappropriating the proprietary rights of others. There are many patents and patent applications filed, and that may be filed, by others relating to drug discovery and development programs that could be determined to be similar, identical or superior to ours or our licensors or collaborators. We may be exposed to future litigation by others based on claims that our drug candidates, technologies or activities infringe the intellectual property rights of others. Numerous United States and foreign issued patents and pending patent applications owned by others exist in the area of GPCRs, including some which purport to allow the patent holder to control the use of all drugs that modulate a particular drug target or GPCR, regardless of whether the infringing drug bears any structural resemblance to a chemical compound known to the patent holder at the time of patent filing. Numerous United States and foreign issued patents and pending patent applications owned by others also exist in the therapeutic areas in, and for the therapeutic targets for, which we are developing drugs. There are also numerous issued patents and patent applications to chemical compounds or synthetic processes that may be necessary or useful to use in our research, development, manufacturing or commercialization activities. These could materially affect our ability to develop our drug candidates or manufacture, import or sell drugs, and our activities, or those of our licensors or collaborators, could be determined to infringe these patents. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our drug candidates or technologies may infringe. There also may be existing patents, of which we are not aware, that our drug candidates or technologies may infringe. Further, there may be issued patents and pending patent applications in fields relevant to our business, of which we are or may become aware, that we believe we do not infringe or that we believe are invalid or relate to immaterial portions of our overall drug discovery, development, manufacturing and commercialization efforts. We cannot assure you that others holding any of these patents or patent applications will not assert infringement claims against us for damages or seeking to enjoin our activities. We also cannot assure you that, in the event of litigation, we will be able to successfully assert any belief we may have as to non-infringement, invalidity or immateriality, or that any infringement claims will be resolved in our favor.
In addition, others may infringe or misappropriate our proprietary rights, and we may have to institute costly legal action to protect our intellectual property rights. We may not be able to afford the costs of enforcing or defending our intellectual property rights against others.
38
Other organizations, companies and individuals are seeking proprietary positions on genomics information that overlap with the government-sponsored project to sequence the human genome. Our activities, or those of our licensors or collaborators, could be affected by conflicting positions that may exist between any overlapping genomics information made available publicly as a result of the government-sponsored project and genomics information that other organizations, companies or individuals consider to be proprietary. There could also be significant litigation and other administrative proceedings in our industry that affect us regarding patent and other intellectual property rights. Any legal action or administrative action against us, or our collaborators, claiming damages or seeking to enjoin commercial activities relating to our drug discovery, development, manufacturing and commercialization activities could:
- •
- require us, or our collaborators, to obtain a license to continue to use, manufacture or market the affected drugs, methods or processes, which may not be available on commercially reasonable terms, if at all;
- •
- prevent us from importing, making, using, selling or offering to sell the subject matter claimed in patents held by others and subject us to potential liability for damages;
- •
- consume a substantial portion of our managerial, scientific and financial resources; or
- •
- be costly, regardless of the outcome.
Furthermore, because of the substantial amount of pre-trial document and witness discovery required in connection with intellectual property litigation, there is risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the trading price of our common stock.
We have been contacted from time to time by third parties regarding their intellectual property rights, sometimes asserting that we may need a license to use their technologies. If we fail to obtain any required licenses or make any necessary changes to our technologies, we may be unable to develop or commercialize some or all of our drug candidates.
We cannot protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our drug discovery technologies and all of our potential drug candidates throughout the world would be prohibitively expensive. Competitors may use our technologies to develop their own drugs in jurisdictions where we have not obtained patent protection. These drugs may compete with our drugs, if any, and may not be covered by any of our patent claims or other intellectual property rights. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, the patent owner has failed to "work" the invention in that country or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our drug candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patents and other intellectual property
39
protection, particularly those relating to biotechnology and/or pharmaceuticals, which makes it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Risks Relating to Our Securities
Our stock price will likely be volatile, and your investment in our stock could decline in value.
Our stock price has fluctuated historically. From January 1, 2006 to February 29, 2008, the market price of our stock was as low as $6.50 per share and as high as $20.68 per share.
Very few drug candidates being tested will ultimately receive FDA approval, and biotechnology or biopharmaceutical companies may experience a significant drop in stock price based on a clinical trial result or regulatory action. Our stock price may fluctuate significantly depending on a variety of factors, including:
- •
- the success or failure of our clinical-stage development programs, or other results or decisions affecting, the development of our drug candidates;
- •
- the timing of the discovery of drug leads and the development of our drug candidates;
- •
- the entrance into a new collaboration or the modification or termination of an existing collaboration;
- •
- the timing and receipt by us of milestone and royalty payments or failing to achieve and receive the same;
- •
- changes in our research and development budget or the research and development budgets of our existing or potential collaborators;
- •
- the introduction of new drug discovery techniques or the introduction or withdrawal of drugs by others that target the same diseases and conditions that we or our collaborators target;
- •
- regulatory actions;
- •
- expenses related to, and the results of, litigation and other proceedings relating to intellectual property rights or other matters;
- •
- financing strategy or decisions; and
- •
- accounting changes.
We are not able to control all of these factors. If our financial or scientific results in a particular period do not meet stockholders' or analysts' expectations, our stock price may decline and such decline could be significant.
Holders of our Series B Preferred can require us to redeem their Series B Preferred.
On December 24, 2003, we completed a private placement of (i) 3,500 shares of our Series B-1 Preferred, (ii) seven-year warrants to purchase 1,486,200 shares of our common stock at an exercise price of $10.00 per share (subject to weighted-average adjustment in certain circumstances) and (iii) unit warrants to purchase $11.5 million of our Series B-2 Preferred and additional seven-year warrants to purchase 450,000 shares of our common stock at an exercise price of $10.00 per share (subject to weighted-average adjustment in certain circumstances). On April 22, 2005, the investors exercised their unit warrants in full.
The holders of our Series B-1 Preferred can require us at any time to redeem all or some of their shares of Series B-1 Preferred at such shares' stated value, plus accrued but unpaid dividends thereon
40
to the date of payment and any applicable penalties. The stated value is the original holder's investment plus any dividends settled by increasing the stated value at the time the dividend is payable. The Series B-1 Preferred, which accrues dividends at 4% annually, had an aggregate redemption price of $41.1 million at December 31, 2007. We expect the aggregate redemption price at the mandatory redemption date of December 24, 2008 to be $42.8 million.
The holders of our Series B-2 Preferred will be entitled to require us to redeem their shares of Series B-2 Preferred at such shares' stated value, plus accrued but unpaid dividends thereon to the date of payment and any applicable penalties if, in the future, the average of the closing prices of our common stock for any 30 consecutive trading days is below $7.00 per share, which is the conversion price for the Series B-2 Preferred. The Series B-2 Preferred, which accrues dividends at 4% annually, had an aggregate redemption price of $12.8 million at December 31, 2007. We expect the aggregate redemption price at the mandatory redemption date of April 22, 2010 to be $14.0 million.
Also, the holders of the Series B-2 Preferred may require us to redeem their shares if we issue common stock or common stock equivalents for an effective net price to us per share less than approximately $5.33 (excluding, among other things, certain common stock and common stock equivalents issued or issuable (i) to our officers, directors, employees or consultants, (ii) in connection with certain strategic partnerships or joint ventures, and (iii) in connection with certain mergers and acquisitions). "Effective net price" is not defined in the Certificate of Designations governing our Series B-2 Preferred. The holders of our Series B-2 Preferred may assert that effective net price should be calculated as the amount we receive after paying any discounts and other expenses related to any such issuance.
At the option of any holder of any Series B Preferred, any Series B Preferred held by such holder may be converted into common stock based on the applicable conversion price then in effect for such shares of Series B Preferred.
In addition to the foregoing redemption rights, at any time following the occurrence of a "Triggering Event," a holder of the Series B Preferred may require us to repurchase all or any portion of the Series B Preferred then held by such holder at a price per share equal to the greater of 115% of the stated value or the market value (as calculated under the Certificate of Designations for the Series B Preferred) of such shares of Series B Preferred plus all accrued but unpaid dividends thereon to the date of payment. "Triggering Event" is specifically defined in the Certificate of Designations for the Series B Preferred, and includes any of the following events (i) immediately prior to a bankruptcy event; (ii) we fail for any reason to timely deliver a certificate evidencing any securities to a purchaser or the exercise or conversion rights of the holders are otherwise suspended for other than a permissible reason; (iii) any of certain events of default (as set forth in the Registration Rights Agreement with the Series B Preferred holders) occur and remain uncured for 60 days; (iv) we fail to make any cash payment required under the Series B Preferred transaction documents and such failure is not timely cured; (v) the issuance of a going concern opinion by our independent registered public accounting firm that is not timely cured; (vi) we breach a section of the Series B Preferred purchase agreement relating to indebtedness and subordination; or (vii) we default in the timely performance of any other obligation under the Series B Preferred transaction documents and such default is not timely cured.
If we are required to redeem all or some of the currently outstanding shares of our Series B Preferred, we may be able to pay all or a portion of the redemption price using shares of our common stock if certain enumerated conditions are satisfied, including:
- •
- we have sufficient number of shares of common stock available for issuance;
- •
- the shares of common stock to be issued are registered under an effective registration statement or are otherwise available for sale under Rule 144(k) under the Securities Act of 1933, as amended, or Securities Act;
41
- •
- our common stock is listed on the Nasdaq Global Market or other eligible market;
- •
- the shares to be issued can be issued without violating the rules of the Nasdaq Global Market or any applicable trading market or a provision of our Certificate of Designations for the Series B Preferred; and
- •
- no bankruptcy event has occurred.
If we are permitted to satisfy all or a portion of a redemption by using shares of our common stock, and if we elect to do so, the number of shares to be issued to holders of Series B Preferred will be determined by dividing their cash redemption price by the lesser of the conversion price or 95% of the average of the volume weighted-average price of our common stock for, depending on the specified circumstances, 10 or 15 consecutive trading days prior to the delivery of the redemption notice or date of the triggering event.
There can be no assurance that if we have to redeem our Series B Preferred, that we will be able to pay a portion of the redemption price using shares of our common stock. If we use common stock to redeem a portion of the Series B Preferred, the ownership interests of the current holders of our common stock may be significantly diluted. If we are required or elect to redeem shares of the Series B Preferred using cash, we may not have sufficient cash to redeem these shares or to continue our planned research and discovery activities. In such event we may try to raise additional capital by issuing new stock, but there can be no assurance that capital will be available on acceptable terms or at all.
There are a substantial number of shares of our common stock eligible for future sale in the public market, and the sale of these shares could cause the market price of our common stock to fall.
There were 73,759,776 shares of our common stock outstanding as of February 29, 2008. The outstanding shares of our Series B-1 Preferred are convertible into up to 5,481,740 shares of common stock at $7.50 per share of common stock. The outstanding shares of our Series B-2 Preferred are convertible into up to 1,829,909 shares of common stock at $7.00 per share of common stock. Holders of Series B Preferred are entitled to receive a 4% annual dividend that is payable by issuing common stock or by increasing the amount of common stock that is issuable upon conversion of the Series B Preferred. In connection with the Series B Preferred financing, we issued warrants to acquire 1,936,200 shares of common stock at an exercise price of $10.00 per share to the two purchasers in our Series B Preferred financing. As of February 29, 2008, 1,106,344 of such warrants were outstanding. Such warrants provide that if the closing price of our common stock is equal to or above $14.00 per share for 30 consecutive trading days, upon 10 trading days' prior written notice, we will have the right to, and the warrant holders will have the right to require us to, call and cancel any unexercised portion of the warrants (subject to certain conditions). Following such a call notice, we would be obligated to issue to the warrant holder an exchange warrant entitling the holder to purchase shares of our common stock equal to the "Call Amount" (as such term is defined in the warrants). This exchange warrant would contain the same terms and conditions as the original warrant, except that the maturity date would be seven years from the date of issuance of such exchange warrant and the exercise price would be equal to 130% of the average of the volume weighted-average price of our common stock for the five trading days preceding the original warrant cancellation date.
On March 31, 2006, following our call notice to one of our two warrant holders, Smithfield Fiduciary LLC, such holder exercised its warrants to purchase 829,856 shares of our common stock. In connection with this exercise in full of its warrants, Smithfield claimed that it was entitled to receive exchange warrants that would include a provision that could require us to issue additional exchange warrants in the future. We disagreed with this interpretation and, on June 30, 2006, we entered into a Settlement Agreement and Release with Smithfield. As part of the Settlement Agreement and Release, (a) Smithfield and we provided each other with a release of any claims relating to (i) Smithfield's demand for, and our non-issuance of, exchange warrants, and (ii) any breach or default under certain
42
of our agreements on account of the foregoing, (b) we issued Smithfield a seven-year warrant to purchase 829,856 shares of our common stock at an initial exercise price of $15.49 per share, and (c) we filed a registration statement covering the sale of the shares of common stock issuable under the new warrant. The new warrant does not contain any right for us, or for the holder to require us, to call the warrant, nor does it provide the holder the right to receive any exchange warrants in the future. As of February 29, 2008, 829,856 of such warrants were outstanding.
In addition, as of February 29, 2008, there were options to purchase 5,551,088 shares of our common stock issued and outstanding under our equity incentive plans at a weighted-average exercise price of $10.41, 1,631,800 performance-based restricted stock unit awards outstanding under our 2006 Long-Term Incentive Plan, as amended, 2,566,532 additional shares of common stock issuable under our 2006 Long-Term Incentive Plan, as amended, 449,594 shares of common stock issuable under our 2001 Employee Stock Purchase Plan, as amended, and 107,919 shares of common stock issuable under our Deferred Compensation Plan. A substantial number of the shares described above, when issued upon exercise, will be available for immediate resale in the public market. The market price of our common stock could decline as a result of such resales due to the increased number of shares available for sale in the market.
Any future equity or debt issuances by us may have dilutive or adverse effects on our existing stockholders.
We have financed our operations, and we expect to continue to finance our operations, primarily by issuing and selling our common stock or securities convertible into or exercisable for shares of our common stock. In light of our need for additional financing, we may issue additional shares of common stock or additional convertible securities that could dilute your ownership in our company and may include terms that give new investors rights that are superior to yours. Moreover, any issuances by us of equity securities may be at or below the prevailing market price of our common stock and in any event may have a dilutive impact on your ownership interest, which could cause the market price of our common stock to decline. The terms of our Series B Preferred limit our ability to engage in certain equity issuances.
We may also raise additional funds through the incurrence of debt, and the holders of any debt we may issue would have rights superior to your rights in the event we are not successful and are forced to seek the protection of bankruptcy laws. The terms of our Series B Preferred limit our ability to incur debt.
Our largest stockholders may take actions that are contrary to your interests, including selling their stock.
A small number of our stockholders hold a significant amount of our outstanding stock. These stockholders may support competing transactions and have interests that are different from yours. Sales of a large number of shares of our stock by these large stockholders or other stockholders within a short period of time could adversely affect our stock price.
We may have disagreements with our warrant holders.
We previously had a disagreement with one of our two warrant holders regarding whether such holder was entitled to receive exchange warrants following the exercise of its warrants in full. Although we entered into a Settlement Agreement and Release with this holder, we may have a similar dispute with the other warrant holder. Moreover, we may be involved with other disagreements with our warrant holders in the future. Such disagreements may lead to litigation which may be expensive and consume management's time, or involve settlements, the terms of which may not be favorable to us.
43
Our rights agreement and certain provisions in our charter documents and Delaware law could delay or prevent a change in management or a takeover attempt that you may consider to be in your best interest.
We have adopted certain anti-takeover provisions, including a stockholders' rights agreement, dated as of October 30, 2002, between us and Computershare Trust Company, Inc., as Rights Agent, as amended. The rights agreement will cause substantial dilution to any person who attempts to acquire us in a manner or on terms not approved by our board of directors.
The rights agreement and Certificate of Designations for the Series B Preferred, as well as other provisions in our certificate of incorporation and bylaws and under Delaware law, could delay or prevent the removal of directors and other management and could make more difficult a merger, tender offer or proxy contest involving us that you may consider to be in your best interest. For example, these provisions:
- •
- allow our board of directors to issue preferred stock without stockholder approval;
- •
- limit who can call a special meeting of stockholders;
- •
- eliminate stockholder action by written consent; and
- •
- establish advance notice requirements for nomination for election to the board of directors or for proposing matters to be acted upon at stockholders meetings.
Item 1B. Unresolved Staff Comments.
None.
Item 2. Properties.
As set forth in the below table, the principal facilities that we occupy include approximately 268,000 square feet of research, development, warehouse and office space located at various addresses in the same business park on Nancy Ridge Drive in San Diego, California and approximately 67,000 square feet of manufacturing, warehouse and office space located in Zofingen, Switzerland.
Location
| | Own/
Lease
| | Description
|
|---|
| 6114 Nancy Ridge Drive | | Lease with option to purchase | | This chemical development facility consists of approximately 40,000 square feet (which includes approximately 18,000 of internal square feet and approximately 22,000 square feet of integrated external space), of which approximately 5,000 square feet is office space. The remaining approximately 35,000 square feet of space is dedicated to process research and scale-up chemistry, the production of intermediates and other compounds for research and development purposes, and the production of active pharmaceutical ingredients to support our clinical trials. We are using this facility for the production of scale-up lots for our internal research programs, safety studies and clinical trials. We commenced cGMP operations in this facility in the second quarter of 2004. In May 2007, we completed a sale and leaseback of this facility, and have an option to purchase it back. |
6118 Nancy Ridge Drive |
|
Lease with option to purchase |
|
This facility of approximately 30,000 square feet consists of approximately 50% laboratory space and 50% office space. In May 2007, we completed a sale and leaseback of this facility, and have an option to purchase it back. |
44
6122-6124-6126 Nancy Ridge Drive |
|
Lease with option to purchase |
|
The portion of this facility we lease consists of approximately 40,000 square feet, of which approximately 24,000 square feet is laboratory space and 16,000 square feet is office space. We sublease to another company approximately 2,000 square feet of office space in this facility. We have assigned our option to purchase the entire facility, which includes approximately 68,000 square feet, and have an option to purchase it back. |
6138-6150 Nancy Ridge Drive |
|
Lease with option to purchase |
|
This facility of approximately 55,000 square feet consists of approximately 33,000 square feet of laboratory space and 22,000 square feet of office space. In December 2003, we completed a sale and leaseback of this facility, and have an option to purchase it back. |
6154 Nancy Ridge Drive |
|
Lease with option to purchase |
|
This facility of approximately 68,000 square feet consists of approximately 56,000 square feet of office space and 12,000 square feet of warehouse space. We are in the process of improving and substantially expanding this facility by approximately 75,000 square feet. In May 2007, we completed a sale and leaseback of this facility, and have an option to purchase it back. |
6162 Nancy Ridge Drive |
|
Own |
|
This facility includes approximately 20,000 square feet of warehouse and office space. We are leasing this facility to another company through April 2008, and the lease can be extended at the tenant's option in monthly increments through July 2008. |
6166 Nancy Ridge Drive |
|
Lease |
|
This facility of approximately 37,000 square feet consists of approximately 23,000 square feet of laboratory space and 14,000 square feet of office space. |
Zofingen, Switzerland |
|
Own |
|
This facility of approximately 67,000 square feet consists of approximately 35,000 square feet of manufacturing space, 25,000 square feet of warehouse space and 7,000 square feet of office space. |
We expect to need additional space depending on the success of our clinical programs and whether we partner or internally develop our programs.
Item 3. Legal Proceedings.
None.
Item 4. Submission of Matters to a Vote of Security Holders.
No matters were submitted to a vote of security holders during the fourth quarter of the fiscal year covered by this Annual Report on Form 10-K.
45
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market information
Our common stock is listed on the NASDAQ Global Market under the symbol "ARNA." The following table sets forth, for the periods indicated, the high and low sale prices for our common stock as reported by the NASDAQ Global Market and its predecessor, the NASDAQ National Market.
| | High
| | Low
|
|---|
| Year ended December 31, 2006 | | | | | | |
| | First Quarter | | $ | 20.68 | | $ | 14.21 |
| | Second Quarter | | $ | 18.19 | | $ | 10.26 |
| | Third Quarter | | $ | 12.97 | | $ | 9.18 |
| | Fourth Quarter | | $ | 17.69 | | $ | 11.93 |
| | High
| | Low
|
|---|
| Year ended December 31, 2007 | | | | | | |
| | First Quarter | | $ | 14.58 | | $ | 9.96 |
| | Second Quarter | | $ | 14.74 | | $ | 10.34 |
| | Third Quarter | | $ | 14.78 | | $ | 10.56 |
| | Fourth Quarter | | $ | 11.39 | | $ | 7.76 |
Holders
As of February 29, 2008, there were approximately 169 stockholders of record of our common stock, one of which is Cede & Co., a nominee for Depository Trust Company, or DTC. Shares of common stock that are held by financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and are considered to be held of record by Cede & Co. as one stockholder.
Dividends
We have never paid cash dividends on our capital stock. We anticipate that we will retain earnings, if any, to support operations and finance the growth and development of our business and, therefore, do not expect to pay cash dividends in the foreseeable future. In addition, we are prohibited from paying cash dividends on any of our capital stock other than our series B redeemable convertible preferred stock without the approval of the holders of our series B redeemable convertible preferred stock.
46
Securities authorized for issuance under equity compensation plans
The following table summarizes our compensation plans under which our equity securities are authorized for issuance as of December 31, 2007:
Plan category
| | Number of securities to be
issued upon exercise of
outstanding options, warrants
and rights
| | Weighted-average
exercise price of
outstanding options,
warrants and rights
| | Number of securities remaining
available for future issuance
under equity compensation plans
(excluding securities reflected in
column (a))
| |
|---|
| | (a)
| | (b)
| | (c)
| |
|---|
| Equity compensation plans approved by security holders* | | 7,149,602 | | $ | 8.05 | | 3,060,452 | ** |
| Equity compensation plans not approved by security holders | | — | | | — | | — | |
| | |
| |
| |
| |
| Total* | | 7,149,602 | | $ | 8.05 | | 3,060,452 | ** |
| | |
| |
| |
| |
- *
- Includes stock options with a per share weighted-average exercise price of $10.43 and performance-based restricted stock unit awards which have no per share weighted-average exercise price.
- **
- Includes 449,594 shares of common stock available for future issuance under our 2001 Employee Stock Purchase Plan, as amended.
In 2003, we set up a deferred compensation plan for our executive officers, whereby executive officers may elect to defer their shares of restricted stock. At December 31, 2007, a total of 107,919 shares of restricted stock were in the plan. All of the shares contributed to this plan were previously granted to executive officers under an equity compensation plan approved by our stockholders.
On March 3, 2008, the Compensation Committee of our board of directors granted 1,155,600 stock options to employees, including executive officers, and directors. On March 3, 2008, the Compensation Committee also granted 371,800 performance-based restricted stock unit awards to certain employees, substantially all of whom were not previously granted performance-based restricted stock unit awards, and none of which were granted to executive officers. The stock options, which generally vest 25% per year over four years and are exercisable for up to 10 years from the date of grant, had an exercise price equal to the fair market value of our stock on the date of grant, which was $6.99 per share.
47
Item 6. Selected Financial Data.
The following Selected Financial Data should be read in conjunction with "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Item 8. Financial Statements and Supplementary Data" included below in this Annual Report on Form 10-K.
| | Years ended December 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2004
| | 2003
| |
|---|
| | (In thousands, except share and per share data)
| |
|---|
| Revenues | | | | | | | | | | | | | | | | |
| Collaborative agreements | | $ | 19,332 | | $ | 30,569 | | $ | 23,233 | | $ | 13,686 | | $ | 12,734 | |
| Collaborative agreements with affiliates | | | — | | | — | | | — | | | — | | | 100 | |
| | |
| |
| |
| |
| |
| |
| | Total revenues | | | 19,332 | | | 30,569 | | | 23,233 | | | 13,686 | | | 12,834 | |
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Research and development | | | 149,524 | | | 103,388 | | | 79,710 | | | 58,579 | | | 52,867 | |
| General and administrative | | | 26,571 | | | 18,466 | | | 13,122 | | | 11,066 | | | 9,808 | |
| Amortization of acquired technology | | | 1,537 | | | 1,537 | | | 1,537 | | | 1,825 | | | 1,621 | |
| | |
| |
| |
| |
| |
| |
| | Total operating expenses | | | 177,632 | | | 123,391 | | | 94,369 | | | 71,470 | | | 64,296 | |
| Interest and other income (expense), net | | | 15,134 | | | 6,574 | | | 3,235 | | | (208 | ) | | 4,403 | |
| | |
| |
| |
| |
| |
| |
| Net loss | | | (143,166 | ) | | (86,248 | ) | | (67,901 | ) | | (57,992 | ) | | (47,059 | ) |
| Dividends on redeemable convertible preferred stock | | | (2,114 | ) | | (2,031 | ) | | (1,813 | ) | | (1,437 | ) | | (27 | ) |
| Accretion of discount on redeemable convertible preferred stock | | | — | | | — | | | (7,372 | ) | | (1,852 | ) | | (36 | ) |
| | |
| |
| |
| |
| |
| |
| Net loss allocable to common stockholders | | $ | (145,280 | ) | $ | (88,279 | ) | $ | (77,086 | ) | $ | (61,281 | ) | $ | (47,122 | ) |
| | |
| |
| |
| |
| |
| |
| Net loss per share allocable to common stockholders, basic and diluted | | $ | (2.31 | ) | $ | (1.89 | ) | $ | (2.24 | ) | $ | (2.40 | ) | $ | (1.74 | ) |
| | |
| |
| |
| |
| |
| |
| Shares used in calculating net loss per share allocable to common stockholders, basic and diluted | | | 62,782,850 | | | 46,750,596 | | | 34,377,693 | | | 25,527,617 | | | 27,159,234 | |
| | |
| |
| |
| |
| |
| |
48
| | As of December 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| | 2004
| | 2003
| |
|---|
| | (In thousands)
| |
|---|
| | Balance Sheet Data: | | | | | | | | | | | | | | | | |
| Cash and cash equivalents | | $ | 386,989 | | $ | 373,044 | | $ | 73,781 | | $ | 58,686 | | $ | 60,472 | |
| Short-term investments, available-for-sale | | | 11,196 | | | 15,781 | | | 54,158 | | | 54,628 | | | 93,545 | |
| Accounts receivable | | | 1,901 | | | 310 | | | 848 | | | 22,590 | | | 28 | |
| Total assets | | | 487,506 | | | 468,465 | | | 198,129 | | | 206,365 | | | 229,898 | |
| Total deferred revenues | | | 4,049 | | | 13,054 | | | 24,144 | | | 30,070 | | | 3,973 | |
| Total lease financing obligations | | | 62,307 | | | 13,678 | | | 13,485 | | | 13,259 | | | 13,000 | |
| Redeemable convertible preferred stock | | | 53,922 | | | 51,808 | | | 49,777 | | | 29,092 | | | 25,776 | |
| Deferred compensation | | | — | | | — | | | (396 | ) | | (780 | ) | | (2,648 | ) |
| Accumulated deficit | | | (479,451 | ) | | (334,171 | ) | | (245,892 | ) | | (168,806 | ) | | (107,525 | ) |
| Total stockholders' equity | | | 336,377 | | | 366,115 | | | 99,540 | | | 126,723 | | | 183,148 | |
49
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis in conjunction with "Item 8. Financial Statements and Supplementary Data" included below in this Annual Report on Form 10-K, or Annual Report. Operating results are not necessarily indicative of results that may occur in future periods.
This discussion and analysis contains forward-looking statements that involve a number of risks, uncertainties and assumptions. Actual events or results may differ materially from our expectations. Important factors that could cause actual results to differ materially from those stated or implied by our forward-looking statements include, but are not limited to, those set forth in "Item 1A. Risk Factors" in this Annual Report. All forward-looking statements included in this Annual Report are based on information available to us as of the time we file this Annual Report and, except as required by law, we undertake no obligation to update publicly or revise any forward-looking statements.
OVERVIEW
We have incurred net losses of $479.5 million from our inception in April 1997 through December 31, 2007, and expect to incur substantial and increasing net losses for the next several years or more as we continue our research and development activities, including our clinical program for our lead drug candidate, lorcaserin hydrochloride, or lorcaserin, for the treatment of obesity, and our clinical programs for APD125 for the treatment of insomnia and APD791 for the treatment of arterial thromboembolic diseases. We expect that the external expenses for our ongoing Phase 3 lorcaserin program, the majority of which we expect will be expensed through the first half of 2009, will be substantial. To date, we have generated cash and funded our operations primarily through the sale of common and preferred stock, payments from collaborators and sale leaseback transactions. From our inception through December 31, 2007, we have generated $1.0 billion in cash from these sources, of which $839.6 million was through sales of stock, $140.0 million was through payments from our current and past collaborators and $61.1 million was from sale leaseback transactions.
Recent 2008 and 2007 highlights include:
- •
- In January 2008, announced the initiation of a Phase 1 clinical trial of a second generation oral niacin receptor agonist intended for the treatment of atherosclerosis under our partnership with Merck & Co., Inc., or Merck.
- •
- In January 2008, entered into strategic cooperation agreements with Siegfried Ltd that are primarily related to the manufacturing of lorcaserin, which is expected to be necessary for our planned New Drug Application submission to the United States Food and Drug Administration, or FDA, and for commercialization of lorcaserin after regulatory marketing approval. The agreements include a long-term supply agreement for our purchase of lorcaserin active pharmaceutical ingredient, or API, the purchase of certain drug product facility assets, including fixtures, equipment, other personal property and real estate assets, a contract manufacturing agreement whereby we will manufacture certain products for Siegfried, and a services agreement.
- •
- In January 2008, reported positive Phase 1a clinical trial results of APD791, our oral, internally discovered drug candidate intended for the treatment and prevention of arterial thromboembolic diseases, and initiated a Phase 1b clinical trial to further evaluate this drug candidate.
- •
- In January 2008, announced that initial clinical trial results for APD668, an oral drug candidate discovered by Arena and investigated for the treatment of type 2 diabetes under our partnership with Ortho-McNeil Pharmaceutical, Inc., a Johnson & Johnson company, or Ortho-McNeil, suggest that the Glucose-Dependent Insulinotropic Receptor, or GDIR, may improve glucose control in patients with type 2 diabetes. Based on the data from those studies, Ortho-McNeil put APD668 on hold and has advanced a potentially more potent Arena-discovered GDIR agonist into preclinical development.
50
- •
- In December 2007, initiated BLOSSOM and BLOOM-DM, the second and third Phase 3 clinical trials evaluating the efficacy and safety of lorcaserin for the treatment of obesity. These one-year, double-blind, randomized and placebo-controlled trials are expected to collectively enroll approximately 3,750 overweight and obese patients. Consistent with our proposal, the FDA has allowed us to eliminate the requirement to perform echocardiographic testing prior to enrolling patients in both of these trials.
- •
- In November 2007, completed a public offering of 11.0 million shares of our common stock at $9.91 per share, resulting in net proceeds of $103.2 million.
- •
- In September 2007, announced positive preliminary results from our Phase 2a clinical trial of APD125 in patients with chronic insomnia. In this Phase 2a clinical trial, APD125 significantly improved endpoints measuring improvements in sleep maintenance with no observations of next day cognitive impairment.
- •
- In September 2007, reported that an independent Echocardiographic Data Safety Monitoring Board, or ESMB, found no reason to stop our ongoing pivotal Phase 3 lorcaserin BLOOM trial following a planned review of unblinded echocardiograms performed after patients completed six months of dosing in the trial. The review confirmed that differences, if any, in the rates of FDA-defined valvulopathy in patients treated with lorcaserin and in the control group did not meet predetermined stopping criteria. The review also confirmed that the rate of FDA-defined valvulopathy is consistent with our statistical powering assumptions used in the design of the Phase 3 clinical trial program to monitor patients for any increased risk of developing valvulopathy.
- •
- In May 2007, completed the sale to an affiliate of BioMed Realty Trust, Inc., or BioMed, of three properties owned and occupied by us and the assignment to BioMed of an option to purchase a fourth property currently leased and primarily occupied by us. We received net proceeds of $48.5 million for the properties and the purchase option. Concurrently with the closing of the transaction, we leased back the three properties sold to BioMed under leases with 20-year terms and two consecutive options to extend such terms for five years each. As part of the transaction, we also retained the option to purchase from BioMed all the properties included in the transaction on the 10th, 15th or 20th anniversary of the execution date of the leases.
- •
- In February 2007, completed patient enrollment in our BLOOM trial, a double-blind, randomized and placebo-controlled trial that enrolled over 3,100 patients at approximately 100 sites in the United States.
We will need to raise a substantial amount of cash to continue to develop our drug candidates and sustain our research efforts. At December 31, 2007, we had $398.2 million in cash, cash equivalents and short-term investments. The drug development process is long, uncertain and expensive, and our ability to achieve our goals depends on numerous factors, many of which are out of our control. We will seek to balance the need to invest heavily in research to find new drugs and in clinical development and manufacturing to advance our drug candidates against the need to sustain our operations long enough for our collaborators or us to commercialize the results of our efforts. As a result, we expect to continue to incur significant and increasing losses over the next several years. We do not expect to generate positive operating cash flows for at least several years and, accordingly, we will need to raise additional funds through equity, debt or other financing, or through partnering one or more of our more advanced programs. Our cash used in operations is expected to increase as we continue our clinical-stage programs and our research efforts, continue to incur general and administrative expenses, including prosecuting patents, and have reached the end of the research funding portion of our collaborations with Ortho-McNeil and Merck in the fourth quarter of 2007. Absent any new collaboration, we expect to recognize no revenues from research funding in 2008 and thereafter.
51
SUMMARY OF REVENUES AND EXPENSES
We are providing the following summary of our revenues and expenses to supplement the more detailed discussion below. The following tables are stated in millions.
Revenues
| | Years ended December 31,
|
|---|
Collaborations
|
|---|
| | 2007
| | 2006
| | 2005
|
|---|
| Ortho-McNeil | | $ | 13.4 | | $ | 18.5 | | $ | 13.4 |
| Merck | | | 5.9 | | | 12.1 | | | 9.8 |
| | |
| |
| |
|
| | Total revenues | | $ | 19.3 | | $ | 30.6 | | $ | 23.2 |
| | |
| |
| |
|
Research and development expenses
| | Years ended December 31,
|
|---|
Type of expense
|
|---|
| | 2007
| | 2006
| | 2005
|
|---|
| External preclinical and clinical study fees and expenses | | $ | 73.5 | | $ | 40.4 | | $ | 30.2 |
| Personnel costs | | | 43.4 | | | 34.0 | | | 25.4 |
| Facility and equipment costs | | | 15.1 | | | 13.3 | | | 11.8 |
| Research supplies | | | 12.3 | | | 12.2 | | | 10.5 |
| Other | | | 5.2 | | | 3.5 | | | 1.8 |
| | |
| |
| |
|
| | Total research and development expenses | | $ | 149.5 | | $ | 103.4 | | $ | 79.7 |
| | |
| |
| |
|
General and administrative expenses
| | Years ended December 31,
|
|---|
Type of expense
|
|---|
| | 2007
| | 2006
| | 2005
|
|---|
| Personnel costs | | $ | 13.1 | | $ | 8.8 | | $ | 6.2 |
| Legal, accounting and other professional fees | | | 8.7 | | | 6.0 | | | 4.0 |
| Facility and equipment costs | | | 3.0 | | | 2.4 | | | 1.9 |
| Other | | | 1.8 | | | 1.3 | | | 1.0 |
| | |
| |
| |
|
| | Total general and administrative expenses | | $ | 26.6 | | $ | 18.5 | | $ | 13.1 |
| | |
| |
| |
|
YEAR ENDED DECEMBER 31, 2007 COMPARED TO YEAR ENDED DECEMBER 31, 2006
Revenues. We recorded revenues of $19.3 million during the year ended December 31, 2007, compared to $30.6 million during the year ended December 31, 2006. All of our revenues recorded during the year ended December 31, 2007 resulted from our collaborations with Ortho-McNeil and Merck, and included $9.5 million in amortization of milestone achievements and technology access and development fees received in prior years, $5.9 million in research funding, and $3.9 million for patent activities. All of our revenues during the year ended December 31, 2006 were also from our collaborations with Ortho-McNeil and Merck, and included a $5.0 million milestone earned under our Ortho-McNeil collaboration and a $4.0 million milestone earned under our Merck collaboration, both of which we recognized immediately in accordance with our revenue recognition policy, $9.6 million in amortization of milestone achievements and technology access and development fees, $8.1 million in research funding, and $3.9 million in additional sponsored research and patent activities.
In October 2004, we extended and expanded the collaboration we entered into with Merck in 2002, and Merck purchased $7.5 million of our stock at a price of $8.00 per share, approximately a 70%
52
premium to the then current market price. We performed an evaluation on this stock purchase and determined that $3.9 million of the $7.5 million purchase price was an upfront payment related to the collaboration extension and expansion. Accordingly, we recognized the $3.9 million upfront payment, as well as the remaining portion of the unamortized upfront payment at October 2004 of $1.3 million, over the extended research portion of the collaboration term of three years. Additionally, in October 2004, we achieved a $1.0 million milestone under this collaboration which was also recognized over the extended term of the research portion of the collaboration because it was reasonably assured to be achieved at the time we extended and expanded the collaboration. In February 2007, we amended the collaboration to reduce the number of Arena research employees funded under the collaboration in exchange for Merck purchasing $1.0 million of our common stock. This equity investment, equal to the reduction in their research funding obligation, was at a price of $24.81 per share, approximately a 70% premium to the then current market price. We performed an evaluation on this stock purchase and determined that $0.5 million of the $1.0 million purchase price was an upfront payment related to the collaboration amendment. Accordingly, we recognized this upfront payment and the unamortized portion of the previously received upfront payments over the remaining term of the research portion of the collaboration. The research portion of this collaboration ended in October 2007.
In December 2004, we entered into our collaboration and license agreement with Ortho-McNeil. This collaboration included a $17.5 million upfront payment, as well as research funding of $2.4 million per year, initially until December 2006 and subsequently extended until December 2007. We amortized this $17.5 million upfront payment over three years. In December 2004, we achieved two milestones of $2.5 million each under this collaboration, which we also recognized over three years because they were reasonably assured to be achieved at the time we entered into the collaboration. The research portion of this collaboration ended in December 2007.
Our collaborators often pay us before we recognize such payments as current revenues and, accordingly, these payments are recorded as deferred revenues until earned. As of December 31, 2007, we had $4.0 million in deferred revenues, all of which is attributable to our license agreement with TaiGen Biotechnology Co., Ltd. and is expected to be recognized as revenue in 2009. Absent any new collaboration, we do not expect to record any revenues from research funding in 2008. Future revenues for research or clinical milestones that have not yet been achieved are difficult to predict, and our revenues may vary significantly from quarter to quarter and year to year. We expect that any significant revenues over the next several years will depend on the clinical success of our partnered programs as well as whether we partner lorcaserin, APD125, APD791 or any of our other current or future drug candidates. Ultimately, we expect our future revenues in the long term to primarily depend upon the regulatory approval and commercialization of our partnered or internally developed drugs.
Research and development expenses. Research and development expenses, which account for the majority of our expenses, consisted primarily of costs associated with external clinical and preclinical study fees, manufacturing costs and other related expenses, and the development of our earlier-stage programs and technologies. Our most significant research and development costs are for clinical trials (including payments to contract research organizations, or CROs), preclinical study fees, personnel costs, research supplies, and facility and equipment costs. We expense research and development costs to operations as they are incurred when these expenditures relate to our research and development efforts and have no alternative future uses. In the fourth quarter of 2007, we expensed $4.6 million of lorcaserin clinical drug supply for which we previously had an alternative future use. As of December 31, 2007, we had no capitalized research and development costs. Other than external expenses for our clinical and preclinical programs, we generally do not track our research and development expenses by project; rather, we track such expenses by the type of cost incurred.
Research and development expenses for the year ended December 31, 2007 increased $46.1 million to $149.5 million, from $103.4 million for the year ended December 31, 2006. The difference was due primarily to (i) a $33.1 million increase in external clinical and preclinical study fees and expenses,
53
including manufacturing costs, as we continued the first of our three Phase 3 clinical trials and initiated the second and third clinical trials for lorcaserin, and completed a Phase 2a clinical trial of APD125 and a Phase 1a clinical trial of APD791, and (ii) an increase in personnel costs of a total of $9.4 million as we increased the number of our research and development employees from 301 at the end of 2006 to 349 at the end of 2007 and recorded an increase of $1.3 million to $4.2 million in non-cash, share-based compensation related to the expensing of share-based compensation under Statement of Financial Accounting Standards, or SFAS, No. 123R, "Share-Based Payment." Included in the $73.5 million in external clinical and preclinical study fees and expenses for the year ended December 31, 2007 was $51.3 million related to our lorcaserin program, $15.7 million related to our APD125 program and $3.1 million related to our APD791 program. Included in the $40.4 million in external clinical and preclinical study fees and expenses for the year ended December 31, 2006 was $30.2 million related to our lorcaserin program, $4.9 million related to our APD125 program and $2.9 million related to our APD791 program. Nearly all of the increase in research and development personnel related to the development of our internal programs, primarily lorcaserin, APD125 and APD791. Assuming favorable results from our month-12 ESMB review of BLOOM, we expect to continue to incur significant research and development expenses as we continue our ongoing and planned clinical development.
Cumulatively through December 31, 2007, we have recorded $106.9 million, $29.7 million and $6.1 million in external clinical and preclinical study fees and other related expenses for lorcaserin, APD125 and APD791, respectively. While expenditures on current and future clinical development programs are expected to be substantial and to increase, they are subject to many uncertainties, including whether we develop our drug candidates with a partner or independently. As a result of such uncertainties, we cannot predict with any significant degree of certainty the duration and completion costs of our research and development projects or whether, when and to what extent we will generate revenues from the commercialization and sale of any of our product candidates. The duration and cost of clinical trials may vary significantly over the life of a project as a result of unanticipated events arising during clinical development and a variety of factors, including:
- •
- the number of trials and studies in a clinical program;
- •
- the number of patients who participate in the trials;
- •
- the number of sites included in the trials;
- •
- the rates of patient recruitment and enrollment;
- •
- the duration of patient treatment and follow-up;
- •
- the costs of manufacturing our drug candidates; and
- •
- the costs, requirements, timing of, and the ability to secure regulatory approvals.
However, based upon our current plans, we expect to incur $114.0 million to $124.0 million in external clinical and preclinical study fees and other related expenses in 2008, which includes $90.0 million, $18.0 million and $3.0 million for lorcaserin, APD125 and APD791, respectively. This assumes that we continue all three of the ongoing Phase 3 clinical trials of lorcaserin, as well as the clinical development of APD125 and APD791 in 2008. We do not expect to receive regulatory approval for any of our drug candidates until late 2010 at the earliest, if at all.
General and administrative expenses. General and administrative expenses for the year ended December 31, 2007 increased $8.1 million to $26.6 million, from $18.5 million for the year ended December 31, 2006. This increase was due primarily to (i) personnel costs increasing by a total of $4.3 million as we increased our general and administrative employees from 54 at the end of 2006 to 68 at the end of 2007 and recorded an increase of $2.5 million to $4.6 million in non-cash, share-based compensation under SFAS No. 123R, and (ii) an increase of $1.9 million in patent costs primarily
54
related to our partnered programs. To the extent our partners reimburse us for patent costs, the reimbursements are classified as revenues. Such reimbursements totaled $3.9 million in 2007 and $2.1 million in 2006. We expect partner reimbursements for patent costs will be significantly lower in 2008 than in 2007. We also expect that our general and administrative expenses will be higher in the future due primarily to increases in the number of personnel, as well as commercialization, marketing and business development expenses.
Amortization of acquired technology. We recorded $1.5 million for amortization of acquired technology for both of the years ended December 31, 2007 and 2006 related to our patented Melanophore technology, our primary screening technology, which we acquired in 2001 for $15.4 million. The Melanophore technology is being amortized over its estimated useful life of 10 years. We expect to recognize $1.5 million in each of the next three years for amortization of this technology.
Interest and other income, net. Interest and other income, net, totaled $15.1 million for the year ended December 31, 2007, compared to $6.6 million for the year ended December 31, 2006. Interest and other income, net, for the year ended December 31, 2007 was comprised primarily of (i) $18.8 million in interest income and (ii) interest expense and financing costs of $3.7 million, which included lease payments accounted for in accordance with SFAS No. 66 "Accounting for Sales of Real Estate" and SFAS No. 98 "Accounting for Leases" on our lease financing obligations. Interest and other income, net, for the year ended December 31, 2006 was comprised primarily of (i) $12.7 million in interest income, (ii) a $4.6 million non-cash charge related to a warrant issued as part of a settlement with one of our warrant holders, and (iii) interest expense and financing costs of $1.8 million. The increased interest income resulting from higher cash balances throughout 2007 was partially offset by increased interest expense recorded in connection with our 2007 lease financing. Due to declining interest rates and lower cash balances due to our ongoing and planned clinical development, we expect our 2008 interest income to be less than 2007.
Dividends on redeemable convertible preferred stock. We recorded a dividend expense of $2.1 million related to our series B redeemable convertible preferred stock, or Series B Preferred, for the year ended December 31, 2007, compared to $2.0 million for the year ended December 31, 2006. The holders of our Series B Preferred are entitled to dividends that accrue at 4% annually. This dividend expense, which may be paid in common stock or by increasing the stated value of the Series B Preferred, increases the net loss allocable to common stockholders. Assuming that the Series B Preferred is held until the applicable mandatory redemption dates, we expect to record dividends on the Series B Preferred of $2.2 million, $0.5 million and $0.2 million for the years ending December 31, 2008, 2009 and 2010, respectively.
YEAR ENDED DECEMBER 31, 2006 COMPARED TO YEAR ENDED DECEMBER 31, 2005
Revenues. We recorded revenues of $30.6 million during the year ended December 31, 2006, compared to $23.2 million during the year ended December 31, 2005. All of our revenues during the year ended December 31, 2006 were from our collaborations with Ortho-McNeil and Merck, and included a $5.0 million milestone earned under our Ortho-McNeil collaboration and a $4.0 million milestone earned under our Merck collaboration, both of which we recognized immediately in accordance with our revenue recognition policy. Also included in our revenues during the year ended December 31, 2006 was $9.6 million in amortization of milestone achievements and technology access and development fees, $8.1 million in research funding, and $3.9 million in additional sponsored research and patent activities. All of our revenues during the year ended December 31, 2005 were also from our collaborations with Ortho-McNeil and Merck, and included a $2.0 million milestone earned under our Merck collaboration and recognized immediately in accordance with our revenue recognition policy, $9.6 million in amortization of milestone achievements and technology access and development
55
fees, $8.1 million in research funding, and $3.5 million in additional sponsored research and patent activities.
Research and development expenses. Research and development expenses for the year ended December 31, 2006 increased $23.7 million to $103.4 million, from $79.7 million for the year ended December 31, 2005. The difference was due primarily to (i) external clinical and preclinical study fees and expenses, including manufacturing costs, increasing by $10.2 million as we initiated our larger and more costly Phase 3 clinical program for lorcaserin during the third quarter of 2006 and continued to advance APD791 closer to clinical development, and (ii) personnel costs increasing by a total of $8.6 million as we increased the number of our research and development employees from 266 at the end of 2005 to 301 at the end of 2006 and recorded an additional $2.9 million in non-cash, share-based compensation related to the expensing of share-based compensation under SFAS No. 123R. Included in the $40.4 million in external clinical and preclinical study fees and expenses for the year ended December 31, 2006 was $30.2 million related to our lorcaserin program, $4.9 million related to our APD125 program and $2.9 million related to our APD791 program. Included in the $30.2 million in external clinical and preclinical study fees and expenses for the year ended December 31, 2005 was $20.2 million related to our lorcaserin program and $6.7 million related to our APD125 program. Nearly all of the increase in research and development personnel related to the development of our internal programs, primarily lorcaserin, APD125 and APD791.
General and administrative expenses. General and administrative expenses for the year ended December 31, 2006 increased $5.4 million to $18.5 million, from $13.1 million for the year ended December 31, 2005. This increase was due primarily to (i) personnel costs increasing by a total of $2.6 million as we increased our general and administrative employees from 48 at the end of 2005 to 54 at the end of 2006 and recorded an additional $2.1 million in non-cash, share-based compensation under SFAS No. 123R, and (ii) an increase of $1.6 million in patent costs related to our partnered programs and our internal programs and technologies. To the extent our partners reimburse us for patent costs, the reimbursements are classified as revenues. Such reimbursements totaled $2.1 million in 2006 and $1.1 million in 2005.
Amortization of acquired technology. We recorded $1.5 million for amortization of acquired technology for both of the years ended December 31, 2006 and 2005 related to our patented Melanophore technology.
Interest and other income, net. Interest and other income, net, totaled $6.6 million for the year ended December 31, 2006, compared to $3.2 million for the year ended December 31, 2005. Interest and other income, net, for the year ended December 31, 2006 was comprised primarily of (i) $12.7 million in interest income, (ii) a $4.6 million non-cash charge related to a warrant issued as part of a settlement with one of our warrant holders, and (iii) interest expense and financing costs of $1.8 million. Interest and other income, net, for the year ended December 31, 2005 was comprised primarily of (i) $4.4 million in interest income, (ii) interest expense and financing costs of $1.8 million, and (iii) a $0.5 million payment received for the termination of our Fujisawa collaboration and classified as other income. The increase in interest income in the year ended December 31, 2006 was the result of both higher cash balances from the two public offerings we completed in 2006 and higher average interest rates in 2006 compared to 2005.
Dividends on redeemable convertible preferred stock. We recorded a dividend expense of $2.0 million related to our Series B Preferred for the year ended December 31, 2006, compared to $1.8 million for the year ended December 31, 2005. In April 2005, we issued an additional $11.5 million in redeemable convertible preferred stock as a result of the preferred stockholders' exercise of their unit warrants.
56
Accretion of discount on redeemable convertible preferred stock. We recorded as an expense accretion of discount and deemed dividend on our redeemable convertible preferred stock in the amount of $7.4 million for the year ended December 31, 2005 in accordance with Emerging Issues Task Force, or EITF, Issue No. 00-27, "Application of Issue No. 98-5 to Certain Convertible Instruments." We allocated the total proceeds received in our preferred stock financing among the series B-1 redeemable convertible preferred stock, or Series B-1 Preferred, and the related warrants and unit warrants, estimating the value of the warrants and unit warrants at $6.5 million using the Black-Scholes method. The fair value of the common stock into which the redeemable convertible preferred stock was convertible into on the date of issuance exceeded the proceeds allocated to the redeemable convertible preferred stock by $2.8 million, resulting in a beneficial conversion feature that we recognized as an increase to paid-in capital and as a deemed dividend to the redeemable convertible preferred stock. As a result of the public offering we completed in February 2005, which resulted in the Series B-1 Preferred becoming immediately redeemable at the option of the holders, we recorded a charge in the first quarter of 2005 of $7.4 million to accrete the remaining unaccreted discount and deemed dividend on the redeemable convertible preferred stock.
LIQUIDITY AND CAPITAL RESOURCES
Short term
Our sources of liquidity include our cash balances and short-term investments. As of December 31, 2007, we had $398.2 million in cash and cash equivalents and short-term investments. In addition to our cash and investments, other potential sources of near-term liquidity include (i) equity, debt or other financing, (ii) the out-licensing of our drug candidates, internal drug programs and technologies, (iii) the sale of facilities that we own, and (iv) milestone payments from our collaborators.
To date, we have generated cash and funded our operations primarily through the sale of common and preferred stock, payments from collaborators and sale leaseback transactions. From our inception through December 31, 2007, we have generated $1.0 billion in cash from these sources, of which $839.6 million was through sales of stock, $140.0 million was through payments from our current and past collaborators and $61.1 million was from sale leaseback transactions.
We anticipate that our research and development expenditures will increase significantly as we continue our Phase 3 program for lorcaserin, initiate a Phase 2b clinical trial of APD125, and continue a Phase 1b clinical trial of APD791. We expect that the external expenses for our Phase 3 lorcaserin program, the majority of which we expect will be expensed through the first half of 2009, will be substantial. A large portion of these external clinical trial expenses are expected to be paid through CROs. Our contracts with the primary CROs for our Phase 3 lorcaserin program can be terminated if, depending on the contract, we give five or 30 days prior written notice, or less in certain circumstances. In addition to costs related to these clinical trials, we expect to incur significant manufacturing and other pre-launch costs for lorcaserin. We estimate that our Phase 3 lorcaserin program will continue in 2009 and could take significantly longer than expected to complete for various reasons including those set forth in "Item 1A. Risk Factors" in this Annual Report.
The research funding we received from our collaborations with Ortho-McNeil and Merck ended in the fourth quarter of 2007 and, absent any new collaborations, we expect no revenues from research funding to be recognized in 2008 or thereafter from our existing collaborators. We expect to recognize, in Swiss francs, CHF 8.2 million, or $7.4 million, in revenues from our contract manufacturing agreement with Siegfried in 2008, and that such revenues will be offset by related costs and expenses.
We believe we have sufficient cash to meet our objectives over at least the next year, including continuing our development programs for lorcaserin, APD125 and APD791, continuing development of our other lead internal programs, discovering and developing additional drug candidates, integrating our Swiss operations, continuing to build our development and manufacturing capabilities, including
57
our manufacturing facilities in Switzerland, and maintaining our research discovery capabilities. We will continue to monitor and evaluate the proper level of research, development and manufacturing expenditures, and may adjust such expenditures based upon a variety of factors, such as our month-12 ESMB and other clinical trial and preclinical results for our drug candidates, as well as our ability to generate cash through financings and collaborative activities. We expect our 2008 capital expenditures will be higher than in 2007 due to the purchase of our Swiss manufacturing facilities in January 2008 and planned purchases of equipment and improvements to our San Diego facilities, including a significant expansion of our property located at 6154 Nancy Ridge Drive that is expected to cost approximately $16.2 million, of which up to $15.0 million is expected to be reimbursed by the owner of the property in early 2009, less applicable commissions.
The holders of our Series B-1 Preferred can require us to redeem all or some of their outstanding shares of Series B-1 Preferred at any time. We will be required to redeem any shares of Series B-1 Preferred that remain outstanding on December 24, 2008 at a price equal to the amount of the original holder's original investment, plus all accrued but unpaid dividends thereon to the date of such payment. The aggregate redemption price of our Series B-1 Preferred at December 31, 2007 was $41.1 million, and we expect the aggregate redemption price at the mandatory redemption date of December 24, 2008 to be $42.8 million. We may be able to satisfy all or a portion of this amount with shares of our common stock. Our ability and decision whether to use cash or stock to satisfy any redemption will depend on, among other factors, the amount of cash we have, our stock price and the amount of common stock then held by our preferred stockholders.
In May 2007, we sold to BioMed three properties that we owned and continue to occupy, and assigned to BioMed an option to purchase a fourth property that we currently lease and primarily occupy for total consideration of $50.1 million, resulting in net proceeds to us of $48.5 million. Concurrently with the closing of the transaction, we leased back the three properties sold to BioMed under leases with 20-year terms and two consecutive options to extend such terms for five years each. Initial base rent for these three properties (net of taxes, insurance and maintenance costs (i.e. triple net) for which we are responsible) is an aggregate of $4.5 million annually, subject to an annual increase of 2.5% and other specified adjustments. If, at our election, we complete certain improvements to the properties sold, BioMed will pay us up to an additional $16.0 million (less applicable commissions) and our lease payments would increase. The amount of such increase would depend on the year in which such improvements are completed, if ever, with the initial amount of such increase for 2007 set at, assuming we receive the full $16.0 million (before applicable commissions), $1.4 million per year and increasing by approximately 2.5% each year. We expect to receive $1.0 million of such additional amount for improvements in the first quarter of 2008, but that we will not receive the remaining $15.0 million until 2009, if ever. Such additional amounts, if any, will be reduced by applicable commissions.
We will continue to lease a portion of the property that is subject to BioMed's purchase option from the current owner through the expiration of the lease with such owner, at which time we expect that BioMed will exercise the purchase option and rent will commence under a lease with BioMed for a term that is concurrent with the leases for the other three properties and at an initial base rent for such property (triple net) of $0.8 million per year, which would be subject to an annual increase of 2.5%. If BioMed is unable to exercise the option due to (i) an amendment to our lease with the current owner of such property that adversely affects such option and such amendment is not consented to by BioMed, or (ii) any casualty loss or proceeding in eminent domain pursuant to which BioMed has a right not to exercise the option in accordance with our agreement of purchase and sale, and BioMed elects not to exercise the option as a consequence of the occurrence of any event described in (i) and (ii) above, we would be required to pay BioMed $12.1 million. If BioMed elects to not exercise the option due to (ii) above, the lease payments on the remaining three properties would be reduced. The amount of such reduction would depend on the year in which BioMed elects to not
58
exercise the option, if ever, with the initial amount of such reduction for 2007 set at $1.1 million per year and increasing by 2.5% each year. In addition, subject to certain restrictions, we will have the option to repurchase all of the properties included in the transaction on the 10th, 15th or 20th anniversary of the execution date of the leases, and earlier if the leases are terminated under certain circumstances.
In January 2008, we entered into strategic cooperation agreements with Siegfried Ltd that are primarily related to the manufacturing of lorcaserin, which is expected to be necessary for our planned New Drug Application, or NDA, submission to the FDA and for commercialization of lorcaserin after regulatory marketing approval. The agreements include an asset purchase agreement for the purchase from Siegfried Ltd of certain drug product facility assets and technology, including fixtures, equipment, other personal property and real estate assets in Zofingen, Switzerland. We paid CHF 21.8 million, or $19.8 million, of the cash purchase price in January 2008, and will pay the remaining cash portion of the purchase price of CHF 10.0 million in three equal installments in the third, fourth and fifth years after closing. This transaction also included a long-term supply agreement, a contract manufacturing agreement and a services agreement.
We will continue to be opportunistic in our efforts to generate cash. We also continue to regularly evaluate potential acquisitions and in-licensing opportunities. Any such transaction may impact our liquidity as well as affect our expenses if, for example, our operating expenses increase as a result of such license or acquisition or we use our cash to finance the license or acquisition.
Long term
We will need to raise or generate significant amounts of cash to achieve our objectives of internally developing drugs, which take many years and potentially several hundreds of millions of dollars to develop, and continuing our research programs. If we decide to market and commercialize lorcaserin or any other drug candidate independently or with a partner, we may need to invest heavily in associated marketing and commercialization costs. Such costs will be substantial and some will need to be incurred prior to receiving marketing approval from the FDA. We do not currently have adequate internal liquidity to meet these objectives in the long term. In order to do so, we will need to continue our out-licensing activities and look to other external sources of liquidity, including the public and private financial markets and strategic partners.
The length of time that our current cash and cash equivalents, short-term investments and any available borrowings will sustain our operations will be based on, among other things, our progress in preclinical and clinical testing, the time and costs related to current and planned clinical trials and regulatory decisions, our research, development and manufacturing costs (including personnel costs), the progress in our collaborations, costs associated with intellectual property, our capital expenditures, and costs associated with securing any in-licensing opportunities. We do not know whether adequate funding will be available to us or, if available, that such funding will be available on acceptable terms. Any significant shortfall in funding could result in the partial or full curtailment of our development and/or research efforts, which, in turn, will affect our development pipeline and ability to generate cash in the future.
In addition to the public and private financial markets, potential sources of liquidity in the long term are milestone and royalty payments from existing and future collaborators and revenues from sales of our drugs.
59
Sources and Uses of Our Cash
Net cash used in operating activities was $128.1 million during the year ended December 31, 2007, and was used primarily to fund our net losses in the period, adjusted for non-cash expenses. Non-cash expenses included $8.8 million in share-based compensation, $7.8 million in depreciation and amortization expense, $1.5 million in amortization of acquired technology, as well as changes in operating assets and liabilities. Net cash used in operating activities was $71.0 million during the year ended December 31, 2006, and was used primarily to fund our net losses in the period, adjusted for non-cash expenses. Non-cash expenses included $7.4 million in depreciation and amortization expense, $5.0 million in share-based compensation, a $4.6 million charge related to a warrant settlement, $1.5 million in amortization of acquired technology, as well as changes in operating assets and liabilities. Net cash used in operating activities during the year ended December 31, 2005 was $42.9 million, and was used primarily to fund our net losses in the period, adjusted for non-cash expenses, including $6.9 million in depreciation and amortization expense, $1.5 million in amortization of acquired technology, $0.4 million in amortization of deferred compensation, as well as changes in operating assets and liabilities. We expect net cash used in operating activities will increase substantially assuming favorable results from the ESMB's month-12 review of echocardiograms in BLOOM and that we continue our clinical development programs and hiring of employees, primarily in clinical development.
Net cash of $12.6 million was used in investing activities during the year ended December 31, 2007, and was primarily the result of $14.2 million used for improvements to our facilities and purchases of equipment and $3.2 million used to purchase a facility on our San Diego campus, partially offset by net proceeds from short-term investments of $5.0 million. Net cash of $25.1 million was provided by investing activities during the year ended December 31, 2006, and was primarily the result of net proceeds from short-term investments of $39.1 million, partially offset by $3.6 million used to purchase a facility on our San Diego campus and $10.6 million used for equipment and improvements to our facilities. Net cash used in investing activities during the year ended December 31, 2005 was $2.9 million, and was primarily the result of $3.6 million used for the purchase of equipment and improvements to our facilities, partially offset by net proceeds from the sale of short-term investments of $0.4 million. We expect our 2008 capital expenditures will be higher than 2007 due to the purchase of our Swiss manufacturing facilities in January 2008 and planned purchases of equipment and improvements to our San Diego facilities, including a major expansion of approximately 75,000 square feet at our facility located at 6154 Nancy Ridge Drive that is expected to cost approximately $16.2 million.
Net cash of $154.7 million was provided by financing activities during the year ended December 31, 2007. This was due primarily to net proceeds of $103.2 million we received in November 2007 from the sale of 11,000,000 shares of our common stock at $9.91 per share, as well as net proceeds of $48.5 million we received in May 2007 from our lease financing transaction and net proceeds of $3.5 million received from option exercises, purchases under our employee stock purchase plan, and from the equity component of the $1.0 million payment we received from Merck in February 2007, which were partially offset by $0.5 million in principal payments on our lease financing obligations. Net cash of $345.2 million was provided by financing activities during the year ended December 31, 2006 due primarily to net proceeds of $165.1 million and $169.0 million we received in December 2006 and February 2006, respectively, from the sale of shares of our common stock, as well as proceeds of $8.3 million from the exercise of warrants to purchase our common stock in March 2006. Net cash of $60.8 million was provided by financing activities during the year ended December 31, 2005, and was due primarily to net proceeds of $48.2 million we received in February 2005 from the sale of shares of our common stock and $11.5 million received in April 2005 from our preferred stockholders' exercise of their unit warrants.
60
Contractual Obligations Table
The following table summarizes our contractual obligations as of December 31, 2007:
| |
| | Payments due by period (in thousands)
|
|---|
Contractual Obligations
| | Total
| | Less than 1
year
| | 1-3
years
| | 3-5
years
| | More than 5
Years
|
|---|
| Series B Preferred | | $ | 53,922 | | $ | 41,113 | | $ | 12,809 | | $ | — | | $ | — |
| Operating leases | | | 5,946 | | | 1,115 | | | 2,427 | | | 2,148 | | | 256 |
| Purchase obligations | | | 449 | | | 449 | | | — | | | — | | | — |
| Financing obligations | | | 131,809 | | | 5,597 | | | 12,659 | | | 13,300 | | | 100,253 |
| | |
| |
| |
| |
| |
|
| | Total | | $ | 192,126 | | $ | 48,274 | | $ | 27,895 | | $ | 15,448 | | $ | 100,509 |
| | |
| |
| |
| |
| |
|
The holders of our Series B-1 Preferred can require us to redeem all or some of their outstanding shares of Series B-1 Preferred at any time at such shares' stated value, which includes dividends that accrue at 4% annually. In addition, if not earlier redeemed, we are required to redeem our Series B-1 Preferred on December 24, 2008. If not previously converted, we are required to redeem the Series B-2 Preferred on April 22, 2010, at such shares' stated value, which includes dividends that accrue at 4% annually. Although we may be able to satisfy all or a portion of these amounts with shares of our common stock if certain criteria are met, the above table includes the full cash redemption price for both series of preferred stock as of December 31, 2007.
We have entered into agreements with CROs to conduct our clinical trials, and expect to continue to enter into such agreements. We will make payments to these sites and organizations primarily based upon the number of subjects enrolled and the length of their participation in the trials.
In determining the amount of our purchase obligations for contracts, we have included only the minimum obligation we have under our contracts (which analysis often assumed that such contracts were terminated on December 31, 2007) and did not include any amount which was previously paid, accrued, expensed or associated with a contingent event, such as a change in control or termination of a key employee.
In December 2003, we completed the sale and leaseback of one of our properties for total consideration of $13.0 million and in May 2007, we completed the sale and leaseback of three of our properties and assigned an option to purchase a fourth property for total consideration of $50.1 million. We have accounted for these transactions in accordance with SFAS No. 66, "Accounting for Sales of Real Estate" and SFAS No. 98, "Accounting for Leases." Our option to repurchase these properties in the future is considered continued involvement under SFAS No. 66 and, therefore, we have applied the financing method under SFAS No. 98. Under the financing method, the book value of the properties and related accumulated depreciation remain on our balance sheet and no sale is recognized. Instead, the sales price of the properties is recorded as a financing obligation and a portion of each lease payment is recorded as interest expense. At December 31, 2007, we expect interest expense over the term of these leases to total $79.5 million. We have included our lease obligations related to these properties in the above table as "financing obligations." At December 31, 2007, in accordance with SFAS No. 98, our total financing obligation for both of these transactions was $62.3 million. The aggregate residual value of the facilities at the end of the lease terms is $10.0 million.
In January 2008, we entered into strategic cooperation agreements with Siegfried Ltd. The agreements include an asset purchase agreement for the purchase from Siegfried Ltd of certain drug product facility assets, including fixtures, equipment, other personal property and real estate assets in Zofingen, Switzerland. We paid CHF 21.8 million, or $19.8 million, of the cash purchase price in January 2008, and will pay the remaining cash portion of the purchase price of CHF 10.0 million in
61
three equal installments in the third, fourth and fifth years after closing. This contractual obligation is not included in the above table.
The following is a summary of our significant collaborations as of December 31, 2007:
Ortho-McNeil Pharmaceutical, Inc.
In December 2004, we entered into a collaboration and license agreement with Ortho-McNeil to further develop compounds for the potential treatment of type 2 diabetes and other disorders. In January 2005, we received a non-refundable $17.5 million upfront payment and two milestone payments of $2.5 million each, and, in February 2006, we received a $5.0 million milestone payment related to Ortho-McNeil's initiation of a Phase 1 clinical trial of the then lead drug candidate, APD668. In September 2006, Ortho-McNeil exercised its option to extend the research portion of the collaboration through December 2007, beyond which date we no longer perform services or have significant involvement. Based on the data from studies of APD668, in January 2008 Ortho-McNeil decided to put APD668 on hold and has advanced a potentially more potent Arena-discovered GDIR agonist into preclinical development. We are eligible to receive a total of $295.0 million in milestone payments for each compound, as well as royalty payments associated with Ortho-McNeil's commercialization of any products discovered under the agreement. These milestones include development and approval milestone payments of up to $132.5 million for the first indication and $62.5 million for the second indication for each compound, and up to $100.0 million in sales milestone payments for each product resulting from the collaboration. From the inception of this collaboration through December 31, 2007, we received $27.5 million from Ortho-McNeil in upfront and milestone payments and $7.2 million in research funding. We recognized the upfront payment ratably over three years, along with the two milestones received in January 2005 as their achievability was reasonably assured at the time we entered into the collaboration.
Our agreement with Ortho-McNeil will continue until the expiration of Ortho-McNeil's payment obligations under the agreement, unless the agreement is terminated earlier by either party. We and Ortho-McNeil each have the right to terminate the agreement early on 60 days prior written notice if the other party commits an uncured material breach of its obligations. Ortho-McNeil may terminate the agreement at any time by providing at least 60 days prior written notice. Upon termination of the agreement, all rights to the compounds developed under the collaboration will revert to us.
For the year ended December 31, 2007, we recognized revenues under the Ortho-McNeil agreement of $13.4 million, which included $7.3 million from amortization of milestones and technology access and development fees received in prior years, $3.8 million for patent activities, and $2.3 million in research funding. For the year ended December 31, 2006, we recognized revenues under the Ortho-McNeil agreement of $18.5 million, which included $7.5 million from amortization of milestones and technology access and development fees received in prior years, $5.0 million from a milestone earned, $2.4 million in research funding, and $3.6 million for additional sponsored research and patent activities. For the year ended December 31, 2005, we recognized revenues under this agreement of $13.4 million, which included $7.5 million from amortization of milestones and technology access and development fees received in prior years, $2.4 million in research funding, and $3.5 million in additional sponsored research and patent activities. At December 31, 2007, there were no deferred revenues remaining under this agreement.
Merck & Co., Inc.
In October 2002, we entered into a research and licensing agreement with Merck to collaborate on three G protein-coupled receptors, or GPCRs, to develop therapeutics for atherosclerosis and related disorders. We believe one or more of these GPCRs plays a role in regulating plasma lipid profiles, including HDL cholesterol, the so-called "good cholesterol," and is responsible for the HDL-raising
62
activity of niacin. In October 2004, we extended and expanded this collaboration, and Merck selected one of our compounds for preclinical development. In February 2007, we amended our Merck collaboration to reduce the number of Arena research employees funded under the collaboration in exchange for Merck making a $1.0 million equity investment in Arena equal to the reduction in their research funding obligation and at approximately a 70% premium to the then current market price.
In September 2006, we announced that Merck completed a Phase 2 clinical trial of MK-0354, a niacin receptor agonist discovered by us and intended for the treatment of atherosclerosis and related disorders. Based on the results of this trial, Merck discontinued development of MK-0354. In January 2008, Merck initiated a Phase 1 clinical trial of a second generation niacin receptor agonist under our partnership for atherosclerosis and other disorders.
From the inception of this collaboration through December 31, 2007, we received $18.0 million from Merck in upfront and milestone payments, and equity investments totaling $8.5 million. We may receive additional milestone payments of up to $28.0 million for Merck's clinical and marketing achievements, as well as royalty payments associated with Merck's commercialization of any products discovered under the agreement. In addition, we received research funding from Merck through October 2007 totaling $27.5 million when, under our amended agreement, Merck's obligation for research funding ended, and beyond which date we no longer perform services or have significant involvement.
Our agreement with Merck will continue until the expiration of all royalty obligations under the agreement, unless the agreement is terminated early by either party. Either Merck or we can terminate our agreement if the other party breaches its material obligations under the agreement by causes and reasons within its control, has not cured such breach within 90 days of receiving a letter requesting such cure, and there is no dispute as to whether such breach has occurred. The non-breaching party in such a termination would receive the rights to continue the program. In addition, Merck can terminate the agreement at anytime by giving 90 days notice, but all milestones and royalties would still be payable as provided in the agreement.
As part of the extension and expansion of our collaboration with Merck in October 2004, Merck purchased $7.5 million of our stock at approximately a 70% premium to the then current market price. We performed an evaluation on this Merck stock purchase and determined that $3.9 million of this $7.5 million purchase price was an upfront payment related to the collaboration extension and expansion. Accordingly, we recognized the $3.9 million upfront payment, as well as the remaining portion of the unamortized upfront payment at October 2004 of $1.3 million, over the extended collaboration term of three years. Additionally, in October 2004, we achieved a $1.0 million milestone under the collaboration which we also recognized over the extended collaboration term of three years because the milestone was reasonably assured to be achieved at the time we extended and expanded this collaboration.
In connection with the February 2007 amendment of the collaborative agreement with Merck, we performed an evaluation on the stock purchase, which was at a purchase price of $24.81 per share, and determined that $0.5 million of the $1.0 million purchase price was an upfront payment related to the collaboration amendment. Accordingly, we recognized this upfront payment and the unamortized portion of the previously received upfront payments over the remaining term of the research portion of the collaboration. Merck's obligation for research funding ended in October 2007, beyond which date we no longer perform services or have significant involvement.
For the year ended December 31, 2007, we recognized revenues under the Merck agreement of $5.9 million, which included $3.6 million in research funding, $2.2 million from amortization of milestones and technology access and development fees received in prior years, and $0.1 million for patent activities. For the year ended December 31, 2006, we recognized revenues under the Merck agreement of $12.1 million, which included $5.7 million in research funding, $4.0 million from a
63
milestone earned, $2.1 million from amortization of milestones and technology access and development fees received in prior years, and $0.3 million for additional sponsored research and patent activities. For the year ended December 31, 2005, we recognized revenues under this agreement of $9.8 million, which included $5.7 million in research funding, $2.1 million from amortization of milestones and technology access and development fees received in prior years, and $2.0 million from a milestone earned. At December 31, 2007, there were no deferred revenues remaining under this agreement.
Recently issued accounting standards
In June 2006, the Financial Accounting Standards Board, or FASB, issued Statement of Financial Accounting Standards, or SFAS, No. 157, "Fair Value Measurements," which defines fair value, establishes a framework for measuring fair value in accordance with United States generally accepted accounting principles, or GAAP, and expands disclosures about fair value measurements. SFAS No. 157 is effective for fiscal years beginning after November 15, 2007, and for interim periods within those fiscal years. We are evaluating the effect, if any, the adoption of SFAS No. 157 will have on our consolidated financial statements.
In February 2007, the FASB issued SFAS No. 159, "The Fair Value Option for Financial Assets and Financial Liabilities—Including an amendment of SFAS No. 115," which allows an entity to voluntarily choose to measure certain financial assets and liabilities at fair value. SFAS No. 159 is effective for fiscal years beginning after November 15, 2007. We are evaluating the effect, if any, the adoption of SFAS No. 159 will have on our consolidated financial statements.
In June 2007, the FASB ratified the consensus reached by the EITF on EITF Issue No. 07-3, "Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities." EITF Issue No. 07-3 requires that nonrefundable advance payments for goods or services that will be used or rendered for future research and development activities be deferred and capitalized. Such amounts should be recognized as an expense as the related goods are delivered or the related services are performed or such time when the entity does not expect the goods to be delivered or services to be performed. EITF Issue No. 07-3 is effective, on a prospective basis, for fiscal years beginning after December 15, 2007. The adoption of EITF Issue No. 07-3 will not have a material effect on our consolidated financial statements.
In December 2007, the FASB issued SFAS No. 141R, "Business Combinations," which establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed and any noncontrolling interest in the acquiree. SFAS No. 141R also establishes disclosure requirements to enable the evaluation of the nature and financial effects of the business combination. SFAS No. 141R applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008, and interim periods within those fiscal years. We are evaluating the effect, if any, the adoption of SFAS No. 141R will have on our consolidated financial statements.
CRITICAL ACCOUNTING POLICIES AND MANAGEMENT ESTIMATES
The SEC defines critical accounting policies as those that are, in management's view, important to the portrayal of our financial condition and results of operations and demanding of management's judgment. Our discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosures. We base our estimates on historical experience and on various assumptions that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of
64
assets and liabilities that are not readily apparent from other sources. Actual results may differ from those estimates.
Our critical accounting policies include:
Clinical trial expenses. We accrue clinical trial expenses based on work performed. In determining the amount to accrue, we rely on estimates of total costs incurred based on the enrollment of subjects, the completion of studies and other events. We follow this method because we believe reasonably dependable estimates of the costs applicable to various stages of a clinical trial can be made. However, the actual costs and timing of clinical trials are highly uncertain, subject to risks and may change depending on a number of factors. Differences between the actual clinical trial costs and the estimated clinical trial costs that we have accrued in any prior period are recorded in the subsequent period in which the actual costs become known. Historically, these differences have not been material and we have not had to make material adjustments in the amounts recorded in a subsequent period; however, material differences could occur in the future.
Revenue recognition. Our revenue recognition policies are in accordance with SEC Staff Accounting Bulletin, or SAB, No. 104, "Revenue Recognition," and EITF Issue No. 00-21, "Revenue Arrangements with Multiple Deliverables," which provide guidance on revenue recognition in financial statements. Some of our agreements contain upfront technology access fees, research funding, milestone achievements and royalties.
Revenue from a milestone achievement is recognized when earned, as evidenced by acknowledgment from our collaborator, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement, (ii) the milestone represents the culmination of an earnings process, (iii) the milestone payment is non-refundable and (iv) our performance obligations after the milestone achievement will continue to be funded by our collaborator at a level comparable to the level before the milestone achievement. If all of these criteria are not met, the milestone achievement is recognized over the remaining minimum period of our performance obligations under the agreement. We defer non-refundable upfront fees under our collaborations and recognize them over the period in which we have significant involvement or perform services, using various factors specific to each collaboration. Amounts we receive for research funding for a specified number of full-time researchers are recognized as revenue as the services are performed. Advance payments we receive in excess of amounts earned are classified as deferred revenues until earned.
Share-based compensation. On January 1, 2006, we adopted SFAS No. 123R using the modified-prospective transition method. Under this method, prior period results are not restated. Compensation expense recognized subsequent to adoption includes: (i) compensation expense for all share-based awards granted prior to, but unvested as of, January 1, 2006, based on the grant-date fair value, estimated in accordance with the original provision of SFAS No. 123 using the Black-Scholes option pricing model, and (ii) compensation expense for all share-based awards granted subsequent to January 1, 2006, based on the grant-date fair value, estimated in accordance with the provisions of SFAS No. 123R using the Black-Scholes option pricing model.
The determination of the grant-date fair value of share-based awards using the Black-Scholes option pricing model is based on the exercise price of the award and our stock price on the date of grant, as well as assumptions for expected volatility, the expected life of options granted and the risk-free interest rate. Changes in the assumptions can have a material impact on the compensation expense we recognize. Expected volatility for awards granted after adoption of SFAS No. 123R is based on a combination of 75% historical volatility of our common stock and 25% market-based implied volatility from traded options on our common stock, with historical volatility being more heavily weighted due to the low volume of traded options on our common stock. Prior to adoption of SFAS No. 123R, our computation of expected volatility was based only on the historical volatility of our
65
common stock. The expected life of options granted under SFAS No. 123R is determined based on historical experience of similar awards, giving consideration to the contractual terms of the share-based awards, vesting schedules and post-vesting cancellations. Prior to the adoption of SFAS No. 123R, an average expected life of five years was used in determining the fair value of option grants based on the vesting period of the options and the short period of time our stock had been publicly traded. The risk-free interest rates are based on the US Treasury yield curve, with a remaining term approximately equal to the expected term used in the option pricing model.
As compensation expense recognized is based on awards ultimately expected to vest, it is reduced for estimated forfeitures. SFAS No. 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. If actual forfeitures do vary from estimates, we will recognize the difference in compensation expense in the period the actual forfeitures occur or when options vest.
For the year ended December 31, 2007, we recorded total non-cash, share-based compensation expense of $8.8 million.
Accounting for lease financing obligations. We have accounted for our sale leaseback transactions in accordance with SFAS No. 66 and SFAS No. 98. Our option to repurchase these properties in the future is considered continued involvement under SFAS No. 66 and, therefore, we have applied the financing method under SFAS No. 98. Under the financing method, the book value of the properties and related accumulated depreciation remain on our balance sheet and no sale is recognized. Instead, the sales price of the properties is recorded as a financing obligation, and a portion of each lease payment is recorded as interest expense. We estimated and apply an incremental borrowing rate to the lease payments to record interest expense.
Intangibles. Purchase accounting requires estimates and judgments to allocate the purchase price to the fair market value of the assets received and liabilities assumed. In February 2001, we acquired Bunsen Rush Laboratories, Inc. for $15.0 million in cash and assumed $0.4 million in liabilities. We allocated $15.4 million to the patented Melanophore technology acquired in such transaction. The Melanophore technology, our primary screening technology, is being amortized over its estimated useful life of 10 years, which was determined based on an analysis, as of the acquisition date, of the conditions in, and the economic outlook for, the pharmaceutical and biotechnology industries and the patent life of the technology. As with any intangible asset, we will continue to evaluate the value of the Melanophore technology. If, in the future, we determine that the Melanophore technology has become impaired or we no longer use it internally as our primary screening technology, we may record a write-down of the carrying value or we will accelerate the amortization if we determine that its life has been shortened.
The above listing is not intended to be a comprehensive list of all of our accounting policies. In many cases, the accounting treatment of a particular transaction is specifically dictated by GAAP. See our audited consolidated financial statements and notes thereto included elsewhere in this Annual Report, which contain additional accounting policies and other disclosures required by GAAP.
INCOME TAXES
As of December 31, 2007, we had $345.0 million of Federal net operating loss carryforwards and $25.3 million of Federal research and development tax credit carryforwards for income tax purposes which expire on various dates beginning in 2012. These amounts reflect different treatment of expenses for financial reporting and for tax purposes. United States tax law contains provisions that may limit our ability to use net operating loss and tax credit carryforwards in any year, including if there has been a significant ownership change.
66
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Our management establishes and oversees the implementation of board-approved policies covering our investments. We manage our market risk in accordance with our investment guidelines which (i) emphasize preservation of principal over other portfolio considerations, (ii) require investments to be placed in US government and agency obligations and in debt instruments that are rated investment grade, (iii) establish guidelines for the diversification of our investment portfolio, and (iv) require investments to be placed with maturities that maintain safety and liquidity. We target our portfolio to have an average duration of no more than four years with no one instrument having a duration exceeding five years and one month. We do not invest in derivative instruments, or any financial instruments for trading purposes. Our primary market risk exposure as it affects our cash equivalents, short-term investments, and securities available-for-sale is interest rate risk. We monitor our interest rate risk on a periodic basis and we ensure that our cash equivalents, short-term investments, and securities available-for-sale are invested in accordance with our investments guidelines. Managing credit ratings and the duration of our financial investments enhances the preservation of our capital.
We model interest rate exposure by a sensitivity analysis that assumes a hypothetical parallel shift downward in the US Treasury yield curve of 100 basis points. Under these assumptions, if the yield curve were to shift lower by 100 basis points from the level existing at December 31, 2007, we would expect future interest income from our portfolio to decline by less than $4.0 million over the next 12 months. As of December 31, 2006, this same hypothetical reduction in interest rates would have resulted in a decline in interest income of less than $3.9 million over the 12 months following December 31, 2006. The difference in these two estimates is due to the difference in our cash and cash equivalents, short-term investments, and securities available-for-sale between the two periods.
The model we use is not intended to forecast actual losses in interest income, but is used as a risk estimation and investment management tool. These hypothetical changes and assumptions are likely to be different from what actually occurs in the future. Furthermore, such computations do not incorporate actions our management could take if the hypothetical interest rate changes actually occur. As a result, the impact on actual earnings will likely differ from those quantified herein.
We have a wholly owned subsidiary in Switzerland, which exposes us to foreign exchange risk. The functional currency of our subsidiary in Switzerland is the Swiss franc. Accordingly, all assets and liabilities of our subsidiary are translated to US dollars based on the applicable exchange rate on the balance sheet date. Expense components are translated to US dollars at weighted-average exchange rates in effect during the period. Gains and losses resulting from foreign currency translation are included as a component of our stockholders' equity. Other foreign currency transaction gains and losses are included in our results of operations and, to date, have not been significant. We have not hedged exposures denominated in foreign currencies or any other derivative financial instrument, but may do so in the future.
67
Item 8. Financial Statements and Supplementary Data.
ARENA PHARMACEUTICALS, INC.
INDEX TO FINANCIAL STATEMENTS
| Report of Independent Registered Public Accounting Firm | | 69 |
| Consolidated Balance Sheets | | 70 |
| Consolidated Statements of Operations | | 71 |
| Consolidated Statements of Stockholders' Equity | | 72 |
| Consolidated Statements of Cash Flows | | 73 |
| Notes to Consolidated Financial Statements | | 74 |
68
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of Arena Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Arena Pharmaceuticals, Inc. as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders' equity, and cash flows for each of the three years in the period ended December 31, 2007. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Arena Pharmaceuticals, Inc. at December 31, 2007 and 2006, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 1 to the consolidated financial statements, Arena Pharmaceuticals, Inc. changed its method of accounting for share-based payments in accordance with Statement of Financial Accounting Standards No. 123 (revised 2004) on January 1, 2006.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Arena Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2007, based on the criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 29, 2008 expressed an unqualified opinion thereon.
| | /s/ Ernst & Young LLP |
San Diego, California
February 29, 2008 |
|
69
ARENA PHARMACEUTICALS, INC.
Consolidated Balance Sheets
(In thousands, except share and per share data)
| | December 31, 2007
| | December 31, 2006
| |
|---|
| Assets | | | | | | | |
| Current assets: | | | | | | | |
| | Cash and cash equivalents | | $ | 386,989 | | $ | 373,044 | |
| | Short-term investments, available-for-sale | | | 11,196 | | | 15,781 | |
| | Accounts receivable | | | 1,901 | | | 310 | |
| | Prepaid expenses and other current assets | | | 9,162 | | | 10,551 | |
| | |
| |
| |
| | | Total current assets | | | 409,248 | | | 399,686 | |
Land, property and equipment, net |
|
|
65,940 |
|
|
56,500 |
|
| Acquired technology, net | | | 4,875 | | | 6,412 | |
| Other non-current assets | | | 7,443 | | | 5,867 | |
| | |
| |
| |
| | | Total assets | | $ | 487,506 | | $ | 468,465 | |
| | |
| |
| |
| Liabilities and Stockholders' Equity | | | | | | | |
| Current liabilities: | | | | | | | |
| | Accounts payable and accrued expenses | | $ | 26,922 | | $ | 20,769 | |
| | Accrued compensation | | | 3,136 | | | 2,178 | |
| | Deferred revenues | | | — | | | 13,054 | |
| | Current portion of lease financing obligations | | | 231 | | | — | |
| | |
| |
| |
| | | Total current liabilities | | | 30,289 | | | 36,001 | |
| Deferred rent | | | 793 | | | 863 | |
| Deferred revenues | | | 4,049 | | | — | |
| Lease financing obligations, less current portion | | | 62,076 | | | 13,678 | |
Commitments |
|
|
|
|
|
|
|
| Series B redeemable convertible preferred stock, $.0001 par value: 4,650 shares authorized, issued and outstanding at December 31, 2007 and 2006; liquidation preference $46,500 at December 31, 2007 and 2006 | | | 53,922 | | | 51,808 | |
Stockholders' equity: |
|
|
|
|
|
|
|
| | Series A preferred stock, $.0001 par value: 350,000 shares authorized at December 31, 2007 and 2006; no shares issued and outstanding at December 31, 2007 and 2006 | | | — | | | — | |
| | Common stock, $.0001 par value: 142,500,000 shares authorized at December 31, 2007 and 2006; 72,260,254 and 60,771,401 shares issued and outstanding at December 31, 2007 and 2006, respectively | | | 8 | | | 6 | |
| | Additional paid-in capital | | | 838,913 | | | 723,363 | |
| | Treasury stock—3,000,000 shares at December 31, 2007 and 2006 | | | (23,070 | ) | | (23,070 | ) |
| | Accumulated other comprehensive loss | | | (23 | ) | | (13 | ) |
| | Accumulated deficit | | | (479,451 | ) | | (334,171 | ) |
| | |
| |
| |
| | | Total stockholders' equity | | | 336,377 | | | 366,115 | |
| | |
| |
| |
| | | Total liabilities and stockholders' equity | | $ | 487,506 | | $ | 468,465 | |
| | |
| |
| |
See accompanying notes.
70
ARENA PHARMACEUTICALS, INC.
Consolidated Statements of Operations
(In thousands, except share and per share data)
| | Years ended December 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| Revenues: | | | | | | | | | | |
| | Total revenues | | $ | 19,332 | | $ | 30,569 | | $ | 23,233 | |
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
| Research and development | | | 149,524 | | | 103,388 | | | 79,710 | |
| General and administrative | | | 26,571 | | | 18,466 | | | 13,122 | |
| Amortization of acquired technology | | | 1,537 | | | 1,537 | | | 1,537 | |
| | |
| |
| |
| |
| | Total operating expenses | | | 177,632 | | | 123,391 | | | 94,369 | |
| | |
| |
| |
| |
| | | Loss from operations | | | (158,300 | ) | | (92,822 | ) | | (71,136 | ) |
Interest and other income (expense): |
|
|
|
|
|
|
|
|
|
|
| Interest income | | | 18,850 | | | 12,691 | | | 4,426 | |
| Interest expense | | | (3,746 | ) | | (1,838 | ) | | (1,838 | ) |
| Non-cash warrant settlement | | | — | | | (4,554 | ) | | — | |
| Loss on sale of investments | | | (116 | ) | | (8 | ) | | (28 | ) |
| Other income, net | | | 146 | | | 283 | | | 675 | |
| | |
| |
| |
| |
| | Total interest and other income (expense), net | | | 15,134 | | | 6,574 | | | 3,235 | |
| | |
| |
| |
| |
| Net loss | | | (143,166 | ) | | (86,248 | ) | | (67,901 | ) |
| Dividends on redeemable convertible preferred stock | | | (2,114 | ) | | (2,031 | ) | | (1,813 | ) |
| Accretion of discount on redeemable convertible preferred stock | | | — | | | — | | | (7,372 | ) |
| | |
| |
| |
| |
| Net loss allocable to common stockholders | | $ | (145,280 | ) | $ | (88,279 | ) | $ | (77,086 | ) |
| | |
| |
| |
| |
| Net loss per share allocable to common stockholders, basic and diluted | | $ | (2.31 | ) | $ | (1.89 | ) | $ | (2.24 | ) |
| | |
| |
| |
| |
| Shares used in calculating net loss per share allocable to common stockholders, basic and diluted | | | 62,782,850 | | | 46,750,596 | | | 34,377,693 | |
| | |
| |
| |
| |
See accompanying notes.
71
ARENA PHARMACEUTICALS, INC.
Consolidated Statements of Stockholders' Equity
(In thousands, except share data)
| | Common Stock
| |
| |
| |
| |
| |
| |
| |
|---|
| | Additional
Paid-In
Capital
| | Treasury
Stock
| | Accumulated Other
Comprehensive
Income (Loss)
| | Deferred
Compensation
| | Accumulated
Deficit
| | Total
Stockholders'
Equity
| |
|---|
| | Shares
| | Amount
| |
|---|
| Balance at December 31, 2004 | | 26,566,419 | | $ | 3 | | $ | 319,540 | | $ | (23,070 | ) | $ | (164 | ) | $ | (780 | ) | $ | (168,806 | ) | $ | 126,723 | |
| Issuance of common stock upon exercise of options | | 75,790 | | | — | | | 405 | | | — | | | — | | | — | | | — | | | 405 | |
| Issuance of common stock under the employee stock purchase plan | | 197,862 | | | — | | | 784 | | | — | | | — | | | — | | | — | | | 784 | |
| Issuance of restricted stock | | 8,000 | | | — | | | 54 | | | — | | | — | | | (54 | ) | | — | | | — | |
| Issuance of common stock in public offering, net of offering costs of $3,599 | | 8,625,000 | | | 1 | | | 48,150 | | | — | | | — | | | — | | | — | | | 48,151 | |
| Amortization of deferred compensation | | — | | | — | | | — | | | — | | | — | | | 438 | | | — | | | 438 | |
| Dividends on redeemable convertible preferred stock | | — | | | — | | | — | | | — | | | — | | | — | | | (1,813 | ) | | (1,813 | ) |
| Accretion of discount and deemed dividend on redeemable convertible preferred stock | | — | | | — | | | — | | | — | | | — | | | — | | | (7,372 | ) | | (7,372 | ) |
| Restricted shares released from deferred compensation plan | | 17,500 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Net loss | | — | | | — | | | — | | | — | | | — | | | — | | | (67,901 | ) | | (67,901 | ) |
| Net unrealized gain on available-for-sale securities and investments | | — | | | — | | | — | | | — | | | 125 | | | — | | | — | | | 125 | |
| | | | | | | | | | | | | | | | | | | | | | |
| |
| Net comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (67,776 | ) |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| Balance at December 31, 2005 | | 35,490,571 | | $ | 4 | | $ | 368,933 | | $ | (23,070 | ) | $ | (39 | ) | $ | (396 | ) | $ | (245,892 | ) | $ | 99,540 | |
| Issuance of common stock upon exercise of options | | 180,364 | | | — | | | 1,184 | | | — | | | — | | | — | | | — | | | 1,184 | |
| Issuance of common stock under the employee stock purchase plan | | 307,086 | | | — | | | 1,649 | | | — | | | — | | | — | | | — | | | 1,649 | |
| Issuance of restricted stock | | 81,000 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Issuance of common stock upon exercise of warrants | | 829,856 | | | — | | | 8,298 | | | — | | | — | | | — | | | — | | | 8,298 | |
| Issuance of common stock in public offering, net of offering costs of $10,809 | | 10,637,524 | | | 1 | | | 168,964 | | | — | | | — | | | — | | | — | | | 168,965 | |
| Issuance of common stock in public offering, net of offering costs of $9,574 | | 13,225,000 | | | 1 | | | 165,127 | | | — | | | — | | | — | | | — | | | 165,128 | |
| Issuance of warrants in settlement | | — | | | — | | | 4,554 | | | — | | | — | | | — | | | — | | | 4,554 | |
| Share-based compensation expense, net of forfeitures | | — | | | — | | | 4,298 | | | — | | | — | | | — | | | — | | | 4,298 | |
| Reclassification of deferred compensation | | — | | | — | | | (396 | ) | | — | | | — | | | 396 | | | — | | | — | |
| Compensation expense related to restricted stock | | — | | | — | | | 752 | | | — | | | — | | | — | | | — | | | 752 | |
| Dividends on redeemable convertible preferred stock | | — | | | — | | | — | | | — | | | — | | | — | | | (2,031 | ) | | (2,031 | ) |
| Restricted shares released from deferred compensation plan | | 20,000 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Net loss | | — | | | — | | | — | | | — | | | — | | | — | | | (86,248 | ) | | (86,248 | ) |
| Net unrealized gain on available-for-sale securities and investments | | — | | | — | | | — | | | — | | | 26 | | | — | | | — | | | 26 | |
| | | | | | | | | | | | | | | | | | | | | | |
| |
| Net comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (86,222 | ) |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| Balance at December 31, 2006 | | 60,771,401 | | $ | 6 | | $ | 723,363 | | $ | (23,070 | ) | $ | (13 | ) | $ | — | | $ | (334,171 | ) | $ | 366,115 | |
| Issuance of common stock upon exercise of options | | 206,571 | | | — | | | 1,230 | | | — | | | — | | | — | | | — | | | 1,230 | |
| Issuance of common stock under the employee stock purchase plan | | 235,726 | | | — | | | 1,862 | | | — | | | — | | | — | | | — | | | 1,862 | |
| Issuance of common stock to Merck | | 40,306 | | | — | | | 480 | | | — | | | — | | | — | | | — | | | 480 | |
| Issuance of common stock in public offering, net of offering costs of $5,847 | | 11,000,000 | | | 2 | | | 103,162 | | | — | | | — | | | — | | | — | | | 103,164 | |
| Share-based compensation expense, net of forfeitures | | — | | | — | | | 8,556 | | | — | | | — | | | — | | | — | | | 8,556 | |
| Compensation expense related to restricted stock | | — | | | — | | | 260 | | | — | | | — | | | — | | | — | | | 260 | |
| Dividends on redeemable convertible preferred stock | | — | | | — | | | — | | | — | | | — | | | — | | | (2,114 | ) | | (2,114 | ) |
| Restricted shares released from deferred compensation plan | | 6,250 | | | — | | | — | | | — | | | — | | | — | | | — | | | — | |
| Net loss | | — | | | — | | | — | | | — | | | — | | | — | | | (143,166 | ) | | (143,166 | ) |
| Net unrealized gain on available-for-sale securities and investments | | — | | | — | | | — | | | — | | | 11 | | | — | | | — | | | 11 | |
| Translation loss | | — | | | — | | | — | | | — | | | (21 | ) | | — | | | — | | | (21 | ) |
| | | | | | | | | | | | | | | | | | | | | | |
| |
| Net comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (143,176 | ) |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| Balance at December 31, 2007 | | 72,260,254 | | $ | 8 | | $ | 838,913 | | $ | (23,070 | ) | $ | (23 | ) | $ | — | | $ | (479,451 | ) | $ | 336,377 | |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
See accompanying notes.
72
ARENA PHARMACEUTICALS, INC.
Consolidated Statements of Cash Flows
(In thousands)
| | Years ended December 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| OPERATING ACTIVITIES | | | | | | | | | | |
| Net loss | | $ | (143,166 | ) | $ | (86,248 | ) | $ | (67,901 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | |
| | Depreciation and amortization | | | 7,848 | | | 7,361 | | | 6,850 | |
| | Amortization of acquired technology | | | 1,537 | | | 1,537 | | | 1,537 | |
| | Amortization of deferred compensation | | | — | | | — | | | 438 | |
| | Non-cash share-based compensation | | | 8,816 | | | 5,050 | | | — | |
| | Non-cash warrant settlement | | | — | | | 4,554 | | | — | |
| | Amortization/accretion of short-term investment premium/discount | | | (398 | ) | | (717 | ) | | 154 | |
| | Amortization of prepaid financing costs | | | 305 | | | — | | | — | |
| | Amortization of lease financing obligations | | | (361 | ) | | — | | | — | |
| | Deferred rent | | | (70 | ) | | (45 | ) | | (24 | ) |
| | Deferred interest expense | | | (677 | ) | | 193 | | | 226 | |
| | Loss on disposal of equipment | | | 114 | | | 8 | | | 19 | |
| | Changes in operating assets and liabilities: | | | | | | | | | | |
| | | Accounts receivable | | | (1,591 | ) | | 538 | | | 21,742 | |
| | | Prepaid expenses and other assets | | | 1,389 | | | (4,830 | ) | | (389 | ) |
| | | Deferred revenues | | | (9,005 | ) | | (11,090 | ) | | (9,497 | ) |
| | | Accounts payable, accrued expenses and accrued compensation | | | 7,111 | | | 12,672 | | | 3,986 | |
| | |
| |
| |
| |
| | | | Net cash used in operating activities | | | (128,148 | ) | | (71,017 | ) | | (42,859 | ) |
INVESTING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
| | Purchases of short-term investments, available-for-sale | | | (60,998 | ) | | (17,976 | ) | | (152,639 | ) |
| | Proceeds from sales/maturities of short-term investments | | | 65,992 | | | 57,096 | | | 153,079 | |
| | Purchases of land, property and equipment | | | (17,423 | ) | | (14,231 | ) | | (3,581 | ) |
| | Proceeds from sale of equipment | | | 21 | | | 1 | | | 69 | |
| | Deposits, restricted cash and other assets | | | (188 | ) | | 166 | | | 186 | |
| | |
| |
| |
| |
| | | | Net cash provided by (used in) investing activities | | | (12,596 | ) | | 25,056 | | | (2,886 | ) |
FINANCING ACTIVITIES |
|
|
|
|
|
|
|
|
|
|
| | Principal payments on lease financing obligations | | | (481 | ) | | — | | | — | |
| | Proceeds from lease financing | | | 48,455 | | | — | | | — | |
| | Proceeds from exercise of warrants | | | — | | | 8,298 | | | — | |
| | Proceeds from issuance of redeemable convertible preferred stock and warrants | | | — | | | — | | | 11,500 | |
| | Proceeds from issuance of common stock | | | 106,736 | | | 336,926 | | | 49,340 | |
| | |
| |
| |
| |
| | | | Net cash provided by financing activities | | | 154,710 | | | 345,224 | | | 60,840 | |
| Effect of exchange rate changes on cash and cash equivalents | | | (21 | ) | | — | | | — | |
| | |
| |
| |
| |
| Net increase in cash and cash equivalents | | | 13,945 | | | 299,263 | | | 15,095 | |
| Cash and cash equivalents at beginning of year | | | 373,044 | | | 73,781 | | | 58,686 | |
| | |
| |
| |
| |
| Cash and cash equivalents at end of year | | $ | 386,989 | | $ | 373,044 | | $ | 73,781 | |
| | |
| |
| |
| |
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | | | | | | | | | | |
| Interest paid | | $ | 4,295 | | $ | 1,499 | | $ | 1,460 | |
| | |
| |
| |
| |
| Unrealized gain on short-term investments, available-for-sale | | $ | 11 | | $ | 26 | | $ | 125 | |
| | |
| |
| |
| |
See accompanying notes.
73
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The Company
Arena Pharmaceuticals, Inc., or the Company, was incorporated on April 14, 1997, and commenced operations in July 1997. The Company operates in one business segment and is a clinical-stage biopharmaceutical company with a pipeline of internally discovered small molecule drug candidates that target G protein-coupled receptors, or GPCRs.
Principles of Consolidation
The consolidated financial statements include the activities of the Company and its wholly owned subsidiaries. All material intercompany accounts and transactions have been eliminated in consolidation.
Financial Statement Preparation
The preparation of financial statements in conformity with United States generally accepted accounting principles, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and highly liquid investments with original maturities of three months or less when purchased.
Short-term Investments, Available-for-Sale
In accordance with Statement of Financial Accounting Standards, or SFAS, No. 115, "Accounting for Certain Debt and Equity Securities," short-term investments are classified as available-for-sale. The Company defines short-term investments as income-yielding securities that can be readily converted to cash. These securities are carried at fair value, with unrealized gains and losses reported as a separate component of accumulated other comprehensive income or loss. The cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion is included in interest income. Realized gains and losses and declines in securities judged to be other than temporary are included in other income or expense. The cost of securities sold is based on the specific identification method. Interest and dividends on available-for-sale securities are included in interest income.
Fair Value of Financial Instruments
Cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities are carried at cost, which management believes approximates fair value due to the short-term maturity of these instruments. Short-term investments are carried at fair value. Based on borrowing rates currently available to the Company for loans with similar terms, management believes the carrying value of the lease financing obligations approximates fair value.
74
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Concentration of Credit Risk and Major Customers
Financial instruments, which potentially subject the Company to concentrations of credit risk, consist primarily of cash, cash equivalents and short-term investments. The Company limits its exposure to credit loss by placing its cash and investments, in accordance with its board-approved investment policy, in US government and agency obligations and in debt instruments that are rated investment grade.
Merck & Co., Inc., or Merck, and Ortho-McNeil Pharmaceutical, Inc., a Johnson & Johnson company, or Ortho-McNeil, accounted for 100% of total revenues for the years ended December 31, 2007, 2006 and 2005. Ortho-McNeil accounted for 98%, 90% and 99% of accounts receivable as of December 31, 2007, 2006 and 2005, respectively.
Property and Equipment
Property and equipment are stated at cost and depreciated over the estimated useful lives of the assets (generally three to seven years) using the straight-line method. Buildings and building improvements are stated at cost and depreciated over an estimated useful life of approximately 20 years using the straight-line method. Leasehold improvements are stated at cost and amortized over the shorter of the estimated useful lives of the assets or the lease term. Capital improvements are stated at cost and amortized over the estimated useful lives of the assets.
Intangible Assets
Purchase accounting requires estimates and judgments to allocate the purchase price to the fair market value of the assets received and liabilities assumed. In February 2001, the Company acquired Bunsen Rush Laboratories, Inc., or Bunsen Rush, for $15.0 million in cash and assumed $0.4 million in liabilities. The Company allocated $15.4 million to the patented Melanophore technology, its primary screening technology, acquired in such transaction. Acquired technology from the Company's acquisition of Bunsen Rush is being amortized over its estimated useful life of 10 years, which was determined based on an analysis, as of the acquisition date, of the conditions in, and the economic outlook for, the pharmaceutical and biotechnology industries and the patent life of the technology. As with any intangible asset, the Company continues to evaluate the value of the Melanophore technology. If, in the future, the Company determines that the technology has become impaired or no longer uses this technology internally as a primary screening technology, the Company may record a write-down of the carrying value or accelerate the amortization if it determines that the technology life has been shortened. Accumulated amortization from acquired technology totaled $10.5 million and $9.0 million at December 31, 2007 and 2006, respectively. As of December 31, 2007, the Company anticipates that total charges of $1.5 million will be recognized from the amortization of acquired technology in each of the next three years.
Long-lived Assets
In accordance with SFAS No. 144, "Accounting for the Impairment or Disposal of Long-Lived Assets," the Company reviews the recoverability of long-lived and finite-lived intangible assets when circumstances indicate that the carrying amount of assets may not be recoverable. This review is based on various analyses, including undiscounted cash flow projections. In the event such analysis indicated an impairment, the Company would record an impairment loss, if any, based on the fair value of the
75
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
assets. The Company did not record any impairments or write-offs of long-lived or finite-lived intangible assets in the years ended December 31, 2007, 2006 or 2005.
Deferred Rent
For financial reporting purposes, rent expense is recognized on a straight-line basis over the term of the lease. The difference between rent expense and amounts paid under lease agreements is recorded as deferred rent in the liability section of the accompanying consolidated balance sheets.
Share-based Compensation
Prior to January 1, 2006, the Company accounted for share-based compensation in accordance with the provisions of Accounting Principles Board, or APB, Opinion No. 25, "Accounting for Stock Issued to Employees" and its related Interpretations, which state that no compensation expense is recorded for stock options or other share-based awards to employees and directors that are granted with an exercise price equal to or above the fair value per share of the Company's common stock on the grant date. In the event that stock options were granted with an exercise price below the fair value of the Company's common stock on the grant date, the difference between the fair value of its common stock and the exercise price of the stock option was recorded as deferred compensation. For stock options granted to its employees and directors, the Company adopted the disclosure-only requirements of SFAS No. 123, "Accounting for Stock-Based Compensation," which allowed compensation expense to be disclosed in the notes to the financial statements based on the fair value of the options granted at the date of the grant. Compensation expense for options granted to non-employees other than directors had been determined in accordance with SFAS No. 123 and Emerging Issues Task Force, or EITF, Issue No. 96-18, "Accounting for Equity Instruments that are Issued to Other than Employees for Acquiring, or in Conjunction with Selling Goods or Services." Such expense was based on the fair value of the options issued using the Black-Scholes option pricing model and was periodically remeasured as the underlying options vested in accordance with EITF Issue No. 96-18.
On January 1, 2006, the Company adopted SFAS No. 123R, "Share-Based Payment," using the modified-prospective transition method. Under this method, prior period results are not restated. Compensation expense recognized subsequent to adoption includes: (i) compensation expense for all share-based awards granted prior to, but unvested as of, January 1, 2006, based on the grant-date fair value, estimated in accordance with the original provisions of SFAS No. 123, and (ii) compensation expense for all share-based awards granted subsequent to January 1, 2006, based on the grant-date fair value, estimated in accordance with the provisions of SFAS No. 123R. Compensation expense related to share-based awards, which is recognized on a straight-line basis over the vesting period, is included in research and development and in general and administrative expenses in the accompanying consolidated statements of operations.
The Company measures the value of restricted stock awards based on the fair value of the stock on the grant date. The restrictions generally lapse in equal annual installments over a vesting period of two, three or four years. Prior to the adoption of SFAS No. 123R, deferred compensation for grants of restricted stock equivalent to the fair value of the shares at the date of grant was recorded as a separate component of stockholders' equity and subsequently amortized to compensation expense over the vesting period of each award. The remaining unamortized deferred compensation of $0.4 million at January 1, 2006 was reclassified to additional paid-in capital upon adoption of SFAS No. 123R. In
76
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
accordance with SFAS No. 123R, stockholders' equity is now credited as compensation expense is recognized over the applicable vesting period.
The Company recorded total share-based compensation expense for all share-based awards of $8.8 million and $5.0 million during the years ended December 31, 2007 and 2006, respectively, and recorded expense of $0.4 million for amortization of deferred compensation during the year ended December 31, 2005.
Revenue Recognition
The Company's revenue recognition policies are in accordance with SEC Staff Accounting Bulletin, or SAB, No. 101, "Revenue Recognition in Financial Statements," as amended by SAB No. 104, "Revenue Recognition," and EITF Issue No. 00-21, "Revenue Arrangements with Multiple Deliverables," which provide guidance on revenue recognition in financial statements. Some of the Company's agreements contain upfront technology access fees, research funding, milestone achievements and royalties. Revenue from a milestone achievement is recognized when earned, as evidenced by acknowledgment from the Company's collaborator, provided that (i) the milestone event is substantive and its achievability was not reasonably assured at the inception of the agreement, (ii) the milestone represents the culmination of an earnings process, (iii) the milestone payment is non-refundable and (iv) the Company's performance obligations after the milestone achievement will continue to be funded by the collaborator at a level comparable to the level before the milestone achievement. If all of these criteria are not met, the milestone is recognized over the remaining minimum period of the Company's performance obligations under the agreement. Non-refundable upfront fees under the Company's collaborations are deferred and recognized over the period in which the Company has significant involvement or performs services, using various factors specific to the collaboration. Amounts received for research funding for a specified number of full-time researchers are recognized as revenue as the services are performed. Advance payments received in excess of amounts earned are classified as deferred revenues until earned.
Research and Development Costs
Research and development expenses, which consist primarily of costs associated with external clinical trial and preclinical study fees, manufacturing costs and other related expenses, and the development of our earlier-stage programs and technologies, are expensed as incurred when these expenditures relate to our research and development efforts and have no alternative future uses.
Clinical Trial Expenses
The Company accrues clinical trial expenses based on work performed. In determining the amount to accrue, the Company relies on estimates of total costs incurred based on the enrollment of subjects, the completion of studies and other events. The Company follows this method because it believes reasonably dependable estimates of the costs applicable to various stages of a clinical trial can be made. However, the actual costs and timing of clinical trials are highly uncertain, subject to risks and may change depending on a number of factors. Differences between the actual clinical trial costs and the estimated clinical trial costs that have been accrued in any prior period are recorded in the subsequent period in which the actual costs become known. Historically, these differences have not been material and the Company has not had to make material adjustments in a subsequent period.
77
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Patent Costs
Costs related to filing and prosecuting patent applications are expensed to general and administrative as incurred as recoverability of such expenditures is uncertain.
Comprehensive Income (Loss)
In accordance with SFAS No. 130, "Reporting Comprehensive Income," all components of comprehensive income (loss), including unrealized gains and losses on investment securities and foreign currency translation adjustment, are reported in the financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources.
Net Loss Per Share
Basic and diluted net loss per share allocable to common stockholders are presented in conformity with SFAS No. 128, "Earnings per Share." In accordance with SFAS No. 128, basic and diluted net loss per share has been computed using the weighted-average number of shares of common stock outstanding during the period, less any shares subject to repurchase or forfeiture.
The total number of shares of common stock outstanding excluded from the calculation of basic and diluted net loss per share because they were subject to repurchase or forfeiture was 99,811, 71,420 and 195,329 for the years ended December 31, 2007, 2006 and 2005, respectively. Had they been dilutive, such shares would have been included in the computation of diluted net loss per share. In addition, the Company has excluded all unvested performance-based restricted stock unit awards, which are subject to forfeiture, outstanding stock options, preferred stock and warrants from the calculation of basic and diluted net loss per share allocable to common stockholders because these securities are antidilutive for all years presented.
Pro Forma Information under SFAS No. 123 for Year Ended December 31, 2005
Prior to adopting the provisions of SFAS No. 123R, the Company provided pro forma disclosures of estimated share-based compensation expense as permitted under SFAS No. 123. For pro forma purposes, the fair value of stock options was estimated at the date of grant using the Black-Scholes option pricing model and amortized to expense over the options' vesting periods using the assumptions stated below. The following table illustrates the pro forma effect on net loss allocable to common stockholders and net loss per share, in thousands except per share data, as if the Company had accounted for its employee and director stock options and stock issued under the 2001 Arena
78
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
Employee Stock Purchase Plan, as amended, or the Purchase Plan, using the fair value method prescribed by SFAS No. 123.
| | Year ended
December 31, 2005
| |
|---|
| Net loss allocable to common stockholders, as reported | | $ | (77,086 | ) |
| Add: Stock-based employee compensation expense included in net loss allocable to common stockholders, as reported, net of related tax effects | | | 438 | |
| Fair value of stock-based employee compensation | | | (4,347 | ) |
| | |
| |
| Pro forma net loss | | $ | (80,995 | ) |
| Net loss per share: | | | | |
| | Basic and diluted—as reported | | $ | (2.24 | ) |
| | Basic and diluted—pro forma | | $ | (2.36 | ) |
Assumptions used for employee stock options: |
|
|
|
|
| | Risk-free interest rate | | | 4.2 | % |
| | Dividend yield | | | 0 | % |
| | Stock price volatility | | | 44 | % |
| | Expected life (years) | | | 4.99 | |
| | Weighted-average estimated fair value per share | | $ | 3.03 | |
Assumptions used for Purchase Plan: |
|
|
|
|
| | Risk-free interest rate | | | 3.8 | % |
| | Dividend yield | | | 0 | % |
| | Stock price volatility | | | 48 | % |
| | Expected life (years) | | | 0.25 | |
| | Weighted-average estimated fair value per share | | $ | 1.78 | |
Effect of New Accounting Standards
In June 2006, the FASB issued SFAS No. 157, "Fair Value Measurements," which defines fair value, establishes a framework for measuring fair value in accordance with GAAP and expands disclosures about fair value measurements. SFAS No. 157 is effective for fiscal years beginning after November 15, 2007, and for interim periods within those fiscal years. The Company is evaluating the effect, if any, the adoption of SFAS No. 157 will have on its consolidated financial statements.
In February 2007, the FASB issued SFAS No. 159, "The Fair Value Option for Financial Assets and Financial Liabilities—Including an amendment of SFAS No. 115," which allows an entity to voluntarily choose to measure certain financial assets and liabilities at fair value. SFAS No. 159 is effective for fiscal years beginning after November 15, 2007. The Company is evaluating the effect, if any, the adoption of SFAS No. 159 will have on its consolidated financial statements.
In June 2007, the FASB ratified the consensus reached by the EITF on EITF Issue No. 07-3, "Accounting for Nonrefundable Advance Payments for Goods or Services Received for Use in Future Research and Development Activities." EITF Issue No. 07-3 requires that nonrefundable advance payments for goods or services that will be used or rendered for future research and development
79
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(1) THE COMPANY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES (Continued)
activities be deferred and capitalized. Such amounts should be recognized as an expense as the related goods are delivered or the related services are performed or such time when the entity does not expect the goods to be delivered or services to be performed. EITF Issue No. 07-3 is effective, on a prospective basis, for fiscal years beginning after December 15, 2007. The adoption of EITF Issue No. 07-3 will not have a material effect on the Company's consolidated financial statements.
In December 2007, the FASB issued SFAS No. 141R, "Business Combinations," which establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed and any noncontrolling interest in the acquiree. SFAS No. 141R also establishes disclosure requirements to enable the evaluation of the nature and financial effects of the business combination. SFAS No. 141R applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008, and interim periods within those fiscal years. The Company is evaluating the effect, if any, the adoption of SFAS No. 141R will have on its consolidated financial statements.
(2) AVAILABLE-FOR-SALE SECURITIES
The following table summarizes the investment categories comprising available-for-sale securities at December 31, 2007 and 2006, in thousands:
December 31, 2007
| | Amortized Cost
| | Gross Unrealized
Gains
| | Gross Unrealized
Losses
| | Estimated Fair
Value
|
|---|
| Federal agency notes | | $ | 1,276 | | $ | 4 | | $ | — | | $ | 1,280 |
| Corporate debt securities | | | 9,922 | | | — | | | (6 | ) | | 9,916 |
| | |
| |
| |
| |
|
| | Total available-for-sale securities | | $ | 11,198 | | $ | 4 | | $ | (6 | ) | $ | 11,196 |
| | |
| |
| |
| |
|
December 31, 2006
| | Amortized Cost
| | Gross Unrealized
Gains
| | Gross Unrealized
Losses
| | Estimated Fair
Value
|
|---|
| Federal agency notes | | $ | 11,905 | | $ | — | | $ | (1 | ) | $ | 11,904 |
| Corporate debt securities | | | 3,877 | | | — | | | — | | | 3,877 |
| | |
| |
| |
| |
|
| | Total available-for-sale securities | | $ | 15,782 | | $ | — | | $ | (1 | ) | $ | 15,781 |
| | |
| |
| |
| |
|
The amortized cost and estimated fair value of available-for-sale securities by contractual maturity at December 31, 2007 are shown below, in thousands:
| | Amortized Cost
| | Estimated Fair Value
|
|---|
| Due in one year or less | | $ | 9,922 | | $ | 9,916 |
| Due after one year through four years | | | 1,276 | | | 1,280 |
| | |
| |
|
| | Total | | $ | 11,198 | | $ | 11,196 |
| | |
| |
|
Proceeds from the sales of available-for-sale securities totaled $66.0 million, $57.1 million and $153.1 million in 2007, 2006 and 2005, respectively.
80
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(3) PROPERTY AND EQUIPMENT
Property and equipment consisted of the following, in thousands:
| | December 31,
| |
|---|
| | 2007
| | 2006
| |
|---|
| Laboratory and computer equipment | | $ | 36,585 | | $ | 30,373 | |
| Furniture, fixtures and office equipment | | | 1,721 | | | 1,503 | |
| Land, building and capital improvements | | | 50,917 | | | 47,311 | |
| Leasehold improvements | | | 16,818 | | | 9,804 | |
| | |
| |
| |
| | | | 106,041 | | | 88,991 | |
| Less accumulated depreciation and amortization | | | (40,101 | ) | | (32,491 | ) |
| | |
| |
| |
| | Net property and equipment | | $ | 65,940 | | $ | 56,500 | |
| | |
| |
| |
Depreciation expense was $7.8 million, $7.4 million and $6.9 million for the years ended December 31, 2007, 2006 and 2005, respectively.
(4) ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Accounts payable and accrued expenses consisted of the following, in thousands:
| | December 31,
|
|---|
| | 2007
| | 2006
|
|---|
| Accounts payable | | $ | 4,161 | | $ | 3,165 |
| Accrued contracts and study fees | | | 19,766 | | | 14,328 |
| Other accrued liabilities | | | 2,995 | | | 3,276 |
| | |
| |
|
| | Total | | $ | 26,922 | | $ | 20,769 |
| | |
| |
|
81
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(5) COMMITMENTS
The following table summarizes the Company's real property leasing arrangements and essential provisions as of December 31, 2007:
Address on
Nancy Ridge Drive,
San Diego, California
| | Own/ Lease
| | Description of Arrangements
|
|---|
| 6166 | | Lease | | In 1997, the Company began leasing this property under a lease that included an option to buy the property for $2.1 million. In 1998, the Company assigned the option to another company in exchange for $0.7 million in cash, and such company exercised the option and leased the property to the Company under a lease that expires in 2013. The $0.7 million is being recognized on a straight-line basis as a reduction in the rent expense on the underlying lease. The Company has two five-year options to extend the lease term. The new lease terms stipulate annual increases in monthly rental payments of 2.75% beginning in April 2000. |
6122-6124-6126 |
|
Lease with option to purchase |
|
In 2002, the Company leased a property located at 6124-6126 Nancy Ridge Drive. Under the terms of this lease, effective April 2003, monthly rental payments increased by 2% and are subject to a 2% annual increase thereafter. In 2005, the Company amended this lease to include additional square footage in a contiguous building, 6122 Nancy Ridge Drive. As discussed in the below section on 6114, 6118, 6154 Nancy Ridge Drive, the Company assigned its option to buy this entire building for $7.9 million when the lease ends in March 2012. |
6138-6150 |
|
Lease with option to purchase |
|
In 2003, the Company completed the sale and leaseback of this property. The sales price for this property was $13.0 million and net proceeds to the Company were $12.6 million. The Company has accounted for this transaction in accordance with SFAS No. 66 "Accounting for Sales of Real Estate" and SFAS No. 98 "Accounting for Leases." The Company's option to repurchase this property in the future is considered continued involvement under SFAS No. 66 and, therefore, the Company has applied the financing method under SFAS No. 98. Under the financing method, the book value of the property and related accumulated depreciation remain on the Company's balance sheet and no sale is recognized. Instead, the sales price of the property is recorded as a financing obligation and a portion of each lease payment is recorded as interest expense. The term of the lease, which became effective in December 2003, is 15 years, with monthly rental payments increasing by 2.5% annually, beginning in January 2005. The Company has the right to repurchase this property through year 14 of the lease. The Company recorded interest expense of $0.4 million in the year ended December 31, 2007 and $1.6 million in each of the years ended December 31, 2006 and 2005 related to this lease. At December 31, 2007, in accordance with SFAS No. 98, the total financing obligation on the accompanying consolidated balance sheets related to this transaction was $12.5 million. |
82
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(5) COMMITMENTS (Continued)
6114, 6118, 6154 |
|
Lease with option to purchase |
|
In May 2007, the Company completed the sale and leaseback of these properties. The total consideration for these properties and the assignment of the option to purchase the property located at 6122-6124-6126 Nancy Ridge Drive was $50.1 million, resulting in net proceeds to the Company of $48.5 million after financing costs and commissions. Concurrently with the closing of the transaction, the Company leased back the three properties under leases with 20-year terms and two consecutive options to extend such terms for five years each. In addition, subject to certain restrictions, the Company has the option to repurchase all of the properties included in the transaction on the 10th, 15th or 20th anniversary of the execution date of the leases, and earlier if the leases are terminated under certain circumstances. The Company has accounted for this transaction in accordance with SFAS No. 66 and SFAS No. 98. The Company's option to repurchase this property in the future is considered continued involvement under SFAS No. 66 and, therefore, the Company has applied the financing method under SFAS No. 98. Initial base rent for the three properties (net of taxes, insurance and maintenance costs (i.e. triple net) for which the Company is responsible) that were purchased as part of this transaction is an aggregate of $4.5 million annually, subject to an annual increase of 2.5% and other specified adjustments. The Company recorded interest expense of $3.1 million in the year ended December 31, 2007 related to this transaction. At December 31, 2007, in accordance with SFAS No. 98, the total financing obligation related to this transaction was $49.8 million. |
In accordance with the terms of two of the above leases, the Company is required to maintain restricted cash balances, which are included in other non-current assets on the accompanying consolidated balance sheets, on behalf of the landlord as rent deposits throughout the term of the lease. In accordance with the terms of one of the leases, the Company has paid a security deposit equal to one month rent as a security deposit, which is also included in other non-current assets on the accompanying consolidated balance sheets. A total of $1.1 million is recorded in other non-current assets related to these three leases.
The Company recognizes rent expense on a straight-line basis over the term of each lease. Rent expense was $1.1 million in each of the years ended December 31, 2007 and 2006 and $1.0 million in the year ended December 31, 2005.
At December 31, 2007 the Company expects interest expense over the terms of the leases related to the facilities accounted for under SFAS No. 66 and SFAS No. 98 to total $79.5 million. As of December 31, 2007, the total financing obligation for these facilities was $62.3 million. The aggregate residual value of the facilities at the end of the lease terms is $10.0 million.
83
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(5) COMMITMENTS (Continued)
Annual future obligations as of December 31, 2007 are as follows, in thousands:
Year ending December 31,
| | Financing
Obligation
| | Operating Leases
|
|---|
| 2008 | | $ | 5,597 | | $ | 1,115 |
| 2009 | | | 6,251 | | | 1,199 |
| 2010 | | | 6,408 | | | 1,228 |
| 2011 | | | 6,568 | | | 1,258 |
| 2012 | | | 6,732 | | | 890 |
| Thereafter | | | 100,253 | | | 256 |
| | |
| |
|
| | Total minimum lease payments | | $ | 131,809 | | $ | 5,946 |
| | |
| |
|
(6) COLLABORATIONS
Ortho-McNeil Pharmaceutical, Inc.
In December 2004, the Company entered into a collaboration and license agreement with Ortho-McNeil to further develop compounds for the potential treatment of type 2 diabetes and other disorders. In January 2005, the Company received a non-refundable $17.5 million upfront payment and two milestone payments of $2.5 million each and, in February 2006, the Company received a $5.0 million milestone payment related to Ortho-McNeil's initiation of a Phase 1 clinical trial of the then lead drug candidate, APD668. In September 2006, Ortho-McNeil exercised its option to extend the research portion of the collaboration through December 2007, beyond which date the Company no longer performs services or has significant involvement. The Company is eligible to receive a total of $295.0 million in milestone payments for each compound, as well as royalty payments associated with Ortho-McNeil's commercialization of any products discovered under the agreement. These milestones include development and approval milestone payments of up to $132.5 million for the first indication and $62.5 million for the second indication for each compound, and up to $100.0 million in sales milestone payments for each product resulting from the collaboration. From the inception of this collaboration through December 31, 2007, the Company received $27.5 million from Ortho-McNeil in upfront and milestone payments and $7.2 million in research funding. The Company recognized the upfront payment ratably over three years, along with the two milestones received in January 2005 as their achievability was reasonably assured at the time the Company entered into the collaboration.
The agreement with Ortho-McNeil will continue until the expiration of Ortho-McNeil's payment obligations under the agreement, unless the agreement is terminated earlier by either party. The Company and Ortho-McNeil each have the right to terminate the agreement early on 60 days prior written notice if the other party commits an uncured material breach of its obligations. Ortho-McNeil may terminate the agreement at any time by providing at least 60 days prior written notice. Upon termination of the agreement, all rights to the compounds developed under the collaboration will revert to the Company.
For the year ended December 31, 2007, the Company recognized revenues under the Ortho-McNeil agreement of $13.4 million, which included $7.3 million from amortization of milestones and technology access and development fees received in prior years, $3.8 million for patent activities, and $2.3 million in research funding. For the year ended December 31, 2006, the Company recognized
84
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(6) COLLABORATIONS (Continued)
revenues under this agreement of $18.5 million, which included $7.5 million from amortization of milestones and technology access and development fees received in prior years, $5.0 million from a milestone earned, $2.4 million in research funding, and $3.6 million for additional sponsored research and patent activities. For the year ended December 31, 2005, the Company recognized revenues under this agreement of $13.4 million, which included $7.5 million from amortization of milestones and technology access and development fees received in prior years, $2.4 million in research funding, and $3.5 million for additional sponsored research and patent activities. At December 31, 2007, there were no deferred revenues remaining under this agreement.
Merck & Co., Inc.
In October 2002, the Company entered into a research and licensing agreement with Merck to collaborate on three G protein-coupled receptors, or GPCRs, to develop therapeutics for atherosclerosis and related disorders. The Company believes one or more of these GPCRs plays a role in regulating plasma lipid profiles, including HDL cholesterol, the so-called "good cholesterol," and is responsible for the HDL-raising activity of niacin. In October 2004, Merck extended and expanded the collaboration and selected one of the Company's compounds for preclinical development. In February 2007, the Company amended the Merck collaboration to reduce the number of the Company's research employees funded under the collaboration in exchange for Merck making a $1.0 million equity investment in the Company equal to the reduction in their research funding obligation and at approximately a 70% premium to the then current market price. In September 2006, the Company announced that Merck completed a Phase 2 clinical trial of MK-0354, a niacin receptor agonist discovered by the Company and intended for the treatment of atherosclerosis and related disorders. From the inception of this collaboration through December 31, 2007, the Company received $18.0 million from Merck in upfront and milestone payments and equity investments totaling $8.5 million. The Company may receive additional milestone payments of up to $28.0 million for Merck's clinical and marketing achievements, as well as royalty payments associated with Merck's commercialization of any products discovered under the agreement.
In addition, the Company received research funding from Merck through October 2007 totaling $27.5 million when, under the Company's amended agreement, Merck's obligation for research funding ended, and beyond which date the Company no longer performs services or has significant involvement.
The agreement with Merck will continue until the expiration of all royalty obligations under the agreement, unless the agreement is terminated early by either party. Either Merck or the Company can terminate the agreement if the other party breaches its material obligations under the agreement by causes and reasons within its control, has not cured such breach within 90 days of receiving a letter requesting such cure, and there is no dispute as to whether such breach has occurred. The non-breaching party in such a termination would receive the rights to continue the program. In addition, Merck can terminate the agreement at anytime by giving 90 days notice, but all milestones and royalties would still be payable as provided in the agreement.
As part of the extension and expansion of the collaboration with Merck in October 2004, Merck purchased $7.5 million of the Company's stock at approximately a 70% premium to the then current market price. The Company performed an evaluation on this stock purchase and determined that $3.9 million of the $7.5 million purchase price was an upfront payment related to the collaboration extension and expansion. Accordingly, the Company recognized the $3.9 million upfront payment, as
85
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(6) COLLABORATIONS (Continued)
well as the remaining portion of the unamortized upfront payment at October 2004 of $1.3 million, over the extended collaboration term of three years. Additionally, in October 2004, the Company achieved a $1.0 million milestone under the collaboration which the Company also recognized over the extended collaboration term of three years because the milestone was reasonably assured to be achieved at the time the Company extended and expanded its collaboration with Merck.
In connection with the February 2007 amendment of the collaborative agreement with Merck, the Company performed an evaluation on the stock purchase, which was at a purchase price of $24.81 per share, and determined that $0.5 million of the $1.0 million purchase price was an upfront payment related to the collaboration amendment. Accordingly, the Company recognized this upfront payment and the unamortized portion of the previously received upfront payments over the remaining term of the research portion of the collaboration. Merck's obligation for research funding ended in October 2007, beyond which date the Company no longer performs services or has significant involvement.
For the year ended December 31, 2007, the Company recognized revenues under the Merck agreement of $5.9 million, which included $3.6 million in research funding, $2.2 million from amortization of milestones and technology access and development fees received in prior years, and $0.1 million for patent activities. For the year ended December 31, 2006, the Company recognized revenues under this agreement of $12.1 million, which included $5.7 million in research funding, $4.0 million from a milestone earned, $2.1 million from amortization of milestones and technology access and development fees received in prior years, and $0.3 million for additional sponsored research and patent activities. For the year ended December 31, 2005, the Company recognized revenues under this agreement of $9.8 million, which included $5.7 million in research funding, $2.1 million from amortization of milestones and technology access and development fees received in prior years, and $2.0 million from a milestone earned. At December 31, 2007, there were no deferred revenues remaining under this agreement.
(7) REDEEMABLE CONVERTIBLE PREFERRED STOCK AND WARRANTS
In December 2003, the Company sold to two institutional investors 3,500 shares of series B-1 redeemable convertible preferred stock, or Series B-1 Preferred, together with (i) seven-year warrants to purchase up to 1,486,200 shares of common stock at an exercise price of $10.00 per share; and (ii) unit warrants giving such investors the right to purchase from the Company for a period of approximately 16 months from December 24, 2003, at their option, up to $11.5 million of series B-2 redeemable convertible preferred stock, or Series B-2 Preferred, and collectively with our Series B-1 Preferred, Series B Preferred, and additional seven-year warrants to purchase up to 450,000 shares of common stock at an initial exercise price of $10.00 per share. The aggregate purchase price was $35.0 million, and the Company received $34.2 million in net cash proceeds after closing costs. In addition, the Company issued 45,000 shares of common stock, valued at $0.3 million based on the fair value of the common stock on the date of the closing of the Series B-1 Preferred, as a finder's fee. In April 2005, the investors exercised their unit warrants in full, resulting in aggregate gross proceeds to the Company of $11.5 million.
In accordance with EITF Issue No. 00-27, "Application of Issue No. 98-5 for Certain Convertible Instruments," the Company allocated the components of the sale of the Series B-1 Preferred between the Series B-1 Preferred, the warrants and the unit warrants on the basis of the relative fair values at the date of issuance using the Black-Scholes model. The aggregate amount allocated to the warrants
86
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(7) REDEEMABLE CONVERTIBLE PREFERRED STOCK AND WARRANTS (Continued)
and unit warrants was $6.5 million. The fair value of the common shares into which the Series B-1 Preferred was convertible on the date of issuance exceeded the proceeds allocated to the Series B-1 Preferred by $2.8 million, resulting in a beneficial conversion feature that was recognized as an increase to paid-in capital and as a deemed dividend to the Series B-1 Preferred.
The Company valued the components of the Series B-1 Preferred as follows, in thousands:
| Series B-1 Preferred | | $ | 25,740 |
| Warrants | | | 4,535 |
| Deemed dividend | | | 2,800 |
| Unit warrants | | | 1,925 |
| | |
|
| | Total | | $ | 35,000 |
| | |
|
The holders of the Company's Series B-1 Preferred can require the Company at any time to redeem all or some of their shares of Series B-1 Preferred at such shares' stated value, plus accrued but unpaid dividends thereon to the date of payment and any applicable penalties. The stated value is the original holder's investment plus any dividends settled by increasing the stated value at the time the dividend is payable. The Company may be able to satisfy all or a portion of any redemption with shares of its common stock. The Series B-1 Preferred is convertible into common stock at a fixed conversion price of $7.50 per share. As a result of the public offering the Company completed in February 2005 which resulted in the Series B-1 Preferred becoming immediately redeemable at the option of the holders, the Company recorded a charge of $7.4 million in 2005 to accrete the remaining unaccreted discount and deemed dividend on the redeemable convertible preferred stock. The Company will be required to redeem any shares of the Series B-1 Preferred that remain outstanding on December 24, 2008 at a price equal to the amount of the original holder's original investment, plus all accrued but unpaid dividends thereon to the date of such payment. The Series B-1 Preferred, which accrues dividends at 4% annually, had an aggregate redemption price of $41.1 million at December 31, 2007.
If not previously converted, the Company must redeem the Series B-2 Preferred on April 22, 2010, or earlier under certain circumstances, at such shares' stated value, plus accrued but unpaid dividends thereon to the date of payment and any applicable penalties. The Series B-2 Preferred, which accrues dividends at 4% annually, had an aggregate redemption price of $12.8 million at December 31, 2007. The Company may be able to satisfy all or a portion of any redemption with shares of its common stock. The Series B-2 Preferred is convertible into common stock at a fixed conversion price of $7.00 per share. Otherwise, the Series B-2 Preferred has substantially identical terms as the Series B-1 Preferred. The holders of the Company's Series B-2 Preferred will be entitled to require the Company to redeem their shares of Series B-2 Preferred at such shares' stated value, plus accrued but unpaid dividends thereon to the date of payment and any applicable penalties if, in the future, the average of the closing price of the Company's common stock for any 30 consecutive trading days is below $7.00 per share, which is the conversion price for the Series B-2 Preferred.
Assuming that the Series B-1 Preferred and the Series B-2 Preferred are held until the applicable mandatory redemption date, the Company expects to record dividends on redeemable convertible preferred stock of $2.2 million, $0.5 million and $0.2 million for the years ending December 31, 2008, 2009 and 2010, respectively.
87
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(7) REDEEMABLE CONVERTIBLE PREFERRED STOCK AND WARRANTS (Continued)
At the option of any holder of Series B Preferred, any Series B Preferred held by such holder may be converted into common stock based on the applicable conversion price then in effect for such shares. If the Company is permitted to satisfy a portion of a redemption by using shares of its common stock, and if the Company elects to do so, the number of shares to be issued to holders of Series B Preferred will be determined by dividing such holder's cash redemption price by the lesser of the fixed conversion price or 95% of the arithmetic average of the volume weighted-average price of the Company's common stock for, depending on the specified circumstances, 10 or 15 consecutive trading days prior to the delivery of the redemption notice or date of the triggering event.
If the Company is required to redeem all or some of the currently outstanding shares of its Series B Preferred, the Company may be able to pay all or a portion of the redemption price using shares of its common stock if certain enumerated conditions are satisfied, including: (i) the Company has sufficient number of shares of common stock available for issuance; (ii) the shares of common stock to be issued are registered under an effective registration statement or are otherwise available for sale under Rule 144(k) under the Securities Act; (iii) the Company's common stock is listed on the NASDAQ Global Market or other eligible market; (iv) the shares to be issued can be issued without violating the rules of the NASDAQ Global Market or any applicable trading market or a provision of the Company's Certificate of Designations governing the Company's Series B Preferred, or Series B Certificate of Designations; and (v) no bankruptcy event has occurred.
Also, the holders of the Series B-2 Preferred may require the Company to redeem their shares if the Company issues common stock or common stock equivalents for an effective net price of less than $5.33 per share (excluding, among other things, certain common stock and common stock equivalents issued or issuable (i) to the Company's officers, directors, employees or consultants, (ii) in connection with certain strategic partnerships or joint ventures, and (iii) in connection with certain mergers and acquisitions). "Effective net price" is not defined in the Series B Certificate of Designations. The holders of the Company's Series B-2 Preferred may assert that effective net price should be calculated as the amount the Company receives after paying any discounts and other expenses related to any such issuance.
In addition to the foregoing redemption rights, at any time following the occurrence of a "Triggering Event," a holder of the Series B Preferred may require the Company to repurchase all or any portion of the Series B Preferred then held by such holder at a price per share equal to the greater of 115% of the stated value (as calculated under the Series B Certificate of Designations) of such shares plus all accrued but unpaid dividends thereon to the date of payment. "Triggering Event" is specifically defined in the Series B Certificate of Designations, and includes any of the following events: (i) immediately prior to a bankruptcy event; (ii) the Company fails for any reason to timely deliver a certificate evidencing any securities to a purchaser or the exercise or conversion rights of the holders are otherwise suspended for other than a permissible reason; (iii) any of certain events of default (as set forth in the Registration Rights Agreement with the Series B Preferred holders) occur and remain uncured for 60 days; (iv) the Company fails to make any cash payment required under the Series B Preferred transaction documents and such failure is not timely cured; (v) the issuance of a going concern opinion by the Company's independent registered public accounting firm that is not timely cured; (vi) the Company breaches a section of the Series B Preferred purchase agreement relating to indebtedness and subordination; or (vii) the Company defaults in the timely performance of any other obligation under the Series B Preferred transaction documents and such default is not timely cured.
88
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(7) REDEEMABLE CONVERTIBLE PREFERRED STOCK AND WARRANTS (Continued)
If the closing price of the Company's common stock is equal to or above $14.00 per share for 30 consecutive trading days, upon 10 trading days' prior written notice, the Company has the right to, and the warrant holders will have the right to require the Company to, call and cancel any unexercised portion of the warrants. Upon exercise of a warrant following such call notice and prior to the warrant cancellation date, the Company will be obligated to issue to the warrant holder an exchange warrant entitling the holder to purchase shares of the Company's common stock equal to the amount of the holder's warrant that was called. This exchange warrant would contain the same terms and conditions as the original warrant, except that the maturity date would be seven years from the date of issuance of such exchange warrant and the exercise price would be equal to 130% of the average of the volume weighted-average price of the Company's common stock for the five trading days preceding the original warrant cancellation date.
On March 31, 2006, following the Company's call notice to one of the two warrant holders, Smithfield Fiduciary LLC, such holder exercised its warrants to purchase 829,856 shares of the Company's common stock, resulting in gross proceeds of $8.3 million. In connection with this exercise in full of its warrants, Smithfield claimed that it was entitled to receive exchange warrants that would include a provision that could require the Company to issue additional exchange warrants in the future. The Company disagreed with this interpretation. On June 30, 2006, the Company entered into a Settlement Agreement and Release with Smithfield. As part of the Settlement Agreement and Release, (a) Smithfield and the Company provided each other with a release of any claims relating to (i) Smithfield's demand for, and the Company's non-issuance of, exchange warrants, and (ii) any breach or default under certain of the agreements on account of the foregoing, (b) the Company issued Smithfield a seven-year warrant to purchase 829,856 shares of the Company's common stock at an initial exercise price of $15.49 per share, and (c) the Company filed a registration statement covering the sale of the shares of common stock issuable under their new warrant. The new warrant does not contain any right for the Company, or for the holder to require the Company, to call the warrant, nor does it provide the holder the right to receive any exchange warrants in the future. The Company recorded a non-cash charge of $4.6 million related to the warrant settlement in the second quarter of 2006. The Company does not know whether it will have a similar dispute with its other warrant holder, Mainfield Enterprises, or, if it does, the likely outcome of the dispute. As such, the Company has not recorded any charges related to the Mainfield warrant.
Each investor agrees that for so long as it holds Series B-1 Preferred and Series B-2 Preferred, it shall vote its shares of Series B-1 Preferred, Series B-2 Preferred and common stock on all matters in which such investor is entitled to vote and on which holders of common stock have the right to vote, in the manner recommended by the Company's board of directors to all of its stockholders unless the Company's board of directors elects to permit the investors to vote such shares in their own discretion.
(8) STOCKHOLDERS' EQUITY
Preferred Stock
In October 2002, and in conjunction with the stockholders' rights plan (see "Stockholders' Rights Plan" below in this note), the Company's board of directors created a series of preferred stock, consisting of 350,000 shares with a par value of $.0001 per share, designated as Series A Junior Participating Preferred Stock, or the Series A Preferred Stock. Such number of shares may be increased or decreased by the Company's board of directors, provided that no decrease shall reduce the number
89
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
of shares of Series A Preferred Stock to a number less than the number of shares then outstanding, plus the number of shares reserved for issuance upon the exercise of outstanding options, rights or warrants or upon the conversion of any outstanding securities issued by the Company convertible into Series A Preferred Stock. As of December 31, 2007, no shares of Series A Preferred Stock were issued or outstanding.
Treasury Stock
In October 2003, Biotechnology Value Fund, L.P. and certain of its affiliates accepted the Company's offer of $23.1 million to purchase from them 3,000,000 shares of the Company's common stock at a cash price of $7.69 per share.
Equity Compensation Plans
In June 2006, the Company's stockholders approved the Company's 2006 Long-Term Incentive Plan, as amended, or the 2006 LTIP, which provides for the grant of up to a total of 6,000,000 shares of common stock (subject to certain adjustments described in the 2006 LTIP) to designated employees, certain consultants and advisors who perform services for the Company, and non-employee members of the Company's board of directors as stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards and performance awards. Effective in June 2006, the Company's Amended and Restated 1998 Equity Compensation Plan, Amended and Restated 2000 Equity Compensation Plan and 2002 Equity Compensation Plan, or the Prior Plans, were terminated. However, notwithstanding such termination, all outstanding awards under the Prior Plans will continue to be governed under the terms of the Prior Plans. The 6,000,000 shares of common stock authorized for issuance under the 2006 LTIP is increased by the number of shares subject to any stock awards under the Prior Plans that are forfeited, expire or otherwise terminate without the issuance of such shares and as otherwise provided in the 2006 LTIP. As of December 31, 2007, a total of 2,610,858 shares of common stock were available for future grant under the 2006 LTIP.
Stock options generally vest 25% per year over four years and are exercisable for up to 10 years from the date of grant. Restricted common stock generally vests over a two, three or four-year period and the recipient, at the date of grant, has all rights of a stockholder, subject to certain restrictions on transferability and a risk of forfeiture. The Company issues new shares of common stock upon the exercise of stock options, for purchases made under the Purchase Plan and for grants of restricted stock.
In the event of termination of service, unvested restricted stock is subject to forfeiture and restricted common stock issued from the exercise of unvested stock options is subject to repurchase at the original purchase price. In the event the Company elects to not buy back any such unvested shares, any related compensation will be expensed immediately. In accordance with SFAS No. 128, the Company has excluded all unvested restricted stock and restricted common stock issued from the exercise of unvested stock options from its calculation of basic and diluted net loss per share.
90
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
In 2003, the Company set up a deferred compensation plan for its executive officers, whereby executive officers elected to contribute their shares of restricted stock into the plan. At December 31, 2007, 2006 and 2005, a total of 107,919, 114,169 and 134,169 shares of restricted stock were contributed to the plan, respectively.
The following table summarizes the Company's stock option activities under the Prior Plans and the 2006 LTIP, or collectively, the Equity Compensation Plans, for the years ended December 31, 2007, 2006 and 2005:
| | Options
| | Weighted-
Average
Exercise Price
| | Weighted-Average
Remaining Contractual
Term
(in years)
| | Aggregate Intrinsic
Value
(in thousands)
|
|---|
| Outstanding at December 31, 2004 | | 2,780,399 | | $ | 8.66 | | | | | |
| Granted | | 1,201,635 | | | 6.73 | | | | | |
| Exercised | | (75,790 | ) | | 5.34 | | | | | |
| Forfeited/cancelled/expired | | (253,413 | ) | | 9.08 | | | | | |
| | |
| | | | | | | | |
| Outstanding at December 31, 2005 | | 3,652,831 | | | 8.07 | | | | | |
| Granted | | 1,139,384 | | | 13.51 | | | | | |
| Exercised | | (180,364 | ) | | 6.56 | | | | | |
| Forfeited/cancelled/expired | | (89,470 | ) | | 10.81 | | | | | |
| | |
| | | | | | | | |
| Outstanding at December 31, 2006 | | 4,522,381 | | | 9.44 | | | | | |
| Granted | | 1,437,787 | | | 13.04 | | | | | |
| Exercised | | (206,571 | ) | | 5.96 | | | | | |
| Forfeited/cancelled/expired | | (239,595 | ) | | 11.25 | | | | | |
| | |
| |
| |
| |
|
| Outstanding at December 31, 2007 | | 5,514,002 | | $ | 10.43 | | 6.90 | | $ | 4,304 |
| | |
| |
| |
| |
|
| Vested and expected to vest at December 31, 2007 | | 5,255,585 | | $ | 10.34 | | 6.81 | | $ | 4,249 |
| | |
| |
| |
| |
|
| Vested and exercisable at December 31, 2007 | | 2,984,227 | | $ | 9.18 | | 5.54 | | $ | 3,516 |
| | |
| |
| |
| |
|
The aggregate intrinsic value in the above table is calculated as the difference between the closing price of the Company's common stock at December 31, 2007 of $7.83 per share and the exercise price of stock options that had strike prices below the closing price. The intrinsic value of all stock options exercised during the years ended December 31, 2007, 2006 and 2005 was $1.2 million, $1.7 million and $0.4 million, respectively.
91
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
The following table summarizes outstanding and exercisable stock options as of December 31, 2007:
| | Options Outstanding
| | Options Exercisable
|
|---|
Range of Exercise Prices
| | Number
Outstanding
| | Weighted-Average
Remaining Contractual
Life (in years)
| | Weighted-Average
Exercise Price
| | Number
Exercisable
| | Weighted-Average
Exercise Price
|
|---|
| $0.20 - $5.99 | | 342,700 | | 4.70 | | $ | 3.24 | | 305,138 | | $ | 3.02 |
| $6.00 - $6.01 | | 669,312 | | 5.89 | | | 6.00 | | 566,486 | | | 6.00 |
| $6.12 - $6.16 | | 763,846 | | 6.83 | | | 6.16 | | 488,155 | | | 6.16 |
| $6.30 - $10.14 | | 572,698 | | 5.99 | | | 8.34 | | 435,199 | | | 8.37 |
| $10.21 - $11.31 | | 720,346 | | 7.50 | | | 10.58 | | 357,589 | | | 10.62 |
| $11.37 - $13.39 | | 584,625 | | 6.15 | | | 12.17 | | 356,440 | | | 12.21 |
| $13.50 - $13.50 | | 1,037,587 | | 9.15 | | | 13.50 | | 75,737 | | | 13.50 |
| $13.60 - $31.34 | | 822,888 | | 6.50 | | | 17.23 | | 399,483 | | | 18.18 |
| | |
| |
| |
| |
| |
|
| $0.20 - $31.34 | | 5,514,002 | | 6.90 | | $ | 10.43 | | 2,984,227 | | $ | 9.18 |
| | |
| |
| |
| |
| |
|
Stock options exercisable pursuant to the terms of the Prior Plans can be exercised prior to vesting; however, unvested shares are subject to repurchase at the original purchase price if a grantee terminates employment prior to vesting. At December 31, 2007, 2006 and 2005, 312, 924 and 1,537 shares of common stock issued upon the exercise of stock options were subject to repurchase at the original purchase price at a weighted-average price of $6.16, $6.11 and $6.10 per share, respectively.
In February 2007, the Company granted 1,690,500 performance-based restricted stock unit awards under the 2006 LTIP. The awards provide employees until February 26, 2012 to achieve four key drug development and strategic performance goals. A fixed number of awards will be earned for each goal that is successfully achieved. Once earned, the awards will remain unvested until the performance period is complete. The awards that have been earned at February 26, 2012 will vest and be settled in shares of the Company's common stock, with the holder receiving one share of common stock for each award earned and vested. Termination of employment prior to vesting will result in the forfeiture of any earned (as well as unearned) awards, except for in limited circumstances such as termination due to death, disability or a change in control. No compensation expense was recognized related to these awards during the year ended December 31, 2007 as management believes achievement of the performance goals is not probable at December 31, 2007. The following table summarizes activity with respect to such awards during the year ended December 31, 2007:
| | Performance
Units
| | Weighted-Average
Grant-Date Fair
Value
|
|---|
| Outstanding at January 1, 2007 | | — | | $ | — |
| Granted | | 1,690,500 | | | 13.50 |
| Vested | | — | | | — |
| Forfeited/cancelled | | (54,900 | ) | | 13.50 |
| | |
| |
|
| Outstanding at December 31, 2007 | | 1,635,600 | | $ | 13.50 |
| | |
| |
|
| Vested at December 31, 2007 | | — | | | — |
| | |
| |
|
92
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
The following table summarizes the Company's unvested restricted stock activity, excluding shares contributed to the Company's deferred compensation plan, during the years ended December 31, 2007, 2006 and 2005:
Unvested Restricted Stock
| | Shares
| | Weighted-Average
Grant-Date Fair
Value
|
|---|
| Unvested at December 31, 2004 | | 249,497 | | $ | 6.45 |
| Granted | | 8,000 | | | 6.78 |
| Vested | | (187,001 | ) | | 6.46 |
| Forfeited | | — | | | — |
| | |
| |
|
| Unvested at December 31, 2005 | | 70,496 | | | 6.47 |
| Granted | | 81,000 | | | 16.80 |
| Vested | | (51,997 | ) | | 6.44 |
| Forfeited | | — | | | — |
| | |
| |
|
| Unvested at December 31, 2006 | | 99,499 | | | 14.89 |
| Granted | | — | | | — |
| Vested | | (41,499 | ) | | 13.19 |
| Forfeited | | — | | | — |
| | |
| |
|
| Unvested at December 31, 2007 | | 58,000 | | $ | 16.11 |
| | |
| |
|
The total grant-date fair value of restricted stock vested during the years ended December 31, 2007, 2006 and 2005 was $0.5 million, $0.3 million and $1.2 million, respectively.
Share-based Compensation
The Company uses the Black-Scholes option pricing model to estimate the grant-date fair value of share-based awards in determining the share-based compensation expense recognized under SFAS No. 123R. The table below sets forth the weighted-average assumptions and estimated fair value of stock options granted under the Equity Compensation Plans during the years ended December 31, 2007 and 2006:
| | December 31,
| |
|---|
| | 2007
| | 2006
| |
|---|
| Risk-free interest rate | | | 4.6 | % | | 4.6 | % |
| Dividend yield | | | 0 | % | | 0 | % |
| Expected volatility | | | 64 | % | | 70 | % |
| Expected life (years) | | | 5.39 | | | 5.19 | |
| Weighted-average estimated fair value of stock options granted | | $ | 7.82 | | $ | 8.35 | |
93
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
The table below sets forth the weighted-average assumptions and estimated fair value of the options to purchase stock granted under the Purchase Plan for multiple offering periods during the years ended December 31, 2007 and 2006:
| | December 31,
|
|---|
| | 2007
| | 2006
|
|---|
| Risk-free interest rate | | 3.8% - 5.3% | | 1.7% - 5.3% |
| Dividend yield | | 0% | | 0% |
| Expected volatility | | 66% - 72% | | 65% - 75% |
| Expected life (years) | | 0.25 - 2.0 | | 0.25 - 2.0 |
| Weighted-average estimated fair value of options granted under the Purchase Plan | | $2.18 to $5.46 | | $1.99 to $7.29 |
Expected volatility for awards granted after adoption of SFAS No. 123R is based on a combination of 75% historical volatility of the Company's common stock and 25% market-based implied volatilities from traded options on its common stock, with historical volatility being more heavily weighted due to low volume of traded options on its common stock. Prior to adoption of SFAS No. 123R, the Company's computation of expected volatility was based only on historical volatility of its common stock. The expected life of options granted under SFAS No. 123R is determined based on historical experience of similar awards, giving consideration to the contractual terms of the share-based awards, vesting schedules and post-vesting terminations. Prior to the adoption of SFAS No. 123R, an average expected life of five years was used in determining the fair value of option grants based on the vesting period of the options due to the short period of time the Company's stock had been publicly traded. The risk-free interest rates are based on the US Treasury yield curve, with a remaining term approximately equal to the expected term used in the option pricing model.
SFAS No. 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Forfeitures of unvested stock options were estimated to be 5.4% and 6.7% for the years ended December 31, 2007 and 2006 based on historical experience. As a result, the Company reduced its share-based compensation expense by $0.4 million for each of the years ended December 31, 2007 and 2006. If actual forfeitures vary from these estimates, the Company will recognize the difference in compensation cost in the period the actual forfeitures occur or when stock options vest. Prior to the adoption of SFAS No. 123R, the Company accounted for forfeitures as they occurred in the pro forma disclosure required under SFAS No. 123.
94
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
The Company recognized share-based compensation expense in accordance with SFAS No. 123R as follows (in thousands, except per share data):
| | December 31,
|
|---|
| | 2007
| | 2006
|
|---|
| Research and development | | $ | 4,190 | | $ | 2,901 |
| General and administrative | | | 4,626 | | | 2,149 |
| | |
| |
|
| Total share-based compensation expense and impact on net loss allocable to common stockholders | | $ | 8,816 | | $ | 5,050 |
| | |
| |
|
| Impact on net loss per share allocable to common stockholders, basic and diluted | | $ | 0.14 | | $ | 0.11 |
| | |
| |
|
At December 31, 2007, total unrecognized estimated compensation cost, excluding estimated forfeitures, related to unvested stock options was $13.6 million, which is expected to be recognized over a weighted-average remaining requisite service period of 2.44 years. At December 31, 2007, total unrecognized estimated compensation cost related to restricted stock was $0.5 million, which is expected to be recognized over a weighted-average remaining requisite service period of 1.09 years.
Cash of $1.2 million was received from stock option exercises during the year ended December 31, 2007. Cash of $1.9 million was received from stock purchases under the Purchase Plan during the year ended December 31, 2007. Tax benefits recognized related to share-based compensation and related cash flow impacts were not material during the year ended December 31, 2007 because the Company is in a net operating loss position.
Employee Stock Purchase Plan
The Purchase Plan qualifies under Section 423 of the Internal Revenue Service and permits substantially all employees to purchase shares of the Company's common stock at a discount to market. Under the Purchase Plan, employees can choose to have up to 15% of their annual compensation withheld to purchase shares of common stock, subject to certain limitations. The shares of common stock may be purchased over an offering period with a maximum duration of two years at 85% of the lower of the fair market value of the common stock on the first day of the applicable offering period or on the last day of the three-month purchase period. In June 2006, the Company's stockholders approved an increase in the aggregate number of shares of common stock that may be issued pursuant to the Purchase Plan from 1,000,000 to 1,500,000. During the years ended December 31, 2007, 2006 and 2005, 235,726, 307,086 and 197,862 shares, respectively, were purchased under to the Purchase Plan. As of December 31, 2007, a total of 1,050,406 shares has been issued under the Purchase Plan.
95
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(8) STOCKHOLDERS' EQUITY (Continued)
Common Shares Reserved for Future Issuance
The following shares of common stock are reserved for future issuance at December 31, 2007:
| Equity Compensation Plans | | 9,760,460 |
| Deferred compensation plan | | 107,919 |
| Warrants | | 1,936,200 |
| Series B-1 Preferred | | 5,481,740 |
| Series B-2 Preferred | | 1,829,909 |
| Payment of dividends | | 1,935,084 |
| Purchase Plan | | 449,594 |
| | |
|
| | Total | | 21,500,906 |
| | |
|
Stockholders' Rights Plan
In October 2002, the Company's board of directors adopted a stockholders' rights plan, or the Rights Agreement, under which all stockholders of record as of November 13, 2002 received rights to purchase shares of the Series A Preferred Stock, or the Rights. Each Right entitles the registered holder to purchase from the Company one one-hundredth of a share of the Series A Preferred Stock at an initial exercise price of $36.00 per share, subject to adjustment. The Rights are not exercisable until the 10th day after such time as a person or group acquires beneficial ownership of 10% or more, or announces a tender offer for 10% or more, of the Company's common stock. At such time, all holders of the Rights, other than the acquiror, will be entitled to purchase shares of the Company's common stock at a 50% discount to the then current market price.
The Rights will trade with the Company's common stock, unless and until they are separated due to a person or group acquiring beneficial ownership of 10% or more, or announcing a tender offer for 10% or more, of the Company's common stock. The Company's board of directors may terminate the Rights Agreement at any time or redeem the Rights prior to the time a person acquires 10% or more of the common stock.
In November 2006, the Rights Agreement was amended to provide, among other things, that the triggering percentage for when a Beneficial Owner (as defined in the Rights Agreement) of the Company's common stock would be an Acquiring Person (as further defined in the Amendment) increased from 10% to 15%.
(9) EMPLOYEE BENEFIT PLAN
The Company has a defined contribution retirement plan that complies with Section 401(k) of the Internal Revenue Code. All employees of the Company are eligible to participate in the plan. The Company matches 100% of each participant's voluntary contributions, subject to a maximum Company contribution of 6% of the participant's compensation. The Company's matching portion, which totaled $1.4 million in each of the years ended December 31, 2007 and 2006 and $1.1 million in the year ended December 31, 2005, vests over a five-year period from the date of hire.
96
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(10) INCOME TAXES
In July 2006, the FASB issued FASB Interpretation No., or FIN, 48, "Accounting for Uncertainty in Income Taxes—An Interpretation of SFAS No. 109," which clarifies the accounting for uncertainty in income taxes recognized in an entity's financial statements in accordance with SFAS No. 109, "Accounting for Income Taxes," and prescribes a recognition threshold and measurement attributes for financial statement disclosure of tax positions taken or expected to be taken on a tax return. Under FIN 48, the impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. FIN 48 is effective for fiscal years beginning after December 15, 2006.
The Company adopted the provisions of FIN 48 on January 1, 2007. The total amount of unrecognized tax benefits as of the date of adoption was $8.6 million. Pursuant to Sections 382 and 383 of the Internal Revenue Code, annual use of the Company's net operating loss and credit carryforwards could be limited in the event of cumulative changes in ownership of more than 50%. Such a change occurred in prior years, and the Company is currently undergoing a Section 382/383 analysis. Until this analysis has been completed, the Company has removed the deferred tax assets for net operating losses of $131.3 million and research and development credits of $36.5 million from its deferred tax asset schedule. As such, the Company has recorded a corresponding decrease to its valuation allowance.
A rollforward of changes in the Company's unrecognized tax benefits is shown below, in thousands:
| Balance at January 1, 2007 | | $ | (8,566 | ) |
| Additions based on tax positions related to the current year | | | — | |
| Additions for tax positions of prior years | | | — | |
| Reductions for tax positions of prior years | | | 8,566 | |
| Settlements | | | — | |
| | |
| |
| Balance at December 31, 2007 | | $ | — | |
| | |
| |
Due to the existence of the valuation allowance, future changes in the Company's unrecognized tax benefits will not likely impact the effective tax rate.
The Company's practice is to recognize interest and/or penalties related to income tax matters in income tax expense. The Company did not have any accrued interest or penalties included in its consolidated balance sheets at December 31, 2007 or 2006, and did not recognize any interest and/or penalties in its consolidated statement of operations during the year ended December 31, 2007.
The Company is subject to income taxation in the United States at the federal and state levels. The Company's tax years for 1997 and later are subject to examination by the United States and California tax authorities due to the carryforward of unutilized net operating losses and research and development credits. The Company is currently not under examination by any taxing authorities.
The adoption of FIN 48 did not impact the Company's financial condition, results of operations or cash flows. At December 31, 2007, the Company had net deferred tax assets of $13.9 million. The deferred tax assets are primarily comprised of deferred revenues, SFAS No. 123R expense, depreciation
97
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(10) INCOME TAXES (Continued)
and capitalized research and development costs. Due to uncertainties surrounding the Company's ability to generate future taxable income to realize these assets, the Company has not recognized these assets and a full valuation allowance has been established to offset the Company's net deferred tax assets. The future utilization of the Company's NOL and R&D credit carryforwards to offset future taxable income may be subject to a substantial annual limitation as a result of ownership changes that may have occurred previously or that could occur in the future. The Company has not yet determined when such an ownership change occurred; however, the Company is in the process of completing a Section 382/383 analysis regarding the limitation of the NOL and R&D credit carryforwards. Until this analysis has been completed, the Company has removed the deferred tax assets associated with these carryforwards from its deferred tax asset schedule and has recorded a corresponding decrease to their valuation allowance. When the Section 382/383 analysis is completed, the Company plans to update its unrecognized tax benefits under FIN 48. The Company expects the Section 382/383 analysis to be completed within the next twelve months.
Significant components of the Company's deferred tax assets at December 31, 2007 and 2006 are shown below, in thousands. A valuation allowance of $13.9 million and $137.0 million has been recognized to offset the net deferred tax assets as of December 31, 2007 and 2006, respectively, as realization of such assets is uncertain. The valuation allowance decreased by $123.1 million in 2007 compared to 2006, primarily due to the removal of the net operating losses and research and development credits from the Company's deferred tax assets.
| | December 31,
| |
|---|
| | 2007
| | 2006
| |
|---|
| Deferred tax assets: | | | | | | | |
| | Net operating loss carryforwards | | $ | — | | $ | 99,913 | |
| | Research and development credits | | | — | | | 27,846 | |
| | Capitalized R&D (state) | | | 1,920 | | | 2,330 | |
| | Deferred revenues | | | 7,426 | | | 5,200 | |
| | Depreciation | | | 2,030 | | | 1,518 | |
| | SFAS No. 123R expense | | | 2,383 | | | 830 | |
| | Other, net | | | 2,065 | | | 1,900 | |
| | |
| |
| |
| Total deferred tax assets | | | 15,824 | | | 139,537 | |
| Deferred tax liabilities: | | | | | | | |
| | Acquired intangible amortization | | | (1,942 | ) | | (2,554 | ) |
| | |
| |
| |
| Total deferred tax liabilities | | | (1,942 | ) | | (2,554 | ) |
| | |
| |
| |
| Net deferred tax assets | | | 13,882 | | | 136,983 | |
| Valuation allowance | | | (13,882 | ) | | (136,983 | ) |
| | |
| |
| |
| Net deferred tax assets | | $ | — | | $ | — | |
| | |
| |
| |
At December 31, 2007, the Company had Federal tax net operating loss carryforwards of $345.0 million that will begin to expire in 2017 unless previously utilized. At the same date, the Company had state tax net operating loss carryforwards of $302.9 million, which does not include $12.8 million which expired in 2007. The California net operating loss carryforwards will begin to expire in 2012. At December 31, 2007, $9.1 million of net operating loss carryforwards related to stock option
98
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(10) INCOME TAXES (Continued)
exercises, which will result in an increase to additional paid-in capital and a decrease in income taxes payable at the time when the tax loss carryforwards are utilized. The Company also had Federal and California research and development tax credit carryforwards of $25.3 million and $17.0 million, respectively. The Federal research and development credit carryforwards will begin to expire in 2012 unless previously utilized. The California research and development credit carryforwards carry forward indefinitely.
The provision for income taxes on earnings subject to income taxes differs from the statutory Federal rate at December 31, 2007, 2006 and 2005, due to the following, in thousands:
| | December 31,
| |
|---|
| | 2007
| | 2006
| | 2005
| |
|---|
| Statutory Federal rate | | $ | (49,395 | ) | $ | (30,014 | ) | $ | (26,209 | ) |
| State income tax, net of Federal benefit | | | (6,391 | ) | | (5,146 | ) | | (4,494 | ) |
| Permanent items and other | | | 3,053 | | | 739 | | | (183 | ) |
| SFAS No. 123R expense | | | 1,589 | | | 1,208 | | | — | |
| Deferred compensation | | | — | | | — | | | 175 | |
| Foreign losses | | | 12,125 | | | — | | | — | |
| Research and development credit | | | (8,321 | ) | | (5,353 | ) | | (5,074 | ) |
| Dividends and accretion on preferred stock | | | 842 | | | 809 | | | 3,658 | |
| Removal of NOL's and R&D credits | | | 169,599 | | | — | | | — | |
| Valuation allowance and other | | | (123,101 | ) | | 37,757 | | | 32,127 | |
| | |
| |
| |
| |
| Provision for income taxes | | $ | — | | $ | — | | $ | — | |
| | |
| |
| |
| |
(11) SUBSEQUENT EVENT
On January 9, 2008, the Company acquired from Siegfried Ltd, or Siegfried, certain drug product facility assets, including fixtures, equipment, other personal property and real estate assets under an Asset Purchase Agreement, between Siegfried and the Company's wholly owned Swiss subsidiary, Arena Pharmaceuticals GmbH. The purchase price under such agreement, in Swiss francs, was CHF 31.8 million in cash and 1,488,482 shares of the Company's common stock, which were issued to Siegfried on January 8, 2008. The Company paid CHF 21.8 million, or $19.8 million, of the cash purchase price at the closing of such transaction, and will pay the remaining CHF 10.0 million in three equal installments in the third, fourth and fifth years after closing. In connection with this transaction, the Company and Siegfried also entered into a long-term supply agreement for the active pharmaceutical ingredient of lorcaserin, a contract manufacturing agreement and a technical services agreement.
99
ARENA PHARMACEUTICALS, INC.
Notes to Consolidated Financial Statements (Continued)
(12) QUARTERLY FINANCIAL DATA (UNAUDITED)
The following table presents quarterly data for the years ended December 31, 2007 and 2006, in thousands, except per share data:
2007
| | Quarter ended
December 31
| | Quarter ended
September 30
| | Quarter ended
June 30
| | Quarter ended
March 31
| | Year ended
December 31
| |
|---|
| Revenues | | $ | 4,569 | | $ | 5,041 | | $ | 4,811 | | $ | 4,911 | | $ | 19,332 | |
| Net loss | | | (40,386 | ) | | (32,278 | ) | | (38,608 | ) | | (31,895 | ) | | (143,166 | ) |
| Net loss allocable to common stockholders | | | (40,926 | ) | | (32,813 | ) | | (39,132 | ) | | (32,409 | ) | | (145,280 | ) |
| Net loss per share allocable to common stockholders, basic and diluted | | $ | (0.60 | ) | $ | (0.54 | ) | $ | (0.64 | ) | $ | (0.53 | ) | $ | (2.31 | ) |
2006
| | Quarter ended December 31
| | Quarter ended September 30
| | Quarter ended June 30
| | Quarter ended March 31
| | Year ended December 31
| |
|---|
| Revenues | | $ | 4,699 | | $ | 4,416 | | $ | 9,328 | | $ | 12,126 | | $ | 30,569 | |
| Net loss | | | (35,889 | ) | | (19,624 | ) | | (18,509 | ) | | (12,226 | ) | | (86,248 | ) |
| Net loss allocable to common stockholders | | | (36,409 | ) | | (20,138 | ) | | (19,013 | ) | | (12,719 | ) | | (88,279 | ) |
| Net loss per share allocable to common stockholders, basic and diluted | | $ | (0.73 | ) | $ | (0.43 | ) | $ | (0.40 | ) | $ | (0.30 | ) | $ | (1.89 | ) |
100
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Based on this evaluation, our principal executive officer and our principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this annual report.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining for us adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Under the supervision and with the participation of our management, including our CEO and VP, Finance and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework inInternal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2007.
The registered public accounting firm that audited the financial statements included in this Annual Report on Form 10-K has issued an attestation report on our internal control over financial reporting, and such report is included below.
Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting during the fourth quarter of the period covered by this Annual Report that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
101
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Arena Pharmaceuticals, Inc.
We have audited Arena Pharmaceuticals, Inc.'s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Arena Pharmaceuticals, Inc.'s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Arena Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2007 of Arena Pharmaceuticals, Inc. and our report dated February 29, 2008 expressed an unqualified opinion thereon.
| | /s/ Ernst & Young LLP |
San Diego, California
February 29, 2008 |
|
102
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
We have adopted a Code of Business Conduct and Ethics that applies to our directors and employees (including our principal executive officer, principal financial officer, principal accounting officer and controller), and have posted the text of the policy on our website (www.arenapharm.com) in connection with "Investor" materials. In addition, we intend to promptly disclose (i) the nature of any amendment to the policy that applies to our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions and (ii) the nature of any waiver, including an implicit waiver, from a provision of the policy that is granted to one of these specified individuals, the name of such person who is granted the waiver and the date of the waiver on our website in the future.
The other information required by this item is incorporated herein by reference from the information under the captions "Election of Directors," "Compensation and Other Information Concerning Executive Officers, Directors and Certain Stockholders" and "Section 16(a) Beneficial Ownership Reporting Compliance" contained in our proxy statement for the annual meeting of stockholders to be held in June 2008, or the Proxy Statement.
Item 11. Executive Compensation.
The information required by this item is incorporated herein by reference from the information under the captions "Compensation and Other Information Concerning Executive Officers, Directors and Certain Stockholders," "Compensation Committee Interlocks and Insider Participation" and "Compensation Committee Report" contained in the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
Information relating to securities authorized for issuance under our equity compensation plans is set forth in "Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities" above in this Annual Report. The other information required by this item is incorporated herein by reference from the information under the caption "Security Ownership of Certain Beneficial Owners and Management" contained in the Proxy Statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is incorporated herein by reference from the information under the captions "Certain Relationships and Related Transactions" and "Election of Directors" contained in the Proxy Statement.
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated herein by reference from the information under the captions "Independent Auditors' Fees" and "Pre-Approval Policies and Procedures" contained in the Proxy Statement.
103
PART IV
Item 15. Exhibits, Financial Statement Schedules.
| (a) | | 1. | | FINANCIAL STATEMENTS. |
|
|
|
|
Reference is made to the Index to Financial Statements under Item 8, Part II hereof. |
|
|
2. |
|
FINANCIAL STATEMENT SCHEDULES. |
|
|
|
|
The Financial Statement Schedules have been omitted either because they are not required or because the information has been included in the financial statements or the notes thereto included in this annual report. |
|
|
3. |
|
EXHIBITS |
EXHIBIT NO.
| | DESCRIPTION
|
|---|
| 2.1* | | Agreement of Purchase and Sale, dated as of March 21, 2007, by and between Arena and BMR-6114-6154 Nancy Ridge Drive LLP (as assignee of BioMed Realty, L.P.) (incorporated by reference to Exhibit 2.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on May 8, 2007, Commission File No. 000-31161) |
| 3.1 | | Fifth Amended and Restated Certificate of Incorporation of Arena (incorporated by reference to Exhibit 3.1 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2002, filed with the Securities and Exchange Commission on August 14, 2002, Commission File No. 000-31161) |
| 3.2 | | Certificate of Amendment of the Fifth Amended and Restated Certificate of Incorporation of Arena (incorporated by reference to Exhibit 4.2 to Arena's registration statement on Form S-8, filed with the Securities and Exchange Commission on June 28, 2006, Commission File No. 333-135398) |
| 3.3 | | Amended and Restated Bylaws of Arena (incorporated by reference to Exhibit 3.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on October 4, 2007, Commission File No. 000-31161) |
| 3.4 | | Certificate of Designations of Series A Junior Participating Preferred Stock of Arena, dated November 4, 2002 (incorporated by reference to Exhibit 3.3 to Arena's quarterly report on Form 10-Q for the quarter ended September 30, 2002, filed with the Securities and Exchange Commission on November 14, 2002, Commission File No. 000-31161) |
| 3.5 | | Certificate of Designations of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock of Arena, dated December 24, 2003 (incorporated by reference to Exhibit 3.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 4.1 | | Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on November 1, 2002, Commission File No. 000-31161) |
| 4.2 | | Amendment No. 1, dated December 24, 2003, to Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
104
| 4.3 | | Amendment No. 2, dated November 16, 2006, to Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.3 to Amendment No. 2 to Arena's Registration Statement on Form 8-A filed with the Securities and Exchange Commission on November 16, 2006, Commission File No. 000-31161) |
| 4.4 | | Form of common stock certificates (incorporated by reference to Exhibit 4.2 to Arena's registration statement on Form S-1, as amended, filed with the Securities and Exchange Commission on July 19, 2000, Commission File No. 333-3594) |
| 10.1** | | 1998 Equity Compensation Plan (incorporated by reference to Exhibit 10.1 to Arena's registration statement on Form S-1, as amended, filed with the Securities and Exchange Commission on June 22, 2000, Commission File No. 333-3594) |
| 10.2** | | Amended and Restated 2000 Equity Compensation Plan (incorporated by reference to Exhibit 10.2 to Arena's annual report on Form 10-K for the year ended December 31, 2001, filed with the Securities and Exchange Commission on March 15, 2002, Commission File No. 000-31161) |
| 10.3 | | 2001 Arena Employee Stock Purchase Plan, as amended (incorporated by reference to Exhibit 10.5 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2006, filed with the Securities and Exchange Commission on August 4, 2006, Commission File No. 000-31161) |
| 10.4** | | 2002 Equity Compensation Plan (incorporated by reference to Exhibit A to Arena's Proxy Statement regarding Arena's June 11, 2002, Annual Stockholders Meeting, filed with the Securities and Exchange Commission on April 23, 2002, Commission File No. 000-31161) |
| 10.5+ | | Research Collaboration and License Agreement, dated effective as of October 21, 2002, by and between Arena and Merck & Co., Inc. (incorporated by reference to Exhibit 10.20 to Arena's annual report on Form 10-K for the year ended December 31, 2002, filed with the Securities and Exchange Commission on March 28, 2003, Commission File No. 000-31161) |
| 10.6+ | | First Amendment to Research Collaboration and License Agreement, dated as of October 20, 2004, by and between Arena and Merck (incorporated by reference to Exhibit 10.19 to Arena's annual report on Form 10-K for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 2, 2005, Commission File No. 000-31161) |
| 10.7 | | Second Amendment to Research Collaboration and License Agreement, dated as of February 20, 2007, by and between Arena and Merck |
| 10.8** | | Form of Termination Protection Agreement, dated December 20, 2002, by and among Arena and the employees listed on Schedule 1 thereto (incorporated by reference to Exhibit 10.1 to Arena's quarterly report on Form 10-Q for quarter ended June 30, 2003, filed with the Securities and Exchange Commission on August 13, 2003, Commission File No. 000-31161) |
| 10.9 | | Securities Purchase Agreement for Arena's Series B Convertible Preferred Stock and warrants dated December 24, 2003, among Arena and the investor signatories thereto (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 10.10 | | Registration Rights Agreement dated December 24, 2003, among Arena and the investor signatories thereto (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
105
| 10.11 | | Form of Warrant dated December 24, 2003 (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 10.12 | | Settlement Agreement and Release, dated as of June 30, 2006, between Arena and Smithfield Fiduciary LLC. (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
| 10.13 | | Amendment to Registration Rights Agreement, dated as of June 30, 2006, between Arena and Smithfield Fiduciary LLC. (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
| 10.14 | | Amendment to Registration Rights Agreement, dated as of June 30, 2006, between Arena and Mainfield Enterprises, Inc. (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
| 10.15 | | Purchase and Sale Agreement and Joint Escrow Instructions, dated December 22, 2003, between Arena and ARE—Nancy Ridge No. 3, LLC (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Securities and Exchange Commission on January 6, 2004, Commission File No. 000-31161) |
| 10.16 | | Lease Agreement, dated December 30, 2003, between Arena and ARE—Nancy Ridge No. 3, LLC (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 6, 2004, Commission File No. 000-31161) |
| 10.17** | | Arena's Deferred Compensation Plan, effective November 11, 2003, between Arena and participating executive officers (incorporated by reference to Exhibit 10.29 to Arena's annual report on Form 10-K for the year ended December 31, 2003, filed with the Securities and Exchange Commission on March 1, 2004, Commission File No. 000-31161) |
| 10.18+ | | Collaboration and License Agreement, dated as of December 20, 2004, by and between Arena and Ortho-McNeil Pharmaceutical, Inc. (incorporated by reference to Exhibit 10.20 to Arena's annual report on Form 10-K for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 2, 2005, Commission File No. 000-31161) |
| 10.19** | | Form of stock option grant for non-employee directors under Arena's 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 21, 2005, Commission File No. 000-31161) |
| 10.20** | | Severance Benefit Plan, providing benefits for specified executive officers, dated effective January 20, 2006 (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 24, 2006, Commission File No. 000-31161) |
| 10.21** | | 2006 Long-Term Incentive Plan, as Amended (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on April 13, 2007, Commission File No. 000-31161) |
| 10.22** | | Form of Stock Option Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
106
| 10.23** | | Form of Stock Option Grant Agreement—Director under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.24** | | Form of Incentive Stock Option Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.25** | | Form of Restricted Stock Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.4 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.26** | | Form of Restricted Stock Unit Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.5 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.27 | | Form of Performance-Based Restricted Stock Grant Agreement for non-executive employees under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on March 1, 2007, Commission File No. 000-31161) |
| 10.28** | | Form of Performance-Based Restricted Stock Grant Agreement for executive officers under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on March 1, 2007, Commission File No. 000-31161) |
| 10.29** | | Form of Indemnification Agreement between Arena and its directors (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.30** | | Form of Indemnification Agreement between Arena and its executive officers (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.31** | | Form of Indemnification Agreement between Arena and individuals serving as its directors and executive officers (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.32 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6114 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.5 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.33 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6118 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.6 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.34 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6122, 6124 and 6126 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.7 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
107
| 10.35 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6154 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.8 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.36** | | Summary of compensation for non-employee directors |
| 10.37** | | 2008 Annual Incentive Plan for Arena's executive officers (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 30, 2008, Commission File No. 000-31161) |
| 10.38* | | Asset Purchase Agreement, dated as of December 18, 2007, by and between Arena Pharmaceuticals GmbH and Siegfried Ltd |
| 10.39* | | Toll Manufacturing Agreement, dated as of January 7, 2008, by and between Arena Pharmaceuticals GmbH and Siegfried Ltd |
| 21.1 | | Subsidiaries of the registrant |
| 23.1 | | Consent of Independent Registered Public Accounting Firm |
| 31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934 |
| 31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934 |
| 32.1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 and Rule 13a-14(B) promulgated under the Securities Exchange Act of 1934 |
- +
- Confidential treatment has been granted for portions of this document.
- *
- Exhibits and schedules to this agreement have been omitted pursuant to the rules of the Securities and Exchange Commission. We will submit copies of such exhibits and schedules to the Securities and Exchange Commission upon request.
- **
- Management contract or compensatory plan or arrangement.
(b) EXHIBITS
See Item 15(a)(3) above.
(c) FINANCIAL STATEMENT SCHEDULES
See Item 15(a)(2) above.
108
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on March 4, 2008.
| | | Arena Pharmaceuticals, Inc.,
a Delaware corporation |
|
|
By: |
/s/ JACK LIEF
Jack Lief
President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities indicated on March 4, 2008.
Signatures
| | Title
|
|---|
|
|
|
|
|
| By: | | /s/ JACK LIEF
Jack Lief | | President, Chief Executive Officer and Director |
By: |
|
/s/ ROBERT E. HOFFMAN
Robert E. Hoffman, CPA |
|
Vice President, Finance and Chief Financial Officer (principal financial and accounting officer) |
By: |
|
/s/ DOMINIC P. BEHAN
Dominic P. Behan, Ph.D. |
|
Director |
By: |
|
/s/ DONALD D. BELCHER
Donald D. Belcher |
|
Director |
By: |
|
/s/ SCOTT H. BICE
Scott H. Bice |
|
Director |
By: |
|
/s/ HARRY F. HIXSON
Harry F. Hixson, Jr., Ph.D. |
|
Director |
By: |
|
/s/ J. CLAYBURN LA FORCE, JR.
J. Clayburn La Force, Jr., Ph.D. |
|
Director |
109
By: |
|
/s/ TINA NOVA BENNETT
Tina Nova Bennett, Ph.D. |
|
Director |
By: |
|
/s/ PHILLIP M. SCHNEIDER
Phillip M. Schneider |
|
Director |
By: |
|
/s/ CHRISTINE A. WHITE, M.D.
Christine A. White, M.D. |
|
Director |
By: |
|
/s/ RANDALL E. WOODS
Randall E. Woods |
|
Director |
110
EXHIBIT INDEX
EXHIBIT NO.
| | DESCRIPTION
|
|---|
| 2.1* | | Agreement of Purchase and Sale, dated as of March 21, 2007, by and between Arena and BMR-6114-6154 Nancy Ridge Drive LLP (as assignee of BioMed Realty, L.P.) (incorporated by reference to Exhibit 2.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on May 8, 2007, Commission File No. 000-31161) |
| 3.1 | | Fifth Amended and Restated Certificate of Incorporation of Arena (incorporated by reference to Exhibit 3.1 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2002, filed with the Securities and Exchange Commission on August 14, 2002, Commission File No. 000-31161) |
| 3.2 | | Certificate of Amendment of the Fifth Amended and Restated Certificate of Incorporation of Arena (incorporated by reference to Exhibit 4.2 to Arena's registration statement on Form S-8, filed with the Securities and Exchange Commission on June 28, 2006, Commission File No. 333-135398) |
| 3.3 | | Amended and Restated Bylaws of Arena (incorporated by reference to Exhibit 3.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on October 4, 2007, Commission File No. 000-31161) |
| 3.4 | | Certificate of Designations of Series A Junior Participating Preferred Stock of Arena, dated November 4, 2002 (incorporated by reference to Exhibit 3.3 to Arena's quarterly report on Form 10-Q for the quarter ended September 30, 2002, filed with the Securities and Exchange Commission on November 14, 2002, Commission File No. 000-31161) |
| 3.5 | | Certificate of Designations of Series B-1 Convertible Preferred Stock and Series B-2 Convertible Preferred Stock of Arena, dated December 24, 2003 (incorporated by reference to Exhibit 3.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 4.1 | | Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on November 1, 2002, Commission File No. 000-31161) |
| 4.2 | | Amendment No. 1, dated December 24, 2003, to Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 4.3 | | Amendment No. 2, dated November 16, 2006, to Rights Agreement, dated October 30, 2002, between Arena and Computershare Trust Company, Inc. (incorporated by reference to Exhibit 4.3 to Amendment No. 2 to Arena's Registration Statement on Form 8-A filed with the Securities and Exchange Commission on November 16, 2006, Commission File No. 000-31161) |
| 4.4 | | Form of common stock certificates (incorporated by reference to Exhibit 4.2 to Arena's registration statement on Form S-1, as amended, filed with the Securities and Exchange Commission on July 19, 2000, Commission File No. 333-3594) |
| 10.1** | | 1998 Equity Compensation Plan (incorporated by reference to Exhibit 10.1 to Arena's registration statement on Form S-1, as amended, filed with the Securities and Exchange Commission on June 22, 2000, Commission File No. 333-3594) |
111
| 10.2** | | Amended and Restated 2000 Equity Compensation Plan (incorporated by reference to Exhibit 10.2 to Arena's annual report on Form 10-K for the year ended December 31, 2001, filed with the Securities and Exchange Commission on March 15, 2002, Commission File No. 000-31161) |
| 10.3 | | 2001 Arena Employee Stock Purchase Plan, as amended (incorporated by reference to Exhibit 10.5 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2006, filed with the Securities and Exchange Commission on August 4, 2006, Commission File No. 000-31161) |
| 10.4** | | 2002 Equity Compensation Plan (incorporated by reference to Exhibit A to Arena's Proxy Statement regarding Arena's June 11, 2002, Annual Stockholders Meeting, filed with the Securities and Exchange Commission on April 23, 2002, Commission File No. 000-31161) |
| 10.5+ | | Research Collaboration and License Agreement, dated effective as of October 21, 2002, by and between Arena and Merck & Co., Inc. (incorporated by reference to Exhibit 10.20 to Arena's annual report on Form 10-K for the year ended December 31, 2002, filed with the Securities and Exchange Commission on March 28, 2003, Commission File No. 000-31161) |
| 10.6+ | | First Amendment to Research Collaboration and License Agreement, dated as of October 20, 2004, by and between Arena and Merck (incorporated by reference to Exhibit 10.19 to Arena's annual report on Form 10-K for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 2, 2005, Commission File No. 000-31161) |
| 10.7 | | Second Amendment to Research Collaboration and License Agreement, dated as of February 20, 2007, by and between Arena and Merck |
| 10.8** | | Form of Termination Protection Agreement, dated December 20, 2002, by and among Arena and the employees listed on Schedule 1 thereto (incorporated by reference to Exhibit 10.1 to Arena's quarterly report on Form 10-Q for quarter ended June 30, 2003, filed with the Securities and Exchange Commission on August 13, 2003, Commission File No. 000-31161) |
| 10.9 | | Securities Purchase Agreement for Arena's Series B Convertible Preferred Stock and warrants dated December 24, 2003, among Arena and the investor signatories thereto (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 10.10 | | Registration Rights Agreement dated December 24, 2003, among Arena and the investor signatories thereto (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 10.11 | | Form of Warrant dated December 24, 2003 (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Securities and Exchange Commission on December 30, 2003, Commission File No. 000-31161) |
| 10.12 | | Settlement Agreement and Release, dated as of June 30, 2006, between Arena and Smithfield Fiduciary LLC. (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
112
| 10.13 | | Amendment to Registration Rights Agreement, dated as of June 30, 2006, between Arena and Smithfield Fiduciary LLC. (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
| 10.14 | | Amendment to Registration Rights Agreement, dated as of June 30, 2006, between Arena and Mainfield Enterprises, Inc. (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on July 6, 2006, Commission File No. 000-31161) |
| 10.15 | | Purchase and Sale Agreement and Joint Escrow Instructions, dated December 22, 2003, between Arena and ARE—Nancy Ridge No. 3, LLC (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Securities and Exchange Commission on January 6, 2004, Commission File No. 000-31161) |
| 10.16 | | Lease Agreement, dated December 30, 2003, between Arena and ARE—Nancy Ridge No. 3, LLC (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 6, 2004, Commission File No. 000-31161) |
| 10.17** | | Arena's Deferred Compensation Plan, effective November 11, 2003, between Arena and participating executive officers (incorporated by reference to Exhibit 10.29 to Arena's annual report on Form 10-K for the year ended December 31, 2003, filed with the Securities and Exchange Commission on March 1, 2004, Commission File No. 000-31161) |
| 10.18+ | | Collaboration and License Agreement, dated as of December 20, 2004, by and between Arena and Ortho-McNeil Pharmaceutical, Inc. (incorporated by reference to Exhibit 10.20 to Arena's annual report on Form 10-K for the year ended December 31, 2004, filed with the Securities and Exchange Commission on March 2, 2005, Commission File No. 000-31161) |
| 10.19** | | Form of stock option grant for non-employee directors under Arena's 2002 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 21, 2005, Commission File No. 000-31161) |
| 10.20** | | Severance Benefit Plan, providing benefits for specified executive officers, dated effective January 20, 2006 (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 24, 2006, Commission File No. 000-31161) |
| 10.21** | | 2006 Long-Term Incentive Plan, as Amended (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on April 13, 2007, Commission File No. 000-31161) |
| 10.22** | | Form of Stock Option Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.23** | | Form of Stock Option Grant Agreement—Director under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.24** | | Form of Incentive Stock Option Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
113
| 10.25** | | Form of Restricted Stock Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.4 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.26** | | Form of Restricted Stock Unit Grant Agreement under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.5 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on August 1, 2006, Commission File No. 000-31161) |
| 10.27 | | Form of Performance-Based Restricted Stock Grant Agreement for non-executive employees under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on March 1, 2007, Commission File No. 000-31161) |
| 10.28** | | Form of Performance-Based Restricted Stock Grant Agreement for executive officers under the Arena 2006 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on March 1, 2007, Commission File No. 000-31161) |
| 10.29** | | Form of Indemnification Agreement between Arena and its directors (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.30** | | Form of Indemnification Agreement between Arena and its executive officers (incorporated by reference to Exhibit 10.2 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.31** | | Form of Indemnification Agreement between Arena and individuals serving as its directors and executive officers (incorporated by reference to Exhibit 10.3 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on June 18, 2007, Commission File No. 000-31161) |
| 10.32 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6114 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.5 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.33 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6118 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.6 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.34 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6122, 6124 and 6126 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.7 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.35 | | Lease agreement between BMR-6114-6154 Nancy Ridge Drive LLC and Arena for 6154 Nancy Ridge Drive, San Diego, California (incorporated by reference to Exhibit 10.8 to Arena's quarterly report on Form 10-Q for the quarter ended June 30, 2007, filed with the Securities and Exchange Commission on August 9, 2007, Commission File No. 000-31161) |
| 10.36** | | Summary of compensation for non-employee directors |
| 10.37** | | 2008 Annual Incentive Plan for Arena's executive officers (incorporated by reference to Exhibit 10.1 to Arena's report on Form 8-K filed with the Securities and Exchange Commission on January 30, 2008, Commission File No. 000-31161) |
114
| 10.38* | | Asset Purchase Agreement, dated as of December 18, 2007, by and between Arena Pharmaceuticals GmbH and Siegfried Ltd |
| 10.39* | | Toll Manufacturing Agreement, dated as of January 7, 2008, by and between Arena Pharmaceuticals GmbH and Siegfried Ltd |
| 21.1 | | Subsidiaries of the registrant |
| 23.1 | | Consent of Independent Registered Public Accounting Firm |
| 31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934 |
| 31.2 | | Certification of Chief Financial Officer pursuant to Rule 13a-14(A) promulgated under the Securities Exchange Act of 1934 |
| 32.1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 and Rule 13a-14(B) promulgated under the Securities Exchange Act of 1934 |
- +
- Confidential treatment has been granted for portions of this document.
- *
- Exhibits and schedules to this agreement have been omitted pursuant to the rules of the Securities and Exchange Commission. We will submit copies of such exhibits and schedules to the Securities and Exchange Commission upon request.
- **
- Management contract or compensatory plan or arrangement.
115
QuickLinks
ARENA PHARMACEUTICALS, INC. TABLE OF CONTENTSINFORMATION RELATING TO FORWARD-LOOKING STATEMENTSPART IPART IIARENA PHARMACEUTICALS, INC. INDEX TO FINANCIAL STATEMENTSREPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRMARENA PHARMACEUTICALS, INC. Consolidated Balance Sheets (In thousands, except share and per share data)ARENA PHARMACEUTICALS, INC. Consolidated Statements of Operations (In thousands, except share and per share data)ARENA PHARMACEUTICALS, INC. Consolidated Statements of Stockholders' Equity (In thousands, except share data)ARENA PHARMACEUTICALS, INC. Consolidated Statements of Cash Flows (In thousands)ARENA PHARMACEUTICALS, INC. Notes to Consolidated Financial StatementsReport of Independent Registered Public Accounting FirmPART IIIPART IVSIGNATURESEXHIBIT INDEX