Exhibit 99.1
|

XP23829 PHASE 2 PSORIASIS TRIAL
PRELIMINARY TOPLINE DATA PRESENTATION
SEPTEMBER 15, 2015
© COPYRIGHT 2015 XENOPORT, INC. ALL RIGHTS RESERVED.
|

SAFE HARBOR DISCLAIMER
These slides and the accompanying oral presentation by XenoPort, Inc. contain forward-looking statements that involve risks and uncertainties, including all statements relating to future clinical and commercial opportunities for XP23829; the suitability of XP23829 as a potential treatment for moderate-to-severe chronic plaque-type psoriasis and/or relapsing forms of multiple sclerosis (MS); XenoPort’s belief that the efficacy of XP23829 is likely to improve with a more extended duration of treatment beyond 12 weeks; XenoPort’s beliefs regarding the potential advantages of
XP23829 and that XP23829 could lead to a differentiated product in psoriasis; the XP23829 development program, including potential future Phase 3 clinical studies and the timing thereof, and potential partnerships that could accelerate the development of XP23829; potential regulatory matters, including XenoPort’s expectation that a single positive Phase 3 clinical trial of XP23829 could support a New Drug Application submission to the U.S. Food and Drug Administration; and the commercial and other value proposition and opportunities for XP23829 and its potential regulatory approval. XenoPort can give no assurance with respect to these statements, and XenoPort assumes no obligation to update them. Further, these forward-looking statements are based upon XenoPort’s current expectations and involve risks and uncertainties. The following important factors, among others, could cause actual results and the timing of events to differ materially from those anticipated in such forward-looking statements: the difficulty and uncertainty of pharmaceutical product development and the uncertain results and timing of clinical trials, including the risk that success in preclinical testing and early clinical trials do not ensure that later clinical trials, such as Phase 3 clinical trials, will be successful, and that the results of clinical trials by other parties may not be indicative of the results in trials that XenoPort may conduct; risks related to XenoPort’s ability to successfully advance XP23829 development and to conduct clinical trials in the anticipated timeframes, or at all; the risk that XenoPort’s belief regarding potential distinguishing attributes of XP23829 may not be observed or validated in future Phase 3 or other clinical testing; that XP23829 will require significant additional clinical testing prior to any possible regulatory approvals and failure could occur at any stage of its development; the uncertainty of the FDA’s review process and other regulatory requirements; XenoPort’s ability to obtain and its dependence on potential future collaborative partners; the availability of resources to develop XP23829 and to support XenoPort’s operations; and the uncertain therapeutic and commercial value of XP23829. These and other risk factors are discussed under the heading “Risk Factors” in XenoPort’s Quarterly Report on Form 10-Q for the quarter ended June 30, 2015 filed with the Securities and Exchange Commission on August 6, 2015.
PAGE 2 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

KEY OBSERVATIONS FROM PRELIMINARY RESULTS OF XP23829 PHASE 2 CLINICAL TRIAL
Met primary endpoint of % change in PASI with 800 QD and 400 BID doses (p£0.001)
Efficacy still improving at end of treatment (week 12) Low incidence of flushing (similar to placebo) Low incidence and severity of lymphocyte reduction with recovery to within normal limits by four to six weeks post treatment First demonstration that a MMF prodrug other than DMF can be effective on a clinical symptom endpoint
PAGE 3 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
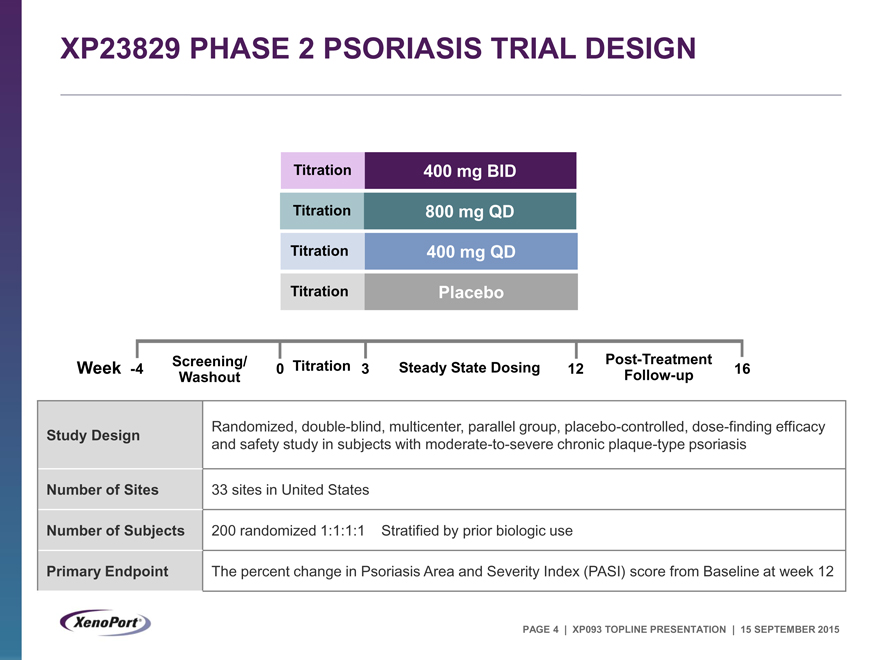
XP23829 PHASE 2 PSORIASIS TRIAL DESIGN
Titration 400 mg BID Titration 800 mg QD Titration 400 mg QD Titration Placebo
Screening/ Post-Treatment Week -4 0 Titration 3 Steady State Dosing 12 16 Washout Follow-up
Study Design Randomized, double-blind, multicenter, parallel group, placebo-controlled, dose-finding efficacy
and safety study in subjects with moderate-to-severe chronic plaque-type psoriasis
Number of Sites 33 sites in United States
Number of Subjects 200 randomized 1:1:1:1 Stratified by prior biologic use
Primary Endpoint The percent change in Psoriasis Area and Severity Index (PASI) score from Baseline at week 12
PAGE 4 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

KEY ELIGIBILITY CRITERIA
Chronic plaque-type psoriasis diagnosed for at least six months Moderate-to-severe psoriasis as defined by:
PASI (Psoriasis Area and Severity Index) score of³12
sPGA (Static Physician’s Global Assessment) score of³3
BSA (body surface area) affected by plaque-type psoriasis of³10%
Candidate for phototherapy and/or systemic therapy
May be either treatment naïve or have a history of previous treatment
Excluded subjects with inadequate response to more than three approved systemic agents for psoriasis
PAGE 5 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
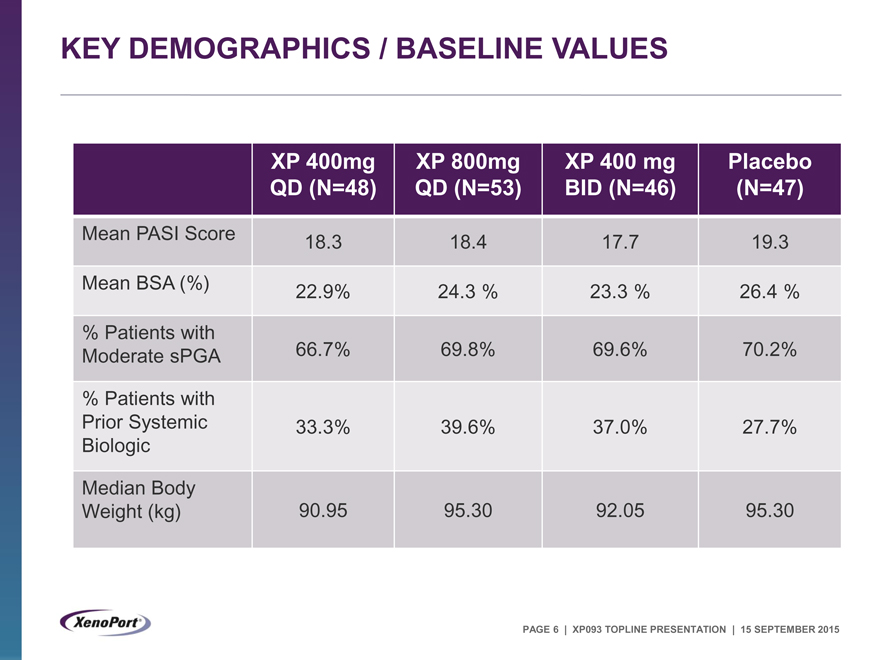
KEY DEMOGRAPHICS / BASELINE VALUES
XP 400mg XP 800mg XP 400 mg Placebo
QD (N=48) QD (N=53) BID (N=46) (N=47)
Mean PASI Score 18.3 18.4 17.7 19.3
Mean BSA (%) 22.9% 24.3 % 23.3 % 26.4 %
% Patients with
Moderate sPGA 66.7% 69.8% 69.6% 70.2%
% Patients with
Prior Systemic 33.3% 39.6% 37.0% 27.7%
Biologic
Median Body
Weight (kg) 90.95 95.30 92.05 95.30
PAGE 6 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
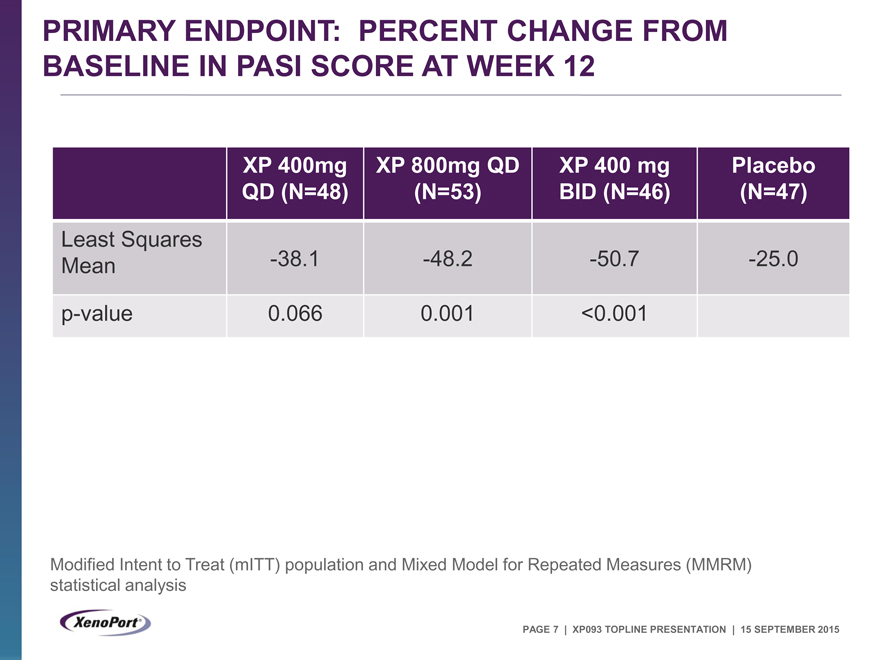
PRIMARY ENDPOINT: PERCENT CHANGE FROM
BASELINE IN PASI SCORE AT WEEK 12
XP 400mg XP 800mg QD XP 400 mg Placebo
QD (N=48) (N=53) BID (N=46) (N=47)
Least Squares
Mean -38.1 -48.2 -50.7 -25.0
p-value 0.066 0.001 <0.001
Modified Intent to Treat (mITT) population and Mixed Model for Repeated Measures (MMRM) statistical analysis
PAGE 7 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

LEAST SQUARES MEAN PERCENT CHANGE FROM BASELINE IN PASI SCORE OVER TIME
mITT population, MMRM
PAGE 8 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
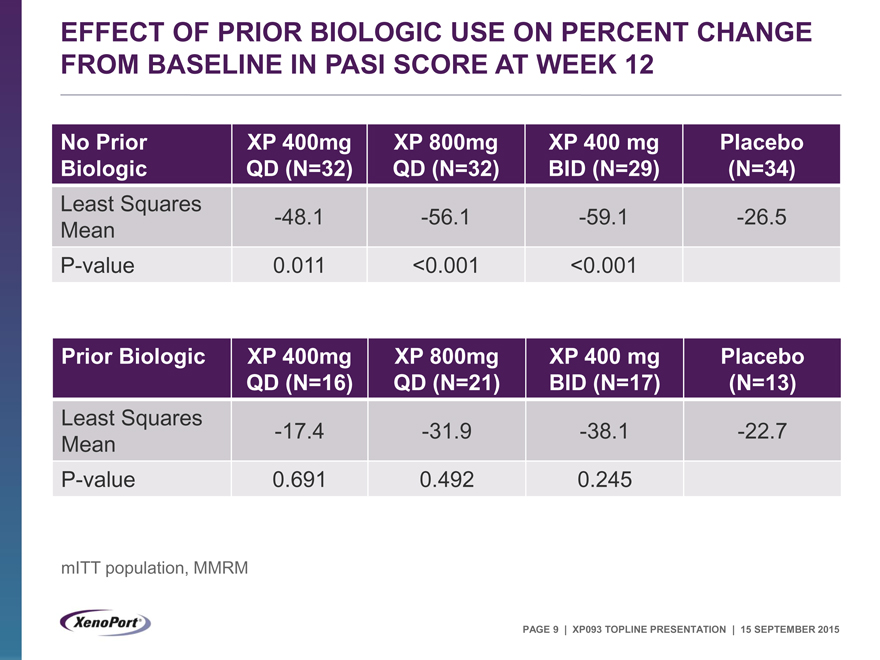
EFFECT OF PRIOR BIOLOGIC USE ON PERCENT CHANGE FROM BASELINE IN PASI SCORE AT WEEK 12
No Prior XP 400mg XP 800mg XP 400 mg Placebo
Biologic QD (N=32) QD (N=32) BID (N=29) (N=34)
Least Squares -48.1 -56.1 -59.1 -26.5
Mean
P-value 0.011 <0.001 <0.001
Prior Biologic XP 400mg XP 800mg XP 400 mg Placebo
QD (N=16) QD (N=21) BID (N=17) (N=13)
Least Squares -17.4 -31.9 -38.1 -22.7
Mean
P-value 0.691 0.492 0.245
mITT population, MMRM
PAGE 9 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
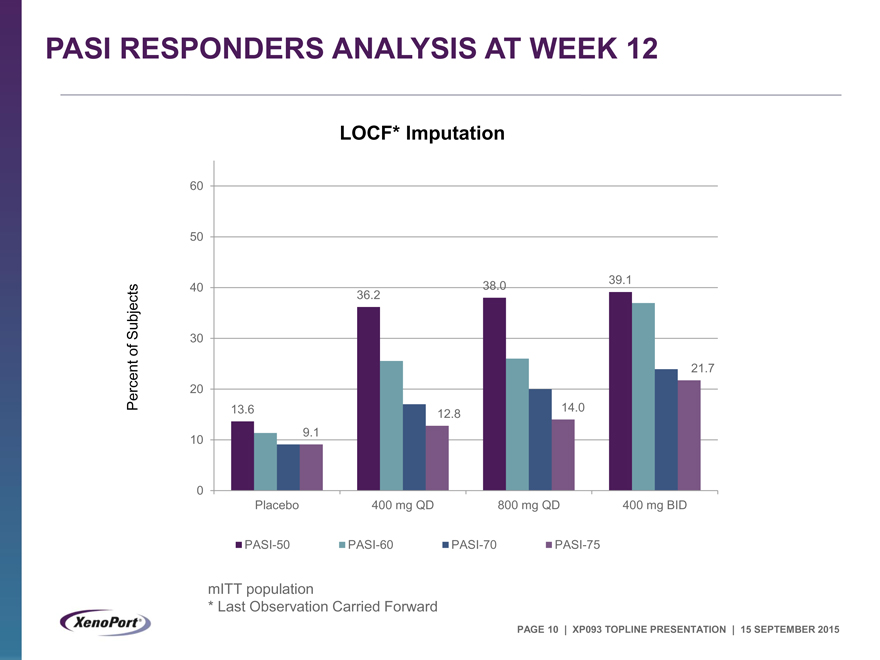
PASI RESPONDERS ANALYSIS AT WEEK 12
LOCF* Imputation
60
50
40 38.0 39.1
Subjects 36.2
30
of
21.7
Percent 20
13.6 12.8 14.0
10 9.1
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
mITT population
* |
| Last Observation Carried Forward |
PAGE 10 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
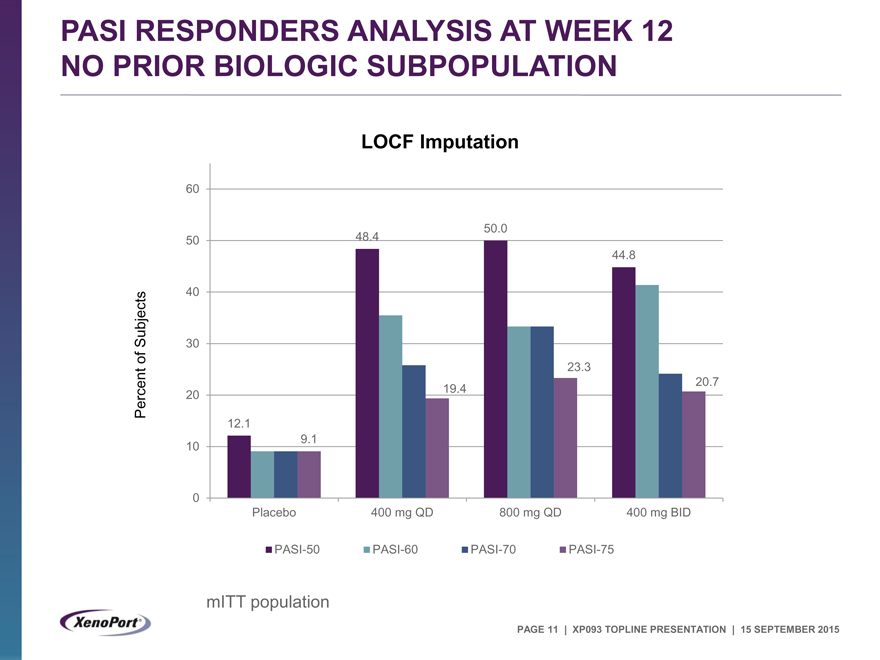
PASI RESPONDERS ANALYSIS AT WEEK 12 NO PRIOR BIOLOGIC SUBPOPULATION
LOCF Imputation
60
50.0
50 48.4
44.8
40
Subjects 30
of 23.3
20.7
Percent 20 19.4
12.1
10 9.1
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
mITT population
PAGE 11 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

SAFETY OVERVIEW
No deaths or life-threatening adverse events (AEs) No subjects met safety discontinuation criteria Majority of treatment emergent adverse events (TEAEs) were non-serious and mild or moderate in severity Three subjects with serious TEAEs – All recovered
One subject with acute cholecystitis; possibly related to XP23829 treatment, per investigator (800 mg QD)
One subject with enterocolitis; possibly related to XP23829 treatment, per investigator (400 mg BID)
One subject with an abdominal hernia; not related to XP23829 treatment, per investigator (400 mg QD)
PAGE 12 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
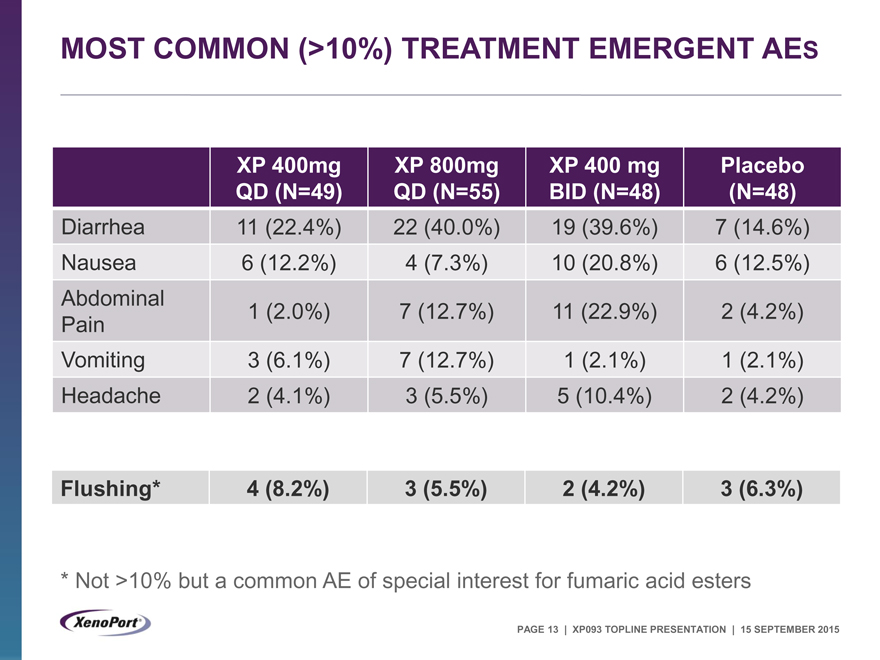
MOST COMMON (>10%) TREATMENT EMERGENT AES
XP 400mg XP 800mg XP 400 mg Placebo
QD (N=49) QD (N=55) BID (N=48) (N=48)
Diarrhea 11 (22.4%) 22 (40.0%) 19 (39.6%) 7 (14.6%)
Nausea 6 (12.2%) 4 (7.3%) 10 (20.8%) 6 (12.5%)
Abdominal
Pain 1 (2.0%) 7 (12.7%) 11 (22.9%) 2 (4.2%)
Vomiting 3 (6.1%) 7 (12.7%) 1 (2.1%) 1 (2.1%)
Headache 2 (4.1%) 3 (5.5%) 5 (10.4%) 2 (4.2%)
Flushing* 4 (8.2%) 3 (5.5%) 2 (4.2%) 3 (6.3%)
* |
| Not >10% but a common AE of special interest for fumaric acid esters |
PAGE 13 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
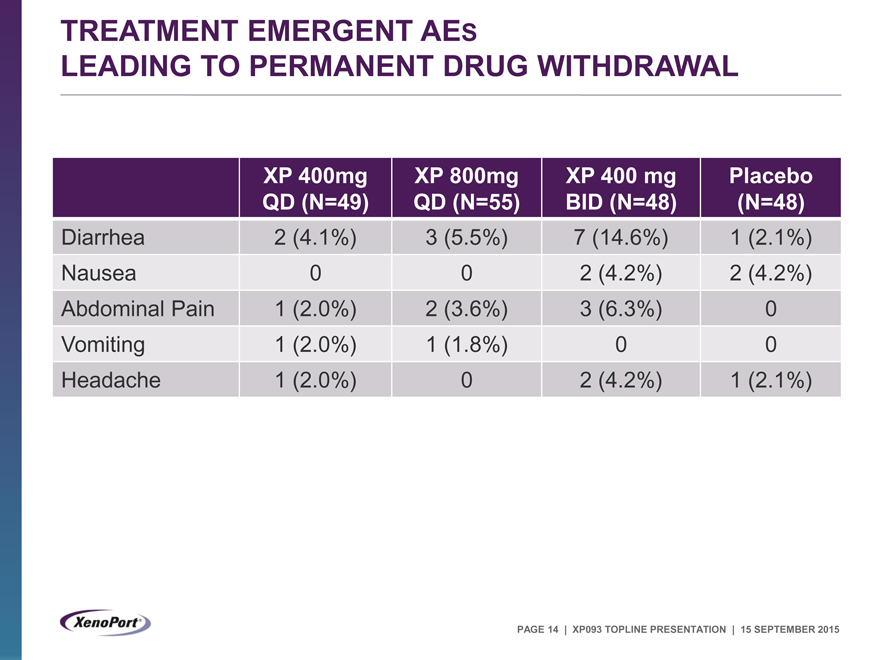
TREATMENT EMERGENT AES
LEADING TO PERMANENT DRUG WITHDRAWAL
XP 400mg XP 800mg XP 400 mg Placebo
QD (N=49) QD (N=55) BID (N=48) (N=48)
Diarrhea 2 (4.1%) 3 (5.5%) 7 (14.6%) 1 (2.1%)
Nausea 0 0 2 (4.2%) 2 (4.2%)
Abdominal Pain 1 (2.0%) 2 (3.6%) 3 (6.3%) 0
Vomiting 1 (2.0%) 1 (1.8%) 0 0
Headache 1 (2.0%) 0 2 (4.2%) 1 (2.1%)
PAGE 14 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
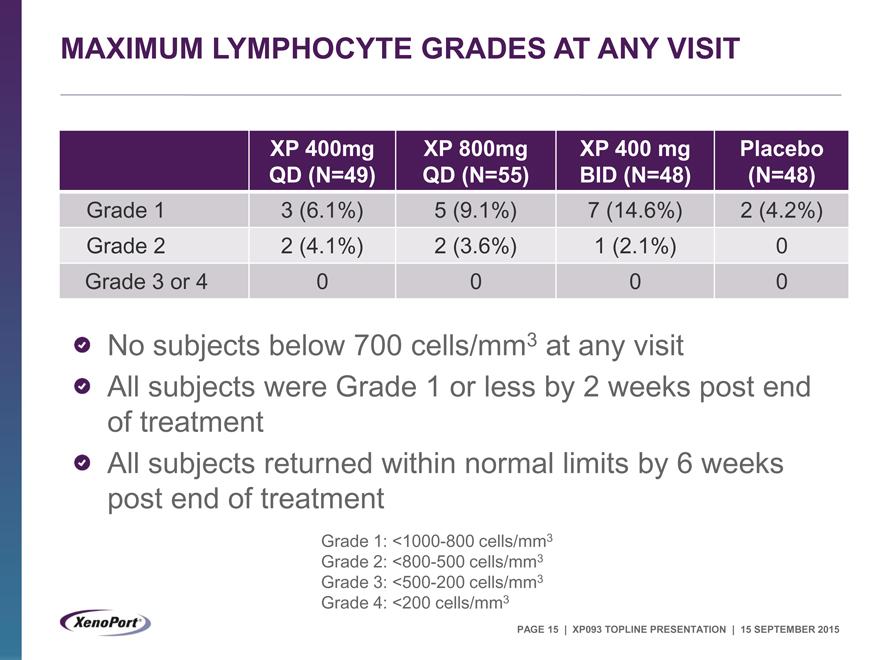
MAXIMUM LYMPHOCYTE GRADES AT ANY VISIT
XP 400mg XP 800mg XP 400 mg Placebo
QD (N=49) QD (N=55) BID (N=48) (N=48)
Grade 1 3 (6.1%) 5 (9.1%) 7 (14.6%) 2 (4.2%)
Grade 2 2 (4.1%) 2 (3.6%) 1 (2.1%) 0
Grade 3 or 4 0 0 0 0
No subjects below 700 cells/mm3 at any visit
All subjects were Grade 1 or less by 2 weeks post end of treatment All subjects returned within normal limits by 6 weeks post end of treatment
Grade 1: <1000-800 cells/mm3
Grade 2: <800-500 cells/mm3
Grade 3: <500-200 cells/mm3
Grade 4: <200 cells/mm3
PAGE 15 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

KEY OBSERVATIONS FROM PRELIMINARY RESULTS OF XP23829 PHASE 2 CLINICAL TRIAL
Met primary endpoint of % change in PASI with 800 QD and 400 BID doses (p£0.001)
Efficacy still improving at end of treatment (week 12) Low incidence of flushing (similar to placebo) Low incidence and severity of lymphocyte reduction with recovery to within normal limits by four to six weeks post treatment First demonstration that a MMF prodrug other than DMF can be effective on a clinical symptom endpoint
PAGE 16 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

THANK YOU
© COPYRIGHT 2015 XENOPORT, INC. ALL RIGHTS RESERVED.
|

Appendix
PAGE 18 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|

HISTORICAL TECFIDERA PHASE 3 PSORIASIS STUDY
Conducted at 10 sites in Europe. Unlikely to have enrolled many patients with prior biologic treatment 16-week study; titration not described but likely 1 week Dose of TECFIDERA was 240 mg TID
Median PASI score reduction at week 12 for TECFIDERA was 59% vs 13% placebo. ~45% for TECFIDERA at week 8. Did not describe any imputation PASI reduction increased from week 12 to 16 PASI responders at week 16 (did not describe imputation)
PASI 50 65% vs 14% placebo
PASI 75 39% vs 1% placebo
Adverse Events
Flushing 42% vs. 9% placebo
Diarrhea 31% vs. 9% placebo
Abdominal Pain 12% vs. 3 % placebo
Headache 11% vs. 6% placebo
Nausea 10% vs. 0% placebo
No indication of drop-out rates
PAGE 19 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
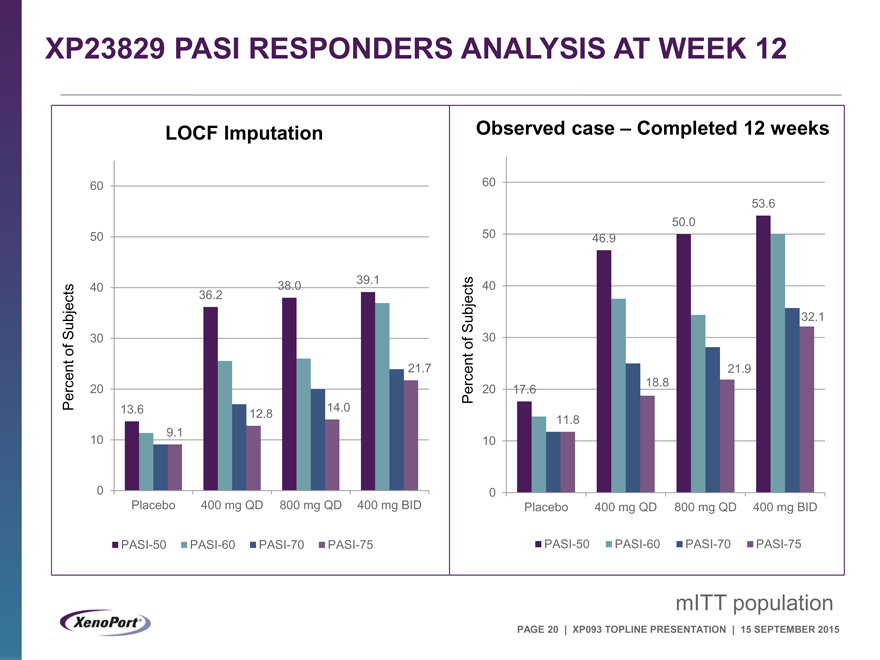
XP23829 PASI RESPONDERS ANALYSIS AT WEEK 12
LOCF Imputation
60
50
40 38.0 39.1
Subjects 36.2
30
of
21.7
Percent 20
13.6 12.8 14.0
10 9.1
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
Observed case – Completed 12 weeks
60
53.6
50.0
50 46.9
40
Subjects 32.1
of 30
21.9
Percent 20 17.6 18.8
11.8
10
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
mITT population
PAGE 20 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015
|
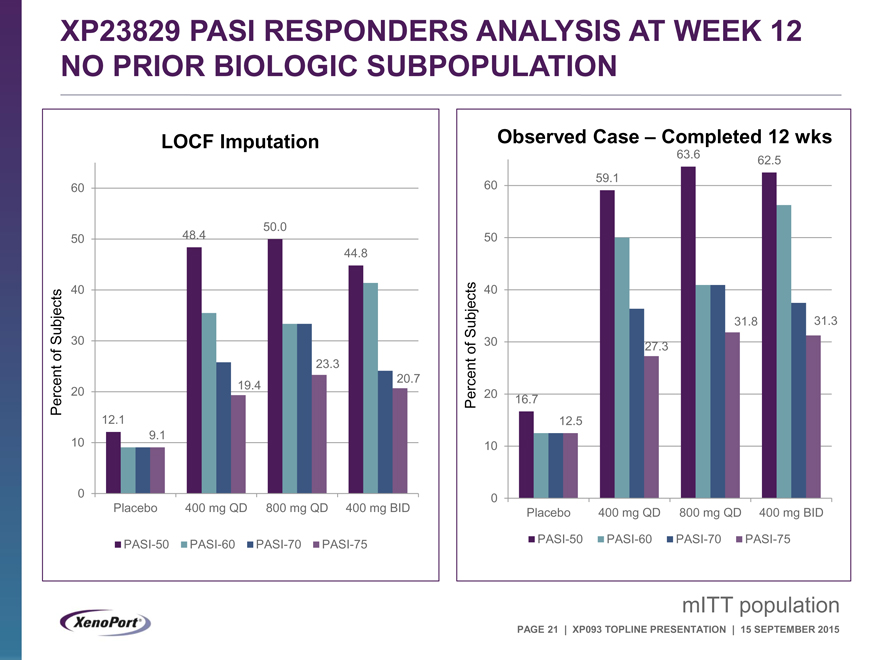
XP23829 PASI RESPONDERS ANALYSIS AT WEEK 12 NO PRIOR BIOLOGIC SUBPOPULATION
LOCF Imputation
60
50.0
50 48.4
44.8
40
Subjects 30
of 23.3
20.7
Percent 20 19.4
12.1
10 9.1
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
Observed Case – Completed 12 wks
63.6 62.5
60 59.1
50
40
Subjects 31.8 31.3
of 30 27.3
Percent 20 16.7
12.5
10
0
Placebo 400 mg QD 800 mg QD 400 mg BID
PASI-50 PASI-60 PASI-70 PASI-75
mITT population
PAGE 21 | XP093 TOPLINE PRESENTATION | 15 SEPTEMBER 2015