Table of Contents
As filed with the Securities and Exchange Commission on June 11, 2007
RegistrationNo. 333-142646
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1
TO
FORM S-1
REGISTRATION STATEMENT
UNDER THE SECURITIES ACT OF 1933
ImaRx Therapeutics, Inc.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 2834 | 86-0974730 | ||
| (State or Other Jurisdiction of Incorporation or Organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification Number) |
1635 East 18th Street
Tucson, AZ 85719
(520) 770-1259
(Address, Including Zip Code, and Telephone Number, Including Area Code, of
Registrant’s Principal Executive Offices)
Bradford A. Zakes
1635 East 18th Street
Tucson, AZ 85719
(520) 770-1259
(Name, Address, Including Zip Code, and Telephone Number,
Including Area Code, of Agent for Service)
Copies to:
| John M. Steel, Esq. | Jody R. Samuels, Esq. | |
Mark F. Hoffman, Esq. | Benjamin M. Alexander, Esq. | |
Heidi M. Drivdahl, Esq. | Richardson & Patel LLP | |
DLA Piper US LLP | 405 Lexington Avenue, 26th Floor | |
701 Fifth Avenue, Suite 7000 | New York, NY 10174 | |
Seattle, WA 98104-7044 | (212) 907-6686 | |
(206) 839-4800 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this Registration Statement.
If any of the securities being registered on this form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
CALCULATION OF REGISTRATION FEE
| Proposed Maximum | ||||||||||||
| Title of Each Class of | Number of Shares | Offering Price | Proposed Maximum | Amount of | ||||||||
| Securities to be Registered | to be Registered | per Share | Aggregate Offering Price(1) | Registration Fee(3) | ||||||||
| Common Stock, par value $0.0001 per share | 3,450,000(2) | $7.50 | $25,875,000 | $794.36 | ||||||||
| (1) | Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(a) under the Securities Act of 1933. |
| (2) | Represents 3,450,000 shares of the registrant’s common stock being offered pursuant to the registrant’s initial public offering, including 450,000 shares subject to the underwriters’ over-allotment option. |
| (3) | A registration fee of $8,025 has been paid previously by ImaRx Therapeutics, Inc. on May 19, 2006 in connection with Registration No.333-134311. Pursuant to Rule 457(p), such previous filing fee offsets the filing fee due herewith. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment that specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933, as amended, or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
| The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted. |
SUBJECT TO COMPLETION, DATED JUNE 11, 2007
PRELIMINARY PROSPECTUS

3,000,000 Shares
Common Stock
$ per share
We are selling 3,000,000 shares of our common stock. This is the initial public offering of our common stock and no public market currently exists for our common stock. We currently expect the initial public offering price to be between $6.50 and $7.50 per share. We have applied to have our common stock approved for listing on The NASDAQ Capital Market under the symbol “IMRX.”
Investing in our common stock involves a high degree of risk. Please read the “Risk Factors” beginning on page 9.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per Share | Total | |||||||
| Public offering price | $ | $ | ||||||
| Underwriting discounts | $ | $ | ||||||
| Proceeds to us (before offering-related expenses) | $ | $ | ||||||
We expect total costs and expenses of this offering to be approximately $1.8 million, which will include a non-accountable expense allowance of 2.0% of the gross proceeds of this offering, or $420,000, payable to the representative of the underwriters. We have granted the underwriters a45-day option to purchase up to 450,000 shares of common stock on the same terms and conditions as set forth above, solely to cover over-allotments, if any. Upon completion of this offering we will issue warrants to purchase up to 210,000 shares of our common stock at an exercise price equal to 115% of the initial public offering price per share to the representative of the underwriters, or representative’s warrants, as additional compensation for its services in connection with this offering.
The underwriters are offering the common stock on a firm commitment basis and expect to deliver the shares to purchasers on or about , 2007.
| Maxim Group LLC | I-Bankers Securities, Inc. |
Sole Bookrunner
The date of this prospectus is , 2007
Table of Contents
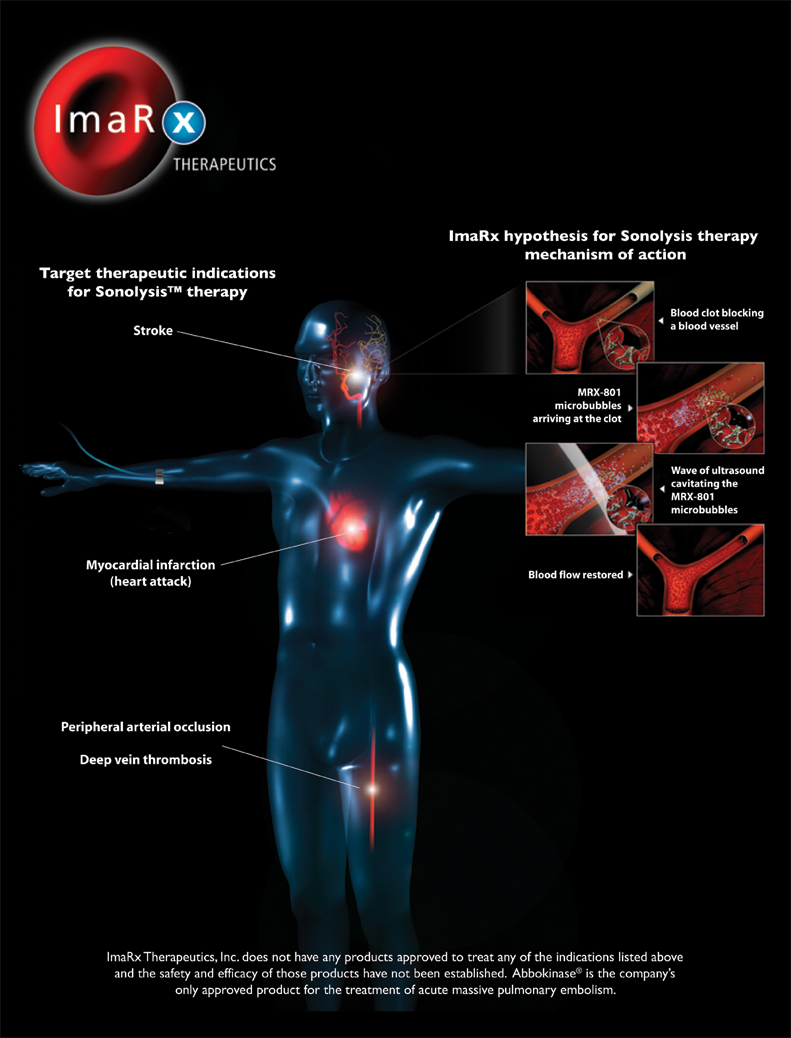
Table of Contents
| Page | ||||||||
| 1 | ||||||||
| 9 | ||||||||
| 28 | ||||||||
| 30 | ||||||||
| 31 | ||||||||
| 32 | ||||||||
| 34 | ||||||||
| 35 | ||||||||
| 36 | ||||||||
| 38 | ||||||||
| 51 | ||||||||
| 73 | ||||||||
| 88 | ||||||||
| 90 | ||||||||
| 93 | ||||||||
| 97 | ||||||||
| 99 | ||||||||
| 102 | ||||||||
| 107 | ||||||||
| 107 | ||||||||
| 107 | ||||||||
| F-1 | ||||||||
| EXHIBIT 1.1 | ||||||||
| EXHIBIT 23.1 | ||||||||
You should rely only on the information contained in this prospectus or any filed issuer free writing prospectus. We have not, and the underwriters have not, authorized anyone to provide you with information different from that contained in this prospectus or any filed issuer free writing prospectus. We are offering to sell, and are seeking offers to buy, shares of common stock only in jurisdictions where offers and sales are permitted. The information contained in this prospectus or any filed issuer free writing prospectus is accurate only as of its date, regardless of its time of delivery or of any sale of the common stock.
Table of Contents
Summary
You should read the entire prospectus carefully before deciding to invest in shares of our common stock.
ImaRx Therapeutics, Inc.
Overview
We are a biopharmaceutical company developing and commercializing therapies for vascular disorders. Our research and development efforts are focused on therapies for stroke and other vascular disorders, using our proprietary microbubble technology to treat vascular occlusions, or blood vessel blockages, as well as the resulting ischemia, which is tissue damage caused by a reduced supply of oxygen. Our commercialization efforts are currently focused on our product approved by the U.S. Food and Drug Administration, or FDA, for the treatment of acute massive pulmonary embolism, or blood clots in the lungs.
Over eight million people in the U.S. are afflicted each year with complications related to blood clots. Approximately 700,000 adults in the U.S., or one every 45 seconds, are afflicted with, and 150,000 die as a result of, some form of stroke each year. Stroke is currently the third leading cause of death, and the leading cause of disability, in the United States. Approximately three million Americans are currently disabled from stroke. The American Stroke Association estimates that approximately $62.7 billion will be spent in the U.S. in 2007 for stroke-related medical costs and disability.
The vast majority of strokes, approximately 87% according to the American Stroke Association, are ischemic strokes, meaning that they are caused by blood clots, while the remainder are the more deadly hemorrhagic strokes caused by bleeding in the brain. Currently available treatment options for ischemic stroke are subject to significant therapeutic limitations. For example, the most widely used treatment for ischemic stroke is a clot-dissolving, or thrombolytic, drug that can be administered only during a narrow time window and poses a risk of bleeding, resulting in 6% or less of ischemic stroke patients receiving such treatment. To facilitate increased administration of stroke therapies, in 2005 the Centers for Medicare and Medicaid Services, or CMS, responded to requests by the American Stroke Association and related groups for higher reimbursement amounts for ischemic stroke patients treated with a thrombolytic drug by approximately doubling the amount of reimbursement provided for such treatment to $11,578 per patient.
In addition to the brain and the lungs, blood clots can block blood flow and cause damage to other tissues in the body such as the heart, in the case of coronary arterial disease, and the legs and other extremities, in the case of peripheral vascular disease. We believe our development and research stage products may address significant unmet medical needs not only for stroke but also for clot-induced damage in tissues other than the brain.
Our Commercial and Development Stage Products
The following table summarizes the status of our commercial product and development stage product candidates:
Product or Candidate | Product Elements | Indication | Development Status | |||
SonoLysistm+tPA therapy | • MRX-801 microbubbles • Ultrasound • tPA | Ischemic stroke | Phase I/II clinical trial in progress | |||
| SonoLysis therapy | • MRX-801 microbubbles | Ischemic stroke | Preclinical | |||
| • Ultrasound | ||||||
Abbokinase® | • Urokinase | Acute massive pulmonary embolism | Approved for marketing | |||
1
Table of Contents
SonoLysis Program. Our SonoLysis program is focused on the development of two product candidates that involve the administration of our proprietary MRX-801 microbubbles and ultrasound, with or without a thrombolytic drug, to break up blood clots and restore blood flow to oxygen deprived tissues. Our MRX-801 microbubbles are a proprietary formulation of a lipid shell encapsulating an inert biocompatible gas. We believe thesub-micron size of our MRX-801 microbubbles allows them to penetrate a blood clot, so that when ultrasound is applied their expansion and contraction, or cavitation, can break the clot into very small particles. We believe that these product candidates have the potential to treat a broad variety of vascular disorders associated with blood clots.
Our initial therapeutic focus for our SonoLysis program is ischemic stroke. The only FDA approved drug for the treatment of ischemic stroke is the thrombolytic drug alteplase, or tPA. The FDA has restricted tPA’s use to patients who are able to begin treatment within three hours of onset of ischemic stroke symptoms and who do not have certain risk factors for bleeding, such as recent surgery or taking medications that prevent clotting. According to Datamonitor, approximately 23% of ischemic stroke patients arrive at a hospital within three hours of onset of symptoms. However, due to the three-hour window for treatment and other limitations, only 1.6% to 2.7% of patients with ischemic stroke in community hospitals, and only 4.1% to 6.3% in academic hospitals or specialized stroke centers are treated with a thrombolytic therapy. Our two SonoLysis product candidates being developed as potential treatments for ischemic stroke are further described below:
| • | SonoLysis+tPA therapyinvolves the administration of our proprietary MRX-801 microbubbles and ultrasound in conjunction with tPA. We believe that this therapeutic approach incorporates two complementary mechanisms of action, mechanical and enzymatic, that together can reduce the time required to dissolve a blood clot and help ensure more rapid and complete restoration of blood flow to at risk brain tissues in patients with ischemic stroke. We are conducting a Phase I/II dose-escalation clinical trial evaluating SonoLysis+tPAtherapy in patients with ischemic stroke. We initiated this trial in January 2007, and intend to enroll a total of 72 patients in various medical centers in the United States and Europe. We anticipate enrollment for this trial will be completed in the first half of 2008 and intend to initiate a Phase II study following completion of the ongoing Phase I/II study. We estimate that if approved by the FDA, over 90,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis+tPAtherapy annually. |
| • | SonoLysis therapyinvolves administration of our MRX-801 microbubbles with ultrasound, but without the administration of a thrombolytic drug. Because SonoLysis therapy does not involve use of a thrombolytic drug and its associated risk of bleeding, we believe SonoLysis therapy may offer advantages over existing treatments for ischemic stroke, including extending the treatment window beyond three hours from onset of symptoms and broadening treatment availability to patients for whom thrombolytic drugs are contraindicated due to risk of bleeding. We have not yet conducted any clinical trials using our proprietary MRX-801 microbubbles with ultrasound to treat blood clot indications without a thrombolytic drug. We are conducting and intend to conduct additional preclinical studies of SonoLysis therapy through the first half of 2008. We expect to initiate a Phase II study to treat patients with ischemic stroke following completion of our SonoLysis+tPAtherapyPhase I/II clinical trial. Because of the preclinical data package as well as our ongoingPhase I/II clinical trial evaluating SonoLysis+tPAtherapy in patients with ischemic stroke, we believe no Phase I study will be required prior to initiating the Phase II study for SonoLysis therapy. We estimate that if approved by the FDA, over 200,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis therapy annually. |
Abbokinase. Our commercially available urokinase product, which we market as Abbokinase, is a thrombolytic drug. Urokinase is a natural human protein primarily produced in the kidneys that stimulates the body’s natural clot-dissolving processes. Abbokinase is FDA approved and marketed for the treatment of acute massive pulmonary embolism. Abbokinase has been administered to over four million patients, and we estimate that approximately 400 acute care hospitals in the U.S. include Abbokinase on their pharmacy formulary today. We acquired Abbokinase, including approximately a four-year supply of inventory, from Abbott Laboratories in April 2006, and began selling Abbokinase in October 2006. We believe Abbokinase sales will provide us with near-term revenue and an opportunity to form relationships with vascular physicians and acute care institutions that regularly administer blood clot therapies. Of the Abbokinase vials that we
2
Table of Contents
expect hospitals to purchase, approximately 64% as of March 31, 2007 will no longer be saleable after October 2007 based on their current expiration dates. In order to facilitate obtaining an extension of current expiration dates, we intend to continue the stability testing program started by Abbott Laboratories, which has been ongoing for over four years. Based on the testing to date, which has shown that the product changes very little from year to year, we believe it is probable that the stability data will support extension of the inventory expiration dates. In connection with our Abbokinase acquisition, we issued a $15.0 million non-recourse promissory note that matures in December 2007. If we are unable to satisfy this debt obligation when due, Abbott Laboratories will have the right to reclaim our remaining inventory of Abbokinase, along with a portion of the cash we have received from our sales of Abbokinase. In April 2007 we sold approximately $9.0 million of Abbokinase, net of discounts and fees, to two of our primary wholesalers, of which, approximately $4.1 million has been placed into an escrow account as security for repayment of our $15.0 million non-recourse promissory note due in December 2007. If the escrowed amount were to be applied to the outstanding balance of principal and accrued interest on that note, the remaining balance due under the note would be approximately $11.9 million as of May 31, 2007.
Our Research Stage Product Candidates
The following table summarizes the status of our research stage product candidates:
| Research | ||||||
Product Candidate | Product Elements | Indication(s) | Status | |||
| SonoLysis therapy | • MRX-801 microbubbles • Ultrasound | Ischemic stroke in pre- hospital setting | Preclinical | |||
SonoLysis+tPAtherapy | • MRX-801 microbubbles • Ultrasound • tPA | Myocardial infarction Peripheral arterial occlusive disease Deep vein thrombosis | Preclinical Preclinical Preclinical | |||
NanO2tm | • MRX-804 emulsion/microbubbles | Hemorrhagic shock Neuroprotection for ischemic stroke | Preclinical Research | |||
| Targeted SonoLysis therapy | • MRX-802 targeted microbubbles | Myocardial infarction and other vascular clots | Research | |||
| Targeted drug delivery | • MRX-803 targeted drug delivery microbubbles | Angiogenic tumors | Research | |||
Additional SonoLysis Opportunities. We believe SonoLysis therapy may be suitable for administration for ischemic stroke in an ambulance before arriving at a hospital because it does not involve use of a thrombolytic drug and its associated risk of bleeding. To pursue an ambulance-based ischemic stroke treatment, we would be required to show either that hemorrhage can be ruled out in an ambulance setting, or that SonoLysis therapy has no detrimental effect on a hemorrhagic stroke. Additionally, we believe that the ability of our SonoLysis+tPAtherapy to reduce the time required to dissolve a blood clot could make this therapy suitable for use in treating a broad variety of vascular disorders beyond ischemic stroke. For example, we believe SonoLysis+tPAtherapy could potentially enable more rapid treatment of recently formed acute clots, such as those that cause myocardial infarction, or heart attack. We also believe SonoLysis+tPA therapy has the potential to treat more establishedsub-acute and chronic clots, such as those in peripheral vascular indications that cannot be effectively treated with thrombolytic therapy alone.
Other Research Stage Opportunities. We are exploring a number of potential future product development opportunities based on our microbubble technology, including:
| • | Oxygen Delivery. We are investigating the potential use of our proprietary MRX-804 emulsion/microbubbles, which we call NanO2, to carry oxygen to parts of the body as a potential treatment for a broad variety of disorders in which reduced blood flow results in oxygen-deprived tissues, such as ischemic stroke, heart attack, and injuries that involve significant blood loss, or hemorrhagic shock. We |
3
Table of Contents
| are working with an academic collaborator who has recently received an approximately $700,000 grant from the U.S. Department of Defense to conduct preclinical animal studies of MRX-804 microbubbles to treat hemorrhagic shock. We believe our NanO2 product candidate may have the ability to be stored at room temperature, which could make it suitable for emergency battlefield or ambulance-based treatments. |
| • | Targeted SonoLysis Therapy. Our research team has developed MRX-802, our next generation SonoLysis microbubbles with targeting technology that causes the microbubbles to bind to blood clots. We believe that our MRX-802 targeted microbubbles will have a greater ability tobreak-up blood clots than non-targeted microbubbles when combined with ultrasound. To further the research on our next generation SonoLysis technology, we have received and are near the mid-point of our work on an approximately $1.2 million grant from the National Institutes of Health, or NIH, to study MRX-802 targeted microbubbles to treat vascular clots. |
| • | Targeted Drug Delivery. We have also developed targeted drug delivery microbubbles, known as MRX-803, which have the potential for selective drug delivery when used in conjunction with ultrasound. We have received an approximately $1.0 million subcontract and have reached the mid-point of our research on an NIH grant to study the use of our proprietary MRX-803 targeted drug delivery microbubbles to treat a variety of tumors. We believe this technology has the potential for broad applications, including delivering drugs to dissolve blood clots or arterial plaque as well as to treat a variety of types of cancer. |
Our Business Strategy
Our goal is to become the leading provider of therapies for stroke and other vascular disorders by developing and marketing products to treat occlusions as well as the resulting ischemia. The key elements of our business strategy are to:
| • | develop and commercialize our SonoLysis product candidates to expand the number of ischemic stroke patients who are eligible for treatment; | |
| • | sell our Abbokinase inventory and benefit from our commercial relationships; | |
| • | leverage our SonoLysis product candidates to accelerate initiation of treatment for ischemic stroke in an ambulance setting and address additional clot disorders in cardiology and peripheral vascular disease; and | |
| • | create a deep pipeline of products based on our microbubble technologies to address additional indications. |
Risks Related to Our Business and Business Strategy
Our business is subject to numerous risks that could prevent us from successfully implementing our business strategy. These risks are highlighted in the section entitled “Risk Factors” immediately following this prospectus summary, and include the following:
| • | we have a history of operating losses, including an accumulated deficit of approximately $65.5 million and an overall stockholders’ deficit of approximately $32.7 million at March 31, 2007, and expect to continue to incur substantial losses for the foreseeable future; | |
| • | we will need substantial additional capital to fund our operations; | |
| • | we may never complete clinical development of our product candidates or have more than one product approved for marketing, and even if approved, our product candidates may never achieve market acceptance; | |
| • | failure to comply with various government regulations in connection with the development, manufacture and commercialization of our product candidates, and post-approval manufacturing and |
4
Table of Contents
| marketing of our products, could result in significant interruptions or delays in our development and commercialization activities; |
| • | we may not be able to sell our inventory of Abbokinase at such times, in such quantities, and at such prices as we anticipate, or at all; |
| • | if we are unable to meet testing specifications for extension of the expiration dates currently applicable to about 64% of our vials of Abbokinase that we expect hospitals to purchase, we will not be allowed to continue selling these vials after October 2007; |
| • | if we fail to satisfy our December 2007 debt obligation to Abbott Laboratories, Abbott Laboratories could reclaim our remaining inventory of Abbokinase, along with the portion of the cash we have received from our sales of Abbokinase that is in an escrow account; and | |
| • | we compete against companies that have longer operating histories, more established products and greater resources than we do. |
In addition, our independent registered public accounting firm has expressed doubt as of May 4, 2007 about our ability to continue as a going concern.
Our Corporate Information
We were organized as an Arizona limited liability company on October 7, 1999, which was our date of inception for accounting purposes. We were subsequently converted to an Arizona corporation on January 12, 2000, and then reincorporated as a Delaware corporation on June 23, 2000. Our principal executive offices are located at 1635 E. 18th St., Tucson, Arizona 85719, and our telephone number at that location is(520) 770-1259. Our corporate website address is www.imarx.com. The information contained in or that can be accessed through our corporate website is not part of this prospectus. Unless the context indicates otherwise, as used in this prospectus, the terms “ImaRx,” “we,” “us” and “our” refer to ImaRx Therapeutics, Inc., a Delaware corporation.
We have rights to use Abbokinase®, which is a U.S. registered trademark owned by Abbott Laboratories. We use SonoLysistm, NanO2tm and the ImaRx Therapeutics logo as trademarks in the U.S. and other countries. All other trademarks and trade names mentioned in this prospectus are the property of their respective owners.
5
Table of Contents
The Offering
| Common stock offered | 3,000,000 shares | |
| Common stock to be outstanding after this offering | 9,054,928 shares | |
| Estimated initial public offering price | Between $6.50 and $7.50 per share | |
| Use of proceeds | To continue the development of our product candidates, including clinical trials, to fund our commercialization efforts, to fund our research and preclinical development activities, and for working capital and other general corporate purposes including a possible partial repayment of debt. See “Use of Proceeds.” |
| Proposed NASDAQ Capital Market symbol | Currently no market for our common stock exists. We have applied to have our common stock listed on The NASDAQ Capital Market under the symbol “IMRX”. |
The number of shares to be outstanding immediately after this offering as shown above is based on 6,054,928 shares outstanding as of May 31, 2007 and excludes:
| • | 550,959 shares of common stock issuable upon the exercise of options outstanding having a weighted average exercise price of $18.43 per share, and 84,433 shares of common stock reserved for future grants, under our 2000 Stock Plan; |
| • | 226,655 shares of common stock issuable upon the exercise of options to be granted under our 2000 Stock Plan upon completion of this offering, having an exercise price equal to the public offering price per share in this offering; | |
| • | 24,997 shares of common stock to be issued pursuant to restricted stock grants under our 2000 Stock Plan upon completion of this offering; | |
| • | 352,324 shares of common stock issuable upon the exercise of warrants outstanding, having a weighted average exercise price of $15.79 per share; |
| • | 210,000 shares of common stock issuable upon the exercise of the representative’s warrant and 496,589 shares of common stock issuable upon the exercise of other warrants to be granted upon completion of this offering, having an exercise price equal to 115% of the public offering price per share in this offering; and |
| • | 850,000 shares of common stock reserved for future issuance under our 2007 Performance Incentive Plan, which will become effective immediately upon the signing of the underwriting agreement for this offering. |
Except as otherwise indicated, all information in this prospectus assumes:
| • | the conversion of all our outstanding shares of preferred stock into 3,448,189 shares of common stock upon the closing of this offering, assuming a1-to-0.84 conversion ratio of our Series F preferred stock. See “Conversion of Series F Preferred Stock”; | |
| • | aone-for-three reverse stock split of our common stock that was effected on May 4, 2007; | |
| • | the filing of our amended and restated certificate of incorporation upon completion of this offering; and | |
| • | no exercise of the underwriters’ over-allotment option. |
6
Table of Contents
Summary Consolidated Financial Data
The following tables summarize certain of our consolidated financial data. We derived the consolidated statements of operations data for the years ended December 31, 2004, 2005 and 2006 from our consolidated audited financial statements included elsewhere in this prospectus. We derived the consolidated statements of operations data for the three months ended March 31, 2006 and 2007, as well as the balance sheet data at March 31, 2007 from our unaudited financial statements included elsewhere in this prospectus. You should read this data together with our financial statements and related notes included elsewhere in this prospectus and the information under “Selected Consolidated Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” (Dollar amounts in thousands, except for per share data.)
| Years Ended December 31, | Three Months Ended March 31, | |||||||||||||||||||
| 2004 | 2005 | 2006 | 2006 | 2007 | ||||||||||||||||
| (Unaudited) | ||||||||||||||||||||
Consolidated Statements of Operations Data: | ||||||||||||||||||||
| Product sales, grant and other revenue | $ | 575 | $ | 619 | $ | 1,327 | $ | 177 | $ | 1,208 | ||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of product sales | — | — | 204 | — | 461 | |||||||||||||||
| Research and development | 2,490 | 3,579 | 8,396 | 1,723 | 1,500 | |||||||||||||||
| General and administrative | 3,183 | 4,142 | 7,371 | 1,618 | 1,098 | |||||||||||||||
| Depreciation and amortization | 186 | 194 | 1,049 | 60 | 363 | |||||||||||||||
| Acquired in-process research and development | — | 24,000 | — | — | — | |||||||||||||||
| Total cost and expenses | 5,859 | 31,915 | 17,020 | 3,401 | 3,422 | |||||||||||||||
| Interest and other income, net | 29 | 122 | 381 | 104 | 41 | |||||||||||||||
| Interest expense | (469 | ) | (587 | ) | (1,515 | ) | (225 | ) | (225 | ) | ||||||||||
| Gain on extinguishment of debt | — | 3,835 | 16,128 | — | — | |||||||||||||||
| Net loss | (5,724 | ) | (27,926 | ) | (699 | ) | (3,345 | ) | (2,398 | ) | ||||||||||
| Accretion of dividends on preferred stock | (301 | ) | (601 | ) | (1,167 | ) | (150 | ) | (433 | ) | ||||||||||
| Net loss attributable to common stockholders | $ | (6,025 | ) | $ | (28,527 | ) | $ | (1,866 | ) | $ | (3,495 | ) | $ | (2,831 | ) | |||||
| Net loss attributable to common stockholders per share — Basic and diluted | $ | (5.37 | ) | $ | (15.11 | ) | $ | (0.72 | ) | $ | (1.35 | ) | $ | (1.09 | ) | |||||
| Weighted average shares outstanding — Basic and diluted | 1,122,881 | 1,888,291 | 2,599,425 | 2,585,315 | 2,605,915 | |||||||||||||||
7
Table of Contents
The following table sets forth a summary of our consolidated balance sheet data at March 31, 2007:
| • | on an actual basis; | |
| • | on a pro forma basis to reflect the conversion of all outstanding shares of preferred stock, valued on our balance sheet at approximately $40.3 million, into 3,448,189 shares of common stock upon the closing of this offering; and | |
| • | on a pro forma as adjusted basis to reflect our receipt of the estimated net cash proceeds from our sale of 3,000,000 shares of common stock in this offering at an assumed initial public offering price of $7.00, the midpoint of the range on the front cover of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
| At March 31, 2007 | ||||||||||||
| Pro Forma | ||||||||||||
| Actual | Pro Forma | as Adjusted | ||||||||||
| (In thousands) | ||||||||||||
| (Unaudited) | ||||||||||||
Consolidated Balance Sheet Data: | ||||||||||||
| Cash and cash equivalents | $ | 2,748 | $ | 2,748 | $ | 20,493 | ||||||
| Working capital(1) | 583 | 583 | 18,328 | |||||||||
| Total assets | 23,384 | 23,384 | 41,129 | |||||||||
| Redeemable convertible preferred stock | 36,297 | — | — | |||||||||
| Total stockholders’ equity (deficit) | $ | (36,676 | ) | $ | 3,621 | $ | 21,366 | |||||
| (1) | Includes $147,000 of deferred financing costs. |
8
Table of Contents
Risk Factors
Investing in our common stock involves a high degree of risk. You should carefully consider the following risk factors and all other information contained in this prospectus before purchasing our common stock. If any of the following events were to occur, our business, financial condition or results of operations could be materially and adversely affected. In these circumstances, the market price of our common stock could decline, and you may lose some or all of your investment.
Risks Relating to Our Business
Unless we are able to generate sufficient product or other revenue, we will continue to incur losses from operations and may never achieve or maintain profitability.
We have a history of net losses and negative cash flow from operations since inception. In the quarter ended March 31, 2007, we generated product revenue of approximately $1.1 million and have funded our operations primarily from private sales of our securities. Net losses attributable to common stockholders for the fiscal years ended December 31, 2004, 2005, and 2006 were approximately $6.0 million, $28.5 million, and $1.9 million, respectively, and for the quarters ended March 31, 2006 and 2007 we had net losses attributable to common stockholders of approximately $3.5 million and $2.8 million, respectively. At March 31, 2007, we had an accumulated deficit of approximately $65.5 million. Except for Abbokinase, which is approved and marketed for the treatment of acute massive pulmonary embolism and which we acquired from Abbott Laboratories in April 2006, we do not have regulatory approval for any of our product candidates. Even if we receive regulatory approval for any product candidates, sales of such products may not generate sufficient revenue for us to achieve or maintain profitability.
Our ability to generate revenue depends on a number of factors, including our ability to:
| • | market and sell our sole commercial product, Abbokinase, or any of our product candidates if we ever obtain regulatory approval for their sale; | |
| • | obtain regulatory approval for SonoLysis+tPAtherapy, SonoLysis therapy, NanO2 and other product candidates; | |
| • | obtain commercial quantities of our products after approval at acceptable cost levels; and | |
| • | enter into strategic partnerships for some of our product candidates. |
We anticipate that our expenses will increase substantially following this offering as a result of:
| • | research and development programs, including significant requirements for clinical trials, preclinical testing, contract manufacturing, and potential regulatory submissions; | |
| • | developing additional infrastructure and hiring additional management and other employees to support the anticipated growth of our development and regulatory activities; | |
| • | regulatory submissions and commercialization activities; | |
| • | additional costs for intellectual property protection and enforcement; and | |
| • | expenses as a result of being a public company. |
Because of the numerous risks and uncertainties associated with developing and commercializing our potential products, we may experience larger than expected future losses and may never become profitable.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
We have received an audit report from our independent registered accounting firm containing an explanatory paragraph stating that our historical recurring losses from operations and net capital deficiency raise substantial doubt about our ability to continue as a going concern. We believe that the completion of this offering will eliminate this doubt and allow us to continue as a going concern at least in the near term. We
9
Table of Contents
estimate that the net proceeds from this offering and our existing cash and cash equivalents will be sufficient to meet our anticipated cash requirements until December 2008, assuming continuing sales of Abbokinase (including the extension of product expiration date) to wholesalers will be adequate to repay the $15.0 million note due to Abbott Laboratories on December 31, 2007. We believe that, based on conversations with our wholesale distributors about the current market demand for Abbokinase, we will sell a sufficient amount of Abbokinase prior to December 31, 2007 to repay the note to Abbott Laboratories. It is possible that the sales of Abbokinase that we expect to occur prior to December 31, 2007 may instead occur in the first quarter of 2008 or later. In such event we would use a portion of the net proceeds of this offering to repay the note on December 31, 2007 and we would replenish our cash resources from subsequent sales of Abbokinase. Alternatively, we may refinance the note using our Abbokinase inventory as collateral. If we are unable to complete this offering, we will need to obtain alternative financing and modify our operational plans to continue as a going concern.
We incurred significant indebtedness in connection with our acquisition of Abbokinase assets from Abbott Laboratories. If we are unable to satisfy this obligation in December 2007, Abbott Laboratories will have a right to reclaim our remaining inventory of Abbokinase, along with a portion of the cash we have received from our sales of Abbokinase.
In connection with our April 2006 acquisition of the remaining inventory of and certain rights related to Abbokinase, we issued to Abbott Laboratories a $15.0 million non-recourse note that is secured by the inventory and rights acquired and matures in December 2007. Although we have commenced selling Abbokinase to obtain near-term revenue that will help fund our cash needs, the asset purchase agreement provides that after we have received initial net revenue of $5.0 million from the sale of Abbokinase, we are then required to deposit 50% of the cash receipts we receive from further sales of Abbokinase into an escrow account to secure the repayment of the note. As of March 31, 2007, our net cash received from sales of Abbokinase to wholesalers and customers totaled approximately $2.6 million and we had not deposited any funds in escrow as security for the note. If the escrow amount is not adequate to repay the note and we are otherwise unable to repay the note by its maturity date, Abbott Laboratories has the right to reclaim our remaining inventory of Abbokinase, along with the portion of the cash we have received from our sales of Abbokinase that is in the escrow account.
We will need substantial additional capital to fund our operations. If we are unable to raise capital when needed, we may be forced to delay, reduce or eliminate our research and development programs or commercialization efforts, and we may be unable to timely pay our debts or may be forced to sell or license assets or otherwise terminate further development of one or more of our programs.
Since our inception, we have financed our operations principally through the private placement of shares of our common and preferred stock and convertible notes and the receipt of government grants. Upon completion of this offering we believe that we will have working capital sufficient to meet our anticipated cash needs through December 2008, assuming our projected sales of Abbokinase to wholesalers occur within a timeframe adequate to repay the $15.0 million note due to Abbott Laboratories on December 31, 2007. We expect our expenses to increase substantially following this offering, and we will require substantial additional financing at various times in the future as we expand our operations and as our debt obligations mature.
Our funding requirements will, however, depend on numerous factors, including:
| • | the timing, scope and results of our preclinical studies and clinical trials; | |
| • | the timing and amount of revenue from sales of Abbokinase; | |
| • | our ability to refinance our $15.0 million secured non-recourse note due to Abbott Laboratories on December 31, 2007, if sales of Abbokinase are insufficient to repay the note; | |
| • | the timing and amount of revenue from grants and other sources; | |
| • | the timing of initiation of manufacturing for our product candidates; |
10
Table of Contents
| • | the timing of, and the costs involved in, obtaining regulatory approvals; |
| • | our ability to establish and maintain collaborative relationships; | |
| • | personnel, facilities and equipment requirements; and | |
| • | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other patent-related costs, including litigation costs, if any, and the result of any such litigation. |
We intend to seek additional funding from a variety of sources, which may include collaborations involving our technology, technology licensing, grants and public or private equity and debt financings. We cannot be certain that any additional funding will be available on terms acceptable to us, or at all. Accordingly, we may not be able to secure the substantial funding that is required to maintain and continue our commercialization and development programs at levels that may be required in the future. We may be forced to accept funds on terms or pricing that are highly dilutive or otherwise disadvantageous to our existing stockholders. We are restricted from granting any additional security interest in our Abbokinase assets that we acquired in 2006. Raising additional funds through debt financing, if available, may involve covenants that restrict our business activities. To the extent that we raise additional funds through collaborations and licensing arrangements, we may have to relinquish valuable rights and control over our technologies, research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to secure adequate financing, we could be required to sell or license assets, delay, scale back or eliminate one or more of our development programs or enter into licenses or other arrangements with third parties to commercialize products or technologies that we would otherwise seek to develop and commercialize ourselves.
We have expanded our business strategy to include the sale of Abbokinase and this exposes us to additional risks which we may not be able to overcome.
Until September 2005, our business strategy focused on the development of microbubbles for the treatment of blood clots and various vascular disorders. In October 2006 we began selling Abbokinase, a thrombolytic drug that we acquired in April 2006. Abbokinase is approved by the FDA for marketing in the U.S. for acute massive pulmonary embolism. We have limited experience in marketing or selling Abbokinase, and we may not be successful in these undertakings. Use of Abbokinase in general involves significant risks, such as bleeding. In addition, adding Abbokinase to our business places additional burdens on our management and technical staff to undertake commercialization activities and may distract them from development activities. Furthermore, our customers may return outdated, short dated or damaged product that is in its original, unopened cartons and received by us prior to 12 months past the expiration date. Finally, the FDA must formally approve the release of each lot of Abbokinase we wish to sell. We must submit a request for each lot we intend to ship to our product wholesalers prior to shipment. If the FDA does not release these lots for shipment in a timely manner or at all, our sales of Abbokinase may be adversely affected.
We may be unable to sell our existing inventory of Abbokinase before product expiration, and even if we are able to sell the existing inventory, the product may be returned prior to use by hospitals and clinics. Additionally, even if we are successful in extending the product expiration dates, we will need to re-brand the product.
In our acquisition of Abbokinase, we received approximately 153,000 vials of Abbokinase manufactured between 2003 and 2005. At the time of our acquisition of Abbokinase, we estimated that hospitals would purchase, and we would thereby recognize revenue for, approximately 111,000 vials, or approximately 72% of the total vials we acquired, which we believe represented approximately a four-year supply of inventory. We also estimated that, due to expiration of the vials or for other reasons, hospitals would not purchase approximately 42,000 vials, or approximately 28% of the vials we acquired. Approximately $16.7 million of the $20.0 million purchase price for Abbokinase was allocated to the vials we expect hospitals to purchase. Of our vials of Abbokinase held in inventory either by us or by our wholesalers as of March 31, 2007, approximately 64% of the vials we expect hospitals to purchase, or approximately $10.7 million in inventory value, will expire by October 2007 based on current stability data. The remaining approximately 36% of the vials we expect to sell to hospitals, or approximately $6.1 million in inventory value, will expire at various
11
Table of Contents
times up to August 2009. We commenced sales of Abbokinase in October 2006. We may or may not be able to sell the entire inventory we acquired before the product expires, and we are not permitted to sell this inventory after its expiration dates. We will continue our ongoing stability program to potentially extend the expiration dates for this inventory. Our license to use the Abbokinase trademark does not cover any inventory with extended expiration dates. Accordingly, if we are successful in demonstrating extended stability and shelf life, we would need to re-brand the inventory to commercialize it. We cannot be certain that we will be successful in establishing an alternate brand name for Abbokinase and obtaining market acceptance. Even if we are able to sell the Abbokinase inventory to wholesalers prior to expiration, the product may be returned to us if outdated or short dated, and our sales could be significantly reduced.
The thrombolytic drug market is highly competitive and dominated by products from Genentech. We have limited sales and marketing capabilities and depend on drug wholesalers to distribute our Abbokinase product.
The market for thrombolytic drugs is currently dominated by thrombolytic drugs offered by Genentech, Inc., in particular alteplase, or tPA, which is approved for treatment of ischemic stroke and pulmonary emboli, among other indications. We cannot be certain that we have sufficient resources to effectively market or sell Abbokinase. We have a limited sales and marketing staff and depend on the efforts of third parties for the sale and distribution of Abbokinase to hospitals and clinics. If we are unable to maintain effective third party distribution on commercially reasonable terms, we may be unable to market and sell Abbokinase in commercial quantities. Drug wholesale companies may be unwilling to continue selling Abbokinase, or we may be forced to accept lower prices or other unfavorable terms or to expend significant additional resources to sell our Abbokinase inventory. Additionally, even if we are able to market and sell Abbokinase in commercial quantities, we do not expect sales of Abbokinase to generate enough revenue for us to achieve profitability.
Our competitors generally are larger than we are, have greater financial resources available to them than we do and may have a superior ability to develop and commercialize competitive products. In addition, if our competitors have products that are approved in advance of ours, marketed more effectively or demonstrated to be safer or more effective than ours, our commercial opportunity will be reduced or eliminated and our business will be harmed.
Our industry sector is intensely competitive, and we expect competition to continue to increase. Many of our actual or potential competitors have substantially longer operating histories and greater financial, research and development and marketing capabilities than we do. Many of them also have substantially greater experience than we have in undertaking preclinical studies and clinical trials, obtaining regulatory approvals and manufacturing and distributing products. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical companies. In addition, academic institutions, government agencies and other public and private research organizations also conduct research, seek patent protection and establish collaborative arrangements for product development and marketing. We may not be able to develop products that are more effective or achieve greater market acceptance than our competitors’ products. Any company that brings competitive products to market before us may achieve a significant competitive advantage.
We believe that the primary competitive factors in the market for treatments of vascular disorders include safety and efficacy, access to and acceptance by leading physicians, cost-effectiveness, physician relationships and sales and marketing capabilities. We may be unable to compete successfully on the basis of any one or more of these factors, which could have a material adverse effect on our business, financial condition and results of operations.
If we are unable to develop, manufacture and commercialize our product candidates, we may not generate sufficient revenue to continue our business.
We currently have only one product, urokinase, currently marketed as Abbokinase, that has received regulatory approval, and we have limited experience commercializing Abbokinase. The process to develop,
12
Table of Contents
obtain regulatory approval for and commercialize potential drug candidates is long, complex and costly. Our proprietary SonoLysis microbubble technology has not been used in clinical trials other than our ongoing Phase I/II clinical trial of our SonoLysis+tPA therapy. We do not expect to have the results of any clinical trials using our proprietary MRX-801 microbubbles until at least 2008. As a result, our business in the near term is substantially dependent upon our ability to sell Abbokinase and to complete development, obtain regulatory approval for and commercialize our SonoLysis product candidates in a timely manner. If we are unable to further develop, commercialize or license our SonoLysis product candidates, we may not be able to earn sufficient revenue to continue our business.
If we want to sell urokinase beyond our existing inventory of Abbokinase, we would need to undertake manufacturing and secure regulatory approval for a new manufacturing process and facility.
As part of our acquisition of Abbokinase, we acquired cell lines that could be used to manufacture urokinase. If we want to sell urokinase beyond our existing inventory of acquired Abbokinase, we would need to undertake manufacturing and to demonstrate that our manufactured material is comparable to the urokinase we purchased from Abbott Laboratories. To demonstrate this, we would need to have our manufacturing process validated by the FDA and may be required to conduct additional preclinical studies, and possibly additional clinical trials, to demonstrate its safety and efficacy. In addition, the manufacturing process for Abbokinase involves a roller bottle production method that is used infrequently today and is available only from a limited number of manufacturers worldwide. We do not currently intend to undertake any efforts required for manufacturing and regulatory approval of additional urokinase in the near term, and even if we were to undertake these efforts in the future, we cannot be certain that we would be able to manufacture and receive regulatory approval for additional sales of urokinase beyond our existing inventory.
We do not plan to manufacture any of our product candidates and will depend on commercial contract manufacturers to manufacture our products.
We do not have our own manufacturing facilities, have no experience in large-scale product manufacturing, and do not intend to develop such facilities or capabilities. Our ability to conduct clinical trials and commercialize our product candidates will depend, in part, on our ability to manufacture our products through contract manufacturers. For all of our product candidates, we or our contract manufacturers will need to have sufficient production and processing capacity to support human clinical trials, and if those clinical trials are successful and regulatory approvals are obtained, to produce products in commercial quantities. Delays in providing or increasing production or processing capacity could result in additional expense or delays in our clinical trials, regulatory submissions and commercialization of our products. In addition, we will be dependent on such contract manufacturers to adhere to the FDA’s current Good Manufacturing Practices, or cGMP, and other regulatory requirements.
Establishing contract manufacturing is costly and time-consuming and we cannot be certain that we will be able to engage contract manufacturers who can meet our quantity and quality requirements in a timely manner and at competitive costs. The manufacturing processes for our product candidates have not yet been tested at commercial levels, and it may not be possible to manufacture such materials in a cost-effective manner. Further, there is no guarantee that the components of our proposed drug product candidates will be available to our manufacturers when needed on terms acceptable to us. If we are unable to obtain contract manufacturing on commercially reasonable terms, we may not be able to conduct or complete planned or necessary clinical trials or commercialize our product candidates.
If our clinical trials are not successful, or if we are unable to obtain regulatory approvals, we will not be able to commercialize our products and we will continue to incur significant operating losses.
Abbokinase is our only product approved for commercial sale. The sale of all of our product candidates in the U.S. requires approval from the FDA and from foreign regulatory agencies for sales outside the U.S. To gain regulatory approval for the commercial sale of our product candidates, we must demonstrate the safety and efficacy of each product candidate in human clinical trials. This process is expensive and can take many
13
Table of Contents
years, and failure can occur at any stage of the testing process. There are many risks associated with our clinical trials. For example:
| • | the only completed clinical trials related to our development of SonoLysis therapy or SonoLysis+tPA therapy have not utilized our proprietary MRX-801 microbubbles and may not be indicative of the safety and effectiveness of our product candidates; | |
| • | if the clinical trial is not conducted in accordance with current Good Clinical Practices, or cGCP, it may not be possible to complete the trial and the FDA may not accept the results of the clinical trial; | |
| • | clinicians, physicians and regulators may not favorably interpret the results of our preclinical studies and clinical trials; | |
| • | some patients in our clinical trials may experience unforeseen adverse medical events related or unrelated to the use of our product candidates; | |
| • | we may be unable to secure a sufficient number of clinical trial sites or patients to enroll in our clinical trials; | |
| • | we may experience delays in securing the services of, or difficulty scheduling, clinical investigators for our clinical trials; | |
| • | third parties who conduct our clinical trials may not fulfill their obligations; | |
| • | we may in the future experience, and have in the past experienced, deviations from the approved clinical trial protocol by our clinical trial investigators; | |
| • | the FDA or the local institutional review board, or IRB, at one or more of our clinical trial sites may interrupt, suspend or terminate a clinical trial or the participation of a particular site in a clinical trial; and | |
| • | the FDA or other regulatory bodies may change the policies and procedures we are required to follow in connection with our clinical trials. |
Any of these or other unexpected events could cause us to delay or terminate our ongoing clinical trials, increase the costs associated with our clinical trials or affect the statistical analysis of the safety and efficacy of our product candidates. If we fail to adequately demonstrate the safety and efficacy of our product candidates, we will not obtain regulatory approval to commercialize our products. Significant delays in clinical development could materially increase our product development costs or impair our competitive position. In addition, any approvals we may obtain may not cover all of the clinical indications for which we seek approval, or an approval may contain significant limitations in the form of narrow labeling and warnings, precautions or contraindications with respect to limitations on use. Accordingly, we may not be able to obtain our desired product registration or marketing approval for any of our product candidates.
We rely on third parties to conduct our clinical trials who may not carry out their contractual duties, with resulting negative impacts on our clinical trials.
We depend on contract research organizations, or CROs, for managing some of our preclinical testing and clinical trials. If we are not able to retain CROs in a timely manner and on commercially reasonable terms, we may not be able to conduct or complete clinical trials or commercialize our product candidates and we do not know whether we will be able to develop or attract partners with such capabilities. We have established relationships with multiple CROs for our existing clinical trials, although there is no guarantee that the CROs will be available for future clinical trials on terms acceptable to us. We may not be able to control the amount and timing of resources that CROs devote to our clinical trials. In the event that we are unable to maintain our relationship with any of our CROs or elect to terminate the participation of any of these CROs, we may lose the ability to obtainfollow-up information for patients enrolled in ongoing clinical trials unless we are able to transfer the care of those patients to another qualified CRO.
14
Table of Contents
Our product candidates may never achieve market acceptance.
We cannot be certain that our products will achieve any degree of market acceptance among physicians and other health care providers and payors, even if necessary regulatory approvals are obtained. We believe that recommendations by physicians and other health care providers and payors will be essential for market acceptance of our products, and we cannot be certain we will ever receive any positive recommendations or reimbursement. Physicians will not recommend our products unless they conclude, based upon clinical data and other factors, that our products are safe and effective. We are unable to predict whether any of our product candidates will ever achieve market acceptance, either in the U.S. or internationally. A number of factors may limit the market acceptance of our products, including:
| • | the timing and scope of regulatory approvals of our products and market entry compared to competitive products; | |
| • | the safety and efficacy of our products, including any inconveniences in administration, as compared to alternative treatments; | |
| • | the rate of adoption of our products by hospitals, doctors and nurses and acceptance by the health care community; | |
| • | the product labeling and marketing claims permitted or required by regulatory agencies for each of our products; | |
| • | the competitive features of our products, including price, as compared to other similar products; | |
| • | the availability of sufficient third party coverage or reimbursement for our products; | |
| • | the extent and success of our sales and marketing efforts; and | |
| • | possible unfavorable publicity concerning our products or any similar products. |
If our products are not commercialized, our business will be materially harmed.
Technological change and innovation in our market sector may cause our products to become obsolete shortly after or even before such products reach the market.
New products and technological development in the pharmaceutical and medical device industries may adversely affect our ability to complete required regulatory requirements and introduce our product candidates into the market or may render our products obsolete. The markets into which we plan to introduce our products are characterized by constant and sometimes rapid technological change, new and improved product introductions, changes in regulatory requirements, and evolving industry standards. Our ability to execute our business plan will depend to a substantial extent on our ability to identify new market trends and develop, introduce and support our candidate products on a timely basis. If we fail to develop and commercialize our product candidates on a timely basis, we may be unable to compete effectively. For example, we are aware of other thrombolytic drugs in development such as ancrod and desmoteplase, which are currently in Phase III clinical trials as treatments for acute ischemic stroke. Since none of our product candidates for treatment of ischemic stroke will be able to achieve regulatory approval for commercial sale in the U.S. any earlier than 2011, if ever, we could by that time find that competitive developments have diminished our product opportunities, which would have an adverse impact on our business prospects and financial condition.
If we are unable to obtain acceptable prices or adequate reimbursement from third-party payors for any product candidates that we seek to commercialize, our revenue and prospects for profitability will suffer.
The commercialization of our product candidates is substantially dependent on whether third-party coverage and reimbursement is available from governmental payors such as Medicare and Medicaid, private health insurers, including managed care organizations and other third-party payors. The U.S. Centers for Medicare and Medicaid Services, health maintenance organizations and other third-party payors in the U.S. and in other jurisdictions are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for new drugs and medical devices and, as a result, they may not cover or
15
Table of Contents
provide adequate payment for our products. Our products may not be considered cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our products to be marketed on a competitive basis. Large private payors, managed care organizations, group purchasing organizations and similar organizations are exerting increasing influence on decisions regarding the use of, and reimbursement levels for, particular treatments. Such third-party payors, including Medicare, are challenging the prices charged for medical products and services, and many third-party payors limit or delay reimbursement for newly approved medical products and indications. Cost-control initiatives could lower the price we may establish for our products which could result in product revenue lower than anticipated. If the prices for our product candidates decrease or if governmental and other third-party payors do not provide adequate coverage and reimbursement levels, our prospects for profitability could suffer.
We intend to rely heavily on third parties to implement critical aspects of our business strategy, and our failure to enter into and maintain these relationships on acceptable business terms, or at all, would materially adversely affect our business.
We intend to rely on third parties for certain critical aspects of our business, including:
| • | manufacturing of our MRX-801 and other proprietary microbubbles; | |
| • | conducting clinical trials; | |
| • | conducting preclinical studies; | |
| • | performing stability and product release testing with respect to Abbokinase; | |
| • | preparing, submitting and maintaining regulatory records sufficient to meet the requirements of the FDA; and | |
| • | customer logistics and distribution of our products. |
We do not currently have many of these relationships in place. Although we use a third party manufacturer to produce MRX-801 microbubbles for our clinical trials on a purchase order basis, that third party does not have the capacity to produce the volume of MRX-801 microbubbles necessary for large-scale clinical trials or commercial sales. We currently have agreements with contract research organizations to manage our clinical trials; audit our clinical trials; help us write protocols and study reports for our clinical trials; store, label, package and distribute our commercial product; and conduct stability and product release testing for our commercialized product. We also have agreements with wholesalers to market and distribute our product, as well as agreements in place with many Group Purchasing Organizations that negotiate prices on behalf of hospitals and clinics. To the extent that we are unable to maintain these relationships or to enter into any one or more of the additional relationships necessary to our business on commercially reasonable terms, or at all, or to eliminate the need for any such relationship by establishing our own capabilities in a particular functional area in a timely manner, we could experience significant delays or cost increases that could have a material adverse effect on our ability to develop and commercialize our product candidates.
We rely on third party products, technology and intellectual property, which could negatively affect our ability to sell our MRX-801 microbubbles or other products commercially or could adversely affect our ability to derive revenue from such products.
Our SonoLysis program may require the use of multiple proprietary technologies, including commercially available ultrasound devices and patented technologies. Manufacturing our products or customizing related ultrasound devices may also require licensing technologies and intellectual property from third parties. Obtaining and maintaining licenses for these technologies may require us to make royalty payments or other payments to several third parties, potentially reducing our revenue or making the cost of our products commercially prohibitive. We cannot be certain that we will be able to establish any or all of the partnering relationships and technology licenses that may be necessary for the pursuit of our business strategy, or, even if such relationships can be established, that they will be on terms favorable to us or that they can be managed in a way that will assist us in executing our business plan.
16
Table of Contents
As a highly specialized scientific business enterprise, our ability to execute our business plan is substantially dependent on certain key members of our scientific and management staff, the loss of any of whom could have a material adverse effect on our business.
A small number of key officers and members of our professional staff are responsible for certain critical areas of our business, such as product research and development, clinical trials, regulatory affairs, manufacturing, intellectual property protection and licensing. The services provided by our key personnel, including: Bradford A. Zakes, our President and Chief Executive Officer; Lynne Weissberger, our Vice President, Regulatory Affairs, Quality Assurance and Regulatory Compliance; Walter Singleton, our Chief Medical Officer; Terry Matsunaga, our Vice President, Research; Rajan Ramaswami, our Vice President, Product Development; Reena Zutshi, our Vice President, Operations; John McCambridge, our Vice President, Sales and Marketing; and Greg Cobb, our Chief Financial Officer, would be difficult to replace. Dr. Singleton recently advised us of his decision to leave the employ of the Company to pursue personal interests. He has entered into a one-year consulting agreement with us. We believe that we will be able to continue our drug development activities as planned. All of our employees are employed at will. Our business and future operating results also depend significantly on our ability to attract and retain qualified management, manufacturing, technical, marketing, regulatory, sales and support personnel for our operations, and competition for such personnel is intense. We cannot be certain that our key executive officers and scientific staff members will remain with us or that we will be able to attract or retain such personnel. If we are unable to retain and continue to attract qualified management and technical staff, this could significantly delay and may prevent the achievement of our research, development and business objectives. We do not maintain key-person life insurance on the lives of any of our executive officers or scientific staff and we do not intend to secure any key-person life insurance after the completion of this offering.
We will need to increase the size of our organization, and we may experience difficulties in managing our growth.
As of May 31, 2007, we had 32 full-time employees. In the future, we will need to expand our managerial, operational, financial, clinical, regulatory and other personnel to manage and expand our operations, undertake clinical trials, manufacture our product candidates, continue our research and development and collaborative activities and commercialize our product candidates. In the next 12 months we anticipate hiring between five and eight new employees at an approximate aggregate cost of between $450,000 and $700,000 annually. Our management and scientific personnel, systems and facilities currently in place will not be adequate to support our planned future growth. Our need to effectively manage our operations, growth and various projects requires that we:
| • | utilize a small sales and marketing organization; | |
| • | identify and manage third party manufacturers for our products; | |
| • | manage our clinical trials effectively; | |
| • | manage our internal research and development efforts effectively while carrying out our contractual obligations to collaborators and other third parties; | |
| • | continue to improve our operational, financial and management controls, reporting systems and procedures under increasing regulatory requirements; and | |
| • | attract and retain sufficient numbers of talented employees. |
We may be unable to implement and manage many of these tasks on a larger scale or in a timely manner and, accordingly, may not achieve our research, development and commercialization goals.
17
Table of Contents
We depend on patents and other proprietary rights, some of which are uncertain and unproven. Further, our patent portfolio and other intellectual property rights are expensive to maintain, protect against infringement claims by third parties, and enforce against third party infringements, and are subject to potential adverse claims.
Because we are developing product candidates that rely on advanced and innovative technologies, our ability to execute our business plan will depend in large part on our ability to obtain and effectively use patents and licensed patent rights, preserve trade secrets and operate without infringing upon the proprietary rights of others. Our Abbokinase product has no patent protection and we have a one-half interest in a patent related to the manufacturing process for Abbokinase. Some of our intellectual property rights are based on licenses that we have entered into with owners of patents.
Although we have rights to 143 issued U.S. patents, plus some foreign equivalents and numerous pending patent applications, the patent position of pharmaceutical, medical device and biotechnology companies in general is highly uncertain and involves complex legal and factual questions. Effective intellectual property protection may also be unavailable or limited in some foreign countries. We have not pursued foreign patent protection in all jurisdictions or for all of our patentable intellectual property. As a result, our patent protection for our intellectual property will likely be less comprehensive if and when we commence international sales.
There are also companies that are currently commercializing FDA approved microbubbles-based products for diagnostic uses. These companies may promote these products for off-label uses which may directly compete with our products when and if approved. Additionally, physicians may prescribe the use of such products for off-label indications which could have the impact of reducing our revenues for our product candidates when and if approved.
In the U.S. and internationally, enforcing intellectual property rights against infringing parties is often costly. Pending patent applications may not issue as patents and may not issue in all countries in which we develop, manufacture or sell our products or in countries where others develop, manufacture and sell products using our technologies. Patents issued to us may be challenged and subsequently narrowed, invalidated or circumvented. We have been notified that, in February 2005, a third party filed an opposition claim to one of our patents in Europe that relates to targeted bubbles for therapeutic and diagnostic use. The third party has agreed to voluntarily dismiss and terminate this claim, but other such conflicts could occur and could limit the scope of the patents that we may be able to obtain or may result in the denial of our patent applications. If a third party were to obtain intellectual property protection for any of the technologies upon which our business strategy is based, we could be required to challenge such protections, terminate or modify our programs that rely on such technologies or obtain licenses for use of these technologies. For example, in July 2003 we received a notice from a third party who owns a patent relating to the administration of ultrasound to break up blood clots indicating that we may need a license to its patent if we intend to administer our therapies according to its patented method. Although we do not intend to administer our therapies according to the third party’s patented method, other similar third party patents, if valid, could require us to seek a license that may not be available on terms acceptable to us or at all, could impose limitations on how we administer our therapies, and may require us to adopt restrictions or requirements as to the manner of administration of our products that we might not otherwise adopt to avoid infringing patents of others. Moreover, we may not have the financial resources to protect our patent and other intellectual property rights and, in that event, our patents may not afford meaningful protection for our technologies or product candidates, which would materially adversely affect our ability to develop and market our product candidates and to generate licensing revenue from our patent portfolio.
Additional risks related to our patent rights and other proprietary rights include:
| • | challenge, invalidation, circumvention or expiration of issued patents already owned by or licensed to us; | |
| • | claims by our consultants, key employees or other third parties that our products or technologies are the result of technological advances independently developed by them and, therefore, not owned by us; |
18
Table of Contents
| • | our failure to pay product development costs, license fees, royalties, milestone payments or other compensation required under our technology license and technology transfer agreements, and the subsequent termination of those agreements; | |
| • | failure by our licensors or licensees to comply with the terms of our license agreements; | |
| • | misrepresentation by technology owners of the extent to which they have rights to the technologies that we purport to acquire or license from them; | |
| • | a potentially shorter patent term as a result of legislation which sets the patent termination date at 20 years from the earliest effective filing date of the patent application instead of 17 years from the date of the grant; and | |
| • | loss of rights that we have licensed due to our failure or decision not to fund further research or failure to achieve required development or commercialization milestones or otherwise comply with our obligations under the license and technology transfer agreements. |
If any of these events occurs, our business may be harmed.
We have limited patent protection for Abbokinase, and third parties likely could develop urokinase without a license from us, which could decrease the market opportunity for Abbokinase.
We own a one-half interest in a patent related to the manufacturing process for Abbokinase. We also have a license to use the “Abbokinase” trademark that expires when our inventory is sold, expires or its expiration date is extended, and trade secrets relating to the manufacturing process for Abbokinase. A third party could acquire or develop a cell line capable of producing urokinase and could devise a manufacturing process that could yield a product consistent with or superior to our Abbokinase product in quality, safety and activity, in each case without a license from us, which could decrease the market opportunity for Abbokinase.
Other companies may claim that we infringe their patents or trade secrets, which could subject us to substantial damages.
A number of third parties, including certain of our competitors, have developed technologies, filed patent applications or obtained patents on technologies and compositions that are related to aspects of our business, including thrombolytic drug therapy, microbubbles and ultrasound. Such third parties may sue us for infringing their patents. If we face an infringement action, defending against such an action could require substantial resources that may not be available to us. In the event of a successful claim of infringement against us, we may be required to:
| • | pay substantial damages; | |
| • | stop using infringing technologies and methods; | |
| • | stop certain research and development efforts; | |
| • | develop non-infringing products or methods; and | |
| • | obtain one or more licenses from third parties. |
Any claims of infringement could cause us to incur substantial costs and could divert management’s attention away from our business in defending against the claim, even if the claim is invalid. A party making a claim could secure a judgment that requires us to pay substantial damages. A claim of infringement could also be used by our competitors to delay market introduction or acceptance of our products. If we are sued for infringement, we could encounter substantial delays in development, manufacture and commercialization of our product candidates. Any litigation, whether to enforce our patent rights or to defend against allegations that we infringe third party rights, will be costly and time consuming and will likely distract management from other important tasks.
19
Table of Contents
Our rights to develop and commercialize certain of our product candidates are subject to the terms and conditions of licenses or sublicenses granted to us by third parties, including other pharmaceutical companies, that contain restrictions that may limit our ability to capitalize on these products.
Our SonoLysis therapy and SonoLysis+tPAtherapy product candidates are based in part on patents and other intellectual property that we license or sublicense from third parties. Our rights to develop and commercialize these product candidates using intellectual property licensed from UNEMED Corporation may terminate, in whole or in part, if we fail to pay royalties to third party licensors, or if we fail to comply with certain restrictions regarding our development activities. In the event of an early termination of any such license or sublicense agreement, rights licensed and developed by us under such agreements may be extinguished, and our rights to the licensed technology may revert back to the licensor. Any termination or reversion of our rights to develop or commercialize any such product candidate may have a material adverse effect on our business.
We are party to an agreement with Bristol-Myers Squibb that restricts us from using our bubble technology for non-targeted diagnostic imaging applications. Bristol-Myers Squibb also has a right of first negotiation should we wish to license to a third party any of our future products or technology related to the use of bubbles for targeted imaging of blood clots, or breaking up blood clots with ultrasound and bubbles. Bristol-Myers Squibb has waived its rights under this agreement with respect to our current generation of MRX-801 microbubbles that we are developing for breaking up blood clots, as well as a new generation of MRX-802 microbubbles that we are developing for breaking up blood clots that include targeting mechanisms to cause the bubbles to attach to blood clots. This right of first negotiation for future technology we may develop in these applications could adversely impact our ability to attract a partner or acquirer for SonoLysis therapy.
In addition, we have been awarded various government funding grants and contracts from The National Institutes of Health and other government agencies. These grants include provisions that provide the U.S. government with the right to use the technologies developed under such grants for certain uses, under certain circumstances. If the government were to exercise its rights, our ability to commercialize such technology would likely be impaired.
We could be exposed to significant product liability claims, which could be time consuming and costly to defend, divert management attention and adversely impact our ability to obtain and maintain insurance coverage. The expense and potential unavailability of insurance coverage for our company or our customers could adversely affect our ability to sell our products, which would negatively impact our business.
We face a risk of product liability exposure related to the testing of our product candidates in clinical trials and will face even greater risks upon any commercialization by us of our product candidates. Thrombolytic drugs are known to involve certain medical hazards, such as risks of bleeding or immune reactions. Our product candidates may also involve presently unknown medical risks of equal or even greater severity. Product liability claims or other claims related to our products, or their off-label use, regardless of their merits or outcomes, could harm our reputation in the industry, and reduce our product sales. Additionally, any lawsuits or product liability claims against us may divert our management from pursuing our business strategy and may be costly to defend. Further, if we are held liable in any of these lawsuits, we may incur substantial liabilities and may be forced to limit or forego further commercialization of one or more of our products. A product liability related claim or recall could be materially detrimental to our business. Our current product liability insurance, which provides us with $10 million of coverage in the aggregate, may be insufficient. We may not be able to obtain or maintain such insurance in adequate amounts, or on acceptable terms, to provide coverage against potential liabilities. The product liability coverage we currently have for our clinical trials may be insufficient to cover fully the costs of any claim or any ultimate damages we may be required to pay. Our inability to obtain or maintain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or limit the commercialization of any products we develop, and could leave us exposed to significant financial losses relating to any products that we do develop and commercialize.
20
Table of Contents
Moreover, Abbokinase is made from human neonatal kidney cells. Products made from human source material may contain infectious agents, such as viruses, that can cause disease. We believe the risk that Abbokinase will transmit an infectious agent has been reduced by changes made by Abbott Laboratories to its tissue acquisition and related manufacturing process that included screening donors for prior exposure to certain viruses, testing donors for the presence of certain current virus infections, testing for certain viruses during manufacturing and inactivatingand/or removing certain viruses. All of our inventory was produced after these changes were made. Despite these measures, Abbokinase may still present a risk of transmitting infectious agents, which could expose us to product liability lawsuits.
If we use hazardous or biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities involve the controlled use of potentially hazardous substances, including toxic chemical and biological materials. Our recent expansion of our business strategy to include the sale of Abbokinase has increased our involvement in the handling and distribution of biological materials. In addition, our operations produce hazardous waste products. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous and biological materials. While we believe that we are currently in compliance with these laws and regulations, continued compliance may be expensive, and current and future environmental regulations may impair our research, development and manufacturing efforts. In addition, if we fail to comply with these laws and regulations at any point in the future, we may be subject to criminal sanctions and substantial civil liabilities and could be required to suspend or modify our operations. Even if we continue to comply with all applicable laws and regulations regarding hazardous materials, we cannot eliminate the risk of accidental contamination or discharge and our resultant liability for any injuries or other damages caused by these accidents. Although we maintain general liability insurance, this insurance may not fully cover potential liabilities for these damages, and the amount of uninsured liabilities may exceed our financial resources and materially harm our business.
The FDA approval process for drugs involves substantial time, effort and financial resources, and we may not receive any new approvals for our product candidates on a timely basis, or at all.
The process required by the FDA before product candidates may be marketed in the U.S. generally involves the following:
| • | preclinical laboratory and animal testing; | |
| • | submission of an IND application which must become effective before clinical trials may begin; | |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of proposed drugs or biologics for their intended use; | |
| • | pre-approval inspection of manufacturing facilities, company regulatory files and selected clinical investigators; and | |
| • | FDA approval of a new drug application, or NDA, or FDA approval of an NDA supplement in the case of a new indication if the product is already approved for another indication. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any new approvals for our product candidates will be granted on a timely basis, if at all. We have failed in the past, and may in the future fail, to make timely submissions of required reports or modifications to clinical trial documents, and such delays as well as possible errors or omissions in such submissions could endanger regulatory acceptance of clinical trial results or even our ability to continue with our clinical trials.
The results of product development, preclinical tests and clinical trials are submitted to the FDA as part of an NDA, or as part of an NDA supplement. The FDA may deny approval of an NDA or NDA supplement if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or an additional pivotal Phase III clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA
21
Table of Contents
or NDA supplement does not satisfy the criteria for approval. The FDA may move to withdraw product approval, once issued, if ongoing regulatory standards are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA may move to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Satisfaction of FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Government regulation may delay or prevent marketing of product candidates for new indications for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our product candidates on a timely basis, if at all. Success in early stage clinical trials does not ensure success in later stage clinical trials. Data obtained from clinical trials is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Even if a product candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain, additional regulatory approvals for our products would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
The FDA’s policies may change and additional government regulations may be enacted, which could prevent or delay regulatory approval of our product candidates or approval of new indications for our product candidates. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or internationally.
If we or our contract manufacturers fail to comply with applicable regulations, sales of our products could be delayed and our revenue could be harmed.
Every medical product manufacturer is required to demonstrate and maintain compliance with cGMP. We and any third party manufacturers or suppliers with whom we enter into agreements will be required to meet these requirements. Our contract manufacturers will be subject to unannounced inspections by the FDA and corresponding foreign and state agencies to ensure strict compliance with cGMP and other applicable government quality control and record-keeping regulations. In addition, transfer of ownership of products triggers a mandatory manufacturing inspection requirement from the FDA. We cannot be certain that we or our contract manufacturers will pass any of these inspections. If we or our contract manufacturers fail one of these inspections in the future, our operations could be disrupted and our manufacturing and sales delayed significantly until we can demonstrate adequate compliance. If we or our contract manufacturers fail to take adequate corrective action in a timely fashion in response to a quality system regulations inspection, the FDA could shut down our or our contract manufacturers’ manufacturing operations and require us, among other things, to recall our products, either of which would harm our business.
Failure to comply with cGMP or other applicable legal requirements can lead to federal seizure of violative products, injunctive actions brought by the federal government, and potential criminal and civil liability on the part of a company and its officers and employees. Because of these and other factors, we may not be able to replace our manufacturing capacity quickly or efficiently in the event that our contract manufacturers are unable to manufacture our products at one or more of their facilities. As a result, the sale and marketing of our products could be delayed or we could be forced to develop our own manufacturing capacity, which would require substantial additional funds and personnel and compliance with extensive regulations.
22
Table of Contents
Our products will remain subject to ongoing regulatory review even if they receive marketing approval. If we fail to comply with applicable regulations, we could lose these approvals, and the sale of our products could be suspended.
Even if we receive regulatory approval to market a particular product candidate, the FDA or foreign regulatory authority could condition approval on conducting additional and costly post-approval clinical trials or could limit the scope of approved labeling. For example, to sell Abbokinase, we are required to continue an ongoing immunogenicity clinical trial that Abbott Laboratories commenced in 2003. Moreover, the product may later cause adverse effects that limit or prevent its widespread use, force us to withdraw it from the market or impede or delay our ability to obtain regulatory approvals in additional countries. In addition, the manufacturer of the product and its facilities will continue to be subject to FDA review and periodic inspections to ensure adherence to applicable regulations. After receiving marketing approval, the FDA imposes extensive regulatory requirements on the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping related to the product. We may not promote or advertise any future FDA-cleared or approved products for use outside the scope of our product’s label or make unsupported promotional claims about the benefits of our products. If the FDA determines that our claims are outside the scope of our label or are unsupported, it could require us to revise our promotional claims, correct any prior statements or bring an enforcement action against us. Moreover, the FDA or other regulatory authorities may bring charges against us or convict us of violating these laws, and we could become subject to third party litigation relating to our promotional practices and there could be a material adverse effect on our business.
If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities or discover previously unknown problems with our products, manufacturers or manufacturing processes, we could be subject to administrative or judicially imposed sanctions, including:
| • | restrictions on the products, manufacturers or manufacturing processes; | |
| • | warning letters; | |
| • | civil or criminal penalties or fines; | |
| • | injunctions; | |
| • | product seizures, detentions or import bans; | |
| • | voluntary or mandatory product recalls and publicity requirements; | |
| • | suspension or withdrawal of regulatory approvals; | |
| • | total or partial suspension of production; and | |
| • | refusal to approve pending applications of marketing approval of new drugs or supplements to approved applications. |
If we were subject to any of the foregoing actions by the FDA, our sales could be delayed, our revenue could decline and our reputation among clinicians, doctors, inventors and research and academic institutions could be harmed.
Marketing and reimbursement practices and claims processing in the pharmaceutical and medical device industries are subject to significant regulation in the U.S.
In addition to FDA restrictions on marketing of pharmaceutical products, several other state and federal laws have been applied to regulate certain marketing practices in the pharmaceutical and medical device industries in recent years, in particular anti-kickback statutes and false claims statutes.
The federal health care program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary
23
Table of Contents
managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from potential liability, the exemptions and safe harbors are drawn narrowly. Practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our future practices may not in all cases meet the criteria for safe harbor protection from anti-kickback liability.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. For example, several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the company’s marketing of the product for unapproved, and thus non-reimbursable, uses. The majority of states also have statutes or regulations similar to the federal anti-kickback and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment.
Because of the breadth of these laws and the limited safe harbors, it is possible that some of our commercial activities in the future could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business.
If we seek regulatory approvals for our products in foreign jurisdictions, we may not obtain any such approvals.
We may market our products outside the U.S., either with a commercial partner or alone. To market our products in foreign jurisdictions, we will be required to obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and jurisdictions and can involve additional testing, and the time required to obtain foreign approvals may differ from that required to obtain FDA approval. We have no experience with obtaining any such foreign approvals. Additionally, the foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. For all of these reasons, we may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. We may not be able to submit applications for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations.
Risks Related to this Offering
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds of this offering, including for any of the purposes described in “Use of Proceeds.” The failure of our management to apply these funds effectively could result in financial losses and materially harm our business, cause the price of our common stock to decline and delay product development.
Our principal stockholders and management own a significant percentage of our stock and will be able to exercise significant influence over our affairs.
Our executive officers, current directors and holders of five percent or more of our common stock, as of the date of this prospectus, beneficially owned approximately 54.7% of our common stock. We expect that upon the closing of this offering, that same group will continue to hold approximately 36.8% of our outstanding common stock. Consequently, even after this offering, these stockholders will likely continue to have significant influence over our operations. The interests of these stockholders may be different than the interests of other stockholders. This concentration of ownership could also have the effect of delaying or
24
Table of Contents
preventing a change in control of our company or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could reduce the price of our common stock.
We will incur increased costs as a public company which may make it more difficult to achieve profitability.
Upon effectiveness of the registration statement for this offering, we will become subject to the reporting obligations set forth in the Securities Exchange Act of 1934, as amended. As a public company, we will incur significant legal, accounting, insurance, investor relations and other expenses that we did not incur as a private company. The disclosures that we will be required to make will generally involve a substantial expenditure of financial resources. In addition, the Sarbanes-Oxley Act of 2002, as well as new rules subsequently implemented by the Securities and Exchange Commission, or SEC, and The NASDAQ Capital Market have required changes in corporate governance practices of public companies. We expect that full compliance with these new rules and regulations will significantly increase our legal and financial compliance costs and make some activities more time-consuming and costly. For example, in connection with becoming a reporting company, we have created additional board committees and will be required to adopt and maintain policies regarding internal controls and disclosure controls and procedures. We plan to retain a consultant to assist us in developing our internal controls to comply with regulatory requirements and may have to retain additional consultants and employees to assist us with other aspects of complying with regulatory requirements applicable to public companies. Such additional reporting and compliance costs may negatively impact our financial results and may make it more difficult to achieve profitability. The rules and regulations imposed by the SEC and as implemented under the Sarbanes-Oxley Act may also make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. To the extent our earnings suffer as a result of the financial impact of our SEC reporting or compliance costs, our business could be harmed.
If you purchase shares of common stock in this offering, you will suffer immediate and substantial dilution of your investment.
Purchasers of common stock in this offering will pay a price per share that substantially exceeds the per share book value of our tangible assets after subtracting our liabilities. Assuming an initial public offering price of $7.00 per share, the midpoint of the range on the front cover of this prospectus, our pro forma as adjusted net tangible book value per share as of March 31, 2007 would have been $2.12. This represents immediate dilution of $4.88 per share to new investors purchasing shares of common stock in this offering at the assumed initial public offering price. See “Dilution.”
There has been no prior public market for our common stock, and an active trading market for our common stock may not develop, potentially lessening the value of your shares and impairing your ability to sell.
Prior to this offering, there has been no public market for our common stock. Although we have applied to have our common stock listed on The NASDAQ Capital Market, an active trading market for our shares may never develop or be sustained following this offering. Accordingly, you may not be able to sell your shares quickly or at the market price if trading in our stock is not active. We will negotiate and determine the initial public offering price with the representative of the underwriters and this price may not be indicative of prices that will prevail in the trading market after the offering. Investors may not be able to sell their common stock at or above the initial public offering price. In addition, there are continuing eligibility requirements for companies listed on The NASDAQ Capital Market. If we are not able to continue to satisfy the eligibility requirements of The NASDAQ Capital Market, then our stock may be delisted. This could result in a lower price of our common stock and may limit the ability of our stockholders to sell our stock, any of which could result in your losing some or all of your investment.
25
Table of Contents
We expect the price of our common stock to be volatile, and if you purchase shares of our common stock you could incur substantial losses if you are unable to sell your shares at or above the offering price.
The price for the shares of our common stock sold in this offering will be determined by negotiation between the representatives of the underwriters and us, but this price may not reflect the market price for our common stock following the offering. In addition, our stock price is likely to be volatile. The stock markets in general and the market for small health care companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the initial public offering price. The price for our common stock may be influenced by many factors, including:
| • | announcements of technological innovations or new products by us or our competitors; | |
| • | announcements of the status of FDA review of our products; | |
| • | the success rate of our discovery efforts, animal studies and clinical trials; | |
| • | developments or disputes concerning patents or proprietary rights, including announcements of infringement, interference or other litigation regarding these rights; | |
| • | the willingness of collaborators to commercialize our products and the timing of commercialization; | |
| • | changes in our strategic relationships which adversely affect our ability to acquire or commercialize products; | |
| • | announcements concerning our competitors or the health care industry in general; | |
| • | public concerns over the safety of our products or our competitors’ products; | |
| • | changes in governmental regulation of the health care industry; | |
| • | changes in the reimbursement policies of third-party insurance companies or government agencies; | |
| • | actual or anticipated fluctuations in our operating results from period to period; | |
| • | variations in our quarterly results; | |
| • | changes in financial estimates or recommendations by securities analysts; | |
| • | changes in accounting principles; and | |
| • | the loss of any of our key scientific or management personnel. |
A decline in the market price of our common stock could cause investors to lose some or all of their investment and may adversely impact our ability to attract and retain employees and raise capital.
A significant portion of our outstanding common stock may be sold into the market in the near future. Substantial sales of common stock, or the perception that such sales are likely to occur, could cause the price of our common stock to decline.
If our existing stockholders sell a large number of shares of common stock or the public market perceives that existing stockholders might sell shares of common stock, the market price of our common stock could decline significantly. All of the shares offered under this prospectus will be freely tradable without restriction or further registration under the federal securities laws, unless purchased by our “affiliates,” as that term is defined in Rule 144 under the Securities Act of 1933. An aggregate of 6,054,928 shares of our common stock outstanding prior to this offering may also be sold pursuant to Rules 144, 144(k) and 701 upon completion of this offering, subject to the expiration oflock-up agreements covering an aggregate of 6,001,621 of these shares.Lock-up agreements covering 5,288,449 shares expire 180 days after the date of this prospectus, andlock-up agreements covering the remaining 713,172 shares expire 12 months after the date of this prospectus.
In addition, as of May 31, 2007, holders of an aggregate of 5,171,298 shares of common stock and warrants to purchase an aggregate of 1,019,530 shares of common stock (including certain warrants to be
26
Table of Contents
issued contingent upon the closing of this offering) have rights with respect to the registration of their shares of common stock with the SEC. See “Description of Capital Stock — Registration Rights.” If we register their shares of common stock following the expiration of thelock-up agreements, they can immediately sell those shares in the public market.
Promptly following this offering, we intend to file a registration statement covering up to a maximum of 1,712,047 shares of common stock that are authorized for issuance under our equity incentive plans. As of May 31, 2007, 550,959 shares were subject to outstanding options, of which 273,452 shares were vested. Once we register these shares, they can be freely sold in the public market upon issuance, subject tolock-up agreements and restrictions on our affiliates. For more information, see the discussion under the caption “Shares Eligible for Future Sale.”
If we fail to develop and maintain an effective system of internal controls, we may not be able to accurately report our financial results or prevent fraud; as a result, current and potential stockholders could lose confidence in our financial reporting, which could harm our business and the trading price of our common stock, should a market for such securities ever develop.
Effective internal controls are necessary for us to provide reliable financial reports and effectively prevent fraud. We have not undertaken any efforts to develop a sophisticated financial reporting system. Section 404 of the Sarbanes-Oxley Act of 2002 will require us, beginning with our fiscal year 2008, to evaluate and report on our internal controls over financial reporting and will require our independent registered public accounting firm annually to attest to such evaluation, as well as issue their own opinion on our internal control over financial reporting. Because we have historically operated as a private company, we have limited experience attempting to comply with public company obligations, including Section 404 of the Sarbanes-Oxley Act. The process of strengthening our internal controls and complying with Section 404 is expensive and time consuming, and requires significant management attention, especially given that we have not previously undertaken any efforts to comply with the requirements of Section 404. We plan to retain a consultant to assist us in developing our internal controls to comply with regulatory requirements and may be required to retain additional consultants or employees to assist us with other aspects of complying with regulatory requirements applicable to public companies in the future. The implementation of compliance efforts with Section 404 will be challenging in the face of our planned rapid growth to support our operations as well as the establishment of infrastructure to support our commercial operations. We cannot be certain that the measures we will undertake will ensure that we will maintain adequate controls over our financial processes and reporting in the future. Furthermore, if we are able to rapidly grow our business, the internal controls that we will need will become more complex, and significantly more resources will be required to ensure our internal controls remain effective. Failure to implement required controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. If we or our auditors discover a material weakness, the disclosure of that fact, even if quickly remedied, could diminish investors’ confidence in our financial statements and harm our stock price. In addition, non-compliance with Section 404 could subject us to a variety of administrative sanctions, including ineligibility for listing on The NASDAQ Capital Market and the inability of registered broker-dealers to make a market in our common stock.
Anti-takeover defenses that we have in place could prevent or frustrate attempts to change our direction or management.
Provisions of our amended and restated certificate of incorporation and bylaws that will become effective upon the closing of this offering and applicable provisions of Delaware law may make it more difficult or impossible for a third party to acquire control of us without the approval of our board of directors. These provisions:
| • | limit who may call a special meeting of stockholders; | |
| • | establish advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted on at stockholder meetings; |
27
Table of Contents
| • | prohibit cumulative voting in the election of our directors, which would otherwise permit holders of less than a majority of our outstanding shares to elect directors; | |
| • | prohibit stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; and | |
| • | provide our board of directors the ability to designate the terms of and issue new series of preferred stock without stockholder approval. |
In addition, Section 203 of the Delaware General Corporation Law generally prohibits us from engaging in any business combination with certain persons who own 15% or more of our outstanding voting stock or any of our associates or affiliates who at any time in the past three years have owned 15% or more of our outstanding voting stock. These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock market in general, and The NASDAQ Capital Market and the market for biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and health care industry factors may materially harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a particular company’s securities, securities class action litigation has often been brought against that company. We may become involved in this type of litigation in the future, regardless of the merits. Litigation often is expensive and diverts management’s attention and resources, which could materially harm our financial condition and results of operations.
We do not intend to pay cash dividends on our common stock in the foreseeable future.
We have never declared or paid any cash dividends on our common stock or other securities, and we currently do not anticipate paying any cash dividends in the foreseeable future. Instruments governing any future indebtedness may also contain various covenants that would limit our ability to pay dividends. Accordingly, our stockholders will not realize a return on their investment unless the trading price of our common stock appreciates. Our common stock may not appreciate in value after the offering and may not even maintain the price at which investors purchased shares.
Forward-looking Statements
This prospectus contains forward-looking statements that are based on our management’s beliefs and assumptions and on information currently available to our management. The forward-looking statements are contained principally in the sections entitled “Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Our Business.” Forward-looking statements include, but are not limited to, statements about:
| • | our ability to conduct and complete our clinical trials and preclinical studies; | |
| • | our expectations with respect to regulatory submissions and approvals; | |
| • | our ability to engage and retain qualified third parties to manufacture our product candidates in a timely and cost-effective manner; | |
| • | our ability to commercialize our product candidates; | |
| • | our ability to market and sell Abbokinase, and the quantities of Abbokinase we may have available for sale; |
28
Table of Contents
| • | our estimates regarding our capital requirements and our need for additional financing; and | |
| • | our expectations with respect to our intellectual property position. |
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “intends,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have included important factors in the cautionary statements included in this prospectus, particularly in the “Risk Factors” section, that could cause actual results or events to differ materially from the forward-looking statements that we make.
You should read this prospectus, any filed issuer free writing prospectus and the documents that we have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward-looking statements.
29
Table of Contents
Use of Proceeds
We estimate that we will receive approximately $17.7 million in net proceeds from this offering, or approximately $20.6 million if the underwriters’ over-allotment option is exercised in full, based upon an assumed initial public offering price of $7.00 per share, the midpoint of the range on the front cover of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses of approximately $1.8 million payable by us, including the non-accountable expense allowance of 2.0% of the gross proceeds of this offering.
A $1.00 increase (decrease) in the assumed initial public offering price of $7.00 per share, the midpoint of the range on the front cover of this prospectus, would increase (decrease) the net proceeds to us from this offering by approximately $2.7 million after deducting underwriting discounts and commissions and estimated offering expenses payable by us, assuming the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same. Regardless of whether there is a decrease of $1.00 in the assumed initial public offering price, we anticipate that the net proceeds from this offering, our existing cash and cash equivalents and continuing sales of Abbokinase will be sufficient to meet our anticipated cash requirements until December 2008.
We plan to utilize the net proceeds from this offering, which we estimate at $17.7 million, or $20.6 million if the underwriters’ over-allotment option is exercised in full, in the following manner:
| • | approximately $12 million to fund development activities in our SonoLysis programs in ischemic stroke, including a Phase I/II clinical trial for SonoLysis+tPAtherapy, preclinical safety studies for our SonoLysis therapy, and manufacturing, additional personnel and material costs related to these development programs; |
| • | approximately $3 million to fund Abbokinase commercialization, including sales and marketing costs, medical affairs activities, continuation of our ongoing product stability studies and related regulatory matters, product storage and labeling, continuation of our ongoing200-patient immunogenicity study, rebranding, additional personnel and exploring the regulatory and commercial feasibility of manufacturing additional Abbokinase inventory; |
| • | approximately $1 million to fund research and preclinical development activities of our SonoLysis programs for additional indications, as well as our NanO2 and other microbubble technologies; and | |
| • | working capital and other general corporate purposes. |
The amounts we actually expend in these areas may vary significantly from our expectations and will depend on a number of factors, including developments relating to scientific, regulatory, competitive and partnering matters. Accordingly, management will retain broad discretion in the allocation of the net proceeds of this offering. A portion of the net proceeds may be used to partially repay our $15.0 million secured non-recourse promissory note maturing in December 2007, which would be reduced to approximately $11.9 million as of May 31, 2007, including accrued interest at the rate of 6% per annum, after applying the escrowed funds associated with our Abbokinase sales in April 2007, if we are unable to secure additional significant sales of Abbokinase to our third party distributors or to refinance the promissory note with Abbott Laboratories. We will use a portion of the net proceeds from this offering to repay the promissory note only if, at the time of such repayment, we anticipate sales of Abbokinase sufficient to replenish our cash resources so as not to affect our planned expenditures under our then-current operating plan. Additionally, a portion of the net proceeds may be used to acquire or invest in complementary businesses, technologies, services or products. We have no current plans, agreements or commitments with respect to any such material acquisition or investment, and we are not currently engaged in any negotiations with respect to any such transaction. Pending such uses, the net proceeds of this offering will be invested in short-term, interest-bearing, investment-grade securities.
30
Table of Contents
Dividend Policy
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any future earnings to finance the growth and development of our business. We do not anticipate paying any cash dividends in the foreseeable future.
31
Table of Contents
Capitalization
The following table sets forth our capitalization as of March 31, 2007:
| • | on an actual basis; | |
| • | on a pro forma basis to reflect the conversion of all outstanding shares of preferred stock, valued on our balance sheet at approximately $40.3 million, into 3,448,189 shares of common stock upon the closing of this offering; and | |
| • | on a pro forma as adjusted basis to reflect our receipt of the estimated net cash proceeds from our sale of 3,000,000 shares of common stock in this offering at an assumed initial public offering price of $7.00, the midpoint of the range on the front cover of this prospectus, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
| At March 31, 2007 | ||||||||||||
| Pro Forma | ||||||||||||
| Actual | Pro Forma | as Adjusted | ||||||||||
| (In thousands) | ||||||||||||
| (Unaudited) | ||||||||||||
| Redeemable convertible preferred stock, $0.0001 par value: 6,443,316 shares issued and outstanding, actual, no shares issued or outstanding, pro forma and pro forma as adjusted | 36,297 | — | — | |||||||||
| Stockholders’ (deficit) equity: | ||||||||||||
| Preferred stock, $0.0001 par value: 30,000,000 shares authorized, actual and pro forma, 5,000,000 shares authorized, pro forma as adjusted; 1,000,000 Series E Preferred shares issued and outstanding, actual, no shares issued or outstanding, pro forma and pro forma as adjusted | 4,000 | — | — | |||||||||
| Common stock, $0.0001 par value: 70,000,000 shares authorized, actual and pro forma, 100,000,000 shares authorized, pro forma as adjusted; 2,606,739 shares issued and outstanding, actual, 6,054,928 shares issued and outstanding, pro forma, and 9,054,928 shares issued and outstanding, pro forma as adjusted | 1 | 1 | 2 | |||||||||
| Additional paid-in capital | 28,783 | 69,080 | 86,824 | |||||||||
| Deficit accumulated during the development stage | (65,460 | ) | (65,460 | ) | (65,460 | ) | ||||||
| Total stockholders’ (deficit) equity | (32,676 | ) | 3,621 | 21,366 | ||||||||
| Total capitalization | $ | 3,621 | $ | 3,621 | $ | 21,366 | ||||||
The pro forma number of shares to be outstanding immediately after this offering as shown above is based on 6,054,928 shares outstanding as of March 31, 2007 and excludes:
| • | 622,709 shares of common stock issuable upon the exercise of options outstanding having a weighted average exercise price of $18.11 per share, and 12,683 shares of common stock reserved for future grants, under our 2000 Stock Plan; | |
| • | 226,655 shares of common stock issuable upon the exercise of options to be granted under our 2000 Stock Plan upon completion of this offering, having an exercise price equal to the public offering price per share in this offering; | |
| • | 24,997 shares of common stock to be issued pursuant to restricted stock grants under our 2000 Stock Plan upon completion of this offering; |
32
Table of Contents
| • | 352,324 shares of common stock issuable upon the exercise of warrants outstanding, having a weighted average exercise price of $15.79 per share; |
| • | 210,000 shares of common stock issuable upon the exercise of the representative’s warrant and 496,589 shares of common stock issuable upon the exercise of other warrants to be granted upon completion of this offering, having an exercise price equal to 115% of the public offering price per share in this offering; and |
| • | 850,000 shares of common stock reserved for future issuance under our 2007 Performance Incentive Plan, which will become effective immediately upon the signing of the underwriting agreement for this offering. |
33
Table of Contents
Dilution
If you invest in our common stock in this offering, the amount you pay per share will be substantially more than the net tangible book value per share of the common stock you purchase.
Our actual net tangible book value as of March 31, 2007 was a deficit of approximately $34.9 million, or approximately $(13.38) per share of common stock. Net tangible book value per share represents total tangible assets less total liabilities, divided by the number of shares of common stock outstanding as of March 31, 2007. Our pro forma net tangible book value as of March 31, 2007 was approximately $1.4 million, or approximately $0.23 per share of common stock. Our pro forma net tangible book value gives effect to the conversion of all outstanding shares of preferred stock, valued on our balance sheet at approximately $40.3 million, into 3,448,189 shares of common stock upon the closing of this offering.
After giving effect, based on an assumed initial public offering price of $7.00 per share, the midpoint of the range on the front cover of this prospectus, to (i) the automatic conversion of our outstanding preferred stock into 3,448,189 shares of common stock in connection with the closing of this offering, and (ii) receipt of the net cash proceeds from the sale of 3,000,000 shares of common stock in this offering, after deducting underwriting discounts and commissions and estimated offering expenses payable by us, our pro forma as adjusted net tangible book value as of March 31, 2007 would have been approximately $19.1 million, or $2.12 per share. See “Conversion of Series F Preferred Stock.” This represents an immediate increase in pro forma as adjusted net tangible book value per share of $1.89 to existing stockholders and an immediate dilution of $4.88 per share to new investors purchasing shares of common stock in this offering at the assumed initial offering price.
The following table illustrates this dilution on a per share basis:
| Assumed initial public offering price per share | $ | 7.00 | ||||||
| Actual net tangible book value (deficit) per share as of March 31, 2007 | $ | (13.38 | ) | |||||
| Increase per share due to pro forma adjustments | 13.61 | |||||||
| Pro forma net tangible book value per share as of March 31, 2007, before this offering | 0.23 | |||||||
| Increase in pro forma net tangible book value per share attributable to this offering | 1.89 | |||||||
| Pro forma as adjusted net tangible book value per share after this offering | 2.12 | |||||||
| Dilution in pro forma net tangible book value per share to new investors in this offering | $ | 4.88 | ||||||
If the underwriters exercise their over-allotment option to purchase 450,000 additional shares from us in this offering, our pro forma as adjusted net tangible book value per share will increase to $2.32 per share, representing an immediate increase to existing stockholders, of $2.09 per share and an immediate dilution of $4.68 per share to new investors assuming conversion of all shares of our preferred stock. If any shares are issued in connection with outstanding options, you will experience further dilution.
The following table summarizes, on the pro forma as adjusted basis described above, as of March 31, 2007, the number of shares of common stock purchased from us, the total consideration paid and the average price per share paid to us by existing stockholders, and to be paid by new investors purchasing shares of common stock for cash in this offering. The table assumes an initial public offering price of $7.00 per share, the midpoint of the range on the front cover of this prospectus, before deducting underwriting discounts and commissions and estimated offering expenses payable by us.
| Total Shares | Total Consideration | Average Price | ||||||||||||||||||
| Number | % | Amount | % | Per Share | ||||||||||||||||
| Existing stockholders | 6,054,928 | 66.9 | % | $ | 53,000,000 | 71.6 | % | $ | 8.75 | |||||||||||
| New investors | 3,000,000 | 33.1 | 21,000,000 | 28.4 | 7.00 | |||||||||||||||
| Total | 9,054,928 | 100.0 | % | 74,000,000 | 100.0 | % | ||||||||||||||
34
Table of Contents
The number of shares to be outstanding immediately after this offering as shown above is based on 6,054,928 shares outstanding as of March 31, 2007 and excludes:
| • | 622,709 shares of common stock issuable upon the exercise of options outstanding having a weighted average exercise price of $18.11 per share, and 12,683 shares of common stock reserved for future grants, under our 2000 Stock Plan; | |
| • | 226,655 shares of common stock issuable upon the exercise of options to be granted under our 2000 Stock Plan upon completion of this offering, having an exercise price equal to the public offering price per share in this offering; | |
| • | 24,997 shares of common stock to be issued pursuant to restricted stock grants under our 2000 Stock Plan upon completion of this offering; | |
| • | 352,324 shares of common stock issuable upon the exercise of warrants outstanding having a weighted average exercise price of $15.79 per share; | |
| • | 210,000 shares of common stock issuable upon the exercise of the representative’s warrant and 496,589 shares of common stock issuable upon the exercise of other warrants to be granted upon completion of this offering, having an exercise price equal to 115% of the public offering price per share in this offering; | |
| • | 850,000 shares of common stock reserved for future issuance under our 2007 Performance Incentive Plan, which will become effective immediately upon the signing of the underwriting agreement for this offering. |
If the underwriters’ over-allotment option is exercised in full, the following will occur:
| • | the percentage of shares of common stock held by existing stockholders will decrease to approximately 66.9% of the total number of shares of common stock outstanding after this offering; and | |
| • | the number of shares held by new investors will increase to 3,000,000, or approximately 33.1%, of the total number of shares of common stock outstanding after this offering. |
Conversion of Series F Preferred Stock
In connection with the closing of this offering, all of our outstanding preferred stock will convert into common stock. The per share conversion rate of our Series F preferred stock is variable and will be determined by dividing $5.00 by the lesser of (a) $25.00 (as adjusted for any stock dividends, combinations, splits, recapitalizations and the like with respect to such shares) or (b) 85% of the price per share paid in this offering. Therefore, depending on the price of the shares sold in this offering, the holders of the Series F preferred stock may receive a greater number of shares of common stock for each share of Series F preferred stock converted in connection with this offering than they would otherwise be entitled to receive. We will not know the conversion rate of our Series F preferred stock until the public offering price is determined.
In this prospectus, we have estimated the number of shares of common stock issuable upon conversion of the Series F preferred stock assuming an initial public offering price of $7.00, the midpoint of the range on the front cover of this prospectus, meaning that we have assumed aone-to-0.84 conversion ratio of our Series F preferred stock (as adjusted for any stock dividends, combinations, splits, recapitalizations and the like with respect to such shares).
Upon completion of this offering, our existing stockholders will continue to have significant influence over the outcome of corporate actions requiring stockholder approval, including the election of directors, any merger, consolidation or sale of all or substantially all of our assets or any other significant corporate transaction. Because only some of our stockholders own Series F preferred stock, changes in our valuation in connection with this offering will impact the conversion ratio of our Series F preferred stock and thus the relative ownership of our common stock upon completion of this offering among our existing stockholders.
35
Table of Contents
Selected Consolidated Financial Data
You should read the following selected consolidated financial data in conjunction with our consolidated financial statements and the related notes appearing elsewhere in this prospectus and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” We have derived the consolidated statements of operations data for the years ended December 31, 2004, 2005 and 2006 and the consolidated balance sheet data at December 31, 2005 and 2006 from our consolidated audited financial statements, which are included elsewhere in this prospectus. We have derived the consolidated statements of operations data for the years ended December 31, 2002 and 2003 and the consolidated balance sheet data as of December 31, 2002, 2003 and 2004 from our audited financial statements, which are not included in this prospectus. The selected consolidated statements of operations data for the three months ended March 31, 2006 and 2007 and the selected consolidated balance sheet data at March 31, 2007 are derived from our unaudited consolidated financial statements which are included elsewhere in this prospectus. Our historical results for any prior period are not necessarily indicative of results to be expected for any future period. (In thousands, except per share data.)
| Three Months Ended | ||||||||||||||||||||||||||||
| Years Ended December 31, | March 31, | |||||||||||||||||||||||||||
| 2002 | 2003 | 2004 | 2005 | 2006 | 2006 | 2007 | ||||||||||||||||||||||
| (Unaudited) | ||||||||||||||||||||||||||||
Consolidated Statements of Operations Data: | ||||||||||||||||||||||||||||
| Product sales, grant and other revenue | $ | 71 | $ | 224 | $ | 575 | $ | 619 | $ | 1,327 | $ | 177 | $ | 1,208 | ||||||||||||||
| Costs and expenses: | ||||||||||||||||||||||||||||
| Costs of product sales | — | — | — | — | 204 | — | 461 | |||||||||||||||||||||
| Research and development | 1,399 | 1,878 | 2,490 | 3,579 | 8,396 | 1,723 | 1,500 | |||||||||||||||||||||
| General and administrative | 1,840 | 1,654 | 3,183 | 4,142 | 7,371 | 1,618 | 1,098 | |||||||||||||||||||||
| Depreciation and amortization | 245 | 209 | 186 | 194 | 1,049 | 60 | 363 | |||||||||||||||||||||
| Acquired in-process research and development(1) | — | — | — | 24,000 | — | — | — | |||||||||||||||||||||
| Total cost and expenses | 3,484 | 3,741 | 5,859 | 31,915 | 17,020 | 3,401 | 3,422 | |||||||||||||||||||||
| Minority interest in loss of consolidated subsidiary | 369 | — | — | — | — | — | — | |||||||||||||||||||||
| Interest and other income, net | 14 | 22 | 29 | 122 | 381 | 104 | 41 | |||||||||||||||||||||
| Interest expense | (170 | ) | (325 | ) | (469 | ) | (587 | ) | (1,515 | ) | (225 | ) | (225 | ) | ||||||||||||||
| Gain on extinguishment of debt(2) | — | — | — | 3,835 | 16,128 | — | — | |||||||||||||||||||||
| Net loss | (3,200 | ) | (3,820 | ) | (5,724 | ) | (27,926 | ) | (699 | ) | (3,345 | ) | (2,398 | ) | ||||||||||||||
| Accretion of dividends on preferred stock | (1,640 | ) | (1,287 | ) | (301 | ) | (601 | ) | (1,167 | ) | (150 | ) | (433 | ) | ||||||||||||||
| Net loss attributable to common stockholders | $ | (4,840 | ) | $ | (5,107 | ) | $ | (6,025 | ) | $ | (28,527 | ) | $ | (1,866 | ) | $ | (3,495 | ) | $ | (2,831 | ) | |||||||
| Net loss attributable to common stockholders per share — Basic and diluted | $ | (8.24 | ) | $ | (8.71 | ) | $ | (5.37 | ) | $ | (15.11 | ) | $ | (0.72 | ) | $ | (1.35 | ) | $ | (1.09 | ) | |||||||
| Weighted average shares outstanding — Basic and diluted | 587,599 | 586,396 | 1,122,881 | 1,888,291 | 2,599,425 | 2,585,315 | 2,605,915 | |||||||||||||||||||||
36
Table of Contents
| At December 31, | At March 31, | |||||||||||||||||||||||
| 2002 | 2003 | 2004 | 2005 | 2006 | 2007 | |||||||||||||||||||
| (In thousands) | (In thousands) | |||||||||||||||||||||||
| (Unaudited) | ||||||||||||||||||||||||
Consolidated Balance Sheet Data: | ||||||||||||||||||||||||
| Cash and cash equivalents | $ | 2,104 | $ | 736 | $ | 1,538 | $ | 8,513 | $ | 4,256 | 2,748 | |||||||||||||
| Working capital (deficit) | 1,568 | (1,440 | ) | 739 | (8,111 | ) | 2,657 | 583 | ||||||||||||||||
| Total assets | 2,908 | 1,298 | 2,122 | 9,516 | 25,293 | 23,384 | ||||||||||||||||||
| Long-term notes payable, less current portion | 3,740 | 4,002 | 4,282 | — | — | — | ||||||||||||||||||
| Redeemable convertible preferred stock | 19,189 | 20,826 | 21,127 | 21,727 | 35,863 | 36,297 | ||||||||||||||||||
| Total stockholders’ deficit | (20,971 | ) | (26,003 | ) | (24,529 | ) | (29,327 | ) | (30,008 | ) | (32,676 | ) | ||||||||||||
| (1) | Research and development expense for the year ended December 31, 2005 includes the purchase of in-process research and development operations valued at $24,000,000 in accordance with an Asset Purchase Agreement entered into with Abbott Laboratories in September 2005 related to our acquisition of certain recombinant thrombolytic drug technologies. In December 2006, our Board of Directors decided not to complete payment for these technologies under the non-recourse debt we had issued to Abbott Laboratories, and instead to allow the acquired technologies to be repossessed by Abbott Laboratories. | |
| (2) | Gain on extinguishment of a debt payable to a development partner in a joint development agreement entered into in 2001 resulted in a gain on extinguishment of note of $3.8 million in March 2005. Extinguishment of the non-recourse debt issued to Abbott Laboratories as partial payment for the 2005 purchase of recombinant thrombolytic drug technologies resulted in a gain on extinguishment of debt of $16.1 million in December 2006. |
37
Table of Contents
Management’s Discussion and Analysis of
Financial Condition and Results of Operations
Financial Condition and Results of Operations
The following discussion and analysis should be read in conjunction with “Selected Consolidated Financial Information” and our consolidated financial statements and related notes included elsewhere in this prospectus. This discussion and analysis and other parts of the prospectus contain forward-looking statements based upon current beliefs, plans and expectations that involve risks, uncertainties and assumptions. Our actual results and the timing of selected events could differ materially from those anticipated in these forward-looking statements as a result of several factors, including those set forth under “Risk Factors” and elsewhere in this prospectus. You should carefully read the “Risk Factors” section of this prospectus to gain an understanding of the important factors that could cause actual results to differ materially from our forward-looking statements. Please also see the section entitled “Forward-looking Statements.”
Overview
We are a biopharmaceutical company developing and commercializing therapies for vascular disorders. Our development efforts are focused on therapies for stroke and other vascular disorders, using our proprietary microbubble technology to treat vascular occlusions, or blood vessel blockages, as well as the resulting ischemia, which is tissue damage caused by a reduced supply of oxygen. Our commercialization efforts are currently focused on our product approved to treat acute massive pulmonary embolism, or blood clots in the lungs.
We were organized as an Arizona limited liability company on October 7, 1999, which was our date of inception for accounting purposes. We were subsequently converted to an Arizona corporation on January 12, 2000, and then reincorporated as a Delaware corporation on June 23, 2000. In the quarter ended March 31, 2007, we generated approximately $1.1 million in net sales of our commercial product Abbokinase. From our inception through March 31, 2007, we accumulated a deficit from operations of approximately $65.5 million. We have funded our operations to date primarily through private placements of our preferred and common stock as well as the sale of convertible notes and the receipt of government grants. Through March 31, 2007, we had received net proceeds of approximately $46.8 million from the issuance of shares of our preferred and common stock and convertible notes.
Since our inception, we have devoted substantially all of our efforts toward planning, conducting and funding the various stages of development for our product candidates, researching potential new product opportunities based upon our proprietary technologies, and acquiring technology and potential products. We expect our operating losses to increase for at least the next several years due to increasing expenses associated with proposed clinical trials, product development, selling, general and administrative costs and regulatory activities.
In September 2005, we acquired the technology and development assets of Abbott Laboratories relating to two recombinant thrombolytic drug candidates. We determined at that time that, since they had not yet received FDA approval and presented no alternative future use, these technologies did not meet established guidelines for technological feasibility sufficiently to be recorded as assets. As a result, the full purchase price consideration of $24.0 million was recorded as acquired in-process research and development expense for the year ended December 31, 2005. In December 2006, we chose not to pursue further development and commercialization of these technologies because we were unable to obtain adequate financing to repay the $15.0 million non-recourse note due December 31, 2006, that we had issued to Abbott Laboratories as partial consideration for the acquisition of these technologies or to pay the costs of such further development and commercialization. Following that decision, Abbott Laboratories indicated its intent to repossess the assets in accordance with its security interest. As a result, we realized a gain of $16.1 million in December 2006 relating to extinguishment of the non-recourse note and accrued interest. We incurred approximately $0.5 million in research and development costs on these products before deciding not to pursue them further.
In April 2006, we also acquired from Abbott Laboratories the assets related to Abbokinase, including the remaining inventory of finished product, all regulatory and clinical documentation, validated cell lines, and intellectual property rights, including trade secrets and know-how relating to the manufacture of urokinase
38
Table of Contents
using the tissue culture method. Since no employees, equipment, manufacturing facilities or arrangements or sales and marketing organization were included in this transaction, we accounted for it as an acquisition of assets rather than as an acquisition of a business, with a purchase price of $20.0 million. The purchase price has been allocated to the assets acquired based upon the fair value assessments. We commenced selling Abbokinase in October 2006. In the quarter ended March 31, 2007, we had recognized revenues of approximately $1.1 million from sales of Abbokinase, and had not been required to deposit any of these sale proceeds in escrow as security for the payment of our $15.0 million non-recourse note payable to Abbott Laboratories, which will mature on December 31, 2007. At March 31, 2007, our remaining inventory of Abbokinase represented approximately a four-year supply based upon market demand statistics from our wholesalers, but our ability to sell such inventory may be limited by product expiration dates and current stability data. Based on current stability data from the ongoing stability program, as of March 31, 2007, approximately 64% of our vials of Abbokinase that we expect hospitals to purchase and that is held in inventory either by us or by our wholesalers will expire between August and October 2007 with the remainder expiring at various times up to August 2009. The next testing point of our ongoing stability program, at which we may obtain data sufficient to extend the expiration dates of our unlabeled inventory, will be completed in the fall of 2007. We will be required to submit this data to the FDA. If the parameters tested are within the specifications previously approved by the FDA, we may then submit a lot release request to the FDA, and upon the FDA’s approval, we may at that time label vials with extended expiration dating to between June and August 2009. We must obtain FDA approval for each lot release of inventory. Inventory is labeled with an expiration date upon approval of a lot release by the FDA. Once labeled, we cannot extend the expiration date of the vials labeled. If we are successful in extending the expiration dates of our unlabeled inventory, we intend to continue the stability program after the fall of 2007 to potentially enable further expiration extensions for future product labeling. Additionally, even if we are successful in extending the product expiration dates, we will need to re-brand the product. We may be unable to sell our existing inventory of Abbokinase before product expiration, and even if we are able to sell the existing inventory the product may be returned prior to use by hospitals and clinics. In order to facilitate obtaining an extension of current expiration dates, we are continuing the stability testing program started by Abbott Laboratories, which has been ongoing for over four years. Based on the testing to date, which has shown that the product changes very little from year to year, we believe it is probable that the stability data will support extension of the inventory expiration dates. As a result, we believe that we will be able to sell this inventory and that we will recover the cost of this inventory. Additionally, if we are successful in extending the product expiration dates, we will need to re-brand the product before selling further inventory.
Product Sales, Grant and Other Revenue
We have generated only a limited amount of revenue to date, primarily by providing research services for projects funded under various government grants and from Abbokinase sales. We commenced sales of Abbokinase in October 2006 and anticipate that we will generate additional revenue from sales of Abbokinase. However, any such revenue is difficult to predict as to both timing and amount, may not be achieved in any consistent or predictable pattern, and in any case will not be sufficient to prevent us from incurring continued and increasing losses from our development and other activities. Additionally, wholesalers and hospitals may return outdated, short dated or damaged Abbokinase product that is in its original, unopened cartons and received by us prior to 12 months past the expiration date. We have a limited product returns history, therefore we recognize revenue only after inventory has shipped from a wholesaler to a hospital. In April 2007, we sold a total of approximately $9.0 million of Abbokinase, net of discounts and fees, to two of our primary wholesalers, of which approximately $4.1 million of the proceeds has been placed into an escrow account as security for repayment of our $15.0 million promissory note due in December 2007. The vials of Abbokinase that we sold have expiration dates ranging from December 2008 to August 2009. If the escrowed amount were to be applied to the outstanding balance of principal and interest on that note, the remaining amount due under the note would be approximately $11.9 million as of May 31, 2007.
All product sales recorded relate to sales of Abbokinase in the United States, which we commenced in October 2006. Due to the lack of returns history, we currently account for these product shipments using a deferred revenue recognition model. We do not recognize revenue upon product shipment to a wholesaler but
39
Table of Contents
rather, we defer the recognition of revenue until the right of return no longer exists or when the product is sold to the end user hospital as is stipulated by SFAS No. 48,Revenue Recognition When the Right of Return Exists. We record product sales net of chargebacks, distributor fees, discounts paid to wholesalers, and administrative fees paid to Group Purchasing Organizations (GPOs). The allowances are based on historical information and other pertinent data. As of March 31, 2007, we had deferred revenue of approximately $0.6 million. A more detailed discussion of our revenue recognition practices is set forth in Note 2 of the Notes to our Consolidated Financial Statements.
Cost of product sales is determined using a weighted-average method and includes the acquisition cost of the inventory as well as additional labeling costs we incur to bring the product to market. Our product pricing is fixed, but could include a variable sales or cash discount depending on the nature of the sale. Our gross margins will be affected by chargebacks, discounts and administrative fees paid to the wholesalers and GPOs.
Research and Development Expenses
We classify our research and development expenses into four categories of activity, namely, research, development, clinical and regulatory. To date, our research and development efforts have been focused primarily on product candidates from our microbubble technology program. Historically we have not tracked research and development expenses by product candidate. However, in the future we intend to separately track expenses related to activities such as manufacturing and preclinical studies or clinical trials for each of our primary product candidates and products. We expect our research and development expenses to increase with the planned continuation of clinical trials for our SonoLysis product candidates. Clinical development timelines, likelihood of commercialization and associated costs are uncertain and therefore vary widely. We anticipate determining which research and development projects to pursue as well as the level of funding available for each project based on the scientific and clinical results of each product candidate. We currently estimate we will complete the current or imminent stage of development for each primary product candidate as follows:
| • | For our SonoLysis program, we intend to conduct the ongoing Phase I/II clinical trial for ischemic stroke evaluating SonoLysis+tPA therapy, and to conduct additional preclinical studies and initiate Phase II clinical trials for both SonoLysis+tPA therapy and SonoLysis therapy. We estimate that these efforts will cost approximately $12.0 million through December 2008. We expect to allocate approximately $12.0 million of the net proceeds from this offering toward development of our SonoLysis program. | |
| • | We intend to maintain the regulatory status of Abbokinase as an FDA-approved product, to continue our ongoing product stability studies and related regulatory matters, product storage and labeling to enable us to seek the extension of the expiration dates of the inventory, to continue our ongoing200-patient immunogenicity study and to explore the feasibility of manufacturing Abbokinase. We estimate that these efforts may cost approximately $3.0 million through December 2008. | |
| • | We intend to further pursue research of our microbubble technologies and estimate that this effort may cost approximately $1.0 million through December 2008. Any new government grants or research collaborations could significantly alter our total research expense depending on the timing and amount of any such awards or agreements. |
At this time, due to the risks inherent in the clinical trial process and the related regulatory process, our development completion dates and costs vary significantly for each product candidate and are very difficult to estimate. The lengthy process of seeking regulatory approvals and the subsequent compliance with applicable regulations require the expenditure of substantial additional resources. Any failure by us to obtain, or any delay in obtaining, regulatory approvals for our product candidates could cause our research and development expenditures to increase and, in turn, have a material adverse effect on our results of operations. We cannot be certain when, if ever, any cash flows from our current product candidates will commence.
40
Table of Contents
General and Administrative Expenses
General and administrative expenses consist primarily of personnel-related expenses and other costs and fees associated with our general corporate activities, such as sales and marketing, administrative support, business development, intellectual property protection, corporate compliance and preparing to become a public reporting company, as well as a portion of our overhead expenses. Our selling expenses have increased and may continue to increase as we expand our infrastructure to support increased commercialization efforts relating to Abbokinase. We also anticipate incurring additional expenses of approximately $1.5 million to $2.0 million per year as a public company following the completion of this offering as a result of additional legal, accounting and corporate governance expenses, including costs associated with tax return preparations, accounting support services, Sarbanes-Oxley compliance expenses, filing annual and quarterly reports with the SEC, directors’ fees, directors’ and officers’ insurance, listing and transfer agent fees, and investor relations expenses.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosed amounts of contingent assets and liabilities and our reported revenue and expenses. Significant management judgment is required to make estimates in relation to clinical trial costs and costs related to public reporting company preparation. We evaluate our estimates, and judgments related to these estimates, on an ongoing basis. We base our estimates of the carrying values of assets and liabilities that are not readily apparent from other sources on historical experience and on various other factors that we believe are reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
We believe that the following accounting policies are critical to a full understanding of our reported financial results. Our significant accounting policies are more fully described in Note 2 of our consolidated financial statements.
Inventory
Inventory is comprised of finished goods and is stated at the lower of cost or market value. We have one commercially available product, marketed as a clot-dissolving, or thrombolytic, urokinase drug called Abbokinase. Abbokinase is FDA approved and marketed for the treatment of acute massive pulmonary embolism. Cost was determined as a result of the purchase price allocation from the acquisition of Abbokinase from Abbott Laboratories in 2006. We estimate that as of March 31, 2007 the vials we expect hospitals to purchase and that are held in inventory either by us or by our wholesalers comprises approximately a four-year supply. We periodically review the composition of inventory in order to identify obsolete, slow-moving or otherwise un-saleable inventory. We will provide a valuation reserve for estimated obsolete or un-saleable inventory in an amount equal to the difference between the cost of the inventory and the estimated market value based upon assumptions about future demand and market conditions. Approximately 64% of our vials of Abbokinase that we expect hospitals to purchase and that are held in inventory either by us or by our wholesalers are not salable after October 2007 based upon their current expiration dates unless the results of an ongoing stability program allows for expiration date extensions. We believe that the expiration dates will be extended. In January 2007, we purchased approximately 1,600 vials of Abbokinase, previously sold by Abbott Laboratories, from one of our wholesale distributors. These vials were placed into inventory that we expect hospitals to purchase.
Clinical Trial Accrued Expenses
We record accruals for clinical trial costs associated with clinical research organizations, investigators and other vendors based upon the estimated amount of work completed on each clinical trial. All such costs are charged to research and development expenses based on these estimates. These estimates may or may not match the actual services performed by the organizations as determined by patient enrollment levels and
41
Table of Contents
related activities. We monitor patient enrollment levels and related activities to the extent possible through internal reviews, correspondence and discussions with our contract research organization and review of contractual terms. However, if we have incomplete or inaccurate information, we may underestimate or overestimate activity levels associated with various clinical trials at a given point in time. In this event, we could record significant research and development expenses in future periods when the actual level of activities becomes known. To date, we have not experienced material changes in these estimates.
Deferred Tax Asset Valuation Allowance
Our estimate of the valuation allowance for deferred tax assets requires us to make significant estimates and judgments about our future operating results. Our ability to realize the deferred tax assets depends on our future taxable income as well as limitations on utilization. A deferred tax asset must be reduced by a valuation allowance if it is more likely than not that some portion or all of the deferred tax asset will not be realized prior to its expiration. The projections of our operating results on which the establishment of a valuation allowance is based involve significant estimates regarding future demand for our products, competitive conditions, product development efforts, approvals of regulatory agencies and product cost. We have recorded a full valuation allowance on our net deferred tax assets as of December 31, 2005 and 2006 due to uncertainties related to our ability to utilize our deferred tax assets in the foreseeable future. These deferred tax assets primarily consist of net operating loss carry forwards and research and development tax credits.
We adopted the Financial Accounting Standards Board’s interpretation No. 48, “Accounting for Uncertainty in Income Taxes, an interpretation of FASB Statement No. 109” (FIN 48), effective January 1, 2007. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in financial statements and requires the impact of a tax position to be recognized in the financial statements if that position is more likely than not to be sustained by the taxing authority. The adoption of FIN 48 had no effect on our consolidated financial position or results of operations.
Revenue Recognition
We provide research services under certain contract and grant agreements, including federal grants from the National Institutes of Health. We recognize revenue for these research services as the services are performed. Revenue from grants is recognized over the contractual period of the related award.
Revenue from product sales is recognized pursuant to Staff Bulletin No. 104 (SAB 104),Revenue Recognition in Financial Statements. Accordingly, revenue is recognized when all four of the following criteria are met: (i) persuasive evidence that an arrangement exists; (ii) delivery of the products has occurred; (iii) the selling price is both fixed and determinable; and (iv) collectibility is reasonably assured. We apply SFAS No. 48,Revenue Recognition When the Right of Return Exists,which among other criteria requires that future returns can be reasonably estimated in order to recognize revenue. The amount of future returns is uncertain due to the lack of returns history data. Due to the uncertainty of returns, we are accounting for these product shipments to wholesale distributors using a deferred revenue recognition model. Under the deferred revenue model, we do not recognize revenue upon product shipment to wholesale distributors; therefore, recognition of revenue is deferred until the product is sold by the wholesale distributor to a hospital or other healthcare provider expected to be the end user.
Our customers consist primarily of large pharmaceutical wholesalers who sell directly to hospitals and other healthcare providers. Provisions for product returns and exchanges, sales discounts, chargebacks, managed care and Medicaid rebates and other adjustments are established as a reduction of product sales revenues at the time such revenues are recognized. These deductions from gross revenue are established by us as our best estimate at the time of sale adjusted to reflect known changes in the factors that impact such reserves.
Stock-Based Compensation
In the first quarter of 2006, we adopted Statement of Financial Accounting Standards, or SFAS, No. 123R,Share-Based Paymentor SFAS 123R, which revises SFAS 123,Accounting for Stock-Based Compensation, and supersedes Accounting Principles Board Opinion, or APB, No. 25,Accounting for Stock Issued to
42
Table of Contents
Employees. SFAS 123R requires that share-based payment transactions with employees be recognized in the financial statements based on their value and recognized as compensation expense over the requisite service period. Prior to SFAS 123R, we disclosed the pro forma effects of SFAS 123 under the minimum value method. We adopted SFAS 123R effective January 1, 2006, prospectively for new equity awards issued subsequent to December 31, 2005. The adoption of SFAS 123R through December 31, 2006 has resulted in the recognition of additional stock-based compensation expense and a reduction in net income of approximately $955,000 for the 12 months ended December 31, 2006 and $163,000 for the quarter ended March 31, 2007. The impact of adopting SFAS 123R was a decrease in basic and diluted earnings per share of $0.37 and $0.06 for the 12 months ended December 31, 2006 and March 31, 2007, respectively.
Under SFAS 123R we calculated the fair value of stock option grants using the Black-Scholes option-pricing model. The weighted average assumptions used in the Black-Scholes model were 7 years for the expected term, 75% for the expected volatility, weighted average risk free rate of 4.90% and 0% dividend yield for the twelve month period ended December 31, 2006. Future expense amounts for any particular quarterly or annual period could be affected by changes in our assumptions or changes in market conditions.
The weighted average expected option term for 2006 reflects the application of the simplified method set out in SEC Staff Accounting Bulletin No., or SAB, 107 which was issued in March 2005. The simplified method defines the life as the average of the contractual term of the options and the weighted average vesting period for all option tranches.
Estimated volatility for fiscal 2006 also reflects the application of SAB 107 interpretive guidance and, accordingly, incorporates historical volatility of similar public entities.
Prior to January 1, 2006, we accounted for employee stock-based compensation in accordance with provisions of APB 25 and FASB Interpretation No. 44,Accounting for Certain Transactions Involving Stock Compensation — an Interpretation of APB No. 25, and comply with the disclosure provisions of SFAS 123 and related SFAS 148,Accounting for Stock-Based Compensation — Transaction and Disclosure. Under APB 25, compensation expense was based on the difference, if any, on the date of the grant, between the fair value of our stock and the exercise price of the option. We amortize deferred stock-based compensation using the straight-line method over the vesting period.
The accounting for and disclosure of employee equity instruments requires judgment by our management on a number of assumptions, including the fair value of the underlying instrument, estimated lives of the outstanding instruments, and the instrument’s volatility. Changes in key assumptions will impact the valuation of such instruments. Because there has been no public market for our stock, our board of directors has determined the fair value of our common stock based on several factors, including, but not limited to, our operating and financial performance and internal valuation analyses considering key terms and rights of the related instruments.
Our board of directors estimated the fair value of common stock for options granted during the two-year period prior to the filing of this registration statement, with input from our management, using the market approach and sales to third parties of our common and preferred stock.
Results of Operations
Quarter Ended March 31, 2006 Compared to 2007
Product Sales, Grant and Other Revenue. Our revenue-producing activities during the first quarter of 2006 and 2007 consisted of providing services under research grants and contracts, and sales of Abbokinase which commenced in October 2006. Our total revenues increased from approximately $0.2 million in the first quarter of 2006 to $1.2 million in the first quarter of 2007, primarily as a result of our commencement of sales of Abbokinase product which accounted for $1.1 million of our revenue in the first quarter of 2007.
Cost of Product Sales. Cost of product sales was approximately $0.5 million in the first quarter of 2007. There was no cost of product sales for the first quarter of 2006 as we acquired the commercial product in
43
Table of Contents
April 2006 and commenced sales in October 2006. The cost of product sales includes the price paid to acquire the asset as well as labeling costs that are directly incurred in bringing the product to market.
Research and Development Expenses. Research and development expenses decreased from approximately $1.7 million to approximately $1.5 million in the first quarter of 2006 and 2007, respectively. This decrease was principally a result of decreased outside contract work performed on grants and pre-clinical studies as well as a decrease in staff and clinical trial expenses.
General and Administrative Expenses. General and administrative expenses decreased from approximately $1.6 million to approximately $1.1 million in the first quarter of 2006 and 2007, respectively. This decrease was principally a result of decreased third party services, mainly consulting and legal.
Interest and Other Income. Interest and other income decreased from approximately $100,000 to approximately $41,000 in the first quarter 2006 and 2007, respectively. This decrease was a result of a lower cash balance throughout the period.
Interest Expense. Interest expense was $0.2 million in both the first quarter of 2006 and 2007 and was due to the interest accrued on a note payable issued in September 2005 and a second note payable issued in April 2006. The note payable issued in 2005, plus interest, was extinguished early in December 2006.
Twelve Months Ended December 31, 2005 Compared to 2006
Product Sales, Grant and Other Revenue. Our revenue-producing activities during 2005 and 2006 consisted of providing services under research grants and contracts, and sales of Abbokinase which commenced in October 2006. Our total revenues increased from approximately $0.6 million in 2005 to $1.3 million in 2006, primarily as a result of our commencement of sales of Abbokinase product which accounted for $0.5 million of our revenue in 2006. Our grant and other revenue increased from approximately $0.6 million in 2005 to approximately $0.8 million in 2006, primarily due to the receipt of an additional grant.
Cost of Product Sales. Cost of product sales was approximately $0.2 million in 2006. There was no cost of product sales for the year ending December 31, 2005 as we did not acquire our commercialized product until April 2006 and did not commence product sales until October 2006. The cost of product sales includes the price paid to acquire the asset as well as labeling costs that are directly incurred in bringing the product to market.
Research and Development Expenses. Research and development expenses increased from approximately $3.6 million in 2005 to approximately $8.4 million in 2006. This increase was principally a result of the Company’s continuing transition from a research organization to a clinical development organization, which required the expansion of both clinical and regulatory departments. The main components of increased cost were: approximately $1.7 million in increased compensation associated with increased headcount; approximately $0.9 million in increased expenses for the initiation of a clinical trial in stroke which began in August 2006 as well as other clinical trial activities; approximately $0.3 million in increased preclinical study costs related to our SonoLysis product candidates; $0.1 million in expense for storing our commercial inventory of Abbokinase and related assets and approximately $1.4 million in increased third party service costs and other expenses. Of this total, approximately $0.5 million were costs related to the recombinant thrombolytic drug assets that we decided to relinquish to Abbott Laboratories in December 2006.
General and Administrative Expenses. General and administrative expenses increased from approximately $4.1 million in 2005 to approximately $7.3 million in 2006. This increase was principally a result of our expansion of financing and selling activities, which required additional headcount and third party services. The main components of increased cost were approximately $2.0 million in increased third party service costs, principally legal and accounting expenses related to financing matters, asset acquisitions and matters associated with becoming a public company; approximately $0.7 million in additional compensation expense to support increased headcount, and stock-based compensation expense including the expense under SFAS 123R; and approximately $0.2 million in third party service costs associated with marketing and sales of Abbokinase.
44
Table of Contents
Interest and Other Income. Interest and other income increased from approximately $0.1 million in 2005 to approximately $0.4 million in 2006, as a result of a higher cash balance throughout the year and higher interest rates.
Interest Expense. Interest expense increased from approximately $0.6 million in 2005 to approximately $1.5 million in 2006, due to the interest accrued on a note payable issued in April 2006 and the payment of a full year of interest in 2006 on a note payable issued in September 2005.
Gain on Extinguishment of Debt. In March 2005, we repurchased a note from a former development partner at a discount. The outstanding principal and accrued interest, totaling approximately $4.3 million, was settled in cash for approximately $0.5 million resulting in a non-recurring gain of approximately $3.8 million. In December 2006, we extinguished a non-recourse debt that had been issued as partial consideration for the acquisition of recombinant thrombolytic drug technologies, resulting in a non-recurring gain of $16.1 million.
Year Ended December 31, 2004 Compared to 2005
Product Sales, Grant and Other Revenue. Our revenue-producing activities during 2004 and 2005 consisted solely of providing services under research grants and contracts and did not include any product sales. Revenue was approximately $0.6 million in both 2004 and 2005.
Research and Development Expenses. Research and development expenses increased from approximately $2.5 million in 2004 to approximately $3.6 million in 2005. This increase was principally a result of beginning the transition from a research-focused organization to a clinical development organization, which required the creation of both clinical and regulatory departments. The main components of increased cost were clinical trial costs, consulting, compensation and cost of hiring and increased overhead. Of the total increase, approximately $0.6 million was for the initiation of our pilot study in stroke which began in March 2005; approximately $0.2 million resulted from increased third party service costs; approximately $0.2 million resulted from increased compensation expense to support increased headcount; and approximately $0.5 million resulted from increased overhead, laboratory chemicals and supplies, travel and other expenses. An offset of approximately $0.4 million was due to timing of preclinical and manufacturing expenses.
General and Administrative Expenses. General and administrative expenses increased from approximately $3.2 million in 2004 to approximately $4.1 million in 2005. This increase resulted primarily from the expenditure of approximately $0.6 million in increased compensation expense to support increased headcount; approximately $0.2 million in increased third party service costs, principally legal and accounting expenses related to financing matters and asset acquisitions; and approximately $0.1 million in increased business development and other expenses.
Interest and Other Income. Interest and other income increased from approximately $29,000 in 2004 to approximately $122,000 in 2005, as a result of higher cash balances and higher interest rates.
Interest Expense. Interest expense increased from approximately $0.5 million in 2004 to approximately $0.6 million in 2005, primarily due to the interest on the promissory note issued in September 2005 and the early extinguishment of the note payable to a former development partner in March 2005.
Gain on Extinguishment of Debt. In April 2004, a development partner experienced financial difficulty and began auctioning portions of its investment portfolio. In March 2005, we repurchased our debt from the development partner at a discount. The outstanding principal and accrued interest, totaling approximately $4.3 million, was settled in cash for approximately $0.5 million, resulting in a non-recurring gain of approximately $3.8 million. No other consideration was paid in connection with the repurchase of the debt.
Liquidity and Capital Resources
Sources of Liquidity
We have incurred losses since our inception. At March 31, 2007 we had an accumulated deficit of approximately $65.5 million. We have historically financed our operations principally through the private placement of shares of our common and preferred stock and convertible notes, government grants, and, more
45
Table of Contents
recently, product sales, which commenced in October 2006. During the years ended December 31, 2004, 2005 and 2006, we received net proceeds of approximately $5.0 million, $17.9 million, $13.0 million, respectively, from the issuance of shares of our common and preferred stock and convertible notes. These amounts do not include the $15.0 million secured non-recourse note and $4.0 million of Series E preferred stock that we issued as partial consideration for an acquisition of recombinant thrombolytic drug technologies in September 2005, or the $15.0 million secured non-recourse note that we issued to acquire Abbokinase and related assets in April 2006.
At March 31, 2007, we had approximately $2.7 million in cash and cash equivalents. The exact timing of future sales of Abbokinase will depend on a number of external factors, such as our ability to obtain an extension of the expiration dates for the bulk of our Abbokinase inventory beyond October 2007, our ability to establish additional sales relationships with current customers for that product, inventory levels of the wholesalers that are currently stocking the product, and other competitive and regulatory factors. Based on annualized Intercontinental Marketing Services, or IMS, sales data, we believe the vials of Abbokinase that we acquired and expect hospitals to purchase represent approximately a four-year supply as of March 31, 2007. Based on current stability data as of March 31, 2007, approximately 64% of our vials of Abbokinase that we expect hospitals to purchase and that is held in inventory either by us or by our wholesalers will no longer be saleable after October 2007 with the remainder expiring at various times up to August 2009. We are not permitted to sell these vials after expiration. We are continuing the current stability testing program started by Abbott Laboratories, which has been ongoing for over four years. The testing to date has shown that the product changes very little from year to year. However, if the results of the stability testing program are outside the limits established by the FDA, we estimate that approximately 64% of our vials of Abbokinase that we expect hospitals to purchase and that are held in inventory either by us or by our wholesalers, or approximately $10.7 million in inventory value out of the total of $16.8 million carried at March 31, 2007, is at risk of being written off. Currently, we believe it is probable that the stability data will support extension of the inventory expiration dates, that we will be able to sell this inventory and that we will recover the cost of this inventory. If the expiration dates of this inventory are extended we will need to re-brand the remaining inventory because our license to use the Abbokinase trademark does not extend beyond the current inventory expiration dates.
We allocated the $20.0 million purchase price for Abbokinase as follows:
Asset | Estimated Value | |||
| Inventory | $ | 16.7 million | ||
| Abbokinase trade name | $ | 0.5 million | ||
| Other identifiable intangibles | $ | 2.8 million | ||
The anticipated carrying value of the inventory does not include a reserve for excess inventory. We anticipate that hospitals will not purchase approximately 28% of the total number of vials of Abbokinase inventory that we acquired from Abbott Laboratories, and, consequently, these vials are carried with zero book value assigned, in effect creating a valuation allowance. We anticipate that these vials will not be sold for a variety of reasons, including expiration of vials that are labeled with a fixed expiration date prior to sale, potential future competition from new products entering the market, and use of some of the vials for our own research purposes. Of the remaining vials of Abbokinase that we expect hospitals to purchase and that are held in inventory either by us or by our wholesalers, we estimate that at March 31, 2007, approximately 36% of these vials, or approximately $6.1 million in inventory value, is available for sale without risk of being written off and approximately 64% of these vials, or approximately $10.7 million in inventory value, is available for sale but may be at risk of being written off. We estimate that the remaining vials with zero inventory value will not be sold. The estimated useful life of the Abbokinase trade name is one year, and the estimated useful life of the other identifiable intangibles is four years from May 2006. While we intend to investigate the requirements for us to manufacture Abbokinase, we currently have no plans to manufacture Abbokinase in the near term. Not manufacturing Abbokinase reduces the period of benefit for the intangible assets to the Company to four years from May 2006, which is directly related to the years of inventory supply.
46
Table of Contents
In April 2007, we sold a total of approximately $9.0 million of Abbokinase, net of discounts and fees, to two of our primary wholesalers, of which approximately $4.1 million of the proceeds has been placed into an escrow account as security for repayment of our $15.0 million promissory note due in December 2007. These vials have expiration dates ranging from December 2008 to August 2009. We expect that these orders will reduce Abbokinase demand in the near term. In addition, we are required to place 50% of the proceeds from all future sales of Abbokinase into the escrow account as required by our escrow agreement with Abbott Laboratories until the $15.0 million note is repaid. If the escrowed amount were to be applied to the outstanding balance of principal and interest on that note, the remaining amount due under the note would be approximately $11.9 million as of May 31, 2007.
Cash Flows
Net Cash Used in Operating Activities. Net cash used in operating activities was approximately $4.1 million, $11.2 million and $16.0 million for the years ended December 31, 2004, 2005 and 2006, respectively, and $2.1 million and $1.2 million for the quarters ended March 31, 2006 and 2007, respectively. The net cash used in each of these periods primarily reflects the net loss for those periods, offset in part by depreciation, amortization of warrant expense and debt discount, and non-cash gain on extinguishment of debt, stock-based compensation and changes in working capital.
Net Cash Used in Investing Activities. Net cash used in investing activities was approximately $0.1 million, $0.6 million and $1.3 million for the years ended December 31, 2004, 2005 and 2006, respectively, and $0.2 million for each of the quarters ended March 31, 2006 and 2007. Net cash used in investing activities primarily reflects purchases of property and equipment, including manufacturing, information technology, laboratory and office equipment.
Net Cash Provided by Financing Activities. Net cash provided by financing activities was approximately $5.0 million, $18.7 million and $13.0 million for the years ended December 31, 2004, 2005 and 2006, respectively, and $0.2 million and $0.1 million for the quarters ended March 31, 2006 and 2007, respectively. Net cash provided by financing activities was primarily attributable to the issuance of common stock totaling approximately $4.4 million net of issuance costs and the issuance of convertible notes totaling approximately $0.6 million in 2004; the issuance of common stock totaling approximately $17.9 million net of issuance costs and the issuance and repayment of secured promissory notes totaling approximately $4.0 million in 2005; and the issuance of Series F preferred stock totaling approximately $13.0 million net of issuance costs in 2006.
Our cash flows for the remainder of 2007 and beyond will depend on a variety of factors, including the anticipated revenue and funding requirements discussed above, as well as the timing of completion of the offering contemplated by this prospectus and our use of offering proceeds as described under “Use of Proceeds” elsewhere in this prospectus. Despite our commencement of sales of our Abbokinase product in October 2006, we expect our net cash outflows to continue increasing as we expand our research and development, manufacturing, regulatory and sales and marketing activities. In addition, for the remainder of 2007, our agreement with Abbott Laboratories calls for 50% of the cash receipts from Abbokinase sales after we have received a total of $5.0 million in cash receipts from the sales of Abbokinase, which we surpassed in April 2007, to be held in an escrow account. The balance of the escrow account is to be used to pay down the $15.0 million note that is due to Abbott Laboratories on December 31, 2007.
Funding Requirements
Based on our existing liquid assets, including the proceeds of our sales of Abbokinase, we believe we have sufficient capital to fund anticipated levels of operations, and pay our debt obligations as they come due, until August 2007, assuming we make no additional sales of Abbokinase to wholesalers or direct to customers. We have received an audit report from our independent registered public accounting firm containing an explanatory paragraph stating that our historical recurring losses and net capital deficiency raise substantial doubt about our ability to continue as a going concern. We believe that the completion of this offering will enable us to continue as a going concern until at least December 2008, assuming sales of Abbokinase are sufficient to repay the $15.0 million note due December 31, 2007. In April 2007 we sold approximately
47
Table of Contents
$9.0 million of Abbokinase, net of discounts and fees, to two of our primary wholesalers, of which approximately $4.1 million of the proceeds has been placed into an escrow account as security for repayment of our $15.0 million non-recourse promissory note due in December 2007. These vials have expiration dates ranging from December 2008 to August 2009. If the escrowed amount were to be applied to the outstanding balance of principal and interest on that note, the remaining amount due under the note would be approximately $11.9 million as of May 31, 2007. If we are unable to complete this offering, we will need to obtain alternative financing and modify our operational plan to continue as a going concern.
Our funding requirements will, however, depend on numerous factors, including:
| • | the timing, scope and results of our preclinical studies and clinical trials; | |
| • | the timing and amount of revenue from sales of Abbokinase; | |
| • | the timing and amount of revenue from grants and other sources; | |
| • | our ability to refinance our $15.0 million secured non-recourse note due to Abbott Laboratories on December 31, 2007, if sales of Abbokinase are insufficient to repay the note; | |
| • | the timing of initiation of manufacturing for our product candidates; | |
| • | the timing of, and the costs involved in, obtaining regulatory approvals; | |
| • | our ability to establish and maintain collaborative relationships; | |
| • | personnel, facilities and equipment requirements; and | |
| • | the costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other patent-related costs, including litigation costs, if any, and the result of any such litigation. |
Until we can consistently generate significant cash from our sales of Abbokinase and other operations, we expect to continue to fund our operations primarily from the proceeds of offerings of our equity securities, including this offering, from revenue or payments received under collaborations, grants, and possibly from debt financing. However, we may not be successful in obtaining additional collaboration agreements or grants, or in receiving milestone or royalty payments under any such agreements. If we do not generate sufficient revenue from collaborations and grants, we may require additional funding sooner than we currently anticipate. We cannot be sure that our existing cash and cash equivalents will be adequate, or that additional financing will be available when needed, or that, if available, financing will be obtained on terms favorable to us or our stockholders. Having insufficient funds may require us to delay, scale back or eliminate some or all of our research or development programs or to relinquish greater or all rights to product candidates at an earlier stage of development or on less favorable terms than we would otherwise choose. Failure to obtain adequate financing may also adversely affect our ability to operate as a going concern. If we raise additional funds by issuing equity securities, substantial dilution to existing stockholders will likely result. If we raise additional funds by incurring debt obligations, the terms of the debt will likely involve significant cash payment obligations as well as covenants and specific financial ratios that may restrict our ability to operate our business.
Contractual Obligations
The following table summarizes our outstanding contractual obligations as of March 31, 2007:
| Payments Due By Period | ||||||||||||||||||||
| Less than | More than | |||||||||||||||||||
Total | Total | 1 Year | 1-3 Years | 3-5 Years | 5 Years | |||||||||||||||
| Operating leases | $ | 118,690 | $ | 64,740 | $ | 53,950 | — | — | ||||||||||||
| Secured non-recourse note | $ | 15,000,000 | $ | 15,000,000 | — | — | — | |||||||||||||
| Total | $ | 15,118,690 | $ | 15,064,740 | $ | 53,950 | — | — | ||||||||||||
48
Table of Contents
We enter into agreements with clinical sites and contract research organizations, or CROs, that conduct our clinical trials. We make payments to these sites and CROs based upon the number of patients enrolled. For the years ended December 31, 2004, 2005 and 2006, we incurred clinical trial expenses of approximately $0.3 million, $0.9 million and $1.6 million, respectively. Due to the variability associated with these agreements, we are unable to estimate with certainty the future patient enrollment costs we will incur and therefore have excluded these costs from the above table. We do, however, anticipate that these costs will increase significantly in future periods as a result of our initiation of multiple clinical trials for ischemic stroke.
We also have contractual payment obligations that are contingent on future events.
| • | If we or our sublicensees sell products or processes that utilize the intellectual property we license from UNEMED Corporation, we will be obligated pay a royalty to UNEMED of 2% of such net sales. | |
| • | If we or our sublicensees sell products or processes that utilize the intellectual property we license from the University of Arkansas, we will be obligated to pay, in addition to a one-time fee of $25,000, royalties to the University of Arkansas of (i) 4% of net sales up to $1.0 million; (ii) 3% of net sales between $1.0 million and $10.0 million; and (iii) 2% of net sales greater than $10.0 million, subject to minimal royalty thresholds and a maximum aggregate royalty of $20.0 million. | |
| • | If we or our sublicensees sell products or processes that utilize the intellectual property that we license from Dr. Schlief, we will be obligated to pay a royalty to Dr. Schlief of 2% of such net sales by us and 3% of any net sales by sublicensees. |
Quantitative and Qualitative Disclosure About Market Risk
Our exposure to market risk is confined to our cash and cash equivalents. We invest in high-quality financial instruments, primarily money market funds, which we believe are subject to limited credit risk. We currently do not hedge interest rate exposure. The effective duration of our portfolio is less than three months and no security has an effective duration in excess of three months. Due to the short-term nature of our investments, we do not believe that we have any material exposure to interest rate risk arising from our investments.
Most of our transactions are conducted in U.S. dollars, although we do have some development and clinical trial agreements with vendors located outside the U.S. Transactions under certain of these agreements are conducted in U.S. dollars while others occur in the local currency. If the exchange rate were to change by ten percent, we do not believe that it would have a material impact on our results of operations or cash flows.
Off-Balance Sheet Transactions
At December 31, 2004, 2005 and 2006, and for the quarters ended March 31, 2006 and 2007, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
Recently Issued Accounting Pronouncements
In September 2006, the FASB issued SAFS No. 157, “Fair Value Measurements.” SFAS 157 defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements. SFAS 157 applies under other accounting pronouncements that require or permit fair value measurements, the FASB having previously concluded in those accounting pronouncements that fair value is the relevant measurement attribute. Accordingly, SFAS 157 does not require any new fair value measurements, but may change current practice for some entities. SFAS 157 is effective for fiscal years beginning after December 15, 2006. The adoption of SFAS No. 157 is not expected to have a material effect on the Company’s financial position or results of operations.
In September 2006, the Securities and Exchange Commission issued Staff Accounting Bulletin, or SAB, No. 108, “Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current
49
Table of Contents
Year Financial Statements,” which provides interpretive guidance on the consideration of the effects of prior year misstatements in quantifying current year misstatements for the purpose of a materiality assessment. SAB No. 108 requires registrants to quantify misstatements using both the balance sheet and income statement approaches and to evaluate whether either approach results in quantifying an error that is material based on relevant quantitative and qualitative factors. The guidance is effective for the first fiscal period ending after November 15, 2006. The Company is currently evaluating the impact of adopting SAB No. 108 on its financial position, results of operations and cash flows.
50
Table of Contents
Our Business
We are a biopharmaceutical company developing and commercializing therapies for vascular disorders. Our research and development efforts are focused on therapies for stroke and other vascular disorders, using our proprietary microbubble technology to treat vascular occlusions, or blood vessel blockages, as well as the resulting ischemia, which is tissue damage caused by a reduced supply of oxygen. Our commercialization efforts are currently focused on our product approved by the U.S. Food and Drug Administration, or FDA, for the treatment of acute massive pulmonary embolism, or blood clots in the lungs.
Over eight million people in the U.S. are afflicted each year with complications related to blood clots. Approximately 700,000 adults in the U.S., or one every 45 seconds, are afflicted with, and 150,000 die as a result of, some form of stroke each year. Stroke is currently the third leading cause of death, and the leading cause of disability, in the United States. Approximately three million Americans are currently disabled from stroke. The American Stroke Association estimates that approximately $62.7 billion will be spent in the U.S. in 2007 for stroke-related medical costs and disability.
The vast majority of strokes, approximately 87% according to the American Stroke Association, are ischemic strokes, meaning that they are caused by blood clots, while the remainder are the more deadly hemorrhagic strokes caused by bleeding in the brain. Currently available treatment options for ischemic stroke are subject to significant therapeutic limitations. For example, the most widely used treatment for ischemic stroke is a clot-dissolving, or thrombolytic, drug that can be administered only during a narrow time window and poses a risk of bleeding, resulting in 6% or less of ischemic stroke patients receiving such treatment. To facilitate increased administration of stroke therapies, in 2005 the Centers for Medicare and Medicaid Services, or CMS, responded to requests by the American Stroke Association and related groups for higher reimbursement amounts for ischemic stroke patients treated with a thrombolytic drug by approximately doubling the amount of reimbursement provided for such treatment to $11,578 per patient.
In addition to the brain and the lungs, blood clots can block blood flow and cause damage to other tissues in the body such as the heart, in the case of coronary arterial disease, and the legs and other extremities, in the case of peripheral vascular disease. We believe our development and research stage products may address significant unmet medical needs not only for stroke but also for clot-induced damage in tissues other than the brain.
Our Commercial and Development Stage Products
The following table summarizes the status of our commercial product and development stage product candidates:
Product or Candidate | Product Elements | Indication | Development Status | |||
SonoLysistm+tPAtherapy | • MRX-801 microbubbles | Ischemic stroke | Phase I/II clinical trial in progress | |||
| • Ultrasound | ||||||
| • tPA | ||||||
| SonoLysis therapy | • MRX-801 microbubbles | Ischemic stroke | Preclinical | |||
| • Ultrasound | ||||||
Abbokinase® | • Urokinase | Acute massive pulmonary embolism | Approved for marketing | |||
SonoLysis Program. Our SonoLysis program is focused on the development of two product candidates that involve the administration of our proprietary MRX-801 microbubbles and ultrasound, with or without a thrombolytic drug to break up blood clots and restore blood flow to oxygen deprived tissues. SonoLysis+tPA therapy involves the administration of MRX-801 microbubbles, ultrasound and the thrombolytic drug alteplase, or tPA. Alteplase is the formulation of tPA that is approved for treatment of acute ischemic stroke. Alteplase is manufactured by Genentech Inc. and is widely available under the trade name Activase. SonoLysis therapy includes the administration of MRX-801 microbubbles and ultrasound without a thrombolytic drug. Our MRX-801 microbubbles are a proprietary formulation of a lipid shell encapsulating an inert biocompatible gas. We believe thesub-micron size of our MRX-801 microbubbles allows them to penetrate a blood clot, so that when
51
Table of Contents
ultrasound is applied their expansion and contraction, or cavitation, can break the clot into very small particles. We believe that these product candidates have the potential to treat a broad variety of vascular disorders associated with blood clots.
Our initial therapeutic focus for our SonoLysis program is ischemic stroke. Although Abbokinase is a thrombolytic drug that we own, it is approved only for the treatment of acute massive pulmonary embolism. We chose to use tPA as the thrombolytic drug in our SonoLysis+tPAtherapy because it is the only FDA approved drug for the treatment of ischemic stroke. The FDA has restricted tPA’s use to patients who are able to begin treatment within three hours of onset of ischemic stroke symptoms and who do not have certain risk factors for bleeding, such as recent surgery or taking medications that prevent clotting. According to Datamonitor, approximately 23% of ischemic stroke patients arrive at a hospital within three hours of onset of symptoms. However, due to the three hour window for treatment and other limitations, only 1.6% to 2.7% of patients with ischemic stroke in community hospitals, and only 4.1% to 6.3% in academic hospitals or specialized stroke centers are treated with a thrombolytic therapy. For these patients who are eligible for treatment with tPA, we believe SonoLysis+tPAtherapy may have advantages over tPA alone, including more rapid and complete restoration of blood flow. In addition, we believe our SonoLysis therapy may have an improved bleeding and safety profile over tPA and therefore may represent a new treatment option for ischemic stroke patients ineligible for treatment with tPA by extending the treatment window beyond three hours from onset of symptoms, as well as broadening treatment availability to patients for whom tPA is contraindicated due to risk of bleeding. Our two SonoLysis product candidates being developed as potential treatments for ischemic stroke are further described below:
| • | SonoLysis+tPA therapyinvolves the administration of our proprietary MRX-801 microbubbles and ultrasound in conjunction with tPA. To administer our SonoLysis+tPAtherapy, MRX-801 microbubbles and tPA are injected intravenously into the bloodstream. The MRX-801 microbubbles are distributed naturally throughout the body including to the site of the blood clot. Ultrasound is then administered to the site of the blood clot, and the energy from the ultrasound causes the MRX-801 microbubbles to expand and contract vigorously, or cavitate. We believe this cavitation both mechanically breaks up the blood clot and helps the administered tPA permeate the clot to facilitate clot dissolving activity. The gas released by the MRX-801 microbubbles is then cleared from the body simply by exhaling, and the lipid shell is metabolized like other fats in the body. We believe that this therapeutic approach incorporates two complementary mechanisms of action, mechanical and enzymatic, that together can reduce the time required to dissolve a blood clot and help ensure more rapid and complete restoration of blood flow to at risk brain tissues in patients with ischemic stroke. We are conducting a Phase I/II dose-escalation clinical trial evaluating SonoLysis+tPAtherapy in patients with ischemic stroke. We initiated this trial in January 2007, and intend to enroll a total of 72 patients in various medical centers in the United States and Europe. Patients will be enrolled into one of four successive cohorts, or groups, that will receive escalating doses of our MRX-801 microbubbles and the standard dose of tPA. A Data and Safety Monitoring Board will review the data from each cohort before granting approval to enroll patients in the succeeding cohort utilizing the next higher dose of MRX-801 microbubbles. We anticipate enrollment for this trial will be completed in the first half of 2008 and intend to initiate a Phase II study following completion of the ongoing Phase I/II study. Our SonoLysis+tPA therapy will likely have the same usage restrictions as tPA. These restrictions include a requirement that treatment be initiated within three hours of onset of symptoms, and a requirement that the patient not have uncontrolled hypertension, has not recently had surgery and is not currently using an anticoagulant drug such as heparin. We estimate that if approved by the FDA, over 90,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis+tPAtherapy annually. |
| • | SonoLysis therapyinvolves administration of our MRX-801 microbubbles with ultrasound, but without the administration of a thrombolytic drug. To administer our SonoLysis therapy, MRX-801 microbubbles are injected intravenously into the bloodstream and disperse naturally throughout the body including to the site of the blood clot. Ultrasound is then administered to the site of the blood clot, and the energy from the ultrasound causes the MRX-801 microbubbles to cavitate. We believe this |
52
Table of Contents
| cavitation mechanically breaks up the blood clot and also helps to enhance the body’s natural clot dissolving processes. The gas released by the MRX-801 microbubbles is then cleared from the body simply by exhaling, and the lipid shell is processed like other fats in the body. Because SonoLysis therapy does not involve use of a thrombolytic drug and its associated risk of bleeding, we believe SonoLysis therapy may offer advantages over existing treatments for ischemic stroke, including extending the treatment window beyond three hours from onset of symptoms and broadening treatment availability to patients for whom thrombolytic drugs are contraindicated due to risk of bleeding. We have not yet conducted any clinical trials using our proprietary MRX-801 microbubbles with ultrasound to treat blood clot indications without a thrombolytic drug. We are conducting and intend to conduct additional preclinical studies of SonoLysis therapy through the first half of 2008. We expect to initiate a Phase II study to treat patients with ischemic stroke following completion of our SonoLysis+tPA therapy Phase I/II clinical trial. Because of the preclinical data package as well as our ongoing Phase I/II clinical trial evaluating SonoLysis+tPA therapy in patients with ischemic stroke, we believe no Phase I study will be required prior to initiating the Phase II study for SonoLysis therapy. Patients ineligible for SonoLysis therapy include those patients who are treated or treatable with SonoLysis+tPA therapy, patients for whom treatment is not initiated within 24 hours of onset of symptoms and patients that have no at-risk but salvageable brain tissue at the time of diagnosis. We estimate that if approved by the FDA, over 200,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis therapy annually. |
Abbokinase. Our commercially available urokinase product, which we market as Abbokinase, is a thrombolytic drug. Urokinase is a natural human protein primarily produced in the kidneys that stimulates the body’s natural clot-dissolving processes. Abbokinase is FDA approved and marketed for the treatment of acute massive pulmonary embolism. Abbokinase has been administered to over four million patients, and we estimate that approximately 400 acute care hospitals in the U.S. include Abbokinase on their pharmacy formulary today. We acquired approximately 153,000 vials of Abbokinase from Abbott Laboratories in April 2006, and began selling Abbokinase in October 2006. At the time of our acquisition of Abbokinase, we estimated that hospitals would purchase, and we would thereby recognize revenue for, approximately 111,000 vials, or approximately 72% of the total vials we acquired, which we believe represented approximately a four-year supply of inventory. We also estimated that hospitals would not purchase approximately 42,000 vials, or approximately 28% of the vials we acquired, due to expiration of labeled vials, potential future competition from new products entering the market, and our own use of some of the vials for research purposes. As of March 31, 2007, we have recognized revenue on approximately 5,800 vials of Abbokinase sold by our wholesalers to hospitals. Data we have obtained from our wholesalers, as well as our current sales data, lead us to believe that we will sell approximately $12 to $14 million of Abbokinase to hospitals per year between 2007 and 2011. Our sales and marketing staff continue to detail the product to physicians and support the product in the marketplace. We believe Abbokinase sales will provide us with near-term revenue and an opportunity to form relationships with vascular physicians and acute care institutions that regularly administer blood clot therapies. Approximately 64% of our Abbokinase vials that we expect hospitals to purchase and that are held in inventory either by us or by our wholesalers as of March 31, 2007 will no longer be saleable after October 2007 based on their current expiration dates. In order to facilitate obtaining an extension of current expiration dates, we are continuing the stability testing program started by Abbott Laboratories, which has been ongoing for over four years. Based on the testing to date, which has shown that the product changes very little from year to year, we believe it is probable that the stability data will support extension of the inventory expiration dates. The next testing point of our ongoing stability program at which we may obtain data sufficient to extend the expiration dates of our unlabeled inventory will be completed in the fall of 2007. If the parameters tested are within the specifications previously approved by the FDA, we may then label vials at that time with extended expiration dating to between June and August 2009. We must obtain FDA approval for each lot release of inventory. Inventory is labeled with an expiration date upon approval of a lot release by the FDA. Once labeled, we cannot extend the expiration date of the vials labeled. If we are successful in extending the expiration dates of our unlabeled inventory, we intend to continue the stability program after the fall of 2007 to potentially enable further expiration extensions for future product labeling. In connection with our Abbokinase acquisition, we issued a $15.0 million non-recourse promissory note that matures in December
53
Table of Contents
2007 and bears interest at the rate of 6% per annum. If we are unable to satisfy this debt obligation when due, Abbott Laboratories will have the right to reclaim our remaining inventory of Abbokinase, along with a portion of the cash we have received from our sales of Abbokinase. In April 2007 we sold approximately $9.0 million of Abbokinase, net of discounts and fees, to two of our primary wholesalers, of which approximately $4.1 million has been placed into an escrow account as security for repayment of our $15.0 million non-recourse promissory note due in December 2007. These vials have expiration dates ranging from December 2008 to August 2009. If the escrowed amount were to be applied to the outstanding balance of principal and accrued interest on that note, the remaining balance due under the note would be approximately $11.9 million as of May 31, 2007. Although we acquired cell lines from Abbott Laboratories that could be used to manufacture urokinase, we do not currently intend to undertake any efforts to manufacture urokinase in the near term. We are evaluating the market opportunity for urokinase, as well as the cost, complexity, time and expertise required to manufacture urokinase.
Our Research Stage Product Candidates
The following table summarizes the status of our research stage product candidates:
| Research | ||||||
Product Candidate | Product Elements | Indication(s) | Status | |||
| SonoLysis therapy | • MRX-801 microbubbles • Ultrasound | Ischemic stroke in pre- hospital setting | Preclinical | |||
SonoLysis+tPAtherapy | • MRX-801 microbubbles • Ultrasound • tPA | Myocardial infarction Peripheral arterial occlusive disease Deep vein thrombosis | Preclinical Preclinical Preclinical | |||
NanO2tm | • MRX-804 emulsion/microbubbles | Hemorrhagic shock Neuroprotection for ischemic stroke | Preclinical Research | |||
| Targeted SonoLysis therapy | • MRX-802 targeted microbubbles | Myocardial infarction and other vascular clots | Research | |||
| Targeted drug delivery | • MRX-803 targeted drug delivery microbubbles | Angiogenic tumors | Research | |||
Additional SonoLysis Opportunities. We believe SonoLysis therapy may be suitable for administration for ischemic stroke in an ambulance before arriving at a hospital because it does not involve use of a thrombolytic drug and its associated risk of bleeding. To pursue an ambulance-based ischemic stroke treatment, we would be required to show either that hemorrhage can be ruled out in an ambulance setting, or that SonoLysis therapy has no detrimental effect on a hemorrhagic stroke. Additionally, we believe that the ability of our SonoLysis+tPAtherapy to reduce the time required to dissolve a blood clot could make this therapy suitable for use in treating a broad variety of vascular disorders beyond ischemic stroke. For example, we believe SonoLysis+tPAtherapy could potentially enable more rapid treatment of recently formed acute clots, such as those that cause myocardial infarction, or heart attack. We also believe SonoLysis+tPAtherapy has the potential to treat more establishedsub-acute and chronic clots, such as those in peripheral vascular indications that cannot be effectively treated with thrombolytic therapy alone. We have used microbubbles and ultrasound with a thrombolytic drug both to conduct preclinical animal studies with academic collaborators to treat myocardial infarction, as well as to conduct clinical proof of concept trials to treat patients with occluded dialysis grafts, peripheral artery occlusive disease and deep vein thrombosis. In many countries, we believe that various risk factors for formation of blood clots such as high blood pressure, obesity, diabetes, aging population, and limited mobility caused by injury, illness or simply long distance air travel are increasing, resulting in a growing unmet medical that we believe our SonoLysis product candidates could address and help reduce the need for angioplasty, stents and vascular surgery.
54
Table of Contents
Other Research Stage Opportunities. We are exploring a number of potential future product development opportunities based on our microbubble technology, including:
| • | Oxygen Delivery. We are investigating the potential use of our proprietary MRX-804 emulsion/microbubbles, which we call NanO2, to carry oxygen to parts of the body as a potential treatment for a broad variety of disorders in which reduced blood flow results in oxygen-deprived tissues, such as ischemic stroke, heart attack, and injuries that involve significant blood loss or hemorrhagic shock. NanO2 is administered intravenously as an emulsion. We believe upon entering the bloodstream it converts from a liquid to a gas, forming microbubbles with a high oxygen carrying capacity. We are working with an academic collaborator who has recently received an approximately $700,000 grant from the U.S. Department of Defense to conduct preclinical animal studies of MRX-804 microbubbles to treat hemorrhagic shock. We believe our NanO2 product candidate may have the ability to be stored at room temperature, which could make it suitable for emergency battlefield or ambulance-based treatments. | |
| • | Targeted SonoLysis Therapy. Our research team has developed MRX-802, our next generation SonoLysis microbubbles with targeting technology that causes the microbubbles to bind to blood clots. We have demonstrated in laboratory experiments that our MRX-802 targeted microbubbles improve binding to blood clots. We believe that our MRX-802 targeted microbubbles will have a greater ability tobreak-up blood clots than non-targeted microbubbles when combined with ultrasound. We have conducted preclinical animal studies with academic collaborators evaluating MRX-802 targeted microbubbles and ultrasound to treat various clot disorders, including myocardial infarction. To further the research on our next generation SonoLysis technology, we have received and are near the mid-point of our work on an approximately $1.2 million grant from the National Institutes of Health, or NIH, to study MRX-802 targeted microbubbles to treat vascular clots. | |
| • | Targeted Drug Delivery. We have also developed targeted drug delivery microbubbles, known as MRX-803, which have the potential for selective drug delivery when used in conjunction with ultrasound. MRX-803 is comprised of a gas core, an oil containing a drug payload and a lipid shell. We have received an approximately $1.0 million subcontract and have reached the mid-point of our research on an NIH grant to study the use of our proprietary MRX-803 targeted drug delivery microbubbles to treat a variety of tumors. We believe this technology has the potential for broad applications, including delivering drugs to dissolve blood clots or arterial plaque as well as to treat a variety of types of cancer. |
Our Business Strategy
Our goal is to become the leading provider of therapies for stroke and other vascular disorders by developing and marketing products to treat occlusions as well as the resulting ischemia. The key elements of our business strategy are to:
| • | Develop and commercialize our SonoLysis product candidates to expand the number of ischemic stroke patients who are eligible for treatment. We believe that our SonoLysis+tPAtherapy has the potential to enable more rapid and complete restoration of blood flow and therefore potentially improve outcomes for ischemic stroke patients eligible to receive tPA. We also believe our SonoLysis therapy may have an improved bleeding and safety profile over tPA and therefore create a new treatment option for ischemic stroke patients ineligible for tPA by extending the treatment window beyond three hours from onset of symptoms as well as broadening treatment availability to patients for whom tPA is contraindicated due to risk of bleeding. | |
| • | Sell our Abbokinase inventory and benefit from our commercial relationships. We commenced selling Abbokinase in October 2006. We are conducting continuing product stability studies, and plan to seek regulatory approval for extension of the expiration dates applicable to our inventory in order to optimize its value. We believe Abbokinase will not only provide us with a source of revenue to help fund our product development programs, but also allow us to establish relationships with numerous vascular physicians and acute care institutions. We believe that direct interactions with these caregivers |
55
Table of Contents
| will afford us improved information as to the unmet needs of the vascular therapy market, which we can use to guide our product development decisions. |
| • | Leverage our SonoLysis product candidates to accelerate initiation of treatment for ischemic stroke in an ambulance setting and address additional clot disorders in cardiology and peripheral vascular disease. Because our SonoLysis therapy does not involve use of a thrombolytic drug and its associated risk of bleeding, we believe SonoLysis therapy may enable the initiation of treatment for ischemic stroke prior to arriving at a hospital in an ambulance setting. To pursue an ambulance-based ischemic stroke treatment, we would be required to show either that hemorrhage can be ruled out in an ambulance setting, or that SonoLysis therapy has no detrimental effect on a hemorrhagic stroke. In addition we believe that our SonoLysis product candidates may be suitable for use in treating a broad variety of vascular disorders beyond ischemic stroke, including myocardial infarction, peripheral artery occlusive disease and deep vein thrombosis. We have used microbubbles and ultrasound with a thrombolytic drug both to conduct preclinical animal studies with academic collaborators to treat myocardial infarction, as well as to conduct clinical proof of concept trials to treat patients with occluded dialysis grafts, peripheral artery occlusive disease and deep vein thrombosis. We believe our SonoLysis product candidates could help reduce the need for angioplasty, stents and vascular surgery. | |
| • | Create a deep pipeline of products based on our microbubble technologies to address additional indications. We intend to continue to explore using our microbubble technology to treat a variety of other vascular disorders. We have ongoing research programs, many of which are grant funded, to use our microbubble technology to deliver oxygen, target clots for SonoLysis therapy, and deliver drugs. We also believe that our microbubble technology could be adapted, by changing the composition and size of the bubbles, to deliver genetic materials to targeted sites in the body, or to identify and treat vascular plaque. |
Industry Background
The formation of a blood clot is a natural process by which blood thickens and coagulates into a mass of blood cells, platelets and strands of fibrin. Thrombosis occurs when a blood clot, or thrombus, begins to block a blood vessel. Formation of a clot is the body’s primary mechanism for obstructing blood flow and curtailing bleeding from wounds or other injuries to blood vessels. Blood clots can be caused by a variety of factors other than injury or trauma, such as the rupture of vulnerable plaque in a vessel. Blood clots can also arise in connection with surgical and other medical procedures, such as catheter-based administration of dialysis or other treatments, which can lead to clotting around the site of an incision or within a penetrated blood vessel. An embolism occurs if all or part of a blood clot breaks away and lodges in another part of the body. When a blood clot blocks normal blood flow within the body, it can have a variety of undesirable effects, such as causing pain and swelling, ischemia or tissue damage, stroke, or even death.
Over eight million people in the U.S. are afflicted each year with complications related to blood clots. Our business is currently focused primarily on two segments of the thrombosis market in which safe and rapid removal of blood clots is essential for patient care, namely ischemic stroke and acute massive pulmonary embolism.
Ischemic Stroke
Approximately 700,000 adults in the U.S., or one every 45 seconds, are afflicted with, and 150,000 die as a result of, some form of stroke each year. Stroke is currently the third leading cause of death, and the leading cause of disability, in the United States. Approximately three million Americans are currently disabled from stroke. The American Stroke Association estimates that approximately $62.7 billion will be spent in the U.S. in 2007 for stroke related medical costs and disability.
The vast majority of strokes, approximately 87% according to the American Stroke Association, are ischemic strokes, meaning that they are caused by blood clots, while the remainder are hemorrhagic strokes, or caused by bleeding in the brain, and are more deadly. However, available treatment options for ischemic stroke are subject to significant therapeutic limitations. For example, the most widely used treatment for
56
Table of Contents
ischemic stroke is alteplase, or tPA, a drug that can be administered only during a narrow time window and poses a risk of bleeding, resulting in 6% or less of ischemic stroke patients receiving such treatment. In 2005, in response to requests by the American Stroke Association and related groups for higher reimbursement amounts for ischemic stroke patients treated with a thrombolytic drug, the Centers for Medicare and Medicaid Services, or CMS, approximately doubled the amount of reimbursement provided for such treatment to $11,578 per patient.
When blood clots block arteries that supply blood to the brain, they reduce the oxygen supply to brain tissues, a condition known as cerebral ischemia which can gradually degrade the oxygen-deprived tissues and result in long-term impairment of brain functions. More than 600,000 Americans have an ischemic stroke each year. Approximately 80% of U.S. ischemic stroke patients reach an emergency room within 24 hours after the onset of stroke symptoms, according to Datamonitor; but by contrast, only about 23% of U.S. ischemic stroke patients reach an emergency room within the FDA-mandatedthree-hour time window for treatment with the currently approved thrombolytic drug, tPA. Due to thisthree-hour treatment window and other limitations, according to Datamonitor only 1.6% to 2.7% of patients with ischemic stroke in community hospitals, and only 4.1% to 6.3% in academic hospitals or specialized stroke centers, are treated with thrombolytic therapy.
Acute Massive Pulmonary Embolism
According to the National Institutes of Health, approximately 600,000 people in the U.S. every year experience a blood clot that lodges in the lungs, known as a pulmonary embolism. A portion of these are classified as acute massive pulmonary emboli, meaning that they involve obstruction of blood flow to a lobe or multiple segments of the lungs. Acute massive pulmonary emboli, which result in nearly 60,000 deaths in the U.S. annually, must be treated quickly, as most of these deaths occur within 30 to 60 minutes after the onset of symptoms.
Existing Blood Clot Therapies and Their Limitations
Various different treatments currently exist for the prevention and treatment of blood clots. Aspirin and other anti-platelets as well as heparin and other anticoagulants are commonly used to prevent or reduce the incidence of blood clots, but have no effect in eliminating such blood clots once they have formed. We focus on the treatment of blood clots once they have formed. Currently available therapeutic approaches for dissolving or otherwise eradicating blood clots before they cause serious medical consequences or death fall into two categories: clot-dissolving drugs, or thrombolytics, and mechanical devices and procedures.
Thrombolytic Drugs
Thrombolytic drugs dissolve blood clots by breaking up fibrin, the protein that provides the structural scaffold of blood clots. The most widely used thrombolytic drug today is a form of tissue plasminogen activator, commonly referred to as tPA. tPA is marketed in several different formulations that are approved for a variety of specific vascular disorders, such as: alteplase for acute ischemic stroke, acute massive pulmonary embolism, central venous catheter clearance and acute myocardial infarction; and reteplase and tenecteplase for acute myocardial infarction. Other thrombolytic agents include urokinase, or Abbokinase, which is approved for treatment of acute massive pulmonary embolism; and streptokinase, which is approved for treatment of acute massive pulmonary embolism, acute myocardial infarction and deep vein thrombosis. Worldwide annual sales of thrombolytic drugs are approximately $500 million.
Thrombolytic drugs involve a variety of risks and potential side effects that can limit their usefulness:
| • | Risk of Bleeding — Thrombolytic drugs dissolve blood clots, including those formed naturally as a protective response to vessel injury, which can result in bleeding. The risk of bleeding increases relative to the dosage and duration of treatment and differs among the various thrombolytic drugs. Patients who are already taking other medications to prevent formation of clots, such as anticoagulants or antiplatelets, also may not be good candidates for the use of thrombolytic drugs, due to the increased difficulty of controlling bleeding. As a result, thrombolytic drugs are approved by the FDA subject to strict limitations on when, how long and in what dosages they can be administered. These limitations |
57
Table of Contents
| deny patients the opportunity to receive thrombolytic treatment in early response settings such as in an ambulance. This delay in the initiation of treatment and the resulting restoration of blood flow is a significant unmet medical need for time sensitive indications such as ischemic stroke and myocardial infarction. |
| • | Time Window for Administration — Due to the risk of bleeding, which increases over time, tPA is only approved for administration to ischemic stroke patients within three hours after the onset of stroke symptoms. Thisthree-hour window is considered to be one of the primary limiting factors in treating ischemic stroke. Approximately 23% of ischemic stroke patients in the U.S. recognize their symptoms and reach an emergency room within thethree-hour window. However, due to other limitations, fewer than 6% of U.S. ischemic stroke patients ultimately receive treatment with a thrombolytic drug. | |
| • | Possible Immune Response — Some patients experience an immune response due to the continued administration of thrombolytic drugs. For example, thrombolytic drugs that are based on non-human biological material, such as streptokinase, which is produced using streptococcus bacteria, may stimulate such an immune reaction. |
Mechanical Devices and Procedures
There are several mechanical means for removing or destroying blood clots. Thrombectomy, or surgical clot removal, is used to treat patients with occluded dialysis grafts and some clots in the peripheral vascular system as well as in acute massive pulmonary embolism. These procedures are invasive and entail delays, costs and risks that accompany any major surgery. Although these procedures are less suitable for removing blood clots from the brain, there are devices approved for these cranial surgical procedures.
In addition, there are some mechanical devices that can be introduced through a catheter-based delivery system to mechanically break up a blood clot, or to ensnare and retract a clot through the vascular system and out of the body. These mechanical devices are generally not found outside of major medical centers, as they require a catheter laboratory and skilled personnel to administer the therapy. While they do not cause the same bleeding risk as thrombolytic drugs, these mechanical interventions pose some risk of damaging other tissues during treatment, as well as a risk of breaking off a piece of the clot that can itself become the cause of a stroke or embolism in some other part of the body.
Our Products and Product Candidates
We are currently pursuing two product candidates to treat vascular disorders: our SonoLysis+tPAtherapy is in the clinical development phase as a treatment for ischemic stroke and our SonoLysis therapy without a thrombolytic drug is in the preclinical development phase as a potential treatment for ischemic stroke. In addition, we are currently selling Abbokinase, which is approved for the treatment of acute massive pulmonary embolism.
SonoLysis Microbubble Technology
Prior to the founding of our company and while employed by ImaRx Pharmaceutical Corp., members of our scientific team invented the microbubble technology that became Definity, a microbubble product that has been administered safely as a diagnostic ultrasound contrast agent since it received regulatory approval in 2001. Definity is marketed by Bristol-Myers Squibb and has been approved by the FDA for diagnostic ultrasound contrast applications in cardiology. We have an agreement with Bristol-Myers Squibb pursuant to which they agreed that they may only sell Definity to the non-targeted, diagnostic ultrasound imaging contrast agent market, and we agreed not to sell our products in the same market. Therefore, we do not believe Definity is a competitor with our product candidates. Our proprietary MRX-801 microbubbles are similar in composition to Definity microbubbles, which we believe may improve the prospects for acceptance of our SonoLysis therapy by physicians, regulators, health care providers and third-party payors. However, we have designed our MRX-801 microbubbles with a proprietary formulation of a lipid shell and an inert biocompatible gas for use as a therapeutic agent that results in approximately three times more microbubbles per unit volume than Definity. We believe thesub-micron size of our MRX-801 microbubbles allows them to
58
Table of Contents
penetrate and disperse within a blood clot, so that their expansion and contraction, or cavitation, can break the clot into very small particles, which we believe reduces the risk that an embolism may occur downstream from the original blood clot. In addition, we have developed a proprietary MRX-801 microbubbles manufacturing process that we believe enables us to reliably and cost-effectively create sterile and stable submicron-sized bubbles from a suspension of lipid nanoparticles. We believe our SonoLysis therapy can be used with or without a thrombolytic drug to treat ischemic stroke and other vascular disorders.
SonoLysis+tPA Therapy
Our most advanced development program, SonoLysis+tPAtherapy, involves the administration of our proprietary MRX-801 microbubbles, ultrasound and tPA as a potential treatment for ischemic stroke. To administer our SonoLysis+tPAtherapy, MRX-801 microbubbles and tPA are injected intravenously into the bloodstream. The MRX-801 microbubbles are distributed naturally throughout the body and are carried to the site of the blood clot. The ultrasound is administered to the site of the blood clot, and the energy from the ultrasound causes the MRX-801 microbubbles to expand and contract vigorously, or cavitate. We believe this cavitation both mechanically breaks up the blood clot and helps the administered tPA permeate the clot to facilitate clot dissolving activity. The gas released by the MRX-801 microbubbles is then cleared from the body by exhaling, and the lipid shell is metabolized like other fats in the body. We believe that this therapeutic approach incorporates two complementary mechanisms of action, mechanical and enzymatic, that can reduce the time required to dissolve a blood clot and help ensure more rapid and complete restoration of blood flow to at risk brain tissues in patients with ischemic stroke.
An independent academic investigator has conducted a clinical trial to evaluate the effects of administering diagnostic microbubbles (not our MRX-801 microbubbles) on the speed and degree of cerebral artery recanalization in combination with ultrasound and tPA. That clinical trial involved 111 patients with acute ischemic stroke who presented within three hours after onset of symptoms. Of the patients in the clinical trial, 38 patients were treated with microbubbles and ultrasound after administration of tPA. The results of the clinical trial indicated that the rate of complete recanalization after two hours in the patients who received microbubbles, ultrasound and tPA was significantly higher, at 54.5%, than a combination of the 40.8% rate in patients who received tPA and ultrasound only, and the 23.9% rate in patients who received tPA alone. These differences were statistically significant, with a p-value of 0.038. A p-value measures the likelihood that a difference between the investigational and control groups is due to random chance. A p-value of less than or equal to 0.05 means the chance that the difference is due to random chance is less than 5.0%, and is a commonly accepted threshold for denoting a meaningful difference between investigational and control groups. Since this clinical trial did not involve our MRX-801 microbubbles, the results of any clinical trials using our proprietary microbubbles may differ from these results.
This data from an independent investigator provided proof of concept support for the Phase I/II clinical trial that we recently initiated using our MRX-801 microbubbles and ultrasound in combination with tPA. This is a Phase I/II dose-escalation trial designed to expand upon the prior work of the academic investigators. We initiated this trial in January 2007, and intend to enroll a total of 72 patients in medical centers in the United States and Europe. Patients will be enrolled in four successive cohorts, or groups. Patients randomized to the treatment arm will receive an increasing dose of our MRX-801 microbubbles and the standard dose of tPA. A Data and Safety Monitoring Board will review the data from each cohort before granting approval to enroll patients in the next successive cohort utilizing the next higher dose of MRX-801 microbubbles. We anticipate enrollment for this trial will be completed in the first half of 2008 and intend to initiate a Phase II study following completion of the ongoing Phase I/II study.
We believe that the synergistic combination of the mechanical action of our proprietary MRX-801 microbubbles and ultrasound, together with the enzymatic activity of tPA, will reduce the time required to dissolve a blood clot, enable the clot to be dissolved more completely and restore blood flow more quickly to at risk brain tissues in patients with ischemic stroke, all without increasing the risk of bleeding associated with tPA. We believe that accelerated restoration of blood flow could improve outcomes for ischemic stroke patients who are currently eligible to receive thrombolytic therapy. We estimate that if approved by the FDA over 90,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis+tPAtherapy annually.
59
Table of Contents
SonoLysis Therapy
Our SonoLysis therapy involves administration of our MRX-801 microbubbles with ultrasound, but without the external administration of a thrombolytic drug. To administer our SonoLysis therapy, MRX-801 microbubbles are injected intravenously into the bloodstream, disperse naturally throughout the body and are carried to the site of the blood clot. Ultrasound is then administered to the site of the blood clot, and the energy from the ultrasound causes the MRX-801 microbubbles to expand and contract vigorously, or cavitate. We believe this cavitation both mechanically breaks up the blood clot and helps to enhance the body’s natural clot dissolving processes. The gas released by the MRX-801 microbubbles is then cleared from the body by exhaling, and the lipid shell is processed like other fats in the body.
We believe SonoLysis therapy represents a new approach to the treatment of ischemic stroke and since it does not involve use of a thrombolytic drug and its associated risk of bleeding, SonoLysis therapy may offer several advantages over other treatments for ischemic stroke, including an extended treatment window, rapid initiation of treatment, and availability for use in patients for whom thrombolytic drugs cannot be used due to risk of bleeding. Recent studies have shown that nearly 25% of ischemic stroke victims still have at-risk but viable brain tissue as long as 24 hours after onset of stroke symptoms. Because SonoLysis therapy does not involve use of an externally administered thrombolytic drug or the associated risk of bleeding, we believe SonoLysis therapy may offer a treatment option for ischemic stroke patients who arrive at a hospital after tPA’s approved three hour window. In addition, we believe SonoLysis therapy also could offer a treatment option to ischemic stroke patients who arrive at a hospital within the three hour window but are ineligible for tPA due to recent surgery, taking certain medications or other factors that increase bleeding risk. This unique treatment approach could enable us to offer an effective therapy to ischemic stroke patients with fewer risks and restrictions, thus potentially affording a treatment option to more patients than can be treated with tPA today.
We have conducted preclinical animal studies to evaluate the safety of our MRX-801 microbubbles as well as to evaluate the ability of our SonoLysis therapy to break up blood clots. We enrolled 24 patients in a proof of concept clinical trial that applied ultrasound to Definity microbubbles as a means for breaking up blood clots in thrombosed dialysis grafts, eleven of which also received tPA. This clinical trial demonstrated improved restoration of blood flow, based on imaging results only. There was one adverse event reported, involving moderate bleeding and oozing at an unspecified site, that may have been related to the treatment. No other adverse events were determined to be related to the treatment. We have not yet conducted any clinical trials using our proprietary MRX-801 microbubbles with ultrasound to treat blood clot indications without a thrombolytic drug. We intend to conduct additional preclinical studies of SonoLysis therapy through the first half of 2008 and initiate a Phase II study to treat patients with ischemic stroke following completion of the SonoLysis+tPA therapy Phase I/II clinical trial. Because of the preclinical data package as well as our ongoing Phase I/II clinical trial evaluating SonoLysis+tPA therapy in patients with ischemic stroke, we believe no Phase I study will be required prior to initiating the Phase II study for SonoLysis therapy.
We believe that our SonoLysis therapy may be used in the treatment of ischemic stroke patients in a variety of settings where tPA is not approved, such as in a hospital but when treatment is initiated after the three hour window, in an ambulance prior to arrival at a hospital, or in a hospital when treatment is initiated within the three hour window but the patient is ineligible for tPA due to recent surgery, taking certain medications or other factors that increase bleeding risk. We estimate that if approved by the FDA over 200,000 ischemic stroke patients in the U.S. could be eligible for SonoLysis therapy annually.
Abbokinase
We are currently selling Abbokinase, which is approved for the treatment of acute massive pulmonary embolism. Abbokinase is a tissue culture form of urokinase, a natural human protein primarily produced in the kidneys that stimulates the body’s natural clot-dissolving processes. Urokinase breaks up blood clots by converting plasminogen, an inactive ingredient in human blood, into the enzyme plasmin, which in turn degrades the fibrin protein strands that are essential to the structural integrity of a clot. Abbokinase is approved by the FDA for the treatment of acute massive pulmonary embolism. Abbokinase has been
60
Table of Contents
administered to over four million patients since its approval, and we estimate that approximately 400 acute care hospitals in the U.S. include Abbokinase on their pharmacy formulary today. We acquired Abbokinase from Abbott Laboratories in April 2006, and commenced selling Abbokinase in October 2006. We believe that Abbokinase sales will provide us with a source of near-term revenue as well as an opportunity to form relationships with vascular physicians and acute care institutions that regularly administer blood clot therapies.
Prior to 1998, Abbokinase was approved by the FDA for acute massive pulmonary embolism, catheter occlusion clearance and acute myocardial infarction. The product was withdrawn from the market in 1998 due to concerns over the manufacturing process, including failure to screen donors and test materials for infectious disease, and inadequate storage and handling of materials to prevent contamination with infectious agents. After revising its manufacturing processes to the FDA’s satisfaction, in 2002 Abbott Laboratories obtained FDA approval to resume commercial sales of Abbokinase for use in treating acute massive pulmonary embolism. In 2004, however, Abbott Laboratories halted its Abbokinase sales and marketing effort when it decided to divest its thrombolytic drug assets. The FDA’s approval of Abbokinase was not withdrawn or suspended in 2004 and has not been withdrawn or suspended at any time since then. Based upon generally recognized industry sales measures, known as IMS data, sales of Abbokinase to end user customers since the product’s relaunch were approximately $4 million, $27 million, $33 million, and $19 million in 2002, 2003, 2004, and 2005, respectively, and based upon data provided by our wholesalers, sales in 2006 were approximately $12 million. In our acquisition of Abbokinase in April 2006, we purchased substantially the entire inventory of Abbokinase in existence, except for that previously sold to and held by wholesalers and end user customers, and we believe we are currently the only company selling the product into the distribution network.
We acquired Abbokinase and related assets from Abbott Laboratories, including the remaining inventory of finished product, all regulatory and clinical documentation, validated cell lines, and intellectual property rights, including trade secrets and know-how relating to the manufacture of urokinase using the tissue culture method. We have limited patent rights associated with Abbokinase, and our right to use the Abbokinase trademark does not extend to additional product that we might manufacture in the future. In our acquisition of Abbokinase, we received approximately 153,000 vials of Abbokinase. At the time of our acquisition we estimated that hospitals would purchase, and we would thereby recognize revenue for, approximately 111,000 vials of Abbokinase, or approximately 72% of the total vials we acquired, which we believe represented approximately a four-year supply of inventory. We also estimated that hospitals would not purchase approximately 42,000 vials, or approximately 28% of the vials we acquired. Approximately $16.7 million of the $20.0 million purchase price for Abbokinase was allocated to the vials we expect hospitals to purchase. Of our vials of Abbokinase held in inventory either by us or by our wholesalers as of March 31, 2007, approximately 64% of the vials we expect hospitals to purchase, or approximately $10.7 million in inventory value, will expire by October 2007 based on current stability data. The remaining approximately 36% of the vials we expect hospitals to purchase, or approximately $6.1 million in inventory value, will expire at various times up to August 2009. We are continuing the ongoing stability testing program initiated by Abbott Laboratories to support a request for extension of the expiration dates of this inventory. The testing to date has shown that the product changes very little from year to year. Currently, we believe it is probable that the stability data will support extension of the inventory expiration dates. We anticipate that the next testing point of our ongoing stability program, at which we may obtain data sufficient to extend the expiration dates of our unlabeled inventory, will be completed in the fall of 2007. We will be required to submit this data to the FDA. If the parameters tested are within the specifications previously approved by the FDA, we may then submit a lot release request to the FDA, and upon approval, label vials with extended expiration dating at that time to between June and August 2009. If the expiration dates of this inventory are extended, we will need to re-brand the remaining inventory because our license to use the Abbokinase trademark does not extend beyond the current inventory expiration dates. In May 2007 we obtained FDA approval of a new product trade name and labeling reflecting the new trade name.
To sell Abbokinase for the treatment of acute massive pulmonary embolism, we are required to continue an ongoing200-patient immunogenicity clinical trial that commenced in 2003. The purpose of this trial is to evaluate the rate and severity of immune, or allergic, response in patients who are treated with Abbokinase.
61
Table of Contents
Since the original approval of Abbokinase, the FDA has changed its requirements for approval of biologic agents and now requires the sponsor to demonstrate in clinical trials that the biologic product does not induce an immune response in the patients treated. This is now one of the routine safety evaluations for all biologic agents. Abbokinase is a biologic agent and, although its original approval pre-dated this requirement, the FDA required this study to be conducted as a condition of re-approval in 2004. We can market and sell our Abbokinase inventory while this trial progresses, subject to existing expiration dates.
Additional SonoLysis Product Opportunities
It is well known that brain tissue degrades quickly when blood flow is cut off or restricted. We believe that SonoLysis therapy may be administered rapidly via intravenous infusion, perhaps even in an ambulance setting, to quickly restore blood flow, to minimize tissue damage, and to improve the odds for full or improved patient recovery. To pursue an ambulance-based ischemic stroke treatment, we would be required to show either that hemorrhage can be ruled out in an ambulance setting, or that SonoLysis therapy has no detrimental effect on a hemorrhagic stroke.
We believe that the ability of our SonoLysis+tPAtherapy to reduce the time required to dissolve a blood clot could make this therapy suitable for use in treating a broad variety of vascular disorders beyond ischemic stroke. For example, we believe SonoLysis+tPAtherapy could potentially enable the more rapid treatment of recently formed acute clots, such as those that cause myocardial infarction, or heart attack. We also believe this therapy has the potential as a treatment for more establishedsub-acute and chronic clots, such as those in peripheral vascular indications that cannot be effectively treated with thrombolytic therapy alone.
Current treatments for blood clots in the heart have limitations. While more than one thrombolytic drug is approved to treat myocardial infarction, these drugs take time to dissolve the clot. Thus, the standard of care for most interventional cardiologists is angioplasty, or using a balloon-tipped catheter to reopen the blood vessel more quickly. A stent is often used as well to help keep the vessel propped open. However, catheter-based therapy requires time to access a specialized catheter lab facility, ensure the availability of highly trained physicians, prepare the patient, and thread the catheter to the site of the clot. We believe that the synergistic combination of the mechanical action of SonoLysis therapy, together with the enzymatic activity of a thrombolytic drug, may reduce the time required to dissolve an acute blood clot in the heart, enable the clot to be dissolved more completely and restore blood flow more quickly to at risk heart tissues, all delivered rapidly in an emergency room setting. We believe our SonoLysis+tPA therapy may restore blood flow in certain patients before they reach the catheter lab, and thus could help reduce the need for more invasive and costly angioplasty and stents, and the associated complications.
Similarly, current treatments for blood clots in the peripheral vascular system also have limitations. Mechanical thrombectomy and vascular surgery is the standard of care for acute cases of peripheral artery occlusive disease and deep vein thrombosis, but some physicians use thrombolytic drugs off label in this area as well. Mechanical thrombectomy and vascular surgery are invasive procedures that take place in a costly operating room setting and have the potential for complications. Thrombolytic therapy requires the placement of a catheter in a catheter lab followed by thrombolytic drug infusion, often over several hours or over night, in an intensive care or physician clinic setting. We believe that the synergistic combination of the mechanical action of SonoLysis therapy, together with the enzymatic activity of a thrombolytic drug, may reduce the time required to dissolve largesub-acute and chronic blood clots often found in the legs with fewer complications than mechanical thrombectomy, vascular surgery or thrombolytic therapy alone. We believe that the speed of our SonoLysis+tPA therapy could enable more patients to be treated in a lower cost, outpatient physician clinic setting. The recent introduction of various intravascular ultrasound devices could be used as a component in this therapy.
We have used microbubbles and ultrasound with a thrombolytic drug both to conduct preclinical animal studies with academic collaborators to treat myocardial infarction, as well as to conduct clinical proof of concept trials to treat patients with occluded dialysis grafts, peripheral artery occlusive disease and deep vein thrombosis. However, we have not conducted any clinical trials in this area with our MRX-801 microbubbles. Currently we intend to focus our SonoLysis development efforts in ischemic stroke, but we intend to further
62
Table of Contents
explore the application of our SonoLysis technology in cardiology and peripheral vascular disorders in the future when additional resources are available or with a development partner.
In many countries worldwide, the risk factors for formation of blood clots are increasing, such as high blood pressure, obesity, diabetes, aging population, and limited mobility caused by injury, illness or simply long distance air travel. We believe there is a growing unmet medical need for improved treatments for blood clot related vascular disorders that our SonoLysis product candidates could address and help reduce the need for angioplasty, stents and vascular surgery.
Research Stage Opportunities
We are exploring a number of potential future product development opportunities based on our microbubble technology. These opportunities include pursuing applications in oxygen delivery, targeted SonoLysis, drug delivery as well as researching new microbubbles product opportunities.
Oxygen Delivery
We are exploring a number of potential future product development opportunities based on our microbubble technology. These opportunities include our NanO2 technology as well as researching new microbubble product opportunities.
Our lead research stage program is our NanO2 program that utilizes our proprietary MRX-804 microbubbles for carrying oxygen to parts of the body afflicted with a reduced supply of blood or oxygen. NanO2 does not involve the application of ultrasound. NanO2 is administered intravenously as an emulsion. We believe upon entering the bloodstream it converts from a liquid to a gas forming microbubbles with a high oxygen carrying capacity. We believe that NanO2 can be used to treat a broad variety of disorders in which reduced blood flow results in oxygen-deprived tissues, such as injuries that involve significant blood loss, ischemic stroke and heart attack. Because MRX-804 microbubbles are smaller than red blood cells, we believe NanO2 will be able to penetrate clots and other blockages of blood vessels and deliver oxygen to tissues beyond the blockage. In addition, we believe that NanO2 has the capacity to carry more oxygen per unit volume than red blood cells, potentially enabling a modest-sized dose to keep tissues alive until normal blood flow can be restored. Unlike human blood, we anticipate that NanO2 may be able to be stored at room temperature, making it mobile for potential battlefield or ambulance environments. We are conducting additional research on the application of our MRX-804 microbubbles in ischemic stroke and conducting preclinical animal studies of NanO2 to treat hemorrhagic shock.
We have not yet conducted any clinical trials using our proprietary MRX-804 microbubbles to treat oxygen-deprived tissues, but we are planning and conducting various preclinical studies using our MRX-804 microbubbles. We are working with academic collaborators who have conducted a variety of preclinical animal studies evaluating MRX-804 microbubbles as an oxygen delivery agent. One group of collaborators has conducted preclinical studies testing the ability of MRX-804 microbubbles to prevent hemorrhagic shock in pigs with significant blood loss. All five of the pigs treated with NanO2 in this study survived, while four of the five control pigs died. These same collaborators have also recently received a $700,000 grant from the U.S. Army to conduct an additional preclinical animal study in rats and pigs evaluating NanO2 as a potential treatment for hemorrhagic shock. ImaRx is participating in this preclinical study and the outcome will impact the type and timing of future clinical trials, if any, for NanO2.
In addition to ischemic stroke and hemorrhagic shock, we believe that NanO2 can be used to treat other ischemic conditions such as myocardial infarction. We also believe that our proprietary MRX-804 microbubbles may have applications beyond oxygen delivery. Because NanO2 has the ability to absorb and transport many different gases, it also has the potential to help remove unwanted gases from tissues, such as anesthesia gases that cause post-operative cognitive disorder, carbon monoxide which can build to toxic or even fatal levels, or nitrogen that can cause decompression sickness. Our academic collaborators have performed preclinical animal studies demonstrating the feasibility of using NanO2 to absorb and transport gases other than oxygen, such as nitrogen.
63
Table of Contents
Other Research Stage Opportunities
Targeted SonoLysis Therapy. Our research team has developed MRX-802, our next generation SonoLysis microbubbles with targeting technology that causes the microbubbles to bind to blood clots. We believe that our MRX-802 targeted microbubbles will have a greater ability tobreak-up blood clots than non-targeted microbubbles when combined with ultrasound. We have demonstrated in laboratory experiments that our MRX-802 targeted microbubbles improved binding to blood clots. We have conducted preclinical animal studies with academic collaborators evaluating MRX-802 targeted microbubbles and ultrasound to treat various clot disorders, including myocardial infarction. To further the research on our next generation SonoLysis technology, we have received and are near the mid-point of our work on an approximately $1.2 million grant from the National Institutes of Health, or NIH, to study MRX-802 targeted microbubbles to treat vascular clots.
Targeted Drug Delivery. In addition to our targeted SonoLysis technology, our research team has demonstrated the ability to add a drug payload to our microbubbles or use a liquid instead of a gas core to createsub-micron sized targeted droplets for drug delivery. When administered, the drug is encapsulated in the microbubbles or droplets and inactive. Ultrasound can then be used to cause the microbubbles or droplets to deliver their payload at a desired location in a patient’s body. Our proprietary targeted drug delivery microbubbles, MRX-803 comprises a gas core and an oil layer containing a drug payload surrounded by a lipid shell. We have received an approximately $1.0 million subcontract and are near the mid-point of our work under that subcontract to study our proprietary MRX-803 targeted drug delivery microbubbles to treat a variety of tumors. We believe this technology has the potential for broad applications, including delivering drugs to dissolve blood clots or arterial plaque as well as treat a variety of cancer types.
Future Microbubbles Research. Our proprietary microbubbles are biocompatible spheres of varying size and composition that we believe could be adapted, by changing their composition and size, for a variety of additional applications. These applications could include the delivery of genetic materials to targeted sites in the body, destroying cancer cells with high intensity focused ultrasound, or HIFU, as well as identifying and treating vulnerable plaque. In addition, we believe that the competitive position of our microbubbles-based therapies will be aided by our broad portfolio of issued patents, patent applications and exclusive licenses relating to the use of microbubbles and ultrasound for a wide variety of applications.
Manufacturing
We currently do not have, and do not intend to establish, our own manufacturing facilities. Instead, we plan to engage third parties to manufacture our products and product candidates, which we believe will allow us to focus on our core research and product development programs. We also believe that the use of experienced manufacturers will give us access to facilities and processes that are qualified under the FDA’s current Good Manufacturing Practices, or cGMP, greater manufacturing specialization and expertise, higher levels of flexibility and responsiveness and faster delivery of products than we might achieve through in-house manufacturing. Specialized manufacturers are often used in the biopharmaceutical industry because they relieve product developers from the infrastructure required to support compliance with applicable cGMP requirements, and other rules and regulations of foreign regulatory authorities.
Our contract manufacturers will be subject to unannounced inspections by the FDA and corresponding foreign and state agencies to ensure strict compliance with cGMP and other applicable governmental quality control and record-keeping regulations. In addition, transfer of ownership of products could trigger a manufacturing inspection requirement from the FDA. We do not have control over and cannot ensure third-party manufacturers’ compliance with these regulations and standards. If one of our manufacturers fails to maintain compliance, the production of our products or product candidates could be interrupted, which could result in substantial delays, additional costs and lost sales.
We have contracted with a third party to produce small quantities of our MRX-801 microbubbles for clinical research purposes. We manufacture MRX-804 internally in small quantities for research and preclinical purposes. Neither we nor any other third party currently manufactures Abbokinase. While we intend to investigate the requirements for us to manufacture Abbokinase, we currently have no plans to
64
Table of Contents
manufacture Abbokinase in the near term. In order to manufacture Abbokinase, we would need to have our manufacturing process validated by the FDA and would likely be required to conduct additional preclinical studies, and possibly additional clinical trials, to demonstrate its comparability, safety and efficacy. The manufacturing process for Abbokinase involves a roller bottle production method that is used infrequently today, is available from a very limited number of manufacturers worldwide, and may be difficult to qualify for necessary regulatory approvals.
Sales and Marketing
We commenced selling Abbokinase in the U.S. in October 2006. Our internal sales and marketing staff, currently consisting of three individuals, manages our relationships with third-party distribution partners and institutional Abbokinase customers, and oversees our related direct and indirect advertising and promotional activities. We intend to focus our sales and marketing activities on servicing the existing demand for Abbokinase through existing distribution channels, and we believe that our current staffing will be sufficient to meet these needs.
For the marketing and sale of potential future products in the U.S., we intend to gradually expand our U.S. sales force and broaden our domestic sales and marketing efforts to the community of vascular physicians and acute care institutions that we believe will be most critical to acceptance and widespread adoption of our products. Outside of the U.S., we intend to rely primarily on distribution partners. We may also enter into strategic relationships with pharmaceutical and other companies for the marketing and distribution of some of our products, and may rely on third parties for advertising and promotion of our products, particularly for markets outside the U.S. We intend to have our distribution partners manage any third-party logistics.
Competition
The market for therapies to treat vascular disorders associated with blood clots is highly competitive. Numerous companies either offer or are developing competing treatments for ischemic stroke and acute massive pulmonary embolism. Many of these competitors have significantly greater financial resources and expertise in development and regulatory matters than we do, as well as more established products, distribution and reimbursement. We expect that our competitors will also continue to develop new or improved treatments for the vascular disorders we are targeting.
To become accepted as treatments for ischemic stroke or acute massive pulmonary embolism, we believe competing therapies must offer a combination of efficacy, safety, rapid effect, ease of administration, approved window of administration and cost-effectiveness. While we believe that our products and product candidates will offer advantages over many of the currently available competing therapies, our business could be negatively impacted if our competitors’ present or future offerings are more effective, safer or less expensive than ours, or more readily accepted by regulators, health care providers or third-party payors.
There are two principal groups of competitors offering treatments to break up or remove blood clots: thrombolytic drug companies, and vendors of mechanical thrombectomy or similar devices.
Thrombolytic Drug Competitors
The U.S. market for thrombolytic drugs is dominated by Genentech, Inc., which manufactures tPA, the most widely used thrombolytic drug. We are not a significant competitor in the sale of thrombolytic drugs, since we recently acquired our only approved product, Abbokinase, which is approved by the FDA only for treatment of acute massive pulmonary embolism. Genentech’s tPA in various formulations is currently the only thrombolytic drug that has been approved by the FDA for treatment of ischemic stroke, and is also approved for acute massive pulmonary embolism, as well as catheter occlusion clearance and myocardial infarction indications. We are aware that other thrombolytic drugs are also under development, such as desmoteplase, which is a recombinant form of a derivative of vampire bat saliva being developed by PAION AG, and ancrod, which is an enzyme derived from Malaysian pit viper venom being developed by Neurobiological Technologies, Inc., both of which are currently in separate Phase III clinical trials for treatment of ischemic stroke. Other companies also offer or are developing thrombolytic drugs for treatment of blood clots
65
Table of Contents
associated with myocardial infarction and peripheral vascular occlusions, but since we view thrombolytic drugs as complementary to our SonoLysis therapy, we do not consider those product offerings or programs to be competitive with our current business strategy.
Device Competitors
We believe that the primary device-based treatment for ischemic stroke clots is the Mechanical Embolus Removal in Cerebral Ischemia retrieval system or the MERCI system, which is an intravascular catheter-based therapy marketed by Concentric Medical, Inc. This device is used to engage the clot and retract it through the catheter and out of the body. Other devices are also approved and marketed for treating blood clots associated with peripheral vascular and coronary indications and with dialysis access grafts, such as the Fogarty Catheter by Edwards Lifesciences, formerly a division of Baxter International, AngioJet by Possis Medical, Inc., Micro-Infusion Catheter by EKOS Corp., and Resolution Endovascular System by OmniSonics Medical Technologies, Inc. A variety of companies also offer catheter-delivery systems for thrombolytic drugs or other drugs used in the treatment of blood clots, but we do not consider these devices to be directly competitive with our current business strategy.
We are unaware of any other companies that are developing bubble technologies for therapeutic use in vascular disorders.
Material Contracts
Following is a summary of our material contracts, other than contracts entered into in the ordinary course of business, to which we are a party:
April 2006 Agreements with Abbott Laboratories
In April 2006, we acquired from Abbott Laboratories the assets related to Abbokinase, including the remaining inventory of finished product, all regulatory and clinical documentation, validated cell lines, and intellectual property rights, including trade secrets and know-how relating to the manufacture of urokinase using the tissue culture method, for consideration consisting of $5.0 million in cash and a 6% non-recourse promissory note for $15.0 million that matures on December 31, 2007. The note is secured by the acquired inventories and related assets and an escrow of 50% of proceeds from our sales of such inventories in excess of $5.0 million, up to a maximum escrow of $15.0 million. As of March 31, 2007, we had received aggregate net proceeds of $2.6 million from the sale of a portion of our Abbokinase inventory, and, since the $5.0 million threshold for the escrow requirement had not yet been met, we had not escrowed any of the proceeds from sales of Abbokinase as of March 31, 2007. As part of this arrangement we entered into a trademark license agreement with Abbott Laboratories in which it granted to us an exclusive, non-transferable license, without any sublicense rights, to use the Abbokinase trademark. We must adhere to certain quality control standards when marketing and selling the Abbokinase inventory under the trademark. This trademark license automatically terminates on the earlier to occur of the completion of our sale of the acquired Abbokinase inventory or the expiration dates that were applicable to our Abbokinase inventory as of the date it was transferred to us. Abbott Laboratories is also entitled to terminate the license if we are in material breach of the agreement and fail to cure such breach within 15 days notice, or if we commit a non-material breach of the agreement and fail to cure it within 30 days.
License Agreement with Bristol-Myers Squibb Medical Imaging, Inc.
We are party to an exclusive, worldwide, royalty-free license agreement with Bristol-Myers Squibb Medical Imaging, Inc. (as successor to DuPont Contrast Imaging, Inc.) dated October 7, 1999 for the use of intellectual property related to targeted and tissue-specific diagnostic ultrasound products, outside the field of contrast enhancement of diagnostic ultrasound imaging. The intellectual property covered by this agreement addresses microbubble compositions, methods of use and manufacturing processes that encompass most of our development and research stage product candidates. Under the agreement, to the extent we develop any products or technology in the area of thrombus imaging or sonothrombolysis, which is the use of ultrasound to
66
Table of Contents
break up blood clots, we must first offer Bristol-Myers the right to negotiate an exclusive license for such product or technology for development and commercialization for a period of 90 days before offering it to any third party for license. This license is indefinite in duration and contains no express termination provisions. On September 1, 2005, Bristol-Myers executed a letter waiving their right of first negotiation in our current MRX-801 microbubbles, and that we have satisfied all of our obligations under the license agreement with respect to our MRX-801 microbubbles, as they existed on that date. This acknowledgement encompasses our proprietary MRX-801 microbubbles together with ultrasound, with or without a thrombolytic drug, currently under development.
License Agreement with UNEMED Corporation
On October 10, 2003, UNEMED Corporation granted us an exclusive, worldwide license, with sublicense rights, to intellectual property and patents relating to the use of microbubbles together with ultrasound for the treatment of thrombosis. The intellectual property covered by this agreement addresses microbubble compositions and methods for treating thrombosis that likely encompass our SonoLysis therapy and SonoLysis+tPA therapy product candidates. We are obligated to pay UNEMED a royalty of 2% on any future net sales of products or processes which utilize the licensed technology, of which there have been no sales to date. We are also obligated to pay license maintenance fees in amounts from $3,000 to $7,000 annually for the life of the agreement. These fees are creditable against any royalty payments owed by us to UNEMED in the applicable calendar year. The license agreement will terminate contemporaneously with the expiration of the licensed patents, or on October 17, 2015. We may terminate the agreement, in our sole discretion, upon 90 days written notice for any reason. UNEMED may terminate the agreement for cause upon either 45 days or 90 days written notice, depending on the cause for termination, or at any time if we fail to meet certain milestones. Upon termination of the license, we would likely be required to change our SonoLysis therapy product development plans.
License Agreement with Dr. med. Reinhard Schlief
On January 4, 2005, Dr. med. Reinhard Schlief granted us an exclusive, worldwide license, with the right to sublicense, to intellectual property and patents relating to methods of destroying cells by applying ultrasound to them in the presence of microbubbles. This intellectual property may encompass our SonoLysis therapy and SonoLysis+tPA therapy product candidates. As consideration for this license, we reimbursed Dr. Schlief for certain pastout-of-pocket costs, such as maintenance fees and patent transfer fees, and also granted Dr. Schlief a five-year warrant to purchase up to 4,000 shares of our common stock at an exercise price of $15.00 per share. We are obligated to pay Dr. Schlief a royalty of 2% of net sales revenue derived from the sale of products that utilize the licensed technology. The license agreement will terminate contemporaneously with the expiration of the licensed patents, or on January 10, 2012. We may terminate the license, with or without cause, upon 60 days written notice and Dr. Schlief may terminate the agreement, with cause, 60 days after notice of the default is provided if the default has not been cured. Upon termination or expiration of the license, our plans for developing our MRX-801 microbubbles would likely not change.
License Agreement with University of Arkansas
On February 14, 2006, the University of Arkansas granted us an exclusive, worldwide license, with the right to sublicense, intellectual property and patents relating to the use of a specific ultrasound device to be used in conjunction with bubbles, a thrombolytic drug, or a combination of bubbles and a thrombolytic drug to break up blood clots. This intellectual property may encompass our SonoLysis therapy and SonoLysis+tPA therapy product candidates. To maintain this license we must meet certain product development milestones. We are obligated to pay the University of Arkansas a one-time fee of $25,000 within 30 days after the first commercial sale of a product incorporating the licensed technology, and varying royalties depending on the amount of net revenue derived from the sale of products using the licensed technology, subject to minimum annual royalties of $5,000 per year commencing February 10, 2007, increasing to $7,000 per year on February 10, 2009, and each year thereafter. The maximum aggregate royalty payable under this license is $20.0 million. We are also obligated to pay a one-time success fee of $250,000 in the first year that net revenue derived from the sale of products using the licensed technology exceeds $10.0 million. The license
67
Table of Contents
agreement will terminate contemporaneously with the expiration of the licensed patents, or on September 8, 2023. In addition, we may terminate this license at any time upon 90 days written notice to the University of Arkansas and the University may terminate the agreement for cause upon 90 days written notice. Upon termination or expiration of the license, our plans for developing our MRX-801 microbubbles would likely not change.
Patents and Proprietary Rights
Our success depends in part on our ability to develop a competitive advantage over potential competitors for the use of bubbles and ultrasound for treatment of blood clots and vascular diseases in various parts of the body. Our ability to obtain intellectual property that protects our MRX-801 microbubbles and ultrasound treatment in the presence or absence of drugs will be important to our success. Our strategy is to protect our proprietary positions by, among other things, filing U.S. and foreign patent applications related to our technology, inventions and improvements that are directed to the development of our business and our competitive advantages. Our strategy also includes developing know-how and trade secrets, and licensing technology related to bubbles and ultrasound from third parties. As of May 31, 2007 we owned 57 issued U.S. patents, 30 U.S. pending patent applications, 41 foreign patents and 72 international or foreign patent applications. In addition, as of May 31, 2007, we have licensed patents from third parties that grant us rights to 86 U.S. patents, at least five U.S. patent applications, and their respective international and foreign patent and patent application counterparts.
The U.S. patents that we own cover certain applications related to bubble compositions and methods of making and using such bubbles with ultrasound for the treatment of blood clots. Patents that cover our core technology expire between 2009 and 2021.
We have several pending patent claims, including allowed claims that have not yet issued, that cover additional elements of our bubble technology. For example, we have pending claims directed to the following aspects of bubble technology:
| • | methods of preparing gas filled bubbles; | |
| • | methods of using gas filled bubbles in combination with ultrasound for eliminating or reducing thrombi or for delivering drug compounds; | |
| • | methods of preparing gas filled bubbles that are targeted to specific cells in the body or that are activated at a specified temperature; and | |
| • | apparatus for preparing gas filled bubbles described above. |
We plan to file additional patent applications on inventions that we believe are patentable and important to our business and intend to aggressively pursue and defend patent protection on our proprietary technologies.
Our ability to operate without infringing the intellectual property rights of others and to prevent others from infringing our intellectual property rights will also be important to our success. To this end, we have reviewed all patents owned by third parties of which we are aware that are related to bubble technology and gas filled vesicles, in the presence or absence of ultrasound, and thrombolysis using gas filled vesicles, and believe that our current products do not infringe any valid claims of the third party patents that we have analyzed. There are a large number of patents directed to therapies for blood clots, and there may be other patents or pending patent applications of which we are currently unaware that may impair our ability to operate. We are currently not aware of any third parties infringing our issued claims.
In July 2003 we received a notice from a third party who owns a patent relating to the administration of ultrasound to break up blood clots indicating that we may need a license to its patent if we intend to administer our therapies according to the methods claimed in its patent.
When appropriate, we actively seek protection for our products, technologies, know-how and proprietary information by licensing intellectual property from third parties. We have obtained rights relating to our product candidates and future development programs from third parties as appropriate.
68
Table of Contents
Government Regulation
We are subject to extensive regulation by the FDA and comparable regulatory agencies in state, local and foreign jurisdictions in connection with the development, manufacture and commercialization of our product candidates.
Categories of Regulation
In the U.S., our marketed product and product candidates are subject to regulation as drugs, biologics, which are drugs derived from a living source, or medical devices. In some cases, our product candidates may fall into multiple categories and require regulatory approval in more than one category. For example, Abbokinase is a biologic, but it is subject to regulation as a drug. Our SonoLysis therapy and our SonoLysis+tPA therapy involve a combination of drug and device, which would require approval as a combination product before we could market either of these therapies. Our proprietary MRX-801 microbubbles, which are injected into the bloodstream, have been designated as a drug by the FDA. Outside the U.S., our product candidates are also subject to regulation as drugs, biologics or medical devices, and must meet similar regulatory hurdles as in the U.S. to gain approval and reach the market.
Drug and Biologics Regulation
The process required by the FDA before drug or biologic product candidates may be marketed in the U.S. generally involves the following:
| • | preclinical laboratory and animal tests; | |
| • | submission and approval of an Investigational New Drug application, or IND application; | |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of proposed drugs for their intended use and safety, purity and potency of biologic products for their intended use; | |
| • | preapproval inspection of manufacturing facilities, company regulatory files and selected clinical investigators; | |
| • | for drugs, FDA approval of a new drug application, or NDA, or FDA approval of an NDA supplement in the case of a new indication if the product is already approved for another indication; and | |
| • | for biologics, FDA approval of a biologics license application, or BLA, or FDA approval of a BLA supplement in the case of a new indication if the product is already approved for another indication. |
Prior to commencing the first human clinical trial, we must submit an IND application to the FDA. The IND application automatically becomes effective 30 days after receipt by the FDA, unless the FDA within such period raises concerns or questions about the preclinical drug testing or nonclinical safety evaluation in animals, or the design or conduct of the first proposed clinical trial. In such a case, the IND application sponsor and the FDA must resolve any outstanding concerns before the clinical trial may begin. A separate submission must be made for each successive clinical trial conducted during product development. The FDA must not object to the submission before each clinical trial may start and continue. Further, an independent Institutional Review Board, or IRB, for investigations in human subjects within each medical center in which an investigator wishes to participate in the clinical trial must review and approve the preclinical drug testing and nonclinical safety evaluation and efficacy in animals or prior human clinical trials as well as the design and goals of the proposed clinical trial before the clinical trial commences at that center. Regulatory authorities, an IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
For purposes of NDA or BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap. Moreover, the objectives of each phase may be split or combined, leading to
69
Table of Contents
Phase I/II and other similar trials that may be used to satisfy the requirements of otherwise separate clinical trials as follows:
Phase I: Phase I clinical trials are usually conducted in normal, healthy volunteers or a limited patient population to evaluate the product candidate for safety, dosage tolerance, absorption, metabolism, distribution and excretion.
Phase II: Phase II clinical trials are conducted in a limited patient population, the population for which the indication applies, to further identify and measure possible adverse effects or other safety risks, to determine the efficacy of the product candidate for the specific targeted disease and to determine dosage tolerance and optimal dosage. Multiple Phase II clinical trials may be conducted to obtain information prior to beginning Phase III clinical trials.
Phase III: When Phase II clinical trials demonstrate that a dose range of the product candidate appears to be effective and has an acceptable safety profile, Phase III clinical trials are undertaken in a larger patient population to confirm clinical efficacy and to further evaluate safety at multiple, and often internationally located, clinical trial sites.
Phase II or III studies of drugs are generally required to be listed in a public clinical trials registry, such aswww.clinicaltrials.gov. The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called Phase IV clinical studies may be made a condition to be satisfied after a drug receives approval. The results of Phase IV clinical studies may confirm the effectiveness of a product and may provide important safety information to augment the FDA’s voluntary adverse drug reaction reporting system.
The results of product development, preclinical testing and clinical trials are submitted to the FDA as part of an NDA or BLA. The submission of an NDA or BLA must be accompanied by a user fee of several hundred thousand dollars, unless a particular waiver applies. The FDA may deny approval of an NDA or BLA if the applicable regulatory criteria are not satisfied or for any other reason, or it may require additional clinical data or an additional Phase III clinical trial. Satisfaction of FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the products. The FDA also closely regulates the marketing and promotion of commercialized products. A company is permitted to make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Medical Device Regulation
The process required by the FDA before medical devices may be marketed in the U.S. pursuant to clearance or approval generally involves FDA review of the following:
| • | product design, development and manufacture; | |
| • | product safety, testing, labeling and storage; | |
| • | preclinical testing in animals and in the laboratory; and | |
| • | clinical investigations in humans. |
Unless an exemption applies, each medical device distributed commercially in the U.S. requires either prior 510(k) clearance or pre-market approval, referred to as a PMA, from the FDA. The FDA classifies medical devices into one of three classes. Class I devices are subject only to general controls, such as establishment registration and device listing, labeling, medical devices reporting, and prohibitions against adulteration and misbranding. Class II medical devices require prior 510(k) clearance before they may be commercially marketed in the U.S. The FDA will clear marketing of a medical device through the 510(k) process if the FDA is satisfied that the new product has been demonstrated to have the same intended use and
70
Table of Contents
is substantially equivalent to another legally marketed device, including a 510(k)-cleared, or predicate, device, and otherwise meets the FDA’s requirements. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a predicate device, are placed in Class III, generally requiring submission of a PMA supported by clinical trial data. Currently we have one shaker device that is a Class I device that we use to form our MRX-801 microbubbles.
To obtain 510(k) clearance, a notification must be submitted to the FDA demonstrating that a proposed device is substantially equivalent to a predicate device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a PMA application. The FDA’s 510(k) clearance process generally takes from three to 12 months from the date the application is submitted, but can take significantly longer. If the FDA determines that the device, or its intended use, is not substantially equivalent to a previously-cleared device or use, the device is automatically placed into Class III, requiring the submission of a PMA. Any modification to a 510(k)-cleared device that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, in connection with safety and effectiveness, a PMA.
Clinical trials are generally required to support a PMA application and are sometimes required for 510(k) clearance. To perform a clinical trial in the U.S. for a significant risk device, prior submission of an application for an IDE to the FDA is required. An IDE amendment must also be submitted before initiating a new clinical study under an existing IDE, such as initiating a pivotal clinical trial following the conclusion of a feasibility clinical trial. The FDA responds to an IDE or an IDE amendment for a new clinical trial within 30 days. The FDA may approve the IDE or amendment, grant an approval with certain conditions, or identify deficiencies and request additional information. It is common for the FDA to require additional information before approving an IDE or amendment for a new clinical trial, and thus final FDA approval on a submission may require more than the initial 30 days. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, and any available data on human clinical experience, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The animal and laboratory testing must meet the FDA’s good laboratory practice requirements.
Clinical trials are subject to extensive recordkeeping and reporting requirements. Our clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. We, the FDA or the IRB may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a clinical trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the U.S. Similarly, in Europe the clinical study must be approved by a local ethics committee and in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
Once a device is in commercial distribution, we or our agents are subject to ongoing regulatory compliance including Quality System Regulation and cGMP compliance, recordkeeping, adverse experience reporting, and conformity of promotion and advertising materials to the approved instructions for use.
Regulatory Enforcement
Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA or state authorities, which may include any of the following sanctions:
| • | warning letters, fines, injunctions, consent decrees and civil penalties; | |
| • | product recalls or market withdrawals; | |
| • | customer notifications, repair, replacement, refunds, recall or seizure of our products; | |
| • | operating restrictions, partial suspension or total shutdown of production; | |
| • | refusal to grant new regulatory approvals; |
71
Table of Contents
| • | withdrawing NDAs, BLAs, 510(k) clearance or PMA that have already been granted; and | |
| • | criminal prosecution. |
Employees
We had 32 full-time employees as of May 31, 2007, of whom 12 were engaged in executive, administrative, business development and intellectual property functions, and 20 were engaged in research, development and clinical or regulatory activities. We anticipate that we will need to recruit additional personnel to manage our expanded research and development programs and manage our planned clinical trials and regulatory applications, in accordance with our business strategy. We believe relations with our employees are generally good. None of our employees is covered by a collective bargaining agreement.
Facilities
Our current facilities are located in three leased buildings in Tucson, Arizona. One facility provides office, storage and laboratory space, is approximately 3,500 square feet, and is subject to a one-year lease at approximately $30,000 per year that terminates December 31, 2007. The second facility serves as our corporate headquarters and principal laboratory facility, is approximately 6,200 square feet, and is subject to a six-year lease at approximately $64,000 per year that terminates on October 31, 2008. This lease may be extended at our option for up to four additional six-year periods. Our headquarters facility is owned by a partnership whose beneficial owners include two of our executive officers and several of our stockholders. Our third facility is a temporary modular office space which is used for our expanded administrative staff, and is subject to amonth-to-month lease, with a minimum two-year term that expires on September 30, 2007. Our annual rent for this facility is approximately $22,000.
Legal Proceedings
From time to time, we may be involved in litigation relating to claims arising out of our operations. We are not currently subject to any material legal proceedings and are also not aware of any pending legal, arbitration or governmental proceedings against us that may have material effects on our financial position or results of operations.
72
Table of Contents
Management
Our executive officers and directors and their respective ages and positions as of June 11, 2007 are as follows:
Name | Age | Position | ||||
| Bradford A. Zakes | 41 | President, Chief Executive Officer and Director | ||||
| Greg Cobb | 37 | Chief Financial Officer, Secretary and Treasurer | ||||
| Terry Matsunaga, Ph.D. | 53 | Vice President, Research | ||||
| John McCambridge | 58 | Vice President, Sales and Marketing | ||||
| Kevin Ontiveros | 46 | Vice President, Legal Affairs and General Counsel | ||||
| Rajan Ramaswami, Ph.D. | 54 | Vice President, Product Development | ||||
| Walter Singleton | 65 | Chief Medical Officer | ||||
| Lynne E. Weissberger, Ph.D. | 59 | Vice President, Regulatory Affairs, Quality Assurance and Regulatory Compliance | ||||
| Reena Zutshi, Ph.D. | 39 | Vice President, Operations | ||||
| Richard Otto(1)(3) | 57 | Director, Chairman of the Board | ||||
| Richard Love(2)(3) | 63 | Director | ||||
| Thomas W. Pew(2)(3) | 68 | Director | ||||
| Philip Ranker(1)(3) | 47 | Director | ||||
| James M. Strickland(1)(2) | 64 | Director | ||||
| (1) | Member of the audit committee. | |
| (2) | Member of the compensation committee. | |
| (3) | Member of the nominating and corporate governance committee. |
Bradford A. Zakeshas served as our President and Chief Executive Officer since October 2006 and as a director since March 2007. From July 2006 to October 2006, Mr. Zakes served as our Chief Operating Officer, and from August 2005 to July 2006, Mr. Zakes served as our Vice President, Business Development. From December 2001 to August 2005, Mr. Zakes served as Director, Business Management at ICOS Corporation, a biotechnology company. From March 1999 to December 2001, Mr. Zakes served as President of Heart Research Centers International, a clinical research organization. Mr. Zakes holds a B.S. in Biology from Oregon State University, an M.S. degree in Toxicology from the American University and an M.B.A. from Duke University’s Fuqua School of Business.
Greg Cobbhas served as our Chief Financial Officer since April 2005. He was a co-founder and Managing Director of Catalyst Partners, LLC, a boutique merger, acquisition and business development firm, from April 2002 to April 2005. Mr. Cobb served as our interim Chief Financial Officer from October 2001 to April 2002. From July 2000 to November 2001, he was a Managing Director of the Arizona Angels Investor Network, Inc. Mr. Cobb holds a B.S. in Computer Engineering from Iowa State University and a J.D. and an M.B.A. from Arizona State University.
Terry Matsunaga, Ph.D. has served as our Vice President, Research since March 2004. From October 1999 to March 2004, he served as our Senior Director, New Product Development. Dr. Matsunaga holds an AB from the University of California, Berkeley, and a Ph.D. in Pharmaceutical Chemistry and a Pharm.D. degree in Clinical Pharmacy from the University of California, San Francisco.
John McCambridgehas served as our Vice President, Sales and Marketing since May 2006. From December 1997 to February 2006, Mr. McCambridge was the President and Chief Operating Officer of MRI Medical, a designer and manufacturer of highly engineered silicone medical devices. From December 1987 to January 1997, Mr. McCambridge was Senior Vice President of Sales and Marketing for Genzyme Tissue Repair, formerly BioSurface Technology, a developer of novel biologic therapeutics for the repair of human
73
Table of Contents
skin and cartilage tissue. Mr. McCambridge holds a B.S. in Business Administration from the University of Delaware.
Kevin Ontiveroshas served as our Vice President, Legal Affairs and General Counsel since March 2007. Prior to joining us he was employed from April 1996 to March 2007 at NPS Pharmaceuticals, Inc. a biopharmaceutical company, where he served in several positions, including Vice President Corporate Law, Associate General Counsel, Assistant Corporate Secretary and Senior Director Corporate Law. Mr. Ontiveros holds a LL.M. Taxation from the University of Florida College of Law and a J.D. from the University of Utah College of Law.
Rajan Ramaswami, Ph.D. has served as our Vice President, Product Development since March 2005. From September 2001 to February 2005, Dr. Ramaswami served as our Vice President, Research and Development, and from October 1999 to September 2001, he served as our Senior Director of Product Development. Dr. Ramaswami holds a MS/Ph.D. in Polymer Chemistry from Carnegie-Mellon University.
Walter Singletonhas served as our Chief Medical Officer since May 2006. Dr. Singleton changed his status with us from full time employee to consultant in June 2007. From August 2005 to April 2006, Dr. Singleton served as a consultant to us and other pharmaceutical and biotechnology companies through New Drug Development Services, a company that he founded in 1996 to advise companies in all areas of drug development and medical affairs. From October 2004 to July 2005, Dr. Singleton served as Vice President, Regulatory Affairs for Inovio, Inc., a biotechnology company. From October 2000 to December 2003, Dr. Singleton was Senior Vice President of New Drug Development at Chugai Pharma U.S.A. (formerly Chugai Biopharmaceuticals, Inc.) a Japanese biotechnology company. Dr. Singleton holds a Masters Degree, B.M. and a B.Ch. degree (equivalent to M.D. in the U.S.) and a Masters Degree in Animal Physiology from Oxford University Medical School.
Lynne E. Weissberger, Ph.D. has served as our Vice President, Regulatory Affairs, Quality Assurance and Regulatory Compliance since February 2006. From January 2004 to December 2005, Dr. Weissberger served as Senior Director at Myogen, Inc., a biotechnology company. From April 1996 to December 2003, Dr. Weissberger served as an Associate Director for G.D. Searle, Pharmacia and Pfizer, which are pharmaceutical companies. Dr. Weissberger holds a Ph.D. in Nutrition and Physiology from Cornell University.
Reena Zutshi, Ph.D. has served as our Vice President, Operations since October 2006. Prior to being appointed to that position she served as Vice President, Program Management from October 2005 to October 2006. From June 2001 to October 2005, Dr. Zutshi held various positions with us, including Director of Research and Development. Dr. Zutshi holds a Ph.D. in Organic Chemistry from Purdue University. She received her postdoctoral training at Yale University, Department of Chemistry.
Richard E. Ottohas served as a director since July 2004 and as Chairman of the Board of Directors since February 2006. From February 2003 to December 2006, Mr. Otto served as President and Chief Executive Officer of Corautus Genetics, Inc., a gene therapy company. Mr. Otto founded Clique Capital, a venture capital company, in January 1999, where he was employed until January 2002. Mr. Otto serves on the board of directors of Medi-Hut Co., Inc. Mr. Otto holds a B.S. in Chemistry and Zoology from the University of Georgia and engaged in graduate studies in Biochemistry at Medical College of Georgia.
Richard L. Lovehas served as a director since March 2006. From January 2005 to January 2006 Mr. Love served as Managing Director of TGEN Accelerator LLC for his employer Translational Genomics Research Institute. From January 2003 to January 2005, Mr. Love served as Chief Operating Officer for Translational Genomics Research Institute, from January 2002 to January 2003 Mr. Love served as a director of Parexel International, a pharmaceutical services company, and ILEX Oncology, Inc., a biotechnology company evaluating cancer therapeutics, and from June 1993 to January 2002 Mr. Love served as Chief Executive Officer and a director of ILEX Oncology, Inc. Mr. Love also serves as a director for Parexel International, Systems Medicine Inc., Medical Consultant Services, Xilas Medical and Molecular Profiling Institute. Mr. Love holds B.S. and M.S. degrees in Chemical Engineering from the Virginia Polytechnic Institute.
74
Table of Contents
Thomas W. Pewhas served as a director since January 2004. Since 1994, Mr. Pew has been a private investor in formative-stage biotechnology companies and currently serves as a director for AGF Pharma. He holds a B.A. in Economics from Cornell University.
Philip Rankerhas served as a director since February 2006. Since August 2004, Mr. Ranker has served as the Chief Financial Officer and Vice President of Finance of Nastech Pharmaceutical Company, Inc. From September 2001 to August 2004, Mr. Ranker served as Director of Finance for ICOS Corporation. From July 1998 to December 2000, Mr. Ranker served as Assistant Controller of Scholastic Corporation. Mr. Ranker holds a B.A. in Accounting from the University of Kansas.
James M. Stricklandhas served as a director since August 2000. Since February 2004, Mr. Strickland has served as the Chief Executive Officer of Thayer Medical Corporation, a medical device company. Since March 1998, Mr. Strickland has served as the General Partner and Managing Director of the Coronado Venture Funds, a group of venture investing partnerships formed in 1988. Mr. Strickland holds B.S. and M.S. degrees in Electrical Engineering from the University of New Mexico and an M.S. in Industrial Administration from Carnegie Institute of Technology (now Carnegie-Mellon University).
Board Composition
Our board of directors is currently composed of six members, all of whom are non-employee members other than Bradford A. Zakes, our President and Chief Executive Officer. There is currently one vacant seat on our board of directors. Upon completion of this offering, our bylaws will be amended and restated to provide that the authorized number of directors may be changed only by resolution of the board of directors.
We believe that the composition of our board of directors meets the requirements for independence under the current requirements of The NASDAQ Capital Market. As required by The NASDAQ Capital Market, we anticipate that our independent directors will meet in regularly scheduled executive sessions at which only independent directors are present. We intend to comply with any governance requirements that are or become applicable to us.
Committees of the Board of Directors
Our board of directors has an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and responsibilities described below.
Audit Committee
Our audit committee is composed of Richard Otto, James Strickland and Philip Ranker, each of whom is a non-employee member of our board of directors. Mr. Otto is the chairperson of the audit committee. Our board of directors has determined that each of Messrs. Otto, Strickland and Ranker is an “audit committee financial expert” as defined under SEC rules and regulations. We believe that the composition of our audit committee meets the requirements for independence and financial sophistication under the current requirements of The NASDAQ Capital Market and SEC rules and regulations. In addition, our audit committee has the specific responsibilities and authority necessary to comply with the current requirements of The NASDAQ Capital Market and SEC rules and regulations.
Our audit committee is responsible for, among other things, overseeing the independent auditors, reviewing the financial reporting, policies and processes, overseeing risk management, related party transactions and legal compliance and ethics and preparing the audit committee reports required by SEC rules.
Compensation Committee
Our compensation committee is composed of James Strickland, Thomas Pew and Richard Love, each of whom is a non-employee member of our board of directors. James Strickland is the chairperson of the compensation committee. We believe that the composition of our compensation committee meets the
75
Table of Contents
requirements for independence under the current requirements of The NASDAQ Capital Market and SEC rules and regulations.
Our compensation committee is responsible for, among other things, reviewing and recommending compensation and annual performance objectives and goals for our Chief Executive Officer, reviewing and making recommendations to the board of directors regarding incentive-based or equity-based compensation plans, employment agreements, severance arrangements, change in control agreements and other benefits, compensations, compensation policies or arrangement for other executive officers and preparing the compensation committee reports required by SEC rules.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee is composed of Richard Otto, Richard Love, Thomas Pew and Philip Ranker. Mr. Otto is the chairperson of the nominating and corporate governance committee. We believe that the composition of our nominating and corporate governance committee meets the requirements for independence under the current requirements of The NASDAQ Capital Market.
Our nominating and corporate governance committee is responsible for, among other things, identifying, evaluating and recommending individuals qualified to become directors, reviewing and making recommendations to the board of directors regarding board of director and committee compensation, committee composition and reviewing compliance with corporate governance principles applicable to our company.
Code of Conduct
Our board of directors has adopted a code of conduct that establishes the standards of ethical conduct applicable to all directors, officers and employees of our company. The code addresses, among other things, conflicts of interest, compliance with disclosure controls and procedures and internal control over financial reporting, corporate opportunities and confidentiality requirements. The audit committee is responsible for applying and interpreting our code of conduct.
Compensation Committee Interlocks and Insider Participation
None of our executive officers currently serves, or served during 2006, on the compensation committee or board of directors of any other entity that has one or more executive officers serving as a member of our board of directors or compensation committee. Prior to establishing the compensation committee, our full board of directors made decisions relating to compensation of our executive officers. No member of our compensation committee has ever been an officer or employee of the company.
Compensation Discussion and Analysis
Role of Board of Directors and Compensation Committee
Our compensation policy is set by the board of directors, with the advice and recommendation of the compensation committee. The board has delegated to the compensation committee the responsibility for implementing, reviewing and continually monitoring adherence with our compensation policy. The compensation committee is responsible for establishing, reviewing, approving and recommending to the board for final approval each of our compensation programs. In addition, the compensation committee is responsible for ensuring that the total compensation paid to our executive officers, including the named executive officers referenced in the Summary Compensation Table below, is consistent with our compensation policy and objectives.
Compensation Policy and Objectives
Our compensation policy consists of three fundamental objectives: (1) to attract, retain and motivate talented and dedicated executives and employees; (2) to align cash incentives and a portion of stock option
76
Table of Contents
vesting to achievement of specified individual and company performance objectives; and (3) to align our executives’ and employees’ interests with those of our stockholders by rewarding short-term and long-term performance. To accomplish our compensation objectives, the compensation committee works together with the chief executive officer and chief financial officer to establish key strategic goals. These goals include developing our product candidates, establishing and maintaining key strategic relationships, managing our cash position and growing our business, as measured by metrics such as achievement of clinical development, regulatory or other milestones by targeted dates, the achievement of specified revenue, cash burn and year-end cash targets.
Elements and Setting of Compensation
We have not retained a compensation consultant to advise us with respect to executive compensation or our compensation programs. Our compensation committee conducts an annual review of the aggregate level of compensation for each of our executives, as well as the principal elements comprising their total compensation. This review is based on input from members of our board of directors and publicly available data relating to the compensation practices and policies of other companies within and outside our industry, while at the same time taking into consideration the geographic location of our company and our financial resources. We believe that gathering this information is an important part of our compensation-related decision-making process. Our compensation committee evaluates both individual executive performance and corporate performance with the goal of setting compensation at levels the committee and the board of directors believe are comparable to executives in other companies of similar size and stage of development operating in the biopharmaceutical industry while taking into account our relative performance, our own strategic goals and our financial metrics at the time.
Executive compensation consists of the following elements:
Base Salary. Base salaries for our executives are established based on the scope of their responsibilities, taking into account compensation paid by other companies in our industry and in our region for similar positions, and our cash resources available for the payment of salaries to executives. Base salaries are reviewed annually, and adjusted from time to time to realign salaries with market levels after taking into account individual responsibilities, performance and experience. For 2007, this review occurred in the first quarter. In general, our executives’ base salaries are below market rates. In addition, nearly all of our employees are eligible for an increase of up to 5% in their base salary each calendar year, based upon their achievement of individual performance goals and the results of a skills assessment conducted annually.
When setting salaries for our executives in 2007, our compensation committee reviewed a salary survey produced by Equilar, Inc. that compared our executive salaries against a group of 33 publicly-traded biotechnology and biopharmaceutical companies that were chosen based on their market capitalization and number of employees. The companies used in this analysis are companies against which we believe we compete for both talent and for stockholder investment. The companies included in the survey were:
| • Aastrom Biosciences, Inc. | • Entremed, Inc. | • Northfield Laboratories Inc. | ||||||
| • Aclara Biosciences, Inc. | • Genta Incorporated | • Novavax, Inc. | ||||||
| • Advanced Magnetics, Inc. | • GTx, Inc. | • Palatin Technologies, Inc. | ||||||
| • Anika Therapeutics, Inc. | • Hemispherx Biopharma, Inc. | • Panacos Pharmaceuticals, Inc. | ||||||
| • Avant Immunotherapeutics, Inc. | • Hollis-Eden Pharmaceuticals, Inc. | • Peregrine Pharmaceuticals, Inc. | ||||||
| • Avigen, Inc | • Inkine Pharmaceutical Company, Inc. | • Sangamo Biosciences, Inc. | ||||||
| • Barrier Therapeutics, Inc. | • Insite Vision Incorporated | • Solexa, Inc. | ||||||
| • Biocryst Pharmaceuticals, Inc. | • Insmed Incorporated | • Sonus Pharmaceuticals, Inc. | ||||||
| • Bioenvision, Inc. | • Kosan Biosciences Incorporated | • StemCells, Inc | ||||||
| • Collateral Therapeutics, Inc. | • Micromet, Inc. | • Tercica, Inc. | ||||||
| • Depomed, Inc. | • Nitromed, Inc. | • Trimeris, Inc. |
77
Table of Contents
Quarterly Cash Bonus. In April 2007 we adopted a quarterly cash incentive bonus plan for all of our employees, including our executive officers. The quarterly cash incentive bonuses are intended to compensate employees for the achievement of both company-wide and departmental quarterly goals. Amounts payable under the quarterly cash incentive bonus plan are calculated as a percentage of the applicable employee’s base salary with the attainable bonus ranging from 5% up to a maximum of 12.5% of the employee’s base salary per quarter, with more senior employees being eligible for bonuses based on higher percentages of their base salaries. The company-wide targets and the departmental objectives are given roughly equal weight in the bonus analysis. The company-wide targets generally include achievement of clinical trial milestones, completion of strategic or partnering transactions, completion of financing transactions, achieving sales targets and advancing in our research programs. Departmental objectives are necessarily tied to the particular area of responsibility of the department and the department’s performance in attaining those objectives relative to external forces, internal resources utilized and overall departmental group effort. The compensation committee determines whether the quarterly cash incentive awards, if any, for all employees were earned for a particular quarter.
Options. Our 2000 Stock Plan authorizes us to grant options to purchase shares of common stock to our employees, directors and consultants. Our board of directors administers the stock option plan. Stock option grants are made at the commencement of employment and, occasionally, following a significant change in job responsibilities or to meet other special retention or performance objectives. The compensation committee recommends to our board of directors for approval stock option awards to executive officers based upon a review of competitive compensation data, its assessment of individual performance, a review of each executive’s existing long-term incentives, and retention considerations. Periodic stock option grants are made at the discretion of the board of directors to eligible employees and, in appropriate circumstances, the compensation committee and the board of directors considers the recommendations of members of management, including our chief executive officer and our chief financial officer. In 2006, certain named executive officers were awarded stock options in the amounts indicated in the section entitled “Grants of Plan Based Awards.” These options were awarded for purposes of retention and to provide continued incentive for the recipients, and were not tied to the recipients’ achievement of specific milestones or objectives. In addition, our board of directors has approved granting options to purchase an aggregate of 226,655 shares to our employees effective and contingent upon the closing of this offering. Historically, stock options granted by us are immediately exercisable, have an exercise price equal to the fair market value of our common stock on the day of grant, typically vest 25% per annum based upon continued employment over a four-year period, and generally expire ten years after the date of grant. The options granted upon the closing of this offering will have an exercise price equal to the price per share in this offering. Incentive stock options also include certain other terms necessary to assure compliance with the Internal Revenue Code of 1986, as amended.
2007 Performance Incentive Plan. Our 2007 Performance Incentive Plan authorizes us to grant incentive stock options, non-qualified stock options, stock appreciation rights, restricted stock awards, restricted stock units, performance shares and units, deferred compensation awards, other cash-based or stock-based awards and non-employee director awards. Our board of directors will administer the plan. Similar to our 2000 Stock Plan, we expect that stock option awards will be made at the commencement of employment and, occasionally, following a significant change in job responsibilities or to meet other special retention or performance objectives. The compensation committee will review and recommend to the board of directors for approval stock option awards to executive officers based upon a review of competitive compensation data, its assessment of individual performance, a review of each executive’s existing long-term incentives, and retention considerations. Periodic stock option awards will continue to be made at the discretion of the board of directors to eligible employees and, in appropriate circumstances, the compensation committee and the board of directors will consider the recommendations of members of management, as discussed above. Our 2007 Performance Incentive Plan will become effective upon the signing of the underwriting agreement for this offering. Following the closing of this offering and the issuance of the options in connection therewith, we will cease to grant additional stock options under our 2000 Stock Plan.
Other Compensation. Our executive officers who were parties to employment agreements prior to this offering will continue, following this offering, to be parties to such employment agreements in their current
78
Table of Contents
form until such time as the compensation committee determines in its discretion that revisions to such employment agreements are advisable. In addition, consistent with our compensation philosophy, we intend to continue to maintain our current benefits and perquisites for our executive officers; however, the compensation committee in its discretion may revise, amend or add to the officers’ executive benefits and perquisites if it deems it advisable. We believe these benefits and perquisites are currently lower than median competitive levels for comparable companies. We currently have no plans to change either the employment agreements or levels of benefits and perquisites provided thereunder.
Role of Executive Officers in Compensation Decisions
Bradford A. Zakes, our President and Chief Executive Officer, Greg Cobb, our Chief Financial Officer, and Kevin Ontiveros, our General Counsel, generally attend all meetings of the compensation committee. Generally, all compensation committee meetings include an executive session at which no company executives are present. Mr. Zakes and Mr. Cobb work together with our compensation committee to develop total compensation recommendations for our named executive officers other than themselves. Mr. Zakes and Mr. Cobb do not participate with the compensation committee in the determination of their respective total compensation. After review and discussion as to the appropriate level of compensation for our employees and executives, the compensation committee will render a final recommendation of the committee for total compensation and then submits its recommendation to our board of directors for consideration and approval.
Impact of Accounting and Tax Treatment
Deductibility of Compensation. Section 162(m) of the Internal Revenue Code limits the deductibility for federal income tax purposes of certain compensation paid in any year by a publicly held corporation to its chief executive officer and its four other most highly compensated officers to $1 million per executive. The $1 million cap does not apply to “performance-based” compensation as defined under Section 162(m) or to compensation paid pursuant to certain plans that existed prior to a corporation becoming publicly held. We believe that awards made under our 2000 Stock Plan and 2007 Performance Incentive Plan will not be subject to the $1 million cap because we will not become a public company until this offering closes. Once theprivate-to-public transition rule is not available, we intend that awards made under our 2007 Performance Incentive Plan will qualify as “performance-based” compensation for purposes of Section 162(m). We believe we can continue to preserve related federal income tax deductions, although individual exceptions may occur.
Accounting for Share-Based Compensation. On January 1, 2006, we adopted the fair value recognition provisions of Statement of Financial Accounting Standards No. 123(R) “Share-Based Payment,” or SFAS 123(R), to account for all stock grants under all of our plans. Prior to adoption of SFAS 123(R), we accounted for share-based compensation pursuant to Accounting Principles Board Opinion No. 25, or APB 25,“Accounting for Stock Issued to Employees.” Due to our prior accounting practices and transition provisions of SFAS 123(R), options granted prior to adopting of SFAS 123(R) retain their prior accounting treatment and previously deferred share-based compensation continues to be recognized on pre-adoption grants.
79
Table of Contents
SUMMARY COMPENSATION TABLE
The following table sets forth information regarding compensation earned by our former Chief Executive Officer, our current Chief Executive Officer, our Chief Financial Officer and our three other most highly compensated executive officers for the fiscal year ended December 31, 2006. We refer to these officers collectively as our “named executive officers.” The compensation described in this table does not include medical, group life insurance or other benefits that are available generally to all of our salaried employees.
2006 Summary Compensation Table
| Non-Equity | ||||||||||||||||||||||||
| Option | Incentive Plan | All Other | ||||||||||||||||||||||
| Salary | Awards(1) | Compensation(2) | Compensation | Total | ||||||||||||||||||||
Name and Principal Position | Year | ($) | ($) | ($) | ($) | ($) | ||||||||||||||||||
| Evan Unger, M.D. | 2006 | 251,936 | — | — | — | 251,936 | ||||||||||||||||||
| President and Chief Executive Officer(3) | ||||||||||||||||||||||||
| Bradford A. Zakes | 2006 | 165,001 | 330,950 | 28,600 | — | 524,551 | ||||||||||||||||||
| President and Chief Executive Officer(4) | ||||||||||||||||||||||||
| Greg Cobb | 2006 | 164,420 | 263,327 | — | — | 427,747 | ||||||||||||||||||
| Chief Financial Officer, Secretary and Treasurer | ||||||||||||||||||||||||
| Rajan Ramaswami, Ph.D. | 2006 | 125,002 | 12,001 | 22,438 | — | 159,441 | ||||||||||||||||||
| Vice President, Product Development | ||||||||||||||||||||||||
| Walter Singleton | 2006 | 148,844 | 648,386 | 18,019 | 52,015 | (5) | 867,264 | |||||||||||||||||
| Chief Medical Officer | ||||||||||||||||||||||||
| Lynne Weissberger, Ph.D. | 2006 | 168,633 | 317,210 | 24,750 | 16,154 | (6) | 526,747 | |||||||||||||||||
| Vice President, Regulatory Affairs, Quality Assurance and Regulatory Compliance | ||||||||||||||||||||||||
| (1) | The amounts in this column represent the compensation expense recognized in 2006 related to stock option awards pursuant to SFAS No. 123(R). A discussion of the valuation assumptions used to determine the expense is included in Note 2 of our audited financial statements filed as part of this prospectus. |
| (2) | The amounts shown in this column constitute the cash bonuses made to each named executive officer based on the attainment of certain pre-established performance criteria established by our board of directors. These awards are discussed in further detail under “Incentive Plan” below. |
| (3) | Dr. Unger resigned as our President and Chief Executive Officer in October 2006, and resigned as a director and chairman of our Scientific Advisory Board in May 2007. |
| (4) | Mr. Zakes was named our President and Chief Executive Officer in October 2006. |
| (5) | Reflects consulting fees of $42,500 paid to Dr. Singleton prior to his employment with us in May 2006, and relocation expenses in the amount of $9,515. |
| (6) | Reflects consulting fees of $10,659 paid to Dr. Weissberger prior to her employment with us in February 2006, and relocation expenses in the amount of $5,495. |
Employment and Related Agreements
We currently have an employment agreement with our President and Chief Executive Officer, Bradford A. Zakes, and our Chief Financial Officer, Greg Cobb. In addition, during 2006 we had employment agreements with our former President and Chief Executive Officer, Evan C. Unger, M.D. Dr. Unger voluntarily terminated his employment with us in October 2006, and we entered into a consulting agreement and a separation agreement with Dr. Unger following termination of his employment. Dr. Unger voluntarily
80
Table of Contents
terminated his consulting agreement in May 2007. Material terms of each of these agreements are described below.
Bradford A. Zakes
We have entered into an employment agreement with Bradford A. Zakes, our President and Chief Executive Officer. Under this agreement, Mr. Zakes is entitled to an annual base salary of $225,000 which, although reviewed annually, may not be decreased without Mr. Zakes’ consent. Mr. Zakes is also eligible for an annual bonus of up to 50% of his base salary based upon the achievement of pre-determined milestones set by our board of directors. In addition, we agreed to grant Mr. Zakes options to purchase up to 50,000 shares of our common stock upon achievement of certain milestones relating to enrollment in a clinical trial, completion of a strategic partnering transaction and completion of financing transactions in 2007 and 2008. In the event that we terminate Mr. Zakes’ employment without cause, or if he resigns for good reason, we will be obligated to continue to pay Mr. Zakes his full base salary for a six-month period and his outstanding options will vest with respect to an additional twelve months. If Mr. Zakes’ employment is terminated without cause or if he resigns for good reason during the twelve months preceding or following a change in control transaction, we will make a lump sum payment to Mr. Zakes equal to 50% of his then current base salary and all of his outstanding options will vest immediately. Receipt of any severance payment is contingent on signing a full general release of all claims. The agreement, including the continued receipt of any severance payments, requires Mr. Zakes to abide by restrictive covenants relating to non-solicitation, non-hire and non-competition for one year following termination of employment.
Greg Cobb
We have entered into an employment agreement with Greg Cobb, our Chief Financial Officer. Under this agreement, Mr. Cobb was originally entitled to an annual base salary of $175,000 which, although reviewed annually, may not be decreased without Mr. Cobb’s consent. In April 2007, Mr. Cobb’s salary was increased to $200,000. Mr. Cobb is also eligible for an annual bonus of up to 50% of his base salary based upon the achievement of pre-determined milestones set by our board of directors. In addition, we agreed to grant Mr. Cobb options to purchase up to 50,000 shares of our common stock upon achievement of certain milestones relating to enrollment in a clinical trial, completion of a strategic partnering transaction and completion of financing transactions in 2007 and 2008. In the event that we terminate Mr. Cobb’s employment without cause, or if he resigns for good reason, we will be obligated to continue to pay Mr. Cobb his full base salary for a six-month period and his outstanding options will vest with respect to an additional twelve months. If Mr. Cobb’s employment is terminated without cause or if he resigns for good reason during the twelve months preceding or following a change in control transaction, we will make a lump sum payment to Mr. Cobb equal to 50% of his then current base salary and all of his outstanding options will vest immediately. Receipt of any severance payment is contingent on signing a full general release of all claims. The agreement, including the continued receipt of any severance payments, requires Mr. Cobb to abide by restrictive covenants relating to non-solicitation, non-hire and non-competition for one year following termination of employment.
Evan C. Unger, M.D.
We entered into an employment agreement with Dr. Unger, our former President and Chief Executive Officer. Under this agreement, Dr. Unger was entitled to an annual base salary of $250,000. Dr. Unger was also eligible to receive an annual bonus award of up to 50% of his base salary, payable quarterly based on annual pre-determined milestones agreed upon by our board of directors and Dr. Unger. In addition, the employment agreement retained the vesting schedule for options previously granted to Dr. Unger. We paid Dr. Unger base salary and bonuses in the aggregate amount of $251,936 in 2006 pursuant to the terms of his employment agreement. Dr. Unger resigned as our President and Chief Executive Officer in October 2006 and entered into a consulting agreement with us, pursuant to which he agreed to serve as the head of our Scientific Advisory Board and to provide other services to us on an as-needed basis, up to a maximum of 35 hours per month. The consulting agreement had a12-month term which originally expired in October 2007 and entitled Dr. Unger to receive an aggregate amount of up to $50,000 payable on a monthly basis upon submission of
81
Table of Contents
appropriate invoices. Pursuant to the consulting agreement, all of the stock options previously granted to Dr. Unger continued to vest in accordance with their original terms during the term of the consulting agreement. We and Dr. Unger mutually agreed to terminate the consulting agreement in May 2007 upon Dr. Unger’s resignation from our board of directors and as chairman of our Scientific Advisory Board. As a result, all outstanding nonvested options have expired, and vested options to purchase 21,250 shares of common stock remain exercisable for three months following the date of his resignation. In addition, in October 2006 we entered into a separation agreement with Dr. Unger following his resignation, pursuant to which we paid Dr. Unger, in exchange for his broad waiver and release of claims against us, the aggregate amount of $250,000.
Incentive Plan
In August 2006, our board of directors adopted an incentive plan, that allowed our executive officers and other senior officers designated by our independent directors to earn quarterly and annual performance-based cash awards in addition to their annual base salary. The quarterly and annual performance goals included achievement of clinical trial milestones, completion of financing transactions, and product development milestones. The maximum annual cash award amount that could have been earned by each individual under the incentive plan was equal to 10% of the individual’s base salary as of the end of each calendar quarter with respect to which a cash award was being determined, and an additional 10% of the individual’s base salary as of the end of the fiscal year, for an aggregate possible annual cash award of up to 50% of the individual’s base salary. The incentive plan terminated on March 31, 2007.
GRANTS OF PLAN-BASED AWARDS
The compensation committee approved awards under our 2000 Stock Plan to certain of our named executive officers in 2006. Our compensation committee has not established guidelines for the grant of plan-based awards for 2007. Set forth below is information regarding awards granted during 2006 to our named executive officers:
2006 Grants of Plan-Based Awards
| Estimated Future | ||||||||||||||||
| Payouts Under | ||||||||||||||||
| Equity Incentive | Exercise Price of | Grant Date Fair | ||||||||||||||
| Plan Awards Target | Option Awards | Value of Option | ||||||||||||||
Name | Grant Date | (#) | ($/Sh) | Awards ($)(1) | ||||||||||||
| Bradford A. Zakes | 12/12/2006 | 30,333 | $ | 15.00 | 205,322 | |||||||||||
| Greg Cobb | 5/16/2006 | 12,000 | $ | 25.00 | 136,293 | |||||||||||
| 12/12/2006 | 4,000 | $ | 15.00 | 27,075 | ||||||||||||
| Rajan Ramaswami, Ph.D. | 12/12/2006 | 1,100 | $ | 15.00 | 7,446 | |||||||||||
| Walter Singleton | 5/16/2006 | 35,000 | $ | 25.00 | 397,522 | |||||||||||
| 12/12/2006 | 700 | $ | 15.00 | 4,738 | ||||||||||||
| Lynne Weissberger, Ph.D. | 2/1/2006 | 20,000 | $ | 20.00 | 180,575 | |||||||||||
| 12/12/2006 | 2,400 | $ | 15.00 | 16,245 | ||||||||||||
| (1) | The amounts in this column represent the fair value of the options granted in 2006 in accordance with SFAS No. 123(R). A discussion of the valuation assumptions used to determine the fair value is included in Note 2 of our audited financial statements filed as part of this prospectus. |
Each stock option may be exercised prior to vesting, subject to repurchase by us at the original exercise price. The repurchase right lapses at the rate of 25% per year over four years. Under certain circumstances in connection with a change of control, the vesting of the option grants may accelerate and become fully vested.
Each stock option was granted with an exercise price equal to or greater than the fair market value of our common stock on the grant date, as determined by our board of directors. Because there was no public market for our common stock prior to this offering, our board of directors determined the fair market value of our
82
Table of Contents
common stock by considering a number of factors, including, but not limited to, aggregate liquidation preference of our preferred stock, status of product development, our financial condition and prospects for future growth.
Benefit Plans
2000 Stock Plan
Our board of directors adopted, and our stockholders approved, our 2000 Stock Plan on January 12, 2000. We have reserved 1,000,000 shares of common stock for issuance under the plan. As of May 31, 2007, options to purchase 550,959 shares of our common stock were outstanding under our 2000 Stock Plan. Under certain circumstances, shares underlying awards granted under the plan may again be available for issuance under the plan. Following the closing of this offering the 2000 Stock Plan will terminate and no additional options will be granted thereunder. Although the 2000 Stock Plan will terminate, all outstanding options will continue to be governed by the 2000 Stock Plan and their existing terms.
Our board of directors administers our 2000 Stock Plan. Our board of directors has the authority to interpret the plan and any agreement entered into under the plan, grant awards and make all other determinations for the administration of the plan. Under our 2000 Stock Plan, our board of directors can grant stock options and stock purchase rights to our employees, consultants and directors.
Our 2000 Stock Plan provides for the grant of both incentive stock options that qualify for favorable tax treatment under Section 422 of the Internal Revenue Code for their recipients and nonqualified stock options. Incentive stock options may be granted only to our employees. The exercise price of incentive stock options must be at least equal to the fair market value of our common stock on the date of grant. The exercise price of incentive stock options granted to 10% stockholders must be at least equal to 110% of the fair market value of our common stock on the date of grant. Nonstatutory stock options granted under the 2000 Stock Plan must have an exercise price not less than 85% of the fair market value of our common stock on the date of grant. Nonstatutory stock options granted under the 2000 Stock Plan to 10% stockholders must have an exercise price not less than 110% of the fair market value of our common stock on the date of the grant. Generally, options vest 25% per year. Upon termination of an option holder’s service with us, he or she may exercise his or her vested options for the period of three months from the termination of service; provided, if termination is due to death or disability, the option will remain exercisable for 12 months after such termination. However, the option may never be exercised later than the expiration of its term. The maximum permitted term of options granted under our 2000 Stock Plan is ten years and the maximum term of options granted to 10% stockholders is five years. Our standard form of option agreement also allows for the early exercise of options. All options exercised early are subject to repurchase by us at the original exercise price. The repurchase right lapses over time, generally at a rate of 25% per year over four years from the date the options are granted. Our 2000 Stock Plan also provides for the grant of stock purchase rights.
In the event we merge with or into another corporation, or sell substantially all of our assets, our 2000 Stock Plan provides that options and stock purchase rights held by current employees, directors and consultants that are not assumed or substituted will immediately vest in full and become exercisable prior to the transaction and all options and stock purchase rights shall expire 15 days following notice. If there is a transaction or event which changes our stock that does not involve our receipt of consideration, such as a stock split, reverse stock split, stock dividend, combination or reclassification, or any other increase or decrease in the number of issued shares of our common stock effected without receipt of consideration by us, our 2000 Stock Plan provides that the number of shares of our common stock covered by each outstanding option and stock purchase right, and the price per share of common stock covered by each such outstanding option and stock purchase right, shall be proportionally adjusted.
2007 Performance Incentive Plan
In March 2007 our board of directors adopted, and in May 2007 our stockholders approved, our 2007 Performance Incentive Plan, or the Incentive Plan. The Incentive Plan will become effective upon the signing of the underwriting agreement for this offering. We will not issue any stock under the Incentive Plan nor grant
83
Table of Contents
any options under the Incentive Plan with an exercise price less than the price at which common stock is offered hereby until public trading of our common stock has started.
The Incentive Plan is intended to make available incentives that will assist us to attract, retain and motivate employees, consultants and members of the board of directors, whose contributions are essential to our success. We may provide these incentives through the grant of stock options, stock appreciation rights, restricted stock awards, restricted stock units, performance shares and units, deferred compensation awards, other cash-based or stock-based awards and non-employee director awards.
A total of 850,000 shares of our common stock are initially authorized and reserved for issuance under the Incentive Plan, plus up to an additional 862,047 shares that are subject to outstanding options under our 2000 Stock Plan as of the date of the plan’s termination and for which such options expire or otherwise terminate without having been exercised in full.
The administrator of our Incentive Plan will generally be the compensation committee of our board of directors, although the board of directors or compensation committee may delegate to one or more of our officers limited authority to grant awards to service providers who are neither officers nor directors. The administrator has the sole authority to construe and interpret the terms of the Incentive Plan and awards granted under it. Subject to the provisions of the Incentive Plan, the administrator has the discretion to determine the persons to whom and the times at which awards are granted, the types and sizes of such awards, and all of their terms and conditions.
Our employees and consultants are eligible to receive grants of nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock units and performance shares or units under the Incentive Plan, while only employees are eligible for incentive stock option awards. For all options granted under the Incentive Plan, the exercise price may not be less than the fair market value of a share of our common stock on the date of grant. Deferred compensation awards may be granted only to officers, directors or members of a select group of highly compensated employees. Non-employee director awards may be granted only to members of the board of directors who are not employees of the company or any affiliate of the company. Non-employee directors may be granted nonstatutory stock options, stock appreciation rights, restricted stock or restricted stock units. Non-employee director awards are limited to no more than 50,000 shares in any fiscal year.
In the event of certain changes in control of the company, stock options and stock appreciation rights outstanding under the Incentive Plan may be assumed or substituted by the successor entity. Any stock options or stock appreciation rights that are not assumed in connection with a change in control or exercised prior to a change in control will terminate without further action by the administrator. However, the administrator may choose to:
| • | accelerate the vesting and exercisability of any or all outstanding options and stock appreciation rights upon such terms as it determines; or | |
| • | cancel each or any outstanding option or stock appreciation right in exchange for a payment to the holder with respect to each share. |
In the event of a change in control, the administrator may also, in certain cases, choose to accelerate the vesting or settlement of any restricted stock award, restricted stock unit award, performance share or performance unit award, deferred compensation award, or cash-based or other stock-based award upon such conditions as it determines. In addition, the vesting of all non-employee director awards will automatically be accelerated in full upon a change in control.
The Incentive Plan will continue in effect until it is terminated by the administrator, provided, however, that all awards will be granted, if at all, within ten years of the effective date of the Incentive Plan. The administrator may amend, suspend or terminate the Incentive Plan at any time, provided, that without stockholder approval, the plan cannot be amended to increase the number of shares authorized, change the class of persons eligible to receive incentive stock options or effect any other change that would require stockholder approval under any applicable law or listing rule. Amendment, suspension or termination of the
84
Table of Contents
Incentive Plan may not adversely affect any outstanding award without the consent of the participant, unless such amendment, suspension or termination is necessary to comply with any applicable law, regulation or rule.
Other Benefits
Our employees, including our executive officers, are entitled to various employee benefits. These benefits include the following: Our employees, including our executive officers, are entitled to various employee benefits. These benefits include the following: medical and dental care plans; flexible spending accounts for healthcare; life, accidental death and dismemberment and disability insurance; a 401(k) plan; and paid time off.
401(k) Plan. We have a 401(k) profit sharing benefit plan covering substantially all of our employees who are at least twenty-one years of age and who provide a certain number of hours of service. Under the terms of the 401(k) Plan, employees may make voluntary contributions, subject to Internal Revenue Code limitations. We match 25% of the employee’s contributions up to a total of 15% of the employee’s gross salary. Our contributions to the 401(k) Plan vest equally over five years.
Outstanding Option Awards at Fiscal Year-End
The following table summarizes the outstanding stock options held by our named executive officers as of December 31, 2006.
Outstanding Equity Awards at Fiscal Year End 2006
| Option Awards | ||||||||||||
| Number of | ||||||||||||
| Securities | ||||||||||||
| Underlying | Option Exercise | |||||||||||
| Unexercised Options | Price | Option Expiration | ||||||||||
Name | (# Exercisable)(1) | ($) | Date | |||||||||
| Evan Unger, M.D. | 80,000 | $ | 15.00 | 2015 | ||||||||
| 13,000 | 20.00 | 2010 | ||||||||||
| Bradford A. Zakes | 24,000 | 15.00 | 2015 | |||||||||
| 4,000 | 20.00 | 2015 | ||||||||||
| 30,333 | 15.00 | 2016 | ||||||||||
| Greg Cobb | 30,000 | 15.00 | 2015 | |||||||||
| 9,000 | 20.00 | 2015 | ||||||||||
| 12,000 | 25.00 | 2016 | ||||||||||
| 4,000 | 15.00 | 2016 | ||||||||||
| Rajan Ramaswami, Ph.D. | 9,000 | 2.50 | 2011 | |||||||||
| 1,700 | 2.50 | 2012 | ||||||||||
| 2,000 | 2.50 | 2013 | ||||||||||
| 1,300 | 20.00 | 2015 | ||||||||||
| 1,100 | 15.00 | 2016 | ||||||||||
| Walter Singleton | 35,000 | 25.00 | 2016 | |||||||||
| 700 | 15.00 | 2016 | ||||||||||
| Lynne Weissberger, Ph.D. | 20,000 | 20.00 | 2016 | |||||||||
| 2,400 | 15.00 | 2016 | ||||||||||
| (1) | All stock options are immediately exercisable, and, when and if exercised, will be subject to a repurchase right held by us, which right lapses in accordance with the respective vesting schedules for such options. |
Option Exercises and Stock Vested
Our named executive officers did not exercise any stock options during fiscal year 2006.
85
Table of Contents
Pension Benefits
None of our named executive officers participates in or has account balances in qualified or non-qualified defined benefit plans sponsored by us.
Nonqualified Deferred Compensation
None of our named executive officers participates in or has account balances in non-qualified defined contribution plans or other deferred compensation plans maintained by us. The compensation committee, which will be comprised solely of “outside directors” as defined for purposes of Section 162(m) of the Internal Revenue Code, may elect to provide our officers and other employees with non-qualified defined contribution or deferred compensation benefits if the compensation committee determines that doing so is in our best interests.
Potential Payments Upon Termination or Change in Control
Employment Agreements. We have entered into employment agreements with each of Bradford A. Zakes, our President and Chief Executive Officer, and Greg Cobb, our Chief Financial Officer, providing for certain payments upon termination of employment or a change in control. We also entered into an employment agreement with Evan C. Unger, M.D., our former President and Chief Executive Officer. Dr. Unger resigned as our President and Chief Executive Officer in October 2006, and we agreed to pay him $250,000 pursuant to a separation agreement and up to an aggregate of $50,000 pursuant to a consulting agreement entered into at the time of Dr. Unger’s resignation. We and Dr. Unger mutually agreed to terminate the consulting agreement in May 2007 upon Dr. Unger’s resignation from our board of directors and as chairman of our Scientific Advisory Board. See “Employment and Related Agreements.”
Change in Control Provisions. Under the terms of our 2000 Stock Plan, upon a change in control the vesting of each option outstanding under such plan would accelerate in full.
Director Compensation
Each non-employee member of our board of directors receives the following compensation:
| • | $1,500 for each board and committee meeting attended in person; | |
| • | $250 for each board and committee meeting attended via teleconference; | |
| • | $15,000 annual retainer for each non-employee director payable in cash if our cash balance exceeds $10 million on the date of payment, or in stock valued at the fair market value on the date of payment; | |
| • | annual grant of an option to purchase 3,333 shares of common stock with an exercise price equal to fair market value of our common stock on the date of grant; and | |
| • | reimbursement of actual, reasonable travel expenses incurred in connection with attending board or committee meetings. |
In addition, the following additional compensation will be paid annually:
| • | $7,500 to the chairman of our audit committee; | |
| • | $2,500 to each audit committee member other than the chairman; | |
| • | $5,000 to the chairman of our compensation committee; | |
| • | $1,500 to each compensation committee member other than the chairman; | |
| • | $5,000 to the chairman of our nomination and governance committee; and | |
| • | $1,500 to each nomination and governance committee member other than the chairman. |
In addition, each non-employee director will receive a one-time grant of shares of common stock under our 2000 Stock Plan, contingent upon the closing of this offering. The number of shares to be granted to each
86
Table of Contents
non-employee director will be determined by dividing $25,000 by the offering price per share in this offering, and multiplying the resulting figure by the ratio of the number of months of service of each individual to the total number of months between our 2006 and 2007 annual stockholder meetings. One of our non-employee directors, Richard Love, will receive an additional one-time grant of shares of common stock under our 2000 Stock Plan, also contingent upon the closing of this offering, to reflect extraordinary service to our company. The number of shares to be granted to Mr. Love in this manner will be determined by dividing $50,000 by the offering price per share in this offering. Assuming an offering price of $7.00, the midpoint of the range on the front cover of this prospectus, the aggregate number of shares of restricted stock to be granted to our non-employee directors upon the closing of this offering will be 24,997 shares.
The following table sets forth a summary of the compensation we paid to our non-employee directors for the fiscal year ended December 31, 2006:
2006 Director Compensation
| Fees Earned or Paid | ||||||||||||
| in Cash | Option Awards(1) | Total | ||||||||||
Name | ($) | ($) | ($) | |||||||||
| Richard Otto | $ | 11,500 | — | $ | 11,500 | |||||||
| James M. Strickland | $ | 20,000 | — | $ | 20,000 | |||||||
| Thomas W. Pew | $ | 15,000 | — | $ | 15,000 | |||||||
| Richard Love | $ | 11,375 | $ | 125,000 | $ | 136,375 | ||||||
| Philip Ranker | $ | 9,375 | $ | 125,000 | $ | 134,375 | ||||||
| (1) | The amounts in this column represent the fair value of the options granted in 2006 in accordance with SFAS No. 123(R). A discussion of the valuation assumptions used to determine the expense is included in Note 2 of our audited financial statements filed as a part of this prospectus. |
Limitation of Liability and Indemnification
Our amended and restated certificate of incorporation, which will become effective upon the closing of this offering, limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
| • | breach of their duty of loyalty to the corporation or its stockholders; | |
| • | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| • | unlawful payment of dividends or redemption of shares; or | |
| • | transaction from which the directors derived an improper personal benefit. |
These limitations of liability do not apply to liabilities arising under federal securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our amended and restated bylaws, which will become effective upon the closing of this offering, provide that we will indemnify our directors and executive officers, and may indemnify other officers, employees and other agents, to the fullest extent permitted by law. Our amended and restated bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to us, regardless of whether our amended and restated bylaws permit such indemnification. We maintain a liability insurance policy pursuant to which our directors and officers may be indemnified against liability incurred for serving in their capacities as directors and officers.
We have entered into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated bylaws. These agreements, among other things, require us to indemnify our directors and executive officers for certain expenses, including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or executive officer in any
87
Table of Contents
action or proceeding arising out of their services as one of our directors or executive officers, or any of our subsidiaries or any other related company or enterprise to which the person provides services at our request.
We believe provisions in our new amended and restated certificate of incorporation and indemnification agreements are necessary to attract and retain qualified persons as directors and officers. The limitation of liability and indemnification provisions in our certificate of incorporation and bylaws may discourage stockholders from bringing a lawsuit against our directors for breach of their fiduciary duty. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Furthermore, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers as required by these indemnification provisions.
At present we are not aware of any pending litigation or proceeding involving any of our directors, officers, employees or agents in their capacity as such, for which indemnification will be required or permitted. In addition, we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification by any director or officer.
Certain Relationships and Related Transactions
Since January 1, 2004, we have engaged in the following transactions involving amounts exceeding $120,000 with our executive officers, directors and holders of 5% or more of our stock. We believe that all of these transactions were on terms as favorable as could have been obtained from related third parties.
Lease
We lease a 6,200 square foot facility located at 1635 E. 18th St., Tucson, Arizona 85719 as our headquarters and laboratory facility for approximately $64,000 per year. This facility is owned by a partnership whose beneficial owners include Evan Unger, a holder of 5% or more of our stock, and a former director and our former President and Chief Executive Officer; Dean Unger, father of Evan Unger and a former director; Rajan Ramaswami, our Vice President Development; and Terry Matsunaga, our Vice President Research. This lease provides for a rental rate of $10.39 per square foot per year,triple-net, and expires in October 2008.
Stock Sales
During 2004, the Evan and Susan Unger Family Trust dated October 24, 1995 purchased convertible secured promissory notes from us in the aggregate principal amount of $25,000. On March 30, 2004, the outstanding principal and accrued interest under these convertible secured promissory notes and certain other convertible promissory notes purchased prior to 2004 converted into 15,643 shares of our common stock at a conversion price of $10.00 per share. In January 2005, the Unger Family Trust also purchased 6,800 shares of our common stock at a purchase price of $15.00 per share pursuant to a private placement conducted by First Montauk Securities Corp. as placement agent. In November 2005, the Unger Family Trust also purchased 250 shares of our common stock at a purchase price of $20.00 per share pursuant to a private placement conducted by First Montauk Securities Corp. as placement agent.
On March 30, 2004, the outstanding principal amount and accrued interest under a convertible promissory note purchased prior to 2004 by Edson Moore Healthcare Ventures, Inc., an entity controlled by John Moore, our former Chairman, Executive Vice President and a former director, converted into 22,463 shares of our common stock at a conversion price of $10.00 per share. On January 30, 2005, Edson Moore Healthcare Ventures purchased 8,000 shares of our common stock pursuant to a private placement conducted by First Montauk Securities as placement agent.
We sold the convertible secured promissory notes and common stock pursuant to securities purchase agreements, under which we made standard representations, warranties, and covenants, and granted certain rights to the purchasers of these securities. The only rights that survive beyond this offering are registration rights. See “Description of Capital Stock — Registration Rights.”
88
Table of Contents
Consulting Agreements
On March 31, 2006 we entered into an agreement with Edson Moore Healthcare Ventures and John Moore, our former Chairman and Executive Vice President, that provides for the payment of $250,000 to Edson Moore Healthcare Ventures in exchange for past consulting services and future financial consulting services on an as-needed basis through March 2008. Mr. Moore has participated in numerous telephone calls and meetings with us and others in connection with the provision of consulting services under this agreement, and we expect Mr. Moore to continue to provide these services through the term of the agreement. There is no provision in the agreement that requires us or Mr. Moore to track time spent in providing consulting services, and no such records have been kept. The agreement also includes, with respect to Edson Moore Healthcare Ventures, a waiver of certain of the protective provisions of the Series B preferred stock and Series C preferred stock set forth in our charter in connection with the Series F preferred stock financing, a mutual nondisparagement provision and our agreement not to provide, after the date that our stock is traded on a national securities exchange or quoted on The Nasdaq Global Market, any non-public information to Edson Moore Healthcare Ventures. The Agreement also includes, with respect to John Moore, Mr. Moore’s resignation as a director, a mutual nondisparagement provision and our agreement not to provide, after the date that our stock is traded on a national securities exchange or quoted on The Nasdaq Global Market, any non-public information to Mr. Moore.
In October 2006, we entered into a consulting agreement with Dr. Evan Unger, who until that time was our CEO and was a member of our board of directors. Pursuant to this agreement, Dr. Unger served as the head of our Scientific Advisory Board and agreed to provide other services to us on an as-needed basis, up to a maximum of 35 hours per month. The consulting agreement had a12-month term which expired in October 2007 and entitled Dr. Unger to receive an aggregate amount of up to $50,000 payable on a monthly basis upon submission of appropriate invoices. All of the stock options previously granted to Dr. Unger continued to vest in accordance with their original terms during the term of the consulting agreement. We and Dr. Unger mutually agreed to terminate the consulting agreement in May 2007 upon Dr. Unger’s resignation from our board of directors and as chairman of our Scientific Advisory Board. In addition, we have entered into a separation agreement with Dr. Unger following his resignation as our CEO. The separation agreement contains Dr. Unger’s waiver and release of claims against us arising out of, or relating in any way to, Dr. Unger’s service as an employee, officer and/or director of ImaRx and any other matter, act, occurrence, or transaction with respect to us by or involving Dr. Unger and us in any way, including but not limited to, Dr. Unger’s ownership of shares of our capital stock, options or warrants to acquire our capital stock. Under the separation agreement, we agreed to pay Dr. Unger the aggregate amount of $250,000, half of which is payable in twelve equal installments in accordance with our regular payroll practices, and the remainder of which will be due and payable in a lump sum on the next regular payroll date thereafter. As of March 31, 2007, we had approximately $151,000 accrued expense related to this liability.
Agreements and Future Agreements with Executive Officers and Directors
We have entered into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated bylaws. See “Management — Limitation of Liability and Indemnification.”
We also have entered into employment and other agreements with some of our executive officers and former executive officers. See “Compensation Discussion and Analysis — Employment Agreements.” All future transactions between us and our officers, directors, principal stockholders and their affiliates will be approved by a majority of our board of directors, including a majority of the independent and disinterested directors in these transactions, or by a committee composed of independent directors.
89
Table of Contents
Principal Stockholders
The following table sets forth, as of May 31, 2007, information regarding beneficial ownership of our capital stock by the following:
| • | each person, or group of affiliated persons, known by us to be the beneficial owner of 5% or more of any class of our voting securities; | |
| • | each of our current directors; | |
| • | each of our named executive officers; and | |
| • | all current directors and officers as a group. |
Beneficial ownership is determined according to the rules of the SEC. Beneficial ownership means that a person has or shares voting or investment power of a security, and includes shares underlying options and warrants that are currently exercisable or exercisable within 60 days after the measurement date. This table is based on information supplied by officers, directors and principal stockholders. Except as otherwise indicated, we believe that the beneficial owners of the common stock listed below, based on the information each of them has given to us, have sole investment and voting power with respect to their shares, except where community property laws may apply.
Options and warrants to purchase shares of our common stock that are exercisable within 60 days after May 31, 2007 are deemed to be beneficially owned by the persons holding these options and warrants for the purpose of computing percentage ownership of that person, but are not treated as outstanding for the purpose of computing any other person’s ownership percentage. Unless otherwise indicated, all footnotes below the table reflect options and warrants exercisable within 60 days after May 31, 2007.
This table lists applicable percentage ownership based on 6,054,928 shares of common stock outstanding as of May 31, 2007, assuming conversion of all outstanding shares of our preferred stock into common stock, including the assumption that our Series F preferred stock converts at a 1-to-0.84 conversion ratio (see “Conversion of Series F Preferred Stock”), but assuming no exercise of outstanding warrants or options, and also lists applicable percentage ownership based on 9,054,928 shares of common stock outstanding after the closing of the offering.
90
Table of Contents
Unless otherwise indicated, the address for each of the stockholders in the table below is c/o ImaRx Therapeutics, Inc., 1635 East 18th Street, Tucson, AZ 85719.
| Shares of | ||||||||||||
| Common Stock | Percent | |||||||||||
| Beneficially | Before | After | ||||||||||
Name and Address of Beneficial Owner | Owned(1) | Offering | Offering(1) | |||||||||
5% Stockholders | ||||||||||||
| Boston Scientific Corporation(2) | 840,336 | 13.9 | % | 9.3 | % | |||||||
| One Boston Scientific Place | ||||||||||||
| Natick, MA 01760 | ||||||||||||
| ITX International Equity Corp.(3) | 504,202 | 8.3 | 5.6 | |||||||||
| c/o ITX International Holdings, Inc. 700 E. El Camino Real, Suite 200 Mountain View, CA 94040 | ||||||||||||
| Berg & Berg Enterprises, LLC(4) | 436,134 | 7.2 | 4.8 | |||||||||
| 10050 Bandley Drive Cupertino, CA 95014 | ||||||||||||
Evan and Susan Unger Family Trust dated10/24/95(5) | 411,398 | 6.8 | 4.5 | |||||||||
| John A. Moore(6) | 365,882 | 6.0 | 4.0 | |||||||||
| c/o Edson Moore Healthcare Ventures, Inc. | ||||||||||||
| 101 Brook Meadow Road Wilmington, DE 19807 | ||||||||||||
Directors and Named Executive Officers | ||||||||||||
| Richard Love(7) | 21,713 | * | * | |||||||||
| Richard Otto(8) | 14,571 | * | * | |||||||||
| Thomas W. Pew(9) | 82,586 | 1.4 | * | |||||||||
| Philip Ranker(10) | 14,571 | * | * | |||||||||
| James M. Strickland(11) | 96,666 | 1.6 | 1.1 | |||||||||
| Evan C. Unger, M.D.(12) | 473,023 | 7.8 | 5.2 | |||||||||
| Bradford A. Zakes(13) | 149,999 | 2.4 | 1.6 | |||||||||
| Greg Cobb(14) | 146,666 | 2.4 | 1.6 | |||||||||
| Rajan Ramaswami, Ph.D.(15) | 44,100 | * | * | |||||||||
| Walter Singleton(16) | 49,033 | * | * | |||||||||
| Lynne E. Weissberger(17) | 35,733 | * | * | |||||||||
| All Directors and Officers as a Group (14 persons) (18) | 765,321 | 11.3 | 7.8 | |||||||||
| * | Less than one percent (1%). |
| (1) | Upon completion of this offering, our existing stockholders will own 6,054,928 shares, representing approximately 67% of our outstanding common stock. |
| (2) | Consists of 840,336 shares of common stock. |
| (3) | Consists of 504,202 shares of common stock. |
| (4) | Consists of 436,134 shares of common stock. Mr. Carl E. Berg is the manager and a member of Berg & Berg Enterprises LLC. Mr. Berg may be deemed to have shared voting and dispositive power with respect to the shares held by such entity. |
| (5) | Consists of 406,366 shares of common stock, 3,128 shares issuable upon exercise of warrants to purchase common stock and 1,904 shares issuable upon exercise of warrants to be issued immediately following and contingent upon the completion of this offering, held by the Evan and Susan Unger Family Trust dated10/24/95, of which Dr. Unger and Susan J. Unger are co-trustees and share voting and dispositive power. |
| (6) | Consists of 14,823 shares of common stock, 526 shares issuable upon exercise of warrants to purchase common stock and 2,693 shares issuable upon exercise of warrants to be issued immediately following and contingent upon the completion of this offering, held by Mr. Moore, 266,186 shares of common stock held by Edson Moore Corp., and 25,463 shares of common stock, 4,492 shares issuable upon |
91
Table of Contents
| exercise of warrants to purchase common stock and 1,699 shares issuable upon exercise of warrants to be issued immediately following and contingent upon the completion of this offering, held by Edson Moore Healthcare Ventures, Inc. Edson Moore Corp. and Edson Moore Healthcare Ventures Inc. are corporations, of which Mr. Moore is president and/or a director, and Mr. Moore may be deemed to share voting and dispositive power over the shares held by such entities. |
| (7) | Consists of 10,713 shares of common stock to be issued to Mr. Love immediately following and contingent upon the completion of this offering and 11,000 shares of common stock issuable upon exercise of options, 8,250 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (8) | Consists of 3,571 shares of common stock to be issued to Mr. Otto immediately following and contingent upon the completion of this offering and 11,000 shares of common stock issuable upon exercise of options, 2,750 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (9) | Consists of 55,326 shares of common stock, 3,571 shares of common stock to be issued to Mr. Pew immediately following and contingent upon the completion of this offering, 3,282 shares of common stock issuable upon exercise of warrants, 9,407 shares issuable upon exercise of warrants to be issued to Mr. Pew immediately following and contingent upon the completion of this offering and 11,000 shares of common stock issuable upon exercise of options, 5,500 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (10) | Consists of 3,571 shares of common stock to be issued to Mr. Ranker immediately following and contingent upon the completion of this offering and 11,000 shares of common stock issuable upon exercise of options, 8,250 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (11) | Consists of 79,095 shares of common stock held by Coronado Venture Fund IV, LP, 2,000 shares of common stock held by Mr. Strickland, 3,571 shares of common stock to be issued to Mr. Strickland immediately following and contingent upon the completion of this offering, 11,000 shares of common stock issuable upon exercise of options held by Mr. Strickland, 5,500 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 1,000 shares issuable upon exercise of warrants to be issued to Mr. Strickland immediately following and contingent upon the completion of this offering. With regard to Coronado Venture Fund IV, LP, Coronado Venture Management LLC is the sole general partner of and may be deemed to have voting and dispositive power over shares held by Coronado Venture Fund IV, LP. Mr. Strickland is a managing director of Coronado Venture Management LLC. Mr. Strickland disclaims beneficial ownership of the shares held by Coronado Venture Fund IV, LP, except to the extent of his direct pecuniary interest therein. |
| (12) | Consists of 406,366 shares of common stock, 3,128 shares of common stock issuable upon exercise of warrants to purchase common stock and 1,904 shares issuable upon exercise of warrants to be issued immediately following and contingent upon the completion of this offering, held by Evan C. and Susan J. Unger Trust dated10/24/95, of which Dr. Unger and Susan J. Unger are co-trustees and share voting and dispositive power, 250 shares of common stock held by Evan C. Unger and Susan J. Unger and 125 shares issuable upon exercise of warrants to be issued to Evan C. Unger and Susan J. Unger immediately following and contingent upon the completion of this offering, and 40,000 shares of common stock, and 21,250 shares issuable upon exercise of stock options. |
| (13) | Consists of 58,333 shares of common stock issuable upon exercise of options, 49,833 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, 41,666 shares of common stock issuable upon exercise of options to be granted to Mr. Zakes immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 50,000 shares of common stock issuable upon exercise of options to be granted to Mr. Zakes contingent upon and following our achievement of certain corporate milestones, which options will be fully vested at the time they are granted (see “Compensation Discussion and Analysis — Employment Agreements”). |
| (14) | Consists of 55,000 shares of common stock issuable upon exercise of options, 33,250 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, 41,666 shares of common |
92
Table of Contents
| stock issuable upon exercise of options to be granted to Mr. Cobb immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 50,000 shares of common stock issuable upon exercise of options to be granted to Mr. Cobb contingent upon and following our achievement of certain corporate milestones, which options will be fully vested at the time they are granted. See “Compensation Discussion and Analysis — Employment Agreements”. |
| (15) | Consists of 19,000 shares of common stock, 15,100 shares of common stock issuable upon exercise of options, 2,575 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 10,000 shares of common stock issuable upon exercise of options to be granted to Dr. Ramaswami immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (16) | Consists of 35,700 shares of common stock issuable upon exercise of options, 23,200 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 13,333 shares of common stock issuable upon exercise of options to be granted to Dr. Singleton immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (17) | Consists of 22,400 shares of common stock issuable upon exercise of options, 13,500 of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007, and 13,333 shares of common stock issuable upon exercise of options to be granted to Dr. Weissberger immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
| (18) | Includes shares described in Footnotes (4) through (14) above, 7,084 shares of common stock held by Terry Matsunaga, 62,600 shares issuable upon exercise of stock options held by Terry Matsunaga, Reena Zutshi and John McCambridge, 33,230 of which, if exercised, are subject to repurchase within 60 days after May 31, 2007, and 39,999 shares of common stock issuable upon exercise of options to be granted to Terry Matsunaga, Reena Zutshi and John McCambridge immediately following and contingent upon the completion of this offering, all of which, if exercised, are subject to repurchase by us within 60 days after May 31, 2007. |
Description of Capital Stock
Upon the effectiveness of this offering and the filing of our amended and restated certificate of incorporation with the Delaware Secretary of State, our authorized capital stock will consist of 100,000,000 shares of common stock, $0.0001 par value per share, and 5,000,000 shares of preferred stock, $0.0001 par value per share. The following description of our capital stock does not purport to be complete and is subject to, and qualified in its entirety by, our amended and restated certificate of incorporation and bylaws, which are exhibits to the registration statement of which this prospectus forms a part.
Common Stock
As of May 31, 2007, 6,054,928 shares of our common stock were outstanding and held of record by 358 stockholders. This amount assumes the conversion of all outstanding shares of our preferred stock into common stock, which will occur immediately prior to the effectiveness of this offering. In addition, as of May 31, 2007, 550,959 shares of our common stock were subject to outstanding options, 352,324 shares of our common stock were subject to outstanding warrants. Upon the closing of this offering, 9,054,928 shares of our common stock will be outstanding, assuming no exercise of outstanding stock options or warrants or the underwriters’ over-allotment option.
Each share of our common stock entitles its holder to one vote on all matters to be voted on by our stockholders. Subject to preferences that may apply to any of our outstanding preferred stock, holders of our common stock will participate equally in all dividends payable with respect to our common stock, if and when declared by our board of directors. If we liquidate, dissolve or wind up, the holders of common stock are entitled to share ratably in all distributions of assets subject to any liquidation rights and preferences of any of
93
Table of Contents
our outstanding preferred stock. Our common stock has no preemptive rights, conversion rights or other subscription rights or redemption or sinking fund provisions. The shares of our common stock to be issued upon the closing of this offering will be fully paid and non-assessable.
Preferred Stock
After the offering, our board of directors will have the authority, without further action by our stockholders, to issue up to 5,000,000 shares of our preferred stock in one or more series. Our board of directors may designate the rights, preferences, privileges and restrictions of our preferred stock, including any qualifications, limitations or restrictions thereon. The issuance of our preferred stock could have the effect of restricting dividends on our common stock, diluting the voting power of our common stock, impairing the liquidation rights of our common stock, or delaying or preventing a change in control. Even the ability to issue preferred stock could delay or impede a change in control. Immediately after the closing of this offering, no shares of our preferred stock will be outstanding, and we currently have no plan to issue any shares of our preferred stock.
Warrants
As of May 31, 2007 the following warrants were outstanding:
| • | Warrant to purchase 2,281 shares of our common stock, at an exercise price of $13.75 per share. This warrant may be exercised at any time prior to the later of either January 16, 2011 or five years after our initial public offering. | |
| • | Warrant to purchase an aggregate of 614 shares of our common stock at an exercise price of $35.00 per share. This warrant may be exercised at any time prior to March 6, 2011. | |
| • | Warrant to purchase an aggregate of 1,000 shares of our common stock at an exercise price of $10.00 per share. This warrant may be exercised at any time prior to October 10, 2013. | |
| • | Warrants to purchase an aggregate of 37,769 shares of our common stock at an exercise price of $10.00 per share issued pursuant to our March 2003 bridge financing. These warrants may be exercised from time to time prior to January 28, 2011. | |
| • | Warrants to purchase an aggregate of 100,000 shares of our common stock at an exercise price of $15.00 per share. These warrants may be exercised at any time prior to March 28, 2009. | |
| • | Warrants to purchase an aggregate of 50,000 shares of our common stock at an exercise price of $10.00 per share. These warrants may be exercised at any prior to October 5, 2008. | |
| • | Warrant to purchase an aggregate of 4,000 shares of our common stock at an exercise price of $15.00 per share. This warrant may be exercised at any time prior to January 3, 2010. | |
| • | Warrants to purchase an aggregate of 46,664 shares of our common stock at an exercise price of $16.50 per share. These warrants may be exercised at any time prior February 27, 2010. | |
| • | Warrants to purchase an aggregate of 20,000 shares of our common stock at an exercise price of $20.00 per share. These warrants may be exercised at any time prior to September 27, 2015. | |
| • | Warrants to purchase an aggregate of 74,996 shares of our common stock at an exercise price of $21.25 per share. These warrants may be exercised at any time prior to October 6, 2012. | |
| • | Warrants to purchase an aggregate of 15,000 shares of our common stock at an exercise price of $20.00 per shares. These warrants may be exercised at any time prior to January 13, 2013. |
All of our outstanding warrants contain provisions for the adjustment of the exercise price and the number of shares issuable upon the exercise of the warrant in the event of stock dividends, stock splits, reorganizations, reclassifications and consolidations. In addition, certain of the warrants contain a net exercise provision.
94
Table of Contents
Contingent Commitments to Issue Equity Securities
As of May 31, 2007, we have made commitments to issue shares of our common stock and options and warrants to purchase shares of our common stock that are contingent upon the closing of this offering. All of such issuances and grants will be made immediately following the completion of this offering. The contingent issuances are as follows:
| • | Warrants to purchase an aggregate of 210,000 shares of our common stock at an exercise price equal to 115% of the public offering price per share in this offering. These warrants may be exercised commencing twelve months from the date of their issuance and for a period of four years thereafter. See “Underwriting.” |
| • | Warrants to purchase an aggregate of 496,589 shares of our common stock at an exercise price equal to 115% of the public offering price per share in this offering. These warrants may be exercised at any time during a period of 5 years following their issuance. | |
| • | Options to purchase an aggregate of 226,655 shares of our common stock pursuant to our 2000 Stock Plan, with an exercise price equal to the public offering price per share in this offering. | |
| • | An aggregate of 24,997 shares of our common stock pursuant our 2000 Stock Plan to our non-employee directors. See “Compensation Discussion and Analysis — Director Compensation.” |
In addition, we have made commitments to issue options to purchase up to an aggregate of 100,000 shares of our common stock pursuant to our 2007 Performance Incentive Plan to our CEO and our CFO, based upon achievement of certain milestones relating to enrollment in our clinical trial, completion of a strategic partnering transaction and completion of financing transactions 2007 and 2008. These options will have an exercise price equal to the fair market value of our common stock on the date such grants are approved by our board of directors following the closing of this offering. See “Compensation Discussion and Analysis — Employment Agreements.”
Registration Rights
At anytime after December 31, 2008, the holders of approximately 3,248,189 shares of our common stock or certain transferees and the holders of approximately 2,281 shares of common stock issuable upon the exercise of an outstanding warrant (calculated based on the assumptions set forth under “The Offering” in this prospectus) will be entitled to require us to register these shares under the Securities Act, subject to limitations and restrictions. In addition, the holders of these shares may require us, at our expense and on not more than one occasion in any twelve month period, to file a registration statement onForm S-3 under the Securities Act, if we become eligible to use such form, covering their shares of our common stock, and we will be required to use our best efforts to have the registration statement declared effective. Also, if at any time, we propose to register any of our securities under the Securities Act, either for our own account or for the account of other security holders, the holders of these shares, the holders of approximately an additional 1,923,109 shares of common stock, and the holders of approximately 807,249 shares of common stock issuable upon the exercise of outstanding warrants and warrants to be issued upon the completion of this offering, will be entitled to notice of the registration and, subject to certain exceptions, will be entitled to include, at our expense, their shares of our common stock in the registration. These rights terminate at various times between five and seven years after the closing of this offering, or, with respect to an individual holder, such time as Rule 144 or another similar exemption under the Securities Act is available for the sale of all of such holder’s shares during a three-month period without registration. These registration rights are subject to conditions and limitations, including the right of the underwriters to limit the number of shares of our common stock included in the registration statement.
Anti-Takeover Provisions
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law, which regulates, subject to some exceptions, acquisitions of publicly held Delaware corporations. In general, Section 203 prohibits us
95
Table of Contents
from engaging in a “business combination” with an “interested stockholder” for a period of three years following the date the person becomes an interested stockholder, unless:
| • | our board of directors approved the business combination or the transaction in which the person became an interested stockholder prior to the date the person attained this status; | |
| • | upon consummation of the transaction that resulted in the person becoming an interested stockholder, the person owned at least 85% of our voting stock outstanding at the time the transaction commenced, excluding shares owned by persons who are directors and also officers and issued under employee stock plans under which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| • | on or subsequent to the date the person became an interested stockholder, our board of directors approved the business combination and the stockholders other than the interested stockholder authorized the transaction at an annual or special meeting of stockholders by the affirmative vote of at least two-thirds of the outstanding stock not owned by the interested stockholder. |
Section 203 defines a “business combination” to include:
| • | any merger or consolidation involving us and the interested stockholder; | |
| • | any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of our assets; | |
| • | in general, any transaction that results in the issuance or transfer by us of any of our stock to the interested stockholder; | |
| • | any transaction involving us that has the effect of increasing the proportionate share of our stock owned by the interested stockholders; and | |
| • | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges, or other financial benefits provided by or through us. |
In general, Section 203 defines an “interested stockholder” as any person who, together with the person’s affiliates and associates, owns, or within three years prior to the time of determination of interested stockholder status did own, 15% or more of a corporation’s voting stock.
Certificate of Incorporation and Bylaw Provisions
Upon completion of this offering, our amended and restated certificate of incorporation and bylaws will include a number of provisions that may have the effect of deterring hostile takeovers or delaying or preventing changes in control or our management. These provisions include the following:
| • | our board of directors can issue up to 5,000,000 shares of preferred stock, with any rights or preferences, including the right to approve or not approve an acquisition or other change in control; |
| • | our bylaws provide that our board of directors may be removed with or without cause by the affirmative vote of a majority of our stockholders; | |
| • | our bylaws limit who may call a special meeting of stockholders to our board of directors, chairman of the board, president and one or more stockholders holding not less than 25% of all shares entitled to be cast on any issue proposed to be considered at that meeting; | |
| • | our bylaws provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide timely advance written notice to us in writing; | |
| • | our bylaws specify requirements as to the form and content of a stockholder’s notice; |
96
Table of Contents
| • | our bylaws provides that, subject to the rights of the holders of any outstanding series of our preferred stock, all vacancies, including newly created directorships, may, except as otherwise required by law, be filled by the affirmative vote of a majority of our directors then in office, even if less than a quorum; | |
| • | our bylaws provide that our board of directors may fix the number of directors by resolution; |
| • | our amended and restated certificate of incorporation provides that all stockholder actions must be effected at a duly called meeting of stockholders and not by written consent; and |
| • | our amended and restated certificate of incorporation does not provide for cumulative voting for our directors. The absence of cumulative voting may make it more difficult for stockholders owning less than a majority of our stock to elect any directors to our board. |
Transfer Agent and Registrar
Registrar and Transfer Company has been appointed as the transfer agent and registrar for our common stock.
Listing
We have applied to have our common stock listed on The NASDAQ Capital Market under the trading symbol “IMRX.”
Material U.S. Federal Tax Consequences
ToNon-U.S. Holders
ToNon-U.S. Holders
The following is a summary of material U.S. federal income and estate tax consequences of the ownership and disposition of our common stock by anon-U.S. holder. For purposes of this discussion, anon-U.S. holder is any beneficial owner that for U.S. federal income tax purposes is not a U.S. person; the term U.S. person means:
| • | an individual citizen or resident of the U.S.; | |
| • | a corporation or other entity taxable as a corporation created or organized in the U.S. or under the laws of the U.S. or any political subdivision thereof; | |
| • | an estate whose income is subject to U.S. federal income tax regardless of its source; or | |
| • | a trust (x) whose administration is subject to the primary supervision of a U.S. court and which has one or more U.S. persons who have the authority to control all substantial decisions of the trust or (y) which has made an election to be treated as a U.S. person. |
An individual may, in certain cases, be treated as a resident of the U.S., rather than a nonresident, among other ways, by virtue of being present in the U.S. on at least 31 days in that calendar year and for an aggregate of at least 183 days during the three-year period ending in that calendar year (counting for such purposes all the days present in the current year, one-third of the days present in the immediately preceding year and one-sixth of the days present in the second preceding year). Residents are subject to U.S. federal income tax as if they were U.S. citizens.
If a partnership, a pass-through entity treated as a partnership for U.S. federal income tax purposes, or an entity treated as a disregarded entity for U.S. federal income tax purposes holds common stock, the tax treatment of an owner of such entity will generally depend on the status of the owner and upon the activities of the entity. Accordingly, we urge such entities which hold our common stock and owners in these entities to consult their tax advisors.
This discussion assumes thatnon-U.S. holders will acquire our common stock pursuant to this offering and will hold our common stock as a capital asset (generally, property held for investment). This discussion does not address all aspects of U.S. federal income taxation that may be relevant in light of anon-U.S. holder’s special tax status or special tax situations. U.S. expatriates, controlled foreign corporations, passive foreign
97
Table of Contents
investment companies, corporations that accumulate earnings to avoid federal income tax, life insurance companies, tax-exempt organizations, dealers in securities or currencies, brokers, banks or other financial institutions, certain trusts, hybrid entities, pension funds and investors that hold common stock as part of a hedge, straddle or conversion transaction are among those categories of potential investors that are subject to special rules not covered in this discussion. This discussion does not consider the tax consequences for partnerships, entities classified as a partnership for tax purposes, or persons who hold their interests through a partnership or other entity classified as a partnership for U.S. federal income tax purposes. This discussion does not address any U.S. federal gift tax consequences, or state or local ornon-U.S. tax consequences. Furthermore, the following discussion is based on current provisions of the Internal Revenue Code of 1986, as amended, and Treasury Regulations and administrative and judicial interpretations thereof, all as in effect on the date hereof, and all of which are subject to change, possibly with retroactive effect.
Dividends
We do not plan to pay any dividends on our common stock for the foreseeable future. However, if we do pay dividends on our common stock, those payments will constitute dividends to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those dividends exceed our current and accumulated earnings and profits, the dividends will constitute a return of capital and will first reduce a holder’s basis, but not below zero, and then will be treated as gain from the sale of stock.
The gross amount of any dividend (out of earnings and profits) paid to anon-U.S. holder of common stock generally will be subject to U.S. withholding tax at a rate of 30% unless the holder is entitled to an exemption from or reduced rate of withholding under an applicable income tax treaty. To receive a reduced treaty rate, prior to the payment of a dividend anon-U.S. holder must provide us with a properly completed IRSForm W-8BEN (or successor form) certifying qualification for the reduced rate.
Dividends received by anon-U.S. holder that are effectively connected with a U.S. trade or business conducted by thenon-U.S. holder (or dividends attributable to anon-U.S. holder’s permanent establishment in the U.S. if an income tax treaty applies) are exempt from this withholding tax. To obtain this exemption, prior to the payment of a dividend anon-U.S. holder must provide us with a properly completed IRSForm W-8ECI (or successor form) properly certifying this exemption. Effectively connected dividends (or dividends attributable to a permanent establishment), although not subject to withholding tax, are subject to U.S. federal income tax at the same graduated rates applicable to U.S. persons, net of certain deductions and credits. In addition, dividends received by a corporatenon-U.S. holder that are effectively connected with a U.S. trade or business of the corporatenon-U.S. holder (or dividends attributable to a corporatenon-U.S. holder’s permanent establishment in the U.S. if an income tax treaty applies) may also be subject to a branch profits tax at a rate of 30% (or such lower rate as may be specified in an income tax treaty).
Anon-U.S. holder who provides us with an IRSForm W-8BEN or an IRSForm W-8ECI will be required to periodically update such form.
Anon-U.S. holder of common stock that is eligible for a reduced rate of withholding tax pursuant to an income tax treaty may obtain a refund of any excess amounts currently withheld if an appropriate claim for refund is timely filed with the IRS.
Gain on Disposition of Common Stock
Anon-U.S. holder generally will not be subject to U.S. federal income or withholding tax on gain realized on the sale or other disposition of our common stock unless:
| • | the gain is effectively connected with a U.S. trade or business of thenon-U.S. holder (or attributable to a permanent establishment in the U.S. if an income tax treaty applies), which gain, in the case of a corporatenon-U.S. holder, must also be taken into account for branch profits tax purposes; |
98
Table of Contents
| • | thenon-U.S. holder is an individual who is present in the U.S. for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met; or | |
| • | our common stock constitutes a U.S. real property interest by reason of our status as a “U.S. real property holding corporation” for U.S. federal income tax purposes at any time within the shorter of the five-year period preceding the disposition or the holder’s holding period for our common stock. We believe that we are not currently, and that we are not likely to become, a “U.S. real property holding corporation” for U.S. federal income tax purposes. |
If we were to become a U.S. real property holding corporation, so long as our common stock is regularly traded on an established securities market and continues to be so traded, anon-U.S. holder would be subject to U.S. federal income tax on any gain from the sale, exchange or other disposition of shares of our common stock, by reason of such U.S. real property holding corporation status, only if suchnon-U.S. holder actually or constructively owned, more than 5% of our common stock at any time during the shorter of the five-year period preceding the disposition or the holder’s holding period for our common stock.
Backup Withholding and Information Reporting
Generally, we must report annually to the IRS the amount of dividends paid, the name and address of the recipient, and the amount, if any, of tax withheld. A similar report is sent to the holder. Pursuant to income tax treaties or other agreements, the IRS may make its reports available to tax authorities in thenon-U.S. holder’s country of residence.
Payments of dividends or of proceeds on the disposition of stock made to anon-U.S. holder may be subject to additional information reporting and backup withholding (currently at a rate of 28%). Backup withholding will not apply if thenon-U.S. holder establishes an exemption, for example, by properly certifying itsnon-U.S. status on an IRSForm W-8BEN (or successor form). Notwithstanding the foregoing, backup withholding may apply if either we or our paying agent has actual knowledge, or reason to know, that the holder is a U.S. person.
Backup withholding is not an additional tax. Rather, the U.S. income tax liability of persons subject to backup withholding will be reduced by the amount of tax withheld. If withholding results in an overpayment of U.S. federal income tax, a credit or refund may be obtained, provided that the required information is furnished to the IRS in a timely manner.
Federal Estate Tax
If an individualnon-U.S. holder is treated as the owner, or has made certain lifetime transfers, of an interest in our common stock then the value thereof will be included in his or her gross estate for U.S. federal estate tax purposes, and such individual’s estate may be subject to U.S. federal estate tax unless an applicable estate tax or other treaty provides otherwise.
This discussion is for general purposes only. Prospective investors are urged to consult their own tax advisors regarding the application of the U.S. federal income and estate tax laws to their particular situations and the consequences under U.S. federal gift tax laws, as well as foreign, state, and local laws and tax treaties.
Shares Eligible for Future Sale
Before this offering, there has been no public market for our common stock. Market sales of shares of our common stock after this offering and from time to time, and the availability of shares for future sale, may reduce the market price of our common stock. While substantially all of our outstanding shares are subject to contractual and legal restrictions on resale for at least 180 days after the date of this prospectus, as described below, sales of substantial amounts of our common stock, or the perception that these sales could occur, could
99
Table of Contents
adversely affect prevailing market prices for our common stock and could impair our future ability to obtain capital, especially through an offering of equity securities.
Upon the closing of this offering, we will have outstanding an aggregate of 9,054,928 shares of our common stock, based upon the number of shares outstanding on May 31, 2007 and assuming no outstanding options or warrants are exercised prior to the closing of this offering and no exercise of the underwriters’ over-allotment option. All shares sold in this offering will be freely tradable without restrictions or further registration under the Securities Act, unless they are purchased by our “affiliates,” as that term is defined in Rule 144 under the Securities Act.
The remaining 6,054,928 shares of common stock outstanding upon the closing of this offering are restricted securities as defined under Rule 144. 6,001,621 of these shares are subject tolock-up agreements. See“Lock-up Agreements” below for a description of theselock-up agreements. Restricted securities may be sold in the U.S. public markets only if registered or if they qualify for an exemption from registration, including by reason of Rule 144, 144(k) or 701 under the Securities Act, which rules are summarized below. These remaining shares will be available for sale as follows (without taking into consideration the effect of thelock-up agreements described below):
| • | 2,176,266 shares of the outstanding common stock will be immediately eligible for sale in the public market without restriction; | |
| • | the remaining 3,878,662 shares of the outstanding common stock will be eligible for sale in the public market under Rule 144 or Rule 701, beginning 90 days after the effective date of the registration statement of which this prospectus is a part, subject to the volume, manner of sale and other limitations under those rules; and |
| • | additionally, approximately 550,959 shares of common stock issuable upon exercise of options outstanding as of May 31, 2007, will be eligible for sale pursuant to Rule 701 beginning 90 days after the date of this prospectus, subject to the vesting provisions that may be contained in individual option agreements. |
Rule 144
In general, under Rule 144 as currently in effect, beginning 90 days after the effective date of this offering, a person who has beneficially owned restricted securities for at least one year, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted shares within any three-month period that does not exceed the greater of:
| • | one percent of the number of shares of our common stock then outstanding, which will equal approximately 90,549 shares immediately after this offering; and | |
| • | the average weekly trading volume of our common stock on The NASDAQ Capital Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale. |
Sales of restricted shares under Rule 144 are also subject to requirements regarding the manner of sale, notice, and the availability of current public information about us. Rule 144 also provides that affiliates relying on Rule 144 to sell shares of our common stock that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement.
Rule 144(k)
Under Rule 144(k), a person who is not and has not been deemed to be our affiliate at any time during the three months preceding a sale and who has beneficially owned the restricted securities proposed to be sold for at least two years, including the holding period of any prior owner other than an affiliate of us, may sell those shares without complying with themanner-of-sale, public information, volume limitation or notice provisions of Rule 144.
100
Table of Contents
Rule 701
Under Rule 701, shares of our common stock acquired upon the exercise of currently outstanding options or pursuant to other rights granted under our stock plans may be resold, to the extent not subject tolock-up agreements, by:
| • | persons other than affiliates, beginning 90 days after the effective date of this offering, subject only to themanner-of-sale provisions of Rule 144; and | |
| • | our affiliates, beginning 90 days after the effective date of this offering, subject to themanner-of-sale, current public information, and filing requirements of Rule 144, in each case, without compliance with the one-year holding period requirement of Rule 144. |
As of May 31, 2007, options to purchase a total of 550,959 shares of common stock were outstanding, of which 273,452 were vested. Of the total number of shares of our common stock issuable under these options, all are subject to contractuallock-up agreements with us.
Form S-8 Registration Statements
We intend to file one or more registration statements onForm S-8 under the Securities Act after the closing of this offering to register the shares of our common stock that are issuable pursuant to our 2007 Performance Incentive Plan and 2000 Stock Plan. These registration statements are expected to become effective upon filing. Shares covered by these registration statements will then be eligible for sale in the public markets, subject to any applicablelock-up agreements and to Rule 144 limitations applicable to affiliates.
Lock-up Agreements
Prior to the effectiveness of the offering, our directors, officers and certain of our stockholders, collectively holding approximately 713,172 shares of our outstanding common stock and options and warrants to purchase an aggregate of approximately 316,161 shares of common stock will have entered into agreements with Maxim Group providing that for a period of 12 months after the date of this prospectus, such holders will not directly or indirectly offer, sell, agree to offer or sell, solicit offers to purchase, grant any call option or purchase any put option with respect to, pledge, borrow or otherwise dispose of any of our securities, and will not establish or increase any “put equivalent position” or liquidate or decrease any “call equivalent position” or otherwise enter into any swap, derivative or other transaction or arrangement that transfers the economic consequences of ownership of our securities. These restrictions will be subject to customary exceptions, and Maxim Group may, in its sole discretion, at any time and without notice, release for sale in the public market all or any portion of the shares subject to thelock-up agreements.
In addition, certain of our stockholders, collectively holding approximately 5,288,449 shares of our outstanding common stock and substantially all of our outstanding options and warrants, are subject to similar contractual lock-up restrictions with us, restricting transfers for a period of 180 days following the date of this prospectus. Therefore, in aggregate, holders of approximately 6,001,621 shares out of the total of 6,054,928 shares of our common stock outstanding as of May 31, 2007, are subject to contractual lock-up restrictions.
101
Table of Contents
UNDERWRITING
Subject to the terms and conditions in the underwriting agreement, dated , 2007, between us and Maxim Group LLC, who is acting as the representative of the underwriters of this offering, each underwriter named below has agreed to purchase from us, on a firm commitment basis, the respective number of shares of common stock shown opposite its name below, at the public offering price, less the underwriting discount set forth on the cover page of this prospectus:
Name | Number of Shares | |||
| Maxim Group LLC | ||||
| I-Bankers Securities, Inc. | ||||
| Total | ||||
The underwriting agreement provides that the underwriters are committed to purchase all of the shares of common stock offered by this prospectus if they purchase any of the shares. This commitment does not apply to the shares of common stock subject to an over-allotment option granted by us to the underwriters to purchase additional shares of common stock in this offering. The underwriting agreement also provides that the obligations of the underwriters to pay for and accept delivery of the shares of common stock are subject to the passing upon of certain legal matters by counsel and certain other conditions.
Pursuant to the underwriting agreement, we have granted to the underwriters an option, exercisable for 45 days after the date of this prospectus, to purchase up to an additional 450,000 shares of common stock from us on the same terms and at the same per share price as the other shares of common stock being purchased by the underwriters from us. The underwriters may exercise the option solely to cover over-allotments, if any, in the sale of shares of common stock that the underwriters have agreed to purchase from us. If the over-allotment option is exercised in full, the total public offering price, underwriting discounts and commissions and proceeds to us before expenses will be $ , $ and $ , respectively.
The following table shows the per share and total underwriting discounts and commissions to be paid by us in connection with this offering. These amounts are shown assuming both no exercise and full exercise of the underwriters’ over-allotment option.
| Total | ||||||||||||
| Per | Without | With | ||||||||||
| Share | Over-Allotment | Over-Allotment | ||||||||||
| Public offering price | $ | $ | $ | |||||||||
| Underwriting discount | $ | $ | $ | |||||||||
| Non-accountable expense allowance(1) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us(2) | $ | $ | $ | |||||||||
| (1) | The non-accountable expense allowance is not payable with respect to the shares of common stock sold upon exercise of the underwriters’ over-allotment option. | |
| (2) | We estimate that the total expenses of this offering excluding the underwriters’ discount and the non-accountable expense allowance, will be approximately $1.8 million. |
We have agreed to sell the shares of common stock to the underwriters at the initial public offering price less the underwriting discount set forth on the cover page of this prospectus. The underwriting agreement also provides that the representatives of the underwriters will be paid a non-accountable expense allowance equal to 2.0% of the gross proceeds from the sale of the shares of common stock offered by this prospectus ($30,000 of which has been previously advanced to Maxim Group LLC), exclusive of any common stock purchased on exercise of the underwriters’ over-allotment option.
We estimate that the total expenses of the offering payable by us, not including underwriting discounts, commissions, the non-accountable expense allowance and not taking into consideration the underwriters’ over-allotment option, will be approximately $1.8 million. These expenses include, but are not limited to, SEC registration fees, NASD filing fees, proposed NASDAQ Capital Market listing fees, accounting fees and
102
Table of Contents
expenses, legal fees and expenses, printing and engraving expenses, transfer agent fees and blue sky fees and expenses.
The underwriters will initially offer the shares of common stock to be sold in this offering directly to the public at the initial public offering price set forth on the cover of this prospectus and to selected dealers at the initial public offering price less a selling concession not in excess of $ per share. The underwriters may allow, and the selected dealers may reallow, a concession not in excess of $ per share on sales to brokers and dealers. After the offering, the underwriters may change the offering price and other selling terms. No change in those terms will change the amount of proceeds to be received by us as set forth on the cover of this prospectus.
We will issue warrants to the representative on the completion of the offering entitling the representative or its assigns to purchase up to 210,000 shares of common stock. These warrants will be exercisable commencing twelve months from the date of their issuance and for a period of four years thereafter at a price per share equal to 115% of the public offering price per share in this offering and will allow for cashless exercise. The warrants will provide for unlimited piggyback registration rights commencing on the effective date and expiring five years thereafter at our expense.
The representative’s warrants will be restricted from sale, transfer, assignment, pledge or hypothecation for a period of six months after the date of this prospectus, except to officers and partners of the representative, co-underwriters, selling group members and their officers and partners. Thereafter, the representative’s warrants will be transferable provided such transfer is made in compliance with the Securities Act.
Prior to this offering, there was no public market for the common stock. The initial public offering price of our common stock was determined by negotiation between us and the underwriters. The principal factors considered in determining the public offering price of the common stock included:
| • | the information in this prospectus and otherwise available to the underwriters; | |
| • | the history and the prospects for the industry in which we will compete; | |
| • | the size of the potential markets that our product candidates may address; | |
| • | the ability of our management; | |
| • | the prospects for our future earnings; | |
| • | the present state of our development and our current financial condition; | |
| • | the general condition of the economy and the securities markets at the time of this offering; and | |
| • | the recent market prices of, and the demand for, publicly traded securities of generally comparable companies. |
We cannot be sure that the initial public offering price will correspond to the price at which the common stock will trade in the public market following this offering or that an active trading market for the common stock will develop and continue after this offering.
Prior to the effectiveness of the offering, our directors, officers and certain of our stockholders, collectively holding approximately 713,172 shares of our common stock, will have entered into agreements with Maxim Group providing that for a period of 12 months after the date of this prospectus, such holders will not directly or indirectly offer, sell, agree to offer or sell, solicit offers to purchase, grant any call option or purchase any put option with respect to, pledge, borrow or otherwise dispose of any of our securities, and will not establish or increase any “put equivalent position” or liquidate or decrease any “call equivalent position” or otherwise enter into any swap, derivative or other transaction or arrangement that transfers the economic consequences of ownership of our securities. These restrictions will be subject to customary exceptions, and Maxim Group may, in its sole discretion, at any time and without notice, release for sale in the public market all or any portion of the shares subject to thelock-up agreements. See “Shares Eligible for Future Sale.”
Maxim has no present intention to waive or shorten thelock-up period; however, the terms of thelock-up agreements may be waived at its discretion. In determining whether to waive the terms of thelock-up agreements, Maxim may base its decision on its assessment of the relative strengths of the securities markets
103
Table of Contents
and small capitalization companies in general, and the trading pattern of, and demand for, our securities in general.
For a period of twelve months from the completion of the offering, we have granted Maxim Group LLC the right of first refusal to manage or co-manage any underwritten public offering or private placement of our debt or equity securities and those of our subsidiaries and any successor to us, except for (i) sales to employees under any stockholder-approved compensation or stock option plan, (ii) shares issued in payment of the consideration for an acquisition, and (iii) conventional banking arrangements and commercial debt financing.
In connection with this offering, the underwriters may distribute prospectuses electronically. No forms of prospectus other than printed prospectuses and electronically distributed prospectuses that are printable in Adobe PDF format will be used in connection with this offering.
The underwriters have informed us that they do not expect to confirm sales of shares of common stock offered by this prospectus to accounts over which they exercise discretionary authority.
Maxim Group LLC acted as one of our placement agents in connection with our private placements completed in April 2006 and May 2006, and they received an aggregate of $36,000 in commissions for such services.
In connection with this offering, our underwriters may engage in stabilizing transactions, over-allotment transactions, covering transactions and penalty bids in accordance with Regulation M under the Securities Exchange Act of 1934, as amended.
| • | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. | |
| • | Over-allotment involves sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase, which creates a short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that it may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any covered short position by either exercising their over-allotment option or purchasing shares in the open market. | |
| • | Covering transactions involve the purchase of common stock in the open market after the distribution has been completed in order to cover short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which it may purchase shares through the over-allotment option. If the underwriters sell more shares than could be covered by the over-allotment option, a naked short position, the position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in this offering. | |
| • | Penalty bids permit the underwriters to reclaim a selling concession from a selected dealer when the common stock originally sold by the selected dealer is purchased in a stabilizing covering transaction to cover short positions. |
These stabilizing transactions, covering transactions and penalty bids may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of our common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. These transactions may be effected on the NASDAQ Capital Market or otherwise and, if commenced, may be discontinued at any time.
The underwriting agreement provides for indemnification between us and the underwriters against specified liabilities, including liabilities under the Securities Act, and for contribution by us and the underwriters to payments that may be required to be made with respect to those liabilities. We have been
104
Table of Contents
advised that, in the opinion of the Securities and Exchange Commission, indemnification liabilities under the Securities Act is against public policy as expressed in the Securities Act, and is therefore, unenforceable.
Foreign Regulatory Restrictions on Purchase of Shares
We have not taken any action to permit a public offering of shares of our common stock outside the United States or to permit the possession or distribution of this prospectus outside the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about and observe any restrictions relating to this offering of shares of our common stock and the distribution of the prospectus outside the United States.
Italy. This offering of shares of common stock has not been cleared by Consob, the Italian Stock Exchanges regulatory agency of public companies, pursuant to Italian securities legislation and, accordingly, no shares may be offered, sold or delivered, nor may copies of this prospectus or of any other document relating to the shares be distributed in Italy, except (1) to professional investors(operatori qualificati);or (2) in circumstances which are exempted from the rules on solicitation of investments pursuant to Decree No. 58 and Article 33, first paragraph, of Consob Regulation No. 11971 of May 14, 1999, as amended. Any offer, sale or delivery of the shares or distribution of copies of this prospectus or any other document relating to the shares in Italy under (1) or (2) above must be (i) made by an investment firm, bank or financial intermediary permitted to conduct such activities in Italy in accordance with the Decree No. 58 and Legislative Decree No. 385 of September 1, 1993, or the Banking Act; and (ii) in compliance with Article 129 of the Banking Act and the implementing guidelines of the Bank of Italy, as amended from time to time, pursuant to which the issue or the offer of securities in Italy may need to be preceded and followed by an appropriate notice to be filed with the Bank of Italy depending,inter alia, on the aggregate value of the securities issued or offered in Italy and their characteristics; and (iii) in compliance with any other applicable laws and regulations.
Germany. The offering of shares of common stock is not a public offering in the Federal Republic of Germany. The shares may only be acquired in accordance with the provisions of the Securities Sales Prospectus Act (Wertpapier-Verkaudfspropsektgestz), as amended, and any other applicable German law. No application has been made under German law to publicly market the shares in or out of the Federal Republic of Germany. The shares are not registered or authorized for distribution under the Securities Sales Prospectus Act and accordingly may not be, and are not being, offered or advertised publicly or by public promotion. Therefore, this prospectus is strictly for private use and the offering is only being made to recipients to whom the document is personally addressed and does not constitute an offer or advertisement to the public. The shares of common stock will only be available to persons who, by profession, trade or business, buy or sell shares for their own or a third party’s account.
France. The shares of common stock offered by this prospectus may not be offered or sold, directly or indirectly, to the public in France. This prospectus has not been or will not be submitted to the clearance procedure of the Autorité des Marchés Financiers, or the AMF, and may not be released or distributed to the public in France. Investors in France may only purchase the shares offered by this prospectus for their own account and in accordance with articles L.411-1, L.441-2 and L.412-1 of the Code Monétaire et Financier and decreeno. 98-880 dated October 1, 1998, provided they are “qualified investors” within the meaning of said decree. Each French investor must represent in writing that it is a qualified investor within the meaning of the aforesaid decree. Any resale, directly or indirectly, to the public of the shares offered by this prospectus may be effected only in compliance with the above mentioned regulations. “Les actions offertes par ce document d’information ne peuvent pas être, directement ou indirectement, offertes ou vendues au public en France. Ce document d’information n’a pas été ou ne sera pas soumis au visa de l’Autorité des Marchés Financiers et ne peut être diffusé ou distribué au public en France. Les investisseurs en France ne peuvent acheter les actions offertes par ce document d’information que pour leur compte propre et conformément aux articles L.411-1, L.441-2 et L.412-1 du Code Monétaire et Financier et du décretno. 98-880 du 1 octobre 1998, sous réserve qu’ils soient des investisseurs qualifiés au sens du décret susvisé. Chaque investisseur doit déclarer par écrit qu’il est un investisseur qualifié au sens du décret susvisé. Toute revente, directe ou indirecte, des actions offertes par ce document d’information au public ne peut être effectuée que conformément à la réglementation susmentionnée.”
105
Table of Contents
Switzerland. This prospectus may only be used by those persons to whom it has been directly handed out by the offeror or its designated distributors in connection with the offer described therein. The shares of common stock are only offered to those personsand/or entities directly solicited by the offeror or its designated distributors, and are not offered to the public in Switzerland. This prospectus constitutes neither a public offer in Switzerland nor an issue prospectus in accordance with the respective Swiss legislation, in particular but not limited to Article 652A Swiss Code Obligations. Accordingly, this prospectus may not be used in connection with any other offer, whether private or public and shall in particular not be distributed to the public in Switzerland.
United Kingdom. In the United Kingdom, the shares of common stock offered by this prospectus are directed to and will only be available for purchase to a person who is an exempt person as referred to at paragraph (c) below and who warrants, represents and agrees that: (a) it has not offered or sold, will not offer or sell, any shares offered by this prospectus to any person in the United Kingdom except in circumstances which do not constitute an offer to the public in the United Kingdom for the purposes of the section 85 of the Financial Services and Markets Act 2000 (as amended) (“FSMA”); and (b) it has complied and will comply with all applicable provisions of FSMA and the regulations made thereunder in respect of anything done by it in relation to the shares offered by this prospectus in, from or otherwise involving the United Kingdom; and (c) it is a person who falls within the exemptions to Section 21 of the FSMA as set out in The Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (“the Order”), being either an investment professional as described under Article 19 or any body corporate (which itself has or a group undertaking has a called up share capital or net assets of not less than £500,000 (if more than 20 members) or otherwise £5 million) or an unincorporated association or partnership (with net assets of not less than £5 million) or is a trustee of a high value trust or any person acting in the capacity of director ,officer or employee of such entities as defined under Article 49(2)(a) to (d) of the Order, or a person to whom the invitation or inducement may otherwise lawfully be communicated or cause to be communicated. The investment activity to which this document relates will only be available to and engaged in only with exempt persons referred to above. Persons who are not investment professionals and do not have professional experience in matters relating to investments or are not an exempt person as described above, should not review nor rely or act upon this document and should return this document immediately. It should be noted that this document is not a prospectus in the United Kingdom as defined in the Prospectus Regulations 2005 and has not been approved by the Financial Services Authority or any competent authority in the United Kingdom.
Israel. The shares of common stock offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority (ISA). The shares may not be offered or sold, directly or indirectly, to the public in Israel. The ISA has not issued permits, approvals or licenses in connection with the offering of the shares or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the shares being offered. Any resale, directly or indirectly, to the public of the shares offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Sweden. Neither this prospectus nor the shares of common stock offered hereunder have been registered with or approved by the Swedish Financial Supervisory Authority under the Swedish Financial Instruments Trading Act (1991:980) (as amended), nor will such registration or approval be sought. Accordingly, this prospectus may not be made available nor may the shares offered hereunder be marketed or offered for sale in Sweden other than in circumstances which are deemed not to be an offer to the public in Sweden under the Financial Instruments Trading Act. This prospectus may not be distributed to the public in Sweden and a Swedish recipient of the prospectus may not in any way forward the prospectus to the public in Sweden.
Norway. This prospectus has not been produced in accordance with the prospectus requirements laid down in the Norwegian Securities Trading Act 1997, as amended. This prospectus has not been approved or disapproved by, or registered with, either the Oslo Stock Exchange or the Norwegian Registry of Business Enterprises. This prospectus may not, either directly or indirectly be distributed to Norwegian potential investors.
Denmark. This prospectus has not been prepared in the context of a public offering of securities in Denmark within the meaning of the Danish Securities Trading Act No. 171 of 17 March 2005, as amended
106
Table of Contents
from time to time, or any Executive Orders issued on the basis thereof and has not been and will not be filed with or approved by the Danish Financial Supervisory Authority or any other public authority in Denmark. The offering of shares of common stock will only be made to persons pursuant to one or more of the exemptions set out in Executive Order No. 306 of 28 April 2005 on Prospectuses for Securities Admitted for Listing or Trade on a Regulated Market and on the First Public Offer of Securities exceeding EUR 2,500,000 or Executive Order No. 307 of 28 April 2005 on Prospectuses for the First Public Offer of Certain Securities between EUR 100,000 and EUR 2,500,000, as applicable.
Legal Matters
The validity of the issuance of the shares of common stock offered by this prospectus will be passed upon for us by our counsel, DLA Piper US LLP, Seattle, Washington. Richardson & Patel LLP, New York, New York, is acting as counsel for the underwriters in connection with this offering.
Experts
The consolidated financial statements of ImaRx Therapeutics, Inc. at December 31, 2005 and 2006, and for each of the three years ended December 31, 2004, 2005 and 2006 appearing in this prospectus and registration statement have been audited by Ernst & Young LLP, an independent registered public accounting firm, as set forth in their report thereon appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
Where You Can Find Additional Information
We have filed with the SEC a registration statement onForm S-1 under the Securities Act that registers the shares of our common stock to be sold in this offering. The registration statement, including the attached exhibits and schedules, contains additional relevant information about us and our capital stock. The rules and regulations of the SEC allow us to omit from this prospectus certain information included in the registration statement. For further information about us and our common stock, you should refer to the registration statement and the exhibits and schedules filed with the registration statement. With respect to the statements contained in this prospectus regarding the contents of any agreement or any other document, in each instance, the statement is qualified in all respects by the complete text of the agreement or document, a copy of which has been filed as an exhibit to the registration statement. In addition, upon the closing of this offering, we will file reports, proxy statements and other information with the SEC under the Securities Exchange Act of 1934, as amended. You may obtain copies of this information by mail from the Public Reference Room of the SEC at 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at1-800-SEC-0330. The SEC also maintains an internet website that contains reports, proxy statements and other information about issuers that file electronically with the SEC. The address of that site is www.sec.gov.
We intend to provide our stockholders with annual reports containing consolidated financial statements that have been examined and reported on, with an opinion expressed by an independent registered public accounting firm, and to file with the SEC quarterly reports containing unaudited consolidated financial data for the first three quarters of each year.
107
ImaRx Therapeutics, Inc.
| F-2 | ||||
| Consolidated Financial Statements | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 |
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders
ImaRx Therapeutics, Inc.
We have audited the accompanying consolidated balance sheets of ImaRx Therapeutics, Inc. as of December 31, 2005 and 2006, and the related consolidated statements of operations, redeemable convertible preferred stock and stockholders’ deficit, and cash flows for each of the three years in the period ended December 31, 2006. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. Our audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of ImaRx Therapeutics, Inc. at December 31, 2005 and 2006, and the results of its operations and its cash flows for the three year period ended December 31, 2006. in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that ImaRx Therapeutics, Inc. will continue as a going concern. As more fully described in Note 2, the Company has recurring losses, which has resulted in a significant accumulated deficit at December 31, 2006. This condition, among others, raises substantial doubt about the Company’s ability to continue as a going concern. Management plans in regards to these matters are also described in Note 2. The financial statements do not include any adjustments to reflect possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
As discussed in Note 2 to the consolidated financial statements, effective January 1, 2006, the Company adopted the provisions of Statement of Financial Accounting Standards No. 123 (revised 2004),Share-Based Payment.
/s/ Ernst & Young LLP
Phoenix, Arizona
May 4, 2007,
except for the second paragraph of Note 15, as to which the date is
May 30, 2007
F-2
Table of Contents
ImaRx Therapeutics, Inc.
Consolidated Balance Sheets
| December 31 | March 31 | |||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (Unaudited) | ||||||||||||
Assets | ||||||||||||
| Current assets: | ||||||||||||
| Cash and cash equivalents | $ | 8,513,387 | $ | 4,256,399 | $ | 2,748,495 | ||||||
| Inventory | — | 16,059,730 | 16,414,412 | |||||||||
| Inventory subject to return | — | 445,245 | 433,753 | |||||||||
| Accounts receivable, less allowances of $20,819 in 2006 and $451 in 2007 | — | 575,610 | 61,699 | |||||||||
| Prepaid expenses and other | 272,486 | 539,048 | 322,598 | |||||||||
| Deferred financing costs | — | — | 146,573 | |||||||||
| Total current assets | 8,785,873 | 21,876,032 | 20,127,530 | |||||||||
| Long-term assets: | ||||||||||||
| Property and equipment, net | 729,961 | 916,966 | 1,056,424 | |||||||||
| Intangible assets, net | — | 2,500,000 | 2,200,000 | |||||||||
| Total assets | $ | 9,515,834 | $ | 25,292,998 | $ | 23,383,954 | ||||||
| Liabilities and stockholders’ deficit | ||||||||||||
| Current liabilities: | ||||||||||||
| Accounts payable | $ | 775,684 | $ | 1,413,032 | $ | 799,835 | ||||||
| Accrued expenses | 893,198 | 1,235,510 | 2,300,803 | |||||||||
| Deferred revenue | — | 955,263 | 603,708 | |||||||||
| Notes payable | 15,227,500 | 15,615,000 | 15,840,000 | |||||||||
| Total current liabilities | 16,896,382 | 19,218,805 | 19,544,346 | |||||||||
| Other long-term liability | 218,856 | 218,856 | 218,856 | |||||||||
| Total liabilities | 17,115,238 | 19,437,661 | 19,763,202 | |||||||||
| Redeemable convertible preferred stock: | ||||||||||||
| Series A 8% Redeemable Convertible Preferred Shares, | ||||||||||||
| $.0001 par, at carrying value including accrued dividends (liquidation value of $8,902,752, $9,406,804 and $9,532,817 at December 31, 2005, 2006 and March 31, 2007 (unaudited), respectively): | ||||||||||||
| Authorized shares — 2,400,000 at December 31, 2005 and 2,302,053 at December 31, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 2,291,144 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | 8,824,695 | 9,328,747 | 9,454,760 | |||||||||
| Series B 7% Mandatorily Redeemable Convertible Preferred Shares, | ||||||||||||
| $.0001 par, at carrying value (liquidation value of $9,491,622 at December 31, 2005, 2006 and March 31, 2007 (unaudited), respectively): | ||||||||||||
| Authorized shares — 800,000 at December 31, 2005 and 593,226 at December 31, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 593,226 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | 9,491,622 | 9,491,622 | 9,491,622 | |||||||||
| Series C Mandatorily Redeemable Convertible Preferred Shares, | ||||||||||||
| $.0001 par, at carrying value (liquidation value of $1,999,998 at December 31, 2005, 2006 and March 31, 2007 (unaudited), respectively): | ||||||||||||
| Authorized shares — 1,700,000 at December 31, 2005 and 285,714 at December 31, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 285,714 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | 1,945,563 | 1,945,563 | 1,945,563 | |||||||||
| Series D 8% Redeemable Convertible Preferred Shares, | ||||||||||||
| $.0001 par, at carrying value including accrued dividends (liquidation value of $1,487,332, $1,583,743 and $1,607,847 at December 31, 2005, 2006 and March 31, 2007 (unaudited), respectively): | ||||||||||||
| Authorized shares — 545,500 at December 31, 2005 and 438,232 at December 31, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 438,232 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | 1,465,593 | 1,562,007 | 1,586,110 | |||||||||
| Series F 8% Redeemable Convertible Preferred Shares, | ||||||||||||
| $.0001 par, at carrying value including accrued dividends (liquidation value of $14,742,000 and $15,025,500 at December 31, 2006 and March 31, 2007(unaudited), respectively): | ||||||||||||
| Authorized shares — 4,000,000 at December 31, 2006 and March 31,2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 2,835,000 at December 31, 2006 and March 31, 2007 (unaudited) | — | 13,535,559 | 13,819,059 | |||||||||
| Total redeemable convertible preferred stock | 21,727,473 | 35,863,498 | 36,297,114 | |||||||||
| Stockholders’ deficit: | ||||||||||||
| Series E Redeemable Convertible Preferred Shares, $.0001 par: | ||||||||||||
| Authorized shares — 1,000,000 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 1,000,000 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | 4,000,000 | 4,000,000 | 4,000,000 | |||||||||
| Common stock, $.0001 par: | ||||||||||||
| Authorized shares — 70,000,000 at December 31, 2005, 2006 and March 31, 2007 (unaudited) | ||||||||||||
| Issued and outstanding shares — 2,584,663 at December 31, 2005 and 2,606,739 at December 31, 2006 and March 31, 2007(unaudited) | 258 | 260 | 260 | |||||||||
| Additional paid-in capital | 27,435,109 | 28,619,883 | 28,782,998 | |||||||||
| Accumulated deficit | (60,762,244 | ) | (62,628,304 | ) | (65,459,620 | ) | ||||||
| Total stockholders’ deficit | (29,326,877 | ) | (30,008,161 | ) | (32,676,362 | ) | ||||||
| Total liabilities and stockholders’ deficit | $ | 9,515,834 | $ | 25,292,998 | $ | 23,383,954 | ||||||
See accompanying notes.
F-3
Table of Contents
ImaRx Therapeutics, Inc.
Consolidated Statements of Operations
| Three Months Ended | ||||||||||||||||||||
| Years Ended December 31 | March 31 | |||||||||||||||||||
| 2004 | 2005 | 2006 | 2006 | 2007 | ||||||||||||||||
| (Unaudited) | ||||||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Product sales, net | $ | — | $ | — | $ | 480,290 | $ | — | $ | 1,085,699 | ||||||||||
| Research and development | 575,014 | 619,046 | 847,442 | 177,338 | 122,004 | |||||||||||||||
| Total operating revenue | 575,014 | 619,046 | 1,327,732 | 177,338 | 1,207,703 | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of product sales (exclusive of amortization shown separately below of $783,333 and $293,750 in 2006 and 2007, respectively) | — | — | 204,135 | — | 461,064 | |||||||||||||||
| Research and development | 2,489,640 | 3,578,703 | 8,396,273 | 1,723,300 | 1,499,634 | |||||||||||||||
| General and administrative | 3,183,850 | 4,142,279 | 7,370,755 | 1,618,281 | 1,097,831 | |||||||||||||||
| Depreciation and amortization | 185,905 | 194,206 | 1,048,586 | 60,063 | 363,251 | |||||||||||||||
| Acquired in-process research and development | — | 24,000,000 | — | — | — | |||||||||||||||
| Total cost and expenses | 5,859,395 | 31,915,188 | 17,019,749 | 3,401,644 | 3,421,780 | |||||||||||||||
| Operating loss | (5,284,381 | ) | (31,296,142 | ) | (15,692,017 | ) | (3,224,306 | ) | (2,214,077 | ) | ||||||||||
| Other income (expense): | ||||||||||||||||||||
| Interest and other income, net | 29,109 | 122,187 | 380,923 | 104,586 | 41,377 | |||||||||||||||
| Interest expense | (468,536 | ) | (587,341 | ) | (1,515,000 | ) | (225,000 | ) | (225,000 | ) | ||||||||||
| Gain on extinguishment of debt | — | 3,834,959 | 16,127,500 | — | — | |||||||||||||||
| Net loss | (5,723,808 | ) | (27,926,337 | ) | (698,594 | ) | (3,344,720 | ) | (2,397,700 | ) | ||||||||||
| Accretion of dividends on preferred stock | (301,031 | ) | (600,452 | ) | (1,167,466 | ) | (150,116 | ) | (433,616 | ) | ||||||||||
| Net loss attributed to common stockholders | $ | (6,024,839 | ) | $ | (28,526,789 | ) | $ | (1,866,060 | ) | $ | (3,494,836 | ) | $ | (2,831,316 | ) | |||||
| Net loss attributed to common stockholders per share | ||||||||||||||||||||
| — Basic and diluted: | ($ | 5.37 | ) | ($ | 15.11 | ) | ($ | 0.72 | ) | ($ | 1.35 | ) | ($ | 1.09 | ) | |||||
| Weighted-average shares outstanding | ||||||||||||||||||||
| — Basic and diluted: | 1,122,881 | 1,888,291 | 2,599,425 | 2,585,315 | 2,605,915 | |||||||||||||||
See accompanying notes.
F-4
Table of Contents
ImaRx Therapeutics, Inc.
Consolidated Statements of Redeemable Convertible Preferred Stock and Stockholders’ Deficit
| Series E | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Redeemable Convertible Preferred Stock | Redeemable | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Series A | Series B | Series C | Series D | Series F | Convertible | Additional | Total | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carrying | Carrying | Carrying | Carrying | Carrying | Preferred Shares | Common Stock | Paid-in | Accumulated | Stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Value | Shares | Value | Shares | Value | Shares | Value | Shares | Value | Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at January 1, 2004 | 2,291,144 | $ | 7,816,591 | 593,226 | $ | 9,791,063 | 285,714 | $ | 1,945,563 | 438,232 | $ | 1,272,773 | — | $ | — | — | $ | — | 587,309 | $ | 58 | $ | 207,280 | $ | (26,210,616 | ) | $ | (26,003,278 | ) | |||||||||||||||||||||||||||||||||||||||
| Issuance of 12,136 warrants with convertible subordinated notes | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 65,325 | — | 65,325 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Discount related to beneficial conversion feature of warrants issued with convertible subordinated notes | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 32,961 | — | 32,961 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | — | — | — | — | — | — | 2,000 | — | 5,000 | — | 5,000 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Common Stock, net of issuance costs of $598,334 | — | — | — | — | — | — | — | — | — | — | — | — | 500,000 | 50 | 4,401,616 | — | 4,401,666 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Conversion of convertible subordinated notes to Common Stock | — | — | — | — | — | — | — | — | — | — | — | — | 206,465 | 21 | 2,064,665 | — | 2,064,686 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 100,000 warrants in conjunction with consulting agreements | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 464,378 | — | 464,378 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 50,000 warrants to placement agent | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 452,269 | — | 452,269 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of undeclared dividends on Series A and D Redeemable Convertible Preferred Stock | — | 504,052 | — | — | — | — | — | 96,420 | — | — | — | — | — | — | — | (600,472 | ) | (600,472 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Contractual reduction of Series B Redeemable Convertible Preferred stock undeclared dividend accrual | — | — | — | (299,441 | ) | — | — | — | — | — | — | — | — | — | — | 299,441 | 299,441 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | — | — | — | — | — | — | 5,266 | 1 | 13,164 | — | 13,165 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (5,723,808 | ) | (5,723,808 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2004 | 2,291,144 | 8,320,643 | 593,226 | 9,491,622 | 285,714 | 1,945,563 | 438,232 | 1,369,193 | — | — | — | — | 1,301,040 | 130 | 7,706,658 | (32,235,455 | ) | (24,528,667 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Common Stock, net of issuance costs of $593,100 | — | — | — | — | — | — | — | — | — | — | — | — | 277,999 | 28 | 3,576,872 | — | 3,576,900 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 4,000 warrants in conjunction with patent licensing | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 35,870 | — | 35,870 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Common Stock, net of issuance costs of $885,931 | — | — | — | — | — | — | — | — | — | — | — | — | 188,664 | 19 | 1,944,043 | — | 1,944,062 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 46,664 warrants to placement agent | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 415,200 | — | 415,200 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Preferred Stock related to acquisition of assets | — | — | — | — | — | — | — | — | — | — | 1,000,000 | 4,000,000 | — | — | — | — | 4,000,000 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 20,000 warrants with subordinated notes | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 273,327 | — | 273,327 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Common Stock, net of issuance costs of $1,487,527 | — | — | — | — | — | — | — | — | — | — | — | — | 531,750 | 53 | 9,147,420 | — | 9,147,473 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Common Stock, net of issuance costs of $1,510,645 | — | — | — | — | — | — | — | — | — | — | — | — | 218,250 | 22 | 2,854,333 | — | 2,854,355 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 74,996 warrants to placement agent | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 943,195 | — | 943,195 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of undeclared dividends on Series A and D Redeemable Convertible Preferred Stock | — | 504,052 | — | — | — | — | — | 96,400 | — | — | — | — | — | — | — | (600,452 | ) | (600,452 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | — | — | — | — | — | — | 3,506 | — | 35,062 | — | 35,062 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | — | — | — | — | — | — | 63,454 | 6 | 296,129 | — | 296,135 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Shared-based compensation | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 207,000 | 207,000 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (27,926,337 | ) | (27,926,337 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2005 | 2,291,144 | 8,824,695 | 593,226 | 9,491,622 | 285,714 | 1,945,563 | 438,232 | 1,465,593 | — | — | 1,000,000 | 4,000,000 | 2,584,663 | 258 | 27,435,109 | (60,762,244 | ) | (29,326,877 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of 15,000 warrants to consultant | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 173,909 | — | 173,909 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of Series F Redeemable Convertible Preferred Stock net of issuance costs of $1,206,441 | — | — | — | — | — | — | — | — | 2,835,000 | 12,968,559 | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Accretion of undeclared dividends on Series A, D and F Redeemable Convertible Preferred Stock | — | 504,052 | — | — | — | — | — | 96,414 | — | 567,000 | — | — | — | — | — | (1,167,466 | ) | (1,167,466 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 954,766 | — | 954,766 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | — | — | — | — | — | — | 22,076 | 2 | 56,099 | — | 56,101 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (698,594 | ) | (698,594 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2006 | 2,291,144 | $ | 9,328,747 | 593,226 | $ | 9,491,622 | 285,714 | $ | 1,945,563 | 438,232 | $ | 1,562,007 | 2,835,000 | $ | 13,535,559 | 1,000,000 | $ | 4,000,000 | 2,606,739 | 260 | $ | 28,619,883 | $ | (62,628,304 | ) | $ | (30,008,161 | ) | ||||||||||||||||||||||||||||||||||||||||
| Accretion of undeclared dividends on Series A, D and F Redeemable Convertible Preferred Stock (unaudited) | — | 126,013 | — | — | — | — | — | 24,103 | — | 283,500 | — | — | — | — | — | (433,616 | ) | (433,616 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Stock-based compensation (unaudited) | — | — | — | — | — | — | — | — | — | — | — | — | — | — | 163,115 | — | 163,115 | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Net loss (unaudited) | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | (2,397,700 | ) | (2,397,700 | ) | |||||||||||||||||||||||||||||||||||||||||||||||||
| Balance at March 31, 2007 (unaudited) | 2,291,144 | $ | 9,454,760 | 593,226 | $ | 9,491,622 | 285,714 | $ | 1,945,563 | 438,232 | $ | 1,586,110 | 2,835,000 | $ | 13,819,059 | 1,000,000 | $ | 4,000,000 | 2,606,739 | 260 | $ | 28,782,998 | $ | (65,459,620 | ) | $ | (32,676,362 | ) | ||||||||||||||||||||||||||||||||||||||||
See accompanying notes.
F-5
Table of Contents
ImaRx Therapeutics, Inc.
Consolidated Statements of Cash Flows
| Years Ended December 31 | Three Months Ended March 31 | |||||||||||||||||||
| 2004 | 2005 | 2006 | 2006 | 2007 | ||||||||||||||||
| (Unaudited) | ||||||||||||||||||||
Operating activities | ||||||||||||||||||||
| Net loss | $ | (5,723,808 | ) | $ | (27,926,337 | ) | $ | (698,594 | ) | $ | (3,344,720 | ) | $ | (2,397,700 | ) | |||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||||||
| Depreciation and amortization | 185,905 | 194,206 | 1,048,586 | 60,063 | 363,251 | |||||||||||||||
| Stock-based compensation | — | 207,000 | 954,766 | 25,510 | 163,115 | |||||||||||||||
| Warrant amortization expense | 916,647 | 35,870 | 173,909 | 173,909 | — | |||||||||||||||
| Amortization of debt discount | 156,688 | 273,327 | — | — | — | |||||||||||||||
| Gain on extinguishments of debt | — | (3,834,959 | ) | (16,127,500 | ) | — | — | |||||||||||||
| Note issued for acquisition of technology expensed to operations | — | 15,000,000 | — | — | — | |||||||||||||||
| Preferred stock issued for acquisition of technology expensed to operations | — | 4,000,000 | — | — | — | |||||||||||||||
| Loss on sale of property and equipment | 2,681 | — | 3,215 | 1,727 | — | |||||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||||||||
| Inventory | 8,494 | 36,251 | (3,872,610 | ) | — | (354,682 | ) | |||||||||||||
| Inventory subject to return | — | — | (107,365 | ) | — | 11,492 | ||||||||||||||
| Accounts receivable | — | — | (575,610 | ) | — | 513,911 | ||||||||||||||
| Prepaid expenses and other | (212,276 | ) | (26,702 | ) | (266,562 | ) | (51,356 | ) | 216,450 | |||||||||||
| Accounts payable | (245,911 | ) | 385,834 | 637,348 | 971,555 | (613,197 | ) | |||||||||||||
| Accrued expenses and other liabilities | 753,930 | 540,548 | 1,857,312 | 23,051 | 1,290,293 | |||||||||||||||
| Deferred revenue | — | — | 955,263 | — | (351,555 | ) | ||||||||||||||
| Net cash used in operating activities | (4,157,650 | ) | (11,114,962 | ) | (16,017,842 | ) | (2,140,261 | ) | (1,158,622 | ) | ||||||||||
Investing activities | ||||||||||||||||||||
| Purchase of property and equipment | (64,719 | ) | (564,202 | ) | (438,806 | ) | (198,333 | ) | (202,709 | ) | ||||||||||
| Purchase of intangibles | — | — | (825,000 | ) | — | — | ||||||||||||||
| Net cash used in investing activities | (64,719 | ) | (564,202 | ) | (1,263,806 | ) | (198,333 | ) | (202,709 | ) | ||||||||||
Financing activities | ||||||||||||||||||||
| Deferred financing costs | 57,909 | (57,909 | ) | — | 170,000 | (146,573 | ) | |||||||||||||
| Principal payments under capital lease obligations | (1,404 | ) | — | — | — | — | ||||||||||||||
| Net change in borrowings under lines of credit | (52,060 | ) | — | — | — | — | ||||||||||||||
| Payment upon extinguishments of note | — | (500,212 | ) | — | — | — | ||||||||||||||
| Proceeds from issuance of common stock | 4,419,831 | 17,853,987 | 56,101 | 4,100 | — | |||||||||||||||
| Proceeds from bridge notes payable | 600,000 | — | — | — | — | |||||||||||||||
| Issuance of promissory note for acquisition of technology | — | 4,000,000 | — | — | — | |||||||||||||||
| Payment of promissory note for acquisition of technology | — | (4,000,000 | ) | — | — | — | ||||||||||||||
| Issuance of warrants | — | 1,358,395 | — | — | — | |||||||||||||||
| Net proceeds from issuance of preferred stock | — | — | 12,968,559 | — | — | |||||||||||||||
| Net cash provided by (used in) financing activities | 5,024,276 | 18,654,261 | 13,024,660 | 174,100 | (146,573 | ) | ||||||||||||||
| Net increase (decrease) in cash and cash equivalents | 801,907 | 6,975,097 | (4,256,988 | ) | (2,164,494 | ) | (1,507,904 | ) | ||||||||||||
| Cash and cash equivalents at the beginning of the period | 736,383 | 1,538,290 | 8,513,387 | 8,513,387 | 4,256,399 | |||||||||||||||
| Cash and cash equivalents at the end of the period | $ | 1,538,290 | $ | 8,513,387 | $ | 4,256,399 | $ | 6,348,893 | $ | 2,748,495 | ||||||||||
Supplemental schedule of cash flow information | ||||||||||||||||||||
| Cash paid during the period for interest | $ | 16,425 | $ | 116,999 | $ | — | $ | — | $ | — | ||||||||||
Supplemental Schedule of Noncash Investing and Financing Activities: | ||||||||||||||||||||
| Accretion of undeclared dividends on Series A/D | ||||||||||||||||||||
| Redeemable Convertible Preferred Stock | $ | 600,472 | $ | 600,452 | $ | 1,167,466 | $ | 150,116 | $ | 433,616 | ||||||||||
| Reversal of undeclared dividends on Series B | ||||||||||||||||||||
| Redeemable Convertible Preferred Stock | (299,441 | ) | — | — | — | — | ||||||||||||||
| Fair value of stock warrants issued for consulting services and placement agreement amendment | 916,647 | — | 173,909 | 173,909 | — | |||||||||||||||
| Fair value of stock warrants issued for patents | — | 35,870 | — | — | — | |||||||||||||||
| Fair value of stock warrants issued for bridge notes | 65,325 | 273,327 | — | — | — | |||||||||||||||
| Fair value of stock warrants issued in connection with private placement | — | 1,358,395 | — | — | — | |||||||||||||||
| Fair value of beneficial conversion feature of stock warrants issued for convertible subordinated notes | 32,961 | — | — | — | — | |||||||||||||||
| Issuance of common stock upon conversion of convertible subordinated notes | 2,064,686 | — | — | — | — | |||||||||||||||
| Note issued for acquisition of technology and related inventory and intangibles | — | 15,000,000 | 15,000,000 | — | — | |||||||||||||||
| Preferred stock issued for acquisition of technology | — | 4,000,000 | — | — | — | |||||||||||||||
See accompanying notes.
F-6
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements
(Information pertaining to March 31, 2007 and the three months
ended March 31, 2006 and 2007 is unaudited)
Notes to Consolidated Financial Statements
(Information pertaining to March 31, 2007 and the three months
ended March 31, 2006 and 2007 is unaudited)
| 1. | Business Description |
ImaRx Therapeutics, Inc. (the Company or ImaRx) is a biopharmaceutical company focused on developing and commercializing therapies for vascular disorders. The Company has devoted substantially all of its efforts towards the research and development of its product candidates and the commercialization of its currently marketed product, Abbokinase®.
ImaRx was organized as an Arizona limited liability company on October 7, 1999, which was the Company’s date of inception for accounting purposes. The Company was subsequently converted to an Arizona corporation on January 12, 2000, and then reincorporated as a Delaware corporation on June 23, 2000.
During September 2006, the Company began selling the Abbokinase product to wholesale distributors and exited the development stage, as defined by Statement of Financial Accounting Standards (SFAS) No. 7,Accounting and Reporting by Development Stage Enterprises.
| 2. | Significant Accounting Policies |
Consolidation
The consolidated financial statements include the accounts of the Company and its consolidated subsidiaries, ImaRx Oncology, Ltd. (IOL) and ImaRx Europe Limited (IEL). On January 19, 2001, the Company acquired an 80.1% ownership interest in IOL, a Bermuda limited liability company formed for the purpose of joint development activities between ImaRx and its development partner. The development partner owned the remaining 19.9% of IOL until October 2, 2002, when a termination agreement was entered into between the parties. The Company acquired the remaining 19.9% interest in IOL in exchange for an interest in future royalties. Since October 2, 2002, IOL has been a wholly owned subsidiary of ImaRx. The dissolution of IOL was completed on March 9, 2007. IEL is a wholly owned subsidiary created in 2005 by the Company to facilitate clinical trials in Europe. It was later determined that the European subsidiary was not required and IEL was dissolved in December 2006 with no activity reported for the period. All significant inter-company accounts and transactions have been eliminated.
Interim Financial Information
The financial statements at March 31, 2007 and for the three months ended March 31, 2007 and 2006 are unaudited. The unaudited financial statements have been prepared on the same basis as the audited financial statements and, in the opinion of management, include all adjustments, consisting only of normal recurring adjustments, necessary to state fairly the financial information therein in accordance with the U.S. generally accepted accounting principles (GAAP). The results of operations for the three months ended March 31, 2007 are not necessarily indicative of the results that may be reported for the year ending December 31, 2007.
Basis of Presentation
The Company will require additional funding in the future and may seek to do so through collaborative arrangementsand/or public or private financings. If the Company is unable to obtain funding on a timely basis, the Company may be required to significantly curtail certain of its sales and marketing efforts, its development efforts with respect to its product candidates and may be required to limit, scale back or cease its operations. The Company’s ability to continue as a going concern depends on the successful future sales of the Abbokinase product acquired in 2006, and the commercialization or licensing of its technologies. For the period from October 7, 1999 (date of inception) to March 31, 2007, the Company has had historical recurring losses, which have resulted in a significant accumulated deficit at March 31, 2007. These conditions, among
F-7
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
others, raise substantial doubt about the Company’s ability to continue as a going concern. The financial statements do not include any adjustments relating to the recoverability and classification of recorded assets, or the amounts and classification of liabilities that might be necessary in the event the Company cannot acquire additional financing. The Company is attempting to sell a sufficient amount of Abbokinase to provide capital to fund operations in the near term future. However, the Company’s ability to continue as a going concern depends on the successful commercialization or licensing of its technologies. In addition, management is seeking to secure additional capital as may be required.
Use of Estimates
The consolidated financial statements have been prepared by the Company in accordance with U.S. generally accepted accounting principles (GAAP). Conformity with GAAP requires the use of estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results may differ from these estimates.
Cash Equivalents
The Company considers all highly liquid investments with a maturity of three months or less when purchased to be cash equivalents. Cash equivalents are recorded at cost, which approximates market value.
Fair Value of Financial Instruments
The Company measures its financial assets and liabilities in accordance with GAAP. The carrying amounts of financial instruments, including cash and cash equivalents, accounts payable, accrued expenses and notes payable, approximate fair value based on the liquidity or on the short-term maturities of these financial instruments.
Accounts Receivable
Accounts receivable consist of amounts due from wholesalers for the purchase of Abbokinase and are recorded net of allowances for sales discounts and prompt payment discounts. To date the Company has not recorded a bad debt allowance due to the fact that the majority of its product revenue comes from sales to a limited number of financially sound wholesale distributors. The need for bad debt allowance is evaluated each reporting period based on our assessment of the creditworthiness of our customers.
Inventory
Inventory is comprised of finished goods and is stated at the lower of cost or market value. The Company has one commercially available product, marketed as a clot-dissolving, or thrombolytic urokinase drug called Abbokinase. Abbokinase is FDA approved and marketed for the treatment of acute massive pulmonary embolism. Cost was determined as a result of the purchase price allocation from the acquisition of Abbokinase from Abbott Laboratories (Abbott) in 2006. Approximately $16.7 million of the $20.0 million purchase price for Abbokinase was allocated to the vials the Company expects hospitals to purchase. Of the vials of Abbokinase held in inventory either by the Company or by its wholesalers as of March 31, 2007, approximately 64% of the vials the Company expects hospitals to purchase, or approximately $10.7 million in inventory value, will expire by October 2007 based on current stability data. The remaining approximately 36% of the vials the Company expects hospitals to purchase, or approximately $6.1 million in inventory value, will expire at various times up to August 2009. The Company has an ongoing stability program to allow for expiration date extensions. The next testing point of the ongoing stability program, at which we may obtain data sufficient to extend the expiration dates of our unlabeled inventory, will be completed in the fall of 2007. If the parameters tested are within the specifications previously approved by the FDA, the Company may then label vials with extended expiration dating at that time to between June and August 2009. The Company must
F-8
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
obtain FDA approval for each lot release of inventory. Inventory is labeled with an expiration date upon approval of a lot release by the FDA. Once labeled, we cannot extend the expiration date of the vials labeled. If we are successful in extending the expiration dates of our unlabeled inventory, we intend to continue the stability program after the fall of 2007 to potentially enable further expiration extensions for future product labeling. The Company periodically reviews the composition of inventory in order to identify obsolete, slow-moving or otherwise un-saleable inventory. The Company will provide a valuation reserve for estimated obsolete or un-saleable inventory in an amount equal to the difference between the cost of the inventory and the estimated market value based upon assumptions about future demand and market conditions. Management believes the expiration dates will be extended. Refer to Note 12. In January 2007, we purchased approximately 1,600 vials of Abbokinase, previously sold by Abbott, from one of our wholesale distributors. These vials were placed into inventory the Company expects to sell.
Inventory Subject to Return
Inventory subject to return is comprised of finished goods, stated at the lower of cost or market value, and represents the amount of inventory that has been sold to wholesale distributors. When product is sold by the wholesale distributor to a hospital or other health care provider, a reduction in this account occurs and cost of sales is recorded.
Property and Equipment
All property and equipment are recorded at cost and depreciated over their estimated useful lives, ranging from three to seven years, using the straight-line method. Leasehold improvements are amortized using the straight-line method over the lesser of the lease term or the estimated useful life.
Intangible Assets
Intangible assets include customer relationships, trade name, contracts and technology and are accounted for based on SFAS No. 142,Goodwill and Other Intangible Assets. Intangible assets with finite useful lives are amortized over the estimated useful lives from the date of acquisition, ranging from one to four years, using the straight-line method. The Abbokinase trade name has an estimated life of one year.
Long-Lived Assets
In accordance with SFAS No. 144,Accounting for the Impairment or Disposal of Long-Lived Assets, if indications of impairment exist, the Company assures the recoverability of the affected long-lived assets by determining whether the carrying value of such assets can be recovered through undiscounted future cash flows. If impairment is indicated, the Company measures the amounts of such impairments by comparing the carrying value of the asset to the present value of the expected cash flows associated with the use of the asset. Although the Company has accumulated losses since inception, the Company believes that future cash flows will exceed the carrying value of the Company’s long-lived assets.
Revenue Recognition
The Company provides research services under certain contract and grant agreements, including federal grants from the National Institutes of Health. The Company recognizes revenue for these research services as the services are performed. Revenue from grants is recognized over the contractual period of the related award.
Revenue from product sales is recognized pursuant to Staff Bulletin No. 104 (SAB 104),Revenue Recognition in Financial Statements. Accordingly, revenue is recognized when all four of the following criteria are met: (i) persuasive evidence that an arrangement exists; (ii) delivery of the products has occurred;
F-9
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
(iii) the selling price is both fixed and determinable; and (iv) collectibility is reasonably assured. The Company applies SFAS No. 48,Revenue Recognition When the Right of Return Exists, which amongst other criteria requires that future returns can be reasonably estimated in order to recognize revenue. The amount of future returns is uncertain due to the lack of returns history data. Due to the uncertainty of returns, the Company is accounting for these product shipments to wholesale distributors using a deferred revenue recognition model. Under the deferred revenue model, the Company does not recognize revenue upon product shipment to wholesale distributors; therefore, recognition of revenue is deferred until the product is sold by the wholesale distributor to a hospital or other health care providers expected to be the end user. The Company’s returns policy allows end users to return product within 12 months after expiration, but current practice by wholesalers and end users is a “just in time” purchasing methodology, meaning that the product is purchased on an as-needed basis, typically on a daily or weekly basis. Based on input from our wholesalers, current purchasing practices and the estimated amount of product in the channel, the Company anticipates immaterial product returns from hospitals.
The Company’s customers consist primarily of large pharmaceutical wholesalers who sell directly to hospitals and other healthcare providers. Provisions for product returns and exchanges, sales discounts, chargebacks, managed care and Medicaid rebates and other adjustments are established as a reduction of product sales revenues at the time such revenues are recognized. These deductions from gross revenue are established by the Company’s management as its best estimate at the time of sale adjusted to reflect known changes in the factors that impact such reserves.
Stock-Based Compensation
The Company maintains performance incentive plans under which incentive and non-qualified stock options are granted primarily to employees and non-employee directors. Prior to January 1, 2006, the Company accounted for stock-based compensation in accordance with Accounting Principles Board Opinion No. 25 (APB No. 25),Accounting for Stock Issued to Employees, SFAS No. 123,Accounting for Stock Based Compensation, and related interpretations. The Company’s policy is to grant all stock options at the fair market value of the underlying stock at the date of grant. Accordingly, no compensation expense was required to be recognized for the stock options at the date of grant prior to January 1, 2006. For non-employee grants issued prior to January 1, 2006, the calculation of expense was determined using the Black-Scholes option pricing model. The Company calculated the expense using the exercise price of the option, the fair market value of the underlying stock at the date of the grant, the expected volatility of the stock price, the life of the option and the risk-free interest rate. The expense was recorded in accordance with the vesting period of the option.
In December 2004, the Financial Accounting Standards Board (FASB) issued SFAS No. 123 (revised 2004) that supersedes APB No. 25. SFAS No. 123(R) requires that the cost of share-based payment transactions (including those with employees and non-employees) be recognized in the financial statements. For non-employees, this expense is recognized as the service is provided, in accordance with guidance in EITF IssueNo. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services. SFAS No. 123(R) applies to all share-based payment transactions in which an entity acquires goods or services by issuing (or offering to issue) its shares, share options, or other equity instruments or by incurring liabilities (1) in amounts based on the price of the entity’s shares or other equity instruments, or (2) that require (or may require) settlement by the issuance of an entity’s shares or other equity instruments.
Effective January 1, 2006, the Company adopted SFAS 123(R), requiring measurement of the cost of employee services received in exchange for all equity awards granted, based on the fair market value of the award as of the grant date. Under this standard, the fair value of each employee stock option is estimated on the date of grant using an option pricing model that meets certain requirements. The Company currently uses
F-10
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
the Black-Scholes option pricing model to estimate the fair value of our share-based payments. The determination of the fair value of share-based payment awards utilizing the Black-Scholes model is affected by our stock price and a number of assumptions, including expected volatility, expected life, risk-free interest rate and expected dividends. The Company uses guideline companies to determine volatility. The expected life of the stock options is based on historical data and future expectations. The risk-free interest rate assumption is based on observed interest rates appropriate for the terms of our stock options. The dividend yield assumption is based on our history and expectation of dividend payouts. Stock-based compensation expense recognized in our financial statements in 2006 and thereafter is based on awards that are ultimately expected to vest. The amount of stock-based compensation expense in 2006 and thereafter will be reduced for estimated forfeitures. Forfeitures are required to be estimated at the time of the grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company will evaluate the assumptions used to value stock awards on a quarterly basis. If factors change and the Company employs different assumptions, stock-based compensation expense may differ significantly from what has previously been recorded. To the extent that the Company grants additional equity securities to employees, the stock-based compensation expense will be increased by the additional compensation resulting from those additional grants. The Company has adopted SFAS 123(R) using the prospective application method of adoption which requires recording compensation cost related to awards granted on or after January 1, 2006 based on the fair value related to stock options at the grant dates.
The weighted-average expected option term for the year ending December 31, 2006 reflects the application of the simplified method set out in SEC Staff Accounting Bulletin No. 107 (SAB 107), which was issued in March 2005. The simplified method defines the life as the average of the contractual term of the options and the weighted-average vesting period for all option tranches. Estimated volatility for period ended March 31, 2007 also reflects the application of SAB 107 interpretive guidance and, accordingly, incorporates historical volatility of similar entities whose share prices are publicly available. Volatility for 2004 and 2005 was based on the minimum value method.
The Company issued no options during the three months ended March 31, 2007. The Company recorded approximately $163,000 in stock-based compensation expense related to options granted prior to January 1, 2007 for the three months ended March 31, 2007 and there was no income tax benefit related to this expense.
The pro forma amounts required by SFAS No. 123 was applied to the stock-based compensation during the years ended December 31, 2004 and 2005. The pro forma effect on net loss was determined as if the fair value of the stock-based compensation had been recognized as compensation expense on a straight-line basis over the vesting period of the stock options in each period.
Pro forma information regarding net loss is required by SFAS No. 123 which requires that the information be determined as if the Company has accounted for its employee stock options granted during the years ended December 31, 2004 and 2005, under the fair value method of SFAS 123. The deemed fair value for options granted was estimated at the date of grant using the minimum value option valuation model, which assumes the stock price has no volatility since the common stock is not publicly traded. The following assumptions were used to calculate the deemed fair value of the option awards at the date of grant: no dividend payout expected, expected option life of five years and a risk-free interest rate averaging 3% for the years ended December 31, 2004 and 2005. The weighted-average estimated fair value of stock options granted with an exercise price equal to the fair value of the underlying common stock on the date of the grant during fiscal years 2004 and 2005 were $0.72 and $1.03 respectively.
The minimum value option valuation model was developed for use in estimating the fair value of traded options that have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of highly subjective assumptions, including the expected life of the option. Because, among other things, changes in the subjective input assumptions can materially affect the fair value estimate, in management’s opinion, the existing models do not necessarily provide a reliable single measure of the fair
F-11
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
value of its stock options. For purposes of pro forma disclosures, the deemed fair value of the options is amortized to expense over the vesting periods.
If compensation for options granted under the plan had been determined based on the deemed fair value at the grant date consistent with the method provided under SFAS 123, then the Company’s net loss would have been as indicated in the pro forma amounts below:
| Years Ended December 31, | ||||||||
| 2004 | 2005 | |||||||
| Net loss attributable to common stockholders: | ||||||||
| As reported | $ | (6,024,839 | ) | $ | (28,526,789 | ) | ||
| Pro forma SFAS No. 123 expense | 56,504 | 141,781 | ||||||
| Pro forma | $ | (6,081,343 | ) | $ | (28,668,570 | ) | ||
| Years Ended December 31, | ||||||||
| 2004 | 2005 | |||||||
| Net loss attributable to common stockholders per share — basic and diluted: | ||||||||
| As reported | ||||||||
| Basic and diluted | $ | (5.37 | ) | $ | (15.11 | ) | ||
| Pro forma loss attributable to common stockholders per share — basic and diluted: | ||||||||
| Basic and diluted | $ | (5.41 | ) | $ | (15.18 | ) | ||
Research and Development Expenses
Research and development costs primarily consist of salaries and related expenses for personnel, fees paid to consultants and outside service providers, facilities costs, and the costs associated with clinical trials and research and development. The Company charges all research and development expenses to operations as incurred.
Shipping and Handling
Costs related to shipping and handling are charged to general and administrative expense as incurred.
Income Taxes
The Company accounts for income taxes under the liability method pursuant to SFAS No. 109,Accounting for Income Taxes. Under the liability method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when the Company determines that it is more likely than not that some portion or all of a deferred tax asset will not be realized.
We adopted the Financial Accounting Standards Board’s Interpretation No. 48, “Accounting for Uncertainty in Income Taxes, an Interpretation of FASB Statement No. 109” (“FIN 48”), effective January 1, 2007, FIN 48 clarifies the accounting for uncertainty in income taxes recognized in financial statements and requires the impact of a tax position to be recognized in the financial statements if that position is more likely than not of being sustained by the taxing authority. The adoption of FIN 48 had no effect on our consolidated financial position or results of operations.
F-12
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Concentration of Credit Risk and Limited Suppliers
The Company has no significant off-balance sheet concentrations of credit risk such as foreign exchange contracts, options contracts or other foreign hedging arrangements. Financial instruments that potentially subject the Company to credit risk consist principally of cash investments and uncollateralized accounts receivable. The Company maintains the majority of its cash balances in the form of cash deposits in bank checking and money market accounts with a highly rated commercial bank.
The Company relies on certain materials used in its research and development processes which are procured from single sources. The failure of any of these suppliers to deliver the materials could delay or interrupt the development timelines and thereby adversely affect the Company’s operating results.
Net Loss Attributable to Common Stockholders per Share
Basic and diluted net loss attributable to common stockholders per share is calculated by dividing the net loss applicable to common stockholders by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is the same as basic net loss per common share for all periods presented. The effects of potentially dilutive securities are antidilutive in the loss periods.
The following potential common shares have been excluded from the computation of diluted net loss per share since their effect would be antidilutive in each of the loss periods presented:
| Years-Ended December 31, | Three Months Ended March 31, | |||||||||||||||
| 2004 | 2005 | 2006 | 2007 | |||||||||||||
| (Unaudited) | ||||||||||||||||
| Convertible preferred stock | 865,796 | 1,065,796 | 3,448,189 | 3,448,189 | ||||||||||||
| Stock options | 393,552 | 534,143 | 630,351 | 622,709 | ||||||||||||
| Warrants | 191,665 | 337,324 | 352,324 | 352,324 | ||||||||||||
Recently Issued Accounting Pronouncements
In September 2006, the FASB issued SAFS No. 157,Fair Value Measurements. SFAS 157 defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements. SFAS 157 applies under other accounting pronouncements that require or permit fair value measurements, the FASB having previously concluded in those accounting pronouncements that fair value is the relevant measurement attribute. Accordingly, SFAS 157 does not require any new fair value measurements, but may change current practice for some entities. SFAS 157 is effective for fiscal years beginning after December 15, 2006. The adoption of SFAS No. 157 is not expected to have a material effect on the Company’s financial position or results of operations.
In September 2006, the SEC issued Staff Accounting Bulletin No. 108,Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements, which provides interpretive guidance on the consideration of the effects of prior year misstatements in quantifying current year misstatements for the purpose of a materiality assessment. SAB No. 108 requires registrants to quantify misstatements using both the balance sheet and income statement approaches and to evaluate whether either approach results in quantifying an error that is material based on relevant quantitative and qualitative factors. The guidance is effective for the first fiscal period ending after November 15, 2006. The adoption of SAB No. 108 is not expected to have any impact on these financial statements.
F-13
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
| 3. | Balance Sheet Data |
Property and Equipment
Property and equipment consist of the following:
| December 31, | At March 31, | |||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (Unaudited) | ||||||||||||
| Leasehold improvements | $ | 622,663 | $ | 627,963 | $ | 627,963 | ||||||
| Laboratory machinery and equipment | 937,697 | 1,557,039 | 1,728,156 | |||||||||
| Computer and communications equipment | 347,856 | 374,104 | 374,104 | |||||||||
| Office furniture and equipment | 184,377 | 223,471 | 227,163 | |||||||||
| Construction in progress | 292,229 | — | 27,900 | |||||||||
| 2,384,822 | 2,782,577 | 2,985,286 | ||||||||||
| Less accumulated depreciation | 1,654,861 | 1,865,611 | 1,928,862 | |||||||||
| $ | 729,961 | $ | 916,966 | $ | 1,056,424 | |||||||
For the years ended December 31, 2004, 2005 and 2006 and the three months ended March 31, 2006 and 2007, the Company recorded depreciation expense of $185,905, $194,206, $248,586, $60,063 and $63,251, respectively.
Intangible Assets
Intangibles consist of the following:
| December 31, | At March 31, | |||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (Unaudited) | ||||||||||||
| Customer lists | $ | — | $ | 2,700,000 | $ | 2,700,000 | ||||||
| Trade name | — | 500,000 | 500,000 | |||||||||
| Cell technology | — | 100,000 | 100,000 | |||||||||
| — | 3,300,000 | 3,300,000 | ||||||||||
| Less accumulated amortization | — | 800,000 | 1,100,000 | |||||||||
| $ | — | $ | 2,500,000 | $ | 2,200,000 | |||||||
For the year ended December 31, 2006 and the three months ended March 31, 2007, the Company recorded amortization expense of $800,000 and $300,000 respectively. As of December 31, 2006, the Company expects the amortization of the intangible assets for the next four years to be as follows:
| 2007 | $ | 700,000 | ||
| 2008 | 700,000 | |||
| 2009 | 700,000 | |||
| 2010 | 400,000 | |||
| $ | 2,500,000 | |||
F-14
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Accrued Expenses
Accrued expenses consist of the following:
| December 31, | At March 31, | |||||||||||
| 2005 | 2006 | 2007 | ||||||||||
| (Unaudited) | ||||||||||||
| Accrued compensation | $ | 118,940 | $ | 250,060 | $ | 392,921 | ||||||
| Accrued contract services | 574,024 | 176,048 | 365,170 | |||||||||
| Accrued chargebacks and administrative fees | — | 319,344 | 345,189 | |||||||||
| Accrual for inventory purchase | — | — | 694,430 | |||||||||
| Other accrued expenses | 200,234 | 490,058 | 503,093 | |||||||||
| $ | 893,198 | $ | 1,235,510 | $ | 2,300,803 | |||||||
| 4. | Income Taxes |
A reconciliation of the U.S. federal statutory income tax rate to the effective rate follows.
| Years Ended December 31, | ||||||||||||
| 2004 | 2005 | 2006 | ||||||||||
| Tax benefit at statutory rate | $ | (1,946,000 | ) | $ | (9,245,000 | ) | $ | (237,000 | ) | |||
| State taxes (net of federal benefit) | (385,000 | ) | (1,328,000 | ) | 57,000 | |||||||
| Foreign rates lower than U.S. statutory rates | 61,000 | — | — | |||||||||
| Net benefit from research and development credits | (91,000 | ) | (129,000 | ) | (23,000 | ) | ||||||
| Stock compensation | — | — | 118,000 | |||||||||
| Other, net | 11,000 | 393,000 | 15,000 | |||||||||
| Valuation allowance | 2,350,000 | 10,309,000 | 70,000 | |||||||||
| Tax benefit at statutory rate | $ | — | $ | — | $ | — | ||||||
F-15
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred tax assets and liabilities are attributed to the following temporary differences:
| December 31, | ||||||||
| 2005 | 2006 | |||||||
| Current deferred tax assets: | ||||||||
| Reserves and accrued liabilities | $ | 26,000 | $ | 55,000 | ||||
| Other | 3,000 | 5,000 | ||||||
| 29,000 | 60,000 | |||||||
| Noncurrent deferred tax assets: | ||||||||
| Property and equipment | 165,000 | 203,000 | ||||||
| Deferred revenue | — | 257,000 | ||||||
| Intangibles | 9,805,000 | 3,501,000 | ||||||
| Research and development credits | 1,507,000 | 1,324,000 | ||||||
| Net operating loss carryforward | 5,865,000 | 12,156,000 | ||||||
| 17,342,000 | 17,441,000 | |||||||
| Total deferred tax assets | 17,371,000 | 17,441,000 | ||||||
| Valuation allowance | (17,371,000 | ) | (17,441,000 | ) | ||||
| Net deferred tax assets | $ | — | $ | — | ||||
In 2005, the Company purchased certain in-process research and development technologies from Abbott Laboratories which was immediately expensed for financial reporting purposes. For income tax purposes, these in-process research and development technologies were capitalized as an intangible asset and amortized over 15 years. Following the guidance set forth in EITFNo. 96-1,Sale of Put Options on Issuer’s Stock That Require or Permit Cash Settlement,the Company recorded a deferred tax asset for this temporary difference. In 2006, the Company returned the same in-process research and development technologies assets to Abbott Laboratories in exchange for the cancellation of $15 million of purchase money debt. The debt forgiveness resulted in income for financial reporting purposes and a partial recovery of basis for income tax purposes. This difference in treatment resulted in a decrease in current year taxable income (i.e., an increase in net operating loss) and a corresponding decrease in the Company’s deferred tax asset. The remaining deferred tax asset will be recovered over 14 years in accordance with IRC Section 197.
At December 31, 2005 and 2006, the Company had net operating loss carryforwards of approximately $15,474,000 and $31,050,000, respectively, for federal tax purposes that begin to expire in the year 2020. For state income tax purposes, the Company had net operating loss carryforwards at December 31, 2006 of $26,450,000 that expire within five years of being incurred and will begin to expire for state purposes in the year 2007. Additionally, the Company has research and development credit carryforwards of approximately $787,000 and $537,000 that begin to expire in 2020 and 2015 for federal and state purposes, respectively.
For financial reporting purposes, a valuation allowance of $17,371,000 and $17,441,000 has been established at December 31, 2005 and 2006, respectively, to offset deferred tax assets relative to the net operating loss carryforwards and other deferred tax assets. The gross deferred tax assets resulted from accumulated net operating loss carryforwards since inception. Pursuant to SFAS 109, the Company has established a valuation allowance against the entire tax asset. As a result, the Company does not recognize any tax benefit until the Company is in a tax paying position, and therefore, more likely to realize the tax benefit. The Company’s valuation allowance changed by $2,350,000, $10,309,000 and $70,000 during the years ended December 31, 2004, 2005 and 2006, respectively. Equity offerings by the Company, and other transactions which may impact the Company’s ownership structure, have triggered IRC Section 382 and 383
F-16
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
provisions, which may limit or eliminate the potential future tax benefit to be realized by the Company from its accumulated net operating loss carryforwards and research and development credits.
For December 31, 2006, the Company realized a tax deduction for stock-based compensation on non-qualified stock options that gave rise to $700,000 tax deduction. The benefit of the deduction will be recognized as an adjustment to paid-in capital at a point in time when a valuation allowance is not required.
| 5. | Investment in ImaRx Oncology, Ltd. |
During 2001, the Company entered into a joint venture agreement with a development partner to form ImaRx Oncology, Ltd. (IOL) for the development of certain patents and technology. Upon the formation of IOL, the Company acquired an 80.1% interest in IOL by purchase of 100% of IOL’s voting common shares for $5,000,000 and 60.2% of IOL’s preferred shares for $3,010,000, representing a total of 80.1% of IOL’s outstanding shares. The development partner acquired the remaining 39.8% of IOL’s preferred shares for $1,990,000, representing a total of 19.9% of IOL’s outstanding shares.
On October 2, 2002, the Company entered into a termination agreement (Termination Agreement) of the joint venture with the development partner whereby the Company acquired the remaining 19.9% interest in IOL in exchange for consideration equal to approximately $56,279 plus future contingent consideration in the form of a net royalty interest in the sale, licensing or other commercialization proceeds, as defined in the Termination Agreement, of all IOL operations. This acquisition cost was expensed to research and development in 2002 at the time the Company entered into the Termination Agreement. IOL received funding pursuant to a convertible promissory note (Development Note) with the development partner for funding of the development partner’s pro rata share of the development costs.
Under the Termination Agreement, the Development Note was amended and restated (Restated Development Note) to provide for funding by the development partner up to a maximum principal amount of $3,610,076. Refer to Note 7 for the terms of the Restated Development Note and its extinguishment in full in March 2005. The Company completed the dissolution of IOL on March 9, 2007.
| 6. | Related Party Transactions |
The Company leases its office facility from a partnership whose beneficial owners include a member of the Board of Directors of the Company. Rent expense related to this lease, which has a remaining life of two years, amounted to approximately $60,000 in 2004 and $64,000 in both 2005 and 2006, and $16,000 for the three months ended March 31, 2007. The Company’s related party rent expense will be approximately $64,000 and 2007 and $54,000 in 2008 for the office facility.
In October 2006, the Company entered into a separation agreement with the former CEO and member of the Board of Directors. The separation agreement provided for a severance payment of $250,000, which was charged to expense.
| 7. | Notes Payable |
Note Payable to Development Partner
On March 6, 2005, the Company executed a Securities Purchase Agreement with its former development partner (refer to Note 5) whereby the outstanding principal and accrued interest totaling $4,335,171 as of that date was purchased by the Company for $500,212, resulting in a gain on the extinguishment of $3,834,959. No other consideration was given by the Company in connection with the Securities Purchase Agreement.
F-17
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Secured Promissory Notes Payable
In September 2005, $4,000,000 in secured promissory notes were issued by the Company for cash. The notes were secured by all assets of the Company other than those represented by research and development stage technologies acquired by the Company during September 2005 from Abbott In October 2005, the Company repaid the notes in full.
Note Payable for Technology Acquisition
In September 2005, the Company entered into an agreement with Abbott to acquire certain assets related to Abbott’s development of recombinant pro-urokinase (rproUK) and recombinant urokinase (rUK) (Abbott Agreement). The total purchase price under the Abbott Agreement of $24,000,000 included a payment of $5,000,000 in cash, a $15,000,000 note payable to Abbott and the issuance of 1,000,000 shares of Series E valued at $4.00 per share.
The purchase of these assets did not constitute the purchase of a business as defined in EITFNo. 98-3,Determining Whether a Nonmonetary Transaction Involves Receipt of Productive Assets or of a Business. Assets included in the purchase, among others, were rproUK and rUK drug substance and drug product inventories, raw materials including master and working cell banks, intellectual property related to these drug products, rights under existing contractual agreements and all related applications and supplements filed with the U.S. Food and Drug Administration (FDA). Although these product candidates may have significant future importance, the Company determined that, since they had not yet received FDA approval and presented no alternative future use, they did not meet established guidelines for technological feasibility sufficiently to be recorded as assets. As a result, the full amount of the purchase price of $24,000,000 was expensed as acquired in-process research and development expense in September 2005.
In November 2006, the Company and its Board of Directors made the decision to return these assets to Abbott. The Company notified Abbott on December 13, 2006 of its intent to default on the note and return the purchased assets. Abbott sent a default notice to ImaRx on December 31, 2006 confirming receipt of the Company’s intent and plan for the return of assets. The outstanding principal and accrued interest as of December 31, 2006 totaling $16,127,500 was written-off by the Company, resulting in an extinguishment of debt. The default had no impact on the Company’s ownership of the inventory and rights also purchased from Abbott for cash and a separate $15,000,000 note.
Note Payable for Asset Acquisition
In connection with an Asset Purchase Agreement dated April 25, 2006 with Abbott for the purchase of inventory and related intangibles, the Company issued a $15,000,000 secured promissory note payable. The note is due December 31, 2007, accrues interest at 6% annually and is secured by the Company’s right, title and interest in the purchased assets. The balance outstanding at December 31, 2006 and the three months ended March 31, 2007 is $15,615,000 and $15,840,000, respectively. Refer to Note 11 for a description of the asset acquisition.
| 8. | Equity Transactions |
Preferred Stock
The Company has authorized a total of 30,000,000 shares as preferred stock. At March 31, 2007, the following series of stock were issued and outstanding.
F-18
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Series A, D and F Preferred Stock
The Company entered into a Series A Preferred Stock Purchase Agreement in August 2000, a Series D Preferred Stock Purchase Agreement in October 2002 and a Series F Preferred Stock Purchase Agreement in April 2006 (collectively, Series A/D/F). The Series A/D/F shares have a par value of $0.0001. In the event of liquidation, the holders of Series A/D/F shares are entitled to receive preference to any distributions of the assets of the Company equal to the original purchase price of the shares and cumulative accrued dividends of 8% per year, less the amount of any dividends actually paid. The shares are convertible into the Company’s common stock based on the original issue price, subject to certain adjustments. In the event dividends are paid on any shares of common stock, the holders of the Series A/D/F shares will be entitled, if and when declared by the Board of Directors of the Company, to dividends based on the number of shares of common stock into which the Series A/D/F shares are convertible. Series A preferred stock (Series A) has a conversion rate of 0.209 or 479,136 shares of common stock as of March 31, 2007. The conversion rate for Series D preferred stock (Series D) is 0.275 or 120,513 shares of common stock at March 31, 2007. The per share conversion rate of Series F preferred stock (Series F) is variable and will be determined by dividing $5.00 by the lesser of (a) $25.00 (as adjusted for any stock dividends, combinations, splits, recapitalizations and the like with respect to such shares) or (b) 85% of the price per share paid in an initial public offering. Therefore, depending on the price of the shares sold in an initial public offering, the holders of the Series F may receive more than one share of common stock for each share of Series F preferred stock converted in connection with an initial public offering. The Company will not know the conversion rate of the Series F until after a public offering price has been determined. The beneficial conversion is a contingent conversion as contemplated by EITF Issue No. 00-27,Application of Issue No. 98-5 to Certain Convertible Instruments. Upon completion of an initial public offering, a deemed dividend would be recorded if the offering price was lower than $29.41, and if the price were to be $7.00, the deemed dividend would be approximately $12,700,000, payable in the form of shares of common stock. The holders of the Series A/D/F shares are also entitled to the number of votes equal to the number of shares of common stock into which the Series A/D/F shares are convertible.
At any time after December 31, 2008, holders of a majority of Series A/D/F shares may elect to redeem, in three annual installments, their shares in cash.
During 2000, the Company issued 1,294,772 shares of Series A and received net proceeds of $3,719,313. Additionally, the Company converted unsecured notes plus accrued interest in the amount of $2,442,205 into 880,075 shares of Series A. During 2001, an additional 116,297 shares of Series A were issued, including certain shares subscribed in 2000, and received net proceeds of $305,905. In October 2002, the Company issued 310,232 shares of Series D and received net proceeds of $833,031. In January 2003, the Company issued an additional 128,000 shares of Series D and received net proceeds of $350,371. In April 2006, the Company issued 2,070,000 shares of Series F and received net proceeds of $9,623,500. In May 2006, the Company issued an additional 765,000 shares of Series F and received net proceeds of $3,345,059.
Cumulative undeclared dividends on Series A, D and F were $2,884,297, $4,051,763 and $4,485,379 at December 31, 2005, 2006 and March 31, 2007, respectively. In connection with consulting services received during the Series A equity financing, in March 2001, warrants were issued for 614 shares of common stock with an exercise price of $35.00 per share. The warrants are exercisable at any time for a period of ten years from the date of issue. The fair value of the warrants was charged to paid-in capital as a cost of issuance of the Series A in the amount of $3,514.
Series B Preferred Stock
In connection with the formation of the IOL joint venture with its development partner in January 2001, the Company entered into a Securities Purchase Agreement (Agreement). Under the Agreement, the Company issued 500,625 shares of Series B preferred stock (Series B) and 285,714 shares of Series C preferred stock
F-19
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
(Series C), each having a par value of $.0001. Net proceeds from the issuance of the Series B shares were $8,010,000. These proceeds were used to acquire the Company’s 80.1% interest in the IOL joint venture.
The holder of Series B is entitled to receive preference to any distributions of the assets of the Company equal to the original purchase price of the shares ($16.00). Mandatory Series B dividends were payable every year beginning July 19, 2002, in shares of Series B at the original issue price per share at the rate of 7% of $8,010,000 compounded annually. In March 2004, the holder of Series B permanently waived its right of payment of accrued and unpaid dividends and its right to the accrual on or payment of any future dividends on the Series B shares in exchange for reduction in the adjusted conversion price of the shares to $45.85. Cumulative undeclared dividends on Series B were $299,441 at December 31, 2003. Additional dividends through the subsequent date of declaration in 2003 of $341,094 were declared and paid in 38,809 shares of Series B in 2003. Upon waiver of the right of payment of accrued and unpaid dividends in March 2004, the cumulative undeclared dividends were reversed.
The Series B shares are convertible into the Company’s common stock at $45.85 per share, subject to certain adjustments, at the option of the holder at any time after January 19, 2003, and before December 31, 2008.
To the extent ImaRx has funds legally available for payment, redemption of the Series B is mandatory on December 31, 2008, as in the amount of the liquidation preference, either in cash or shares of common stock of ImaRx (if registered pursuant to a public offering), or in shares of Series C, at the option of the Company.
Series C Preferred Stock
Net proceeds of $1,945,563 were received from the sale of the Series C preferred stock (Series C), also in connection with the formation of the IOL joint venture in January 2001.
The holder of the Series C is entitled to receive preference to any distributions of the assets of the Company equal to the $7.00 original purchase price of the shares. The Series C is convertible into the Company’s common stock at a conversion price of $33.80 per share, subject to certain adjustments, which price reflects an adjustment to the Series C conversion price made in conjunction with amendments to the terms of the Series B in 2004. The conversion privilege at the option of the holder is exercisable at any time before December 31, 2008.
To the extent ImaRx has funds legally available for payment, redemption of the Series C is mandatory on December 31, 2008, in the amount of the liquidation preference either in cash or shares of common stock (if registered pursuant to a public offering), at the option of the Company.
Series E Preferred Stock
In September 2005, the Company entered into an Asset Purchase Agreement (September Abbott Agreement) with Abbott to acquire certain assets. As partial consideration related to the September Abbott Agreement, the Company issued 1,000,000 shares of Series E preferred stock (Series E) valued at $4.00 per share. The Series E has a par value of $.0001. The Series E is convertible into the Company’s common stock at a conversion price of $20.00 per share, subject to certain adjustments. The holders of Series E participate equally in all dividends payable to common stockholders, if and when declared by the Board of Directors of the Company, based on the number of shares of common stock into which the Series E are convertible.
The Series E has been classified as equity as this series of preferred stock carries neither dividend preferences nor mandatory redemption rights. The redemption is contingent on the following: (i) the Company has not become subject to the public reporting requirements of the Securities Exchange Act of 1934 (1934 Act) before September 30, 2007 and (ii) the Company has sold substantially all of the technologies purchased from Abbott under the Abbott Agreement. The redemption price is $10.00 per share in cash. On
F-20
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
December 13, 2006, the Company notified Abbott of its intent to return the technologies purchased under the Abbott Agreement. Refer to Note 7 for a description of the purchase transaction.
All shares of each series of preferred stock will automatically convert into shares of common stock in connection with the closing of an initial public offering.
Common Stock
In March 2004, the Company issued 500,000 shares of common stock in a private placement at $10.00 per share and received net proceeds of $4,401,666. Additionally, the Company converted convertible subordinated notes plus accrued interest in the amount of $2,064,686 into 206,465 shares of common stock. The holders of the common stock issued in the offering and conversion of the notes are entitled to certain additional rights and privileges, among them (i) the right to put the shares back to the Company at $15.00 per share under certain circumstances in the event of a merger or consolidation or sale of substantially all of the assets of the Company where the Company is not the surviving company, (ii) the right to receive additional shares of common stock of the Company should the Company not become a reporting company under the 1934 Act by September 1, 2007, equal to (A) 10% of the original ($10.00 per share) investment amount and (B) thereafter 5% of the original investment amount each quarter until the reporting requirement is met, (iii) the right to receive additional shares of common stock of the Company should shares subsequently be issued at less than $10.00 per share, based on a formula that takes into account both the reduced price and the number of shares issued at such reduced price, and (iv) “piggy-back” registration rights.
In January and February 2005, the Company completed two closings of a $7,000,000 private placement offering at $15.00 per share for the issuance of 466,663 shares of common stock. These common stock shares include certain additional rights and privileges, among them (i) the right to put the shares back to the Company at $22.50 per share under certain circumstances in the event of a merger or consolidation or sale of substantially all of the assets of the Company where the Company is not the surviving company, (ii) the right to receive additional shares of common stock of the Company should the Company not become a reporting company under the 1934 Act by September 1, 2007, equal to (A) 10% of the original ($15.00 per share) investment amount and (B) thereafter 5% of the original investment amount each quarter until the reporting requirement is met, (iii) the right to receive additional shares of common stock of the Company should shares subsequently be issued at less than $15.00 per share, based on a formula that effectively reduces the per share price paid for the shares of common stock to such reduced price, and (iv) “piggy-back” registration rights. Net proceeds to the Company were $5,520,962, including offset of the value of warrants issued to the placement agent in the offering of $415,200.
In October and November 2005, the Company completed two closings of a private placement of $15,000,000 at $20.00 per share for the issuance of 750,000 shares of common stock. These shares of common stock include certain additional contractual rights and privileges, among them (i) the right to put the shares back to the Company at $30.00 per share under certain circumstances in the event of a merger or consolidation or sale of substantially all of the assets of the Company where the Company is not the surviving company, (ii) the right to receive additional shares of common stock of the Company should the Company not become a reporting company under the 1934 Act, as amended, by November 8, 2007 equal to (A) 10% of the original ($20.00 per share) investment amount and (B) thereafter 5% of the original investment amount each quarter until the reporting requirement is met, (iii) the right to receive additional shares of common stock of the Company should shares subsequently be issued at less than $20.00 per share, based on a formula that effectively reduces the per share price paid for the shares of common stock to such reduced price, and (iv) “piggy-back” registration rights. Net proceeds to the Company were $12,001,828, including offset of the value of warrants issued to the placement agent in the offering of $943,195.
The Company has recorded these shares of stock as equity since redemption is triggered only by a merger, consolidation or sale of assets, which events are within the control of the company. In addition, should
F-21
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
the Company be required to issue additional shares of common stock because it does not become a 1934 Act reporting company by one or more of the dates specified, it will be required to issue additional unregistered shares of common stock. According to SFAS No. 5,Accounting for Contingencies,should it become probable that the Company will be required to issue these additional shares, a liability will be recorded for the fair value of the shares to be delivered with a corresponding charge to earnings. Upon issuance of these additional shares, the liability will be reclassified to permanent equity. Presently, the Company does not believe it is probable that any additional shares must be issued. However, the Company will continue to evaluate facts and circumstances that may influence its assessment of becoming subject to the reporting requirements of the 1934 Act.
Warrants to Purchase Common and Preferred Stock
In January 2001, the Company entered into an equipment line of credit agreement with a bank for $500,000. In connection with the line of credit, the Company issued preferred stock warrants at an exercise price of $2.75 per share to purchase 21,818 shares of Series A. The warrants (determined to have a value of $32,220) were recorded as a loan discount. The warrants were reduced to 10,909 in January 2002, with a fair value of $16,110. The fair value of the warrants was recorded as a loan discount during the term of the outstanding loan. Both the final payments of 8.5% of the advances and the estimated fair value of the warrants were amortized over the term of the equipment line to interest expense. The line of credit expired in 2002, and the Company has no obligations thereunder.
A warrant to purchase 614 shares of common stock with a fair value of $3,514 at date of issue was issued in connection with a consulting agreement in March 2001. The exercise price of the warrant is $35.00 per share and has not been exercised. In addition, a warrant to purchase 2,000 shares of common stock with a fair value of $2,689 at the date of issue was issued in connection with a consulting agreement with a related party on December 1, 2001. The warrant was exercised in March 2004 at a price of $2.50 per share. A warrant to purchase 10,909 shares of Series A with an exercise price of $2.75 per share, was issued in connection with a line of credit in January 2001. The fair value of the warrant, $16,110, was recorded as a loan discount during the term of the loan, and the warrant has not yet been exercised. A warrant to purchase 1,000 shares of common stock with a fair value of $2,688 at date of issue was issued in connection with a licensed patent in October 2003. The exercise price of this warrant is $10.00 per share, and this warrant has not been exercised. A warrant to purchase 4,000 shares of common stock with a fair value of $35,870 at date of issue was issued in connection with a licensed patent in January 2005. The exercise price of this warrant is $15.00 per share and has not been exercised.
In connection with the issuance of the convertible subordinated notes during 2003 and 2004, the Company issued warrants to purchase 29,157 and 12,136 shares of common stock, respectively. The warrants are exercisable at any time for a period of seven years at a price of $10.00 per share. Of these warrants, 3,506 were exercised in October 2005. The value of the warrants was recorded as debt discount and amortized to interest expense using the interest rate method over the maturity of the notes. The fair values of the warrants issued in 2003 and 2004 were $70,000 and $135,325, respectively. On March 30, 2004, the conversion date of the notes, all remaining unamortized debt discount was expensed to interest. In addition, debt discount related to the beneficial conversion feature of the warrants in the amount of $32,961 was expensed to interest at the date of conversion of the notes.
In addition to the warrants for the purchase of shares of common stock issued with the convertible subordinated notes, in January 2004, the Company engaged the services of a financial advisor and an investor relations consultant pursuant to consulting agreements, which provided that each of the two consultants received a warrant to purchase 50,000 shares of the Company’s common stock at an exercise price of $15.00 per share. The warrants are exercisable at any time for a term of five years. The fair value of the warrants totaling $464,378 was charged to expense in 2004.
F-22
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
In October 2004, the Company issued a warrant for the purchase of 50,000 shares of common stock at an exercise price of $10.00 per share to the placement agent of the offering of 500,000 shares of common stock closed in March 2004. The Company issued the warrant and agreed to pay $170,000 upon or immediately prior to closing of a public offering in exchange for the placement agent’s permanent waiver of its right of first refusal to undertake public offerings on behalf of the Company. The warrant is exercisable at any time on a cashless basis for a term of four years. The fair value of the warrant at the date of issue totaling $452,269 was charged to expense in 2004.
A warrant to purchase 46,664 shares of common stock exercisable at any time at $16.50 per share on a cashless basis was issued on February 28, 2005, for a term of five years as provided in the placement agent agreement relative to the $7,000,000 common stock offering. The fair value of the warrant at the date of issue of $415,200 was offset against proceeds of the offering.
In September 2005, warrants to purchase 20,000 shares of common stock were issued in connection with the issuance of $4,000,000 in secured promissory notes. The warrants are exercisable at any time for a period of ten years at a price of $20.00 per share. The value of the warrants were recorded as debt discount and amortized to interest expense over the expected maturity of the notes. The fair value of the warrants outstanding at September 30, 2005 of $273,327 was amortized to interest as the expected maturity date of the notes was less than one month. The warrants were fully expensed in October 2005.
A warrant to purchase 74,996 shares of common stock exercisable at any time at $21.25 per share on a cashless basis was issued on November 8, 2005, for a term of seven years as provided in the placement agent agreement relative to the $15,000,000 common stock offering. The fair value of the warrant at the date of issue of $943,195 was offset against proceeds of the offering.
In connection with a consulting agreement, warrants to purchase 15,000 shares of common stock were issued on February 1, 2006. The warrants are exercisable at any time at $20.00 per share on a cashless basis for a term of seven years. The fair value of the warrant at the date of issue of $173,909 has been recorded as expense.
The following table summarizes the warrants that were outstanding as of March 31, 2007:
Warrants Issued
| Weighted-Average | ||||||||||||
| Warrants | Remaining Life | Warrants | ||||||||||
Exercise Price | Outstanding | in Years | Exercisable | |||||||||
| $10.00 | 88,769 | 2.29 | 88,769 | |||||||||
| 13.74 | 2,281 | 3.79 | 2,281 | |||||||||
| 15.00 | 104,000 | 2.02 | 104,000 | |||||||||
| 16.50 | 46,664 | 2.92 | 46,664 | |||||||||
| 20.00 | 35,000 | 7.36 | 35,000 | |||||||||
| 21.25 | 74,996 | 5.61 | 74,996 | |||||||||
| 35.00 | 614 | 3.93 | 614 | |||||||||
| 352,324 | 3.52 | 352,324 | ||||||||||
Reverse Stock Split
The Company’s Board of Directors and stockholders approved in September 2006 a reverse stock split. On September 12, 2006, asix-for-ten reverse stock split of the Company’s common stock became effective. All common shares, per share and stock option data information in the accompanying financial statements and notes thereto has been retroactively restated for all periods to reflect the reverse stock split.
F-23
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
| 9. | Stock Options |
A summary of activity under the Company’s 2000 Stock Plan is as follows:
| Exercise Price | Weighted-Average | |||||||||||
| Options | Per Share | Exercise Price | ||||||||||
| Balance at January 1, 2004 | 215,450 | $ | 2.50 | $ | 2.50 | |||||||
| Granted | 230,208 | 10.00-15.00 | 13.59 | |||||||||
| Exercised | (5,266 | ) | 2.50 | 2.50 | ||||||||
| Canceled | (46,840 | ) | 2.50 | 2.50 | ||||||||
| Balance at December 31, 2004 | 393,552 | 2.50-15.00 | 9.00 | |||||||||
| Granted | 350,978 | 15.00-20.00 | 16.22 | |||||||||
| Exercised | (63,454 | ) | 2.50-15.00 | 4.66 | ||||||||
| Canceled | (146,933 | ) | 2.50-15.00 | 14.34 | ||||||||
| Balance at December 31, 2005 | 534,143 | 2.50-20.00 | 13.11 | |||||||||
| Granted | 210,772 | 15.00-30.00 | 21.64 | |||||||||
| Exercised | (22,076 | ) | 2.50-27.50 | 2.53 | ||||||||
| Canceled | (92,488 | ) | 2.50-30.00 | 16.64 | ||||||||
| Balance at December 31, 2006 | 630,351 | $ | 2.50-30.00 | $ | 18.15 | |||||||
| Granted | — | — | — | |||||||||
| Exercised | — | — | — | |||||||||
| Canceled | (7,642 | ) | 2.50-27.50 | 21.78 | ||||||||
| Balance at March 31, 2007 | 622,709 | $ | 2.50-27.50 | $ | 18.11 | |||||||
| Available for grant at March 31, 2007 | 264,334 | |||||||||||
Below is a summary of stock option grant activity and related fair value information for the 12 months ended March 31, 2007.
| Fair Value of | ||||||||||||
| Options | Exercise | Common Stock on | ||||||||||
2006 Grants | Granted | Price | Date of Grant | |||||||||
| May | 94,000 | 25.00 | 25.00 | |||||||||
| July | 17,400 | 27.50 | 27.50 | |||||||||
| August | 4,600 | 30.00 | 30.00 | |||||||||
| December | 60,272 | 15.00 | 15.00 | |||||||||
| Subtotal | 176,272 | |||||||||||
F-24
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
All outstanding options are currently exercisable. The following table summarizes information relating to currently outstanding and vested options at March 31, 2007:
| Weighted-Average | ||||||||||||
| Options | Remaining Life | Options | ||||||||||
Range of Exercise Prices | Outstanding | (Years) | Vested | |||||||||
| $ 2.50 | 79,425 | 4.28 | 75,825 | |||||||||
| 2.51-10.00 | 50,000 | 7.00 | 50,000 | |||||||||
| 10.01-15.00 | 287,712 | 8.60 | 77,440 | |||||||||
| 15.01-20.00 | 105,072 | 8.74 | 33,812 | |||||||||
| 20.01-25.00 | 92,000 | 9.11 | 5,000 | |||||||||
| 25.01-27.50 | 5,500 | 9.30 | 1,250 | |||||||||
| 27.51-30.00 | 3,000 | 9.39 | — | |||||||||
| Total | 622,709 | 8.03 | 243,327 | |||||||||
The Company provides a stock option plan for employees, directors and consultants. Under the plan, options to purchase common stock of the Company are granted to certain employees and directors at the estimated fair value of the underlying common stock at the date of grant. The options generally have a term of 10 years and generally vest over four years commencing on the date of the grant. During 2005, the Company’s Board of Directors and stockholders approved an amendment to the stock option plan to increase the number of shares that can be issued under the plan by 400,000 shares of common stock to a total of 1,000,000 shares. As of March 31, 2007, the total compensation cost related to non-vested options not yet recognized is approximately $1,704,000, which will be charged to expense using the method of calculation prescribed by SFAS No. 123(R).
In July 2005, the Company entered into a consulting agreement with a physician and approved an option grant as part of the compensation for consulting services. The option provided that 9,000 of the shares subject to the option would be immediately vested, and the remaining shares would vest in accordance with milestone achievements. The charge that resulted was approximately $82,000 and was charged to expense in 2005. The remaining shares under the option were treated as performance-based awards and will be expensed at the time the milestones are achieved. In March 2006, the performance-based vesting applied to these options was modified and converted to time-based vesting. This modification resulted in a $167,133 and $27,856 charge to expense in the year ended December 31, 2006 and the three months ended March 31, 2007, respectively. The remaining estimated compensation will be charged to expense as the services are provided.
In August 2005, the Company approved the acceleration of vesting in stock option awards previously granted to the Company’s former and retired Chief Financial Officer, who retired on April 27, 2005. The Board of Directors approved the acceleration of the vesting in the two option grants made previously effective as of the date of her retirement. Furthermore, the Board of Directors extended the post-termination of employment/service exercise period from July 26, 2005 (90 days after termination of employment/service) to April 27, 2006. The Company recorded a charge of $125,000 on the new measurement in 2005. In April 2006, these options were exercised, resulting in an additional charge of $130,000 to expense in the year ending December 31, 2006.
In August 2005, the Company issued a performance-based option grant to an employee. The shares originally vested in accordance with milestone achievements. In March 2006, the performance-based vesting applied to these options was modified and converted to time-based vesting. This modification resulted in a remeasurement of the option’s fair value and resulted in a $41,267 and $7,738 charge to expense for the year ended December 31, 2006 and the three months ended March 31, 2007, respectively. The remaining cost will be charged to expense over the vesting period of the option.
F-25
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
In November 2006, the Company entered into consulting agreements with two former employees. All previously granted options continued to vest pursuant to the awards original terms. In accordance withEITF 96-18, the options were subject to a remeasurement at the time of the change in status from employee to non-employee. Remeasurement at March 31, 2007 resulted in a reduction of the original expense booked in 2006 of approximately $12,000. The options will be remeasured and any change in fair value will be recorded to expense at each subsequent reporting period.
| 10. | Benefit Plan |
The Company has a 401(k) profit sharing benefit plan (401(k) Plan) covering substantially all employees who are at least 21 years of age and provide a certain number of hours of service. Under the terms of the 401(k) Plan, employees may make voluntary contributions, subject to Internal Revenue Code limitations. The Company matches 25% of the employee’s contributions up to a total of 15% of the employee’s gross salary. The Company’s contributions to the 401(k) Plan vest equally over five years. Company contributions to the 401(k) Plan were $22,466, $24,476, $32,936 and $14,799, for 2004, 2005, 2006 and the three months ended March 31, 2007, respectively.
| 11. | Asset Acquisition |
In April 2006, we acquired from Abbott Laboratories the assets related to Abbokinase, including the remaining inventory of finished product, all regulatory and clinical documentation, validated cell lines, and intellectual property rights, including trade secrets andknow-how relating the manufacture of urokinase using the tissue culture method, for a total purchase price of $20,000,000. The purchase price is comprised of $5,000,000 in cash and a $15,000,000 secured promissory note. The note is due December 31, 2007, accrues interest at 6% annually and is secured by the Company’s right, title and interest in the purchased assets. The purchase of these assets did not constitute the purchase of a business as defined in EITFNo. 98-3,Determining Whether a Nonmonetary Transaction Involves Receipt of Productive Assets or of a Business, since no employees, equipment, manufacturing facilities or arrangements, or sales and marketing organization were included in the transaction. Since the purchase was not a business, the purchase price has been allocated based upon fair value assessments as follows: inventory $16,700,000, Abbokinase trade name $500,000 and other identifiable intangibles $2,800,000. The Company commenced selling Abbokinase in October 2006. Of the total number of vials of Abbokinase inventory that we acquired from Abbott, it is estimated that 28% of such vials will not be sold and, consequently, these vials are carried with no book value assigned. Under the purchase agreement, after the Company has received cash proceeds of $5,000,000 from the sale of Abbokinase, the Company is required to deposit 50% of the cash received from sales of Abbokinase into an escrow account securing the repayment of the $15,000,000 promissory note. If the promissory note is not repaid by its maturity date, Abbott has the right to the amount held in the escrow account and to reclaim any remaining inventory of Abbokinase and related rights.
| 12. | Abbokinase Inventory |
In the acquisition of Abbokinase, the Company received approximately 153,000 vials of Abbokinase manufactured between 2003 and 2005. At the time of the transaction the Company estimated that hospitals would purchase, and the Company would thereby recognize revenue for, approximately 111,000 vials, or approximately 72% of the total vials we acquired. The Company also estimated that hospitals would not purchase approximately 42,000 vials, or approximately 28% of the vials it acquired, and it assigned zero inventory value to these vials. The Company may or may not be able to sell the entire inventory acquired before the product expires, and the Company is not permitted to sell this inventory after expiration. Moreover, even if the Company were able to sell the Abbokinase inventory to wholesalers prior to expiration, unless the product is sold on to hospitals and administered prior to expiration, the product may be returned to the Company and deferred revenue could be significantly reduced. The Company is continuing the current
F-26
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
stability testing program started by Abbott, which has been ongoing for over four years. The testing to date has shown that the product changes very little from year to year. However, of the vials of Abbokinase held in inventory either by the Company or by our wholesalers as of March 31, 2007, approximately 64% of the vials the Company expects hospitals to purchase, or approximately $10.7 million in inventory value, will expire by October 2007 based on current stability data. The remaining approximately 36% of the vials the Company expects hospitals to purchase, or approximately $6.1 million in inventory value, will expire at various times up to August 2009. Currently, the Company believes it is probable that the stability data will support extension of the inventory expiration dates, that the Company will be able to sell this inventory and that the Company will recover the cost of this inventory.
| 13. | Commitments and Contingencies |
Lease Commitments
Total rent expense was $89,000, $91,000, $121,000 and $30,300 in 2004, 2005, 2006 and the three months ended March 31, 2007, respectively. Payments under noncancelable operating leases are $64,000 for the year 2007, and $54,000 in 2008.
Clinical Research Agreement
On December 11, 2006, the Company entered into a clinical research and related services agreement with INC RESEARCH, Inc., (INC), pursuant to which INC will assist the Company in conducting and managing clinical trials as requested from time to time. The Company will be obligated to pay fees and to reimburse INC for direct and indirect costs incurred by them under the agreement within 30 days after the Company’s receipt of invoices provided from time to time by INC. The agreement will terminate upon completion of the study, unless earlier terminated by either party upon 30 days’ written notice to the other party. The Company made a non-refundable payment to INC of $200,000 that was expensed.
| 14. | Licensing Agreements |
License Agreement with UNEMED Corporation
On October 10, 2003, UNEMED Corporation granted the Company an exclusive, worldwide license, with sublicense rights, to intellectual property and patents relating to the use of a thrombolytic agent together with microbubbles for the treatment of thrombosis. The Company is obligated to pay UNEMED a royalty on any future net sales of products or processes which utilize the licensed technology, of which there have been no sales to date. The Company is also obligated to pay maintenance fees and expenses related to the maintenance of one of the patents covered by the license. The license agreement will terminate contemporaneously with the expiration of the licensed patents. Warrants were issued for the purchase of 4,000 shares of common stock at $10.00 per share with a fair value of $3,000 to acquire these rights.
License Agreement with Dr. med. Reinhard Schlief
On January 4, 2005, Dr. med. Reinhard Schlief granted the Company an exclusive, worldwide license, with the right tosub-license, to intellectual property and patents relating to methods of destroying cells by applying ultrasound to them in the presence of microbubbles. The Company is obligated to pay Dr. Schlief a royalty of 2% of net sales revenue derived from the sale of products that utilize the licensed technology. The license agreement will terminate contemporaneously with the expiration of the licensed patents. Warrants were issued for the purchase of 4,000 shares of common stock at $15.00 per share with a fair value of $37,500 to acquire these rights.
F-27
Table of Contents
ImaRx Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
License Agreement with University of Arkansas
On February 14, 2006, the University of Arkansas granted the Company an exclusive, worldwide license, with the right to sublicense, intellectual property and patents relating to the use of a specific ultrasound device to be used in conjunction with bubbles, a thrombolytic, or a combination of bubbles and a thrombolytic to break up blood clots. To maintain this license, the Company must meet certain product development milestones. The Company is obligated to pay the University of Arkansas a one-time fee of $25,000 within 30 days after the first commercial sale of a product incorporating the licensed technology, and varying royalties depending on the amount of net revenue derived from the sale of products using the licensed technology, of which there have been no sales to date. The Company is also obligated to pay a one-time success fee of $250,000 in the first year that net revenue derived from the sale of products using the licensed technology exceeds $10.0 million. The license will terminate upon expiration of the last patent to which it relates.
| 15. | Subsequent Events |
The Company’s Board of Directors and stockholders approved a second reverse stock split. On May 4, 2007, aone-for-three reverse stock split of the Company’s common stock will become effective. All common shares, per share and stock option data information in the accompanying financial statements and notes thereto has been retroactively restated for all periods to reflect the reverse stock split.
On May 24, 2007, the Company was notified by BRACCO International (Bracco) that a liability related to patent prosecution expenses recorded in prior years of approximately $219,000 was no longer due to Bracco. The extinguishment of this liability will result in a gain of $219,000.
F-28
Table of Contents

Table of Contents
3,000,000 Shares
Common Stock
PRELIMINARY PROSPECTUS
, 2007
Through and including , 2007 (25 days after the date of this prospectus), all dealers that buy, sell or trade our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
Maxim Group LLC
Sole Bookrunner
I-Bankers Securities, Inc.
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution |
The following table sets forth all expenses, other than the underwriting discounts and commissions, payable by the registrant in connection with the sale of the common stock being registered. All the amounts shown are estimates except the registration fee, the NASD filing fee and the NASDAQ Capital Market initial listing fee. We intend to pay all expenses of registration, issuance and distribution.
| SEC registration fee | $ | 8,025 | ||
| NASD filing fee | 8,000 | |||
| NASDAQ Capital Market initial listing fee | 75,000 | |||
| Blue sky qualification fees and expenses | 5,000 | |||
| Printing and engraving expenses | 160,000 | |||
| Legal fees and expenses | 800,000 | |||
| Accounting fees and expenses | 250,000 | |||
| Transfer agent and registrar fees and expenses | 8,000 | |||
| Underwriter non-accountable fees | 420,000 | |||
| Miscellaneous | 55,975 | |||
| Total | $ | 1,790,000 |
| * | To be provided by amendment. |
| Item 14. | Indemnification of Officers and Directors |
The registrant is a Delaware corporation. Section 145 of the Delaware General Corporation Law, or the DGCL, provides that a corporation may indemnify any person who is or was a director, officer, employee or agent of a corporation of an enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative, by reason of being or having been in any such capacity, if he acted in good faith in a manner reasonably believed to be in or not opposed to the best interest of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful, except that with respect to an action brought by or in the right of the corporation, such indemnification is limited to expenses (including attorneys’ fees). Under the DGCL, Section 145 is not exclusive of other rights to which those seeking indemnification may be entitled under any bylaw, agreement, vote of stockholders or disinterested directors or otherwise.
In addition, Section 102(b)(7) of the DGCL permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except for liability for any breach of the director’s duty of loyalty to the corporation or its stockholders, for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, for unlawful payments of dividends or unlawful stock repurchases, redemptions or other distributions, or for any transaction from which the director derived an improper personal benefit.
The registrant’s amended and restated certificate of incorporation includes a provision that eliminates the personal liability of its directors for monetary damages for breach of fiduciary duty as a director to the fullest extent permitted by the DGCL. The registrant’s amended and restated certificate of incorporation requires indemnification of its directors and officers to the fullest extent permissible under the DGCL and the registrant’s amended and restated bylaws provide for indemnification of officers and directors to the fullest extent authorized by the DGCL.
II-1
Table of Contents
The registrant maintains a liability insurance policy pursuant to which its directors and officers may be indemnified against liability incurred for serving in their capacities as directors and officers.
Prior to the completion of this offering, the registrant intends to enter into stockholder-approved indemnification agreements with each of its directors and officers and we intend to enter into indemnification agreements with any new directors and officers in the future. The indemnification agreements set forth certain procedures that will apply in the event of a claim for indemnification thereunder. At present, no litigation or proceeding is pending that involves a director or officer of the registrant regarding which indemnification is sought, nor is the registrant aware of any threatened litigation that may result in claims for indemnification.
The form of underwriting agreement filed as an exhibit to this registration statement provides for indemnification under certain circumstances by the underwriters of the registrant, its directors, certain of its officers and its controlling persons for certain liabilities arising under the Securities Act or otherwise.
The Second Amended and Restated Investors’ Rights Agreement between the registrant and certain investors provides for cross-indemnification in connection with registration of the registrant’s common stock on behalf of such investors.
See also the undertakings set out in response to Item 17.
Reference is made to the following documents filed as exhibits to this registration statement regarding relevant indemnification provisions described above and elsewhere herein:
Exhibit Document | Number | |||
| Form of Underwriting Agreement | 1.1 | |||
| Registrant’s Amended and Restated Certificate of Incorporation, to be effective upon closing of this offering† | 3.4 | |||
| Registrant’s Amended and Restated Bylaws, to be effective upon closing of this offering† | 3.6 | |||
| Form of Indemnification Agreement† | 10.1 | |||
| Second Amended and Restated Investors’ Rights Agreement, dated April 14, 2006† | 10.2 | |||
† Previously filed
| Item 15. | Recent Sales of Unregistered Securities |
Since January 1, 2004, the registrant has sold the following securities that were not registered under the Securities Act:
1. We sold an aggregate of 90,796 shares of our common stock to certain of our employees, directors and consultants for cash consideration in the aggregate amount of $365,401 upon the exercise of stock options granted under our 2000 Stock Plan, none of which have been repurchased by us.
2. We granted stock options to certain employees, directors and consultants under our 2000 Stock Plan covering an aggregate of 791,958 shares of common stock, at exercise prices ranging from $10.00 to $30.00 per share. Of these, options covering an aggregate of 309,388 shares were canceled without being exercised.
3. In March 2004, we issued a warrant to each of Bridge Ventures, Inc. and Saggi Capital Corp., each of which is an accredited investor, as partial consideration for annual consulting services. Each warrant is for the purchase of 50,000 shares of our common stock at an exercise price of $15.00 per share.
4. In March 2004, we sold 500,000 shares of our common stock to accredited investors at a purchase price of $10.00 per share pursuant to a private placement in which First Montauk Securities Corp. served as our exclusive placement agent. In connection with this private placement, the outstanding principal amount and accrued interest under previously issued convertible promissory notes was automatically converted into 206,465 shares of our common stock at a conversion price of $10.00 per share.
II-2
Table of Contents
5. In October 2004, we issued a warrant to First Montauk Securities Corp. and certain executive officers of First Montauk Securities Corp., each of whom is an accredited investor, to purchase up to an aggregate of 50,000 shares of common stock at an exercise price of $10.00 per share.
6. Between October 2004 and February 2005, we sold an aggregate of 466,663 shares of common stock to accredited investors at a purchase price of $15.00 per share pursuant to a private placement in which First Montauk Securities Corp. served as our exclusive placement agent. In connection with this offering and as partial consideration for entering into the placement agency agreement, First Montauk Securities Corp. and certain executive officers of First Montauk Securities Corp. also received warrants to purchase up to an aggregate of 46,664 shares of our common stock at an exercise price of $16.50 per share.
7. In January 2005, as partial consideration for a patent license, we granted Dr. med. Reinhard Schlief a warrant to purchase up to an aggregate of 4,000 shares of common stock at an exercise price of $15.00 per share.
8. In September 2005, we sold 1,000,000 shares of Series E preferred stock, valued at $4.0 million, to Abbott Laboratories, an accredited investor, as partial consideration for our acquisition of certain technologies from Abbott Laboratories pursuant to an Asset Purchase Agreement dated September 30, 2005. In connection with this Asset Purchase Agreement, we also issued Abbott Laboratories a secured 6% of promissory note in the principal amount of $15,000,000. No underwriters were involved in this sale of securities.
9. In September 2005, we issued secured 6% promissory notes in the aggregate principal amount of $4,000,000 and warrants for the purchase of an aggregate of 20,000 shares of our common stock at an exercise price of $20.00 per share to accredited investors. No underwriters were involved in this sale of securities. The secured promissory notes issued in this offering were repaid in full in October 2005 with proceeds from the private placement offering described below.
10. In October 2005 and November 2005, we sold an aggregate of 750,000 shares of our common stock to accredited investors at a purchase price of $20.00 per share in a private placement in which First Montauk Securities Corp. served as our exclusive placement agent. In connection with its placement agency agreement, First Montauk Securities Corp. and certain executive officers of First Montauk Securities Corp. received warrants to purchase up to 74,996 shares of our common stock at an exercise price of $21.25 per share.
11. In February 2006 we issued warrants to purchase an aggregate of up to 15,000 shares of our common stock at an exercise price of $20.00 per share to consultants.
12. In April 2006 and May 2006, we sold an aggregate of 2,835,000 shares of Series F preferred stock to accredited investors at a price of $5.00 per share pursuant to a private placement in which First Albany Capital, First Montauk Securities Corp. and Maxim Group LLC served as our placement agents.
13. In April 2006, we issued Abbott Laboratories a secured 6% promissory note in the principal amount of $15,000,000 in partial consideration for assets we acquired. No underwriters were involved in this sale of securities.
The sales of the above securities described in items (1) and (2) above were exempt from registration under the Securities Act in reliance on Rule 701 promulgated under the Securities Act as transactions pursuant to compensatory benefit plans and contracts relating to compensation as provided under Rule 701. The recipients of securities in each of these transactions represented their intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the share certificates and instruments issued in such transactions. All recipients had adequate access, through their relationship with the registrant, to information about the registrant.
The sale of securities described in items (4), (6), (9), (10) and (12) above were exempt from registration under the Securities Act in reliance on Rule 506 of Regulation D promulgated thereunder as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions were
II-3
Table of Contents
sophisticated entities, all of whom are “accredited investors,” as such term is defined in Rule 501 promulgated under the Securities Act, and all of whom had adequate access, through their relationship with us, to information about us.
The sale of securities described in items (3), (5), (7), (8), (11) and (13) above were exempt from registration under Section 4(2) of the Securities Act as transactions by an issuer not involving a public offering. The recipients of securities in each of these transactions represented their intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the share certificates and instruments issued in such transactions. All recipients had adequate access, through their relationship with the registrant, to information about the registrant.
No underwriters were involved in the foregoing sales of securities.
| Item 16. | Exhibits and Financial Statement Schedules |
(a) Exhibits.
| Exhibit | ||||
Number | Description of Document | |||
| 1 | .1 | Form of Underwriting Agreement | ||
| 3 | .1† | Fourth Amended and Restated Certificate of Incorporation of the registrant | ||
| 3 | .2† | Amendment to Certificate of Incorporation of the registrant to effect asix-for-ten reverse stock split | ||
| 3 | .3† | Second Amendment to Certificate of Incorporation of the registrant to effect aone-for-three reverse stock split | ||
| 3 | .4† | Form of Amended and Restated Certificate of Incorporation of the registrant, to be effective following this offering | ||
| 3 | .5† | Bylaws of the registrant, as amended | ||
| 3 | .6† | Form of Amended and Restated Bylaws of the registrant, to be effective following this offering | ||
| 4 | .1† | Specimen certificate evidencing shares of common stock | ||
| 5 | .1* | Opinion of DLA Piper US LLP | ||
| 10 | .1† | Form of Indemnification Agreement entered into between the registrant and each of its directors and officers | ||
| 10 | .2† | Second Amended and Restated Investors’ Rights Agreement, dated April 14, 2006, by and among the registrant and certain stockholders | ||
| 10 | .3† | 2000 Stock Plan and related agreements | ||
| 10 | .4† | 2007 Performance Incentive Plan and related agreements | ||
| 10 | .5† | Bonus Plan | ||
| 10 | .6† | License Agreement, dated January 4, 2005, between the registrant and Dr. med. Reinhard Schlief | ||
| 10 | .7† | Exclusive Sublicense Agreement, dated October 10, 2003, between the registrant and UNEMED Corporation | ||
| 10 | .8† | Assignment, Assumption and License Agreement, dated October 7, 1999, between the registrant and Bristol-Myers Squibb Medical Imaging, Inc. (as successor to DuPont Contrast Imaging, Inc.) dated October 7, 1999, and amendments thereto | ||
| 10 | .9† | �� | License Agreement, dated February 10, 2006, between the registrant and the University of Arkansas for Medical Sciences | |
| 10 | .10† | Asset Purchase Agreement, dated April 10, 2006, between the registrant and Abbott Laboratories, and amendments thereto | ||
| 10 | .11† | Escrow Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .12† | Inventory Trademark License Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .13† | Security Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
II-4
Table of Contents
| Exhibit | ||||
Number | Description of Document | |||
| 10 | .14† | Secured Promissory Note, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .15† | Second Amended Executive Employment Agreement, dated May 15, 2006, between the registrant and Evan C. Unger | ||
| 10 | .16† | Consulting Agreement, dated October 20, 2006, between the registrant and Evan C. Unger | ||
| 10 | .17† | Confidential Separation Agreement and Mutual General Release of All Claims, dated November 28, 2006, between the registrant and Evan C. Unger | ||
| 10 | .18† | Consulting Agreement, dated April 11, 2005, between the registrant and Greg Cobb | ||
| 10 | .19† | Amended Executive Employment Agreement, dated February 1, 2007, between the registrant and Greg Cobb | ||
| 10 | .20† | Amended Executive Employment Agreement, dated February 1, 2007, between the registrant and Bradford A. Zakes | ||
| 10 | .21† | Agreement, dated March 31, 2006, by and among the registrant, John A. Moore and Edson Moore Healthcare Ventures | ||
| 10 | .22† | Subscription Agreement and Investor Questionnaire, dated March 2004, between the registrant and each of the signatory investors, offering price $2.00 per share | ||
| 10 | .23† | Subscription Agreement and Investor Questionnaire, dated December 2004, between the registrant and each of the signatory investors, offering price $3.00 per share | ||
| 10 | .24† | Subscription Agreement and Investor Questionnaire, dated September and October 2004, between the registrant and each of the signatory investors, offering price $4.00 per share | ||
| 10 | .25† | Commercial Lease — Triple Net, dated November 1, 2002, between the registrant and ImaRx Investments L.L.C. | ||
| 10 | .26† | Standard Commercial — Industrial Lease, dated December 30, 1997, between the registrant and Tucson Tech Park and addenda thereto | ||
| 21 | .1† | Subsidiaries of the registrant | ||
| 23 | .1 | Consent of Ernst & Young LLP | ||
| 23 | .2* | Consent of DLA Piper US LLP (included in Exhibit 5.1) | ||
| 24 | .1† | Power of Attorney | ||
| * | To be filed by amendment. |
| † | Previously filed. |
All schedules are omitted because they are not required, are not applicable or the information is included in the financial statements or notes thereto.
| Item 17. | Undertakings |
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers, and controlling persons of the registrant pursuant to the foregoing provisions or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
II-5
Table of Contents
The undersigned registrant undertakes that:
(1) for purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective,
(2) for the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fideoffering thereof,
(3) for purposes of determining any liability under the Securities Act, if the registrant is subject to Rule 430C, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use, and
(4) for purposes of determining any liability under the Securities Act, in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
II-6
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Amendment No. 1 to Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of Tucson, in the County of Pima, State of Arizona, on the 11th day of June, 2007.
IMARX THERAPEUTICS, INC.
| By: | /s/ Bradford A. Zakes |
Bradford A. Zakes
President and Chief Executive Officer
Pursuant to the requirements of the Securities Act of 1933, as amended, this Amendment No. 1 to Registration Statement has been signed below by the following persons in the capacities and on the dates indicated.
Signature | Title | Date | ||||
/s/ Bradford A. Zakes Bradford A. Zakes | President, Chief Executive Officer and Director (principal executive officer) | June 11, 2007 | ||||
/s/ Greg Cobb Greg Cobb | Chief Financial Officer (principal financial and accounting officer) | June 11, 2007 | ||||
/s/ Richard Love* Richard Love | Director | June 11, 2007 | ||||
/s/ Richard Otto* Richard Otto | Director | June 11, 2007 | ||||
/s/ Thomas W. Pew* Thomas W. Pew | Director | June 11, 2007 | ||||
/s/ Philip Ranker* Philip Ranker | Director | June 11, 2007 | ||||
/s/ James M. Strickland* James M. Strickland | Director | June 11, 2007 | ||||
| *By: | /s/ Greg Cobb Greg Cobb Attorney-in-Fact | |||||
II-7
Table of Contents
EXHIBIT INDEX
| Exhibit | ||||
Number | Description of Document | |||
| 1 | .1 | Form of Underwriting Agreement | ||
| 3 | .1† | Fourth Amended and Restated Certificate of Incorporation of the registrant | ||
| 3 | .2† | Amendment to Certificate of Incorporation of the registrant to effect asix-for-ten reverse stock split | ||
| 3 | .3† | Second Amendment to Certificate of Incorporation of the registrant to effect aone-for-three reverse stock split | ||
| 3 | .4† | Form of Amended and Restated Certificate of Incorporation of the registrant, to be effective following this offering | ||
| 3 | .5† | Bylaws of the registrant, as amended | ||
| 3 | .6† | Form of Amended and Restated Bylaws of the registrant, to be effective following this offering | ||
| 4 | .1† | Specimen certificate evidencing shares of common stock | ||
| 5 | .1* | Opinion of DLA Piper US LLP | ||
| 10 | .1† | Form of Indemnification Agreement entered into between the registrant and each of its directors and officers | ||
| 10 | .2† | Second Amended and Restated Investors’ Rights Agreement, dated April 14, 2006, by and among the registrant and certain stockholders | ||
| 10 | .3† | 2000 Stock Plan and related agreements | ||
| 10 | .4† | 2007 Performance Incentive Plan and related agreements | ||
| 10 | .5† | Bonus Plan | ||
| 10 | .6† | License Agreement, dated January 4, 2005, between the registrant and Dr. med. Reinhard Schlief | ||
| 10 | .7† | Exclusive Sublicense Agreement, dated October 10, 2003, between the registrant and UNEMED Corporation | ||
| 10 | .8† | Assignment, Assumption and License Agreement, dated October 7, 1999, between the registrant and Bristol-Myers Squibb Medical Imaging, Inc. (as successor to DuPont Contrast Imaging, Inc.) dated October 7, 1999, and amendments thereto | ||
| 10 | .9† | License Agreement, dated February 10, 2006, between the registrant and the University of Arkansas for Medical Sciences | ||
| 10 | .10† | Asset Purchase Agreement, dated April 10, 2006, between the registrant and Abbott Laboratories, and amendments thereto | ||
| 10 | .11† | Escrow Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .12† | Inventory Trademark License Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .13† | Security Agreement, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .14† | Secured Promissory Note, dated April 14, 2006, between the registrant and Abbott Laboratories | ||
| 10 | .15† | Second Amended Executive Employment Agreement, dated May 15, 2006, between the registrant and Evan C. Unger | ||
| 10 | .16† | Consulting Agreement, dated October 20, 2006, between the registrant and Evan C. Unger | ||
| 10 | .17† | Confidential Separation Agreement and Mutual General Release of All Claims, dated November 28, 2006, between the registrant and Evan C. Unger | ||
| 10 | .18† | Consulting Agreement, dated April 11, 2005, between the registrant and Greg Cobb | ||
| 10 | .19† | Amended Executive Employment Agreement, dated February 1, 2007, between the registrant and Greg Cobb | ||
| 10 | .20† | Amended Executive Employment Agreement, dated February 1, 2007, between the registrant and Bradford A. Zakes | ||
| 10 | .21† | Agreement, dated March 31, 2006, by and among the registrant, John A. Moore and Edson Moore Healthcare Ventures | ||
II-8
Table of Contents
| Exhibit | ||||
Number | Description of Document | |||
| 10 | .22† | Subscription Agreement and Investor Questionnaire, dated March 2004, between the registrant and each of the signatory investors, offering price $2.00 per share | ||
| 10 | .23† | Subscription Agreement and Investor Questionnaire, dated December 2004, between the registrant and each of the signatory investors, offering price $3.00 per share | ||
| 10 | .24† | Subscription Agreement and Investor Questionnaire, dated September and October 2004, between the registrant and each of the signatory investors, offering price $4.00 per share | ||
| 10 | .25† | Commercial Lease — Triple Net, dated November 1, 2002, between the registrant and ImaRx Investments L.L.C. | ||
| 10 | .26† | Standard Commercial — Industrial Lease, dated December 30, 1997, between the registrant and Tucson Tech Park and addenda thereto | ||
| 21 | .1† | Subsidiaries of the registrant | ||
| 23 | .1 | Consent of Ernst & Young LLP | ||
| 23 | .2* | Consent of DLA Piper US LLP (included in Exhibit 5.1) | ||
| 24 | .1† | Power of Attorney | ||
| * | To be filed by amendment. |
| † | Previously filed. |
II-9