UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): January 20, 2015
Arena Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 000-31161 | 23-2908305 | ||
(State or other jurisdiction of incorporation) | (Commission File Number) | (I.R.S. Employer Identification No.) |
6154 Nancy Ridge Drive, San Diego, California 92121
(Address of principal executive offices) (Zip Code)
858.453.7200
(Registrant’s telephone number, including area code)
N/A
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
In this report, “Arena Pharmaceuticals,” “Arena,” “Company,” “we,” “us” and “our” refer to Arena Pharmaceuticals, Inc., and/or one or more of our wholly owned subsidiaries, unless the context otherwise provides. Arena Pharmaceuticals® and Arena® are registered service marks of Arena Pharmaceuticals, Inc. BELVIQ® is a registered trademark of our wholly owned subsidiary, Arena Pharmaceuticals GmbH.
Item 2.02 Results of Operations and Financial Condition.
On January 20, 2015, we filed with the Securities and Exchange Commission, or SEC, a prospectus supplement pursuant to Rule 424(b)(5) of the Securities Act of 1933, as amended, in which we disclosed that our cash and cash equivalents were approximately $163.2 million as of December 31, 2014.
Item 8.01 Other Events.
We are filing the following information with the SEC for the purpose of updating certain aspects of our publicly disclosed descriptions of our business, intellectual property and risk factors.
ARENA PHARMACEUTICALS, INC.
We are a biopharmaceutical company focused on discovering, developing and commercializing novel drugs that target G protein-coupled receptors, or GPCRs, to address unmet medical needs. Our US operations are located in San Diego, California, and our operations outside of the United States, including our commercial manufacturing facility, are located in Zofingen, Switzerland. BELVIQ® (lorcaserin HCl), our internally discovered drug approved by the US Food and Drug Administration, or FDA, for chronic weight management as an adjunct to reduced calorie diet and increased physical activity in adults who are overweight with a comorbidity or obese, is our first and only drug approved by any regulatory agency for marketing. BELVIQ was made available by prescription in the United States in June 2013.
Our collaborators have pending applications for the regulatory approval of BELVIQ for marketing in a number of additional countries. We also have a pipeline of drug candidates and compounds at various stages of research and development. Our most advanced drug candidates include BELVIQ for smoking cessation, in a combination therapy and in a once-daily formulation; ralinepag for vascular diseases; APD334 for autoimmune diseases; APD371 for pain; and temanogrel for thrombotic diseases. Our pipeline also includes numerous earlier-stage programs.
The key elements of our strategy are as follows:
| • | Make BELVIQ Available to Patients for Chronic Weight Management.We have agreements with pharmaceutical companies (including Eisai Inc. and Eisai Co., Ltd., for most territories worldwide; Ildong Pharmaceutical Co., Ltd., or Ildong, for South Korea; CY Biotech Company Limited for Taiwan; and Teva Pharmaceutical Industries Ltd.’s local Israeli subsidiary for Israel) that provide them rights and responsibilities to seek regulatory approval and commercialize BELVIQ for chronic weight management. Our Swiss subsidiary, Arena Pharmaceuticals GmbH, manufactures and supplies BELVIQ for these pharmaceutical companies to commercialize in their respective territories. |
| • | Pursue Additional BELVIQ Opportunities.We will explore with our collaborators or independently additional indications, formulations and combinations for BELVIQ. |
| • | Advance our Pipeline and GPCR Research.Our technologies, infrastructure and integrated approach to research and development have allowed us to identify and develop an FDA-approved drug and a pipeline of novel drug candidates and preclinical compounds. We will advance our pipeline of drug candidates independently and through collaborations with pharmaceutical companies, as well as continue our research and development efforts to discover and advance new compounds. |
1
Following is a summary of our internally discovered GPCR portfolio:
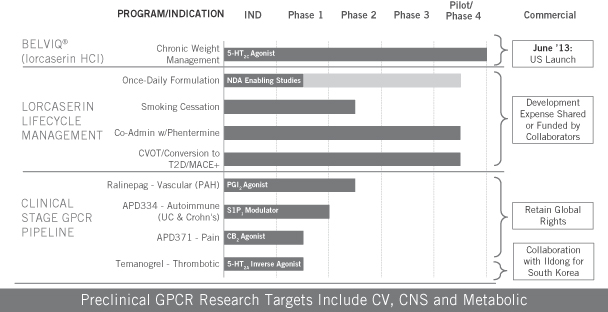
We have commercial rights for our programs and drug candidates, except for our collaborators’ rights with respect to BELVIQ and Ildong’s rights with respect to temanogrel.
Recent Clinical Developments
APD334
In January 2015, we announced top-line results from a Phase 1b multiple ascending dose clinical trial for APD334, an oral drug candidate that targets the sphingosine 1-phosphate subtype 1, or S1P1, receptor for the potential treatment of autoimmune diseases. In the Phase 1b clinical trial, APD334 demonstrated a dose-dependent effect on lymphocyte count lowering in blood, with mean decreases from baseline of up to 69%. Lymphocyte counts, on average, recovered to baseline within one week of conclusion of dosing. There were no clinically significant safety findings with respect to heart rate or rhythm or pulmonary function, and no clinically significant elevations in liver enzyme tests. The most common treatment-emergent adverse events were mild or moderate contact dermatitis, headache, constipation and diarrhea, with none being clearly drug related. There were no discontinuations for adverse events, and no serious adverse events were observed. The randomized, double-blind, placebo-controlled Phase 1b clinical trial evaluated the safety, tolerability, pharmacodynamics and pharmacokinetics of multiple-ascending doses of APD334. In five different dosing cohorts, a total of 50 healthy volunteers received APD334 and 10 received placebo for 21 days. We plan to advance APD334 into Phase 2 clinical trials by around the middle of 2015 for ulcerative colitis and Crohn’s disease.
Ralinepag
In January 2015, we initiated patient dosing in a 22-week, randomized, double-blind and placebo-controlled Phase 2 clinical trial of ralinepag, an orally available agonist of the prostacyclin, or IP, receptor, for the treatment of Pulmonary Arterial Hypertension, or PAH. The trial will seek to evaluate the hemodynamic and exercise tolerance effects, safety and tolerability of multiple-ascending doses of ralinepag in up to 60 patients with PAH.
INTELLECTUAL PROPERTY
BELVIQ Intellectual Property
As of January 1, 2015, we owned issued patents that cover compositions of matter for the BELVIQ new chemical entity and related compounds, and methods of treatment utilizing BELVIQ and related compounds in 69 jurisdictions, including the United States, Japan, China, Germany, France, Italy, the United Kingdom, Spain, Canada, Russia, India, Australia, and South Korea, and had applications pending in two other jurisdictions, of which the one with the largest pharmaceutical market was Brazil. Based on sales statistics provided by IMS Health, the jurisdictions where BELVIQ patents have been issued accounted for more than 92% of global pharmaceutical sales in 2013, while other jurisdictions where BELVIQ patents remain pending accounted for more than 3% of global pharmaceutical sales in that same year. The patents on BELVIQ issued by the US Patent and Trademark Office have serial numbers US 6,953,787; US 7,514,422; US 7,977,329; US 8,207,158; US 8,273,734; US 8,575,149; and US 8,546,379, while the corresponding patent granted by the European Patent Office has serial number EP 1 411 881 B1. Other of our BELVIQ issued patents and patent applications including those directed to the HCl salt of BELVIQ (e.g., US 8,367,657), the hemihydrate of the HCl salt of BELVIQ as well as its crystalline forms (e.g., US 8,168,624; US 8,697,686; and EP 1 838 677 B1), and synthetic routes and intermediates useful in the manufacturing of BELVIQ, are all present in a lesser number of commercially important jurisdictions. The earliest priority date for the patents on BELVIQ is 2002. The terms of the new chemical entity patents are capable of continuing into 2023 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications. With respect to the United States, we have filed applications for patent extension, which, if granted, will extend the patent term for one of our BELVIQ composition of matter patents into 2026 and potentially into 2027.
As of January 1, 2015 , we owned registered trademarks on the use of the name BELVIQ in Class 5 for the sale and marketing of pharmaceutical preparations for weight management, weight loss, the treatment of obesity and the maintenance of weight loss in 121 jurisdictions, including the United States, Japan, China, Germany, France, Italy,
2
United Kingdom, Spain, Russia, India, Australia and South Korea, and had trademark applications pending in 29 other jurisdictions, of which the two with the largest pharmaceutical markets were Brazil and Canada. The trademark on the name BELVIQ registered by the US Patent and Trademark Office has serial number US 4,080,253, while the corresponding trademark registered by the European Union’s Office for Harmonization in the Internal Market has serial number CTM 010224905. Other of our BELVIQ registered trademarks and trademark applications, including those in classes 9, 16, 41 and 44 for downloadable publications, publications, educational services and medical services, respectively, directed to weight management, weight loss and the maintenance of weight loss are all present in a lesser number of commercially important jurisdictions. As of January 1, 2015, we have also filed trademark applications in Class 5 on one or more transliterations of the name BELVIQ in the local character set or alphabet of 24 jurisdictions, including Japan, China, Russia and South Korea.
Relinepag (APD811) Intellectual Property
As of January 1, 2015, we owned issued patents covering compositions of matter for APD811 and related compounds and methods of treatment utilizing APD811 and related compounds, synthetic routes, and various solid state forms of APD811, in 55 jurisdictions, including the United States, Japan, China, Germany, France, Italy, United Kingdom, Spain, Russia, and Australia, and we had applications pending in 9 other jurisdictions, of which the ones with the largest pharmaceutical markets were Brazil, Canada, India, and South Korea. Based on sales statistics provided by IMS Health, the jurisdictions where APD811 patents have been issued accounted for more than 85% of global pharmaceutical sales in 2013, while other jurisdictions where APD811 patents remain pending accounted for more than 9% of global pharmaceutical sales in that same year. The patent on APD811 issued by the US Patent and Trademark Office has serial number US 8,895,776, while the corresponding patent granted by the European Patent Office has serial number EP 2 280 696 B2. Other of our APD811 patent applications, including those directed to formulations, synthetic processes, and dosage regimens of APD811, have been filed. The earliest priority date for the patents on APD811 is 2008. The terms of these patents are capable of continuing into 2029 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
APD334 Intellectual Property
As of January 1, 2015, we owned issued patents that cover compositions of matter for APD334 and related compounds, methods of treatment utilizing APD334 and related compounds, and various salts of APD334 and crystalline forms thereof in 16 jurisdictions, including the United States, Japan, China, and Russia, and had applications pending in 8 other jurisdictions, of which the largest pharmaceutical markets were Europe, Brazil, Canada, India, Russia, Australia, and South Korea. Based on sales statistics provided by IMS Health, the jurisdictions where APD334 patents have been issued accounted for more than 58% of global pharmaceutical sales in 2013, while other jurisdictions where APD334 patents remain pending accounted for more than 34% of global pharmaceutical sales in that same year. The patent on APD334 issued by the US Patent and Trademark Office has serial number US 8,580,841. Other of our APD334 pending patent applications, including those directed to synthetic routes and intermediates useful in the manufacturing of APD334 have all been filed in a lesser number of commercially important jurisdictions. The earliest priority date for the patents on APD334 is 2008. The terms of any patents that may issue from these patent applications should be capable of continuing into 2029 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
APD371 Intellectual Property
As of January 1, 2015, we owned issued patents covering compositions of matter for APD371 and related compounds in 4 jurisdictions, including the United States, and we had applications pending in 19 other jurisdictions, of which the ones with the largest pharmaceutical markets were Europe, Japan, China, Brazil, Canada, Russia, India, Australia, and South Korea. Based on sales statistics provided by IMS Health, the jurisdictions where ADP371 patents have been issued accounted for more than 39% of global pharmaceutical sales in 2013, while other jurisdictions where APD371 patents remain pending accounted for more than 57% of global pharmaceutical sales in that same year. The patent on APD371 issued by the US Patent and Trademark Office has serial number US 8,778,950. Other of our APD371 patent applications, including those directed to various solid state forms of APD371, have all been filed in a similar number of commercially important jurisdictions. The earliest priority date for the patents on APD371 is 2009. The terms of these patents are capable of continuing into 2030 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
3
Temanogrel Intellectual Property
As of January 1, 2015, we owned issued patents that cover compositions of matter for temanogrel and related compounds and methods of treatment utilizing temanogrel and related compounds in 84 jurisdictions, including the United States, Japan, China, Germany, France, Italy, the United Kingdom, Spain, Canada, Russia, Australia, and South Korea, and had applications pending in 15 other jurisdictions, of which the largest pharmaceutical markets were Brazil and India. Based on sales statistics provided by IMS Health, the jurisdictions where temanogrel patents have been issued accounted for more than 91% of global pharmaceutical sales in 2013, while other jurisdictions where temanogrel patents remain pending accounted for more than 7% of global pharmaceutical sales in that same year. The patent on temanogrel issued by the US Patent and Trademark Office has serial number US 7,884,101, while the corresponding patent granted by the European Patent Office has serial number EP 1 833 799 B1. Other of our temanogrel issued patents and patent applications, including those directed to the temanogrel HCl salt as well as its crystalline forms, synthetic routes and intermediates useful in the manufacturing of temanogrel, and the active metabolites of temanogrel have all been filed in a lesser number of commercially important jurisdictions. The earliest priority date for the patents on temanogrel is 2004. The terms of these patents are capable of continuing into 2025 in most jurisdictions without taking into account any patent term adjustment or extension regimes of any country or any additional term of exclusivity we might obtain by virtue of the later filed patent applications.
RISK FACTORS
You should consider carefully the following information about the risks described below, together with all of the other information included in this Current Report and in our other filings with the SEC, before making any investment decisions regarding our securities. If any of the following risks actually occurs, our business, financial condition, results of operations and future growth prospects would likely be materially and adversely affected. In these circumstances, the market price of our common stock would likely decline, and you may lose all or part of the money you paid to buy our securities.
Risks Relating to Our Business
Our prospects are highly dependent on the success of BELVIQ, our first and only FDA-approved drug. To the extent BELVIQ is not commercially successful, our business, financial condition and results of operations may be materially adversely affected and the price of our common stock may decline.
Our prospects are highly dependent on the success of BELVIQ, which has been approved for chronic weight management by the US Food and Drug Administration, or FDA, and is our first and only drug approved by any regulatory agency. We believe our prospects are highly dependent on, and a significant portion of the value of our company relates to, the successful commercialization of BELVIQ in the United States and potentially in additional territories. We have granted rights to commercialize BELVIQ to collaborators for most of the territories in the world, and are highly dependent on our collaborators for obtaining marketing approval and commercializing BELVIQ. In this regard, we are particularly dependent on Eisai Inc. and Eisai Co., Ltd. (collectively with Eisai Inc., Eisai) as Eisai has commercialization and other rights to BELVIQ for the United States and the vast majority of all other territories. We do not know whether or when BELVIQ will be approved for sale or commercialized in any territories outside of the United States, and BELVIQ may not receive marketing approval from any other regulatory agency or be commercialized in any other territories.
We expect that revenues generated by BELVIQ will constitute the majority of our revenues over the next several years, which will substantially depend on product sales of BELVIQ and the achievement of milestones, and potentially on the development, approval and commercialization of other BELVIQ products, if any. We cannot guarantee future product sales or achievement of any other milestones. In addition, any of our collaborations for BELVIQ may be terminated early in certain circumstances, which may result in us not receiving additional milestone or other payments under the terminated agreement.
4
With respect to the United States, the degree of market acceptance and commercial success of BELVIQ will depend on a number of factors, including the following, as well as risks identified in other risk factors:
| • | the number of patients eligible to receive BELVIQ, the number of patients treated with BELVIQ and the results achieved by such patients; |
| • | market acceptance and use of BELVIQ, which may depend on the public’s view of BELVIQ, economic changes, national and world events, potentially seasonal and other fluctuations in demand, the timing and impact of current or new competition, and BELVIQ’s perceived advantages or disadvantages over alternative treatments (including relative convenience, ease of administration, and prevalence and severity of any adverse events, including any unexpected adverse events); |
| • | the actual and perceived safety and efficacy of BELVIQ on both a short- and long-term basis among actual or potential patients, healthcare providers and others in the medical community, regulatory agencies and insurers and other payers, including related decisions by any such entity or individual; |
| • | incidence and severity of any side effects, including as a result of off-label use or in combination with one or more drugs; |
| • | new data relating to BELVIQ, including as a result of additional studies, trials or analyses of BELVIQ (such as BELVIQ for a different indication, in a different formulation or in combination with another drug) or related drugs or drug candidates; |
| • | physicians may not prescribe, and patients may not take, BELVIQ until at least results from our required postmarketing studies are available or other long-term efficacy and safety data exists; |
| • | the claims, limitations, warnings and other information in BELVIQ’s current or future labeling; |
| • | the current or future scheduling designation for BELVIQ by the US Drug Enforcement Administration, or DEA; |
| • | Eisai’s maintenance of an effective sales force, marketing team, strategy and program and medical affairs group and related functions, as well as its sales, marketing and other representatives accurately describing BELVIQ consistent with its approved labeling; |
| • | BELVIQ’s price and perceived cost-effectiveness, including as compared to possible alternatives; |
| • | the placement of BELVIQ on third-party payer formularies, and the ability of patients and physicians and other providers to obtain and maintain coverage and adequate reimbursement, if any, by third-party payers, including government payers; |
| • | the ability and desire of group purchasing organizations, or GPOs, including distributors and other network providers, to sell BELVIQ to their constituencies; |
| • | introduction of counterfeit or unauthorized versions of BELVIQ; |
| • | the development of the market for weight-management medications; |
| • | to the extent BELVIQ is approved and marketed in a jurisdiction with a significantly lower price than in another jurisdiction, the impact of the lower pricing in the higher-priced territory, including on the pricing of reimbursement, if available, and by the diversion of lower-priced BELVIQ into the higher-priced territory; and |
| • | the maintenance of adequate commercial manufacturing capabilities ourselves or through third-party manufacturers, our ability to meet commercial demand for BELVIQ, and supply chain issues. |
If BELVIQ does not achieve sufficient market acceptance in the United States, and ultimately in other territories, the revenues we generate from sales of BELVIQ will be limited, our collaborators may negatively change marketing strategies or resources, our collaborations may be modified or terminated and we may not be profitable.
In addition, with respect to the United States and other territories, if the results or timing of regulatory filings, the regulatory process, regulatory developments, clinical trials or preclinical studies, or other activities, actions or decisions related to BELVIQ do not meet our, your, analysts’ or others’ expectations, the market price of our common stock could decline significantly.
5
BELVIQ or any of our future drugs may not be commercially successful if not widely covered and adequately reimbursed by third-party payers, and we may depend on others to obtain and maintain third-party payer access; inadequate third-party coverage and reimbursement could make entering into agreements with pharmaceutical companies to collaborate or commercialize our drugs more difficult and diminish our revenues.
Our and our collaborators’ ability to successfully commercialize any of our drugs that have been or may be approved will depend, in part, on government regulation and the availability of coverage and adequate reimbursement from third-party payers, including private health insurers and government payers, such as the Medicaid and Medicare programs, increases in government-run, single-payer health insurance plans and compulsory licenses of drugs. We expect government and third-party payers will continue their efforts to contain healthcare costs by limiting coverage and reimbursement levels for new drugs. In addition, many countries outside of the United States have nationalized healthcare systems in which the government pays for all such products and services and must approve product pricing. A government or third-party payer decision not to approve pricing, or provide adequate coverage and reimbursements, for our drugs, if any, could limit market acceptance of and demand for our drugs.
It is increasingly difficult to obtain coverage and adequate reimbursement levels from third-party payers, and significant uncertainty exists as to the coverage and reimbursement of newly approved prescription drug products. We or our collaborators also face competition in negotiating for coverage from pharmaceutical companies and others with competitive drugs or other treatment, and these competitors may have significantly more negotiating leverage or success with respect to individual payers than we or our collaborators may have.
In the United States, even if a third-party payer ultimately elects to cover and reimburse for BELVIQ, most payers will not reimburse 100% of the cost, but rather require patients to pay a portion of the cost through a co-payment. Thus, even if reimbursement is available, the percentage of drug cost required to be borne by the patients may make use of BELVIQ financially undesirable, difficult or impossible for certain patients, which would have a negative impact on sales of BELVIQ, including related revenues. For example, payers may approve coverage for BELVIQ in tiers requiring unacceptably high patient co-payments or only as a second- or later-line treatment. Several third-party payers have approved coverage for BELVIQ with limitations, including co-payments that may be unacceptably high for certain patients, regardless of the availability of any coupon, voucher or other discount program. In addition, even if a payer approves coverage for BELVIQ, individual employers or others may not opt to select a plan that provides such coverage. Failure to improve coverage or the reduction or loss of coverage could materially harm the ability to successfully market BELVIQ. Achieving coverage and acceptable reimbursement levels typically involves negotiating with individual payers and is a time-consuming and costly process. In addition, Medicare explicitly excludes coverage for drugs for weight loss.
We expect that the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, PPACA, as well as other federal and state healthcare reform measures that have and may be implemented in the future, may result in more rigorous coverage criteria, more limited coverage and downward pressure on the price that we may receive for any approved product, which could seriously decrease our future revenues. Any reduction in reimbursement from Medicare, Medicaid or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may also limit our commercial opportunities by reducing the amount a potential collaborator is willing to pay to license our programs or drug candidates in the future, which may prevent us from being able to generate revenue, attain profitability, commercialize our products or establish and maintain collaborations.
Forecasting of BELVIQ sales will be difficult, and if BELVIQ projections are inaccurate, our business may be harmed and our stock price may be adversely affected.
Our business planning requires us to forecast demand and revenues for BELVIQ despite numerous uncertainties, which may be increased because we rely to a large extent on our collaborators, particularly Eisai, conducting commercial activities and providing us with accurate and timely information. Actual results may deviate materially from projected results for various reasons, including the following, as well as risks identified in other risk factors:
| • | the rate of adoption in the United States, including fluctuations in demand for various reasons, such as fluctuations related to economic changes, national and world events, holidays and seasonal changes; |
6
| • | pricing (including discounting or other promotions), reimbursement, product returns or recalls, competition, labeling, DEA scheduling, adverse events and others items that impact commercialization; |
| • | lack of patient and physician familiarity with BELVIQ; |
| • | lack of patient use and physician prescribing history; |
| • | lack of commercialization experience with BELVIQ, in particular, and weight loss or management drugs, in general; |
| • | actual sales to patients may significantly differ from expectations based on sales to wholesalers; |
| • | our collaborators control the commercialization of BELVIQ in most of the world, including related strategy and their allocation of resources, and we expect that any future collaborators for BELVIQ will similarly control the commercialization in the applicable territory; and |
| • | uncertainty relating to when BELVIQ may become commercially available to patients and rate of adoption in other territories. |
We expect that our revenues from BELVIQ will continue to be based in part on estimates, judgment and accounting policies, and incorrect estimates or regulators’ or others’ disagreement regarding such estimates or accounting policies may result in changes to guidance, projections or previously reported results. For example, with respect to the commercialization of BELVIQ in the United States, our revenues are based on information we receive from Eisai, including their estimates of deductions for certain items, such as taxes, credits, allowances, discounts, rebates, chargebacks and returns, which are subject to significant judgment and may change from time to time. We expect to continue to recognize revenues upon Eisai’s sales to wholesalers. As BELVIQ is sold through to patients, if the actual level of deductions differ materially from Eisai’s estimates, this could have a material impact on our revenues. In addition, expected and actual product sales and quarterly and other results may greatly fluctuate, including in the near-term, and such fluctuations can adversely affect the market price of our common stock, perceptions of our ability to forecast demand and revenues, and our ability to maintain and fund our operations.
Data generated or analyzed with respect to product use in the market or required postmarketing or other studies or trials may result in decreased demand, lower sales, product recall or regulatory action.
A New Drug Application, or NDA, holder is responsible for assessing and monitoring the safety of a drug that has been approved for marketing. Eisai is the NDA holder of BELVIQ, and we expect that Eisai and other of our collaborators will hold the BELVIQ regulatory approvals, if any, in territories outside of the United States. Eisai, we and, potentially, our other collaborators will assess and monitor the safety of BELVIQ in the marketplace, and will receive reports of adverse safety events. In addition, we expect that, from time to time, we or others will conduct additional studies or trials or analyze new or previous data related to BELVIQ, including with respect to required postmarketing studies and in connection with seeking regulatory approval of BELVIQ outside of the United States, in combination with other drugs, for other indications or using different formulations. For example, as a condition to obtaining FDA approval of BELVIQ, the FDA required the conduct of postmarketing studies, including evaluation of the effect of long-term treatment with BELVIQ on the incidence of major adverse cardiovascular events in overweight and obese subjects with cardiovascular disease or multiple cardiovascular risk factors (otherwise known as the cardiovascular outcomes trial, or CVOT). The FDA-required portion of the trial is designed to evaluate BELVIQ’s effect on the incidence of major adverse cardiovascular events, or MACE, (non-fatal myocardial infarction, non-fatal stroke and cardiovascular death) compared to placebo, with a non-inferiority margin for the hazard ratio of 1.4. The trial will also include FDA-required echocardiographic assessments. Along with the FDA-required portion of the trial, we expect that the trial may include the non-FDA required evaluation of whether lorcaserin reduces the incidence of conversion to type 2 diabetes in patients without type 2 diabetes at baseline and the incidence of MACE+ (MACE or hospitalization for unstable angina or heart failure, or any coronary revascularization), both as compared to placebo. We expect that the trial (including the non-FDA required portion) will run approximately five years. The FDA is also requiring as a postmarketing commitment the assessment of the safety and efficacy of BELVIQ for weight management in obese pediatric patients.
New data relating to BELVIQ, including from adverse event reports, required postmarketing and other studies and trials in the United States, and registration and other studies and trials in territories outside the United States, may result in label changes, may adversely affect sales or result in withdrawal of BELVIQ from the market, and may adversely affect prospects of developing or commercializing BELVIQ in combination with other drugs, for
7
other indications or using different formulations. In addition, analyses of previous data can have similar risks. Eisai and we expect to continue to generate data from new studies and trials, as well as to continue analyzing existing data from previously conducted studies and trials, including for potential use in applications for the marketing approval of BELVIQ. Foreign regulatory agencies may consider the new data or analyses in reviewing marketing applications for BELVIQ in their territories or impose post-approval requirements that require significant additional expenditures. Furthermore, the discovery of significant problems with a product or class of products similar to BELVIQ could have an adverse effect on the BELVIQ program, including commercialization.
New data, analyses or other information, including information about product misuse, may lead government agencies, professional societies, practice management groups or organizations involved in various diseases to publish guidelines or recommendations related to the use of BELVIQ or place greater restrictions on sales. Such guidelines or recommendations may lead to lower sales of BELVIQ.
We will need to further collaborate or obtain additional funds to conduct our planned research, development and commercialization efforts; we may not be able to further collaborate or obtain adequate funds, your ownership may be substantially diluted if we do obtain additional funds, and you may not agree with the manner in which we allocate our available resources; and we may not be profitable.
We have accumulated a large deficit since inception that has primarily resulted from the significant research and development expenditures we have made with respect to BELVIQ and in seeking to identify and validate new drug targets and develop other compounds that could become marketed drugs. We expect that our losses and operating expenses will continue to be substantial for at least the short term.
Cash we may generate in the future from sales of BELVIQ or otherwise is uncertain and difficult to predict. All of our other programs are in the research or development stage, and we may not have adequate funds to develop our compounds into marketed drugs. We intend to explore BELVIQ’s therapeutic potential for other indications, in combination with other drugs or using different formulations, and from time to time we expect to collaborate with Eisai or others, or, possibly, to work independently, on related studies and trials. We also intend to advance other of our drug candidates and preclinical compounds in our pipeline. It takes many years and potentially hundreds of millions of dollars to successfully develop a drug candidate or preclinical compound into a marketed drug, and our efforts may not result in any additional marketed drugs.
We cannot assure you that any additional amounts paid to us or others for BELVIQ will be sufficient to fund our planned research and development and other activities. We may enter into collaborative agreements to research, develop and commercialize other drug candidates in our pipeline, and we may not be able to enter into any such agreement on terms that we or third parties, including investors or analysts, view as favorable, if at all.
Our ability to enter into new collaborations for any of our programs or drug candidates may depend on the outcomes of additional preclinical and clinical testing or regulatory applications for marketing approval. We do not control these outcomes.
We may seek to obtain additional funding from the capital markets or we may eliminate, scale back or delay some or all of our research or development programs. Any such additional funding may dilute or otherwise negatively impact your ownership interest, and any such reductions or failure to apply our resources effectively may narrow, slow or otherwise adversely impact the development and commercialization of our pipeline, which we believe may reduce our opportunities for success and have a material adverse effect on our business and prospects.
We may allocate our resources in ways that do not improve our results of operations or enhance the value of our assets, and our stockholders and others may also not agree with the manner in which we choose to allocate our resources or obtain additional funding. Any failure to apply our resources effectively, how we obtain additional funding and the related views of stockholders or others could have a material adverse effect on our business or the development of our drug candidates and cause the market price of our common stock to decline. In addition, we cannot assure you that we will be profitable or, if we are profitable for any particular time period, that we will be profitable in the future.
If BELVIQ is not approved for marketing in any additional territories, or if any such approval is significantly delayed or limited, our results of operations and business may be materially adversely affected and our stock price may decline; if BELVIQ is approved in any additional territories, commercializing BELVIQ in such territory will carry risks.
8
We and our collaborators have filed applications for regulatory approval for BELVIQ outside of the United States, and we expect our collaborators will seek regulatory approval for BELVIQ in additional territories in the future. The FDA’s approval of a drug does not assure or predict with any certainty that any other regulatory authority will grant marketing approval for such drug. For example, as described below, we withdrew our MAA for BELVIQ in the European Union. We cannot assure or predict with any certainty that BELVIQ will be approved in any additional territories or the expected timeframe of any such approval. The review and potential approval of BELVIQ carries many risks and uncertainties, and our or others’ BELVIQ regulatory submissions may not be satisfactory to the applicable regulatory authorities, including with regard to demonstrating adequate safety and efficacy for regulatory approval. We have made, and expect to make in the future, assumptions, estimations, calculations and decisions as part of our analyses of data and regulatory submissions, and the applicable regulatory authorities may not accept or agree with our assumptions, estimations, calculations, decisions or analyses, may interpret or weigh the importance of data differently or require additional information for approval.
Furthermore, as was the case with FDA approval, other regulatory approvals, even if obtained, may be limited to specific indications, limit the type of patients in which the drug may be used, or otherwise require specific warning or labeling language, any of which might reduce the commercial potential of BELVIQ. As with the FDA’s approval of BELVIQ, regulatory authorities in other territories may condition BELVIQ marketing approval on the conduct of specific postmarketing studies to further evaluate safety and efficacy, in either particular or general patient populations or both. The results of these studies, discovery of previously unknown issues involving safety or efficacy or failure to comply with post-approval regulatory requirements, including requirements with respect to manufacturing practices, reporting of adverse effects, advertising, promotion and marketing, may result in restrictions on the marketing of BELVIQ or the withdrawal of BELVIQ from the market.
With respect to the European Union, in 2013, the EMA’s CHMP identified major objections related to nonclinical and clinical issues, including tumors in rats, valvulopathy and psychiatric events, and the CHMP requested that we further justify BELVIQ’s overall benefit-risk balance taking these issues into consideration. The major objections needed to be addressed before the CHMP could have recommended BELVIQ for marketing approval in the European Union. We did not believe we could resolve the major objections related to the results of nonclinical studies prior to the time we expected the CHMP to issue its final opinion, and, therefore, we withdrew the BELVIQ MAA for the European Union. We also previously received feedback with respect to regulatory applications in other territories that included major objections. We expect Eisai to submit for regulatory approval of BELVIQ in Europe and in other territories in the future, but such submissions not occur when expected or ever. With respect to activities related to regulatory efforts and strategy, Eisai and we expect to continue to generate data from new studies and trials, as well as to continue analyzing existing data from previously conducted studies and trials, including for potential use in applications for the marketing approval of BELVIQ in Europe and other territories. As part of such efforts, Eisai and we expect to continue analyzing data from one of our long-term preclinical carcinogenicity studies for BELVIQ. While Eisai and we believe that such studies and analysis may be helpful with respect to regulatory applications, it is unknown whether any new data, or the results of such analysis, will be viewed favorably or if any data or results will positively or negatively impact any regulatory approvals, applications or strategy.
We cannot assure you that our collaborators’ or our past or any future responses or submissions will be sufficient to the applicable regulatory authority or others, that the applicable regulatory authority or others will consider our BELVIQ program or data, including with regard to BELVIQ’s efficacy or safety, as sufficient, or that any other regulatory authority will ever approve BELVIQ.
If BELVIQ is not approved or commercialized in additional territories, the potential revenues we will receive for BELVIQ will be limited and any related regulatory actions may negatively impact the approval or commercialization of BELVIQ in any territories in which it is approved.
If BELVIQ is approved in any additional territories, the degree of market acceptance and commercial success of BELVIQ in such territory, as well as our resulting revenues, will depend on similar factors as in the United States, as well as territory-specific risks.
Our commercialization and continuing development of BELVIQ may be adversely impacted by cardiovascular side effects associated with drugs used for the treatment of obesity.
We developed BELVIQ to more selectively stimulate the serotonin 2C receptor than did fenfluramine or dexfenfluramine because we believe this may avoid the cardiovascular side effects associated with fenfluramine and
9
dexfenfluramine (often used in combination with phentermine, the combination of which was commonly referred to as “fen-phen”). These two drugs were serotonin-releasing agents and non-selective serotonin receptor agonists, and were withdrawn from the market in 1997 after reported incidences of heart valve disease and pulmonary hypertension associated with their usage. Inin vitrostudies examining affinity, activity and serotonin receptor subtype specificity, BELVIQ demonstrated affinity for, and activity at, serotonin 2A, 2B and 2C receptors, but demonstrated greater affinity, activity and selectivity for the serotonin 2C receptor than for the serotonin 2A and 2B receptors. Activation of the latter two receptors has been associated with undesirable effects. Activation of the 2A receptor has been associated with central nervous system, or CNS, effects, including altered perception, mood and abuse potential, and activation of the 2B receptor has been associated with cardiac valvulopathy.
We may not be correct in our belief that more selectively stimulating the serotonin 2C receptor will avoid these undesired side effects, or BELVIQ’s selectivity profile may not be adequate to avoid these side effects. BELVIQ’s selectivity profile and the potential relationship between the activity of BELVIQ and the activity of fenfluramine and dexfenfluramine may result in increased FDA or other regulatory scrutiny of the safety of BELVIQ, may raise potential adverse publicity and may affect enrollment of any future clinical trials or product sales. In addition, we cannot guarantee that any other regulatory authority will find our safety data to be sufficient to approve BELVIQ for marketing outside of the United States.
We are dependent on marketing and supply agreements for BELVIQ and the failure to maintain such agreements, or poor performance under such agreements, could negatively impact our business.
Our collaborators have primary responsibility for the regulatory approval and, ultimately, marketing and distribution of BELVIQ in the territory or territories under the applicable collaboration. We have limited or no control over the amount and timing of resources that any of these collaborators will dedicate to such activities. In addition, they are responsible for compliance with certain regulatory requirements.
We are subject to a number of other risks associated with our dependence on our collaborative agreements for BELVIQ, including:
| • | our collaborators may not comply with applicable regulatory guidelines with respect to BELVIQ, which could adversely impact the commercialization or development of BELVIQ; |
| • | there could be disagreements regarding the agreements or the study or development of BELVIQ that delay or terminate the commercialization, research, study or development of BELVIQ, delay or eliminate potential payments under the agreements or increase our costs under or outside of the agreements; |
| • | our collaborators may not effectively allocate adequate resources or otherwise support BELVIQ or may have limited experience in a particular territory; and |
| • | our collaborators may not perform as expected, including with regard to making any required payments, and the agreements may not provide adequate protection or may not be effectively enforced. |
We and our collaborators have the right to terminate our agreements in certain circumstances. We could also agree with a collaborator to amend the terms of our agreement, and we or others, including investors and analysts, may not view any amendments as favorable. If any of our marketing and supply agreements for BELVIQ is terminated early, we may not be able to find another company to further develop and commercialize BELVIQ in the covered territory on acceptable terms, if at all, and even if we elected to pursue further development or commercialization of BELVIQ on our own, we might not have the funds or otherwise be able to do so successfully.
We may enter into additional agreements for the commercialization of BELVIQ or one or more of our drug candidates, and may be similarly dependent on the performance of third parties with similar and potentially company-specific risks.
We are responsible for supplying BELVIQ under our marketing and supply agreements, including for commercial sale. We rely on other companies, including third-party manufacturers and sole-source suppliers, and we or such other companies may encounter failures or difficulties or not receive or provide adequate supply, which could adversely affect the commercial production of BELVIQ or the clinical development or regulatory approval of our drug candidates.
10
Under each of our marketing and supply agreements for BELVIQ, we are the exclusive supplier of BELVIQ. Our drug product manufacturing facility in Switzerland is currently our only source for finished drug product of BELVIQ. Without this facility, we would need to rely on third-party manufacturers for such production or develop or acquire such facilities, which, in either case, would require substantial time and funds. With respect to BELVIQ, we are in the process of securing a second supplier for the finished drug product, but we estimate that it will take two years or longer from commencement and a substantial amount of financial and other resources to secure a second source. We may not be successful in securing such second, or any other, source for the finished drug product for BELVIQ.
In addition, we do not own or operate manufacturing facilities that can produce active pharmaceutical ingredient, or API, intermediates and other material required to make BELVIQ and our drug candidates, or finished drug product for all of our drug candidates. Instead, we currently contract with other companies to supply API, intermediates and other materials. Certain of these materials are available from only one or a small number of suppliers, and using a new supplier, if available, could result in substantial delay and greater cost. We expect Siegfried AG, or Siegfried, will be the only source of API for BELVIQ for at least the short term. Our dependence on one source of finished drug product and API, as well as our dependence on other third parties in the supply chain, may adversely affect our ability to develop and deliver drug products on a timely and competitive basis, or at all.
Any performance failure on the part of us or a third-party manufacturer could delay or otherwise adversely affect the sales of BELVIQ or the clinical development or regulatory approval of BELVIQ or one or more of our drug candidates. We or third-party manufacturers may encounter difficulties involving production yields, regulatory compliance, lot release, quality control and quality assurance, as well as shortages of qualified personnel. For example, in December 2014, Eisai and we discovered that a small number of bottles of BELVIQ in a limited number of lots had a missing or incomplete label, and, as a precautionary measure, Eisai voluntarily initiated a recall from wholesalers of the involved lots for inspection.
The ability to adequately and timely manufacture and supply drug product is dependent on the uninterrupted and efficient operation of the manufacturing facilities, which is impacted by many manufacturing variables, including:
| • | availability or contamination of raw materials and components used in the manufacturing process, particularly those for which we have no other source or supplier; |
| • | capacity of our facilities or those of our contract manufacturers; |
| • | having the ability to adjust to changes in actual or anticipated use of the facility, including with respect to having sufficient capacity and a sufficient number of qualified personnel; |
| • | facility contamination by microorganisms or viruses or cross contamination; |
| • | compliance with regulatory requirements, including inspectional notices of violation and warning letters; |
| • | maintenance and renewal of any required licenses or certifications; |
| • | changes in actual or forecasted demand; |
| • | timing and number of production runs; |
| • | production success rates and bulk drug yields; and |
| • | timing and outcome of product quality testing. |
In addition, we or our third-party manufacturers may encounter delays and problems in manufacturing our drug candidates or drugs for a variety of reasons, including accidents during operation, failure of equipment, delays in receiving materials, natural or other disasters, political or governmental unrest or changes, social unrest, intentional misconduct or other factors inherent in operating complex manufacturing facilities. Commercially available starting materials, reagents and excipients may be or become scarce or more expensive to procure, and we may not be able to obtain favorable terms in agreements with subcontractors. We or our third-party manufacturers may not be able to operate our respective manufacturing facilities in a cost-effective manner or in a time frame that is consistent with our expected future manufacturing needs. If we or our third-party manufacturers cease or interrupt production or if our third-party manufacturers and other service providers fail to supply materials, products or services to us for any reason, such interruption could delay progress on our programs, or interrupt the commercial supply, with the potential for additional costs and lost revenues. If this were to occur, we may also need to seek alternative means to fulfill our manufacturing needs.
11
We may not be able to enter into or maintain agreements for the manufacture of BELVIQ or one or more of our drug candidates with manufacturers whose facilities and procedures comply with applicable law. Manufacturers are subject to ongoing periodic inspection (which may be unannounced) by the FDA, the DEA, corresponding state and foreign authorities and other regulatory authorities to ensure strict compliance with Current Good Manufacturing Practices, or cGMPs, regulations and other applicable government regulations and corresponding foreign standards. We do not have control over a third-party manufacturer’s compliance with these regulations and standards. In addition, we have contracted with Siegfried to provide to us certain business and technical services, including safety, health and environmental services. We are, therefore, relying at least in part on Siegfried’s judgment, experience and expertise. We intend to reduce or eliminate our dependence on Siegfried for such business and technical services, and any changes may result in increased cost, additional risk or otherwise negatively impact our operations. If we or one of our manufacturers fail to maintain compliance or otherwise experience setbacks, we or they could be subject to civil or criminal penalties, the production of BELVIQ or one or more of our drug candidates could be interrupted or suspended, or our product could be recalled or withdrawn, resulting in delays, additional costs and potentially lost revenues.
Negative US and global economic conditions may pose challenges to our business strategy, which relies on funding from collaborators or the financial markets, and creates other financial risks for us.
Negative conditions in the US or global economy, including financial markets, may adversely affect our business and the business of our current and prospective collaborators, distributors and licensees, which we sometimes refer to generally as our collaborators, and others with which we do or may conduct business. The duration and severity of these conditions is uncertain. If negative economic conditions persist or worsen, we may be unable to secure funding to sustain our operations or to find suitable collaborators to advance our internal programs, even if we achieve positive results from our research and development or business development efforts. Such negative conditions could also impact commercialization of BELVIQ or any other drugs we develop as well as our financial condition.
From time to time, we may maintain a portfolio of investments in marketable debt securities, which are recorded at fair value. Although we have established investment guidelines relative to diversification and maturity with the objectives of maintaining safety of principal and liquidity, we rely on credit rating agencies to help evaluate the riskiness of investments, and such agencies may not accurately predict such risk. In addition, such agencies may reduce the credit quality of our individual holdings, which could adversely affect their value. Lower credit quality and other market events, such as changes in interest rates and further deterioration in the credit markets, may have an adverse effect on the fair value of our investment holdings and cash position.
We and certain of our current and former employees and directors have been named as defendants in litigation that could result in substantial costs and divert management’s attention.
Beginning in September 2010, a number of lawsuits were filed against us and certain of our employees and directors on behalf of certain purchasers of our common stock. The lawsuits in general include allegations that we and certain of our employees and directors violated laws by making materially false and misleading statements regarding our BELVIQ trials, thereby artificially inflating the price of our common stock. The plaintiffs are seeking unspecified monetary damages and other relief.
There is no guarantee that we will be successful in defending these lawsuits. Also, our insurance coverage may be insufficient, our assets may be insufficient to cover any amounts that exceed our insurance coverage, and we may have to pay damage awards or otherwise may enter into settlement arrangements in connection with such claims. A settlement of any of these lawsuits could involve the issuance of common stock or other equity, which may dilute your ownership interest. Any payments or settlement arrangements could have material adverse effects on our business, operating results, financial condition or your ownership interest. Even if the plaintiffs’ claims are not successful, this litigation could result in substantial costs and significantly and adversely impact our reputation and divert management’s attention and resources, which could have a material adverse effect on our business, operating results or financial condition. In addition, such lawsuits may make it more difficult to finance our operations, obtain certain types of insurance (including directors’ and officers’ liability insurance), and attract and retain qualified executive officers, other employees and directors.
12
Our business may be negatively impacted based on the clinical trials and preclinical studies of, and decisions affecting, BELVIQ or one or more of our drug candidates.
The results and timing of clinical trials and preclinical studies, as well as related decisions, can affect our stock price. Preclinical studies include experiments performed in test tubes, in animals, or in cells or tissues from humans or animals. These studies, which are sometimes referred to as nonclinical studies, include all drug studies except those conducted in human subjects, and may occur before or after initiation of clinical trials for a particular compound. Results of clinical trials and preclinical studies, as well as related analyses of such results, of BELVIQ or one or more of our drug candidates (including development programs related to BELVIQ) may not be viewed favorably by us or third parties, including investors, analysts, current or potential collaborators, the academic and medical communities, and regulators. The same may be true of decisions regarding the focus and prioritization of our research and development efforts, how we design individual studies, trials and development programs of BELVIQ as well as for any of our drug candidates, and regulatory decisions (including by us or regulatory authorities) affecting our programs. Stock prices of companies in our industry have declined significantly when such results and decisions were unfavorable or perceived negatively or when a drug candidate or product did not otherwise meet expectations.
From time to time we have drug programs in clinical trials. In addition to successfully completing clinical trials, to conduct long-term clinical trials and gain regulatory approval to commercialize drug candidates, regulatory authorities require that all drug candidates complete short- and long-term preclinical toxicity and carcinogenicity studies. These preclinical, animal studies are required to help us and regulatory authorities assess the potential risk that drug candidates may be toxic or cause cancer in humans. The results of clinical trials and preclinical studies are uncertain and subject to different interpretations, and the design of these trials and studies (which may change significantly and be more expensive than anticipated depending on results and regulatory decisions) may also be viewed negatively by us, regulatory authorities or other third parties and adversely impact the development and opportunities for regulatory approval and commercialization of our drug candidates and those under collaborative agreements.
Certain countries in the European Union have denied Eisai’s application to conduct the CVOT in their countries until the major objections identified in the MAA for BELVIQ for weight management that was withdrawn from the European Medicines Agency have been addressed. We may be similarly restricted in additional territories in the future, and restrictions may cause delay or otherwise negatively impact our ability to conduct and complete clinical trials for BELVIQ. Unfavorable results or delays with respect to studies, trials or analyses for BELVIQ could negatively impact market acceptance of BELVIQ, limit the revenues we generate from sales, negatively impact regulatory agencies’ views or restrictions on BELVIQ, result in BELVIQ’s withdrawal from the market, and preclude us from being profitable.
We may not be successful in initiating or completing our studies or trials or advancing our programs on our projected timetable, if at all. Any failure to initiate or delays in our studies, trials or development programs, or unfavorable results or decisions or negative perceptions regarding any of our programs, could cause our stock price to decline significantly. This is particularly the case with respect to BELVIQ (including related development programs).
We may report top-line data from time to time, which is based on a preliminary analysis of then-available efficacy and safety data, and such findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. In addition, we make assumptions, estimations and calculations as part of our analyses of data, and others, including regulatory agencies, may not accept or agree with our assumptions, estimations, calculations or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular drug candidate or drug and our company in general.
We depend on our collaborators for commercializing BELVIQ, and, without collaborators, our lack of corporate experience and resources may negatively impact our ability to commercialize BELVIQ independently.
We expect our collaborators to commercialize BELVIQ, subject to any applicable regulatory approval. We may not be able to maintain our marketing and supply agreements for BELVIQ or enter into new agreements for BELVIQ on acceptable terms, if at all. If we are unable to maintain or enter into agreements to commercialize BELVIQ and we develop or acquire our own capabilities to commercialize BELVIQ in any territory independently,
13
we may require additional capital to develop such capabilities, and the marketing and sale of BELVIQ in such territory may be delayed or otherwise impeded by our lack of resources. We may not be successful in developing the requisite capabilities to commercialize BELVIQ without a collaborator. Even if we were able to do so, we have not previously commercialized a drug, and our limited experience may make us less effective at commercial planning, marketing and selling than a more experienced pharmaceutical company. Our lack of corporate experience and adequate resources may impede our efforts to successfully commercialize BELVIQ independently.
If our competitors have commercialization arrangements with companies who allocate substantially greater resources than we allocate (or, with respect to commercializing BELVIQ in a territory under one of our agreements, than our collaborator allocates) to the respective drugs, our competitors may be more successful in marketing and selling their drugs, and our ability to successfully commercialize BELVIQ will be limited.
Our drug candidates are subject to extensive regulation, and we may not receive required regulatory approvals, or timely approvals, for any of our drug candidates.
The preclinical and clinical development, manufacturing, labeling, packaging, storage, recordkeeping, advertising, promotion, export, marketing and distribution, and other possible activities relating to BELVIQ and our drug candidates are, and any other resulting drugs will be, subject to extensive regulation by the FDA and other regulatory agencies. We are subject to periodic inspections (which may be unannounced) by the FDA, the DEA and other regulatory agencies, including inspections at Arena GmbH by the FDA and other regulatory agencies. Failure to comply with applicable regulatory requirements may, either before or after product approval, subject us to administrative or judicially imposed sanctions that may negatively impact the commercialization of BELVIQ or approval of one or more of our drug candidates or otherwise negatively impact our business. Regulatory agencies have in the past inspected certain aspects of our business in the United States and Switzerland, and we were provided with observations of objectionable conditions or practices with respect to our business in the United States. We believe we satisfactorily addressed such observations, but there is no assurance that regulatory agencies will not provide us with observations in future inspections or that we satisfactorily addressed observations provided to us in past inspections.
Neither collaborators nor we are permitted to market a drug candidate in the United States until the particular drug candidate is approved for marketing by the FDA. Specific preclinical data, chemistry, manufacturing and controls data, a proposed clinical trial protocol and other information must be submitted to the FDA as part of an investigational new drug, or IND, application, and clinical trials may commence only after the IND application becomes effective. To market a new drug in the United States, we must submit to the FDA and obtain FDA approval of an NDA. An NDA must be supported by extensive clinical and preclinical data, as well as extensive information regarding chemistry, manufacturing and controls to demonstrate the safety and effectiveness of the drug candidate. Following its review of an NDA or a response to a Complete Response Letter, or CRL, the FDA may approve the NDA or issue a CRL.
Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. As part of the Prescription Drug User Fee Act, or PDUFA, the FDA has a goal to review and act on a percentage of all submissions in a given time frame. The FDA’s review goals are subject to change, and it is unknown whether any particular FDA review will be completed within the FDA’s review goals or will be delayed. Moreover, the duration of the FDA’s review may depend on the number and types of other submissions made to the FDA around the same time period.
As with BELVIQ, any drug that acts on the CNS has the potential to be scheduled as a controlled substance by the DEA. DEA scheduling is a separate process that can delay when a drug may become available to patients beyond the issuance of an NDA approval letter, and the timing and outcome of such DEA process is uncertain. For example, the FDA approved the NDA for BELVIQ in June 2012, subject to the final scheduling of BELVIQ by the DEA. The DEA’s final rule placing BELVIQ into Schedule IV of the Controlled Substances Act was not effective until June 2013. The scheduling designation can also change after it has been finalized. DEA scheduling ranges from I to V, with I being the most tightly controlled category. If BELVIQ were to be rescheduled into a different category, such scheduling could negatively impact the ability or willingness to prescribe or dispense BELVIQ, the likelihood that patients will use it and other aspects of our and Eisai’s ability to commercialize it.
Regulatory approval of an NDA is not guaranteed. The number and types of preclinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to target and the regulations applicable to any particular drug candidate. Despite the time and expense exerted in preclinical and clinical studies, failure can occur at any stage, and we could encounter
14
problems that cause us to abandon clinical trials or to repeat or perform additional preclinical studies and clinical trials. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including:
| • | a drug candidate may not be deemed adequately safe and effective; |
| • | FDA officials may not find the data from preclinical studies and clinical trials sufficient; |
| • | the FDA’s interpretation and our interpretation of data from preclinical studies and clinical trials may differ significantly; |
| • | our or our contractors’ or collaborators’ failure to comply with applicable FDA and other regulatory requirements, including those identified in other risk factors; |
| • | the FDA may not approve the manufacturing processes or facilities; |
| • | the FDA may change its approval policies or adopt new regulations; or |
| • | the FDA may not accept an NDA or other submission due to, among other reasons, the content or formatting of the submission. |
Even if approved, drug candidates may not be approved for all indications requested and such approval may be subject to limitations on the indicated uses for which the drug may be marketed, restricted distribution methods or other limitations, such as those required by a Risk Evaluation and Mitigation Strategies, or REMS.
Our preclinical and clinical data, other information and procedures relating to a drug candidate may not be sufficient to support approval by the FDA or any other US or foreign regulatory authority, or regulatory interpretation of these data and procedures may be unfavorable. Our business and reputation may be harmed by any failure or significant delay in receiving regulatory approval for the sale of any drugs resulting from our drug candidates. As a result, we cannot predict when or whether regulatory approval will be obtained for any drug we or our collaborators develop.
To market any drugs outside of the United States, we and our current or future collaborators must comply with numerous and varying regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks associated with FDA approval as well as additional risks, some of which may be unanticipated.
For example, the EMA guidelines provide that clinical trials assessing drug candidates intended for weight control should subject patients to a weight reducing diet run-in period, and our Phase 3 clinical trials of BELVIQ did not include a run-in period. Such EMA guidelines also provide primary and alternative primary efficacy criteria for weight loss drug candidates. We believe BELVIQ will satisfy the EMA’s alternative primary efficacy criterion, which is the proportion of responders achieving more than 10% weight loss at the end of a 12-month period. However, we do not believe BELVIQ meets the more stringent EMA primary efficacy criterion, which requires demonstrating weight loss of at least 10% of baseline weight that is also at least 5% greater than that associated with placebo. Also, with respect to our previously filed MAA for BELVIQ in the European Union, the EMA raised questions regarding the dropout rate in our clinical trials and how this affects the analysis of efficacy in those trials. We also previously received feedback with respect to regulatory applications in other territories that included major objections.
We cannot assure you that our collaborator’s or our past or any future responses or submissions will be sufficient to the applicable regulatory authority or others, that the applicable regulatory authority or others will consider our BELVIQ program or data, including with regard to BELVIQ’s efficacy or safety, as sufficient, or that any other regulatory authority will ever approve BELVIQ.
Regulatory approval of a drug in one territory does not ensure additional regulatory approval in such territory (such as approval of the drug in combination with other drugs, for other indications or using different formulations) or regulatory approval in another territory, but a failure or delay in obtaining regulatory approval may negatively impact other regulatory processes. Failure to obtain regulatory approval in a territory, any delay or setback in obtaining such approval, or our regulatory strategy or decisions could adversely affect the regulatory approval or commercialization of our drug candidates in other territories, including that our drug candidates may not be approved for all indications requested, that such approval may be subject to limitations on the indicated uses for which the drug may be marketed, and with regard to the pricing or reimbursement of any approved drugs.
15
Our drugs will still be subject to extensive postmarketing regulation if approved.
Following regulatory approval of any of our drug candidates, we and our collaborators will be subject to ongoing obligations and continued regulatory review from the FDA and other applicable regulatory agencies, such as continued adverse event reporting requirements. As with BELVIQ, there may also be additional postmarketing obligations imposed by the FDA or other regulatory agencies. These obligations may result in significant expense and limit the ability to commercialize such drugs.
The FDA or other regulatory agencies may also require that the sponsor of the NDA or foreign equivalent, as applicable, conduct additional clinical trials to further assess approved drugs after approval under a post-approval commitment. Such additional studies may be costly and may impact the commercialization of the drug. For example, as part of the approval of BELVIQ, the FDA required the conduct of the CVOT described above as well as postmarketing studies to assess the safety and efficacy of BELVIQ for weight management in obese pediatric patients. Along with being costly and time consuming, a delay or unfavorable results from these trials could negatively impact market acceptance of BELVIQ; limit the revenues we generate from sales; result in BELVIQ’s withdrawal from the market; negatively impact the potential approval of BELVIQ in other territories for weight management, for other indications, in combination with other drugs or using different formulations; and preclude us from being profitable.
The FDA or other regulatory agencies may also impose significant restrictions on the indicated uses for which a drug may be marketed. Additionally, the FDA may require a REMS, including in connection with a drug’s approval, to help ensure that the benefits of the drug outweigh its risks. A REMS may be required to include various elements, such as a medication guide or patient package insert, a communication plan to educate healthcare providers of the drug’s risks, limitations on who may prescribe or dispense the drug, requirements that patients enroll in a registry or undergo certain health evaluations or other measures that the FDA deems necessary to ensure the safe use of the drug.
With regard to BELVIQ and any of our drug candidates that receive regulatory approval, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the drug will be subject to extensive regulatory requirements. We and the manufacturers of our products are also required to comply with cGMP regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our products, and these facilities are subject to ongoing regulatory inspections. In addition, regulatory agencies subject a drug, its manufacturer and the manufacturer’s facilities to continual review and inspections. The subsequent discovery of previously unknown problems with a drug, including adverse events of unanticipated severity or frequency, or problems with the facility where the drug is manufactured, may result in restrictions on the marketing of that drug, up to and including withdrawal of the drug from the market. In the United States, the DEA and comparable state-level agencies also heavily regulate the manufacturing, holding, processing, security, recordkeeping and distribution of drugs that are considered controlled substances, and the DEA periodically inspects facilities for compliance with its rules and regulations.
If our manufacturing facilities or those of our suppliers fail to comply with applicable regulatory requirements, such noncompliance could result in regulatory action and additional costs to us. Failure to comply with applicable FDA and other regulatory requirements may, either before or after product approval, if any, subject us to administrative or judicially imposed sanctions, including:
| • | issuance of inspectional notices of violation or warning letters by any regulatory agency; |
| • | imposition of fines and other civil penalties; |
| • | criminal prosecutions; |
| • | injunctions, suspensions or revocations of regulatory approvals; |
| • | suspension of any ongoing clinical trials; |
| • | total or partial suspension of manufacturing; |
16
| • | delays in commercialization; |
| • | refusal by any regulatory agency to approve pending applications or supplements to approved applications filed by us or collaborators; |
| • | refusals to permit drugs or related materials to be imported into or exported from the United States or other countries; |
| • | restrictions on operations, including costly new manufacturing requirements; and |
| • | product recalls or seizures. |
The FDA’s and other regulatory agencies’ policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates or further restrict or regulate post-approval activities. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we or our collaborators might not be permitted to market our drugs and our business could suffer.
Our ability to generate revenues from BELVIQ or any of our drug candidates that receive regulatory approval will be subject to a variety of risks, many of which are out of our control.
BELVIQ or any of our drug candidates that may be approved for marketing may not gain market acceptance among patients, healthcare providers, healthcare payers or the medical community. We believe that the degree of market acceptance and our ability to generate revenues from such products will depend on a number of factors, including:
| • | timing of market introduction of our drugs and competitive drugs and alternative treatments; |
| • | actual and perceived efficacy and safety of our drug candidates; |
| • | incidence and severity of any side effects; |
| • | potential or perceived advantages or disadvantages as compared to alternative treatments; |
| • | strength of sales, marketing and distribution support; |
| • | price of our future products, both in absolute terms and relative to alternative treatments; |
| • | the general marketplace for the particular drug; |
| • | the effect of current and future healthcare laws on our drug candidates; |
| • | availability of coverage and adequate reimbursement from government and other third-party payers; and |
| • | product labeling or product insert requirements of the FDA or other regulatory authorities. |
If our approved drugs fail to achieve market acceptance, we may not be able to generate significant revenues to be profitable.
Drug development programs are expensive, time consuming, uncertain and susceptible to change, interruption, delay or termination.
Drug development programs are very expensive, time consuming and difficult to design and implement. Our drug candidates are in various stages of research and development and are prone to the risks of failure inherent in drug development. In addition, the FDA or other regulatory authority may require us to, or we or others may decide to, conduct additional research and development of any of our approved drugs. Clinical trials and preclinical studies are needed to demonstrate that drug candidates are safe and effective to the satisfaction of the FDA and similar non-US regulatory authorities. These trials and studies are expensive and uncertain processes that may take years to complete. Failure can occur at any stage of the process, and successful early preclinical studies or clinical trials do not ensure that later studies or trials will be successful. In addition, the commencement or completion of our planned preclinical studies or clinical trials could be substantially delayed or prevented by several factors, including the following:
17
| • | limited number of, and competition for, suitable patients required for enrollment in our clinical trials or animals to conduct our preclinical studies; |
| • | limited number of, and competition for, suitable sites to conduct our clinical trials or preclinical studies; |
| • | delay or failure to obtain approval or agreement from the applicable regulatory authority to commence a clinical trial or approval of a study protocol; |
| • | delay or failure to obtain sufficient supplies of drug candidates, drugs or other materials for the trial or study; |
| • | delay or failure to reach agreement on acceptable agreement terms or protocols; and |
| • | delay or failure to obtain institutional review board, or IRB, approval to conduct a clinical trial at a prospective site. |
Even if the results of our development programs are favorable, the development programs of our most advanced drug candidates, including those being developed by collaborators, may take significantly longer and cost more than expected to complete. In addition, the FDA, other regulatory authorities, collaborators, or we may suspend, delay or terminate our development programs at any time for various reasons, including:
| • | lack of effectiveness of any drug candidate during clinical trials; |
| • | side effects experienced by study participants or other safety issues; |
| • | slower than expected rates of patient recruitment and enrollment or lower than expected patient retention rates; |
| • | delays or inability to manufacture or obtain sufficient quantities of materials for use in clinical trials; |
| • | inadequacy of or changes in our manufacturing process or compound formulation; |
| • | delays in obtaining regulatory approvals to commence a study, or “clinical holds,” or delays requiring suspension or termination of a study by a regulatory authority, such as the FDA, after a study is commenced; |
| • | changes in applicable regulatory policies and regulations; |
| • | delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites; |
| • | uncertainty regarding proper dosing; |
| • | unfavorable results from ongoing clinical trials or preclinical studies; |
| • | failure of our clinical research organizations to comply with all regulatory and contractual requirements or otherwise perform their services in a timely or acceptable manner; |
| • | scheduling conflicts with participating clinicians and clinical institutions; |
| • | failure to design appropriate clinical trial protocols; |
| • | insufficient data to support regulatory approval; |
| • | termination of clinical trials by one or more clinical trial sites; |
| • | inability or unwillingness of medical investigators to follow our clinical protocols; |
| • | difficulty in maintaining contact with subjects during or after treatment, which may result in incomplete data; |
| • | lack of sufficient funding to continue clinical trials or preclinical studies; or |
| • | changes in business priorities or perceptions of the value of the program. |
There is typically a high rate of attrition from the failure of drug candidates proceeding through clinical trials, and many companies have experienced significant setbacks in advanced development programs even after promising results in earlier studies or trials. We have experienced setbacks in our internal and partnered development programs
18
and expect to experience additional setbacks from time to time in the future. If we or our collaborators abandon or are delayed in our development efforts related to BELVIQ or any drug candidate, we may not be able to generate sufficient revenues to continue our operations at the current level or be profitable, our reputation in the industry and in the investment community would likely be significantly damaged, additional funding may not be available to us or may not be available on terms we or others believe are favorable, and our stock price may decrease significantly.
The results of preclinical studies and completed clinical trials are not necessarily predictive of future results, and our current drug candidates or any approved drugs may not be further developed or have favorable results in later studies or trials.
Preclinical studies and Phase 1 and Phase 2 clinical trials are not primarily designed to test the efficacy of a drug candidate, but rather to test safety, to study pharmacokinetics and pharmacodynamics, and to understand the drug candidate’s side effects at various doses and schedules. Favorable results in early studies or trials may not be repeated in later studies or trials, including continuing preclinical studies and large-scale clinical trials, and our drug candidates or drugs in later-stage trials may fail to show desired safety and efficacy despite having progressed through earlier-stage trials. Unfavorable results from ongoing preclinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials, or abandonment of a program. Preclinical and clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated; a program to be abandoned; or negatively impact a related marketed drug.
Many of our research and development programs are in the discovery or preclinical stage of development. The process of discovering compounds with therapeutic potential is expensive, time consuming and unpredictable. Similarly, the process of conducting preclinical studies of compounds that we discover requires the commitment of a substantial amount of our technical and financial resources and personnel. We may not discover additional compounds with sufficient therapeutic potential, and any of our preclinical compounds may not result in the commencement of clinical trials. We cannot be certain that results sufficiently favorable to justify commencement of Phase 1 clinical trials will be obtained in these preclinical investigations or that we will further develop a drug candidate at any stage of development. Even if favorable results are obtained from preclinical studies or trials, our financial resources may not allow us to advance a compound or drug candidate. If we are unable to identify and develop new drug candidates, we may not be able to maintain a clinical development pipeline or generate additional revenues.
We may participate in new strategic transactions that could impact our liquidity, increase our expenses, present significant distractions to our management and be viewed as unfavorable.
From time to time we consider strategic transactions, such as out-licensing or in-licensing of compounds or technologies, acquisitions of companies and asset purchases. Additional potential transactions we may consider include a variety of different business arrangements, such as strategic collaborations, joint ventures, spin-offs, restructurings, divestitures, business combinations and investments. In addition, another entity may pursue us as an acquisition target. Any such transaction may be viewed as unfavorable by our stockholders or others and may require us to incur non-recurring or other charges, may create potential liabilities, may increase our near- and long-term expenditures and may pose significant integration challenges, require additional expertise or disrupt our management or business, which could harm our operations and financial results.
As part of an effort to enter into significant transactions, we conduct business, legal and financial due diligence with the goal of identifying and evaluating material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, might not realize the intended advantages of the transaction. If we fail to realize the expected benefits from any transaction we may consummate, whether as a result of unidentified risks, integration difficulties, regulatory setbacks or other events, our business, results of operations and financial condition could be adversely affected.
Drug discovery and development is intensely competitive in the therapeutic areas on which we focus. If our competitors increase or they develop treatments that are approved faster, marketed better, less expensive or demonstrated to be more effective or safer than our drugs or drug candidates, our commercial opportunities will be reduced or eliminated.
19
Many of the drugs we or our collaborators are or may attempt to discover and develop may compete with existing therapies in the United States and other territories. In addition, many companies are pursuing the development of new drugs that target the same diseases and conditions that we target.
For example, with regard to BELVIQ, VIVUS and Orexigen Therapeutics, Inc., announced the US market availability of their drugs for chronic weight management in September 2012 and October 2014, respectively, and, in December 2014, the FDA approved Novo Nordisk’s drug candidate for the treatment of obesity. We also face competition from other drugs that may be indicated or used off label or otherwise for weight loss and from other approaches for weight loss, including behavior modification (such as diet and exercise), surgical approaches (such as gastric bypass surgery and gastric banding), and herbal or other supplements. With respect to future weight-loss treatments, we expect that companies and others may allocate resources to discover and develop additional drugs, additional drug candidates may be approved and competition may increase. For example, in December 2014, Orexigen Therapeutics, Inc., announced that the CHMP has adopted a positive opinion recommending the granting of a centralized marketing authorization for the approval of its drug candidate for chronic weight management in the European Union; and, in December 2013, Novo Nordisk announced that it has filed a submission for regulatory approval in Europe of its drug candidate for the treatment of obesity.
Our competitors, particularly large pharmaceutical companies, may have substantially greater research, development and marketing capabilities and greater financial, scientific and human resources than we do. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before we do for the same indication may achieve a significant competitive advantage, including certain patent and marketing exclusivity rights. In addition, our competitors’ drugs may have fewer side effects, more desirable characteristics (such as efficacy, route of administration or frequency of dosing), or be viewed more favorably by patients, healthcare providers, healthcare payers, the medical community, the media or others than our drug candidates or drugs, if any, for the same indication. Our competitors may also market generic or other drugs that compete with our drugs at a lower price than our drugs, which may negatively impact our drug sales, if any. Any results from our research and development efforts, or from our joint efforts with our existing or any future collaborators, may not compete successfully with existing or newly discovered products or therapies.
We have obtained orphan drug designation from the FDA for ralinepag for the treatment of pulmonary arterial hypertension, or PAH, but we may be unable to maintain the benefits associated with orphan drug designation, including the potential for market exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is defined as one occurring in a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a drug that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the drug is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the drug with orphan drug exclusivity or where the manufacturer is unable to assure sufficient drug quantity.
Even though ralinepag has been granted orphan drug status for the treatment of PAH, we may not be the first to obtain marketing approval for a drug candidate for this orphan-designated indication due to the uncertainties associated with developing drugs. In addition, exclusive marketing rights in the United States may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the drug to meet the needs of patients with the rare disease or condition. Further, even if we obtain orphan drug exclusivity for a drug, that exclusivity may not effectively protect the drug from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan drug is approved, the FDA can subsequently approve the same drug with the same active moiety for the same condition if the FDA concludes that the later drug is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
20
Collaborative relationships may lead to disputes and delays in drug development and commercialization, and we may not realize the full commercial potential of our drug candidates or drugs.
We may have conflicts with our prospective, current or past collaborators, such as conflicts concerning rights and obligations under our agreements, the interpretation of preclinical or clinical data, the achievement of milestone or other payments, the ownership of intellectual property, or research and development, regulatory, commercialization or other strategy. Collaborators may stop supporting our drug candidates or drugs, including if they no longer view the program as in their best financial or other interests or they develop or obtain rights to competing drug candidates or drugs. In addition, collaborators may fail to effectively develop, obtain approval for or commercialize our drugs, which may result in us not realizing their full commercial potential. If any conflicts arise with any of our current, past or prospective collaborators, the other party may act in a manner that is adverse to our interests. Any such disagreement could result in one or more of the following, each of which could delay, or lead to termination of, development or commercialization of our drug candidates or drugs, and in turn prevent us from generating revenues:
| • | unwillingness on the part of a collaborator to pay for studies or other research, milestones, royalties or other payments that we believe are due to us under a collaboration; |
| • | uncertainty regarding ownership of intellectual property rights arising from our collaborative activities, which could prevent us from entering into additional collaborations; |
| • | unwillingness on the part of a collaborator to keep us informed regarding the progress of its development, regulatory, commercialization, pharmacovigilance or other activities or to permit public disclosure of the results of those activities; |
| • | slowing or cessation of a collaborator’s research, development, regulatory or commercialization efforts with respect to our drug candidates or drugs; or |
| • | litigation or arbitration. |
Setbacks and consolidation in the pharmaceutical and biotechnology industries could make entering into agreements with pharmaceutical companies to collaborate or commercialize our drugs more difficult and diminish our revenues.
Setbacks in the pharmaceutical and biotechnology industries, such as those caused by safety concerns relating to drugs or drug candidates, as well as competition from generic drugs, litigation and industry consolidation, may have an adverse effect on us, including by making it more difficult to enter into agreements with pharmaceutical companies to collaborate or commercialize our drugs and diminishing our revenues. For example, the FDA may be more cautious in approving our drug candidates based on safety concerns relating to these or other drugs or drug candidates, or pharmaceutical companies may be less willing to enter into new collaborations or continue existing collaborations if they are integrating a new operation as a result of a merger or acquisition or if their therapeutic areas of focus change following a merger.
We and our collaborators may from time to time rely on third parties to conduct clinical trials and preclinical studies. If those parties do not comply with regulatory and contractual requirements, successfully carry out their contractual duties or meet expected deadlines, our drug candidates may not advance in a timely manner or at all.
In the course of our discovery, preclinical testing and clinical trials, we and our collaborators may from time to time rely on third parties, including laboratories, investigators, clinical research organizations and manufacturers, to perform critical services. For example, we rely on third parties to conduct our clinical trials and many of our preclinical studies. Clinical research organizations are responsible for many aspects of the trials, including finding and enrolling subjects for testing and administering the trials. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as Good Clinical Practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may
21
not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner, and we may need to enter into new arrangements with alternative third parties and our preclinical studies or clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. In addition, if such third parties fail to perform their obligations in compliance with regulatory requirements and our protocols, our preclinical studies or clinical trials may not meet regulatory requirements or may need to be repeated. As a result of our dependence on third parties, we may face delays or failures outside of our direct control. These risks also apply to the development activities of collaborators, and we do not control their research and development, clinical trial or regulatory activities.
Our efforts will be seriously jeopardized if we are unable to retain and attract key and other employees.
Our success depends on the continued contributions of our principal management, development and scientific personnel, and the ability to hire and retain key and other personnel. We face competition for such personnel, and we believe that risks and uncertainties related to our business, including the timing and risk associated with research, development and commercialization, the regulatory process, our available and anticipated cash resources, litigation involving us, and the volatility of our stock price, may impact our ability to hire and retain key and other personnel. The loss of services of any principal member of our management or scientific staff or other personnel, particularly Jack Lief, our President and Chief Executive Officer, and Dominic P. Behan, Ph.D., D.Sc., our Executive Vice President and Chief Scientific Officer, or a combination of different key employees, could adversely impact our operations and ability to generate or raise additional capital. To our knowledge, neither Mr. Lief nor Dr. Behan plans to leave, retire or otherwise disassociate with us in the near future.
We may incur substantial liabilities for any product liability claims or otherwise as a drug product manufacturer.
We develop, test, manufacture and expect to commercialize drugs for use by humans. We face an inherent risk of product liability exposure related to the testing of our drug candidates in clinical trials, and face an even greater risk with the commercialization of BELVIQ as well as any other drug that may be approved for marketing. In addition, under the marketing and supply agreement with Eisai, Arena GmbH and Eisai will, in general, share equally in losses resulting from third-party product liability claims, with certain limited exceptions.
Whether or not we are ultimately successful in any product liability or related litigation, such litigation would consume substantial amounts of our financial and managerial resources, and might result in adverse publicity, all of which would impair our business. In addition, damages awarded in a product liability action could be substantial and could have a negative impact on our financial condition.
An individual may bring a liability claim against us if one of our drugs or drug candidates causes, or merely appears to have caused, an injury. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for our drug; |
| • | injury to our reputation; |
| • | increased difficulty to attract, or withdrawal of, clinical trial subjects; |
| • | costs of related litigation; |
| • | substantial monetary awards to subjects or other claimants; |
| • | loss of revenues; and |
| • | the inability to commercialize our drug candidates. |
We will have limited product liability insurance that covers our clinical trials and products. We may not be able to maintain or obtain insurance coverage at a reasonable cost, and we may not have insurance coverage that will be adequate to satisfy any liability that may arise, which could have an adverse effect on our results of operations and financial condition.
We expect that Arena GmbH will manufacture BELVIQ for commercialization and, from time to time, for clinical trials or other studies. Arena GmbH also manufactures certain generic drug products for Siegfried. Arena GmbH is subject to liability for non-performance, product recalls and breaches of the agreements with Siegfried and our collaborators under our marketing and supply agreements.
22
We have significant contractual obligations, which may adversely affect our cash flow, cash position and stock price.
We have long-term leases on real properties and other contractual obligations. If we are unable to generate cash from operations sufficient to meet financial obligations, we will need to obtain additional funds from other sources, which may include one or more financings. However, we may be unable to obtain sufficient additional funds when we need them on favorable terms or at all. The sale of equity or convertible debt securities in the future may be dilutive to our stockholders, and debt-financing arrangements may require us to enter into covenants that would further restrict certain business activities or our ability to incur additional indebtedness, and may contain other terms that are not favorable to our stockholders or us.
Also, if we are unable to generate cash from operations or obtain additional funds from other sources sufficient to meet our contractual obligations, or we need to use existing cash to fund our contractual obligations, we may have to delay or curtail some or all of our research, development and commercialization programs, or sell or license some or all of our assets on terms that you or others may view as unfavorable. Our contractual obligations could have significant additional negative consequences, including, without limitation:
| • | increasing our vulnerability to general adverse economic conditions; |
| • | limiting our ability to obtain additional funds; and |
| • | placing us at a possible competitive disadvantage to less leveraged competitors and competitors that have better access to capital resources. |
We may be subject, directly or indirectly, to federal and state healthcare laws, including but not limited to fraud and abuse and false claims laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties and prosecution.
In the United States, drug manufacturers and marketers are subject to various state and federal fraud and abuse laws, including, without limitation, the Federal Anti-Kickback Statute and Federal False Claims Act. There are similar laws in other countries. These laws may impact, among other things, the research, manufacturing, sales, marketing and education programs for our drugs.
The Federal Anti-Kickback Statute prohibits persons and entities from knowingly and willingly soliciting, offering, receiving or providing any remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the purchase, lease, order or the furnishing or arranging for, a good, item, facility or service, for which payment may be made, in whole or in part, under a federal healthcare program such as the Medicare and Medicaid programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The Federal Anti-Kickback Statute is broad and, despite a series of narrow statutory exceptions and regulatory safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. Moreover, the PPACA, among other things, amended the intent requirement of the Federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of these statutes or specific intent to violate them. The PPACA also provides that the government may assert that a claim including items or services resulting from a violation of the Federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the Federal Civil False Claims Act. Many states have also adopted laws similar to the Federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare items or services reimbursed by any source, not only the Medicare and Medicaid programs.
The Federal Civil False Claims Act prohibits, among other things, persons or entities from knowingly presenting, or causing to be presented, a false claim to, or the knowing use of false statements to obtain payment from the federal government. Suits filed under the Federal Civil False Claims Act can be brought by any individual on behalf of the government, known as “qui tam” actions, and such individuals, commonly known as “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The filing of qui tam actions has caused a number of pharmaceutical, medical device and other healthcare companies to have to
23
defend a Federal Civil False Claims Act action. When an entity is determined to have violated the Federal Civil False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim, in addition to other penalties that may apply. Various states have also enacted laws modeled after the Federal Civil False Claims Act, some of which are broader in scope and may apply regardless of payer.
The Federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Additionally, the civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The Federal Physician Payment Sunshine Act, created under the PPACA, and its implementing regulations requires certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the US Department of Health and Human Services, or HHS, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit the required information may result in civil monetary penalties up to an aggregate of $150,000 per year (and up to an aggregate of $1 million per year for “knowing failures”) for all payments, transfers of value or ownership or investment interests not reported in an annual submission, and may result in liability under other federal laws or regulations.
We may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, impose specified requirements relating to the privacy, security and transmission of individually identifiable health information.
Additionally, other legislative changes have been proposed and adopted since the PPACA was enacted. For example, the Drug Supply Chain Security Act imposes new obligations on manufacturers of pharmaceutical products, among others, related to product tracking and tracing. Among the requirements of this new legislation, manufacturers will be required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, label drug product with a product identifier, and keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers will eventually be required to be done electronically. Manufacturers will also be required to verify that purchasers of the manufacturers’ products are appropriately licensed. Further, under this new legislation, manufacturers will have drug product investigation, quarantine, disposition, and notification responsibilities related to counterfeit, diverted, stolen, and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
We are unable to predict whether we could be subject to actions under any of these fraud and abuse or other laws, or the impact of such actions. If we are found to be in violation of any of the laws described above and other applicable state and federal fraud and abuse laws, we may be subject to penalties, including civil, criminal and/or administrative penalties, damages, fines, individual imprisonment, disgorgement, possible exclusion from government healthcare reimbursement programs and the curtailment or restructuring of our operations, all of which could have a material adverse effect on our business and results of operations.
We may not be able to effectively integrate or manage our international operations and such difficulty could adversely affect our stock price, business operations, financial condition and results of operations.
The headquarters of our operations outside of the United States is in Switzerland. Activities conducted at this location include manufacturing, quality control, quality assurance, development of manufacturing processes, qualifying suppliers and otherwise managing aspects of the supply chain, regulatory compliance, distribution of
24
finished products, alliance management, and strategic planning and development. There are significant risks associated with foreign operations, including, but not limited to, compliance with local laws and regulations, the protection of our intellectual property, the ability to integrate our corporate culture with local customs and cultures, the distraction to our management, foreign currency exchange rates and the impact of shifts in the US and local economies on those rates, and integration of our policies and procedures, including disclosure controls and procedures and internal control over financial reporting, with our international operations.
We use biological materials, hazardous materials, chemicals and radioactive compounds.
Our research, development and manufacturing activities involve the use of potentially harmful biological materials, as well as materials, chemicals and various radioactive compounds that could be hazardous to human health and safety or the environment. These materials and various wastes resulting from their use are stored at our facility pending ultimate use and disposal. We cannot completely eliminate the risk of contamination, which could cause:
| • | interruption of our research, development or manufacturing efforts; |
| • | injury to our employees and others; |
| • | environmental damage resulting in costly clean up; and |
| • | liabilities under domestic or foreign laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. |
In such an event, we may be held liable for any resulting damages, and any such liability could exceed our resources. Although we carry insurance in amounts and type that we consider commercially reasonable, we cannot be certain that the coverage or coverage limits of our insurance policies will be adequate and we do not have insurance coverage for losses relating to an interruption of our research and development efforts caused by contamination.
Our business and operations might be adversely affected by business disruptions and security breaches, including any cybersecurity incidents.
Our US operations, including laboratories, offices and a chemical development facility, are located in the same business park in San Diego. We also have a drug product manufacturing facility in Zofingen, Switzerland, and we expect that, at least for the near-term, this facility will be the sole location for the manufacturing of BELVIQ finished drug product. We depend on our facilities and on collaborators, contractors and vendors for the continued operation of our business, some of whom are located in Europe and Asia. Natural disasters or other catastrophic events, including interruptions in the supply of natural resources, political and governmental changes, disruption in transportation networks or delivery services, severe weather conditions, wildfires and other fires, explosions, actions of animal rights activists, terrorist attacks, earthquakes and wars could disrupt our operations or those of our collaborators, contractors and vendors.
We depend on the efficient and uninterrupted operation of our computer and communications systems, which we use for, among other things, sensitive company data, including our financial data, intellectual property and other proprietary business information. We maintain the information technology, or IT, infrastructure for our San Diego campus and our manufacturing facility in Switzerland.
While certain of our operations have business continuity and disaster recovery plans and other security measures intended to prevent and minimize the impact of IT-related interruptions, our IT infrastructure and the IT infrastructure of our current and any future collaborators, contractors and vendors are vulnerable to damage from cyberattacks, computer viruses, unauthorized access, electrical failures and natural disasters or other catastrophic events. We could experience failures in our information systems and computer servers, which could result in an interruption of our normal business operations and require substantial expenditure of financial and administrative resources to remedy. System failures, accidents or security breaches can cause interruptions in our operations and can result in a material disruption of our research and development programs, manufacturing or commercialization activities and other business operations. The loss of data from completed or future studies or clinical trials could result in delays in our research, development or regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Similarly, we rely on third parties to supply materials for the manufacture of BELVIQ and our drug candidates, conduct studies and clinical trials of our drug candidates and warehouse, market and
25
distribute BELVIQ, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the development of any of our other drug candidates and the commercialization of BELVIQ could be delayed or otherwise adversely affected.
Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we might suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors’ insurance policies or for which we or our contractors do not have coverage. For example, we are not insured against a terrorist attack. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. Moreover, any such event could delay our research and development programs and adversely affect, which may include stopping, our commercial production.
Our executive officers and directors may sell shares of their stock, and these sales could adversely affect our stock price.
Sales of our stock by our executive officers and directors, or the perception that such sales may occur, could adversely affect the market price of our stock. Our executive officers and directors may sell stock in the future, either as part, or outside, of trading plans under SEC Rule 10b5-1.
Currency fluctuations may negatively affect our financial condition.
We primarily spend and generate cash in US dollars, and present our consolidated financial statements in US dollars. However, a portion of our expected and potential payments and receipts under our agreements are in foreign currencies, including Swiss francs. For example, payments and receipts under our agreements with Siegfried are required to be paid in Swiss francs. A fluctuation of the exchange rates of foreign currencies versus the US dollar may, thus, adversely affect our financial results, including cash balances, expenses and revenues. We may in the future enter into hedging transactions to try to reduce our foreign currency exposure, but there is no assurance that such transactions will occur or be successful.
Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers.
Laws and regulations affecting public companies, including rules adopted by the SEC and by NASDAQ, as well as the laws and regulations of foreign governments, may result in increased costs to us, particularly as we continue to develop the required capabilities in the United States and abroad to commercialize our products. These laws, rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including directors’ and officers’ liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on our board committees or as executive officers. We cannot estimate accurately the amount or timing of additional costs we may incur to respond to these laws, rules and regulations.
Risks Relating to Our Intellectual Property
Our success is dependent on intellectual property rights held by us and third parties and our interest in these rights is complex and uncertain.
Our success will depend on our own and on current or future collaborators’ abilities to obtain, secure and defend patents. In particular, the patents directed to BELVIQ and our drug candidates are important to commercializing drugs. We have numerous US and foreign patent applications pending for our technologies. There is no assurance that any of our patent applications will issue, or that any of the patents will be enforceable or will cover a drug or other commercially significant technology or method, or that the patents will be held to be valid for their expected terms.
The procedures for obtaining a patent in the United States and in most foreign countries are complex. These procedures require an analysis of the scientific technology related to the invention and many sophisticated legal issues. Obtaining patent rights outside the United States often requires the translation of highly technical documents
26
and an improper translation may lead to the loss of, or otherwise jeopardize, the patent protection of our inventions. Ensuring adequate quality of translators and foreign patent attorneys is often very challenging. Consequently, the process for having our pending patent applications issue as patents will be difficult, complex and time consuming. Our patent position is very uncertain and we do not know when, or if, we will obtain additional patents for our technologies, or if the scope of the patents obtained will be sufficient to protect our drugs, or be considered sufficient by parties reviewing our patent positions pursuant to a potential marketing, licensing or financing transaction.
In addition, other entities may challenge the validity or enforceability of our patents and patent applications in litigation or administrative proceedings. Even the issuance of a patent is not conclusive as to its validity or enforceability. We cannot make assurances as to how much protection, if any, will be given to our patents if we attempt to enforce them or they are challenged. It is possible that a competitor or a generic pharmaceutical provider may successfully challenge our patents and those challenges may result in reduction or elimination of our patents’ coverage.
We also rely on confidentiality agreements and trade secrets to protect our technologies. However, such information is difficult to protect. We require our employees to contractually agree not to improperly use our confidential information or disclose it to others, but we may be unable to determine if our employees have conformed or will conform to their legal obligations under these agreements. We also enter into confidentiality agreements with prospective collaborators, collaborators, service providers and consultants, but we may not be able to adequately protect our trade secrets or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of this information. Many of our employees and consultants were, and many of them may currently be, parties to confidentiality agreements with other pharmaceutical and biotechnology companies, and the use of our technologies could violate these agreements. In addition, third parties may independently discover our trade secrets or proprietary information.
Some of our academic institution licensors, research collaborators and scientific advisors have rights to publish data and information to which we have rights. We generally seek to prevent our collaborators from disclosing scientific discoveries before we have the opportunity to file patent applications on such discoveries. In some of our collaborations, we do not control our collaborators’ ability to disclose their own discoveries under the collaboration and in some of our academic collaborations we are limited to relatively short periods to review a proposed publication and file a patent application. If we cannot maintain the confidentiality of our technologies and other confidential information in connection with our collaborations, our ability to receive patent protection or protect our proprietary information will be impaired.
We believe that the United States is by far the largest single market for pharmaceuticals in the world. Because of the critical nature of patent rights to our industry, changes in US patent laws could have a profound effect on our future profits, if any. It is unknown which, if any, patent laws will change, how changes to the patent laws will ultimately be enforced by the courts and the impact on our business. For example, in September 2011, the America Invents Act was signed into US law, which changes include, among others, the awarding of a patent to the first inventor to file a patent as opposed to the first inventor to make an invention and the creation of new administrative procedures for challenging US patents. It may be several years before the impact of the America Invents Act on patent law is understood, and we cannot predict with certainty whether or to what extent the changes may impair our business.
A dispute regarding the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be costly and result in delays or termination of our future research, development, manufacturing and sales activities.
Our commercial success depends upon our ability to develop and manufacture our drugs and drug candidates, market and sell drugs, and conduct our research and development activities without infringing or misappropriating the proprietary rights of others. There are many patents and patent applications filed, and that may be filed, by others relating to drug discovery and development programs that could be determined to be similar, identical or superior to ours or our licensors or collaborators. We may be exposed to future litigation by others based on claims that our drugs, drug candidates, technologies or activities infringe the intellectual property rights of others. Numerous US and foreign issued patents and pending patent applications owned by others exist in the area of G protein-coupled receptors, or GPCRs, including some which purport to allow the patent holder to control the use of all drugs that modulate a particular drug target or GPCR, regardless of whether the infringing drug bears any structural
27
resemblance to a chemical compound known to the patent holder at the time of patent filing. Numerous US and foreign issued patents and pending patent applications owned by others also exist in the therapeutic areas in, and for the therapeutic targets for, which we are developing drugs. There are also numerous issued patents and patent applications to chemical compounds or synthetic processes that may be necessary or useful to use in our research, development, manufacturing or commercialization activities. These could materially affect our ability to develop our drug candidates or manufacture, import or sell drugs, and our activities, or those of our licensors or collaborators, could be determined to infringe these patents. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our drugs, drug candidates or technologies may infringe. There also may be existing patents, of which we are not aware, that our drug candidates or technologies may infringe. Further, there may be issued patents or pending patent applications in fields relevant to our business, of which we are or may become aware, that we believe (i) are invalid, unenforceable, or we do not infringe; (ii) relate to immaterial portions of our overall drug discovery, development, manufacturing and commercialization efforts; or (iii) in the case of pending patent applications, the resulting patent would not be granted or, if granted, would not likely be enforced in a manner that would materially impact such efforts. We cannot assure you that others holding any of these patents or patent applications will not assert infringement claims against us for damages or seek to enjoin our activities. We also cannot assure you that, in the event of litigation, we will be able to successfully assert any belief we may have as to non-infringement, unenforceability, invalidity or immateriality, or that any infringement claims will be resolved in our favor.
In addition, others may infringe or misappropriate our proprietary rights, and we may have to institute costly legal action to protect our intellectual property rights. We may not be able to afford the costs of enforcing or defending our intellectual property rights against others.
There could be significant litigation and other administrative proceedings in our industry that affect us regarding patent and other intellectual property rights. Any legal action or administrative action against us, or our collaborators, claiming damages or seeking to enjoin commercial activities relating to our drug discovery, development, manufacturing and commercialization activities could:
| • | require us, or our collaborators, to obtain a license to continue to use, manufacture or market the affected drugs, methods or processes, which may not be available on commercially reasonable terms, if at all; |
| • | prevent us from importing, making, using, selling or offering to sell the subject matter claimed in patents held by others and subject us to potential liability for damages; |
| • | consume a substantial portion of our managerial, scientific and financial resources; or |
| • | be costly, regardless of the outcome. |
Furthermore, because of the substantial amount of pre-trial document and witness discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the trading price of our common stock.
We are aware of third-party patents, as well as third-party patent applications, that could adversely affect the potential commercialization of APD334. For example, we are aware of a third-party patent, as well as third-party patent applications, with broad claims to administrating an S1P1 receptor agonist by starting with a lower dose and then increasing to a higher, standard daily dose. While we do not believe that any claims of such patent that would cover the potential commercialization of APD334 are valid and enforceable, we may be incorrect in this belief. In addition, other patents may issue from third-party patent applications with respect to certain dosing regimens, which could also adversely affect the potential commercialization of APD334, if APD334 is approved with a specific dosing regimen. We are also aware of pending third-party patent applications with claims to broad generic structural formulas, which claims if issued in their broadest form could adversely affect the potential commercialization of APD334.
We have been contacted from time to time by third parties regarding their intellectual property rights, sometimes asserting that we may need a license to use their technologies. For example, a third party has communicated that it believes its issued US patents include patent claims that cover BELVIQ or its use. We do not believe such patent claims are valid or, even if they were held valid, that they cover BELVIQ or its use. If we fail to obtain any required licenses or make any necessary changes to our technologies, we may become involved in expensive and time-consuming litigation or we may be unable to develop or commercialize some or all of our drugs or drug candidates.
28
We cannot protect our intellectual property rights throughout the world.
Filing, prosecuting, defending and enforcing patents on all of our drug discovery technologies and all of our potential drug candidates throughout the world would be prohibitively expensive. Competitors may use our technologies to develop their own drugs in jurisdictions where we have not obtained patent protection. These drugs may compete with our drugs, if any, and may not be covered by any of our patent claims or other intellectual property rights. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, the patent owner has failed to “work” the invention in that country or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our drug candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which makes it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Risks Relating to Our Securities
Our stock price will likely be volatile, and your investment in our stock could decline in value.
Our stock price has fluctuated historically. From January 1, 2013, to January 16, 2015, the market price of our stock was as low as $3.26 per share and as high as $11.00 per share.
Very few drug candidates being tested will ultimately receive regulatory approval, and companies in our industry sometimes experience significant volatility in their stock price. Our stock price may fluctuate significantly depending on a variety of factors, including:
| • | regulatory actions or decisions or legislation affecting BELVIQ, including decisions of regulatory authorities relating to BELVIQ, or other drugs or drug candidates, including those of our competitors; |
| • | the commercial availability and success or failure of BELVIQ (including perceptions of prescription trends or other information) or any of our drug candidates; |
| • | the entrance into, or failure to enter into, a new collaboration or the modification or termination of an existing collaboration or other material transaction; |
| • | the timing and receipt by us of milestone and other payments or failing to achieve and receive the same; |
| • | fluctuation in prescriptions, sales or financial results (including with respect to revenue recognition) or inaccurate sales or cash forecasting; |
| • | accounting restatements and changes; |
| • | supply chain or manufacturing issues; |
| • | discussions or recommendations affecting our drugs or drug candidates by FDA advisory committees or other reviewers of preclinical or clinical data or other information related to BELVIQ, drug candidates or other drugs; |
| • | results or decisions affecting the development or commercialization of BELVIQ or any of our drug candidates, including the results of studies, trials and other analyses; |
29
| • | the development and implementation of our continuing development and research plans, including outcome studies and other research and development for BELVIQ (including related development programs); |
| • | the timing of the discovery of drug leads and the development of our drug candidates; |
| • | changes in our research and development budget or the research and development budgets of our existing or potential collaborators; |
| • | the introduction, development or withdrawal of drug candidates or drugs by others that target the same diseases and conditions that we or our collaborators target or the introduction of new drug discovery techniques; |
| • | the success, failure or setbacks of our or a perceived competitor’s drugs or drug candidates; |
| • | expenses related to, and the results of, litigation, other disputes and other proceedings; |
| • | financing strategy or decisions; |
| • | developments in intellectual property rights or related announcements; and |
| • | capital market conditions. |
We are not able to control many of these factors. If our financial or scientific results in a particular period do not meet stockholders’ or analysts’ expectations, our stock price may decline and such decline could be significant.
There are a substantial number of shares of our common stock that may become eligible for future sale in the public market, and the sale of our common stock could cause the market price of our common stock to fall.
As of January 16, 2015, we had outstanding a warrant to purchase 1,965,418 shares of our common stock at an exercise price of $4.34 per share that expires on August 14, 2015. Such warrant was adjusted as a result of certain equity sales following its issuance to decrease the exercise price and increase the number of shares issuable upon exercise of the warrant. Certain future equity issuances below the pre-defined warrant adjustment price may result in additional adjustments to such warrant to the extent then outstanding.
Along with our outstanding warrant, as of January 16, 2015, there were (i) options to purchase 15,664,405 shares of our common stock outstanding under our equity incentive plans at a weighted-average exercise price of $5.26 per share, (ii) 676,284 restricted stock unit awards outstanding under our equity incentive plans, (iii) performance restricted stock unit awards outstanding under our equity incentive plans targeted at 1,475,000 shares (however, the actual number of shares that may be awarded ranges from 0% to 200% of such amount), (iv) 21,451,043 additional shares of common stock remaining issuable under our 2013 Long-Term Incentive Plan, (v) 596,574 shares of common stock remaining issuable under our 2009 Employee Stock Purchase Plan, as amended, and (vi) 79,169 shares of common stock remaining issuable under our Deferred Compensation Plan.
Once issued, the shares described above will be available for immediate resale in the public market. The market price of our common stock could decline as a result of such resales due to the increased number of shares available for sale in the market. As of January 16, 2015, there were 220,463,035 shares of our common stock outstanding.
Any future equity or debt issuances by us may have dilutive or adverse effects on our existing stockholders.
We have been opportunistic in our efforts to obtain cash, and we expect to continue to evaluate various funding alternatives from time to time. We have primarily financed our operations, and we may continue to finance our operations, by issuing and selling our common stock or securities convertible into or exercisable for shares of our common stock. We may issue additional shares of common stock or convertible securities that could dilute your ownership in our company and may include terms that give new investors rights that are superior to yours. Moreover, any issuances by us of equity securities may be at or below the prevailing market price of our common stock and in any event may have a dilutive impact on your ownership interest, which could cause the market price of our common stock to decline. In addition, we may also raise additional funds through the incurrence of debt, and the holders of any debt we may issue would have rights superior to your rights in the event we are not successful and are forced to seek the protection of bankruptcy laws.
30
The holders of our common stock and other securities may take actions that are contrary to your interests, including selling their stock.
A small number of stockholders may hold or acquire a significant amount of our outstanding stock. From time to time, there is a large short interest in our stock. These holders of such stock or positions may seek control of us, support transactions that we or you do not believe are favorable, and have interests that are different from yours. In addition, sales of a large number of shares of our stock by these large stockholders or other stockholders within a short period of time could adversely affect our stock price.
We may also be involved in disagreements with the holders of our stock, warrants or other securities in the future. Such disagreements may lead to proxy contests or litigation, which may be expensive and consume management’s time, involve settlements, the terms of which may not be favorable to us, or result in other negative consequences to our business.
Certain of our agreements, provisions in our charter documents, possible future agreements and Delaware law could delay or prevent a change in management or a takeover attempt that you may consider to be in your best interests.
There is a standstill provision in our marketing and supply agreement with Eisai, and we may enter into agreements with similar provisions. In addition, we may in the future adopt a stockholders’ rights agreement, which would cause substantial dilution to any person who attempts to acquire us in a manner or on terms not approved by our board of directors. These provisions or agreements, as well as other provisions in our certificate of incorporation and bylaws and under Delaware law, could delay or prevent the removal of directors and other management and could make more difficult a merger, tender offer or proxy contest involving us that you may consider to be in your best interests. For example, our charter provisions:
| • | allow our board of directors to issue preferred stock without stockholder approval; |
| • | limit who can call a special meeting of stockholders; |
| • | eliminate stockholder action by written consent; and |
| • | establish advance notice requirements for nomination for election to the board of directors or for proposing matters to be acted upon at stockholders’ meetings. |
Forward-Looking Statements
This Current Report includes forward-looking statements that involve a number of risks, uncertainties and assumptions. These forward-looking statements can generally be identified as such because the context of the statement will include words such as “may,” “will,” “intend,” “plan,” “believe,” “anticipate,” “expect,” “estimate,” “predict,” “potential,” “continue,” “likely,” or “opportunity,” the negative of these words or other similar words, though not all forward-looking statements contain these words. Similarly, statements that describe our plans, strategies, intentions, expectations, objectives, goals or prospects and other statements that are not historical facts are also forward-looking statements. For such statements, we claim the protection of the Private Securities Litigation Reform Act of 1995. Readers of this Current Report are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the time this Current Report was filed with the SEC. These forward-looking statements are based largely on our expectations and projections about future events and future trends affecting our business, and are subject to risks and uncertainties that could cause actual results to differ materially from those anticipated in the forward-looking statements. These risks and uncertainties include, without limitation, the risk factors identified in our SEC reports, including this Current Report. We can give no assurances that any of the events anticipated by the forward-looking statements will occur or, if any of them do, what impact they will have on our results of operations and financial condition. Except as required by law, we undertake no obligation to update publicly or revise our forward-looking statements.
31
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| Date: January 20, 2015 | Arena Pharmaceuticals, Inc.
| |||
| By: | /s/ Steven W. Spector | |||
| Steven W. Spector | ||||
| Executive Vice President, General Counsel and Secretary | ||||
32