Exhibit 99.4
ANNUAL INFORMATION FORM
FOR FISCAL YEAR ENDED SEPTEMBER 30, 2007
DECEMBER 28, 2007
TABLE OF CONTENTS
INTERPRETATION | 3 |
DOCUMENTS INCORPORATED BY REFERENCE | 3 |
CAUTION REGARDING FORWARD-LOOKING INFORMATION | 3 |
PART I – CORPORATE STRUCTURE | 5 |
1. | Jurisdiction of Incorporation and Articles | 5 |
2. | Current Organization | 5 |
PART II – DESCRIPTION OF THE BUSINESS | 6 |
1. | Overview | 6 |
2. | Growth Strategy | 6 |
3. | Customers | 7 |
4. | Significant Customers | 7 |
PART III - GENERAL DEVELOPMENT OF THE BUSINESS | 8 |
1. | Recent Industry Developments | 8 |
2. | Change in Wholesaler’s Business Models | 8 |
3. | Financial Developments | 9 |
4. | License and Development Agreement | 9 |
PART IV - BUSINESS OF AXCAN | 10 |
1. | Main Marketed Products | 10 |
2. | Products in Development | 16 |
3. | Regulatory Affairs and Quality Assurance | 19 |
4. | Regulation | 19 |
5. | Patents and Proprietary Rights | 21 |
6. | Employees | 24 |
7. | Environmental Protection, Health and Safety | 25 |
8. | Facilities | 25 |
PART V - RISK FACTORS | 26 |
PART VI - SELECTED CONSOLIDATED FINANCIAL INFORMATION | 42 |
PART VII - MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND OPERATING RESULTS | 43 |
PART VIII - DIVIDEND POLICY | 43 |
PART IX – DESCRIPTION OF CAPITAL STRUCTURE | 44 |
1. | Common Shares | 44 |
2. | Preferred Shares | 44 |
PART X - MARKET FOR SECURITIES | 45 |
1. | Market for Securities | 45 |
2. | Trading History | 45 |
PART XI - DIRECTORS AND OFFICERS | 46 |
1. | Directors | 46 |
2. | Senior Officers | 46 |
PART XII - ADDITIONAL DISCLOSURE FOR DIRECTORS AND EXECUTIVE OFFICERS | 46 |
PART XIII - LEGAL PROCEEDINGS | 48 |
PART XIV – TRANSFER AGENTS AND REGISTRARS | 48 |
PART XV - MATERIAL CONTRACTS | 49 |
PART XVI - INTERESTS OF EXPERTS | 49 |
PART XVII - ADDITIONAL INFORMATION | 49 |
SCHEDULE A - GLOSSARY OF TECHNICAL TERMS | 50 |
ii
INTERPRETATION
In this Annual Information Form (“AIF”), “we”, “us”, “our”, “Axcan”, and “the Company” are used to refer to Axcan Pharma Inc., its predecessors and its direct and indirect subsidiaries and their predecessors, collectively, unless the context otherwise requires.
In this AIF, all references to specific years are references to the fiscal year ended September 30. Unless otherwise stated, all dollar amounts appearing in this AIF are stated in U.S. dollars, and all financial data included in this AIF has been prepared in accordance with U.S. generally accepted accounting principles.
Unless otherwise stated, all market size information appearing in this AIF has been provided by IMS Health Ltd., a widely accepted provider of information services specializing in medical research information.
Certain terms and abbreviations used in this AIF are defined in “Schedule A – Glossary of Technical Terms”.
DOCUMENTS INCORPORATED BY REFERENCE
The following documents are incorporated by reference into this AIF:
1. The audited consolidated financial statements of Axcan Pharma Inc. for the years ended September 30, 2007, September 30, 2006, and September 30, 2005, reported on by Raymond Chabot Grant Thornton Chartered Accountants (the “2007 Financial Statements”); and
2. The Management’s Discussion and Analysis of financial condition and results of operations of Axcan Pharma Inc. (the “2007 MD&A”).
Please note that both of these documents are available on SEDAR on www.sedar.com.
CAUTION REGARDING FORWARD-LOOKING INFORMATION
From time to time, we make written or oral forward-looking statements, as defined under applicable securities laws. We may make such statements in this document, in other filings with Canadian regulators or the United States Securities and Exchange Commission, in reports to shareholders or in other communications. These forward-looking statements include, among others, statements with respect to our objectives for 2008, our medium-term goals, and strategies to achieve those objectives and goals, as well as statements with respect to our beliefs, plans, objectives, expectations, anticipations, estimates and intentions. The words “may”, “could”, “should”, “would”, “suspect”, “outlook”, “believe”, “plan”, “anticipate”, “estimate”, “expect”, “intend”, “forecast”, “objective”, and words and expressions of similar import are intended to identify forward-looking statements.
By their very nature, forward-looking statements involve inherent risks and uncertainties, both general and specific, which give rise to the possibility that predictions, forecasts, projections, and other forward-looking statements will not be achieved. You should not place undue reliance on forward-looking statements since they involve known and unknown risks, uncertainties and other factors which are, in some cases, beyond our control and which could materially affect actual results, levels of activity, performance or achievements. Factors that may cause actual results to differ materially from current expectations include, but are not limited to: (i) our ability to obtain and maintain United States Food and Drug Administration (“FDA”) and other regulatory approvals for our products for the indications and on the timetables specified herein, (ii) our ability to obtain and protect our intellectual property, including market exclusivity (where applicable) related to our products and product candidates; (iii) our potential for future growth and the development of our product pipeline, including our ability to identify and acquire, on commercially attractive terms, additional products, (iv) our ability to form and maintain collaborative relationships to develop and commercialize our product candidates; and (v) general economic and business conditions.
3
Other factors that may cause actual results to differ materially from current expectations include, but are not limited to, management of credit, market, liquidity and funding and operational risks; the strength of the Canadian and United States economies and the economies of other countries in which we conduct business; the impact of the movement of the Canadian dollar relative to other currencies, particularly the US dollar and the Euro; the effects of changes in monetary policy, including changes in interest rate policies of the Bank of Canada and the Board of Governors of the Federal Reserve System in the United States; the effects of competition in the markets in which we operate; the impact of changes in the laws and regulations and enforcement thereof; judicial judgments and legal proceedings; our ability to obtain accurate and complete information from or on behalf of our customers and counterparties; our ability to successfully realign our organization, resources and processes; our ability to complete strategic acquisitions and to integrate our acquisitions successfully; changes in accounting policies and methods we use to report our financial condition, including uncertainties associated with critical accounting assumptions and estimates; operational and infrastructure risks; other factors that may affect future results including changes in trade policies, timely development and introduction of new products, changes in our estimates relating to reserves and allowances, changes in tax laws, natural disasters such as hurricanes, the possible impact on our businesses from public health emergencies, international conflicts and other developments including those relating to the war on terrorism; and our success in anticipating and managing the foregoing risks.
We caution that the foregoing list of important factors that may affect future results is not exhaustive. Axcan undertakes no obligation to update or revise any forward-looking statement, unless obligated to do so pursuant to applicable securities laws and regulations. When relying on our forward-looking statements to make decisions with respect to the Company, investors and others should carefully consider the foregoing factors and other uncertainties and potential events. We do not undertake to update any forward-looking statement, whether written or oral, that may be made from time to time by us or on our behalf.
4
PART I – CORPORATE STRUCTURE
1. JURISDICTION OF INCORPORATION AND ARTICLES
Axcan was incorporated under the Canada Business Corporations Act on May 6, 1982 under the name 115391 Canada Inc. On February 14, 1983, Axcan changed its name to Interfalk Canada Inc. and on October 1, 1993, it amalgamated with Axcan Holdings Ltd., its parent corporation, under the name Interfalk Canada Inc., which was changed to Axcan Pharma Inc. on July 12, 1994. On October 30, 1995, the Company’s Articles were amended to delete the private company restrictions, re-designate the existing Class “A” shares and Class “B” shares as common shares and preferred shares, respectively, and consolidate the common shares on a 0.44 for one basis. On June 6, 2000, the Company’s Articles were amended again to create 14,175,000 Series A preferred shares and 12,000,000 Series B preferred shares.
The head office of Axcan, and its principal place of business is located at 597, Laurier Boulevard, Mont-Saint-Hilaire, Quebec, J3H 6C4, Canada.
2. CURRENT ORGANIZATION
Significant operating subsidiaries are defined as those companies that contribute 10% or more of the consolidated revenues or consolidated operating income of the Company or account for 10% or more of the consolidated total assets of the Company. The following chart shows the jurisdictions of incorporation of Axcan and those principal operating subsidiaries. All of the outstanding shares of such operating subsidiaries or associated corporations are owned directly or indirectly by Axcan.
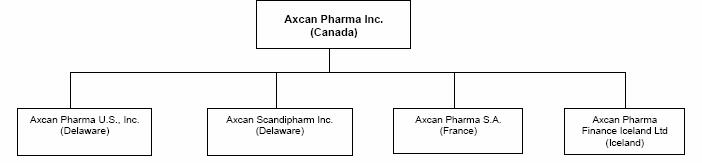
5
PART II – DESCRIPTION OF THE BUSINESS
1. OVERVIEW
Axcan is a leading specialty pharmaceutical company concentrating in the field of gastroenterology, with operations in North America and Europe. Axcan markets and sells pharmaceutical products used in the treatment of a variety of gastrointestinal diseases and disorders. The Company’s main products include pancreatic enzymes (ULTRASE, PANZYTRAT and VIOKASE) for the treatment of pancreatic insufficiency; bile acid (URSO/URSO 250, URSO FORTE/URSO DS and DELURSAN) for the treatment of certain cholestatic liver diseases; mesalamine (SALOFALK and CANASA) for the treatment of certain inflammatory bowel diseases; a Helicobacter pylori eradication drug (PYLERA) for the eradication of the cause of gastric and duodenal ulcers; and sucralfate (CARAFATE and SULCRATE) for the treatment of gastric and duodenal ulcers. Axcan also has a number of projects in all phases of clinical development, further described in the section “Products in Development” of this AIF.
In addition to its marketing activities, Axcan carries out research and development activities on products at various stages of development. These activities are carried out primarily with respect to product candidates, which are acquired or licensed from third parties. By combining its marketing expertise with its research and development experience, Axcan distinguishes itself from specialty pharmaceutical companies that focus solely on distribution of products and offers licensors the prospect of rapidly expanding the potential market for their products. As a result, Axcan is presented with opportunities to acquire or in-license products that have been advanced to the later stages of development by other companies. This focus on products in late-stage development enables Axcan to reduce risks and expenses associated with new drug development.
2. GROWTH STRATEGY
Given the highly competitive industry in which the Company operates, Axcan is pursuing a number of strategic options to drive sustainable growth, including 1) growing its base business; 2) launching new products; 3) making strategic acquisitions; 4) advancing its research and development portfolio; and 5) expanding internationally.
Base Business and New Products: | | Axcan’s intention is to grow sales by building on its solid base business and introducing new products on to the market. The Company’s strategy brings together its focus on product differentiation and a competitive time-to-market for its products, combined with astute portfolio management and a heightened sales focus. |
| | |
Strategic Acquisitions: | | Axcan’s goal is to build on its proven acquisition strategy and continue to aggressively seek products and/or companies to fuel future growth. By capitalizing on its current business model, the Company seeks to leverage its strengths and capabilities in order to develop and acquire products that have significant market potential and complement its area of focus. |
| | |
Research and Development: | | Axcan’s products continue to provide great benefit to patients. The Company will sustain investment into research and development, in order to develop the next generation of products to address unmet needs in gastroenterology. |
| | |
International Expansion: | | Axcan’s current infrastructure, both in North America and Europe, will form the basis of the Company’s international expansion, as management believes that it will serve as a springboard to increase Axcan’s multinational sales and marketing footprint. |
6
3. CUSTOMERS
Axcan’s revenue has historically been and continues to be principally derived from sales of pharmaceutical products to large pharmaceutical wholesalers. Axcan utilizes a “pull-through” marketing approach that is typical of pharmaceutical companies. Under this approach, Axcan’s sales representatives demonstrate the features and benefits of its products to physicians, in particular, gastroenterologists who may write their patients prescriptions for Axcan’s products. The patients, in turn, take the prescriptions to pharmacies to be filled. The pharmacies then place orders with the wholesalers, to whom Axcan sells its products.
4. SIGNIFICANT CUSTOMERS
The following table identifies external customers that accounted for 10% or more of the Company’s total revenue:
| | Percentage
of Total Revenue | |
| | 2007 | | 2006 | | 2005 | |
| | % | | % | | % | |
McKesson Corporation | | 41.2 | | 39.0 | | 30.2 | |
Cardinal Health | | 25.0 | | 24.4 | | 20.9 | |
Amerisource Bergen | | 11.1 | | 11.4 | | 11.0 | |
TOTAL | | 77.3 | | 74.8 | | 62.1 | |
7
PART III - GENERAL DEVELOPMENT OF THE BUSINESS
1. RECENT INDUSTRY DEVELOPMENTS
The pharmaceutical industry is highly competitive and subject to rapid and significant changes.
These changes include:
1. rising healthcare costs;
2. intensifying cost containment pressures;
3. rapid growth in demand for services due to aging population bases;
4. rapid innovation in technology, increasing the availability of sophisticated treatment options;
5. growing consumer awareness of healthcare choices; and
6. growing awareness within emerging and developing countries of the benefits of adequate healthcare systems and the improving ability to pay for improved healthcare solutions.
Our products face competition from both branded and generic products, sold by other pharmaceutical companies. Many of these competitors have greater financial resources and marketing capabilities than Axcan. Our competitors in the United States and abroad are numerous and include major pharmaceutical companies, and some of the manufacturers of our products. We believe that our focus on gastroenterology, combined with our strategy of funding and controlling all or most aspects of our business, will provide the cost savings, efficiencies in product development and acceleration of regulatory filings necessary for us to compete effectively with such companies.
Our competitors, however, may succeed in developing products that are as, or more, clinically or cost-effective than any that are being developed or licensed by Axcan, or that would render our products obsolete or uncompetitive. In addition, certain of our competitors have greater experience than our Company in clinical testing and human clinical trials of pharmaceutical products and in obtaining FDA and other regulatory approvals.
2. CHANGE IN WHOLESALER’S BUSINESS MODELS
Historically, wholesalers’ business models in the United States were dependent on drug price inflation. Their profitability and gross margins were directly tied to the speculative purchasing of pharmaceutical products at pre-increase prices, and the selling of their product inventory to their customers at the increased price. This inventory price arbitrage accounted for a predominant portion of wholesalers’ compensation for their distribution services and had a dramatic effect on wholesaler buying patterns, as they invested in inventories in anticipation of generating higher gross margins from manufacturer price increases. More recently, pharmaceutical manufacturers have not been increasing drug prices as frequently, and the percentage increases have been lower. For these and other reasons, some wholesalers have changed their business model to a fee-for-service arrangement, whereby manufacturers pay wholesalers a fee for inventory management and other services. These fees typically are a percentage of the wholesaler’s purchases from the manufacturer or a fixed charge per item or per unit. The fee-for-service approach results in wholesalers’ compensation being more stable and volume-based as opposed to price-increase based.
As a result of the move to a fee-for-service business model, many wholesalers are no longer investing in inventory ahead of anticipated price increases and are reducing their inventories from their historical levels. Under the new model, the consequence of manufacturers using wholesalers is that they now realize the benefit of price increases more rapidly in return for paying wholesalers for the services they
8
provide, on a fee-for-service basis. This change in wholesalers’ business models negatively affected Axcan’s revenue since fiscal 2005, as the resulting distribution services agreement (“DSA”) fees are deducted from gross sales and as wholesale distributors reduce inventory levels.
We entered into a DSA with Cardinal Health in 2005, one of our pharmaceutical wholesale distributors. We anticipate that we will enter into DSA’s with McKesson Corporation and Amerisource Bergen, our two other main pharmaceutical wholesale distributors. Once our DSA’s are put in place, it is possible that such pharmaceutical wholesale distributors could reduce their inventory levels (as DSA’s normally provide for lower targeted inventory levels). Such a reduction in inventory levels by a pharmaceutical wholesale distributor could result in a one-time reduction in our sales to such wholesale distributor for the period during which the reduction occurs, and could have a negative effect on our results of operations for this period.
3. FINANCIAL DEVELOPMENTS
On March 5, 2003, Axcan completed a $125.0 million 4.25% Notes (the “Notes”) financing due 2008. During the third quarter of fiscal 2007, the Company called for redemption all of its Notes and the holders of all of the Notes exercised their right to convert their Notes, in lieu of redemption, by June 28, 2007. The Company completed the conversion of the Notes by issuing an aggregate of 8,924,080 common shares. Long-term debt was consequently reduced by $125.0 million and capital stock increased by the same amount.
The Company has a standby 5 year credit facility with a syndicate of lenders led by the National Bank of Canada for an amount of up to $125.0 million. The amount of this facility may be drawn-down by the Company, subject to fulfilment of certain conditions specified in the credit agreement with its syndicate of lenders, until 2012. The availability of funding may be extended annually with the consent of a specified majority of Lenders and the facility matures on April 30, 2012. Under certain conditions, the amount of the credit can be increased up to an additional $100.0 million and the maturity date extended.
4. LICENSE AND DEVELOPMENT AGREEMENT
On September 30 2007, Axcan entered into an exclusive license and development agreement with Cellerix SL (“Cellerix”) of Spain, for the North American (United States, Canada and Mexico) rights to Cx401, an innovative biological product in development for the treatment of perianal fistulas. Cx401 uses non-embryonic stem-cells to treat perianal fistulas in Crohn’s and non-Crohn’s diseased patients. A Phase II clinical trial has demonstrated the efficacy and confirmed the safety of Cx401 in the treatment of perianal fistulas. Patent applications have been submitted, which, if granted, could provide protection until 2025. Under the terms of the agreement, Axcan made a $10.0 million upfront payment to Cellerix (which was charged to expense in the fourth quarter of 2007), and will make regulatory milestone payments that could total up to $30.0 million. Furthermore, Axcan will pay scaled royalties based on the net sales of Cx401. Axcan has also agreed to make an equity investment of up to $5.0 million in Cellerix, should Cellerix complete its initial public offering by September 30, 2010.
9
PART IV - BUSINESS OF AXCAN
Axcan’s focus is in the field of gastroenterology. Our current portfolio of commercial products includes a number of gastrointestinal products, for the treatment of a range of gastrointestinal diseases and disorders such as inflammatory bowel disease, irritable bowel syndrome, cholestatic liver diseases and complications related to pancreatic insufficiency.
According to IMS Health Ltd., a widely accepted provider of information services specializing in medical research information, the U.S. market for gastrointestinal products was valued at approximately $20 billion in 2006, of which approximately $14 billion was represented by proton-pump inhibitors.
While our business focus is to sell products in the United States, Canada and Europe, several of our products have been commercialized globally through licensing agreements with strategic marketing partners with expertise in their local markets.
We also have various products in development, both in North America and Europe. A discussion on these projects and on the regulatory process follows under the headings “Products in Development” and “Regulatory Environment”.
1. MAIN MARKETED PRODUCTS
The majority of the products that Axcan markets do not benefit from extensive patent protection. The Company believes however that certain of its products benefit from other barriers to the entry of competitors or generics. The Company’s products nevertheless remain subject to competition and generic product entries into their respective markets, which could significantly and negatively affect Axcan’s revenues including, in the case of generics, since they are typically sold at a significant discount to reference drug prices. Intellectual property protection, regulatory exclusivities and certain other barriers to entry are discussed below on a product-by-product basis. As of September 30, 2007, Axcan’s main products are ULTRASE, CANASA, URSO 250/URSO FORTE, PYLERA and CARAFATE in the United States, SALOFALK, SUCRALFATE and URSO in Canada and LACTEOL, DELURSAN and PANZYTRAT in Europe. Since the end of fiscal 2007, the patent covering URSO 250/URSO FORTE’s use in the treatment of Primary Biliary Cirrhosis (“PBC”) has expired in the United States (November 19, 2007) and the market exclusivity obtained on CANASA pursuant to the clinical investigation exclusivity provisions of the Hatch-Waxman Act, covering a change in the formulation of this drug from a 500mg formulation to a 1,000mg suppository formulation, has expired (November 5, 2007).
Axcan’s main marketed products, their sales, prescriptions and patent/regulatory protection are discussed below.
PANCREATIC ENZYMES
ULTRASE
Axcan markets under the trademark ULTRASE certain pancreatic enzyme microsphere (ULTRASE MS) and mini-tablet (ULTRASE MT) formulations designed to help patients with exocrine pancreatic insufficiency, (including pancreatic insufficiency associated with Cystic Fibrosis) better digest food. ULTRASE is marketed in the United States, Canada and a few export markets but this product is actively promoted by Axcan solely in the United States.
For the year ended September 30, 2007, Axcan reported sales of $47.9 million ($39.1 million in 2006 and $36.0 million in 2005) for ULTRASE. ULTRASE competes with a number of pancreatic enzyme formulations including PANCREASE® (Ortho-McNeil Pharmaceutical), CREON® (Solvay Pharmaceuticals, Inc.) and PANCRECARB® (Digestive Care, Inc.), as well as with various other unbranded products.
10
ULTRASE is licensed exclusively from Eurand International S.p.A (“Eurand”) under an exclusive license and supply agreement. This agreement, which was entered into in 2000, initially was for a period of ten years with automatic renewals for subsequent periods of two years. This agreement was amended in 2007 and extended to 2015. Axcan has paid Eurand licensing fees totalling $3.5 million, and Axcan agreed to pay to Eurand royalties of 6% on annual net sales.
In April 2004, the FDA formally notified manufacturers of pancreatic insufficiency products that these drugs, which include ULTRASE, must receive regulatory approval under a New Drug Application (“NDA”), before April 2008, in order to remain on the market. The FDA recently extended this deadline to April 2010. Axcan completed the submission of its NDA for ULTRASE MT in the fourth quarter of fiscal 2007, which filing was accepted by the FDA in December 2007.
ULTRASE MT was also granted a priority review or accelerated approval (as defined by the FDA) designation by the FDA. Under the Prescription Drug User Fee Act, the file must be reviewed by the FDA by a target date of April 2008.
ULTRASE does not currently benefit from patent protection. The Company believes that it will be entitled to receive New Product Exclusivity pursuant to the Hatch-Waxman Act upon approval of its NDA. This exclusivity is expected to prohibit the FDA from approving abbreviated new drug applications for products containing the same active moiety, for a period of five years. Further in its guidelines (“Guidance for Industry - Exocrine Pancreatic Insufficiency Drug Products - Submitting NDAs”; April 2006), the FDA has stated that because of their complexity, pancreatic enzyme extract products are not likely to be appropriate for Abbreviated NDA filings (“ANDAs”).
Other manufacturers currently pursuing approval for porcine derived pancreatic enzyme products are Eurand International S.p.A. (“Eurand”) and Solvay Pharmaceuticals, Inc. Eurand announced on December 20, 2007 that it had completed its NDA submission for its ZENTASE® product line and was granted priority review. Solvay Pharmaceuticals submitted an NDA for its CREON® Product Line in 2003. Solvay reported that it received a letter from the FDA in August 2007 citing certain chemistry, manufacturing and control data work and clinical concerns. Solvay has not reported further on its application.
Of the other pancreatic enzyme products currently on the market PANCRECARB® (Digestive Care, Inc.) and PANCREASE® (Ortho-McNeil Pharmaceutical), we are not aware that any NDA filings were made for these products. Digestive Care Inc. has initiated a Phase III clinical trial for PANCRECARB® (completion was expected June 2007). Although, Johnson & Johnson of which Ortho-McNeil Pharmaceutical is a subsidiary had completed a Phase II for PANCREASE® in 2006, we are not aware of a Phase III ongoing at this time. We are currently not aware of any clinical trial activities being conducted by any generic manufacturers.
VIOKASE
Axcan markets under the brand name VIOKASE non-enteric coated pancreatic replacement enzymes for the treatment of exocrine pancreatic insufficiency. VIOKASE, although not actively promoted, is sold in the United States and Canada.
For the year ended September 30, 2007, Axcan reported sales of $11.2 million ($7.6 million in 2006 and $7.8 million in 2005) for VIOKASE. VIOKASE is the only branded non-enteric coated replacement enzyme and competes with a number of generic products.
In April 2004, the FDA formally notified manufacturers of pancreatic insufficiency products that these drugs, which include VIOKASE, must receive regulatory approval under a New Drug Application (“NDA”), before April 2008, in order to remain on the market. The FDA recently extended this deadline to April 2010. Axcan is currently advancing the completion of its NDA for VIOKASE.
11
VIOKASE does not currently benefit from patent protection. The Company believes that it will be entitled to receive New Product Exclusivity pursuant to the Hatch-Waxman Act upon approval of its NDA. This exclusivity prohibits the FDA from approving abbreviated new drug applications for products containing the same active moiety, for a period of five years. Further in its guidelines, entitled “Guidance for Industry - Exocrine Pancreatic Insufficiency Drug Products - Submitting NDAs” dated April 2006, the FDA stated that because of their complexity, pancreatic enzyme extract products are not likely to be appropriate for ANDA filings.
The Company believes that VIOKASE is currently the only branded uncoated pancreatic enzyme formulation commercially available in the United States.
PANZYTRAT
PANZYTRAT consists of enteric coated microtablets for use in the treatment of exocrine pancreatic insufficiency and pancreatic enzyme deficiency. PANZYTRAT is marketed in several countries, mainly Germany and the Netherlands as well as a few export markets. For the year ended September 30, 2007, Axcan reported sales of $14.8 million ($12.1 million in 2006 and $14.8 million in 2005) for PANZYTRAT, including $1.4 million ($0.8 million in 2006 and $2.9 million in 2005) in export markets. Sales in Germany and the Netherlands represent 80% of the total sales of PANZYTRAT and the main competitor in these territories is CREON® (Solvay Pharmaceuticals, Inc.).
PANZYTRAT does not benefit from any patent protection, nor any other form of regulatory exclusivity in the main countries in which it is marketed, namely Germany and the Netherlands.
URSODIOL
URSO 250 and URSO FORTE - (United States)
In the United States, Axcan has been marketing URSO 250, a 250-milligram ursodiol tablet for the treatment of PBC, since May 1998. URSO FORTE, a 500-milligram ursodiol tablet, was launched in November 2004.
For the year ended September 30, 2007, Axcan reported sales of $68.1 million ($49.3 million in 2006 and $36.0 million in 2005) for URSO250 / URSO FORTE in the United States.
In the United States, there is currently no therapy specifically approved to be marketed for the treatment of PBC other than URSO 250/URSO FORTE. However, other ursodiol products, approved and prescribed for gallstone dissolution as associated with active weight loss, are sometimes used for the treatment of various liver diseases, including PBC. These products include ACTIGALL™ (Watson Pharmaceuticals) and generic versions of ACTIGALL™.
In addition, on November 19, 2007 the FDA Orange book listed patent covering the use of URSO 250/USO FORTE, expired. URSO 250/URSO FORTE does not benefit from any other patent protection or other form of regulatory exclusivity in the United States. As a result, if a generic ursodiol tablet were to be launched, it could have a significant negative impact on sales of URSO 250/ URSO FORTE in the United States.
In order to minimize the impact of any potential generic competition for URSO in the United States, the Company has undertaken, and plans to continue to undertake, certain strategic and tactical measures, including the sequential modification and improvements to URSO’s attributes and specifications. For example in July 2007, the Company introduced a scored tablet version of URSO FORTE (which differentiates URSO from its competitors by giving physicians more flexible dosing options).
The Company believes that these sequential modifications and improvements coupled with URSO’s recognized brand name, its specific approval for the treatment of PBC, its similar pricing compared to
12
comparable generics of ACTIGALL™, and its targeted marketing approach aimed at hepatology specialists, provide a competitive advantage to URSO 250 and URSO FORTE in the United States.
URSO and URSO DS - (Canada)
In Canada, Axcan markets URSO (250 mg) and URSO DS (500 mg) for the treatment of cholestatic liver diseases, which include PBC and Primary Sclerosing Cholangitis (“PSC”). URSO/URSO DS were covered by a patent relating to the use ursodiol for the treatment of PBC in Canada, which was to expire in 2010. However, in 2006, the generic product manufacturer, Pharmascience Inc. successfully challenged the validity of this patent under the Notice of Compliance Regulation procedures of Health Canada. In May 2006, generic versions of URSO and URSO DS received approval for sale in Canada and were launched in fiscal 2007. The launch of these generic products has had a negative impact on sales of URSO/URSO DS in the second half of fiscal 2007 and is expected to continue to negatively impact sales going forward.
URSO/URSO DS does not benefit from any other patent protection or other form of regulatory exclusivity in Canada.
For the year ended September 30, 2007, Axcan reported sales of $9.0 million ($11.4 million in 2006 and $11.1 million in 2005) for URSO/URSO DS in Canada.
DELURSAN
DELURSAN is an ursodiol preparation marketed in France and indicated for the treatment of cholestatic liver diseases, including PBC, PSC and liver disorders related to Cystic Fibrosis.
DELURSAN does not have any patent protection or any regulatory exclusivity in France. For the year ended September 30, 2007, Axcan reported sales of $16.7 million ($13.9 million in 2006 and $13.1 million in 2005) for DELURSAN. As a result, if a generic ursodiol preparation were to be launched, it could have a significant negative impact on sales of DELURSAN in France.
In France, DELURSAN currently competes mainly with URSOLVAN® (Sanofi-Aventis S.A.).
MESALAMINE
CANASA
CANASA is a mesalamine suppository sold by Axcan in the United States for the treatment of distal ulcerative proctitis (UP). The Company believes that CANASA currently is the only commercially available mesalamine suppository in the United States.
For the year ended September 30, 2007, Axcan reported sales of $65.1 million ($53.1 million in 2006 and $28.7 million in 2005) for CANASA in the United States.
CANASA competes with topical corticosteroid enemas and suppositories, as well as mesalamine enemas.
CANASA does not have any patent protection in the United States. On November 5, 2007 the previously granted clinical investigation exclusivity pursuant to the Hatch-Waxman Act, covering a change in the formulation of this drug from a 500mg formulation to a 1,000mg suppository formulation, expired. As a result, if a generic mesalamine suppository were to be launched, it could have a significant negative impact on sales of CANASA in the United States.
However, in June 2007, the FDA published draft guidance on the requirements it expects manufacturers seeking approval of a mesalamine suppository to meet, in order to obtain approval for such a product. The guidance specifies, among other things, that a request of approval must be supported by placebo
13
and reference-drug controlled clinical studies demonstrating efficacy in patients with UP.
The Company believes that it is unlikely that generic competitors will be able to meet the requirements of this FDA guidance, since ethical review boards in the United States may not allow companies to conduct the FDA required placebo-controlled studies on their UP patients. However, the Company cannot provide assurances that any potential generic competitor will not be able to meet such requirements.
Moreover, on July 27, 2007, the Company filed a Citizen’s Petition asking the FDA to require manufacturers seeking approval of mesalamine rectal suppositories to perform an adequate and well-controlled clinical safety and efficacy trial that provides substantial evidence that the generic product demonstrates therapeutic equivalence to CANASA in patients with UP, and to perform a pharmacokinetic study to evaluate the systemic exposure in patients with UP. This petition has not yet been heard. The Company believes this request to be consistent with the standards the FDA has previously applied to other topically acting or non-systemically absorbed drugs and with the above referenced draft guidance published in June, 2007; but cannot provide assurances that its request will be granted.
SALOFALK
SALOFALK is a mesalamine-based product line (tablets, suspensions and suppositories) sold by Axcan in Canada, for the treatment of certain inflammatory bowel diseases, such as ulcerative colitis, ulcerative proctitis and Crohn’s Disease. In Canada, SALOFALK does not have any patent protection, or any regulatory exclusivity.
For the year ended September 30, 2007, Axcan reported sales of $19.3 million ($16.5 million in 2006 and $14.5 million in 2005) for SALOFALK.
Several products containing mesalamine in controlled-release tablets or capsules are available on the Canadian market, including ASACOLTM (The Proctor & Gamble Company) and DIPENTUMTM (UCB Pharma).
SUCRALFATE
CARAFATE / SULCRATE
CARAFATE / SULCRATE line of products is indicated for the treatment of gastric and duodenal ulcers. CARAFATE is sold in the United States as oral tablets and an oral suspension and SULCRATE oral suspensions in Canada, where it is not actively promoted.
For the year ended September 30, 2007, Axcan reported combined sales of $52.2 million ($43.1 million in 2006 and $37.7 million in 2005) for CARAFATE / SULCRATE. CARAFATE / SULCRATE do not have any patent protection or any regulatory exclusivity in their respective markets. Patent protection for CARAFATE in fact lapsed in 2001. Currently, no generic versions of CARAFATE / SULCRATE oral suspension are available in the United States or Canada. If a generic version of these drugs were to be launched, it could have a significant negative impact on sales of CARAFATE / SULCRATE oral suspension in the United States and Canada.
The Company believes, but cannot provide assurances, that due to the mode of absorption of CARAFATE oral suspension, the FDA will require manufacturers seeking an ANDA approval for a generic version of CARAFATE oral suspension to conduct clinical trials in order to demonstrate therapeutic equivalence, which would likely be a barrier to the introduction of a competing generic product in the United States.
14
HELICOBACTER PYLORI ERADICATION
PYLERA
Since May 7, 2007, Axcan has been marketing under the trademark PYLERA a 3-in-1 capsule therapy for the eradication of Helicobacter pylori, a bacterium recognized as being the main cause of gastric and duodenal ulcers. PYLERA is protected by patent claims covering triple and quadruple therapies for Helicobacter pylori eradication. These claims cover the treatment of duodenal ulcer disease (and in some countries reflux esophagitis and gastric ulcer) through the eradication of Helicobacter pylori using a bismuth compound together with two or more antibiotics. The expiry dates of these patents vary depending on the jurisdiction. In the United States, they expire in March 2010. The double capsule formulation of this product and its use in multiple therapies is also covered by patent in a number of countries. The US patent expires in December 2018.
Since its launch in May 2007, sales of PYLERA in the United States have amounted to $1.8 million for the year ended September 30, 2007.
However, as PYLERA is considered by the FDA as pre-1997 antibiotic, the U.S. patent claims covering triple and quadruple therapies for Helicobacter pylori eradication and claims covering the capsule formulation, are not listable in the FDA’s Orange Book and may thus not be eligible to benefit from the automatic stay provisions, nor market exclusivities pursuant to the Hatch-Waxman Act.
PYLERA competes with PREVPAC® (TAP Pharmaceutical Products Inc.), the market leader and HELIDAC® (Prometheus, Inc.)
OTHER PRODUCTS
LACTEOL
LACTEOL is a product containing a specific proprietary strain of Lactobacillus Acidophilus in a lyophilized powder form. It is available in a number of dosage forms, including capsules, and is primarily indicated for the treatment of diarrhoea. LACTEOL, which is mainly sold in France and in over 40 export markets, does not have any patent protection, or any regulatory exclusivity. However, the product is derived from a proprietary strain of bacterium for which the ultimate parent organism is protected by a number of security safeguards, such that access by third parties seeking to reproduce it for competitive or other purposes is limited and controlled.
For the year ended September 30, 2007, Axcan reported sales of $16.3 million ($18.1 million in 2006 and $20.3 million in 2005) for LACTEOL, including $9.9 million ($9.1 million in 2006 and $8.8 million in 2005) outside of France. Sales significantly decreased in the last two years since, as part of new measures designed to reduce healthcare cost associated with the reimbursement of prescription drugs, the French government decided to stop the reimbursement of approximately 200 drugs, including LACTEOL.
LACTEOL competes with a number of generic products on most markets. As LACTEOL has been marketed for several decades, the brand name LACTEOL constitutes a definite marketing advantage wherever the product is sold.
PHOTOFRIN / PHOTOBARR
Axcan markets (directly or through distributors) PHOTOFRIN / PHOTOBARR in North America, Europe, and other selected markets, for the treatment of High-Grade Dysplasia associated with Barrett’s Esophagus, obstructing esophageal cancer and non small cell lung cancer, as well as certain types of gastric cancers and cervical dysplasia. PHOTOFRIN / PHOTOBARR is a photo-sensitizer approved for use in photodynamic therapy, an innovative medical therapy based on the use of light-activated drugs. PHOTOFRIN / PHOTOBARR is covered by a number of patents claiming compositions (including product by process claims), methods of use in approved indications, methods of manufacture, as well as patents
15
for certain devices used in connection with treatment with these products. In the United States, this product’s main market, patent expiries range from June 2009 to May 2016.
For the year ended September 30, 2007, Axcan reported sales of $5.9 million ($5.5 million in 2006 and $7.7 million for 2005) for PHOTOFRIN / PHOTOBARR.
To the Company’s knowledge, there are currently no other photo-sensitizers approved as drugs in Canada, the United States and Europe for the treatment of High-Grade Dysplasia associated with Barrett’s Esophagus, obstructing esophageal cancer and lung cancer, gastric cancer or cervical dysplasia.
2. PRODUCTS IN DEVELOPMENT
Our Scientific Affairs team leverages its expertise in the field of gastroenterology to develop high-value products as well as enhancements and modifications to new and existing products, consistent with our research and development growth strategy. We mainly consider the development of late-stage (Phase II and beyond) novel molecules and innovative product candidates that provide an acceptable risk/return ratio.
We also seek to enhance and extend exclusivity through the staged introduction of product enhancements. These may include improvements in the frequency of administration of drug products, improvements in the convenience of administration, reduction in dose, reduction in side effects (improved tolerability), or improved therapeutic effect/benefit.
Our staff of research scientists has expertise in all aspects of the drug-development process, from pre-formulation studies and formulation development, to scale-up and manufacturing. In fiscal 2007, our development efforts resulted in the approval of PYLERA in the United States, for the eradication of the Helicobacter pylori bacterium.
As part of our business strategy, we enter into licensing agreements with companies that are developing compounds and innovative products in the field of gastroenterology. These compounds and products are typically in-licensed with some combination of upfront payments, development milestone payments and/or royalty payments. In some cases, we have an option to acquire an ownership position in the company.
We currently have development efforts ongoing for 8 products that we believe may, upon regulatory approval, provide clinically meaningful benefits to patients.
Our pipeline products are in various stages of development. Despite the reduced risk profile of our pipeline programs (relative to New Chemical Entities), they do carry development risk, and as such, we do not anticipate the commercialization of all of these products. In addition, we routinely review and prioritize our pipeline as new product candidates are added, which can result in the discontinuation or delay in other ongoing development programs which offer, in our estimation, a less attractive risk/return profile. This is normal practice in the pharmaceutical industry.
Given that the successful development of any pipeline program is dependent on a number of variables, it is difficult to accurately predict timelines for regulatory approval and accordingly clinical development expenses. In fiscal 2007 our research-and-development expenses were approximately $28.6 million or 8.2% of our total revenues (excluding acquired research-and-development expenses from the Cellerix acquisition).
The following is a description of our active and disclosed pipeline projects. Intellectual Property around each of this project is discussed in section “Licensing and Intellectual Property Protection”.
PANCREATIC ENZYMES
16
ULTRASE and VIOKASE
In April 2004, the FDA formally notified manufacturers of pancreatic insufficiency products that these drugs, which include ULTRASE and VIOKASE, must receive NDA approval before April 2008, in order to remain on the market. This deadline was recently extended to April 2010. The FDA has also published final guidelines aimed at assisting manufacturers of exocrine pancreatic insufficiency drug products in preparing and submitting these New Drug Applications. Axcan completed all the clinical trial and the chemistry, manufacturing and control data work required for the submission of its NDA for ULTRASE MT in the fourth quarter of fiscal 2007, which filing was accepted by the FDA in the first quarter of fiscal 2008. The work required to submit VIOKASE is currently ongoing.
We believe that some of the manufacturers of pancreatic enzyme insufficiency products that are currently on the market in the United States may not be able to satisfy the FDA’s requirements for NDAs for these products, which could create a significant growth opportunity for Axcan.
NMK 150
Axcan is developing NMK 150, a new high protease pancrelipase preparation developed for the relief of pain in small duct chronic pancreatitis, which represents an unmet medical need. A dose-ranging, animal study assessing the toxicity of NMK 150, which paid special attention to duodenal irritation, confirmed the safety profile of this compound. A Phase I, ascending, multiple-dose clinical study was also completed and confirmed the safety and tolerability of this compound alone and in combination with a PPI. Results of the pharmacodynamics part of the studies are currently being analyzed.
URSODIOL
SUDCA (Ursodiol Disulfate)
Axcan is currently studying the use of SUDCA, a new ursodiol derivative, in the prevention of the recurrence of colorectal adenomateous polyps, considered to be a pre-cancerous stage of colorectal cancer.
The Company had previously completed a Phase II study of the effectiveness of URSO 250 in preventing the recurrence of colorectal adenomateous polyps in the United States and Canada, for which 792 patients were randomized. The final analysis confirmed a trend in the sub-group of patients suffering from early stage colorectal cancer at baseline: a 26% reduction of polyp recurrence rate occurred in the URSO 250 group when compared to placebo, both in terms of mean number of recurring polyps at one year (0.67 vs. 0.90) and the average size of the polyps (0.23 vs. 0.31). However, in the overall study population, no statistically significant difference between the two groups was observed in terms of mean number of recurring polyps (0.97 vs. 1.04) and average polyp size (0.30 vs. 0.28).
Preliminary results of studies conducted with SUDCA showed that ursodiol disulfate reduces the number of aberrant crypts in a rat model of colon cancer. Aberrant crypts are considered to be early abnormal changes in the intestinal lining that are precursors to colon cancer. In a small pilot study, where rats were injected with the carcinogen azoxymethane, a 56% reduction in the total number of aberrant crypts in the colon was observed after four weeks in those animals treated with this new ursodiol formulation compared to control models. Ursodiol disulfate alone fed to rats had no adverse effects on the appearance of the lining of the colon. Long-term animal studies are ongoing to determine the effect of ursodiol disulfate on the timing of appearance, the number, and the size of colonic tumours in the azoxymethane rat model of chemically-induced colon cancer.
In addition to these animal studies, a single, ascending-dose Phase I clinical study was completed in early 2006, and a multiple, ascending-dose Phase I study was completed in September 2006, to evaluate the safety, tolerability and preliminary pharmacokinetics of SUDCA. Both studies confirmed the safety and tolerability of this compound. Results of the pharmacokinetics part of the studies are currently being analyzed.
17
MESALAMINE
CANASA Max-002
Axcan has initiated the CANASA MAX-002 program, a Phase III clinical trial to evaluate the efficacy and safety of a novel, high-concentration, 1-gram mesalamine suppository for the treatment of ulcerative proctitis.
Ulcerative proctitis is a subgroup of ulcerative colitis, one of the most common inflammatory bowel diseases. For approximately 30% of patients with ulcerative colitis, the illness begins as ulcerative proctitis where bowel inflammation is limited to the rectum. Currently, it is estimated that there are 1 million cases of inflammatory bowel disease in the U.S., with approximately 400,000 new cases every year.
In June 2007, the FDA issued draft guidance on the type of clinical program required for the approval of mesalamine suppositories. Based on this guidance, Axcan temporarily suspended the recruitment of this trial and expects to resume it in the first half of calendar 2008, upon completion of ongoing discussions with the FDA.
HELICOBACTER PYLORI ERADICATION
PYLERA
Axcan intends to initiate a Phase III clinical program with PYLERA in Europe to obtain approval to market this therapy for the eradication of Helicobacter pylori. This Phase III clinical trial in Europe is expected to be conducted on approximately 400 patients and is intended to compare Axcan’s PYLERA regimen given in combination with omeprazole, to the widely used OAC triple therapy (20 mg of omeprazole, 1 g of amoxicillin and 500 mg of clarithromycin, all given twice a day). The trial is expected to be completed in the second half of calendar 2009. PYLERA was successfully launched in the United States in fiscal 2007.
OTHERS
AGI-010
Axcan and AGI Therapeutics, plc (“AGI”) are co-developing AGI-010, a delayed/controlled release formulation of the Proton Pump Inhibitor (“PPI”) drug omeprazole, which is being developed for the treatment of gastro-esophageal reflux disease (“GERD”), and in particular to address the control of night-time gastric acidity, known as Nocturnal Acid Breakthrough (“NAB”). NAB remains a significant unmet medical need, and is estimated to occur in more than 50% of GERD patients on a PPI therapy.
Development of the final formulation for this compound is ongoing and once completed, will allow both companies to make a decision on the most appropriate development and filing strategy for this product.
CX401
On September 30, 2007, Axcan entered into an exclusive license and development agreement with Cellerix of Spain, for the North American (United States, Canada and Mexico) rights to Cx401, an innovative biological product in development for the treatment of perianal fistulas.
A Phase II trial conducted in 50 patients in Europe demonstrated the efficacy and safety of Cx401. This randomized, open-label, parallel assignment study evaluated the safety and efficacy of Cx401 in the treatment of perianal fistulas in Crohn’s and non-Crohn’s Disease patients. The primary endpoint for this study was photographically assessed complete closure and healing, and showed a 71% response rate in the acute phase, both in Crohn’s and non-Crohn’s Disease patients. Results of this study were presented at Digestive Disease Week in May 2007 (Garci-Olmo D. et al., “Expanded Adipose-Derived Stem Cells (Cx401) for the Treatment of Complex Perianal Fistula. A Phase II Clinical Trial” (Digestive Disease Week 2007; Abstract: 492)) and are pending publication.
18
Axcan will be responsible for the development of this product in North America. The Company expects to initiate a Phase IIb study in North America in fiscal 2008, and details of this study are expected to be communicated to the market upon its initiation. Depending on the outcome of further discussions with the FDA, filing could occur as early as 2011.
3. REGULATORY AFFAIRS AND QUALITY ASSURANCE
Our Regulatory Affairs Department is involved in the development and registration of all products and has prepared product submissions for regulatory agencies in the United States, Canada and Europe. This group also coordinates all data and document management for submissions, including amendments, supplements and adverse events reporting. Our Quality Assurance Departments seek to ensure that all stages of product development and manufacturing fully comply with applicable good clinical, laboratory and manufacturing practices.
4. REGULATION
The research and development, manufacture, and marketing of pharmaceutical products are subject to regulation by U.S., Canadian and foreign governmental authorities and agencies. Such national agencies and other federal, state, provincial and local entities regulate the testing, manufacturing, safety and promotion of our products. The regulations applicable to our products may be subject to change as regulators acquire additional experience in the specific area.
UNITED STATES REGULATIONS
New Drug Application
With the exception of the pancreatic enzyme products which require an NDA approval before April 2010, we are required by the FDA to comply with NDA procedures for our branded products prior to commencement of marketing. New drug compounds and new formulations for existing drug compounds which cannot be filed as ANDAs are subject to NDA procedures. These procedures include: (1) preclinical laboratory and animal toxicology tests; (2) submission of an Investigational New Drug Application (‘‘IND’’), and its required acceptance by FDA before any human clinical trials can commence; (3) adequate and well-controlled replicate human clinical trials to establish the safety and efficacy of a drug for its intended indication; (4) the submission of an NDA to the FDA; and (5) FDA approval of an NDA prior to any commercial sale or shipment of the product, including pre-approval and post-approval inspections of its manufacturing and testing facilities. When all data in a product application are owned by the applicant, the FDA will issue its approval.
Abbreviated New Drug Application
In certain cases, where the objective is to develop a generic version of an approved product already on the market, an ANDA may be filed in lieu of filing an NDA. Under the ANDA procedure, the FDA waives the requirement to submit complete reports of preclinical and clinical studies of safety and efficacy, and instead, requires the submission of bioequivalency data, that is, demonstration that the generic drug produces the same blood levels of drug in the body as its brand-name counterpart. It is mandatory that it have a comparable pharmacokinetic profile, or change in blood concentration over time.
505(b)(2) Application Process In certain cases, pharmaceutical companies may also submit a 505(b)(2) NDA application for marketing approval of a drug. This mechanism essentially relies upon the same FDA conclusions that would support the approval of an ANDA available to an applicant who develops a modification of a drug that is not supported by a suitability petition. Relative to normal regulatory requirements for a 505(b)(1) NDA, regulation may permit a 505(b)(2) applicant to forego costly and time-consuming drug development studies by relying on the FDA’s finding of safety and efficacy for a previously approved drug product.
19
Under some circumstances the extent of this reliance approaches that permitted under the generic drug approval provisions. This approach is intended to encourage innovation in drug development without requiring duplicative studies to demonstrate what is already known about a drug, while protecting the patent and exclusivity rights for the approved drug.
CANADIAN REGULATIONS
The requirements for selling pharmaceutical drugs in Canada are substantially similar to those of the U.S. described above, with the exception of the 505(b)(2) route, and marketing exclusivity under the Hatch-Waxman provisions of the FDA in the U.S.
EUROPEAN ECONOMIC AREA (“EEA”)
A medicinal product may only be placed on the market in the EEA, composed of the 27 EU member states, plus Norway, Iceland and Lichtenstein, when a marketing authorization has been issued by the competent authority of a member state. There are essentially three community procedures created under prevailing European pharmaceutical legislation that, if successfully completed, allow an applicant to place a medicinal product on the market in the EEA.
Centralized Procedure
Centralized procedure exists when a marketing authorization is granted by the European Commission, acting in its capacity as the European Licensing Authority on the advice of The European Medicines Agency. That authorization is valid throughout the entire community and directly or indirectly (Norway, Iceland and Liechtenstein) allows the applicant to place the product on the market in all member states of the EEA. The European Medicines Agency is the administrative body responsible for coordinating the existing scientific resources available in the member states for evaluation, supervision and pharmacovigilance of medicinal products.
Mutual Recognition and Decentralized Procedures
The competent authorities of the member states are responsible for granting marketing authorizations for medicinal products that are placed on their markets. If the applicant for a marketing authorization intends to market the same medicinal product in more than one member state, the applicant may seek an authorization progressively in the community under the mutual recognition or decentralized procedure.
Mutual Recognition
Mutual recognition is used if the medicinal product has already been authorized in one member state. In this case, the holder of this marketing authorization requests the member state where the authorization has been granted to act as reference member state by preparing an updated assessment report that is then used to facilitate mutual recognition of the existing authorization in the other member states in which approval is sought (the so-called concerned member state(s)). The reference member state must prepare an updated assessment report which, together with the approved Summary of Product Characteristics, or SmPC (which sets out the conditions of use of the product), and a labelling and package leaflet are sent to the concerned member states for their consideration. The concerned member states are required to approve the assessment report, the SmPC and the labelling and package leaflet within 90 days of receipt of these documents.
The decentralized procedure
This procedure is used in cases where the medicinal product has not received a marketing authorization in the EU at the time of application. The applicant sends its application simultaneously to all concerned member states and requests a member state of its choice to act as reference member state to prepare an assessment report that is then used to facilitate agreement with the concerned member states and the
20
grant of a national marketing authorization in all of these member states. In this procedure, the reference member state must prepare, for consideration by the concerned member states, the draft assessment report, a draft SmPC and a draft of the labelling and package leaflet within 120 days after receipt of a valid application. As in the case of mutual recognition, the concerned member states are required to approve these documents within 90 days of their receipt.
INTERNATIONAL REGULATIONS
Sales of our products outside the United States, Canada and Europe are subject to local regulatory requirements governing the testing, registration and marketing of pharmaceuticals, which vary widely from country to country.
In addition to the regulatory approval process, pharmaceutical companies are subject to regulations under provincial, state and federal laws, including requirements regarding occupational safety, laboratory practices, environmental protection and hazardous substance control, and may be subject to other present and future local, provincial, state, federal and foreign regulations, including possible future regulations of the pharmaceutical industry. We believe that we are in compliance in all material respects with such regulations as are currently in effect.
5. PATENTS AND PROPRIETARY RIGHTS
We believe that trademark protection is an important part of establishing product and brand recognition. We own a number of registered trademarks and trademark applications and have acquired the rights to several trademarks by license. Axcan maintains trademarks registrations in a number of jurisdictions where it sells its products. In the United States, trademark registrations remain in force for ten years and may be renewed every ten years after issuance, provided the mark is still being used in commerce.
A patent is a statutory private right that grants to the patentee exclusive rights to exclude others from using the patented invention during the term of the patent. A patent is territorial and may be sought in many jurisdictions. In the United States, as in most other countries, the term of patent protection is 20 years from the date the patent application was filed. An invention may be patentable if it meets the criteria of being “new,” “useful” and “non-obvious.” Depending upon whether a particular drug is patentable and the relative cost associated with obtaining its issuance in a given jurisdiction, an inventor will either apply for a patent in order to protect the invention or maintain the confidentiality of the information to rely on the common law protection afforded to trade secrets.
A company may also enter into licensing agreements with third-party licensors in order to obtain the right to make, use and sell certain products, thereby gaining access to know-how, secret formulas and patented technology. The value of a license is generally enhanced by the existence of one or more patents. A license gives the licensee access to developed and, in many cases, tested technology and provides the licensee faster and often less expensive entry into the market. Licensing also establishes relationships, which may provide access to additional products or technology or may lead to joint ventures or alliances affording the licensor and the licensee an opportunity to evaluate each other’s products and technology. This is also true, to a lesser extent, for distribution relationships.
Pursuant to license agreements with third parties, we have acquired rights to manufacture, use or market certain of our existing products, as well as many of our development products and technologies. Such agreements typically contain provisions requiring us to use commercially reasonable efforts or otherwise exercise diligence in pursuing market development for such products in order to maintain the rights granted under the agreements, and may be cancelled upon our failure to perform our payment or other obligations.
21
We further rely and expect to continue to rely upon unpatented proprietary know-how and technological innovation in the development and manufacture of many of our principal products. Our policy is to require all our employees, consultants and advisors to enter into confidentiality agreements with us.
Axcan has entered into several of these types of collaborative agreements with licensors, licensees and other third parties and will continue to do so. The following agreements have been entered into in connection with products marketed by Axcan or under development.
ULTRASE
Axcan is the owner of the trademark ULTRASE and markets certain pancreatic enzyme based microspheres and mini-tablets under the ULTRASE brand in North and Latin America, under an exclusive development, license and supply agreement with Eurand International S.p.A. This agreement was entered into in 2000, and was initially for a period of ten years with automatic renewals for subsequent periods of two years. This agreement was recently amended in 2007, including to provide for an extension of its term to 2015. Axcan has paid Eurand licensing fees totalling $3.5 million, and Axcan agreed to pay to Eurand royalties of 6% on its annual net sales of ULTRASE.
URSODIOL
Axcan developed URSO for the Canadian market in collaboration with Falk Pharma GmbH (“Falk”) and acquired the rights to manufacture, use and market URSO in the United States on March 23, 1993 through the acquisition of the shares of Axcan Pharma U.S., Inc. that it did not already own. In April 1999, Axcan entered into two agreements with Sanofi-Synthélabo S.A. of France (now, Sanofi-Aventis) that secured Axcan’s right to manufacture, use and market URSO for the treatment of PBC in Canada and the United States. For the United States, Axcan licensed the rights to the treatment of PBC under a patent, which expired on November 19, 2007. For Canada, Axcan acquired full ownership of the patent relating to the use of ursodiol for the treatment of PBC, which expires in 2010. However, in May 2006, the Canadian PBC patent was held to be invalid for the purposes of opposing the issuance of a Notice of Compliance for a generic version of URSO 250mg, under proceedings pursuant to the Canadian Patented Medicines (Notice of Compliance) Regulations.
In October 2000, Axcan entered into a licensing agreement with the Children’s Hospital Research Foundation, an operating division of Children’s Hospital Medical Centre of Cincinnati, Ohio, for a series of patented sulfated derivatives of ursodeoxycholic acid compounds (“SUDCA” or “ursodiol disulfate”). According to the terms of this agreement, Axcan has exclusive worldwide rights to commercially exploit formulations of SUDCA under the licensed patents and know-how developed by Children’s Hospital Research Foundation.
PHOTOFRIN
In May 2000, Axcan purchased from QLT Inc. (“QLT”) the trademark “PHOTOFRIN” for the United States, Canada and all other countries where it has been registered as a trademark or used in marketing. Axcan also purchased, licensed or sublicensed from QLT, as the case may be, the worldwide rights of other assets and intellectual property related to PHOTOFRIN. As part of the transaction, Axcan acquired a European subsidiary of QLT, which holds the European regulatory approvals for PHOTOFRIN. The last of the patents, which form part of the acquired assets, expires in April 2013. As part of the acquisition, Axcan agreed to assume QLT’s obligation to pay royalties of up to 5% on net sales of PHOTOFRIN to Health Research Inc. (“Health Research”), pursuant to arrangements under which Axcan is a sub-licensee of the technology that QLT licensed from Health Research. Pursuant to the terms of the transaction between Axcan and QLT, Axcan has paid to QLT milestone cash payments of CAN $20.0 million (representing the maximum potential aggregate amount of milestone cash payments under the terms of the transaction).
PANZYTRAT
In December 2002, Axcan acquired from an affiliate of Abbott Laboratories (‘‘Abbott’’) certain assets related to the distribution, marketing and sale of a line of pancreatic enzyme products used to enhance the digestion of fats. This pancreatic enzyme product line is commonly marketed under the trademark
22
PANZYTRAT. The product line is subject to patents assigned to Axcan directly from Abbott, the last of which expires in 2008. The know-how and trade secrets associated with these products and their manufacture are the object of a perpetual unrestricted license from Abbott. The PANZYTRAT and related trademark portfolio, was assigned directly from Abbott to Axcan’s French subsidiary, Axcan Pharma S.A. This portfolio contains trademarks associated with the product line for a number of countries throughout the world.
PYLERA
In January 2000, Axcan entered into a worldwide (excluding Australia and New Zealand) licensing agreement (which was amended in November 2000 and August 2001) with Exomed Australia PTY Limited, Gastro Services PTY Limited, Ostapat PTY Limited, and Capability Services PTY Ltd. This agreement, as amended, provides Axcan with exclusive rights in a number of countries, including Canada and the United States, to a series of patents covering triple and quadruple therapies for Helicobacter pylori eradication. These patents cover the treatment of duodenal ulcer disease (and in some countries reflux esophagitis and gastric ulcer) through the eradication of Helicobacter pylori using a bismuth compound together with two (2) or more antibiotics. Axcan paid approximately $1.64 million cash for the license and will pay a 5.5% royalty based on sales. The expiry dates of these patents vary depending on the jurisdiction and expire in March 2010 in the United States.
In May 1999, Axcan acquired the rights to a double capsule delivery technology to be used for PYLERA from Gephar S.A. (“Gephar”), in an asset swap transaction, whereby Axcan sold to Gephar its interest in Axcan Ltd., a manufacturer and distributor of the PROTECTAID™ contraceptive sponge. This patent expires in December 2018 in the United States.
LACTEOL and LACTEOL FORT
In April 2002, Axcan acquired all of the shares of Laboratoire du Lactéol du Docteur Boucard which is the owner of all of the intellectual property rights to the antibacterial composition marketed by Le Laboratoire du Lactéol under different trademarks, including the trademark LACTEOL. The antibacterial composition is also subject to a patent in France and to an international patent application. These patents rights are owned by Axcan and a French research institute.
AGI-010
On September 25, 2006, the Company entered into a license and co-development agreement with AGI. Pursuant to this agreement, AGI and the Company will co-develop a controlled release omeprazole product (“AGI-010”) for the treatment of Gastro-esophageal Reflux Disease and a better control of Nocturnal Acid Breakthrough. Under the agreement, the Company has licensed exclusive rights to patent applications and know-how related to AGI-010 and will retain rights to any improvements to the product for North America and, if extended by Axcan, to other territories. However, the Company cannot provide assurances that such patent will be granted. The Company and AGI have further agreed to share certain development expenses, and the Company paid a $1.5 million upfront fee (which was charged to expense in fiscal 2006) and may be required to make specified milestone payments that could total up to $17.5 million at various stages of development, up to and including regulatory approvals. The Company also agreed to pay royalties on net sales.
23
Cx401
Under the terms of the agreement signed with Cellerix on September 30, 2007, Axcan made a $10.0 million upfront payment to Cellerix (which was charged to expense in the fourth quarter of 2007), and will make regulatory milestone payments that could total up to $30.0 million. In addition, patent applications were submitted, which, if granted, could provide protection until 2025. However, the Company cannot provide assurances that such patent will be granted.
In addition, under the terms of the agreement with Cellerix, Axcan will pay scaled royalties based on the net sales of Cx401. Axcan has also agreed to make an equity investment of up to $5.0 million in Cellerix, should Cellerix complete its initial public offering by September 30, 2010.
6. EMPLOYEES
As at September 30, 2007, the end of Axcan’s most recently completed fiscal year, Axcan employed 480 persons. Of these employees, 154 are located in the United States, 169 are located in Canada and 157 are located in Europe. In Canada, Axcan is a party to a collective bargaining agreement, which was recently renewed and expires March 24, 2011. This agreement covers 60 employees, all of whom are non-management employees. In France, Axcan’s employees are subject to the Convention Collective Nationale de l’Industrie Pharmaceutique, a collective bargaining agreement which applies to the entire pharmaceutical industry. Axcan believes that relations with both its unionized and non-unionized employees are good.
In the United States, Axcan sells its products to most major wholesale drug companies and distributors, which in turn distribute Axcan’s products to chain and independent pharmacies, hospitals and mail order organizations. Our 83 sales representatives, 10 regional sales managers (managed by 2 zone directors) and 5 national account managers in our managed care group, all located in the United States, call on high-volume prescribing gastroenterology physicians, cystic fibrosis centres, hepatologists and transplant centres, potential and current PHOTOFRIN centres as well as third-party payors, clinical pharmacists and formularies administrators. Since the launch of PYLERA, in May 2007, sales representatives also visit general practitioners known to be high-prescribers of Helicobacter pylori eradication therapies.
Increasingly, in North America, third-party payors, such as private insurance companies and drug plan benefit managers, aim to rationalize the use of pharmaceutical products and medical treatments, in order to ensure that prescribed products are necessary for the patients’ conditions. Moreover, large drug store chains now account for an increasing portion of the retail sales of prescription medicines. The pharmacists and store managers of such retail outlets are under pressure to reduce the number of items in inventory in order to reduce costs.
In Canada, Axcan sells its products to hospitals and wholesale drug companies, which in turn distribute Axcan’s products to pharmacies. Axcan’s major products are included in most provincial drug benefit formularies and are promoted by Axcan’s 10 sales representatives, under the supervision of the regional sales manager, to gastroenterologists and internal medicine specialists with a particular interest in gastrointestinal diseases, as well as to colorectal surgeons.
In France, Axcan sells its products to distributors, which in turn distribute them to wholesale drug companies, which in turn distribute them to pharmacies. The 43 sales representatives under the supervision of 5 regional sales directors regularly visit high-prescribing physicians to promote Axcan’s other products.
This international sales structure is complemented by Axcan’s sponsorship of high-level international medical meetings on topics related to Axcan’s products and research activities. These events are recognized by leading institutions, and continuing medical education credits are awarded to attendees. As a consequence, Axcan is recognized not only as a supplier of quality products, but also as an important link in the continuous medical education process.
24
7. ENVIRONMENTAL PROTECTION, HEALTH AND SAFETY
Axcan is committed to environmental protection and the promotion of environmental issues. This includes a safe, healthy and secure environment for its employees, subcontractors and customers and the community in which the Company operates.
The Company has established a series of policies to facilitate compliance with applicable environmental laws and regulations. These policies require that business units conduct regular environmental assessments of Company activities, establish remedial and contingency plans to deal with any incidents, and establish regular processes to review and report to senior corporate management on the environmental status of the Company and its subsidiaries.
Axcan generates a small amount of hazardous waste that is disposed of by certified third-party carriers. Axcan believes its approach to environmental compliance meets applicable regulatory requirements and it is not expected that its policies or practices will have a significant impact on capital expenditures, consolidated earnings or our competitive position.
8. FACILITIES
We own and lease space for manufacturing, warehousing, research, development, sales, marketing, and administrative purposes. All these facilities are inspected on a regular basis by regulatory authorities, and our own internal auditing team ensures compliance on an ongoing basis with such standards.
Axcan owns 107,000 square feet of office space, manufacturing facilities and warehousing in Mont-Saint-Hilaire, Quebec, the location of its corporate headquarters. The building houses administrative, marketing and pharmaceutical manufacturing operations as well as the research and development activities of Axcan. Axcan further owns property next to its corporate headquarters, which could be used to expand. The building and real estate owned by Axcan is subject to security granted in favour of its lenders, pursuant to the credit facilities described under “Selected Consolidated Financial Information - Liquidity and Capital Resources”.
Axcan owns 606,837 square feet of office space, manufacturing facilities and land in Houdan, France.
Axcan leases approximately 20,000 square feet of office space in Birmingham, Alabama under a lease expiring in December 2008.
Axcan leases approximately 3,650 square feet of office space in Bridgewater, New Jersey, under a lease expiring in August, 2009.
We believe that we have sufficient facilities to conduct our operations during fiscal 2008 and that our facilities are in satisfactory condition and are suitable for their intended use, although investments to improve and expand our manufacturing and other related facilities are contemplated, as our business requires.
25
PART V - RISK FACTORS
This AIF contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed in this form. Factors that could cause or contribute to these differences include, but are not limited to, those discussed below and elsewhere in this form and in any documents incorporated in this form by reference.
If any of the following risks, or other risks not presently known to us or that we currently believe to not be significant, develop into actual events, then our business, financial condition, results of operations or prospects could be materially adversely affected. If that happens, the market price of our common stock could decline, and stockholders may lose all or part of their investment.
We currently depend on four groups of products for a large portion of our revenues; any material decline in the sales of any of them would have an adverse impact on our business.
Any factor that adversely affects the sale or price of our key products could significantly decrease our sales and profits. Pancreatic enzymes (ULTRASE, VIOKASE and PANZYTRAT), ursodiol (URSO 250/URSO DS/URSO FORTE and DELURSAN), mesalamine (CANASA and SALOFALK) and sucralfate (CARAFATE and SULCRATE) accounted for 21.2%, 26.7%, 24% and 17% respectively, of our total revenues for the year ended September 30, 2007. We believe that sales of these products will continue to constitute a significant portion of our total revenues until we launch additional products. Any significant setback with respect to any one of these products, including shipping, manufacturing, product safety, marketing, government licenses and approvals, intellectual property rights problems, or generic or other forms of competition, could have a material adverse effect on our financial position, cash flows or overall trends in results of operations.
If we are unable to obtain FDA approval for ULTRASE before April 2010, we would no longer be able to sell such product in the United States; this could have a material adverse effect on our revenues, results of operations and liquidity.
In April 2004, the FDA formally notified manufacturers of pancreatic enzyme products (“PEPs”) that these drugs had to be approved by the FDA in order to remain on the market. Under the new requirements, manufacturers are required to file an IND for their PEPs by April 2008, to file an NDA by April 2009 and to obtain FDA approval by April 2010. The FDA decided to require these approvals for all pancreatic extract drug products after reviewing data that showed substantial variation among currently marketed PEPs.
ULTRASE, our enteric coated pancreatic enzyme formulation, accounted for approximately 13.7 %, or $47.9 million, of our revenues in fiscal 2007. If we are unable to obtain FDA approval to market ULTRASE prior to April 2010, we would no longer be able to sell ULTRASE in the United States, which would impair our results of operations and liquidity.
We recently filed an NDA for the mini-tablet formulation of ULTRASE in the fourth quarter of fiscal 2007, which filing was accepted for review in the first quarter of fiscal 2007 by the FDA. However, there is no guarantee that we will succeed in obtaining approval for such product from the FDA prior to April 2010 or at all.
Despite the FDA’s announcement of its intention to remove from the market those PEPs that have not filed an IND by April 2008, filed an NDA by April 2009 and/or obtained approval by April 2010, its position is non-binding. As a result, the agency may not pursue regulatory action against companies that fail to meet these deadlines.
If we are successful in obtaining FDA approval for ULTRASE and the FDA does not enforce its stated position, the level of competition that our drug ULTRASE currently faces in the market may not decline and could increase in the future.
26
We no longer have patent protection for our URSO drug in the United States and Canada. This could result in competition from generic products leading to a significant reduction in sales of this drug.
On November 19, 2007 our U.S. patent covering URSO 250/ URSO FORTE’s use in the treatment of PBC expired. In addition, the validity of our Canadian patent for the similar use of URSO and URSO DS in Canada was successfully challenged recently by Pharmascience Inc., a generic product manufacturer, under the Notice of Compliance Regulation procedures of Health Canada. Therefore, these products no longer benefit from any patent protection or other form of regulatory protection or exclusivity in the United States or Canada.
As a result, competition from generic products that treat the same conditions may increase. For example, sales of generic products in Canada have already had a negative sales impact on our Canadian sales of URSO / URSO DS; this trend is expected to continue going forward.
The loss of intellectual property protection in the United States for our URSO 250 / URSO FORTE, which made up 19.5% of our total revenues in fiscal 2007, could have a material adverse effect on our results of operations and financial condition.
In order to minimize the impact of any potential generic competition in the United States, we have undertaken, and plan to continue to undertake, certain strategic measures, including the sequential modification of and improvements to URSO’s attributes and specifications. See “Part IV – Business of Axcan—Main Marketed Products—Ursodiol—URSO 250 and URSO FORTE.” There is no assurance that any of these measures will minimize the generic competition we face with respect to URSO and generic sales could have a significant negative impact on our sales of URSO.
We no longer have clinical investigation exclusivity in the United States for our CANASA drug. This could result in competition from generic products leading to a significant reduction in sales of this drug.
Although our CANASA drug does not have any patent protection in the United States, we previously had clinical investigation exclusivity from the FDA covering a change in the formulation of this drug from a 500mg formulation to a 1,000mg formulation. This clinical investigation exclusivity effectively precluded the FDA from approving a competitor’s ANDA relating to the protected formulation change for a period of three years. However, pursuant to the Hatch-Waxman Act, the clinical investigation exclusivity that we previously had covering the change of formulation of this product to a 1,000mg formulation, recently expired.
The lack of patent protection and the loss of this clinical investigation exclusivity for this product in the United States, could give rise to competition from generic products or therapeutically substitutable products. A significant reduction in the sales for this product, which along with SALOFALK made up 24% of our revenues for the year ended September 30, 2007 (a substantial majority of which were derived from sales of CANASA in the United States), could have a material adverse effect on our results of operations and financial condition.
Although we believe that recently published FDA draft guidance make it more difficult for potential competitors to obtain the FDA approval required to sell competing mesalamine suppository in the United States, there can be no assurance that a generic or therapeutically substitutable mesalamine suppository will not be approved by the FDA, which could significantly negatively impact our sales of CANASA in the future. The Company believes that generic competitors may have difficulty meeting the requirements of this FDA guidance, since ethical review boards in the United States may not allow companies to conduct such FDA required placebo-controlled studies on their UP patients. However, the Company cannot provide assurances that any potential generic competitor will not be able to meet such requirements.
27
Some of our key products face generic competition.
Some of our key products, such as ULTRASE, URSO and CANASA, currently face competition from products which are subject to being therapeutically substituted for our products (such as ACTIGALL and its generics in the case of URSO, other branded and unbranded PEP’s in the case of ULTRASE and other rectal dosage forms of mesalamine in the case of CANASA), and from their respective generic versions. Third-party payors and pharmacists can substitute generics for our products even if physicians prescribe our products by name. Particularly in the United States, government agencies and third-party payors often put pressure on patients to purchase generic products instead of brand-name products as a way to reduce healthcare costs.
Products which are no longer protected by a marketing exclusivity or a patent are subject to generic competition. Generic competition against any of our products would lower prices and unit sales and could therefore have a material adverse effect on our results of operations and financial condition.
The concentration of our product sales to only a few wholesale distributors increases the risk that we will not be able to effectively distribute our products if we need to replace any of these customers, which would cause our sales to decline.
The majority of our sales are to a small number of pharmaceutical wholesale distributors, which in turn sell our products primarily to retail pharmacies, which ultimately dispense our product to the end consumers. In fiscal 2007, sales to McKesson Corporation accounted for 41.2 % of our total revenue, sales to Cardinal Health accounted for 25.0% of our total revenue, and sales to Amerisource Bergen accounted for 11.1 % of our total revenue.
If any of these customers cease doing business with us or materially reduce the amount of product they purchase from us and we cannot conclude agreements with replacement wholesale distributors on commercially reasonable terms, we might not be able to effectively distribute our products through retail pharmacies. The possibility of this occurring is exacerbated by the recent significant consolidation in the wholesale drug distribution industry, including through mergers and acquisitions among wholesale distributors and the growth of large retail drugstore chains. As a result, a small number of large wholesale distributors control a significant share of the market.
Wholesaler buying patterns may change, and this could have a detrimental effect on our future revenue and financial condition.
Wholesalers, on which we largely depend for revenue generation, have historically practiced inventory price arbitrage. For example, in the past prior to any routine increase in prices for drugs by manufacturers, many of our wholesalers have purchased excess inventory at lower prices and resold the inventory after the price increase, thereby profiting. This inventory price arbitrage was predominantly how wholesalers were previously compensated for the distribution services they provided and had a dramatic effect on wholesaler buying patterns as wholesalers invested in inventories in anticipation of generating higher gross margins from price increases from manufacturers.
Recently, pharmaceutical manufacturers have not been able to increase drug prices as frequently as they used to, and the percentage of any such increases has been lower. The reduction in the size and frequency of price increases has contributed to wholesalers turning to fee-for-service arrangements under which their fees are expressed as a percentage of the wholesaler’s purchases from the manufacturer or as an amount per unit. As a result, wholesalers’ buying patterns have shifted from large pre-price-increase purchases to more periodic purchasing based on volume activity. Overall, wholesalers that have entered into these fee-for-service arrangements carry lower levels of inventory. This change in wholesaler’s business model has and may in the future negatively impact our revenues..
We entered into a DSA with Cardinal Health in 2005, one of our pharmaceutical wholesale distributors. We anticipate that we will enter into DSA’s with McKesson Corporation and Amerisource Bergen, our two other main pharmaceutical wholesale distributors. Once our DSA’s are put in place, it is possible that such pharmaceutical wholesale distributors could reduce their inventory levels (as DSA’s normally provide
28
for lower targeted inventory levels). Such a reduction in inventory levels by a pharmaceutical wholesale distributor could result in a one-time reduction in our sales to such wholesale distributor for the period during which the reduction occurs, and could have a negative effect on our results of operations for this period.
We have recently begun to deduct these DSA fees, which totaled approximately $2.7 million and $3.6 million for the years ended September 30, 2006 and 2007, respectively, from our total revenues. For example, in the quarter ended September 30, 2006, we chose to adopt the classification of these fees as a deduction from sales. Previously, these fees were included in selling and administrative expenses. As a result, our sales were reduced by approximately $1.9 million in the fourth quarter of fiscal 2006. Fees associated with these agreements incurred in 2005 were not material. It is possible that changes in wholesaler buying patterns may occur in the future, and these changes could result in a reduction in our revenues and could have a detrimental effect on our overall financial condition or results of operations.
Our quarterly results may fluctuate.
Our quarterly operating results have fluctuated in the past and may continue to fluctuate in the future. Factors, many of which are not within our control, that could cause quarterly operating results to decline include, for example, the size and timing of product orders, which can be affected by customer budgeting and buying patterns, or the size and timing of expenses associated with our development and marketing programs. As a result, if customer buying patterns cause revenue shifts from one fiscal quarter to another, or if the development and marketing of our products causes our expenses to change from one fiscal quarter to another, our net quarterly income may not meet the market’s expectations.
Our business depends on key scientific, sales and managerial personnel for continued success.
Much of our success to date has resulted from the skills of certain of our officers, scientific personnel and sales force. If these individuals were no longer employed by us, we might not be able to attract or retain employees with similar skills or carry out our business plan.
We use third parties to manufacture and perform analytical testing for almost all of our products and product candidates. This may increase the risk that we will not have sufficient quantities of our products or product candidates or such quantities at an acceptable cost, which could result in clinical development and commercialization of our product candidates being delayed, prevented or impaired.
We currently rely, and expect to continue to rely, on third parties for (i) the supply and testing of the active pharmaceutical ingredients in our products and product candidates, and (ii) the manufacture and testing of the finished forms of these drugs and their packaging. The current manufacturers of our products and product candidates are, and any future third party manufacturers that we enter into agreements with will likely be, our sole suppliers of our product candidates for a significant period of time. These manufacturers are commonly referred to as single source suppliers. As a result of these exclusive arrangements, some of our contracts prohibit us from using alternative manufacturers or suppliers for the products supplied under these contracts, which prevents us from diversifying manufacturing and supply sources. In addition, we currently purchase clinical supplies for many of our product candidates from third party manufacturers on a purchase order basis under short-term supply agreements. If any of these manufacturers should become unavailable to us for any reason, we may be unable to conclude agreements with replacements on favorable terms, if at all, and may be delayed in identifying and qualifying such replacements. In any event, identifying and qualifying a new third party manufacturer could involve significant costs associated with the transfer of the active pharmaceutical ingredient or finished product manufacturing process. A change in manufacturer requires formal approval by the FDA before the new manufacturer may produce commercial supplies of our products. This approval process typically takes a minimum of 12 to 18 months and, during that time we may face a shortage of supply of our products.
Reliance on third party manufacturers entails risks, to which we would not be subject if we manufactured products or product candidates ourselves, including:
29
· reliance on the third party for regulatory compliance and quality assurance;
· the possible breach of the manufacturing agreement by the third party because of factors beyond our control; and
· the possible termination or non-renewal of the agreement by the third party, based on its own business priorities, at a time that is costly or inconvenient for us.
Our products and product candidates may compete with other products and product candidates for access to manufacturing facilities. There are a limited number of manufacturers that operate under current good manufacturing practice regulations and that are both capable of manufacturing for us and willing to do so. If the third parties that we engage to manufacture and/or test a product for commercial sale or for our clinical trials should cease to continue to do so for any reason, we likely would experience delays in obtaining sufficient quantities of our products for us to meet commercial demand or in advancing our clinical trials while we identify and qualify replacement suppliers. If for any reason we are not able to obtain adequate supplies of our product candidates or the drug substances used to manufacture them, it will be more difficult for us to develop our product candidates and compete effectively.
Our current and anticipated future dependence upon others for the manufacture of our products and product candidates may adversely affect our profit margins and our ability to develop product candidates and commercialize any additional products that receive regulatory approval on a timely and competitive basis.
We rely on our third party manufacturers for compliance with applicable regulatory requirements. This may increase the risk of sanctions being imposed on us or on a manufacturer of our products or product candidates, which could result in our inability to obtain sufficient quantities of these products or product candidates.
Our manufacturers may not be able to comply with current good manufacturing practice regulations or other regulatory requirements or similar regulatory requirements outside the United States. Our manufacturers are subject to unannounced inspections by the FDA, state regulators and similar regulators outside the United States. Our failure, or the failure of our third party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including:
· fines;
· injunctions;
· civil penalties;
· failure of regulatory authorities to grant marketing approval of our product candidates;
· delays, suspension or withdrawal of approvals;
· suspension of manufacturing operations;
· license revocation;
· seizures or recalls of products or product candidates;
· operating restrictions; and
· criminal prosecutions.
Any of these sanctions could significantly and adversely affect supplies of our products and product candidates and may adversely affect our profit margins and our ability to develop product candidates and commercialize any additional products that receive regulatory approval on a timely and competitive basis.
30
We rely on third parties to conduct, supervise and monitor our clinical trials, and those third parties may perform in an unsatisfactory manner, such as by failing to meet established deadlines for the completion of such trials.
We rely on third parties such as contract research organizations, or CROs, medical institutions and clinical investigators to enroll qualified patients and conduct, supervise and monitor our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Our reliance on these third parties, however, does not relieve us of our regulatory responsibilities, including ensuring that our clinical trials are conducted in accordance with good clinical practice regulations, and the investigational plan and protocols contained in the relevant regulatory application, such as the investigational new drug application. In addition, such third parties may not complete activities on schedule, or may not conduct our preclinical studies or clinical trials in accordance with regulatory requirements or our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for, and to commercialize, our product candidates may be delayed or prevented.
We may not be able to acquire or successfully integrate new products or businesses.
Our products are maturing and therefore a significant component of our strategy is growth through acquisitions of products or businesses. However, we cannot be certain that we will be able to identify appropriate acquisition candidates. And even if an acquisition candidate is identified, there can be no assurance we will be able to successfully negotiate the terms of any such acquisition on favorable terms or at all, finance such acquisition or integrate such acquired product or business into our existing business. We face significant competition from other pharmaceutical companies for acquisition candidates, which makes it more difficult to find attractive transaction opportunities for products or companies on acceptable terms. Furthermore, the negotiation of potential acquisitions and the integration of such acquired products or businesses could divert management’s time and resources and require significant financial resources to consummate. Failure to acquire new products may diminish our rate of growth and adversely affect our competitive position.
Acquisitions that we may undertake will involve a number of inherent risks, any of which could cause us not to realize the anticipated benefits.
Acquisition transactions involve various inherent risks, such as assessing the value, strengths, weaknesses, contingent and other liabilities and potential profitability of the acquisition; the potential loss of key personnel of an acquired business; the ability to achieve identified operating and financial synergies anticipated to result from an acquisition and unanticipated changes in business, industry or general economic conditions that affect the assumptions underlying the acquisition. Any one or more of these factors could cause us not to realize the anticipated benefits from the acquisition of businesses or products.
If our products fail in clinical studies or if we fail, or encounter difficulties, in obtaining regulatory approval for our new products or new indications of existing products, we will have expended significant resources for no return.
If our products are not successful in clinical trials or if we do not obtain such regulatory approvals, we will have expended significant resources for no return. For example in 2005 our clinical trials of itopride hydrochloride (“ITAX”), a proposed treatment for functional dyspepsia, were not successful and we terminated the program. Our ongoing clinical studies might be delayed or halted or additional studies might be required, for various reasons, including if:
· our products are shown not to be effective;
· we do not comply with requirements concerning the investigational NDAs or New Drug Submissions (‘‘NDSs’’) for the protection of the rights and welfare of human subjects;
· patients experience unacceptable side effects or die during clinical trials;
31
· patients do not enroll in the studies at the rate we expect;
· a drug is modified during testing; or
· product supplies are delayed or are not sufficient to treat the patients participating in the studies.
If we cannot obtain regulatory approvals for the products we are developing or may seek to develop in the future, our rate of sales growth and competitive position will suffer. This risk is most acute for products that are currently in the development of the final formulation, such as the AGI-010 compound.
Approval might entail ongoing requirements for post-marketing studies. Even if regulatory approval is obtained, labeling and promotional activities are subject to continual scrutiny by the regulatory agencies, in particular the FDA, and, in some circumstances, the Federal Trade Commission. FDA enforcement policy prohibits the marketing of approved products for unapproved, or off-label, uses. These regulations and the FDA’s interpretation of them might impair our ability to effectively market our products.
We and our third-party manufacturers are also required to comply with the applicable FDA current Good Manufacturing Practices regulations, which include requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation. Furthermore,
manufacturing facilities must be approved by regulatory authorities before they can be used to manufacture our products, and they are subject to additional inspections. If we or any of our manufacturers or suppliers fails to comply with any of the FDA’s or other relevant foreign counterparts continuing regulations, we could be subject to sanctions, including:
· delays, warning letters and fines;
· product recalls or seizures and injunctions on sales;
· refusal to review pending applications;
· total or partial suspension of production;
· withdrawals of previously approved marketing applications, and
· civil penalties and criminal prosecutions.
In addition, identification of side effects after a drug is on the market or the occurrence of manufacturing problems could cause subsequent withdrawal of the product from the market, reformulation of the drug, additional testing or changes in labeling of the product.
Our approved products and pipeline products remain subject to ongoing regulatory requirements. If we fail to comply with these requirements, we could lose these approvals, and the sales of any such approved commercial products could be suspended.
After receipt of initial regulatory approval, each product remains subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. Furthermore, even if we receive regulatory approval to market a particular product, such product will remain subject to the same extensive regulatory requirements. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, which could reduce our revenues, increase our expenses and render the approved product not commercially viable. In addition, as clinical experience with a drug expands after approval because it is typically used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials or other studies.
32
Our business could suffer as a result of adverse drug reactions to the products we market.
Unexpected adverse drug reactions by patients to any of our products could negatively impact utilization or market availability of our product.
Absence of long-term safety data may also limit the approved uses of our products, if any. If we fail to comply with the regulatory requirements of the FDA and other applicable regulatory authorities, or if
previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including:
· restrictions on the products, manufacturers or manufacturing processes;
· warning letters and untitled letters;
· civil penalties and criminal prosecutions and penalties;
· fines;
· injunctions;
· product seizures or detentions;
· import or export bans or restrictions;
· voluntary or mandatory product recalls and related publicity requirements;
· suspension or withdrawal of regulatory approvals;
· total or partial suspension of production; and
· refusal to approve pending applications for marketing approval of new products or of supplements to approved applications.
We may find it difficult to achieve market acceptance of products in our product pipeline.
The commercial success of any of our product candidates for which we obtain marketing approval from the FDA, Health Canada’s Therapeutic Products Directorate (“TPD”) or other regulatory authorities will depend upon the acceptance of these products by the medical community, including physicians, patients and healthcare payors. The degree of market acceptance of any of our approved products will depend on a number of factors, including:
· demonstration of clinical safety and efficacy compared to other products;
· the relative convenience and ease of administration;
· the prevalence and severity of any adverse side effects;
· limitations or warnings contained in a product’s FDA or TPD approved labeling;
· availability of alternative treatments for the indications we target;
· pricing and cost effectiveness compared to competing products, particularly generic products;
· the effectiveness of our or any future collaborators’ sales and marketing strategies;
· the effectiveness of our manufacturing and distribution plans;
· our ability to obtain sufficient third-party coverage or reimbursement; and
· the willingness of patients to pay out-of-pocket in the absence of third-party coverage.
33
If any of our product candidates are approved but do not achieve an adequate level of acceptance by physicians, healthcare payors and patients, we may not generate sufficient revenue from these products, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
The publication of negative results of studies or clinical trials may adversely impact our sales revenue.
From time to time, studies or clinical trials on various aspects of pharmaceutical products are conducted by academics or others, including government agencies. The results of these studies or trials, when published, may have a dramatic effect on the market for the pharmaceutical product that is the subject of the study. The publication of negative results of studies - or clinical trials related to our products or the therapeutic areas in which our products compete - - could adversely affect our sales, the prescription trends for our products and the reputation of our products. In the event of the publication of negative results of studies or clinical trials related to our products or the therapeutic areas in which our products compete, our business, financial condition, results of operation and cash flows could be materially adversely affected.
There is no assurance that we will continue to be successful in our licensing and marketing operations.
Certain of our products are marketed by third parties. Such third-party arrangements may not be successfully negotiated in the future. Any such arrangements may not be available on commercially reasonable terms. Even if acceptable and timely marketing arrangements are available, the products we develop may not be accepted in the marketplace, and even if such products are initially accepted, sales
may thereafter decline. Additionally, our clients or marketing partners may make important marketing and other commercialization decisions with respect to products we develop that are not within our control. As a result, many of the variables that may affect our revenues, cash flows and net income are not exclusively within our control.
The success of our strategic investments, partnerships or development alliances (like our co-development of AGI-010 or Cx401) depends upon the performance of the companies in which we invest, or with which we partner or co-develop products.
Economic, governmental, industry and internal company factors outside our control affect each of the companies in which we may invest or with which we may partner or co-develop products, like AGI Therapeutics Research Ltd. or Cellerix S.L. Some of the material risks relating to such companies include:
· the ability of these companies to successfully develop, manufacture and obtain necessary governmental approvals for the products which serve as the basis for our investments in, or relationship with, such companies;
· the ability of competitors of these companies to develop similar or more effective products, making the drugs developed by these companies difficult or impossible to market;
· the ability of these companies to adequately secure patents for their products and protect their proprietary information;
· the ability of these companies to enter the marketplace without infringing upon competitors’ patents;
· the ability of these companies to remain technologically competitive; and
· the dependence of these companies upon key scientific and managerial personnel.
We may have limited or no control over the resources that any such company may devote to develop the products for which we collaborate with them. Any such company may not perform as expected. These
34
companies may breach or terminate their agreements with us or otherwise fail to conduct product discovery and development activities successfully, or in a timely manner. If any of these events occurs, it could have a material adverse effect on our business and our financial results.
The pharmaceutical industry is highly competitive and is subject to rapid and significant change, which could render our products obsolete or uncompetitive.
Our products face competition from other pharmaceutical and generic companies. We compete with companies in North America and internationally, including major pharmaceutical and generic companies. Many of our competitors have greater financial resources and selling and marketing capabilities, have greater experience in clinical testing and human clinical trials of pharmaceutical products, and have greater experience in obtaining approvals from the FDA, the TPD (the Canadian federal authority that regulates pharmaceutical drugs and medical devices for human use), or other applicable regulatory approvals. Our competitors may succeed in developing products that are more effective or less expensive to use than any that we may develop or license. These developments could render our products obsolete or uncompetitive, which would have a material adverse effect on our business and financial results.
We may not be able to obtain adequate third-party reimbursement for the cost of our products and related treatment.
Our ability to successfully commercialize our products and product candidates - even if FDA or TPD approval is obtained - depends, in part, on whether appropriate reimbursement levels for the cost of the products and related treatments are obtained from government authorities and private health insurers and other organizations, such as Health Maintenance Organizations (‘‘HMOs’’), Managed-Care Organizations (‘‘MCOs’’) and provincial formularies.
Third-party payors increasingly challenge the pricing of pharmaceutical products. In addition, the trend toward managed health-care in the U.S., the growth of organizations such as HMOs and MCOs, and legislative proposals to reform health-care and government insurance programs, could significantly influence the purchase of pharmaceutical products, resulting in lower prices and a reduction in product demand. Such cost-containment measures and health-care reform could affect our ability to sell our products and may have a material adverse effect on our business, financial condition, cash flows and results of operations.
Uncertainty also exists regarding the reimbursement status of certain newly-approved pharmaceutical products and reimbursement may not be available for some of our products. Any reimbursements granted may not be maintained or limits on reimbursements available from third-party payors may reduce the demand for, or negatively affect the price of, these products. If our products do not qualify for reimbursement, if reimbursement levels diminish, or if reimbursement is denied, our sales and profitability would be adversely affected.
Our relationships with customers and payors are subject to applicable fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, and diminished profits and future earnings.
Healthcare providers, physicians and others play a primary role in the recommendation and prescription of our products. Our arrangements with third party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products. Applicable U.S. federal and state healthcare laws and regulations, include, but are not limited to, the following:
· The federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal healthcare programs such as Medicare and Medicaid.
35
· The federal False Claims Act imposes criminal and civil penalties, including civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government.
· The federal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services.
· Analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third party payors, including private insurers, and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government.
· There are similar laws in countries outside the United States.
Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations could be costly. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our past or present operations, including activities conducted by our sales team or agents, are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, exclusion from third party payor programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we do business are found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Many aspects of these laws have not been definitively interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of subjective interpretations, which increases the risk of potential violations. In addition, these laws and their interpretations are subject to change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
Recently enacted legislation may make it more difficult and costly for us to obtain regulatory approval of our product candidates and to produce, market and distribute our existing products.
On September 27, 2007, President Bush signed the Food and Drug Administration Amendments Act of 2007 (the “FDAAA”), into law. The FDAAA grants a variety of new powers to the FDA, many of which are aimed at improving drug safety and assuring the safety of drug products after approval. Under the FDAAA, companies that violate the new law are subject to substantial civil monetary penalties. While we expect the FDAAA to have a substantial effect on the pharmaceutical industry, the extent of that effect is not yet known. As the FDA issues regulations, guidance and interpretations relating to the new legislation, the impact on the industry, as well as our business, will become clearer. The new requirements and other changes that the FDAAA imposes may make it more difficult, and likely more costly, to obtain approval of new pharmaceutical products and to produce, market and distribute existing products.
Changes in laws and regulations could affect our results of operations, financial position or cash flows.
Our future operating results, financial position or cash flows could be adversely affected by changes in laws and regulations such as (i) changes in the FDA or TPD (or their foreign equivalents) approval processes that may cause delays in, or prevent the approval of, new products, (ii) new laws, regulations
36
and judicial decisions affecting product marketing, promotion or the healthcare field generally, (iii) new laws or judicial decisions affecting intellectual property rights and (iv) changes in the application of tax principles, including tax rates, new tax laws, or revised interpretations of existing tax laws and precedents, which result in shift of taxable earnings between tax jurisdictions.
We may be subject to investigations or other inquiries concerning our compliance with reporting obligations under federal healthcare program pharmaceutical pricing requirements.
Under federal healthcare programs, some state governments and private payors investigate and have filed civil actions against numerous pharmaceutical companies alleging that the reporting of prices for pharmaceutical products has resulted in false and overstated Average Wholesale Price, or AWP, which in turn may be alleged to have improperly inflated the reimbursements paid by Medicare, private insurers, state Medicaid programs, medical plans and others to healthcare providers who prescribed and administered those products or pharmacies that dispensed those products. These same payors may allege that companies do not properly report their “best prices” to the state under the Medicaid program. Suppliers of outpatient pharmaceuticals to the Medicaid program are also subject to price rebate agreements. Failure to comply with these price rebate agreements may lead to federal or state investigations, criminal or civil liability, exclusion from federal healthcare programs, contractual damages, and otherwise harm our reputation, business and prospects.
Pricing pressures from third-party payors, including managed care organizations, government sponsored health systems and regulations relating to Medicare and Medicaid, healthcare reform, pharmaceutical reimbursement and pricing in general could decrease our U.S. revenues.
Our commercial success in producing, marketing and selling products in the United States (which generates a majority of our revenues) depends, in part, on the availability of adequate reimbursement from third-party healthcare payors, such as managed care organizations, and government bodies and agencies for the cost of the products and related treatment. The market for our products in the United States may be limited by actions of third-party payors.
Managed care organizations and other third-party payors in the United States try to negotiate the pricing of medical services and products to control their costs, including by developing formularies to encourage plan beneficiaries to utilize preferred products for which the plans have negotiated favorable terms. Exclusion of a product from a formulary, or placement of a product on a disfavored formulary tier, can lead to sharply reduced usage in the managed care organization patient population. If our products are not included within an adequate number of formularies or if adequate reimbursement levels are not provided, or if reimbursement policies increasingly favor generic products, our market share and business could be negatively affected.
Recent reforms in Medicare added an out-patient prescription drug reimbursement beginning 2006 for all Medicare beneficiaries. The U.S. federal government and private plans contracting with the government to deliver the benefit, through their purchasing power under these programs, are demanding discounts from pharmaceutical companies that may implicitly create price controls on prescription drugs. These reforms may decrease our future revenues from products such as CARAFATE, URSO, ULTRASE and CARAFATE that are covered by the Medicare drug benefit. Further, a number of other legislative and regulatory proposals aimed at changing the healthcare system have been proposed. While we cannot predict whether any such proposals will be adopted or the effect such proposals may have on our business, the existence of such proposals, as well as the adoption of any proposal, may increase industry-wide pricing pressures, thereby adversely affecting our results or operations and cash flows.
Future litigation and of the outcome of current litigation may harm our business.
In general, and subject to the terms of each specific agreement, we have agreed to indemnify our licensors and certain of our contract manufacturers for product liability claims and there is a risk that we will be subject to product liability claims and claims for indemnification from licensors. A substantial portion of our revenues are derived and will continue to be derived from activities in the United States, where pharmaceutical companies are exposed to a higher risk of litigation than in other jurisdictions.
37
Currently in the United States, we maintain claims-based product liability insurance coverage, in respect of all our products marketed in the United States. We cannot be certain that existing or future insurance coverage available to us will be adequate to satisfy any or all future product liability claims and defense costs in the United States.
Exposure relating to product liability claims may harm our business.
We face an inherent business risk of exposure to product liability and other claims in the event that the use of our products results, or is alleged to have resulted, in adverse effects. While we have taken, and will continue to take, what we believe are appropriate precautions, there can be no assurance that we will avoid significant product liability claims. Although we currently carry product liability insurance that we believe is appropriate for the risks that we face, there can be no assurance that we have sufficient coverage, or can in the future obtain sufficient coverage at a reasonable cost. An inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the growth of our business or the number of products we can successfully market. Our obligation to pay indemnities, the withdrawal of a product following complaints, or a product-liability claim could have a material adverse effect on our business, results of operations, cash flows and financial condition.
Our business is subject to limitations imposed by government regulations.
Governmental agencies in the countries in which we conduct business regulate pharmaceutical products intended for human use. These regulations normally require extensive clinical trials and other testing in addition to governmental review and final approval before products can be marketed. Governmental authorities in such countries also regulate the research and development, manufacture, distribution, promotion, testing and safety of products, and therefore, the cost of complying with governmental regulations can be substantial.
Requirements for approval can vary widely from country to country. A product must normally be approved by regulatory authorities in each country in which we intend to market it, prior to the commencement of marketing in such country. There can be long delays in obtaining required clearances from regulatory authorities in many countries after applications are filed. Therefore, there can be no assurance that we will obtain regulatory approvals in such countries or that we will not incur significant costs in obtaining or maintaining such regulatory approvals. Moreover, the regulations applicable to our existing and future products may change.
Government regulations require the detailed inspection and control of research and laboratory procedures, clinical studies, manufacturing procedures and marketing and distribution methods, all of which significantly increase the level of difficulty and the costs involved in obtaining and maintaining the regulatory approval for marketing new and existing products. Moreover, regulatory measures adopted by governments provide for the possible withdrawal of products from the market and in certain cases, suspension or revocation of the required approvals for their production and sale.
Failure to obtain necessary regulatory approvals; the restriction, suspension or revocation of existing approvals; or any other failure to comply with regulatory requirements would restrict or impair our ability to market our products and carry on our business.
We rely on the intellectual property of others and may not be able to protect our own intellectual property.
Our continued success will depend, in part, on our ability to obtain, protect and maintain intellectual property rights and licensing arrangements for our products. Proprietary rights in some of our products are held by third parties, which require us to obtain licenses for the use of such products. Despite these licenses there can be no guarantees that the rights or patents used by us will not be challenged by third parties. Third party patents may cover the materials of methods of treatment related to our products and third parties may make allegations of infringement, regardless of merit.
38
To protect our own intellectual property, we have historically relied on patents and trade secrets, know-how and other proprietary information, as well as requiring our employees and other vendors and suppliers to sign confidentiality agreements which prohibit them from taking our proprietary information and technology or from using or disclosing proprietary information to third parties except in specified circumstances. However, confidentiality agreements may be breached despite all precautions taken, and we may not have adequate remedies for any breach. Some of our employees may have been employed by other pharmaceutical or biotechnology companies who may allege violations of their trade secrets by these individuals, irrespective of the steps that we may take. Third parties may gain access to our proprietary information or may independently develop substantially equivalent proprietary information.
Our inability to protect and maintain intellectual property rights in our products may impair our competitive position and adversely affect our growth. If a lawsuit is commenced with respect to any alleged intellectual property infringement by us, the uncertainties inherent in such litigation make the outcome difficult to predict and the costs that we may incur as a result may have an adverse effect on our profitability. Such litigation would involve significant expense and would be a substantial diversion of the efforts of our scientific and our management team. Any such lawsuit may result in the award of monetary damages to the intellectual property holder and payment by us of any such damages, or an injunction prohibiting all of our business activities that infringe the intellectual property or may require us to obtain licenses from third parties on terms that may not be commercially acceptable to us, which would in each case adversely affect our profitability and our business. We or our collaborators may be restricted or prevented from developing and commercializing our products if judicial or administrative proceedings against us or our collaborators results in an adverse determination. If so, we may attempt to redesign our processes, products or technologies so that they do not infringe, but that might not be possible.
If we need to initiate proceedings to enforce our proprietary rights against infringement by third parties, this could involve significant expense and divert substantial employee resources from our business, with no guarantee of success. If we fail to effectively enforce our proprietary rights against others, our business will be harmed.
Our credit agreement imposes certain restrictions on us and on our operations.
Our credit agreement requires us to maintain specified financial ratios. The credit agreement also contains customary covenants relating to our ability to incur additional indebtedness, make future acquisitions, enter into certain related party transactions, consummate asset dispositions, incur capital expenditures and make restricted payments. All of these restrictions may limit our ability to expand, pursue our business strategies and obtain additional funds. Our ability to meet these financial ratios and to comply with these covenants may be affected by changes in business conditions or results of operations, adverse regulatory developments and other events beyond our control. We cannot assure that we will meet these financial ratios or continue to comply with these covenants. Failure to comply with these restrictions may result in the occurrence of an event of default under the credit agreement. Upon the occurrence of an event of default, the lenders may terminate the credit agreement and demand immediate payment of all amounts borrowed by us under that agreement.
Additional financing will cause increased dilution, an increase in debt levels, or both.
The research, development, manufacturing and marketing of products and the acquisition of new products and companies requires the application of considerable financial resources, while the revenues that are generated from such products may not be realized for a number of years. If we require additional capital to fund such activities, we may seek additional debt or equity financing or both. If we increase our debt levels, we may be restricted in our ability to raise additional capital and may be subject to additional financial and restrictive covenants, which may impede our ability to grow or carry on our business. If we issue additional equity, holders of common shares will suffer dilution of their ownership interest, which could be substantial. There can be no assurance that we will be successful in securing such financing.
39
Our common shares are subject to market price volatility.
Market prices for the securities of pharmaceutical companies, including our common shares, have historically been highly volatile, and from time to time, the market has experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. In addition, factors such as fluctuations in operating results which do not correspond to market expectations, future acquisitions, public announcements, concerns as to the safety of drugs, timing and approvals of product candidates, and general market conditions may have an adverse effect on the market price of our common shares.
We might be impacted by unfavorable decisions in any possible procedures related to any future tax assessment.
We operate in a number of jurisdictions and are from time to time subject to audits and reviews by taxation authorities in the relevant jurisdictions with respect to income, consumption, payroll and other taxes and remittances, for current as well as past filings. Accordingly, we are subject to future tax assessments by relevant authorities. Any future tax assessment or any amount we might be required to pay in connection with an assessment may have a material adverse effect on our financial position, cash flows or overall in results of operations. There is the possibility of a material adverse impact on the results of operations of the period in which the matter is ultimately resolved, if it is resolved unfavorably, or in the period in which an unfavorable outcome becomes probable and reasonably estimable.
We may not have sufficient cash and may be limited in our ability to access financing for future capital requirements, which may prevent us from expanding our business and our portfolio of products.
We may in the future need to incur additional debt or issue equity to satisfy working capital and capital expenditure requirements, as well as to make acquisitions and other investments. To the extent we are unable to renew our existing credit facility or raise new capital, we may be unable to expand our business. If we raise funds through the issuance of debt or equity, any debt securities or preferred shares issued will have rights and preferences and privileges senior to those of holders of our common shares. The terms of the debt securities may impose restrictions on our operations that have an adverse impact on our financial condition. If we raise funds through the issuance of equity, the proportional ownership interests of our shareholders could be diluted.
We are exposed to risks relating to foreign currencies.
We operate internationally, but a majority of our revenue and expense activities and capital expenditures are denominated in U.S. dollars. The other currencies in which we engage in significant transactions are Canadian dollars and Euros. We face foreign currency exposure on the translation of our operations in Canada and Europe from their local currencies to the U.S. dollar. Currently, we do not utilize forward contracts to hedge against foreign currency risk on an ongoing basis. A significant change in foreign currency exchange rates may have a material effect on our consolidated results of operations, financial position or cash flows.
40
We are exposed to risks if we are unable to comply with changes to laws affecting public companies, including the Sarbanes-Oxley Act of 2002, and also to increased costs associated with complying with such laws.
Recently enacted and any future changes to the laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002 in the U.S. and Part XXIII.1 of the Securities Act (Ontario) and related rules, will cause us to incur increased costs as we evaluate the implications of new rules and respond to new requirements. Delays, or a failure to comply with the new laws, rules and regulations could result in enforcement actions, the assessment of other penalties and civil suits. The new laws and regulations make it more expensive for us under indemnities provided by us to our officers and directors and may make it more difficult for us to obtain certain types of insurance, including liability insurance for directors and officers; as such, we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our Board of Directors, or as executive officers. We may be required to hire additional personnel and utilize additional outside legal, accounting and advisory services — all of which could cause our general and administrative costs to increase beyond what we currently have planned. We are presently evaluating and monitoring developments with respect to these laws, rules and regulations, and we cannot predict or estimate the amount of the additional costs we may incur or the timing of such costs.
Rising insurance costs could adversely impact our business.
The cost of insurance — including insurance for directors and officers, worker’s compensation, property, product-liability and general liability insurance — rose significantly in the past year and may continue to increase in future years. In response, we may increase deductibles and/or decrease certain coverages to mitigate these costs. These increases, and our increased risk due to increased deductibles and reduced coverages, could have an adverse impact on our results of operations, financial condition and cash flows.
Our operations could be disrupted if our information systems fail or if we are unsuccessful in implementing necessary upgrades.
Our business depends on the efficient and uninterrupted operation of our computer and communications systems and networks, hardware and software systems and our other information technology. If our systems were to fail or we are unable to successfully expand the capacity of these systems, or we are unable to integrate new technologies into our existing systems, our operations and financial results could suffer.
41
PART VI - SELECTED CONSOLIDATED FINANCIAL INFORMATION
(in thousands of U.S. dollars, except share related data)
Last three fiscal years (audited)
| | Fiscal Year, Ended September 30 | |
| | 2007 | | 2006 | | 2005 | |
| | $ | | $ | | $ | |
Statement of Operations Data: U.S. GAAP | | | | | | | |
Revenue | | 348,947 | | 292,317 | | 251,343 | |
Cost of goods sold | | 83,683 | | 72,772 | | 71,534 | |
Selling and administrative expenses | | 101,273 | | 93,338 | | 85,997 | |
Research and development expenses | | 28,655 | | 39,789 | | 31,855 | |
Depreciation and amortization | | 22,494 | | 22,823 | | 21,532 | |
Acquired in-process research | | 10,000 | | — | | — | |
Partial write-down of intangible assets | | — | | 5,800 | | — | |
| | 246,105 | | 234,522 | | 210,918 | |
Operating income | | 102,842 | | 57,795 | | 40,425 | |
| | | | | | | |
Financial expenses | | 4,825 | | 6,988 | | 7,140 | |
Interest income | | (11,367 | ) | (5,468 | ) | (1,340 | ) |
Loss (gain) on foreign currency | | 2,352 | | (1,110 | ) | (213 | ) |
| | (4,190 | ) | 410 | | 5,587 | |
Income before income taxes | | 107,032 | | 57,385 | | 34,838 | |
Income taxes | | 35,567 | | 18,266 | | 8,413 | |
| | | | | | | |
Net income | | 71,465 | | 39,119 | | 26,425 | |
42
| | Fiscal Year, Ended September 30, | |
| | 2007 | | 2006 | | 2005 | |
| | $ | | $ | | $ | |
U.S. GAAP | | | | | | | |
Net income | | 71,465 | | 39,119 | | 26,425 | |
Income Per Common Share(1) | | | | | | | |
Basic | | 1.47 | | 0.86 | | 0.58 | |
Diluted | | 1.33 | | 0.79 | | 0.56 | |
| | | | | | | |
Balance Sheet Data: U.S. GAAP | | | | | | | |
Total assets | | 832,611 | | 695,817 | | 641,407 | |
Total liabilities | | 142,414 | | 228,393 | | 223,803 | |
Total shareholders’ equity | | 690,197 | | 467,424 | | 417,604 | |
(1) Based on the weighted average number of shares outstanding during the year.
PART VII - MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND OPERATING RESULTS
Please refer to the 2007 MD&A that is incorporated herein by reference.
PART VIII - DIVIDEND POLICY
Axcan currently intends to retain future earnings to finance the development of its business and, accordingly, does not anticipate paying dividends on its common shares in the near future. In addition, the terms of Axcan’s existing credit facilities restrict its ability to declare and pay dividends to 10% of consolidated net earnings for the preceding fiscal year.
43
PART IX — DESCRIPTION OF CAPITAL STRUCTURE
The authorized share capital of the Company consists of an unlimited number of Common Shares, 14,175,000 Series A preferred shares and 12,000,000 Series B preferred shares. None of such Preferred Shares are issued and outstanding.
1. COMMON SHARES
The common shares are entitled to receive dividends as and when declared by the Board of Directors and, unless otherwise provided by legislation, are entitled to one vote per common share on all matters to be voted on at all meetings of shareholders. Upon the voluntary or involuntary liquidation, dissolution or winding-up of the Company, the holders of Common Shares are entitled to share rateably in the remaining assets available for distribution, after payment of liabilities.
2. PREFERRED SHARES
The preferred shares are issuable from time to time in one or more series. The Board of Directors is authorized to fix before issue the number of, the consideration per share of, the designation of, and the provisions attaching to, the shares of each series. The preferred shares of each series rank on a parity with the preferred shares of every other series and are entitled to preference over the common shares and any other shares ranking junior to the preferred shares with respect to the payment of dividends and distribution of assets in the event of liquidation, dissolution or winding up of the Company. If any cumulative dividends or amounts payable on a return of capital are not paid in full, the preferred shares of all series participate rateably in accordance with the amounts that would be payable on such shares on the return of capital if all amounts so payable were paid in full, as the case may be.
44
PART X - MARKET FOR SECURITIES
1. MARKET FOR SECURITIES
Since December 27, 1995, Axcan’s common shares are listed for trading on the Toronto Stock Exchange (“TSX”) under the trading symbol “AXP” and, since June 28, 2000, on the NASDAQ Global Select MarketSM (“NASDAQ”) under the trading symbol “AXCA”.
2. TRADING HISTORY
The following table set forth the price ranges and volume of Axcan’s common shares traded on the TSX and on NASDAQ for each month of fiscal 2007.
TORONTO STOCK EXCHANGE
Period | | High (CAN) | | Low (CAN) | | Close (CAN) | | Volume | |
| | | | | | | | | |
October 2006 | | 16.71 | | 15.14 | | 16.55 | | 3,174,555 | |
November 2006 | | 17.49 | | 16.45 | | 16.93 | | 2,071,133 | |
December 2006 | | 17.06 | | 15.70 | | 16.58 | | 2,728,610 | |
January 2007 | | 18.02 | | 15.88 | | 17.89 | | 3,313,388 | |
February 2007 | | 19.58 | | 17.60 | | 18.56 | | 2,309,743 | |
March 2007 | | 16.45 | | 17.79 | | 19.13 | | 2,835,541 | |
April 2007 | | 16.69 | | 18.80 | | 19.30 | | 1,188,152 | |
May 2007 | | 20.26 | | 18.92 | | 20.15 | | 1,264,403 | |
June 2007 | | 21.50 | | 19.28 | | 20.65 | | 1,971,137 | |
July 2007 | | 21.25 | | 19.50 | | 19.50 | | 2,059,603 | |
August 2007 | | 21.00 | | 15.65 | | 20.34 | | 3,517,765 | |
September 2007 | | 21.16 | | 19.60 | | 20.55 | | 2,801,995 | |
NASDAQ Global Market
Period
(Fiscal 2007) | | High
(USD) | | Low
(USD) | | Close
(USD) | | Volume | |
| | | | | | | | | |
October 2006 | | 14.86 | | 13.49 | | 14.72 | | 3,093,459 | |
November 2006 | | 15.37 | | 14.47 | | 14.85 | | 3,242,832 | |
December 2006 | | 14.84 | | 13.61 | | 14.24 | | 2,455,170 | |
January 2007 | | 15.28 | | 13.50 | | 15.19 | | 2,894,208 | |
February 2007 | | 16.50 | | 14.92 | | 15.92 | | 6,804,209 | |
March 2007 | | 16.77 | | 15.14 | | 16.51 | | 6,924,702 | |
April 2007 | | 17.61 | | 16.27 | | 17.41 | | 4,306,477 | |
May 2007 | | 18.86 | | 17.02 | | 18.79 | | 6,609,755 | |
June 2007 | | 20.06 | | 18.19 | | 19.33 | | 5,912,007 | |
July 2007 | | 20.36 | | 18.31 | | 18.31 | | 4,818,889 | |
August 2007 | | 19.83 | | 14.85 | | 19.08 | | 11,170,319 | |
September 2007 | | 20.94 | | 19.02 | | 20.77 | | 4,281,551 | |
45
PART XI - DIRECTORS AND OFFICERS
1. DIRECTORS
Each director of the Company is elected to serve until the next annual meeting of the Company or until his or her successor is elected or appointed. The disclosure under the heading “Election of Directors” in the Company’s Management Proxy Circular dated January 22, 2007 contains information about each director of the Company and is incorporated herein by reference.
2. SENIOR OFFICERS
The company’s Management Team currently comprises the following individuals:
The following tables set forth the name, municipality of residence, and principal position of each of Axcan’s senior officers:
Name | | Position with Axcan | | Province or State and/or
Country of Residence |
| | | | |
Frank A.G.M. Verwiel | | President and Chief Executive Officer | | Quebec, Canada |
David W. Mims | | Executive Vice President and Chief Operating Officer | | Alabama, USA |
Nicholas Franco | | Senior Vice President, International Commercial Operations | | France |
Steve Gannon | | Senior Vice President and Chief Financial Officer | | Quebec, Canada |
Martha D. Donze | | Vice President, Corporate Administration | | Alabama, USA |
Richard Tarte | | Vice President, Corporate Development and General Counsel | | Quebec, Canada |
Michael E. Thiel | | Vice President, North American Marketing Operations | | New Jersey, USA |
Darcy Toms | | Vice President, Business Development | | New Jersey, USA |
Jean Vézina | | Vice President, Finance | | Quebec, Canada |
During the past five years, each of the foregoing directors and officers has held the same or a similar position with the entities indicated above or a related entity, except for (1) Dr. Frank Verwiel, who prior to July 2005 held the position of vice president, Hypertension, Worldwide Human Health Marketing with Merck & Co., Inc., while concurrently serving as a member of Merck’s Worldwide Hypertension Business Strategy Team. Prior to joining Merck, from 1988 to 1996, Dr. Verwiel worked with Servier in Europe in various executive positions; (2) Mr. Nicholas Franco who, prior to joining Axcan in June 2007, was Head of Market Access Region Europe for Novartis Pharma AG in Basel, Switzerland, a company where he has held various management positions since 1991; (3) Mr. Steve Gannon who, prior to February 2006 held the position of Chief Financial Officer at CryoCath Technologies Inc.; and; (4) Mr. Darcy Toms who, prior to January 2007, was Senior Director, Business Development with Biovail Pharmaceuticals and prior to 2005 held various management positions with Aventis Inc.
PART XII - ADDITIONAL DISCLOSURE FOR DIRECTORS AND EXECUTIVE OFFICERS
As at December 1, 2007, the directors and senior officers of Axcan beneficially own as a group an aggregate of 3,775,276 common shares, which represents approximately 6.82% of the issued and outstanding shares of Axcan.
No director or officer of Axcan or shareholder of Axcan holding a sufficient number of securities of Axcan to affect materially the control of Axcan (a “control person”); is, or has been within the past ten years, a
46
director or officer of any other company that, while such person was acting in that capacity; was the subject of a cease trade or similar order or an order that denied such company access to any exemptions under Canadian securities legislation for a period of more than 30 consecutive days; or was declared bankrupt or made a voluntary assignment in bankruptcy; made a proposal under any legislation relating to bankruptcy or insolvency; or was subject to or instituted any proceedings, arrangement or compromise with creditors or had a receiver, receiver manager or trustee appointed to hold its assets.
No director, officer or control person of Axcan has been subject to any penalties or sanctions imposed by a court relating to Canadian securities legislation or by a Canadian securities regulatory authority or has entered into a settlement agreement with a Canadian securities regulatory authority; nor has any director, officer or control person of Axcan been subject to any other penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable investor in making an investment decision.
No director, officer or control person of Axcan, nor any personal holding company of any such person, has within the past ten years, been declared bankrupt or made a voluntary assignment in bankruptcy; made a proposal under any legislation relating to bankruptcy or insolvency; been subject to or instituted any proceedings, arrangement or compromise with creditors; or had a receiver, receiver manager or trustee appointed to hold the assets of that individual.
Conflicts of interest may arise as a result of the directors and officers of Axcan also holding positions as directors and/or officers of other companies. Some of the directors and officers have been and will continue to be engaged in the identification and evaluation of assets and businesses, with a view to potential acquisition of interests in businesses and companies on their own behalf and on behalf of other companies, and situations may arise where the directors and officers will be in direct competition with Axcan. Conflicts, if any, will be subject to the procedures and remedies under the Canada Business Corporations Act.
47
PART XIII - LEGAL PROCEEDINGS
One of our subsidiaries, Axcan Scandipharm, has been a party to several legal proceedings related to the product line it markets under the trademark ULTRASE. These lawsuits were filed and claims were asserted against Axcan Scandipharm and certain other companies, including the product’s manufacturer, stemming from allegations that, among other things, Axcan Scandipharm’s enzyme products caused colonic strictures. In November 2006, a complaint alleging a claim for damages related to these products was received in the Supreme Court of the Sate of New York. This complaint is in its initial preliminary stage such that the Company is not currently able to assess its merit. Of the 12 other previous lawsuits, the last of which was filed in 2001, Axcan Scandipharm was dismissed from one, non-suited in another and settled ten. At this time, it is difficult to predict the number of potential cases and because of the young age of the patients involved, Axcan Scandipharm’s product liability exposure for this issue in the United States will remain for a number of years. Axcan Scandipharm’s insurance carriers have defended the lawsuits to date and Axcan expects them to continue to defend Axcan Scandipharm (to the extent of its product liability insurance) should other lawsuits be filed in the future.
In December 2003 and May 2004, Pharmascience Inc. (“PMS”) served the Company two notices of allegation in accordance with Section 5 of the Patented Medicines (Notice of Compliance) Regulations (‘‘Regulations’’). The first notice of allegation indicated that PMS was seeking a regulatory approval to market a generic version of URSO 250 in the Canadian market (‘‘PMS Product’’) for use in gallstone dissolution, an indication for which URSO 250 was formerly approved for and marketed in Canada. The second notice of allegation dealt with the validity of Axcan’s Canadian PBC use patent. Axcan opposed both these notices of allegation by seeking prohibition orders. In September 2005, with respect to the first notice of allegation, the Federal Court of Canada held that, for purposes of the Regulations, the PMS Product did not infringe the patent that Axcan owns for the use of URSO in the treatment of PBC. In May 2006, the second application seeking a prohibition order was dismissed as Axcan’s Canadian PBC use patent was held to be invalid for purposes of the Regulations. PMS has more recently filed a claim against Axcan pursuant to section 8 of the Regulations seeking recovery of alleged damages sustained by reason of the delay to commercialize its product sustained by reason of Axcan’s opposition.
In June 2007, the Company initiated litigation against KV Pharmaceutical Company, Ethex Corporation (a wholly owned subsidiary of KV), Impax Laboratories, Inc. and Global Pharmaceuticals (the generic marketing division of Impax) related to the improper substitution of their products (Pangestyme UL and Lipram UL, respectively) for ULTRASE MT. The Company alleges that the defendants falsely advertised and promoted their products as generic equivalents to and substitutes for ULTRASE MT in order to encourage pharmacists to dispense their products to patients as generic substitutes for ULTRASE MT. Axcan seeks injunctive relief to stop the defendants from continued false advertising and marketing as well as damages resulting from the defendants’ unlawful conduct.
Axcan is involved in other routine litigation matters that Axcan believes not to be material.
PART XIV — TRANSFER AGENTS AND REGISTRARS
The transfer agent and registrar for Axcan’s common shares on both the TSX and the NASDAQ is Computershare Trust Company of Canada, 1500 University Street, Suite 700, Montreal, Quebec Canada H3A 3S8. The Company’s register is held in Montreal, Quebec, Canada.
48
PART XV - MATERIAL CONTRACTS
The following are the only material contracts, other than contracts entered into in the ordinary course of business, which have been entered into by Axcan during the last financial year or that are still in effect:
(a) The exclusive license and development agreement with Cellerix;
(b) The exclusive license and supply agreement with Eurand International S.p.A, as amended;
(c) The licensing agreement with Exomed Australia Limited;
(d) The license and co-development agreement with AGI Therapeutics Research Ltd; and
(e) The amended and restated credit Agreement with National Bank of Canada dated September 22, 2004, as amended.
PART XVI - INTERESTS OF EXPERTS
The Company’s auditors are Raymond Chabot Grant Thornton LLP located at 600, de La Gauchetière Street West, Office 1900, Montreal, Quebec H3B 4L8. The auditors have confirmed to the Company that they are independent in accordance with the applicable rules of professional conduct of each of the provinces of Canada and that they have complied with the rules on auditor independence of the United States Securities and Exchange Commission.
PART XVII - ADDITIONAL INFORMATION
Additional information about Axcan is available on the Company’s web site at www.axcan.com, on SEDAR (System for Electronic Document Analysis and Retrieval) at www.sedar.com, and on the U.S. Securities and Exchange web site at www.sec.gov.
Additional information, including directors’ and executive officers’ remuneration and indebtedness, principal holders of the Company’s securities and securities authorized for issuance under equity compensation plans is contained in the Management Proxy Circular dated as of January 22, 2007 prepared in connection with the Company’s Annual Meeting of Shareholders which was held on February 28, 2007.
Additional financial information is provided in the 2007 Financial Statements and the 2007 MD&A for its fiscal year ended September 30, 2007.
Copies of this AIF, as well as copies of the 2007 Financial Statements and the 2007 MD&A of Axcan for the year ended September 30, 2007 and the Management Proxy Circular dated January 22, 2007 are available on SEDAR at www.sedar.com, and may be obtained from:
Isabelle Adjahi
Senior Director, Investor Relations and Communications
Axcan Pharma Inc.
597, Laurier Blvd.
Mont-Saint-Hilaire, QC J3H 6C4
Canada
Tel: (450) 467-2600 or 1 (877) 982-2600 ext. 2000
Fax: (450) 464-9979
E-mail: iadjahi@axcan.com
The names AXCAN, AXCAN PHARMA, BENTYL, BENTYLOL, CANASA, CARAFATE, DELURSAN, FLUTTER, HELIZIDE, HEPENAX, ITAX, LACTEOL, MODULON, PHOTOBARR, PHOTOFRIN, PANZYTRAT, PROCTOSEDYL, PYLERA, SALOFALK, SCANDICAL, SCANDISHAKE, SULCRATE, TAGAMET, TRANSITOL, TRANSULOSE, ULTRASE, URSO, URSO DS, URSO FORTE, VIATOL and VIOKASE appearing in this AIF are trademarks of Axcan or one of its subsidiaries.
49
SCHEDULE A - GLOSSARY OF TECHNICAL TERMS
The text following the technical terms reproduced in this glossary is explanatory only and does not in any way modify the meanings of such terms.
Bile Ducts: Channels that collect bile from the liver and deliver it to the intestine.
Cervical Dysplasia: Modification of the size, shape and organization of cells in the cervix. Dysplasia is generally considered to be a pre-cancerous condition.
Cholestatic Diseases of the Liver: Conditions in which the bile flow from the liver is impaired.
Cholestasis: Combined conditions causing the reduction of bile flow and the retention of bile acids.
Cirrhosis: Disease of the liver, originating from many causes and characterized by a progressive replacement of liver cells by scarring tissue.
Colorectal Adenomateous Polyps: Polyps which are considered precursors to colorectal cancer.
Crohn’s Disease (CD): Inflammatory bowel disease that affects the wall of the gastrointestinal tract. CD can affect any part of the gastrointestinal tract but mostly affects the ileum, the last portion of the small bowel and the colon.
Cystic Fibrosis (CF): Congenital disease characterized by excessive secretions of certain glands, resulting in pancreatic insufficiency and pulmonary disorders. The average lifespan of CF patients is approximately 32 years.
Distal: The part of the colon closest to the rectum.
Duodenum: Part of the small intestine attached to the end of the stomach.
Exocrine Pancreatic Insufficiency: Decreased production and release of the enzymes produced in the pancreas, which leads to digestive problems.
Fistula: Abnormal connection or passageway between organs or vessels that normally do not connect.
Gastric Cancer: Cancer of the cell lining of the gastric mucosa.
Gastro-esophageal reflux disease (GERD): Symptoms and/or mucosal damage produced by the abnormal reflux of gastric contents into the esophagus, causing heartburn or acid regurgitation.
Helicobacter Pylori (Hp): Bacterium with a spiral tail which lives under the gastric mucosa layer. The presence of Hp is correlated with gastritis, as well as gastric and duodenal ulcers. Hp is considered to be the most important factor in the cause of peptic ulceration and is formally classified as a Category 1 human carcinogen by the World Health Organization. Once a diagnosis of Hp infection has been established, a therapy should be prescribed to eradicate the bacterium in all peptic ulcer patients, to reduce the rate of ulcer recurrence.
Hepatitis: Inflammation of the liver due to infection or toxins.
High-Grade Dysplasia: As associated with Barrett’s Esophagus, High-Grade Dysplasia is a condition that results from prolonged acid reflux (heartburn) which causes the lining of the esophagus to be converted into tissue similar to that which lines the stomach. This transformation makes the esophageal tissue more susceptible to cancer.
Inflammatory Bowel Diseases (IBDs): Chronic diseases of unknown cause characterized by inflammation of portions of the gastrointestinal tract. Ulcerative colitis, ulcerative proctitis (a distal form of ulcerative colitis) and Crohn’s Disease constitute the group of illnesses referred to as idiopathic inflammatory bowel diseases. IBDS follow a pattern of acute attacks followed by periods of remission. There are no cures for IBDS and the goals of therapy are to reduce symptoms during acute attacks and to maintain remission when the disease is under control.
Irritable Bowel Syndrome (IBS): Functional bowel disorder which primarily affects gastrointestinal motility.
50
Liver: Organ located in the top right portion of the abdominal cavity connected to the digestive tract. It secretes bile that is excreted in the duodenum, thus facilitating digestion of food in the small intestine. The liver plays a key role in the processing and storage of various products of absorption.
Mesalamine: five-aminosalicylic acid (5-ASA).
Moiety: One of two or more parts into which something may be divided, such as the various parts of a vitamin or molecule.
Motility: Ability of the gastrointestinal tract to undergo rhythmic muscular contractions.
Mucosa: Thin sheets of tissue that cover or line various parts of the body such as the mouth or digestive tract.
Nocturnal Acid Breakthrough (NAB): The presence of at least 60 continuous minutes of intragastric pH less than 4 during the night, in patients taking a proton pump inhibitor twice daily before meals.
Pancreas: Abdominal gland located behind the stomach and connected to the gastrointestinal tract that secretes pancreatic juice to aid digestion (pancreatic enzymes) and insulin, an essential hormone for the metabolism of sugars.
Pancreatic Juice: Alkaline secretion of the pancreas containing enzymes that aid in the digestion of protein, carbohydrates, and fats.
Pancreatitis: Inflammation of the pancreas.
Perianal Fistula: Abnormal connection that connects the rectum or other ano-rectal area to the skin surface.
Polyp: Small tumor-like growth that projects from a mucus membrane surface (i.e. colon or rectum).
Primary Biliary Cirrhosis (PBC): A chronic cholestatic liver disease that progresses slowly towards a terminal phase characterized by jaundice, signs of decompensated cirrhosis, ascites and variceal bleeding. The prognosis averages seven to 12 years from diagnosis to death or liver transplant.
Primary Sclerosing Cholangitis (PSC): A liver disorder characterized by an inflammatory and sclerosing process leading to a progressive reduction in the diameter of the bile ducts. Its progressive course generally leads to liver cirrhosis, portal hypertension and often death, as the bile that normally flows out of the liver instead accumulates there, resulting in an alteration of liver cells. The average survival rate is four to ten years following diagnosis.
Proton pump inhibitor (PPI): Proton-pump inhibitors reduce the production of acid by blocking the enzyme in the wall of the stomach that produces acid. The reduction of acid prevents ulcers and allows any ulcers that exist in the esophagus, stomach and duodenum to heal.
Ulcer: Necrotic lesion characterized by a crater-like erosion of the wall of the stomach (gastric ulcer) or the duodenum (duodenal ulcer), often associated with painful symptoms.
Ulcerative Colitis/Proctitis: Chronic inflammatory disease which affects the inner mucus membrane of the colon, particularly the distal portions of the colon (i.e., the rectum and sigmoid).
Ursodiol (ursodeoxycholic acid): Naturally occurring bile acid present as a minor fraction of the total human bile acids, and in greater concentrations in the bile of certain animal species such as bears. Ursodiol is also a drug indicated for the treatment of different diseases such as dissolution of gallstones, primary biliary cirrhosis and other cholestatic liver diseases.
51