Table of Contents
Registration No. 333-116303
As filed with the Securities and Exchange Commission on June 10, 2004
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
Amendment No. 1
to
Form F-1
Registration Statement
Under the Securities Act of 1933
GPC BIOTECH AG
(Exact Name of Registrant as Specified in its Charter)
| Germany | 2834 | 04-3158193 | ||
| (State or Other Jurisdiction of Incorporation or Organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification No.) |
Fraunhoferstrasse 20 D-82152 Martinsried/Munich, Germany Tel: 011 49 89 8565 2600 | Brent Hatzis-Schoch Vice President – Legal Affairs GPC Biotech Inc. 101 College Road East Princeton, New Jersey 08540 Tel: (609) 524-1000 | |
(Address and telephone number of Registrant’s principal executive offices) | (Name, address and telephone number of agent for service) |
Copies to:
Danielle Carbone Shearman & Sterling LLP 599 Lexington Avenue New York, NY 10022 Tel: (212) 848-4000 Fax: (212) 848-7179 | Michael Leppert Shearman & Sterling LLP Gervinusstrasse 17 D-60322 Frankfurt am Main, Germany Tel: 011 49 69 9711 1000 Fax: 011 49 69 9711 1100 | Wolfgang Feuring David B. Rockwell Sullivan & Cromwell LLP Neue Mainzer Strasse 52 Frankfurt, Germany 60311 Tel: 011 49 69 4272 520 Fax: 011 49 69 4272 5210 |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box.¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earliest effective registration statement for the same offering.¨
If delivery of the registration statement is expected to be made pursuant to Rule 434, please check the following box.¨
CALCULATION OF REGISTRATION FEE
Title of each class of securities to be registered(1) | Amount to be registered(2) | Proposed maximum offering price per security(3) | Proposed maximum aggregate offering price(3) | Amount of registration fee(4) | |||||||
Ordinary Bearer Shares with no par value | 8,579,000 | $ | 16.40 | $ | 140,695,600 | $ | 17,826 | ||||
| (1) | American depositary receipts evidencing American Depositary Shares issuable on deposit of the shares registered hereby are being registered pursuant to a separate Registration Statement on Form F-6 (Registration No. 333-116306). |
| (2) | Includes (a) shares that may be purchased pursuant to the underwriters’ over-allotment option and (b) shares initially offered and sold outside the United States that may be resold from time to time in the United States. The shares are not being registered for the purpose of sales outside the United States. See “Plan of Distribution.” |
| (3) | Estimated pursuant to Rule 457(c) solely for the purposes of calculating the registration fee based on the average of the high and low price per ordinary bearer share as reported by the Frankfurt Stock Exchange on June 2, 2004 translated into U.S. dollars based on the noon buying rate on June 4, 2004 of $1.23 per euro. |
| (4) | This amount was previously paid. |
The registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the Registration Statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell nor does it seek an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion. Dated June 10, 2004.
7,460,000 Shares

GPC BIOTECH AG
Ordinary Bearer Shares, with no par value,
in the form of shares or American Depositary Shares
This is an initial public offering of ordinary bearer shares of GPC Biotech AG, a German stock corporation, in the form of shares or American Depositary Shares, or ADSs. Each ADS represents one ordinary share. GPC Biotech AG is offering 7,160,000 of the shares or ADSs to be sold in the offering. The selling shareholders identified in this prospectus are offering an additional 300,000 shares. GPC Biotech AG will not receive any of the proceeds from the sale of the shares being sold by the selling shareholders.
Prior to this offering, there has been no public market in the United States for our shares or ADSs. Our shares are listed on the Frankfurt Stock Exchange under the symbol “GPC”, and application has been made for the quotation of the ADSs on the Nasdaq National Market under the symbol “GPCB”. On June 8, 2004, the closing price of our shares on the Frankfurt Stock Exchange was€ 13.40 per share.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| Per Share | Per ADS | Total | ||||||
Initial public offering price | € | $ | $ | |||||
Underwriting discount and commissions | € | $ | $ | |||||
Proceeds, before expenses, to GPC Biotech | € | $ | $ | |||||
Proceeds, before expenses, to selling shareholders | € | $ | $ | |||||
To the extent that the underwriters sell more than 7,460,000 shares or ADSs, the underwriters have the option to purchase up to an additional 1,119,000 shares or ADSs from GPC Biotech at the offering price less the underwriting discount and commissions.
The underwriters expect to deliver the shares or ADSs against payment in New York, New York on or as soon as practicable after July 2, 2004.
Goldman, Sachs & Co. oHG
Global Coordinator
Goldman, Sachs & Co.
Lehman Brothers
Pacific Growth Equities, LLC | WestLB AG |
Prospectus dated June , 2004.
Table of Contents
This summary does not contain all of the information that you should consider before buying shares or ADSs in this offering and highlights information contained elsewhere in this prospectus that we believe is most important to understanding our business. You should read the entire prospectus carefully, including the Risk Factors section and our consolidated financial statements and the related notes included elsewhere in this prospectus, before making an investment decision.
GPC Biotech AG
GPC Biotech is a biopharmaceutical company discovering and developing new drugs to treat cancer. Our lead product candidate is satraplatin, an oral platinum-based compound intended for use as a chemotherapy drug. Satraplatin is being evaluated in a Phase 3 registrational trial for second-line treatment of hormone refractory prostate cancer, or HRPC. Patients suffering from HRPC are those who have suffered relapse after having been treated with hormone ablation therapy. See “Business—Our Product Pipeline—Prostate Cancer and Existing Treatment Regimens.” Second-line treatments are those implemented when primary, or first-line, treatment fails. In Phase 3 registrational trials, the drug is tested on patients in a controlled randomized trial comparing the investigational new drug to a placebo or an approved form of therapy, if there is one. Phase 3 registrational trials are usually the final clinical evaluation to support a new drug application, or NDA. “Randomized” means that patients are randomly assigned to receive the drug candidate or a placebo. Our goal is to file an NDA, with the U.S. Food and Drug Administration, or FDA, in this disease setting in 2006.
Our second most advanced product candidate is 1D09C3, a monoclonal antibody. Assuming successful and timely completion of preclinical activities and timely approval by the relevant regulatory authorities and ethics committees, we anticipate initiating clinical trials for 1D09C3 in 2004. A monoclonal antibody is an immune system related protein that binds preferentially to one type of foreign substance and may thereby stimulate a biological response. Our third development program involves inhibitors of the cell division cycle, or cell cycle. The cell cycle is the process by which a cell grows, duplicates its DNA and divides into two cells. RGB-286199, our lead compound in this program, is currently in preclinical development. Preclinical development includes work to demonstrate a drug candidate’s efficacy and safety in cell culture and animal models prior to testing in humans. Our goal is to complete preclinical development work on RGB-286199 in the first half of 2005. We also have a number of research programs focused on the discovery of new anticancer drugs.
Cancer is the second leading cause of death in the United States after cardiovascular disease. Prostate cancer is the most common cancer among men and the second leading cause of cancer deaths in men in the United States. Despite progress in understanding cancer and the resulting development of new therapies, there is still a lack of effective treatments, resulting in a substantial unmet medical need.
Our Product Candidates
Satraplatin. Satraplatin is in a Phase 3 registrational trial as second-line treatment of HRPC. Satraplatin is an oral platinum-based compound intended for use as a chemotherapy drug. Platinum-based drugs have been clinically proven to be one of the most effective classes of anticancer therapies and are used to treat a wide range of cancers. Unlike the currently marketed platinum-based drugs, satraplatin is administered orally. We licensed satraplatin in September 2002 and, in September 2003, initiated a Phase 3 registrational trial of satraplatin as a second-line chemotherapy treatment for HRPC, with sites in the United States, Europe and South America. We modelled our registrational trial, in which we expect over 900 patients to be enrolled, on a successful 50-patient study of satraplatin in
1
Table of Contents
the first-line chemotherapy treatment of HRPC, conducted by others. There is currently no approved second-line chemotherapy treatment regimen for patients who fail first-line chemotherapy treatment for HRPC. We were therefore able to obtain fast-track designation from the FDA for satraplatin in this disease setting. The FDA’s fast track program is intended to facilitate the development and to expedite the review of drugs that are intended for the treatment of a serious or life-threatening condition for which there is no approved treatment. Our goal is to complete the filing of an NDA with the FDA for satraplatin as a treatment of HRPC in 2006. Based on existing clinical data, we believe that satraplatin may have application in a number of cancers, both as a single agent and in combination with other therapies.
1D09C3. 1D09C3 is a monoclonal antibody intended for the treatment of selected leukemias and lymphomas, including non-Hodgkin’s lymphoma. Preclinical activities that need to be completed prior to initiating clinical trials include a toxicological evaluation and the development of certain tests for monitoring drug levels and drug responses in the blood of treated patients. We expect to complete these preclinical activities during 2004. Assuming successful and timely completion of these preclinical activities and timely approval by the relevant regulatory authorities and ethics committees, we anticipate initiating clinical trials for this product candidate in 2004.
RGB-286199. RGB-286199 is our lead compound in a family of small molecule compounds that inhibit the cell cycle, which is essential for tumor growth. RGB-286199 is currently in preclinical development. Our goal is to complete preclinical development work on RGB-286199 in the first half of 2005 and, if successful, to advance this product candidate into clinical trials thereafter.
Our Research Programs
Our research is focused on new anticancer drug discovery opportunities. To date, our research activities have led us to the discovery of 1D09C3 and RGB-286199, our two product candidates in preclinical development. Our current approach to new drug discovery programs is to start with existing small molecules or small molecule classes that are either known to have anticancer activity or known to act on selected targets relevant for cancer drug research. We design our research programs to discover product candidates that have potentially broad application in the treatment of various cancers. Most of our research programs are directed toward discovering cytotoxic drugs, which are drugs that kill cancer cells rather than prevent their growth. Our current research programs can be classified in two areas, inducers of programmed cell death and inhibitors of protein kinases, which play a role in signalling between and within cells.
Our Drug Discovery Technologies
We also have a portfolio of technologies in the fields of proteomics (the systematic study of proteins and their functions) and genomics (the systematic study of genes) that we use in our internal drug discovery efforts and that we have licensed to, or used in collaborations with, various pharmaceutical companies. In early 2003, we expanded our technology portfolio through the introduction of LeadCodeTM, our proprietary cell-based drug-protein interaction screening technology. LeadCode enables us to improve our understanding of the potential efficacy and side effects of drugs and drug candidates. We believe that these technologies allow us to prioritize and optimize drug candidates in our pipeline, identify new drug uses and potentially reduce the risk of failure in drug development.
In 2001, we entered into an agreement with ALTANA Pharma AG, which is our most significant technology collaboration to date. Under this contract, we are assisting ALTANA Pharma through 2007 with its establishment of a research institute in the United States. This agreement includes a research
2
Table of Contents
collaboration as well as a transfer of technologies. Effective January 2003, we have also licensed LeadCode to ALTANA Pharma under a separate agreement. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding over the term of the agreements. As of December 31, 2003, we had received an aggregate of€37.4 million from ALTANA Pharma under these agreements, of which€27.4 million have been recognized as revenue.
Our Strategy
Our goals are to discover, develop and commercialize new anticancer drugs. We are pursuing these goals through the following strategies:
| • | Enhance the commercial value of satraplatin to us; |
| • | Broaden the potential range of applications of satraplatin; |
| • | Build and maintain a balanced portfolio of oncology drugs; |
| • | Use our research expertise, technologies and external network to build our product candidate pipeline; |
| • | Use our internal expertise to find the fastest path to market approval for our product candidates; and |
| • | Generate revenue through technology and research collaborations. |
Our Management and Scientific Team
We have assembled a management and scientific team, which we believe has the expertise and experience required to discover, develop and commercialize oncology product candidates. Our clinical development team is highly experienced in designing and conducting successful clinical trial programs for oncology drugs. Members of this group have a combined career track record of obtaining more than 20 marketing approvals with the FDA, including those for TAXOL® (paclitaxel), a widely used chemotherapy drug, and PARAPLATIN® (carboplatin), one of the most well-established platinum-based chemotherapy drugs. Furthermore, we have an extensive external network of scientists and experts in the oncology field, which helps us to identify and evaluate opportunities to license drug candidates from third parties.
Risks of Drug Development
All of our product candidates are undergoing clinical trials or are in earlier stages of development, and failure is common and can occur at any stage of development. To date, we have not obtained regulatory approval for the commercial sale of any drug product, and we have not received any revenues from the commercial sale of drug products. For the year ended December 31, 2003, our total revenues were€21.6 million, our net loss was€26.8 million and, as of that date, we had an accumulated deficit of€127.3 million.
Company Information
GPC Biotech AG was originally formed as a limited liability company (Gesellschaft mit beschränkter Haftungor GmbH) under German law in January 1997, and was named RM 102 Vermôgensverwaltungs GmbH. In August 1997, all of the company’s shares were sold and transferred to members of the company’s management team and scientific advisory board, and its name was
3
Table of Contents
changed to Genome Pharmaceuticals Corporation GmbH. In February 1998, Genome Pharmaceuticals Corporation GmbH was transformed into a stock corporation (Aktiengesellschaft orAG) under German law and its name was changed to GPC Aktiengesellschaft Genome Pharmaceuticals Corporation. On March 3, 2000, the company changed its name to GPC Biotech AG.
In March 2000, we acquired Mitotix, Inc., a U.S. drug discovery and development company focused on treatments for cancer and fungal infections. The total purchase price for the acquisition was approximately€31.2 million, which we paid for with our ordinary shares. After the acquisition, Mitotix, Inc. changed its name to GPC Biotech Inc.
Historically, we have been focused on applying genomic, proteomic and drug-discovery technologies to accelerate the identification and validation of novel drug targets for the development of mechanism-based drugs in oncology, infectious diseases and immunology.
In September 2002, we licensed satraplatin, our late stage chemotherapy product candidate for the treatment of HRPC, from Spectrum Pharmaceuticals, Inc. We are now focusing our efforts on our oncology drug discovery and development.
Trademarks
We own rights in the following trademarks: GPC Biotech, the GPC Biotech design, the globe design, LeadCode, ExpressCode, PathCode, UbiCode and KeyCode. All other trademarks or tradenames referred to in this prospectus are the property of their respective owners.
Executive Offices
Our principal executive offices are located at Fraunhoferstrasse 20, D-82152 Martinsried/Munich, Germany and our telephone number is 011 49 89 8565 2600.
Presentation of Financial Information
In this prospectus, references to “€” are to euro and references to “USD”, “U.S.$”, “$” and “U.S. dollars” are to United States dollars. We publish our financial statements in euro.
In this prospectus, unless otherwise provided, references to “GPC Biotech”, “the company”, “we”, “us” and “our” refer to GPC Biotech AG and its wholly owned subsidiary, GPC Biotech Inc.
4
Table of Contents
The Combined Offering
The shares being offered by this prospectus are part of a combined offering of up to 8,279,000 newly issued shares offered by the Company (including an over-allotment option of up to 1,119,000 new shares) and 300,000 existing shares offered by selling shareholders. The combined offering consists of a rights offering to existing holders of our shares and a global offering consisting of public offerings in Germany and the United States and private placements with institutional investors in certain other jurisdictions. We are offering up to 7,152,047 newly issued shares in the rights offering. To avoid a fractional subscription ratio, we have excluded shareholders’ subscription rights for a residual amount of 7,953 newly issued shares in accordance with German law and our Articles of Association. Those new shares as to which subscription rights have been excluded or not exercised during the subscription period will be offered in the global offering. In addition, the global offering will include 300,000 existing shares to be sold by certain management shareholders. See “Principal and Selling Shareholders”. All the shares offered in the rights offering and the global offering will be offered through the underwriters. The combined offering can be terminated under some circumstances, even with respect to rights that have been effectively exercised. See “The Combined Offering”, “Plan of Distribution” and “The Rights Offering” below.
The Rights Offering | Existing holders of our shares will have the right to subscribe for one new share for every three shares held at the subscription price. As of the evening of June 14, 2004, Clearstream Banking AG will automatically credit the subscription rights for shares held in collective custody to the depositary banks. |
Subscription Price | We will determine the subscription price at the latest by 12:00 p.m. (noon) Frankfurt time on June 25, 2004. The subscription price will be between or at (x) the volume-weighted average price of the shares from the beginning of the subscription period until the determination of the subscription price and (y) the current market price of our shares at the time of the determination of the subscription price. In determining the subscription price, we will, among other things, consider the current market conditions and the preliminary results of the bookbuilding process for the shares to be offered in the global offering. |
We will only determine the subscription price during the subscription period. Therefore, we advise you to inform yourself about the subscription price via the various media set forth below before exercising your subscription rights. See also “The Rights Offering—Important Notices”. |
The subscription price will be published through electronic media (Reuters) immediately after being determined and in the electronic version of the German Federal Gazette (elektronischer Bundesanzeiger) on the first practicable date after determination, at the latest on June 25, 2004. As soon as practicable after determination, the subscription price will also be published in the Frankfurter Allgemeine Zeitung and made available on GPC Biotech’s website at http://www.gpc-biotech.com.
5
Table of Contents
Existing holders and investors should note that the subscription price in the rights offering may be higher than the initial offer price of the shares to be sold in the global offering. See “The Combined Offering—Important Notices” and “The Right Offering—Important Notices”. |
The subscription rights expire at 11:59 p.m. (Frankfurt time) on June 28, 2004. If you subscribe for new shares in the rights offering, you will receive them as soon as practicable on or after July 2, 2004.
The Global Offering | Those new shares as to which shareholders’ subscription rights have been excluded and any new shares not subscribed for in the rights offering, will be offered by GPC Biotech, together with the shares offered by certain management shareholders, through the underwriters in the global offering. We plan to offer these shares through the underwriters to the public in Germany and the United States, and to institutional investors in certain other jurisdictions. |
Offer Price | The initial offer price of those new shares as to which shareholders’ subscription rights have been excluded, any new shares not subscribed for in the rights offering and the shares offered by certain management shareholders will be determined by GPC Biotech and Goldman, Sachs & Co. oHG at the end of a bookbuilding period. The bookbuilding period will begin on June 15 and is expected to end on or about June 29, 2004. |
The initial offer price of the shares to be sold in the global offering to the public will be published through electronic media (Reuters) immediately after being determined on or about June 29, 2004. The initial offer price to the public will be published in the Frankfurter Allgemeine Zeitung and The Wall Street Journal on or about July 1, 2004.
Over-Allotment Option | As part of the global offering, the underwriters have an option to purchase up to 1,119,000 additional new shares from GPC Biotech to cover over-allotments; shareholders’ subscription rights with respect to these shares will be excluded. |
Shares Outstanding | 29,735,140 (assuming that all the new shares are sold in the combined offering and that the underwriters exercise their over-allotment option in full). |
American Depositary Shares | Investors in the United States may also purchase shares in the form of ADSs. Each ADS, evidenced by an American Depositary Receipt, represents an ownership interest in one share. We are offering ADSs in addition to shares so that our company can be quoted on the Nasdaq National Market and investors will be able to trade our securities and receive dividends on them in U.S. dollars if they wish. |
6
Table of Contents
Voting Rights | Each share entitles the holder to cast one vote on all matters submitted to a vote of our shareholders. Holders of ADSs must follow specific procedures to exercise the voting rights of the shares underlying the ADSs and will not be able to exercise those rights unless we request The Bank of New York, as ADS depositary, to solicit voting instructions from ADS holders. We discuss these voting rights and procedures further in the sections of this prospectus entitled “Description of Share Capital—Shareholders’ Meeting and Voting Rights” and “Description of American Depositary Receipts—Voting Rights”. |
Dividends | All shares offered in the combined offering are or will be entitled to full dividend rights for all periods commencing January 1, 2003. We have, however, not paid any dividends on our shares in the past and do not intend to pay dividends on our shares for the foreseeable future. |
Lock-Up | We will agree with the underwriters that, on or before December 27, 2004 we will, subject to certain exceptions, (a) not directly or indirectly, offer, sell or otherwise dispose of any shares of our capital stock or any other securities which are convertible into or exchangeable for shares of our capital stock, (b) not exercise any authorization pursuant to our articles of association to increase our capital other than for the purpose of issuing ordinary shares to the beneficiaries of our existing stock option plans and convertible bond programs upon the exercise of option or conversion rights, and (c) not propose a capital increase to our shareholders other than a proposal for authorized capital (genehmigtes Kapital) or for conditional capital (bedingtes Kapital), in each case except with the prior written consent of Goldman, Sachs & Co. oHG. Certain exceptions apply for share issuances directly to strategic partners in the pharmaceutical or biotech sector in connection with partnering or licensing transactions or in connection with an acquisition or joint venture. See “Plan of Distribution”. |
All Management Board members will individually agree with the underwriters that, except with the prior written consent of Goldman, Sachs & Co. oHG, on or before December 27, 2004 they will not sell their shares or enter into similar transactions to this effect or propose, or vote in favor of, any increase in our share capital unless GPC Biotech is or would be permitted to propose such resolution. These lock-up restrictions do not apply to up to 150,000 cash settled stock options, which two of the Company’s Management Board members intend to exercise before the end of the combined offering but after the publication of the initial offer price (see “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”).
Listing and Quotation | Our shares are listed on the Frankfurt Stock Exchange under the symbol GPC (International Securities Identification Number (ISIN) DE0005851505; German securities code (WKN) 585 |
7
Table of Contents
150). Application has been made for quotation of the ADSs on the Nasdaq National Market under the symbol “GPCB.” |
Application has been made for admission of the new shares offered in the combined offering to the Frankfurt Stock Exchange, which is expected to be granted on June 28, 2004. Trading of the new shares on the Frankfurt Stock Exchange is expected to commence on June 30, 2004. |
The subscription rights (ISIN DE000A0AYYP6) will be admitted for trading on the Frankfurt Stock Exchange. The subscription rights will not be listed on a U.S. exchange or quoted on the Nasdaq National Market. Since we will only determine the subscription price during the subscription period, a price for the traded subscription rights may not be determinable or, to the extent that a price for the traded subscription rights can be determined, any such price may not constitute the arithmetic value of the subscription rights. See “The Rights Offering—Important Notices.”
Stabilization | Goldman, Sachs & Co. oHG is authorized for a period beginning on the date of publication of the initial offer price and ending 30 days after allocation of the shares in the global offering to perform either by itself or through any of its affiliates certain stabilization measures for the purpose of keeping the market price of the shares or ADSs at a level that may not otherwise prevail. Any stabilization may be discontinued at any time. Stabilization measures will be performed in Germany and elsewhere in accordance with applicable laws. |
Use of Proceeds | We intend to use the net proceeds from this offering to continue to fund clinical trials of satraplatin, to make regulatory filings for marketing approval of satraplatin, and to fund our other clinical, preclinical and research programs, including our monoclonal antibody program (1D09C3) and cell cycle inhibitor program (RGB-286199). We currently estimate that from the net proceeds of this offering we will spend between 50% and 60% to continue to fund clinical trials of satraplatin, to broaden the potential anticancer applications of this drug candidate, and to make regulatory filings for marketing approval of satraplatin; between 20% and 30% to fund our other clinical, preclinical and research programs, including our monoclonal antibody program (1D09C3) and cell cycle inhibitor program (RGB-286199), and to invest in new capabilities relating to our research and development activities; and between 15% and 25% for general corporate purposes, which may include the licensing of other product candidates, technologies or intellectual property. We will not receive any proceeds from the sale of the shares by the selling shareholders. See “Use of Proceeds”. |
8
Table of Contents
SUMMARY CONSOLIDATED FINANCIAL INFORMATION
The following table presents summary of consolidated financial information of GPC Biotech. We prepare our consolidated financial statements in accordance with accounting principles generally accepted in the United States of America, or U.S. GAAP. You should read the following summary of consolidated financial information in conjunction with the section of this prospectus entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes contained elsewhere in this prospectus.
| Three months ended March 31, | Year ended December 31, | ||||||||||||||||||||
| 2004 | 2003 | 2003 | 2002 | 2001 | 2000 | 1999 | |||||||||||||||
| € | € | € | € | € | € | € | |||||||||||||||
| (amounts in thousands, except share and per share data) | |||||||||||||||||||||
Statement of operations data: | |||||||||||||||||||||
Revenues | 3,959 | 6,112 | 21,594 | 21,511 | 13,889 | 5,936 | 4,264 | ||||||||||||||
Research and development expenses | 8,682 | 8,323 | 37,741 | 38,053 | 30,644 | 20,404 | 5,280 | ||||||||||||||
Impairment of goodwill | — | — | — | 7,314 | — | — | — | ||||||||||||||
Acquired in-process research and development | — | — | — | — | — | 15,974 | — | ||||||||||||||
Total operating expenses | 11,569 | 11,112 | 51,068 | 57,907 | 45,239 | 46,013 | 6,802 | ||||||||||||||
Operating loss | (7,610 | ) | (5,000 | ) | (29,474 | ) | (36,396 | ) | (31,350 | ) | (40,077 | ) | (2,538 | ) | |||||||
Net loss | (6,925 | ) | (4,162 | ) | (26,831 | ) | (32,934 | ) | (26,217 | ) | (34,783 | ) | (3,305 | ) | |||||||
Per share data: | |||||||||||||||||||||
Basic and diluted loss per share | (0.33 | ) | (0.20 | ) | (1.29 | ) | (1.59 | ) | (1.42 | ) | (2.39 | ) | (0.54 | ) | |||||||
Shares used in computing basic and diluted loss per share | 21,085,710 | 20,724,174 | 20,731,535 | 20,688,515 | 18,509,398 | 14,572,917 | 6,118,750 | ||||||||||||||
| At March 31, | At December 31, | |||||||||||
| 2004 | 2003 | 2002 | 2001 | 2000 | 1999 | |||||||
| € | € | € | € | € | € | |||||||
| (amounts in thousands) | ||||||||||||
Balance sheet data: | ||||||||||||
Cash and cash equivalents(1) | 18,201 | 34,947 | 39,947 | 91,245 | 55,108 | 4,787 | ||||||
Marketable securities and short-term investments | 63,783 | 54,221 | 74,935 | 48,885 | 53,573 | 14,928 | ||||||
Total assets | 97,117 | 101,564 | 132,333 | 167,860 | 132,739 | 22,341 | ||||||
Long-term debt, including capital lease obligations | 835 | 959 | 1,528 | 1,680 | 1,952 | 9,253 | ||||||
Total shareholders’ equity | 77,168 | 81,879 | 107,270 | 139,093 | 124,516 | 10,341 | ||||||
| Three months ended March 31, | Year ended December 31, | |||||||||||||||||||
| 2004 | 2003 | 2003 | 2002 | 2001 | 2000 | 1999 | ||||||||||||||
| € | € | € | € | € | € | € | ||||||||||||||
| (amounts in thousands) | ||||||||||||||||||||
Other financial data: | ||||||||||||||||||||
Net cash provided by (used in) operating activities | (8,268 | ) | (5,390 | ) | (22,974 | ) | (23,537 | ) | 2,372 | (17,581 | ) | (150 | ) | |||||||
Net cash provided by (used in) investing activities | (9,783 | ) | (14,084 | ) | 18,723 | (26,771 | ) | 1,477 | (39,878 | ) | (14,797 | ) | ||||||||
Net cash provided by (used in) financing activities | 1,176 | (102 | ) | 42 | (255 | ) | 35,019 | 107,709 | 19,381 | |||||||||||
Depreciation, amortization and impairment of tangible, intangible assets and goodwill | 560 | 678 | 3,827 | 10,840 | 6,616 | 4,923 | 570 | |||||||||||||
| (1) | Does not include restricted cash of€2.5 million,€3.0 million and€3.5 million at December 31, 2003, 2002 and 2001, respectively and€2.6 million at March 31, 2004. |
9
Table of Contents
An investment in our shares or ADSs involves a high degree of risk. In addition to the other information contained in this prospectus, you should consider carefully the specific risk factors described below, together with all of the other information in this prospectus, before making a decision to invest in our shares or ADSs. The trading price of our shares and ADSs could decline as a result of any of these risks, and you may lose part or all of your investment.
Risks Related to Our Business
We are substantially dependent on the success of our lead product candidate, satraplatin. If we do not successfully complete our current Phase 3 registrational trial, receive regulatory approval or achieve market acceptance for satraplatin, we may be unable to commercialize satraplatin within the timeframe we planned, or at all.
Satraplatin is our only product candidate currently in clinical trials. We have expended significant time, money and effort developing satraplatin, which entered a Phase 3 registrational trial for the second-line chemotherapy treatment of HRPC in late 2003. Before we can market and sell satraplatin, we will need to demonstrate in adequate and well controlled clinical trials that the drug is safe and effective. We will need to obtain necessary approvals from the FDA and similar regulatory agencies in Europe and elsewhere.
Although our current Phase 3 registrational trial as a second-line chemotherapy for HRPC is modelled on a successful earlier 50-patient study as a first-line chemotherapy for HRPC conducted by others, the results from the earlier study may not accurately predict the results of our Phase 3 trial. If our satraplatin Phase 3 registrational trial is significantly delayed or fails to demonstrate that satraplatin is safe or effective, regulatory approval of satraplatin would be significantly delayed or may not be obtainable. In this event, the overall costs of the program will increase and the time at which we can introduce the drug into the marketplace and begin to generate product revenues will also be delayed. We expect that our Phase 3 trial will be completed in 2006 and the FDA’s review and approval of the application for marketing approval of satraplatin could then occur in 2007. If regulatory approval is significantly delayed, competing therapies could be developed, which could decrease the market potential for satraplatin.
Even if we ultimately receive regulatory approval for satraplatin, satraplatin may not gain market acceptance. Furthermore, the availability of less expensive or more effective alternative treatments may affect our ability to successfully commercialize satraplatin.
Our two other product candidates are both in preclinical development. We may not successfully develop these product candidates. Even if their further development is successful, it will take several more years before we can file for regulatory approval of these product candidates. Therefore, if we fail to commercialize satraplatin, our ability to achieve profitability will be significantly delayed and our business prospects will be seriously limited.
We have a history of losses and our future profitability is uncertain.
We have incurred losses in each year since our inception in 1997 because our research and development and general and administrative expenses exceeded our revenues. Our net loss for the years ended December 31, 2003, 2002 and 2001 was€26.8 million,€32.9 million and€26.2 million, respectively. As of December 31, 2003, we had an accumulated deficit of€127.3 million.
10
Table of Contents
To attain profitability, we will need to develop and bring products to market. We have never generated revenue from the commercialization of our product candidates, and there is no guarantee that we will be able to do so in the future. If satraplatin fails to show positive results in our ongoing Phase 3 registrational trial and we do not receive regulatory approval for satraplatin, or if satraplatin does not achieve market acceptance even if approved, we will not become profitable in the foreseeable future. Furthermore, if our other product candidates do not advance to clinical trials or fail in clinical trials, we may not be able to achieve or maintain profitability. If we fail to become or remain profitable, or if we are unable to fund our continuing losses, we may be unable to continue our research and development programs, and you could lose all or part of your investment.
We expect to make substantial additional expenditures and incur substantial losses in the future as we continue our clinical trials of satraplatin, file for regulatory approvals and commercialize satraplatin, if and when we receive regulatory approvals. We also expect to make substantial expenditures to advance 1D09C3, our monoclonal antibody, and RGB-286199, our cell cycle inhibitor, into clinical trials and to continue our other research activities. As our product candidates advance, we will continue to need resources for research and development activities, for regulatory approval filings and for commercialization of any approved drugs.
We currently depend on our agreements with ALTANA Pharma for substantially all of our revenues.
We derive a significant portion of our revenues from our agreements with ALTANA Pharma. Our revenues from these agreements accounted for approximately 94% of our total revenue in 2003 and 81% of our total revenue in 2002. ALTANA Pharma accounted for 100% of our revenues in the three months ended March 31, 2004. In 2001, we entered into an agreement with ALTANA Pharma AG, which is our most significant technology collaboration to date. Under this contract, we are assisting ALTANA Pharma through 2007 with its establishment of a research institute in the United States. This agreement includes a research collaboration as well as a transfer of technologies. Effective January 2003, we have also licensed LeadCode™ to ALTANA Pharma under a separate agreement. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding over the term of the agreements. As of December 31, 2003, we had received an aggregate of€37.4 million from ALTANA Pharma under these agreements, of which€27.4 million have been recognized as revenue. Revenues from ALTANA Pharma are expected to constitute a substantial portion of our revenues in future periods through 2007. Our operating results would be adversely affected if this collaboration were to terminate early. After expiration of our agreements with ALTANA Pharma, if we have not commercialized satraplatin or developed alternative sources of revenues, our revenues could be significantly lower than in prior periods.
If our competitors develop and market products that are more effective, safer or more affordable than ours, or obtain marketing approval before we do, our commercial opportunities will be more limited.
Competition in the biotechnology and pharmaceutical industries is intense and continues to increase, particularly in the area of cancer treatment. Our competitors include other biotechnology companies, pharmaceutical companies, as well as academic institutions and other research institutions engaged in the discovery and development of anticancer drugs and therapies. Many of our competitors have significantly greater research and development capabilities, greater experience in obtaining regulatory approvals, manufacturing and marketing, or greater financial and management resources than we have. Our competitors may succeed in developing products that are more effective, safer or more affordable than the ones we have under development or that render our product candidates or technologies noncompetitive or obsolete. Moreover, competitors that are able to achieve patent protection, obtain regulatory approvals and commence commercial sales of their products before we do, and competitors that have already done so, may enjoy a significant competitive advantage.
11
Table of Contents
Satraplatin, our lead product candidate, will face significant competition from other drugs that are either marketed or that may be developed for treating prostate cancer, as well as from other platinum-based compounds and other chemotherapy drugs.
In the prostate cancer market, currently approved drugs include EMCYT® (Pfizer, Inc.), NOVANTRONE® (OSI Pharmaceuticals Inc./Serono S.A.), QUADRAMET® (Schering AG/CYTOGEN Corporation), METASTRON® (Amersham Health/Medi-Physics, Inc.) and TAXOTERE® (Aventis S.A.). Two of these drugs (NOVANTRONE and QUADRAMET) are injectable pharmaceuticals approved for use in treating bone pain in cancer patients, and EMCYT is an oral drug used to relieve symptoms of advanced prostate cancer. The most recent approved of these prostate cancer drugs, TAXOTERE, is approved in the United States, in combination with prednisone (a commonly used synthetic steroid), for the treatment of patients with advanced prostate cancer. TAXOTERE has been shown to prolong survival of patients with HRPC.
In addition to the drugs mentioned above, there are other drugs in development for both advanced HRPC and earlier stages of prostate cancer, which may compete with satraplatin. The other agents of which we are aware include the drugs which are listed in the following table.
Drug | Company | Stage of development for | ||
| Atrasentan | Abbott | · Not a marketed drug · Phase 3 trials have been completed for various stages of prostate cancer | ||
| Calcitriol | Novacea | · Not a marketed drug · Phase 2/3 trial in combination with TAXOTERE for first line HRPC is on-going | ||
| Provenge | Dendreon | · Not a marketed drug · Phase 3 trials for first line HRPC on-going | ||
| Ixabepilone | Bristol-Myers Squibb | · Not a marketed drug · Phase 2 data have been reported for first line HRPC | ||
There are currently three marketed platinum-based drugs in the United States and in Europe. These are cisplatin, carboplatin and oxaliplatin. All three agents are administered intravenously and are not approved for the treatment of prostate cancer. In addition to these, there are other platinum-based compounds approved and/or marketed in the Asian markets such as lobaplatin (China), nedaplatin (Japan) and eptaplatin (South Korea). These drugs are not approved, however, for the treatment of prostate cancer. All three of these are also administered intravenously. Another platinum-based drug, which is not currently on the market, is AMD-473. AMD-473, which is administered intravenously, has shown activity for HRPC in a Phase 2 clinical trial. There are no reported clinical trials for an oral formulation of AMD-473. We are aware that other companies may be developing orally bioavailable platinum-based compounds. We are not aware, however, of any other orally bioavailable platinum-based compounds that are approved or are in Phase 3 clinical trials.
If 1D09C3 is approved and commercialized, it will face significant competition. Currently marketed antibodies for the treatment of non-Hodgkin’s lymphoma are RITUXAN® (Biogen Idec, Inc./Genentech, Inc./Roche Holdings AG), ZEVALIN® (Biogen Idec, Inc/Schering AG), and BEXXAR® (Corixa/
12
Table of Contents
GlaxoSmithKline). CAMPATH® (Berlex Laboratories) is approved for chronic lymphatic leukemia. In addition, there are a number of other antibodies and other drugs in development for the treatment of lymphoma and leukemia.
1D09C3 could also be developed for the treatment of leukemias and melanoma. There is, and will continue to be, significant competition in these markets from both large molecule drugs (antibodies) and small molecule drugs.
If RGB-286199 is approved and commercialized, it will face significant competition. There are no currently marketed cell cycle inhibitors. Competitors with development programs for cell cycle inhibitors include Aventis S.A. (flavopiridol), Cyclacel Limited (CYC 202) and Bristol-Myers Squibb (BMS-387032). Aventis and Cyclacel have advanced their cell cycle inhibitors to Phase 2 clinical trials, however, Aventis announced in February 2004 that development of flavopiridol had been terminated. Bristol-Myers Squibb has advanced its cell cycle inhibitor to Phase 1 clinical trials.
Even if our product candidates are approved for marketing, they may not be competitive with established drugs and therapies or may not be able to supplant established products and therapies in the disease settings that we target, thereby reducing the commercial value of our products.
Our operating results may fluctuate considerably on a quarterly basis. These fluctuations could have an adverse effect on the price of our shares and ADSs.
Our results of operations may fluctuate significantly in the future on a quarterly basis as a result of a number of factors, many of which are beyond our control. Although many companies may encounter this problem, it is particularly relevant to us because of our relatively small size, the fact that we do not have any marketed products and the dynamics of the biotechnology industry in which we operate. Factors that could cause our results of operations to fluctuate include, among others:
| • | timing of clinical trial expenses; |
| • | failure to achieve milestones under collaborative arrangements; |
| • | regulatory events; |
| • | new product introductions by us or our competitors; |
| • | variations in the demand for products we may introduce; |
| • | litigation involving patents, licenses or other intellectual property; and |
| • | product failures or product liability lawsuits. |
Any of the foregoing factors could cause us to fail to meet the expectations of securities analysts or investors, which could cause the trading price of our shares and ADSs to decline.
Currency fluctuations may expose us to increased costs and revenue decreases.
Our business is affected by fluctuations in foreign exchange rates between the U.S. dollar and the euro. A significant portion of our revenues are denominated in U.S. dollars but are reported in euros. Therefore currency fluctuations could cause our revenues to decline. Historically, the majority of our expenses were denominated in euros, but, on a going forward basis we expect that the majority of our expenses will be denominated in U.S. dollars. The majority of our cash and cash equivalents are denominated in euros. In addition, we conduct clinical trials in many different countries, which exposes us to cost increases if the euro declines in value compared to the currencies of those countries.
13
Table of Contents
We will need substantial additional funding and may be unable to raise capital when needed, which could force us to delay, reduce or eliminate our drug discovery, development and commercialization activities.
We may need to raise additional capital to fund our operations and clinical trials, to continue our research and development activities and to commercialize future product candidates.
We believe the net proceeds from this offering, together with our cash on hand, as well as future payments we expect to receive from our collaboration with ALTANA Pharma and interest earned on our investments are sufficient to fund our anticipated operating requirements for at least the next two years. However, we may to need to raise additional funds in the future. Our ability to raise additional funds will depend on financial, economic, market conditions and other factors, many of which are beyond our control. If necessary funds are not available to us on satisfactory terms, we may have to reduce expenditures for research and development and clinical trials, which could delay, reduce or eliminate our drug discovery, development and commercialization activities. Any delay in our development activities could delay our ability to commercialize a product.
Our success depends on recruiting and retaining key personnel and if we fail to do so, it may be more difficult for us to execute our business strategy.
We depend on key members of our management team and scientific personnel. The loss of key managers or scientists could delay the advancement of our research and development activities. We do not maintain any key man insurance. The implementation of our business strategy and our future success will also depend in large part on our continued ability to attract and retain other highly qualified scientific, technical and management personnel, as well as personnel with expertise in clinical testing and regulatory approval.
We face competition for personnel from other companies, universities, public and private research institutions and other organizations. The process of hiring suitably qualified personnel is often lengthy. If our recruitment and retention efforts are unsuccessful in the future, it may be more difficult for us to execute our business strategy.
The members of the Management Board and of the management team identified in the Section “Directors, Senior Management and Employees” comprise our key employees. The members of our Management Board have service agreements for a term of four years each, except for Dr. Seizinger who has a service agreement for a term of five years. The Germany-based members of the management team, Drs. Bancroft, Hombeck, and Nagy all have standard German employment contracts of indefinite duration subject to termination in accordance with applicable law and upon reaching the age of 65. In the United States, only Mr. Gregory Hamm (our Vice President, Corporate Integration and Princeton Site Head) has an employment contract as a result of his having been our first US-based employee. Pursuant to Mr. Hamm’s agreement, we must provide Mr. Hamm 60 days’ written notice of any termination without cause and must continue paying Mr. Hamm’s base salary and other benefits for six months after such termination, unless Mr. Hamm sooner commences new employment, in which case these severance benefits will terminate on that earlier date. No other US-based member of the management team has an employment contract, and their employment relationship with us is terminable at will.
We expect to expand our research and clinical development capabilities and, as a result, may encounter difficulties in managing our growth, which could disrupt our operations.
As our research and development programs continue to advance, we expect that the number of our employees and the scope of our operations will grow. To manage our anticipated future growth, we
14
Table of Contents
must continue to improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Because we are a relatively small company, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations could increase our costs significantly and may divert our management and business development resources. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend, in part, on our ability to manage future growth effectively.
We depend on intellectual property licensed from third parties, and termination of any of these licenses could result in the loss of significant rights, which would harm our business.
We hold licenses granted by Spectrum Pharmaceuticals for satraplatin, by MorphoSys for our antibody product candidate, 1D09C3, and by Bristol-Myers Squibb related to our cell cycle inhibitor, RGB-286199. In addition, because the field of antibody therapeutics is characterized by a large amount of intellectual property, we also have a number of other licenses in addition to our license from MorphoSys related to the discovery or the production and commercialization of 1D09C3. We also hold licenses to third party patents important for our research technologies. Any termination of these licenses could result in the loss of significant rights and could harm our ability to commercialize our drug candidates or technologies. Our ownership of patents relating to some or all of our products will not reduce our reliance on these and other third party patents.
Our agreement with Spectrum Pharmaceuticals is a sublicense under a license agreement between Spectrum Pharmaceuticals and Johnson Matthey. We must therefore rely on Spectrum Pharmaceuticals to enforce its rights and Johnson Matthey’s obligations under their license agreement. If Spectrum Pharmaceuticals were to become insolvent or go into receivership, we would have limited access to assets related to satraplatin and limited ability to enforce directly any of Spectrum Pharmaceuticals’ rights or Johnson Matthey’s obligations under such agreement. As a result, our development of satraplatin could be significantly delayed or prevented.
When we license intellectual property from third parties, those parties generally retain most or all of the obligations to maintain, as well as the rights to assert, that intellectual property. We generally have no rights to require our licensors to apply for new patents, except to the extent that we actually assist in the creation or development of patentable intellectual property. With respect to intellectual property that we license, we are generally also subject to all of the same risks with respect to its protection as we are for intellectual property that we own, which are described below.
We rely on third parties to supply the active pharmaceutical ingredients in our product candidates. If they do not supply materials of satisfactory quality, in a timely manner, in sufficient quantities or at an acceptable cost, clinical development and commercialization of our product candidates could be delayed.
We will depend on supply agreements for compounds that are essential for the development and commercialization of our product candidates, including satraplatin. We believe that we have obtained sufficient amounts of satraplatin from Johnson Matthey to conduct our current Phase 3 registrational trial of satraplatin. If we are unable, however, to secure a supply of satraplatin in sufficient quantity and of satisfactory quality in a timely manner for any future clinical trials or commercial sale of satraplatin, after regulatory approval, our ability to further develop and commercialize satraplatin will suffer. Johnson Matthey has been our sole supplier of satraplatin to date. We are currently discussing the manufacture of satraplatin with selected manufacturers who have experience in producing platinum-based drugs. We estimate that identifying and contracting with an alternative supplier for satraplatin will cost approximately $1 million.
15
Table of Contents
Our sole supplier of 1D09C3, our monoclonal antibody product candidate, has been Icos Corporation. We estimate that, under current regulations, if we had to replace a manufacturer of 1D09C3, the costs of verifying both quality and equivalence as compared to the previously produced material could be in excess of $5 million.
Our supply of the active ingredient for satraplatin and other product candidates may become limited, be interrupted or become restricted in certain geographic regions, and our suppliers may not perform their contractual obligations, for reasons including a shortage of raw materials or adverse regulatory actions. In this event, because we have not yet completed negotiations with alternate suppliers, we may not be able to obtain materials of acceptable quality from other manufacturers, or at prices, on terms or in quantities acceptable to us. Any inability to obtain alternate suppliers, including an inability to obtain approval of an alternate supplier from the FDA and other regulatory agencies, would delay or prevent the clinical development and commercialization of satraplatin and our other product candidates.
Although we believe we have sufficient amounts of 1D09C3 for our preclinical studies and initial planned clinical trials from a third party, we will need to procure additional supplies of 1D09C3 to continue its development should early clinical trials be successful or should our current supplies be damaged or lost.
We rely on third parties to manufacture our product candidates. If they do not manufacture our product candidates of satisfactory quality, in a timely manner, in sufficient quantities or at an acceptable cost, clinical development and commercialization of our product candidates could be delayed.
We do not currently own or operate manufacturing facilities and rely and expect to continue to rely on third parties for the production of clinical and commercial quantities of our product candidates. Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our ability to develop and commercialize any product candidates on a timely and competitive basis. We may not be able to maintain or renew our existing manufacturing arrangements on acceptable terms, if at all.
To date, our product candidates have been manufactured in small quantities for preclinical studies or clinical trials. If any of our product candidates is approved by the FDA or other regulatory agencies for commercial sale, we will need to have it manufactured in commercial quantities. We may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or economic manner or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA and other regulatory agencies must review and approve. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed, or there may be a shortage of supply, which could negatively affect our product sales.
We rely on third parties to conduct clinical trials and assist with preclinical activities. If they do not perform as contractually required or expected, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We do not have the ability to independently conduct clinical trials for our product candidates, and we rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to conduct our clinical trials. In particular, with regard to our Phase 3 registrational trial for satraplatin, we have contracts with two contract research organizations, PharmaNet LLC and Cvitkovic Associes Consultants S.A. In the event that one of these clinical research organizations were to terminate its agreement with us, we would shift the full responsibility for
16
Table of Contents
conducting the trial to the remaining clinical research organization. In the event that both of our clinical research organizations were to terminate their agreements with us and, as a result, we needed to transfer responsibilities to a new contract research organization, we believe we could engage one or more new clinical research organizations with sufficient qualifications and international capabilities. Any of the foregoing events could, however, cause significant delays, disruptions and cost increases in our satraplatin trials.
In addition, we rely on third parties to assist us with our preclinical development of product candidates. Our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, if:
| • | these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines; |
| • | these third parties need to be replaced; |
| • | or the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons. |
If any of these events occur, we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates.
We depend in part on the continued availability of outside scientific collaborators, including researchers at clinical research organizations and universities, in areas relevant to our research and product development. The competition for these relationships is intense, and we cannot assure you that we will be able to maintain such relationships on acceptable terms, if at all. In addition, these outside relationships may be terminated by the collaborator at any time. Our scientific collaborators are not employees of GPC Biotech. As a result, we have limited control, if any, over their activities and can expect that only limited amounts of their time will be dedicated to GPC Biotech activities. In addition, we may not control any intellectual property resulting from their activities.
We are, and expect to continue to be, dependent upon collaborative arrangements to complete the development and commercialization of some of our product candidates. These collaborative arrangements may place the development and commercialization of our product candidates outside of our control, may require us to relinquish important rights or may otherwise be on terms unfavorable to us.
We have limited experience in selling, marketing or distributing products and have no internal sales force to do so. If we receive regulatory approval to commence commercial sales of any of our product candidates, we will either have to establish a sales and marketing organization with appropriate technical expertise and supporting distribution capability, or engage one or more collaboration partners, such as a pharmaceutical or other healthcare company with an existing distribution network and direct sales and marketing organization.
We may not be successful in entering into collaborative arrangements with third parties. Any failure to enter into additional collaborative arrangements on favorable terms could delay or hinder our ability to develop and commercialize our product candidates and could increase our costs of development and commercialization. Dependence on collaborative arrangements will subject us to a number of risks, including:
| • | we may not be able to control the amount or timing of resources that our collaborators may devote to the product candidates; |
| • | we may be required to relinquish important rights, including intellectual property, marketing and distribution rights; |
17
Table of Contents
| • | should a collaborator fail to develop or commercialize one of our compounds or product candidates, we may not receive any future milestone payments or royalties; |
| • | a collaborator could separately move forward with a competing product candidate developed either independently or in collaboration with others, including our competitors; |
| • | our collaborators may experience financial difficulties; |
| • | business combinations or significant changes in a collaborator’s business strategy may also adversely affect a collaborator’s willingness or ability to complete its obligations under any arrangement; and |
| • | collaborative research and development arrangements are often terminated or allowed to expire, which would delay the development and may increase the cost of developing our product candidates. |
The primary patents covering satraplatin in the United States will expire in 2008 and 2010, and in 2009 in most other countries. If we are unable to extend these patents, we may be subject to competition from third parties with products with the same active pharmaceutical ingredients as satraplatin.
Even if our product candidates and technologies are covered by valid and enforceable patents, the patents will provide protection only for a limited amount of time. For example, the primary patents covering the active pharmaceutical ingredient and anticancer use of satraplatin will expire in 2008 and 2010 in the United States, respectively, and in 2009 in most other countries. Thereafter, we will have no direct means to prevent third parties from making, selling, using or importing satraplatin in the United States, Europe or Japan. Instead, we expect to rely upon the U.S. Drug Price Competition and Patent Term Restoration Act of 1984, commonly known as the Hatch-Waxman Act, and comparable foreign legislation, to seek product exclusivity for satraplatin. While we believe that satraplatin will meet the Hatch-Waxman criteria for patent extension, delays in the completion of our Phase 3 registrational trial or in obtaining regulatory approval may jeopardize our ability to obtain a timely patent extension or a patent extension may ultimately not be granted. The terms of the Hatch-Waxman Act, or similar foreign statutes, could be amended to our disadvantage. If we do not qualify for marketing exclusivity for satraplatin, the competition we will face would increase significantly, reducing our potential revenues and harming our ability to achieve profitability.
Under the provisions of the Hatch-Waxman Act, we may also have to defend one or more of our patents, if challenged. Although we are currently not involved in any litigation concerning our intellectual property related to satraplatin and we are not currently aware of any threats or challenges with respect to our product candidates, the risk of a challenge increases as our product candidates progress toward commercialization. Information about the patents covering drug products in the United States is published by the FDA in a publicly available database, Approved Drug Products with Therapeutic Equivalance Evaluations, also known as the Orange Book. A competitor (usually a generic drug company) seeking to market a competing or generic version of our drug products in the United States may notify us that its competing drug product does not infringe one or more patents listed in the Orange Book covering our product, or may challenge the validity or enforceability of one or more of our listed patents covering our product. Once we are so notified we would have 45 days in which to file a lawsuit claiming patent infringement based on the competitor’s assertion about the characteristics of its proposed product. If we file such a lawsuit within 45 days, the FDA is required to delay, or stay, final approval of the competing product for up to 30 months. If a court determines that the patent would be infringed by the product proposed in the competitor’s drug application, the FDA will not approve the application until the patent expires. If, however, the court decides that the patent would not be infringed, or is invalid, the FDA may approve the competitor’s drug application when that decision occurs. The FDA may approve the application at the thirty-month date, even if the litigation is ongoing. If litigation is pending and the FDA
18
Table of Contents
approves the application at the end of the thirty-month period, the competitor may launch a competing product. Under the provisions of the recently enacted Medicare Prescription Drug Improvement and Modernization Act of 2003, we are limited to only a single 30-month stay per competing or generic drug application.
Risks Related to Our Industry
Early-stage drug discovery is subject to a high degree of failure.
Although we devote significant resources to the discovery of new anticancer drugs and employ advanced technologies in our efforts to identify promising drug candidates to advance into preclinical studies, the risk that all or any one of our early-stage product candidates will fail is high. According to pharmaceutical industry statistics published in 2001 by the Pharmaceutical Research and Manufacturers of America, only 1 in 1000 early stage drug discovery compounds is tested in clinical trials, and only 1 in 5 compounds that enters clinical trials receives FDA approval for marketing as a prescription drug. Moreover, the results from preclinical studies and early clinical trials may not accurately predict the results obtained in later-stage clinical trials required for regulatory approval. Because there is no prior experience in treating humans with early stage product candidates, we cannot assure you that early-stage product candidates will prove in clinical testing to be effective and safe for use in humans. If our early-stage product candidates do not prove to be effective in such tests, regulatory approval for such products would be delayed or may not be obtainable.
Our product candidates must undergo rigorous clinical testing, the results of which are uncertain and could substantially delay or prevent us from bringing these products to market.
Before we can obtain regulatory approval for a product candidate, we must undertake extensive clinical testing in humans to demonstrate the product’s safety and effectiveness. These clinical trials are expensive, time-consuming and often take years to complete. According to pharmaceutical industry statistics published in 2001 by the Pharmaceutical Research and Manufacturers of America, an average drug candidate receiving approval for marketing as a prescription drug required 6.5 years of clinical testing prior to submission of a request to the FDA for marketing approval.
In connection with clinical trials, we face risks that:
| • | the product candidate may not be efficacious; |
| • | the product candidate may cause harmful side effects or patients may die; |
| • | the results may not confirm the results of earlier trials; or |
| • | the results may not meet the level of statistical significance required by the FDA or other regulatory agencies. |
Any of these events could cause a trial to fail and may significantly delay or prevent us from obtaining regulatory approval for a product candidate.
Difficulties in enrolling patients in our clinical trials may increase costs and negatively affect the timing and outcome of our trials.
Completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients, which is a function of many factors, including:
| • | the nature of the clinical protocol; |
| • | the therapeutic endpoints chosen for evaluation; |
| • | the eligibility criteria for the trial that is related to the protocol; |
| • | the size of the patient population required for analysis of the trial’s therapeutic endpoints; |
19
Table of Contents
| • | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| • | competition for patients by clinical trial programs for other treatments; |
| • | the proportion of patients leaving the study before reaching an endpoint; and |
| • | the availability of adequate insurance. |
We may experience difficulties in enrolling patients in our clinical trials, which could increase the costs or affect the timing or outcome of these trials. This is particularly true with respect to diseases with relatively small patient populations, such as HRPC, which is the indication for which our product candidate satraplatin is currently being evaluated in a Phase 3 registrational trial.
We are subject to significant regulatory approval requirements, which could delay, prevent or limit our ability to market our product candidates.
Our research and development activities, preclinical studies, clinical trials and the anticipated manufacturing and marketing of our product candidates are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in Europe and elsewhere. We require the approval of the relevant regulatory authorities before we may commence commercial sales of our product candidates in a given market. The regulatory approval process is expensive and time-consuming, and the timing of receipt of regulatory approval is difficult to predict.
Our product candidates could require a significantly longer time to gain regulatory approval than expected, or may never gain approval. A delay or denial of regulatory approval could delay our ability to generate product revenues and to achieve profitability.
We have had an “End-of-Phase 2 Meeting” with the FDA and completed a Special Protocol Assessment under which the FDA has evaluated our registrational approach for satraplatin to assess whether it is adequate to meet scientific and regulatory requirements in the United States. We have also received a Scientific Advice Letter from the European Agency for the Evaluation of Medicinal Products, or EMEA, relating to our registrational approach for satraplatin in the European Union. A successful “End-of-Phase 2 Meeting”, Special Protocol Assessment and a Scientific Advice Letter, however, do not guarantee that satraplatin will receive regulatory approval and, in any event, are subject to further developments in the medical and regulatory field. The Scientific Advice Letter from the EMEA also identifies specific issues to be addressed in the clinical trial program and indicates that if the clinical data are not sufficiently convincing, then one Phase 3 trial will be insufficient to obtain approval.
Changes in the regulatory approval policy during the development period of any of our product candidates, changes in, or the enactment of, additional regulations or statutes, or changes in regulatory review practices for a submitted product application may cause a delay in obtaining approval or result in the rejection of an application for regulatory approval.
Regulatory approval, if obtained, may be made subject to limitations on the indicated uses for which we may market a product. These limitations could adversely affect our potential product revenues and our ability to achieve profitability. Regulatory approval may also require costly post-marketing follow-up studies. In addition, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record-keeping related to the product will be subject to extensive ongoing regulatory requirements. Furthermore, for any marketed product, its manufacturer and its manufacturing facilities will be subject to continual review and periodic inspections by the FDA and other regulatory authorities. Failure to comply with applicable regulatory requirements may, among
20
Table of Contents
other things, result in fines, suspensions of regulatory approvals, product recalls, product seizures, operating restrictions and criminal prosecution.
Our ability to commercialize our product candidates successfully will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish appropriate reimbursement levels relating to any products we may eventually sell.
Third-party payors are increasingly challenging the prices charged for medical products and services. Also, the trend toward managed health care in the United States, which could significantly influence the purchase of health care services and products, as well as legislative proposals to reform health care or reduce government insurance programs, may result in lower prices for any drug products we may eventually sell or exclusion of our drug products from reimbursement programs. The U.S. Medicare Prescription Drug, Improvement and Modernization Act of 2003 was signed into law in December 2003. This new law has not yet been implemented and it is difficult to predict how it will impact the reimbursement of cancer therapies. The cost containment measures that health care payors and providers are instituting and the effect of any health care reform could adversely affect our potential product revenues and our ability to achieve profitability.
Obtaining and maintaining reimbursement status is time consuming and costly. There is significant uncertainty as to the reimbursement status of newly approved medical products. Third party payors may not reimburse for treatments with our drugs or they may delay making the decision to reimburse. Either of these would reduce the commercial value of the drug to us.
In certain foreign markets, including Germany and other markets we may seek to target, the pricing, and especially the reimbursement under social security systems, of prescription pharmaceuticals is subject to government control. These governments or related bodies may deny reimbursement status or establish prices for products we may introduce at levels that are too low to enable us to realize an appropriate return on our investment in product development.
We have no control over our key manufacturers’ and suppliers’ compliance with manufacturing regulations and their failure to comply could result in an interruption in the supply of our product candidates.
The manufacturing process of pharmaceutical products is highly regulated. Our present or future manufacturers and suppliers may not be able to comply with FDA mandated current Good Manufacturing Practices, or cGMP, other FDA regulatory requirements or similar regulatory requirements outside the United States. Any failure by our third-party manufacturers or suppliers to comply with applicable regulations could result in sanctions being imposed on them (including fines, injunctions and civil penalties), the failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecution. Any of these events would likely cause an interruption in the supply of our product candidate and cost increases.
We are dependent on patents and proprietary technology, both our own and those licensed from others. If we or our licensors fail to adequately protect this intellectual property or if we do not have exclusivity for the marketing of our products, our ability to commercialize products could suffer.
Our success depends in part on our ability, and the ability of our licensors, to obtain patent protection for technologies, products and processes, to preserve trade secrets, to defend and enforce rights against infringement and to operate without infringing the proprietary rights of third parties, in the
21
Table of Contents
United States, Europe and elsewhere. The validity and breadth of claims in medical or pharmaceutical technology and biotechnology or life science patents involve complex legal and factual questions and, therefore, may be highly uncertain. For example, the value of our intellectual property rights depend on whether:
| • | we, the co-owners of our intellectual property rights or our licensors were the first to make the inventions, or the first to file patent applications covering the intellectual property important for our business; |
| • | we will develop, co-develop or license additional technologies or product candidates that are patentable; |
| • | the scope of any patent protection we, the co-owners of our intellectual property rights or our licensors receive will exclude competitors or provide us with competitive advantages; |
| • | any of the patents that have been or may be issued to us, the co-owners of our intellectual property rights or our licensors will provide protection for commercially viable products, or be held valid if subsequently challenged; |
| • | patent authorities will grant patents to our competitors or others that restrict our business based on applications they have filed or may file; |
| • | we will be able to detect, or if detected, defend in an effective manner against infringement of any patent we, the co-owners of our intellectual property rights or our licensors receive; |
| • | others claim rights in or ownership of the patents and other proprietary rights that we hold or license; |
| • | any patent that we, the co-owners of our intellectual property rights or our licensors receive will be eligible under, and benefit from, any laws or regulations governing patent term extension; |
| • | the patents of others have an adverse effect on our business; or |
| • | others have developed or will develop similar technologies, product candidates, products or processes, duplicate any of those, or design around any patents that have been or may be issued to us, the co-owners of our intellectual property rights or our licensors, particularly in relation to satraplatin, 1D09C3 or RGB-286199. |
We try to protect our proprietary position by generally filing U.S. and foreign patent applications related to those of our proprietary technologies, inventions and improvements that are important to our business, including those related to the development of our product candidates. Our ability to obtain patents is, however, highly uncertain because, to date, some legal principles remain unresolved and there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States and elsewhere. Moreover, the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. The policies governing biotechnology patents outside the United States, especially in Germany, are even more uncertain. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection.
Patents, if issued, may be challenged, invalidated or circumvented. United States patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings. Foreign patents may be subject to opposition or comparable proceedings. Moreover, the Federal Food, Drug, and Cosmetic Act and FDA regulations and policies provide incentives to manufacturers to challenge patent validity or create modified, non-infringing versions of a drug in order to facilitate the approval of abbreviated new drug applications for generic
22
Table of Contents
substitutes. Such proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination and opposition proceedings may be costly and time-consuming, and even if we were to prevail, would distract management. Although we are not currently facing any threats of these types of legal actions with respect to our product candidates, the risk of these legal actions increases as our product candidates progress toward commercialization.
Any patents or patent applications that we own, co-own or license from others may not provide any protection against competitors. Our pending patent applications, those we may file in the future, or those we have licensed or may license from third parties, may not result in patents being issued. If issued, the patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology, products or processes. Furthermore, others may independently develop similar technologies, products or processes or duplicate any of those that we have developed.
We depend on third parties, such as patent-annuity payment companies, to pay the annuity, renewal and other fees required to maintain our patents and patent applications. Non-payment or delay in the payment of these fees is likely to result in irrevocable loss of patents or patent rights important to our business.
We, the co-owners of our intellectual property rights or our licensors may face difficulties in protecting intellectual property in countries other than the United States, which may diminish the value of our intellectual property in those countries.
The laws of some foreign jurisdictions do not protect intellectual property rights to the same extent as in the United States and Europe, and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we, the co-owners of our intellectual property rights or licensors encounter such difficulties in protecting, or are otherwise precluded from effectively protecting, in foreign jurisdictions the intellectual property rights important for our business, the value of these rights may be diminished and we may face additional competition from others in these jurisdictions.
Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (if, for example, the patent owner has failed to “work” the invention in that country, or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent.
Those ideas, developments, discoveries and inventions made by employees and consultants working under German employment law are subject to the provisions of the German Act on Employees’ Inventions (Gesetz über Arbeitnehmererfindungen), which regulates the ownership of, and compensation for, inventions made by employees. For such inventions, we face the risk that disputes can occur between employer and employee, ex-employee, or employers of our consultants pertaining to alleged non-adherence to the provisions of this act. Even if we, the co-owners of our intellectual property rights or licensors prevailed in any such dispute, such action could result in substantial costs and be a distraction to management. If we fail in such dispute, in addition to paying money claims, we may lose valuable intellectual property rights.
23
Table of Contents
Claims that we infringe a third party’s intellectual property may give rise to burdensome litigation, result in potential liability for damages or stop our development and commercialization efforts.
The pharmaceutical, biotechnology and other life sciences industries are characterized by the existence of a large number of patents and frequent litigation based upon allegations of patent infringement. The owners or licensees of these and other patents may file one or more infringement actions against us or our collaborators or licensees. Patent litigation can involve complex factual and legal questions, and its outcome is uncertain. Any claim relating to infringement of patents that is successfully asserted against us may result in us paying substantial damages. Even if we were to prevail, any litigation could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. Furthermore, as a result of a patent infringement suit brought against us or our collaborators or licensees, we or our partners or licensees may be forced to stop or delay developing, manufacturing or selling products that are claimed to infringe a third party’s intellectual property unless that party grants us or our collaborators or licensees rights to use its intellectual property. In these cases, we or our collaborators or licensees may be required to obtain licenses to patents or proprietary rights of others in order to continue to commercialize our products. However, we or our collaborators or licensees may not be able to obtain any licenses required under any patents or proprietary rights of third parties on acceptable terms, or at all. Even if our collaborators, licensees or we were able to obtain rights to the third party’s intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we or our collaborators or licensees may be unable to commercialize some of our products or may have to discontinue development or use of a technology or product candidate or cease some of our or their business operations as a result of patent infringement claims, which could severely harm our business.
United States and foreign patents have been issued to third parties in the same fields as some of our technologies and product candidates and in fields that relate to the development and manufacture of our product candidates. We are aware of an issued patent covering a drug-protein interaction technology similar to our LeadCode technology that describes a “biotin” based drug-fusion molecule. Biotin is a naturally occurring molecule that binds to certain proteins enabling them to perform their function. There are also numerous patent filings claiming various genetic sequences, such as protein coding sequences and regulatory sequences that may be useful if used in our research programs and technologies, including our LeadCode technology. Additionally, a large number of patents have been issued with respect to methods of discovering, producing, and other aspects of therapeutic antibodies, and we are aware of issued patents held by third parties that relate to the production of recombinant antibodies, such as patents covering production in single cells by the independent expression of the two protein chains that make up the antibody, or more generally to the production of recombinant proteins. We are also aware that a third party has an issued patent relating to treating plasmacytoma/multiple myeloma, Hodgkin’s lymphoma (also known as Hodgkin’s disease), non-Hodgkin’s lymphoma and B cell leukemias by using monoclonal antibodies that specifically react with certain MHC class II molecules. We have not attempted to obtain licenses to any of these patents. If we decide to obtain licenses to these patents, we cannot guarantee that we would be able to obtain such licenses on commercially reasonable terms, or at all. Should we not obtain one or more appropriate licenses and infringement claims be brought against us, we may be unable to convince a court that the allegedly infringed patent was invalid or otherwise unenforceable against our product candidates, research programs or technologies, and we may be prevented from practicing or marketing our product candidates, research programs or technologies in countries in which the patent is in force.
While our product candidates are in clinical trials, and prior to commercialization, we believe these activities fall within the scope of the exemptions from patent infringement provided by 35 U.S.C. Section 271(e)(1) in the United States, which covers activities related to developing information for submission to the FDA. As our product candidates progress toward commercialization, the possibility
24
Table of Contents
of an infringement claim against us increases. Analogous provisions may not exist or may not exempt from patent infringement those clinical trials we conduct in other countries.
Our competitive position depends on trade secrets and other forms of non-patented intellectual property protection. If we are unable to protect our trade secrets, other companies may be able to compete more effectively against us, and our business could suffer.
We rely on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. We try to protect this information by entering into confidentiality agreements with parties that have access to it, such as our collaborators, licensees, employees and consultants. Any of these parties may breach these agreements and disclose our confidential information, or our competitors might learn of the information in some other way. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our ability to generate revenues from our product candidates could be severely damaged.
We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors, or at universities. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could delay or prevent our ability to commercialize one or more of our product candidates.
We are subject to significant environmental, health and safety regulation, compliance with which can be expensive.
We are subject to a variety of health, safety and environmental laws and regulations in the United States, Germany and other countries. These laws and regulations govern, among other things, waste water discharge, air emissions and waste management. We have incurred, and will continue to incur, capital and operating expenditures and other costs in the ordinary course of our business in complying with these laws and regulations. Because we produce small amounts of experimental compounds and operate laboratory facilities, some risk of environmental liability is inherent in our business. Additionally, material costs of environmental compliance may arise in the future, increasing the overall costs of regulatory compliance.
Our research activities require access to tissue samples and other biological material. If we lose access to these materials, our ability to discover and develop new product candidates could be impaired.
Our research activities require access to normal and diseased human tissue samples, other biological materials and related clinical or other information, which may be in limited supply. We may not be able to obtain or maintain access to these materials or information on commercially acceptable terms or at all. In addition, government regulations in the United States or Europe could result in restricted access to, or use of, human or other tissue samples or related information. If we lose access to sufficient numbers or sources of tissue samples, or if tighter restrictions are imposed on our use of the information generated from tissue samples, we may not be able to conduct our research activities as we have planned and may have to incur additional development costs.
25
Table of Contents
We may become exposed to costly and damaging product liability claims and may not be able to maintain sufficient product liability insurance to cover claims against us. Even in the absence of product liability lawsuits, unforeseen side effects could harm sales of our products.
We face the risk of substantial liability for damages in the event a patient experiences adverse side effects during clinical trials or after any drugs we may develop are marketed. If any of our products were to cause adverse side effects, substantial losses in excess of our insurance coverage could result, which could negatively impact our financial condition, results of operations and cash flows. Our products are intended to be used to treat cancer cases, in which the patient and physician may conclude that the therapeutic benefits of the drug outweigh the potential risk of side effects. Patients who suffer complications may attribute these complications to the drug and, as a consequence, bring product liability actions against us.
Although the clinical trial process is designed to identify and assess potential side effects, it is always possible that a drug, even after approval, may exhibit unforeseen side effects. Such side effects could affect the safety profile of the product. Even if these side effects are not so serious as to warrant withdrawing the product from use, they could reduce the product’s competitive advantage, especially if alternative products offer comparable therapeutic benefits with less severe potential side effects.
We maintain limited product liability insurance for our product candidates when used in clinical trials. In nine countries which have legally mandated amounts of coverage, we have purchased insurance to meet these requirements, which varies widely from€1.0 million per study in Hungary to€50.0 million in Germany. For other countries, such as the United States, where there is no such legal mandate, our insurance coverage is limited to $20 million per occurence and in the aggregate. We expect to obtain more extensive product liability insurance for any products that we may eventually commercialize when it is economical to do so, given the level of premiums and the risk and magnitude of potential liability. We may not be able to obtain insurance on acceptable terms or at all or the amount of insurance we have may not be adequate to cover potential claims or losses. Uninsured losses would adversely affect our profitability.
We may not be able to conduct, or contract others to conduct, animal testing in the future, which could harm our research and development activities.
Certain laws and regulations relating to drug development require us to test our product candidates on animals before initiating clinical trials involving humans. Animal testing activities have been the subject of controversy and adverse publicity. Animal rights groups and other organizations and individuals have attempted to stop animal testing activities by pressing for legislation and regulation in these areas and by disrupting these activities through protests and other means. To the extent the activities of these groups are successful, our research and development activities may be interrupted or delayed.
Risks Related to This Offering
The price of our shares is highly volatile and could decline significantly.
The market price of our ordinary shares historically has been, and we expect will continue to be, subject to significant fluctuations over short periods of time. These fluctuations may be due to factors specific to us, to changes in analysts’ recommendations and earnings estimates, or to factors affecting the biopharmaceutical industry or the securities markets in general. For example, during fiscal year 2003, the closing price per share of our ordinary shares ranged from a low of€ 2.57 on January 28, 2003 to a high of€ 9.25 on October 9, 2003. During the period from January 1, 2004 to June 8, 2004, the price per share of our ordinary shares ranged from€ 7.90 to€ 16.36. We may experience a material decline in the market price of our shares, regardless of our operating performance. Therefore you may not be able to sell your ADSs at or above the offering price.
26
Table of Contents
Price declines in our shares could result from a variety of factors, including many outside our control. These factors include:
| • | adverse results or delays in our clinical trial programs, especially our current Phase 3 registrational trial for satraplatin; |
| • | FDA or other regulatory actions; |
| • | failure of any of our product candidates, if approved, to achieve commercial success; |
| • | announcements of the introduction of new products by us or our competitors; |
| • | market conditions in the pharmaceutical and biotechnology sectors; |
| • | developments or litigation concerning patents, licenses and other intellectual property rights; |
| • | litigation or public concern about the safety of our potential products; |
| • | changes in recommendations or earnings estimates by securities analysts; |
| • | actual and anticipated fluctuations in our quarterly operating results; |
| • | deviations in our operating results from the estimates of securities analysts; |
| • | rumors relating to us or our competitors; |
| • | additions or departures of key personnel; |
| • | changes in third party reimbursement policies; and |
| • | developments concerning current or future strategic alliances or acquisitions. |
In the past, following periods of volatility in the market price of a particular company’s securities, securities class action litigation has often been brought against that company. We may become involved in this type of litigation in the future. Litigation of this type is often extremely expensive and diverts management’s attention and the company’s resources.
There has been no prior market for our shares or ADSs in the United States, and the trading market may not be liquid.
Prior to this offering, there has been no public market for our shares or ADSs in the United States. Our shares are quoted on the Frankfurt Stock Exchange, and we have applied for approval for the quotation of our ADSs on the Nasdaq National Market. We cannot assure you that an active and liquid trading market for our ADSs will develop or will be sustained in the United States. Nor can we assure you that future market prices for our shares or ADSs will equal or exceed the offering price set forth on the cover page of this prospectus. The market prices of securities of biopharmaceutical companies similar to GPC Biotech have historically been highly volatile.
We will have broad discretion in how we use the proceeds of this offering, and we may not use these proceeds effectively, which could affect our results of operations and cause the price of our shares and ADSs to decline.
Our management will have considerable discretion in the application of the net proceeds of this offering. We currently intend to use the net proceeds of this offering to fund clinical trials of satraplatin and to make regulatory filings for marketing approval of satraplatin, as well as to fund our other clinical trials, preclinical studies and research programs and for other corporate purposes. However, our plans may change and we could spend the proceeds in ways that do not necessarily improve our results of operations or enhance the value of our ordinary shares. The failure of our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business. We may use the net proceeds for corporate purposes that do not yield a significant return or any return at all for our shareholders.
27
Table of Contents
We have never paid dividends on our shares, and we do not anticipate paying dividends in the foreseeable future.
We have never paid dividends on our shares. Under German Corporate law, we currently have no ability to pay dividends because of our past losses. We do not expect to have any annual net income for the forseeable future. We currently plan to retain our future annual net income, if any, to fund our product development activities and internal growth. As a result, capital appreciation, if any, of our shares will be your sole potential source of gain for the foreseeable future.
The price of our ADSs and the U.S. dollar value of any dividends will be affected by fluctuations in the U.S. dollar/euro exchange rate.
Our ADSs will trade in U.S. dollars. Fluctuations in the exchange rate between the euro and the U.S. dollar are likely to affect the market price of the ADSs. This could adversely affect the price at which the ADSs trade on the U.S. securities markets. Additionally, any dividend we might pay in the future would be denominated in euro. A decline in the value of the euro against the U.S. dollar would reduce the dollar equivalent of any such dividend.
As a U.S. holder of ordinary shares or ADSs you may not be able to exercise pre-emptive rights and may lose the value of these rights.
Under German law, shareholders generally have pre-emptive rights to subscribe on a pro rata basis for issuances of new shares or other securities giving rights, directly or indirectly, to acquire additional shares or such securities. U.S. holders of ordinary shares or ADSs may not be able to exercise pre-emptive rights for shares unless a registration statement under the U.S. Securities Act of 1933, as amended, is effective with respect to the shares to which the rights relate or an exemption from the registration requirements of that Act is available. We intend to evaluate at the time of any future rights offering the costs and potential liabilities associated with any such registration statement. We also intend to evaluate the indirect benefits of enabling ADS holders to exercise the pre-emptive rights associated with the shares underlying their ADSs, and any other factors we consider appropriate at the time, and then make a decision as to whether to file such a registration statement. If we do not file a registration statement or a registration statement, if filed, is not declared effective in a timely manner and no exemption from the registration requirement is available, you will not be able to exercise your premptive rights.
If pre-emptive rights cannot be exercised by an ADS holder, the depositary will, if possible, sell the holder’s pre-emptive rights and distribute the net proceeds of the sale to the holder. If the depositary determines, in its discretion, that it cannot sell the rights, the depositary may allow the rights to lapse. In either case, ADS holders’ interest in us will be diluted and, if the depositary allows the rights to lapse, ADS holders will not realize any value from the pre-emptive rights.
ADS holders may be subject to additional risks related to holding ADSs rather than shares.
Because ADS holders do not hold their shares directly, they are subject to the following additional risks:
| • | You may instruct the depositary to vote the shares underlying your ADSs, but only if we ask the depositary to ask for your instructions. Otherwise, you will not be able to control voting of shares unless you surrender your ADSs and withdraw the shares from the deposit facility. However, you may not have enough advance notice to withdraw the shares. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that your voting instructions might |
28
Table of Contents
not be carried out and there may be nothing you can do if shares are not voted as you requested. |
| • | ADS holders may not receive copies of all reports from us or the depositary. You may have to go to the depositary’s offices to inspect any reports issued. |
| • | We and the depositary may amend or terminate the deposit agreement without the ADS holders’ consent in a manner that could prejudice ADS holders. |
Future sales of our shares in the public market could adversely affect the price of our shares and ADSs.
The market price of our shares and ADSs could drop due to sales of a large number of our shares in the market or the perception that such sales could occur. These sales could also make it more difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
As of June 4, 2004, members of our Supervisory Board and Management Board together held approximately 4.74% of our outstanding shares. In connection with this offering, we and members of our Management Board will enter into lock-up agreements. Under these agreements, until December 27, 2004, neither we nor members of our Management Board will, subject to certain customary exceptions, issue, offer, sell, contract to sell, pledge or otherwise dispose of any shares or securities convertible into or exchangeable for shares. After the expiration of the lock-up period, we and members of our Management Board will be free to issue, offer, sell or otherwise dispose of shares. Furthermore, this lock-up restriction will not apply to options granted by us to our employees, members of our Supervisory Board and Management Board, pursuant to existing option plans described in this prospectus. These lock-up restrictions also do not apply to up to 150,000 cash settled stock options, which two of the Company’s Management Board members intend to exercise before the end of the combined offering but after the publication of the initial offer price. See “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”.
Your rights as a shareholder in a German corporation may differ from your rights as a shareholder in a U.S. corporation.
We are organized as a stock corporation (Aktiengesellschaft) under the laws of Germany, and by participating in the global offering you will become a shareholder in a German corporation. You should be aware that the rights of shareholders under German law differ in important respects from those of shareholders in a U.S. corporation. These differences include, in particular:
| · | All public offers for the acquisition of shares, securities comparable to shares and certificates representing shares, such as ADSs, as well as bids for other types of securities representing the right to acquire shares, securities comparable to shares and certificates representing shares are subject to the provisions of German takeover laws. In particular, the creation or transfer, directly or indirectly, of 30% or more of the voting rights in a public German corporation generally triggers the obligation to launch a mandatory bid for all outstanding shares in that German corporation. |
| · | Under German law, certain important resolutions, such as resolutions relating to changes in the articles of association of a German corporation, any increases or decreases in a German corporation’s share capital, the issuance of convertible bonds or bonds with warrants attached and the dissolution of the German corporation apart from insolvency and certain other proceedings, require a 75% majority of the votes present or represented at the shareholders’ meeting. Therefore, the holder or holders of a blocking minority of 25% or, depending on the attendance level at the shareholders’ meeting, the holder or holders of a |
29
Table of Contents
smaller percentage of the shares in a German corporation may be able to block any such votes, possibly to the detriment of the German corporation and the other shareholders. |
| · | As a general rule under German law, a shareholder has no direct recourse against the members of the management board or supervisory board of a German corporation in the event that it is alleged they have breached their duty of loyalty and care to the German corporation. Apart from insolvency or other special circumstances, only the German corporation itself has the right to claim damages from members of either board. A German corporation may waive or settle these damages only if at least three years have passed and the shareholders approve the waiver or settlement at the shareholders’ meeting with a simple majority of the votes cast, provided that a minority holding, in the aggregate, 10% or more of the German corporation’s share capital does not have their opposition formally noted in the minutes maintained by a German notary. |
In addition, as a non-U.S. corporation, we are not subject to some of the provisions of U.S. securities laws that apply to U.S. issuers. We are not subject to the proxy rules promulgated under Section 14 of the U.S. Securities Exchange Act of 1934, or Exchange Act. Additionally, short-swing profit and recovery provisions under Section 16 of the Exchange Act do not apply to us or the members of our Management Board and Supervisory Board. Furthermore, as a foreign private issuer, we are not required to file quarterly reports on Form 10-Q or current reports on Form 8-K with the U.S. Securities and Exchange Commission. In accordance with German law and the rules and regulations promulgated by the Frankfurt Stock Exchange, GPC Biotech’s historical practice, however, has been to publish its three-, six- and nine-month interim financial information. We expect to continue this practice, and to file this information with the U.S. Securities and Exchange Commission in the United States on Form 6-K.
For more information, we have provided summaries of relevant provisions of German law and of our Articles of Association in “Directors, Senior Management and Employees” and “Description of Share Capital.”
We may become a passive foreign investment company, which could result in adverse U.S. tax consequences to U.S. investors.
Depending on various factors, we could be classified, for United States income tax purposes, as a “passive foreign investment company,” or PFIC. This characterization could result in adverse U.S. federal income consequences for any U.S. holder of our shares or ADSs. We could be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after taking into account the income and assets of our subsidiary, either:
| • | at least 75% of our gross income is passive income, or |
| • | at least 50% of the average value of our assets is attributable to assets that produce or are held to produce passive income. |
If we are treated as a PFIC for any taxable year U.S. investors in our shares or ADSs may desire to make an election to treat us as a “qualified electing fund” with respect to shares or ADSs owned (a “QEF election”), in which case U.S. investors will be required to take into account a pro rata share of our earnings and net capital gain for each year, regardless of whether we make any distributions. As an alternative to the QEF election, a U.S. investor may be able to make an election to “mark-to-market” our shares each taxable year and recognize ordinary income pursuant to such election based upon increases in the value of our shares.
30
Table of Contents
Absent an election described above, a U.S. investor of a PFIC would be subject to adverse tax consequences, including:
| • | U.S. federal income tax at ordinary income tax rates in respect of gain derived from a disposition of our shares, as well as some distributions by us; |
| • | interest charges on taxes allocable to prior periods; and |
| • | no increase in the tax basis for our shares or ADSs to fair market value upon the death of an individual shareholder. |
We do not believe that we are currently a PFIC. The determination of whether we are a PFIC is made on an annual basis and will depend on factors such as the composition of our income and assets from time to time as well as our share price. Therefore, it is possible that we could be classified as a PFIC for any particular year.
31
Table of Contents
This prospectus includes forward-looking statements. Forward-looking statements may be, but are not necessarily, identified by words like “believe”, “anticipate”, “intend”, “expect”, “target”, “goal”, “estimate”, “plan”, “assume”, “may”, “will”, “could” and similar expressions. Forward-looking statements include, but are not limited to, statements about:
| • | the progress, timing and completion of our research, development and preclinical studies and clinical trials for our product candidates; |
| • | our ability to market, commercialize, achieve market acceptance for and sell our product candidates; |
| • | our ability to adequately protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; and |
| • | our estimates regarding anticipated operating losses, future revenues, capital requirements and our needs for additional financing. |
We have based these forward-looking statements on our current expectations and projections about future events. These forward-looking statements are subject to risks, uncertainties and assumptions about us, including those listed in the sections of this prospectus entitled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this prospectus.
In light of these risks, uncertainties and assumptions, the forward-looking events discussed in this prospectus might not occur. Except as required by law, we do not undertake any obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
32
Table of Contents
The shares being offered by this prospectus are part of a combined offering of up to 8,279,000 newly issued shares offered by the Company (including an over-allotment option of up to 1,119,000 new shares) and 300,000 existing shares offered by selling shareholders. The combined offering consists of a rights offering to existing holders of our shares and a global offering consisting of public offerings in Germany and the United States and private placements with institutional investors elsewhere.
We are offering up to 7,152,047 newly issued shares in the rights offering to existing holders of our shares (see “Plan of Distribution”). To avoid a fractional subscription ratio, we have excluded shareholders’ subscription rights for a residual amount of 7,953 newly issued shares in accordance with German law and our Articles of Association. We intend to offer those new shares as to which subscription rights have been excluded or not exercised during the subscription period for sale through the underwriters in the global offering. In addition, the global offering will include 300,000 existing shares to be sold by certain management shareholders. See “Principal and Selling Shareholders”.
You should bear in mind that the combined offering may be terminated at any time before the signing of the underwriting agreement and the registration of the capital increase from authorized capital (see “Description of Share Capital—Authorized Capital”) with the commercial register (Handelsregister) for GPC Biotech AG relating to the combined offering, even with respect to rights that have been exercised. You should also bear in mind that if the combined offering is terminated, acquirors of rights in the secondary market may suffer a loss upon the rights becoming worthless. See “—Important Notices”.
We expect the underwriting agreement to be signed on or about June 28, 2004 and the capital increase to be registered with the commercial register on June 28, 2004. After the underwriting agreement is signed, the combined offering may be terminated only if all parties to the underwriting agreement so agree or if certain conditions are met. After the capital increase is registered, shareholders who have effectively exercised their rights will be entitled to receive the related shares.
The Rights Offering
Our existing shareholders will receive transferable rights to subscribe for one new share for every three shares they hold. Clearstream Banking AG will automatically credit the subscription rights for shares that are held in collective custody as of the evening of June 14, 2004 to the relevant depositary banks for further credit to the accounts of holders. The subscription period will begin on June 15, 2004 and end on June 28, 2004 (both dates inclusive).
We will determine the subscription price at the latest by 12:00 p.m. (noon)(Frankfurt time) on June 25, 2004. The subscription price will lie between or at (x) the volume-weighted average price of the shares from the beginning of the subscription period until the determination of the subscription price and (y) the current market price of our shares at the time of the determination of the subscription price. In determining the subscription price, we will, among other things, consider current market conditions and the preliminary results of the bookbuilding process contemplated for the shares to be offered in the global offering.
The subscription price will be published through electronic media (Reuters) immediately after being determined and in the electronic version of the German Federal Gazette (elektronischer Bundesanzeiger) on the first practicable date after determination, at the latest on June 25, 2004.
33
Table of Contents
As soon as practicable after determination, the subscription price also will be published in the Frankfurter Allgemeine Zeitung and made available on GPC Biotech’s website at http://www.gpc-biotech.com.
The subscription rights (ISIN DE000A0AYYP6) will be traded on the Frankfurt Stock Exchange from June 15, 2004 to June 24, 2004 (both dates inclusive). If shareholders who hold their ordinary shares through a custodian bank have not exercised their rights by delivering a letter to their custodian bank at or about 10:00 a.m. Frankfurt time (depending on the respective custodian bank’s deadline) on June 24, 2004 the respective custodian bank will, in accordance with the general terms and conditions of the custody arrangements as standardized in Germany, sell the rights allocated to these shareholders for their accounts on a best-efforts basis.
If you are a shareholder and are interested in subscribing for new shares in the rights offering, the procedures for doing so are described below under the heading “The Rights Offering”. That section also contains information about how you may sell your subscription rights. If you are interested in purchasing rights, whether or not you currently hold GPC Biotech shares, you will also find important information under the heading “The Rights Offering”, including specific notices relating to the risks for existing holders and investors resulting from the determination of the subscription price during the subscription period.
Shareholders who exercise subscription rights will only receive shares and not ADSs. Shareholders may, however, elect to deposit their shares (including new shares acquired in the rights offering) with the depositary for our ADSs to hold their shares in the form of ADSs.
The Global Offering
The global offering will consist of public offerings in Germany and the United States and private placements with institutional investors in certain other jurisdictions. The residual share amount as to which shareholders’ subscription rights have been excluded and any new shares not subscribed to in the rights offering will be offered by GPC Biotech, together with shares offered by management shareholders, through the underwriters in the global offering. The number of shares offered in the global offering and the initial offer price to public will be determined at the end of the bookbuilding period. The bookbuilding period will begin on June 15, 2004 and is expected to end on or about June 29, 2004.
The initial offer price to public in the global offering will be published through electronic media (Reuters) immediately after being determined on or about June 29, 2004. The initial offer price to the public will be published in the Frankfurter Allgemeine Zeitung and The Wall Street Journal on or about July 1, 2004.
As part of the global offering, we will grant an option to the underwriters to purchase up to 1,119,000 additional new shares to cover over-allotments for which shareholders’ subscription rights will be excluded.
Important Notices
As described above, the subscription price in the rights offering is to be determined at the latest by June 25, 2004. The initial offer price in the global offering is expected to be determined on June 29, 2004 on the basis of a bookbuilding process. Depending on the development of our share price and the result of the bookbuilding process, the subscription price may therefore be higher than the initial offer price in the global offering. Our existing shareholders should be aware that they may not be able to acquire shares by exercising their subscription rights in the rights offering for a lesser price than the price that they would have to pay when participating in the global offering or purchasing our shares in the market.
34
Table of Contents
The underwriters reserve the right to terminate the underwriting agreement for certain reasons or to delay the completion of the combined offering by up to one week. These reasons include, without limitation, material adverse changes in the financial position or results of operations or shareholders’ equity of GPC Biotech and its subsidiary, taken together, material restrictions on exchange trading or on banking business, the eruption or escalation of hostilities that have, or can be expected to have, a material adverse effect on the financial markets, and the failure to register the implementation of the capital increase with the commercial register by 12:00 p.m. (noon) (Frankfurt time) on June 28, 2004.
In the event of a termination of the underwriting agreement prior to the registration of the implementation of the capital increase in the commercial register, the subscription rights become void. If this were to occur, brokerage institutions generally would not reverse any subscription rights trading transactions. Accordingly, investors who would have acquired subscription rights on a stock exchange would suffer a loss. To the extent that the underwriters terminate the underwriting agreement after the registration of the implementation of the capital increase in the commercial register, existing shareholders who have exercised their subscription rights will receive the new shares at the subscription price.
Form and Settlement
The shares offered in the combined offering will be represented by global share certificates with global dividend coupons attached. The global shares will be deposited with Clearstream Bank AG, or Clearstream Frankfurt, as share depositary. The shares purchased in the combined offering may be credited at the option of investors either to the account of a German bank with Clearstream Frankfurt for the account of such investor or to the accounts of participants with Euroclear or Clearstream Luxembourg. According to our Articles of Association, shareholders have no right to receive physical share certificates. See “Description of Share Capital Form, Certification and Transferability of the Shares.”
It is expected that delivery of the shares purchased pursuant to subscription rights will be made against payment therefor on the fourth business day following the expiration of the rights offering. Under Rule 15c6-1 of the Securities Exchange Act of 1934, as amended, trades in the secondary market generally are required to settle in three business days, unless the parties to a trade expressly otherwise agree.
The International Securities Identification Code, or ISIN, for the shares is DE0005851505. The German Securities Code (Wertpapier-Kenn-Nummer, or WKN) for the shares is 585150, and the Common Code for the shares is 011060854.
35
Table of Contents
We estimate that the net proceeds to us from the combined offering will be approximately€86.9 million, or€101.0 million if the underwriters’ over-allotment option is exercised in full, after deducting underwriting discounts and commissions of approximately€6.2 million, or approximately€7.2 million if the underwriters’ over-allotment option is exercised in full, and estimated offering expenses payable by us, of approximately€2.8 million (based on an assumed offering price of€13.40 per share, which was the closing market price on the XETRA electronic trading platform of the Frankfurt Stock Exchange for the shares on June 8, 2004 and an assumed exchange rate of $1.23/€).
We intend to use these proceeds to continue to fund clinical trials of satraplatin, to make regulatory filings for marketing approval of satraplatin, and to fund our other clinical, preclinical and research programs, including our monoclonal antibody program (1D09C3) and cell cycle inhibitor program (RGB-286199).
We currently estimate that from the net proceeds of this offering we will spend between 50% and 60% to continue to fund clinical trials of satraplatin, to broaden the potential anticancer applications of this drug candidate, and to make regulatory filings for marketing approval of satraplatin; between 20% and 30% to fund our other clinical, preclinical and research programs, including our monoclonal antibody program (1D09C3) and cell cycle inhibitor program (RGB-286199), and to invest in new capabilities relating to our research and development activities; and between 15% and 25% for general corporate purposes, which may include the licensing of other product candidates, technologies, or intellectual property.
The amounts and timing of our actual expenditure will depend on numerous factors, including the timing and success of our Phase 3 registrational trial for satraplatin, the timing and success of any clinical trials and preclinical studies we may commence in the future, the timing of regulatory submissions, the status of our research and development efforts, the amounts of proceeds actually raised in this offering, the timing and scope of potential partnering and licensing activities for our product candidates and technologies as well as for new product candidates and technologies and the amount of cash generated by our operations. Because we operate in a very dynamic and highly competitive industry, the actual use of proceeds may differ substantially from the ranges indicated above. Our management will have broad discretion to allocate the net proceeds from this offering.
Pending our use of the net proceeds, we intend to invest them in short-term and medium-term interest-bearing instruments.
We will not receive any proceeds from the sale of shares by the selling shareholders.
36
Table of Contents
DIVIDEND POLICY AND LIQUIDATION PROCEEDS
We have never paid any dividends on our ordinary bearer shares. Under German corporate law, we currently have no ability to pay dividends because of our past losses. We do not expect to have any annual net income. If we were to earn annual net income, we currently plan to retain such annual net income for the foreseeable future, if any, to finance business development and internal growth in the foreseeable future. We therefore do not anticipate to pay dividends in the foreseeable future.
Under German law, GPC Biotech may pay dividends only from retained earnings (Bilanzgewinn)reflected in its unconsolidated financial statements (as opposed to the consolidated financial statements for GPC Biotech and its subsidiary) prepared in accordance with the principles set forth in the German Commercial Code (Handelsgesetzbuch) and as adopted and approved by the Management Board (Vorstand) and the Supervisory Board (Aufsichtsrat). In determining the retained earnings that may be distributed as dividends, under German law, the Management Board may allocate to earnings reserves (Gewinnrücklagen) up to 50% of GPC Biotech’s remaining net income for the fiscal year after deducting amounts to be allocated to legal and statutory reserves and losses carried forward. The Management Board may also increase retained earnings when preparing the financial statements with funds withdrawn from earnings reserves.
Our shareholders, in their resolution on the appropriation of retained earnings, may carry forward distributable retained earnings in part or in full and may allocate additional amounts to earnings reserves. Profits carried forward will be automatically incorporated in the retained earnings of the next fiscal year. Amounts allocated to the earnings reserves are available for dividends only if and to the extent the earnings reserves have been dissolved by the Management Board when preparing the financial statements, thereby increasing the retained earnings.
Our shareholders may declare dividends at an ordinary general shareholders’ meeting, which must be held within the first eight months of each fiscal year. Dividends approved at an ordinary general shareholders’ meeting are payable promptly after the meeting, unless otherwise decided at the meeting. Because all of our shares are in book-entry form represented by one or more global certificates deposited with Clearstream Banking AG in Frankfurt am Main, Germany, shareholders receive dividends through Clearstream Frankfurt for credit to their deposit accounts.
Apart from liquidation as a result of insolvency proceedings, GPC Biotech may be liquidated only with a majority of three-quarters of the share capital present or represented at a shareholders’ meeting at which the vote is taken. In accordance with the German Stock Corporation Act (Aktiengesetz), upon a liquidation of GPC Biotech, any liquidation proceeds remaining after paying off all of GPC Biotech’s liabilities would be distributed among the shareholders in proportion to the number of shares held by each shareholder.
Dividends are subject to German withholding tax. See “German Taxation—Withholding Tax”.
37
Table of Contents
Prior to the offering, there has been no public market in the United States for our shares or the ADSs. We have applied for the quotation of the ADSs on the Nasdaq National Market under the symbol “GPCB”.
Our shares are currently traded on the Frankfurt Stock Exchange under the symbol “GPC”. Our shares are also included in the TecDAX index, which includes certain large companies in the technology segment of the Frankfurt Stock Exchange.
The table below sets forth, for the periods indicated, the high and low closing prices in euro of our shares as reported by the Frankfurt Stock Exchange Xetra trading system:
| High | Low | |||
2000 | €70.00 | €22.78 | ||
2001 | 30.50 | 7.50 | ||
2002: | ||||
First quarter | 13.47 | 8.90 | ||
Second quarter | 9.95 | 4.49 | ||
Third quarter | 6.20 | 2.36 | ||
Fourth quarter | 3.71 | 2.17 | ||
2003: | ||||
First quarter | 3.15 | 2.57 | ||
Second quarter | 5.98 | 2.91 | ||
Third quarter | 7.98 | 4.32 | ||
Fourth quarter | 9.25 | 7.52 | ||
2004: | ||||
First quarter | 16.36 | 7.90 | ||
Previous six months: | ||||
December 2003 | 8.32 | 7.78 | ||
January 2004 | 11.80 | 7.90 | ||
February 2004 | 16.36 | 12.81 | ||
March 2004 | 15.70 | 13.26 | ||
April 2004 | 13.29 | 11.20 | ||
May 2004 | 12.50 | 10.68 | ||
The average daily volume of our shares traded on the Frankfurt Stock Exchange both on the XETRA electronic trading platform and on the stock exchange floor for the years 2003, 2002 and 2001 was 163,764, 86,495 and 82,327, respectively. In the first quarter of 2004, the average daily volume of our shares traded on the Frankfurt Stock Exchange was 398,697.
On June 8, 2004, the closing price of our shares on the Frankfurt Stock Exchange was€13.40.
38
Table of Contents
The following table shows, for the years and dates indicated, certain information concerning the rate of exchange of euro per U.S. dollar based on the Noon Buying Rates quoted by the Federal Reserve Bank of New York for euros expressed in U.S. dollar for one euro. No representation is made that the euro or the U.S. dollar amounts referred to herein could have been or could be converted into U.S. dollars or euros, as the case may be, at any particular rate.
| U.S. dollar for one euro | ||||||||
Year | High | Low | Period Average(1) | Period End | ||||
2003 | 1.2597 | 1.0361 | 1.1411 | 1.2597 | ||||
2002 | 1.0485 | 0.8594 | 0.9495 | 1.0485 | ||||
2001 | 0.9520 | 0.8370 | 0.8909 | 0.8901 | ||||
2000 | 1.0335 | 0.8270 | 0.9207 | 0.9388 | ||||
1999 | 1.1812 | 1.0016 | 1.0588 | 1.0071 | ||||
| (1) | The average of the Noon Buying Rates on the last business day of each full month during the relevant period. |
The high and low exchange rates for the euro for each month during the previous six months is set forth below:
| U.S. dollar for one euro | ||||
Month | High | Low | ||
December 2003 | 1.2597 | 1.1956 | ||
January 2004 | 1.2853 | 1.2389 | ||
February 2004 | 1.2848 | 1.2426 | ||
March 2004 | 1.2431 | 1.2088 | ||
April 2004 | 1.2358 | 1.1830 | ||
May 2004 | 1.2274 | 1.1801 | ||
The Noon Buying Rate for the euro on June 8, 2004 was quoted by the Federal Reserve Bank of New York at 1.2273 U.S. dollars for one euro.
39
Table of Contents
The following table sets forth our capitalization at March 31, 2004, on an actual basis and as adjusted to reflect the sale of 7,160,000 shares and ADSs in the combined offering, at an assumed offering price of€13.40 per share (based on the closing market price on the XETRA electronic trading platform of the Frankfurt Stock Exchange for the shares on June 8, 2004), and after deducting underwriting discounts and commissions and estimated offering expenses of approximately€9.0 million payable by us.
At March 31, 2004, we had cash and cash equivalents of€18.2 million, marketable securities and short-term investments of€63.8 million and restricted cash of€2.6 million.
| At March 31, 2004 | ||||||
| Actual | As Adjusted | |||||
| € | € | |||||
| (amounts in thousands, except per share data) | ||||||
Long-term debt, net of current portion | 575 | 575 | ||||
Capital lease obligations, net of current portion | 260 | 260 | ||||
Shareholders’ equity: | ||||||
Ordinary shares, with no par value (notional par value of€1 per share), (21,334,229 shares issued and outstanding at March 31, 2004; 28,494,229 shares issued and outstanding as adjusted for this offering of 7,160,000 new shares)(1) | 21,334 | 28,494 | ||||
Additional paid-in-capital | 191,566 | 271,350 | ||||
Subscribed shares | 315 | 315 | ||||
Accumulated other comprehensive loss | (1,799 | ) | (1,799 | ) | ||
Accumulated deficit | (134,248 | ) | (134,248 | ) | ||
Total shareholders’ equity | 77,168 | 164,112 | ||||
Total capitalization | 78,003 | 164,947 | ||||
| (1) | The actual number of shares shown as issued and outstanding excludes 4,344,327 ordinary shares issuable upon the exercise of stock options, convertible bonds and warrants outstanding as of March 31, 2004, with exercise prices ranging from€1.16 to€59.74. The actual number of shares does not include 121,911 shares issued upon the exercise of stock options and convertible bonds in April 2004. |
40
Table of Contents
Our net tangible book value at March 31, 2004 was€75.7 million or€3.55 per share. Based on an assumed offering price per share of€13.40 (based on the closing market price on the XETRA electronic trading platform of the Frankfurt Stock Exchange for the shares on June 8, 2004), purchasers of shares in the offering will experience an immediate dilution of€7.69 or 57% per share. This information has not been adjusted to give effect to the exercise of options and other convertible securities that occurred between April 1, 2004 and June 8, 2004 for an aggregate of 121,911 shares.
| • | “Net tangible book value per share” is determined by dividing our consolidated net tangible book value by the number of our shares outstanding. |
| • | “Net tangible book value” is calculated by subtracting goodwill and other intangible assets, total liabilities and minority interests from total assets. |
| • | “Dilution per share” is the difference between the price per share paid by purchasers in this offering and our pro forma consolidated net tangible book value per share after the offering. |
The following table illustrates this dilution per share or ADS.
| Per share or ADS (€) | ||
Offering price(1) | 13.40 | |
Consolidated net tangible book value at March 31, 2004 | 3.55 | |
Dilution in consolidated net tangible book value | 7.69 | |
Dilution if over-allotment option exercised in full | 7.43 |
| (1) | Based on the closing market price of the shares on June 8, 2004. |
The following table summarizes, on an adjusted basis to give effect to the offering and to give effect to all options and convertible bonds exerciseable as of March 31, 2004, the number of shares purchased from us or granted by us, the total consideration paid and the average price per share paid (before deducting underwriting discount and estimated offering expenses and based on an assumed offering price of€13.40 per share and ADS):
| Number of Shares | Total Consideration | |||||||||
| Number(1) | Percent(2) | Amount(3) (€ in thousands) | Average price per share or ADS (€) | |||||||
Members of the Management Board and Supervisory Board | 1,955,400 | (4) | 6.6 | 9,270 | 4.74 | |||||
Purchasers of new shares in this offering | 7,160,000 | 25.1 | 95,944 | 13.40 | (5) | |||||
| (1) | This amount includes shares for which options or convertible bonds were not in fact exerciseable on March 31, 2004 due to minimum market price requirements pursuant to the respective stock option plan or convertible bond program. |
| (2) | These percentages are based on the number of shares outstanding as of March 31, 2004, adjusted for the offering of 7,160,000 new shares, and, in the case of the percentage of shares of the members of the Supervisory Board and the Management Board, also adjusted to include shares issuable upon the exercise of options and convertible bonds exercisable as of March 31, 2004. |
| (3) | This amount includes purchases in U.S. dollars which have been converted into euro on the basis of the exchange rate as of March 31, 2004. |
| (4) | This amount includes 937,800 shares issuable upon the exercise of all options and convertible bonds exerciseable as of March 31, 2004. |
| (5) | Based on the closing market price of the shares on June 8, 2004. |
The table above assumes no exercise of the underwriters’ over-allotment option.
41
Table of Contents
SELECTED CONSOLIDATED FINANCIAL INFORMATION
The following table presents our selected consolidated financial information. We prepare our consolidated financial statements in accordance with U.S. GAAP. The selected financial data for the three years ended December 31, 2003 have been derived from our consolidated financial statements, which have been audited by Ernst & Young AG, Wirtschaftsprüfungsgesellschaft, independent registered public accounting firm. The selected financial data for the two years ended December 31, 2000 have been derived from our consolidated financial statements, which have been audited by Ernst & Young Deutsche Allgemeine Treuhand AG, Wirtschaftsprüfungsgesellschaft, independent auditors, in accordance with auditing standards generally accepted in Germany. The consolidated balance sheets at December 31, 2003, 2002 and 2001 and the related consolidated statements of operations and cash flows for each of the three years in the period ended December 31, 2003 and notes thereto appear elsewhere in this prospectus.
The financial data for the three month periods ended March 31, 2004 and 2003 are derived from unaudited interim consolidated financial statements. The unaudited interim consolidated financial statements include all adjustments, consisting of normal recurring accruals, which we consider necessary for a fair presentation of the financial position and the results of operations for these periods.
Operating results for the three months ended March 31, 2004 are not necessarily indicative of the results that may be expected for the entire year ending December 31, 2004. The data should be read in conjunction with the consolidated financial statements, related notes and other financial information included in this prospectus.
You should read the following selected consolidated financial information in conjunction with the section of this prospectus entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the notes thereto contained elsewhere in this prospectus.
| Three months ended March 31, | Year ended December 31, | ||||||||||||||||||||
| 2004 | 2003 | 2003 | 2002 | 2001 | 2000 | 1999 | |||||||||||||||
| € | € | € | € | € | € | € | |||||||||||||||
| (amounts in thousands, except share and per share data) | |||||||||||||||||||||
Statement of operations data: | |||||||||||||||||||||
Total revenues | 3,959 | 6,112 | 21,594 | 21,511 | 13,889 | 5,936 | 4,264 | ||||||||||||||
Research and development expenses | 8,682 | 8,323 | 37,741 | 38,053 | 30,664 | 20,404 | 5,280 | ||||||||||||||
Impairment of goodwill | — | — | — | 7,314 | — | — | — | ||||||||||||||
Acquired in-process research and development | — | — | — | — | — | 15,974 | — | ||||||||||||||
Total operating expenses | 11,569 | 11,112 | 51,068 | 57,907 | 45,239 | 46,013 | 6,802 | ||||||||||||||
Operating loss | (7,610 | ) | (5,000 | ) | (29,474 | ) | (36,396 | ) | (31,350 | ) | (40,077 | ) | (2,538 | ) | |||||||
Net loss | (6,925 | ) | (4,162 | ) | (26,831 | ) | (32,934 | ) | (26,217 | ) | (34,783 | ) | (3,305 | ) | |||||||
Per share data: | |||||||||||||||||||||
Basic and diluted loss per share | (0.33 | ) | (0.20 | ) | (1.29 | ) | (1.59 | ) | (1.42 | ) | (2.39 | ) | (0.54 | ) | |||||||
Shares used in computing basic and diluted loss per share | 21,085,710 | 20,724,174 | 20,731,535 | 20,688,515 | 18,509,398 | 14,572,917 | 6,118,750 | ||||||||||||||
42
Table of Contents
| At March 31, 2004 | At December 31, | |||||||||||||||||||
| 2003 | 2002 | 2001 | 2000 | 1999 | ||||||||||||||||
| € | € | € | € | € | € | |||||||||||||||
| (amounts in thousands) | ||||||||||||||||||||
Balance sheet data: | ||||||||||||||||||||
Cash and cash equivalents(1) |
| 18,201 | 34,947 | 39,947 | 91,245 | 55,108 | 4,787 | |||||||||||||
Marketable securities and short-term investments |
| 63,783 | 54,221 | 74,935 | 48,885 | 53,573 | 14,928 | |||||||||||||
Total assets |
| 97,117 | 101,564 | 132,333 | 167,860 | 132,739 | 22,341 | |||||||||||||
Long term debt, including capital lease obligations |
| 835 | 959 | 1,528 | 1,680 | 1,952 | 9,253 | |||||||||||||
Total shareholders’ equity |
| 77,168 | 81,879 | 107,270 | 139,093 | 124,516 | 10,341 | |||||||||||||
| Three months ended March 31, | Year ended December 31, | |||||||||||||||||||
| 2004 | 2003 | 2003 | 2002 | 2001 | 2000 | 1999 | ||||||||||||||
| € | € | € | € | € | € | € | ||||||||||||||
| (amounts in thousands) | ||||||||||||||||||||
Other financial data: | ||||||||||||||||||||
Net cash provided by (used in) operating activities | (8,268 | ) | (5,390 | ) | (22,974 | ) | (23,537 | ) | 2,372 | (17,581 | ) | (150 | ) | |||||||
Net cash provided by (used in) investing activities | (9,783 | ) | (14,084 | ) | 18,723 | (26,771 | ) | 1,477 | (39,878 | ) | (14,797 | ) | ||||||||
Net cash provided by (used in) financing activities | 1,176 | (102 | ) | 42 | (255 | ) | 35,019 | 107,709 | 19,381 | |||||||||||
Depreciation, amortization and impairment of tangible and intangible assets and goodwill | 560 | 678 | 3,827 | 10,840 | 6,616 | 4,923 | 570 | |||||||||||||
| (1) | Does not include restricted cash of€2.5 million,€3.0 million and€3.5 million at December 31, 2003, 2002 and 2001, respectively and€2.6 million at March 31, 2004. |
43
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
You should read the following discussion of our financial condition and results of operations in conjunction with the consolidated financial statements and the notes to our financial statements included elsewhere in this prospectus. This discussion contains forward-looking statements that involve risks, uncertainties and assumption. As a result of many factors, including those set forth under the section entitled “Risk Factors” and elsewhere in this prospectus, our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
GPC Biotech is a biopharmaceutical company discovering and developing new drugs to treat cancer. Our lead product candidate is satraplatin, an oral platinum-based compound intended for use as a chemotherapy drug. Satraplatin is currently being tested in a Phase 3 registrational trial as a second-line chemotherapy treatment for hormone refractory prostate cancer, or HRPC. We licensed satraplatin in 2002 and, in September 2003, initiated a Phase 3 registrational trial of satraplatin as a second-line chemotherapy treatment for HRPC in the United States, Europe and South America. Our goal is to complete the filing of an NDA with the FDA for satraplatin as a treatment for HRPC in 2006.
Our second most advanced product candidate is 1D09C3, a monoclonal antibody that is in late preclinical development and is intended for the treatment of selected leukemias and lymphomas. We plan to complete a series of preclinical toxicology evaluations and the development of certain tests for monitoring drug levels and drug responses in the blood of treated patients in 2004. Assuming successful and timely completion of these preclinical tests and timely approval of the relevant regulatory authorities and ethics committees, we anticipate initiating clinical trials for this product candidate in 2004. Our third development program involves small molecule cell cycle inhibitors, from which we have identified a lead compound, RGB-286199, that is currently in preclinical development. Our goal is to complete preclinical development work on RGB-286199 in the first half of 2005.
We also have a portfolio of proteomics and genomics technologies, which we use in our internal drug discovery efforts and which we have licensed to or used in collaborations with various pharmaceutical companies. In 2001, we entered into an agreement with ALTANA Pharma to assist ALTANA Pharma through 2007 in the establishment of a research institute in the United States. This alliance includes a research collaboration as well as a transfer of technologies. We have also licensed LeadCode to ALTANA Pharma as of January 2003, under a separate agreement. ALTANA Pharma is our largest customer, accounting for 94% of our total revenues in 2003, 81% of our total revenues in 2002, and 58% of our total revenues in 2001. As of June 4, 2004, an affiliate of ALTANA Pharma owned 7.88% of our outstanding share capital.
Since our inception in August 1997, we have incurred significant net losses. As of December 31, 2003, we had an accumulated deficit of€127.3 million. We expect to incur substantial and increasing losses for the next several years:
| • | as satraplatin advances through Phase 3 development and commercialization; |
| • | if we initiate additional clinical trials involving satraplatin; |
| • | as our monoclonal antibody (1D09C3) and cell cycle inhibitor (RGB-286199) advance through preclinical studies and into clinical development; |
| • | if we advance other product candidates discovered by us in research programs into preclinical studies and clinical development; |
| • | as we expand our research programs and further develop our proprietary drug discovery technologies to broaden our pipeline of preclinical product candidates; and |
| • | if we license additional anticancer product candidates. |
44
Table of Contents
Over the past five years, we have funded our operations primarily through the sale of equity securities, payments received under our agreements with ALTANA Pharma and other pharmaceutical companies, interest earned on investments, government grants, capital lease financings and long-term debt. We expect to continue to fund our operations over the next several years primarily through the net proceeds of this offering, our existing cash resources, future payments received from ALTANA Pharma and interest earned on investment.
At December 31, 2003, we held€34.9 million of cash and cash equivalents and€54.2 million of short-term investments and marketable securities. In addition, we also had€2.5 million in restricted cash at that date.
Revenue
All of our product candidates are currently in development, and we therefore do not expect to generate any direct revenue from drug product sales for at least the next several years. We will be required to successfully complete clinical trials for satraplatin, our most advanced product candidate, and obtain required regulatory approvals before we can market satraplatin. We do not expect to file an NDA for satraplatin until 2006. Our other two product candidates are in preclinical studies. Assuming successful and timely completion of preclinical tests and timely approval by the relevant regulatory authorities and ethics committees, we expect to advance one of these product candidates into clinical trials in 2004 and to complete preclinical development of the other in the first half of 2005. Any delay in obtaining or failure to obtain regulatory approval for satraplatin will materially adversely affect our ability to generate revenues from drug sales in the next several years.
A substantial portion of our historical and current revenues are generated from our alliance with ALTANA Pharma. ALTANA Pharma accounted for 94% of our total revenues in 2003, 81% of our total revenues in 2002 and 58% of our total revenues in 2001. As of December 31, 2003 we had received a total of€59.3 million in payments under various arrangements with ALTANA Pharma. In 2002 and 2001, Boehringer Ingelheim accounted for 11% and 15%, respectively of our total revenues. In 2001, Aventis accounted for 13% of total revenues, and theBundesministerium für Bildung und Forschung (the German Federal Ministry for Education and Research, or BMBF) accounted for 11% of total revenues. No other customer accounted for more than 10% of our total revenues in 2003, 2002 and 2001.
We have entered into various collaborations with ALTANA Pharma. Our current collaboration with ALTANA Pharma, which we entered into in 2001, is our most significant technology collaboration to date. This agreement, which expires in June 2007, includes a research collaboration, which expires in 2005 as well as a transfer of technologies. Effective January 2003, we entered into another agreement with ALTANA Pharma pursuant to which we licensed LeadCode, our drug-protein interaction technology. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees and research funding, over the term of the agreements. As of December 31, 2003 we have received a total of€37.4 million under these agreements, of which€11.1 million were upfront payments. Of the total of€37.4 million received,€27.4 million have been recognized as revenue. ALTANA Pharma is also obligated to make certain milestone payments to us in connection with the development and commercialization of products resulting from our research collaborations. We are also entitled to receive royalties on the net sales of any products resulting from these research collaborations that receive regulatory approval and are marketed by ALTANA Pharma. Past collaborations include research collaborations in the field of anti-infectives in 1998 and oncology in 2001, both of which expired in 2003.
We expect that our future revenues will be derived primarily from licensing various rights to our anticancer drugs under development and from royalties on sales from drugs developed by us and licensed to other biotechnology or pharmaceutical companies, as well as from direct product sales of our drugs and, to a lesser and decreasing extent, collaboration arrangements and technology licensing.
45
Table of Contents
Research and Development Expenses
We incur development expenses related to our clinical and preclinical drug development programs. We also incur research expenses associated with both partnered and unpartnered research activities, as well as the development and maintenance of our drug discovery technologies.
The expenses we incur in connection with our research, preclinical and clinical development activities consist primarily of costs of contract research and consultants, costs of employee compensation, costs of clinical drug supplies, other supplies and materials, intellectual property, travel and facilities costs. We also incur licensing fees, milestone achievement fees and royalties in the case of several of our technologies and drug discovery and development programs.
Development timelines and costs for our drug discovery programs and for development of our product candidates are difficult to estimate and may vary significantly for each drug candidate and from quarter to quarter. Research and development costs totalled€37.7 million,€38.1 million and€30.6 million for the years ended December 31, 2003, 2002 and 2001, respectively.
The following table summarizes the costs of significant projects and reconciling items to arrive at total research and development expenses for the periods shown (in thousands of€):
| Three months ended March 31, | Year ended December 31, | |||||
Project Costs: | 2004 | 2003 | 2002 | |||
Satraplatin | 2,509 | 9,819 | 2,581 | |||
1D09C3 | 809 | 1,401 | 4,148 | |||
RGB-286199 | 472 | 1,654 | 3,890 | |||
Cost of performing research and development for others | 432 | 3,565 | 3,724 | |||
Other projects | 854 | 4,069 | 5,944 | |||
Total project cost | 5,076 | 20,508 | 20,287 | |||
Other costs to arrive at total research and development expenses: | ||||||
Benefits and other salaries | 1,152 | 6,466 | 6,234 | |||
Stock option compensation | 108 | 1,290 | 2,675 | |||
Building and facilities | 1,147 | 4,060 | 2,731 | |||
Depreciation | 331 | 1,361 | 1,904 | |||
Intellectual property expenses | 127 | 845 | 1,620 | |||
Other expenses | 741 | 3,211 | 2,602 | |||
Total research and development expenses | 8,682 | 37,741 | 38,053 | |||
There were no costs related to satraplatin in 2001. Other project costs in 2001 cannot be presented in a meaningful manner on a project basis as we did not have our project systems fully implemented at all of our sites in that year. Project costs shown in the table above include direct project costs plus the salaries of the employees who directly charged time to the project and do not include overhead and other unallocated costs. Salaries do not include an allocation of benefits or compensation costs for stock option plans and convertible bonds. Other salaries relate to departmental salaries not directly allocated to a project.
In 2003, a significant portion of our research and development expenses for satraplatin related to payments to two contract research organizations. We expect to spend an additional€30 to€35 million to complete our Phase 3 registrational trial over the period from 2004 to 2006. This estimate excludes costs for salaries and milestones. If our satraplatin phase 3 registrational trial is significantly delayed or fails to demonstrate that satraplatin is safe or effective, or we are unable to obtain necessary approvals from the
46
Table of Contents
FDA and similar regulatory agencies in Europe and elsewhere, we may be unable to meet our development timelines. If we fail to commercialize satraplatin on schedule, our ability to achieve profitability will be significantly delayed as the overall costs of the program will increase and the timing of revenue generation will be delayed. Although we currently intend to fully fund our Phase 3 registrational trial for satraplatin ourselves, we intend to enter into one or several strategic partnerships with pharmaceutical companies for sales and marketing of satraplatin.
Additionally, assuming continued successful preclinical development and receipt of necessary approvals, we intend to advance our monoclonal antibody product candidate into clinical trials in 2004 and to complete preclinical development work on RGB-286199 in the first half of 2005. Even if their further development is successful, it will take several more years before we can file for regulatory approval of these product candidates, and we may be unable to obtain the necessary approvals. If we do not receive regulatory approvals, we will not be able to generate product revenues from these product candidates. Because these product candidates are in very early stages of development, and we have not selected an indication for which we intend to conduct clinical trials, we cannot provide a meaningful estimate of future development costs.
As we continue our clinical trial of satraplatin and advance the development of our other product candidates, we expect to incur higher levels of research and development expenses in future periods. This future increase might also result, among other things, from milestone payments for satraplatin upon the acceptance of an NDA filing and the European equivalent and the approval of commercial sales in the United States and Europe for a total of $15.0 million. All of our product candidates are currently in development, and we therefore do not expect to generate any direct revenue from drug product sales for at least the next several years. For satraplatin, we estimate that we may be able to receive marketing approval in 2007. However, with respect to our other product candidates, we are currently unable to determine the period, if any, during which we will be able to generate revenue from them given the early stage of their current development.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including finance, business development, investor relations, information technology and human resources. Other significant costs in this category include facilities costs and professional fees for accounting and legal services, travel, insurance premiums and depreciation. After completion of the offering, we anticipate increases in expenses relating to insurance, legal and accounting services, investor relations and other internal resource requirements arising from additional compliance and reporting obligations imposed by the Nasdaq and the U.S. federal securities laws.
Stock Compensation
We grant stock options to our Management Board, senior management, employees and consultants. We have recognized compensation costs relating to stock options granted in accordance with SFAS No. 123,Accounting for Stock-Based Compensation, since our inception. We recognize compensation expense as a charge to operations over the relevant vesting period of the options, which generally is four years.
We also grant convertible bonds to members of our Supervisory Board, Management Board and senior management. The fair value of our convertible bonds is recognized as a compensation expense that is charged to general and administrative expenses over the relevant vesting period of the convertible bonds, which generally is two to four years.
The aggregate estimated fair value for options and convertible bonds issued during the year ended December 31, 2003 was approximately€2.2 million, which is being recognized over the vesting periods. Total compensation expense recorded related to options and convertible bonds during the
47
Table of Contents
year ended December 31, 2003, was approximately€2.5 million. From inception to December 31, 2003, we have incurred cumulative compensation expense related to stock options and convertible bonds of approximately€12.0 million.
Foreign Currency
We publish our consolidated financial statements in euros. Most of our revenues are denominated in U.S. dollars. Historically, most of our expenses have been denominated in euros, however, beginning in 2003 the majority of our expenses were denominated in U.S. dollars. In recent years, particularly in 2003, the U.S. dollar has depreciated significantly against the euro. On December 31, 2002, the exchange rate was€1.00 = $1.05 and at December 31, 2003, the exchange rate was€1.00 = $1.26. Because most of our revenues in 2003 were denominated in U.S. dollars, this depreciation has had the effect of decreasing the reported euro amount of our revenues from annual license fees, milestone payments, and FTE services. Moreover, the depreciation of the U.S. dollar in 2003 also had the effect of decreasing the reported euro amount of those of our expenses denominated in U.S. dollars. These primarily consist of all of the expenses incurred by our subsidiary, GPC Biotech, Inc., and some external research and development expenses and legal expenses incurred by GPC Biotech AG. Any substantial future appreciation of the U.S. dollar against the euro could have the effect of reversing these trends, in the whole or in part.
The table below summarizes the impact of currency fluctuations on the significant components of the statements of operations for 2003 compared to 2002, 2002 compared to 2001, and for the three months ended March 31, 2004 compared to the same period in 2003. The amounts presented represent the amounts by which each component decreased as a result of foreign currency fluctuations as compared to the comparative period.
| Three months ended March 31, | Twelve months ended December 31, | ||||||||
2004 compared to | 2003 compared to 2002 | 2002 compared to 2001 | |||||||
| (in€ thousands) | |||||||||
Total revenues | (561 | ) | (1,639 | ) | (398 | ) | |||
Research and development expenses | (715 | ) | (3,621 | ) | (1,057 | ) | |||
General and administrative expenses | (177 | ) | (977 | ) | (215 | ) | |||
Restructuring Costs
In October 2003, we announced a restructuring plan to realign resources to focus on our oncology drug discovery and development programs. The restructuring included a staff reduction of 42 positions, mainly in the technology research staff. Total restructuring costs incurred for the year ended December 31, 2003 were approximately€0.5 million. These costs mainly related to employee termination costs and legal and consulting fees. The restructuring plan was completed on December 31, 2003. At December 31, 2003,€0.1 million was accrued relating to remaining severance payments.
Income Taxes
As a result of the net losses we have incurred in each year since inception, no provision for income taxes has been recorded. At December 31, 2003, we had net operating loss carry-forwards for German corporation and trade tax purposes of€33.3 million. For fiscal years ending in 2004 and beyond, a new law is in effect restricting the use of net operating loss carry-forwards for German corporation tax and trade tax purposes. Our maximum net operating loss carry-forward that may be utilized in any one year is restricted to 60% of annual taxable income above€1 million. These net operating loss carry-forwards do not expire.
48
Table of Contents
Additionally, we have U.S. net operating loss carry-forwards of€66.2 million. As a result of an ownership change in 2000, we have a limitation of€1.4 million on the amount of income that is available to be offset by net operating loss carry-forwards generated in the U.S. We have€32.3 million of net operating losses carried forward that are subject to this limitation. Net operating loss carry-forwards generated in the U.S. expire at various times between the years 2007 and 2023.
In addition, we had research and development credits of approximately€3.3 million, at December 31, 2003.
At December 31, 2003, 2002 and 2001, we had deferred tax assets representing the benefit of accumulated tax net operating loss carry-forwards. We have provided a full valuation allowance on our deferred tax assets because we believe it is more likely than not that our deferred tax assets will not be realized.
German Tax Treatment of Loss Carry Forwards
Under current German tax law, a corporation is generally permitted to set-off loss carry forwards (Verlustvorträge) against annual taxable income up to€1 million. The remaining loss carry forwards can be set-off against up to 60% of the taxable income of the individual tax assessment period. Any set-off against loss carry forwards requires that the offsetting entity is economically identical with the loss incurring entity. According to the German Corporate Income Tax Act (Kôrperschaftsteuergesetz), the offsetting entity would not be economically identical to the loss incurring entity if:
| • | at any time more than fifty percent (in aggregate) of the shares in a corporation are transferred, which, according to the German tax authorities, includes the subscription of new shares issued in the course of a capital increase to new shareholders, and |
| • | predominantly new assets are injected into such corporation within a period of five years thereafter. |
Any loss carry forwards existing at the time these two conditions occur will no longer be available for set-off against any of the corporation’s current and future taxable income. Thereafter, a corporation would generally be permitted to set-off only newly incurred losses against€1 million per year of its taxable income of the following years. The remaining loss carry forwards can be set-off against up to 60% of the taxable income of the individual tax assessment period.
We expect that, as a result of the combined offering and the related injection of new assets, we will not be able to set-off our accumulated tax loss carry forwards against any future taxable income for German tax purposes.
Results of Operations
Three months ended March 31, 2004 compared with the three months ended March 31, 2003
Total revenues
Revenues for the three months ended March 31, 2004 decreased by 34.4%, to€4.0 million, from€6.1 million for the three months ended March 31, 2003. ALTANA Pharma accounted for 100% of our revenues in the three months ended March 31, 2004.
During the three months ended March 31, 2004, we recognized€1.5 million of revenues due to the amortization of deferred upfront payments and annual license fees, compared with€2.0 million in the same period in 2003, a decrease of€0.5 million or 25.0%. A decrease of€0.4 million in the three months ended March 31, 2004 relates to the completion of one collaboration with ALTANA Pharma, at the end of 2003. During the three months ended March 31, 2004, our revenue related to deferred upfront payments under the ALTANA Pharma agreements, and annual license fees, decreased by€0.2 million due to foreign currency differences on annual license fees. This decrease was partially
49
Table of Contents
offset by an increase of€0.1 million under the 3-hybrid agreement with ALTANA Pharma, which commenced after the first quarter of 2003 and under which additional annual license and sublicense fees were payable.
During the three months ended March 31, 2004, we also recorded€0.9 million of revenues through the sale of FTE services to collaboration partners, compared with€2.4 million during the same period in 2003, a decrease of€1.5 million or 62.5%. A decrease in revenue of€1.0 million related to the completion of two collaborations with ALTANA Pharma by June and December of 2003. An additional decrease of€0.4 million related to a planned decrease in the number of FTEs in the ongoing collaboration with ALTANA Pharma as well as the decline of the U.S. dollar compared to the euro as these payments were U.S. dollar-denominated. The remaining decline of€0.1 million resulted from the completion of the FTE support to PanTherix and Aventis in 2003.
During the three months ended March 31, 2004, we generated€1.6 million in milestone payments under our agreements with ALTANA Pharma. This compares to€1.5 million in milestone payments in the same period during 2003 from ALTANA Pharma.
During the three months ended March 31, 2004, we generated no grant revenues, compared with€0.2 million during the same period in 2003. The decline was related to the completion of two governmental sponsored research programs in 2003.
Total revenues from ALTANA Pharma amounted to€4.0 million for the three-month period ended March 31, 2004, compared with€5.8 million in the same period in 2003. Of the 2004 total revenues,€1.5 million related to the amortization of deferred upfront payments and annual license fees,€0.9 million related to the sale of FTE services, and€1.6 million represented milestone payments. No other customer accounted for more than 10% of our total revenues in the three-month period ended March 31, 2004.
Research and development expenses
Research and development expenses were€8.7 million for the three months ended March 31, 2004, compared with€8.3 million for the three months ended March 31, 2003, an increase of€0.4 million or 4.8%. For the three months ended March 31, 2004 and 2003,€2.5 million and€0.8 million, respectively, of our research and development expenses related to the development of satraplatin.
With the initiation of our Phase 3 registration trial for satraplatin in 2003, we entered into contracts with two contract research organizations to perform clinical trials. In the three months ended March 31, 2004, the expense related to these agreements was€1.1 million. None of those costs were incurred during the same period in 2003. We expect costs for contract research organizations to increase in future periods. Additionally, in relation to the administration of the satraplatin trial, travel expenses increased by€0.1 million.
The costs for external research and development increased from€0.7 million during the three months ended March 31, 2003 to€1.6 million during the same period in 2004. This increase was attributable to the achievement of a milestone related to our monoclonal antibody program. This event triggered a milestone payment of€0.5 million during the first quarter of 2004. No such milestone was reached in the three months ended March 31, 2003. The remaining increase of external research and development was attributable to additional preclinical development costs incurred in the first three months ended March 31, 2004 related to the toxicity studies for satraplatin.
Other research and development expenses increased by€0.1 million, from€1.8 million during the three months ended March 31, 2003 to€1.9 million during the same period in 2004. This increase relates primarily to operating costs of our facilities.
50
Table of Contents
Employee compensation including stock option expenses allocated to research and development decreased by€1.1 million, to€2.8 million in the three months ending March 31, 2004 compared with€3.9 million during the same period in 2003. This decline was mainly due to a decrease in payroll costs of€0.8 million as a result of our October 2003 restructuring and workforce reduction of approximately 21% of our staff, mainly in technology research activities. The restructuring was aimed at reallocating resources to reflect our increasing focus on oncology drug discovery and development. A further decrease of€0.3 million related to a decrease in stock option compensation costs due to a lower number of options granted.
During the three months ended March 31, 2004, we incurred costs for raw materials and consumables totalling€0.5 million, as compared with€1.3 million during the same period in 2003, a decrease of€0.8 million.€0.5 million of this decrease was attributable to decreased expenses in the three months ended March 31, 2004 for contract manufacturing of antibodies for our monoclonal antibody drug development program in preparation for clinical testing. These expenses were mostly incurred in 2002 and to a lesser extent, in 2003. The remaining€0.3 million of this decrease was attributable to a shift in our focus toward later-stage drug development and a lower number of research employees, which resulted in fewer expenses related to research laboratory materials.
Clinical development timelines, likelihood of success and total completion costs vary significantly for each product candidate and are difficult to estimate. We anticipate that we will make determinations as to which research programs to pursue and how much funding to direct to each program on an ongoing basis in response to the scientific and clinical success of each product candidate. The lengthy process of seeking regulatory approvals, and the subsequent compliance with applicable regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals could cause our research and development expenditures to increase, which would have a material adverse effect on our results of operations.
General and administrative expenses
General and administrative expenses increased by 7.7%, to€2.8 million for the three months ended March 31, 2004, compared with€2.6 million during the same period in 2003. This increase was mainly due to an increase of€0.1 million for higher compensation costs for convertible bonds resulting from convertible bonds issued in 2002 and 2003.
Amortization of intangible assets acquired in a business combination
Amortization expenses of intangible assets acquired in a business combination decreased€0.1 million to€0.1 million for the three months ended March 31, 2004, compared with€0.2 million during the same period in 2003. The decrease in amortization expenses is due to a lower cost basis of capitalized patent portfolio as a result of the impairment charges related to the discontinuation in 2003 of specific programs that Mitotix had in progress at the time of the acquisition. The impairment charge was recognized in the fourth quarter of 2003.
Interest income
Interest income was€0.6 million during the three months ended March 31, 2004, compared with€0.9 million during the same period in 2003. The decrease of€0.3 million was attributable to a decrease in interest rates and lower average balances of cash, cash equivalents, marketable securities and short-term investments in 2004.
Other income and expense, net
Other income (expense), net was€0.1 million for the three months ended March 31, 2004, compared with€(0.1) million for the same period during 2003. This decrease in expense related to foreign currency gains and losses on accounts receivable, accounts payable and cash.
51
Table of Contents
2003 Compared With 2002
Total revenues
Revenues for the year ended December 31, 2003 increased by approximately 1%, to€21.6 million, from€21.5 million for the year ended December 31, 2002, despite the depreciation of the U.S. dollar, in which a substantial portion of our revenue is denominated, against the euro.
During 2003, we recognized€8.1 million of revenues from the amortization of deferred upfront payments and annual license fees, compared with€9.4 million in 2002, a decrease of€1.3 million or 13.8%. A decrease of€0.6 million in 2003 related to the completion of our collaboration with Boehringer Ingelheim at the end of 2002. An additional decrease of€0.5 million was attributable to the amortization of a deferred upfront payment made by Sankyo to us related to an antifungal program. We recognized this payment as revenue during 2002. This amount was received in prior years and deferred until 2002, when all the criteria for revenue recognition were met. During 2003, our revenue related to deferred upfront payments and annual license fees under the ALTANA Pharma agreements, which decreased by€0.1 million.
During the year ended December 31, 2003, we also recorded€8.1 million of revenues through the sale of FTE services to collaboration partners, compared to€9.7 million during 2002, a decrease of€1.6 million or 16.5%. A decrease in revenue of€1.4 million, or 14.4%, related to the completion of our collaboration with Boehringer Ingelheim by the end of 2002. A decrease of€0.5 million, or 5.2%, related to the completion by the end of the second quarter of 2003 of an earlier collaboration with ALTANA Pharma for research relating to anti-infectives. An increase of€0.3 million related to the execution of a research collaboration agreement with Eli Lilly & Company.
We generated€4.6 million in milestone payments under our agreements with ALTANA Pharma and under our agreement with Aventis related to osteoarthritis during 2003. This figure compares to€1.4 million in 2002 from ALTANA Pharma and from Boehringer Ingelheim under our agreement with them related to virology research.
During the year ended December 31, 2003, we generated€0.8 million in grant revenues, compared with€1.0 million during the year ended December 31, 2002. The decline was related to the completion of a large governmental sponsored research program in 2002.
Total revenues from ALTANA Pharma amounted to€20.2 million in 2003, compared with€17.6 million in 2002. Of the 2003 total revenues,€8.1 million related to the amortization of deferred upfront payments and annual license fees,€7.7 million related to the sale of FTE services, and€4.4 million represented milestone payments. No other customer accounted for more than 10% of our total revenues in 2003.
Research and development expenses
Research and development expenses were€37.7 million for the year ended December 31, 2003, compared with€38.1 million for the year ended December 31, 2002, a decrease of€0.4 million or 1.0%. For the years ended December 31, 2003 and 2002,€9.8 million and€2.6 million, respectively, of our research and development expenses related to the development of satraplatin.
In September 2002, we licensed satraplatin from a third party, which resulted in a non-recurring license payment of $2.0 million or€2.0 million. Under this license, an additional payment of $2.0 million or€1.7 million became due upon the treatment of the first patient in our Phase 3 registrational trial for satraplatin in September 2003. Of the $2.0 million payment, $0.9 million was for an equity investment in Spectrum Pharmaceuticals and $1.1 million or€0.9 million was recorded as research and development expense, a decrease of€1.1 million from the license expense related to satraplatin in 2002.
52
Table of Contents
Employee compensation including stock option expenses allocated to research and development decreased by€1.6 million, to€15.1 million in 2003 compared with€16.7 million in 2002. This decline was due mainly to a decrease in stock option compensation costs, which decreased by€1.4 million due to a decrease in the number of options granted as well as a lower fair market value of the shares. U.S. dollar based payroll costs increased due to additional hiring in our clinical and preclinical development department, but were offset by favorable changes in the U.S. dollar-euro exchange rate. In October 2003, we undertook a restructuring and workforce reduction of approximately 21% of our staff, mainly in technology research activities. The restructuring was aimed at reallocating resources to reflect our increasing focus on oncology drug discovery and development. The restructuring did not result in significant cost savings in 2003 because the reduction in personnel costs was off-set by the restructuring charges.
The costs for external research and development decreased from€4.2 million during 2002 to€3.7 million during 2003. The decrease of€0.5 million was mainly attributable to non-recurring preclinical development costs incurred in 2002 related to our cell cycle inhibitor program and our monoclonal antibody program.
We entered into contracts in 2003 with two contract research organizations to perform clinical trials. In 2003, the expense related to these agreements was€3.2 million. No such costs were incurred in 2002. We expect costs for contract research organizations to increase going forward.
During 2003, we incurred costs for raw materials and consumables totalling€4.6 million, as compared with€6.2 million in 2002, a decrease of€1.6 million.€0.9 million of this decrease was attributable to a shift in our focus toward later-stage drug development, which resulted in fewer expenses related to research laboratory materials. The remaining€0.7 million of this decrease was attributable to decreased expenses in 2003 for the contract manufacturing of antibodies for our monoclonal antibody drug development program in preparation for clinical testing. These expenses were mostly incurred in 2002.
During the year ended December 31, 2003, we incurred costs for research and development facilities, primarily related to rent expenses, of€4.1 million, compared with€2.7 million during 2002. The increase of€1.4 million is due to the expansion of our leased space at our Waltham, Massachusetts facility at the beginning of the fourth quarter of 2002.
The costs for depreciation of property and equipment allocated to research and development declined by€0.5 million to€1.4 million during the year ended December 31, 2003 from€1.9 million during 2002. As we have been shifting from early-stage research activities to later-stage drug development, our business has become less equipment intensive. Therefore, we invested less cash in property and equipment in 2003 as compared with 2002. In addition, older property and equipment was fully depreciated at the end of 2002 and beginning of 2003.
Clinical development timelines, likelihood of success and total completion costs vary significantly for each product candidate and are difficult to estimate. We anticipate that we will make determinations as to which research programs to pursue and how much funding to direct to each program on an ongoing basis in response to the scientific and clinical success of each product candidate. The lengthy process of seeking regulatory approvals, and the subsequent compliance with applicable regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals could cause our research and development expenditures to increase, which would have a material adverse effect on our results of operations.
General and administrative expenses
General and administrative expenses increased by 2.7%, to€11.5 million for the year ended December 31, 2003, compared with€11.2 million for the year ended December 31, 2002. An increase
53
Table of Contents
of€0.6 million was due to increased salary and benefit costs, and to the addition of three general and administrative personnel, mainly in the area of strategic marketing and business development. Furthermore, while compensation costs for stock options decreased by���0.3 million, compensation costs for convertible bonds increased by€0.7 million due to additional grants of convertible bonds during 2003. These increases were offset by the impact on expenses incurred in U.S. dollars and translated into euros resulting from the weakening of the U.S. dollar against the euro.
Impairment of goodwill
During the year ended December 31, 2002, we recorded a goodwill impairment charge of€7.3 million. The goodwill related to the purchase of Mitotix, Inc. (now GPC Biotech Inc.) in 2000 and resulted from the excess of purchase price over the estimated fair market value of net tangible assets acquired that was not allocated to specific intangible assets.
On January 1, 2002, we adopted SFAS No. 142,Goodwill and Other Intangible Assets under which goodwill is reviewed for impairment annually, or more frequently if certain events or changes in circumstances indicate that the carrying value may not be recoverable. In addition, we were required to perform a transitional goodwill impairment test related to the carrying value of goodwill as of January 1, 2002. During the quarter ended June 30, 2002, we completed the transitional impairment test and determined that there was no impairment as of the January 1, 2002 implementation date. We performed our annual impairment test on the carrying value of goodwill as of December 31, 2002. As we determined that we have one reporting unit under SFAS 142, we determined the fair value of the reporting unit based on our market capitalization for the purposes of the first step of our goodwill impairment test. During 2002, we experienced a significant decline in market capitalization due to a decline in our share price. Based on this decline in market capitalization, we determined that our carrying value was in excess of our market capitalization. We determined as a result of the first step of the impairment analysis that goodwill was fully impaired. Consequently, we recognized a goodwill impairment charge of€7.3 million in 2002, representing the entire balance of goodwill. No such impairments occurred in 2003.
Amortization, including impairment of intangible assets acquired in a business combination
During the year ended December 31, 2003, we recorded amortization expense, including an impairment charge related to intangible assets acquired in a business combination, of€1.8 million compared with€1.3 million during 2002. In 2003, these expenses consisted of amortization expense of€0.6 million and impairment charges of€1.2 million. The 2002 expenses included amortization expenses of€0.8 million and impairment charges of€0.5 million.
The intangible assets consisted of a capitalized patent portfolio and partnered technology related to the acquisition of Mitotix, Inc. in 2000. Impairment charges related to the patent portfolio and resulted from the discontinuation in 2002 of specific programs, and of additional programs in 2003, that Mitotix had at the time of the acquisition.
Interest income
Interest income was€2.9 million during the year ended December 31, 2003, compared with€4.4 million during 2002. The decrease of€1.5 million was attributable to a decrease in interest rates and lower average balances of cash, cash equivalents, marketable securities and short-term investments in 2003.
During the year ended December 31, 2003, interest expense was€0.1 million, compared with€0.2 million during 2002.
54
Table of Contents
Other income and expenses, net
Other income (expense), net was€(0.1) million for the year ended December 31, 2003, compared with€(0.7) million for the year ended December 31, 2002. A significant portion of other income and expense relates to foreign currency gains and losses on accounts receivable, accounts payable and cash.
2002 Compared With 2001
Total revenues
Revenues for the year ended December 31, 2002 increased by€7.6 million or 55% to€21.5 million, from€13.9 million for the year ended December 31, 2001.
During the year ended December 31, 2002, we generated€9.4 million in revenues through the amortization of deferred upfront payments and annual license fees compared with€4.0 million in 2001, an increase of€5.4 million.€5.2 million of the increase was due to a collaboration and license agreement with ALTANA Pharma that was signed in late 2001. An additional increase of€0.5 million was attributable to the amortization of a deferred upfront payment from Sankyo that was recognized as revenue during 2002. This amount was received in prior years and deferred until 2002 when all the criteria for revenue recognition were met. A decrease of€0.3 million is related to the main part of the collaboration with Aventis, which was completed by the end of 2001.
During the year ended December 31, 2002, we also recorded revenues amounting to€9.7 million from the sale of FTE services to collaboration partners, compared with€7.2 million during 2001, an increase of€2.5 million or 34.7%. An increase of€4.0 million related to the collaboration and license agreement with ALTANA Pharma. The increase was offset by a decrease of€1.2 million in 2002 related to the completion in 2001 of the main part of our collaboration with Aventis related to osteo-arthritis.
We generated€1.4 million in milestone payments from ALTANA Pharma and Boehringer Ingelheim during 2002, compared with€1.1 million in 2001 from ALTANA Pharma and Aventis.
During the year ended December 31, 2002, we generated€1.0 million in grant revenues compared with€1.6 million during the year ended December 31, 2001. A decline of€0.7 million is related to the completion of one governmental sponsored research program in early 2002. An increase of€0.1 million was due to higher grant revenues from another grant program.
Total revenues from ALTANA Pharma amounted to€17.5 million in 2002, as compared with€8.0 million in 2001. Of the 2002 total revenues,€8.3 million related to the amortization of deferred upfront payments and annual license fees,€8.2 million related to the sale of FTE services, and€1.1 million represented milestone payments.
In 2002 and 2001, Boehringer Ingelheim accounted for€2.3 million and€2.1 million, respectively, of our total revenues. Of the 2002 total revenue,€0.6 million related to the amortization of deferred upfront payments and annual license fees,€1.4 million related to the sale of FTE services, and€0.3 million represented milestone payments.
Research and development expenses
Research and development expenses were€38.1 million for the year ended December 31, 2002, compared with€30.6 million for the year ended December 31, 2001, an increase of€7.5 million or 24.5%.
55
Table of Contents
Employee compensation relating to our research and development activities, including stock option expenses, increased by€1.9 million, from€14.8 million in 2001 to€16.7 million in 2002. An increase of€2.1 million, due to salary and benefits expenses resulting from additional hiring of 32 employees, was partially offset by a decrease in stock compensation expense of€0.2 million. The decrease in compensation expense was due to fewer stock options issued and a lower average share price.
The costs for external research and development increased€2.9 million, or 223.1%, to€4.2 million for the year ended December 31, 2002, from€1.3 million during 2001. In 2002, we incurred significant costs related to our cell cycle inhibitor program and our monoclonal antibody program that were not incurred in 2001.
During 2002, we incurred costs for raw materials and consumables expenses of€6.2 million compared with€4.7 million in 2001, an increase of€1.5 million or 31.9%. We incurred significant costs in 2002 related to the contract manufacturing of antibodies for our monoclonal antibody drug development program in preparation for preclinical testing, resulting in an increase of€1.7 million in 2002 over 2001.
In September of 2002, we licensed satraplatin from a third party, resulting in a non-recurring licensing payment of $2.0 million. As a result, we incurred total in-licensing expenses in 2002 of€2.0 million. No comparable expenses were incurred in 2001.
A decrease in intellectual property expenses from€2.6 million to€1.9 million was primarily caused by charges for acquired patents of€0.5 million in 2001. No such charges accrued in 2002.
During the year ended December 31, 2002, we incurred costs for research and development facilities, related primarily to rent expense, of€2.7 million compared with a similar amount in 2001.
The costs for depreciation of property and equipment remained stable at€1.9 million during the year ended December 31, 2002, compared to€1.8 million during 2001.
General and administrative expenses
General and administrative expenses were€11.2 million for the year ended December 31, 2002 compared with€9.9 million for the year ended December 31, 2001, an increase of€1.3 million or 13.1%. An increase of€1.0 million was primarily due to increased salary and benefit costs and due to additional hiring of nine general and administrative personnel in 2002. We also experienced decreases in legal and consulting expenses of€1.0 million in 2002 compared with 2001. The additional expenses in 2001 related to due diligence costs. An increase in facilities expenses of€0.7 million to€1.3 million was caused by increased rent and facility operating expense after moving our offices from Cambridge, Massachusetts to Waltham, Massachusetts in 2001.
Impairment of goodwill
On January 1, 2002, we adopted SFAS No. 142, under which goodwill is reviewed for impairment annually, or more frequently if certain events or changes in circumstances indicate that the carrying value may not be recoverable. In addition, we were required to perform a transitional goodwill impairment test related to the carrying value of goodwill as of January 1, 2002. During the quarter ended June 30, 2002, we completed the transitional impairment test and determined that there was no impairment as of the January 1, 2002 implementation date. We also performed our annual impairment test on the carrying value of goodwill as of December 31, 2002. As we determined that we have one reporting unit under SFAS 142, we determined the fair value of the reporting unit based on our market
56
Table of Contents
capitalization for the purposes of the first step of our goodwill impairment test. During 2002, we experienced a significant decline in market capitalization due to a decline in our share price. Based on this decline in market capitalization, we determined that our carrying value was in excess of our market capitalization. We determined as a result of the first step of the impairment analysis that goodwill was fully impaired. Consequently, we recognized a goodwill impairment charge of€7.3 million in 2002. No such impairments occurred in 2001.
Amortization, including impairment of intangible assets acquired in a business combination
Amortization, including impairment of intangible assets acquired in a business combination was€1.3 million for the year ended December 31, 2002 compared to€4.7 million for the year ended December 31, 2001, a decrease of€3.4 million or 72.3%. The 2001 expense includes€3.8 million related to amortization of goodwill and assembled workforce. On January 1, 2002, we adopted SFAS No. 142 under which goodwill is no longer amortized, but reviewed annually for impairment. In addition, assembled workforce was classified to goodwill and no longer amortized. Consequently, there was no amortization of goodwill or assembled workforce in 2002.
The decrease in amortization, including impairment of intangible assets was partially offset by impairment charges of€0.5 million in 2002. The intangible assets consisted of a capitalized patent portfolio and partnered technology related to the acquisition of Mitotix in 2000. Impairment charges relate to the patent portfolio and resulted from the discontinuation in 2002 of specific programs that Mitotix was pursuing at the time of the acquisition.
Interest income
Interest income was€4.4 million during the year ended December 31, 2002, compared with€5.1 million during 2001. The decrease of€0.7 million or 13.7% was attributable to a decrease in interest rates and lower average balances of cash, cash equivalents, marketable securities and short-term investments in 2002.
During the years ended December 31, 2002 and 2001, interest expense remained flat at€0.2 million.
Other income and expenses, net
Other income (expense), net was€(0.7) million for the year ended December 31, 2002 compared with€0.2 million for the year ended December 31, 2001. A significant portion of other income and expense relates to foreign currency gains and losses on accounts receivable, accounts payable, and cash.
Liquidity and Capital Resources
Our cash, cash equivalents and marketable securities and short-term investments totaled€82.0 million, and our restricted cash totaled€2.6 million, at March 31, 2004. Over the last five years, we have financed our operations primarily through the sale of equity securities, cash payments (including licensing fees) from several collaboration partners, interest earned on investments, government grants, capital lease financings and long-term debt.
57
Table of Contents
The following tables summarizes our sources of funding over the last five years (in thousands of €):
Year | From the sale of equity securities | From cash payments from collaboration partners | From capital lease financings | From long- term debt | From interest on investments | From government grants | Total | |||||||
2003 | 292 | 16,383 | — | — | 3,276 | 807 | 20,758 | |||||||
2002 | 290 | 15,814 | 863 | — | 3,866 | 1,052 | 21,885 | |||||||
2001 | 35,765 | 30,171 | 537 | — | 5,076 | 1,388 | 72,937 | |||||||
2000 | 115,488 | 3,815 | 273 | 1,023 | 3,020 | 1,219 | 124,838 | |||||||
1999 | 13,336 | 4,268 | 711 | — | 134 | 988 | 19,437 |
Net cash used in operating activities was€8.3 million for the three months ended March 31, 2004, primarily reflecting the net loss for this period of€6.9 million, adjusted for non-cash depreciation and amortization, non-cash compensation expense for stock options and convertible bonds and changes in accounts payable, other current and non-current assets, deferred revenue and other liabilities and accrued expenses. Net cash used in investing activities for the three months ended March 31, 2004 amounted to€9.8 million, including purchases of property and equipment in the amount of€0.5 million and net purchases of marketable securities and short term investments of€9.3 million. Net cash provided by financing activities was€1.2 million for the three months ended March 31, 2004, mainly reflecting proceeds from the exercise of stock options and convertible bonds of€1.3 million partially offset by payments under capital lease obligations in the amount of€0.1 million and principal payments on loans totaling€0.1 million.
Net cash used in operating activities was€23.0 million and€23.5 million for the years ended December 31, 2003 and 2002, respectively. Net cash provided by operating activities was€2.4 million for the year ended December 31, 2001. These amounts primarily reflect net losses of€26.8 million,€32.9 million, and€26.2 million for the years ended December 31, 2003, 2002 and 2001, respectively, adjusted for non-cash depreciation and amortization, non-cash compensation expense for stock options and convertible bonds and changes in accounts payable, other current assets, deferred revenue and other liabilities and accrued expenses. In 2002, cash used in operating activities was significantly less than the net loss because of a€7.3 million non-cash charge related to an impairment of goodwill. In 2001, the amount of cash provided by operating activities was primarily due to our receipt of€20.6 million upfront payments (including license fees) from ALTANA Pharma and our receipt of a€1.3 million upfront payment from Boehringer Ingelheim. These payments are recognized as revenue ratably over the term of the respective collaboration. Other collaborations yielded additional cash resources of€4.2 million and€4.8 million in 2003 and 2002. Other major sources of cash are derived from the sale of FTE services, milestone and grant revenues. In 2003, 2002 and 2001, cash from the sale of FTE services was€7.8 million,€9.7 million and€7.0 million, respectively. Cash from milestones reached during 2003, 2002 and 2001 was€ 4.4 million,€ 1.4 million and€ 1.3 million, respectively. Grant revenues provided cash of€ 0.8 million,€ 1.1 million and€ 1.4 million during the years ended December 31, 2003, 2002 and 2001, respectively.
Net cash provided by investing activities consisted of€18.7 million for the year ended December 31, 2003. Net cash used in investing activities amounted to€26.8 million for the year ended December 31, 2002. Net cash provided by investing activities totaled€1.5 million for the year ended December 31, 2001. Purchases of property, equipment and licenses amounted to€1.7 million,€1.6 million and€3.0 million for the years ended December 31, 2003, 2002 and 2001, respectively. Cash was primarily provided when our investments matured or when we sold investments to meet liquidity needs. We used cash in investing activities to fund purchases of our investments and, to a lesser extent, to fund purchases of property, equipment and licenses for operations.
58
Table of Contents
Net cash provided by financing activities was€42,000 for the year ended December 31, 2003, reflecting the issuance of our ordinary bearer shares upon the exercise of vested stock options held by our employees. Net cash used in financing activities was€0.3 million for the year ended December 31, 2002. This amount mainly represented principal payments under capital lease obligations and long term debt, partially offset by proceeds from the issuance of convertible bonds and from the issuance of our ordinary bearer shares upon exercise of vested stock options. Net cash provided by financing activities was€35.0 million for the year ended December 31, 2001, of which€34.2 million was attributable to the sale of 1,690,000 of our ordinary bearer shares to ALTANA Technology Projects GmbH. The remainder relates to the issuance of additional ordinary bearer shares upon the exercise of stock options and convertible bonds.
Contractual Obligations
Set forth below is a description of our contractual obligations as of December 31, 2003 (in thousands of€):
| Payments due by period | ||||||||||
Contractual Obligations | Total | Less than 1 year | 1-3 years | 3-5 years | More than 5 years | |||||
Operating leases | 31,271 | 4,572 | 9,046 | 8,277 | 9,376 | |||||
Long-term debt | 767 | 128 | 256 | 256 | 127 | |||||
Convertible bonds | 884 | — | — | 40 | 844 | |||||
Capital leases | 678 | 342 | 336 | — | — | |||||
Research obligations | 836 | 60 | 517 | 259 | — | |||||
Total | 34,436 | 5,102 | 10,155 | 8,832 | 10,347 | |||||
Our long-term commitments under operating leases shown above consist of payments relating to our facility leases in Munich, Germany, Waltham, Massachusetts and Princeton, New Jersey. They all expire at various times through 2011. Aggregate future minimum rentals to be received under non-cancellable subleases with ALTANA Pharma and another company as of December 31, 2003 were approximately€3.5 million. During the year ended December 31, 2003, income from subleases amounted to€1.4 million. We have investigated additional office space expansion opportunities to support our development requirements beyond 2004, since we expect that as we continue to execute our strategy, we may require additional space. As of the date of this prospectus, we have made no formal commitments or plans to lease any material additional space.
We have a loan agreement with IKB Deutsche Industriekreditbank AG in the amount of€1.0 million, all of which was borrowed in July 2000. The term of the loan is 10 years with semi-annual repayments beginning in March 2002. The loan is unsecured and bears interest at 4.25% per annum. The remaining principal outstanding as of December 31, 2003 was€0.8 million.
The contractual obligations table does not include amounts for milestone payments to various collaboration partners resulting from the achievement of defined contractual milestones, as the payments are contingent on the achievement of defined contractual milestones.
As of March 31, 2004, we had one unused line of credit totaling€0.2 million available for capital leases of equipment in the United States. Total credit available under this agreement is limited to $1.0 million and is secured by the assets purchased under this line of credit.
We expect to incur substantial costs as we continue to expand our discovery programs and development activities. Development expenses for our unpartnered drug discovery and development programs consist primarily of employee compensation, supplies and materials, milestone payments,
59
Table of Contents
costs to maintain intellectual property and license rights, costs for consultants and contract research, facilities costs and depreciation of equipment. We expect to incur significant development expenses to complete our Phase 3 registrational trial for satraplatin. We further expect to incur significant development expenses to initiate clinical trials for our monoclonal antibody product candidate and cell cycle inhibitor product candidate.
Our future capital uses and requirements depend on numerous forward-looking factors. These factors include, but are not limited to the following:
| • | the initiation, progress, timing and completion of preclinical research, development, and clinical trials for our product candidates and potential product candidates; |
| • | our ability to establish strategic alliances and successfully complete activities required for commercialization of our potential drugs; |
| • | the timing and extent of partnering or out-licensing one or more of our current and potential product candidates; |
| • | the time and costs involved in obtaining regulatory approvals; |
| • | delays that may be caused by evolving requirements of regulatory agencies; |
| • | the number of existing or new product candidates we pursue; |
| • | the costs involved in filing patent applications and enforcing or defending patent claims; |
| • | the level of funding that we may provide for future product candidates; |
| • | the acquisition or in-licensing of products, other business opportunities or technologies that require financial commitments; |
| • | our revenues, if any, from successful development of our product candidates and commercialization of potential drugs; and |
| • | our plans or ability to establish sales, marketing or manufacturing capabilities and to achieve market acceptance for potential drugs. |
We believe that the net proceeds of this offering, our existing cash resources, future payments we expect to receive from ALTANA Pharma and other collaboration partners and interest earned on investments will be sufficient to meet our projected operating requirements for at least the next two years. If, at any time, our ability to internally finance our research programs changes, we may decide to reduce research and development expenses by delaying, discontinuing or reducing our funding of development of one or more product candidates. Alternatively, we might raise funds through public or private financings, collaborations or other arrangements. We cannot assure you that the funding, if needed, will be available on attractive terms, or at all. Furthermore, any additional equity financing may be dilutive to stockholders, and debt financing, if available, may involve restrictive covenants. Similarly, financing obtained through future co-development or co-marketing arrangements may require us to forego certain commercial rights to, and revenues from, current or potential product candidates. Our failure to raise capital as and when needed could have a negative impact on our business, financial condition, operating results and ability to pursue our business strategy.
Off-Balance Sheet Transactions
As of December 31, 2003, 2002 and 2001, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to
60
Table of Contents
any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships. We do not have relationships or transactions with persons or entities that derive benefits from their non-independent relationship with us or our related parties.
Critical Accounting Policies
The discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our consolidated financial statements included elsewhere in this prospectus, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We recognize revenue in accordance with Securities and Exchange Commission (SEC) Staff Accounting Bulletin, or SAB, No. 101,“Revenue Recognition in Financial Statements,” as updated by SAB No. 104. SAB No. 101 requires that four basic criteria be met before revenue can be recognized: (i) persuasive evidence that an arrangement exists; (ii) delivery has occurred or services rendered; (iii) the fee is fixed and determinable; and (iv) collectibility is reasonably assured. Determination of whether these criteria have been met are based on management’s judgments regarding the fixed nature of the fee charged for research performed and milestones met, and the collectibility of those fees. Should changes in conditions cause management to determine that these criteria are not met for certain future transactions, revenue recognized for any reporting period could be adversely affected.
Our revenues consist of fees earned from research and development collaboration agreements, license agreements and grant revenues. Revenues from research and development collaboration agreements generally consist of licensing fees and/or technology access fees, annual research fees, fees for research support, as well as milestone and royalty payments. Revenues from grants generally consist of reimbursements of a portion of expenses incurred in performing research in specified projects. We do not begin recognizing revenue until an agreement is entered into and authorized by both parties. Our policy is to require contractual payments to be received upfront. Revenue related to arrangements in which payments are deferred or extended for more than 30 days is deferred until payment is received, and therefore, collectibility is assured.
The Company generally receives non-refundable licensing and technology access and initiation fees upfront upon signing of a research and collaboration agreement. In addition, the Company generally receives fixed non-refundable annual licensing fees upfront each year for the duration of the agreement. In accordance with Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, all fees received or to be received under these arrangements are recognized on a straight line basis over the term of the agreements, as this is the period in which research is expected to be performed.
Fees for research support are received from collaboration partners for research services performed under collaboration agreements. Fees are generally stated at our standard yearly flat-fee rate per research scientist (FTE). The number of FTEs is generally stated in the collaboration agreements, and research partners are contractually obliged to pay for a specified number of FTEs
61
Table of Contents
over the life of the agreement. Rates for FTEs are intended to approximate our anticipated costs. Fees are generally billed on a monthly or quarterly basis in advance, and payment is immediately due. Amounts received in advance of services performed are recorded as deferred income and recognized as collaborative revenues as the contract research is performed.
Milestone revenues are derived from the achievement of predetermined goals, which are defined in the collaboration agreements. These milestones are subject to significant contingencies and therefore, the related contingent revenue is not realized until the milestone has been reached and customer acceptance has been obtained. Payments received that relate to the achievement of substantive milestones that were contingent at the outset of the arrangement are recorded as revenue when the milestones are achieved. The reaching of a milestone is evidenced by a milestone confirmation letter that is signed and dated by both parties. In the absence of such milestone confirmation, no milestone revenue is recognized.
Royalty revenues generally result from sales of products based on research performed under the collaboration agreements. Royalties are based on the volume of product sold and are recognized as revenue upon notification of sales from the collaboration partner. We have not yet received or recognized any royalty revenue payments from any of our past or current collaborative agreements.
Grant revenues from governmental agencies for the support of specific research and development projects are recorded as revenue to the extent the related expenses have been incurred and research is performed. The grant agreements consist of a budget that specifies the amount and nature of expenses allowed under the agreement for the entire grant term. Expenses incurred under the grants are calculated according to agreed-upon terms on a quarterly basis, filed with the grant partner, and recorded as revenue. The grant partner makes payments to us based on these calculations. The calculation of the amount and nature of the expenses incurred under the grants involves the judgment of management. We recognize grant revenues based on estimated calculations of expenses incurred under the grants. If these estimated calculations change, then we will adjust grant revenues in the subsequent period. We believe that our calculations are based on the agreed-upon terms as stated in the grant agreements. However, the grant partner has the right to audit our calculations. If the grant partner disagrees with our calculations, then the amount of grant revenue recognized could change. Our calculations have not historically been challenged by the grant partner, and we do not anticipate any subsequent changes in our revenues.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate certain expenses and make appropriate accruals based on these estimates. This process involves identifying services that third parties have performed on our behalf, and estimating the level of services performed and the associated cost incurred for these services as of each balance sheet date in our financial statements. Examples of estimated accrued expenses include contract service fees, such as amounts paid to contract research organizations, professional service fees, such as amounts paid to attorneys and accountants and investigators in conjunction with preclinical and clinical trials, and fees paid to contract manufacturers in conjunction with the production of materials related to our product candidates. In connection with these service fees, our estimates are affected by our understanding of the status and timing of services provided relative to the actual level of services performed by the service providers. In the event that we under- or over-estimate the level of services or the costs of such services, our reported expenses for a reporting period could be overstated or understated. The date on which certain services commence, the level of services performed on or before a given date, and the cost of services are often subject to our judgment. We make these judgments based upon the facts and circumstances known to us at the date of the Financial Statements.
62
Table of Contents
In connection with our internal research and development programs, we may incur milestone payments to other parties. As our product candidates have not received regulatory approval, we expense milestone payments when we incur the liability, which is when the milestones are achieved.
Stock-Based Compensation
We grant stock options to our Management Board, senior management, employees and consultants. Under the terms of our stock option plans, options are exercisable within ten years of the date of grant, subject to an initial two-year non-exercise period as required by German law. The exercise price equals the average of the market price of our ordinary bearer shares over a five-day period prior to the date of grant. We account for our stock option plans in accordance with the provisions of SFAS No. 123, “Accounting for Stock-Based Compensation”. SFAS No. 123 defines a “fair value” based method of accounting for an employee stock option or similar equity investment. We determine the fair value of the options we grant using the Black-Scholes option pricing model and recognize this as compensation expense as a charge to operations over the relevant vesting period of the options, which is generally four years. The expense is recognized on a graded vesting basis as opposed to a straight-line basis. Graded vesting results from portions of the grant vesting at different times. Our options vest at 25% each over four years. This means that one quarter vests over year one, one quarter over year one and two, one quarter over year one, two and three, and the last quarter over years one, two, three and four. As such, the amortized compensation expense is higher in the first years than in the later years.
We grant convertible bonds to our Supervisory Board, Management Board and senior management. The fair value of our convertible bonds is calculated using the same vesting schedule and assumptions as those used for our stock options, and is recognized as compensation expense over the relevant vesting period of the convertible bonds, generally two to four years.
Compensation expense is determined by multiplying the number of options and convertible bonds granted times the fair value of each option or convertible bond. We calculate the fair value of options as of the date of grant and do not make any adjustments for subsequent changes in share price or volatility. To determine the fair value, the Black-Scholes model requires management to make estimates regarding the inputs of the model, namely for share price volatility, risk-free interest rate, future dividends paid by us and life of the option or convertible bond. We estimate share price volatility based on actual historical share prices over the estimated life of the option. We revise the estimate of the share price volatility at the beginning of each year and use the same volatility for all options and convertible bonds issued during the year, unless the market for our shares fluctuates significantly during a period, in which case the calculation is updated quarterly. The risk-free interest rate used in the calculation is the interest on debt issued by the German government with a maturity similar to the estimated life of the stock options or convertible bonds. We estimate that we will not pay dividends in the foreseeable future. We estimate the life of the option and convertible bonds to be equal to the vesting period of the option or convertible bond. Any changes in estimates of volatility, risk-free interest rate, future dividends and life of the options and convertible bonds would result in increases or decreases in the amount of compensation expense recognized.
Impairment of Long-Lived Assets
Our long-lived assets at December 31, 2003 consist of intangible assets such as licenses and acquired patents and partnered technology purchased in connection with our acquisition of Mitotix, Inc. in a transaction accounted for under the purchase method and fixed assets such as laboratory equipment, computer and other office equipment, furniture and fixtures, and leasehold improvements. In accordance with SFAS No. 144, “Accounting for the Impairment or Disposal of Long-Lived Assets”, we review long-lived assets for impairment when impairment indicators exist and, if necessary,
63
Table of Contents
recognize an impairment charge equal to the difference between the carrying amount of the asset and the fair value as determined by discounted cash flows.
Indicators that result in reviewing our long-lived assets for impairment are, among others, termination of research programs, restructuring programs, equipment obsolescence, idle equipment, or vacating of facilities. During the fourth quarter 2002, we reviewed our long-lived assets to determine whether an impairment charge was required. Tangible assets were not determined to be impaired, as these assets are used in our daily activities and are depreciated using reasonable useful lives, which result in book values that approximate their fair values at year end. However, after a review of the intangible assets, the patent portfolio was identified as having impairment indicators. We reviewed the programs that use the patents and determined that some of the programs were discontinued, and/or the probabilities of success of the program had changed. The fair value of the patent portfolio was determined using the estimated discounted cash flows of each program that used the patents, and we recorded an impairment charge of€ 0.5 million at December 31, 2002. During the fourth quarter of 2003, we again reviewed our patents for impairment, as management decided during 2003 to discontinue additional programs, and also obtained additional information regarding the probability of success of the remaining program. An impairment charge of€ 1.2 million was recorded at December 31, 2003, based on the updated discounted cash flow analysis.
The fair value of the patent portfolio is calculated using the relief-from-royalty approach. This approach estimates the royalty that we would have to pay if we had to license our patents from a third party. Our fair value model uses inputs and assumptions to calculate the fair value of the patent portfolio based on discounted cash flows. As with all estimations of fair value, the estimation of fair value of the patent portfolio involves significant judgments of management. Our fair value model uses the following inputs: identified research programs, estimated revenues of each program, probabilities of success of each program, implied royalty rates and risk-adjusted discount factor. The estimated cash flows of each program are adjusted for the probability of success of the program. An implied royalty rate is applied to the probability adjusted cash flows to arrive at the implied royalties. The implied royalties are discounted at a risk-adjusted rate of return.
Program revenues are one of the most subjective of the assumptions used in the discounted cash flows analysis. This assumption requires us to forecast the peak sales expected to be achieved through sales of our product. We have estimated revenues based on market research performed, which provided a range of peak sales related to this type of technology. We have used this market data, and our internal projections, to arrive at a peak sales number that we believe reasonable. Actual peak sales could differ significantly, resulting in a different fair value for our patent portfolio.
The probability of success of each program is based on the most recent scientific information available to management regarding the progress of the program at a given point in time. Programs are continually evaluated for their viabilities and whether our resources will continue to be used in pursuing the programs. Several programs were discontinued during 2002 and 2003, while others were reevaluated in terms of their probabilities. As of December 31, 2003, only one program acquired as part of the Mitotix, Inc. acquisition, our cell cycle inhibitor program, is still being pursued. The remaining programs have been discontinued. These factors were incorporated into the discounted cash flow analysis.
Royalty rates are based on actual royalty rates stated in our license and/or collaboration agreements with collaboration partners. The rates used in the discounted cash flow analysis take into consideration the similarities of the technology.
The risk-adjusted discount factor considers the risk free rate, the cost of equity, the cost of debt, and a market risk. The rate used to discount the cash flows takes into consideration the risks identified
64
Table of Contents
and already adjusted for in the royalty rate and the probability rate. We have used a risk-adjusted discount rate of 20%, which is specific to our company, but which is based on comparable rates and data used by other biotechnology companies.
The inputs of the model, including the program revenues, probability of success of the programs, implied royalty rates and the risk-adjusted discount rate, are highly subjective and their estimation involves significant judgment of management as to the amounts and timing of the revenues. We have selected values for these inputs that we believe to be fair and supportable. Changes in these estimates would result in a change in the estimated fair value of the intangible assets and thus a change in the impairment charge recognized.
Recent Accounting Pronouncements
In January 2003, FASB issued Interpretation No. 46,Consolidation of Variable Interest Entities(FIN 46). FIN 46 amended Accounting Research Bulletin No. 51,Consolidated Financial Statements, and established standards for determining under what circumstances a variable interest entity (VIE) should be consolidated with its primary beneficiary. FIN 46 also requires disclosures about VIEs that an entity is not required to consolidate, but in which it has a significant variable interest. On December 17, 2003, the FASB deferred the effective date of FIN 46 to no later than the end of the first reporting period that ends after March 15, 2004, however, for special purpose entities, public companies are required to apply FIN 46 by no later than December 31, 2003. We do not believe that any entities will be consolidated with GPC Biotech AG as a result of FIN 46.
In December 2003, the SEC published Staff Accounting Bulletin (SAB) No. 104, “Revenue Recognition”. SAB 104 updates portions of the SEC staff’s interpretive guidance provided in SAB 101. SAB 104 deletes interpretive material no longer necessary, and conforms the interpretive material retained to current authoritative accounting and auditing guidance and SEC rules and regulations, to reflect pronouncements issued by the FASB and EITF on various revenue recognition topics. The adoption of SAB 104 did not have any impact on the results of operation or the financial position of the Company.
65
Table of Contents
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our main exposure to market risk is limited to interest rate and currency exchange rate risks.
Our investment portfolio is subject to interest rate risk and its value will decrease if market interest rates increase. The primary objective of our investment activities is to preserve capital. To minimize risk, we maintain a portfolio of cash, cash equivalents, and short-term, long-term and restricted investments in a variety of interest-bearing instruments, including a bond fund, high-grade European and United States fixed and variable rate corporate bonds and money market funds. As a general policy, we do not invest in equity securities for cash management purposes. Any equity securities in other companies that we own were acquired as part of licensing agreements. Since our investments are short term in duration, we believe that an immediate 10% change in market interest rates would not be material to our financial condition or results of operations.
Our results of operations and financial condition are also subject to exchange rate risks. Fluctuations between the euro and the U.S. dollar can affect our financial results. The U.S. dollar denominated proportion of our revenues and operating costs can vary from year to year. In 2003, a significant amount of our revenues and expenses was denominated in U.S. dollars. Additionally, we hold a small amount of cash in the form of U.S. dollars and our investment portfolio includes some marketable securities denominated in U.S. dollars to fund our U.S. operations. Accordingly, any appreciation of the euro against the dollar would have the effect of reducing our reported revenues and reducing our reported expenses. We do not, however, hold any derivative financial instruments to protect us from the exchange rate risk associated with the U.S. dollar and the euro. A significant portion of other income and expense relates to foreign currency gains and losses on accounts receivables, accounts payable and cash. Other income (expense), net was€(0.1) million for the year ended December 31, 2003 compared with€(0.7) million for the year ended December 31, 2002.
66
Table of Contents
Overview
GPC Biotech is a biopharmaceutical company discovering and developing new drugs to treat cancer. Our lead product candidate, currently in a Phase 3 registrational trial, is satraplatin, a platinum-based compound intended for use as a chemotherapy drug. Platinum-based drugs have been clinically proven to be one of the most effective classes of anticancer therapies and are used to treat a wide range of cancers. Unlike currently marketed platinum-based drugs, satraplatin is administered orally. We licensed satraplatin in 2002 and, in September 2003, initiated a Phase 3 registrational trial of satraplatin as a second-line chemotherapy treatment for hormone refractory prostate cancer, HRPC, with sites in the United States, Europe and South America. We refer to this trial as the “SPARC” (Satraplatin and Prednisone Against Refractory Cancer) Trial. We modelled the SPARC Trial, in which we expect over 900 patients to be enrolled, on a successful 50-patient study of satraplatin as a first-line chemotherapy treatment of HRPC conducted by others. There is currently no approved second-line chemotherapy treatment regimen for patients who fail first-line chemotherapy treatment for HRPC. We were therefore able to obtain fast-track designation from the FDA for satraplatin in this disease setting. Our goal is to complete the filing of a new drug application, or NDA, with the FDA for satraplatin as a treatment of HRPC in 2006. Based on existing clinical data, we believe that satraplatin may have application in a number of cancers, both as a single agent and in combination with other therapies.
Our second most advanced product candidate is 1D09C3, a monoclonal antibody that is in late preclinical development and is intended for the treatment of selected leukemias and lymphomas, including non-Hodgkin’s lymphoma. A monoclonal antibody is an immune system related protein that binds preferentially to one type of foreign substance, potentially stimulating a biological response. We plan to complete a series of preclinical toxicology evaluations and the development of certain tests for monitoring drug levels and drug responses in the blood of treated patients during 2004. If these activities are completed successfully and if we receive approval of the relevant regulatory authorities and ethics committees on a timely basis, we would be able to advance 1D09C3 into Phase 1 clinical trials during 2004. Our third development program involves small molecule compounds that inhibit the cell division cycle, which is essential for tumor growth. The cell division cycle, or cell cycle, is the process by which a cell grows, duplicates its DNA and divides into two cells. We have identified a lead compound, RGB-286199, that is currently in preclinical development. Our goal is to complete preclinical development work on RGB-286199 in the first half of 2005. In addition, we have several distinct research programs to discover new anticancer drug candidates.
We also have a portfolio of technologies in the fields of proteomics (the systematic study of proteins and their functions) and genomics (the systematic study of genes). In early 2003, we expanded our technology portfolio through the introduction of LeadCodeTM, our proprietary cell-based drug-protein interaction screening technology. LeadCode enables us to improve our understanding of the potential efficacy and side effects of drugs and drug candidates. We believe that these technologies allow us to prioritize and optimize drug candidates in our pipeline, identify new drug uses and potentially reduce the risk of failure in drug development.
In 2001, we entered into an agreement with ALTANA Pharma, which is our most significant technology collaboration to date. Under this contract, we are assisting ALTANA Pharma through 2007 with its establishment of a research institute the United States. This agreement includes a research collaboration as well as a transfer of technologies. Effective January 2003, we have also licensed LeadCode to ALTANA Pharma under a separate agreement. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding over the term of the agreements. As of December 31, 2003, we had received an aggregate€ 37.4 million from ALTANA Pharma under these agreements, of which€27.4 million have been recognized as revenue.
67
Table of Contents
Our Strategy
We are focused on oncology because this is an area in which, despite progress in understanding cancer and the resulting development of new therapies, there is still a lack of effective treatments. Also, due to the relatively short timeframe and low number of patients needed for oncology clinical trials, the costs of clinical development of anticancer drugs are significantly lower than those for many other diseases. We have assembled a highly experienced team of scientists and executives specialized in the fields of oncology drug discovery, development and commercialization. We believe that the oncology market’s rapid growth and substantial unmet medical need, combined with our focused expertise, position us to compete effectively in the oncology drug market.
Our goals are to discover, develop and commercialize new anticancer drugs. We are pursuing these goals through the following strategies:
| • | Enhance the commercial value of satraplatin to us. We believe that we have the internal expertise and, after this offering, will have the financial resources to conduct our planned clinical trials for satraplatin without a partner. We believe that we will be able to achieve greater value from satraplatin by advancing its development on our own. We expect, however, to seek a strategic partner to assist us in the sales and marketing of satraplatin. A sales and marketing partnership would allow us to benefit from the expertise, marketing capabilities and resources of such partner to increase both speed and depth of market penetration. |
| • | Broaden the potential range of applications of satraplatin. Clinical trials with satraplatin conducted by others have demonstrated activity in prostate cancer, as well as in other cancers, such as ovarian cancer and lung cancer, and have also demonstrated beneficial effects when combined with other chemotherapy drugs or when combined with radiation therapy. As part of our satraplatin development program, we intend to conduct additional clinical trials for other cancers with satraplatin as a single agent, in combination with other chemotherapy drugs and in combination with radiation therapy. |
| • | Build and maintain a balanced portfolio of oncology drugs. To diversify the inherent risk associated with the development of anticancer drugs, we work on a diverse group of anticancer drug candidates. For example, we seek to diversify our risk profile by developing product candidates that have known mechanisms of action, such as satraplatin, as well as developing product candidates that have novel mechanisms of action. Additionally, we diversify our risk by working with biologic agents, such as our human monoclonal antibody, as well as synthetic small molecules, such as our cell cycle inhibitor. |
| • | Use our research expertise, technologies and external network to build our product candidate pipeline. We seek to leverage our internal expertise and external network of scientists and experts in the oncology field to identify and evaluate in-licensing opportunities. This strategy has already proven successful in the in-licensing of satraplatin, and we continue to evaluate numerous opportunities on an ongoing basis. Additionally, we plan to use our internal research expertise and our drug discovery technologies to help us identify promising new drug discovery projects and to initiate new drug discovery programs. For example, we believe that our technologies, particularly our LeadCode technology, will allow us to discover and subsequently enhance the activity profiles of small molecule classes known to have anticancer activity. |
| • | Use our internal expertise to find the fastest path to market approval for our product candidates. When planning our approach for obtaining regulatory approval for a new anticancer drug candidate, we seek to minimize the time to market and maximize the likelihood of approval. We do this by designing registrational trials targeted at disease settings where no effective treatment alternatives exist. Additionally, we discuss our clinical trial plans |
68
Table of Contents
with the relevant regulatory authorities prior to initiating clinical trials to ensure that our trial designs will be acceptable to these regulatory authorities. For our lead product candidate, satraplatin, we have obtained fast track approval from the FDA, participated in an “End-of-Phase 2 Meeting” with the FDA and completed a Special Protocol Assessment thereby obtaining a meaningful review of our overall registrational approach by the FDA. Additionally, we have obtained a Scientific Advice Letter from the EMEA providing the EMEA’s perspective on our registrational approach for satraplatin. We expect to take a similar approach for our other product candidates. We have assembled a clinical development team that is highly experienced in designing and conducting successful clinical trial programs. Members of this group have a combined career track record of obtaining more than 20 marketing approvals with the FDA, including those for TAXOL® (paclitaxel), a widely used chemotherapy drug, and PARAPLATIN® (carboplatin), one of the most well-established platinum-based chemotherapy drugs. |
| • | Generate revenue through technology and research collaborations. While we ultimately plan to generate revenues from the sale of new anticancer drugs, we have a strong history of generating revenues through technology and research collaborations with pharmaceutical partners across a broad range of disease areas. By leveraging our technologies to generate substantially all of our past and current revenues, we have been able to internally finance a significant portion of our own drug discovery and development activities. Our alliance with ALTANA Pharma is expected to continue to provide us with revenues through 2007. In 2001, we entered into an agreement with ALTANA Pharma, which is our most significant technology collaboration to date. Under this contract, we are assisting ALTANA Pharma through 2007 with its establishment of a research institute in the United States. Effective January 2003, we have also licensed LeadCode to ALTANA Pharma under a separate agreement. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding over the term of the agreements. As of December 31, 2003, we had received an aggregate of€ 37.4 million from ALTANA Pharma under these agreements, of which€ 27.4 million have been recognized as revenue. We continue to seek opportunities to generate revenues by licensing our technologies. |
Our Product Pipeline
Our product pipeline currently contains three product candidates intended for the treatment of cancer. Cancer is the second leading cause of death in the United States after cardiovascular disease. The American Cancer Society estimates that in 2004 there will be 1,368,000 new cases of cancer diagnosed and 536,700 deaths attributable to cancer in the United States. According to GLOBOCAN 2000, in the year 2000, there were a total of 1,296,000 new cases of cancer and 789,000 deaths attributable to cancer in the United Kingdom, Germany, France, Italy and Spain.
Many different approaches are used in treating cancer, including surgery, radiation therapy, drugs or a combination of these approaches. Drugs used to treat cancer include chemotherapeutics, hormones and immune-based therapies. According to the MattsonJack CancerMetric database, over 1 million cancer patients were treated with anticancer drugs in 2003 in the United States. According to Reuters Business Insight estimates, the worldwide sales of anticancer drugs totaled over $14 billion in 2002 and are forecast to reach $20 billion in 2008.
69
Table of Contents
The following table summarizes the development status of our product candidates and research programs:
Product Candidate | Indication | Development Status | ||
Satraplatin | Second-line chemotherapy of HRPC | Phase 3—registrational trial ongoing | ||
| Combination with radiation therapy in various cancers | Phase 2—study protocol under review | |||
| Combination with TAXOL® in lung cancer | Phase 2—study protocol in preparation | |||
| Combination with TAXOTERE® in various cancers | Phase 1—study protocol in preparation | |||
Human Monoclonal Antibody (1D09C3) | Refractory non-Hodgkin’s lymphoma | Preclinical studies—ongoing | ||
| Combination therapy with RITUXAN® in non-Hodgkin’s lymphoma | Preclinical studies—ongoing | |||
Cell Cycle Inhibitor (RGB-286199) | Various refractory cancers | Preclinical studies—ongoing | ||
Research Programs | ||||
Apoptosis Inducers | Various cancers | Research—ongoing | ||
Novel Kinase Inhibitors | Various cancers | Research—ongoing | ||
Satraplatin
Our lead product candidate is satraplatin, a member of the platinum family of drugs. Platinum-based drugs represent one of the mainstays of modern chemotherapy and are used to treat a wide variety of cancers, with worldwide sales of these drugs in 2003 exceeding $1.7 billion. We licensed the exclusive commercial rights to satraplatin, including the right to sublicense, in the field of treating cancer, from Spectrum Pharmaceuticals, Inc. (formerly NeoTherapeutics, Inc.) at the end of the third quarter of 2002.
Market Opportunity. Three platinum-based drugs are currently marketed in the United States and Europe: carboplatin, cisplatin and oxaliplatin. These platinum-based drugs are used to treat more than 34 different cancers; in many cases regimens containing a platinum-based drug represent the current standard of care. Unlike these platinum-based drugs, which are administered intravenously, satraplatin is administered orally in the form of a capsule. Satraplatin has been studied in a variety of adult cancers in clinical trials that in total have enrolled over 600 patients. In randomized clinical trials, satraplatin has shown significant anticancer activity in HRPC, and is the only platinum-based compound for which such clinical data have been generated. Additional Phase 2 trials have been successfully completed by others in ovarian cancer and small cell lung cancer. Our goal is to develop satraplatin not only for the treatment of prostate cancer, but also for other cancers that are known to respond to treatment with platinum-based drugs.
Prostate Cancer and Existing Treatment Regimens. Prostate cancer is the most common cancer among men and the second leading cause of cancer deaths in men in the United States. Of the
70
Table of Contents
700,000 new cases of cancer that are anticipated in American men in 2004, the American Cancer Society estimates that approximately 230,000 of those cases will be men diagnosed with prostate cancer. Moreover, of the approximately 291,000 men in the United States who are expected to die of cancer in 2004, approximately 30,000 of those men are expected to die of prostate cancer. Since the risk of prostate cancer increases with age, the aging of the overall population over the next several decades is expected to further increase the number of prostate cancer patients.
During the early stages of prostate cancer, when the tumor is still confined within the prostate and adjacent tissues, treatment may involve surgery, radiation therapy or a combination of the two. Surgery aims at completely removing the tumor from the body. Radiation therapy aims at killing prostate cancer cells, thereby shrinking tumors or preventing cancer cells from dividing and spreading to remote sites in the body, a process known as metastasis. In many cases, however, the cancer progresses despite these treatments.
Once the cancer spreads outside the prostate gland, hormone ablation therapy is often used to try to control the cancer by limiting the supply of hormones that this type of cancer needs to grow. Hormone ablation therapy may be accomplished by surgical castration or by chemical castration, which involves the administration of drugs to prevent the production of or to block the action of testosterone and other male hormones.
While most prostate cancer patients initially respond to hormone ablation therapy, the vast majority eventually relapse, becoming refractory to hormone treatment, typically after 18 to 24 months of treatment. According to the estimates of Datamonitor plc, approximately 70,000 men with prostate cancer in the United States were hormone refractory in 2002. For these HRPC patients, subsequent treatment involves a limited number of options, including chemotherapy and radiation therapy. There are currently two chemotherapy drugs approved for such first-line chemotherapy of HRPC. While these two drugs are effective in reducing the severe pain that these patients experience, they have not been shown to delay the progression of this disease. Because these drugs have not been shown to change the overall course of the patient’s cancer, physicians do not frequently use currently approved chemotherapies in this disease setting.
In 2002 and 2003, published Phase 2 clinical trial results using TAXOTERE® (docetaxel), which is a chemotherapy drug that has recently been approved by the FDA for the first-line treatment of HRPC and is also approved for treatment of other adult cancers, demonstrated that—unlike existing drugs approved for first-line chemotherapy treatment of HRPC—it may delay disease progression when used as a first-line chemotherapy treatment of HRPC. Although these Phase 2 studies demonstrated a slowing in the progression of the cancer in HRPC patients, these patients ultimately became refractory to treatment with TAXOTERE and their conditions deteriorated, which often happens after a number of courses of this chemotherapy treatment. Several large Phase 3 trials of TAXOTERE for first-line treatment of HRPC were recently completed. We expect the results of these trials to be reported at medical meetings to be held in mid-2004.If the results of these trials are positive, TAXOTERE could become the new standard of care for the first-line chemotherapy treatment of HRPC. We believe that the use of TAXOTERE to treat HRPC will increase the number of HRPC patients who receive chemotherapy and also create the need for a second-line chemotherapy treatment of this cancer. We believe that satraplatin is the only product candidate currently in a Phase 3 registrational trial as a second-line chemotherapy treatment of HRPC.
History and Prior Clinical Trials of Satraplatin. Satraplatin was invented and synthesized by scientists at Johnson Matthey as part of a collaboration with Bristol-Myers Squibb to find new platinum-based chemotherapy drugs. Unlike any of the marketed platinum-based drugs, satraplatin showed anticancer activity when administered orally.
71
Table of Contents
Between 1992 and 1999, Bristol-Myers Squibb initiated 29 clinical trials of satraplatin in a variety of cancers. A total of 615 patients were enrolled in these trials. Satraplatin demonstrated anticancer activity in various cancers in these trials, including prostate cancer, ovarian cancer and small cell lung cancer. Additionally, satraplatin demonstrated anticancer activity in cancers of the head and neck when given in combination with radiation therapy.
The following chart summarizes selected clinical studies involving satraplatin that were sponsored by Bristol-Myers Squibb.
Indication | Clinical Status | Date Initiated | ||
First-line chemotherapy of HRPC | 50-patient randomized Phase 3 study conducted by the European Organization for Research and Treatment of Cancer (EORTC) | 1998 | ||
Second-line chemotherapy of ovarian cancer | Randomized Phase 2 study | 1995 | ||
First-line chemotherapy of small cell lung cancer | Phase 2 study | 1994 | ||
Combination with paclitaxel in various cancers | Phase 1 study | 1996 | ||
Combination with radiation therapy in various cancers | Phase 1 study | 1997 | ||
Combination with etoposide in various cancers | Phase 1 study | 1994 |
The most important of the clinical trials initiated during this period was a randomized Phase 3 trial comparing outcomes in a group of HRPC patients who received satraplatin plus prednisone to outcomes in another group that received prednisone alone. Bristol-Myers Squibb initiated this clinical trial in 1998 with the intention of enrolling 380 patients. The trial was conducted by the EORTC. In 1999, Bristol-Myers Squibb terminated its satraplatin program as part of a strategic realignment of its development portfolio. At that time the EORTC had entered 50 patients into the trial. The EORTC investigators continued to treat these 50 patients until either their cancer progressed or they died. The EORTC investigators reported the results at the 2003 Annual Meeting of the American Society of Clinical Oncology. The major finding from the study was a significant prolongation of time during which patients lived without their cancer progressing (progression-free survival) in the group treated with satraplatin plus prednisone, as indicated in the following diagram.
72
Table of Contents
Progression-Free Survival
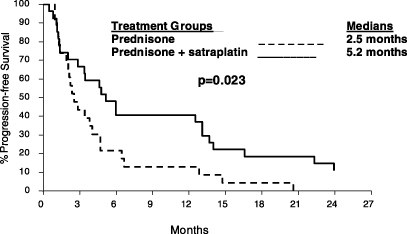
Note: “p” is a measure of statistical significance. A “p” value of less than 0.05 indicates that the results of this comparison were considered significant.
The results also showed that a statistically greater proportion of patients treated with satraplatin plus prednisone experienced a decline of more than 50% of prostate-specific antigen in their blood as compared with pre-treatment levels. The blood of prostate cancer patients usually contains elevated levels of prostate-specific antigen. Many clinicians use this protein in the diagnosis of prostate cancer, in measuring response to treatment and in monitoring the recurrence and progression of this cancer.
Comparison of the median overall survival of the two groups in this trial showed a numerical trend favoring the satraplatin plus prednisone group, but this difference was not considered significant, as indicated in the following diagram.
Overall Survival
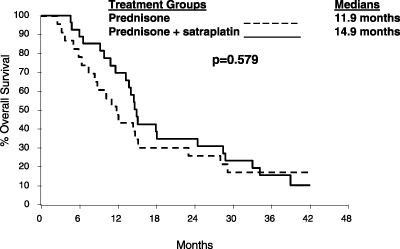
Note: “p” is a measure of statistical significance. A “p” value of greater than 0.05 indicates that the results of this comparison were not considered significant.
73
Table of Contents
We believe that the inability to show a statistically significant difference in median overall survival in this trial may be due to the enrollment of only 50 patients instead of the initially planned total of 380. We believe the number of patients was probably too small to yield a significant difference in overall survival between the two treatment groups. However, it may be that no significant difference would have been observed had this trial been completed as initially planned.
Finally, a review of the clinical laboratory data and the adverse events experienced in both groups of patients in this study, together with the clinical observations reported by the study physicians, indicated that satraplatin plus prednisone was a well tolerated chemotherapy regimen in this setting and warranted further study in the treatment of HRPC.
Current Clinical Trials. Shortly after we licensed satraplatin in 2002, we began discussions with the FDA that culminated in an official “End-of-Phase 2 Meeting” with the FDA in July 2003. The purposes of this meeting included assessing the safety of the drug in earlier trials, evaluating our Phase 3 registrational trial plan, and identifying any additional information that would be needed to support an NDA. Additionally, we requested a review of our Phase 3 registrational trial protocol under the FDA’s Special Protocol Assessment program. The combination of the “End-of-Phase 2 Meeting” and the Special Protocol Assessment provided us the opportunity to hold meaningful discussions with the FDA regarding our overall registrational approach. As a result, the FDA has confirmed its agreement with us that successful completion of the SPARC trial will form the primary basis for an efficacy claim for our NDA for satraplatin. This agreement becomes part of the administrative record and may only be changed by mutual agreement of the parties or if the FDA identifies a substantial scientific issue relevant to safety or efficacy after the trial has begun. The FDA has also granted fast track designation to satraplatin as a second-line chemotherapy treatment for patients with HRPC. The FDA’s fast track program is intended to facilitate the development and expedite the review of drugs that treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs. The fast track designation enables us to file sections of the NDA on a rolling submission basis, submitting sections as they become available.
We have also received a Scientific Advice Letter from the EMEA relating to our Phase 3 clinical trial. Although this letter is not required for the initiation of a Phase 3 clinical trial in Europe, it is helpful because it allowed the EMEA to comment on our overall registrational approach before implementation.
In September 2003, we initiated the SPARC Trial. The SPARC Trial is a multinational, multicenter, randomized, double-blind Phase 3 registrational trial to test satraplatin plus prednisone versus a placebo plus prednisone in patients with HRPC whose disease has progressed on first-line chemotherapy. “Randomized” means that patients are randomly assigned to receive the drug candidate or a placebo. To encourage participation, we have set the randomization ratio at 2:1 in favor of active treatment. “Double-blind” means that neither the physician nor the patient knows whether the patient has received the drug candidate or a placebo. “Progressed on first-line chemotherapy” means that a patient with HRPC has shown further advancement of their cancer while being treated with a regimen that includes a chemotherapy drug. The SPARC Trial is designed to determine the efficacy and evaluate the safety of satraplatin plus prednisone in slowing the progression of cancer in this patient population. According to the criteria we have discussed with the FDA and the EMEA, the trial will be considered positive if the treatment group has a 30% or greater increase in the period of time required for progression of disease to occur, as compared with the placebo group.
During the course of the SPARC Trial an independent data monitoring board, established in accordance with guidelines provided by the FDA, will meet periodically to review the results of the trial and evaluate the safety and/or efficacy of satraplatin in the study population. At the time of any of these
74
Table of Contents
interim analyses, the data monitoring board can recommend that we either stop or continue the clinical trial based on safety, efficacy or ethical reasons. Under our clinical trial plan, the data monitoring board may communicate the results of one or more interim analyses to us. There can be no assurances whether or when the data monitoring board will do so.
The SPARC Trial is underway at over 100 clinical centers in the United States, Europe and South America and is expected to enroll over 900 patients. We are working together with two contract research organizations to conduct the SPARC trial. Patient recruitment is proceeding at a rate that should allow us to complete the filing of an NDA in 2006. There can be no assurance, however, that we will be able to complete the NDA on this schedule or at all.
Planned Additional Clinical Trials. We intend to conduct additional clinical trials to further explore satraplatin’s usefulness for the treatment of other cancers, such as ovarian and small cell lung cancer, where completed Phase 2 clinical trials have shown evidence of efficacy. Additionally, we plan to conduct clinical trials using satraplatin in combination with other chemotherapeutic drugs, such as TAXOL and TAXOTERE. When used in combination with a taxane drug, of which TAXOL or TAXOTERE are examples, other platinum-based drugs, such as cisplatin or carboplatin, have demonstrated synergistic effects in a number of clinical settings. For example, a combination therapy of TAXOL and carboplatin is the current standard of care for first-line chemotherapy treatment of non-small cell lung cancer. We intend to determine whether similar clinical benefits may be realized by combining satraplatin with a taxane drug.
We also plan to conduct clinical trials using satraplatin in combination with radiation therapy to explore satraplatin’s potential to enhance the efficacy of radiation therapy. Promising clinical results have been observed for satraplatin and other platinum-based drugs administered in combination with radiation therapy. We believe that the oral administration of satraplatin may provide an important advantage over currently marketed platinum-based drugs when used in combination with radiation therapy. In practice, it has been difficult to provide intravenously administered platinum-based drugs to patients continuously during the course of radiation therapy due to the need for different safety precautions for the two treatments.
Manufacturing. We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of satraplatin. Johnson Matthey manufactured the material that we plan to use in our clinical trials of satraplatin. We have a contract with Johnson Matthey for additional supplies of the active pharmaceutical ingredient for future clinical trials. Johnson Matthey has been a major manufacturer of platinum-based pharmaceuticals for many years. We believe we currently have a sufficient supply of satraplatin to complete our Phase 3 registrational trial. We do not expect to develop our own manufacturing capabilities; therefore, if regulatory approval of satraplatin is obtained, we will be required to contract with a third party for commercial amounts of the active pharmaceutical ingredient.
1D09C3—Monoclonal Antibody
Our second most advanced product candidate, 1D09C3 is a monoclonal antibody that we are developing with the intent of treating selected leukemias and lymphomas. 1D09C3 is currently in late preclinical development. 1D09C3 has shown anticancer activity in several preclinical tests involving animal models of these cancers. Our monoclonal antibody functions through a mechanism of action that we believe is novel and may represent a significant opportunity in treating a variety of cancers. We hold exclusive rights in a field that includes human therapeutic use to patent applications covering 1D09C3, which include composition of matter and medical use applications in the United States, Europe and other markets.
75
Table of Contents
Market Opportunity. The initial indication for which we intend to develop 1D09C3 is non-Hodgkin’s lymphoma. Lymphomas are a group of cancers that arise from cells in the lymph nodes, bone marrow or spleen and often spread throughout the body. There are two major types of lymphoma: non-Hodgkin’s lymphoma and Hodgkin’s disease. The American Cancer Society estimates that, in 2004, there will be more than 54,000 new cases of non-Hodgkin’s lymphoma and more than 19,000 deaths attributable to non-Hodgkin’s lymphoma in the United States.
Anticancer drugs currently used to treat non-Hodgkin’s lymphoma include monoclonal antibodies and chemotherapy drugs such as doxorubicin. Currently, there are three monoclonal antibodies approved for the treatment of non-Hodgkin’s lymphoma in the United States: RITUXAN®, ZEVALIN® and BEXXAR®. CAMPATH® is approved for chronic lymphatic leukemia. Approximately 50% of patients initially treated with RITUXAN, the most widely used of these drugs, fail to respond. Of those that do respond, many relapse and become refractory after several treatment cycles. As a result, there is currently an unmet medical need for treatment of patients who have failed RITUXAN therapy.
Based on our preclinical research, 1D09C3 may also prove useful for the treatment of other cancers, including leukemias and melanoma. These cancers present additional significant unmet medical needs because effective treatment options are currently limited.
Scientific Overview. Lymphomas and leukemias are characterized by uncontrolled proliferation of certain cells of the immune system. Studies in recent years have shown that immune system cells, known as B-cells and T-cells, may respond to external signals by committing cellular suicide (programmed cell death). This response is thought to play a role in the natural process of regulating immune responses.
Monoclonal antibodies are manufactured antibodies that share characteristics of naturally occurring antibodies. Naturally occurring antibodies are proteins that are an essential component of the human immune system. They are produced in response to the presence of foreign substances, or antigens, in the body and are extremely specific. Each antibody binds preferentially to one particular type of antigen presented on a cell and can interfere with that cell’s activity or cause cell death. Monoclonal antibodies may be used alone or in combination with other anticancer drugs or radiation therapy to attack cancer cells in several ways.
The aim of our antibody research program was to develop a monoclonal antibody that binds to MHC class II molecules (immune system-related proteins found on the surface of cells) and that can also induce programmed cell death in selected lymphomas and leukemias. In 1999, we entered into a collaboration with MorphoSys AG, a German biotechnology company specialized in producing novel monoclonal antibodies based on human genes. We developed our monoclonal antibody product candidate, 1D09C3, from our collaboration with MorphoSys.
1D09C3 is being developed with the intent to treat patients that have failed treatment with RITUXAN. This monoclonal antibody has several characteristics that suggest that it may be suitable for use in cancer patients, including that:
| • | it binds to an antigen (MHC class II molecules) that is different from that targeted by RITUXAN, ZEVALIN and BEXXAR, thereby reducing the likelihood of failure after pre-treatment with RITUXAN, ZEVALIN or BEXXAR; |
| • | it does not require immune system function, which is frequently compromised in those patients pre-treated with chemotherapy and/or RITUXAN; and |
| • | in preclinical animal studies, when combined with RITUXAN, it shows greater efficacy than either treatment alone. |
76
Table of Contents
Results of Preclinical Studies. 1D09C3 has been evaluated in numerous preclinical studies and has shown anticancer activity in animal models, including models of non-Hodgkin’s and Burkitts lymphoma, multiple myeloma, chronic lymphatic leukemia and hairy-cell leukemia. In a non-Hodgkin’s lymphoma model in mice, we showed that the combination of our antibody product candidate with RITUXAN produced greater anticancer activity than with either antibody alone, as indicated in the following diagram.
Combination Therapy of Non-Hodgkin’s Lymphoma
Mouse Model with 1D09C3 and Rituxan
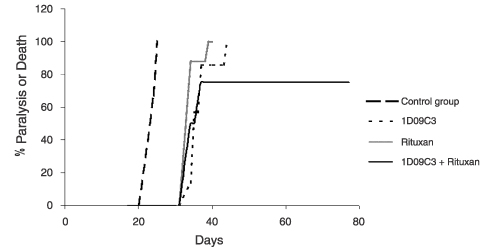
There can be no assurance that similar results will be observed in clinical trials with humans.
We have not yet initiated clinical trials of 1D09C3, as we are currently completing advanced preclinical development. Preclinical activities that need to be completed prior to initiating clinical trials include a toxicological evaluation and the development of certain tests for monitoring drug levels and drug responses in the blood of treated patients. In a preclinical study involving repeat dosing, we observed anaphylactoid reactions in two animals. Anaphylactoid reactions, which are immediate allergic reactions of the immune system, are not uncommon in antibody treatments and can frequently be avoided by simultaneously administering antihistamines or steroid hormones, or both. Antihistamines and steroid hormones are commonly used to treat the symptoms of allergic reactions. We plan to further investigate anaphylactoid reactions as a potential side effect of 1D09C3 in the preclinical toxicological evaluation before initiating clinical trials.
We have met with the relevant regulatory authority in Europe to discuss the design of these preclinical tests. Assuming successful and timely completion of these preclinical tests and timely approval of the relevant regulatory authorities and ethics committees, we anticipate initiating clinical trials for this product candidate in 2004 in patients that have failed RITUXAN treatment. There can be no assurance, however, that we will initiate clinical trials in this timeframe or at all.
Manufacturing. We do not currently own or operate manufacturing facilities for the production of clinical or commercial quantities of 1D09C3. We believe we have obtained a sufficient amount of 1D09C3 for use in our preclinical and initial planned clinical trials from a third party, manufactured to our specifications.
RGB-286199—Cell Cycle Inhibitor
Our third most advanced product candidate, RGB-286199, is a small molecule drug that inhibits the cell cycle, which is essential for the spread of cancer. Although no effective cell cycle inhibitor
77
Table of Contents
drugs have yet been approved, the cell cycle has been recognized for its importance in cancer development for many years. This product candidate, which is currently in preclinical development, resulted from a research program initiated by Mitotix, Inc., which we acquired in 2000.
Market Opportunity. Cancers are characterized by uncontrolled cell proliferation. The cell cycle is a key cellular process that fails to function normally in cancer development. Studies on a wide range of human cancers have shown that mutations in genes that regulate the cell cycle are found in almost all cases. Therefore, drugs that inhibit the cell cycle could find application in a large number of cancers. The clinical use of a cell cycle inhibitor drug will, however, be conditioned on its stability and distribution in the body after administration and limited by its potential side-effects, making it unlikely that any particular cell cycle inhibitor drug would be used in all or nearly all cancers.
Numerous cell cycle inhibitors have been discovered and explored in early development stages. Of those still in development, few have shown compelling anticancer activity in the clinic. Many of these drug candidates are known as narrow-spectrum cell cycle inhibitors because they target only a few components regulating the cell cycle. Recent scientific publications indicate that to achieve long-lasting inhibition of the cell cycle, a broader spectrum of inhibition may be required. Our cell cycle inhibitor has shown this broader spectrum of inhibition in preclinical tests. We believe that this may translate into greater anticancer activity than has been achieved with most other drug candidates of this class.
Scientific Overview. Cyclin-dependent kinases (CDKs) are proteins that are involved in the regulation of the cell cycle. At least four different CDKs control various key events during the cell cycle. Certain of these also appear to regulate survival pathways in cancer cells. A survival pathway is a biochemical signaling process inside a particular cell by which its survival is regulated. RGB-286199 is a potent inhibitor of all of these CDKs, inhibiting cell division and inducing cell death in cancer cells. Furthermore, we have shown that RGB-286199 inhibits non-cyclin dependent kinases that are associated with cancer.
Preclinical Studies. We have shown that RGB-286199 is active against a broad range of cultured cancer cells, including cancer cells that are not actively dividing, while having comparatively little effect on normal cultured cells that are not actively dividing. We believe these data suggest that RGB-286199 may have potential for treating solid tumors in which a large percentage of tumor cells do not actively divide. We have also shown that growth of a variety of human cancer cells inoculated into mice is substantially inhibited. When evaluated 120 days later, the animals treated with RGB-286199 frequently showed no evidence of disease. In contrast, control groups of animals that did not receive anticancer treatment all showed progressive tumor growth by day 20 of the study. We have shown anticancer activity in animal models of ovarian, prostate and colon cancer. There can be no assurance that similar results will be observed in clinical trials with humans.
We have not yet initiated clinical trials of RGB-286199. We are currently planning additional toxicity studies as well as studies to determine appropriate dosing and administration schedules. Our goal is to complete preclinical development work on RGB-286199 in the first half of 2005 and, if successful, we anticipate advancing the drug candidate into clinical trials thereafter. There can be no assurance, however, that we will complete preclinical development or initiate clinical trials in this timeframe or at all. Given the potentially broad application of this drug candidate in the treatment of cancer, we expect to conduct early clinical trials in patients with a variety of cancer types. We plan to use the results from these early clinical trials to guide us to those cancer indications that represent the fastest path of development for this drug.
Manufacturing. We intend to use a third party contract manufacturer for the production to our specifications of RGB-286199 for use in preclinical studies and clinical trials. We are currently seeking a third party manufacturer for RGB-286199.
78
Table of Contents
Our Research Programs
Our research is focused on new anticancer drug discovery opportunities. To date, our research activities have led us to the discovery of 1D09C3 and RGB-286199, two product candidates that are now in preclinical development. Our current approach to new drug discovery programs is to start with existing small molecules or small molecule classes that are either known to have anticancer activity or known to act on selected targets relevant for cancer drug research. We design our research programs to discover drug candidates that have potentially broad application in the treatment of various cancers. Most of our research programs are directed toward discovering cytotoxic drugs, which are drugs that kill cancer cells rather than prevent their growth.
The nature of drug discovery is that programs are continuously initiated, pursued and frequently discontinued, unless progress is satisfactory or potential for identifying a development candidate remains sufficiently high to merit further investment of resources. For example, most recently we have discontinued our bryologs and bryostatin-1 programs.
Our current research projects can be classified into two main areas:
| • | Inducers of Programmed Cell Death. Most cytotoxic drugs currently used in cancer therapy work by inducing programmed cell death, or apoptosis, in cancer cells. The mechanisms by which many types of molecules induce apoptosis in cancer cells remain unknown. We are using our technological expertise, specifically our LeadCode technology, to study these mechanisms of action. We believe that elucidating the mechanism of action of apoptosis-inducing molecules will allow us to modify their structure and improve their activity, making them more suitable for development as anticancer drugs. |
We are currently researching the mechanism of action of four molecules with known anticancer activity. These are at various stages of research, and our goal is to advance at least one of them into early preclinical development in the first half of 2005. There can be no assurance, however, that we will be able to do so on this schedule or at all.
| • | Inhibitors of Protein Kinases. Protein kinases represent a large family of proteins that play an important role in signaling between and within cells. We believe that the inhibition of kinases may represent an important and widely applicable approach to the treatment of cancers. There are more than 500 protein kinases encoded by the human genome, and many of these kinases have been implicated in cancer development. Two novel kinase inhibitors, GLEEVEC® and IRESSA®, have been introduced into the market in the last three years for the treatment of chronic myeloid leukemia and lung cancer, respectively. |
There are several classes of small molecules that have been shown to inhibit kinases. We are currently working on the generation of several new kinase inhibitors based on these known classes of kinase inhibitors. We hope that these new kinase inhibitors will have profiles suitable for development as novel anticancer drugs.
Licensing to Others. From time to time we consider licensing to others research programs and related technologies that we are no longer developing. In 2002, we granted a license providing worldwide rights to our antibacterial program to PanTherix, Ltd. Because PanTherix was not fulfilling its obligations under the license agreement, we terminated the license in 2003 and the program was returned to us. We are currently seeking to license our immunology programs and anti-infectives programs to others, including the antibacterial program previously licensed by PanTherix, and immunology research programs, which are not aligned with our strategic focus on oncology drugs.
Our Drug Discovery Technologies
From our inception in 1997, we have developed and licensed access to technologies that facilitate drug discovery research. We have applied these technologies in research alliances with
79
Table of Contents
pharmaceutical companies, including ALTANA Pharma, Aventis, Bayer, Boehringer Ingelheim and Eli Lilly. These alliances have been a major source of revenue to date, and our ongoing alliance with ALTANA Pharma continues to provide a significant portion of current and future expected revenues. We have also applied these technologies to advance our internal drug discovery programs.
Our technologies, some of which we have developed internally and some of which we have licensed from third parties, are based on genomics and proteomics approaches. These allow us to gain a better understanding of the molecular basis of disease and of drug mechanisms of action. Specifically, we have a gene expression technology, known as ExpressCodeTM, two technologies for studying protein-protein interactions, known as PathCodeTM and UbiCodeTM, as well as two technologies for finding new approaches to controlling cell growth, known as KeyCodeTM and MaRXTM.
In early 2003, we introduced LeadCode, our proprietary drug-protein interaction screening technology. LeadCode identifies and analyzes the interaction of a drug with human proteins. LeadCode uses yeast cells containing fragments of human proteins to test whether any of these proteins bind to a drug molecule of interest. LeadCode enables us to improve our understanding of the mechanisms of action of drug candidates. This understanding helps us in the evaluation of the potential efficacy and side effects of drug candidates. This technology thereby allows us to prioritize and optimize drug candidates in our pipeline, identify new drug uses and, we believe potentially reduce the risk of failure in drug development.
To augment our ability to study drug-protein interactions, which has become central to our drug discovery approach, we employ a technology called MAPPIT. This technology was originally designed to detect protein-protein interactions in mammalian cells. We have re-engineered this technology to detect drug-protein interactions in mammalian cells. We believe that the combination of LeadCode and MAPPIT provides us with a powerful platform for studying drug mechanisms of action.
We have applied these technologies, either internally or in alliances, in various areas including antibacterial, antiviral, anticancer, osteoarthritis and immunology research. Currently, our internal applications of these technologies are primarily in our anticancer drug discovery efforts. In addition to the technologies described above, we also have internal expertise in target validation, test development, screening, biochemistry, medicinal chemistry, analytical chemistry and animal pharmacology. We believe that these capabilities, taken together, permit us to conduct innovative research in oncology drug discovery and will provide further drug discovery and development opportunities.
Our Licensing Activities
We continue to evaluate opportunities to acquire licenses on an ongoing basis to enhance our current product pipeline. We consider a variety of oncology compounds with different mechanisms of action as licensing candidates and thereby seek to balance our portfolio of products under development. In 2002, we successfully licensed satraplatin, our current Phase 3 product candidate. Over the past year we have evaluated a large number of oncology compounds representing numerous small and large molecules with a variety of proposed mechanisms of action. Our efforts to date have focused on evaluating compounds that have demonstrated anticancer activity in animal models or in clinical trials.
Strategic Alliances and License Agreements
Spectrum Pharmaceuticals
In September 2002, we entered into a license agreement with Spectrum Pharmaceuticals, Inc. (formerly known as Neotherapeutics, Inc.), under which we received an exclusive license, with the right to grant sublicenses, for the commercialization of satraplatin in the field of treating cancer in humans.
80
Table of Contents
This license includes a sublicense of all patent rights and other rights previously licensed to Spectrum Pharmaceuticals by Johnson Matthey plc, the company that invented satraplatin. In the event that the license agreement between Spectrum Pharmaceuticals and Johnson Matthey is terminated, Johnson Matthey has agreed with Spectrum Pharmaceuticals that our sublicense will be automatically assigned to Johnson Matthey, which will receive all rights and have all obligations currently held by Spectrum Pharmaceuticals. However, we have limited ability to enforce directly any Spectrum Pharmaceuticals rights or Johnson Matthey’s obligations in their agreement.
Under the agreement with Spectrum Pharmaceuticals, we made an upfront payment to Spectrum Pharmaceuticals of $2.0 million in 2002 and a further payment of $2.0 million in September 2003 upon the first dosing of the first patient in the first registrational clinical trial of satraplatin. Of the 2003 payment, $1.0 million was made in the form of an equity investment by us in Spectrum Pharmaceuticals common stock (representing approximately 2% of Spectrum Pharmaceuticals’ outstanding common stock as of December 31, 2003). In addition, we are obligated to make milestone payments to Spectrum Pharmaceuticals upon the occurrence of specific events in the development of satraplatin beginning with the acceptance by the FDA of an NDA for satraplatin. These milestone payments would total up to $18 million if all milestones were to be achieved. We also have established a joint development committee, comprised of three representatives from each company, responsible for the planning and oversight of the development of satraplatin. The chairperson of the committee, a representative of GPC Biotech, has a tie-breaking vote in the event the committee cannot reach a unanimous decision.
If we market and sell satraplatin ourselves, Spectrum Pharmaceuticals will receive royalties on our net sales of products, commencing on the date of first commercial sale and running until the expiration of the last-to-expire valid claim of a licensed patent or patent application. In the event we decide to market satraplatin without a partner in the United States, Spectrum Pharmaceuticals will also have the right to elect to co-promote the product with us on terms to be negotiated. Spectrum Pharmaceuticals is also entitled to receive a share of sublicense fees and royalties received by us if we enter into a marketing arrangement with another company. The term of the agreement ends with the expiration of our obligation to pay Spectrum Pharmaceuticals royalties on sales of products although some obligations, such as provisions relating to confidentiality and indemnification, survive termination. In addition, the agreement may be terminated earlier by either party, based upon material breach or the commencement of bankruptcy or insolvency proceedings involving the other, or by us upon six months’ notice to Spectrum Pharmaceuticals.
ALTANA Pharma
In November 2001, we entered into a collaboration and license agreement with ALTANA Pharma AG, a subsidiary of ALTANA AG, which is our most significant technology collaboration to date. Under this contract, we formed a strategic alliance with ALTANA Pharma in connection with the establishment by ALTANA Pharma of the ALTANA Research Institute, ALTANA Pharma’s U.S. research institute based in Waltham, Massachusetts. This alliance encompasses a technology transfer component as well as a research collaboration. Pursuant to the technology transfer component of the agreement, we agreed to transfer certain of our genomic and proteomic technologies to ALTANA Pharma for use by the ALTANA Research Institute. Under the terms of the agreement, we have granted ALTANA Pharma a non-exclusive license, with limited sublicense rights, to utilize these technologies and any improvements to these technologies in its research and development activities. Improvements made to our technologies by either party are owned by us.
The alliance also includes a research collaboration to identify and validate targets in various disease areas. Subject to the payment of exclusivity fees by ALTANA Pharma, we have granted ALTANA Pharma an exclusive license, with the right to grant sublicenses, to perform research on drug targets discovered in the collaboration, as selected by ALTANA Pharma, and to commercialize any products resulting from the use of these drug targets. We retain our intellectual property rights to any
81
Table of Contents
drug targets not selected by ALTANA Pharma. Under certain circumstances, rights to drug targets selected by ALTANA Pharma revert to us. The duration of the portion of the agreement pursuant to which we assist ALTANA Pharma with the establishment of ALTANA Research Institute runs through June 2007, while the term of the research collaboration is through April 2005.
Effective January 2003, we entered into another collaboration agreement with ALTANA Pharma pursuant to which we licensed LeadCode. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding over the term of the collaboration and license agreement and the Lead Code collaboration agreement. ALTANA Pharma is also obligated to make certain milestone payments totaling approximately $15 million (subject to reduction in certain circumstances) to us in connection with the development and commercialization of each product resulting from the research collaborations. For any of these products that receive regulatory approval and are marketed by ALTANA Pharma, we will also be entitled to royalties on net sales of such products. ALTANA’s obligation to pay royalties to us expires on a country-by-country basis on the later of the date of expiration of the last-to-expire patent having certain valid claims covering a particular product and the date that is ten years from the date of the first commercial sale of such product. These agreements terminate with the expiration of ALTANA Pharma’s obligation to pay us royalties. The agreement can be terminated earlier by either us or ALTANA Pharma in the case of material breach, by the other party, a change of control of GPC Biotech or the bankruptcy, insolvency, dissolution or winding up of either party. As of December 31, 2003, we had received€37.4 million under these two agreements, of which€27.4 million have been recognized as revenue. Of the€37.4 million received as of December 31, 2003,€35.7 million comprised payments under the collaboration and license agreement of November 2001 and€1.7 million comprised payments under the collaboration and license agreement of January 2003 related to LeadCode.
Under the collaboration and license agreement, ALTANA Pharma may negotiate with us for additional non-exclusive licenses to our technology from time to time. We granted a non-exclusive license to ALTANA Pharma with respect to our MaRX technology effective in June 2002. In addition, as of December 31, 2003, we have subleased to ALTANA Pharma 23,500 square feet of space in our facilities in Waltham, Massachusetts for the ALTANA Research Institute. The term of this sublease runs until June 30, 2007, with an option for ALTANA Pharma to extend the term for one year.
Prior to the current ALTANA Pharma alliance, we entered into two collaborations with ALTANA Pharma in the fields of oncology and infectious disease. These collaborations have been successfully concluded, and we are entitled to milestone payments and royalties on products that may be derived from these collaborations.
An affiliate of ALTANA AG, ALTANA Technology Projects GmbH, owned 1,690,000 of our ordinary shares, which are currently subject to lock-up, representing 7.88% of our outstanding shares at June 4, 2004.
MorphoSys
In April 1999, we entered into a collaboration and license agreement with MorphoSys AG, under which we collaborated with MorphoSys in the discovery and generation of monoclonal antibodies directed at specified targets. MorphoSys identified these antibodies using its proprietary HuCal® technology. MorphoSys has granted us exclusive rights, with the right to grant sublicenses, to develop and commercialize antibody product candidates resulting from the collaboration in a field that includes human therapeutic use. Our license agreement with MorphoSys includes a number of sublicenses related to enabling technologies. Since the initiation of the collaboration, MorphoSys has identified more than a dozen antibodies, from which we selected our product candidate known as 1D09C3.
82
Table of Contents
The research term of the collaboration expired in February 2001. However, there are milestone payments due to MorphoSys and the licensors of enabling technologies upon the occurrence of specified events in the development and commercialization of licensed products. If all milestones were achieved, these milestone payments would total approximately€10 million. MorphoSys and the licensors of the enabling technologies are also entitled to receive royalties on net sales of products by us. In addition, if we enter into an agreement with a partner for the commercialization of the product, instead of paying the milestones and royalties specified in the agreement, we would be obligated to pay MorphoSys various percentages of sublicense fees, milestone payments and royalties received from such partner. Unless sooner terminated, our obligation to pay MorphoSys and certain third party licensors royalties on sales of a product expires on a country-by-country basis on the later of the last-to-expire licensed patents having a valid claim covering a particular product and a date that is no later than twelve years from the date of the first commercial sale. Either party may terminate the agreement based on material breach or the commencement of bankruptcy or insolvency proceedings involving the other, and we may also terminate the agreement for any reason upon fifteen days notice to MorphoSys. The obligations under certain provisions of the agreement, including provisions relating to confidentiality and ownership of intellectual property, survive termination.
Bristol-Myers Squibb
Through our acquisition of Mitotix, Inc. in March 2000, we are party to a research, development and marketing agreement with Bristol-Myers Squibb, as successor to DuPont Pharmaceuticals, in the field of cyclin-dependent kinase inhibitors, of which RGB-286199 is one. Pursuant to an amendment to the agreement in April 2000, the research collaboration was terminated, although various provisions of the agreement including some sublicenses and milestone and royalty obligations remained in effect. We are entitled to milestone payments, totaling $16 million for the first product and totaling $8 million for any subsequent product if all milestones are achieved, and royalties on net sales for any products developed and commercialized by Bristol-Myers Squibb based upon lead compounds identified during the collaboration. Both we and Bristol-Myers Squibb also retain exclusive rights to develop and commercialize certain other selected compounds tested during the collaboration. Each party is obligated to make milestone and royalty payments to the other for any product incorporating any of these selected compounds. Aggregate milestones payable by Bristol-Myers Squibb for any product that incorporates one of these selected compounds are the same as discussed above. For the first product developed and commercialized by us that incorporates one of these selected compounds we would owe Bristol-Myers Squibb milestone payments totaling $19.5 million and totaling approximately $5 million for any subsequent product, in both cases if all milestones are achieved.
In addition, both we and Bristol-Myers Squibb have co-exclusive rights to utilize the intellectual property resulting from the collaboration for the identification, research, development and commercialization of new cyclin-dependent kinase inhibitors. We are each obligated to make royalty payments to the other for any product incorporating a compound independently identified during a specified time period through the use of this co-exclusive intellectual property. In addition, Bristol-Myers Squibb would owe us milestone payments as described above for any product incorporating a compound identified through the use of this intellectual property during the same specified time period. The royalty term expires on a country-by-country basis on the later of the date of expiration of the last-to-expire licensed patent having a valid claim covering a given product and the date that is ten years from the date of the first commercial sale. Either party may terminate the agreement based on material breach or the commencement of bankruptcy or insolvency proceedings involving the other, and we may also terminate the license to Bristol-Myers Squibb upon six months’ notice for failure to diligently pursue the discovery and development of products. The obligations under certain provisions of the agreement, including provisions relating to confidentiality and non-solicitation of employees, survive termination.
83
Table of Contents
Commercialization
We do not currently have any sales and marketing capabilities. In order to commercialize any products that are approved for commercial sale, we must either develop a sales and marketing infrastructure or collaborate with third parties that have sales and marketing experience. We believe that the medical oncology market is readily accessible by a limited sales and marketing presence due to the concentration of prescribing physicians.
We intend to seek strategic partners in major international markets, potentially including the United States and Europe, for the commercialization of satraplatin. We do not believe we need these partnerships to complete our application for marketing approval for satraplatin in the United States or in Europe. An appropriate strategic partner could, however, facilitate broader market awareness and enhance the overall market acceptance of satraplatin. Our current plan for our other product candidates is to develop these independently while pursuing the fastest path to marketing approval. We may seek a strategic partner to assist in demonstrating the breadth of application of these drugs in the treatment of a broad range of cancers. Additionally, we are likely to seek a strategic partner for marketing and sales of our other product candidates.
Sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors in the disease settings in which they are used. Significant uncertainty exists as to the reimbursement status of newly approved drugs. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of satraplatin. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Intellectual Property
We actively seek, when appropriate, protection for our products, technologies and proprietary information that is commercially important to the development of our business through filing for, prosecuting, maintaining or licensing United States, European and/or foreign patents, including German utility models (Gebrauchsmuster). In addition, we rely upon trade secrets and contractual arrangements to protect proprietary information that may be important to the development of our business.
We aim to file for, prosecute, maintain or license those patents and patent applications that we believe are relevant to the strategic needs of our business, and hence the exact number and scope of issued patents and/or pending patent applications to which we have rights at any given time may increase, decrease or otherwise change in the future from those shown here.
In particular, under our exclusive sub-license from Spectrum Pharmaceuticals, Inc., we have access to intellectual property owned by Johnson Matthey that is important to our ability to develop and commercialize our product candidate, satraplatin.
We have various exclusive and non-exclusive rights under certain patents, patent applications and technologies relevant to the discovery, production, or commercialization of 1D09C3 under our Collaboration and License Agreement with MorphoSys.
Under our Collaborative Research, Development and Marketing Agreement with Bristol-Myers Squibb, we have certain co-exclusive rights to intellectual property owned by Bristol-Myers Squibb relevant to our ability to develop and commercialize cell cycle inhibitor products, including RGB-286199.
84
Table of Contents
We also have certain exclusive or co-exclusive rights to other patents and patent applications relevant to:
| • | the technologies we use in our business; |
| • | other discovery programs; |
| • | certain genes, proteins and uses thereof in the area of cell-cycle regulation; |
| • | and other areas. |
These rights have been licensed or sub-licensed from various academic institutions and pharmaceutical and biopharmaceutical companies.
As of June 4, 2004, we owned or co-owned more than 75 patents and patent applications representing over twenty patent families, including:
| • | more than fifteen United States patents; |
| • | more than twenty United States patent applications; |
| • | two EPC patents or patents directly issued in an EU state; |
| • | more than ten EPC applications or applications filed directly in an EU state; |
| • | one other foreign patent; and |
| • | more than thirty other international patent applications. |
These patents and patent applications relate to our technologies and our drug discovery and development programs (both current and past). In particular, we own or co-own patent applications that are important to our ability to develop and commercialize our product candidates 1D09C3 and RGB-286199.
Of those families of patents and patent applications for which we have rights either as owner, co-owner, licensee or sublicensee, the table below summarizes those families with published patents or applications that we believe are most relevant to our product candidates and drug-protein interaction screening technologies.
Patent Families
Program | Subject matter | Earliest | Status | GPC Biotech rights | Earliest | |||||
Satraplatin | Platinum (IV) complexes and their uses as anti-tumor agents | February 2, 1988 | Issued in US(1), European Patent (EP), CA, JP and several other countries | Exclusive rights to patents owned by Johnson Matthey Inc. for the treatment of cancer in humans | US: late-2008(1) EP: early-2009 | |||||
Satraplatin | Improvements in platinum complexes(2) | April 25, 1994 | Issued in US, EP; pending in other countries | Exclusive rights to patents owned by Johnson Matthey Plc for the treatment of cancer in humans | US: mid-2015 EP: mid-2015 | |||||
1D09C03 | Human polypeptides causing or leading to the killing of cells including lymphoid tumor cells | May 12, 2000 | Pending worldwide | Co-owned with exclusive rights to Morphosys’ rights for therapeutic and prophylactic use | US: mid-2021 EP: mid-2020(3) | |||||
85
Table of Contents
Program | Subject matter | Earliest | Status | GPC Biotech rights | Earliest | |||||
1D09C03 | Immunomodulatory human MHC class II antigen-binding polypeptides | May 12, 2000 | Pending worldwide | Co-owned with exclusive rights to Morphosys’ rights for therapeutic and prophylactic use | US: mid-2021 EP: mid-2021 | |||||
RGB-286199 | Inhibitors of cyclin-dependent-kinases, compositions and uses related thereto | October 15, 2001 | Pending worldwide | Owned | US: late-2022(4)EP: late-2022(4) | |||||
RGB-286199 | Various patent families covering structures of small molecule compounds having CDK inhibitory activity, and uses thereof | Various, earliest being April 21, 1998 | Issued in US(5), pending worldwide | Co-exclusive (with BMS) for certain therapeutic purposes | US: mid-2019 EP: mid-2019 | |||||
LeadCode | Three hybrid testing system | March 2, 2001 | Pending worldwide | Owned | US: early-2022 EP: early-2022 | |||||
LeadCode | Yeast three-hybrid system | April 26, 1996 | Issued in US, EP & Germany(6), pending CA and JP | Non-exclusive rights to patents owned by Massachusetts Institute of Technology in US, co-exclusive elsewhere for internal use of technology and for the benefit of collaborators | US: mid-2017 EP: mid-2017 | |||||
LeadCode | Various patent families covering regulated transcription of targeted genes and other biological events | Various, earliest being February 12, 1993 | Issued in US, pending worldwide | Non-exclusive rights to patents owned by Stanford University and Harvard College(7) for research and development of therapeutic or prophylactic products | US: mid-2015 EP: early-2014 | |||||
MAPPIT | Receptor-based interaction trap | May 22, 2000 | Pending worldwide | Co-exclusive rights to patents owned by Vlaams Interuniversitar Instituut voor Biotechnologie to uses for discovering, identifying or analyzing protein-protein interactions and drug-protein interactions, non-exclusive for other uses | US: mid-2021 EP: mid-2021 | |||||
| (1) | Represented by two issued U.S. patents: a composition of matter patent (expires December 10, 2008) and a method of use patent (expires September 14, 2010). |
| (2) | Covers certain platinum complexes other than satraplatin, including certain metabolites of satraplatin. Recent decisions of U.S., English and German courts make it unlikely that such metabolite patents will provide us with material intellectual property protection, if at all, for satraplatin. |
| (3) | A second EP application is pending (expires May 14, 2021). |
| (4) | An unpublished Patent Cooperation Treaty application is pending (if nationalized would expire in US/EP mid 2024). |
86
Table of Contents
| (5) | Represented by four issued U.S. patents covering structures and therapeutic uses of various classes of cell cycle inhibitors. |
| (6) | German utility model. |
| (7) | Sublicensed to us by Ariad Pharmaceuticals Inc. |
Generally, patents expire 20 years after their earliest nonprovisional filing date. However, U.S. patents issuing from patent applications filed before June 8, 1995 will expire after the longer of 17 years after issuance or 20 years after the earliest nonprovisional filing date. U.S. patents may also be extended in limited circumstances by a period representing delay by the United States Patent and Trademark Office, or USPTO, and hence the exact expiry date of currently pending United States applications can not be predicted. The protection of certain patents that cover pharmaceutical products may be extended by up to five years under provisions of the Hatch-Waxman Act in the United States and analogous provisions in other countries See “—Government Regulation—Regulation in the United States—Drug Price Competition and Patent Term Restoration Act of 1984”. Any patent or patent application will expire earlier than the terms described above, if appropriate renewal or annuity fees are not paid timely to the appropriate patent office or if prosecution actions or responses are not filed with the relevant patent office according to applicable regulations.
Satraplatin. Our lead product candidate satraplatin is protected by two issued U.S. patents, a patent issued under the European Patent Convention, or EPC, and a number of patents issued in other national jurisdictions including other EU countries, Japan, Canada and Australia. These patents are owned by Johnson Matthey, and we hold an exclusive sublicense, with the right to sublicense, under these patents through our Co-development and License Agreement with Spectrum Pharmaceuticals, Inc. in the field of treating cancer in humans. These patents cover the composition of matter and anticancer uses of various platinum-based compounds, including satraplatin. The two U.S. patents expire in 2008 and 2010, respectively, and in 2009 in most other countries. Under provisions such as the Hatch-Waxman Act in the United States and analogous regulations in Europe, there are opportunities to extend the patent life of a patent to cover any approved drug product in a given jurisdiction. These regulations provide for the possibility of a patent term extension of up to five years to only one of the U.S. patents related to satraplatin and a Supplementary Protection Certificate in European countries to extend the patent term of the European patent, and any appropriate national equivalents in the European Union, by up to five years. We plan on applying for such extended protection, if applicable, for the fullest term and extent appropriate. While we believe that satraplatin will meet the Hatch-Waxman criteria for patent extension, delays in the completion of our Phase 3 registrational trial or in obtaining regulatory approval may jeopardize our ability to obtain a timely patent extension; in addition there can be no assurance that we will ultimately be granted this type of extension or an analogous extension for satraplatin in any specific country, or at all.
1D09C3—Monoclonal Antibody. Our product candidate 1D09C3 is covered by a number of patent applications in major market countries, including the United States, Japan, Canada, Australia and those states contracted to the European Patent Convention at the date of filing, or EPC States. These published patent applications are co-owned by us and MorphoSys and are governed by the terms of our Collaboration and License Agreement with MorphoSys. Under this agreement, we have exclusive rights to develop and commercialize antibody products covered by such patent applications, including 1D09C3, including in the field of human therapeutic use. These patent applications and resulting patents, if issued and maintained, would expire in 2021 in most major markets. Through our collaboration with MorphoSys, we have secured certain rights to a number of enabling patents, patent applications and technologies relevant to the discovery, production, and commercialization of 1D09C3.
RGB-286199—Cell Cycle Inhibitor. Several patent applications cover our cell cycle inhibitor, RGB-286199, in major market countries including the United States, EPC States, Japan, Canada and Australia. These published patent applications are owned by GPC Biotech or by Bristol-Myers Squibb
87
Table of Contents
and result from research conducted by us or Bristol-Myers Squibb as part of and subsequent to the parties’ Collaborative Research, Development and Marketing Agreement. We have exclusive rights to the patent applications owned by us, and under the terms of our agreement with Bristol-Myers Squibb, co-exclusive rights to inventions made or conceived by employees of Bristol-Myers Squibb during and as a result of the collaboration between us and Bristol-Myers Squibb.
We intend to continue using our scientific expertise to pursue and seek patent protection for new developments with respect to compositions, methods of use and other aspects of such developments that may enhance our position in the field of anticancer drugs. In particular, we have rights to certain unpublished patent applications related to our product candidates. Patents, if issued, may be challenged, invalidated or circumvented. Thus, any patent that we own, co-own or license from third parties may not provide adequate protection against competitors. Our pending patent applications, those we may file in the future, or those we may license from third parties may not result in issued patents. Also, patents may not provide us with adequate proprietary protection or advantages against competitors with similar or competing technologies. In particular, we are aware that a third party has an issued patent relating to treating plasmacytoma/multiple myeloma, Hodgkin’s lymphoma (also known as Hodgkin’s disease), non-Hodgkin’s lymphoma and B cell leukemias by using monoclonal antibodies which specifically react with certain MHC class II molecules. We are aware that numerous patent applications owned by third parties have been filed and are currently pending and expect that others may be filed in the future in fields similar to our technologies or product candidates. For example, a large number of patents have been issued with respect to methods of discovering, producing, and other aspects of therapeutic antibodies. We are aware of issued patents held by third parties that relate to the production of recombinant antibodies, such as patents covering production in single cells by the independent expression of the two protein chains that make up the antibody, or more generally to the production of recombinant proteins. There are also numerous patent filings claiming various genetic sequences, such as protein coding sequences and regulatory sequences that may be useful if used in our research programs and technologies, including our LeadCode technology. We are also aware of an issued patent covering a drug-protein interaction technology related to our LeadCode technology that describes a “biotin” based drug-fusion molecule. Biotin is a naturally occurring molecule that binds to certain proteins enabling them to perform their function. We have not attempted to obtain licenses to any of these patents. Due to these factors and the inherent uncertainty in conducting patent searches, we may violate third-party patent rights that we have not yet identified, or fully or correctly investigated. As a result of potential conflicts with the proprietary rights of others, we or our collaborators or licensees may in the future have to prove that we or our collaborators or licensees are not infringing the patent rights of others or be required to obtain one or more licenses to third party patents. We do not know whether such a license would be available on commercially reasonable terms, or at all. If we or our collaborators or licensees are unable to secure such licenses, we or our collaborators or licensees may be required by a court to cease further development, use or commercialization of one or more of our technologies or product candidates.
The technologies we use in our business comprise a complicated set of individual components or techniques, some of which are protected by intellectual property owned by third parties. We have entered into certain licenses to intellectual property covering particular enabling components or techniques that comprise parts of our technologies. Under the terms of certain of these licenses we will owe milestone and royalty payments to the licensors for the commercialization of the licensed intellectual property or products derived therefrom. Under such licenses, we are also obliged to grant back rights to improvements and certain other intellectual property to the licensor. In the future, we may be required to take additional licenses to other intellectual property that is necessary to some of our technologies or products at additional cost to us.
Much of our technology and many of our processes depend upon the knowledge, experience and skills of our scientific and technical personnel. To protect rights to our trade secrets, proprietary know-how and technology, we require all employees, contractors, consultants, advisors and
88
Table of Contents
collaborators, as well as potential collaborators, to enter into confidentiality agreements or other obligations of confidentiality that prohibit the disclosure of confidential information and require disclosure of all ideas, developments, discoveries and inventions related to our proprietary know-how, trade secrets or technology. Under these agreements, our employees and most of our consultants are generally obligated to assign to us all rights to such ideas, developments, discoveries and inventions, and to reasonably assist us in any further prosecution of patent applications filed to cover such inventions. Those ideas, developments, discoveries and inventions made by employees and consultants working under German employment law are subject to the provisions of the German Act on Employees’ Inventions (Gesetz über Arbeitnehmererfindungen—ArbNErfG), which regulates the ownership of, and compensation for, inventions made by employees. It is possible, however, that these parties may breach those agreements, and we may not have adequate remedies for any breach. It is also possible that our trade secrets or unpatentable know-how will otherwise become known or be independently developed by competitors, for which there most likely will be no remedy available to us. Additionally, we or our co-owners or licensors may not have complied, or may not in the future comply, with the provisions of theArbNErfG, and may become subject to disputes relating to alleged non-adherence to the provisions of theArbNErfG.
Some of our advisors and consultants are currently employed by universities or other commercial entities. Most of these individuals are parties to agreements pursuant to which some of the work product created by these individuals belongs to their respective employers. While we and these individuals try to maintain records that make it clear that the work these individuals do for us is not subject to their agreements with their employer, it is always possible that an employer such as a university will assert an ownership claim to the work of one or more of these individuals. Our advisors and consultants employed under German employment law will be subject to the provisions of theArbNErfG under which their employer, rather than we, has first rights to inventions made by them.
Competition
We operate in the highly competitive segment of the pharmaceutical market composed of pharmaceutical and biotechnology companies that research, develop and commercialize products designed to treat cancer. Many of our competitors have significantly greater financial, manufacturing, marketing and product development resources than we do. Large pharmaceutical companies in particular have extensive experience in clinical testing and in obtaining regulatory approval for drugs. These companies also have significantly greater research capabilities than we do. In addition, many universities and private and public research institutes are active in cancer research, some in direct competition with us. We also must compete with these organizations to recruit scientists and clinical development personnel.
Satraplatin. If satraplatin is approved and commercialized, it will face significant competition. Competition for satraplatin may include other drugs either marketed or being developed for prostate cancer, as well as other platinum-based compounds and other chemotherapy drugs for other cancers.
In the prostate cancer market, currently approved drugs include EMCYT (Pfizer, Inc.), NOVANTRONE (OSI Pharmaceuticals, Inc./Serono S.A.), QUADRAMET (Schering AG/CYTOGEN Corporation), METASTRON (Amersham Health/Medi-Physics, Inc.) and TAXOTERE (Aventis S.A.). Two of these drugs (NOVANTRONE and QUADRAMET) are injectable pharmaceuticals approved for use in treating bone pain in cancer patients, and EMCYT is an oral drug used to relieve symptoms of advanced prostate cancer. Although these drugs are effective in relieving the pain of patients with advanced prostate cancer, with the exception of TAXOTERE, they have not been shown to delay the progression of the cancer. We do not believe drugs that only provide relief of symptoms will be significant competitors to satraplatin. The most recently approved of these prostate cancer drugs, TAXOTERE, is approved in the United States, in combination with prednisone, for the treatment of
89
Table of Contents
patients with advanced prostate cancer. TAXOTERE has been shown to prolong survival of patients with HRPC. TAXOTERE has also been used in combination with EMCYT for patients with advanced prostate cancer. Since the initial indication for satraplatin will be second-line treatment of HRPC, and therefore, satraplatin will be used in patients who have already failed TAXOTERE or one of the other cytotoxic drugs, we do not believe that TAXOTERE will be a significant competitor. Also, we believe that the use of TAXOTERE will increase the need for second-line treatment of HRPC, because Phase 2 studies for TAXOTERE have shown that HRPC patients ultimately became refractory to treatment with TAXOTERE. If we subsequently develop satraplatin for the first-line treatment of HRPC, we believe it will most likely be developed in a combination regimen with TAXOTERE, since the two drugs may have complementary mechanisms of action.
In addition to the drugs already mentioned, there are other agents in development for both advanced HRPC and earlier stages of prostate cancer, which may compete with satraplatin. These other agents include the drugs which are listed in the following table.
Drug | Company | Stage of development for | ||
| Atrasentan | Abbott | · Not a marketed drug · Phase 3 trials have been completed for various stages of prostate cancer | ||
| Calcitriol | Novacea | · Not a marketed drug · Phase 2/3 trial in combination with TAXOTERE for first line HRPC is on-going | ||
| Provenge | Dendreon | · Not a marketed drug · Phase 3 trials for first line HRPC on-going | ||
| Ixabepilone | Bristol-Myers Squibb | · Not a marketed drug · Phase 2 data have been reported for first line HRPC | ||
We believe that satraplatin is the only product candidate currently in a Phase 3 registrational trial as a second-line chemotherapy treatment of HRPC. The drugs listed in the above table are being developed for first line treatment of HRPC and so will not compete directly with satraplatin in the second line setting. All of these drugs also represent complementary mechanisms of action compared to satraplatin. It is common practice for oncologists to treat patients with combination chemotherapy regimens, combining drugs with complementary mechanisms of action. It is therefore expected that satraplatin in the long run will be developed and used in combination with one or more of these drugs.
There are currently three marketed platinum-based drugs in the United States and in Europe. These are cisplatin, carboplatin and oxaliplatin. All three agents are administered intravenously and are not indicated for the treatment of prostate cancer. In addition to these, there are other platinum-based compounds approved and/or marketed in the Asian markets such as lobaplatin (China), nedaplatin (Japan) and eptaplatin (South Korea). These drugs are not approved, however, for the treatment of prostate cancer. All three of these are also administered intravenously. Another platinum-based drug, which is not currently on the market, is AMD-473. AMD-473, administered intravenously, has shown activity for HRPC in a Phase 2 clinical trial. There are no reported clinical trials for an oral formulation of AMD-473. We are aware that other companies may be developing orally bioavailable platinum-based compounds. We are not aware, however, of any other orally bioavailable platinum-based
90
Table of Contents
compounds that are approved or are in Phase 3 clinical trials. We anticipate that satraplatin will be able to compete successfully with other platinum-based drugs because:
| • | Satraplatin is the only platinum-based drug to have demonstrated efficacy in a randomized trial for the treatment of hormone refractory prostate cancer. |
| • | Satraplatin is the only agent currently in Phase 3 development for second line treatment of HRPC. |
| • | Satraplatin is the only agent that has undergone clinical development with an orally bioavailable formulation. |
| • | Satraplatin is relatively well tolerated; in clinical trials to date, significant nephro-, neuro- and ototoxicities have not been observed with satraplatin. |
Satraplatin could also be developed for the treatment of other cancers, either as a single agent or in combination with radiation therapy or other drugs. In these other clinical settings, it will also face competition from a variety of other anticancer drugs.
1D09C3—Monoclonal Antibody. If 1D09C3 is approved and commercialized, it will face significant competition. Currently marketed antibodies for the treatment of non-Hodgkin’s lymphoma are RITUXAN (Biogen Idec, Inc./Genentech, Inc./Roche Holdings AG), ZEVALIN (Biogen Idec, Inc/Schering AG) and BEXXAR (Corixa/GlaxoSmithKline). CAMPATH (Berlex Laboratories) is approved for chronic lymphatic leukemia. In addition, there are a number of other antibodies and other drugs in development for the treatment of lymphoma and leukemia.
1D09C3 could also be developed for the treatment of leukemias and melanoma. There is, and will continue to be, significant competition in these markets from both large molecule drugs (antibodies) and small molecule drugs.
RGB-286199—Cell Cycle Inhibitor. If RGB-286199 is approved and commercialized, it will face significant competition. There are no currently marketed CDK inhibitors. Competitors with development programs for CDK inhibitors include Aventis S.A. (flavopiridol), Cyclacel Limited (CYC 202) and Bristol-Myers Squibb (BMS-387032). Aventis and Cyclacel have advanced their CDK inhibitors to Phase 2 clinical trials, however, Aventis announced in Februrary 2004 that development of flavopiridol had been terminated. Bristol-Myers Squibb has advanced its CDK inhibitor to Phase 1 clinical trials.
Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the development, manufacture and marketing of pharmaceutical products and in ongoing research and development activities. All of our products will require regulatory approval by governmental agencies prior to commercialization. In particular, pharmaceuticals are subject to rigorous preclinical testing and clinical trials and other pre-marketing approval requirements by the FDA and regulatory authorities in other countries.
Regulation in the United States
In the United States, drugs are subject to rigorous regulation by the FDA. The Federal Food, Drug, and Cosmetic Act and other federal and state statutes and regulations govern, among other things, the research, development, testing, safety, effectiveness, manufacture, quality control, storage, record keeping, labeling, promotion, marketing and distribution of pharmaceutical products. The failure to comply with the applicable regulatory requirements may subject a company to a variety of
91
Table of Contents
administrative or judicially imposed sanctions and/or the inability to obtain or maintain required approvals or to market approved drug products.
In addition, the lengthy process of seeking required approvals and the continuing need for compliance with applicable statutes and regulations require the expenditure of substantial resources. Regulatory approval, when and if obtained for any of our products, may be limited in scope, which may significantly limit the indicated uses for which our products may be marketed. Furthermore, approved drugs and manufacturers are subject to ongoing review and discovery of previously unknown problems that may result in restrictions on their manufacture, sale or use or in their withdrawal from the market.
The steps ordinarily required before a new drug product may be marketed in the United States include preclinical laboratory tests, animal tests and formulation studies, the submission to the FDA of a notice of claimed exemption for an investigational new drug, or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. The following paragraphs provide a general overview of the approval process for a new drug.
Preclinical Testing. Preclinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of preclinical tests, together with manufacturing information and analytical data, are submitted as part of an IND application to the FDA.
Investigational new drug application. If a company wants to test a new drug in human patients, an IND must be prepared and filed with the FDA to request FDA authorization to begin human testing of the drug. The IND application automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about adequacy of the preclinical studies, the preclinical product characterization and/or the proposed conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The submission of an IND may not result in FDA authorization to commence a clinical trial. A separate supplemental submission to an existing IND must also be made for each successive clinical trial conducted during product development, and the FDA must review each supplemental IND before each clinical trial can begin. Furthermore, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center, and the IRB must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive Good Clinical Practice, or GCP, regulations and regulations for obtaining informed consent from the study subjects.
Clinical Trials. Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, Phases 1, 2 and 3, with Phase 4 studies conducted after marketing approval. Phase 4 trials are generally required for products that receive accelerated approval. These phases may be compressed, may overlap or may be omitted in some circumstances.
| • | Phase 1 Clinical Trials: After an IND becomes effective, Phase 1 human clinical trials can begin. These studies are initially conducted in a limited population to evaluate a drug candidate’s safety profile, and the range of safe dosages that can be administered to the patient, including the maximum tolerated dose that can be given to a patient with the target disease. Phase 1 studies also determine how a drug candidate is absorbed, distributed, metabolized and excreted by the body, and its duration of action. In some cases, particularly in cancer trials, a sponsor may decide to run what is referred to as a “Phase 1b” evaluation, |
92
Table of Contents
which is a second safety-focused Phase 1 clinical trial typically designed to evaluate the impact of the drug candidate in combination with currently approved drugs. |
| • | Phase 2 Clinical Trials: Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, to determine the efficacy of the drug candidate for specific targeted indications and to determine dose tolerance and optimal dosage. Phase 2 clinical trials of cancer drugs typically are designed to evaluate the potential effectiveness of the drug on patients with specific types and stages of cancer and to further ascertain the safety of the drug at the dosage given in a larger patient population. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. In some cases, a sponsor may decide to run what is referred to as a “Phase 2b” evaluation, which is a second, confirmatory Phase 2 clinical trial that could, if positive and accepted by the FDA, serve as a registrational clinical trial in the approval of a drug candidate. |
| • | Phase 3 ClinicalTrials: These are commonly referred to as registrational (or pivotal) studies, and are undertaken when Phase 2 clinical trials suggest that a dose range of the drug candidate is effective and has an acceptable safety profile. In Phase 3 clinical trials, the drug is usually tested in a controlled randomized trial comparing the investigational new drug to an approved form of therapy in an expanded and well defined patient population and at multiple clinical sites. The goal of these studies is to obtain definitive statistical evidence of safety and efficacy of the investigational new drug regimen as compared to an approved standard treatment in defined patient populations with a given disease and stage of illness. |
In the case of products for life-threatening diseases such as cancer, the initial human testing is often conducted in patients with the target disease rather than in healthy volunteers. These studies may provide initial evidence of efficacy traditionally obtained in Phase 2 clinical trials, and so these trials are frequently referred to as Phase 1/2 trials.
In addition, a company may hold an “End-of-Phase 2 Meeting” with the FDA to assess the safety of the drug regimen to be tested in the Phase 3 clinical trial, in part to evaluate the Phase 3 plan, and identifying any additional information that will be needed to support a new drug application. If the Phase 3 clinical trials had been the subject of discussion at an “End-of-Phase 2 Meeting”, the company is eligible for a Special Protocol Assessment, or SPA by the FDA, a process by which the FDA must evaluate within 45 days protocols and issues relating to the protocols to assess whether they are adequate to meet scientific and regulatory requirements identified by the sponsor. The satraplatin Phase 3 registrational trial protocol has now successfully completed both the “End-of-Phase 2 Meeting” and this SPA process.
Success in early stage clinical trials does not necessarily assure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be subject to alternative interpretations that could delay, limit or even prevent regulatory approval.
New Drug Application. After successful completion of the required clinical testing of a drug candidate an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the United States. The NDA must include the results of extensive clinical and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial. Under Federal law, the submission of NDAs are additionally subject to substantial application user fees, currently exceeding $500,000, and the manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees, currently exceeding $30,000 per product and $200,000 per establishment. These fees are typically increased annually.
93
Table of Contents
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the agency’s threshold determination that the NDA is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the Prescription Drug User Fee Act, or PDUFA, the FDA has agreed to specific performance goals in the review of NDAs. Most such applications for non-priority drug products are reviewed within ten months. The review process is often significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee.
If FDA evaluations of the NDA and the manufacturing facilities are favorable, the FDA may issue an approval letter, or, in some cases, an approvable letter followed by an approval letter. An approvable letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require post-approval testing and surveillance to monitor the drug’s safety or efficacy and may impose other conditions, including labeling restrictions which can materially impact the potential market and profitability of the drug. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA may refuse to approve the NDA or issue a not approvable letter. A “not approvable” letter outlines the deficiencies in the submission and often requires additional testing or information in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. With limited exceptions, FDA may withhold approval of an NDA regardless of prior advice it may have provided or commitments it may have made to the sponsor.
Fast Track Designation. FDA’s fast track program is intended to facilitate the development and to expedite the review of drugs that are intended for the treatment of a serious or life-threatening condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug candidate may request the FDA to designate the drug candidate for a specific indication as a fast track drug concurrent with or after the filing of the IND for the drug candidate. The FDA must determine whether the drug candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
If fast track designation is obtained, the FDA may initiate review of sections of an NDA before the application is complete. This rolling review is available if the applicant provides and the FDA approves a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the time period specified in the PDUFA, which governs the time period goals the FDA has committed to reviewing an application, does not begin until the complete application is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
94
Table of Contents
In some cases, a fast track designated drug candidate may also qualify for one or more of the following programs:
| • | Priority Review. Under FDA policies, a drug candidate is eligible for priority review, or review within a six-month time frame from the time a complete NDA is accepted for filing, if the drug candidate provides a significant improvement compared to marketed drugs in the treatment, diagnosis or prevention of a disease. A fast track designated drug candidate would ordinarily meet the FDA’s criteria for priority review. We cannot guarantee any of our drug candidates will receive a priority review designation, or if a priority designation is received, that review or approval will be faster than conventional FDA procedures, or that the FDA will ultimately grant drug approval. |
| • | Accelerated Approval. Under the FDA’s accelerated approval regulations, the FDA is authorized to approve drug candidates that have been studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit to patients over existing treatments based upon either a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than patient survival. In clinical trials, surrogate endpoints are alternative measurements of the symptoms of a disease or condition that are substituted for measurements of observable clinical symptoms. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to validate the surrogate endpoint or confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or to validate a surrogate endpoint or confirm a clinical benefit during post-marketing studies, will allow the FDA to withdraw the drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA. |
Although we have obtained a fast track designation from the FDA for our development of satraplatin to treat HRPC patients who have failed prior treatment with chemotherapy, we cannot guarantee a faster development process, review process or approval compared to conventional FDA procedures. We have elected to seek approval under the accelerated approval process for satraplatin. Under the terms of the Special Protocol Assessment, the primary endpoint of the Phase 3 registrational trial for accelerated approval by the FDA will be the time to disease progression. Our strategy and timing for seeking regulatory approval of this drug may change depending on the results of our studies.
Orphan Drug Designation. Orphan drug designation is designed to encourage manufacturers to develop drugs intended for a rare disease or condition. A rare disease or condition is statutorily defined as one affecting less than 200,000 individuals in the United States, or one that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available the drug for the disease or condition will be recovered from sales of the drug in the United States. Orphan drug designation qualifies a company for tax credits and marketing exclusivity for seven years following the date of the drug’s marketing approval if granted by the FDA.An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product.A drug becomes an “orphan” when it receives orphan designation from the Office of Orphan Products Development, or OOPD, at the FDA based on acceptable confidential requests made under the regulatory provisions. The drug must then go through the new drug approval process like any other drug. Orphan drug designations are decided solely by the OOPD staff, but the Office occasionally will request opinions from the Center for Drug Evaluation and Research, especially when dealing with issues such as the appropriateness of the requested indication or the scientific rationale described by the sponsor.
A sponsor may request orphan drug designation of a previously unapproved drug, or of a new orphan indication for an already marketed drug. In addition, a sponsor of a drug that is otherwise the
95
Table of Contents
same drug as an already approved orphan drug may seek and obtain orphan drug designation for the subsequent drug for the same rare disease or condition if it can present a plausible hypothesis that its drug may be clinically superior to the first drug. More than one sponsor may receive orphan drug designation for the same drug for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
The period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for which the drug has been designated. The FDA could approve a second application for the same drug for a different use or a second application for a clinically superior version of the drug for the same use. The FDA cannot, however, approve the same drug made by another manufacturer for the same indication during the marketing exclusivity period unless it has the consent of the sponsor or the sponsor is unable to provide sufficient quantities.
We have applied for orphan drug designation for satraplatin and have recently responded to an initial letter from the FDA with additional information that we believe the FDA reviewers require to determine whether the characteristics of satraplatin limit its use to HRPC patients such that an orphan drug designation is appropriate. There can be no assurances that such a designation will be made, and if we receive such designation, we have additionally undertaken to petition the FDA to petition for cancellation of our orphan drug status if we apply for and receive approval to market satraplatin for earlier stage prostate cancer.
Drug Price Competition and Patent Term Restoration Act of 1984
The Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act, established a regulatory framework designed to balance the incentives for innovative drug research with the opportunities for market entry of generic manufacturers. In order to achieve this balance, the Hatch-Waxman Act provided for the restoration of patent terms based on the regulatory review period of a drug product, and data exclusivity periods following the FDA approval of an NDA, while allowing for the submission of simplified drug applications by generic manufacturers. The simplified drug applications created by the Hatch-Waxman Act include two kinds of applications: an abbreviated new drug application, or ANDA, which can rely on FDA’s previous finding of safety and effectiveness for the referenced innovator drug product, and a new drug application for which the sponsor must submit full reports of clinical studies, some of which the sponsor does not own or have a legal right of reference (also known as a Section 505(b)(2) application after its authorizing statutory provision). The patent and exclusivity status of the innovator drug product has implications for the review and approval of both ANDAs and Section 505(b)(2) applications.
A key element of the Hatch-Waxman Act is the extension of the life of a patent to compensate the innovator drug company for marketing time lost while developing the product and awaiting regulatory approval. The Act added Section 156 to the Patent Act permitting patent term extensions for patents on products (or processes for making or using the same) including, but not limited to, drug products used to treat humans. The Hatch-Waxman Act allows only partial recovery of the patent term lost to regulatory approval requirements. In addition, the statute imposes caps on term extension. The term of the patent eligible for extension equals one half of the IND testing phase and the full NDA review phase of testing required under the Federal Food, Drug, and Cosmetic Act. The IND testing phase is measured as the time between the effective date of an IND and the date the FDA receives the NDA; the NDA review phase is the time between the FDA receives the NDA and approval of the NDA. However, any testing conducted prior to patent issuance is not considered for patent extension. The maximum total patent term remaining after term extension is capped at fourteen years. Similarly, absolute caps limit the duration of term extension to five years. Furthermore, a patent is only eligible for one term extension. This patent term extension is only available for the first commercial marketing of a
96
Table of Contents
given active ingredient. In addition, the product must have been subject to regulatory review before its commercial marketing or use, and the resulting permission for commercial marketing or use must be the first granted. As a practical consequence generally, only one patent may be extended per approved product. Also, the original patent must still be in force when the application for term extension is filed, and the application must be filed by the patent owner of record or its agent. The application for patent term extension is subject to approval by the USPTO. The FDA, however, determines the length of the product’s regulatory review period at the request of the USPTO. In some instances, the term of the patent for which a patent term extension is being requested may expire before such an extension is granted.
The Hatch-Waxman Act also provides for data exclusivity for the data demonstrating safety and efficacy of a drug product as submitted in an NDA: five-year new chemical entity, or NCE, exclusivity and three-year new clinical study exclusivity. Five-year NCE exclusivity is granted to those drugs for which the active ingredient is an active moiety (i.e., the molecule or ion responsible for physiological or pharmacological action, excluding appended portions that would cause the drug to be an ester, salt, or other noncovalent derivative of the molecule) not previously approved by the FDA. Five-year NCE exclusivity prohibits the FDA from accepting an ANDA or Section 505(b)(2) application for a drug product containing the same active moiety for a five-year period beginning from the date of approval of the NDA. The only exception to this prohibition on the FDA’s acceptance of an ANDA or Section 505(b)(2) application is if a generic competitor challenges patents listed in the Orange Book for the drug product at the end of four years. The five-year exclusivity provision, however, does not prohibit the FDA from accepting another full NDA, for example from a competitor, if the sponsor of the second application has done all the work itself. The FDA can accept the second application, review it, and approve it; NCE exclusivity only prohibits the agency from accepting a 505(b)(2) application or an ANDA.
The Hatch-Waxman Act requires an applicant for an ANDA to submit a certification for each patent listed in the Orange Book. This certification requirement also extends to Section 505(b)(2) applications. One of four certifications must be made: 1) that the drug has not been patented; 2) that the patent has already expired; 3) the date on which the patent will expire, and that the generic drug will not go on the market until that date passes; and 4) that the patent is not infringed or is invalid. Those certifications are now referred to as paragraph I, II, III, or IV certifications. Whereas the first three certifications are relatively straightforward, the paragraph IV certification presents added requirements.
When an ANDA contains a paragraph IV certification, the applicant is required to notify the innovator company that it has filed the ANDA with the FDA, and describe the reasons it believes the patent will not be infringed, is invalid, or is unenforceable. The only exception to this rule is if a company is not seeking approval for one of the drug’s uses. In that case, an applicant may submit a “Section 8” statement that the company is not seeking approval for a particular use. Once the innovator company receives notice that a generic application has been filed and its patent is being challenged, the innovator drug company has 45 days in which to file a lawsuit claiming patent infringement based on the generic drug company’s assertion about the characteristics of its proposed product. The filing of a lawsuit as a result of the paragraph IV notice has a substantial effect on the time of approval of the ANDA or 505(b)(2) application. If a lawsuit is brought by the innovator drug company, the FDA’s final approval is stayed for 30 months. If the patent court determines that the patent would be infringed by the product proposed in the ANDA or 505(b)(2) application, the FDA will not approve the application until the patent expires. If the court decides that the patent will not be infringed, or is invalid, the FDA may approve the generic application when that decision occurs. The FDA may approve the application at the thirty-month date, even if the litigation is ongoing. If litigation is pending and the agency approves an ANDA at the end of the 30-month period, most generic drug companies seem unwilling to risk liability for damages by bringing a generic drug product onto the market before the patent litigation is resolved. A generic applicant who is the first to challenge a listed patent using a paragraph IV
97
Table of Contents
certification is granted a 180-day exclusivity period with respect to other generic applicants. This exclusivity period provides generic applicants with an incentive by which to challenge listed patents for the innovator drug product.
Under the provisions of the recently enacted Medicare Prescription Drug Improvement and Modernization Act of 2003, the patent owner and the NDA holder have the opportunity to trigger only a single 30-month stay per abbreviated new drug application or Section 505(b)(2) application.
Three-year clinical study exclusivity is granted for certain changes in a drug product, for which the NDA or supplement contains reports of new clinical studies in humans conducted by the sponsor that are essential to approval. This exclusivity covers only the change in the product supported by the new clinical studies. If there are other indications not covered by any patent or exclusivity, and available for competition, generic drugs can be approved for those indications. A grant of three years of exclusivity to a drug product means FDA cannot approve a section 505(b)(2) application or an ANDA for the same product for three years. Unlike the five-year exclusivity, the agency can accept an application and review it during this time period. Like NCE exclusivity, this exclusivity will not bar approval of a full NDA where the applicant has done the work to support the same change for the drug product. Exclusivities are published in the Orange Book.
Post-Marketing Studies. As a condition of NDA approval, the FDA may require post-marketing “Phase 4” clinical trials to confirm that the drug is safe and effective for its intended uses. Where drugs are approved under accelerated approval regulations or the FDA otherwise requests, additional studies will likely be required to document a clinical benefit and monitor the long-term effects of the therapy. We expect that for any product for which a single pivotal clinical trial is authorized for approval, we will be required to conduct extended Phase 4 clinical trials to monitor the long-term effects of the therapy.
Other Regulatory Requirements. Any products we manufacture or distribute under FDA approvals are subject to pervasive and continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the products. Drug manufacturers and their subcontractors are required to register with the FDA and, where appropriate, state agencies, and are subject to periodic unannounced inspections by the FDA and state agencies for cGMPs, which impose procedural and documentation requirements upon us and any third-party manufacturers we utilize. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug.
FDA Regulation of Post-Approval Marketing and Promotion. The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label uses.
98
Table of Contents
From time to time, including presently, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be.
Regulation in the European Union
Clinical trials in Europe proceed in much the same manner as they do in the United States, requiring Phase 1, 2, and 3 trials, as well as Phase 4 trials that take place after market approval in order to gather data on an approved product.
Pursuant to the recent Clinical Trials Directive, a new system is being implemented for the approval of clinical trials in the EU. The Clinical Trials Directive must be enacted through national legislation of the Member States of the EU by the beginning of May 2004, and these rules amend or replace existing national procedures. Similar to the IND system in the United States, under this new system, approval must be obtained from the national regulatory agency of an EU Member State in which the study is planned to be conducted. For this clinical trial application, an Investigational Medicinal Product Dossier must be submitted. While the Clinical Trial Directive permits a maximum review period of 60 days, this period is shorter in some Member States. In Germany for example, for most drugs, the national agency will probably have 30 days to raise questions about the application. Approval is deemed to be given if notice of objection is not given within the relevant time limit, but for certain products, including biotechnology compounds such as monoclonal antibodies written approval may be required under the national rules, and for other types of products, such as gene and cell therapy, written approval is always needed. In addition, approval must also be obtained from the responsible ethics committees (equivalent to the IRB in the United States).
Drug approval in the Member States of the European Union generally proceeds under one of two approval procedures: a centralized approval procedure and a decentralized procedure, also known as the Mutual Recognition Procedure. The centralized approval procedure is mandatory for biotechnology products and is becoming mandatory for certain other high-technology products, such as new cancer drugs. The London-based European Agency for the Evaluation of Medicinal Products, or EMEA, and the European Commission in Brussels govern the centralized drug registration and approval process. Under this centralized procedure, an approval of a new drug application by the European Commission allows a company to market its drug product in all Member States of the European Union, without having to obtain separate approvals from each Member State. However, marketing remains subject to national pricing and reimbursement rules that often delay marketing and can sometimes effectively prevent it.
In contrast, a company may pursue a decentralized procedure to obtain mutual recognition of a new drug by the Member States. Under the decentralized procedure, an applicant can go directly to a national marketing authority to obtain permission to market its product in the Member State and then seek to have other Member States accept the marketing approval of the first Member State. National pricing and reimbursement rules will also apply to companies following the decentralized procedure, and therefore marketing delays may occur.
Under the centralized approval procedure, the EMEA’s Committee for Medicinal Products for Human Use, composed of experts nominated by each Member State’s national marketing authority, serves as the scientific committee that renders opinions about the safety, efficacy, and quality of human drug products. Each Member State of the European Union has two members on the committee.
99
Table of Contents
Once an application is submitted to the EMEA, the EMEA ensures that the application is complete, and then selects two of its members, known as rapporteurs, to perform independent scientific evaluations of the safety, efficacy, and quality of the drug product candidate. The rapporteurs can draw on two sources of European Union-wide scientific expertise in forming their review teams—experts from the national marketing authorities of Member States and any of over 1,000 outside experts located at universities and institutions throughout Europe. Once the rapporteurs have completed their respective evaluations, they present the case to the EMEA Committee, which then must render an opinion within 210 days after the application was submitted (subject to certain administrative delays). This deadline is postponed when the reviewers request additional information from the applicant and under new rules, further extensions are possible. If the EMEA Committee opinion is favorable, it is transferred to the applicant, all Member States, and the European Commission. The European Commission uses the EMEA Committee’s opinion to prepare a draft decision. This draft decision is then finalized in cooperation with the Standing Committee (composed of representatives of the Member States), which generally agrees to the draft.
Products approved via the centralized registration procedure receive a data protection period of 10 years, which is being replaced by a period of 8 plus 2 years. Under the new rules, no third party may reference the preclinical and clinical data of the originator during the first 8 years, but can only market a generic version after 10 years have lapsed. For products approved via the decentralized procedure, data exclusivity ranges from 6 to 10 years (or 6 years but limited to patent life) in individual Member States, but this is being replaced by a similar 8 plus 2-year protection period. The protection period under both procedures can under the new rules be extended by another year in case of a new therapeutic indication that is of significant benefit. If satraplatin is approved in the European Union, it is likely that it will be covered by the new rules but no assurances can be given that this will be the case in any or all EU Member States.
A drug may also qualify for an orphan medicinal product, or OMP, designation in the European Union. An application for designation as OMP must be submitted prior to submission of an application for marketing approval. OMP designation qualifies a drug for ten years of market exclusivity once the drug is approved. OMP designations are issued by the European Commission, acting on the advice of an expert committee, with representatives of the Member States, patient organizations, and other interests. OMP designation will be granted if the product meets either of two tests: one based on prevalence criteria (the disease or condition must affect no more than five per ten thousand persons in the European Union); and the other based on a determination that it would be infeasible economically to develop the product without orphan drug incentives. The second condition allows the orphan medicine status to be given to products for diseases for which treatments are not likely to be commercially viable. In addition, it must be shown that there is no satisfactory authorized method for diagnosis, prevention or treatment of the respective disease in the European Union. However, if a medicinal product is deemed orphan and if there is a satisfactory alternative already approved in a European Member State, then the product is eligible for an OMP designation if it is of significant benefit to patients. During the development and regulatory review phase, the orphan drug status can be lost if the designation criteria are not met anymore, for instance because a new treatment for the disease in question is approved.
Although orphan drug exclusivity in the EU is granted for ten years, at the end of the fifth year, any member state can initiate proceedings to restrict that period to six years if it believes that the criteria for orphan designation no longer apply (e.g., because the prevalence of the disease has increased or the manufacturer is earning a sufficient profit not to maintain the exclusivity). In addition, competitive products can be approved during the marketing exclusivity period, for example, if they are not “similar” to the original product or are safer, more effective, or otherwise clinically superior to it.
100
Table of Contents
Regulation in Other Countries
Approval of a product by comparable regulatory authorities may be necessary in foreign countries prior to the commencement of marketing of the product in those countries, whether or not FDA or EMEA approval has been obtained. The approval procedure varies among countries and can involve requirements for additional testing. The time required may differ from that required for FDA or EMEA approval. Although there are some procedures for unified filings for some European countries with the sponsorship of the country that first granted marketing approval, in general each country has its own procedures and requirements, many of which are time consuming and expensive. Thus, there can be substantial delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed. Several years ago, representatives of the regulators in the United States, the European Union and Japan launched the International Conference on Harmonization (“ICH”), a collaborative effort with the goal of streamlining the development and registration of medicinal products by harmonizing the applicable procedures in the three regions. For the foreseeable future, however, we will have to seek separate approval in each region.
Hazardous Materials
Our research and development processes involve the controlled use of hazardous materials, chemicals and radioactive materials and produce waste products. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We do not expect the cost of complying with these laws and regulations to be material.
Legal Proceedings
We are not currently, and since January 1, 2002, have not been a party to any material legal proceedings.
Facilities
Our headquarters are located in Martinsried/Planegg, Germany. We lease approximately 3,500 square meters (37,674 square feet) in this facility, in which we house our corporate offices and laboratories under an operating lease expiring on December 31, 2007. This lease extends automatically on December 31st of each year thereafter for one additional year unless terminated by either party with twelve months notice. Additionally, we may terminate the lease on six months notice to the end of the year. We lease 8,700 square feet (800 square meters) of office space in a facility in Princeton, New Jersey under an operating lease expiring on June 30, 2005. We also lease 85,430 square feet (7,937 square meters) of office and laboratory space located in Waltham, Massachusetts under an operating lease expiring in 2011 with an option to extend the lease for two consecutive five-year periods. We believe our existing facilities are sufficient to meet our needs for the foreseeable future and, if needed, additional space will be available in the near term at a reasonable cost to us.
In 2002, we entered into a sublease agreement with ALTANA Pharma pursuant to which ALTANA Pharma subleased space at our Waltham facility through June 30, 2007, with a restricted option for one additional year. As of December 31, 2003, the amount of space subject to the sublease agreement was 23,500 square feet.
Employees
As of December 31, 2003, we had181 employees worldwide, compared to 208 as of December 31, 2002, a decrease of 13%. Part of this change resulted from a restructuring in October 2003, which affected a total of 42 positions or 21% of the work force as of that date. The affected employees were
101
Table of Contents
mainly in the technology research staff. As of December 31, 2003, 47% of our employees were based in Munich, Germany and 53% were based in our Waltham, Massachusetts and Princeton, New Jersey facilities. Of our 181 employees at December 31, 2003, 135.5 are engaged in research and development activities and 45.5 are involved in general administration. None of our employees are covered by labor unions or covered by a collective bargaining agreement, nor have we experienced any work stoppages at our sites in the past. We believe that we have good relations with our employees. We draw on a pool of consultants from time to time for advice on certain matters in which we have not developed internal expertise.
Our Subsidiary
The following table presents selected information relating to our sole subsidiary as of December 31, 2003 (in thousands of€, except for share of capital).
Name and location of the entity | Primary Activity | Common Stock | Share of % | Equity | Net (loss) | Liabilities owed by us | |||||||
GPC Biotech Inc., Waltham, Massachusetts, USA and Princeton, New Jersey, USA | research and development | 31,696 | 100 | 9,327 | (10,417 | ) | 2,699 |
Capital Expenditures
The following table sets forth our capital expenditures, including purchases under capital lease obligations in fiscal 2003, 2002 and 2001 and first quarter 2004:
Three | Fiscal Year ended December 31, | |||||||
| 2003 | 2002 | 2001 | ||||||
Property, equipment and licenses | 492 | 1,669 | 2,498 | 3,522 | ||||
Intangibles | — | — | — | — | ||||
Total | 492 | 1,669 | 2,498 | 3,522 | ||||
We finance our current capital expenditures out of our cash resources. Of our total capital expenditures in the first quarter 2004 and fiscal years 2003, 2002 and 2001, 24.2%, 54.5%, 59.6% and 64.1%, respectively, were invested in the United States.
102
Table of Contents
DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
Overview
In accordance with the German Stock Corporation Act (Aktiengesetz), we have two separate boards of directors. These are theVorstand, or Management Board, and theAufsichtsrat, or Supervisory Board. The two boards are separate, and generally no individual may simultaneously be a member of both boards.
The Management Board is responsible for the day-to-day management of our business in accordance with applicable law, our Articles of Association (Satzung) and the internal rules of procedure (Geschäftsordnung) adopted for it by the Supervisory Board. The Management Board represents GPC Biotech in its dealings with third parties. The principal function of the Supervisory Board is to supervise the Management Board. It is also responsible for appointing and removing the members of the Management Board and representing GPC Biotech in connection with transactions between a member of the Management Board and the company. The Supervisory Board is not itself permitted to make management decisions, but, in accordance with German law and in addition to its statutory responsibilities, our Supervisory Board has determined that the following matters, among others, require its prior consent: (1) the conclusion, amendment and termination of license, know-how, patent and collaboration agreements above specified thresholds, (2) the acquisition, sale and encumbrance of real property, (3) the commencement, acquisition, discontinuance and sale of businesses, business units or branch offices, (4) the granting of guarantees, sureties and other collateral (other than product warranties/guarantees) above a specified threshold, (5) our fiscal and financial planning and budgeting, (6) the granting and termination of profit participation rights, and (7) the purchase and sale of equity interest.
The members of both the Management Board and the Supervisory Board are solely responsible for and manage their own areas of competency (Kompetenztrennung); therefore, neither board may make decisions that are the responsibility of the other board pursuant applicable law, our Articles of Association or the internal rules of procedure. Members of both boards owe a duty of loyalty and care to GPC Biotech. In exercising their duties, the applicable standard of care is that of a diligent and prudent businessperson. Members of both boards must take into account a broad range of considerations when making decisions, including the interests of GPC Biotech and its shareholders as well as those of employees and creditors.
As a general rule under German law, a shareholder has no direct recourse against the members of the Management Board or the Supervisory Board in the event that they are believed to have breached their duty of loyalty and care to GPC Biotech. Apart from insolvency or other special circumstances, only GPC Biotech has the right to claim damages from members of either board. We may waive these damages or settle these claims only if at least three years have passed and the shareholders approve the waiver or settlement at the shareholders’ meeting with a simple majority of the votes cast, provided that a minority holding, in the aggregate, ten percent or more of our share capital does not have their opposition formally noted in the minutes maintained by a German notary.
Our Supervisory Board has comprehensive monitoring functions. To ensure that these functions are carried out properly, our Management Board must, among other things, regularly report to the Supervisory Board with regard to current business operations and future business planning (including financial, investment and personnel planning). The Supervisory Board may, at any time, request special reports regarding the affairs of GPC Biotech, the legal or business relations of GPC Biotech and our subsidiaries and the affairs of any of our subsidiaries to the extent that the affairs of such subsidiary may have a significant impact on GPC Biotech.
The Management Board is required to ensure that adequate risk management and internal monitoring systems exist within the company to detect risks relating to our business activities at the earliest stage possible.
103
Table of Contents
Supervisory Board
Our Supervisory Board generally consists of six members, all of whom are elected by our shareholders in our annual general meeting in accordance with the provisions of the German Stock Corporation Act. Due to a resignation by one member, there is currently one vacancy on our Supervisory Board that will be filled at the next annual general meeting. Unless otherwise determined by our shareholders, under our Articles of Association, the term of a Supervisory Board member expires at the end of the annual general meeting in which the shareholders discharge the Supervisory Board member from liability in respect of the second fiscal year following the year in which the member was elected. The term of office of a member of our Supervisory Board ends at the annual general meeting after he or she has attained the age of 75. None of the present members of our Supervisory Board will turn 75 during their current terms.
Any member so elected by our shareholders may be removed by a majority of three quarters of the votes cast by the shareholders in a general meeting. In addition, any member of the Supervisory Board may, at any time, resign by written notice to the Management Board. According to our Articles of Association and the internal rules of procedure of the Supervisory Board, the Supervisory Board has a quorum when all members were invited or requested to participate in a decision and a majority, but in no event less than three, of the members of the Supervisory Board participated. Unless not required by law or by the Articles of Association of GPC Biotech, resolutions of the Supervisory Board are passed by simple majority of the votes cast. In the case of a deadlock, the Chairman of the Supervisory Board, or in his absence, the Vice Chairman, has the deciding vote. The Supervisory Board meets at least twice each half-year.
The following table shows the current members of the Supervisory Board, the years in which they were first elected, the end of their current terms and their principal occupations.
Name | Year first elected | End of term(1) | Principal occupation | |||
Jürgen Drews, M.D., Ph.D. (Chairman) | 1998 | 2005 | Managing partner of Bear Stearns Health Innoventure | |||
Michael Lytton, J.D. (Vice Chairman) | 2001 | 2006 | General partner of Oxford Bioscience Partners | |||
Metin Colpan, Ph.D. | 1998 | 2005 | Founder and member of the supervisory board of Qiagen N.V. | |||
Prabhavathi B. Fernandes, Ph.D. | 2003 | 2005 | President and chief executive officer of DarPharma, Inc. | |||
Peter Preuss | 2001 | 2006 | President of Preuss Foundation for Brain Tumor Research; Regent of the University of California | |||
Helmut Schühsler, Ph.D.(2) | 1998 | — | Managing Partner of TVM Techno Venture Management GmbH & Co. KG |
| (1) | Term ends upon the adjournment of the annual general meeting held in the year indicated. |
| (2) | Helmut Schühsler resigned as a member and Vice Chairman of our Supervisory Board effective December 31, 2003. We will propose to our shareholders at the next annual general meeting that a new member be elected to the Supervisory Board. |
The members of our Supervisory Board may be contacted at the address of GPC Biotech.
Jürgen Drews. Chairman of the Supervisory Board since 2001. Professor Drews is Managing Partner at Bear Stearns Health Innoventures, the former President of Global Research at Hoffmann-La Roche, and a former member of the Board of Directors and a Partner of BIOMEDICINE, International Biomedicine Management Partners. Until 1998, he served as the Chairman of EuropaBio, the
104
Table of Contents
European Association for Bioindustries, which has more than 500 individual member companies. Professor Drews also serves on the boards of several biotech companies worldwide, including Human Genome Sciences, Rockville, MD, MorphoSys, Munich, Germany, Protein Design Labs, Fremont, CA and TeGenero Würzburg, Germany. He is the chairman of the board of directors of Genaissance Pharmaceuticals, New Haven, CT.
Michael Lytton. Vice Chairman of the Supervisory Board since January 2004. Mr. Lytton has been a General Partner at Oxford Bioscience Partners since 2001. Previously, he was Partner, Chairman of the Technology Group and member of the Executive Committee of Palmer & Dodge LLP. In this capacity, he advised GPC Biotech for several years. Mr. Lytton is a graduate of Princeton University and recipient of a Fulbright Scholarship for study at the University of London. He received his J.D. from Harvard Law School and an M.Sc. in Epidemiology and Medical Statistics from the London School of Hygiene and Tropical Medicine. Mr. Lytton also serves on the boards of several companies worldwide, including Acambis, Cambridge, UK, Alantos Pharmaceuticals, Heidelberg, Germany, Graffinity Pharmaceuticals, Heidelberg, Germany, Rib-X Pharmaceuticals, New Haven, CT and VaxInnate Pharmaceuticals, New Haven, CT. He is the chairman of the board of directors of Descartes Therapeutics, Waltham, MA.
Metin Colpan. Dr. Colpan is co-founder of Qiagen and served as Qiagen’s Chief Executive Officer and President from 1985 through 2003. Qiagen has been listed on Nasdaq since 1996 and on the Frankfurt Stock Exchange since 1997. He obtained his Ph.D. and M.Sc. in Organic Chemistry and Chemical Engineering from the Darmstadt Institute of Technology, Darmstadt, Germany, in 1983. Prior to founding Qiagen, Dr. Colpan was an Assistant Investigator at the Institute for Biophysics at the University of Düsseldorf, Germany. Dr. Colpan has a wide range of experience in separation techniques, particularly in the separation and purification of nucleic acids, and has filed many patents in the field. Dr. Colpan serves on the boards of several biotechnology companies worldwide, including Ingenium Pharmaceuticals, Munich, Germany, Omnitron, Griesheim, Germany and MorphoSys AG, Munich, Germany, and is a member of the board of Qiagen, Venlo, The Netherlands. In addition, he is active as a consultant for the federal and state governments of Germany.
Prabhavathi B. Fernandes. Dr. Fernandes currently serves as President and Chief Executive Officer of DarPharma Inc. Dr. Fernandes has over 25 years of experience in drug discovery and development, having held leadership positions at several biotechnology and pharmaceutical companies. During these years she was directly involved in the development of many drugs, of which four have been approved and one had many years of sales in the billions of dollars. She has served as Chief Executive Officer of Ricerca Biosciences LLC. Prior to joining Ricerca Biosciences, Dr. Fernandes was founder and Chief Executive Officer of Small Molecule Therapeutics. Prior to founding Small Molecule Therapeutics, Dr. Fernandes worked for many years at major pharmaceutical companies, including as Vice President Biomolecular Screening Drug Discovery at Bristol-Myers Squibb, Pharmaceutical Research Institute. She also worked for Abbott Laboratories and The Squibb Institute for Medical Research. Dr. Fernandes served on the United States Congressional Panel for Assessment of Impact of Antibiotic Resistant Bacteria and on the American Society for Microbiology Advisory Panel for Antibiotic Resistance. She was awarded a post-doctoral fellowship to Fox Chase Cancer Center from the National Institutes of Health and received her Ph.D. in Microbiology from Thomas Jefferson University, Philadelphia, PA. Dr. Fernandes has authored numerous publications and has received many awards for her achievements.
Peter Preuss. Peter Preuss is President and founder of the Preuss Foundation for Brain Tumor Research, and is currently serving a 12-year term as a Regent of the University of California. He is a former member of the Advisory Committee to the Director of the National Institutes of Health. Mr. Preuss was founder and Chief Executive Officer of Integrated Software Systems Corporation, one of the world’s first software companies. His extensive business expertise and experience has been recognized
105
Table of Contents
with numerous awards, honors and memberships in Honorary Societies. Recently, Mr. Preuss received the Woodrow Wilson award for public service. He has been on the boards of various biotech and high-tech companies, both public and private, and is currently a member of the board of Overland Storage, Inc., San Diego, CA. Mr. Preuss holds an advanced degree in mathematics from the University of California, San Diego, CA.
Helmut Schühsler. Resigned as member and Vice Chairman of the Supervisory Board effective December 31, 2003. Dr. Schühsler is a Managing Partner of TVM Techno Venture Management and resigned to devote more time to his role at TVM in advising and financing early stage, private companies.
Supervisory Board Practices
Decisions are generally made by the Supervisory Board as a whole. The meeting agendas of the Supervisory Board are determined by the Chairman of the Supervisory Board. The other members of the Supervisory Board receive in advance of Supervisory Board meetings materials allowing them to prepare for the handling of the items on the agenda. Furthermore, the Supervisory Board conducts an annual self-evaluation of the effectiveness of its performance as a whole.
To assist the Supervisory Board in carrying out its duties, the following committees have been created in accordance with our Articles of Association and the internal rules of procedure of the Supervisory Board: the Finance Committee, the Compensation Committee and the Audit Committee (the “Board Committees”). The Board Committees may, to the extent legally possible, additionally be charged with decision-making powers. The granting of loans to members of our Supervisory Board pursuant to § 115 of the German Stock Corporation Act and the entering into by GPC Biotech of advisory contracts or service agreements with members of our Supervisory Board pursuant to § 114 of the German Stock Corporation Act, however, requires the approval of the Supervisory Board as a whole. The Supervisory Board may, in its own discretion, establish, permanently or temporarily, other committees and charge them with decision-making power.
Finance Committee. The current members of the Finance Committee are Professor Jürgen Drews and Michael Lytton. The Finance Committee oversees the preparation of the annual general meeting as well as the execution of major transactions approved by the Supervisory Board as a whole, such as mergers and acquisitions, divestitures, and financing transactions.
Compensation Committee. The current members of the Compensation Committee are Professor Jürgen Drews, Michael Lytton and Peter Preuss. The Compensation Committee reviews and approves our compensation policies and programs, including stock option programs and similar incentive based compensation. It is responsible for reviewing and approving the compensation paid to the members of our Management Board and oversees ongoing personnel matters of the members of the Management Board, including their membership on boards of other companies.
Audit Committee. The current members of our Audit Committee are Dr. Metin Colpan (Chairman), Michael Lytton and Dr. Prabhavathi Fernandes. The Supervisory Board has assured itself that the members of the Audit Committee have sufficient experience and ability in finance and matters of compliance to enable them to adequately discharge their responsibilities.
Our Audit Committee is directly responsible for:
| • | accounting and risk management matters; |
| • | ensuring the independence of our external auditors; |
| • | the external audit scope and the engagement of our external auditors as elected by our shareholders in annual general meetings; and |
| • | the determination of specific key aspects of the external audit and the compensation of our external auditors. |
106
Table of Contents
Management Board
Our Management Board currently consists of four members. The Supervisory Board determines the size of the Management Board, which, under our Articles of Association, may consist of only one member even though our share capital is in excess of€3 million. Members of the Management Board are appointed by the Supervisory Board for a maximum term of five years and are eligible for reappointment or extension after the completion of their term in office. Under certain circumstances, such as a serious breach of duty or a vote of no confidence by the shareholders in an annual meeting, a member of the Management Board may be removed from office by the Supervisory Board prior to the expiration of his term. Furthermore, the term of office of a Management Board member ends three months after he or she has attained the age of 65.
Under our Articles of Association, if the Management Board consists of only one member, GPC Biotech is legally represented by that sole member. If the Management Board consists of two or more members, GPC Biotech is legally represented by two members of the Management Board acting together, or by one member of the Management Board together with a person possessing a special power of attorney authorizing him or her to legally represent GPC Biotech (Prokura). The Supervisory Board may additionally authorize any member of our Management Board to legally represent GPC Biotech alone. To date, none of the members of our Management Board have been so authorized.
A member of the Management Board may not deal with, or vote on, matters relating to proposals, arrangements or contractual agreements between himself and GPC Biotech and may be liable to us if he has a material interest in any contractual agreement between GPC Biotech and a third party which was not disclosed to, and approved by, the Supervisory Board.
If not otherwise required by law, the Management Board makes decisions by a simple majority of the votes cast. In case of deadlock, the resolution is not adopted.
The following table shows the members of the Management Board, their ages, current areas of responsibility, the years in which they were initially appointed, the end of their current terms and the membership in statutory supervisory boards in Germany or abroad.
Name (age) | Year first appointed | End of | Responsibility | Other board | ||||
Bernd R. Seizinger, M.D., Ph.D. (47) | 1998 | 6/2008 | President and Chief Executive Officer | ALTANA Pharma AG, Graffinity Pharmaceuticals AG and BioXell SpA | ||||
Elmar Maier, Ph.D. (38) | 1998 | 2/2008 | Senior Vice President Business Development and Chief Operating Officer (Munich, Germany) | GATC Biotech AG | ||||
Mirko Scherer, Ph.D. (35) | 1999 | 8/2007 | Senior Vice President and Chief Financial Officer | — | ||||
Sebastian Meier-Ewert, Ph.D. (36) | 1998 | 2/2008 | Senior Vice President, Chief Scientific Officer and Chief Operating Officer (Waltham/ Boston, MA) | Immatics GmbH |
The members of our Management Board may be contacted at the address of GPC Biotech.
107
Table of Contents
Bernd R. Seizinger. President and Chief Executive Officer of GPC Biotech. Dr. Seizinger joined GPC Biotech in 1998 from Genome Therapeutics Corporation of Waltham/Boston, MA, where he was Executive Vice President and Chief Scientific Officer from 1996 to 1998. From 1992 to 1996, Dr. Seizinger was at Bristol-Myers Squibb, Pharmaceutical Research Institute, where he held the posts of Vice President of Oncology Drug Discovery and, in parallel, Vice President of Corporate and Academic Alliances. From 1984 to 1992, Dr. Seizinger served both as Associate Professor of Neuroscience at Harvard Medical School and Associate Geneticist and Director of the Molecular Neuro-Oncology Laboratory at Massachusetts General Hospital. Dr. Seizinger also held a visiting professorship at the Department of Molecular Biology at Princeton University. He was awarded his M.D. from the Ludwig Maximilians University and his Ph.D. from the Max Planck Institute of Psychiatry, both in Munich. He is the recipient of a number of scientific awards and has authored over 100 publications.
Elmar Maier. Co-founder of GPC Biotech and Senior Vice President Business Development (joined the company in 1997). He is also Chief Operating Officer of the Munich facility. Dr. Maier has successfully negotiated multiple pharmaceutical and biotech alliances. Previously, Dr. Maier held an executive position at a consulting firm commercializing biotech know-how. He was also Group Leader and Project Manager at the Max Planck Institute for Molecular Genetics in Berlin, where he arranged collaborations with pharmaceutical and biotech companies. Dr. Maier has received numerous awards, including fellowships for his research in the field of genomics technologies at the Imperial Cancer Research Fund in London. Dr. Maier holds a degree in Chemistry and obtained his Ph.D. in Biology from the University of Konstanz in Germany.
Mirko Scherer. Co-founder of GPC Biotech and Senior Vice President and Chief Financial Officer (joined the company in 1997). Previously, Dr. Scherer worked for The Boston Consulting Group in Munich, where he was a management consultant to several industries, including banking. He graduated with a degree in Business Administration from the University of Mannheim in Germany and obtained his MBA from the Harvard University Graduate School of Business Administration. Dr. Scherer received his Ph.D. in Finance from the European Business School in Oestrich-Winkel in Germany.
Sebastian Meier-Ewert. Co-founder of GPC Biotech and Senior Vice President and Chief Scientific Officer (joined the company in 1997). He is also Chief Operating Officer of the Waltham facility. Prior to co-founding GPC Biotech, Dr. Meier-Ewert established and led a team of scientists working on gene expression analysis and bioinformatics at the Max Planck Institute for Molecular Genetics in Berlin. He also co-founded a consulting firm specializing in biotechnology know-how and technologies. Dr. Meier-Ewert studied Biochemistry at University College, London. He completed his postgraduate training at the Imperial Cancer Research Fund in London and received his Ph.D. from the University of London.
108
Table of Contents
Management Team
Our Management Board is supported by a highly qualified and internationally experienced management team.
Name (age) | Position | |
David R. Bancroft, Ph.D.(36) | Vice President Intellectual Property | |
Colin Freund (34) | Vice President Business Development (U.S.) | |
Gregory H. Hamm (53) | Vice President Corporate Integration, Vice President Bioinformatics and Information Technology, Site Head Princeton | |
Brent Hatzis-Schoch, J.D. (39) | Vice President Legal Affairs | |
Torsten Hombeck, Ph.D. (34) | Vice President Finance | |
Nikolai Kley, Ph.D. (42) | Vice President Research, Waltham | |
Arthur Kluge, Ph.D. (60) | Vice President Drug Discovery | |
Thomas J. McKearn, M.D., Ph.D. (55) | Vice President Medical Affairs | |
Edward F. McNiff, Ph.D. (51) | Vice President Pharmaceutical Development | |
Zoltan Nagy, DVM, Ph.D. (63) | Vice President Research, Munich | |
Michael E. Petrone, M.D. (53) | Vice President Clinical Operations | |
John P. Richard (46) | Senior Business Advisor | |
Marcel Rozencweig, M.D. (58) | Senior Vice President Drug Development | |
Hemanshu Shah, Ph.D. (43) | Vice President Strategic Marketing |
David R. Bancroft. Co-founder of GPC Biotech and Vice President Intellectual Property. Prior to founding GPC Biotech in 1997, Dr. Bancroft was responsible for development and implementation of new automated technologies at the Max Planck Institute for Molecular Genetics in Berlin, Germany. His previous research positions include those at the Imperial Cancer Research Fund, London, UK, and at Trinity College, Dublin, Ireland, as a Royal Society European Visiting Fellow. Dr. Bancroft was awarded a Ph.D. in Molecular Population Genetics from the University of Cambridge, UK, for his research within a large-scale and multidisciplinary project. Dr. Bancroft is an inventor on several pending patent applications in the fields of molecular biology and automation.
Colin Freund. Vice President Business Development (U.S.). Prior to joining GPC Biotech in 2002, he held the position of Vice President Business Development at Double Twist, Inc., Oakland, CA, where he was responsible for creating alliances with a variety of genomics and proteomics companies. Prior to joining Double Twist, Mr. Freund was a project manager at the Boston Consulting Group, San Francisco, CA, and London, UK, where he managed assignments in the healthcare and high technology practice areas. Mr. Freund received his BA in Economics and Management Studies from the University of Cambridge, UK, and his MBA from Stanford University, CA.
Gregory H. Hamm. Vice President Corporate Integration, Vice President Bioinformatics and Information Technology and Site Head Princeton, New Jersey. Prior to joining GPC Biotech in 1999, he was Vice President Bioinformatics at Genome Therapeutics Corporation, Waltham/Boston, MA, where he was responsible for all computational aspects of the company’s scientific program. Before 1995, Mr. Hamm was Director of the Molecular Biology Computing Laboratory at Rutgers University, NJ, and Principal Consultant at Systems & Bioinformatics, Highland Park, NJ. From 1980 to 1986, Mr. Hamm was the founder of EMBL Data Library and Head of Computing at the European Molecular Biology Laboratory (EMBL) in Heidelberg, Germany. Mr. Hamm received his BS from Harvey Mudd College, Claremont, CA.
Brent Hatzis-Schoch. Vice President Legal Affairs. Mr. Hatzis-Schoch joined GPC Biotech in 2003 as Vice President Legal Affairs. Prior to joining GPC Biotech, Mr. Hatzis-Schoch was Associate General Counsel for Global Research & Development at Pharmacia Corporation, and earlier was legal
109
Table of Contents
counsel to Baxter Healthcare in Europe. He also spent several years in private practice in the United States and Germany. Mr. Hatzis-Schoch received his J.D. from George Washington University and was also the recipient of a Fulbright Scholarship for study in Germany.
Torsten Hombeck. Vice President Finance. Dr. Hombeck joined GPC Biotech in 1999 as Director Finance. He previously held positions in the area of Corporate Finance and Controlling at Beiersdorf AG, Hamburg, Germany, where he was responsible for South America. Dr. Hombeck graduated with a Degree in Business Administration from the European Business School in Oestrich-Winkel, Germany, where he also received his Ph.D. in Finance.
Nikolai Kley. Vice President Research, Waltham. Prior to joining GPC Biotech in 1999, Dr. Kley held positions as Director and Vice President Functional Genomics at Genome Therapeutics Corporation, Waltham/Boston, MA. While there, he was instrumental in establishing numerous functional technology platforms and research programs, including a collaborative oncology program with Princeton University, Princeton, New Jersey. Prior to 1997, Dr. Kley held posts as Senior Scientist and Group Leader/Principal Scientist within Oncology Drug Discovery at Bristol-Myers Squibb Company, Pharmaceutical Research Institute. He also spent several years at Harvard Medical School and the Massachusetts General Hospital, Boston, MA, working in the fields of cancer genetics and biology. Dr. Kley retains his adjunct faculty position as Research Professor at the Department of Molecular Biology at Princeton University.
Arthur Kluge. Vice President Drug Discovery. Prior to joining GPC Biotech in 2000, Dr. Kluge served as Vice President Drug Discovery at Mitotix Inc. since 1996, prior to which he had held the post of Vice President Chemistry at Cubist Pharmaceuticals. He has also worked at Syntex Research in Palo Alto, CA, where he served as Vice President and Director of the Institute of Organic Chemistry, with responsibility for the direction of a multinational drug discovery and development group. Dr. Kluge brings over 25 years of management experience in the pharmaceutical industry to GPC Biotech and is the inventor of two currently marketed drugs: ToradolTM (ketoroalc), CattlystTM (laidlomycin propionate). Dr. Kluge received his BA in chemistry from Park College, his MS and Ph.D. from the University of Massachusetts and his MBA from the University of Santa Clara, CA.
Thomas J. McKearn. Vice President Medical Affairs. Dr. McKearn is responsible for the design and implementation of our clinical and regulatory development strategy for new lead candidates as they enter development through eventual registration and commercialization. His responsibilities include oversight of all clinical study protocols, selection of investigators, and the review and analysis of study results. His position also serves as the primary point of contact with worldwide regulatory agencies and health authorities. Furthermore, Dr. McKearn will play a key role in our compound in-licensing strategy. Prior to joining GPC Biotech in 2002, Dr. McKearn held several executive positions both in biotech and in the pharmaceutical industry. He was a founder and Chief Executive Officer of Cytogen Corporation. His most recent position was Executive Director Strategic Science & Medicine at Bristol-Myers Squibb Company, Pharmaceutical Research Institute.
Edward F. McNiff. Vice President Pharmaceutical Development. Prior to joining GPC Biotech in 2002, Dr. McNiff held several positions at Bristol-Myers Squibb Company. His most recent position was Vice President Analytical Research and Development at Bristol-Myers Squibb Company, Pharmaceutical Research Institute, supporting pharmaceutical development activities spanning the identification of new drug candidates through registration, launch and life-cycle activities. Dr. McNiff is responsible for the design and implementation of our pharmaceuticals development strategy for new lead candidates as they enter development through eventual registration and commercialization. These responsibilities include analytical and process chemistry, dosage form development, quality assurance, metabolism and pharmacokinetics.
110
Table of Contents
Zoltan Nagy. Vice President Research, Munich, Germany. Dr. Nagy, a world-renowned immunologist, joined GPC Biotech in 1998 from Hoffmann-La Roche where he was Department Head of Immunology at the company’s Nutley, New Jersey, facility. He previously held positions as Project Leader in Preclinical Research at Novartis, Basle, Switzerland, Deputy Director of the Max Planck Institute for Biology, Tübingen, Germany, and as a Scientific Member of the Basle Institute of Immunology. Dr. Nagy’s research into the major histocompatibility complex has resulted in over 140 papers in peer-reviewed journals.
Michael E. Petrone. Vice President Clinical Operations. Dr. Petrone is responsible for project planning and clinical outsourcing. He has more than ten years of experience in the pharmaceutical and biotechnology industry in the areas of strategic planning, product development, and clinical research from the initiation of clinical trials to marketing approval. Dr. Petrone has extensive expertise and leadership in new drug development and Clinical Research Organization management. Prior to joining GPC Biotech in 2002, he was with Roberts Pharmaceutical Corporation as Vice President Clinical Research and Medical Affairs, and more recently with the Genaera Corporation as Vice President Clinical Research.
John P. Richard. Senior Business Advisor. John P. Richard joined GPC Biotech in 1999. Previously, he was Executive Vice President, Business Development at SEQUUS Pharmaceuticals, where he was responsible for negotiating the acquisition of SEQUUS by ALZA Corporation in 1999. Prior to SEQUUS, Mr. Richard headed business development for VIVUS and Genome Therapeutics Corporation, where he was responsible for establishing numerous pharmaceutical alliances. He was also co-founder and the original Chief Executive Officer of IMPATH Inc. Mr. Richard currently serves on the board of directors of Altus Biologics Inc., Cambridge, MA, Targacept Inc, Winston-Salem, NC and Zygogen LLC, Atlanta, GA. Mr. Richard received his MBA from Harvard Business School and his BS from Stanford University.
Marcel Rozencweig. Senior Vice President Drug Development. Prior to joining GPC Biotech in 2001, Dr. Rozencweig held several senior leadership positions in drug development and strategic planning at Bristol-Myers Squibb Company, where his most recent position was Vice President Strategic Planning and Portfolio Management. Over his 18-year career, Dr. Rozencweig has established himself as an expert in several areas, including pharmaceutical R&D, worldwide clinical R&D, strategic planning, portfolio management and development, particularly focused in oncology, immunology and infectious diseases. Among his many R&D achievements are significant contributions leading to the successful FDA approval of numerous anticancer treatments including TAXOL (paclitaxel) and PARAPLATIN (carboplatin). Dr. Rozencweig has authored or co-authored more than 200 papers.
Hemanshu Shah. Vice President Strategic Marketing. Dr. Shah joined GPC Biotech in 2003. Previously, he was Global Commercial Leader for ZARNESTRA and PROCRIT in the Oncology Global Marketing Group at Johnson & Johnson. Prior to joining Johnson & Johnson, he was Director of Marketing for Bristol-Myers Squibb Oncology with significant responsibility for marketing TAXOL and PARAPLATIN. Dr. Shah’s earlier experience at Bristol-Myers Squibb included various positions in Business Development, and Research & Development. Dr. Shah received his Ph.D. in Pharmaceutical Sciences from Rutgers University, and his MBA from SUNY Buffalo, NY.
None of the above members of our Management Board, Supervisory Board or management team have any family relationship with any other member of the Management Board, Supervisory Board or management team. None of the above members of the Management Board, Supervisory Board or management team were appointed pursuant to an agreement or understanding between such member of the Management Board, Supervisory Board or management team and any third party.
111
Table of Contents
Compensation of Management Board and Supervisory Board Members
Compensation of Members of the Management Board
We have entered into service agreements with all current members of our Management Board. These agreements generally provide for a base salary and an annual bonus. For a discussion of the stock option and similar plans in which the members of our Management Board participate, see “—Equity-Based Plans”. In addition to these fixed and variable remuneration components, under the terms of their service agreements, the members of our Management Board are entitled to specific insurance benefits (including accident and D&O insurance) and reimbursement of necessary and reasonable disbursements. In the event of a change in control and termination of employment within a specified period thereof, each member of our Management Board is entitled to severance benefits in the amount of 175 percent (250 percent in the case of the Chairman of our Management Board) of the sum of their respective highest annual salary on or after the date of the change in control and the average of their respective last two annual bonuses received prior to the change in control. In addition, subject to the term of the stock options and convertible bonds under the terms and conditions of the relevant plan or program (see “—Equity Based Plans”), stock options and convertible bonds granted to the members of our Management Board may no longer be terminated upon a change in control. A change in control will have occurred if, as a result of any takeover, exchange or other transfer a single shareholder or a group of shareholders acting in concert acquire more than 50 percent of the outstanding voting rights in GPC Biotech or, if as a result of a merger or reverse merger we cease to own more than 50 percent of the outstanding voting shares in the merged entity. Furthermore, Dr. Seizinger, the Chairman of our Management Board, is entitled to severance benefits in the amount of 200 percent of the sum of his last annual salary and the average of his last two annual bonuses in the event that our Supervisory Board determines to terminate his appointment as Chairman of the Management Board and not to extend his service agreement beyond June 2008. The other members of our Management Board are entitled to severance benefits in the amount of 100 percent of their respective last annual salaries in the event that our Supervisory Board determines not to renew their respective service agreements beyond their current term.
We believe that the service agreements between GPC Biotech and the members of our Management Board provide for payments and benefits (including upon termination of employment) that are in line with customary market practices.
2003 Summary Compensation Table
| Annual compensation | All other compensation (€)(1) | Total compensation(€)(2) | Long-term compensation | |||||||||||
Name | Salary (€) | Cash bonus (€) | Stock options (number) | Convertible bonds (number)(3) | ||||||||||
Bernd R. Seizinger, M.D., Ph.D. | 429,995 | (4) | 263,317 | 56,516 | (5) | 749,828 | — | 75,000 | ||||||
Elmar Maier, Ph.D. | 247,914 | 100,000 | 7,774 | 355,688 | — | 36,000 | ||||||||
Mirko Scherer, Ph.D. | 265,623 | 100,000 | 7,734 | 373,357 | — | 76,000 | ||||||||
Sebastian Meier-Ewert, Ph.D. | 271,413 | 110,000 | 7,494 | 388,907 | — | 130,500 | ||||||||
Total | 1,214,945 | 573,317 | 79,518 | 1,867,780 | — | 317,500 | ||||||||
| (1) | Amounts include, among others, GPC Biotech’s contributions to accident/D&O insurance. |
| (2) | Excludes long-term compensation. |
| (3) | Each convertible bond entitles the holder thereof to purchase one ordinary bearer share subject to adjustment in the event of changes to our capitalization. |
| (4) | Includes US$166,430 which has been converted into euros at the US$/euro exchange rate of US$1.00 =€ 0.8856 (average exchange rate during fiscal year 2003). |
| (5) | This amount includes GPC Biotech’s contributions to accident/D&O insurance, and€31,881 in double household allowances and€17,140 in forgiven loans (see “—Loans Extended and Guarantees Provided”). |
112
Table of Contents
Compensation of Members of the Supervisory Board
Our Articles of Association provide that the annual compensation for each Supervisory Board member be fixed by our shareholders in annual general meetings. At our annual general meeting on May 21, 2003, our shareholders resolved that the Chairman of our Supervisory Board receive an annual fixed remuneration of€35,000, that the Vice Chairman receive an annual fixed remuneration of€25,000 and that all other members of our Supervisory Board receive an annual fixed remuneration of€15,000. Members of our Supervisory Board who serve on the Audit Committee receive an additional€10,000 annually. Our shareholders additionally resolved that the Chairman of the Supervisory Board receive 10,000 convertible bonds in each year for which he was elected to serve on the Supervisory Board in this capacity, that the Vice Chairman receive 8,000 convertible bonds in each year for which he was elected to serve on the Supervisory Board in this capacity and that each other member of our Supervisory Board receive 5,000 convertible bonds in each year for which he or she was elected to serve on the Supervisory Board. In addition, each member who serves on a Board Committee receives an additional 2,500 convertible bonds annually. Each convertible bond entitles the holder thereof to purchase one of our ordinary bearer shares (subject to changes in our capitalization). The bonds have a maximum term of five years and vest (i) in full in respect of Supervisory Board members whose term ends upon the adjournment of the annual general meeting in 2004, at the end of their term, (ii) in respect of Supervisory Board members whose term ends upon the adjournment of the annual general meeting in 2005 to 50% upon adjournment of the annual general meeting in 2004 and in full at the end of their term and (iii) in respect of Supervisory Board members whose term ends upon the adjournment of the annual general meeting in 2006 one-third upon adjournment of the annual general meeting in 2004 and 2005, respectively, and in full at the end of their term. The vested bonds may only be exercised if the annual general meeting discharges the relevant Supervisory Board members in the relevant annual general meeting and if our share price (as quoted in the XETRA closing auction on the Frankfurt Stock Exchange) exceeds the conversion price for the registered convertible bonds on five successive stock exchange trading days during the last month prior to the exercise of the conversion rights by a certain percentage (which percentage is ten percent for the first year after expiry of the vesting period and is increased by five percent for each year thereafter). To the extent that a member of our Supervisory Board resigns prior to the end of the term for which he or she was elected by our shareholders, any unvested convertible bonds granted to him or her will be cancelled; however, any convertible bonds granted for the year during which a member of the Supervisory Board resigns will remain exercisable, prorated to the date of resignation. Furthermore, we reimburse our Supervisory Board members for out-of-pocket expenses incurred in connection with their services and, to the extent applicable, any value added tax on this compensation.
We have no contracts with any of our Supervisory Board members that would provide for benefits upon termination of service on the Supervisory Board.
113
Table of Contents
2003 Summary Compensation Table
Name | Salary (€) | Total compensation(1)(€) | Convertible bonds (number) | |||
Jürgen Drews, M.D., Ph.D.(2)(4)(5) | 23,411 | 27,418 | 12,500 | |||
Michael Lytton, J.D.(3)(4)(5)(6) | 17,274 | 21,281 | 31,500 | |||
Metin Colpan, Ph.D.(7) | 11,137 | 15,144 | 10,000 | |||
Prabhavathi B. Fernandes, Ph.D.(8) | 11,137 | 15,144 | 10,000 | |||
Peter Preuss(4) | 11,137 | 15,144 | 22,500 | |||
Helmut Schühsler, Ph.D.(9) | 23,411 | 27,418 | 10,500 | |||
Total | 97,507 | 121,549 | 97,000 |
| (1) | Amounts include, among others, GPC Biotech’s contributions to D&O insurance; excludes convertible bonds. |
| (2) | Chairman of the Supervisory Board. |
| (3) | Vice Chairman of the Supervisory Board. |
| (4) | Member of the Compensation Committee. |
| (5) | Member of the Finance Committee. |
| (6) | Member of the Audit Committee. |
| (7) | Chairman of the Audit Committee since March 5, 2004. |
| (8) | Member of the Audit Committee since March 5, 2004. |
| (9) | Helmut Schühsler resigned as a member and Vice Chairman of our Supervisory Board effective December 31, 2003. Until then, Dr. Schühsler also served as Chairman of the Audit Committee. A total of 4,027 convertible bonds initially granted to Dr. Schühsler were cancelled effective December 31, 2003 as a result of his resignation. We will propose to our shareholders at the next annual general meeting that a new member be elected to the Supervisory Board. |
Compensation of Former Management Board and Supervisory Board Members
Our Management Board has not changed since our incorporation; hence, we have not paid any compensation to former members of our Management Board or their surviving dependents. Members of our Supervisory Board are not eligible for compensation once their service has been terminated.
Equity-Based Plans
As of December 31, 2003, we had granted a total of 2,473,780 stock options and convertible bonds to purchase our ordinary bearer shares to members of our Management Board and a total of 215,800 stock options and convertible bonds to the members of our Supervisory Board. The following tables set forth all option and convertible bond grants to our Management Board as well as to our Supervisory Board by year of grant.
Supervisory Board Members:
Grant year | Options/convertible bonds granted (number) | Conversion rate | Exercise price (€) | Term life (years) | |||||
1998 | 14,400 | 1:1 | 1.16 | 10 | |||||
1999 | 14,400 | 1:1 | 1.16 | 10 | |||||
2000 | 15,000 25,000 | 1:1 1:1 | 6.62 17.89 | 10 10 | | ||||
2001 | 40,000 | 1:1 | 9.55 | 5 | (1) | ||||
| 10,000 | 1:1 | 16.38 | 10 | ||||||
2002 | — | — | — | — | |||||
2003 | 97,000 | 1:1 | 5.77 | 5 | (2) | ||||
Total | 215,800 |
| (1) | As of the date of the resignation as a member of the Supervisory Board, not exceeding one year. |
| (2) | As of the date of the resignation as a member of the Supervisory Board, not exceeding two years. |
114
Table of Contents
Management Board Members:
Grant year | Options/convertible bonds granted (number) | Conversion rate | Exercise price (€) | Term life (years) | |||||
1998 | 445,440 | (1) | 1:1 | 1.16 | 10 | ||||
1999 | 148,480 111,360 | (1)(2) (1) | 1:1 1:1 | 1.16 3.98 | 10 10 | ||||
2000 | 306,000 530,000 | | 1:1 1:1 | 6.62 17.89 | 10 10 | ||||
2001 | 250,000 50,000 | | 1:1 1:1 | 9.69 13.34 | 10 10 | ||||
2002 | 100,000 215,000 | | 1:1 1:1 | 5.24 4.36 | 10 10 | ||||
2003 | 125,000 48,574 143,926 | | 1:1 1:1 1:1 | 2.70 4.87 4.96 | 10 10 10 | ||||
Total | 2,473,780 |
| (1) | A total of 705,280 stock options initially granted to Dr. Seizinger in 1998 and 1999 were transferred by him to a financial institution in connection with cash settled share option agreements dated July and December 2001, respectively. Upon any exercise by him of the options granted in the agreement of July 2001, Dr. Seizinger will receive cash, not shares, from the financial institution. On any exercise by him of the options in the agreement of December 2001, Dr. Seizinger will, provided that he has paid to the financial institution the amounts required by the agreement and that certain other conditions are satisfied, be entitled to receive cash, not shares, from the financial institution (see also “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”). |
| (2) | Dr. Scherer and Dr. Maier each transferred 12,000 stock options initially granted to them in 1999 to family members. |
We have established the following equity compensation plans for members of our Management Board, employee directors of our U.S. subsidiary and selected employees of GPC Biotech as well as consultants and members of our Supervisory Board:
German Equity-Based Plans
2002 Stock Option Plan, 2001 Stock Option Plan and 2000 Stock Option Plan
Our annual general meetings held on June 11, 2002, June 5, 2001 and May 3, 2000, respectively, approved the creation of several specific authorized and conditional capitals and the implementation in general of the 2002 Stock Option Plan, the 2001 Stock Option Plan and the 2000 Stock Option Plan, respectively. For a description of the specific authorized and conditional capitals underlying the stock options pursuant to the plans, see “Description of Share Capital—Authorized Capital—Authorized Capital I” and “Description of Share Capital—Conditional Capital”.
Our Management Board, together with our Supervisory Board, and, to the extent that our Management Board is concerned, the Supervisory Board alone, may determine that, in lieu of the issuance of new shares out of the specifically created conditional capitals, own shares acquired by GPC Biotech through market purchases or otherwise in accordance with applicable German stock corporation law may be issued to optionees upon exercise of the stock options. In respect of the 2000 Stock Option Plan, our Management Board, together with our Supervisory Board, and, to the extent that our Management Board is concerned, the Supervisory Board alone, may additionally determine that, in lieu of the issuance of new ordinary shares out of the specifically created authorized and conditional capitals, new ordinary shares issued out of additional authorized or conditional capitals may be issued to optionees upon exercise of the stock options or that stock options may be settled in cash.
115
Table of Contents
Under the 2002 Stock Option Plan, the members of our Management Board and selected employees of GPC Biotech are eligible to acquire our ordinary shares pursuant to stock options. Under the 2001 Stock Option Plan, selected employees of GPC Biotech, members of our Management Board and members of the management bodies of our affiliates (verbundene Unternehmen) are eligible to acquire our ordinary shares pursuant to stock options. Under the 2000 Stock Option Plan, selected employees of GPC Biotech, members of our Management Board, members of the management bodies of our affiliates as well as members of the Supervisory Board and consultants are eligible to acquire our ordinary shares pursuant to stock options.
The following is a general description of the material features of the 2002 Stock Option Plan, the 2001 Stock Option Plan and the 2000 Stock Option Plan.
Stock options may be granted under the plans at an exercise price determined on the basis of the average closing price for our ordinary shares as quoted in the XETRA closing auction on the Frankfurt Stock Exchange during the last five stock exchange trading days prior to the date of grant. In addition, under the 2000 Stock Option Plan, the exercise price for the stock options granted to optionees under this plan prior to our initial public offering in 2000, was determined on the basis of the fair market value of our shares. The exercise price is subject to adjustment upon changes in our capitalization and must at all times be at least equal to the notional nominal value (the amount attributable to each of our ordinary shares) of our ordinary shares (currently€1.00 per ordinary share). The Management Board, and, to the extent that our Management Board is concerned, the Supervisory Board, selects the eligible persons to whom options will be granted and determines the grant date, amounts, exercise price, vesting periods and other relevant terms of the stock option grants (including acceleration provisions) in accordance with the provisions of the respective plans. The details of any grants of stock options to members of our Supervisory Board under the terms of the 2000 Stock Option Plan were approved by our shareholders and any shares to be issued upon exercise of these stock options will be issued out of authorized capital.
A stock option granted under the 2002 Stock Option Plan, the 2001 Stock Option Plan or the 2000 Stock Option Plan may not be exercised until the occurrence of each of the following events: (i) the stock option satisfies a two-year waiting requirement (measured from the date of grant); (ii) the stock option vests; and (iii) the price of our ordinary bearer shares exceeds the exercise price of the stock option on five successive stock exchange trading days (10 days in the case of the 2000 Stock Option Plan) within one month prior to exercise by a certain percentage (which percentage is ten percent for the first year after the expiration of the two-year waiting period and is increased by five percent for each year thereafter). In addition, pursuant to the 2002 Stock Option Plan and the 2001 Stock Option Plan, a stock option may only be exercised within a period of six weeks after publication of our quarterly reports or annual financial statements, and may not be exercised during a subscription rights offering by GPC Biotech prior to the commencement of “ex subscription rights” trading on the Frankfurt Stock Exchange and during the period from December 24 to December 31 of each calendar year. Furthermore, pursuant to the 2000 Stock Option Plan, a stock option may only be exercised during four designated exercise periods lasting one month each that begin on the third bank working day after publication of our quarterly reports; and stock options may not be exercised during a subscription rights offering by GPC Biotech prior to the commencement of “ex subscription rights” trading on the Frankfurt Stock Exchange.
In the event of the termination of service of an optionee, the unvested portion of any stock option grant is generally forfeited, and the vested and exercisable portion is generally terminated 130 business days after a termination of service due to the death of the optionee or 30 business days (90 business days in the case of the 2002 stock option plan) after termination of employment for any other reason. In extraordinary circumstances (such as maternity leave, permanent disability or early retirement and similar circumstances), our Management Board, and, to the extent that our Management Board is concerned, the Supervisory Board, may abstain from terminating the stock option grants.
116
Table of Contents
Notwithstanding the foregoing, a stock option may not be exercised following the expiration of its term, which under the plans cannot exceed ten years from the date of grant. A stock option is generally not transferable during the life of the optionee, but is inheritable upon the death of the optionee.
In the event of our merger with or into another entity (Verschmelzung), a change in our legal form (Umwandlung), a change in the notional nominal value of our ordinary shares and similar measures leading to the cancellation or conversion of the shares underlying the stock options, each outstanding stock option will be substituted by the right to purchase at the exercise price a specific number of shares or other interests (calculated on the basis of the fair market value of our ordinary bearer shares at the time of any such change) substituting our ordinary shares.
1999 and 1997 Stock Option Plans
Our annual general meetings held on May 12, 1999 and December 8, 1997, respectively, approved the creation of several specific authorized and conditional capitals and the implementation in general of each of the 1999 and 1997 Stock Option Plans. For a description of the specific authorized and conditional capitals underlying the stock options pursuant to the plans see “Description of Share Capital—Authorized Capital—Authorized Capital I” and “Description of Share Capital—Conditional Capital”.
Under these plans, the members of our Management Board and Supervisory Board as well as selected consultants are eligible to acquire our ordinary shares pursuant to stock options.
The terms of the 1999 and 1997 Stock Option Plans are substantially similar to the terms of the stock option plans discussed in “—2002 Stock Option Plan, 2001 Stock Option Plan and 2000 Stock Option Plan” above, with the following exceptions:
| • | the exercise price for the stock options granted to optionees under the 1999 and 1997 Stock Option Plans was determined on the basis of the fair market value of our ordinary shares; |
| • | a stock option granted under the 1999 and 1997 Stock Option Plans may only be exercised if the price of our ordinary bearer shares exceeds the exercise price of the stock option on five successive bank working days within one month prior to exercise by a certain percentage (which percentage is 15 percent for the first year after the expiration of the two-year waiting period and is increased by five percent for each year thereafter); |
| • | a stock option granted under the 1999 and 1997 Stock Option Plans is generally transferable during the life of the optionee; |
| • | the periods during which stock options may be exercised deviate slightly from those under the 2002 Stock Option Plan and the 2001 Stock Option Plan; |
| • | in the event of a termination of the stock options granted under the 1999 and 1997 Stock Option Plans, the optionees have to be compensated in an amount equal to the lower of (1) the fair value of the terminated stock options and (2) the amount of income tax paid at the time of grant; and |
| • | stock options issued under the 1999 Stock Option Plan may be settled in cash. |
As of June 4, 2004, a total of 729,280 stock options have been transferred under the 1999 and 1997 Stock Option Plans.
U.S. Equity-Based Plans
On the basis of the resolutions of our shareholders in our annual general meetings held on June 11, 2002, June 5, 2001, May 3, 2000 and May 12, 1999, respectively, we adopted our 2002 Incentive Stock Option Plan, the 2001 Incentive Stock Option Plan, the 2000 Incentive Stock Option
117
Table of Contents
Plan and the 1999 Incentive Stock Option Plans. For a description of the specific authorized and conditional capitals underlying the stock options pursuant to the plans, see “Description of Share Capital—Authorized Capital—Authorized Capital I” and “Description of Share Capital—Conditional Capital”.
Under the 2002 Incentive Stock Option Plan and the 2001 Incentive Stock Option Plan, selected employees of GPC Biotech and employee directors of our U.S. subsidiary and members of our Management Board are eligible to acquire our ordinary shares pursuant to stock options. Under the 2000 Incentive Stock Option Plan and the 1999 Incentive Stock Option Plan, selected employees and employee directors of our U.S. subsidiary, members of our Management Board and consultants of GPC Biotech, as well as members of the Supervisory Board residing in the United States, are eligible to acquire our ordinary shares pursuant to stock options.
Stock options granted under these plans may be incentive stock options, or ISOs, which are intended to qualify for favorable U.S. federal income tax treatment under the provisions of Section 422 of the U.S. Internal Revenue Code of 1986, as amended, or U.S. Internal Revenue Code, or non-qualified stock options, or NSOs, which do not so qualify. The aggregate fair market value of the ordinary shares represented by any given optionee’s ISOs that become exercisable for the first time in any calendar year may not exceed US$100,000. Stock options in excess of this limit are treated as NSOs.
The Management Board, and, to the extent that our Management Board is concerned, the Supervisory Board, selects the eligible persons to whom stock option, will be granted and determines the grant date, amounts, exercise price, vesting periods and other relevant terms of the stock option grants (including acceleration provisions) in accordance with the provisions of the respective plans, including whether the options are intended to be ISOs or NSOs. Under the plans, employees are eligible to receive grants of ISOs and NSOs, while directors of our U.S. subsidiary and members of our Management Board and consultants are eligible to receive grants of NSOs only. Stock option grants to members of the Management Board require the approval of the Supervisory Board. The details of any grants of stock options to members of our Supervisory Board under the terms of the 2000 Incentive Stock Option Plan and the 1999 Incentive Stock Option Plan were approved by our shareholders. The exercise price of options granted under the plans can be determined by the Management Board, and, to the extent that our Management Board is concerned, the Supervisory Board within specific parameters fixed by our shareholders in the respective resolutions relating to the several plans.
A stock option granted under the 2002 Incentive Stock Option Plan, the 2001 Incentive Stock Option Plan, the 2000 Incentive Stock Option Plan or the 1999 Incentive Stock Option Plan may not be exercised until the occurrence of each of the following events: (i) the stock option satisfies a two-year waiting requirement (measured from the date of grant); (ii) the stock option vests; and (iii) the price of our ordinary shares exceeds the exercise price of the stock option on five successive stock exchange trading days, or bank working days in the case of the 2000 Incentive Stock Option Plan and the 1999 Incentive Stock Option Plan, during the one month prior to exercise by a certain percentage (which percentage is ten percent, 15 percent in the case of stock options granted under the 1999 Stock Option Plan, for the first year after the expiration of the two-year waiting period and is in each case increased by five percent for each year thereafter). In addition, pursuant to the 2002 Incentive Stock Option Plan and the 2001 Incentive Stock Option Plan, a stock option may only be exercised within a period of six weeks after publication of our quarterly reports or annual financial statements, and may not be exercised during a subscription rights offering by GPC Biotech and during the period from December 24 to December 31 of each calendar year. Furthermore, pursuant to the 2000 Incentive Stock Option Plan and the 1999 Incentive Stock Option Plan, a stock option may only be exercised during four designated exercise periods lasting one month each that begin on the third bank working day after publication of our annual report and our quarterly reports; and stock options may not be exercised during a subscription rights offering by GPC Biotech.
118
Table of Contents
In the event of the termination of service of an optionee, the unvested portion of a stock option is generally forfeited, and the vested and exercisable portion generally terminates eighteen months after a termination of service due to the death of the optionee, twelve months after a termination of service due to the permanent disability of the optionee or three months after termination of employment for any other reason. Notwithstanding the foregoing, a stock option may not be exercised following the expiration of its term, which under the plans cannot exceed ten years from the date of grant. A stock option is generally not transferable during the life of the optionee, but is inheritable upon the death of the optionee.
In the event of our dissolution, our liquidation, a sale of all or substantially all of our assets, a merger or reverse merger (in which we are not the surviving corporation), each outstanding stock option may be assumed or an equivalent stock option may be substituted by the successor corporation. In the event that no such substitution or assumption occurs, subject to the expiry of the two-year waiting period the outstanding stock options will automatically vest and become exercisable as of a specified time prior to the event and will terminate immediately prior to the consummation of such event. Notwithstanding the foregoing, if, upon consummation of such dissolution, liquidation, sale, merger or reverse merger, our shareholders will receive a cash payment for each share surrendered, the Management Board may instead provide that all outstanding stock options will terminate upon consummation of such event and that each optionee will receive in exchange for the cancelled stock options a cash payment equal to the amount by which the acquisition price for the ordinary shares exceeds the exercise price of the stock options.
Shares Reserved and Options Outstanding Under the German and U.S. Equity-Based Plans
At the time of adoption of the 2002 Stock Option Plan and the 2002 Incentive Stock Option Plan, an aggregate maximum of 810,000 ordinary shares were reserved for issuance under these plans. At the time of adoption of the 2001 Stock Option Plan and the 2001 Incentive Stock Option Plan, an aggregate maximum of 700,000 ordinary shares were reserved for issuance under these plans. At the time of adoption of the 2000 Stock Option Plan and the 2000 Incentive Stock Option Plan, an aggregate maximum of 1,201,000 ordinary shares were reserved under these plans. The aggregate maximum number of shares reserved under each of these plans is subject to adjustment upon changes to our capitalization.
At the time of adoption, an aggregate maximum of 840,000 ordinary shares were reserved for issuance under the 1999 Incentive Stock Option Plan and the 1999 Stock Option Plan, in each case subject to adjustment upon changes to our capitalization. At the time of adoption, an aggregate maximum of 1,212,000 ordinary shares were reserved for issuance under the 1997 Stock Option Plan, subject to adjustment upon changes to our capitalization.
As of June 4, 2004, options to purchase a total of 239,125, 363,486 and 721,800 shares, respectively, were outstanding under the 2002 Stock Option Plan, the 2001 Stock Option Plan and the 2000 Stock Option Plan. As of June 4, 2004, options to purchase a total of 429,425, 314,850, 297,175 and 32,000 ordinary shares, respectively, were outstanding under the 2002 Incentive Stock Option Plan, the 2001 Incentive Stock Option Plan, the 2000 Incentive Stock Option Plan and the 1999 Incentive Stock Option Plan. As of June 4, 2004, options to purchase a total of 584,840 and 449,530 shares, respectively, were outstanding under each of the 1999 and 1997 Stock Option Plans.
We may continue to issue stock options under each of the 2002, 2001 and 2000 German and U.S. Equity-Based Plans. No additional grants will be made under the 1999 Incentive Stock Option Plan and the 1999 and 1997 Stock Option Plans.
Mitotix, Inc. 1996 Equity Incentive Plan
In connection with the acquisition of Mitotix, we assumed the outstanding stock options under the Mitotix, Inc. 1996 Equity Incentive Plan. The former employees, officers, directors and consultants of
119
Table of Contents
Mitotix were eligible to participate in the plan. The plan was terminated as ofFebruary 24, 2000, and the optionees are now eligible to participate in our equity plans. As of June 4, 2004, stock options to purchase58,777 of our ordinary shares were outstanding made under the Mitotix, Inc. 1996 Equity Incentive Plan. Ordinary shares underlying the assumed stock options will be created out of authorized capital which our shareholders approved on May 3, 2000 (see “Description of Share Capital—Authorized Capital—Authorized Capital I”).
2003 Convertible Bond Program and 2002 Convertible Bond Program
On May 21, 2003, and on June 11, 2002, respectively, our shareholders approved the implementation of the 2003 Convertible Bond Program and the 2002 Convertible Bond Program. Under these programs, members of our Management Board, members of the management bodies and other employees with management functions of our affiliates and consultants are eligible to acquire our ordinary shares pursuant to convertible bonds, with a nominal value€1.00 per convertible bond. The May 21, 2003 annual general meeting of GPC Biotech approved the creation of conditional capitals in the amount of up to€200,000, to cover the convertible bonds to be issued pursuant to the 2003 Convertible Bond Program, and the June 11, 2002 annual general meeting of GPC Biotech approved the creation of a conditional capital in the amount of up to€500,000 to cover the convertible bonds to be issued pursuant to the 2002 Convertible Bond Program, in each case subject to adjustment upon changes to our capitalization. The Management Board, together with the Supervisory Board, and, to the extent that our Management Board is concerned, the Supervisory Board alone, determines whether the ordinary shares underlying the convertible bonds will be made available from these conditional capitals or from a program to repurchase our ordinary shares. As of June 4, 2004, there were 700,000 ordinary shares subject to outstanding convertible bonds under the programs and no additional grants will be made under either of these programs. As of June 4, 2004, no holder of convertible bonds has exercised his or her conversion rights.
The Management Board, and to the extent that our Management Board is concerned, the Supervisory Board, selects the eligible persons to whom convertible bonds will be offered and determines the amounts, vesting periods and other relevant terms (including acceleration provisions) of the convertible bonds.
Convertible bonds issued pursuant to the 2003 Convertible Bond Program and the 2002 Convertible Bond Program may be exercised by the bondholder, after expiry of the two-year waiting period from the date of issuance, to obtain one ordinary share for each convertible bond upon payment of the conversion price and bear interest at 3.5 percent per annum starting from the date of issuance. The convertible bonds may be offered to the eligible persons for subscription within the last 15 working days of each calendar month. The time to maturity of the convertible bonds is ten years from the date of issuance, after which the convertible bonds lapse without compensation to the bondholder (other than repayment of the nominal amount of the convertible bond plus accrued interest). Convertible bonds are generally not transferable during the life of the bondholder, except to a credit institution specified by GPC Biotech following the expiration of the two-year waiting period or of the cancellation period described below.
Under the 2003 Convertible Bond Program and the 2002 Convertible Bond Program, ordinary shares issued upon exercise of a convertible bond—provided that they are issued before the beginning of our annual general meeting that resolves on the allocation of retained earnings—are entitled to dividends from the beginning of the previous fiscal year. In the event that the ordinary shares are issued after our general meeting, such ordinary shares are entitled to dividends in the fiscal year in which they were issued.
Convertible bonds issued under the 2003 Convertible Bond Program and the 2002 Convertible Bond Program generally vest and become uncancellable in one-quarter installments on each of the
120
Table of Contents
first four anniversaries of the date of issuance. A convertible bond may not be exercised until the occurrence of each of the following events: (i) satisfaction of a two-year waiting requirement (measured from the date of issuance); (ii) vesting; and (iii) the achievement of a share price performance goal on the date of exercise. With respect to convertible bonds issued pursuant to the 2003 Convertible Bond Program, the price of our ordinary shares on the Frankfurt Stock Exchange on the date of exercise exceeds the share price on May 21, 2003 by 100 percent. With respect to convertible bonds issued pursuant to the 2002 Convertible Bond Program, the price of our ordinary shares on the Frankfurt Stock Exchange must exceed the exercise price of the convertible bonds on five successive trading days within one month prior to exercise by a certain percentage (which percentage is ten percent for the first year after the expiration of the two-year waiting period and is increased by five percent for each year thereafter). In addition, pursuant to the 2003 Convertible Bond Program and the 2002 Convertible Bond Program, a convertible bond may only be exercised within a period of six weeks after publication of our quarterly reports or annual financial statement, and may not be exercised during a subscription rights offering by GPC Biotech and during the period from December 24 to December 31 of each calendar year. Notwithstanding the foregoing, convertible bonds may not be exercised following the expiration of their respective terms, which under the plans cannot exceed ten years from the date of grant. The convertible bonds are generally not transferable during the life of the bondholder, but are inheritable upon the death of the bondholder.
In the event of the termination of service of a bondholder, the unvested portion of a convertible bond may be cancelled without compensation (other than repayment of the nominal amount of the convertible bond plus accrued interest), and the vested portion is generally cancelled 12 months after the termination of service. In addition, GPC Biotech has the right to cancel a bondholder’s convertible bonds in the event insolvency proceedings are instituted against the assets of such bondholder or the convertible bonds are attached by a creditor of such bondholder for more than six months. A bondholder may cancel his or her convertible bonds with three months’ notice at the end of a quarter.
In the event of our merger with and into another company, our reorganization, a change in the nominal value of our ordinary bearer shares or similar changes to our capitalization or corporate structure, the convertible bonds will be replaced by the right to purchase, at the base price, an equivalent number, respectively, of ordinary shares, equity interests or other interests in GPC Biotech or the successor corporation, the value of which corresponds to the market value of the ordinary shares on the date on which such event occurred.
Loans Extended and Guarantees Provided
Under the provisions of the US Sarbanes-Oxley Act enacted in July 2002, no new loans or guarantees may be extended or provided to the members of our Management Board or the members of the Supervisory Board. In December 1998 and January and September 1999, we made loans in an aggregate amount of approximately€86,000 to Dr. Seizinger, our Chief Executive Officer. Each of these loans has a five-year term and bears interest at the rate of 6 percent per annum. For so long as Dr. Seizinger serves as the Chairman of our Management Board, principal and accumulated interest of the loans are forgiven in equal installments over the term of the respective loan. See “—Compensation of Management Board and Supervisory Board Members—Compensation of Members of the Management Board—2003 Summary Compensation Table.” As of December 31, 2003 and January 31, 2004, respectively, the loans granted to Dr. Seizinger in December 1998 and January 1999 had been forgiven in full. On March 15, 2004, Dr. Seizinger made a payment to the Company in the amount of€6,783.21 in full repayment of all principal and accrued interest outstanding under the loan granted to Dr. Seizinger in September 1999.
Furthermore, in July 2001, our U.S. subsidiary made a loan in the aggregate amount of $200,000 to Dr. Kley, our Vice President Research Waltham. The loan terms were amended in July 2003. The amended loan has a four-year term and bears interest at the rate of 5.5 percent per annum.
121
Table of Contents
For so long as Dr. Kley remains employed by our U.S. subsidiary, principal and accumulated interest of the loan is forgiven in equal installments over the term of the loan. The loan agreement additionally provides that the loan will be forgiven in full in specific circumstances following a change in control of GPC Biotech.
In July 2002, we made a loan in the aggregate amount of€100,000 to Dr. Scherer, our Chief Financial Officer. This loan had a five-year term and bore interest at the rate of 6 percent per annum. Following partial repayment of this loan in fiscal year 2003, Dr. Scherer repaid the remaining principal and accrued interest of this loan in full on March 4, 2004.
All of these loans were made for personal financial purposes.
The aggregate outstanding balance of the loans, including accrued interest, to Dr. Seizinger and Dr. Scherer as of December 31, 2003 was approximately€79,000. Dr. Seizinger and Dr. Scherer have repaid this balance in full as of March 31, 2004. The aggregate outstanding balance of the loan, including accrued interest, to Dr. Kley as of December 31, 2003 was approximately $205,500.
Share Ownership by Members of Our Supervisory Board and Management Board
Supervisory Board
The following table provides information with respect to ownership of our ordinary bearer shares, options and convertible bonds for each of our members of the Supervisory Board as of June 4, 2004, based on an aggregate of 21,456,140 shares outstanding.
Name | Shares | % of Outstanding Shares | Options or | Exercise Price (€) | Expiration Date | |||||
Jürgen Drews, M.D., Ph.D. | — | — | 12,500 convertible bonds | 9.55 | 10/9/2006 | |||||
| 12,500 convertible bonds | 5.77 | 5/30/2008 | ||||||||
| 14,400 options | 1.16 | 12/31/2008 | ||||||||
| 14,400 options | 1.16 | 01/04/2009 | ||||||||
| 10,000 options | 17.89 | 05/04/2010 | ||||||||
Michael Lytton, J.D. | — | — | 7,500 convertible bonds | 9.55 | 10/9/2006 | |||||
| 31,500 convertible bonds | 5.77 | 5/30/2008 | ||||||||
| 10,000 options | 16.38 | 03/13/2011 | ||||||||
Metin Colpan, Ph.D. | 14,400 | * | 5,000 convertible bonds | 9.55 | 10/9/2006 | |||||
| 10,000 convertible bonds | 5.77 | 05/30/2008 | ||||||||
| 10,000 options | 17.89 | 05/4/2010 | ||||||||
Prabhavathi B. Fernandes, Ph.D. | — | — | 10,000 convertible bonds | 5.77 | 5/30/2008 | |||||
Peter Preuss. | 80,000 | * | 7,500 convertible bonds | 9.55 | 10/9/2006 | |||||
| 22,500 convertible bonds | 5.77 | 5/30/2008 | ||||||||
Total | 94,400 | * | 119,000 convertible bonds | |||||||
| 58,800 options |
| * | Less than one percent of class. |
122
Table of Contents
Management Board
The following table provides information with respect to ownership of our ordinary bearer shares, options and convertible bonds for each of our members of the Management Board as of June 4, 2004, based on an aggregate of 21,456,140 shares outstanding.
Name | Shares | % of Outstanding Shares | Options or Convertible Bonds(1) | Exercise Price (€) | Expiration Date | |||||
Bernd R. Seizinger, M.D., Ph.D.(2) | — | — | 148,480 options | 1.16 | 11/11/2008 | |||||
| 49,490 options | 1.16 | 1/08/2009 | ||||||||
| 111,360 options | 3.98 | 9/30/2009 | ||||||||
| 99,000 options | 6.62 | 3/31/2010 | ||||||||
| 300,000 options | 17.89 | 5/04/2010 | ||||||||
| 90,000 options | 9.69 | 8/31/2011 | ||||||||
| 125,000 convertible bonds | 4.36 | 8/31/2012 | ||||||||
| 18,925 convertible bonds | 4.87 | 5/20/2013 | ||||||||
| 56,075 convertible bonds | 4.96 | 5/21/2013 | ||||||||
Elmar Maier, Ph.D.(3) | 416,000 | 1.94 | 69,000 options | 6.62 | 3/31/2010 | |||||
| 70,000 options | 17.89 | 5/04/2010 | ||||||||
| 50,000 options | 9.69 | 8/31/2011 | ||||||||
| 50,000 options | 5.24 | 7/31/2012 | ||||||||
| 65,000 convertible bonds | 4.36 | 8/31/2012 | ||||||||
| 9,084 convertible bonds | 4.87 | 5/20/2013 | ||||||||
| 26,916 convertible bonds | 4.96 | 5/21/2013 | ||||||||
Sebastian Meier-Ewert, Ph.D. . | 483,200 | 2.25 | 69,000 options | 6.62 | 3/31/2010 | |||||
| 70,000 options | 17.89 | 5/04/2010 | ||||||||
| 60,000 options | 9.69 | 8/31/2011 | ||||||||
| 50,000 options | 5.24 | 7/31/2012 | ||||||||
| 85,000 convertible bonds | 2.70 | 1/31/2013 | ||||||||
| 34,019 convertible bonds | 4.96 | 5/21/2013 | ||||||||
| 11,481 convertible bonds | 4.87 | 5/20/2013 | ||||||||
Mirko Scherer, Ph.D(4). | 24,000 | * | 69,000 options | 6.62 | 3/31/2010 | |||||
| 90,000 options | 17.89 | 5/04/2010 | ||||||||
| 50,000 options | 13.34 | 3/30/2011 | ||||||||
| 50,000 options | 9.69 | 8/31/2011 | ||||||||
| 25,000 convertible bonds | 4.36 | 8/31/2012 | ||||||||
| 40,000 convertible bonds | 2.70 | 1/31/2013 | ||||||||
| 9,084 convertible bonds | 4.87 | 5/20/2013 | ||||||||
| 26,916 convertible bonds | 4.96 | 5/21/2013 | ||||||||
Total | 923,200 | 4.30 | 1,545,330 options | |||||||
| 532,500 convertible bonds |
| * | Less than one percent of class. |
| (1) | Numbers exclude grants of 300,000, 50,000, 50,000 and 50,000 stock options, which our Supervisory Board has approved to Dr. Seizinger, Dr. Scherer, Dr. Meier-Ewert and Dr. Maier, respectively, subject only to creation of new conditional capital underlying these grants at our 2004 annual general meeting. |
| (2) | Dr. Seizinger transferred a total of 705,280 stock options initially granted to him in 1998 and 1999 to a financial institution in connection with cash settled share option agreements dated July and December 2001, respectively. Upon any exercise by him of the options granted in the agreement of July 2001, Dr. Seizinger will receive cash, not shares, from the financial institution. On any exercise by him of the options in the agreement of December 2001, Dr. Seizinger will, provided that he has paid to the financial institution the amounts required by the agreement and that certain other conditions are satisfied, be entitled to receive cash, not shares, from the financial institution. Of the 705,280 options, a total of 395,950 were exercised by the financial institution in July 2001 (see also “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”). |
| (3) | Dr. Maier transferred 12,000 stock options not included in the amount listed in this table to family members. Additionally, the amount of ordinary shares held by Dr. Maier listed in this table does not include 66,266 ordinary shares held by family members. |
| (4) | In July 2001, Dr. Scherer transferred 96,000 ordinary bearer shares to a financial institution in connection with a cash settled share option agreement (see also “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”). On any exercise of his option under this agreement, Dr. Scherer will receive cash, not shares, from the financial institution. This table does not include 3,770 shares held by family members of Dr. Scherer. |
As of June 4, 2004, the members of our Management Board held an aggregate of 923,200 shares of GPC Biotech, while the members of our Supervisory Board held an aggregate of 94,400 shares. The aggregate amount of our shares owned by current Management Board and Supervisory
123
Table of Contents
Board members amounts to approximately 4.74% of our outstanding share capital. Of this amount, Dr. Elmar Maier held 416,000 shares, amounting to 1.94% of our share capital, Dr. Sebastian Meier-Ewert held 483,200 shares, amounting to 2.25% of our share capital, Dr. Mirko Scherer held 24,000 shares, amounting to 0.11% of our share capital, Dr. Metin Colpan held 14,400 shares, amounting to 0.07% of our share capital and Peter Preuss held 80,000 shares, amounting to 0.37% of our share capital.
Employees
As of December 31, 2003, we had a total of 181 employees. The following tables show the breakdown of the total year-end and average numbers of our workforce by main category of activity and geographic area for the past three years.
As of December 31, 2003 | Research & Development | General & Administration | Total | |||
Germany | 64.5 | 21.5 | 86 | |||
United States | 71 | 24 | 95 | |||
As of December 31, 2002 | ||||||
Germany | 74 | 25 | 99 | |||
United States | 83 | 26 | 109 | |||
As of December 31, 2001 | ||||||
Germany | 68 | 27 | 95 | |||
United States | 63 | 20 | 83 | |||
Average during the year ended December 31, 2003 | ||||||
Germany | 67.25 | 22 | 89.75 | |||
United States | 83 | 25.83 | 108.83 | |||
Average during the year ended December 31, 2002 | ||||||
Germany | 72.42 | 23.42 | 95.82 | |||
United States | 71.58 | 24.92 | 96.50 | |||
Average during the year ended December 31, 2001 | ||||||
Germany | 67.08 | 24.33 | 91.42 | |||
United States | 55.33 | 18.58 | 73.92 | |||
We consider the developments in the numbers of employees in our various departments and geographic regions to be in line with our expectations for growth. We have undergone a reduction in our workforce in October 2003. This involved a reduction in research capabilities not in line with our increased focus on oncology drug discovery and development (areas in which the staff has expanded in the past year). Employees were affected primarily in Munich and Waltham. The reductions in numbers are as follows:
October 2003 | US | Munich | Total | |||
Research | 18 | 20 | 38 | |||
G&A | 2 | 2 | 4 | |||
Total | 20 | 22 | 42 | |||
None of our employees work under any collective bargaining agreements and we have no relations with any labor unions.
124
Table of Contents
Corporate Governance
The Management Board and the Supervisory Board adopted the company’s current corporate governance principles on September 9, 2003. Our principles are based on the German Corporate Governance Code and supplement the obligations of the Management Board and the Supervisory Board prescribed by law, in our Articles of Association and in the internal rules of procedure. The principles are intended to ensure that GPC Biotech is managed and monitored in a responsible and value-driven manner. They include the protection of shareholders’ rights, comprehensive disclosure and transparency requirements and rules governing conflicts of interest. We are committed to continue to adapt our corporate governance principles to best-practice developments.
Our compliance declaration dated September 9, 2003 states the following:
“GPC Biotech complied in the past with the recommendations of the “Commission German Corporate Governance Code” as of November 7, 2002 and will comply with the recommendations of the “Commission German Corporate Governance Code” as of May 21, 2003.”
Upon listing, we expect to be in compliance with the corporate governance requirements of the Nasdaq and applicable U.S. law with the following exceptions for which we have been granted waivers by the Nasdaq National Market and in which we continue to apply the practices of our home country, Germany:
| • | The exact terms and conditions of our equity compensation plans are not the subject of a shareholder vote at the annual general meeting. Instead, our shareholders only vote on the creation of underlying capital to service the equity awards and certain other general terms prescribed by German law and authorize the Management Board or, in respect of awards to be made to the members of our Management Board, the Supervisory Board, to determine the details of our equity compensation plans. It is intended that all such plans be established within the policies and programs approved by the Compensation Committee. |
| • | Neither German law nor the rules and regulations promulgated by the Frankfurt Stock Exchange, the primary market for our shares, require a specific quorum for annual general meetings; therefore, our Articles of Incorporation do not provide for a quorum. |
Our shareholders appoint our external auditors at our annual general meeting as required by German law. This practice is not in violation of the corporate governance requirements of the Nasdaq and applicable U.S. law.
125
Table of Contents
PRINCIPAL AND SELLING SHAREHOLDERS AND EXERCISE OF CASH SETTLED STOCK OPTIONS
The following tables set forth certain information regarding the ownership of our share capital as of June 4, 2004, of our principal shareholders, the shares to be sold by certain selling shareholders and as adjusted to reflect the issuance of 8,279,000 shares in the combined offering (assuming that all new shares are sold in the combined offering and that the underwriters exercise their over-allotment option in full).
Ownership of Stock and Voting Rights
Prior to the combined offering, a total 21,456,140 shares were outstanding, with 21,456,140 voting rights. Ownership of the stock and voting rights was as follows, according to the information available to us on the basis of notifications according to the German Securities Trading Act (WpHG). See also “Information about the share capital of GPC Biotech AG—Notification and Disclosure Obligations”.
| June 4, 2004 | After the offering(1) | |||||||||||
Shareholder | Number of shares | % of share capital | % of voting rights | Number of shares | % of share capital | % of voting rights | ||||||
Management Board and Supervisory Board as a group | 1,017,600 | 4.74 | 4.74 | 717,600 | 2.41 | 2.41 | ||||||
ALTANA Technology Projects GmbH(2) | 1,690,000 | 7.88 | 7.88 | 2,253,333 | 7.58 | 7.58 | ||||||
ROI Verwaltungsgesellschaft mbH(3) | not available | at least 5.0 | at least 5.0 | not available | not available | not available | ||||||
| (1) | Assuming that the underwriters fully exercise their option to purchase 1,119,000 additional shares for the purpose of covering over-allotments. See “Plan of Distribution”. |
| (2) | Assuming that ALTANA Technology Projects GmbH fully exercises its subscription rights. The voting rights of ALTANA Technology Projects GmbH are attributed to ALTANA Pharma AG and Susanne Klatten. |
| (3) | On February 19, 2004, ROI Verwaltungsgesellschaft mbH notified us that it owned 5% of our share capital, or 1,066,712 ordinary shares. Since then, we have not received any notice that ROI Verwaltungsgesellschaft mbH owns less than 5% of our share capital. The voting rights of ROI Verwaltungsgesellschaft mbH are attributed to Roland Oetker. |
To the knowledge of our management, no other shareholder owns directly or indirectly more than five percent of our equity or voting rights. Under German law, shareholders in a public company are required to notify the company and the German Federal Financial Supervisory Authority(Bundesanstalt für Finanzdienstleistungsaufsicht) of the number of shares they own when their percentage ownership reaches, exceeds or falls below certain threshold levels. German law does not require a shareholder to update this information unless it again reaches, exceeds or falls below a notification threshold. As a result, we cannot be certain whether the number of shares owned by the shareholders listed above is accurate. See “Description of Share Capital—Notification and Disclosure Obligations.”
126
Table of Contents
Selling Shareholders
The following table sets forth information concerning the ownership of our shares by each of the selling shareholders immediately before and after the completion of the combined offering and indicates the amounts to be offered by each in the combined offering.
| June 4, 2004 | Shares to be sold in this offering | After the offering(2) | ||||||||||
Shareholder | Number of Shares | % of share capital | Number of Shares | % of share capital(1) | Number of Shares | % of share capital | ||||||
Sebastian Meier-Ewert, Ph.D. | 483,200 | 2.25 | 150,000 | 0.70 | 333,200 | 1.12 | ||||||
Elmar Maier, Ph.D. | 416,000 | 1.94 | 150,000 | 0.70 | 266,000 | 0.89 | ||||||
| (1) | These percentages were calculated based on the number of shares outstanding prior to the combined offering. |
| (2) | Assuming that the underwriters fully exercise their option to purchase 1,119,000 additional shares for the purpose of covering over-allotments. |
For more details about the global offering and the conditions under which the selling shareholders are offering these shares, see “The Combined Offering—The Global Offering” and “Plan of Distribution”.
Exercise of Cash Settled Stock Options
Dr. Seizinger transferred a total of 705,280 stock options initially granted to him in 1998 and 1999 to a financial institution in connection with cash settled share option agreements dated July and December 2001, respectively. The exercise of the options under the option agreement dated December 2001 is subject to several conditions, in particular to the payment of specified amounts to the financial institution in accordance with the option agreement. In July 2001, Dr. Scherer has also transferred 96,000 ordinary bearer shares to a financial institution in connection with a cash settled share option agreement. Upon any exercise of the options granted in the agreements, Dr. Seizinger and Dr. Scherer will receive cash based on the current share price of the Company’s shares, but not shares, from the financial institution. See also “Directors, Senior Management and Employees—Share Ownership by Members of Our Supervisory Board and Management Board”.
Dr. Seizinger and Dr. Scherer will agree with the underwriters not to sell any shares or enter into any transaction with an equivalent economic result before December 27, 2004 without the prior written consent of the Global Coordinator (see also “Plan of Distribution”). This includes the exercise of the above mentioned cash settled stock options, except for 120,000 cash settled stock options of Dr. Seizinger and 30,000 cash settled stock options of Dr. Scherer that Dr. Seizinger and Dr. Scherer will exercise in accordance with the above mentioned option agreements with the financial institution before the closing of the Combined Offering, but after publication of the initial offer price. Dr. Seizinger and Dr. Scherer will determine the timing of the exercise in coordination with Goldman, Sachs & Co. oHG.
It is possible that, in connection with the exercise of these cash settled stock options, the financial institution will sell shares of the Company in the market. Neither the Company nor the underwriters nor Dr. Seizinger or Dr. Scherer have any influence on the decisions of the financial institution. Such sale of shares by the financial institution in the market could have a negative impact on the Company’s share price.
127
Table of Contents
Changes in Principal Shareholders During the Past Three Fiscal Years
On November 16, 2001, ALTANA Technology Projects GmbH notified GPC Biotech that its shareholdings amounted to 8.3% of GPC Biotech’s voting shares. The shareholdings are attributed to ALTANA Pharma AG and Susanne Klatten.
On April 1, 2002, BB Bioventures LP notified GPC Biotech that its shareholdings amounted to 12.16% of GPC Biotech’s voting shares.
On May 8, 2003, BB Bioventures LP notified GPC Biotech that its shareholdings amounted to 9.91% of GPC Biotech’s voting shares.
On May 27, 2003, BB Bioventures LP notified GPC Biotech that its shareholdings amounted to 4.81% of GPC Biotech’s voting shares.
On February 19, 2004, ROI Verwaltungsgesellschaft mbH notified GPC Biotech that its shareholdings amounted to 5.00% of GPC Biotech’s voting shares. The shareholdings are attributed to Roland Oetker.
128
Table of Contents
We have, from time to time, entered into agreements with our shareholders and affiliates. We describe the principal transactions entered into since 2001 below.
In November 2001, we formed a strategic alliance with ALTANA Pharma AG in connection with the establishment by ALTANA Pharma of the ALTANA Research Institute, ALTANA Pharma’s U.S. research operation based in Waltham, Massachusetts. This alliance encompasses a technology transfer component as well as research collaboration. Pursuant to the agreement with ALTANA Pharma, we are collaborating with ALTANA Pharma in the creation of the ALTANA Research Institute through the transfer of certain of our genomic and proteomic technologies to the ALTANA Research Institute. Effective January 2003, we entered into another collaboration agreement with ALTANA Pharma pursuant to which we licensed LeadCode to ALTANA Pharma. We are entitled to receive a total of $60 million in payments from ALTANA Pharma, including upfront payments, license and technology transfer fees, and research funding under these agreements. Under each of these agreements, ALTANA Pharma is also obligated to make certain milestone payments totaling approximately $15 million (subject to reduction in certain circumstances) to us in connection with the development and commercialization of each product resulting from the research collaborations. As of December 31, 2003, we had received€37.4 million under these agreements, of which€27.4 million have been recognized as revenue. Of the€37.4 million received as of December 31, 2003,€35.7 million comprised payments under the collaboration and license agreement of November 2001 and€1.7 million comprised payments under the collaboration and license agreement of January 2003 related to LeadCode.
ALTANA Pharma is our largest customer, accounting for 94% of our total revenues in 2003, 81% of our total revenues in 2002, and 58% of our total revenues in 2001. Our collaboration agreement with ALTANA Pharma terminates in June 2007. As of June 4, 2004, an affiliate of ALTANA Pharma owned 7.88% of our outstanding share capital.
In addition, Dr. Seizinger, our President and Chief Executive Officer, is a member of the Supervisory Board of ALTANA Pharma AG, which is a subsidiary of ALTANA AG.
Professor Drews, the Chairman of our Supervisory Board, is also the Vice Chairman of the supervisory board of MorphoSys AG. Additionally, Dr. Metin Colpan, a member of our Supervisory Board, is also a member of the supervisory board of MorphoSys AG. In April 1999, we entered into a collaboration and license agreement with MorphoSys. Since the initiation of the collaboration, MorphoSys has identified more than a dozen antibodies, from which we selected our product candidate known as 1D09C3. MorphoSys is entitled to milestone payments upon the occurrence of specified events in the development and commercialization of antibody product candidates resulting from the collaboration. If all milestones were achieved, these milestone payments would total approximately€10 million. MorphoSys is also entitled to receive royalties on net sales of any of these antibody products by us. If we enter into an agreement with a partner for the commercialization of any of these antibody products, we would be obligated to pay MorphoSys a percentage of sublicense fees, milestone payments and royalties.
Mr. Preuss, a member of our Supervisory Board, is also a Regent of the University of California. In February, 2004, we entered into a research agreement with The Regents of the University of California as sponsor of a research project entitled“Characterization of JM-118 Resistance” (the “Project”). We support the Project by a grant of $100,005.
129
Table of Contents
The following description is a summary of certain information relating to our share capital as well as certain provisions of our Articles of Association and German stock corporation law. This summary does not purport to be complete and speaks as of the date of this prospectus.
Objects and Purposes
As described in Section 2 of our Articles of Association, our business purpose is the research, development and application of technologies and procedures to study biological material, the functional analysis of identified gene products and their commercial use to develop diagnostics and pharmaceutical products, the rendering of services related thereto and the granting of licenses as well as the manufacturing and sale of pharmaceuticals. We may engage in all activities that are suitable to directly or indirectly promote our business purpose. We may establish branch offices and/or subsidiaries both in Germany and abroad and may acquire shareholdings in other companies.
Share Capital
Our share capital amounts to€21,456,140 and is divided into 21,456,140 no par-value ordinary bearer shares (auf den Inhaber lautende Stückaktien ohne Nennbetrag), each with a notional nominal value (the proportional amount of the share capital attributable to each share) of€1.
Form, Certification and Transferability of the Shares
The form and contents of our share certificates, dividend and renewal coupons are determined by our Management Board with the consent of our Supervisory Board. We may combine individual shares into share certificates that represent multiple shares (global shares, global certificates). Shareholders have no right to receive individual share certificates and all shares are represented by one or more global bearer share certificate.
All of our outstanding shares are bearer shares. If a resolution regarding a capital increase does not specify whether such increase will be in bearer or registered form, the new shares resulting from such capital increase will be bearer shares by default. Any resolution regarding a capital increase may determine the profit participation of the new shares resulting from such capital increase in deviation from § 60(2)(3) of the German Stock Corporation Act.
Our shares are freely transferable, with the transfer of ownership governed by the rules of the relevant clearing system.
General Information on Capital Measures
An increase of our share capital requires a resolution passed at our shareholders’ meeting with a majority of three quarters of the share capital, represented at the relevant shareholders’ meeting. Moreover, the shareholders at such meeting may authorize our Management Board to increase our share capital with the consent of our Supervisory Board, within a period of five years by issuing shares for a certain total amount (genehmigtes Kapitalor “authorized capital”). Finally, our shareholders may resolve to create conditional capital; however, they may do so only to issue conversion or subscription rights to holders of convertible bonds, in preparation for a merger with another company or to issue subscription rights to employees and members of the management of our company or of an affiliated company by way of a consent or authorization resolution (bedingtes Kapitalor “conditional capital”). Any resolution pertaining to the creation of authorized or conditional capital requires a majority of three quarters of the share capital represented at the relevant shareholders’ meeting.
130
Table of Contents
The aggregate nominal amount of the authorized capital created by the shareholders may not exceed one half of the share capital existing at the time of registration of the authorized capital with the commercial register.
The aggregate nominal amount of the conditional capital created by the shareholders may not exceed one half of the share capital existing at the time of the annual general meeting adopting such resolution. The aggregate nominal amount of the conditional capital created for the purpose of granting subscription rights to employees and members of the management of our company or of an affiliated company may not exceed ten percent of the share capital existing at the time of the annual general meeting adopting the resolution.
Any resolution relating to a reduction of our share capital requires a majority of at least three quarters of the share capital represented at the relevant shareholders’ meeting.
Changes in Our Share Capital During the Last Three Fiscal Years
As of January 1, 2001, our share capital as registered with the commercial register amounted to€17,667,274. An additional 104,620 shares were issued in 2000 out of conditional capital but registered on March 12, 2001 (in case of a conditional capital increase, registration with the commercial register only has a notational function, while the shares come into existence, and the share capital is increased, upon their issuance). Since January 1, 2001, our share capital has changed as follows (the dates listed refer to the dates of registration of the capital measure with the commercial register):
| • | increase out of authorized capital from€17,667,274 by€26,820 to€17,694,094, registered on January 15, 2001; |
| • | registration on March 12, 2001 of the increase out of conditional capital during 2000 by 104,620 shares, from€17,694,094 by€104,620 to€17,798,714; |
| • | increase out of authorized capital from€17,798,714 by€97,614 to€17,896,328, registered on April 25, 2001; |
| • | increase out of authorized capital from€17,896,328 by€17,072 to€17,913,400, registered on July 25, 2001; |
| • | increase out of authorized capital from€17,913,400 by€52,486 to€17,965,886, and by a further€1,690,000 to€19,655,886, registered on November 13, 2001; |
| • | registration on March 4, 2002 of the increase out of conditional capital during 2001 from€19,655,886 by€957,440 to€20,613,326; |
| • | increase out of authorized capital from€20,613,326 by€55,108 to€20,668,434, registered on April 16, 2002; |
| • | increase out of authorized capital from€20,668,434 by€21,946 to€20,690,380, registered on September 11, 2002; |
| • | registration on February 4, 2003 of the increase out of conditional capital during 2002 from€20,690,380 by€29,400 to€20,719,780; |
| • | increase out of authorized capital from€20,719,780 by€5,285 to€20,725,065, registered on February 12, 2003; |
| • | increase out of authorized capital from€20,725,065 by€7,090 to€20,732,155, registered on October 30, 2003; |
| • | increase out of authorized capital from€20,732,155 by€54,630 to€20,786,785, registered on January 19, 2004; |
131
Table of Contents
| • | registration on February 9, 2004 of the increase out of conditional capital during 2003 from€20,786,785 by€21,920 to€20,808,705; |
| • | increase out of authorized capital from€20,808,705 by€219,524 to€21,028,229, registered on March 16, 2004; and |
| • | increase out of authorized capital from€21,028,229 by€91,052 to€21,119,281, registered on April 22, 2004. |
The capital increase out of authorized capital in an amount of€1,690,000, registered on November 13, 2001, was subscribed by ALTANA Technology Projects GmbH. All capital increases related to share issuances resulting from the exercise of stock options were covered by either authorized or conditional capital. In January and March 2004, a total of 336,859 conversion rights attached to convertible bonds and stock options covered by conditional capital have been exercised. Thus, our share capital currently amounts to€21,456,140. We have applied for the notional registration of the related capital increases with the commercial register and registration is expected to occur prior to June 28, 2004.
Authorized Capital
Our Articles of Association reflect two shareholders’ resolutions creating authorized capital, passed at the annual general meetings held on May 3, 2000 and June 11, 2002, respectively, which can be summarized as follows (the numbers are shown as adjusted by the Supervisory Board following previous partial uses of such authorized capital):
Authorized Capital I
Our Management Board is authorized to increase our share capital, with the consent of the Supervisory Board, on one or more occasions until April 30, 2005 by an aggregate amount of up to€3,223,229 by issuing up to 3,223,229 new ordinary shares against contributions in cash or in kind. The Management Board is authorized to determine, with the consent of the Supervisory Board, the rights pertaining to the new shares as well as other conditions of their issuance.
Shareholders’ subscription rights are excluded in the event of a partial use of such authorized capital for one of the following purposes:
| • | in an amount of up to€159,560 by issuing up to 159,560 new shares against cash contributions, to the extent necessary to cover stock options issued to Supervisory Board members or consultants pursuant to a shareholder’s resolution passed on May 12, 1999; |
| • | in an amount of up to€567,000 by issuing up to 567,000 new shares against cash contributions, to the extent necessary to cover stock options issued to Management Board members, members of the management of subsidiaries, or employees of our company, pursuant to a shareholder’s resolution passed on May 12, 1999; |
| • | in an amount of up to€1,201,000 by issuing up to 1,201,000 new shares against cash contributions, to the extent necessary to cover stock options issued to Management Board members, members of the management of subsidiaries, Supervisory Board members, consultants, or employees of our company pursuant to a shareholder’s resolution passed on May 3, 2000; |
| • | in an amount of up to€277,709 by issuing up to 277,709 new shares against contributions in cash or in kind to holders of shares, options or warrants of the former Mitotix, Inc. who are entitled to receive shares in our company as a result of the merger in March 2000; or |
| • | to the extent the capital increase excluding shareholders’ subscription rights does not exceed ten percent of our share capital and the issue price of the new shares is not significantly lower than the exchange trading price. |
Furthermore, the Management Board may exclude shareholders’ subscription rights with the consent of the Supervisory Board
| • | for fractional amounts; or |
132
Table of Contents
| • | to the extent the new shares are issued in a capital increase against contribution in kind for the transformation or acquisition of companies or parts of companies. |
Authorized Capital II
Our Management Board is further authorized to increase our share capital, with the consent of the Supervisory Board, on one or more occasions until June 1, 2007 by an aggregate amount of up to€6,708,000 by issuing up to 6,708,000 new ordinary shares against contributions in cash or in kind. The Management Board is authorized to determine, with the consent of the Supervisory Board, the rights pertaining to the new shares as well as other conditions of their issuance.
The Management Board may, with the consent of the Supervisory Board, exclude shareholders’ subscription rights on one or more occasions
| • | if the new shares are issued against contributions in kind; |
| • | for fractional amounts; or |
| • | in the event of a capital increase against cash contributions, if the amount of the capital increase does not exceed ten percent of the share capital existing at the time of effectiveness of this authorization and the issue price is not significantly lower than the exchange trading price. New shares or bonds issued by excluding shareholders’ subscription rights in accordance with § 186(3)4 of the German Stock Corporation Act pursuant to § 5(5) of our Articles of Association or pursuant to an authorization granted by the shareholders in their meeting held on June 11, 2002 shall count towards this ten percent threshold. |
Conditional Capital
Our Articles of Association reflect shareholders’ resolutions creating ten different conditional capitals which can be summarized as follows (the numbers show the remaining amounts reflecting previous share issuances out of the respective conditional capital):
Conditional Capital I
Our share capital is conditionally increased by up to€454,720, divided into up to 454,720 ordinary shares, to cover stock options issued pursuant to a general authorization by the shareholders passed at the annual general meeting held on December 8, 1997. The conditional capital increase will only be implemented to the extent that the bondholders exercise their conversion rights. The new shares are entitled to profit participation as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights.
Conditional Capital II
Our share capital is conditionally increased by up to€306,000, divided into up to 306,000 ordinary shares, to cover convertible bonds issued pursuant to an authorization by the shareholders passed at the annual general meeting held on December 8, 1997. The conditional capital increase will only be implemented to the extent the bondholders exercise their conversion rights. The new shares are entitled to profit participation as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights. In January 2004, 306,000 conversion rights attaching to convertible bonds covered by this conditional capital have been exercised. Thus, this conditional capital has been depleted and will be cancelled with the commercial register upon (notional) registration of the capital increase, which is expected to happen prior to June 28, 2004.
133
Table of Contents
Conditional Capital III
Our share capital is conditionally increased by up to€49,000, divided into up to 49,000 ordinary registered shares, to cover stock options granted pursuant to a shareholders’ resolution passed at the annual general meeting held on May 3, 2000. The conditional capital increase will only be implemented to the extent such stock options are actually issued and the option holders exercise their option rights. The new shares are entitled to profit participation as from the beginning of the fiscal year in which they are issued. The Management Board is authorized to determine, with the consent of the Supervisory Board, further details of the implementation of the conditional capital increase.
Conditional Capital IV
Our share capital is conditionally increased by up to€690,541, divided into up to 690,541 ordinary shares, to cover stock options granted to members of our Management Board, members of the management of affiliated companies and employees of our company or of affiliated companies pursuant to a shareholders’ resolution passed at the annual general meeting held on June 5, 2001. The conditional capital increase will only be implemented to the extent the option holders exercise their option rights. The new shares are entitled to profit participation as from the beginning of the fiscal year in which they are issued by the exercise of option rights. The Management Board, or the Supervisory Board to the extent the Management Board is concerned, is authorized to determine the further conditions of the implementation of the conditional capital increase.
Conditional Capital V
Our share capital is conditionally increased by up to€42,500, divided into up to 42,500 ordinary shares, to cover convertible bonds issued to Supervisory Board members pursuant to a shareholders’ resolution passed at the annual general meeting held on June 5, 2001. The new shares are entitled to profit participation as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights.
Conditional Capital VI
Our share capital is conditionally increased by up to€810,000, divided into up to 810,000 ordinary shares, to cover stock options granted to members of the Management Board, employees of our company or employees of affiliated companies, in each case in Germany or abroad, pursuant to a shareholders’ resolution passed at the annual general meeting held on June 11, 2002. The conditional capital increase will only be implemented to the extent the option holders exercise their option rights. The new shares are entitled to profit participation as from the beginning of the previous fiscal year if they are issued by the exercise of option rights prior to the annual general meeting, otherwise as from the beginning of the fiscal year in which they are issued by the exercise of option rights. The Management Board, or the Supervisory Board to the extent the Management Board is concerned, is authorized to determine the further conditions of the implementation of the conditional capital increase.
Conditional Capital VII
Our share capital is conditionally increased by up to€500,000, divided into up to 500,000 ordinary shares, to cover conversion rights granted to members of our Management Board, to members of the management bodies and other employees with management functions of our company and our subsidiaries, in each case in Germany or abroad, or to consultants pursuant to a shareholders’ resolution passed at the annual general meeting held on June 11, 2002. The conditional capital increase will only be implemented to the extent the holders of conversion rights exercise their
134
Table of Contents
conversion rights. The new shares are entitled to profit participation as from the beginning of the previous fiscal year if they are issued by the exercise of conversion rights prior to the annual general meeting, otherwise as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights. The Management Board, or the Supervisory Board to the extent the Management Board is concerned, is authorized to determine the further conditions of the implementation of the conditional capital increase.
Conditional Capital VIII
Our share capital is conditionally increased by up to€5,000,000, divided into up to 5,000,000 ordinary shares. The conditional capital increase will only be implemented to the extent the holders or creditors of conversion or exercise rights attached to convertible bonds or bonds with warrants attached issued by us pursuant to an authorization granted by the shareholders at the annual general meeting held on June 11, 2002 exercise their conversion or option rights. The new shares are entitled to profit participation as from the beginning of the previous fiscal year if they are created by the exercise of conversion or option rights prior to the annual general meeting, otherwise as from the beginning of the fiscal year in which they are issued by the exercise of conversion or option rights. The Management Board is authorized to determine the further conditions of the implementation of the conditional capital increase.
Conditional Capital IX
Our share capital is conditionally increased by up to€121,500, divided into up to 121,500 ordinary shares, to cover conversion rights granted to Supervisory Board members pursuant to a shareholders’ resolution passed at the annual general meeting held on May 21, 2003. The new shares are entitled to profit participation as from the beginning of the previous fiscal year if they are issued by the exercise of conversion rights prior to the annual general meeting, otherwise as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights.
Conditional Capital X
Our share capital is conditionally increased by up to€200,000, divided into up to 200,000 ordinary shares, to cover conversion rights granted to members of the Management Board, members of the management bodies or employees with management functions of our company or subsidiaries, in each case in Germany or abroad, or to consultants pursuant to a shareholders’ resolution passed at the annual general meeting held on May 21, 2003. The conditional capital increase will only be implemented to the extent the holders of conversion rights exercise their conversion rights. The new shares are entitled to profit participation as from the beginning of the previous fiscal year if they are issued by the exercise of conversion rights prior to the annual general meeting, otherwise as from the beginning of the fiscal year in which they are issued by the exercise of conversion rights. The Management Board, or the Supervisory Board to the extent the Management Board is concerned, is authorized to determine the further conditions of the implementation of the conditional capital increase.
Authorization to Acquire Our Own Shares
By resolution of the annual general meeting held on May 21, 2003, we are authorized in accordance with § 71(1) No. 8 of the German Stock Corporation Act to acquire our own shares until November 20, 2004, up to an aggregate amount of ten percent of the share capital existing at the time of this annual general meeting. The authorization may be exercised, in whole or in part, once or several times within the scope of these limitations. The total number of shares acquired, together with other treasury shares held by or attributable to us pursuant to §§ 71a et seq. of the German Stock Corporation Act, may not exceed ten percent of the share capital at any time. In the discretion of our
135
Table of Contents
Management Board, shares may be acquired on a stock exchange or by means of a public tender offer to all shareholders. Shares may only be acquired on a stock exchange if the per share consideration (not including ancillary fees) does not exceed or fall below the average share price (final auction price during XETRA trading and/or a comparable successor system) on the five exchange trading days prior to the acquisition by more than ten percent.
If the shares are acquired by way of a public offering, the per share consideration (not including ancillary fees) may not exceed or fall below the average share price (final auction price during XETRA trading and/or a comparable successor system) on the five exchange trading days prior to the publication of the offer by more than ten percent. If after the publication of the public offer, material deviations in the relevant price occur, the offer may be adjusted accordingly. In this case, the price will be based on the average price on the last five exchange trading days prior to the public announcement of any such adjustment.
If the offer and/or invitation to tender shares is oversubscribed, the acceptance will take place on a pro rata basis. Preferential acceptance may be provided for small lots of up to 150 shares per shareholder. The public offer can stipulate further conditions. Public offers must comply with the requirements of the German Securities Acquisition and Takeover Act (Wertpapiererwerbs- und Übernahmegesetz) if and to the extent applicable.
The Management Board, with the consent of the Supervisory Board, is authorized to sell the company’s shares so acquired also by other means than over the exchange or by way of an offer to all shareholders, if the shares are sold at a cash price that is not significantly less than the market price of shares of our company that carry the same rights at the time of the sale. In this case, the shares so to be sold together with any shares or bonds issued by excluding shareholders’ subscription rights pursuant to § 5(5) and (6) of our Articles of Association pursuant to the authorization by the shareholders granted at the annual general meeting held on June 11, 2002 are applied against such threshold.
Moreover, the Management Board, with the consent of the Supervisory Board, is authorized to sell the company’s shares so acquired also by other means than over the exchange or by way of an offer to all shareholders, if the shares are offered to third parties in connection with a merger or acquisition or if the shares are disposed of to cover subscription or option rights granted pursuant to the authorizations by the shareholders granted at the annual general meetings held on May 12, 1999, May 3, 2000, June 5, 2001 and June 11, 2002. To the extent shares are disposed of to cover subscription rights granted to members of the Management Board, the responsibility lies with the Supervisory Board.
To date, the Management Board has not made use of this authorization to acquire our own shares. We do not currently hold any treasury shares.
Notification and Disclosure Obligations
The German Securities Trading Act requires every shareholder whose equity participation in a company with a registered office in Germany, and that is listed for trading on an organized market in a member state of the European Union or a country that is a party to the Treaty on the European Economic Area, reaches, exceeds, or falls below thresholds of 5%, 10%, 25%, 50%, or 75% of the voting rights of such company to inform the company and the German Federal Financial Supervisory Authority (Bundesanstalt für Finanzdienstleistungsaufsichtor BaFin) without undue delay and, in any case, no later than seven days after reaching, exceeding or falling below these thresholds in writing. In the context of this requirement, the German Securities Trading Act contains various rules that are meant to ensure that share ownership is attributed to the person that actually controls the voting rights
136
Table of Contents
pertaining to such shares. As long as the shareholder fails to make such notification, he may generally not exercise any rights pertaining to these shares (including voting rights and dividend rights). Upon receipt of any such shareholder notification, the German company is required to immediately publish the notification in a supra-regional newspaper that has been designated by the Frankfurt Stock Exchange for official notices.
In addition, the German Securities Trading Act requires the members of the management board and the supervisory board, their spouses and close relatives, who purchase or sell shares, or other types of securities representing the right to acquire shares, including convertible bonds and bonds with warrants attached, issued by a company whose shares have been admitted to trading on a German stock exchange in excess of a diminutive number, to immediately notify the issuer and the BaFin of such purchases or sales. Upon receipt of such notice, the issuer is required to publish this notification by, among other things, posting it on its website.
German Foreign Exchange Controls
In accordance with UN and EU laws and regulations, Germany currently does not restrict capital movements between Germany and other countries except for capital transactions with Iraq, Burma/Myanmar, Zimbabwe and institutions of the Taliban party in Afghanistan, Osama bin Laden and Al Qaida.
For statistical purposes, subject to certain exceptions, each corporation or person domiciled in Germany is required to report to the Deutsche Bundesbank each payment received from or made to a corporation or person not domiciled in Germany in excess of€12,500 (or an equivalent amount in a foreign currency). Moreover, all claims and liabilities of a corporation or person domiciled in Germany against or towards a corporation or person not domiciled in Germany in excess of€5 million (or an equivalent amount in a foreign currency), individually or in the aggregate, are required to be reported to the Deutsche Bundesbank.
General Meetings, Resolutions and Voting Rights
General meetings are held at our registered office or in a German city that is the seat of a stock exchange. In general, annual general meetings are convened by our Management Board. The Supervisory Board is additionally required to convene a general meeting if this is in the best interest of GPC Biotech. Both our Management Board and our Supervisory Board may convene extraordinary general meetings. In addition, shareholders who, individually or as a group, own at least five percent of our share capital may request our Management Board to convene a general meeting. If our Management Board does not convene a general meeting when so requested, the shareholders may petition the competent German court to be authorized to convene a general meeting.
We must publish the invitation to a general meeting and the agenda thereof in the electronic version of the German Federal Gazette (elektronischer Bundesanzeiger) at least one month before the last day on which shares may be deposited in order for shareholders to exercise their voting rights. Shareholders who, individually or as a group, own at least five percent or€500,000 of our share capital may require that modified or additional items be added to the agenda of the general meeting and that these items be published before the general meeting takes place.
Our annual general meeting must take place within the first eight months of each fiscal year. Among other things, the annual general meeting is required to decide on the following issues:
| • | appropriation and use of annual net income; |
| • | discharge or ratification of the actions taken by the members of our Management Board and our Supervisory Board; |
137
Table of Contents
| • | the election of our statutory auditors; |
| • | increases or decreases in our share capital; |
| • | the election of Supervisory Board members; and |
| • | to the extent legally required, the approval of GPC Biotech’s unconsolidated financial statements. |
Unless otherwise prescribed by the German Stock Corporation Act, the resolutions of our general meeting are passed with a simple majority of the votes cast. Where the German Stock Corporation Act requires a majority of the share capital present or represented at the meeting for adoption of a resolution, a simple majority of the share capital present or represented at the meeting is sufficient. Neither the German Stock Corporation Act nor our Articles of Association provide for a minimum participation for a quorum for general meetings. Under the German Stock Corporation Act, resolutions of fundamental importance require a simple majority of the votes cast and a majority of at least three quarters of the share capital present or represented at the meeting at the time of adoption of the resolution. Resolutions of fundamental importance include, in particular, capital increases with exclusion of preemptive rights, capital decreases, the creation of authorized or conditional share capital, the dissolution of GPC Biotech, a merger into or with another company, split-offs and split-ups, the transfer of all or substantially all of our assets, the conclusion of inter-company agreements (Unternehmensverträge) in particular control agreements (Beherrschungsverträge) and profit and loss transfer agreements (Ergebnisabführungsverträge), and a change of the legal form of GPC Biotech.
Our Articles of Association provide that, to the extent permitted by applicable law, our general meetings may be transmitted audio-visually via electronic media by a method and in a scope determined by our Management Board, with the consent of the Supervisory Board, provided that this possibility was announced in the invitation to the general meeting.
Each share entitles its holder to one vote. Shareholders are entitled to attend general meetings and to exercise their voting rights if, by the seventh day before the day of the general meeting, they deposit their shares with a depositary designated in the invitation, a German notary or at a central depositary for securities until the end of the general meeting (the “deposit deadline”). The deposit deadline for attending general meetings is published concurrently with the notice convening the general meeting in the electronic version of the German Federal Gazette. The depositing with a depositary shall also be in proper form if shares are kept in a blocked securities account with a credit institution with the depositary’s consent until the end of the general meeting. If the shares are deposited with a German notary, a central depositary for securities or with any depositary designated in the invitation, the certificate to be issued by the notary, a central depositary for securities or the designated depositary must be submitted to us no later than the business day (other than a Saturday) following the deposit deadline. Proxies require written authorization, which may be given electronically, as per further instructions determined by our Management Board. These instructions will be published concurrently with the notice convening the general meeting in the electronic version of the German Federal Gazette. The Chairman of our Supervisory Board, or, in his absence, another member of our Supervisory Board appointed by the Chairman, will chair general meetings.
138
Table of Contents
DESCRIPTION OF AMERICAN DEPOSITARY RECEIPTS
The following is a summary of the material provisions of the deposit agreement to be entered into among us, The Bank of New York as depositary,and the registered holders and beneficial owners of American Depositary Receipts, referred to as ADRs. As this is a summary, it does not contain all of the information that may be important to you. For more complete information, you should read the entire deposit agreement including the form of ADR, copies of which have been filed as an exhibit to the Registration Statement of which this prospectus is a part.
Directions on how to obtain copies of these documents from the Securities and Exchange Commission are provided in the section entitled “Where You Can Find More Information.” You may also inspect a copy of the deposit agreement at the depositary’s office, currently located at 101 Barclay Street, New York, New York 10286, and at the principal Frankfurt offices of the agent of the depositary, Dresdner Bank AG, the custodian.
American Depositary Receipts
The depositary will deliver the GPC Biotech American Depositary Shares, referred to as ADSs, which you will be entitled to receive in this offering. Each ADS will represent one of our shares which we will deposit with the custodian, as agent of the depositary, under the deposit agreement. In the future, each ADS will also represent any other securities, cash or other property deposited with the depositary but which it has not distributed directly to you. Your American depositary shares will be evidenced by ADRs.
You may hold ADSs either directly or indirectly through your broker or other financial institution. If you hold ADSs directly, by having ADSs registered in your name on the books of the depositary, you are an ADR holder. This description assumes you hold your ADSs directly. If you hold the ADSs through your broker or financial institution nominee, you must rely on the procedures of such broker or financial institution to assert the rights of an ADR holder described in this section. You should consult with your broker or financial institution to determine what those procedures are.
As an ADR holder, we will not treat you as one of our shareholders and you will not have shareholder rights. German law governs shareholder rights. The depositary will be the holder of the shares underlying your ADSs. As a holder of ADRs, you will have ADR holder rights. The deposit agreement sets out ADR holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADRs.
Share dividends and other distributions
How will I receive dividends and other distributions on the shares underlying my ADSs?
We may make various types of distributions with respect to our securities. The depositary may pay to you the cash dividends or other distributions it or the custodian receives on shares or other deposited securities, after deducting or upon payment of its fees and expenses as described below. You will receive these distributions in proportion to the number of underlying shares that your ADSs represent.
Except as stated below, to the extent the depositary is legally permitted, it will deliver such distributions to ADR holders in proportion to their interests in the following manner:
Cash. The depositary will convert any cash dividend or other cash distribution we pay on the shares into U.S. dollars, if it can do so on a reasonable basis and can transfer the U.S. dollars to the United States. If that is not possible or if any government approval is needed and cannot be obtained,
139
Table of Contents
the deposit agreement allows the depositary to distribute the foreign currency only to those ADR holders to whom it is possible to do so. It will hold the foreign currency it cannot convert for the account of the ADR holders who have not been paid. It will not invest the foreign currency and it will not be liable for any interest. Before making a distribution, the depositary will deduct any withholding taxes that must be paid and deduct its fees and expenses. See “German Taxation” and “United States Federal Income Taxation—U.S. Information Reporting and Backup Withholding”.
Shares. In the case of a free distribution of shares, the depositary may deliver additional ADSs to represent such shares. Only whole ADSs will be issued. The depositary will try to sell any shares which would result in fractional ADSs and will distribute the net proceeds to the ADR holders entitled thereto. If the depositary does not distribute additional ADSs, the outstanding ADSs will also represent the new shares.
Rights to receive additional shares. In the case of a distribution of rights to subscribe for additional shares, if we provide satisfactory evidence that the depositary may lawfully distribute such rights, the depositary may arrange for ADR holders to instruct the depositary as to the exercise of such rights. However, if we do not furnish such evidence or if the depositary determines it is not practical to distribute such rights, the depositary may:
| • | sell such rights if practicable and distribute the net proceeds as cash; or |
| • | allow such rights to lapse, in which case ADR holders will receive nothing. |
We have no obligation to file a registration statement under the U.S. Securities Act of 1933 in order to make any rights available to ADR holders.
Other Distributions. In the case of a distribution of securities or property other than those described above, the depositary may either (i) distribute such securities or property in any manner it deems equitable and practicable, (ii) to the extent the depositary deems distribution of such securities or property not to be equitable and practicable, sell such securities or property and distribute any net proceeds in the same way it distributes cash or (iii) hold the distributed property, in which case the ADSs will also represent the distributed property.
Any U.S. dollars will be distributed by checks drawn on a bank in the United States for whole U.S. dollars and cents (fractional cents will be rounded to the nearest whole cent).
The depositary may choose any practical method of distribution for any specific ADR holder, including the distribution of foreign currency, securities or property, or it may retain such items, without paying interest on or investing them, on behalf of the ADR holder as deposited securities.
The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADR holders.
There can be no assurances that the depositary will be able to convert any currency at a specified exchange rate, or at all, or sell any property, rights, shares or other securities at a specified price, or at all, nor that any of such transactions can be completed within a specified time period.
Deposit, withdrawal and cancellation
How are ADSs issued?
The depositary will deliver ADSs if you or your broker deposit GPC Biotech shares or evidence of rights to receive GPC Biotech shares with the custodian.
140
Table of Contents
Shares deposited in the future with the custodian must be accompanied by certain documents, including instruments showing that such shares have been properly transferred or endorsed to the person on whose behalf the deposit is being made.
The custodian will hold all deposited shares (including those being deposited by us or on our behalf in connection with this offering) for the account of the depositary.
Upon each deposit of shares, receipt of related delivery documentation and compliance with the other provisions of the deposit agreement, including the payment of the fees and charges of the depositary and any taxes or other fees or charges owing, the depositary will deliver ADSs registered in the name of the person entitled thereto. ADRs will be delivered at the depositary’s office or any other location that it may designate as its transfer office.
How do ADR holders cancel ADSs and obtain deposited securities?
When you turn in your GPC Biotech ADSs at the depositary’s office, the depositary will, upon payment of certain applicable fees, charges and taxes, and upon receipt of proper instructions, deliver the underlying shares to an account designated by you and maintained by us, in the case of shares in registered form, or transfer them to an account of an accredited financial institution on your behalf in the case of shares in bearer form. At your risk, expense and request, the depositary may deliver deposited securities at such other place as you may request.
The depositary may only restrict the withdrawal of deposited securities in connection with:
| • | temporary delays caused by closing our transfer books or those of the depositary or the deposit of shares in connection with voting at a shareholders’ meeting, or the payment of dividends; |
| • | the payment of fees, taxes and similar charges; or |
| • | compliance with any U.S. or foreign laws or governmental regulations relating to the ADRs or to the withdrawal of deposited securities. |
This right of withdrawal may not be limited by any other provision of the deposit agreement.
Voting rights
How do I vote?
If you are an ADR holder and the depositary asks you to provide it with voting instructions, you may instruct the depositary how to exercise the voting rights for the shares which underlie your ADSs. After receiving voting materials from us, if we request the depositary to act, the depositary will notify the ADR holders of any shareholder meeting or solicitation of consents or proxies. This notice will describe how you may instruct the depositary to exercise the voting rights for the shares which underlie your ADSs. For instructions to be valid, the depositary must receive them on or before the date specified. The depositary will try, as far as is practical, subject to the provisions of and governing the underlying shares or other deposited securities, to vote or to have its agents vote the shares or other deposited securities as you instruct. The depositary will only vote or attempt to vote as you instruct. The depositary will not itself exercise any voting discretion. Furthermore, neither the depositary nor its agents are responsible for any failure to carry out any voting instructions, for the manner in which any vote is cast or for the effect of any vote.
There is no guarantee that you will receive voting materials in time to instruct the depositary to vote, and it is possible that you, or persons who hold their ADSs through brokers, dealers or other third parties, will not have the opportunity to exercise a right to vote.
141
Table of Contents
Record dates
The depositary may fix record dates for the determination of the ADR holders who will be entitled:
| • | to receive a dividend, distribution or rights, or |
| • | to give instructions for the exercise of voting rights at a meeting of holders of ordinary shares or other deposited securities, |
| • | be responsible for charges assessed by the depositary, |
all subject to the provisions of the deposit agreement.
Reports and other communications
Will I be able to view your reports?
The depositary will make available for inspection by ADR holders any written communications from us which are both received by the depositary as a holder of deposited securities and made generally available to the holders of deposited securities. We will furnish these communications in English when so required by any rules or regulations of the U.S. Securities and Exchange Commission.
Additionally, if we request, the depositary will mail copies of them to ADR holders.
Fees and expenses
What fees and expenses will I be responsible for paying?
ADR holders will be charged a fee for each issuance of ADSs, including issuances resulting from distributions of shares, rights and other property, and for each surrender of ADSs in exchange for deposited securities, including if the depositary agreement terminates. The fee in each case is $5.00 for each 100 ADSs (or any portion thereof) issued or surrendered.
The following additional charges shall be incurred by the ADR holders, by any party depositing or withdrawing shares or by any party surrendering ADRs or to whom ADRs are issued (including, without limitation, issuance pursuant to a stock dividend or stock split declared by GPC Biotech or an exchange of stock regarding the ADRs or the deposited securities or a distribution of ADRs), whichever is applicable:
| • | a fee of $0.02 or less per ADS (or portion thereof) for any cash distribution made pursuant to the deposit agreement; |
| • | stock transfer or other taxes and other governmental charges; |
| • | cable, telex and facsimile transmission and delivery charges incurred at your request; |
| • | transfer or registration fees for the registration of transfer of deposited securities on any applicable register in connection with the deposit or withdrawal of deposited securities; |
| • | expenses of the depositary in connection with the conversion of foreign currency into U.S. dollars; |
| • | a fee for distribution of securities distributed to holders of deposited securities which are distributed by the depositary to ADR holders equivalent to the fee that would be payable if securities distributed to you had been shares and the shares had been deposited for issuance of ADSs; |
142
Table of Contents
| • | $0.02 (or less) per ADS per calendar year for depositary services to the extent the depositary has not collected a cash distribution fee of $0.02 per ADS during that year); and |
| • | any charges incurred by the depositary or its agents for servicing the deposited securities. |
We will pay all other charges and expenses of the depositary and any agent of the depositary (except the custodian) pursuant to agreements from time to time between us and the depositary. The fees described above may be amended from time to time.
Payment of taxes
ADR holders must pay any tax or other governmental charge payable by the custodian or the depositary on any ADSs, deposited security or distribution. If an ADR holder owes any tax or other governmental charge, the depositary may (i) deduct the amount thereof from any cash distributions or (ii) sell deposited securities and deduct the amount owing from the net proceeds of such sale. In either case, the ADR holder remains liable for any shortfall. Additionally, if any tax or governmental charge is unpaid, the depositary may also refuse to effect any registration, registration of transfer, split-up or combination of ADRs or withdrawal of deposited securities (except under limited circumstances mandated by securities regulations). If any tax or governmental charge is required to be withheld on any non-cash distribution, the depositary may sell the distributed property or securities to pay such taxes and distribute any remaining net proceeds to the ADR holders entitled thereto.
Reclassifications, Recapitalizations and Mergers
If we take certain actions that affect the deposited securities, including (i) any change in par value, split-up, consolidation, cancellation or other reclassification of deposited securities or (ii) any recapitalization, reorganization, merger, consolidation, liquidation, receivership, bankruptcy or sale of all or substantially all of our assets, then the depositary may choose to:
| • | amend the form of American depositary receipt; |
| • | distribute additional or amended ADRs; |
| • | distribute cash, securities or other property it has received in connection with such actions; |
| • | sell any securities or property received and distribute the proceeds as cash; or |
| • | none of the above. |
If the depositary does not choose any of the above options, any of the cash, securities or other property it receives will constitute part of the deposited securities and each ADS will then represent a proportionate interest in such property.
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the ADRs without your consent for any reason. ADR holders must be given at least 30 days’ notice of any amendment that imposes or increases any fees or charges (other than stock transfer or other taxes and other governmental charges, transfer or registration fees, cable, telex or facsimile transmission costs, delivery costs or other such expenses), or affects any substantial existing right of ADR holders. If an ADR holder continues to hold ADRs after being so notified, such ADR holder is deemed to agree to such amendment.
143
Table of Contents
No amendment will impair your right to surrender your ADSs and receive the underlying securities.
How may the deposit agreement be terminated?
The depositary will terminate the deposit agreement if we ask it to do so. The depositary may also terminate the deposit agreement if the depositary has told us that it would like to resign and we have not appointed a new depositary bank within 60 days. In either case, the depositary must notify you at least 30 days before termination. After termination, the depositary’s only responsibility will be (i) to deliver deposited securities to ADR holders who surrender their ADRs and (ii) to hold or sell distributions received on deposited securities. At any time after the expiration of one year from the termination date, the depositary may sell the deposited securities which remain and hold the net proceeds of such sales, without liability for interest, in trust for the ADR holders who have not yet surrendered their ADRs. After making such sale, the depositary shall have no obligations except to account for such proceeds and other cash. The depositary will not be required to invest such proceeds or pay interest on them.
Limitations on obligations and liability to ADR holders
Limits on our obligations and the obligations of the depositary; limits on liability to ADR holders and holders of ADSs
The deposit agreement expressly limits the obligations and liability of the depositary, ourselves and our respective agents. Neither we nor the depositary nor any such agent will be liable if:
| • | law, regulation, the provisions of or governing any deposited securities, act of God, war or terrorism or other circumstance beyond its control prevents, delays or subjects to any civil or criminal penalty any act which the deposit agreement or the ADRs provide must be done or performed by it; |
| • | it exercises or fails to exercise discretion under the deposit agreement; |
| • | it performs its obligations without negligence or bad faith; |
| • | it takes any action or inaction in reliance upon the advice of or information from legal counsel, accountants, any person presenting shares for deposit, any registered holder of ADRs, or any other person believed by it to be competent to give such advice or information; or |
Neither we nor our agents the depositary nor its agents have any obligation to appear in, prosecute or defend any action, suit or other proceeding in respect of any deposited securities or the ADRs.
The depositary may own and deal in deposited securities and in ADSs.
Disclosure of interest in ADSs
From time to time we may request you and other holders and beneficial owners of ADSs to provide information as to:
| • | the capacity in which you and other holders and beneficial owners own or owned ADSs; |
| • | the identity of any other persons then or previously interested in such ADSs; and |
| • | the nature of such interest and various other matters. |
144
Table of Contents
You must provide any information requested by us or the depositary pursuant to the deposit agreement. The depositary has agreed to use reasonable efforts to comply with written instructions received from us requesting that it forward any such requests to you and other holders and beneficial owners and to forward to us any responses to such requests to the extent permitted by applicable law.
Requirements for depositary actions
We, the depositary or the custodian may refuse to:
| • | deliver, register or transfer ADRs; |
| • | effect a split-up or combination of ADRs; |
| • | deliver distributions on any ADRs; or |
| • | permit the withdrawal of deposited securities (unless the deposit agreement provides otherwise), until the following conditions have been met: |
| – | the holder has paid all taxes, governmental charges, and fees and expenses as required in the deposit agreement; |
| – | the holder has provided the depositary with any information it may deem necessary or proper, including, without limitation, proof of identity and the genuineness of any signature; and |
| – | the holder has complied with such regulations as the depositary may establish under the deposit agreement. |
The depositary may also suspend the deposit of shares or the registration of transfer of ADRs if the register for ADRs or any deposited securities is closed or if we or the depositary decide it is advisable to do so.
Books of Depositary
The depositary or its agent will maintain a register for the registration, registration of transfer, combination and split-up of ADRs. You may inspect such records at the depositary’s office during regular business hours, but solely for the purpose of communicating with other holders in the interest of business matters relating to the deposit agreement. The depositary will maintain facilities to record and process the issuance, cancellation, combination, split-up and transfer of ADRs. These facilities may be closed from time to time.
Pre-release of American Depositary Shares
The depositary may deliver ADSs prior to the deposit with the custodian of shares (or rights to receive shares). This is called a pre-release of the ADSs. The depositary may also deliver shares upon cancellation of pre-released ADRs (even if the ADRs are canceled before the pre-release transaction has been closed out). A pre-release is closed out as soon as the underlying shares (or other ADSs) are delivered to the depositary.
The depositary may pre-release ADRs only under the following conditions:
| • | before or at the time of the pre-release, the person to whom the pre-release is being made represents to the depositary in writing that it or its customer owns the shares or ADRs to be deposited; |
145
Table of Contents
| • | the pre-release is fully collateralized with cash or other collateral that the depositary considers appropriate; and |
| • | the depositary must be able to close out the pre-release on not more than five business days’ notice. |
In general, the number of shares represented by pre-released ADSs will not exceed thirty percent of total deposited shares at any given time. However, the depositary may change or disregard such limit from time to time as it deems appropriate. The depositary may retain for its own account any earnings on collateral for pre-released ADSs and its charges for issuance thereof.
146
Table of Contents
The following discussion is a summary of the material German tax consequences for beneficial owners of shares or ADSs who are (i) not German residents for German income tax purposes (i.e., persons whose residence, habitual abode, statutory seat or place of effective management and control is not located in Germany) and (ii) whose shares do not form part of the business property of a permanent establishment or fixed base in Germany. Throughout this section we refer to these owners as “non-German Holders”. This summary is based on German tax laws and typical tax treaties to which Germany is a party, as they are in effect on the date hereof, and is subject to changes in German tax laws or such treaties. The following discussion does not purport to be a comprehensive discussion of all German tax consequences that may be relevant for non-German Holders.
Prospective purchasers of shares or ADSs are advised to consult their tax advisers about the tax consequences of the purchase, holding, disposal, or other transfer of shares or ADSs and the procedures for obtaining a possible refund of German withholding tax paid.
Taxation of Corporations in Germany
Profits earned by a German resident corporation are generally subject to a corporate income tax rate of 25% plus 5.5% solidarity surcharge thereon, amounting to a total of 26.375%. German corporations are also subject to profit-related trade tax on income, the exact amount of which depends on the municipality in which the corporation maintains its business establishment(s). The trade income tax rates range between 15% and 20%. Trade tax on income is a deductible item in computing the corporation’s tax base for corporate income and trade tax purposes.
Dividends Received by Corporations
Dividend distributions received by corporations are generally 95% exempt from corporate income tax. Dividend distributions by a German resident corporation are generally 95% exempt from trade income tax, if the corporation holds, as of the beginning of the relevant assessment period, at least 10% of the share capital of the distributing corporation.
Withholding Tax
Dividends are subject to withholding tax at a tax rate of 20% of the gross amount of the dividend. In addition, the solidarity surcharge of 5.5% on the withholding tax will be retained, resulting in a total withholding from dividends of 21.1%. The withholding tax rate may be reduced for non-German Holders by an applicable double tax treaty. Under most double tax treaties the withholding tax rate is reduced to 15%. To reduce the withholding to the applicable treaty rate of 15%, a non-German Holder may apply for a refund of withholding taxes paid. The application for refund must be filed with the German Federal Tax Office (Bundesamt für Finanzen, Friedhofstrasse 1, D-53221 Bonn, Germany). The relevant forms can be obtained from the German Federal Tax Office or from German embassies and consulates.
Special Tax Rules for U.S. Holders
Under the United States-German Convention for the Avoidance of Double Taxation (the “Treaty”), the withholding tax rate is reduced to 15% of the gross amount of the dividends for U.S. holders (as defined below in “United States Federal Income Taxation”) that are eligible for the benefits of the Treaty and that hold less than 10% of the voting shares in GPC Biotech. For eligible U.S. holders that hold
147
Table of Contents
10% of the voting shares or more the withholding tax rate is reduced to 5%. An eligible U.S. holder is entitled to a refund from the German tax authorities of 6.1% of the gross amount of the dividend. An eligible U.S. holder will generally not be liable for German tax on capital gains realized or accrued on the sale or other disposition of shares or ADSs. However, under certain circumstances, the Treaty does allow Germany a limited right to tax former German resident holders of a substantial interest (at least 25%) in GPC Biotech on such capital gains.
Dividend Refund Procedure for U.S. Holders
For shares and ADSs kept in custody with The Depositary Trust Company (“DTC”) in New York or one of its participating banks, the German tax authorities have introduced a collective procedure for the refund of German withholding tax and the solidarity surcharge thereon, on a trial basis. Under this procedure, DTC may submit claims for refunds payable to eligible U.S. holders under the Treaty collectively to the German tax authorities on behalf of the holders. The German Federal Tax Office will pay the refund amounts on a preliminary basis to DTC, which will redistribute these amounts to the eligible U.S. holders. The Federal Tax Office may review whether the refund was made in accordance with the law within four years after making the payment to DTC. Details of this collective procedure are available from DTC.
Alternatively, a newly introduced simplified collective refund procedure based on electronic data exchange (Datenträgerverfahren) may be available. Financial institutions which deal with dividend distributions of GPC Biotech (for example, custodian banks or clearing offices) or GPC Biotech itself may apply to participate in this procedure at the German Federal Tax Office. Upon acceptance, the participant may electronically file collective refund claims with the German Federal Tax Office.
Individual claims for refund may be made on a special German form, which must be filed with the German Federal Tax Office, Friedhofstrasse 1, 53221 Bonn, Germany. Copies of the required form may be obtained from the German Federal Tax Office at the same address or from the Embassy of the Federal Republic of Germany, 4645 Reservoir Road, N.W., Washington, D.C. 20007-1998. The form can also be downloaded from the following website: www.bff-online.de/. Claims must be filed within a four-year period from the end of the calendar year in which the dividend was received.
As part of the individual refund claim, an eligible U.S. holder must submit to the German tax authorities the original bank voucher (or certified copy thereof) issued by the paying entity documenting the tax withheld, and an official certification on IRS Form 6166 of its last United States federal income tax return. IRS Form 6166 may be obtained by filing a request with the Internal Revenue Service Center in Philadelphia, Pennsylvania, Foreign Certification Request, P.O. Box 16347, Philadelphia, PA 19114-0447. Requests for certifications must include the eligible U.S. holder’s name, Social Security Number or Employer Identification Number, tax return form number, and tax period for which the certification is requested. Requests for certification can include a request to the Internal Revenue Service to send the certification directly to the German tax authorities. If no such request is made, the Internal Revenue Service will send a certification on IRS Form 6166 to the eligible U.S. holder, who then must submit this document with its claim for refund.
Capital Gains
Half of the gains from a disposal of shares or ADSs held by a non-German individual Holder may be subject to German individual income tax, if the individual held, directly or indirectly, at any time during the five years preceding the disposal at least 1% of the share capital of GPC Biotech. Half of the losses resulting from the disposal of shares may be deductible for tax purposes, subject to further requirements.
148
Table of Contents
Many double tax treaties, however, provide complete exemption from German taxation of gains derived from the disposal of shares or ADSs if and to the extent the shares or ADSs are not held as assets attributable to a permanent establishment in Germany.
Individuals owning at least 25% of the voting interest of GPC Biotech who gave up residence in Germany and have become residents in the United States could be liable for German tax on capital gains under the Treaty if certain prerequisites are met.
Gains from the disposal of shares or ADSs held by a non-German corporate Holder are generally exempt from trade income tax and corporate income tax. If the shareholder held, directly or indirectly, at any time during the five years preceding the disposal, at least 1% of the share capital of the corporation, only 95% of the gain from the disposal of the shares or ADSs may be exempt from corporate income tax, the solidarity surcharge and trade income tax. Losses deriving from the disposal of shares or ADSs are not tax-deductible.
Inheritance and Gift Tax
The transfer of shares or ADSs to other persons by way of gift or inheritance is only subject to German inheritance and gift tax, if
| (1) | the testator, donor, heir, donee or any other beneficiary had his domicile or residence in Germany or has not been living abroad as a German citizen for more than five years without having a domicile in Germany; or |
| (2) | the shares or ADSs belonged to assets attributable to a permanent establishment or a permanent representative of the testator or of the donor in Germany; or |
| (3) | the testator or donor, either alone or with other closely related persons, held at the time of the inheritance or donation, directly or indirectly at least 10% of the share capital of the corporation. |
The few presently applicable inheritance and gift taxation treaties Germany is a party to generally provide that German inheritance or gift tax is levied in case (1) and, with certain restrictions, in case (2) above.
Other Taxes
Upon the acquisition, sale or other disposal of shares or ADSs, no German stock exchange transfer tax, value added tax, stamp duty or other tax is levied. Under special circumstances it is possible that entrepreneurs elect for a value added tax duty of otherwise value added tax exempt turnovers. Net wealth tax is, at present, not levied in Germany.
Tax Law Changes
Since January 1, 2004, the German thin capitalization rules have been tightened, for example in the case that loans are secured by substantial shareholders or parties related to such shareholders. This could lead to interest being taxed as a constructive dividend in case of intragroup financing. This could lead to an increased tax burden for GPC Biotech for German income tax purposes.
149
Table of Contents
UNITED STATES FEDERAL INCOME TAXATION
The following discussion is a summary of material U.S. federal income tax considerations applicable to the ownership and disposition of shares or ADSs by you, if you are a U.S. holder. In general you will be a “U.S. holder” if:
| • | you are the beneficial owner of shares or ADSs; |
| • | you are either (i) an individual resident or citizen of the United States, (ii) a corporation or any other entity treated as a corporation for U.S. federal income tax purposes created in or organized under the laws of the United States or any state thereof, (iii) an estate whose income is subject to U.S. federal income taxation regardless of its source, or (iv) a trust if a U.S. court can exercise primary supervision over the administration of the trust and one or more U.S. persons are authorized to control all substantial decisions of the trust; |
| • | you own our shares or ADSs as capital assets; |
| • | you own directly or indirectly less than 10% of our outstanding voting stock; |
| • | you are fully eligible for benefits under the Limitations on Benefits article of the Income Tax Treaty between the United States of America and the Germany, signed 29 August 1989 (the “Treaty”); and |
| • | you are not also a resident of Germany for German tax purposes. |
The Treaty benefits discussed below generally are not available to holders who hold shares or ADSs in connection with the conduct of business through a permanent establishment, or the performance of personal services through a fixed base, in Germany.
If a partnership holds shares or ADSs, the tax treatment of a partner generally will depend upon the status of the partner and the activities of the partnership. If you are a partner in a partnership that holds shares or ADSs, you are urged to consult your own tax advisor regarding the specific tax consequences of owning and disposing of your shares or ADSs.
The summary does not purport to be a comprehensive description of all of the tax considerations that may be relevant to any particular holder, including tax considerations that arise from rules of general application or that are generally assumed to be known by U.S. holders. This summary is based on provisions of the Internal Revenue Code of 1986, as amended (the “Code”), existing and proposed U.S. Treasury Regulations, rulings, administrative pronouncements and judicial decisions in effect as of the date of this prospectus. All of the authorities are subject to change, possibly with retroactive effect, and to differing interpretations. In addition, this summary does not discuss all aspects of U.S. federal income taxation that may be applicable to investors in light of their particular circumstances or to U.S. holders who are subject to special treatment under U.S. federal income tax law, including insurance companies, dealers in stocks or securities, financial institutions, tax-exempt organizations, persons subject to the alternative minimum tax, and persons having a functional currency other than the U.S. dollar.
U.S. holders are urged to consult with their own tax advisors regarding the tax consequences of the ownership and disposition of shares or ADSs, including the effects of federal, state, local, foreign and other tax laws with respect to their particular circumstances.
For U.S. federal income tax purposes, if you own ADSs represented by ADRs, you generally will be treated as the owner of the shares represented by such ADRs.
150
Table of Contents
Dividends
Subject to the passive foreign investment company rules discussed below, if we make any distributions of cash or other property to you, you generally will be required to include in gross income the amount of any distributions (including the amount of any German taxes withheld in respect of such distribution as described above in the German Taxation section), to the extent that such distributions are paid out of our current or accumulated earnings and profits as determined for U.S. federal income tax purposes. Distributions in excess of our earnings and profits will be applied against and will reduce your tax basis in your shares or ADSs and, to the extent in excess of such tax basis, will be treated as gain from a sale or exchange of such shares or ADSs. Dividends paid by us will not be eligible for the dividends received deduction applicable in some cases to U.S. corporations.
Any dividend paid in Euro, including the amount of any German taxes withheld therefrom, will be includible in your gross income in an amount equal to the U.S. dollar value of the Euro calculated by reference to the spot rate of exchange in effect on the date the dividend is received by you, in the case of shares, or by the Depositary, in the case of ADSs, regardless of whether the Euros are converted into U.S. dollars. Any gain or loss resulting from currency exchange fluctuations during the period from the date the dividend is includible in your gross income to the date such payment is converted into U.S. dollars will be treated as U.S. source ordinary income or loss.
Any dividends paid by us to you with respect to shares or ADSs will be treated as foreign source income and will be characterized as “passive income” or, in the case of some U.S. holders, “financial services income” for U.S. foreign tax credit purposes. Subject to limitations, you may elect to claim a foreign tax credit against your U.S. federal income tax liability for German income tax withheld from dividends received in respect of shares or ADSs. The rules relating to the determination of the foreign tax credit are complex. Accordingly, you should consult your own tax advisor to determine whether and to what extent you would be entitled to the credit. The United States Treasury has expressed concerns that parties to whom ADSs are pre-released may be taking actions that are inconsistent with the claiming of foreign tax credits by U.S. holders of ADSs. Accordingly, the discussion above regarding the creditability of German withholding tax on dividends could be affected by future actions that may be taken by the United States Treasury. If you do not elect to claim a foreign tax credit, you may instead claim a deduction for German income tax withheld, but only for a year in which you elect to do so with respect to all foreign income taxes.
Under the Jobs and Growth Tax Relief Reconciliation Act of 2003 (the “2003 Act”), for capital assets held for over one year and sold or exchanged in taxable years beginning before January 1, 2009, the maximum rate of tax generally will be 15%. In addition, “qualified dividend income” received by non-corporate U.S. holders in taxable years beginning before January 1, 2009 generally will be taxed at the same rates (i.e., a maximum rate of 15%) rather than the rates applicable to other items of ordinary income. For this purpose, “qualified dividend income” generally includes dividends paid on shares of U.S. corporations as well as dividends paid on shares of certain non-U.S. corporations if, among other things, (i) the shares of the non-U.S. corporation (including ADRs backed by such shares) are readily tradable on an established securities market in the United States, or (ii) the non-U.S. corporation is eligible with respect to substantially all of its income for the benefits of a comprehensive income tax treaty with the United States which contains an exchange of information program (the Treaty has been identified by the U.S. Treasury Department as a qualifying treaty). Dividends paid by us with respect to the shares or ADSs should constitute “qualified dividend income” for U.S. federal income tax purposes unless we qualify as a PFIC (as discussed below) in which case they would be taxed at the higher rates applicable to other items of ordinary income.
Sale or Exchange of Shares or ADSs
Subject to the passive foreign investment company rules discussed below, upon the sale or other disposition of shares or ADSs, you generally will recognize capital gain or loss equal to the difference
151
Table of Contents
between the amount realized on the disposition and your adjusted tax basis in your shares or ADSs. Gain or loss upon the disposition of shares or ADSs generally will be U.S. source gain or loss, and will be treated as long-term capital gain or loss if, at the time of the disposition, your holding period for the shares or ADSs exceeds one year. If you are a non-corporate U.S. holder, any capital gains recognized before January 1, 2009, generally will be subject to U.S. federal income tax at a maximum rate of 15% if you have a holding period greater than one year. The deductibility of capital losses is subject to significant limitations.
The surrender of ADSs in exchange for shares pursuant to the Deposit Agreement governing the ADSs will not be a taxable event for U.S. federal income tax purposes. Accordingly, you will not recognize any gain or loss upon such surrender.
Passive Foreign Investment Company Status
We do not currently believe that we will be a passive foreign investment company, or PFIC, for the year ended December 31, 2004. However, because the determination of whether a company is a PFIC must be made annually, and because there are uncertainties in the application of the relevant rules, there can be no assurance that we will not be classified as a PFIC for any particular year.
A non-U.S. corporation will be classified as a PFIC in any taxable year in which, after taking into account the income and assets of certain subsidiaries, either (i) at least 75% of its gross income is passive income or (ii) at least 50% of the average value of its assets is attributable to assets that produce or are held to produce passive income. Whether or not we will be classified as a PFIC in any taxable year is a factual determination and will depend upon our assets (including proceeds of the Offering), the market value of the share and our activities in each year, and is therefore subject to change.
If we are a classified as a PFIC for any taxable year, the so-called “interest charge regime” of Code section 1291 will apply to any U.S. holder of shares or ADSs that does not make a “mark-to-market” election (as described below). Under the interest charge regime, (i) any gain you realize on the sale or other disposition of the shares or ADSs (possibly including a gift, exchange in a corporate reorganization, or grant as security for a loan) and any “excess distribution” that we make to you (generally, any distributions to you in respect of the shares or ADSs during a single taxable year that are greater than 125% of the average annual distributions received by you in the three preceding years, or if shorter, your holding period for the shares or ADSs), will be treated as ordinary income that was earned ratably over each day in your holding period for the shares or ADSs, (ii) the portion of such gain or distribution that is allocable to prior taxable years will, with certain exceptions, be subject to tax at the highest rate applicable to ordinary income for the relevant taxable years, regardless of the tax rate otherwise applicable to you, and (iii) the interest charge generally applicable to underpayments of tax will be imposed in respect of the tax attributable to each such year.
The interest charge regime would not apply to you if you were eligible for and timely make a valid “qualifying electing fund” election, in which case you would be required to include in income on a current basis your pro rata share of our ordinary income and net capital gains. We do not currently intend to complete the actions necessary for U.S. holders to make a qualifying electing fund election in the event that we are considered a passive foreign investment company for any taxable year.
If we are a PFIC and our shares or ADSs are treated as “marketable security” under applicable Treasury regulations, you may avoid the interest charge regime by making a valid “mark-to-market” election with respect to the shares or ADSs. If a “mark-to-market” election is made, the electing U.S. holder generally (i) will be required to recognize as ordinary income an amount equal to the difference, if any, between the fair market value of the shares or ADSs and the holder’s adjusted tax basis in such
152
Table of Contents
shares or ADSs at the close of each taxable year, and (ii) if the U.S. holder’s adjusted tax basis in the shares or ADSs exceeds their fair market value, will be allowed to deduct the excess as an ordinary loss, but only to the extent of the net amount of income previously included as a result of the mark-to-market election. A U.S. holder’s adjusted basis in the shares or ADSs will be adjusted to reflect the amounts included or deducted with respect to the mark-to-market election, and any gain or loss on the disposition of shares or ADSs will generally be ordinary income, or, to the extent of previously included mark-to-market inclusions, ordinary loss. The mark-to-market election is made on a shareholder-by-shareholder basis and, once made, cannot be revoked without the consent of the United States Internal Revenue Service unless the shares or ADSs cease to be marketable. Under applicable Treasury regulations, marketable stock includes stock of a PFIC that is “regularly traded” on a qualified exchange or other market. Because our shares are traded on the Frankfurt Stock Exchange and our ADSs will be traded on Nasdaq, we expect the shares and ADSs to be treated as “regularly traded”, and a U.S. Holder should be able to make a mark-to-market election, but no assurance can be given.
Special rules apply with respect to the calculation of the amount of the foreign tax credit with respect to excess distributions by a PFIC. In general, these rules allocate creditable foreign taxes over the U.S. holder’s holding period for shares or ADSs and otherwise coordinate the foreign tax credit limitation rules with the PFIC rules.
In addition to the special PFIC tax regime, dividends paid on shares of a PFIC are not eligible for the reduced (maximum 15%) rate of taxation of dividends received by non-corporate U.S. holders on shares of qualifying corporations for purposes the 2003 Act and would be taxed at the higher rates applicable to other items of ordinary income. If we are a PFIC for any taxable year, U.S. holders who acquire shares or ADSs from decedents could be denied the step-up in the tax basis for such shares or ADSs, which would otherwise have been available.
If you own shares or ADSs during any year in which we are a PFIC, you must file IRS Form 8621 with your annual United States federal income tax return for each year in which you own shares or ADSs, even if we subsequently would not be considered a PFIC.
U.S. holders should consult their own tax advisors regarding the application of the PFIC rules to the shares or ADSs and the availability and advisability of making an election to avoid the adverse U.S. federal income tax consequences of the interest charge regime should we be classified as a PFIC for any taxable year.
U.S. Information Reporting and Backup Withholding
Dividend payments with respect to shares or ADSs and proceeds from the sale, exchange, redemption, or other disposition of shares or ADSs may be subject to information reporting to the IRS and possible U.S. backup withholding. Certain exempt recipients (such as corporations) are not subject to these information reporting requirements. Backup withholding will also not apply to a holder who furnishes a correct taxpayer identification number or certificate of foreign status and makes any other required certification. U.S. persons who are required to establish their exempt status generally must provide IRS Form W-9 (Request for Taxpayer Identification Number and Certification).
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a holder’s U.S. federal income tax liability, and a holder may obtain a refund of any excess amounts withheld by filing the appropriate claim for refund with the IRS and furnishing any required information.
153
Table of Contents
We expect to enter into an agreement with Goldman, Sachs & Co. oHG and the other underwriters for the combined offering on or about June 11, 2004, pursuant to which the underwriters will agree, subject to certain conditions, to offer shares for subscription to shareholders in the rights offering. For these purposes, the underwriters, the selling management shareholders and GPC Biotech intend to enter into an underwriting agreement on or about June 28, 2004, pursuant to which the underwriters will agree to subscribe for the new shares in order to satisfy the subscription rights of existing holders who are exercising their rights and to offer for sale in the combined offering the shares to be sold by the selling shareholders together with the new shares as to which shareholders’ subscription rights have been excluded and any new shares not subscribed in the rights offering.
If the underwriting agreement is not signed, or if the underwriters terminate the underwriting agreement prior to the registration of the implementation of the capital increase relating to the new shares with the commercial register (Handelsregister) for GPC Biotech AG at the local court (Amtsgericht) in Munich, the subscription rights will become void. If this were to occur, brokerage institutions generally would not reverse any subscription rights trading transactions. Accordingly, investors who have acquired subscription rights on a stock exchange would suffer a loss. To the extent that the underwriters terminate the underwriting agreement after the registration of the implementation of the capital increase relating to the new shares in the commercial register, existing shareholders who have exercised their subscription rights are entitled to subscribe for the new shares as to which they have exercised their subscription rights at the subscription price.
In the event of a termination of the Underwriting Agreement after the end of the subscription period (June 28, 2004), which is possible until July 2, 2004, the termination would apply to unsubscribed shares only. The purchase agreements for unsubscribed shares are therefore contingent. To the extent that you have already sold short any new shares to be acquired in the global offering prior to the cancellation of the settlement, you will bear the risk of being unable to settle such sales by delivering new shares to be purchased in the global offering.
All of the underwriters may sell shares in the global offering. Each underwriter may offer and sell shares anywhere in the world where it is legally permitted to do so. Sales of shares in the United States will be effected through a U.S. selling group. There are no minimum or maximum limits on how many shares may be offered or sold in any particular country or region. Subject to certain conditions, each of the underwriters named below is expected to agree severally to offer new shares to existing holders in the rights offering and to agree severally to purchase shares for sale in the combined offering as follows (not including shares to be sold following the exercise of the over-allotment option and excluding shares to be purchased from the selling shareholders).
Underwriters | ||
Goldman, Sachs & Co. oHG | ||
Lehman Brothers International (Europe) | ||
WestLB AG | ||
Pacific Growth Equities, LLC | ||
Total |
Goldman, Sachs & Co. oHG is the global coordinator of the combined offering.
The underwriters may choose to take some or all of their shares in the global offering in the form of ADSs. The members of the U.S. selling group are: Goldman, Sachs & Co., Lehman Brothers Inc. and Pacific Growth Equities, LLC.
154
Table of Contents
The respective underwriting commitments of the underwriters, the per share and total underwriting discounts and commissions and the reallowance to dealers will be set forth in a supplement to this prospectus.
The underwriting agreement will provide that the obligations of the underwriters to purchase shares may be terminated under certain circumstances, by or before 9:00 a.m. (Frankfurt time) on July 2, 2004, including upon the occurrence of certainforce majeure events. The underwriting agreement will further provide that the obligations of the underwriters may be terminated if registration with the commercial register for GPC Biotech AG at the local court in Munich of the capital increase does not occur by 11:59 p.m. (Frankfurt time) on June 28, 2004.
If the underwriters sell more shares than the total number offered in the combined offering, the underwriters will have an option to purchase up to an additional 1,119,000 new shares from GPC Biotech to cover such sales. They may exercise that option until July 30, 2004. If any shares are purchased pursuant to this option, the underwriters will severally purchase shares in approximately the same proportion purchased in the combined offering. The underwriters may choose to take some or all of their additional shares in the form of ADSs.
For the purpose of enabling settlement to be made of over-allotments prior to the exercise of the option described in the paragraph above, Goldman, Sachs & Co. oHG will also enter into a share lending agreement with ALTANA Technology Projects GmbH pursuant to which ALTANA Technology Projects GmbH will agree with Goldman, Sachs & Co. oHG, acting in its own name but for the account of the several underwriters, to lend, and Goldman, Sachs & Co. oHG will agree to borrow from ALTANA Technology Projects GmbH, up to an aggregate of 1,119,000 shares of the company. Assuming that the underwriters exercise their over-allotment option only at the end of the thirty-day exercise period, the share loan is expected to end in the beginning of August, depending on the registration of the capital increase for the additional shares. The exact date of the end of the share loan will depend on the date of registration of the capital increase for the optional shares with the commercial register for GPC Biotech AG at the local court in Munich.
Shares sold by the underwriters in the rights offering will be offered at the subscription price determined as described in “The Combined Offering” above and “The Rights Offering” below. Shares sold by the underwriters in the global offering will initially be offered at the initial offer price of the shares to be sold in the global offering to the public to be determined by GPC Biotech, the selling shareholders and Goldman, Sachs & Co. oHG at the end of the bookbuilding period.
The initial offer price to public in the global offering will be published through electronic media (Reuters) immediately after being determined on or about June 29, 2004. The initial offer price to the public will be published in the Frankfurter Allgemeine Zeitung and The Wall Street Journal on or about July 1, 2004. If the shares are not all sold at the initial offer price to public in the global offering, the global coordinator may change the offering price and the other selling terms.
For reasons of German law, the global coordinator will initially subscribe for all of the shares on behalf of the underwriters, at an issue price equal to the notional value of€ 1.00 (geringster Ausgabebetrag) per share. This issue price will be credited against the amount due from the underwriters at the closing.
In the underwriting agreement, GPC Biotech will agree with the underwriters, subject to certain exceptions, that on or before December 27, 2004 it will not, directly or indirectly, offer, sell or otherwise dispose of any shares of GPC Biotech or any other securities which are convertible into or exchangeable for shares of GPC Biotech; or exercise any authorization pursuant to its articles of association to increase its capital other than for the purpose of issuing ordinary shares to the
155
Table of Contents
beneficiaries of our existing stock option plans and convertible bond programs upon the exercise of options or conversion rights; or propose a capital increase to its shareholders other than a proposal for authorized capital (genehmigtes Kapital) or for conditional capital (bedingtes Kapital). The same restrictions apply to derivatives transactions that would have an economic effect similar to that of a sale of GPC Biotech’s shares. These restrictions will not apply to shares issued to strategic partners in the pharmaceutical or biotech sector in connection with partnering or licensing transactions or in connection with an acquisition or joint venture. All Management Board members will also individually agree with the underwriters that on or before December 27, 2004 they will not sell their shares or enter into similar transactions to this effect or propose, or vote in favor of, any increase in GPC Biotech’s share capital unless the Company is or would be permitted to propose such resolution. These lock-up restrictions do not apply to up to 150,000 cash settled stock options, which two of the Company’s Management Board members intend to exercise before the end of the combined offering but after the publication of the initial offer price (see “Principal and Selling Shareholders and Exercise of Cash Settled Stock Options—Exercise of Cash Settled Stock Options”). In each case, these restrictions may be waived if Goldman, Sachs & Co. oHG consents to the proposed transaction in writing.
Goldman, Sachs & Co. oHG as stabilization manager is authorized for a period beginning on the date of the publication of the initial offer price and ending 30 days after allocation of the shares in the global offering to perform either by itself or through any of its affiliates certain stabilization measures. To the extent permitted under German law, these transactions may include short sales, stabilizing transactions and purchases to cover positions created by short sales. Short sales involve the sale by the underwriters of a greater number of shares than they are required to purchase in the offering. Stabilizing transactions consist of certain bids or purchases made for the purpose of preventing or retarding a decline in the market price of the shares or ADSs.
The underwriters also may impose a penalty bid. This occurs when a particular underwriter repays to the underwriters a portion of the underwriting discount received by it because the representatives have repurchased shares sold by or for the account of such underwriter in stabilizing or short covering transactions.
These activities by the underwriters may stabilize, maintain or otherwise affect the market price of the shares or ADSs. As a result, the price of these securities may be higher than the price that otherwise might exist in the open market. If these activities are commenced, they may be discontinued by the underwriters at any time. These transactions may be effected on any of the exchanges where the shares or ADSs are listed, in the over-the-counter market or otherwise.
GPC Biotech estimates that its share of the total expenses of the combined offering, excluding underwriting discounts and commissions and the total net proceeds to the selling shareholders, will be approximately€2.8 million.
GPC Biotech and the selling shareholders have agreed to indemnify the several underwriters against certain liabilities, including liabilities under the Securities Act of 1933.
Some of the underwriters have from time to time performed services for GPC Biotech and have normal banking relationships with GPC Biotech in the ordinary course of their business.
156
Table of Contents
If you hold GPC Biotech shares you are entitled to subscribe for new shares. Existing holders of our shares will have the right to subscribe at the subscription price for one new share for every three shares held. We will determine the subscription price at the latest by 12:00 p.m. (noon)(Frankfurt time) on June 25, 2004. The subscription price will lie between or at (x) the volume-weighted average price of the shares from the beginning of the subscription period until the determination of the subscription price and (y) the current market price of our shares at the time of the determination of the subscription price. In determining the subscription price, we will, among other things, consider current market conditions and the preliminary results of the bookbuilding process contemplated for the shares to be offered in the global offering. WestLB AG is entitled to take appropriate measures in order to organize an orderly trading of subscription rights.
GPC Biotech expects to publish the subscription offer on June 14, 2004, among others, in the electronic version of the German Federal Gazette and in the Frankfurter Allgemeine Zeitung.
The procedures for you to subscribe for new shares and information about the purchase and sale of rights to subscribe for new shares are summarized below.
Timetable
This timetable lists some dates that may be important to you if you hold GPC Biotech shares and want to participate in the rights offering:
Crediting of the subscription rights based on status of collective custody accounts | June 14, 2004 | |||
Beginning of the period during which you can subscribe for new shares | June 15, 2004 | |||
Start of trading of subscription rights on the Frankfurt Stock Exchange | June 15, 2004 | |||
Last time for delivery to your custodian bank of instructions that you want to participate in the rights offering | at or about 10:00 a.m. | |||
| Frankfurt time (depending on the respective custodian bank’s deadline) | June 24, 2004 | |||
Trading of subscription rights ends | at or about 12:30 p.m. Frankfurt time | June 24, 2004 | ||
Latest date when subscription price is determined and announced | June 25, 2004 | |||
End of the period during which you can subscribe for | 11:59 p.m. Frankfurt time | June 28, 2004 | ||
Date by which the bookbuilding for the global offering ends | June 29, 2004 | |||
Latest date for pricing of the global offering | June 29, 2004 | |||
First day of trading of the new shares on the Frankfurt Stock Exchange | June 30, 2004 | |||
Payment date for the new shares | July 2, 2004 | |||
New shares purchased in the rights offering and the global offering delivered on or as soon as practicable after | July 2, 2004 |
Subscription for New Shares
If you hold GPC Biotech shares you may subscribe for new shares as follows:
157
Table of Contents
Entitlement and Rights Trading.Each outstanding ordinary share will entitle you to one subscription right to subscribe for new shares. If you have three rights, you will be entitled to subscribe for one new share at the subscription price as described above. Subscription rights will be listed and traded on the Frankfurt Stock Exchange from June 15, 2004 through approximately 12:30 p.m. Frankfurt time on June 24, 2004.
Clearstream Banking AG will automatically credit the subscription rights for shares held in collective custody as of the evening of June 14, 2004 to the relevant depositary bank for further credit to the accounts of existing holders.
If you are a rights holder, your custodian bank in Germany will, at your request, purchase additional rights for you to give you enough rights to subscribe for one or more whole new shares. You may subscribe for whole new shares only. GPC Biotech will not issue any fractional new shares. You may otherwise use your rights as you see fit.
Subscription Price.We will determine the subscription price at the latest by 12:00 p.m.(noon) Frankfurt time on June 25, 2004. The subscription price will lie between or at (x) the volume-weighted average price of the shares from the beginning of the subscription period until the determination of the subscription price and (y) the current market price of our shares at the time of the determination of the subscription price. In determining the subscription price, we will, among other things, consider the current market conditions and the preliminary results of the bookbuilding process for the shares to be offered in the global offering.
Method of Subscription and Payment.You will receive from your custodian bank necessary information relating to the subscription for new shares, a statement telling you the number of rights you are entitled to exercise and a letter requesting instructions about whether you want to exercise or sell your rights. You may exercise your rights by delivering a properly completed and signed instruction letter to the custodian bank by mail or by hand. Deposit in the mail is not enough.If you fail to return the instruction letter to your custodian bank at or about 10:00 a.m. Frankfurt time (depending on the respective custodian bank’s deadline) on June 24, 2004, the custodian bank will, in accordance with the general terms and conditions of the custody arrangements as standardized in Germany, sell your rights for you on a best-efforts basis unless you have made other arrangements with the custodian bank. It will remit the proceeds from the sale to your account.
You may pay the subscription price in euros by delivering (by mail or by hand) a certified check to that custodian bank, by money transfer using a money transfer order to a bank account of that custodian bank or, if you have a bank account with that custodian bank, by authorizing that custodian bank to deduct the full amount of the subscription price from your bank account at the custodian bank.In all cases the custodian bank must receive payment of the full subscription price on or before July 2, 2004. You must draw certified checks and money transfer orders on a euro account at a bank or savings institution in the European Union. You should make your certified check or money transfer order payable to or name as payee the custodian bank. You will not receive interest if you pay before July 2, 2004.
You will elect the method of delivering instruction letters and paying the subscription price, and you will bear any risk associated with it. If you send instruction letters or payments by mail, you should use registered mail, properly insured, with return receipt requested, and allow enough time to ensure delivery of the instruction letters before the rights expire and delivery and clearance of payment before the payment date for the new shares.
The underwriters will determine all questions about the timeliness, validity, form and eligibility of exercising rights. Their determinations will be final and binding. The underwriters will offer the new shares to existing shareholders in accordance with § 186(5) of the German Stock Corporation Act (Aktiengesetz). GPC Biotech may decide to waive a defect or irregularity, or permit you to correct a defect or irregularity within the time it determines. Instruction letters will not be considered received or
158
Table of Contents
accepted until GPC Biotech has waived all irregularities or you have cured them in time. Neither GPC Biotech nor the custodian bank has to notify you of any defect or irregularity in submitting instruction letters. They will not incur any liability for failing to do so.
Partial Exercise of Rights. If you hold shares and wish to subscribe for only a portion of the new shares you may do so by giving appropriate instructions in the instruction letter to be returned to your custodian bank. You may not, however, subscribe for any fractional shares.
If you deliver a completed and properly signed instruction letter you will be irrevocably exercising the related rights. You may not cancel or modify the exercise. By delivering the instruction letter you will be promising to pay the subscription price by July 2, 2004.
Purchase and Sale of Rights. You may exercise, sell or transfer rights to others through banks in Germany and through banks or brokers in other jurisdictions outside the United States where ordinary shares trade. GPC Biotech ordinary shares trade on the Frankfurt Stock Exchange.
You may place an order with custodian bank either:
| 1. | To purchase any number of rights for your account, whether or not you are exercising a subscription, or |
| 2. | To sell rights for your account, whether or not you are exercising a subscription. |
The custodian bank must receive your order before approximately 10:00 a.m. Frankfurt time (depending on the respective custodian bank’s deadline) on June 24, 2004. You must instruct your custodian bank in writing to purchase or sell rights. You may, before the date on which the rights expire, instruct the custodian in writing to exercise rights.
Unexercised Rights. If you do not deliver a letter to your custodian bank by approximately 10:00 a.m. Frankfurt time (depending on the respective custodian bank’s deadline) on June 24, 2004 the custodian bank will, in accordance with the general terms and conditions of the custody arrangements as standardized in Germany, sell your rights for you on a best-efforts basis. It will remit the proceeds from the sale to your account. The underwriters will sell new shares which remain unsubscribed for after the time the rights expire in the global offering.
Delivery of New Shares. You should receive delivery of the new shares you subscribed for through a credit of the new shares to your securities custody account on or about July 2, 2004. New shares will rank equally in all respects with existing shares.
Important Notices
We will only determine the subscription price during the subscription period, possibly as late as 12:00 p.m. (noon) Frankfurt time on June 25, 2004. Therefore, we advise you to inform yourself about the subscription price via the various media set forth below before exercising your subscription rights.
In addition, since we will only determine the subscription price during the subscription period, a price for the traded subscription rights may not be determinable or, to the extent that a price for the traded subscription rights can be determined, any such price may not constitute the arithmetic value of the subscription rights.
Moreover, the initial offer price in the global offering is only expected to be determined on June 29, 2004 on the basis of the bookbuilding process. Depending on the development of our share price and the result of the bookbuilding process, the subscription price may by higher than the initial offer price in the global offering. Our existing shareholders should be aware that they may not be able to acquire shares by exercising their subscription rights in the rights offering for a lesser price than the price that they would have to pay when participating in the global offering or purchasing our shares in the market.
159
Table of Contents
The validity of the shares and certain other legal matters will be passed upon by Shearman & Sterling LLP, Frankfurt, Germany and New York, New York, counsel for GPC Biotech AG, and Sullivan & Cromwell LLP, Frankfurt, Germany, counsel for the underwriters.
Ernst & Young AG Wirtschaftsprüfungsgesellschaft, independent registered public accounting firm, have audited our consolidated financial statements at December 31, 2003, 2002 and 2001, and for each of the three years in the period ended December 31, 2003, as set forth in their report. We have included our financial statements in the prospectus and elsewhere in the registration statement in reliance on Ernst & Young AG Wirtschaftsprüfungsgesellschaft’s report, given on their authority as experts in accounting and auditing.
The address of the independent registered public accounting firm is Arnulfstrasse 126, D-80636 Munich, Germany.
ENFORCEABILITY OF CIVIL LIABILITIES
We are organized under the laws of Germany. Our and our subsidiary’s directors and officers, as well as certain of the experts named in this prospectus, are non-U.S. residents, and a substantial portion of our assets and the assets of our directors and officers and these experts are and will be located outside the United States. As a result, you may not be able to effect service of process within the United States upon these persons or to enforce, in U.S. courts, against these persons judgments of U.S. courts predicated upon any civil liability provisions of the U.S. federal or state securities laws. In addition, you may not be able to enforce certain civil liabilities predicated upon U.S. federal or state securities laws in Germany against us, our directors and officers and the persons named in this prospectus.
EXPENSES RELATING TO THIS OFFERING
The following table sets forth the estimated costs and expenses, other than the underwriting discounts and commissions, payable by us in connection with this offering (all amounts are estimated):
U.S. Securities and Exchange Commission registration fee | $ | 19,000 | |
Nasdaq National Market listing fee | 5,000 | ||
NASD | 14,700 | ||
Legal fees and expenses | 1,000,000 | ||
Accounting fees and expenses | 580,000 | ||
Printing costs | 300,000 | ||
Miscellaneous | 1,481,000 | ||
Total | $ | 3,399,700 | |
160
Table of Contents
WHERE YOU CAN FIND MORE INFORMATION
We have filed a Registration Statement on Form F-1 with the U.S. Securities and Exchange Commission (the “SEC”) of which this prospectus forms a part. This prospectus does not contain all of the information included in the Registration Statement, certain parts of which have been omitted in accordance with the rules and regulations of the SEC. For further information about us and our shares and ADSs, you should refer to our Registration Statement and its exhibits. This prospectus summarizes the content of contracts and other documents that we refer you to. Since this prospectus may not contain all of the information that is important to you, you should review the full text of these documents. We have included copies of these documents as exhibits to our Registration Statement.
As a result of this offering, we will become subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, and file reports, including annual reports on Form 20-F, and other information with the SEC. However, as we are a foreign private issuer, we and our shareholders are exempt from some of the Exchange Act reporting requirements. The reporting requirements that do not apply to us or our shareholders include the proxy solicitation rules and Section 16 short-swing profit reporting for our officers and directors and for holders of more than 10% of our shares. In addition, we are not required to file annual, quarterly or current reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. However, we will file with the SEC, as long as we are required to do so, within 180 days after the end of each fiscal year, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm. We also intend to file periodic reports on Form 6-K. You may read and obtain copies, at the prescribed rate, of any document we file with the SEC at its public reference rooms at 450 Fifth Street, N.W., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for more information on the public reference rooms and their copy charges. Our SEC filings, including the prospectus, are also available to you on the SEC’s website at www.sec.gov.
161
Table of Contents
CONSOLIDATED FINANCIAL STATEMENTS
Contents
| F-2 | ||
| F-3 | ||
| F-4 | ||
| F-5 | ||
| F-6 | ||
| F-7 | ||
| F-32 | ||
| F-33 | ||
| F-34 | ||
Unaudited Interim Consolidated Statements of Changes in Shareholders’ Equity | F-35 | |
Notes to the Unaudited Interim Consolidated Financial Statements | F-36 |
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Supervisory Board and Shareholders of GPC Biotech AG:
We have audited the accompanying consolidated balance sheets of GPC Biotech AG as of December 31, 2003, 2002 and 2001, and the related consolidated statements of operations, changes in shareholders’ equity, and cash flows for each of the three years in the period ended December 31, 2003. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of GPC Biotech AG at December 31, 2003, 2002 and 2001, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2003 in conformity with accounting principles generally accepted in the United States.
As discussed in Note 2 to the consolidated financial statements, effective January 1, 2002, the Company adopted Statement of Financial Accounting Standards No. 142, “Goodwill and Other Intangible Assets.”
As discussed in Note 3, the accompanying consolidated balance sheets of GPC Biotech AG as of December 31, 2003, 2002 and 2001, and the related consolidated statements of operations, changes in shareholders’ equity, and cash flows for each of the three years in the period ended December 31, 2003 have been restated.
Munich, March 5, 2004
except for Note 3, as to which the date is June 8, 2004
Ernst & Young AG
Wirtschaftsprüfungsgesellschaft
/s/ Dr. E. Napolitano | /s/ D. Gallowsky | |
Dr. E. Napolitano | D. Gallowsky | |
German Public Auditor | German Public Auditor |
F-2
Table of Contents
GPC BIOTECH AG
| December 31, | |||||||||
| 2003 | 2002 | 2001 | |||||||
| (in thousand€, except share and per share data) | |||||||||
Assets | |||||||||
Current assets: | |||||||||
Cash and cash equivalents | 34,947 | 39,947 | 91,245 | ||||||
Marketable securities and short-term investments | 54,221 | 74,935 | 48,885 | ||||||
Accounts receivable | 755 | 726 | 731 | ||||||
Prepaid expenses | 1,540 | 906 | 1,414 | ||||||
Other current assets | 2,042 | 2,181 | 2,966 | ||||||
Total current assets | 93,505 | 118,695 | 145,241 | ||||||
Property and equipment, net | 3,264 | 5,128 | 5,425 | ||||||
Goodwill, net | — | — | 8,008 | ||||||
Intangible assets acquired in a business combination, net | 613 | 2,676 | 5,308 | ||||||
Other assets, non-current | 1,679 | 2,860 | 388 | ||||||
Restricted cash | 2,503 | 2,974 | 3,490 | ||||||
Total assets | 101,564 | 132,333 | 167,860 | ||||||
Liabilities and shareholders’ equity | |||||||||
Current liabilities: | |||||||||
Accounts payable | 629 | 387 | 2,196 | ||||||
Accrued expenses and other current liabilities | 6,499 | 4,873 | 4,993 | ||||||
Current portion of deferred revenue, related party | 5,238 | 7,236 | 7,398 | ||||||
Current portion of deferred revenue | 165 | 590 | 1,205 | ||||||
Current portion of long-term debt | 128 | 128 | 128 | ||||||
Current portion of capital lease obligations | 308 | 648 | 634 | ||||||
Total current liabilities | 12,967 | 13,862 | 16,554 | ||||||
Capital lease obligations, net of current portion | 320 | 761 | 785 | ||||||
Long-term debt, net of current portion | 639 | 767 | 895 | ||||||
Deferred revenue, related party, net of current portion | 4,875 | 6,826 | 10,447 | ||||||
Deferred revenue, net of current portion | — | 2,480 | — | ||||||
Convertible bonds | 884 | 367 | 86 | ||||||
Shareholders’ equity: | |||||||||
Ordinary shares,€ 1 non-par, notional value; shares authorized: 39,225,630 at December 31, 2003, 38,934,130 at December 31, 2002 and 29,276,130 at December 31, 2001 shares issued and outstanding: 20,754,075 at December 31, 2003, 20,719,780 at December 31, 2002 and 20,615,496 at December 31, 2001 | 20,754 | 20,720 | 20,615 | ||||||
Additional paid-in capital | 190,335 | 187,781 | 184,046 | ||||||
Subscribed shares | 215 | — | — | ||||||
Accumulated other comprehensive income (loss) | (2,102 | ) | (739 | ) | 1,990 | ||||
Accumulated deficit | (127,323 | ) | (100,492 | ) | (67,558 | ) | |||
Total shareholders’ equity | 81,879 | 107,270 | 139,093 | ||||||
Total liabilities and shareholders’ equity | 101,564 | 132,333 | 167,860 | ||||||
See accompanying notes to consolidated financial statements.
F-3
Table of Contents
GPC BIOTECH AG
CONSOLIDATED STATEMENTS OF OPERATIONS
| Year ended December 31, | |||||||||
| 2003 | 2002 | 2001 | |||||||
| (in thousand€, except share and per share data) | |||||||||
Collaborative revenues(a) | 20,821 | 20,499 | 12,314 | ||||||
Grant revenues | 773 | 1,012 | 1,575 | ||||||
Total revenues | 21,594 | 21,511 | 13,889 | ||||||
Research and development expenses | 37,741 | 38,053 | 30,644 | ||||||
General and administrative expenses | 11,526 | 11,191 | 9,903 | ||||||
Impairment of goodwill | — | 7,314 | — | ||||||
Amortization, including impairment of intangible assets acquired in a business combination | 1,801 | 1,349 | 4,692 | ||||||
Total operating expenses | 51,068 | 57,907 | 45,239 | ||||||
Operating loss | (29,474 | ) | (36,396 | ) | (31,350 | ) | |||
Other income | 1,289 | 764 | 1,057 | ||||||
Interest income | 2,892 | 4,350 | 5,076 | ||||||
Other expenses | (1,402 | ) | (1,479 | ) | (835 | ) | |||
Interest expense | (136 | ) | (173 | ) | (165 | ) | |||
| 2,643 | 3,462 | 5,133 | |||||||
Net loss | (26,831 | ) | (32,934 | ) | (26,217 | ) | |||
Basic and diluted loss per share in euro | (1.29 | ) | (1.59 | ) | (1.42 | ) | |||
Shares used in computing basic and diluted loss per share | 20,731,535 | 20,688,515 | 18,509,398 | ||||||
| (a) | Revenues from related party |
Collaborative revenues | 20,192 | 17,525 | 8,009 |
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
GPC BIOTECH AG
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year ended December 31, | |||||||||
| 2003 | 2002 | 2001 | |||||||
| (in thousand€) | |||||||||
Cash flows from operating activities: | |||||||||
Net loss | (26,831 | ) | (32,934 | ) | (26,217 | ) | |||
Adjustments to reconcile net loss to net cash provided by (used in) operating activities: | |||||||||
Depreciation | 1,820 | 2,177 | 1,924 | ||||||
Amortization including impairment of intangible assets | 2,007 | 1,349 | 4,692 | ||||||
Goodwill impairment | — | 7,314 | — | ||||||
Compensation cost for stock option plan and convertible bonds | 2,511 | 3,550 | 3,662 | ||||||
Accrued interest income on short term investments | (60 | ) | (792 | ) | — | ||||
Bond premium amortization | 444 | 308 | — | ||||||
Loss (gain) on sale of property and equipment | 151 | (56 | ) | — | |||||
Loss (gain) on sale of marketable securities and short-term investments | (32 | ) | 21 | (101 | ) | ||||
Changes in operating assets and liabilities: | |||||||||
Accounts receivable | (32 | ) | (12 | ) | (455 | ) | |||
Other assets, current and non-current | (1,201 | ) | 975 | (1,621 | ) | ||||
Accounts payable | 249 | (1,407 | ) | 623 | |||||
Deferred revenue, related party | (3,924 | ) | (3,767 | ) | 17,538 | ||||
Deferred revenue | (276 | ) | (582 | ) | 319 | ||||
Other liabilities and accrued expenses | 2,200 | 319 | 2,008 | ||||||
Net cash provided by (used in) operating activities | (22,974 | ) | (23,537 | ) | 2,372 | ||||
Cash flows from investing activities: | |||||||||
Purchases of property, equipment and licenses | (1,669 | ) | (1,635 | ) | (2,985 | ) | |||
Proceeds from the sale of marketable securities and short-term investments | 121,076 | 100,105 | 155,245 | ||||||
Purchases of marketable securities and short-term investments | (100,684 | ) | (125,241 | ) | (150,783 | ) | |||
Net cash provided by (used in) investing activities | 18,723 | (26,771 | ) | 1,477 | |||||
Cash flows from financing activities: | |||||||||
Principal payments under capital lease obligations | (639 | ) | (698 | ) | (746 | ) | |||
Proceeds from issuance of convertible bonds | 517 | 281 | — | ||||||
Proceeds from issuance of ordinary shares to related party | — | — | 34,223 | ||||||
Proceeds from exercise of stock options, convertible bonds and warrants | 77 | 290 | 1,542 | ||||||
Cash received for subscribed shares | 215 | — | — | ||||||
Principal payment of loans | (128 | ) | (128 | ) | — | ||||
Net cash provided by (used in) financing activities | 42 | (255 | ) | 35,019 | |||||
Effect of exchange rate changes on cash | (769 | ) | (693 | ) | 759 | ||||
Restricted cash | (22 | ) | (42 | ) | (3,490 | ) | |||
Net increase (decrease) in cash | (5,000 | ) | (51,298 | ) | 36,137 | ||||
Cash and cash equivalents at beginning of year | 39,947 | 91,245 | 55,108 | ||||||
Cash and cash equivalents at end of year | 34,947 | 39,947 | 91,245 | ||||||
Supplemental information: | |||||||||
Cash paid for interest | 136 | 173 | 168 | ||||||
Non-cash investing and financing activities: | |||||||||
Purchase of property and equipment under capital lease obligations | — | 863 | 537 | ||||||
Deferred revenue related to sale of research program in exchange for convertible note | — | 2,666 | — | ||||||
Write off of shares received from conversion of note, including accrued interest against deferred revenue | 2,630 | — | — | ||||||
See accompanying notes to consolidated financial statements.
F-5
Table of Contents
GPC BIOTECH AG
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
(in thousand€, except share data)
| Ordinary Shares | Additional | Subscribed | Accumulated | Accumulated | Total | ||||||||||||
| Shares | Amount | ||||||||||||||||
Balance at December 31, 2000 | 17,798,714 | 17,798 | 147,436 | — | (41,341 | ) | 623 | 124,516 | |||||||||
Components of comprehensive income (loss): | |||||||||||||||||
Net loss | — | — | — | — | (26,217 | ) | — | (26,217 | ) | ||||||||
Change in unrealized gain on available-for-sale securities | — | — | — | — | — | (111 | ) | (111 | ) | ||||||||
Accumulated translation adjustments | — | — | — | — | — | 1,478 | 1,478 | ||||||||||
Total comprehensive loss | — | — | — | — | — | — | (24,850 | ) | |||||||||
Sale of ordinary shares | 1,690,000 | 1,690 | 32,533 | — | — | — | 34,223 | ||||||||||
Exercise of stock options, convertible bonds and warrants | 1,126,782 | 1,127 | 415 | — | — | — | 1,542 | ||||||||||
Compensation cost, stock options and convertible bonds | — | — | 3,662 | — | — | — | 3,662 | ||||||||||
Balance at December 31, 2001 | 20,615,496 | 20,615 | 184,046 | — | (67,558 | ) | 1,990 | 139,093 | |||||||||
Components of comprehensive income (loss): | |||||||||||||||||
Net loss | — | — | — | — | (32,934 | ) | — | (32,934 | ) | ||||||||
Change in unrealized gain on available-for-sale securities | — | — | — | — | — | 502 | 502 | ||||||||||
Accumulated translation adjustments | — | — | — | — | — | (3,231 | ) | (3,231 | ) | ||||||||
Total comprehensive loss | — | — | — | — | — | — | (35,663 | ) | |||||||||
Exercise of stock options, convertible bonds and warrants | 104,284 | 105 | 185 | — | — | — | 290 | ||||||||||
Compensation cost, stock options and convertible bonds | — | — | 3,550 | — | — | — | 3,550 | ||||||||||
Balance at December 31, 2002 | 20,719,780 | 20,720 | 187,781 | — | (100,492 | ) | (739 | ) | 107,270 | ||||||||
Components of comprehensive income (loss): | |||||||||||||||||
Net loss | — | — | — | — | (26,831 | ) | — | (26,831 | ) | ||||||||
Change in unrealized gain on available-for-sale securities | — | — | — | — | — | 30 | 30 | ||||||||||
Accumulated translation adjustments | — | — | — | — | — | (1,393 | ) | (1,393 | ) | ||||||||
Total comprehensive loss | — | — | — | — | — | — | (28,194 | ) | |||||||||
Exercise of stock options and convertible bonds | 34,295 | 34 | 43 | 215 | — | — | 292 | ||||||||||
Compensation cost, stock options and convertible bonds | — | — | 2,511 | — | — | — | 2,511 | ||||||||||
Balance at December 31, 2003 | 20,754,075 | 20,754 | 190,335 | 215 | (127,323 | ) | (2,102 | ) | 81,879 | ||||||||
See accompanying notes to consolidated financial statements.
F-6
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of Business and Organization
GPC Biotech AG, a company incorporated in the Federal Republic of Germany, has its registered offices in Martinsried/Planegg (hereafter referred to as GPC Biotech or the “Company”). In its internal drug discovery and development programs, the Company focuses on oncology and has programs in various stages of development. The Company’s lead product candidate—satraplatin—entered a Phase 3 clinical trial as a second-line chemotherapy treatment in hormone-refractory prostate cancer in the fall of 2003. Other anticancer programs in development include a monoclonal antibody with a novel mechanism-of-action and a cell cycle inhibitor. The Company is leveraging its drug discovery technologies to support the growth of its drug pipeline. The Company has successfully formed collaborations with a number of pharmaceutical and biotechnology firms.
The Company has one wholly owned subsidiary, GPC Biotech Inc., with two locations, Waltham (MA, USA) and Princeton (NJ, USA). The Waltham site is primarily engaged in oncology drug discovery activities, in addition to activities related to the collaboration with the ALTANA Research Institute (a research center of ALTANA Pharma AG), also located in Waltham. The Princeton location is the headquarters for the Company’s drug development activities.
2. Significant Accounting Policies
Basis of Financial Statement Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”).
Cash Equivalents, Marketable Securities and Short-term Investments
Highly-liquid investments with maturities of three months or less at the date of purchase are considered to be cash equivalents; those with maturities greater than three months are included in marketable securities and short-term investments. In accordance with Statement of Financial Accounting Standards (SFAS) No. 115 “Accounting for Certain Investments in Debt and Equity Securities”, the Company has classified all marketable securities and short-term investments as available-for-sale and, as a result, classifies them as current assets and carries such amounts at fair value. Unrealized gains and losses are included in accumulated other comprehensive income (loss) in shareholders’ equity until realized. Realized gains and losses on sales of all such securities are reported in earnings and computed using the specific identification cost method. Realized gains or losses and charges for other-than-temporary declines in value, if any, on available-for-sale securities are reported in other income or expense as incurred.
As of December 31, 2003, 2002 and 2001,€ 1,200,000,€ 2,700,000 and€ 2,500,000, respectively, of short-term investments were pledged as security for a bank guarantee related to a line of credit for leased assets.
As a general policy the Company does not invest in equity securities for cash management purposes. Any equity securities owned by the Company were acquired as part of licensing agreements.
F-7
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Unused Lines of Credit
As of December 31, 2003, the Company had one unused line of credit totaling€ 212,000 that is used for capital leases of equipment in the United States. Total credit available under this agreement is limited to $1,000,000 (€ 796,700) and is secured by the assets purchased under the lease line.
Restricted Cash
Restricted cash represents amounts held in an interest bearing escrow account as a security deposit related to a lease.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to credit risk consist primarily of cash and cash equivalents and marketable securities and short-term investments. The risk is minimized by the Company’s policies, which limit investments to those that have relatively short maturities and that are placed with highly rated issuers.
The Company’s largest customer, ALTANA Pharma AG, accounted for 94% of total revenues in 2003 representing€ 20,192,000, 81% of total revenues in 2002 representing€ 17,525,000 and 58% of the total revenues in 2001 representing€ 8,009,000. In addition, Boehringer Ingelheim accounted for 11% of total revenues in 2002 and 15% in 2001. In 2001, Aventis Pharma Deutschland GmbH accounted for 13% of total revenues, and the “Bundesministerium für Bildung und Forschung” (the German Federal Ministry for Education and Research, or BMBF) accounted for 11% of total revenues. No other customer accounted for more than 10% of total revenues in 2003, 2002 and 2001.
Segment Information
SFAS No. 131, “Disclosures about the Segments of an Enterprise and Related Information”, establishes standards for the way that public business enterprises report information about operating segments in annual financial statements and requires that those enterprises report selected information about operating segments in interim financial reports. SFAS No. 131 also establishes standards for related disclosures about the products and services, geographic areas and major customers.
The Company operates in one business segment, which primarily focuses on discovery and development of anticancer drugs. The Company’s revenue is derived primarily from research collaborations with pharmaceutical companies. Additional revenue is derived from governmental grants for specific research and development programs. The results of operations are reported to the Company’s chief operating decision-makers on an aggregate basis.
The total book value of long-lived assets located outside of the Company’s home location of Germany was€ 2,326,000,€ 5,413,000 and€ 16,107,000 at December 31, 2003, 2002 and 2001, respectively. All of these assets were located in the United States of America.
Revenues from external customers attributed to the Company’s home location of Germany were€ 21,197,000,€ 20,466,000 and€ 13,889,000 in the years ended December 31, 2003, 2002 and 2001, respectively. Revenues from external customers attributed to locations outside of Germany were€ 397,000,€ 1,045,000 and€ 0 in the years ended December 31, 2003, 2002 and 2001, respectively.
F-8
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Revenues are attributed to countries based on the location of the Company’s legal entity that is party to the underlying contract.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets as follows:
Estimated Useful Life | ||
Computer equipment and software costs | 3 years | |
Office equipment | 10 years | |
Laboratory equipment | 5 years | |
Furniture and fixtures | 5 years | |
Leasehold improvements | Lesser of useful life or life of lease | |
Equipment under capital lease | Lesser of useful life or life of lease |
Goodwill and Intangible Assets Acquired in a Business Combination
Intangible assets consist of specifically identified intangible assets. Goodwill is the excess of any purchase price over the estimated fair market value of net tangible and intangible assets acquired. Amortization of intangible assets is computed using the straight-line method over the useful lives of the respective assets.
| Estimated Useful Life | ||
Patents | 10 years | |
Acquired partnered technology | 4 years |
The Company accounts for goodwill and indefinite-lived intangible assets in accordance with SFAS No. 142, “Goodwill and Other Intangible Assets.” Under SFAS No. 142, goodwill and indefinite-lived intangible assets are not amortized, but are reviewed annually for impairment, or more frequently if impairment indicators arise. The Company adopted SFAS No. 142 effective January 1, 2002 and reclassified amounts to goodwill that were previously allocated to assembled workforce. Upon adoption, the Company ceased amortizing goodwill.
F-9
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following proforma adjusted net loss and net loss per ordinary share have been prepared as if SFAS No. 142 had been applied retroactively:
| Year ended December 31, | |||||||||
| 2003 | 2002 | 2001 | |||||||
| (in thousand€, except per share amounts) | |||||||||
Net loss | (26,831 | ) | (32,934 | ) | (26,217 | ) | |||
Add back: Goodwill amortization | — | — | 3,522 | ||||||
Add back: Assembled workforce amortization | — | — | 284 | ||||||
Adjusted net loss | (26,831 | ) | (32,934 | ) | (22,411 | ) | |||
Net loss per ordinary share, basic and diluted: | |||||||||
Net loss | (1.29 | ) | (1.59 | ) | (1.42 | ) | |||
Add back: Goodwill amortization | — | — | 0.19 | ||||||
Add back: Assembled workforce amortization | — | — | 0.02 | ||||||
Adjusted net loss per share | (1.29 | ) | (1.59 | ) | (1.21 | ) | |||
Impairment of Long-lived Assets
In accordance with SFAS No. 144, “Accounting for the Impairment or Disposal of Long-Lived Assets”, the Company reviews long-lived assets, including definite-lived intangible assets, for impairment when impairment indicators exist and, if necessary, recognizes an impairment expense equal to the difference between the carrying amount of the asset and the fair value as determined by discounted cash flows.
Fair Value of Financial Instruments
The carrying value of financial instruments such as cash and cash equivalents, accounts receivable and accounts payable approximate their fair value based on the short-term maturities of these instruments. The estimated fair values of the Company’s long-term debt approximate their carrying value based upon the current rates offered to the Company for similar type arrangements.
Revenue Recognition
The Company’s revenues consist of fees earned from research and development collaborations and grant revenues. Revenues from research and development agreements generally consist of licensing fees and/or technology access fees, fees for research support, as well as milestone and royalty payments. Revenues from grants generally consist of reimbursements of a portion of expenses incurred in performing research in specified projects.
The Company generally receives non-refundable licensing and technology access and initiation fees upfront upon signing of a research and collaboration agreement. In addition, the Company generally receives fixed non-refundable annual licensing fees upfront each year for the duration of the agreement. In accordance with Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, all fees received or to be received under these arrangements are recognized on a straight line basis over the term of the agreements, as this is the period in which research is expected to be performed.
F-10
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Fees for research support are received from collaboration partners for research services performed under the agreements. Fees are generally stated at a yearly flat fee rate per research scientist. Fees are generally billed on a monthly or quarterly basis in advance. The Company recognizes research support revenue as the services are performed. Amounts received in advance of services performed are recorded as deferred revenue until earned.
Milestone revenues are derived from the achievement of predetermined goals, which are defined in the collaboration agreements. These milestones are subject to significant contingencies and, therefore, the related contingent revenue is not realized until the milestone has been reached and customer acceptance has been obtained. Payments received that relate to the achievement of substantive milestones that were contingent at the outset of the arrangement are recorded as revenue when the milestones are achieved.
Royalty revenues generally result from sales of products based on research performed under collaboration agreements. Royalties are based on the volume of product sold and are recognized as revenue upon notification of sales from the collaboration partner. The Company has not yet received or recognized any royalty payments from collaboration agreements.
Grant revenues from governmental agencies for the support of specific research and development projects are recorded as revenue to the extent the related expenses have been incurred.
At the November 21, 2002 meeting of the FASB Emerging Issues Task Force (“EITF”), a final consensus was reached on EITF Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (EITF 00-21). EITF 00-21 addresses how to account for arrangements that may involve the delivery or performance of multiple products, services and/or rights to use assets. The consensus uses a transaction model that divides arrangements into separate units of accounting based on certain criteria. Additionally, consideration given should be allocated to the different units of accounting based on their fair value. Revenue recognition should be considered separately for each accounting unit. The Company adopted EITF 00-21 for all revenue arrangements after July 1, 2003. There were no new arrangements entered into since adoption of the consensus. The adoption of EITF 00-21 did not have a material impact on the Company’s results of operations, financial position or cash flows.
Research and Development
The Company enters into research and development agreements with third parties consisting of technology licensing fees and technology access fees, as well as, milestone and royalty payments. In addition, the Company purchases chemical compounds and the right to develop the compounds in perpetuity and on an exclusive basis. GPC Biotech also enters into contracts with contract research organizations to conduct clinical trials, which are expensed as incurred.
Technology license fees and technology access fees relate to technology licenses that the Company pays in connection with its research and development arrangements. These licenses have alternative future uses and are used in various research and development programs and some may be sublicensed to third parties. If the licenses do not have an alternative future use they are expensed immediately.
The Company treats payments for licenses that have alternative future uses as intangible assets in accordance with SFAS No. 2, “Accounting for Research and Development Costs”, (“SFAS No. 2”) and amortizes them to research and development expense over the period of the license.
F-11
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Technology license fees, which are typically paid upfront annually and relate to licenses where the Company typically has the option to renew for a subsequent year at the end of the license term, are recorded as prepaid expenses.
Technology access fees, which relate to upfront, one-time fees paid to access technology in connection with obtaining technology licenses with terms in excess of one year, are classified as other assets, non-current.
On January 31, 2003, the Company entered into a license agreement for technology to be used in research and development and to be sublicensed to collaboration partners with terms in excess of one year. This license contained payment terms for technology access fees and technology license fees. The gross carrying value of technology access fees was€ 1,175,000 as of December 31, 2003. These access fees are amortized on a straight line basis over the license term of five years. At December 31, 2003, the accumulated amortization was€ 207,000 and the remaining estimated useful life was 49 months. The carrying value of technology access fees included in other assets, non-current amounted to€ 968,000 at December 31, 2003. Amortization related to these fees is recorded in research and development expenses. In the consolidated statement of cash flows, the technology access fee is classified as an investing activity and is included in purchases of property, equipment and licenses.
Milestone payments to other parties are expensed when the liability is incurred, which is when the milestones are achieved.
Fees paid to license chemical compounds and for the right to further develop those compounds are generally paid upfront and expensed as incurred in accordance with SFAS No. 2.
Income Tax
The Company provides for income taxes in accordance with SFAS No. 109, “Accounting for Income Taxes”. Under SFAS No. 109, the liability method is used in accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. Deferred tax assets are reduced by a valuation allowance to reflect the uncertainty associated with their ultimate realization.
Foreign Currency
The functional currency of GPC Biotech AG is the euro whereas the functional currency of GPC Biotech Inc. is the U.S. Dollar. All balance sheet items have been translated into euro using the exchange rate at the balance sheet date and all items in the statement of operations have been converted into euro at average rates during the year. The effect of the translation is shown as a separate component of shareholders’ equity.
Gains and losses resulting from foreign currency transactions denominated in a currency other than the functional currency are recorded at the approximate rate of exchange at the transaction date. The gross amounts of foreign currency gains and losses are shown in other income and other expenses, respectively in the statement of operations.
For the years ended December 31, 2003, 2002 and 2001, the Company recorded a net foreign currency loss of€ 215,000, a net loss of€ 936,000 and a net gain of€ 195,000, respectively.
F-12
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiary, GPC Biotech Inc. All significant intercompany investments, accounts and transactions have been eliminated.
On July 1, 2002, the Company merged its two wholly owned subsidiaries, GPC Biotech Inc. and GPC Biotech USA, Inc., to create one wholly owned subsidiary, GPC Biotech Inc., with two locations.
Stock-Based Compensation
The Company accounts for its stock option plans and convertible bonds in accordance with the provisions of SFAS No. 123, “Accounting for Stock-Based Compensation.” Convertible bonds are issued as compensation to Management Board, senior management and Supervisory Board members. The fair value of options and convertible bonds granted is established as of the date of grant and recognized as compensation expense over the vesting life of the option or convertible bond. Compensation expense recognized on options and convertible bonds that do not vest due to termination of employment is reversed in the period of forfeiture. Compensation cost for stock options and convertible bonds is included in research and development expenses and general and administrative expenses in the statement of operations, which is allocated based on the department of the recipient.
Use of Estimates
The preparation of consolidated financial statements in accordance with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities and the reported amounts of revenues and expenses at the date of financial statements. Actual results could differ from those estimates.
Reclassifications
Certain prior year amounts in the statement of cash flows have been reclassified to conform with the current year presentation.
New Accounting Standards
In January 2003, the Financial Accounting Standards Board (FASB) issued Interpretation No. 46,Consolidation of Variable Interest Entities(FIN 46). FIN 46 amended Accounting Research Bulletin No. 51,Consolidated Financial Statements, and established standards for determining under what circumstances a variable interest entity (VIE) should be consolidated with its primary beneficiary. FIN 46 also requires disclosures about VIEs that an entity is not required to consolidate, but in which it has a significant variable interest. On December 17, 2003, the FASB deferred the effective date of FIN 46 to no later than the end of the first reporting period that ends after March 15, 2004, however, for special purpose entities, public companies are required to apply FIN 46 by no later than December 31, 2003. The Company does not believe that any entities will be consolidated with GPC Biotech AG as a result of FIN 46.
In December 2003, the Securities and Exchange Commission (SEC) published Staff Accounting Bulletin (SAB) No. 104, “Revenue Recognition”. SAB 104 updates portions of the SEC staff’s
F-13
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
interpretive guidance provided in SAB 101. SAB 104 deletes interpretive material no longer necessary, and conforms the interpretive material retained to current authoritative accounting and auditing guidance and SEC rules and regulations, to reflect pronouncements issued by the FASB and EITF on various revenue recognition topics. The adoption of SAB 104 did not have any impact on the results of operations, financial position, or cash flows of the Company.
3. Restatement
In connection with internal research and development programs the Company may be required to make milestone payments to other parties. Historically, the Company accrued for the milestone payments when management determined that the achievement of the milestone was probable, and the amount was estimable. The milestone payments were accrued ratably over the period beginning from the time the milestone was determined to be probable and estimable through the expected date of achievement of the milestone.
In connection with the review of its registration statement by the United States Securities and Exchange Commission, the Company has revised its accounting policy related to milestone payments to make its policy consistent with U.S. GAAP. In accordance with this policy, the Company expenses milestone payments when the liability is incurred, which is when the milestones are achieved.
The Company determined that the restatement of its financial statements for the years ended December 31, 2003, 2002 and 2001 is necessary and appropriate to reflect the application of this revised accounting policy. The following table reflects the impact of the revised accounting policy on amounts as previously reported and as restated resulting from the restatement:
| As of and for the Years Ended December 31, | |||||||||||||||||||||||||||
2003 (in thousand€, | 2002 (in thousand€, | 2001 (in thousand€, | |||||||||||||||||||||||||
| As Reported | As Restated | Adjustment | As Reported | As Restated | Adjustment | As Reported | As Restated | Adjustment | |||||||||||||||||||
Accrued expenses and other current liabilities | 8,522 | 6,499 | (2,023 | ) | 9,458 | 4,873 | (4,585 | ) | 8,195 | 4,993 | (3,202 | ) | |||||||||||||||
Total shareholders’ equity | 79,856 | 81,879 | 2,023 | 102,685 | 107,270 | 4,585 | 135,891 | 139,093 | 3,202 | ||||||||||||||||||
Research and development expenses | 35,630 | 37,741 | 2,111 | 39,831 | 38,053 | (1,778 | ) | 33,840 | 30,644 | (3,196 | ) | ||||||||||||||||
Net loss | (24,351 | ) | (26,831 | ) | (2,480 | ) | (34,490 | ) | (32,934 | ) | 1,556 | (29,413 | ) | (26,217 | ) | 3,196 | |||||||||||
Basic and diluted loss per share in€ | (1.17 | ) | (1.29 | ) | (0.12 | ) | (1.67 | ) | (1.59 | ) | 0.08 | (1.59 | ) | (1.42 | ) | 0.17 | |||||||||||
F-14
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
4. Marketable Securities and Short-term Investments
The following is a summary of marketable securities at December 31, 2003, 2002 and 2001 (in thousand€), all of which are available-for-sale:
| 2003 | |||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Estimated Fair Value | ||||||
Variable rate corporate bonds | 4,998 | 16 | — | 5,014 | |||||
Fixed rate corporate bonds | 26,397 | 105 | (14 | ) | 26,488 | ||||
Equity securities | 781 | 75 | — | 856 | |||||
Total | 32,176 | 196 | (14 | ) | 32,358 | ||||
| 2002 | |||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Estimated Fair Value | ||||||
Variable rate corporate bonds | 20,003 | 1 | (6 | ) | 19,998 | ||||
Fixed rate corporate bonds | 32,185 | 127 | — | 32,312 | |||||
Total | 52,188 | 128 | (6 | ) | 52,310 | ||||
| 2001 | |||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Estimated Fair Value | ||||||
Variable rate corporate bonds | 24,991 | — | (84 | ) | 24,907 | ||||
Fixed rate corporate bonds | 2,476 | — | (12 | ) | 2,464 | ||||
Total | 27,467 | — | (96 | ) | 27,371 | ||||
The fair market values of marketable securities are based on quoted market prices. All bonds included in marketable securities have maturities of 3 years or less.
The equity securities above represent 128,370 unregistered shares of Spectrum Pharmaceuticals, Inc. purchased in accordance with a licensing agreement (see Note 17).
F-15
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following is a summary of the short-term investments at December 31, 2003, 2002 and 2001 (in thousand€), all of which are available-for-sale:
| 2003 | ||||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Accrued Interest Receivable | Estimated Fair Value | ||||||
Bond fund | 21,334 | 469 | — | 60 | 21,863 | |||||
Total | 21,334 | 469 | — | 60 | 21,863 | |||||
| 2002 | ||||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Accrued Interest Receivable | Estimated Fair Value | ||||||
Bond fund | 21,334 | 499 | — | 792 | 22,625 | |||||
Total | 21,334 | 499 | — | 792 | 22,625 | |||||
| 2001 | ||||||||||
| Cost | Gross Unrealized | Gross Unrealized Losses | Accrued Interest Receivable | Estimated Fair Value | ||||||
Bond fund | 21,221 | 293 | — | — | 21,514 | |||||
Total | 21,221 | 293 | — | — | 21,514 | |||||
The realized gain on available-for-sale marketable securities and short-term investments in 2003, 2002 and 2001 was€ 8,000,€ 20,000 and€ 171,000, respectively. The realized loss on available-for-sale marketable securities and short-term investments in 2003, 2002 and 2001 amounted to€ 1,000,€ 41,000 and€ 70,000, respectively.
The aggregate unrealized losses on available-for-sale marketable securities and short-term investments at December 31, 2003 was€ 14,000. The aggregate fair value of securities with unrealized losses at December 31, 2003 was€ 10,180,000 and consisted of fixed-rate corporate bonds with remaining maturities of less than 2 years. These securities were in a loss position for less than 12 months.
F-16
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
5. Property and Equipment
Property and equipment consisted of the following at December 31, 2003, 2002 and 2001 (in thousand€):
| 2003 | 2002 | 2001 | |||||||
Leasehold improvements | 934 | 1,261 | 735 | ||||||
Office and laboratory equipment | 5,421 | 6,469 | 6,515 | ||||||
Computer equipment and related software | 2,567 | 2,654 | 2,235 | ||||||
Furniture and fixtures | 347 | 478 | 374 | ||||||
| 9,269 | 10,862 | 9,859 | |||||||
Less accumulated depreciation | (6,005 | ) | (5,734 | ) | (4,434 | ) | |||
Property and equipment, net | 3,264 | 5,128 | 5,425 | ||||||
Depreciation expense amounted to€ 1,820,000,€ 2,177,000 and€ 1,924,000 in 2003, 2002 and 2001, respectively.
6. Intangible Assets Acquired in a Business Combination
The Company has recorded intangible assets acquired in a business combination based on their fair values, as determined by independent valuations at the date of acquisition, in connection with its acquisition of Mitotix, Inc. (now GPC Biotech, Inc.) in March 2000. These assets consisted of the following:
| December 31, 2003 | |||||
Gross Amount | Accumulated Amortization | ||||
| (in thousand€) | |||||
Amortized intangible assets: | |||||
Patents | 534 | — | |||
Partnered Technology | 1,282 | (1,203 | ) | ||
Total | 1,816 | (1,203 | ) | ||
| December 31, 2002 | |||||
| Gross Carrying Amount | Accumulated Amortization | ||||
| (in thousand€) | |||||
Amortized intangible assets: | |||||
Patents | 2,196 | — | |||
Partnered Technology | 1,536 | (1,056 | ) | ||
Total | 3,732 | (1,056 | ) | ||
F-17
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| December 31, 2001 | |||||
| Gross Carrying Amount | Accumulated Amortization | ||||
| (in thousand€) | |||||
Amortized intangible assets: | |||||
Patents | 4,411 | (772 | ) | ||
Assembled workforce-in-place | 1,149 | (503 | ) | ||
Partnered Technology | 1,818 | (795 | ) | ||
Total | 7,378 | (2,070 | ) | ||
During the fourth quarter 2003, the Company reviewed its intangible assets acquired in a business combination for impairment and recorded an impairment charge at December 31, 2003 relating to the patent portfolio in the amount of€ 1,163,000, thereby creating a new cost basis of€ 534,000. Indicators of impairment arose when management decided in 2003 to discontinue several of the research programs that used these patents. The fair value of the patent portfolio was determined using estimated discounted cash flows of each program that uses the patents. From these cash flows an implied royalty rate, success probabilities and discounting factor were used to determined the fair value of the underlying patents. The impairment charge is included in operating expenses as amortization, including impairment of intangible assets acquired in a business combination.
During the fourth quarter 2002, in conjunction with its annual review of goodwill for impairment, the Company reviewed its acquired intangible assets for impairment and recorded an impairment charge at December 31, 2002 in the amount of€ 507,000 relating to the patent portfolio, thereby creating a new cost basis of€ 2,196,000. Further development work on the programs that used these patents provided additional information regarding the probabilities of success of each of the continuing programs. As a result, some of the programs were discontinued during 2002 or probabilities of success were revised. The fair value of the patent portfolio was determined using estimated discounted cash flows of each program that used the patents. The impairment charge is included in operating expenses as amortization, including impairment of intangible assets acquired in a business combination.
The amortization expense for intangibles acquired in a business combination, including impairment expenses, for the years ended December 31, 2003, 2002 and 2001 was€ 1,801,000,€ 1,349,000 and€ 1,170,000, respectively. The estimated amortization expense for intangibles acquired in a business combination for the next five years is as follows (in thousand€):
For the year ending December 31, | ||
2004 | 133 | |
2005 | 53 | |
2006 | 53 | |
2007 | 53 | |
2008 | 53 |
F-18
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
7. Goodwill
The Company recorded goodwill in connection with its acquisition of Mitotix, Inc. (now GPC Biotech Inc.) in March 2000 in the amount of $ 12.6 million.
The changes in the carrying amount of goodwill for the year ended December 31, 2002 and 2001, are as follows (in thousand€):
Balance as of December 31, 2000 | 10,882 | ||
Amortization | (3,522 | ) | |
Foreign currency translation effects | 648 | ||
Balance as of December 31, 2001 | 8,008 | ||
Reclassification from intangibles upon adoption of SFAS No. 142 | 646 | ||
Impairment charge | (7,314 | ) | |
Foreign currency translation effects | (1,340 | ) | |
Balance as of December 31, 2002 | — | ||
In accordance with SFAS No. 142, the Company is required to allocate its net assets, including goodwill, to components in order to measure impairment of goodwill. The Company has determined that it has a single component and that fair value of the component will be determined primarily based on its traded share price. During 2002, the Company experienced a significant decline in market capitalization due to a decline in its share price. Based on this decline in market capitalization, a goodwill impairment charge of€ 7,314,000 was recognized in 2002. The fair value of the Company was estimated using the value of the market capitalization.
8. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities at December 31, 2003, 2002 and 2001 consisted of the following (in thousand€):
| 2003 | 2002 | 2001 | ||||
Accrued external R&D obligations | 2,108 | 836 | 459 | |||
Accrued personnel expenses | 2,305 | 1,930 | 1,836 | |||
Accrued legal, patent and advisory services | 552 | 659 | 1,096 | |||
Other accruals | 1,262 | 1,341 | 1,273 | |||
Payroll liabilities | 272 | 107 | 329 | |||
| 6,499 | 4,873 | 4,993 | ||||
9. Leases
The Company has entered into several lease arrangements for office, laboratory and storage space as well as for laboratory and office equipment.
The Company incurred rental expenses of€ 5,067,000,€ 5,272,000 and€ 2,760,000 in the years 2003, 2002 and 2001, respectively. Income from subleases amounted to€ 1,393,000,€ 3,051,000 and€ 822,000 in the years 2003, 2002 and 2001, respectively.
The Company has entered into lease agreements for office and laboratory space that expire at different times through 2011. One agreement calls for incremental increases in rental payments in
F-19
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
2004 and 2008. The Company records its rental expenses and sublease rental income using the straight-line method in accordance with SFAS No. 13, “Accounting for Leases”. The remaining balance of accrued rental payments was€ 431,000 at December 31, 2003. Additionally, this lease requires $3,000,000 to be held in escrow as security for credit.
In 2003, the Company did not enter into any new capital leases. During 2002 and in prior years, the Company entered into various capital lease contracts. At December 31, 2003, 2002 and 2001, the gross value of assets under capital leases included in fixed assets was€ 2,259,000,€ 3,357,000 and€ 4,077,000, respectively, and related accumulated amortization was€ 1,763,000,€ 2,176,000 and€ 2,066,000, respectively. Amortization of assets recorded under capital leases is recorded in research and development expenses and general and administrative expenses. All capital lease obligations are collateralized by leased assets.
Future minimum lease commitments for all non-cancelable operating and capital leases for the years ending December 31 are as follows (in thousand€):
| Capital | Operating | ||||
2004 | 342 | 4,572 | |||
2005 | 228 | 4,563 | |||
2006 | 108 | 4,483 | |||
2007 | — | 4,483 | |||
2008 | — | 3,794 | |||
Thereafter | — | 9,376 | |||
Total minimum lease payments | 678 | 31,271 | |||
Less amounts representing interest | (50 | ) | |||
Present value of minimum lease payments | 628 | ||||
Less amounts due within one year | (308 | ) | |||
Obligations under capital lease | 320 | ||||
Aggregate future minimum rentals to be received under non-cancelable subleases as of December 31, 2003 were€ 3,496,000.
10. Loans Receivable from Members of the Management Board and Employees
At December 31, 2003, 2002 and 2001, loans receivable from two members of the Management Board amounted to€ 79,000,€ 126,000 and€ 41,000, respectively, and were included in other current and non-current assets. At December 31, 2003, the amount related to three different loans with original terms of five years each. Two loans are being forgiven in equal installments. The remaining loan will be repaid in full at the end of the loan term.
At December 31, 2003, 2002 and 2001, loans receivable from employees of the Company amounted to€ 181,000,€ 283,000 and€ 297,000, respectively, and are included in other current and non-current assets. At December 31, 2003, the amount related to three different loans with original terms between two and four years. The loans are being forgiven or repaid in equal installments.
Forgiveness of loans, if applicable, is included in compensation expense in the period they are forgiven and is contingent upon continued employment of the employee.
All loans made to members of the Management Board and employees bear interest at the rate of 5.5% to 6% per annum and are made for personal financial purposes.
F-20
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
11. Related Party Disclosures
In 2003, 2001, 2000 and 1998, the Company entered into collaboration agreements with ALTANA Pharma AG (“ALTANA Pharma”, formerly known as Byk Gulden Lomberg Chemische Fabrik GmbH) as described in Note 15. ALTANA Pharma is a related party, since the Company’s Chairman (“Vorstandsvorsitzender”), Dr. Bernd Seizinger, is a member of the Supervisory Board of ALTANA Pharma.
At December 31, 2003 and 2002, there were no accounts receivable from ALTANA Pharma. At December 31, 2001, accounts receivable from ALTANA Pharma were€27,000. All accounts receivable from ALTANA Pharma have been settled for cash at amounts stipulated in the various contracts with ALTANA Pharma. Collaborative revenues and deferred revenues related to ALTANA Pharma have been classified as related party on the statements of operations and balance sheets, respectively.
In addition, the Company subleases part of its facilities to ALTANA Pharma. Of the total sublease income disclosed in Note 9,€ 1,051,000,€ 601,000 and€ 0 for the years ended December 31, 2003, 2002 and 2001, respectively, relates to the subleases with ALTANA Pharma.
12. Financing
Long-term Debt
During September 1999, the Company entered into a loan agreement with IKB Deutsche Industriekreditbank AG in the amount of€ 1,023,000, all of which was received in July 2000. The term of the loan is 10 years with semi-annual repayments beginning in March 2002. The loan is unsecured and bears interest at 4.25% per annum. Interest expenses for the years ended December 31, 2003, 2002 and 2001 were€ 35,000,€ 41,000 and€ 43,000, respectively, all of which was paid.
Future minimum principal payments on the loan for the next five years are as follows (in thousand €):
For the year ending December 31, | ||
2004 | 128 | |
2005 | 128 | |
2006 | 128 | |
2007 | 128 | |
2008 | 128 | |
Thereafter | 127 | |
Total | 767 | |
13. Convertible Bonds and Warrants
Convertible bonds are issued as compensation to members of Management Board, senior management and the Supervisory Board. All convertible bonds are valued at fair value using the Black-Scholes option pricing model using the same assumptions as those used for stock options (see Note 14) and recognized in the statement of operations over their future vesting periods. Total compensation cost related to convertible bonds included in general and administration expenses for the years ended December 31, 2003, 2002 and 2001 was€ 890,000,€ 205,000 and€ 37,000,
F-21
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
respectively. The aggregate fair value of convertible bonds issued in 2003, 2002 and 2001 was€ 1,541,000,€ 692,000 and€ 141,000, respectively.
The Company also issued 676,000 convertible bonds in 1998. These convertible bonds entitled the holder to purchase ordinary shares of the Company at a premium of€ 1.03 above the nominal value of€ 0.13 per convertible bond. The nominal value of the convertible bonds must be repaid by the Company for all unexercised convertible bonds upon expiration in October 2007. The convertible bonds bear interest at the rate of 5% per annum. At December 31, 2003, 370,000 convertible bonds had been exercised and the remaining 306,000 convertible bonds are exercisable.
During 2001, the Company also issued 47,500 convertible bonds at a nominal value of€ 1.00 each to the members of the Supervisory Board. These convertible bonds vest over two years, and half can be exercised at each closing of the ordinary shareholders’ meetings in 2002 and 2003, respectively. The convertible bond holders are entitled to purchase 47,500 ordinary shares of the Company at€ 8.55 per share above the nominal value. The convertible bonds bear interest at the rate of 3.5% per annum. The total amount of interest is payable upon exercise or expiration. At December 31, 2003, 47,500 of the convertible bonds were exercisable.
In August 2002, the Company issued 280,000 convertible bonds at a nominal value of€ 1.00 each to certain members of senior management. These convertible bonds vest over four years and can be first exercised in September 2004. The convertible bond holders are entitled to purchase 280,000 ordinary shares of the Company at€ 3.36 per share above nominal value. The convertible bonds bear interest at the rate of 3.5% per annum. None of the convertible bonds were exercisable at December 31, 2003.
In January 2003, the Company issued 152,500 convertible bonds at a nominal value of€ 1.00 each to certain members of senior management. These convertible bonds vest over four years and can be first exercised in February 2004. The convertible bond holders are entitled to purchase 152,500 ordinary shares of the Company at€ 1.70 per share above nominal value. The convertible bonds bear interest at a rate of 3.5% per annum. None of the convertible bonds were exercisable at December 31, 2003.
In May 2003, the Company issued 364,500 convertible bonds at a nominal value of€ 1.00 each to Supervisory Board members and certain members of senior management. These convertible bonds vest over two years for Supervisory Board members and over four years for the members of senior management. The warrants can be first exercised in June 2005. The convertible bond holders are entitled to purchase 364,500 ordinary shares of the Company at€ 3.87 to€ 4.77 per share above nominal value. The convertible bonds bear interest at the rate of 3.5% per annum. None of the convertible bonds were exercisable at December 31, 2003.
The following table summarizes the status of the various convertible bonds at December 31, 2003 (in thousand€ except for number):
| Number | Nominal amount | |||
Issued | 1,520,500 | 931 | ||
Exercised | 370,000 | 47 | ||
Expired | — | — | ||
Outstanding | 1,150,500 | 884 | ||
Exercisable | 353,500 | 87 |
F-22
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In connection with a 1992 equipment lease financing of Mitotix, Inc. (now GPC Biotech Inc.), the Company issued warrants entitling the holder to purchase up to 24,420 ordinary shares at€ 7.67 per share, exercisable through May 29, 2005. The Company also issued warrants in 1997 entitling the holder to purchase up to 2,320 ordinary shares at€ 29.91 per share, exercisable through May 29, 2005. At December 31, 2003, 4,760 warrants were outstanding.
14. Stock Options Issued to Employees and Consultants
The Company grants stock options to the employees, the Management Board and to certain consultants.
According to the stock option plans, the options give the right to exercise the option within ten years. The respective strike prices equal the five-day average of the Company’s ordinary shares prior to the respective date of grants. The vesting period is three years for options granted through March 30, 2000 and four years for options granted from April 1, 2000 onwards, with graded vesting of the options over the vesting period. According to German Law (§ 193 II, No. 3 AktG), the rights can be exercised two years after the grant, at the earliest.
The fair value of the options was determined following the Black-Scholes options pricing model as proposed by SFAS No. 123. The following table summarizes the assumptions used in calculating the fair value:
Date Granted | Before (Pre-IPO) | From June 2000 to December 2001 | 2002 to 2003 | ||||||
Risk-free rate | 5.65 | % | 5.65 | % | 3.24 | % | |||
Dividend yield | 0 | % | 0 | % | 0 | % | |||
Volatility | 0.01 | % | 69.00 | % | 91.02 | % | |||
Estimated life (years) | 4.00 | 4.00 | 4.00 |
F-23
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
For options issued during 2003, 2002 and 2001, compensation cost was€ 636,000,€ 2,122,000 and€ 5,913,000, respectively, which is being recognized over the vesting period of those options. Compensation cost related to stock options included in the statements of operations was€ 1,621,000,€ 3,345,000 and€ 3,625,000 in 2003, 2002 and 2001, respectively. The following table presents the stock option activity for the years ended December 31, 2003, 2002 and 2001:
| 2003 | 2002 | 2001 | |||||||||||||
| Options | Weighted- Average Exercise Price | Options | Weighted- Average Exercise Price | Options | Weighted- Average Exercise Price | ||||||||||
Outstanding at January 1 | 4,112,859 | € 8.80 | 3,597,300 | € 9.59 | 3,507,762 | € 7.39 | |||||||||
Granted | 177,000 | € 5.44 | 783,700 | € 4.78 | 895,978 | €11.84 | |||||||||
Exercised | (88,925 | ) | € 3.29 | (104,284 | ) | € 2.77 | (739,462 | ) | € 1.49 | ||||||
Forfeited | (365,476 | ) | € 8.86 | (163,857 | ) | €10.61 | (66,978 | ) | €14.02 | ||||||
Expired | — | — | — | — | — | — | |||||||||
Outstanding at December 31 | 3,835,458 | € 8.77 | 4,112,859 | € 8.80 | 3,597,300 | € 9.59 | |||||||||
Vested at December 31 | 2,575,250 | € 8.70 | 2,093,046 | € 7.79 | 1,350,775 | € 7.67 | |||||||||
Exercisable at December 31 | 2,383,250 | € 9.02 | 1,867,386 | € 7.19 | 873,030 | € 2.65 | |||||||||
Weighted-average fair value per options granted during the year | € 3.59 | € 2.71 | € 6.60 | ||||||||||||
The following table represents weighted-average price and contractual life information regarding significant option groups outstanding at December 31, 2003.
Range of Exercise Prices in€ | Number Outstanding | Weighted-Average Exercise Price | Weighted-Average Remaining Contractual Life (Years) | Number Exercisable | ||||
1.00 – 3.98 | 1,341,073 | 2.90 | 6.46 | 951,961 | ||||
4.36 – 7.44 | 857,800 | 6.00 | 6.84 | 453,625 | ||||
7.61 – 10.02 | 569,195 | 9.40 | 6.77 | 272,721 | ||||
11.78 – 18.92 | 966,050 | 16.64 | 6.65 | 634,331 | ||||
24.60 – 34.02 | 91,640 | 28.60 | 6.98 | 63,332 | ||||
58.12 – 59.74 | 9,700 | 59.57 | 6.72 | 7,280 | ||||
| 3,835,458 | 6.65 | 2,383,250 | ||||||
F-24
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
15. Income Taxes
The Company’s loss before income taxes for the years ended December 31, arose in the following jurisdictions (in thousand€):
| 2003 | 2002 | 2001 | ||||
Germany | 16,122 | 8,996 | 6,818 | |||
Other | 10,709 | 23,938 | 19,399 | |||
Loss before income taxes | 26,831 | 32,934 | 26,217 | |||
Deferred tax liabilities and assets are comprised of the following at December 31 (in thousand€):
| 2003 | 2002 | 2001 | |||||||
Deferred tax assets: | |||||||||
Net operating loss carryforwards | 39,854 | 42,945 | 41,208 | ||||||
Research and development tax credits | 3,262 | 2,914 | 2,391 | ||||||
Accrued expenses | 542 | (775 | ) | (466 | ) | ||||
Intangible assets | 170 | 203 | 263 | ||||||
Plant and equipment | 138 | 185 | 223 | ||||||
Other | 816 | 759 | 87 | ||||||
Valuation allowance | (44,578 | ) | (45,730 | ) | (43,043 | ) | |||
| 204 | 501 | 663 | |||||||
Deferred tax liabilities: | |||||||||
Marketable securities and short-term investments | (204 | ) | (501 | ) | (663 | ) | |||
Net deferred taxes | — | — | — | ||||||
The reconciliation of income tax computed at the statutory rate applicable to the Company’s income tax expense for the years ended December 31 is as follows (in thousands of€, except percent):
| 2003 | �� | 2002 | 2001 | |||||||||||||||
| Amount | Percent | Amount | Percent | Amount | Percent | |||||||||||||
German tax benefit | (10,062 | ) | 37.5 | % | (11,856 | ) | 36.0 | % | (9,438 | ) | 36.0 | % | ||||||
Non-deductible expenses | 3,626 | (13.5 | )% | 4,242 | (12.9 | )% | 3,354 | (12.8 | )% | |||||||||
Change in tax rate | 240 | (0.9 | )% | — | (0.0 | )% | (715 | ) | 2.7 | % | ||||||||
Research and development tax credit | (922 | ) | 3.4 | % | (993 | ) | 3.0 | % | — | — | ||||||||
Different tax rate in other countries | (278 | ) | 1.0 | % | (1,571 | ) | 4.8 | % | — | — | ||||||||
Change in valuation allowance | 7,396 | (27.6 | )% | 10,178 | (30.9 | )% | 6,799 | (25.9 | )% | |||||||||
Income tax expense | — | — | % | — | — | % | — | — | % | |||||||||
As a result of the net losses incurred by the Company in each year since inception, the Company is in a net deferred tax asset position. Realization of deferred tax assets is dependent upon future taxable income, if any, the timing and amount of which is uncertain. Accordingly, a valuation allowance has been established against the net deferred tax assets as it is more likely than not that they will not be utilized in the near future.
F-25
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
For fiscal years ending in 2004 and beyond, a new law is in effect restricting the use of net operating loss (NOL) carryforwards for German corporation tax and trade tax purposes. The Company’s maximum NOL carryforward that may be utilized in any one year is restricted to 60% of the annual taxable income above€ 1 million. For fiscal years ending before 2004, a NOL carryforward could be utilized without any restrictions to reduce the annual corporate income and trade tax taxable income to€ 0. The amount of NOL carryforwards for German corporation and trade tax purposes is€ 33,274,000 and€ 33,306,000, respectively. These NOL’s do not expire.
As a result of an ownership change in 2000, the Company has a limitation of€ 1,396,000, on the amount of income that is available to be offset by NOL carryforwards generated in the U.S. The Company has€ 32,289,000 of net operating losses carried forward that are subject to this limitation. Generally, net operating losses generated in the U.S. can be carried back 2 years and forward 20 years. For net operating losses in tax years beginning before August 6, 1997, taxpayers can carry back a net operating loss 3 years and forward 15 years. The Company has€ 45,245,000 of NOL carryforwards subject to a 20 year carryforward and€ 20,939,000 subject to a 15 year carryforward. NOL carryforwards generated in the U.S. expire at various times between the years 2007 and 2023.
The Company’s U.S. subsidiary had research and development credits of€ 3,262,000 at December 31, 2003.
Because the Company has historically incurred net operating losses, current and deferred tax expense for the years ending December 31, 2003, 2002 and 2001 was€ 0.
No income taxes are provided for on undistributed earnings of the foreign subsidiary where those earning are considered to be permanently invested. Total undistributed earnings in such foreign subsidiaries amounted to approximately€ 0. A 5% German income tax could be incurred on certain distributions made from the foreign subsidiary to its parent. If distribution of those earnings would be made the income tax would be€ 542,000.
16. Collaboration Agreements
Over the last years, the Company has formed several alliances with ALTANA Pharma, Aventis Pharma Deutschland GmbH (“Aventis”) and Boehringer Ingelheim International GmbH (“Boehringer Ingelheim”). These agreements include alliances based on the transfer and/or non-exclusive licenses of technology platforms, alliances which combine a technology license with a focus on a specific disease or therapeutic approach, and disease-focused programs under which the Company conducts research funded by its partners. The Company’s technology-based alliances are generally structured as research collaborations. Under these arrangements, the Company performs research in a specific disease area aimed at discoveries leading to novel pharmaceutical products. These alliances generally provide research funding over an initial period, with renewal provisions, varying by agreement. Under these agreements, the Company’s partners may make up-front payments, license payments, ongoing research funding, additional payments upon the achievement of specific research and product development milestones and/or pay royalties or, in some cases, profit-sharing payments to the Company based upon any product sales resulting from the collaboration.
Total revenue recognized under collaboration and license agreements amounted to€ 20,821,000,€ 20,499,000 and€ 12,314,000 for the years ended December 31, 2003, 2002 and 2001, respectively. Total cash received from collaboration agreements amounted to€ 16,383,000,€ 15,814,000 and€ 30,171,000 for the years ended December 31, 2003, 2002 and 2001, respectively. Total costs incurred under research collaborations and license agreements amounted to€ 3,565,000,€ 3,724,000 and€ 2,393,000 for the years ended December 31, 2003, 2002 and 2001, respectively.
F-26
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Significant Agreements Beginning in 2003
Effective January 31, 2003, the Company entered into an agreement with ALTANA Pharma to license the Company’s proprietary drug-protein interaction technology, LeadCode™, to evaluate compounds in the public domain, as well as certain proprietary compounds of ALTANA Pharma. Under the terms of this agreement, the Company will receive additional funding for the license, transfer and implementation of LeadCode™ for use at the ALTANA Research Institute. In addition, the Company is eligible to receive over $15 million in milestone payments, in addition to royalties, for each product that is a direct result of the collaboration.
Significant Agreements Beginning in 2002
No significant collaborations agreements were initiated during 2002.
Significant Agreements Beginning in 2001 and Earlier
Effective November 1, 2001, the Company entered into an alliance with ALTANA Pharma to establish a U.S. Research Institute in Waltham, MA (USA) to use the Company’s genomic and proteomic technologies. ALTANA Pharma and the Company entered into three agreements under which the Company receives funding under a collaboration and license agreement in the form of an upfront payment, technology license and implementation fees, as well as research and technology transfer funding. The Company is non-exclusively licensing selected genomics, proteomics, automation and bioinformatics technologies to ALTANA Pharma and is collaborating with ALTANA Pharma on two drug discovery programs. The Company receives further funding under a sublease agreement. Furthermore, the Company received additional funding until March 31, 2003 under a separate service agreement. In addition, the Company is eligible to receive implementation, research, preclinical and clinical milestone payments and royalty payments for each product resulting from the two collaborative drug discovery programs. The collaboration term expires in June 2007.
Effective December 29, 2000, ALTANA Pharma and the Company entered into a target identification and drug discovery collaboration in oncology. Under this agreement, GPC Biotech utilized its proprietary functional genomics and proteomics platform for ALTANA Pharma to identify tumor-specific targets whose inhibition selectively drives cancer cells, but not normal cells, into programmed cell death (apoptosis). GPC Biotech received an upfront payment and received research funding and was eligible to receive research and clinical milestones, which were subject to achieving specific research and development goals. In addition, under the terms of the agreement the Company will receive royalties on products from drug targets resulting from the alliance sold by ALTANA Pharma and will have exclusive rights to develop products for drug targets resulting from the alliance that are not selected by ALTANA Pharma. This agreement terminated on December 29, 2003.
Effective December 29, 2000, Boehringer Ingelheim and the Company entered into a collaboration to develop and commercialize a new generation of products for the prevention and treatment of human papillomavirus (HPV)-related human diseases. Under this agreement, GPC Biotech used its integrated technology platform to identify novel targets and their molecular pathways associated with high and low risk human HPV infections for Boehringer Ingelheim´s research center in Laval, Quebec (Canada). The research and license agreement included up-front license fees, research funding as well as research and clinical development milestones to the Company. In addition, under the terms of the agreement Boehringer Ingelheim will pay GPC Biotech royalties on sales of products discovered as a result of the collaboration. This agreement terminated on December 29, 2002.
F-27
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Effective June 18, 1998, ALTANA Pharma and the Company entered into a 5-year research agreement to identify and validate new targets for diagnostic and therapeutic development for the treatment ofHelicobacter pyloriandClamydia pneumonia infections. A separate license and support agreement included an up-front fee for a technology license, and the research agreement included research funding over the term of the collaboration as well as research and clinical development milestone payments to the Company. This agreement terminated on June 18, 2003.
Effective January 1, 1999, the Company entered into a 3-year collaboration and license agreement with Aventis to identify and validate new targets for diagnostic and therapeutic development for the treatment of osteoarthritis. Aventis will consider the resulting target genes for drug discovery and development and is granted commercialization license rights for targets licensed from GPC Biotech within the collaboration. Aventis funded the research program and is to pay additional milestone payments to the Company if certain research and clinical milestones have been reached. Additionally, under the terms of the agreement Aventis will pay the Company royalties based on the sales of products derived from compounds resulting from the research collaboration. On December 17, 2001, the collaboration and license agreement was extended for a 6-month period until June 30, 2002. On July 1, 2002, the collaboration and license agreement was further extended for a 12-month period until June 30, 2003. On July 1, 2003, the collaboration and license agreement was amended and extended for another 12-month period until June 30, 2004. However, the only obligation of the Company under this extension is the participation in the joint steering committee meetings which decide on the achievement of milestones by Aventis thus triggering further payments to the Company.
17. Licensing Activities
On September 30, 2002, the Company signed an agreement with Spectrum Pharmaceuticals, Inc. (“Spectrum”) (formerly NeoTherapeutics, Inc.) granting GPC Biotech an exclusive worldwide license to satraplatin, in the field of treating cancer in humans; an orally administered anticancer agent in Phase 3 clinical development. An initial licensing fee of $2 million was paid to Spectrum upon signing. In October 2003, another licensing fee was paid upon the dosing of the first patient in a registrational Phase 3 study in the amount of $2 million (approximately€ 1,725,000), consisting of a $1 million cash payment and a $1 million equity investment. The equity investment in Spectrum represents 128,370 shares of Spectrum. Under the terms of the agreement, GPC Biotech also will pay milestones totaling up to $18 million, the first of which is anticipated to be the acceptance of the NDA (New Drug Application) for filing by the U.S. Food and Drug Administration. Upon commercialization of satraplatin, Spectrum will also receive royalties based on net sales. Under the terms of the agreement, GPC Biotech will fully fund development and commercialization expenses for satraplatin. GPC Biotech and Spectrum have formed a joint development committee, headed by GPC Biotech, to govern the development of satraplatin.
On June 6, 2002, the Company licensed worldwide rights to the antibacterial program to PanTherix, Ltd. (“PanTherix”), a privately held company located in Glasgow, Scotland. Under the agreement PanTherix obtained targets developed by the Company. As consideration for the license, the Company accepted a convertible note receivable in the amount of $2.5 million. The revenue on this, including accrued interest, was deferred due to the uncertainty surrounding the collectibility of the note receivable. Due to PanTherix not fulfilling its diligence obligations, the license agreement was terminated during 2003 and the program was returned to GPC Biotech. The convertible note receivable and associated accrued interest were converted into shares of PanTherix, now a non-operational company. Due to the non-operational status, the Company determined that the shares have a net carrying value of€ 0 and wrote off the shares against deferred revenue. Other than payments for 1
F-28
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
full-time equivalent (FTE) for one year, no revenue was ever recognized related to the licensing of the program or the conversion of the note into shares.
18. Loss per Share
Basic loss per ordinary share is based on the weighted-average number of ordinary shares outstanding. For the years ended December 31, 2003, 2002 and 2001, diluted net loss per ordinary share was the same as basic net loss per ordinary share as the inclusion of weighted-average ordinary shares issuable upon exercise of stock options and convertible bonds would be antidilutive. The number of potentially dilutive shares excluded from the loss per share calculation due to their antidilutive effect was 1,196,101, 1,092,473 and 1,727,511 for the years ended December 31, 2003, 2002 and 2001, respectively.
The following table sets forth the computation of basic and diluted loss per share (in thousand€, except share and per share data):
| 2003 | 2002 | 2001 | |||||||
Numerator: | |||||||||
Net loss | (26,831 | ) | (32,934 | ) | (26,217 | ) | |||
Denominator: | |||||||||
Denominator for basic and diluted earnings per | 20,731,535 | 20,688,515 | 18,509,398 | ||||||
Basic and diluted loss per share in euro | (1.29 | ) | (1.59 | ) | (1.42 | ) | |||
19. Shareholders’ Equity
Ordinary Shares
The holders of the Company’s ordinary shares are entitled to one vote for each share held at all meetings of shareholders. Dividends and distribution of assets of the Company in the event of liquidation are subject to the rights of any then-outstanding ordinary shares. The Company has never declared or paid dividends on any of its ordinary shares and does not expect to do so in the foreseeable future.
Authorized Shares
To assist management in undertaking strategic activities and to service the stock option plans and convertible bonds, the shareholders of the Company have authorized the future issuance of ordinary shares in specific circumstances with the permission of the Supervisory Board. At December 31, 2003, the total number of ordinary shares potentially issuable was 18,501,555. The number of ordinary shares authorized for exercise of stock options and convertible bonds was 4,598,045 and 1,177,500, respectively. The number of ordinary shares authorized for the purpose of potential merger and acquisition activities is 7,726,010. The number of ordinary shares authorized for the purposes of potential convertible debt issuances is 5,000,000.
Exercise of Stock Options and Convertible Bonds
During 2003, 2002 and 2001, members of the Management Board, consultants and employees of the Company exercised some of their fully vested options and convertible bonds, receiving 34,295, 104,284 and 1,126,728 ordinary shares, respectively.
F-29
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
At December 31, 2003, consultants and employees of the Company had subscribed to 54,630 ordinary shares with a total value of€ 214,660, which has been included in shareholders’ equity. The subscribed shares represent amounts paid for exercises of stock options for which ordinary shares have not been issued at December 31, 2003. The ordinary shares were subsequently registered on January 19, 2004.
Sale of Ordinary Shares
In October 2001, GPC Biotech sold 1,690,000 ordinary shares to ALTANA Technology Projects GmbH, an affiliate of ALTANA Pharma, for€ 34,223,000 or€ 20.25 per share. The transaction resulted in an increase in ordinary shares of€ 1,690,000 and an increase in additional paid-in capital of€ 32,533,000.
Accumulated Other Comprehensive Income (Loss)
The balance of the components of accumulated other comprehensive income (loss) as of December 31, 2003, 2002 and 2001 were as follows (in thousand€):
| December 31, | ||||||||
| 2003 | 2002 | 2001 | ||||||
Unrealized gain on available-for-sale securities | 651 | 621 | 119 | |||||
Accumulated translation adjustments | (2,753 | ) | (1,360 | ) | 1,871 | |||
Total accumulated other comprehensive income (loss) | (2,102 | ) | (739 | ) | 1,990 | |||
20. Employee Benefit Plan
GPC Biotech Inc. has a defined contribution plan qualified under the provisions of the Internal Revenue Code section 401(k). All individuals who are employed by GPC Biotech Inc. are immediately eligible for enrollment in the plan. GPC Biotech Inc. contributes 25 % of the first 4 % of compensation contributed by each participant. Contributions are fully vested immediately. Costs of the plan, including the Company’s matching contribution, charged to operating expenses amounted to€ 88,000,€ 64,000 and€ 71,000 in 2003, 2002 and 2001, respectively.
21. Restructuring costs
Beginning January 1, 2003, the Company adopted SFAS No. 146 “Accounting for Costs Associated with Exit or Disposal Activities.” SFAS No. 146 requires a company to set up a liability for a cost associated with an exit or disposal activity to be recognized when the liability is incurred.
In October 2003, the Company announced a restructuring plan to realign resources to accelerate its oncology drug discovery and development programs. The restructuring included a staff reduction of 42 employees, mainly in the technology research staff.
The restructuring is part of the Company’s ongoing strategy to focus on oncology drug discovery and clinical development and accelerate the building of a broad anticancer drug pipeline. The restructuring plan was completed on December 31, 2003 with completion of the employee terminations.
F-30
Table of Contents
GPC BIOTECH AG
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Restructuring costs incurred were mainly employee termination costs, legal and consultant fees. Total costs incurred were approximately€ 490,000. All costs related to the restructuring were incurred in 2003 and included in research and development expenses and general and administrative expenses in the statement of operations for the year ending December 31, 2003. At December 31, 2003€ 94,000 was accrued related to remaining severance payments.
22. Contingencies
The Company may be party to certain legal proceedings and claims which arise during the ordinary course of business. In the opinion of management, the ultimate outcome of these matters will not have material adverse effects on the Company’s financial position, results of operations or cash flows.
23. Subsequent Events
During January 2004, 306,000 convertible bonds with a nominal value of€39,113 were exercised resulting in the issuance of 306,000 ordinary shares.
During January and February 2004, 219,524 fully vested stock options were exercised, resulting in the issuance of 219,524 ordinary shares with a nominal value of€219,524.
F-31
Table of Contents
GPC BIOTECH AG
UNAUDITED INTERIM CONSOLIDATED BALANCE SHEETS
| March 31, 2004 | December 31, 2003 | |||||
| (in thousand€, except share and per share data) | | |||||
Assets | ||||||
Current assets: | ||||||
Cash and cash equivalents | 18,201 | 34,947 | ||||
Marketable securities and short-term investments | 63,783 | 54,221 | ||||
Accounts receivable | 518 | 755 | ||||
Accounts receivable, related party | 1,540 | — | ||||
Prepaid expenses | 1,014 | 1,540 | ||||
Other current assets | 2,792 | 2,042 | ||||
Total current assets | 87,848 | 93,505 | ||||
Property and equipment, net | 3,351 | 3,264 | ||||
Intangible assets acquired in a business combination, net | 528 | 613 | ||||
Other assets, non-current | 2,806 | 1,679 | ||||
Restricted cash | 2,584 | 2,503 | ||||
Total assets | 97,117 | 101,564 | ||||
Liabilities and shareholders’ equity | ||||||
Current liabilities: | ||||||
Accounts payable | 1,374 | 629 | ||||
Accrued expenses and other current liabilities | 7,469 | 6,499 | ||||
Current portion of deferred revenue, related party | 4,454 | 5,238 | ||||
Current portion of deferred revenue | 165 | 165 | ||||
Current portion of long-term debt | 128 | 128 | ||||
Current portion of capital lease obligations | 303 | 308 | ||||
Total current liabilities | 13,893 | 12,967 | ||||
Capital lease obligations, net of current portion | 260 | 320 | ||||
Long-term debt, net of current portion | 575 | 639 | ||||
Deferred revenue, related party, net of current portion | 4,388 | 4,875 | ||||
Convertible bonds | 833 | 884 | ||||
Shareholders equity: | ||||||
Ordinary shares,€ 1 non-par, notional value | 21,334 | 20,754 | ||||
Additional paid-in capital | 191,566 | 190,335 | ||||
Subscribed shares | 315 | 215 | ||||
Accumulated other comprehensive loss | (1,799 | ) | (2,102 | ) | ||
Accumulated deficit | (134,248 | ) | (127,323 | ) | ||
Total shareholders’ equity | 77,168 | 81,879 | ||||
Total liabilities and shareholders’ equity | 97,117 | 101,564 | ||||
See accompanying notes to unaudited interim consolidated financial statements.
F-32
Table of Contents
GPC BIOTECH AG
UNAUDITED INTERIM CONSOLIDATED STATEMENTS OF OPERATIONS
| Three months ended March 31, | ||||||
| 2004 | 2003 | |||||
| (in thousand €, except share and per share data) | ||||||
Collaborative revenues(a) | 3,959 | 5,890 | ||||
Grant revenues | — | 222 | ||||
Total revenues | 3,959 | 6,112 | ||||
Research and development expenses | 8,682 | 8,323 | ||||
General and administrative expenses | 2,786 | 2,621 | ||||
Amortization of intangible assets acquired in a business combination | 101 | 168 | ||||
Total operating expenses | 11,569 | 11,112 | ||||
Operating loss | (7,610 | ) | (5,000 | ) | ||
Other income | 234 | 75 | ||||
Interest income | 608 | 936 | ||||
Other expenses | (131 | ) | (144 | ) | ||
Interest expense | (26 | ) | (29 | ) | ||
| 685 | 838 | |||||
Net loss | (6,925 | ) | (4,162 | ) | ||
Basic and diluted loss per share in euro | (0.33 | ) | (0.20 | ) | ||
Shares used in computing basic and diluted loss per share | 21,085,710 | 20,724,174 | ||||
(a) Revenues from related party | ||||||
| Collaborative revenues | 3,959 | 5,820 | ||||
See accompanying notes to unaudited interim consolidated financial statements.
F-33
Table of Contents
GPC BIOTECH AG
UNAUDITED INTERIM CONSOLIDATED STATEMENTS OF CASH FLOWS
| Three months ended March 31, | ||||||
| 2004 | 2003 | |||||
| (in thousand €) | ||||||
Cash flows from operating activities | ||||||
Net loss | (6,925 | ) | (4,162 | ) | ||
Adjustment to reconcile net loss to net cash used in operating activities: | ||||||
Depreciation | 433 | 486 | ||||
Amortization | 127 | 192 | ||||
Compensation cost for stock option plan and convertible bonds | 388 | 588 | ||||
Accrued interest income on marketable securities and short-term investments | (258 | ) | (225 | ) | ||
Bond premium amortization | 107 | 106 | ||||
Loss on disposal of property and equipment | 23 | — | ||||
Changes in operating assets and liabilities: | ||||||
Accounts receivable, related party | (1,540 | ) | — | |||
Accounts receivable | 236 | (329 | ) | |||
Other assets, current and non-current | 10 | (1,266 | ) | |||
Accounts payable | 740 | (14 | ) | |||
Deferred revenue, related party | (1,274 | ) | (2,017 | ) | ||
Deferred revenue | — | (47 | ) | |||
Other liabilities and accrued expenses | (335 | ) | 1,298 | |||
Net cash used in operating activities | (8,268 | ) | (5,390 | ) | ||
Cash flows from investing activities: | ||||||
Purchases of property, equipment and licenses | (492 | ) | (888 | ) | ||
Proceeds from the sale of marketable securities and short-term investments | 13,213 | 17,506 | ||||
Purchases of marketable securities and short-term investments | (22,504 | ) | (30,702 | ) | ||
Net cash used in investing activities | (9,783 | ) | (14,084 | ) | ||
Cash flows from financing activities: | ||||||
Principal payments under capital lease obligations | (81 | ) | (203 | ) | ||
Proceeds from issuance of convertible bonds | — | 153 | ||||
Payments for cancellation of convertible bonds | (4 | ) | — | |||
Proceeds from exercise of stock options and convertible bonds | 1,170 | 12 | ||||
Cash received for subscribed shares | 155 | — | ||||
Principal payment of loans | (64 | ) | (64 | ) | ||
Net cash provided by (used in) financing activities | 1,176 | (102 | ) | |||
Effect on exchange rate changes on cash | 133 | (6 | ) | |||
Restricted cash | (4 | ) | (5 | ) | ||
Net decrease in cash | (16,746 | ) | (19,587 | ) | ||
Cash and cash equivalents at beginning of period | 34,947 | 39,947 | ||||
Cash and cash equivalents at end of period | 18,201 | 20,360 | ||||
Supplemental information: | ||||||
Cash paid for interest | 20 | 29 | ||||
Non-cash investing and financing activities: | ||||||
Accrual of costs incurred in connection with planned equity offering and NASDAQ listing | 1,200 | — | ||||
Amounts receivable for subscribed shares | 152 | — | ||||
See accompanying notes to the unaudited interim consolidated financial statements.
F-34
Table of Contents
GPC BIOTECH AG
UNAUDITED INTERIM CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
(in thousand€, except share data)
| Ordinary shares | |||||||||||||||||
| Shares | Amount | Additional Paid-in Capital | Subscribed Shares | Accumulated Deficit | Accumulated Other | Total Shareholders’ Equity | |||||||||||
Balance at December 31, 2002 | 20,719,780 | 20,720 | 187,781 | — | (100,492 | ) | (739 | ) | 107,270 | ||||||||
Components of comprehensive loss: | |||||||||||||||||
Net loss | — | — | — | — | (4,162 | ) | — | (4,162 | ) | ||||||||
Change in unrealized gain on available-for-sale securities | — | — | — | — | — | (32 | ) | (32 | ) | ||||||||
Accumulated translation adjustments | — | — | — | — | — | (160 | ) | (160 | ) | ||||||||
Total comprehensive loss | — | — | — | — | — | — | (4,354 | ) | |||||||||
Exercise of stock options and convertible bonds | 5,285 | 5 | 7 | — | — | — | 12 | ||||||||||
Compensation cost, stock options and convertible bonds | — | — | 588 | — | — | — | 588 | ||||||||||
Balance at March 31, 2003 | 20,725,065 | 20,725 | 188,376 | — | (104,654 | ) | (931 | ) | 103,516 | ||||||||
Balance at December 31, 2003 | 20,754,075 | 20,754 | 190,335 | 215 | (127,323 | ) | (2,102 | ) | 81,879 | ||||||||
Components of comprehensive loss: | |||||||||||||||||
Net loss | — | — | — | — | (6,925 | ) | — | (6,925 | ) | ||||||||
Change in unrealized gain on available-for-sale securities | — | — | — | — | — | 120 | 120 | ||||||||||
Accumulated translation adjustments | — | — | — | — | — | 183 | 183 | ||||||||||
Total comprehensive loss | — | — | — | — | — | — | (6,622 | ) | |||||||||
Exercise of stock options and convertible bonds | 580,154 | 580 | 843 | 100 | — | — | 1,523 | ||||||||||
Compensation cost, stock options and convertible bonds | — | — | 388 | — | — | — | 388 | ||||||||||
Balance at March 31, 2004 | 21,334,229 | 21,334 | 191,566 | 315 | (134,248 | ) | (1,799 | ) | 77,168 | ||||||||
See accompanying notes to unaudited interim consolidated financial statements.
F-35
Table of Contents
GPC BIOTECH AG
NOTES TO THE UNAUDITED INTERIM CONSOLIDATED FINANCIAL STATEMENTS
1. Basis of Presentation
The accompanying unaudited consolidated financial statements of GPC Biotech AG (the “Company”) have been prepared in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”) for interim financial information. Accordingly, they do not include all of the information and footnotes required by U.S. GAAP for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation have been included. Operating results for the three-month period ended March 31, 2004 are not necessarily indicative of results to be expected for the full year ending December 31, 2004. The balance sheet at December 31, 2003 has been derived from the audited consolidated financial statements at that date, but does not include all of the information required by U.S. GAAP for complete financial statements. For further information, refer to the consolidated financial statements and footnotes thereto for the year ended December 31, 2003.
2. Restatement
In connection with internal research and development programs the Company may be required to make milestone payments to other parties. Historically, the Company accrued for the milestone payments when management determined that the achievement of the milestone was probable, and the amount was estimable. The milestone payments were accrued ratably over the period beginning from the time the milestone was determined to be probable and estimable through the expected date of achievement of the milestone.
In connection with the review of its registration statement by the United States Securities and Exchange Commission, the Company has revised its accounting policy related to milestone payments to make its policy consistent with U.S. GAAP. In accordance with this policy, the Company expenses milestone payments when the liability is incurred, which is when the milestones are achieved.
The Company determined that the restatement of our financial statements for the three months ended March 31, 2004, and 2003 is necessary and appropriate to reflect the application of this revised accounting policy. The following table reflects the impact of the revised accounting policy on amounts as previously reported and as restated resulting from the restatement:
| As of and for the Three Months Ended March 31, | ||||||||||||||||||
2004 (in thousand€, | 2003 (in thousand€, | |||||||||||||||||
| As Reported | As Restated | Adjustment | As Reported | As Restated | Adjustment | |||||||||||||
Accrued expenses and other current liabilities | 9,037 | 7,469 | (1,568 | ) | 11,074 | 6,103 | (4,971 | ) | ||||||||||
Other non-current liabilities | 704 | — | (704 | ) | — | — | — | |||||||||||
Total shareholders’ equity | 74,896 | 77,168 | 2,272 | 98,545 | 103,516 | 4,971 | ||||||||||||
Research and development expenses | 8,875 | 8,682 | (193 | ) | 8,742 | 8,323 | (419 | ) | ||||||||||
Net loss | (7,174 | ) | (6,925 | ) | 249 | (4,581 | ) | (4,162 | ) | 419 | ||||||||
Basic and diluted loss per share in€ | (0.34 | ) | (0.33 | ) | 0.01 | (0.22 | ) | (0.20 | ) | 0.02 | ||||||||
3. Other Assets, Non-current
On April 2, 2004, the Company announced that it is planning a global equity offering and a NASDAQ listing. As of March 31, 2004, the Company has deferred certain costs of€ 1.2 million incurred related to this share offering. Upon the completion of the offering these costs will be offset against the proceeds from the offering and recorded in shareholders’ equity. These costs are recorded in other assets at March 31, 2004.
F-36
Table of Contents
GPC BIOTECH AG
NOTES TO THE UNAUDITED INTERIM CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
4. Loss per Share
Basic loss per common share is computed using the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed using the weighted average number of common and dilutive common equivalent shares from stock options, warrants and convertible debt using the treasury stock method. For all periods presented, diluted net loss per share is the same as basic net loss per share, as the inclusion of weighted average shares of common stock issuable upon the exercise of stock options, warrants and convertible debt would be antidilutive.
5. Comprehensive Loss
Comprehensive loss was€ 6,622,000 and€ 4,354,000 for the three months ended March 31, 2004 and 2003, respectively. Comprehensive loss is composed of net loss, unrealized gains and losses on marketable securities and cumulative foreign currency translation adjustments. Accumulated other comprehensive loss at March 31, 2004 and 2003 reflected€ 771,000 and€ 589,000 of unrealized gains on marketable securities and short-term investments, and€ 2,570,000 and€ 1,520,000 of cumulative foreign currency translation loss adjustments, respectively.
6. Loans Receivable from Members of the Management Board
As of March 31, 2004, the aggregate amount of outstanding loans to members of the Management Board and Supervisory Board was€ 0. During the three months ended March 31, 2004, loans receivable from two members of the Management Board were fully repaid in the amount of € 79,000.
7. Shareholders’ Equity
During the three months ended March 31, 2004, employees and advisors of the Company exercised some of their fully vested options, receiving 274,154 new ordinary shares of the Company.
During the three months ended March 31, 2004, convertible bondholders of the Company converted some of their fully vested bonds, receiving 306,000 new ordinary shares of the Company.
During the three months ended March 31, 2004, a total of 54,630 subscribed shares were registered and issued resulting in a reclassification of€ 215,000 from subscribed shares to ordinary shares and additional paid-in capital.
At March 31, 2004, consultants, employees and convertible bond holders of the Company had subscribed to 115,078 shares with a total value of€ 315,000, which has been included in shareholders’ equity. The subscribed shares represent amounts paid for exercises of stock options and convertible bonds for which ordinary shares have not been issued at March 31, 2004. This amount includes€ 152,000 from the exercise of 82,210 options, which was not yet paid in by the option holder. This amount was included in other current assets at March 31, 2004, and subsequently collected on April 5, 2004.
F-37
Table of Contents
No dealer, salesperson or other person is authorized to give any information or to represent anything not contained in this prospectus. You must not rely on any unauthorized information or representations. This prospectus is an offer to sell only the shares offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. The information contained in this prospectus is current only as of its date.
| Page | ||
| 1 | ||
| 10 | ||
| 32 | ||
| 33 | ||
| 36 | ||
| 37 | ||
| 38 | ||
| 39 | ||
| 40 | ||
| 41 | ||
| 42 | ||
Management’s Discussion and Analysis of Financial Condition and Results of Operations | 44 | |
| 66 | ||
| 67 | ||
| 103 | ||
Principal and Selling Shareholders and Exercise of Cash Settled Stock Options | 126 | |
| 129 | ||
| 130 | ||
| 139 | ||
| 147 | ||
| 150 | ||
| 154 | ||
| 157 | ||
| 160 | ||
| 160 | ||
| 160 | ||
| 160 | ||
| 161 | ||
| F-1 |
Through and including , 2004 (the 25th day after the date of this prospectus) , all dealers that effect transactions in our ordinary bearer shares and ADSs, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to a dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to an unsold allotment or subscription.
GPC Biotech AG
7,460,000 ordinary shares
No par value
Ordinary Bearer Shares
in the form of shares or
American Depositary Shares
Goldman, Sachs & Co.
Lehman Brothers
Pacific Growth Equities, LLC
WestLB AG
Table of Contents
PART TWO
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 6. Indemnification of Directors and Officers
We provide customary liability insurance for our directors and officers. Our Articles of Association do not provide for any indemnification of members of our Supervisory Board and our Management Board for any liabilities they may incur.
Item 7. Recent Sales of Unregistered Securities
Our securities that were sold by us within the past three years and not registered under the Securities Act of 1933 are described below. Unless otherwise indicated below, all of the securities described below were offered and issued outside the United States to individuals or entities that to the best of our knowledge were neither citizens nor residents of the United States. Accordingly, the offering and issuance of such securities were not subject to the registration requirements of the Securities Act. No underwriters were involved in the securities issuances described below.
Date of Issuance | Securities | Aggregate Offering Price(€) | Purchasers | |||
November 2001 | 1,690,000 Ordinary Bearer Shares | 34,223,000 | ALTANA Technology Projects GmbH | |||
| Between January 1, 2001 and December 31, 2003 | 1,856,678 Stock Options(1) | — | Members of our Supervisory Board, members of our Management Board, consultants and employees(2) | |||
| Between January 1, 2004 and June 4, 2004 | 41,500 Stock Options(1) | — | Members of our Supervisory Board, members of our Management Board, consultants and employees(2) | |||
| Between January 1, 2001 and December 31, 2003 | 844,500(5) Convertible Bonds(3) | 844,500 | Members of our Supervisory Board, members of our Management Board and employees(2) | |||
| Between January 1, 2004 and June 4, 2004 | 0 Convertible Bonds(3) | — | Members of our Supervisory Board, members of our Management Board and employees(2) | |||
| Between January 1, 2001 and December 31, 2003 | 1,265,361 Ordinary Bearer Shares(4) | 1,861,000 | Members of our Supervisory Board, members of our Management Board, consultants and employees(2) | |||
| Between January 1, 2004 and June 4, 2004 | 647,435 Ordinary Bearer Shares(4) | 1,561,821 | Members of our Supervisory Board, members of our Management Board, consultants and employees(2) | |||
| (1) | We issued these stock options pursuant to compensatory benefit plans. |
| (2) | Some members of our Supervisory Board and Management Board and some of our consultants and employees are U.S. persons. For issuances of our securities to these persons, we have relied on an exemption from registration under the Securities Act of 1933 provided by Rule 701. |
| (3) | We issued these convertible bonds pursuant to compensatory benefit plans. |
| (4) | We issued these shares upon exercises of stock options and or upon conversion of convertible bonds. |
| (5) | 4,027 convertible bonds have been cancelled as of December 31, 2003 as a result of Dr. Schühsler’s resignation as a member and Vice Chairman of our Supervisory Board. |
II-1
Table of Contents
Item 8. Exhibits and Financial Statement Schedules
The following exhibits are filed as part of this Prospectus:
| Exhibit Number | Description | |
| 1.1* | Form of Pre-Underwriting Agreement. | |
| 1.2* | Form of Underwriting Agreement. | |
| 3* | Articles of Association of GPC Biotech AG (English translation). | |
| 4* | Form of Deposit Agreement between GPC Biotech, The Bank of New York, as depositary, and all registered holders and beneficial owners from time to time of American Depositary Receipts issued thereunder, including the form of American Depositary Receipt. | |
| 5 | Opinion of Shearman & Sterling LLP as to the validity of the shares. | |
| 8* | Opinion of Shearman & Sterling LLP with regard to certain tax matters. | |
| 10.1* | Co-Development and License Agreement, dated September 30, 2002, by and between NeoTherapeutics, Inc. and GPC Biotech AG.† | |
| 10.2* | Collaboration and License Agreement, dated April 15, 1999, by and between GPCAG-Genome Pharmaceuticals Corporation and MorphoSys AG.† | |
| 10.3* | Amendment to Collaboration and License Agreement, dated December 4, 2000, by and between GPC Biotech AG (formerly GPCAG-Genome Pharmaceuticals Corporation) and MorphoSys AG.† | |
| 10.4* | Amendment to Collaboration and License Agreement, dated February 23, 2004, by and between GPC Biotech AG (formerly GPCAG-Genome Pharmaceuticals Corporation) and MorphoSys AG.† | |
| 10.5* | Amended and Restated Collaborative, Research, Development and Marketing Agreement, dated June 2, 1997, by and between Mitotix, Inc. and the DuPont Merck Pharmaceuticals Company.† | |
| 10.6* | Amendment to Amended and Restated Collaborative, Research, Development and Marketing Agreement, dated April 3, 2000, by and between Mitotix, Inc. and The DuPont Pharmaceuticals Company.† | |
| 10.7* | Amendment to Amended and Restated Collaborative, Research, Development and Marketing Agreement, dated July 30, 2000, by and between Mitotix, Inc. and The DuPont Pharmaceuticals Company. | |
| 10.8* | Amendment to Amended and Restated Collaborative, Research, Development and Marketing Agreement, dated October 1, 2000, by and between Mitotix, Inc. and The DuPont Pharmaceuticals Company. | |
| 10.9* | Amendment to Amended and Restated Collaborative, Research, Development and Marketing Agreement, dated December 28, 2000, by and between Mitotix, Inc. and Bristol-Myers Squibb Pharma Company.† | |
| 10.10* | Collaboration and License Agreement, dated as of November 1, 2001, by and between BYK Gulden Lomberg Chemische Fabrik GMBH and GPC Biotech AG.† | |
| 10.11* | Amendment to Collaboration and License Agreement, dated as of June 30, 2002, by and between BYK Gulden Lomberg Chemische Fabrik GMBH and GPC Biotech AG.† | |
| 10.12* | Amendment to Collaboration and License Agreement, dated January 31, 2003, by and between ALTANA Pharma AG (formerly BYK Gulden Lomberg Chemische Fabrik GMBH) and GPC Biotech AG.† | |
II-2
Table of Contents
| 10.13* | 3-Hybrid Collaboration and License Agreement dated as of January 31, 2003, by and between ALTANA Pharma AG and GPC Biotech AG.† | |
| 10.14* | Service Agreement (Vorstandsvertrag) with Dr. Bernd Seizinger dated June 1, 2003 (English translation). | |
| 10.15* | Addendum to Service Agreement with Dr. Bernd Seizinger dated May 1, 2003 relating to revised severance benefits upon a change in control of GPC Biotech AG (English translation). | |
| 10.16* | Service Agreement (Vorstandsvertrag) with Dr. Mirko Scherer dated September 1, 2003 (English translation). | |
| 10.17* | Addendum to Service Agreement with Dr. Mirko Scherer dated May 1, 2003 relating to revised severance benefits upon a change in control of GPC Biotech AG (English translation). | |
| 10.18* | Service Agreement (Vorstandsvertrag) with Dr. Sebastian Meier-Ewert dated March 5, 2004 (English translation). | |
| 10.19* | Addendum to Service Agreement with Dr. Sebastian Meier-Ewert dated May 1, 2003 relating to revised severance benefits upon a change in control of GPC Biotech AG (English translation). | |
| 10.20* | Service Agreement (Vorstandsvertrag) with Dr. Elmar Maier dated March 5, 2004 (English translation). | |
| 10.21* | Addendum to Service Agreement with Dr. Elmar Maier dated May 1, 2003 relating to revised severance benefits upon a change in control of GPC Biotech AG (English translation). | |
| 10.22* | Stock Option Plan 2002 (English translation). | |
| 10.23* | Stock Option Plan 2001 (English translation). | |
| 10.24* | Stock Option Plan 2000 (English translation). | |
| 10.25* | Option Terms dated September 1999 (English translation). | |
| 10.26* | Option Terms for 1997 (English translation). | |
| 10.27* | 2002 Incentive Stock Option Plan. | |
| 10.28* | 2001 Incentive Stock Option Plan. | |
| 10.29* | 2000 Incentive Stock Option Plan. | |
| 10.30* | 1999 U.S. Incentive Stock Option Plan. | |
| 10.31* | Convertible Bonds Terms and Conditions for Managers of the Company and Management Bodies and Second-Tier Domestic and Management of Foreign Affiliated Enterprises as well as for Consultants dated May 21, 2003. | |
| 10.32* | Convertible Bonds Terms and Conditions for Managers of the Company and Management Bodies and Managers of Second-Tier Domestic and Foreign Affiliated Enterprises as well as for Consultants dated May 20, 2003. | |
| 10.33* | Convertible Bonds Terms and Conditions for Members of the Supervisory Board dated October 9, 2001 (English translation). | |
| 10.34* | Convertible Bonds Terms and Conditions for Members of the Supervisory Board dated May 26, 2003 (English translation). | |
II-3
Table of Contents
| 10.35* | Transfer Agreement between Credit Suisse First Boston International, Dr. Bernd Seizinger and GPC Biotech AG dated November 15, 2001. | |
| 21* | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Ernst & Young AG, Wirtschaftsprüfungsgesellschaft, independent registered public accounting firm. | |
| 23.2 | Consent of Shearman & Sterling LLP (included in its opinion filed as Exhibit 5). | |
| 23.3* | Consent of Shearman & Sterling LLP (included in its opinion filed as Exhibit 8). | |
| 24.1* | Power of attorney (included on the signature page to our registration statement on Form F-1 filed on June 9, 2004). | |
† Request for confidential treatment.
* Previously filed.
Item 9. Undertakings
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions described under “Indemnification of Directors and Officers” above, or otherwise, the registrant has been advised that, in the opinion of the Securities and Exchange Commission, such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable.In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
| (1) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this Registration Statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this Prospectus as of the time it was declared effective. |
| (2) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new Prospectus relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fide offering thereof. |
| (3) | The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser. |
The undersigned registrant hereby undertakes to supplement the prospectus, after the expiration of the subscription period, to set forth the results of the subscription offer, the transactions by the underwriters during the subscription period, the amount of unsubscribed securities to be purchased by the underwriters, and the terms of any subsequent reoffering thereof. If any public offering by the underwriters is to be made on terms differing from those set forth on the cover page of the prospectus, a post-effective amendment will be filed to set forth the terms of such offering.
II-4
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-1 and has duly caused this Prospectus to be signed on its behalf by the undersigned, thereunto duly authorized, in Martinsried, Germany on June 9, 2004.
| GPC BIOTECH AG | ||
By: | /s/ Bernd R. Seizinger | |
Name: Title: | Bernd R. Seizinger President and Chief Executive Officer | |
By: | /s/ Mirko Scherer | |
Name: Title: | Mirko Scherer Senior Vice President and Chief Financial Officer | |
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ Bernd R. Seizinger Bernd R. Seizinger, M.D., Ph.D. | Member of Management Board—President and Chief Executive (principal executive officer) | June 9, 2004 | ||
* Elmar Maier, Ph.D. | Member of Management Board—Senior Vice President and Chief Operating Officer | June 9, 2004 | ||
/s/ Mirko Scherer Mirko Scherer, Ph.D. | Member of Management Board—Senior Vice President and Chief Financial Officer (principal financial and accounting officer) | June 9, 2004 | ||
* Sebastian Meier-Ewert, Ph.D. | Member of Management Board—Senior Vice President, Chief Scientific Officer and Chief Operating Officer | June 9, 2004 | ||
*By: | /s/ Brent Hatzis-Schoch | |
Brent Hatzis-Schoch Attorney-in-Fact | ||
II-5