UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| | | |
þ | | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the fiscal year ended December 31, 2005 |
or |
o | | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the transition period from to |
Commission File Number:000-51709
Iomai Corporation
(exact name of registrant as specified in its charter)
| | | |
Delaware | | 52-2049149 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. Employer
Identification No.) |
20 Firstfield Road, Suite 250, Gaithersburg, Maryland 20878
(Address of principal executive offices including zip code)
Registrant’s telephone number, including area code:
(301) 556-4500
Securities registered pursuant to Section 12(b) of the Act:
| | | |
None | | None |
| (Title of each Class) | | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $.01 Par Value Per Share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes þ No o
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes o No þ
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” inRule 12b-2 of the Exchange Act.
| | |
| Large accelerated filer o | Accelerated Filer o | Non-accelerated filer þ |
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Exchange Act). Yes o No þ
As of June 30, 2005, the last business day of the registrant’s most recently completed second quarter, the registrant’s Common Stock was not listed on the Nasdaq National Market and there was no other established trading market therefor. There were 11,897,386 shares of the registrant’s Common Stock outstanding as of March 3, 2006.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the registrant’s 2006 Annual Meeting of Stockholders to be held on May 16, 2006, which definitive proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year of December 31, 2005, are incorporated by reference into Part III of this Annual Report onForm 10-K.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This annual report onForm 10-K contains forward-looking statements. All statements contained in this annual report other than statements of historical fact are forward-looking statements. Forward-looking statements include statements regarding our future financial position, business strategy, budgets, projected costs, plans and objectives of management for future operations. The words “may,” “continue,” “estimate,” “intend,” “plan,” “will,” “believe,” “project,” “expect,” “seek,” “anticipate” and similar expressions may identify forward-looking statements, but the absence of these words does not necessarily mean that a statement is not forward-looking. These forward-looking statements include, among other things, statements about:
| | |
| | • | the implementation of our corporate strategy; |
| |
| | • | our future financial performance and projected expenditures; |
| |
| | • | our ability to enter into future collaborations with pharmaceutical, biopharmaceutical and biotechnology companies and academic institutions or to obtain funding from government agencies; |
| |
| | • | our product research and development activities, including the timing and progress of our clinical trials, and projected expenditures; |
| |
| | • | our technology’s potential efficacy, advantages over current approaches to vaccination and broad applicability to infectious diseases; |
| |
| | • | our ability to receive regulatory approvals to develop and commercialize our product candidates; |
| |
| | • | our ability to increase our manufacturing capabilities for our product candidates; |
| |
| | • | our ability to develop or obtain funding for our immunostimulant patch for pandemic flu applications; |
| |
| | • | our projected markets and growth in markets; |
| |
| | • | our product formulations and patient needs and potential funding sources; |
| |
| | • | our staffing needs; |
| |
| | • | our use of the proceeds from this offering; and |
| |
| | • | our plans for sales and marketing. |
The forward-looking statements in this annual report are subject to risks and uncertainties that may cause actual results to differ materially. These risks and uncertainties include those described in Item 1A. “Risk factors.” In light of these risks and uncertainties, the forward-looking events and circumstances discussed in this annual report may not occur as contemplated, and actual results could differ materially from those anticipated or implied by the forward-looking statements.
You should not unduly rely on these forward-looking statements, which speak only as of the date of this annual report. Unless required by law, we undertake no obligation to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise. You should, however, review the factors and risks we describe in the reports we file from time to time with the SEC after the date of this annual report. See “Availability of Periodic SEC Reports” in Item 1. “Business.”
Iomai®, Transcutaneous Immunizationtm, TCItm and the Iomai logo are trademarks or service marks of Iomai Corporation. All other trademarks and service marks appearing in this prospectus are the property of their respective holders.
ii
PART I
OVERVIEW
We are a biopharmaceutical company focused on the discovery, development and commercialization of vaccines and immunostimulants delivered to the skin. Our proprietary technology, known as transcutaneous immunization, or TCI, is founded on the biological insight that the skin is a highly attractive immune environment for vaccination and immunostimulation. TCI targets Langerhans cells, a specialized component of the immune system located in the outer layers of the skin, for the delivery of vaccines and adjuvantsand/or antigens to stimulate a robust immune response. Our approach to vaccination and immunostimulation has the potential to enhance vaccine efficacy, expand the market for existing vaccines and enable the development of new vaccines.
We are developing two distinct product applications for our TCI technology. The first application is a needle-free vaccine patch that is applied to the skin and delivers both pathogen-specific antigens and an adjuvant. We are using this application to develop needle-free vaccines to replace currently injectable vaccines as well as novel vaccines that cannot be delivered by injection. The second application is an immunostimulant, or IS, patch that contains only an adjuvant that is delivered to the skin by the patch. In this second application, our IS patch is placed over the vaccine injection site in order to stimulate a stronger immune response to that vaccine. Initially we expect to target our IS patch for use in patient populations with weakened or impaired immune systems, such as the elderly, for whom existing vaccines often do not elicit an adequate immune response.
We currently have three product candidates for flu in clinical and preclinical development and one product candidate for travelers’ diarrhea in clinical development:
| | |
| | • | Needle-free flu vaccine patch. We are developing a needle-free flu vaccine patch that combines flu antigens with an adjuvant in a single patch. We completed a Phase 1 study of our needle-free flu vaccine patch in Europe during the fourth quarter of 2004 and are working to further optimize our formulation and product design. We plan to commence a Phase 1/2 trial during the third quarter of 2006, with the goal of stimulating an immune response equal to or greater than that of existing injectable vaccines. We plan to conduct additional Phase 2 trials after completing the Phase 1/2 trial. In the development of our needle-free flu vaccine patch, we are also actively seeking to collaborate with suppliers of commercial flu antigens because the manufacture of flu antigens is not part of our business strategy. Each flu season, approximately 15 million to 60 million people in the United States become ill with influenza. This infection rate results in approximately 36,000 deaths and approximately 200,000 hospitalizations in the United States annually. Approximately 80 million people in the United States are vaccinated against the flu annually. |
| |
| | • | IS patch for elderly receiving flu vaccines. We are developing our IS patch to improve the immune response of the elderly to existing injectable influenza vaccines. We have completed a number of Phase 1 studies in the United States and Phase 1 and Phase 2 studies in Europe. We are planning to commence a Phase 2 study in the United States in 2006. Approximately 40,000 individuals aged 65 and over die from influenza, influenza-related pneumonia and related respiratory complications in the United States each year, making influenza one of the leading causes of death among the elderly. According to the United States Centers for Disease Control and Prevention, or CDC, over 65% of the elderly population in the United States receive influenza vaccination, and public health officials have stated the goal of achieving 90% vaccination rates by 2010. Our IS patch, when administered in combination with existing commercially available injectable flu vaccines distributed by different flu vaccine manufacturers, which will require individual testing and FDA approval with our IS patch, is designed to improve the immune response of the elderly to these flu vaccines. |
| |
| | • | IS patch for pandemic flu preclinical program. We are developing an IS patch to achieve dose-sparing when administered in combination with pandemic flu vaccines. While there are currently pandemic flu vaccines under development by different manufacturers, no pandemic flu vaccines have been approved for use to date. We have conducted preclinical studies in mice, the results of which suggest that a10-fold reduction in vaccine dose may be achievable when our IS patch is applied to the injection site of pandemic flu |
1
| | |
| | | vaccine that is delivered into muscle tissue, and a 100-fold reduction in vaccine dose may be achievable when our IS patch is applied to the injection site of pandemic flu vaccine that is delivered just below the skin surface. In January 2005, the NIH awarded us a two-year, $2.9 million grant for further development of our IS patch technology for pandemic flu applications. We are continuing to conduct preclinical studies of our IS patch for pandemic flu and continue to seek additional government funding for these studies. If our IS patch for pandemic flu receives regulatory approval, we expect to sell our IS patches to the United States government rather than to the public. While it is difficult to estimate the ultimate size of the market for our IS patch for pandemic flu, the United States government recently announced a $7.1 billion initiative to prevent the spread of pandemic flu. |
| | |
| | • | Needle-free travelers’ diarrhea vaccine patch. We are also developing a needle-free travelers’ diarrhea vaccine patch product candidate, consisting of a new vaccine and patch delivery system, for which we completed a double-blind, placebo-controlled safety and efficacy trial in the United States during the first quarter of 2005. The results of this study demonstrated the product candidate’s ability to induce a strong immune response that diminished the severity and delayed the onset of the most common form of travelers’ diarrhea. In a recent Phase 1/2 travelers’ diarrhea trial comparing our current dry patch formulation to a wet patch, interim data indicates that our current dry patch formulation provided equal or greater immune responses to vaccination than the wet patch. We are currently preparing for additional Phase 2 trials designed to determine the final dry patch formulation, number and timing of doses, and duration of patch wear. If we successfully complete these Phase 2 trials on our currently anticipated timeline of completion by early 2007, we expect to begin enrollment in Phase 3 clinical trials in mid-2007. Travelers’ diarrhea usually is contracted by the ingestion of food or water contaminated with EnterotoxigenicE. colibacteria (ETEC). The CDC estimates that 20% to 50% of international travelers contract traveler’s diarrhea. Currently, in the United States there are no approved vaccines against traveler’s diarrhea caused by ETEC. |
We expect that each of our product candidates, consisting of a patch and one or more active ingredients, will be treated together as a single investigational drug by the FDA, even if any active ingredient is part of an existing approved product. None of the active ingredients in our product candidates have been approved by the FDA for commercial sale in any product.
We believe that TCI technology is broadly applicable and may provide the means to prevent or mitigate many other diseases. For example, we have compiled Phase 1 data supporting the ability of TCI-based vaccines to stimulate an immune response against anthrax exposure. On the basis of our preclinical and clinical findings to date, we believe products based on our TCI technology have the potential to address major unmet needs in preventing infectious diseases, and would represent a fundamental change in the way vaccines are administered.
MARKET OVERVIEW
The immune system and vaccines
The immune system is the body’s natural defense mechanism for fighting disease caused by infectious pathogens, which are organisms such as bacteria, viruses and other microbes. The skin is one of the body’s first lines of defense against pathogens. In addition to presenting a barrier to entry, the skin contains specialized immune surveillance cells called Langerhans cells, a subset of the body’s main class of antigen-presenting cells, that serve an important early-warning function. The role of Langerhans cells is to detect antigens embodied in pathogens and present them to other components of the immune system, which in turn produce antibodies that bind to and destroy the invading pathogen or render it harmless. Once produced, these antibodies remain in the body to ward off future infections by the same pathogen, causing the individual to become immune for some period of time after initial exposure.
An immune response may be induced by natural exposure to a pathogen or by a controlled exposure to the antigens embodied in a pathogen by using a vaccine. A vaccine contains a dead or weakened form, or a derivative, of an infectious pathogen, including the antigens that allow the body to recognize the pathogen. Exposure to a vaccine induces the immune system to destroy the pathogen and prevent it from subsequently infecting the body. Several doses of a vaccine may be needed for a full immune response. In addition, the immunity provided by some vaccines
2
is not lifelong. In such cases, the immune response may decrease over time, requiring subsequent doses of a vaccine, or boosters, to restore or increase immunity.
In order to increase the effectiveness of the body’s immune response to a vaccine, vaccines are often administered in combination with an adjuvant. Adjuvants are substances that act as danger signals to the immune system and increase the ability of vaccines to stimulate the immune system. The use of adjuvants in combination with a vaccine has a variety of advantages over the use of a vaccine by itself. For instance, adjuvants may be used to help generate a meaningful immune response in groups of individuals whose immune systems may not react effectively to a vaccine alone, such as the elderly and individuals whose immune systems have been weakened by disease. Adjuvants may also be used to lower the required dose of a vaccine necessary to stimulate an effective immune response in healthy individuals.
Our initial target markets
Influenza
Influenza, commonly called the flu, is an infection of the respiratory tract caused by an influenza virus. Influenza is highly contagious and occurs mainly in the late fall, winter and early spring. Influenza affects all age groups and commonly causes moderate to severe illness that results in absences from school and work and lost productivity. Some individuals, however, develop serious complications, such as pneumonia, that require hospitalization and may even result in death. Flu-related complications can occur at any age; however, the elderly and people with weakened or impaired immune systems are much more likely to develop serious complications than are younger, healthier people.
Each flu season approximately 15 million to 60 million people in the United States become ill with influenza. This infection rate results in approximately 36,000 deaths and approximately 200,000 hospitalizations in the United States annually. Approximately 80 million people in the United States are vaccinated against the flu annually. The CDC has indicated that it wants to increase this figure to 185 million annually by 2010. According to a study conducted by Kalorama Information, the worldwide market for influenza vaccines was estimated to be in excess of $1.4 billion in 2004 and is expected to grow to $1.8 billion in 2006. In addition, Kalorama estimates that the worldwide market for influenza vaccines has the potential to reach approximately $3 billion by 2010. This market growth is expected to be driven principally by expanded supply, heightened public awareness and increased advocacy for vaccination by public health organizations, particularly for the elderly and young children, as well as higher prices. We believe that this market can be further expanded by providing an alternate vaccine delivery method, such as a needle-free flu vaccine patch, that is more convenient and less painful than a hypodermic needle.
Influenza in the elderly
The CDC has highlighted the relatively low effectiveness of existing flu vaccines for persons aged 65 and over. Studies show that the efficacy of flu vaccines decreases from the 70% to 90% range observed in healthy individuals who are less than 65 years of age, down to the 30% to 40% range for individuals 65 years of age and over. Most of the elderly population receives a flu shot annually, yet in the United States influenza is one of the leading causes of death in persons 65 years of age and older, and approximately 40,000 people in this age group die from influenza, influenza-related pneumonia and related respiratory complications each year. This significant number of deaths reaffirms the need for a more efficacious vaccine. As a result, we believe there is an unmet need for immunostimulants that can be delivered in conjunction with a flu vaccine to help persons with weakened or impaired immune systems, such as the elderly, develop adequate immune responses.
Pandemic flu
When a new, highly infectious and dangerous strain of the influenza virus appears, the general lack of immunity to this strain in the general population can lead to a pandemic outbreak that quickly spreads. Three influenza pandemics have occurred in the 20th century, the most recent of which occurred in 1968. By United States government estimates, pandemic flu has a greater potential to cause more deaths and illnesses than virtually any other natural health threat. Signs of a possible pandemic flu have emerged in Southeast Asia, as lethal infections of poultry and humans with avian influenza virus continue. This virus is now endemic in bird populations, which
3
increases the likelihood of continued human exposure and the risk of a pandemic flu outbreak. In response, the United States government has been actively trying address the risk, as President Bush recently announced a $7.1 billion initiative to prevent the spread of pandemic flu, and the United States government is also trying to induce companies to expand the current United States vaccine production capacity, but this may take many years to accomplish. Potentially compounding the strain on manufacturing capacity, a candidate pandemic flu vaccine was recently tested in a Phase 1 trial by the United States National Institutes of Health, or NIH, and the NIH reported that a large two-dose regimen was required to achieve what experts believe to be protective levels of immunity.
The manufacture of flu vaccine is a complex and time consuming process. The vaccine is manufactured well before the relevant flu season. If there is an outbreak of pandemic flu, vaccine manufacturers using current techniques will likely not be able to rapidly scale up production to meet increased demand. Because of these manufacturing limitations, governments are exploring dose-sparing strategies by which a smaller dose of vaccine could produce an effective immune response. With pressure on governments to develop dose-sparing strategies, we believe that there may be an important role for an IS patch, when administered in combination with injected pandemic flu vaccines being developed by different manufacturers, either to reduce the required dose of, or improve the timing or magnitude of the immune response to, a pandemic flu vaccine. Pandemic flu vaccines are still being developed by third parties, and no pandemic flu vaccines have been approved for use by the FDA.
In January 2005, the NIH awarded us a two-year, $2.9 million grant for further development of our IS technology for pandemic flu applications. The U.S. Department of Health and Human Services also issued in March 2006 a request for proposals, or RFP, to award one or more multi-year contracts to develop antigen sparing approaches to administering flu vaccines, to increase overall influenza vaccine capacity by utilizing adjuvants or alternate administration routes. We believe that we are well positioned to take advantage of this opportunity to respond to the RFP as currently outlined in this announcement, although there can be no assurances that we will be awarded any part of this contract.
Travelers’ diarrhea
Travelers’ diarrhea is a disease caused by a bacterial, viral or other microbial infection contracted by the ingestion of contaminated food or water while abroad. According to the United States Centers for Disease Control and Prevention, or CDC, travelers’ diarrhea is the most common illness affecting travelers, with an estimated 20% to 50% of international travelers contracting travelers’ diarrhea.
EnterotoxigenicE. colibacteria (ETEC) is believed to be the most common cause of travelers’ diarrhea. Ingested ETEC bacteria move to the small intestine where they grow and secrete toxins which induce massive fluid secretion from the lining of the small intestine. Common symptoms associated with ETEC-induced diarrhea include frequent loose or watery bowel movements, nausea, vomiting, abdominal cramping, bloating and fever. A majority of affected individuals tend to have six or more loose stools per day. Severe infections that cause large fluid loss may cause dehydration and require hospitalization in order to administer intravenous fluids. ETEC secretes one or both of two different toxins: heat labile toxin (LT) and heat stabile toxin (ST). LT, either acting alone or in combination with ST, is responsible for approximately two-thirds of all cases of ETEC disease. Each year more than 50 million individuals from the United States, Western Europe and Japan/Australasia visit regions where infection by ETEC is common, resulting in an estimated 10 million cases of travelers’ diarrhea caused by LT-secreting ETEC strains. Our travelers’ diarrhea program focuses on ETEC disease caused by LT-secreting strains.
Currently, in the United States there are no approved vaccines against travelers’ diarrhea caused by ETEC infection. Travelers’ diarrhea, consequently, is generally treated with antibiotics, either prophylactically or following onset, or withover-the-counter products that alleviate symptoms but do not treat the underlying infection. Use of antibiotics either as a treatment or as a prophylaxis can be effective but has many drawbacks. Multi-drug resistance in ETEC infection is common, especially in the developing world, and prophylactic antibiotic use is implicated in causing patterns of widespread resistance. In addition, patients must take many doses over several days or weeks for antibiotics to be effective and this poses potential compliance problems. Antibiotics also destroy beneficial resident intestinal bacteria that normally keep in check any ingested disease-causing bacteria. Following antibiotic treatment, these toxic bacteria may grow rapidly leading to severe intestinal inflammation and
4
additional episodes of diarrhea. For these reasons, health authorities generally prefer vaccination to treatment with antibiotics for diseases preventable by vaccine.
We believe a vaccine that reduces the risk of contracting travelers’ diarrhea caused by ETEC, or that reduces the severity of the symptoms associated with such an infection, would be appealing to travelers. According to a recent study, approximately 25% of United States travelers already seek medications and vaccinations for other diseases, such as hepatitis A and B, prior to traveling internationally. Additional studies estimate that the worldwide market for a vaccine to combat travelers’ diarrhea caused by ETEC infection is approximately $450 million.
THE TCI SOLUTION
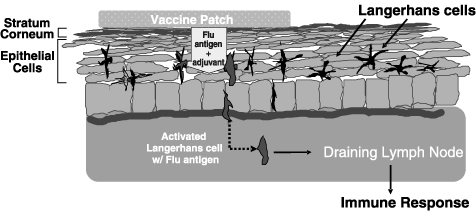
TCI is a proprietary technology designed to trigger an immune response by targeting Langerhans cells. After contact with an infectious pathogen and its associated antigens, Langerhans cells leave the skin and migrate to the nearest lymph node via the lymphatic system. Upon arrival, the Langerhans cells present the antigens in the lymph node, which triggers a potent and highly specific immune response that persists to combat future attacks by the invading pathogen. Because Langerhans cells are evenly distributed throughout the human skin surface, we believe that TCI-based immunizations may be delivered through a patch to a variety of locations on the body.
Our research to date suggests that the immune response to TCI-delivered vaccines may be improved by applying adjuvants along with antigens to the skin. Specifically, we are exploring the use of the LT toxin from ETEC as an adjuvant. While LT is not currently approved by the FDA for any indication nor as a component of any product approved by the FDA, LT has a long history of experimental use as an adjuvant because of its potency as a general immune system stimulant and has been used preclinically with a wide variety of vaccine formulations, including those utilizing peptides, proteins, conjugates, whole killed cells and live vectors. Other means of delivery such as injection, intranasal and oral delivery methods act systemically, preventing the use of LT as an adjuvant without the occurrence of significant side effects. TCI avoids these side effects by delivering LT to the skin, where the LT is sequestered, rather than delivered systemically.
The diagram below highlights the use of TCI in a needle-free vaccine patch.
Our needle-free vaccine patch is designed to work as follows:
| | |
| | • | Following pretreatment that involves a mild abrasion of the skin at the application point to disrupt the outer dead layer of skin, known as the stratum corneum, a patch containing an adjuvant and one or more antigens from the target pathogen (the three antigens associated with the flu vaccine each year, for example) is applied. |
| |
| | • | After permeating the disrupted stratum corneum, the adjuvant and antigens reach and activate the underlying Langerhans cells. |
| |
| | • | The Langerhans cells take up the adjuvant and antigens and migrate through the skin and the lymphatic system to the nearest lymph node, where they stimulate a systemic immune response. |
5
We believe that our needle-free vaccine patch offers the following potential advantages over traditional, injection-based approaches to vaccination:
| | |
| | • | Increased efficacy with reduced risk of side effects. Because the adjuvant and antigens delivered by our needle-free vaccine patch cannot penetrate further than the outer layers of the skin, neither the adjuvant nor the antigens circulate systemically as they would if delivered through injection. Preventing a systemic distribution of adjuvants and antigens reduces the risk of side effects. As a result, our needle-free vaccine patch permits the use of more potent adjuvants relative to other systemic means of delivery (such as injection, intranasal and oral delivery), which increases the overall level of immune response. We and our collaborators have conducted over 21 trials, including three double-blind, placebo-controlled trials, with over 1,600 volunteers receiving the LT adjuvant, and no vaccine-related serious adverse events from applying this adjuvant to the skin were observed. |
| |
| | • | Increased compliance. We believe needle-free administration has the potential to increase acceptance by patients who dislike the use of needles and desire a more convenient and less painful means of vaccination. |
| |
| | • | Safer administration and disposal. Needle-free administration eliminates the risk to the health care provider of inadvertent needle sticks and avoids the need to dispose of contaminated needles. |
| |
| | • | Reduced dosages. By targeting the skin and eliciting a stronger immune response, we believe the amount of antigen required for immunization may be reduced. |
| |
| | • | Cheaper transport and storage. Most vaccines must be stored in refrigerated environments. We believe our vaccines will be stable at room temperature, making both transport and storage without refrigeration simple and inexpensive. |
Our IS patch uses TCI-administered adjuvants to boost the immune response to traditional, injectable vaccines in specific patient populations. Our IS patch would work similarly to our needle-free vaccine patch, except that a needle would be used to inject the pathogenic antigens and a patch containing only an adjuvant would be applied to the skin over the injection site. We believe our IS patch could be applied to boost the immune response in individuals who might not otherwise achieve immunity through the injected vaccine alone. In a Phase 2 trial, elderly subjects who received an IS patch in combination with an injected flu vaccination generated antibody levels that were significantly higher than the antibody level generated with an injected flu vaccine alone. We also believe our IS patch could be used to lower the injected vaccine dosage necessary to immunize individuals with healthy immune responses.
PRODUCTS IN DEVELOPMENT
We are developing two distinct product applications for our TCI technology. The first application is a needle-free vaccine patch applied to the skin that contains both pathogen-specific antigens and an adjuvant. We are using this application to develop needle-free vaccines to replace currently injectable vaccines as well as novel vaccines that cannot be delivered by injection. The second application is an immunostimulant, or IS, patch for use in combination with an injected vaccine and through which only an adjuvant is delivered by the patch. In this second application, we expect that our IS patch would be placed over the injection site of an existing injectable vaccine in order to stimulate a stronger immune response to that vaccine. Initially we expect to target our IS patch for use in patient populations with weakened or impaired immune systems, such as the elderly, for whom existing vaccines often do not elicit an adequate immune response or possibly in pandemic flu applications.
We are focusing our current product development efforts on the product candidates set forth in the chart below.
| | | | | |
Product Candidates | | Clinical Status | |
| |
| Needle-free flu vaccine patch | | | Phase 1 | |
| IS patch for elderly receiving flu vaccines | | | Phase 2 | |
| IS patch for pandemic flu | | | Preclinical | |
| Needle-free travelers’ diarrhea vaccine patch | | | Phase 2 | |
6
We view our IS patches and our needle-free travelers’ diarrhea vaccine patch as distinct products. Although both patches contain the same active ingredient, they will have distinct formulations and require separate regulatory approvals.
Needle-free flu vaccine patch
Our needle-free flu vaccine patch product candidate combines flu antigens and an adjuvant in a single patch. We believe this approach offers the potential for effective flu vaccination without the use of needles.
We completed a Phase 1 study of our needle-free flu vaccine patch in Europe during the fourth quarter of 2004. The data from our Phase 1 trial demonstrated that the needle-free flu vaccine patch stimulated an immune response to antigens from all three flu strains recommended by the World Health Organization for the 2004 flu season and demonstrated a positive effect from using our adjuvant, although the observed immune response was not as great as that of existing injectable flu vaccines. We have been working to further optimize our formulation and product design in an effort to achieve an immune response equal to or greater than that of existing injectable vaccines, which is one of the requirements necessary in order to achieve regulatory approval for this product candidate. We did not observe any systemic vaccine-related side effects in this Phase 1 trial. Based on new preclinical studies, we believe we have reformulated this product candidate in a manner that will result in a greater immune response than that observed in the 2004 Phase 1 study. We are currently conducting toxicology studies on this new formulation. Assuming the results of the toxicology studies are favorable, we plan to commence a Phase 1/2 clinical trial in the United States in the third quarter of 2006. We plan to conduct additional Phase 2 trials after completing the Phase 1/2 trial.
Because we do not manufacture our own flu antigen, we conducted our previous clinical trial for this program, and intend to conduct our contemplated Phase 1/2 trial, using flu antigen supplied to us by a flu vaccine manufacturer under a material transfer agreement. We plan to work with companies who can provide a source of vaccine antigen and complementary commercial capabilities, such as sales and marketing expertise. We expect that we will not advance our needle-free flu vaccine product candidate into additional clinical trials after the planned Phase 1/2 trial until we enter into a long-term supply arrangement for flu antigen.
Immunostimulant (IS) patch for elderly receiving flu vaccines
We are currently investigating ways to improve the immune response in the elderly to standard influenza vaccination through the use of adjuvants. We believe that the placement of an IS patch over the injection site of an injected flu vaccine will provide a stronger immune response in elderly recipients of the flu vaccine. In 2002, we completed a Phase 1 clinical study in which elderly individuals who were vaccinated with an injected flu vaccine in combination with our IS patch had a higher antibody response, on average, than those receiving the flu vaccine without an IS patch.
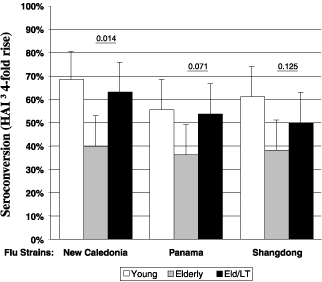
7
The following chart describes the immune response achieved in the Phase 1 study in the elderly using an IS patch in addition to a standard flu injection. In our study, three groups of approximately 55 volunteers were vaccinated with a standard injected flu vaccine consisting of three strains of flu antigen. The first group of volunteers consisted of healthy young adults. The second and third groups consisted of elderly (over 65 years old) volunteers. In the third group, the volunteers were given both the injected vaccine and an IS patch. In this trial, elderly individuals who were vaccinated with a standard flu vaccine in combination with our IS patch had a higher antibody response, on average, than those receiving the flu vaccine without a patch. In addition, we did not observe any systemic vaccine-related side effects in this Phase 1 trial. We conducted a subsequent study in which we utilized a dry patch formulation and were unable to demonstrate this same effect. After analysis, we concluded that we were not able to replicate the previous results due to a change in formulation, which we believe caused the active agent, our LT adjuvant, to remain in the patch and, therefore, not to be delivered to the skin. Based on data from animal studies and data from a recent travelers’ diarrhea clinical trial that indicated that our current dry patch formulation provided equal or greater immune responses to vaccination as compared to the wet patch used in our prior clinical trials, we expect our new dry patch formulations will be at least equivalent to the wet patches; however, we will not know whether these dry patch formulations will perform adequately to boost immune response in the elderly to standard influenza vaccination until they are tested in humans.
Enhanced Immune Responses In Elderly After IS Patch Application
We are planning a confirmatory Phase 2 study in the second half of 2006 designed to demonstrate that our IS patch may represent an effective strategy for improving response rates to influenza vaccines in the elderly. Following this trial, we expect to conduct additional Phase 2 trials to evaluate dose ranging andtime-of-patch-wear. We intend to request discussions with regulatory agencies in the United States and Europe prior to initiating any Phase 3 studies in order to finalize endpoints and design for our Phase 3 studies, which we expect to commence in 2007.
IS patch for pandemic flu
We anticipate that the IS patch could play a role in containing pandemic flu by helping governments execute dose-sparing strategies by which a smaller dose of injected vaccine could produce an effective immune response. We believe our IS patch, when administered in combination with an injected vaccine, may be able to achieve this either by reducing the required dose of injected vaccine, or improving the timingand/or magnitude of the immune response to an injected pandemic flu vaccine. We believe that our IS patch for pandemic flu presents a particularly attractive dose-sparing alternative because it has the potential to be used with pandemic flu vaccines being developed by different manufacturers without requiring different formulations or packaging. In January 2005, the
8
NIH awarded us a two-year, $2.9 million grant for further development of our IS patch technology for pandemic flu applications. Because pandemic flu vaccines are still being developed by third parties and no pandemic flu vaccines have been approved for use by the FDA, and because our IS patch for pandemic flu is in the preclinical development stage and extensive additional preclinical and clinical testing would be required before approval, this product candidate may not be available for the potential treatment of pandemic flu in the next few years, if ever.
We have conducted preclinical studies in mice that highlight the dose-sparing potential of our IS patch. The goal of these preclinical studies is to demonstrate that the use of our IS patch, if confirmed in larger scale human studies, could effectively expand the nation’s vaccine supply quickly. In one study, we examined the effects of our IS patch when used in combination with an avian flu antigen delivered either intramuscularly, into the muscle tissue, or intradermally, just below the skin surface. In this study, we immunized mice intradermally with injections of an avian flu strain isolated from Vietnam (H5N1) to look at the effect of immunizing with declining flu doses with and without our IS patch. Accordingly, the first group received 1.5 ug of vaccine and the other two groups received doses ten times lower (0.15 ug) and 100 times lower (0.015 ug), and half of each of these three groups had an IS patch placed over the injection site. After two weeks, we then measured antibody titers to the avian flu strain. As indicated in the following chart, the group receiving the 0.015 ug dose with our IS patch had an antibody response (47,491) comparable to the group receiving the 1.5 ug dose intradermally alone (20,142). This data suggests that a100-fold reduction in vaccine may be achievable when the vaccine is delivered intradermally in conjunction with our IS patch. Data that we collected in the same study suggests that a ten-fold reduction in vaccine may be achievable when the vaccine is delivered intramuscularly, which is the current standard of care, in conjunction with our IS patch. While intramuscular administration of flu vaccines is the current standard of care, recent studies have shown that flu vaccines administered intradermally can achieve comparable immune response by using less vaccine.
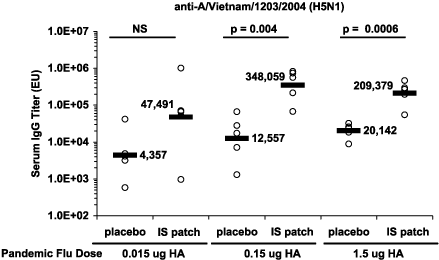
The U.S. Department of Health and Human Services issued in March 2006 a request for proposals, or RFP, to award one or more multi-year contracts to develop antigen sparing approaches to administering flu vaccines, to increase overall influenza vaccine capacity by utilizing adjuvants or alternate administration routes. Given our encouraging preclinical results and experience with the IS patch route of administration, we believe that we are well positioned to take advantage of this opportunity as currently outlined in this announcement, although there can be no assurances that we will be awarded any part of this government contract.
Needle-free travelers’ diarrhea vaccine patch
Our traveler’s diarrhea program is designed to reduce the risk of contracting travelers’ diarrhea or reduce the severity of symptoms in the event of infection. Our vaccine candidate targets disease caused byLT-secreting ETEC. The LT toxin alone or in conjunction with the ST toxin is the pathogenic agent in approximately two-thirds of the cases of ETEC infection. Immunity against LT has been shown to mitigate travelers’ diarrhea caused by ETEC infection, which we believe makes LT an excellent vaccine candidate. For our needle-free travelers’ diarrhea
9
vaccine patch, LT acts both as the antigen and the adjuvant. Because the LT toxin is sequestered in the skin, it is not delivered systemically in a way that might cause diarrhea.
We have conducted several clinical studies of our travelers’ diarrhea vaccine patch. In our initialproof-of-principle trial, a simple gauze patch containing a 500ug LT dose was applied to the skin for six hours with no pretreatment. Three such doses resulted in robust immune responses with no systemic side effects. This initial finding, robust immunity without systemic effects, has been replicated in 21 trials using a patch containing LT with approximately 1,600 healthy volunteers across all of our clinical programs.
In several clinical studies, we have explored skin pretreatment strategies to disrupt the stratum corneum to improve efficiency of delivering LT. In one study we used prep pads, normally used for the preparation of skin for an electrocardiogram, to pretreat the patch application site. In this study, a 50ug LT dose applied to the pretreated skin at days 0 and 21 resulted in an approximately 50-fold rise in anti-LT antibody, or IgG. This result is in comparison to an approximately 30-fold rise found in patients who did not receive a pretreatment step prior to the administration of a 400ug dose.
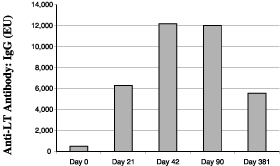
In other trials we have studied the effects of multiple doses and the durability of response. For example, in a study involving 60 volunteers where a needle-free travelers’ diarrhea vaccine patch was given on the first testing day and again 21 days later, test subjects experienced a 12-fold rise in antibody levels on the day the second patch was administered and a 23-fold rise in antibody levels 21 days after the second patch was administered (see figure, below). The subjects did not experience any vaccine-related systemic adverse events. Ninety-three percent of subjects experienced a mild rash which was limited to the skin that came in direct contact with the patch. The incidence of this mild rash decreased during the second application. Three hundred sixty days after receiving the second patch, 52 of the 53 returning test subjects retained LT antibodies in their systems, suggesting that the vaccine effects will last for at least one year.
Antibody Response to Toxin After Vaccination
During the first quarter of 2005, we completed a clinical study designed to test whether high antibody responses to LT delivered in a patch could prevent or reduce the severity of travelers’ diarrhea caused by oral ingestion of ETEC bacteria. We conducted the trial with the assistance of investigators at the Johns Hopkins School of Public Health. Forty-seven volunteers participated in this double-blind, placebo controlled clinical trial. Twenty-seven of the volunteers were vaccinated with a “wet patch” formulation of our needle-free travelers’ diarrhea vaccine patch and 20 were given placebos. At the end of the vaccination period, the volunteers from both vaccinated and unvaccinated groups ingested approximately one-half billion ETEC organisms. Although experts estimate that between 1,000 and 100,000 bacteria can cause an ETEC infection, we chose a considerably larger amount of ETEC organisms to ensure exposure in substantially all of our volunteers to an infectious dose. In this trial we also gave our volunteers live bacteria that secreted both the LT and ST toxins. This combination of high doses of ETEC bacteria and the introduction of two toxins resulted in a severe test of our vaccine candidate. After ingestion of the ETEC organisms, the volunteers were carefully monitored for diarrhea. The volunteers that received the needle-free travelers’ diarrhea vaccine patch experienced a delayed onset of ETEC disease and were found to have less severe diarrheal symptoms (measured by frequency and weight of diarrheal stools) as compared to volunteers that received the placebo. Recipients of the LT patch also required less treatment in terms of administration of intravenous fluids
10
and antibiotics to overcome the effects of their ETEC infections. Although prevention of travelers’ diarrhea was not possible in the clinical study due to the use of the very large challenge dose of ETEC bacteria that secreted both LT and ST toxins, our LT patch vaccine demonstrated its ability to induce a strong immune response that diminished the severity of the disease.
We are conducting a Phase 1/2 travelers’ diarrhea human clinical trial involving 160 subjects in the United States. In this study, we are comparing the dry and wet formulations of our patches with several skin pretreatment regimens. We are testing a total of eight groups, each with 20 subjects. 100 subjects have been dosed using a dry formulation and 60 subjects have been dosed using a wet formulation. All but one group received two doses of LT, the first on the first day and the second 21 days later. The final group received just one dose of LT, which was administered on the first day. The study was designed to ascertain whether immune responses after wearing the dry patch formulation were equal to those obtained with wet patches, and the protocol was designed to allow for analysis of immune responses at 21 days after the initial vaccination. Interim analysis of the data for all subjects as of day 21 indicates that our current dry patch formulation provided equal or greater immune responses than the wet patch formulation. In thehead-to-head groups the dry patch test subjects experienced higher responses as measured by fold rise in antibody levels in one group (27.0 fold rise in antibody levels for dry patch v. 9.3 fold rise in antibody levels for wet patch, p=0.03) and was equivalent in the other groups compared to the wet patch at day 21 (p> 0.05). Under the protocol we will continue to follow the immune responses for each group over a period of 200 days.
We are currently preparing for additional Phase 2 trials designed to determine the final dry patch formulation, number and timing of doses, and duration of patch wear. If we successfully complete these Phase 2 trials on our currently anticipated timeline, we expect to begin enrollment in Phase 3 clinical trials in mid-2007. We expect to meet and correspond with regulatory authorities in the United States and Europe over the course of the next year prior to initiating Phase 3 studies. We also are evaluating several potential Phase 3 field sites in Mexico and Central America, which have known ETEC disease rates, established infrastructure, extensive field trial experience and are readily accessible from the United States.
Additional indications
While we are currently focusing our resources on the four programs described above, we believe that our needle-free vaccine patch and IS patch technologies may be broadly applicable. If we have adequate resources, we plan to explore application of our technology to anthrax and other infectious diseases, as well as to cancer, allergic diseases and other indications. We have compiled Phase 1 data supporting the ability of TCI-based vaccines to stimulate an immune response against anthrax exposure. Anthrax is a rare, acute infectious disease caused by bacteria that is contracted through the skin, lungs or gastrointestinal system. The anthrax bioterrorism attacks of 2001 heightened the urgency to develop a safe and effective anthrax vaccine for use in the general population. The only anthrax vaccine currently approved by the FDA requires six injections over 18 months, has limited availability and distribution, and is only indicated for at-risk individuals, such as members of the military. We intend to seek a government grant to fund the development of a vaccine patch candidate. At this time, the United States government is not offering such a grant. If we are successful, we would hope to have the vaccine developed to the stage where it would be eligible for purchase under the biodefense stockpile program.
OUR STRATEGY
Our goal is to become a leader in the discovery, development and commercialization of vaccines and immunostimulants delivered to the skin. The key elements of our strategy are as follows:
| | |
| | • | Seek a strategic collaboration for the development, marketing and commercialization of our needle-free flu vaccine patch. As we do not intend to manufacture the flu antigens for our needle-free flu vaccine patch, we are actively exploring collaborations with large pharmaceutical companies in the United States and Europe that would provide the flu antigens and financial resources to develop and, if approved, commercialize our needle-free flu vaccine patch. To date, we have retained all of our rights to this product, and we believe this will enable us to obtain advantageous terms in any potential collaboration. |
| |
| | • | Commercialize our IS patch for elderly receiving flu vaccines. We intend to advance our IS patch for elderly receiving flu vaccines into and through Phase 3 clinical trials ourselves, and then into |
11
| | |
| | | commercialization, possibly with a partner. To date, we have retained all of our rights to this product candidate, and we believe this will enable us to obtain advantageous terms in any potential collaboration. |
| | |
| | • | Develop our IS patch for pandemic flu applications. We plan to utilize the $2.9 million grant from NIH to fund the further development of our IS patch technology for pandemic flu applications. We also plan to respond to the request for proposals to be issued by the U.S. Department of Health and Human Services for contracts to develop antigen sparing approaches to administering flu vaccines, to increase overall influenza vaccine capacity by utilizing adjuvants or alternate administration routes. |
| |
| | • | Commercialize our needle-free travelers’ diarrhea vaccine patch. We intend to advance our needle-free travelers’ diarrhea vaccine patch into and through Phase 3 clinical trials ourselves, and then into commercialization, possibly with a partner. To date, we have retained all of our rights to this product candidate, and we believe this will enable us to obtain advantageous terms in any potential collaboration. |
| |
| | • | Continue to develop and expand our manufacturing capabilities to retain manufacturing rights. We intend to retain manufacturing rights for our adjuvant and patch technologies. To the extent practicable, we intend to continue to develop and expand our manufacturing capabilities to meet our expected demands. In 2005, we completed the build-out and validation of the major systems of a pilot manufacturing facility, and we believe we currently have the capacity to produce anticipated needs through initial commercialization of our first product. |
| |
| | • | Leverage our broad technology platform to develop additional product candidates. As our TCI technology has the potential to be useful with many different types of vaccines, other companies developing vaccines represent possible partners with which to develop new products. For example, we are working with a large pharmaceutical company to evaluate three vaccine candidates in preclinical studies. We intend to continue to evaluate and develop additional product candidates to expand our pipeline where we perceive a significant unmet medical need and commercial potential. |
INTELLECTUAL PROPERTY AND PROPRIETARY TECHNOLOGY
Protection of our intellectual property and proprietary technology is a strategic priority for our business. We rely on a combination of patent, trademark, copyright and trade secret laws along with institutional know-how and continuing technological advancement to develop and maintain our competitive position. Our ability to protect and use our intellectual property rights in the continued development and commercialization of our technologies and products, operate without infringing the proprietary rights of others, and prevent others from infringing our proprietary rights, is crucial to our potential success. We will be able to protect our products and technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, trademarks or copyrights, or are effectively maintained as trade secrets, know-how or other proprietary information.
Our policy is to seek US and international patent protection for technological developments that we believe will enhance the market position of our product candidates and methods of using our product candidates. As of February 28, 2006, we held exclusive rights, either through our license from the Walter Reed Army Institute of Research (WRAIR) or on our own, to a patent portfolio consisting of 68 issued patents or patent applications as follows:
| | |
| | • | 3 issued US patents; |
| |
| | • | 14 US non-provisional patent applications; |
| |
| | • | 20 issued foreign patents; |
| |
| | • | 29 foreign patent applications that are in various national stages of prosecution; and |
| |
| | • | 2 foreign patent applications not at the national stage. |
The three issued United States patents will expire between November 2016 and February 2019. The issued foreign patents will expire between November 2017 and February 2019. Our core patents claim the composition of matter of the skin patch system as well as methods for inducing an antigen specific immune response from applying antigen and adjuvant to the skin. The first of these patents expires in November 2016, but subsequent patents
12
provide additional coverage until February 2019. In addition, we are prosecuting patent applications relating to the use of different antigen and adjuvant combinations on the skin and other improvements to our TCI technology, such as methods for inducing an immune response by applying antigens and adjuvants to the skin in dry form.
We license substantially all of our TCI technology from WRAIR. Pursuant to a license agreement dated April 6, 2001, WRAIR has granted us an exclusive, worldwide license to use the TCI technology in all fields of use, including the right to grant sublicenses subject to WRAIR’s approval. Each of our current product candidates is dependent on TCI technology subject to this license.
The terms of the WRAIR license require us to pay WRAIR royalties equal to a percentage of:
| | |
| | • | net sales of licensed products by us and by our affiliates; and |
| |
| | • | revenues received by us from the licensed TCI technology and by our affiliates under any sublicenses of the licensed TCI technology. |
| |
| | • | In addition, we are required to pay to WRAIR: |
| | |
| | • | a minimum annual payment of $15,000 as a non-recoverable advance against royalty and milestone payments; |
| |
| | • | milestone payments upon the achievement of specified regulatory approval based milestones, which may total up to $1,560,000 in the aggregate if we secure regulatory approval for six or more products; and |
| |
| | • | all costs and expenses relating to prosecution of patent protection for the licensed TCI technology. |
To date, we have paid WRAIR a license issue fee of $150,000, minimum annual advances against royalties and milestone payments totalling $60,000 in the aggregate, and milestone payments totalling $30,000. We have also issued to WRAIR 57,500 shares of our common stock valued at $34.125 per share on the date these shares were issued. The common stock issued to WRAIR was subject to a put option, which expired by its terms on February 1, 2006, the date of our initial public offering. The common shares issued to WRAIR are held in trust for WRAIR by MdBio, Inc. pursuant to a voting trust and escrow agreement dated April 6, 2001. Under the terms of this agreement, we are required to pay MdBio an annual service fee of $5,000.
Our license is subject to a non-exclusive, non-transferable, royalty-free right of the United States government to practice the TCI technology for research and other governmental purposes on behalf of the United States and on behalf of any foreign government or international organization pursuant to an existing or future treaty or agreement with the United States. Additionally, WRAIR reserves the right to require us to grant sublicenses to third parties if WRAIR determines that:
| | |
| | • | such sublicenses are necessary to fulfill public health and safety needs that we are not reasonably addressing; |
| |
| | • | such sublicenses are necessary to meet requirements for public use specified by applicable United States government regulations with which we are not reasonably in compliance; or |
| |
| | • | we are not manufacturing our products substantially in the United States. |
The license agreement with WRAIR will automatically terminate upon the expiration of the last licensed patent which, based on our current portfolio of issued patents and our assumption that such patents will not be invalidated, will occur no earlier than February 2019. WRAIR may unilaterally terminate or modify the license if we:
| | |
| | • | do not expend reasonable efforts and resources to carry out the development and marketing of the licensed TCI technology and do not manufacture, use or operate products that use the TCI technology by April 2008 (subject to extension based upon a showing of reasonable diligence in developing the technology); |
| |
| | • | do not continue to make our TCI-based products available to the public on commercially reasonable terms after we have developed such products; |
| |
| | • | misuse the licensed TCI technology or permit any of our affiliates or sub-licensees to do so; |
13
| | |
| | • | fail to pay royalties or meet our other payment or reporting obligations under the license; |
| |
| | • | become bankrupt; or |
| |
| | • | otherwise materially breach our obligations under the license. |
As we do not expect to have regulatory approval for any of our TCI-based product candidates by April 2008, we may need to seek an extension of the April 2008 clinical milestone.
We retain the right to terminate the WRAIR license with respect to any or all countries covered by the license at any time, and for any reason upon 60 days prior written notice. Upon termination of the WRAIR license, other than by expiration, we will be permitted to sell our existing inventory of licensed products for a period of 180 days, after which time we will no longer retain the right to make, use or sell products licensed under the WRAIR license. Our right to make, use or sell products subject to the WRAIR license will end immediately upon expiration of the WRAIR license.
In June 2005, we licensed from DowPharma, on a non-exclusive, world-wide basis, rights to the proprietary expression system that we plan to use to manufacture the LT to be used in each of our current product candidates. Pursuant to an existing letter agreement for services with DowPharma, DowPharma has provided us with technological assistance, as well as the master and working cell banks of the expression system that we plan to use to produce our LT.
In exchange for these licensed rights and such technical assistance as DowPharma has and is expected to continue to provide, the terms of the DowPharma license require us to pay DowPharma:
| | |
| | • | royalties equal to a percentage of net sales of products incorporating LT produced under the license; |
| |
| | • | milestone payments based upon regulatory approval of products incorporating LT produced under the license, which may total up to $500,000 for each approved product; and |
| |
| | • | time-based fees for any continuing technical assistance we may request at DowPharma’s prevailing hourly rate, with fees for the first 500 hours of technical assistance due only upon the date the first milestone payment, if any, is due. |
Our payment obligations under the license do not go into effect until our first product receives regulatory approval. As a result, we have not made any payments to DowPharma under the license. As of December 31, 2005, we had paid DowPharma $124,165 for services relating to technology transfer under our existing letter agreement. DowPharma has agreed to defer payment of the remaining $45,700 due under the letter agreement until we receive regulatory approval of our first product that utilizes DowPharma’s expression system.
The DowPharma license also requires us to grant DowPharma and its affiliates an irrevocable, world-wide, non-exclusive, royalty-free license to any improvements to the expression technology that we or our permitted sublicensees may create during the term of the license.
The DowPharma license will automatically terminate upon the earlier of (i) ten years from the date we first sell a product incorporating LT produced under the license and (ii) the end of the term of the last to expire patent subject to the license which, assuming the relevant DowPharma patent applications are issued and the resulting patents are not invalidated, is expected to occur no earlier than November 19, 2024. After expiration of the DowPharma license, we will retain an irrevocable and fullypaid-up license to produce LT using DowPharma’s proprietary expression system. DowPharma may unilaterally terminate the license agreement prior to expiration if we:
| | |
| | • | at any time during the term of the license agreement, reasonably agree with Dow that there are material health and safety risks associated with products produced under the license which create a material risk of liability to DowPharma; |
| |
| | • | fail to meet our payment obligations under the license; |
| |
| | • | become bankrupt; or |
| |
| | • | otherwise materially breach our obligations under the license. |
14
In the event DowPharma unilaterally terminates the license as described above, we will not have any continuing right to make, use or sell LT produced using DowPharma’s proprietary expression technology.
We also rely on trade secrets and know-how, which are not protected by patents, to maintain our competitive position. We have implemented trade secret protection policies to actively protect and secure the technology and proprietary information that we develop. We require our officers, employees and consultants to execute confidentiality agreements upon commencement of their relationships with us. These agreements also provide that all inventions developed by the individual on behalf of us must be assigned to us and that the individual will cooperate with us to secure patent protection for these inventions if we wish to pursue such protection. There can be no assurance, however, that these agreements will provide meaningful protection for our inventions, trade secrets or other proprietary information in the event of unauthorized use or disclosure of such information.
We also execute confidentiality agreements with outside collaborators. However, disputes may arise as to the ownership of proprietary rights to the extent that outside collaborators apply technological information developed independently by them or others to projects involving our technology and there can be no assurance that any such disputes would be resolved in our favor. In addition, any of these parties may breach the agreements and disclose our confidential information or our competitors might learn of the information in some other way. If any of our trade secrets, know-how or other proprietary information that is not protected by a patent were to be disclosed to or independently developed by a competitor, our business, results of operations and financial condition could be adversely affected.
MANUFACTURING AND COMMERCIAL SUPPLY
Prior to 2005, third parties supplied all of the LT and the formulated patches that we used in our clinical trials. For most of our history, we had one supplier of LT and relied on two contract manufacturers to produce our formulated patches. At this time, we have no further agreements with these firms to provide us with any further components or services.
To maintain control over the manufacture of our product candidates during development and possibly into commercialization, we constructed a pilot production plant in which we expect to manufacture the LT and the finished patch formulations for our clinical products. In connection with the construction of our pilot plant, we engaged DowPharma, a business unit within The Dow Chemical Company, to assist us with the development of our own LT strain and associated manufacturing process. In June 2005, we licensed from DowPharma a proprietary expression system for our LT strain. As part of the technology transfer under the license, DowPharma provided us with technological assistance, as well as the master and working cell banks of the expression system that we plan to use to produce our LT in our pilot plant.
We have substantially completed the validation of our pilot plant’s major systems, and we have completed initial engineering runs for the fermentation and purification of our LT, as well as the manufacture of certain patch formulations. Our LT requires precise, high quality manufacturing that is subject to the FDA’s Good Manufacturing Practices. Beginning in the fourth quarter of 2005, we began to manufacture batches of our LT for use in clinical trials in accordance with GMP standards. During the first half of 2006, we intend to initiate a clinical trial to assess the safety and immunogenicity of our LT. In addition, we are currently manufacturing certain patch formulations for our next clinical trials. We believe that our pilot plant has adequate capacity to meet our currently anticipated clinical trial needs, and we currently intend to use this facility for all future clinical production of LT adjuvant and formulated patches. To the extent practicable, we also intend to use this facility for initial commercial launch of our needle-free travelers’ diarrhea vaccine patch, if we obtain marketing approval.
The only product components that we currently intend to manufacture at our pilot plant are our LT and formulated patches. We currently intend to rely on either a collaborator or third-party supplier to produce other product components, such as the flu antigens contained in our needle-free flu vaccine patch. A flu vaccine manufacturer is currently supplying flu antigens for our contemplated Phase 1/2 clinical trial for our needle-free flu vaccine patch under a material transfer agreement. As variability in product specifications and characteristics generally is not acceptable to regulatory authorities, our primary sourcing strategy will be to establish long-term, single-source relationships with suppliers of these other components. For this reason, we expect that we will need to
15
enter into a long-term supply arrangement for flu antigen with a flu vaccine manufacturer before advancing our needle-free flu vaccine patch product candidate into additional clinical trials. If there are no existing suppliers or collaborators able or willing to meet our selection criteria, then we will consider contracting the manufacture of key components that we do not manufacture ourselves to a contract manufacturer. At this time, we have not entered into any agreements that provide us assurance of continued supply of any of these other product components.
COMPETITION
The pharmaceutical, biopharmaceutical and biotechnology industries are intensely competitive and are characterized by rapid and significant technological progress. Our competitors include large integrated pharmaceutical and biotechnology companies, universities, and public and private research institutions which currently engage in, have engaged in or may engage in efforts related to the discovery and development of new biopharmaceuticals and vaccines, some of which may be competitive. Almost all of these entities have substantially greater research and development capabilities and financial, scientific, manufacturing, marketing and sales resources than we do, as well as more experience in research and development, clinical trials, regulatory matters, manufacturing, marketing and sales.
If approved for marketing, our product candidates may compete with existing drugs and vaccines. For example, there are multiple influenza vaccines approved for sale in both the United States and Europe. These vaccines are marketed by companies that include Chiron Corporation, GlaxoSmithKline plc, MedImmune, Inc., Sanofi-Aventis S.A. and Solvay S.A. In addition, we know of multiple flu vaccine candidates that incorporate adjuvants to enhance immune responses, particularly in the elderly. Some of these flu vaccines with adjuvants are being developed by pharmaceutical companies that have substantial resources and well-established reputations among physicians and patients, such as Sanofi-Aventis S.A., GlaxoSmithKline plc and Chiron Corporation. These flu vaccines with adjuvants would compete against our IS patch for improved flu vaccination in the elderly.
While there are no vaccines against ETEC infection that have been approved for sale in the United States, we are aware of several companies with ETEC vaccine product candidates that are under development, which, if approved, would compete against our needle-free travelers’ diarrhea vaccine patch. Those companies with potential ETEC vaccine candidates include Avant Immunotherapeutics, Inc., Cambridge Biostability Ltd., Berna Biotech AG, SBL Vaccin AB and Chiron Corporation. In the absence of vaccines, travelers’ diarrhea is generally treated, either prophylactically or following onset, with antibiotics orover-the-counter products that alleviate symptoms. Some of these antibiotics and OTC products are marketed by pharmaceutical companies, such as Bayer. In addition, Salix Pharmaceuticals, Ltd. has announced that it has completed a Phase 3 study to evaluate the efficacy and safety of an antibiotic specifically designed to be taken prophylactically for travelers’ diarrhea.
We are aware of companies that are developing alternative methods to the syringe for delivering vaccines and biopharmaceutical products. These alternative methods include microneedles, electroporation, microporation, jet injectors, nasal sprays and oral delivery. For example, 3M Company, Alza Corporation and Becton, Dickinson and Company are developing microneedles and intradermal devices to deliver drugs to the skin. PowderMed Limited has developed a hand-held jet injector unit, MedImmune, Inc. has a nasal flu spray that has been approved for sale in the United States and ID Biomedical Corporation has a nasal flu spray under development. There are also a number of companies developing drug delivery technologies that make microchannels in the skin, including Altea Corporation, TransPharma Medical Ltd. and Sontra Medical Corporation.
We believe that the principal competitive factors in the markets for our product candidates will include:
| | |
| | • | safety and efficacy profile; |
| |
| | • | product price; |
| |
| | • | ease of application; |
| |
| | • | length of time to receive regulatory approval; |
| |
| | • | product supply; |
16
| | |
| | • | enforceability of patent and other proprietary rights; and |
| |
| | • | marketing and sales capability. |
Because our product candidates are in early stages of development, our relative competitive position in the future is difficult to predict precisely.
GOVERNMENT REGULATION
The testing, approval, manufacturing, labeling, advertising and marketing, post-approval safety reporting, and export, among other things, of our product candidates are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates biopharmaceutical products under the Federal Food, Drug, and Cosmetic Act, or FDCA, and other laws, including, in the case of biologics, the Public Health Service Act. Each of our product candidates consists of a patch and one or more active ingredients, such as vaccines or adjuvants, which we expect will be treated together as an investigational vaccine by the FDA, even if any of its active ingredients is part of an existing approved product. None of the active ingredients in our product candidates have been approved by the FDA for commercial sale in any product. We believe that most of our product candidates will be regulated by the FDA as biologics. We cannot market a biologic until we have submitted a Biologics License Application, or BLA, to the FDA, and the FDA has approved it. Both before and after approval is obtained, violations of regulatory requirements may result in various adverse consequences, including the FDA’s suspension of clinical studies, delay in approving or refusal to approve a product, suspension or withdrawal of an approved product from the market, operating restrictions, and the imposition of civil or criminal penalties.
The steps required before the FDA may approve a product for marketing in the United States generally include:
| | |
| | • | preclinical laboratory tests and animal tests; |
| |
| | • | the submission to the FDA of an investigational new drug application, or IND, for human clinical testing, which must become effective before human clinical trials may begin; |
| |
| | • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its specific intended use(s); |
| |
| | • | the submission to the FDA of a BLA; |
| |
| | • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is made to assess compliance with current good manufacturing practices, or cGMP; and |
| |
| | • | FDA review and approval of the BLA. |
Preclinical tests include laboratory evaluation of the product candidate, as well as animal studies to assess the potential safety and efficacy of the product candidate. We must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of an IND, which must become effective before we may commence human clinical trials. The IND will automatically become effective 30 days after its receipt by the FDA, unless the FDA before that time raises concerns or questions about the conduct of the trials as outlined in the IND. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed. We cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend such trials.
Clinical trials involve the administration of the product candidate to healthy volunteers or patients under the supervision of principal investigators, generally physicians who are not employed by or under the trial sponsor’s control. An institutional review board must review and approve each clinical study. Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. In Phase 1, the initial introduction of the drug into human subjects, the drug is usually tested for safety (adverse effects), dosage tolerance, and pharmacologic action. Phase 2 usually involves studies in a limited patient population to:
| | |
| | • | evaluate preliminarily the efficacy of the drug for specific, targeted conditions; |
17
| | |
| | • | determine dosage tolerance and appropriate dosage; and |
| |
| | • | identify possible adverse effects and safety risks. |
Phase 3 trials generally further evaluate clinical efficacy and test further for safety within an expanded patient population. We or the FDA may suspend clinical trials at any time on various grounds, including a finding that subjects are being exposed to an unacceptable health risk.
The results of the preclinical and clinical studies, together with other detailed information, including information on the manufacture and composition of the product candidate, are submitted to the FDA in the form of a BLA requesting approval to market the product. If the BLA contains all pertinent information and data, the FDA will “file” the application and begin review. The FDA may “refuse to file” the BLA if it does not contain all pertinent information and data. In that case, the applicant may resubmit the BLA when it contains the missing information and data. Before approving a BLA, the FDA will inspect the facilities at which the product candidate is manufactured, and will not approve the product candidate unless cGMP compliance is satisfactory. The FDA may refuse to approve a BLA if applicable regulatory criteria are not satisfied, require additional testing or information, limit the indications for useand/or require postmarketing testing and surveillance to monitor the safety or efficacy of a product.
The testing and approval process require substantial time, effort and financial resources, and we cannot be sure that any of our product candidates will be approved on a timely basis, if at all. The results of preclinical studies and initial clinical trials are not necessarily predictive of the results from large-scale clinical trials, and clinical trials may be subject to additional costs, delays or modifications due to a number of factors, including difficulty in obtaining enough patients, investigators or product candidate supply. Failure by us or our collaborators, licensors or licensees to obtain, or any delay in obtaining, regulatory approvals or in complying with requirements could adversely affect the commercialization of product candidates and our ability to receive product or royalty revenues. Also, if regulatory approval is granted, it is granted for use of the product in a specific dosage form for a specific indication. If a BLA holder wishes to market a product for a different dosage form or indication, it is generally required to submit and have approved a supplementary application.
Once the FDA approves a product, the BLA holder and product’s manufacturers are required to comply with a number of post-approval requirements. For example, we may be required to conduct postmarketing testing to monitor the safety and the efficacy of the marketed product, and we will be required to report certain adverse reactions to the FDA and to comply with certain requirements concerning advertising and promotional labeling for our products. Also, quality control and manufacturing procedures must continue to conform to cGMP regulations after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, the manufacturer must continue to expend time, monies and effort in the area of production and quality control to maintain cGMP compliance. We expect to rely on third parties to manufacture some of our product candidates after FDA approval, which will also be required to comply with cGMP. In addition, discovery of problems may result in marketing restrictions on a product. Also, new federal, state or local government requirements may be established that could delay or prevent regulatory approval of our product candidates or impose other additional restrictions on our business.
Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to commencement of marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. In those countries, we also are subject to foreign regulatory requirements governing clinical trials, labeling, promotion and marketing, post-approval safety reporting, export, pricing and reimbursement, which also vary greatly from country to country.
We are also subject to various federal, state and local laws, rules, regulations and policies relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances used in connection with our research work. Although we have developed safety procedures for handling and disposing of such materials, the risk of accidental injury or contamination from these materials cannot be entirely eliminated.
18
SALES AND MARKETING
We currently have no sales, marketing or distribution capability. In order to commercialize any of our product candidates, we must either internally develop sales, marketing and distribution capabilities or make arrangements with a third party to perform these services. We believe that by presenting a distinctive route of administration, TCI technology offers marketing and branding opportunities for us and our potential strategic partners. We intend to sell, market and distribute some products directly and rely on relationships with third parties to sell, market and distribute other products. To market any of our products directly, we must develop a marketing and sales force with technical and regulatory expertise and with supporting distribution capabilities.
EMPLOYEES
As of December 31, 2005, we directly employed 66 people full-time, of whom one has an M.D. degree, 15 have Ph.D. degrees, and two have DVM degrees. Of our workforce, 55 employees are engaged in research and development and 11 are engaged in business development, finance and administration. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
CORPORATE INFORMATION
We were incorporated in Delaware on May 14, 1997. Our address is 20 Firstfield Road, Suite 250, Gaithersburg, Maryland 20878, U.S.A. Our telephone number is(301) 556-4500.
AVAILABILITY OF PERIODIC SEC REPORTS
Our Internet website address iswww.iomai.com. We make available free of charge through our website our annual reports onForm 10-K, quarterly reports onForm 10-Q, current reports onForm 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC. The contents of our website are not part of, or incorporated into, this document. The reports and other information we file with the SEC can be read and copied at the SEC’s Public Reference Room at 100 F Street, N.E., Washington D.C. 20549. Copies of these materials can be obtained at prescribed rates from the SEC’s Public Reference Room at such address. You may obtain information regarding the operation of the public reference room by calling1-800-SEC-0330. The SEC also maintains a web site (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
Our future operating results may differ materially from the results described in this annual report due to the risks and uncertainties related to our business and our industry, including those discussed below. In addition, these factors represent risks and uncertainties that could cause actual results to differ materially from those implied by forward-looking statements in this report. We refer you to our “Cautionary Statement Regarding Forward-Looking Statements,” at the beginning of this report, which identifies forward-looking statements in this report. The risks described below are not the only risks we face. Additional risk and uncertainties not currently known to us or that we currently deem to be immaterial may also materially and adversely affect our business, financial condition or results of operations.
Risks Relating to Our Business
| |
| | We are a biopharmaceutical company with a limited operating history and have generated no revenue from product sales. As a development stage company, we face many risks inherent in our business. If we do not overcome these risks, our business will not succeed. |
Biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We commenced operations in May 1997, and since that time we have been engaged in research and development activities in connection with our product candidates. We have never generated any revenue from
19
product sales. All of our current product candidates are at an early stage of development, and we do not expect to realize revenue from product sales for several more years, if at all. In addition, we are not currently receiving any significant funding for our research and development programs from third parties through collaborations or grants. We are seeking to create a business based upon new technology that is intended to change existing practices of vaccine delivery. As such, we are subject to all the risks incident to the creation of new products and may encounter unforeseen expenses, difficulties, complications and delays and other unknown factors. You also should consider that we will need to:
| | |
| | • | obtain sufficient capital to support our efforts to develop our technology and commercialize our product candidates; |
| |
| | • | complete and continue to enhance the product characteristics and development of our product candidates; and |
| |
| | • | attempt to transition from a development stage company to a company capable of supporting commercial activities. |
| |
| | We have a history of operating losses and may never be profitable. |
We have incurred substantial losses since our inception, and we expect to continue to incur substantial losses for the foreseeable future. Our net loss for the twelve months ended December 31, 2005 was $18.0 million. As of December 31, 2005, we had an accumulated deficit of approximately $68.6 million. These losses have resulted principally from costs incurred in our research and development programs and from our general and administrative costs. We expect to incur additional operating losses in the future, and we expect these losses to increase significantly, whether or not we generate revenue, as we expand our product development and clinical trial efforts. These losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and stockholders’ equity.
To date, we have generated no revenue from product sales or royalties. We do not expect to achieve any revenue from product sales or royalties unless and until we receive regulatory approval and begin commercialization of our product candidates. We are not certain of when, if ever, that will occur.
Even if the regulatory authorities approve any of our product candidates and we commercialize these candidates, we may never be profitable. Even if we achieve profitability for a particular period, we may not be able to sustain or increase profitability.
| |
| | We will need additional funding, and we cannot guarantee that we will find adequate sources of capital in the future. |
As of December 31, 2005, we had approximately $5.2 million in cash, cash equivalents and marketable securities. We have incurred negative cash flows from operations since inception and have expended, and expect to continue to expend, substantial funds to conduct our research and development programs. On February 6, 2006, we closed our initial public offering (IPO), in which we raised net proceeds, before expenses, of $32,853,574. We estimate that our IPO final expenses will be approximately $2.0 million, so our total net proceeds after expenses will be approximately $30.8 million. We believe that our current working capital and reimbursement of expenses under our existing grants will be sufficient to fund our operating expenses and capital requirements through early 2007, although we cannot assure you that we will not require additional funds before then. We have based this estimate on assumptions that may prove to be wrong. Our future capital requirements will depend on many factors, including:
| | |
| | • | the number, size and complexity of our clinical trials; |
| |
| | • | our progress in developing our product candidates; |
| |
| | • | the timing of and costs involved in obtaining regulatory approvals; |
| |
| | • | costs of manufacturing our product candidates; |
20
| | |
| | • | costs to maintain, expand and protect our intellectual property portfolio; and |
| |
| | • | costs to develop our sales and marketing capability. |
We expect to seek to raise additional funds in advance of when we exhaust our cash resources, which, if we raise additional funds by issuing equity securities, will result in further dilution to our stockholders. Any equity securities issued also may provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise additional funds by issuing debt securities, these debt securities would have rights, preferences and privileges senior to those of holders of our common stock, and the terms of the debt securities issued could impose significant restrictions on our operations. If we raise additional funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technology, product candidates or products that we would otherwise seek to develop or commercialize ourselves, or to grant licenses on terms that are not favorable to us.
We do not know whether additional financing will be available on commercially acceptable terms when needed. If adequate funds are not available or are not available on commercially acceptable terms, we may need to downsize or halt our operations and may be unable to continue developing our products.
The success of some programs, such as our needle-free flu vaccine patch, may depend on licensing biologics from, and entering into collaboration arrangements with, third parties. We cannot be certain that our licensing or collaboration efforts will succeed or that we will realize any revenue from them.
The success of our business strategy is, in part, dependent on our ability to enter into multiple licensing and collaboration arrangements and to manage effectively the numerous relationships that will result. For example, we currently do not intend to manufacture any product components other than LT adjuvant and formulated patches. Therefore, we will need to negotiate agreements to acquire biologics, such as the flu antigens contained in our needle-free flu vaccine patch, and other product components from third parties in order to develop some of our vaccine candidates (other than our needle-free travelers’ diarrhea vaccine patch and IS patch). We are seeking a collaboration with respect to our needle-free flu patch. A failure to enter into a collaboration could delay development of this product candidate. Also, we may not establish a direct sales force for our products and, therefore, may need to establish marketing arrangements with third parties or major pharmaceutical companies.
Our ability to enter into agreements with commercial partners depends in part on convincing them that our TCI technology can help achieve and accelerate their goals or efforts. This may require substantial time and effort on our part. While we anticipate expending substantial funds and management effort, we cannot assure you that a strategic relationship will result or that we will be able to negotiate additional strategic agreements in the future on acceptable terms, if at all. Furthermore, we may incur significant financial commitments to collaborators in connection with potential licenses and sponsored research agreements. Generally, we will not be able to directly control the areas of responsibility undertaken by our strategic partners and will be adversely affected should these partners prove unable to carry the product candidates forward to full commercialization or should they lose interest in dedicating the necessary resources toward developing such products quickly.
Third parties may terminate our licensing and other strategic arrangements if we do not perform as required under these arrangements. Generally, we expect that agreements for rights to develop technologies will require us to exercise diligence in bringing product candidates to market and will require us to make milestone and royalty payments that, in some instances, are substantial. Our failure to exercise the required diligence or make any required milestone or royalty payment could result in the termination of the relevant license agreement, which could have a material adverse effect on us and our operations. In addition, these third parties may also breach or terminate their agreements with us or otherwise fail to conduct their activities in connection with our relationships in a timely manner. If we or our partners terminate or breach any of our licenses or relationships, we:
| | |
| | • | may lose our rights to develop and market our product candidates; |
| |
| | • | may lose patentand/or trade secret protection for our product candidates; |
| |
| | • | may experience significant delays in the development or commercialization of our product candidates; |
21
| | |
| | • | may not be able to obtain any other licenses on acceptable terms, if at all; and |
| |
| | • | may incur liability for damages. |
Licensing arrangements and strategic relationships in our industry can be very complex, particularly with respect to intellectual property rights. Disputes may arise in the future regarding ownership rights to technology developed by or with other parties. These and other possible disagreements between us and third parties with respect to our licenses or our strategic relationships could lead to delays in the research, development, manufacture and commercialization of our product candidates. These disputes could also result in litigation or arbitration, both of which are time-consuming and expensive. These third parties also may pursue alternative technologies or product candidates either on their own or in strategic relationships with others in direct competition with us.
None of our product candidates has been approved for commercial sale, and we may never receive such approval.
All of our product candidates are in early pre-commercial stages, and we do not expect our product candidates to be commercially available for several years, if at all. We expect that each of our product candidates, consisting of a patch and one or more active ingredients, will be treated together as a single investigational product by the FDA, even if any active ingredient is part of an existing approved product. None of the active ingredients in our product candidates have been approved by the FDA for commercial sale in any product. Our product candidates are subject to stringent regulation by regulatory authorities in the United States and in other countries. We cannot market any product candidate until we have completed our clinical trials and have obtained the necessary regulatory approvals for that product candidate. We do not know whether regulatory authorities will grant approval for any of our product candidates.
Conducting clinical trials and obtaining regulatory approvals are uncertain, time consuming and expensive processes. Our product candidates must complete rigorous preclinical testing and clinical trials. It will take us many years to complete our testing, and failure could occur at any stage of testing. Results of early trials frequently do not predict results of later trials, and acceptable results in early trials may not be repeated.
Even if we complete preclinical and clinical trials successfully, we may not be able to obtain regulatory approvals. Data obtained from preclinical and clinical studies are subject to varying interpretations that could delay, limit or prevent regulatory approval, and failure to observe regulatory requirements or inadequate manufacturing processes are examples of other problems that could prevent approval. In addition, we may encounter delays or rejections due to additional government regulation from future legislation, administrative action or changes in FDA policy. Even if the FDA approves a product, the approval will be limited to those indications encompassed in the approval; marketing the product for a different indication would require a supplementary application.
Outside the United States, our ability to market any of our potential products is contingent upon receiving marketing authorizations from the appropriate regulatory authorities. These foreign regulatory approval processes include all of the risks associated with the FDA approval process described above.
If our research and testing is not successful, or if we cannot show that our product candidates are safe and effective, we will be unable to commercialize our product candidates, and our business may fail.
Our technology is unproven and presents challenges for our product development efforts.
Our TCI technology represents a novel approach to stimulating the body’s immune system. There is no precedent for the successful commercialization of product candidates based on our technology. Developing biopharmaceuticals based on new technologies presents inherent risks of failure, including:
| | |
| | • | research could demonstrate that the scientific basis of our technology is incorrect or less sound than we had believed; |
| |
| | • | unexpected research results could reveal side effects or other unsatisfactory conditions that either will add cost to development of our product candidates or jeopardize our ability to complete the necessary clinical trials; and |
22
| | |
| | • | time and effort required to solve technical problems could sufficiently delay the development of product candidates such that any competitive advantage that we may enjoy is lost. |
All of our product candidates currently in development rely on LT, a naturally occurring bacterial toxin, as an adjuvant. LT has been shown to cause undesirable side effects when delivered through conventional mechanisms such as injection, intranasal and oral delivery methods. LT cannot be administered orally as an adjuvant and was linked to an increase in the risk of Bell’s palsy, a temporary paralysis of the facial muscles, when used in a nasal flu vaccine that was on the market in Europe during the 2000/2001 flu season. If we find that LT is not safe when administered to patients through the skin using our TCI technology, we will not be able to use LT as an adjuvant in our products. This would have a material adverse effect on our product development efforts.
Successful commercialization of our TCI technology will require integration of multiple dynamic and evolving components, such as antigens, adjuvants and a delivery mechanism, into finished product candidates. This complexity will likely increase the number of technical problems that we can expect to confront in the clinical and product development processes and, therefore, add to the cost and time required to commercialize each product candidate.
As part of our product development efforts, we may make significant changes to our product candidates. These changes may not yield the benefits that we expect.
We have made changes in the design or application of our product candidates in response to preclinical and clinical trial results and technological changes, and we may make additional changes in the future before we are able to commercialize our products. These and other changes to our product candidates may not yield the benefits that we expect and may result in complications and additional expenses that delay the development of our product candidates. In addition, changes to our product candidates may not be covered by our existing patents and patent applications, and may not qualify for patent protection, which could have a material adverse effect on our ability to commercialize our product candidates. See the risk factor entitled “If we are unable to protect our intellectual property, we may not be able to operate our business profitably.”
We recently began testing a new formulation for our TCI patch technology. If this new formulation does not perform as well as we expect, we will need to seek an alternate formulation for our patch, which would delay, or prevent, the development of our TCI products.
In order to accelerate our product development efforts, we have conducted many of our clinical trials to date using a “wet patch,” which entails topically administering antigens and adjuvants to the skin in liquid form and covering the area with a gauze bandage. In order to successfully commercialize our products, we believe we will need to, and we intend to, administer our needle-free vaccine patches and IS patches using a “dry patch.” In a dry patch, antigens and adjuvants would be incorporated into the patch through the manufacturing process. In a Phase 2 clinical study of our IS patch using a dry patch formulation, we were unable to replicate the clinical data obtained in a similar prior trial using a wet patch formulation in the elderly indicating that adding an adjuvant-containing patch to a standard flu injection could result in an improved immune response. After analysis, we concluded that we were not able to replicate the previous results due to a change in formulation, which we believe caused the active agent, our LT adjuvant, to remain in the patch and, therefore, not to be delivered to the skin. Based on data from animal studies and the data from a recent travelers’ diarrhea clinical trial that indicated that our current dry patch formulation provided equal or greater immune responses to vaccination as compared to the wet patch, we expect that these new dry patch formulations will be at least equivalent to the wet patches used in our prior clinical trials; however, we will not know whether these dry patch formulations will perform adequately until they are tested repeatedly in humans. If they do not perform as we expect, we would need to reformulate them again. If we are unable to produce our products in a dry patch formulation, it could significantly reduce the commercial attractiveness of our products. In that case, we may have to repeat studies, which would increase our development costs and delay our clinical development programs.
23
We may be required to conduct clinical trials of our IS patch with flu vaccines developed by different manufacturers, which may lead to added cost and delay in approval and commercialization of our IS patch.
In order for our IS patch to gain approval from the FDA and to be used with commercially available injectable flu vaccines, we may need to conduct multiple clinical trials of our IS patch with multiple flu vaccines developed by different manufacturers. This may substantially expand the cost and time required for the clinical trials of our IS patch, which could delay its potential approval by the FDA and its commercialization.
We recently began developing and manufacturing heat labile toxin which may not perform comparably to the heat labile toxin we have purchased for use in previous clinical trials.
Until recently, we have obtained all of the heat labile toxin (LT) used as an adjuvant in our product candidates (and also as an antigen in our needle-free travelers’ diarrhea vaccine patch) from a contract manufacturer. We believe that it is important for us to have control over the manufacture and development of the LT used in our product candidates. As a result, we have terminated our relationship with our contract manufacturer and have developed and are manufacturing our own strain of LT. Because LT is produced in biological processes, there can be no assurance of equivalence between the materials manufactured by our previous supplier and materials manufactured by us in our pilot plant in terms of purity, safety, potency and concentration. While we believe our LT strain is substantially similar to the LT provided by the contract manufacturer and the two strains appear to perform comparably in animal studies, we will not know if there are any differences in humans until we perform comparability studies in humans. We intend to initiate a clinical trial during the first half of 2006 to assess the safety and immunogenicity of our LT.
The skin pretreatment methods used in connection with our product candidates may make our products less attractive to consumers.
We have observed in our product development efforts that the delivery of antigens and adjuvants to Langerhans cells through the skin is significantly improved by pretreating the skin to disrupt the outer dead layer of skin, known as the stratum corneum. Based on animal studies, we believe that the method and level of skin pretreatment may differ from one product candidate to another because of the different physical properties of the active ingredients. Therefore, more than one pretreatment method may be required for our different products to be effective. In recent clinical studies for our needle-free travelers’ diarrhea vaccine patch and IS patch, we have tested simple abrasive materials to disrupt the stratum corneum and are now working to design and test improved embodiments of these materials for use in our upcoming clinical trials for our product candidates. We believe our pretreatments will be simple to perform and will not involve discomfort to the patient. However, to the degree that our pretreatments are not perceived to be simple to perform or involve patient discomfort, it could reduce the attractiveness of our products since alternative vaccines or treatments are available for many of the indications that our product candidates under development seek to address.
If any of our products is approved for commercial sale, we may be subject to penalties if we fail to comply with post-approval regulatory requirements or experience unanticipated problems with any approved products.
If we obtain marketing approval for a product, that product, along with the associated manufacturing processes, any post-approval clinical data and the advertising and promotional activities for the product will be subject to stringent ongoing regulation including review and periodic inspection by the FDA and other regulatory bodies. Product approval may be subject to limitations on the indicated uses for which the product may be marketed or to other restrictive conditions of approval. Furthermore, any approval may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products or failure to comply with regulatory requirements, may result in:
| | |
| | • | product recalls; |
| |
| | • | revocation of previously granted approvals; |
24
| | |
| | • | the need to conduct additional clinical trials; and |
| |
| | • | fines and other censures. |
We may be slow or unable to adapt to changes in existing regulatory requirements or the adoption of new regulatory requirements or policies.
If physicians and patients do not accept our products, we may be unable to generate significant revenue.
Even if any of our vaccine candidates obtain regulatory approval, they still may not gain market acceptance among physicians, patients and the medical community, which would limit our ability to generate revenue and would adversely affect our results of operations. Physicians will not recommend products developed by us or our collaborators until clinical data or other factors demonstrate the safety and efficacy of our products as compared to other available treatments. Even if the clinical safety and efficacy of our products is established, physicians may elect not to recommend these products for a variety of factors, including the reimbursement policies of government and third-party payors. There are other established treatment options for the diseases that many of our product candidates target, such as travelers’ diarrhea and the flu. In order to successfully launch a product based on our TCI technology, we must educate physicians and patients about the relative benefits of our products. If our products are not perceived as easy and convenient to use, for example as compared to an injectable vaccine or antibiotics, or are perceived to present a greater risk of side effects or are not perceived to be as effective as other available treatments, physicians and patients might not adopt our products. A failure of our technology to gain commercial acceptance would have a material adverse effect on our business. We expect that, if approved for commercialization, our needle-free travelers’ diarrhea vaccine patch will be paid for by patients out of pocket. Our needle-free travelers’ diarrhea vaccine patch is not designed to protect against all forms of travelers’ diarrhea, rather it is designed to protect against only those cases of travelers’ diarrhea caused by ETEC and in which the LT toxin is present. We estimate this to be approximately one-third of all cases of travelers’ diarrhea. This could limit commercial acceptance of this product. Because our needle-free flu vaccine patch and IS patch candidates are targeted at preventing or ameliorating the effects of flu infection, if the launch of these products for a particular flu season fails, we may not receive significant revenues from that product until the next season, if at all. See the risk factors set forth below entitled “Our competitors in the biopharmaceutical industry may have superior products, manufacturing capability or marketing expertise,” “Our ability to generate revenues will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products from third-party payors,” and “We have no experience in sales, marketing and distribution and will depend on the sales and marketing efforts of third parties.”
Our IS patch for pandemic flu program relies on government health organizations for funding clinical development and for ultimately procuring the product, if approved.
We currently rely upon funding from the United States government for the preclinical development of our IS patch for pandemic flu and plan to seek additional funding from the United States government for this program. The procurement of government funding is a time consuming process and is subject to government appropriations and other political risks. As a result, the receipt by us of funds from the United States government to fund our research and development of our IS patch for pandemic flu cannot be assured. In addition, if approved, we expect that United States and foreign governments would be the entities procuring any pandemic flu products, not individual consumers. While there has recently been increased public awareness of the risks of pandemic flu, government health organizations may not devote significant resources to the prevention of an outbreak and may not seek to procure pandemic flu products from us. The acceptance of our product candidate for pandemic flu may depend on whether government health organizations adopt a dose-sparing strategy to prevent pandemic flu and whether a competing technology for dose sparing is adopted. If a dose-sparing strategy is not endorsed or a competing technology for dose sparing is adopted, our product candidate will not yield any revenues.
The development and clinical testing of our IS patch for pandemic flu product candidate will likely take several years. Even if our IS patch for pandemic flu obtains regulatory approval, by that time the threat of a pandemic flu outbreak may be reduced or government health organizations may have adequate stockpiles of flu vaccine or have adopted other technologies or strategies to prevent or limit outbreaks.
25
Even if our IS patch for pandemic flu gains regulatory approval and governmental health organizations choose to stockpile the product, we may be not be able to produce enough of the product to fulfill public health and safety needs.
Our license to use TCI technology from The Walter Reed Army Institute of Research, or WRAIR, is critical to our success. Under some circumstances, WRAIR may modify or terminate our license or sublicense the TCI technology to third parties which could adversely affect our business.
We license substantially all of our patented and patentable TCI technology through an exclusive license from WRAIR, a federal government entity. This license will terminate automatically on the expiration date of the last to expire patent subject to the license, which, based on currently issued patents and our assumption that such patents will not be invalidated, would be February 2019. WRAIR may unilaterally modify or terminate the license if we, among other things:
| | |
| | • | do not expend reasonable efforts and resources to carry out the development and marketing of the licensed TCI technology and do not manufacture, use, or operate products that use the TCI technology by April 2008 (subject to extension based upon a showing of reasonable diligence in developing the technology); |
| |
| | • | do not continue to make our TCI-based products available to the public on commercially reasonable terms after we have developed such products; |
| |
| | • | misuse the licensed TCI technology or permit any of our affiliates or sub-licensees to do so; |
| |
| | • | fail to pay royalties or meet our other payment or reporting obligations under the license; |
| |
| | • | become bankrupt; or |
| |
| | • | otherwise materially breach our obligations under the license. |
As we do not expect to have regulatory approval for any of our TCI-based product candidates by April 2008, we may need to seek an extension of the April 2008 milestone. If we violate the terms of the WRAIR license, or otherwise lose our licensed rights to the TCI technology, we would likely be unable to continue to develop our products. WRAIR and third parties may dispute the scope of our rights to the TCI technology under the license. Additionally, WRAIR may breach the terms of its obligations under the license or fail to prevent infringement or fail to assist us to prevent infringement by third parties of the patents underlying the licensed TCI technology. Loss or impairment of the WRAIR license for any reason could materially harm our financial condition and operating results.
In addition to WRAIR’s termination and modification rights described above, our license is subject to a non-exclusive, non-transferable, royalty-free right of the United States government to practice the licensed TCI technology for research and other governmental purposes on behalf of the United States and on behalf of any foreign government or international organization pursuant to any existing or future treaty or agreement with the United States. WRAIR also reserves the right to require us to grant sublicenses to third parties if WRAIR determines that:
| | |
| | • | such sublicenses are necessary to fulfill public health and safety needs that we are not reasonably addressing; |
| |
| | • | such sublicenses are necessary to meet requirements for public use specified by applicable United States government regulations with which we are not reasonably in compliance; or |
| |
| | • | we are not manufacturing our products substantially in the United States. |
Although we are currently the only parties licensed to actively develop the TCI technology, we cannot assure you that WRAIR will not in the future require us to sublicense the TCI technology. Any action by WRAIR to force us to issue such sublicenses or development activities instituted by the United States government pursuant to its reserved rights in the TCI technology would erode our ability to exclusively develop products based on the TCI technology and could materially harm our financial condition and operating results.
Licenses of technology owned by agencies of the United States government, including the WRAIR license, require that licensees — in this case, us — and our affiliates and sub-licensees agree that products covered by the license will be manufactured substantially in the United States. This may restrict our ability to contract for manufacturing facilities, if we attempt to do so, outside the United States and we may risk losing our rights under the WRAIR license, which could materially harm our financial condition and operating results.
26
If we are unable to protect our intellectual property, we may not be able to operate our business profitably.
We base our TCI technology in large part on innovations for which WRAIR has sought protection under the United States and certain foreign patent laws. We consider patent protection of our TCI technology to be critical to our business prospects. As of December 31, 2005, there are three issued United States patents, 20 issued foreign patents and 45 United States and foreign patent applications relating to TCI and improvements on the technology. The three issued United States patents will expire between November 2016 and February 2019. The issued foreign patents will expire between November 2017 and February 2019. While we have filed several patent applications relating to the use of our IS patch technology in conjunction with existing injectable vaccines, no patent relating to that technology has been issued. Under our license agreement with WRAIR, we bear financial responsibility for the preparation, filing, prosecution and maintenance of any and all patents and patent applications licensed. With respect to enforcement, we have the right to bring actions to enforce patents licensed under the WRAIR license agreement, subject to WRAIR’s continuing right to intervene, and WRAIR maintains the right to bring enforcement actions if we fail to do so.
Patent protection in the field of biopharmaceuticals is highly uncertain and involves complex legal and scientific questions and has recently been the subject of much litigation. We cannot control when or if any patent applications will result in issued patents. Even if issued, our patents may not afford us protection against competitors marketing similar products. Neither the US Patent and Trademark Office nor the courts have a consistent policy regarding the breadth of claims allowed or the degree of protection afforded under many biopharmaceutical patents. The claims of our existing US patents and those that may issue in the future, or those licensed to us, may not offer significant protection of our TCI technology and other technologies. Our patents on transcutaneous immunization, in particular, are broad in that they cover the delivery of antigens and adjuvants to the skin to induce an immune response. While our TCI technology is covered by issued patents and we are not aware of any challenges, patents with broad claims tend to be more vulnerable to challenge by other parties than patents with more narrowly written claims. Patent applications in the United States and many foreign jurisdictions are typically not published until 18 months following their priority filing date, and in some cases not at all. In addition, publication of discoveries in scientific literature often lags significantly behind actual discoveries. Therefore, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. In addition, changes in either patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. Furthermore, our competitors may independently develop similar technologies or duplicate technology developed by us in a manner that does not infringe our patents or other intellectual property.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patented technology, we rely upon unpatented proprietary technology, processes and know-how. We and our licensors seek to protect this information in part by confidentiality agreements with employees, consultants and third parties. These agreements may be breached, and there may not be adequate remedies for any such breach. In addition, our trade secrets may otherwise become known or be independently developed by competitors. If we are unable to protect the confidentiality of our proprietary information and know-how, competitors may be able to use this information to develop products that compete with our products, which could adversely impact our business.
If the use of our technology conflicts with the intellectual property rights of third parties, we may incur substantial liabilities, and we may be unable to commercialize products based on this technology in a profitable manner, if at all.
Our competitors or others may have or acquire patent rights that they could enforce against us. In addition, we may be subject to claims from others that we are misappropriating their trade secrets or confidential proprietary information. If our technology conflicts with the intellectual property rights of others, they could bring legal action
27
against us or our licensors, licensees, suppliers, customers or collaborators. If a third party claims that we infringe upon its proprietary rights, any of the following may occur:
| | |
| | • | we may become involved in time-consuming and expensive litigation, even if the claim is without merit; |
| |
| | • | we may become liable for substantial damages for past infringement if a court decides that our technology infringes upon a competitor’s patent; |
| |
| | • | a court may prohibit us from selling or licensing our product without a license from the patent holder, which may not be available on commercially acceptable terms, if at all, or which may require us to pay substantial royalties or grant cross licenses to our patents; and |
| |
| | • | we may have to redesign our product so that it does not infringe upon others’ patent rights, which may not be possible or could be very expensive and time-consuming. |
If any of these events occurs, our business will suffer and the market price of our common stock will likely decline.
We may be involved in lawsuits to protect or enforce our patents or the patents of our collaborators or licensors, which could be expensive and time consuming.
Competitors may infringe our patents or the patents of our collaborators or licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings brought by the US Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patent applications or those of our collaborators or licensors. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and be a distraction to our management. We may not be able, alone or with our collaborators and licensors, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
We depend on our key personnel, the loss of whom would adversely affect our operations. If we fail to attract and retain the talent required for our business, our business will be materially harmed.
We are a small company with 66 employees as of December 31, 2005, and we depend to a great extent on principal members of our management and scientific staff. If we lose the services of any key personnel, in particular, Stanley Erck, our President and Chief Executive Officer, or Gregory Glenn, our Chief Scientific Officer, it could significantly impede the achievement of our research and development objectives and could delay our product development programs and approval and commercialization of our product candidates. We do not currently have any key man life insurance policies. While we have entered into agreements with certain of our executive officers relating to severance after a change of control and these officers have agreed to restrictive covenants relating to non-competition and non solicitation, we have not entered into employment agreements with members of our senior management team other than Stanley Erck. Our agreement with Stanley Erck does not ensure that we will retain his services for any period of time in the future. Our success depends on our ability to attract and retain highly qualified scientific, technical and managerial personnel and research partners. Competition among biopharmaceutical and biotechnology companies for qualified employees is intense, and we may not be able to retain existing personnel or
28
attract and retain qualified staff in the future. If we fail to hire and retain personnel in key positions, we will be unable to develop or commercialize our product candidates in a timely manner.
We rely on third parties to conduct our clinical trials, and those third-parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such trials.
We do not have the ability to independently conduct clinical trials for our vaccine candidates, and we rely on third parties such as contract research organizations, medical institutions and clinical investigators to enroll qualified patients and conduct our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for and commercialize our vaccine candidates may be delayed or prevented.
Our competitors may have superior products, manufacturing capability or marketing expertise.
Our business may fail because it faces intense competition from major pharmaceutical companies, specialized biopharmaceutical and biotechnology companies and drug development companies engaged in the development and production of vaccines, vaccine delivery technologies and other biopharmaceutical products. Several companies are pursuing programs that target the same markets we are targeting. In addition to pharmaceutical, biopharmaceutical and biotechnology companies, our competitors include academic and scientific institutions, government agencies and other public and private research organizations. Many of our competitors have greater financial and human resources and more experience. Our competitors may:
| | |
| | • | develop products or product candidates earlier than we do; |
| |
| | • | form collaborations before we do, or preclude us from forming collaborations with others; |
| |
| | • | obtain approvals from the FDA or other regulatory agencies for such products more rapidly than we do; |
| |
| | • | develop and validate manufacturing processes more rapidly than we do; |
| |
| | • | obtain patent protection or other intellectual property rights that would limit our ability to use our technologies or develop our product candidates; |
| |
| | • | develop products that are safer or more effective than those we develop or propose to develop; or |
| |
| | • | implement more effective approaches to sales and marketing. |
Alternative competitive technologies and products could render our TCI technology and our product candidates based on this technology obsolete and non-competitive. Presently, there are a number of companies developing alternative methods to the syringe for delivering vaccines and other biopharmaceutical products. These alternative methods include microneedles, electroporations, microporations, jet injectors, nasal sprays and oral delivery, and several of these delivery mechanisms are in late stage clinical trials.
While there are no vaccines against infection by enterotoxigenicE. coli bacteria(ETEC) that have been approved for sale in the United States, we are aware of several companies with ETEC vaccine product candidates that are in development, which, if approved, would compete against our needle-free travelers’ diarrhea vaccine patch. Those companies with potential ETEC vaccine candidates include Avant Immunotherapeutics, Inc., Berna Biotech AG, Cambridge Biostability, Limited, SBL Vacin AB and Emergent BioSolutions, Inc. One of our competitors, SBL Vacin AB, has announced the results from a study of an ETEC vaccine indicating the vaccine may be effective in preventing diarrhea caused by ETEC. In the absence of vaccines, travelers’ diarrhea is generally treated, either prophylactically or following onset, with antibiotics orover-the-counter, or OTC, products that alleviate symptoms. Some of these OTC products and antibiotics, such as Cipro, are marketed by pharmaceutical companies with substantial resources and enjoy widespread acceptance among physicians and patients. In addition, Salix Pharmaceuticals, Ltd. has announced that it has completed a Phase 3 study to evaluate the efficacy and safety of an antibiotic specifically designed to be taken prophylactically for the prevention of travelers’ diarrhea.
29
There are multiple influenza vaccines approved for sale in both the United States and Europe. In many cases, these products are manufactured and distributed by pharmaceutical companies with substantial resources, such as Chiron Corporation, GlaxoSmithKline plc, Sanofi-Aventis SA, Solvay SA and MedImmune, Inc. FluMist, a nasal flu vaccine, has received marketing approval from the FDA and would compete against our needle-free flu vaccine. We are also aware of other flu vaccine candidates to be delivered by alternative methods, such as nasal spray and skin delivery, which, if approved, would compete against our needle-free flu vaccine. In addition, we know of multiple flu vaccine candidates that incorporate adjuvants to enhance immune responses, particularly in the elderly. Some of these adjuvanted flu vaccines are being developed by pharmaceutical companies with substantial resources, such as Sanofi-Aventis SA, GlaxoSmithKline plc and Chiron Corporation. These adjuvanted vaccines would compete against our IS patch for the elderly.
Our ability to generate revenues will be diminished if we fail to obtain acceptable prices or an adequate level of reimbursement for our products.
We expect that most patients will rely on private health insurers, Medicare and Medicaid and other third party payors to pay for any products that we or our collaborators may market, other than our needle-free travelers’ diarrhea vaccine patch for which we expect patients will payout-of-pocket. The continuing efforts of government and third party payors to contain or reduce the costs of health care through various means may limit our commercial opportunity. For example, in some foreign markets, pricing and profitability of prescription biopharmaceuticals are subject to government control. In the United States, we expect that there will continue to be a number of federal and state proposals to implement similar government controls. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of biopharmaceutical products. Cost control initiatives could decrease the price that we would receive for any products in the future.
Our ability to commercialize biopharmaceutical product candidates, alone or with third parties, could be adversely affected by cost control initiatives and also may depend in part on the extent to which reimbursement for the product candidates will be available from:
| | |
| | • | government and health administration authorities; |
| |
| | • | private health insurers; and |
| |
| | • | other third party payors. |
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Third party payors, including Medicare, are challenging the prices charged for medical products and services. Government and other third party payors increasingly attempt to contain health care costs by limiting both coverage and the level of reimbursement for new biopharmaceutical products and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted labeling approval. In the United States, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to market and sell our product candidates profitably. In particular, in December 2003, President Bush signed into law new Medicare prescription drug coverage legislation which went into effect on January 1, 2006. Under this legislation, the Centers for Medicare and Medicaid Services, or CMS, the agency within the Department of Health and Human Services that administers Medicare and is responsible for reimbursement of the cost of drugs, has asserted the authority of Medicare to elect not to cover particular drugs if CMS determines that the drugs are not “reasonable and necessary” for Medicare beneficiaries or to elect to cover a drug at a lower rate similar to that of drugs that CMS considers to be “therapeutically comparable.” Changes in reimbursement policies or health care cost containment initiatives that limit or restrict reimbursement for our products may cause our potential revenues to decline. Third party insurance coverage may not be available to patients for any product candidates we discover and develop, alone or through our strategic relationships. If government and other third party payors do not provide adequate coverage and reimbursement levels for our product candidates, the market acceptance of these product candidates may be reduced.
30
We may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities by us and third party manufacturers for preclinical and clinical trials. If any of our product candidates is approved by the FDA or other regulatory agencies for commercial sale, we will need to manufacture it in larger quantities. We or our third party manufacturers may not be able to successfully increase the manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our product candidates require precise, high quality manufacturing that is subject to the FDA’s Good Manufacturing Practices. Our failure or the failure of our third party manufacturers to achieve and maintain these high manufacturing standards and comply with the FDA’s Good Manufacturing Practices, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business. In addition, our manufacturing facilities will be subject to unannounced inspections by the FDA, and significantscale-up of manufacturing may require additional validation studies, which the FDA must review and approve.
We have no experience in sales, marketing and distribution and will depend on the sales and marketing efforts of third parties.
We plan to establish marketing arrangements with third parties or major pharmaceutical companies and do not expect to establish direct sales capability for several years. However, these types of marketing arrangements might not be available on acceptable terms, or at all. In the future, to market any of our product candidates directly, we will need to develop a marketing and sales force with technical expertise and distribution capability. To the extent that we enter into marketing or distribution arrangements, any revenues we receive will depend upon the efforts of third parties. We cannot assure you that we will be successful in gaining market acceptance for any products we may develop.
Our business exposes us to potential product liability claims.
Our proposed products could be the subject of product liability claims. A failure of our product candidates to function as anticipated, whether as a result of the design of these products, unanticipated health consequences or side effects, or misuse or mishandling by third parties of such products, could result in injury. Claims also could be based on failure to immunize as anticipated. Tort claims could be substantial in size and could include punitive damages. We cannot assure you that any warranty disclaimers provided with our proposed products would be successful in protecting us from product liability exposure. Damages from any such claims could be substantial and could affect our financial condition.
Our business exposes us to potential product liability risks that are inherent in the testing, manufacturing and marketing of biopharmaceutical products. We have obtained clinical trial liability insurance for our clinical trials in the aggregate amount of $10 million. We cannot be certain that we will be able to maintain adequate insurance for our clinical trials. We also intend to seek product liability insurance in the future for products approved for marketing, if any. However, we may not be able to acquire or maintain adequate insurance at a reasonable cost. Any insurance coverage may not be sufficient to satisfy any liability resulting from product liability claims. A successful product liability claim or series of claims could have a material adverse impact on our operations.
We deal with hazardous materials that may cause injury to others and are regulated by environmental laws that may impose significant costs and restrictions on our business.
Our research and development programs and manufacturing operations involve the controlled use of potentially harmful biological materials such as toxins fromE. coliand other hazardous materials. We cannot completely eliminate the risk of accidental contamination or injury to others from the use, manufacture, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. We are
31
also subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling or disposal of hazardous materials and waste products. If we fail to comply with these laws and regulations or with the conditions attached to our operating licenses, then our operating licenses could be revoked, we could be subjected to criminal sanctions and substantial liability and we could be required to suspend, modify or terminate our operations. We may also have to incur significant costs to comply with future environmental laws and regulations. We do not currently have a pollution and remediation insurance policy.
Until recently, we have operated as a private company and as a result, we have limited experience attempting to comply with public company obligations. Attempting to comply with these requirements will increase our costs and require additional management resources, and we still may fail to comply.
On February 6, 2006, we closed our initial public offering (IPO). Previously, as a private company, we maintained a small finance and accounting staff. While we expect to expand our staff, we may encounter substantial difficulty attracting qualified staff with requisite experience due to the high level of competition for experienced financial professionals.
We will face increased legal, accounting, administrative and other costs and expenses as a public company that we did not incur as a private company. Compliance with the Sarbanes Oxley Act of 2002, as well as other rules of the SEC, the Public Company Accounting Oversight Board and The Nasdaq National Market will result in a significant initial cost to us as well as an ongoing increase in our legal, audit and financial compliance costs. As a public company, we will become subject to Section 404 of the Sarbanes Oxley Act relating to internal control over financial reporting. We have only recently begun a formal process to evaluate our internal controls for purposes of Section 404, and we cannot assure that our internal control over financial reporting will prove to be effective.
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, stockholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of our common stock.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports and, together with adequate disclosure controls and procedures, are designed to prevent fraud. If we cannot provide reliable financial reports or prevent fraud, our operating results could be harmed. We have only recently begun a formal process to evaluate our internal control over financial reporting. Given the status of our efforts, coupled with the fact that guidance from regulatory authorities in the area of internal controls continues to evolve, substantial uncertainty exists regarding our ability to comply by applicable deadlines. Any failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our common stock.
Investment Risks
We expect that our stock price will fluctuate significantly, which may adversely affect holders of our stock and our ability to raise capital.
The stock market, particularly in recent years, has experienced significant volatility particularly with respect to pharmaceutical, biopharmaceutical and biotechnology stocks. The volatility of pharmaceutical, biopharmaceutical and biotechnology stocks often does not relate to the operating performance of the companies represented by the shares. Factors that could cause volatility in the market price of our common stock include:
| | |
| | • | the timing and the results from our clinical trial programs; |
| |
| | • | FDA or international regulatory actions; |
| |
| | • | failure of any of our product candidates, if approved, to achieve commercial success; |
| |
| | • | announcements of clinical trial results or new product introductions by our competitors; |
| |
| | • | market conditions in the pharmaceutical, biopharmaceutical and biotechnology sectors; |
32
| | |
| | • | developments concerning intellectual property rights; |
| |
| | • | litigation or public concern about the safety of our potential products; |
| |
| | • | actual and anticipated fluctuations in our quarterly operating results; |
| |
| | • | deviations in our operating results from the estimates of securities analysts; |
| |
| | • | additions or departures of key personnel; |
| |
| | • | third party reimbursement policies; and |
| |
| | • | developments concerning current or future strategic alliances. |
These and other factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock and our ability to raise capital.
If the price and volume of our common stock experience extreme fluctuations, then we could face costly litigation.
In the past, companies that experience volatility in the market price of their securities have often faced securities class action litigation. Whether or not meritorious, if any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management and harm our ability to grow our business.
Our directors and management will exercise significant control over our company.
Our directors and executive officers and their affiliates collectively control approximately 40.0% of our outstanding common stock. These stockholders, if they act together, may be able to influence our management and affairs and all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. The concentration of ownership may have the effect of delaying or preventing a change in control of our company and might affect the market price of our common stock.
We may not achieve our projected development goals in the time frames we announce and expect.
We set goals for and make public statements regarding timing of the accomplishment of objectives material to our success, such as the commencement and completion of clinical trials. The actual timing of these events can vary dramatically due to factors such as delays or failures in our clinical trials, the uncertainties inherent in the regulatory approval process and delays in achieving manufacturing or marketing arrangements sufficient to commercialize our products. There can be no assurance that our clinical trials will be completed, that we will make regulatory submissions or receive regulatory approvals as planned or that we will be able to adhere to our current schedule for the launch of any of our products. If we fail to achieve one or more of these milestones as planned, the market price of our shares could decline.
Provisions of Delaware law or our charter documents could delay or prevent an acquisition of our company, even if the acquisition would be beneficial to our stockholders, and could make it more difficult for you to change management.
Provisions of our certificate of incorporation and bylaws may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. This is because these provisions may prevent or frustrate attempts by stockholders to replace or remove our current management or members of our board of directors.
These provisions include:
| | |
| | • | a staggered board of directors; |
| |
| | • | a prohibition on stockholder action through written consent; |
33
| | |
| | • | a requirement that special meetings of stockholders be called only by the chairman of the board of directors, the chief executive officer, or the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors; |
| |
| | • | advance notice requirements for stockholder proposals and nominations; and |
| |
| | • | the authority of the board of directors to issue preferred stock with such terms as it may determine. |
As a result, these provisions and others available under Delaware law could limit the price that investors are willing to pay in the future for shares of our common stock.
Because we do not expect to pay dividends in the foreseeable future, you must rely on stock appreciation for any return on your investment.
We have paid no cash dividends on any of our capital stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, we do not expect to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Furthermore, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Accordingly, the success of your investment in our common stock will likely depend entirely upon any future appreciation. There is no guarantee that our common stock will appreciate in value after you purchase them or even maintain the price at which you purchased your shares, and you may not realize a return on your investment in our common stock.
| |
| Item 1B. | Unresolved Staff Comments |
Not applicable.
We currently lease approximately 34,800 square feet of research, manufacturing and administrative space in Gaithersburg, Maryland for our principal laboratories, pilot manufacturing facility and corporate offices. This lease expires in May 2013 and has a five-year renewal option. In February 2006, we entered into a one-year, full-service sublease for an additional 5,572 square feet of laboratory and office space in our current building. This sublease expires in February 2007.
| |
| Item 3. | Legal Proceedings |
We are not party to any material legal proceedings.
| |
| Item 4. | Submission of Matters to a Vote of Security Holders |
On December 1, 2005, our stockholders authorized by written consent the following:
| | |
| | • | amendment of our Second Amended and Restated Certificate of Incorporation to effect a1-for-13 reverse stock split of all issued and outstanding Common Stock; |
| |
| | • | election of M. James Barrett and Stanley C. Erck as Class I directors, to serve until the annual meeting of stockholders in 2006, election of Jeff Himawan and R. Gordon Douglas as Class II directors, to serve until the annual meeting of stockholders in 2007, election of James Young and Richard Douglas as Class III directors, to serve until the annual meeting of stockholders in 2008; |
| |
| | • | approval and adoption of our Third Amended and Restated Certificate of Incorporation, which became effective concurrently with the closing of the initial public offering of our Common Stock; |
| |
| | • | approval and adoption of our Second Amended and Restated By-laws, which became effective immediately upon approval by the stockholders; |
34
| | |
| | • | approval and adoption of our 2005 Incentive Plan, which became effective concurrently with the closing of the initial public offering of our Common Stock; and |
| |
| | • | approval and adoption of our 2006 Employee Stock Purchase Plan, which became effective concurrently with the closing of the initial public offering of our Common Stock. |
All such actions were effected pursuant to an action by written consent of our stockholders in compliance with Section 228 of the Delaware General Corporation Law. The written consent was adopted by holders of 114,444,230 shares of our capital stock out of 154,638,591 shares issued and outstanding as of the date above, including 112,717,650 shares out of 129,590,034 shares of our Series C preferred stock issued and outstanding.
Executive Officers of the Registrant
The following table sets forth the names and ages of our executive officers as of December 31, 2005.
| | | | | | | |
Name | | Age | | | Position(s) |
| |
| Stanley C. Erck | | | 57 | | | President, Chief Executive Officer, Treasurer and Director |
| Gregory M. Glenn, M.D. | | | 51 | | | Senior Vice President and Chief Scientific Officer |
| Russell P. Wilson | | | 46 | | | Senior Vice President, Chief Financial Officer, General Counsel and Secretary |
Stanley C. Erck. Mr. Erck has served as President, Chief Executive Officer, Treasurer and Director since May 2000. Mr. Erck has 30 years of management experience in healthcare and biotechnology. Mr. Erck has worked at Baxter International, Procept, and Integrated Genetics. Mr. Erck has a B.S. from the University of Illinois and an M.B.A. from the University of Chicago.
Gregory M. Glenn, M.D. Dr. Glenn has served as Senior Vice President and Chief Scientific Officer since September 1997 and was a Director from February 1998 through May 2000. Dr. Glenn is the co-discoverer of the TCI technology and a co-founder of Iomai. He has been responsible for the conception, implementation and development of the basic science and early clinical trials relating to TCI, and has multiple patents, publications and book chapters describing TCI. Dr. Glenn is a pediatrician who completed the Medical Research Fellowship at the Walter Reed Army Institute of Research, or WRAIR, where he continued his research in vaccine delivery while on active duty. He received a B.A. from Whitman College and his M.D. from Oral Roberts University School of Medicine, where he received the Pediatrics Award and Dean’s Award for Academic Excellence.
Russell P. Wilson. Mr. Wilson has served as Senior Vice President since May 2005, Chief Financial Officer since June 2002, General Counsel since March 2000 and Secretary since May 2000, and served as Vice President, Business Development from March 2000 to June 2002. Mr. Wilson received a B.A. from Princeton University and holds a joint M.B.A./J.D. degree from the University of Virginia.
We have adopted a code of ethics, included within the Iomai Corporation Code of Business Conduct and Ethics, that applies to all directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer and others performing similar functions. Copies of the Iomai Corporation Code of Business Conduct and Ethics are available, without charge, by contacting Iomai Investor Relations at(301) 556-4478.
PART II
| |
| Item 5. | Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Our common stock has been traded on The NASDAQ National Market under the symbol “IOMI” since February 1, 2006.
35
The following table sets forth, for the periods indicated, the high and low sale prices per share of our common stock as reported on the NASDAQ National Market.
| | | | | | | | | |
| | | High | | | Low | |
| |
| 2006 | | | | | | | | |
| First Quarter (through March 15, 2006) | | $ | 7.34 | | | $ | 5.22 | |
As of March 17, 2006, there were approximately 150 registered holders and approximately 892 beneficial owners of our common stock.
We have never declared or paid any cash dividends on our capital stock and we do not currently anticipate declaring or paying cash dividends on our capital stock in the foreseeable future. We currently intend to retain all of our future earnings, if any, to finance operations. Any future determination relating to our dividend policy will be made at the discretion of our board of directors and will depend on a number of factors, including future earnings, capital requirements, financial conditions, future prospects, contractual restrictions and covenants and other factors that our board of directors may deem relevant.
Securities Authorized For Issuance Under Equity Compensation Plans
The following table summarizes the number of securities issuable under our incentive compensation plans as of December 31, 2005.
| | | | | | | | | | | | | |
| | | | | | | | | Number of Securities
| |
| | | Number of Securities
| | | | | | Remaining Available for
| |
| | | to Be Issued
| | | Weighted-average
| | | Future Issuance Under
| |
| | | Upon Exercise of
| | | Exercise Price of
| | | Equity Compensation Plan
| |
| | | Outstanding Options,
| | | Outstanding Options,
| | | (Excluding Securities
| |
| | | Warrants and Rights(1)
| | | Warrants and Rights
| | | Reflected in Column(a))(2)
| |
Plan Category | | (a) | | | (b) | | | (c) | |
| |
| Equity compensation plans approved by security holders | | | 1,893,264 | | | $ | 1.64 | | | | 1,187,074 | |
| Equity compensation plans not approved by security holders | | | — | | | | | | | | — | |
| | | | | | | | | | | | | |
Total | | | 1,893,264 | | | | | | | | 1,187,074 | |
| | | | | | | | | | | | | |
| | |
| (1) | | Includes (i) outstanding options to purchase 50,380 shares of common stock under the 1998 Plan, of which 50,380 have been vested; and (ii) outstanding awards to purchase 1,842,884 shares of common stock under the 1999 Plan, of which 734,368 have been vested. Options granted under the 1999 Plan to purchase 38,169 shares of common stock were exercised prior to December 31, 2005. |
| | |
| (2) | | Includes (i) up to 26,543 shares of common stock that may be issued under the 1998 Plan; (ii) up to 120,531 shares of common stock that may be issued under the 1999 Plan; and (iii) up to 1,040,000 shares of common stock that may be issued under the 2005 Plan. Does not include up to 80,000 shares that may be issued under the 2006 Employee Stock Purchase Plan which became effective upon our initial public offering. |
Use of proceeds from registered securities
On February 6, 2006, we closed our initial public offering (IPO) of 5,000,000 shares of common stock pursuant to our Registration Statement onForm S-1/A (FileNo. 333-128765), which was declared effective by the Securities and Exchange Commission on February 1, 2006. The net proceeds from our IPO, before expenses, were $32,853,574. We estimate that our IPO final expenses will be approximately $2.0 million, so our total net proceeds after expenses will be approximately $30.8 million. We expect to use the net proceeds from our IPO to fund our clinical trials and for other general corporate purposes.
36
| |
| Item 6. | Selected Financial Data |
The following table sets forth selected financial data that is qualified in its entirety by and should be read in conjunction with Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our financial statements and notes thereto appearing elsewhere in this annual report onForm 10-K. The financial data for the fiscal years ended December 31, 2005, 2004 and 2003 are derived from our audited financial statements appearing elsewhere in this document. The financial data for the fiscal years ended December 31, 2002 and 2001 are derived from our audited financial statements not included in this document.
| | | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | |
Statement of Operations Data: | | 2001 | | | 2002 | | | 2003 | | | 2004 | | | 2005 | |
| | | (In thousands, except share and per share data) | |
| |
Revenues: | | | | | | | | | | | | | | | | | | | | |
| License, option agreements and grants | | $ | | | | $ | 995 | | | $ | 1,601 | | | $ | 2,345 | | | $ | 2,371 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total revenues | | | — | | | | 995 | | | | 1,601 | | | | 2,345 | | | | 2,371 | |
Cost and expenses: | | | | | | | | | | | | | | | | | | | | |
| Research and development | | | 6,463 | | | | 7,354 | | | | 13,332 | | | | 14,349 | | | | 16,529 | |
| General and administrative | | | 4,666 | | | | 2,411 | | | | 3,348 | | | | 3,206 | | | | 3,780 | |
| Purchased in-process research and development | | | — | | | | 2,517 | | | | — | | | | — | | | | — | |
| Reimbursed by related party | | | (5,736 | ) | | | (426 | ) | | | — | | | | — | | | | — | |
| Loss on investments in related entities | | | 4,905 | | | | — | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Total costs and expenses | | | 10,298 | | | | 11,856 | | | | 16,680 | | | | 17,555 | | | | 20,309 | |
| Loss from operations | | | (10,298 | ) | | | (10,861 | ) | | | (15,079 | ) | | | (15,210 | ) | | | (17,938 | ) |
Other (expense) income: | | | | | | | | | | | | | | | | | | | | |
| Gain on discharge of convertible notes payable | | | — | | | | 11,219 | | | | — | | | | — | | | | — | |
| Interest income | | | 66 | | | | 57 | | | | 953 | | | | 620 | | | | 392 | |
| Interest expense | | | (1,352 | ) | | | (2,120 | ) | | | (128 | ) | | | (265 | ) | | | (467 | ) |
| Other expense, net | | | — | | | | (75 | ) | | | (448 | ) | | | (225 | ) | | | (17 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Total other (expense) income, net | | | (1,286 | ) | | | 9,081 | | | | 377 | | | | 130 | | | | (92 | ) |
| Net loss | | | (11,584 | ) | | | (1,780 | ) | | | (14,702 | ) | | | (15,080 | ) | | | (18,030 | ) |
| Dividends on and accretion of convertible preferred stock | | | — | | | | (380 | ) | | | (5,466 | ) | | | (5,525 | ) | | | (5,562 | ) |
| Carrying value of preferred stock in excess of fair value transferred | | | — | | | | 11,092 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Net income (loss) available to common stockholders | | $ | (11,584 | ) | | $ | 8,932 | | | $ | (20,168 | ) | | $ | (20,605 | ) | | $ | (23,592 | ) |
| Net income (loss) per share of common stock — basic and diluted | | $ | (15.29 | ) | | $ | 11.79 | | | $ | (26.43 | ) | | $ | (26.90 | ) | | $ | (30.14 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Weighted average shares of common stock outstanding — basic and diluted | | | 757,402 | | | | 757,402 | | | | 763,075 | | | | 765,945 | | | | 782,715 | |
| | | | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | The pro forma basic and diluted net loss per share reflects the weighted effect of the assumed conversion of convertible preferred stock. See Note 2 to our financial statements for information regarding computation of basic and diluted net loss per share and pro forma basic and diluted net loss per share. |
37
| | | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, | |
Balance Sheet Data: | | 2001 | | | 2002 | | | 2003 | | | 2004 | | | 2005 | |
| | | (In thousands) | |
| |
| Cash, cash equivalents and marketable securities | | $ | 1,718 | | | $ | 49,493 | | | $ | 38,213 | | | $ | 22,397 | | | $ | 5,190 | |
| Working capital | | | 309 | | | | 47,263 | | | | 34,945 | | | | 18,878 | | | | 270 | |
| Total assets(1) | | | 4,991 | | | | 51,398 | | | | 41,511 | | | | 28,942 | | | | 11,861 | |
| Debt and capital lease obligations | | | 12,522 | | | | 706 | | | | 2,966 | | | | 5,273 | | | | 4,230 | |
| Redeemable convertible preferred stock and Series C warrant | | | 9,750 | | | | 52,435 | | | | 59,555 | | | | 65,357 | | | | 70,386 | |
| Common stock subject to put right | | | 1,958 | | | | 1,958 | | | | 1,958 | | | | 1,958 | | | | 1,958 | |
| Total stockholders’ deficit | | | (21,157 | ) | | | (6,533 | ) | | | (26,038 | ) | | | (45,993 | ) | | | (68,458 | ) |
| | |
| (1) | | As of December 31, 2005, we had net operating loss (NOL) and research and development credit carryforwards of approximately $56.6 million. Tax benefits may arise from these carryforwards in the future in the event that we realize U.S. taxable income. Potential tax benefits arising from these carryforwards are not reflected in our total assets. Despite the NOL carryforward, we may have an income tax liability in future years due to the application of the alternative minimum tax rules. The NOL may also be limited in its ability to offset future losses in the event that there is a change in the stock ownership as defined by federal tax regulations. |
| |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion of our financial condition and results of operations should be read in conjunction with our financial statements and the notes to those financial statements appearing elsewhere in this annual report onForm 10-K. This discussion contains forward-looking statements, which are identified under our “Cautionary Statement Regarding Forward-Looking Statements” at the beginning of this report, that involve significant risks and uncertainties. As a result of many factors, such as those set forth under “Risk factors” in Item 1A and elsewhere in this annual report onForm 10-K, our actual results may differ materially from those anticipated in these forward-looking statements.
OVERVIEW
We are a biopharmaceutical company focused on the discovery, development and commercialization of vaccines and immune system stimulants delivered to the skin via a novel, needle-free technology called transcutaneous immunization (TCI). TCI exploits the unique benefits of a major group of antigen-presenting cells found in the outer layers of the skin (Langerhans cells) to generate an enhanced immune response. TCI has the potential to enhance the efficacy of existing vaccines, enable new vaccines that are viable only through transcutaneous administration and expand the global vaccine market. We are developing two distinct product applications: (1) a needle-free vaccine patch and (2) an immunostimulant, or IS, patch. We are actively pursuing three product candidates in clinical development. Our three product candidates are a needle-free flu vaccine patch, an IS patch intended to boost the immune response of the elderly to the standard flu vaccine and a needle-free travelers’ diarrhea vaccine patch. We are also pursuing preclinical development of an IS patch to either reduce the required dose of, or improve the timing or magnitude of the immune response to, pandemic flu vaccines. None of our product candidates has been approved for commercial sale by the FDA or any comparable foreign agencies.
We commenced operations in May 1997 and our principal business activities since that time have included research and development, intellectual property prosecution, and conducting clinical trials. We have generated limited revenue from government grants and have had no revenue from product sales. As a result, we have incurred significant losses since our inception. As of December 31, 2005, we had an accumulated deficit of $68.6 million. We expect to incur substantial expenses over the next several years as we:
| | |
| | • | continue the clinical development of our needle-free flu vaccine patch and IS patch for elderly receiving flu vaccines; |
| |
| | • | expand the clinical trial program for our needle-free travelers’ diarrhea vaccine patch; |
38
| | |
| | • | continue our existing preclinical development programs, including our IS patch for pandemic flu vaccines, and commence new programs; |
| |
| | • | increase our manufacturing capabilities for product candidates; and |
| |
| | • | expand our research and development activities. |
We anticipate that a substantial portion of our efforts in 2006 will be focused on conducting additional development for our three product candidates in clinical development, as well as building our infrastructure to support our operations as a public company.
Financing history
Through December 31, 2005, we have financed the majority of our operations through the private placement of equity securities and debt financings, as well as through equipment and leasehold improvement financing. From inception through December 31, 2005, we raised approximately $87.3 million through the issuance of equity securities and convertible debt financings.
Since December 2002, we have funded our operations through a series of private placements of our Series C Redeemable Convertible Preferred Stock (Series C Preferred Stock) in which we raised $57.3 million. In December 2002, we raised gross proceeds of $54.3 million through the sale of shares of Series C Preferred Stock. In 2003, we issued additional shares of Series C Preferred Stock for gross proceeds of $2.1 million. In June 2004, we issued additional shares of Series C Preferred Stock for gross proceeds of approximately $842,000.
Prior to the initial sale of Series C Preferred Stock, we funded our operations principally through a collaboration with affiliates of Elan Corporation, plc, or Elan, which included a joint venture with Elan to develop our TCI technology. In connection with the closing on the Series C Preferred Stock in 2002, we restructured our collaboration with Elan so that all of Elan’s debt and equity interests in the joint venture and us were either paid down or converted into shares of Series B Preferred Stock. On January 5, 2006, Elan sold all of the shares of Series B Preferred Stock to certain holders of our Series C Preferred Stock pursuant to a contractual right of first refusal. Elan now has no other interest in us, except for an option for an exclusive license to our TCI technology in the area of Alzheimer’s disease vaccines and adjuvants, which expires in December 2009.
To maintain control over the manufacture and development of our products, we have constructed a pilot production plant in which we expect to manufacture the LT adjuvant and the finished patch formulations for our clinical trials, including our needle-free flu and travelers’ diarrhea vaccine patches and our IS patches. During 2003, we leased in stages approximately 10,000 additional square feet of contiguous space to accommodate the build-out of the pilot plant. The construction spanned from September 2003 to mid-2004, and while we have completed validation of the pilot plant’s major systems, we will have to validate our process manufacturing systems during 2006 and 2007 to support our clinical development timelines. We financed the build-out of the pilot plant with a combination of landlord financing of tenant improvements and asset-backed financing from a finance company that specializes in equipment leasing to biotechnology companies. As of December 31, 2005, we had drawn down in total approximately $4.8 million of our $5 million line of credit with the finance company to fund this project and $1.45 million of the tenant improvement allowance made available to us by our landlord. The landlord participated in the Series C financing and, therefore, is a related party. Accordingly, the landlord’s financing of our leasehold improvements has been recorded on the balance sheet as notes payable from related party.
In October 2005, we amended our lease to, among other things, expand our administrative space in our facility by approximately 8,000 additional square feet and to extend our lease term by seven years through May 2013. In connection with the lease amendment, the landlord provided us with additional tenant improvement allowances totaling approximately $134,000 for our existing space and approximately $120,000 for the expansion space. The landlord has also committed to provide up to $280,000 of additional financing to apply toward alterations to the expansion space on the same terms and conditions as our existing landlord financing. At this time, we expect to utilize the tenant improvement allowances in full and to draw down the entire $280,000 in additional landlord financing.
39
Prior to 2005, third parties supplied all of the LT adjuvant and the formulated patches that we used in our clinical trials. Berna Biotech AG was our primary supplier of LT adjuvant, and we have relied on two contract manufacturers to produce our formulated patches, including Elan during our collaboration. At this time, we have no further agreements with these firms to provide us with any further components or services. Over the past few years, in parallel with our relationship with Berna Biotech, we also engaged DowPharma, a business unit within The Dow Chemical Company, to assist us with the development of our own LT strain and associated manufacturing process. In June 2005, we licensed from DowPharma a proprietary expression system for our LT strain. As part of the technology transfer under the license, DowPharma provided us with technological assistance, as well as the master and working cell banks of the expression system that we use to produce LT in our pilot plant. Our payment obligations under the license do not go into effect until our first product receives regulatory approval. As a result, we have not made any payments to DowPharma under the license. As of December 31, 2005, we had paid DowPharma $124,165 for services relating to technology transfer, and DowPharma has agreed to defer payment of the remaining $45,700 due until we receive regulatory approval of our first product that utilizes DowPharma’s expression system.
As of December 31, 2005, we had approximately $5.2 million in cash, cash equivalents and marketable securities. On February 6, 2006, we closed our initial public offering (IPO), in which we raised approximately $30.8 million in net proceeds. We believe that our current working capital and reimbursement of expenses under our existing government grants will be sufficient to fund our operating expenses and capital requirements through early 2007. However, we expect to attempt to raise additional funds through the issuance of additional equity or the incurrence of debt in advance of depleting our current funds. The timing of future capital raises will depend on our product development progress, corporate partnering activity, and capital market conditions. Satisfying our long-term liquidity needs will require the successful commercialization of one or more of our product candidates and will require substantial additional capital.
FINANCIAL OPERATIONS REVIEW
Revenues
Our revenues to date have principally been limited to amounts we have received under U.S. federal grant programs. From January 1, 2003 to December 31, 2005, we have received $6.3 million in reimbursement under various grants. In January 2005, the National Institutes of Health, or NIH, awarded us a two-year, $2.9 million grant for further development of our IS technology for pandemic flu applications. In the first quarter of 2006, the NIH awarded us the $1.4 million remaining to be received under this grant for 2006. This amount includes potential reimbursement for our employees’ time and benefits and other expenses related to performance under the various contracts. Funding of government grants beyond the U.S. government’s current fiscal year are subject to annual congressional appropriations.
Research and development expenses
Our research and development expenses consist primarily of:
| | |
| | • | salaries and related expenses for personnel; |
| |
| | • | fees paid to consultants and clinical research organizations in conjunction with their monitoring our clinical trials and acquiring and evaluating data in conjunction with our clinical trials; |
| |
| | • | consulting fees paid to third parties in connection with other aspects of our product development efforts; |
| |
| | • | fees paid to research organizations in conjunction with preclinical animal studies; |
| |
| | • | costs of materials used in research and development; |
| |
| | • | depreciation of facilities and equipment used to develop our product candidates; and |
| |
| | • | milestone payments, license fees, and royalty payments for technology licenses. |
40
We expense both internal and external research and development costs as incurred, other than those capital expenditures that have alternative future uses, such as the build-out of our pilot plant. Due to the risks inherent in the clinical trial process and the early stage of development of our product candidates, we do not currently track our internal research and development costs by program and cannot state precisely the costs incurred for each of our research and development programs. However, the following table shows our estimate of the total costs that have been incurred for our lead product candidates for the period from January 1, 2003 to December 31, 2005:
| | | | | |
| | | Estimated Total Costs Incurred
| |
| | | From January 1, 2003
| |
| | | to December 31,
| |
Product Candidate | | 2005 | |
| | | (In thousands) | |
| |
| Needle-free flu vaccine patch | | $ | 7,204 | |
| IS patch for elderly receiving flu vaccines | | | 14,554 | |
| Needle-free travelers’ diarrhea vaccine patch | | | 10,201 | |
| Other programs, including pandemic flu | | | 12,251 | |
| | | | | |
| TOTAL: | | $ | 44,210 | |
| | | | | |
We expect our research and development costs will continue to be substantial and that they will increase as we advance our current portfolio of product candidates through clinical trials and move other product candidates into preclinical and clinical trials.
General and administrative expense
General and administrative expense consists primarily of compensation for employees in executive and operational functions, including finance and accounting, business development, and corporate development. Other significant costs include facilities costs and professional fees for accounting and legal services. With the completion of our IPO, we anticipate our general and administrative expenses will increase due to increased costs for insurance, professional fees, public company reporting requirements, Sarbanes-Oxley compliance, and investor relations costs associated with operating as a publicly-traded company. These increases will also likely include the hiring of additional personnel.
RESULTS OF OPERATIONS
Comparison of the years ended December 31, 2005 and 2004
Revenues. We recognized revenues from government grants of approximately $2.4 million during 2005, as compared with $2.3 million during 2004. The increase in grant revenues is principally the result of revenues from the NIH grant awarded in January 2005 for further development of our IS patch technology for pandemic flu applications, which were offset by a decline in reimbursable activities in 2005 under other grants for our needle-free patch programs.
Research and Development Expenses. Our research and development expenses were approximately $16.5 million during 2005, as compared to $14.3 million during 2004. The $2.2 million increase in research expenditures was driven by three major factors associated with supporting our clinical and product development programs: (1) increased preclinical study costs as a result of higher charges for services and continuing development of preclinical models; (2) increased depreciation and amortization costs associated with our pilot manufacturing facility, which was put into service in July 2004; and (3) higher payroll costs associated with a 25% increase in headcount and expensing of stock options. These increased costs were partially offset by lower clinical trial costs and contract manufacturing expenses, both of which were higher during 2004 as we initiated five clinical trials during 2004.
General and Administrative Expenses. General and administrative expenses were approximately $3.8 million during 2005, as compared to $3.2 million during 2004. The increase in general and administrative expenses was principally due to consulting expenses associated with evaluating potential corporate collaborations, higher legal
41
costs associated with prosecuting our patent portfolio, and higher payroll costs associated with expensing stock options.
Interest Income (Expense) and Other — Net. Our net interest and other expense was approximately $91,000 for 2005, as compared to net interest and other income of approximately $130,000 for 2004. The net interest and other income reflects the interest received on our cash and marketable securities, offset by interest expense on financing to purchase equipment and leasehold improvements. The decrease in net interest and other income was a result of lower interest income because of declining cash balances and higher interest expense associated funds borrowed to finance the construction of our pilot manufacturing facility.
Net Loss. Our net loss for 2005 was approximately $18.0 million, and our net loss for 2004 was approximately $15.0 million. The increase in our net loss was principally a result of increased research and development expenses in 2005.
Comparison of the years ended December 31, 2004 and 2003
Revenues. We recognized revenues from government grants of approximately $2.3 million during 2004, as compared with $1.6 million during 2003. The principal source of this increase in grant revenues for 2004 was the award of the two grants by the NIH in the first half of 2004 for our needle-free anthrax vaccine patch and needle-free flu vaccine patch programs.
Research and Development Expenses. Our research and development expenses were approximately $14.3 million during 2004, as compared to $13.3 million during 2003. The $1.0 million increase in research expenditures was primarily attributable to the following factors associated with our clinical and product development programs: (1) payroll costs increased by $800,000 as we hired additional employees in 2004 to support our clinical and product development efforts; (2) facilities costs increased by $200,000 in 2004 because we leased in stages during 2003 approximately 10,000 square feet of additional space in our building to accommodate the build-out for our pilot plant; and (3) depreciation and amortization expense increased in 2004 by $1.2 million as a result of bringing our pilot manufacturing facility into service in July 2004. These increased costs were partially offset in 2004 by lower expenses for regulatory and consulting services and a decrease in contract manufacturing costs of approximately $1.2 million. During 2003, our contract manufacturing expenses were higher due to costs associated with the purchase LT adjuvant from Berna Biotech and formulating and manufacturing patches to support clinical trials.
General and Administrative Expense. General and administrative expenses were approximately $3.2 million during 2004 and $3.3 million during 2003. The $0.1 million decrease in these expenses was principally a result of higher outside consulting, legal and patent expenses in 2003, and a one-time write-off of $135,000 of bad debt expense in 2003 for amounts deemed uncollectible. These higher costs in 2003 were offset partially by higher payroll costs in 2004.
Interest Income (Expense) and Other — Net. Our net interest and other income was approximately $130,000 during 2004, as compared to net interest and other income of approximately $377,000 during 2003. Our net interest and other income reflects the interest received on our cash and marketable securities, offset by interest expense on financing to purchase equipment and leasehold improvements. The decrease in net interest and other income was principally a result of lower interest income because of declining cash balances and higher interest expense associated with drawing down funds to finance the construction of our pilot manufacturing facility.
Net Loss. Our net loss for 2004 was approximately $15.1 million, and our net loss for 2003 was approximately $14.7 million. The increase in our net loss in 2004 was principally a result of increased research and development expenses, partially offset by higher grant revenues.
42
RESEARCH AND DEVELOPMENT PROGRAMS
Due to the risks inherent in the clinical trial process and the early stage of development of our product candidates, we do not currently track the internal research and development costs for each of our research and development programs. We use our research and development resources, including employees and our TCI technology, across multiple product development programs. As a result, we cannot state precisely the costs incurred for each of our research and development programs or our clinical and preclinical product candidates. However, the table below presents our estimate of our total research and development costs allocable to our leading research and development programs for the periods indicated. We have allocated direct and indirect costs to each program based on certain assumptions and our review of the status of each program, payroll related expenses, and other overhead costs based on estimated usage by each program.
| | | | | | | | | | | | | |
| | | Years Ended December 31, | |
Research and Development Program | | 2003 | | | 2004 | | | 2005 | |
| | | (In thousands) | |
| |
| Needle-free flu vaccine patch | | $ | 194 | | | $ | 2,116 | | | $ | 4,895 | |
| IS patch for elderly receiving flu vaccines | | | 8,576 | | | | 4,434 | | | | 1,545 | |
| Needle-free travelers’ diarrhea vaccine patch | | | 1,670 | | | | 2,890 | | | | 5,641 | |
| Other programs | | | | | | | | | | | | |
| Pandemic flu | | | — | | | | 4 | | | | 2,409 | |
| Other research, excluding pandemic flu | | | 2,892 | | | | 4,905 | | | | 2,039 | |
| | | | | | | | | | | | | |
| Subtotal other programs | | | 2,892 | | | | 4,909 | | | | 4,448 | |
| Total Research and Development Expenses | | $ | 13,332 | | | $ | 14,349 | | | $ | 16,529 | |
| | | | | | | | | | | | | |
Our lead programs are in various stages of completion as described below. Significant additional expenditures will be required as we complete clinical trials, start new trials, apply for regulatory approvals, continue development of our technologies, expand our operations, and attempt to bring product candidates to market. The eventual total cost of each clinical trial is dependent on a number of uncertain variables such as trial design, the length of the trial, the number of clinical sites, and the number of subjects. The process of obtaining and maintaining regulatory approvals for new therapeutic products is lengthy, expensive, and uncertain. We anticipate that we will determine which of our early stage product candidates is best suited for further development, as well as how much funding to direct to each program, on an on-going basis in response to the scientific and clinical results and the commercial potential of each product candidate. Because of these and other uncertainties, we cannot reliably estimate completion dates, completion costs, and capital requirements for our lead programs, and, therefore, we cannot reliably estimate when we might receive material net cash inflows from our research and development projects.
Needle-Free Flu Vaccine Patch. Our needle-free flu vaccine patch combines flu antigens and an adjuvant. We believe this approach offers the potential for effective flu vaccination without the use of needles.
In 2004, we completed a Phase 1 study of our needle-free flu vaccine patch in Europe. The data from this Phase 1 trial demonstrated that the needle-free flu vaccine patch stimulated an immune response to antigens from all three flu strains recommended by the World Health Organization for the 2004 flu season and demonstrated a positive effect from using our adjuvant, although the observed immune response was not as great as that of existing injectable flu vaccines. We have been working to further optimize our formulation and product design in an effort to achieve an immune response equal to or greater than that of existing injectable vaccines, which is one of the requirements necessary in order to obtain regulatory approval for this product candidate. Based on recently-completed pre-clinical studies, we believe we have reformulated this product candidate in a manner that will result in a greater immune response than that observed in the 2004 Phase 1 study. We are currently conducting toxicology studies on this new formulation. Assuming the results of the toxicology studies are favorable, we plan to commence a Phase 1/2 clinical trial in the United States in the third quarter of 2006. We plan to conduct additional Phase 2 trials after completing the Phase 1/2 trial.
We expect to spend approximately $5 million on this program through the completion of the Phase 1/2 trial. We currently rely on third-party suppliers to provide the flu antigens contained in our needle-free flu vaccine patch.
43
IS Patch for elderly receiving flu vaccines. We are developing an IS patch to improve the immune response of the elderly to existing injectable flu vaccines.
We are planning a confirmatory Phase 2 study in 2006 designed to demonstrate that our IS patch may represent an effective strategy for improving response rates to influenza vaccines in the elderly. Following this trial, we expect to explore dose ranging andtime-of-patch-wear studies leading to Phase 3 pivotal trials beginning in 2007. At this time, we expect to incur third-party costs to conduct the remaining Phase 2 trials, including costs for clinical trial enrollment, conduct, and monitoring and statistical analysis, of between $2 and $4 million. These clinical costs could vary significantly based on the results from each of these follow-on trials, which could impact development timelines for future trials, as well as their design in terms of number of subjects, dosing, and endpoints. As a result, we cannot be certain when and under what conditions we will undertake future clinical trials. Upon completion of our Phase 2 studies, we will request discussions with regulatory agencies in the United States and Europe prior to initiating any Phase 3 studies in order to finalize endpoints and design for our Phase 3 studies. The costs required to complete development of our IS patch will be largely dependent on the results of our Phase 2 trials and our discussions with the regulatory authorities, and these costs cannot be precisely estimated at this time.
Needle-free travelers’ diarrhea vaccine patch. Our needle-free travelers’ diarrhea vaccine patch is targeting infection by LT-secreting ETEC, the leading cause of travelers’ diarrhea. Our vaccine candidate is designed to reduce the risk of contracting travelers’ diarrhea or reduce the severity of diarrhea in the event of infection. We have conducted several Phase 1 and 2 ETEC trials. During the first quarter of 2005, we completed a clinical study in which volunteers were infected with live ETEC organisms. In November 2005, we commenced a Phase 1/2 trial which compares our current dry patch formulation to a wet patch, and interim data indicates that our current dry patch formulation provided equal or greater immune responses to vaccination than the wet patch.
We are currently planning a series of additional Phase 2 trials designed to determine the final dry patch formulation, number and timing of doses, and duration of patch wear. At this time, we expect to incur third-party costs to conduct these follow-on Phase 2, including costs for clinical trial enrollment, conduct, and monitoring and statistical analysis, trials of between $8 and $10 million. These clinical costs could vary significantly based on our results from each of these follow-on trials, which could impact development timelines for future trials, as well as their design in terms of number of subjects, dosing, and endpoints. As a result, we cannot be certain when and under what conditions we will undertake future clinical trials. Upon completion of our Phase 2 studies, we intend to request discussions with regulatory agencies in the United States and Europe prior to initiating any Phase 3 studies in order to finalize endpoints and design for our Phase 3 studies. The costs required to complete development of our needle-free travelers’ diarrhea vaccine patch will be largely dependent on the results of our Phase 2 trials and our discussions with the regulatory authorities, and these costs cannot be precisely estimated at this time.
We intend to manufacture the patches for the clinical trials for our product candidates in-house in our pilot plant. Our programs are dependent, in large part, on the cost and efficiency of our manufacturing process, as well as our ability to deliver clinical materials in a timely fashion. We estimate that our expenses for the needle-free travelers’ diarrhea vaccine patch and the IS patch for elderly receiving flu vaccines programs, other than third-party clinical trial expenses, through the Phase 2 trials and prior to initiation of Phase 3 trials will be approximately $22 to $26 million. As our needle-free travelers’ diarrhea vaccine patch and IS patch both contain the same active ingredient and will likely have similar patch configurations, many of the development activities for these two programs, other than clinical trial expenses, are shared.
Liquidity and capital resources
We have incurred annual operating losses since inception, and, as of December 31, 2005, we had an accumulated deficit of $68.6 million. We expect to incur increasing and significant losses over the next several years as we continue our clinical trials, apply for regulatory approvals, continue development of our technologies, and expand our operations. Since our inception, we have financed our operations primarily through the sale of equity securities, interest income earned on cash, cash equivalents, and short-term investment balances, and debt. We have also generated funds from collaborative partners.
As of December 31, 2005, we had approximately $5.2 million in cash, cash equivalents, and marketable securities. On February 1, 2006, we completed our IPO in which we raised approximately $30.9 million in net
44
proceeds. We have used cash primarily to finance our research operations, including clinical trials. We invest in cash equivalents and US government and agency obligations. Our investment objectives are, primarily, to assure liquidity and preservation of capital and, secondarily, to obtain investment income. All of our marketable securities are classified asavailable-for-sale. These securities are carried at fair value, plus any accrued interest.
We expect that we will be able to fund our capital expenditures and growing operations with our current working capital, including the net proceeds from our IPO, through early 2007. In order to fund our needs subsequently, we will need to raise additional money and may seek to do so by: (1) out-licensing technologies or product candidates to one or more corporate partners, (2) completing an outright sale of assets, (3) securing debt financing,and/or (4) selling additional equity securities. Our ability to successfully enter into any such arrangements is uncertain, and, if funds are not available, or not available on terms acceptable to us, we may be required to revise our planned clinical trials, other development activities, capital expenditure requirements, and the scale of our operations. We expect to attempt to raise additional funds in advance of depleting our existing cash balances; however, we may not be able to raise funds or raise amounts sufficient to meet the long-term needs of the business. Satisfying long-term needs will require the successful commercialization of our product candidates and, at this time, we cannot reliably estimate if or when that will occur.
Our future cash requirements include, but are not limited to, supporting our clinical trial efforts and continuing our other research and development programs. We have entered into various agreements with institutions and clinical research organizations to conduct and monitor our current clinical studies. Under these agreements, subject to the enrollment of patients and performance by the applicable institution of certain services, we have estimated our potential payments to be $4.2 million over the term of currently ongoing studies. Through December 31, 2005, approximately $2.5 million of this amount has been expensed as research and development expenses and $2.3 million has been paid related to these clinical studies. The timing of our expense recognition and future payments related to these agreements are subject to the enrollment of patients and performance by the applicable institutions of certain services. The actual amounts we pay out, if any, will depend on a range of factors outside of our control, including the success of our pre-clinical and clinical development efforts with respect to any products being developed, the content and timing of decisions made by the FDA and other regulatory authorities, and other factors affecting future operating results. As we expand our clinical studies, we plan to enter into additional agreements.
Net cash used in operating activities for 2005 and 2004 was $14.3 million and $13.5 million, respectively. There was only a $0.8 million change in net cash used in operations principally because the $2.9 million increase in the 2005 net loss was offset by (1) a $1.0 million increase in the depreciation and amortization expense as the pilot plant was placed into service as of July 2004, (2) a $0.4 million increase in stock option expense, and (3) the $1.3 millionyear-to-year increase in accounts payable and accrued expenses. As we develop our technologies and further our clinical trial programs, we expect to increase our spending. Our future ability to generate cash from operations will depend on achieving regulatory approval of our products, market acceptance of such products, and our ability to enter into collaborations.
Net cash provided by investing activities for 2005 was $14.1 million as compared to $8.9 million for 2004. The increase is principally the result of using investments as they matured to finance our operations. During 2005, we invested $6.8 million of our available cash in marketable securities and received proceeds from the maturity of such investments of $22.1 million. Additionally, for the year ended December 31, 2004, we invested $5.1 million in the purchase of equipment, furniture, fixtures, and validation services primarily for the build-out of our pilot manufacturing facility, as compared to $1.3 million of such purchases in 2005.
Net cash used in financing activities was $1.5 million for 2005 as compared to net cash provided by financing activities of $3.1 million for 2004. During 2005 and 2004, net proceeds from sales of equity and exercises of stock options totaled approximately $21,000 and $0.8 million, respectively. During 2005 and 2004, net proceeds from debt borrowings totaled $0.5 and $3.4 million, respectively, as we financed the build-out of our pilot manufacturing facility. During 2005, we repaid $1.5 million of our debt, as compared to $1.0 million during 2004.
45
The following summarizes our long-term contractual obligations as of December 31, 2005:
| | | | | | | | | | | | | | | | | | | | | |
| | | Payments Due by Period | |
| | | | | | Less Than
| | | | | | | | | More Than
| |
Contractual Obligations | | Total | | | 1 Year | | | 1-3 Years | | | 3-5 Years | | | 5 Years | |
| | | (In thousands) | |
| |
| Long-Term Debt(1) (2) | | $ | 5,175 | | | $ | 1920 | | | $ | 1,979 | | | $ | 619 | | | $ | 657 | |
| Capital Lease Obligations | | | 6 | | | | 5 | | | | 1 | | | | — | | | | — | |
| Operating Lease Obligations(2) | | | 4,682 | | | | 571 | | | | 1,188 | | | | 1,270 | | | | 1,653 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total | | $ | 9,863 | | | $ | 2,496 | | | $ | 3,168 | | | $ | 1,889 | | | $ | 2,310 | |
| | | | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | Includes interest payable in the period. |
| |
| (2) | | In October 2005, we amended our lease agreement to increase our rented research, manufacturing, and administrative space in Gaithersburg, Maryland to approximately 34,800 square feet, with the lease term extending until May 2013. We also extended our payment terms for the indebtedness we have with our landlord. Our obligations under the amended lease and these revised debt repayment terms are reflected in the table as long term debt and operating lease obligations. See discussion of amended lease terms below. |
Under our existing license agreements, we could be required to pay up to a total of $800,000 for each product candidate in milestone payments through product approval, in addition to sales milestones, and royalties on commercial sales, if any occur.
In October 2005, we amended our lease to extend its term by seven years from June 2006 to May 2013. In connection with the lease extension, we renegotiated with the landlord the terms of the financing for the build-out of our pilot plant, which is recorded as notes payable to related party on our balance sheet, and we agreed to lease approximately 8,000 square feet of additional space in the building. Effective as of June 1, 2006, the then-remaining balance owed to the landlord under this debt financing, which we expect will total $1,342,719, will be amortized over the seven-year extension period at a rate of 10.5%. This change will result in a monthly payment of approximately $22,639 through May 2013, as opposed to the current monthly payment of approximately $44,689, which was being amortized through May 2009. In addition, we now have the right to prepay this debt obligation at any time upon four months’ written notice to the landlord.
Also, effective June 1, 2006, our annual base rent for our existing space will be reduced from approximately $500,000 to approximately $455,000. With respect to the expansion space of approximately 8,000 square feet, we will pay approximately $27,000 during the first 5 months of 2006 and then beginning June 1, 2006, the annual base rent will adjust to approximately $116,000. All base rents will be subject to minimum rent escalation of 3.5% per year.
As part of the amended lease, the landlord has provided us with tenant improvement allowances totaling approximately $134,000 for the existing space and approximately $120,000 for the expansion space. The landlord has also provided us with up to $280,000 of additional financing to apply toward alterations to the expansion space on the same terms and conditions as our existing landlord financing. At this time, we expect to make alterations to the expansion space during the first quarter of 2006 totaling approximately $400,000, and we expect to finance the majority of these alterations by utilizing in full the approximately $120,000 in tenant improvement allowance and by drawing down the full $280,000 in additional landlord financing.
In addition to the alterations to the expansion space, we currently expect to spend in 2006 approximately $2.0 million in additional capital expenditures, primarily for additional equipment for pilot plant and related validation costs, which we may fund in whole or in part through equipment financing.
Our future capital uses and requirements depend on numerous forward-looking factors. These factors include but are not limited to the following:
| | |
| | • | the progress and costs of preclinical development and laboratory testing and clinical trials; |
| |
| | • | the time and costs involved in obtaining regulatory approvals; |
46
| | |
| | • | delays that may be caused by evolving requirements of regulatory agencies; |
| |
| | • | our ability to establish, enforce, and maintain collaborations required for product commercialization; |
| |
| | • | the number of product candidates we pursue; |
| |
| | • | the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; |
| |
| | • | our plans to establish sales, marketing,and/or manufacturing capabilities; |
| |
| | • | the acquisition of technologies, products, and other business opportunities that require financial commitments; and |
| |
| | • | our revenues, if any, from successful development and commercialization of our products. |
As of December 31, 2003, 2004 and 2005, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to any financing, liquidity, market, or credit risk that could arise if we had engaged in these relationships.
Critical accounting policies and estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our financial statements included in this annual report, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
Revenue Recognition. We recognize revenue when all terms and conditions of the agreements have been met, including persuasive evidence of an arrangement, services have been rendered, price is fixed or determinable, and collectability is reasonably assured. For reimbursable cost research grants, we recognize revenue as costs are incurred. Provisions for estimated losses on research grant projects and any other contracts are made in the period such losses are determined.
Research and Development Costs. We expense our research and development costs as incurred.
Stock-Based Compensation. We have four stock-based employee compensation plans, described more fully in Note 7 to the Financial Statements, and we record compensation expense based upon the fair value of stock-based awards. Effective January 1, 2003, we adopted the fair value recognition provisions of SFAS No. 123,Accounting for Stock-Based Compensation(SFAS 123). Under the prospective method of adoption selected by us under the provisions of FASB Statement No. 148,Accounting for Stock-Based Compensation — Transition and Disclosure, an amendment of FASB Statement No. 123(SFAS 148), the recognition provisions have been applied to all employee awards granted, modified, or settled after January 1, 2003.
Equity instruments issued to nonemployees are accounted for under the provisions of SFAS 123 and Emerging Issues Task Force IssueNo. 96-18,Accounting for Equity Instruments That are Issued to Other Than Employees for Acquiring, or in conjunction with, Selling, Goods and Services. Accordingly, the estimated fair value of the equity instrument is recorded on the earlier of the performance commitment date or the date the services required are completed.
Prior to our IPO, our board of directors determined the fair value of our common stock for options to acquire shares of our common stock given the lack of an active public market for our common stock. Our board of directors
47
made contemporaneous determinations of fair value and did not employ a third party valuation firm to determine fair value.
Prior to our IPO in the absence of a public trading market, and as a clinical-stage company with no significant revenues, the board of directors believed that it was appropriate to consider a range of factors to determine the fair value of the common stock at each option grant date. These factors included: (1) the achievement of clinical and operational milestones, (2) the status of strategic relationships with collaborators, (3) the significant risks associated with our early stage of development of a new technology, (4) capital market conditions for life science companies, particularly similarly situated privately-held, early-stage life science companies, (5) our available cash, financial condition, and results of operations, (6) the most recent sales of the our preferred stock and (7) the preferential rights of the outstanding preferred stock.
In connection with the preparation of the financial statements for the IPO, we reassessed our estimate of the fair value for financial reporting purposes of our common stock following its stock option grants in March 2004. This valuation was done retrospectively by management, a related party, and we did not obtain contemporaneous valuations from an independent valuation specialist. Based on this reassessment, we determined that there were five periods between March 2004 and October 2005 in which our reassessment of the fair value of our common stock ranged from $2.75 to $9.00 per share during the period options were granted.
As a result of this reassessment of the fair value of our common stock for financial reporting purposes, we anticipate recognizing approximately $1.8 million of compensation expense over future periods as options outstanding as of December 31, 2005 vest in accordance with their terms, less any amounts for options forfeited or cancelled.
Based on the initial public offering price of $7.00 per share, the intrinsic value of options outstanding at December 31, 2005 was $10.1 million, of which $3.6 million related to vested options and $6.5 million related to unvested options.
Recent accounting pronouncements
On December 16, 2004, the Financial Accounting Standards Board (FASB) issued FASB Statement No. 123 (revised 2004),Share-Based Payment, which is a revision of FASB Statement No. 123,Accounting for Stock-Based Compensation. Statement 123(R) supersedes APB Opinion No. 25,Accounting for Stock Issued to Employees, and amends FASB Statement No. 95,Statement of Cash Flows. Generally, the approach in Statement 123(R) is similar to the approach described in Statement 123. However, Statement 123(R) requires all share-based payments to employees, including grants of employee stock options, to be recognized in the income statement based on their fair values. Pro forma disclosure is no longer an alternative.
Statement 123(R) originally required adoption no later than July 1, 2005. In April 2005, the Securities and Exchange Commission (“SEC”) issued a release that amends the compliance dates for Statement 123(R). Under the SEC’s new rule, the Company will be required to apply Statement 123(R) as of January 1, 2006.
The Company adopted thefair-value-based method of accounting for share-based payments effective January 1, 2003 using the prospective method described in FASB Statement No. 148,Accounting for Stock-Based Compensation — Transition and Disclosure. Currently, the Company uses the Black-Scholes-Merton formula to estimate the value of stock options granted to employees and expects to continue to use this option valuation model upon the required adoption of Statement 123(R) on January 1, 2006. Statement 123(R) also requires the benefits of tax deductions in excess of recognized compensation cost to be reported as a financing cash flow rather than as an operating cash flow as required under current literature. This requirement will reduce net operating cash flows and increase net financing cash flows in periods after adoption; however, the Company cannot estimate what those amounts will be in the future (because they depend on, among other things, when employees exercise stock options). Since the Company has accounted for stock options in accordance with Statement No. 123 since January 2003, management believes that the adoption of Statement 123(R) will not have a material impact on its financial statements going forward.
48
| |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because the majority of our investments are in short-term US government and agency debt securities. The market value of these investments fluctuates with changes in current market interest rates. In general, as rates increase, the market value of a debt investment would be expected to decrease. Likewise, as rates decrease, the market value of a debt investment would be expected to increase. To minimize such market risk, we generally hold these instruments to maturity at which time they are redeemed at their stated or face value. Due to the nature of our short-term investments, we believe that we are not subject to any material market risk exposure.
The interest rates on our debt obligations are fixed so repayment of these obligations is not subject to any material market risk exposure.
We do not have any foreign currency or other derivative financial instruments.
| |
| Item 8. | Financial Statements and Supplementary Data |
Financial Statements and Supplementary Data are submitted as a separate section of this report commencing onPage F-1.
| |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
Not Applicable.
| |
| Item 9A. | Controls and Procedures |
Disclosure Controls and Procedures
We currently have in place systems relating to disclosure controls and procedures (as defined inRules 13a-15(e) and15d-15(e) of the Securities Exchange Act of 1934). Our principal executive officer and our principal financial officer evaluated the effectiveness of these disclosure controls and procedures as of the end of our 2005 fiscal year in connection with the preparation of this annual report. They concluded that the controls and procedures are effective and adequate at that time.
Changes in Internal Controls Over Financial Reporting
There have been no significant changes in our internal control over financial reporting during the quarter ended December 31, 2005, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| |
| Item 9B. | Other Information |
None.
PART III
The information required by Item 10 — Directors and Executive Officers of the Registrant; Item 11 — Executive Compensation; Item 12 — Security Ownership of Certain Beneficial Owners and Management; Item 13 — Certain Relationships and Related Transactions; and Item 14 — Principal Accounting Fees and Services is incorporated into Part III of this Annual Report onForm 10-K by reference to our Proxy Statement for the Annual Meeting of Stockholders scheduled to be held on May 16, 2006, except that the information required by Item 10 pertaining to our executive officers is contained in Part I of this report.
49
PART IV
| |
| Item 15. | Exhibits, Financial Statement Schedules |
(a) 1.Consolidated Financial Statements
The consolidated financial statements are listed under Item 8 of this report.
2. Consolidated Financial Statement Schedules
The consolidated financial statement schedules required under this Item and Item 8 are omitted because they are not applicable or the required information is shown in the consolidated financial statements or the footnotes thereto.
3. Exhibits
The exhibits are listed below under Part IV Item 15(b).
(b) Exhibits
Exhibit Index
| | | | | | | | | |
Exhibit No. | | Description | | Location |
| |
| | 3 | .1.3 | | Third Amended and Restated Certificate of Incorporation | | | (1) | |
| | 3 | .2.3 | | Third Amended and Restated By-laws | | | (2) | |
| | 4 | .1 | | Specimen Stock Certificate | | | (1) | |
| | 4 | .2 | | Grant of Put Option dated April 6, 2001 by Iomai Corporation to the Walter Reed Army Institute of Research as representative of the United States of America. | | | (1) | |
| | 4 | .3 | | Option Agreement dated December 4, 2002 by and between Iomai Corporation and Elan Corporation, plc. | | | (1) | |
| | 4 | .4 | | Stock Purchase Warrant dated December 4, 2002 issued by Iomai Corporation to Friedman Billings Ramsey & Co., Inc. | | | (1) | |
| | 4 | .5.1 | | Investor Rights Agreement dated December 4, 2002 between Iomai Corporation and the individuals specified in Exhibit A thereto. | | | (1) | |
| | 4 | .5.2 | | Amendment to the Investor Rights Agreement dated March 27, 2003 among Iomai Corporation and the Purchasers listed on the signature pages thereto. | | | (1) | |
| | 4 | .5.3 | | Amendment to Investor Rights Agreement and Consent dated May 30, 2003 among Iomai Corporation and the Purchasers listed on the signature pages thereto. | | | (1) | |
| | 4 | .5.4 | | Amendment to Investor Rights Agreement and Consent dated December 1, 2005 among Iomai Corporation and the purchasers listed on the signature pages thereto. | | | (1) | |
| | 4 | .6 | | Registration Rights Agreement dated April 6, 2001 by and between Iomai Corporation and MdBio, Inc. as trustee for and on behalf of the Walter Reed Army Institute of Research, a representative of the United States of America. | | | (1) | |
| | 4 | .7 | | Registration Rights Agreement dated March 30, 1999 by and between Iomai Corporation and Maryland Health Care Product Development Corporation. | | | (1) | |
| | 4 | .8 | | Registration Rights Agreement dated July 18, 2000 by and between Iomai Corporation and Eiicha Ida. | | | (1) | |
| | 4 | .9 | | Registration Rights Agreement dated July 18, 2000 by and between Iomai Corporation and Yuichi Suzuki. | | | (1) | |
| | 4 | .10 | | Registration Rights Agreement dated July 18, 2000 by and between Iomai Corporation and Toshiro Osoegawa. | | | (1) | |
| | 4 | .11 | | Registration Rights Agreement dated August 15, 2000 by and between Iomai Corporation and CZ Venture Operations, Inc. | | | (1) | |
50
| | | | | | | | | |
Exhibit No. | | Description | | Location |
| |
| | 4 | .12 | | Registration Rights Agreement dated January 4, 2001 by and between Iomai Corporation and Alexandria Real Estate Equities, L.P. | | | (1) | |
| | 9 | .1 | | Voting Trust and Escrow Agreement dated April 6, 2001 between Iomai Corporation and MdBio, Inc., as trustee for and on behalf of Walter Reed Army Institute of Research, a representative of the United States of America. | | | (1) | |
| | 10 | .1 | | Employment Agreement dated May 18, 2002 between Iomai Corporation and Stanley Erck, as amended on October 25, 2002.** | | | (1) | |
| | 10 | .1.1 | | Amendment No. 2, dated December 1, 2005, to the Employment Agreement between Iomai Corporation and Stanley Erck.** | | | (1) | |
| | 10 | .2 | | 1998 Stock Option Plan, as amended January 16, 2002.** | | | (1) | |
| | 10 | .3.1 | | 1999 Stock Incentive Plan | | | (1) | |
| | 10 | .3.2 | | Form of Incentive Stock Option Agreement.** | | | (1) | |
| | 10 | .3.3 | | Form of Nonqualified Stock Option Agreement.** | | | (1) | |
| | 10 | .4 | | 2005 Incentive Plan.** | | | (1) | |
| | 10 | .5 | | 2006 Employee Stock Purchase Plan.** | | | (1) | |
| | 10 | .6 | | Terms of Non-Employee Director Compensation.** | | | (1) | |
| | 10 | .7 | | Amended and Restated License Agreement dated April 6, 2001 by and between Iomai Corporation and the Walter Reed Army Institute of Research as representative of the United States of America.+ | | | (1) | |
| | 10 | .8 | | Form of Subordinated Convertible Promissory Note issued by Iomai Corporation to the Walter Reed Army Institute of Research as representative of the United States of America. | | | (1) | |
| | 10 | .9 | | Commercial License Agreement dated June 30, 2005 by and between Iomai Corporation and Dow Global Technologies Incorporated.+ | | | (1) | |
| | 10 | .10.1 | | Master Security Agreement dated September 26, 2003 by and between Iomai Corporation and Oxford Finance Corporation. | | | (1) | |
| | 10 | .10.2 | | Form of Promissory Note issued by Iomai Corporation to Oxford Finance Corporation together with schedule identifying dates, principal amounts and interest rates of all outstanding promissory notes. | | | (1) | |
| | 10 | .11.1 | | Lease Agreement dated December 18, 2000 by and between Iomai Corporation and ARE-20/22/1300 Firstfield Quince Orchard, LLC. | | | (1) | |
| | 10 | .11.2 | | First Amendment to Lease dated November 29, 2001 by and between Iomai Corporation and ARE-20/22/1300 Firstfield Quince Orchard, LLC. | | | (1) | |
| | 10 | .11.3 | | Second Amendment to Lease dated April 14, 2003 by and between Iomai Corporation and ARE-20/22/1300 Firstfield Quince Orchard, LLC. | | | (1) | |
| | 10 | .11.4 | | Third Amendment to Lease dated August 28, 2003 by and between Iomai Corporation and ARE-20/22/1300 Firstfield Quince Orchard, LLC. | | | (1) | |
| | 10 | .11.5 | | Fourth Amendment to Lease dated October 26, 2005 by and between Iomai Corporation and ARE 20/22/1300 Firstfield Quince Orchard, LLC. | | | (1) | |
| | 10 | .11.6 | | Letter Agreement amending Fourth Amendment to Lease dated January 3, 2006 by and between Iomai Corporation and ARE 20/22/1300 Firstfield Quince Orchard, LLC. | | | | |
| | 10 | .11.7 | | Sublease Agreement dated February 28, 2006 by and between Geomet Technologies, LLC and Iomai Corporation. | | | | |
| | 10 | .12 | | Form of change in Control Agreement, between Iomai Corporation and certain officers.** | | | (1) | |
| | 23 | .1 | | Consent of Independent Registered Accounting Firm. | | | | |
| | 31 | .1 | | Certification of Chief Executive Officer pursuant toRules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as amended. | | | | |
51
| | | | | | | | | |
Exhibit No. | | Description | | Location |
| |
| | 31 | .2 | | Certification of Chief Financial Officer pursuant toRules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as amended. | | | | |
| | 32 | .1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | | | | |
| | |
| ** | | Indicates a management contract or compensatory plan. |
| |
| + | | Confidential Treatment Requested Under 17 C.F.R. §§ 200.80(b)(4) and 230.406. The confidential portions of this exhibit have been omitted and are marked by an asterisk. |
| |
| (1) | | Previously filed as an exhibit to the Company’sForm S-1/A (FileNo. 333-128765) and incorporated herein by reference thereto. |
| |
| (2) | | Previously filed as an exhibit to the Company’sForm 8-K filed March 24, 2006 and incorporated herein by reference thereto. |
52
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
IOMAI CORPORATION
Stanley C. Erck
Name: Stanley C. Erck
Title:President, Chief Executive Officer,
Treasurer and Director
Dated: March 23, 2006
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant in the capacities indicated as of March 23, 2006.
| | | | | |
Signature | | Title |
| |
/s/ Stanley C. Erck
Stanley C. Erck | | Chief Executive Officer, President,
Treasurer and Director
(Principal Executive Officer) |
| | | |
/s/ Russell P. Wilson
Russell P. Wilson | | Senior Vice President, Chief Financial Officer,
General Counsel and Secretary
(Principal Financial and Accounting Officer) |
| | | |
/s/ M. James Barrett
M. James Barrett, Ph.D. | | Chairman of the Board of Directors |
| | | |
/s/ R. Gordon Douglas
R. Gordon Douglas, M.D. | | Director |
| | | |
/s/ Richard Douglas
Richard Douglas, Ph.D. | | Director |
| | | |
/s/ Jeff Himawan
Jeff Himawan, Ph.D. | | Director |
| | | |
/s/ James Young
James Young, Ph.D. | | Director |
53
INDEX TO FINANCIAL STATEMENTS
| | | | | |
| | | Page |
| |
| | | F-2 | |
| | | F-3 | |
| | | F-4 | |
| | | F-5 | |
| | | F-6 | |
| | | F-7 | |
F-1
Report of Independent Registered Public Accounting Firm
Board of Directors and Shareholders
Iomai Corporation
We have audited the accompanying balance sheets of Iomai Corporation as of December 31, 2005 and 2004 and the related statements of operations, changes in redeemable preferred stock and stockholders’ deficit, and cash flows for each of the three years in the period ended December 31, 2005. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Iomai Corporation at December 31, 2005 and 2004, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2005 in conformity with U.S. generally accepted accounting principles.
/s/ Ernst & Young LLP
McLean, Virginia
February 27, 2006
F-2
IOMAI CORPORATION
BALANCE SHEETS
| | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | |
| |
ASSETS |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 5,190,457 | | | $ | 6,941,752 | |
| Marketable securities | | | — | | | | 15,455,613 | |
| Accounts receivable | | | 11,515 | | | | 73,452 | |
| Prepaid expenses and other current assets | | | 257,647 | | | | 137,444 | |
| | | | | | | | | |
| Total current assets | | | 5,459,619 | | | | 22,608,261 | |
| Property and equipment, net | | | 4,465,042 | | | | 5,538,314 | |
| Restricted cash | | | 49,150 | | | | 62,638 | |
| Restricted marketable securities | | | 589,740 | | | | 595,987 | |
| Deferred financing costs | | | 1,108,230 | | | | — | |
| Other noncurrent assets | | | 188,847 | | | | 136,460 | |
| | | | | | | | | |
| Total assets | | $ | 11,860,628 | | | $ | 28,941,660 | |
| | | | | | | | | |
LIABILITIES AND STOCKHOLDERS’ DEFICIT |
| Current liabilities: | | | | | | | | |
| Accounts payable | | | 2,011,400 | | | $ | 997,322 | |
| Accrued expenses | | | 1,611,108 | | | | 1,267,740 | |
| Notes payable, current portion | | | 1,333,369 | | | | 1,119,418 | |
| Notes payable to related party, current portion | | | 229,226 | | | | 332,877 | |
| Capital lease obligation, current portion | | | 4,840 | | | | 13,074 | |
| | | | | | | | | |
| Total current liabilities | | | 5,189,943 | | | | 3,730,431 | |
| Notes payable, long-term portion | | | 1,396,828 | | | | 2,308,155 | |
| Notes payable to related party, long-term portion | | | 1,264,457 | | | | 1,493,683 | |
| Capital lease obligation, long-term portion | | | 1,239 | | | | 6,080 | |
| Deferred rent | | | 121,648 | | | | 80,388 | |
| | | | | | | | | |
| Total liabilities | | | 7,974,115 | | | | 7,618,737 | |
| Common stock subject to put right | | | 1,958,450 | | | | 1,958,450 | |
| Warrant to purchase Series C preferred stock | | | 23,100 | | | | 35,100 | |
| Series C convertible redeemable preferred stock, $0.01 par value; 150,000,000 shares authorized; 129,590,034 shares issued and outstanding at December 31, 2005 and 2004; liquidation preference in the aggregate of $113,823,648 at December 31, 2005 and $92,589,013 at December 31, 2004 | | | 70,363,054 | | | | 65,322,000 | |
| Stockholders’ equity (deficit): | | | | | | | | |
| Series B convertible preferred stock; $0.01 par value; 15,200,000 shares authorized; 14,734,578 shares issued and outstanding at December 31, 2005 and 2004; liquidation preference in the aggregate of $8,117,554 at December 31, 2005 and $7,595,949 at December 31, 2004 | | | 147,346 | | | | 147,346 | |
| Common stock, $0.01 par value; 220,000,000 shares authorized; 795,519 and 771,921 shares issued and outstanding at December 31, 2005 and 2004 | | | 7,955 | | | | 7,719 | |
| Additional paid-in capital | | | — | | | | 3,847,734 | |
| Accumulated other comprehensive income (loss) | | | 298 | | | | (25,612 | ) |
| Accumulated deficit | | | (68,613,690 | ) | | | (49,969,814 | ) |
| | | | | | | | | |
| Total stockholders’ equity (deficit) | | | (68,458,091 | ) | | | (45,992,627 | ) |
| | | | | | | | | |
| Total liabilities and stockholders’ equity (deficit) | | $ | 11,860,628 | | | $ | 28,941,660 | |
| | | | | | | | | |
See accompanying notes.
F-3
IOMAI CORPORATION
STATEMENTS OF OPERATIONS
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2005 | | | 2004 | | | 2003 | |
| |
| Revenues | | $ | 2,371,460 | | | $ | 2,344,773 | | | $ | 1,601,037 | |
| Cost and expenses: | | | | | | | | | | | | |
| Research and development | | | 16,529,376 | | | | 14,349,229 | | | | 13,331,968 | |
| General and administrative | | | 3,780,514 | | | | 3,205,658 | | | | 3,347,853 | |
| | | | | | | | | | | | | |
| Total costs and expenses | | | 20,309,890 | | | | 17,554,887 | | | | 16,679,821 | |
| | | | | | | | | | | | | |
| Loss from operations | | | (17,938,430 | ) | | | (15,210,114 | ) | | | (15,078,784 | ) |
| Other income (expense) | | | | | | | | | | | | |
| Interest income | | | 392,232 | | | | 620,382 | | | | 952,749 | |
| Interest expense | | | (466,927 | ) | | | (265,688 | ) | | | (127,797 | ) |
| Other expense, net | | | (16,603 | ) | | | (224,860 | ) | | | (448,178 | ) |
| | | | | | | | | | | | | |
| Total other income (expense), net | | | (91,298 | ) | | | 129,834 | | | | 376,774 | |
| | | | | | | | | | | | | |
| Net loss | | | (18,029,728 | ) | | | (15,080,280 | ) | | | (14,702,010 | ) |
| Dividends on and accretion of convertible preferred stock | | | (5,562,657 | ) | | | (5,525,314 | ) | | | (5,466,280 | ) |
| | | | | | | | | | | | | |
| Net income (loss) available to common stockholders | | $ | (23,592,385 | ) | | $ | (20,605,594 | ) | | $ | (20,168,290 | ) |
| | | | | | | | | | | | | |
| Net income (loss) available to common stockholders per share of common stock — basic and diluted | | $ | (30.14 | ) | | $ | (26.90 | ) | | $ | (26.43 | ) |
| | | | | | | | | | | | | |
| Weighted-average number of shares of common stock — basic and diluted | | | 782,715 | | | | 765,945 | | | | 763,075 | |
| | | | | | | | | | | | | |
See accompanying notes.
F-4
IOMAI CORPORATION
STATEMENTS OF CHANGES IN REDEEMABLE PREFERRED STOCK AND STOCKHOLDERS’ DEFICIT
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | Stockholders’ Deficit | |
| | | Series C Convertible
| | | | | | | | | | | | | | | | | | Accumulated
| | | | | | | |
| | | Redeemable
| | | Series B Convertible
| | | | | | | | | Additional
| | | Other
| | | | | | Total
| |
| | | Preferred Stock | | | Preferred Stock | | | Common Stock | | | Paid-In
| | | Comprehensive
| | | Accumulated
| | | Stockholders’
| |
| | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Capital | | | Income (Loss) | | | Deficit | | | Deficit | |
| |
| Balance at December 31, 2002 | | | 122,935,926 | | | | 52,375,850 | | | | 14,734,578 | | | | 147,346 | | | | 757,402 | | | | 7,574 | | | | 13,499,397 | | | | — | | | | (20,187,524 | ) | | | (6,533,207 | ) |
| Issuance of Series C convertible redeemable preferred stock, net of issuance costs of $21,598 | | | 4,750,056 | | | | 2,078,402 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Amortization of Series C warrants | | | — | | | | 12,000 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Accretion of issuance costs | | | — | | | | 441,089 | | | | — | | | | — | | | | — | | | | — | | | | (441,089 | ) | | | — | | | | — | | | | (441,089 | ) |
| Accrual of dividends | | | — | | | | 4,491,587 | | | | — | | | | — | | | | — | | | | — | | | | (4,491,587 | ) | | | — | | | | — | | | | (4,491,587 | ) |
| Refund of issuance costs, net | | | — | | | | 109,322 | | | | — | | | | — | | | | — | | | | — | | | | 55,490 | | | | — | | | | — | | | | 55,490 | |
| Issuance of warrants on notes payable | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 1,062 | | | | — | | | | — | | | | 1,062 | |
| Stock option compensation | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 48,580 | | | | — | | | | — | | | | 48,580 | |
| Exercise of options | | | — | | | | — | | | | — | | | | — | | | | 6,923 | | | | 69 | | | | 6,231 | | | | — | | | | — | | | | 6,300 | |
| Other comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | �� | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (14,702,010 | ) | | | (14,702,010 | ) |
| Unrealized gain on investments | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 18,732 | | | | — | | | | 18,732 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (14,683,278 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2003 | | | 127,685,982 | | | | 59,508,250 | | | | 14,734,578 | | | | 147,346 | | | | 764,325 | | | | 7,643 | | | | 8,678,084 | | | | 18,732 | | | | (34,889,534 | ) | | | (26,037,729 | ) |
| Issuance of Series C convertible redeemable preferred stock, net of issuance costs of $31,741 | | | 1,904,052 | | | | 810,040 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Amortization of Series C warrants | | | — | | | | 12,000 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Accretion of issuance costs | | | — | | | | 437,740 | | | | — | | | | — | | | | — | | | | — | | | | (437,740 | ) | | | — | | | | — | | | | (437,740 | ) |
| Accrual of dividends | | | — | | | | 4,553,970 | | | | — | | | | — | | | | — | | | | — | | | | (4,553,970 | ) | | | — | | | | — | | | | (4,553,970 | ) |
| Issuance of Common Stock warrants | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 818 | | | | — | | | | — | | | | 818 | |
| Stock option compensation | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 153,705 | | | | — | | | | — | | | | 153,705 | |
| Exercise of options | | | — | | | | — | | | | — | | | | — | | | | 7,596 | | | | 76 | | | | 6,837 | | | | — | | | | — | | | | 6,913 | |
| Other comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (15,080,280 | ) | | | (15,080,280 | ) |
| Unrealized loss on investments | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (44,344 | ) | | | — | | | | (44,344 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (15,124,624 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2004 | | | 129,590,034 | | | | 65,322,000 | | | | 14,734,578 | | | | 147,346 | | | | 771,921 | | | | 7,719 | | | | 3,847,734 | | | | (25,612 | ) | | | (49,969,814 | ) | | | (45,992,627 | ) |
| Amortization of Series C warrants | | | — | | | | 12,000 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Accretion of issuance costs | | | — | | | | 441,567 | | | | — | | | | — | | | | — | | | | — | | | | (331,173 | ) | | | — | | | | (110,394 | ) | | | (441,567 | ) |
| Accrual of dividends | | | — | | | | 4,587,487 | | | | — | | | | — | | | | — | | | | — | | | | (4,083,733 | ) | | | — | | | | (503,754 | ) | | | (4,587,487 | ) |
| Issuance of Common Stock warrants | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 8,678 | | | | — | | | | — | | | | 8,678 | |
| Stock option compensation | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 538,144 | | | | — | | | | — | | | | 538,144 | |
| Cash in lieu of fractional common shares | | | — | | | | — | | | | — | | | | — | | | | (60 | ) | | | (1 | ) | | | (944 | ) | | | | | | | | | | | (945 | ) |
| Exercise of options | | | — | | | | | | | | — | | | | — | | | | 23,658 | | | | 237 | | | | 21,294 | | | | — | | | | — | | | | 21,531 | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (18,029,728 | ) | | | (18,029,728 | ) |
| Unrealized gain on investments | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 25,910 | | | | | | | | 25,910 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Comprehensive loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (18,003,818 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2005 | | | 129,590,034 | | | $ | 70,363,053 | | | | 14,734,578 | | | $ | 147,346 | | | | 795,719 | | | $ | 7,955 | | | $ | — | | | $ | 298 | | | $ | (68,613,690 | ) | | $ | (68,458,091 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes.
F-5
IOMAI CORPORATION
STATEMENTS OF CASH FLOWS
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2005 | | | 2004 | | | 2003 | |
| |
Cash flows from operating activities | | | | | | | | | | | | |
| Net loss | | $ | (18,029,728 | ) | | $ | (15,080,280 | ) | | $ | (14,702,010 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
| Depreciation and amortization | | | 2,670,444 | | | | 1,626,768 | | | | 365,909 | |
| Stock-based compensation expense | | | 538,144 | | | | 153,705 | | | | 48,580 | |
| Non-cash interest expense and amortization of premium/discount of marketable securities | | | 28,366 | | | | 228,615 | | | | 272,911 | |
| Deferred rent | | | (54,411 | ) | | | (35,695 | ) | | | (18,631 | ) |
| Provision for doubtful accounts | | | — | | | | (135,000 | ) | | | 135,000 | |
| Loss on disposal of property and equipment | | | 3,896 | | | | — | | | | 5,076 | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | |
| Accounts receivable | | | 61,937 | | | | 264,753 | | | | 31,178 | |
| Prepaid expenses and other current assets | | | (40,358 | ) | | | 135,050 | | | | 27,079 | |
| Other noncurrent assets | | | (45,434 | ) | | | 29,010 | | | | (59,833 | ) |
| Accounts payable | | | 420,096 | | | | (470,894 | ) | | | 625,422 | |
| Accrued expenses | | | 143,825 | | | | (217,771 | ) | | | (230,097 | ) |
| | | | | | | | | | | | | |
| Net cash used in operating activities | | | (14,303,223 | ) | | | (13,501,739 | ) | | | (13,499,416 | ) |
Cash flows from investing activities | | | | | | | | | | | | |
| Purchases of property and equipment | | | (1,338,752 | ) | | | (5,074,855 | ) | | | (1,264,678 | ) |
| Restricted cash and marketable securities | | | 35,076 | | | | (3,685 | ) | | | (598,086 | ) |
| Sales/maturities of marketable securities | | | 22,150,000 | | | | 32,450,000 | | | | 35,438,000 | |
| Purchases of marketable securities | | | (6,790,305 | ) | | | (18,509,757 | ) | | | (65,453,165 | ) |
| | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | 14,056,019 | | | | 8,861,703 | | | | (31,877,929 | ) |
Cash flows from financing activities | | | | | | | | | | | | |
| Proceeds from the exercise of stock options | | | 21,531 | | | | 6,913 | | | | 6,300 | |
| Cash in lieu of fractional common stock | | | (945 | ) | | | — | | | | — | |
| Proceeds from issuance of Series C convertible redeemable preferred stock, net | | | — | | | | 810,040 | | | | 2,078,402 | |
| Deferred stock issuance costs | | | (481,350 | ) | | | — | | | | (2,585 | ) |
| Proceeds from notes payable | | | 468,367 | | | | 1,902,386 | | | | 2,472,920 | |
| Principal payments on notes payable | | | (1,165,743 | ) | | | (819,746 | ) | | | (127,986 | ) |
| Proceeds from notes payable to related party | | | — | | | | 1,450,500 | | | | — | |
| Principal payments on notes payable to related party | | | (332,877 | ) | | | (207,440 | ) | | | (67,371 | ) |
| Payments under capital lease obligations | | | (13,074 | ) | | | (18,025 | ) | | | (18,176 | ) |
| | | | | | | | | | | | | |
| Net cash provided by (used in) financing activities | | | (1,504,091 | ) | | | 3,124,628 | | | | 4,341,504 | |
| | | | | | | | | | | | | |
| Net decrease in cash and cash equivalents | | | (1,751,295 | ) | | | (1,515,408 | ) | | | (41,035,841 | ) |
| Cash and cash equivalents at beginning of year | | | 6,941,752 | | | | 8,457,160 | | | | 49,493,001 | |
| | | | | | | | | | | | | |
| Cash and cash equivalents at end of year | | $ | 5,190,457 | | | $ | 6,941,752 | | | $ | 8,457,160 | |
| | | | | | | | | | | | | |
Supplemental schedule of noncash investing and financing activities | | | | | | | | | | | | |
| Cash paid for interest | | $ | 496,784 | | | $ | 424,088 | | | $ | 117,546 | |
| | | | | | | | | | | | | |
See accompanying notes.
F-6
IOMAI CORPORATION
NOTES TO FINANCIAL STATEMENTS
Iomai Corporation (the Company or Iomai) was incorporated in May 1997 as a Delaware corporation to discover and develop vaccines and immune system stimulants, delivered to the skin via a novel, needle-free technology called transcutaneous immunization (TCI). TCI exploits the unique benefits of a major group of antigen-presenting cells found in the outer layers of the skin (Langerhans cells) to generate an enhanced immune response. TCI has the potential to enhance the efficacy of existing vaccines, develop new vaccines that are viable only through transcutaneous administration and expand the global vaccine market. The Company currently has four product candidates in development: three targeting influenza and pandemic flu and one to preventE. coli-related travelers’ diarrhea.
| |
| 2. | SIGNIFICANT ACCOUNTING POLICIES |
Use of estimates
The preparation of the financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and cash equivalents
The Company considers demand deposits and all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents. As of December 31, 2005 and 2004, the Company’s cash equivalents consisted of money market accounts, an overnight repurchase agreement and a savings account.
Marketable securities
All marketable securities, consisting of U.S. government obligations and U.S. agency obligations, are classified asavailable-for-sale. These securities are carried at fair value, plus accrued interest. Any unrealized holding gains and losses are reported as accumulated other comprehensive income (loss), which is a separate component of stockholders’ deficit. Realized gains and losses are reported in operations, and gains and losses on sales of securities are computed using the specific-identification method.
The Company periodically reviews its marketable securities to determine whether a decline in fair value below the carrying value exists and isother-than-temporary. This evaluation consists of a review of several factors, including but not limited to: the length of time and extent that a security has been in an unrealized loss position, the existence of an event that would impair the issuer’s future earnings potential, the near-term prospects for recovery of the market value of a security, and the intent and ability of the Company to hold the security until the market value recovers. Declines in value below cost for debt securities are not assumed to beother-than-temporary where: it is considered probable that all contractual terms of the security will be satisfied, the decline is due primarily to changes in interest rates (and not because of increased credit risk), or the Company intends and has the ability to hold the investment for a period of time sufficient to allow a market recovery. Unrealized losses related to debt securities greater than and less than one year are not significant.
If management determines that such an impairment exists, the carrying value of the investment will be reduced to the current fair value of the investment and the Company will recognize a charge in the statement of operations equal to the amount of the carrying value reduction.
F-7
Restricted cash and marketable securities
Restricted cash at December 31, 2005 and 2004 relates to a deposit held as collateral for a letter of credit acting as a security deposit on a facility operating lease. The interest rate is the bank’s prime rate plus one percent per annum and the fee is one percent annually.
Restricted marketable securities at December 31, 2005 and 2004 include marketable securities with a face value of $600,000 pledged as collateral to secure payment of promissory notes issued to finance, in part, the build-out of the Company’s facilities (see Note 4 — Long-Term Debt).
Accounts receivable
Accounts receivable that management has the intent and ability to hold until payment are reported in the balance sheets at outstanding amounts, less the allowance for doubtful accounts. The Company writes off uncollectible receivables when the likelihood of collection is remote. The Company maintains an allowance for doubtful accounts, which is determined based on historical experience and management’s expectations of future losses. There was no allowance for doubtful accounts in either 2004 or 2005.
Unbilled accounts receivable consist principally of expenses incurred on reimbursable research grants prior to year-end that have not yet been billed to the contracting agent.
Concentration of credit risk and fair value of financial instruments
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash and cash equivalents, marketable securities, accounts receivable and notes payable. The Company places its cash and cash equivalents with financial institutions, and its marketable securities consist of U.S. government obligations and U.S. agency obligations. Management believes that the financial risks associated with its cash and cash equivalents and marketable securities are minimal. Accounts receivable consist of amounts due from government agencies under government grants.
The carrying amount of current assets and liabilities approximates their fair values due to their short-term maturities. The fair value of notes payable approximates their carrying amount as of December 31, 2005 and 2004 based on rates currently available to the Company for debt with similar terms and remaining maturities.
Property and equipment
Property and equipment are stated at cost. Depreciation is computed using the straight-line method over the estimated useful lives of three to seven years. Leasehold improvements are amortized over the shorter of the life of the lease or the related asset. Maintenance and repairs are charged to expense as incurred.
Property and equipment consist of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | |
| |
| Furniture and equipment | | $ | 769,186 | | | $ | 695,404 | |
| Lab equipment | | | 3,327,189 | | | | 2,164,042 | |
| Leasehold improvements | | | 5,296,983 | | | | 4,907,502 | |
| Construction-in-progress | | | 208,197 | | | | 355,645 | |
| | | | | | | | | |
| Subtotal | | | 9,601,555 | | | | 8,122,593 | |
| Accumulated depreciation | | | (5,136,513 | ) | | | (2,584,279 | ) |
| | | | | | | | | |
| Property and equipment, net | | $ | 4,465,042 | | | $ | 5,538,314 | |
| | | | | | | | | |
Accumulated amortization for equipment under capital leases was $14,051 and $23,203 at December 31, 2005 and 2004, respectively. The Company recognized amortization expense for assets under capital lease obligations of approximately $6,813, $9,810 and $11,172 for the years ended December 31, 2005, 2004, and 2003, respectively. These amounts are included in depreciation and amortization. For the year ended December 31, 2005 and 2004, the
F-8
Company capitalized interest expense of $25,772 and $175,336, respectively. There was no interest capitalized for the year ended December 31, 2003.
Income taxes
Income taxes are accounted for using the liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities, and their respective tax bases and operating loss carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
Valuation allowances are established when necessary to reduce net deferred tax assets to the amount expected to be realized. Income tax expense is the tax payable for the period and the change during the period in deferred tax assets and liabilities.
Revenue recognition
The Company recognizes revenue when all terms and conditions of the agreements have been met, including persuasive evidence of an arrangement, services have been rendered, price is fixed or determinable, and collectability is reasonably assured. For reimbursable cost research grants, the Company recognizes revenue as costs are incurred. Provisions for estimated losses on research grant projects and any other contracts are made in the period such losses are determined.
Research and development costs
The Company expenses its research and development costs as incurred; however, equipment and facilities that are acquired or constructed for research and development activities that have alternative future uses (in research and development projects or otherwise) are capitalized and depreciated as tangible assets.
Comprehensive loss
Statement of Financial Accounting Standards (SFAS) No. 130,Reporting Comprehensive Income(SFAS No. 130), requires the presentation of the comprehensive loss and its components as part of the financial statements. Comprehensive loss is comprised of net loss and other changes in equity that are excluded from net loss. The Company includes unrealized holding gains and losses onavailable-for-sale securities in accumulated other comprehensive loss on its balance sheets and statements of changes in redeemable preferred stock and stockholders’ deficit.
Stock-based compensation
The Company has four stock-based employee compensation plans, described more fully in Note 7 — Stockholders’ Deficit. Prior to 2003, the Company accounted for these plans under Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees(APB 25), and related Interpretations. Effective January 1, 2003, the Company adopted the fair value recognition provisions of SFAS No. 123,Accounting for Stock-Based Compensation(SFAS No. 123). Under the prospective method of adoption selected by the Company under the provisions of FASB Statement No. 148,Accounting for Stock-Based Compensation — Transition and Disclosure,an amendment of FASB Statement No. 123 (SFAS No. 148), the recognition provisions have been applied to all employee awards granted, modified, or settled after January 1, 2003.
Equity instruments issued to nonemployees are accounted for under the provisions of SFAS No. 123 and Emerging Issues Task Force IssueNo. 96-18,Accounting for Equity Instruments That are Issued to Other Than Employees for Acquiring, or in conjunction with, Selling, Goods and Services. Accordingly, the estimated fair value of the equity instrument is recorded on the earlier of the performance commitment date or the date the services required are completed.
F-9
Given the lack of an active public market for our common stock prior to the Company’s initial public offering (see Note 12 — Subsequent Events), our board of directors determined fair value of our common stock for options to acquire shares of our common stock. Our board of directors made contemporaneous determinations of fair value and did not employ a third party valuation firm to determine fair value. In the absence of a public trading market prior to the Company’s initial public offering, and as a clinical-stage company with no significant revenues, the Company believes that it was appropriate to consider a range of factors to determine the fair value market value of the common stock at each option grant date. These factors included: (1) the achievement of clinical and operational milestones by the Company, (2) the status of strategic relationships with collaborators, (3) the significant risks associated with the Company’s early stage of development of a new technology, (4) capital market conditions for life science companies, particularly similarly situated privately-held, early-stage life science companies, (5) the Company’s available cash, financial condition and results of operations, (6) the most recent sales of the Company’s preferred stock and (7) the preferential rights of the outstanding preferred stock.
In connection with the preparation of the financial statements for the Company’s initial public offering, the Company reassessed its estimate of the fair value for financial reporting purposes of its common stock following its stock option grants in March 2004. This valuation was done retrospectively by management, a related party, and the Company did not obtain contemporaneous valuations from an independent valuation specialist. Based on this reassessment, the Company determined that there were five periods between March 2004 and October 2005 in which the Company’s reassessment of the fair value of its common stock ranged from $2.75 to $9.00 per share during the period options were granted. In determining the reassessed fair value of the common stock as of each grant date, the factors described in the previous paragraph were taken into account. In reassessing the value of common stock in 2004 and 2005, the Company considered the price it received in June 2004 for its Series C preferred stock of $0.4421 per share (or approximately $5.75 upon conversion into Common Stock). During the period between March 2004 and September 2004, the Company received positive data from a clinical trial of its needle-free travelers’ diarrhea vaccine patch and initiated two Phase 1 clinical trials of its anthrax and TCI flu patch product candidates, resulting in a adjusted fair value of common stock of $2.75 per share, approximately a 50% discount to the price of the Series C preferred stock. The Company kept this value constant until it received preliminary data from its Phase 1 clinical trial of the needle-free flu vaccine patch in late September 2004. Based on these results and observed capital market conditions for early-stage life science companies, the Company adjusted the estimated fair value for financial reporting purposes for the period of late September 2004 to February 2005 to $4.40 per share, approximately a 25% discount to the price of the Series C preferred stock. From March 2005 until July 2005, the Company increased the estimated fair value for financial reporting purposes to $6.50 per share, approximately a 13% discount to the price of the Series C preferred stock. This was primarily based on positive results from its Phase 2 clinical trial of the needle-free travelers’ diarrhea patch and the license of the gene expression system from DowPharma, a business unit within The Dow Chemical Company, to allow the production of the LT adjuvant. In August 2005, the Company increased the estimated fair value for financial reporting purposes to $7.50 per share. This was primarily based on positive developments in the capital markets for early stage life science companies. From September to October 2005, the Company increased the estimated fair value for financial reporting purposes to $9.00 per share. This was primarily based on positive developments in the capital markets for early stage life science companies and the Company’s engagement of underwriters to pursue an initial public offering. This valuation method was selected because the Company believes it reflected the change in value held by the common stockholders that would result from a successful public offering, which included the conversion of our preferred stock into common stock and thereby eliminated the preferences and rights attributable to the preferred stock. Furthermore, the Company believes this valuation approach was consistent with valuation methodologies applied to other similar companies pursuing an initial public offering.
As a result of this reassessment of the fair value of its common stock for financial reporting purposes, the Company anticipates recognizing approximately $1.8 million of compensation expense ratably over future periods as stock options outstanding as of December 31, 2005 vest in accordance with their terms, less any amounts for options forfeited or cancelled.
F-10
Basic and diluted net income (loss) attributable to common stockholders per share of common stock
Basic net loss attributable to common stockholders per share of common stock excludes dilution for potential common stock issuances and is computed by dividing net income (loss) attributable to common stockholders by the weighted-average number of shares outstanding for the period. Diluted net income (loss) attributable to common stockholders per share reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted into common stock. Mandatorily redeemable convertible preferred stock, stock options and warrants were not considered in the computation of diluted net income (loss) attributable to common stockholders per share for the years ended December 31, 2005, 2004 and 2003 as their effect is antidilutive.
Recent accounting pronouncements
On December 16, 2004, the Financial Accounting Standards Board (FASB) issued FASB Statement No. 123 (revised 2004),Share-Based Payment, which is a revision of FASB Statement No. 123,Accounting for Stock-Based Compensation. Statement 123(R) supersedes APB Opinion No. 25,Accounting for Stock Issued to Employees, and amends FASB Statement No. 95,Statement of Cash Flows. Generally, the approach in Statement 123(R) is similar to the approach described in Statement 123. However, Statement 123(R) requires all share-based payments to employees, including grants of employee stock options, to be recognized in the income statement based on their fair values. Pro forma disclosure is no longer an alternative.
Statement 123(R) originally required adoption no later than July 1, 2005. In April 2005, the Securities and Exchange Commission (“SEC”) issued a release that amends the compliance dates for Statement 123(R). Under the SEC’s new rule, the Company will be required to apply Statement 123(R) as of January 1, 2006.
The Company adopted thefair-value-based method of accounting for share-based payments effective January 1, 2003 using the prospective method described in FASB Statement No. 148,Accounting for Stock-Based Compensation — Transition and Disclosure. Currently, the Company uses the Black-Scholes-Merton formula to estimate the value of stock options granted to employees and expects to continue to use this option valuation model upon the required adoption of Statement 123(R) on January 1, 2006. Statement 123(R) also requires the benefits of tax deductions in excess of recognized compensation cost to be reported as a financing cash flow rather than as an operating cash flow as required under current literature. This requirement will reduce net operating cash flows and increase net financing cash flows in periods after adoption; however, the Company cannot estimate what those amounts will be in the future (because they depend on, among other things, when employees exercise stock options). Since the Company has accounted for stock options in accordance with Statement No. 123 since January 2003, management believes that the adoption of SFAS 123(R) will not have a material impact on its financial statements going forward.
Reverse stock split
All share and per share amounts have been retroactively adjusted to give effect to a1-for-13 reverse stock split effected on December 2, 2005.
F-11
Following is a summary of theavailable-for-sale marketable securities at December 31, 2005 and 2004:
| | | | | | | | | | | | | | | | | |
| | | December 31, 2005 | |
| | | | | | Gross
| | | Gross
| | | | |
| | | | | | Unrealized
| | | Unrealized
| | | | |
| | | Amortized Cost | | | Gains | | | Losses | | | Fair Value | |
| |
Available for Sale | | | | | | | | | | | | | | | | |
| U.S. Treasury and agency obligations | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Accrued interest | | | — | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | |
| Total — Marketable securities | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | | |
Restricted Marketable Securities | | | | | | | | | | | | | | | | |
| U.S. Treasury and agency obligations | | $ | 589,442 | | | $ | 298 | | | $ | — | | | $ | 589,740 | |
| Accrued interest | | | — | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | |
| Total — Restricted marketable securities | | $ | 589,442 | | | $ | 298 | | | $ | — | | | $ | 589,740 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | December 31, 2004 | |
| | | | | | Gross
| | | Gross
| | | | |
| | | | | | Unrealized
| | | Unrealized
| | | | |
| | | Amortized Cost | | | Gains | | | Losses | | | Fair Value | |
| |
Available for Sale | | | | | | | | | | | | | | | | |
| U.S. Treasury and agency obligations | | $ | 15,399,148 | | | $ | 315 | | | $ | (23,676 | ) | | $ | 15,375,787 | |
| Accrued interest | | | 79,826 | | | | — | | | | — | | | | 79,826 | |
| | | | | | | | | | | | | | | | | |
| Total — Marketable securities | | $ | 15,478,974 | | | $ | 315 | | | $ | (23,676 | ) | | $ | 15,455,613 | |
| | | | | | | | | | | | | | | | | |
Restricted Marketable Securities | | | | | | | | | | | | | | | | |
| U.S. Treasury and agency obligations | | $ | 598,219 | | | $ | — | | | $ | (2,251 | ) | | $ | 595,968 | |
| Accrued interest | | | 19 | | | | — | | | | — | | | | 19 | |
| | | | | | | | | | | | | | | | | |
| Total — Restricted marketable securities | | $ | 598,238 | | | $ | — | | | $ | (2,251 | ) | | $ | 595,987 | |
| | | | | | | | | | | | | | | | | |
The contractual maturities of the Company’s marketable securities and restricted marketable securities were less than one year at December 31, 2005 and 2004. Actual maturities may differ from contractual maturities because the issuers of the securities may have the right to prepay obligations without prepayment penalties.
The Company’s gross proceeds from maturities of its marketable securities for the years ended December 31, 2005, 2004 and 2003 were $22,150,000, $32,450,000 and $35,438,000, respectively. For these periods, the Company did not realize any gains or losses related to its marketable securities.
Notes payable
In September 2003, the Company entered into a loan arrangement with an equipment leasing company to finance up to $5 million for the build-out of the Company’s facility to accommodate (1) a GMP pilot plant for biological and patch manufacturing to support clinical trials and to advance patch formulation and development, and (2) laboratories to support patch formulation and development, as well as small-scale biological research. Under the terms of the loan, the Company was permitted to draw down up to $5 million. The facility was extended through September 2005.
In connection with the loan agreement, the Company entered into a master security agreement under which the Company pledged to the equipment leasing company as collateral (i) a security interest in existing equipment then having a net book value of approximately $416,000, (ii) a security interest in all laboratory/scientific and related manufacturing equipment, together with furniture and computer hardware, financed under the facility, and
F-12
(iii) marketable securities with a face value of $600,000. As of December 31, 2005 and 2004, the Company had drawn down in total $4,843,672 and $4,375,305, respectively, on the loan agreement. The lender has the right to accelerate repayment of the indebtedness upon a default, including if the lender determines in good faith that there has been a material adverse change in the Company’s business plan which would materially impair the ability of the Company to perform its obligations or of the lender to enforce the indebtedness or realize upon the collateral.
The components of notes payable are as follows:
| | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | |
| |
| Promissory Note dated September 26, 2003; 8.93%, maturing March 26, 2007 | | $ | 546,625 | | | $ | 942,201 | |
| Promissory Note dated September 26, 2003; 9.03%, maturing September 26, 2007 | | | 34,884 | | | | 52,487 | |
| Promissory Note dated December 30, 2003; 9.24%, maturing June 30, 2007 | | | 464,660 | | | | 740,644 | |
| Promissory Note dated February 26, 2004; 9.03%, maturing February 26, 2008 | | | 123,971 | | | | 173,530 | |
| Promissory Note dated April 30, 2004; 9.48%, maturing April 30, 2008 | | | 212,569 | | | | 290,277 | |
| Promissory Note dated June 30, 2004; 10.06%, maturing December 30, 2007 | | | 62,945 | | | | 89,992 | |
| Promissory Note dated June 30, 2004; 10.03%, maturing June 30, 2008 | | | 519,611 | | | | 693,743 | |
| Promissory Note dated September 30, 2004; 9.58%, maturing March 30, 2008 | | | 19,732 | | | | 27,231 | |
| Promissory Note dated September 30, 2004; 9.40%, maturing September 30, 2008 | | | 135,321 | | | | 176,508 | |
| Promissory Note dated November 30, 2004; 9.72%, maturing November 30, 2008 | | | 187,837 | | | | 240,960 | |
| Promissory Note dated March 30, 2005; 10.46%, maturing March 30, 2009 | | | 59,321 | | | | — | |
| Promissory Note dated March 30, 2005; 10.70%, maturing September 30, 2008 | | | 20,482 | | | | — | |
| Promissory Note dated July 27, 2005; 10.32%, maturing July 27, 2009 | | | 172,913 | | | | — | |
| Promissory Note dated July 27,2005; 10.66%, maturing January 27, 2009 | | | 20,896 | | | | — | |
| Promissory Note dated September 30, 2005; 10.31%, maturing September 30, 2009 | | | 148,430 | | | | — | |
| | | | | | | | | |
| Total notes payable | | | 2,730,197 | | | | 3,427,573 | |
| Less current portion of notes payable | | | (1,333,369 | ) | | | (1,119,418 | ) |
| | | | | | | | | |
| Long-term portion of notes payable | | $ | 1,396,828 | | | $ | 2,308,155 | |
| | | | | | | | | |
F-13
The Company has long-term obligations to a related party (see Note 5 — Related Party Transactions). Related-party notes payable consist of the following at December 31, 2005 and 2004:
| | | | | | | | | |
| | | 2005 | | | 2004 | |
| |
| Total related-party notes payable, with interest rates ranging from 14.0% to 10.5%, maturing May 31, 2013(1) | | | 1,493,683 | | | | 1,826,560 | |
| Less current portion of related-party notes payable | | | (229,226 | ) | | | (332,877 | ) |
| | | | | | | | | |
| Long-term portion of related-party notes payable | | $ | 1,264,457 | | | $ | 1,493,683 | |
| | | | | | | | | |
| | |
| (1) | | On October 26, 2005, the Company amended its lease agreement to, among things, to extend the maturity of the related-party notes payable from May 2009 until May 2013. In connection with this lease amendment, the then-remaining balance owed to the landlord as of May 31, 2006 under the related-party notes payable, which the Company expects will total $1,342,719, will then be amortized over the seven-year period until May 2013 at a revised rate of 10.5%. See Note 5 — Related Party Transactions. |
As of December 31, 2005, scheduled principal repayments on notes payable are as follows:
| | | | | | | | | | | | | | | | | |
| | | | | | Notes Payable from
| | | | | | | |
| | | Notes Payable | | | Related Party(1) | | | Total | | | | |
| |
| Year ending: | | | | | | | | | | | | | | | | |
| 2006 | | $ | 1,333,369 | | | $ | 229,226 | | | $ | 1,562,595 | | | | | |
| 2007 | | | 930,663 | | | | 145,785 | | | | 1,076,448 | | | | | |
| 2008 | | | 393,110 | | | | 161,852 | | | | 554,962 | | | | | |
| 2009 | | | 73,055 | | | | 179,688 | | | | 252,743 | | | | | |
| 2010 and thereafter | | | | | | | 777,132 | | | | 777,132 | | | | | |
| | | | | | | | | | | | | | | | | |
| | | $ | 2,730,197 | | | $ | 1,493,683 | | | $ | 4,223,880 | | | | | |
| | | | | | | | | | | | | | | | | |
| | |
| (1) | | On October 26, 2005, the Company amended its lease agreement to, among things, to extend the lease from May 2009 until May 2013. In connection with this lease amendment, the then-remaining balance owed to the landlord as of May 31, 2006 under the related-party notes payable, which the Company expects will total $1,342,719, will then be amortized over the seven-year period until May 2013 at a rate of 10.5%. The revised debt repayment terms are reflected in the table in long term debt and operating lease obligations. See discussion of amended lease terms below. See Note 5 — Related Party Transactions. |
| |
| 5. | RELATED PARTY TRANSACTIONS |
The landlord for the Company’s laboratory and office facilities participated in the Series C financing in 2002 and, therefore, is a related party. Accordingly, the landlord’s financing of the Company’s leasehold improvements has been recorded on the balance sheets as notes payable to related party.
When the Company entered into its original laboratory and office facilities lease agreement with the landlord in 2001, the Company also entered into a participation agreement with the landlord. The agreement provides the landlord with the right to purchase any capital stock of Iomai or any options, warrants, or other securities convertible into Iomai capital stock in the Company’s next two rounds of equity financing that raise gross proceeds of at least $1.0 million (subject to certain limitations). The Series C Preferred Stock financing constituted the first of these two rounds. In the Series C financing, the landlord purchased 565,483 shares of Series Preferred Stock for $250,000 in the aggregate (see Note 6 — Redeemable Preferred Stock). In connection with the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events), the landlord agreed to terminate this participation agreement and its shares of Series C Preferred Stock were automatically converted into 43,498 shares of Common Stock (see Note 6 — Redeemable Preferred Stock).
Under the terms of the original lease agreement, the Company leased its laboratory and office facilities from June 2001 through May 31, 2006, and the Company had a three-year renewal option to extend the term through May 31, 2009. In June 2001, the landlord financed leasehold improvements of approximately $740,000, which
F-14
amount was repayable monthly through May 31, 2009 with imputed interest rate of 14% (assuming exercise of the three-year renewal option). In 2003, the Company amended its lease agreement to take over the balance of the second floor not already leased by the Company in its building. In conjunction with the lease amendment, the landlord provided the Company with a tenant improvement allowance of $29,010 and agreed to finance a portion of any additional leasehold improvements up to approximately $1.4 million. During 2004, the Company drew down from the landlord $1,450,500 to finance leasehold improvements for the build-out of its pilot plant. The repayment commenced in June 2004 and was repayable monthly through May 2009 (assuming exercise of three-year renewal option) under the following terms: (1) $870,300 of principal at an imputed interest rate of 12%, and (2) $580,200 of principal at an imputed interest rate of 10.5%.
In October 2005, the Company amended its lease to extend its term by seven years from June 2006 to May 2013. In connection with the lease extension, the Company renegotiated with the landlord the terms of the financing for the build-out of its pilot plant and agreed to lease approximately 8,000 square feet of additional space in the same building. Effective as of June 1, 2006, the then-remaining balance owed to the landlord under all of its existing debt financing, which the Company expects will total $1,342,719, will be amortized over the seven-year extension period at a rate of 10.5%. This change will result in a monthly payment of approximately $22,639 through May 2013, as opposed to the prior monthly payment of approximately $44,689, which was being amortized through May 2009. In addition, the Company now has the right to prepay this debt obligation at any time upon four months’ written notice to the landlord.
As part of the amended lease, the landlord has provided the Company with tenant improvement allowances totaling approximately $134,000 for the existing space and approximately $120,000 for the expansion space. As of December 31, 2005, the Company utilized $95,671 of these allowances. Both tenant improvement allowances are classified as other current assets in the accompanying 2005 balance sheet. The landlord has also provided us with up to $280,000 of additional financing to apply toward alterations to the expansion space on the same terms and conditions as the existing landlord financing (that is, amortized from date of draw down through May 2013 at a rate of 10.5%). The Company expects to draw down the full $280,000 in additional financing during the first half of 2006.
| |
| 6. | REDEEMABLE PREFERRED STOCK |
Series C convertible redeemable preferred stock
In December 2002, the Company issued 122,935,926 shares of Series C Redeemable Convertible Preferred Stock (Series C Preferred Stock) for $0.4421 per share for net proceeds of $52,093,856, which includes $60,000 of Series C Preferred Stock issued to an investor for professional fees. In 2003, the Company issued an additional 4,750,056 shares of Series C Preferred Stock at $0.4421 per share for gross proceeds of $2,100,000. In June 2004, the Company issued an additional 1,904,052 shares of Series C Preferred Stock at $0.4421 per share for gross proceeds of $841,781.
The Series C Preferred Stock bears a cumulative annual dividend at a rate of $0.0354 per share, prior and in preference to the Series B Preferred Stock, common stock and any other junior classes of stock.
The Series C Preferred Stock has a liquidation preference equal to the greater of (i) $0.4421 per share increased by an annual implicit growth factor of 20% compounded annually, subject to certain adjustments, plus any declared or accrued and unpaid dividends, and (ii) the amount per share that would have been payable had each such share of Series C Preferred Stock been converted into common stock immediately prior to a liquidation, dissolution, winding up, sale or merger. This liquidation preference is senior to the Series B Preferred Stock, common stock and any other junior class of stock. As of December 31, 2005 and 2004, the aggregate per share liquidation preference of the outstanding shares of Series C Preferred Stock was $0.8783 and $0.7145, respectively, which includes accrued and unpaid dividends of $13,940,759 and $9,353,272, respectively.
Holders of Series C Preferred Stock can elect at any time to convert all or part of the Series C Preferred Stock into shares of the Company’s common stock. The conversion rate is one-thirteenth ( 1/13) of one share of common stock for each share of Series C Preferred Stock, which is adjustable for certain dilutive events, as defined. In addition, upon proper notice to the Company, holders of Series C Preferred Stock may require the Company to
F-15
redeem at December 4, 2007, 2008 and 2009 the shares of Series C Preferred Stock then outstanding (prior to any conversion into common stock) for an amount equal to the purchase price of $0.4421 per share, subject to certain adjustments, plus any declared or accrued and unpaid dividends. All shares of Series C Preferred Stock were automatically converted by their terms into 9,968,443 shares of Common Stock upon closing of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events).
Each share of Series C Preferred Stock is entitled to the number of votes equal to the number of shares of common stock into which such shares are then convertible.
Series C preferred stock warrants
In connection with the Series C Preferred Stock financing, the Company issued to its placement agent warrants to purchase 250,000 shares of Series C Preferred Stock at an exercise price of $0.4421 per share. The fair value of the warrants of $60,000 was recorded as a reduction to the Series C preferred stock. The fair value was estimated using the Black-Scholes option-pricing model assuming a risk-free interest rate of 2.79%, dividend yield of 0%, volatility of 60.0%, and expected term of approximately five years. The Company accreted $12,000 of the Series C warrants to the Series C Preferred Stock in 2005 and 2004, and will continue accretion through the redemption date. The conversion rate is one-thirteenth ( 1/13) of one share of common stock for each share of Series C Preferred Stock, which is adjustable for certain dilutive events, as defined. With completion of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events), the warrant holder can now purchase up to 19,231 shares of Common Stock at an exercise price of $5.7473 per share.
Series B convertible preferred stock
In connection with the closing of the Series C Preferred Stock financing, the Company issued 14,734,578 shares of Series B Convertible Preferred Stock (Series B Preferred Stock) to a subsidiary of Elan Corporation plc (Elan) as part of the unwinding of a pre-existing joint venture. On January 5, 2006, Elan sold all of the 14,734,578 shares of our Series B Preferred Stock held by it to certain of the holders of Series C Preferred Stock pursuant to a contractual right of first refusal.
The Series B Preferred Stock bears a cumulative annual dividend at a rate of $0.0354 per share, prior and in preference to the common stock and any other junior classes of stock.
The Series B Preferred Stock has a liquidation preference equal to the greater of (i) $0.4421 per share, subject to certain adjustments, plus any declared or accrued and unpaid dividends, and (ii) the amount per share that would have been payable had each such share of Series B Preferred Stock been converted into common stock immediately prior to a liquidation, dissolution, winding up, sale or merger. This liquidation preference is prior and in preference to the common stock and any other junior classes of stock. As of December 31, 2005 and 2004, the aggregate per share liquidation preference of the outstanding shares of Series B Preferred Stock was $0.4421, plus accrued and unpaid dividends of $1,603,397 and $1,081,793, respectively.
Holders of Series B Preferred Stock can elect at any time to convert all or part of the Series B Preferred Stock into shares of the Company’s common stock. The initial conversion rate is one-thirteenth (1/13) of one share of common stock for each share of Series B Preferred Stock, which is adjustable for certain dilutive events, as defined. All Series B Preferred Stock were automatically converted into 1,133,424 shares of common stock upon closing of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events).
Each share of Series B Preferred Stock is entitled to the number of votes equal to the number of shares of common stock into which such shares are then convertible.
Stock Options
Effective January 1, 2003, the Company adopted the fair value recognition provisions of SFAS No. 123, as amended by SFAS No. 148, using the prospective adoption method (see Note 2). Under the prospective method of adoption selected by the Company under the provisions of SFAS 148, the recognition provisions were applied to all
F-16
employee awards granted, modified or settled after January 1, 2003. Therefore, the Company began to recognize compensation based on the fair value of the options at the date of grant using the Black-Scholes model.
1998 Stock Option Plan
In 1998, the Company adopted a stock option plan (the 1998 Plan) and reserved one million shares of common stock pursuant to the 1998 Plan. Incentive stock awards are granted with an exercise price equal to the fair market value of the Company’s stock on the date of grant. Nonstatutory options are granted with an exercise price at not less than 85% of the fair market value of the Company’s stock on the date of grant. The options expire 10 years from the date of grant.
1999 Stock Option Plan
In August 1999, the Company adopted the 1999 Stock Incentive Plan (the 1999 Plan). The 1999 Plan currently provides for the issuance of restricted common stock, stock options or the direct award of common stock, for up to 2,001,584 shares. Incentive stock awards are required to be granted with an exercise price equal to the estimated fair market value of the Company’s stock on the date of grant. Nonstatutory options are granted with an exercise price at not less than 85% of the fair market value of the Company’s stock on the date of grant. The 1999 Plan options expire 10 years from the date of grant.
In January 2003, the 1999 Plan was amended to increase the number of shares of Common Stock available for issuance under the 1999 Plan from 423,076 shares to 1,476,923 shares. In September 2003, the 1999 Plan was further amended to increase the number of shares of Common Stock available for issuance under the 1999 Plan by an additional 524,661 shares to 2,001,584 shares, in the aggregate.
2005 Stock Option Plan
Under the 2005 Incentive Plan (the 2005 Plan), a total of 1,040,000 shares of common stock has been reserved for issuance upon the exercise of awards granted under the 2005 Plan. The 2005 Plan provides for the grant of awards consisting of any or a combination of stock options, stock appreciation rights, restricted stock, unrestricted stock, stock unit, performance and cash awards. The compensation committee may make grants to key employees of Iomai and its affiliates, board members, including outside directors, and members of an affiliate’s board of directors, and consultants, advisors or other independent contractors who perform services for us or any of our affiliates. The maximum number of shares of stock for which options and stock appreciation rights may be granted to any particular participant in a calendar year shall be, in each case, 780,000, and the maximum number of shares of stock subject to other awards that may be delivered to any particular participant in any calendar year shall be 140,000. These limits, and the total number of shares reserved under the 2005 Plan, are subject to adjustment in the case of stock dividends and other transactions affecting the common stock. As of December 31, 2005, there were no awards outstanding under the 2005 Plan to purchase shares of common stock.
2006 Employee Stock Purchase Plan
Under the 2006 Employee Stock Purchase Plan (the ESPP), a total of 80,000 shares of common stock has been reserved for issuance under the ESPP. The ESPP will permit eligible employees to purchase shares of our common stock at a discount from fair market value. The Company intends for the ESPP to meet the requirements for an “employee stock purchase plan” under Section 423 of the Internal Revenue Code.
The ESPP did not go into effect until completion of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events), so as of December 31, 2005, no shares of Common Stock were issued under the ESPP. The ESPP will be implemented by a series of offering periods, each of which has a duration of approximately six months, commencing generally on the third Sunday of February and August of each year. The first offering will commence on February 19, 2006 and will end on August 19, 2006. The compensation committee, or a duly appointed administrator, will determine the frequency and duration of individual offerings under the ESPP and the dates when employees may purchase stock.
F-17
Eligible employees participate voluntarily and may withdraw from any offering at any time before they purchase stock. Participation terminates automatically upon termination of employment. The purchase price per share of common stock in an offering will equal 85% of the fair market value of the common stock at the first trading day of the offering period or at the last trading day of the offering period, whichever is less. Employees will pay through payroll deductions.
Additional information with respect to all stock option plan activity is summarized as follows:
| | | | | | | | | |
| | | | | | Weighted-Average
| |
| | | Shares | | | Exercise Price | |
| |
| Outstanding at December 31, 2002 | | | 235,309 | | | $ | 36.89 | |
| Granted | | | 1,279,257 | | | | 0.91 | |
| Exercised | | | (6,922 | ) | | | 0.91 | |
| Forfeited | | | (186,664 | ) | | | 40.11 | |
| | | | | | | | | |
| Outstanding at December 31, 2003 | | | 1,320,980 | | | | 1.78 | |
| | | | | | | | | |
| Exercisable at December 31, 2003 | | | 181,112 | | | | 7.26 | |
| | | | | | | | | |
| Granted | | | 277,466 | | | | 0.92 | |
| Exercised | | | (7,591 | ) | | | 0.91 | |
| Forfeited | | | (28,072 | ) | | | 0.91 | |
| | | | | | | | | |
| Outstanding at December 31, 2004 | | | 1,562,783 | | | | 1.65 | |
| | | | | | | | | |
| Exercisable at December 31, 2004 | | | 469,920 | | | | 3.36 | |
| | | | | | | | | |
| Granted | | | 433,637 | | | | 1.43 | |
| Exercised | | | (23,656 | ) | | | 0.91 | |
| Forfeited | | | (79,500 | ) | | | 0.91 | |
| | | | | | | | | |
| Outstanding at December 31, 2005 | | | 1,893,264 | | | | 1.64 | |
| | | | | | | | | |
| Exercisable at December 31, 2005 | | | 784,748 | | | | 2.38 | |
| | | | | | | | | |
Stock options granted to directors and employees generally vest in equal installments over a four-year period. The following table summarizes information about stock options outstanding under the Company’s option plans at December 31, 2005.
| | | | | | | | | | | | | | | | | | | | | |
| | | Options Outstanding | | | | | | | |
| | | | | | Weighted-Average
| | | | | | Options Exercisable | |
Range of
| | Number
| | | Remaining Contractual
| | | Weighted-Average
| | | Number
| | | Weighted-Average
| |
Exercise Prices | | Outstanding | | | Life — Years | | | Exercise Price | | | Exercisable | | | Exercise Price | |
| |
| $0.91 | | | 1,727,939 | | | | 7.5 | | | | 0.91 | | | | 735,334 | | | | 0.91 | |
| $2.86 | | | 115,911 | | | | 9.7 | | | | 2.86 | | | | — | | | | 2.86 | |
| 5.85 | | | 769 | | | | 8.9 | | | | 5.85 | | | | 769 | | | | 5.85 | |
| 16.25-22.75 | | | 32,690 | | | | 2.4 | | | | 20.84 | | | | 32,690 | | | | 20.84 | |
| 28.99-34.19 | | | 15,955 | | | | 3.9 | | | | 32.15 | | | | 15,955 | | | | 32.15 | |
| | | | | | | | | | | | | | | | | | | | | |
| | | | 1,893,264 | | | | | | | | | | | | 748,748 | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
F-18
The weighted-average fair value of options granted during the years ended December 31, 2005, 2004, and 2003 was $4.62, $0.77, and $0.49, respectively, utilizing the following weighted-average assumptions:
| | | | | | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | | | 2003 | |
| |
| Expected dividend yield | | | 0 | % | | | 0 | % | | | 0 | % |
| Expected volatility | | | 60 | % | | | 56 | % | | | 55 | % |
| Risk-free interest rate | | | 3.89 | % | | | 3.03 | % | | | 2.88 | % |
| Expected average life of options | | | 5 years | | | | 5 years | | | | 4.8 years | |
On January 23, 2003, the Company repriced options exercisable into 186,203 shares of common stock with a weighted average exercise price of $40.12. At the date of modification, these options were repriced to the then estimated fair value of the Company’s common stock of $0.91 per share. Under SFAS 123, a repricing is treated no differently than any other modification. That is, incremental compensation cost is recognized for the excess of fair value of the modified award over the value of the original option immediately before its terms are modified. The incremental cost from the repricing was $72,315, of which $47,212 was recognized immediately as this amount related to fully vested options on the repricing date.
In June 2003, the Company issued a consultant options to purchase 3,230 shares of common stock for services to be performed over a three-year period ending May 31, 2006. The fair value of the options on the date of grant was $2,100. The expense that the Company ultimately will recognize is contingent upon the value of the underlying common stock.
In November 2004, the Company issued a consultant options to purchase 769 shares of common stock for services to be performed over a four-month period ending March 15, 2005. The fair value of the options on the date of grant was $200. The expense that the Company ultimately will recognize is contingent upon the value of the underlying common stock. .
The Company recognized the following stock option compensation expense for the years ended:
| | | | | | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | | | 2003 | |
| |
| Employees and directors | | $ | 508,575 | | | $ | 152,928 | | | $ | 48,172 | |
| Non-employee consultants | | | 29,569 | | | | 777 | | | | 408 | |
| | | | | | | | | | | | | |
| | | $ | 538,144 | | | $ | 153,705 | | | $ | 48,580 | |
| | | | | | | | | | | | | |
Stock warrants
In connection with a financing arrangement with Oxford to finance the build-out of the Company’s facility (see Note 4 — Long-Term Debt), the Company issued to Oxford warrants to purchase up to 16,529 shares of common stock over a six-year period at a per share exercise price of $5.75. The exact number of shares will be equal to 1.9% of the maximum principal amount loaned to the Company by Oxford divided by the exercise price; however, in no event shall the aggregate number of warrants exceed 16,529 shares. For the years ended December 31, 2005, 2004 and 2003, the Company issued warrants of 1,548, 6,289 and 8,175, with fair values of $8,678, $818 and $1,063, respectively. In connection with the closing of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events), Oxford exercised these warrants in accordance with its terms in a cashless exercise for 2,865 shares of Common Stock.
| |
| 8. | COMMITMENTS AND CONTINGENCIES |
The Company leases laboratory and office facilities and scientific equipment under operating lease agreements. For the years ended December 31, 2005, 2004, and 2003, rent expense was $624,310, $601,391 and $482,718, and, respectively. On October 26, 2005, the Company amended its lease, among other things, to extend the term by seven years from May 2006 to May 2013. In connection with the lease extension, the Company’s annual base rent for its existing space will be reduced effective June 1, 2006 from approximately $500,000 to
F-19
approximately $455,000. With respect to the expansion space of approximately 8,000 square feet, the Company will pay approximately $27,000 during the first 5 months of 2006 and then beginning June 1, 2006, the annual base rent for this space will adjust to approximately $116,000. All base rents will be subject to minimum rent escalation of 3.5%.
Future minimum payments under capital leases and noncancelable operating lease obligations at December 31, 2005 are as follows:
| | | | | | | | | | | | | |
| | | Capital | | | Operating | | | Total | |
| |
| 2006 | | $ | 5,408 | | | $ | 570,632 | | | $ | 576,040 | |
| 2007 | | | 1,352 | | | | 584,517 | | | | 585,869 | |
| 2008 | | | — | | | | 603,607 | | | | 603,607 | |
| 2009 | | | — | | | | 624,087 | | | | 624,087 | |
| Thereafter | | | — | | | | 2,298,830 | | | | 2,297,830 | |
| | | | | | | | | | | | | |
| Total minimum lease payments | | | 6,760 | | | $ | 4,681,673 | | | $ | 4,688,433 | |
| | | | | | | | | | | | | |
| Less: amount representing interest | | | (680 | ) | | | | | | | | |
| | | | | | | | | | | | | |
| Present value of minimum lease payments | | | 6,080 | | | | | | | | | |
| Less: current portion of capital lease obligation | | | (4,840 | ) | | | | | | | | |
| | | | | | | | | | | | | |
| Long-term portion | | $ | 1,240 | | | | | | | | | |
| | | | | | | | | | | | | |
At December 31, 2005, and 2004, the Company had commitments for capital expenditures relating primarily to the expansion of the Company’s facilities of approximately $341,000 and $0, respectively.
License agreement with the Walter Reed Army Institute of Research (WRAIR)
On April 6, 2001, the Company entered into an Amended and Restated License Agreement with WRAIR (the WRAIR Agreement). The WRAIR Agreement grants an exclusive license to Iomai for its TCI technology as of December 15, 1997. The Company paid a license issuance fee of $150,000, annual license fees of $15,000 and additional fees upon certain milestones.
The Company may terminate the WRAIR Agreement at any time after 60 days’ notice to WRAIR without WRAIR’s consent. The Company did not expense any milestone payments to WRAIR in 2005, 2004, or 2003. No royalty payments have been made to WRAIR under the license.
In conjunction with the WRAIR Agreement, the Company issued 57,500 shares of common stock to WRAIR in 2001. These shares are held in escrow by an unrelated third party under a Voting Trust and Escrow Agreement, which provides economic benefit to WRAIR but removes WRAIR from any voting control or direct influence over the Company’s operations. The fair value of the shares of common stock was charged to research and development upon issuance.
In addition, the Company issued a put option (Put Option) to WRAIR related to the 57,500 shares of common stock. The put price will be the greater of $34.19 per share or the then fair market value at the date of exercise (Put Price). The Put Option may be exercised immediately should the Company fail to pay any amounts under the WRAIR Agreement or at any time after April 6, 2004. In the event that the Company is unable to purchase the shares once the Put Option is exercised by WRAIR, the Company will deliver a subordinated convertible promissory note (the Convertible Note) for the amount of the aggregate Put Price. The outstanding principal amount due under the Convertible Note would be due in full on the fifth anniversary of the issue date. Interest would accrue at prime rate plus 3% per annum. The Put Option will remain in effect as long as the 57,500 shares of common stock remain in trust or until a public offering of the Company’s common stock, as defined. Upon the closing of the Company’s initial public offering in February 2006 (see Note 12 — Subsequent Events), the Put Option terminated in accordance with its terms without further financial obligation to the company.
F-20
For the years ending December 31, 2005, 2004, and 2003, there is no current provision for federal or state income taxes. The deferred tax benefit has been entirely offset by valuation allowances.
The Company’s net deferred tax asset consists of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2004 | |
| |
| NOL carryforward | | $ | 21,238,530 | | | $ | 14,889,823 | |
| Research and development credit carryforward | | | 1,648,323 | | | | 1,149,824 | |
| Deferred temporary differences | | | 1,198,211 | | | | 283,867 | |
| Valuation allowance | | | (24,085,064 | ) | | | (16,323,514 | ) |
| | | | | | | | | |
| Net deferred tax asset | | $ | — | | | $ | — | |
| | | | | | | | | |
The net operating loss carryforwards of approximately $55.0 million will begin to expire in the year 2017 if unused. The use of the Company’s net operating loss carryforwards may be restricted due to changes in Company ownership. The Company paid no income taxes in 2002, 2003, and 2004.
The provision for income taxes differs from the amount of taxes determined by applying the U.S. federal statutory rate to loss before provision for income taxes as a result of the following:
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2005 | | | 2004 | | | 2003 | |
| |
| Federal tax at statutory rates | | | 34.0 | % | | | 34.0 | % | | | 34.0 | % |
| State taxes, net of federal benefit | | | 4.6 | | | | 4.6 | | | | 4.6 | |
| Research and development credit | | | 2.8 | | | | 2.5 | | | | 1.7 | |
| Permanent difference | | | (1.2 | ) | | | (0.1 | ) | | | (0.1 | ) |
| Change in valuation allowance | | | (40.2 | ) | | | (41.0 | ) | | | (40.2 | ) |
| | | | | | | | | | | | | |
| Provision for income taxes | | | 0.0 | % | | | 0.0 | % | | | 0.0 | % |
| | | | | | | | | | | | | |
| |
| 10. | EMPLOYEE BENEFIT PLAN |
The Company maintains a defined contribution plan (the 401(k) Plan) qualified under Section 401(k) of the Internal Revenue Code. The 401(k) Plan covers substantially all employees. Under the 401(k) Plan, employees may make elective salary deferrals and the Company provides for a 50% matching of qualified deferrals, up to a maximum of 6%. During the years ended December 31, 2005, 2004, and 2003, the Company made matching contributions of approximately $132,999, $122,883 and $101,988, respectively.
F-21
| |
| 11. | QUARTERLY FINANCIAL INFORMATION (UNAUDITED) |
Quarterly financial information for the years ended December 31, 2003 and 2004 is presented in the following tables:
| | | | | | | | | | | | | | | | | |
| | | 1st Quarter | | | 2nd Quarter | | | 3rd Quarter | | | 4th Quarter | |
| |
| Year ended December 31, 2004 Revenue | | | 676,404 | | | | 1,036,097 | | | | 531,671 | | | | 100,601 | |
| Income (loss) from operations | | | (3,494,362 | ) | | | (2,837,814 | ) | | | (4,316,508 | ) | | | (4,561,430 | ) |
| Net income (loss) | | | (3,400,363 | ) | | | (2,737,780 | ) | | | (4,353,170 | ) | | | (4,588,967 | ) |
| Net income (loss) per share, basic and diluted | | $ | (4.45 | ) | | $ | (3.58 | ) | | $ | (5.68 | ) | | $ | (5.97 | ) |
| | | | | | | | | | | | | | | | | |
| Year ended December 31, 2005 Revenue | | $ | 970,895 | | | $ | 855,604 | | | $ | 503,705 | | | $ | 41,256 | |
| Income (loss) from operations | | | (3,648,934 | ) | | | (3,681,057 | ) | | | (4,342,698 | ) | | | (6,265,741 | ) |
| Net income (loss) | | | (3,669,210 | ) | | | (3,694,688 | ) | | | (4,364,552 | ) | | | (6,301,277 | ) |
| Net income (loss) per share, basic and diluted | | $ | (4.75 | ) | | $ | (4.78 | ) | | $ | (5.52 | ) | | $ | (7.94 | ) |
| | | | | | | | | | | | | | | | | |
Initial Public Offering
On February 1, 2006, the Company’s common stock began trading on The NASDAQ National Market under the symbol “IOMI.” The Company raised $35,000,000 in gross proceeds in its initial public offering by selling 5,000,000 shares of its common stock at a public offering price of $7.00 per share. Upon completion of this offering, the Company had 16,900,251 shares of Common Stock outstanding after giving effect to the automatic conversion of the Company’s Series B and C Preferred Stock, the cashless exercise of the warrant to purchase common stock for 2,865 shares by the equipment financing company, and the issuance of 5,000,000 shares of common stock in the initial public offering.
Our net proceeds, before expenses, were $32,853,574 and underwriting discounts and commissions were $2,146,426. We estimate that the Company’s expenses for the initial public offering, including legal, accounting and printing fees, will be approximately $2.0 million.
Facility Sublease
In February 2006, the Company entered into a full-service sublease for an additional 5,572 square feet of laboratory and office space in the Company’s building. The sublease commenced in mid-March and expires February 28, 2007, with total rent cost and expenses during the term of approximately $107,000.
F-22