EXHIBIT 1

RENEWAL ANNUAL INFORMATION FORM
for the Year Ended December 31, 2006
March 2, 2007
Item 7A (1/46)
TABLE OF CONTENTS
| Page | |||
|---|---|---|---|
CORPORATE STRUCTURE | 4 | ||
| GENERAL DEVELOPMENT OF THE BUSINESS | 4 | ||
| General | 4 | ||
| Clinical Trials | 5 | ||
| Manufacturing | 10 | ||
| Financings and Other Distributions | 10 | ||
| Shareholdings in Other Issuers | 10 | ||
| Publications and Presentations | 10 | ||
| Patents | 11 | ||
| Recent Developments | 11 | ||
| Future Developments | 12 | ||
| NARRATIVE DESCRIPTION OF THE BUSINESS | 13 | ||
| Our Business | 13 | ||
| Scientific Background | 14 | ||
| The Potential Cancer Product | 15 | ||
| Repayable Grants | |||
| Business Strategy | 15 | ||
| Regulatory Requirements | 16 | ||
| Market and Competition | 17 | ||
| Product Marketing Strategy | 17 | ||
| Third Party Advisor, Collaborators and Scientific Advisory Board | 18 | ||
| Intellectual Property Policy | 18 | ||
| Patent and Patent Application Summary | 19 | ||
| Acquisition of all of the Shares of Oncolytics Biotech Inc by SYNSORB | 22 | ||
| Employees | 22 | ||
| Research and Development Expenditures | 22 | ||
| Dividend Policy | 22 | ||
| MARKET FOR SECURITIES | 23 | ||
| Market for Common Shares | 23 | ||
| Description of Common Shares | 23 | ||
| DIRECTORS AND OFFICERS | 23 | ||
| AUDIT COMMITTEE MATTERS | 27 | ||
| Mandate of the Audit Committee | 27 | ||
| Composition of the Audit Committee | 33 | ||
| RISK FACTORS | 34 | ||
| ADDITIONAL INFORMATION | 41 | ||
| Legal Proceedings | 41 | ||
| Interest of Management and Others in Material Transactions | 41 | ||
| Transfer Agent and Registrar | 41 | ||
-2-
| Page | |||
|---|---|---|---|
| Material Contracts | 41 | ||
| Interests of Experts | 41 | ||
| External Auditor Service Fees | 41 | ||
| Audit Fees | 42 | ||
| Audit-Related Fees | 42 | ||
| Tax Fees | 42 | ||
| All Other Fees | 42 | ||
| Other Additional Information | 42 | ||
| GLOSSARY | 44 | ||
This Annual Information Form contains forward-looking statements, including statements that may contain words such as “anticipate,” “estimate,” expect,” “project,” “intend,” “plan,” “believe” and similar expressions and statements relating to matters that are not historical facts. Forward-looking statements, including our belief as to the potential of REOLYSIN® as a cancer therapeutic and our expectations as to the success of our research and development and manufacturing programs in 2007 and beyond, future financial position, business strategy and plans for future operations, and statements that are not historical facts, involve known and unknown risks and uncertainties, which could cause our actual results to differ materially from those in the forward-looking statements. Such risks and uncertainties include, among others, the need for and availability of funds and resources to pursue research and development projects, the efficacy of REOLYSIN® as a cancer treatment, the success and timely completion of clinical studies and trials, our ability to successfully commercialize REOLYSIN®, uncertainties related to the research, development and manufacturing of pharmaceuticals, uncertainties related to competition, changes in technology, the regulatory process and general changes to the economic environment. Investors should consult our quarterly and annual filings with the Canadian and U.S. securities commissions for additional information on risks and uncertainties relating to the forward-looking statements. Forward-looking statements are based on assumptions, projections, estimates and expectations of management at the time such forward-looking statements are made, and such assumptions, projections, estimates and/or expectations could change or prove to be incorrect or inaccurate. Investors are cautioned against placing undue reliance on forward-looking statements. We do not undertake to update these forward-looking statements.
CORPORATE STRUCTURE
Oncolytics Biotech Inc. was incorporated pursuant to the provisions of the ABCA on April 2, 1998 as 779738 Alberta Ltd. On April 8, 1998, we amended our articles and changed our name to Oncolytics Biotech Inc. On July 29, 1999, we further amended our articles by removing the private company restrictions and subdividing our issued and outstanding 2,222,222 common shares to create 6,750,000 common shares. Our head office and principal place of business is located at 210, 1167 Kensington Crescent N.W., Calgary, Alberta T2N 1X7. Our registered office is located at 4500 Bankers Hall East, 855 – 2nd Street S.W., Calgary, Alberta T2P 4K7.
GENERAL DEVELOPMENT OF THE BUSINESS
General
We focus on the discovery and development of oncolytic viruses for the treatment of cancers that have not been successfully treated with conventional therapeutics. Recent scientific advances in oncology, virology, and molecular biology have created opportunities for new approaches to the treatment of cancer. The product we are presently developing may represent a novel treatment for Ras-mediated cancers which can be used as an alternative to existing cytotoxic or cytostatic therapies or as an adjuvant therapy to conventional chemotherapy, radiation therapy, or surgical resections. It could also potentially be used to treat certain cellular proliferative disorders for which no current therapy exists.
Our technologies are based primarily on discoveries in the Department of Microbiology and Infectious Diseases at the University of Calgary in the 1990s. Oncolytics Biotech Inc. was formed in 1998 to explore the natural oncolytic capability of the reovirus, a virus that preferentially replicates in cells with an activated Ras pathway.
The lead product being developed by us may represent a novel treatment for certain tumour types and some cellular proliferative disorders. Our lead product is a virus that is able to replicate specifically in, and hence kill, certain tumour cells both in tissue culture as well as in a number of animal models without damaging normal cells. See “Narrative Description of the Business – Our Business; Scientific Background.”
We are also assessing the potential opportunities for product candidates resulting from issued patents received for Ras-targeted adenovirus and herpes virus.
Item 7A (4/46)
-5-
Clinical Trials
On December 22, 2006, we reported that we had received a letter of approval from the U.K. Medicines and Healthcare products Regulatory Agency (“MHRA”) for our Clinical Trial Application to begin a clinical trial using intravenous administration of REOLYSIN® in combination with paclitaxel and carboplatin in patients with advanced cancers including melanoma, lung, and ovarian. The combination of paclitaxel and carboplatin chemotherapy is used in cancer patients with ovarian and lung cancers, and is also used widely in the treatment of many other types of cancer.
In studies conducted by the U.S. National Cancer Institute (“NCI”), the combination of REOLYSIN® and paclitaxel was uniformly synergistic against six non-small cell lung cancer cell lines examined, including cell lines resistant to paclitaxel or REOLYSIN®. Preclinical studies conducted at Cornell University also found that REOLYSIN® in combination with platinum drugs enhanced the cytotoxicity of the chemotherapeutic agents.
This trial has two components. The first is an open-label, dose-escalating, non-randomized study of REOLYSIN® given intravenously with paclitaxel and carboplatin every three weeks. Standard dosages of paclitaxel and carboplatin will be delivered with escalating dosages of REOLYSIN® intravenously. A maximum of three cohorts will be enrolled in the REOLYSIN® dose escalation portion. The second component of the trial will immediately follow and will include the enrolment of a further 12 patients at the maximum dosage of REOLYSIN® in combination with standard dosages of paclitaxel and carboplatin.
Eligible patients include those who have been diagnosed with advanced or metastatic solid tumours such as melanoma, lung and ovarian cancers that are refractory (have not responded) to standard therapy or for which no curative standard therapy exists. The primary objective of the trial is to determine the maximum tolerated dose (“MTD”), dose limiting toxicity (“DLT”), recommended dose and dosing schedule and safety profile of REOLYSIN® when administered in combination with carboplatin and paclitaxel. Secondary objectives include the evaluation of immune response to the drug combination, the body’s response to the drug combination compared to chemotherapy alone and any evidence of anti-tumour activity.
On December 22, 2006, we commenced patient enrolment in our U.K. Phase II clinical trial to evaluate the anti-tumour effects of direct injection of REOLYSIN® in combination with low-dose radiation in patients with advanced cancers.
On November 9, 2006, we reported additional clinical results from our first systemic administration study. Results of our UK Phase I systemic administration clinical trial investigating the use of REOLYSIN® to treat patients with advanced cancers indicated that REOLYSIN® can be delivered systemically to various tumour types and cause virus-mediated tumour responses. A total of 33 patients were treated in the trial to a maximum daily dose of 1x1011 TCID50. A maximum tolerated dose was not reached and the treatment appears to have been well tolerated by the patients.
Toxicities possibly related to REOLYSIN® treatment in this trial have generally been mild (grade 1 or 2) and have included chills, fever, headache, cough, runny nose, sore throat and fatigue. Transient grade 3 toxicities include lymphopenia, neutropenia and troponin I. These symptoms were more frequently observed from day two of treatment and usually lasted less than six hours.
Of the patients assessed, anti-tumour activity was noted in seven patients. Patients were assessed with CT scans, and where possible tumour marker assessment, and histopathology of tumour biopsies. Two patients with colorectal cancer had tumour stabilization (one for three months, the other classified as stable disease for six months) and had CEA tumour marker reduction of 27% and 60% respectively. One
-6-
patient with metastatic prostate cancer had stable disease for four months, had a 50% decrease in PSA, and had extensive product-induced necrosis with associated intratumoural viral replication in metastatic lesions in the lymph nodes. One patient with metastatic bladder cancer had stable disease for four months and had a minor tumour response in a metastatic lesion in a lymph node (reduction from 2.5 to 1.9 cm). A patient with pancreatic cancer and a patient with lung cancer had stable disease for four months. A patient with endometrial cancer had stable disease for five months
On August 23, 2006, we reported that we had completed patient enrolment in our Phase I U.S. clinical trial investigating the systemic delivery of REOLYSIN® to treat patients with advanced cancers. A total of 18 patients were treated in the Phase I trial with REOLYSIN® at escalating dosages of 1x108, 3x108, 1x109, 3x109, 1x1010 or 3x1010 TCID50. A maximum tolerated dose was not reached and the treatment appears to have been well tolerated by the patients.
On July 18, 2006, we reported that we had received a letter of approval from the MHRA for our Clinical Trial Application to begin a Phase II clinical trial to evaluate the anti-tumour effects of intratumoural administration of REOLYSIN® in combination with low-dose radiation in patients with advanced cancers.
The trial is an open-label, single-arm, multi-centre Phase II study of REOLYSIN® delivered via intratumoural injection to patients during treatment with low-dose radiotherapy. Up to 40 evaluable patients, including approximately 20 patients with head and neck and esophageal cancers, and approximately 20 patients with other advanced cancers, will be treated with two intratumoural doses of REOLYSIN® at 1x1010 TCID50 with a constant localized radiation dose of 20 Gy in five consecutive daily fractions. Eligible patients include those who have been diagnosed with advanced or metastatic cancers including head, neck and esophageal tumours that are refractory (have not responded) to standard therapy or for which no curative standard therapy exists.
The primary objective of this trial is to assess the anti-tumour activity of the combination of REOLYSIN® and low dose radiotherapy in treated and untreated lesions. Secondary objectives include the evaluation of viral replication, immune response to the virus and to determine the safety and tolerability of intratumoural administration of REOLYSIN® in patients with advanced cancers who are receiving radiation treatment.
On July 11, 2006, we began patient enrolment in our clinical trial to investigate the use of REOLYSIN® for patients with recurrent malignant gliomas in the U.S. This clinical trial is an open-label dose escalation Phase I/II trial in which a single dose of REOLYSIN® is administered by infusion to patients with recurrent malignant gliomas that are refractory to standard therapy. The administration involves the stereotactically-guided placement of a needle into the tumour, through which REOLYSIN® is administered or infused into the tumour mass and surrounding tissue using a pump.
The primary objective of the study is to determine the MTD, DLT and safety profile of REOLYSIN®. Secondary objectives include the evaluation of viral replication, immune response to the virus and any evidence of anti-tumour activity.
On July 10, 2006, we commenced patient enrolment in our Phase Ib U.K. clinical trial investigating REOLYSIN® in combination with radiation therapy as a treatment for patients with advanced cancers. The Phase Ib trial will treat patients with a range of two to six intratumoural doses of REOLYSIN® at 1x1010 TCID50 with a constant radiation dose of 36 Gy in 12 fractions.
The primary objective of the Phase Ib trial is to determine the MTD, DLT, and safety profile of REOLYSIN® when administered intratumourally to patients receiving radiation treatment. A secondary objective is to examine any evidence of anti-tumour activity. Eligible patients include those who have been diagnosed with advanced or metastatic solid tumours that are refractory (have not responded) to
-7-
standard therapy or for which no curative standard therapy exists. An additional group of patients is planned to be treated at the maximum dose regimen reached in the Ib trial.
On June 20, 2006, we reported that we had completed patient enrolment in our Phase Ia U.K. clinical trial investigating the use of REOLYSIN® in combination with radiation to treat patients with advanced cancers. A total of 11 patients were treated in the Phase Ia trial with two intratumoural treatments of REOLYSIN® at dosages of 1x108, 1x109, or 1x1010 TCID50 with a constant localized radiation dose of 20 Gy in five fractions. A maximum tolerated dose was not reached and the combination treatment appears to have been well tolerated by the patients.
On June 5, 2006, positive interim and final results for two Phase I clinical trials were reported at poster sessions at the American Society of Clinical Oncology (“ASCO”) Annual Meeting in Atlanta, Georgia. The data covered interim results of our Phase I systemic administration trial being conducted in the U.K. and final results of our Phase I recurrent malignant glioma trial conducted in Canada.
Interim results of our UK Phase I systemic administration clinical trial investigating the use of REOLYSIN® to treat patients with advanced cancers indicated that REOLYSIN�� can be delivered systemically to various tumour types and cause virus-mediated tumour responses. A total of 30 patients had been treated in the escalating frequency and dosage portion of the trial to a maximum daily dose of 1x1011 TCID50. A maximum tolerated dose was not reached and the treatment appears to have been well tolerated by the patients. Toxicities possibly related to the REOLYSIN® treatment in this trial were generally mild (grade 1 or 2) and included chills, fever, headache, cough, runny nose, sore throat and fatigue. Transient grade 3 toxicities included lymphopenia, neutropenia and troponin I. These symptoms were more frequently observed from day two of treatment and usually lasted less than six hours.
Of the cohorts whose patients had completed treatment (seven), anti-tumour activity was noted in patients with colorectal, prostate, pancreatic, bladder, and lung cancer. Patients were assessed with CTR scans, and where possible tumour marker assessment, and histopathology of tumour biopsies. Two patients with colorectal cancer had tumour stabilization (one for three months, the other classified as stable disease at six months) and had CEA tumour marker reduction of 27% and 60% respectively. One patient with metastatic prostate cancer had stable disease at four months, had a 50% decrease in PSA, and had extensive product-induced necrosis with associated intratumoural viral replication in metastatic lesions in the lymph nodes. One patient with metastatic bladder cancer had stable disease at four months and had a minor tumour response in a metastatic lesion in a lymph node (reduction from 2.5 to 1.9 cm). A patient with pancreatic cancer and a patient with lung cancer had stable disease at four months.
As well, final results of our Canadian Phase I trial for recurrent malignant glioma indicated that intratumoural administration of REOLYSIN® was well tolerated by the patients and a maximum tolerated dose was not reached. A total of 12 patients were treated with a single, intratumoural injection of REOLYSIN® at dosages of 1x107, 1x108, and 1x109 TCID50 in a delivery volume of 0.9 ml.
Toxicities possibly related to the REOLYSIN® treatment in this trial were generally mild (grade 1 and 2) and included fever, headache and neutropenia. A transient grade 3 elevation in GGT was noted. Three patients lived longer than one year and one of these patients was still alive approximately 45 months post-treatment.
On April 4, 2006, we reported interim results of our Phase I combination REOLYSIN®/radiation clinical trial. The interim results indicated that the combination of intratumoural REOLYSIN® and radiation was well tolerated and that both local clinical responses and early indications of systemic effects were observed. Preliminary observations in the first seven patients showed that the combination of intratumoural REOLYSIN® and radiation have been well-tolerated. Most toxicities have been mild, generally grade 1 and 2, and include fever, sweating and skin erythema. One patient in the second cohort developed grade 3 fatigue and grade 2 flu-like symptoms and could not receive the second REOLYSIN®
-8-
injection. There has been no evidence that the REOLYSIN® injections exacerbated the acute reactions expected from the radiation. There was also no evidence of viral shedding in the blood, urine, stool or sputum on day eight post-REOLYSIN® injection. Interim analysis also showed evidence of local responses and an indication of systemic effects. Amongst the first five patients that completed treatment, three patients had partial tumour responses. At the time of these interim results, there was one case of progressive disease at one month, one case of stable disease at one month, two cases of partial responses at one, two and three months and one case of stable disease at one and two months, which became a pathological partial response at three months. CT scans from the treated lymph node tumour in the first patient in the trial clearly show the partial response, which as of date of the interim results, had lasted for over eight months. A metastatic tumour in this patient that was outside the radiation field also showed a partial response.
On January 18, 2006, we reported that the Cancer Therapy Evaluation Program (“CTEP”), part of the U.S. NCI, had issued a solicitation for Letters of Intent with respect to the conduct of two human clinical trials using REOLYSIN®. The first trial is a Phase II study of REOLYSIN® administered systemically in patients with melanoma and the second is a Phase I/II study of REOLYSIN® co-administered both systemically and intraperitoneally in patients with ovarian cancer. The NCI initially approved REOLYSIN® for collaborative development after an analysis of preclinical, GLP toxicology and clinical data.
On January 17, 2006, we reported that the six-month follow up period had concluded for patients in our Phase I Canadian recurrent malignant glioma clinical trial. As reported in October 2005, intratumoural administration of reovirus was well tolerated by the patients and a maximum tolerated dose was not reached. The study examined the use of a single, intratumoural injection of REOLYSIN®, delivered using imaging-guided surgery, in patients with malignant gliomas that had recurred despite other treatments, including surgery and radiation therapy. A total of 12 patients were treated in the study at dosages of 107, 108, and 109 TCID50 in a delivery volume of 0.9 ml.
On November 14, 2005, we reported results in conjunction with a poster presentation at the AACR-NCI-EORTC conference in the U.S. by our principal investigator for our Phase I systemic delivery clinical trial in the U.K. We reported on 22 patients who made up seven of nine potential cohorts with each cohort receiving escalating dose levels of REOLSYIN®. Our principal investigator reported that REOLYSIN® is well tolerated when administered intravenously with minimal toxicity observed and that reovirus replication in tumours has been identified with evidence of tumor necrosis. The principal investigator also reported that there have been encouraging hints of activity in prostate and colorectal cancer.
On October 19, 2005, we announced that we had ended patient treatment in our Canadian Phase I clinical trial investigating the use of REOLYSIN® to treat patients with recurrent malignant glioma. A total of twelve patients were treated in the study at dosages of 107, 108, and 109 TCID50. A maximum tolerated dose was not reached over this dosage range and REOLYSIN® appears to have been well tolerated by the patients. Determination of the safety of REOLYSIN® was the primary purpose of this Canadian Phase I study. The study examined the use of a single, intratumoural injection of REOLYSIN®, delivered using imaging-guided surgery, in patients with malignant gliomas that had recurred despite other treatments, including surgery and radiation therapy. After treatment with REOLYSIN®, the Phase I patients were monitored and evaluated for safety for a period of six months.
On April 7, 2005, we announced that we received clearance from the US FDA to begin a Phase I clinical trial to investigate the systemic delivery of REOLYSIN® as a treatment for patients with advanced or metastatic solid tumours. This clinical trial is an open-label, dose-escalation Phase I study in which a single dose of REOLYSIN® was administered intravenously to patients diagnosed with selected advanced or metastatic solid tumours that are refractory (have not responded) to standard therapy or for which no
-9-
curative standard therapy exists. The primary objective of the study was to determine the MTD, DLT and safety profile of REOLYSIN®. Secondary objectives included the evaluation of viral replication, immune response to the virus and any evidence of anti-tumour activity. The enrolment in this study, which commenced on September 28, 2005, was expected to be up to 36 evaluable patients and depended in part upon the number of cohorts tested.
On February 28, 2005, we announced that we had received clearance from the US FDA to begin a Phase I/II clinical trial to investigate the use of REOLYSIN® to treat patients with recurrent malignant gliomas. This clinical trial is an open-label dose escalation Phase I/II study in which a single dose of REOLYSIN® is administered by infusion to patients with recurrent malignant gliomas that are refractory to standard therapy. The administration involves the stereotactically-guided placement of a needle into the tumour, through which REOLYSIN® is administered or infused into the tumour mass and surrounding tissue using a pump. The primary objective of the study is to determine the MTD, DLT and safety profile of REOLYSIN®. Secondary objectives include the evaluation of viral replication, immune response to the virus and any evidence of antitumour activity. The enrolment in this study is expected to be up to 30 evaluable patients in the dose escalation phase with up to an additional 14 patients added at the MTD.
On February 18, 2005, we announced that we received a letter of approval from the U.K. MHRA for our Clinical Trial Application (“CTA”) to begin a Phase I clinical trial to evaluate the feasibility, safety and anti-tumour effects of intratumoural administration of REOLYSIN® in combination with radiation in patients with advanced cancers. The trial is a Phase I open-label, dose-escalation study of REOLYSIN® combined with two different radiation dosages/schedules. The enrolment in this study is expected to be approximately 30 evaluable patients and will depend upon the number of dose levels tested. Up to an additional 15 patients are expected to be treated at the MTD. The primary objective of the study is to determine the MTD, DLT, and safety profile of REOLYSIN® when administered intratumourally to patients receiving radiation treatment. A secondary objective is to examine any evidence of anti-tumour activity. Patients who have been diagnosed with advanced or metastatic solid tumours that are refractory (have not responded) to standard therapy or for which no curative standard therapy exists will be eligible. Patient enrolment commenced on July 26, 2005.
On February 27, 2004, we announced that we received approval to commence a Phase I clinical trial to investigate the systemic delivery of REOLYSIN® as a treatment for patients with advanced or metastatic solid tumours from the MHRA in the United Kingdom. The principal investigator for the study is Dr. J. de Bono of the Royal Marsden Hospital in London, England. This clinical trial is the first to examine the systemic delivery of REOLYSIN®, which is expected to result in delivery of the virus throughout the body to both the primary tumour and metastatic disease sites. The trial is an open-label, dose escalation Phase I study in which REOLYSIN® is administered intravenously to patients diagnosed with advanced or metastatic solid tumours that are refractory (have not responded) to standard therapy or for which no curative standard therapy exists. The primary objective of the study is to determine the MTD, DLT and safety profile of REOLYSIN®. Secondary objectives include the evaluation of viral replication, immune response to the virus and any evidence of anti-tumour activity. The enrolment in this study is expected to be up to 40 evaluable patients and will depend upon the number of dose levels tested.
On February 27, 2004, we announced the final update on our technical clinical study for T2 prostate cancer. On March 23, 2003, we previously reported results from an interim assessment of this clinical trial. These results were presented by Dr. Don Morris, from the Alberta Cancer Board, the principal investigator for the trial. Dr. Morris reported that there was evidence of viral activity in five of six patients and there were no safety concerns, from either a clinical or histopathological perspective, in all six patients reported upon. The data in four of the six patients, showed clear histopathological evidence of apoptotic tumour cell death (one measure of viral activity). In a fifth patient, the PSA level dropped by 53% and the prostate gland shrunk by 67% from the period of time prior to treatment to the time of surgical removal. There was no evidence of viral activity in the sixth patient. In all six patients, there was
-10-
no histopathological evidence of any viral effect on healthy prostate tissue. This trial, which was approved by Health Canada on October 11, 2001, was primarily a technical study designed to allow us to measure overall tumour response and examine changes or effects inside the tumour and in surrounding normal tissue, as part of a human clinical trial.
Manufacturing
We engage a toll manufacturer, Cobra Biomanufacturing Plc for the production of reovirus for human clinical trials and animal toxicology studies. The product is produced in compliance with current regulatory requirements and the manufacturer will confirm biosafety testing. See “Risk Factors – Manufacturing.”
Financings and Other Distributions
Since inception we have raised net cash proceeds of $84,139,357 through public offerings, private placements and the exercise of warrants and options.
Shareholdings in Other Issuers
During 2006, we sold no common shares of BCY LifeSciences Inc. (2005 – 120,000; 2004 – 697,945) receiving no cash proceeds (2005 – $7,965; 2004 – $133,609) with no reported gain on sale of shares in 2006 (2005 – $765; 2004 – $34,185). As at December 31, 2006, our remaining ownership in BCY LifeSciences Inc. was 80,000 common shares.
Publications and Presentations
During 2006, in conjunction with our various collaborators, we reported the results of a number of research collaborations. A poster at the British Society of Gene Therapy 3rd annual conference in London U.K was presented. Our investigator concluded that immune interventions which prolong local viral replication, and/or enhance levels of tumour specific T cells, should have significant therapeutic impacts both against the local, injected tumour and against systemic metastatic disease not accessible to direct viral injection.
As well, a poster by Dr. E. Anders Kolb was presented at the AACR annual meeting in Washington D.C. The investigators tested reovirus against various pediatric sarcoma cell lines in vitro and in vivo. In all tumour lines evaluated, the reovirus exhibited significant antitumour activity. The investigators concluded that REOLYSIN® demonstrates excellent anti-tumor activity in vitro and in vivo in childhood sarcoma cell lines, and that these promising results suggest that a clinical trial of systemic reovirus in pediatric solid tumours is warranted.
Finally, in the fourth quarter of 2006, Dr. Shizuko Sei of SAIC-Frederick, Inc., prime contractor to the U.S. NCI at Frederick presented a poster at the 18th EORTC-NCI-AACR symposium on Molecular Targets and Cancer Therapeutics. The research focused on work conducted by the NCI with reovirus in combination with a number of common chemotherapeutic agents. In general, the combination of reovirus with cisplatin, gemcitabine, mitomycin or vinblastine was synergistic against lung cancer cell lines sensitive to anti-cancer drugs. Of particular interest to the researchers, the combination of reovirus and paclitaxel was uniformly synergistic in all six cell lines examined, including in those with high-level resistance to paclitaxel or reovirus. The data suggest that the combination of reovirus and paclitaxel may help in promoting cell-death signaling, resulting in a more efficient and synergistic anti-cancer effect against these cell lines than using each agent on its own.
-11-
On November 15, 2005, a poster entitled “Reovirus enhances radiation cytotoxicity in vitro and in vivo” was presented by Dr. Kevin J. Harrington of the Targeted Therapy Laboratory, The Institute of Cancer Research, Cancer Research UK Centre for Cell and Molecular Biology, UK.
On April 20, 2005, a poster by Errington et al. entitled “Potential role for reovirus in the treatment of melanoma: Targeted killing and immune stimulation” was presented at the 96th Annual Meeting of the American Association for Cancer Research in Anaheim, CA. USA. This research supports the use of reovirus as a targeted therapy for melanomas. The research also showed that activation of certain classes of immune cells by the reovirus may enable additional anti-tumour activity beyond that of tumour cell death caused directly by the reovirus.
On December 15, 2004, “Efficacy and Safety Evaluation of Human Reovirus Type 3 in Immunocompetent Animals” was published in Clinical Cancer Research Vol. 10. This publication concluded that the data showed the efficacy and safety of reovirus when it is used in the treatment of gliomas in immunocompetent hosts. As well, inoculation of reovirus into the brain of nonhuman primates did not produce significant toxicities.
On September 30, 2004, a poster was presented at the 16th EORTC-NCI-AACR 2004 Symposium on ‘Molecular Targets and Cancer Therapeutics’ in Geneva, Switzerland entitled “The oncolytic reovirus, Reolysin, augments the anticancer effects of cytotoxic agents in vitro against the ras-mutated human colon cancer cell line HCT 116.” The researchers were able to show that REOLYSIN® enhances the cytotoxicity of chemotherapeutic agents including 5-FU, gemcitabine, doxorubicin and cisplatin.
Patents
We secured four U.S. patents and two Canadian patents in 2006. These patents enhance and expand intellectual property coverage in several areas, including manufacturing, methods of identifying patients who may respond well to reovirus treatment, using additional molecular mechanisms that can lead to reovirus susceptibility in cancer cells, and manipulation of the immune system that may enhance the effectiveness of REOLYSIN® treatment. Currently, our patent portfolio includes 18 U.S. patents, five Canadian patents and three European patents. In addition, we have a number of other patents under application, both in the United States, and through filings under the Patent Cooperation Treaty. See “Narrative Description of the Business – Patent and Patent Application Summary.”
Recent Developments
U.K. REOLYSIN® in Combination with Docetaxel
On January 3, 2007, we received a letter of approval from the U.K. MHRA for our CTA to begin a clinical trial using intravenous administration of REOLYSIN® in combination with docetaxel (Taxotere®) in patients with advanced cancers including bladder, prostate, lung and upper gastro-intestinal. The principal investigator is Professor Hardev Pandha of The Royal Surrey Hospital, U.K. Docetaxel is used in patients with lung, breast and prostate cancers, and is also used widely in the treatment of many other types of cancers.
The trial has two components. The first is an open-label, dose-escalating, non-randomized study of REOLYSIN® given intravenously with docetaxel every three weeks. A standard dosage of docetaxel will be delivered with escalating dosages of REOLYSIN® intravenously. A maximum of three cohorts will be enrolled in the REOLYSIN® dose escalation portion. The second component of the trial will immediately follow and will include the enrolment of a further 12 patients at the maximum dosage of REOLYSIN® in combination with a standard dosage of docetaxel.
-12-
Eligible patients include those who have been diagnosed with advanced or metastatic solid tumours such as bladder, lung, prostate or upper gastro-intestinal cancers that are refractory to standard therapy or for which no curative standard therapy exists. The primary objective of the trial is to determine the MTD, DLT, recommended dose and dosing schedule and safety profile of REOLYSIN® when administered in combination with docetaxel. Secondary objectives include the evaluation of immune response to the drug combination, the body’s response to the drug combination compared to chemotherapy alone and any evidence of anti-tumour activity.
U.K. REOLYSIN® in Combination with Gemcitabine
On January 3, 2007, we received a letter of approval from the U.K. MHRA for our CTA to begin a clinical trial using intravenous administration of REOLYSIN® in combination with gemcitabine (Gemzar®) in patients with advanced cancers including pancreatic, lung and ovarian. The principal investigators are Dr. Johann de Bono of The Royal Marsden NHS Foundation Trust and The Institute of Cancer Research, London and Professor Jeff Evans of the University of Glasgow and the Beatson Oncology Centre in Glasgow, Scotland. Gemcitabine is used in patients with lung, pancreatic and ovarian cancers and is also used widely in the treatment of many other types of cancers.
This trial has two components. The first is an open-label, dose-escalating, non-randomized study of REOLYSIN® given intravenously with gemcitabine every three weeks. A standard dosage of gemcitabine will be delivered with escalating dosages of REOLYSIN® intravenously. A maximum of three cohorts will be enrolled in the REOLYSIN® dose escalation portion. The second component of the trial will immediately follow and will include the enrolment of a further 12 patients at the maximum dosage of REOLYSIN® in combination with a standard dosage of gemcitabine.
Eligible patients include those who have been diagnosed with advanced or metastatic solid tumours including pancreatic, lung and ovarian cancers that are refractory to standard therapy or for which no curative standard therapy exists. The primary objective of the trial is to determine the MTD, DLT, recommended dose and dosing schedule and safety profile of REOLYSIN® when administered in combination with gemcitabine. Secondary objectives include the evaluation of immune response to the drug combination, the body’s response to the drug combination compared to chemotherapy alone and any evidence of anti-tumour activity.
Financing Activity
On February 22, 2007, we issued 4,000,000 units at a price of $3.00 per unit for net proceeds of approximately $10,590,000. Each whole common share purchase warrant shall entitle the holder thereof to acquire one common share upon payment of $3.50 expiring on February 22, 2010. As well, we granted an over-allotment option of 600,000 units which can be exercised, in whole or in part, prior to March 24, 2007 for additional cash proceeds of $1,800,000. The net proceeds from this offering will be used for our clinical trial program, manufacturing activities in support of the clinical trial program and for general corporate purposes.
Future Developments
We anticipate that many important activities related to our clinical trial program, our product manufacturing and our intellectual property development and protection will occur in 2007 and beyond. We presently intend to continue to focus our clinical trial program on the evaluation of the effectiveness of REOLYSIN® as a potential treatment for various cancer indications. This will include continuing our clinical trial program presently underway, expanding into other areas including utilizing our association with the NCI to broaden our clinical trial program. We also intend to expand human clinical trials to determine the safety and effectiveness of systemic delivery of REOLYSIN® as a cancer therapeutic.
-13-
Various forms of cancer are being assessed and we intend to evaluate and select one or more forms of cancer that appear to provide the best opportunity for timely approval.
We plan to continue our focus on establishing strategic relationships with potential partners who can provide expertise in marketing and distribution, as well as assistance with research and development.
Except for historical information, this review contains statements which by their nature are forward-looking and which involve known and unknown risks, delays, uncertainties and other factors not under our control. Any of these factors may cause our actual results, performance or achievement to be materially different from the results, performance or expectations implied by these forward-looking statements. These factors include, but are not limited to, results of current or pending clinical trials, actions by regulatory authorities such as the FDA in the United States, the HPB in Canada, or MHRA in the UK as well as those factors detailed in our regulatory filings.
NARRATIVE DESCRIPTION OF THE BUSINESS
Our Business
Our potential product for human use, REOLYSIN®, is developed from the reovirus. This virus has been demonstrated to replicate specifically in tumour cells bearing an activated Ras pathway. Activating mutations of Ras occur in approximately 30% of all human tumours directly, but considering its central role in signal transduction, activation of the Ras pathway has been shown to play a role in approximately two-thirds of all tumours.
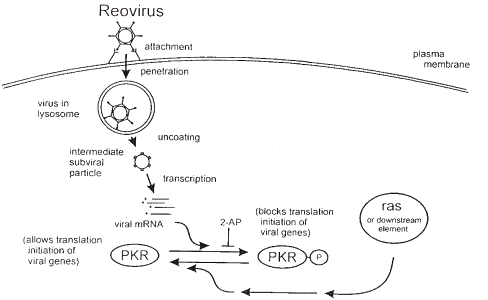
The functionality of the product is based upon the finding that tumours bearing an activated Ras pathway are deficient in their ability to activate the anti-viral response mediated by the host cellular protein, PKR. Since PKR is responsible for preventing reovirus replication, tumour cells lacking the activity of PKR are susceptible to reovirus infections. As normal cells do not possess Ras activations, these cells are able to thwart reovirus infections by the activity of PKR. In a tumour cell with an activated Ras pathway, reovirus is able to freely replicate and hence kill the host tumour cell. The result of this replication is progeny viruses that are then free to infect surrounding cancer cells. This cycle of infection, replication and cell death is believed to be repeated until there are no longer any tumour cells carrying an activated Ras pathway available.
The following schematic illustrates the molecular basis of how the reovirus kills cancer cells.
-14-
Scientific Background
The Ras protein is a key regulator of cell growth and differentiation. It transmits signals from the cell’s surface, via growth factor receptors, to downstream elements, which are in turn relayed to the nucleus. This transmission of signals from the cell surface to the cell’s nucleus is collectively referred to as “signal transduction.” The transmission of these signals results in cell growth, division, and in some instances cellular differentiation. In normal cells, cell growth occurs only in the presence of factors stimulating the cells to grow. Mutations in Ras itself, or any of the elements along the Ras pathway, often lead to activation of the pathway in the absence of the appropriate growth stimuli, leading to the uncontrolled growth of these cells and ultimately to the development of a cancerous state. In fact, approximately 30% of all cancers are known to be due to mutations in Ras itself. The frequency of these Ras mutations, as well as their etiology in a given tumour is however, tissue specific. Activating mutations in Ras are found in many types of human malignancies but are highly represented in pancreatic (90%), sporadic colorectal (50%), lung carcinomas (40%), and myeloid leukemia (30%). Because Ras is a regulator of key mitogenic signals, aberrant function of upstream elements such as receptor tyrosine kinases (RTKs) can also result in Ras activation in the absence of mutations in Ras itself. Indeed, over-expression of these RTKs such as HER2/neu/ErbB2 or the epidermal growth factor receptor is common in breast cancer (25-30%), and over-expression of the platelet-derived growth factor receptor (“PDGFR”) is common in glioblastomas and gliomas, all of which are tumour types in which Ras mutations are relatively rare. Although activating mutations of Ras itself are thought to occur in only about 30% of all tumours it is expected that approximately two-thirds of all tumours have activated Ras signaling pathways as a result of mutations in genes that lie upstream of Ras. With this in mind, Ras becomes a significant therapeutic target in oncology.
All available scientific evidence developed or reviewed by us to date supports the premise that the reovirus only actively infects and replicates in cells with an activated Ras pathway. This naturally occurring virus is believed to cause only mild infections of the respiratory and gastrointestinal tract and in general, reovirus infections in humans are asymptomatic and usually sub-clinical. Research has indicated this virus replicates in, and therefore kills, only cancer cells (i.e. cancer cells with an activated Ras pathway), but does not replicate in normal cells. It has been demonstrated that reovirus replication is restricted in “normal” cells due to the activation of the double stranded RNA-activated protein kinase (“PKR”). PKR is a crucial element in protecting cells from reovirus infection and is capable of blocking viral protein translation. Activated Ras (or an activated element of the Ras pathway) prevents PKR
-15-
activation, and thus allows viral replication to ensue only in this subset of cancer cells. To prove that reovirus could be used as a potential cancer therapeutic, a number of animal models were developed. Experiments using this virus to treat mouse tumours, expanded animal models as well as human brain, breast, and prostate tumours implanted in immuno-compromised mice have yielded promising results. In animals where tumour regression was noted, a single injection of reovirus is often enough to cause complete tumour regression. More importantly, it was demonstrated that this treatment is effective in causing tumour regression in immune competent animals. We believe that the nature of this virus, combined with its selective replication makes it an attractive candidate as a cancer therapy.
We also believe that this research may have broad utility in the treatment of tumours with an activated Ras pathway as well as a potential use as an adjuvant therapy following surgical tumour resection or as an adjuvant therapy to conventional chemotherapeutic or radiation therapies.
The Potential Cancer Product
Cancer is a group of related diseases characterized by the aberrant or uncontrolled growth of cells and the spread of these cells to other sites in the body. These cancer cells eventually accumulate and form tumours that can disrupt and impinge on normal tissue and organ function. In many instances, cells from these tumours can break away from the original tumour and travel through the body to form new tumours through a process referred to as metastasis.
Our cancer product is a potential therapeutic for tumours possessing an activated Ras pathway. In tumour cells with this type of activation, the virus is cytotoxic but may have no effect on the surrounding normal tissue. Activating mutations of Ras are believed to account for approximately 30% of all human tumours directly. It is also possible to activate Ras through mutation of proteins that control its activity rather than through direct mutations of Ras itself. This suggests that approximately two thirds of tumours may respond to this treatment.
Repayable Grants
Pursuant to the Technology Commercialization Agreement with the Alberta Heritage Foundation, we received $150,000 to offset the REOLYSIN® development costs. Under the Technology Commercialization Agreement, we agreed to repay the amount of the grant from our gross sales. We agreed to repay the Alberta Heritage Foundation in annual installments in an amount equal to the lesser of: (a) 5% of gross sales; or (b) $15,000 per annum until the entire grant has been paid in full.
In accordance with the Clinical Trial Agreement with the ACB, we received funding and overhead support from the ACB to offset the REOLYSIN® clinical trial expenditures. Under the Clinical Trial Agreement, we agreed to repay the amount of the grant together with a royalty, to a combined maximum amount of $400,000 plus an overhead repayment of $100,000, upon sales of product. We agreed to repay the ACB in annual installments from the date of commencement of sales in an amount equal to the lesser of: (a) 5% of gross sales of REOLYSIN®; or (b) $100,000 per annum.
Business Strategy
Our business strategy is to develop and market REOLYSIN® in an effective and timely manner, and access additional technologies at a time and in a manner that we believe is best for our development. We intend to achieve our business strategy by focusing on these key areas:
• | Develop REOLYSIN® by initiating toxicology and manufacturing programs and progress the product through a clinical setting to assess its safety and efficacy in human subjects. |
-16-
• | Establish collaborations with experts to assist us with scientific and clinical developments of this new potential pharmaceutical product. |
• | Implement strategic alliances with selected pharmaceutical and biotechnology companies and selected laboratories, at a time and in a manner where such alliances may complement and expand our research and development efforts on the product and provide sales and marketing capabilities. |
• | Utilize our broadening patent base and collaborator network as a mechanism to meet our strategic objectives. |
• | Develop relationships with companies that could be instrumental in assisting us to access other innovative therapeutics. |
Our business strategy is based on attaining a number of commercial objectives, which, in turn, are supported by a number of product development goals. Our new product development presently being conducted is primarily of a research and development nature. In the context of this Annual Information Form, statements of our “belief” are based primarily upon our results derived to date from our research and development program with animals, and early stage human trials, and upon which we believe that we have a reasonable scientific basis to expect the particular results to occur. It is not possible to predict, based upon studies in animals, or early stage human trials, whether a new therapeutic will ultimately prove to be safe and effective in humans. There are no assurances that the particular result expected by us will occur. See “Risk Factors.”
At this time we do not intend to become a fully integrated pharmaceutical company with substantial in-house research and development, marketing and distribution or manufacturing capabilities. We are pursuing a strategy of establishing relationships with larger companies as strategic partners. We intend to partner or joint venture with larger pharmaceutical companies that have existing and relevant marketing capability for our products. It is anticipated that future clinical development into large international or pivotal trials would generally occur in conjunction with a strategic partner or partners, who would contribute expertise and financial assistance. In exchange for certain product rights and commitments to market our products, the strategic partners would be expected to share in gross proceeds from the sale of our product or products. The proceeds generated from partnering or joint venturing projects are expected to be distributed on the basis of relative risk taken and resources contributed by each party to the partnership or joint venture.
Regulatory Requirements
The development of new pharmaceuticals is strongly influenced by a country’s regulatory environment. The drug approval process in Canada is regulated by Health Canada. The primary regulatory body in the United States is the FDA and in the UK is the MHRA. Similar processes are conducted in other countries by equivalent regulatory bodies. Regulations in each jurisdiction require the licensing of manufacturing facilities and mandate strict research and product testing standards. Companies must establish the safety and efficacy of their products, comply with current Good Manufacturing Practices and submit marketing materials before being allowed to market pharmaceutical products. While we plan to pursue or support the pursuit of the approval of our product, success in acquiring regulatory approval for any product is not assured.
In order to market our pharmaceutical product in Canada, the United States, Europe and other jurisdictions, we must successfully meet the requirements of those jurisdictions. The requirements of the Appropriate Regulatory Authority will generally include the following stages as part of the regulatory process:
-17-
Pre-Pharmacological Studies - Pre-Pharmacological studies involve extensive testing on laboratory animals to determine if a potential therapeutic product has utility in an in vivo disease model and has any adverse toxicology in a disease model.
Investigational New Drug Application - An Investigational New Drug (“IND”) Submission, or the equivalent, must be submitted to the appropriate regulatory authority prior to conducting Pharmacological Studies.
Pharmacological Studies (or Phase I Clinical Trials) - Pharmacological studies are designed to assess the potential harmful or other side effects that an individual receiving the therapeutic compound may experience. These studies, usually short in duration, are often conducted with healthy volunteers or actual patients and use up to the maximum expected therapeutic dose.
Therapeutic Studies (or Phase II and III Clinical Trials) - Therapeutic studies are designed primarily to determine the appropriate manner for administering a drug to produce a preventive action or a significant beneficial effect against a disease. These studies are conducted using actual patients with the condition that the therapeutic is designed to remedy.
Prior to initiating these studies, the organization sponsoring the program is required to satisfy a number of requirements via the submission of documentation to support the approval for a clinical trial.
New Drug Submission - After all three phases of a clinical trial have been completed, the results are submitted with the original IND Submission to the appropriate regulatory authority for marketing approval. Once marketing approval is granted, the product is approved for commercial sales.
Market and Competition
According to estimates for 2006 from the American Cancer Society, 1.4 million Americans are expected to be diagnosed with cancer in the year, and 565,000 Americans are forecast to die of cancer. In the United States cancer accounts for 25% of all deaths, second only to heart disease. In the United States, the relative lifetime risk of a male developing cancer is 1 in 2, while for women, this risk is 1 in 3.
The costs of this disease state are also significant. In the United States, the National Institute of Health estimates that the overall annual costs for cancer treatment are $263.3 billion. Of this figure, $78.2 billion can be attributed to direct patient costs.
It has been estimated that approximately 30% of all tumours are a result of activating mutations of Ras itself. Since Ras can be activated by mechanisms other than direct mutations it is believed that the number of tumours with activated Ras (either through direct activating mutation or mutation or over-expression of elements upstream of Ras) is approximately two thirds.
We are aware of large pharmaceutical companies developing small molecule programs for the development of therapeutics to treat Ras mediated tumours. In addition, there are numerous companies, both big and small, that are working in the field of cancer therapeutics including some companies developing other oncolytic viruses. See “Risk Factors.”
Product Marketing Strategy
The markets for the cancer product being developed by us may be large and could require substantial sales and marketing capability. Before or upon successful completion of the development of a cancer product, we intend to enter into one or more strategic partnerships or other collaborative arrangements with a pharmaceutical company or other company with marketing and distribution expertise to address this need. If necessary, we will establish arrangements with various partners for different geographical
-18-
areas or specific applications at various times in the development process. Our management and consultants have relevant experience with the partnering process.
Third Party Advisor, Collaborators and Scientific Advisory Board
We use various third party advisors, scientific collaborators and our scientific advisory board to assist us with the advancement of REOLYSIN®. We typically report on the activity of these groups once their work is completed.
Scientific Advisory Board
Our Scientific Advisory Board is comprised of Ramon Alemany, Ph.D., Richard Gorlick, M.D., Alan Tuchman, M.D., and Frank Tufaro, Ph.D.
Ramon Alemany, Ph.D., is a recognized expert on the development of antitumoural agents based on the adenovirus. During an eight year period in the United States he held progressively more senior positions in gene therapy laboratories at the MD Anderson Cancer Center, Baxter Healthcare Corporation and the University of Alabama at Birmingham. In 2001, he was appointed Director of the Gene and Viral Therapy Group at the Institut Catala d’Oncologia in Barcelona. Dr. Alemany is currently collaborating with us to develop modified adenoviruses that are selective for Ras mediated cancers.
Richard Gorlick, M.D., is the Section Chief of Hematology/Oncology in the department of pediatrics at the Children’s Hospital at Montefiore in New York. He is actively involved in the national pediatric cooperative group, the Children’s Oncology Group, for which he serves as the Chairman of the subcommittee on Bone Tumour Biology. Dr. Gorlick is known for his research work on molecular pharmacology of antifolate resistance and developing new therapeutic approaches for osteosarcoma.
Alan Tuchman, M.D., works in private practice and is Clinical Professor of Neurology at New York Medical College. He is also the Principal of NeuroPhysics Corporation, a healthcare and neuroscience consulting firm. From 1997 to 2001 Dr. Tuchman was the Senior Vice President of Equity Research for Oscar Gruss & Son, where he conducted investment research and helped develop marketing strategies for healthcare companies. He also held senior neurology positions at New York Medical College and Lincoln Medical and Mental Health Center.
Frank Tufaro, Ph.D., has extensive experience with biotech firms and was one of the founders of NeuroVir Inc., a Vancouver-based biotech company, which is now merged with MediGene AG to develop Herpes Simplex virus-based oncolytic vectors for cancer therapy. Under Dr. Tufaro’s direction, NeuroVir and then MediGene Inc. were able to initiate and complete the first Phase I/II U.S. clinical trials of two herpes-based oncolytic viruses for the treatment of malignant brain tumours, and the treatment of colorectal cancer metastatic to the liver. He currently serves on scientific advisory boards for several biotech companies.
Intellectual Property Policy
At the end of 2006, we had 17 patents issued in the United States, five issued in Canadian and three issued in Europe with additional applications in process. In addition, we have numerous patent issuances in other jurisdictions throughout the world. All potentially valuable intellectual property is identified by the inventory, and classified by us in terms of its sensitivity. All sensitive documentation related to the intellectual property is protected and kept in secure areas. All employees execute agreements containing
-19-
confidentiality clauses, which assign any new intellectual property to us. We believe that we apply our intellectual property protection policy consistently. Where appropriate, and consistent with management’s objective, patents are pursued as soon as the concepts have been validated through appropriate laboratory work. To that end, patents will continue to be sought on components or concepts that we perceive to be essential.
We believe that one of the best intellectual property control policies is a strong human resources policy to ensure that technical leaders with access to proprietary intellectual property do not consider leaving us for other employment. We intend that all staff be compensated through competitive salaries and participation in our stock option program.
Patent and Patent Application Summary
Where a patent is filed in the United States there is an option to file a Patent Cooperation Treaty (“PCT”) application. The PCT application process is a means for technology patented in one of the PCT signatory countries to receive protection in other PCT countries. The PCT includes over 100 countries. Within one year of filing a patent in the United States, the applicant files for PCT coverage in all PCT countries. Approximately 18 months after the PCT filing, the applicant must pay individual filing fees in designated PCT countries and at that time the applicant may wish to restrict coverage to a subset of countries which have potential for the technology. At the time of filing the PCT application the applicant designates which of the member countries are to be covered by the application. The PCT application allows the applicant to defer national filings in the various designated countries for a period of up to 30 months from the original PCT application filing date. After the PCT application deferral period, the applicant must file for separate national or regional patents in one or more designated countries, depending on which specific markets the applicant intends to target.
The following table sets forth certain select patent issuances:
| Title | Ownership | Inventors | Status of Patent | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patent Number U.S. 6,110,461 | Oncolytics Biotech Inc. | Dr. Patrick W.K. Lee | Filing date: | Aug. 13, 1997 | |||||
| Reovirus for the Treatment of | Dr. James E. Strong | Issued: | Aug. 29, 2000 | ||||||
| Neoplasia | Dr. Matthew C. Coffey | ||||||||
| Patent Number U.S. 6,136,307 | Oncolytics Biotech Inc. | Dr. Patrick W.K. Lee | Filing date: | Feb. 24, 1999 | |||||
| Reovirus for the Treatment of | Dr. James E. Strong | Issued: | Oct. 24, 2000 | ||||||
| cellular proliferative disorders | Dr. Matthew C. Coffey | ||||||||
| Patent Number U.S. 6,261,555 | Oncolytics Biotech Inc. | Dr. Patrick W .K. Lee | Filing date: | Aug. 12, 1998 | |||||
| Reovirus for the treatment of | Dr. James Strong | Issued: | July 17, 2001 | ||||||
| Neoplasia | Dr. Matthew C. Coffey | ||||||||
| Patent Number U.S. 6,344,195 | Oncolytics Biotech Inc. | Dr. Patrick W. K. Lee | Filing date: | May 12, 2000 | |||||
| Reovirus for the treatment of | Dr. James Strong | Issued: | Feb. 5, 2002 | ||||||
| Neoplasia | Dr. Matthew C. Coffey | ||||||||
| European Patent Number 1003534 | Oncolytics Biotech Inc. | Dr. Patrick W. K. Lee | Filing date: | Aug 12, 1998 | |||||
| Reovirus for the treatment of | Dr. James Strong | Issued: | March 6, 2002 | ||||||
| Neoplasia | Dr. Matthew Coffey | ||||||||
-20-
| Title | Ownership | Inventors | Status of Patent | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Patent Number U.S. 6,455,038 | Oncolytics Biotech Inc. | Dr. Patrick L. Lee | Filing date: | June 15, 2000 | |||||
| Reovirus for the treatment of | Dr. James E. Strong | Issued: | Sept. 24, 2002 | ||||||
| Cellular Proliferative Disorders | Dr. Matthew C. Coffey | ||||||||
| Patent Number U.S. 6,528,305 | Oncolytics Biotech Inc. | Dr. Bradley G. Thompson | Filing date: | Aug. 2, 2001 | |||||
| Method of Producing Infectious | Dr. Matthew C. Coffey | Issued : | March 4, 2003 | ||||||
| Reovirus | |||||||||
| Patent Number U.S. 6,565,831 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Aug. 10, 2000 | |||||
| Methods of Preventing Reovirus | Dr. Bradley G. Thompson | Issued: | May 20, 2003 | ||||||
| Recognition for the Treatment | |||||||||
| for Cellular Proliferative | |||||||||
| Disorders | |||||||||
| Patent Number U.S. 6,576,234 | Oncolytics Biotech Inc. | Dr. Patrick L. Lee | Filing date: | May 10, 2001 | |||||
| Reovirus for the Treatment of | Dr. James E. Strong | Issued: | June 10, 2003 | ||||||
| Neoplasia | Dr. Matthew C. Coffey | ||||||||
| Patent Number U.S. 6,596,268 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Nov. 9, 2000 | |||||
| Viruses for the Treatment of | Dr. Bradley G. Thompson | Issued: | July 22, 2003 | ||||||
| Cellular Proliferative | |||||||||
| Disorders (1) | |||||||||
| Patent Number U.S. 6,649,157 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Sept. 28, 2001 | |||||
| Viruses for the Treatment of | Dr. Bradley G. Thompson | Issued: | Nov 18, 2003 | ||||||
| Cellular Proliferative | |||||||||
| Disorders (2) | |||||||||
| Patent Number U.S. 6,703,232 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | January 8, 2003 | |||||
| Methods of Producing Infectious | Dr. Bradley G. Thompson | Issued: | March 9, 2004 | ||||||
| Reovirus | |||||||||
| Patent Number U.S. 6,808,916 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | March 14, 2002 | |||||
| Method of Extracting Virus from | Dr. Bradley G. Thompson | Issued: | Oct. 26, 2004 | ||||||
| Cell Culture | |||||||||
| Patent Number U.S. 6,811,775 | Oncolytics Biotech Inc. | Dr. Patrick L. Lee | Filing date: | August 15, 2002 | |||||
| Reovirus for the Treatment of | Dr. James E. Strong | Issued: | Nov. 2, 2004 | ||||||
| Cellular Proliferative Disorders | Dr. Matthew C. Coffey | ||||||||
| European patent number 1,309,672 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | July 20, 2001 | |||||
| Method of Producing Infectious | Dr. Bradley G. Thompson | Issued: | June 29, 2005 | ||||||
| Reovirus | |||||||||
| Canadian patent number 2,374,388 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | March 4, 2002 | |||||
| The use of Ribozymes in the | Issued: | July 12, 2005 | |||||||
| Detection of Adventitious Agents | |||||||||
-21-
| Title | Ownership | Inventors | Status of Patent | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Canadian patent number 2,428,206 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Oct 26, 2001 | |||||
| Methods for the Treatment of | Issued: | Sept. 27, 2005 | |||||||
| Cellular Proliferative Disorders | |||||||||
| Canadian patent number 2,283,280 | Oncolytics Biotech Inc. | Dr. Patrick L. Lee | Filing date: | Aug. 12, 1998 | |||||
| Reovirus for the Treatment of | Dr. James E. Strong | Issued: | Oct. 18, 2005 | ||||||
| Neoplasia | Dr. Matthew C. Coffey | ||||||||
| Canadian patent number 2,437,962 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Mar 11, 2002 | |||||
| Method of Extracting Virus From | Dr. Bradley G. Thompson | Issued: | Nov. 15, 2005 | ||||||
| Cell Culture | |||||||||
| U.S. patent number 6,994,858 | Oncolytics Biotech Inc. | Dr. Don Morris | Filing date: | May 3, 2001 | |||||
| Reovirus Clearance of Ras-Mediated | Dr. Bradley G. Thompson | Issued: | Feb. 7, 2006 | ||||||
| Neoplastic Cells from Mixed | Dr. Matthew C. Coffey | ||||||||
| Cellular Compositions | |||||||||
| U.S. patent number 7,014,847 | Oncolytics Biotech Inc. | Dr. Bradley G. Thompson | Filing date: | Mar. 28, 2003 | |||||
| Methods for Preventing Reovirus | Dr. Matthew C. Coffey | Issued: | Mar. 21, 2006 | ||||||
| Recognition for the Treatment of | |||||||||
| Cellular Proliferative | |||||||||
| U.S. patent number 7,049,127 | Oncolytics Biotech Inc. | Dr. Bradley G. Thompson | Filing date: | Dec. 11, 2003 | |||||
| Method of Producing Infectious | Dr. Matthew C. Coffey | Issued: | May 23, 2006 | ||||||
| Reovirus | |||||||||
| U.S. patent number 7,052,832 | Oncolytics Biotech Inc. | Dr. Matthew C. Coffey | Filing date: | Nov. 6, 2001 | |||||
| Methods for the Treatment of | Issued: | May 30, 2006 | |||||||
| Cellular Proliferative Disorders | |||||||||
| U.S. patent number 7,163,678 | Oncolytics Biotech Inc. | Dr. Patrick W.K. Lee | Filing date: | Nov. 6, 2003 | |||||
| Reovirus for the Treatment of | Dr. Kara L. Norman | Issued: | Jan. 16, 2007 | ||||||
| Ral-Mediated Cellular | |||||||||
| Proliferative Disorders | |||||||||
| Canadian patent number 2,415,750 | Oncolytics Biotech Inc. | Dr. Bradley G. Thompson | Filing date: | July 20, 2001 | |||||
| Methods for Preventing Reovirus | Dr. Matthew C. Coffey | Issued: | Mar. 28, 2006 | ||||||
| Recognition for the Treatment of | |||||||||
| Cellular Proliferative Disorders | |||||||||
| European patent number: 1,498,129 | Oncolytics Biotech Inc. | Dr. Ramon Alemany | Filing date: | Mar. 25, 2003 | |||||
| Use of Adenoviruses Mutated in the | Dr. Manel M. Cascallo | Issued: | Nov. 16, 2005 | ||||||
| VA Genes for Cancer Treatment | |||||||||
-22-
Notes:
(1) | Claims cover the treatment of Ras mediated tumours using modified adenoviruses. |
(2) | Claims cover the treatment of Ras mediated tumours using modified herpes viruses. |
Other patent applications have been filed by us, but have yet to be published or approved.
Acquisition of all of the Shares of Oncolytics Biotech Inc. by SYNSORB
In April 1999, Oncolytics Biotech Inc., the Vendors and SYNSORB entered into the Share Purchase Agreement whereby SYNSORB acquired all of our then outstanding common shares for a share and cash exchange valued at $2,500,000 paid primarily in common shares of SYNSORB, four milestone payments payable to the Vendors valued, in the aggregate, at up to $4,000,000 and a royalty commitment. Pursuant to an assignment dated July 29, 1999, the obligation to make the milestone and certain royalty payments was assigned from SYNSORB to us. We thereby agreed to indemnify and save harmless SYNSORB from all actions, suits, demands, claims, costs, losses, expenses, charges and damages brought against SYNSORB in relation to the payment or non-payment of such obligations; however such assignment did not affect or release SYNSORB from its liabilities and responsibilities under the terms of the Share Purchase Agreement. As at the date hereof, we have made three milestone payments totaling $3,000,000. The final milestone payment is $1.0 million payable within 90 days of the first receipt, in any country, from the Appropriate Regulatory Authority, for marketing approval to sell REOLYSIN® to the public or the approval of a new drug application for REOLYSINÒ. In addition to the milestone payments, royalty payments payable to the Vendors will become due and payable in accordance with the Share Purchase Agreement upon realization of sales of REOLYSIN®.
In 2004, we reached an agreement that cancelled a portion of our future contingent obligation for consideration of $400,000 consisting of $250,000 cash and 21,459 common shares valued at $150,000. As a result, our future contingent obligations were reduced to 11.75% (formerly 14.25%) of royalty payments or other payments received as a result of entering into partnerships or other arrangements for the development of the reovirus technology. Alternatively, if we develop the reovirus treatment to the point where it may be marketed at a commercial level, the payments referred to in the foregoing sentence will be amended to equal a royalty payment of 2.35% (formerly 2.85%) of net sales received by us for such products.
Employees
As of December 31, 2006, we had 11 employees. The majority of our activities are conducted under contract with third party service providers.
Research and Development Expenditures
For the period ended December 31, 2006, we incurred research and development expenditures of $10,535,689 representing approximately 67.8% of our total expenses for the year. See “Management’s Discussion and Analysis – Results of Operations – Research and Development Expenses”.
Dividend Policy
To date, we have not paid any dividends on our outstanding common shares. The future payment of dividends will be dependent upon our financial requirements to fund future growth, our financial condition and other factors which our Board of Directors may consider appropriate in the circumstances. It is unlikely that dividends will be paid in the foreseeable future.
-23-
MARKET FOR SECURITIES
Market for Common Shares
Our outstanding common shares are listed and posted for trading on the Toronto Stock Exchange under the trading symbol “ONC” and on the Nasdaq Capital Market under the trading symbol “ONCY”. The
following table sets forth the market price ranges and the aggregate volume of trading of the common shares on the Toronto Stock Exchange and Nasdaq Small Cap Market for the periods indicated:
| Toronto Stock Exchange | Nasdaq Capital Market | ||||||
| High | Low | Close | Volume | High | Low | Close | Volume |
Period |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2006 |
|
|
|
|
|
|
|
|
January | 5.49 | 4.68 | 4.90 | 665,987 | 4.79 | 4.12 | 4.35 | 1,128,094 |
February | 5.47 | 4.60 | 5.20 | 774,307 | 4.75 | 3.98 | 4.51 | 879,890 |
March | 6.05 | 5.20 | 6.05 | 849,957 | 5.25 | 4.57 | 5.16 | 1,527,008 |
April | 6.09 | 4.53 | 4.55 | 1,011,013 | 5.23 | 3.94 | 4.07 | 1,317,432 |
May | 4.60 | 3.69 | 3.72 | 569,967 | 4.23 | 3.22 | 3.39 | 944,610 |
June | 4.03 | 3.18 | 3.19 | 582,377 | 3.65 | 2.80 | 2.85 | 872,172 |
July | 3.40 | 2.25 | 2.54 | 834,167 | 2.96 | 2.14 | 2.26 | 878,257 |
August | 3.29 | 2.50 | 3.20 | 945,281 | 2.99 | 2.20 | 2.89 | 739,891 |
September | 3.73 | 3.01 | 3.05 | 610,426 | 3.38 | 2.69 | 2.76 | 710,165 |
October | 3.09 | 2.64 | 2.65 | 725,084 | 2.80 | 2.36 | 2.36 | 673,207 |
November | 2.85 | 2.19 | 2.23 | 873,397 | 2.55 | 1.91 | 1.94 | 1,202,946 |
December | 2.75 | 2.04 | 2.39 | 1,272,395 | 2.38 | 1.80 | 2.09 | 1,635,947 |
Description of Common Shares
The holders of our common shares are entitled to one vote per share at meetings of shareholders, to receive such dividends as declared by us and to receive our remaining property and assets upon dissolution or wind up. Our common shares are not subject to any future call or assessment and there are no pre-emptive, conversion or redemption rights attached to such shares. As at December 31, 2006, we have outstanding stock options and common share purchase warrants as set forth in Note 9 and 10 of our audited financial statements.
DIRECTORS AND OFFICERS
Our directors are elected by the shareholders at each Annual General Meeting and typically hold office until the next Annual General Meeting at which time they may be re-elected or replaced. Casual vacancies on the board are filled by the remaining directors and the persons filling those vacancies hold office until the next Annual General Meeting at which time they may be re-elected or replaced. The officers are appointed by the Board of Directors and hold office indefinitely at the pleasure of the Board of Directors.
The following table sets forth the names and municipalities of residence of all our directors and officers as at the date hereof, as well as the positions and offices held by such persons and their principal occupations.
-24-
Name and Municipality of Residence | Position with the Corporation | Principal Occupation | Director of the Corporation Since |
|
|
|
|
Bradley G. Thompson | President, Chief Executive Officer and Executive Chairman of the Board | Executive Chairman of the Board, President and Chief Executive Officer since April 1999. | April 21, 1999 |
Douglas A. Ball C.A. | Chief Financial Officer and Director | Chief Financial Officer since May 2000. Mr. Ball was Vice President, Finance and Chief Financial Officer of SYNSORB from June 1997 to May 2000. Prior to this, he was the Vice President, Finance and Administration and Chief Financial Officer of ECL Group of Companies Ltd. Mr. Ball held this position from December 1995 until May 1997. Prior to ECL, he was Controller and then Vice President and Controller of Canadian Airlines International Ltd. from June 1993 until August 1995. | April 21, 1999 |
William A. Cochrane, OC, M.D. (2),(3) Calgary, Alberta | Director | President of W.A. Cochrane & Associates, Inc. (a consulting company) since 1989 and Chairman of Resverlogix Corp. (a public biopharmaceutical company) and University Technologies International Inc. (UTI) at the University of Calgary since 2000, and is a director of Sernova Corp., and Medicure Inc. Dr. Cochrane is an Officer of the Order of Canada and a 2002 recipient of the Queens Golden Jubilee Medal. Dr. Cochrane also served as the Deputy Minister of Health Services for the Province of Alberta from 1973 to 1974 and President of the University of Calgary from 1974 to 1978. | October 31, 2002 |
Matthew C. Coffey Ph.D. | Chief Scientific Officer | Chief Scientific Officer of the Corporation since December 2004, Vice-President of Product Development from July 1999 to December 2004 and Chief Financial Officer from September 1999 to May 2000. | N/A |
George M. Gill, M.D. Washington, D.C.
| Senior Vice President, Clinical and Regulatory Affairs | Dr. Gill has been a consultant in clinical research and regulatory affairs to the pharmaceutical and biotechnology industries since he retired from Ligand Pharmaceuticals in 1999. During his 38 years in the industry, he also served in senior executive positions with ICI Pharmaceuticals (now AstraZeneca), Bristol-Myers Squibb, and Hoffmann-La Roche. Dr. Gill holds a B.Sc. in chemistry from Dickinson College in Pennsylvania and an M.D. from the School of Medicine of the University of Pennsylvania in Philadelphia. | N/A |
Robert B. Schultz, F.C.A. (1) | Lead Director | Chairman and Director of Rockwater Capital Corporation formerly McCarvill Corporation (a financial services company) since June 2001. Chairman and Chief Executive Officer of Merrill Lynch Canada from August 1998 until his retirement on May 1, 2000. Prior to this appointment, Mr. Schultz was Chief Executive Officer at Midland Walwyn since 1990. Since joining the investment industry in 1971, Mr. Schultz has held a variety of senior positions, and has participated on various industry-related boards and committees including Director and Chairman of the Investment Dealers Association of Canada. | June 30, 2000 |
-25-
Name and Municipality of Residence | Position with the Corporation | Principal Occupation | Director of the Corporation Since |
|
|
|
|
Fred A. Stewart, Q.C.(1)(2), | Director | President of Fred Stewart & Associates Inc. (a government and corporate relations consulting company) since March 1996. Prior to that, Mr. Stewart was an associate with Milner Fenerty, Barristers and Solicitors from June 1993 to March 1996. Mr. Stewart served as Member of the Legislative Assembly of the Province of Alberta from 1986 to 1993. | August 27, 1999 |
J. Mark Lievonen C.A.(3) | Director | President of Sanofi Pasteur Limited, a vaccine development, manufacturing and marketing company, since October 1998 and holding various positions with Sanofi Pasteur Limited and its predecessors since 1983. Mr. Lievonen serves on a number of industry and community boards and councils including BIOTECanada, the Ontario Genomics Institute, the Ontario Institute for Cancer Research, and York University.
| April 5, 2004 |
Karl Mettinger, M.D., Ph.D | Chief Medical Officer | Dr. Mettinger has been involved in clinical and regulatory affairs with various pharmaceutical companies since 1985. Prior to joining Oncolytics, he was Senior Vice President and Chief Medical Officer with SuperGen Inc. Prior to that, he was Executive Director, Clinical Research at IVAX/Baker Norton, the new drug subsidiary of IVAX Corporation. He began his career in the industry as a Medical Director with KABI in Sweden. Dr. Mettinger holds an MD from the University of Lund in Sweden and a PhD (hematology/stroke) from the Karolinska Institute/Karolinska Hospital in Stockholm, Sweden, where he was a physician and an Associate Professor. He has overseen the global development and approval of a number of products including several in oncology.
| N/A |
Jim Dinning(1) Calgary, Alberta | Director | Chair of Western Financial Group since September 2004. Mr. Dinning was Executive Vice President of TransAlta Corporation (power generation and wholesale marketing company) from 1997 to 2004 and served as Member of the Legislative Assembly of the Province of Alberta from 1986 to 1997. Mr. Dinning is a director of Finning International Inc. and Shaw Communications Inc as well as other public and private companies. | March 24, 2004 |
-26-
Name and Municipality of Residence | Position with the Corporation | Principal Occupation | Director of the Corporation Since |
|
|
|
|
Ger van Amersfoort,(2) | Director | President and Chief Executive Officer of Novartis Canada, a pharmaceutical company with in excess of $1 billion in annual sales and a workforce of 1,500, until his retirement in 2001. Before joining Novartis, he was President and Chief Executive Officer of the U.K. SmithKline Beecham operations from 1997 until managing the merger with Novartis in 1999. From 1990 to 1997, Mr. van Amersfoort headed up SmithKline Beecham operations in Canada as President and Chief Executive Officer. Prior to that, he held managing director positions with Beecham and The Boots Company, and sales positions with Bristol Myers in Holland. He is a recipient of the Paul Harris Medal and the Commemorative Medal of the Queen for outstanding services to the community. He has served on the Board of the Pharmaceutical Manufacturers Association of Canada (now Rx and D) for more than nine years, serving as chairman in 1996. | June 15, 2006 |
Ed Levy, Ph.D, (3) | Director | Adjunct professor at the W. Maurice Young Centre for Applied Ethics at the University of British Columbia since retiring from QLT Inc. in late 2002. From 1988 to 2002, Dr. Levy was with Vancouver-based biotechnology company QLT Inc., most recently as Senior Vice President from 1998. In these roles, he was primarily responsible for negotiating and managing QLT’s strategic alliances, led strategic planning and oversaw the company’s intellectual property. Dr. Levy served on the board of BIOTECanada from 1999-2002, and he has served on the boards of several technology companies and not-for-profits. Dr. Levy holds a PhD in the History and Philosophy of Science from Indiana University and taught philosophy of science at UBC from 1967-1988. | May 17, 2006 |
Mary Ann Dillahunty, | Vice President, Intellectual Property | Ms. Dillahunty was a principal in the law firm of Fish & Richardson, a leading intellectual property firm in the U.S. In 1992, she joined the law firm of Burns, Doane, Swecker & Mathis (now part of Buchanan Ingersoll & Rooney), and subsequently became a partner in the firm. During 1996-1997, Ms. Dillahunty held the position of patent counsel to the Implant Division of ALZA Corporation. Before joining Burns Doane, she was a patent agent and law clerk with the law firm of Heller, Ehrman, White & McAuliffe. Prior to focusing her career on patent law, Ms. Dillahunty held numerous positions in the biotechnology, pharmaceutical and medical device industries, including responsibilities in regulatory affairs and research science. Ms. Dillahunty holds a B.S. in Microbiology from Michigan State University, an MBA from George Washington University, and a JD degree from Stanford Law School. | N/A |
Notes:
(1) | These persons are members of the Audit Committee. Mr. Stewart is the Chair of the Audit Committee. |
(2) | These persons are members of the Compensation Committee. Mr. Stewart is the Chair of the Compensation Committee. |
(3) | These persons are members of the Corporate Governance and Nominating Committee. Mr. Lievonen is the Chair of the Corporate Governance and Nominating Committee. |
As at the date hereof, the directors and senior officers as a group beneficially owned, directly or indirectly, 672,201 of our common shares, representing 1.7% of the issued and outstanding common shares.
-27-
Certain of our directors are associated with other companies, which may give rise to conflicts of interest. In accordance with the ABCA, directors who have a material interest in any person who is a party to a material contract or a proposed material contract with us are required, subject to certain exceptions, to disclose that interest and abstain from voting on any resolution to approve that contract. In addition, the directors are required to act honestly and in good faith with a view to the best interests of Oncolytics Biotech Inc.
AUDIT COMMITTEE MATTERS
Mandate of the Audit Committee
1. | Policy Statement |
It is the policy of Oncolytics Biotech Inc. (the “Corporation”) to establish and maintain an Audit Committee, composed entirely of independent directors, to assist the Board of Directors (the “Board”) in carrying out their oversight responsibility for the Corporation’s internal controls, financial reporting and risk management processes. The Audit Committee will be provided with resources commensurate with the duties and responsibilities assigned to it by the Board including administrative support. If determined necessary by the Audit Committee, it will have the discretion to institute investigations of improprieties, or suspected improprieties within the scope of its responsibilities, including the standing authority to retain special counsel or experts.
2. | Composition of the Committee |
| (a) | The Audit Committee shall consist of a minimum of three (3) directors, at least half of whom shall be resident Canadians. The Board shall appoint the members of the Audit Committee and may seek the advice and assistance of the Corporate Governance and Nominating Committee in identifying qualified candidates. The Board shall appoint one member of the Audit Committee to be the Chair of the Audit Committee, or delegate such authority to appoint the Chair of the Audit Committee to the Audit Committee. |
| (b) | The Chair of the Committee shall be responsible for leadership of the Committee, including preparing or approving the agenda, presiding over the meetings, and making committee assignments. |
| (c) | Each director appointed to the Audit Committee by the Board shall be an outside director who is unrelated. An outside, unrelated director is a director who meets the requirements of NASDAQ Rule 4200 and Multilaterial Instrument 52-110. A director appointed to the audit committee shall also meet the requirements of NASDAQ Rule 4350 (d)(2)(A)(ii) and Exchange Act Rule 10A-3(b)(1). Such director shall be independent of management and free from any interest, any business or other relationship which could, or could reasonably be perceived, to materially interfere with the director’s ability to act with a view to the best interests of the Corporation, other than interests and relationships arising from shareholding. In determining whether a director is independent of management, the Board shall make reference to the abovementioned rules and any applicable revisions thereto, and any additional relevant then current legislation, rules, policies and instruments of applicable regulatory authorities. |
-28-
| (d) | Each member of the Audit Committee shall be financially literate. In order to be financially literate, a director must be, at a minimum, able to read and understand basic financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Corporation’s financial statements. At least one member shall have accounting or related financial management expertise, meaning the ability to analyze and interpret a full set of financial statements, including the notes attached thereto, in accordance with generally accepted accounting principles. In determining whether a member of the Audit Committee is financially literate or has accounting or related financial expertise, reference shall be made to the then current legislation, rules, policies and instruments of applicable regulatory authorities, which for further clarification, shall include but not be limited to the definition of “financial expert” as defined by the U.S. Securities and Exchange Commission rule. |
| (e) | A director appointed by the Board to the Audit Committee shall be a member of the Audit Committee until replaced by the Board or until his or her resignation. |
3. | Meetings of the Committee |
| (a) | The Audit Committee shall convene a minimum of four times each year at such times and places as may be designated by the Chair of the Audit Committee and whenever a meeting is requested by the Board, a member of the Audit Committee, the auditors, or senior management of the Corporation. Scheduled meetings of the Audit Committee shall correspond with the review of the year-end and quarterly financial statements and management discussion and analysis. |
| (b) | Notice of each meeting of the Audit Committee shall be given to each member of the Audit Committee and to the auditors, who shall be entitled to attend each meeting of the Audit Committee and shall attend whenever requested to do so by a member of the Audit Committee. |
| (c) | Notice of a meeting of the Audit Committee shall: |
| (i) | be in writing, including by electronic communication facilities; |
| (ii) | state the nature of the business to be transacted at the meeting in reasonable detail; |
| (iii) | to the extent practicable, be accompanied by copies of documentation to be considered at the meeting; and |
| (iv) | be given at least two business days prior to the time stipulated for the meeting or such shorter period as the members of the Audit Committee may permit. |
| (d) | A quorum for the transaction of business at a meeting of the Audit Committee shall consist of a majority of the members of the Audit Committee. However, it shall be the practice of the Audit Committee to require review, and, if necessary, approval of certain important matters by all members of the Audit Committee. |
| (e) | A member or members of the Audit Committee may participate in a meeting of the Audit Committee by means of such telephonic, electronic or other communication facilities, as permits all persons participating in the meeting to communicate adequately with each |
-29-
|
| other. A member participating in such a meeting by any such means is deemed to be present at the meeting. |
| (f) | In the absence of the Chair of the Audit Committee, the members of the Audit Committee shall choose one of the members present to be Chair of the meeting. In addition, the members of the Audit Committee shall choose one of the persons present to be the Secretary of the meeting. |
| (g) | A member of the Board, senior management of the Corporation and other parties may attend meetings of the Audit Committee; however the Audit Committee (i) shall, at each meeting, meet with the external auditors independent of other individuals other than the Audit Committee and (ii) may meet separately with management. |
| (h) | Minutes shall be kept of all meetings of the Audit Committee and shall be signed by the Chair and the Secretary of the meeting. |
4. | Duties and Responsibilities of the Committee |
| (a) | The Audit Committee’s primary duties and responsibilities are to: |
| (i) | identify and monitor the management of the principal risks that could impact the financial reporting of the Corporation ; |
| (ii) | monitor the integrity of the Corporation’s financial reporting process and system of internal controls regarding financial reporting and accounting compliance; |
| (iii) | monitor the independence and performance of the Corporation’s external auditors. This will include receipt, review and evaluation, at least annually, of a formal written statement from the independent auditors confirming their independence, and qualifications, including their compliance with the requirements of the relevant oversight boards ; |
| (iv) | deal directly with the external auditors to pre-approve external audit plans, other services (if any) and fees; |
| (v) | directly oversee the external audit process and results (in addition to items described in Section 4(d) below); |
| (vi) | provide an avenue of communication among the external auditors, management and the Board; and |
| (vii) | carry out a review designed to ensure that an effective “whistle blowing” procedure exists to permit stakeholders to express any concerns regarding accounting, internal controls, auditing matters or financial matters to an appropriately independent individual. |
| (viii) | Pre-approve any related party transactions to be entered into by the Company, and ensure appropriate disclosure thereof. |
| (ix) | Ensure financial disclosure incorporates inclusion of any material correcting adjustments required by the external auditors. |
-30-
| (x) | Require and ensure that the external auditors are directly responsible to the Audit Committee, to whom they report |
| (b) | The Audit Committee shall have the authority to: |
| (i) | inspect any and all of the books and records of the Corporation and its affiliates; |
| (ii) | discuss with the management of the Corporation and its affiliates, any affected party and the external auditors, such accounts, records and other matters as any member of the Audit Committee considers necessary and appropriate; |
| (iii) | engage independent counsel and other advisors as it determines necessary to carry out its duties; and |
| (iv) | to set and pay the compensation for any advisors employed by the Audit Committee. |
| (c) | The Audit Committee shall, at the earliest opportunity after each meeting, report to the Board the results of its activities and any reviews undertaken and make recommendations to the Board as deemed appropriate. |
| (d) | The Audit Committee shall: |
| (i) | review the audit plan with the Corporation’s external auditors and with management; |
| (ii) | Review with the independent auditors the matters required to be discussed relating to the conduct of the audit, including (a) the proposed scope of their examination, with emphasis on accounting and financial areas where the Committee, the independent auditors or management believes special attention should be directed; (b) the results of their audit, including their audit findings report and resulting letter, if any, of recommendations for management; (c) their evaluation of the adequacy and effectiveness of the Company’s internal controls over financial reporting; (d) significant areas of disagreement, if any, with management; (e) co-operation received from management in the conduct of the audit; (f) significant accounting, reporting, regulatory or industry developments affecting the Company; and (g) review any proposed changes in major accounting policies or principles proposed or contemplated by the independent auditors or management, the presentation and impact of material risks and uncertainties and key estimates and judgements of management that may be material to financial reporting; |
| (iii) | review with management and with the external auditors material financial reporting issues arising during the most recent fiscal period and the resolution or proposed resolution of such issues; |
| (iv) | review any problems experienced or concerns expressed by the external auditors in performing an audit, including any restrictions imposed by management or material accounting issues on which there was a disagreement with management; |
| (v) | review with senior management the process of identifying, monitoring and reporting the principal risks affecting financial reporting; |
-31-
| (vi) | review audited annual financial statements (including management discussion and analysis) and related documents in conjunction with the report of the external auditors and obtain an explanation from management of all material variances between comparative reporting periods. Without restricting the generality of the foregoing, the committee will discuss with management and the independent auditors to the extent required, any issues and disclosure requirements regarding (a) the use of “pro forma” or “adjusted” non-GAAP information, as well as financial information and earnings guidance provided to analysts and rating agencies, (b) any off balance sheet arrangements, and (c) any going concern qualification. |
| (vii) | consider and review with management, the internal control memorandum or management letter containing the recommendations of the external auditors and management’s response, if any, including an evaluation of the adequacy and effectiveness of the internal financial controls of the Corporation and subsequent follow-up to any identified weaknesses; |
| (viii) | review with financial management and the external auditors the quarterly unaudited financial statements and management discussion and analysis before release to the public; |
| (ix) | before release, review and if appropriate, recommend for approval by the Board, all public disclosure documents containing audited or unaudited financial information, including any prospectuses, annual reports, annual information forms, management discussion and analysis and press releases; and |
| (x) | oversee, any of the financial affairs of the Corporation or its affiliates, and, if deemed appropriate, make recommendations to the Board, external auditors or management. |
| (e) | The Audit Committee shall: |
| (i) | evaluate the independence and performance of the external auditors and annually recommend to the Board the appointment of the external auditor or the discharge of the external auditor when circumstances are warranted and monitor the audit partners’ rotation as required by law.; |
| (ii) | consider the recommendations of management in respect of the appointment of the external auditors; |
| (iii) | pre-approve all non-audit services to be provided to the Corporation or its subsidiary entities by its external auditors’, or the external auditors of affiliates of the Corporation subject to the over-riding principle that the external auditors not being permitted to be retained by the Corporation to perform specifically listed categories of non-audit services as set forth by the Securities and Exchange Commission as well as internal audit outsourcing services, financial information systems work and expert services. Notwithstanding, the foregoing the pre-approval of non-audit services may be delegated to a member of the Audit Committee, with any decisions of the member with the delegated authority reporting to the Audit Committee at the next scheduled meeting; |
-32-
| (iv) | approve the engagement letter for non-audit services to be provided by the external auditors or affiliates, together with estimated fees, and considering the potential impact of such services on the independence of the external auditors; |
| (v) | when there is to be a change of external auditors, review all issues and provide documentation related to the change, including the information to be included in the Notice of Change of Auditors and documentation required pursuant to the then current legislation, rules, policies and instruments of applicable regulatory authorities and the planned steps for an orderly transition period; and |
| (vi) | review all reportable events, including disagreements, unresolved issues and consultations, as defined by applicable securities policies, on a routine basis, whether or not there is to be a change of external auditors. |
| (f) | The Audit Committee shall enquire into and determine the appropriate resolution of any conflict of interest in respect of audit or financial matters, which are directed to the Audit Committee by any member of the Board, a shareholder of the Corporation, the external auditors, or senior management. |
| (g) | The Audit Committee shall periodically review with management the need for an internal audit function. |
| (h) | The Audit Committee shall review the Corporation’s accounting and reporting of costs, liabilities and contingencies. |
| (i) | The Audit Committee shall establish and maintain procedures for: |
| (i) | the receipt, retention and treatment of complaints received by the Corporation regarding accounting controls, or auditing matters; and |
| (ii) | the confidential, anonymous submission by employees of the Corporation or concerns regarding questionable accounting or auditing matters. |
| (j) | The Audit Committee shall review and approve the Corporation’s hiring policies regarding employees and former employees of the present and former external auditors. |
| (k) | The Audit Committee shall review with the Corporation’s legal counsel, on no less than an annual basis, any legal matter that could have a material impact on the Corporation’s financial statements, and any enquiries received from regulators, or government agencies. |
| (l) | The Audit Committee shall assess, on an annual basis, the adequacy of this Mandate and the performance of the Audit Committee. |
5. | Date of Mandate |
This Mandate was initially approved by the Board on September 3, 1999. Subsequent to that date, the Board has amended and restated this Mandate on each of December 13, 2002 April 23, 2003, March 5, 2004 and December 8, 2004. This Mandate is effective from and after December 14, 2006.
-33-
Composition of the Audit Committee
The following table sets forth the name of each of the current members of the Audit Committee, whether such member is independent, whether such member is financially literate and the relevant education and experience of such member.
Name | Independent | Financially Literate | Relevant Education and Experience |
|
|
|
|
Fred A. Stewart, Q.C. | Yes | Yes | Mr. Stewart graduated with Bachelor of Commerce (Saskatchewan) and Bachelor of Laws (Toronto) and practised corporate and commercial law for over 20 years, receiving his Queen’s Counsel designation in 1980. Mr. Stewart served as a Member of Cabinet in the Government of Alberta. In addition, Mr. Stewart has acquired significant financial experience and exposure to accounting and financial issues as a founding partner of his law firm, as a Member of the Treasury Board of the Government of Alberta and while serving as a director and audit committee member of both private and public companies. |
|
|
|
|
Jim Dinning | Yes | Yes | Mr. Dinning is currently Chair of the Board of Western Financial Group. He graduated with a bachelor of commerce honours degree and a master’s degree in public administration from Queen’s University. Mr. Dinning served as a Member of the Legislative Assembly of the Province of Alberta from 1986 to 1997. Mr. Dinning has acquired significant financial experience and exposure to accounting and financial issues while serving as a director and audit committee chair/member for other publicly traded companies and as Provincial Treasurer for the Alberta Provincial Government. |
-34-
RISK FACTORS
All of our potential products, including REOLYSIN®, are in the research and development stage and will require further development and testing before they can be marketed commercially.
Prospects for companies in the biotechnology industry generally may be regarded as uncertain given the nature of the industry and, accordingly, investments in biotechnology companies should be regarded as speculative. We are currently in the research and development stage on one product, REOLYSIN®, for human application, the riskiest stage for a company in the biotechnology industry. It is not possible to predict, based upon studies in animals and early stage human clinical trials whether REOLYSIN® will prove to be safe and effective in humans. REOLYSIN® will require additional research and development, including extensive additional clinical testing, before we will be able to obtain the approvals of the relevant regulatory authorities in applicable countries to market REOLYSIN® commercially. There can be no assurance that the research and development programs we conducted will result in REOLYSIN® or any other products becoming commercially viable products, and in the event that any product or products result from the research and development program, it is unlikely they will be commercially available for a number of years.
To achieve profitable operations we, alone or with others, must successfully develop, introduce and market our products. To obtain regulatory approvals for products being developed for human use, and to achieve commercial success, human clinical trials must demonstrate that the product is safe for human use and that the product shows efficacy. Unsatisfactory results obtained from a particular study relating to a program may cause us to abandon our commitment to that program or the product being tested. No assurances can be provided that any current or future animal or human test, if undertaken, will yield favourable results. If we are unable to establish that REOLYSIN® is a safe, effective treatment for cancer, we may be required to abandon further development of the product and develop a new business strategy.
There are inherent risks in pharmaceutical research and development.
Pharmaceutical research and development is highly speculative and involves a high and significant degree of risk. The marketability of any product we develop will be affected by numerous factors beyond our control, including but not limited to:
| • | the discovery of unexpected toxicities or lack of sufficient efficacy of products which make them unattractive or unsuitable for human use; |
| • | preliminary results as seen in animal and/or limited human testing may not be substantiated in larger, controlled clinical trials; |
| • | manufacturing costs or other production factors may make manufacturing of products ineffective, impractical and non-competitive; |
| • | proprietary rights of third parties or competing products or technologies may preclude commercialization; |
| • | requisite regulatory approvals for the commercial distribution of products may not be obtained; and |
-35-
| • | other factors may become apparent during the course of research, up-scaling or manufacturing which may result in the discontinuation of research and other critical projects. |
Our products under development have never been manufactured on a commercial scale, and there can be no assurance that such products can be manufactured at a cost or in a quantity to render such products commercially viable. Production and utilization of our products may require the development of new manufacturing technologies and expertise. The impact on our business in the event that new manufacturing technologies and expertise are required to be developed is uncertain. There can be no assurance that we will successfully meet any of these technological challenges, or others that may arise in the course of development.
Pharmaceutical products are subject to intense regulatory approval processes.
The regulatory process for pharmaceuticals, which includes preclinical studies and clinical trials of each compound to establish its safety and efficacy, takes many years and requires the expenditure of substantial resources. Moreover, if regulatory approval of a drug is granted, such approval may entail limitations on the indicated uses for which it may be marketed. Failure to comply with applicable regulatory requirements can, among other things, result in suspension of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Further, government policy may change, and additional government regulations may be established that could prevent or delay regulatory approvals for our products. In addition, a marketed drug and its manufacturer are subject to continual review. Later discovery of previously unknown problems with the product or manufacturer may result in restrictions on such product or manufacturer, including withdrawal of the product from the market.
The U.S. FDA and similar regulatory authorities in other countries may deny approval of a new drug application if required regulatory criteria are not satisfied, or may require additional testing. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. The FDA and similar regulatory authorities in other countries may require further testing and surveillance programs to monitor the pharmaceutical product that has been commercialized. Non-compliance with applicable requirements can result in fines and other judicially imposed sanctions, including product withdrawals, product seizures, injunction actions and criminal prosecutions.
In addition to our own pharmaceuticals, we may supply active pharmaceutical ingredients and advanced pharmaceutical intermediates for use in our customers’ drug products. The final drug products in which the pharmaceutical ingredients and advanced pharmaceutical intermediates are used, however, are subject to regulation for safety and efficacy by the FDA and other jurisdictions, as the case may be. Such products must be approved by such agencies before they can be commercially marketed. The process of obtaining regulatory clearance for marketing is uncertain, costly and time consuming. We cannot predict how long the necessary regulatory approvals will take or whether our customers will ever obtain such approval for their products. To the extent that our customers do not obtain the necessary regulatory approvals for marketing new products, our product sales could be adversely affected.
The FDA and other governmental regulators have increased requirements for drug purity and have increased environmental burdens upon the pharmaceutical industry. Because pharmaceutical drug manufacturing is a highly regulated industry, requiring significant documentation and validation of manufacturing processes and quality control assurance prior to approval of the facility to manufacture a specific drug, there can be considerable transition time between the initiation of a contract to manufacture a product and the actual initiation of manufacture of that product. Any lag time in the initiation of a contract to manufacture product and the actual initiation of manufacture could cause us to lose profits or incur liabilities.
-36-
The pharmaceutical regulatory regime in Europe and other countries is, by and large, generally similar to that of the United States. We could face similar risks in these other jurisdictions, as the risks described above.
Our operations and products may be subject to other government manufacturing and testing regulations.
Securing regulatory approval for the marketing of therapeutics by the FDA in the United States and similar regulatory agencies in other countries is a long and expensive process, which can delay or prevent product development and marketing. Approval to market products may be for limited applications or may not be received at all.
The products we anticipate manufacturing will have to comply with the FDA’s current Good Manufacturing Practices (“cGMP”) and other FDA, and local government guidelines and regulations, including other international regulatory requirements and guidelines. Additionally, certain of our customers may require the manufacturing facilities contracted by us to adhere to additional manufacturing standards, even if not required by the FDA. Compliance with cGMP regulations requires manufacturers to expend time, money and effort in production, and to maintain precise records and quality control to ensure that the product meets applicable specifications and other requirements. The FDA and other regulatory bodies periodically inspect drug-manufacturing facilities to ensure compliance with applicable cGMP requirements. If the manufacturing facilities contracted by us fail to comply with the cGMP requirements, the facilities may become subject to possible FDA or other regulatory action and manufacturing at the facility could consequently be suspended. We may not be able to contract suitable alternative or back-up manufacturing facilities on terms acceptable to us or at all.
The FDA or other regulatory agencies may also require the submission of any lot of a particular product for inspection. If the lot product fails to meet the FDA requirements, then the FDA could take any of the following actions: (i) restrict the release of the product; (ii) suspend manufacturing of the specific lot of the product; (iii) order a recall of the lot of the product; or (iv) order a seizure of the lot of the product.
We are subject to regulation by governments in many jurisdictions and, if we do not comply with healthcare, drug, manufacturing and environmental regulations, among others, our existing and future operations may be curtailed, and we could be subject to liability.
In addition to the regulatory approval process, we may be subject to regulations under local, provincial, state, federal and foreign law, including requirements regarding occupational health, safety, laboratory practices, environmental protection and hazardous substance control, and may be subject to other present and future local, provincial, state, federal and foreign regulations.
The biotechnology industry is extremely competitive and we must successfully compete with larger companies with substantially greater resources.
Technological competition in the pharmaceutical industry is intense and we expect competition to increase. Other companies are conducting research on therapeutics involving the Ras pathway as well as other novel treatments or therapeutics for the treatment of cancer which may compete with our product. Many of these competitors are more established, benefit from greater name recognition and have substantially greater financial, technical and marketing resources than us. In addition, many of these competitors have significantly greater experience in undertaking research, preclinical studies and human clinical trials of new pharmaceutical products, obtaining regulatory approvals and manufacturing and marketing such products. In addition, there are several other companies and products with which we may compete from time to time, and which may have significantly better and larger resources than us.
-37-
Accordingly, our competitors may succeed in manufacturing and/or commercializing products more rapidly or effectively, which could have a material adverse effect on our business, financial condition or results of operations.
We anticipate that we will face increased competition in the future as new products enter the market and advanced technologies become available. There can be no assurance that existing products or new products developed by our competitors will not be more effective, or be more effectively manufactured, marketed and sold, than any that may be developed or sold by us. Competitive products may render our products obsolete and uncompetitive prior to recovering research, development or commercialization expenses incurred with respect to any such products.
We rely on patents and proprietary rights to protect our technology.
Our success will depend, in part, on our ability to obtain patents, maintain trade secret protection and operate without infringing the rights of third parties. We have patents in the United States, Canada and Europe and have filed applications for patents in the United States and under the PCT, allowing us to file in other jurisdictions. See “Narrative Description—Patent and Patent Application Summary”. Our success will depend, in part, on our ability to obtain, enforce and maintain patent protection for our technology in Canada, the United States and other countries. We cannot be assured that patents will issue from any pending applications or that claims now or in the future, if any, allowed under issued patents will be sufficiently broad to protect our technology. In addition, no assurance can be given that any patents issued to or licensed by us will not be challenged, invalidated, infringed or circumvented, or that the rights granted thereunder will provide continuing competitive advantages to us.
The patent positions of pharmaceutical and biotechnology firms, including us, are generally uncertain and involve complex legal and factual questions. In addition, it is not known whether any of our current research endeavours will result in the issuance of patents in Canada, the United States, or elsewhere, or if any patents already issued will provide significant proprietary protection or will be circumvented or invalidated. Since patent applications in the United States and Canada may be maintained in secrecy until at least 18 months after filing of the original priority application, and since publication of discoveries in the scientific or patent literature tends to lag behind actual discoveries by several months, we cannot be certain that we or any licensor were the first to create inventions claimed by pending patent applications or that we or the licensor were the first to file patent applications for such inventions. Loss of patent protection could lead to generic competition for these products, and others in the future, which would materially and adversely affect our financial prospects for these products.
Similarly, since patent applications filed before November 29, 2000 in the United States may be maintained in secrecy until the patents issue or foreign counterparts, if any, publish, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor were the first to file patent applications for such inventions. There is no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Accordingly, we may not be able to obtain and enforce effective patents to protect our proprietary rights from use by competitors, and the patents of other parties could require us to stop using or pay to use certain intellectual property, and as such, our competitive position and profitability could suffer as a result.
In addition, we may be required to obtain licenses under patents or other proprietary rights of third parties. No assurance can be given that any licenses required under such patents or proprietary rights will be available on terms acceptable to us. If we do not obtain such licenses, we could encounter delays in introducing one or more of our products to the market while we attempt to design around such patents, or
-38-
could find that the development, manufacture or sale of products requiring such licenses could be foreclosed. In addition, we could incur substantial costs in defending ourselves in suits brought against us on such patents or in suits in which our attempts to enforce our own patents against other parties.
Our products may fail or cause harm, subjecting us to product liability claims.
Use of our product during current clinical trials may entail risk of product liability. We maintain clinical trial liability insurance; however, it is possible this coverage may not provide full protection against all risks. Given the scope and complexity of the clinical development process, the uncertainty of product liability litigation, and the shrinking capacity of insurance underwriters, it is not possible at this time to assess the adequacy of current clinical trial coverage, nor the ability to secure continuing coverage at the same level and at reasonable cost in the foreseeable future. While we carry, and intend to continue carrying amounts believed to be appropriate under the circumstances, it is not possible at this time to determine the adequacy of such coverage.
In addition, the sale and commercial use of our product entails risk of product liability. We currently do not carry any product liability insurance for this purpose. There can be no assurance that we will be able to obtain appropriate levels of product liability insurance prior to any sale of our pharmaceutical products. An inability to obtain insurance on economically feasible terms or to otherwise protect against potential product liability claims could inhibit or prevent the commercialization of products developed by us. The obligation to pay any product liability claim or a recall of a product could have a material adverse effect on our business, financial condition and future prospects.
We have limited manufacturing experience and intend to rely on third parties to commercially manufacture our products, if and when developed.
To date, we have relied upon a contract manufacturer to manufacture small quantities of REOLYSIN®. The manufacturer may encounter difficulties in scaling up production, including production yields, quality control and quality assurance. Only a limited number of manufacturers can supply therapeutic viruses and failure by the manufacturer to deliver the required quantities of REOLYSIN® on a timely basis at a commercially reasonable price may have a material adverse effect on us. We have completed a program for the development of a commercial process for manufacturing REOLYSIN® and have filed a number of patent applications related to the process. There can be no assurance that we will successfully obtain sufficient patent protection related to our manufacturing process.
New products may not be accepted by the medical community or consumers.
Our primary activity to date has been research and development and we have no experience in marketing or commercializing products. We will likely rely on third parties to market our products, assuming that they receive regulatory approvals. If we rely on third parties to market our products, the commercial success of such product may be outside of our control. Moreover, there can be no assurance that physicians, patients or the medical community will accept our product even if it proves to be safe and effective and is approved for marketing by Health Canada, the FDA and other regulatory authorities. A failure to successfully market our product would have a material adverse effect on our revenue.
Our technologies may become obsolete.
The pharmaceutical industry is characterized by rapidly changing markets, technology, emerging industry standards and frequent introduction of new products. The introduction of new products embodying new technologies, including new manufacturing processes, and the emergence of new industry standards may render our products obsolete, less competitive or less marketable. The process of developing our products is extremely complex and requires significant continuing development efforts and third party
-39-
commitments. Our failure to develop new technologies and products and the obsolescence of existing technologies could adversely affect our business.
We may be unable to anticipate changes in our potential customer requirements that could make our existing technology obsolete. Our success will depend, in part, on our ability to continue to enhance our existing technologies, develop new technology that addresses the increasing sophistication and varied needs of the market, and respond to technological advances and emerging industry standards and practices on a timely and cost-effective basis. The development of our proprietary technology entails significant technical and business risks. We may not be successful in using our new technologies or exploiting our niche markets effectively or adapting our businesses to evolving customer or medical requirements or preferences or emerging industry standards.
We are highly dependent on third party relationships for research and clinical trials.
We rely upon third party relationships for assistance in the conduct of research efforts, pre-clinical development and clinical trials, and manufacturing. In addition, we expect to rely on third parties to seek regulatory approvals for and to market our product. Although we believe that our collaborative partners will have an economic motivation to commercialize our product included in any collaborative agreement, the amount and timing of resources diverted to these activities generally is expected to be controlled by the third party. Furthermore, if we cannot maintain these relationships, our business may suffer.
We have no operating revenues and a history of losses.
To date, we have not generated operating revenues to offset our research and development costs and accordingly have not generated positive cash flow or made an operating profit. As of December 31, 2006, we had an accumulated deficit of approximately $65 million. We have incurred net losses of approximately $14.3 million, $12.8 million and $13.0 million for the years ended December 31, 2006, 2005, and 2004, respectively. We anticipate that we will continue to incur significant losses during 2007 and in the foreseeable future. We will not reach profitability until after successful commercialization of one or more of our products. Even if one or more of our products are profitably commercialized, the initial losses incurred by us may never be recovered.
We may need additional financing in the future to fund the research and development of our products and to meet our ongoing capital requirements.
As of December 31, 2006, we had cash and cash equivalents (including short-term investments) of $27.6 million and working capital of approximately $25.7 million. On February 22, 2007, we received gross proceeds of approximately $10,590,000 for the issuance of 4,000,000 units whereby each unit consisted of one common share and one half of one common share purchase warrant (see – “Recent Developments”). We anticipate that we may need additional financing in the future to fund research and development and to meet our ongoing capital requirements. The amount of future capital requirements will depend on many factors, including continued scientific progress in our drug discovery and development programs, progress in our pre-clinical and clinical evaluation of drug candidates, time and expense associated with filing, prosecuting and enforcing our patent claims and costs associated with obtaining regulatory approvals. In order to meet such capital requirements, we will consider contract fees, collaborative research and development arrangements, and additional public or private financings (including the incurrence of debt and the issuance of additional equity securities) to fund all or a part of particular programs as well as potential partnering or licensing opportunities. There can be no assurance that additional funding will be available or, if available, that it will be available on acceptable terms. If adequate funds are not available on terms favorable to us, we may have to reduce substantially or eliminate expenditures for research and development, testing, production and marketing of our proposed product, or obtain funds through arrangements with corporate partners that require us to relinquish rights
-40-
to certain of our technologies or product. There can be no assurance that we will be able to raise additional capital if our current capital resources are exhausted.
The cost of director and officer liability insurance may increase substantially and may affect our ability to retain quality directors and officers.
We carry liability insurance on behalf of our directors and officers. Given a number of large director and officer liability insurance claims in the U.S. equity markets, director and officer liability insurance has become increasingly more expensive with increased restrictions. Consequently, there is no assurance that we will continue to be offered this insurance or be able to obtain adequate coverage. The inability to acquire the appropriate insurance coverage may limit our ability to attract and maintain directors and officers as required to conduct our business.
We are dependent on our key employees and collaborators.
Our ability to develop the product will depend, to a great extent, on our ability to attract and retain highly qualified scientific personnel and to develop and maintain relationships with leading research institutions. Competition for such personnel and relationships is intense. We are highly dependent on the principal members of our management staff as well as our advisors and collaborators, the loss of whose services might impede the achievement of development objectives. The persons working with us are affected by a number of influences outside of our control. The loss of key employees and/or key collaborators may affect the speed and success of product development.
We presently carry key man insurance in the amounts of $1,500,000, $1,000,000 and $500,000 for Dr. Thompson, Dr. Coffey and Mr. Ball, respectively.
Our share price may be highly volatile.
Market prices for securities of biotechnology companies generally are volatile. This increases the risk of securities litigation. Factors such as announcements (publicly made or at scientific conferences) of technological innovations, new commercial products, patents, the development of proprietary rights, results of clinical trials, regulatory actions, publications, quarterly financial results, our financial position, public concern over the safety of biotechnology, future sales of shares by us or our current shareholders and other factors could have a significant effect on the market price and volatility of the common shares.
We incur some of our expenses in foreign currencies and therefore we are exposed to foreign currency exchange rate fluctuations.
We incur some of our manufacturing, clinical, collaborative and consulting expenses in foreign currencies, primarily the U.S. dollar and the Great British pound (“GBP”). Over the past few years the Canadian dollar has appreciated relative to the U.S. dollar and the GBP thereby decreasing the Canadian dollar equivalent. However, if this trend reverses, our Canadian dollar equivalent costs will increase. Also, as we expand to other foreign jurisdictions there may be an increase in our foreign exchange exposure.
We earn interest income on our excess cash reserves and are exposed to changes in interest rates.
We invest our excess cash reserves in investment vehicles that provide a rate of return with little risk to principal. As interest rates change the amount of interest income we earn will be directly impacted.
-41-
ADDITIONAL INFORMATION
Legal Proceedings
We are not aware of any material legal proceedings nor are we aware of any such proceedings being contemplated.
Interest of Management and Others in Material Transactions
Other than as discussed herein, there are no material interests, direct or indirect, of directors, executive officers, senior officers, or any direct or indirect shareholder of ours who beneficially owns, or who exercises control over, more than 10% of our outstanding common shares or any known associate or affiliate of such persons, in any transaction within the three most recently completed financial years or during the current financial year that has materially affected or will materially affect us.
Transfer Agent and Registrar
The transfer agent and registrar for our common shares is Computershare Trust Company of Canada at its principal offices in Calgary, Alberta and Toronto, Ontario.
Material Contracts
Other than as discussed herein, there are no material contracts, other than contracts entered into in the ordinary course of business, that are material to us that were entered into within the most recently completed financial year, or before the most recently completed financial year but are still in effect.
Interests of Experts
Ernst & Young LLP, Chartered Accountants, have audited our financial statements for the year ended December 31, 2006, as set forth in our Annual Report.
As at the date hereof, the partners and associates of Ernst & Young LLP, Chartered Accountants, our independent auditors, as a group did not beneficially own any of our outstanding common shares.
External Auditor Service Fees
During the financial years ended December 31, 2006, 2005, and 2004, Ernst & Young LLP received the following fees:
| December 31, |
| |||
Item | 2006 $ | 2005 $ | 2004 $ | ||
Audit fees | 79,900 | 63,500 | 60,460 | ||
Audit-related fees (1),(3), (4) | 32,260 | 20,250 | 33,354 | ||
Tax fees (2) | 8,214 | 9,048 | 22,150 | ||
All other fees | — | — | — | ||
-42-
Notes:
(1) | Includes review of interim financial statements, accounting consultations and subscription to on-line accounting. |
(2) | Comprised of tax return preparation, scientific research and development return and other tax consultation fees. |
(3) | Includes fees associated with matters relating to the prospectus offerings in 2004. |
(4) | Includes fees associated with preliminary internal control work for Sarbanes Oxley Section 404. |
Audit Fees
Audit fees were for professional services rendered by Ernst & Young, LLP for the audit of our annual financial statements and services provided in connection with statutory and regulatory filings or engagements.
Audit-Related Fees
Audit-related fees were for assurance and related services reasonably related to the performance of the audit or review of the annual statements and are not reported under the heading Audit Fees above. These services consisted of accounting consultations, assistance with prospectus filings and assistance with preparations for compliance with section 404 of the Sarbanes-Oxley Act of 2002.
Tax Fees
Tax fees were for tax compliance and professional tax consultations.
All Other Fees
There were no fees paid to Ernst & Young, LLP that would be considered All Other Fees in 2004 or 2003. Fees to be disclosed under this category would be for products and services other than those described under the headings Audit Fees, Audit-Related Fees and Tax Fees above.
Other Additional Information
Additional information, including information as to our directors’ and officers’ remuneration and indebtedness, principal holders of our securities, options to purchase securities and interests of insiders in material transactions is contained in our Information Circular for our most recent annual meeting of shareholders that involved the election of directors, which is incorporated herein by reference and forms an integral part of this Annual Information Form.
Additional financial information is contained in our financial statements for the year ended December 31, 2006 and under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our 2006 Annual Report, which are incorporated herein by reference and form an integral part of this Annual Information Form.
The documents referred to above as well as additional information relating to us is available through the Internet on the Canadian System for Electronic Document Analysis and Retrieval (“SEDAR”) at www.sedar.com.
Alternatively, a request for any documents referred to above may be made to the Chief Financial Officer, Oncolytics Biotech Inc., Suite 210, 1167 Kensington Crescent N.W., Calgary, Alberta, Canada, T2N 1X7 or by telecopier at (403) 283-0858.
-43-
GLOSSARY
In this Annual Information Form, unless the context otherwise requires, the following words and phrases shall have the meaning set forth below:
ABCA - Business Corporations Act (Alberta), as amended.
ACB - Alberta Cancer Board.
Activating mutations – a type of genetic mutation that results in a particular protein being active in the absence of an appropriate stimuli. This type of mutation typically leads to the development of a cancerous transformation of a cell.
Adjuvant therapy - a form of therapy that is to be used in conjunction with one or more addition therapies.
Alberta Heritage Foundation – the Alberta Heritage Foundation for Medical Research.
Animal model - a human disease given to an animal which exhibits similar or identical characteristics to this disease in humans.
Appropriate Regulatory Authority – means (a) Health Canada, (b) the Food and Drug Administration in the United States, or (c) the comparable authorities in the following countries or areas: United Kingdom, France, Germany, Japan, Benelux.
Asymptomatic – without any signs or symptoms.
Cancer - a heterogeneous group of diseases that is characterized by the uncontrolled or aberrant growth of cells. In addition to the uncontrolled growth of these tumour cells, these cells are able to invade and colonize other sites in the body; by definition these tumours are malignant.
Carcinomas – a type of cancer that arises from epithelial tissue.
CEA – Carcinoembryonic antigen – a substance that is sometimes found in an increased amount in the blood of people who have certain cancers, other diseases, or who smoke. It is used as a tumor marker for colorectal cancer.
Cellular proliferative disorder - a heterogeneous group of disease characterized by the uncontrolled or aberrant growth of cells; is distinct from cancer in that it does not necessarily imply a malignant state.
Clinical Trial Agreement - the agreement among Oncolytics Biotech Inc., Dr. Don Morris and the ACB dated May 1, 1999, providing for, among other things, a repayable grant of $200,000 to us to offset the future REOLYSIN® clinical trial expenditures. (this also refers to our NCI arrangement)
CT – computed tomography
Cytostatic - any drug or agent that is capable of preventing a cell’s growth and division.
Cytotoxic - any drug or agent that is capable of causing cell death.
Differentiation – a form of growth; a process whereby a cell develops different or more advanced processes than were possessed by the cell before.
-44-
DLT – Dose-Limiting Toxicity
Epidermal growth factor - a compound that promotes the growth of cells.
Epidermal growth factor receptor – the cellular receptor that interacts with the epidermal growth factor; a particular family of receptor tyrosine kinase.
Epithelial - the tissue that forms the outer layer of the body surface or the tissue that lines the gut or other hollow structure.
Etiology – the reason or causation of an illness, disease or disorder.
FDA – the U.S. Food and Drug Administration
GAAP – Generally Accepted Accounting Principles
Gastrointestinal tract - within the digestive system including the stomach, intestine, and all accessory organs.
Glioblastoma - a specific form of cancer derived from brain tissue.
Gliomas - a specific form of cancer derived from brain tissue.
GGT – Gamma-Glutamyl Transpeptidase - is a liver enzyme. It is involved in the transfer of amino acids across the cellular membrane and in glutathione metabolism. GGT is found in high concentrations in the liver, bile ducts and kidney.
GLP – Good Laboratory Practice
Good Manufacturing Practices - the current regulatory requirements and standards regarding quality assurance procedures to be adhered to in the manufacturing of therapeutic products established and monitored by various governments including Canada and the United States.
Growth factor receptor – a form of receptor that interacts with growth factors.
Gy – a measurement of dosage of radiation.
HER2/neu/ErbB2 – a form of receptor tyrosine kinase that is frequently overexpressed in breast cancers.
Immune competent - an animal with a fully functional immune system; an animal that can mount a response to a foreign or infectious agent.
Immuno-compromised - an animal that is lacking an immune system.
Investigational New Drug Submission (“IND”) - documentation filed with government agencies responsible for evaluating and licensing pharmaceutical drugs. This documentation is necessary for the initiation of clinical trials.
In Vivo - in the living body.
Lesion - a morbid change in the functioning or texture of an organ or tissue.
-45-
Metastasis - the process whereby a tumour cell is able to leave the original tumour mass and spread to secondary sites in the body forming additional tumour sites.
MHRA - Medicines and Healthcare products Regulatory Agency (U.K.)
Mitogenic - a drug or agent that promotes cellular division or growth.
MTD – Maximum Tolerated Dose
Neoplasia – a group of diseases characterized by uncontrolled cell growth, including, but not limited to, cancer.
Nucleus - an organelle in the cell that contains genetic material.
Oncology - the study and treatment of cancer and tumours.
Patent Cooperation Treaty or PCT - an international patent treaty, of which Canada is a signatory, whereby a single international patent application can be filed in the applicant’s or inventor’s home country for possible protection of intellectual property in over 100 PCT member countries.
PKR (or double stranded RNA dependent protein kinase) - a host protein that plays a key role in mediating the cell’s antiviral activity.
Platelet-derived growth factor receptor (PDGFR) - the cellular receptor that interacts with the platelet-derived growth factor; a particular family of receptor tyrosine kinase.
PSA – Prostate Specific Antigen is a protein produced by the cells of the prostate gland.
Ras - a cellular protein that is a key relay in the transmission of growth signals from the outside of the cell to the cell’s nucleus. In a noncancerous cell, Ras is activated in the presence of an appropriate growth signal.
Receptor - a cellular structure, usually found on the cell surface, that can interact with a certain compound to elicit a specific type of cellular response.
Receptor tyrosine kinase (RTK) - a type of host receptor that uses a particular residue for cellular signaling to the nucleus. Mutation or overexpression of this type of receptor is frequently seen in the development of a variety of cancers.
REOLYSIN® - is our trademark for the human reovirus for the treatment of a specific disease.
Reovirus - a double stranded RNA virus first identified in 1959. The name is an acronym for Respiratory Enteric Orphan virus. The virus is given the designate of orphan virus since it is not associated with a known disease state. For the purpose of this document, all reference to reovirus is to reovirus type III Dearing.
Research Contract - an agreement between Oncolytics Biotech Inc. and the Governors of the University of Calgary, providing for the aggregate sum of $102,000 to be paid by us for a research project under the direction and supervision of Dr. Patrick Lee.
Share Purchase Agreement - the share purchase agreement among the Vendors, SYNSORB and Oncolytics Biotech Inc. dated April 21, 1999 providing for the purchase by SYNSORB of all of our issued and outstanding shares.
-46-
Signal Transduction - The transmission of signals from the cell surface to the cell’s nucleus.
SYNSORB - SYNSORB Biotech Inc. (now Iteration Energy Ltd. by name change formerly Hawker Resources Inc.), a public Company incorporated under the ABCA.
Technology Commercialization Agreement - the agreement between Oncolytics Biotech Inc. and the Alberta Heritage Foundation dated February 9, 1999 providing for a repayable grant of $150,000 to us to offset reovirus clinical trial expenditures.
TCID50 – a measure of viral particle concentration.
Toxicology - the scientific determination of the quantity of a substance that is required to act adversely in the body.
Tumour - an abnormal growth of tissue whether benign or malignant.
Vendors - Dr. Patrick Lee, Dr. James Strong, Dr. Matthew Coffey, Dr. Bradley Thompson and University Technologies International Inc.