Exhibit 10.1
AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT | 1. CONTRACT ID CODE | PAGE OF PAGES | ||||||||
1 | 2 | |||||||||
2. AMENDMENT/MODIFICATION NO. 0004 | 3. EFFECTIVE DATE See Block 16C | 4. REQUISITION/PURCHASE REQ. NO. | 5. PROJECT NO. (If applicable) | |||||||
6. ISSUED BY | CODE | ASPR-BARDA | 7. ADMINISTERED BY (if other than Item 6) | CODE | ASPR–BARDA01 | |||||
ASPR-BARDA 200 Independence Ave., S.W. Room 640-G Washington DC 20201 |
| ASPR-BARDA 330 Independence Ave, SW, Rm G644 Washington DC 20201 |
| |||||||
8. NAME AND ADDRESS OF CONTRACTOR (No., street, county. State and ZIP Code) CYTORI THERAPEUTICS, INC 1386447 CYTORI THERAPEUTICS, INC. 3020 3020 CALLAN RD SAN DIEGO CA 921211109 | (x) | 9A. AMENDMENT OF SOLICITATION NO. | ||||||||
| ||||||||||
9B. DATED (SEE ITEM 11) | ||||||||||
X | 10A. MODIFICATION OF CONTRACT/ORDER NO. HHS0100201200008C | |||||||||
10B. DATED (SEE ITEM 13) 09/28/2012 | ||||||||||
CODE 1386447 | FACILITY CODE | |||||||||
11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS | ||||||||||
o The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers o is extended. o is not extended, Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning ________ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or telegram which Includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGEMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by telegram or letter, provided each telegram or letter makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. | ||||||||||
12. ACCOUNTING AND APPROPRIATION DATA (If required) See Schedule | ||||||||||
13. THIS ITEM ONLY APPLIES TO MODIFICATION OF CONTRACTS/ORDERS. IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. | ||||||||||
CHECK ONE | A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. | |||||||||
| ||||||||||
| B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc.) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). | |||||||||
X | C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: FAR 52.243-2 Alternate 1 (APR 1987) Changes - cost-reimbursement and Mutual agreement of the parties | |||||||||
| D. OTHER (Specify type of modification and authority) | |||||||||
E. IMPORTANT: Contractor oIs not. x Is required to sign this document and return 1 copies to the issuing office. | ||||||||||
14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.)
Tax ID Number: 33-0827593
DUNS Number: 111029179
Proof of Concept for Use of the Celution System as a Medical Countermeasure for Thermal Burn
A. The purpose of this modification is to revise the SOW for Proof of Concept for Use of the Celution System as a Medical Countermeasure for Thermal Burn.
B. This is a bilateral, supplemental agreement no-cost modification. The total contract amount and all other terms and conditions remain the same.
Continued ...
Except as provided herein, all terms and conditions of the document referenced in Item 9A or 10A, as heretofore changed, remains unchanged and in full force and effect.
15A. NAME AND TITLE OF SIGNER (Type or print) TIAGO M.GIKAO CFO | 16A. NAME AND TITLE OF CONTRACTING OFFICER (Type or print) WENDELL CONYERS | |||
15B. CONTRACTOR/OFFEROR /s/ TIAGO M.GIKAO CFO | 15C. DATE SIGNED 3/30/16 | 16B. UNITED STATES OF AMERICA /s/ WENDELL CONYERS | 16C. DATE SIGNED 4-1-2016 | |
(Signature of person authorized to sign) | (Signature of Contracting Officer) | |||
NSN 7540-01-152-8070 Previous edition unusable |
| STANDARD FORM 30 (REV. 10-83) Prescribed by GSA FAR (48 CFR) 53.243 | ||
CONTINUATION SHEET | REFERENCE NO. OF DOCUMENT BEING CONTINUED HHS0100201200008C/0004 | PAGE | OF | |||||
2 | 2 | |||||||
NAME OF OFFEROR OR CONTRACTOR CYTORI THERAPEUTICS, INC 1386447 | ||||||||
ITEM NO. (A) | SUPPLIES/SERVICES (B) | QUANTITY (C) | UNIT (D) | UNIT PRICE (E) | AMOUNT (F) | |||
| Period of Performance: 09/28/2012 to 09/27/2016 |
|
|
|
| |||
NSN 7540-01-152-8067 |
|
|
|
| OPTIONAL FORM 336 (4-86) Sponsored by GSA FAR (48 CFR) 53.110 | |||
Contract No. | Continuation Sheet | Page 3 of 15 |
HHS0100201200008C | Block 14 |
|
Modification #04 |
|
|
SUMMARY OF CHANGES
Revised Statement of Work for Option 1
Preface
Independently and not as an agent of the Government, the Contractor shall be required to furnish all the necessary services, qualified personnel, material, equipment, and facilities, not otherwise provided by the Government, as needed to perform the Statement of Work submitted in response to Broad Agency Announcement (BAA) BARDA CBRN BAA 11-100-SOL-00009.
Introduction
The goal of this project is to develop a countermeasure for thermal burn injury that requires minimal to no stockpiling and that is effective in the treatment of both thermal burn injury and thermal burn injury that is complicated by concomitant radiation exposure. The issue of stockpiling will be addressed by development of a countermeasure that is effectively pre–deployed through regular commercially viable use. Ideally, this commercial use is in both thermal burn–thereby ensuring availability within the burn center of persons trained in operation and use of the countermeasure in burn–and outside of burn, thereby bolstering wider commercial viability.
Page 1
Timeline
A flowchart of the tasks within this CLIN is shown below.
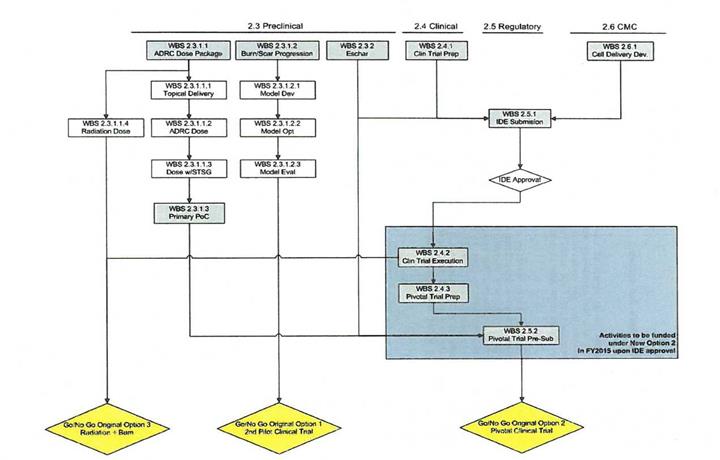
Page 2
Tasks
Updated Project Overview
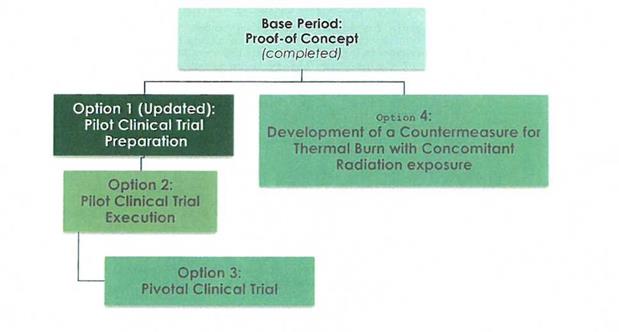
· | Option 1, as amended from the original Statement of Work, includes research and development, regulatory, clinical, and other tasks required for preparation for a Pilot Clinical Trial of the Celution System in thermal burn injury. Activities include those needed to obtain FDA approval to execute the trial. The Option also includes development of a system and process suitable for delivering ADRCs to thermal burn wounds within the clinical trial as well as preclinical activities dedicated to increasing understanding of the countermeasure in thermal burn. |
· | Option 2, is to be funded in FY16 or FY17 upon FDA approval to initiate the Pilot Clinical Trial. Option 2 (New) includes tasks needed to execute and complete the Pilot Clinical Trial, those needed to prepare trial data for submission to FDA within a Pre-Submission Meeting Package in support of a proposed Pivotal Trial, and, potentially, support for ongoing development of CT-X2. |
| · | Option 2 is to be triggered by FDA approval to execute the study |
· | Option 3 includes a Pivotal Clinical Trial leading to FDA licensure for use of the Celution System in thermal burn injury. Given the likely timing of the start of activities under Option 2 it is probable that this activity will be executed under a new contract rather than as an Option of the current contract. |
| · | Option 3 will be triggered by the FDA's response to the proposed Pivotal Study Design and Clinical and Preclinical Support Data submitted in a Pre-Submission Meeting Package. Specifically, the Option will be triggered if the FDA indicates adequacy of the study design and preclinical and clinical data set with regard to moving forward to the proposed clinical trial. Given the likely timing of the start of activities under Option 3 it is probable that this activity will be executed under a new contract rather than as an Option of the current contract. |
· | Option 4 includes studies to optimize the treatment for thermal burn injury with concomitant radiation exposure. Ideally these activities would lead to development of FDA Emergency Use Instructions |
| · | The triggers for Option 4 remain unchanged from the original submission. Specifically, it will be triggered if the following three parameters are met: (1) autologous ADRCs mediate improved healing of thermal burn injury in an irradiated animal; (2) the cell output of the CT-X2 is equivalent or superior to that of the Celution 800; and (3) ADRCs can be obtained from patients with thermal burn injury. Studies executed within WBS 2.3.1.1.5 may further inform this decision. Given the likely timing of the start of activities under Option 4 it is probable that this activity will be executed under a new contract rather than as an Option of the current contract. |
· | The current CLIN includes activities in WBS 2.3.1.2 (Burn Wound/Scar Progression). Demonstration of efficacy and practicability of ADRC treatment to reduce scar progression following burn wounds to a meaningful degree would trigger consideration of a further new component of support which would fund a Pilot Clinical Trial in Burn Wound Progression. |
| · | Specifically, such an Option would be triggered if the application of ADRCs led to a reduction of more than approximately 33% in the incidence or severity of scarring. |
Page 3
Base Period
The Base Period obtained proof of concept data for use of the Celution System as a medical countermeasure for combined injury involving radiation exposure and thermal burn injury. Specifically, in the absence of radiation exposure, autologous ADRCs improved healing parameters including increased burn wound re- epithelialization. The same improved healing was also observed in animals subjected to total body irradiation sufficient to induce profound, transient myelosuppresion. Viable, functional ADRCs were reliably and reproducibly obtained from patients with severe full thickness thermal burn injury. Finally, a prototype of Cytori’s next generation Celution System (CT-X2) showed cell processing capabilities that were equivalent or superior to those of the current generation system, Celution 800.
Option 1: Pilot Clinical Trial Preparation
Specific Objectives and Scope
As proposed herein, the Contractor intends to design, execute, and complete robust preclinical and to design a clinical study that meet two objectives: (1) obtain FDA approval to execute a pilot clinical trial of the countermeasure in thermal burn injury wherein said trial will inform and support development of a pivotal clinical trial to be funded by a future CLIN/contract option; and (2) execute preclinical studies that will expand understanding of the countermeasure with respect to cell dose, route of administration, and efficacy in arresting or slowing progression of indeterminate thickness thermal burn injury or in addressing scarring following thermal burn.
Start Date: Q4, FY14
WBS 2.1 Technical and Project Management: Original and Supplement
Project-wide Activities
Purpose
Execution of activities throughout this project will require that meetings, site visits, In-Process Reviews, and related activities are properly coordinated and that the outcome of said activities be communicated to BARDA and other stakeholders in an efficient, timely manner. The purpose of these project-wide activities is to facilitate this coordination and communication.
Description
Examples of activities performed to facilitate meetings, site visits, In- Process Reviews and similar activities include: scheduling meetings, timely distribution of an agenda in advance of the meeting, and timely distribution of meeting minutes, action items, and other deliverables after the meeting.
Deliverable: | a. Ensure proper coordination of all meetings, site visits, In-Process Reviews |
b. Disposition of meeting minutes and action items and all deliverables under this Statement of Work to BARDA
Success Criterion: CO and PO deem meeting communications are managed satisfactorily
Timing: Full duration of project (including all options)
2.1.1.1 | Kick-off Meeting: Unchanged from Original |
Following a kickoff meeting with BARDA, Cytori will update the project schedule and provide an updated Task and Deliverables list to the Contract Officer.
Deliverable: Updated Task and Deliverables Document
Success Criterion: Includes updates of tasks and deliverables as discussed with CO and PO during kickoff meeting
Timing: Q2, FY15
2.1.1.2 | Complete new hiring needed for execution of contract activities Unchanged from Previous |
Execute hiring of new staff needed for execution of CLIN activities
Deliverable: Report showing that key positions have been filled and added to contract provided within bi-weekly meetings
Success Criteria: Positions identified during negotiations have been filled by qualified persons
Timing: Q3, FY15
Page 4
2.1.2 | Maintain Subcontractor Management Plan: Unchanged from Previous |
Maintain Subcontractor Management Plan
Deliverable: Updated Subcontractor Management Plan
Success Criteria: Identifies key interactions between prime contractor and subcontractor with regard to progress updates and risk management
Timing: Semi-annually starting six months after award of CLIN
2.1.3 | Maintain Risk Management Plan: Unchanged from Previous |
Maintain Risk Management Plan
Deliverable: Updated Risk Management Plan
Success Criteria: Identifies key risks, assesses mitigations, contingencies, and impact as well as update process
Timing: Semi-annually starting six months after award of CLIN
2.1.4 | EVMS Systems Report: Unchanged from Previous |
EVMS Systems Report
Deliverable: EVMS Systems Report
Success Criteria: Acceptance by BARDA Contract Officer that the accounting and related systems are EVMS-compliant
Timing: Q2, FY15
2.2 | Non-Clinical Toxicology |
Not applicable
2.3 | Preclinical |
2.3.1 | Porcine Studies: |
2.3.1.1 | ADRC Dose Package |
Objective: To determine the minimally effective dose of ADRCs in thermal burn injury
Rationale: In the Base Period Cytori has evaluated a narrow range of ADRC doses range limited at the upper end by the amount of adipose tissue easily obtainable from an individual mini-pig. The effectiveness of lower doses has not been assessed. The total dose of cells available for treatment is largely dependent on the volume of adipose tissue processed. Larger volumes require more collection and processing time and can increase risk. In order to appropriately balance risk and benefit it is important to determine the optimal cell dose so that the amount of tissue collected is adequate, but not excessive, for the size of the injury. This will be assessed by local delivery into the wound.
2.3.1.1.1 | ADRC Dose: Additional Evaluation of Previously Collected Biopsies: Unchanged from Original, Studies Completed |
Objective: To evaluate value of additional evaluations
Rationale: Studies in WBS 1.3.1 executed within the Base Period collected biopsies that were subjected to a number of histologic and immunohistologic stains (for example; Masson's trichrome and immunostaining for Ki67 and CD31). Data from these stains was informative. As is often the case in research, the information obtained raised new questions that can be addressed by application of additional histologic approaches. However, there was insufficient time and insufficient funds to apply these approaches during the Base Period. These approaches have the capacity to be informative in the studies to be executed under Option 1. The activities to be executed under this WBS element will develop and validate selected additional markers on biopsies collected during the Base Period in anticipation of applying said stains in ADRC dose studies to be conduced under WBS 2.3.1.1 (Dose Package) and other activities in WBS 2.3.1.
Page 5
Approach: Biopsies and sections have already been prepared and several core analyses have been performed. Additional digital imaging and analysis including immunohistochemical and molecular assessment of parameters associated with both normal healing and with scarring, particularly hypertrophic scarring, will be performed.
Description:
For each group multiple wound healing parameters will be assessed. Candidate stains include: Movat’s stain (a pentachrome stain that is recommended by UTMB Galveston for analysis of hypertrophic scarring), alpha smooth muscle actin (for contraction-inducing myofibroblasts), decorin (reduced in hypertrophic scarring), and type III collagen. Similarly, commercially available molecular arrays targeted for wound healing may be used to interrogate existing biopsy samples. All stained slides will be digitally scanned prior analysis. Using software such as the ImageScope software, each slide can be annotated to identify superficial and mid/deep regions within the wound tissue. The percent of positive staining can be quantified using the Deconvolution analysis algorithm (Aperio). This software algorithm makes use of a deconvolution method to separate the red and blue stain of the Masson's Trichrome. Epithelial thickness can be determined by histomorphometric analysis on digital slides using the Image Scope software. 3-6 measurements throughout the wound site (at edges and the center) can be performed to determine the thickness of the stratified and cornified layer of the neo-epidermis.
Deliverable: Interim and Final Study Reports
Success Criteria: Acceptance of reports by BARDA Program
Timing: Q2, FY15
2.3.1.1.2 | ADRC Dose: Topical Delivery: Unchanged from Original, Study Completed |
Objective: To determine the efficacy of topical delivery of ADRCs in thermal burn injury
Rationale: In the Base Period Cytori has demonstrated that delivery of ADRCs by direct injection into the base of the wound leads to increased epithelialization. Cytori's Burn Science Advisory Board has recommended that we evaluate mechanisms that might be easier and potentially faster to perform, in particular, topical delivery such as a spray, drip, or paint approach. A brief series of in vitro and in vivo studies similar to those executed in the base Period for other delivery routes is indicated in order to evaluate this approach.
Approach: These studies will use an approach selected during in vitro testing to apply viable ADRCs to burn wound following escharectomy.
Description:
Animals will receive thermal burn injury induced according to parameters optimized during the Base Period. Autologous ADRCs will be delivered on the same day as escharectomy. Different groups of animals will receive topical delivery of ADRCs with contralateral wounds used as matched control (treated with vehicle only). Healing will be evaluated by planimetry and histology at appropriate time points selected on the basis of results obtained in the Base Period. Each group will comprise a sufficient number of animals (for example, 4 or 6) .
Deliverable: Interim and Final Study Reports
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Q2, FY15
2.3.1.1.3 | ADRC Dose: Dose Finding: Unchanged from Original, Study In Progress |
Objective: To determine the minimally effective dose of ADRCs in thermal burn injury
Rationale: In the Base Period Cytori has evaluated a narrow range of ADRC doses range limited at the upper end by the amount of adipose tissue easily obtainable from an individual mini-pig. In order to maximize the likelihood of seeing a difference between treated and control wounds the first arm will apply dosing in wounds that are not treated with a split thickness skin graft (STSG) where data obtained in the Base Period demonstrate a robust signal to noise ratio for ADRC-induced increased epithelial migration. Once a dose has been determined in this model it will be applied in follow-up studies that include STSG (WBS 2.3.1.1.4 below).
Approach: These studies will use the approach applied in the Base Period to assess effectiveness of ADRCs when delivered at different doses by either local (that is, injection or topical as assessed by WBS 2.3.1.1.2) or intravenous delivery. For the local injection arm of the study, each animal may act as its own control using a paired wound model in which wounds on one side of the animal will receive vehicle alone (control) while the matching wounds on the other side receive ADRC treatment. Given the clinical relevance of hypertrophic scarring and the relative resistance of Gottingen minipigs and Yorkshire farm swine to this phenomenon these studies may be extended to include the Red Duroc strain of pigs (in place of or in addition to other strains) which is known to have a native susceptibility to hypertrophic scarring that is more similar to that of humans.
Page 6
Description:
Animals will receive thermal burn injury induced according to parameters optimized during the Base Period. Autologous ADRCs will be delivered on the same day as escharectomy. Different groups of animals will receive local injection of ADRCs. A suitable number (for example, four) of different ADRC doses will be evaluated. Doses selected will cover a wide range (for example, 250,000 ADRCs/cm2; 125,000 ADRCs /cm2; 50,000 ADRCs /cm2; and Control = no ADRCs). Healing will be evaluated by planimetry, histologic, and molecular analysis at appropriate time points selected on the basis of results obtained in the Base Period. Each group will comprise a sufficient number of animals (for example, 4 or 6).
Deliverable: Interim and Final Study Reports
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Ql, FY16
2.3.1.1.4 | ADRC Dose: Confirmation with STSG: Deleted |
2.3.1.1.5 | ADRC Dose: Confirmation with Higher Radiation Dose: Deleted |
2.3.1.2 | Burn/Scar Progression |
Objective: To obtain preclinical data that will allow assessment of feasibility of use of the Celution System as a treatment for burn wound progression or scarring.
Rationale: Burn wound progression is the pathophysiologic process by which a partial thickness thermal burn evolves over the first few days after injury to become a full thickness injury requiring a skin graft. This process occurs through vascular damage leading to ischemia: reperfusion injury and to the inflammatory response1. The same mechanisms that are proposed to be behind the efficacy of ADRCs observed in the Base Period (angiogenesis, modulation of inflammation, etc.) have the potential to mitigate burn progression. Similarly, the healing process following thermal burn injury can lead to scarring that is both disfiguring and that limits function, for example, limits range of motion of a joint. Interventions that impact progression of scar development and maturation have the potential to significantly improve burn care.
Approach:
The overall approach selected is taken from that applied in the Base Period for full thickness injury in which a porcine model of vertical burn wound progression or scarring taken from the published literature2'3 is adapted, optimized, and validated and then used to assess efficacy of autologous ADRC treatment.
2.3.1.2.1Model Development: Unchanged from Original, Study Completed
Objective: To develop an animal model that will allow assessment of the effects of treatment with autologous ADRCs to treat burn wound progression.
Rationale: While a suitable animal model has been described in the literature2, it has not previously been executed by this team. Pilot activities are needed to establish the basic model.
Approach: Porcine models widely used for evaluation of thermal burn injury and have also been used for evaluation of burn and scar progression. The approach to be applied is essentially identical to that used in the Base Period for creation of a full thickness burn wound with the exception that the progression model must create a wound that is only partial thickness at the time of application but which progresses to deep/full thickness over a period of 3-4 days after injury whereas a model of hypertrophic scarring will require longer follow-up after injury.
1 Shupp, J.W., et al., A review of the local pathophysiologic bases of burn wound progression. J Burn Care Res, 2010. 31(6): p. 849-73
2 Singer, A.J., et al., Validation of a vertical progression porcine burn model. J Burn Care Res, 2011. 32(6): p. 638-46
3 Harunari, N., et al., Histology of the thick scar on the female, red Duroc pig: Final similarities to human hypertrophic scar. Burns, 2006. 32(6): p669-677
Page 7
Description:
Each experimental animal will be subjected to thermal burn injury using the device developed for this purpose during the Base Period. These activities demonstrated that application of the device at a temperature of 200°C and contact pressure of 0.4kg/cm2, for 60 seconds created a reproducible full thickness injury. A published study2 has shown that application of a similar device at a temperature of 80°C and contact pressure of 0.32kg/cm2 for 20-30 seconds creates a partial thickness burn that progresses to full thickness. In these studies parameters of time, temperature, and contact pressure will be managed to determine the precise combination that generates a wound that reproducibly progresses from partial to full thickness over ~3-4 days after injury. The same approach may be used to generate a burn that develops to hypertrophic scarring in an appropriate pig strain.
Thermal burn wounds will be created in a suitable number of animals as described above and assessed by histology of biopsies performed at the time of injury and at suitable intervals (for example, 6 hours, 24 hours, 48 hours, 72 hours and 96 hours) after injury to assess burn depth and progression over the ~96 hour timeframe with the different burn induction parameters. For evaluation of scarring a similar approach will be applied using the Red Duroc strain that develops hypertrophic scarring that is similar to that of humans.
Deliverable: Interim and Final Study Reports
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Q1, FY15
2.3.1.2.2Model Optimization: Study in Progress
Objective: To optimize an animal model that will allow assessment of the effects of treatment with autologous ADRCs to treat burn wound/scar progression.
Rationale: Assesses reproducibility of the model established in WBS 2.3.1.2.1 above.
Approach: The parameters defined to provide the intended model in WBS 2.3.1.2.1 will be assessed for reproducibility.
Description:
A series of wounds will be induced in a cohort of animals (for example, six animals per arm) using the approach determined in Model Development above (WBS 2.3.1.2.1). Animals will also undergo lipectomy for ADRC isolation for treatment of wounds assigned for treatment. Wounds will receive treatment with ADRCs or with vehicle control. The study will assess ability of ADRCs to modify the development of scarring and to treat a scar that has already developed or has started to develop (for example, six months after injury). Progression of scarring will be assessed by histologic and molecular analysis of biopsies taken at suitable intervals (for example, 6 hours, 24 hours, 48 hours, 72 hours and 96 hours) after injury or later times (for example, two weeks, two months, six months) to assess scarring.
Deliverable: Interim and Final Study Reports
Page 8
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Q3, FY16
2.3.1.2.3 | Model Evaluation: Deleted |
2.3.1.3 | Primary Proof of Concept: Modified |
Objective: To demonstrate proof of concept for use of ADRCs in enhancing healing of thermal burn injury
Rationale:
These activities will obtain safety, efficacy, and mechanism of action data that can be included in the investigational Device Exemption application to be submitted to FDA under WBS 2.5.1
Approach: Conditions defined in the studies described above will be applied to evaluate healing when ADRCs are delivered following thermal burn injury.
Description
Animals will receive thermal burn injury induced according to parameters optimized in the studies described above. Wounds will be treated with either control or ADRCs. ADRCs applied at the time of the initial treatment will be applied locally (for example directly into or onto the wound) and/or by systemic administration (for example, by intravenous injection). Each group will comprise a sufficient number of animals (for example, 4 or 5). Healing will be evaluated at time points selected on the basis of results obtained in the studies described above (for example, at the time of application of the STSG and two weeks after application of STSG). Assessment of fibrosis and hypertrophic scarring at the treatment site may be performed a suitable time after injury (for example, six months). These in vivo activities may be supplemented by in vitro studies that examine mechanisms underlying in vivo observations, for example, prior studies have shown an effect of ADRCs on inflammatory cell infiltration and blood vessel density. These phenomena can be assessed by in vitro activities assessing endothelial cells and leukocytes.
Deliverable: Interim and Final Study Reports
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Q4, FY16
2.3.2 | Human Thermal Burn ADRC Characterization: Eschar: Complete: No further activity needed |
Objective: To characterize the ADRC population obtained from patients with thermal burn injury
Rationale: As currently proposed, the pilot clinical trial includes use of adipose tissue obtained by excision of adipose tissue exposed during tangential or fascial excision of eschar. Studies performed in the Base Period have provided evidence that the yield, viability, function, and composition of ADRCs obtained from material obtained from fascial excision escharectomy are all within the range seen when processing adipose tissue obtained by liposuction from healthy donors. The current studies are needed to expand on this preliminary data by collecting and evaluating additional specimens and by extending the study to include tissue obtained by tangential excision. In addition, optimal enzymatic digestion of the adipose tissue requires that the excised tissue be morselized into fragments creating a surface area-to-volume ratio that allows efficient extraction of ADRCs.
Approach: Adipose tissue from patients with thermal burn injury will be obtained following informed consent and processed to prepare ADRCs. The number and function of these cells will be assessed using approaches such as cell viability and cell characterization methods that are used routinely by Cytori for evaluation of cells from conventional sources (liposuction) as applied in the Base Period. This will include development of a rapid, efficient, and validated method by which the excised tissue is morselized.
Page 9
Description
Human adipose tissue will be obtained following informed consent from a sufficient number of patients (for example, 20) undergoing treatment for thermal burn injury. Research subjects will be drawn from burn programs located in geographic proximity to the Contractor's research facility (for example, at the University of California at San Diego Burns Center located approximately five miles from the Cytori laboratories) and the University of California at Irvine Burn Center (located approximately 80 miles from Cytori laboratories). The tissue will be processed to prepare morselized adipose tissue that will then be digested to prepare ADRCs. Cell yield and viability will be determined using a Nuclecounter™ device in accordance with standard practices at Cytori. Other examples of tools for characterization include multicolor flow cytometry to evaluate cell ADRC cellular composition, and molecular probes to evaluate the population as a whole.
Deliverable: Monthly Updates (during bi-weekly calls); Report for IDE Submission, and written Interim and Final Study Reports
Success Criteria: Achieves success parameter defined in study protocol agreed to by BARDA and the Contractor prior to initiation of studies
Timing: Monthly updates for enrollment reporting; Ql, FY15 for IDE Submission; Monthly reports to Q4 FY16; Q4, FY16 for interim and final reports.
2.4 | Clinical Tasks: Unchanged from Original |
2.4.1.1 | Pilot Clinical Trial Preparation: Unchanged from Original |
Objective: To perform the groundwork necessary for FDA approval of a Pilot clinical trial of the use of the Celution System in thermal burn injury and for expedited start of enrollment in the trial following FDA approval.
Rationale: The Celution System is regulated as a device within the Center for Biologics Evaluation and Research (CBER). CBER has approved the use of the Celution System in three prior IDE clinical trials. In the course of discussions with the FDA regarding these studies the Agency communicated to Cytori that they strongly preferred the study design to include evaluation of cell dose. The proposed study will obtain the safety and feasibility of a pilot study with additional information of cell dose and assessment of secondary outcomes associated with efficacy. These data may allow determination of sample size in any Pivotal Trial to follow (as described in Option 2). Prior to filing the IDE package for this Pilot Trial, the clinical team must develop a detailed clinical protocol and Investigator's Brochure (IB). These and related activities associated with assessment and selection of potential clinical trial sites and CROs will be executed under WBS 2.4.1.4.
Additional tasks must be performed in order to minimize the delay between FDA approval and start of the trial itself. These include selection of a qualified Clinical research Organization and of clinical sites that have the capability to enroll patients and execute the study with the level of quality required to achieve study goals.
Approach: With the assistance of a Scientific Advisory Board, the team will develop the protocol and IB as for past Cytori IDE filings. The team will also execute clinical site evaluation, CRO evaluation, and an preliminary assessment of contractual matters to ensure that the budget for proposed Option 2 is accurate.
Purpose
To provide the Regulatory team with the documentation needed to obtain FDA approval to initiate the specified clinical trial.
Description
Activities to be conducted include: development of the clinical trial protocol; assessment and selection of Clinical Contract Research Organization (CRO); and assessment, selection, and qualification of clinical trial sites. These activities will be executed with input from Cytori's Thermal Burn Scientific Advisory Board.
Deliverables: Monthly Updates (during bi-weekly calls) Clinical Trial Protocol and Investigator's Brochure for IDE Submission Q1 FY16 Site Initiation Readiness Report Q4 FY16
Success Criteria: FDA approval of trial
Timing: Clinical Trial Protocol and Investigator's Brochure for IDE Submission Q4 FY16 Site Initiation Readiness Report Q6 FY16
Page 10
2.5 | Regulatory Tasks |
2.5.1 | Pilot Clinical Trial IDE Approval |
Purpose
The FDA must grant approval before clinical use of an Investigational Device can be initiated. In the case of the Celution System in Thermal Burn Injury this will require approval under the Investigational Device Exemption mechanism.
Description
Cytori's regulatory team will prepare and submit a package of documents. Contents will be based upon the clinical trial protocol, data obtained in studies described above, and feedback received from the Agency in a Pre-IDE Meeting.
The content of the IDE package will largely mirror that used in prior submissions of this kind to the Agency. Contents will include relevant study reports from activities executed during the Base Period and Option 1. In the event that the FDA has additional questions to be answered following review of the initial package, the Regulatory team will develop, collate, and submit responses to said questions.
2.5.1 | Pre-Submission Meeting |
Deliverable: Presubmission Package and Meeting Minutes
Success Criteria: Clarity on path to successful IDE submission
Timing: Q3, FY16
2.5.2 | IDE Approval |
Deliverable: IDE Package and Responses (as required) to FDA Questions
Success Criteria: IDE Approval Granted by FDA
Timing: Q4, FY16
2.6 | CMC |
2.6.1 | ADRC Delivery System |
Objective: To develop a system capable of preparing adipose tissue obtained by excision rather than by liposuction for processing within the CT-X2 System and subsequent rapid, reproducible delivery to a thermal burn injury.
2.6.1.1 | Tissue Pre-Processing: Unchanged |
Objective: To develop a system capable of preparing adipose tissue obtained by excision rather than by liposuction for processing.
Rationale: Cytori methods have been optimized for preparing ADRCs from tissue collected by liposuction. During this process the suction force combined with the geometry of the liposuction cannula cuts the adipose tissue into small fragments. The conditions for enzymatic digestion of this material are based upon the standard surface area to volume ratio of tissue prepared by liposuction. Tissue prepared by excision will have a different surface area to volume ratio and, hence, will not be processed optimally without pre- processing. Activities within WBS 2.6.1.1 are designed to develop a standard, reproducible, and clinically acceptable pre-processing method that will prepare excisional samples for optimal processing. Cytori has developed approaches for pre-processing the tissue that are acceptable for the laboratory, but not for the clinic. The activities to be performed in WBS 2.6.1.1 are intended to address this deficiency.
Approach: Human adipose tissue will be pre-processed by two methods; (1) Cytori's standard laboratory approach and (2) using tools and supplies commonly available in the operating room. Tissue will then be processed. The yield, viability, and composition of the ADRCs derived will be evaluated using methods described above (WBS 2.3.2).
Page 11
Description: Human adipose tissue (see WBS 2.3.2) will be obtained following escharectomy. Adipose tissue will be excised from the sample and then sliced into fragments that approximate the size of fragments obtained by liposuction. Tissue slicing will be performed by two methods; (1) Cytori's standard laboratory approach and (2) using tools and supplies commonly available in the operating room. Tissue will then be processed. The yield, viability, and composition of the ADRCs derived will be evaluated using methods described above (WBS 2.3.2). Once the optimal approach using Operating Room materials has been developed, the approach will then be validated.
Deliverable: Validated Standard Operating Procedure with Validation Data
Success Criteria: Protocol accepted by FDA in IDE Submission
Timing: Q4, FY16
2.6.1.2 | Cell Delivery Mechanism: Unchanged |
Objective: To develop a system capable of reproducibly and conveniently delivery ADRCs to a thermal wound following escharectomy.
Rationale: The current clinical protocol, based on preclinical data, specifies delivery ADRCs into the wound bed a single injection indicated for each 10cm2 of treatment area. There is currently no off-the-shelf approach available that achieves this delivery without unnecessarily prolonging surgical time.
Approach: Injection of ADRCs into the wound could, for example, take the form of a powered dosing syringe delivering a specified volume of material (ADRCs) into the wound bed at each touch of the button. This markedly reduces strain in the Surgeon's hand for large wounds requiring many injections. Examples of this approach were presented to the FDA at the Pre-Submission meeting where they met with general approval with the natural proviso that full review in the IDE Submission would be required. For example, FDA indicated that they would require data showing that the output of the injection system was consistent over time. Another possible approach is a topical spray similar to that already used in burn care for application of fibrin glue used to help secure skin grafts. The activities performed herein will continue development of a suitable approach in order to complete the information needed for the IDE submission. Additional technical support will be required in the early phase of the clinical trial for matters such as set-up and training.
Description: Cytori engineers have already identified candidate powered syringe and spray systems that may be suitable for this purpose. These devices will be brought in-house. The convenience, time, and reproducibility of injection may be assessed by, for example, injecting ADRCs into surrogate materials, for example, porcine skin, human skin obtained from patients undergoing elective cosmetic procedures (eg: “tummy tuck”) and/or into sample collection vials.
Deliverable: Interim and Final Reports
Success Criteria: Protocol accepted by FDA in IDE Submission
Timing: Q4, FY16
2.6.1.3 | ADRC Delivery System and Process: Unchanged but renamed “WBS 2.6.2 Verification and Validation” |
Objective: To obtain data and reports that will allow FDA to assess the safety and suitability of the ADRC Delivery System and Process for use in the proposed clinical trial.
Rationale: Robust testing, verification, and validation of the system and process used for ADRC preparation and delivery is a necessary component of the package to be submitted for FDA review as part of obtaining approval to execute the proposed clinical trial.
Approach: FDA requirements and guidance documents specify a range of testing that must be performed on systems such as that proposed herein before said systems can be used in a clinical trial. Small companies like Cytori invariably find it more efficient to outsource much of this specialized testing to vendors with specific expertise. For this reason, certain aspects of the work proposed for WBS 2.6.1.3 will be executed by subcontractors.
Description
CMC testing on hardware, consumable, and software elements of the Cell Delivery System and Process.
Page 12
Deliverables:
| 1. | Consumable component mold verification report |
| 2. | CMC Test Report of Cell Delivery System and Process circuit board element |
| 3. | Electromagnetic compatibility testing report |
| 4. | Consumables for use in testing to be executed by Cytori in WBS 2.3.1, WBS 2.3.2, WBS 2.4.1, WBS 2.5.1, and WBS 2.6.1 |
| 5. | Software Verification and Validation report |
Success Criterion: Reports accepted by FDA in IDE Submission
Timing: Q4 2016
Page 13